Область техники
Настоящее изобретение относится к применению новых соединений, которые ингибируют Btk и полезны для лечения онкологических, аутоиммунных и воспалительных заболеваний, вызванных нарушением активации В-клеток.
Предшествующий уровень техники
Протеинкиназы составляют одно из наиболее больших семейств ферментов человека и регулируют многие различные сигнальные процессы за счет добавления фосфатных групп к белкам (Т. Hunter, Cell 1987 50:823-829). В частности, тирозинкиназы фосфорилируют белки по фенольной группе тирозиновых остатков. Семейство тирозинкиназ включает членов, которые контролируют клеточный рост, миграцию и дифференциацию. Ненормальная активность киназы отмечена при различных заболеваниях человека, включая рак, аутоиммунные и воспалительные заболевания. Поскольку протеинкиназы являются ключевыми регуляторами клеточного сигналинга, они представляют собой мишень для регулирования клеточной функции при помощи небольших молекулярных ингибиторов киназ и таким образом являются хорошей целью для дизайна лекарственных средств. Кроме того, для лечения киназ-опосредованных заболеваний также полезны селективные и эффективные ингибиторы киназной активности для исследования процессов клеточного сигналинга и идентификации других клеточных мишеней, представляющих интерес для терапии.
Существуют хорошие данные о том, что В-клетки играют ключевую роль в патогенезе аутоиммунных и/или воспалительных заболеваний. Лекарственные препараты, основанные на белковой основе, которые истощают В-клетки, такие как Ритуксан, являются эффективным против воспалительных заболеваний, обусловленных аутоантителами, таких как ревматоидный артрит (Rastetter et al. Annu Rev Med 2004 55:477). Потому ингибиторы протеинкиназ, которые играют роль в активации В-клеток, должны быть полезными для лечения заболеваний, обусловленных В-клеточной патологией, такой как продукция аутоантител.
Сигналинг через В-клеточный рецептор (BCR) контролирует ряд В-клеточных ответов, включая пролиферацию и дифференциацию в зрелые клетки, продуцирующие антитела. BCR является ключевой регуляторной точкой для В-клеточной активности и нарушенный сигналинг может вызывать дерегулированную пролиферацию В-клеток и образование патогенетически значимых аутоантител, которые приводят к множественным аутоиммунным и/или воспалительным заболеваниям. Тирозинкиназа Брутона (Btk) представляет собой киназу, не ассоциированную с BCR, является мембранопроксимальной и расположено непосредственно ниже BCR. Показано, что утрата Btk блокирует BCR сигналинг, и поэтому ингибирование Btk может быть полезным терапевтическим подходом к блокированию процессов В-клеточно-опосредованных заболеваний.
Btk является членом семейства Тес-тирозинкиназ, и показана как критический регулятор раннего развития В-клеток, активации зрелых В-клеток и выживания (Khan et al. Immunity 1995 3:283; Ellmeier et al. J. Exp. Med. 2000 192:1611). Мутация Btk у людей приводит к состоянию агаммаглобулинемии, сцепленной с Х-хромосомой (XLA) (рассмотрено в Rosen et al. New Eng. J. Med. 1995 333:431 and Lindvall et al. Immunol. Rev. 2005 203:200). Данные пациенты имеют ослабленную иммунную систему и проявляют нарушенное созревание В-клеток, пониженные уровни иммуноглобулина и периферических В-клеток, уменьшенные Т-независимые иммунные ответы, а также сниженную кальциевую мобилизацию вслед за стимуляцией BCR.
Данные о роли Btk при аутоиммунных и воспалительных заболеваниях также получены на Btk-дефицитных мышиных моделях. В преклинических мышиных моделях с системной красной волчанкой (SLE), Btk-дефицитные мыши показали заметное улучшение прогресса заболевания. Кроме того, Btk-дефицитные мыши устойчивы к коллаген-индуцированному артриту (Jansson and Holmdahl Clin. Exp. Immunol. 1993 94:459). Селективный ингибитор Btk продемонстрировал дозозависимую эффективность на мышиной модели артрита (Z. Pan et al., Chem. Med Chem. 2007 2:58-61).
Btk также экспрессируется клетками другими чем В-клетки, которые могут быть вовлечены в процесс заболевания. Например, Btk экспрессируется тучными клетками, и Btk-дефицитные тучные клетки, полученные из костного мозга, демонстрируют нарушенную антиген-индуцированную дегрануляцию (Iwaki et al. J. Biol. Chem. 2005 280:40261). Это показывает, что Btk может быть полезна для лечения патологических ответов тучных клеток, таких как аллергия или астма. Также моноциты от пациентов с XLA, в которых отсутствует активность Btk, показывают пониженную продукцию TNF-альфа после стимуляции (Horwood et al. J Exp Med 197:1603, 2003). Поэтому TNF-альфа опосредованное воспаление может быть модулировано при помощи небольших молекулярных ингибиторов Btk. Также Btk играет роль в апаптозе (Islam and Smith Immunol. Rev. 2000 178:49) и, таким образом, ингибиторы Btk будут полезны для лечения определенных В-клеточных лимфом и лейкемий (Feldhahn et al. J. Exp. Med. 2005 201:1837).
Краткое описание изобретения
Настоящая заявка предлагает соединения-ингибиторы Btk формулы I, способы их применения, как описано в данном документе ниже:
Заявка предлагает соединение формулы I,

где:
R1 представляет собой Н или гало;
R2 представляет собой Н, гало, или циано;
R3 представляет собой R4 или R5;
R4 представляет собой гало или циано;
R5 представляет собой фенил, гетероарил, -C(=O)R5, низший алкил, или бензил, возможно замещенный одним или более R5';
R5' представляет собой низший алкил, циано, гидроксил, гетероциклоалкил, фенил, амино, алкиламино, диалкиламино или низший алкокси; и
X представляет собой низший алкил или гало;
или его фармацевтически приемлемую соль.
Авторы заявки предлагают способ лечения воспалительного и/или аутоиммунного заболевания, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I.
Авторы заявки предлагают фармацевтическую композицию, содержащую соединение формулы I, с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
Подробное описание изобретения
Определения
Фраза "а" или "an" существительное, как употреблено в данном документе, относится к одному или более из таких существительных; например, соединение относится к одному или более соединениям или по меньшей мере одному соединению. В связи с этим термины "а" (или "an"), "один или более", и "по меньшей мере один" могут быть употреблены в данном документе взаимозаменяемо.
Фраза "как определено в данном документе выше" относится к наиболее широкому определению для каждой группы, как предложено в Кратком описании изобретения самой широкой формуле изобретения. Во всех других воплощениях, предложенных ниже, заместители, которые могут присутствовать в каждом воплощении и, которые не четко определены, сохраняют наиболее широкое определение, предложенное в Кратком описании изобретения.
Как употреблено в данном описании, будь то в переходной фразе или в теле формулы изобретения, термины "содержит(ат)" и "содержащий" следует интерпретировать как неограниченное значение. Другими словами, термины следует интерпретировать синонимично с фразами "имеющий по меньшей мере" или "включающий по меньшей мере". В случае употребления контекста способа, термин "содержащий" означает, что способ включает по меньшей мере изложенные этапы, но может включать дополнительные этапы. В случае употребления в контексте соединения или композиции, термин «содержащий» означает, что соединение или композиция включают по меньшей мере изложенные свойства или компоненты, но могут также включать дополнительные свойства или компоненты.
Как употреблено в данном документе, если специально не указано иное, слово "или" употреблено во включающем смысле "и/или" и не "исключающем" смысле "тот или иной/или".
Термин "независимо" при употреблении в данном документе показывает, что переменную применяют в каком-либо примере безотносительно наличия или отсутствия переменной, имеющей такое же или иное определение внутри одного и того же соединения. Таким образом, в соединении, в котором присутствует два R'' и определены как «независимо углерод или азот», оба R'' могут представлять собой углерод, оба R'' могут представлять собой азот, или один R'' может представлять собой углерода, а другой азот.
В случае, когда переменная присутствует более чем один раз в любой группе или формуле, изображенного или описываемого соединения, применяемого или приведенного в формуле настоящего изобретения, ее определение в каждом случае является независимым от ее определения в каждом другом случае. Также комбинации заместителей и/или переменных допустимы только если такие соединения приводят к стабильным соединениям.
Символы "*" в конце связи или " ", изображенные через связь, каждый относятся к точке прикрепления функциональной группы или другой химической группы к остатку молекулы, частью которой он является. Таким образом, например:
", изображенные через связь, каждый относятся к точке прикрепления функциональной группы или другой химической группы к остатку молекулы, частью которой он является. Таким образом, например:
MeC(=O)OR4 где  или
или  .
.
Связь, изображенная в кольцевой системе (в отличие от присоединенной в дискретной точке) показывает, что связь может быть прикреплена к любому приемлемому атому кольца.
Термин "возможный" или "возможно", как употреблено в данном документе означает, что последовательно описываемое событие или обстоятельство может, но не обязательно, происходить и что описание включает случаи, где событие или обстоятельство возникает и случаи, в которых не возникает. Например, «возможно замещенный» означает, что возможно замещенная группа может включать атом водорода или заместитель.
Фраза «возможная связь» означает, что связь может присутствовать или нет, и что описание включает единичную, двойную или тройную связи. Если заместитель обозначен как «связь» или «отсутствие», атомы, связанные с заместителями непосредственно соединены.
Термин "приблизительно" при употреблении в данном документе означает приблизительно в районе, около или примерно. В случае, когда термин "приблизительно" применяют в сочетании с числовым диапазоном, он модифицирует данный диапазон за счет распространения границ выше и ниже указанных численных значений. Обычно, термин «приблизительно» применяют в данном документе для модификации численного значения выше или ниже установленной величины посредством вариации на 20%.
Конкретные соединения формулы I могут проявлять таутомеризм. Таутомерные соединения могут существовать как две или более взаимоконвертируемые формы. Прототропные таутомеры возникают в результате миграции ковалентно связанного атома водорода между двумя атомами. Таутомеры обычно существуют в равновесии и попытки выделить отдельные таутомеры обычно дают смесь, химические и физические свойства которой совмещены со смесью соединений. Позиция равновесия зависит от химических свойств молекулы. Например, во многих алифатических альдегидах и кетонах, таких как ацетальдегид, кетоформы преобладают; в фенолах, преобладает енольная форма. Обычные прототропные таутомеры включают кето/енол (-С(=O)-СН-  -С(-ОН)=СН-), амид/имидокислоту (-C(=O)-NH- -C(-OH)=N-) и амидин (-C(=NR)-NH- -С(-NHR)=N-) таутомеры. Последние два имеют частичное сходство в гетероарильных и гетероциклических кольцах и настоящее изобретение охватывает все таутомерные формы соединений.
-С(-ОН)=СН-), амид/имидокислоту (-C(=O)-NH- -C(-OH)=N-) и амидин (-C(=NR)-NH- -С(-NHR)=N-) таутомеры. Последние два имеют частичное сходство в гетероарильных и гетероциклических кольцах и настоящее изобретение охватывает все таутомерные формы соединений.
Технические и научные термины, примененные в данном документе, имеют значение, обычно понимаемое специалистами в области техники, которых касается настоящее изобретение, если не указано иное. В данном документе сделаны ссылки на различные способы и материалы, известные специалистам в области техники. Стандартные ссылки по основным принципам фармакологии включают Goodman and Gilman's The Pharmacological Basis of Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Любые приемлемые материалы и/или способы, известные специалистам в области техники, могут быть применены при осуществлении настоящего изобретения. Однако описаны предпочтительные материалы и способы. Материалы, реагенты и т.п., на которые дана ссылка в следующем описании и примерах, доступны из коммерческих источников, если не указано иное.
Определения, описанные в данном документе, могут быть представлены в виде химически значимых комбинаций, таких как "гетероалкиларил", "галоалкилгетероарил", "арилалкилгетероциклил", "алкилкарбонил", "алкоксиалкил" и т.п. В случае, когда термин "алкил" применяют в качестве суффикса, следующего за другим термином, как в "фенилалкил", или "гидроксиалкил", он предназначен для ссылки на алкильную группу, как определено выше, замещенную одним-двумя заместителями, выбранными из другой специально названной группы. Таким образом, например "фенилалкил" относится к алкильной группе, имеющей один-два фенильных заместителя, и таким образом включает бензил, фенилэтил, и бифенил. "Алкиламиноалкил" представляет собой алкильную группу, имеющую один-два алкиламино заместителя. "Гидроксиалкил" включает 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и т.п. Соответственно, как применено в данном документе, термин "гидроксиалкил" применяют для определения набора гетероалкильных групп, определенных ниже. Термин -(ар)алкил относится или к незамещенному алкилу или к аралкильной группе. Термин (гетеро)арил или (гет)арил относится или к арильной или к гетероарильной группе.
Термин "спироциклоалкил", как употреблено в данном документе, означает спироциклическую циклоалкильную группу, такую как, например, спиро[3,3]гептан Термин спирогетероциклоалкил, как употреблено в данном документе, означает спироциклический гетероциклоалкил, такой как, например, 2,6-диаза спиро[3,3]гептан.
Термин "ацил", как употреблено в данном документе означает группу формулы -C(=O)R, где R представляет собой водород или низший алкил, как определено в данном документе. Термин или "алкилкарбонил", как употреблено в данном документе, означает группу формулы C(=O)R, где R представляет собой алкил, как определено в данном документе. Термин C1-6 ацил относится к группе -C(=O)R, содержащей 6 атомов углерода. Термин "арилкарбонил" как употреблено в данном документе, означает группу формулы C(=O)R, где R представляет собой арильную группу; термин "бензоил", как употреблено в данном документе "арилкарбонильную" группу, где R представляет собой фенил.
Термин "эфир", как употреблено в данном документе, означает группу формулы -C(=O)OR, где R представляет собой низший алкил, как определено в данном документе.
Термин "алкил", как употреблено в данном документе, означает неразветвленную или разветвленную цепь, насыщенного, моновалентного углеводородного остатка, содержащего 1-10 атомов углерода. Термин "низший алкил" означает прямую или разветвленную цепь углеводородного остатка, содержащего 1-6 атомов углерода. "С1-10 алкил", как употреблено в данном документе относится к ал килу, состоящему из 1-10 атомов углерода. Примеры алкильных групп включают, но не ограничены, низшие алкильные группы, включая метил, этил, пропил, i-пропил, н-бутил, i-бутил, t-бутил или фенил, изопентил, неопентил, гексил, гептил и октил.
В случае когда термин "алкил" применяют в качестве суфикса вслед за другим термином, как "фенилалкил," или "гидроксиалкил," он предназначен для ссылки на алкильную группу, как определено выше, замещенную одним или двумя заместителями, выбранными из другой специально названной группы. Таким образом, например, "фенилалкил" означает радикал R'R''-, где R' представляет собой фенильный радикал, и R'' представляет собой алкиленовый радикал, как определено в данном документе, при условии, что точка прикрепления фенилалкильной группы будет на алкиленовом радикале. Примеры арилалкильных радикалов включают, но не ограничены, бензил, фенилэтил, 3-фенилпропил. Термины "арилалкил" или "аралкил" интерпретируются сходно, исключая R' представляющий собой арильный радикал. Термины "(гет)арилалкил" или "(гет)аралкил" интерпретируются сходно, исключая R' который возможно представляет собой арильный или гетероарильный радикал.
Термины "галоалкил" или "гало-низший алкил" или "низший галоалкил" относятся к углеводородному остатку с прямой или разветвленной цепью, содержащему 1-6 атомов углерода, где один или более атомов углерода замещены одним или более атомами галогена.
Термин "алкилен" или "алкиленил", как употреблено в данном документе, означает дивалентный насыщенный линейный углеводородный радикал из 1-10 атомов углерода (например, (СН2)n) или разветвленный насыщенный дивалентный углеводородный радикал из 2-10 атомов углерода (например, -СНМе- или -СН2СН(i-Pr)СН2-), если не указано иное. Исключая случай метилена, открытые валентности алкиленовой группы не прикреплены к такому же атому. Примеры алкиленовых радикалов включают, но не ограничены, метилен, этилен, пропилен, 2-метил-пропилен, 1,1-диметил-этилен, бутилен, 2-этилбутилен.
Термин "алкокси", как применено в данном документе, означает -О-алкильную группу, где алкил определен выше, такой как метокси, этокси, н-пропилокси, изо-пропилокси, н-бутилокси, изо-бутилокси, трет-бутилокси, пентилокси, гексилокси, включая их изомеры. "Низший алкокси" как применено в данном документе означает алкокси группу с "низшей алкильной" группой, как определено ранее. "С1-10 алкокси" как применено в данном документе относится к О-алкилу, где алкил представляет собой С1-10.
Термин "PCy3" относится к фосфину тризамещенному тремя циклическими группами.
Термины "галоалкокси" или "гало-низший алкокси" или "низший галоалкокси" относится к низшей алкокси группе, где один или более атомов замещены одним или более атомами галогена.
Термин "гидроксиалкил" как применено в данном документе означает алкильный радикал, как определено в данном документе, где один-три атома водорода на различных атомах углерода замещен(ы) гидроксильными группами.
Термины "алкилсульфонил" и "арилсульфонил" как применено в данном документе, относятся к группе формулы -S(=O)2R, где R представляет собой алкил или арил соответственно и алкил и арил определены в данном документе. Термин "гетероалкилсульфонил" как применено в данном документе относится к группе формулы -S(=O)2R, где R представляет собой "гетероалкил" как определено в данном документе.
Термины "алкилсульфониламино" и "арилсульфониламино" как применено в данном документе относятся к группе формулы -NR'S(=O)2R, где R представляет собой алкил или арил соответственно, R' представляет собой водород или C1-3 алкил, и алкил и арил определены в данном документе.
Термин "циклоалкил" как применено в данном документе относится к насыщенному карбоциклическому кольцу, содержащему 3-8 атомов углерода, т.е. циклопропил, циклобутил, циклопентил, циклогексил, циклогептил или циклооктил. "С3-7 циклоалкил" как применено в данном документе относится к циклоалкилу, состоящему из 3-7 атомов углерода в карбоциклическом кольце.
Термин "карбокси-алкил", как применено в данном документе, относится к алкильной группе, где один атом водорода замещен карбоксилом с учетом того, что точка прикрепления гетероалкильного радикала находится на атоме углерода. Термин "карбокси" или "карбоксил" относится к группе -CO2H.
Термин "гетероарил" или "гетероароматический" как применено в данном документе означает моноциклический или бициклический радикал из 5-12 кольцевых атомов, имеющих по меньшей мере одно ароматическое или частично ненасыщенное кольцо, содержащее четыре - восемь атомов на кольцо, включающее один или более гетероатомов N, О или S, оставшиеся кольцевые атомы представляют собой углерод, при этом, точка прикрепления гетероарильного радикала будет на ароматическом или частично ненасыщенном кольце. Как известно специалистам в области техники гетероарильные кольца имеют менее ароматические свойства, чем их только углеродные аналоги. Таким образом, в целях данного изобретения гетероарильная группа должна иметь некоторую степень ароматических свойств. Примеры гетероарильных групп включают моноциклические ароматические гетероциклы, имеющие 5-6 кольцевых атомов и 1-3 гетероатомов, включают, но не ограничены пиридинил, пиримидинил, пиразинил, оксазинил, пирролил, пиразолил, имидазолил, оксазолил, 4,5-дигидро-оксазолил, 5,6-дигидро-4Н-[1,3]оксазолил, изооксазолил, тиазол, изотиазол, триазолин, тиадиазол и оксадиаксолин, который может быть замещен одним или более, предпочтительно одним или двумя заместителями, выбранными из гидрокси, циано, алкил, алкокси, тио, низший галоалкокси, алкилтио, гало, низший галоалкил, алкилсульфинил, алкилсульфонил, галоген, амино, алкиламино, диалкиламино, аминоалкил, алкиламиноалкил и диалкиламиноалкил, нитро, алкоксикарбонил и карбамоил, алкилкарбамоил, диалкилкарбамоил, арилкарбомоил, алкилкарбониламино и арилкарбониламино. Примеры бициклических групп включают, но не ограничены, хинолинил, изохинолинил, бензофурил, бензотиопентил, бензоксазол, бензизоксазол, бензотиазол, нафтиридинил, 5,6,7,8-тетрагидро-[1,6]нафтиридинил, и бензизотиазол. Бициклические группы могут быть замещены по кольцу. Однако точка прикрепления расположена на кольце, содержащем гетероатом.
Термин "гетероциклил", "гетероциклоалкил" или "гетероцикл" как применено в данном документе, означает моновалентный насыщенный циклический радикал, состоящий из одного или более колец, предпочтительно одного-двух колец, включая спироциклические кольцевые системы, три-восемь атомов на кольцо, включая один или более кольцевых гетероатомов (выбранных из N, O или S(O)0-2), и который может быть независимо замещен одним или более, предпочтительно одним или двумя заместителями, выбранными из гидрокси, оксо, циано, низшего алкила, низшего алкокси, алкилтио, гало, низшего галоалкила, гидроксиалкила, нитро, алкоксикарбонила, амино, алкиламино, алкилсульфонила, арилсульфонила, алкиламиносульфонила, ариламиносульфонила, алкилсульфониламино, алкиламинокарбонил, ариламинокарбонила, алкилкарбониламино, и их ионных форм, если не указано иное. Примеры гетероциклических радикалов включают, но не ограничены, морфолинил, пиперазинил, пиперидинил, азетидинил, пирролидинил, гексагидроазепенил, оксэтанил, тетрагидрофуранил, тетрагидротиофенил, оксазолидинил, тиазолидинил, изоксазолидинил, тетрагидропиранил, тиоморфолинил, хинуклидинил и имидазолинил, и их ионные формы. Примеры могут также быть бициклическими, такими как, например, 3,8-диаза-бицикло[3.2.1]октан, 2,5-диаза-бицикло[2.2.2]октан или октагидро-пиразино[2,1-с][1,4]оксазин.
Ингибиторы Btk
Заявка предлагает соединение формулы I,

где:
R1 представляет собой Н или гало;
R2 представляет собой Н, гало или циано;
R3 представляет собой R4 или R5;
R4 представляет собой гало или циано;
R5 представляет собой фенил, гетероарил, -C(=O)R5', низший алкил или бензил, возможно замещенный одним или более R5';
R5' представляет собой низший алкил, циано, гидроксил, гетероциклоалкил, фенил, амино, алкиламино, диалкиламино или низший алкокси; и
X представляет собой низший алкил или гало;
или его фармацевтически приемлемую соль.
Заявка предлагает соединение формулы I, где X представляет собой метил.
Заявка предлагает соединение формулы I, где X представляет собой гало.
Заявка предлагает соединение формулы I, где X представляет собой метил и R5 представляет собой гетероарил, возможно замещенный одним или более R5'.
Заявка предлагает соединение формулы I, где R5 представляет собой тиофенил, возможно замещенный одним или более R5'.
Заявка предлагает соединение формулы I, где R5 представляет собой пиридинил, возможно замещенный одним или более R5'.
Заявка предлагает любое из вышеупомянутых соединений формулы I, где R1 представляет собой F и R2 представляет собой F.
Заявка альтернативно предлагает любое из вышеупомянутых соединений формулы I, где R1 представляет собой Н и R2 представляет циано.
Заявка предлагает соединение формулы I, где R5 представляет собой -C(=O)R5'.
Заявка предлагает соединение формулы I, где R5' представляет собой морфолинил, пиперидинил, низший алкил, пиперидинил или низший алкокси.
Заявка предлагает соединение формулы I, где R1 представляет собой F и R2 представляет собой F.
Заявка предлагает соединение формулы I, где R1 представляет собой Н и R2 представляет собой циано.
Заявка предлагает соединение формулы I, где R5 представляет собой фенил или бензил, возможно замещенный одним или более R5'.
Заявка предлагает соединение формулы I, где R5 представляет собой низший алкилен, возможно замещенный одним или более R5'.
Заявка предлагает любое из вышеупомянутых соединений формулы I, где R1 представляет собой F и R2 представляет собой F.
Заявка альтернативно предлагает любое из вышеупомянутых соединений формулы I, где R1 представляет собой Н и R2 представляет собой циано.
Заявка предлагает соединение формулы I, выбранное из группы, состоящей из:
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-(4-бром-1H-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-фенил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-3-ил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиразол-1-ил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-(4-тиофен-2-ил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-[4-(морфолин-4-карбонил)-1Н-индол-2-ил]-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-[4-(1-метил-1Н-пиразол-4-ил)-1Н-индол-2-ил]-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-(4-пиридин-2-ил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бензил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-[4-(1Н-пиразол-4-ил)-1H-индол-2-ил]-метанона;
3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразоол-4-карбонил}-1Н-индол-4-ил)-бензонитрила;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(3-хлор-фенил)-1Н-индол-2-ил]-метанона;
3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1H-индол-4-илметил)-бензонитрила;
3-(4-{5-амино-4-[4-(1Н-пиразол-4-ил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила;
3-(4-{5-амино-4-[4-(морфолин-4-карбонил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила;
3-(4-{5-амино-4-[4-(4-метил-пиперазин-1-карбонил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила;
3-(4-{5-амино-4-[4-(3-метокси-бензил)-1H-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенкоси)-бензонитрила;
Метилового эфира 2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-карбоново кислоты;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-(4-морфолин-4-илметил-1Н-индол-2-ил)-метанона;
3-{4-[5-амино-4-(4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила; и
Метиламида 2-{5-амино-1-[4-(3-циано-фенокси)-2-метил-фенил]-1H-пиразол-4-карбонил}-1H-индол-4-карбоновой кислоты.
Заявка предлагает способ лечения воспалительного и/или аутоиммунного состояния, содержащий введение пациенту, нуждающемуся в том, терапевтически эффективного количества соединения формулы I.
Заявка предлагает способ лечения ревматоидного артрита, содержащий введение пациенту, нуждающемуся в том, терапевтически эффективного количества соединения формулы I.
Заявка предлагает способ лечения астмы, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I.
Заявка предлагает способ лечения рака, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I.
Заявка предлагает фармацевтическую композицию соединения формулы I.
Заявка предлагает фармацевтическую композицию, содержащую соединение формулы I, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
Заявка предлагает применение соединения формулы I в качестве терапевтически активного вещества.
Заявка предлагает применение соединения формулы I при производстве лекарственного препарата для лечения воспалительного заболевания.
Заявка предлагает применение соединения формулы I при производстве лекарственного препарата для лечения аутоиммунного заболевания.
Заявка предлагает применение соединения формулы I при производстве лекарственного препарата для лечения ревматоидного артрита.
Заявка предлагает применение соединения формулы I при производстве лекарственного препарата для лечения астмы.
Заявка предлагает применение соединения, как описано выше, для лечения воспалительного и/или аутоиммунного заболевания.
Заявка предлагает применение соединения, как описано выше, для лечения ревматоидного артрита.
Заявка предлагает применение соединения, как описано выше, для лечения астмы.
Заявка предлагает применение соединения, как описано выше, для лечения воспалительного и/или аутоиммунного состояния.
Заявка предлагает применение соединения, как описано выше, для лечения ревматоидного артрита.
Заявка предлагает применение соединения, как описано выше, для лечения астмы.
Заявка предлагает соединение, как описано выше, для применения с целью лечения воспалительного и/или аутоиммунного состояния.
Заявка предлагает соединение, как описано выше, для применения для лечения ревматоидного артрита.
Заявка предлагает соединение, как описано выше, для применения при лечении астмы.
Заявка предлагает соединение, способ или композицию, как описано в данном документе.
Соединения и препараты
Примеры репрезентативных соединений, охватываемых настоящим изобретением и находящиеся внутри объема изобретения, предложены в таблице. Данные примеры и препараты, которые предложены в дальнейшем, позволят специалистам в области техники более точно понять и практически применить настоящее изобретение. Их не следует рассматривать как ограничивающие объем изобретения, а только в качестве его иллюстрации и представления.
Обычно, номенклатура, применяемая в данной заявке, основана на AUTONOMTM v.4.0, компьютеризированной системы института Бельштейна для получения номенклатуры согласно системе ИЮПАК. В случае несоответствия между изображенной структурой и названием, данным этой структуре, изображенная структура является более значимой. Кроме того, в случае, когда стереохимия структуры или части структуры не обозначена, например, при помощи жирных или пунктирных линий, структура или часть структуры должна быть интерпретирована как охватывающая все ее стереоизомеры.
Таблица I изображает примеры соединений согласно общей формуле I:

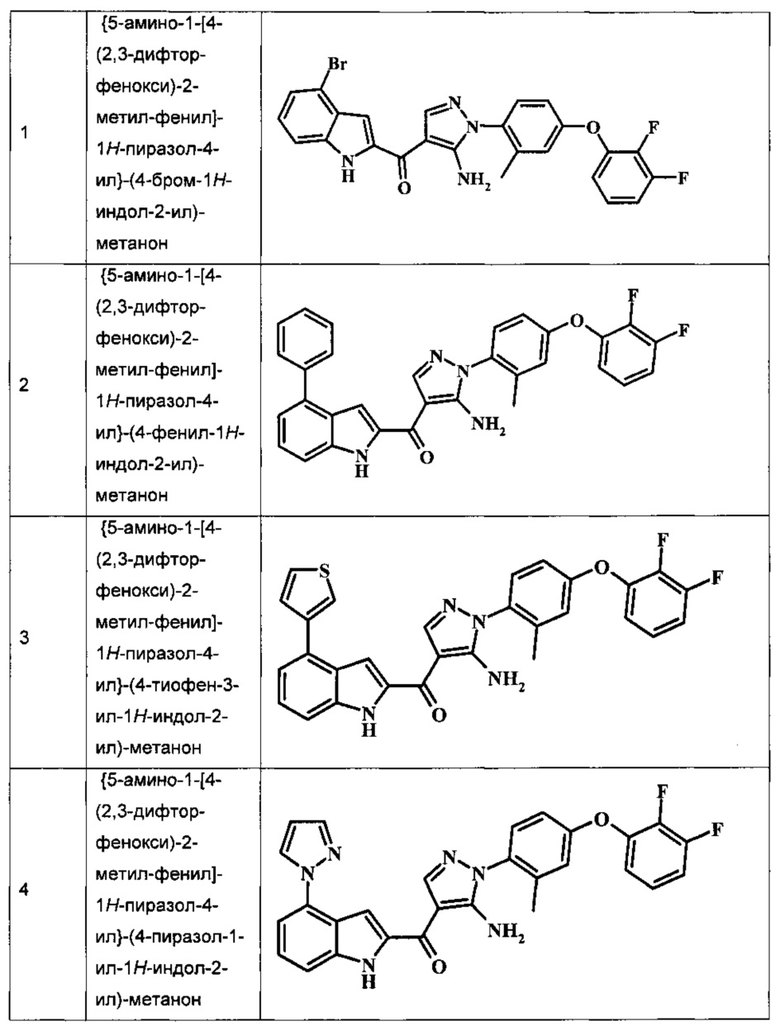

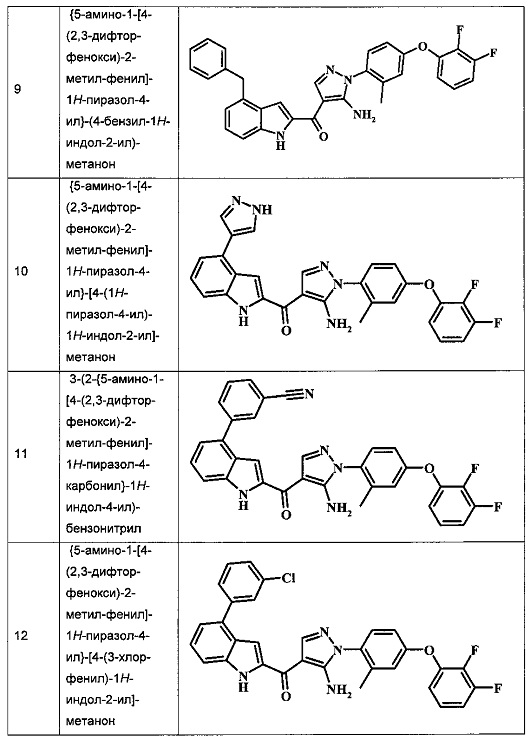
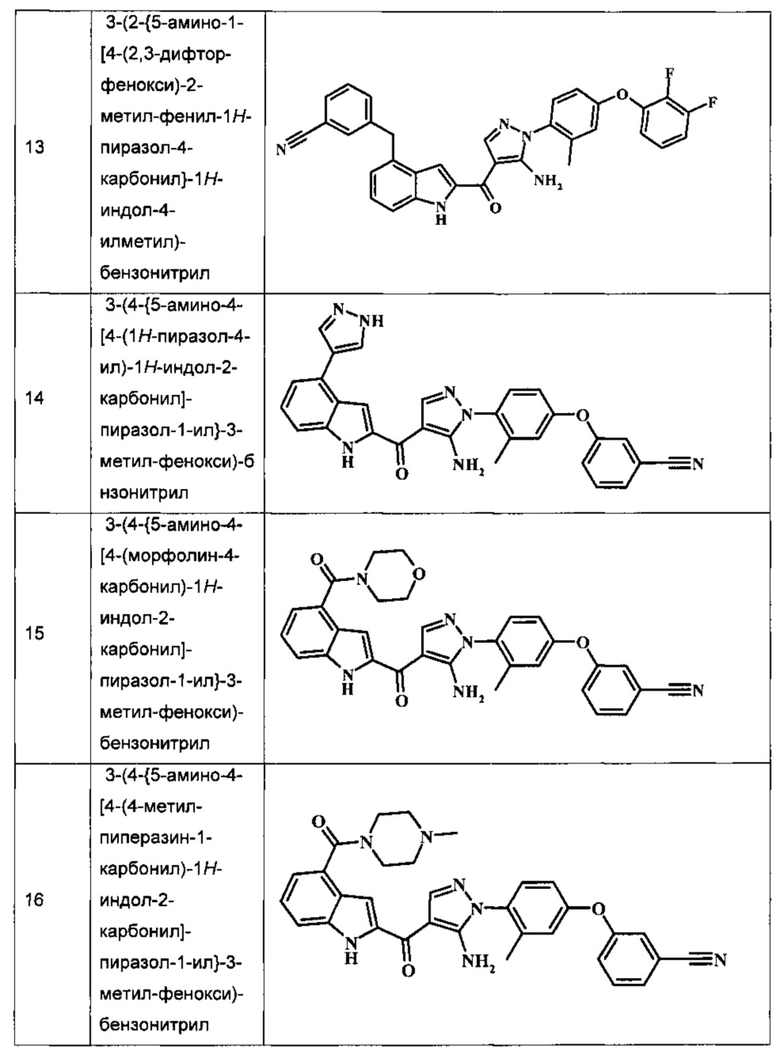

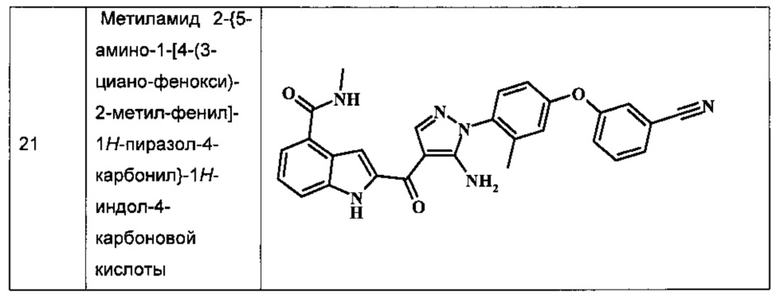
Обычные схемы синтеза
Соединения по настоящему изобретению могут быть получены посредством любых обычных способов. Приемлемые способы синтеза данных соединений предложены в примерах. Обычно, соединения по изобретению могут быть получены согласно схемам, приведенным ниже.
Схема 1
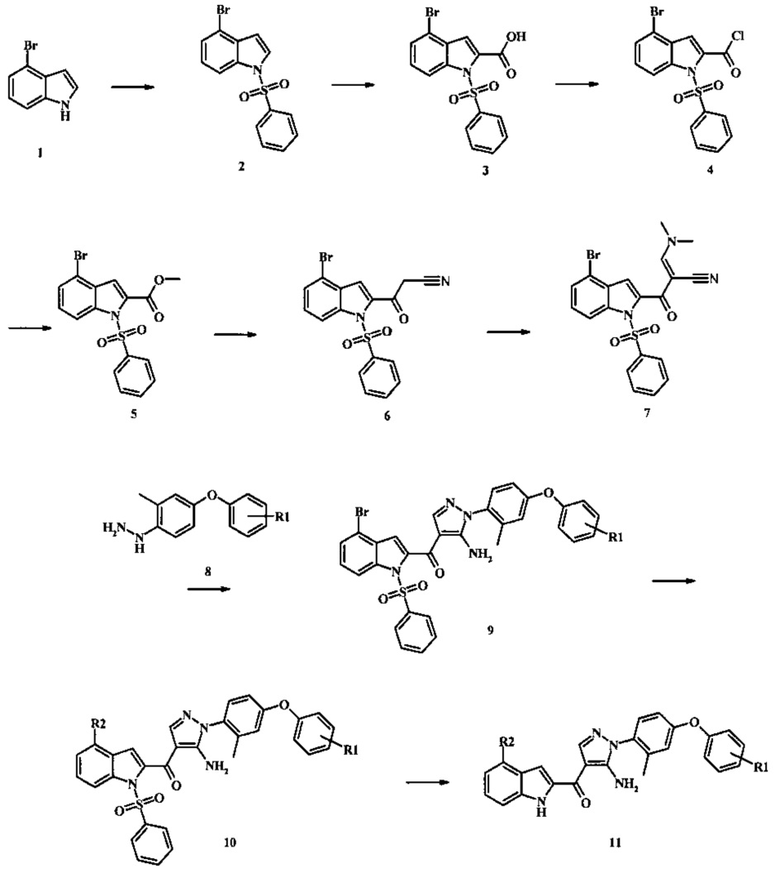
Соединения формулы 11, где R1 и R2 являются таковыми описанными выше в виде формулы I, могут быть получены при помощи пути, обозначенного на схеме 1. Согласно данному способу соединение формулы 1, 4-броминдол, который является коммерчески доступным, может быть переведено в фенилсульфонамид формулы 2. Обработка сильным основанием и двуокисью углерода обеспечивает карбоновую кислоту 3, которую переводят в метиловый эфир 5 через хлорангидрид 4. Затем эфир 5 может реагировать с производным ацетонитрила с получением производного цианоацетила формулы 6. Реакция с диметилформамид диметилацеталем обеспечивает производное акрилонитрила 7 и оно реагирует с производным фенилгидразина формулы 8 с получением аминопиразола формулы 9. Затем группа R2 может быть введена при помощи реакции конденсации, катализируемой переходным металлом, или реакцией нуклеофильного ароматического замещения, как показано ниже, до получения соединения формулы 10. Последующее удаление фенилсульфонильной защитной группы обеспечивает соединение по изобретению формулы 11.
4-броминдол, соединение формулы 1, может быть беспрепятственно обработано основанием таким, как гидрид натрия в инертном растворителе таком, как тетрагидрофуран при температуре приблизительно 0°С для получения соответствующего аниона. Он может быть обработан бензолсульфонилхлоридом и затем смесь перемешивают при комнатной температуре в течение приблизительно часа для получения производного бензолсульфониламида формулы 2.
Соединение формулы 2 затем может быть обработано н-бутиллитием в тетрагидрофуране при низкой температуре такой, как приблизительно -78°С, и соответствующий анион обработан излишком твердой двуокиси углерода для получения карбоновой кислоты формулы 3.
Превращение карбоновой кислоты формулы 3 в метиловый эфир формулы 5 может быть осуществлено при помощи одного из различных способов, хорошо известных средним специалистам в области техники органического синтеза. Многие приемлемые способы перечислены в Greene's Protective Groups in Organic Synthesis [Wuts, P.G.M and Greene, T.W., 4th Edition, Wiley-lnterscience, New York, 2006, pages 553 et seq.]. Например, тарнсформация может быть беспрепятственно проведена за счет обработки карбоновой кислоты формулы 3 хлорирующим агентом таким, как тионилхлорид или в чистом или в инертном растворителе таком, как бензол, при температуре между приблизительно 50°С и приблизительно температурой рефлюкса. Полученный хлорид кислоты формулы 4 затем может быть обработан метанолом в присутствии основания такого, как триэтиламин или диизопропилэтиламин или пиридинового эфира, метанолом в качестве растворителя или в инертном растворителе, таком как тетрагидрофуран при приблизительно комнатной температуре.
Конкретные условия получения соединения формулы 5 могут быть найдены в литературе, в Mahboobi, S. et al. J. Med. Chem. 2006, 49, 3101-3115.
Соединение формулы 5 может быть беспрепятственно переведено в производное цианоацетила формулы 6 за счет обработки его смесью ацетонитрила и сильного основания такого, как диизопропиламид лития или гексаметилдисилазид лития в растворителе таком, как тетрагидрофуран при низкой температуре, такой как приблизительно -78°С. Условия для такой реакции могут быть найдены в патентной литературе, например в Taka, N. et al. US 20120208811 Page 163.
Соединение формулы 6 может быть переведено в производное акрилонитрила формулы 7 за счет обработки N,N-диметилформамид диметилацеталем в инертном растворителе, таком как ароматический углеводород (например, толуол) или тетрагидрофуране при приблизительно комнатной температуре. Условия для такой реакции могут быть найдены в патентной литературе, например, в Taka, N. et al. US 20120208811 page 132.
Производное акрилонитрила формулы 7 может быть переведено в производное аминопиразола формулы 9 за счет обработки промежуточным соединением формулы 8, где R1 является таковым, описанным выше, в виде формулы I, в спиртовом растворителе, таком как метанол или этанол или изопропанол, при приблизительно температуре рефлюкса растворителя. Условия такой реакции могут быть найдены в патентной литературе, например, в Taka, N. et al. US 20120208811 Page 94.
Реакция соединения формулы 9 с соединением формулы R2-X, где X представляет собой бориновую кислоту, боронатный эфир, трифторборат калия, триметилтин или три-н-бутил-тин для получения соединения формулы 10, может быть осуществлена при помощи условий конденсации сузуки или Стила или Негиши, которые хорошо известны специалистам в области техники. Например, при реакции Сузуки, реакция может быть беспрепятственно проведена при помощи реакции соединения формулы 9 с соединением формулы R2-B(OH)2, в обычном инертном растворителе таком, как полярный апротонный растворитель (например, N,N-диметилформамид) или эфире (например, диоксан) или воде, или даже смеси таких растворителей, в присутствии каталитического количества предшественника палладия (0) (например палладий(II)ацетат или бис(трифенилфосфин)палладий(II)хлорид), при возможном дополнительном присутствии каталитического количества фосфинового лиганда, например, три-о-толилфосфина или три-трет-бутилфосфина или альтернативно в присутствии подготовленного комплекса палладия(0) с фосфиновым лигандом, такого как бис(три-циклогексилфосфин)палладий, тетракис(трифенилфосфин)-палладий(0) или [1,1-бис(дифенилфосфино)ферроцен]дихлорпалладий(II)), и также в присутствии неорганического основания, например, щелочного металла, карбоната щелочного металла, бикарбоната или фосфата (например, фосфата калия или карбоната натрия) при температуре между приблизительно комнатной температурой и приблизительно 100 градусами, и предпочтительно между приблизительно комнатной температурой и приблизительно 50 градусами. Реакция Сузуки известна специалистам в области техники органического синтеза, и была описана несколько раз, в частности в Miyaura, N.; Suzuki, A. Chem. Rev. 1995, 95, 2457-2483 и недавно в Alonso, F.; Beletskaya, I.P.; Yus, M. Tetrahedron 2008, 64, 3047-3101. Примеры конкретных условий, применяемых для Сузуки конденсации, могут быть найдены во многих ссылках в литературе, включая: Tiede, S. et al. Angew. Chem. Intl. Edn. 2010, 49, 3972-3975; Schmidt, A. and Rahimi, A. Chem. Commun. 2010, 46, 2995-2997; Lee, S.H. et al. US 20100063281; и Tobisu, M. et al. J. Org. Chem. 2010, 75, 4835-4840 (Supporting Information). Реакция Стилла хорошо известна специалистам в области органического синтеза, и может быть применена в качестве альтернативной реакции Сузуки, примеры условий которой приведены выше. Реакция Стилла была описана, в том числе в Farina, V. et al. Org. Reactions 1997, 50, 1-652. Примеры конкретных условий, которые применяют для реакция Стилла, могут быть найдены в литературе, например, в Littke, A.F. et al. J. Am. Chem. Soc. 2002, 124, 5343-6348; в Alberati-Giani, D. et al. US 7,462,617; и в Robl, J.A. US 5,072,023. Например, реакция может быть проведена за счет обработки соединения формулы 9 с соединением формулы R2-SnA3, где А представляет собой низшую алкильную группу, такую как метил или н-бутил, в обычном инертном растворителе, таком как полярный апротонный растворитель (например, N,N-диметилформамид) или ароматическом углеводороде (например, толуоле) или ацетонитриле или диметоксиэтане, в присутствии каталитического количества палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий(0) или бис(трифенилфосфин)палладий(II) хлорид или бис(ацетато)бис(трифенилфосфин)палладий(0) или трис(дибензилиденацетон)дипалладий(0) при температуре между приблизительно 80°С и приблизительно 180°С.
Соединения формулы 10, в которых R2 представляет собой карбоксамид или сложноэфирную функциональную группу с атомом углерода, прикрепленным к индольному кольцу, могут быть получены при помощи карбонилативной реакции конденсации, катализируемой переходным металлом. Согласно данному способу соединение формулы 9 нагревают с амином (для получения карбоксамидного продукта) или низшим спиртом (для получения продукта карбоксилатного сложного эфира) в присутствии окиси углерода и каталитического количества палладиевого катализатора, такого как тетракис(трифенилфосфин)палладий(0) или комбинации палладиевого катализатора, такого как бис(бензонитрил)палладий(II) дихлорид с лигандом, таким как 1,1'-бис(дифенилфосфино)ферроцен в растворителе, таком как тетрагидрофуран или толуол в запаянном сосуде при температуре между приблизительно 80°С и приблизительно 100°С. Примеры конкретных условий, которые могут быть применены для такой реакции, могут быть найдены в литературе, например, в Kumar, K. et al. Org. Letters 2004, 6, 7-10.
Соединения формулы 10, в которых R2 представляет собой аралкильную группу, такую как бензильная группа или замещенная бензильная группа, могут быть беспрепятственно получены при помощи реакции конденсации, катализируемой переходным металлом, такой как реакция Негиши. Согласно данном способу соединение формулы 9 нагревают с реагентом аралкилцинк в присутствии каталитического количества палладиевого катализатора, такого как палладий(II) ацетат с лигандом, таким как 2-дициклогексилфосфино-2',6'-диметоксибифенил (S-Phos) или при помощи тетракис(трифенилфосфин)-палладия(0) в растворителе таком, как тетрагидрофуран или толуол, при температуре между приблизительно 50°С и приблизительно 90°С. Примеры конкретных условий, которые могут быть применены для такой реакции, могут быть найдены в литературе, например, в Ellsworth, В.A. et al. US 20110082165 page 51.
Соединения формулы 10, в которой R2 представляет собой цианометильную группу и могут быть беспрепятственно получены при реакции конденсации, катализируемой переходным металлом, с трибутилстананил-ацетонитрилом или низшим алкилцианоацетатом. Реакция с трибутилстананил-ацетонитрилом может быть проведена посредством обработки соединения формулы 9 трибутилстананил-ацетонитрилом в присутствии палладиевого катализатора, такого как бис(трифенилфосфино)палладий(II) дихлорид или бис(три-о-толилфосфин)палладий(II) дихлорид в толуоле или ксилоле при температуре между приблизительно 110°С и приблизительно 130°С. Условия для такой реакции могут быть найдены в литературе, например, в Ettaoussi, М. et al. Eur. J. Med. Chem. 2012, 49, 310-323 and in Song, D. et al. WO 2011117211 page 118. Реакция с низшим алкилцианоацетатом может быть проведена посредством обработки соединения формулы 9 низшим алкилцианоацетатом в присутствии палладиевого катализатора, такого как бис(три-трет-бутилфосфин)палладий(0), или смеси палладиевого катализатора, такого как трис-(дибензелиденацетон)дипалладий(0) или бис(дибензилиденацетон)палладий(0) и лиганда, такого как трет-бутилфосфин, и основания, такого как тринатрийфосфат в растворителе, таком как толуол в запаянном сосуде при температуре между приблизительно 70°С и приблизительно 100°С. В случае, когда низший алкилцианоацетат представляет собой трет-бутилцианоацетат, трет-бутильная группа может подвергаться реакции декарбоксилирования при условиях реакции для получения желаемого соединения формулы 10, где R2 представляет собой цианометильную группу. В случае, когда низший алкилцианоацетат представляет собой метилцианоацетат или этилцианоацетат, необходим дополнительный этап гидролиза и его обычно проводят посредством нагревания продукта палладий-катализируемой конденсации с 3 М соляной кислотой в диметилсульфоксиде при приблизительно 70°С в течение нескольких часов. Примеры условий, которые могут быть применены для такой реакции, могут быть найдены в литературе, например, в Alargova, R.G. et al. US 20120015999 page 10.
Конверсия соединения формулы 10 в соединение по изобретению формулы 11 может быть осуществлена посредством любого обычного способа. Например, реакция может быть проведена посредством обработки соединения формулы 10 смесью основания, такого как карбонат цезия с низшим спиртом, таким как метанол в растворителе, таком как тетрагидрофуран при температуре между приблизительно комнатной температурой и приблизительно температурой рефлюкса смеси. Примеры условий, которые могут быть применены для такой реакции могут, быть найдены в литературе, например, в Zhang, В and Wee. A.G.Н. Org. Biomol. Chem. 2012, 10, 4597-4608 Supplementary Information; в Alam, M. et al. US 20110071150 page 54; and in Taka, N. et al. US 20120208811 Page 55.
Схема 2

Специалистам в области техники органического синтеза будет очевидно, что как показано на схеме 2, многие соединения формулы 11 также легко доступны при снятии защитной группы с соединения формулы 9 скорее, чем с соединения формулы 10.
Согласно данному способу, реакция снятия защитной группы может быть проведена посредством обработки соединения формулы 9 смесью основания, такого как карбонат цезия и низшего спирта, такого как метанол, в растворителе, таком как тетрагидрофуран при температуре между приблизительно комнатной температурой и приблизительно температурой рефлюкса смеси. Примеры условий, которые могут быть применены для такой реакции могут быть найдены в литературе, например, в Zhang, В and Wee. A.G.Н. Org. Biomol. Chem. 2012, 10, 4597-4608 Supplementary Information; в Alam, M. et al. US 20110071150 page 54; and in Taka, N. et al. US 20120208811 Page 55.
Полученное соединение формулы 12 затем может быть обработано соединением формулы R2-B(OH)2, при условиях, описанных выше, для реакции Сузуки соединения формулы 9 для получения желаемого соединения формулы 11.
Соединения формулы 11, в которых R2 представляет собой N-связанный гетероцикл, такой как пиразол-1-ил может быть обычно получено при помощи Схемы 2. Согласно данному способу соединение формулы 12 обрабатывают N-связанным гетероциклом, таким как пиразол, в присутствии основания, такого как карбонат калия, в присутствии медного катализатора, например, йодида меди(I) и в присутствии L-пролина, в инертном растворителе, таком как диметилсульфоксил при температуре приблизительно 100°С и приблизительно 130°С. Примеры условий, которые могут быть применены для такой реакции, могут быть найдены в литературе, например, в Sun, X. et al. Bioorg. Med. Chem. Lett. 2011, 21, 3671-3675 Supplemental Information; and in Yokotani, J. et al. US 20110275797 page 31.
Схема 3

Соединения формулы 21, где R3 представляет собой вторичную аминогруппу, такую как диалкиламино (например, диметиламино или диэтиламино) или циклическую вторичную аминогруппу, такую как пирролидино, пиперидино, морфолин-4-ил, 1-метил-пиперазин-4-ил или т.п. могут быть получены при помощи способа, показанного на схеме 3. Согласно данному способу 1-бензолсульфонил-1Н-индол-4-метанол, соединение формулы 13 (которое представляет собой известное соединение, которое может быть получено согласно способу, приведенному в Castro Pineiro, J.L. et al. US 6187805 Column 15, или при помощи способов, описанных в нижеприведенных примерах) превращают в соответствующее производное хлорметилиндола формулы 14. Его подвергают реакции замещения с амином для получения соединения формулы 15. Последовательность карбоксилирования, этерификации и реакции с анионом ацетонитрила, затем дает производное цианоацетила формулы 18. Реакция с диметилформамид диметилацеталем дает производное акрилонитрила формулы 19, которое подвергается реакции с арилгидразином формулы 8 для получения производного аминопиразола формулы 20. Удаление фенилсульфонильной защитной группы затем дает соединение по изобретению формулы 21.
Соединение формулы 13 может быть обработано метансульфонилхлоридом в присутствии основания такого, как триэтиламин или диизопропилэтиламин в растворителе таком, как тетрагидрофуран при приблизительно комнатной температуре для получения соединения формулы 14.
Соединение формулы 14 может быть обработано вторичным амином, таким как диалкиламин (например, диметиламин гидрохлорид или диэтиламин) или циклической вторичной аминогруппой, такой как пирролидин, пиперидин, морфолин, или 1-метил-пиперазин, в присутствии неорганического основания, такого как карбонат калия или карбонат цезия, в инертном растворителе, таком как ацетонитрил, при температуре между приблизительно 50°С и приблизительно 80°С для получения амина формулы 15.
Соединение формулы 15 затем может быть обработано сильным основанием, таким как диизопропиламид лития или гексаметилдисилазид лития в тетрагидрофуране при низкой температуре, такой как при приблизительно -78°С, и соответствующий анион, обработанный излишком твердой двуокиси углерода для получения карбоновой кислоты формулы 16.
Конверсия карбоновой кислоты формулы 16 в метиловый эфир формулы 17 может быть осуществлена при помощи одного из ряда способов, хорошо известных специалистам в области техники органического синтеза. Многие приемлемые способы приведены в Greene's Protective Groups in Organic Synthesis [Wuts, P.G.M and Greene, T.W., 4th Edition, Wiley-lnterscience, New York, 2006, pages 553 et seq.] например, трансформация может быть беспрепятственно проведена посредством обработки карбоновой кислоты формулы 16 хлорирующим агентом, таким как тионилхлорид или чистым или в инертном растворителе, таком как бензол при температуре между приблизительно 50°С и приблизительно температурой рефлюкса. Полученный хлорид кислоты затем может быть обработан метанолом в присутствии основания такого, как триэтиламин или диизопропилэтиламин или пиридин или при помощи метанола в качестве растворителя или в инертном растворителе, таком как тетрагидрофуран при приблизительно комнатной температуре для получения сложного эфира формулы 17.
Соединение формулы 17 может быть беспрепятственно превращено в производное цианоацетила формулы 18 посредством его обработки смесью ацетонитрила и сильного основания, такого как диизопропиламид лития или гексаметилдисилазид лития в растворителе таком, как тетрагидрофуран при низкой температуре, такой как при приблизительно -78°С. Условия данной реакции могут быть найдены в патентной литературе, например, в Taka, N. et al. US 20120208811 Page 163.
Соединение формулы 18 может быть превращено в производное акрилонитрила формулы 19 за счет обработки N,N-диметилформамид диметилацеталем в инертном растворителе, таком как ароматический углеводород (например, толуол) или тетрагидрофуране при приблизительно комнатной температуре. Условия для такой реакции могут быть найдены в патентной литературе, например, в Taka, N. et al. US 20120208811 page 163.
Производное акрилонитрила формулы 19 может быть превращено в производное аминопиразола формулы 20 за счет обработки промежуточным соединением формулы 8, где R1 представляет собой описанное выше в виде формулы I, в алкогольном растворителе, таком как метанол или этанол или изопропанол, при приблизительно температуре рефлюкса растворителя. Условия для такой реакции могут быть найдены в патентной литературе, например, в Taka, N. et al. US 20120208811, страница 94.
Конверсия соединения формулы 20 в соединение формулы 21 может быть проведена при помощи обычного способа. Например, реакция может быть осуществлена за счет обработки соединения формулы 20 смесью основания, такого как карбонат цезия, и низшего спирта, такого как метанол. В растворителе, таком как тетрагидрофуран при температуре между приблизительно комнатной температурой и приблизительно температурой рефлюкса смеси. Примеры условий, которые могут быть применены для такой реакции, можно найти в литературе, например, в Zhang, В and Wee. A.G.Н. Org. Biomol. Chem. 2012, 10, 4597-4608 Supplementary Information; в Alam, M. et al. US 20110071150 page 54; and in Taka, N. et al. US 20120208811 Page 55.
Схема 4

Промежуточные соединения формулы 8, где R1 представляет собой описанное выше в виде формулы I, могут быть получены согласно схеме 4. Соединение формулы 22, 4-хлор-2-метил-1-нитро-бензол подвергают реакции нуклеофильного ароматического замещения с производным фенола формулы 23 с получением соединения формулы 24. Восстановление нитро группы в соединение формулы 24, с последующим диазотированием и восстановлением дает производное арил-гидразина формулы 8.
4-хлор-2-метил-1-нитро-бензол (22) может быть обработан фенолом формулы 23 в присутствии основания, такого как карбонат калия или карбонат цезия в инертном растворителе, таком как диметилформамид при температуре между приблизительно 100°С и приблизительно 150°С, возможно под действием микроволнового излучения, для получения нитро соединения формулы 24. Примеры конкретных условий, которые могут быть применены для данной реакции, могут быть найдены в литературе, например, в Chee, G.-L et al. US 20040266738 Page 5; and in Cui, S.-L. et al. Synlett 2004, 1829-1831.
Восстановление нитрогруппы в соединении формулы 24 может быть осуществлено при помощи различных способов хорошо известных специалистам в области техники органического синтеза. Многие из данных способов приведены в Larock, R.С. Comprehensive Organic Transformations John Wiley & Sons Inc. NY 1999, pp. 823 et seq. Один обычный способ заключается в обработке соединения формулы 24 газообразным водородом в присутствии катализатора благородного металла такого как палладий на углеродном носителе в растворителе таком как спирт (например, метанол или этанол) при давлении между приблизительно одной атмосферой водорода и приблизительно тремя атмосферами водорода при приблизительно комнатной температуре. Примеры конкретных условий, которые могут быть применены для такой реакции, могут быть найдены в литературе, например, в Chee, G.-L et al. US 20040266738 Page 5; and in Schoenafinger, K. et al. US 20030236288 Page 18.
Диазотирование и восстановление анилиновой группы в соединении формулы 25 может быть осуществлено при помощи обычного способа. Например, реакцию обычно проводят при помощи обработки соединения формулы 25 нитритом натрия в водном растворе в присутствии неорганической кислоты, такой как соляная кислота при температуре ниже приблизительно 5°С и предпочтительно ниже приблизительно 0°С, с последующим добавлением восстановителя, такого как хлорид олова(II) или дитионит натрия при приблизительно одинаковых температурах. Примеры конкретных условий, которые могут быть применены для такой реакции могут быть найдены в литературе, например в Wipf, P. and Qiming, J. WO 2012078859 page 47; в Rewolinski, М.V. et al. WO 2009055721 page 82; и в Schoenafinger, K. et al. US 20030236288 page 18.
Фармацевтические композиции и введение
Соединения по настоящему изобретению могут быть получены в виде широкого ряда дозированных форм для орального введения и носителей. Оральное введение может быть в виде таблеток, покрытых таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропов или суспензий. Соединения по настоящему изобретению являются действенными и в случае других путей введения, включая непрерывное (внутривенная капельница) местное, парентеральное, внутримышечное, внутривенное, подкожное, чрескожное (которое может включать агент, усиливающий проникновение), буккальное, назальное, ингаляционное и суппозиторное введение, среди других путей введения. Предпочтительным способом введения обычно является оральное применение с обычным ежедневным режимом дозирования, который может быть скорректирован согласно степени заболевания и ответа пациента на активный ингредиент.
Соединение или соединения по настоящему изобретению, а также их фармацевтически приемлемые соли, вместе с одним или более обычными эксципиентами, носителями или разбавителями, могут быть помещены в виде фармацевтических композиций и единичных дозированных форм. Фармацевтические композиции и единичные дозированные формы могут состоять из обычных ингредиентов в обычных пропорциях, с или без дополнительных активных соединений или основ, и единичные дозированные формы могут содержать любое приемлемое эффективное количество активного ингредиента сопоставимого с назначенным к применению ежедневным диапазоном дозирования. Фармацевтические композиции могут быть применены в качестве твердых веществ, таких как таблетки или заполненные капсулы, полутвердых порошков, препаратов замедленного высвобождения или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры или заполненные капсулы для орального применения; или в виде суппозиторий для ректального или вагинального введения; или в виде стерильных растворов для парентерального применения. Местный препарат будет содержать от приблизительно 5% до приблизительно 95% активного соединения или соединений (масс./масс.). Термин "препарат" или "дозированная форма" предназначен для включения как твердых так и жидких препаратов активного соединения и специалистам в области техники очевидно, что активный ингредиент может существовать в различных препаратах в зависимости от мишенного органа или ткани и желаемой дозы и фармакокинетических параметров.
Термин "эксципиент" при применении в данном документе относится к соединению, которое является полезным при получении фармацевтической композиции, обычно безопасным, нетоксичным и не обладающим нежелательными биологическими или иными свойствами, и включает эксципиенты, которые приемлемы для ветеринарного применения, а также фармацевтического применения у человека. Соединения данного изобретения могут быть введены сами по себе или в смеси с одним или более приемлемыми фармацевтическими эксципиентами, разбавителями или носителями, выбранными с учетом назначенного пути введения и стандартной фармацевтической практики.
"Фармацевтически приемлемый" означает такой, который полезен при получении фармацевтической композиции, которая обычно является безопасной, нетоксичной и не обладает нежелательными биологическими или иными свойствами, и включение которого является приемлемым для ветеринарного применения, а также применения для людей.
"Фармацевтически приемлемая солевая" форма активного ингредиента также может изначально придавать желаемые фармакокинетические свойства активному ингредиенту, которые отсутствуют у несолевой формы, и даже может положительно влиять на фармакодинамику активного ингредиента в отношении его терапевтической активности в теле. Фраза "фармацевтически приемлемая соль" соединения означает соль, которая является фармацевтически приемлемой и которая обладает желаемой фармакологической активностью исходного соединения. Такие соли включают: (1) соли добавления кислот, образованные при помощи неорганических кислот, таких как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, ортофосфорная кислота и т.п.; или образованные при помощи органических кислот, таких как уксусная кислота, пропионовая кислота, капроновая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этан-дисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталенсульфоновая кислота, 4-толуолсульфоновая кислота, камфорасульфокислота, 4-метилбицикло[2.2.2]-окт-2-ене-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутил уксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и т.п.; или (2) соли образованные, в случае, когда кислый протон, присутствующий в исходном соединении или замещают ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; или координируют с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и т.п.
Твердые формы препаратов включают порошки, таблетки, пилюли, капсулы, саше, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или более вещество, которое также может действовать в качестве разбавителей, ароматизаторов, солюбилизаторов, лубрикантов, суспендирующих агентов, связующих веществ, консервантов, агентов, дезинтегрирующих таблетки, или инкапсулирующий материал. В порошках, носитель обычно представляет собой окончательно разделенное твердое вещество, которое представляет собой смесь с окончательно разделенным активным компонентом. В таблетках, активный компонент обычно смешивают с носителем, имеющим необходимую способность связывания в приемлемых пропорциях и компактизуют до желаемой формы и размера. Приемлемые носители включают, но не ограничены, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, карбоксиметилцеллюлозу натрия, легкоплавкий воск, какао масло и т.п. препараты твердых форм могут содержать в добавление к активному компоненту, красители, ароматизаторы, стабилизаторы, буферы, искусственные и натуральные подсластители, дисперсанты, загустители, солюбилизаторы и т.п.
Жидкие препараты также приемлемые для орального введения включают жидкий препарат, включающий эмульсии, сиропы, эликсиры, водные растворы, водные суспензии. Они включают препараты твердых форм, которые предназначены для превращения в препараты жидких форм непосредственно перед применением. Эмульсии могут быть получены в виде растворов, например, в водных растворах пропиленгликоля или могут содержать эмульгаторы, такие как лецитин, сорбитанмоноолеат или акацию. Водные растворы могут быть получены посредством растворения активного компонента в воде и добавления приемлемых красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии могут быть получены за счет диспергирования окончательно разделенного активного компонента в воде с вязким материалом, таким как натуральные или синтетические камеди, смолы, метилцеллюлоза, карбоксиметилцеллюлоза натрия и другие хорошо известные суспендирующие агенты.
Соединения по настоящему изобретению могут быть получены для парентерального введения (например, посредством инъекции, например болюсной инъекции или непрерывной инфузии) и может быть представлено в единичной дозированной форме в ампулах, предварительно наполненных шприцах, инфузии небольшого объема или в многодозовых контейнерах с добавленным консервантом. Композиции могут иметь такие формы как суспензии, растворы или эмульсии в масляном или водном носителях, например, растворы в водном полиэтиленгликоле. Примеры масляных или неводных носителей, разбавителей, растворителей или носителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например, оливковое масло), и инъецируемые органические сложные эфиры (например, этилолеат), и могут содержать вспомогательные вещества, такие как консерванты, увлажнители, эмульгирующие или суспендирующие агенты, стабилизаторы и/или диспергирующие агенты. Альтернативно активный ингредиент может быть в порошковой форме, полученной посредством асептического выделения стерильного твердого вещества или посредством лиофилизации из раствора для составления перед применением с приемлемым носителем, например, стерильной апирогенной водой.
Соединения по настоящему изобретению могут быть получены для местного введения в эпидермис в виде мазей, кремов или лосьонов, или в виде трансдермального пластыря. Мази и крема могут, например, быть получены при помощи водной или масляной основы с добавлением приемлемого загустителя и/или гелеобразующих агентов. Лосьоны могут быть получены при помощи водного или масляного основания и обычно также будут содержать один или более эмульгатор, стабилизатор, диспергирующий агент, суспендирующий агент, загуститель или краситель. Препараты, приемлемые для местного введения мыши включают таблетки для рассасывания, содержащие активные агенты во вкусовой основе, обычно сахарозу, акацию или трагакант; пастилки, содержащие активные агенты в инертной основе, такой как желатин и глицерин или сахарозе ил акации; и жидкости для полоскания рта, содержащие активный ингредиент в приемлемом жидком носителе.
Соединения по настоящему изобретению могут быть получены для введения в виде суппозиторий. Легкоплавкий воск, такой как смесь глицеридов жирных кислот или какао-масла сначала плавят и активный компонент гомогенно диспергируют, например, посредством перемешивания. Расплавленную гомогенную смесь затем заливают в формы подходящего размера, оставляют остывать и отвердевать.
Соединения по настоящему изобретению могут быть получены для вагинального введения. Маточные кольца, тампоны, крема, гели, пасты, мыла или спреи, содержащие в дополнение к активному ингредиенту такие носители хорошо известны в области техники.
Соединения по настоящему изобретению могут быть получены для назального введения. Растворы или суспензии вводят непосредственно в носовую полость обычными способами, например, при помощи капельницы, пипетки или спрея. Препараты могут быть предложены в единичной или многодозовой форме. В последнем случае капельницы или пипетки, он может быть введен пациенту в соответствующем заранее определенном объеме раствора или суспензии. В случае спрея это может быть достигнуто, например, посредством способа дозирования при помощи насоса для распыления.
Соединения по настоящему изобретению могут быть получены для аэрозольного введения, в частности в респираторный тракт и включая интраназальное введение. Соединение обычно обладает небольшим размером частиц, например, порядка пяти микрон (5) или менее. Такой размер частиц может быть получен посредством способов известных в области техники, например, микронизации. Активный ингредиент предложен в упаковке под давлением с приемлемым газом-вытеснителем, таким как хлорфторкарбон (CFC), например, дихлордифторметан, трихлорфторметан, или дихлортетрафторэтан, или двуокись углерода или другой приемлемый газ. Аэрозоль может обычно также содержать поверхностно-активное вещество, такое лецитин. Доза лекарственного средства может контролироваться посредством насадки распылителя. Альтернативно активные ингредиенты могут быть предложены в форме сухого порошка, например, порошковой смеси соединения в приемлемом порошковом основании таком, как лактоза, крахмал, производные крахмала, таком как гидроксипропилметил целлюлоза и поливинилпиролидон (ПВП). Порошковый носитель будет формировать гель в носовой полости. Порошковая композиция может быть представлена в единичной дозированной форме, например, в капсулах или картриджах, например, желатиновые или блистерные упаковки из которых порошок может быть введен посредством ингалятора.
При желании препараты могут быть получены при помощи таблеток в кишечнорастворимой оболочке, адаптированной для введения с замедленным или контролируемым высвобождением активного ингредиента. Например, соединения по настоящему изобретению могут быть получены в устройствах чрескожной или подкожной доставки лекарственного средства. Данные системы доставки являются преимуществом, когда необходимо замедленное высвобождение соединения и в случае, когда исполнительность пациентом режима дозирования является критичной. Соединения в трансдермальных системах доставки часто прикреплены к твердой подложке, адгезирующей к коже. Интересующее соединение также может быть скомбинировано с агентом, усиливающим проникновение, например, Азон (1-додецилаза-циклогептан-2-он). Системы замедленного высвобождения вводят подкожно в субдермальный слой при помощи операции или инъекции. Субдермальные импланты инкапсулируют соединение в жирорастворимой мембране, например, силиконовой смоле, или биодеградируемом полимере, например, полилактидной кислоте.
Приемлемые препараты наряду с фармацевтическими носителями, разбавителями и эксципиентами описаны в Remington: The Science and Practice of Pharmacy 1995, edited by E.W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania. Разработчики препаратов могут модифицировать препараты в пределах описания для обеспечения различных препаратов для конкретного пути введения без нарушения стабильности композиции по изобретению или ее терапевтической активности.
Модификация настоящих соединений для придания им большей растворимости в воде или другом носителе, например, может быть легко выполнена за счет минорных модификаций (образования соли, этерификации и т.д.), которые хорошо известны специалистам в области техники. Также специалистам в области техники известна модификация пути введения и режим дозирования конкретного соединения с целью управления фармакокинетикой настоящих соединений для максимально полезного эффекта у пациентов.
Термин "терапевтически эффективное количество", как применено в данном документе, означает количество, необходимое для уменьшения симптомов заболевания у индивидуума. Дозу подбирают соответственно индивидуальным требованиям в каждом конкретном случае. Такая дозировка может варьировать внутри широких пределов в зависимости от различных факторов, таких как серьезность заболевания, подлежащего лечению, возраст и общее состояние здоровья пациента, другие лекарственные препараты, которые принимает пациент, путь и форма введения и предпочтения и опыт лечащих врачей. Для орального введения должна быть уместна ежедневная дозированная форма от приблизительно 0,01 и до приблизительно 1000 мг/кг массы тела в день в монотерапии и/или в комбинированной терапии. Предпочтительная дозировка составляет между приблизительно 0,1 и приблизительно 500 мг/кг массы тела, более предпочтительно 0,1 и приблизительно 100 мг/кг массы тела и наиболее предпочтительно между 1,0 и приблизительно 10 мг/кг массы тела в день. Таким образом, для введения индивидууму массой 70 кг, диапазон дозирования должен составлять приблизительно от 7 мг до 0,7 г в день. Ежедневная доза может быть введена в виде единичной дозы или в виде отдельных доз, обычно между 1 и 5 дозами в день. Обычно лечение начинают с небольших доз, которые являются меньше оптимальной дозы соединения. После этого, дозу повышают посредством небольших увеличений до достижения оптимального эффекта для отдельного индивидуума. Специалисты при лечении заболеваний, описанных в данном документе, без лишних экспериментов и полагаясь на личные знания, опыт и раскрытие данной заявки, будут способны определить терапевтически эффективное количество соединений по настоящему изобретению для данного заболевания и пациента.
Предпочтительны фармацевтические препараты в виде единичных дозированных форм. В такой форме, препарат подразделен на единичные дозы, содержащие соответствующие количества активного компонента. Единичная дозированная форма может представлять собой упакованный препарат, упаковку, содержащую дискретные количества препарата, такие как упакованные таблетки, капсулы и порошки в сосудах или ампулах. Также единичная дозированная форма может представлять собой капсулу, таблетку, саше или пастилку для рассасывания сами по себе или он может представлять собой соответствующее число любых из них в упакованном виде.
Показания и способы лечения
Соединения общей формулы I ингибируют тирозинкиназу Брутона (Btk). Активация Btk за счет вышерасположенных киназ приводит к активации фосфолипазы С, которая, напротив, стимулирует высвобождение провоспалительных медиаторов. Соединения формулы I полезны при лечении артрита и других антивоспалительных и аутоиммунных заболеваний. Соединения формулы I соответственно полезны при лечении артрита. Соединения формулы I полезны для ингибирования Btk в клетках и для модулирования развития В-клеток. В дальнейшем настоящее изобретение содержит фармацевтические композиции, содержащие соединения формулы I, смешанные с фармацевтически приемлемым носителем, эксципиентами или разбавителями.
Соединения, описанные в данном документе, представляют собой ингибиторы киназ, в частности ингибиторы Btk. Данные ингибиторы могут быть полезны при лечении одного или более заболеваний, чувствительных к ингибированию киназы, включая заболевания чувствительные к ингибированию Btk и/или ингибированию В-клеточной пролиферации у млекопитающих. Без связи с какой-либо конкретной теорией предположительно взаимодействие соединений по изобретению с Btk приводит к ингибированию активности Btk и таким образом находит фармацевтическое применение данных соединений. Соответственно изобретение включает способ лечения млекопитающего, например, человека, имеющего заболевание, чувствительное к ингибированию активности Btk и/или ингибированию В-клеточной пролиферации, содержащий введение млекопитающему, имеющему такое заболевание, эффективного количества по меньшей мере одного химического соединения, предложенного в данном документе. Эффективная концентрация может быть определена экспериментально, например, за счет оценки концентрации соединения в крови, или теоретически, за счет вычисления биодоступности. Другие киназы, на которые можно повлиять дополнительно к Btk, включают, но не ограничены, другие тирозинкиназы и серин/треонинкиназы.
Киназы играют существенные роли в сигнальных путях, контролирующих фундаментальные клеточные процессы, такие как пролиферация, дифференциация и клеточная гибель (апаптоз). Ненормальная киназная активность влечет за собой широкий диапазон заболеваний, включая множественный рак, аутоиммунные и/или воспалительные реакции. Многостороння роль киназ в ключевых путях клеточного сигналинга обеспечивает существенный потенциал для идентификации новых лекарственных средств, нацеленных на киназы и сигнальные пути.
Воплощение включает способ лечение пациента, имеющего аутоиммунное и/или воспалительное заболевание, или острую воспалительную реакцию, чувствительную к ингибированию активности Btk и/или В-клеточной пролиферации.
Аутоиммунные и/или воспалительные заболевания, на которые может быть оказано воздействие при помощи соединений и композиций по изобретению, включают, но не ограничены: псориаз, аллергию, болезнь Крона, синдром раздраженного кишечника, синдром  , реакцию отторжения трансплантированного органа и сверхострого отторжения трансплантированного органа, астму, системную красную волчанку (и ассоциированный гломерулонефрит), дерматомиозит, множественный склероз, склеродермию, васкулит (ANCA-ассоциированные и другие васкулиты), аутоиммунные гемолитические и тромбоцитопинические состояния, синдром Гудпасчера (и ассоциированный гломерулонефрит и легочное кровотечение), атеросклероз, ревматоидный артрит, хроническую идиопатическую тромбоцитопеническую пурпуру (ITP), болезнь Аддисона, болезнь Паркинсона, болезнь Альцгеймера, диабет, септический шок и миастению гравис.
, реакцию отторжения трансплантированного органа и сверхострого отторжения трансплантированного органа, астму, системную красную волчанку (и ассоциированный гломерулонефрит), дерматомиозит, множественный склероз, склеродермию, васкулит (ANCA-ассоциированные и другие васкулиты), аутоиммунные гемолитические и тромбоцитопинические состояния, синдром Гудпасчера (и ассоциированный гломерулонефрит и легочное кровотечение), атеросклероз, ревматоидный артрит, хроническую идиопатическую тромбоцитопеническую пурпуру (ITP), болезнь Аддисона, болезнь Паркинсона, болезнь Альцгеймера, диабет, септический шок и миастению гравис.
Включенное в данный документ представляет собой способы лечения, при которых по меньшей мере одно химическое соединение, предложенное в данном документе, вводят в комбинации с противовоспалительным агентом. Противовоспалительные агенты включают, но не ограничены, НПВС, неспецифические и СОХ-2 специфические ингибиторы циклооксигеназы, соединения золота, кортикостероиды, метотрексат, рецепторы-антагонисты рецепторов фактора некроза опухолей (TNF), иммуносупрессанты и метотрексат.
Примеры НПВС включают, но не ограничены, ибупрофен, флурбипрофен, напроксен и напроксен натрия, диклофенак, комбинации диклофенака натрия и мизопростола, сулиндак, оксапрозин, дифлюнизал, пироксикам, индометацин, этодолак, фенопрофен кальция, кетопрофен, натрий набуметон, сульфасалазин, толметин натрия и гидроксихлорохин. Примеры НПВС также включают xamples СОХ-2 специфичные ингибиторы, такие как целекоксиб, вальдекоксиб, люмиракоксиб и/или эторикоксиб.
В некоторых воплощениях противовоспалительный агент представляет собой салицилат. Салицилаты включают, но не ограничены, ацетилсалициловую кислоту или аспирин, салицилат натрия и холин и салицилаты магния.
Противовоспалительный агент также может представлять собой кортикостероид. Например, кортикостероид может представлять собой кортизон, дексаметазон, метилпреднизолон, преднизолон, преднизолон натрий фосфат или преднизон.
В дополнительных воплощениях противовоспалительный агент представляет собой соединение золота, такое как ауротиомалат натрия или ауранофин.
Изобретение также включает воплощения, в которых противовоспалительный агент представляет собой метаболический ингибитор, такой как ингибитор дигидрофолат редуктазы, такой как метотрексат или ингибитор дигидрооротат дегидрогеназы, такой как лефлуномид.
Другие воплощения изобретения относятся к комбинациям, в которых по меньшей мере одно противовоспалительное соединение представляет собой моноклональное антитело к С5 (такое как экулизумаб или пекселизумаб), антагонист TNF, такой как энтанерцепт или инфликсимаб, который представляет собой моноклональное антитело к TNF альфа.
Еще одно воплощение изобретения относится к комбинациям, в которых по меньшей мере один активный агент представляет собой соединение иммуносупрессант, такое как соединение иммуносупрессант, выбранное из метотрексата, лефлуномида, циклоспорина, такролимуса, азатиоприна и мофетил микофенолата.
В-клетки и предшественники В-клеток, экспрессирующие ВТК, задействованы в патологии В-клеточных злокачественных новообразований, включая, но не ограничено, В-клеточную лимфому, лимфому (включая лимфому Ходжкина и неходжкинскую лимфому), лимфому "волосатых клеток", множественную миелому, хроническую и острую гранулоцитарную лейкемию. ВТК показана как ингибитор Fas/APO-1 (CD-95) сигнального комплекса, индуцирующего апоптоз (DISC) в В-линии лимфоцитов. Направление развития лимфомных/лейкемийных клеток может находится в балансе между противоположными проапоптотическими эффектами каспаз, активированных DISC, и вышерасположенным противоапоптотическим регуляторным механизмом, вовлекающим ВТК и/или ее субстраты (Vassilev et al., J. Biol. Chem. 1998, 274, 1646-1656).
Также было открыто, что ингибиторы ВТК полезны в качестве хемосенситизирующих агентов и таким образом, полезны в комбинации с другими хемотерапевтическими лекарственными средствами, в частности, лекарственными средствами, которые индуцируют апоптоз. Примеры других хемотерапевтических лекарственных средств, которые могут быть применены в сочетании с хемосенситизирующими ингибиторами ВТК, включают ингибиторы топоизомеразы I (камптотецин или топотекан), ингибиторы топоизомеразы II (например, дауномицин и этопозид), алкилирующие агенты (например, циклофосфамид, мелфалан и BCNU), агенты, направленные на тубулин (например, таксол и винбластин), и биологические агенты (например, антитела, такие как антитело к CD20, IDEC 8, иммунотоксины и цитокины).
Btk активность также ассоциирована с некоторыми лейкемиями, экспрессирующими слитый ген bcr-abl, являющийся результатом транслокации частей хромосомы 9 и 22. Данное нарушение обычно наблюдается при хронической миелогенной лейкемии. Btk постоянно фосфорилирована киназой bcr-abl, которая инициирует нижерасположенные сигналы выживания, которые ингибируют апоптоз в bcr-abl клетках. (N. Feldhahn et al. J. Exp. Med. 2005 201(11): 1837-1852).
Способы лечения
Заявка предлагает способ лечения воспалительного и/или аутоиммунного состояния, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I.
Заявка предлагает способ лечения воспалительного состояния, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I.
Заявка предлагает способ лечения ревматоидного артрита, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I.
Заявка предлагает способ лечения астмы, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I.
Заявка предлагает способ лечения воспалительного и/или аутоиммунного состояния, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества Btk ингибиторного соединения формулы I.
Заявка предлагает способ лечения артрита, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества Btk ингибиторного соединения формулы I.
Заявка предлагает способ лечения астмы, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества Btk ингибиторного соединения формулы I.
Заявка предлагает способ лечения рака, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества Btk ингибиторного соединения формулы I.
Заявка предлагает способ ингибирования В-клеточной пролиферации, содержащий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества Btk ингибиторного соединения формулы I.
Заявка предлагает способ ингибирования активности Btk, содержащий введение Btk ингибиторного соединения любой из формул I, где Btk ингибиторное соединение проявляет IC50 50 микромоль или менее в in vitro биохимическом исследовании активности Btk.
В одном варианте вышеупомянутого способа, Btk ингибиторное соединение проявляет IC50 100 наномоль или менее в in vitro биохимическом исследовании активности Btk.
В другом варианте вышеупомянутого способа, соединение проявляет IC50 10 наномоль или менее в in vitro биохимическом исследовании активности Btk.
Заявка предлагает способ лечения воспалительного состояния, содержащий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективного количества противовоспалительного соединения в сочетании с Btk ингибиторным соединением формулы I.
Заявка предлагает способ лечения артрита, содержащий совместное введение пациенту, нуждающемуся в этом, терапевтически эффективного количества противовоспалительного соединения в сочетании с Btk ингибиторным соединением формулы I.
Заявка предлагает способ лечения лимфомы или BCR-ABL1+ лейкемических клеток посредством введения пациенту, нуждающемуся в этом, терапевтически эффективного количества Btk ингибиторного соединения формулы I.
Примеры
Обычные сокращения
Обычно применяемые сокращения включают: ацетил (Ас), азо-бис-изобутирилнитрил (AIBN), атмосферы (Atm), 9-борбицикло[3.3.1]нонан (9-BBN или BBN), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (BINAP), трет-бутоксикарбонил (Boc), ди-трет-бутил пирокарбонат или boc-ангидрид (BOC2O), бензил (Bn), бутил (Bu), уникальный численный идентификатор химических соединений (CASRN), бензилоксикарбонил (CBZ или Z), карбонилдиимидазол (CDI), 1,4-диазабицикло[2.2.2]октан (DABCO), трифторид диметиламиносеры (DAST), дибензилиденацетон (dba), 1,5-диазабицикло[4.3.0]нон-5-ен (DBN), 1,8-диазабицикло[5.4.0]ундек-7-ен (DBU), N,N'-дициклогексилкарбодиимид (DCC), 1,2-дихлорэтан (DCE), дихлорметан (DCM), 2,3-дихлор-5,6-дициано-1,4-бензохинон (DDQ), диэтил азодикарбоксилат (DEAD), ди-изо-пропилазодикарбоксилат (DIAD), ди-изо-бутилалюминийгидрид (DIBAL или DIBAL-H), ди-изо-пропилэтиламин (DIPEA), N,N-диметилацетамид (DMA), 4-N,N-диметиламинопиридин (DMAP), N,N-диметилформамид (DMF), диметилсульфоксид (DMSO), 1,1'-бис-(дифенилфосфино)этан (dppe), 1,1'-бис-(дифенилфосфино)ферроцен (dppf), 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид (EDCl), 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (EEDQ), этил (Et), этилацетат (EtOAc), этанол (EtOH), этиловый эфир 2-этокси-2Н-хинолин-1-карбоновой кислоты (EEDQ), диэтиловый эфир (Et2O), этилизопропиловый эфир (EtOiPr), O-(7-азабензотриазол-1-ил)-N,N,N'N'-тетраметилуроний гексафторфосфат уксусной кислоты (HATU), уксусная кислота (НОАс), 1-N-гидроксибензотриазол (HOBt), высокоэффективная жидкостная хроматография (ВЭЖХ), изо-пропанол (IPA), хлорид изопропилмагния (iPrMgCl), гексаметилдисилазан (HMDS), жидкостная хромато-масс-спектрометрия (LCMS), литий гексаметил дисилазан (LiHMDS), мета-хлорпероксибензойная кислота (m-СРВА), метанол (МеОН), температура плавления (mp), MeSO2- (мезил или Ms), метил (Me), ацетонитрил (MeCN), m-хлорбензойная кислота (МСРВА), масс-спектр (ms), метил-трет-бутиловый эфир (МТВЕ), метилтетрагидрофуран (MeTHF), N-бромсукцинимид (NBS), н-бутиллитий (nBuLi), N-карбоксиангидрид (NCA), N-хлорсукцинимид (NCS), N-метилморфолин (NMM), N-метилпирролидон (NMP), пиридинийхлорхромат (РСС), дихлор-((бис-дифенилфосфино)ферроценил)палладий(II) (Pd(dppf)Cl2), палладий(II) ацетат (Pd(OAc)2), трис(дибензилиденацетон)дипалладий(0) (Pd2(dba)3), пиридиний дихромат (PDC), фенил (Ph), пропил (Pr), изо-пропил (i-Pr), фунты на квадратный дюйм (psi), пиридин (pyr), 1,2,3,4,5-пентафенил-1'-(ди-трет-бутилфосфино)ферроцен (Q-Phos), комнатная температура (температура окружающей среды, кт или КТ), втор-бутиллитий (sBuLi), трет-бутилдиметилсилил или f-BuMe2Si (TBDMS), тетра-н-бутиламмоний фторид (TBAF), триэтиламин (TEA или Et3N), 2,2,6,6-тетраметилпиперидин 1-оксил (TEMPO), триметилсилилэтоксиметил (SEM), трифлат или CF3SO2- (Tf), трифторуксусная кислота (TFA), 1,1'-бис-2,2,6,6-тетраметилгептан-2,6-дион (TMHD), O-бензотриазол-1-ил-N,N,N',N'-тетраметилуроний тетрафторборат (TBTU), тонкослойная хроматография (TLC), тетрагидрофуран (THF), триметилсилил или Me3Si (TMS), моногидрат п-толуолсульфоновой кислоты (TsOH или pTsOH), 4-Me-C6H4SO2- или тозил (Ts), и N-уретан-N-карбоксиангидрид (UNCA). Обычная номенклатура, включающая суффиксы нормальный (н), изо (изо), вторичный (втор-), третичный (трет-) и нео имеют свое обычное значение, в случае применения в отношении алкильной группы (J. Rigaudy and D.P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford.).
Обычные условия
Соединения по настоящему изобретению могут быть получены, начиная с коммерчески доступных исходных материалов посредством применения обычных способов синтеза и способов, известных специалистам в области техники. Нижеприведенное описание представляет собой схемы реакций, приемлемые для получения таких соединений, below are reaction schemes suitable for preparing such compounds. Дальнейшие пояснения могут быть найдены в конкретных примерах.
Примеры получения
Специальные сокращения
Общие детали эксперимента
Реагенты были закуплены у Aldrich, Oakwood, Matrix или других поставщиков и применены без дополнительной очистки. Реакции с применением микроволнового излучения для нагревания проводили при помощи или Personal Chemistry Emrys Optimizer System или СЕМ Discovery System. Очистку в масштабах от миллиграмм до грамм проводили при помощи способов, известных специалистам в области техники, таких как элюция через флэш-колонку с силикагелем; препаративные очистки на флэш-колонке также осуществляли в некоторых случаях за счет применения предварительно упакованных мультиграммовых колонок с силикагелем (RediSep), элюируемых при помощи системы CombiFlash Biotage™ и ISCO™ также представляют собой флэш-колонки, которые могут быть применены в данном изобретении для очистки промежуточных соединений.
С целью идентификации соединения и определения его чистоты регистрировали LC/MS (жидкостная хроматография/масс-спектроскопия) спектр при помощи следующей системы. Для измерения масс-спектра система состоит из спектрометра Micromass Platform II: ES ионизация положительным способом (диапазон масс: 150 -1200). Одновременного хроматографического разделения достигали при помощи следующей ВЭЖХ системы: ES Industries Chromegabond WR С-18 3u 120 (3,2×30 мм) картридж-колонки; Мобильная фаза А: Вода (0,02% TFA) и Фаза В: ацетонитрил (0,02% TFA); градиент от 10% В до 90% В в течение 3 минут; время уравновешивания 1 минута; скорость тока 2 мл/минуту.
(3,2×30 мм) картридж-колонки; Мобильная фаза А: Вода (0,02% TFA) и Фаза В: ацетонитрил (0,02% TFA); градиент от 10% В до 90% В в течение 3 минут; время уравновешивания 1 минута; скорость тока 2 мл/минуту.
Многие соединения формулы 1 также очищали при помощи обращенно-фазовой ВЭЖХ, при помощи способов, хорошо известных специалистам в области техники. В некоторых случаях препаративную ВЭЖХ-очистку проводили при помощи масс-спектрометра РЕ Sciex 150 EX Mass Spec, контролирующего коллектор Gilson 215, прикрепленный к препаративной ВЭЖХ-системе Shimadzu и автосэмплеру Leap. Соединения собирали из потока элюата при помощи LC/MS детекции при детекции положительных ионов: Элюцию соединений с колонок С-18 (2,0×10 см элюировали при 20 мл/мин) осуществляли при помощи соответствующего линейного градиента в течение 10 минут Растворитель (А) 0,05% TFA/H2O и Растворитель (В) 0,035% TFA/ацетонитрил. Для инъекции в системы ВЭЖХ, неочищенные образцы растворяли в смесях метанол, ацетонитрил и ДМСО.
1Н-ЯМР характеристику проводили при помощи ЯМР спектрометров Bruker или Varian 300 или 400 МГц.
Соединения по настоящему изобретению могут быть синтезированы согласно следующим способам. Следующие примеры и ссылки предложены для облегчения понимания настоящего изобретения. Примеры не являются ограничивающими изобретение, реальный объем которого определен формулой изобретения. Названия конечных продуктов в примерах образованы при помощи Isis AutoNom 2000.
Получение промежуточных соединений
Промежуточное соединение 1
[4-(2,3-дифтор-фенокси)-2-метил-фенил]-гидразин

Этап 1: 4-(2,3-дифтор-фенокси)-2-метил-1-нитро-бензол
Смесь 4-хлор-2-метил-1-нитро-бензола (5 г, 29,1 ммоль), 2,3-дифторфенола (4,55 г, 35,0 ммоль) и Cs2CO3 (14,2 г, 43,7 ммоль) в ДМФ (10 мл) нагревали в запаянной пробирке в микроволновой печи при 150°С в течение 30 мин. EtOAc (300 мл) добавляли и смесь промывали водой (150 мл) и солевым раствором. Органический слой высушивали (Na2SO4), фильтровали и выпаривали. Неочищенный материал очищали посредством флэш-хроматографии (силикагель, 10% этилацетат в гексанах) для получения 4-(2,3-дифтор-фенокси)-2-метил-1-нитробензола (5,6 г, 72%) в виде светло-желтого масла.
Этап 2: 4-(2,3-дифтор-фенокси)-2-метил-фениламин

Смесь 4-(2,3-дифтор-фенокси)-2-метил-1-нитро-бензола (5,28 г, 19,9 ммоль) и палладия на углеродной подложке (587 мг) в МеОН (55 мл) перемешивали при 30 psi водорода в шейкере Parr в течение 4 часов. Смесь фильтровали через Celite и Celite промывали МеОН. Фильтрат выпаривали до получения 4-(2,3-дифтор-фенокси)-2-метил-фениламина (4,56 г, 97%), который применяли на следующем этапе без дополнительной очистки.
Этап 3: [4-(2,3-дифтор-фенокси)-2-метил-фенил]-гидразин

Смесь 4-(2,3-дифтор-фенокси)-2-метил-фениламина (546 мг, 2,32 ммоль) и конц. HCl (0,6 мл) в МеОН (1 мл) и воды (2 мл) охлаждали на ледяной бане. Раствор NaNO2 (168 мг, 2,44 ммоль) в воде (0,4 мл) медленно добавляли и смесь перемешивали в течение 30 мин. Реакционную смесь переносили пипеткой в перемешанный раствор олово(II) хлориддигидрата (2,25 г, 10 ммоль) в конц. HCl (5 мл) и смесь перемешивали в течение 4 ч. МеОН (4 мл) добавляли, с последующим добавлением 10 М NaOH до достижения рН 7-8. растворитель выпаривали при пониженном давлении и осадок высушивали под вакуумом в течение 1 ч. Осадок растирали в порошок с 10% MeOH/CH2Cl2, и твердое вещество фильтровали и промывали 10% MeOH/CH2Cl2. Фильтрат выпаривали при пониженном давлении для получения [4-(2,3-дифтор-фенокси)-2-метил-фенил]-гидразина (495 мг, 68%) в виде желтого масла. Данный материал применяли на следующем этапе без дополнительной очистки.
Промежуточное соединение 2
3-(4-гидразино-3-метил-фенокси)-бензонитрил

Смесь 3-(4-амино-3-метил-фенокси)-бензонитрила (которая может быть получена как описано в Akama, Т. et al. Bioorg. Med. Chem. Lett. 2009, 19, 2129-2132; 2,2 г, 9,8 ммоль) и конц. HCl (3,5 мл) в МеОН (7 мл) и воды (5 мл) охлаждали на ледяной бане. Раствор NaNO2 (1,35 г, 19,6 ммоль) в воде (6 мл) медленно добавляли и смесь перемешивали в течение 30 мин. Реакционную смесь переносили пипеткой в перемешанный раствор олово(II) хлориддигидрата (8,85 г, 39,2 ммоль) в конц. HCl (7 мл) и смесь перемешивали в течение 30 мин. МеОН (10 мл) добавляли с последующим добавлением 10 М NaOH до достижения рН 7-8. Добавляли воду (100 мл) и смесь экстрагировали EtOAc (500 мл). Органический слой промывали солевым раствором (Na2SO4), фильтровали и выпаривали до получения 3-(4-гидразино-3-метил-фенокси)-бензонитрила (2,2 г, 94%) в виде масла. Данный материал применяли на следующем этапе без дополнительной очистки.
Пример I-1
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанон

Этап 1: метиловый эфир 1-бензолсульфонил-4-бром-1Н-индол-2-карбоновой кислоты

DIPEA (2,6 мл, 14,8 ммоль) и МеОН (3 мл, 74,2 ммоль) добавляли в раствор 1-бензолсульфонил-4-бром-1Н-индол-2-карбонилхлорида (который может быть получен как описано в Mahboobi, S. et al. J. Med. Chem. 2006, 49, 3101-3115; 1,97 г, 4,94 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, и растворитель выпаривали при пониженном давлении. Осадок растирали в порошок с 15% EtOAc/гексаны, фильтровали, промывали 15% EtOAc/гексаны, и высушивали под вакуумом в течение ночи для получения метилового эфира 1-бензолсульфонил-4-бром-1Н-индол-2-карбоновой кислоты (1,78 г, 91%).
Этап 2: 3-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-3-оксо-пропионитрил
Раствор метил-4-бром-1-(фенилсульфонил)-1Н-индол-2-карбоксилата (970 мг, 2,46 ммоль) и ацетонитрила (770 мкл, 14,8 ммоль) в ТГФ (25 мл) охлаждали до -78°С. LDA (2М/ТГФ) (2,5 мл, 5 ммоль) добавляли медленно в течение 5 мин. Реакционную смесь перемешивали при -78°С в течение 30 мин и затем добавляли насыщенный раствор NH4Cl (40 мл). Добавляли воду (150 мл) и смесь экстрагировали EtOAc (500 мл). Органический слой промывали солевым раствором, высушивали (Na2SO4), фильтровали и выпаривали. Осадок очищали посредством хроматографии (силикагель, 30% EtOAc/гексаны) до получения 3-(4-бром-1-(фенилсульфонил)-1Н-индол-2-ил)-3-оксопропаннитрила (650 мг, 66%) в виде пены.
Этап 3: (Е)-2-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-3-диметиламино-акрилонитрил

N,N-диметилформамиддиметилацеталь (465 мг, 3,9 ммоль) добавляли к раствору 3-(4-бромо-1-(фенилсульфонил)-1Н-индол-2-ил)-3-оксопропаннитрила (1,21 г, 3,00 ммоль) в толуоле (20 мл), и смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 70% EtOAc/гексаны) до получения (Е)-2-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-3-диметиламино-акрилонитрила (1,06 г, 77%) в виде желтой пены.
Этап 4: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанон
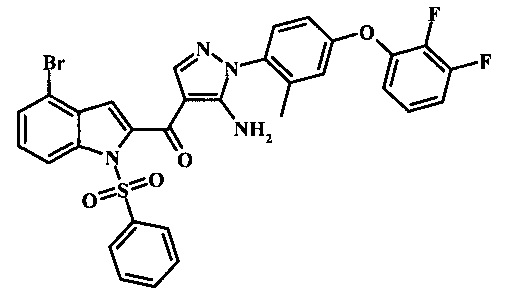
Смесь [4-(2,3-дифтор-фенокси)-2-метил-фенил]-гидразин (который может быть получен как описано для промежуточного соединения 1; 816 мг, 3,26 ммоль), (Е)-2-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-3-диметиламино-акрилонитрила (650 мг, 1,42 ммоль) и EtOH (25 мл) нагревали при рефлюксе в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали Осадок очищали при помощи хроматографии (силикагель, 30% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (820 мг, 87%) в виде пены.
Этап 5: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанон

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (275 мг, 0,414 ммоль), Cs2CO3 (540 мг, 1,66 ммоль), ТГФ (10 мл) и МеОН (5 мл) перемешивали при комнатной температуре в течение ночи, растворитель удаляли при пониженном давлении. Осадок очищали посредством хроматографии (силикагель, 40% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанона (210 мг, 97%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 12,10 (s, 1Н), 8,28 (s, 1Н), 7,51 (d, J=8,3 Гц, 1Н), 7,25-7,42 (m, 4Н), 7,15-7,23 (m, 3Н), 7,09 (t, J=7,5 Гц, 1Н), 7,03 (d, J=8,5 Гц, 1Н), 6,96 (br. s., 2Н), 2,09 (s, 3Н). MS вычисленное для C25H18BrF2N4O2 [(М+Н)+] 523, наблюдаемое 522,9.
Пример I-2
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-фенил-1Н-индол-2-ил)-метанон
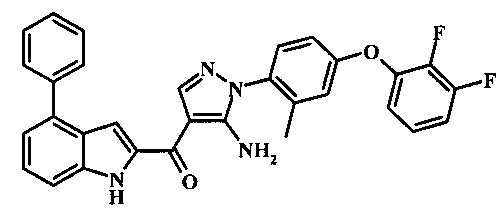
Этап 1: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-фенил-1Н-индол-2-ил)-метанон

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 Этапа 4; 30 мг, 0,045 ммоль), фенилборной кислоты (11 мг, 0,09 ммоль), Pd(PPh3)4 (5,2 мг, 0,004 ммоль), K2CO3 (25 мг, 0,18 ммоль), воды (0,5 мл), толуола (1 мл), и EtOH (1 мл) нагревали при 90°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Органический слой выпаривали. Осадок очищали при помощи хроматографии (силикагель, 30% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-фенил-1Н-индол-2-ил)-метанона (28 мг, 94%) в виде масла.
Этап 2: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-фенил-1Н-индол-2-ил)-метанон

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-фенил-1Н-индол-2-ил)-метанона (28 мг, 0,04 ммоль), Cs2CO3 (41,4 мг, 0,13 ммоль), ТГФ (2 мл) и МеОН (1 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-фенил-1Н-индол-2-ил)-метанона (20 мг, 86%) в виде светло-желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,65 (s, 1Н), 8,17 (s, 1Н), 7,72 (dd, J=8,4, 1,4 Гц, 2Н), 7,48-7,56 (m, 3Н), 7,37-7,46 (m, 3Н), 7,31 (d, J=8,6 Гц, 1Н), 7,26 (s, 1Н), 7,23 (dd, J=6,8, 1,4 Гц, 1Н), 7,07 (d, J=5,5 Гц, 2Н), 6,99 (d, J=2,7 Гц, 1Н), 6,92 (dt, J=8,1, 2,6 Гц 2Н), 2,17 (s, 3Н). MS вычисленный для C31H23F2N4O2 [(М+Н)+] 521, наблюдаемый 521,1.
Пример I-3
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-3-ил-1Н-индол-2-ил)-метанон

Этап 1: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-тиофен-3-ил-1Н-индол-2-ил)-метанон
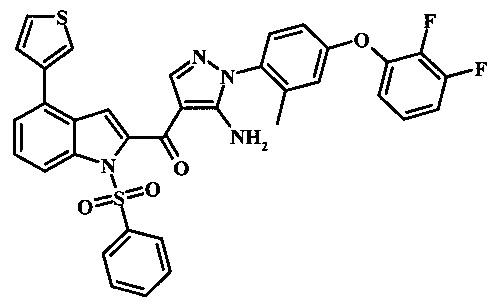
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 Этапа 4; 95 мг, 0,14 ммоль), тиофен-3-бориновой кислоты (37 мг, 0,29 ммоль), Pd(PPh3)4 (16,5 мг, 0,014 ммоль), K2CO3 (79,2 мг, 0,57 ммоль), воды (1,5 мл), толуола (3 мл), и EtOH (3 мл) нагревали при 90°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Органический слой выпаривали. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-тиофен-3-ил-1Н-индол-2-ил)-метанона (94 мг, 92%) в виде масла.
Этап 2: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-3-ил-1Н-индол-2-ил)-метанон
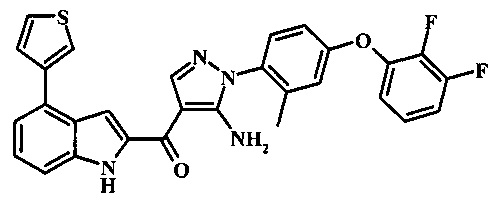
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-тиофен-3-ил-1Н-индол-2-ил)-метанона (94 мг, 0,14 ммоль), Cs2CO3 (137 мг, 0,42 ммоль), ТГФ (6 мл) и МеОН (3 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-3-ил-1Н-индол-2-ил)-метанона (70 мг, 88%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,40 (s, 1Н), 8,20 (s, 1Н), 7,59-7,61 (m, 1Н), 7,54 (dd, J=2.3, 0,8 Гц, 1Н), 7,48-7,53 (m, 2Н), 7,40-7,44 (m, 1Н), 7,35-7,40 (m, 1Н), 7,27-7,34 (m, 2Н), 7,03-7,13 (m, 2Н), 7,00 (d, J=2,7 Гц, 1Н), 6,89-6,96 (m, 2Н), 2,17 (s, 3Н). MS, вычисленный для C29H21F2N4O2S [(М+Н)+] 527, наблюдаемый 527.
Пример I-4
(5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиразол-1-ил-1Н-индол-2-ил)-метанон

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 5; 100 мг, 0,15 ммоль), пиразола (20,5 мг, 0,3 ммоль), L-пролина (7 мг, 0,06 ммоль), йодида меди(I) (6 мг, 0,03 ммоль), и K2CO3 (62,5 мг, 0,45 ммоль) в ДМСО (2 мл) продували аргоном. Смесь нагревали при 130°С в течение 40 ч и затем очищали при помощи препаративной ВЭЖХ до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиразол-1-ил-1Н-индол-2-ил)-метанона (6 мг, 8%) в виде желтого порошка. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,44 (br. s., 1Н), 8,29 (s, 1Н), 8,08 (dd, J=2,5, 0,5 Гц, 1Н), 7,83-7,93 (m, 2Н), 7,71 (s, 1Н), 7,39-7,48 (m, 2Н), 7,31-7,37 (m, 2Н), 7,05-7,17 (m, 2Н), 7,03 (d, J=2,8 Гц, 1Н), 6,91-7,00 (m, 2H), 6,57-6,59 (m, 1H), 2,20 (s, 3H). MS вычисленный для C28H21F2N6O2 [(M+H)+] 511, наблюдаемый 511.
Пример I-5
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-2-ил-1Н-индол-2-ил)-метанон

Этап 1: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-тиофен-2-ил-1Н-индол-2-ил)-метанон
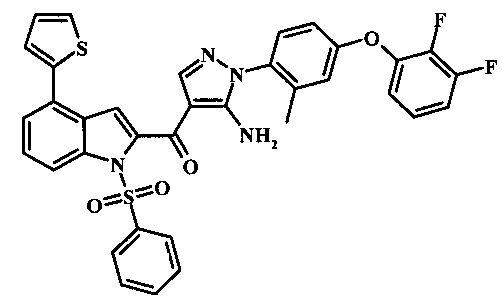
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 4; 60 мг, 0,09 ммоль), 2-(трибутилстаннил)тиофена (67,5 мг, 0,18 ммоль), Pd(PPh3)4 (10,4 мг, 0,009 ммоль), и толуола (2 мл) нагревали при 100°С в течение ночи. Растворитель выпаривали. Осадок очищали при помощи хроматографии (силикагель, 35% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензол сульфонила-4-тиофен-2-ил-1Н-индол-2-ил)-метанона (46 мг, 76%) в виде масла.
Этап 2: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-2-ил-1Н-индол-2-ил)-метанон
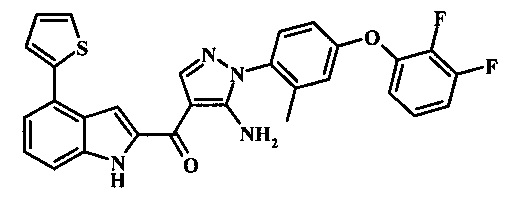
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-тиофен-2-ил-1Н-индол-2-ил)-метанона (46 мг, 0,07 ммоль), Cs2CO3 (67,4 мг, 0,21 ммоль), ТГФ (2 мл) и МеОН (1 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-2-ил-1Н-индол-2-ил)-метанона (32 мг, 88%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,41 (br. s., 1Н), 8,24 (s, 1Н), 7,73 (dd, J=2,0, 0,8 Гц, 1Н), 7,49 (dd, J=3,5, 1,2 Гц, 1Н), 7,40-7,44 (m, 2Н), 7,36-7,39 (m, 2Н), 7,30-7,36 (m, 2Н), 7,20 (dd, J=5,1, 3,5 Гц, 1Н), 7,02-7,13 (m, 2Н), 7,00 (d, J=2,7 Гц, 1Н), 6,88-6,96 (m, 2Н), 2,18 (s, 3Н). MS вычисленный для C29H21F2N4O2S [(M+H)+] 527, наблюдаемый 526,9.
Пример I-6
трифторуксуснокислая соль {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(морфолин-4-карбонил)-1Н-индол-2-ил]-метанона
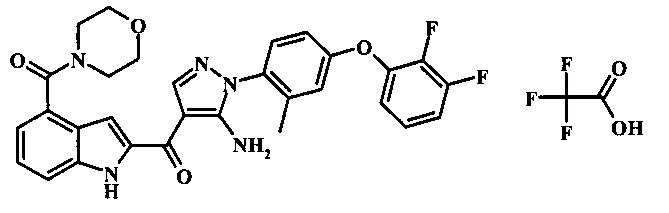
Газообразную окись углерода барботировали через смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 5; 94 мг, 0,18 ммоль), морфолина (313 мг, 3,6 ммоль), Pd(PPh3)4 (62,3 мг, 0,054 ммоль), и ТГФ (10 мл) в запаянной пробирке в течение 5 мин. Пробирку запаивали и нагревали при 90°С в течение 2 ч. Растворитель выпаривали и осадок очищали при помощи препаративной ВЭЖХ до получения трифторуксуснокислой соли {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(морфолин-4-карбонил)-1Н-индол-2-ил]-метанона (24 мг, 24%) в виде светло-желтого порошка. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,52 (s, 1Н), 8,23 (s, 1Н), 7,51 (d, J=8,2 Гц, 1Н), 7,30-7,38 (m, 3Н), 7,18 (dd, J=7,2, 1,0 Гц, 1Н), 7,02-7,15 (m, 2Н), 7,00 (d, J=2,7 Гц, 1Н), 6,88-6,96 (m, 2Н), 3,41-4,00 (m, 8Н), 2,17 (s, 3Н). MS вычисленный для C30H26F2N5O4 [(M+H)+] 558, наблюдаемый 558.
Пример I-7
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(1-метил-1Н-пиразол-3-ил)-1Н-индол-2-ил]-метанон
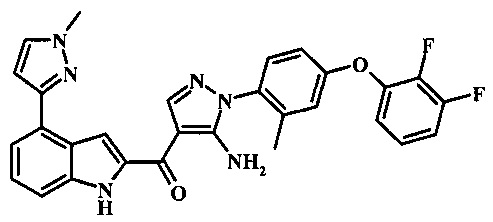
Этап 1: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[1-бензолсульфонил-4-(1-метил-1Н-пиразол-3-ил)-1Н-индол-2-ил]-метанон
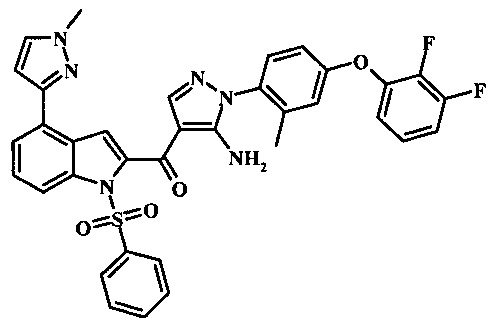
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 4; 65 мг, 0,098 ммоль), гидрохлорида 1-метил-1Н-пиразол-4-бориновой кислоты (32 мг, 0,196 ммоль), Pd(PPh3)4 (11,3 мг, 0,01 ммоль), K2CO3 (54,2 мг, 0,39 ммоль), воды (1 mL), толуола (2 мл), и EtOH (2 мл) нагревали при 90°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Органический слой выпаривали. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[1-бензолсульфонил-4-(1-метил-1Н-пиразол-3-ил)-1Н-индол-2-ил]-метанона (31 мг, 48%) в виде масла.
Этап 2: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(1-метил-1Н-пиразол-3-ил)-1Н-индол-2-ил]-метанон
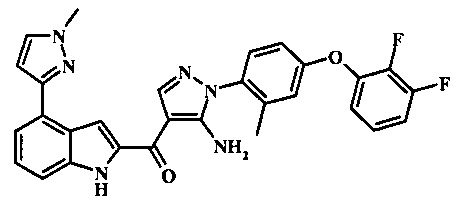
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[1-бензолсульфонил-4-(1-метил-1Н-пиразол-3-ил)-1Н-индол-2-ил]-метанон (31 мг, 0,046 ммоль), Cs2CO3 (60,8 мг, 0,19 ммоль), ТГФ (1,5 мл) и МеОН (0,75 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 90% EtOAc/гексаны) до получения (5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(1-метил-1Н-пиразол-3-ил)-1Н-индол-2-ил]-метанона (21 мг, 86%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,29 (s, 1Н), 8,21 (в, 1Н), 7,93 (s, 1Н), 7,78 (s, 1Н), 7,47-7,50 (m, 1Н), 7,30-7,37 (m, 3Н), 7,22 (dd, J=6,4, 1,8 Гц, 1Н), 7,02-7,14 (m, 2Н), 7,00 (d, J=2,7 Гц, 1Н), 6,86-6,96 (m, 2Н), 4,05 (s, 3Н), 2,18 (s, 3Н). MS вычисленный для C29H23F2N6O2 [(М+Н)+] 525, наблюдаемый 525.
Пример I-8
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиридин-2-ил-1Н-индол-2-ил)-метанон

Этап 1: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-пиридин-2-ил-1Н-индол-2-ил)-метанон
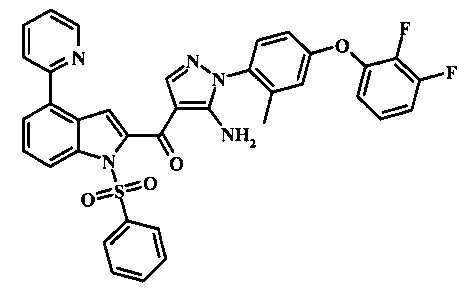
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 4; 64 мг, 0,097 ммоль), 2-(трибутилстаннил)пиридина (71 мг, 0,19 ммоль), Pd(PPh3)4 (11,1 мг, 0,01 ммоль), и толуола (2 мл) нагревали при 100°С в течение ночи. Растворитель выпаривали. Осадок очищали при помощи хроматографии (силикагель, 45% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-пиридин-2-ил-1Н-индол-2-ил)-метанона (27 мг, 42%) в виде масла.
Этап 2: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиридин-2-ил-1Н-индол-2-ил)-метанон
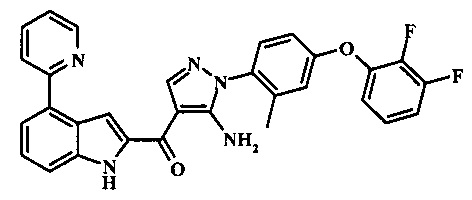
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-пиридин-2-ил-1Н-индол-2-ил)-метанона (27 мг, 0,041 ммоль), Cs2CO3 (53,2 мг, 0,16 ммоль), ТГФ (2 мл) и МеОН (1 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 50% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиридин-2-ил-1Н-индол-2-ил)-метанона (19 мг, 79%; чистота 88%) в виде грязно-белого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,37 (br. s., 1Н), 8,87 (d, J=4,8 Гц, 1Н), 8,29 (s, 1Н), 7,91 (d, J=18,1 Гц, 3Н), 7,44-7,65 (m, 3Н), 7,35 (d, J=8,5 Гц, 2Н), 7,04-7,18 (m, 2Н), 7,01-7,04 (m, 1Н), 6,88-6,99 (m, 2Н), 2,20 (s, 3Н). MS вычисленный для C30H22F2N5O2 [(М+Н)+] 522, наблюдаемый 522.
Пример I-9
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бензил-1Н-индол-2-ил)-метанон
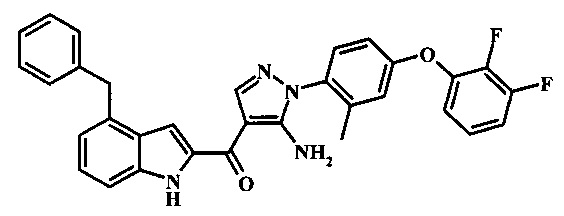
Этап 1: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бензил-1Н-индол-2-ил)-метанон

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (которую получают как описано в примере I-1 этапа 4; 65 мг, 0,098 ммоль), Pd(OAc)2 (2,8 мг, 0,012 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенила (Aldrich; 9 мг, 0,022 ммоль), и ТГФ (2 мл) продували аргоном в течение 3 мин и затем перемешивали при комнатной температуре в течение 5 мин. Добавляли бензилцинк(II) бромид (Aldrich; 0,5 М в ТГФ; 0,3 мл; 0,15 ммоль) и смесь нагревали при 75°С в течение 2 ч. Растворитель выпаривали. Осадок очищали при помощи хроматографии (силикагель, 35% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бензил-1Н-индол-2-ил)-метанон (66 мг, 82%) в виде масла.
Этап 2: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бензил-1Н-индол-2-ил)-метанон

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бензил-1Н-индол-2-ил)-метанона (63 мг, 0,093 ммоль), Cs2CO3 (122 мг, 0,37 ммоль), ТГФ (5 мл) и МеОН (2,5 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бензил-1Н-индол-2-ил)-метанона (42 мг, 84%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,26 (br. s., 1Н), 8,06 (s, 1Н), 7,30-7,42 (m, 8Н), 7,25 (dd, J=2,1, 0,9 Гц, 2Н), 7,05-7,17 (m, 2Н), 6,91-7,04 (m, 4Н), 4,36 (s, 2Н), 2,19 (s, 3Н). MS вычисленный для C32H25F2N4O2 [(М+Н)+] 535, наблюдаемый 536.
Пример 1-10
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(1Н-пиразол-4-ил)-1Н-индол-2-ил]-метанон

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 5; 55 мг, 0,105 ммоль), трет-бутоксикарбонил-1Н-пиразол-4-бориновой кислоты (44,6 мг, 0,21 ммоль), Pd(PPh3)4 (12,1 мг, 0,011 ммоль), K2CO3 (58,1 мг, 0,42 ммоль), воды (0,75 мл), толуола (1,5 мл), и EtOH (1,5 мл) нагревали при 90°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Органический слой выпаривали. Осадок очищали при помощи хроматографии (силикагель, 60% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(1Н-пиразол-4-ил)-1Н-индол-2-ил]-метанона (30 мг, 56%) в виде масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 13,04 (br. s., 1Н), 11,78 (s, 1Н), 8,39-8,47 (m, 2Н), 8,07 (s, 1Н), 7,23-7,49 (m, 7Н), 7,17 (d, J=2,8 Гц, 1Н), 6,99-7,13 (m, 2Н), 6,90 (s, 2Н), 2,10 (s, 3Н). MS вычисленный для C28H21F2N6O2 [(М+Н)+] 511, наблюдаемый 511.
Пример I-11
3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-ил)-бензонитрил

Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 5; 55 мг, 0,105 ммоль), 3-цианофенилбориновой кислоты (30,9 мг, 0,21 ммоль), Pd(PPh3)4 (12,1 мг, 0,011 ммоль), K2CO3 (58,1 мг, 0,42 ммоль), воды (0,75 мл), толуола (1,5 мл), и EtOH (1,5 мл) нагревали при 90°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Органический слой выпаривали. Осадок очищали при помощи хроматографии (силикагель, 35% EtOAc/гексаны) до получения 3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-ил)-бензонитрил (38 мг, 66%) в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,43 (s, 1Н), 8,15 (s, 1Н), 7,96-8,01 (m, 2Н), 7,73-7,78 (m, 1Н), 7,64-7,70 (m, 1Н), 7,53-7,57 (m, 1Н), 7,42-7,49 (m, 1Н), 7,32-7,38 (m, 2Н), 7,23 (dd, J=7,2, 0,9 Гц, 1Н), 7,05-7,17 (m, 2Н), 7,02 (d, J=3,0 Гц, 1Н), 6,91-6,99 (m, 2Н), 2,20 (s, 3Н). MS вычисленный для C32H22F2N5O2 [(М+Н)+] 546, наблюдаемый 546.
Пример I-12
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(3-хлор-фенил)-1Н-индол-2-ил]-метанон
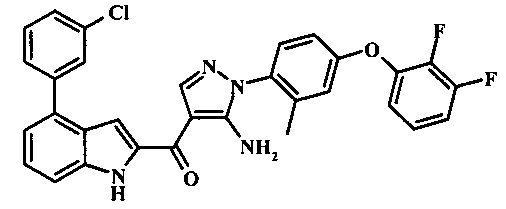
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанон (который может быть получен как описано в примере I-1 этапа 5; 55 мг, 0,105 ммоль), 3-хлорфенилбориновой кислоты (32,9 мг, 0,21 ммоль), Pd(PPh3)4 (12,1 мг, 0,011 ммоль), K2CO3 (58,1 мг, 0,42 ммоль), воды (0,75 мл), толуола (1,5 мл), и EtOH (1,5 мл) нагревали при 90°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Органический слой выпаривали. Осадок очищали при помощи хроматографии (силикагель, 35% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(3-хлор-фенил)-1Н-индол-2-ил]-метанона (54 мг, 87%) в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,41 (br. s., 1Н), 8,17 (s, 1Н), 7,71 (t, J=1,8 Гц, 1Н), 7,63 (dt, J=7,5, 1,4 Гц, 1Н), 7,48-7,53 (m, 2Н), 7,41-7,47 (m, 3Н), 7,34 (d, J=8,5 Гц, 1Н), 7,24 (dd, J=7,3, 1,0 Гц, 1Н), 7,05-7,16 (m, 2Н), 7,02 (d, J=2,5 Гц, 1Н), 6,90-6,99 (m, 2Н), 2,20 (s, 3Н). MS вычисленный для C31H22CIF2N4O2 [(М+Н)+] 555, наблюдаемый 555.
Пример I-13
3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-илметил)-бензонитрил

Этап 1: 3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1-бензолсульфонил-1Н-индол-4-илметил)-бензонитрил
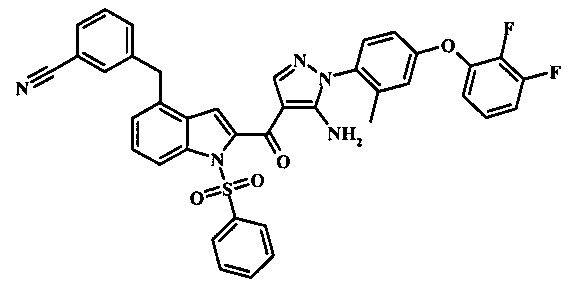
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 4; 65 мг, 0,098 ммоль), Pd(OAc)2 (2,8 мг, 0,012 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенил (Aldrich; 9 мг, 0,022 ммоль), и ТГФ (2 мл) продували аргоном в течение 3 мин и затем перемешивали при комнатной температуре в течение 5 мин. (3-циано-бензил)цинк(II) бромид (Aldrich; 0,5 М в ТГФ; 0,3 мл; 0,15 ммоль) добавляли и смесь нагревали при 75°С в течение 2 ч. Добавляли воду (3 мл) и смесь экстрагировали EtOAc (2×5 мл). Комбинированные органические слои выпаривали. Осадок очищали при помощи хроматографии (силикагель, 35% EtOAc/гексаны) до получения 3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1-бензолсульфонил-1Н-индол-4-илметил)-бензонитрила (64 мг, 93%) в виде масла.
Этап 2: 3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-илметил)-бензонитрил
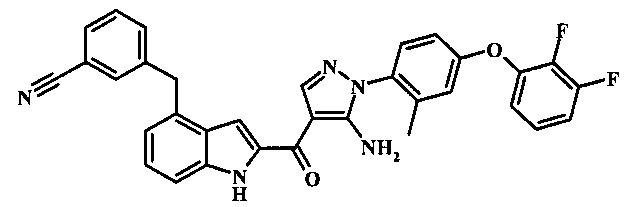
Смесь 3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1-бензолсульфонил-1Н-индол-4-илметил)-бензонитрил (64 мг, 0,092 ммоль), Cs2CO3 (119 мг, 0,37 ммоль), ТГФ (5 мл) и МеОН (2,5 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения 3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-илметил)-бензонитрила (40 мг, 78%) в виде желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,30 (br. s., 1Н), 8,07 (s, 1Н), 7,50-7,60 (m, 3Н), 7,43 (dd, J=11,2, 8,2 Гц, 2Н), 7,30-7,36 (m, 2Н), 7,20 (s, 1Н), 7,10 (ddd, J=15,2, 8,7, 5,9 Гц, 2Н), 7,02 (d, J=2,5 Гц, 1Н), 6,90-6,98 (m, 3Н), 4,39 (s, 2Н), 2,19 (s, 3Н). MS вычисленный для C33H24F2N5O2 [(М+Н)+] 560, наблюдаемый 560,1.
Пример I-14
3-(4-{5-амино-4-[4-(1Н-пиразол-4-ил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил

Этап 1: 3-{4-[5-амино-4-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил

Смесь 3-(4-гидразино-3-метил-фенокси)-бензонитрила (который может быть получен как описано для промежуточного соединения 2; 789 мг, 3,3 ммоль), (Е)-2-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-3-диметиламино-акрилонитрила (который может быть получен как описано в примере I-1 этапа 3; 510 мг, 1,11 ммоль) и EtOH (30 мл) нагревали при рефлюксе в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 30% EtOAc/гексаны) до получения 3-{4-[5-амино-4-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (598 мг, 82%) в виде пены.
Этап 2: 3-{4-[5-амино-4-(4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил
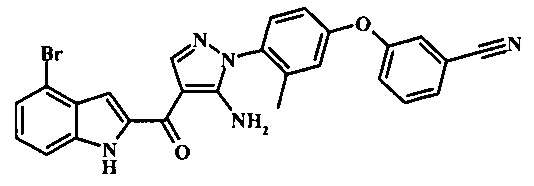
Смесь 3-{4-[5-амино-4-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (450 мг, 0,69 ммоль), Cs2CO3 (899 мг, 2,76 ммоль), ТГФ (15 мл) и МеОН (7,5 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения 3-{4-[5-амино-4-(4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила.
Этап 3: 3-(4-{5-амино-4-[4-(1Н-пиразол-4-ил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил
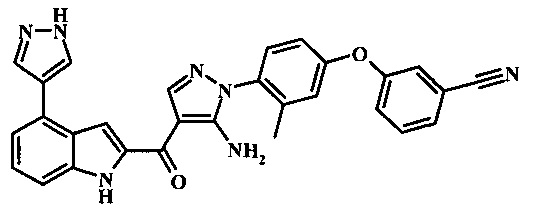
Смесь 3-{4-[5-амино-4-(4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (55 мг, 0,107 ммоль), 1-трет-бутоксикарбонил-1Н-пиразол-4-бориновой кислоты (Combi-Blocks Inc., 7949 Silverton Avenue, Suite 915, San Diego, CA 92126, USA; 45,5 мг, 0,22 ммоль), Pd(PPh3)4 (12,4 мг, 0,011 ммоль), K2CO3 (59,3 мг, 0,43 ммоль), воды (0,75 мл), толуола (1,5 мл), и EtOH (1,5 мл) нагревали при 90°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Органический слой выпаривали, осадок очищали при помощи хроматографии (силикагель, 80% EtOAc/гексаны) до получения 3-(4-{5-амино-4-[4-(1Н-пиразол-4-ил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила (30 мг, 56%) в виде масла. 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 13,04 (br. s., 1Н), 11,78 (s, 1Н), 8,38-8,49 (m, 2H), 8,08 (br. s., 1H), 7,56-7,71 (m, 3H), 7,35-7,52 (m, 4H), 7,19-7,30 (m, 3H), 7,08 (dd, J=8,5, 2,8 Гц, 1H), 6,92 (s, 2H), 2,11 (s, 3H). MS вычисленный для C29H22N7O2 [(M+H)+] 500, наблюдаемый 500.
Пример I-15
3-(4-{5-амино-4-[4-(морфолин-4-карбонил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил

Газообразную окись углерода продували через смесь 3-{4-[5-амино-4-(4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (который может быть получен как описано в примере I-14 этапа 2; 53 мг, 0,10 ммоль), морфолина (180 мг, 2,1 ммоль), Pd(PPh3)4 (35,9 мг, 0,031 ммоль), и ТГФ (10 мл) в запаянной пробирке в течение 5 мин. Пробирку запаивали и нагревали при 90°С в течение ночи. Растворитель выпаривали и осадок очищали при помощи препаративной ВЭЖХ до получения 3-(4-{5-амино-4-[4-(морфолин-4-карбонил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила (21 мг, 37%) в виде светло-желтого порошка. MS, вычисленный для C31H27N6O4 [(М+Н)+] 547, наблюдаемый 546,9.
Пример I-16
3-(4-{5-амино-4-[4-(4-метил-пиперазин-1-карбонил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил
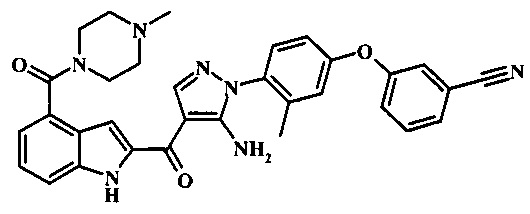
Газообразную окись углерода продували через смесь 3-{4-[5-амино-4-(4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (который может быть получен как описано в примере I-14 этапа 2; 53 мг, 0,10 ммоль), 1-метилпиперазина (155 мг, 1,55 ммоль), Pd(PPh3)4 (35,9 мг, 0,031 ммоль) и ТГФ (10 мл) в запаянной пробирке в течение 5 мин. Пробирку запаивали и нагревали при 90°С в течение ночи. Растворитель выпаривали и осадок очищали при помощи препаративной ВЭЖХ до получения 3-(4-{5-амино-4-[4-(4-метил-пиперазин-1-карбонил)-1Н-индол-2-карюонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила (32 мг, 55%) в виде грязно-белой пены. MS вычисленный для C32H30N7O3 [(М+Н)+] 560, наблюдаемый 560.
Пример I-17
3-(4-{5-амино-4-[4-(3-метокси-бензил)-1Н-индол-2-карбонил]-пиразол-1-иил}-3-метил-фенокси)-бензонитрил
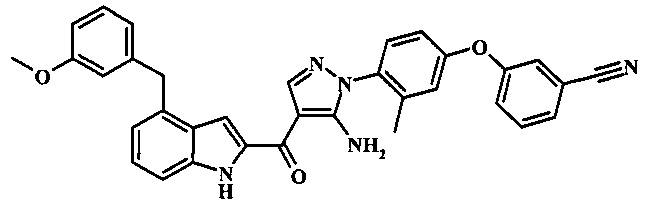
Этап 1: 3-(4-{5-амино-4-[1-бензолсульфонил-4-(3-метокси-бензил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил

Смесь 3-{4-[5-амино-4-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (который может быть получен как описано в примере I-14 этапа 1; 65 мг, 0,10 ммоль), Pd(OAc)2 (2,8 мг, 0,012 ммоль), 2-дициклогексилфосфино-2',6'-диметоксибифенил (Aldrich; 9 мг, 0,022 ммоль), и ТГФ (2 мл) продували аргоном в течение 5 мин и затем перемешивали при комнатной температуре в течение 5 мин. (3-метокси-бензил)цинк(II) бромид (Aldrich; 0,5 М в ТГФ; 0,3 мл; 0,15 ммоль) добавляли и смесь нагревали при 75°С в течение 2 ч. Добавляли воду (3 мл) и смесь экстрагировали EtOAc (2×5 мл). Комбинированные органические слои выпаривали. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения 3-(4-{5-амино-4-[1-бензолсульфонил-4-(3-метокси-бензил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила (62 мг, 90%) в виде масла.
Этап 2: 3-(4-{5-амино-4-[4-(3-метокси-бензил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил
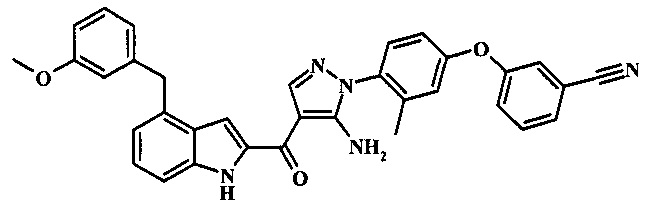
Смесь 3-(4-{5-амино-4-[1-бензолсульфонил-4-(3-метокси-бензил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила (62 мг, 0,089 ммоль), Cs2CO3 (116 мг, 0,36 ммоль), ТГФ (5 мл) и МеОН (2,5 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения 3-(4-{5-амино-4-[4-(3-метокси-бензил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила (41 мг, 83%) в виде желтой пены. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,28 (s, 1Н), 8,11 (s, 1Н), 7,44-7,56 (m, 2Н), 7,37-7,42 (m, 2Н), 7,30-7,37 (m, 4 Н), 7,25 (d, J=8,0 Гц, 1Н), 6,91-7,08 (m, 5Н), 6,77-6,87 (m, 2Н), 4,34 (s, 2Н), 3,78 (s, 3Н), 2,22 (s, 3Н). MS вычисленный для C34H28N5O3 [(М+Н)+] 554, наблюдаемый 554,1.
Пример I-18
метиловый эфир 2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-карбоновой кислоты
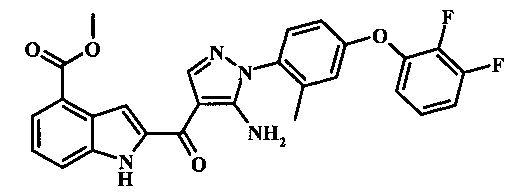
Газообразную окисб углерода продували через смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанона (который может быть получен как описано в примере I-1 этапа 5; 200 мг, 0,38 ммоль), МеОН (2,5 мл, 62 ммоль), Pd(PPh3)4 (132 мг, 0,115 ммоль), и ТГФ (20 мл) в запаянной пробирке в течение 5 мин. Пробирку запаивали и нагревали при 90°С в течение ночи. Растворитель выпаривали и осадок очищалли при помощи хроматографии (силикагель, 45% EtOAc/гексаны) до получения метилового эфира 2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-карбоновой кислоты (101 мг, 45%; 85% чистота) в виде масла. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,52 (br. s., 1Н), 8,26 (s, 1Н), 7,87-7,95 (m, 2Н), 7,60 (d, J=8,3 Гц, 2Н), 7,43 (d, J=5,5 Гц, 2Н), 7,23-7,35 (m, 3Н), 7,19 (s, 2Н), 7,11 (d, J=7,5 Гц, 2Н), 6,96-7,05 (m, 3Н), 6,94 (d, J=2,8 Гц, 1Н), 6,79-6,90 (m, 2Н), 3,96 (s, 3Н), 2,11 (s, 3Н). MS вычисленный для C27H21F2N4O4 [(M+H)+] 503, наблюдаемый 502,9.
Пример I-19
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-морфолин-4-илметил-1Н-индол-2-ил)-метанон
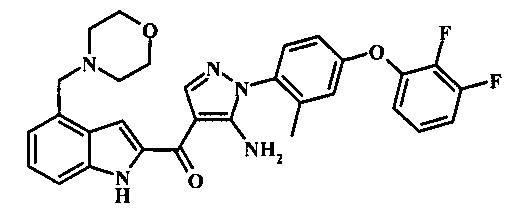
Этап 1: (1-бензолсульфонил-1Н-индол-4-ил)-морфолин-4-ил-метанон

Запаянную пробирку, содержащую 1-бензолсульфонил-4-бром-1Н-индол (который может быть получен как описано в Bell, I. М. et al. WO 2007061694 Page 103; 5,18 г, 59,5 ммоль), Pd(PPh3)4 (1,37 г, 1,19 ммоль) и ТГФ (125 мл) нагнетали 40 psi окиси углерода. Смесь нагревали при 95°С в течение ночи, и затем растворитель выпаривали. Осадок очищали при помощи хроматографии (силикагель, 80% EtOAc/гексаны) до получения (1-бензолсульфонил-1Н-индол-4-ил)-морфолин-4-ил-метанона (2,67 г, 61%) в виде пены.
Этап 2: (1-бензолсульфонил-1Н-индол-4-ил)-метанол

Раствор (1-бензолсульфонил-1Н-индол-4-ил)-морфолин-4-ил-метанона (2,07 г, 5,59 ммоль) в ТГФ (50 мл) охлаждали до -20°С и LiAlH4 (2 M в ТГФ; 6,2 мл, 12,4 ммоль) добавляли по капле. Смесь перемешивали в течение 30 мин и добавляли МеОН (20 мл). Добавляли NaBH4 (634 мг, 16,8 ммоль) и смесь перемешивали в течение 30 мин. Добавляли EtOAc (300 мл) и смесь промывали солевым раствором. Органический слой высушивали (Na2SO4), фильтровали и выпаривали до получения (1-бензолсульфонил-1Н-индол-4-ил)-метанола (1,41 г, 88%) в виде масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 3: 1-бензолсульфонил-4-хлорметил-1Н-индол
DIPEA (3,17 г, 24,5 ммоль) и метансульфонилхлорид (1,69 г, 14,7 ммоль) добавляли к раствору (1-бензолсульфонил-1Н-индол-4-ил)-метанола (1,41 г, 4,91 ммоль) в ТГФ (40 мл). Смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 20% EtOAc/гексаны) до получения 1-бензолсульфонил-4-хлорметил-1Н-индола (1,4 г, 93%) в виде масла.
Этап 4: 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол

Смесь 1-бензолсульфонил-4-хлорметил-1Н-индола (1,4 г, 4,58 ммоль), морфолина (1,2 мл, 13,7 ммоль), K2CO3 (3,16 г, 22,9 ммоль) и CH3CN (60 мл) нагревали при 65°С в течение ночи. EtOAc (250 мл) добавляли и смесь промывали водой и солевым раствором, высушивали (Na2SO4), фильтровали и выпаривали. Осадок очищали при помощи хроматографии (силикагель, 60% EtOAc/гексаны) до получения 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол (1,45 г, 89%) в виде масла.
Этап 5: 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбоновая кислота
Раствор 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол (600 мг, 1,68 ммоль) в ТГФ (12 мл) охлаждали до -78°С. LDA (2М в ТГФ; 1,7 мл, 3,4 ммоль) добавляли и смесь перемешивали в течение 1 ч. Добавляли излишек твердого СО. После 30 мин при -78°С, реакционную смесь нагревали до комнатной температуры и добавляли 2 N HCl. Смесь экстрагировали CH2Cl2 (3×150 мл). Органический слой высушивали (Na2SO4), фильтровали и выпаривали до получения 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты (670 мг, 99%) в виде пены, которую применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 6: метиловый эфир 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты
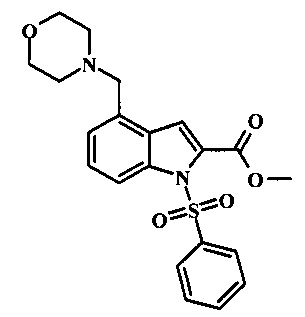
Смесь 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбоновой икслоты (670 мг, 1,67 ммоль) и SOCl2 (10 мл, 137 ммоль) нагревали при рефлюксе в течение 1 ч. Растворитель выпаривали при пониженном давлении и осадок высушивали под вакуумом в течение 1 ч. Добавляли ТГФ (20 мл), затем МеОН (1,5 мл) и реакционную смесь перемешивали при комнатной температуре в течение 1 ч. Растворитель выпаривали при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 60% EtOAc/гексаны) до получения метилового эфира 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты (486 мг, 70%) в виде масла.
Этап 7: 3-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-ил)-3-оксо-пропионитрил
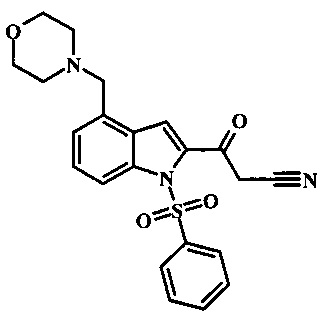
Смесь метилового эфира 1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты (486 мг, 1,17 ммоль), CH3CN (0,37 мл, 7,0 ммоль) и ТГФ (10 мл) охлаждали до -78°С. Добавляли LDA (2М в ТГФ; 1,2 мл, 2,4 ммоль) и смесь перемешивали при -78°С в течение 30 мин. Насыщенный водный раствор NH4Cl (10 мл) и затем добавляли воду (50 мл). Смесь экстрагировали EtOAc (350 мл). Органический слой промывали солевым раствором, высушивали (Na2SO4), фильтровали и выпаривали до получения 3-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-ил)-3-оксо-пропионитрил (506 мг, 93%) в виде масла, которое применяли непосредственно на следующем этапе без дополнительной очистки.
Этап 8: (Е)-2-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбонил)-3-диметиламино-акрилонитрил

Смесь 3-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-ил)-3-оксо-пропионитрила (506 мг, 1,17 ммоль), N,N-диметилформамиддиметилацеталя (0,21 мл, 1,55 ммоль), и толуола (10 мл) перемешивали при комнатной температуре течение ночи. Растворитель выпаривали. Осадок очищали при помои хроматографии (силикагель, 100% EtOAc) до получения (Е)-2-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-карбонил)-3-диметиламино-акрилонитрил (285 мг, 50%) в виде желтой пены.
Этап 9: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-ил)-метанон

Смесь [4-(2,3-дифтор-фенокси)-2-метил-фенил]-гидразин (который может быть получен как описано для промежуточного соединения 1; 130 мг, 0,52 ммоль), (Е)-2-(1-бензолсульфонил-4-морфолин-4-ил метил-1Н-индол-2-карб6нил)-3-диметиламино-акрилонитрил (60 мг, 0,125 ммоль), и EtOH (10 мл) нагревали при рефлюксе в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 50% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-ил)-метанона (56 мг, 65%) в виде масла.
Этап 10: {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-морфолин-4-илметил-1Н-индол-2-ил)-метанон
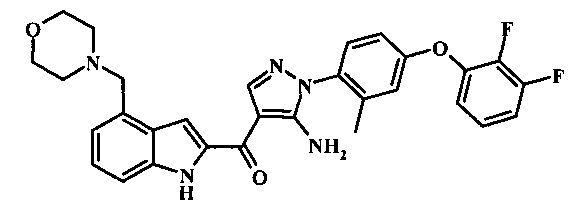
Смесь {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-бензолсульфонил-4-морфолин-4-илметил-1Н-индол-2-ил)-метанона (56 мг, 0,082 ммоль), Cs2CO3 (133 мг, 0,41 ммоль), ТГФ (4 мл) и МеОН (2 мл) перемешивали при комнатной температуре в течение. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 70% EtOAc/гексаны) до получения {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-морфолин-4-илметил-1Н-индол-2-ил)-метанона (35 мг, 79%) в виде светло-желтого масла. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,27 (br. s., 1Н), 8,27 (s, 1Н), 7,62 (s, 1Н), 7,39 (d, J=8,0 Гц, 1Н), 7,35 (d, J=8,5 Гц, 1Н), 7,30 (d, J=7,3 Гц, 1Н), 3,86 (br. s., 2Н), 3,75 (br. s., 4Н), 2,54 (br. s., 3H), 2,20 (s, 3H). MS вычисленный для C30H28F2N5O3 [(М+Н)+] 544, наблюдаемый 544,1.
Пример I-20
3-{4-[5-амино-4-(4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил
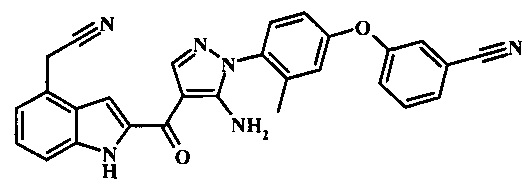
Этап 1: 3-{4-[5-амино-4-(1-бензолсульфонил-4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил

Смесь 3-{4-[5-амино-4-(1-бензолсульфонил-4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (который может быть получен как описано в примере I-14 этапа 1; 107 мг, 0,164 ммоль), трет-бутилцианоацетата (81 мг, 0,57 ммоль), бис(три-трет-бутилфосфин)палладия(0) (Strem Chemicals; 29,5 мг, 0,058 ммоль), тринатрий фосфата (121 мг, 0,74 ммоль) и толуола (2 мл) нагревали в запаянной пробирке при 100°С в течение ночи. Добавляли воду (2 мл) и смесь экстрагировали EtOAc (2×5 мл). Комбинированные органические слои выпаривали. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения 3-{4-[5-амино-4-(1-бензолсульфонил-4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (34 мг, 34%) в виде масла.
Этап 2: 3-{4-[5-амино-4-(4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил

Смесь 3-{4-[5-амино-4-(1-бензолсульфонил-4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (34 мг, 0,056 ммоль), Cs2CO3 (81,4 мг, 0,25 ммоль), ТГФ (4 мл) и МеОН (2 мл) перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Осадок очищали при помощи хроматографии (силикагель, 40% EtOAc/гексаны) до получения 3-{4-[5-амино-4-(4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила (11 мг, 42%) в виде светло-желтого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,38 (br. s., 1Н), 8,31 (s, 1Н), 7,46-7,58 (m, 4Н), 7,31-7,45 (m, 7Н), 7,22-7,26 (m, 1Н), 6,99-7,11 (m, 3Н), 4,10 (s, 2Н), 2,25 (s, 4Н). MS вычисленный для C28H21N6O2 [(М+Н)+] 473, наблюдаемый 473.
Пример I-21
Метиламидтрифторуксуснокислая соль 2-{5-амино-1-[4-(3-циано-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-карбоновой кислоты

Запаянную пробирку, содержащую 3-{4-[5-амино-4-(4-бром-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил (который может быть получен как описано в примере I-14 этапа 2; 160 мг, 0,31 ммоль), Pd(PPh3)4 (108 мг, 0,094 ммоль), метиламин (2 M в ТГФ; 2 мл, 4 ммоль) и ТГФ (20 мл) нагнетали 30 psi окиси углерода. Смесь нагревали при 95°С в течение ночи и затем растворитель выпаривали. Осадок очищали при помощи препаративной ВЭЖХ до получения метиламид трифторуксуснокислой соли 2-{5-амино-1-[4-(3-циано-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1Н-индол-4-карбоновой кислоты (73 мг, 48%) в виде грязно-белого лиофилизированного порошка. 1Н ЯМР (400 МГц, CDCl3) δ ppm 9,43 (s, 1Н), 8,36 (s, 1Н), 7,89 (dd, J=2,1, 0,9 Гц, 1Н), 7,60-7,64 (m, 1Н), 7,44-7,55 (m, 3Н), 7,38-7,44 (m, 2Н), 7,31-7,37 (m, 2Н), 7,07 (d, J=2,8 Гц, 1Н), 7,01 (dd, J=8,4, 2,4 Гц, 1Н), 3,15 (d, J=4,8 Гц, 3Н), 2,22 (s, 3Н). MS вычисленный для C28H21F2N6O2 [(М+Н)+] 511, наблюдаемый 511.
Биологические примеры
Анализ ингибирования тирозинкиназы Брутона (Btk)
Анализ представляет собой ловушка радиоактивного 33Р фосфорилированного продукта при помощи фильтрования. Взаимодействия Btk, биотинилированного SH2 пептидного субстрата (Src гомология) и АТР приводит к фосфорилированию пептидного субстрата. Биотинилированный продукт связывается стрептавидин сефарозными гранулами. Все связи, радиоактивно-меченные продукты детектируют при помощи сцинцилляционного счетчика.
Анализируемые плашки представляют собой 96-луночные полипропиленовые (Greiner) и 96-луночные 1,2 мкм гидрофильные PVDF фильтровальные пластины (Millipore). Концентрации, приведенные в данном документе, представляют собой конечные концентрации для анализа: 10-100 мкМ соединений в ДМСО (Burdick and Jackson), 5-10 нМ фермента Btk (His-меченный, полноразмерный), 30 мкМ пептидного субстрата (Biotin-Aca-AAAEEIYGEI-NH2), 100 мкМ ATP (Sigma), 8 мМ имидазол (Sigma, рН 7,2), 8 мМ глицерол-2-фосфат (Sigma), 200 мкМ EGTA (Roche Diagnostics), 1 мМ MnCl2 (Sigma), 20 мМ MgCl2 (Sigma), 0,1 мг/мл БСА (Sigma), 2 мМ DTT (Sigma), 1 μCi 33P ATP (Amersham), 20% стрептавидин сефарозные гранулы (Amersham), 50 мМ ЭДТА (Gibco), 2 М NaCl (Gibco), 2 M NaCl масс./ 1% фосфорной кислоты (Gibco), microscint-20 (Perkin Elmer).
IC50 определения вычисляли исходя из 10 данных точек на соединение, используя данные, полученные из анализа со стандартными 96-луночными планшетами. Одно контрольное соединение и семь неизвестных ингибиторов тестировали в каждом планшете и каждый планшет анализировали дважды. Обычно соединения разводили в поллогарифма начиная с 100 мкМ и заканчивая 3 нМ. Контрольное соединение представляло стауроспорин. Фон подсчитывали в отсутствии пептидного субстрата. Общую активность определяли в присутствии пептидного субстрата. Следующий протокол применяли для определения ингибирования Btk.
1) Получение препарата: тестируемые соединения разводили при шаге в пол логарифма в аналитическом буфере (имидазол, глицерол-2-фосфат, EGTA, MnCl2, MgCl2, БСА).
2) Получение гранул
a.) промывка гранул посредством центрифугирования при 500 g
b.) восстановление гранул PBS и ЭДТА до получения 20% суспензии гранул
3) Преинкубирование реакционной смеси без субстрата (аналитический буфер, DTT, ATP, 33Р АТР) и смесь с субстратом (аналитический буфер, DTT, ATP 33P ATP пептидный субстрат) 30°С в течение 15 мин.
4) Начало анализа, преинкубирование 10 мкл Btk в ферментном буфере (имидазол, глицерол-2-фосфат, БСА) и 10 мкл тестируемых соединений в течение 10 мин при КТ.
5) Добавление 30 мкл реакционной смеси без или с субстратом для Btk и соединениями.
6) Инкубирование 50 мкл общей анализируемой смеси в течение 30 мин при 30°С.
7) Перенос 40 мкл анализируемого образца в 150 мкл суспензии гранул на фильтровальной пластине для остановки реакции.
8) Промывка фильтровальной пластины спустя 30 мин, с последующими этапами
а. 3×250 мкл NaCl
b. 3×250 мкл NaCl, содержащего 1% фосфорной кислоты
c. 1×250 мкл H2O
9) Высушивание пластины в течение 1 ч при 65°С или в течение ночи при КТ
10) Добавление 50 мкл microscint-20 и подсчет 33Р ц/мин на сцинтилляционном счетчике.
Вычисление процента активности исходя из первичных данных в ц/мин
Процент активности = (образец - bkg) / (общая активность - bkg) × 100
Вычисление IC50 из процента активности, применение односайтовой сигмоидальной модели дозы-ответа
y=А+((В-А)/(1+((x/C)D))))
х - концентрация соединения, у - % активности, А - мин, В - max, С - IC50, D - 1 (угловой коэффициент Хилла)
TR-FRET (FRET анализ с разрешением по времени) анализ ингибирования тирозинкиназы Брутона (ВТК)
Данный ВТК конкурентный анализ измеряет активность соединения (IC50) в отношении инактивирования тирозинкиназы Брутона при помощи технологии FRET (резонансный перенос энергии флуорисценции/Фестера, Forster/Flouresence Resonance Energy Transfer). Комплекс ВТК - Eu инкубировали на льду один час перед применением при начальной концентрации 50 нМ BTK-BioeaseTm : 10 нМ Eu-стрептавидин (Perkin- Elmer Catalog* AD0062). Аналитический буфер состоял из 20 мМ HEPES (рН 7,15), 0,1 мМ DTT, 10 мМ MgCl2, 0,5 мг/мл БСА с 3% стабилизатора киназы (Fremont Biosolutions, Catalog # STB-K02). Спустя 1 ч, вышеупомянутую реакционную смесь разводили в десять раз в аналитическом буфере для получения 5 нМ ВТК: 1 нМ комплекса Eu-Стрептавидин (донор флуорофора). 18 мкл смеси 0,11 нМ ВТК-Eu и 0,11 нМ киназная метка 178 (Invitrogen, Catalog # PV5593,) с только ВТК-Eu в качестве отрицательного контроля, затем наносили в 384-луночные плоскодонные планшеты (Greiner, 784076). Соединения, тестируемые в ходе анализа, получали в виде 10х концентраций и серийное разведение с шагом в поллогарифма выполняли в ДМСО так, чтобы получить кривую по 10 точкам. Для инициации реакции FRET соединения, полученные в виде 10-кратного стокового раствора в ДМСО добавляли в планшеты и планшеты инкубировали в течение от 18 до 24 ч при.
После инкубации планшеты анализировали на планшетном ридере BMG Pherastar Fluorescent plate reader (или эквивалент) и применяли для измерения энергии эмиссии донора флуорофора европия (620 нм эмиссия) и FRET (665 нм эмиссия). Значения лунок отрицательного контроля усредняли для получения среднего минимума. Лунка положительного контроля ("без ингибирования") усредняли для получения среднего максимума. Процент максимального FRET вычисляли при помощи следующей формулы:
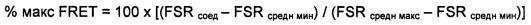
где FSR = FRET соотношение сигнала. Кривые % макс FRET получали в Activity Base (Excel) и IC50 (%), угловой коэффициент Хилла, z' и % CV определяли. Среднее IC50 и стандартное отклонение получали из дупликатов кривых (синглетные кривые ингибирования из двух независимых разведений) при помощи Microsoft Excel.
Репрезентативные данные соединений данного анализа представлены ниже в таблице II.
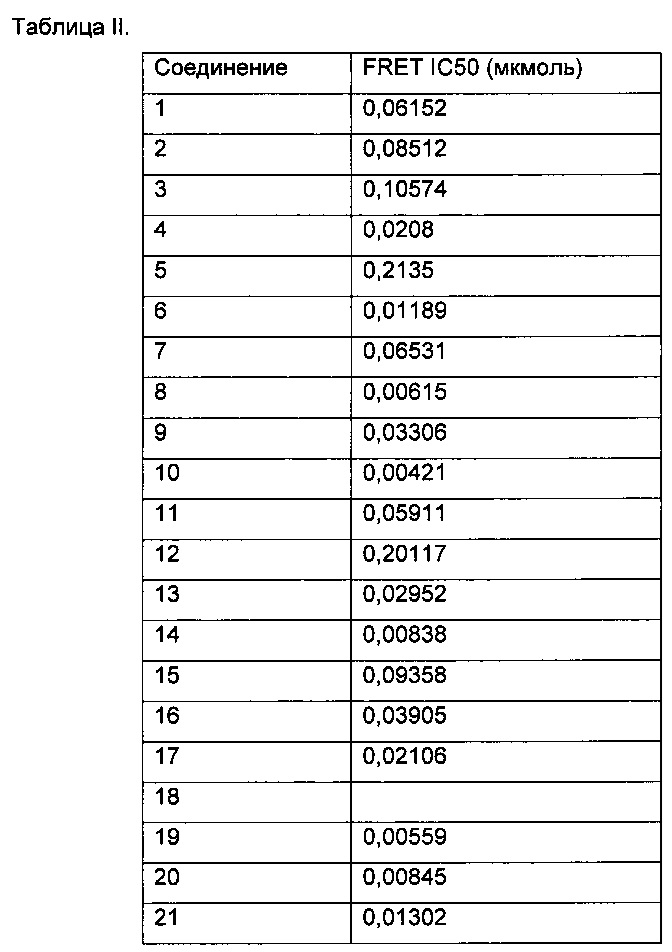
Ингибирование В-клеточной активации в цельной крови, измеренное за счет экспрессии CD69
Способ оценки способности Btk ингибиторов подавлять активацию В-клеток, опосредованную В-клеточным рецептором в крови человека представляет собой следующее:
Цельную кровь человека (HWB) получали от здоровых волонтеров со следующими ограничениями: 24 часа после приема лекарственного средства, некурящие. Кровь собирали при помощи венипункции в пробирки Vacutainer, содержащие антикоагулянт гепарин натрия. Тестируемые соединения разводили в десять раз до желаемой начальной концентрации лекарственного средства в PBS (20х), с последующими трехкратными серийными разведениями в 10% ДМСО в PBS для получения кривой дозовой зависимости по девяти точкам. 5,5 мкл каждого разведения соединения добавляли в дупликатах к 2 мл 96-луночным планшетам с V-образным дном (Analytical Sales and Services, #59623-23); 5,5 мкл 10% ДМСО в PBS добавляли в контрольные лунки и лунки, в которых не было стимулирования. HWB (100 мкл) добавляли к каждой лунке и после перемешивания планшеты инкубировали при 37°С, 5% CO2, 100% влажности в течение 30 минут. Козьи F(ab')2 антитела к IgM человека (Southern Biotech, #2022-14) (10 мкл 500 мкг/мл раствор, 50 мкг/мл конечной концентрации) добавляли к каждой лунке (исключая лунки без стимулирования) с перемешиванием и планшеты инкубировали в течение дополнительных 20 часов.
После 20-часовой инкубации образцы инкубировали с флуоресцентно-меченными антителами (15 мкл РЕ Mouse anti-Human CD20, BD Pharmingen, #555623, и/или 20 мкл АРС Mouse anti-Human CD69, BD Pharmingen #555533) в течение 30 минут, при 37°С, 5% CO2, 100% влажности. Включенное представляло собой индуцированный контроль, неокрашенный и единичные окраски для компенсационной корректировки и установки первичного напряжения. Образцы затем лизировали при помощи 1 мл 1Х Pharmingen Lyse Buffer (BD Pharmingen # 555899), и планшеты центрифугировали при 1800 rpm в течение 5 минут. Супернатанты удаляли отсасывателем и оставшиеся осадки лизировали снова 1 мл свежего 1Х Pharmingen Lyse Buffer, и планшеты центрифугировали, как описано ранее. Супернатанты удаляли и оставшиеся осадки промывали в буфере FACs (PBS плюс 1% FBS). После окончательного центрифугирования супернатанты удаляли и осадки ресуспендировали в 180 мкл буфера FACs. Образцы переносили в 96 луночный планшет, приемлемый для работы с HTS 96-луночной системой на проточном цитометре BD LSR II.
Для флуорофоров применяли соответствующие длины волн возбуждения и излучения, данные регистрировали и значения процента положительных клеток получали при программного обеспечения Cell Quest Software, результаты изначально анализировали при помощи программного обеспечения FACS analysis software (Flow Jo). IC50 для тестируемых соединений определяли в виде концентрации, которая снижалась на 50% процентов у CD69-положительных клеток, которые также являются CD20-положительными после стимулирования антителами к IgM (среднее по 8 контрольным лункам, после вычитания среднего по 8 лункам для нестимулированного фона). Значения IC50 вычисляли при помощи программного обеспечения XLfit версии 3, equation 201.
Ингибирование В-клеточной активации - FLIPR анализ В-клеток в клетках линии Ramos
Ингибирование активации В-клеток при помощи соединений по настоящему изобретению продемонстрировано за счет влияния тестируемых соединений на В-клеточные ответы, стимулированные антителами к IgM.
FLIPR анализ В-клеток представляет собой клеточный функциональный метод определения эффекта потенциальных ингибиторов внутриклеточного повышения кальция в результате стимулирования антителами к IgM. Клетки линии Ramos (клеточная линия лимфомы Беркита человека. ATCC-No. CRL-1596) культивировали в ростовой среде (описана ниже). За один день до анализа клетки линии Ramos ресуспендировали в свежей ростовой среде (такой же как упомянута выше) и помещали при концентрации 0,5×106/мл во флаконы для культуры тканей. В день анализа клетки подсчитывали и помещали при концентрации 1×106/мл в ростовую среду, дополненную 1 М FLUO-ЗАМ (TefLabs Cat-No. 0116, полученным в безводном ДМСО и 10% плюрониловой кислоте) во флаконах для культивирования тканей, и инкубировали при 37°С (4% CO2) в течение 1 ч. Для удаления внеклеточного красителя клетки собирали при помощи центрифугирования (5 мин, 1000 rpm), ресуспендировали в буфере FLIPR (описан ниже) при 1×106 клеток/мл и затем помещали в 96-луночные планшеты, покрытые поли-D-лизином темно/прозрачные планшеты (BD Cat-No. 356692) в концентрации 1×105 клеток на лунку. Тестируемые соединения добавляли при различных концентрациях, варьируя от 100 мкМ до 0,03 мкМ (7 концентраций, детали приведены ниже), и инкубировали с клетками в течение 30 мин при КТ. Са2+ сигналинг клеток линии Ramos стимулировали при помощи добавления 10 мкг/мл антител к IgM (Southern Biotech, Cat-No. 2020-01) и измеряли на FLIPR (Molecular Devices, получая изображения 96-луночных планшет при помощи ПЗС-камеры с аргоновым лазером при возбуждении 480 нМ).
Среды/Буферы:
Ростовая среда: RPMI 1640 среда с L-глутамином (Invitrogen, Cat-No. 61870-010), 10% фетальная бычья сыворотка (FBS, Summit Biotechnology Cat-No. FP-100-05); 1 мМ пируват натрия (Invitrogen Cat. No. 11360-070).
буфер FLIPR: HBSS (Invitrogen, Cat-No. 141175-079), 2 мМ CaCl2 (Sigma Cat-No. C-4901), HEPES (Invitrogen, Cat-No. 15630-080), 2,5 мМ пробенецид (Sigma, Cat-No. P-8761), 0,1% БСА (Sigma, Cat-No.A-7906), 11 мМ глюкоза (Sigma, Cat-No.G-7528).
Детали разведения соединений:
С целью получения наиболее высокой конечной концентрации 100 мкМ, 24 мкл 10 мМ стокового раствора соединения (получен в ДМСО) добавляли непосредственно к 576 мкл буфера FLIPR. Тестируемые соединения разводили в буфере FLIPR (при помощи Biomek 2000 автоматической пипетки) получая следующие разведения: носитель, 1,00×10-4 М, 1,00×10-5, 3,16×10-6, 1,00×10-6, 3,16×10-7, 1,00×10-7, 3,16×10-8.
Исследование и анализ:
Внутриклеточное повышение кальция регистрировали при помощи статистики max - min statistic (вычитание исходных данных из пика, вызванного добавлением стимулирующего антитела при помощи Molecular Devices FLIPR control и статистического программного обеспечения). IC50 определяли при помощи подбора нелинейной кривой (программное обеспечение GraphPad Prism).
Коллаген-индуцированный артрит у мышей (mCIA)
На день 0 вводили в основание хвоста или в некоторые места на спине эмульсию коллагена типа II (внутрикожно) в полном адъюванте Фрейнда (CFA). После иммунизации коллагеном у животных развивался артрит на приблизительно 21-35 день. Начало артрита синхронизировано (бустировано) системным введением коллагена в неполном адъюванте Фрейнда) (IFA; внутрикожно) на день 21. Животных исследовали после 20 дня в отношении появления умеренного артрита (баллы 1 или 2; см. описание баллов ниже), который является сигналом для бустирования. После бустирования мышей оценивали и вводили дозы кандидатов на терапевтические агенты в течение заданного времени (обычно от 2 до 3 недель) и частоты дозирования, ежедневно (QD) или дважды в день (BID).
Коллаген-индуцированный артрит у крыс (rCIA)
На день 0, крысам вводили эмульсию бычьего коллагена типа II в неполном адъюванте Фрейнда (IFA) внутрикожно в нескольких местах на спине. Бусте-инъекцию эмульсии коллагена проводили приблизительно на день 7, (внутрикожно) в основание хвоста или альтернативные участки на спине. Артрит обычно наблюдали на 12-14 день после начальной инъекции коллагена. Животных оценивали в отношении развития артрита как описано ниже (Оценка артрита) начиная с 14 дня. Животным вводили дозы кандидатов на терапевтические агенты в профилактической форме начиная с времени вторичного введения и до заданного времени (обычно от 2 до 3 недель) и частоты дозирования, ежедневно (QD) или дважды в день (BID).
Оценка артрита:
В обеих моделях развитие воспаления в суставах конечностей оценивали при помощи системы баллов, которая включает оценку 4 лап согласно следующим критериям, описанным ниже:
Баллы:
1 - опухание и/или покраснение лапы или одного пальца.
2 - опухание двух иди более суставов.
3 - сильное опухание лапы с вовлечением более чем двух суставов.
4 - сильный артрит целой лапы и пальцев.
Оценку проводили на день 0 для получения исходной инфорации и начинали снова при первых признаках или опухании до трех раз в неделю до конца эксперимента. Индекс артрита для каждой мыши получали посредством сложения четырех баллов по каждой отдельной лапе, что давало максимальное число 16 баллов на животное.
In Vivo модель астмы у крыс
Самцов серых крыс сенсибилизировали интраперитонеально 100 мкг овальбумина в 0,2 мл алюминии один раз каждую неделю в течение трех недель (день 0, 7 и 14). На день 21 (одна неделя после сенсибилизации), крысам вводили один раз в день или носитель или препарат соединения подкожно за 0,5 часа до OA введение аэрозоля (1% OA в течение 45 минут) и заканчивали за 4 или 24 часа после введения. При умервщлении сыворотку и плазму собирали у всех животных на серологию и фармакокинетику соответственно. Трахеальную канюлю вводили и легкие промывали три раза PBS. Жидкость бронхоальвеолярного лаважа анализировали на общее число лейкоцитов и лейкоцитарную формулу. Общее число лейкоцитов в аликвоте клеток (20-100 мкл) определяют при помощи Coulter Counter. Для лейкоцитарной формулы, 50-200 мкл образца центрифугируют посредством Cytospin и полученное стекло окрашивают при помощи Diff-Quik. Соотношения моноцитов, эозинофилов, нейтрофилов и лимфоцитов подсчитывают под световым микроскопом применяя стандартные морфологические критерии, выражая в процентах. Репрезентативные ингибиторы Btk показали пониженное общее число лейкоцитов в BAL OA сенсибилизированных и крыс после введения по сравнению с контрольными уровнями.
Вышеизложенное изобретение описано в более детально при помощи иллюстраций и примеров для ясности и понимания. Специалистам в области техники будет очевидно. Что изменения и модификации могут быть сделаны внутри объема прилагаемой формулы изобретения. Поэтому будет понятно, что вышеприведенное описание предназначено для иллюстрации, а не ограничения. Объем изобретения, таким образом, должен быть определен не со ссылкой на вышеприведенное описание, а со ссылкой на последующую прилагаемую формулу изобретения, наряду с полным объемом эквиваленте, на которые распространяется данная формула изобретения.
Все патенты, заявки на патент и публикации, процитированные в данной заявке, включены в данный документ посредством ссылки во всей их полноте до той степени, как если бы каждый отдельный патент, заявка на патент или публикация были упомянуты отдельно.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2648236C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2653500C2 |
| ПРОИЗВОДНОЕ АМИНОПИРАЗОЛА | 2010 |
|
RU2580543C9 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| ЗАМЕЩЕННЫЕ [1,2,4]ТРИАЗОЛО[1,5-a]ПИРИМИДИН-7-ИЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE2 | 2015 |
|
RU2659070C9 |
| ПРОИЗВОДНЫЕ ИНДОЛ-3-ИЛ-КАРБОНИЛ-ПИПЕРИДИН-БЕНЗИМИДАЗОЛА В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРОВ V1А | 2006 |
|
RU2415139C2 |
Изобретение относится к соединениям общей формулы I или их фармацевтически приемлемым солям. В общей формуле I R1 представляет собой Н или гало; R2 представляет собой Н, гало или циано; R3 представляет собой R4 или R5; R4 представляет собой гало; R5 представляет собой фенил или бензил, возможно замещенные циано, низшим алкокси или гало; 5-6-членный гетероарил, содержащий 1-2 гетероатома, выбранных из азота и серы, возможно замещенный низшим алкилом; -C(=O)R5'; низший алкил, замещенный циано или морфолинилом; R5' представляет собой 6-членный гетероциклоалкил, содержащий два гетероатома, выбранных из азота и кислорода, возможно замещенный низшим алкилом, амино, (низший алкил)амино, ди(низший алкил)амино, или низший алкокси; X представляет собой низший алкил. Соединения формулы I ингибируют Btk (тирозинкиназу Брутона). Соединения формулы I полезны для модулирования активности Btk и лечения заболеваний, ассоциированных с излишней активностью Btk. Соединения полезны для лечения онкологических, аутоиммунных и воспалительных заболеваний, вызванных нарушенной активацией В-клеток. Изобретение также относится к фармацевтическим композициям, содержащим соединения формулы I и, по меньшей мере, один носитель, разбавитель или эксципиент. 10 н. и 14 з.п. ф-лы, 2 табл., 21 пр.
. 
1. Соединение формулы I,
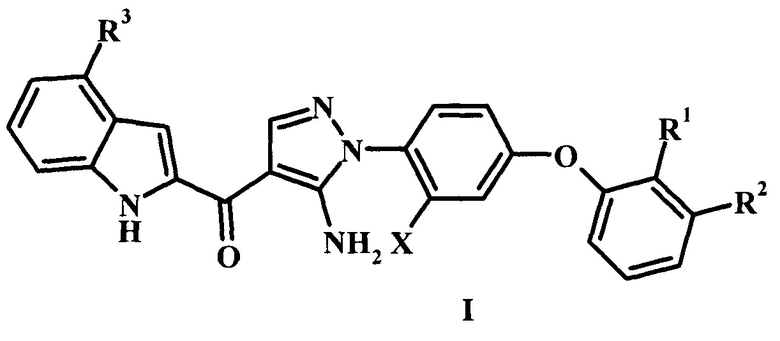
где:
R1 представляет собой Н или гало;
R2 представляет собой Н, гало или циано;
R3 представляет собой R4 или R5;
R4 представляет собой гало;
R5 представляет собой фенил или бензил, возможно замещенные циано, низшим алкокси или гало; 5-6-членный гетероарил, содержащий 1-2 гетероатома, выбранных из азота и серы, возможно замещенный низшим алкилом; -C(=O)R5'; низший алкил, замещенный циано или морфолинилом;
R5' представляет собой 6-членный гетероциклоалкил, содержащий два гетероатома, выбранных из азота и кислорода, возможно замещенный низшим алкилом, амино, (низший алкил)амино, ди(низший алкил)амино, или низший алкокси;
X представляет собой низший алкил;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где X представляет собой метил и R5 представляет собой 5-6-членный гетероарил, содержащий 1-2 гетероатома, выбранных из азота и серы, возможно замещенный низшим алкилом.
3. Соединение по п. 1, где R5 представляет собой тиофенил, возможно замещенный низшим алкилом.
4. Соединение по п. 1, где R5 представляет собой пиридил, возможно замещенный низшим алкилом.
5. Соединение по п. 1, где R1 представляет собой F и R2 представляет собой F.
6. Соединение по п. 1, где R1 представляет собой Н и R2 представляет собой циано.
7. Соединение по п. 1, где R5 представляет собой -C(=O) R 5'.
8. Соединение по п. 7, где R5' представляет собой морфолинил, пиперазинил, (низший алкил)пиперазинил или низший алкокси.
9. Соединение по п. 8, где R1 представляет собой F и R2 представляет собой F.
10. Соединение по п. 8, где R1 представляет собой Н и R2 представляет собой циано.
11. Соединение по п. 1, где R5 представляет собой фенил или бензил, возможно замещенные циано, низшим алкокси или гало.
12. Соединение по п. 1, где R5 представляет собой низший алкил, замещенный циано или морфолинилом.
13. Соединение по п. 11, где R1 представляет собой F и R2 представляет собой F.
14. Соединение по п. 11, где R1 представляет собой Н и R2 представляет собой циано.
15. Соединение, выбранное из группы, состоящей из:
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-(4-бром-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-фенил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-(4-тиофен-3-ил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиразол-1-ил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-тиофен-2-ил-1H-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(морфолин-4-карбонил)-1Н-индол-2-ил]-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[4-(1-метил-1Н-пиразол-4-ил)-1H-индол-2-ил]-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-пиридин-2-ил-1H-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-бензил-1Н-индол-2-ил)-метанона;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-[4-(1Н-пиразол-4-ил)-1Н-индол-2-ил]-метанона;
3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1H-индол-4-ил)-бензонитрила;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1H-пиразол-4-ил}-[4-(3-хлор-фенил)-1Н-индол-2-ил]-метанона;
3-(2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1-индол-4-илметил)-бензонитрила;
3-(4-{5-амино-4-[4-(1Н-пиразол-4-ил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила;
3-(4-{5-амино-4-[4-(морфолин-4-карбонил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила;
3-(4-{5-амино-4-[4-(4-метил-пиперазин-1-карбонил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила;
3-(4-{5-амино-4-[4-(3-метокси-бензил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрила;
метилового эфира 2-{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1H-индол-4-карбоновой кислоты;
{5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(4-морфолин-4-илметил-1H-индол-2-ил)-метанона;
3-{4-[5-амино-4-(4-цианометил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила; и
метиламида 2-{5-амино-1-[4-(3-циано-фенокси)-2-метил-фенил]-1Н-пиразол-4-карбонил}-1H-индол-4-карбоновой кислоты.
16. Способ лечения воспалительного и/или аутоиммунного состояния, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
17. Способ лечения воспалительного состояния, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
18. Способ лечения ревматоидного артрита, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
19. Способ лечения астмы, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
20. Фармацевтическая композиция, ингибирующая тирозинкиназу Брутона (Btk), включающая соединение по любому из пп. 1-15, смешанное с, по меньшей мере, одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
21. Применение соединения по любому из пп. 1-15 в качестве ингибитора тирозинкиназы Брутона (Btk).
22. Применение соединения по любому из пп. 1-15 для лечения воспалительного и/или аутоиммунного состояния.
23. Соединение по любому из пп. 1-15 для применения при лечении воспалительного и/или аутоиммунного состояния.
24. Применение соединения по любому из пп. 1-15 для производства лекарственного препарата для лечения воспалительного и/или аутоиммунного состояния.
| YAN LOU ET AL, "Bruton's Tyrosine Kinase Inhibitors: Approaches to Potent and Selective Inhibition, Preclinical and Clinical Evaluation for Inflammatory Diseases and B Cell Malignancies", JOURNAL OF MEDICINAL CHEMISTRY, 2012, vol | |||
| Устройство двукратного усилителя с катодными лампами | 1920 |
|
SU55A1 |
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| Устройство для регулирования дуговых ламп, в особенности для прожекторов | 1925 |
|
SU4539A1 |
| US 20120208811 A1, 16.08.2012 | |||
| ЕА 201270553 А, 12.10.2010. | |||

