ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет по временной патентной заявке США № 61/697882, поданной 7 сентября 2012 года.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к области прицельной трансформации растений, нацеливания генов, направленного встраивания в геном и экспрессии белков в растениях. В предпочтительном варианте осуществления настоящее изобретение относится к сконструированной способами инженерии платформе для встраивания трансгена (ETIP), которую можно встраивать случайным образом или в заданные положения в геномах растений.
УРОВЕНЬ ТЕХНИКИ
Чтобы удовлетворить возрастающую глобальную потребность в продуцировании продуктов питания, типичные подходы для улучшения сельскохозяйственной производительности (например, увеличенный выход или сообщенная способами инженерии устойчивость к вредителям) основаны либо на мутационной селекции, либо на внесении новых генов в геномы сельскохозяйственных культур путем трансформации. Эти процессы по своему существу являются неспецифическими и относительно неэффективными. Например, общепринятые способы трансформации растений доставляют экзогенную ДНК, которая встраивается в геном в случайных положениях. Таким образом, для идентификации и отделения трансгенных линий растений с желаемыми признаками, необходимо осуществить сотни уникальных случайных событий встраивания на конструкцию, а затем провести скрининг в отношении желаемых особей. В результате, общепринятые способы инженерии признаков растений являются трудоемкой, времязатратной и непредсказуемой задачей. Более того, случайная природа этих встраиваний затрудняет прогнозирование того, возникнут ли плейотропные эффекты вследствие непреднамеренного разрушения генома.
Случайная природа современных способов трансформации требует осуществления сотен событий встраивания для идентификации и селекции кандидатов с трансгенным событием (трансформация и скрининг события являются скорость-лимитирующими в отношении генов-кандидатов, идентифицированных в функциональных геномных исследованиях). Кроме того, в зависимости от положения встраивания в геноме экспрессирующая кассета для гена может экспрессироваться на различных уровнях в результате эффекта положения в геноме. Этот эффект положения в геноме делает результаты сравнения влияния различных регуляторных элементов и конструкций трансгенов при случайном встраивании в геном с использованием общепринятого способа трансформации в высокой степени вариабельными. В результате, получение, выделение и охарактеризация линий растений с встроенными способами инженерии генами или признаки является чрезвычайно трудоемким и дорогостоящим процессом с низкой вероятностью успеха.
Прицельная модификация генов решает логистические проблемы общепринятых практик в растительных системах, и она является давно существующей целью ведущих исследователей растений и сельскохозяйственных биотехнологов. Однако, за исключением "нацеливания генов" посредством положительной-отрицательной селекции с лекарственным средством в рисе или использования предварительно встроенных способами инженерии участков рестрикции, направленная модификация во всех видах растений, как модельных, так и возделываемых, до недавнего времени оставалась труднодостижимой. Terada et al. (2002) Nat Biotechnol 20(10): 1030; Terada et al. (2007) Plant Physiol 144(2):846; D'Halluin et al. (2008) Plant Biotechnology J. 6(1):93.
Недавно были описаны способы и композиции для направленного расщепления геномной ДНК. Такие события направленного расщепления можно использовать, например, для индукции направленного мутагенеза или направленных делеций клеточных последовательностей ДНК, или для способствования направленной рекомбинации в заданном хромосомном локусе. См., например, публикации патентов США 2003/0232410, 2005/0208489, 2005/0026157, 2005/0064474 и 2006/0188987, и международную публикацию WO 2007/014275.
В публикации патента США №2008/0182332 описано применение неканонических нуклеаз с цинковыми пальцами (ZFN) для направленной модификации геномов растений. В патентной заявке США №12/284888 описано опосредуемое ZFN направленное встраивание в локус EPSPS растений. Однако все еще остается потребность в обнаружении композиций и способов для идентификации, селекции и быстрого осуществления стабильного направленного встраивания в конкретные положения в геноме растений.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Вариант осуществления настоящего изобретения относится к способу получения трансгенной клетки растения. В следующем варианте осуществления предусматривается клетка растения, имеющая геномную ДНК, содержащую поддающуюся нацеливанию молекулу нуклеиновой кислоты. Следующие варианты осуществления включают поддающуюся нацеливанию молекулу нуклеиновой кислоты, содержащую: по меньшей мере один участок распознавания сайт-специфической нуклеазой; первый фрагмент первого маркерного гена и второй фрагмент второго маркерного гена. В другом варианте осуществления клетка растения трансформирована донорной молекулой нуклеиновой кислоты и молекулой нуклеиновой кислоты сайт-специфической нуклеазы. В следующем варианте осуществления геномная ДНК клетки растения расщепляется по меньшей мере в одном участке расщепления сайт-специфической нуклеазой. Дополнительный вариант осуществления включает встраивание донорной молекулы нуклеиновой кислоты в поддающуюся нацеливанию молекулу нуклеиновой кислоты, где встраивание в поддающуюся нацеливанию молекулу нуклеиновой кислоты включает встраивание по меньшей мере одного функционального маркерного гена, с получением трансгенной клетки растения, причем трансгенная клетка растения содержит поддающуюся нацеливанию молекулу нуклеиновой кислоты и встроенную донорную молекулу нуклеиновой кислоты, содержащую по меньшей мере один функциональный маркерный ген.
В другом варианте осуществления настоящее изобретение относится к хромосомному участку-мишени в Brassica napus, выбранному из группы, состоящей из от нуклеотидов 1-579 SEQ ID NO: 431 до нуклеотидов 166-732 SEQ ID NO: 432, от нуклеотидов 1-550 SEQ ID NO: 433 до нуклеотидов 190-653 SEQ ID NO: 434, от нуклеотидов 1-298 SEQ ID NO: 435 до 51-644 SEQ ID NO: 436, от нуклеотидов 1-536 SEQ ID NO: 437 до нуклеотидов 146-545 SEQ ID NO: 438, от нуклеотидов 1-431 SEQ ID NO: 439 до нуклеотидов 167-685 SEQ ID NO: 440, от нуклеотидов 1-599 SEQ ID NO: 441 до нуклеотидов 116-521 SEQ ID NO: 442, от нуклеотидов 1-298 SEQ ID NO: 443 дод нуклеотидов 193-775 SEQ ID NO: 444, и от нуклеотидов 1-651 SEQ ID NO: 445 до нуклеотидов 120-578 SEQ ID NO: 446.
В следующем варианте осуществления настоящее изобретение относится к способу получения трансгенной клетки растения. Следующие варианты осуществления включают поддающуюся нацеливанию молекулу нуклеиновой кислоты, содержащую: по меньшей мере один участок распознавания сайт-специфической нуклеазой; первый фрагмент первого маркерного гена; и второй фрагмент второй некодирующей полинуклеотидной последовательности. В другом варианте осуществления клетка растения трансформирована донорной молекулой нуклеиновой кислоты и молекулой нуклеиновой кислоты сайт-специфической нуклеазы. В следующем варианте осуществления геномная ДНК клетки растения расщепляется по меньшей мере в одном участке распознавания сайт-специфической нуклеазой. Дополнительный вариант осуществления включает встраивание донорной молекулы нуклеиновой кислоты в поддающуюся нацеливанию молекулу нуклеиновой кислоты, где встраивание в поддающуюся нацеливанию молекулу нуклеиновой кислоты включает встраивание по меньшей мере одного функционального маркерного гена, с получением трансгенной клетки растения, причем трансгенная клетка растения содержит поддающуюся нацеливанию молекулу нуклеиновой кислоты и встроенную донорную молекулу нуклеиновой кислоты, содержащую по меньшей мере один функциональный маркерный ген.
Указанные выше и другие признаки станут более понятными из представленного ниже подробного описания нескольких вариантов осуществления, которое предоставлено с отсылкой на прилагаемые чертежи.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1A-1E: представлено выравнивание последовательностей гена FAD2, проведенное с использованием AlignX®.
Фиг. 2: представлено филогенетическое дерево для последовательностей гена FAD2, полученное с использованием Jalview™ v2.3 на основе расстояний, полученных методом ближайшего связывания.
Фиг. 3A-3M': представлено выравнивание последовательностей гена FAD3, проведенное с использованием AlignX®.
Фиг. 4: представлено филогенетическое дерево последовательностей гена FAD3, полученное с использованием Jalview™ v2.3 на основе расстояний, полученных методом ближайшего связывания. Обозначенные последовательности соответствуют следующему: FAD3A'/A'' описана в настоящем описании как FAD3A'; гаплотип 2 описан в настоящем описании как FAD3C'; гаплотип 1 описан в настоящем описании как FAD3C'' и гаплотип 3 описан в настоящем описании как FAD3A''.
Фиг. 5: представлена карта плазмиды pDAB104010 и представлена репрезентативная экспрессирущая кассета для нуклеаз с цинковыми пальцами (ZFN). План этой конструкции сходен с другими экспрессирующими кассетами для ZFN, где домены цинковых пальцев 24828 и 24829 заменены альтернативными доменами цинковых пальцев, которые описаны ниже.
Фиг. 6: представлен иллюстративный график с множеством кривых, демонстрирующий количество считанных последовательностей с делециями в заданном участке ZFN на 10000 считанных последовательностей. На оси X на графике представлено количество удаленных оснований, на оси Y представлено количество считанных последовательностей, и на оси Z представлено кодируемое цветом обозначение образца, как описано справа на графике. Конкретный представленный пример представляет собой пример локуса 1 семейства генов FAD2, которое содержит 3 участка-мишени для ZFN: A, B и C, с четырьмя представителями семейства генов и двумя контрольными трансфекциями, оцениваемыми в качестве контрольных образцов A и B. Линии, приведенные сверху вниз (от A-контроль_FADA' сверху легенды до C_образец_FAD2C снизу легенды) представлены на графике от наиболее близкой к обозначенной оси X (A_контроль_FADA') до наиболее удаленной от обозначенной оси X (C_образец_FAD2C).
Фиг. 5: представлены данные нацеливания ZFN на локус 4 семейства генов FAD2. Локус содержит два участка ZFN и две необходимые контрольные трансфекции. Фиг. 7B: конкретный контекст последовательности (SEQ ID NO: 471-480), окружающий участок-мишень для ZFN, где обозначены FAD2A и C, содержащие тринуклеотидные повторы C, T и G, что приводит к наблюдаемому увеличению однонуклеотидных делеций при секвенировании локусов FAD2A и C.
Фиг. 8: представлена карта плазмиды pDAS000130.
Фиг. 9: представлена карта плазмиды pDAS000271.
Фиг. 10: представлена карта плазмиды pDAS000272.
Фиг. 11: представлена карта плазмиды pDAS000273.
Фиг. 12: представлена карта плазмиды pDAS000274.
Фиг. 13: представлена карта плазмиды pDAS000275.
Фиг. 14: представлена карта плазмиды pDAS000031.
Фиг. 15: представлена карта плазмиды pDAS000036.
Фиг. 16: представлена карта плазмиды pDAS000037.
Фиг. 17: представлены конфигурации ETIP и нуклеиновой кислоты-груза, а также продукт направленного встраивания груза в участок ETIP в геноме клетки растения.
Фиг. 18: проиллюстрирована трансформация клеток протопластов с последующей селекцией с помощью FACS нацеленной ДНК груза в ETIP в линии-хозяине с использованием реконструирования укороченных поддающихся оценке и селективных маркеров как на 3'-, так и на 5'-конце.
Фиг. 19A-B: проиллюстрирована направляемая гомологией репарация события ETIP в каноле, которое является результатом двухцепочечного разрыва ДНК геномного локуса нуклеазой с цинковыми пальцами (pDAS000074 или pDAS000075) и последующего встраивания донора Ds-red (pDAS000068, pDAS000070 или pDAS000072) в локус ETIP хромосомы канолы. Встраивания донора в геномный локус приводит к полностью функциональному трансгену Ds-red с высокой экспрессией.
Фиг. 20: представлена сортировка способом FACS протопластов канолы и вычисленная эффективность трансфекции протопластов канолы, которые были трансфицированы pDAS000031 ("pDAS31"). Кроме того, предоставлены результаты сортировки способом FACS нетрансформированных протопластов канолы в качестве отрицательного контроля.
Фиг. 21: представлена сортировка способом FACS протопластов канолы и вычисленная эффективность трансфекции для событий ETIP протопластов канолы, которые были трансфицированы pDAS000064/pDAS000074 (верхний график) и pDAS000064/pDAS000075 (нижний график).
Фиг. 22: представлена сортировка способом FACS протопластов канолы и вычисленная эффективность трансфекции для событий ETIP в протопластах канолы, трансформированных pDAS000068/pDAS000074 (верхний график) и pDAS000068/pDAS000075 (нижний график).
Фиг. 23: представлена сортировка способом FACS протопластов канолы и вычисленная эффективность трансфекции для событий ETIP в протопластах канолы, трансформированных pDAS000070/pDAS000074 (верхний график) и pDAS000070/pDAS000075 (нижний график).
Фиг. 23: представлена сортировка способом FACS протопластов канолы и вычисленная эффективность трансфекции для событий ETIP в протопластах канолы, трансформированных pDAS000072/pDAS000074 (верхний график) и pDAS000072/ pDAS000075 (нижний график).
Фиг. 25: представлена карта плазмиды pDAS000074.
Фиг. 26: представлена карта плазмиды pDAS000075.
Фиг. 27: представлена карта плазмиды pDAS000064.
Фиг. 28: представлена карта плазмиды pDAS000068.
Фиг. 29: представлена карта плазмиды pDAS000070.
Фиг. 30: представлена карта плазмиды pDAS000072.
Фиг. 31: представлена схема, демонстрирующая участки связывания праймеров и зонда на трансгене-мишени для анализа в целях оценки числа копий трансгена.
Фиг. 32: представлен файл Sequencher, демонстрирующий домен ZFN, распознающий ДНК FAD2A (bc12075_Fad2a-r272a2 и bc12075_Fad2a-278a2) и участки связывания специфических праймеров для ZFN (FAD2A.UnE.F1 и FAD2A.UnE.R1) и эндогенных праймеров (FAD2A/2C.RB.UnE.F1 и FAD2A/2C.RB.UnE.R1).
Фиг. 33: представлена схема, демонстрирующая участки связывания праймеров на эндогенной и трансгенной мишени, используемых для обнаружения встраивания трансгенов FAD2A с помощью полной HDR.
Фиг. 34: представлена схема, где участки рестрикции эндонуклеазой pn1 встречаются в полностью отредактированном локусе FAD2A и где связываются зонды для саузерн-блоттинга FAD2a 5', hph и FAD2A 3'.
Фиг. 35: представлены репрезентативные данные оценки числа копий с помощью q-ПЦР. В левом столбце представлены данные, полученные для известного растения T0 с одной случайной вставкой, которое использовали в качестве калибровочного образца, относительно которого все другие образцы "нормализовывали". В правом столбце представлено известное трансгенное растение T0 с 5 вставками трансгенов. Число встраиваемых копий для обоих растений определяли с использованием саузерн-анализа. В остальных столбцах предоставлены оценки числа копий для предполагаемых трансгенных растений. Столбцы обозначены слева направо фигуры как; контроль с 1 копией, 310420, 311819, 311821, 311822, 311823, 311824, 311827, 312524, 312525, 312526, 312527, 312529, 312530, 312532, 313810, 313811, 313905, 313941, 313942, 313944 и контроль с 5 копиями. Столбцы можно использовать для определения оцененного числа копий каждого трансгенного растения. При использовании программного обеспечения для оценки числа копий, растения дикого типа, нетрансформированные растения и контроли только с плазмидой не дают результата для числа копий, поскольку они не обладают Cq как для мишени hph, так и для мишени HMG 7/7.
Фиг. 36: представлена карта плазмиды pDAB105855, содержащей последовательность ДНК-мишень, содержащую; последовательность RB7 MAR/участок связывания v1 eZFN4, промотор OsUbi3/Phi YFP/ZmPer5 3'UTR v2/участок связывания eZFN1/ELP1 HR2 v2, промотор v8 ZmUbi1/Cry34Ab1 v2/StPinII 3' UTR v2, промотор v3 TaPer/Cry35Ab1 v5/StPinII 3' UTR v2, SCBVv2/AAD-1v3/ZmLip 3' UTR v1 между границами T-ДНК.
Фиг. 37: представлена карта плазмиды pDAB105941, содержащей кодирующую последовательность ZFN1 под контролем экспрессии промотора убиквитина 1 с интроном 1 кукурузы (промотор v2 ZmUbi1) и ZmPer5 3'UTR v2.
Фиг. 38: представлена карта плазмиды pDAB112366, содержащей интрон без промотора (интрон rubi3) промотора убиквитина 3 риса (OsUbi3), за которым следует ген устойчивости к гербициду (фосфинотрицинацетилтрансфераза (PAT)) и ZmLip 3'UTR.
Фиг. 39: представлена схема использования участка ETIP, встроенного в геном, для нацеливания донорного полинуклеотида, содержащего экспрессирующую ген кассету. Как представлено на схеме, донор способен экспрессировать трансген, если встраивание происходит в локусе ETIP. Случайное встраивание донора не приводит к экспрессии трансгена, поскольку, как правило, отсутствуют промоторные элементы, которые могут контролировать экспрессию.
ПОДРОБНОЕ ОПИСАНИЕ
В настоящем описании описаны способы и композиции для получения сконструированной способами инженерии платформы для встраивания трансгена (ETIP), содержащего поддающуюся нацеливанию молекулу нуклеиновой кислоты, которая стабильно встроена в геном клетки растения и служит в качестве зародышевой плазмы-хозяина для трансформации и встраивания донорной молекулы нуклеиновой кислоты. Изобретение относится к области прицельной трансформации растений, нацеливания генов, направленного встраивания в геном и экспрессии белков в растениях. В предпочтительном варианте осуществления ETIP можно встраивать случайным образом или в заданные положения в геномах растений для облегчения быстрой селекции и обнаружения одного или нескольких представляющих интерес генов (GOI), которые были нацелены (как 3'-, так и 5'-концы) в положение ETIP в геноме. В некоторых вариантах осуществления в ETIP можно вносить специфические двухцепочечные разрывы. В других вариантах осуществления описан способ селекции и увеличения содержания выделенных клеток или протопластов после нацеливания, за которым следует регенерация фертильных растений с использованием проточной цитометрии и FACS.
Конкретные способы включают получение трансгенной клетки растения, имеющей стабильную наследуемую генетическую модификацию в растении и его потомстве, а также состав трансгенного растения. В определенных вариантах осуществления ETIP описана для применения в системе для прицельной трансформации растений, где ETIP, содержащая встраиваемую нуклеиновую кислоту, стабильно встраивается в зародышевую плазму хозяина. Другие варианты осуществления относятся к способам получения трансгенной клетки растения так, чтобы геномная ДНК клетки растения включала по меньшей мере один стабильно встроенный функциональный маркерный ген. В некоторых вариантах осуществления способ включает использование поддающейся нацеливанию молекулы нуклеиновой кислоты, содержащей один или несколько участков распознавания нацеливающей сайт-специфической нуклеазой, первый фрагмент первого маркерного гена и второй фрагмент второго маркерного гена. В другом варианте осуществления донорная молекула нуклеиновой кислоты содержит представляющую интерес нуклеотидную последовательность и две последовательности нуклеиновой кислоты, фланкирующие представляющую интерес последовательность нуклеиновой кислоты. В другом варианте осуществления две последовательности нуклеиновой кислоты, фланкирующие представляющую интерес последовательность нуклеиновой кислоты, представляют собой последовательность нуклеиновой кислоты первого и второго участков гомологии. В других вариантах осуществления для трансформации клетки растения используют донорную молекулу нуклеиновой кислоты. Последовательности нуклеиновой кислоты участка гомологии могут быть гомологичны областям ETIP или поддающейся нацеливанию последовательности молекулы нуклеиновой кислоты, и они могут быть гомологичны первому и второму фрагменту первого и второго маркерных генов в поддающейся нацеливанию молекуле нуклеиновой кислоты.
Также описаны композиции и способы для получения трансгенных растений, где донорная молекула нуклеиновой кислоты экспрессирует один или несколько продуктов экзогенной последовательности нуклеиновой кислоты (например, белок или молекула РНК), которая стабильно встроилась в ETIP или в поддающуюся нацеливанию молекулу нуклеиновой кислоты в клетке растения. В некоторых вариантах осуществления в ETIP или поддающейся нацеливанию молекуле нуклеиновой кислоты используются участки связывания нуклеазами с цинковыми пальцами или сайт-специфической нуклеазы, содержащей белок, проявляющий активность нуклеазы с цинковыми пальцами. В альтернативных вариантах осуществления для сайт-специфической нуклеазы используются дополнительные технологии нацеливания, такие как мегануклеазы, TAL, РНК-направляемые CRISPR-Cas9 или лейциновые молнии. В конкретных вариантах осуществления ETIP представляет собой поддающуюся нацеливанию молекулу нуклеиновой кислоты, которая облегчает исследование генов-кандидатов и конструкций экспрессирующих векторов растений ранних фаз процесса разработки трансгенного растения. В некоторых вариантах осуществления трансгенная клетка растения включает встраиваемую молекулу нуклеиновой кислоты, имеющую полинуклеотидную последовательность с одним или несколькими участками распознавания нацеливающей сайт-специфической нуклеазой и первый и второй фрагменты первого и второго маркерных генов. Понятно, что фрагменты маркерных генов могут не кодировать функциональный продукт экспрессии маркерного гена. Более того, в качестве одного варианта осуществления маркерные гены могут содержать последовательность нуклеиновой кислоты интрона. В других вариантах осуществления маркерные гены могут содержать последовательность нуклеиновой кислоты участка гомологии.
В некоторых вариантах осуществления одна или две, или более областей донорной молекулы нуклеиновой кислоты лишены гомологии последовательностей с геномной ДНК (экзогенной) растения. В дополнительных вариантах осуществления донорная молекула нуклеиновой кислоты может содержать последовательность нуклеиновой кислоты участка гомологии. Последовательности нуклеиновой кислоты участков гомологии в донорной молекуле нуклеиновой кислоты могут быть встроены в области, фланкирующие участок расщепления сайт-специфической нуклеазой в поддающейся нацеливанию молекуле нуклеиновой кислоты. В определенных вариантах осуществления последовательности участка гомологии могут иметь длину от 50 п.н. до 3 т.п.н.
В конкретных вариантах осуществления донорная молекула нуклеиновой кислоты может содержать экзогенные последовательности нуклеиновой кислоты, которые обеспечивают нацеливание на поддающуюся нацеливанию молекулу нуклеиновой кислоты ETIP и селекцию на 5'- и 3'-концах точно нацеленных событий. Продукты экзогенных последовательностей нуклеиновой кислоты могут содержать, например, один или несколько генов или молекул кДНК, или любой тип кодирующей или некодирующей нуклеотидной последовательности, а также один или несколько регуляторных элементов генов (например, промоторов). Конструкция донорной молекулы нуклеиновой кислоты обеспечивает вырезание кодирующей и некодирущей ДНК, включая, но не ограничиваясь ими, селективные маркеры, после селективного встраивания донорной ДНК в ETIP.
В конкретных вариантах осуществления поддающаяся нацеливанию молекула нуклеиновой кислоты ETIP может включать первый фрагмент первого маркерного гена, и второй фрагмент второго маркерного гена, и донорную молекулу нуклеиновой кислоты, содержащую соответствующий второй фрагмент первого маркерного гена и первый фрагмент второго маркерного гена. Первый фрагмент первого маркерного гена может быть расположен на 5'-конце поддающейся нацеливанию молекулы нуклеиновой кислоты, а второй фрагмент второго маркерного гена может быть расположен на 3'-конце поддающейся нацеливанию молекулы нуклеиновой кислоты, так чтобы стабильное встраивание донорной молекулы нуклеиновой кислоты, содержащей второй фрагмент первого маркерного гена и первый фрагмент второго маркерного гена, в геном клетки растения формировало функциональный первый маркерный ген и второй маркерный ген. В некоторых вариантах осуществления эти маркерные гены можно использовать для селекции стабильного встраивания ETIP. В некоторых вариантах осуществления подходящие маркерные гены могут включать PMI, Xyl(A), YFP, DSR, GFP, GUS, NPTII, AAD-1, AAD-12, DGT-28, AHAS, PAT, DSM-2, HYG, BAR и гены флуоресцентных белков. В других вариантах осуществления маркерный ген представляет собой маркерный ген, поддающийся визуальному скринингу, присутствие которого можно определять путем мониторинга изменения цвета клетки. В других вариантах осуществления маркерный ген представляет собой ген селективного маркера (например, ген, кодирующий устойчивость к гербициду или антибиотику), селекцию присутствия которого проводят с использованием гербицида или антибиотика, который уменьшает рост клеток. В следующих вариантах осуществления маркерный ген представляет собой маркерный ген для положительной селекции.
В выбранный экспрессирующий вектор можно включать различные селективные маркеры, также описанные в качестве репортерных генов, для обеспечения идентификации и селекции трансформированных растений ("трансформантов"). Для подтверждения экспрессии селективных маркеров в трансформированных растений доступно множество способов, включая, например секвенирование ДНК и ПЦР (полимеразная цепная реакция), саузерн-блоттинг, РНК-блоттинг, иммнологические способы для обнаружения белка, экспрессируемого с вектора, например, преципитированного белка, который опосредует устойчивость к фосфотрицину, или визуального наблюдения других белков, таких как репортерные гены, кодирующие β-глюкуронидазу (GUS), люциферазу, зеленый флуоресцентный белок (GFP), желтый флуоресцентный белок (YFP), DsRed, β-галактозидаза, хлорамфениколацетилтрансфераза (CAT), щелочная фосфатаза и т.п. (см. Sambrook, et al., Molecular Cloning: A Laboratory Manual, Third Edition, Cold Spring Harbor Press, N.Y., 2001).
Гены селективных маркеров используют для селекции трансформирвоанных клеток или тканей. Гены селективных маркеров включают гены, кодирующие устойчивость к антибиотикам, такие как гены, кодирующие неомицинфосфотрансферазу II (NEO) и гигромицинфосфотрансферазу (HPT), а также гены, сообщающие устойчивость к гербицидным соединениям. Гены устойчивости к гербицидам кодируют модифицированный белок-мишень, нечувствительный к гербициду, или фермент, который деградирует или детоксифицирует гербицид в растении до того, как он сможет подействовать. Например, устойчивости к глифосату можно достигать с использованием генов, кодирующих мутантный фермент-мишень - 5-енолпирувилшикимат-3-фосфатсинтазу (EPSPS). Гены и мутанты EPSPS хорошо известны и дополнительно описаны ниже. Устойчивость к глюфосинату аммония, бромксинилу и 2,4-дихлорфеноксиацетату (2,4-D) достигают с использованием бактериальных генов, кодирующих pat или DSM-2, гена нитрилазы, генов aad-1 или aad-12, которые детоксифицируют соответствующие гербициды.
В одном варианте осуществления гербициды могут ингибировать конус нарастания или меристему, включая имидазолинон или сульфонилмочевину, и хорошо известны гены синтазы ацетогидроксикислот (AHAS) и ацетолактатсинтазы (ALS) для устойчивости/толерантности к этим гербицидам. Гены устойчивости к глифосату включают мутантные гены 5-енолпирувилшикимат-3-фосфатсинтазы (EPSP) и dgt-28 (посредством встраивания рекомбинантных нуклеиновых кислот и/или различных форм мутагенеза нативных генов EPSP in vivo), гены aroA и гены глифосатацетилтрансферазы (GAT), соответственно). Гены устойчивости к другим фосфоносоедениниям включают гены bar из видов Streptomyces, включая Streptomyces hygroscopicus и Streptomyces viridichromogenes, и гены устойчивости к пиридокси или феноксипропионовым кислотам и циклогексонам (гены, кодирующие ингибитор ACC-азы). Иллюстративные гены, сообщающие устойчивость к циклогександионам и/или арилоксифеноксипропионовой кислоте (включая галоксифоп, диклофоп, феноксипроп, флуазифоп, квизалофоп) включают гены ацетилкофермент-A-карбоксилазы (ACC-аза): Acc1-S1, Acc1-S2 и Acc1-S3. В одном варианте осуществления гербициды могут ингибировать фотосинтез и включают триазин (гены psbA и 1s+) или бензонитрил (ген нетрилазы).
В одном варианте осуществления гены селективных маркеров включают, но не ограничиваются ими, гены, кодирующие: неомицинфосфотрансферазу II; цианамидгидратазу; аспартаткиназу; дигидродипиколинатсинтазу; триптофандекарбоксилазу; дигидродипиколинатсинтазу и десенсибилизированную аспартаткиназу; продукт гена bar; триптофандекарбоксилазу; неомицинфосфотрансферазу (NEO); гигромицинфосфотрансферазу (HPT или HYG); дигидрофолатредуктазу (DHFR); фосфинотрицинацетилтрансферазу; дегалогеназу 2,2-дихлорпропионовой кислоты; синтазу ацетогидроксикислоты; 5-енолпирувилшикиматфосфатсинтазу (aroA); галоарилнитрилазу; ацетилкофермент-A-карбоксилазу; дигидроптероатсинтазу (sulI) и полипептид фотосистемы II массой 32 кДа (psbA).
Один вариант осуществления также включает гены, кодирующие устойчивость к: хлорамфениколу; метотрексату; гигромицину; спектиномицину; бромксинилу; глифосату и фосфинотрицину.
Подразумевается, что описанный выше перечень генов селективных маркеров не является ограничивающим. Настоящее изобретение охватывает любой репортерный ген или ген селективного маркера.
В одном варианте осуществления поддающаяся нацеливанию полинуклеотидная молекула ETIP встроена в геномных локус. В одном примере геномные локусы FAD2, FAD3 и IPK1 могут быть выбраны в качестве мишеней для встраивания ETIP и последующего встраивания донорной полинуклеотидной молекулы. Было показано, что разрушение эндогенных генов FAD2 и FAD3 не оказывает неблагоприятного влияния на агрономические или качественные свойства клетки растения. Таким образом, когда участки FAD2 и FAD3 разрушены, не происходит ухудшения агрономических или качественных свойств, ассоциированных с функциональностью растения или растительного продукта (канола, соя, кукуруза и т.д.), и группирование признаков, ассоциированных с конкретными характеристиками качества масла, ускоряет интрогрессию, и наблюдают развитие новой зародышевой плазмы. В конкретных вариантах осуществления донорная молекула нуклеиновой кислоты может быть связана с регуляторным элементом, таким как промотор, интрон, 5'-UTR или 3'-UTR.
Встраивание донорной молекулы нуклеиновой кислоты в ETIP можно облегчать с помощью направленного двухцепочечного расщепления сайт-специфической нуклеазой. Сайт-специфическая нуклеаза может быть расположена в поддающейся нацеливанию молекуле нуклеиновой кислоты ETIP. Более того, сайт-специфическая нуклеаза может быть расположена в подающейся нацеливанию молекуле нуклеиновой кислоты ETIP, содержащей сконструированную способами инженерии подушку для встраивания (ELP), см. патент США №20110191899. Более того, сайт-специфическая нуклеаза может быть расположена в или вблизи ELP в выбранной ETIP. Расщепление можно нацеливать на ETIP с использованием слитых белков, содержащих ДНК-связывающий домен, такой как ДНК-связывающий домен мегануклеазы, ДНК-связывающий домен РНК-направляемых CRISPR-Cas9, ДНК-связывающий домен в виде лейциновой молнии, ДНК-связывающий домен TAL, белок с цинковыми пальцами (ZFP), нуклеаза с цинковыми пальцами или химерные комбинации вышеуказанных, которые сконструированы так, чтобы они связывали встроенную способами инженерии последовательность в выбранной ETIP. Такое расщепление стимулирует встраивание экзогенных полинуклеотидных последовательностей груза или донорной нуклеиновой кислоты в или вблизи участка расщепления в ETIP. Встраивание донорных молекул нуклеиновой кислоты с использованием описанных способов может происходить как по зависимым от гомологии, так и по независимым от гомологии механизмам, причем селекцию направленных событий встраивания осуществляют путем скрининга новых поддающихся селекции и/или оценке маркеров, которые являются функциональными при направленных событиях встраивания как в 3'-, так и в 5'-областях ETIP. Настоящее изобретение демонстрирует применение новых сконструированных способами инженерии участков связывания цинковых пальцев и ZFN для обеспечения селективных двухцепочечных разрывов в ETIP.
В альтернативных вариантах осуществления новые модифицированные способами инженерии ДНК-связывающие домены (например, ZFP, мегануклеазы, лейциновые молнии, TAL, РНК-направляемые CRISPR-Cas9) связываются с одним или несколькими участками-мишенями в ETIP, которые не существуют в нативном геноме клетки растения. ДНК-связывающий домен(ы) может включать, например, любой из модифицированных способами инженерии ДНК-связывающих доменов с цинковыми пальцами, содержащих распознающие спирали, такие как описаны в заявке США № 12/931096. В некоторых вариантах осуществления любой из ДНК-связывающих доменов, описанных в настоящем описании, может дополнительно содержать функциональный домен, например, домен расщепления или половинный домен расщепления. В других вариантах осуществления половинный домен расщепления может происходить из эндонуклеазы рестрикции типа IIS, такой как Fokl или Stsl. Следующие варианты осуществления домена расщепления могут включать хоминг-эндонуклеазу, например, такую как хоминг-эндонуклеаза с модифицированным ДНК-связывающим доменом.
Варианты осуществления настоящего изобретения включают применение ДНК-связывающего белка с цинковыми пальцами. ДНК-связывающий белок с цинковыми пальцами, "ZFP" (или связывающий домен) представляет собой белок или домен в более крупном белке, который связывает ДНК специфическим для последовательности образом через один или несколько цинковых пальцев, которые представляют собой области аминокислотной последовательности в связывающем домене, структура которых стабилизирована координационной связью с ионом цинка. Термин "ДНК-связывающий белок с цинковыми пальцами" часто сокращенно обозначают как белок с цинковыми пальцами или ZFP. Связывающие домены цинковых пальцев могут быть "модифицированы способами инженерии" так, чтобы они связывались с заданной нуклеотидной последовательностью. Неограничивающие примеры модификации способами инженерии белков с цинковыми пальцами являются конструирование и селекция. Сконструированный белок с цинковыми пальцами представляет собой белок, не встречающийся в природе, конструкция/композиция которого в основном является результатом рациональных критериев. Рациональные критерии для конструирования включают использование правил замены и компьютерных алгоритмов для обработки информации в базе данных, хранящей информацию о существующих конструкциях ZFP и данные о связывании. См., например, патенты США 6140081, 6453242, 6534261 и 6785613; также см. WO 98153058, WO 98153059, WO 98153060, WO 021016536 и WO 031016496 и патенты США 6746838, 6866997 и 7030215.
В конкретных вариантах осуществления настоящего изобретения можно использовать нуклеазу с цинковыми пальцами (ZFN). ZFN может представлять собой любую нуклеазу с цинковыми пальцами, которая может быть доставлена в клетку растения в соответствии с настоящим изобретением. Например, ZFN могут включать слитые белки, содержащие домен расщепления (или половинный домен расщепления) и связывающий домен цинковых пальцев, полинуклеотиды, кодирующие эти белки, и комбинации полипептидов и кодирующих полипептиды полинуклеотидов. Связывающий домен цинковых пальцев может включать один или несколько цинковых пальцев (например, 2, 3, 4, 5, 6, 7, 8, 9 или более цинковых пальцев), и он может быть модифицирован способами инженерии так, чтобы он связывался с любой представляющей интерес областью. Таким образом, путем идентификации представляющей интерес области-мишени, в которой является желательным расщепление или рекомбинация, способами, описанными в настоящем описании, можно сконструировать один или несколько слитых белков, содержащих домен расщепления (или половинный домен расщепления) и домен цинкового пальца, модифицированный способами инженерии так, чтобы он распознавал последовательность-мишень в указанной представляющей интерес области. Присутствие такого слитого белка (или белков) в клетке приводит к связыванию слитого белка(ов) с его (их) участком(ами) связывания и расщепление в или вблизи указанной представляющей интерес области. Более того, если в такой клетке также присутствует экзогенный полинуклеотид, гомологичный представляющей интерес области, между нуклеотидной последовательностью с двухцепочечным разрывом и экзогенным полинуклеотидом гомологичная рекомбинация происходит с высокой частотой. Для целей настоящего изобретения "гомологичная рекомбинация" относится к специализированной форме такого обмена, которая происходит, например, в ходе репарации двухцепочечных разрывов в клетках по механизмам направляемой гомологией репарации. Для этого процесса требуется гомология нуклеотидных последовательностей, в нем используется "донорная" молекула для репарации по матрице молекулы-"мишени" (т.е. молекулы, которая подверглась двухцепочечному разрыву), и он широко известен как "конверсия генов без кроссинговера" или "конверсия генов по короткому участку", поскольку он приводит к переносу генетической информации из донора в мишень. Без связи с какой-либо конкретной теорией, такой перенос может вовлекать коррекцию несоответствий оснований в гетеродуплексной ДНК, которая формируется между мишенью с разрывом и донором, и/или "зависимый от синтеза отжиг цепей", в котором донор используется для ресинтеза генетической информации, которая станет частью мишени, и/или сходные процессы. Такая специализированная гомологичная рекомбинация часто приводит к изменению последовательности молекулы-мишени такому, чтобы часть или вся последовательность донорного полинуклеотида включалась в полинуклеотид-мишень.
В способах по изобретению одна или несколько нацеленных сайт-специфических нуклеаз, как описано в настоящем описании, вносят двухцепочечный разрыв в последовательность-мишень (например, клеточный хроматин) в заданном участке, и "донорный" полинуклеотид, обладающий гомологией с нуклеотидной последовательностью в области разрыва, может быть введен в клетку. Было показано, что наличие двухцепочечного разрыва способствует встраиванию донорной последовательности. Донорная последовательность может физически встраиваться или альтернативно донорный полинуклеотид используется в качестве матрицы для репарации разрыва посредством гомологичной рекомбинации, приводящей к внесению всей или части нуклеотидной последовательности, соответствующей донорной последовательности, в хроматин клетки. Таким образом, первая последовательность в клеточном хроматине может быть изменена и в определенных вариантах осуществления может быть преобразована в последовательность, присутствующую в донорном полинуклеотиде. Таким образом, использование терминов "заменить" или "замена" может подразумевать замену одной нуклеотидной последовательности другой (т.е. замену последовательности в значении замены информации), и она не обязательно требует физической или химической замены одного полинуклеотида другим.
В некоторых вариантах осуществления сайт-специфическое встраивание можно проводить с использованием факторов, которые способны распознавать и связываться с конкретными нуклеотидными последовательностями, например, в геноме организма хозяина. Например, многие белки содержат полипептидные домены, которые способны распознавать и связывать ДНК сайт-специфическим образом. Последовательность ДНК, которую распознает ДНК-связывающий полипептид, может быть обозначена как последовательность-"мишень". Полипептидные домены, которые способны распознавать и связывать ДНК сайт-специфическим образом, как правило, правильно сворачиваются и функционируют независимо, связывая ДНК сайт-специфическим образом, даже при экспрессии в полипептиде, отличном от белка, из которого домен был первоначально выделен. Аналогично, последовательности-мишени для распознавания и связывания ДНК-связывающими полипептидами, как правило, могут распознаваться и связываться такими полипептидами, даже когда они присутствуют в крупных структурах ДНК (например, в хромосоме), в частности, когда участок, где расположена последовательность-мишень, представляет собой участок, о котором известно, что он является доступным для растворимых клеточных белков (например, ген).
В то время как ДНК-связывающие полипептиды, идентифицированные в белках, которые существуют в природе, как правило, связываются с определенной нуклеотидной последовательностью или мотивом (например, консенсусная последовательность распознавания), в данной области существуют и известны способы модификации многих таких ДНК-связывающих полипептидов так, чтобы они распознавали отличающуюся нуклеотидную последовательность или мотив ДНК-связывающих полипептидов, которые включают, например, но не ограничиваясь ими: ДНК-связывающие домены с цинковыми пальцами; лейциновые молнии; ДНК-связывающие домены UPA; GAL4; TAL; LexA; РНК-направляемые CRISPR-Cas9; репрессор Tet; LacR и рецептор стероидных гормонов.
В некоторых примерах ДНК-связывающий полипептид представляет собой цинковый палец. Индивидуальные мотивы в виде цинковых пальцев можно конструировать для нацеливания и специфического связывания с любым из большого диапазона участков. Канонические полипептиды с цинковыми пальцами Cys2His2 (а также неканонические Cys3His) связывают ДНК путем встраивания α-спирали в большую бороздку двойной спирали ДНК-мишени. Распознавание ДНК цинковым пальцем является модульным; каждый палец контактирует в основном с тремя последовательно расположенными парами оснований в мишени, и распознавание опосредуют несколько ключевых остатков в полипептиде. Путем включения нескольких ДНК-связывающих доменов с цинковыми пальцами в нацеливающую эндонуклеазу, специфичность связывания ДНК у нацеливающей эндонуклеазы может быть дополнительно увеличена (и, таким образом, также может быть увеличена специфичность любых сообщаемых ей эффектов регуляции генов). См., например, Urnov et al. (2005) Nature 435:646-51. Таким образом, один или несколько ДНК-связывающих полипептидов с цинковыми пальцами можно модифицировать способами инженерии и использовать таким образом, чтобы нацеливающая эндонуклеаза, введенная в клетку-хозяина, взаимодействовала с последовательностью ДНК, которая является уникальной в геноме клетки-хозяина.
Предпочтительно, белок с цинковыми пальцами не является встречающимся в природе, а является сконструированным для связывания выбранного участка-мишени. См., например, Beerli et al. (2002) Nature Biotechnol. 20:135-141; Pabo et al. (2001) Ann. Rev. Biochem. 70:313-340; Isalan et al. (2001) Nature Biotechnol. 19:656-660; Segal et al. (2001) Curr. Opin. Biotechnol. 12:632-637; Choo et al. (2000) Curr. Opin. Struct. Biol. 10:411-416; патенты США №6453242; 6534261; 6599692; 6503717; 6689558; 7030215; 6794136; 7067317; 7262054; 7070934; 7361635; 7253273; и публикации патентов США №2005/0064474; 2007/0218528; 2005/0267061.
Сконструированный связывающий домен с цинковыми пальцами может иметь новую специфичность связывания по сравнению со встречающимся в природе белком с цинковыми пальцами. Способы конструирования включают, но не ограничиваются ими, рациональное конструирование и различные типы селекции. Рациональное конструирование включает, например, использование баз данных, содержащих триплетные (или квадруплетные) нуклеотидные последовательности и индивидуальные аминокислотные последовательности цинковых пальцев, в которых каждая триплетная или квадруплетная нуклеотидная последовательность ассоциирована с одной или несколькими аминокислотными последовательностями цинковых пальцев, которые связывают конкретную триплетную или квадруплетную последовательность. См., например, патенты США того же заявителя 6453242 и 6534261, включенные в настоящее описание в качестве ссылок в полном объеме.
Иллюстративные способы селекции, включая фаговый дисплей и двухгибридные системы, описаны в патентах США 5789538; 5925523; 6007988; 6013453; 6410248; 6140466; 6200759 и 6242568; а также в WO 98/37186; WO 98/53057; WO 00/27878; WO 01/88197 и GB 2338237. Кроме того, усиление специфичности связывания у связывающих доменов с цинковыми пальцами описано, например, в WO 02/077227 того же заявителя.
Кроме того, как описано в этих и других источниках литературы, домены цинковых пальцев и/или белки с несколькими цинковыми пальцами могут быть связаны вместе с использованием любых подходящих линкерных последовательностей, включая, например, линкеры длиной 5 или более аминокислот. Также см. патенты США №№6479626, 6903185 и 7153949 для иллюстративных линкерных последовательностей длиной 6 или более аминокислот. Белки, описанные в настоящем описании, могут включать любую комбинацию подходящих линкеров между отдельными цинковыми пальцами белка.
Выбор участков-мишеней; ZFP и способы моделирования и конструирования слитых белков (и полинуклеотидов, кодирующих их) известны специалистам в данной области и подробно описаны в патентах США №61400815; 789,538; 6453242; 6534261; 5925523; 6007988; 6013453; 6200759; WO 95/19431; WO 96/06166; WO 98/53057; WO 98/54311; WO 00/27878; WO 01/60970 WO 01/88197; WO 02/099084; WO 98/53058; WO 98/53059; WO 98/53060; WO 02/016536 и WO 03/016496.
Кроме того, как описано в этих и других источниках литературы, домены цинковых пальцев и/или белки с несколькими цинковыми пальцами могут быть связаны вместе с использованием любых подходящих линкерных последовательностей, включая, например, линкеры длиной 5 или более аминокислот. Также см. патенты США №6479626, 6903185 и 7153949 для иллюстративных линкерных последовательностей длиной 6 или более аминокислот. Белки, описанные в настоящем описании, могут включать любую комбинацию подходящих линкеров между отдельными цинковыми пальцами белка.
В некоторых примерах ДНК-связывающий полипептид представляет собой ДНК-связывающий домен из GAL4. GAL4 представляет собой модульный трансактиватор в Saccharomyces cerevisiae, однако он также функционирует в качестве трансактиватора во многих других организмах. См., например, Sadowski et al. (1988) Nature 335:563-4. В этой регуляторной системе экспрессия генов, кодирующих ферменты метаболического пути галактозы в S. cerevisiae, строго регулируются доступным источником углерода. Johnston (1987) Microbiol. Rev. 51: 458-76. Контроль транскрипции этих метаболических ферментов опосредуется взаимодействием между положительным регуляторным белком, GAL4 и симметричной последовательностью ДНК размером 17 п.н., с которой GAL4 специфически связывается (UAS).
Нативный GAL4 состоит из 881 аминокислотного остатка с молекулярной массой 99 кДа. GAL4 содержит функционально автономные домены, совокупная активность которых обеспечивает активность GAL4 in vivo. Ma and Ptashne (1987) Cell 48:847-53); Brent and Ptashne (1985) Cell 43(3 Pt 2):729-36. N-концевые 65 аминокислот GAL4 содержат ДНК-связывающий домен GAL4. Keegan et al. (1986) Science 231: 699-704; Johnston (1987) Nature 328:353-5. Специфическое связывание последовательности требует присутствия двухвалентного катиона, координируемого 6 остатками Cys, присутствующими в ДНК-связывающем домене. Скоординированный катионсодержащий домен взаимодействует и распознает консервативный триплет CCG на каждом конце UAS из 17 п.н. посредством прямых контактов с большой бороздкой спирали ДНК. Marmorstein et al. (1992) Nature 356:408-14. Функция связывания ДНК у белка обеспечивает расположение C-концевых доменов активации транскрипции вблизи промотора, так чтобы активирующие домены могли контролировать транскрипцию.
Дополнительные ДНК-связывающие полипептиды, которые можно использовать в определенных вариантах осуществления, включают, например, но не ограничиваясь ими, связывающую последовательность из AVRBS3-индуцируемого гена; консенсусную связывающую последовательность из AVRBS3-индуцируемого гена или синтетическую связывающую последовательность, сконструированную из них (например, ДНК-связывающий домен UPA); TAL; LexA (см., например, Brent & Ptashne (1985), выше); LacR (см., например, Labow et al. (1990) Mol. Cell. Biol. 10:3343-56; Baim et al. (1991) Proc. Natl. Acad. Sci. USA 88(12):5072-6); рецептор стероидных гормонов (Ellliston et al. (1990) J. Biol. Chem. 265:11517-121); репрессор Tet (патент США 6271341) и мутантный репрессор Tet, который связывается с последовательностью оператора tet в присутствии, но не в отсутствие тетрациклина (Tc); ДНК-связывающий домен NF-κβ; и компоненты регуляторной системы, описанной в Wang et al. (1994) Proc. Natl. Acad. Sci. USA 91(17):8180-4, в которой используется слияние GAL4, рецептора гормона и VP16.
В определенных вариантах осуществления ДНК-связывающий домен одной или нескольких нуклеаз, используемых в способах и композициях, описанных в настоящем описании, содержит встречающийся в природе или сконструированный (не встречающийся в природе) ДНК-связывающий домен эффектора TAL. См., например, публикацию патента США №20110301073. Известно, что патогенные для растений бактерии рода Xanthomonas вызывают множество заболеваний в важных сельскохозяйственных культурах. Патогенность Xanthomonas зависит от консервативной системы секреции типа III (T3S), которая инъецирует более 25 различных эффекторных белков в растительную клетку. Среди этих инъецируемых белков находятся эффекторы, подобные активаторам транскрипции (TAL), которые имитируют активаторы транскрипции растений и манипулируют транскриптомом растений (см. Kay et al (2007) Science 318:648-651). Эти белки содержат ДНК-связывающий домен и домен активации транскрипции. Одним из наиболее хорошо охарактеризованных эффекторов TAL является AvrBs3 из Xanthomonas campestgris pv. Vesicatoria (см. Bonas et al (1989) Mol Gen Genet 218: 127-136 и WO 2010079430). Эффекторы TAL содержат централизованный домен из тандемных повторов, причем каждый повтор содержит приблизительно 34 аминокислоты, которые являются ключевыми для специфичности этих белков в отношении связывания ДНК. Кроме того, они содержат последовательность ядерной локализации и кислый домен активации транскрипции (для обзора см. Schornack S, et al (2006) J Plant Physiol 163(3): 256-272). Кроме того, в фитопатогенных бактериях Ralstonia solanacearum было выявлено два гена, обозначаемых brg11 и hpx17, которые гомологичны семейству AvrBs3 Xanthomonas в R. solanacearum биоваре 1 штамма GMI1000 и в биоваре 4 штамма RS1000 (См. Heuer et al (2007) Appl and Envir Micro 73(13): 4379-4384). Эти гены на 98,9% идентичны друг другу по нуклеотидной последовательности, но отличаются делецией 1575 п.н. в домене повтора hpx17. Однако продукты обоих генов имеют менее чем 40% идентичность последовательности с белками семейства AvrBs3 Xanthomonas. См., например, патенты США №8420782 и 8440431 и публикацию патента США №20110301073.
В других вариантах осуществления нуклеаза содержит систему CRISPR/Cas. Локус CRISPR (кластеризованные регулярно расположенные короткие палиндромные повторы), который кодирует РНК-компоненты системы, и локус cas (ассоциированный с CRISPR), который кодирует белки (Jansen et al., 2002. Mol. Microbiol. 43: 1565-1575; Makarova el al., 2002. Nucleic Acids Res. 30: 482-496; Makarova et al., 2006. Biol. Direct 1: 7; Haft et al., 2005. PLoS Comput. Biol. 1: e60) формируют последовательности генов системы нуклеаз CRISPR/Cas. Локусы CRISPR в микробных хозяевах содержат комбинацию ассоциированных с CRISPR (Cas) генов, а также некодирующие РНК-элементы, способные программировать специфичность опосредуемого CRISPR расщепления нуклеиновых кислот.
CRISPR типа II представляет собой одну из наиболее охарактеризованных систем, и она осуществляет направленный разрыв двухцепочечной ДНК за четыре последовательных стадии. Во-первых, с локуса CRISPR транскрибируются две некодирующих РНК: матрица пре-crRNA и tracrRNA. Во-вторых, tracrRNA гибридизуется с областями повторов пре-crRNA и опосредует процессинг пре-crRNA в зрелые crRNA, содержащие индивидуальные спейсерные последовательности. В-третьих, зрелый комплекс crRNA:tracrRNA направляет Cas9 к ДНК-мишени через образование пар по принципу Уотсона-Крика между спейсером на crRNA и протоспейсером на ДНК-мишени рядом с соседним с протоспейсером мотивом (PAM), что является дополнительным требованием для распознавания мишени. Наконец, Cas9 опосредует расщепление ДНК-мишени, создавая двухцепочечный разрыв в протоспейсере. Активность Cas-системы CRISPR включает три стадии: (i) встраивание чужеродных последовательностей ДНК в матрицу CRISPR для предупреждения будущих атак в процессе, называемом "адаптацией", (ii) экспрессия соответствующих белков, а также экспрессия и процессинг матрицы, за которой следует (iii) РНК-опосредуемая интерференция в отношении чужеродной нуклеиновой кислоты. Таким образом, в бактериальной клетке несколько так называемых белков "Cas" вовлечено в природную функцию системы CRISPR/Cas и участвуют в таких функциях, как встраивание чужеродной ДНК и т.д.
В определенных вариантах осуществления белок Cas может представлять собой "функциональное производное" встречающегося в природе белка Cas. "Функциональное производное" полипептида с нативной последовательностью представляет собой соединение, обладающее качественным биологическим свойством, являющимся общим с полипептидом с нативной последовательностью. "Функциональные производные" включают, но не ограничиваются ими, фрагменты полипептида с нативной последовательностью и производные полипептида с нативной последовательностью и их фрагменты при условии, что они обладают общей биологической активностью с соответствующим полипептидом с нативной последовательностью. Биологическая активность, предусматриваемая в рамках настоящего изобретения, представляет собой способность функционального производного гидролизовать ДНК-субстрат на фрагменты. Термин "производное" охватывает как варианты аминокислотной последовательности полипептида, так и ковалентные модификации, и его слитые конструкции. Подходящие производные полипептида Cas или его фрагмента включают, но не ограничиваются ими, мутанты, слитые конструкции, ковалентные модификации белка Cas или его фрагмента. Белок Cas, который включает белок Cas или его фрагмент, а также производные белка Cas или его фрагмента, можно получать из клетки или синтезировать химически или путем комбинирования этих двух действий. Клетка может представлять собой клетку, которая в естественным путем продуцирует белок Cas, или клетку, которая естественным путем продуцирует белок Cas и модифицирована способами генной инженерии для продуцирования эндогенного белка Cas на более высоком уровне экспрессии или для продуцирования белка Cas с экзогенно введенной нуклеиновой кислоты, которая кодирует Cas, который является таким же или отличается от эндогенного Cas. В некоторых случаях клетка не продуцирует естественным образом белок Cas и она модифицирована способами генной инженерии для продуцирования белка Cas.
В конкретном варианте осуществления, в котором вносят по меньшей мере два двухцепочечных разрыва, репарация двухцепочечных разрывов может включать удаление материала между двухцепочечными разрывами и обратное соединение концов нуклеотидной последовательности, чтобы вырезались последовательности между двухцепочечными разрывами. В вариантах осуществления вырезанные последовательности могут, но не ограничиваясь этим, включать последовательности, кодирующие всю или часть нуклеотидной последовательности, кодирующей высоко, более высоко, очень высоко или наиболее высоко экспрессируемый белок. В следующих вариантах осуществления вырезанные последовательности могут, но не ограничиваясь этим, включать регуляторные последовательности, обеспечивающие экспрессию высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка. В таких вариантах осуществления экспрессия высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка снижается относительно уровней до расщепления.
В альтернативных вариантах осуществления, в которых вносят по меньшей мере два двухцепочечных разрыва, репарация двухцепочечных разрывов может включать удаление материала между двухцепочечными разрывами с встраиванием вместо него его донорной последовательности, чтобы заменить последовательности между двухцепочечными разрывами донорной последовательностью. В других вариантах осуществления удаленные последовательности могут, но не ограничиваясь этим, включать последовательности, кодирующие всю или часть нуклеотидной последовательности, кодирующей высоко, более высоко, очень высоко или наиболее высоко экспрессируемый белок. В следующих вариантах осуществления удаленные последовательности могут, но не ограничиваясь этим, включать регуляторные последовательности, обеспечивающие экспрессию высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка. В таких вариантах осуществления экспрессия высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка снижается относительно уровней до расщепления.
В вариантах осуществления, в которых вносят один двухцепочечный разрыв, репарация двухцепочечного разрыва может включать встраивание донорной последовательности в или на концах двухцепочечного разрыва. В определенных вариантах осуществления донорную последовательность можно встраивать в кодирующую последовательность высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка. В вариантах осуществления встраивание такой последовательности может нарушать транскрипцию кодирующей последовательности высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка посредством, в качестве неограничивающего примера, присутствия стоп-кодона в рамке считывания. В следующих вариантах осуществления донор может, но не ограничиваясь этим, нарушать функцию регуляторных последовательностей, обеспечивающих экспрессию высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка. В вариантах осуществления экспрессия высоко, более высоко, очень высоко или наиболее высоко экспрессируемого белка снижается относительно уровней экспрессии до расщепления.
В других вариантах осуществления донорная последовательность может кодировать представляющий интерес белок. В следующих вариантах осуществления экспрессия представляющего интерес белка с донорной последовательности может контролироваться, регулироваться или может быть функционально связана с регуляторными последовательностями, присутствующими в донорной последовательности, и/или регуляторными последовательностями, присутствующими в последовательности, в которую встроена донорная последовательность. В дополнительных вариантах осуществления последовательность нуклеиновой кислоты, кодирующая представляющий интерес белок, может быть предоставлена клетке отдельно или вместе с донорной последовательностью. В некоторых вариантах осуществления донорная последовательность может содержаться в той же молекуле нуклеиновой кислоты, в которой содержится представляющая интерес последовательность, кодирующая белок.
В других вариантах осуществления нуклеотидная последовательность, кодирующая высоко, более высоко, очень высоко или наиболее высоко экспрессируемый белок, может быть расположена, в качестве неограничивающего примера, в геноме, плазмиде, космиде, искусственной хромосоме, эписоме или другой нуклеотидной структуре в клетке.
В одном аспекте в рамках настоящего изобретения описаны двухцепочечные донорные полинуклеотиды для встраивания в эндогенный выбранный локус после расщепления in vivo донора с использованием по меньшей мере одной нуклеазы. Донорные нуклеотиды включают экзогенную последовательность (трансген), подлежащую встраиванию в эндогенный локус, и содержат по меньшей мере один участок-мишень для нуклеазы. Донорные нуклеотиды могут включать области гомологии (например, участки гомологии), фланкирующие последовательность трансгена. Гомология с хромосомой, присутствующая в донорной последовательности, может располагаться в участке связывания нуклеазой. В определенных вариантах осуществления, в которых нуклеаза используется для расщепления донора, нуклеаза не является такой же, как и нуклеаза, используемая для расщепления хромосомы, и возможно, что между хромосомой и донорной последовательностью будет отсутствовать гомология. В других вариантах осуществления донорные молекулы встраиваются в эндогенный локус посредством независимых от гомологии механизмов (например, NHEJ). В других вариантах осуществления двухцепочечный донор содержит трансген длиной по меньшей мере 1 т.п.н. и участок(участки)-мишень для нуклеазы, располагающийся с 3'- и/или 5'-стороны от трансгена, для расщепления in vivo. Донорная молекула может представлять собой, например, плазмиду. В определенных вариантах осуществления донор встраивается после опосредуемого нуклеазой расщепления эндогенного локуса. При любом опосредуемом нуклеазой встраивании донорной молекулы, одна или несколько нуклеаз, используемых для расщепления донора, может быть такой же, как одна или несколько нуклеаз, используемых для расщепления эндогенного локуса. Альтернативно одна или несколько из нуклеаз, используемых для расщепления донора, могут отличаться от одной или нескольких нуклеаз, используемых для расщепления эндогенного локуса.
В некоторых вариантах осуществления донор содержится на плазмиде. Донор может встраиваться после опосредуемого нуклеазой расщепления, где донор фланкируется в плазмиде по меньшей мере двумя участками расщепления нуклеазой. В определенных вариантах осуществления последовательность участков расщепления нуклеазой в донорной плазмиде является такой же, как и последовательность участка расщепления нуклеазой в хромосомном локусе, содержащем ETIP, подлежащую нацеливанию. В других вариантах осуществления участки расщепления нуклеазами, фланкирующие донор, на содержащей донор плазмиде отличаются от участка расщепления в ETIP хромосомы. В дополнительных вариантах осуществления участки расщепления нуклеазой, фланкирующее донор, в содержащей донор плазмиде, могут не быть одинаковыми, и также могут отличаться от участка расщепления нуклеазой в хромосоме. В следующих вариантах осуществления донор может содержаться в плазмиде, фланкируемой по меньшей мере двумя участками расщепления нуклеазой, и он может встраиваться в участок делеции в ETIP на хромосоме, внесенный действием двух нуклеаз. В таких вариантах осуществления участки расщепления нуклеазой, фланкирующие донор, на плазмиде и участки расщепления нуклеазой в ETIP на хромосоме либо могут быть одинаковыми, либо могут различаться. В других вариантах осуществления донор представляет собой плазмиду, содержащую только один участок расщепления нуклеазой, и участок расщепления нуклеазой в ETIP в хромосоме либо может быть таким же, либо может отличаться.
Представляющая интерес последовательность донорной молекулы может содержать одну или несколько последовательностей, кодирующих функциональный полипептид (например, кДНК), с промотором или без него. В определенных вариантах осуществления последовательность нуклеиновой кислоты содержит последовательность, кодирующую антитело, антиген, фермент, фактор роста, рецептор (клеточной поверхности или ядерный), гормон, лимфокин, цитокин, репортер, функциональные фрагменты любого из вышеуказанных и комбинации вышеуказанных. В вариантах осуществления, в которых функциональный полипептид, кодирующий последовательности, не имеет промотора, экспрессия встроенной последовательности обеспечивается транскрипцией, запускаемой эндогенным промотором или другим элементом контроля в представляющей интерес области. В других вариантах осуществления в выбранный участок может встраиваться тандемную кассету следующим образом: первый компонент кассеты содержит последовательность без промотора, как описано выше, за которой следует последовательность терминации транскрипции и вторая последовательность, кодирующая автономную экспрессирующую кассету. В донорную молекулу могут быть включены дополнительные последовательности (кодирующие или некодирующие последовательности) между участками гомологии. В дополнительных вариантах осуществления донорная нуклеиновая кислота содержит последовательности, кодирующие функциональные РНК, например, микроРНК (miRNA) или кшРНК (shRNA).
Описанные способы и композиции для направленного расщепления можно использовать для индукции мутаций в геномной последовательности. Направленное расщепление также можно использовать для осуществления нокаута или нокдауна генов (например, функциональная геномика или подтверждение мишени) и для облегчения направленного встраивания последовательности в геном (т.е. нокин последовательности). Встраивание можно осуществлять через замену хромосомных последовательностей посредством, в качестве неограничивающего примера, гомологичной рекомбинации или направленного встраивания, при котором новую последовательность (т.е. последовательность, не присутствующую в представляющей интерес области) встраивают в заданный участок-мишень. В определенных примерах такие новые последовательности могут быть фланкированы последовательностями, гомологичными представляющей интерес области в хромосоме. Те же способы также можно использовать для замены последовательности дикого типа мутантной последовательностью или для преобразования одного аллеля в другой аллель.
Описанные способы продуцирования путем направленной рекомбинации представляющего интерес белка можно использовать для замены любой геномной последовательности неидентичной последовательностью. Например, мутантную геномную последовательность можно заменять ее аналогом дикого типа, тем самым обеспечивая способы лечения заболеваний растений; обеспечивая устойчивость к патогенам растений; увеличивая урожай культуры и т.д. Аналогично, с использованием способов направленной рекомбинации, описанных в настоящем описании, один аллель гена можно заменять другим аллелем.
Во многих из этих случаев представляющая интерес область содержит мутацию и донорный полинуклеотид содержит соответствующую последовательность дикого типа. Аналогично, геномную последовательность дикого типа можно заменять мутантной последовательностью, если это желательно. Например, сверхэкспрессию онкогена можно обращать вспять либо путем мутации гена, либо путем замены его последовательностей контроля последовательностями, которые обеспечивают более низкий непатогенный уровень экспрессии. Действительно, любую патологию, зависящую от конкретной геномной последовательности любым образом можно корректировать или ослаблять с использованием способов и композиций, описанных в настоящем описании.
Направленное расщепление, встраивание, вырезание и/или рекомбинацию также можно использовать для изменения некодирующих последовательностей (например, регуляторные последовательности, такие как промоторы, энхансеры, инициаторы, терминаторы, участки сплайсинга) для изменения уровней экспрессии продукта гена. Такие способы можно использовать, например, для терапевтических целей, функциональной геномики и/или исследований для подтверждения мишени.
Направленную модификацию структуры хроматина можно использовать для облегчения связывания слитых белков с клеточным хроматином. В дополнительных вариантах осуществления можно использовать одну или несколько слитых конструкций между связывающим доменом цинковых пальцев и рекомбиназой (или ее функциональным фрагментом), в дополнение к или вместо слитых конструкций цинковый палец-домен расщепления, описанных в настоящем описании, для облегчения направленной рекомбинации. См., например, патент США №6534261 того же заявителя и Akopian et al. (2003) Proc. Natl. Acad. Sci. USA 100:8688-8691. В дополнительных вариантах осуществления описанные способы и композиции используют для предоставления слитых конструкций связывающих доменов ZFP с доменами активации или репрессии транскрипции, для активности которых требуется димеризация (либо гомодимеризация, либо гетеродимеризация). В этих случаях слитый полипептид содержит связывающий домен цинковых пальцев и мономер функционального домена (например, мономер из димерного домена активации или репрессии транскрипции). Связывание двух таких слитых полипептидов с надлежащим образом расположенными участками-мишенями обеспечивает димеризацию для восстановления функционального домена активации или репрессии транскрипции.
Более того, как описано выше, способы и композиции, описанные в настоящем описании, можно использовать для направленного встраивания экзогенных последовательностей в представляющую интерес область в геноме клетки, например, где расщепление усиливает встраивание по зависимым от гомологии механизмам (например, встраивание донорной последовательности, содержащей экзогенную последовательность, вместе с одной или несколькими последовательностями, которые либо являются идентичными, либо являются гомологичными, но не идентичными, заданной геномной последовательности (т.е. участку-мишени)).
Донорная последовательность может обладать достаточной гомологией в областях, фланкирующих экзогенную последовательность, для обеспечения направляемой гомологией репарации (HDR) двухцепочечного разрыва в геномной последовательности, и, тем самым, встраивания экзогенной последовательности в геномный участок-мишень. Таким образом, донорная нуклеиновая кислота может иметь любой размер, достаточный для обеспечения встраивания экзогенной последовательности по механизмам зависимой от гомологии репарации (например, гомологичная рекомбинация). Без связи с какой-либо конкретной теорией полагают, что области гомологии, фланкирующие экзогенную последовательность, обеспечивают на разорванных концах хромосомы матрицу для ресинтеза генетической информации в участке двухцепочечного разрыва. В определенных вариантах осуществления присутствуют две идентичных последовательности или две гомологичных, но не идентичных последовательности (или по одной из них), фланкирующие экзогенную последовательность. Экзогенная последовательность (или экзогенная нуклеиновая кислота или экзогенный полинуклеотид) представляет собой последовательность, которая содержит нуклеотидную последовательность, которая обычно не присутствует в представляющей интерес области.
Понятно, что конструкция, содержащая ген ZFN под контролем индуцибельного промотора вместе с его соответствующей последовательностью распознавания, может стабильно встраиваться в арабидопсис, и показано, что она вносит направленные мутации вследствие негомологичного соединения концов в участке распознавания. Например, в международной публикации патента № WO/2008/021207 описан способ прицельного встраивания трансгенов через опосредуемую ZFN гомологичную рекомбинацию. Напротив, когда белок ZFN может быть экспрессирован и очищен вне организма-мишени, а затем доставлен в растительные клетки-мишени, хирургически специфические мутация/нокаут гена могут быть индуцированы путем негомологичного соединения концов (NHEJ). Таким образом, настоящее изобретение может обеспечивать нетрансгенное генетически модифицированное растение, которое будет обходить ограничения для трансгенных культур и процесс направленного редактирования генов станет возможен без необходимости в трансгенном подходе.
Направленное встраивание гена, как правило, проводят путем трансфекции гена селективного маркера, фланкированного существенным количеством ДНК, гомологичной локусу-мишени. В локусе-мишени формируются спонтанные двухцепочечные разрывы (DSB), вероятно, вследствие заклинивающих репликативных вилок ДНК. Несмотря на обычно безошибочную репарацию путем направляемой репарацией гомологии (HDR) с сестринской хромосомой в качестве матрицы, HDR вместо этого может использовать гомологичную донорную ДНК для заживления разрыва. Когда дополнительную последовательность ДНК встраивают между двумя областями гомологии в донорной плазмиде, клеточный аппарат репарации ДНК невольно копирует эту генетическую информацию в хромосому. Поскольку это нацеливание на основе гомологии основано на перехвате очень редких DSB в области гомологии с донором, необходима обширная гомология с локусом-мишенью для достижения направленного встраивания с полезной частотой.
В альтернативных вариантах осуществления сходная способность встраивания генов может быть предоставлена типам клеток, лишенным эффективной репарации ДНК на основе гомологии, через NHEJ. Такой подход может оказаться особенно полезным для встраивания генов в первичных неделящихся клетках, которые предпочтительно используют каскад репарации ДНК NHEJ. Встраивание гена через NHEJ также может быть пригодным для неотсеквенированных геномов в качестве донорной конструкции без геномной последовательности, которая требует трудного предварительного клонирования и секвенирования. Понятно, что неспецифическая ДНК может захватываться в участке опосредуемой NHEJ репарации DSB. В конкретном варианте осуществления информацию, присутствующую в одноцепочечных выступающих концах, создаваемых при расщеплении ZFN, можно использовать для проведения направленного встраивания ДНК с использованием аппарата репарации ДНК NHEJ.
В других вариантах осуществления способность к встраиванию гена может быть обеспечена как эффективной репарацией ДНК на основе гомологии через NHEJ, так и направляемой гомологией репарацией. В таком варианте осуществления один конец донорной последовательности встраивают в хромосомную мишень через NHEJ, а другой конец донорной последовательности встраивают в хромосомную мишень посредством направляемой гомологией репарации.
Определенные варианты осуществления включают способы продуцирования донорной ДНК или ДНК груза, которая экспрессирует один или несколько продуктов экзогенной последовательности нуклеиновой кислоты (т.е. белок или молекула РНК), которая стабильно встроилась в ETIP в клетке растения (Фиг. 17).
Альтернативные варианты осуществления изобретения включают встраивание маркерных генов и последующую экспрессию маркерного белка, который встроился в поддающуюся нацеливанию молекулу нуклеиновой кислоты ETIP. В некоторых вариантах осуществления в способах селекции направленного встраивания донорной ДНК в локус ETIP используется сортировка и селекция по экспрессии маркерных генов, встроенных на один или оба из 5'- и 3'-концов ETIP (Фиг. 18). Оказывается, что встраивание ETIP в или вблизи локуса FAD2, FAD3 или IPK2, или встраивание ETIP в случайных положениях в геноме растения не нарушает способности растения-хозяина регенерировать, цвести, продуцировать семена, и обеспечивает селекцию, регенерацию и наследственную передачу последующих продуктов экзогенной последовательности нуклеиновой кислоты, доставляемых донорной полинуклеотидной молекулой, которая нацелена на ETIP, через поколения.
Более того, ETIP можно использовать для уменьшения количества трансгенных событий, которые необходимо осуществить в традиционных экспериментах по интрогрессии трансгенных событий, поскольку гены-кандидаты и конструкции экспрессирующих векторов растений встраиваются в одно и то же эндогенное положение генома растения. Технологию ETIP можно использовать для всех видов растений, включая возделываемые и модельные виды, включая кукурузу, сою, рис, канолу, пшеницу, ячмень, подсолнечник, томат, арабидопсис, хлопок, картофель, сорго, кормовые травы, виды brassica (включая, но не ограничиваясь ими, B. napus, B. rapa, B. oleracea, B. nigra, B. juncea, B. carrinata), сахарный тростник, сахарную свеклу, брахиподию и люцерну).
Конкретный вариант осуществления включает трансгенное растение или трансгенную клетку растения, полученную с использованием способов, описанных в настоящем описании. В частности, в одном варианте осуществления трансгенная клетка растения включает молекулу нуклеиновой кислоты, которая имеет нуклеотидную последовательность (которая может быть геномной), содержащую по меньшей мере одну поддающуюся нацеливанию молекулу нуклеиновой кислоты, где поддающаяся нацеливанию молекула нуклеиновой кислоты содержит участок распознавания сайт-специфической нуклеазой и фрагмент по меньшей мере одного маркерного гена, где фрагмент не кодирует функциональный продукт экспрессии маркерного гена. Конкретные варианты осуществления поддающейся нацеливанию молекулы нуклеиновой кислоты могут включать первый фрагмент первого маркерного гена и второй фрагмент второго маркерного гена. Другие варианты осуществления поддающейся нацеливанию молекулы нуклеиновой кислоты могут включать одну или несколько экспрессирующих кассет для генов. В следующих вариантах осуществления первый и второй фрагменты могут фланкировать участок распознавания сайт-специфической нуклеазой.
В некоторых вариантах осуществления донорная нуклеотидная последовательность может быть функционально связана с регуляторным элементом, таким как промотор, 5'-UTR, 3'-UTR, интрон или MAR. В других вариантах осуществления нуклеотидная последовательность может содержать фрагменты двух маркерных генов.
Система ETIP представляет собой платформенную технологию, которая обеспечивает быструю селекцию направленного встраивания донорной последовательности в геномы растений. В определенных вариантах осуществления технологию ETIP можно использовать в культурах клеток растений, таких как системы водорослей, мхов, грибов и культуры клеток млекопитающих, такие как NK-1, CHO и т.д. Система ETIP формирует основу для разработки системы для прицельной трансформации растений в различных сельскохозяйственных культурах для обеспечения высокопроизводительного исследования генов и конструкций в ходе процесса выведения признака.
Все ссылки, включая публикации, патенты и патентные заявки, цитированные в настоящем описании, включены в настоящее описание в качестве ссылок, до той степени, чтобы они не были противоречащими настоящему изобретению, и, таким образом, они включены в той же степени, как если было отдельно и конкретно указано, что каждая ссылка включена в качестве ссылки и была представлена в настоящем описании в полном объеме Ссылки, рассмотренные в настоящем описании, предоставлены только для их раскрытия до даты подачи настоящей заявки. Ничто в настоящем документе не следует истолковывать как допущение того, что это изобретение дает право на противопоставление факта создания изобретения с более ранним приоритетом.
ПРИМЕРЫ
Представленные ниже примеры предоставлены для иллюстрации определенных конкретных признаков и/или аспектов. Примеры не следует истолковывать как ограничивающие раскрытие конкретных описанных признаков или аспектов.
ПРИМЕР 1: ИДЕНТИФИКАЦИЯ ПАРАЛОГИЧНЫХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ-МИШЕНЕЙ В FAD2 И FAD3 ИЗ БИБЛИОТЕКИ БАКТЕРИАЛЬНЫХ ИСКУССТВЕННЫХ ХРОМОСОМ
КОНСТРУИРОВАНИЕ BAC
Библиотеку бактериальных искусственных хромосом (BAC) получали от коммерческого поставщика (Amplicon Express, Pullman, WA). Библиотека BAC состояла из 110592 клонов BAC, содержащих высокомолекулярные фрагменты геномной ДНК (гДНК), выделенные из Brassica napus L. сорта DH10275. гДНК расщепляли ферментом рестрикции либо BamΗΙ, либо HindIII. Выделенные фрагменты гДНК размером приблизительно 135 т.п.н. лигировали в вектор pCC1BAC (Epicentre, Madison, WI) и трансформировали в Escherichia coli штамма DH10B (Invitrogen). Библиотека BAC состояла из равного количества клонов BAC, которые были сконструированы с использованием двух различных ферментов рестрикции. По существу, сконструированная с помощью HindIII библиотека BAC состояла из 144 индивидуальных 384-луночных планшетов. Аналогично, сконструированная с помощью BamHI библиотека BAC состояла из 144 индивидуальных 384-луночных планшетов. Всего 110592 клонов BAC было выделено и распределено в 288 индивидуальных 384-луночных планшетах. Каждый из 288 индивидуальных 384-луночных планшетов был предоставлен поставщиком в качестве единичного экстракта ДНК для быстрого скрининга на основе ПЦР. Полученная библиотека BAC охватывает приблизительно 15 м.п.н. гДНК, что соответствует 12-кратному охвату генома Brassica napus L. сорта DH10275 (согласно оценке геном Brassica napus L. составляет приблизительно 1,132 м.п.н., как описано в Johnston et al. (2005) Annals of Botany 95:229-235).
АНАЛИЗ ПОСЛЕДОВАТЕЛЬНОСТИ КОДИРУЮЩИХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ FAD2, ВЫДЕЛЕННЫХ ИЗ БИБЛИОТЕКИ BAC
Сконструированную библиотеку BAC использовали для выделения кодирующих последовательностей гена FAD2. Эксперименты по секвенированию проводили для идентификации конкретных последовательностей генов для четырех паралогов гена FAD2 из Brassica napus L. сорта DH10275.
Последовательность гена FAD2 первоначально идентифицировали в модельном виде Arabidopsis thaliana. Последовательность гена приведена в Genbank под обозначением локуса: At3g12120. Ранее была описана сравнительная геномная взаимосвязь между модельным видом растений Arabidopsis thaliana и диплоидным Brassica rapa, одним из предшественников тетраплоидного Brassica napus. (Schranz et al. (2006) Trends in Plant Science 11(11):535-542). В отношении конкретно гена FAD2 сравнительный анализ спрогнозировал, что в диплоидном геноме Brassica может встречаться 3-4 копии гена. Дополнительные исследования по генетическому картированию были осуществлены Scheffler et al. (1997) Theoretical and Applied Genetics 94; 583-591. Результаты этих исследований по генетическому картированию показали, что в Brassica napus присутствует четыре копии гена FAD2.
Анализ путем секвенирования библиотеки BAC, которая была сконструирована из B. napus L. сорта DH12075, привел к выделению четырех последовательностей BAC (SEQ ID NO: 1, SEQ ID NO: 2, SEQ ID NO: 3 и SEQ ID NO: 4), в которых были определены кодирующие последовательности для генов FAD2A (SEQ ID NO: 5), FAD2-1 (SEQ ID NO: 6), FAD2-2 (SEQ ID NO: 7) и FAD2-3 (SEQ ID NO: 8). Последовательности генов FAD2A, FAD2-1, FAD2-2 и FAD2-3 были идентифицированы и генетически картированы. Анализ последовательностей четырех генов FAD2 проводили с использованием программы для выравнивания последовательностей и дерева, полученного по методу ближайшего связывания, с использованием процента идентичности. Выравнивание последовательностей проводили с помощью программы AlignX® из компьютерной программы Vector NTI Advance 11.0 (Life Technologies, Carlsbad, CA), и оно представлено на Фиг. 1. В AlignX® используется модифицированный алгоритм Clustal W для проведения множественного выравнивания либо белковых последовательностей, либо последовательностей нуклеиновых кислот, для сравнения сходства и для проведения аннотирования.
Дерево, полученное по методу ближайшего связывания, создавали с помощью программного обеспечения Jalview v2.3®, и оно представлено на Фиг. 2. (Waterhouse et al. (2009) Bioinformatics 25 (9) 1189-1191). Как показано на Фиг. 2, анализ выделенных последовательностей показал, что последовательности FAD2A и FAD2-3 обладали высокими уровнями сходства последовательностей и что, аналогично, FAD2-1 и FAD2-2 обладали высокими уровнями сходства последовательностей. Эти четыре последовательности могут быть подразделены на два клада, где FAD2A и FAD2-3 составляют первый клад, и FAD2-1 и FAD2-2 составляют второй клад.
Далее, вновь выделенные последовательности FAD2 из Brassica napus использовали для проведения BLAST геномных библиотек, выделенных из геномной библиотеки BAC Brassica rapa и получения данных о геномной последовательности Brassica oleracea с помощью метода стохастических геномных фрагментов. Как Brassica rapa, и так и Brassica oleracea, являются диплоидными предшественники Brassica napus, который является амфиплоидным видом (геном AC, n=19). Brassica napus произошел в результате недавнего события гибридизации между Brassica rapa (субгеном A, n=10) и Brassica oleracea (субгеном C, n=9). Диплоидные последовательности-предшественники сравнивали с четырьмя различными кодирующими последовательностями FAD2, выделенными из Brassica napus, с использованием анализа BLASTn. Этот анализ последовательностей идентифицировал специфические аннотированные последовательности генов из Brassica rapa и Brassica oleracea, которые обладали наибольшим сходством последовательностей с вновь выявленными последовательностями FAD2 Brassica napus. В таблице 1 приведена вновь идентифицированная кодирующая последовательность FAD2, и соответствующий номер доступа эталонной последовательности предшественника, и организм-источник.
Гены FAD2 существуют в геноме Brassica napus в качестве двух копий каждого гена на субгеном. Одна копия каждого гена расположена на субгеноме A, и, аналогично, одна копия каждого гена расположена на субгеноме C. Новые соглашения о наименованиях описаны для указания на то, на каком субгеноме расположен каждый ген. Более высокие уровни сходства последовательностей между четырьмя различными кодирующими последовательностями FAD2, выделенными из библиотеки геномной ДНК BAC Brassica napus, и данные о последовательностях-предшественниках указывают на то, что FAD2-3 является дубликатом последовательности FAD2 из субгенома C, и он может быть переобозначен как FAD2C; FAD2-1 является дубликатом последовательности FAD2 из субгенома A и, таким образом, он может быть обозначен как FAD2A'; и, наконец, FAD2-2 является второй копией, которая является дубликатом последовательности FAD2 субгенома С, и он может быть обозначен как FAD2C.
АНАЛИЗ ПОСЛЕДОВАТЕЛЬНОСТИ КОДИРУЮЩИХ ПОСЛЕДОВАТЕЛЬНОСТЕЙ FAD3, ВЫДЕЛЕННЫХ ИЗ БИБЛИОТЕКИ BAC
Сконструированную библиотеку BAC использовали для выделения кодирующих последовательностей гена FAD3. Эксперименты по секвенированию проводили для идентификации конкретных последовательностей генов для пяти паралогов гена FAD3 из Brassica napus L. сорта DH10275.
Последовательность гена FAD3 первоначально идентифицировали в модельном виде Arabidopsis thaliana. Последовательность гена приведена в Genbank под обозначением локуса: At2g29980. Ранее была описана сравнительная геномная взаимосвязь между модельным видом растений Arabidopsis thaliana и диплоидным Brassica rapa, одним из предшественников тетраплоидного Brassica napus. (Schranz et al. (2006) Trends in Plant Science 11(11):535-542). В отношении конкретно гена FAD2 сравнительный анализ спрогнозировал, что в диплоидном геноме Brassica может встречаться 3-4 копии гена. Дополнительные исследования по генетическому картированию были осуществлены Scheffler et al. (1997) Theoretical and Applied Genetics 94; 583-591. Результаты этих исследований по генетическому картированию показали, что в Brassica napus присутствует шесть копий гена FAD3.
Предшествующие попытки секвенирования, сфокусированные на генах FAD3 из Brassica napus, идентифицировали и генетически картировали копи, специфические как для генома A, так и для генома C (Hu et al., (2006) Theoretical and Applied Genetics, 113(3): 497-507). Ранее Andrew Sharpe из Agriculture and Agri-food Canada, 107 Science Place, Saskatoon, Saskatchewan, был сконструирован и отсеквенирован набор последовательностей EST из специфических библиотек кДНК семян из линии растений DH12075. Набор EST из полноразмерных последовательностей генов растения канолы DH12075 с удвоенным гаплоидным набором не был доступен, более того также не были доступны данные о качестве последовательности и достоверности обозначения нуклеотидов. Следовательно, варьирование последовательности между различными считанными последовательностями гена FAD не могли быть однозначно отнесены к различным копиям гена различных паралогов семейства генов FAD3, а также не была доступна геномная последовательность. Однако, когда проводили комбинированный анализ последовательности с EST, а также двумя полноразмерными последовательностями генов FAD3A и FAD3C, описанными Hu et al., (2006), были идентифицированы EST, которые совпадали в обоих генах, а также 3 дополнительных гаплотипа. В результате, было идентифицировано всего шесть уникальных гаплотипов FAD3. После объединения всех доступных данных для различных гаплотипов FAD3, были идентифицированы высокие уровни дивергенции экзонных последовательностей в экзоне 1. Дивергенция последовательности FAD3 в экзоне 1 была идентифицирована в качестве возможности, которую можно использовать для конструирования ген/аллель-специфических праймеров для ПЦР. Кроме того, были идентифицированы экзоны, которые либо минимально различались между гаплотипами (например, экзоны 5, 6, 7 и 8 имели 1-3 п.н., которые различались между FAD3A и FAD3C), либо были лишены варьирования последовательности (например, экзоны 2 и 3).
Анализ путем секвенирования библиотеки BAC, которая была сконструирована из B. napus L. сорта DH12075, привел к выделению шести последовательностей BAC (SEQ ID NO: 9, SEQ ID NO: 10, SEQ ID NO: 11, SEQ ID NO: 12, SEQ ID NO: 13 и SEQ ID NO: 14), в которых были определены кодирующие последовательности для генов FAD3A (SEQ ID NO: 15), FAD3A' (SEQ ID NO: 16), FAD3A" (SEQ ID NO: 17), FAD3C (SEQ ID NO: 18), FAD3C" (SEQ ID NO: 19) и FAD3C' (SEQ ID NO: 20). Последовательности генов FAD3A, FAD3A', FAD3A", FAD3C, FAD3C" и FAD3C' были идентифицированы и генетически картированы.
Анализ последовательностей шести генов FAD2 проводили с использованием программы для выравнивания последовательностей и дерева, полученного по методу ближайшего связывания, с использованием процента идентичности. Выравнивание последовательностей проводили с помощью программы AlignX® из компьютерной программы Vector NTI Advance 11.0 (Life Technologies, Carlsbad, CA), и оно представлено на Фиг. 3. В AlignX® используется модифицированный алгоритм Clustal W для проведения множественного выравнивания либо белковых последовательностей, либо последовательностей нуклеиновых кислот, для сравнения сходства и для проведения аннотирования. Дерево, полученное по методу ближайшего связывания, создавали с помощью программного обеспечения Jalview v2.3®, и оно представлено на Фиг. 4. (Waterhouse et al. (2009) Bioinformatics 25 (9) 1189-1191). Контиги, идентифицированные как содержащие гены FAD3, использовали в качестве последовательностей запроса в BLASTn против базы данных генов Arabidopsis thaliana. Область каждого из 6 контигов, содержавших ген FAD3, была идентифицирована путем сравнения с геном FAD3 Arabidopsis thaliana (номер доступа Genbank № At2g29980). Затем контиги FAD3 ориентировали таким образом, чтобы все гены FAD3 находились в ориентации от 5' к 3'. Контиги FAD3 укорачивали так, чтобы они содержали не более не более 2 вышележащих (5') и 1 нижележащий (3') гены Arabidopsis thaliana. После ориентирования полную кодирующую область генов FAD3 извлекали из каждого контига и использовали для создания дерева, полученного по методу ближайших соседей, чтобы отобразить взаимосвязь между различными представителями семейства генов FAD3. 6 представителей семейства FAD3 были выровнены в 3 пары генов FAD3 (Фиг. 4).
СКРИНИНГ НА ОСНОВЕ ПЦР
Для скрининга упомянутой выше библиотеки BAC была сконструирована группа праймеров для ПЦР. Праймеры были сконструированы либо в качестве универсальных праймеров, которые амплифицируют всех представителей данного семейства генов, либо в качестве специфических генных праймеров для направленной амплификации аллеля. Праймеры для ПЦР были сконструированы так, чтобы они имели длину 20 п.н. (+/-1 п.н.) и имели содержание G/C 50% (+/-8%). В таблице 2 и в таблице 3 приведены праймеры, которые были сконструированы и синтезированы. Клоны библиотеки BAC были объединены и подвергнуты скринингу с помощью полимеразной цепной реакции (ПЦР).
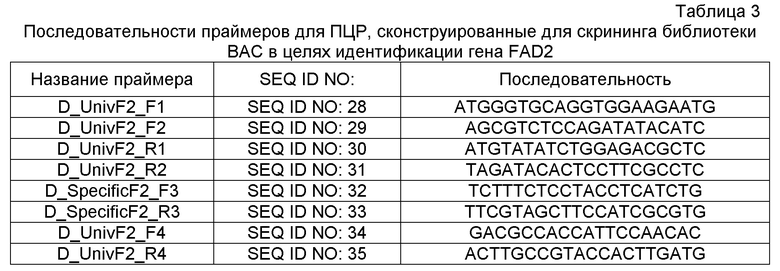
Для полимеразных цепных реакций (ПЦР) использовали два различных набора условий. Первая серия реакций ПЦР включала: 1X буфер для ПЦР (содержащий dNTP); 1,5 мМ MgCl2; 200 мкМ ДНК-полимеразы Immolase® с 0,25 Е (Bioline, London, Великобретания); 250 нМ каждого праймера и приблизительно 5-10 нг ДНК-матрицы. Вторую серию реакций ПЦР разрабатывали для амплификации геномной ДНК, и она включала: 5-10 нг геномной ДНК, 1× буфер для ПЦР, 2 мМ dNTP, 0,4-мкМ прямой и обратный праймер и 0,25 Е ДНК-полимеразы Immolase® (Bioline, London, Великобритания). Реагенты для амплификации объединяли в конечном объеме 13 мкл и амплифицировали с использованием термоциклера MJ PTC200® (BioRad, Hercules, CA) или ABI 9700 Gene Amp System® (Life Technologies, Carlsbad, CA). Скрининг на основе ПЦР в специальных планшетах проводили с использованием подхода 4-мерного скрининга на основе системы скрининга, описанной Bryan et al. (Scottish Crops Research Institute annual report: 2001-2002) с описанными выше условиями ПЦР. После скрининга на основе ПЦР объединенных библиотек BAC, амплифицированный продукт ПЦР секвенировали с использованием способа прямого секвенирования Сэнгера. Амплифицированные продукты очищали этанолом, ацетатом натрия и EDTA в соответствии с протоколом BigDye® v3.1 (Applied Biosystems) и проводили электрофорез на автоматической платформе для капиллярного электрофореза ABI3730x1®.
После скрининга на основе ПЦР и подтверждающего секвенирования по методу Сэнгера идентифицировали набор планшетов, который содержал различных представителей семейств генов FAD2 и FAD3. Было идентифицировано всего четыре уникальных последовательности паралогов генов FAD2 и FAD3 (таблица 4 и таблица 5). Всего два планшета на каждую последовательность паралога гена FAD2 и FAD3 было выбрано для проведения скрининга планшета в целях идентификации конкретной лунки и клона в планшете, которые содержали ген FAD2 и FAD3 (таблица 4 и таблица 5). Конкретные лунки идентифицировали для обоих планшетов и для каждого из представителей семейств генов FAD2 и FAD3 выбирали индивидуальный клон.
Единичный клон BAC для каждого идентифицированного представителя семейства генов FAD далее анализировали секвенированием. ДНК выделяли для клона BAC и подготавливали для секвенирования с использованием набора Large Construct kit® (Qiagen, Valencia, CA) в соответствии с инструкциями изготовителя. Экстрагированную ДНК BAC подготавливали для секвенирования с использованием GS-FLX Titanium Technology® (Roche, Indianapolis, IΝ) в соответствии с инструкциями изготовителя. Реакции секвенирования проводили с использованием физически секторированного планшета GS-FLX TI Pico-titer plate®, где BAC были объединены парами для оптимального получения данных. BAC объединяли парами, причем ген FAD2 объединяли в пару с геном FAD3. Все полученные данные о последовательности объединяли в Newbler v2.0.01.14® (454 Life Sciences, Branford, CT). Собранные контиги оценивали вручную в отношении присутствия соответствующего гена FAD с использованием Sequencher v3.7® (GeneCodes, Ann Arbor, MI).
После идентификации и полной охарактеризации полной геномной последовательности всех четырех генов FAD2 и шести генов FAD3, конструировали нуклеазы с цинковыми пальцами, чтобы они связывались с последовательностями каждого конкретного представителя семейства генов.
ПРИМЕР 2: КОНСТРУИРОВАНИЕ СВЯЗЫВАЮЩИХ ДОМЕНОВ С ЦИНКОВЫМИ ПАЛЬЦАМИ, СПЕЦИФИЧНЫХ К ГЕНАМ FAD2
Белки с цинковыми пальцами, направленные против последовательностей ДНК, кодирующих различные функциональные последовательности локуса генов FAD2, конструировали, как описано ранее. См., например, Urnov et al. (2005) Nature 435:646-651. Иллюстративная последовательность-мишень и распознающие спирали представлены в таблице 6 и в таблице 7 (модели распознающих спиральных областей) и в таблице 8 и в таблице 9 (участки-мишени). В таблице 8 и в таблице 9 нуклеотиды в участке-мишени, которые контактируют с распознающими спиралями ZFP, указаны прописными буквами; неконтактирующие нуклеотиды указаны строчными буквами.
Участки-мишени нуклеазы с цинковыми пальцами (ZFN) конструировали так, чтобы они связывали пять участков-мишеней FAD2A и семь участков-мишеней FAD3. Конструкции цинковых пальцев для FAD2 и FAD3 включали в экспрессирующие векторы для цинковых пальцев, кодирующие белок, имеющий по меньшей мере один палец со структурой CCHC. См., публикацию патента США №2008/0182332. В частности, последний палец в каждом белке имел остов CCHC для распознающей спирали. Неканонические последовательности, кодирующие цинковые пальцы, подвергали слиянию с нуклеазным доменом фермента рестрикции FokI типа IIS (аминокислоты 384-579 последовательности Wah et al, (1998) Proc. Natl. Acad. Sci. USA 95:10564-10569) через линкер ZC из четырех аминокислот и сигнал ядерной локализации opaque-2, происходящий из Zea mays, с получением нуклеаз с цинковыми пальцами (ZFN) для FAD2A. Экспрессия слитых белков контролировалась относительно сильным конститутивным промотором, таким как промотор, происходящий из промотора вируса мозаики прожилок маниоки (CsVMV) и фланкировались 3'-нетранслируемой областью ORF23 Agrobacterium tumefaciens (3'UTR v1 AtuORF23). Между двумя слитыми белками нуклеаз с цинковыми пальцами, которые были клонированы в конструкцию, добавляли самогидролизующуюся нуклеотидную последовательность, кодирующую 2A, из вируса Thosea asigna (Szymczak et al, 2004). Иллюстративные векторы описаны ниже.
Активность расщепления у оптимальных цинковых пальцев подтверждали с использованием системы на основе почкования дрожжей, для которой ранее было показано, что она идентифицирует активные нуклеазы. См., например, публикацию патента США №20090111119; Doyon et al. (2008) Nat Biotechnol. 26:702-708; Geurts et al. (2009) Science 325:433. Цинковые пальцы для различных функциональных доменов выбирали для применения in-vivo. Среди многочисленных ZFN, которые были сконструированы, продуцированы и исследованы в отношении связывания с предполагаемыми геномными полинуклеотидными участками-мишенями FAD, были идентифицированы ZFN в качестве имеющих активность in vivo на высоких уровнях, и выбрано для дальнейшего экспериментирования. Эти ZFN были охарактеризованы как способные эффективно связывать и расщеплять уникальные геномные полинуклеотидные участки-мишени FAD2 в растениях.

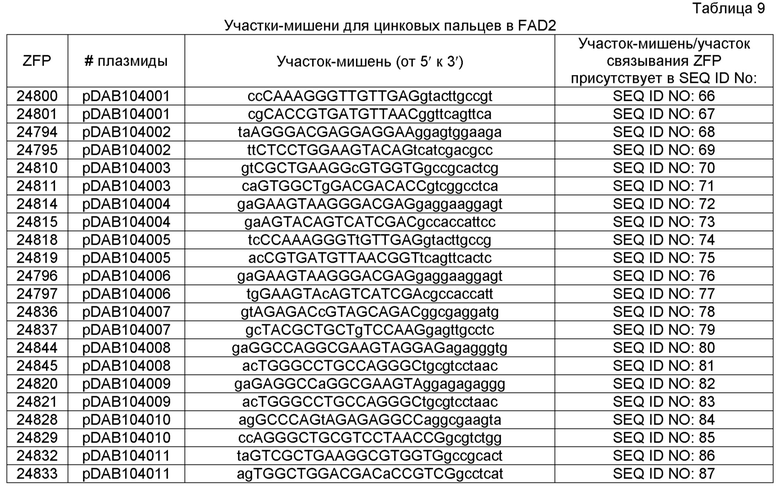
ПРИМЕР 3: ОЦЕНКА РАСЩЕПЛЕНИЯ НУКЛЕАЗАМИ С ЦИНКОВЫМИ ПАЛЬЦАМИ ГЕНОВ FAD2
СБОРКА КОНСТРУКЦИИ
Плазмидные векторы, содержащие конструкции для экспрессии иллюстративных нуклеаз с цинковыми пальцами ZFN, которые были идентифицированы с использованием анализа на дрожжах, как описано в примере 2, моделировали и получали с использованием навыков и способов, широко известных в данной области. Каждую последовательность, кодирующую цинковый палец, подвергали слиянию с последовательностью, кодирующей сигнал ядерной локализации opaque-2 (Maddaloni et al. (1989) Nuc. Acids Res. 17(18):7532), который был расположен выше нуклеазы с цинковыми пальцами.
Далее формировали пару из слитой последовательности сигнал ядерной локализации opaque-2 :: нуклеаза с цинковыми пальцами с комплементарной слитой последовательностью сигнал ядерной локализации opaque-2 :: нуклеаза с цинковыми пальцами. По существу, каждая конструкция состояла из одной открытой рамки считывания, состоящей из двух слитых последовательностей сигнал ядерной локализации opaque-2 :: нуклеаза с цинковыми пальцами, разделенных последовательностью 2A из вируса Thosea asigna (Mattion et al. (1996) J. Virol. 70:8124-8127). Экспрессия слитых белков контролировалась относительно сильным конститутивным промотором, таким как промотор, происходящий из промотора вируса мозаики прожилок маниоки (CsVMV), и фланкировалась 3'-нетранслируемой областью ORF23 Agrobacterium tumefaciens (3'UTR AtuORF23).
Векторы собирали с использованием IΝ-FUSION™ Advantage Technology (Clontech, Mountain View, CA). Эндонуклеазы рестрикции получали от New England BioLabs (NEB; Ipswich, MA) и для лигирования ДНК использовали ДНК-лигазу T4 (Invitrogen). Получение плазмид проводили с использованием набора NUCLEOSPIN® Plasmid Kit (Macherey-Nagel Inc., Bethlehem, PA) или набора Plasmid Midi Kit (Qiagen) в соответствии с инструкциями поставщиков. Фрагменты ДНК выделяли с использованием набора QIAquick Gel Extraction Kit™ (Qiagen) после электрофореза в агарозном Tris-ацетатном геле. Колонии всех собранных плазмид сначала подвергали скринингу с помощью рестрикционного расщепления ДНК Miniprep. Плазмидную ДНК выбранных клонов секвенировали в коммерческой организации, осуществляющей секвенирование (Eurofins MWG Operon, Huntsville, AL). Данные о последовательности собирали и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI). Перед доставкой в протопласты B. napus, плазмидную ДНК получали из культур E. coli с использованием Pure Yield Plasmid Maxiprep System® (Promega Corporation, Madison, WI) или Plasmid Maxi Kit® (Qiagen, Valencia, CA) в соответствии с инструкциями поставщиков.
Полученные одиннадцать плазмидных конструкций: pDAB104008 (содержащая конструкции ZFN24845 и ZFN24844), pDAB104009 (содержащая конструкции ZFN24820 и ZFN24821), pDAB104010 (содержащая конструкции ZFN24828 и ZFN24829) (Фиг. 3), pDAB104003 (содержащая конструкции ZFN24810 и ZFN24811), pDAB104011 (содержащая конструкции ZFN24832 и ZFN24833), pDAB104002 (содержащая конструкции ZFN24794 и ZFN24795), pDAB104006 (содержащая конструкции ZFN24796 и ZFN24797), pDAB104004 (содержащая конструкции ZFN24814 и ZFN24815), pDAB104001 (содержащая конструкции ZFN24800 и ZFN24801), pDAB104005 (содержащая конструкции ZFN24818 и ZFN24819) и DAB104007 (содержащая конструкции ZFN24836 и ZFN24837) подтверждали посредством расщепления ферментами рестрикции и секвенирования ДНК. В таблице 10 приведены различные конструкции и конкретная последовательность FAD, для расщепления и связывания которой была сконструирована каждая ZFN.
Полученные плазмидные конструкции pDAB107824 (ZFN 28025-2A-28026), pDAB107815 (ZFN 27961-2A-27962), pDAB107816 (ZFN 27969-2A-27970), pDAB107817 (ZFN 27973-2A-27974), pDAB107825 (ZFN 28035-2A-28036), pDAB107826 (ZFN 28039-2A-28040), pDAB107818 (ZFN 27987-2A-27988), pDAB107827 (ZFN 28051-2A-28052), pDAB107821 (ZFN 28004-2A-28005), pDAB107819 (ZFN 27989-2A-27990), pDAB107828 (ZFN 28053-2A-28054), pDAB107829 (ZFN 28055-2A-28056), pDAB107820 (ZFN 27991-2A-27992), pDAB107822 (ZFN 28021-2A-28022)и pDAB107823 (ZFN 28023-2A-28024) подтверждали путем расщепления ферментом рестрикции и секвенирования ДНК.
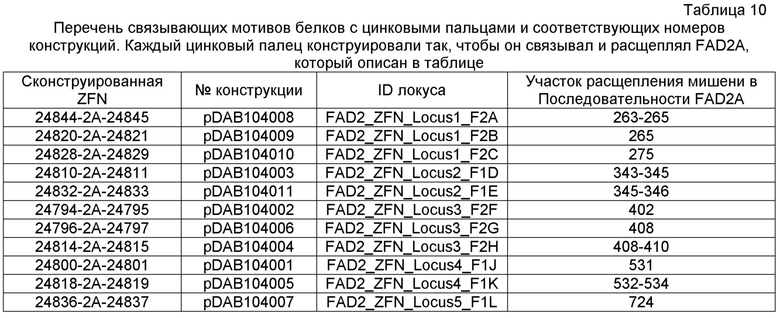
ПОЛУЧЕНИЕ ДНК ДЛЯ ТРАНСФЕКЦИИ
Плазмидную ДНК описанных выше векторов стерилизовали путем осаждения и промывания в 100% (об./об.) этаноле и сушили в вытяжном шкафу с ламинарным потоком. Осадок ДНК суспендировали в 30 мкл стерильной дважды дистиллированной воды в конечной концентрации 0,7 г/л для трансфекции в клетки протопластов, как описано ниже. Получение плазмидной ДНК проводили так, чтобы получить сверхскрученную плазмидную ДНК для временной трансфекции и линеаризованную плазмидную ДНК для стабильной трансфекции. Добавление ДНК-носителя (например ДНК спермы рыб) к трансформирующей плазмиде не требовалось для временной трансфекции протопластов клеток. Для временных исследований использовали приблизительно 30 мкг плазмидной ДНК на 106 протопластов на трансформацию.
ТРАНСФЕКЦИЯ
Трансфекцию Brassica napus L. сорта DH10275 проводили, как описано в Spangenberg et al, (1986) Plant Physiology 66: 1-8, составы сред описаны в Spangenberg G. and Protrykus I. (1995) Polyethylene Glycol-Mediated Direct Gene Transfer in Tobacco Protoplasts., Gene Transfer to Plants. (Protrykus I. and Spangenberg G. Eds.) Springer-Verlag, Berlin. Семена Brassica napus подвергали поверхностной стерилизации в 70% этаноле. Семена погружали в 12 мл 70% раствора этанола и перемешивали путем осторожного качания смеси в течение 10 минут. 70% раствор этанола удаляли путем декантации раствора и заменяли раствором для стерилизации семян, состоявшим из 1% масс./об. гипохлорита кальция и 0,1% об./об. Tween-20. Семена погружали в раствор для стерилизации семян и перемешивали путем осторожного качания смеси в течение 25 минут. Раствор для стерилизации семян декантировали и стерилизованные семена промывали три раза в 50 мл стерильной воды. Наконец, семена переносили на стерильный 80-мм фильтрующий бумажный диск Whatman filter paper disc® (Fisher-Scientific, St. Louis, MO), который был помещен в чашку Петри, и семена слегка насыщали стерильной водой. Чашку Петри накрывали с помощью Parafilm® (Fisher-Scientific, St. Louis, MO) и планшеты инкубировали при 25°C в условиях полной темноты в течение от одних до двух суток. После того, как наблюдали появление признаков прорастания семян, проростки переносили в чашку Петри, содержавшую затвердевшую среду GEM, для стимуляции дальнейшего прорастания семян. Проростки инкубировали на среде GEM при 25°C в течение от четырех до пяти суток.
Объем жидкой среды PS (приблизительно 10 мл) декантировали в стерильную чашку Петри. С использованием стерильных щипцов и скальпеля, надземную часть проростков в возрасте от четырех до пяти суток на стадии роста и развития 4 листков, удаляли и отбрасывали. Было определено, что сегменты гипоктиля длиной 20-40 мм образуют наиболее высокую популяцию небольших богатых цитоплазмой протопластов. Сегменты гипоктиля вырезали в асептических условиях и переносили в жидкую среду PS. Вырезанные сегменты гипоктиля группировали вместе и нарезали поперечно на сегменты размером 5-10 мм. Далее сегменты гипоктиля переносили в свежую среду PS и инкубировали при комнатной температуре в течение 1 часа. Подвергнутые плазмолизу гипоктили переносили в чашку Петри, содержавшую раствор фермента. Обращали внимание на то, чтобы погрузить в раствор все сегменты гипоктиля. Чашки Петри накрывали Parafilm® и инкубировали в течение от шестнадцати до восемнадцати часов при 20-22°C при осторожном качании.
Из сегментов гипоктиля высвобождали протопласты клеток. Продукты расщепления гипоктиля в течение ночи осторожно встряхивали для высвобождения протопластов в раствор фермента. Чашку Петри наклоняли немного, чтобы облегчить перенос расщепляющей суспензии, которая состояла из раствора фермента и растительного дебриса. С использованием 10-мл пипетки расщепляющую суспензию переносили в стерилизованный элемент для фильтрации протопластов (фильтр с размером сита 100 микрометров) для дальнейшего отделения протопластов от растительного дебриса. По фильтрационному элементу постукивали осторожно для высвобождения избытка жидкости, который остался в сите. Суспензию протопластов объемом приблизительно от 8 до 9 мл осторожно перемешивали и распределяли в 14-мл стерильные пластмассовые центрифужные пробирки с круглым дном. На каждую суспензию наслаивали 1,5 мл раствора W5. Раствор W5 осторожно распределяли над суспензией протопластов под углом и распределяли капельно с минимальным встряхиванием. Добавление раствора W5 к суспензии протопластов приводило к образованию обогащенной протопластами поверхности контакта. Этот слой поверхности контакта собирали с использованием пипетки. Далее собранные протопласты переносили в новую 14-мл центрифужную пробирку и осторожно перемешивали. Выход полученных протопластов определяли с использованием счетной камеры для определения количества протопластов на миллилитр. Способ повторяли, где ткань листьев расщепляли с получением протопластов мезофилла.
Далее, добавляли раствор W5 до объема 10 мл и протопласты осаждали при 70 g, а затем раствор W5 удаляли. Оставшуюся суспензию протопластов ресупендировали путем осторожного качания. Каждую пробирку, содержавшую суспензию протопластов, заполняли 5 мл раствора W5 и инкубировали при комнатной температуре в течение от одного до четырех часов. Суспензии протопластов осаждали при 70 g и весь раствор W5 удаляли. Далее в каждую из осажденных суспензий протопластов, содержавших выделенные протопласты добавляли 300 мкл буфера для трансформации. В каждой из пробирок к суспензиям протопластов добавляли 10 мкг плазмидной ДНК. Плазмидная ДНК состояла из конструкций нуклеаз с цинковыми пальцами, описанных выше (например, pDAB104010). Далее к суспензии протопластов добавляли 300 мкл предварительно нагретого раствора PEG 4000 и проводили осторожное постукивание по пробиркам. Суспензиям протопластов и смеси для трансформации позволяли инкубироваться при комнатной температуре в течение пятнадцати минут без какого-либо встряхивания. В каждую пробирку добавляли дополнительные 10 мл раствора W5 последовательными аликвотами объемом 1 мл, 1 мл, 1 мл, 2 мл, 2 мл и 3 мл с осторожным переворачиванием пробирок между добавлениями раствора W5 в каждом случае. Протопласты осаждали путем центрифугирования в центрифуге при 70g. Весь раствор W5 удаляли, оставляя чистую суспензию протопластов.
Далее к осажденным протопластам клеток добавляли 0,5 мл среды K3 и клетки ресуспендировали. Ресуспендированные протопласты клеток помещали в центр чашки Петри и добавляли 5 мл K3 и 0,6 мл агарозы Sea Plaque™ (Cambrex, East Rutherford, NJ) в концентрации 1:1. Чашки Петри встряхивали одним осторожным вращающим движением и оставляли инкубироваться в течение 20-30 минут при комнатной температуре. Чашки Петри накрывали Parafilm® и протопласты культивировали в течение двадцати четырех часов в полной темноте. После инкубации в темноте чашки Петри культивировали в течение шести суток при тусклом освещении (белые трубки Osram L36 W/21 Lumilux с 5 мкМоль м-2 с-1). После стадии культивирования использовали стерильный шпатель для разделения содержавших агарозу протопластов на квадранты. Отделенные квадранты помещали в 250-мл пластмассовую емкость для культивирования, содержавшую 20 мл среды A, и инкубировали на ротационном устройстве для встряхивания при 80 об./мин. и смещении 1,25 см при 24°C при постоянном тусклом освещении в течение 14 суток, а затем анализировали для определения уровня активности каждой конструкции нуклеазы с цинковыми пальцами.
ВЫДЕЛЕНИЕ ГЕНОМНОЙ ДНК ИЗ ПРОТОПЛАСТОВ КАНОЛЫ
Трансфицированные протопласты предоставляли в индивидуальных 1,5- или 2,0-мл микроцентрифужных пробирках. Клетки осаждали на дне пробирки в буферном растворе. Экстракцию ДНК проводили путем быстрого замораживания клеток в жидком азоте с последующей лиофилизацией клеток в течение приблизительно 48 часов в Labconco Freezone 4.5® (Labconco, Kansas City, MO) при -40°C и давлении приблизительно 133 × 10-3 мБар. Лиофилизированные клетки подвергали экстракции ДНК с использованием набора для растений DNeasy® (QIAGEN, Carlsbad, CA) в соответствии с инструкцией изготовителя, за исключением того, что разрушение ткани не требовалось и протопласты клеток добавляли непосредственно в лизирующий буфер.
ИССЛЕДОВАНИЕ ZFN ДЛЯ FAD2A и FAD3 В ОТНОШЕНИИ РАСЩЕПЛЕНИЯ ПОСЛЕДОВАТЕЛЬНОСТИ ГЕНОМНОЙ ДНК В ПРОТОПЛАСТАХ КАНОЛЫ
Смоделированные участки-мишени для ZFN в локусах FAD2A и FAD3 кластеризовали, так чтобы сконструировать множество пар ZFN для перекрывания участков-мишеней. Кластеризация участков-мишеней для ZFN обеспечила конструирование праймеров для ПЦР, которые амплифицируют окружающую геномную последовательность всех представителей семейства генов FAD2A и FAD3 в пределах окна размером 100 п.н., чтобы охватить все перекрывающиеся участки-мишени для ZFN. По существу, для оценки целостности участка-мишени для ZFN в трансфицированных протопластах можно использовать технологию короткого считывания последовательностей Illumina. Кроме того, необходимо, чтобы конструкции праймеров для ПЦР включали конкретные нуклеотидные основания, которые позволят соотнести данные считывания последовательностей с конкретным представителем семейств FAD2A и FAD3. Таким образом, требуется, чтобы все праймеры для ПЦР связывались на удалении 5-10 нуклеотидов от участка-мишени для расщепления посредством ZFN, поскольку известно, что активность негомологичного соединения концов (NHEJ) вызывает небольшие делеции, которые могут удалять участок затравки, ингибировать амплификацию и, таким образом, исказить оценку активности NHEJ.
Праймеры конструировали так, чтобы они связывались со всеми локусами-мишенями для ZFN из семейств генов FAD2A и FAD3 (таблица 11), и их эмпирически исследовали в отношении амплификации всех представителей семейства с помощью секвенирования продуктов амплификации способом ПЦР по методу Сэнгера. В нескольких случаях нельзя было разработать праймеры, которые различают всех представителей семейства генов (таблица 12 и таблица 13), однако во всех случаях последовательности гена-мишени FAD2A и FAD3, можно было различить. После конструирования праймеров для ПЦР в праймеры для ПЦР включали специализированные последовательности ДНК штрихового кода, которые использовали для различения различных локусов-мишеней для ZFN и идентификации конкретных считанных последовательностей для трансфекции и ZFN (таблицы 11, 12 и 13).

После экстракции ДНК протопластов канолы, трансфицированных ZFN, проводили амплификацию способом ПЦР локусов-мишеней для ZFN, чтобы получить необходимые специфичные к локусам молекулы ДНК в правильном формате для технологии секвенирования на основе IIlumina посредством синтеза. Каждый анализ оптимизировали для функционирования на 25 нг исходной ДНК (приблизительно 12500 клеточных эквивалентов генома Brassica napus). Проводили множество реакций на образец для обеспечения охвата, требуемого для оценки эффективности и специфичности NHEJ на соответствующем уровне: приблизительно шестнадцать реакций ПЦР, эквивалентных 200000 копиям генома Brassica napus, полученным из индивидуальных протопластов. Основные смеси для амплификации способом ПЦР получали для всех образцов, подлежащих исследованию с помощью одного и того же анализа, и анализировали одну реакцию, проведенную в трех экземплярах, с использованием способа количественной ПЦР, который использовали для определения оптимального количества циклов для проведения исследования на заданной ткани, чтобы гарантировать, что амплификация ПЦР не будет ограничиваться реагентом и все еще будет находится на стадии экспоненциальной амплификации. Экспериментирование с необходимыми отрицательными контрольными реакциями проводили в 96-луночном формате с использованием термоциклера MX3000P thermocycler® (Stratagene, LaJolla, CA). Из данных, полученных с помощью платформы количественной ПЦР, относительное увеличение флуоресценции наносили на график от цикла к циклу и определяли количество циклов на анализ, которое обеспечивало достаточную амплификацию, одновременно не позволяя реакции стать ограниченной реагентом, в попытках снизить избыточное циклирование и амплификацию общих транскриптов или молекул. Неиспользованную основную смесь, остававшуюся на льду до тех пор, пока не завершился анализ способом количественной ПЦР и не было определено количество циклов, распределяли аликвотами в желаемое количество реакционных пробирок (приблизительно 16 на анализ ZFN) и проводили реакцию ПЦР.
После амплификации образцы для одного локуса ZFN объединяли вместе и 200 мкл объеденного продукта на ZFN очищали с использованием набора MinElute PCR purification kit® (Qiagen) в соответствии с инструкциями изготовителя. Для обеспечения секвенирования образца с использованием технологии короткого считывания Illumina требовалось связывание дополнительных парных концевых праймеров путем амплификации с полученными фрагментами. Это обеспечивали амплификацией способом ПЦР с использованием праймеров, которые являются частично комплементарными последовательности, добавленной в первом раунде амплификации, но также содержат требуемую парную концевую последовательность. Оптимальное количество циклов проводимых ПЦР, которое добавит парные концевые последовательности к матрице без избыточной амплификации общих фрагментов, вновь определяли с использованием подвергания образца количественному анализу циклов ПЦР, как описано ранее.
После амплификации способом ПЦР полученный продукт очищали с использованием колонки MinElute column® (Qiagen) в соответствии с инструкциями изготовителя и разделяли на 2,5% агарозном геле. Фрагменты ДНК, визуализированные с использованием Syber® Safe (Life Technologies, Carlsbad, CA), в качестве полос правильного размера подвергали экстракции из геля для удаления каких-либо остаточных образовавшихся в ходе ПЦР димеров праймеров или других случайных фрагментов, ДНК экстрагировали из среза геля с использованием набора MinElute gel extraction kit® (Qiagen) в соответствии с инструкциями изготовителя. После завершения экстракции из геля проводили дополнительную очистку ДНК с использованием магнитных гранул AMPure magnetic beads® (Beckman-Coulter, Brea, CA) с соотношением ДНК и гранул 1:1,7. Затем ДНК оценивали в отношении концентрации с использованием набора для количественного определения библиотеки на основе количественной ПЦР, чтобы провести секвенирование с помощью Illumina (KAPA) с разведением 1/40000 и 1/80000 и с проведением реакции в трех экземплярах. Исходя из результатов количественной ПЦР ДНК разбавляли до стандартной концентрации 2 нМ и все библиотеки объединяли для секвенирования. Образцы приготавливали для секвенирования с использованием набора cBot cluster generation kit® (Illumina, San Diego, CA) и их секвенировали на Illumina GA2x® со считыванием в секвенировании с парными концами по 100 п.н. в соответствии с инструкциями изготовителя.
СПОСОБ АНАЛИЗА ДАННЫХ ДЛЯ ОБНАРУЖЕНИЯ НЕГОМОЛОГИЧНОГО СОЕДИНЕНИЯ КОНЦОВ В УЧАСТКАХ-МИШЕНЯХ ЦИНКОВЫХ ПАЛЬЦЕВ
После завершения реакции секвенирования и первичного сбора данных, проведенного с использованием биоинформатической системы Illumina для основного анализа, проводили полный анализ для идентификации удаленных оснований в участке-мишени для ZFN в каждом случае. Был разработан специализированный сценарий PERL для компьютерного извлечения и сортировки штриховых кодов из последовательностей ДНК по перечню входящих последовательностей. Штриховой код должен был совпадать с эталонной последовательностью с приемлемым показателем Phred более 30 для снижения неправильного сопоставления считанных последовательностей. После сортировки считанных последовательностей на две различных группы штриховых кодов, которые были использованы, все последовательности пропускали через фильтр качества. Фильтр качества был втором специально разработанным сценарием PERL. Считанные данные о последовательностях исключали, если существовало более трех оснований, обозначенных как "N", или если срединный показатель Phred составлял менее 20, или если 3 последовательно расположенных основания имели показатель Phred менее 20, или если считанная последовательность имела длину короче 40 п.н. Оставшиеся последовательности объединяли, где обе из считанных парных последовательностей были доступны, с использованием пакета программ NextGENe® (SoftGenetics, State College, PA). Затем оставшиеся объединенные считанные последовательности сводили к коллекции уникальных считанных последовательностей с использованием третьего специализированного сценария PERL с количеством избыточных последовательностей, которые были идентифицированы, записанным на конце оставшегося идентификатора последовательности. Затем уникальные считанные последовательности выравнивали с эталонной последовательностью FAD2 и FAD3 с использованием программного обеспечения NextGENe®, которое создавало файл выравнивания с пропусками FASTA.
С использованием файла с пропусками FASTA проводили преобразование положения оснований с пропусками во входной эталонный сигнал с использованием четвертого специализированного сценария PERL. Это обеспечило идентификацию оснований, которые различают различных представителей семейства генов (варьирование либо гомологичных, либо паралогичных последовательностей между различными представителями семейства генов) в собранных данных. После проведения преобразования нумерации оснований стало возможным получить отчет о гаплотипе для каждой из уникальных считанных последовательностей и отнести считанные последовательности к конкретным представителям семейства генов. После группировки считанных последовательностей по генам было идентифицировано окно из 10 п.н., и было оценено, что оно окружало участок-мишень для ZFN. Регистрировали количество последовательностей с делециями на ген вместе с количеством отсутствующих оснований.
Затем данные графически отображались в качестве графика с множеством кривых с количеством последовательностей с делецией с 1 по 10 основания на участок-мишень для ZFN на 10000 считанных последовательностей (Фиг. 6). Этот анализ проводили для всех трансфекций ZFN вместе с контрольными трансфекциями. В нескольких случаях повторы в нативной последовательности ДНК приводили к увеличению ошибок секвенирования в участке-мишени для ZFN, такую ошибку обычно можно увидеть в качестве увеличения распространенности делеций единичных оснований, которые были сообщены для всех образцов, как для трансфицированных ZFN, так и для контролей (Фиг. 7).
Из этих результатов наиболее высокий уровень активности ZFN в участке-мишени FAD2 при определении по наибольшей активности NHEJ, был идентифицирован в локусе E. ZFN, которые кодировались на плазмиде pDAB104010 (т.е., ZFN24828 и 24829), отбирали для нацеливания в растениях на сконструированную способами инженерии платформу для встраивания трансгена (ETIP), учитывая их характеристики значительной активности расщепления геномной ДНК и минимальной активности вне мишени.
ПРИМЕР 4: КОНСТРУКЦИИ ДНК ДЛЯ СКОНСТРУИРОВАННОЙ СПОСОБАМИ ИНЖЕНЕРИИ ПЛАТФОРМЫ ДЛЯ ВСТРАИВАНИЯ ТРАНСГЕНА (ETIP) В ЛИНИЯХ РАСТЕНИЙ КАНОЛЫ
Конструкции плазмидных векторов, описанные ниже, конструировали с использованием способов и технологий, широко известных специалистам в данной области. Использование конкретных реагентов и способов, описанных в этом разделе, хорошо известно специалистам в данной области, и они могут быть без труда заменены другими реагентами и способами для достижения желаемой цели конструирования конструкций плазмидных векторов. Эндонуклеазы рестрикции приобретали от New England BioLabs (NEB; Ipswich, MA). Лигирование осуществляли с помощью ДНК-лигазы T4 (Invitrogen, Carlsbad, CA). Реакции Gateway проводили с использованием смеси ферментов GATEWAY® LR CLONASE® (Invitrogen) для сбора одного исходного вектора в единый конечный вектор. Реакции IN-FUSIONTM проводили с использованием технологии IN-FUSIONTM Advantage Technology (Clontech, Mountain View, CA) для сбора одного исходного вектора в единый конечный вектор. Получение плазмид проводили с использованием набора NUCLEOSPEN® Plasmid Kit (Macherey-Nagel Inc., Bethlehem, PA) или Plasmid Midi Kit® (Qiagen) в соответствии с инструкциями поставщиков. Фрагменты ДНК выделяли с использованием набора QIAquick Gel Extraction Kit™ (Qiagen) после электрофореза в агарозном Tris-ацетатном геле. Колонии всех собранных плазмид первоначально подвергали скринингу посредством рестрикционного расщепления ДНК Miniprep. Плазмидную ДНК выбранных клонов секвенировала коммерческая организация, осуществляющая секвенирование (Eurofins MWG Operon, Huntsville, AL). Данные о последовательности собирали и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes Corp., Ann Arbor, MI).
ВЕКТОРЫ ДЛЯ ПРЯМОЙ ДОСТАВКИ ДЛЯ ПРИЦЕЛЬНОГО ВСТРАИВАНИЯ ETIP В ЛОКУС FAD2A КАНОЛЫ
Для конструирования содержащих ΕTIΡ векторов pDAS000130 (Фиг. 6, вставка T-цепи в качестве SEQ ID NO: 61) для специфического встраивания в ген FAD2A B. napus использовали стандартные способы клонирования. Эту конструкцию конструировали для доставки в протопласты канолы конструкции нуклеазы с цинковыми пальцами pDAB104010. Конструкция нуклеазы с цинковыми пальцами расщепляет локус FAD2A, а затем конструкция pDAS000130 встраивается в геном конолы посредством механизма направляемой гомологией репарации, и ее можно модифицировать путем удаления фланкирующих областей гомологии для опосредуемого NHEJ встраивания. ETIP состоит из четырех экспрессирующих кассет (две неполных), разделенных дополнительными последовательностями распознавания ZFN и сконструированной способами инженерии подушкой для встраивания (ELP), содержащей другие последовательности распознавания ZFN. Дополнительные последовательности распознавания ZFN являются уникальными, и они были сконструированы для нацеливания встраивания полинуклеотидных последовательностей во вставки трансгенов ETIP и ELP. Аналогично, последовательности распознавания ZFN можно использовать для вырезания полинуклеотидных последовательностей. Первая экспрессирующая ген кассета представляла собой неполную экспрессирующую кассету dsRED, и она содержала промотор, 5'-нетранслируемую область и интрон из гена полиубиквитина 10 (промотор AtUbi) Arabidopsis thaliana (Callis, et al., (1990) J. Biol. Chem., 265: 12486-12493), за которыми следовали 210 п.н. гена dsRed из рифовых кораллов Discosoma sp. (Clontech, Mountain View, CA), кодон-оптимизированного для экспрессии в двудольных растениях (dsRED (оптимизированный для двудольных растений) экзон 1), за которыми следовали интрон из тиоредуктаза-подобного гена Arabidopsis thaliana (интрон 1 из тиоредуктазы At: номер доступа №: NC_00374) и 3'-нетранслируемая область, содержащая терминатор транскрипции и участок полиаденилирования гена Viviparous-1 (Vp1) Zea mays (терминатор Zmlip: Paek et al., (1998) Molecules and Cells, 8(3): 336-342). Вторая экспрессирующая кассета содержала промотор 19S, включая 5'-UTR из вируса мозаики цветной капусты (CaMV 19S: Cook and Penon (1990) Plant Molecular Biology 14(3): 391-405), за которым следовал ген hph из E. coli, кодон-оптимизированный для экспрессии в двудольных растениях (hph(HygR): Kaster et al, (1983) Nucleic Acids Research 11(19): 6895-691 1) и 3-'UTR, содержащая терминатор транскрипции и участок полиаденилирования открытой рамки считывания 1 A. tumefaciens pTi15955 (терминатор At-ORF1: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Третья экспрессирующая кассета представляла собой неполную экспрессирующую кассету PAT, и она содержала первый интрон из 4-кумарил-CoA-синтазы Arabidopsis (интрон #2 4-кумарил-CoA-синтазы v: номер доступа № At3g21320/NC003074), за которым следовали последние 256 п.н. синтетической оптимизированной для растений версии гена фосфинотрицинацетилтрансферазы, выделенной из Streptomyces viridochromogenes, которые кодируют белок, который сообщает устойчивость к ингибиторам глутаминсинтетазы, включающим фосфинотрицин, глюфосинат и биалафос (3'-конец PAT(v6): Wohlleben et al, (1988) Gene 70(1): 25-37). Эта кассета терминировалась 3'-UTR, содержащей терминатор транскрипции и участки полиаденилирования открытой рамки считывания 23 pTi15955 A tumefaciens (терминатор AtuORF23: Barker et al, (1983), Plant Molecular Biology 2(6): 335-50). Четвертая экспрессирующая кассета представляла собой кассету с геном ipt, и она содержала укороченную версию промотора размером 588 п.н. и 5'-UTR из гена MYB32 ДНК-связывающего белка Arabidopsis (U26933) (промотор AtMYB32(T): Li et al., (1999) Plant Physiology 121:313), за которыми следовал ген изопентилтрансферазы (ipt) из A. tumefaciens и терминатор 35s, содержащий терминатор транскрипции и участки полиаденилирования из вируса мозаики цветной капусты (терминатор CaMV 35S: Chenault et al, (1993) Plant Physiology 101 (4): 1395-1396). Для доставки в FAD2A, каждый конец последовательности ETIP был фланкирован 1 т.п.н. геномной последовательности FAD2A с каждой стороны от положения двухцепочечного разрыва, индуцируемого доставкой ZFN, кодируемой в pDAB104010, в гене FAD2A B. napus.
Последовательность ETIP синтезировала коммерческая организация, осуществляющая синтез генов (GeneArt, Life Technologies). Сегменты размером 1 т.п.н. геномной последовательности FAD2A амплифицировали с геномной ДНК, очищенной из ткани листьев B. napus DH12075, с использованием набора Qiagen DNeasy plant mini kit® (Qiagen, Hilden) в соответствии с инструкциями, предоставленными поставщиком. Последовательности FAD2A размером 1 т.п.н. лигировали в вектор ΕΤIΡ с использованием лигазы T4 (NEB, Ipswich, MA). Колонии всех собранных плазмид первоначально подвергали скринингу путем рестрикционного расщепления ДНК Miniprep. Эндонуклеазы рестрикции получали от New England BioLabs (NEB, Ipswich, MA) и Promega (Promega Corporation, WI). Получение плазмид проводили с использованием набора QIAprep Spin Miniprep Kit® (Qiagen) или системы Pure Yield Plasmid Maxiprep System® (Promega Corporation, WI) в соответствии с инструкциями поставщиков. Плазмидную ДНК выбранных клонов секвенировали с использованием секвенирования по методу Сэнгера ABI и протокола секвенирования Big Dye Terminator v3.1 cycle sequencing protocol® (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes Corp., Arm Arbor, MI).
ВЕКТОРЫ ДЛЯ ПРЯМОЙ ДОСТАВКИ ДЛЯ ПРИЦЕЛЬНОГО ВСТРАИВАНИЯ ETIP В ЛОКУС FAD3 КАНОЛЫ
Для конструирования содержащих ΕTIΡ векторов pDAS000271 (Фиг. 9, вставка T-цепи в виде SEQ ID NO: 142), pDAS000272 (Фиг. 10, вставка T-цепи в виде SEQ ID NO: 143), pDAS000273 (Фиг. 11, вставка T-цепи в виде SEQ ID NO: 144), pDAS000274 (Фиг. 12, вставка T-цепи в виде SEQ ID NO: 145) и pDAS000275 (Фиг. 13, вставка T-цепи в виде SEQ ID NO: 146) для специфического встраивания в локус генов FAD3A или FAD3C B. napus использовали стандартные способы клонирования. Эту конструкцию конструировали для доставки в протопласты канолы конструкции нуклеазы с цинковыми пальцами pDAB107828 или pDAB107829. Конструкция специфической нуклеазы с цинковыми пальцами расщепляет локус FAD3A, а затем конструкция pDAS000273 или pDAS275 встраивается в геном конолы посредством механизма направляемой гомологией репарации. Аналогично, конструкция специфической нуклеазы с цинковыми пальцами расщепляет локус FAD3С, а затем конструкция pDAS000271, pDAS000272 или pDAS000274 встраивается в геном конолы посредством механизма направляемой гомологией репарации. ETIP состоит из четырех экспрессирующих кассет (две неполных), разделенных дополнительными последовательностями распознавания ZFN и сконструированной способами инженерии подушкой для встраивания (ELP), содержащей другие последовательности распознавания ZFN. Дополнительные последовательности распознавания ZFN являются уникальными, и они были сконструированы для нацеливания встраивания полинуклеотидных последовательностей во вставки трансгенов ETIP и ELP. Аналогично, последовательности распознавания ZFN можно использовать для вырезания полинуклеотидных последовательностей. Первая экспрессирующая ген кассета представляла собой неполную экспрессирующую кассету dsRED, и она содержала промотор, 5'-нетранслируемую область и интрон из гена полиубиквитина 10 (промотор AtUbi) Arabidopsis thaliana (Callis, et al., (1990) J. Biol. Chem., 265: 12486-12493), за которыми следовали 210 п.н. гена dsRed из рифовых кораллов Discosoma sp. (Clontech, Mountain View, CA), кодон-оптимизированного для экспрессии в двудольных растениях (dsRED (оптимизированный для двудольных растений) экзон 1), за которыми следовали интрон из тиоредуктаза-подобного гена Arabidopsis thaliana (интрон 1 из тиоредуктазы At: номер доступа №: NC_00374) и 3'-нетранслируемая область, содержащая терминатор транскрипции и участок полиаденилирования гена Viviparous-1 (Vp1) Zea mays (терминатор Zmlip: Paek et al., (1998) Molecules and Cells, 8(3): 336-342). Вторая экспрессирующая кассета содержала промотор 19S, включая 5'-UTR из вируса мозаики цветной капусты (CaMV 19S: Cook and Penon (1990) Plant Molecular Biology 14(3): 391-405), за которым следовал ген hph из E. coli, кодон-оптимизированный для экспрессии в двудольных растениях (hph(HygR): Kaster et al, (1983) Nucleic Acids Research 11(19): 6895-691 1) и 3'-UTR, содержащая терминатор транскрипции и участок полиаденилирования открытой рамки считывания 1 A. tumefaciens pTi15955 (терминатор At-ORF1: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Третья экспрессирующая кассета представляла собой неполную экспрессирующую кассету PAT, и она содержала первый интрон из 4-кумарил-CoA-синтазы Arabidopsis (интрон #2 4-кумарил-CoA-синтазы v: номер доступа № At3g21320/NC003074), за которым следовали последние 256 п.н. синтетической оптимизированной для растений версии гена фосфинотрицинацетилтрансферазы, выделенной из Streptomyces viridochromogenes, которые кодируют белок, который сообщает устойчивость к ингибиторам глутаминсинтетазы, включающим фосфинотрицин, глюфосинат и биалафос (3'-конец PAT(v6): Wohlleben et al, (1988) Gene 70(1): 25-37). Эта кассета терминировалась 3'-UTR, содержащей терминатор транскрипции и участки полиаденилирования открытой рамки считывания 23 pTi15955 A tumefaciens (терминатор AtuORF23: Barker et al, (1983), Plant Molecular Biology 2(6): 335-50). Четвертая экспрессирующая кассета представляла собой кассету с геном ipt, и она содержала укороченную версию промотора размером 588 п.н. и 5'-UTR из гена MYB32 ДНК-связывающего белка Arabidopsis (U26933) (промотор AtMYB32(T): Li et al., (1999) Plant Physiology 121:313), за которыми следовал ген изопентилтрансферазы (ipt) из A. tumefaciens и терминатор 35s, содержащий терминатор транскрипции и участки полиаденилирования из вируса мозаики цветной капусты (терминатор CaMV 35S: Chenault et al, (1993) Plant Physiology 101 (4): 1395-1396). Для доставки в FAD3A или FAD3C, каждый конец последовательности ETIP был фланкирован 1 т.п.н. геномной последовательности FAD3A или FAD3C с каждой стороны от положения двухцепоченого разрыва, индуцируемого доставкой ZFN, кодируемой в FAD3A или FAD3c B. napus.
Последовательность ETIP синтезировала коммерческая организация, осуществляющая синтез генов (GeneArt, Life Technologies). Сегменты размером 1 т.п.н. геномной последовательности FAD3A и FAD3C амплифицировали с геномной ДНК, очищенной из ткани листьев B. napus DH12075, с использованием набора Qiagen DNeasy plant mini kit® (Qiagen, Hilden) в соответствии с инструкциями, предоставленными поставщиком. Последовательности FAD3A или FAD3C размером 1 т.п.н. лигировали в вектор ΕΤIΡ с использованием лигазы T4 (NEB, Ipswich, MA). Колонии всех собранных плазмид первоначально подвергали скринингу путем рестрикционного расщепления ДНК Miniprep. Эндонуклеазы рестрикции получали от New England BioLabs (NEB, Ipswich, MA) и Promega (Promega Corporation, WI). Получение плазмид проводили с использованием набора QIAprep Spin Miniprep Kit® (Qiagen) или системы Pure Yield Plasmid Maxiprep System® (Promega Corporation, WI) в соответствии с инструкциями поставщиков. Плазмидную ДНК выбранных клонов секвенировали с использованием секвенирования по методу Сэнгера ABI и протокола секвенирования Big Dye Terminator v3.1 cycle sequencing protocol® (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали с использованием программного обеспечения SEQUENCHER™ (Gene Codes Corp., Arm Arbor, MI).
КОНТРОЛЬНЫЕ ВЕКТОРЫ
Контрольный вектор использовали для разработки способа сортировки клеток на основе активированной флуоресценцией сортировки клеток (FACS). Для конструирования контрольного вектора pDAS000031 (Фиг. 14: вставка T-цепи в качестве SEQ ID NO: 147), состоящего из двух экспрессирующих кассет для генов, использовали стандартные способы клонирования. Первая экспрессирующая кассета для гена содержала промотор вируса мозаики цветной капусты 19s (промотор CaMV 19S; Shillito, et al., (1985) Bio/Technology 3; 1099-1103) :: ген устойчивости к гигромицину (hph(HygR); патент США №4727028) :: и 3'-нетранслируемую область открытой рамки считывания 1 Agrobacterium tumefaciens (терминатор AtORF1; Huang et al, (1990) J. Bacteriol. 1990 172:1814-1822). Вторая экспрессирующая кассета для гена содержала промотор убиквитина 10 Arabidopsis thaliana (промотор AtUbi10; Callis, et al., (1990) J. Biol Chem., 265: 12486-12493):: dsRED (dsRED(D); патент США № 6852849) и интрон из Arabidopsis (интрон #1; GenBank: AB025639.1) :: 3'-нетранслируемую область открытой рамки считывания 23 Agrobacterium tumefaciens (терминатор AtORF23; патент США №5428147) в качестве слитой в рамке считывания конструкции с транс-ориентацией (например, ориентация "голова к голове"). Плазмидный вектор собирали с использованием технологии IΝ-FUSION™ Advantage Technology (Clontech, Mountain View, CA).
КОНСТРУИРОВАНИЕ БИНАРНЫХ ВЕКТОРОВ ДЛЯ СЛУЧАЙНОГО ВСТРАИВАНИЯ ETIP В КАНОЛЕ
Конструировали два бинарных вектора для случайного встраивания последовательности T-цепи ETIP в геном Brassica napus. При конструировании содержащих ETIP векторов pDAS000036 (Фиг. 15, вставка T-цепи в виде SEQ ID NO: 148) и pDAS000037 (Фиг. 16, вставка T-цепи в виде SEQ ID NO: 149) использовали стандартные способы клонирования. Векторы ETIP состоят из четырех экспрессирующих кассет (две неполных экспрессирующих кассеты), разделенных последовательностями распознавания ZFN и сконструированной способами инженерии подушкой для встраивания (ELP), содержащей другие последовательности распознавания ZFN. Дополнительные последовательности распознавания ZFN являются уникальными, и они были сконструированы для нацеливания встраивания полинуклеотидных последовательностей во вставки трансгенов ETIP и ELP. Аналогично, последовательности распознавания ZFN можно использовать для вырезания полинуклеотидных последовательностей. Первая экспрессирующая ген кассета представляла собой неполную экспрессирующую кассету dsRED, и она содержала промотор, 5'-нетранслируемую область и интрон из гена полиубиквитина 10 (промотор AtUbi) Arabidopsis thaliana (Norris et al., (1993) Plant Molecular Biology, 21(5): 895-906), за которыми следовали 210 п.н. гена dsRed из рифовых кораллов Discosoma sp. (Clontech, Mountain View, CA), кодон-оптимизированного для экспрессии в двудольных растениях, за которыми следовали интрон из тиоредуктаза-подобного гена Arabidopsis thaliana (номер доступа №: NC_00374) и 3'-нетранслируемая область, содержащая терминатор транскрипции и участок полиаденилирования гена Viviparous-1 (Vp1) Zea mays (терминатор Zmlip: Paek et al., (1998) Molecules and Cells, 8(3): 336-342). Вторая экспрессирующая кассета содержала промотор 19S, включая 5'-UTR из вируса мозаики цветной капусты (CaMV 19S: Cook and Penon (1990) Plant Molecular Biology 14(3): 391-405), за которым следовал ген hph из E. coli, кодон-оптимизированный для экспрессии в двудольных растениях (hph(HygR): Kaster et al, (1983) Nucleic Acids Research 11(19): 6895-691 1) и 3-'UTR, содержащая терминатор транскрипции и участок полиаденилирования открытой рамки считывания 1 A. tumefaciens pTi15955 (терминатор At-ORF1: Barker et al., (1983) Plant Molecular Biology 2(6): 335-50). Третья экспрессирующая кассета представляла собой неполную экспрессирующую кассету PAT, и она содержала первый интрон из 4-кумарил-CoA-синтазы Arabidopsis (номер доступа № At3g21320/NC003074), за которым следовали последние 256 п.н. синтетической оптимизированной для растений версии гена фосфинотрицинацетилтрансферазы, выделенной из Streptomyces viridochromogenes, которые кодируют белок, который сообщает устойчивость к ингибиторам глутаминсинтетазы, включающим фосфинотрицин, глюфосинат и биалафос (PATv6 (экзон 2): Wohlleben et al, (1988) Gene 70(1): 25-37). Эта кассета терминировалась 3'-UTR, содержащей терминатор транскрипции и участки полиаденилирования открытой рамки считывания 23 pTi15955 A tumefaciens (терминатор AtuORF23: Barker et al, (1983), Plant Molecular Biology 2(6): 335-50). Четвертая экспрессирующая кассета представляла собой кассету с геном ipt, и она содержала укороченную версию промотора размером 588 п.н. и 5'-UTR из гена MYB32 ДНК-связывающего белка Arabidopsis (U26933) (промотор AtMYB32(T): Li et al., (1999) Plant Physiology 121:313), за которыми следовал ген изопентилтрансферазы (ipt) из A. tumefaciens и терминатор 35s, содержащий терминатор транскрипции и участки полиаденилирования из вируса мозаики цветной капусты (терминатор CaMV 35S: Chenault et al, (1993) Plant Physiology 101 (4): 1395-1396).
Экспрессирующие кассеты и ELP с участками Multi-Gateway синтезировала коммерческая организация, осуществляющая синтез генов (GeneArt, Life Technologies). Исходные клоны конструировали для каждой экспрессирующей кассеты и ELP с использованием смеси ферментов BP clonase II enzyme mix™ (Invitrogen, Life Technologies) и pDONR221 vector suite™ (Invitrogen, Life Technologies). Затем исходные клоны использовали в реакции Multi-Gateway с предоставленным Gateway бинарным вектором с использованием смеси ферментов LR Clonase II Plus Enzyme mix™ (Invitrogen, Life Technologies). Колонии всех собранных плазмид первоначально подвергали скринингу путем рестрикционного расщепления ДНК Miniprep. Эндонуклеазы рестрикции получали от New England BioLabs (NEB; Ipswich, MA)и Promega (Promega Corporation, WI). Получение плазмид проводили с использованием набора QIAprep Spin Miniprep Kit™ (Qiagen, Hilden) или системы Pure Yield Plasmid Maxiprep System™ (Promega Corporation, WI) в соответствии с инструкциями поставщиков. Плазмидную ДНК выбранных клонов секвенировали с использованием секвенирования по методу Сэнгера ABI и протокола секвенирования Big Dye Terminator v3.1 cycle sequencing protocol™ (Applied Biosystems, Life Technologies). Данные о последовательностях собирали и анализировали с использованием программного обеспечения Sequencher™ (Gene Codes Corporation, Ann Arbor, MI).
ПРИМЕР 5: ПОЛУЧЕНИЕ ЛИНИЙ РАСТЕНИЙ КАНОЛЫ С ETIP
ТРАНСФОРМАЦИЯ Brassica napus
Конструкции ETIP (pDAS000036, pDAS000037), контрольная конструкция DS-Red (pDAS000031) и сайт-специфические конструкции FAD2A, FAD3A и FAD3C (pDAS000130 и pDAS000271-pDAS000275) и соответствующие нуклеазы с цинковыми пальцами (pDAB104010, pDAB10728 и pDAB10729) описаны в примере 4. Бинарные векторы трансформировали в Agrobacterium tumefaciens штамма GV3101: PM90. Трансформацию клеток протопластов Brassica napus осуществляли с использованием протокола трансфекции, описанного в примере 3, с некоторой модификацией.
Модификации протокола включали использование альгината натрия вместо агарозы Sea Plaque™. Эксперименты по трансфекции, в которых как конструкцию нуклеазы с цинковыми пальцами, так и конструкцию ETIP совместно доставляли в клетки протопластов Brassica napus, осуществляли в концентрациях плазмидной ДНК, составляющих молярное соотношение 5:1. Другие конструкции ETIP и контрольные плазмидные конструкции трансформировали в концентрациях 30 мкг плазмидной ДНК. По существу, pDAS000130 имела концентрацию 27,8 мкг плазмидной ДНК и pDAB104010 имела концентрацию 2,2 мкг плазмидной ДНК. Другие конструкции ETIP и контрольные плазмидные конструкции трансформировали в концентрации 30 мкг плазмидной ДНК.
Дополнительные модификации протокола включали восстановление целых растений из трансформированных протопластов клеток в среде, содержавшей 1,5 мг/мл гигромицина. Восстановление целых растений требовало, чтобы среду A заменяли каждые две недели и проводили мониторинг роста происходящих из протопластов колоний. После вырастания происходящих из протопластов колоний до размера приблизительно 2-3 мм в диаметре колонии переносили в индивидуальные ячейки планшета с 12 ячейками Costar® (Fisher Scientific, St. Louis, MO), содержавшие застывшую среду MS morpho. Планшеты инкубировали в течение от одной до двух недель при 24°C при непрерывном тусклом освещении до тех пор, пока каллюсы не пролиферировали до размера 8-10 мм в диаметре.
После достижения протопластами клеток диаметра 1-2 см протопласты клеток переносили в индивидуальные 250-мл емкости для культивирования, содержавшие среду MS morpho. Емкости инкубировали при 24°C в условиях 16 ч на свету (белые трубки Osram L36 W/21 Lumilux с 20 мкМоль м-2 с-1) и 8 ч в темноте. В течение от одной до двух недель становилось видно множество побегов. Побеги переносили в 250-мл емкости для культивирования, содержавшие среду MS, после того, как они достигали длины 3-4 см. 250-мл емкости для культивирования инкубировали при 24°C в условиях 16 ч на свету (белые трубки Osram L36 W/21 Lumilux с 20 мкМоль м-2 с-1) и 8 ч в темноте. Побеги поддерживали в емкостях для культивирования до тех пор, пока они не развивались в проростки, и в это время их переносили в теплицу для выращивания до зрелого состояния.
ПРИМЕР 6: МОЛЕКУЛЯРНОЕ ПОДТВЕРЖДЕНИЕ ВСТРАИВАНИЯ T-ДНК, СОДЕРЖАЩИХ ETIP, В КАНОЛЕ
МОЛЕКУЛЯРНОЕ ПОДТВЕРЖДЕНИЕ СЛУЧАЙНОГО ВСТРАИВАНИЯ ETIP В КАНОЛЕ
Геномную ДНК экстрагировали из тканей листьев всех предполагаемых трансгенных растений с использованием набора для экстракции ДНК DNeasy 96 Plant DNA extraction kit™ или набора DNeasy Plant Mini Kit™ (Qiagen). Геномную ДНК из каждого растения анализировали способом ПЦР с использованием праймеров, сконструированных для амплификации virC: pTiC58 Forward (SEQ ID NO: 150 CGAGAACTTGGCAATTCC) и pTiC58 Reverse (SEQ ID NO: 151 TGGCGATTCTGAGATTCC) для исследования устойчивости в A.tumfaciens, праймеров, сконструированных для амплификации актина из B. napus: Actin Forward (SEQ ID NO: 152 GACTCATCGTACTCTCCCTTCG) и Actin Reverse (SEQ ID NO: 153 GACTCATCGTACTCTCCCTTCG) для проверки качества геномной ДНК. Конструировали праймеры для амплификации гена hph: HPH Forward (SEQ ID NO: 154 TGTTGGTGGAAGAGGATACG) и HPH Reverse (SEQ ID NO: 155 ATCAGCAGCAGCGATAGC), кодируемого ETIP. Растения, которые не давали продукта с праймеров virC, но из которых амплифицировались продукты правильного размера с помощью праймеров к актину и hph, классифицировали как трансгенные.
Проводили второй скрининг, где гДНК из каждого геномного растения анализировали способом ПЦР с использованием пяти наборов праймеров, сконструированных для амплификации бинарного вектора вне области T-ДНК [(1F SEQ ID NO: 156 ATGTCCACTGGGTTCGTGCC; 1R SEQ ID NO: 157 GAAGGGAACTTATCCGGTCC) (2F SEQ ID NO: 158 TGCGCTGCCATTCTCCAAAT; 2R SE ID NO:159 ACCGAGCTCGAATTCAATTC) (3F SEQ ID NO: 160 CCTGCATTCGGTTAAACACC; 3R SEQ ID NO: 161 CCATCTGGCTTCTGCCTTGC) (4F SEQ ID NO: 162 ATTCCGATCCCCAGGGCAGT; 4R SEQ ID NO: 163 GCCAACGTTGCAGCCTTGCT) (5F SEQ ID NO: 164 GCCCTGGGATGTTGTTAAGT; 5R SEQ ID NO: 165 GTAACTTAGGACTTGTGCGA)]. Растения, из которых амплифицировались продукты ПЦР правильного и ожидаемого размера с наборов праймеров 3 и 4, считали имеющими встраивание в остов.
ДНК из растений без встраивания в остов очищали из 20 г ткани листьев с использованием модифицированного способа CTAB (Maguire et al,. (1994) Plant Molecular Biology Reporter, 12(2): 106-109). Выделенную гДНК расщепляли с помощью нескольких ферментов рестрикции и 10 мкг гДНК разделяли электрофорезом на агарозном геле и переносили на мембрану с использованием стандартного протокола саузерн-блоттинга. Мембраны обрабатывали зондами с использованием системы DIG Easy Hyb System™ (Roche, South San Francisco, CA) в соответствии с инструкциями изготовителя. Зонды для каждой экспрессирующей кассеты, для ELP и для эндогенного контрольного гена актина амплифицировали с конструкции ETIP с использованием следующих праймеров: (IPT-F SEQ ID NO: 166 TCTCTACCTTGATGATCGG; IPT-R SEQ ID NO: 167 AACATCTGCTTAACTCTGGC; dsRED-F SEQ ID NO: 168 ATGGCTTCATCTGAGAACG; dsRED-R SEQ ID NO: 169 TTCCGTATTGGAATTGAGG; PAT-F SEQ ID NO: 170 TTGCTTAAGTCTATGGAGGCG; PAT-R SEQ ID NO: 171 TGGGTAACTGGCCTAACTGG; ELP-F SEQ ID NO: 172 ATGATATGTAGACATAGTGGG; ELP-R SEQ ID NO: 173 AGGGTGTAAGGTACTAGCC; Hph-F SEQ ID NO: 174 TGTTGGTGGAAGAGGATACG; Hph-R SEQ ID NO: 175 ATCAGCAGCAGCGATAGC; actin-F SEQ ID NO: 176 GTGGAGAAGAACTACGAGCTACCC; actin-R SEQ ID NO: 177 GACTCATCGTACTCTCCCTTCG).
Последовательность ETIP амплифицировали и секвенировали из всех растений, содержавших только одну копию ETIP. Последовательность каждой вставки T-ДНК анализировали путем прямого секвенирования продуктов ПЦР с использованием ABI3730xI™ (Applied Biosystems, Life Teachnologies). Вставку T-ДНК амплифицировали из геномной ДНК с использованием Phusion Hot Start II Polymerase™ (Finnzymes, Thermo Fisher Scientific). Реакции амплификации T-ДНК проводили с несколькими парами праймеров для амплификации перекрывающихся последовательностей длиной приблизительно 2 т.п.н. Каждый продукт ПЦР секвенировали с помощью множества праймеров для обеспечения полного охвата. Перед реакцией секвенирования с помощью ПЦР реакционные смеси ПЦР обрабатывали щелочной фосфатазой креветок и экзонуклеазой I (Applied Biosystems, Life Technologies) для инактивации избытка праймеров. Последовательности, фланкирующие вставку T-ДНК каждой единичной копии ETIP, идентифицировали путем расщепления геномной ДНК восемью эндонуклеазами рестрикции с последующим лигированием двухцепочечных адаптеров, специфичных к выступающим концам, внесенным с помощью эндонуклеаз рестрикции. После этой стадии лигирования ПЦР проводили с биотинилированным праймером либо к 3'-, либо к 5'-концу ETIP, и праймером к каждому адаптору. Продукты ПЦР улавливали и очищали на гранулах Ampure Solid Phase Reversible Immobilization (SPRI) beads™ (Agencourt Bioscience Corporation, Beckman Coulter Company). Проводили гнездовую ПЦР, и все продукты секвенировали с использованием секвенирования по методу Сэнгера ABI и протокола секвенирования Big Dye Terminator v3.1 cycle™ (Applied Biosystems, Life Technologies). Данные о последовательностях объединяли и анализировали с использованием программного обеспечения Sequencher™ (Gene Codes Corp., Ann Arbor, MI).
АНАЛИЗ С ИСПОЛЬЗОВАНИЕМ САУЗЕРН-БЛОТТИНГА
Выбирали специфические ферменты рестрикции для расщепления образцов гДНК перед обработкой зондами для саузерн-анализа. Предполагаемые трансгенные растения анализировали путем расщепления геномной ДНК посредством EcoRI и SwaI. Далее расщепленную гДНК и неразрезанные образцы гДНК обрабатывали любым из полинуклеотидных фрагментов, содержащих элементы генов PATv6, IPT или ELP, поскольку эти полинуклеотидные фрагменты-зонды обеспечили различение множества вставок в продуктах расщепления EcoRI, а также в продуктах расщепления SwaI. Затем идентифицированные трансгенные линии растений с единичными копиями далее анализировали с помощью шести зондов для идентификации присутствия всех необходимых элементов встроенного вектора.
Таким образом, проводили взятие образцов для 67 независимых событий трансформации ETIP-pDAS000036, и их исследовали в отношении присутствия трансгена (hph) и присутствия остова вектора. Было выявлено, что из 67 исследованных растений 47 имеют трансген, встроенный в геном. Было выявлено, что из 47 трансгенных растений 17 растений содержат остов вектора (таблица 14). Из оставшихся 30 растений, которые не содержали значительной части остова вектора (отсутствие Ori или SpecR) брали образцы для саузерн-анализа. В качестве общего правила, растения сначала подвергали скринингу с использованием зонда IPT, и линии растений, идентифицированные в качестве предполагаемых линий с одной копией, далее исследовали с помощью зондов, содержащих элементы генов dsRED, PAT, ELP и hph, для подтверждения присутствия всей кассеты.
Аналогично, проводили взятие образцов для 52 независимых событий трансформации ETIP-pDAS000037 с выживанием в почве и проводили их исследование в отношении присутствия трансгена (hph) и присутствия остова вектора. Было выявлено, что из 52 исследованных растений 48 имеют трансген, встроенный в геном. Было выявлено, что из 48 трансгенных растений 23 растения также содержат остов вектора и 3 растения не исследовали (таблица 14). Для остальных 22 растений, которые не содержали значительной части остова вектора (отсутствие Ori или SpecR) проводили взятие образцов для саузерн-анализа. Эти трансгенные растения первоначально подвергали скринингу с использованием зонда IPT, и линии растений, идентифицированные в качестве предполагаемых линий с одной копией, далее исследовали с помощью зондов для dsRED, PAT, ELP и hph, для подтверждения присутствия всей кассеты. После идентификации 5 независимых линий с одной копией саузерн-анализ завершали для остальных растений. Всего саузерн-анализ проводили для 11 линий ETIP-pDAS000037.
РЕЗУЛЬТАТЫ ДЛЯ КАНОЛЫ С ТРАНСГЕННЫМ ETIP, ТРАНСФОРМИРОВАННОЙ pDAS000036 и pDAS000037
Трансгенные события Brassica napus, которые осуществляли путем трансформации pDAS000036 и pDAS000037, приводили к образованию полноразмерных вставок T-цепи в одной копии. От трех до четырех событий для каждого растения полностью охарактеризовывали и картировали предположительно на конкретных хромосомах в геноме Brassica napus. Хотя в ходе встраивания T-цепи происходили единичные перестройки пар оснований, выбранные события включали полноразмерные экспрессирующие кассеты, которые способны обеспечивать устойчивую экспрессию трансгена. Выбранные растения с событиями T0 выращивали до стадии развития T1. T1 подвергали повторному скринингу с использованием описанных выше анализов способом ПЦР для определения зиготности встроенной T-цепи. После скрининга события классифицировали как гомозиготные, гемизиготные или нулевые.
Последовательность ETIP амплифицировали и секвенировали с всех трансгенных событий, содержащих только одну копию встроенной последовательности ETIP. Последовательность каждой вставки T-ДНК анализировали путем прямого секвенирования продуктов ПЦР. Вставку T-ДНК амплифицировали с геномной ДНК с использованием полимеразы Phusion Hot Start II PolymeraseTM (Finnzymes, Thermo Fisher Scientific). Далее T-ДНК амплифицировали с помощью нескольких пар праймеров для амплификации перекрывающихся последовательностей длиной приблизительно 2 т.п.н. Каждый продукт ПЦР секвенировали с помощью нескольких праймеров для обеспечения полного охвата. Перед реакцией секвенирования с помощью ПЦР реакционные смеси ПЦР обрабатывали щелочной фосфатазой креветок и экзонуклеазой I (Applied Biosystems, Life Technologies) для инактивации избытка праймеров.
Последовательности, фланкирующие вставку T-ДНК каждой единичной копии ETIP, идентифицировали путем расщепления геномной ДНК восемью эндонуклеазами рестрикции с последующим лигированием двухцепочечных адаптеров, специфичных к выступающим концам, внесенным с помощью эндонуклеаз рестрикции. После этой стадии лигирования ПЦР проводили с биотинилированным праймером либо к 3'-, либо к 5'-концу ETIP, и праймером к каждому адаптору. Продукты ПЦР улавливали и очищали на гранулах Ampure Solid Phase Reversible Immobilization (SPRI) beads™ (Agencourt Bioscience Corporation, Beckman Coulter Company). Проводили гнездовую ПЦР, и все продукты секвенировали с использованием секвенирования по методу Сэнгера ABI и протокола секвенирования Big Dye Terminator v3.1 cycle™ (Applied Biosystems, Life Technologies). Данные о последовательностях объединяли и анализировали с использованием программного обеспечения Sequencher™ (Gene Codes Corp., Ann Arbor, MI). Было идентифицировано восемь линий с ETIP, и они были отобраны для анализа фланкирующей последовательности (таблица 15). Левая и правая фланкирующие последовательности (также описанные как пограничные последовательности или последовательности области соединения) предоставлены в качестве SEQ ID NO: 431 - SEQ ID NO: 446, подчеркнутые последовательности указывают на плазмидный вектор, неподчеркнутые последовательности указывают на геномную фланкирующую последовательность.
Детали события pDAS000036: em02-5788-1-1, фланкирующее левую границу (SEQ ID NO: 431)
TCGAGATTGTGCTGAAGTAAACCATTTTACTTCAAATCTATTTTTAACTATTTACTTTTATTAAGGAGAGAAACTTTGCTGATTAATTCAAATTAGTGATCATTAAGATTCCAAAGATTCCGATTTAGAAAAGTCAAAGATTCAAAGAACAAGTCTAGGTCCTCATGGCTCATGTTGCATCCGATTCACCATCCACTCATCTTTCATATCTTCCTCCACTGTCTCTCTAGAAACAACTCATTTAATTTAGAAAACTCCTTTTTCAATTTAGAAATATTAAAGTTTATCACAATGTATCAATTAAATATTATCCGATGACTCATTCATAGTCAGGACCTTGCTGTCTGTGTCGTCCGTAATTATTATTTCAATACAAAACAAATATATGTTCACTCAGAAAATTACGGCGCAATCATCTAATTTTGTGGACCAAAATAAATAGCGTAGCTTCGAGATTTCAAAGTTGTGTTCAAATTTAATTTTGATTTCCGTTCCTCGTATACTCTTTTATGTATAGAAAATAATAATATCCACTACTAGTAGTTGATAACTACATTACATATATTAAAATTATGATGTCACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGCTTGAGCTTGGATC
em02-5788-1-1, фланкирующее левую границу (SEQ ID NO: 432)
CATAACCACCATCTCAAACAATAGAACTTCCTAAGTGAAGCAATGACTTCAAATCTACTTGAAGGCATGGAGTATAAGCCATGTTCCTTTCAGAGGGGACTGTACTTCTGTAGATTACTTTCCCTCATTAACCAGATCTGGCCGGCCTACCCAGCTTTCTTGTACACATAGCGACCGAGCTCGAGCCGAAGTTCGGTCGCTGTTTCACTGTTTGGGAAAGCATCAGTAACGCAGAAGACATAATTAAAAATTAATTATATATGGTAGTTTTTCTAGATTCTCCACTATACCTCATTGTGATTGAAAAACAACTATATATATATATATATATGTATTTAAAATTAAGAAATCATTAAATCGTACCATAATGCAGAAAACTTTATAAGTTCCTATTCTTTTGTCAAGATGAGTAGATGACACATCATGTACCACATAACATAACCATAATGGTGCGATACCATACCAAAACTTAATTTAGAAACTAATTAAAATTTTGGTAGTTTAAGATTCCTCCTAATGGTTGTCAAAAAAAAAAAAAGATTCCTCCTAATACATGGTAGAATATATTTTTGAGTTAAAATGAAAGTAAAATCTTCAAATTAGTCATAAGGAAATTCTAAAAAACATCGACTTTCTTTATAAAGATCCCATTGTAATTTTAGATGATTAATTTTATCCCAATCCAATTAAGAATTGTACACATCGGCCTCTATATATATCAAACACCTAAAA
Детали события pDAS000036: ad58-5784-2-1, фланкирующее левую границу (SEQ ID NO: 433)
CGATTTGCAGCTATAATCAATCACACCTTATCGTTCTTTCAAAGAAAAATCGAAAGTTGTAAACTTTATCAGCCTGTGTAGTGATTATTTCAATTTGATAAAGAAAAAAAAAGGCTTAGCTTTATTTGGTTTTTTGTTACAATCTTGATTAATTTTAGATTAGCACTCTGATTCTAGCGGAACATGAGAGTGGTTCCATCAAACCTCAGACAGTGAGCACAGTGGTTGCAGCAAACCATTTGGGTGAGAGCTCTTCAGTTTCTTTGCTATTAGCTGGTTCTGGCTCTTCTCTTCAAGAAGCTGCTTCTCAAGCTGCCTCTTGTCATCCTTCTGTTTCCGAGGTAACTAACTACATTCTTCATTTGTCCTTTTTTCTTGTGGGTCTTAAATGTTYGTGCTTTTCTTTATAGGTACTTGTTGCTGATTCAGATAGATTTGAGTACCCTTTAGCTGAACCTTGGGCTAAACTGGTTGACTTTGTTCGCCAACAAAGAGATTACTCTCACATCCTTGCCTTCCTCTAGCTCATTTGGCAAGAACATACTTCCTCTAGACAACTTAATAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTC
ad58-5784-2-1, фланкирующее правую границу (SEQ ID NO: 434)
CCTGTCATAACCACCATCTCAAACAATAGAACTTCCTAAGTGAAGCAATGACTTCAAATCTACTTGAAGGCATGGAGTATAAGCCATGTTCCTTTCAGAGGGGACTGTACTTCTGTAGATTACTTTCCCTCATTAACCAGATCTGGCCGGCCTACCCAGCTTTCTTGTACAAAGTGGTGATAAACTATCGCCGGCCTACCTCGCGTTGCTGCTCTTTTAGATGTCTCTCCTGTTACTGATGTTGTCAAAATCTTAGGATCCAATGAGTTTATCAGGTATACTTCTATCATGTATTGCTTGAGATTTTGGAGTGTTAGTAAAGATTTCAATAAAAGAATTTTTTCAAACAAAATTTTGGGGGCTTGAAGCTAATGTTTGGAAATATGTAACGGAGTTTAAATCTTTTGGCAGGCCTATATATGCGGGAAACGCCTTGTGTAGAGTTCGCTATACTGGTGCTGGTCCTTGTGTGTTGACTATTAGAACTACATCTTTTCCTGTTACCCCAATAACTGAGTCAAAGAAAGCTACTATCTCTCAGATTGATCTCTCGAAATTCAAAGAAGGTTTGTTTAGTATTATTCTCTTGTGCATAGCCTTTTTGCTTTTTTTTTTTTTATAAAAAAAGTTGAGTATGCTTATTGCCCATTGCA
Детали события pDAS000036: ad58-5898-10-1, фланкирующее левую границу (SEQ ID NO: 435)
TAATTTCATTTTCGTCATTTTGGTAAAGTAACAAAAGACAGAATATTGGTTAGTCGTGTTGGTTAGGAATAAAATAAAGAACGTGGACATCGTGGAATAAAAATATTCAGACAAGGAAACTAACAATAAAACAGTAATGAACATGGTTCTGAATCTCATCTTTGTGTATCTCCAATGGAATCCACCGCCACGAATCAGACTCCTTCTCCAAGCTCCACCGTCGACGATGACAATGGCGACGGTGTCTATACCGACGAATTTACCAAACTACCCCCGGACCCGGGGATCCTCTAGAGTCAACAAATTGACGCTTAGACAACTTAATAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGCTTGAGCTTGGATCAGATTGTC
ad58-5898-10-1, фланкирующее правую границу (SEQ ID NO: 436)
CGGCCTACCCAGCTTTCTTGTACAAAGTGGTGATAAACTATCAGTGTTTGATTAAAGATAAAATTTGATTTTTCATTACATAATAATCCATTAATTTTCACGCACGTGGAACCCATTTGGTGTACTTCCACGTCCTCTAAGAGAGCACTGACCCACTCATCAAAAAGATACATCTTTATAAGCCCCTTCATCGTCACACAGACACACACTTCCCTCTCAATTATTCCATATTCTCCTAATTTCTAATTGTTACACCTCAACACATTAGATTCGTACCAAACAAAAACGATTAGCTCCCAAGCCTAAGCTTTTATTTCCTTATAATTTTTCTTGGGTTTCTCTCTATAAAGAATGCAAATGACTGAGAGAGGCCGAGCCATGTGGCACACGTCCCTAGCCTCGGCATTCCGCACAGCTCTAGCTTGCACAATCGTTGGTGCGGCTACGCTCTACGGACCCGAGTGGATCCTCCGTTTTGTGGCATTCCCGGCGTTTTCTTACGTCACGGTCATTCTCATCATTACGGACGCCACGCTAGGCGACACACTACGTGGCTGCTGGCTAGCCCTTTACGCCACATGTCAGAGCGTTGCACCGGCTATCATTACACTAAGGCTTATAGGACCAGCTCGGCTCACGGCCGG
Детали события pDAS000037: lf31-6139-2-3, фланкирующее левую границу (SEQ ID NO: 437)
TTCTTTCCATCAGTTCTTCGGCACCTTCCTGGCTCTGCGTCTATCTTTCTTTCCATCCCGGCCCATCTCGTACACATTCCACCCAACTACACAAACACGTTATAGTCTTTTACATTATGACCAAATCAACCCTAAAGATACAGCCTTTATAAAAAATATAGGGGTCAAAGCAAAGAAGAGAAAGTTTGCTTACAGTGTGGAGAAAAGAAGTTGGAAGATGAGGAGTAAGAAGAAGAAGAGAAGAGAAAGGGTCTTCTGATGAGGAATAGGAGATAAGGTGGAACTGGAATGTTTGCCGCTAAATACTTGAAGACAAGAGCTTGATTTTCAAGCTCTTCCCATTGTGACTCAGTGAAAGGGATCCTAGTCGTCATGAAGAGATAGAGAAAATTGATGATGAAGGAGGTCTGATGAAGAGAAGAGAGAGAGAGAGATATTACACAAAGAGGGTTGTATTGCAAAATTGAAAGTGTAGAGAGAGTAGTAGGTAAGTTTTTATTAATAATGTTGTTCACACCTGCAGTCTGCAGAATATTGTGGTGTGAACAAATTGACGCTTAGACAACTTAATAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTA
lf31-6139-2-3, фланкирующее правую границу (SEQ ID NO: 438)
TTCAATCTACTTGAAGGCATGGAGTATAAGCCATGTTCCTTTCAGAGGGGACTGTACTTCTGTAGATTACTTTCCCTCATTAACCAGATCTGGCCGGCCTACCCAGCTTTCTTGTACAAAGTGGTGATAAACTATCAGTGTTTGAACATATATATACGCATAATATTCTCAGAACCCGACCCATTGGTTGACTCGGATCAAGATCGACCCGATCCGACCCGGTTAAAAGCACGTCGTCTCCTTTGGTTCGCCCCTTATATTCGACGAGTGAGTTCGATTGGATCGCTGTTTGTCATTTCTCACTACCTTAAGAAAAAAAAAAAGGTGCGTCTCTCTCTCACCTTTACCGCTCACTTACCTCTCAGATCTGACATCGATTTTCCAAATCTTC:TCCAGGTACTCTCTCTCCTGGCCGTTGACGGTCCCGTCCCGGCCGTGGATCTGATTTCGCCGCGATCTGAGGTCAAGCATGGCGGCAGCTAACGCCCCCATCACGATGAAGGAGGTCCTAACGGTGAGTCCCGCCTCCATTTTTAGTAACATA
Детали события pDAS000037: bm56-6315-1-1, фланкирующее левую границу (SEQ ID NO: 439)
TACATCGCGATTCATCCTGGTTTGATTAGAATGACGAGGAAGTTGTCATATTCCCAAACAGGAAAATTGGGATCGCCTTATTTGAAAGTGGGATAACTTCTTCATCTTAATTCTTATGAGAATTATTCCACTTCCTGGTGATTCTCCACTACTTTTTGTATATAAATACAGCTTCTTACATCGCGATTCATCCTGATTTGATTAGAATGACGAGAAAGTTGTCATATTCCCAAACAGGAAAACTGGGATCACCTGATTTGAAAGTGGGATAACTTCTTCATCCTAATTCTTATGAGATTTATTCCACTTCCTGGTGATTCTCCACTTCTTTATGTATCCAAATACATCTTCTTACATCGCGATTCATCCTGGTTTGATTAGAATGACGAGGAAGTTGTCATATTCCCAAACATGAAAACTGGGAAAGTGGGATTGACGCTTAGACAACTTAATAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGCTTGAGCTTGGAT
bm56-6315-1-1, фланкирующее праву границу (SEQ ID NO: 440)
CCACCATCTCAAACAATAGAACTTCCTAAGTGAAGCAATGACTTCAAATCTACTTGAAGGCATGGAGTATAAGCCATGTTCCTTTCAGAGGGGACTGTACTTCTGTAGATTACTTTCCCTCATTAACCAGATCTGGCCGGCCTACCCAGCTTTCTTGTACAAAGTGACGATAAACTATCAGTCTTTCAAAGCGCATCTATGGCTAGTCATCACGGTTTTTAACTGTTTTACGAAGTCGCGGAGAGGCTCGTCTTCTCCCTAGGATAAACTGCAGAGGTCGACATCGGAGGTTTCTCGATCTATGAACATAGAGTACTGTTTGAGAAACTCCGAGGCGAGTTGGCGGAAACTCCCTATAGAGTTTTTCTTTAGACAAGAGAACCACTCAAGCGCTGCTTCGCGGAGGTTCTCGACGAAGAGGCGGCATTCGCCGGCATCTCTTTCTCTATCTTTAAACTTGGCTCTTCCCATCGCGATCTGGAAAGCCTGCAAGTGTGCCTTAGGATCGGTTGTACCATCGTAAGGAGCCACTTTGACTTTATCAGGATCCGAAACCGTAGTTTCGGTGATGCGGGTGGTGAAGGGCGTTTTTCGAGACTCCTCGAGGAGTAGGGTGATATCGAGTGCAGTACTAGTAGCGTGATGATTTGGGATTTCACCGCTCTAACCTCCGCAGCCGTTTTCA
Детали события pDAS000037: ad58-6372-1-1, фланкирующее левую границу (SEQ ID NO: 441)
TTTTACAGTGTTAGAAGAAGTGGATGAAGCTGAAATTGAATTTTCAAACTCGTTCAGCTTGACTAGAGGTGGGAGAGAGTCAAAGCCTCCCATCAAGTACCAAAACATGGAATAGAAGACAGTCCGAGGGAGAGGAAACCGTGGCCGACGAGGCCGTGGCTCCTATCATTAACTAGTGTCTTTCTTACTATTAATGGTTTATTAAGTCTCAGCCTTTGTTGTTTCATTGGTTTGAGATTCACTCATACATGAAACTTGTTTCATTCCAGCTTTTCCAAACTATAAGAATATTTCCAATCTTATCTTGTAATAGTTTAAGTTTTAAATTGAAAGCCCTTAGTTCAAAAAACAAAAAAAAAAAATTAGCCCTTGAATTTATATATAATCACGACGGCCATATTTGGCAGCTACACTGATATGTTTTCAATTGGCTGACAGGCCTTGAGCAGGGTTTGCTGGGTATATTGGTAGGAAGATGTGTTGCGAGGTTGAAGCCTCATTTAGGCAATATAAACATGATCATTAGCGTTATGTCATTAGTTATACTTATACGTAGACTAAGTAACCCACTAAAGGTTGCTGATTCCTTTTGTATCGACTAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGCTTGAGCTTGGATCAGATTGTCGTTTCCCGC
ad58-6372-1-1, фланкирующее правую границу (SEQ ID NO: 442) CATGTTCCTTTCAGAGGGGACTGTACTTCTGTAGATTACTTTCCCTCATTAACCAGATCTGGCCGGCCTACCCAGCTTTCTTGTACAAAGTGGTGATAAACTATCAGTGTTTGACTGAATTTTAATTTCTAATTTTTGTAAAAAATTTGTATAACCTCAAATTATTAAAAGGCGGATTTTATTAGAATTATAACTAAATTATCTATAACTCCAAAATTTTGACAATCAATCATGTCTATATCTTTATTTTTTTGCTAAATTATCATGTCTATATCTTTTCTTTCTTCCAAACTTACTTGAGACTAAAAGTCTTTATAAATTTTGATAGGAGTTCCACACACAAACAAAAACAAAACAAATATTTTTCATCAAGGGATACTTATTTAACATCACGGATTCACAGTTTATTAACAAAAATCCAAACAAAGACTGAAAGACAGAAGATTCAATCTAACAATAGTCGGCAAACACCAGTGATTAACTAACGAAATAAATTAACAAGTGGTCAGATCTTCGGGAAA
Детали события pDAS000037: ad58-6620-4-1, фланкирующее левую границу (SEQ ID NO: 443)
TAATTTCATTTTCGTCATTTTGGTAAAGTAACAAAAGACAGAATATTGGTTAGTCGTGTTGGTTAGGAATAAAATAAAGAACGTGGACATCGTGGAATAAAAATATTCAGACAAGGAAACTAACAATAAAACAGTAATGAACATGGTTCTGAATCTCATCTTTGTGTATCTCCAATGGAATCCACCGCCACGAATCAGACTCCTTCTCCAAGCTCCACCGTCGACGATGACAATGGCGACGGTGTCTATACCGACGAATTTACCAAACTACCCCCGGACCCGGGGATCCTCTAGAGTCAACAAATTGACGCTTAGACAACTTAATAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGCTTGAGCTTGGATCAGATTGTC
ad58-6620-4-1, фланкирующее правую границу (SEQ ID NO: 444)
CATAACCACCATCTCAAACAATAGAACTTCCTAAGTGAAGCAATGACTTCAAATCTACTTGAAGGCATGGAGTATAAGCCATGTTCCTTTCAGAGGGGACTGTACTTCTGTAGATTACTTTCCCTCATTAACCAGATCTGGCCGGCCTACCCAGCTTTCTTGTACAAAGTGGTGATAAACTATCAGTGTTTGAAATAATCGGATATTTAATTTTCTTAGACAGTTCATTAGTAGTTGATCTTAAACATTCACGTTTTATTTTCTTTTCTTTTCGAATGCTAGACTCTAGTTTGGTACCCATAGGATTCGAGTTACATGAAGCTATTGACACTGGGAGTGCTTCAAGATCTGTAAGAGGCAAAGATTCACAAACAGAACGTGATTTCTTGGATAGTGATGTGGAGATTGTGATAAGAACCAGCATGAGTATTACTTTTACTGCCCCTGCTGTGGTGAAGACATCACCAAAACAGTCAAGCTCGTGAAGAAATCAGATATTCAACCCGCAAAAAAATCTGACAATGCAAATAAACCTATTGACACTAAGAATGGTTCAAGATCCGAAGACAAGAAGACAAAAAATTTGTCCTGGCTCCCTGCTTATCTCCAGAAGCTGTTTCTTTCTGTTTATGGCCACATCAAAGACAAAGGTACCCTTTCTTTTGGTGCCTTTGGTGTGGACAGGTGTGATTGACTTTGGTTTCTTGGCAGATTCAGGCAAGATAGAGGTTGATTCAAAGTCAACTAACAATGATCTTGGTACCAATAGTGAGGA
Детали события pDAS000037: ad58-6620-17-1, фланкирующее левую границу (SEQ ID NO: 445)
CCGCCTTGAACAACCGCTCCGCCGTTGCTCAGTACATTATCGAGGTTACCAAAAAGTTCAATCCTTTATGCTTGTTTTGGCGTGTGATTCGTTACGAATGAGTAAAATTGATTTGGTTTTTTTCTTTGAACAGCATGGTGGAGATGTGAATGCGACGGATCATACGGGGCAGACTGCGTTGCATTGGAGTGCGGTTCGTGGTGCGGTGCAAGTTGCGGAGCTTTTGCTTCAAGAGGGTGCAAGGGTTGATGCTACGGATATGTATGGATATCAGGTTCTAACCACTTCCTCTTTCTTTGTGGAGATTGTCTTTTTGTTTCAATGCTAGTCAACTTCTTTCTTTCTTTCACAAAACTAAGTAGTATTGCTTGTTTTGTTGTCGTTGCATTTTTTCTTATGGCTGTGTTCTGAGGTTCATGTAGGTATATAAGCACTTCGTACTTTGCCACTTGTTTCATTTAGGCAACATTGTGCACATATCTAAGTAGTTGGTCTTTGTAAAATTAGTTTGTTTGTCTTCAAAGTATATTGAGCAGTTTCATGACTCATTATTCAAAGGTTTGTCTAAATTAGAGAGAACTTTCATTTTGCCTGGATTTAATCAGCATTTAGAATGTTTATAGCGATATCATTTTTAGTTGAAAAAATCTCAACAAATTGACGCTTAGACAACTTAATAACACATTGCGGACGTTTTTAATGTACTGAATTAACGCCGAATTGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGCTTAGCTTGAGCTTGGATCAGATTGTCGTTTC
ad58-6620-17-1, фланкирующее правую границу (SEQ ID NO: 446) GTATAAGCCATGTTCCTTTCAGAGGGGACTGTACTTCTGTAGATTACTTTCCCTCATTAACCAGATCTGGCCGGCCTACCCAGCTTTCTTGTACAAAGTGGTGATAAACTATCAGTGTTAGATCCCCGACCGACCGCCCATCCTGGACGGCCTCGTGCATGCTGATGTTGTCAAAATCTTAGGATCCAATGAGTTTATCAGGTATACTTCTATCATGTATTGCTTGAGATTTTGGAGTGTTAGTAAAGATTTCAATAAAAGAATTTTTTCAAACAAAATTTTGGGGGCTTGAAGCTAATGTTTGGAAATATGTAACGGAGTTTAAATCTTTTGGCAGGCCTATATATGCGGGAAACGCCTTGTGTAGAGTTCGCTATACTGGTGCTGGTCCTTGTGTGTTGACTATTAGAACTACATCTTTTCCTGTTACCCCAATAACTGAGTCAAAGAAAGCTACTATCTCTCAGATTGATCTCTCGAAATTCAAAGAAGGTTTGTTTAGTATTATTCTCTTGTGCATAGCCTTTTTGSTTTTTTTTTTTTTATAAAAAAAGTTGAGTATGCTTATTGCCCATTGC
КАРТИРОВАНИЕ ETIP
Для трансгенного события, содержащего вставку одной копии ETIP, фланкирующую последовательность после сборки вручную использовали в качестве последовательности запроса в локальном анализе BLAST. Всего было восемь растений, которые имели встраивания одной копии, идентифицированные с помощью этого процесса (таблица 16 и таблица 17). Коллекцию из 595478 происходящих из генома стохастических последовательностей из Brassica oleracea загружали из базы данных NCBI GSS и форматировали в качестве нуклеотидной базы данных BLAST. Затем фланкирующие последовательности ETIP сравнивали с помощью BLASTn с базой данных и все совпадения исследовали вручную. Затем наиболее значительные совпадения последовательностей с фланкирующей последовательностью ETIP из базы данных B. oleracea выравнивали против доступной через Интернет геномной последовательности Brassica rapa (http://brassicadb.org/brad/blastPage.php), где также выявляли положение в геноме, которое имело наиболее существенное совпадение последовательностей. В случаях, когда только 5'- или 3'-фланкирующие последовательности обеспечивали значительные совпадения с геномными последовательностями B. oleracea, предполагали, что либо невыровненная или несовпавшая последовательность имела идентифицированную отсутствующую в базе данных последовательность, либо во время встраивания ETIP происходили значительные перестройки генома. Для образцов, которые обеспечили значительные совпадения в BLASTn в анализе фланкирующей последовательностей ETIP, наиболее значительную совпадающую последовательность B. oleracea из GSS вместе с наиболее значительной совпавшей последовательностью из генома B. rapa выравнивали вручную с помощью программного обеспечения SequencherTM v5.0 (Gene Codes Corp., Ann Arbor, MI) для каждого из восьми растений с одной копией ETIP. Затем три последовательности сравнивали и для наиболее сходной последовательности из любого диплоидного вида Brassica при сравнении с фланкирующим ETIP устанавливали геном, в котором ETIP была расположена. Для большинства образцов существовало значительное варьирование между двумя последовательностями диплоидного генома Brassica и происходящая из B. napus фланкирующая последовательность ETIP продемонстрировала преобладающую ассоциацию с одной или другой из диплоидных последовательностей. Однако были случаи, когда варьирование последовательностей между диплоидными организмами было недостаточным, и могло быть возможным установление группы сцепления, однако субгеномное определение было невозможным. Затем прогнозировали конкретное положение в геноме, исходя из положения в геномной последовательности Brassica rapa. В случаях, когда ETIP идентифицировали как встроенный в геном C B. oleracea, сравнительную сентению между диплоидными геномами Brassica, описанными в Parkin et al. (Genetics 2005, 171: 765-781) использовали для экстраполяции геномного положения на субгеном C rassica napus. Кроме того, идентифицированные последовательности сравнивали с помощью BLASTn с геномами Arabidopsis thaliana, кодирующими последовательности (TAIR 9 CDS, загруженная с http://arabidopsis.org/index.jsp) и идентифицировали природу любых разрушенных последовательностей генов, а также проводили подтверждение геномного положение после синтении с Arabidopsis Brassica, описанной в Schranz et al. (Trend in Plant Science 2006, 11.11: 535-542).
Гомозиготные события используют для получения протопластов описанным ранее способом. Затем протопласты котрансформируют нуклеазой с цинковыми пальцами, которая сконструирована для нацеливания на участок связывания цинковых пальцев, который включен в последовательность ETIP, и донорной плазмидой, которая обладает гомологией с конкретными областями ETIP. Нуклеаза с цинковыми пальцами расщепляет локус ETIP и донорная плазмида встраивается в геном клеток Brassica napus через направляемую гомологией репарацию. В результате встраивания донорной плазмиды частичный трансген DS-red репарируется в полноразмерный трансген DS-red. Экспрессию теперь полностью функционального трансгена DS-red используют для сортировки протопластов клеток способом FACS. Предполагаемые трансгенные растения сортируют с использованием способа FACS, описанного в примере 7, и выделенные протопласты регенерируют в зрелые растения. Встраивание донорной плазмиды подтверждают в растениях ETIP с использованием способом молекулярного подтверждения. По существу, локус ETIP служит в качестве сайт-специфического локуса для направленного встраивания донорной полинуклеотидной последовательности.
Геномные положения для нацеливания обеспечивают положения в геноме, которые не изменяют нормальный фенотип растений. Полученные события, где трансген встроен в ETIP, не обеспечивают агрономически значимых ненамеренных отличий, когда события ETIP сравнивают с контрольными растениями. Кроме того, белки трансгенов, встроенных в локус ETIP, устойчиво экспрессируются и являются постоянными и стабильными в множестве геномных положений. Описанные геномные последовательности с SEQ ID NO: 431 по SEQ ID NO: 446 обеспечивают геномные положения в геноме brassica, которые поддаются нацеливанию для встраивания экспрессирующих гены кассет, содержащих трансген.
НАЦЕЛИВАНИЕ DS-RED НА ЛИНИИ С ETIP С ПОМОЩЬЮ ОПОСРЕДУЕМОЙ ZFN ГОМОЛОГИЧНОЙ РЕКОМБИНАЦИИ
Получали линию канолы, содержащую вставку T-цепи из pDAS000036, и с помощью молекулярной охарактеризации подтверждали, что она содержит полноразмерную единичную копию T-цепи. Эти события в каноле обозначили как pDAS000036-88 и их использовали для получения протопластов описанным выше способом. Протопласты выделяли, а затем ~50000 протопластов клеток растений котрансформировали нуклеазой с цинковыми пальцами, либо pDAS000074 (Фиг. 25) либо pDAS000075 (Фиг. 26), которые были сконструированы для нацеливания на участки связывания цинковых пальцев, включенные в последовательность ETIP, и донорной плазмидой pDAS000064, pDAS000068, pDAS000070 или pDAS000072 (Фиг. 27, Фиг. 28, Фиг. 29 и Фиг. 30, соответственно), которая обладает гомологией с конкретными областями ETIP. На Фиг. 19 и Фиг. 20 представлены иллюстрации направляемой гомологией репарации, которая приводит к сайт-специфическому встраиванию трансгена Ds-red посредством опосредуемой нуклеазой с цинковыми пальцами гомологичной рекомбинации. Нуклеаза с цинковыми пальцами была сконструирована для расщепления локуса ETIP в определенной последовательности связывания цинковыми пальцами, и для создания тем самым двухцепочечного разрыва в геноме. Далее донорную плазмиду встраивали в геном протопластов клеток Brassica napus посредством направляемой гомологией репарации. Области интрона-1 и интрона-2 донорной плазмиды обладают гомологией с соответствующими областями интрона-1 и интрона-2 локуса ETIP. В результате встраивания донорной плазмиды частичный трансген DS-red репарировался до полноразмерного трансгена DS-red с высокой экспрессией. Экспрессию полностью функционального трансгена DS-red использовали для сортировки протопластов клеток с помощью описанного выше способа FACS. По существу, локус ETIP служит в качестве сайт-специфического локуса для направленного встраивания донорной полинуклеотидной последовательности. Наконец, выделенные протопласты можно сортировать и регенерировать в зрелые растения. Встраивание донорной плазмиды можно подтверждать в растениях с нацеливанием на ETIP с использованием способов молекулярного подтверждения.
Донорную плазмидную ДНК и плазмидную ДНК ZFN смешивали в различных концентрациях, использованных для трансфекции протопластов клеток канолы, содержащих событие pDAS000036-88, и трансгенные протопласты клеток сортировали с использованием трансфекции FACS, которая описана ранее. В таблице 18 описаны различные эксперименты по трансфекции и концентрации ДНК, которые использовали для трансфекции протопластов канолы, содержащих событие pDAS000036-88. ДНК ZFN и донорную плазмидную ДНК выделяли и подготавливали для трансфекции описанными выше способами.
После завершения экспериментов по трансфекции протопласты инкубировали при комнатной температуре в течение 48 часов и хранили с использованием описанного выше протокола FACS. Сортировку для каждого эксперимента проводили независимо и опосредуемую цинковыми пальцами интрогрессию трансгена подтверждали посредством идентификации индивидуальных событий, в случае которых экспрессировался трансген DS-red. На Фиг. 21-24 представлены результаты сортировки FACS. Как указывают результаты, представленные на графиках, было получено множество событий, которые содержали интактный полностью встроенный трансген DS-red. Эти множественные события Ds-Red были результатом опосредуемого цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP. Это сайт-специфическое встраивание обеспечивало полную копию трансгена Ds-Red с высокой экспрессией. Частота экспрессии трансгена Ds-Red находилась в диапазоне приблизительно 0,03-0,07% от всех протопластов клеток канолы (~50000). Однако частота эффективности трансфекции для опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP была значительно более высокой и находилась в диапазоне приблизительно 0,07-0,64%.
На Фиг. 21 представлены результаты трансфекций, при которых котрансфицировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000064 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 26 мкг к 4 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,03% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,22-0,07%. Аналогично, на нижнем графике, когда донор, pDAS000064 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 26 мкг к 4 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,03% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,26-0,08%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
На Фиг. 22 представлены результаты трансфекций, при которых котрансформировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000068 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,03% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,22-0,07%. Аналогично, на нижнем графике, когда донор, pDAS000068 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,04% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,38-0,12%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
На Фиг. 23 представлены результаты трансфекций, при которых котрансформировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000070 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,07% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,64-0,21%. Аналогично, на нижнем графике, когда донор, pDAS000070 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,04% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,34-0,11%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
На Фиг. 24 представлены результаты трансфекций, при которых котрансформировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000072 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,07% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,62-0,20%. Аналогично, на нижнем графике, когда донор, pDAS000072 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,05% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,44-0,14%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
Выбранные эксплантаты переносили и культивировали на среде для регенерации, содержавшей фосфотриноцин. После периода культивирования выжившие эксплантаты переносили в среду для удлинения и среду для индукции укоренения и развития растения. Целые растения, которые состояли из развитых структур корней и побегов, переносили в почву и далее выращивали в теплице. В процессе культивирования тканей использовали среды и условия культивирования, описанные ранее. Результаты для растений, полученных в процессе культивирования тканей, представлены в таблице 19, ниже.
МОЛЕКУЛЯРНОЕ ПОДТВЕРЖДЕНИЕ ВСТРАИВАНИЯ ETIP В FAD2A В КАНОЛЕ
Геномную ДНК экстрагировали из ткани листьев всех предполагаемых трансгенных растений с использованием набора DNeasy Plant Mini Kit™ (Qiagen) в соответствии с инструкциями изготовителя, за исключением того, что ткань извлекали в 80 мкл буфера AE. Тридцать миллиграмм ткани молодых листьев из регенерировавших растений быстро замораживали в жидком азоте, а затем растирали в порошок.
Молекулярную охарактеризацию локуса FAD2A проводили с использованием трех независимых анализов. Анализы были разработаны и оптимизированы с использованием следующих контролей: охарактеризованные трансгенные события, содержащие один случайным образом встроенный трансген, охаратеризованное трансгенное событие с пятью случайным образом встроенными трансгенными, растения канолы дикого типа сорта DH12075 и контрольные реакции без матрицы. Результаты трех описанных ниже молекулярных анализов рассматривали вместе для получения доказательства встраивания ΕΤIΡ в FAD2A посредством HDR.
ИДЕНТИФИКАЦИЯ ВСТРАИВАНИЯ ТРАНСГЕНА С ПОМОЩЬЮ ПОЛИМЕРАЗНОЙ ЦЕПНОЙ РЕАКЦИИ В РЕАЛЬНОМ ВРЕМЕНИ
Четыре реплики для каждого растения анализировали с использованием праймеров, специфичных к гену-мишени hph (также описанному как hpt) (SEQ ID NO: 447, hpt F791 5' CTTACATGCTTAGGATCGGACTTG 3'; SEQ ID NO: 448, hpt R909 5' AGTTCCAGCACCAGATCTAACG 3'; SEQ ID NO: 449, hpt Taqman 872 5' CCCTGAGCCCAAGCAGCATCATCG 3' FAM) (Фиг. 31) и эталонному гену, кодирующему белок группы высокой подвижности I/Y (HMG I/Y) (SEQ ID NO: 450, F 5' CGGAGAGGGCGTGGAAGG 3'; SEQ ID NO: 451, R 5' TTCGATTTGCTACAGCGTCAAC 3'; SEQ TD NO: 452, зонд 5' AGGCACCATCGCAGGCTTCGCT 3' HEX). Реакционные смеси амплифицировали с использованием следующих условий: 95°C в течение 10 минут, затем 40 циклов из 95°C в течение 30 секунд, 60°C в течение 1 минуты, причем данные амплификации получали в конце каждой стадии отжига. Количество копий вычисляли с использованием способа ACq, где ACq=Cq(ген-мишень) - Cq(эталонный ген). Растения с амплификацией hph и HMG I/Y и количеством копий 0,5 или более считали трансгенными, в то время как растения с количеством копий ≥0,5 и ≤1,2 оценивали как предположительно имеющие одну копию. Амплификацию проводили на системе для детекции BioRad CFX96 Touch™ Real-Time PCR Detection System с FastStart Universal Probe Master (ROX) (Roche, Basel, Швейцария).
ОБНАРУЖЕНИЕ РАЗРУШЕННОГО УЧАСТКА ZFN В FAD2A
Каждое растение анализировали в отношении присутствия или отсутствия эндогенной мишени в исследовании разрушенного локуса, которое является основным анализом. Анализ представляет собой анализ SYBR® Green I q-ПЦР и в моноплексном формате, но при этом каждую реакцию проводят одновременно на одном и том же планшете для ПЦР, он нацелен на эндогенный локус (FAD2A/2C.RB.UnE.F1, SEQ ID NO: 453, 5' CTTCCACTCCTTCCTCCTCGT*C 3' и FAD2A 2C.RB.UnE.R1, 5' SEQ ID NO: 454, GCGTCCCAAAGGGTTGTTGA*G 3') и локус ZFN (локус, с которым связывается ZFN pDAB104010 и расщепляет геном) (FAD2A.UnE.F1, SEQ ID NO: 455, 5' TCTCTACTGGGCCTGCCAGGG*C 3' и FAD2A.UnE.Rl, SEQ ID NO: 456, 5' CCCCGAGACGTTGAAGGCTAAGTACAA*A 3') (Фиг. 32). Обе пары праймеров амплифицировали с использованием следующих условий: 98°C в течение 30 секунд, затем 35 циклов (98°C в течение 10 секунд, 65°C в течение 20 секунд, 72°C в течение 90 секунд), за которыми следуют 95°C в течение 10 секунд, а затем анализ плавления от 50°C до 95°C с приращением 0,5°C в течение 0,05 секунд и считыванием планшета при каждом приращении. Условия реакции приведены в таблице 20.
Растения, которые имели амплификацию эндогенной мишени, однако не имели амплификации мишени ZFN, оценивали как положительные в испытании разрушения локуса, и их считали имеющими разрушенный локус ZFN. Этот анализ считали положительным, когда участок связывания ZFN на обоих аллелях в локусе FAD2A был разрушен.
ОБНАРУЖЕНИЕ СПОСОБОМ ПЦР ВСТРАИВАНИЯ ТРАНСГЕНА В FAD2A ПОСРЕДСТВОМ НАПРАВЛЯЕМОЙ ГОМОЛОГИЕЙ РЕПАРАЦИИ
Каждое предполагаемое трансформированное растение анализировали с использованием конечного результата, достигаемого с праймерами для ПЦР, сконструированными для амплификации трансгена-мишени hph (hph_ExoDigPC_F1, SEQ ID NO: 457, 5' TTGCGCTGACGGATTCTACAAGGA 3' и hph_ExoDigPC_R1, SEQ ID NO: 458, 5' TCCATCAGTCCAAACAGCAGCAGA 3'), эндогенным локусом FAD2A (FAD2A.Out.F1, SEQ ID NO: 459, 5' CATAGCAGTCTCACGTCCTGGT*C 3' и FAD2A.Out.Rvs3, SEQ ID NO: 460, 5' GGAAGCTAAGCCATTACACTGTTCA*G 3'), областью, охватывающей 5'-конец трансгена, встроенного в локус FAD2A посредством HDR и область выше трансгена в локусе FAD2A (FAD2A.Out.F1, SEQ ID NO: 461, 5' CATAGCAGTCTCACGTCCTGGT*C 3' и QA520, SEQ ID NO: 462, 5' CCTGATCCGTTGACCTGCAG 3') и областью, охватывающей 3' конце любого трансгена, встроенного в локус FAD2A посредством HDR и область ниже трансгена в локусе FAD2A (QA558, SEQ ID NO: 463, 5' GTGTGAGGTGGCTAGGCATC 3' и FAD2A.Out.Rvs3, SEQ ID NO: 464, 5' GGAAGCTAAGCCATTACACTGTTCA*G 3') (Фиг. 33). Все пары праймеров амплифицировали с использованием следующих условий: 98°C в течение 30 секунд, затем 35 циклов (98°C в течение 10 секунд, 65°C в течение 20 секунд, 72°C в течение 90 секунд). Условия и реагенты реакции описаны в таблице 21.
Амплификация участка 5'-трансген-геном, фланкирующий мишень, и/или амплификация участка 3'-трансген-геном, фланкирующий мишень, указывала на предполагаемое событие встраивания. Необходимо отметить, что вследствие наличия участков гомологии FAD2A размером приблизительно 1000 п.н. в кассете pDAS000130 (содержащей полинуклеотидные последовательности со 100% идентичностью последовательности с областями FAD2A непосредственно выше участка расщепления ZFN), в реакциях ПЦР происходила ложно-положительная амплификация продукта ПЦР вследствие химеризма ПЦР из-за амплификации событий встраивания вне мишени ETIP. Амплификация мишени hph подтвердила, что произошло встраивание трансгена. Амплификация мишени FAD2A указывает на то, что локус FAD2A является интактным или содержит только частичную вставку. Вследствие размера ETIP (11462 п.н. для кассет ETIP или 13472 п.н., включая гомологичные участки FAD2A и кассеты ETIP) ожидается, что праймеры для FAD2A не будут амплфицировать продукт, когда интактный ETIP встроен в локус FAD2A.
ОБНАРУЖЕНИЕ С ПОМОЩЬЮ САУЗЕРН-АНАЛИЗА РЕДАКТИРОВАНИЯ FAD2A
Растения, которые имели либо амплификацию продукта в виде участка 5'-трансген-геном, фланкирующий мишень, и/либо амплификацию участка 3'-трансген-геном, фланкирующий мишень, или отсутствие амплификации локуса-мишени ZFN, или оба из них, подвергали саузерн-анализу для обнаружения встраивания трансгена в локус FAD2A. Геномную ДНК из 5 г ткани листьев с использованием модифицированного способа CTAB (Maguire, T.L., G.G. Collins, and M. Sedgley A modified CTAB DNA extraction procedure for plants belonging to the family proteaceae. Plant Molecular Biology Reporter, 1994. 12(2): p. 106-109). Далее 12 мкг геномной ДНК расщепляли Kpn1-F (New England BioLabs) и фрагменты расщепления разделяли электрофорезом на 0,8% агарозном геле, а затем переносили на мембрану с использованием стандартного протокола саузерн-блоттинга. Праймеры для 5'-области-мишени FAD2A (F, SEQ ID NO: 465, 5' AGAGAGGAGACAGAGAGAGAGT 3' и R, SEQ ID NO: 466, 5' AGACAGCATCAAGATTTCACACA 3'), 3'-области мишени FAD2A (F, SEQ ID NO: 467, 5' CAACGGCGAGCGTAATCTTAG 3' и R, SEQ ID NO: 468, 5' GTTCCCTGGAATTGCTGATAGG 3') и hph (F, SEQ ID NO: 469, 5' TGTTGGTGGAAGAGGATACG 3' и R, SEQ ID NO: 470, 5' ATCAGCAGCAGCGATAGC 3') использовали для получения зондов для обнаружения присутствия ETIP в локусе FAD2A с использованием системы DIG Easy Hyb System® (Roche, South San Francisco, CA) в соответствии с инструкциями изготовителя (Фиг. 34). Гибридизацию проводили при 42°C для 5'-области FAD2A, 45°C для 3'-области FAD2A и 42°C для обнаружения hph.
Связанную с мембраной геномную ДНК исследовали с помощью зондов в конкретном порядке; сначала исследовали с помощью зондов 5'-последовательности FAD2A, затем исследовали с помощью зондов 3'-последовательности FAD2A, и, наконец, исследовали с помощью зондов последовательности hph (Фиг. 34). Обоснование для этого является следующим. Первый зонд (FAD2A 5') представляет собой диагностический зонд, и, если ETIP встроился в FAD2A посредством полной HDR, на мембране будет виден фрагмент размером 5321 п.н. Полученный размер полосы легко дифференцировать в процессе электрофореза, и он будет расположен вблизи фрагментов размером 5148 п.н. в меченном DIG маркере молекулярной массы ДНК Roche DNA Molecular Weight Marker III® (Roche, Indianapolis, IN). Второй зонд мембраны представляет собой зонд FAD2A 3' и растение после редактирования будет иметь фрагмент размером 22433 п.н., в то время как растение без редактирования будет иметь фрагмент размером 16468 п.н. Тот же фрагмент размером 22433 п.н., идентифицированный с помощью зонда FAD2A 3', также должен связываться и идентифицироваться зондом hph. Эти фрагменты трудно различить на геле, поскольку они являются чрезвычайно крупными и может быть трудно определить какое-либо отличие между фрагментом, находящимся выше или ниже наиболее крупного фрагмента размером 21226 п.н. в меченном DIG маркере молекулярной массы ДНК Roche DNA Molecular Weight Marker III®. По существу, эти зонды обеспечивают доказательства, которые могут подтвердить идентификацию встраивания ETIP в FAD2A через направляемую гомологией репарацию (HDR) путем визуализации фрагмента размером 5 т.п.н. с использованием зонда FAD2A 5'. Фермент рестрикции Kpn1 был единственной подходящей эндонуклеазой рестрикции для применения в этом анализе, поскольку участки Kpn1 встречались в одном локусе расщепления кассеты ETIP, и они присутствовали в двух участках локуса ZFN для FAD2A. Один участок был расположен выше, а второй участок был расположен ниже участков гомологии FAD2A. Кроме того, Kpn1 не является чувствительной к метилированию и доступна в качестве рекомбинантного фермента с повышенной точностью (New England Biolabs).
Результаты молекулярного анализа и саузерн-анализа
После трансфекции, культивирования и селекции трансгенные растения переносили в почву. В этом процессе 139 растений выжило и для них проводили взятие образцов тканей для экстракции и анализа гДНК. Все 139 растений анализировали для оценки количества копий. Среди этих 139 растений, 56 были положительными в отношении ΕΤIΡ и 11 из 56 положительных растений имели предполагаемое встраивание одной копии (Фиг. 35) (таблица 22). Из 56 растений, которые были положительными в отношении встраивания ETIP, амплификация 5'-геномной фланкирующей трансген последовательности FAD2A происходила в 7 растениях. Амплификация 3'-геномной фланкирующей трансген последовательности FAD2A не происходила ни в одном из 56 растений, которые были положительными по встраиванию ETIP. Кроме того, среди 56 растений, которые были положительными по встраиванию трансгена, 11 растений были положительными в исследовании с помощью q-ПЦР разрушения локуса. Четырнадцать растений, которые были положительными по амплификации FAD2A 5' геномной фланкирующей трансген последовательности и/или положительными в исследовании с помощью q-ПЦР разрушения локуса, подвергали саузерн-анализу с 3 зондами, описанными выше. Среди 14 растений, для которых проводили саузерн-анализ, все растения продемонстрировали частичное встраивание в локус FAD2A, однако ни одно из этих растений не продемонстрировало доказательств полноразмерного встраивания ETIP в локус FAD2A посредством HDR, когда проводили обработку зондом FAD2A 5', зондом FAD2A 3' и зондом hph. Не было выявлено полос, которые оказались i) большими, чем WT, и ii) идентичными полосам, выявленным для этих образцов, когда проводили обработку зондом FAD2A 3' (таблица 11).
РЕЗУЛЬТАТЫ ДЛЯ ТРАНСГЕННОЙ КАНОЛЫ С ETIP, ТРАНСФОРМИРОВАННОЙ PDAS000130 И PDAB104010
Трансгенные события Brassica napus, которые получены путем трансформации pDAS000130 и DAB104010, приводят к встраиванию единичной копии полноразмерной вставки T-цепи полинуклеотидной последовательности ETIP из pDAS000130 в локус FAD2A. От трех до четырех событий полностью охарактеризовывают и подтверждают, что они содержат встроенный ETIP. Подтверждение осуществляют способом амплификации ПЦР внутри-снаружи, и далее подтверждают с помощью саузерн-блоттинга. Выбранные растения с событиями T0 выращивают до стадии развития T1. Растения T1 повторно подвергают скринингу для определения зиготности встроенной T-цепи. Подвергнутые скринингу события распределяют на категории в качестве гомозиготных, гемизиготных или нулевых.
Гомозиготные события используют для получения протопластов посредством ранее описанного способа. Затем протопласты котрансформируют нуклеазой с цинковыми пальцами, которая сконструирована для нацеливания на участок связывания цинковых пальцев, который включен в последовательность ETEP, и донорной плазмидой, которая обладает гомологией с конкретными областями ETIP, где донор встраивается в ETIP посредством механизма HDR. Аналогично, протопласты впоследствии котрансформируют нуклеазой с цинковыми пальцами, которая сконструирована для нацеливания на участок связывания цинковыми пальцами, который включен в последовательность ETIP, и донорной плазмидой, которая не обладает гомологией с конкретными областями ETIP, где донор встраивается в ETIP посредством механизма негомологичного соединения концов. Нуклеаза с цинковыми пальцами расщепляет локус ETIP и донорная плазмида встраивается в геном клеток Brassica napus посредством направляемой гомологией репарации или негомологичного соединения концов.
В результате встраивания донорной плазмиды, частичный трансген DS-red репарируется до полноразмерного трансгена DS-red. Экспрессию теперь полностью функционального трансгена DS-red используют для сортировки протопластов клеток способом FACS. Предполагаемые трансгенные растения сортируют с использованием способа FACS, описанного в примере 7, и выделенные протопласты регенерируют в зрелые растения. Встраивание донорной плазмиды подтверждают в растениях с нацеливанием на ETIP с использованием способов молекулярного подтверждения. По существу, локус ETIP служит в качестве сайт-специфического локуса для направленного встраивания гена донорной полинуклеотидной последовательности.
РЕЗУЛЬТАТЫ ДЛЯ ТРАНСГЕННОЙ КАНОЛЫ С ETIP, ТРАНСФОРМИРОВАННОЙ НУКЛЕАЗОЙ С ЦИНКОВЫМИ ПАЛЬЦАМИ И КОНСТРУКЦИЯМИ ETIP PDAS000271-PDAS000275
Трансгенные события Brassica napus, которые получены путем трансформации ETIP и конструкций нуклеаз с цинковыми пальцами, приводят к встраиванию единичной копии полноразмерной вставки T-цепи полинуклеотидной последовательности ETIP из pDAS000273 или pDAS275 в локус FAD3A. От трех до четырех событий полностью охарактеризовывают и подтверждают, что они содержат встроенный ETIP. Подтверждение осуществляют способом амплификации ПЦР внутри-снаружи, и далее подтверждают с помощью саузерн-блоттинга. Выбранные растения с событиями T0 выращивают до стадии развития T1. Растения T1 повторно подвергают скринингу для определения зиготности встроенной T-цепи. Подвергнутые скринингу события распределяют на категории в качестве гомозиготных, гемизиготных или нулевых.
Гомозиготные события используют для получения протопластов посредством ранее описанного способа. Затем протопласты котрансформируют нуклеазой с цинковыми пальцами, которая сконструирована для нацеливания на участок связывания цинковых пальцев, который включен в последовательность ETEP, и донорной плазмидой, которая обладает гомологией с конкретными областями. Нуклеаза с цинковыми пальцами расщепляет локус ETIP и донорная плазмида встраивается в геном клеток Brassica napus посредством направляемой гомологией репарации. В результате встраивания донорной плазмиды, частичный трансген DS-red репарируется до полноразмерного трансгена DS-red. Экспрессию теперь полностью функционального трансгена DS-red используют для сортировки протопластов клеток способом FACS. Предполагаемые трансгенные растения сортируют с использованием способа FACS, описанного в примере 7, и выделенные протопласты регенерируют в зрелые растения. Встраивание донорной плазмиды подтверждают в растениях с нацеливанием на ETIP с использованием способов молекулярного подтверждения. По существу, локус ETIP служит в качестве сайт-специфического локуса для направленного встраивания гена донорной полинуклеотидной последовательности.
ПРИМЕР 7: СОРТИРОВКА НА ОСНОВЕ FACS КЛЕТОК ПРОТОПЛАСТОВ
Протопласты Brassica napus, которые были трансфицированы контрольной конструкцией DS-Red pDAS000031, сортировали с помощью опосредуемой FACS сортировки клеток с использованием сортера BD Biosciences Influx-Cell sorterTM (San Jose, CA). Протопласты клеток выделяли и трансфицировали, как описано в примере 3. После трансфекции клеток pDAS000031, клетки сортировали с использованием сортера FACS с условиями, описанными в таблице 23.
Протопласты, которые экспрессировали трансген DS-red, сортировали и выделяли. Выделенные с помощью FACS протопласты подсчитывали с использованием сортера. Приблизительно от 1×105 до 1,8×105 клеток помещали в лунку 24-луночного микропланшета для титрования на первые сутки после выделения с помощью FACS. Клетки переносили в гранульную культуру на от 5 до 20 суток. Исследовали сходные условия, где приблизительно 1×104 клеток помещали в ячейку микропланшета для титрования с 2 или 4 ячейками на вторые сутки после выделения с помощью FACS. Различные условия, которые исследовали, привели к выделению клеток с жизнеспособностью 95-98% от всех выделенных протопластов клеток. Отсортированные с помощью FACS протопласты клеток переносили в гранульную культуру на 3-20 суток. Отсортированные с помощью FACS протопласты клеток регенерировали в растения на среде, которая содержала 1,5 мг/мл гигромицина, с использованием описанного выше протокола. Было подтверждено, что предполагаемые трансгенные растения содержат интактную вставку T-цепи из pDAS000031 с помощью протоколов молекулярного подтверждения.
НАЦЕЛИВАНИЕ DS-RED НА ЛИНИИ С ETIP С ПОМОЩЬЮ ОПОСРЕДУЕМОЙ ZFN ГОМОЛОГИЧНОЙ РЕКОМБИНАЦИИ
Получали линию канолы, содержащую вставку T-цепи из pDAS000036, и с помощью молекулярной охарактеризации подтверждали, что она содержит полноразмерную единичную копию T-цепи. Эти события в каноле обозначили как pDAS000036-88 и их использовали для получения протопластов описанным выше способом. Протопласты выделяли, а затем ~50000 протопластов клеток растений котрансформировали нуклеазой с цинковыми пальцами, либо pDAS000074 (Фиг. 25) либо pDAS000075 (Фиг. 26), которые были сконструированы для нацеливания на участки связывания цинковых пальцев, включенные в последовательность ETIP, и донорной плазмидой pDAS000064, pDAS000068, pDAS000070 или pDAS000072 (Фиг. 27, Фиг. 28, Фиг. 29 и Фиг. 30, соответственно), которая обладает гомологией с конкретными областями ETIP. На Фиг. 19 и Фиг. 20 представлены иллюстрации направляемой гомологией репарации, которая приводит к сайт-специфическому встраиванию трансгена Ds-red посредством опосредуемой нуклеазой с цинковыми пальцами гомологичной рекомбинации. Нуклеаза с цинковыми пальцами была сконструирована для расщепления локуса ETIP в определенной последовательности связывания цинковыми пальцев, и тем самым для создания двухцепочечного разрыва в геноме. Далее донорную плазмиду встраивали в геном протопластов клеток Brassica napus посредством направляемой гомологией репарации. Области интрона-1 и интрона-2 донорной плазмиды обладают гомологией с соответствующими областями интрона-1 и интрона-2 локуса ETIP. В результате встраивания донорной плазмиды частичный трансген DS-red репарировался до полноразмерного трансгена DS-red с высокой экспрессией. Экспрессию полностью функционального трансгена DS-red использовали для сортировки протопластов клеток с помощью описанного выше способа FACS. По существу, локус ETIP служит в качестве сайт-специфического локуса для направленного встраивания донорной полинуклеотидной последовательности. Наконец, выделенные протопласты можно сортировать и регенерировать в зрелые растения. Встраивание донорной плазмиды можно подтверждать в растениях с нацеливанием на ETIP с использованием способов молекулярного подтверждения.
Донорную плазмидную ДНК и плазмидную ДНК ZFN смешивали в различных концентрациях, использованных для трансфекции протопластов клеток канолы, содержащих событие pDAS000036-88, и трансгенные протопласты клеток сортировали с использованием трансфекции FACS, которая описана ранее. В таблице 24 описаны различные эксперименты по трансфекции и концентрации ДНК, которые использовали для трансфекции протопластов канолы, содержащих событие pDAS000036-88. ДНК ZFN и донорную плазмидную ДНК выделяли и подготавливали для трансфекции описанными выше способами.
После завершения экспериментов по трансфекции протопласты инкубировали при комнатной температуре в течение 48 часов и хранили с использованием описанного выше протокола FACS. Сортировку для каждого эксперимента проводили независимо и опосредуемую цинковыми пальцами интрогрессию трансгена подтверждали посредством идентификации индивидуальных событий, в случае которых экспрессировался трансген DS-red. На Фиг. 21-24 представлены результаты сортировки FACS. Как указывают результаты, представленные на графиках, было получено множество событий, которые содержали интактный полностью встроенный трансген DS-red. Эти множественные события Ds-Red были результатом опосредуемого цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP. Это сайт-специфическое встраивание обеспечивало полную копию трансгена Ds-Red с высокой экспрессией. Частота экспрессии трансгена Ds-Red находилась в диапазоне приблизительно 0,03-0,07% от всех протопластов клеток канолы (~50000). Однако частота эффективности трансфекции для опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP была значительно более высокой и находилась в диапазоне приблизительно 0,07-0,64%.
На Фиг. 21 представлены результаты трансфекций, при которых котрансфицировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000064 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 26 мкг к 4 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,03% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,22-0,07%. Аналогично, на нижнем графике, когда донор, pDAS000064 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 26 мкг к 4 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,03% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,26-0,08%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
На Фиг. 22 представлены результаты трансфекций, при которых котрансформировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000068 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,03% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,22-0,07%. Аналогично, на нижнем графике, когда донор, pDAS000068 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,04% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,38-0,12%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
На Фиг. 23 представлены результаты трансфекций, при которых котрансформировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000070 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,07% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,64-0,21%. Аналогично, на нижнем графике, когда донор, pDAS000070 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,04% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,34-0,11%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
На Фиг. 24 представлены результаты трансфекций, при которых котрансформировали донорную плазмиду и плазмиду ZFN. На верхнем графике представлено, что, когда донор, pDAS000072 и нуклеазу с цинковыми пальцами pDAS000074 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,07% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансформировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,62-0,20%. Аналогично, на нижнем графике, когда донор, pDAS000072 и нуклеазу с цинковыми пальцами pDAS000075 котрансформировали в соотношении 28 мкг к 2 мкг плазмидной ДНК, это приводило к опосредуемому нуклеазой с цинковыми пальцами встраиванию донорной конструкции ДНК в геномный локус ETIP с частотой рекомбинации приблизительно 0,05% в ~50000 протопластах клеток канолы. В действительности, частота рекомбинации является значительно более высокой. Среди ~50000 протопластов клеток канолы, которые были предоставлены в ходе эксперимента по трансфекции, в действительности трансфицировались только приблизительно 10-30% этих протопластов клеток канолы. По существу, фактическая эффективность трансфекции опосредуемого нуклеазой с цинковыми пальцами встраивания донорной конструкции ДНК в геномный локус ETIP находится в диапазоне приблизительно 0,44-0,14%. Результаты для опосредуемой цинковыми пальцами направляемой гомологией репарации являются значительно более высокими, чем для экспериментов с отрицательным контролем, где был идентифицирован только один протопласт из ~50000, который имел красную флуоресценцию, что, тем самым, указывает на частоту рекомбинации 0,00%, как показано на Фиг. 20.
Способ сортировки FACS прямо применим для скрининга любой флуоресцентной последовательности трансгена, и его используют для выделения части протопластов клеток Brassica napus, на которую нацелился флуоресценоный трансген через опосредуемую гомологией репарацию в конкретном участке области ETIP в геномном локусе.
ПРИМЕР 8: ДОПОЛНИТЕЛЬНЫЕ КОНСТРУКЦИИ ETIP
КОНСТРУИРОВАНИЕ ВЕКТОРОВ ДЛЯ ТРАНСФОРМАЦИИ РАСТЕНИЙ
Исходный и конечный векторы Gateway® (INVITROGEN) конструировали стандартными способами молекулярного клонирования и использовали для создания окончательных экспрессирующих векторов. Исходный вектор (pDAB105852) содержал последовательность RB7 MAR (Hall et al., 1991) и участок связывания v1 eZFN4 (публикация патента США №20110191899). Другой исходный вектор (pDAB105853) содержал экспрессирующую кассету, содержащую промотор убиквитина риса 3 (OsUbi3) (Sivamani, E., Qu, R., (2006) Plant Molecular Biology 60; 225-239), кодирующую область маркерного гена желтого флуоресцентного белка (Phi YFP) (Shagin, D. A., (2004) Mol Biol Evol. 21;841-50) (Evrogen, Moscow Russia), содержащую интрон ST-LS1 (Vancanneyt, G., (1990) Mol Gen Genet. 220;245-50), за которым следует фрагмент, содержащий 3'-нетранслируемую область (UTR) из гена пероксидазы 5 кукурузы (ZmPer5 3'UTR v2; патент США №6699984), участок связывания eZFN1 и сконструированную способами инженерии подушку для встраивания (ELP1 HR2 v2) (публикация патента США №20110191899). Следующий исходный вектор (pDAB105850) содержал кодирующую область белка Cry34Ab1 (патент США №8273535) под контролем экспрессии копии промотора убиквитина 1 с интроном 1 кукурузы (промотор v8 ZmUbi1) (Christensen, A. H., Quail, P. H., (1996) Transgenic Research 5;213-218; Christensen, A.H., (1992) Plant Molecular Biology 18;675-689) и фрагмент, содержащий 3'UTR StPinII из картофеля (An, G., (1989) Plant Cell. 1; 115-22). Дополнительный исходный вектор (pDAB105851) содержал промотор пероксидазы пшеницы (TaPer) (Hertig, C., (1991) Plant Mol Biol. 16; 171-4) и кодирующую область белка Cry35Ab1 (публикация патента США №20110191899), за которой следует фрагмент, содержащий 3'UTR StPinII из картофеля.
Трансформирующие/экспрессирующие векторы для опосредуемой Agrobacterium трансформации зародышей кукурузы конструировали с использованием стандартных спосбов клонирования и реакций рекомбинации Gateway® в которых используется типичный конечный бинарный вектор (pDAB109805) и исходные векторы, как описано выше. Бинарный конечный вектор pDAB109805 содержал ген устойчивости к гербициду (арилоксиалканоатдиоксигеназа (AAD-1); (патент США №7838733) под контролем экспрессии промотора палочковидного вируса сахарного тростника (SCBV); по существу как описано в патенте США №6093569. Для терминации транскрипции мРНК AAD-1 использовали фрагмент, содержавший 3'-UTR из гена липазы кукурузы (3'-UTR ZmLip, патент США №7179902). Экспрессия AAD-1 сообщает устойчивость к гербицидным соединениям, таким как галоксифоп и квизалофоп. Реакцию рекомбинации Gateway® использовали для рекомбинации исходных векторов pDAB105852, pDAB105853, pDAB105850 и pDAB105851 с конечным вектором pDAB109805 с получением экспрессирующего вектора pDAB105855 (Фиг. 36), который использовали в качестве целевого вектора. Плазмида ETIP содержит pDAB105855 заданную последовательность ДНК, содержажщую: последовательность RB7 MAR/участок свяызвания v1 eZFN4, промотор OsUbi3/Phi YFP/3'UTR v2 ZmPer5/участок связывания eZFN1/ELP1 HR2 v2, промотор v8 ZmUbi1/Cry34Ab1 v2/3'-UTR v2 StPinII, промотор v3 TaPer/3'-UTR v2 Cry35Ab1 v5/StPinII, SCBVv2/AAD-1v3/3'-UTR v1 ZmLip между границами T-ДНК. Плазмида pDAB105855 представляет собой плазмиду ETIP и ее использовали для встраивания в геном кукурузы.
Вектор нуклеазы с цинковыми пальцами (ZFN1) (pDAB105941; Фиг. 37) содержал кодирующую последовательность ZFN1 под контролем экспрессии промотора убиквитина 1 с интроном 1 кукурузы (промотор v2 ZmUbi1) и 3'UTR v2 ZmPer5. Донорный вектор (pDAB112366; Фиг. 38) содержал интрон без промотора (интрон rubi3), промотор убиквитина 3 риса (OsUbi3), за которым следовал ген устойчивости к гербициду (фосфинотрицинацетилтрансфераза (PAT); Wehrmann et al. (1996) Nature Biotechnology 14:1274-1278), и 3'UTR ZmLip. Эта конфигурация донора без активного промотора не продуцирует матричную РНК гена PAT для селекции с гербицидом. После 3'UTR ZmLip в донорном векторе следуют участки связывания eZFN4 и eZFN8 и ELP1 HR2 v2, как описано в публикации патента США №20110191899. Последовательности 5'-интрона rubi3 и 3'-ELP1 HR2 v2 доноре (pDAB112366) используют для 5'- и 3'-гомологии с мишенью (pDAB105855) для нацеливания гена для точной замены последовательности Phi YFP в мишени последовательностью PAT донора с получением функционального PAT, контролируемого промотором убиквитина риса 3 (OsUbi3), обеспечивающего положительную селекцию нацеливания гена.
Конструировали положительный контрольный вектор PAT (pDAB112364), который содержал промотор убиквитина 3 риса (OsUbi3), контролирующий ген устойчивости к гербициду (фосфинотрицинацетилтрансфераза (PAT), за которым следовала 3'UTR ZmLip, и его использовали в последующих экспериментах.
Трансформация Agrobacterium tumefaciens
Бинарные экспрессирующие векторы трансформировали в Agrobacterium tumefaciens тройного штамма DAt13192 (международная публикация патента № WO 2012016222). Бактериальные колонии выделяли и бинарную плазмидную ДНК выделяли и подтверждали посредством расщепления ферментом рестрикции.
Опосредуемая Agrobacterium трансформация
Опосредуемую Agrobacterium трансформацию использовали для стабильного встраивания описанных выше трансгенов в геном растений и, таким образом, для получения трансгенных клеток, тканей и растений кукурузы, которые продуцируют AAD-1. Способы трансформации кукурузы с использованием супербинарных или бинарных векторов для трансформации известны в данной области, как описано, например, в международной публикации PCT № WO 2010/120452. Трансформированные ткани отбирали по их способности расти на среде, содержащей галоксифоп или биалафос.
ИНИЦИАЦИЯ КУЛЬТУРЫ AGROBACTERIUM
Исходные культуры в глицерине проектных векторов были предоставлены в штамме-хозяине DAt13192 Agrobacterium tumefaciens (WO 2012/016222A2). Культуры Agrobacterium наносили штрихами из исходных культур в глицерине на минимальную среду AB и инкубировали при 20°C в темноте в течение 3 суток в присутствии соответствующих антибиотиков. Затем культуры наносили штрихами на чашку со средой YEP с антибиотиками и инкубировали при 20°C в темноте в течение 1 суток.
В день эксперимента приготавливали смесь среды для инокуляции и ацетосирингона (Frame et al. (2011) Methods in Molecular Biology 710:327-341) в объеме, соответствующем количеству конструкций в эксперименте, и переносили пипеткой в стерильную одноразовую 250-мл колбу. Среда для инокуляции содержит: 2,2 г/л солей MS; 1X модифицированные ISU витамины MS (Frame et al., там же), 68,4 г/л сахарозы; 36 г/л глюкозы; 115 мг/л L-пролина и 100 мг/л миоинозитола; при pH 5,4.). Ацетосирингон добавляли в колбу, содержавшую среду для инокуляции, до конечной концентрации 200 мкМ из 1 M исходного раствора в 100% диметилсульфоксиде.
Для каждой конструкции 1 или 2 полных инокулирующих петель Agrobacterium из чашки YEP суспендировали в 15 мл смеси среда для инокуляции/ацетосирингон внутри стерильной одноразовой 50-мл центрифужной пробирки и измеряли оптическую плотность при 550 нм (OD550) в спектрофотометре. Затем суспензию разбавляли до OD550 от 0,3 до 0,4 с использованием дополнительной смеси среда для инокуляции/ацетосирингон. Затем пробирку с суспензией Agrobacterium помещали горизонтально на устройство для встряхивания с платформой, установленное приблизительно на 75 об./мин. при комнатной температуре и встряхивали в течение 1-4 часов пере применением.
СТЕРИЛИЗАЦИЯ ПОЧАТКОВ И ВЫДЕЛЕНИЕ ЗАРОДЫШЕЙ
Початки Zea mays сорта B104 получали в теплице и собирали через 10-12 суток после опыления. Собранные початки очищали и подвергали поверхностной стерилизации путем погружения в 20% раствор коммерческого отбеливателя (Ultra Clorox® Germicidal Bleach, 6,15% гипохлорит натрия; с двумя каплями TWEEN 20) в течение 20 минут, а затем проводили три промывания в стерильной деионизированной воде в ламинарном вытяжном шкафу. Незрелые зиготические зародыши (длиной 1,8-2,2 мм) в асептических условиях из каждого початка и распределяли в одну или несколько микроцентрифужных пробирок, содержавших 2,0 мл суспензии Agrobacterium, в которую было добавлено 2 мкл 10% поверхностно-активного вещества BREAK-THRU® S233 (Evonik Industries; Essen, Германия).
СОКУЛЬТИВИРОВАНИЕ С АГРОБАКТЕРИЯМИ
После выделения зародышей помещали на платформу качалки на 5 минут. Затем содержимое пробирки выливали на чашку со средой для сокультивирования, которая содержала 4,33 г/л солей MS; 1× модифицированные ISU витамины MS; 30 г/л сахарозы; 700 мг/л L-пролина; 3,3 мг/л дикамбы в KOH (3,6-дихлор-о-анисовая кислота или 3,6-дихлор-2-метоксибензойная кислота); 100 мг/л миоинозитола; 100 мг/л ферментативного гидролизата казеина; 15 мг/л AgNO3; 200 мкМ ацетосирингон в DMSO;и 3 г/л агара (SIGMA-ALDRICH, протестированный для культуры клеток растений) при pH 5,8. Жидкую суспензию Agrobacterium удаляли стерильной одноразовой пипеткой для переноса и чашку для сокультивирования, содержавшую зародыши, помещали в заднюю часть ламинарного вытяжного шкафа с приоткрытой крышкой на 30 минут, после чего зародышей ориентировали щитком вверх с использованием стерильного пинцета с помощью микроскопа. Чашку возвращали в заднюю часть ламинарного вытяжного шкафа с приоткрытой крышкой на дополнительные 15 мин. Затем чашку закрывали, герметизировали медицинской лентой 3M™ Micropore™ и помещали в инкубатор при 25°C при постоянном освещении с интенсивностью освещения приблизительно 60 мкЭм-2 с-1.
СЕЛЕКЦИЯ КАЛЛЮСОВ И РЕГЕНЕРАЦИЯ ТРАНСГЕННЫХ СОБЫТИЙ
После периода сокультивирования зародышей переносили в среду покоя, которая состояла из 4,33 г/л солей MS; 1× модифицированных ISU витаминов MS; 30 г/л сахарозы; 700 мг/л L-пролина; 3,3 мг/л дикамбы в KOH; 100 мг/л миоинозитол; 100 мг/л ферментативного гидролизата казеина; 15 мг/л AgNO3; 0,5 г/л MES (моногидрат 2-(N-морфолино)этансульфоновой кислоты; PhytoTechnologies Labr.; Lenexa, KS); 250 мг/л цефотаксима и 7,0 г/л агара при pH 5,8. В каждую чашку перемещали не более 36 зародышей. Чашки оборачивали лентой MicroporeTM и инкубировали при 27°C при постоянном освещении при интенсивности освещения приблизительно 50 мкмоль м-2 с-1 в течение от 7 до 10 суток. Затем каллюсы зародышей (<18/чашка) переносили в среду для селекции I, которая состояла из среды покоя (выше), но содержала только 6,5 г/л агара и либо с 100 нМ R-галоксифоп кислоты (0,0362 мг/л; для селекции трансформантов, содержащих ген AAD-1), либо 5,0 мг/л биалафоса (для селекции трансформантов, содержащих ген PAT), в соответствующих случаях. Биалафос был предоставлен в качестве Herbiace®. Планшеты оборачивали пленкой MicroporeTM и инкубировали при 27°C при непрерывном освещении с интенсивностью освещения приблизительно 50 мкЭм-2 с-1 в течение 7 суток. Затем каллюсы зародышей (<12/чашка) переносили в среду для селекции II, которая состояли из среды покоя (выше), но только с 6,5 г/л агара, и либо с 50 нМ R-галоксифоп кислоты (0,0181 мг/л), либо 5,0 мг/л биалафоса, в соответствующих случаях. Чашки оборачивали и инкубировали при 27°C при непрерывном освещении с интенсивностью освещения приблизительно 50 мкЭм-2 с-1 в течение 14 суток.
На этой стадии устойчивые каллюсы (<9/чашка) переносили в предрегенерационную среду. Предрегенерационная среда содержит 4,33 г/л солей MS; 1× модифицированные ISU витамины MS; 45 г/л сахарозы; 350 мг/л L-пролина; 100 мг/л миоинозитол; 50 мг/л ферментативного гидролизата казеина; 1,0 мг/л AgNO3; 0,5 г/л MES; 0,5 мг/л нафталинуксусной кислоты в NaOH; 2,5 мг/л абсцизовой кислоты в этаноле; 1 мг/л 6-бензиламинопурина; 250 мг/л цефотаксима; 5,5 г/л агара и либо 50 нМ R-галоксифоп кислоту, либо 3,0 мг/л биалафоса, в соответствующих случаях; при pH 5,8. Планшеты оборачивали и инкубировали при 27°C при непрерывном освещении с интенсивностью света приблизительно 50 мкЭм-2 с-1 в течение 7 суток. Затем регенерирующие каллюсы (<6/чашка) переносили в регенерационную среду в лотках PhytatrayTМ (sigma-aldrich) и инкубировали при 28°C с 16 часов на свету/8 часов в темноте в сутки при интенсивности освещения приблизительно 150 мкмоль м-2 с-1 в течение 14 суток или до развития побегов. Регенерационная среда содержит 4,33 г/л солей MS; 1× модифицированных ISU витаминов MS; 60 г/л сахарозы; 0,50 г/л MES; 125 мг/л цефотаксима; 5,5 г/л агара и либо 50 нМ R-галоксифоп кислоты, либо 3,0 мг/л биалафоса, в соответствующих случаях; при pH 5,8. Затем небольшие побеги с первичными корнями выделяли и переносили без селекции в среду для удлинения (т.е. регенерационная среда без R-галоксифоп кислоты или биалафоса) для дальнейшего роста. Укоренившиеся ростки длиной приблизительно 6 см или более переносили в почву и перемещали для закаливания в камеру для выращивания.
ПЕРЕНОС И ВЫРАЩИВАНИЕ РАСТЕНИЙ T0 В ТЕПЛИЦЕ ДЛЯ АНАЛИЗА и ПРОДУЦИРОВАНИЯ СЕМЯН
Трансформированные ткани растений, подвергнутые селекции по их способности расти на среде, содержащей либо галоксифоп, либо биалафос, в соответствующих случаях, пересаживали из лотков Phytatray™ в небольшие горшки (T. O. Plastics, 3,5'' SVD), заполненные средой для выращивания (ProMix BX; Premier Tech Horticulture), накрывали куполами для увлажнения (Arco Plastics Ltd.), а затем закаливали в помещении для выращивания (28°C днем/24°C ночью, 16-часовой световой период, 50-70% RH, интенсивность освещения 200 мкЭм-2 c-1). Когда растения достигали стадии V3-V4, их пересаживали в почвенную смесь Sunshine Custom Blend 160 и выращивали до цветения в теплице (тип освещения: световой или ассимиляция; верхний предел освещения: 1200 PAR; продолжительность дня 16 часов; 27°C днем/24°C ночью). Предполагаемые трансгенные ростки анализировали в отношении количества копий трансгена с помощью количественного анализа ПЦР в реальном времени с использованием праймеров, сконструированных для детекции относительных количеств копий трансгенов и растения с однокопийными событиями, отобранные для выращивания, пересаживали в горшки объемом 5 галлонов (18,9 см3). Наблюдения проводили периодически для отслеживания каких-либо аномальных фенотипов.
ПОЛУЧЕНИЕ ГЕМИЗИГОТНЫХ ЗАРОДЫШЕЙ T1S1 В ТЕПЛИЦЕ ДЛЯ БОМБАРДИРОВКИ ЧАСТИЦАМИ
Растения, трансгенные по последовательности-мишени, подвергали самоопылению с получением семян T1. Семена T1 высевали и растения анализировали в отношении зиготности трансгена-мишени с использованием способа q-ПЦР. Растения, гомозиготные по трансгену-мишени, далее выращивали для получения пыльцы. Пыльцу гомозиготных по мишени растений использовали для обратного скрещивания растений кукурузы B104 с получением гемизиготных незрелых зародышей T1S1. Незрелые зародыши использовали для бомбардировки частицами с донорной ДНК и ДНК ZFN1 для исследования нацеливания на ген.
НАЦЕЛИВАНИЕ НА НЕЗРЕЛЫЕ ЗАРОДЫШИ КУКУРУЗЫ ПОСРЕДСТВОМ БОМБАРДИРОВКИ МИКРОЧАСТИЦАМИ
За трое суток до бомбардировки микрочастицами зародыши размером 1,5-2,2 мм выделяли из подвергнутых поверхностной стерилизации початков и помещали (щитком вверх) на базальную среду N6 и витамины (Phytotechnology Laboratories, Shawnee Mission, KS) с 2,0 мг/л 2,4-D, 2,8 г/л пролина, 30 г/л сахарозы, 100 мг/л ферментативного гидролизата казеина, 100 мг/л миоинозитола и 4,25 мг/л нитрата серебра, отвержденную 2,5 г/л Gelzan (Phytotechnology Laboratories, Shawnee Mission, KS). За четыре часа до бомбардировки микрочастицами ~35-40 зародышей помещали (щитком вверх) в центр чашки Петри размером 100×15 мм, содержавшей ту же среду с добавлением 36,4 г/л сорбита и 36,4 г/л маннита.
Золотые микрочастицы (0,6 микрометров, BioRad, Hercules, CA,) подготавливали для осаждения ДНК путем отвешивания 15 мг в стерильную силиконизированную 1,7-мл микроцентрифужную пробирку (Sigma-Aldrich, St. Louis, MO, T3406) и медленно добавляли 500 мкл ледяного 100% этанола. После обработки ультразвуком в течение 15 секунд на ультразвуковой водяной бане FS-14 (Fisher Scientific, Nazareth, PA), золоту позволяли осесть в течение 30 минут при комнатной температуре, а затем проводили центрифугирование при 3000 об./мин. в течение 60 секунд с использованием MiniSpin (Eppendorf, Hauppauge, NY). После удаления супернатанта добавляли 1 мл ледяной стерильной воды, перемешивали пипеткой вверх и вниз и позволяли осесть в течение 3-5 минут, а затем проводили центрифугирование при 3000 в течение 60 секунд. Стадию промывания повторяли еще один раз, а затем золото суспендировали в 500 мкл ледяной стерильной воды. Затем промытое золото распределяли аликвотами в отдельные 1,7-мл стерильные силиконизированные микроцентрифужные пробирки (50 мкл на пробирку), обращая внимание на то, чтобы поддерживать золото хорошо перемешанным путем пипетирования в пробирках вверх-вниз. Затем промытое золото (~1,5 мг на 50 мкл) хранили при -20°C пока не потребуется.
Для преципитации ДНК одну пробирку, содержавшую ~1,5 мг золота в 50 мкл воды, размораживали для каждой из 10 мишеней, подлежащих бомбардировке, и обрабатывали на ультразвуковой водяной бане в течение 15 секунд, а затем помещали на лед. Плазмидную ДНК (0,6 мкг ZFN + 4,4 мкг донора) предварительно смешивали в 0,6-мл микроцентрифужных пробирках (Fisher Scientific, Nazareth, PA) и добавляли в суспензию золота, осторожно пипетируя вверх и вниз несколько раз для тщательного перемешивания. Добавляли пятьдесят микролитров (50 мкл) ледяного 2,5 M хлорида кальция и осторожно перемешивали путем пипетирования вверх и вниз несколько раз. Затем добавляли двадцать микролитров (20 мкл) холодного 0,1 M спермидина и осторожно перемешивали путем пипетирования вверх и вниз несколько раз. Затем пробирку закрывали и помещали в Vortex Genie 2 (Scientific Instruments Inc., Bohemia, NY) и перемешивали (с установкой на "shake 2") в течение 10 минут, после чего смеси позволяли осесть в течение 3-5 минут. После центрифугирования в течение 15 секунд при 5000 об./мин., супернатант осторожно извлекали и добавляли 250 мкл ледяного 100% этанола, пробирку закрывали и энергично перемешивали рукой для смещения осадка. После второго центрифугирования в течение 15 секунд при 5000 об./мин. добавляли 120 мкл ледяного 100% этанола, пробирку закрывали и энергично перемешивали рукой для смещения осадка.
Для бомбардировки микрочастицами стерилизованные макроносители (BioRad, Hercules, CA) помещали в штативы из нержавеющей стали (BioRad, Hercules, CA) и автоклавировали. Десять микролитров (10 мкл) суспензии золото/ДНК равномерно распределяли в центре макроносителя, обращая внимание на то, чтобы пипетировать вверх и вниз, чтобы поддерживать хорошее перемешивание, а затем помещали на кусок стерильной фильтровальной бумаги 125-мм Whatman #4 (GE Healthcare, Buckinghamshire, UK) на слое Drierite (W.A Hammond Drierite Co., Xenia, OH) калибра 8 в 140×25 мм стеклянной чашке Петри. Золоту/ДНК позволяли полностью высохнуть в течение приблизительно 10 минут. Разрывные диски (650 фунт./кв. дюйм (4,5 кПа), BioRad, Hercules, CA) стерилизовали путем погружения на несколько минут в изопропиловый спирт, а затем помещали на колпачок клапана устройства для бомбардировки микрочастицами (PDS-1000, BioRad, Hercules, CA). Автоклавированный останавливающий экран (BioRad, Hercules, CA) и загруженный макроноситель помещали в исходную сборку, навинчивали крышку и вставляли в камеру для бомбардировки непосредственно перед форсункой. Чашку Петри, содержавшую покрытый экраном являющийся мишенью лист, открывали и помещали в камеру для бомбардировки на 6 см ниже форсунки. Применяли вакуум (-0,9 бар) и устройство включали.
На следующие сутки (16-20 часов после бомбардировки), бомбардированные зародыши переносили (щитком вверх) в базальную среду N6 и витамины (Phytotechnology Laboratories, Shawnee Mission, KS) с 2,0 мг/л 2,4-D, 2,8 г/л пролина, 30 г/л сахарозы, 100 мг/л ферментативного гидролизата казеина, 100 мг/л миоинозитола.
ОПТИМИЗАЦИЯ PAT ДЛЯ БОМБАРДИРОВКИ ЧАСТИЦАМИ с использованием ПОЛОЖИТЕЛЬНОГО КОНТРОЛЬНОГО ВЕКТОРА PAT (pDAB112364)
Контрольный вектор PAT (pDAB112364) использовали для стандартизации условий кукльтивирования тканей зародышей кукурузы после бомбардировки частицами. Донорный вектор (pDAB112366) исследовали в отношении наличия или отсутствия вектора ZFN1 (pDAB105941) для потенциальной первоначальной селекции. Как показано в таблице 25 ниже, с использованием контрольного вектора PAT (pDAB112364) достигали частоты трансфорамации 9%. Эти данные демонстрируют, что промотор убиквитина 3 риса (OsUbi3) запускает устойчивую экспрессию гена PAT для обеспечения селекции для трансформации растений с использованием бомбардировки частицами незрелых зародышей кукурузы B104. Было получено очень небольшое количество (1-2) растений на селективной среде для PAT для незрелых зародышей, подвергнутых бомбардировке с использованием донорного вектора (pDAB112366) с вектором ZFN1 (pDAB105941) или без него. Эти результаты подтверждают, что донорный вектор (pDAB112366), который содержит интрон rubi3 без вышележащего промотора, не обеспечивает селекции трансформантов по гену PAT.
Бомбардировка частицами незрелых зародышей, трансгенных по мишени pDAB105855, с использованием донора и ZFN1 для нацеливания гена
Незрелые зародыши кукурузы, гемизиготные по мишени pDAB105855, обрабатывали для бомбардировки частицами с использованием предварительно полученной смеси ZFN1 и донорной ДНК (pDAB105941/112366) для обеспечения нацеливания гена с использованием положительной селекции по PAT. В таблице 17 показано целевое событие и количество зародышей, которые использовали для каждого события. Также в таблице 26 указано количество растений, которые успешно регенерировали на селективной среде PAT для каждого целевого события. Всего 68 растений регенерировали с 13563 незрелых обработанных зародышей, охватывающих все события. Небольшое количество растений, полученное на селективной среде PAT, демонстрирует, что большинство ненацеленных случайных вставок донора PAT было устранено.
АНАЛИЗ СПОСОБОМ ПЦР НАЦЕЛИВАНИЯ ГЕНА В ETIP
Q-ПЦР проводили для обнаружения кодирующей последовательности YFP и PAT в 67 растениях всего из 68 растений, которые регенерировали на селективной среде для PAT. Результаты обобщенно представлены в таблице 27. Как указано в таблице 27, с использованием анализа q-ПЦР было обнаружено, что 50 растений являются положительными по PAT, в то время как 24 растения были отрицательными по кодирующей YFP последовательности. Данные указывают на то, что 17 растений, полученных при селекции с PAT, ускользнули от селективного агента, и экспрессия ZFN1 разрушила локус-мишень YFP по меньшей мере в 24 растениях.
Кроме того, диагностическую ПЦР внутри/снаружи проводили для 24 отрицательных по YFP событий для измерения нацеливания гена. В 5' ПЦР внутри/снаружи используется специфичный к ДНК-мишени 5'-олигонуклеотид, который гибридизуется с промотором OsUbi3 и специфичный к донорной ДНК 3'-олигонуклеотид, который гибридизуется с кодирующей последовательностью PAT, для получения ожидаемого продукта ПЦР размером 1838 п.н. Ожидаемый продукт ПЦР служит доказательством точного нацеливания гена в 5'-области локуса-мишени. Результаты ПЦР подтвердили, что ожидаемый продукт размером 1838 п.н. амплифицировался в случае 21 события.
Аналогично проводили 3' ПЦР внутри-снаружи. В способе используется специфичный к донорной ДНК 5'-олигонуклеотид, который гибридизуется с кодирующей последовательностью PAT, и специфичный к донорной ДНК 3'-олигонуклеотид, который гибридизуется с последовательностью промотора ZmUbi1, для получения ожидаемого продукта ПЦР размером 2184 п.н. Ожидаемый продукт ПЦР служит доказательством точного нацеливания гена в 3'-области локуса-мишени. Результаты ПЦР подтвердили, что ожидаемый ампликон размером 2184 п.н. продуцировался в случае 16 событий.
Данные ПЦР внутри/снаружи как для 5'-области, так и для 3'-областис локуса-мишени обобщенно представлены в таблице 28. Данные указывают на то, что 15 событий привели к ожидаемым продуктам ПЦР, что показало приблизительно 30% частоту нацеливания генов среди всех положительных по PAT регенерировавших растений.
АНАЛИЗ С ИСПОЛЬЗОВАНИЕМ САУЗЕРН-БЛОТТИНГА НАЦЕЛИВАНИЯ ГЕНОВ
Охарактеризация целевых событий с использованием анализа посредством саузерн-блоттинга выявила укорочение трансгена-мишени в случае события #12. Всего 12 событий анализировали для саузерн-блоттинга. Данные подтвердили, что ожидаемый характер полос был идентифицирован в 5 растениях (3 для события #12 и 2 других события). Эти результаты показали, что на конструкцию ETIP, которая была встроена в геном, впоследствии можно проводить повторное нацеливание донорных последовательностей, и что ETIP могут служить в качестве платформы для нацеливания. На Фиг. 39 представлена схема использования участка ETIP, встроенного в геном, для нацеливания донорного полинуклеотида, который содержит экспрессирующую кассету для гена. Как представлено на схеме, донор способен экспрессировать трансген, если встраивание происходит в локус ETIP. Случайное встраивание донорного полинуклеотида не приводит к экспрессии трансгена, поскольку отсутствуют промоторные элементы, которые могут запускать экспрессию, когда донорный полинуклеотид случайным образом встраивается в геном. Нацеливание донора на участок ETIP приводит к функциональному маркерному гену, который можно выявлять и по которому можно проводить селекцию.
Хотя определенные иллюстративные варианты осуществления описаны в настоящем описании, специалистам в данной области будет известно и понятно, что можно проводить множество дополнений, удалений или модификаций иллюстративных вариантов осуществления без отклонения от объема нижеследующей формулы изобретения. Кроме того, признаки одного варианта осуществления можно комбинировать с признаками другого варианта осуществления.
| название | год | авторы | номер документа |
|---|---|---|---|
| ОБОГАЩЕНИЕ АКТИВИРУЕМОЙ ФЛУОРЕСЦЕНЦИЕЙ СОРТИРОВКИ КЛЕТОК (FACS) ДЛЯ СОЗДАНИЯ РАСТЕНИЙ | 2013 |
|
RU2679510C2 |
| ЛОКУСЫ ФУНКЦИОНАЛЬНОСТИ FAD2 И СООТВЕТСТВУЮЩИЕ СПЕЦИФИЧНЫЕ К УЧАСТКУ-МИШЕНИ СВЯЗЫВАЮЩИЕ БЕЛКИ, СПОСОБНЫЕ ИНДУЦИРОВАТЬ НАПРАВЛЕННЫЕ РАЗРЫВЫ | 2013 |
|
RU2656159C2 |
| ЛОКУСЫ FAD3 ДЛЯ ВЫПОЛНЕНИЯ ОПЕРАЦИЙ И СООТВЕТСТВУЮЩИЕ СВЯЗЫВАЮЩИЕСЯ СО СПЕЦИФИЧЕСКИМИ САЙТАМИ-МИШЕНЯМИ БЕЛКИ, СПОСОБНЫЕ К ВЫЗОВУ НАПРАВЛЕННЫХ РАЗРЫВОВ | 2013 |
|
RU2665811C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ВСТРАИВАНИЯ ЭКЗОГЕННОЙ ПОСЛЕДОВАТЕЛЬНОСТИ В ГЕНОМ РАСТЕНИЙ | 2014 |
|
RU2723130C2 |
| ФУНКЦИОНАЛЬНЫЕ ЛОКУСЫ FAD2 И СООТВЕТСТВУЮЩИЕ СПЕЦИФИЧНЫЕ ДЛЯ САЙТА-МИШЕНИ СВЯЗЫВАЮЩИЕСЯ БЕЛКИ, СПОСОБНЫЕ ИНДУЦИРОВАТЬ НАПРАВЛЕННЫЕ РАЗРЫВЫ | 2013 |
|
RU2656158C2 |
| СПОСОБЫ И КОМПОЗИЦИИ ДЛЯ ОПОСРЕДОВАННОЙ НУКЛЕАЗОЙ НАПРАВЛЕННОЙ ИНТЕГРАЦИИ ТРАНСГЕНОВ | 2013 |
|
RU2650819C2 |
| УНИВЕРСАЛЬНАЯ ДОНОРНАЯ СИСТЕМА ДЛЯ НАПРАВЛЕННОГО ВОЗДЕЙСТВИЯ НА ГЕНЫ | 2014 |
|
RU2687369C2 |
| БЫСТРЫЙ НАПРАВЛЕННЫЙ АНАЛИЗ СЕЛЬСКОХОЗЯЙСТВЕННЫХ КУЛЬТУР ДЛЯ ОПРЕДЕЛЕНИЯ ДОНОРНОЙ ВСТАВКИ | 2014 |
|
RU2668819C2 |
| ОПТИМАЛЬНЫЕ ЛОКУСЫ СОИ | 2014 |
|
RU2719150C2 |
| СПОСОБЫ ОБНАРУЖЕНИЯ ДНК ДЛЯ САЙТ-СПЕЦИФИЧЕСКОЙ НУКЛЕАЗНОЙ АКТИВНОСТИ | 2013 |
|
RU2678001C2 |























































































































































































































































































































































































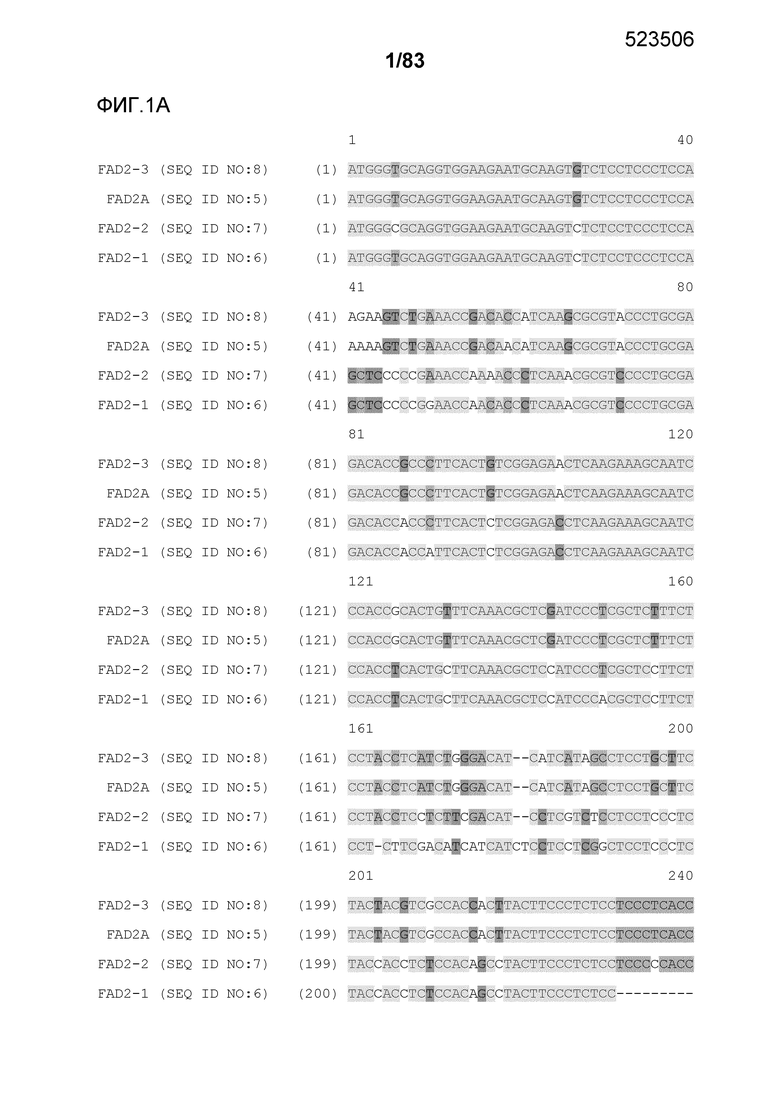




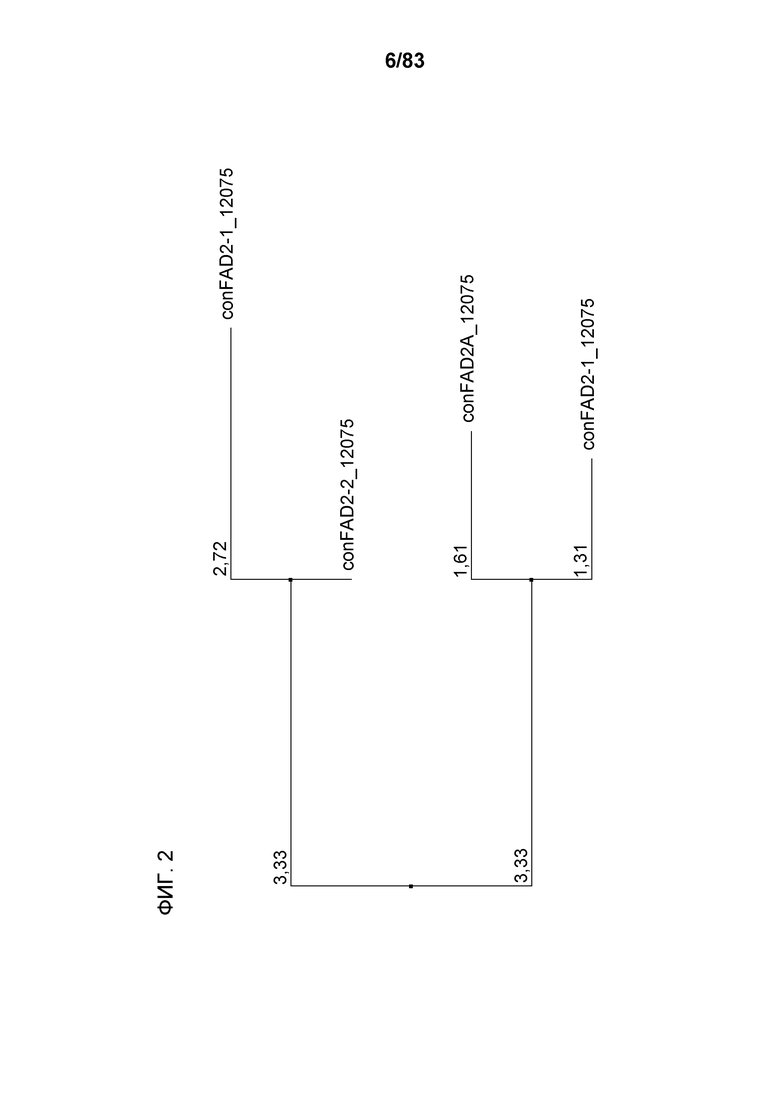



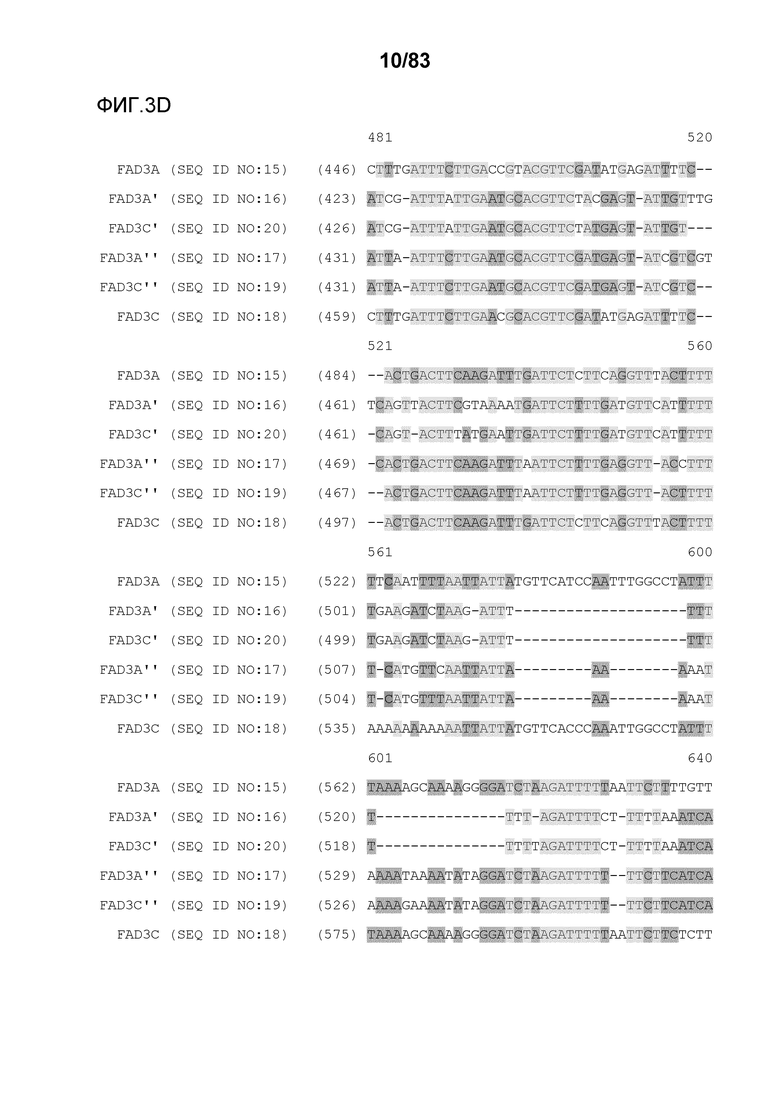





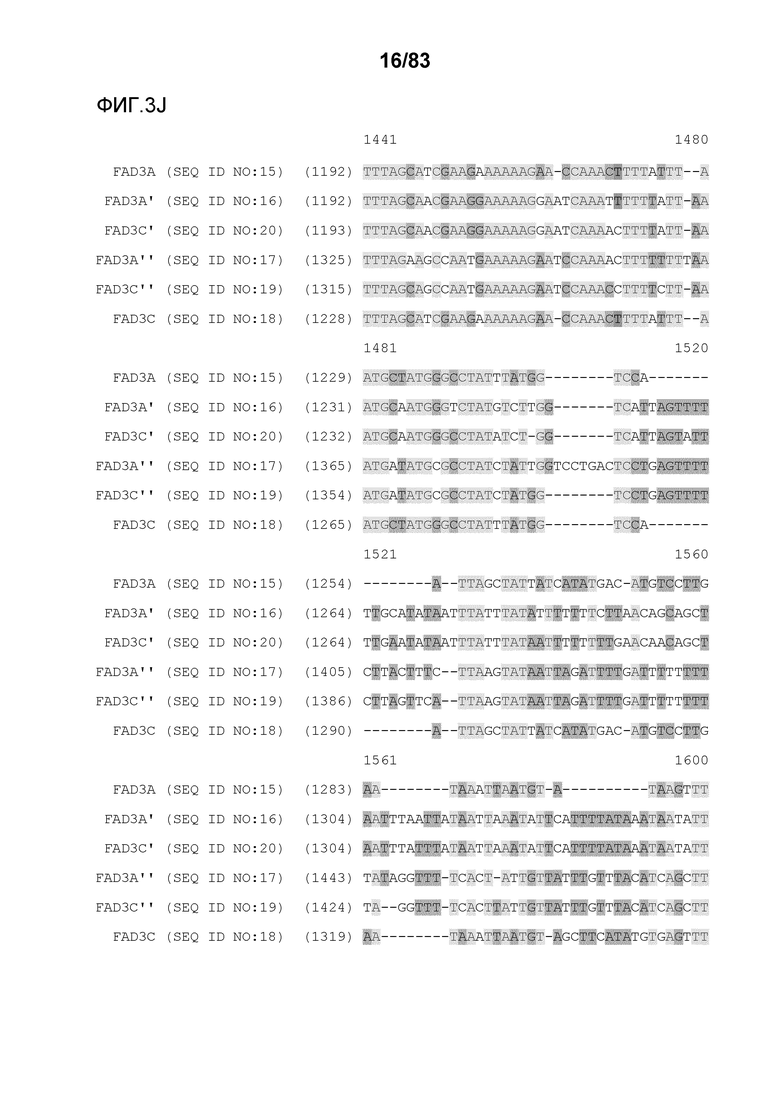


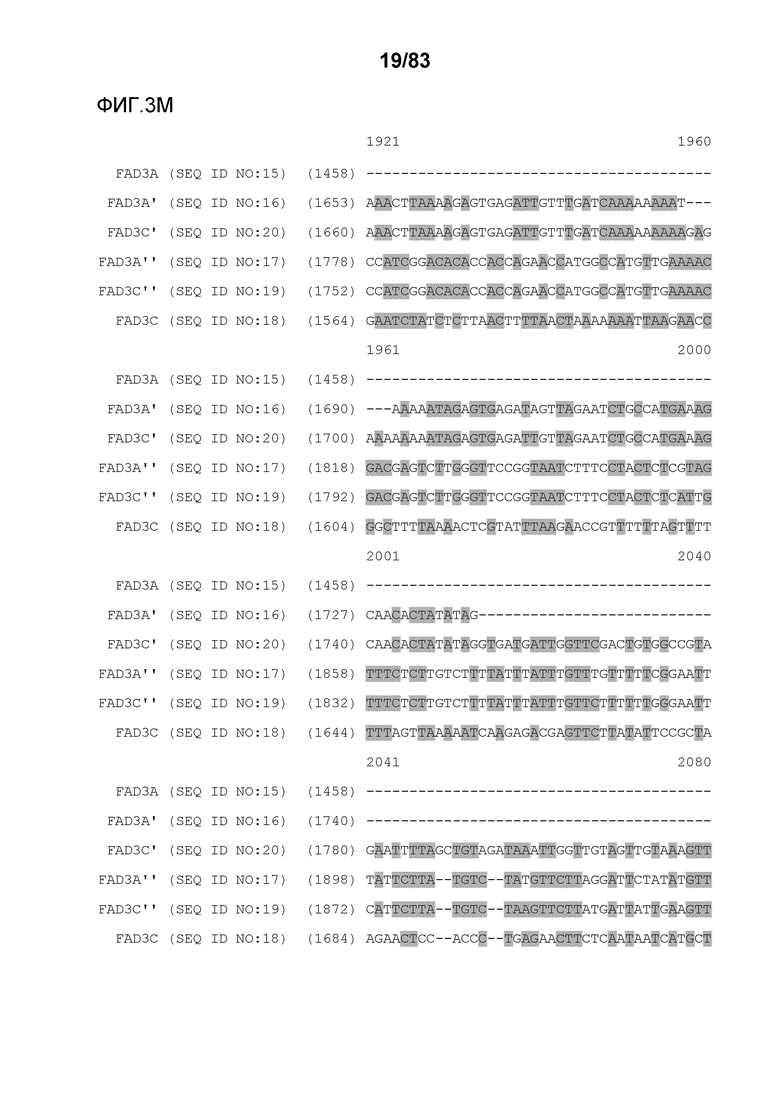






















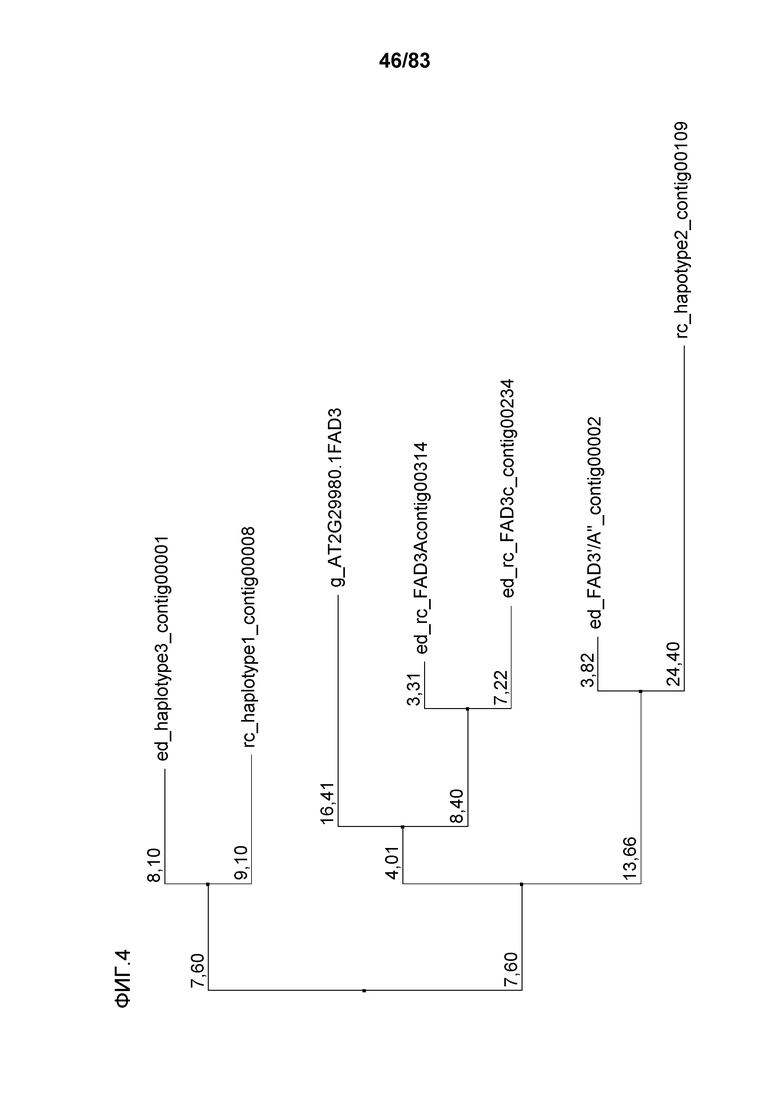
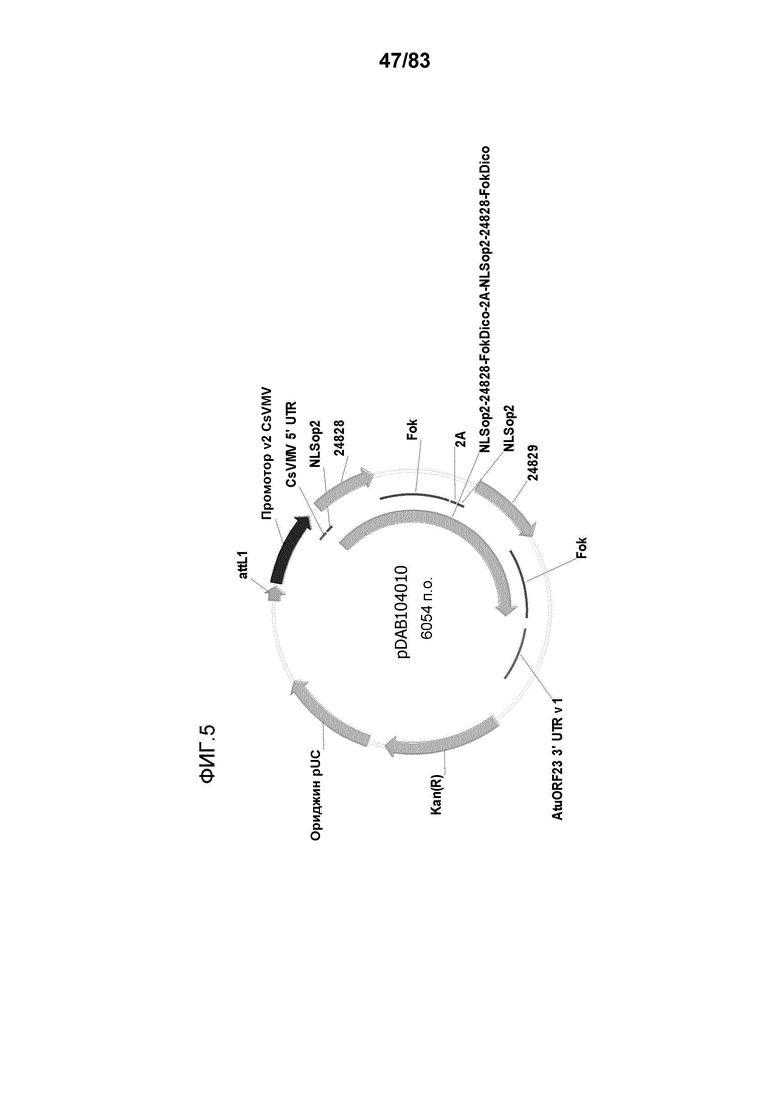
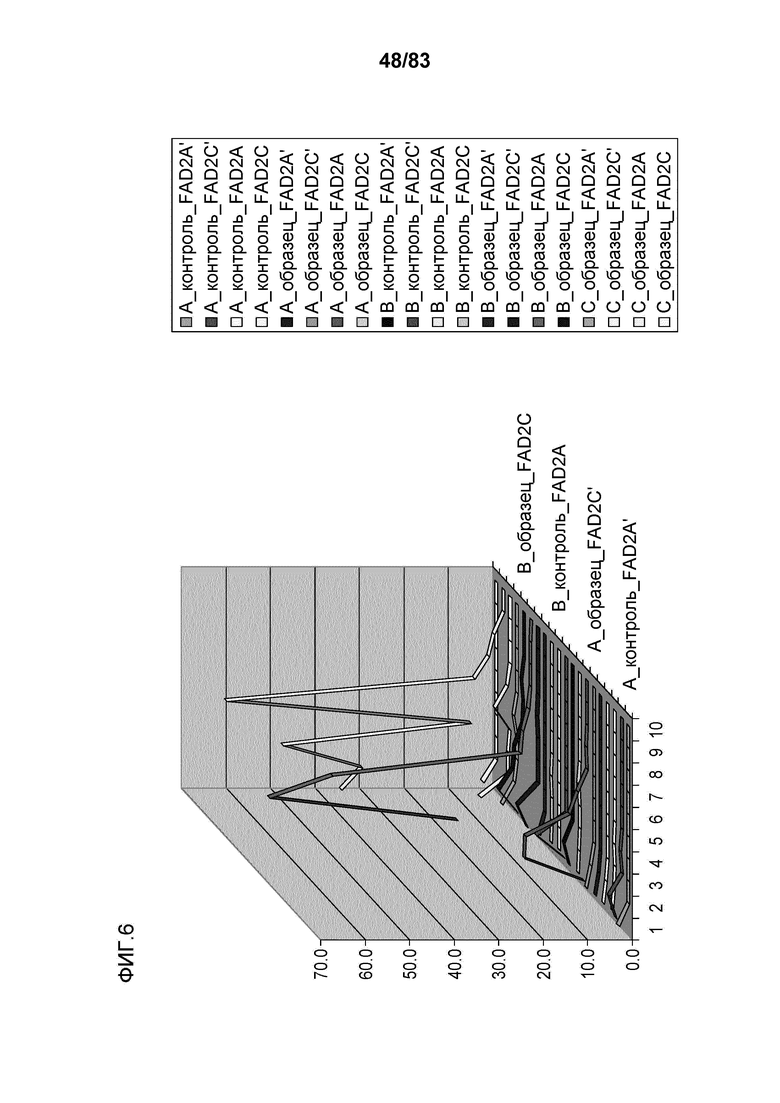

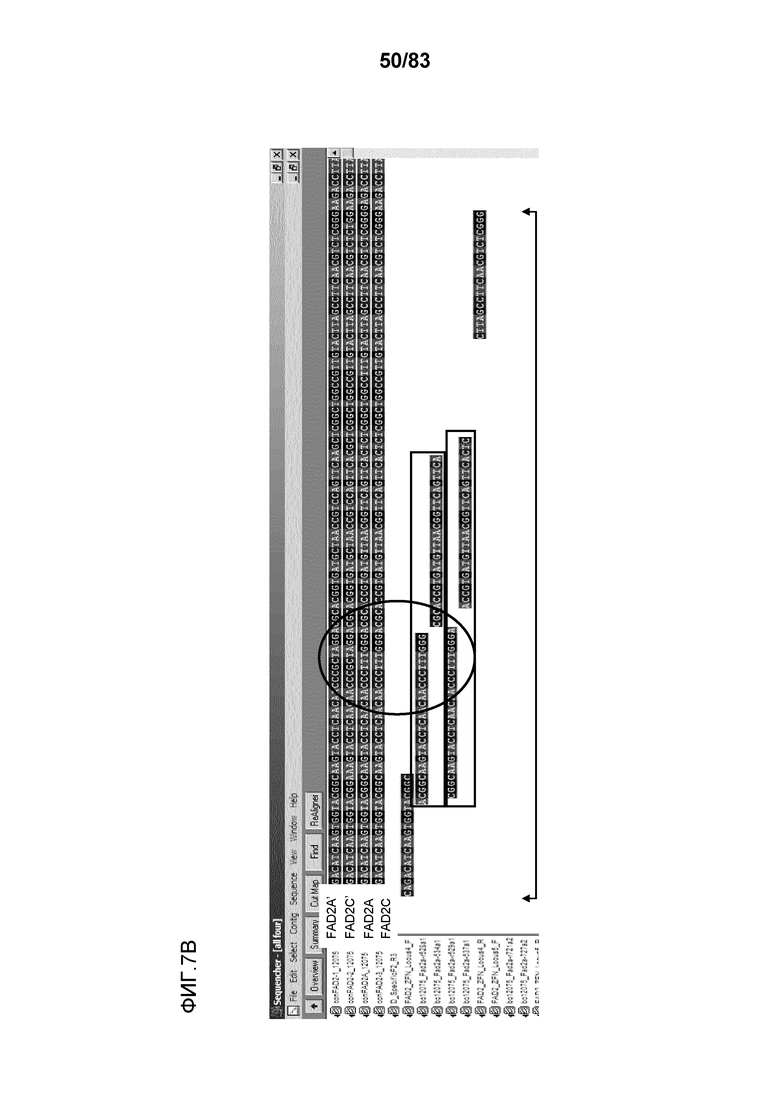
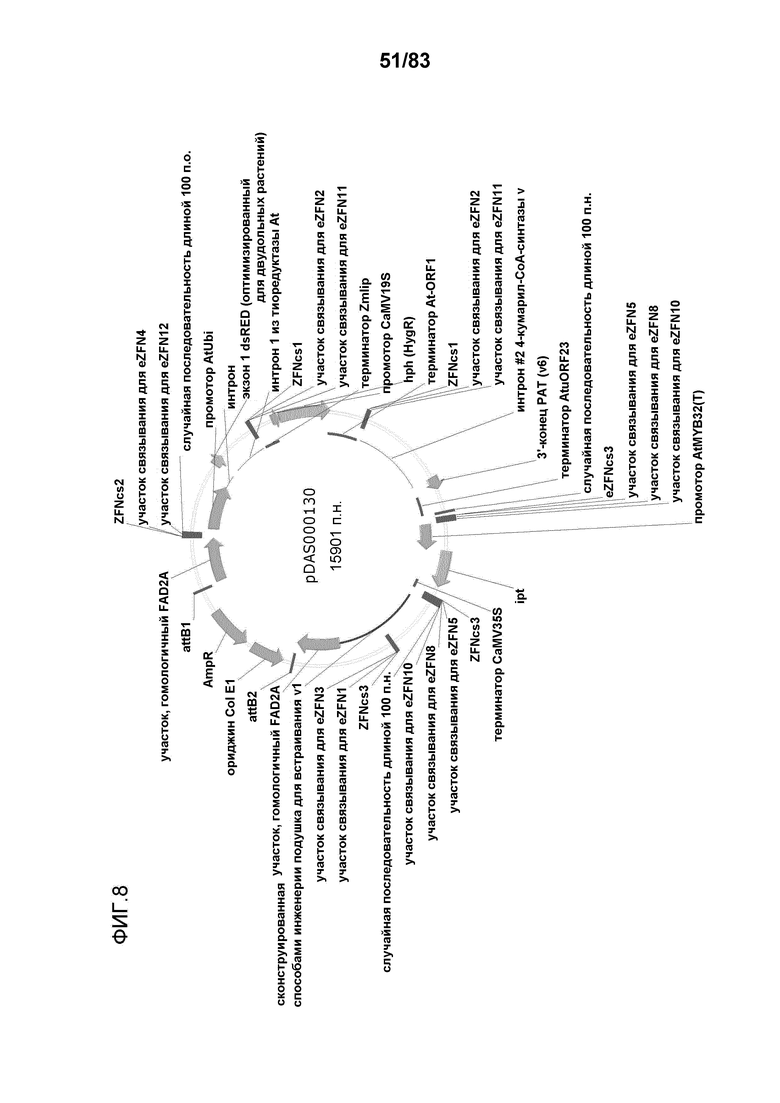
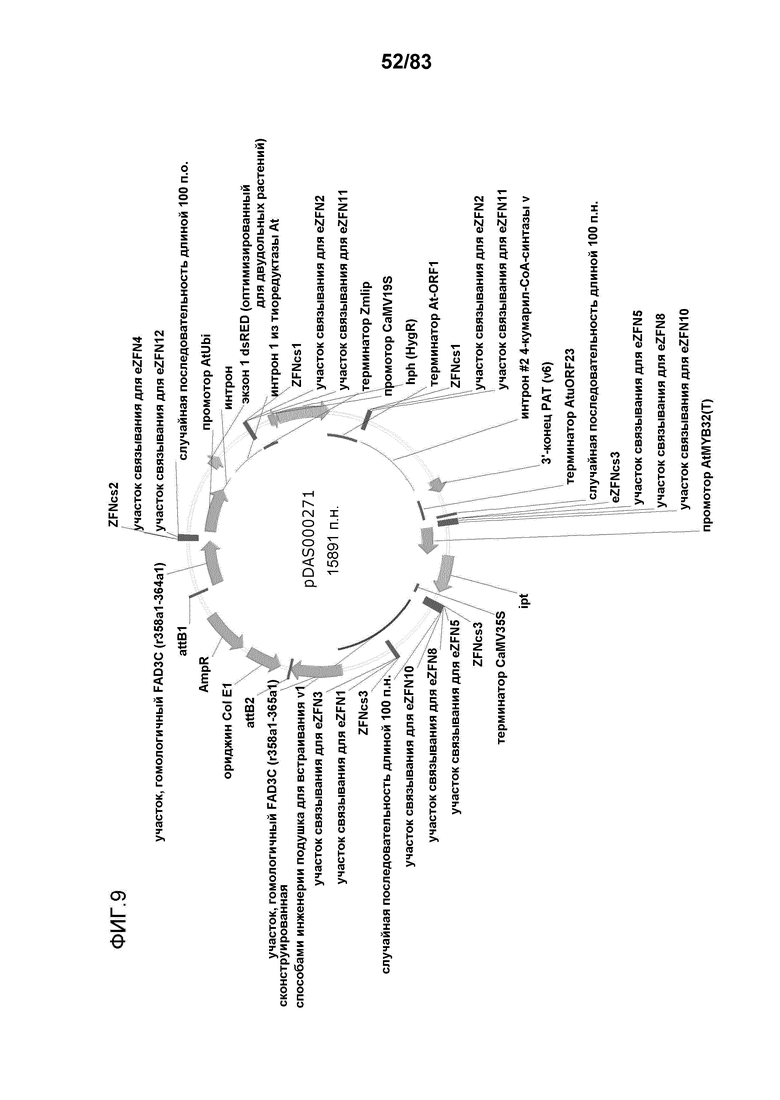
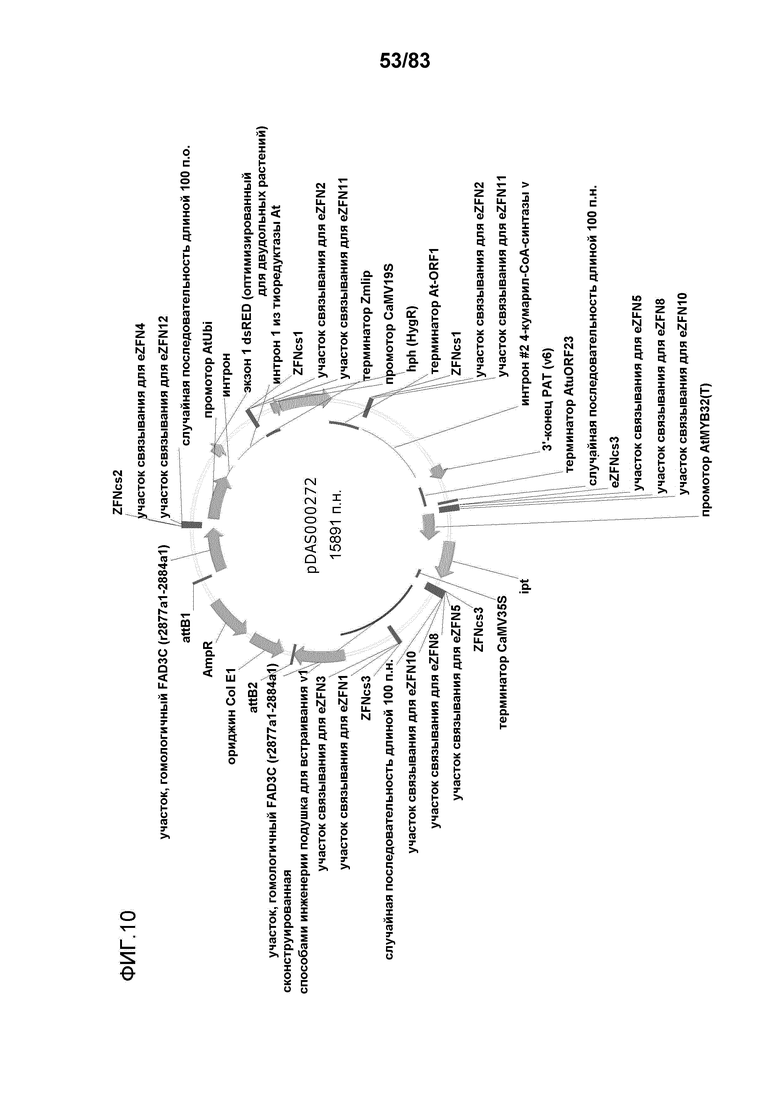
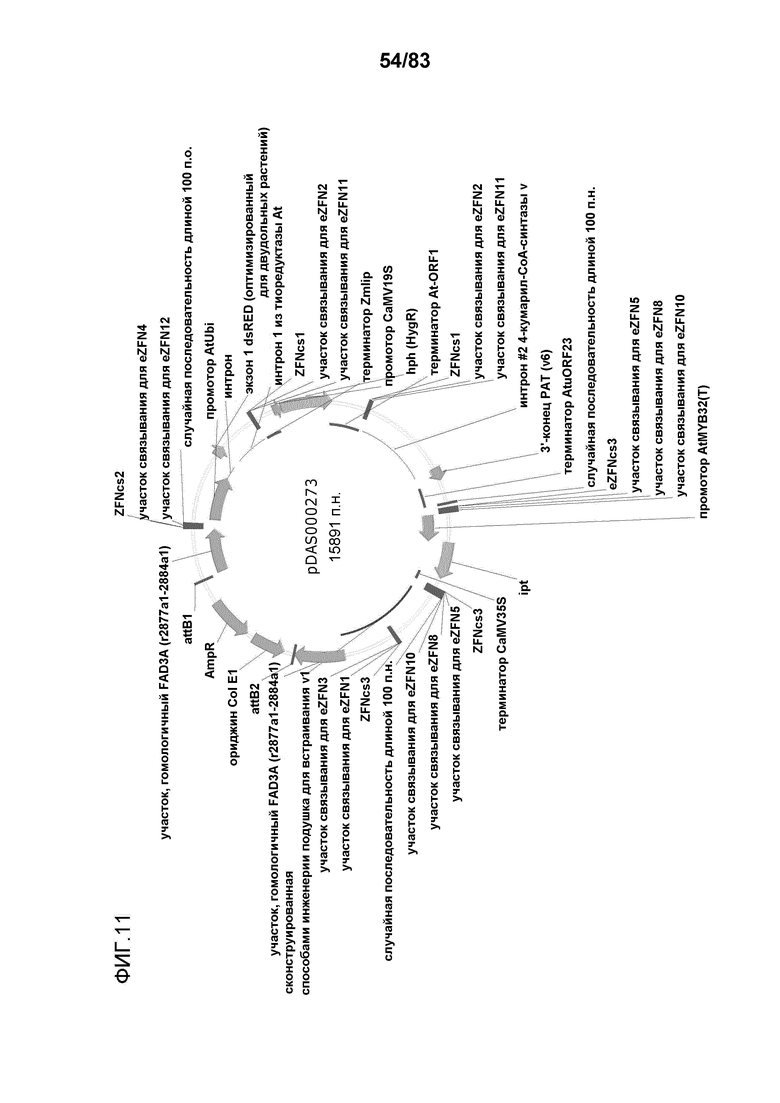
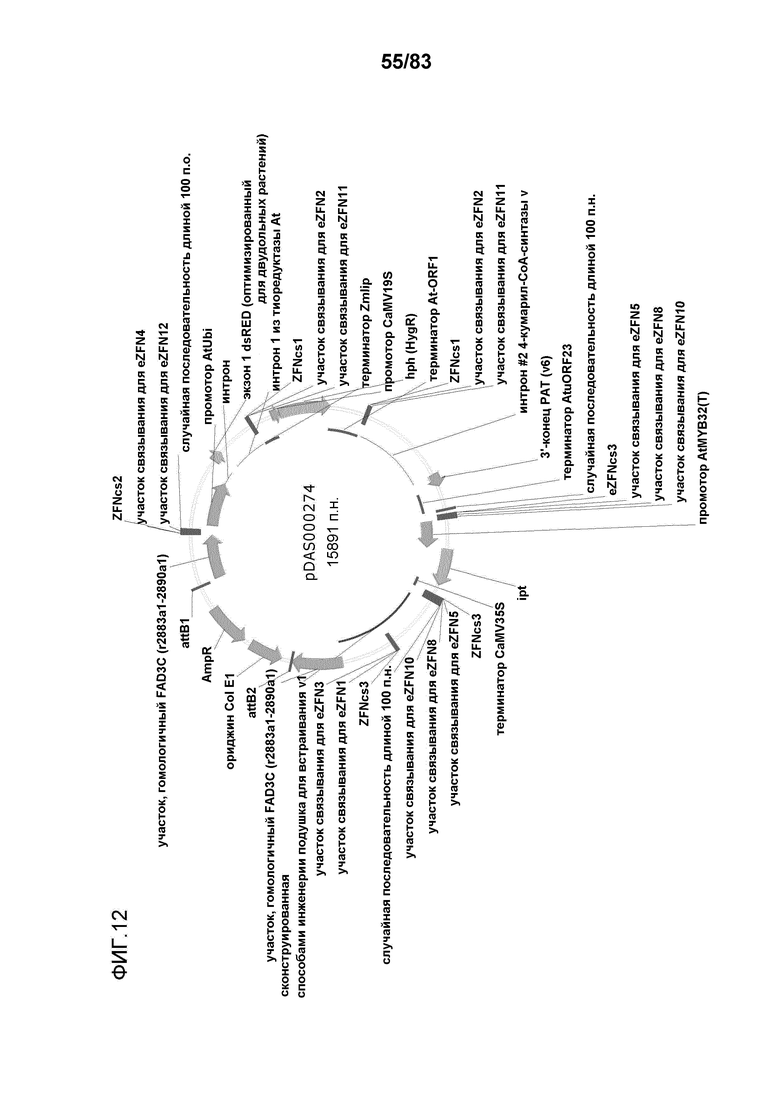
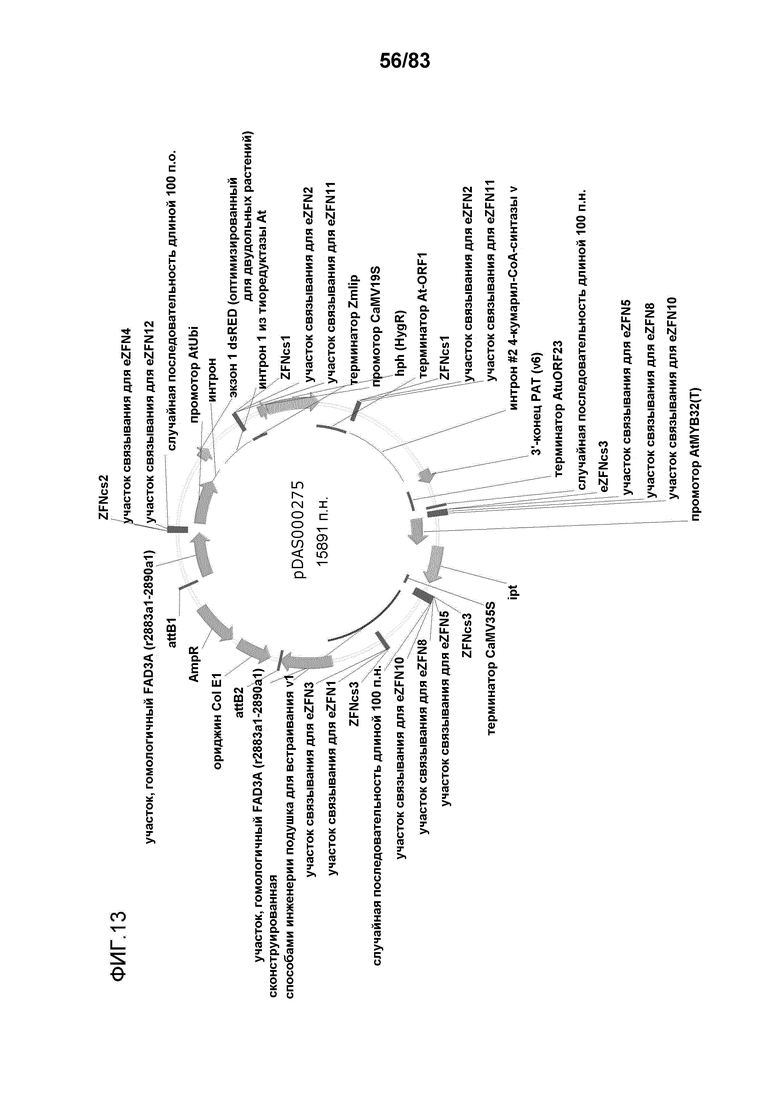
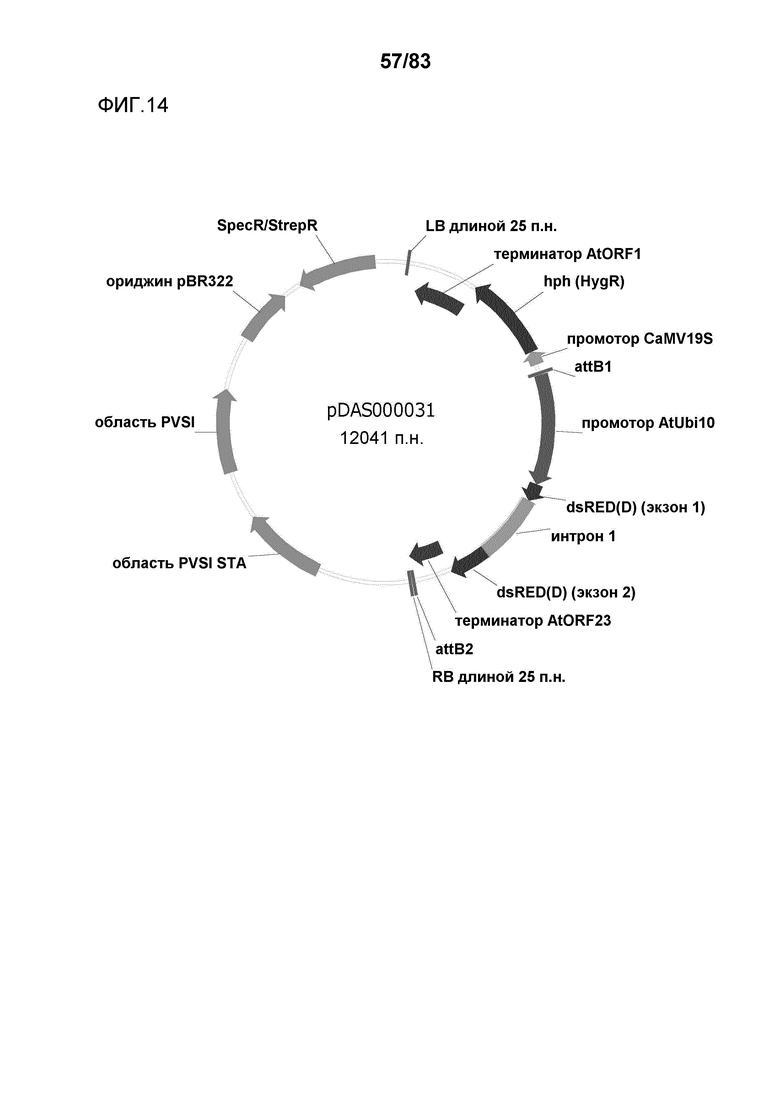
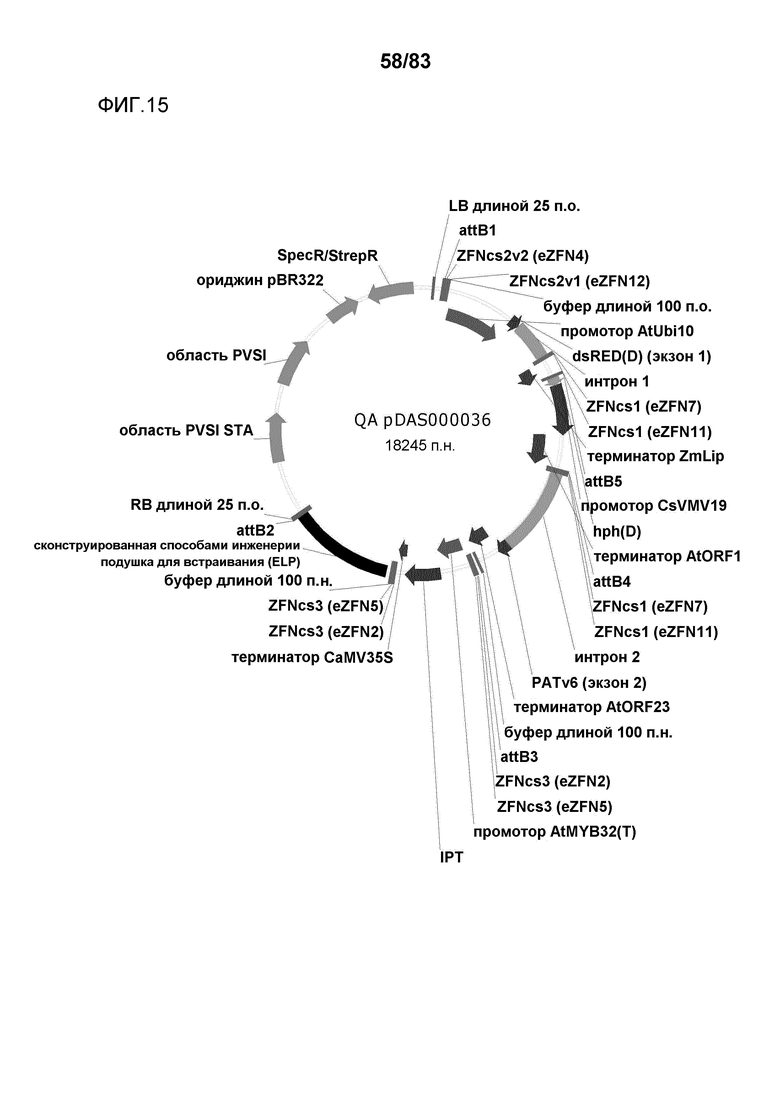
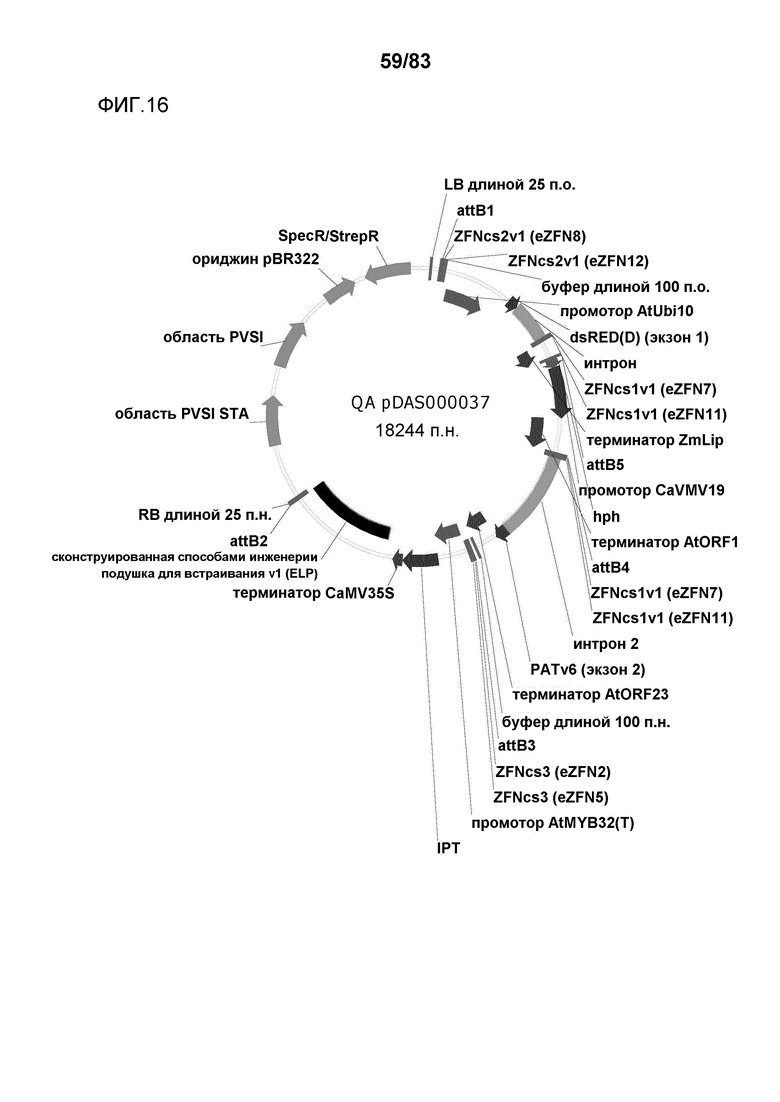
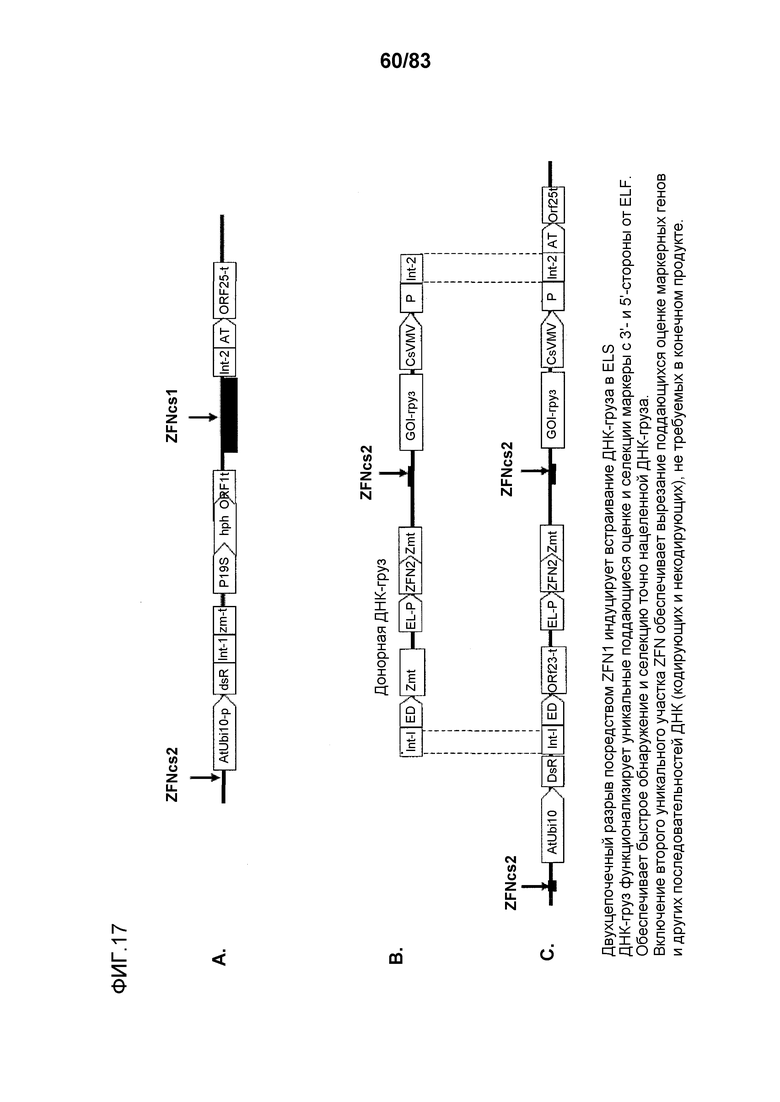
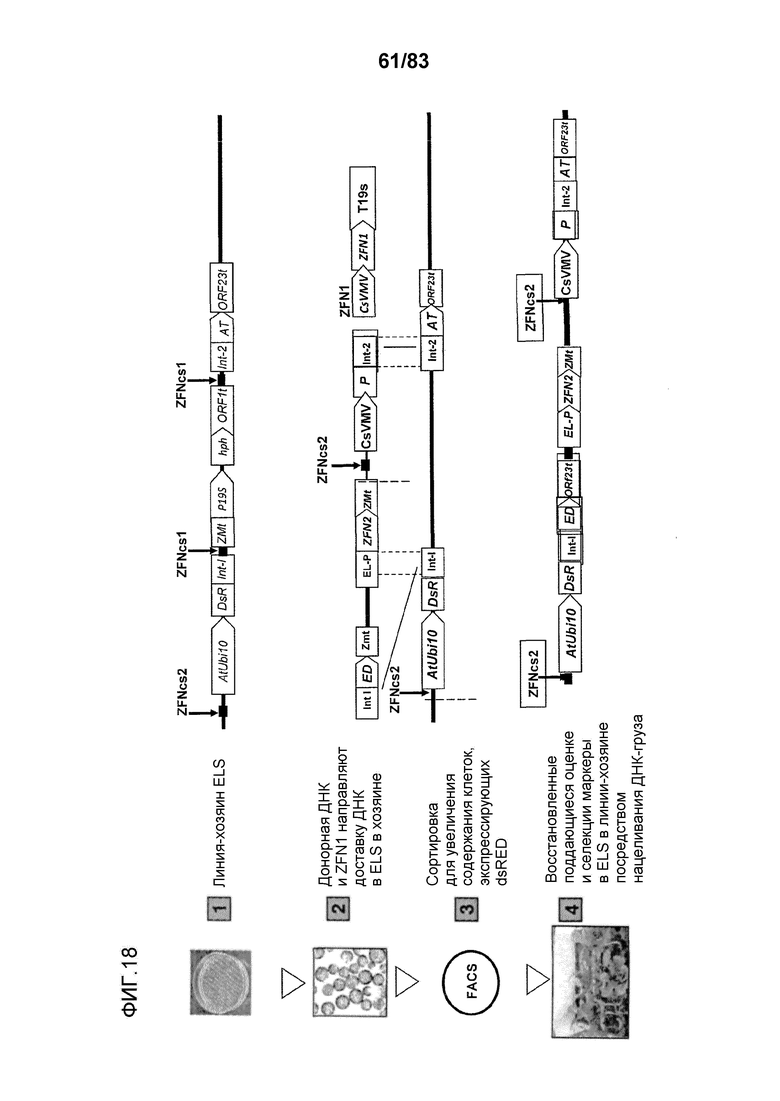
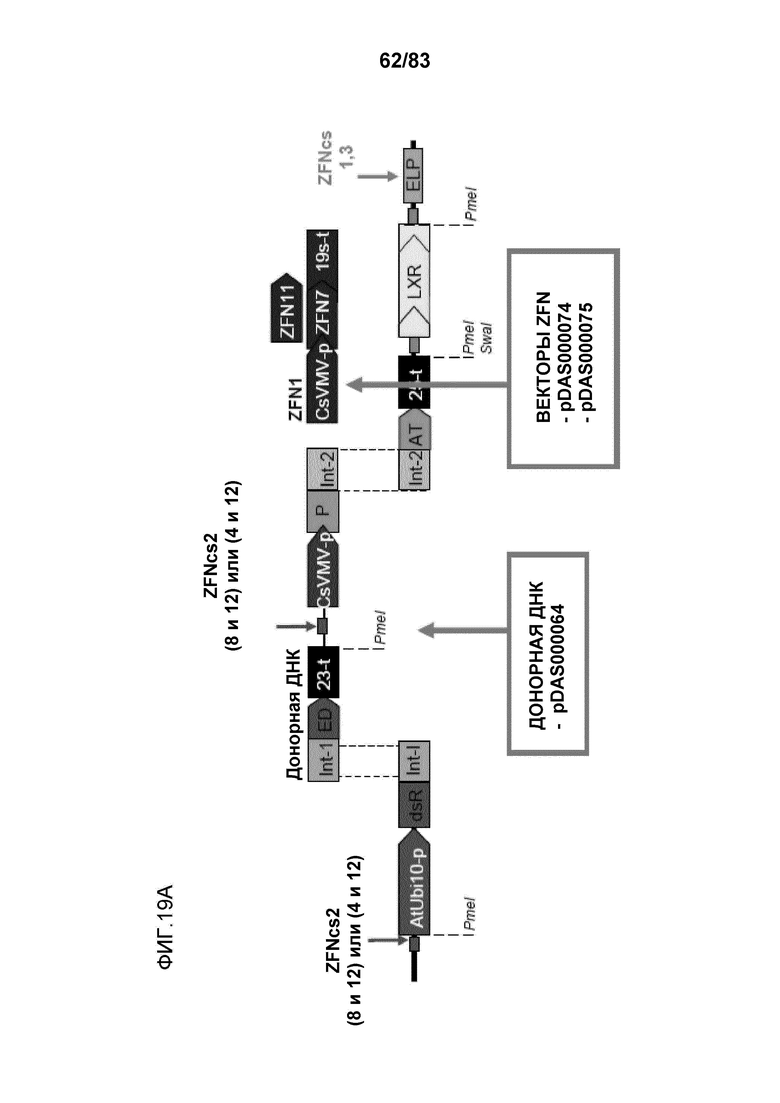
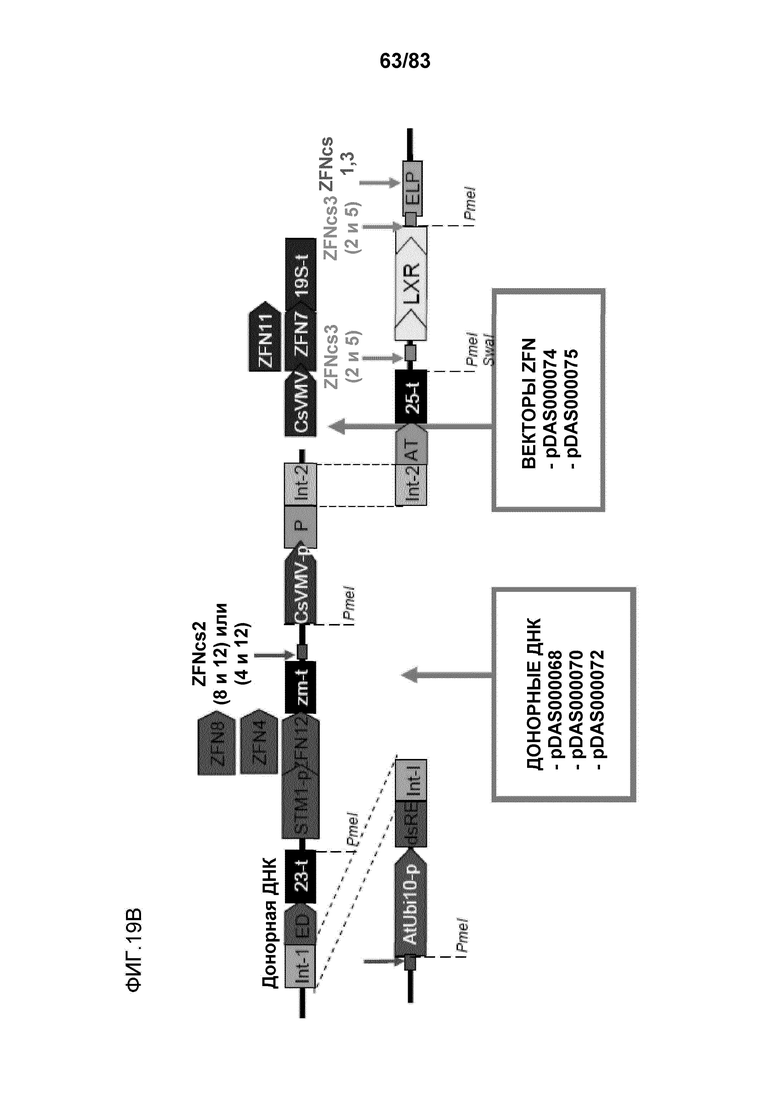
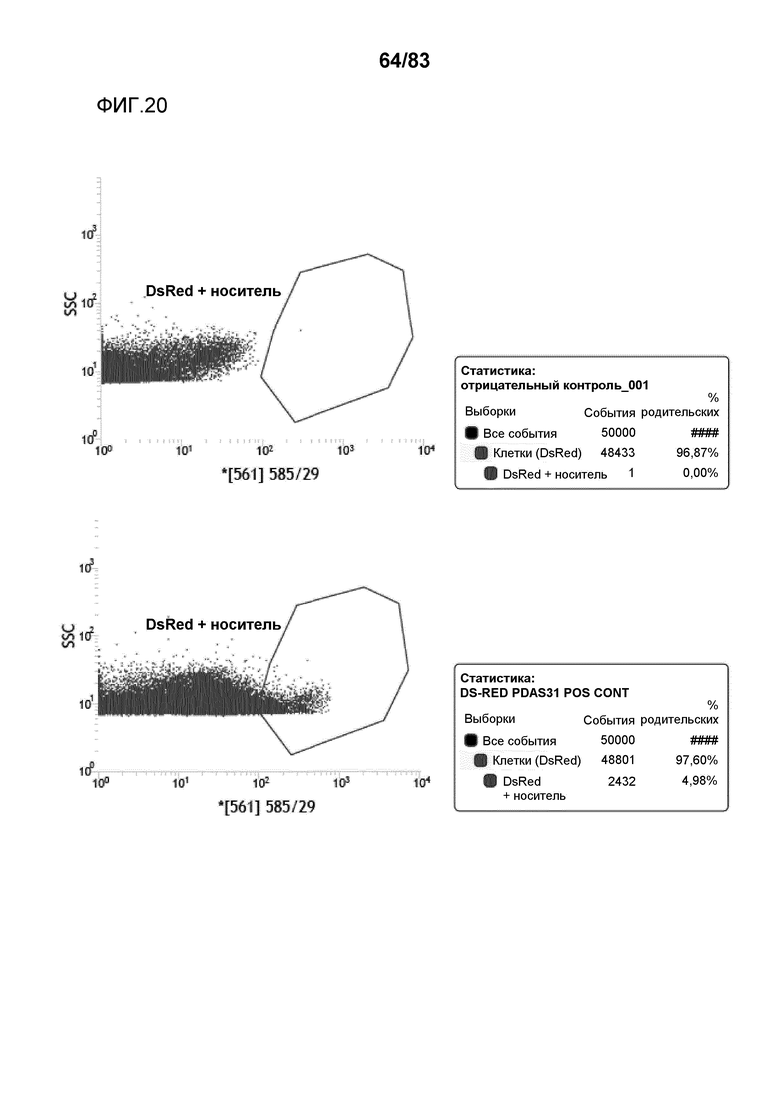
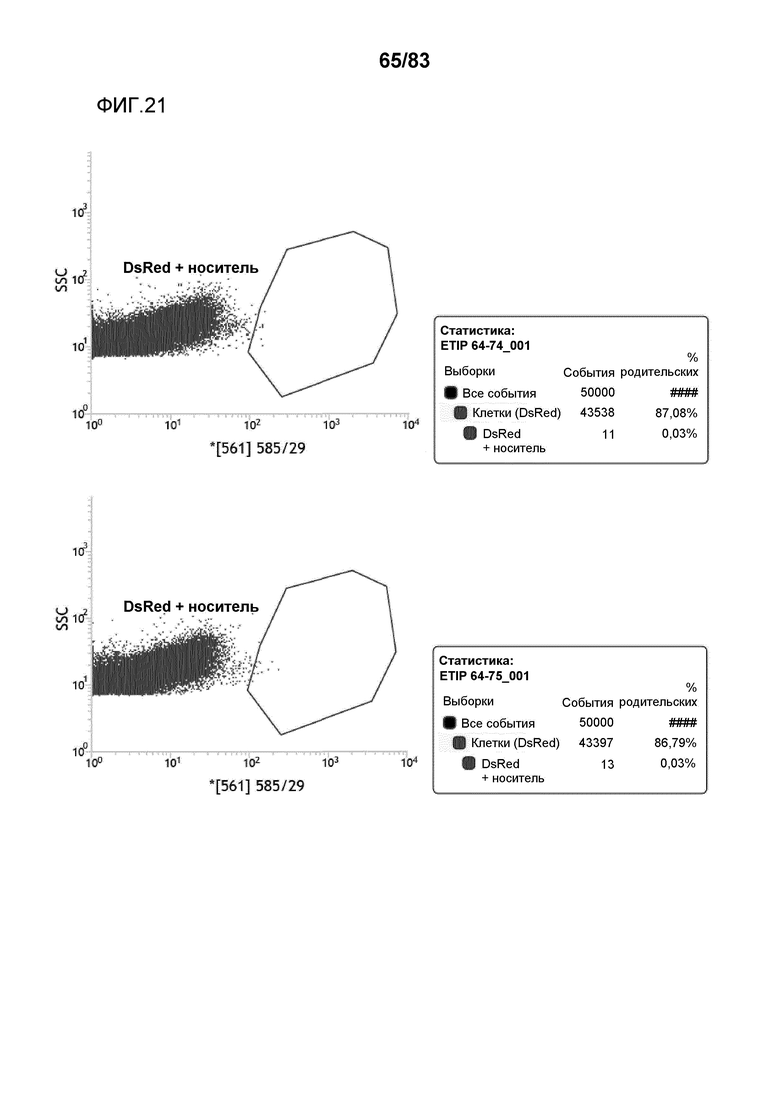
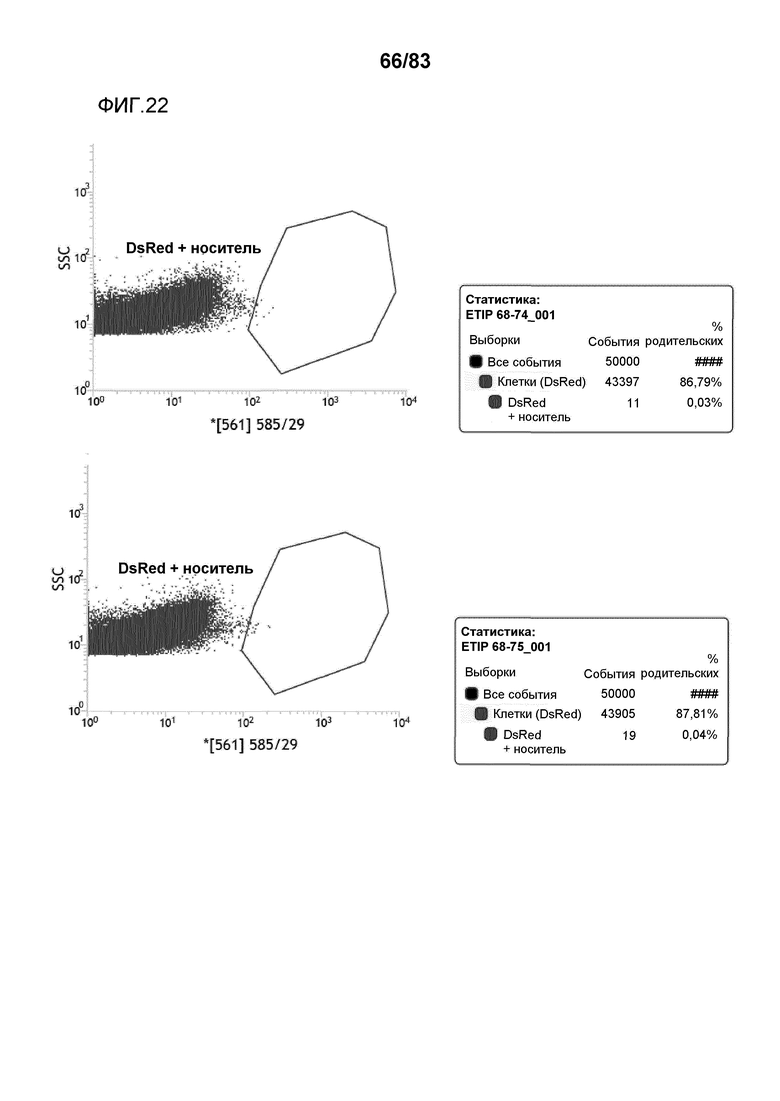
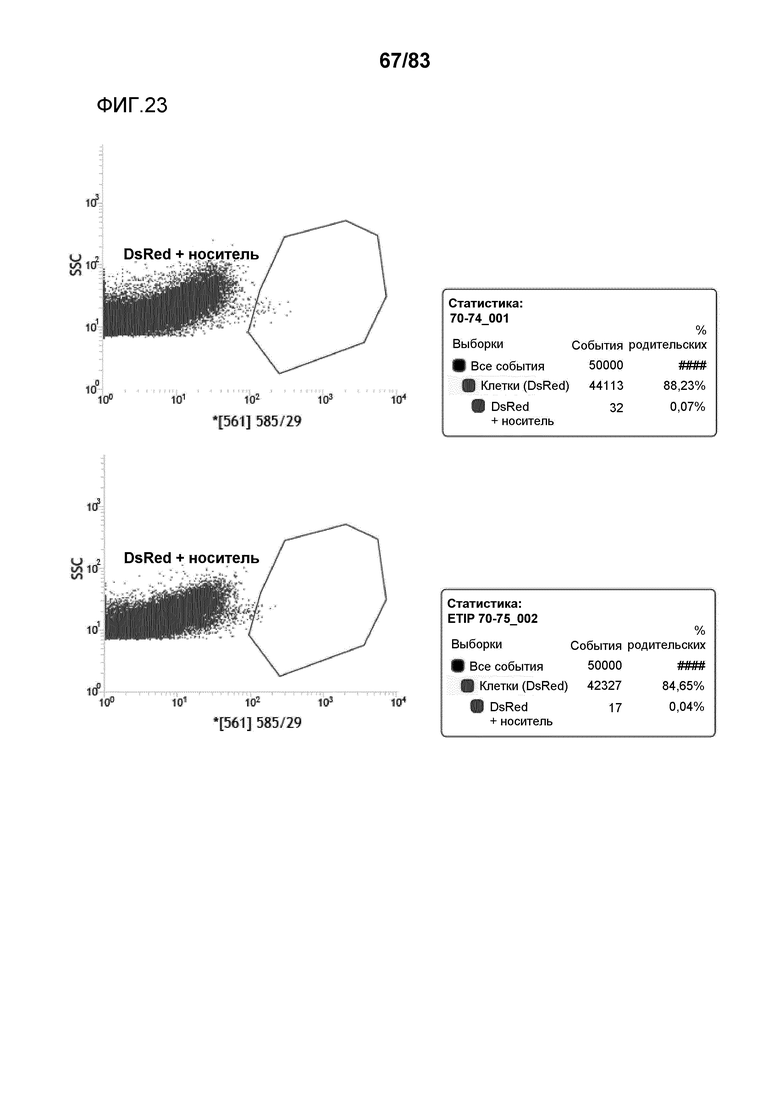
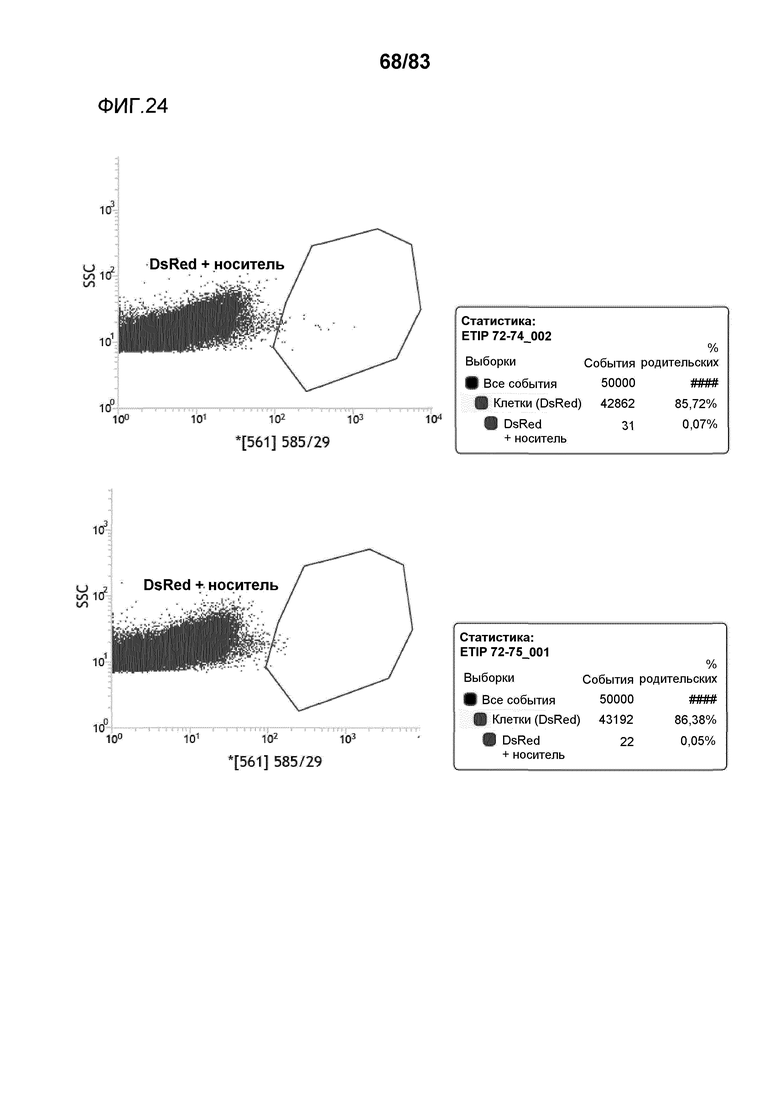
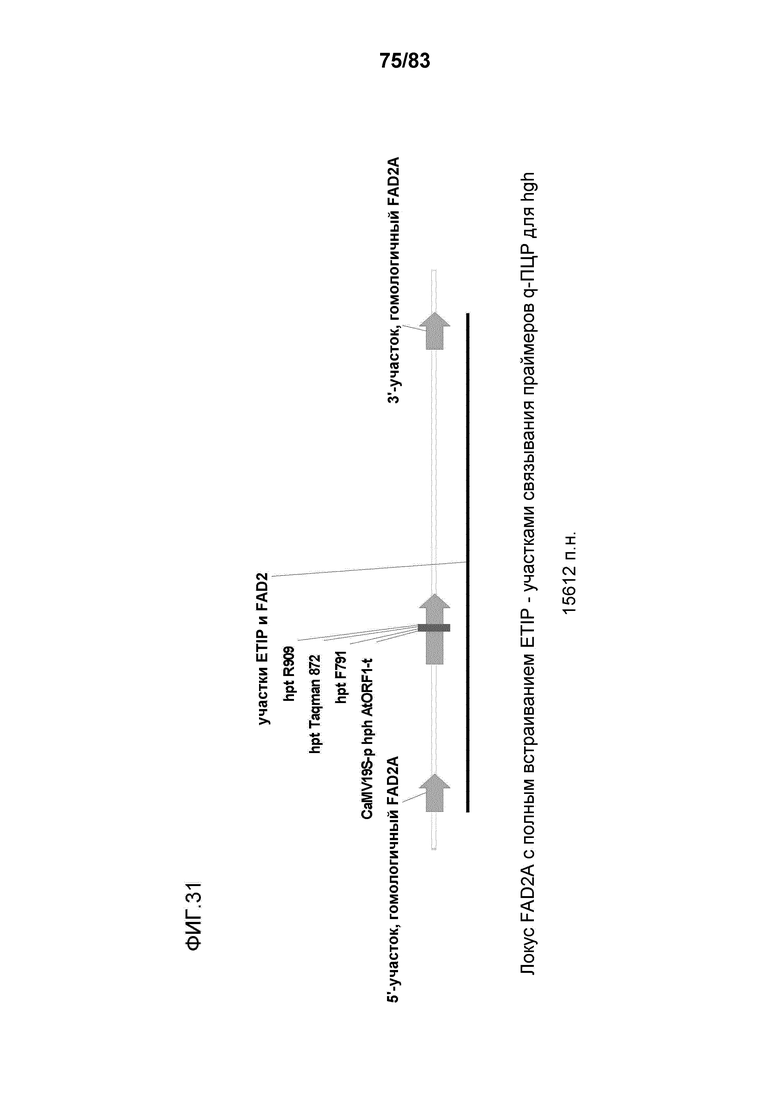
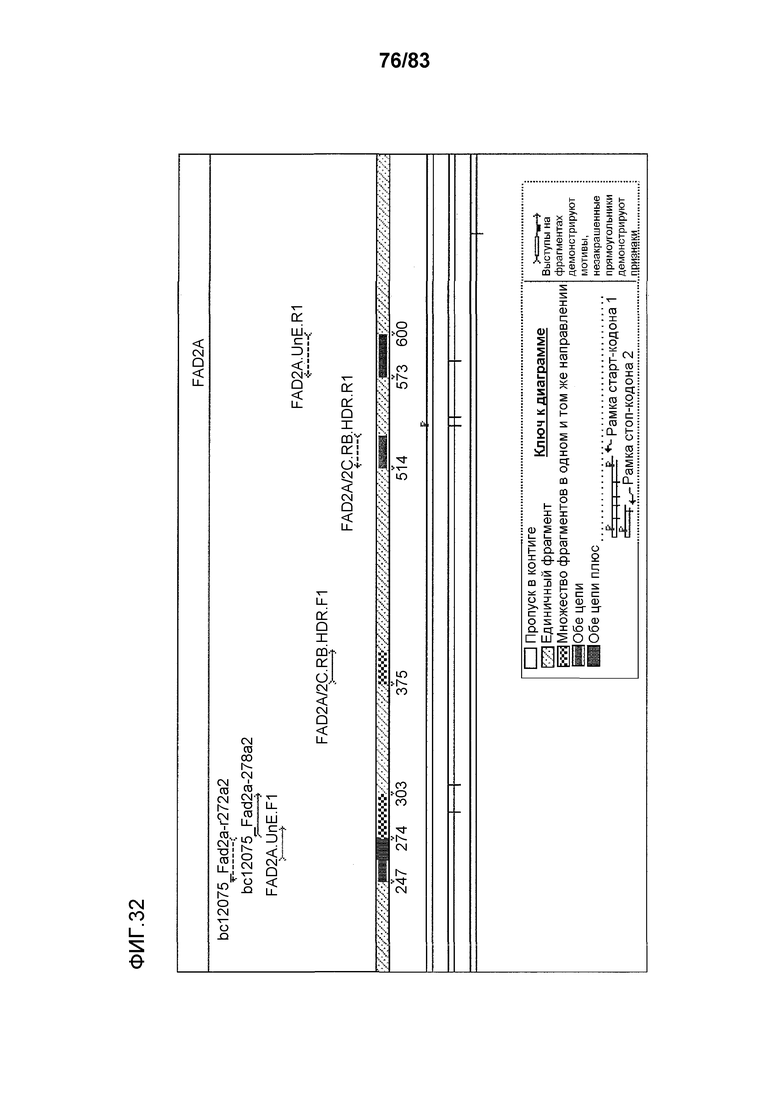
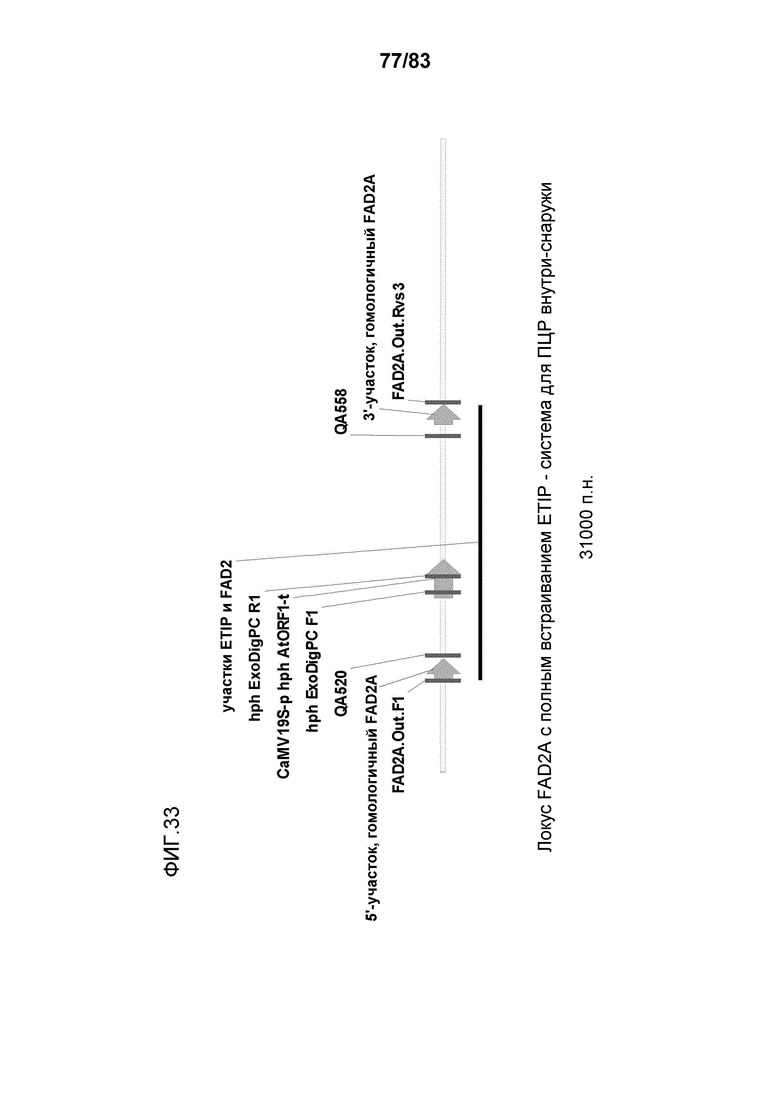
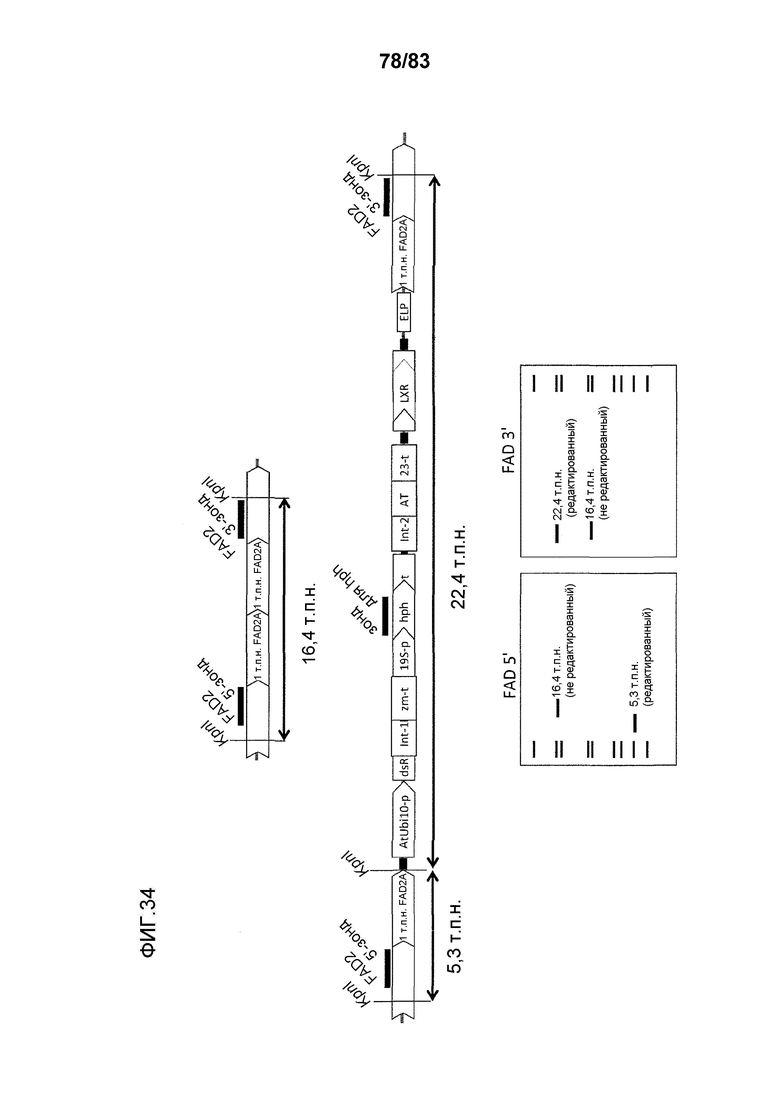
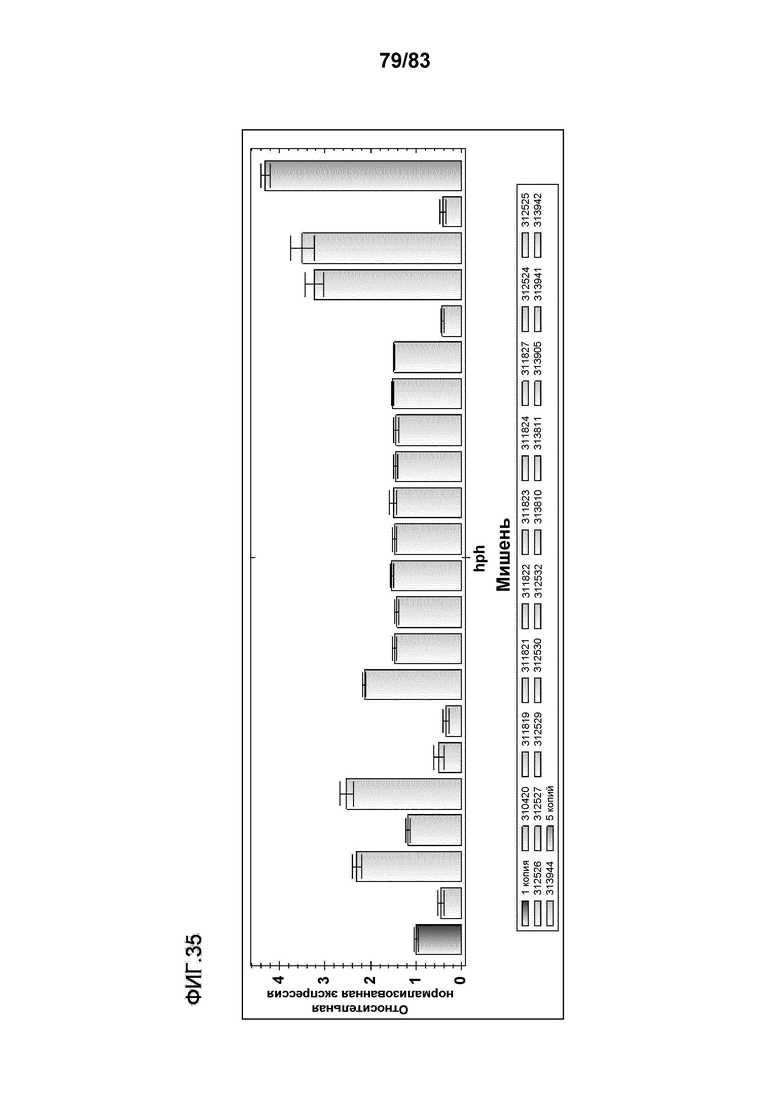
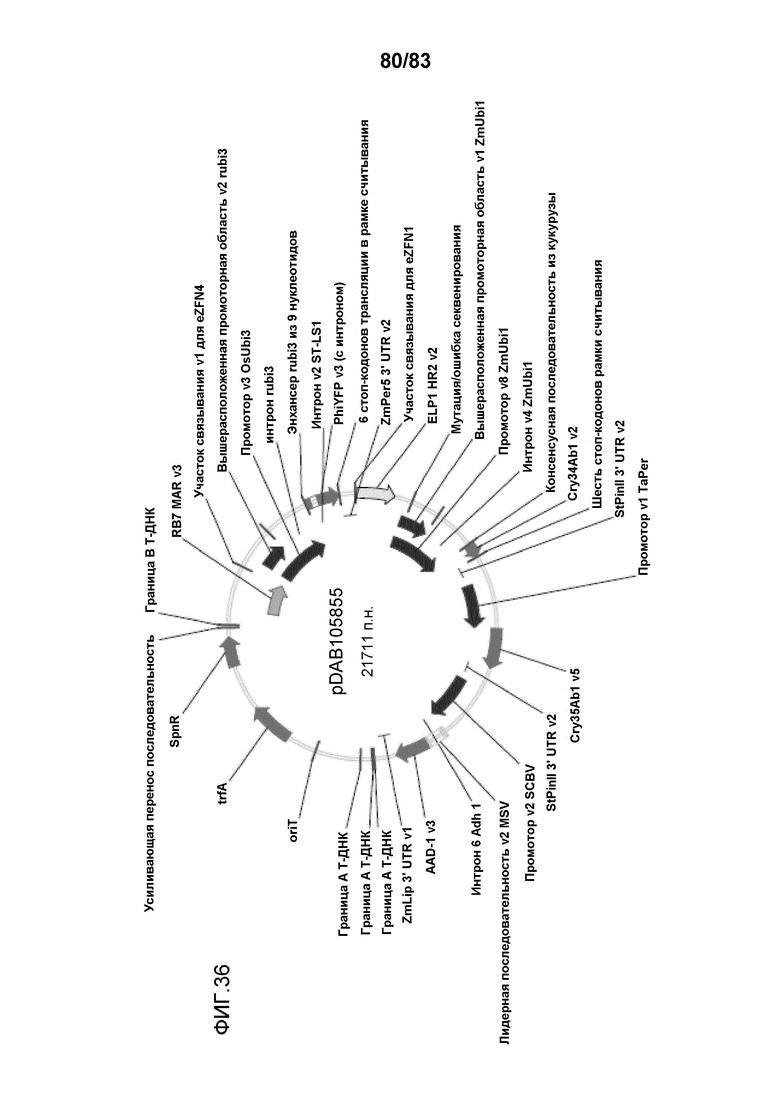
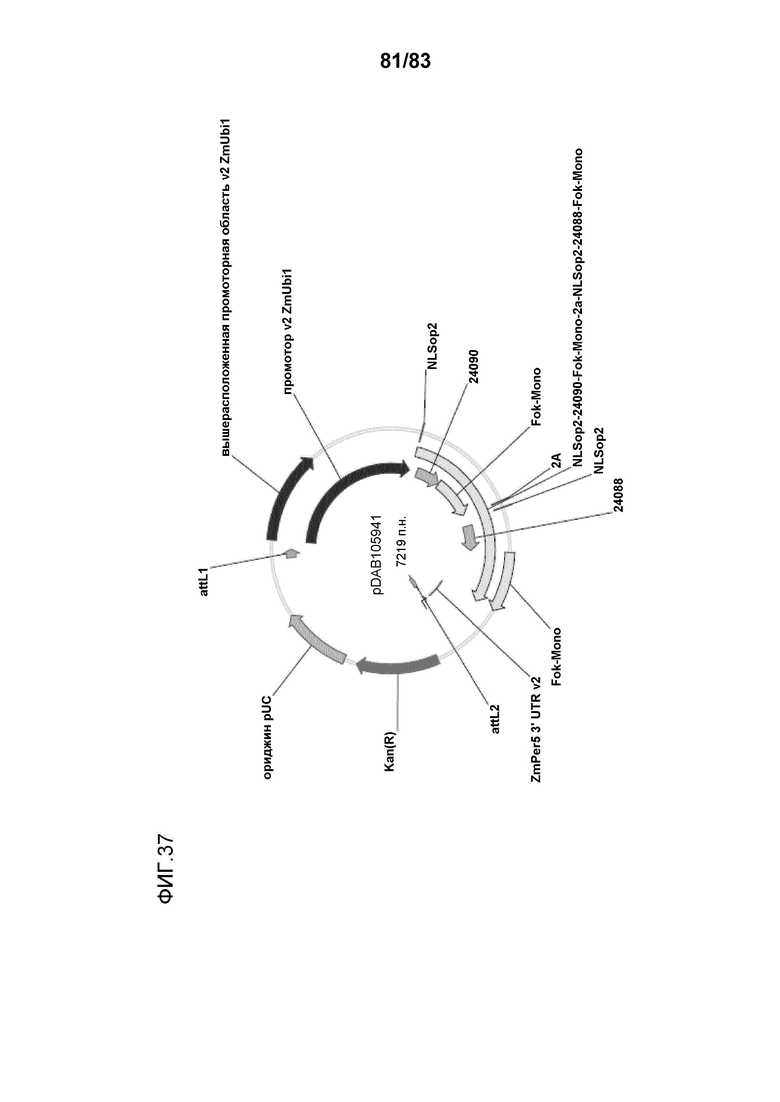
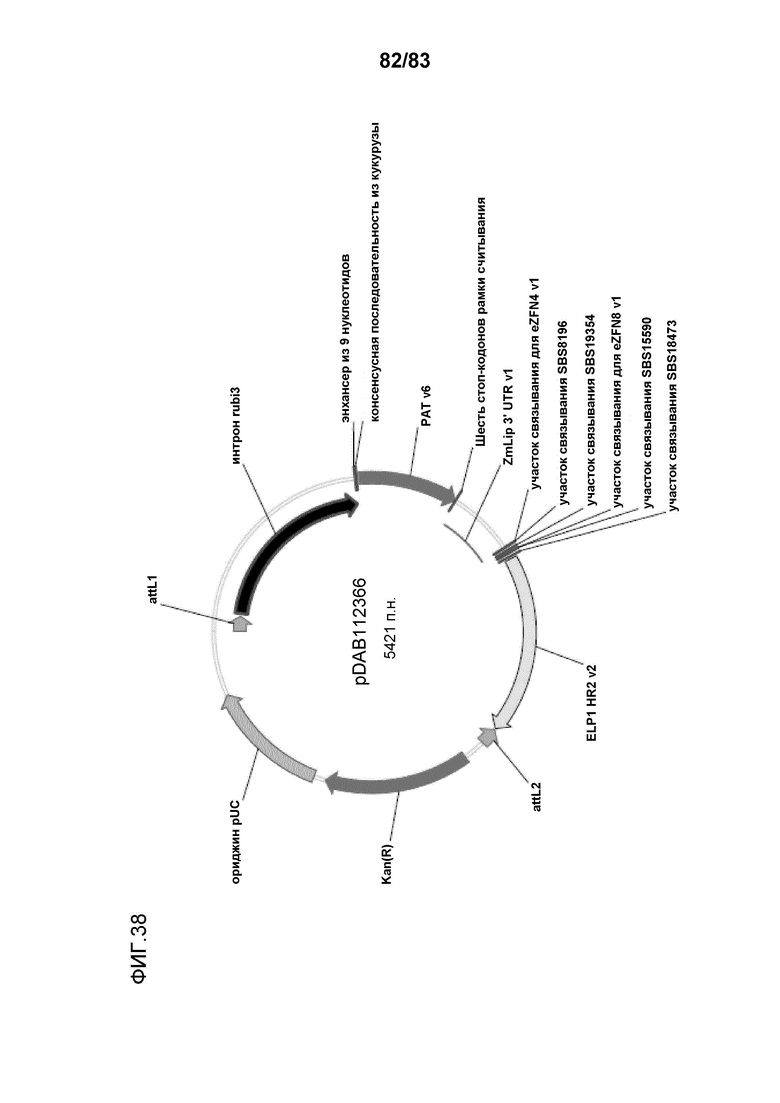
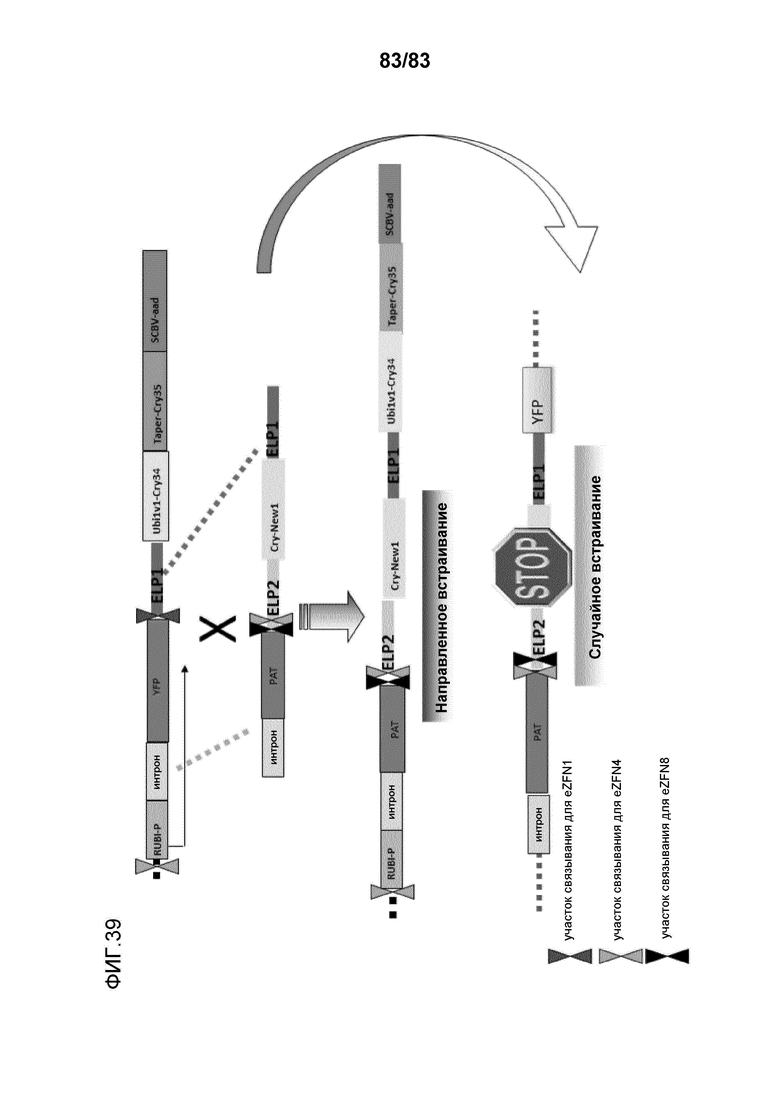
Изобретение относится к области биохимии, в частности к способу получения трансгенной клетки растения. При этом способ включает трансформацию клетки растения донорной молекулой нуклеиновой кислоты, которая встраивается в геномный локус, выбранный из группы, состоящей из FAD2, FAD3 и IPK1, с использованием сайт-специфической нуклеазы. Изобретение позволяет эффективно получать трансгенные клетки растений. 20 з.п. ф-лы, 28 табл., 83 ил., 8 пр.
1. Способ получения трансгенной клетки растения, включающий:
трансформацию клетки растения донорной молекулой нуклеиновой кислоты и молекулой нуклеиновой кислоты, кодирующей сайт-специфическую нуклеазу, где клетка растения содержит полинуклеотид, встроенный в локус, выбранный из группы, состоящей из геномного локуса FAD2, геномного локуса FAD3 и геномного локуса IPK1, в геномной ДНК клетки растения, причем полинуклеотид содержит в направлении 5'-3':
первый промотор,
первый нефункциональный фрагмент первого функционального маркерного гена,
по меньшей мере один участок распознавания сайт-специфической нуклеазы и
второй нефункциональный фрагмент второго функционального маркерного гена, где первый промотор функционально связан с первым нефункциональным фрагментом первого функционального маркерного гена,
где донорная молекула нуклеиновой кислоты содержит донорный полинуклеотид, содержащий в направлении 5'-3' второй фрагмент первого функционального маркерного гена, представляющую интерес нуклеотидную последовательность, второй промотор и первый фрагмент второго функционального маркерного гена, где второй промотор функционально связан с первым фрагментом второго функционального маркерного гена,
вследствие чего экспрессию сайт-специфической нуклеазы для введения двухцепочечного разрыва в геномную ДНК клетки растения в участке распознавания сайт-специфической нуклеазы так, что восстановление двухцепочечного разрыва приводит к встраиванию донорного полинуклеотида в локус для получения трансгенной клетки растения,
где трансгенная клетка растения содержит в локусе в направлении 5'-3':
первый промотор,
первый функциональный маркерный ген,
представляющую интерес нуклеотидную последовательность,
второй промотор и
второй функциональный маркерный ген, в котором первый промотор функционально связан с первым функциональным маркерным геном, и где второй промотор функционально связан со вторым функциональным маркерным геном.
2. Способ по п.1, где полинуклеотид, встроенный в локус в геномной ДНК клетки растения, содержит в направлении 5'-3':
первый фрагмент первого маркерного гена,
нуклеотидную последовательность первого участка гомологии,
участок распознавания сайт-специфической нуклеазы,
нуклеотидную последовательность второго участка гомологии, и
второй фрагмент второго маркерного гена,
где донорный полинуклеотид содержит в направлении 5'-3':
нуклеотидную последовательность первого участка гомологии,
второй фрагмент первого маркерного гена,
представляющую интерес нуклеотидную последовательность,
первый фрагмент второго маркерного гена и
нуклеотидную последовательность второго участка гомологии,
так, что донорный полинуклеотид встраивается в локус посредством механизма направляемой гомологией репарации двухцепочечного разрыва.
3. Способ по п.2, где нуклеотидная последовательность первого или второго участка гомологии представляет собой интрон.
4. Способ по п.2, где нуклеотидные последовательности первого и второго участков гомологии обладают менее чем 50% идентичностью последовательностей.
5. Способ по п.2, где нуклеотидные последовательности первого и второго участков гомологии имеют длину от 50 п.н. до 3 т.п.н.
6. Способ по п.1, где клетка растения содержится в ткани растения или в целом растении.
7. Способ по п.1, где представляющая интерес нуклеотидная последовательность содержит экспрессирующую ген кассету.
8. Способ по п.7, где экспрессирующая ген кассета содержит по меньшей мере один трансген.
9. Способ по п.8, где трансген выбран из группы, состоящей из трансгена устойчивости к насекомым, трансгена устойчивости к гербициду, трансгена эффективности утилизации азота, трансгена эффективности утилизации воды, трансгена питательного качества, трансгена, кодирующего ДНК-связывающий белок, и трансгена, кодирующего селективный маркер.
10. Способ по п.1, где сайт-специфическая нуклеаза представляет собой слитый белок, содержащий ДНК-связывающий домен и домен расщепления или половинный домен расщепления.
11. Способ по п.10, где ДНК-связывающий домен выбран из группы, состоящей из ДНК-связывающего домена мегануклеазы, ДНК-связывающего домена РНК-направляемых CRISPR-Cas9, ДНК-связывающего домена лейциновой молнии, ДНК-связывающего домена белка, подобного активатору транскрипции (TAL), ДНК-связывающего домена рекомбиназы, ДНК-связывающего домена белка с цинковыми пальцами и химерных комбинаций любого из указанных выше, и, где домен расщепления или половинный домен расщепления выбран из группы, состоящей из половинного домена расщепления сайт-специфической нуклеазы рестрикции типа IIS, половинного домена расщепления из сайт-специфической нуклеазы FokI, половинного домена расщепления из сайт-специфической нуклеазы StsI и сайт-специфической хоминг-нуклеазы.
12. Способ по п.10, где ДНК-связывающий домен представляет собой ДНК-связывающий домен с цинковыми пальцами.
13. Способ по п.12, где домен расщепления или половинный домен расщепления выбран из группы, состоящей из половинного домена расщепления сайт-специфической нуклеазы рестрикции типа IIS, половинного домена расщепления из сайт-специфической нуклеазы FokI и половинного домена расщепления из сайт-специфической нуклеазы StsI.
14. Способ по п.1, дополнительно включающий скрининг на экспрессию первого функционального маркерного гена или скрининг на экспрессию второго функционального маркерного гена в трансгенной клетке растения.
15. Способ по п.1, дополнительно включающий скрининг на экспрессию первого функционального маркерного гена и второго функционального маркерного гена.
16. Способ по п.1, где первый функциональный маркерный ген или второй функциональный маркерный ген выбран из группы, состоящей из PMI, YFP, Xyl(A), RFP, DSR, GFP, GUS, NPTII, AAD-1, AAD-12, AHAS, PAT, HPH и BAR.
17. Способ по п.1, дополнительно включающий выделение трансгенной клетки растения с использованием проточной цитометрии.
18. Способ по п.2, где нуклеотидная последовательность первого или второго участка гомологии представляет собой интрон.
19. Способ по п.1, дополнительно включающий регенерацию ткани трансгенного растения или трансгенного растения из трансгенной клетки растения.
20. Способ по п.1, в котором в скрининге используют активируемую флуоресценцией сортировку клеток (FACS).
21. Способ по п.1, в котором локус в геномной ДНК клетки растения содержит:
SEQ ID NO: 431 и SEQ ID NO: 432;
SEQ ID NO: 433 и SEQ ID NO: 434;
SEQ ID NO: 435 и SEQ ID NO: 436;
SEQ ID NO: 437 и SEQ ID NO: 438;
SEQ ID NO: 439 и SEQ ID NO: 440;
SEQ ID NO: 441 и SEQ ID NO: 442;
SEQ ID NO: 443 и SEQ ID NO: 444 или
SEQ ID NO: 445 и SEQ ID NO: 446.
| US 20080182332 A1, 31.07.2008 | |||
| WO 2007014275 A2, 01.02.2007 | |||
| ЛУТОВА Л | |||
| А., Биотехнология высших растений, Издательство С | |||
| - Петербургского университета, 2010, стр | |||
| Способ крашения тканей | 1922 |
|
SU62A1 |

