Область изобретения
Настоящее изобретение относится к применению новых соединений, которые ингибируют Btk (от англ. "Bruton's tyrosine kinase" - тирозинкиназа Брутона) и являются полезными в лечении онкологических, аутоиммунных и воспалительных заболеваний, вызванных аберрантной активацией В-клеток.
Предшествующий уровень техники
Протеинкиназы составляют одно из самых больших семейств ферментов человека и регулируют множество разных процессов передачи сигналов, добавляя фосфатные группы к белкам (Т. Hunter, Cell 1987 50: 823-829). А именно, тирозинкиназы фосфорилируют белки по фенольной группе тирозиновых остатков. Семейство тирозинкиназ включает члены, которые регулируют рост, миграцию и дифференциацию клеток. Аномальная киназная активность наблюдается в целом ряде заболеваний человека, включая рак, аутоиммунные и воспалительные заболевания. Поскольку протеинкиназы входят в число ключевых регуляторов клеточной передачи сигнала, они обеспечивают мишень для модулирования клеточной функции с помощью низкомолекулярных ингибиторов киназы, и таким образом обеспечивают хорошие мишени для разработки лекарственных препаратов. В дополнение к лечению опосредованных киназой болезненных процессов селективные и эффективные ингибиторы активности киназы также полезны для исследования процессов клеточной передачи сигналов и установления других клеточных мишеней, представляющих терапевтический интерес.
Существует надежное доказательство того, что В-клетки играют ключевую роль в патогенезе аутоиммунного и/или воспалительного заболевания. Белковые терапевтические средства, которые сокращают количество В-клеток, такие как ритуксан, эффективны в отношении управляемых аутоантителами воспалительных заболеваний, таких как ревматоидный артрит (Rastetter et al. Annu Rev Med 2004 55: 477). Следовательно, ингибиторы протеинкиназ, которые играют роль в активации В-клетки, должны быть полезными терапевтическими средствами в отношении опосредованной В-клетками патологии заболевания, такой как продуцирование аутоантител.
Передача сигнала В-клеточным рецептором (BCR, от англ. "B-cell receptor") регулирует ряд В-клеточных откликов, включая пролиферацию и дифференциацию в зрелых продуцирующих антитела клетках. BCR является ключевой регулирующей точкой активности В-клеток, и аберрантная передача сигнала может вызывать нерегулируемую В-клеточную пролиферацию и образование патогенных аутоантител, которые приводят к множественным аутоиммунным и/или воспалительным заболеваниям. Тирозинкиназа Брутона (Btk) является киназой, несвязанной с BCR, которая является ближайшей к мембране и находится непосредственно ниже BCR. Недостаток Btk, как было показано, блокирует передачу сигнала BCR, и, следовательно, ингибирование Btk могло бы быть полезным терапевтическим подходом для блокировки болезненных процессов, опосредованных В-клетками.
Btk является членом семейства Тес тирозинкиназ, и как было показано, является решающим регулятором раннего развития В-клеток и активации и выживаемости зрелых В-клеток (Khan et al. Immunity 1995 3:283; Ellmeier et al. J. Exp .Med. 2000 192: 1611). Мутация Btk у людей приводит к состоянию Х-сцепленной агаммаглобулинемии (XLA, от англ. "X-linked agammaglobulinemia") (обзоры в Rosen et al. New Eng. J. Med. 1995 333: 431 и Lindvall et al. Immunol. Rev. 2005 203: 200). Эти пациенты являются иммунокомпрометированными и показывают замедленное созревание В-клеток, пониженные иммуноглобулин и периферийные уровни В-клеток, уменьшенные независимые от Т-клеток иммунные ответы, а также ослабленную мобилизацию кальция вследствие стимуляции BCR.
Доказательство роли Btk в отношении аутоиммунных и воспалительных заболеваний также приведено на мышиных моделях с дефицитом Btk. В предклинических мышиных моделях системной красной волчанки (СКВ) мыши с дефицитом Btk показывают заметное улучшение развития заболевания. Кроме того, мыши с дефицитом Btk устойчивы к коллаген-индуцированному артриту (Jansson and Holmdahl Clin. Exp. Immunol. 1993 94:459). Селективный ингибитор Btk показал зависимую от дозы эффективность на мышиной модели артрита (Z. Pan et al., Chem. Med Chem. 2007 2: 58-61).
Btk также экспрессируется клетками, отличными от В-клеток, которые могут участвовать в болезненных процессах. Например, Btk экспрессируется тучными клетками, и тучные клетки костного мозга с дефицитом Btk демонстрируют ослабленную антиген-индуцированную дегрануляцию (Iwaki et al. J. Biol. Chem. 2005 280: 40261). Это показывает, что Btk мог бы быть полезным в лечении патологических ответов тучных клеток, таких как аллергия и астма. Также моноциты у пациентов с XLA, у которых отсутствует активность Btk, показывают пониженное продуцирование ФНО-альфа (фактор некроза опухолей альфа) после стимуляции (Horwood et al. J. Exp. Med. 2003 197:1603). Следовательно, опосредованное ФНО-альфа воспаление может модулироваться низкомолекулярными ингибиторами Btk. Также сообщалось, что Btk играет роль в апоптозе (Islam and Smith Immunol. Rev. 2000 178: 49), и таким образом ингибиторы Btk могут быть полезны в лечении некоторых В-клеточных лимфом и лейкемий (Feldhahn et al. J. Exp. Med. 2005 201: 1837).
Сущность изобретения
В настоящей заявке предложены ингибирующие Btk соединения Формулы I, способы их применения, как описано в данном документе ниже: В заявке предложено соединение Формулы I
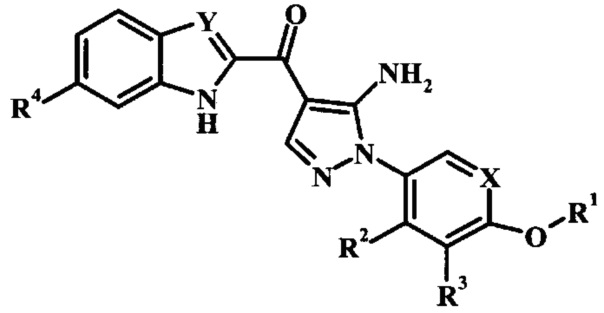 ,
,
где:
R1 представляет собой низший алкил, фенил, циклоалкил или пиридил, возможно замещенный одним или более чем одним R1';
каждый R1' независимо представляет собой низший алкил, галоген, -C(=O)NH2 или циано;
R2 отсутствует, представляет собой галоген, низший алкокси, гидрокси или низший алкил;
R3 отсутствует, представляет собой галоген, низший алкокси, гидрокси или низший алкил;
R4 отсутствует или представляет собой гетероциклоалкил низший алкиленил;
X представляет собой CH или N; и
Y представляет собой CH или N;
или его фармацевтически приемлемая соль.
В заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложена фармацевтическая композиция, включающая соединение Формулы I, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
Подробное описание изобретения
Определения
Фраза об объекте, используемая в данном документе, относится к одному или более чем одному объекту, например, соединение относится к одному или более чем одному соединению, или по меньшей мере одному соединению. Таким образом, термины "один", "один или более чем один" и "по меньшей мере один" могут использоваться в данном документе взаимозаменяемо.
Фраза "как определено в данном документе выше" относится к самому широкому определению каждой группы, как приведено в сущности изобретения или самой широкой формуле изобретения. Во всех других воплощениях, приведенных ниже, заместители, которые могут находиться в каждом воплощении, и которые четко не определены, сохраняют самое широкое определение, приведенное в сущности изобретения.
Как используется в данном описании, либо в промежуточной фразе, либо в пункте формулы изобретения, термины "включает" и "включающий" следует толковать как имеющие неограниченное значение. То есть, термины следует толковать как синонимы фраз "имеющий по меньшей мере" или "включающий по меньшей мере". При использовании в отношении способа термин "включающий" означает, что способ включает по меньшей мере перечисленные стадии, но может включать и дополнительные стадии. При использовании в отношении соединения или композиции термин "включающий" означает, что соединение или композиция включает по меньшей мере перечисленные элементы или компоненты, но также может включать дополнительные элементы или компоненты.
Как используется в данном документе, если не указано иное, слово "или" используется во "включающем" значении "и/или", а не в "исключающем" значении "или/или".
Термин "независимо" используется в данном документе для обозначения того, что переменная группировка используется в любом примере независимо от присутствия или отсутствия переменной группировки, имеющей такое же или отличное значение, в пределах одного соединения. Таким образом, в соединении, в котором R'' встречается дважды и определен как "независимо представляет собой углерод или азот", оба R'' могут представлять собой углерод, оба R" могут представлять собой азот, или один R" может представлять собой углерод и другой - азот.
Когда переменная встречается более чем один раз в любой группировке или формуле, изображающей и описывающей соединения, используемые или заявленные в настоящем изобретении, ее определение в каждом случае не зависит от ее определения в каждом другом случае. Также сочетания заместителей и/или переменных допустимы, только если такие соединения дают в результате стабильные соединения.
Символы "*" на конце связи или "------", проходящая через связь, относятся к точке присоединения функциональной группы или другой химической группировки к остальной части молекулы, частью которой она является. Таким образом, например:

Связь, уходящая в кольцевую систему (в отличие от присоединенной к определенной вершине) указывает на то, что связь может быть присоединена к любому из подходящих кольцевых атомов.
Термин "возможный" или "возможно", как используется в данном документе, означает, что впоследствии описанное событие или обстоятельство может произойти, но не требуется, и что описание включает случаи, когда событие или обстоятельство происходит, и случаи, при которых оно не происходит. Например "возможно замещенный" означает, что возможно замещенная группировка может включать атом водорода или заместитель.
Фраза "возможная связь" означает, что связь может присутствовать или может отсутствовать, и что описание включает одинарную, двойную или тройную связи. Если обозначено, что заместитель является "связью" или "отсутствует", тогда атомы, соединенные с заместителями, непосредственно связаны.
Термин "приблизительно" используется в данном документе для обозначения около, в пределах, ориентировочно или вблизи. Когда термин "приблизительно" используется в сочетании с диапазоном числовых значений, он изменяет этот диапазон, расширяя границы выше и ниже установленных численных значений. В общем, термин "приблизительно" используется в данном документе для изменения численного значения выше и ниже заявленного значения в пределах 20%.
Определенные соединения Формулы I могут проявлять таутомерию. Таутомерные соединения могут существовать в виде двух или более взаимно превращающихся молекул. Прототропные таутомеры получаются в результате перемещения ковалентносвязанного атома водорода между двумя атомами. Как правило, таутомеры находятся в равновесии и попытки выделить отдельные таутомеры обычно дают смесь, чьи химические и физические свойства соответствуют смеси соединений. Положение равновесия зависит от химических элементов внутри молекулы. Например, во многих алифатических альдегидах и кетонах, таких как ацетальдегид, преобладает кето-форма, в то время как в фенолах преобладает енольная форма. Обычные прототропные таутомеры включают кето/енольные (-С(=O)-СН- ↔ -С(-ОН)=СН-), амид/имидокислотные (-C(=O)-NH- ↔ -C(-OH)=N-) и амидиновые (-C(=NR)-NH- ↔ -C(-NHR)=N-) таутомеры. Последние два особенно распространены среди гетероарильных и гетероциклических колец, и настоящее изобретение охватывает все таутомерные формы соединений.
Технические и научные термины, используемые в данном документе, имеют значения, обычно понимаемые под ними квалифицированным специалистом в области техники, к которой относится настоящее изобретение, если не указано иное. Для разных методик и веществ, известных квалифицированным специалистам в данной области техники, в этом документе даются ссылки. Авторитетные справочники, излагающие общие принципы фармакологии, включают Goodman and Gilman's The Pharmacological Basis of Therapeutics, 10th Ed., McGraw Hill Companies Inc., New York (2001). Любые подходящие вещества и/или способы, известные квалифицированным специалистам, могут быть использованы при выполнении настоящего изобретения. Однако предпочтительные вещества и способы описаны. Вещества, реагенты и подобные, ссылки на которые даны в следующем описании и примерах, имеются в продаже, если не указано иное.
Определения, описанные в данном документе, могут быть присоединены с образованием химически уместных сочетаний, таких как "гетероалкиларил", "галоалкилгетероарил", "арилалкилгетероциклил", "алкилкарбонил", "алкоксиалкил" и подобных. Когда термин "алкил" используется в качестве суффикса после другого термина, как в "фенилалкиле" или "гидроксиалкиле", подразумевается, что он относится к алкильной группе, как определено выше, замещенной одним - двумя заместителями, выбранными из других конкретно названных групп. Таким образом, например, "фенилалкил" относится к алкильной группе, имеющей от одного до двух фенильных заместителей, и таким образом включает бензил, фенилэтил и бифенил. "Алкиламиноалкил" представляет собой алкильную группу, имеющую от одного до двух заместителей алкиламино. "Гидроксиалкил" включает 2-гидроксиэтил, 2-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 2,3-дигидроксибутил, 2-(гидроксиметил), 3-гидроксипропил и т.д. Следовательно, как используется в данном документе, термин "гидроксиалкил" используется для определения подмножества гетероалкильных групп, определенных ниже. Термин -(ар)алкил относится либо к незамещенной алкильной, либо аралкильной группе. Термин (гетеро)арил или (гет)арил относится либо к арильной, либо гетероарильной группе.
Термин "спироциклоалкил", как используется в данном документе, означает спироциклическую циклоалкильную группу, такую как, например, спиро[3.3]гептан. Термин спирогетероциклоалкил, как используется в данном документе, означает спироциклический гетероциклоалкил, такой как, например, 2,6-диазаспиро[3.3]гептан.
Термин "ацил", как используется в данном документе, обозначает группу формулы -C(=O)R, где R представляет собой водород или низший алкил, как определено в этом документе. Термин "алкилкарбонил", как используется в данном документе, обозначает группу формулы C(=O)R, где R представляет собой алкил, как определено в этом документе. Термин C1-6-ацил относится к группе -C(=O)R, содержащей 6 атомов углерода. Термин "арилкарбонил", как используется в данном документе, означает группу формулы C(=O)R, где R представляет собой арильную группу; термин "бензоил", как используется в данном документе, означает "арилкарбонильную" группу, где R представляет собой фенил.
Термин "эфир", как используется в данном документе, обозначает группу формулы -C(=O)OR, где R представляет собой низший алкил, как определено в этом документе.
Термин "алкил", как используется в данном документе, обозначает неразветвленный или разветвленный насыщенный одновалентный углеводородный остаток, содержащий от 1 до 10 атомов углерода. Термин "низший алкил" обозначает неразветвленный или разветвленный углеводородный остаток, содержащий от 1 до 6 атомов углерода. "C1-10 алкил", как используется в данном документе, относится к алкилу, состоящему из от 1 до 10 углеродов. Примеры алкильных групп включают, но не ограничиваются этим, низшие алкильные группы, включая метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил или пентил, изопентил, неопентил, гексил, гептил и октил.
Когда термин "алкил" используется в качестве суффикса после другого термина, как в "фенилалкиле" или "гидроксиалкиле", то имеют в виду, что он относится к алкильной группе, как определено выше, замещенной одним - двумя заместителями, выбранными из другой особо названной группы. Таким образом, например, "фенилалкил" обозначает радикал R'R''-, где R' представляет собой радикал фенила, и R'' представляет собой радикал алкилена, как определено в данном документе, исходя из предположения, что точка присоединения фенилалкильной группировки будет находиться на алкиленовом радикале. Примеры радикалов арилалкила включают, но не ограничиваются этим, бензил, фенилэтил, 3-фенилпропил. Термины "арилалкил" или "аралкил" трактуют одинаковым образом с той разницей, что R' представляет собой арильный радикал. Термины "(гет)арилалкил" или "(гет)аралкил" трактуют одинаковым образом с той разницей, что R' возможно представляет собой арильный или гетероарильный радикал.
Термины "галоалкил" или "гало-низший алкил", или "низший галоалкил" относятся к неразветвленному или разветвленному углеводородному остатку, содержащему от 1 до 6 атомов углерода, где один или более атомов углерода замещены одним или более чем одним атомом галогена.
Термин "алкилен" или "алкиленил", как используется в данном документе, обозначает двухвалентный насыщенный линейный углеводородный радикал от 1 до 10 атомов углерода (например (CH2)n) или разветвленный насыщенный двухвалентный углеводородный радикал от 2 до 10 атомов углерода (например -СНМе- или -CH2CH(i-Pr)CH2-), если не указано иное. За исключением случая с метиленом, открытые валентности на алкиленовой группе не закреплены на одном атоме. Примеры алкиленовых радикалов включают, но не ограничиваются этим, метилен, этилен, пропилен, 2-метил-пропилен, 1,1-диметил-этилен, бутилен, 2-этилбутилен.
Термин "алкокси", как используется в данном документе, означает -O-алкильную группу, где алкил является таким, как определено выше, такую как метокси, этокси, н-пропилокси, изопропилокси, н-бутилокси, изобутилокси, трет-бутилокси, пентилокси, гексилокси, включая их изомеры. "Низший алкокси", как используется в данном документе, обозначает алкоксигруппу с "низшей алкильной" группой, как определено прежде. "C1-10 алкокси", как используется в данном документе, относится к -O-алкилу, где алкил представляет собой C1-10.
Термин "PCy3" относится к фосфину, трехзамещенному тремя циклическими группировками.
Термины "галоалкокси" или "гало-низший алкокси", или "низший галоалкокси" относятся к низшей алкоксигруппе, где один или более атомов углерода замещены одним или более чем одним атомом галогена.
Термин "гидроксиалкил", как используется в данном документе, обозначает алкильный радикал, как определено в этом документе, где от одного до трех атомов водорода на разных атомах углерода замещены гидроксильными группами.
Термины "алкилсульфонил" и "арилсульфонил", как используются в данном документе, относятся к группе формулы -S(=O)2R, где R представляет собой алкил или арил, соответственно, и алкил и арил являются такими, как определено в этом документе. Термин "гетероалкилсульфонил", как используется в данном документе, обозначает в этом документе группу формулы -S(=O)2R, где R представляет собой "гетероалкил", как определено в этом документе.
Термины "алкилсульфониламино" и "арилсульфониламино", как используются в данном документе, относятся к группе формулы -NR'S(=O)2R, где R представляет собой алкил или арил, соответственно, R' представляет собой водород или C1-3 алкил, и алкил и арил являются такими, как определено в этом документе.
Термин "циклоалкил", как используется в данном документе, относится к насыщенному карбоциклическому кольцу, содержащему от 3 до 8 атомов углерода, т.е. циклопропилу, циклобутилу, циклопентилу, циклогексилу, циклогептилу или циклооктилу. "C3-7 циклоалкил", как используется в данном документе, относится к циклоалкилу, содержащему от 3 до 7 углеродов в карбоциклическом кольце.
Термин «карбоксиалкил», как используется в данном документе, относится к алкильной группировке, где один атом водорода заменен карбоксильной группой, исходя из предположения, что точка присоединения гетероалкильного радикала проходит через атом углерода. Термин "карбокси" или "карбоксил" относится к группировке -CO2H.
Термин "гетероарил" или "гетероароматический", как используется в данном документе, означает моноциклический или бициклический радикал от 5 до 12 кольцевых атомов, имеющий по меньшей мере одно ароматическое или частично ненасыщенное кольцо, содержащий от четырех до восьми атомов в кольце, включая один или более гетероатомов N, О или S, остальные кольцевые атомы представляют собой углерод, исходя из предположения, что точка присоединения гетероарильного радикала будет находиться на ароматическом или частично ненасыщенном кольце. Как хорошо известно квалифицированным специалистам в данной области техники, гетероарильные кольца имеют менее ароматический характер, чем их полностью углеродные аналоги. Таким образом, для целей изобретения требуется только, чтобы гетероарильная группа имела некоторую степень ароматичности. Примеры гетероарильных группировок включают моноциклические ароматические гетероциклы, имеющие от 5 до 6 кольцевых атомов и от 1 до 3 гетероатомов, включая, но не ограничиваясь этим, пиридинил, пиримидинил, пиразинил, оксазинил, пирролил, пиразолил, имидазолил, оксазолил, 4,5-дигидро-оксазолил, 5,6-дигидро-4Н-[1,3]оксазолил, изоксазол, тиазол, изотиазол, триазолин, тиадиазол и оксадиаксолин, которые возможно замещены одним или более чем одним, предпочтительно одним или двумя заместителями, выбранными из гидрокси, циано, алкила, алкокси, тио, низшего галоалкокси, алкилтио, гало, низшего галоалкила, алкилсульфинила, алкилсульфонила, галогена, амино, алкиламино, диалкиламино, аминоалкила, алкиламиноалкила и диалкиламиноалкила, нитро, алкоксикарбонила и карбамоила, алкилкарбамоила, диалкилкарбамоила, арилкарбамоила, алкилкарбониламино и арилкарбониламино. Примеры бициклических группировок включают, но не ограничиваются этим, хинолинил, изохинолинил, бензофурил, бензотиофенил, бензоксазол, бензизоксазол, бензотиазол, нафтиридинил, 5,6,7,8-тетрагидро-[1,6]нафтиридинил и бензизотиазол. Бициклические группировки могут быть замещены по обоим кольцам, однако точка присоединения расположена на кольце, содержащем гетероатом.
Термин "гетероциклил", "гетероциклоалкил" или "гетероцикл", как используется в данном документе, обозначает одновалентный насыщенный циклический радикал, содержащий одно или более колец, предпочтительно одно-два кольца, включая спироциклические кольцевые системы, от трех до восьми атомов в кольце, включая один или более кольцевых гетероатомов (выбранных из N, О или S(O)0-2), и который возможно независимо замещен одним или более чем одним, предпочтительно одним или двумя заместителями, выбранными из гидрокси, оксо, циано, низшего алкила, низшего алкокси, низшего галоалкокси, алкилтио, гало, низшего галоалкила, гидроксиалкила, нитро, алкоксикарбонила, амино, алкиламино, алкилсульфонила, арилсульфонила, алкиламиносульфонила, ариламиносульфонила, алкилсульфониламино, арилсульфониламино, алкиламинокарбонила, ариламинокарбонила, алкилкарбониламино, арилкарбониламино, и его ионные формы, если не указано иное. Примеры гетероциклических радикалов включают, но не ограничиваются этим, морфолинил, пиперазинил, пиперидинил, азетидинил, пирролидинил, гексагидроазепинил, оксетанил, тетрагидрофуранил, тетрагидротиофенил, оксазолидинил, тиазолидинил, изоксазолидинил, тетрагидропиранил, тиоморфолинил, хинуклидинил и имидазолинил, и их ионные формы. Примеры также могут быть бициклическими, такими как, например, 3,8-диаза-бицикло[3.2.1]октан, 2,5-диаза-бицикло[2.2.2]октан или октагидро-пиразино[2,1-с][1,4]оксазин.
Ингибиторы Btk
В заявке предложено соединение Формулы I
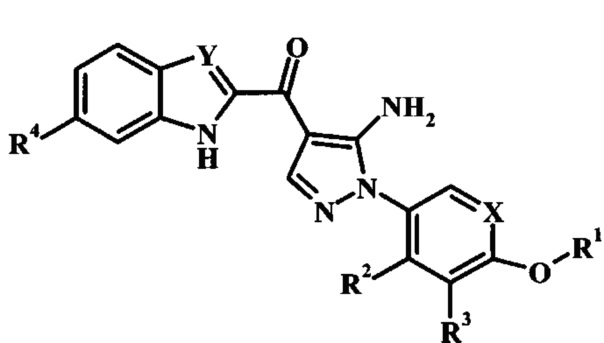 ,
,
где:
R1 представляет собой низший алкил, фенил, циклоалкил или пиридил, возможно замещенный одним или более чем одним R1';
каждый R1' независимо представляет собой низший алкил, галоген, -C(=O)NH2 или циано;
R2 отсутствует, представляет собой галоген, низший алкокси, гидрокси или низший алкил;
R3 отсутствует, представляет собой галоген, низший алкокси, гидрокси или низший алкил;
R4 отсутствует или представляет собой гетероциклоалкил низший алкиленил;
X представляет собой CH или N; и
Y представляет собой CH или N;
или его фармацевтически приемлемая соль.
В заявке предложено соединение Формулы I, где Y представляет собой СН.
В заявке предложено одно из двух вышеприведенных соединений Формулы I, где X представляет собой N.
Альтернативно в заявке предложено одно из двух вышеприведенных соединений Формулы I, где X представляет собой СН.
В заявке предложено любое из вышеприведенных соединений Формулы I, где R4 представляет собой морфолинил метилен.
Альтернативно в заявке предложено любое из вышеприведенных соединений Формулы I, где R4 отсутствует.
В заявке предложено любое из вышеприведенных соединений Формулы I, где R2 отсутствует.
Альтернативно в заявке предложено любое из вышеприведенных соединений Формулы I, где R2 представляет собой галоген.
Альтернативно в заявке предложено любое из вышеприведенных соединений Формулы I, где R2 представляет собой низший алкил.
Альтернативно в заявке предложено любое из вышеприведенных соединений Формулы I, где R2 представляет собой низший алкокси.
Альтернативно в заявке предложено любое из вышеприведенных соединений Формулы I, где R2 представляет собой гидрокси.
В заявке предложено любое из вышеприведенных соединений Формулы I, где R3 представляет собой галоген, низший алкокси или гидрокси.
В заявке предложено любое из вышеприведенных соединений Формулы I, где R1 представляет собой фенил, возможно замещенный одним или более чем одним R1'.
Альтернативно в заявке предложено любое из вышеприведенных соединений Формулы I, где R1 представляет собой низший алкил, циклоалкил или гетероарил, возможно замещенный одним или более чем одним R1'.
В заявке предложено соединение Формулы I, выбранное из группы, состоящей из:
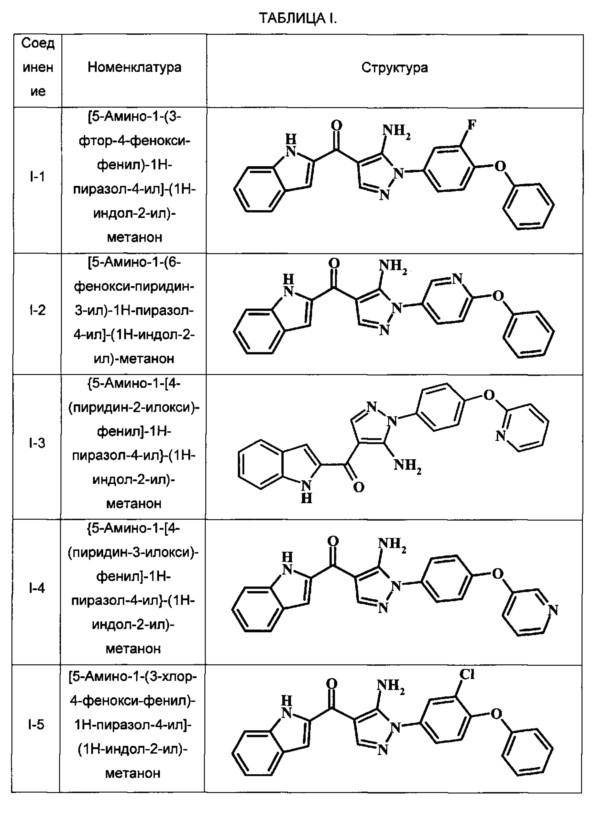
[5-Амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
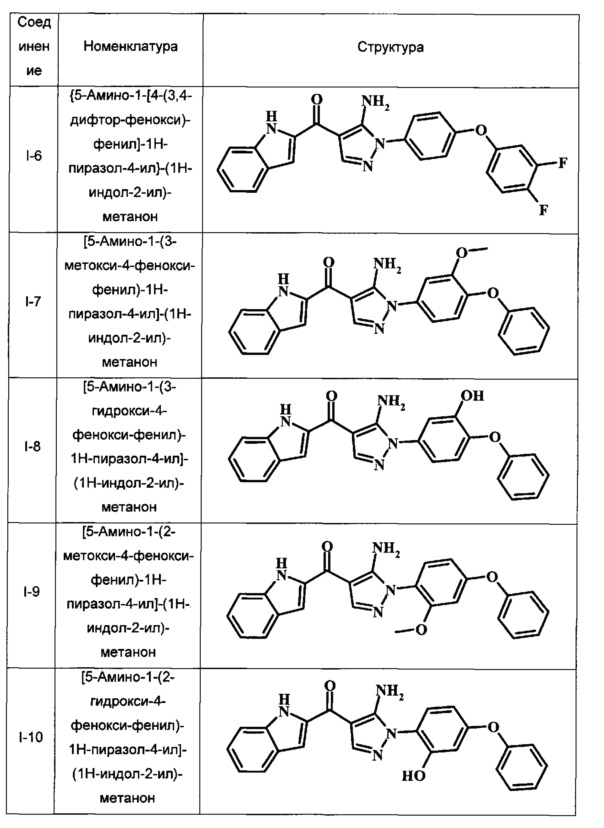
{5-Амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
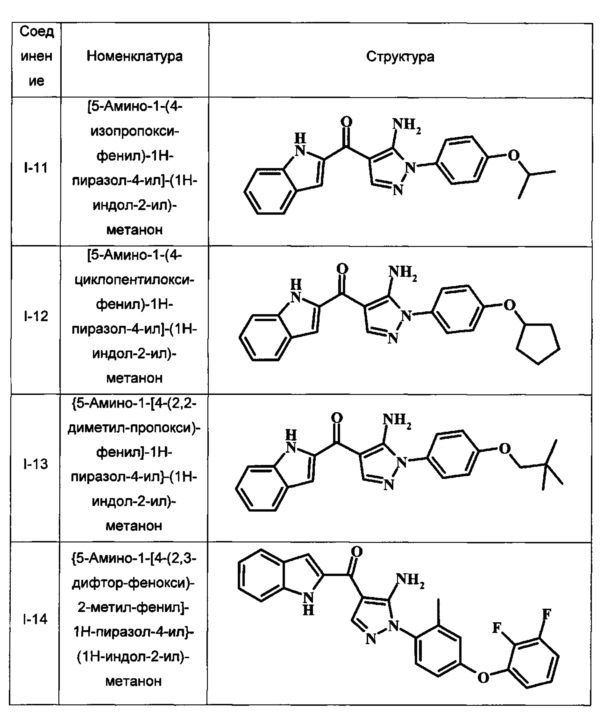
[5-Амино-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
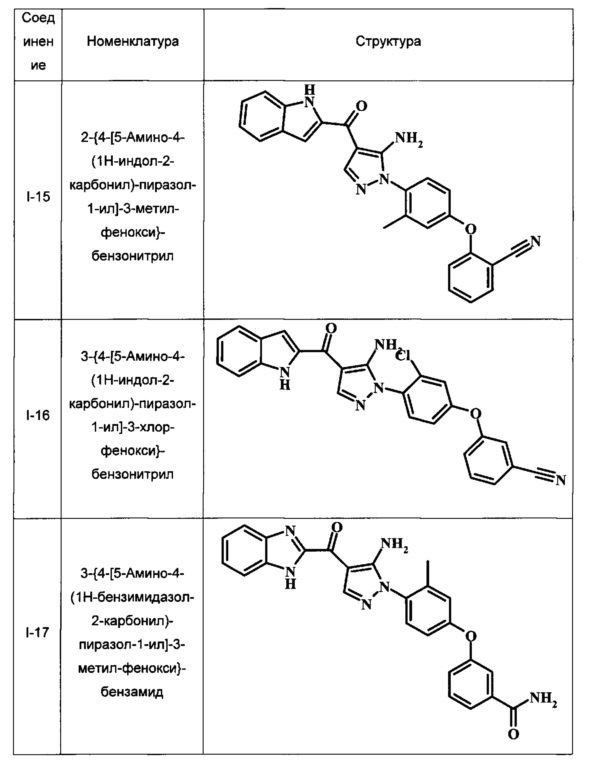
2-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила;
3-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрила;
3-{4-[5-Амино-4-(1Н-бензимидазол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензамида;
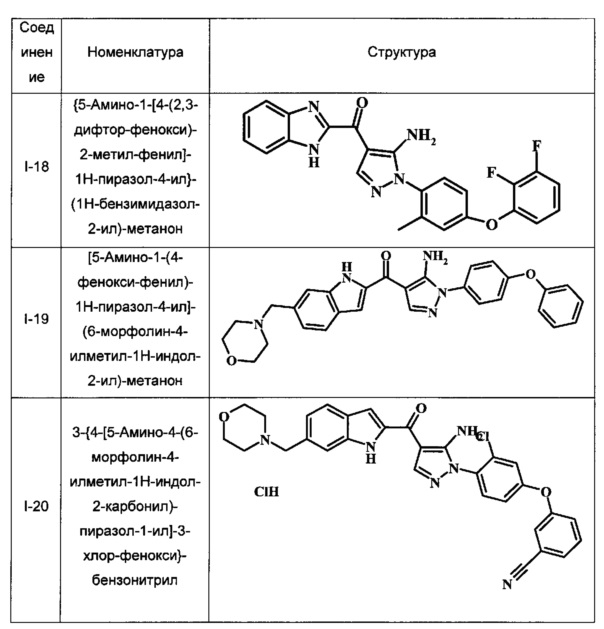
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-бензимидазол-2-ил)-метанона;
[5-Амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-(6-морфолин-4-илметил-1Н-индол-2-ил)-метанона и
3-{4-[5-Амино-4-(6-морфолин-4-илметил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрила.
В заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложен способ лечения ревматоидного артрита, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложен способ лечения астмы, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложен способ лечения рака, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложена фармацевтическая композиция, включающая соединение Формулы I.
В заявке предложена фармацевтическая композиция, включающая соединение Формулы I, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
В заявке предложено применение соединения Формулы I в изготовлении лекарства для лечения воспалительного заболевания.
В заявке предложено применение соединения Формулы I в изготовлении лекарства для лечения аутоиммунного заболевания.
В заявке предложено применение соединения Формулы I в изготовлении лекарства для лечения ревматоидного артрита.
В заявке предложено применение соединения Формулы I в изготовлении лекарства для лечения астмы.
В заявке предложено применение соединения, как описано выше, в лечении воспалительного и/или аутоиммунного состояния.
В заявке предложено применение соединения, как описано выше, в лечении ревматоидного артрита.
В заявке предложено применение соединения, как описано выше, в лечении астмы.
В заявке предложено соединение, как описано выше, для применения в лечении воспалительного и/или аутоиммунного состояния.
В заявке предложено соединение, как описано выше, для применения в лечении ревматоидного артрита.
В заявке предложено соединение, как описано выше, для применения в лечении астмы.
В заявке предложено соединение, способ или композиция, как описано в данном документе.
В заявке предложено изобретение, как описано в данном документе выше.
Соединения и получение
Примеры репрезентативных соединений, охватываемых настоящим изобретением и находящихся в объеме изобретения, приведены в следующей таблице. Эти примеры и способы получения приведены, чтобы позволить квалифицированным специалистам в данной области техники более четко понять и применить на практике настоящее изобретение. Их не следует рассматривать как ограничивающие объем изобретения, а только лишь как иллюстрирующие и представляющие его.
В общем, номенклатура, используемая в этой заявке, основывается на AUTONOMTM v.4.0, автоматизированной системе института Бельштейн для создания систематической номенклатуры ИЮПАК. Если есть расхождение между изображенной структурой и приведенным названием этой структуры, то изображенная структура должна иметь больший вес. Кроме того, если стереохимия структуры или части структуры не указана, например, с помощью жирной или пунктирной линий, то следует считать, что структура или часть структуры охватывает все ее стереоизомеры.
В таблице I показаны примеры соединений согласно общей Формуле I:
Общие схемы синтеза
Соединения по настоящему изобретению можно получить традиционными способами. Подходящие способы синтеза этих соединений приведены в примерах. В общем, соединения по изобретению можно получить согласно схемам ниже.
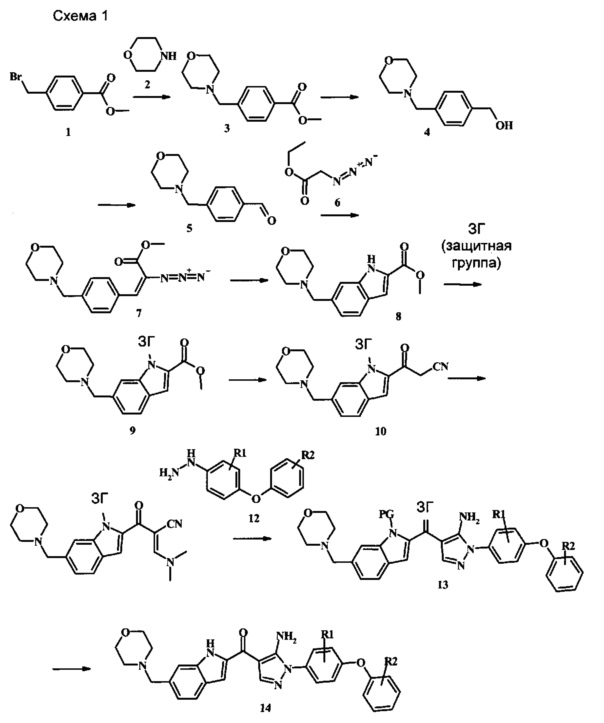
Соединения формулы 14, где R1 и R2 являются такими, как описано выше в общей формуле I, можно получить, используя способ, изображенный на схеме 1. Согласно этой методике соединение формулы 1, метиловый эфир 4-бромметил-бензойной кислоты, который имеется в продаже, можно превратить в бензил-амин, получая соединение формулы 2. Восстановление эфира дает производное бензилового спирта 4, которое можно окислить до альдегидного производного 5. Реакция с этиловым эфиром азидо-уксусной кислоты дает производное метилового эфира акриловой кислоты 7, которое может циклизоваться до соответствующего индольного производного 8. Защита индольного кольца стандартными защитными группами, такими как тозил (Ts) или 2-(триметилсилил)этоксиметил (СЭМ) дает соединение формулы 9. Затем эфир 9 можно подвергнуть взаимодействию с анионом, полученным из ацетонитрила, что дает цианоацетильное производное формулы 10. Реакция с диметилформамида диметилацеталем дает производное акрилонитрила 11, и оно взаимодействует с производным фенилгидразина формулы 12, что дает аминопиразол формулы 13. Удаление защитной группы затем дает соединение по изобретению формулы 14.
Метиловый эфир 4-бромметил-бензойной кислоты, соединение формулы 1, можно удобно обработать основанием, таким как диизопропилэтиламин, в инертном растворителе, таком как тетрагидрофуран, при температуре около 0°C в присутствии морфолина. Смесь можно перемешивать при комнатной температуре в течение времени реакции между одним часом и несколькими часами. Условия для такой реакции можно найти в литературе, например у Moore, Jason L. et al. Arkivoc, 2005, 6, 287-292.
Соединение формулы 3 можно удобно превратить в производное бензилового спирта формулы 4 в ходе взаимодействия его с восстановителем, таким как боргидрид натрия, в смеси растворителей, таких как тетрагидрофуран и метанол. Смесь можно перемешивать при нагревании с обратным холодильником в течение времени реакции от двух часов до нескольких часов.
Соединение формулы 4 можно удобно превратить в альдегидное производное формулы 5 в ходе взаимодействия его с окислителем, таким как диоксид марганца, в растворителе, таком как дихлорметан. Смесь можно перемешивать при комнатной температуре в течение нескольких часов.
Реакцию конденсации между альдегидом 5 и этиловым эфиром азидо-уксусной кислоты 6 можно проводить при температуре около 0°C в присутствии метилата натрия, используя растворитель, такой как метанол. Смесь можно перемешивать в течение времени реакции между 30 минутами или несколькими часами.
Образование индола 8 можно выполнить, используя синтез Hemetsberger-Knittel, начиная с производного метилового эфира акриловой кислоты 7. Используя растворитель, такой как ксилол или толуол, реакционную смесь нагревают при высоких температурах (90°C или выше) в течение нескольких часов. Другие способы выполнения циклизации имеются в литературе. Смотрите Stokes et al., J. Am. Chem. Soc. 2007, 129, 7500-7501, описывающий мягкую процедуру при использованииперфторбутирата родия (II) в качестве катализатора. Также смотрите Tetrahedron Letters 2009, 50, 1708-1709. Альтернативно, индольное кольцо можно синтезировать, используя разные методы синтеза. Для обзора смотрите Chem. Rev. 2006, 106, 2875-2911.
Соединение формулы 8 можно удобно обработать основанием, таким как гидрид натрия, в инертном растворителе, таком как тетрагидрофуран, при температуре около 0°C, чтобы получить соответствующий анион. Его можно обработать защитной группой, такой как тозилхлорид или 2-(триметилсилил)этоксиметилхлорид, и смесь перемешивают при комнатной температуре в течение приблизительно часа, получая производное формулы 9.
Соединение формулы 9 можно удобно превратить в производное цианоацетила формулы 10 при обработке его смесью ацетонитрила и сильного основания, такого как диизопропиламид лития или гексаметилдисилазид лития, в растворителе, таком как тетрагидрофуран, при низкой температуре, такой как приблизительно -78°C. Условия для такой реакции можно найти в патентной литературе, например у Taka, N. et al. US 20120208811, страница 163.
Соединение формулы 10 можно превратить в производное акрилонитрила формулы 11 при обработке N,N-диметилформамида диметилацеталем в инертном растворителе, таком как ароматический углеводород (например толуол) или тетрагидрофуран, при приблизительно комнатной температуре. Условия для такой реакции можно найти в патентной литературе, например у Taka, N. et al. US 20120208811, страница 132.
Производное акрилонитрила формулы 11 можно превратить в производное аминопиразола формулы 13 при обработке промежуточным соединением формулы 12, где R1 и R2 являются такими, как описано выше в общей формуле I, в спиртовом растворителе, таком как метанол или этанол, или изопропанол, приблизительно при температуре кипения растворителя. Условия для такой реакции можно найти в патентной литературе, например у Taka, N. et al. US 20120208811, страница 94.
Превращение соединения формулы 13 в соединение по изобретению формулы 14 можно выполнить, используя любой общепринятый способ. Например, в случае тозильной защитной группы реакцию можно проводить, обрабатывая соединение формулы 13 смесью основания, такого как карбонат цезия, и низшего спирта, такого как метанол, в растворителе, таком как тетрагидрофуран, при температуре между приблизительно комнатной температурой и приблизительно температурой кипения смеси. Примеры условий, которые можно использовать для такой реакции, можно найти в литературе, например у Zhang, В and Wee. A. G. Н. Org. Biomol. Chem. 2012, 10, 4597-4608, дополнительная информация; у Alam, M. et al. US 20110071150, страница 54; и у Taka, N. et al. US 20120208811, страница 55. Например, в случае защитной группы СЭМ реакцию можно проводить, обрабатывая соединение формулы 13 смесью тетрабутиламмония фторида и этилендиамина в растворителе, таком как тетрагидрофуран или диметилформамид, при температуре между приблизительно 50°C и приблизительно температурой кипения смеси. Примеры условий, которые можно использовать для такой реакции, можно найти в литературе, например у Barrett, Т.D. et al. WO 2004007463, страница 182; у Kerns, J. К. et al. WO 2007062318, страница 47; и у Degnan, А.P. et al. US 20090018132, страница 119. Альтернативно, соединение формулы 14 можно обработать концентрированной хлористоводородной кислотой в спиртовом растворителе (таком как метанол, этанол или изопропанол) или в тетрагидрофуране при нагревании с обратным холодильником, получая соединение по изобретению формулы 14. Примеры условий, которые можно использовать для такой реакции, можно найти в литературе, например у Muneau, Y. et al. US 20080262020, страница 24.
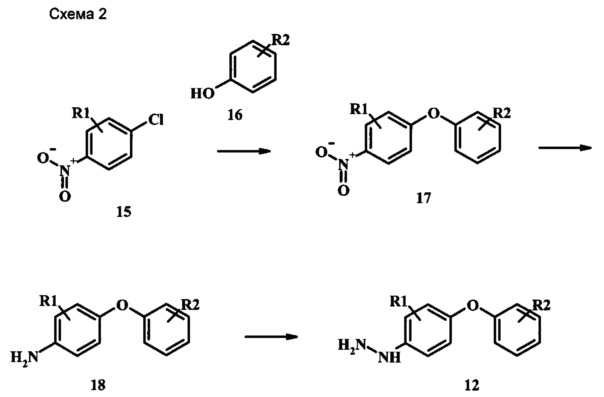
Промежуточные соединения формулы 12, где R1 и R2 являются такими, как описано выше в общей формуле I, можно получить согласно схеме 2. Соединение формулы 15 подвергают реакции нуклеофильного ароматического замещения с производным фенола формулы 16, получая соединение формулы 17. Восстановление нитрогруппы в соединении формулы 18 с последующими диазотированием и восстановлением дает производное арил-гидразина формулы 12.
Производные 4-хлор-1-нитро-бензола, такие как соединение 15, можно обработать фенолом формулы 16 в присутствии основания, такого как карбонат калия или карбонат цезия, в инертном растворителе, таком как диметилформамид, при температуре между приблизительно 100°C и приблизительно 150°C, возможно в условиях микроволнового излучения, что дает нитросоединение формулы 17. Примеры конкретных условий, которые можно использовать для такой реакции, можно найти в литературе, например у Chee, G.-L et al. US 20040266738, страница 5; и у Cui, S.-L. et al. Synlett 2004, 1829-1831.
Восстановление нитрогруппы в соединении формулы 17 можно выполнить, используя целый ряд методик, хорошо известных любому среднему специалисту в области органического синтеза. Многие из этих методик изложены у Larock, R. С.Comprehensive Organic Transformations John Wiley & Sons Inc. NY 1999, pp. 823 et seq. Один из подходящих подходов состоит в обработке соединения формулы 17 газообразным водородом в присутствии катализатора на основе благородного металла, такого как палладий на угле, в растворителе, таком как спирт (например метанол или этанол), при давлении между приблизительно одной атмосферой водорода и приблизительно тремя атмосферами водорода при приблизительно комнатной температуре. Примеры конкретных условий, которые можно использовать для такой реакции, можно найти в литературе, например у Chee, G.-L et al. US 20040266738, страница 5; и у Schoenafinger, К. et al. US 20030236288, страница 18.
Диазотирование и восстановление группы анилина в соединении формулы 17 можно проводить, используя любую общепринятую методику. Например, реакцию удобно выполнять, обрабатывая соединение формулы 18 нитритом натрия в водном растворе в присутствии неорганической кислоты, такой как хлористоводородная кислота, при температуре ниже приблизительно 5°C и предпочтительно ниже приблизительно 0°C, затем добавляя восстановитель, такой как хлорид олова (II) или дитионит натрия, приблизительно при той же температуре. Примеры конкретных условий, которые можно использовать для такой реакции, можно найти в литературе, например у Wipf, P. and Qiming, J. WO 2012078859, страница 47; у Rewolinski, М. V. et al. WO 2009055721, страница 82; и у Schoenafinger, К. et al. US 20030236288, страница 18.
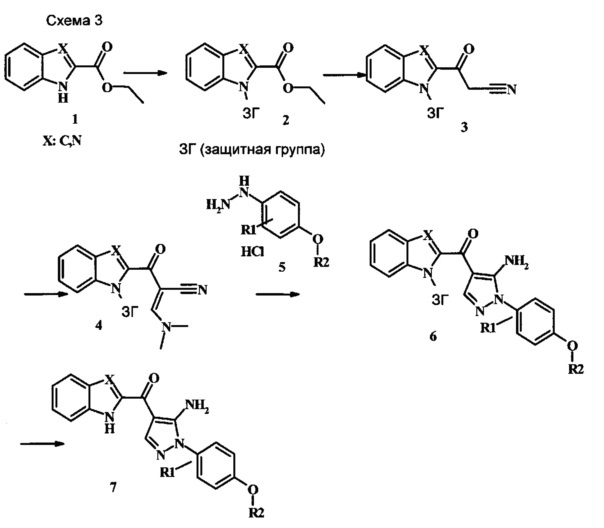
Соединения формулы 7, где R1 и R2 являются такими, как описано выше в общей формуле I, можно получить, используя способ, изображенный на схеме 1. Согласно этой методике соединение формулы 1, которое имеется в продаже, можно защитить, используя защитную группу, такую как тозил (Ts) или 2-(триметилсилил)этоксиметил (СЭМ), получая соединение формулы 2. Затем эфир 2 можно подвергнуть взаимодействию с анионом, полученным из ацетонитрила, получая цианоацетильное производное формулы 3. Реакция с диметилформамида диметилацеталем дает производное акрилонитрила 4, и оно взаимодействует с производным фенилгидразина формулы 5, что дает аминопиразол формулы 6. Удаление защитной группы затем дает соединение по изобретению формулы 7.
Этил 1Н-индол-2-карбоксилат и этил 1Н-бензо[d]имидазол-2-карбоксилат, соединения формулы 1, можно удобно обработать основанием, таким как гидрид натрия, в инертном растворителе, таком как тетрагидрофуран, при температуре около 0°C, получая соответствующий анион. Его можно обработать тозилхлоридом или 2-(триметилсилил)этоксиметилхлоридом, и смесь перемешивают при комнатной температуре в течение приблизительно часа, получая производное формулы 2.
Соединение формулы 2 можно удобно превратить в цианоацетильное производное формулы 3, обрабатывая его смесью ацетонитрила и сильного основания, такого как диизопропиламид лития или гексаметилдисилазид лития, в растворителе, таком как тетрагидрофуран, при низкой температуре, такой как приблизительно -78°C. Условия такой реакции можно найти в патентной литературе, например у Taka, N. et al. US 20120208811, страница 163.
Соединение формулы 3 можно превратить в производное акрилонитрила формулы 4 при обработке N,N-диметилформамида диметилацеталем в инертном растворителе, таком как ароматический углеводород (например толуол) или тетрагидрофуран, при приблизительно комнатной температуре. Условия для такой реакции можно найти в патентной литературе, например у Taka, N. et al. US 20120208811, страница 132.
Производное акрилонитрила формулы 4 можно превратить в производное аминопиразола формулы 6 при обработке промежуточным соединением формулы 5, где R1 и R2 являются такими, как описано выше в общей формуле I, в спиртовом растворителе, таком как метанол или этанол, или изопропанол, приблизительно при температуре кипения растворителя. Условия для такой реакции можно найти в патентной литературе, например у Taka, N. et al. US 20120208811, страница 94.
Превращение соединения формулы 6 в соединение по изобретению формулы 7 можно выполнить, используя любую общепринятую методику. Например, в случае тозильной защитной группы реакцию можно проводить, обрабатывая соединение формулы 6 смесью основания, такого как карбонат цезия, и низшего спирта, такого как метанол, в растворителе, таком как тетрагидрофуран, при температуре между приблизительно комнатной температурой и приблизительно температурой кипения смеси. Примеры условий, которые можно использовать для такой реакции, можно найти в литературе, например у Zhang, В and Wee. A.G.Н. Org. Biomol. Chem. 2012, 10, 4597-4608, дополнительная информация; у Alam, М. et al. US 20110071150, страница 54; и у Taka, N. et al. US 20120208811, страница 55. Например, в случае защитной группы СЭМ реакцию можно проводить, обрабатывая соединение формулы 6 смесью тетрабутиламмония фторида и этилендиамина в растворителе, таком как тетрагидрофуран или диметилформамид, при температуре между приблизительно 50°C и приблизительно температурой кипения смеси. Примеры условий, которые можно использовать для такой реакции, можно найти в литературе, например у Barrett, Т. D. et al. WO 2004007463, страница 182; у Kerns, J. К. et al. WO 2007062318, страница 47; и у Degnan, А. P. et al. US 20090018132, страница 119. Альтернативно, соединение формулы 7 можно обработать концентрированной хлористоводородной кислотой в спиртовом растворителе (таком как метанол, этанол или изопропанол) или в тетрагидрофуране при нагревании с обратным холодильником, получая соединение по изобретению формулы 7. Примеры условий, которые можно использовать для такой реакции, можно найти в литературе, например у Muneau, Y. et al. US 20080262020, страница 24.
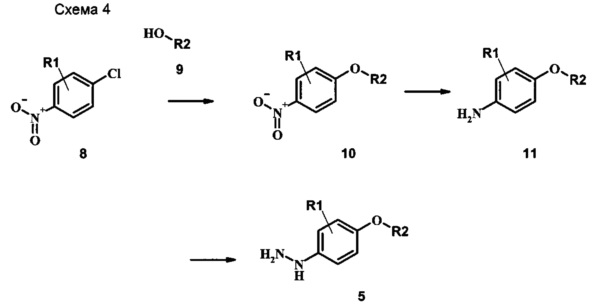
Промежуточные соединения формулы 5, где R1 и R2 являются такими, как описано выше в общей формуле I, можно получить согласно схеме 2. Соединение формулы 8 подвергают реакции нуклеофильного ароматического замещения со спиртовым производным формулы 9, получая соединение формулы 10. Восстановление нитрогруппы в соединении формулы 11 с последующими диазотированием и восстановлением дает производное арил-гидразина формулы 5.
Производные 4-хлор-1-нитро-бензола, такие как соединение 8, можно обработать спиртом (фенолом или алкиловым спиртом) формулы 9 в присутствии основания, такого как карбонат калия или карбонат цезия, в инертном растворителе, таком как диметилформамид, при температуре между приблизительно 100°C и приблизительно 150°C, возможно в условиях микроволнового излучения, что дает нитросоединение формулы 10. Примеры конкретных условий, которые можно использовать для такой реакции, можно найти в литературе, например у Chee, G.-L et al. US 20040266738, страница 5; и y Cui, S.-L. et al. Synlett 2004, 1829-1831.
Восстановление нитрогруппы в соединении формулы 10 можно выполнить, используя целый ряд методик, хорошо известных любому среднему специалисту в области органического синтеза. Многие из этих методик изложены у Larock, R.С. Comprehensive Organic Transformations John Wiley & Sons Inc. NY 1999, pp. 823 et seq. Один из подходящих подходов состоит в обработке соединения формулы 10 газообразным водородом в присутствии катализатора на основе благородного металла, такого как палладий на угле, в растворителе, таком как спирт (например метанол или этанол), при давлении между приблизительно одной атмосферой водорода и приблизительно тремя атмосферами водорода при приблизительно комнатной температуре. Примеры конкретных условий, которые можно использовать для такой реакции, можно найти в литературе, например у Chee, G.-L et al. US 20040266738, страница 5; и y Schoenafinger, К. et al. US 20030236288, страница 18.
Диазотирование и восстановление группы анилина в соединении формулы 11 можно проводить, используя любую общепринятую методику. Например, реакцию удобно выполнять, обрабатывая соединение формулы 11 нитритом натрия в водном растворе в присутствии неорганической кислоты, такой как хлористоводородная кислота, при температуре ниже приблизительно 5°C и предпочтительно ниже приблизительно 0°C, затем добавляя восстановитель, такой как хлорид олова(II) или дитионит натрия, приблизительно при той же температуре. Примеры конкретных условий, которые можно использовать для такой реакции, можно найти в литературе, например у Wipf, P. and Qiming, J. WO 2012078859, страница 47; у Rewolinski, М.V. et al. WO 2009055721, страница 82; и у Schoenafinger, К. etal. US 20030236288, страница 18.
Фармацевтические композиции и введение
Соединения по настоящему изобретению могут быть получены в виде большого числа лекарственных форм и носителей для перорального введения. Пероральное введение может быть осуществлено в форме таблеток, таблеток, покрытых оболочкой, драже, твердых и мягких желатиновых капсул, растворов, эмульсий, сиропов или суспензий. Соединения по настоящему изобретению являются эффективными при введении с помощью других способов введения, включая непрерывное (внутривенное вливание) местное парентеральное, внутримышечное, внутривенное, подкожное, трансдермальное (которое может включать усиливающий проникание агент), буккальное, назальное, ингаляцию и введение суппозиториев, наряду с другими способами введения. Предпочтительный способ введения, как правило, представляет собой пероральное введение при использовании подходящего суточного режима дозирования, который может быть установлен в зависимости от степени тяжести и реакции пациента на активный компонент.
Соединение или соединения по настоящему изобретению, а также их фармацевтически используемые соли вместе с одним или более чем одним традиционным эксципиентом, носителем или разбавителем могут быть помещены в форму фармацевтических композиций и стандартные лекарственные формы. Фармацевтические композиции и стандартные лекарственные формы могут включать традиционные компоненты в обычных пропорциях, в присутствии или отсутствии дополнительных активных соединений или элементов, и стандартные лекарственные формы могут содержать любое подходящее эффективное количество активного компонента в соответствии с предполагаемым суточным диапазон дозировок, который будет использован. Фармацевтические композиции могут быть использованы в виде твердых веществ, таких как таблетки или заполненные капсулы, полутвердых веществ, порошков, препаратов с замедленным высвобождением или жидкостей, таких как растворы, суспензии, эмульсии, эликсиры, или заполненных капсул для перорального применения; или в форме суппозиториев для ректального или вагинального введения; или в форме стерильных растворов для инъекций для парентерального применения. Обычный препарат будет содержать от приблизительно 5% до приблизительно 95% активного соединения или соединений (масс/масс.). Подразумевается, что термин "препарат" или "лекарственная форма" включает как твердые, так и жидкие составы активного соединения, и квалифицированному специалисту в данной области техники понятно, что активный компонент может находиться в разных препаратах в зависимости от органа или ткани, являющихся мишенью, требуемой дозы и фармакокинетических параметров.
Термин "эксципиент", как используется в данном документе, относится к соединению, которое является полезным при получении фармацевтической композиции, как правило, безопасной, нетоксичной и не биологически, не иным образом нежелательной, и включает эксципиенты, которые являются приемлемыми для применения как в ветеринарии, так и для фармацевтического использования человеком. Соединения по этому изобретению могут быть введены отдельно, но обычно вводятся в смеси с одним или более чем одним подходящим фармацевтическим эксципиентом, разбавителем или носителем, выбранным с учетом предполагаемого способа введения и стандартной фармацевтической практики.
"Фармацевтически приемлемый" означает то, что является полезным при получении фармацевтической композиции, которая является, как правило, безопасной, нетоксичной и не биологически, не иным образом нежелательной, и включает то, что является приемлемым как для ветеринарии, так и фармацевтического использования человеком.
Форма "фармацевтически приемлемой соли" активного компонента также может с самого начала придать желательное фармакокинетическое свойство активному компоненту, которое отсутствует в несолевой форме, и даже может положительно влиять на фармакодинамику активного компонента в отношении его терапевтической активности в организме. Фраза "фармацевтически приемлемая соль" соединения означает соль, которая является фармацевтически приемлемой и которая обладает требуемой фармакологической активностью исходного соединения. Такие соли включают: (1) соли присоединения кислоты, образованные неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные; или образованные органическими кислотами, такими как уксусная кислота, пропионовая кислота, гексановая кислота, циклопентанпропионовая кислота, гликолевая кислота, пировиноградная кислота, молочная кислота, малоновая кислота, янтарная кислота, яблочная кислота, малеиновая кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, 3-(4-гидроксибензоил)бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, 1,2-этан-дисульфоновая кислота, 2-гидроксиэтансульфоновая кислота, бензолсульфоновая кислота, 4-хлорбензолсульфоновая кислота, 2-нафталинсульфоновая кислота, 4-толуолсульфоновая кислота, камфорсульфоновая кислота, 4-метилбицикло[2.2.2]-окт-2-ен-1-карбоновая кислота, глюкогептоновая кислота, 3-фенилпропионовая кислота, триметилуксусная кислота, трет-бутилуксусная кислота, лаурилсерная кислота, глюконовая кислота, глутаминовая кислота, гидроксинафтойная кислота, салициловая кислота, стеариновая кислота, муконовая кислота и подобные; или (2) соли, образованные, когда кислотный протон, присутствующий в исходном соединении, замещается либо металлическим ионом, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координирует с органическим основанием, таким как этаноламин, диэтаноламин, триэтаноламин, трометамин, N-метилглюкамин и подобные.
Препараты твердой формы включают порошки, таблетки, пилюли, капсулы, облатки, суппозитории и диспергируемые гранулы. Твердый носитель может представлять собой одно или более веществ, которые также могут служить разбавителями, ароматизаторами, солюбилизаторами, скользящими веществами, суспендирующими веществами, связующими веществами, консервантами, разрыхлителями таблеток или инкапсулирующим веществом. В порошках носитель обычно представляет собой тонкоизмельченное твердое вещество, которое является смесью с тонкоизмельченным активным компонентом. В таблетках активный компонент обычно смешан с носителем, имеющим необходимую связывающую способность, в подходящих пропорциях и спрессован в требуемую форму и размер. Подходящие носители включают, но не ограничиваются этим, карбонат магния, стеарат магния, тальк, сахар, лактозу, пектин, декстрин, крахмал, желатин, трагакант, метилцеллюлозу, карбоксиметилцеллюлозу натрия, легкоплавкий воск, масло какао и подобные. Препараты твердой формы могут содержать в дополнение к активному компоненту красители, ароматизаторы, стабилизаторы, буферы, искусственные и натуральные подсластители, диспергирующие вещества, загустители, солюбилизаторы и подобные.
Жидкие препараты, также подходящие для перорального введения, включают жидкий состав, включая эмульсии, сиропы, эликсиры, водные растворы, водные суспензии. Они включают препараты твердой формы, которые предназначены для перевода в препараты жидкой формы непосредственно перед использованием. Эмульсии могут быть получены в растворах, например, в водных растворах пропиленгликоля, или могут содержать эмульгаторы, такие как лецитин, сорбитана моноолеат или гуммиарабик. Водные растворы могут быть получены путем растворения активного компонента в воде и добавления подходящих красителей, ароматизаторов, стабилизаторов и загустителей. Водные суспензии могут быть получены путем диспергирования тонкоизмельченного активного компонента в воде с вязким веществом, таким как природные или синтетические камеди, смолы, метилцеллюлоза, карбоксиметилцеллюлоза натрия, и другими хорошо известными суспендирующими веществами.
Соединения по настоящему изобретению могут быть приготовлены для парентерального введения (например путем инъекции, например болюсной инъекции или непрерывной инфузии) и могут находиться в стандартной лекарственной форме в ампулах, предварительно заполненных шприцах, инфузиях небольшого объема или в многодозовых контейнерах с добавлением консерванта. Композиции могут принимать такие формы, как суспензии, растворы или эмульсии в масляных или водных наполнителях, например растворы в водном полиэтиленгликоле. Примеры масляных или неводных носителей, разбавителей, растворителей или наполнителей включают пропиленгликоль, полиэтиленгликоль, растительные масла (например оливковое масло) и инъекционные органические сложные эфиры (например этилолеат), и могут содержать вспомогательные вещества, такие как консервирующие, увлажняющие, эмульгирующие или суспендирующие, стабилизирующие и/или диспергирующие агенты. Альтернативно, активный компонент может находиться в форме порошка, полученного путем асептического выделения стерильного твердого вещества или путем лиофилизации из раствора, для разбавления перед использованием подходящим наполнителем, например стерильной апирогенной водой.
Соединения по настоящему изобретению могут быть приготовлены для местного введения в эпидермис в виде мазей, кремов или лосьонов или в виде трансдермального пластыря. Мази и крема могут быть приготовлены, например, на водной или масляной основе с добавлением подходящих загустителей и/или гелеобразующих агентов. Лосьоны могут быть приготовлены на водной или масляной основе и обычно также содержат один или более чем один эмульгатор, стабилизатор, диспергирующий агент, суспендирующий агент, загуститель или краситель. Препараты, подходящие для местного введения в полость рта, включают леденцы, содержащие активные агенты в ароматизированной основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, содержащие активный компонент в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик; и жидкости для полоскания рта, содержащие активный компонент в подходящем жидком носителе.
Соединения по настоящему изобретению могут быть приготовлены для введения в виде суппозиториев. Легкоплавкий воск, такой как смесь глицеридов жирных кислот или масло какао, сначала расплавляют и активный компонент гомогенно диспергируют, например, путем перемешивания. Расплавленную гомогенную смесь затем выливают в формы подходящего размера, дают остыть и затвердеть.
Соединения по настоящему изобретению могут быть приготовлены для вагинального введения. Подходящими являются пессарии, тампоны, крема, гели, пасты, пены или спреи, содержащие в дополнение к активному компоненту такие носители, которые известны в данной области техники.
Соединения по настоящему изобретению могут быть приготовлены для назального введения. Растворы или суспензии вводят непосредственно в полость носа обычными способами, например с помощью капельницы, пипетки или спрея. Препараты могут быть представлены в форме однократной или многократной дозы. В последнем случае с помощью капельницы или пипетки это можно осуществить путем введения пациенту соответствующего заранее установленного объема раствора или суспензии. В случае спрея это можно осуществить, например, посредством дозирующего распыляющего насоса.
Соединения по настоящему изобретению могут быть приготовлены для аэрозольного введения, в частности в дыхательные пути, и включая интраназальное введение. Соединение обычно имеет небольшой размер частиц, например порядка пяти (5) микрон или меньше. Такой размер частиц может быть получен известными в данной области техники способами, например путем микронизации. Активный компонент находится в упаковке под давлением с подходящим пропеллентом, таким как хлорфторуглерод (ХФУ), например дихлордифторметан, трихлорфторметан или дихлортетрафторэтан, или диоксид углерода, или другой подходящий газ. Аэрозоль может также в целях удобства содержать поверхностно-активное вещество, такое как лецитин. Дозу лекарства можно регулировать дозирующим клапаном. Альтернативно, активные компоненты могут находиться в форме сухого порошка, например порошкообразной смеси соединения в подходящей порошкообразной основе, такой как лактоза, крахмал, производные крахмала, такие как гидроксипропилметилцеллюлоза, и поливинилпирролидин (ПВП). Порошкообразный носитель образует гель в носовой полости. Порошкообразная композиция может быть представлена в стандартной лекарственной форме, например в капсулах или картриджах, например желатиновых или блистерных упаковках, из которых порошок может быть введен с помощью ингалятора.
При желании препараты могут быть приготовлены с кишечнорастворимыми оболочками, приспособленными для замедленного введения или введения с контролируемым высвобождением активного компонента. Например, соединения по настоящему изобретению могут быть приготовлены в трансдермальных или подкожных устройствах доставки лекарственных средств. Эти системы доставки предпочтительны, когда требуется замедленное высвобождение соединения и когда соблюдение пациентом схемы лечения имеет решающее значение. Соединения в трансдермальных системах доставки часто присоединяют к прилипающей к коже твердой подложке. Представляющее интерес соединение также можно объединить с веществом, способствующим проникновению, например Azone (1-додецилаза-циклогептан-2-оном). Системы доставки с замедленным высвобождением вводятся подкожно в субдермальный слой хирургическим путем или инъекцией. Соединение инкапсулируют в субдермальные имплантаты в жирорастворимой мембране, например силиконовом каучуке, или биоразлагаемом полимере, например полимолочной кислоте.
Подходящие препараты наряду с фармацевтическими носителями, разбавителями и эксципиентами описаны в Remington: The Science and Practice of Pharmacy 1995, edited by E.W. Martin, Mack Publishing Company, 19th edition, Easton, Pennsylvania. Квалифицированный специалист по разработке лекарств способен изменять препараты в рамках методов описания изобретения, получая многочисленные препараты для конкретного способа введения, не делая композиции по настоящему изобретению неустойчивыми или не нарушая их терапевтическую активность.
Изменение соединений по настоящему изобретению для того, чтобы улучшить их растворимость в воде или другом наполнителе, может быть, например, легко выполнено путем незначительных модификаций (солеобразование, этерификация и т.д.), которые известны среднему специалисту в данной области техники. Также среднему специалисту в данной области техники известно, что изменяя способ введения и схему приема конкретного соединения, можно управлять фармакокинетикой соединений по настоящему изобретению для достижения максимального положительного эффекта у пациентов.
Термин "терапевтически эффективное количество", как используется в данном документе, означает количество необходимое для ослабления симптомов заболевания у человека. Доза устанавливается с учетом индивидуальных потребностей в каждом конкретном случае. Данная дозировка может изменяться в широких пределах в зависимости от многих факторов, таких как тяжесть заболевания, подлежащего лечению, возраст и общее состояние здоровья пациента, другие лекарственные средства, которые пациент употребляет, способ и форма введения и предпочтения и опыт лечащего врача. Для перорального введения суточная доза между приблизительно 0,01 и приблизительно 1000 мг/кг массы тела в день должна быть подходящей при монотерапии и/или при комбинированном лечении. Предпочтительная суточная доза находится между приблизительно 0,1 и приблизительно 500 мг/кг массы тела, более предпочтительно 0,1 и приблизительно 100 мг/кг массы тела и наиболее предпочтительно 1,0 и приблизительно 10 мг/кг массы тела в день. Таким образом, для введения человеку весом 70 кг диапазон доз будет составлять приблизительно от 7 мг до 0,7 г в день. Суточная доза может быть введена в виде однократной дозы или в виде дробных доз, обычно от 1 до 5 доз в день. Как правило, лечение начинают с малых доз, которые меньше чем оптимальная доза соединения. Затем дозу увеличивают небольшими приращениями до достижения оптимального эффекта у конкретного пациента. Средний специалист в области лечения описанных в данном документе заболеваний без неуместных экспериментов и на основе личных знаний, опыта и раскрытия данной заявки способен установить терапевтически эффективное количество соединений по настоящему изобретению для конкретного заболевания и пациента.
Фармацевтические препараты предпочтительно находятся в стандартных лекарственных формах. В такой форме препарат разделен на однократные дозы, содержащие соответствующие количества активного компонента. Стандартная лекарственная форма может представлять собой упакованный препарат, где упаковка содержит отдельные количества препарата, такие как упакованные таблетки, капсулы и порошки во флаконах или ампулах. Также стандартная лекарственная форма может представлять собой капсулу, таблетку, облатку или леденец, или может представлять собой их соответствующее количество в любой из этих форм упаковки.
Показания и способы лечения
Соединения общей Формулы I ингибируют тирозинкиназу Брутона (Btk). Активация Btk под действием киназ против хода транскрипции приводит к активации фосфолипазы-Cy, которая в свою очередь стимулирует высвобождение провоспалительных медиаторов. Соединения Формулы I являются полезными в лечении артрита и других противовоспалительных и аутоиммунных заболеваний. Таким образом, соединения Формулы I являются полезными в лечении артрита. Соединения Формулы I являются полезными для ингибирования Btk в клетках и для модулирующего развития В-клеток. Кроме того настоящее изобретение включает фармацевтические композиции, содержащие соединения Формулы I, смешанные с фармацевтически приемлемыми носителем, эксципиентами или разбавителями.
Соединения, описанные в данном документе, являются ингибиторами киназы, в частности ингибиторами Btk. Эти ингибиторы могут быть полезны в лечении одного или более заболеваний, чувствительных к ингибированию киназы, включая заболевания, чувствительные к ингибированию Btk и/или ингибированию пролиферации В-клеток, у млекопитающих. Не желая быть связанными с какой-то конкретной теорией, полагают, что взаимодействие соединений по изобретению с Btk приводит в результате к ингибированию активности Btk и таким образом к фармацевтической пользе этих соединений. Следовательно, изобретение включает способ лечения млекопитающего, например человека, имеющего заболевание, реагирующее на ингибирование активности Btk и/или ингибирование пролиферации В-клеток, согласно которому вводят млекопитающему, имеющему такое заболевание, эффективное количество по меньшей мере одного химического соединения, приведенного в данном документе. Эффективную концентрацию можно установить экспериментально, например, анализируя концентрацию соединения в крови, или теоретически, вычисляя биодоступность. Другие киназы, на которые возможно влиять в дополнение к Btk, включают, но не ограничиваются этим, другие тирозинкиназы и серин/треонинкиназы.
Киназы играют значительную роль в сигнальных путях, контролирующих фундаментальные клеточные процессы, такие как пролиферация, дифференциация и гибель (апоптоз). Аномальная киназная активность наблюдается при большом числе заболеваний, включая многие виды рака, аутоиммунные и/или воспалительные заболевания, и острые воспалительные реакции. Многогранная роль киназ в ключевых путях клеточной сигнализации обеспечивает благоприятную возможность для установления новых лекарственных средств, нацеленных на киназы и сигнальные пути.
Воплощение включает способ лечения пациента, имеющего аутоиммунное и/или воспалительное заболевание, или острую воспалительную реакцию, реагирующую на ингибирование активности Btk и/или пролиферации В-клеток.
Аутоиммунные и/или воспалительные заболевания, на которые возможно влиять, используя соединения и композиции по изобретению, включают, но не ограничиваются следующими заболеваниями: псориаз, аллергическая реакция, болезнь Крона, синдром раздраженного кишечника, синдром Шегрена, отторжение тканевого трансплантата и сверхострое отторжение трансплантированных органов, астму, системную красную волчанку (и связанный гломерулонефрит), дерматомиозит, множественный склероз, склеродермию, васкулит (связанные с АНЦА (антинейтрофильными цитоплазматическими антителами) и другие васкулиты), аутоиммунные гемолитические и тромбоцитопенические состояния, синдром Гудпасчера (и связанные гломерулонефрит и легочное кровотечение), атеросклероз, ревматоидный артрит, хроническую идиопатическую тромбоцитопеническую пурпуру (ИТП), болезнь Аддисона, болезнь Паркинсона, болезнь Альцгеймера, диабет, септический шок и миастению гравис.
В данный документ включены способы лечения, в которых по меньшей мере одно химическое соединение, приведенное в данном документе, вводят в сочетании с противовоспалительным агентом. Противовоспалительные агенты включают, но не ограничиваются этим, НПВС (нестероидные противовоспалительные средства), неспецифические и специфические ингибиторы фермента циклооксигеназа ЦОГ-2, соединения золота, кортикостероиды, метотрексат, антагонисты рецептора фактора некроза опухоли (ФНО), иммунодепрессанты и метотрексат.
Примеры НПВС включают, но не ограничиваются этим, ибупрофен, флурбипрофен, напроксен и напроксен натрия, диклофенак, сочетания диклофенака натрия и мизопростола, сулиндак, оксапрозин, дифлунизал, пироксикам, индометацин, этодолак, фенопрофен кальция, кетопрофен, набуметон натрия, сульфасалазин, толметин натрия и гидроксихлорохин. Примеры НПВС также включают специфические ингибиторы ЦОГ-2, такие как целекоксиб, вальдекоксиб, лумиракоксиб и/или эторикоксиб.
В некоторых воплощениях противовоспалительный агент представляет собой салицилат. Салицилаты включают, но не ограничиваются этим, ацетилсалициловую кислоту или аспирин, салицилат натрия, и салицилаты холина и магния.
Противовоспалительный агент также может представлять собой кортикостероид. Например, кортикостероид может представлять собой кортизон, дексаметазон, метилпреднизолон, преднизолон, преднизолона натрия фосфат или преднизон.
В дополнительных воплощениях противовоспалительный агент представляет собой соединение золота, такое как ауротиомалат натрия или ауранофин.
Изобретение также включает воплощения, в которых противовоспалительный агент представляет собой метаболический ингибитор, такой как ингибитор дигидрофолатредуктазы, такой как метотрексат, или ингибитор дигидрооротатдегидрогеназы, такой как лефлуномид.
Другие воплощения изобретения относятся к сочетаниям, в которых по меньшей мере одно противовоспалительное соединение представляет собой анти-С5 моноклональное антитело (такое как экулизумаб или пекселизумаб), антагонист ФНО, такой как этанерцепт, или инфликсимаб, который представляет собой анти-ФНО альфа моноклональное антитело.
Еще другие воплощения изобретения относятся к сочетаниям, в которых по меньшей мере один активный агент представляет собой иммунодепрессивное соединение, такое как соединение иммунодепрессанта, выбранное из метотрексата, лефлуномида, циклоспорина, такролимуса, азатиоприна и микофенолата мофетила.
В-клетки и предшественники В-клеток, экспрессирующие ВТК, участвуют в патологии В-клеточных злокачественных новообразований, включая, но не ограничиваясь этим, В-клеточную лимфому, лимфому (включая лимфому Ходжкина и неходжкинскую лимфому), волосатоклеточную лимфому, множественную миелому, хронический и острый миелобластный лейкоз и хронический и острый лимфоцитарный лейкоз.
Было показано, что ВТК представляет собой ингибитор сигнального комплекса, индуцирующего смерть клетки, Fas/APO-1 (CD-95) (DISC, от англ. "death inducing signaling complex") в В-лимфоцитах. Выживаемость клеток лейкоза/лимфомы может находиться в равновесии между противоположными проапоптозными эффектами каспаз, активированных DISC, и антиапоптотическим регулирующим механизмом против хода транскрипции, включающем ВТК и/или ее субстраты (Vassilev et al., J. Biol. Chem. 1998, 274, 1646-1656).
Также обнаружено, что ингибиторы ВТК являются полезными в качестве химиосенсибилизаторов, и таким образом являются полезными в сочетании с другими химиотерапевтическими лекарствами, в частности, лекарствами, которые вызывают апоптоз. Примеры других химиотерапевтических лекарств, которые можно использовать в сочетании с химиосенсибилизирующими ингибиторами ВТК, включают ингибиторы топоизомеразы I (камптотецин или топотекан), ингибиторы топоизомеразы II (например дауномицин и этопозид), алкилирующие агенты (например циклофосфамид, мелфалан и БХНМ (бисхлорэтилнитрозомочевина)), тубулин-направленные агенты (например таксол и винбластин), и биологические агенты (например антитела, такие как антитело против CD20, IDEC 8, иммунотоксины и цитокины).
Активность Btk также связана с некоторыми видами лейкоза, экспрессирующими гибридный ген bcr-abl, возникающий в результате транслокации частей хромосомы 9 и 22. Такая аномалия обычно наблюдается при хроническом миелобластном лейкозе. Btk конститутивно фосфорилируется под действием bcr-abl киназы, которая вызывает нисходящие сигналы выживаемости, которая обходит апоптоз в bcr-abl клетках (N. Feldhahn et al. J. Exp. Med. 2005 201 (11): 1837-1852).
Способы лечения
В заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложен способ лечения воспалительного состояния, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложен способ лечения ревматоидного артрита, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложен способ лечения астмы, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество соединения Формулы I.
В заявке предложен способ лечения воспалительного и/или аутоиммунного состояния, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество ингибирующего Btk соединения Формулы I.
В заявке предложен способ лечения артрита, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество ингибирующего Btk соединения Формулы I.
В заявке предложен способ лечения рака, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество ингибирующего Btk соединения Формулы I.
В заявке предложен способ лечения астмы, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество ингибирующего Btk соединения Формулы I.
В заявке предложен способ ингибирования пролиферации В-клеток, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество ингибирующего Btk соединения Формулы I.
В заявке предложен способ ингибирования активности Btk, согласно которому вводят ингибирующее Btk соединение любой из Формул I, где ингибирующее Btk соединение проявляет IC50 (от англ. half maximal inhibitory concentration - концентрация полумаксимального ингибирования) 50 микромоль или менее в биохимическом анализе in vitro активности Btk.
В одном варианте описанного выше способа ингибирующее Btk соединение проявляет IC50 100 наномоль или менее в биохимическом анализе in vitro активности Btk.
В другом варианте описанного выше способа соединение проявляет IC50 10 наномоль или менее в биохимическом анализе in vitro активности Btk.
В заявке предложен способ лечения воспалительного состояния, согласно которому совместно вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество противовоспалительного соединения в сочетании с ингибирующим Btk соединением Формулы I.
В заявке предложен способ лечения артрита, согласно которому совместно вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество противовоспалительного соединения в сочетании с ингибирующим Btk соединением Формулы I.
В заявке предложен способ лечения лимфомы или лейкозных клеток BCR-АВL1+, согласно которому вводят пациенту, нуждающемуся в этом, терапевтически эффективное количество ингибирующего Btk соединения Формулы I.
Примеры
Общие аббревиатуры
Общепринятые аббревиатуры включают: ацетил (Ас), азо-бис-изобутирилнитрил (АИБН), атмосфера (атм), 9-борабицикло[3.3.1]нонан (9-ББН или ББН), 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (БИНАФ), трет-бутоксикарбонил (Boc), ди-трет-бутилпирокарбонат или boc-ангидрид (BOC2O), бензил (Bn), бутил (Bu), регистрационный номер в журнале Chemical Abstracts (CAS №), бензилоксикарбонил (CBZ или Z), карбонилдиимидазол (КДИ), 1,4-диазабицикло[2.2.2]октан (DABCO), диэтиламиносеры трифторид (ДАСТ), дибензилиденацетон (dba), 1,5-диазабицикло[4.3.0]нон-5-ен (ДБН), 1,8-диазабицикло[5.4.0]ундек-7-ен (ДБУ), N,N'-дициклогексилкарбодиимид (ДЦК), 1,2-дихлорэтан (ДХЭ), дихлорметан (ДХМ), 2,3-дихлор-5,6-дициано-1,4-бензохинон (ДДХ), диэтилазодикарбоксилат (ДЭАД), диизопропилазодикарбоксилат (ДИАД), диизобутилалюмогидрид (ДИБАЛ или ДИБАЛ-Н), диизопропилэтиламин (ДИПЭА), N,N-диметилацетамид (ДМА), 4-N,N-диметиламинопиридин (ДМАП), N,N-диметилформамид (ДМФА), диметилсульфоксид (ДМСО), 1,1'-бис-(дифенилфосфино)этан (dppe), 1,1'-бис-(дифенилфосфино)ферроцен (dppf), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), 2-этокси-1-этоксикарбонил-1,2-дигидрохинолин (ЭЭДХ), этил (Et), этилацетат (EtOAc), этанол (EtOH), этиловый эфир 2-этокси-2Н-хинолин-1-карбоновой кислоты (EEDQ), диэтиловый эфир (Et2O), этилизопропиловый эфир (EtOiPr), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат уксусная кислота (HATU), уксусная кислота (НОАс), 1-N-гидроксибензотриазол (HOBt), высокоэффективная жидкостная хроматография (ВЭЖХ), изопропанол (ИПС), изопропилмагнийхлорид (iPr MgCl), гексаметилдисилазан (ГМДС), жидкостная хроматография масс-спектрометрия (ЖХМС), гексаметилдисилазан лития (LiМДС), мета-хлорпероксибензойная кислота (м-ХПБК), метанол (МеОН), температура плавления (т.пл.), MeSO2- (мезил или Ms), метил (Me), ацетонитрил (MeCN), мета-хлорнадбензойная кислота (МХНБК), масс-спектр (мс), метил-трет-бутиловый (МТБЭ), метил тетрагидрофуран (МеТГФ), N-бромсукцинимид (NBS), н-бутиллитий (HBuLi), N-карбоксиангидрид (NCA), N-хлорсукцинимид (NCS), N-метилморфолин (NMM), N-метилпирролидон (NMP), пиридиния хлорхромат (ПХХ), дихлор-((бис-дифенилфосфино)ферроценил)палладий (II) (Pd (dppf)Cl2), ацетат палладия(II) (Pd(OAc)2), трис(дибензилиденацетон)дипалладий (0) (Pd2(dba)3), пиридиния дихромат (ПДХ), фенил (Ph), пропил (Pr), изопропил (i-Pr), фунт на квадратный дюйм (пси), пиридин (pyr), 1,2,3,4,5-пентафенил-1'-(ди-трет-бутилфосфино)ферроцен (Q-Phos), комнатная температура (температура окружающей среды, к.т.или КТ), втор-бутиллитий (sBuLi), трет-бутилдиметилсилил или t-BuMe2Si (ТБДМС), тетра-н-бутиламмония фторид (ТБАФ), триэтиламин (ТЭА или Et3N), 2,2,6,6-тетраметилпиперидин-1-оксил (ТЕМПО), триметилсилилэтоксиметил (СЭМ), трифлат или CF3SO2- (Tf), трифторуксусная кислота (ТФК), 1,1'-бис-2,2,6,6-тетраметилгептан-2,6-дион (ТМГД), О-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU), тонкослойная хроматография (ТСХ), тетрагидрофуран (ТГФ), триметилсилил или Me3Si (ТМС), пара-толуолсульфоновой кислоты моногидрат (TsOH или pTsOH), 4-Ме-C6H4SO2-или тозил (Ts) и N-ypeтан-N-карбоксиангидрид (UNCA). Общепринятая номенклатура, включая приставки нормальный (н), изо (i-), вторичный (втор-), третичный (трет-) и нео, имеет обычное значение, когда используется с алкильной группировкой (J. Rigaudy and D.P. Klesney, Nomenclature in Organic Chemistry, IUPAC 1979 Pergamon Press, Oxford).
Общие условия
Соединения по настоящему изобретению можно получить, исходя из имеющихся в продаже исходных веществ, используя общие синтетические способы и методики, известные квалифицированным специалистам в данной области техники. Приведенные ниже изображения являются реакционными схемами, подходящими для получения таких соединений. Дополнительные пояснения можно найти в конкретных примерах.
Специальные аббревиатуры
Общие детали эксперимента
Реагенты покупали у Aldrich, Oakwood, Matrix или других поставщиков и использовали без дополнительной очистки. Реакции с использованием микроволнового излучения для нагревания проводили, используя либо Personal Chemistry Emrys Optimizer System, либо СЕМ Discovery System. Очистку на уровне мультимиллиграммов до мультиграммов осуществляли способами, известными квалифицированным специалистам в данной области техники, такими как элюирование на флэш-колонке с силикагелем; препаративные очистки на флэш-колонке также применяли в некоторых случаях, используя удаляемые предварительно упакованные мультиграммовые силикагелевые колонки (RediSep), элюируемые с помощью системы CombiFlash. Biotage™ и ISCO™ также являются устройствами с флэш-колонками, которые можно использовать в данном изобретении для очистки промежуточных соединений.
В целях определения подлинности и чистоты соединения спектры ЖХ/МС (жидкостной хроматографии/масс-спектрометрии) регистрировали, используя следующую систему. Для измерения масс-спектра система состоит из спектрометра Micromass Platform II: ЭС (электроспрей) ионизация в положительном режиме (диапазон масс: от 150 до 1200). Одновременное хроматографическое разделение достигали с помощью следующей системы ВЭЖХ: ES Industries Chromegabond WR С-18 3u 120 (3,2×30 мм) картридж для колонки; подвижная фаза А: вода (0,02% ТФК), и фаза В: ацетонитрил (0,02% ТФК); градиент 10% В до 90% В в течение 3 минут; время установления равновесия 1 минута; скорость потока 2 мл/минута.
(3,2×30 мм) картридж для колонки; подвижная фаза А: вода (0,02% ТФК), и фаза В: ацетонитрил (0,02% ТФК); градиент 10% В до 90% В в течение 3 минут; время установления равновесия 1 минута; скорость потока 2 мл/минута.
Многие соединения Формулы 1 также очищали с помощью обращенно-фазовой ВЭЖХ, используя способы, хорошо известные квалифицированным специалистам в данной области техники. В некоторых случаях очистку с помощью препаративной ВЭЖХ проводили, используя масс-спектрометр РЕ Sciex 150 EX, регулирующий коллектор Gilson 215, присоединенный к системе препаративной ВЭЖХ Shimadzu и автодозатору Leap. Соединения собирали из элюируемого потока, используя детектирование ЖХ/МС при детектировании положительных ионов: Элюирование соединений из колонок С-18 (2,0×10 см, элюируемых при 20 мл/мин) осуществляли, используя соответствующий режим линейного градиента в течение 10 минут растворитель (А) 0,05% ТФК/H2O и растворитель (В) 0,035% ТФК/ацетонитрил. Для ввода в системы ВЭЖХ неочищенные образцы растворяли в смесях метанола, ацетонитрила и ДМСО.
Исследование 1Н-ЯМР (ядерного магнитного резонанса) проводили, используя 300 или 400 МГц ЯМР-спектрометры Bruker или Varian.
Соединения по настоящему изобретению можно синтезировать согласно известным методикам. Следующие примеры и ссылки приведены, чтобы помочь понять настоящее изобретение. Однако не предполагается, что примеры ограничивают изобретение, верный объем которого установлен прилагаемыми пунктами формулы изобретения. Названия конечных продуктов в примерах получали при использовании Isis AutoNom 2000.
Промежуточное соединение 1
(Е)-3-Диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрил
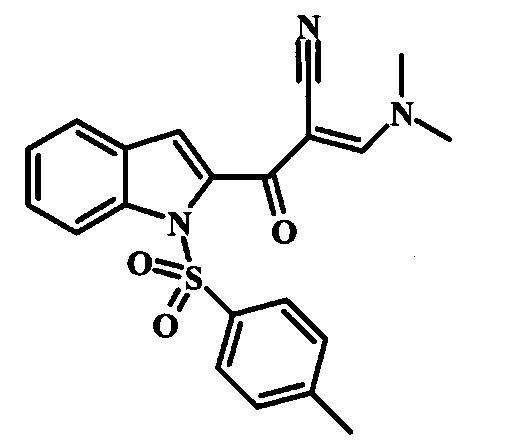
Стадия 1)
Этиловый эфир 1-(толуол-4-сульфонил)-1Н-индол-2-карбоновой кислоты
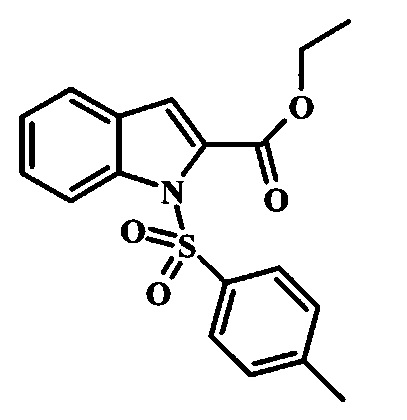
К перемешиваемому раствору этилового эфира 1Н-индол-2-карбоновой кислоты (1 г, 5,29 ммоль) в безводном ТГФ при 0°C добавляли гидрид натрия в 60% масляной дисперсии (0,423 г, 10,58 ммоль) и перемешивали в течение 30 минут. К этой смеси добавляли п-толуолсульфонилхлорид (2 г, 10,58 ммоль) и перемешивали в течение 12 часов. Смесь гасили насыщенным хлоридом аммония, экстрагировали этилацетатом, сушили над безводным сульфатом натрия, выпаривали при пониженном давлении и неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 5% EtOAc/гексаны), получая этиловый эфир 1-(толуол-4-сульфонил)-1Н-индол-2-карбоновой кислоты в виде белого твердого вещества (1,1 г, 61%). МС рассч. для C18H17NO4S [(М+Н)+] 344, наблюд. 344,1.
Стадия 2)
3-Оксо-3-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-пропионитрил
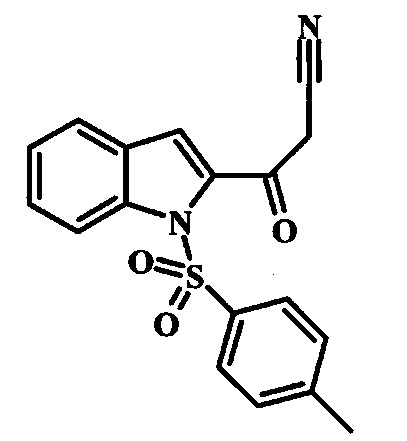
К перемешиваемому раствору этилового эфира 1-(толуол-4-сульфонил)-1Н-индол-2-карбоновой кислоты (8 г, 23,3 ммоль) и ацетонитрила (3,8 мл, 93,3 ммоль) в безводном ТГФ (40 мл) при -78°C добавляли по каплям ЛДА (47,3 мл, 46,64 ммоль) [полученный при добавлении H-BuLi (25,6 мл, 46,64 ммоль) к раствору диизопропиламина (6,7 мл, 46,6 ммоль) в безводном ТГФ (15 мл) при -78°C и перемешивании в течение 30 минут в атмосфере аргона]. Перемешивали в течение 20 минут, гасили хлоридом аммония (10 мл), концентрировали, экстрагировали этилацетатом, сушили над безводным сульфатом натрия, выпаривали при пониженном давлении и очищали с помощью колоночной хроматографии (силикагель, 30% EtOAc/гексаны), получая 3-оксо-3-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-пропионитрил в виде вязкой коричневой жидкости (5,7 г, 72%). МС рассч. для C18H14N2O3S [(М+Н)+] 339, наблюд. 339,0.
Стадия 3)
(Е)-3-Диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрил
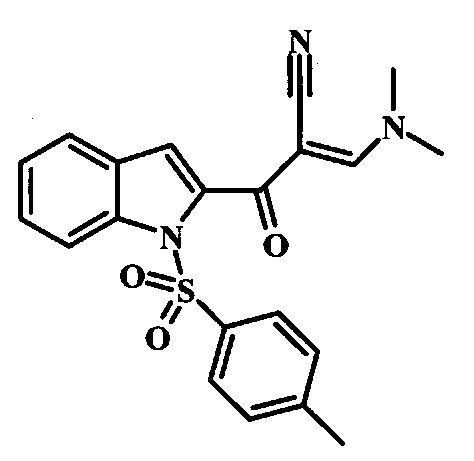
ДМФА-ДМА (0,72 г, 7,09 ммоль) добавляли к перемешиваемому раствору 3-оксо-3-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-пропионитрила (2 г, 5,91 ммоль) в толуоле (30 мл) при к.т. и перемешивали в течение 15 часов. Растворитель выпаривали при пониженном давлении и неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 30% EtOAc/гексаны), получая (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрил в виде желтого твердого вещества (1,2 г, 52%). МС рассч. для C21H19N3O3S [(М+Н)+] 394, наблюд. 394,3.
Промежуточное соединение 2
(Е)-3-Диметиламино-2-[1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-акрилонитрил
Стадия 1)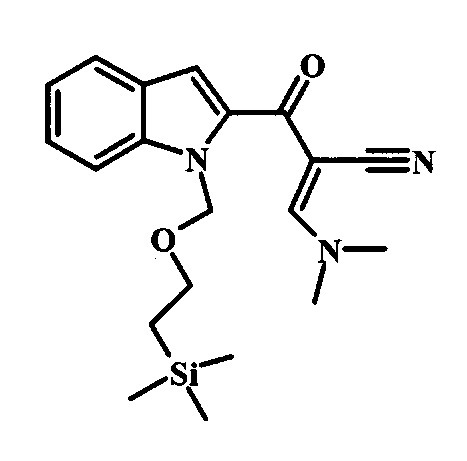
Этиловый эфир 1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбоновой кислоты
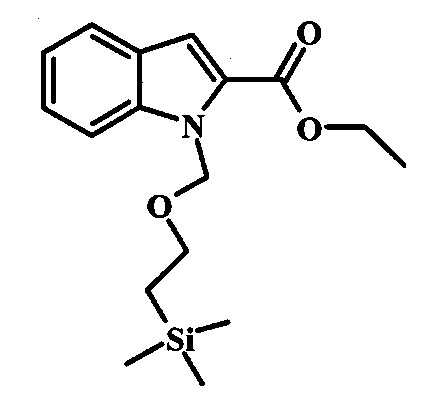
В 250 мл круглодонной колбе объединяли этил 1Н-индол-2-карбоксилат (5 г, 26,4 ммоль), СЭМ-CI (5,29 г, 5,62 мл, 31,7 ммоль) и NaH в 60% масляной дисперсии (1,27 г, 31,7 ммоль) при 4°C с ТГФ (40 мл) и ДМФА (20 мл). Реакционную смесь перемешивали и оставляли нагреваться до комнатной температуры в течение 5 часов. Реакционную смесь гасили МеОН, разбавляли водой и EtOAc. Раствор промывали солевым раствором, объединенные органические фазы сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 40% EtOAc/гексаны), получая этиловый эфир 1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбоновой кислоты (6,8 г, 81%) в виде оранжевого масла. 1Н ЯМР (300 МГц, ДМСО-d6) δ 7,69 (t, J равно 7,63 Гц, 2Н), 7,31-7,41 (m, 2Н), 7,17 (t, J равно 7,50 Гц, 1Н), 5,96 (s, 2Н), 4,32 (q, J равно 7,16 Гц, 2Н), 3,43 (t, J равно 7,82 Гц, 2Н), 1,33 (t, J равно 7,06 Гц, 3Н), 0,76 (t, J равно 7,82 Гц, 2Н), -0,14 (s, 9Н).
Стадия 2)
3-Оксо-3-[1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-ил]-пропионитрил
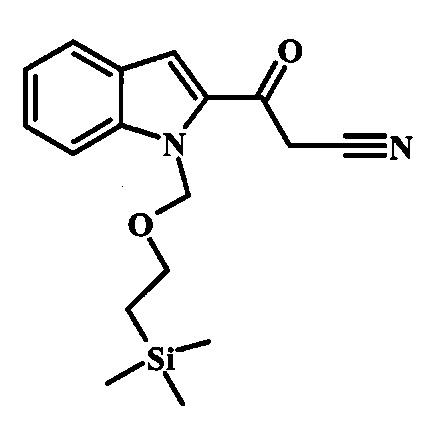
В 500 мл круглодонной колбе объединяли этил 1-((2-(триметилсилил)этокси)метил)-1Н-индол-2-карбоксилат (5 г, 15,7 ммоль), ацетонитрил (3,93 г, 5 мл, 95,7 ммоль) с ТГФ (75 мл) при -78°C в атмосфере азота, получая оранжевый раствор. Добавляли 2 М ЛДА в ТГФ (10 мл, 20,0 ммоль) при -78°C, и реакционную смесь перемешивали и оставляли нагреваться до комнатной температуры. Реакция завершалась через 2 часа 30 минут. Реакционную смесь разбавляли водой и EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 0-40% EtOAc/гексаны), получая 3-оксо-3-[1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-ил]-пропионитрил (3,9 г, 79%) в виде масла. 1Н ЯМР (300 МГц, ДМСО-d6) δ 7,77 (d, J равно 7,91 Гц, 1Н), 7,65-7,72 (m, 2Н), 7,44 (t, J равно 7,63 Гц, 1Н), 7,21 (t, J равно 7,44 Гц, 1Н), 5,93 (s, 2Н), 4,75 (s, 2Н), 1,17 (t, J равно 7,16 Гц, 2Н), 0,78 (t, J равно 7,91 Гц, 2Н), -0,12 (s, 9Н).
Стадия 3)
(Е)-3-Диметиламино-2-[1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-акрилонитрил
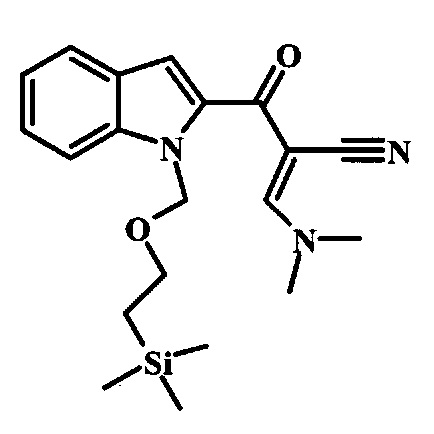
В 250 мл круглодонной колбе объединяли 3-оксо-3-(1-((2-(триметилсилил)этокси)метил)-1Н-индол-2-ил)пропионитрил (3,8 г, 12,1 ммоль) и ДМФА-ДМА (7 мл, 52,3 ммоль) с толуолом (30 мл), получая светло-желтый раствор. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 0-100% EtOAc/гексаны), получая (Е)-3-диметиламино-2-[1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-акрилонитрил (3,5 г, 78%) в виде оранжевой пены. 1Н ЯМР (300 МГц, ДМСО-d6) δ 8,03 (s, 1Н), 7,69 (d, J равно 7,91 Гц, 1Н), 7,63 (d, J равно 8,29 Гц, 1Н), 7,32 (t, J равно 7,25 Гц, 1Н), 7,12-7,19 (m, 2Н), 5,78 (s, 2Н), 3,40 (s, 3Н), 3,31-3,35 (m, 2Н), 3,30 (s, 3Н), 0,75 (t, J равно 8,01 Гц, 2Н), -0,13 (s, 9Н).
Промежуточное соединение 3
(Е)-3-Диметиламино-2-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-карбонил]-акрилонитрил
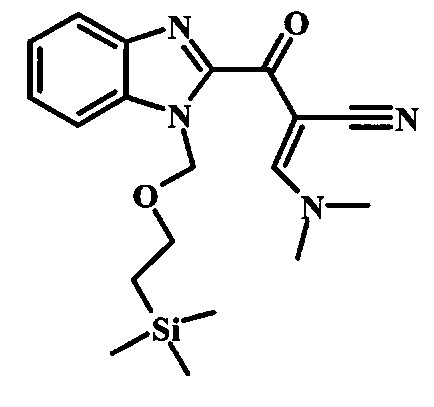
Стадия 1)
Этиловый эфир 1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-карбоновой кислоты
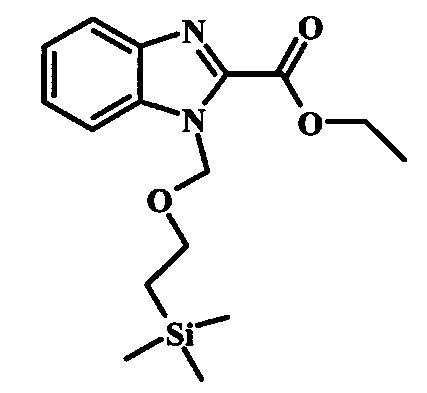
В 250 мл круглодонной колбе объединяли этил 1Н-бензо[d]имидазол-2-карбоксилат (3 г, 15,8 ммоль), СЭМ-CI (3,36 мл, 18,9 ммоль) и NaH в 60% масляной дисперсии (760 мг, 31,7 ммоль) с ТГФ (25 мл) и ДМФА (10 мл), получая белую суспензию. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакцию гасили водой. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 0-40% EtOAc/гексаны), получая этиловый эфир 1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-карбоновой кислоты (2,7 г, 53%) в виде масла. ЖХ/МС: m/z рассчитанная для C16H24N2O3Si([М+Н]+): 321,4. Найденная: 321,0.
Стадия 2)
3-Оксо-3-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-ил]-пропионитрил
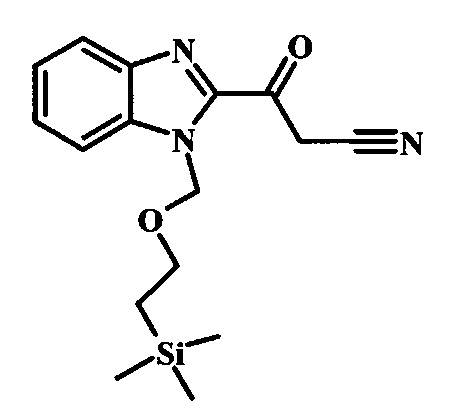
В 100 мл круглодонной колбе объединяли этил 1-((2-(триметилсилил)этокси)метил)-1Н-бензо[d]имидазол-2-карбоксилат (2 г, 6,24 ммоль) и ацетонитрил (2,56 г, 3,26 мл, 62,4 ммоль) с ТГФ (40 мл) при -78°C в атмосфере азота, получая светло-коричневый раствор. Добавляли 2 М ЛДА в ТГФ (5 мл, 10,0 ммоль) при -78°C, и реакционную смесь перемешивали и оставляли нагреваться до комнатной температуры. Реакционную смесь перемешивали при этой температуре в течение 4 часов, затем реакционную смесь выливали в раствор насыщ. водного NH4Cl и экстрагировали EtOAc. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 0-40% EtOAc/гексаны, получая 3-оксо-3-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-ил]-пропионитрил (0,6 г, 31%) в виде масла. 1Н ЯМР (300 МГц, ДМСО-d6) δ 7,88 (d, J равно 8,10 Гц, 1Н), 7,78-7,84 (m, 1Н), 7,49-7,57 (m, 1Н), 7,38-7,47 (m, 1Н), 5,87-6,09 (m, 2Н), 4,86 (s, 2Н), 3,41-3,71 (m, 2Н), 0,68-0,94 (m, 2Н), -0,1 (s, 9Н).
Стадия 3)
(Е)-3-Диметиламино-2-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-карбонил]-акрилонитрил
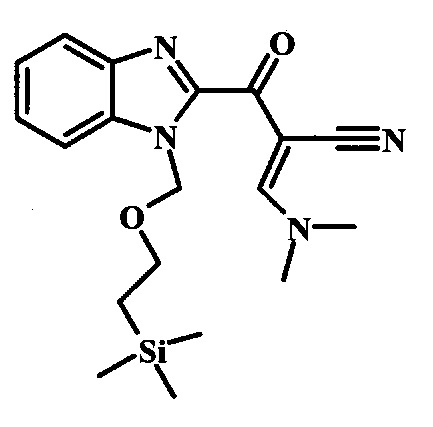
В 20 мл сцинтилляционном флаконе объединяли 3-оксо-3-(1-((2-(триметилсилил)этокси)метил)-1Н-бензо[d]имидазол-2-ил)пропионитрил (500 мг, 1,59 ммоль) и ДМФА-ДМА (0,85 мл, 6,34 ммоль) с толуолом (5 мл), получая желтый раствор. Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 0-100% EtOAc/гексаны), получая (Е)-3-диметиламино-2-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-карбонил]-акрилонитрил (0,26 г, 44%) в виде оранжевой пены. ЖХ/МС: m/z рассчитанная для C19H26N4O2Si ([М+Н]+): 371,5. Найденная: 371,0.
Общая методика A: Нуклеофильное ароматическое замещение
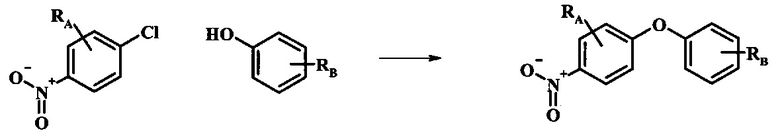
Cs2CO3 (1,5 эквивалента) добавляли к перемешиваемому раствору нитросоединения (1 эквивалент) и производных фенола или алкилового спирта (1,2 эквивалента) в безводном ДМФА. Смесь нагревали в закрытой пробирке при 150°C в течение 24 часов. Реакционную смесь фильтровали, и фильтрат выпаривали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, получая продукт.
Общая методика B: Восстановление нитрогруппы

10% Палладий на угле (10% по массе) добавляли к перемешиваемому раствору нитросоединения в этаноле в атмосфере азота. Реакционную смесь перемешивали в атмосфере водорода в течение 12 часов, фильтровали через воронку с фильтром из спеченного стекла и выпаривали при пониженном давлении, получая соответствующий амин.
Общая методика С-1: Получение арилгидразинов
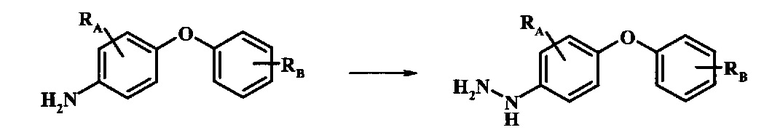
Раствор нитрита натрия (1,5 эквивалента) в воде добавляли к перемешиваемому раствору аминоароматического соединения (1 эквивалент) в хлористоводородной кислоте при -5°C и смесь перемешивали при -5°C в течение 45 минут. Раствор хлорида олова(II) (5 эквивалентов) в хлористоводородной кислоте добавляли, и смесь перемешивали в течение 30 минут. Смесь подщелачивали, добавляя водный раствор NaOH, и смесь экстрагировали EtOAc. Органический экстракт сушили (Na2SO4), фильтровали и выпаривали, получая продукт, который непосредственно использовали на следующей стадии.
Общая методика С-2: Получение арилгидразинов
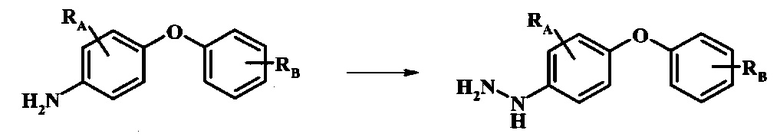
Раствор нитрита натрия (1,5 эквивалента) в воде добавляли к перемешиваемому раствору аминоароматического соединения (1 эквивалент) в хлористоводородной кислоте при -5°C и смесь перемешивали при -5°C в течение 45 минут. Раствор хлорида олова (II) (5 эквивалентов) в хлористоводородной кислоте добавляли, и смесь перемешивали в течение 30 минут. Смесь фильтровали и сушили на воздухе, получая продукт, который непосредственно использовали на следующей стадии.
Общая методика D: Образование кольца пиразола
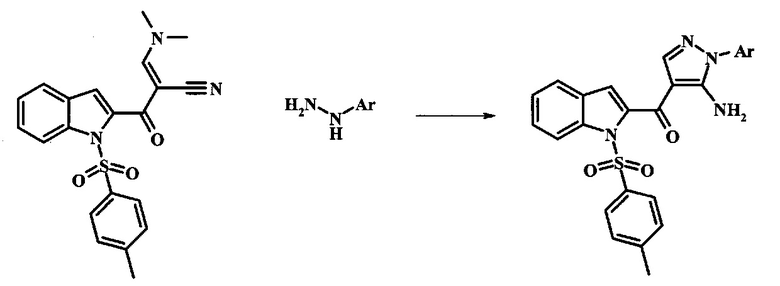
Смесь арилгидразина формулы Ar-NH-NH2 (2 эквивалента) и (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрила (который может быть получен, как описано для промежуточного соединения 1 на стадии 3; 1 эквивалент) в EtOH нагревали с обратным холодильником в течение 16 часов. Растворитель выпаривали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, получая продукт.
Общая методика Е: Удаление защитной группы Ts
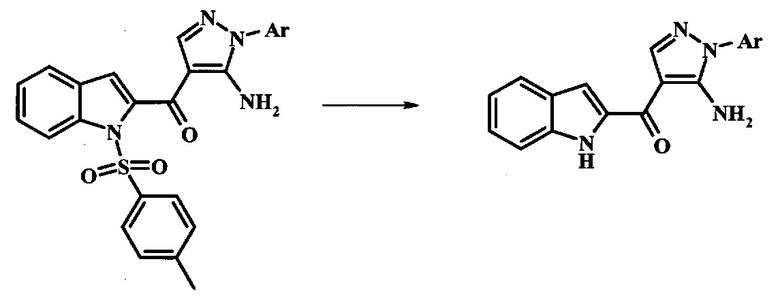
К перемешиваемому раствору Ts-защищенного индола (1 эквивалент) в ТГФ:МеОН (7:3) добавляли карбонат цезия (2 эквивалента). Смесь перемешивали при комнатной температуре в течение 18 часов. Растворитель выпаривали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле, получая продукт.
Пример 1
[5-Амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
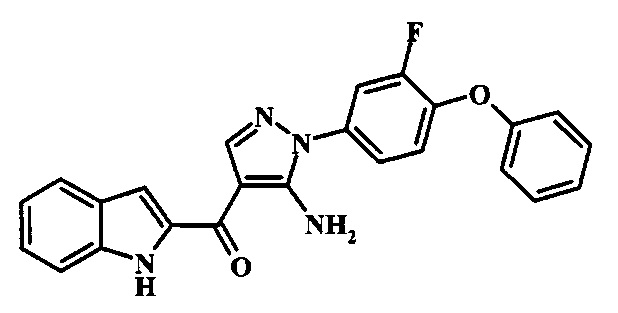
Стадия 1)
2-Фтор-4-нитро-1-фенокси-бензол
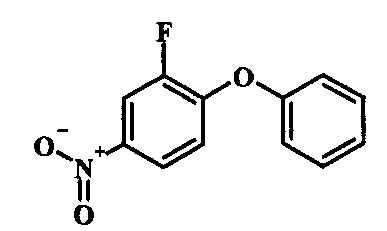
1-Бром-2-фтор-4-нитробензол подвергали взаимодействию с фенолом, используя условия, изложенные в общей методике A, получая 2-фтор-4-нитро-1-фенокси-бензол.
Стадия 2)
3-Фтор-4-фенокси-фениламин
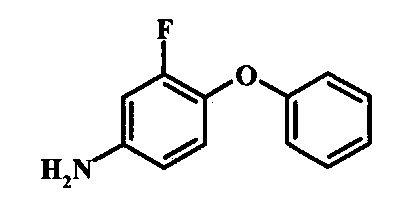
2-Фтор-4-нитро-1-фенокси-бензол восстанавливали, используя условия, изложенные в общей методике B, получая 3-фтор-4-фенокси-фениламин. МС рассч. для C12H11FNO [(М+Н)+] 204, наблюд. 204,2.
Стадия 3)
(3-Фтор-4-фенокси-фенил)-гидразин
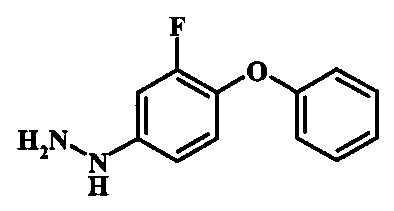
3-Фтор-4-фенокси-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая (3-фтор-4-фенокси-фенил)-гидразин.
Стадия 4)
[5-Амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
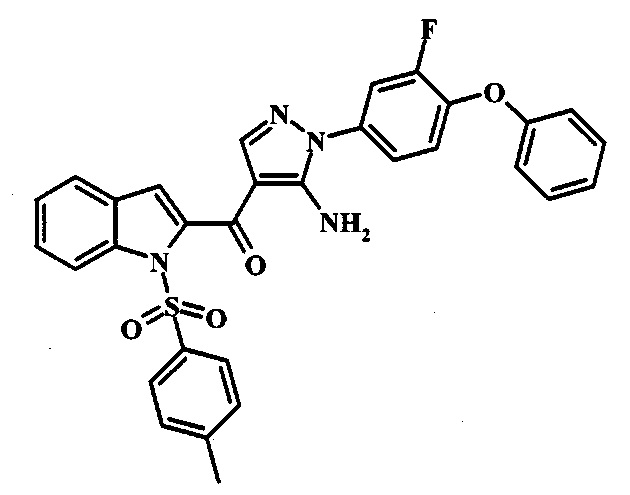
(3-Фтор-4-фенокси-фенил)-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая [5-амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C31H23FN4O4S [(М+Н)+] 567, наблюд. 567,1.
Стадия 5)
[5-Амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
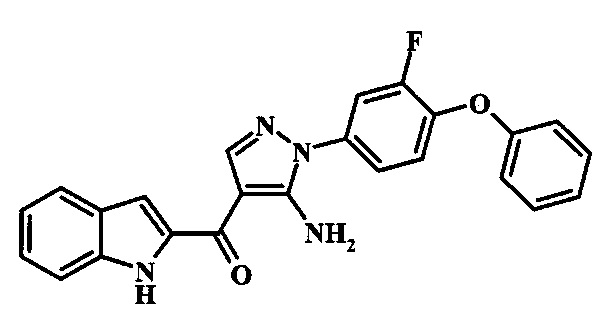
[5-Амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C24H17FN4O2 [(М+Н)+] 413, наблюд. 413,1.
Пример 2
[5-Амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
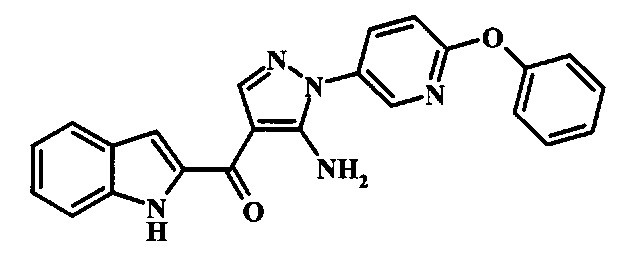
Стадия 1)
5-Нитро-2-фенокси-пиридин
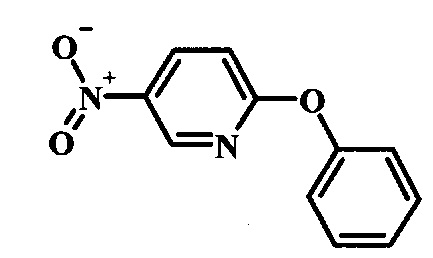
2-Хпор-5-нитро-пиридин подвергали взаимодействию с фенолом, используя условия, изложенные в общей методике A, получая 5-нитро-2-фенокси-пиридин.
Стадия 2)
6-Фенокси-пиридин-3-иламин
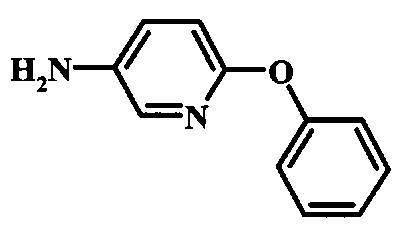
5-Нитро-2-фенокси-пиридин восстанавливали, используя условия, изложенные в общей методике В, получая 6-фенокси-пиридин-3-иламин. МС рассч. для C11H10N2O [(М+Н)+] 187, наблюд. 187,1.
Стадия 3)
(6-Фенокси-пиридин-3-ил)-гидразин
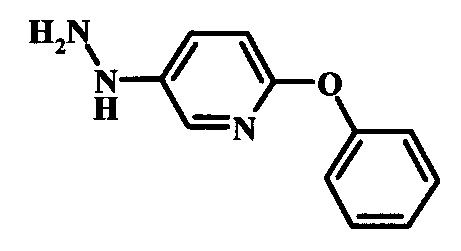
6-Фенокси-пиридин-3-иламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая (6-фенокси-пиридин-3-ил)-гидразин.
Стадия 4)
[5-Амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
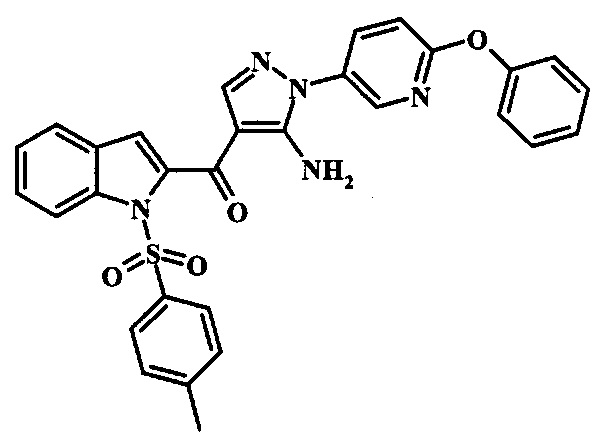
(6-Фенокси-пиридин-3-ил)-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая [5-амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C30H23N5O4S [(М+Н)+] 550, наблюд. 550,2.
Стадия 5)
[5-Амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
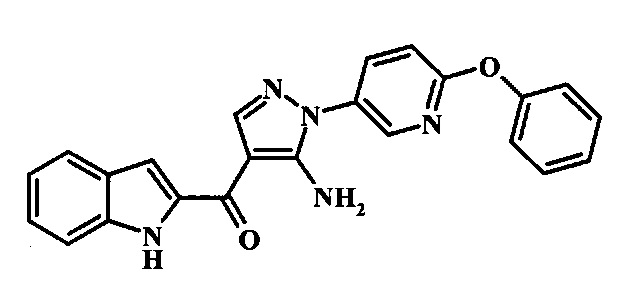
[5-Амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C23H17N5O2S [(М+Н)+] 396, наблюд. 396,1.
Пример 3
{5-Амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
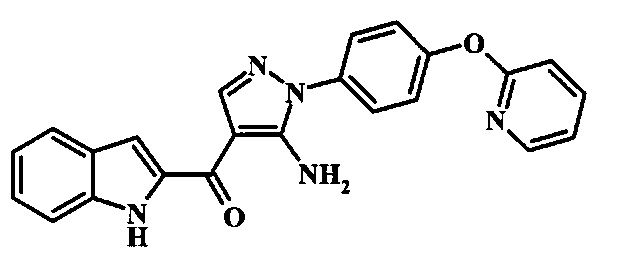
Стадия 1)
2-(4-Нитро-фенокси)-пиридин
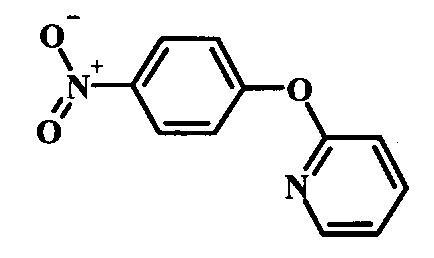
1-Хлор-4-нитро-бензол подвергали взаимодействию с пиридин-2-олом, используя условия, изложенные в общей методике А, получая 2-(4-нитро-фенокси)-пиридин. МС рассч. для C11H8N2O3 [(М+Н)+] 217, наблюд. 217,1.
Стадия 2)
4-(Пиридин-2-илокси)-фениламин
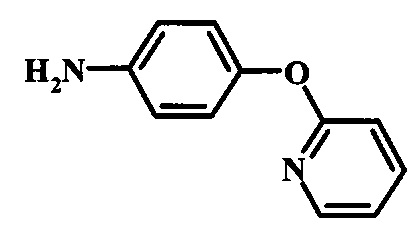
2-(4-Нитро-фенокси)-пиридин восстанавливали, используя условия, изложенные в общей методике В, получая 4-(пиридин-2-илокси)-фениламин. МС рассч. для C11H10N2O [(М+Н)+] 187, наблюд. 187,3.
Стадия 3)
[4-(Пиридин-2-илокси)-фенил]-гидразин
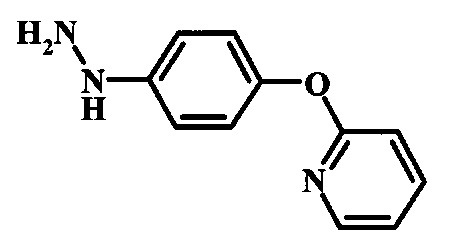
4-(Пиридин-2-илокси)-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая [4-(пиридин-2-илокси)-фенил]-гидразин.
Стадия 4)
{5-Амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
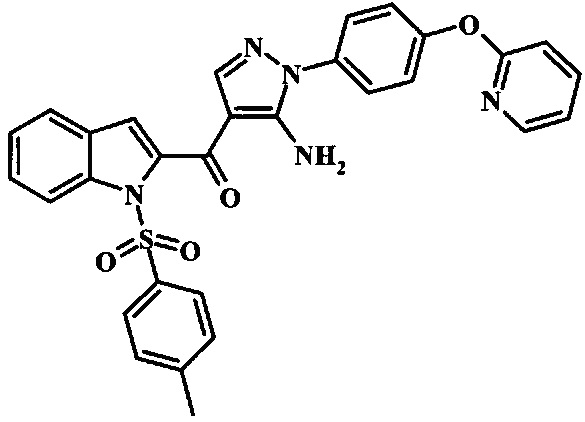
[4-(Пиридин-2-илокси)-фенил]-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая {5-амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C30H23N5O4S [(М+Н)+] 550, наблюд. 550,0.
Стадия 5)
{5-Амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
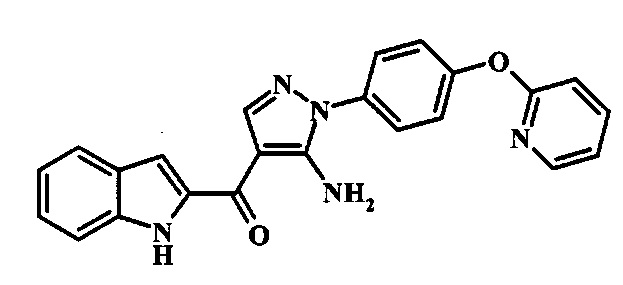
{5-Амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая {5-амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон. МС рассч. для C23H17N5O2S [(М+Н)+] 396, наблюд. 395,9.
Пример 4
{5-Амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
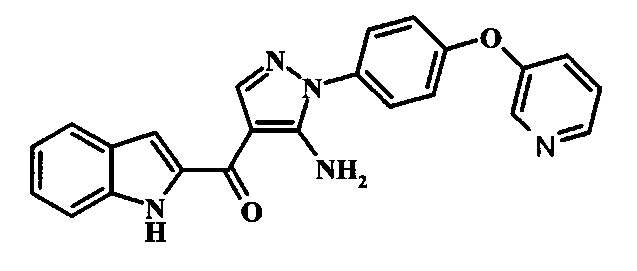
Стадия 1)
3-(4-Нитро-фенокси)-пиридин
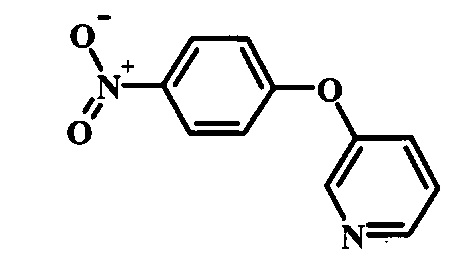
1-Хлор-4-нитро-бензол подвергали взаимодействию с пиридин-3-олом, используя условия, изложенные в общей методике А, получая 3-(4-нитро-фенокси)-пиридин. МС рассч. для C11H8N2O3 [(М+Н)+] 217, наблюд. 217,2.
Стадия 2)
4-(Пиридин-3-илокси)-фениламин
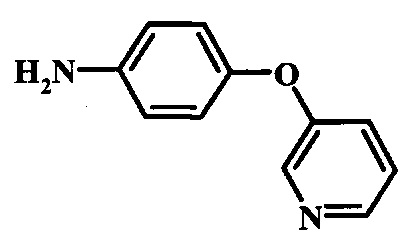
3-(4-Нитро-фенокси)-пиридин восстанавливали, используя условия, изложенные в общей методике В, получая 4-(пиридин-3-илокси)-фениламин. МС рассч. для C11H10N2O [(М+Н)+] 187, наблюд. 187,4.
Стадия 3)
[4-(Пиридин-3-илокси)-фенил]-гидразин
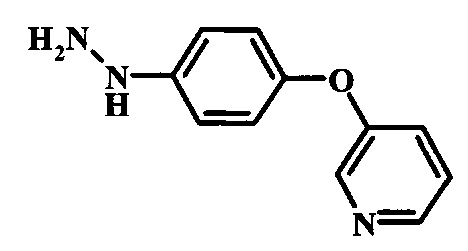
4-(Пиридин-3-илокси)-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая [4-(пиридин-3-илокси)-фенил]-гидразин.
Стадия 4)
{5-Амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
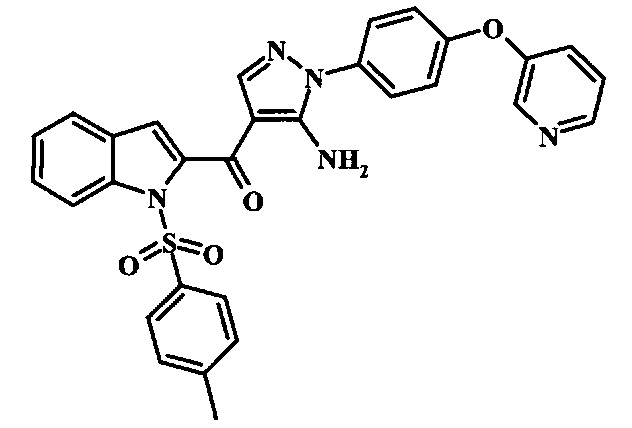
[4-(Пиридин-3-илокси)-фенил]-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая {5-амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C30H23N5O4S [(М+Н)+] 550, наблюд. 550,3.
Стадия 5)
{5-Амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
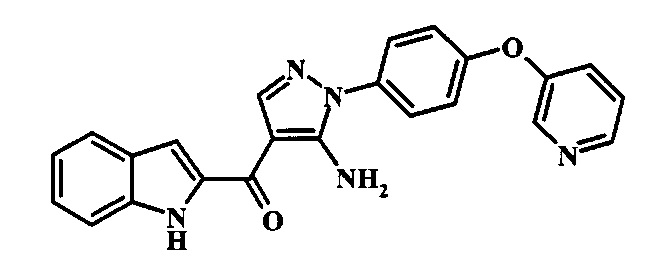
{5-Амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая {5-амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон. МС рассч. для C23H17N5O2S [(М+Н)+] 396, наблюд. 396,4.
Пример 5
5-Амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
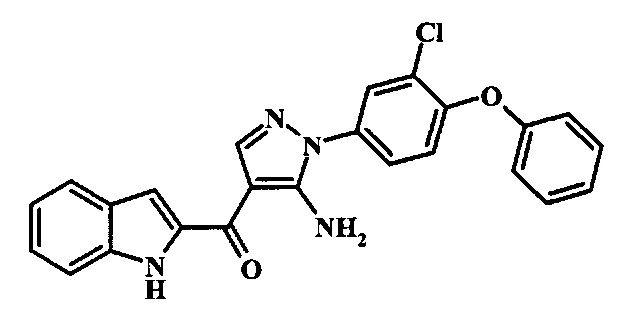
Стадия 1)
2-Хлор-4-нитро-1-фенокси-бензол
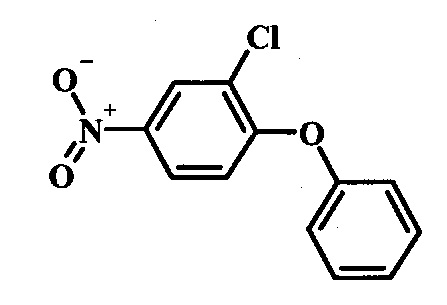
1,2-Дихлор-4-нитро-бензол подвергали взаимодействию с фенолом, используя условия, изложенные в общей методике А, получая 2-хлор-4-нитро-1-фенокси-бензол. МС рассч. для C12H8ClNO3 [(М+Н)+] 250, наблюд. 249,9.
Стадия 2)
3-Хлор-4-фенокси-фениламин
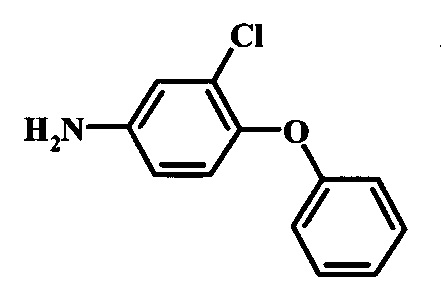
2-Хлор-4-нитро-1-фенокси-бензол восстанавливали, используя условия, изложенные в общей методике B, получая 3-хлор-4-фенокси-фениламин.
Стадия 3)
(3-Хлор-4-фенокси-фенил)-гидразин
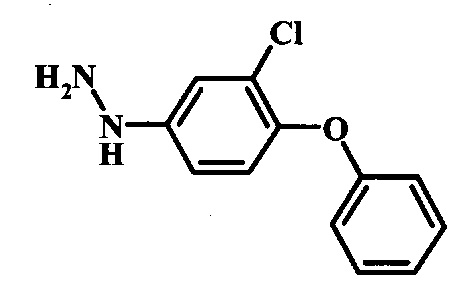
3-Хлор-4-фенокси-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая (3-хлор-4-фенокси-фенил)-гидразин.
Стадия 4)
[5-Амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
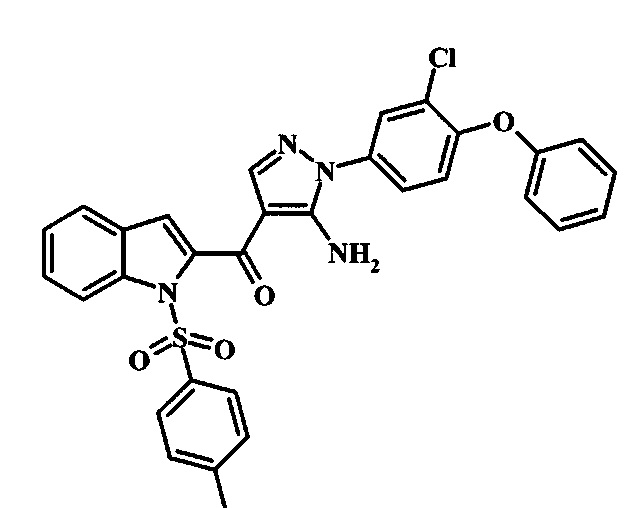
(3-Хлор-4-фенокси-фенил)-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая [5-амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C31H23ClN4O4S [(М+Н)+] 584, наблюд. 585,2.
Стадия 5)
[5-Амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
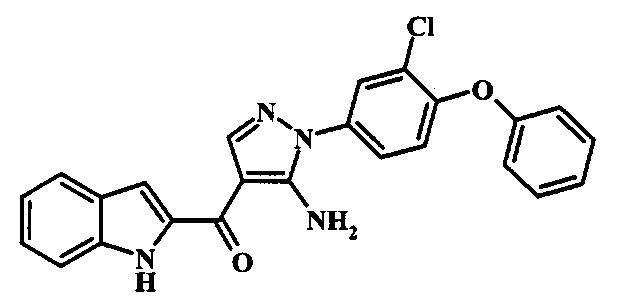
[5-Амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C24H17ClN4O2 [(М+Н)+] 429, наблюд. 429,4.
Пример 6
{5-Амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
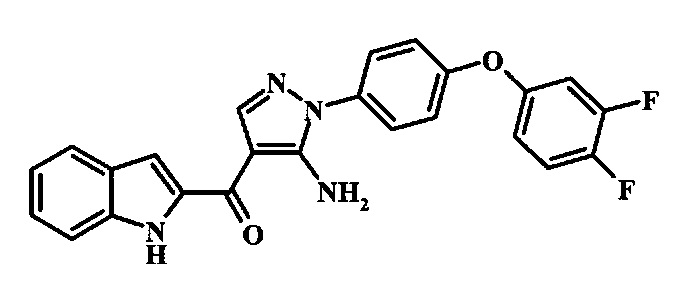
Стадия 1)
3,4-Дифтор-3-(4-нитро-фенокси)-бензол
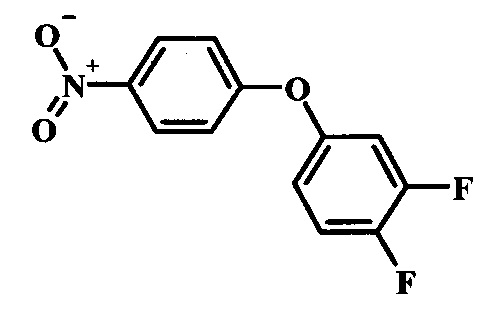
1-Хлор-4-нитро-бензол подвергали взаимодействию с 3,4-дифтор-фенолом, используя условия, изложенные в общей методике А, получая 3,4-дифтор-3-(4-нитро-фенокси)-бензол. МС рассч. для C12H7F2NO3 [(М+Н)+] 252, наблюд. 252,0.
Стадия 2)
4-(3,4-Дифтор-фенокси)-фениламин
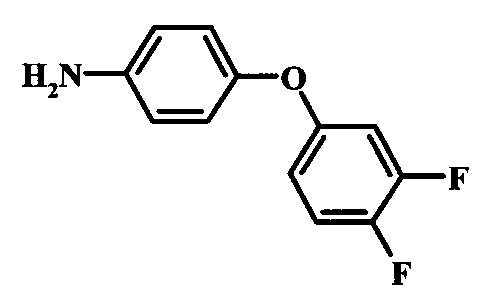
1,2-Дифтор-3-(4-нитро-фенокси)-бензол восстанавливали, используя условия, изложенные в общей методике В, получая 4-(3,4-дифтор-фенокси)-фениламин.
Стадия 3)
[4-(3,4-Дифтор-фенокси)-фенил]-гидразин
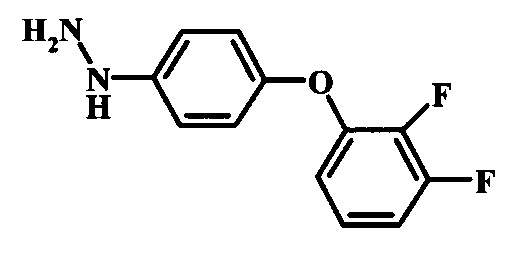
4-(2,3-Дифтор-фенокси)-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая [4-(3,4-дифтор-фенокси)-фенил]-гидразин.
Стадия 4)
{5-Амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
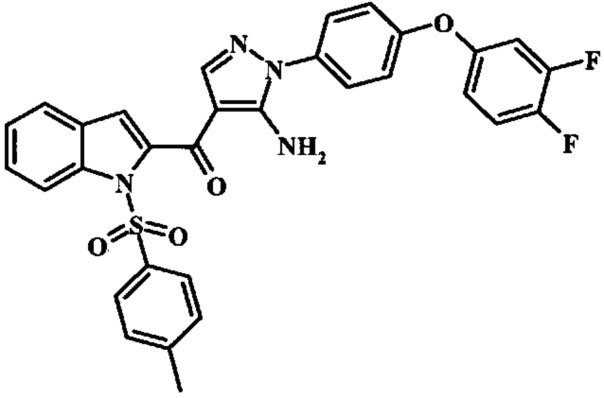
[4-(3,4-Дифтор-фенокси)-фенил]-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая {5-амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон.
Стадия 5)
{5-Амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
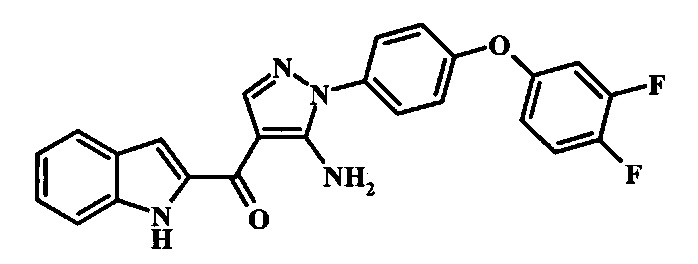
{5-Амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая {5-амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон. МС рассч. для C24H16F2N4O2 [(М+Н)+] 431, наблюд. 431,3.
Пример 7
[5-Амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
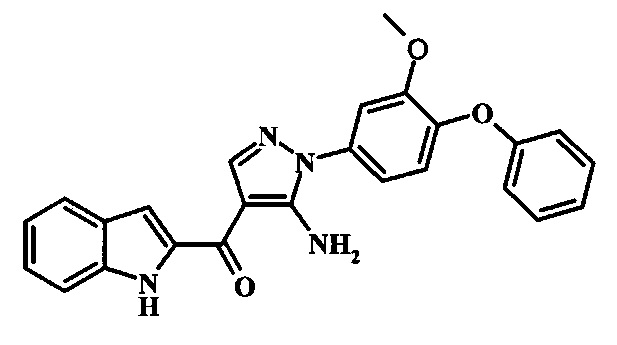
Стадия 1)
2-Метокси-4-нитро-1-фенокси-бензол
1-Хлор-2-метокси-4-нитро-бензол подвергали взаимодействию с фенолом, используя условия, изложенные в общей методике А, получая 2-метокси-4-нитро-1-фенокси-бензол. МС рассч. для C13H11NO4 [(М+Н)+] 246, наблюд. 246,0.
Стадия 2)
3-Метокси-4-фенокси-фениламин
2-Метокси-4-нитро-1-фенокси-бензол восстанавливали, используя условия, изложенные в общей методике B, получая 3-метокси-4-фенокси-фениламин.
Стадия 3)
(3-Метокси-4-фенокси-фенил)-гидразин
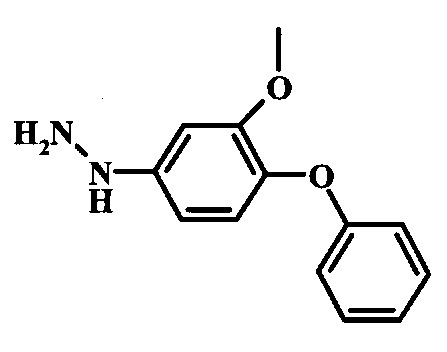
3-Метокси-4-фенокси-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая (3-метокси-4-фенокси-фенил)-гидразин.
Стадия 4)
[5-Амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
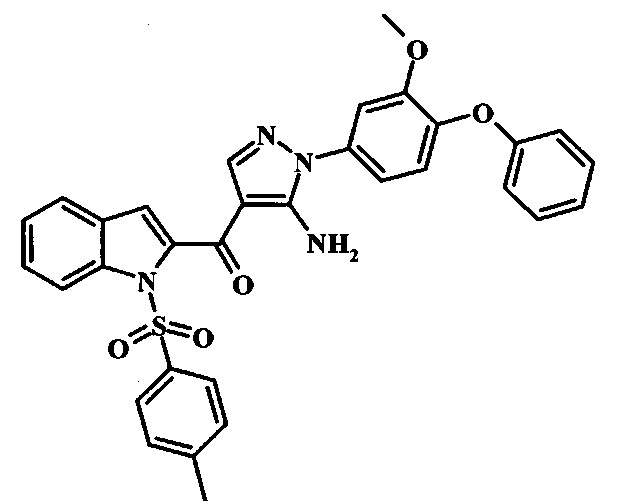
(3-Метокси-4-фенокси-фенил)-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая [5-амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C32H26N4O5S [(М+Н)+] 579, наблюд. 579,1.
Стадия 5)
[5-Амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
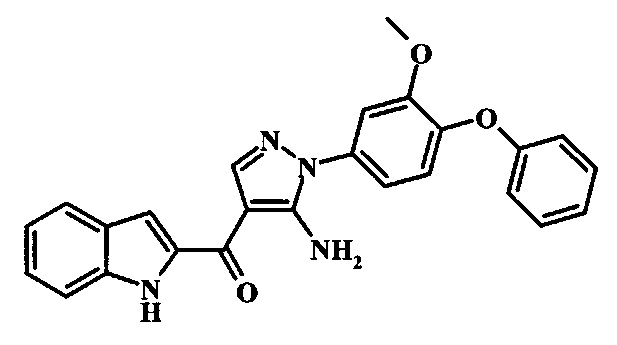
[5-Амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C25H20N4O3 [(М+Н)+] 425, наблюд. 425,3.
Пример 8
[5-Амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
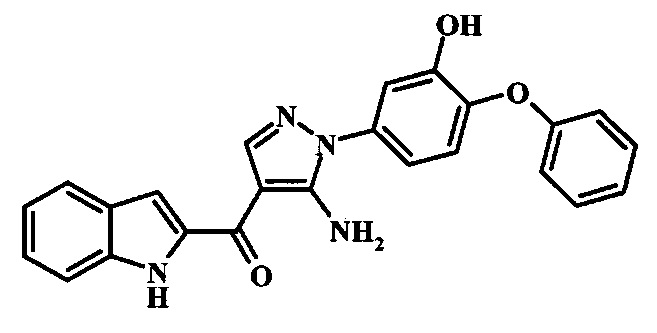
Стадия 1)
[5-Амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
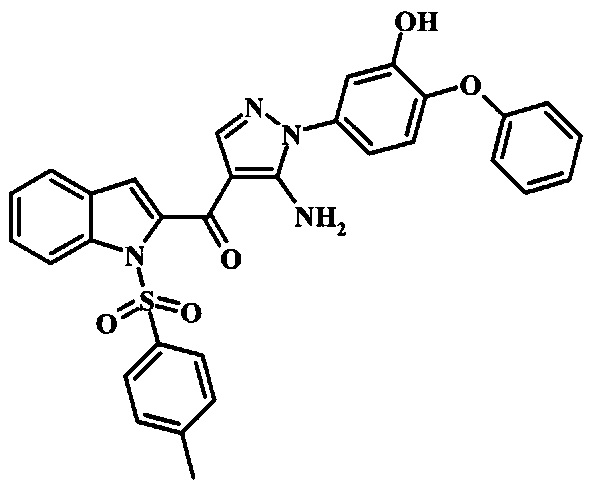
К раствору [5-амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанона (150 мг, 0,296 ммоль) в ДХМ (8 мл) добавляли BBr3 (0,103 мл, 1,04 ммоль) при 0°C и перемешивали в течение 30 минут. ТСХ показала полный расход исходного вещества, затем смесь гасили водным раствором NaOH и экстрагировали ДХМ. Органический слой промывали водой, сушили над безводным сульфатом натрия и концентрировали. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 25% EtOAc/гексаны), получая [5-амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон (100 мг, 68%) в виде желтоватого твердого вещества. МС рассч. для C31H24N4O5S [(М+Н)+] 565, наблюд. 565,3.
Стадия 2)
[5-Амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
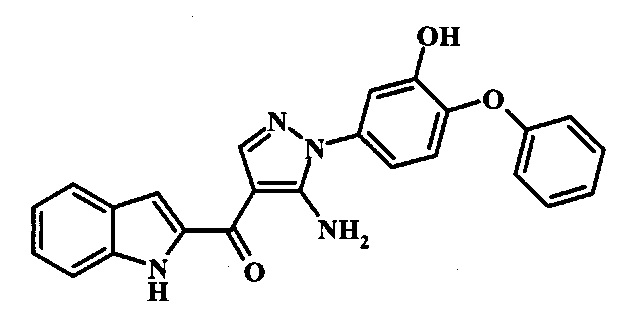
[5-Амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C24H18N4O3 [(М+Н)+] 411, наблюд. 411,2.
Пример 9
[5-Амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
Стадия 1)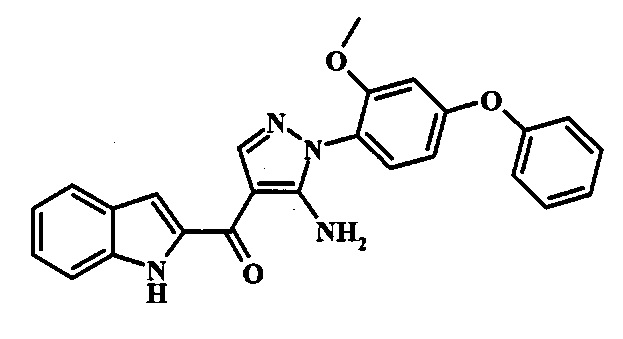
2-Метокси-1-нитро-4-фенокси-бензол
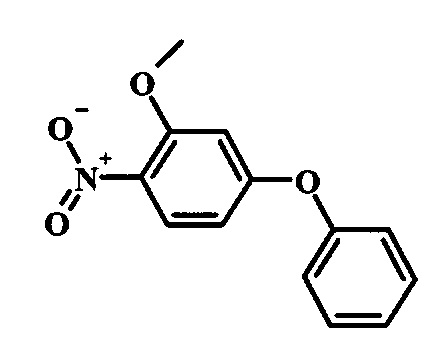
1-Хлор-3-метокси-4-нитро-бензол подвергали взаимодействию с фенолом, используя условия, изложенные в общей методике A, получая 2-метокси-1-нитро-4-фенокси-бензол. МС рассч. для C13H11NO4 [(М+Н)+] 246, наблюд. 246,0.
Стадия 2)
2-Метокси-4-фенокси-фениламин
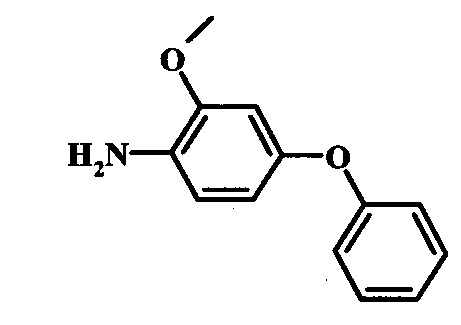
2-Метокси-1-нитро-4-фенокси-бензол восстанавливали, используя условия, изложенные в общей методике B, получая 2-метокси-4-фенокси-фениламин.
Стадия 3)
(2-Метокси-4-фенокси-фенил)-гидразин
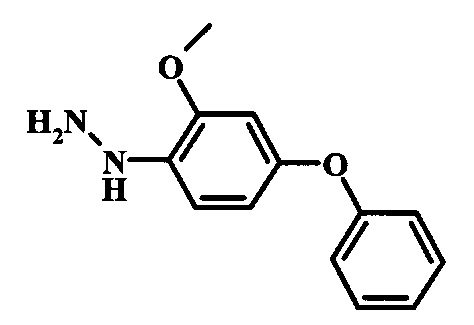
2-Метокси-4-фенокси-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая (2-метокси-4-фенокси-фенил)-гидразин.
Стадия 4)
[5-Амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
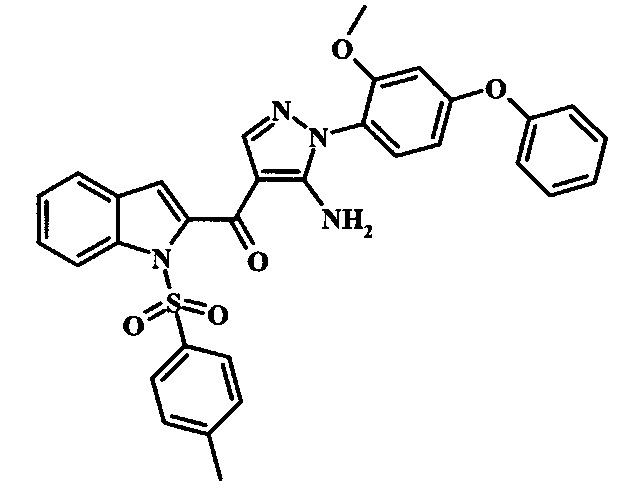
(2-Метокси-4-фенокси-фенил)-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая [5-амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C32H26N4O5S [(М+Н)+] 579, наблюд. 579,1.
Стадия 5)
[5-Амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
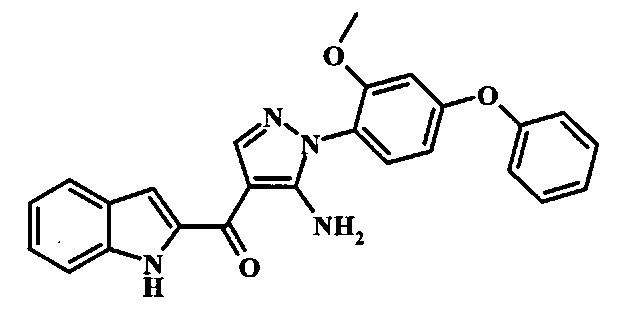
[5-Амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C25H20N4O3 [(М+Н)+] 425, наблюд. 425,3.
Пример 10
[5-Амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
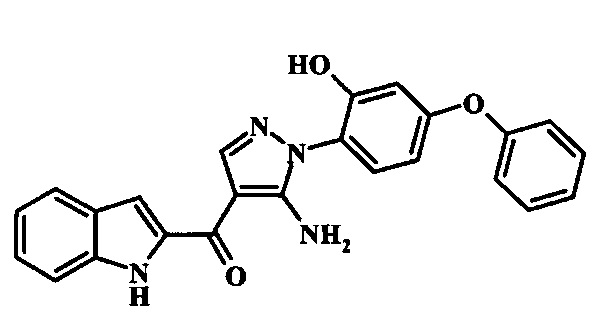
Стадия 1)
[5-Амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
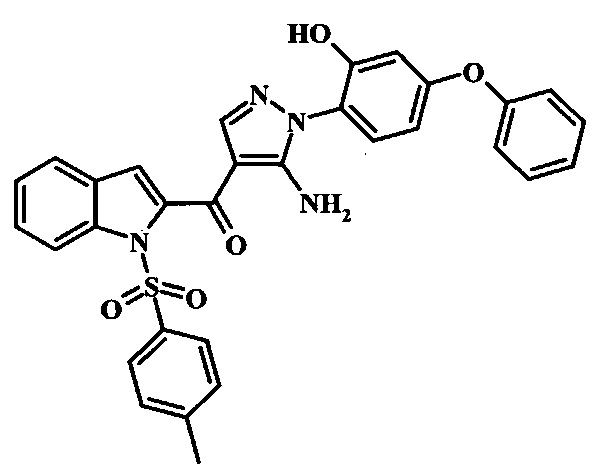
К раствору [5-амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанона (150 мг, 0,296 ммоль) в ДХМ (8 мл) добавляли BBr3 (0,103 мл, 1,04 ммоль) при 0°C и перемешивали в течение 30 минут. ТСХ показала полный расход исходного вещества, затем смесь гасили водным раствором NaOH и экстрагировали ДХМ. Органический слой промывали водой, сушили над безводным сульфатом натрия и концентрировали. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 25% EtOAc/гексаны), получая [5-амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон (100 мг, 68%) в виде желтоватого твердого вещества. МС рассч. для C31H24N4O5S [(М+Н)+] 565, наблюд. 565,3.
Стадия 2)
[5-Амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
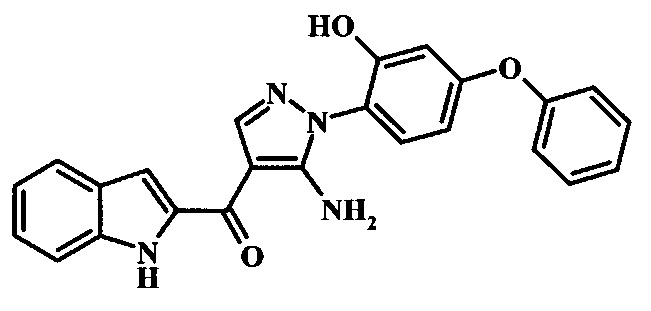
[5-Амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C24H18N4O3 [(М+Н)+] 411, наблюд. 411,3.
Пример 11
[5-Амино.-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
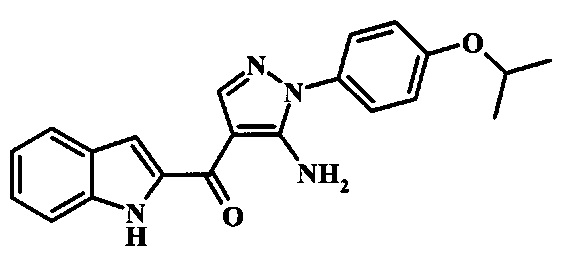
Стадия 1)
1-Изопропокси-4-нитро-бензол
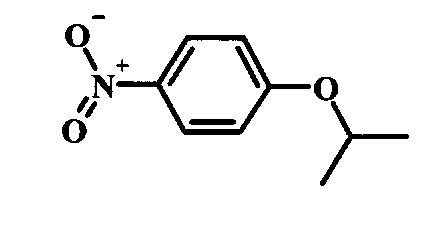
1-Хлор-4-нитро-бензол подвергали взаимодействию с изопропанолом, используя условия, изложенные в общей методике А, получая 1-изопропокси-4-нитро-бензол. МС рассч. для C9H11NO3 [(М+Н)+] 182, наблюд. 182,2.
Стадия 2)
4-Изопропокси-фениламин
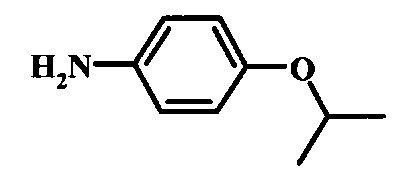
1-Изопропокси-4-нитро-бензол восстанавливали, используя условия, изложенные в общей методике B, получая 4-изопропокси-фениламин.
Стадия 3)
(4-Изопропокси-фенил)-гидразин
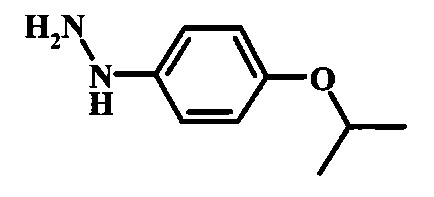
4-Изопропокси-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая (4-изопропокси-фенил)-гидразин.
Стадия 4)
[5-Амино-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
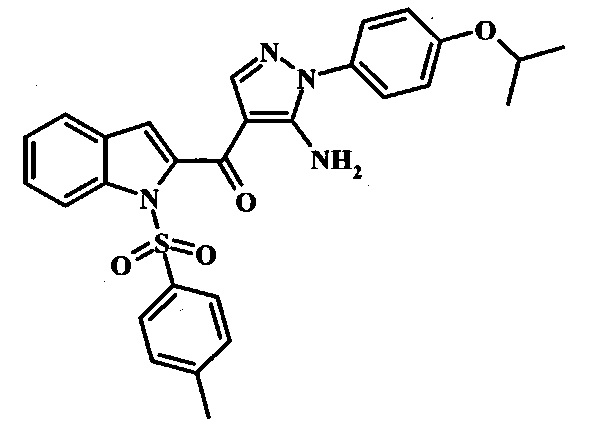
(4-Изопропокси-фенил)-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая [5-амино-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C28H26N4O4S [(М+Н)+] 515, наблюд. 515,2.
Стадия 5)
[5-Амино-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
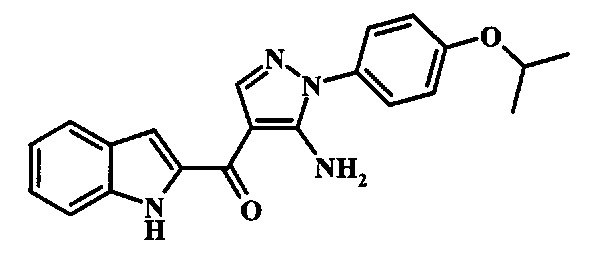
[5-Амино-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C21H20N4O2 [(М+Н)+] 361, наблюд. 361,2.
Пример 12
[5-Амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
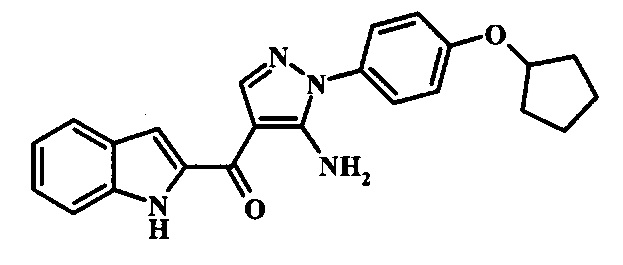
Стадия 1)
1-Циклопентилокси-4-нитро-бензол
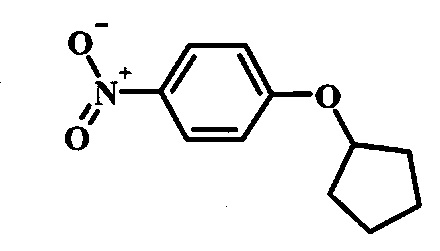
1-Хлор-4-нитро-бензол подвергали взаимодействию с циклопентанолом, используя условия, изложенные в общей методике A, получая 1-циклопентилокси-4-нитро-бензол.
Стадия 2)
4-Циклопентилокси-фениламин
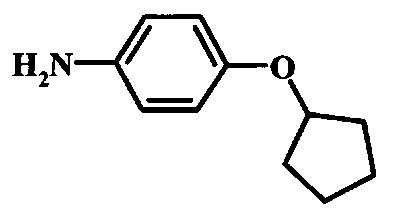
1-Циклопентилокси-4-нитро-бензол восстанавливали, используя условия, изложенные в общей методике B, получая 4-циклопентилокси-фениламин.
Стадия 3)
(4-Циклопентилокси-фенил)-гидразин
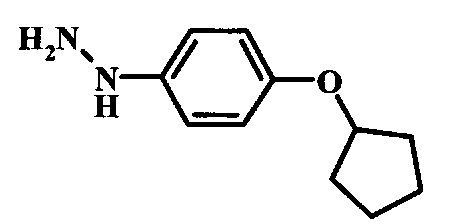
4-Циклопентилокси-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая (4-циклопентилокси-фенил)-гидразин.
Стадия 4)
[5-Амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
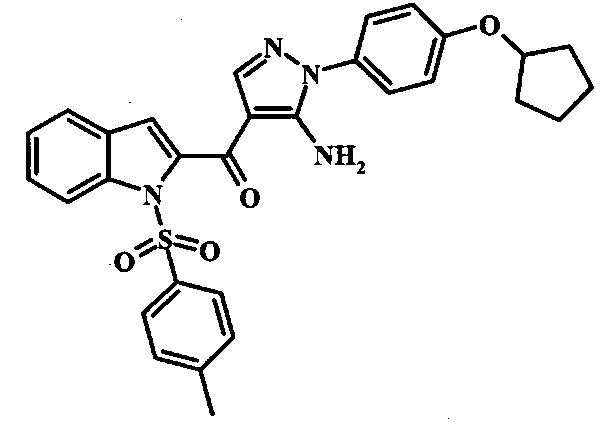
(4-Циклопентилокси-фенил)-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая [5-амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C30H28N4O4S [(М+Н)+] 541, наблюд. 541,3.
Стадия 5)
[5-Амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон
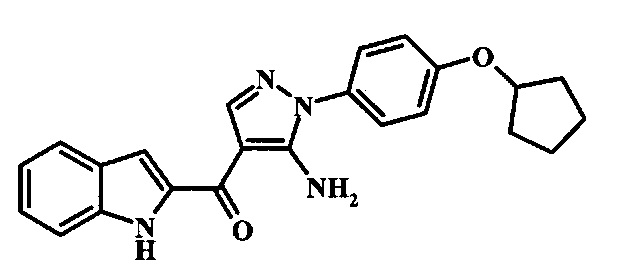
[5-Амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая [5-амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанон. МС рассч. для C23H22N4O2 [(М+Н)+] 387, наблюд. 387,4.
Пример 13
{5-Амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
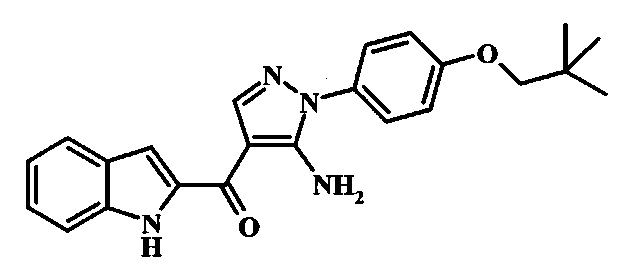
Стадия 1)
1-(2,2-Диметил-пропокси)-4-нитро-бензол
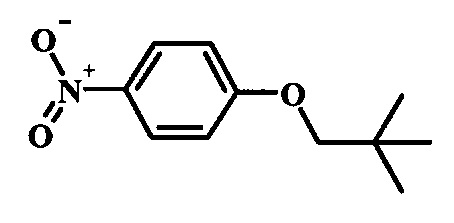
1-Хлор-4-нитро-бензол подвергали взаимодействию с 2,2-диметил-пропан-1-олом, используя условия, изложенные в общей методике A, получая 1-циклопентилокси-4-нитро-бензол. МС рассч. для C11H15NO3 [(М+Н)+] 210, наблюд. 210,2.
Стадия 2)
4-(2,2-Диметил-пропокси)-фениламин
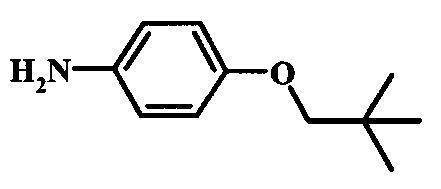
1-(2,2-Диметил-пропокси)-4-нитро-бензол восстанавливали, используя условия, изложенные в общей методике В, получая 4-(2,2-диметил-пропокси)-фениламин.
Стадия 3)
[4-(2,2-Диметил-пропокси)-фенил]-гидразин
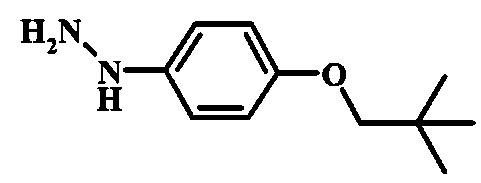
4-(2,2-Диметил-пропокси)-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-1, получая [4-(2,2-диметил-пропокси)-фенил]-гидразин.
Стадия 4)
{5-Амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
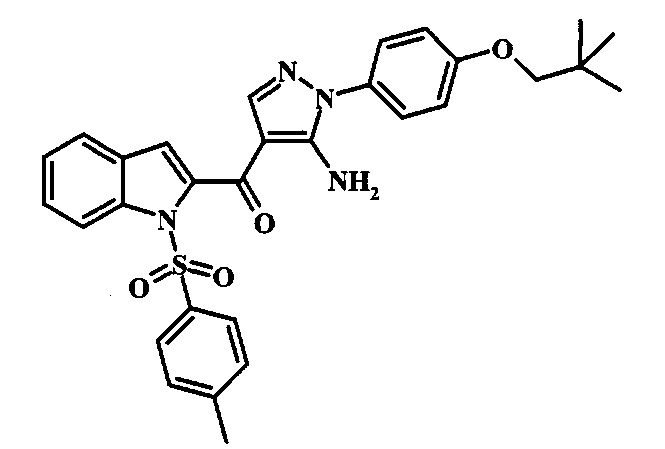
[4-(2,2-Диметил-пропокси)-фенил]-гидразин подвергали взаимодействию с (Е)-3-диметиламино-2-[1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрилом (который может быть получен, как описано для промежуточного соединения 1, стадия 3), используя условия, изложенные в общей методике D, получая {5-амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон. МС рассч. для C30H30N4O4S [(М+Н)+] 543, наблюд. 543,2.
Стадия 5)
{5-Амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
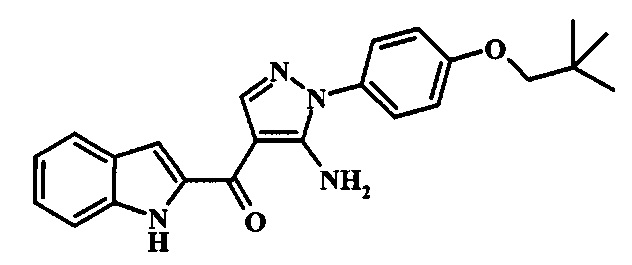
{5-Амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-[1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон подвергали взаимодействию с карбонатом цезия, используя условия, изложенные в общей методике Е, получая {5-амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон. МС рассч. для C23H24N4O2 [(М+Н)+] 389, наблюд. 389,2.
Пример 14
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
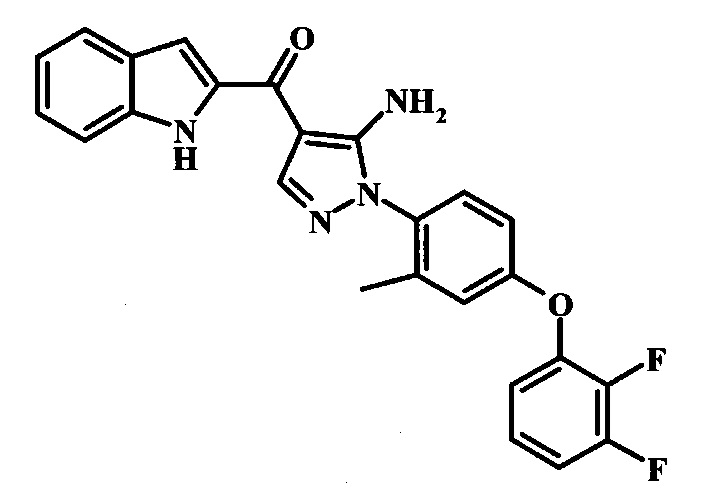
Стадия 1: 2-Метил-1-нитро-4-(2,3-дифтор)-фенокси-бензол
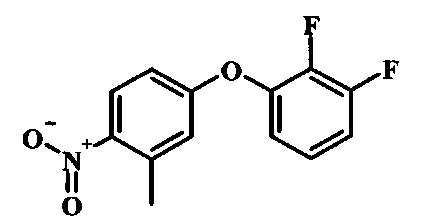
4-Хлор-2-метил-нитробензол подвергали взаимодействию с 2,3-дифтор-фенолом, используя условия, изложенные в общей методике А, получая 2-метил-1-нитро-4-(2,3-дифтор)-фенокси-бензол. МС рассч. для C13H10F2NO3 [(М+Н)+] 266, наблюд. 266,2.
Стадия 2: 4-(2,3-Дифтор-фенокси)-2-метил-фениламин
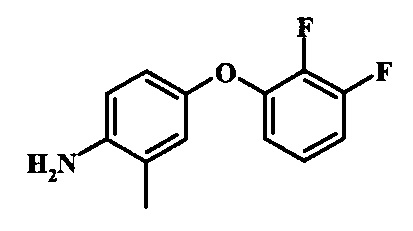
2-Метил-1-нитро-4-(2,3-дифтор)-фенокси-бензол восстанавливали, используя условия, изложенные в общей методике B, получая 4-(2,3-дифтор-фенокси)-2-метил-фениламин. МС рассч. для C13H12F2NO [(М+Н)+] 236, наблюд. 235,8.
Стадия 3: [4-(2,3-Дифтор-фенокси)-2-метил-фенил]-гидразин, гидрохлорид
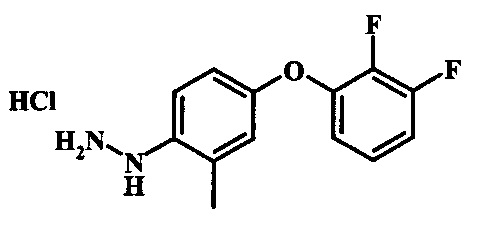
4-(2,3-Дифтор-фенокси)-2-метил-фениламин диазотировали и восстанавливали, используя условия, изложенные в общей методике С-2, получая [4-(2,3-дифтор-фенокси)-2-метил-фенил]-гидразина гидрохлорид.
Стадия 4)
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-этоксиметил-1Н-индол-2-ил)-метанон
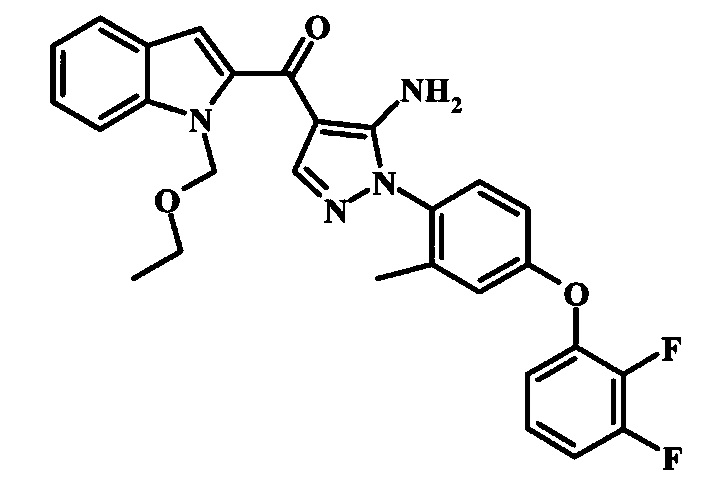
В 20 мл сцинтилляционном флаконе объединяли (Е)-3-(диметиламино)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-индол-2-карбонил)акрилонитрил (промежуточное соединение 2, стадия 3) (200 мг, 0,541 ммоль), (4-(2,3-дифторфенокси)-2-метилфенил)гидразин (271 мг, 1,08 ммоль) и карбонат калия (224 мг, 1,62 ммоль) с EtOH (4 мл), получая желтую суспензию. Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, EtOAc/гексаны от 0 до 100%), получая {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1-этоксиметил-1Н-индол-2-ил)-метанон (165 мг, 79%) в виде масла. ЖХ/МС: m/z рассчитанная для C28H24F2N4O3([M+H]+): 503,5. Найденная: 503,1.
Стадия 5)
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон
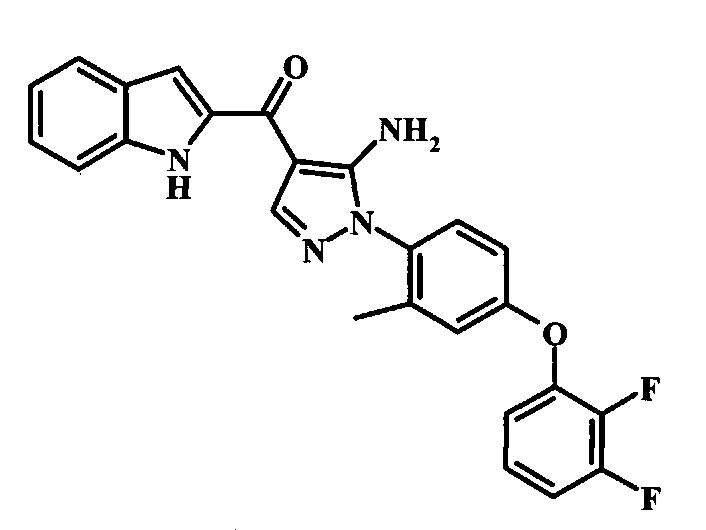
В 20 мл сцинтилляционном флаконе объединяли (5-амино-1-(4-(2,3-дифторфенокси)-2-метилфенил)-1Н-пиразол-4-ил)(1-(этоксиметил)-1Н-индол-2-ил)метанон (100 мг, 0,199 ммоль) с EtOH (4 мл) и водн. 10% HCl (2 мл). Реакционную смесь нагревали при 80°C в течение 2 часов. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 70% EtOAc-гексаны), получая {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанон (36 мг, 41%) в виде масла. ЖХ/МС: m/z рассчитанная для C25H18F2N4O2 ([M+H]+): 445,4. Найденная: 444,9.
Пример 15
2-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил
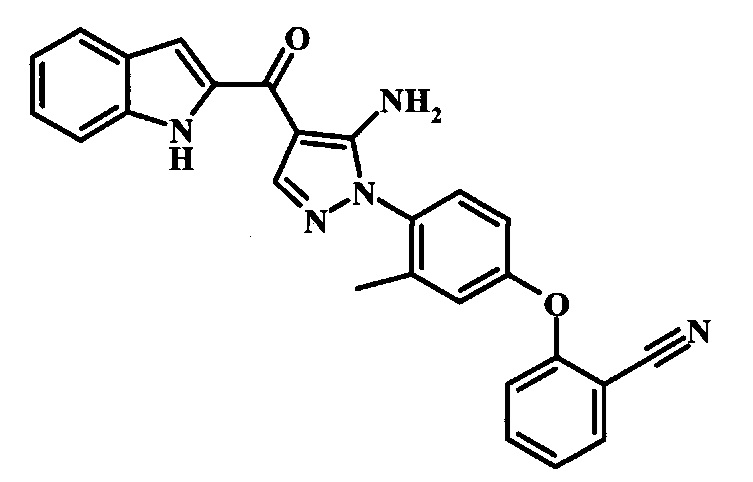
Стадия 1)
2-(3-Метил-4-нитро-фенокси)-бензонитрил
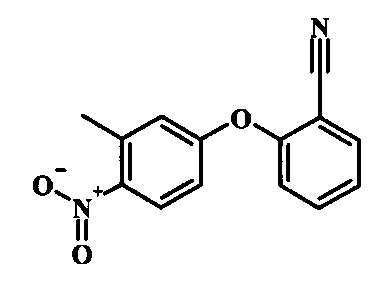
Смесь 4-фтор-2-метил-1-нитро-бензола (13,1 г, 84 ммоль), 2-гидроксибензонитрила (10 г, 84 ммоль) и K2CO3 (23,5 г, 168 ммоль) в ацетоне (100 мл) нагревали при 70°C в течение ночи. Добавляли воду и смесь экстрагировали EtOAc. Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и выпаривали. Добавляли МеОН и смесь оставляли при комнатной температуре на выходные дни. Твердое вещество фильтровали, получая 2-(3-метил-4-нитро-фенокси)-бензонитрил (7,5 г, 35%) двумя порциями в виде желтого твердого вещества. 1Н ЯМР (300 МГц, CDCl3) δ ppm 8,10 (d, J равно 8,9 Гц, 1Н), 7,75 (dd, J равно 7,7, 1,5 Гц, 1Н), 7,56-7,68 (m, 1Н), 7,33 (td, J равно 7,6, 0,9 Гц, 1Н), 7,09 (d, J равно 8,5 Гц, 1Н), 6,88-7,00 (m, 2 Н), 2,64 (s, 3 Н). МС рассч. для C14H11N2O3 [(М+Н)+] 255, наблюд. 255,0.
Стадия 2)
2-(4-Амино-3-метил-фенокси)-бензонитрил
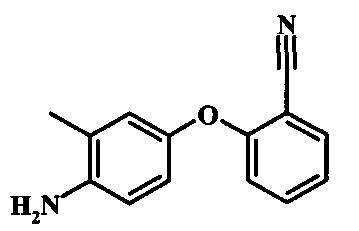
Смесь хлорида олова(II) (18,6 г, 98,3 ммоль), 2-(3-метил-4-нитро-фенокси)-бензонитрила (5 г, 19,7 ммоль), МеОН (60 мл), ТГФ (100 мл) и воды (30 мл) нагревали при 70°C в течение 4 часов. Смесь выпаривали, и остаток делали основным, добавляя 10 н NaOH. Полученную в результате смесь экстрагировали EtOAc. Органический слой сушили (Na2SO4), фильтровали и выпаривали. Остаток очищали с помощью хроматографии (силикагель, от 0 до 30% EtOAc/гексаны), получая 2-(4-амино-3-метил-фенокси)-бензонитрил (3,5 г, 79%) в виде твердого вещества. МС рассч. для C14H13N2O [(М+Н)+] 225, наблюд. 224,9.
Стадия 3)
2-(4-Гидразино-3-метил-фенокси)-бензонитрила гидрохлорид
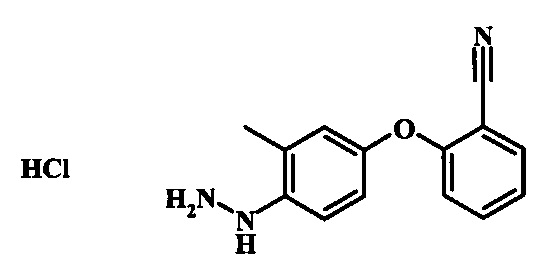
Раствор NaNO2 (1,23 г, 17,8 ммоль) в воде (10 мл) добавляли к смеси 2-(4-амино-3-метил-фенокси)-бензонитрила (2 г, 8,92 ммоль), HCl (10 мл), воды (20 мл) и МеОН (15 мл) при 4°C. Смесь перемешивали при 4°C в течение 45 минут. Раствор хлорида олова(II) (10 г, 44,6 ммоль) в конц. HCl (10 мл) добавляли и смесь перемешивали в течение 4 часов. Смесь фильтровали, получая неочищенный 2-(4-гидразино-3-метил-фенокси)-бензонитрила гидрохлорид (600 мг, 28%) в виде желтой соли. Это вещество использовали на следующей стадии без дополнительной очистки.
Стадия 4)
2-(4-{5-Амино-4-[1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил
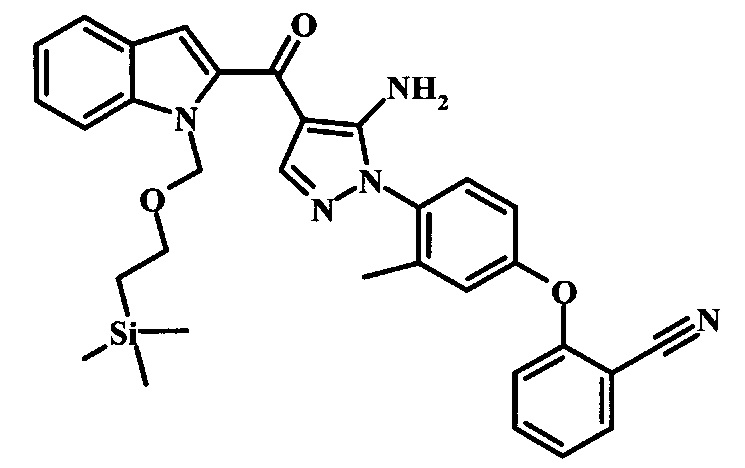
В 20 мл сцинтилляционном флаконе объединяли (Е)-3-(диметиламино)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-индол-2-карбонил)акрилонитрил (промежуточное соединение 2, стадия 3) (200 мг, 0,541 ммоль), 2-(3-гидразинил-2-метилфенокси)бензонитрила гидрохлорид (200 мг, 0,836 ммоль) и карбонат калия (300 мг, 2,17 ммоль) с EtOH (4 мл), получая темно-красную суспензию. Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 70% EtOAc-гексаны), получая 2-(4-{5-амино-4-[1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил (160 мг, 52%) в виде масла. ЖХ/МС: m/z рассчитанная для C32H33N5O3Si ([М+Н]+): 564,7. Найденная: 564,2. Стадия 5)
2-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил
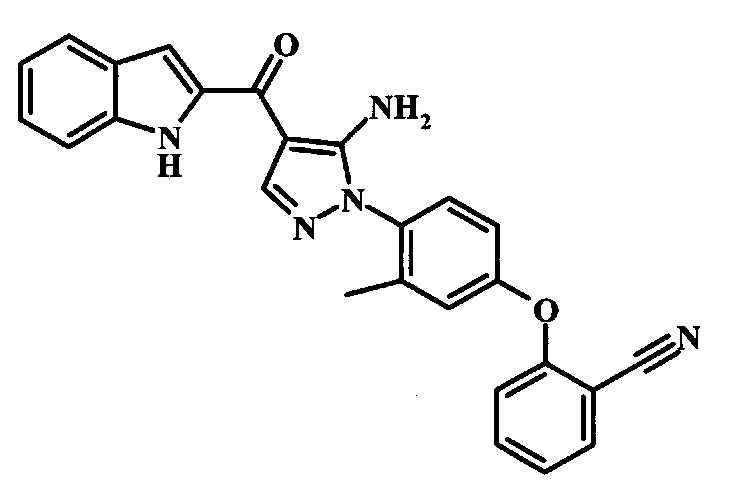
В 20 мл сцинтилляционном флаконе объединяли 2-(4-(5-амино-4-(1-((2-(триметилсилил)этокси)метил)-1Н-индол-2-карбонил)-1Н-пиразол-1-ил)-3-метилфенокси)бензамид (160 мг, 0,275 ммоль, экв.: 1,00), 1 М ТБАФ в ТГФ (5,5 мл, 5,5 ммоль, экв.: 20) и этан-1,2-диамин (165 мг, 2,75 ммоль, экв.: 10). Реакционную смесь нагревали при 70°C в течение ночи. Через 18 часов реакция не завершалась. Добавляли еще 4 мл 1 М ТБАФ в ТГФ и перемешивали при 70°C в течение дополнительных 2 часов. Растворитель удаляли при пониженном давлении. Неочищенное вещество разбавляли EtOAc и промывали насыщенным водным раствором NH4Cl. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 100% EtOAc/гексаны), получая 2-{4-[5-амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрил (50 мг, 42%) в виде твердого вещества. ЖХ/МС: m/z рассчитанная для C26H19N5O2 ([М+Н]+): 434,4. Найденная: 434,0.
Пример 16
3-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил
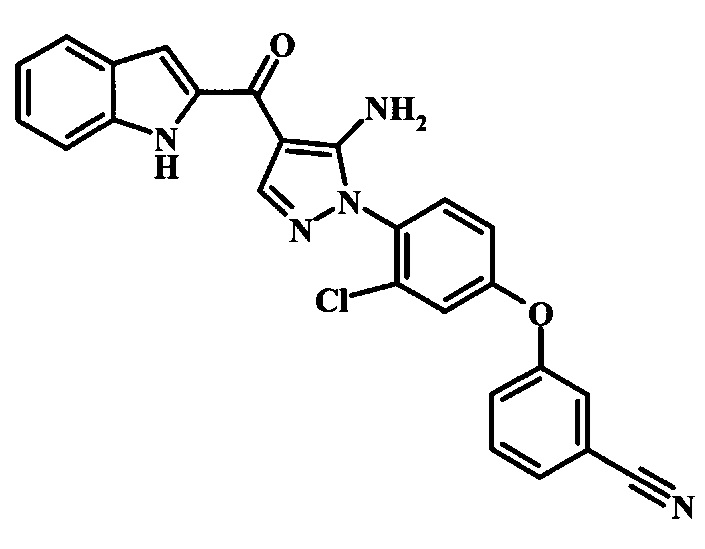
Стадия 1)
3-(3-Хлор-4-нитро-фенокси)-бензонитрил
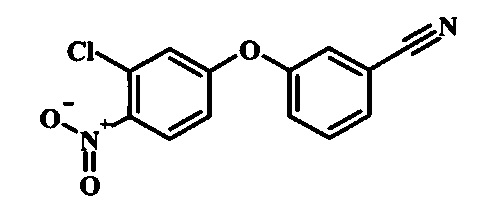
Смесь 2-хлор-4-фтор-1-нитро-бензола (15 г, 85 ммоль), 3-гидроксибензонитрила (10,1 г, 85 ммоль) и Cs2CO3 (30,4 г, 93,5 ммоль) в ДМФА (100 мл) нагревали при 120°C в течение 1 часа. Добавляли EtOAc и смесь промывали водой и солевым раствором. Органический слой сушили (Na2SO4), фильтровали и выпаривали, получая 3-(3-хлор-4-нитро-фенокси)-бензонитрил (23 г, 99%) в виде желтого твердого вещества.
Стадия 2)
3-(4-Амино-3-хлор-фенокси)-бензонитрил
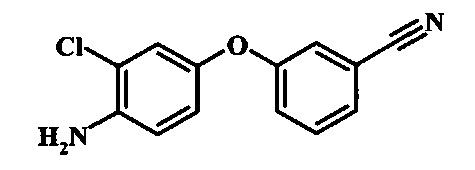
Раствор хлорида олова(II) дигидрата (75,4 г, 335 ммоль) в HCl (50 мл) добавляли к раствору 3-(3-хлор-4-нитро-фенокси)-бензонитрила (23 г, 83,9 ммоль) в МеОН (500 мл) и смесь перемешивали при комнатной температуре в течение 6 часов. Смесь делали основной, добавляя 2 н NaOH, и полученную в результате смесь экстрагировали EtOAc. Органический слой промывали солевым раствором, сушили (Na2SO4), фильтровали и выпаривали. Остаток очищали с помощью хроматографии (силикагель, от 0 до 30% EtOAc/гексаны), получая 3-(4-амино-3-хлор-фенокси)-бензонитрил (13,8 г, 67%) в виде желтого масла.
Стадия 3)
3-(3-Хлор-4-гидразино-фенокси)-бензонитрила гидрохлорид
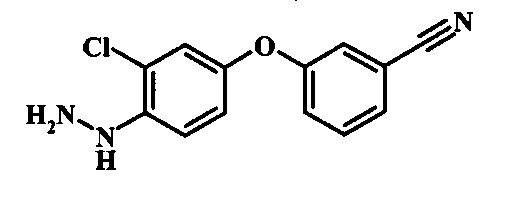
Смесь 3-(4-амино-3-хлор-фенокси)-бензонитрила (5 г, 20,4 ммоль) и конц. HCl (30 мл) в МеОН (30 мл) охлаждали до -5°C. Раствор NaNO2 (1,72 г, 24,5 ммоль) в воде (2 мл) добавляли, и смесь перемешивали в течение 40 минут при -5°C. Раствор хлорида олова(II) дигидрата (23,1 г, 102 ммоль) в HCl (20 мл) добавляли, и смесь перемешивали в течение 1 часа. Смесь выпаривали, и твердое вещество отфильтровывали и сушили в вакууме, получая 3-(3-хлор-4-гидразино-фенокси)-бензонитрила гидрохлорид (5,8 г, 96%). Это вещество использовали на следующей стадии без дополнительной очистки.
Стадия 4)
3-{4-[5-Амино-4-(1-этоксиметил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил
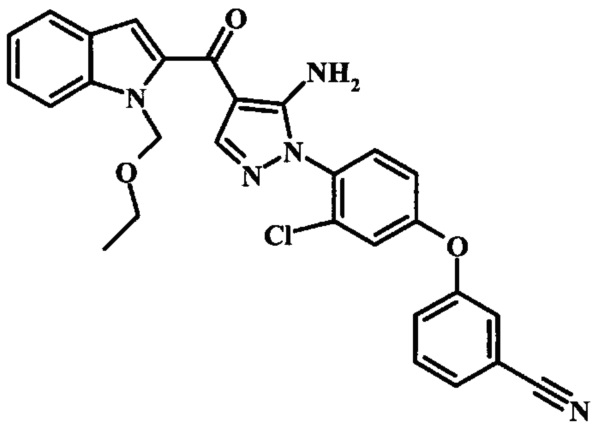
В 20 мл сцинтилляционном флаконе объединяли (Е)-3-(диметиламино)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-индол-2-карбонил)акрилонитрил (промежуточное соединение 2, стадия 3) (100 мг, 0,271 ммоль), карбонат калия (112 мг, 0,812 ммоль) и 3-(3-хлор-4-гидразинилфенокси)бензонитрил (217 мг, 0,836 ммоль) с EtOH (4 мл), получая желтую суспензию. Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 100% EtOAc/гексаны), получая 3-{4-[5-амино-4-(1-этоксиметил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил (95 мг, 69%) в виде масла. ЖХ/МС: m/z рассчитанная для C28H22ClN5O3 ([М+Н]+): 512,9. Найденная: 511,9.
Стадия 5)
3-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил
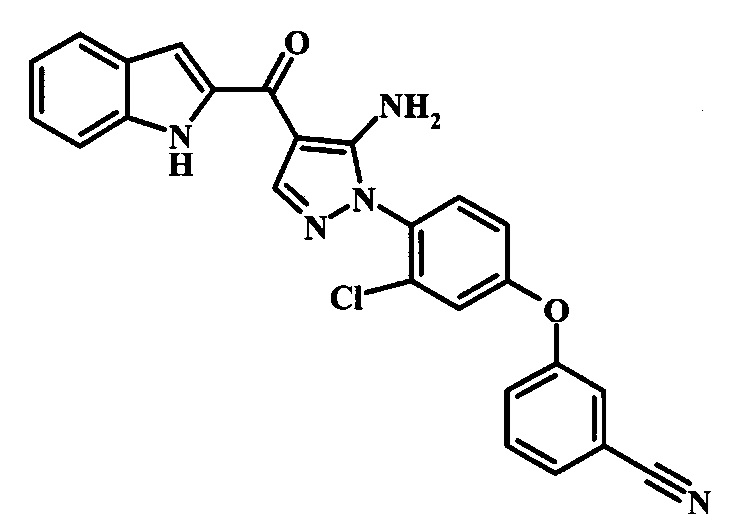
В 20 мл сцинтилляционном флаконе объединяли 3-(4-(5-амино-4-(1-(этоксиметил)-1Н-индол-2-карбонил)-1Н-пиразол-1-ил)-3-хлорфенокси)бензонитрил (60 мг, 0,117 ммоль) с EtOH (4 мл) и водн. 10% HCl (2 мл). Реакционную смесь нагревали при 80°C в течение 3 часов. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 70% EtOAc/гексаны), получая 3-{4-[5-амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил (18 мг, 34%) в виде светло-коричневого твердого вещества. ЖХ/МС: m/z рассчитанная для C25H16ClN5O2 ([M+H]+): 454,8. Найденная: 454,0.
Пример 17
3-{4-[5-Амино-4-(1Н-бензимидазол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензамид
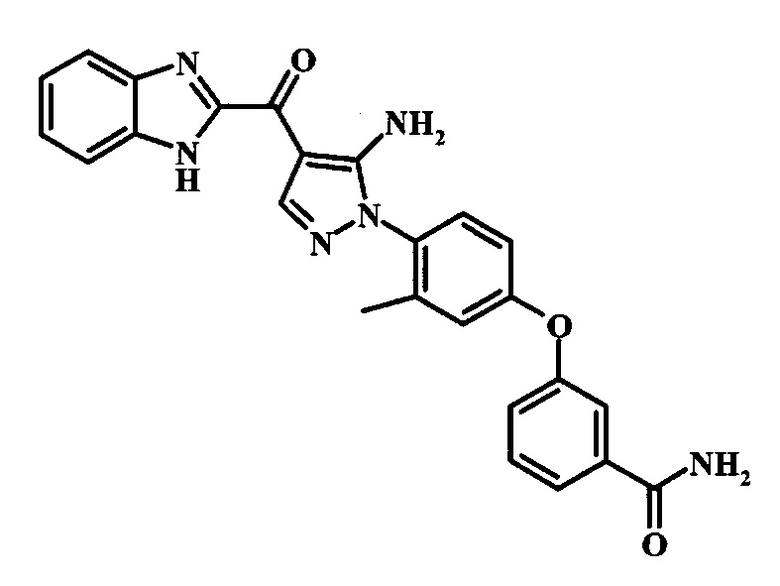
Стадия 1)
3-(3-Метил-4-нитро-фенокси)-бензонитрил
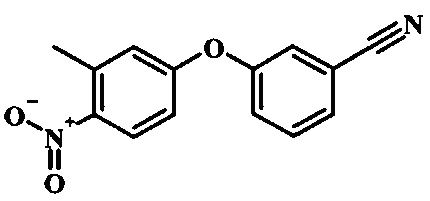
Смесь 4-фтор-2-метил-1-нитро-бензола (13,1 г, 84 ммоль), 3-гидроксибензонитрила (10 г, 84 ммоль) и Cs2CO3 (30,1 г, 92 ммоль) в ДМФА (100 мл) нагревали при 120°C в течение 3 часов. Добавляли Et2O и смесь промывали водой и солевым раствором. Органический слой сушили (Na2SO4), фильтровали и выпаривали, получая 3-(3-метил-4-нитро-фенокси)-бензонитрил (19,2 г, 90%). 1Н ЯМР (400 МГц, ДМСО-d6) δ ppm 8,09 (d, J равно 8,8 Гц, 1Н), 7,71-7,77 (m, 2 Н), 7,62-7,70 (m, 1Н), 7,48-7,55 (m, 1Н), 7,16 (d, J равно 2,8 Гц, 1Н), 7,04 (dd, J равно 8,9, 2,9 Гц, 1Н). МС рассч. для C14H11N2O3 [(М+Н)+] 255, наблюд. 254,9.
Стадия 2)
3-(4-Амино-3-метил-фенокси)-бензонитрил
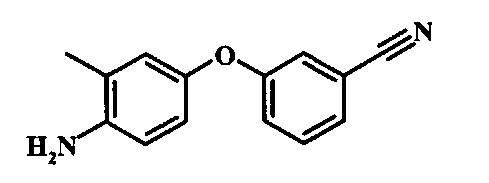
Раствор хлорида олова(II) дигидрата (34,9 г, 155 ммоль) в конц. HCl (35 мл) добавляли к раствору 3-(3-метил-4-нитро-фенокси)-бензонитрила (9,85 г, 38,7 ммоль) в МеОН (300 мл) и смесь перемешивали в течение ночи. Смесь выпаривали, и остаток делали основным, добавляя 2 н NaOH. Полученную в результате смесь экстрагировали четыре раза EtOAc. Органический слой промывали водой и солевым раствором, сушили (Na2SO4), фильтровали и выпаривали. Остаток очищали с помощью хроматографии (силикагель, от 0 до 30% EtOAc/гексаны), получая 3-(4-амино-3-метил-фенокси)-бензонитрил (4,6 г, 53%) в виде черной жидкости. МС рассч. для C14H13N2O ([М+Н]+): 225, наблюд. 225,0.
Стадия 3)
3-(4-Гидразино-3-метил-фенокси)-бензонитрил
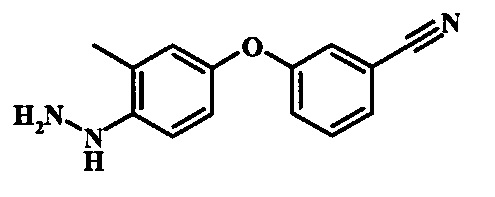
Раствор NaNO2 (340 мг, 4,9 ммоль) в воде (1 мл) добавляли к смеси 3-(4-амино-3-метил-фенокси)-бензонитрила (1 г, 4,46 ммоль), конц. HCl (2 мл), воды (4 мл) и МеОН (2 мл) при 4°C. Смесь перемешивали в течение 30 минут. Раствор хлорида олова(II) (4,23 г, 22 ммоль) в конц. HCl (10 мл) медленно добавляли, и смесь перемешивали в течение 3 часов. Добавляли МеОН (10 мл), затем 10 М NaOH (осторожно: экзотермический эффект). Смесь охлаждали, и добавляли EtOAc и 10 М NaOH. Органическую фазу промывали солевым раствором и выпаривали, получая неочищенный 3-(4-гидразино-3-метил-фенокси)-бензонитрил (600 мг, 56%) в виде красно-коричневого масла. Это вещество использовали на следующей стадии без дополнительной очистки. МС рассч. для C14H14N3O ([М+Н]+): 240, наблюд. 222,9.
Стадия 4)
3-(4-{5-Амино-4-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил
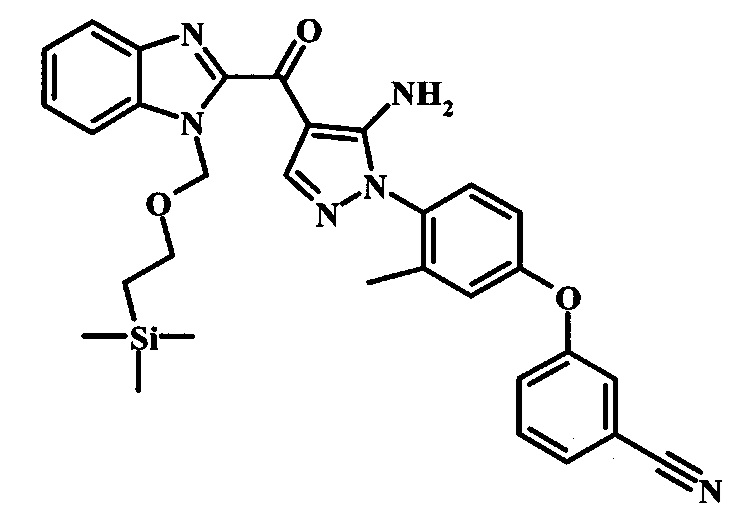
В 20 мл сцинтилляционном флаконе объединяли (Е)-3-(диметиламино)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-бензо[d]имидазол-2-карбонил)акрилонитрил (промежуточное соединение 3, стадия 3) (200 мг, 0,540 ммоль), 3-(4-гидразинил-3-метилфенокси)бензонитрил (200 мг, 0,836 ммоль) и карбонат калия (300 мг, 2,17 ммоль) с EtOH (4 мл), получая оранжевую суспензию. Реакционную смесь нагревали при 80°C в течение ночи. Реакция не завершалась. Добавляли 3-(4-гидразинил-3-метилфенокси)бензонитрил (200 мг, 0,836 ммоль, экв.: 1,55) и реакционную смесь нагревали при 80°C в течение дополнительного часа. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 70% EtOAc/гексаны), получая 3-(4-{5-амино-4-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-карбонил]-пиразол-1-ил}-3-метил-фенокси)-бензонитрил (30 мг, 10%) в виде масла. ЖХ/МС: m/z рассчитанная для C31H32N6O3Si ([М+Н]+): 565,7. Найденная: 565,1.
Стадия 5)
3-{4-[5-Амино-4-(1Н-бензимидазол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензамид
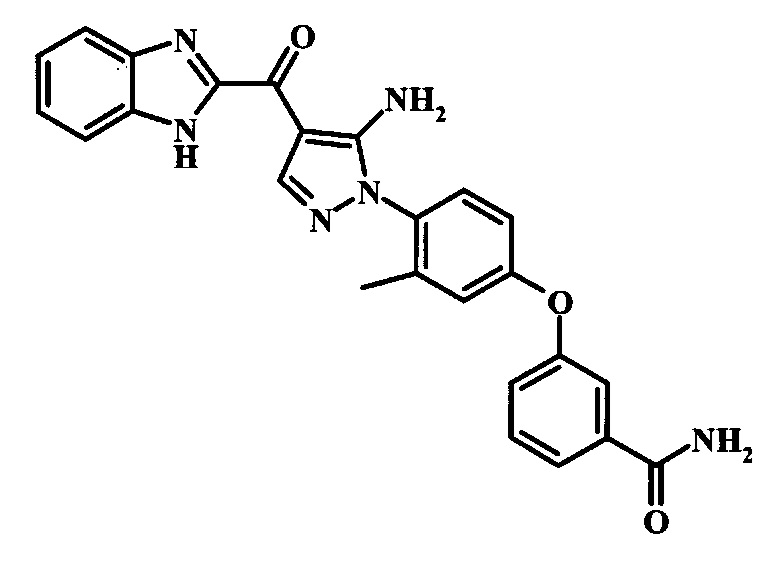
В 20 мл сцинтилляционном флаконе объединяли 3-(4-(5-амино-4-(1-((2-(триметилсилил)этокси)метил)-1Н-бензо[d]имидазол-2-карбонил)-1Н-пиразол-1-ил)-3-метилфенокси)бензонитрил (30 мг, 0,053 ммоль), влажный 1 М ТБАФ в ТГФ (13,9 мг, 0,053 ммоль) и этан-1,2-диамин (32 мг, 0,53 ммоль) с ТГФ (1 мл). Реакционную смесь нагревали при 70°C в течение 24 часов. Растворитель удаляли при пониженном давлении. Неочищенное вещество разбавляли EtOAc и промывали насыщенным водным раствором NH4Cl. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 100% EtOAc/гексаны), получая 3-{4-[5-амино-4-(1Н-бензимидазол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензамид (13 мг, выход 54%) в виде твердого вещества. ЖХ/МС: m/z рассчитанная для C25H20N6O3 ([М+Н]+): 453,4. Найденная: 452,9.
Пример 18
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-бензимидазол-2-ил)-метанон
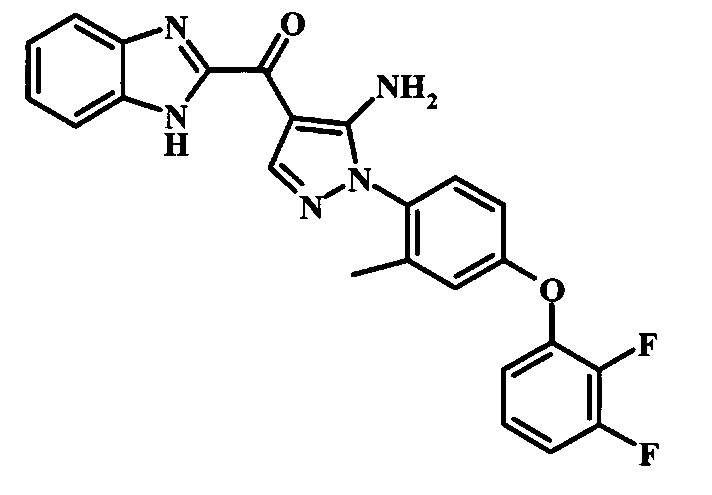
Стадия 1)
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-ил]-метанон
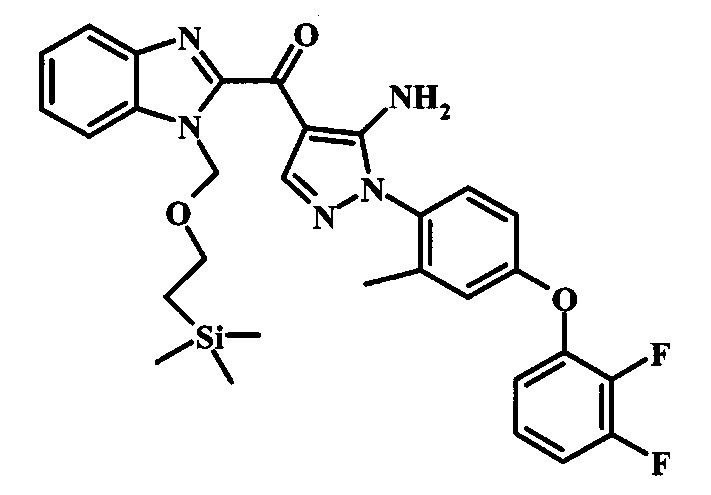
В 20 мл сцинтилляционном флаконе объединяли (Е)-3-(диметиламино)-2-(1-((2-(триметилсилил)этокси)метил)-1Н-бензо[d]имидазол-2-карбонил)акрилонитрил (промежуточное соединение 3, стадия 3) (200 мг, 0,540 ммоль), (4-(2,3-дифторфенокси)-2-метилфенил)гидразин (пример 14, стадия 3) (261 мг, 1,04 ммоль) и карбонат калия (224 мг, 1,62 ммоль) с EtOH (5 мл), получая желтую суспензию. Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь разбавляли EtOAc и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 70% EtOAc/гексаны), получая (5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-[1-(2-триметилсиланил-этоксиметил)-1Н-бензимидазол-2-ил]-метанон (40 мг, выход 13%) в виде масла. ЖХ/МС: m/z рассчитанная для C30H31N5O3Si ([М+Н]+): 576,7. Найденная: 576,1.
Стадия 2)
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-бензимидазол-2-ил)-метанон
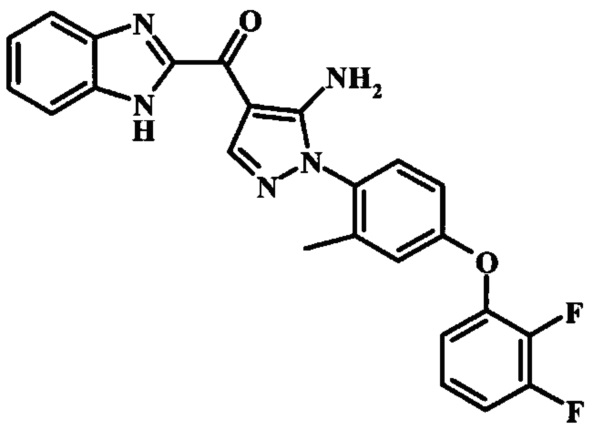
В 20 мл сцинтилляционном флаконе объединяли (5-амино-1-(4-(2,3-дифторфенокси)-2-метилфенил)-1Н-пиразол-4-ил)(1-((2-(триметилсилил)этокси)метил)-1Н-бензо[d]имидазол-2-ил)метанон (40 мг, 0,070 ммоль), этан-1,2-диамин (42 мг, 0,695 ммоль) и 1 М ТБАФ в ТГФ (0,9 мл, 1,8 ммоль) с ТГФ (1 мл), получая светло-красный раствор. Реакционную смесь нагревали при 70°C в течение 24 часов. Растворитель удаляли при пониженном давлении. Неочищенное вещество разбавляли EtOAc и промывали насыщенным водным раствором NH4CI. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 100% EtOAc/гексаны), получая {5-амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-бензимидазол-2-ил)-метанон (20 мг, выход 65%) в виде твердого вещества. ЖХ/МС: m/z рассчитанная для C24H17F2N5O2 ([М+Н]+): 446,4. Найденная: 445,9.
Пример 19
[5-Амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-(6-морфолин-4-илметил-1Н-индол-2-ил)-метанон
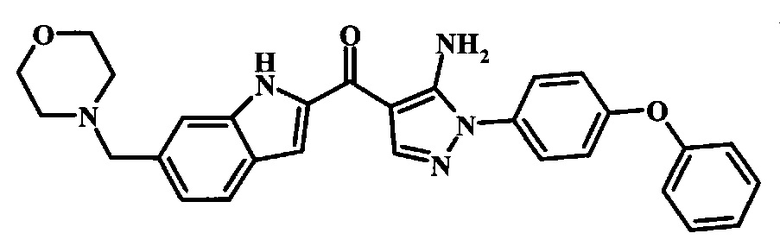
Стадия 1) Метиловый эфир 4-морфолин-4-илметил-бензойной кислоты
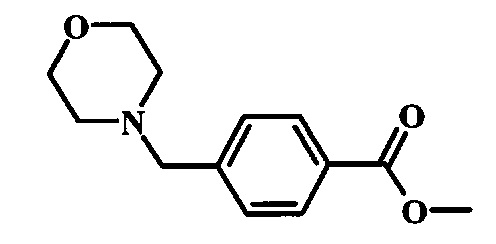
Диизопропилэтиламин (15,2 мл, 87,3 ммоль) добавляли к перемешиваемому раствору морфолина (4,2 мл, 48 ммоль) в безводном ТГФ (50 мл) при 0°C. Добавляли метиловый эфир 4-бромметил-бензойной кислоты (10 г, 87,3 ммоль) в безводном ТГФ (70 мл) и перемешивали в течение 12 часов. Растворитель удаляли при пониженном давлении и разбавляли водой. Полученный в результате раствор экстрагировали этилацетатом. Объединенные органические фазы сушили над безводным сульфатом натрия, выпаривали при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 30% EtOAc/гексаны), получая метиловый эфир 4-морфолин-4-илметил-бензойной кислоты (10,5 г, 93%) в виде твердого вещества белого оттенка. МС рассч. для C13H17NO3 [(М+Н)+] 236, наблюд. 236,1.
Стадия 2) (4-Морфолин-4-илметил-фенил)-метанол
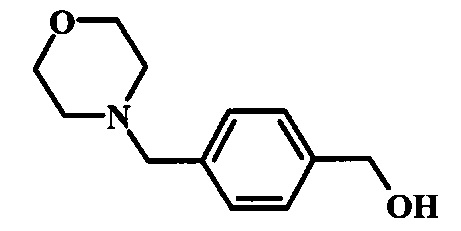
Боргидрид натрия (2,57 г, 68 ммоль) добавляли к перемешиваемому раствору метилового эфира 4-морфолин-4-илметил-бензойной кислоты (2 г, 8,5 ммоль) в смеси ТГФ:МеОН (8:1) (56 мл) при 0°C в атмосфере азота. Реакционную смесь нагревали с обратным холодильником в течение 2 часов. Гасили насыщенным раствором хлорида аммония при 0°C и перемешивали в течение 30 минут. Экстрагировали этилацетатом (20 мл×2), сушили над безводным сульфатом натрия, выпаривали при пониженном давлении и очищали с помощью колоночной хроматографии (силикагель, 2% МеОН/ДХМ), получая (4-морфолин-4-илметил-фенил)-метанол (1,3 г, 74%) в виде белого твердого вещества. МС рассч. для C12H17NO2 [(М+Н)+] 208, наблюд. 208,1.
Стадия 3) 4-Морфолин-4-илметил-бензальдегид
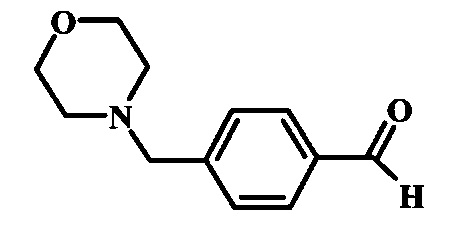
Диоксид марганца (3,28 г, 37,67 ммоль) добавляли к перемешиваемому раствору (4-морфолин-4-илметил-фенил)-метанола (1,3 г, 6,28 ммоль) в дихлорметане (120 мл) при к.т. и перемешивали в течение 72 часов. Реакционную смесь фильтровали через воронку с фильтром из спеченного стекла, используя целит, и промывали дихлорметаном (50 мл). Растворитель выпаривали при пониженном давлении и очищали с помощью колоночной хроматографии (силикагель, 20% EtOAc/гексаны), получая 4-морфолин-4-илметил-бензальдегид (1,2 г, 93%) в виде белого твердого вещества. МС рассч. для C12H15NO2 [(М+Н)+] 206, наблюд. 206,3.
Стадия 4) Метиловый эфир (Е)-2-азидо-3-(4-морфолин-4-илметил-фенил)-акриловой кислоты
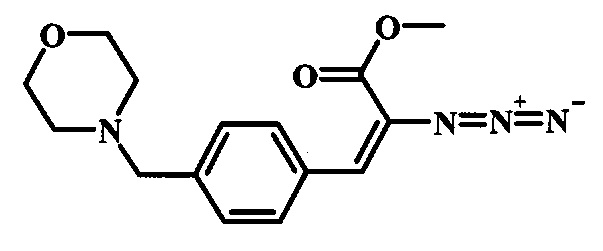
Смесь 4-морфолин-4-илметил-бензальдегида (6,8 г, 33,17 ммоль) и этилового эфира азидо-уксусной кислоты (7,11 г, 132,68 ммоль) в метаноле (18 мл) добавляли к перемешиваемому раствору метилата натрия (5,37 г, 99,51 ммоль) в метаноле (30 мл) при 0°C и перемешивали в течение 30 минут. Реакционную смесь фильтровали через воронку с фильтром из спеченного стекла и промывали водой, получая метиловый эфир (Е)-2-азидо-3-(4-морфолин-4-илметил-фенил)-акриловой кислоты (7 г, 70%) в виде белого твердого вещества. МС рассч. для C15H18N4O3 [(М+Н)+] 302, наблюд. 303,3.
Стадия 5) Метиловый эфир 6-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты
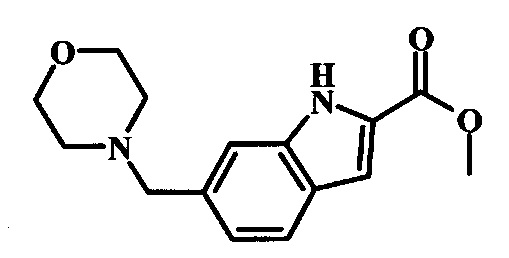
Метиловый эфир (Е)-2-азидо-3-(4-морфолин-4-илметил-фенил)-акриловой кислоты (3,8 г, 12,58 ммоль) в ксилоле (90 мл) нагревали при 90°C в течение 2 часов, охлаждали до к.т. и перемешивали при к.т. в течение 12 часов. Ксилол выпаривали при пониженном давлении и очищали с помощью колоночной хроматографии (силикагель, 2% МеОН/ДХМ), получая метиловый эфир 6-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты (1,9 г, 55%) в виде белого твердого вещества. МС рассч. для C15H18N2O3 [(М+Н)+] 275, наблюд. 275,2.
Стадия 6) Метиловый эфир 6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-карбоновой кислоты
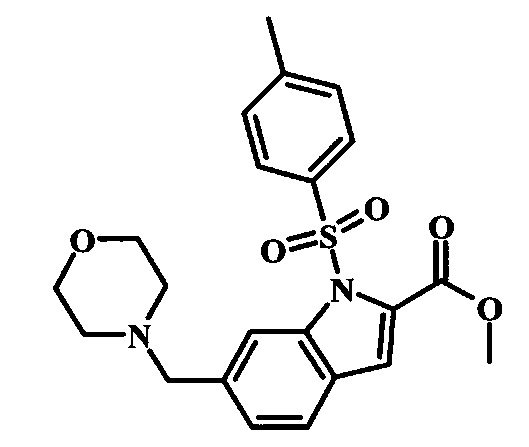
Метиловый эфир 6-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты (3,6 г, 13,13 ммоль) в безводном ТГФ (10 мл) добавляли к перемешиваемому раствору гидрида натрия в 60% масляной дисперсии (1,3 г, 32,84 ммоль) в безводном ТГФ (20 мл) при 0°C и перемешивали в течение 30 минут. Добавляли пара-толуолсульфонилхлорид (5 г, 26,27 ммоль) в безводном ТГФ (10 мл). Реакционную смесь перемешивали при к.т. в течение 12 часов. Гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом, сушили над безводным сульфатом натрия, выпаривали при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 1% МеОН/ДХМ), получая метиловый эфир 6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-карбоновой кислоты (3,5 г, 93%) в виде белого твердого вещества. МС рассч. для C22H24N2O5S [(М+Н)+] 429, наблюд. 428,9.
Стадия 7) 3-[6-Морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-ил]-3-оксо-пропионитрил
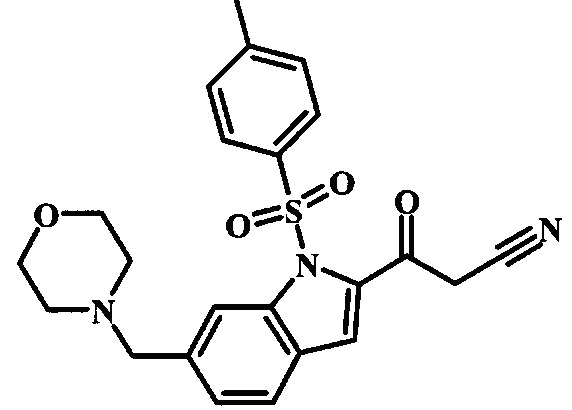
Бутиллитий (15 мл, 13,55 ммоль) добавляли к перемешиваемому раствору диизопропиламина (1,95 мл, 13,55 ммоль) в безводном ТГФ (20 мл) при -78°C и перемешивали в течение 30 минут. Добавляли к перемешиваемому раствору метилового эфира 6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-карбоновой кислоты (2,9 г, 6,77 ммоль) и ацетонитрила (1,4 мл, 27 ммоль) в безводном ТГФ (20 мл) при -78°C и перемешивали в течение 30 минут. Реакционную смесь гасили насыщенным раствором хлорида аммония и экстрагировали этилацетатом (20 мл×2). Объединенные органические фазы сушили над безводным сульфатом натрия, выпаривали при пониженном давлении и очищали с помощью колоночной хроматографии (силикагель, 2% МеОН/ДХМ), получая 3-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-ил]-3-оксо-пропионитрил (1,8 г, 90%) в виде желтого твердого вещества. МС рассч. для C23H23N3O4S [(М+Н)+] 438, наблюд. 438,1.
Стадия 8) (Е)-3-Диметиламино-2-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрил
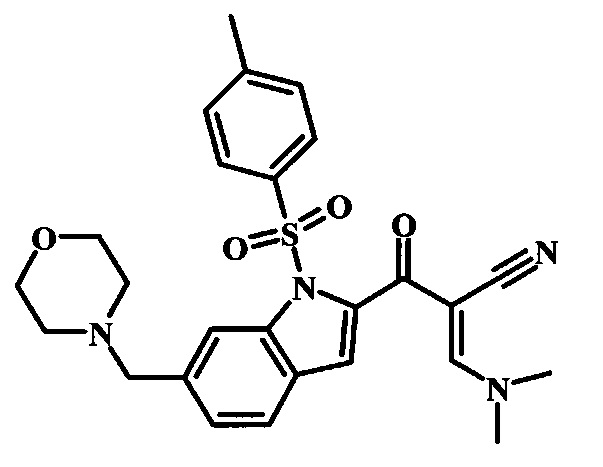
ДМФА-ДМА (3,9 мл, 29,3 ммоль) добавляли к перемешиваемому раствору 3-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-ил]-3-оксо-пропионитрила (3,2 г, 7,32 ммоль) в безводном толуоле (50 мл) при к.т. и перемешивали в течение 12 часов. Толуол выпаривали при пониженном давлении и очищали с помощью колоночной хроматографии (силикагель, 2% МеОН/ДХМ), получая (Е)-3-диметиламино-2-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрил (2,5 г, 97%) в виде желтого твердого вещества. МС рассч. для C26H28N4O4S [(М+Н)+] 493, наблюд. 493,3.
Стадия 9) [5-Амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон
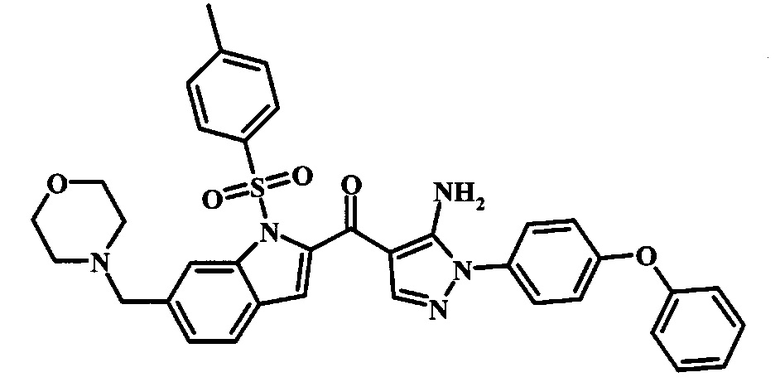
(Е)-3-Диметиламино-2-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-карбонил]-акрилонитрил (0,15 г, 0,305 ммоль) добавляли к перемешиваемому раствору (4-фенокси-фенил)-гидразина (0,091 г, 0,457 ммоль) в этаноле и нагревали с обратным холодильником в течение 16 часов. Этанол выпаривали при пониженном давлении и неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 2% МеОН/ДХМ), получая [5-амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанон (0,14 г, 71%) в виде желтого твердого вещества. МС рассч. для C36H33N5O5S [(М+Н)+] 648, наблюд. 648,2.
Стадия 10) [5-Амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-(6-морфолин-4-илметил-1Н-индол-2-ил)-метанон
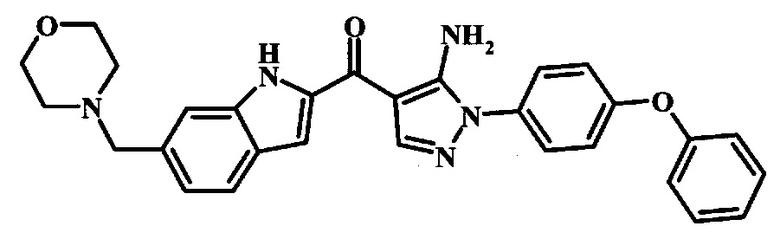
Карбонат цезия (0,21 г, 0,65 ммоль) добавляли к перемешиваемому раствору [5-амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-[6-морфолин-4-илметил-1-(толуол-4-сульфонил)-1Н-индол-2-ил]-метанона (0,14 г, 0,216 ммоль) в смеси ТГФ:МеОН (7:3) (6 мл) при к.т. и перемешивали в течение 18 часов. Растворитель выпаривали при пониженном давлении и неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 5% МеОН/ДХМ), получая [5-амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-[6-морфолин-4-илметил-1Н-индол-2-ил]-метанон (0,055 г, 52%) в виде желтого твердого вещества. МС рассч. для C29H27N5O3 [(М+Н)+] 494, наблюд. 494,4.
Пример 20
3-{4-[5-Амино-4-(6-морфолин-4-илметил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил
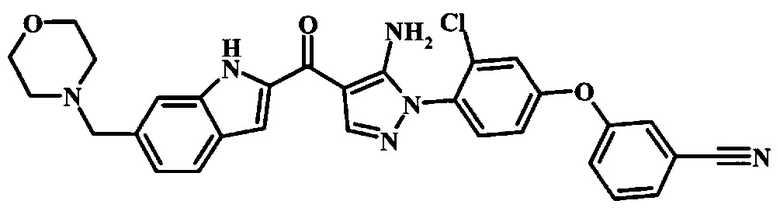
Стадия 1) 3-(3-Хлор-4-нитро-фенокси)-бензонитрил
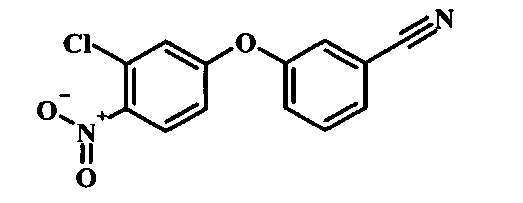
Смесь 2-хлор-4-фтор-1-нитро-бензола (15 г, 85 ммоль), 3-гидроксибензонитрила (10,1 г, 85 ммоль) и Cs2CO3 (30,4 г, 93,5 ммоль) в ДМФА (100 мл) нагревали при 120°C в течение 1 часа. EtOAc добавляли и смесь промывали водой и солевым раствором. Органический слой сушили (Na2SO4), фильтровали и выпаривали, получая 3-(3-хлор-4-нитро-фенокси)-бензонитрил (23 г, 99%) в виде желтого твердого вещества.
Стадия 2) 3-(4-Амино-3-хлор-фенокси)-бензонитрил
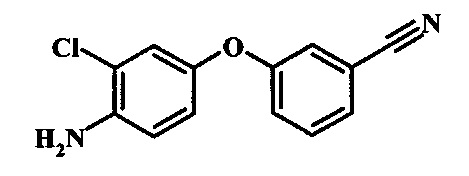
Раствор хлорида олова(II) дигидрата (75,4 г, 335 ммоль) в HCl (50 мл) добавляли к раствору 3-(3-хлор-4-нитро-фенокси)-бензонитрила (23 г, 83,9 ммоль) в МеОН (500 мл) и смесь перемешивали при комнатной температуре в течение 6 часов. Смесь делали основной, добавляя 2 н NaOH, и полученную в результате смесь экстрагировали EtOAc. Органический слой промывали солевым раствором, сушили (Na2SO4), фильтровали и выпаривали. Остаток очищали с помощью хроматографии (силикагель, от 0 до 30% EtOAc/гексаны), получая 3-(4-амино-3-хлор-фенокси)-бензонитрил (13,8 г, 67%) в виде желтого масла.
Стадия 3) 3-(3-Хлор-4-гидразино-фенокси)-бензонитрила гидрохлорид
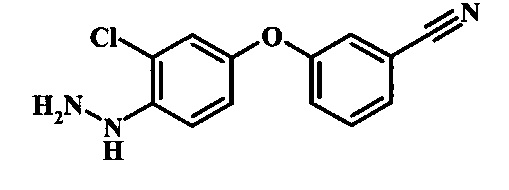
Смесь 3-(4-амино-3-хлор-фенокси)-бензонитрила (5 г, 20,4 ммоль) и конц. HCl (30 мл) в МеОН (30 мл) охлаждали до -5°C. Раствор NaNO2 (1,72 г, 24,5 ммоль) в воде (2 мл) добавляли и смесь перемешивали в течение 40 минут при -5°C. Раствор хлорида олова (II) дигидрата (23,1 г, 102 ммоль) в HCl (20 мл) добавляли, и смесь перемешивали в течение 1 часа. Смесь выпаривали, и твердое вещество отфильтровывали и сушили в вакууме, получая 3-(3-хлор-4-гидразино-фенокси)-бензонитрила гидрохлорид (5,8 г, 96%). Это вещество использовали на следующей стадии без дополнительной очистки.
Стадия 4) Метиловый эфир 6-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты
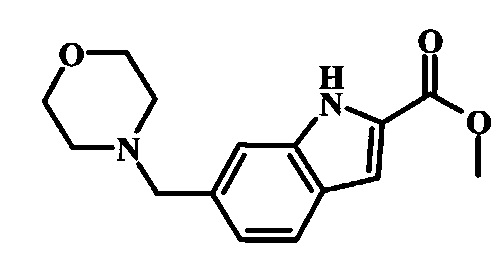
Метиловый эфир (Е)-2-азидо-3-(4-морфолин-4-илметил-фенил)-акриловой кислоты (303,34 мг, 1,00 ммоль), Rh2(pfb)4 (диродий(II) тетракис(перфторбутират)) (52,8 мг, 0,05 ммоль) растворяли в толуоле (758 мкл) и затем нагревали до 60°C в течение 2 дней и затем при 90°C в течение 2 часов. Реакционную смесь разбавляли EtOAc и промывали водой, солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении, и неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, 2% МеОН/ДХМ), получая метиловый эфир 6-морфолин-4-илметил-1Н-индол-2-карбоновой кислоты (0,41 г, 50%) в виде белого твердого вещества. МС рассч. для C15H18N2O3 [(М+Н)+] 275, наблюд. 275,2.
Стадия 5) Метиловый эфир 6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбоновой кислоты
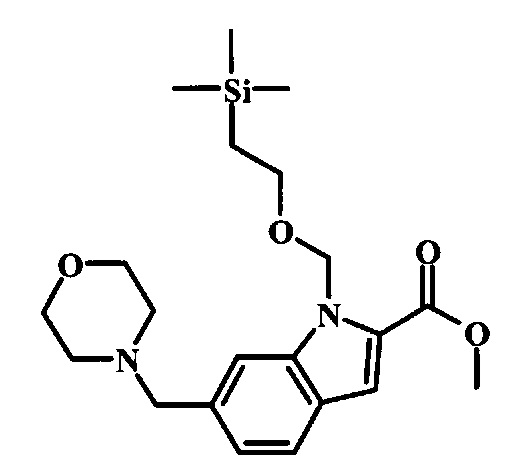
NaH в 60% масляной дисперсии (244 мг, 6,1 ммоль) добавляли небольшими порциями к перемешиваемому раствору этил 6-(морфолинометил)-1Н-индол-2-карбоксилата (1,6 г, 5,5 ммоль) в безводном ТГФ (6 мл) и ДМФА (2,5 мл) при 0°C. Реакционную смесь перемешивали в течение 30 минут при 0°C. Добавляли (2-(хлорметокси)этил)триметилсилан (925 мг, 5,55 ммоль) при 0°C и затем перемешивали при к.т. в течение 1 часа. Реакционную смесь разбавляли EtOAc, промывали водой и солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении, и неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 50% EtOAc/гексаны), получая метиловый эфир 6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбоновой кислоты в виде желтого масла. МС рассч. для C21H32N2O4Si [(М+Н)+] 405, наблюд. 405,2.
Стадия 6) 3-[6-Морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-ил]-3-оксо-пропионитрил
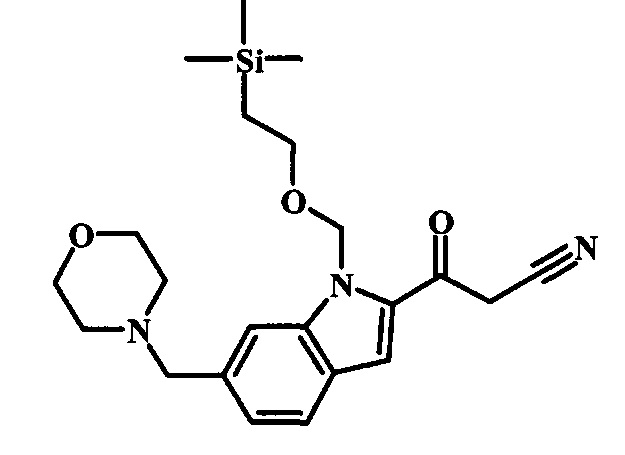
В 100 мл круглодонной колбе объединяли метиловый эфир 6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбоновой кислоты (1,6 г, 3,82 ммоль) и MeCN (941 мг, 1,2 мл, 22,9 ммоль) с ТГФ (20 мл), получая темно-коричневый раствор. После охлаждения до -78°C медленно добавляли ЛДА (2М/ТГФ) (3,82 мл, 7,64 ммоль) в течение 5 минут. Реакционную смесь перемешивали при -78°C в течение 30 минут. Реакцию гасили насыщ. NH4Cl (10 мл). Реакционную смесь разбавляли водой (150 мл), экстрагировали EtOAc (100 мл) и промывали солевым раствором. Объединенные органические фазы сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 40% EtOAc/гексаны), получая 3-[6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-ил]-3-оксо-пропионитрил (0,15 г, 10%) в виде желтого масла. МС рассч. для C22H31N3O3Si [(М+Н)+] 414, наблюд. 414,3.
Стадия 7) (Е)-3-Диметиламино-2-[6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-акрилонитрил
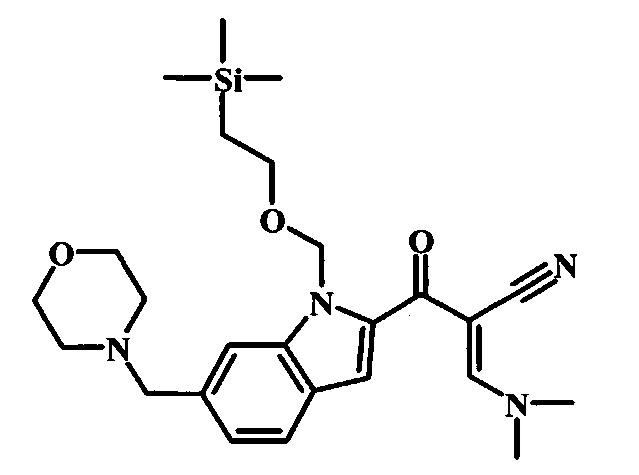
В 25 мл круглодонной колбе объединяли 3-[6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-ил]-3-оксо-пропионитрил (145 мг, 351 мкмоль) с толуолом (1,46 мл), получая светло-желтый раствор. N,N-диметилформамида диметилацеталь (60,5 мкл, 456 мкмоль) добавляли и перемешивали при комнатной температуре в течение 30 минут. Растворитель удаляли при пониженном давлении. Неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 50% EtOAc/гексаны), получая (Е)-3-диметиламино-2-[6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-акрилонитрил (0,12 г, 73%) в виде желтого масла. МС рассч. для C25H36N4O3Si [(М+Н)+] 469, наблюд. 469,2.
Стадия 8) 3-(4-{5-Амино-4-[6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-хлор-фенокси)-бензонитрил
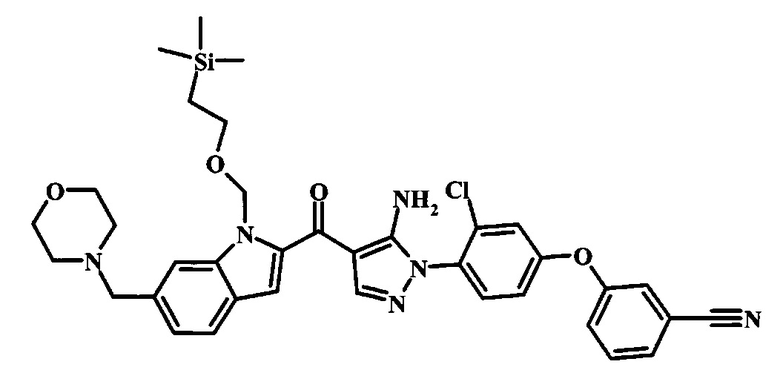
(Е)-3-Диметиламино-2-[6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-акрилонитрил (115 мг, 245 мкмоль), 3-(3-хлор-4-гидразино-фенокси)-бензонитрила гидрохлорид (218 мг, 736 мкмоль) и K2CO3 (170 мг, 1,23 ммоль) в EtOH (2 мл) нагревали при 80°C в течение 2 часов. Реакционную смесь разбавляли EtOAc и промывали водой, солевым раствором и сушили над безводным сульфатом натрия. Растворитель удаляли при пониженном давлении и неочищенное вещество очищали с помощью колоночной хроматографии (силикагель, от 0 до 50% EtOAc/гексаны), получая 3-(4-{5-амино-4-[6-морфолин-4-ил метил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-хлор-фенокси)-бензонитрил (0,103 г, 61%) в виде желтого масла. МС рассч. для C36H39ClN6O4Si [(М+Н)+] 683, наблюд. 683,2.
Стадия 10) 3-{4-[5-Амино-4-(6-морфолин-4-илметил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил
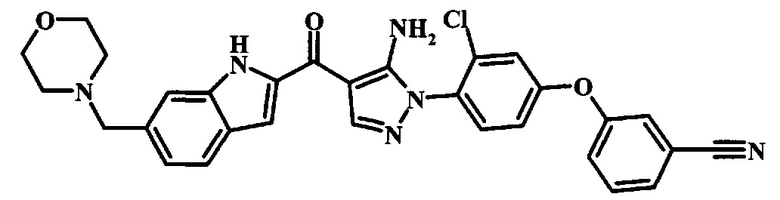
3-(4-{5-Амино-4-[6-морфолин-4-илметил-1-(2-триметилсиланил-этоксиметил)-1Н-индол-2-карбонил]-пиразол-1-ил}-3-хлор-фенокси)-бензонитрил (80 мг, 117 мкмоль) растворяли в EtOH (4 мл) и добавляли 10% HCl (2 мл, 20,0 ммоль), и нагревали до 80°C в течение 10 минут. Растворитель удаляли при пониженном давлении. Неочищенное вещество растирали с эфиром, получая 3-{4-[5-амино-4-(6-морфолин-4-илметил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрил (80 мг, выход 94%). 1Н ЯМР (ДМСО-d6) δ: 12,13 (s, 1Н), 8,45 (s, 1Н), 7,89 (d, J равно 8,3 Гц, 1Н), 7,72-7,86 (m, 6Н), 7,58-7,70 (m, 3Н), 7,42 (d, J равно 8,3 Гц, 1Н), 7,36 (dd, J равно 8,7, 2,6 Гц, 1Н), 7,25 (br, s„ 1Н), 4,57 (d, J равно 4,8 Гц, 2Н), 3,77-4,14 (m, 4Н), 3,16-3,45 (m, 4Н).
Биологические примеры
Анализ на ингибирование тирозинкиназы Брутона (Btk)
Анализ основан на захвате радиоактивного 33Р фосфорилированного продукта в ходе фильтрации. Взаимодействия Btk, биотинилированного SH2 пептидного субстрата (гомология Src) и АТФ (аденозинтрифосфат) приводят к фосфорилированию пептидного субстрата. Биотинилированный продукт связывается со стрептавидин-сефарозными гранулами. Все связавшиеся меченные радиоактивным изотопом продукты детектируют с помощью сцинтилляционного счетчика.
Анализируемые планшеты представляют собой 96-луночные полипропиленовые (Greiner) и 96-луночные с 1,2 мкм гидрофильным ПВДФ (поливинилиденфторид) фильтровальные планшеты (Millipore). Приводимые в данном документе концентрации являются конечными аналитическими концентрациями: от 10 до 100 мкМ соединения в ДМСО (Burdick and Jackson), от 5 до 10 нМ фермент Btk (His-меченый, полной длины), 30 мкМ пептидный субстрат (Biotin-Aca-AAAEEIYGEI-NH2), 100 мкМ АТФ (Sigma), 8 мМ имидазол (Sigma, рН 7,2), 8 мМ глицерин-2-фосфат (Sigma), 200 мкМ ЭГТА (этиленгликольтетрауксусная кислота) (Roche Diagnostics), 1 мМ MnCl2 (Sigma), 20 мМ MgCl2 (Sigma), 0,1 мг/мл БСА (бычий сывороточный альбумин) (Sigma), 2 мМ ДТТ (дитиотреитол) (Sigma), 1 мкКи 33Р АТФ (Amersham), 20% стрептавидин-сефарозные гранулы (Amersham), 50 мМ ЭДТА (этилендиаминтетрауксусная кислота) (Gibco), 2 М NaCl (Gibco), 2 М NaCl вес./1% фосфорной кислоты (Gibco), microscint-20 (Perkin Elmer).
IC50 рассчитывают по 10 точкам измерения на соединение, используя данные, полученные из стандартной аналитической матрицы для 96-луночного планшета. Одно контрольное соединение и семь неизвестных ингибиторов тестировали на каждом планшете, и каждый планшет анализировали дважды. Обычно соединения разбавляли на половину-log, начиная с 100 мкМ и заканчивая 3 нМ. Контрольным соединением являлся стауроспорин. Фон рассчитывали при отсутствии пептидного субстрата. Общую активность определяли в присутствии пептидного субстрата. Следующий протокол использовали для определения ингибирования Btk.
Получение образца: Исследуемые соединения разбавляли с возрастанием на половину-log аналитическим буфером (имидазол, глицерин-2-фосфат, ЭГТА, MnCl2, MgCl2, БСА).
Получение гранул
1) Промывают гранулы, центрифугируя при 500 g.
2) Восстанавливают гранулы с помощью ФБС (фосфатно-буферного солевого раствора) и ЭДТА с получением 20% взвеси гранул.
3) Предварительно инкубируют реакционную смесь без субстрата (аналитический буфер, ДТТ, АТФ, 33Р АТФ) и смешивают с субстратом (аналитический буфер, ДТТ, АТФ, 33Р АТФ, пептидный субстрат) при 30°C в течение 15 минут.
4) Для начала анализа предварительно инкубируют 10 мкл Btk в ферментном буфере (имидазол, глицерин-2-фосфат, БСА) и 10 мкл исследуемых соединений в течение 10 минут при к.т.
5) Добавляют 30 мкл реакционной смеси в отсутствии или в присутствии субстрата к Btk и соединениям.
6) Инкубируют 50 мкл общей аналитической смеси в течение 30 минут при 30°C.
7) Переносят 40 мкл аналитической смеси в 150 мкл взвеси гранул на фильтровальный планшет для остановки реакции.
8) Промывают фильтровальный планшет через 30 минут в ходе следующий стадий:
3×250 мкл NaCl,
3×250 мкл NaCl, содержащих 1% фосфорной кислоты,
1×250 мкл H2O.
9) Сушат планшет в течение 1 часа при 65°C или в течение ночи при к.т.
10) Добавляют 50 мкл microscint-20 и считают 33Р cpm (от англ. "counts per minute" - число импульсов в минуту) на сцинтилляционном счетчике.
Рассчитывают процентную активность из первичных данных в cpm:
процентная активность = (образец - bkg) / (общая активность - bkg)×100.
Рассчитывают IC50 из процентной активности, используя одностороннюю дозозависимую сигмоидальную модель:
y=А+((В-А)/(1+((х/С)D))))
x - конц. соед., y - % активности, А - мин, В - max, С - IC50, D - 1 (угловой коэффициент Хилла).
Анализ TR-FRET (от англ. "time-resolved fluorescence resonance energy transfer" - резонансный перенос энергии флуоресценции с временным разрешением) на ингибирование тирозинкиназы Брутона
В ходе этого конкурентного анализа на ВТК измеряют эффективность соединения (IC50) для инактивированного состояния тирозинкиназы Брутона, используя методику FRET ( Resonance Energy Transfer). Комплекс ВТК - Eu инкубировали на льду в течение одного часа до использования при исходной концентрации 50 нМ BTK-BioeaseTm: 10 нМ Eu-стрептавидин (Perkin-Elmer, № по каталогу AD0062). Аналитический буфер состоял из 20 мМ ГЭПЭС (4-(2-гидроксиэтил)-1-пиперазин-этансульфоновая кислота) (рН 7,15), 0,1 мМ ДТТ, 10 мМ MgCl2, 0,5 мг/мл БСА с 3% стабилизатора киназы (Fremont Biosolutions, № по каталогу STB-K02). Через 1 час вышеприведенную реакционную смесь разбавляли в 10 раз аналитическим буфером, получая комплекс 5 нМ ВТК: 1 нМ Eu-стрептавидин (донор флуорофора). Затем 18 мкл смеси 0,11 нМ ВТК-Eu и 0,11 нМ Kinase Tracer 178 (Invitrogen, №по каталогу PV5593), только с ВТК-Eu не как отрицательная контрольная проба, распределяли на 384-луночных плоскодонных планшетах (Greiner, 784076). Соединения, которые исследовали в ходе анализа, получали в виде 10х концентраций и последовательное разбавление с возрастанием на половину-log осуществляли в ДМСО, чтобы получить 10 точечные кривые. Для инициации реакции FRET соединения, полученные в виде 10x растворов в ДМСО, добавляли в планшеты, и планшеты инкубировали в течение от 18 до 24 часов при 14°C.
Resonance Energy Transfer). Комплекс ВТК - Eu инкубировали на льду в течение одного часа до использования при исходной концентрации 50 нМ BTK-BioeaseTm: 10 нМ Eu-стрептавидин (Perkin-Elmer, № по каталогу AD0062). Аналитический буфер состоял из 20 мМ ГЭПЭС (4-(2-гидроксиэтил)-1-пиперазин-этансульфоновая кислота) (рН 7,15), 0,1 мМ ДТТ, 10 мМ MgCl2, 0,5 мг/мл БСА с 3% стабилизатора киназы (Fremont Biosolutions, № по каталогу STB-K02). Через 1 час вышеприведенную реакционную смесь разбавляли в 10 раз аналитическим буфером, получая комплекс 5 нМ ВТК: 1 нМ Eu-стрептавидин (донор флуорофора). Затем 18 мкл смеси 0,11 нМ ВТК-Eu и 0,11 нМ Kinase Tracer 178 (Invitrogen, №по каталогу PV5593), только с ВТК-Eu не как отрицательная контрольная проба, распределяли на 384-луночных плоскодонных планшетах (Greiner, 784076). Соединения, которые исследовали в ходе анализа, получали в виде 10х концентраций и последовательное разбавление с возрастанием на половину-log осуществляли в ДМСО, чтобы получить 10 точечные кривые. Для инициации реакции FRET соединения, полученные в виде 10x растворов в ДМСО, добавляли в планшеты, и планшеты инкубировали в течение от 18 до 24 часов при 14°C.
После инкубации планшеты считывали на флуоресцентном спектрофотометре для считывания планшетов BMG Pherastar (или эквивалентном) и использовали для измерения энергии излучения от донора флуорофора европия (излучение при 620 нм) и FRET (излучение при 665 нм). Значения лунок с отрицательной контрольной пробой усредняли, чтобы получить средний минимум. Лунки с положительной "без ингибитора" контрольной пробой усредняли, чтобы получить средний максимум. Процент максимального FRET рассчитывали, используя следующее уравнение:
% макс FRET = 100×[(FSRсоед - FSRсредний мин) / (FSRсредний макс - FSRсредний мин)],
где FSR - отношение сигналов FRET. Кривые % максимума FRET строили в Activity Base (Excel) и определяли IC50 (%), угловой коэффициент Хилла, z' и %CV (от англ. "coefficient of variation" - коэффициент вариации). Среднюю IC50 и среднеквадратичное отклонение получают из дублирующих кривых (одиночные кривые ингибирования от двух независимых разбавлений) при использовании Microsoft Excel.
Данные репрезентативных соединений для этого анализа приведены ниже в Таблице II.
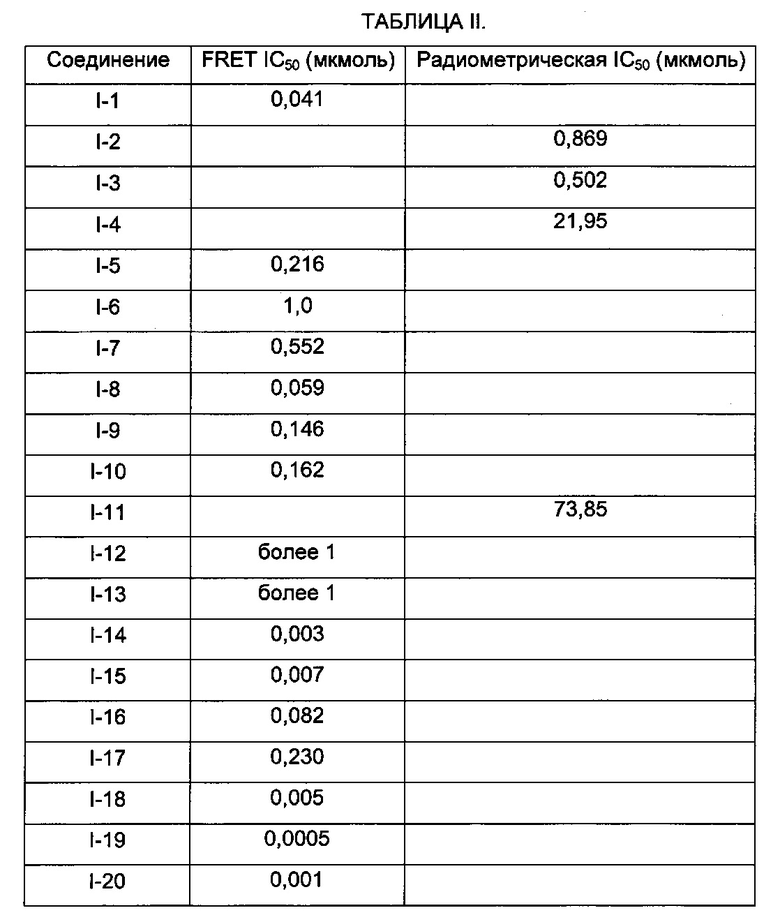
Ингибирование В-клеточной активации на цельной крови, измеренное с помощью экспрессии CD69
Методику для определения способности ингибиторов Btk подавлять В-клеточную опосредованную рецептором активацию В-клеток в крови человека осуществляют следующим образом.
Цельную кровь человека (HWB, от англ. "human whole blood") получают от здоровых волонтеров со следующими ограничениями: 24 часа не принимают лекарственные препараты, некурящие. Кровь собирают из вены в пробирки Vacutainer, антикоагулированные гепарином натрия. Исследуемые соединения разбавляют в десять раз от требуемой начальной концентрации лекарственного препарата ФБС (20×), затем трехкратно серийными разбавлениями 10% ДМСО в ФБС для получения девятиточечной дозозависимой кривой. Добавляют по 5,5 мкл каждого разбавленного соединения в двух параллельных опытах в 2 мл 96-луночный планшет с V-образным дном (Analytical Sales and Services, #59623-23); добавляют по 5,5 мкл 10% ДМСО в ФБС в контрольные и нестимулированные лунки. Добавляют HWB (100 мкл) в каждую лунку и после смешивания планшеты инкубируют при 37°C, 5% CO2, 100% влажности в течение 30 минут. Добавляют F(ab')2 анти-человеческий IgM козы (Southern Biotech, #2022-14) (10 мкл 500 мкг/мл раствора, 50 мкг/мл конечная концентрация) в каждую лунку (за исключением нестимулированных лунок) при перемешивании и планшеты инкубируют в течение дополнительных 20 часов.
В конце 20-часовой инкубации образцы инкубируют с меченными флуоресцентной пробой антителами (15 мкл РЕ античеловеческие CD20 мыши, BD Pharmingen, #555623, и/или 20 мкл АРС античеловеческие CD69 мыши, BD Pharmingen #555533) в течение 30 минут при 37°C, 5% CO2, 100% влажности. Включены индуцированный контроль, неокрашенные и отдельные окрашивания для компенсационных корректировок и начальные установки напряжения. Образцы затем подвергают лизису с 1 мл 1Х буфера для лизиса Pharmingen (BD Pharmingen # 555899), и планшеты центрифугируют при 1800 об/мин в течение 5 минут.
Надосадочные жидкости удаляют отсасыванием, и оставшиеся осадки снова подвергают лизису с еще 1 мл 1Х буфера для лизиса Pharmingen, и планшеты центрифугируют, как описано выше. Надосадочные жидкости отсасывают, и оставшиеся осадки промывают буфером FACs (ФБС+1% ЭБС (эмбриональная бычья сыворотка)). После конечного вращения надосадочные жидкости удаляют, и остатки повторно суспендируют в 180 мкл буфера FACs. Образцы переносят в 96-луночный планшет, подходящий для анализа на HTS 96-луночной системе в проточном цитометре BD LSR II.
Используя подходящие длины волн возбуждения и излучения для используемых флуорофоров, собирают данные, и процентные положительные клеточные значения получают, используя программное обеспечение Cell Quest. Результаты сначала анализируют с помощью программного обеспечения для анализа FACS (Flow Jo). IC50 для исследуемых соединений определяют как концентрацию, которая снижает на 50% процентное содержание CD69-положительных клеток, которые также являются CD20-положительными после стимуляции с помощью анти-IgM (среднее значение 8 контрольных лунок после вычитания среднего значения 8 лунок для нестимулированного фона). Значения IC50 рассчитывают, используя программное обеспечение XLfit версии 3, уравнение 201.
Ингибирование В-клеточной активации - анализ FLIPR (от англ. "fluorescence imaging plate reader" - спектрофотометр для прочтения планшетов для визуализации флуоресценции) В-клеток в клетках Ramos
Ингибирование В-клеточной активации соединениями по настоящему изобретению показано при определении влияния исследуемых соединений на анти-IgM стимулированные В-клеточные отклики.
Анализ FLIPR В-клеток является функциональным способом на основе клеток для определения воздействия потенциальных ингибиторов повышения внутриклеточного кальция в ответ на стимулирование анти-IgM антителом. Клетки Ramos (клеточная линия лимфомы Беркитта человека. ATCC-No. CRL-1596) выращивали в среде для роста (описана ниже). За один день до анализа клетки Ramos повторно суспендировали в свежей среде для роста (такой же, как приведена выше) и помещали в концентрации 0,5×106 клеток/мл в колбы для тканевых культур. В день анализа клетки считывают и помещают в концентрации 1×106 клеток/мл в среду для роста, дополненную 1 мкМ FLUO-3AM (TefLabs, № по каталогу 0116, полученный в безводном ДМСО и 10% плюрониловой кислоте), в колбу для тканевых культур, и инкубируют при 37°C (4% СО) в течение 1 часа. Для удаления внеклеточного красителя клетки собирали центрифугированием (5 мин, 1000 об/мин), повторно суспендировали в буфере FLIPR (описан ниже) при 1×106 клеток/мл и затем распределяли в 96-луночные покрытые поли-D-лизином черные/прозрачные планшеты (BD, № по каталогу 356692) в концентрации 1×105 клеток на лунку. Исследуемые соединения добавляли в разных концентрациях в диапазоне от 100 мкМ до 0,03 мкМ (7 концентраций, подробности ниже) и оставляли инкубироваться с клетками в течение 30 минут при к.т. Передачу сигнала Са2+ клетками Ramos стимулировали, добавляя 10 мкг/мл анти-IgM (Southern Biotech, №по каталогу 2020-01), и измеряли на FLIPR (Molecular Devices, получение изображений 96-луночных планшетов, используя камеру на ПЗС (приборах с зарядовой связью) с аргоновым лазером при возбуждении 480 нМ).
Среда/Буферы:
Среда для роста: среда RPMI 1640 (от англ. "Roswell Park Memorial Institute") с L-глутамином (Invitrogen, № по каталогу 61870-010), 10% эмбриональная бычья сыворотка (ЭБС, Summit Biotechnology, № по каталогу FP-100-05); 1 мМ пируват натрия (Invitrogen, № по каталогу 11360-070).
Буфер FLIPR: HBSS (от англ. "Hank's Balanced Salt Solution" - сбалансированный солевой раствор Хэнка) (Invitrogen, № по каталогу 141175-079), 2 мМ CaCl2 (Sigma, № по каталогу С-4901), ГЭПЭС (Invitrogen, № по каталогу 15630-080), 2,5 мМ пробенецид (Sigma, № по каталогу Р-8761), 0,1% БСА (Sigma, № по каталогу А-7906), 11 мМ глюкоза (Sigma, № по каталогу G-7528).
Подробности разбавления соединения:
Чтобы достигнуть наибольшей конечной аналитической концентрации в 100 мкМ, 24 мкл 10 мМ маточного раствора соединения (полученного в ДМСО) добавляют непосредственно к 576 мкл буфера FLIPR. Исследуемые соединения разбавляют буфером FLIPR (используя автоматизированный пипеточный дозатор Biomek 2000), получая в результате следующую схему разбавления: наполнитель, 1,00×10-4 М, 1,00×10-5, 3,16×10-6, 1,00×10-6, 3,16×10-7, 1,00×10-7, 3,16×10-8.
Проба и анализ
Внутриклеточные повышения кальция регистрировали, используя статистику макс - мин (вычитая остаточную базовую линию из пика, вызванного добавлением стимулирующего антитела), с помощью контрольного устройства FLIPR Molecular Devices и статистического экспортирующего программного обеспечения. IC50 определяли, используя подбор нелинейных кривых (программное обеспечение GraphPad Prism).
Вызванный коллагеном артрит у мыши (mCIA, от англ. "Mouse Collagen-induced arthritis")
В 0 день мышам вводят в основание хвоста или в несколько участков на спине эмульсию коллагена типа II (внутрикожно) в полном адъюванте Фрейнда (CFA, от англ. "Complete Freund's adjuvant"). После иммунизации коллагеном у животных развивается артрит приблизительно через от 21 до 35 дней. Появление артрита синхронизируют (поддерживают) систематическим введением коллагена в неполном адъюванте Фрейнда (IFA, от англ. "Incomplete Freund's adjuvant"; внутрикожно) на 21 день. Животных исследуют ежедневно после 20 дня на любое проявление умеренного артрита (оценка 1 или 2; смотрите описание оценок ниже), что является сигналом для бустер-инъекции. После бустер-инъекции мышей отмечают и дозируют потенциальные терапевтические агенты в течение заданного времени (обычно от 2 до 3 недель) и с частотой дозировок один раз в день (QD, от лат."Quaque in Die") или два раза в день (BID, от лат."Bis in Die").
Вызванный коллагеном артрит у крыс (rCIA, от англ. "Rat Collagen-induced arthritis")
В 0 день крысам вводят эмульсию бычьего коллагена типа II в неполном адъюванте Фрейнда (IFA) внутрикожно (в/к) в несколько мест на спине. Бустер-инъекцию эмульсии коллагена вводят примерно на 7 день (в/к) в основание хвоста или в альтернативные участки на спине. Артрит обычно наблюдается на от 12 до 14 день после первой инъекции коллагена. Животные могут быть оценены на развитие артрита, как описано ниже (оценка артрита) с 14 дня и далее. Животным дозируют потенциальные терапевтические агенты предупредительным образом, начиная со времени вторичного введения и в течение заданного времени (обычно от 2 до 3 недель) и с частотой дозировок один раз в день (QD) или два раза в день (BID).
Оценка артрита:
В обеих моделях развитие воспаления лапок и суставов конечностей количественно определяют, используя систему оценок, которая включает оценку 4 лапок согласно критерию, описанному ниже:
Оценка: 1 соответствует опухание и/или покраснение лапки или одного пальца.
2 соответствует опухание двух или более суставов.
3 соответствует сильное опухание лапки с более чем двумя суставами.
4 соответствует тяжелый артрит всей лапы и пальцев.
Оценки проводят на 0 день для измерения базовой линии и начинают снова при первых признаках или опухании вплоть до трех раз в неделю до окончания эксперимента. Артритный индекс для каждой мыши получают складыванием четырех оценок отдельных лапок, получая максимальную оценку 16 для животного.
Модель in vivo астмы у крыс
Самцам крыс Brown-Norway вводят внутрибрюшинно 100 мкг OA (овальбумин) в 0,2 мл квасцах один раз в неделю в течение трех недель (день 0, 7 и 14). На 21 день (через одну неделю после последней сенсибилизации) крысам дозируют один раз в день либо наполнитель, либо соединение подкожно за 0,5 часа до аэрозольного введения OA (1% OA в течение 45 минут), и заканчивают через 4 или 24 часа после введения. В момент умерщвления собирают сыворотку и плазму у всех животных на серологию и ФК (фармакокинетику), соответственно. Вводят трахеальную канюлю и легкие промывают 3Х ФБС. Жидкость БАЛ (бронхоальвеолярный лаваж) анализируют на общее количество лейкоцитов и дифференцированный подсчет лейкоцитов. Общее количество лейкоцитов в аликвоте клеток (от 20 до 100 мкл) определяют с помощью счетчика Коултера. Для дифференциального подсчета лейкоцитов от 50 до 200 мкл образца центрифугируют в Cytospin и предметное стекло окрашивают Diff-Quik. Доли моноцитов, эозинофилов, нейтрофилов и лимфоцитов определяют с помощью световой микроскопии, используя стандартные морфологические критерии, и выражают в процентах. Репрезентативные ингибиторы Btk показывают снижение общего количества лейкоцитов в БАЛ OA активированных и обработанных крыс по сравнению с контрольными уровнями.
Вышеизложенное изобретение описано более подробно с помощью иллюстраций и примеров с целью ясности и понимания. Специалисту в данной области техники очевидно, что изменения и модификации могут быть проведены в объеме прилагаемых пунктов формулы изобретения. Следовательно, следует понимать, что приведенное выше описание предназначено для иллюстрации и не является ограничивающим. Объем изобретения, следовательно, должен определяться не ссылкой на вышеприведенное описание, а напротив должен определяться ссылкой на следующие прилагаемые пункты формулы изобретения, наряду с полным объемом эквивалентов, на которые такие пункты формулы изобретения распространяются.
Таким образом, все патенты, заявки на патент и публикации, приведенные в данной заявке, включены сюда полностью путем ссылки на них для всех целей в той степени, как если бы каждый отдельный патент, заявка на патент или публикация были бы по отдельности обозначены.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2648236C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2014 |
|
RU2653504C2 |
| ПРОИЗВОДНОЕ АМИНОПИРАЗОЛА | 2010 |
|
RU2580543C9 |
| ПРОИЗВОДНЫЕ ГЕТЕРОАРИЛПИРРОЛИДИНИЛ- И ПИПЕРИДИНИЛКЕТОНА | 2007 |
|
RU2479575C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| НОВЫЕ СОЕДИНЕНИЯ И КОМПОЗИЦИИ ДЛЯ ИНГИБИРОВАНИЯ FASN | 2014 |
|
RU2737434C2 |
| ПЕРВИЧНЫЕ КАРБОКСАМИДЫ В КАЧЕСТВЕ ИНГИБИТОРОВ BТK | 2014 |
|
RU2708395C2 |
| ИНГИБИТОРЫ ЦИТОЗОЛЬНОЙ ФОСФОЛИПАЗЫ А2 | 2002 |
|
RU2337906C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ MKK4 ДЛЯ СТИМУЛЯЦИИ РЕГЕНЕРАЦИИ ПЕЧЕНИ ИЛИ УМЕНЬШЕНИЯ ИЛИ ПРЕДОТВРАЩЕНИЯ ГИБЕЛИ ГЕПАТОЦИТОВ | 2019 |
|
RU2788000C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
Изобретение относится к соединениям общей формулы (I) или к их фармацевтически приемлемым солям. В общей формуле (I) R1 представляет собой низший алкил, фенил, возможно замещенный одним или более чем одним R1', циклоалкил или пиридил; каждый R1' независимо представляет собой низший алкил, галоген, -C(=O)NH2 или циано; R2 отсутствует или представляет собой галоген, низший алкокси, гидрокси или низший алкил; R3 отсутствует или представляет собой галоген, низший алкокси, гидрокси или низший алкил; R4 отсутствует или представляет собой морфолинил-низший алкиленил; X представляет собой СН или N; и Y представляет собой СН или N. Соединения формулы (I) ингибируют Btk (от англ. "Bruton's tyrosine kinase" - тирозинкиназа Брутона). Соединения формулы (I) полезны для модулирования активности Btk и лечения заболеваний, связанных с чрезмерной активностью Btk. Соединения являются полезными в лечении онкологических, аутоиммунных и воспалительных заболеваний, вызванных аберрантной активацией В-клеток. Изобретение также относится к фармацевтическим композициям, содержащим соединения формулы (I) и, по меньшей мере, один носитель, разбавитель или эксципиент. 10 н. и 15 з.п. ф-лы, 2 табл., 20 пр.
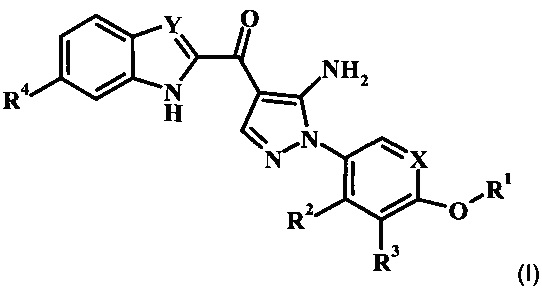
1. Соединение Формулы I,
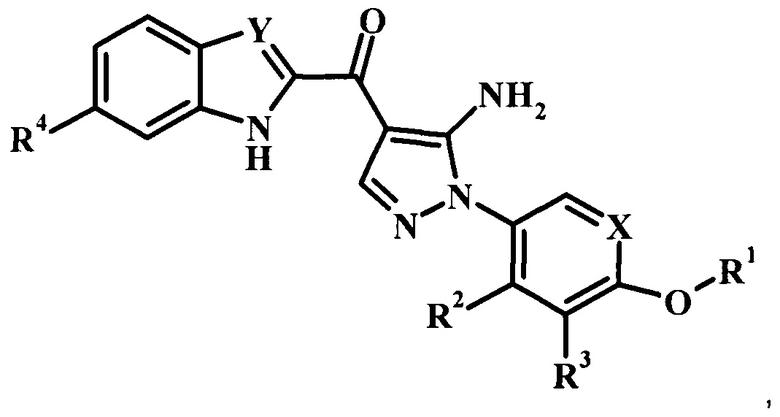
где:
R1 представляет собой низший алкил, фенил, возможно замещенный одним или более чем одним R1', циклоалкил или пиридил;
каждый R1' независимо представляет собой низший алкил, галоген, -C(=O)NH2 или циано;
R2 отсутствует или представляет собой галоген, низший алкокси, гидрокси или низший алкил;
R3 отсутствует или представляет собой галоген, низший алкокси, гидрокси или низший алкил;
R4 отсутствует или представляет собой морфолинил-низший алкиленил;
X представляет собой СН или N; и
Y представляет собой СН или N;
или его фармацевтически приемлемая соль.
2. Соединение по п. 1, где Y представляет собой СН.
3. Соединение по п. 1, где X представляет собой N.
4. Соединение по п. 1, где X представляет собой СН.
5. Соединение по п. 1, где R4 представляет собой морфолинил-метилен.
6. Соединение по п. 1, где R4 отсутствует.
7. Соединение по п. 1, где R2 отсутствует.
8. Соединение по п. 1, где R2 представляет собой галоген.
9. Соединение по п. 1, где R2 представляет собой низший алкил.
10. Соединение по п. 1, где R2 представляет собой низший алкокси.
11. Соединение по п. 1, где R2 представляет собой гидрокси.
12. Соединение по п. 1, где R3 представляет собой галоген, низший алкокси или гидрокси.
13. Соединение по п. 1, где R1 представляет собой фенил, возможно замещенный одним или более чем одним R1'.
14. Соединение по п. 1, где R1 представляет собой низший алкил, циклоалкил или пиридил.
15. Соединение, выбранное из группы, состоящей из:
[5-Амино-1-(3-фтор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(6-фенокси-пиридин-3-ил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(пиридин-2-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(пиридин-3-илокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(3-хлор-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(3,4-дифтор-фенокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(3-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(3-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(2-метокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(2-гидрокси-4-фенокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(4-изопропокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
[5-Амино-1-(4-циклопентилокси-фенил)-1Н-пиразол-4-ил]-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(2,2-диметил-пропокси)-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-индол-2-ил)-метанона;
2-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензонитрила;
3-{4-[5-Амино-4-(1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрила;
3-{4-[5-Амино-4-(1Н-бензимидазол-2-карбонил)-пиразол-1-ил]-3-метил-фенокси}-бензамида;
{5-Амино-1-[4-(2,3-дифтор-фенокси)-2-метил-фенил]-1Н-пиразол-4-ил}-(1Н-бензимидазол-2-ил)-метанона;
[5-Амино-1-(4-фенокси-фенил)-1Н-пиразол-4-ил]-(6-морфолин-4-илметил-1Н-индол-2-ил)-метанона; и
3-{4-[5-Амино-4-(6-морфолин-4-илметил-1Н-индол-2-карбонил)-пиразол-1-ил]-3-хлор-фенокси}-бензонитрила.
16. Способ лечения воспалительного и/или аутоиммунного состояния, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
17. Способ лечения воспалительного состояния, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
18. Способ лечения ревматоидного артрита, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
19. Способ лечения астмы, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
20. Способ лечения рака, включающий введение пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-15.
21. Фармацевтическая композиция, ингибирующая тирозинкиназу Брутона (Btk), включающая соединение по любому из пп. 1-15, смешанное с по меньшей мере одним фармацевтически приемлемым носителем, эксципиентом или разбавителем.
22. Соединение по любому из пп. 1-15 для применения в качестве ингибитора тирозинкиназы Брутона (Btk).
23. Соединение по любому из пп. 1-15 для применения в лечении воспалительного и/или аутоиммунного состояния.
24. Применение соединения по любому из пп. 1-15 для изготовления лекарственного средства для лечения воспалительного и/или аутоиммунного состояния.
25. Применение соединения по любому из пп. 1-15 для лечения воспалительного и/или аутоиммунного состояния.
| RU 2012108164 A, 20.09.2013 | |||
| RU 2011140064 A, 10.04.2013 | |||
| YAN LOU ET AL, Bruton's Tyrosine Kinase Inhibitors: Approaches to Potent and Selective Inhibition, Preclinical and Clinical Evaluation for Inflammatory Diseases and B Cell Malignancies, Journal of Medicinal Chemistry, 2012, Vol:55, Nr 10, pages 4539-4550. |

