Область техники, к которой относится настоящее изобретение
Настоящее изобретение относится к области молекулярной биологии и генной инженерии, а именно, к получению очищенных из исходного раствора смеси целевых и нецелевых двухцепочечных молекул ДНК препаратов целевых молекул ДНК. В частности, настоящее изобретение относится к способам очистки фрагментов двухцепочечной рекомбинантной ДНК, получаемых в результате расщепления плазмидной ДНК, от векторных последовательностей.
Предшествующий уровень техники настоящего изобретения
Получение очищенных из исходного раствора смеси целевых и нецелевых двухцепочечных молекул ДНК препаратов целевых молекул ДНК является рутинной процедурой в области молекулярной биологии. Одной из областей, где часто требуется разделение и очистка целевого фрагмента ДНК, является+ генная инженерия, например, при создании рекомбинантных ДНК с заданными свойствами.
Плазмидные векторы рутинно используются в генной инженерии как инструмент для манипуляций с целевыми фрагментами ДНК, обеспечивая их пропагацию в клетках E.coli. Однако векторная часть плазмиды в данном случае выполняет служебную функцию, и для манипуляций с целевыми фрагментами ДНК и их использования в некоторых приложениях требуется отделение векторных последовательностей от целевого фрагмента ДНК. Технически это реализуется с помощью ферментов (эндонуклеаз рестрикции), которые позволяют физически разделить кольцевую молекулу плазмиды на линейные фрагменты, в том числе физически отделить целевой фрагмент ДНК от вектора, с последующим разделением продуктов расщепления и очисткой целевого фрагмента ДНК от фрагмента, содержащего последовательность вектора. Очистка целевого фрагмента ДНК после расщепления плазмидной ДНК используется при проведении генно-инженерных работ, а также в области трансгенеза животных. В последнем случае при получения трансгенных животных для инъекции эмбрионов используют линейные фрагменты ДНК, содержащие трансген, свободный от векторных последовательностей, присутствие которых может негативно влиять на экспрессию трансгена (Kjer-Nielsen et al., 1992).
Концептуально разделение гетерогенной смеси молекул ДНК на гомогенные фракции и селективный отбор целевых молекул ДНК из смеси осуществляться на основании различий в свойствах молекул ДНК в смеси. Эти свойства могут быть как эндогенными, так и привнесенными.
При манипуляции с молекулами ДНК in vitro, в частности, при генно-инженерных манипуляциях, селекцию целевых молекул ДНК преимущественно осуществляют на основании различий в их молекулярной массе, что эквивалентно их длине в нуклеотидах. Разделение молекул ДНК на основании их длин может осуществляться различными способами, такими как гель-фильтрационная хроматография, центрифугирование в градиенте плотности, гель-электрофорез. Разделение первыми двумя упомянутыми способами основано на различии молекулярных масс молекул ДНК различной длины, в то время как последний способ также использует различия в суммарном заряде молекул, который пропорционален длине молекулы ДНК. Общим для методов разделения молекул ДНК, основанных на различия в длинах, является отсутствие при разделении селективности в отношении нуклеотидных последовательностей разделяемых молекул ДНК, то есть две молекулы ДНК с различными последовательностями, но имеющие одинаковую длину в нуклеотидах, такими методами разделены быть не могут, а при уменьшении различий в длинах разделяемых фрагментов ДНК качество разделения будет снижаться, особенно в случае использования методов, в которых разделение происходит на основании только молекулярных масс, в то время как гель-электрофорез, в котором на процесс разделения дополнительно оказывает влияние заряд молекул, позволяет более качественно разделять молекулы ДНК различной длины, чем гель-фильтрационная хроматография или центрифугирование в градиенте плотности.
В области молекулярной биологии часто используются методы разделения молекул ДНК, основанные на наличии/отсутствии уникальных признаков. Данные методы позволяют сепарировать молекулы ДНК вне зависимости от их длины, однако требуют предварительной модификации целевых молекул ДНК, что в последующем после смешивания в рамках проведения эксперимента позволяет сепарировать их от немодифицированных молекул ДНК. Примером часто используемой для этих целей модификаций является биотин, ковалентное присоединение которого к молекулам ДНК позволяет селективно сепарировать их от немодифицированных молекул с использованием иммобилизованного на твердом носителе стрептавидина, например, после проведения гибридизации при создании библиотек вычитания (Коробко и др., 1997). Ограничением для этого способа сепарации молекул ДНК является необходимость предварительной модификации очищенных целевых молекул ДНК in vitro.
Перечисленные выше способы разделения молекул ДНК также различаются по необходимым процедурам по очистке целевых молекул ДНК после проведения селекции.
При разделении молекул с помощью гель-фильтрационной хроматографии целевой фрагмент находится в хроматографическом буфере и, в зависимости от его состава, может не требовать дальнейшей очистки или требовать минимальной очистки путем переосаждения ДНК и растворения в буфере необходимого состава для получения очищенного препарата ДНК с минимальным присутствием нежелательных примесей.
При разделении молекул с помощью центрифугирования в градиенте плотности, фракция, содержащая целевые молекулы ДНК, требует очистки от веществ, использованных для создания градиента плотности (например, часто используемого для создания градиента плотности хлорида цезия). Присутствие ионов цезия негативно влияет на многие дальнейшие аппликации препарата ДНК, что требует тщательной очистки от хлорида цезия, осуществляемого, например, с помощью повторных диализов.
При разделении молекул ДНК с помощью гель-электрофореза, который является рутинным и наиболее часто используемым для этих целей методом, целевая фракция молекул ДНК находится в физически ограниченном объеме геля, из которого ДНК должна быть экстрагирована, что может осуществляться различными способами. Одним из способов экстракции ДНК из геля является пассивная элюция из фрагмента геля. Данный способ малоэффективен и характеризуется существенными потерями материала, а также требует последующей очистки от следовых количеств компонентов геля, присутствие которых может негативно влиять на дальнейшее целевое использование изолированной ДНК. Другой способ экстракции ДНК из геля, электроэлюция, состоит в продолжении процесса гель-электрофореза после разделения смеси молекул ДНК в геле, в результате которого целевая фракция молекул ДНК сорбируется на положительно заряженный бумажный носитель (бумага DEAE), врезанный в гель перед областью, содержащей целевые молекулы ДНК; далее иммобилизованная на бумаге DEAE ДНК элюируется в раствор. Альтернативно, молекулы ДНК могут быть переведены в раствор буфера для гель-электрофореза из вырезанного фрагмента геля, помещенного в специальную камеру, объем которой ограничен диализной пленкой, препятствующей выходу молекул ДНК из камеры в ходе электрофореза. В обоих случаях, помимо трудоемкости, полученные препараты молекул целевой ДНК в растворе требуют дальнейшей очистки от примесей компонентов геля, присутствие которых может негативно влиять на дальнейшее использование изолированной целевой ДНК.
Наиболее часто и рутинно используемым способом очистки молекул ДНК сегодня является экстракция ДНК из геля с использованием его химического разрушения, с последующей очисткой ДНК из полученного раствора на аффинном носителе, специфически сорбирующем молекулы ДНК. Данный способ позволяет получать препараты целевых молекул ДНК достаточной для большинства аппликаций чистоты, однако при существенной длине целевых молекул ДНК выход очищенного фрагмента после процесса очистки падает, и могут происходить физические разрывы длинных молекул ДНК в результате механического воздействия во время нахождения молекулы ДНК в связанном состоянии на сорбенте.
Существенными преимуществами перед методами, основанными на сепарации молекул ДНК на основании их размера, обладают методы, использующие модифицированные молекулы ДНК для их отделения от немодифицированных молекул, которые обладают высокой селективностью, не зависящей от длин разделяемых молекул ДНК. Эти методы позволяют избежать присутствия нежелательных примесей, таких как компоненты геля, а также - в случае негативной селекции (то есть истощения из раствора модифицированных молекул ДНК и получения раствора целевой немодифицированной ДНК) исключают риск повреждения целевой ДНК в результате механического воздействия. Однако данные методы имеют ограниченную область применения, поскольку требуют предварительной модификации пула молекул ДНК in vitro, то есть априори сначала требуют выделения молекул ДНК для их модификации.
Таким образом, оптимальным методом селекции целевых фрагментов ДНК стал бы метод, позволяющий разделять молекулы ДНК по принципу наличия у них уникальных характеристик, который бы не требовал предварительных манипуляций in vitro для их внесения в молекулы ДНК. Такими характеристиками могут являться специфические последовательности ДНК, присутствие или отсутствие которых потенциально может быть использовано как основа для разделения гетерогенной смеси молекул ДНК.
Действительно, на сегодняшний день известно несколько подходов к практической реализации разделения или очистки молекул ДНК, основанных на присутствии в молекулах ДНК специфических нуклеотидных последовательностей. К ним относятся методы, основанные на образовании последовательность-специфичных триплексных структур с иммобилизованным на твердом носителе олигонуклеотиде (Ito et al., 1992), или основанные на аффинном взаимодействии белков со специфическими последовательностями в целевой ДНК - Lac-penpeccopa (или его ДНК-связывающего фрагмента) с включенной во фрагмент ДНК нуклеотидной последовательностью оператора LacO (Darby & Hine, 2005; Darby et al., 2007; Mayrhofer et al., 2008), домена цинковых пальцев фактора транскрипции со специфической последовательностью для его узнавания (Woodgate et al., 2002) или металлорегуляторного бактериального белка MerR с его сайтом узнавания, включенным в молекулы ДНК (Kostal et al., 2004). При использовании белков для селекции молекул ДНК за счет связывания специфической последовательности ДНК, белок, как и олигонуклеотид, может быть иммобилизован на твердом носителе, что позволяет отбирать связавшиеся молекулы ДНК из раствора. В частности, для иммобилизации белка на твердом носителе возможно использование носителя с ковалентно связанным глутатионом, в случае, когда сам белок представляет собой химеру специфически взаимодействующего с ДНК белка (или его части, достаточной для такого взаимодействия) с глутатион-S-трансферазой, которая, за счет взаимодействия с глутатионом, обеспечивает иммобилизацию на твердом носителе (Woodgate et al., 2002).
Метод очистки, основанный на образовании триплексных структур, использует способность гомопиримидиновых нуклеотидов формировать такие структуры с гомопуриновыми последовательностями двухцепочечной ДНК (Ito et al., 1992). Очевидно, что присутствие гомопуриновых последовательностей часто может не являться специфическим признаком одного пула молекул ДНК, который необходимо сепарировать от другого, из-за случайного присутствия сходных по составу последовательностей в молекулах ДНК другого пула, вероятность чего возрастает с увеличением длины молекул ДНК. Кроме того, данный метод характеризуется медленной кинетикой образования комплексов, что непосредственно влияет на его эфективность.
Использование Lac-репрессора для аффинной очистки молекул ДНК, содержащих узнаваемую им нуклеотидную последовательность LacO-оператора, было предложено как способ очистки свободной от вектора ДНК - суперспирализованных мини-кольцевых молекул с целью их применения в области генной терапии (Mayrhofer et al., 2008). Однако для целей сепарации линейных или кольцевых релаксированных молекул ДНК данный подход не является оптимальным, поскольку аффинность Lac-penpeccopa к ZacO-сайтам ослабляется при нахождении ДНК в релаксированном, а не сверхскрученном состоянии (Whitson et al., 1987а; Whitson et al., 1987b), что будет негативно сказываться на эффективности аффинной сорбции.
Два других упомянутых выше известных подхода, в которых используются домен цинковых пальцев и белок MerR, также основаны на принципе селекции молекул ДНК при наличии в их составе специфических нуклеотидных последовательностей, узнаваемых этими транскрипционными факторами. Оба метода были разработаны для очистки интактных молекул плазмидной ДНК из клеток E.coli по принципу положительной селекции, когда специфические последовательности включены в состав последовательности вектора.
Следует отметить, что все вышеперечисленные методы основаны на принципе положительной селекции и требуют присутствия в целевых молекулах ДНК специфических нуклеотидных последовательностей. В то же время, включение таких последовательностей в целевые молекулы ДНК для возможности их селекции при очистке может потенциально оказывать влияние на их целевые функции, и присутствие таких дополнительных последовательностей в целом является нежелательным.
Как изложено выше, для основанной на принципе присутствия специфической нуклеотидной последовательности очистке молекул ДНК в качестве лиганда возможно применение белков, способных узнавать такие последовательности. В частности, приведенные примеры включают использование белков-регуляторов транскрипции (транскрипционных факторов), которые специфически узнают определенные последовательности ДНК, вводимые в целевые молекулы ДНК. Помимо факторов транскрипции, к белкам, способным узнавать определенные последовательности ДНК и связываться с ними, относятся эндонуклеазы рестрикции и сайт-специфические рекомбиназы. Однако, в отличие от факторов транскрипции, которые не модифицируют молекулу ДНК при связывании, взаимодействие эндонуклеаз рестрикции и сайт-специфических рекомбиназ с ДНК приводит к расщеплению ДНК в месте связывания или рекомбинационному событию, соответственно, ковалентно изменяя структуру молекулы ДНК.
Cre является одной из известных сайт-специфических рекомбиназ, кодируемой геномом бактериофага Р1. Сайт рекомбинации для рекомбиназы Cre, LoxP, представляет собой последовательность из двух инвертированных повторов длинною 13 пар нуклеотидов (п.н.), разделенных спейсером в 8 п. н. (Sadowski, 1986). Cre-рекомбиназа связывается с ZoxP-сайтом, после чего за счет белок-белковых взаимодействий формируется структура, называемая синапсом, состоящая из двух сближенных LoxP-сайтов со связанными с ними молекулами Cre, после чего происходит расщепление цепей ДНК и обмен одной или двумя цепями с последующей диссоциацией молекул рекомбиназы с ДНК (Ghosh et al., 2007; Ringrose et at., 1998). Существенным отличием рекомбиназы Cre от ряда других рекомбиназ является то, что ее эффективное связывание с сайтами узнавания не зависит от наличия сверхспирализации в молекуле ДНК (Sadowski, 1986), что позволяет эффективно взаимодействовать с ZoxP-сайтом не только в замкнутых кольцевых сверхспирализованных молекулах ДНК, но и в релаксированных кольцевых и линейных молекулах ДНК, в которых отсутствует сверхспирализация.
Раскрытие настоящего изобретения
Настоящее изобретение относится к новому способу разделения молекул ДНК, позволяющему проводить очистку целевых молекул ДНК от нецелевых молекул ДНК из их смеси в растворе. В частности, настоящее изобретение позволяет осуществлять очистку целевых двухцепочечных молекул ДНК от иных молекул двухцепочечной ДНК, как кольцевых, так и линейных, непосредственно в растворе, что исключает необходимость проведения дополнительных манипуляций, таких как рутинно используемое для этих целей разделение продуктов расщепления с использованием гель-электрофореза.
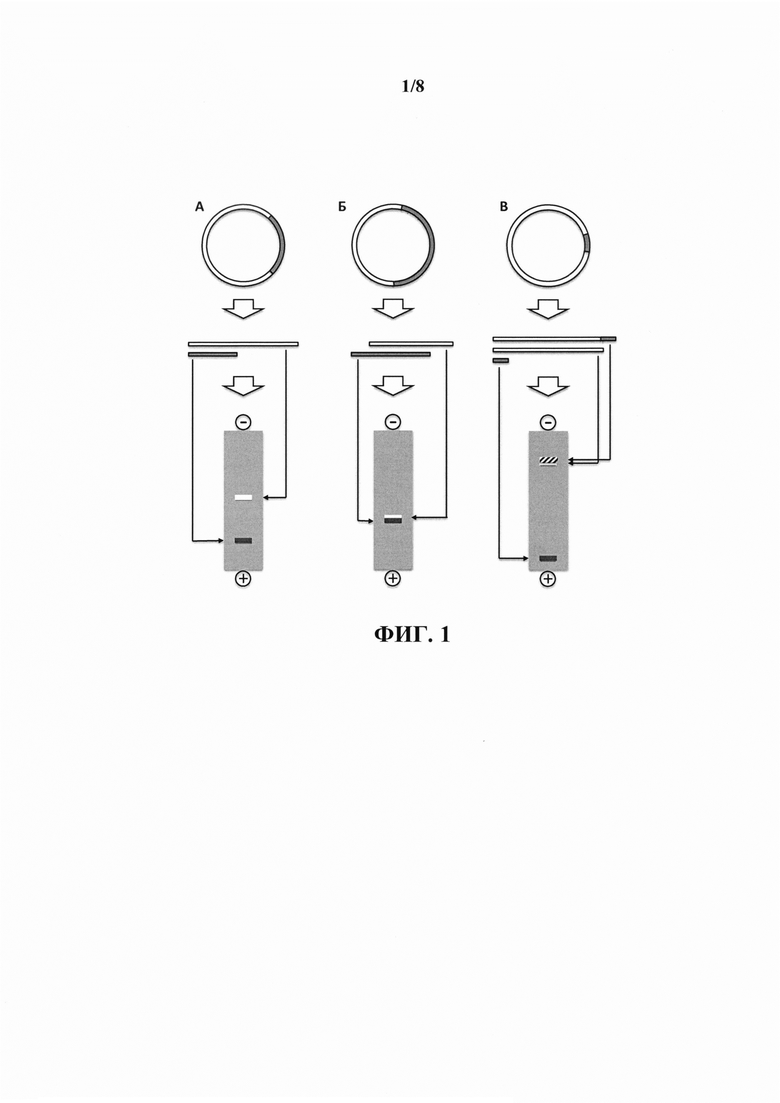
В настоящем изобретении, в отличие от иных способов очистки целевых молекул ДНК, селекция молекул ДНК реализуется не на основании различия в их длинах. Отсутствие необходимости селекции целевых молекул ДНК от нецелевых на основании различий в их размерах, в частности, позволяет исключить невозможность (при использовании, например, гель-электрофореза) очистки целевого фрагмента при его длине, равной или близкой к длине нецелевого фрагмента. При необходимости очистки фрагмента-вставки плазмидного вектора после расщепления плазмиды в случае, когда длина вектора мала по сравнению с длинной целевого фрагмента, предложенный способ также позволяет исключить контаминацию целевого фрагмента линейными продуктами неполного расщепления плазмидной ДНК, содержащей последовательности целевого фрагмента и вектора, из-за невозможности их качественного разделения в результате гель-электрофореза или иными методами, основанными на различиях в длинах молекул ДНК, что обусловлено малыми различиями в их длинах. Ситуации, в которых затруднительно или невозможно получение неконтаминированного иными продуктами расщепления целевого фрагмента ДНК после расщепления плазмидной ДНК с помощью разделения на основании различий в длинах молекул ДНК, проиллюстрированы на фиг. 1.
В настоящем изобретении для разделения молекул ДНК используется принцип негативной селекции. Это позволяет избежать необходимости иммобилизации целевого фрагмента ДНК на твердом носителе в процессе очистки, на чем основаны многие традиционные методы разделения и очистки молекул ДНК и что может приводить к механическим повреждениям длинных молекул ДНК (разрывам). Кроме того, исключение процесса электрофореза в геле, широко используемого при разделении молекул ДНК, исключает риск контаминации целевого фрагмента ДНК компонентами геля, что может негативно влиять на результаты его дальнейшего использования.
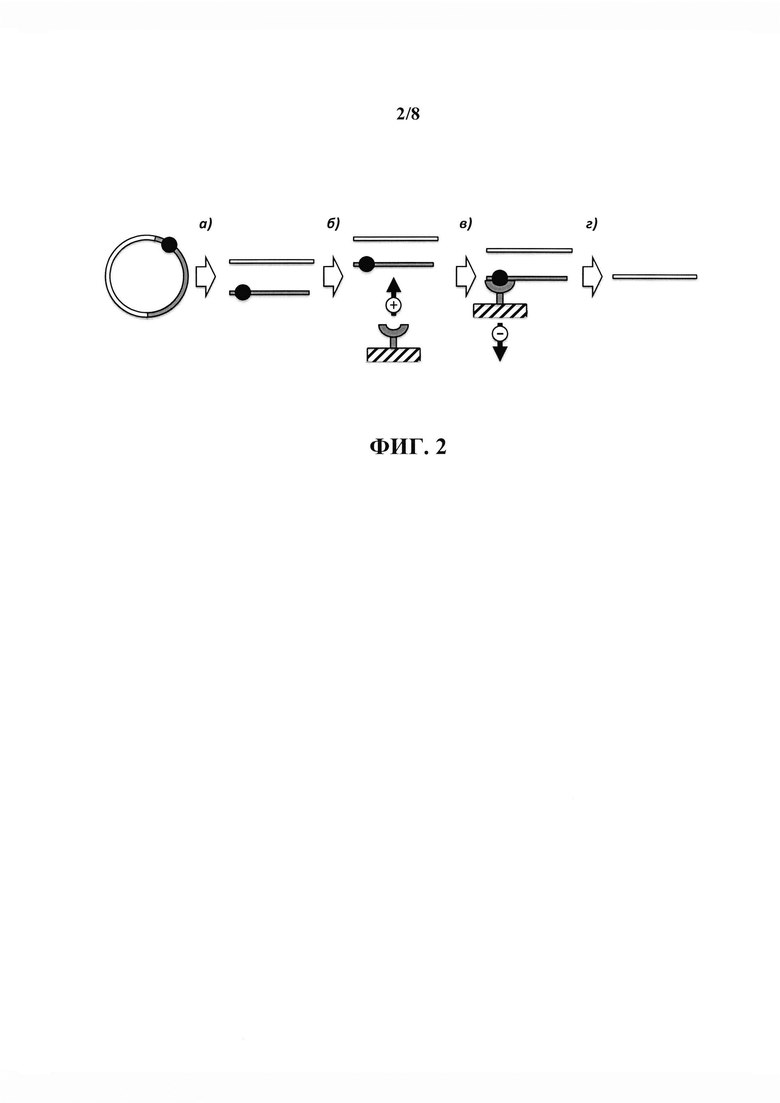
В настоящем изобретении, как и в известных из уровня техники аналогах (Darby & Hine, 2005; Darby et al., 2007; Mayrhofer et al., 2008), (Woodgate et al., 2002), (Kostal et al., 2004), (Ito et al., 1992), разделение молекул ДНК осуществляется с использованием агента, способного взаимодействовать и связывать молекулы ДНК, имеющие в своем составе специфические нуклеотидные последовательности. Благодаря способности агента быть иммобилизованным на твердой фазе или быть переведенным в твердую фазу, становится возможным иммобилизация таких молекулы ДНК, связанных с агентом, из жидкой фазы на твердую фазу, тем самым осуществляя разделение молекул ДНК, имеющих в своем составе специфическую нуклеотидную последовательность, и не имеющую ее. Однако в приведенных выше существующих аналогах целевые молекулы ДНК должны содержать в своем составе дополнительную последовательность нуклеотидов, обеспечивающих их селективное взаимодействие с агентом для связывания ДНК. В отличие от существующих аналогов, в настоящем изобретении описан способ разделения целевых и нецелевых молекул ДНК, не требующий внесения дополнительных последовательностей в целевые молекулы ДНК, что достигается за счет использования принципа негативной селекции. Для этого специфическая нулеотидная последовательность, обеспечивающая селективное взаимодействие с агентом для перевода фрагмента ДНК на твердую фазу из раствора, включается в последовательность нецелевых молекул ДНК, что позволяет селективно деплицировать такие молекулы из исходного раствора ДНК, содержащего смесь целевых и нецелевых молекул ДНК, тем самым позволяя получить раствор, содержащий только целевые молекулы ДНК без примеси нецелевых молекул ДНК. Реализованный в настоящем изобретении принцип очистки целевых молекул ДНК проиллюстрирован на фиг. 2.
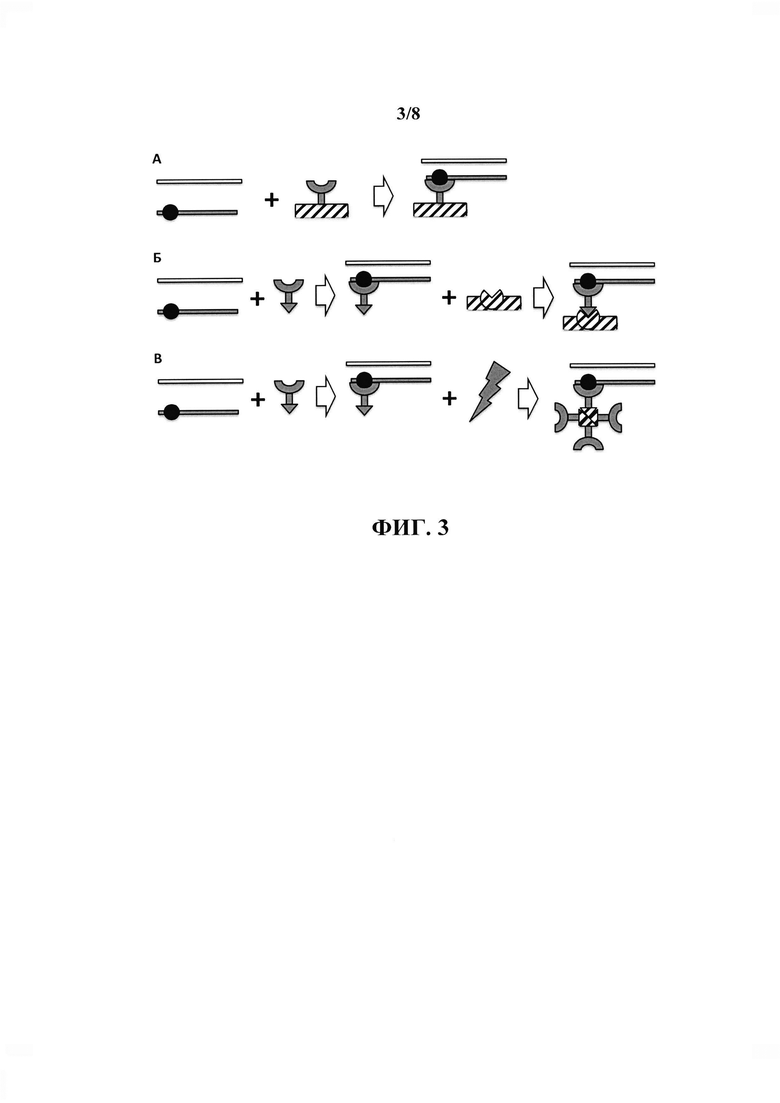
Агент, способный связываться с нецелевыми молекулами ДНК, должен обеспечивать их перевод из жидкой фазы на твердую фазу. Специалисту в данной области очевидно, что это может быть реализовано двумя способами - до проведения связывания агента с молекулами ДНК или после него. При этом при иммобилизации на твердой фазе агента до связывания с ДНК, иммобилизация может обеспечиваться как с помощью ковалентной сшивки молекул агента с твердым носителем (в качестве примера можно привести ковалентную кросс-сшивку агента, являющегося белковой молекулой, с CNBr-активированной сефарозой; специалисту в данной области очевидно, что приведенный пример не является единственным и не исчерпывает все возможные способы ковалентной иммобилизации агентов различной природы на твердом носителе) или обеспечиваться за счет нековалентного взаимодействия (в качестве примера можно привести взаимодействие агента, в состав которого входит биотин, с предварительно иммобилизованным на твердой фазе белком стрептавидином; специалисту в данной области очевидно, что приведенный пример не является единственным и не исчерпывает все возможные способы нековалентной иммобилизации агентов различной природы на твердом носителе). Специалисту также очевидно, что технически процесс сорбции нецелевых молекул ДНК на твердой фазе может осуществляться как в формате batch, так и в форме хроматографии на колонке с аффинным носителем. Также из существующего уровня техники известно, что перевод агента в твердую фазу может достигаться за счет присоединения к нему специфических белковых последовательностей, обеспечивающих фазовый переход при изменении температуры (Kostal et al., 2004). Возможные принципиальные варианты иммобилизации агента и связанных с ним молекул ДНК на твердой фазе проиллюстрированы на фиг. 3.
В качестве агентов, обеспечивающих взаимодействие с молекулами ДНК, имеющими в своем составе специфические нуклеотидные последовательности, могут быть использованы белки, обладающие способностью узнавать и связываться с такими последовательностями. Из существующего уровня техники известно, что роль таких белков могут играть белки, являющиеся транскрипционными факторами, которые участвуют в регуляции транскрипции и специфически взаимодействуют со своими цис-действующими элементами, или домены таких белков, достаточные для такого взаимодействия ((Darby & Hine, 2005; Darby et al., 2007; Mayrhofer et al., 2008), (Woodgate et al., 2002), (Kostal et al., 2004)). Данный тип агентов, способных связывать специфические последовательности ДНК, не приводит к модификации ДНК, и позволяет реализовать эффективное связывание молекул ДНК, имеющих в своем составе последовательности, узнаваемые транскрипционным фактором, как это известно из существующего уровня техники. Соответственно, в одном из своих воплощений в настоящем изобретении для специфического связывания ДНК могут использоваться в качестве агентов белки из числа транскрипционных факторов, а также их части, способные специфически взаимодействовать со своими цис-действующими элементами, и специфические нуклеотидные последовательности, узнаваемые транскрипционным фактором, которые включены в состав нецелевых молекул ДНК.
Помимо транскрипционных факторов, еще некоторые типы белков способны узнавать специфические нуклеотидные последовательности. Однако, в отличие от известных их настоящего уровня техники примеров реализации разделения молекул ДНК, основанных на взаимодействии белков со специфическими нуклеотидными последовательностями, в которых используются белки, не способные к внесению изменений в ковалентную структуру ДНК после связывания, эти белки после связывания вносят ковалентные изменения в ДНК, после чего диссоциируют с молекулы ДНК. Очевидно, что использование таких белков в качестве агента может быть неэффективно из-за процесса диссоциации. Кроме того, при использовании таких агентов, на эффективность истощения нецелевых молекул ДНК может негативно влиять происходящие в процессе разделения ковалентные модификации молекул ДНК. Поэтому настоящее изобретение в одном из своих воплощений предусматривает возможность использование в качестве агентов белков, способных узнавать специфические нуклеотидные последовательности и вносить ковалентных модификации в ДНК, которые, однако, несут мутации, не влияющие на их способность связывать специфические нуклеотидные последовательности, но нарушающие их энзиматическую активность в отношении внесения ковалентных модификаций в ДНК. Применение таких мутантных версий останавливает цикл работы фермента «связывание - модификация - диссоциация» на стадии связывания, обеспечивая эффективное взаимодействие агента с ДНК и исключая потенциальное влияние появления ковалентных модификаций ДНК на этот процесс.
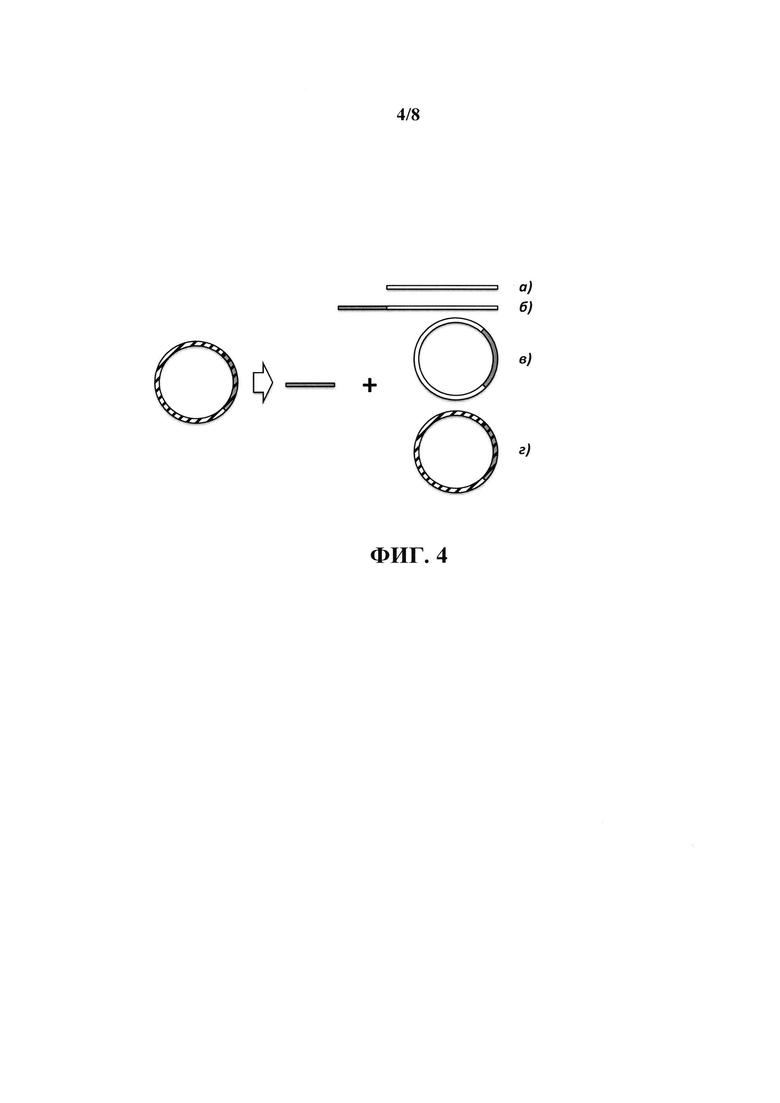
Известно, что ДНК-связывающие белки, обладающие способностью взаимодействовать со специфическими нуклеотидными последовательностями, могут изменять свою аффинность при изменении степени сверхспирализации ДНК. Примером такого белка, использованного для последовательность-специфичной очистки молекул ДНК, является Lac-репрессор, аффинность которого к LacO-сайтам ослабляется при нахождении ДНК в релаксированном, а не сверхспирализованном состоянии (Whitson et al., 1987а; Whitson et al., 1987b). В одном из применений настоящего изобретения может возникать необходимость очистки целевых молекул ДНК из смеси, содержащей как релаксированные кольцевые, линейные, так и сверхспирализованные нецелевые молекулы ДНК. Примером такого применения является очистка целевого фрагмента ДНК после расщепления плазмидной ДНК, которая должна обеспечивать истощение всех возможных продуктов расщепления плазмиды, в том числе и продуктов неполного расщепления, которые, в том числе, могут содержать сверхспирализованные кольцевые молекулы ДНК (см. фиг. 4). Поэтому предпочтительно, чтобы используемый агент, обеспечивающий опосредованной специфической нуклеотидной последовательностью взаимодействие с молекулами ДНК, обладал неизменной аффинностью к ДНК вне зависимости от состояния ее сверхспирализации, что обеспечит одинаково эффективное связывание всех возможных нецелевых молекул ДНК.
Настоящее изобретение предполагает присутствие в составе нецелевых молекул ДНК, от которых осуществляется очистка, специфической нуклеотидной последовательности, обеспечивающей взаимодействие с агентом. В одном из своих воплощений настоящее изобретение предусматривает включение одной копии такой последовательности в одну молекулу ДНК, что будет обеспечивать ее взаимодействие с агентом. При этом каждая конкретная пара «агент - специфическая нуклеотидная последовательность» характеризуется определенной константой связывания, которая определяет, в зависимости от их концентраций, остаточную концентрацию нецелевых молекул ДНК в растворе в равновесном состоянии. Специалисту в данной области очевидно, что включение в состав молекулы ДНК более чем одной копии специфической нуклеотидной последовательности при прочих равных условиях будет сдвигать равновесное состояние в сторону уменьшения концентрации предназначенных для сорбции и истощения молекул ДНК, тем самым обеспечивая более полную и эффективную очистку от нецелевых молекул ДНК. Таким образом, в предпочтительном варианте осуществления настоящее изобретение предусматривает включение в нецелевые молекулы ДНК, от которых производится отделение целевых молекул ДНК, более чем одной копии специфической нуклеотидной последовательности, с которой взаимодействует агент.
В одном из своих воплощений в настоящем изобретений в качестве агента для связывания ДНК может использоваться сайт-специфическая рекомбиназа Cre, специфически взаимодействующая с достаточно высокой константой связывания (Ringrose et al., 1998) с последовательностью нуклеотидов, называемой LoxP-сайтом, представляющую собой последовательность из 34 нуклеотидов (нт), состоящую из двух инвертированных повторов длинною 13 нт, разделенных спейсером в 8 нт. Поскольку рекомбиназа Cre относится к ДНК-связывающим белкам, ковалентно модифицирующим ДНК (в результате процесса рекомбинации), то для применения рекомбиназы Cre в настоящем изобретении возникает необходимость использования ее мутантов, сохраняющих способность специфически взаимодействовать с ZoxP-сайтами, но дефектных по рекомбинационной активности.
Процесс рекомбинации, опосредованный Cre-рекомбиназой, включает в себя несколько стадий. Первой является взаимодействие молекул рекомбиназы с ZoxP-сайтами в составе молекулы ДНК. Далее происходит образование синаптических структур, или синапсов, состоящих из двух ZoxP-сайтов, за счет взаимодействия между субъединицами рекомбиназы Cre, что предшествует процессу рекомбинации и последующей диссоциации молекул Cre с ДНК (Ghosh et al., 2007). Известно, что мутанты рекомбиназы Cre, дефектные по рекомбинационной активности, но сохраняющие способность взаимодействовать с ZoxP-сайтами, могут как сохранять способность образовывать синапсы, так и быть лишенной этой способности. Поскольку ключевым требованием к агенту является сохранение способности взаимодействовать со специфической нуклеотидной последовательностью, оба типа мутантов могут быть использованы при практической реализации настоящего изобретения. В одном из своих воплощений, такими мутантами могут быть белки Cre, содержащие замену аминокислотного остатка лизина в позиции 201 полипептидной цепи на аланин (Cre(К201 А)) или замену аминокислотного остатка тирозина в позиции 324 полипептидной цепи на фенилаланин (Cre(Y324F). При этом известно, что мутант Cre(К201А) сохраняет способность образовывать синапсы, в то время как второй мутант, Cre(Y324F), дефектен не только по каталитической активности, но и по способности образовывать синаптические структуры (Ghosh et al., 2007). Специалисту в настоящей области очевидно, что потенциально возможно получение других мутантов белка Cre, сохраняющих способность специфически взаимодействовать с LoxP-сайтами, но дефектных по рекомбинационной активности, которые могут использоваться при осуществлении настоящего изобретения.
Специалисту в настоящей области очевидно, что при практической реализации настоящего изобретения при применении в качестве ДНК-связывающего агента мутантных версий рекомбиназы Cre в качестве узнаваемой ими последовательностей ДНК могут быть использованы не только ZoxP-сайты, но и их измененные версии, сохраняющие способность взаимодействовать с рекомбиназой Cre, например, описанные в (Kolb, 2001; Langer et al., 2002).
Специалисту в настоящей области также очевидно, что при практической реализации настоящего изобретения могут быть использованы варианты рекомбиназы Cre, специфичность взаимодействия которых с молекулами ДНК изменена в результате мутаций, в совокупности с ZoxP-сайтами с соответствующими нуклеотидными заменами, обеспечивающими взаимодействия с измененным белком Cre, например, описанные в (Hartung М & Kisters-Woike, 1998).
В одном из своих воплощений, для иммобилизации взаимодействующего с молекулами ДНК, содержащих ZoxP-сайты, белка Cre на твердой фазе могут использоваться химерные белки, полипептидные цепи которых включают в себя полипептидную цепь мутантного белка Cre и полипептидную цепь белка глутатион-S-трансферазы, обеспечивающего нековалентную сорбцию на твердом носителе глутатион-сефарозе.
В одном из предпочтительных вариантов осуществления настоящее изобретение предусматривает выбор агента из числа белков, взаимодействие которых со специфической последовательностью ДНК не зависит от наличия сверхспирализации в молекуле ДНК. В варианте осуществления изобретения, предусматривающем использование мутантных вариантов белка Cre в качестве ДНК-связывающего агента, это предпочтение выполняется, поскольку известно, что взаимодействие Cre с ДНК не зависит от наличия сверхспирализации ДНК (Hoess et al., 1984).
Краткое описание фигур
Далее изобретение будет более подробно раскрыто со ссылкой на отдельные иллюстративные фигуры.
На фиг. 1 проиллюстрированы ситуации, в которых затруднительно или невозможно получение неконтаминированного иными продуктами расщепления целевого фрагмента ДНК после расщепления плазмидной ДНК с помощью их разделения на основании различия длин молекул ДНК. На панеле А в результате расщепления плазмидной ДНК целевой фрагмент ДНК (полоса белого цвета) по своей длине существенно отличается от нецелевого фрагмента ДНК (полоса серого цвета), что позволяет эффективно разделить их в результате гель-электрофореза (внизу). На панеле Б целевой и нецелевой фрагменты ДНК имеют незначительное различие в длинах, что не позволяет эффективно разделить их в результате гель-электрофореза. На панеле В проиллюстрирована ситуация, когда нецелевой фрагмент ДНК существенно меньше по длине целевого фрагмента, что не позволяет отделить целевой фрагмент от продукта неполного расщепления плазмидной ДНК (полоса со штриховкой на схематическом изображении геля внизу).
На фиг. 2 представлена принципиальная схема очистки целевых фрагментов ДНК (полоса белого цвета) от нецелевых фрагментов ДНК (полоса серого цвета), имеющих в своем составе специфическую нуклеотидную последовательность (круг черного цвета), получаемых в результате расщепления плазмидной ДНК (а)). К раствору фрагментов ДНК, получаемых в результате расщепления плазмиды, добавляется иммобилизованный на твердом носителе агент (полукольцо серого цвета на схеме), способный взаимодействовать со специфической нуклеотидной последовательностью (б)). После опосредованного специфической нуклеотидной последовательностью связывания нецелевых фрагментов ДНК агентом носитель с иммобилизованными на твердой фазе комплексами агента и нецелевых фрагментов ДНК удаляется (в)), оставляя в растворе только целевой фрагмент ДНК (г)).
На фиг. 3 проиллюстрированы возможные принципиальные варианты иммобилизации агента (полукольцо серого цвета) и связанных с ним нецелевых молекул ДНК (полоса серого цвета), содержащих специфическую нуклеотидную последовательность (круг черного цвета) на твердой фазе (обозначена штриховкой). А - агент может быть изначально иммобилизован на твердом носителе, который добавляется к раствору смеси нецелевых и целевых (полоса белого цвета) молекул ДНК, после чего происходит связывание агентом нецелевых молекул ДНК и их иммобилизация на твердой фазе. Связывание нецелевых молекул ДНК может осуществляться агентом, находящимся в растворе, после чего иммобилизация комплекса агента с нецелевым фрагментом ДНК на твердой фазе осуществляется за счет специфического взаимодействия агента с добавляемым твердым носителем (Б) или за счет индуцируемого внешним воздействием (молния на схеме) фазового перехода агента (В).
На фиг. 4 схематически изображены возможные нецелевые молекулы ДНК после расщепления плазмиды, содержащей целевой фрагмент ДНК (полоса белого цвета): продукт полного расщепления - линейный фрагмент ДНК, содержащий векторную последовательность (полоса серого цвета) (а)), и продукты неполного расщепления - расщепление только по одному сайту (б)), кольцевые релаксированная (без штриховки) (в)) и сверхспирализованная (со штриховкой) (г)) молекулы плазмидной ДНК.
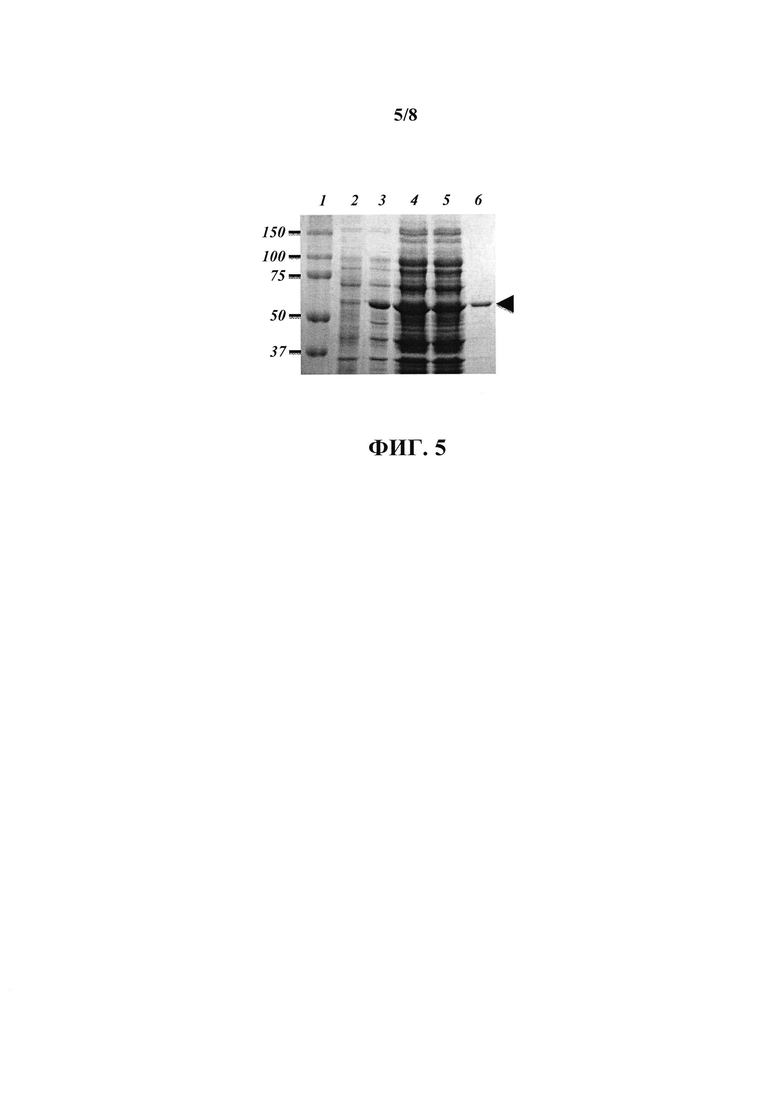
На фиг. 5 представлены репрезентативные результаты очистки и иммобилизации на глутатион-сефарозе рекомбинантного белка GST-Cre(Y324F). Суммарные лизаты клеток E.coli, трансформированных плазмидой pGEX-4T-1 с клонированной в нее кДНК мутантного варианта рекомбиназы Cre, Cre(Y324F), для экспрессии химерного белка GST-Cre(Y324F)) до (дорожка 2) и после (дорожка 3) индукции экспрессии рекомбинантного белка, полученные в результате обработки ультразвуком лизаты тех же клеток E.coli после индукции экспрессии рекомбинантного белка до (дорожка 4) и после (дорожка 5) сорбции рекомбинантного белка на глутатион-сефарозе, и белки, элюированные с помощью кипячения в буфере для нанесения белков на гель с 1 мкл глутатион-сефарозы после сорбции рекомбинантного белка (дорожка 6), разделяли в 10% полиакриламидном геле в присутствии додецилсульфата натрия. После разделения белки в геле окрашивались красителем Bio-Safe Coomassie (Bio-Rad,). Положение рекомбинантного белка GST-Cre(Y324F) в геле показано стрелкой. В дорожке 1 нанесены маркеры молекулярных масс белков, с указанием молекулярных масс компонентов маркера (в кДа) слева.
На фиг. 6 приведены схематические изображения модельных плазмид pBl-NeoR, pBl-LoxP-NeoR и pBl-LoxP-NeoR-LoxP и продуктов их расщепления эндонуклеазами рестрикции EcoRI и SalI. Векторная последовательность плазмиды pBluescript SKII(-) длинною около 3 тысяч пар оснований изображена белым цветом, кДНК неомицин фосфотрансферазы II (~1000 пар нуклеотидов) изображена серым цветом. ZoxP-сайты в плазмидах pBl-LoxP-NeoR, pBl-LoxP-NeoR-LoxP и продуктах их расщепления изображены в виде кругов черного цвета. В плазмидах также отмечены положения сайтов узнавания эндонуклеаз рестрикции EcoRI и SalI.
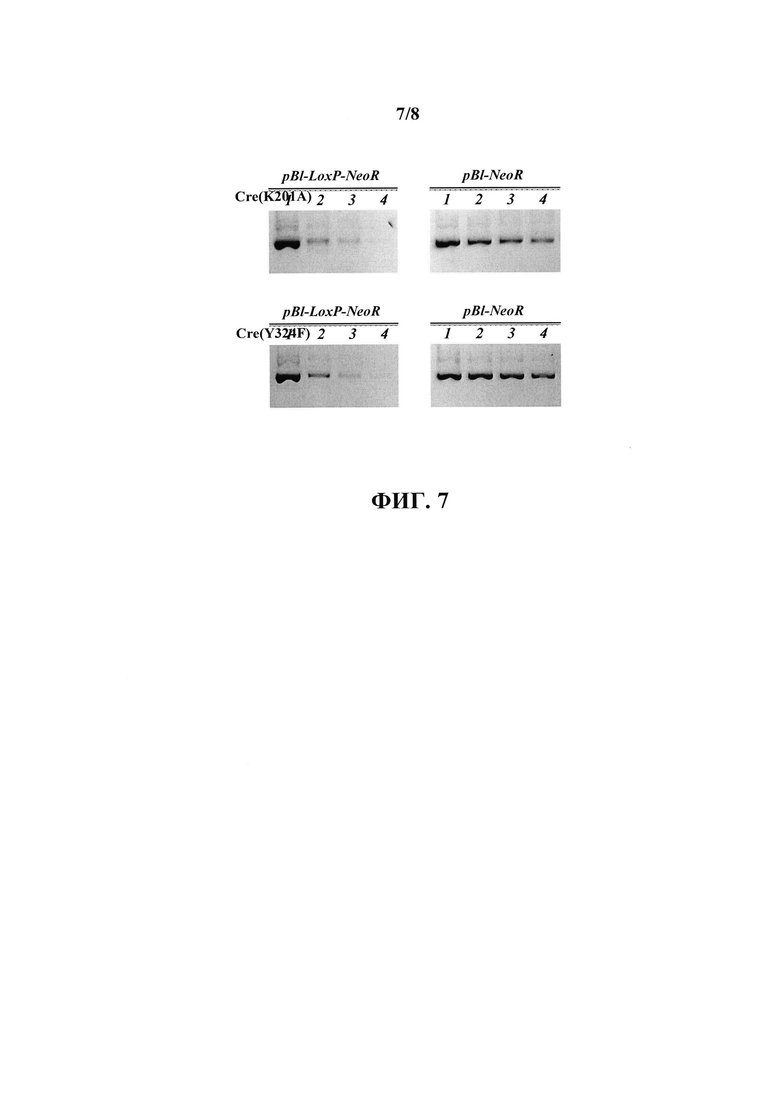
На фиг. 7 приведены результаты агарозного гель-электрофореза указанных плазмидных ДНК (pBl-NeoR и pBl-LoxP-NeoR), присутствующих в растворе до (дорожки 7) и после первой (дорожки 2), второй (дорожки 3) и третьей (дорожки 4) инкубации с глутатион-сефарозой с иммобилизованными белками GST-Cre(K201A) (Cre (201А)) или GST-Cre(Y324F) (Cre(Y324F)).
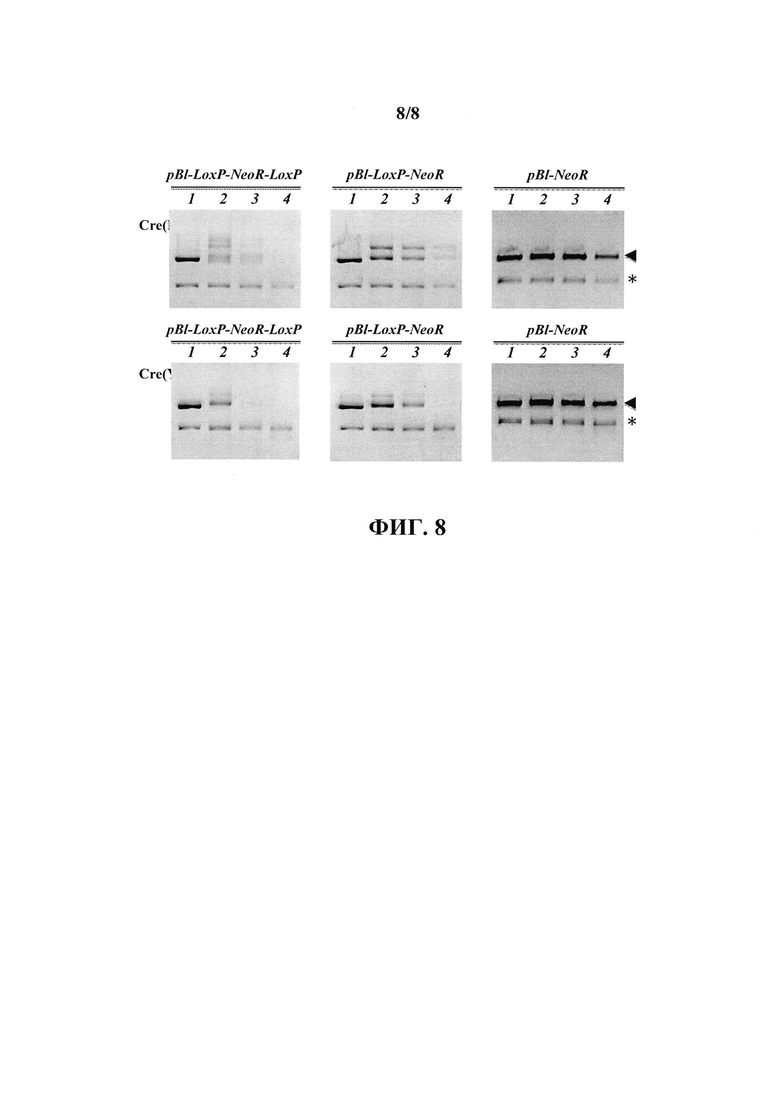
На фиг. 8 приведены результаты агарозного гель-электрофореза продуктов расщепления указанных плазмидных ДНК (pBl-NeoR, pBl-LoxP-NeoR и pBl-LoxP-NeoR-LoxP) рестрикционными эндонуклеазами EcoRI и SalI, присутствующих в растворе до (дорожки 1) и после первой (дорожки 2), второй (дорожки 3) и третьей (дорожки 4) инкубации с глутатион-сефарозой с иммобилизованными белками GST-Cre(K201 А) (Cre(201А)) или GST-Cre(Y324F) (Cre(Y324F)). Фрагмент ДНК, являющийся кДНК NeoR, отмечен звездочкой; стрелкой показан фрагмент ДНК, соответствующий вектору.
Описание конкретных примеров осуществления изобретения
Пример 1. Получение иммобилизованных на твердой фазе дефектных по рекомбинационной активности белков Cre
кДНК рекомбиназы Cre была амплифицирована на матрице плазмиды pBl-hTERT-Surv269-Cre-3DNMT1 с праймерами 5'-agaattcgagctccccaagaagaagaggaag-3' and 5'-agtcgacctaatcgccatcttccag-3', содержащими сайты узнавания эндонуклеаз рестрикции EcoRI и SalI, соответственно, и клонирована в вектор для клонирования ПЦР-продуктов pJET1 (Fermentas).
Для получения мутантных версий рекомбиназы Cre, дефектных по рекомбинационной активности (замена лизина-201 на аланин, К201 А, и замена тирозина-324 на фенилаланин, Y324F (Ghosh et al., 2007)), в кДНК Cre в векторе pJET1 были внесены соответствующие нуклеотидные замены с использованием набора QuickChange site-directed mutagenesis kit (Agilent Technologies) и олигонуклеотидов 5'-atccatattggcagaacggcaacgctggttagcaccgc-3' и 5'-gcggtgctaaccagcgttgccgttctgccaatatggat-3' для мутанта K201 А, и 5'-caatgtaaatattgtcatgaactttatccgtaacctggatagtgaaa-3, and 5'-tttcactatccaggttacggataaagttcatgacaatatttacattg-31 для мутанта Y324F в соответствии с протоколом производителя. Полученные кДНК мутантных вариантов рекомбиназы Cre были клонированы в вектор pGEX-4T-1 (GE Healthcare) для экспрессии мутантов Cre как химер с глутатион-S-трансферазой (GST) (белки GST-Cre(K201 А) и GST-Cre(Y324F)).
Рекомбинантные белки GST-Cre(K201A), GST-Cre(Y324F) и GST экспрессировали в клетках E.coli, штамм Rosetta-gami DE3pLysS, и очищали на глутатион-сефарозе (GE Healthcare) в соответствии с протоколом производителя, причем инкубацию глутатион-сефарозы с клеточным лизатом, содержащим рекомбинантный белок, проводили в избытке рекомбинантного белка по отношению к емкости носителя для насыщения сайтов связывания глутатион-сефарозы белком GST, GST-Cre(K201A) или GST-Cre(Y324F). Глутатион-сефарозу со связавшимся белком промывали фосфатно-солевым буфером и хранили в фосфатно-солевым буфером с добавлением азида натрия (0.02%) при температуре +4°С. На фиг. 5 представлены репрезентативные результаты очистки и иммобилизации на глутатион-сефарозе рекомбинантных белков на примере белка GST-Cre(Y324F).
Пример 2. Получение модельных молекул ДНК
Фрагмент ДНК, содержащий LoxP-стт, был вырезан из плазмиды pCI-CMV-LoxP-NeoR-LoxP-FCU1 с помощью эндонуклеаз рестрикции EcoRI и XhoI и субклонирован в вектор pBK-CMV (Stratagene) по сайтам узнавания рестрикционных эндонуклеаз EcoRI и XhoI, после чего сайт узнавания рестрикционной эндонуклеазы EcoRI был нарушен в результате расщепления плазмиды рестрикционной эндонуклеазы EcoRI с последующей обработкой продуктов расщепления фрагментом Кленова и лигированием (плазмида рВК-LoxP). Плазмида pBK-LoxP была использована в качестве матрицы для амплификации фрагмента ДНК с Z-oxP-сайтом с использованием праймеров 5'-agaattcccgataatattcaa-3' и 5'-aagatctaaactcctcttcagacc-3' с его последующим клонированием в вектор pJET1 (Fermentas) (плазмида pJET1-LoxP). Для получения плазмиды pBl-LoxP, EcoRI-BglII фрагмент ДНК из плазмиды pJET1-LoxP, содержащий LoxP-сайт, был субклонирован в вектор pBluescript SKII(-) (Stratagene) по сайтам узнавания рестрикционных эндонуклеаз EcoRI и BamHI. Для получения плазмиды pBl-LoxP-NeoR, кДНК неомицин фосфотрансферазы II (~1000 пар нуклеотидов) была субклонирована из плазмиды pCI-CMV-LoxP-NeoR-LoxP-FCU1 в плазмиду pBl-LoxP между сайтами узнавания эндонуклеаз рестрикции EcoRI и SalI. Для получения плазмиды pBl-LoxP-NeoR-LoxP, KpnI-SalI фрагмент из плазмиды pBK-LoxP, содержащий ZoxP-сайт, был субклонирован между сайтами узнавания эндонуклеаз рестрикции KpnI и SalI в плазмиду pBl-LoxP-NeoR. Для получения контрольной плазмиды pBl-NeoR, содержащей кДНК неомицин фосфотрансферазы II без ZoxP-сайтов, EcoRI-SalI фрагмент ДНК из плазмиды pCI-CMV-LoxP-NeoR-LoxP-FCU1, содержащий кДНК неомицин фосфотрансферазы II, был клонирован в вектор pBluescript SKII(-) между сайтами узнавания соответствующих эндонуклеаз рестрикции.
Эндонуклеазы рестрикции EcoRI и SalI использовались для расщепления плазмид pBl-NeoR, pBl-LoxP-NeoR и pBl-LoxP-NeoR-LoxP с получением фрагмента кДНК неомицин фосфотрансферазы II, длинною около 1000 пар нуклеотидов, не содержащей в своем составе последовательности ZoxP-сайта, и фрагмента вектора pBluescript SKII(-) длинною около 3000 пар оснований, не содержащего, содержащего один или два LoxP-сайта, соответственно.
Схематические изображения модельных плазмид pBl-NeoR, pBl-LoxP-NeoR и pBl-LoxP-NeoR-LoxP и продуктов их расщепления эндонуклеазами рестрикции EcoRI и SalI приведены на фиг. 6.
Пример 3. Селективная сорбция иммобилизованными на твердой фазе дефектными по рекомбинационной активности белков Cre сверхспирализованных молекул ДНК, содержащих последовательность LoxP-сайта
Для связывания с иммобилизованными на глутатион-сефарозе мутантами белка Cre готовили раствор плазмидных ДНК pBl-NeoR или pBl-LoxP-NeoR в концентрации 50 нг/мкл в буфере для связывания, содержащим 10 мМ Tris-HCl, рН8.0, 400 мМ NaCl, 10 мМ MgCl2 и 1 мМ дитиотриэтола (DTT). Глутатион-сефарозу с иммобилизованным рекомбинантным белком GST, GST-Cre(K201 А) или GST-Cre(Y324F) два раза промывали буфером для связывания, затем к 20 мкл (объем носителя) глутатион-сефарозы с иммобилизованным рекомбинантным белком добавляли 25 мкл раствора плазмидной ДНК и инкубировали с перемешиванием в течение 30 мин. при комнатной температуре. После инкубации носитель осаждали центрифугированием (1000 об./мин., 1 мин.) на дно пробирки. Далее связывание повторяли для супернатанта с новой аликвотой глутатион-сефарозы с иммобилизованным рекомбинантным белком, оставив 2 мкл супернатанта для последующего анализа. Суммарно для каждого образца проводили 3 последовательные инкубации с аликвотами глутатион-сефарозы с иммобилизованным рекомбинантным белком.
Исходный раствор плазмидной ДНК (2 мкл), а также аликвоты супернатанта после каждой инкубации анализировали в агарозном геле в присутствии бромистого этидия. В результате было установлено, что инкубация с глутатион-сефарозы с иммобилизованным рекомбинантным белком GST не приводит к истощению плазмидной ДНК (данные не приведены), что свидетельствует об отсутствии неспецифического связывания ДНК с глутатион-сефарозой и белком GST в выбранных условиях.
При инкубации плазмиды pBl-LoxP-NeoR, имеющей в своей последовательности ZoxP-сайт, с иммобилизованным на глутатион-сефарозе рекомбинантном белке GST-Cre(К201 А) или GST-Cre(Y324F) наблюдалось выраженное истощение плазмидной ДНК из раствора, вплоть до практически полного отсутствия ДНК в растворе после третьей инкубации (фиг. 7). В то же время, при инкубации с идентичной плазмидой pBl-NeoR, в которой, однако, отсутствует ZoxP-сайт, истощение плазмидной ДНК было существенно менее выражено (для мутанта К201А) или практически отсутствовало (для мутанта Y324F) (фиг. 7). Полученные результаты свидетельствуют о специфическом взаимодействии плазмидной ДНК с ZoxP-сайтом с иммобилизованными на твердом носителе белками GST-Cre(K201A) и GST-Cre(Y324F), которое опосредуется ZoxP-сайтом в составе плазмидной ДНК.
Данный пример наглядно демонстрирует, что используемые в качестве агента иммобилизованные на твердой фазе дефектные по рекомбинационной активности белки Cre, в частности, мутанты К201А и Y324F, иммобилизованные на глутатион-сефарозе при экспрессии в виде химер с белком GST, способны селективно взаимодействовать с кольцевыми сверхспирализованными молекулами ДНК, содержащими последовательность LoxP-сайта, и, соответственно, могут применяться для очистки целевых молекул ДНК от таких молекул.
Пример 4. Использование иммобилизованных на твердой фазе дефектных по рекомбинационной активности белков Cre для очистки целевого фрагмента ДНК, полученного в результате расщепления плазмидной ДНК, от фрагмента вектора
Плазмиды pBl-NeoR и идентичная ей плазмида pBl-LoxP-NeoR, имеющая в своем составе LoxP-сайт, были подвергнуты расщеплению эндонуклеазами рестрикции EcoRI и SalI, в результате чего образовывается фрагмент ДНК, представляющий собой кДНК NeoR, не имеющий в своем составе ZoxP-сайта и одинаковый в случае расщепления обоих плазмид, и линейный фрагмент вектора pBluescript SKII(-), который в случае расщепления плазмиды pBl-LoxP-NeoR имеет в своем составе LoxP-сайт (см. фиг. 6). Расщепление проводили в реакции объемом 30 мкл, содержащей 2 мкг плазмидной ДНК в 1х буфере «О» для эндонуклеаз рестрикции (Fermentas). После расщепления к реакции добавляли 10 мкл раствора, содержащего 10 мМ MgCl2, 1.27 М NaCl и 4 мМ DTT для получения 40 мкл раствора расщепленной плазмидной ДНК в концентрации 50 нг/мкл в буфере для связывания (см. Пример 3). Инкубацию расщепленной плазмидной ДНК с иммобилизованными на глутатион-сефарозе мутантами белка Cre проводили, как описано в Примере 3.
В результате анализа фрагментов ДНК, остающихся в растворе после инкубации, было установлено, что при наличии в составе фрагмента ДНК вектора ZoxP-сайта происходит его селективная сорбция из раствора, в то время как фрагмент вставки, не имеющий в своей последовательности ZoxP-сайта, остается в растворе. В то же время, в контрольных инкубациях с продуктами расщепления плазмиды pBl-NeoR, которые не имеют в своих последовательностях ZoxP-сайта, селективного истощения одного из фрагментов не наблюдалось (фиг. 8).
Данный пример наглядно демонстрирует, что иммобилизованные на твердой фазе дефектные по рекомбинационной активности белки Cre, в частности, мутанты К201А и Y324F, иммобилизованные на глутатион-сефарозе при экспрессии в виде химер с белком GST, могут применяться для очистки целевых молекул ДНК от нецелевых молекул ДНК, имеющих в своем составе ZoxP-сайт, в частности, для очистки фрагмента ДНК, полученного в результате расщепления плазмидной ДНК, от фрагмента вектора, последовательность которого содержит ZoxP-сайт.
Пример 5. Увеличение числа ZaxP-сайтов увеличивает эффективность сорбции содержащих их молекул ДНК на твердую фазу с иммобилизованными дефектными по рекомбинационной активности белками Cre
Помимо очистки фрагмента кДНК NeoR, получаемого в результате расщепления плазмиды pBl-LoxP-NeoR, от фрагмента вектора, имеющего в своем составе один ZoxP-сайт (см. Пример 4), нами была проведена очистка фрагмента кДНК NeoR, получаемого в результате расщепления плазмиды pBl-LoxP-NeoR-LoxP, от фрагмента вектора, идентичному получаемому при расщеплении плазмиды pBl-LoxP-NeoR, но имеющего в своем составе не один, а два ZoxP-сайта (см. фиг. 6). Расщепление плазмидных ДНК и инкубации с иммобилизованными на глутатион-сефарозе мутантами белка Cre проводили, как описано в Примере 4.
В результате анализа фрагментов ДНК, остающихся в растворе после инкубаций, было установлено, что при наличии в составе фрагмента ДНК вектора двух, а не одного ZoxP-сайта полное истощение по фрагменту вектора в случае мутанта Cre(Y324F) происходит уже после второй инкубации, в то время как присутствие одной копии ZoxP-сайта в последовательности фрагмента вектора обеспечивает аналогичный результат только после третьей инкубации, а после второй инкубации наблюдается присутствие остаточных количеств фрагмента вектора в растворе (фиг. 8). При использовании мутанта Cre(К201А) наличие одного ZoxP-сайта в последовательности фрагмента вектора не позволило добиться его полного истощения в результате трех последовательных инкубации, однако введение второго ZoxP-сайта позволило полностью сорбировать из раствора фрагмент вектора в таких же условиях (фиг. 8).
Настоящий пример наглядно демонстрирует, что увеличения числа копий специфически узнаваемых ДНК-связывающим агентом нуклеотидных последовательностей в нецелевых молекулах ДНК, в частности, числа копий ZoxP-сайтов при использовании дефектных по рекомбинационной активности белков Cre в качестве агента, в частности, мутантов Cre К201А и Y324F, иммобилизованных на глутатион-сефарозе при экспрессии в виде химер с белком GST, увеличивает эффективность перевода нецелевых молекул ДНК на твердую фазу, тем самым увеличивая эффективность очистки целевых фрагментов ДНК.
Список цитированной литературы
1. Коробко ИВ, Кабишев АА, Киселев СЛ. Идентификация новой протеинкиназы, специфически транскрибирующейся в опухолях мыши с высоким метастатическим потенциалом. Докл. Акад. Наук, 1997, 354: 554-556.
2. Darby RA, Forde GM, Slater NK, Hine AV. Affinity purification of plasmid DNA directly from crude bacterial cell lysates. Biotechnol Bioeng. 2007 Dec 1; 98(5):1103-8.
3. Darby RA, Hine AV. LacI-mediated sequence-specific affinity purification of plasmid DNA for therapeutic applications. FASEB J. 2005 May; 19(7):801-3.
4. Ghosh K, Guo F, Van Duyne GD. Synapsis of loxP sites by Cre recombinase. J Biol Chem. 2007 Aug 17; 282(33):24004-16.
5. Hartung M, Kisters-Woike B. Cre mutants with altered DNA binding properties. J Biol Chem. 1998 Sep 4; 273(36):22884-91.
6. Hoess R, Abremski K, Sternberg N. The nature of the interaction of the PI recombinase Cre with the recombining site loxP. Cold Spring Harb Symp Quant Biol. 1984; 49:761-8.
7. Ito T, Smith CL, Cantor CR. Sequence-specific DNA purification by triplex affinity capture. Proc Natl Acad Sci USA. 1992 Jan 15; 89(2):495-8.
8. Kjer-Nielsen L, Holmberg K, Perera JD, McCluskey J. Impaired expression of chimaeric major histocompatibility complex transgenes associated with plasmid sequences. Transgenic Res. 1992 Jul; 1(4): 182-7.
9. Kolb AF. Selection-marker-free modification of the murine beta-casein gene using a lox2272 [correction of lox2722] site. Anal Biochem. 2001 Mar; 290(2):260-71.
10. Kostal J, Mulchandani A, Chen W. Affinity purification of plasmid DNA by temperature-triggered precipitation. Biotechnol Bioeng. 2004 Feb 5; 85(3):293-7.
11. Langer SJ, Ghafoori AP, Byrd M, Leinwand L. A genetic screen identifies novel non-compatible loxP sites. Nucleic Acids Res. 2002 Jul 15; 30(14):3067-77.
12. Mayrhofer P, Blaesen M, Schleef M, Jechlinger W. Minicircle-DNA production by site specific recombination and protein-DNA interaction chromatography. J GeneMed. 2008 Nov; 10(11):1253-69.
13. Ringrose L, Lounnas V, Ehrlich L, Buchholz F, Wade R, Stewart AF. Comparative kinetic analysis of FLP and cre recombinases: mathematical models for DNA binding and recombination. J Mol Biol. 1998 Nov 27; 284(2):363-84.
14. Sadowski P. Site-specific recombinases: changing partners and doing the twist. J Bacteriol. 1986 Feb; 165(2):341-7.
15. Whitson PA, Hsieh WT, Wells RD, Matthews KS. Influence of supercoiling and sequence context on operator DNA binding with lac repressor. J Biol Chem. 1987a Oct 25; 262(30): 14592-9.
16. Whitson PA, Hsieh WT, Wells RD, Matthews KS. Supercoiling facilitates lac operator-repressor-pseudooperator interactions. J Biol Chem. 1987b Apr 15; 262(11):4943-6.
17. Woodgate J, Palfrey D, Nagel DA, Hine AV, Slater NK. Protein-mediated isolation of plasmid DNA by a zinc finger-glutathione S-transferase affinity linker. Biotechnol Bioeng. 2002 Aug 20; 79(4):450-6.

Изобретение относится к молекулярной биологии. Предложен способ разделения молекул ДНК, предусматривающий негативную селекцию целевых молекул ДНК в растворе от нецелевых молекул ДНК, предусматривающий взаимодействие нецелевых молекул ДНК, содержащих сайт узнавания рекомбиназы Cre бактериофага P1 LoxP, с рекомбиназой Cre бактериофага Р1, дефектной по способности ковалентно модифицировать ДНК, которое обеспечивает перевод нецелевых молекул ДНК из раствора на твердую фазу. Применение принципа негативной селекции позволяет исключить необходимость присутствия дополнительных нуклеотидных последовательностей в целевых молекулах ДНК. Кроме того, предложенный способ не требует наличия минимальных различий в длинах целевых и нецелевых молекул ДНК для их эффективного разделения. 14 з.п. ф-лы, 8 ил., 5 пр.
1. Способ разделения молекул ДНК, предусматривающий негативную селекцию целевых молекул ДНК в растворе от нецелевых молекул ДНК, предусматривающий взаимодействие нецелевых молекул ДНК, содержащих сайт узнавания рекомбиназы Cre бактериофага P1 LoxP, с рекомбиназой Cre бактериофага Р1, дефектной по способности ковалентно модифицировать ДНК, которое обеспечивает перевод нецелевых молекул ДНК из раствора на твердую фазу.
2. Способ по п. 1, в котором специфической нуклеотидной последовательностью является сохраняющий способность взаимодействовать с рекомбиназой Cre LoxP-сайт с измененной нуклеотидной последовательностью.
3. Способ по п. 1, в котором рекомбиназа Cre бактериофага Р1, дефектная по способности ковалентно модифицировать ДНК, содержит аминокислотные замены, изменяющие специфичность ее взаимодействия с LoxP-сайтом (вариант рекомбиназы Cre), а специфической нуклеотидной последовательностью является LoxP-сайт с нуклеотидными заменами, обеспечивающими специфическое взаимодействие с вариантом рекомбиназы Cre.
4. Способ по одному из пп. 1, 2 или 3, в котором осуществляется очистка целевого фрагмента ДНК, представляющего собой вставку в плазмидном векторе и полученного в результате расщепления плазмиды, от фрагмента ДНК, являющегося вектором и содержащего сайт узнавания рекомбиназы Cre бактериофага P1 LoxP или LoxP-сайт с нуклеотидными заменами, обеспечивающими специфическое взаимодействие с вариантом рекомбиназы Cre, обеспечивающую его негативную селекцию, а также негативную селекцию продуктов неполного расщепления плазмидной ДНК.
5. Способ по одному из пп. 1, 2 или 3, при котором нецелевые молекулы ДНК взаимодействуют с рекомбиназой Cre (вариантом рекомбиназы Cre), дефектной по способности ковалентно модифицировать ДНК, предварительно иммобилизованной на твердой фазе.
6. Способ по одному из пп. 1, 2 или 3, при котором нецелевые молекулы ДНК взаимодействуют с рекомбиназой Cre (вариантом рекомбиназы Cre), дефектной по способности ковалентно модифицировать ДНК, в растворе с последующим переводом рекомбиназы Cre (варинта рекомбиназы Cre), дефектной по способности ковалентно модифицировать ДНК, на твердую фазу.
7. Способ по п. 5, при котором рекомбиназа Cre (вариант рекомбиназы Cre), дефектная по способности ковалентно модифицировать ДНК, ковалентно связана с твердой фазой.
8. Способ по п. 5, при котором рекомбиназа Cre (вариант рекомбиназы Cre), дефектная по способности ковалентно модифицировать ДНК, иммобилизована на твердой фазе за счет нековалентных взаимодействий.
9. Способ по п. 6, при котором рекомбиназа Cre (вариант рекомбиназы Cre), дефектная по способности ковалентно модифицировать ДНК, переводится на твердую фазу в результате специфического нековалентного взаимодействия с твердым носителем.
10. Способ по одному из пп. 1, 2 или 3, в котором каждая молекула нецелевой ДНК содержит более одной копии сайта узнавания рекомбиназы Cre бактериофага Р1 LoxP или LoxP-сайта с нуклеотидными заменами, обеспечивающими специфическое взаимодействие с рекомбиназой Cre.
11. Способ по одному из пп. 1, 2 или 3, в котором рекомбиназа Cre (вариант рекомбиназы Cre), дефектная по способности ковалентно модифицировать ДНК, используется как химера с глутатион-8-трансферазой, а для ее иммобилизации на твердой фазе используется твердый носитель с иммобилизованным глутатионом.
12. Способ по одному из пп. 1, 2 или 3, в котором рекомбиназа Cre (вариант рекомбиназы Cre), дефектная по способности ковалентно модифицировать ДНК, сохраняет способность образовывать синапсы.
13. Способ по п. 12, в котором в качестве рекомбиназы Cre, дефектной по способности ковалентно модифицировать ДНК, используется мутант K201A с заменой остатка лизина-201 на остаток аланина.
14. Способ по одному из пп. 1, 2 или 3, в котором рекомбиназа Cre, дефектная по способности ковалентно модифицировать ДНК, не способна образовывать синапсы.
15. Способ по п. 14, в котором в качестве рекомбиназы Cre, дефектной по способности ковалентно модифицировать ДНК, используется мутант Y324F с заменой остатка тирозина-324 на остаток фенилаланина.
| Способ и приспособление для нагревания хлебопекарных камер | 1923 |
|
SU2003A1 |
| GOGOS J.A | |||
| et al., "Binding site selection analysis of protein-DNA interactions via solid phase sequencing of oligonucleotide mixtures." Nucleic acids research, 1991; 19(7): 1449-1453 | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| FAN HSIU-FANG, "Real-time single-molecule tethered particle motion experiments reveal the kinetics and mechanisms of Cre-mediated site-specific recombination." Nucleic acids research, 2012; 40(13): 6208-6222 | |||
| FRANGIONI J.V., NEEL B.G | |||
| "Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins." Analytical biochemistry, 1993; 210(1): 179-187. | |||

