Область изобретения
Настоящее изобретение относится к новым соединениям, фармацевтическим композициям, содержащим такие соединения, и к их применению в терапии.
Предшествующий уровень техники
Геномы эукариотических организмов высоко организованы в ядре клетки. Длинные цепи двухцепочечной ДНК накручены вокруг октамера гистоновых белков (чаще всего содержащих две копии гистонов Н2А, Н2В, Н3 и Н4 с образованием нуклеосомы. Эта основная единица затем дополнительно уплотняется посредством агрегации и укладки нуклеосом с образованием высококонденсированной структуры хроматина. Возможен целый ряд различных состояний конденсации, и плотность этой структуры варьирует в течение клеточного цикла, становясь наиболее сжатой во время процесса деления клетки. Структура хроматина играет важную роль в регулировании транскрипции генов, которая не может совершаться эффективно из высококонденсированного хроматина. Структура хроматина контролируется рядом посттрансляционных модификаций в гистоновых белках, особенно в гистонах Н3 и Н4, и чаще всего в гистоновых хвостах, которые выходят за границы коровой структуры нуклеосомы. Эти модификации включают ацетилирование, метилирование, фосфорилирование, убиквитинирование, сумоилирование. Эти эпигенетические отметки наносятся и стираются специальными ферментами, которые размещают метки на отдельных остатках в гистоновом хвосте, формируя посредством этого эпигенетический код, который затем расшифровывается клеткой с разрешением геноспецифической регуляции структуры хроматина и посредством этого транскрипции.
Ацетилирование гистонов чаще всего ассоциируется с активацией транскрипции генов, так как эта модификация ослабляет взаимодействие ДНК и гистонового октамера за счет изменения электростатики. В дополнение к этому физическому изменению, специфические белки распознают и связываются с ацетилированными остатками лизина в гистонах для считывания эпигенетического кода. Бромодомены представляют собой небольшие (порядка 110 аминокислот) отдельные домены в белках, которые связываются с ацетилированными остатками лизина в большинстве случаев применительно к гистонам, но не только к ним. Это семейство из приблизительно 50 белков, как известно, содержит бромодомены, и имеет целый ряд функций в клетке.
ВЕТ-семейство белков, содержащих бромодомены, включает 4 белка (BRD2, BRD3, BRD4 и BRDT), содержащие тандемные бромодомены, способные связываться с двумя ацетилированными остатками лизина, расположенными в непосредственной близости друг от друга, увеличивая специфичность взаимодействия. При нумерации от N-конца каждого ВЕТ-белка, тандемные бромодомены обычно обозначают связывающим доменом 1 (BD1) и связывающим доменом 2 (BD2) (Chung et al, J Med. Chem. 2011, 54, 3827-3838).
Был обнаружен новый класс соединений, которые ингибируют связывание бромодоменов с узнаваемыми ими ацетилированными белками, более конкретно, класс соединений, которые ингибируют связывание бромодоменов ВЕТ-семейства с ацетилированными остатками лизина, еще более конкретно, класс соединений, которые селективно ингибируют связывание и функционирование бромодоменов ВЕТ-семейства посредством связывающего домена 1 (BD1). Такие соединения в дальнейшем будут называться "ингибиторами бромодоменов".
Краткое изложение сущности изобретения
В первом аспекте настоящего изобретения предложено соединение формулы (I) или его соль, более конкретно соединение формулы (I)
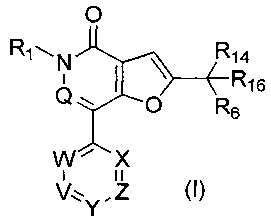
или его фармацевтически приемлемая соль.
Во втором аспекте настоящего изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I), или его фармацевтически приемлемую соль, и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
В третьем аспекте настоящего изобретения предложено соединение формулы (I), или его фармацевтически приемлемая соль для использования в терапии, в частности в лечении заболеваний или состояний, для которых показан ингибитор бромодомена.
В четвертом аспекте настоящего изобретения предложен способ лечения заболеваний или состояний, для которых показан ингибитор бромодомена, у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
В пятом аспекте настоящего изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний или состояний, для которых показан ингибитор бромодомена.
Подробное описание изобретения
Настоящее изобретение относится к соединению формулы (I):
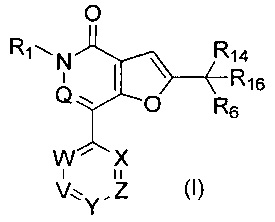
где:
V представляет собой N или C-R2
W представляет собой N или C-R8;
X представляет собой N, СН или С(СН3);
Y представляет собой N или C-R5;
Z представляет собой N или C-R15;
Q представляет собой N или СН;
R1 представляет собой С1-4 алкил или дейтерированный С1-4 алкил;
R2, когда присутствует, представляет собой Н, ОН, С1-4алкил, галоген, -CF3, -NH2, -ОС1-4алкил, -NHC(O)H, -NHC(O)С1-4алкил, -N(СН3)С(O)С1-4алкил, -NHC(O)NH2, -NHC(O)С1-4алкиленNH2, -N(CH3)C(O)NH2, -N(CH3)C(O)C1-4алкиленNH2, -NHC2-4алкиленОСН3, -N(СН3)С2-4алкиленOСН3, -ОС2-4алкиленОСН3, -ОС2-4алкиленОН или
R2 представляет собой группу, выбранную из -G-CH2CH(R3)(R4), -G-CH(R3)(R4) и -G-R3, в которой
G представляет собой NH, N(CH3), О, C(O)NH или NHC(O);
R3 представляет собой фенил, пиридинил, С3-7циклоалкил или гетероцикл, возможно замещенный =O; и
R4 представляет собой Н или С1-4 алкил;
R5, когда присутствует, представляет собой Н, С1-4алкил, галоген, -CF3, CN, ОН, -ОС1-4алкил, -CH2NH2, -OCF3, -SO2CH3, -С(O)NHC1-4алкил или -CO2H;
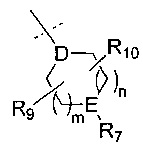
R6 представляет собой -NR11R12 или группу 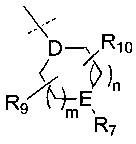 ;
;
D представляет собой СН или N;
Е представляет собой N, О, СН или SO2;
R7, когда присутствует, представляет собой Н, ОН, С1-4алкил, -NH2, -SO2C1-4алкил, -SO2фенил, -SO2бензил, -SO2N(CH3)2, -NHSO2CH3, -С(O)С1-4алкил, -С(O)фенил;
R8, когда присутствует, представляет собой Н, С1-4алкил, галоген, -CF3, CN, ОН, -ОС1-4алкил, -ОС2-4алкиленОС1-4алкил, -OCF3, -ОС1-4алкиленF, -ОС1-4алкиленCHF2, -ОС2-4алкиленОН, -Офенил, -ОС1-4алкиленфенил, -NHC3-7циклоалкил, -NHC1-4алкиленС3-7циклоалкил, -ОС3-7циклоалкил, -ОС1-4алкиленС3-7циклоалкил, -NHC4-6гетероцикл, -NHC1-4алкиленС4-6гетероцикл, -ОС4-6гетероцикл или -ОС1-4алкиленС4-6гетероцикл, где каждый С3-7циклоалкил или С4-6гетероцикл возможно замещен одним или двумя заместителями, независимо выбранными из галогена, ОН, оксо, С1-4алкила и -NH2; или
R8 и R2 вместе с атомами углерода, к которым они присоединены, образуют гетероцикл, возможно замещенный оксо;
R9 представляет собой Н, С1-4алкил, -C(O)NH2, -CO2CH3, -CF3, галоген, ОН, -ОС1-4алкил, -СН2ОН, -C(O)NHCH3, -C(O)NH(CH3)2, -СН2ОС1-4алкил или -СН2ОСН2С3-7циклоалкил;
R10 представляет собой Н, С1-4алкил, -C(O)NH2, -CO2CH3, -CF3, галоген, ОН, -ОС1-4алкил или оксо;
R11 представляет собой Н, С1-4алкил или SO2CH3;
R12 представляет собой Н, С1-4алкил, C2-4aлкилeнNHR13, SO2CH3, гетероцикл или гетероцикл, содержащий SO2;
R13 представляет собой Н или SO2CH3;
R14 представляет собой Н или С1-4алкил;
R15 представляет собой Н, С1-4алкил или NHC(O)С1-4алкил;
R16 представляет собой Н или С1-4алкил: и
каждый из n и m представляет собой целое число, независимо выбранное из 0, 1 и 2; при условии, что не более 2 из V, W, X, Y и Z представляют собой N; или его соль.
В одном воплощении V представляет собой C-R2. В другом воплощении V представляет собой N.
В одном воплощении W представляет собой C-R8. В другом воплощении W представляет собой N.
В одном воплощении R8 представляет собой Н, ОН, -ОС1-4алкил, -ОС2-4алкиленОСН3, -ОС1-4алкиленF, -ОС1-4алкиленCHF2, -ОС2-4алкиленОН, NHCH2C3-7циклоалкил, -ОС3-7циклоалкил, -ОСН2С3-7циклоалкил, -O-С4-6гетероцикл, -ОСН2С4-6гетероцикл или -ОСН2СН2С4-6гетероцикл, где С3-7циклоалкил или С4-6гетероцикл каждый, возможно, замещен одним или двумя заместителями, независимо выбранными из фтора и оксо. В другом воплощении R8 представляет собой Н, ОН, -ОСН2СН3, -ОСН(СН3)2, -ОСН2СН2ОСН3, -ОСН2СН(СН3)ОСН3, -ОСН(СН3)СН2ОСН3, -OCH2CH2F, -OCH2CHF2, -ОСН2СН2ОН, -NHСН2циклопропил, -Оциклопропил, -ОСН2циклопропил, -Отетрагидрофуранил, -Ооксетанил, -ОСН2тетрагидрофуранил, -ОСН2оксетанил или -ОСН2СН2пирролидинил, где каждый С3-7циклоалкил или С4-6гетероцикл возможно, замещен одним или двумя заместителями, независимо выбранными из фтора и оксо. В одном воплощении R8 представляет собой Н, С1-4алкил или -ОСН2С3-7циклоалкил. В другом воплощении R8 представляет собой Н, метил, этил или -ОСН2циклопропил. В другом воплощении R8 представляет собой Н. В другом воплощении R8 представляет собой -ОСН2циклопропил. В другом воплощении R8 представляет собой -ОСН2оксетан. В еще одном воплощении R8 представляет собой (R)-ОСН2-2-оксетан.
В одном воплощении R8 и R2 вместе с атомами углерода, к которым они присоединены, образуют гетероцикл, возможно замещенный оксо. В другом воплощении R8 и R2 вместе с атомами углерода, к которым они присоединены, образуют 2,3-дигидро-1H-пирроло[2,3-b]пиридин, возможно замещенный оксо.
В одном воплощении X представляет собой СН. В другом воплощении X представляет собой С(СН3). В еще одном воплощении X представляет собой N.
В одном воплощении Y представляет собой C-R5. В другом воплощении Y представляет собой N.
В одном воплощении R5 представляет собой Н, -СН3, -СН2СН3, галоген, -CF3, CN, ОН, -ОСН3, -CH2NH2, -OCF3, -SO2CH3, -C(O)NHCH2CH3 или -CO2H. В другом воплощении R5 представляет собой Н, -CF3, CN, -ОСН3, -CH2NH2 или -SO2CH3. В другом воплощении R5 представляет собой Н, -ОСН3 или -CH2NH2. B еще одном воплощении R5 представляет собой Н или -ОСН3.
В одном воплощении Z представляет собой N. В другом воплощении Z представляет собой C-R15.
В одном воплощении R15 представляет собой Н.
В одном воплощении Q представляет собой СН. В другом воплощении Q представляет собой N.
В одном воплощении R1 представляет собой метил или этил. В другом воплощении R1 представляет собой метил.
В одном воплощении R2 представляет собой Н, -NH2, -ОС1-4алкил, -NHC(O)С1-4алкил, -N(СН3)С(O)С1-4алкил, -NHC(O)C1-4алкиленNH2 или -ОС2-4алкиленОСН3. В другом воплощении R2 представляет собой Н, ОН, метил, фтор, хлор, -CF3, -NH2, -ОСН3, -ОСН(СН3)2, -NHC(O)H, -NHC(O)Me, -NCH(CH3)CH2OCH3, -N(CH3)CH2CH2OCH3, -OCH2CH2OCH3, -OCH2CH2CH2OH или -OCH(CH3)CH2OCH3. В другом воплощении R2 представляет собой Н, -ОСН3, -ОСН(СН3)2, -NHC(O)Me, -NCH(CH3)CH2OCH3 или -N(CH3)CH2CH2OCH3. В другом воплощении R2 представляет собой Н, -ОС1-4алкил, -NHC(O)С1-4алкил или --N(СН3)С(O)С1-4алкил. В другом воплощении R2 представляет собой Н, -ОСН3, -NHC(O)CH3, -NHC(O)CH2CH3 или -N(CH3)C(O)CH3. В другом воплощении R2 представляет собой Н, -NH2, -ОСН3, -NHC(O)CH3, -NHC(O)CH2CH3, -N(CH3)C(O)CH3, -NHC(O)CH2CH2CH2CH2NH2 или -OCH2CH2OCH3. В другом воплощении R2 представляет собой Н. В другом воплощении R2 представляет собой -NHC(O)CH3. В другом воплощении R2 представляет собой группу -G-CH(R3)(R4). В другом воплощении R2 представляет собой группу -G-CH2CH(R3)(R4). В еще одном воплощении R2 представляет собой группу -G-R3.
В одном воплощении G представляет собой NH, N(CH3), О или NHC(O). В другом воплощении G представляет собой NH, О или NHC(O). В другом воплощении G представляет собой N(CH3). В другом воплощении G представляет собой NHC(O). В другом воплощении G представляет собой NH. В еще одном воплощении G представляет собой О.
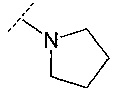
В одном воплощении R3 представляет собой фенил, пиридинил, циклопропил, тетрагидропиранил, пирролидинил или пирролидинил, замещенный =O. В другом воплощении R3 выбран из
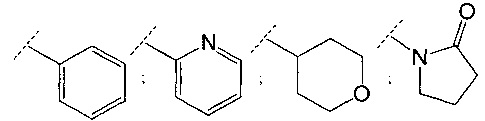 ;
;
 и
и 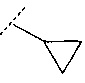 .
.
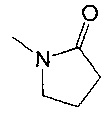
В еще одном воплощении R3 выбран из
 и
и 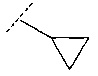 .
.
В одном воплощении R4 представляет собой Н или метил. В другом воплощении R4 представляет собой Н. В еще одном воплощении R4 представляет собой метил.
В одном воплощении R6 представляет собой -NR11R12.
В одном воплощении R11 представляет собой Н или метил. В другом воплощении R11 представляет собой Н.
В одном воплощении R12 представляет собой -CH2CH2CH2NH2, -CH2CH2NH2, -CH2CH2NHSO2CH3, -SO2CH3, -СН3 или 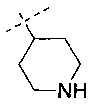 . В другом воплощении R12 представляет собой -CH2CH2NHR13 и R13 представляет собой Н.
. В другом воплощении R12 представляет собой -CH2CH2NHR13 и R13 представляет собой Н.
В одном воплощении R6 представляет собой группу
 .
.
В одном воплощении D представляет собой СН. В другом воплощении D представляет собой N.
В одном воплощении Е представляет собой N, О или СН. В другом воплощении Е представляет собой N. В другом воплощении Е представляет собой О. В другом воплощении Е представляет собой СН. В еще одном воплощении Е представляет собой SO2.
В одном воплощении R7 представляет собой Н, метил, этил, изопропил, -SO2CH3, -SO2CH2CH3 или -С(O)фенил. В другом воплощении R7 представляет собой Н или -SO2CH3. В еще одном воплощении R7 представляет собой -SO2CH3.
В одном воплощении R9 представляет собой Н, метил, этил, бутил, -CONH2, -CO2CH3, -CF3, фтор, ОН или -ОСН3. В другом воплощении R9 представляет собой Н, метил или фтор. В другом воплощении R9 представляет собой Н или метил.
В одном воплощении R10 представляет собой Н, метил или фтор. В другом воплощении R10 представляет собой Н или фтор.
В одном воплощении R9 и R10 присоединены к одному и тому же атому. В другом воплощении R9 и R10 присоединены к разным атомам.
В одном воплощении R14 представляет собой Н. В другом воплощении R14 представляет собой С1-4алкил. В еще одном воплощении R14 представляет собой -СН3.
В одном воплощении R16 представляет собой Н. В другом воплощении R16 представляет собой С1-4алкил. В еще одном воплощении R16 представляет собой -СН3.
В одном воплощении n равен 1 или 2. В другом воплощении n равен 0. В другом воплощении n равен 1. В еще одном воплощении n равен 2.
В одном воплощении m равен 1 или 2. В другом воплощении m равен 0. В другом воплощении m равен 1. В еще одном воплощении m равен 2.
В одном воплощении как n, так и m равны 1. В другом воплощении n равен 1, и m равен 2.
В одном воплощении R6 представляет собой группу, выбранную из:
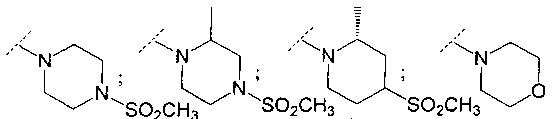 ;
;
 ;
;
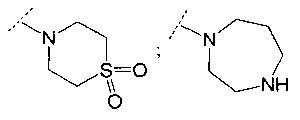 и
и 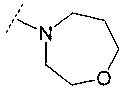 .
.
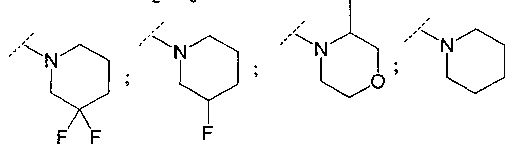
В другом воплощении R6 представляет собой группу, выбранную из:
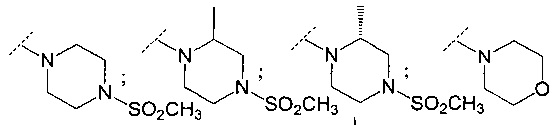 ;
;
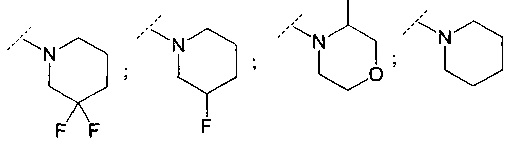 ;
;
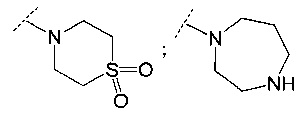 и
и 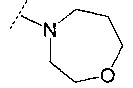 .
.
В другом воплощении R6 представляет собой группу, выбранную из:
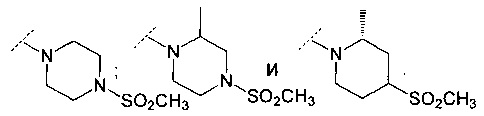
В еще одном воплощении R6 представляет собой группу, выбранную из:
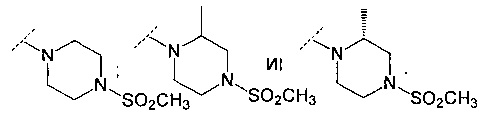
В одном воплощении настоящее изобретение относится к соединению формулы (IC):
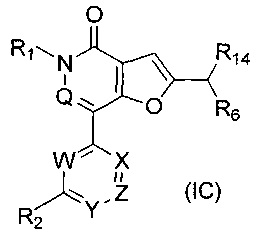
где:
W представляет собой N или C-R8;
X представляет собой N, СН или С(СН3);
Y представляет собой N или C-R5;
Z представляет собой N или C-R15;
Q представляет собой N или СН;
R1 представляет собой С1-4алкил;
R2 представляет собой Н, ОН, С1-4алкил, галоген, -CF3, -NH2, -ОС1-4алкил, -NHC(O)H, -NHC(O)С1-4алкил, -N(СН3)С(O)С1-4алкил, -NHCH(CH3)CH2OCH3, -N(CH3)CH2CH2OCH3, -ОСН2СН2ОСН3, -ОСН2СН2СН2ОН, -ОСН(СН3)СН2ОСН3, или
R2 представляет собой группу, выбранную из -G-CH2CH(R3)(R4), -G-CH(R3)(R4) и -G-R3, в которой
G представляет собой NH, N(CH3), О, C(O)NH или NHC(O);
R3 представляет собой фенил, пиридинил, С3-7циклоалкил или гетероцикл, возможно замещенный =O; и
R4 представляет собой Н или С1-4алкил;
R5 представляет собой Н, С1-4алкил, галоген, -CF3, CN, ОН, -OC1-4 алкил, -CH2NH2, -OCF3 или -SO2CH3;
R6 представляет собой -NR11R12 или группу 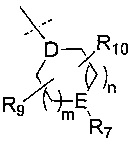 ;
;
D представляет собой СН или N;
Е представляет собой N, О, СН или SO2;
R7, когда присутствует, представляет собой Н, ОН, С1-4алкил, -NH2, -SO2C1-4алкил, -SO2фенил, -SO2бензил, -SO2N(CH3)2, -NHSO2CH3, -С(O)С1-4алкил, -С(O)фенил;
R8 представляет собой Н, С1-4алкил, галоген, -CF3, CN, ОН, -ОС1-4алкил, -OCF3, -ОСН2фенил, -NHCH2C3-7циклоалкил или -ОСН2С3-7циклоалкил;
R9 представляет собой Н, С1-4 алкил, -C(O)NH2, -CO2CH3, -CF3, галоген, ОН, -ОС1-4алкил, -СН2ОН, -C(O)NHCH3 или -C(O)NH(CH3)2, -СН2ОС1-4алкил или -CH2OCH2C3-7циклоалкил;
R10 представляет собой Н, С1-4алкил, -C(O)NH2, -CO2CH3, -CF3, галоген, ОН или -ОС1-4алкил;
R11 представляет собой Н, С1-4алкил или SO2CH3;
R12 представляет собой Н, С1-4алкил, C1-4алкиленNHR13, SO2CH3, гетероцикл или гетероцикл, содержащий SO2;
R13 представляет собой Н или SO2CH3;
R14 представляет собой Н или С1-4алкил;
R15 представляет собой Н, С1-4алкил или NHC(O)C1-4алкил; и
каждый из n и m представляет собой целое число, независимо выбранное из 0, 1 и 2; с оговоркой, что не более 2 из W, X, Y и Z представляют собой N; или его соль.
В одном воплощении соединение формулы (I) представляет собой соединение формулы (IA)
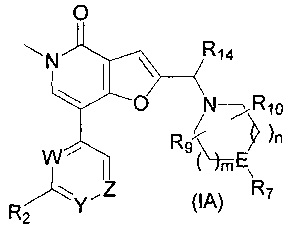
где:
W представляет собой C-R8;
Y представляет собой N или C-R5;
Z представляет собой N или СН;
R2 представляет собой Н, -ОСН3, -NHC(O)CH3, -NHC(O)CH2CH3, -N(CH3)C(O)CH3, или
R2 представляет собой группу, выбранную из -G-CH2CH(R3)(R4) и -G-CH(R3)(R4), в которых
G представляет собой NH, О или NHC(O);
R3 представляет собой фенил, пиридинил, циклопропил или гетероцикл, возможно замещенный =O; и
R4 представляет собой Н или метил;
R5 представляет собой Н, -ОСН3 или -CH2NH2;
E представляет собой N, О, СН или SO2;
R7, когда присутствует, представляет собой Н или -SO2CH3;
R8 представляет собой Н, -NHCH2циклопропил или -ОСН2циклопропил;
R9 представляет собой Н, метил или фтор;
R10 представляет собой Н или фтор;
R14 представляет собой Н или метил; и
каждый из n и m представляет собой целое число, независимо выбранное из 1 и 2; или его соль.
В другом воплощении соединение формулы (I) представляет собой соединение формулы (IB):
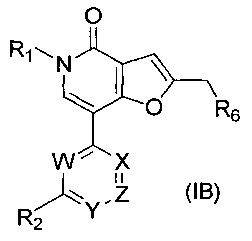
где:
W представляет собой N или C-R8;
каждый из X и Z независимо представляет собой N или СН;
Y представляет собой N или C-R5;
R1 представляет собой С1-4 алкил;
R2 представляет собой Н, ОН, С1-4алкил, галоген, -CF3, -NH2, -ОС1-4алкил, -NHC(O)H, -NHC(O)Me, -NHC(CH3)CH2OCH3, -N(CH3)CH2OCH3, -N(CH3)CH2CH2OCH3, -OCH2CH2OCH3, -OCH2CH2CH2OH, -OCH(CH3)CH2OCH3, или
R2 представляет собой группу -G-CH(R3)(R4), в которой
G представляет собой NH, NCH3, О или C(O)NH;
R3 представляет собой фенил, пиридинил, С3-7циклоалкил или гетероцикл; и
R4 представляет собой Н или С1-4 алкил;
R5 представляет собой Н, С1-4алкил, галоген, -CF3, CN, ОН, -ОС1-4алкил, -CH2NH2, -OCF3 или -SO2CH3;
R6 представляет собой -NR11R12 или группу 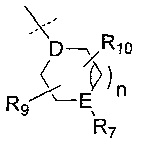 ;
;
D представляет собой СН или N;
Е представляет собой N, О, СН или SO2;
R7, когда присутствует, представляет собой Н, ОН, С1-4алкил, -NH2, -SO2C1-4алкил, -SO2фенил, -SO2бензил, -SO2N(CH3)2, -NHSO2CH3, -С(O)С1-4алкил или -С(O)фенил;
R8 представляет собой Н, С1-4алкил, галоген, -CF3, CN, ОН, -ОС1-4алкил, -OCF3 или -ОСН2фенил;
R9 представляет собой Н, С1-4алкил, -CONH2 или -CO2CH3;
R10 представляет собой Н, С1-4алкил, -CONH2 или -CO2CH3;
R11 представляет собой Н или С1-4алкил;
R12 представляет собой Н, С1-4алкил, C1-4aлкилeнNHR13, SO2CH3 или гетероцикл;
R13 представляет собой Н или SO2CH3; и
n равен 0, 1 или 2; или представляет собой их соль.
Следует понимать, что настоящее изобретение распространяется на все комбинации замещающих групп, описанных в данном описании изобретения.
Соединения по изобретению включают соединения примеров 1-114 и их соли.
В другом воплощении соединения по изобретению включают соединения примеров 1-81б и их соли. В другом воплощении соединения по изобретению включают соединения примеров 1-36 и 37-40 и их соли.
В одном воплощении соединение формулы (I) выбрано из
7-[3,4-бис(метилокси)фенил]-5-метил-2-{[4-(метилсульфонил)-1-пиперазинил]метил}фуро[3,2-с]пиридин-4(5Н)-она;
(R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
7-(3-(бензилокси)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3-(бензиламино)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
2-(((2-аминоэтил)амино)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-она;
7-(4-(аминометил)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3,4-диметоксифенил)-5-метил-2-(пиперидин-1-илметил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3,4-диметоксифенил)-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-7-(3,4-диметоксифенил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
2-((1,4-диазепан-1-ил)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(5-(1-фенилэтокси)пиридин-3-ил)фуро[3,2-с]пиридин-4(5Н)-она;
2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((1-фенилэтил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((1-фенилэтил)амино)фенил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3,4-диметоксифенил)-5-метил-2-((3-метилморфолино)метил)фуро[3,2-с]пиридин-4(5Н)-она;
N-(4-(2-((3-фторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
2-((3-фторпиперидин-1-ил)метил)-7-(4-метоксифенил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(морфолинометил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(S)-5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
2-((1,4-оксазепан-4-ил)метил)-5-метил-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-7-(2-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)циклопропанкарбоксамида;
(R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)пропионамида;
(R)-7-(2-(2-метоксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(пирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
2-((1,1-диоксидотиоморфолино)метил)-5-метил-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[2,3-d]пиридазин-4(5Н)-она;
(R)-N-метил-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3,4-диметоксифенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[2,3-d]пиридазин-4(5Н)-она;
2-((1,4-диазепан-1-ил)метил)-5-метил-7-(2-((пиридин-2-илметил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(2-оксопирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(S)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-N-(5-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)ацетамида;
7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((S)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(циклопропилметокси)пиридин-2-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-((2-метилпиперазин-1-ил)метил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она гидрохлорида;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-3-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
2-((4-ацетил-2-метилпиперазин-1-ил)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она гидрохлорида;
N-(3-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)фенил)ацетамида;
(R)-7-(2-((циклопропилметил)амино)пиридин-3-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-аминофенил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
2-((1,4-диазепан-1-ил)метил)-5-метил-7-(3-((пиридин-2-илметил)амино)фенил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-этоксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
N-(3-(2-((1,4-диазепан-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)фенил)пиколинамида;
(R)-N-(6-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
2-((1,4-диазепан-1-ил)метил)-7-(3-(циклопропилметокси)пиридин-4-ил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-метил-1,4-диазепан-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-метил-1,4-диазепан-1-ил)метил)-7-(3-((пиридин-2-илметил)амино)фенил)фуро[3,2-с]пиридин-4(5Н)-она гидрохлорида;
(R)-7-(3-изопропоксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-7-(2-аминопиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-7-(3-гидроксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3-((2,2-дифторциклопропил)метокси)пиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-N-(4-(5-(2Н3)метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(2-(2-оксопирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-5-амино-N-(3-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)фенил)пентанамида;
N-(4-(5-метил-2-(2-(4-(метилсульфонил)пиперазин-1-ил)пропан-2-ил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-7-(3-(2-метоксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
3-(циклопропилметокси)-4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензойной кислоты;
3-(циклопропилметокси)-N-этил-4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензамида;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-3-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-2-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((3-оксопиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(2-фторэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-7-(3-циклопропоксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(2,2-дифторэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(2-гидроксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-метил-3-оксопиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-оксо-2,3-дигидро-1Н-пирроло[2,3-b]пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
7-(3-(2-метоксипропоксипиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3-((1-метоксипропан-2-ил)окси)пиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-3-илокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-3-ил)окси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(((S)-тетрагидрофуран-3-ил)окси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она; и
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(((R)-тетрагидрофуран-3-ил)окси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
N-(4-(5-метил-4-оксо-2-((5-оксо-1,4-диазепан-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,4-оксазепан-4-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метилпиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метил-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,4-диазепан-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,1-диоксидотиоморфолино)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-((3-оксопиперазин-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонамидо)пиперидин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(S)-N-(4-(2-((3-гидроксипиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-(пиперазин-1-илметил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-этил-3-оксопиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонил)пиперидин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-(((1,1-диоксидотетрагидро-2Н-тиопиран-3-ил)амино)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-(((1,1-диоксидотетрагидротиофен-3-ил)амино)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((2,5-диметил-4-(метилсульфонил)пиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-этил-2-метилпиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метил-5-оксо-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((7-метил-5-оксо-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((2-метил-5-оксо-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,1-диоксидо-1,4-тиазепан-4-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((3-метил-1,1-диоксидотиоморфолино)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((2-метил-1,1-диоксидотиоморфолино)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-ацетил-1,4-диазепан-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-(морфолинометил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-ацетил-2-метилпиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метил-3-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(S)-N-(4-(5-метил-2-((3-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонил)-2-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-этилпиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-(пирролидин-1-илметил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида
N-(4-(5-метил-4-оксо-2-(пиперидин-1-илметил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-((3-оксо-1,4-диазепан-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-N-(4-(5-метил-2-((2-метил-3-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
или их соли.
В другом воплощении соединение формулы (I) выбирают из:
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она; и
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((S)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она; или их соли.
В еще одном воплощении соединение формулы (I) представляет собой:
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он; или его соль.
Термин "С1-4алкил" обозначает прямой или разветвленный алкил, содержащий по меньшей мере один и не более чем четыре атома углерода. Примеры "C1-C4алкила" при использовании в данном описании изобретения включают метил, этил, н-пропил, н-бутил, изобутил, изопропил и трет-бутил, но не ограничиваются ими.
Термин "дейтерированный С1-4алкил" обозначает С1-4алкил, где один или более атомов водорода заменены на дейтерий.
Термин "С1-4алкилен" обозначает прямую или разветвленную алкильную цепь, содержащую по меньшей мере один и не более чем четыре атома углерода. Примеры "С1-4алкилена" при использовании в данном описании изобретения включают метилен, этилен, пропилен и бутилен, но не ограничиваются ими.
Термин "С2-4алкилен" обозначает прямую или разветвленную алкильную цепь, содержащую по меньшей мере два и не более чем четыре атома углерода. Примеры "С2-4алкилена" при использовании в данном описании изобретения включают этилен, пропилен и бутилен, но не ограничиваются ими.
Термин "С3-7циклоалкил" используют для описания неароматического карбоциклического кольца, содержащего по меньшей мере три и не более чем семь атомов углерода. Примеры С3-7циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "гетероцикл" относится к 5- или 6-членному насыщенному кольцу, которое включает одно или более (например, 2) кольцевых гетероатомов, выбранных из азота, кислорода и серы. Примеры насыщенных гетероциклических групп включают тетрагидропиран, тетрагидрофуран, тетрагидротиофен, пиперидин, пиперазин, морфолин, 1,4-диоксан, тиоморфолин, 1,4-оксатиан и 1,4-дитан, но не ограничиваются ими. Точка присоединения к остальной части молекулы может быть представлена любым подходящим атомом углерода или азота.
Термин "С4-6гетероцикл" относится к 4-, 5- или 6-членному насыщенному кольцу, которое включает один или более (например, 2) кольцевых гетероатомов, выбранных из азота, кислорода и серы. Примеры насыщенных С4-6гетероциклических групп включают оксетан, азетидин, тиетиан, тетрагидропиран, тетрагидрофуран, тетрагидротиофен, пиперидин, пиперазин, морфолин, 1,4-диоксан, тиоморфолин, 1,4-оксатиан и 1,4-дитан, но не ограничиваются ими. Точка присоединения к остальной части молекулы может быть представлена любым подходящим атомом углерода или азота.
Термин "гетероцикл, содержащий SO2," относится к 5- или 6-членному насыщенному кольцу, которое включает один или более (например, 2) кольцевых гетероатомов, выбранных из азота, кислорода, серы и диоксида серы, где по меньшей мере один из гетероатомов представляет собой диоксид серы. Примеры гетероциклических групп, содержащих SO2, включают тетрагидротиофенил-1,2-диоксид и тетрагидро-2Н-тиопиранил-1,1-диоксид, но не ограничиваются ими.
Термин "галоген" при использовании в данном описании изобретения относится к фтору, хлору, брому или йоду.
Термин "фармацевтически приемлемый" относится к тем соединениям, веществам, композициям и лекарственным формам, которые с медицинской точки зрения являются подходящими для применения в контакте с тканями людей и животных без чрезмерной токсичности, болезненной чувствительности или другой проблемы или осложнения, в соответствии с допустимым соотношением польза/риск.
При использовании в данном описании изобретения символы и условные обозначения, используемые в этих способах, схемах и примерах, соответствуют символам и условным обозначениям, используемым в современной научной литературе, например в Journal of the American Chemical Society или в Journal of Biological Chemistry. Если не указано иное, все исходные вещества получают от коммерческих поставщиков и используют без дополнительной очистки.
Соединения формулы (I) могут содержать хиральной атом, так что могут быть образованы оптические изомеры, например энантиомеры. Таким образом, настоящее изобретение охватывает все изомеры соединений формулы (I), или в виде отдельных изомеров, выделенных так, что они по существу не содержат другого изомера (то есть чистых), или в виде смесей (то есть рацематов и рацемических смесей). Отдельный изомер, выделенный так, что он по существу не содержит другой изомер (то есть чистый), может быть выделен так, чтобы присутствовало менее 10%, в частности менее примерно 1%, например, менее примерно 0,1% другого изомера.
Разделение изомеров может быть достигнуто посредством общепринятых способов, известных специалисту в данной области техники, например, путем фракционной кристаллизации, хроматографии или HPLC.
Следует понимать, что настоящее изобретение охватывает соединения формулы (I) в виде свободного основания и в виде их солей, например, в виде их фармацевтически приемлемой соли. В одном воплощении изобретение относится к соединениям формулы (I) в форме свободного основания. В одном воплощении изобретение относится к соединениям формулы (I) или их фармацевтически приемлемой соли.
Ввиду их возможного применения в медицине, соли соединений формулы (I) желательно являются фармацевтически приемлемыми. Подходящие фармацевтически приемлемые соли могут включать соли присоединения кислоты. Для обзора подходящих фармацевтически приемлемых солей см. Berge et al., J. Pharm. Sci., 66:1-19, (1977). Обычно фармацевтически приемлемую соль можно легко получить путем использования желаемой кислоты или основания соответствующим образом. Полученную соль могут осадить из раствора и собрать посредством фильтрации, или могут восстановить путем выпаривания растворителя.
Фармацевтически приемлемая соль присоединения кислоты может быть образована путем взаимодействия соединения формулы (I) с подходящей неорганической или органической кислотой (такой как бромистоводородная, соляная, серная, азотная, ортофосфорная, янтарная, малеиновая, уксусная, пропионовая, фумаровая, лимонная, винная, молочная, бензойная, салициловая, глутаминовая, аспарагиновая, пара-толуолсульфоновая, бензолсульфоновая, метансульфоновая, этансульфоновая, нафталинсульфоновая, такая как 2-нафталинсульфоновая, или капроновая кислота), возможно, в подходящем растворителе, таком как органический растворитель, с получением соли, которую обычно выделяют, например, кристаллизацией и фильтрацией или упариванием с последующим растиранием. Фармацевтически приемлемая соль присоединения кислоты соединения формулы (I) может содержать или представлять собой, например, гидробромидную, гидрохлоридную, сульфатную, нитратную, фосфатную, сукцинатную, малеатную, ацетатную, пропионатную, фумаратную, цитратную, тартратную, лактатную, бензоатную, салицилатную, глутаматную, аспартатную, пара-толуолсульфонатную, бензолсульфонатную, метансульфонатную, этансульфонатную, нафталинсульфонатную (например, 2-нафталинсульфонатную) или гексаноатную соль. В одном воплощении фармацевтически приемлемая соль соединения формулы (I) представляет собой гидрохлорид.
Другие фармацевтически неприемлемые соли, например формиаты, оксалаты или трифторацетаты, могут использоваться, например, в выделении соединений формулы (I) и включены в объем данного изобретения.
В объем изобретения включены все возможные стехиометрические и нестехиометрические формы солей соединений формулы (I).
Следует понимать, что многие органические соединения могут образовывать комплексы с растворителями, в которых их приводят во взаимодействие или из которых их осаждают или кристаллизуют. Эти комплексы известны как "сольваты". Например, комплекс с водой известен как "гидрат". Растворители с высокими точками кипения и/или способные к образованию водородных связей, такие как вода, ксилен, N-метил пирролидинон, МеОН и EtOH, можно использовать для образования сольватов. Способы идентификации сольватов включают ЯМР и микроанализ, но не ограничиваются ими. Сольваты соединений формулы (I) входят в объем изобретения.
Изобретение включает в свой объем все возможные стехиометрические и нестехиометрические формы сольватов соединений формулы (I).
Изобретение охватывает все пролекарства соединений формулы (I) или его фармацевтически приемлемой соли, которые при введении реципиенту способны обеспечивать (непосредственно или опосредовано) соединение формулы (I) или его фармацевтически приемлемую соль, или активный метаболит или его остаток. Такие производные распознаваемы специалистом в данной области техники без проведения излишних экспериментов. Тем не менее, делается ссылка на руководство Burger's Medicinal Chemistry and Drug Discovery, 5th Edition, Vol 1: Principles и Practice, которое включено в данное описание изобретение посредством ссылки в пределах изучения таких производных.
Соединения формулы (I) могут находиться в кристаллической или аморфной форме. Кроме того, некоторые из кристаллических форм соединений формулы (I) могут существовать в виде полиморфов, которые включены в объем настоящего изобретения. Полиморфные формы соединений формулы (I) могут быть охарактеризованы и дифференцированы с использованием ряда общепринятых аналитических методов, которые включают картины дифракции рентгеновских лучей на порошке (XRPD), инфракрасные (ИК) спектры, рамановские спектры, дифференциальную сканирующую калориметрию (ДСК), термогравиметрический анализ (ТГА) и твердотельный ядерно-магнитный резонанс (ттЯМР), но не ограничиваются ими.
Из вышесказанного будет ясно, что включенными в объем изобретения являются сольваты, изомеры и полиморфные формы соединений формулы (I) и их солей.
Соединения формулы (I) или их соли могут быть изготовлены посредством ряда способов, включая стандартные химические методы. Иллюстративные общие способы синтеза представлены ниже, и затем получение конкретных соединений формулы (I) и их фармацевтически приемлемых солей представлено в примерах.
Соединения формулы (I) могут быть получены, как описано на любой из схем 1-6 ниже:
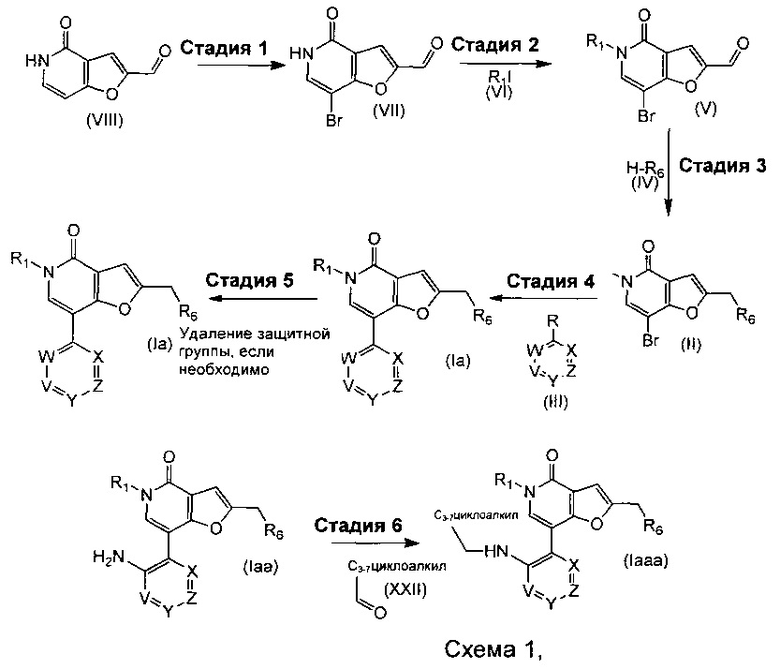
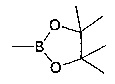
где R1, R6, V, W, X, Y и Z такие, как определено для соединения формулы (I); и R выбирают из -В(ОН)2, -BF3K и
 .
.
Если R6 представляет собой -NR11R12 и R12 представляет собой С1-4алкиленNHR13, то R12 защищают посредством подходящей защитной группы, такой как ВОС, FMOC или бензил, которую удаляют на стадии 5 синтеза. H-R6 представляет собой соединение формулы (IVa) или соединение формулы (IVb)
 ,
,
где m, n, R7, R9, R10, R11, R12 и E такие, как определено для соединения формулы (I).
В одном воплощении V представляет собой C-R2, где R2 такой, как определено для соединения формулы (I).
Что касается стадий, показанных на Схеме 1, то можно использовать следующие реакционные условия.
Стадию 1 могут осуществлять посредством обработки при помощи подходящего бромирующего агента, такого как н-бромсукцинимид или Br2, в подходящем растворителе, таком как THF, АсОН или CH3CN, при подходящих температуре и периоде времени, такой как комнатная температура при использовании NBS в течение, например, 6 часов или при температуре дефлегмации, если используют Br2, в течение, например, 30 минут.
Стадию 2 могут осуществлять в присутствии подходящего основания, например, Cs2CO3, K2CO3 или NaH, в подходящем растворителе, таком как THF или DMF, при подходящей температуре, такой как комнатная температура, в течение периода времени, например, в течение ночи.
Стадию 3 могут осуществлять при помощи подходящего восстановителя, такого как триацетоксиборгидрид натрия, комплекс 2-пиколин-боран или цианоборгидрид натрия, в присутствии подходящей кислоты, такой как уксусная кислота, в присутствии подходящего растворителя, такого как метанол, DCM, 1,2-DCE, хлороформ, THF или диэтиловый эфир, при подходящей температуре, такой как комнатная температура, 40°С или 50°С в течение всего периода взаимодействия 1-72 часов, который включает период 0-4 ч перед добавлением восстановителя.
Стадию 4 могут осуществлять при помощи подходящего палладиевого катализатора, такого как PdCl2(PPh3)2, Pd2(dba)3, PdCl2(dppf), Pd(OAc)2 или Pd(PPh3)4, подходящего фосфинового лиганда, при необходимости, такого как BrettPhos, DavePhos, Xantphos или BINAP, подходящего основания, такого как NaOtBu, KOtBu, Na2CO3, Cs2CO3 или K2CO3, в подходящем растворителе, таком как вода 1,2-DME, ЕtOН в толуоле, толуол, THF или 1,4-диоксан, при подходящей температуре, такой как 80°С, в микроволновой печи, в течение подходящего периода времени, такого как 20 минут.
Стадию 5a (где защитная группа представляет собой ВОС) могут осуществлять при помощи подходящей кислоты, такой как HCl, в 1,4-диоксане или TFA в DCM, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 5b (где защитная группа представляет собой FMOC) могут осуществлять при помощи раствора пиперидина, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 5c (где защитная группа представляет собой Cbz или бензил) могут осуществлять посредством гидрирования в присутствии Pd/C и Н2, в подходящем растворителе, таком как метанол, этанол или вода, при подходящей температуре, такой как 21°С, в течение периода времени, например, 16 часов.
Стадию 6 могут осуществлять при помощи подходящего восстановителя, такого как триацетоксиборгидрид натрия, комплекс 2-пиколин-боран или цианоборгидрид натрия, в присутствии подходящей кислоты, такой как уксусная кислота, в присутствии подходящего растворителя, такого как метанол, DCM, 1,2-DCE, хлороформ, THF или диэтиловый эфир, при подходящей температуре, такой как комнатная температура, 40°С или 50°С, в течение всего периода взаимодействия 1-72 часов, который включает период 0-4 часа перед добавлением восстановителя.
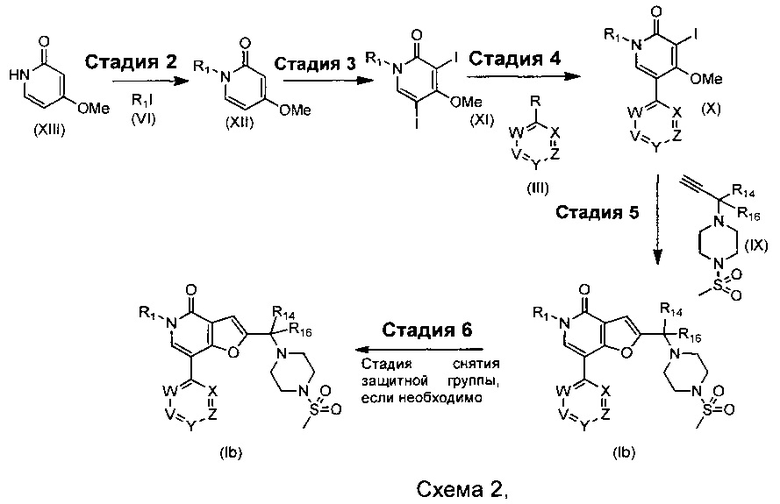
где R1, R14, R16, V, W, X, Y и Z такие, как определено для соединения формулы (I); R выбирают из -В(ОН)2, -BF3K и  .
.
В одном воплощении V представляет собой C-R2, где R2 такой, как определено для соединения формулы (I).
Что касается стадий, показанных на схеме 2, можно использовать следующие реакционные условия.
Стадию 1 могут осуществлять в присутствии меди и хлорида меди в подходящем растворителе, таком как водный Et2O, при подходящей температуре, такой как комнатная температура, в течение периода времени, например, 16 часов.
Стадию 2 могут осуществлять в присутствии подходящего основания, например, Cs2CO3, K2CO3 или NaH, в подходящем растворителе, таком как DMF или THF, при подходящей температуре, такой как комнатная температура или 60°С, в течение периода времени, например, в течение ночи.
Стадию 3 могут осуществлять посредством обработки при помощи 1-йодпирролидин-2,5-диона, йодсукцинимида или I2, в присутствии MeCN или CHCl3, в присутствии кислоты, такой как TFA, АсОН или HNO3, при подходящей температуре, такой как комнатная температура, в течение периода времени, например, 2 часов.
Стадию 4 могут осуществлять при помощи подходящего палладиевого катализатора, такого как Pd(OAc)2, PdCl2(PPh3)2, Pd2(dba)3, PdCl2(dppf) или Pd(PPh3)4, подходящего фосфинового лиганда, при необходимости, такого как TPPTS, BrettPhos, DavePhos, Xantphos или BINAP, подходящего основания, такого как DIPEA, NaOtBu, Cs2CO3 или K2CO3, в подходящем растворителе, таком как водный CH3CN, водный 1,2-DME, EtOH в толуоле, толуол, THF или 1,4-диоксан, при подходящей температуре, такой как 60°С, в течение подходящего периода времени, такого как в течение ночи.
Стадию 5 могут осуществлять при помощи подходящего палладиевого катализатора, такого как PdCl2(PPh3)2, Pd2(dba)3, PdCl2(dppf), Pd(OAc)2 или Pd(PPh3)4, подходящего фосфинового лиганда, при необходимости, такого как PPh3 или Р(2-фурил)3, йодида меди, подходящего основания, такого как триэтиламина, Cs2CO3 или K2CO3, в подходящем растворителе, таком как ACN, толуол, THF, DMF, NMP или 1,4-диоксан, при подходящей температуре, такой как 80°С, в течение подходящего периода времени, такого как 4 суток, или при подходящей температуре, такой как 120°С, в течение подходящего периода времени, такого как 6 часов в микроволновой печи.
Стадию 6a могут осуществлять при помощи подходящей кислоты, такой как HCl в 1,4-диоксане или TFA в DCM, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 6b (где защитная группа представляет собой FMOC) могут осуществлять при помощи раствора пиперидина, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 6c (где защитная группа представляет собой бензил) могут осуществлять посредством гидрирования в присутствии Pd/C и Н2, в подходящем растворителе, таком как метанол, этанол или вода, при подходящей температуре, такой как 21°С, в течение периода времени, например, 16 часов.
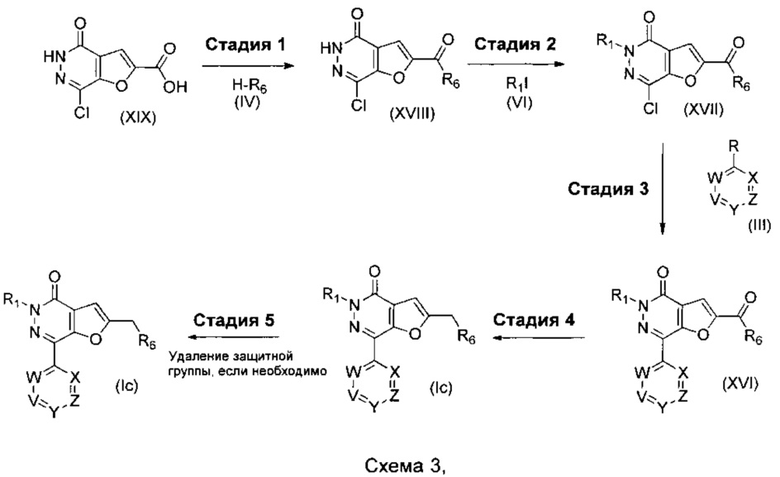
где R1, R6, V, W, X, Y и Z такие, как определено для соединения формулы (I); и R выбирают из -В(ОН)2, -BF3K и  .
.
Если R6 представляет собой -NR11R12 и R12 представляет собой C1-4алкиленNHR13, то R12 защищают посредством подходящей защитной группы, такой как ВОС, FMOC или бензил, которую удаляют на Стадии 5 синтеза. Н-R6 представляет собой соединение формулы (IVa) или соединение формулы (IVb)
 ,
,
где m, n, R7, R9, R10, R11, R12 и E такие, как определено для соединения формулы (I).
В одном воплощении V представляет собой C-R2, где R2 такой, как определено для соединения формулы (I).
Что касается стадий, показанных на схеме 3, можно использовать следующие реакционные условия.
Стадию 1 можно осуществлять посредством обработки при помощи подходящего агента сочетания, такого как HOBt, HOAt или HATU, в присутствии подходящего основания, например Et3N или DIPEA, в подходящем растворителе, таком как THF или DMF, при подходящей температуре, такой как комнатная температура, в течение периода времени, например, 16 часов.
Стадию 2 можно осуществлять в присутствии подходящего основания, например, Cs2CO3, K2CO3 или NaH, в подходящем растворителе, таком как THF или DMF, при подходящей температуре, такой как комнатная температура, в течение периода времени, например, 16 часов.
Стадию 3 можно осуществлять при помощи подходящего палладиевого катализатора, такого как Pd(OAc)2, PdCl2(PPh3)2, Pd2(dba)3, PdCl2(dppf), или Pd(PPh3)4, подходящего фосфинового лиганда, при необходимости, такого как TPPTS, BrettPhos, DavePhos, Xantphos или BINAP, подходящего основания, такого как DIPEA, NaOtBu, Cs2CO3 или K2CO3, в подходящем растворителе, таком как водный CH3CN, водный 1,2-DME, EtOH в толуоле, толуол, THF или 1,4-диоксан, при подходящей температуре, такой как 60°С, в течение подходящего периода времени, такого как в течение ночи.
Стадию 4 можно осуществлять при помощи подходящего восстановителя, такого как комплекс боран-THF или LiAlH4, в подходящем растворителе, таком как THF или Et2O, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, такого как 16 часов.
Стадию 5a можно осуществлять при помощи подходящей кислоты, такой как HCl в 1,4-диоксане или TFA в DCM, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 5b (где защитная группа представляет собой FMOC) можно осуществлять при помощи раствора пиперидина, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 5c (где защитная группа представляет собой бензил) можно осуществлять посредством гидрирования в присутствии Pd/C и Н2, в подходящем растворителе, таком как метанол, этанол или вода, при подходящей температуре, такой как 21°С, в течение периода времени, например, 16 часов.
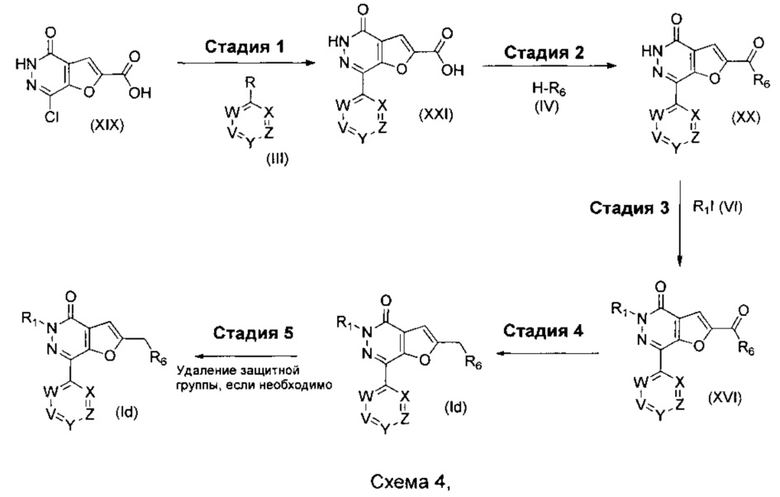
где R1, R6, V, W, X, Y и Z такие, как определено для соединения формулы (I); и R выбирают из -В(ОН)2, -BF3K и
 .
.
Если R6 представляет собой -NR11R12 и R12 представляет собой С1-4алкиленNHR13, то R12 защищают посредством подходящей защитной группы, такой как ВОС, FMOC или бензил, которую удаляют на Стадии 5 синтеза. H-R6 представляет собой соединение формулы (IVa) или соединение формулы (IVb)
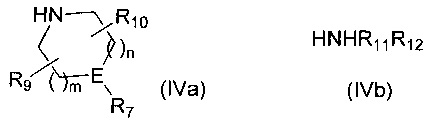
где m, n, R7, R9, R10, R11, R12 и E такие, как определено для соединения формулы (I).
В одном воплощении V представляет собой C-R2, где R2 такой, как определено для соединения формулы (I).
Что касается стадий, показанных на схеме 4, можно использовать следующие реакционные условия.
Стадию 1 можно осуществлять при помощи подходящего палладиевого катализатора, такого как Pd(OAc)2, PdCl2(PPh3)2, Pd2(dba)3, PdCl2(dppf) или Pd(PPh3)4, подходящего фосфинового лиганда, при необходимости, такого как TPPTS, BrettPhos, DavePhos, Xantphos или BINAP, подходящего основания, такого как DIPEA, NaOtBu, Cs2CO3 или K2CO3, в подходящем растворителе, таком как водный CH3CN, водный 1,2-DME, этанол в толуоле, толуол, THF или 1,4-диоксан, при подходящей температуре, такой как 60°С, в течение подходящего периода времени, такого как в течение ночи.
Стадию 2 можно осуществлять посредством обработки при помощи подходящего агента сочетания, такого как HOBt, HOAt или HATU, в присутствии подходящего основания, например Et3N или DIPEA, в подходящем растворителе, таком как THF или DMF, при подходящей температуре, такой как комнатная температура, в течение периода времени, например, 16 часов.
Стадию 3 можно осуществлять в присутствии подходящего основания, например, Cs2CO3, K2CO3 или NaH, в подходящем растворителе, таком как THF или DMF, при подходящей температуре, такой как комнатная температура, в течение периода времени, например, 16 часов.
Стадию 4 можно осуществлять при помощи подходящего восстановителя, такого как комплекс боран-THF или LiAlH4 в подходящем растворителе, таком как THF или Et2O, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, такого как 16 часов.
Стадию 5a можно осуществлять при помощи подходящей кислоты, такой как HCl в 1,4-диоксане или TFA в DCM, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 5b (где защитная группа представляет собой FMOC) можно осуществлять при помощи раствора пиперидина, при подходящей температуре, такой как комнатная температура, в течение подходящего периода времени, например, 1 часа.
Стадию 5c (где защитная группа представляет собой Cbz или бензил) можно осуществлять посредством гидрирования в присутствии Pd/C и Н2 в подходящем растворителе, таком как метанол, этанол или вода, при подходящей температуре, такой как 2°С, в течение периода времени, например, 16 часов.
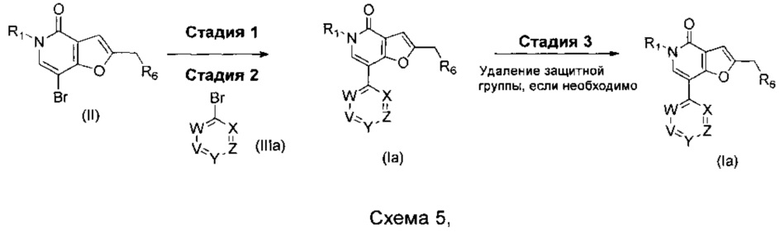
где R1, R6, V, W, X, Y и Z такие, как определено для соединения формулы (I).
В одном воплощении V представляет собой C-R2, где R2 такой, как определено для соединения формулы (I).
Что касается стадий, показанных на схеме, 5 можно использовать следующие реакционные условия.
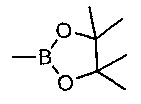
Стадию 1 можно осуществлять посредством обработки при помощи 4,4,5,5-тетраметил-1,3,2-диоксаборолана в присутствии подходящего катализатора, например PEPPSI-SIPr, PEPPSI-IPr или Pd(PPh3)4, в присутствии подходящего основания, например, триэтиламина, в подходящем растворителе, таком как 1,4-диоксан, при подходящей температуре, такой как 100°С, в течение подходящего периода времени, такого как 3 ч.
Стадию 2 можно осуществлять при помощи подходящего палладиевого катализатора, такого как PdCl2(PPh3)2, Pd2(dba)3, PdCl2(dppf).DCM, Pd(OAc)2, Pd(PPh3)4 PEPPSI-IPr или PEPPSI-SIPr, подходящего основания, такого как NaOtBu, KOtBu, Na2CO3, Cs2CO3 или K2CO3, в подходящем растворителе, таком как 1,2-DME, водный 1,2-DME, метанол в толуоле, этанол в толуоле, толуол, THF, водный THF, водный изопропанол, DMF или водный 1,4-диоксан, при подходящей температуре, такой как 80-150°С, возможно в микроволновом реакторе, в течение подходящего периода времени, такого как от 20 мин до 20 ч.
Когда защитная группа представляет собой ВОС, тогда стадию 3 можно осуществлять в присутствии подходящей кислоты, такой как трифторуксусная кислота или соляная кислота, в подходящем растворителе, таком как DCM или 1,4-диоксан, при подходящей температуре, такой как комнатная температура и в течение подходящего периода времени, такого как от 1 до 24 ч.
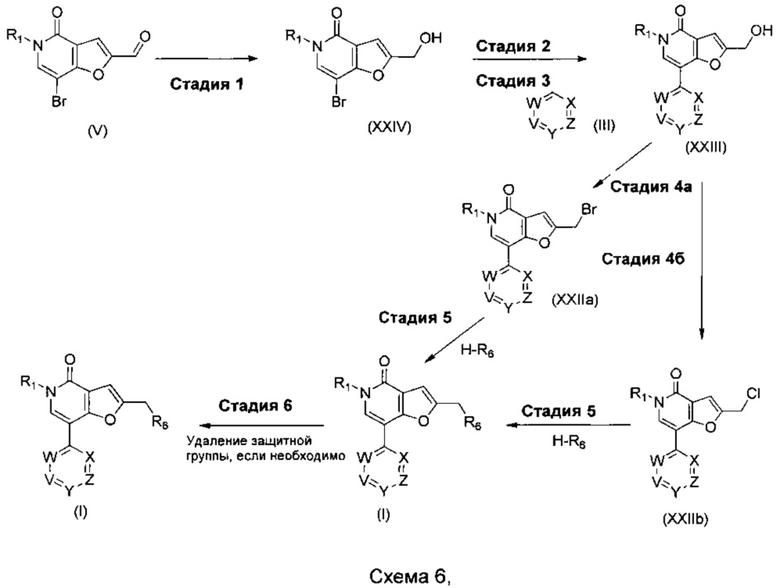
где R1, R6, V, W, X, Y и Z такие, как определено для соединения формулы (I); и R выбирают из -В(ОН)2, -BF3K и
 .
.
Если R6 представляет собой -NR11R12 и R12 представляет собой С1-4алкиленNHR13, то R12 защищают посредством подходящей защитной группы, такой как ВОС, FMOC или бензил, которую удаляют на стадии 6 синтеза. H-R6 представляет собой соединение формулы (IVa) или соединение формулы (IVb)
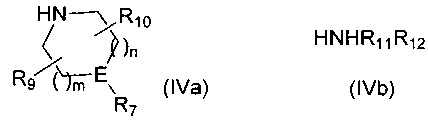 ,
,
где m, n, R7, R9, R10, R11, R12 и E такие, как определено для соединения формулы (I).
В одном воплощении V представляет собой C-R2, где R2 такой, как определено для соединения формулы (I).
Что касается стадий, показанных на схеме 6, можно использовать следующие реакционные условия.
Стадию 1 можно осуществлять посредством обработки при помощи подходящего восстановителя, такого как боргидрид натрия, в подходящем растворителе, таком как этанол, при подходящей температуре, такой как комнатная температура и в течение подходящего периода времени, например, 2 часов.
Стадию 2 можно осуществлять посредством обработки при помощи 4,4,5,5-тетраметил-1,3,2-диоксаборолана в присутствии подходящего катализатора, например, PEPPSI-SIPr, PEPPSI-IPr или Pd(PPh3)4, в присутствии подходящего основания, например, триэтиламина, в подходящем растворителе, таком как 1,4-диоксан, при подходящей температуре, такой как 100°С, в течение подходящего периода времени, такого как 3-18 ч.
Стадию 3 можно осуществлять при помощи подходящего палладиевого катализатора, такого как PdCl2(PPh3)2, Pd2(dba)3, PdCl2(dppf).DCM, Pd(OAc)2, Pd(PPh3)4 PEPPSI-IPr или PEPPSI-SIPr, подходящего основания, такого как NaOtBu, KOtBu, Na2CO3, Cs2CO3 или K2CO3, в подходящем растворителе, таком как 1,2-DME, водный 1,2-DME, метанол в толуоле, этанол в толуоле, толуол, THF, водный THF, водный изопропанол, DMF или водный 1,4-диоксан, при подходящей температуре, такой как 80-150°С, возможно в микроволновом реакторе, в течение подходящего периода времени, такого как от 20 мин до 20 ч.
Стадию 4a можно осуществлять посредством обработки при помощи подходящей бромирующего агента, такого как РВr3, в подходящем растворителе, таком как 1,4-диоксан, при подходящей температуре, такой как 40-60°С и в течение подходящего периода времени, например, от 2 ч до периода ночи.
Стадию 4b можно осуществлять посредством обработки при помощи метансульфонилхлорида, в подходящем растворителе, таком как DCM, при помощи подходящего основания, такого как пиридин, при подходящей температуре, такой как комнатная температура и в течение подходящего периода времени, например, в течение ночи.
Стадию 5 можно осуществлять в присутствии подходящего основания, такого как DIPEA, K2CO3 или NaH, в подходящем растворителе, таком как DMSO или DMF, при подходящей температуре, такой как 110°С, в микроволновом реакторе, или при комнатной температуре и в течение подходящего периода времени, например от 30 мин до 3 ч.
Когда защитная группа представляет собой ВОС, стадию 6 можно осуществлять в присутствии подходящей кислоты, такой как трифторуксусная кислота или соляная кислота, в подходящем растворителе, таком как DCM или 1,4-диоксан, при подходящей температуре, такой как комнатная температура и в течение подходящего периода времени, такого как от 1 часа до 24 часов.
Таким образом, в одном воплощении изобретения предложен способ получения соединения формулы (I), включающий взаимодействие соединения формулы (II)
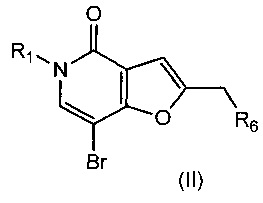 ,
,
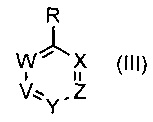
где R1 и R6 такие, как определено выше, с соединением формулы (III)
 ,
,
где V, W, X, Y и Z такие, как определено выше и R выбирают из В(ОН)2, BF3K и
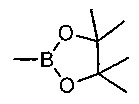 ; возможно с последующей стадией снятия защиты, при необходимости. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
; возможно с последующей стадией снятия защиты, при необходимости. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (II), включающий взаимодействие соединения формулы (V)
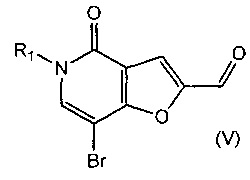 ,
,
где R1 такой, как определено выше, с амином формулы (IVa) или формулы (IVb)
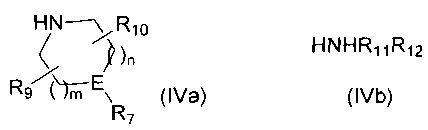 ,
,
где R7, R9, R10, R11, R12, m, n и E такие, как определено выше, и если R12 представляет собой С1-4алкиленNHR13, то R12 защищают посредством подходящей защитной группы, такой как ВОС, FMOC Cbz или бензил.
В другом воплощении изобретения предложен способ получения соединения формулы (V), включающий взаимодействие соединения формулы (VII)
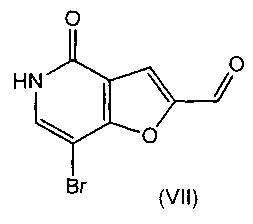
с соединением формулы (VI)
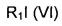 ,
,
где R1 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (VII), включающий взаимодействие соединения формулы (VIII)
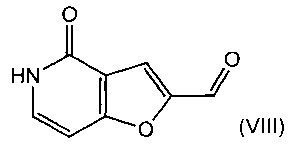
с бромирующим агентом, например с н-бромсукцинимидом.
В другом воплощении изобретения предложен способ получения соединения формулы (I), включающий взаимодействие соединения формулы (X)
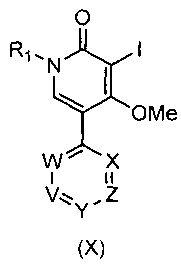 ,
,
где R1, V, W, X, Y и Z такие, как определено выше, с соединением формулы (IX)
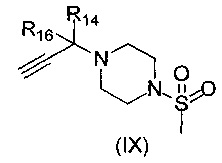 ,
,
где R14 и R16 такие, как определено выше; возможно с последующей стадией снятия защиты, при необходимости. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (X), включающий взаимодействие соединения формулы (XI)
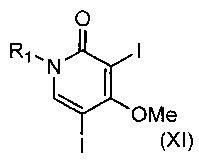 ,
,
где R1 такой, как определено выше, с соединением формулы (III)
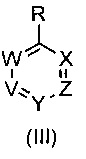 ,
,
где V, W, X, Y и Z такие, как определено выше; и R выбирают из В(ОН)2, BF3K и
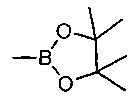 . В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
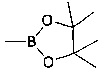
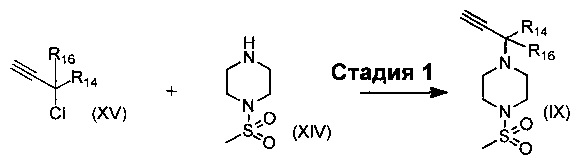
В еще одном воплощении изобретения предложен способ получения соединения формулы (IX), включающий взаимодействие соединения формулы (XIV)
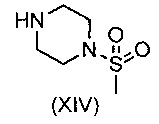
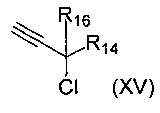
с соединением формулы (XV)
 ,
,
где R14 и R16 такие, как определено выше.
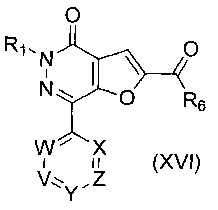
В другом воплощении изобретения предложен способ получения соединения формулы (I), включающий взаимодействие соединения формулы (XVI)
 ,
,
где R1, R6, V, W, X, Y и Z такие, как определено выше, с восстановителем; возможно с последующей стадией снятия защиты, при необходимости. В одном воплощении восстановитель представляет собой комплекс боран-THF или LiAlH4. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
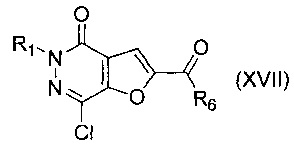
В другом воплощении изобретения предложен способ получения соединения формулы (XVI), включающий взаимодействие соединения формулы (XVII)
 ,
,
где R1 и R6 такие, как определено выше, с соединением формулы (III)
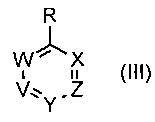 ,
,
где V, W, X, Y и Z такие, как определено выше; и R выбирают из В(ОН)2, BF3K и
 . В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (XVII), включающий взаимодействие соединения формулы (XVIII)
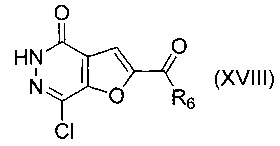 ,
,
где R6 такой, как определено выше, с соединением формулы (VI)
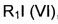 ,
,
где R1 такой, как определено выше.
В другом воплощении изобретения предлагается способ получения соединения формулы (XVIII), включающий взаимодействие соединения формулы (XIX)
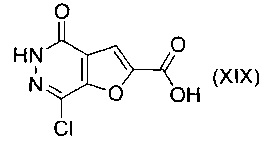
с амином формулы (IVa) или формулы (IVb)
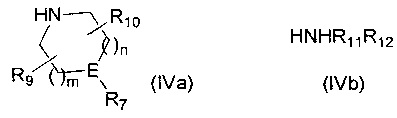 ,
,
где R7, R9, R10, R11, R12, m, n и E такие, как определено выше, и если R12 представляет собой С1-4алкиленNHR13, то R12 защищают подходящей защитной группой, такой как ВОС, FMOC, Cbz или бензил.
В другом воплощении изобретения предложен способ получения соединения формулы (XVI), включающий взаимодействие соединения формулы (XX)
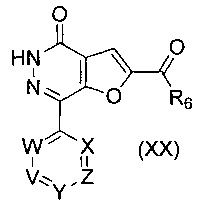 ,
,
где R6, V, W, X, Y и Z такие, как определено выше; с соединением формулы (VI)
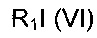 ,
,
где R1 такой, как определено выше. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (XX), включающий взаимодействие соединения формулы (XXI)
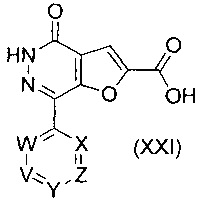 ,
,
где V, W, X, Y и Z такие, как определено выше; с амином формулы (IVa) или формулы (IVb)
 ,
,
где R7, R9, R10, R11, R12, m, n и Е такие, как определено выше, и если R12 представляет собой С1-4алкиленNHR13, то R12 защищают подходящей защитной группой, такой как ВОС, FMOC, Cbz или бензил. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В еще одном воплощении изобретения предложен способ получения соединения формулы (XXI), включающий взаимодействие соединения формулы (XIX)
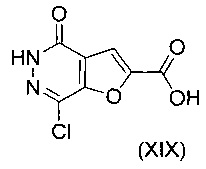
с соединением формулы (III)
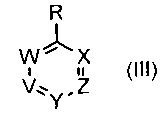 ,
,
где V, W, X, Y и Z такие, как определено выше; и R выбирают из В(ОН)2, BF3K и
 . В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (I), включающий взаимодействие соединения формулы (II)
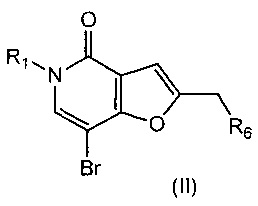 ,
,
где R1 и R6 такие, как определено выше, с помощью промежуточного соединения боронатного сложного эфира, с соединением формулы (IIIa)
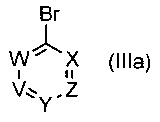 ,
,
где V, W, X, Y и Z такие, как определено выше; возможно с последующей стадией снятия защиты, при необходимости. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (I), включающий взаимодействие соединения формулы (XXIIa) или (XXIIb)
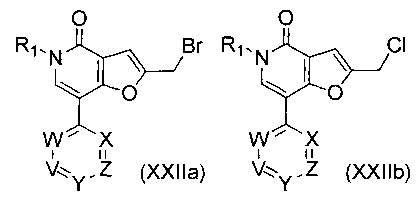 ,
,
где R1, V, W, X, Y и Z такие, как определено выше, с амином формулы (IVa) или формулы (IVb)
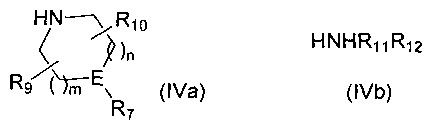 ,
,
где R7, R9, R10, R11, R12, m, n и E такие, как определено выше, и если R12 представляет собой С1-4алкиленNHR13, то R12 защищают подходящей защитной группой, такой как ВОС, FMOC Cbz или бензил. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (XXIIa) или (XXIIb), включающий взаимодействие соединения формулы (XXIII)
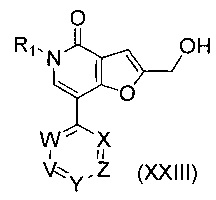 ,
,
где R1, V, W, X, Y и Z такие, как определено выше, в условиях, подходящих для галогенирования. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предложен способ получения соединения формулы (XXIII), включающий взаимодействие соединения формулы (XXIV)
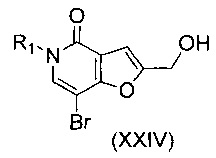 ,
,
где R1 такой, как определено выше, с соединением формулы (III)
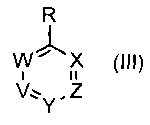 ,
,
где V, W, X, Y и Z такие, как определено выше, и R выбирают из В(ОН)2, BF3K и
 . В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
. В одном воплощении V представляет собой C-R2, где R2 такой, как определено выше.
В другом воплощении изобретения предлагается способ получения соединения формулы (XXIV), включающий взаимодействие соединения формулы (V)
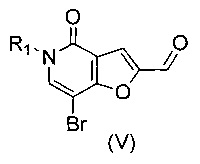
где R1 такой, как определено выше, с подходящим восстановителем.
Соединения формул (III), (IIIa), (IV), (VI), (VIII), (XIII), (XIV), (XV) и (XIX) имеются в продаже или могут быть легко синтезированы посредством известных способов, например как сообщается Suzuki в Chem. Rev., 1995, vol. 95, p 2457-2483.
Специалисту в данной области техники будет ясно, что может быть выгодным защищать одну или более функциональных групп соединений, описанных выше. Примеры защитных групп и средства для их удаления можно найти в Т.W. Greene 'Protective Group in Organic Synthesis (4th edition, J. Wiley and Sons, 2006). Подходящие амино-защитные группы включают ацил (например, ацетил, карбамат (например, 2',2',2'-трихлорэтоксикарбонил, бензилоксикарбонил или трет-бутоксикарбонил) и арилалкил (например, бензил), которые могут быть удалены посредством гидролиза (например, при использовании кислоты, такой как соляная кислота в 1,4-диоксане или трифторуксусная кислота в DCM) или путем восстановления (например, гидрогенолизом бензильной или бензилоксикарбонильной группы или восстановительным удалением 2',2',2'-трихлорэтоксикарбонильной группы, при использовании цинка в уксусной кислоте), при необходимости. Другие подходящие амино-защитные группы включают трифторацетил (-COCF3), который может быть удален путем катализируемого основанием гидролиза.
Понятно, что в любом из путей, описанных выше, точный порядок стадий синтеза, посредством которых различные группы и группировки вводят в молекулу, можно варьировать. В квалификации специалиста в данной области техники обеспечить, чтобы группы или группировки, введенные на одной стадии процесса, не подвергались последующим трансформациям и взаимодействиям, и выбрать порядок стадий синтеза соответствующим образом.
Некоторые промежуточные соединения, описанные выше, образуют еще один аспект изобретения.
Соединения формулы (I) и его соли являются ингибиторами бромодоменов, и, следовательно, как полагают, потенциально полезны в лечении заболеваний или состояний, для которых показан ингибитор бромодомена.
Таким образом, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для использования в терапии. Соединение формулы (I) или его фармацевтически можно применять в лечении заболеваний или состояний, для которых показан ингибитор бромодомена.
Таким образом, в настоящем изобретении предложено соединение формулы (I) или его фармацевтически приемлемая соль для использования в лечении любых заболеваний или состояний, для которых показан ингибитор бромодомена. В одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для использования в лечении острых или хронических аутоиммунных и/или воспалительных состояний. В другом воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для использования в лечении заболеваний или состояний, в которые включают воспалительные ответы на инфекции, вызванные бактериями, вирусами, грибами, паразитами или их токсинами. В другом воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для использования в лечении вирусных инфекций. В еще одном воплощении предложено соединение формулы (I) или его фармацевтически приемлемая соль для использования в лечении рака.
Также предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний или состояний, для которых показан ингибитор бромодомена. В одном воплощении предлагается применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения острых или хронических аутоиммунных и/или воспалительных состояний. В другом воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения заболеваний или состояний, в которые включены воспалительные ответы на инфекции, вызванные бактериями, вирусами, грибами, паразитами или их токсинами. В другом воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения вирусных инфекций. В другом воплощении предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения рака.
Также предложен способ лечения заболеваний или состояний, для которых показан ингибитор бромодомена, у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В одном воплощении предложен способ лечения острых или хронических аутоиммунных и/или воспалительных состояний у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В другом воплощении предложен способ лечения заболеваний или состояний, в которые включены воспалительные ответы на инфекции, вызванные бактериями, вирусами, грибами, паразитами или их токсинами, у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В другом воплощении предложен способ лечения вирусных инфекций у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли. В еще одном воплощении предложен способ лечения рака у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Подходящий субъект, нуждающийся в лечении, является млекопитающим, в частности, человеком.
При использовании в данном описании изобретения термин "эффективное количество" означает, что количество лекарственного средства или фармацевтического агента, который будет вызывать биологический или медицинский ответ ткани, системы или субъекта (например человека), которого добивается, например, исследователь или клиницист. Кроме того, термин "терапевтически эффективное количество" обозначает любое количество, которое, по сравнению с аналогичным субъектом, не получающим такое количество, имеет результатом улучшенное лечение, исцеление, предупреждение или уменьшение интенсивности симптомов заболевания, расстройства или побочного эффекта, или снижение скорости прогрессирования заболевания или расстройства. Термин также включает в свои рамки количества, эффективные для усиления нормальной физиологической функции.
Полагают, что ингибиторы бромодоменов являются полезными в лечении целого ряда заболеваний или состояний, связанных с системным воспалением или воспалением тканей, воспалительными ответами на инфекцию или гипоксию, клеточной активацией и пролиферацией, метаболизмом липидов, фиброзом, и в предупреждении и лечении вирусных инфекций.
Ингибиторы бромодоменов могут быть полезными в лечении широкого спектра острых или хронических аутоиммунных и/или воспалительных состояний, таких как ревматоидный артрит, остеоартрит, острая подагра, псориаз, системная красная волчанка, рассеянный склероз, воспалительная болезнь кишечника (болезнь Крона и неспецифический язвенный колит), астма, хроническое обструктивное заболевание дыхательных путей, пневмонит, миокардит, перикардит, миозит, экзема, дерматит (включая атопический дерматит), алопеция, витилиго, буллезные заболевания кожи, нефрит, васкулит, гиперхолестеринемия, атеросклероз, болезнь Альцгеймера, депрессия, синдром Шегрена, сиаладенит, окклюзия центральной вены сетчатки, окклюзия разветвленный вены сетчатки, синдром Ирвина-Гасса (после катаракты и послеоперационный), пигментный ретинит, парспланит, дробьевидная ретинохороидопатия, эпиретинальная мембрана, кистозный макулярный отек, парафовеальная телеангиэктазия, тракционные макулопатии, витреомакулярный тракционный синдром, отслойка сетчатки, нейроретинит, идиопатический макулярный отек, ретинит, сухость глаз (сухой кератоконъюнктивит), весенний кератоконъюнктивит, атопический кератоконъюнктивит, увеит (такой как передний увеит, панувеит, задний увеит, ассоциированный с увеитом макулярный отек), склерит, диабетическая ретинопатия, диабетический макулярный отек, возрастная макулярная дистрофия, гепатит, панкреатит, первичный биллиарный цирроз, склерозирующий холангит, болезнь Аддисона, гипофизит, тиреоидит, диабет I типа, гигантоклеточный артериит, нефрит, включая волчаночный нефрит, васкулит с поражением органов, такой как гломерулонефрит, васкулит, включая гигантоклеточный артериит, гранулематоз Вегенара, узелковый полиартериит, болезнь Бехчета, болезнь Кавасаки, синдром Такаясу, гангренозная пиодермия, васкулит с поражением органов и острое отторжение трансплантированных органов.
В одном воплощении острое или хроническое аутоиммунное и/или воспалительное состояние представляет собой расстройство метаболизма липидов посредством регуляции АРО-А1, такое как гиперхолестеринемия, атеросклероз и болезнь Альцгеймера.
В другом воплощении острое или хроническое аутоиммунное и/или воспалительное состояние представляет собой респираторное расстройство, такое как астма или хроническое обструктивное заболевание дыхательных путей.
В другом воплощении острое или хроническое аутоиммунное и/или воспалительное состояние представляет собой системное воспалительное расстройство, такое как ревматоидный артрит, остеоартрит, острая подагра, псориаз, системная красная волчанка, рассеянный склероз или воспалительное заболевание кишечника (болезнь Крона и неспецифический язвенный колит).
В другом воплощении острое или хроническое аутоиммунное и/или воспалительное состояние представляет собой рассеянный склероз.
В еще одном воплощении острое или хроническое аутоиммунное и/или воспалительное состояние представляет собой диабет I типа.
Ингибиторы бромодоменов могут быть полезными в лечении заболеваний или состояний, в которые включены воспалительные ответы на инфекции, вызванные бактериями, вирусами, грибами, паразитами или их токсинами, таких как сепсис, острых сепсис, септический синдром, септический шок, эндотоксемия, синдром системного воспалительного ответа (ССВО), синдром мультиорганной дисфункции, синдром токсического шока, острое повреждение легких, РДСВ (респираторный дистресс-синдром взрослых), острая почечная недостаточность, фульминантный гепатит, ожоги, острый панкреатит, послеоперационные синдромы, саркоидоз, реакции Герксхаймера, энцефалит, миелит, менингит, малярия и ССВО, ассоциированный с вирусными инфекциями, такими как грипп, опоясывающий герпес, простой герпес и коронавирус. В одном воплощении заболевание или состояние, которое включает воспалительный ответ на инфекцию, вызванную бактериями, вирусом, грибами, паразитом или их токсинами, представляет собой острый сепсис.
Ингибиторы бромодоменов могут быть полезными в лечении состояний, ассоциированных с ишемическим-реперфузионным повреждением, таким как инфаркт миокарда, сосудисто-мозговая ишемия (инсульт), острый коронарный синдром, реперфузионное повреждение почек, трансплантация органа, шунтирование коронарной артерии, процедуры сердечно-легочного шунтирования, легочная, почечная, печеночная эмболия, эмболия сосудов желудочно-кишечного тракта или периферическая эмболия конечности.
Ингибиторы бромодоменов могут быть полезными в лечении фиброзных состояний, таких как идиопатический фиброз легких, фиброз почек, послеоперационное сужение сосудов, образование келоидных рубцов, склеродермия (включая кольцевидную склеродермию) и фиброз сердца.
Ингибиторы бромодоменов могут быть полезными в лечении вирусных инфекций, таких как инфекции и реактивации простого герпеса, герпетические лихорадки, инфекции и реактивации опоясывающего герпеса, ветряная оспа, опоясывающий лишай, вирус папилломы человека (ВПЧ), вирус иммунодефицита человека (ВИЧ), цервикальная неоплазия, аденовирусные инфекции, включая острое респираторное заболевание, поксвирусные инфекции, такие как коровья оспа и натуральная оспа и вирус африканской лихорадки свиней. В одном воплощении вирусная инфекция представляет собой ВПЧ-инфекцию кожи или цервикального эпителия. В другом воплощении вирусная инфекция представляет собой латентную ВИЧ-инфекцию.
Ингибиторы бромодоменов могут быть полезными в лечении раковых заболеваний, включая гематологический рак (такой как лейкемия, лимфома и множественная миелома), эпителиальный рак, включая карциномы легкого, молочной железы и толстой кишки, карциномы средней линии, мезенхимальные, печеночные, почечные и неврологические опухоли.
Ингибиторы бромодоменов могут быть полезными в лечении одного или более раковых заболеваний, выбранных из рака головного мозга (глиомы), глиобластом, синдрома Баньяна-Зонана, синдрома Коудена, болезни Лермитт-Дуклос, рака молочной железы, воспалительного рака молочной железы, колоректального рака, опухоли Вильмса, саркомы Юинга, рабдомиосаркомы, эпендимомы, медуллобластомы, рака толстой кишки, рака головы и шеи, рака почки, рака легкого, рака печени, меланомы, сквамозной карциномы, рака яичника, рака поджелудочной железы, рака предстательной железы, саркомы, остеосаркомы, гигантоклеточной опухоли кости, рака щитовидной железы, лимфобластного Т-клеточного лейкоза, хронического миелоидного лейкоза, хронической лимфоцитарного лейкоза, волосатоклеточного лейкоза, острого лимфобластного лейкоза, острого миелоидного лейкоза, хронического нейтрофильного лейкоза, острого лимфобластного Т-клеточного лейкоза, плазмоцитомы, иммунобластного крупноклеточного лейкоза, мантийноклеточного лейкоза, множественной миеломы, мегакариобластного лейкоза, острого мегакариоцитарного лейкоза, промиелоцитарного лейкоза, лейкоза смешанного происхождения, эритролейкоза, злокачественной лимфомы, лимфомы Ходжкина, неходжкинской лимфомы, лимфобластной Т-клеточной лимфомы, лимфомы Беркитта, фолликулярной лимфомы, нейробластомы, рака мочевого пузыря, уротелиального рака, рака вульвы, цервикального рака, эндометриального рака, рака почки, мезотелиомы, рака пищевода, рака слюнной железы, печеночно-клеточного рака, рака желудка, рака носоглотки, рака щеки, рака полости рта, GIST (желудочно-кишечная стромальная опухоль), среднелинейной карциномы NUT и рака яичек.
В одном воплощении рак представляет собой лейкоз, например лейкоз, выбранный из острого моноцитарного лейкоза, острого миелоидного лейкоза, хронического миелоидного лейкоза, хронического лимфоцитарного лейкоза и лейкоза смешанного происхождения (MLL). В другом воплощении рак представляет собой среднелинейную карциному NUT. В другом воплощении рак представляет собой множественную миелому. В другом воплощении рак представляет собой рак легкого, такой как мелкоклеточный рак легкого (SCLC). В другом воплощении рак представляет собой нейробластому. В другом воплощении рак представляет собой лимфому Беркитта. В другом воплощении рак представляет собой цервикальный рак. В другом воплощении рак представляет собой рак пищевода. В другом воплощении рак представляет собой рак яичника. В другом воплощении рак представляет собой рак молочной железы. В другом воплощении рак представляет собой колоректальный рак.
В одном воплощении заболевание или состояние, для которого показан ингибитор бромодомена, выбирают из заболеваний, ассоциированных с синдромом системного воспалительного ответа, таких как сепсис, ожоги, панкреатит, обширная травма, кровоизлияние и ишемия. В этом воплощении ингибитор бромодомена следует вводить в диагностированное место для снижения степени: ССВО, начала шока, синдрома мультиорганной дисфункции, который включает начало острого повреждения легких, РДСВ, острого повреждения почки, печени, сердца или желудочно-кишечного тракта и летальности. В другом воплощении ингибитор бромодомена следует вводить до операционных или других процедур, ассоциированных с высоким риском сепсиса, кровоизлияния, обширного повреждения тканей, ССВО или MODS (синдрома мультиорганной дисфункции). В конкретном воплощении заболевание или состояние, для которого показан ингибитор бромодомена, представляет собой сепсис, септический синдром, септический шок и эндотоксемию. В другом воплощении ингибитор бромодомена показан для лечения острого или хронического панкреатита. В другом воплощении бромодомен показан для лечения ожогов.
При использовании в данном описании изобретения ссылка на "лечение" конкретного заболевания или состояния включает предупреждении или профилактику такого заболевания или состояния.
Термин "заболевания или состояния, для которых показан ингибитор бромодомена", подразумевает включение всех или каждого из вышеуказанных заболеваний или состояний.
В изобретении также предложен способ ингибирования бромодомена, включающий контактирование бромодомена с соединением формулы (I) или его фармацевтически приемлемой солью.
Хотя возможно, что для использования в терапии соединение формулы (I), а также его фармацевтически приемлемые соли могут быть введены в виде необработанного химического продукта, они обычно представляют собой активный ингредиент в составе фармацевтической композиции.
Таким образом, в настоящем изобретении предложена в еще одном аспекте фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент. Соединения формулы (I) и их фармацевтически приемлемые соли такие, как описано выше. Носитель(и), разбавитель(и) или эксципиент(и) должны быть приемлемыми, в смысле являться совместимыми с другими ингредиентами композиции и не опасными для ее реципиента. Согласно другому аспекту изобретения также предложен способ изготовления фармацевтической композиции, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или более чем одним фармацевтически приемлемым носителем, разбавителем или эксципиентом. Фармацевтическую композицию можно применять в лечении любого из состояний, описанных в данном описании изобретения.
Так как соединения формулы (I) предназначены для использования в фармацевтических композициях, нетрудно понять, что каждое из них предпочтительно предлагается по существу в чистой форме, например, чистое по меньшей мере на 85%, в особенности чистое по меньшей мере на 98% (% на основе масс./масс.).
Фармацевтические композиции могут быть представлены в форме одиночных доз, содержащих предварительно определенные количества активного ингредиента на одиночную дозу. Предпочтительные композиции с одиночной дозой представляют собой композиции, содержащие суточную дозу или субдозу, или ее соответствующую фракцию активного ингредиента. Такие одиночные дозы, следовательно, могут быть введены более одного раза в сутки. Предпочтительные композиции с одиночной дозой представляют собой композиции, содержащие суточную дозу или субдозу (для введения более одного раза в сутки), как изложено выше в данном описании изобретения, или ее соответствующую фракцию активного ингредиента.
Фармацевтические композиции могут быть адаптированы для введения любым подходящим путем, например, посредством перорального (включая трансбуккальный или сублингвальный), ректального, ингаляционного, интраназального, местного (включая трансбуккальный, сублингвальный или чрескожный), окулярного (включая местный, внутриглазной, подконъюнктивальный, эписклеральный, субтеноновый), вагинального или парентерального (включая подкожный, внутримышечный, внутривенный или внутрикожный) пути. Такие композиции могут быть получены посредством любого способа, известного в области фармации, например путем приведения активного ингредиента в ассоциацию с носителем(яи) или эксципиентом(ами).
В одном воплощении фармацевтическая композиция адаптирована для парентерального введения, в частности внутривенного введения.
В одном воплощении фармацевтическая композиция адаптирована для перорального введения.
В одном воплощении фармацевтическая композиция адаптирована для местного введения.
Фармацевтические композиции, адаптирование для парентерального введения, включают водные и неводные стерильные растворы для инъекций, которые могут содержать антиоксиданты, буферы, бактериостаты и растворенные вещества, которые делают композицию изотоничной крови предназначенного реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие агенты и загустители. Композиции могут быть представлены в однодозовых или многодозовых контейнерах, например, в запаянных ампулах и флаконах, и могут храниться после сушки из замороженного состояния (в лиофилизированном состоянии), требуя только добавления стерильного жидкого носителя, например воды для инъекций, непосредственно перед применением. Приготовленные для немедленного применения растворы для инъекций и суспензии могут быть получены из стерильных порошков, гранул и таблеток.
Фармацевтические композиции, адаптированные для перорального введения, могут быть представлены в виде отдельных дозированных единиц, таких как капсулы или таблетки; порошков или гранул; растворов или суспензий в водных или неводных жидкостях; пищевых пенок или пищевой взбитой массы; или жидких эмульсий масло-в-воде или жидких эмульсий вода-в-масле.
Например, для перорального введения в форме таблетки или капсулы, активный лекарственный компонент может быть объединен с пероральным, нетоксичным фармацевтически приемлемым инертным носителем, таким как EtOH, глицерин, вода и тому подобное. Порошки, подходящие для включения в таблетки или капсулы, могут быть получены путем измельчения соединения до подходящего мелкого размера (например, посредством микронизации) и смешивания с аналогичным образом полученным фармацевтическим носителем, таким как пищевой углевод, например, крахмал или маннит. Ароматизатор, консервант, диспергирующий агент и краситель также могут быть представлены.
Капсулы могут быть изготовлены путем получения смеси порошков, как описано выше, и заполнением сформированных желатиновых оболочек. Скользящие вещества и смазывающие вещества, такие как коллоидный диоксид кремния, тальк, стеарат магния, стеарат кальция или твердый полиэтиленгликоль, могут быть добавлены к смеси порошков перед процессом заполнения. Разрыхляющий или солюбизирующий агент, такой как агар-агар, карбонат кальция или карбонат натрия, также может быть добавлен для улучшения доступности лекарственного средства, когда капсулу принимают внутрь.
Кроме того, если желательно или необходимо, подходящие связующие вещества, скользящие вещества, смазывающие вещества, подсластители, ароматизаторы, разрыхляющие агенты и красители также могут быть включены в смесь. Подходящие связующие вещества включают крахмал, желатин, натуральные сахара, такие как глюкоза или бета-лактоза, сахаристые вещества из кукурузы, натуральные и синтетические камеди, такие как гуммиарабик, трагакант или альгинат натрия, карбоксиметилцеллюлоза, полиэтиленгликоль, воски и тому подобные. Смазывающие вещества, используемые в этих лекарственных формах, включают олеат натрия, стеарат натрия, стеарат магния, бензоат натрия, ацетат натрия, хлорид натрия и тому подобные. Разрыхлители включают крахмал, метилцеллюлозу, агар, бентонит, ксантановую камедь и тому подобные. Таблетки изготовляют, например, путем получения порошковой смеси, гранулирования или брикетирования, добавления смазывающего вещества и разрыхлителя и прессованием в таблетки. Порошковую смесь получают смешиванием соединения, соответствующим образом измельченного, с разбавителем или основанием, как описано выше, и возможно, со связующим веществом, таким как карбоксиметилцеллюлоза, альгинат, желатин, или поливинилпирролидон, ретардантом раствора, таким как парафин, ускорителем ресорбции, таким как четвертичная соль и/или абсорбирующим агентом, таким как бентонит, каолин или дикальция фосфат. Порошковую смесь можно гранулировать путем смачивания при помощи связующего вещества, такого как сироп, крахмальная паста, клейкое вещество акации или растворы целлюлозных или полимерных веществ, и эструдирования через решето. В качестве альтернативы гранулированию порошковую смесь можно пропустить через таблеточную машину и результатом являются неполностью образовавшиеся брикеты, раздробленные на гранулы. Гранулы могут быть смазаны для предотвращения прилипания к формообразующим штампам таблеток посредством добавления стеариновой кислоты, стеаратной соли, талька или минерального масла. Смазанную смесь затем спрессовывают в таблетки. Соединения формулы (I) и его фармацевтически приемлемые соли также могут быть объединены с сыпучим инертным носителем и спрессованы в таблетки непосредственно, без прохождения через стадии гранулирования или брикетирования. Может быть предложено прозрачное или матовое защитное покрытие, состоящее из водонепроницаемого слоя шеллака, покрытие из сахара или полимерного вещества и глянцевое покрытие из воска. В эти покрытия могут быть добавлены красители, чтобы различать разные стандартные дозировки.
Пероральные жидкости, такие как раствор, сиропы и эликсиры могут быть приготовлены в форме единичной дозы так, чтобы заданная доза содержала предварительно определенное количество соединения. Сиропы могут быть приготовлены путем растворения соединения в подходящем ароматизированном водном растворе, в то время как эликсиры готовят посредством применения нетоксического спиртового носителя. Суспензии могут быть приготовлены путем диспергирования соединения в нетоксическом носителе. Также могут быть добавлены солюбилизаторы и эмульгаторы, такие как этоксилированные изостеариловые спирты и полиоксиэтиленсорбитоловые эфиры, консерванты, вкусовые добавки, такие как масло мяты перечной, или натуральные подсластители, или сахарин или другие искусственные подсластители, и тому подобные.
Композиции для перорального введения могут быть предназначены для получения модифицированного профиля высвобождения, так чтобы замедлять или иным образом контролировать высвобождение терапевтически активного агента.
Когда это целесообразно, композиции дозированных единиц для перорального введения можно микроинкапсулировать. Композиция может быть приготовлена, чтобы продлевать или замедлять высвобождение, например, путем покрытия или внедрения вещества из частиц в полимеры, воск или тому подобное.
Соединения формулы (I) и его фармацевтически приемлемые соли также могут быть введены в форме липосомальных систем доставки, таких как небольшие однослойные везикулы, большие однослойные везикулы и многослойные везикулы. Липосомы могут быть образованы из целого ряда фосфолипидов, таких как холестерин, стеариламин или фосфатидилхолины.
Фармацевтические композиции, адаптированные для местного введения, могут быть приготовлены в виде мазей, кремов, суспензий, эмульсий, лосьонов, порошков, растворов, паст, гелей, пенок, спреев, аэрозолей или масел. Такие фармацевтические композиции могут включать общепринятые добавки, которые включают консерванты, растворители для содействия прониканию лекарственного средства, сорастворители, смягчающие вещества, пропелленты, модифицирующие вязкость агенты (гелеобразующие агенты), поверхностно-активные вещества и носители, но не ограничиваются ими. В одном воплощении предложена фармацевтическая композиция, адаптированная для местного введения, содержащая от 0,01 до 10%, или от 0,01 до 1% соединения формулы (I) или его фармацевтически приемлемой соли, по массе композиции.
Для лечения глаз или других наружных тканей, например полости рта и кожи композиции предпочтительно применяют в виде местной мази, крема, геля, спрея или пенки. При изготовлении в мази активный ингредиент может применяться либо с парафиновой, либо смешиваемой с водной основой мази. Альтернативно, активный ингредиент может быть приготовлен в креме с основой масло-в-воде или с основой вода-в-масле.
Фармацевтические композиции, адаптированные для местного введения в глаз, включают глазные капли, где активный ингредиент растворен или суспендирован в подходящем носителе, в особенности в водном растворителе. Композиции для введения в глаз будут иметь офтальмологически совместимые pH и осмоляльность. Один или более чем один офтальмологически приемлемый агент для регулирования pH и/или буферизующий агент может быть включен в композицию по изобретению, включая кислоты, такие как уксусная, борная, лимонная, молочная, ортофосфорная и соляная кислоты; основания, такие как гидроксид натрия, фосфат натрия, борат натрия, цитрат натрия, ацетат натрия и лактат натрия; и буферы, такие как цитрат/декстроза, бикарбонат натрия и хлорид аммония. Такие кислоты, основания и буферы могут быть включены в количестве, необходимом для поддержания pH композиции в офтальмологически приемлемом диапазоне. Одна или более чем одна офтальмологически приемлемая соль может быть включена в композицию в количестве, достаточном для приведения осмоляльности композиции в офтальмологически приемлемый диапазон. Такие соли включают соли имеющие катионы натрия, калия или аммония и анионы хлорида, цитрата, аскорбата, бората, фосфата, бикарбоната, сульфата, тиосульфата или бисульфита.
Устройство для доставки в глаз может предназначено для контролируемого высвобождения одного или более чем одного терапевтического агента с различными определенными скоростями высвобождения и замедленными кинетическими показателями для дозы и проницаемостью. Контролируемое высвобождение может быть получено посредством создания полимерных матриц, включающих различные варианты и свойства биоразлагаемых/биоэродируемых полимеров (например, поли(этиленвинил)ацетат (EVA), супергидролизованный PVA (поливиниловый спирт)), гидроксиалкилцеллюлозы (НРС), метилцеллюлозы (МС), гидроксипропилметилцеллюлозы (НРМС), поликапролактона, поли(гликолевой) кислоты, поли(молочной) кислоты, полиангидрида, молекулярные массы полимеров, кристалличность полимеров, соотношения сополимеров, условия обработки, обработку поверхности, геометрическую форму, добавление эксципиента и полимерные покрытия, которые будут усиливать диффузию, эрозию, растворение и осмос лекарственного средства.
Фармацевтические композиции для доставки в глаз также включают способную к образованию геля in situ водную композицию. Такая композиция содержит гелеобразующий агент в концентрации, эффективной для стимулирования гелеобразования при контакте с глазом или со слезной жидкостью. Подходящие гелеобразующие агенты включают термореактивные полимеры, но не ограничиваются ими. Термин "способный к образованию геля in situ" при использовании в данном описании изобретения включает не только жидкости с низкой вязкостью, которые образуют гели при контакте с глазом или со слезной жидкостью, но также включает более вязкие жидкости, такие как полужидкие и тиксотропные гели, которые обладают существенно повышенной вязкостью или сгущенностью геля при введении в глаз. Смотреть, например, Ludwig (2005) Adv. Drug Deliv. Rev. 3; 57:1595-639, включенную в данное описание изобретения посредством ссылки для целей изучения примеров полимеров для применения в доставке лекарственного средства в глаз.
Лекарственные формы для назального или ингаляционного введения легко могут быть приготовлены в виде аэрозолей, растворов, суспензий, гелей или сухих порошков.
Для композиций, подходящих и/или адаптированных для ингаляционного введения, предпочтительно, чтобы соединение формулы (I) или его фармацевтически приемлемая соль находилась в форме с уменьшенным размером частиц, получаемой, например, посредством микронизации. Предпочтительный уменьшенный размер (например, микронизированный) частиц соединения или соли определяется величиной D50 от примерно 0,5 до примерно 10 микрон (например, при измерении с использованием лазерной дифракции).
Аэрозольные композиции, например, для ингаляционного введения, могут содержать раствор или мелкозернистую суспензию активного вещества в фармацевтически приемлемом водном или неводном растворителе. Аэрозольные композиции могут быть представлены в однодозовом или многодозовых количествах в стерильной форме в герметично закрытом контейнере, который может принимать форму картриджа или запасного блока для применения с распыляющим устройством или ингалятором. Альтернативно, герметично закрытый контейнер может представлять собой единичное дозирующее устройство, такое как однодозовый назальный ингалятор или дозатор аэрозоля, оборудованный дозирующим клапаном (дозирующий ингалятор), который предназначен для утилизации, как только содержимое контейнера будет полностью израсходовано.
Когда лекарственная форма содержит дозатор аэрозоля, она предпочтительно содержит подходящий пропеллент под давлением, такой как сжатый воздух, диоксид углерода или органический пропеллент, такой как гидрофторуглерод (HFC). Подходящие HFC-пропелленты включают 1,1,1,2,3,3,3-гептафторпропан и 1,1,1,2-тетрафторэтан. Аэрозольные лекарственные формы также могут принимать форму атомайзера с помпой. Находящийся под давлением аэрозоль может содержать раствор или суспензию активного соединения. Это может потребовать включение дополнительных эксципиентов, например, сорастворителей и/или поверхностно-активных веществ, для улучшения характеристик дисперсии и гомогенности композиций в форме суспензии. Композиции в растворе также могут требовать добавления сорастворителей, таких как EtOH.
Для фармацевтических композиций, подходящих и/или адаптированных для ингаляционного введения, фармацевтическая композиция может представлять собой сухую порошковую ингалируемую композицию. Такая композиция может содержать порошковую основу, такую как лактоза, глюкоза, трегалоза, маннит или крахмал, соединение формулы (I) или его фармацевтически приемлемую соль (предпочтительно в форме с уменьшенным размером частиц, например, в микронизированной форме) и, возможно, модификатор характеристик, такой как L-лейцин или другая аминокислоту и/или металлическую соль стеариновой кислоты, такую как стеарат магния или кальция. Предпочтительно, сухая порошковая ингалируемая композиция содержит сухую порошковую смесь лактозы, например моногидрата лактозы, и соединения формулы (I) или его соли. Такие композиции можно вводить пациенту, используя подходящее устройство, такое как устройство DISKUS®, продаваемое GlaxoSmithKline, которое описано, например, в GB 2242134 А.
Соединения формулы (I) и его фармацевтически приемлемые соли могут быть приготовлены в виде жидкой композиции для доставки из дозатора жидкости, например, дозатора жидкости, имеющего дозирующий наконечник или дозирующее сопло, через которое отмеренную дозу жидкой композиции распыляют при приложении пользователем силы к механизму помпы дозатора жидкости. Такие дозаторы жидкости обычно обеспечиваются резервуаром многократных отмеренных доз жидкой композиции, где дозы распыляются при последовательных срабатываниях помпы. Дозирующему наконечнику или соплу может быть придана форма для введения в ноздри пользователя при распылении спрея жидкой композиции в носовую полость. Дозатор жидкости вышеупомянутого типа описан и иллюстрирован в WO-A-2005/044354.
Терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли будет зависеть от ряда факторов, включая, например, возраст и массу субъекта, определенного состояния, требующего лечения, и его степени тяжести, природы композиции и пути введения и, в конечном счете, от выбора лечащего врача или ветеринара. В фармацевтической композиции каждая дозированная единица для перорального или парентерального введения предпочтительно содержит от 0,01 до 3000 мг, более предпочтительно от 0,5 до 1000 мг соединения формулы (I) или его фармацевтически приемлемой соли, как вычислено для формы свободного основания. Каждая дозированная единица для назального или ингаляционного введения предпочтительно содержит от 0,001 до 50 мг, более предпочтительно от 0,01 до 5 мг соединения формулы (I) или его фармацевтически приемлемой соли, как вычислено для формы свободного основания.
Фармацевтически приемлемые соединения формулы (I) и их фармацевтически приемлемые соли можно вводить в суточной дозе (для взрослого пациента), например, в пероральной или парентеральной дозе от 0,01 мг до 3000 мг в сутки, от 0,5 до 1000 мг в сутки или от 100 мг до 2500 мг в сутки; или в назальной или ингаляционной дозе от 0,001 до 50 мг в сутки или от 0,01 до 5 мг в сутки соединения формулы (I) или его фармацевтически приемлемой соли, как вычислено для формы свободного основания. Это количество может быть дано в одной дозе в сутки или, чаще, в ряде (таком как две, три, четыре, пять или шесть) субдоз в сутки так, чтобы суммарная суточная доза была такой же самой. Эффективное количество его соли может быть определено в виде пропорции от эффективного количества соединения формулы (I) самого по себе.
Соединения формулы (I) и их фармацевтически приемлемые соли можно применять одни или в комбинации с другими терапевтическими агентами. Комбинированные терапии согласно настоящему изобретению, таким образом, включают введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли и применение по меньшей мере одного другого терапевтически активного агента. Предпочтительно, комбинированные терапии согласно настоящему изобретению включают введение по меньшей мере одного соединения формулы (I) или его фармацевтически приемлемой соли и по меньшей мере одного другого терапевтически активного агента. Соединение(я) формулы (I) и его(их) фармацевтически приемлемые соли и другой(ие) терапевтически активный(е) агент(ы) можно вводить вместе в одной фармацевтической композиции или раздельно и, в случае раздельного введения, оно может происходить одновременно или последовательно, в любом порядке. Количества соединения(й) формулы (I) и его(их) фармацевтически приемлемых солей и другого(их) терапевтически активного(ых) агента(ов) и относительная временная синхронизация введения будет выбираться для того, чтобы достичь желательного комбинированного терапевтического эффекта. Таким образом, в еще одном аспекте предложена комбинация, содержащая соединение формулы (I) или его фармацевтически приемлемую соль вместе одним или более чем одним другим терапевтически активным агентом.
Таким образом, в одном аспекте соединение формулы (I) или его фармацевтически приемлемая соль и фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль, согласно изобретению могут применяться в комбинации с одним или более чем одним другим терапевтическим агентом или включать один или более чем один других терапевтический агент, например, выбранный из антибиотиков, противовирусных препаратов, глюкокортикостероидов, мускариновых антагонистов, бета 2-агонистов и аналогов витамина D3. В еще одном воплощении соединение формулы (I) или его фармацевтически приемлемую соль можно применять в комбинации с дополнительным терапевтическим агентом, который является подходящим для лечения рака. Примеры таких дополнительных терапевтических агентов описаны в Cancer Principles and Practice of Oncology by V.T. Devita and S. Hellman (editors), 6th edition (2001), Lippincott Williams & Wilkins Publishers. Специалист в данной области техники сможет распознать комбинации агентов, которые могут быть полезными, на основании конкретных характеристик лекарственных средств и вовлеченного ракового заболевания. Дополнительные терапевтические агенты для использования в комбинации с соединением формулы (I) или его фармацевтически приемлемой солью включают антимикротубулярные агенты (такие как дитерпеноиды и алкалоиды винка); координационные комплексы платины; алкилирующие агенты (такие как азотистый иприт, оксазафосфорины, алкилсульфонаты, нитрозомочевина и триазены); антибиотики (такие как антрациклины, актиномицины и блеомицины); ингибиторы топоизомеразы II (такие как эпиподофиллотоксины); антиметаболиты (такие как аналоги пурина и пиримидина и антифолатные соединения); ингибиторы топоизомеразы I (такие как камптотицины; гормоны и аналоги гормонов); ингибиторы путей сигнальной трансдукции (такие как ингибиторы тирозиновых рецепторов); нерецепторные тирозинкиназные ингибиторы ангиогенеза; иммунотерапевтические агенты; проапоптотические агенты; эпигенетические модуляторы или модуляторы транскрипции (такие как ингибиторы гистондеацетилазы) и ингибиторы сигналинга клеточного цикла, но не ограничиваются ими.
Следует понимать, что, когда соединение формулы (I) или его фармацевтически приемлемую соль вводят в комбинации с другими терапевтическими агентами, вводимыми обычно посредством ингаляционного, внутривенного, перорального или интраназального пути, то полученную фармацевтическую композицию можно вводить посредством таких же путей. Альтернативно, отдельные компоненты композиции можно вводить посредством разных путей.
Одно воплощение изобретения охватывает комбинации, содержащие один или два других терапевтических агента.
Специалисту в данной области техники будет ясно, что, когда это целесообразно, другой терапевтический(е) ингредиент(ы) можно применять в форме солей, например в виде солей щелочного металла или аминных солей, или в виде солей присоединения кислоты или пролекарственных средств, или в виде сложных эфиров, например, эфиров низших алкилов, или в виде сольватов, например, гидратов, для оптимизации активности и/или стабильности, и/или физических характеристик, таких как растворимость, терапевтического ингредиента. Также будет ясно, что, когда это целесообразно, терапевтические ингредиенты можно применять в оптически чистой форме.
Комбинации, которые указаны выше, легко могут быть представлены для использования в форме фармацевтической композиции и, таким образом, фармацевтические композиции, содержащие комбинацию, как определено выше, вместе с фармацевтически приемлемым разбавителем или носителем, представляют собой еще один аспект изобретения.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть получены посредством способов, описанных ниже, или посредством аналогичных способов. Таким образом, следующие промежуточные соединения и примеры служат для иллюстрации получения соединений формулы (I) и их фармацевтически приемлемых солей, и не рассматриваются в качестве ограничения объема изобретения.
Основные подробности экспериментов
Все температуры указаны в °C.
Названия следующих соединений были получены, используя программу именования соединений "ACD Name Pro 6.02" или ChemDraw Ultra 12.0.
Сокращения
Методика LCMS
Способ с муравьиной кислотой
Условия LC
Анализ UPLC проводили на колонке Acquity UPLC ВЕН С18 (50 мм × 2,1 мм, в.д. (внутренний диаметр) 1,7 мкм диаметр набивки) при 40°С.
Используемые растворители:
А=0,1% об./об. раствор муравьиной кислоты в воде
В=0,1% об./об. раствор муравьиной кислоты в ацетонитриле
Используемый градиент:
УФ-детектирование представляло собой суммарный сигнал, полученный при длине волны от 210 нм до 350 нм.
Условия MS

Способ с муравьиной кислотой (прогон 2,5 минуты)
Анализ LCMS проводили на колонке Agilent 1200-6110 LCMS, Halo-C18 (4,6 мм × 50 мм, 2,7 мкм диаметр набивки) при 40°С.
Используемые растворители:
А=0,05% об./об. раствор муравьиной кислоты в воде
В=0,05% об./об. раствор муравьиной кислоты в ацетонитриле
Используемый градиент: суммарное время 2,5 мин
УФ-детектирование представляло собой суммарный сигнал, полученный при длине волны от 214 нм до 254 нм.
Условия MS

Способ с высоким pH
Условия LC
Анализ UPLC проводили на колонке Acquity UPLC ВЕН С18 (50 мм × 2,1 мм, внутр.д. 1,7 мкм диаметр набивки) при 40°С.
Используемые растворители:
А = 10 мМ гидрокарбонат аммония в воде, доводили до pH 10 при помощи раствора аммиака
В = ацетонитрил
Используемый градиент:
УФ-детектирование представляло собой суммарный сигнал, полученный при длине волны от 210 нм до 350 нм.
Условия MS

ЯМР
Спектры получали на ЯМР-спектрометре с частотой 400 или 600 МГц при 302 K или для спектров VT при 392-393 K.
Промежуточное соединение 1: 7-бром-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегид
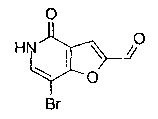
N-Бромсукцинимид (3,93 г, 22,07 ммоль) добавляли порциями к перемешиваемой суспензии 4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (3,0 г, 18,4 ммоль) в безводном тетрагидрофуране (50 мл). Реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Растворитель выпаривали, и остаток растирали с диэтиловым эфиром. Твердое вещество отфильтровали, промывали диэтиловым эфиром и сушили с получением 7-бром-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (3,96 г, 16,36 ммоль, выход 89%) в виде коричневого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,57 мин, МН+ 242/244.
Промежуточное соединение 2: 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегид
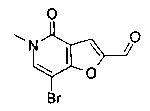
Йодметан (4,63 г, 2,04 мл, 32,6 ммоль) добавляли к перемешиваемой суспензии 7-бром-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть Промежуточное соединение 1, 3,95 г, 16,32 ммоль) и карбоната цезия (15,95 г, 49,0 ммоль) в сухом тетрагидрофуране (100 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Добавляли дополнительную порцию йодметана (4,63 г, 2,04 мл, 32,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение последующих 4 часов. Растворитель выпаривали. Остаток суспендировали в воде (100 мл) и перемешивали в течение 30 минут. Твердое вещество отфильтровали, тщательно промывали при помощи воды и сушили с получением 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (4,00 г, 15,62 ммоль, выход 96%) в виде желтого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,63 мин, МН+ 256/258.
Промежуточное соединение 3: 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
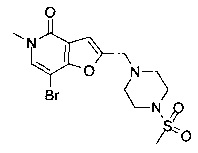
1-(Метилсульфонил)пиперазин (242,09 мг, 1,474 ммоль) добавляли к перемешиваемому раствору 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 251,54 мг, 0,982 ммоль) в МеОН (9 мл) и уксусной кислоте (1 мл). Смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли комплекс 2-пиколин-боран (115,43 мг, 1,079 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель выпаривали. Насыщенный раствор бикарбоната натрия (20 мл) медленно добавляли - выделялся газ. Смесь экстрагировали при помощи DCM (3×20 мл). Объединенные органические вещества сушили, используя сульфат натрия, и растворитель выпаривали. Неочищенное вещество очищали посредством хроматографии на силикагеле, используя градиент 0-6% МеОН в DCM. Соответствующие фракции объединяли и упаривали с получением 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (284 мг, 0,702 ммоль, выход 71,5%) в виде желтого масла. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,50 мин, МН+ 404/406.
Промежуточное соединение 4: (R)-3-метил-1-(метилсульфонил)пиперазин
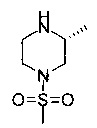
Раствор метансульфонилхлорида (6 г, 52,4 ммоль) в THF (50 мл) добавляли в течение 2 мин в раствор (R)-2-метилпиперазина (5 г, 49,9 ммоль) в NaOH (2 М) (50 мл) и THF (50 мл) при 0°С. Реакционную смесь перемешивали при 0°С в течение 1 часа, затем оставляли нагреваться до комнатной температуры в течение ночи и вливали в 2 М соляную кислоту (100 мл). Смесь промывали этилацетатом (3×50 мл). Водную фазу отделяли и ощелачивали (pH=12) путем добавления твердого гидроксида натрия. Смесь экстрагировали при помощи DCM (2×100 мл). Объединенные экстракты сушили и упаривали с получением (R)-3-метил-1-(метилсульфонил)пиперазина (1,781 г, 9,99 ммоль выход, 20,02%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 3.32 (m, 2Н), 2.9 (m, 1Н), 2.83 (s, 3Н), 2.7-2.54 (m, 3Н), 2.3 (уширенный s, 1Н), 2.2 (t, 1Н), 0.97 (d, 3Н)
Промежуточное соединение 4а: (R)-трет-бутил-2-метил-4-(метилсульфонил)пиперазин-1-карбоксилат
(R)-Трет-бутил-2-метилпиперазин-1-карбоксилат (49 г, 245 ммоль) помещали в THF (150 мл) и 2М NaOH (220 мл, 440 ммоль), и охлаждали в ледяной бане. Реакционную смесь интенсивно перемешивали при медленном добавлении метансульфонилхлорида (20,97 мл, 269 ммоль) в THF (150 мл). Реакционную смесь оставляли перемешиваться в течение ночи. Дополнительно добавляли метансульфонилхлорид (2 мл) и реакционную смесь перемешивали в течение 4 ч. Реакционную смесь вливали в 2 н. HCl (400 мл) и лед (~200 мл) и экстрагировали при помощи EtOAc (2×500 мл). Объединенные органические вещества промывали 2 н. NaOH (400 мл) и рассолом (500 мл), затем сушили с помощью Na2SO4, фильтровали и концентрировали в вакууме с получением (R)-трет-бутил-2-метил-4-(метилсульфонил)пиперазин-1-карбоксилата (62,673 г, 214 ммоль, выход 87%) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 4.27 (m, 1H), 3.84 (m, 1H), 3.48 (m, 1H), 3.31 (m, 1H), 3.03 (m, 1H), 3.86 (s, 3H), 2.84 (m, 1H), 2.67 (m, 1H), 1.41 (s, 9H), 1.13 (d, 3H).
Промежуточное соединение 4b: (R)-3-метил-1-(метилсульфонил)пиперазина гидрохлорид
(R)-Трет-бутил-2-метил-4-(метилсульфонил)пиперазин-1-карбоксилат (относительно получения смотреть промежуточное соединение 4а, 58,506 г, 210 ммоль) помещали в DCM и охлаждали в ледяной бане. Добавляли HCl (400 мл, 1600 ммоль) и реакционную смесь оставляли нагреваться и перемешиваться при комнатной температуре в течение выходных дней. Образовался густой осадок. Реакционную смесь концентрировали в вакууме и остаток азеотропно перегоняли с Et2O с получением (R)-3-метил-1-(метилсульфонил)пиперазина гидрохлорида (49,835 г) в виде белого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 9.18 (br. s, 2Н), 3.63 (m, 2Н), 3.38 (m, 2Н), 3.08 (m, 2Н), 3.00 (s, 3Н), 2.87 (m, 1Н), 1.26 (d, 3Н).
Промежуточное соединение 5: (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-c]пиридин-4(5H)-он
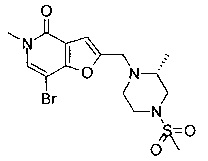
(R)-3-Метил-1-(метилсульфонил)пиперазин (относительно получения смотреть промежуточное соединение 4, 522 мг, 2,93 ммоль) добавляли к суспензии 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 500 мг, 1,953 ммоль) в смеси метанола (18 мл) и уксусной кислоты (2 мл), и перемешивали в течение 15 мин при комнатной температуре. Добавляли комплекс 2-пиколин-боран (1,592 г, 14,88 ммоль) и смесь оставляли перемешиваться при комнатной температуре в течение ночи. Добавляли дополнительные порции смеси комплекса 2-пиколин-боран (104 мг, 0,977 ммоль) и (R)-3-метил-1-(метилсульфонил)пиперазина (70 мг, 0,393 ммоль), и реакционную смесь перемешивали в течение 2 суток. Растворитель выпаривали и добавляли бикарбонат натрия (20 мл). Водную фазу экстрагировали при помощи DCM (3×15 мл), фильтровали через гидрофобную фритту и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 2-4% МеОН в градиенте DCM. Соответствующие фракции концентрировали в вакууме с получением (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (416 мг, 0,994 ммоль, выход 50,9%) в виде белого твердого вещества.
LCMS (2 мин, муравьиная кислота): Rt=0,53 мин, МН+ 418/420
Альтернативное получение
7-Бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегид (относительно получения смотреть промежуточное соединение 2, 45 г, 176 ммоль) и (R)-3-метил-1-(метилсульфонил)пиперазина гидрохлорид (относительно получения смотреть промежуточное соединение 4b, 71,7 г, 334 ммоль) суспендировали в 2-MeTHF (2192 мл) в атмосфере азота. Добавляли Et3N (61,2 мл, 439 ммоль) и смесь перемешивали в течение 10 мин. Триацетоксиборгидрид натрия (74.5 г, 351 ммоль) добавляли частями в течение порядка 10 мин и смесь перемешивали при комнатной температуре. Через 2 ч реакционную смесь концентрировали в вакууме. Остаток осторожно гасили при помощи насыщенного NaHCO3 (1000 мл) и затем экстрагировали при помощи DCM (2×1000 мл). Объединенные органические вещества сушили при помощи Na2SO4, фильтровали и концентрировали в вакууме с получением оранжевого твердого вещества. Его объединяли с порцией неочищенного продукта для получения количества от 5 г для очистки. Попытка поглотить объединенный продукт минимальным количеством DCM не удалась, так как присутствовало немного нерастворенного вещества, поэтому смесь фильтровали. Фильтрат концентрировали до порядка 200 мл DCM, который загружали на картридж SNAP 1500 g и элюировали при помощи 0% 2 М NH3 в метаноле в DCM в количестве 1,6 CV, затем при помощи 0-5% 2 М NH3 в метаноле для 10,6 CV, затем выдерживали в 5% при 3 CV; пороговая величина сбора 15 мА; 400 мл фракции. Соответствующие фракции концентрировали в вакууме с получением твердого вещества кремового цвета. Смешанные фракции объединяли и концентрировали в вакууме, затем повторно пропускали через колонку, используя картридж SNAP 340 g и те же самые условия; фракции по 51 мл. Соответствующие фракции концентрировали в вакууме с получением твердого вещества кремового цвета. Эти две порции объединяли в минимальном количестве DCM и концентрировали в вакууме с получением (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (64,055 г)) в виде твердого вещества кремового цвета. 1Н ЯМР (400 МГц, CDCl3) δ-млн-1 7.37 (1Н, s), 6.88 (1Н, s), 3.92 (2Н, АВ d), 3.63 (3Н, s), 3.53 (2Н, m), 2.97 (2Н, m), 2.78 (3Н, s), 2.64 (3Н, m), 1.26 (3Н, d). LCMS (2 мин, Высокий pH): Rt=0,77 мин, МН+ 418/420.
Промежуточное соединение 5а: (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5H)-он
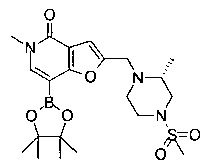
Раствор (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (относительно получения смотреть Промежуточное соединение 5, 30 г, 71,7 ммоль) в 1,4-диоксане (100 мл) обрабатывали триэтиламином (60,1 мл, 430 ммоль), 4,4,5,5-тетраметил-1,3,2-диоксабороланом (62,4 мл, 430 ммоль), затем Pd(PPh3)4 (8,29 г, 7,17 ммоль) и смесь продували азотом, затем нагревали при 100°С в течение 18 ч.
Смесь охлаждали в ледяной бане и гасили при помощи изо-PrOH (30 мл), который добавляли очень осторожно, каплями, из-за сильного вспенивания при добавлении. Полученную суспензию переносили в круглодонную колбу и упаривали. Добавляли изо-PrOH (100 мл), получая прозрачный коричневый раствор, который при перемешивании в течение 30 мин давал густой бежевый осадок. Его собирали посредством фильтрации и промывали изопропанолом (30 мл), затем сушили в вакуумном сушильном шкафу. Продукт суспендировали в изо-PrOH (100 мл) и перемешивали в течение 30 мин, затем фильтровали и твердое вещество промывали при помощи изо-PrOH с получением (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5H)-она (32 г, 68,8 ммоль, выход 96%) в виде серого твердого вещества. 1Н ЯМР (400 МГц, CDCl3) δ-млн-1 7.67 (1Н, s), 6.77 (1Н, s), 3.94 (2Н, АВ d), 3.65 (3Н, s), 3.54 (2Н, m), 2.95 (2Н, m), 2.78 (3Н, s), 2.66 (3Н, m), 1.38 (12Н, s), 1.29 (3Н, d). Использовали на следующей стадии без дополнительной очистки.
Альтернативное получение
Азот барботировали через смесь (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 5, 230 мг, 0,550 ммоль), триэтиламина (0,307 мл, 2,199 ммоль) и PEPPSI-IPr (37,5 мг, 0,055 ммоль) в 1,4-диоксане (3 мл) в течение 2 мин. 4,4,5,5-тетраметил-1,3,2-диоксаборолан (0,479 мл, 3,30 ммоль) добавляли каплями и азот барботировали через раствор, который затем нагревали до 100°С в течение 3 ч в микроволновой печи. Реакционную смесь разбавляли этилацетатом и фильтровали через целит, затем концентрировали в вакууме до получения полутвердого вещества желтого цвета (522 мг, 234%).
LCMS показала присутствие (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5Н)-она (4%), Rt=0,70 мин, МН+ 466; (R)-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бороновой кислоты (36%), Rt=0,38 мин, МН+ 384; и (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (50%), Rt=0,40 мин, МН+ 340. Превращение в целевые промежуточные соединения предполагалось равным 40% (36% бороновой кислоты + 4% боронатного эфира). Теоретический максимальный выход 223 мг, поэтому вещество использовали неочищенным в следующей стадии, предполагающей чистоту 17% масс./масс.
Промежуточное соединение 6: трет-бутил (2-(((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)амино)этил)карбамат
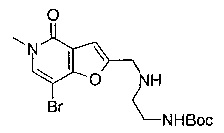
Трет-бутил-(2-аминоэтил)карбамата гидрохлорид (115 мг, 0,586 ммоль) добавляли к раствору 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 100 мг, 0,391 ммоль) в смеси МеОН (7 мл) и уксусной кислоты (0,778 мл). Смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли комплекс 2-пиколин-боран (46,4 мг, 0,434 ммоль) и смесь перемешивали в течение 2 часов. Растворитель выпаривали и добавляли насыщенный раствор бикарбоната натрия (15 мл). Смесь экстрагировали при помощи DCM (3×15 мл) и объединенные органические вещества сушили и упаривали. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 0-5% градиента MeOH/DCM с получением трет-бутил-(2-(((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)амино)этил)карбамата (38 мг, 0,095 ммоль, выход 24,31%). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,59 мин, МН+ 400/402.
Промежуточное соединение 7: трет-бутил (2-(((7-(3,4-диметоксифенил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)амино)этил)карбамат
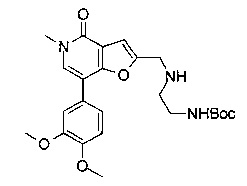
Смесь трет-бутил-(2-(((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)амино)этил)карбамата (относительно получения смотреть промежуточное соединение 6, 34 мг, 0,085 ммоль), (3,4-диметоксифенил)бороновой кислоты (22,85 мг, 0,126 ммоль), карбоната калия (58,7 мг, 0,425 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (5,96 мг, 8,49 мкмоль) в смеси EtOH (2 мл) и толуола (2 мл) нагревали в микроволновой печи при 100°С в течение 1 часа. Смесь разбавляли этилацетатом, фильтровали и концентрировали в вакууме. Остаток очищали посредством MDAP. Фракции, содержащие продукт, объединяли и концентрировали в вакууме с получением трет-бутил-(2-(((7-(3,4-диметоксифенил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)амино)этил)карбамата (25 мг, 0,055 ммоль, выход 64,3%) в виде бесцветной смолы. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,69 мин, МН+ 458.
Промежуточное соединение 8: трет-бутил 4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензилкарбамат
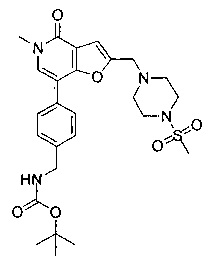
Смесь 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (относительно получения смотреть промежуточное соединение 3, 280 мг, 0,693 ммоль), (4-(((трет-бутоксикарбонил)амино)метил)фенил)бороновой кислоты (257 мг, 1,024 ммоль), карбоната калия (479 мг, 3,46 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (23,33 мг, 0,033 ммоль) в толуоле (2 мл) и EtOH (2 мл) нагревали в микроволновой печи при 80°С в течение 20 минут. Охлажденную реакционную смесь разбавляли этилацетатом (50 мл), сушили при помощи Na2SO4, фильтровали и упаривали в вакууме с получением коричневого масла. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи градиента 0-4% МеОН в DCM. Соответствующие фракции объединяли и упаривали в вакууме с получением трет-бутил-4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензилкарбамата (160,54 мг, 0,303 ммоль, выход 43,7%) в виде коричневого масла.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,76 мин, МН+ 531.
Дополнительные фракции, содержащие продукт, объединяли и упаривали. Растирание с диэтиловым эфиром и DCM давало трет-бутил 4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензилкарбамат (94 мг, 26%) в виде желто-коричневой пены.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,77 мин, МН+ 531.
Промежуточное соединение 9: 7-бром-5-метил-2-(пиперидин-1-илметил)фуро[3,2-с]пиридин-4(5H)-он
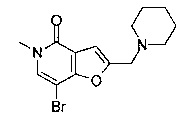
К перемешиваемому раствору 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 200 мг, 0,781 ммоль) в МеОН (18 мл) и уксусной кислоте (2 мл), добавляли пиперидин (200 мг, 2,343 ммоль) и реакционную смесь оставляли перемешиваться в течение 1 часа при комнатной температуре. Добавляли комплекс 2-пиколин-боран (251 мг, 2,343 ммоль) и реакционную смесь оставляли перемешиваться в течение 48 часов. Растворитель выпаривали при пониженном давлении. Добавляли насыщенный раствор бикарбоната натрия (15 мл) и смесь экстрагировали при помощи DCM (3×15 мл). Объединенные органические вещества сушили и упаривали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (25 г), элюируя при помощи градиента 0-6% МеОН в DCM. Соответствующие фракции концентрировали в вакууме с получением 7-бром-5-метил-2-(пиперидин-1-илметил)фуро[3,2-с]пиридин-4(5Н)-она (40 мг, 15,8%) в виде не совсем белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,47 мин, МН+ 325/327.
Промежуточное соединение 10: 7-бром-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5Н)-он
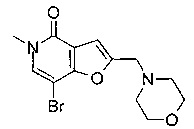
Смесь 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 150 мг, 0,59 ммоль), морфолина (77 мг, 76 мкл, 0,88 ммоль) и уксусной кислоты (0,5 мл) в МеОН (9,5 мл) перемешивали в течение 15 минут. Добавляли комплекс 2-пиколин-боран (188 мг, 1,76 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали. Насыщенный раствор бикарбоната натрия (20 мл) добавляли к остатку. Смесь экстрагировали при помощи DCM (2×20 мл). Объединенные органические вещества сушили и упаривали. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи градиента 0-5% MeOH/DCM. Растирание с диэтиловым эфиром давало 7-бром-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5H)-он (90 мг, 0,275 ммоль, выход 47,0%) в виде бледно-желтого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,4 мин, МН+ 327/329.
Промежуточное соединение 11: трет-бутил 4-((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилат
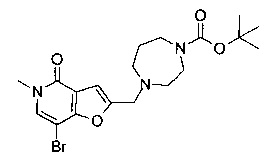
7-Бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегид (относительно получения смотреть промежуточное соединение 2, 100 мг, 0,391 ммоль) и трет-бутил 1,4-диазепан-1-карбоксилат (0,114 мл, 0,586 ммоль) растворяли в уксусной кислоте (1 мл) и МеОН (9 мл). Добавляли комплекс 2-пиколин-боран (45,9 мг, 0,430 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь упаривали в вакууме. Добавляли насыщенный раствор бикарбоната натрия (15 мл). Смесь экстрагировали при помощи DCM (3×15 мл). Органические слои объединяли, сушили над сульфатом магния и упаривали. Остаток очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали с получением желтого твердого вещества. Неочищенное твердое вещество растворяли в МеОН и вносили на колонку SCX (5 g), и элюировали при помощи МеОН и затем NH3/MeOH. Фракции NH3/MeOH упаривали с получением трет-бутил-4-((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилата (52 мг, 0,118 ммоль, выход 30,2%) в виде желтого масла.
LCMS (2 мин, способ с высоким pH): Rt=1,05 мин, МН+ 440/442.
Промежуточное соединение 12: трет-бутил 4-((7-(3,4-диметоксифенил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилат
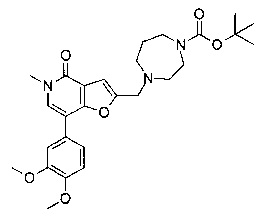
Трет-бутил-4-((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилат (относительно получения смотреть промежуточное соединение 11, 52 мг, 0,118 ммоль), (3,4-диметоксифенил)бороновую кислоту (32,2 мг, 0,177 ммоль), карбонат калия (82 мг, 0,590 ммоль) и бис(трифенилфосфин)палладия(II) хлорид (4,14 мг, 5,90 мкмоль) растворяли в EtOH (2 мл) и толуоле (2 мл), и нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли этилацетат (20 мл) и смесь сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Остаток очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали с получением трет-бутил 4-((7-(3,4-диметоксифенил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилата (50 мг, 0,100 ммоль, выход 85%) в виде желтого масла. LCMS (2 мин, способ с высоким pH): Rt=1,05 мин, МН+ 498.
Промежуточное соединение 13: 7-бром-2-((3,3-дифторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-он
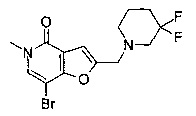
7-Бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегид (относительно получения смотреть промежуточное соединение 2, 200 мг, 0,781 ммоль) и 3,3-дифторпиперидин (185 мг, 1,172 ммоль) растворяли в МеОН (20 мл) и уксусной кислоте (2 мл). Добавляли комплекс 2-пиколин-боран (92 мг, 0,859 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь упаривали в вакууме. Добавляли насыщенный раствор бикарбоната натрия (35 мл) и смесь экстрагировали при помощи DCM (3×35 мл). Органические слои объединяли, сушили над сульфатом магния и упаривали. Неочищенный продукт очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали с получением 7-бром-2-((3,3-дифторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она (150 мг, 53%) в виде белого порошка. LCMS (2 мин, способ с высоким pH): Rt=0,94 мин, МН+ 361/363.
Промежуточное соединение 14: 7-бром-5-метил-2-((3-метилморфолино)метил)фуро[3,2-с]пиридин-4(5H)-он
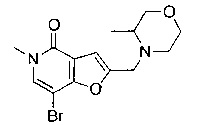
3-Метилморфолин (79 мг, 89 мкл, 0,78 ммоль) добавляли к перемешиваемому раствору 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 100 мг, 0,39 ммоль) в смеси МеОН (5 мл) и уксусной кислоты (0,5 мл). Смесь перемешивали при комнатной температуре в течение 15 минут. Добавляли комплекс 2-пиколин-боран (125 мг, 1,17 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Растворитель выпаривали. Насыщенный раствор бикарбоната натрия (10 мл) добавляли к остатку. Смесь экстрагировали при помощи DCM (3×10 мл). Объединенные экстракты сушили и упаривали. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 0-2% MeOH/DCM. Продукт растворяли в МеОН и загружали на колонку SCX. Его элюировали при помощи МеОН с последующим элюированием посредством 2 М аммиака в МеОН. Фракцию аммиака в МеОН упаривали с получением 7-бром-5-метил-2-((3-метилморфолино)метил)фуро[3,2-с]пиридин-4(5H)-она (56 мг, 0,164 ммоль, выход 42,0%) в виде желтого масла.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,43 мин, МН+ 341/343.
Промежуточное соединение 15: 7-бром-2-((3-фторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5H)-он
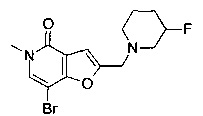
7-Бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегид (относительно получения смотреть промежуточное соединение 2, 200 мг, 0,781 ммоль) и 3-фторпиперидин (169 мг, 1,172 ммоль) растворяли в МеОН (20 мл) и уксусной кислоте (2 мл). Добавляли комплекс 2-пиколин-боран (92 мг, 0,859 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 48 ч. Реакционную смесь упаривали в вакууме. Добавляли насыщенный раствор бикарбоната натрия (35 мл) и смесь экстрагировали при помощи DCM (3×35 мл). Органические слои объединяли, сушили над сульфатом магния и упаривали. Неочищенный продукт очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали с получением 7-бром-2-((3-фторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она (165 мг, 62%).
LCMS (2 мин, способ с высоким pH): Rt=0,89 мин, МН+ 343/345.
Промежуточное соединение 16: 2-((1,4-оксазепан-4-ил)метил)-7-бром-5-метилфуро[3,2-с]пиридин-4(5H)-он
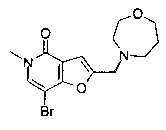
Смесь 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 150 мг, 0,59 ммоль), гомоморфолин гидрохлорида (121 мг, 0,88 ммоль) и уксусной кислоты (0,5 мл) в МеОН (9,5 мл) перемешивали в течение 15 минут. Добавляли комплекс 2-пиколин-боран (188 мг, 1,76 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворитель выпаривали. Насыщенный раствор бикарбоната натрия (20 мл) добавляли к остатку. Смесь экстрагировали при помощи DCM (2×20 мл). Объединенные органические вещества сушили и упаривали. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи градиента 0-5% MeOH/DCM. Полученный остаток растирали с диэтиловым эфиром с получением 2-((1,4-оксазепан-4-ил)метил)-7-бром-5-метилфуро[3,2-с]пиридин-4(5Н)-она (40 мг, 20%) в виде бледно-желтого твердого вещества. Вещество использовали неочищенным в следующей стадии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,43 мин, МН+ 341/343. Продукт содержит порядка 30% 7-бром-2-(гидроксиметил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она (Rt=0,56 мин).
Промежуточное соединение 17: 4-метокси-1-метилпиридин-2(1Н)-он
Раствор 4-метоксипиридин-2(1Н)-она (2 г, 15,98 ммоль) в DMF (40 мл) обрабатывали карбонатом калия (4,42 г, 32,0 ммоль) с последующей обработкой йодметаном (1,499 мл, 23,98 ммоль), и реакционную смесь нагревали при 60°С в течение 16 ч. Реакционную смесь концентрировали при пониженном давлении досуха. Полученное твердое вещество очищали посредством хроматографии на силикагеле (100 г), используя градиент 0-33% (20% 2 М аммиака в MeOH)/DCM. Соответствующие фракции объединяли и концентрировали при пониженном давлении с получением 4-метокси-1-метилпиридин-2(1Н)-она (1,8 г, 81%) в виде желтого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,43 мин, МН+ 140.
Промежуточное соединение 18: 3,5-дийод-4-метокси-1-метилпиридин-2(1Н)-он
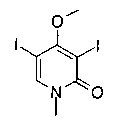
Раствор 4-метокси-1-метилпиридин-2(1H)-она (относительно получения смотреть промежуточное соединение 17, 1,8 г, 12,94 ммоль) в ацетонитриле (50 мл) обрабатывали 1-йодпирролидин-2,5-дионом (2,91 г, 12,94 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 2 ч. Дополнительный 1-йодпирролидин-2,5-дион (4,37 г) добавляли к реакционной смеси с последующим добавлением TFA (0,997 мл, 12,94 ммоль), и реакционную смесь оставляли перемешиваться в атмосфере азота в течение 16 ч. Смолу очищали посредством хроматографии на силикагеле, элюируя при помощи 0-80% циклогексан/EtOAc. Соответствующие фракции объединяли и концентрировали при пониженном давлении с получением 3,5-дийод-4-метокси-1-метилпиридин-2(1Н)-она (5,1 г, выход >99%) в виде желтого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,79 мин, МН+ 392.
Промежуточное соединение 19: 5-йод-4-метокси-1-метил-2'-((тетрагидро-2Н-пиран-4-ил)метокси)-[3,4'-бипиридин]-6(1H)-он
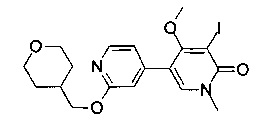
Раствор TPPTS (20 мг, 0,035 ммоль), 3,5-дийод-4-метокси-1-метилпиридин-2(1Н)-она (относительно получения смотреть промежуточное соединение 18, 180 мг, 0,460 ммоль), 2-((тетрагидро-2H-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (BoroPharm Inc) (191 мг, 0,599 ммоль) и диацетоксипалладия (3 мг, 0,013 ммоль) в ацетонитриле (4 мл) и воде (1,333 мл) обрабатывали при помощи DIPEA (0,105 мл, 0,599 ммоль). Реакционную смесь нагревали при 60°С в атмосфере азота в течение 16 ч. Добавляли дополнительный диацетоксипалладий (3 мг, 0,013 ммоль), с последующим добавлением дополнительного 2-((тетрагидро-2Н-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (55 мг) и реакционную смесь перемешивали в течение 4 ч при 60°С в атмосфере азота.
Реакционную смесь концентрировали при пониженном давлении, и полученное неочищенное вещество распределяли между DCM (20 мл) и водой (20 мл). Водный слой экстрагировали при помощи DCM (2×20 мл). Объединенные органические слои пропускали через гидрофобную фритту и концентрировали при пониженном давлении. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя при помощи градиент 0-100% EtOAc/циклогексан. Соответствующие фракции объединяли и концентрировали при пониженном давлении с получением 5-йод-4-метокси-1-метил-2'-((тетрагидро-2Н-пиран-4-ил)метокси)-[3,4'-бипиридин]-6(1Н)-она (90 мг, 43%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,89 мин, МН+ 457.
Промежуточное соединение 20: 1-(бут-3-ин-2-ил)-4-(метилсульфонил)пиперазин
Смесь 3-хлорбут-1-ина (1 г, 11,29 ммоль), 1-(метилсульфонил)пиперазина (3,71 г, 22,59 ммоль), меди (0,014 г, 0,226 ммоль), и хлорида меди(1) (0,022 г, 0,226 ммоль) в диэтиловом эфире (15 мл) и воде (5 мл) перемешивали при комнатной температуре в атмосфере азота в течение 16 ч. Добавляли воду (40 мл) и диэтиловый эфир (40 мл). Органический слой экстрагировали, и водный слой повторно экстрагировали при помощи диэтилового эфира (2×40 мл). Объединенные органические слои пропускали через гидрофобную фритту и концентрировали при пониженном давлении. Неочищенное вещество очищали посредством хроматографии на силикагеле, элюируя при помощи градиента 80-100% EtOAc/циклогексан. Фракции, содержащие продукт (как обнаружено путем внесения KMnO4), объединяли и концентрировали при пониженном давлении с получением 1-(бут-3-ин-2-ил)-4-(метилсульфонил)пиперазина (1,6 г, 66%) в виде прозрачный желтой смолы, которая затвердевала до воскообразного твердого вещества при стоянии.
1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 3.58 (1Н, m), 3.21 (1Н, d), 3.15-3.09 (4Н, m), 2.86 (3Н, s), 2.64-2.60 (2Н, m), 2.51-2.44 (2Н, m), 1.25 (3Н, d).
Промежуточное соединение 21: 1-(бензилокси)-3-бромбензол
Смесь 3-бромфенола (2,60 г, 1,59 мл, 15 ммоль), бромистого бензила (2,82 г 1,96 мл, 16,5 ммоль) и карбоната калия (2,07 г, 15 ммоль) в ацетоне (25 мл) нагревали с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали до комнатной температуры. Смесь фильтровали и растворитель выпаривали из фильтрата. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 0-25% этилацетат/гексан. Соответствующие фракции объединяли и упаривали. Загрязненный продукт повторно очищали посредством хроматографии на силикагеле, элюируя при помощи 0-10% этилацетат/гексан с получением 1-(бензилокси)-3-бромбензола (2,25 г, 8,55 ммоль, выход 57,0%) в виде белого твердого вещества.
1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 7.46-7.32 (m, 5Н), 7.27-7.22 (m, 2Н), 7.13 (m, 1Н), 7.03 (m, 1Н), 5.13 (s, 2Н).
Промежуточное соединение 22: 2-(3-(бензилокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан
Раствор 1-(бензилокси)-3-бромбензола (относительно получения смотреть промежуточное соединение 21, 263 мг, 1,0 ммоль) в 1,4-диоксане (8 мл) добавляли к смеси бис(пинаколато)дибора (1,27 г, 5,0 ммоль), ацетата калия (392 мг, 4,0 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II), (PdCl2(dppf)) (37 мг, 5% мол.). Реакционную смесь нагревали в микроволновой печи при 110°С в течение 60 минут. Охлажденную реакционную смесь разбавляли этилацетатом (20 мл) и фильтровали через целит. Фильтрат сушили и упаривали с получением 2-(3-(бензилокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолана (310 мг, 1,000 ммоль, выход 100%). Количественный выход предполагался. Вещество использовали неочищенным в следующей стадии без дополнительной очистки. LCMS (2 мин, способ с муравьиной кислотой): Rt=1,42 мин, МН+ 311.
Промежуточное соединение 23: N-бензила-3-броманилин
3-Броманилин (3,10 г, 18,0 ммоль) добавляли к перемешиваемому раствору бензальдегида (1,59 г, 15,0 ммоль) в МеОН (18 мл) и ледяной уксусной кислоте (2 мл). Смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли комплекс 2-пиколин-боран (1,765 г, 16,5 ммоль) порциями в течение 5 минут (экзотермическая реакция). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, затем оставляли при комнатной температуре в течение выходных. Растворитель выпаривали. Добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали при помощи DCM (3×50 мл). Объединенные экстракты сушили и упаривали. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 10-30% этилацетат/гексан с получением N-бензила-3-броманилина (3,52 г, 13,43 ммоль, выход 90%) в виде бесцветного масла. LCMS (2 мин, способ с муравьиной кислотой): Rt=1,27 мин, МН+ 262/264.
Промежуточное соединение 24: N-бензила-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин
Раствор N-бензила-3-броманилина (относительно получения смотреть промежуточное соединение 23, 262 мг, 1,0 ммоль) в 1,4-диоксане (8 мл) добавляли к смеси бис(пинаколато)дибора (1,27 г, 5,0 ммоль), ацетата калия (392 мг, 4,0 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II) (37 мг, 5% мол). Реакционную смесь нагревали в микроволновой печи при 110°С в течение 30 минут. Охлажденную реакционную смесь разбавляли этилацетатом (20 мл) и фильтровали через целит. Фильтрат сушили и упаривали с получением N-бензила-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (309 мг, 0,999 ммоль, выход 100%). Количественный выход предполагался. Вещество использовали неочищенным в следующей стадии без дополнительной очистки. LCMS (2 мин, способ с муравьиной кислотой): Rt=1,31 мин, МН+ 310.
Промежуточное соединение 25: 4-бром-N-(1-фенилэтил)пиридин-2-амин
1-Фенилэтанамин (7,38 мл, 57,3 ммоль) и 4-бром-2-хлорпиридин (3,177 мл, 28,6 ммоль) растворяли в EtOH (10 мл). Добавляли N-этил-N-изопропилпропан-2-амин (7,25 мл, 42,9 ммоль) и реакционную смесь нагревали до температуры в течение 24 ч. LCMS показала отсутствие взаимодействия. Растворитель удаляли в вакууме и смесь переносили во флакон с NMP (5 мл) для микроволновой печи. Реакционную смесь нагревали в микроволновой печи при 180°С в течение 1 ч. Реакционную смесь очищали посредством хроматографии на силикагеле (120 г), элюируя при помощи 10-50% EtOAc в циклогексане. Соответствующие фракции объединяли и упаривали с получением 4-бром-N-(1-фенилэтил)пиридин-2-амина (787 мг, 2,84 ммоль, выход 9,92%). LCMS (2 мин, высокий pH): Rt=1,21 мин, МН+ 277/279.
Промежуточное соединение 26: N-(1-фенилэтил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин
4-Бром-N-(1-фенилэтил)пиридин-2-амин (относительно получения смотреть Промежуточное соединение 25, 250 мг, 0,902 ммоль), бис(пинаколато)дибор (1145 мг, 4,51 ммоль), ацетат калия (354 мг, 3,61 ммоль) и PdCl2(dppf) (66,0 мг, 0,090 ммоль) растворяли в 1,4-диоксане (8 мл) и нагревали в микроволновой печи при 110°С в течение 1 ч. Добавляли дополнительные порции 6ис(линаколато)дибора (1145 мг, 4,51 ммоль) и PdCl2(dppf) (66,0 мг, 0,090 ммоль), реакционную смесь нагревали в микроволновой печи при 110°С в течение 2 ч. Охлажденную реакционную смесь разбавляли этилацетатом (20 мл) и фильтровали через целит. Фильтрат сушили и упаривали с получением N-(1-фенилэтил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (292 мг, 0,902 ммоль, выход 100%). Количественный выход предполагался. Вещество использовали неочищенным в следующей стадия без дополнительной очистки.
LCMS (2 мин, способ с высоким pH): Rt=0,59 мин, МН+ 243 (наблюдаемая ионная масса соответствует бороновой кислоте).
Промежуточное соединение 27: 3-бром-5-(1-фенилэтокси)пиридин
Карбонат калия (1,59 г, 11,5 ммоль) добавляли к перемешиваемому раствору 5-бромпиридин-3-ола (1,0 г, 5,75 ммоль) в DMF (10 мл). Реакционную смесь перемешивали при 60°С в течение 30 минут, затем добавляли (1-бромэтил)бромистый бензил (1,12 г, 824 мкл, 6,05 ммоль). Реакционную смесь перемешивали при 60°С в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом (50 мл) и водой (25 мл). Органическую фазу отделяли, промывали водой и рассолом, сушили и упаривали. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи DCM с получением 3-бром-5-(1-фенилэтокси)пиридина (890 мг, 3,20 ммоль, выход 55,7%) в виде желтого масла.
LCMS (2 мин, муравьиная кислота): Rt=1,18 мин, МН+ 278/280.
Промежуточное соединение 28: 3-(1-фенилэтокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
Раствор 3-бром-5-(1-фенилэтокси)пиридина (относительно получения смотреть промежуточное соединение 27, 278 мг, 1,0 ммоль) в 1,4-диоксане (8 мл) добавляли к смеси бис(пинаколато)дибора (1,27 г, 5,0 ммоль), ацетата калия (392 мг, 4,0 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II), (PdCl2(dppf)) (37 мг, 5% мол). Реакционную смесь нагревали в микроволновой печи при 110°С в течение 30 минут. Охлажденную реакционную смесь разбавляли этилацетатом (20 мл) и фильтровали через целит. Фильтрат сушили и упаривали с получением 3-(1-фенилэтокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (325 мг, 0,999 ммоль, выход 100%). Количественный выход предполагался. Вещество использовали неочищенным в следующей стадия без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,62 мин, МН+ 244 (наблюдаемая ионная масса соответствует бороновой кислоте бороновой кислоте).
Промежуточное соединение 29: 3-бром-N-(1-фенилэтил)анилин
3-Броманилин (3,10 г, 18,0 ммоль) добавляли к перемешиваемому раствору ацетофенона (1,80 г, 15,0 ммоль) в МеОН (18 мл) и ледяной уксусной кислоте (2 мл). Смесь перемешивали при комнатной температуре в течение 15 минут, затем добавляли порциями комплекс 2-пиколин-боран (1,765 г, 16,5 ммоль) в течение 5 минут. Реакционную смесь перемешивали при комнатной температуре в течение выходных. Растворитель выпаривали. Добавляли насыщенный раствор бикарбоната натрия (30 мл). Смесь экстрагировали при помощи DCM (3×50 мл). Объединенные экстракты сушили и упаривали. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 10-30% этилацетат/гексан с получением 3-бром-N-(1-фенилэтил)анилина (2,93 г, 10,61 ммоль, выход 70,7%, присутствует порядка 30% примесей согласно ЯМР) в виде бледно-желтого масла.
1Н ЯМР (400 МГц, CDCl3) δ-млн-1 7.34-7.28 (m, 4Н), 7.22 (m, 1Н), 6.9 (t, 1Н), 6.73 (m, 1Н), 6.66 (t, 1Н), 6.38 (m, 1Н), 4.44 (q, 1Н), 1.49 (d, 3Н).
Промежуточное соединение 30: N-(1-фенилэтил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин
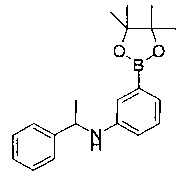
Раствор 3-бром-N-(1-фенилэтил)анилина (относительно получения смотреть промежуточное соединение 29, 276 мг, 1,0 ммоль) в 1,4-диоксане (8 мл) добавляли к смеси бис(пинаколато)дибора (1,27 г, 5,0 ммоль), ацетата калия (392 мг, 4,0 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия(II), (PdCl2(dppf)) (37 мг, 5% мол). Реакционную смесь нагревали в микроволновой печи при 110°С в течение 30 минут. Охлажденную реакционную смесь разбавляли этилацетатом (20 мл) и фильтровали через целит. Фильтрат сушили и упаривали с получением N-(1-фенилэтил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (323 мг, 0,999 ммоль, выход 100%). Количественный выход предполагался. Вещество использовали неочищенным в следующей стадии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=1,34 мин, МН+ 324 и Rt=0,76 мин, МН+ 242 (наблюдаемая ионная масса соответствует бороновой кислоте).
Промежуточное соединение 31: 4-бром-2-(циклопропилметокси)пиридин
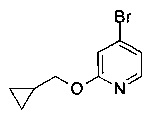
К перемешиваемой суспензии циклопропилметанола (0,493 мл, 6,24 ммоль) в THF (10 мл) в атмосфере азота добавляли порциями гидрид натрия (249 мг, 6,24 ммоль). Через примерно 30 мин добавляли 4-бром-2-хлорпиридин (0,346 мл, 3,12 ммоль). Реакционную смесь перемешивали в течение 3 суток. Смесь разбавляли диэтиловым эфиром (30 мл) и водой (30 мл). Водную фазу повторно экстрагировали диэтиловым эфиром (2×30 мл). Объединенные органические слои промывали рассолом (30 мл) и концентрировали в вакууме с получением желтого масла (685 мг, 96%, чистота посредством LCMS порядка 59%). Вещество использовали неочищенным в следующей стадии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=1,21 мин, МН+ 228/230.
Промежуточное соединение 32: 2-(циклопропилметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
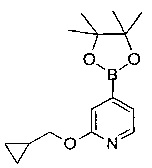
К перемешиваемой суспензии 4-бром-2-(циклопропилметокси)пиридина (относительно получения смотреть промежуточное соединение 31, 679 мг, 2,98 ммоль) в 1,4-диоксане (8 мл) добавляли 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (1512 мг, 5,95 ммоль), ацетат калия (876 мг, 8,93 ммоль) и PdCl2(dppf) (218 мг, 0,298 ммоль). Это закрывали герметично во флаконе для микроволновой печи и нагревали до 100°С в микроволновой печи в течение 60 минут. Смесь растворяли в этилацетате и элюировали через картридж с целитом при помощи этилацетата. Растворитель выпаривали при пониженном давлении с получением 2-(циклопропилметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (2,18 г, 333%) в виде коричневой смолы. Этот продукт брался для следующей стадии синтеза в виде неочищенного вещества, предполагалось 100% превращение, следовательно, максимальная чистота неочищенного вещества составляет 30%.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,61 мин, МН+ 194 (наблюдаемая ионная масса соответствует бороновой кислоте).
Промежуточное соединение 33: N-(4-бромпиридин-2-ил)циклопропанкарбоксамид
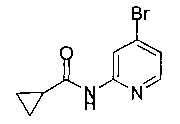
Пиридин (0,467 мл, 5,78 ммоль) добавляли к раствору 4-бромпиридин-2-амина (500 мг, 2.89 ммоль) в DCM (10 мл) и перемешивали при КТ в течение 20 мин. Добавляли циклопропанкарбонил хлорид (0,302 мл, 3,32 ммоль) и раствор перемешивали в течение 4 часов. Добавляли дополнительные порции циклопропанкарбонил хлорида (0,302 мл, 3,32 ммоль) и пиридина (0,234 мл, 2,89 ммоль) и реакционную смесь оставляли перемешиваться в течение 5 часов. Добавляли воду и продукт экстрагировали при помощи DCM. Органический слой промывали рассолом, фильтровали через гидрофобную фритту и концентрировали в вакууме. Остаток растворяли в МеОН и пропускали через аминопропиловую колонку (10 g), элюируя при помощи МеОН. Растворитель выпаривали с получением N-(4-бромпиридин-2-ил)циклопропанкарбоксамида (610 мг, 2,53 ммоль, выход 88%) в виде белого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,84 мин, МН+ 241/243.
Промежуточное соединение 34: N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)циклопропанкарбоксамид
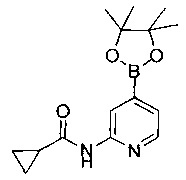
Смесь N-(4-бромпиридин-2-ил)циклопропанкарбоксамида (относительно получения смотреть промежуточное соединение 33, 504 мг, 2,091 ммоль), бис(пинаколато)дибора (1062 мг, 4,18 ммоль), PdCl2(dppf) (153 мг, 0,209 ммоль) и ацетата калия (616 мг, 6,27 ммоль) в 1,4-диоксане (5 мл) нагревали в микроволновой печи при 100°С в течение 30 мин. Реакционную смесь разбавляли этилацетатом и фильтровали через колонку с целитом. Фильтрат упаривали с получением N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)циклопропанкарбоксамида в виде коричневого остатка. Выход предполагали равным 100% (602 мг, 2,089 ммоль). Вещество использовали неочищенным в следующее реакции без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,37 мин, МН+ 207 (наблюдаемая ионная масса согласуется с гидролизом до бороновой кислоты в условиях LCMS).
Промежуточное соединение 35: N-(4-бромпиридин-2-ил)пропионамид
Пиридин (0,374 мл, 4,62 ммоль) добавляли к раствору 4-бромпиридин-2-амина (400 мг, 2,312 ммоль) в DCM (10 мл) и перемешивали при КТ в течение 20 мин. Добавляли пропионилхлорид (0,232 мл, 2,66 ммоль) и раствор перемешивали при КТ в течение ночи. Добавляли воду и продукт экстрагировали при помощи DCM. Органический слой промывали рассолом, сушили посредством гидрофобной фритты и концентрировали в вакууме. Остаток растворяли в МеОН и пропускали через аминопропиловую колонку (10 g), элюируя при помощи МеОН. Растворитель выпаривали с получением N-(4-бромпиридин-2-ил)пропионамид (464 мг, 2,026 ммоль, выход 88%) в виде белого твердого вещества.
LCMS (2 мин, способ с высоким pH): Rt=0,84 мин, МН+ 229/231.
Промежуточное соединение 36: N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пропионамид
Смесь N-(4-бромпиридин-2-ил)пропионамида (относительно получения смотреть промежуточное соединение 35, 200 мг, 0,873 ммоль), бис(пинаколато)дибора (665 мг, 2,62 ммоль), PdCl2(dppf) (63,9 мг, 0,087 ммоль) и ацетата калия (257 мг, 2,62 ммоль) в 1,4-диоксане (5 мл) нагревали в микроволновой печи при 100°С в течение 30 мин. Реакционную смесь разбавляли этилацетатом и фильтровали через колонку с целитом. Фильтрат упаривали с получением N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пропионамида в виде коричневого остатка. Выход предполагали равным 100% (241 мг, 0,873 ммоль). Вещество использовали неочищенным в следующей стадии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,34 мин, МН+ 195 (наблюдаемая ионная масса согласуется с гидролизом до бороновой кислоты в условиях LCMS).
Промежуточное соединение 37: 4-бром-2-(2-метоксиэтокси)пиридин
2-Метоксиэтанол (0,820 мл, 10,39 ммоль) растворяли в THF (30 мл) и в атмосфере азота добавляли гидрид натрия (60% масс./масс.) (0,416 г, 10,39 ммоль) и оставляли перемешиваться при КТ в течение 15 мин. Добавляли 4-бром-2-хлорпиридин (0,577 мл, 5,20 ммоль) и реакционную смесь оставляли перемешиваться при КТ в течение ночи. Дополнительный 2-метоксиэтанол (0,410 мл, 5,19 ммоль) растворяли в 1,2-DME (10 мл) и добавляли гидрид натрия (60% масс./масс.) (0,208 мг, 5,19 ммоль). После 15 мин при КТ в атмосфере азота эту смесь добавляли к реакционной смеси и оставляли перемешиваться при КТ в течение 3 суток. Добавляли воду (40 мл) и органический продукт экстрагировали при помощи этилацетата. Органический слой промывали рассолом, сушили посредством гидрофобной фритты и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 0-20% этилацетата в циклогексане. Фракции, содержащие продукт, концентрировали в вакууме с получением 4-бром-2-(2-метоксиэтокси)пиридина (314 мг, 1,353 ммоль, выход 26,0%) в виде бесцветной жидкости. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,93 мин, МН+ 232/234.
Промежуточное соединение 38: 2-(2-метоксиэтокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
Смесь 4-бром-2-(2-метоксиэтокси)пиридина (относительно получения смотреть промежуточное соединение 37, 306 мг, 1,319 ммоль), бис(пинаколато)дибора (1004 мг, 3,96 ммоль), PdCl2(dppf) (96 мг, 0,132 ммоль) и ацетата калия (388 мг, 3,96 ммоль) в 1,4-диоксане (10 мл) нагревали в микроволновой печи при 100°С в течение 30 мин. Реакционную смесь разбавляли этилацетатом и фильтровали через колонку с целитом. Фильтрат упаривали с получением 2-(2-метоксиэтокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина в виде коричневого остатка. Выход предполагали равным 100% (368 мг, 1,319 ммоль). Вещество использовали неочищенным в следующей стадии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,44 мин, МН+ 198 (наблюдаемая ионная масса согласуется с гидролизом до бороновой кислоты в условиях LCMS).
Промежуточное соединение 39: 4-бром-2-(2-(пирролидин-1-ил)этокси)пиридин
2-(Пирролидин-1-ил)этанол (1,240 мл, 10,39 ммоль) растворяли в THF (30 мл) и в атмосфере азота добавляли частями гидрид натрия (60% масс./масс.) (0,416 г, 10,39 ммоль). Эту смесь оставляли перемешиваться в течение 15 минут при КТ перед добавлением 4-бром-2-хлорпиридина (0,577 мл, 5,20 ммоль). Реакционную смесь перемешивали при КТ в течение 3 суток. Добавляли воду (40 мл) и органический продукт извлекали при помощи этилацетата. Органический слой промывали рассолом, сушили посредством гидрофобной фритты и концентрировали в вакууме. Остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 0-5% MeOH/NH3 в DCM. Фракции, содержащие продукт, объединяли и концентрировали в вакууме с получением 4-бром-2-(2-(пирролидин-1-ил)этокси)пиридина (314 мг, 22%). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,54 мин, МН+ 271/273.
Промежуточное соединение 40: 2-(2-(пирролидин-1-ил)этокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
Смесь 4-бром-2-(2-(пирролидин-1-ил)этокси)пиридина (относительно получения смотреть промежуточное соединение 39, 400 мг, 1,475 ммоль), бис(пинаколато)дибора (1124 мг, 4,43 ммоль), PdCl2(dppf) (108 мг, 0,148 ммоль) и ацетата калия (434 мг, 4,43 ммоль) в 1,4-диоксане (10 мл) нагревали в микроволновой печи при 100°С в течение 30 мин. Реакционную смесь разбавляли этилацетатом и фильтровали через колонку с целитом. Фильтрат упаривали с получением 2-(2-(пирролидин-1-ил)этокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина в виде коричневого остатка. Выход предполагали равным 100% (469 мг, 1,474 ммоль). Вещество использовали неочищенным в следующей стадии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,33 мин, МН+ 237 (наблюдаемая ионная масса согласуется с гидролизом до бороновой кислоты в условиях LCMS).
Промежуточное соединение 41: 7-бром-2-((1,1-диоксидотиоморфолино)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-он
К смеси 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 200 мг, 0,781 ммоль) и тиоморфолин-1,1-диоксида (317 мг, 2,343 ммоль) в МеОН (10 мл) добавляли цианоборгидрид натрия (196 мг, 3,12 ммоль). Реакционную смесь перемешивали при 20°С в течение ночи. Смесь экстрагировали при помощи DCM и промывали рассолом. Органический слой упаривали в вакууме. Неочищенный продукт очищали посредством хроматографии на силикагеле, элюируя при помощи DCM/MeOH. Соответствующие фракции объединяли и упаривали с получением 7-бром-2-((1,1-диоксидотиоморфолин)метил)-5-метилфуро[3,2-с]пиридин-4(5H)-она (215 мг, 0,516 ммоль, выход 66,0%) в виде белого твердого вещества. LCMS: МН+ 375.
Промежуточное соединение 42: 7-хлор-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-он
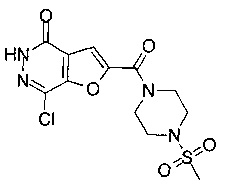
К триэтиламину (0,908 мл, 6,52 ммоль) в NMP (2 мл) добавляли 7-хлор-4-оксо-4,5-дигидрофуро[2,3-d]пиридазин-2-карбоновую кислоту (Peakdale) (250 мг, 1,165 ммоль), 1-(метилсульфонил)пиперазин (265 мг, 1,614 ммоль), 1Н-бензо[d][1,2,3]триазол-1-ола гидрат (215 мг, 1,404 ммоль) и гексафторфосфат 2-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (V) (134 мг, 0,352 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение ночи. Реакционную смесь очищали посредством MDAP. Соответствующие фракции объединяли и удаляли растворитель с получением 7-хлор-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-она (208 мг, 42%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,59 мин, МН+ 361.
Промежуточное соединение 43: 7-хлор-5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5Н)-он
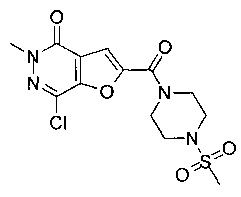
К 7-хлор-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5Н)-ону (относительно получения смотреть промежуточное соединение 42, 100 мг, 0,277 ммоль) в DMF (3 мл) добавляли гидрид натрия (11,09 мг, 0,277 ммоль) и йодистый метил (11 мкл, 0.176 ммоль). Смесь перемешивали в атмосфере азота при комнатной температуре в течение ночи. Добавляли дополнительный гидрид натрия (11,09 мг, 0.277 ммоль) и йодистый метил (11 мкл, 0,176 ммоль) и реакционную смесь перемешивали в течение 4 часов при комнатной температуре в атмосфере азота. Добавляли воду и удаляли растворитель. Остаток растворяли в смеси DCM/вода и распределяли (×2). Объединенные органические слои промывали водой, удаляли растворитель и сушили в глубоком вакууме в течение ночи с получением 7-хлор-5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5Н)-она (106 мг, 95%) в виде желтого твердого вещества. LCMS (2 мин, муравьиная кислота): Rt=0,7 мин, МН+ 375.
Промежуточное соединение 44: 5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[2,3-d]пиридазин-4(5H)-он
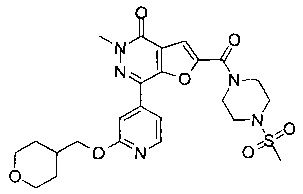
К 7-хлор-5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-ону (относительно получения смотреть промежуточное соединение 43, 106 мг, 0,283 ммоль) в смеси 1,4-диоксана (6 мл) и воды (3 мл) добавляли карбонат натрия (120 мг, 1,131 ммоль), тетракис(трифенилфосфин)палладий(0) (32,7 мг, 0,028 ммоль) и 2-((тетрагидро-2H-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (108 мг, 0,339 ммоль). Смесь нагревали с обратным холодильником при 110°С в атмосфере азота в течение 2 часов. После охлаждении удаляли растворитель и остаток очищали посредством MDAP. Соответствующие фракции объединяли и растворитель удаляли. Остаток сушили в глубоком вакууме в течение 3 часов и растирали с МеОН-d4 с получением 5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[2,3-d]пиридазин-4(5H)-она (50 мг, 30%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,87 мин, МН+ 532.
Промежуточное соединение 45: N-(4-бромпиридин-2-ил)ацетамид
Пиридин (0,374 мл, 4,62 ммоль) добавляли к раствору 4-бромпиридин-2-амина (Princeton BioMolecular Research) (400 мг, 2,312 ммоль) в DCM (10 мл) и перемешивали при КТ в течение 20 мин. Добавляли ацетилхлорид (0,190 мл, 2,66 ммоль) и раствор перемешивали при КТ в течение ночи. Добавляли воду и продукт экстрагировали при помощи DCM. Органический слой промывали рассолом, фильтровали через гидрофобную фритту и концентрировали в вакууме. Остаток растворяли в МеОН и пропускали через 10 g аминопропиловую колонку, элюируя при помощи МеОН. Растворитель выпаривали, получая указанное в заголовке соединение в виде белого твердого вещества (404 мг, 1,879 ммоль, выход 81%).
LCMS (2 мин, способ с высоким pH): Rt=0,65 мин, МН+ 215/217.
Промежуточное соединение 46: N-(4-бромпиридин-2-ил)-N-метилацетамид
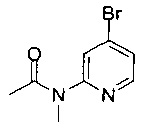
N-(4-Бромпиридин-2-ил)ацетамид (относительно получения смотреть промежуточное соединение 45, 406 мг, 1,888 ммоль) растворяли в DMF и охлаждали до 0°С. Добавляли гидрид натрия (60% масс./масс.) (91 мг, 2,266 ммоль) и смесь перемешивали в течение 15 минут. Добавляли йодметан (142 мкл, 2,266 ммоль) при КТ и реакционную смесь перемешивали в течение 2 ч. Добавляли воду и продукт извлекали диэтиловым эфиром (×4). Объединенные органические вещества упаривали до получения остатка, который очищали путем колоночной хроматографии на силикагеле (50-100% EtOAc/циклогексан). Фракции, содержащие продукт, объединяли и концентрировали в вакууме с получением указанного в заголовке соединения в виде бесцветного масла (252 мг, 1,100 ммоль, выход 58,3%). LCMS (2 мин, способ с высоким pH): Rt=0,69 мин, МН+ 229/231.
Промежуточное соединение 47: N-метил-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамид
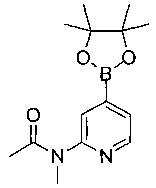
Смесь N-(4-бромпиридин-2-ил)-N-метилацетамида (относительно получения смотреть промежуточное соединение 46, 248 мг, 1,083 ммоль), бис(пинаколато)дибора (779 мг, 3,07 ммоль), PdCl2(dppf) (79 мг, 0,108 ммоль) и ацетат калия (319 мг, 3,25 ммоль) в 1,4-диоксане (5 мл) нагревали в микроволновой печи при 100°С в течение 30 мин. Реакционную смесь разбавляли этилацетатом и фильтровали через колонку с целитом. Фильтрат упаривали с получением N-метил-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамида в виде коричневого остатка. Превращение предполагали равным 100% (299 мг, 1,083 ммоль, выход 100%). Вещество использовали неочищенным в следующем взаимодействии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,37 мин, МН+=195 (наблюдаемая ионная масса согласуется с гидролизом до бороновой кислоты в условиях LCMS).
Промежуточное соединение 48: 4-бром-3-(циклопропилметокси)пиридин
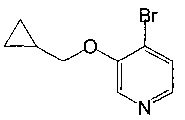
Гидрид натрия (60% масс./масс.) (110 мг, 2,76 ммоль) добавляли к 4-бромпиридин-3-олу (400 мг, 2,299 ммоль) в DMF (15 мл) при 0°С и оставляли перемешиваться в течение 30 минут. Добавляли (бромметил)циклопропан (0.268 мл, 2,76 ммоль) и реакционную смесь нагревали до КТ и перемешивали в течение ночи. Добавляли воду (40 мл) и органический продукт извлекали при помощи EtOAc и промывали рассолом, сушили посредством гидрофобной фритты и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии на силикагеле [0-5% MeOH/NH3 в DCM] и фракции, содержащие продукт, объединяли и концентрировали в вакууме с получением 4-бром-3-(циклопропилметокси)пиридина (234 мг, 1,026 ммоль, 44,6% выход) в виде коричневого масла. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,87 мин, МН+=228/230.
Промежуточное соединение 49: 3-(циклопропилметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
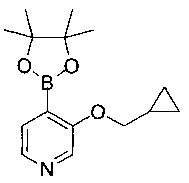
Смесь 4-бром-3-(циклопропилметокси)пиридина (относительно получения смотреть промежуточное соединение 48, 228 мг, 1,000 ммоль), бис(пинаколато)дибора (508 мг, 1,999 ммоль), PdCl2(dppf) (73,1 мг, 0,100 ммоль) и ацетата калия (294 мг, 3,00 ммоль) в 1,4-диоксане (5 мл) нагревали в микроволновой печи при 100°С в течение 30 мин. Реакционную смесь разбавляли этилацетатом и фильтровали через колонку с целитом. Фильтрат упаривали с получением 3-(циклопропилметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина в виде коричневого остатка. Превращение предполагали равным 100% (275 мг, 0,999 ммоль, выход 100%). Вещество использовали неочищенным в следующем взаимодействии без дополнительной очистки.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,35 мин, МН+ 194 (наблюдаемая ионная масса согласуется с гидролизом до бороновой кислоты в условиях LCMS).
Промежуточное соединение 50: 7-(3,4-диметоксифенил)-4-оксо-4,5-дигидрофуро[2,3-d]пиридазин-2-карбоновая кислота
К смеси 7-хлор-4-оксо-4,5-дигидрофуро[2,3-d]пиридазин-2-карбоновой кислоты (Peakdale) (220 мг, 1,025 ммоль) и (3,4-диметоксифенил)бороновой кислоты (187 мг, 1,025 ммоль) в 1,4-диоксане (3 мл) и воде (1,5 мл) добавляли карбонат натрия (435 мг, 4,10 ммоль) и тетракис(трифенилфосфин)палладий(0) (110 мг, 0,095 ммоль). Реакционную смесь нагревали в микроволновой печи при 80°С в течение 2 часов и при 100°С в течение следующих 2 часов. Растворитель удаляли и остаток суспендировали в DMSO, фильтровали и очищали посредством MDAP. Соответствующие фракции объединяли и удаляли растворитель с получением 7-(3,4-диметоксифенил)-4-оксо-4,5-дигидрофуро[2,3-d]пиридазин-2-карбоновой кислоты (25 мг, 7%) в виде желтого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,57 мин, МН+ 317.
Осадок на фильтре промывали при помощи DCM и потом промывали при помощи 1 М HCl и сушили фильтрацией под вакуумом с получением 7-(3,4-диметоксифенил)-4-оксо-4,5-дигидрофуро[2,3-d]пиридазин-2-карбоновой кислоты (79 мг, 24%) в виде желтого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,57 мин, МН+ 317.
Промежуточное соединение 51: 7-(3,4-диметоксифенил)-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-он
К 7-(3,4-диметоксифенил)-4-оксо-4,5-дигидрофуро[2,3-d]пиридазин-2-карбоновой кислоте (относительно получения смотреть промежуточное соединение 50, 25 мг, 0,079 ммоль) в NMP (2 мл) добавляли 1Н-бензо[d][1,2,3]триазол-1-ола гидрат (24,.21 мг, 0.158 ммоль), гексафторфосфат (V) 2-(3Н-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония (60,1 мг, 0,158 ммоль) и триэтиламин (0,033 мл, 0,237 ммоль), с последующим добавлением 1-(метилсульфонил)пиперазина (19,47 мг, 0,119 ммоль). Смесь перемешивали в течение ночи в атмосфере азота при комнатной температуре. Реакционную смесь очищали посредством MDAP. Соответствующие фракции объединяли и растворитель удаляли с получением 7-(3,4-диметоксифенил)-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-она (45 мг, >100%) в виде не совсем белого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,7 мин, МН+ 463.
Промежуточное соединение 52: 7-(3,4-диметоксифенил)-5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5Н)-он
К 7-(3,4-диметоксифенил)-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-ону (относительно получения смотреть промежуточное соединение 51, 0,062 мл, 0,097 ммоль) в DMF (3 мл) добавляли гидрид натрия (60% дисперсия в минеральном масле) (7,78 мг, 0,195 ммоль) и йодистый метил (0,030 мл, 0,487 ммоль). Смесь перемешивали в течение ночи в атмосфере азота при комнатной температуре. Добавляли воду и удаляли растворитель. Остаток растворяли в смеси DCM/вода и распределяли (×2). Объединенные органические слои промывали водой и растворитель удаляли с получением желтого твердого вещества. Его очищали посредством MDAP. Соответствующие фракции объединяли, упаривали и сушили в глубоком вакууме в течение 3 часов с получением 7-(3,4-диметоксифенил)-5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-она (18 мг, 39%).
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,8 мин, МН+ 477.
Промежуточное соединение 53: трет-бутил 4-((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-c]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилат
Смесь 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 500 мг, 1,953 ммоль), трет-бутил-1,4-диазепан-1-карбоксилата (782 мг, 3,91 ммоль) и АсОН (0,011 мл, 0,195 ммоль) в МеОН (30 мл) перемешивали при 25°С в течение 3 часов. Добавляли цианоборгидрид натрия (245 мг, 3,91 ммоль) и реакционную смесь перемешивали в течение ночи. Добавляли воду (60 мл) и смесь экстрагировали при помощи DCM (2×100 мл). Органическую фазу промывали рассолом (100 мл) и концентрировали в вакууме, и очищали посредством хроматографии на силикагеле (DCM:MeOH = 8:1) с получением трет-бутил 4-((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилата (500 мг, 1,136 ммоль, выход 58,2%) в виде желтого масла.
LCMS: МН+ 440/442.
Промежуточное соединение 54: 4-бром-N-(пиридин-2-илметил)пиридин-2-амин
К раствору 4-бром-2-фторпиридина (3,58 г, 20,34 ммоль) в NMP (17 мл) добавляли пиридин-2-илметанамин (2 г, 18,49 ммоль). Реакционную смесь перемешивали при 100°С в течение 1 часа, затем охлаждали до КТ и распределяли между DCM (100 мл) и водой (100 мл). Органическую фазу промывали насыщенным рассолом (50 мл), сушили над сульфатом натрия и концентрировали в вакууме с получением указанного в заголовке соединения (6 г, 3,25 ммоль, выход 17,56%) в виде желтого масла (содержащего NMP), которое использовали без дополнительной очистки на следующей стадии. LCMS: МН+ 264.
Промежуточное соединение 55: N-(Пиридин-2-илметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин
К суспензии неочищенного 4-бром-N-(пиридин-2-илметил)пиридин-2-амина (относительно получения смотреть промежуточное соединение 54, 5,8 г, 3,07 ммоль), 4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (3,90 г, 15,37 ммоль) и ацетата калия (0,905 г, 9,22 ммоль) в 1,4-диоксане (30 мл) перемешивали в атмосфере азота при КТ, добавляли комплекс 1,1'-бис(дифенилфосфино)ферроцен-палладия(II) дихлорид-DCM (0,251 г, 0,307 ммоль). Реакционную смесь перемешивали при 100°С в течение 2 ч. Реакционную смесь охлаждали до КТ и фильтровали. Фильтрат концентрировали и остаток очищали посредством комбинированной флэш-хроматографии на силикагеле (40 г), элюируя при помощи смеси EtOAc/петролейный эфир (0~100% в течение 40 мин, 100% в течение 40 мин) с получением неочищенного продукта в виде коричневого твердого вещества. Твердое вещество дополнительно очищали путем перекристаллизации со смесью эфир/гексан (1:30, 1 мл/30 мл) с получением указанного в заголовке соединения (700 мг, 2.249 ммоль, выход 73,2%) в виде коричневого твердого вещества.
LCMS: M/Z 230 указывает на гидролиз боронатного эфира в условиях LCMS.
1Н ЯМР (400 МГц, CDCl3) δ-млн-1 8.56 (1Н, d), 8.14 (1Н, d), 7.61 (1Н, t), 7.31 (1Н, d), 7.17 (1Н, t), 6.89 (2Н, m), 6.67 (1Н, br.s), 4.69 (2Н, d), 1.31 (12Н, s).
Промежуточное соединение 56: трет-бутил 4-((5-метил-4-оксо-7-(2-((пиридин-2-илметил)амино)пиридин-4-ил)-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилат
К раствору трет-бутил-4-((7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилата (относительно получения смотреть промежуточное соединение 53, 400 мг, 0,908 ммоль) в 1,4-диоксане (24 мл) добавляли N-(пиридин-2-илметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин (относительно получения смотреть промежуточное соединение 55, 424 мг, 1,363 ммоль) и Pd(Ph3P)4 (52,5 мг, 0,045 ммоль). Из реакционной смеси откачивали воздух и продували ее азотом с последующим добавлением Cs2CO3 (592 мг, 1,817 ммоль) и воды (6 мл). Смесь нагревали до 100°С в течение ночи. Реакционную смесь охлаждали до КТ. Органический слой удаляли и водный слой извлекали при помощи EtOAc (3×60 мл). Объединенные органические экстракты промывали рассолом (100 мл) и концентрировали в вакууме. Неочищенный продукт очищали посредством хроматографии на силикагеле (DCM:MeOH = 8:1) с получением трет-бутил 4-((5-метил-4-оксо-7-(2-((пиридин-2-илметил)амино)пиридин-4-ил)-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилата (220 мг, 0,404 ммоль, выход 44,5%) в виде коричневого масла. LCMS: МН+ 545.
Промежуточное соединение 57: 1-(2-((4-бромпиридин-2-ил)окси)этил)пирролидин-2-он
Перемешанную суспензию 1-(2-гидроксиэтил)пирролидин-2-она (0,324 мл, 2,87 ммоль), трифенилфосфина (904 мг, 3,45 ммоль) и 4-бромпиридин-2-ола (500 мг, 2,87 ммоль) в THF (10 мл) продували азотом и охлаждали в ледяной бане в течение 15 мин перед добавлением DIAD порциями (0,670 мл, 3,45 ммоль). Смесь оставляли перемешиваться в течение 1 ч. Смесь разбавляли при помощи EtOAc (20 мл) и воды (20 мл). Два слоя отделяли и водный слой дополнительно экстрагировали при помощи EtOAc (2×20 мл). Органические экстракты упаривали досуха и оставшееся желтое твердое вещество растворяли в DCM и очищали посредством хроматографии на силикагеле (100 д), элюируя при помощи градиента 5% МеОН в EtOAc. Соответствующие фракции объединяли и уменьшали их объем в вакууме с получением 1-(2-((4-бромпиридин-2-ил)оксиэтил)пирролидин-2-она в виде прозрачного масла (490 мг, 59,8%).
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,82 мин, МН+ 285/287.
Промежуточное соединение 58: 1-(2-((4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксиэтил)пирролидин-2-он
К перемешиваемой суспензии 1-(2-((4-бромпиридин-2-ил)оксиэтил)пирролидин-2-она (относительно получения смотреть промежуточное соединение 57, 490 мг, 1,718 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (873 мг, 3,44 ммоль) и ацетата калия (506 мг, 5,16 ммоль) добавляли PdCl2(dppf) (126 мг, 0,172 ммоль). Смесь помещали во флакон для микроволновой печи и нагревали в микроволновой печи при 100°С в течение 1 ч. Смесь разбавляли в этилацетате и фильтровали через картридж с целитом (10 г). Растворитель выпаривали с получением коричневого масла (1,35 г, 4,06 ммоль, 236%). Это неочищенное вещество использовали в следующей стадии без дополнительной очистки: предполагалось 100% превращение, поэтому максимальная чистота неочищенного вещества равна 42%.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,46 мин, МН+=251 соответствует гидролизу до бороновой кислоты в условиях LCMS.
1Н ЯМР (400 МГц, CDCl3) δ-млн-1 8.15 (1Н, d), 7.19 (1Н, d), 7.12 (1Н, s), 4.42 (2Н, t), 3.67 (2Н, t), 3.53 (2Н, t), 2.38 (2Н, t), 2.00 (2Н, m), 1.34 (12Н, s).
Промежуточное соединение 59: N-(5-йод-4-метокси-1-метил-6-оксо-1,6-дигидро-[3,4'-бипиридин]-2'-ил)ацетамид
Раствор TPPTS (0,022 г, 0,039 ммоль), 3,5-дийод-4-метокси-1-метилпиридин-2(1Н)-она (относительно получения смотреть промежуточное соединение 18, 0,2 г, 0,512 ммоль), N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамида (Milestone Pharma Tech) (0,174 г, 0.665 ммоль) и диацетоксипалладия (6,89 мг, 0,031 ммоль) в ацетонитриле (2 мл) и воде (0,667 мл) обрабатывали при помощи DIPEA (0,116 мл, 0,665 ммоль). Реакционную смесь нагревали при 60°С в атмосфере азота в течение 16 ч.
Смесь концентрировали при пониженном давлении, суспендировали в DCM (нерастворима) и очищали посредством хроматографии на силикагеле (100 г), элюируя при помощи градиента 0-20% 2 н. аммиака в MeOH/DCM. Фракции, содержащие продукт, объединяли и концентрировали при пониженном давлении с получением вещества с чистотой<70%. Неочищенное вещество растворяли в МеОН и загружали в предварительно обработанную при помощи МеОН колонку для SCX (10 g), и элюировали при помощи МеОН с последующим элюированием при помощи 2 н. аммиака в МеОН. УФ-активное вещество элюировалось в первой фракции аммиачной промывки, его концентрировали при пониженном давлении с получением N-(5-йод-4-метокси-1-метил-6-оксо-1,6-дигидро-[3,4'-бипиридин]-2'-ил)ацетамида (80 мг, 39%) в виде прозрачной бесцветной смолы.
LCMS (2 мин, способ с высоким pH): Rt=0,67 мин, МН+ 400.
Промежуточное соединение 60: N-(5-бромпиридин-3-ил)ацетамид
К перемешиваемой суспензии 5-бромпиридин-3-амина (500 мг, 2,89 ммоль) в DCM (10 мл) добавляли пиридин (0,467 мл, 5,78 ммоль). Смесь перемешивали в течение 20 мин и добавляли ацетилхлорид (0,236 мл, 3,32 ммоль). Смесь перемешивали в течение 3 ч.
Смесь разбавляли водой (20 мл) и DCM (20 мл). Слои разделяли и водный слой повторно экстрагировали при помощи DCM (3×20 мл). Объединенные органические вещества промывали рассолом (20 мл), сушили с использованием гидрофобной фритты и концентрировали в вакууме с получением N-(5-бромпиридин-3-ил)ацетамида в виде оранжевого твердого вещества (563 мг, 91%).
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,59 мин, МН+ 215/217.
Промежуточное соединение 61: N-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)ацетамид
К суспензии N-(5-бромпиридин-3-ил)ацетамида (относительно получения смотреть промежуточное соединение 60, 563 мг, 2,62 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (1330 мг, 5,24 ммоль) и ацетата калия (771 мг, 7,85 ммоль) в 1,4-диоксане (8 мл) во флаконе для микроволновой печи добавляли PdCl2(dppf) (192 мг, 0,262 ммоль). Флакон закрывали герметично и смесь нагревали в микроволновой печи при 100°С в течение 1 ч. Смесь растворяли в этилацетате и фильтровали через картридж с целитом (10 г). Растворитель выпаривали при пониженном давлении с получением N-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)ацетамида в виде коричневого масла (1,77 г, 258%). Это неочищенное вещество использовали в следующей стадии без дополнительной очистки: предполагалось 100% превращение, поэтому максимальная чистота неочищенного вещества равна 39%. LCMS (2 мин, способ с высоким pH): Rt=0,47 мин, МН+ 263.
Промежуточное соединение 62: трет-бутил (4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)карбамат
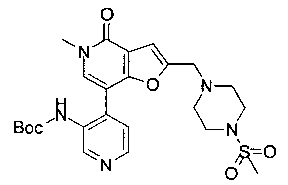
(3-((трет-бутоксикарбонил)амино)пиридин-4-ил)бороновую кислоту (Peptech) (100 мг, 0,420 ммоль), 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 3, 170 мг, 0,420 ммоль), тетракис(трифенилфосфин)палладий(0) (24,27 мг, 0,021 ммоль) и водный раствор карбоната натрия (1,680 мл, 3,36 ммоль) смешивали в 1,2-DME (3 мл) и нагревали в микроволновой печи в течение 2 часов при 120°С. Реакционную смесь разбавляли этилацетатом и водой и фильтровали. Органический слой отделяли и концентрировали в вакууме. Полученный остаток очищали посредством хроматографии на силикагеле (25 г), элюируя при помощи 0-5% МеОН в DCM. Соответствующие фракции объединяли и концентрировали в вакууме с получением трет-бутил (4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)карбамата (8 мг, 0,015 ммоль, выход 3,68%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,55 мин, МН+ 518.
Дополнительные фракции, содержащие продукт, объединяли и концентрировали в вакууме с получением трет-бутил-(4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)карбамата (74 мг, 0,143 ммоль, выход 34,0%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,55 мин, МН+ 518.
Промежуточное соединение 63: 7-(3-аминопиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он
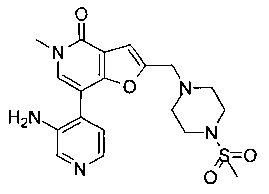
трет-Бутил-(4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)карбамат (относительно получения смотреть промежуточное соединение 62, 74 мг, 0,143 ммоль) суспендировали в DCM (2 мл) и добавляли TFA (2 мл, 26,0 ммоль). Полученный раствор перемешивали при КТ в течение 30 мин. Растворитель выпаривали и полученное желтое твердое вещество растворяли в МеОН и элюировали через аминопропиловый картридж (50 г) при помощи МеОН. Фракции концентрировали в вакууме с получением 7-(3-аминопиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (72 мг, 0,138 ммоль, выход 97%) в виде желтого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,33 мин, МН+ 418.
Промежуточное соединение 64: (R)-трет-бутил (4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)карбамат
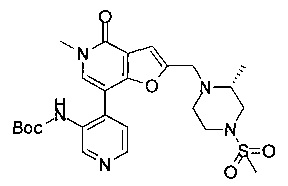
(3-((трет-Бутоксикарбонил)амино)пиридин-4-ил)бороновую кислоту (Peptech) (100 мг, 0,420 ммоль), (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он (относительно получения смотреть промежуточное соединение 5, 176 мг, 0,420 ммоль), тетракис(трифенилфосфин)палладий(0) (24,27 мг, 0,021 ммоль) и водный раствор карбоната натрия (1,680 мл, 3,36 ммоль) смешивали в 1,2-DME (3 мл) и нагревали в микроволновой печи в течение 2 часов при 120°С. Реакционную смесь разбавляли этилацетатом и водой. Органический слой отделяли и концентрировали в вакууме. Остаток очищали посредством колоночной хроматографии на силикагеле (25 г), элюируя при помощи 0-5% МеОН в DCM. Фракции, содержащие продукт, объединяли и концентрировали в вакууме с получением (R)-трет-бутил-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)карбамата (87 мг, 0,164 ммоль, выход 39,0%) в виде белого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,56 мин, МН+ 532.
Промежуточное соединение 65: (R)-7-(3-аминопиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
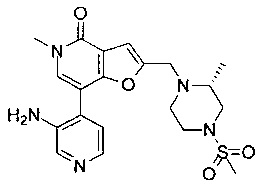
(R)-трет-бутил-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)карбамат (относительно получения смотреть промежуточное соединение 64, 87 мг, 0,164 ммоль) суспендировали в DCM (2 мл) и добавляли TFA (2 мл, 26,0 ммоль). Полученный раствор перемешивали при КТ в течение 30 мин. Растворитель выпаривали и полученное желтое твердое вещество растворяли в МеОН и элюировали через аминопропиловый картридж (50 г) при помощи МеОН. Растворитель концентрировали в вакууме с получением (R)-7-(3-аминопиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (81 мг, 0,160 ммоль, выход 97%) в виде желтого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,34 мин, МН+ 432.
Промежуточное соединение 66: Оксетан-2-илметил метансульфонат
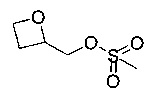
К раствору оксетан-2-илметанола (0,370 мл, 4,54 ммоль, ex.TCI) в DCM (15 мл) при 0°С, добавляли триэтиламин (1,898 мл, 13,62 ммоль) и метансульфонилхлорид (0,387 мл, 4,99 ммоль). Смесь перемешивали при 0°С в течение 1 ч. Смесь разбавляли водой (20 мл) и распределяли при помощи DCM (20 мл). Водную фазу повторно извлекали при помощи DCM (3×20 мл). Органическую фазу сушили с использованием гидрофобной фритты и концентрировали в вакууме с получением желтого масла (572 мг, 76%), которое использовали неочищенным в следующей стадии.
Промежуточное соединение 67: 4-Бром-3-(оксетан-2-илметокси)пиридин
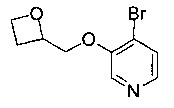
К перемешиваемой суспензии 4-бромпиридин-3-ола (300 мг, 1,724 ммоль) в DMF (11 мл) добавляли гидрид натрия (83 мг, 60% масс./масс., 2,069 ммоль). Все это охлаждали до 0° С и оставляли перемешиваться в течение 30 мин, после чего добавляли неочищенный оксетан-2-илметил-метансульфонат (относительно получения смотреть промежуточное соединение 66, 344 мг, 2,069 ммоль) и смесь снова оставляли перемешиваться в течение 4 ч. Все исходное вещество осталось, поэтому добавляли дополнительный гидрид натрия (60%) (83 мг, 2,069 ммоль) и смесь охлаждали до 0°С в течение 30 мин перед добавлением дополнительного неочищенного оксетан-2-илметил-метансульфоната (228 мг). Реакционную смесь оставляли перемешиваться в течение ночи, затем нагревали до 60°С и перемешивали в течение ночи. Смесь разбавляли при помощи EtOAc (20 мл) и распределяли при помощи воды (20 мл). Два слоя разделяли и водную фазу повторно экстрагировали при помощи EtOAc (3×20 мл). Органическую фазу сушили с использованием гидрофобной фритты и концентрировали в вакууме с получением желтой жидкости. Ее разбавляли при помощи 10% раствора LiCl и распределяли при помощи EtOAc (20 мл). Два слоя разделяли и водную фазу повторно экстрагировали при помощи EtOAc (3×20 мл). Органическую фазу сушили с использованием гидрофобной фритты и концентрировали в вакууме с получением 4-бром-3-(оксетан-2-илметокси)пиридина в виде желтого масла (708 мг, 168%). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,60 мин, МН+ 244/246. Чистоту оценивали как 59,5% и вещество использовали неочищенным в следующей стадии.
Промежуточное соединение 68: (R)-4-бром-3-(оксиран-2-илметокси)пиридин
4-Бромпиридин-3-ол (36,9 г, 212 ммоль) помещали в DMF (667 мл) в атмосфере азота. Карбонат цезия (189 г, 579 ммоль) добавляли одной порцией и смесь перемешивали в течение 10 мин. (R)-оксиран-2-илметил-3-нитробензолсульфонат (50 г, 193 ммоль) добавляли в течение порядка 10 мин и реакционную смесь оставляли перемешиваться при комнатной температуре в течение ночи. Реакционную смесь охлаждали в ледяной бане и медленно добавляли воду (1000 мл) (экзотермическая реакция). Раствор экстрагировали при помощи EtOAc (2×500 мл). Водный слой разбавляли рассолом (1000 мл) и затем повторно экстрагировали при помощи EtOAc (2×1000 мл). Объединенные органические вещества промывали водой (2×2000 мл) и 5% LiCl (2000 мл), и затем сушили при помощи Na2SO4, фильтровали и концентрировали в вакууме с получением (R)-4-бром-3-(оксиран-2-илметокси)пиридина (39,4 г) в виде оранжевого масла. 1Н ЯМР (400 МГц, CDCl3) δ-млн-1 8.27 (1Н, s), 8.10 (1Н, d), 7.52 (1Н, d), 4.44 (1Н, dd) 4.15 (1Н, dd), 3.43 (1Н, m), 2.95 (1Н, m), 2.87 (1Н, dd). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,60 мин, МН+ 230/232. Масло сразу же помещали в tBuOH (100 мл) для применения в следующем взаимодействии.
Промежуточное соединение 69: (R)-4-Бром-3-(оксетан-2-илметокси)пиридин
Калия трет-бутоксид (26,2 г, 234 ммоль) помещали в трет-BuOH (450 мл) в атмосфере азота. Триметилсульфоксония йодид (34,3 г, 156 ммоль) добавляли одной порцией и реакционную смесь нагревали до 80°С в течение 10 мин, затем охлаждали до комнатной температуры. Добавляли (R)-4-бром-3-(оксиран-2-илметокси)пиридин (относительно получения смотреть промежуточное соединение 68, 39,4 г, 171 ммоль) в виде раствора в tBuOH (100 мл) и смесь перемешивали при 60°С в течение 4 ч. Вначале образовывалась белая суспензия, которая становилась оранжевой после добавления оксирана. Через 6 часов реакционную смесь оставляли охлаждаться и стоять при комнатной температуре в течение ночи. Реакционную смесь концентрировали до порядка 1/3 объема и распределяли между водой (500 мл) и EtOAc (500 мл). Водную фазу повторно экстрагировали при помощи EtOAc (3×500 мл). Объединенные органические вещества промывали водой (1000 мл) и рассолом (1000 мл), и затем сушили при помощи MgSO4, фильтровали и концентрировали в вакууме с получением (R)-4-бром-3-(оксетан-2-илметокси)пиридина (18,8 г) в виде оранжевого масла. 1Н ЯМР (400 МГц, CDCl3) δ-млн-1 8.30 (1Н, s), 8.09 (1Н, d), 7.52 (1Н, d), 5.17 (1Н, m), 4.74 (2Н, m) 4.34 (1Н, dd), 4.25 (1Н, dd), 2.85 (2Н, m). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,62 мин, МН+ 244/246.
Промежуточное соединение 70: 4-бром-3-(оксетан-3-илметокси)пиридин
Указанное в заголовке соединение получали из 4-бром-3-пиридинола и 3-(бромметил)оксетана способом, аналогичным описанному для промежуточного соединения 48.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,57 мин, МН+=244/246.
Промежуточное соединение 71: 3-(оксетан-3-илметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
К суспензии 4-бром-3-(оксетан-3-илметокси)пиридина (относительно получения смотреть промежуточное соединение 70, 300 мг, 1,229 ммоль), 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолана) (624 мг, 2,458 ммоль), ацетата калия (362 мг) в 1,4-диоксана (10 мл) во флаконе для микроволновой печи добавляли PdCl2(dppf) (90 мг). Флакон закрывали герметично и смесь нагревали в микроволновой печи Biotage initiator при 100°С в течение 1 часа с нормальной абсорбцией. Добавляли дополнительные 4,4,4',4',5,5,5',5'-октаметил-2,2'-би(1,3,2-диоксаборолан) (624 мг), ацетат калия (362 мг) и PdCl2(dppf) (90 мг) и реакционную смесь нагревали в микроволновой печи в течение следующего 1 часа. Смесь разбавляли этилацетатом и фильтровали через картридж с целитом (5 г). Объем растворителя уменьшали в вакууме с получением коричневого масла (1,72 г, 481%). Это неочищенное вещество использовали в следующей стадии без дополнительной очистки: предполагалось 100% превращение, поэтому максимальная чистота неочищенного вещества представляла собой 21%.
Промежуточное соединение 72: 4-бром-3-((2,2-дифторциклопропил)метокси)пиридин
Указанное в заголовке соединение получали из 4-бром-3-пиридинола и 2-(бромметил)-1,1-дифторциклопропана аналогичным описанному для промежуточного соединения 48 способом.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,87 мин, МН+=264/266.
Промежуточное соединение 73: 4-бром-3-(2-метоксиэтокси)пиридин
Указанное в заголовке соединение получали из 4-бром-3-пиридинола и 1-бром-2-метоксиэтана аналогичным описанному для Промежуточного соединения 48 способом.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,61 мин, МН+=232/234.
Промежуточное соединение 74: 3-(2-метоксиэтокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин
Указанное в заголовке соединение получали из промежуточного соединения 73 аналогичным описанному для промежуточного соединения 71 способом и использовали неочищенным в следующей стадии.
Промежуточное соединение 75: 4-бром-3-((тетрагидрофуран-3-ил)метокси)пиридин
Указанное в заголовке соединение получали из 4-бром-3-пиридинола и 3-(бромметил)тетрагидрофурана аналогичным описанному для промежуточного соединения 48 способом.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,69 мин, МН+=258/260.
Промежуточное соединение 76: 4-бром-2-((тетрагидрофуран-3-ил)метокси)пиридин
Указанное в заголовке соединение получали из 4-бром-3-пиридинола и 2-(бромметил)тетрагидрофурана аналогичным описанному для Промежуточного соединения 48 способом.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,71 мин, МН+=258/260.
Промежуточное соединение 77: 1-(2-метилбут-3-ин-2-ил)-4-(метилсульфонил)пиперазин
Смесь 3-хлор-3-метилбут-1-ина (1 г, 9,75 ммоль), меди (12 мг, 0,195 ммоль) и хлорида меди (I) (19 мг, 0,195 ммоль) и 1-(метилсульфонил)пиперазина (4,32 г, 26,3 ммоль) в диэтиловом эфире (15 мл) и воде (5 мл) перемешивали при комнатной температуре в атмосфере азота в течение 16 часов. Добавляли воду (150 мл) и диэтиловый эфир (150 мл) и органический слой выделяли. Водный слой повторно экстрагировали в диэтиловом эфире (2×150 мл). Объединенные органические слои пропускали через гидрофобную фритту, затем концентрировали при пониженном давлении. Неочищенное вещество очищали посредством колоночной хроматографии на силикагеле, элюируя при помощи 80-100% EtOAc/циклогексан. Соответствующие фракции объединяли, затем концентрировали при пониженном давлении с получением 1-(2-метилбут-3-ин-2-ил)-4-(метилсульфонил)пиперазина в виде белого твердого вещества (1,76 г, 78%). 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 3.22 (1Н, s), 3.11 (4Н, m), 2.86 (3Н, s), 2.61 (4Н, m), 1.31 (6Н, s).
Промежуточное соединение 78: 7-бром-2-(гидроксиметил)-5-метилфуро[3,2-с]пиридин-4(5H)-он
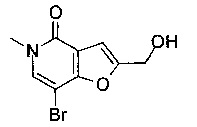
Боргидрид натрия (2,216 г, 58,6 ммоль) добавляли к суспензии 7-бром-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-карбальдегида (относительно получения смотреть промежуточное соединение 2, 5 г, 19,53 ммоль) в этаноле (100 мл) при 0°С. Смесь перемешивали в течение 2 ч, затем гасили путем осторожного, по каплям, добавления насыщенного раствора хлорида аммония (100 мл). Полученное твердое вещество собирали посредством фильтрации и промывали водой (2×30 мл) с получением 7-бром-2-(гидроксиметил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она (3.3 г, 12,79 ммоль, выход 65,5%) в виде бежевого порошка. 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 8.00 (1Н, s), 6.86 (1Н, s), 5.51 (1Н, s), 4.53 (2Н, s), 3.50 (3Н, s). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,58 мин, МН+ 258/260.
Промежуточное соединение 79: N-(4-бромпиридин-2-ил)ацетамид
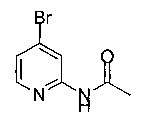
4-Бромпиридин-2-амин (10 г, 57,8 ммоль) помещали в DCM (75 мл) и пиридин (75 мл) в атмосфере азота. Добавляли уксусный ангидрид (8,18 мл, 87 ммоль) и оставляли стоять в течение ночи, и затем концентрировали в вакууме. Остаток сушили в вакуумном сушильном шкафу с получением N-(4-бромпиридин-2-ил)ацетамида (12,4 г, 54,8 ммоль, выход 95%) в виде твердого вещества кремового цвета. 1Н ЯМР (400 МГц, CDCl3) δ-млн-1 8.48 (1Н, s), 8.14 (1Н, br. s), 8.09 (1Н, d), 7.23 (1Н, dd), 2.23 (3Н, s). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,65 мин, МН+ 215/217.
Промежуточное соединение 80: N-(4-(2-(Гидроксиметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
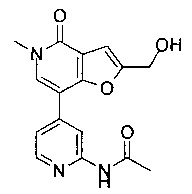
4,4,5,5-Тетраметил-1,3,2-диоксаборолан (11,13 мл, 77 ммоль) добавляли к смеси 7-бром-2-(гидроксиметил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 78, 3,3 г, 12,79 ммоль) и триэтиламина (10,71 мл, 77 ммоль) в 1,4-диоксане (20 мл), и смесь перемешивали в течение 5 мин в атмосфере азота. Добавляли Pd(PPh3)4 (1,478 г, 1,279 ммоль) и смесь нагревали до 100°С в течение 18 часов. Смесь охлаждали в ледяной бане, затем добавляли изопропанол (20,00 мл), вначале очень осторожно, с последующим добавлением воды (10 мл), карбоната калия (5,30 г, 38,4 ммоль), PEPPSI-SIPr (0,871 г, 1,279 ммоль) и N-(4-бромпиридин-2-ил)ацетамида (относительно получения смотреть промежуточное соединение 79, 3,02 г, 14,07 ммоль). Смесь нагревали до 80°С в течение 2 часов в атмосфере азота, затем оставляли стоять в течение выходных. Смесь разбавляли эфиром (100 мл) и перемешивали в течение 10 мин, затем фильтровали и твердое вещество промывали водой (50 мл) и сушили в вакууме в течение 10 мин с получением неочищенного продукта. Полученное твердое вещество нагревали в метаноле (50 мл) до температуры дефлегмации, затем охлаждали в ледяной бане и твердый продукт собирали путем фильтрации с получением N-(4-(2-(гидроксиметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (2,76 г, 8,81 ммоль, выход 68,9%) в виде бесцветного твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 10.53 (1Н, s), 8.50 (1Н, s), 8.38 (1Н, d), 8.13 (1Н, s), 7.48 (1Н, m), 6.85 (1Н, s), 5.46 (1Н, m), 4.55 (2Н, d), 3.61 (3Н, s), 2.13 (3Н, s). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,55 мин, МН+ 314.
Промежуточное соединение 81: N-(4-(2-(Бромметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
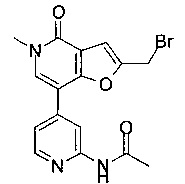
N-(4-(2-(Гидроксиметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид (относительно получения смотреть промежуточное соединение 80, 1,4 г, 4,47 ммоль) суспендировали в 1,4-диоксане (50 мл) и нагревали до 60°С, добавляли затем PBr3 (2,107 мл, 22,34 ммоль) небольшими порциями и суспензию нагревали в течение ночи при 60°С. Суспензию разбавляли при помощи DCM (100 мл) и фильтровали, и желтое твердое вещество промывали при помощи DCM и сушили в вакуум-сушильном шкафу с получением N-(4-(2-(бромметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-c]пиридин-7-ил)пиридин-2-ил)ацетамида (1,9 г, 5,05 ммоль, выход 113%), который использовали неочищенным в следующей стадии без дополнительной очистки. 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 11.48 (1Н, s), 8.48 (1Н, s), 8.41 (1Н, d), 8.31 (1Н, s), 7.72 (1Н, m), 7.17 (1Н, s), 4.94 (2Н, s), 3.64 (3Н, s), 2.24 (3Н, s).
Промежуточное соединение 82: N-(4-(2-(хлорметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
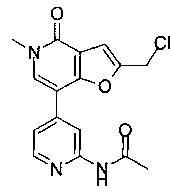
Метансульфонилхлорид (2,75 мл, 35,2 ммоль) добавляли к суспензии N-(4-(2-(гидроксиметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (относительно получения смотреть промежуточное соединение 80, 2,76 г, 8,81 ммоль) в DCM (20 мл) и Et3N (5,53 мл, 39,6 ммоль), и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали и твердое вещество промывали при помощи DCM (20 мл), сушили с получением N-(4-(2-(гидроксиметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (1,4 г, 4,47 ммоль, выход 50,7%), извлеченного исходного вещества. Фильтрат выпаривали в вакууме и остаток повторно растворяли в смеси DCM (200 мл) и метанола (30 мл), промывали водой (2×100 мл) и рассолом (100 мл), сушили и упаривали с получением коричневой смолы. Ее очищали посредством колоночной хроматографии на силикагеле, элюируя при помощи 0-10% MeOH/DCM и продукт, содержащий фракции, упаривали в вакууме с получением N-(4-(2-(хлорметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (0,20 г, 0,603 ммоль, выход 6,84%) в виде коричневого твердого вещества. 1Н ЯМР (400 МГц, DMSO-d6) s-млн-1 10.54 (1Н, s), 8.54 (1Н, s), 8.39 (1Н, d), 8.21 (1Н, s), 7.46 (1Н, dd), 7.13 (1Н, s), 4.97 (2Н, s), 3.62 (3Н, s), 2.13, (3Н, s). Вещество использовали в следующей стадии без дополнительной очистки.
Пример 1: 7-[3,4-бис(метилоксифенил]-5-метил-2-{[4-(метилсульфонил)-1-пиперазинил]метил}фуро[3,2-с]пиридин-4(5H)-он
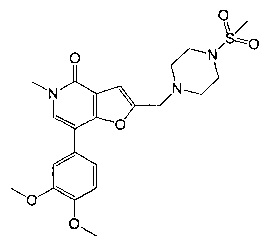
Смесь 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 3, 52 мг, 0,129 ммоль), (3,4-диметоксифенил)бороновой кислоты (46,8 мг, 0,257 ммоль), карбоната калия (53,3 мг, 0,386 ммоль) и бис(трифенилфосфин)палладия(Н) хлорида (9,03 мг, 0.013 ммоль) в смеси воды (0,5 мл) и 1,2-DME (1,5 мл) нагревали в микроволновой печи при 120°C в течение 20 минут. Охлажденную реакционную смесь разбавляли этилацетатом, и раствор сушили над MgSO4 и упаривали. Остаток очищали посредством MDAP с получением 7-[3,4-бис(метилоксифенил]-5-метил-2-{[4-(метилсульфонил)-1-пиперазинил]метил}фуро[3,2-с]пиридин-4(5Н)-она (31 мг, 52%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,59 мин, МН+ 462.
Пример 2: (R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
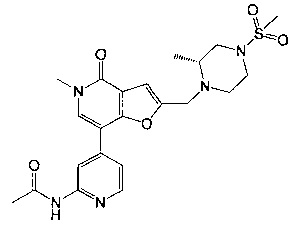
Смесь (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 5, 345 мг, 0,825 ммоль), N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамида (Milestone Pharma tech USA) (432 мг, 1,650 ммоль), карбоната калия (456 мг, 3,30 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (57,9 мг, 0,082 ммоль) в смеси воды (1,5 мл) и 1,2-DME (4,5 мл) нагревали в микроволновой печи при 120°С в течение 20 минут. Охлажденную реакционную смесь разбавляли этилацетатом и раствор сушили над MgSO4 и упаривали. Остаток очищали посредством MDAP с получением (R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (96 мг, 25%) в виде вязкого светло-желтого масла.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,46 мин, МН+ 474.
Пример 3: 7-(3-(бензилокси)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он
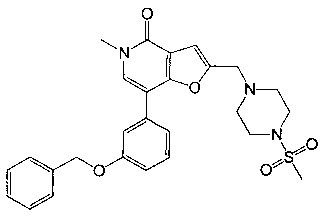
Смесь карбоната калия (273 мг, 1,979 ммоль), бис(трифенилфосфин)палладия(II) хлорида (13,33 мг, 0,019 ммоль), 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (относительно получения смотреть промежуточное соединение 3, 160 мг, 0,396 ммоль), 2-(3-(бензилокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолаан (относительно получения смотреть промежуточное соединение 22, 310 мг, 0,999 ммоль) в EtOH (2 мл) и толуоле (2 мл) нагревали в микроволновой печи при 80°С в течение 20 мин. Охлажденную реакционную смесь разбавляли в 10 мл этилацетата и упаривали. Остаток разбавляли этилацетатом (20 мл) и промывали водой (20 мл). Водный слой экстрагировали при помощи этилацетата (2×20 мл). Объединенные органические слои промывали рассолом (2×20 мл), сушили и концентрировали в вакууме. Остаток растворяли в МеОН, загружали на колонку SCX 20 g и элюировали при помощи МеОН с последующим элюированием 2 М метанольным аммиаком. Основные фракции концентрировали в вакууме с получением 7-(3-(бензилокси)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (15 мг, 7%) в виде черной смолы.
LCMS (2 мин, способ с высоким pH): Rt=1,10 мин, МН+ 508.
Пример 4: 7-(3-(бензиламино)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
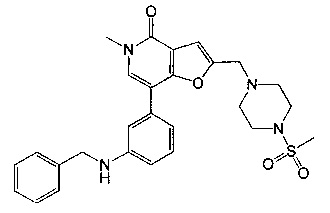
Смесь 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 3, 85,2 мг, 0,211 ммоль), N-бензила-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилина (относительно получения смотреть промежуточное соединение 24, 102,1 мг, 0,330 ммоль), карбоната калия (146,7 мг, 1,061 ммоль), транс-дихлор(трифенилфосфин)палладия(II) (7,7 мг, 10,97 мкмол) в EtOH (2 мл) и толуоле (2 мл) нагревали при перемешивании в герметично закрытом флаконе в микроволновом реакторе в течение 20 мин при 80°С, и затем при 100°С в течение следующих 20 мин. Растворители выпаривали в потоке азота. Твердое вещество промывали раствором бикарбоната натрия (5 мл) и экстрагировали при помощи DCM (4×5 мл). DCM выпаривали. Коричневый твердый остаток очищали посредством MDAP. Соответствующие фракции собирали и упаривали в потоке азота с получением 7-(3-(бензиламино)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (18,3 мг, 17%) в виде бледно-коричневой смолы.
LCMS (2 мин, способ с высоким pH): Rt=1,04 мин, МН+=507.
Пример 5: 2-(((2-аминоэтил)амино)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-он дигидрохлорид
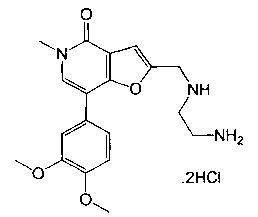
трет-Бутил(2-(((7-(3,4-диметоксифенил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)амино)этил)карбамат (относительно получения смотреть промежуточное соединение 7, 25 мг, 0,055 ммоль) растворяли в EtOH (2 мл) и добавляли HCl (4 М в 1,4-диоксане) (2 мл, 8,00 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 1 часа, получая белый осадок. Продукт концентрировали в вакууме с получением 2-(((2-аминоэтил)амино)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-она дигидрохлорида (19 мг, 81%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,48 мин, МН+=358.
Пример 6: 7-(4-(аминометил)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он гидрохлорид
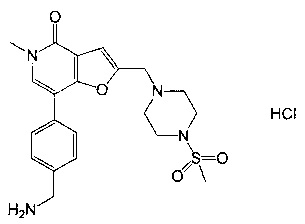
1,4-Диоксан (1 мл) и HCl (4 M в 1,4-диоксан) (1,435 мл, 5,74 ммоль) добавляли к трет-бутил-4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензилкарбамату (относительно получения смотреть промежуточное соединение 8, 152,31 мг, 0,287 ммоль) и реакционную смесь перемешивали в течение ночи. Летучие компоненты удаляли при пониженном давлении. Осуществляли растирание с диэтиловым эфиром с получением белого/желтого порошка. Его затем сушили в сушильном шкафу при 40°С в течение 1 ч с получением 7-(4-(аминометил)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она гидрохлорида (89 мг, 66,4%) в виде белого/желтого порошка.
LCMS (2 мин, способ с высоким pH): Rt=0,65 мин, МН+=431.
Пример 7: 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
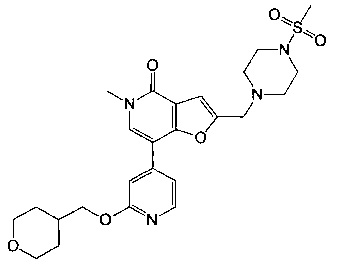
Смесь 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (относительно получения смотреть промежуточное соединение 3, 165 мг, 0,408 ммоль), 2-((тетрагидро-2Н-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (BoroPharm Inc) (261 мг, 0,816 ммоль), карбоната калия (169 мг, 1,224 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (28,6 мг, 0,041 ммоль) в воде (1,5 мл) и 1,2-DME (4,5 мл) нагревали в микроволновой печи при 120°С в течение 20 минут. Охлажденную реакционную смесь разбавляли этилацетатом, и раствор сушили над MgSO4 и упаривали. Остаток очищали посредством MDAP с получением 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она (59 мг, 28%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,64 мин, МН+ 517.
Пример 8: 7-(3,4-диметоксифенил)-5-метил-2-(пиперидин-1-илметил)фуро[3,2-с]пиридин-4(5H)-он
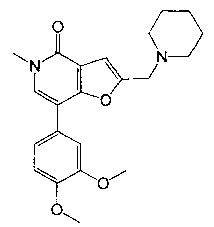
Смесь 7-бром-5-метил-2-(пиперидин-1-илметил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 9, 35 мг, 0,108 ммоль), (3,4-диметоксифенил)бороновой кислоты (29 мг, 0,159 ммоль), карбоната калия (75 мг, 0,543 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (8 мг, 0,011 ммоль) в EtOH (2 мл) и толуоле (2 мл) нагревали в микроволновой печи при 100°С в течение 1 часа. Смесь сушили с использованием гидрофобной фритты и упаривали при пониженном давлении с получением желтого масла. Остаток очищали посредством MDAP. Соответствующие фракции объединяли и концентрировали в вакууме с получением беловатой смолы (40,5 мг). Ее растворяли в МеОН и загружали на аминопропиловый картридж (10 г). Продукт элюировали при помощи МеОН и концентрировали в вакууме с получением 7-(3,4-диметоксифенил)-5-метил-2-(пиперидин-1-илметил)фуро[3,2-с]пиридин-4(5Н)-она (24.2 мг, 58%) в виде белой смолы. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,6 мин, МН+=383.
Пример 9: 7-(3,4-диметоксифенил)-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5H)-он
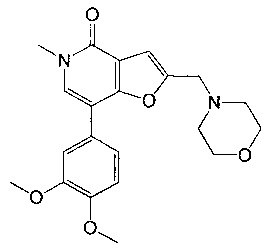
Смесь 7-бром-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 10, 54 мг, 0,165 ммоль), карбоната калия (113 мг, 0,818 ммоль), бис(трифенилфосфин)палладия(II) хлорида (6 мг, 8,55 мкмоль) и (3,4-диметоксифенил)бороновой кислоты (46 мг, 0,253 ммоль) в толуоле (2 мл) и EtOH (2 мл) нагревали в микроволновой печи при 80°С в течение 20 минут. LCMS (2 мин, способ с муравьиной кислотой) показала, что продукт образовался, но осталось большое количество исходного вещества.
Бис(трифенилфосфин)палладия(II) хлорид (8 мг, 0,011 ммоль) добавляли к смеси и реакционную смесь нагревали в микроволновой печи при 80°С в течение 20 минут. LCMS (2 мин, способ с муравьиной кислотой) показала присутствие все еще не прореагировавшего исходного вещества.
Добавляли дополнительные порции (3,4-диметоксифенил)бороновой кислоты (46 мг, 0,253 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (8 мг, 0,011 ммоль) и смесь нагревали в микроволновой печи при 80°С в течение 20 минут. LCMS (2 мин, способ с муравьиной кислотой) показала, что остается непрореагировавшее исходное вещество.
Добавляли дополнительную порцию бис(трифенилфосфин)палладия(II) хлорида (8 мг, 0,011 ммоль) к смеси и все нагревали в микроволновой печи при 80°С в течение 40 минут. LCMS (2 мин, способ с муравьиной кислотой) показала, что остается непрореагировавшее исходное вещество.
Добавляли дополнительную порцию бис(трифенилфосфин)палладия(II) хлорида (8 мг, 0,011 ммоль) к смеси и все нагревали в микроволновой печи при 80°С в течение 30 минут. LCMS (2 мин, способ с муравьиной кислотой) показала, что конечный продукт образовался и остается немного исходного вещества. Реакционную смесь разбавляли этилацетатом (10 мл), сушили (Na2SO4), фильтровали и выпаривали с получением желтого твердого вещества (123 мг). Неочищенное желтое твердое вещество очищали посредством хроматографии на силикагеле, элюируя при помощи градиента 0-10% MeOH/DCM. Соответствующие фракции объединяли и упаривали с получением 7-(3,4-диметоксифенил)-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5Н)-она (8 мг, выход 11%) в виде оранжевого масла.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,55 мин, МН+ 385.
Пример 10: (R)-7-(3,4-диметоксифенил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
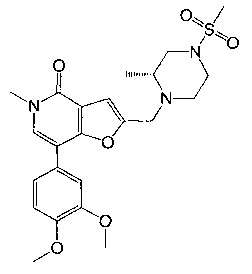
Смесь (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 5, 64 мг, 0,153 ммоль), (3,4-диметоксифенил)бороновой кислоты (57,6 мг, 0,317 ммоль), бис(трифенилфосфин)палладия(II) хлорида (5,15 мг, 7,34 мкмль) и карбоната калия (106 мг, 0,765 ммоль) в толуоле (2 мл) и EtOH (2 мл) нагревали в микроволновой печи при 80°С в течение 20 мин. Смесь разбавляли этилацетатом (10 мл), фильтровали и упаривали в вакууме. Полученный остаток очищали посредством хроматографии на силикагеле, используя градиент 2-4% DCM-MeOH. Соответствующие фракции объединяли и продували в потоке азота с получением (R)-7-(3,4-диметоксифенил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (51 мг, 70%) в виде беловатого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,63 мин, МН+=476.
Пример 11: 2-((1,4-диазепан-1-ил)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-он
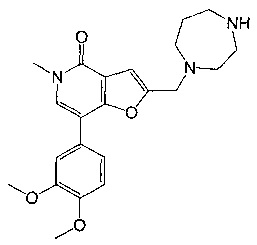
трет-Бутил-4-((7-(3,4-диметоксифенил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилат (относительно получения смотреть промежуточное соединение 12, 50 мг, 0,100 ммоль) растворяли в 1,4-диоксане (1 мл) и добавляли 4 М HCl в 1,4-диоксане (0,5 мл). Реакционную смесь перемешивали в течение 2 часов. Добавляли диэтиловый эфир (10 мл) к осадку продукта. Надосадочную жидкость удаляли. Остаток растирали затем с диэтиловым эфиром (10 мл) и сушили в вакууме с получением 2-((1,4-диазепан-1-ил)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она (30 мг, 75%) в виде белого порошка. LCMS (2 мин, способ с высоким pH): Rt=0,72 мин, МН+ 398.
Пример 12: 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(5-(1-фенилэтокси)пиридин-3-ил)фуро[3,2-с]пиридин-4(5H)-он
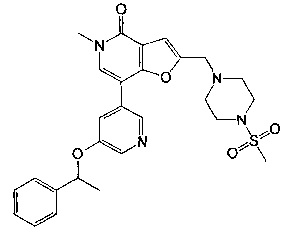
7-Бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 3, 100 мг, 0,247 ммоль), 3-(1-фенилэтокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (относительно получения смотреть промежуточное соединение 28, 80 мг, 0,247 ммоль), карбонат калия (171 мг, 1,237 ммоль) и бис(трифенилфосфин)палладия(II) хлорид (8,68 мг, 0,012 ммоль) растворяли в EtOH (2 мл) и толуоле (2 мл), и нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли этилацетат (20 мл). Смесь сушили над MgSO4, фильтровали и растворитель удаляли в вакууме. Продукт очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали в вакууме с получением 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(5-(1-фенилэтокси)пиридин-3-ил)фуро[3,2-с]пиридин-4(5Н)-она (36 мг, 28%) в виде желтого масла. LCMS (2 мин, способ с высоким pH): Rt=0,97 мин, МН+ 523.
Пример 13: 2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
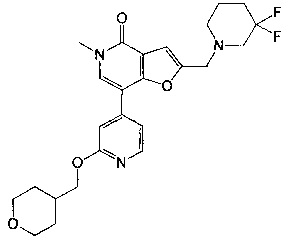
7-Бром-2-((3,3-дифторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 13, 75 мг, 0,208 ммоль), 2-((тетрагидро-2H-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (BoroPharm Inc) (99 мг, 0,311 ммоль), карбонат калия (143 мг, 1,038 ммоль) и бис(трифенилфосфин)палладия(II) хлорид (7,29 мг, 10,38 мкмоль) растворяли в ЕtOН (2 мл) и толуоле (2 мл), и нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли этилацетат (15 мл). Смесь сушили над MgSO4, фильтровали и растворитель выпаривали в вакууме. Остаток очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали в вакууме с получением 2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она (62 мг, 59%) в виде оранжевого масла.
LCMS (2 мин, способ с высоким pH): Rt=1,02 мин, МН+ 474.
Пример 14: 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((1-фенилэтил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
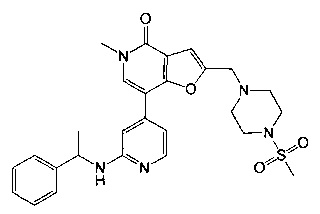
7-Бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он (относительно получения смотреть промежуточное соединение 3, 100 мг, 0,247 ммоль), N-(1-фенилэтил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин (относительно получения смотреть промежуточное соединение 26, 292 мг, 0,901 ммоль), карбонат калия (171 мг, 1,237 ммоль) и бис(трифенилфосфин)палладия(М) хлорид (8,68 мг, 0,012 ммоль) растворяли в EtOH (5 мл) и толуоле (5 мл), и нагревали в микроволновой печи в течение 20 мин при 120°С. Дополнительную порцию бис(трифенилфосфин)палладия(II) хлорида (16 мг, 0,024 ммоль) добавляли к реакционной смеси и нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли этилацетат (20 мл) и смесь сушили над MgSO4, фильтровали и растворитель выпаривали в вакууме. Остаток очищали посредством хроматографии на силикагеле, используя 0-10% градиент MeOH/DCM. Соответствующие фракции объединяли и упаривали в вакууме с получением коричневого масла. Неочищенное масло затем очищали посредством MDAP. Соответствующие фракции объединяли и упаривали с получением 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((1-фенилэтил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она (16 мг, 12%) в виде ярко-оранжевого порошка. LCMS (2 мин, способ с высоким pH): Rt=0,94 мин, МН+ 521.
Пример 15: 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((1-Фенилэтил)амино)фенил)фуро[3,2-с]пиридин-4(5H)-он
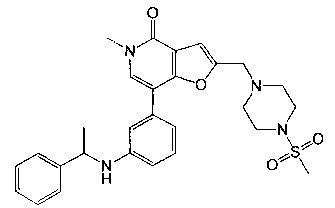
7-Бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 3, 101 мг, 0,.250 ммоль), бис(трифенилфосфин)палладия(II) хлорид (8,42 мг, 0,012 ммоль) и карбонат калия (173 мг, 1,249 ммоль) помещали во флакон для микроволновой печи. В него добавляли N-(1-фенилэтил)-3-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)анилин (относительно получения смотреть промежуточное соединение 30, 323 мг, 1 ммоль) в EtOH (5 мл) и толуоле (5 мл). Реакционный сосуд закрывали герметично и нагревали в микроволновой печи при 80°С в течение 20 минут. LCMS (2 мин, способ с муравьиной кислотой): показала отсутствие взаимодействия. Добавляли дополнительную порцию бис(трифенилфосфин)палладия(II) хлорида (8,42 мг, 0,012 ммоль) и реакционную смесь нагревали в микроволновой печи при 80°С в течение 20 минут. LCMS (2 мин, способ с муравьиной кислотой) показала отсутствие взаимодействия. Газ азот барботировали через реакционную смесь и добавляли дополнительную порцию бис(трифенилфосфин)палладия(II) хлорида (19 мг). Реакционную смесь нагревали в микроволновой печи при 90°С в течение 20 минут. LCMS (2 мин, способ с муравьиной кислотой) показала отсутствие взаимодействия. Реакционную смесь нагревали дополнительно в микроволновой печи при 120°С в течение 20 минут. LCMS (2 мин, способ с муравьиной кислотой) показала, что имело место отсутствие взаимодействия.
Реакционную смесь фильтровали через картридж с целитом и упаривали в вакууме. Остаток растворяли в толуоле (2 мл) и EtOH (2 мл), и добавляли карбонат калия (186 мг) и бис(трифенилфосфин)палладия(II) хлорид (8,7 мг). Реакционный сосуд закрывали герметично и нагревали в микроволновой печи при 80°С в течение 20 минут. Смесь разбавляли этилацетатом (10 мл), сушили при помощи сульфата натрия, фильтровали и упаривали в вакууме.
Остаток очищали посредством хроматографии на силикагеле (25 г), элюируя при помощи градиента 0%-10% MeOH/DCM. Соответствующие фракции объединяли и упаривали. Полученный загрязненный остаток повторно очищали посредством MDAP с получением 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((1-фенилэтил)амино)фенил)фуро[3,2-с]пиридин-4(5Н)-она (11,4 мг, 9%).
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,89 мин, МН+ 522.
Пример 16: 7-(3,4-диметоксифенил)-5-метил-2-((3-метилморфолино)метил)фуро[3,2-с]пиридин-4(5Н)-он
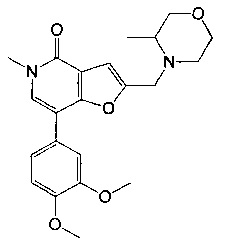
Смесь 7-бром-5-метил-2-((3-метилморфолино)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 14, 22 мг, 0,064 ммоль), (3,4-диметоксифенил)бороновой кислоты (17,60 мг, 0,097 ммоль), карбоната калия (44,6 мг, 0,322 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (4 мг, 5,70 мкмоль) в толуоле (1 мл) и EtOH (1 мл) нагревали в микроволновой печи при 80°С в течение 20 минут. Охлажденную реакционную смесь разбавляли этилацетатом (10 мл), сушили и упаривали. Остаток очищали посредством MDAP. Соответствующие фракции объединяли и упаривали. Остаток загружали на колонку SCX 5 g и элюировали при помощи МеОН (10 мл) с последующим элюированием посредством 2М MeOH/NH3 (10 мл). Основную фракцию упаривали с получением 7-(3,4-диметоксифенил)-5-метил-2-((3-метилморфолино)метил)фуро[3,2-с]пиридин-4(5Н)-она (8 мг, 26%) в виде твердого вещества кремового цвета. LCMS (2 мин, способ с высоким pH): Rt=0,82 мин, МН+ 399.
Пример 17: N-(4-(2-((3-фторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
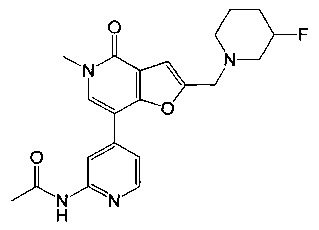
7-Бром-2-((3-фторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 15, 82,5 мг, 0,240 ммоль), N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамид (Milestone Pharm tech USA) (95 мг, 0,361 ммоль), карбонат калия (166 мг, 1,202 ммоль) и бис(трифенилфосфин)палладия(II) хлорид (8,44 мг, 0,012 ммоль) растворяли в EtOH (2 мл) и толуоле (2 мл), и нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли этилацетат (15 мл) и смесь сушили над MgSO4, фильтровали и упаривали. Остаток очищали посредством хроматографии на силикагеле, используя градиент 0-10% MeOH/DCM. Соответствующие фракции объединяли и упаривали в вакууме с получением N-(4-(2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (55 мг, 64%) в виде ярко-желтого твердого вещества LCMS (2 мин, способ с высоким pH): Rt=0,73 мин, МН+ 399.
Пример 18: N-(4-(2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
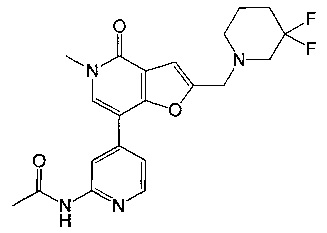
7-Бром-2-((3,3-дифторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 13, 75 мг, 0,208 ммоль), N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамид (Milestone Pharm tech USA) (82 мг, 0,311 ммоль), карбонат калия (143 мг, 1,038 ммоль) и бис(трифенилфосфин)палладия(П) хлорид (7,29 мг, 10,38 мкмоль) растворяли в EtOH (2 мл) и толуоле (2 мл) и нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли этилацетат (15 мл) и смесь сушили над MgSO4, фильтровали и упаривали. Остаток очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали в вакууме с получением N-(4-(2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамит (55 мг, 64%) в виде полупрозрачного масла. LCMS (2 мин, способ с высоким pH): Rt=0,78 мин, МН+ 417.
Пример 19: 2-((3-фторпиперидин-1-ил)метил)-7-(4-метоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-он гидрохлорид
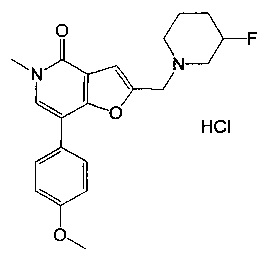
7-Бром-2-((3-фторпиперидин-1-ил)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 15, 82,5 мг, 0,240 ммоль), (4-метоксифенил)бороновой кислоты (54,8 мг, 0,361 ммоль), карбонат калия (166 мг, 1,202 ммоль) и бис(трифенилфосфин)палладия(II) хлорид (8,44 мг, 0,012 ммоль) растворяли в EtOH (2 мл) и толуоле (2 мл), и нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли дополнительные порции (4-метоксифенил)бороновой кислоты (36,5 мг) и бис(трифенилфосфин)палладия(II) хлорида (8,44 мг, 0,012 ммоль), и реакционную смесь нагревали в микроволновой печи в течение 20 мин при 120°С. Добавляли этилацетат (15 мл) и смесь сушили над MgSO4, фильтровали и упаривали. Остаток очищали посредством хроматографии на силикагеле, используя градиент 0-5% MeOH/DCM. Соответствующие фракции объединяли и упаривали, и затем очищали посредством MDAP. Соответствующие фракции объединяли и упаривали с получением коричневой смолы. Эту смолу растирали с диэтиловым эфиром (10 мл) и добавляли HCl в эфире (0,5 мл) для осаждения продукта. Надосадочную жидкость удаляли. Остаток растирали с диэтиловым эфиром и сушили с получением 2-((3-фторпиперидин-1-ил)метил)-7-(4-метоксифенил)-5-метилфуро[3,2-с]пиридин-4(5Н)-он гидрохлорида (54 мг, 0,133 ммоль, выход 55,2%) в виде белого твердого вещества.
LCMS (2 мин, способ с высоким pH): Rt=1,0 мин, МН+ 371.
Пример 20: 5-метил-2-(морфолинометил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
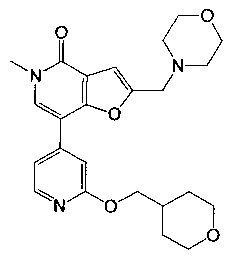
Смесь 7-бром-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5H)-она (относительно получения смотреть промежуточное соединение 10, 80 мг, 0,24 ммоль), 2-((тетрагидро-2Н-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (BoroPharm Inc) (156 мг, 0,49 ммоль), карбоната калия (169 мг, 1,22 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (17 мг, 10% мол.) в толуоле (2 мл) и EtOH (2 мл) нагревали в микроволновой печи при 130°С в течение 45 минут. Охлажденную реакционную смесь разбавляли этилацетатом (25 мл), сушили над сульфатом натрия и фильтровали. Растворитель выпаривали из фильтрата и остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 2% MeOH/DCM. Соответствующие фракции объединяли и упаривали. Остаток растирали с диэтиловым эфиром с получением 5-метил-2-(морфолинометил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она (71 мг, 66%) в виде коричневого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,6 мин, МН+ 440.
Пример 21: 5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он формиат
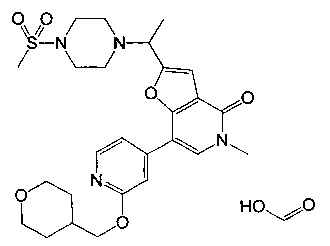
Раствор 5-йод-4-метокси-1-метил-2'-((тетрагидро-2Н-пиран-4-ил)метокси)-[3,4'-бипиридин]-6(1Н)-она (относительно получения смотреть промежуточное соединение 19, 90 мг, 0,197 ммоль) в ацетонитриле (4 мл) обрабатывали при помощи 1-(бут-3-ин-2-ил)-4-(метилсульфонил)пиперазина (относительно получения смотреть промежуточное соединение 20, 68 мг, 0,236 ммоль), йодида меди(I) (4 мг, 0,021 ммоль), бис(трифенилфосфин)палладия(II) дихлорида (7 мг, 9,97 мкмоль) и триэтиламина (4 мл, 28,7 ммоль). Реакционную смесь нагревали при 80°С в атмосфере азота в течение 4 суток, к этому времени смесь высыхала досуха с образованием коричневой смолы, которая отвердевала при охлаждении до комнатной температуры.
Реакционную смесь распределяли между EtOAc (30 мл) и водой (30 мл). Водный слой повторно экстрагировали при помощи EtOAc (30 мл × 2). Объединенные органические слои пропускали через гидрофобную фритту и концентрировали при пониженном давлении. Остаток очищали посредством MDAP. Фракцию, содержащую продукт, концентрировали при пониженном давлении с получением 5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он формиата (6 мг, 6%) в виде прозрачной желтой смолы.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,67 мин, МН+ 531.
Примеры 21а и Пример 21b: (S)-5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он и (R)-5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он;
Смесь 5-йод-4-метокси-1-метил-2'-((тетрагидро-2Н-пиран-4-ил)метокси)-[3,4'-бипиридин]-6(1Н)-она (относительно получения смотреть промежуточное соединение 19, 250 мг, 0,548 ммоль), 1-(бут-3-ин-2-ил)-4-(метилсульфонил)пиперазина (относительно получения смотреть промежуточное соединение 20, 356 мг, 1,644 ммоль), йодида меди(I) (25,04 мг, 0,131 ммоль) и бис(трифенилфосфин)палладия(II) дихлорида (15,00 мг, 0,021 ммоль) в DMF (0,5 мл) нагревали при 120°С в течение 6 часов, используя микроволновую печь.
Взаимодействие повторяли, как указано выше, со второй партией реагентов. Две реакционные смеси объединяли и концентрировали при пониженном давлении и очищали посредством колоночной хроматографии на силикагеле (100 г), элюируя при помощи 0-1% 2М аммиака в МеОН/EtOAc с последующим элюированием посредством 0-10% 2 М аммиака в МеОН/EtOAc. Целевой продукт обнаружили при элюировании в последних нескольких фракциях второго градиента элюирования. Соответствующие фракции объединяли и концентрировали при пониженном давлении с получением неочищенного продукта (порядка 260 мг). Его очищали посредством MDAP с получением 5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она (60 мг, 10%) в виде бледно-желтой смолы.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,67 мин, МН+ 531.
Это вещество разделяли на два составляющих его энантиомера посредством препаративной хиральной HPLC.
Приблизительно 60 мг рацемата растворяли в смеси 1 мл EtOH и 2 мл гептана. Порции по 1 мл раствора вводили в колонку Chiralpak AD-H 30 мм × 25 см. Колонку элюировали при помощи 50% EtOH/гептан, скорость потока = 30 мл/мин, длины волны 215 нм. Соответствующие фракции объединяли и упаривали с получением двух энантиомеров:
Пример 21а: 20 мг, белое твердое вещество. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,67 мин, МН+ 531. Энантиомерная чистота при хиральной HPLC = >99% э.ед.
Пример 216: 19 мг, белое твердое вещество. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,67 мин, МН+ 531. Энантиомерная чистота при хиральной HPLC = 98% э.ед.
Абсолютную стереохимию не устанавливали.
Пример 22: 2-((1,4-оксазепан-4-ил)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
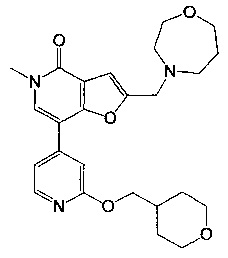
Смесь 2-((1,4-оксазепан-4-ил)метил)-7-бром-5-метилфуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 16, 40 мг, 0,12 ммоль), 2-((тетрагидро-2Н-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (BoroPharm Inc) (75 мг, 0,24 ммоль), карбоната калия (81 мг, 0,57 ммоль) и бис(трифенилфосфин)палладия(II) хлорида (8 мг, 10% мол.) в толуоле (2 мл) и EtOH (2 мл) нагревали в микроволновой печи при 130°С в течение 45 минут. Охлажденную реакционную смесь разбавляли этилацетатом (25 мл). Смесь сушили над сульфатом натрия и фильтровали. Растворитель выпаривали из фильтрата и остаток очищали посредством хроматографии на силикагеле, элюируя при помощи 2% MeOH/DCM с получением неочищенного продукта. Его повторно очищали посредством MDAP с получением 2-((1,4-оксазепан-4-ил)метил)-5-метил-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она (28 мг, 53%) в виде бесцветного стекла.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,6 мин, МН+ 454.
Пример 23: (R)-7-(2-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он
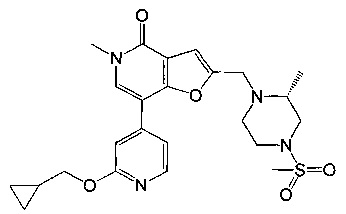
К перемешиваемой суспензии 2-(циклопропилметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (относительно получения смотреть промежуточное соединение 32, 438 мг, 0,478 ммоль) в 1,2-DME (4 мл), добавляли карбонат калия (99 мг, 0,717 ммоль), (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он (относительно получения смотреть промежуточное соединение 5, 100 мг, 0,239 ммоль) и тетракис(трифенилфосфин)палладий(0) (11 мг, 9,52 мкмоль). Это содержимое закрывали герметично во флаконе для микроволновой печи и нагревали при 120°С в течение 2 ч. Смесь растворяли в этилацетате (20 мл) и воде (20 мл), и слои разделяли. Водный слой дополнительно экстрагировали при помощи этилацетата (3×20 мл). Растворитель концентрировали в вакууме с получением коричневого масла. Его очищали посредством MDAP. Соответствующие фракции объединяли и концентрировали в вакууме с получением (R)-7-(2-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (17 мг, 15%) в виде коричневого масла.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,74 мин, МН+ 487.
Пример 24: (R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)циклопропанкарбоксамид
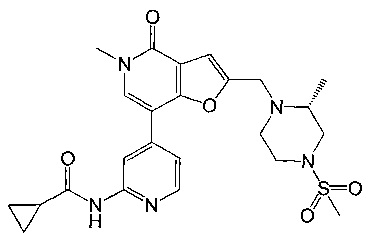
N-(4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)циклопропанкарбоксамид (относительно получения смотреть промежуточное соединение 34, 85 мг, 0,294 ммоль), (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он (относительно получения смотреть промежуточное соединение 5, 100 мг, 0,239 ммоль), тетракис(трифенилфосфин)палладий(0) (13,81 мг, 0,012 ммоль) и водный раствор карбоната натрия (0,956 мл, 1,912 ммоль) смешивали в 1,2-DME (3 мл) и нагревали в микроволновой печи в течение 2 часов при 120°С. Реакционную смесь разбавляли этилацетатом и водой и фильтровали через хлопковую вату. Органический слой отделяли и концентрировали в вакууме. Неочищенное вещество очищали посредством MDAP с получением (R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)циклопропанкарбоксамида (80 мг, 67%) в виде коричневого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,56 мин, МН+=500.
Пример 25: (R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)пропионамид
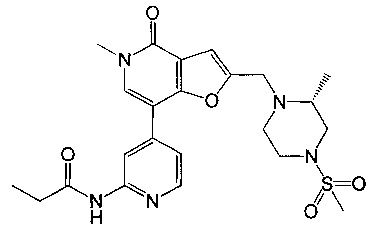
N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пропионамид (относительно получения смотреть промежуточное соединение 36, 80 мг, 0,291 ммоль), (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 5, 100 мг, 0,239 ммоль), тетракис(трифенилфосфин)палладий(0) (13,81 мг, 0,012 ммоль) и водный раствор карбоната натрия (0,956 мл, 1,912 ммоль) смешивали в 1,2-DME (2 мл) и нагревали в микроволновой печи в течение 2 часов при 120°С. Реакционную смесь разбавляли этилацетатом и водой и фильтровали. Органический слой отделяли и концентрировали в вакууме. Неочищенный продукт очищали посредством MDAP с получением (R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)пропионамида (28 мг, 24%) в виде коричневого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,53 мин, МН+=488.
Пример 26: (R)-7-(2-(2-метоксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он
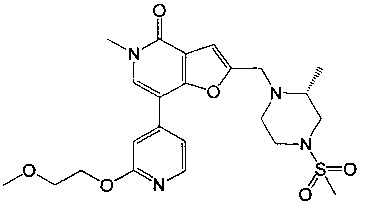
2-(2-Метоксиэтокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (относительно получения смотреть промежуточное соединение 38, 81 мг, 0,289 ммоль), (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 5, 100 мг, 0,239 ммоль), тетракис(трифенилфосфин)палладий(0) (13,81 мг, 0,012 ммоль) и карбонат натрия (2 М) (0,956 мл, 1,912 ммоль) смешивали в 1,2-DME (3 мл) и нагревали в микроволновой печи в течение 2 часов при 120°С. Реакционную смесь разбавляли этилацетатом и водой и фильтровали через хлопковую вату. Органический слой отделяли и концентрировали в вакууме. Неочищенный продукт очищали посредством MDAP с получением (R)-7-(2-(2-метоксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (36 мг, 31%) в виде коричневого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,61 мин, МН+=491.
Пример 27: (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(пирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он
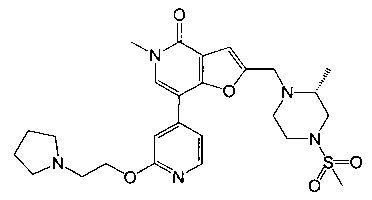
2-(2-(Пирролидин-1-ил)этокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (относительно получения смотреть промежуточное соединение 40, 70,3 мг, 0,221 ммоль), (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он (относительно получения смотреть промежуточное соединение 5, 80 мг, 0,191 ммоль), тетракис(трифенилфосфин)палладий(0) (11,05 мг, 9,56 мкмоль) и карбонат натрия (2 М) (0,765 мл, 1,530 ммоль) смешивали в 1,2-DME (2 мл) и нагревали в микроволновой печи в течение 2 часов при 120°С. Реакционную смесь разбавляли этилацетатом и водой и фильтровали через хлопковую вату. Органический слой отделяли и концентрировали в вакууме. Неочищенный продукт очищали посредством MDAP с получением (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(пирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она (36 мг, 36%) в виде коричневого твердого вещества. LCMS (2 мин, муравьиная кислота): Rt=0,47 мин, МН+=530.
Пример 28: 2-((1,1-диоксидотиоморфолино)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он
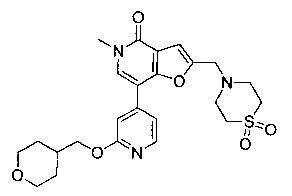
Смесь 7-бром-2-((1,1-диоксидотиоморфолино)метил)-5-метилфуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 41, 200 мг, 0,533 ммоль), 2-((тетрагидро-2Н-пиран-4-ил)метокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (BoroPharm Inc) (204 мг, 0,640 ммоль) и K2CO3 (147 мг, 1,066 ммоль) в 1,4-диоксане (100 мл) дегазировали при помощи N2 и затем добавляли тетракис(трифенилфосфин)палладий(0) (61,6 мг, 0,053 ммоль). Полученную реакционную смесь нагревали до 100°С в течение 2 ч. Остатки распределяли между EtOAc (100 мл) и водой (100 мл). Водную фазу повторно экстрагировали при помощи EtOAc (100 мл). Объединенные органические вещества промывали рассолом (200 мл), сушили при помощи Na2SO4, фильтровали и концентрировали с получением неочищенного твердого вещества. Неочищенный продукт очищали посредством колоночной хроматографии на силикагеле, элюируя при помощи DCM/MeOH с получением целевого продукта 2-((1,1-диоксидотиоморфолино)метил)-5-метил-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она (125 мг, 0,256 ммоль, выход 48,1%).
LCMS: МН+ 488.
Пример 29: 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[2,3-с]пиридазин-4(5H)-он
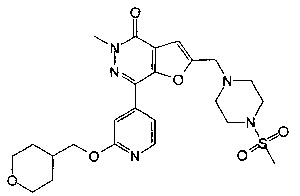
К 5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[2,3-d]пиридазин-4(5H)-ону (относительно получения смотреть промежуточное соединение 44, 45 мг, 0,085 ммоль) добавляли комплекс боран-THF (5 мл, 5,00 ммоль, 1 М раствор в THF), и смесь перемешивали в течение ночи в атмосфере азота при комнатной температуре. Добавляли метанол и удаляли растворитель. Остаток очищали посредством MDAP. Соответствующие фракции объединяли и удаляли растворитель с получением 5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2Н-пиран-4-ил)метокси)пиридин-4-ил)фуро[2,3-d]пиридазин-4(5Н)-она (2,0 мг, 5%) в виде белого твердого вещества
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,78 мин, МН+ 518.
Пример 30: (R)-N-метил-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
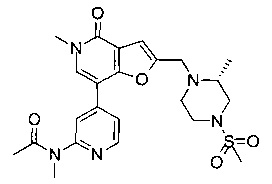
(R)-7-Бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 5, 95 мг, 0,227 ммоль), N-метил-N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамид (относительно получения смотреть промежуточное соединение 47, 71,2 мг, 0,258 ммоль), тетракис(трифенилфосфин)палладий(0) (13,12 мг, 0,011 ммоль) и водный раствор карбоната натрия (2 М) (0,908 мл, 1,817 ммоль) в 1,2-DME (3 мл) нагревали в течение 2 часов при 120°С в микроволновой печи. Реакционную смесь разбавляли этилацетатом и водой и фильтровали через хлопковую вату. Органический слой отделяли и концентрировали в вакууме. Неочищенное вещество очищали посредством MDAP и фракции, содержащие продукт, концентрировали в вакууме. Неочищенный продукт дополнительно очищали посредством MDAP и концентрировали в вакууме. Полученный продукт растворяли в МеОН и элюировали через аминопропиловый картридж (1 г). Растворитель выпаривали с получением (R)-N-метил-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (34 мг, 0,070 ммоль, выход 30,7%) в виде белого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,51 мин, MH+ 488.
Пример 31: 7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
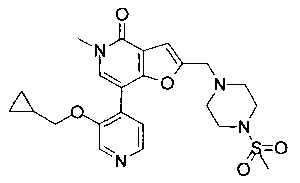
7-Бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он (относительно получения смотреть промежуточное соединение 3, 90 мг, 0,223 ммоль), 3-(циклопропилметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (относительно получения смотреть промежуточное соединение 49, 73.2 мг, 0,266 ммоль), тетракис(трифенилфосфин)палладий(0) (12,86 мг, 0,011 ммоль) и водный раствор карбоната натрия (2 М) (0,890 мл, 1,781 ммоль) в 1,2-DME (3 мл) нагревали в течение 2 часов при 120°С в микроволновой печи. Реакционную смесь разбавляли этилацетатом и водой и фильтровали через хлопковую вату. Органический слой отделяли и концентрировали в вакууме. Неочищенное вещество очищали посредством MDAP и фракции, содержащие продукт, объединяли и концентрировали в вакууме. Неочищенный продукт дополнительно очищали посредством MDAP и фракции, содержащие продукт, объединяли и концентрировали в вакууме. Полученный продукт растворяли в МеОН и элюировали через аминопропиловый картридж (1 г) при помощи МеОН. Растворитель выпаривали с получением 7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (49 мг, 0,104 ммоль, выход 46,6%) в виде белого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,53 мин, МН+ 473.
Пример 32: (R)-7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
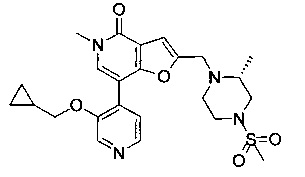
(R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 5, 95 мг, 0,227 ммоль), 3-(циклопропилметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин (относительно получения смотреть промежуточное соединение 49, 73,2 мг, 0,266 ммоль), тетракис(трифенилфосфин)палладий(0) (13,12 мг, 0,011 ммоль) и водный раствор карбоната натрия (2 М) (0,908 мл, 1,817 ммоль) в 1,2-DME (3 мл) нагревали в течение 2 часов при 120°С в микроволновой печи. Реакционную смесь разбавляли этилацетатом и водой и фильтровали через хлопковую вату. Органический слой отделяли и концентрировали в вакууме. Неочищенное вещество очищали посредством MDAP и фракции, содержащие продукт, концентрировали в вакууме. Полученный продукт растворяли в МеОН и элюировали через аминопропиловый картридж (5 г) при помощи МеОН. Растворитель выпаривали с получением (R)-7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (54 мг, 0,111 ммоль, выход 48,9%) в виде белого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,55 мин, МН+ 487.
Пример 33: 7-(3,4-диметоксифенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[2,3-d]пиридазин-4(5H)-он
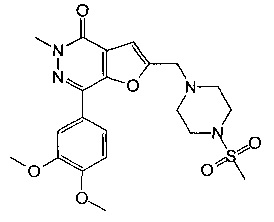
К 7-(3,4-диметоксифенил)-5-метил-2-(4-(метилсульфонил)пиперазин-1-карбонил)фуро[2,3-d]пиридазин-4(5H)-ону (относительно получения смотреть промежуточное соединение 52, 0,025 мл, 0,038 ммоль) добавляли комплекс боран-THF (4 мл, 4,00 ммоль, 1 М раствор в THF) и смесь перемешивали в течение ночи в атмосфере азота при комнатной температуре. Добавляли метанол и растворитель удаляли. Остаток очищали посредством MDAP. Соответствующие фракции объединяли и удаляли растворитель. Остаток сушили в глубоком вакууме в течение 2 часов с получением 7-(3,4-диметоксифенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[2,3-d]пиридазин-4(5Н)-она (10 мг, 57%) в виде мелкозернистого белого твердого вещества. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,7 мин, МН+ 463.
Пример 34: 2-((1,4-диазепан-1-ил)метил)-5-метил-7-(2-((пиридин-2-илметил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он гидрохлорид
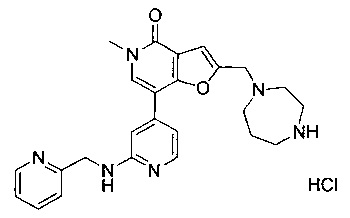
К раствору трет-бутил-4-((5-метил-4-оксо-7-(2-((пиридин-2-илметил)амино)пиридин-4-ил)-4,5-дигидрофуро[3,2-с]пиридин-2-ил)метил)-1,4-диазепан-1-карбоксилата (относительно получения смотреть промежуточное соединение 56, 130 мг, 0,239 ммоль) в DCM (5 мл) добавляли TFA (0,919 мл, 11,93 ммоль). Смесь перемешивали при 25°С в течение 2 ч. Смесь концентрировали в вакууме. Остаток объединяли с другой партией неочищенного продукта (120 мг), который получали, применяя те же самые условия. Неочищенное вещество концентрировали в вакууме и очищали посредством MDAP, 1 М HCl (0,5 мл) добавляли к фракции, содержащей продукт. Соответствующие фракции объединяли и упаривали с получением 2-((1,4-диазепан-1-ил)метил)-5-метил-7-(2-((пиридин-2-илметил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она гидрохлорида (100 мг, 0,225 ммоль, выход 39% от объединенных реакционных смесей) в виде белого твердого вещества. LCMS: МН+ 445.
Пример 35: (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(2-оксопирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
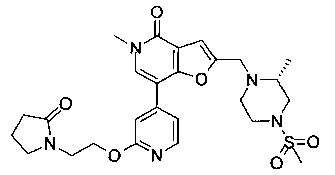
1-(2-((4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)оксиэтил)пирролидин-2-он (относительно получения смотреть промежуточное соединение 58, 376 мг, 42% масс./масс., 0,475 ммоль), (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 5, 100 мг, 0,239 ммоль), тетракис(трифенилфосфин)палладий(0) (13,81 мг, 0,012 ммоль) и карбонат калия (99 мг, 0,717 ммоль) растворяли в 1,2-DME (3 мл) во флаконе для микроволновой печи. Смесь нагревали до 120°С в течение 2 часов в микроволновом реакторе. Смесь разбавляли этилацетатом (20 мл) и водой (20 мл). Слои разделяли и водную фазу экстрагировали при помощи этилацетата (3×20 мл). Объединенные органические вещества сушили с использованием гидрофобной фритты и концентрировали в вакууме. Остаток очищали посредством MDAP. Соответствующие фракции объединяли и отгоняли в вакууме. Остаток растворяли в метаноле и загружали на предварительно обработанный (при помощи метанола) аминопропиловый картридж (1 г) и элюировали при помощи метанола. Соответствующие фракции объединяли и уменьшали по объему в вакууме с получением (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(2-оксопирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она (11 мг, 8,46%) в виде желтого масла. LCMS (2 мин, способ с высоким pH): Rt=0,73 мин, МН+ 544.
Пример 36: N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид, соль муравьиной кислоты
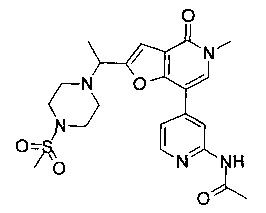
Смесь 1-(бут-3-ин-2-ил)-4-(метилсульфонил)пиперазина (относительно получения смотреть промежуточное соединение 20, 130 мг, 0,601 ммоль), N-(5-йод-4-метокси-1-метил-6-оксо-1,6-дигидро-[3,4'-бипиридин]-2'-ил)ацетамида (относительно получения смотреть промежуточное соединение 59, 80 мг, 0,200 ммоль), йодида меди(1) (8 мг, 0,042 ммоль), триэтиламина (0,999 мл, 7,17 ммоль) и бис(трифенилфосфин)палладия(II) дихлорида (14,07 мг, 0,020 ммоль) в DMF (0.333 мл) нагревали при 120°С в течение 6 часов, используя микроволновую печь. Реакционную смесь концентрировали при пониженном давлении и очищали посредством MDAP. Фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением желтой смолы. Загрязненное вещество повторно очищали посредством MDAP. Фракцию, содержащую целевой продукт, концентрировали при пониженном давлении с получением соли муравьиной кислоты N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (3 мг, 2,9%) в виде желтой смолы. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,48 мин, МН+ 474.
Пример 36а и 36b: (R)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид и (S)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
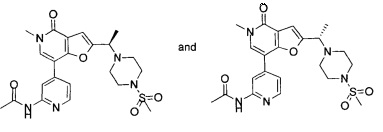
Образец N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (пример 36) очищали посредством хиральной колоночной хроматографии, используя колонку Chiralpak ID 250 мм × 30 мм, 5 микрон. Образец (39 мг) растворяли в смеси 3/7 этанол/метанол (10 мл) и партии по 2-2,5 мл вводили в колонку. Образец элюировали, используя 0,2% об./об. изопропиламин в метаноле при скорости потока 55 мл/мин с УФ-детектором DAD (280 нм, ширина полосы частот 140 нм, эталон 400 нм (ширина полосы частот 20 нм)). Соответствующие фракции объединяли и концентрировали с получением двух энантиомеров:
(R)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида и (S)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида. Абсолютную стереохимию не устанавливали для каждого образца.
Первый изомер элюирования (18 мг), 99,8% одиночного энантиомера, полученного посредством хиральной аналитической HPLC. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,49 мин, МН+ 474.
Второй изомер элюирования (19 мг), 99,6% одиночного энантиомера, полученного посредством хиральной аналитической HPLC. LCMS (2 мин, муравьиная кислота): Rt=0,48 мин, МН+ 474.
Аналитической способ: колонка_Chiralpak ID3 50 мм × 4,6 мм, 3 микрона. Подвижная фаза: 0,2% об./об. изопропиламина в метаноле при скорости потока 1 мл/мин с УФ-детектором DAD (280 нм, ширина полосы частот 140 нм, для референсного образца 400 нм (ширина полосы частот 20 нм)).
Пример 37: N-(4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
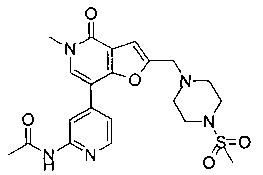
К перемешиваемой суспензии 7-бром-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 3, 100 мг, 0,247 ммоль), N-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)ацетамида (Milestone Pharma Tech) (104 мг, 0,396 ммоль), карбоната калия (103 мг, 0,742 ммоль) в 1,2-DME (3 мл) во флаконе для микроволновой печи добавляли тетракис(трифенилфосфин)палладий(0) (14,29 мг, 0,012 ммоль). Флакон для микроволновой печи закрывали герметично и нагревали в микроволновой печи при 120°С в течение 2 ч. Смесь растворяли в метаноле и концентрировали в вакууме. Остаток повторно растворяли в метаноле и загружали на картридж SCX (1 г) и элюировали при помощи 2 М аммиака в метаноле. Соответствующие фракции объединяли и концентрировали в вакууме. Остаток очищали посредством MDAP. Соответствующие фракции объединяли и концентрировали в вакууме с получением N-(4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (45 мг, 39,6%) в виде белого твердого вещества.
LCMS (2 мин, способ с высоким pH): Rt=0,64 мин, МН+ 460.
Пример 38: (R)-N-(5-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)ацетамид
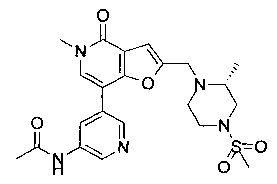
К перемешиваемой суспензии (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (относительно получения смотреть промежуточное соединение 5, 100 мг, 0,239 ммоль), N-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)ацетамида (относительно получения смотреть промежуточное соединение 61, 258 мг, 39% масс./масс., 0,382 ммоль), карбоната калия (99 мг, 0,717 ммоль) в 1,2-DME (3 мл) во флаконе для микроволновой печи добавляли тетракис(трифенилфосфин)палладий(0) (13,81 мг, 0,012 ммоль). Флакон для микроволновой печи закрывали герметично и нагревали в микроволновой печи при 120°С в течение 2 ч. Смесь растворяли в этилацетате и фильтровали через картридж с целитом. Растворитель выпаривали при пониженном давлении, и остаток очищали посредством MDAP. Соответствующие фракции объединяли и концентрировали в вакууме. Остаток растворяли в метаноле и элюировали через аминопропиловый картридж (500 мг). Соответствующие фракции отгоняли в вакууме. Остаток объединяли с другой партией неочищенного продукта, который получали, используя те же самые условия. Неочищенное вещество очищали посредством MDAP. Соответствующие фракции объединяли и концентрировали в вакууме с получением (R)-N-(5-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)ацетамида (5,9 мг, выход 2,6% от объединенных реакционных смесей) в виде белой смолы. LCMS (2 мин, способ с высоким pH): Rt=0,62 мин, МН+ 474.
Пример 39: 7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
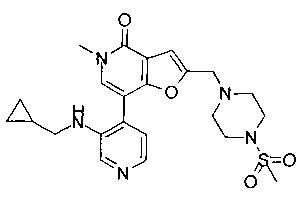
7-(3-Аминопиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он (относительно получения смотреть промежуточное соединение 63, 72 мг, 0,138 ммоль) добавляли к суспензии циклопропанкарбальдегида (10,31 мкл, 0.138 ммоль) в смеси метанола (9 мл) и уксусной кислоты (1 мл) и перемешивали в течение 30 мин при КТ. Добавляли комплекс 2-пиколин-боран (22,14 мг, 0,207 ммоль) и смесь оставляли перемешиваться при комнатной температуре в течение ночи. Растворитель выпаривали и добавляли насыщенный водный раствор натрия бикарбоната (10 мл). Продукт извлекали при помощи DCM (2×10 мл), фильтровали через гидрофобную фритту и концентрировали в вакууме. Остаток очищали посредством MDAP с получением коричневого твердого вещества. Его повторно очищали посредством MDAP с получением 7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (21 мг, 0,045 ммоль, выход 32,3%) в виде белого твердого вещества. LCMS (2 мин, способ с высоким pH): Rt=0,76 мин, МН+ 472.
Пример 40: (R)-7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он
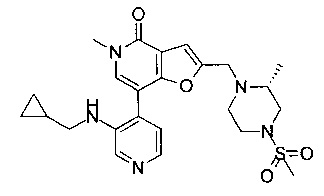
(R)-7-(3-Аминопиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 65, 81 мг, 0,160 ммоль) добавляли к суспензии циклопропанкарбальдегида (0,012 мл, 0,160 ммоль) в смеси метанола (9 мл) и уксусной кислоты (1 мл) и перемешивали в течение 30 мин при КТ. Добавляли комплекс 2-пиколин-боран (25,6 мг, 0,239 ммоль) и смесь оставляли перемешиваться при комнатной температуре в течение ночи. Растворитель выпаривали и добавляли насыщенный водный раствор бикарбоната натрия (20 мл). Продукт экстрагировали при помощи DCM (2×10 мл), фильтровали через гидрофобную фритту и концентрировали в вакууме. Остаток очищали посредством MDAP. Полученный неочищенный продукт дополнительно очищали посредством MDAP. Фракции, содержащие продукт, концентрировали в вакууме с получением желтого масла. Его растворяли в МеОН и элюировали через аминопропиловый картридж (1 г) при помощи МеОН. Растворитель концентрировали в вакууме с получением (R)-7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (22 мг, 0,045 ммоль, выход 28,4%) в виде белого твердого вещества.
LCMS (2 мин, способ с муравьиной кислотой): Rt=0,48 мин, МН+ 486.
Пример 41: 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он
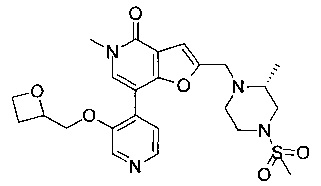
Стадия 1
К суспензии (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (относительно получения смотреть промежуточное соединение 5, 120 мг, 0,287 ммоль), 4,4,5,5-тетраметил-1,3,2-диоксаборолана (0,250 мл, 1,721 ммоль) и триэтиламина (0,160 мл, 1,147 ммоль) в 1,4-диоксане (5 мл) добавляли PEPPSI-SIPr (17 мг, 0,025 ммоль). Смесь нагревали с обратным холодильником при 100°С в течение 3 часов. Реакционную смесь разбавляли при помощи EtOAc и фильтровали через целит, затем концентрировали в вакууме с получением неочищенного (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5H)-она в виде желтого масла (391 мг). Чистоту оценивали в 22,3% и использовали непосредственно на стадии 2 без дополнительной очистки.
Стадия 2
К перемешиваемой суспензии неочищенного (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5H)-она из стадии 1 (391 мг, 22,3% масс./масс., 0,187 ммоль), неочищенного 4-бром-3-(оксетан-2-илметокси)пиридина (относительно получения смотреть промежуточное соединение 67, 80 мг, 59,5% масс./масс., 0,195 ммоль) и 2 М карбоната натрия в воде (0,749 мл, 1,499 ммоль) во флаконе для микроволновой печи с 1,2-DME (3 мл), добавляли тетракис(трифенилфосфин)палладий(0) (12 мг, 10,38 мкмоль). Флакон для микроволновой печи закрывали герметично и помещали в микроволновую печь Biotage initiator и нагревали при 120°С в течение 30 мин. Смесь разбавляли при помощи EtOAc (25 мл) и воды (25 мл). Два слоя разделяли и водную фазу повторно экстрагировали при помощи EtOAc (5×25 мл). Органическую фазу сушили с использованием гидрофобной фритты и концентрировали в вакууме. Остаток растворяли в DMSO (4 мл) и очищали посредством MDAP. Соответствующие фракции объединяли и концентрировали в вакууме с получением желтого масла. Остаток растворяли в МеОН и загружали на аминопропиловый картридж (2 г), который был предварительно обработан при помощи МеОН. Картридж элюировали при помощи МеОН и соответствующие фракции объединяли и концентрировали в вакууме с получением 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она в виде желтого масла (32,2 мг, 34,2%). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,44 мин, МН+ 503.
Пример 42: 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
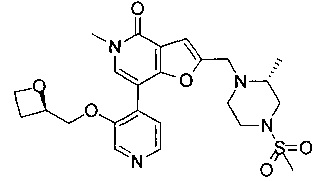
и
Пример 43: 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((S)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
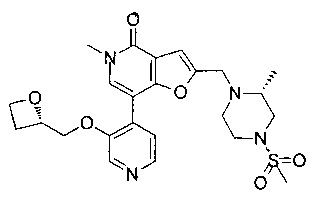
5-Метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он (пример 41) очищали посредством хиральной HPLC, используя колонку Chiralpak IA 30 мм × 25 см. Образец (120 мг) растворяли до 20 мг/мл и партии по 2 мл вводили в колонку. Образец элюировали, используя 40% EtOH/гексан со скоростью 30 мл/мин с детектированием при 215 нм. Соответствующие фракции объединяли и концентрировали с получением:
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-она (пример 42) (32 мг). Хиральная HPLC - чистота 98,8% (колонка 4,6 мм × 25 см Chiralpak IA, 40% EtOH/гептан, скорость потока 1,0 мл/мин, длина волны 215 нм). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,42 мин, МН+ 503.
Также получили:
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((S)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он (пример 43) (36 мг). Хиральная HPLC чистота 98,4% (колонка 4,6 мм × 25 см Chiralpak IA, 40% EtOH/гептан, скорость потока 1,0 мл/мин, длина волны 215 нм). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,42 мин, МН+ 503.
Пример 42: 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он
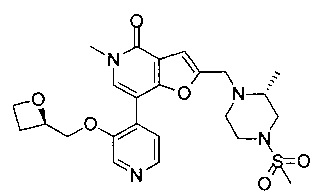
Альтернативное получение
(R)-4-бром-3-(оксетан-2-илметокси)пиридин (относительно получения смотреть промежуточное соединение 69, 19,83 г, 81 ммоль) смешивали с карбонатом цезия (37,8 г, 116 ммоль), толуолом (200 мл) и метанолом (60 мл) и дегазировали барботированием азота через реакционную смесь в течение 20 мин. Добавляли Pd(PPh3)4 (6,70 г, 5,80 ммоль) и (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5Н)-он (относительно получения смотреть промежуточное соединение 5а, 30 г, 58,0 ммоль), и нагревание продолжали в течение 18 часов. Смесь охлаждали и упаривали в вакууме, и остаток распределяли между DCM (300 мл) и водой (500 мл). Водный слой извлекали при помощи DCM (300 мл) и объединенные органические вещества сушили и упаривали в вакууме с получением бледно-желтой пены. Неочищенный продукт загружали на колонку с диоксидом кремния 750 г и элюировали при помощи 10 объемов ацетона, затем при помощи 30% смеси метанол/ацетон. Соответствующие фракции упаривали в вакууме с получением бледно-желтой пены, которую растворяли в DCM (300 мл) и обрабатывали при помощи тиоуреидопропил-диоксида кремния (Aldrich, 30 г). Смесь перемешивали в течение 30 мин, затем фильтровали. Диоксид кремния промывали при помощи DCM (200 мл) и фильтрат упаривали в вакууме указанного в заголовке соединения (11,1 г, 22,9 ммоль, выход 38,1%) в виде бежевой пены. Ее объединяли с дополнительными партиями для конечной очистки посредством хиральной HPLC, используя колонку ChilalPak 1А 20 мкм 5×20 см. Подвижная фаза представляла собой метанол, скорость потока 118 мл/мин и детектирование при 230 нм. Соответствующие фракции объединяли и выпаривали метанол. Остаток повторно упаривали от DCM и EtOAc с получением бледно-желтой пены, которую сушили в вакуум-сушильном шкафу с получением указанного в заголовке соединения. 1Н ЯМР (400 МГц, CDCl3) δ-млн-1 8.46 (1Н, s), 8.38 (1Н, d), 7.88 (1Н, s), 7.59 (1Н, d), 6.88 (1Н, s), 5.13 (1Н, m), 4.68 (1Н, m), 4.43 (1Н, m), 4.30 (2Н, d), 3.90 (2Н, s), 3.68 (3Н, s), 3.50 (2Н, m), 2.93 (2Н, m), 2.73 (3Н, s), 2.74 (1Н, m), 2.60 (4Н, m), 1.22 (3Н, d). LCMS (2 мин, муравьиная кислота): Rt=0,44 мин, МН+ 503. Чистота, определенная посредством хиральной HPLC, >99%.
Дополнительные соединения формулы (I), которые были получены, включают:
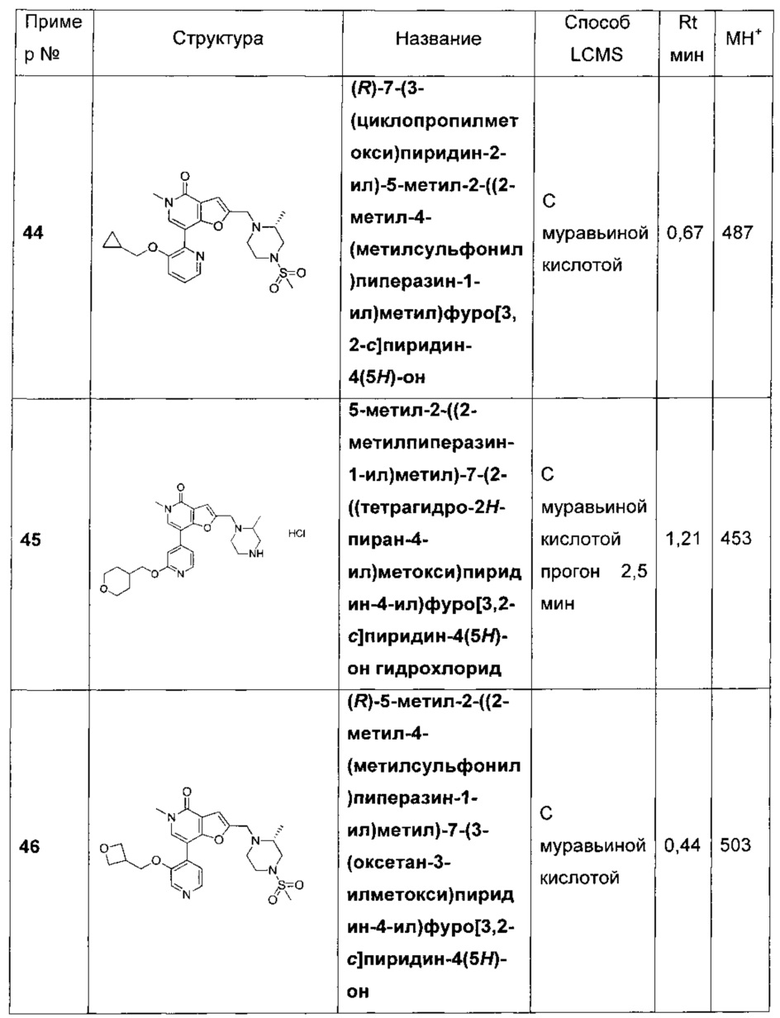
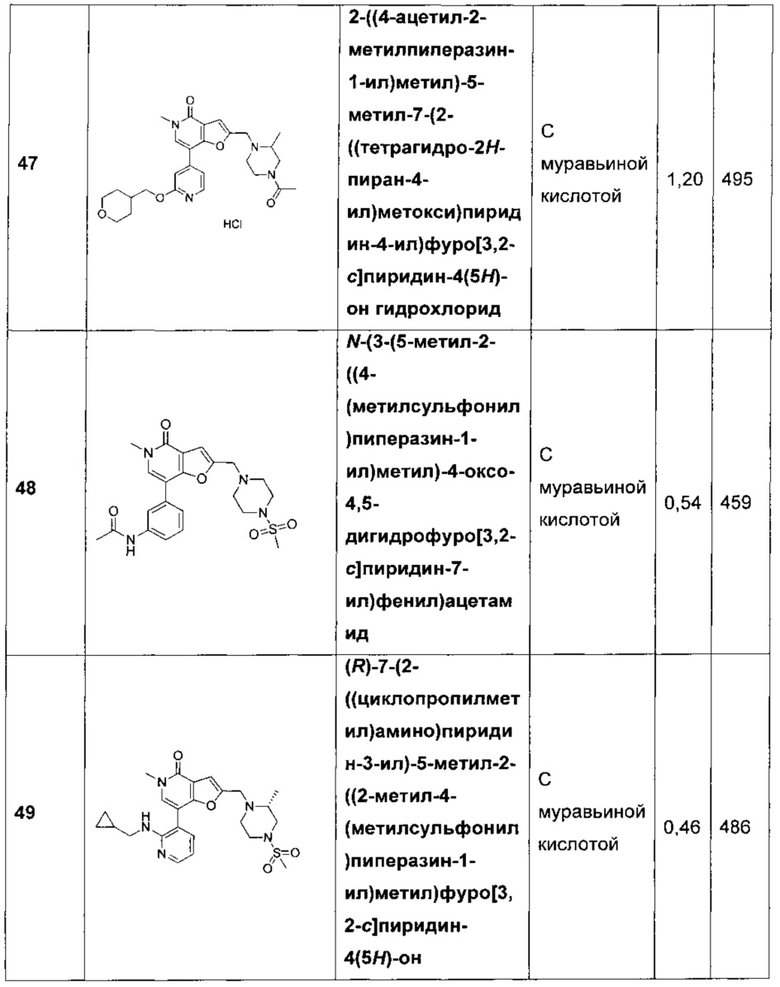
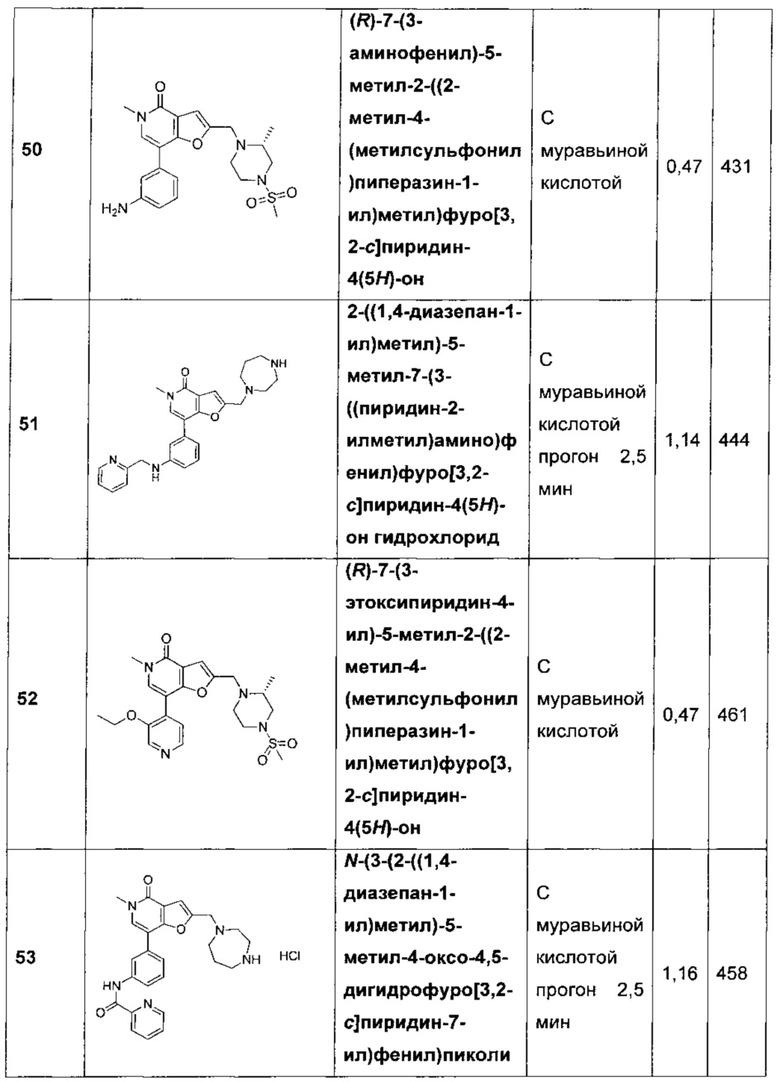
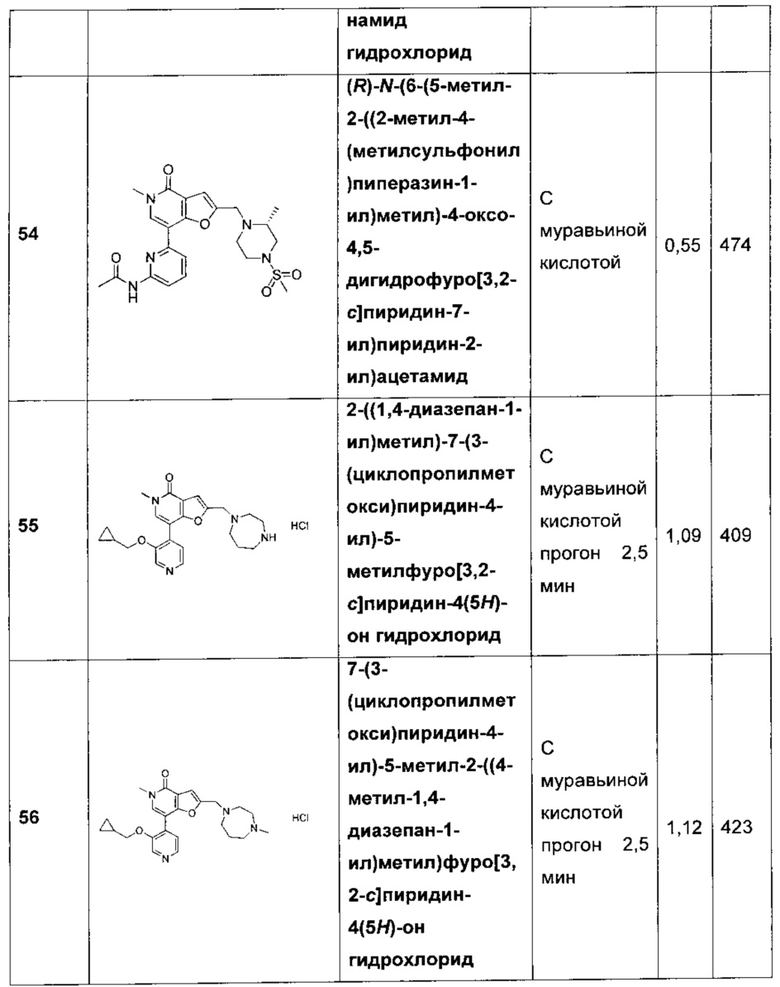
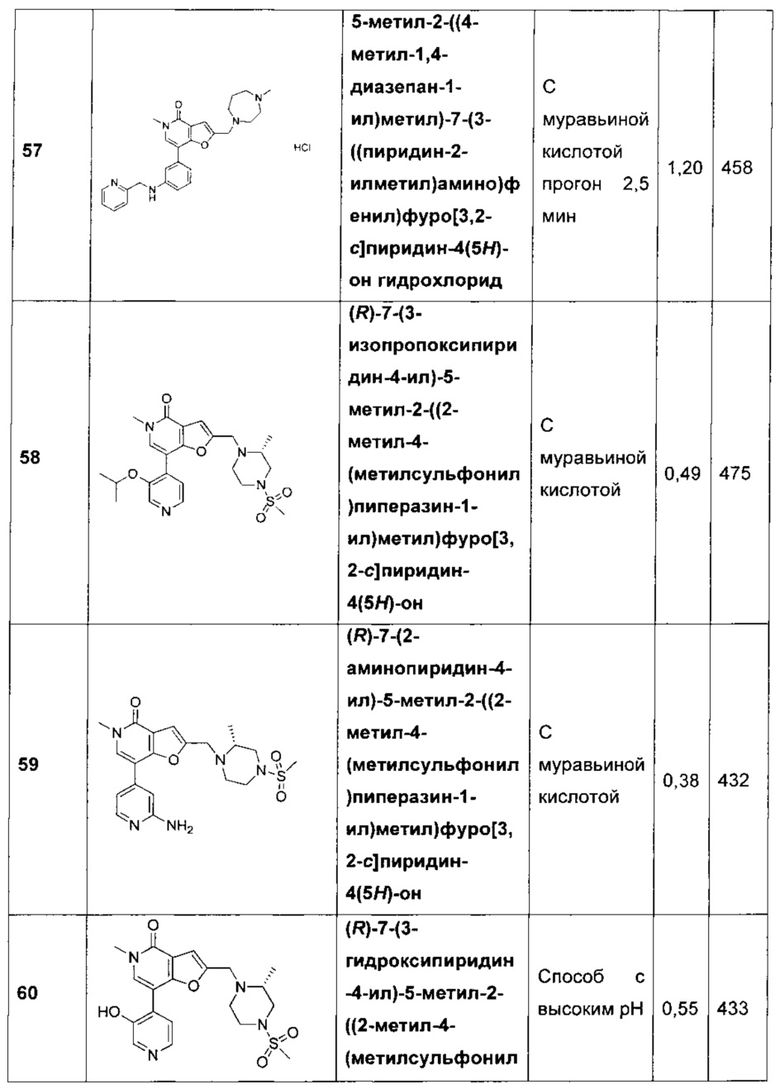
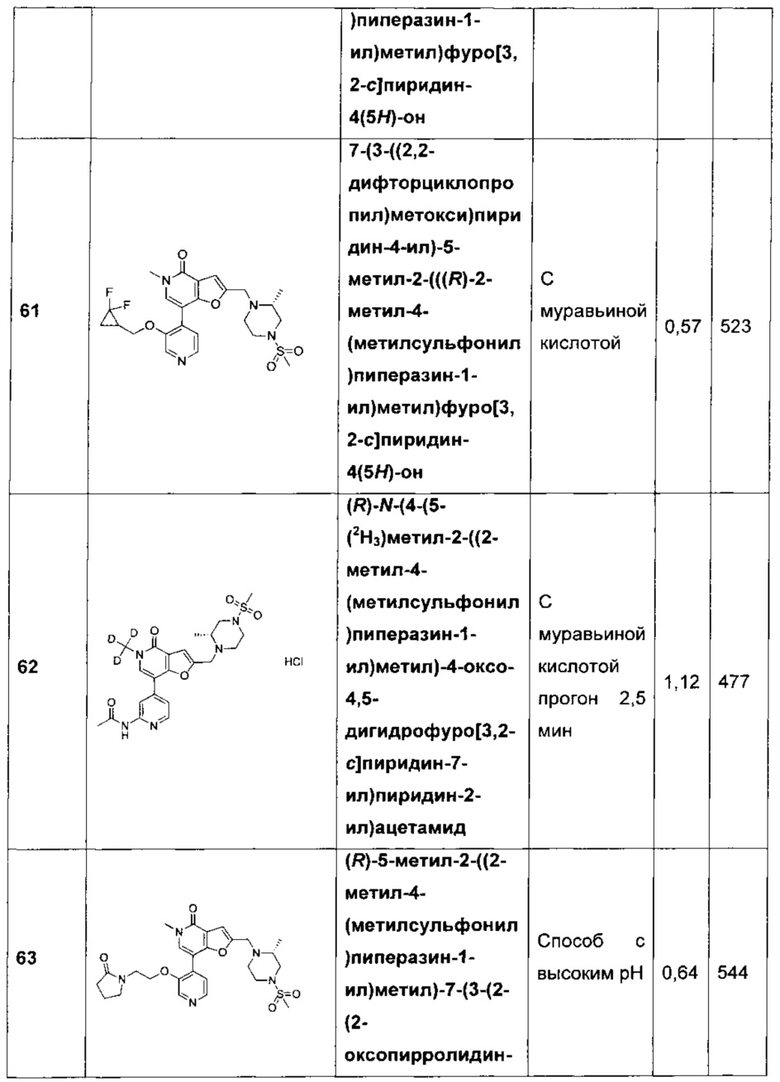
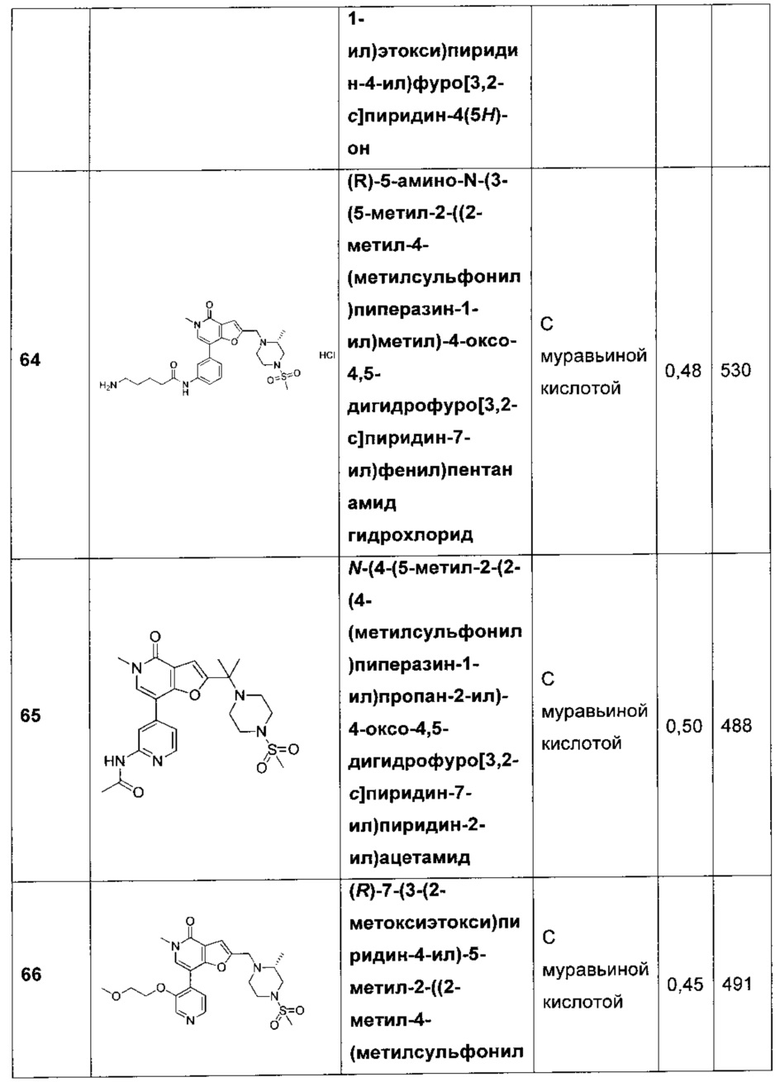
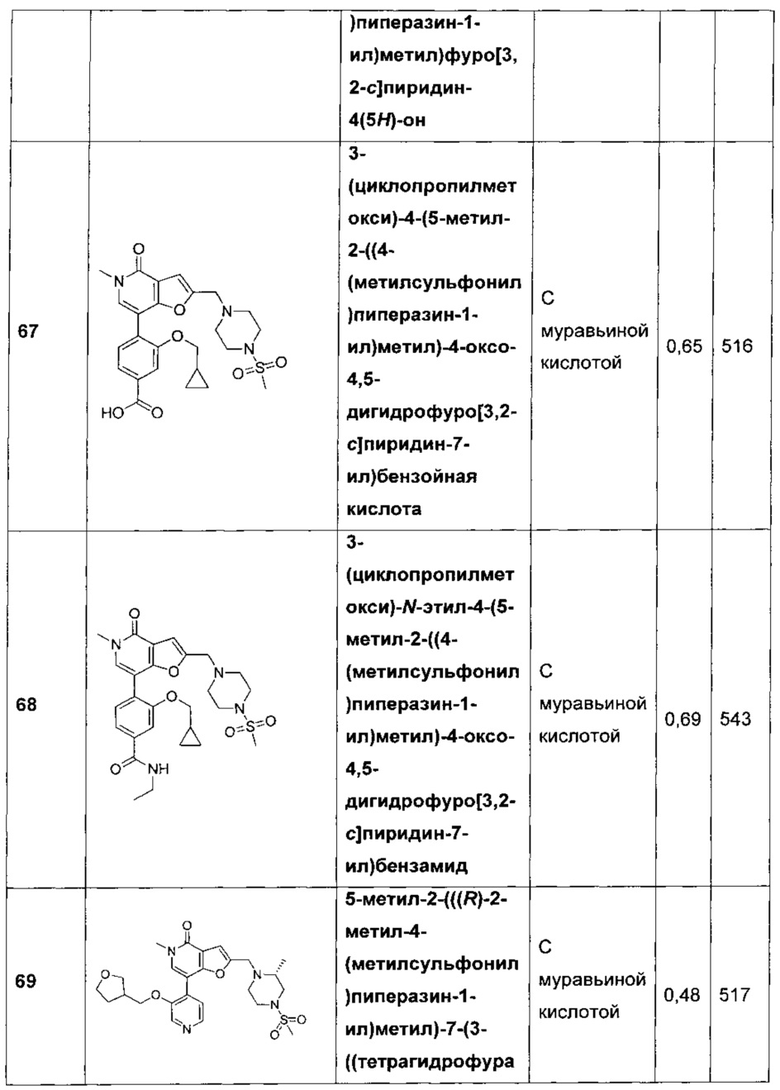
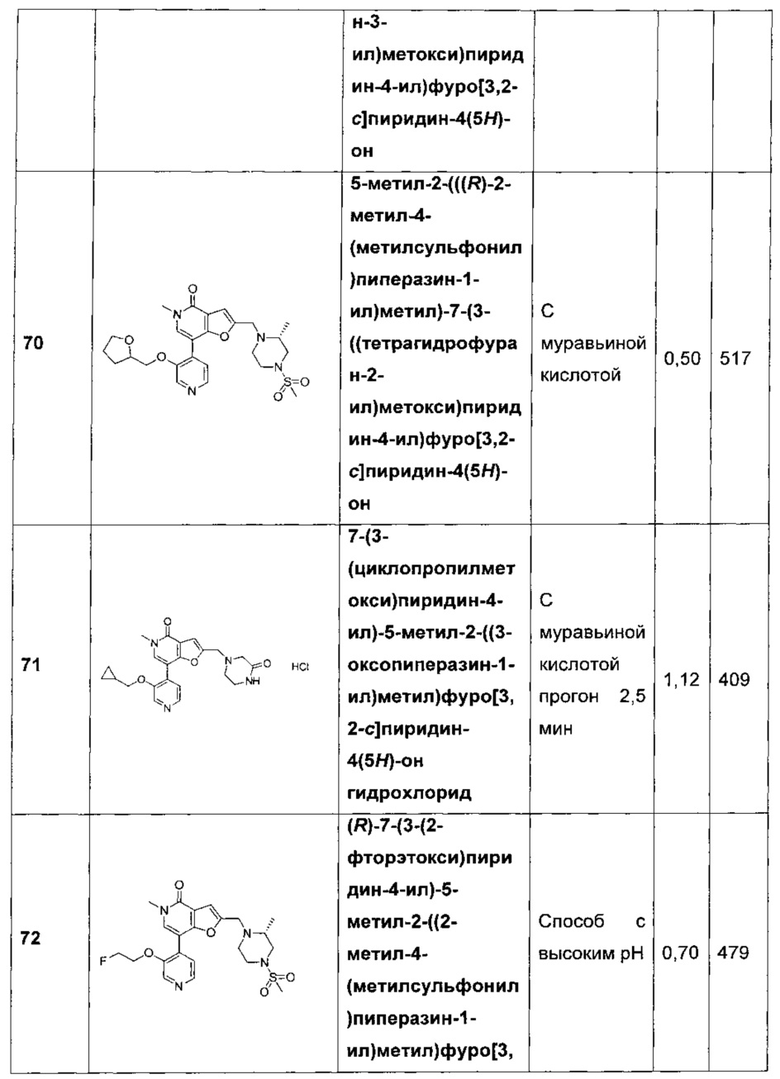
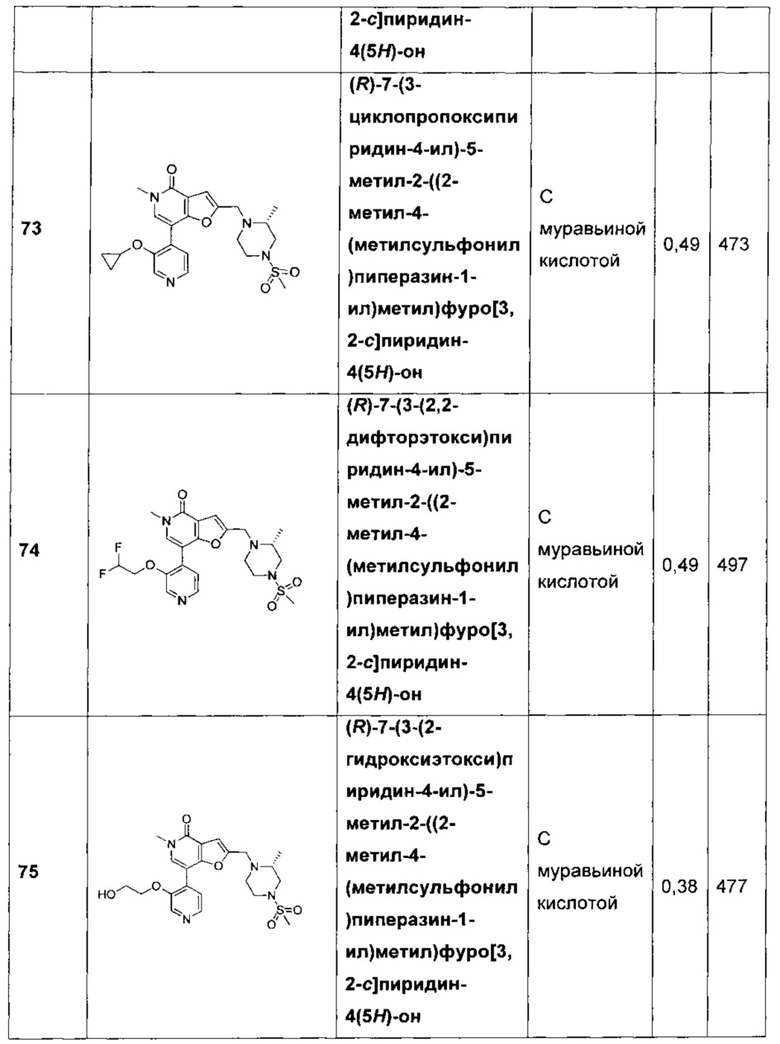
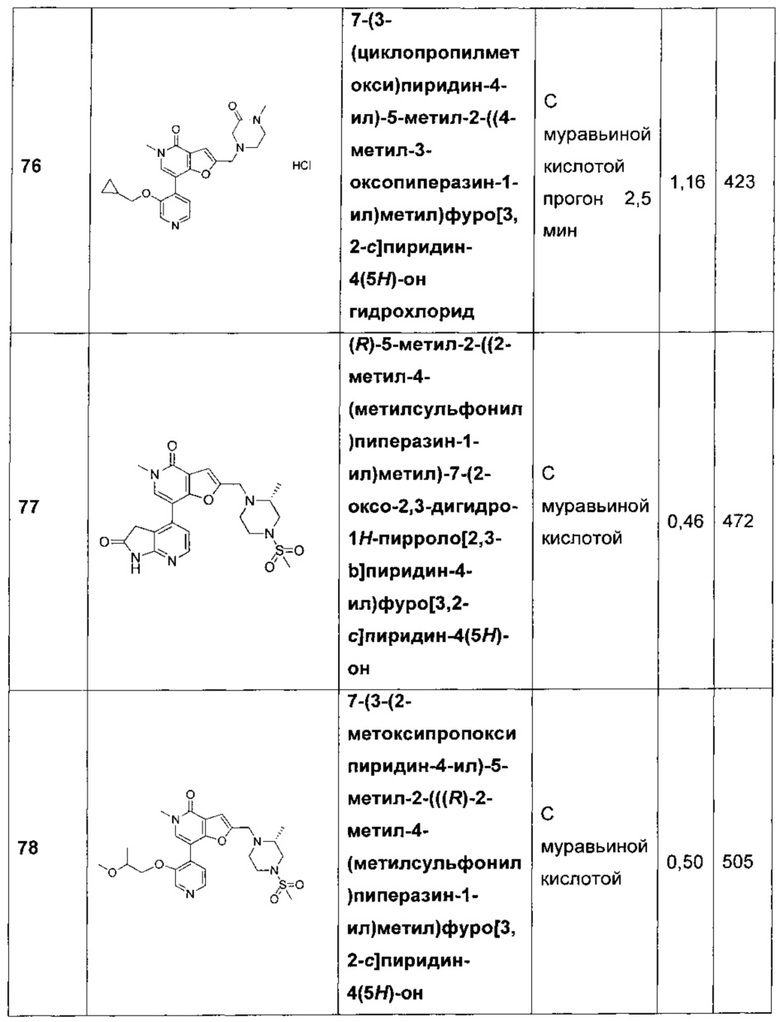
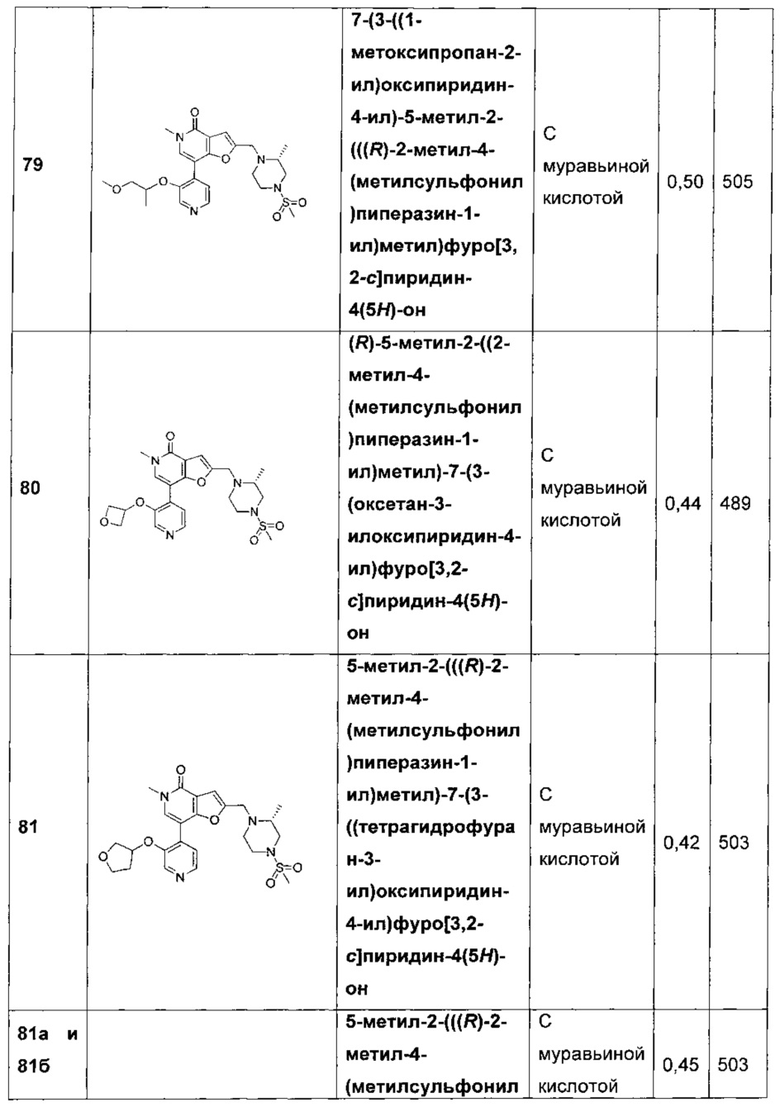
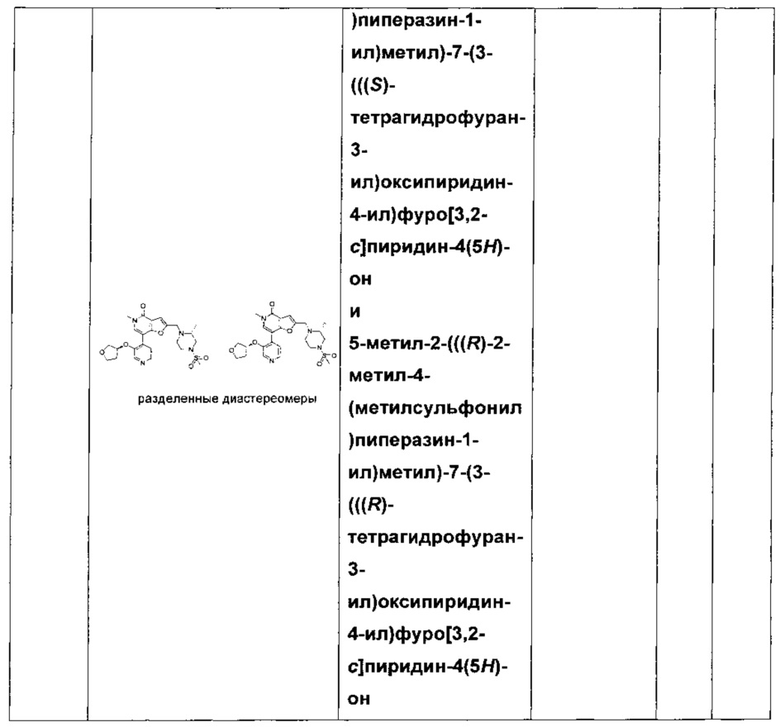
Пример 46: (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-3-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он
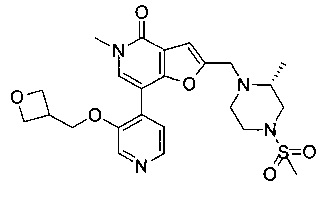
К перемешиваемой суспензии (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть промежуточное соединение 5, 110 мг, 0,263 ммоль), 3-(оксетан-3-илметокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (относительно получения смотреть промежуточное соединение 71, 589 мг, 0,421 ммоль), карбоната калия (109 мг, 0,789 ммоль) в 1,2-DME (3 мл) во флаконе для микроволновой печи добавляли Pd(PPh3)4 (15,19 мг, 0,013 ммоль). Флакон для микроволновой печи закрывали герметично и помещали в микроволновую печь Biotage initiator при 120°С на 2 часа при нормальной абсорбции. Смесь разбавляли этилацетатом (25 мл) и водой (25 мл). Два слоя разделяли, и водный слой повторно экстрагировали этилацетатом (4×25 мл). Органическую фазу концентрировали в вакууме. Остаток растворяли в 3 мл DMSO и очищали посредством MDAP. Соответствующие фракции объединяли и концентрировали в вакууме. Полученное желтое масло растворяли в метаноле и загружали на предварительно обработанный (при помощи метанола) аминопропиловый картридж (1 г), и продукт элюировали при помощи метанола. Соответствующие фракции объединяли и концентрировали в вакууме с получением (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-3-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она в виде желтого масла (18 мг, 13,6%). 1Н ЯМР (600 МГц, MeOH-d4) δ-млн-1 8.47 (1Н, s), 8.30 (1Н, d), 7.97 (1Н, s), 7.65 (1Н, d), 6.91 (1Н, s), 4.76 (2Н, ddd), 4.48 (2Н, td), 4.36 (2Н, d), 3.93-3.99 (1Н, m), 3.88-3.93 (1Н, m), 3.68 (3Н, s), 3.48 (1Н, m), 3.40-3.44 (1Н, m), 3.35-3.40 (1Н, m), 2.90-2.97 (1Н, m), 2.89-2.96 (1Н, m), 2.79 (3Н, s), 2.61-2.67 (1Н, m), 2.52-2.59 (1Н, m), 2.48-2.54 (1Н, m), 1.20 (3Н, d). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,44 мин, МН+ 503.
Пример 61: 7-(3-((2,2-дифторциклопропил)метокси)пиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-он
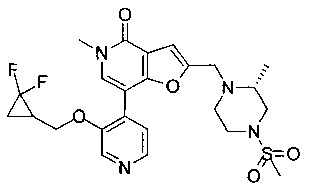
(R)-5-Метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5Н)-он (из альтернативного получения промежуточного соединения 5а, 522 мг, 17% масс./масс., 0,191 ммоль), 4-бром-3-((2,2-дифторциклопропил)метокси)пиридин (относительно получения смотреть промежуточное соединение 72, 179 мг, 0,407 ммоль), тетракис(трифенилфосфин)палладий (0) (22,03 мг, 0,019 ммоль) и водный раствор карбоната натрия (0,763 мл, 1,525 ммоль) смешивали в 1,2-DME (3 мл) и нагревали в микроволновой печи до 120°С в течение 30 мин. Раствор разбавляли водой и этилацетатом. Органический продукт экстрагировали этилацетатом (3×20 мл) и органические слои объединяли, фильтровали через гидрофобную фритту и концентрировали в вакууме с получением неочищенного продукта, который очищали при помощи MDAP с муравьиной кислотой. Фракции, содержащие продукт, объединяли и концентрировали в вакууме, затем растворяли в МеОН и элюировали при помощи МеОН через аминопропиловый картридж (2 г). Растворитель выпаривали в вакууме до получения коричневого твердого вещества. Его повторно очищали посредством MDAP с высоким pH и фракции, содержащие соединение, объединяли и концентрировали в вакууме с получением 7-(3-((2,2-дифторциклопропил)метокси)пиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-она в виде белого твердого вещества (20 мг, 0,038 ммоль, выход 20,07%). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,55 мин, МН+ 523.
Пример 65: N-(4-(5-метил-2-(2-(4-(метилсульфонил)пиперазин-1-ил)пропан-2-ил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
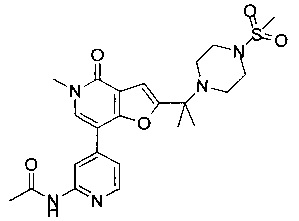
Смесь N-(5-йод-4-метокси-1-метил-6-оксо-1,6-дигидро-[3,4'-бипиридин]-2'-ил)ацетамида (относительно получения смотреть промежуточное соединение 59, 263 мг, 0,659 ммоль), 1-(2-метилбут-3-ин-2-ил)-4-(метилсульфонил)пиперазина (относительно получения смотреть промежуточное соединение 77, 455 мг, 1,977 ммоль), йодида меди(I) (30,1 мг, 0,158 ммоль) и PdCl2(PPh3)2 (26 мг, 0,037 ммоль) в DMF (1 мл) нагревали при 120°С в течение 6 часов, используя микроволновую печь. Реакционную смесь концентрировали при пониженном давлении, затем неочищенное вещество очищали посредством колоночной хроматографии на силикагеле, элюируя при помощи градиента 0-10% 2 М аммиака в МеОН/EtOAc. Соответствующие фракции объединяли, затем концентрировали при пониженном давлении с получением неочищенного продукта (порядка 140 мг). Неочищенный продукт очищали посредством MDAP с получением N-(4-(5-метил-2-(2-(4-(метилсульфонил)пиперазин-1-ил)пропан-2-ил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (25 мг, 8%). 1Н ЯМР (400 МГц, МеОН-d4) δ-млн-1 8.70 (1Н, s), 8.36 (1Н, d), 8.08 (1Н, s), 7.49 (1Н, dd), 6.88 (1Н, s), 3.75 (3Н, s), 3.22 (4Н, m), 2.82 (3Н, s), 2.71 (4Н, m), 2.22 (3Н, s), 1.61 (6Н, s). LCMS (2 мин, Муравьиная кислота): Rt=0,50 мин, МН+ 488.
Пример 66: (R)-7-(3-(2-метоксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5Н)-он
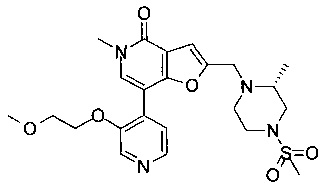
Указанное в заголовке соединение получали из (R)-7-бром-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она (промежуточное соединение 5) и 3-(2-метоксиэтокси)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридина (промежуточное соединение 74), с помощью способа, аналогичного способу, описанному для примера 46. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,45 мин, МН+ 491.
Пример 69: 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-3-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5Н)-он
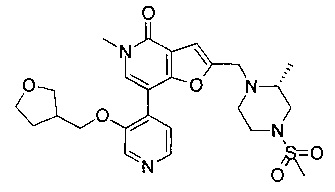
Указанное в заголовке соединение получали из (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть альтернативное получение промежуточного соединения 5а) и 4-бром-3-((тетрагидрофуран-3-ил)метокси)пиридина (промежуточное соединение 75) с помощью способа, аналогичного способу, описанному для примера 61. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,48 мин, МН+ 517.
Пример 70: 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-2-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он
Указанное в заголовке соединение получали из (R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фуро[3,2-с]пиридин-4(5Н)-она (относительно получения смотреть альтернативное получение промежуточного соединения 5а) и 4-бром-2-((тетрагидрофуран-3-ил)метокси)пиридина (промежуточное соединение 76) с помощью способа, аналогичного способу, описанному для примера 61. LCMS (2 мин, способ с муравьиной кислотой): Rt=0,50 мин, МН+ 517.
Пример 82: N-(4-(5-метил-4-оксо-2-((5-оксо-1,4-диазепан-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
Получали основной раствор N-(4-(2-(бромметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (относительно получения смотреть промежуточное соединение 81) (300 мг растворенного/суспендированного в DMF (3 мл)). Аликвоту (0,5 мл) дозировали во флакон для микроволновой печи, содержащий 1,4-диазепан-5-он (18 мг, 0,159 ммоль). Затем добавляли DIPEA (51,5 мг, 0,399 ммоль) и реакционный сосуд закрывали герметично и нагревали в микроволновой печи Anton Parr при 600 Вт до 110°С в течение 30 мин. Во время охлаждения добавляли DMF (0,5 мл) и реакционную смесь очищали посредством MDAP с получением N-(4-(5-метил-4-оксо-2-((5-оксо-1,4-диазепан-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (13,9 мг, выход 23%). LCMS (2 мин, способ с муравьиной кислотой): Rt=0,39 мин, МН+ 410.
Примеры в следующей таблице, примеры 83-106, получали способом, аналогичным способу, описанному для примера 82.
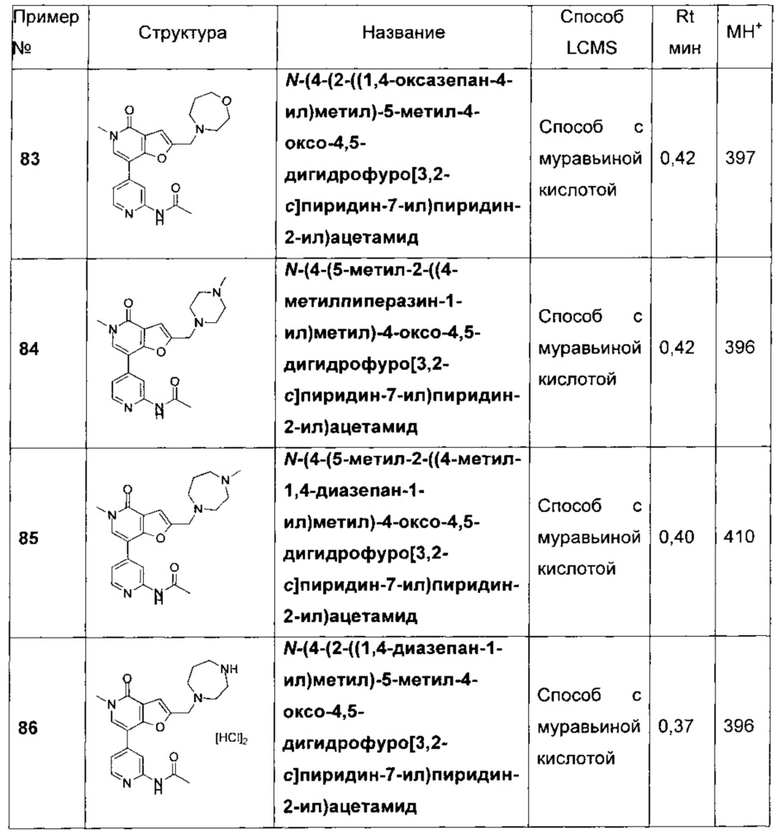
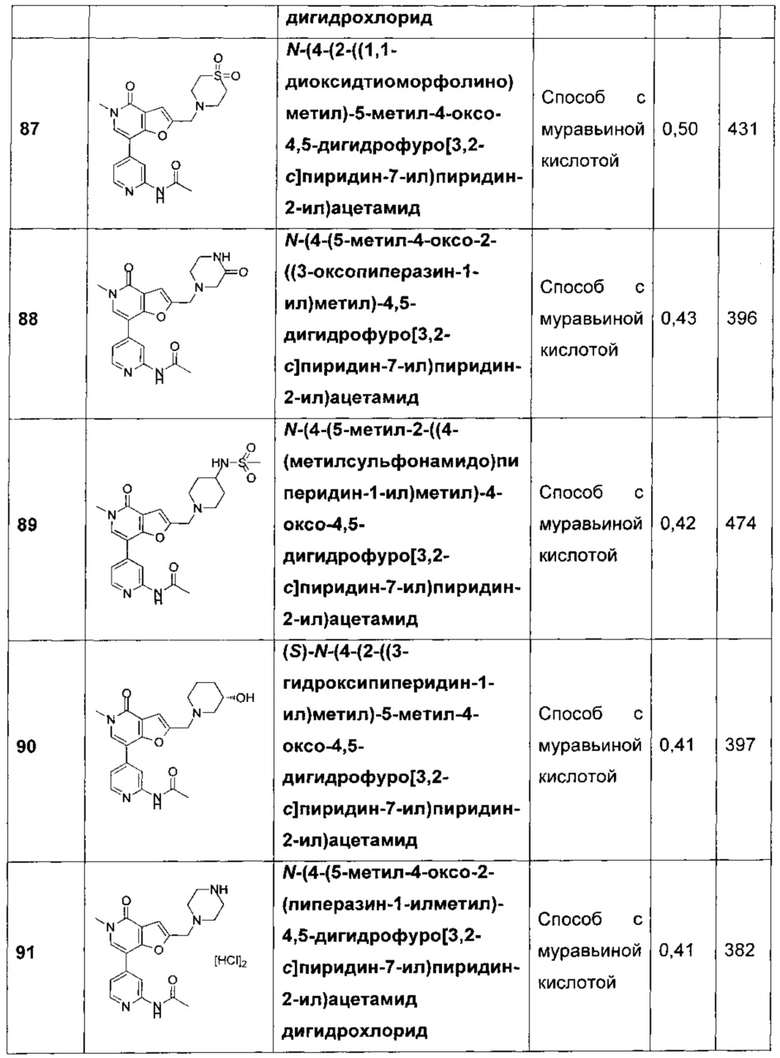

Пример 107: N-(4-(5-метил-2-((4-метил-3-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
N-(4-(2-(Хлорметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид (относительно получения смотреть промежуточное соединение 82, 50 мг, 0,151 ммоль) растворяли в DMF (5 мл), добавляли карбонат калия (62,5 мг, 0,452 ммоль) с последующим добавлением 1-метилпиперазин-2-она (17,20 мг, 0,151 ммоль). Смесь перемешивали в течение 3 ч при комнатной температуре. Смесь разбавляли водой (10 мл) и экстрагировали при помощи смеси метанола (3 мл) и DCM (15 мл), органический слой сушили и упаривали в вакууме и остаток очищали посредством MDAP с получением N-(4-(5-метил-2-((4-метил-3-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (25 мг, 0,061 ммоль, выход 40,5%). LCMS (2 мин, способ с высоким pH): Rt=0,58 мин, МН+ 410.
Пример 108: (S)-N-(4-(5-метил-2-((3-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
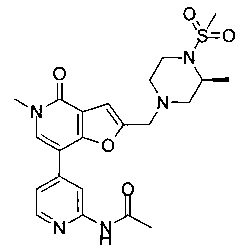
Указанное в заголовке соединение получали из N-(4-(2-(хлорметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (относительно получения смотреть промежуточное соединение 82) и (S)-2-метил-1-(метилсульфонил)пиперазина способом, аналогичным способу, описанному для примера 107. LCMS (2 мин, способ с высоким pH): Rt=0,68 мин, МН+ 474.
Пример 109: N-(4-(5-метил-2-((4-(метилсульфонил)-2-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид
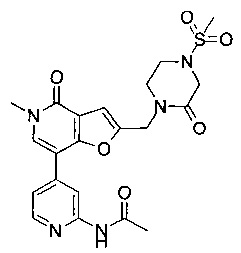
4-(Метилсульфонил)пиперазин-2-он (193 мг, 1,085 ммоль) растворяли в DMF (3 мл), добавляли NaH (43,4 мг, 60% масс./масс., 1,085 ммоль) и смесь перемешивали в течение 10 мин, затем добавляли к раствору N-(4-(2-(хлорметил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида (относительно получения смотреть промежуточное соединение 82, 180 мг, 0,543 ммоль) в DMF (3 мл) при комнатной температуре. Смесь перемешивали в течение 1 ч, затем гасили путем добавления уксусной кислоты (2 капли) и упаривали в вакууме. Неочищенный продукт очищали посредством MDAP с получением указанного в заголовке соединения N-(4-(5-метил-2-((4-(метилсульфонил)-2-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид (15 мг, 0,032 ммоль, выход 5,84%). 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 10.56 (1Н, s), 8.56 (1Н, s), 8.37 (1Н, d), 8.20 (1Н, s), 7.45 (1Н, dd), 6.94 (1Н, s), 4.72 (2Н, s), 3.88 (2Н, s), 3.61 (3Н, s), 3.57 (2Н, m), 3.48 (2Н, m), 2.99 (3Н, s), 2.13 (3Н, s). LCMS (2 мин, способ с высоким pH): Rt=0,60 мин, МН+ 474.
Также получали побочный продукт N-(4-(2,5-диметил-3-(4-(метилсульфонил)-2-оксопиперазин-1-ил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамид (18 мг, выход 7%). 1Н ЯМР (400 МГц, DMSO-d6) δ-млн-1 10.55 (1Н, s), 8.58 (1Н, s), 8.38 (1Н, d), 8.17 (1Н, s), 7.46 (1Н, dd), 4.02 (3Н, m), 3.61 (3Н, m), 3.59 (3Н, s), 3.10 (3Н, s), 2.30 (3Н, s), 2.13 (3Н, s). LCMS (2 мин, способ с высоким pH): Rt=0,65 мин, МН+ 474.
Также получали следующие соединения формулы (I):
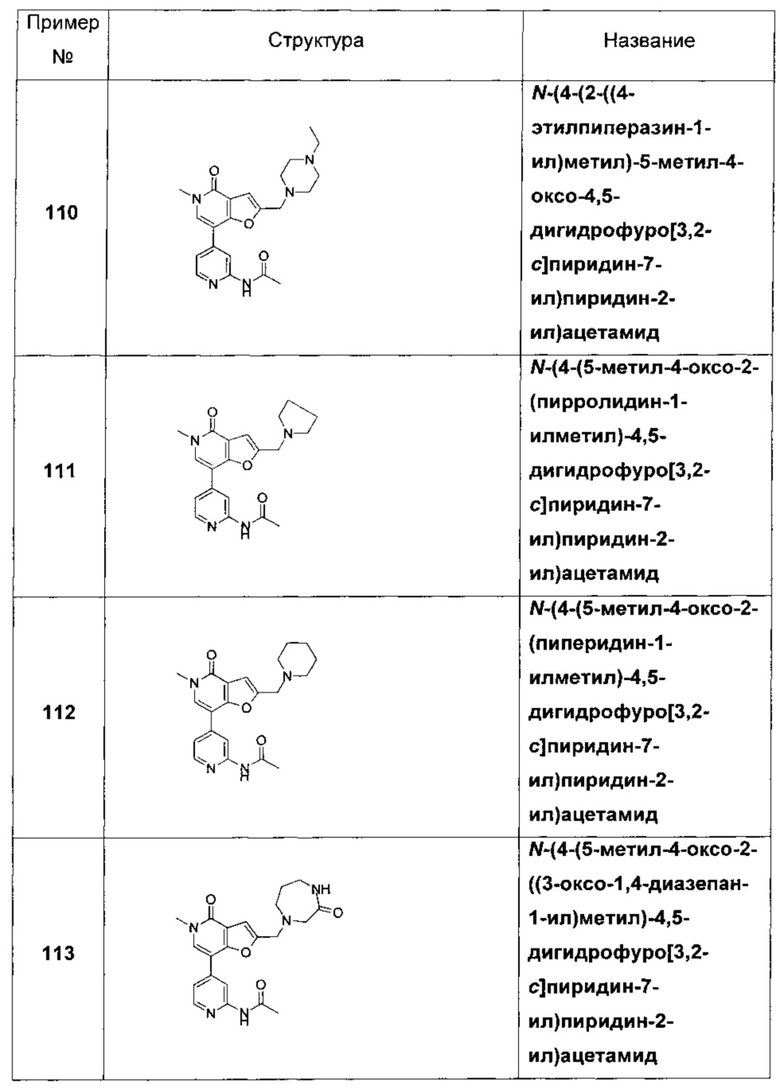
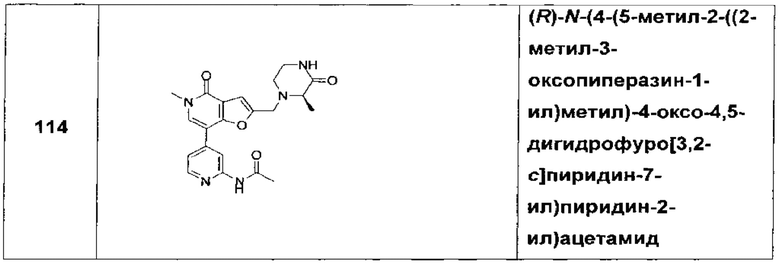
Биологические способы тестирования
Соединения формулы (I) могут быть протестированы в ходе одного или более из следующих анализов:
Анализ методом резонансного переноса энергии флуоресценции с временным разрешением (TR-FRET)
Связывание оценивали с помощью анализа резонансного переноса энергии флуоресценции с временным разрешением. В нем используется метка очистки 6His на N-конце белков в качестве эпитопа для антитела против-6His, меченого хелатом европия (PerkinElmer AD0111), что делает возможным связывание европия с белками, которые действуют как доноры флуорофора. Небольшую молекулу, вещество с высокой аффиностью к бромодоменам BRD2, BRD3, BRD4 и BRDT, метили при помощи Alexa Fluor647 (контрольное соединение X) и оно действовало в качестве акцептора в паре FRET.
Контрольное соединение X: 4-((Z)-3-(6-((5-(2-((4S)-6-(4-хлорфенил)-8-метокси-1-метил-4H-бензо[f][1,2,4]триазоло[4,3-а][1,4]диазепин-4-ил)ацетамидо)пентил)амино)-6-оксогексил)-2-((2Е,4Е)-5-(3,3-диметил-5-сульфо-1-(4-сульфобутил)-3H-индол-1-ий-2-ил)пента-2,4-диен-1-илиден)-3-метил-5-сульфоиндолин-1-ил)бутан-1-сульфонат)
К раствору N-(5-аминопентил)-2-((4S)-6-(4-хлорфенил)-8-метокси-1-метил-4Н-бензо[f][1,2,4]триазоло[4,3-а][1,4]диазепин-4-ил)ацетамида (относительно получения смотреть контрольное соединение J, WO 2011/054848 A1, 1,7 мг, 3,53 мкмоль) в DMF (40 мкл) добавляли раствор AlexaFluor647-ONSu (2,16 мг, 1,966 мкмоль) также в DMF (100 мкл). Смесь ощелачивали при помощи DIPEA (1 мкл, 5,73 мкмоль) и перемешивали в течение ночи на вихревой мешалке.
Реакционную смесь упаривали досуха. Твердое вещество растворяли в смеси ацетонитрил/вода/уксусная кислота (5/4/1, <1 мл), фильтровали и вносили в препаративную колонку Phenomenex Jupiter С18 и элюировали при помощи следующего градиента (А = 0,1% трифторуксусной кислоты в воде, В = 0,1% TFA/90% ацетонитрила/10% воды): Скорость потока = 10 мл/мин, AU = 20/10 (214 нм):
5-35%, t = 0 мин: В = 5%; t = 10 мин: В = 5%; t = 100 мин: В = 35%; t = 115 мин: В = 100% (градиент разделения: 0,33%/мин).
Основной компонент элюировали в диапазоне 26-28% В, но оказалось, что он состоял из двух пиков. Среднюю фракцию (F1.26), которая должна содержать "оба" компонента, анализировали посредством аналитической HPLC (Spherisorb ODS2, 1-35% в течение 60 мин): единственный компонент, элюируемый при 28% В.
Фракции F1.25/26&27 объединяли и упаривали досуха. Переносили с помощью DMF, упаривали досуха, растирали с безводным этиловым эфиром и голубое твердое вещество сушили в течение ночи при давлении ниже 0,2 мбар (20 Па): 1,54 мг.
Аналитическая HPLC (Sphersisorb ODS2, 1-35% В в течение 60 мин): MSM10520-1: [М+Н]+ (наблюдаемое): 661,8/- соответствует М-29. Это приравнивается к [(М+2Н)/2]+ для вычисленной массы 1320,984, являющегося М-29. Это является нормальным явлением в присутствии красителя Alexa Fluor 647 и представляет собой теоретическую потерю двух метиленовых групп в условиях масс-спектрометра.
Принцип анализа: в отсутствие конкурирующего соединения возбуждение европия заставляет донора излучать при λ618 нм, что возбуждает меченое Alexa соединение, связывающее бромодомен, приводя к повышенному переносу энергии, который измеряется при λ647 нм. В присутствии достаточной концентрации соединения, которое может связывать эти белки, взаимодействие прерывают, что приводит к количественно измеримому падению резонансного переноса энергии флуоресценции.
Связывание соединений формулы (I) с бромодоменами BRD2, BRD3, BRD4 и BRDT оценивали, применяя мутантные белки для обнаружения дифференциального связывания либо со связывающим доменом 1 (BD1), либо со связывающим доменом 2 (BD2) на бромодомене. Эти однонаправленные мутации остатка в ацетил-лизин-связывающем-кармане значительно снижают аффинность флуоресцирующего лиганда (контрольное соединение X) в отношении мутированного домена (>1000-кратно в сравнении с немутированным доменом). Поэтому в условиях конечного анализа связывание флуоресцирующего лиганда с мутировавшим доменом не может быть обнаружено, и, как следствие, анализ является подходящим для определения связывания соединений с одним немутированным бромодоменом.
Продуцирование белка: рекомбинантные человеческие бромодомены [(BRD2 (1-473) (Y113A) и (Y386A), BRD3 (1-435) (Y73A) и (Y348A) BRD4 (1-477) (Y97A) и (Y390A) и BRDT (1-397) (Y66A) и (Y309A)] экспрессировали в клетках Е. coli (в векторе pET15b для BRD2/3/4 и в векторе рЕТ28а для BRDT) при помощи метки 6-His на N-конце. Осадок His-меченного бромодомена ресуспендировали в смеси 50 мМ HEPES (pH 7,5), 300 мМ NaCl, 10 мМ имидазола и 1 мкл/мл ингибитора протеазы и экстрагировали из клеток Е. coli, используя обработку ультразвуком, и очищали, используя высокопроизводительную колонку с никель сефарозой, белки промывали и затем элюировали в линейном градиенте 0-500 мМ имидазола с буфером 50 мМ HEPES (pH 7,5), 150 мМ NaCl, 500 мМ имидазола в объеме, эквивалентном 20 объемам колонки. Конечную очистку завершали с помощью колонки для препаративной гель-фильтрации Superdex 200. Очищенный белок хранили при -80°С в 20 мМ HEPES pH 7,5 и 100 мМ NaCl. Идентичность белка подтверждали посредством способа идентификации белков по «отпечаткам пептидных масс» и предсказанную молекулярную массу подтверждали посредством масс-спектрометрии.
Протокол для анализов мутантов бромодомена BRD2, 3, 4 и Т, BD1 + BD2: все компоненты для анализа растворяли в буферной композиции из 50 мМ HEPES pH7,4, 50 мМ NaCl, 5% глицерина, 1 мМ DTT и 1 мМ CHAPS. Конечные концентрации белков бромодомена представляли собой 10 нМ, а концентрация лиганда Alexa Fluor647 была равна константе диссоциации. Эти компоненты предварительно смешивали и по 5 мкл этой реакционной смеси добавляли во все лунки, содержащие 50 нл тестируемого соединения в различных концентрациях или носителя DMSO (конечная концентрация 0,5% DMSO), в 384-луночных малообъемных микротитрационных планшетах черного цвета Greiner и инкубировали в темноте в течение 30 минут при комнатной температуре. По 5 мкл детектируемой смеси, содержащей меченого хелатом европия антитела анти-6His в конечной концентрации 1,5 нМ, добавляли во все лунки и выполняли дополнительную инкубацию в темноте в течение по меньшей мере 30 минут. Затем планшеты считывали на планшет-ридере Envision, (λex = 317 нМ, донор λem = 615 Нм, акцептор λem = 665 нМ; Dichroic LANCE dual). Измерения интенсивности флуоресценции с временным разрешением производили при обеих длинах волн излучения, вычисляли соотношение акцептор/донор и использовали для анализа данных. Все данные приводили к среднему значению от 16 высоких (контроль-ингибитор - пример 11 WO 2011/054846 А1) и 16 низких (DMSO) значений контрольных лунок на каждом планшете. Затем использовали четырехпараметрическую кривую для подбора следующего вида:
y=a+((b-a)/(1+(10^x/10^c)^d)
Где 'а' представляет собой минимум, 'b' представляет собой угловой коэффициент, 'с' представляет собой pIC50 и 'd' представляет собой максимум.
Каждый из примеров 1-81b тестировали по меньшей мере в одном из анализов BRD2, BRD3, BRD4 или BRDT, BD1 или BD2, описанных выше, и по меньшей мере в одном анализе обнаруживали, что они имеют pIC50≥5,0.
Было обнаружено, что примеры 1-7, 9-15, 20-81б, 82, 83, 85-89, 94-96, 98, 100-104, 106, 108 и 109 в анализе BRD4 BD1 имели pIC50≥6,0.
Было обнаружено, что примеры 1-4, 7, 10, 12, 14, 15, 21, 21а, 21б и 23-27, 30-32 и 34-44, 46-50, 52, 54-62, 64-74, 76, 78, 80-816, 82, 87, 96, 100, 102 и 108 в анализе BRD4 BD1 имели pIC50≥7,0.
Было обнаружено, что примеры 32, 35, 61 и 67 в анализе BRD4 BD1 имели pIC50≥8,0.
Вычисление селективности в отношении BRD4 BD1 по сравнению с BRD4 BD2
Селективность в отношении BRD4 BD1 по сравнению с BRD4 BD2 вычисляли следующим образом:
Селективность = BRD4 BD1 pIC50 - BRD4 BD2 pIC50.
Значения pIC50 выражали в виде единиц log10.
Было обнаружено, что примеры 1-7, 9-16, 20-81б и 82-109 имеют селективность в отношении BRD4 BD1 по сравнению с BRD4 BD2≥1 логарифмической единицы в анализах TR-FRET, описанных выше, значит они являются по меньшей мере 10-кратно более селективными в отношении BRD4 BD1 по сравнению с BRD4 BD2.
Было обнаружено, что примеры 1-4, 7, 10, 12, 14, 15, 21, 21а, 21б, 23-28, 30-32, 34-44, 46, 48-52, 54, 56-67, 69, 70, 72-75, 77, 78, 80-81б, 82, 87, 96, 98, 100-103 и 108 имеют селективность в отношении BRD4 BD1 по сравнению с BRD4 BD2≥2 логарифмических единиц в анализах TR-FRET, описанных выше, значит они являются по меньшей мере 100-кратно более селективными в отношении BRD4 BD1 по сравнению с BRD4 BD2.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛО[3, 4-b]ПИРИДИНА, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ (ВАРИАНТЫ), ПРИМЕНЕНИЕ (ВАРИАНТЫ), КОМПОЗИЦИЯ (ВАРИАНТЫ) | 2003 |
|
RU2357967C2 |
| ПРОИЗВОДНЫЕ ПИРИДАЗИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ RORc | 2017 |
|
RU2757571C2 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
| ПРОТИВОВИРУСНЫЕ СРЕДСТВА ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ВИЧ ИНФЕКЦИИ | 2021 |
|
RU2780103C1 |
| КОНДЕНСИРОВАННОЕ ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2005 |
|
RU2389731C2 |
| ИНГИБИТОРЫ ФУРИНА | 2019 |
|
RU2799824C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| МОДУЛЯТОРЫ ПИРУВАТКИНАЗЫ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2797518C2 |
| Гетероароматические соединения и их применение в качестве допаминовых D1 лигандов | 2013 |
|
RU2617842C2 |
| БИЦИКЛИЧЕСКИЕ ЛАКТАМЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2827714C1 |
Изобретение относится к новым производным фуропиридина формулы I
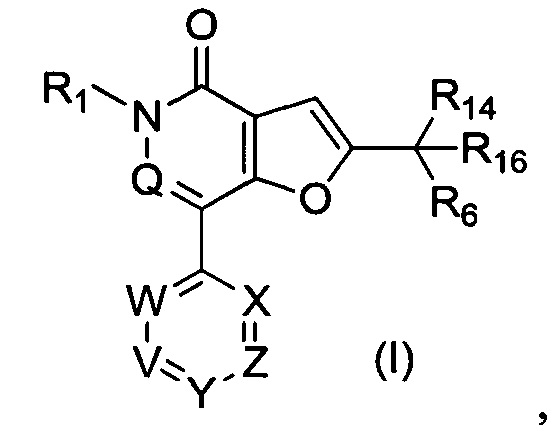
где: V представляет собой N или C-R2; W представляет собой N или C-R8; X представляет собой N, СН или С(СН3); Y представляет собой N или C-R5; Z представляет собой N или C-R15; Q представляет собой N или СН; R1 представляет собой С1-4алкил или дейтерированный С1-4алкил; R2, когда присутствует, представляет собой Н, ОН, -NH2, -ОС1-4алкил, -NHC(O)H, -NHC(O)С1-4алкил, -Н(СН3)С(O)С1-4алкил, -NHC(O)NH2, -NHC(O)С1-4алкиленNH2, -N(CH3)C(O)NH2, -N(СН3)С(O)С1-4алкиленNH2, -ОС2-4алкиленОСН3, -ОС2-4алкиленОН или R2 представляет собой группу, выбранную из -G-CH2CH(R3)(R4), -G-CH(R3)(R4) и -G-R3, в которой G представляет собой NH, N(CH3), О, C(O)NH или NHC(O); R3 представляет собой фенил, пиридинил, С3-7циклоалкил или 5-6-членный гетероцикл, содержащий атом кислорода или азота и возможно замещенный =O; и R4 представляет собой Н или С1-4алкил; R5, когда присутствует, представляет собой Н, ОН, -OC1-4 алкил, -CH2NH2, -OCF3, -С(O)NHC1-4алкил или -CO2H;
R6 представляет собой -NR11R12 или группу 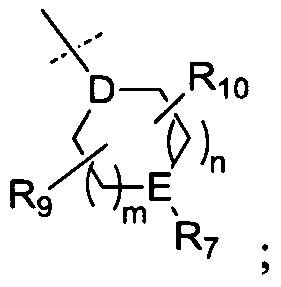
D представляет собой СН или N; Е представляет собой N, О, СН или SO2; R7, когда присутствует, представляет собой Н, ОН, С1-4алкил, -SO2C1-4алкил, -NHSO2CH3, -С(O)С1-4алкил; R8, когда присутствует, представляет собой Н, С1-4алкил, ОН, -ОС1-4алкил, -ОС2-4алкиленОС1-4алкил, -OCF3; -ОС1-4алкиленF, -ОС1-4алкиленCHF2, -ОС2-4алкиленОН, -Офенил, -ОС1-4алкиленфенил, -NHC3-7циклоалкил, -NHC1-4алкиленС3-7циклоалкил, -ОС3-7циклоалкил, -ОС1-4алкиленС3-7циклоалкил, -NHC4-6гетероцикл, -NHC1-4алкиленС4-6гетероцикл, -ОС4-6гетероцикл или -ОС1-4алкиленС4-6гетероцикл, содержащий в качестве гетероатома N или О, где каждый С3-7циклоалкил или С4-6гетероцикл возможно замещен одним или двумя заместителями, независимо выбранными из галогена и оксо; или R8 и R2 вместе с атомами углерода, к которым они присоединены, образуют гетероцикл, содержащий в качестве гетероатома N и возможно замещенный оксо; R9 представляет собой Н, С1-4алкил, галоген, ОН, -ОС1-4алкил, -СН2ОН, -C(O)NHCH3, -C(O)NH(CH3)2; R10 представляет собой Н, С1-4алкил, галоген, ОН, -ОС1-4алкил или оксо; R11 представляет собой Н, С1-4алкил или SO2CH3; R12 представляет собой Н, С1-4алкил, C2-4aлкилeнNHR13, SO2CH3, 5-6-членный гетероцикл, содержащий SO2; R13 представляет собой Н или SO2CH3; R14 представляет собой Н или С1-4алкил; R15 представляет собой Н, С1-4алкил или NHC(O)С1-4алкил; R16 представляет собой Н или С1-4алкил; и каждый из n и m представляет собой целое число, независимо выбранное из 0, 1 и 2; при условии, что не более 2 из V, W, X, Y и Z представляют собой N; или его солям. Изобретение также относится к фармацевтической композиции, обладающей активностью ингибитора бромодоменов, на основе этих соединений применению их в изготовлении лекарственного средства для использования в лечении заболеваний или состояний, для которых показан ингибитор бромодомена, и способу лечения таких заболеваний или состояний. 6 н. и 12 з.п. ф-лы, 2 табл., 109 пр.
1. Соединение формулы (I):
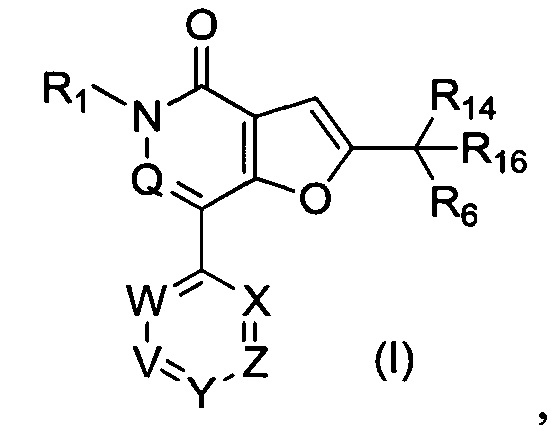
где:
V представляет собой N или C-R2;
W представляет собой N или C-R8;
X представляет собой N, СН или С(СН3);
Y представляет собой N или C-R5;
Z представляет собой N или C-R15;
Q представляет собой N или СН;
R1 представляет собой С1-4алкил или дейтерированный С1-4алкил;
R2, когда присутствует, представляет собой Н, ОН, -NH2, -ОС1-4алкил, -NHC(O)H, -NHC(O)С1-4алкил, -Н(СН3)С(O)С1-4алкил, -NHC(O)NH2, -NHC(O)С1-4алкиленNH2, -N(CH3)C(O)NH2, -N(СН3)С(O)С1-4алкиленNH2, -ОС2-4алкиленОСН3, -ОС2-4алкиленОН или
R2 представляет собой группу, выбранную из -G-CH2CH(R3)(R4), -G-CH(R3)(R4) и -G-R3, в которой
G представляет собой NH, N(CH3), О, C(O)NH или NHC(O);
R3 представляет собой фенил, пиридинил, С3-7циклоалкил или 5-6-членный гетероцикл, содержащий атом кислорода или азота и возможно замещенный =O; и
R4 представляет собой Н или С1-4алкил;
R5, когда присутствует, представляет собой Н, ОН, -OC1-4 алкил, -CH2NH2, -OCF3, -С(O)NHC1-4алкил или -CO2H;
R6 представляет собой -NR11R12 или группу 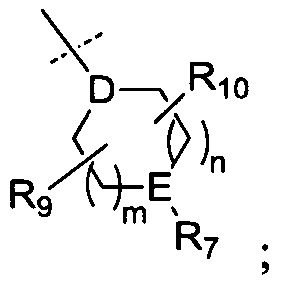
D представляет собой СН или N;
Е представляет собой N, О, СН или SO2;
R7, когда присутствует, представляет собой Н, ОН, С1-4алкил, -SO2C1-4алкил, -NHSO2CH3, -С(O)С1-4алкил;
R8, когда присутствует, представляет собой Н, С1-4алкил, ОН, -ОС1-4алкил, -ОС2-4алкиленОС1-4алкил, -OCF3; -ОС1-4алкиленF, -ОС1-4алкиленCHF2, -ОС2-4алкиленОН, -Офенил, -ОС1-4алкиленфенил, -NHC3-7циклоалкил, -NHC1-4алкиленС3-7циклоалкил, -ОС3-7циклоалкил, -ОС1-4алкиленС3-7циклоалкил, -NHC4-6гетероцикл, -NHC1-4алкиленС4-6гетероцикл, -ОС4-6гетероцикл или -ОС1-4алкиленС4-6гетероцикл, содержащий в качестве гетероатома N или О, где каждый С3-7циклоалкил или С4-6гетероцикл возможно замещен одним или двумя заместителями, независимо выбранными из галогена и оксо; или
R8 и R2 вместе с атомами углерода, к которым они присоединены, образуют гетероцикл, содержащий в качестве гетероатома N и возможно замещенный оксо;
R9 представляет собой Н, С1-4алкил, галоген, ОН, -ОС1-4алкил, -СН2ОН, -C(O)NHCH3, -C(O)NH(CH3)2;
R10 представляет собой Н, С1-4алкил, галоген, ОН, -ОС1-4алкил или оксо;
R11 представляет собой Н, С1-4алкил или SO2CH3;
R12 представляет собой Н, С1-4алкил, C2-4aлкилeнNHR13, SO2CH3, 5-6-членный гетероцикл, содержащий SO2;
R13 представляет собой Н или SO2CH3;
R14 представляет собой Н или С1-4алкил;
R15 представляет собой Н, С1-4алкил или NHC(O)С1-4алкил;
R16 представляет собой Н или С1-4алкил; и
каждый из n и m представляет собой целое число, независимо выбранное из 0, 1 и 2; при условии, что не более 2 из V, W, X, Y и Z представляют собой N;
или его соль.
2. Соединение или его соль по п. 1, где V представляет собой C-R2 и W представляет собой C-R8.
3. Соединение или его соль по п. 1, где R8 представляет собой Н, ОН, -ОСН2СН3, -ОСН(СН3)2, -ОСН2СН2ОСН3, -ОСН2СН(СН3)ОСН3, -ОСН(СН3)СН2ОСН3, -OCH2CH2F, -OCH2CHF2, -ОСН2СН2ОН, -NHCH2циклопропил, -О-циклопропил, -ОСН2циклопропил, -О-тетрагидрофуранил, -О-оксетанил, -ОСН2тетрагидрофуранил, -ОСН2оксетанил или -ОСН2СН2пирролидинил, где каждый С3-7циклоалкил или С4-6гетероцикл возможно замещен одним или двумя заместителями, независимо выбранными из фтора и оксо.
4. Соединение или его соль по п. 1, где X представляет собой СН; Y представляет собой N; Z представляет собой СН и Q представляет собой СН.
5. Соединение или его соль по п. 1, где R1 представляет собой метил.
6. Соединение или его соль по п. 1, где R2 представляет собой Н, -ОС1-4алкил, -NHC(O)С1-4алкил или -N(СН3)С(O)С1-4алкил.
7. Соединение или его соль по п. 1, где R6 представляет собой группу 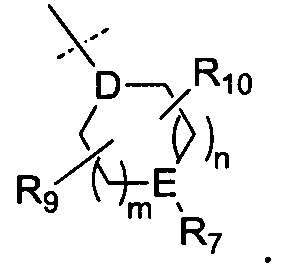
8. Соединение или его соль по п. 7, где D представляет собой N и Е представляет собой N, О или СН.
9. Соединение или его соль по п. 7, где R7 представляет собой -SO2CH3.
10. Соединение, выбранное из:
7-[3,4-бис(метилокси)фенил]-5-метил-2-{[4-(метилсульфонил)-1-пиперазинил]метил}фуро[3,2-с]пиридин-4(5H)-она;
(R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
7-(3-(бензилокси)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3-(бензиламино)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
2-(((2-аминоэтил)амино)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-она;
7-(4-(аминометил)фенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3,4-диметоксифенил)-5-метил-2-(пиперидин-1-илметил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3,4-диметоксифенил)-5-метил-2-(морфолинометил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3,4-диметоксифенил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
2-((1,4-диазепан-1-ил)метил)-7-(3,4-диметоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(5-(1-фенилэтокси)пиридин-3-ил)фуро[3,2-с]пиридин-4(5H)-она;
2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((1-фенилэтил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((1-фенилэтил)амино)фенил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3,4-диметоксифенил)-5-метил-2-((3-метилморфолино)метил)фуро[3,2-с]пиридин-4(5H)-она;
N-(4-(2-((3-фторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((3,3-дифторпиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
2-((3-фторпиперидин-1-ил)метил)-7-(4-метоксифенил)-5-метилфуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(морфолинометил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(S)-5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
2-((1,4-оксазепан-4-ил)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(2-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)циклопропанкарбоксамида;
(R)-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)пропионамида;
(R)-7-(2-(2-метоксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(пирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
2-((1,1-диоксидотиоморфолино)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[2,3-d]пиридазин-4(5H)-она;
(R)-N-метил-N-(4-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3,4-диметоксифенил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[2,3-d]пиридазин-4(5H)-она;
2-((1,4-диазепан-1-ил)метил)-5-метил-7-(2-((пиридин-2-илметил)амино)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-(2-(2-оксопирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(S)-N-(4-(5-метил-2-(1-(4-(метилсульфонил)пиперазин-1-ил)этил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-N-(5-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-3-ил)ацетамида;
7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-((циклопропилметил)амино)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((S)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(циклопропилметокси)пиридин-2-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((2-метилпиперазин-1-ил)метил)-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она гидрохлорида;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-3-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
2-((4-ацетил-2-метилпиперазин-1-ил)метил)-5-метил-7-(2-((тетрагидро-2H-пиран-4-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она гидрохлорида;
N-(3-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)фенил)ацетамида;
(R)-7-(2-((циклопропилметил)амино)пиридин-3-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-аминофенил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
2-((1,4-диазепан-1-ил)метил)-5-метил-7-(3-((пиридин-2-илметил)амино)фенил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-этоксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
N-(3-(2-((1,4-диазепан-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)фенил)пиколинамида;
(R)-N-(6-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
2-((1,4-диазепан-1-ил)метил)-7-(3-(циклопропилметокси)пиридин-4-ил)-5-метилфуро[3,2-с]пиридин-4(5H)-она;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-метил-1,4-диазепан-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-((4-метил-1,4-диазепан-1-ил)метил)-7-(3-((пиридин-2-илметил)амино)фенил)фуро[3,2-с]пиридин-4(5H)-она гидрохлорида;
(R)-7-(3-изопропоксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(2-аминопиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-гидроксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3-((2,2-дифторциклопропил)метокси)пиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-N-(4-(5-(2Н3)метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(2-(2-оксопирролидин-1-ил)этокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-амино-N-(3-(5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)фенил)пентанамида;
N-(4-(5-метил-2-(2-(4-(метилсульфонил)пиперазин-1-ил)пропан-2-ил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-7-(3-(2-метоксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
3-(циклопропилметокси)-4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензойной кислоты;
3-(циклопропилметокси)-N-этил-4-(5-метил-2-((4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)бензамида;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-3-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-2-ил)метокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((3-оксопиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(2-фторэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-циклопропоксипиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(2,2-дифторэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-7-(3-(2-гидроксиэтокси)пиридин-4-ил)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3-(циклопропилметокси)пиридин-4-ил)-5-метил-2-((4-метил-3-оксопиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(2-оксо-2,3-дигидро-1H-пирроло[2,3-b]пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3-(2-метоксипропокси)пиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
7-(3-((1-метоксипропан-2-ил)окси)пиридин-4-ил)-5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)фуро[3,2-с]пиридин-4(5H)-она;
(R)-5-метил-2-((2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(оксетан-3-илокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((тетрагидрофуран-3-ил)окси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(((S)-тетрагидрофуран-3-ил)окси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она; и
5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-(((R)-тетрагидрофуран-3-ил)окси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-она;
N-(4-(5-метил-4-оксо-2-((5-оксо-1,4-диазепан-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,4-оксазепан-4-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метилпиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метил-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,4-диазепан-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,1-диоксидотиоморфолино)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-((3-оксопиперазин-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонамидо)пиперидин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(S)-N-(4-(2-((3-гидроксипиперидин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-(пиперазин-1-илметил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-этил-3-оксопиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонил)пиперидин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-(((1,1-диоксидотетрагидро-2H-тиопиран-3-ил)амино)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-(((1,1-диоксидотетрагидротиофен-3-ил)амино)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((2,5-диметил-4-(метилсульфонил)пиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-этил-2-метилпиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метил-5-оксо-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((7-метил-5-оксо-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((2-метил-5-оксо-1,4-диазепан-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((1,1-диоксидо-1,4-тиазепан-4-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((3-метил-1,1-диоксидотиоморфолино)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((2-метил-1,1-диоксидотиоморфолино)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-ацетил-1,4-диазепан-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-(морфолинометил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-ацетил-2-метилпиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-метил-3-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(S)-N-(4-(5-метил-2-((3-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-2-((4-(метилсульфонил)-2-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(2-((4-этилпиперазин-1-ил)метил)-5-метил-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-(пирролидин-1-илметил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-(пиперидин-1-илметил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
N-(4-(5-метил-4-оксо-2-((3-оксо-1,4-диазепан-1-ил)метил)-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
(R)-N-(4-(5-метил-2-((2-метил-3-оксопиперазин-1-ил)метил)-4-оксо-4,5-дигидрофуро[3,2-с]пиридин-7-ил)пиридин-2-ил)ацетамида;
или его соли.
11. Соединение, которое представляет собой 5-метил-2-(((R)-2-метил-4-(метилсульфонил)пиперазин-1-ил)метил)-7-(3-((R)-оксетан-2-илметокси)пиридин-4-ил)фуро[3,2-с]пиридин-4(5H)-он или его соль.
12. Соединение по любому из пп. 1-11 или его фармацевтически приемлемая соль.
13. Фармацевтическая композиция, обладающая активностью ингибиторов бромодоменов, которая содержит соединение формулы (I) или его фармацевтически приемлемую соль по п. 12 и один или более фармацевтически приемлемых носителей, разбавителей или эксципиентов.
14. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 12 для использования в качестве лекарственного средства, обладающего активностью ингибиторов бромодоменов.
15. Соединение формулы (I) или его фармацевтически приемлемая соль по п. 12 для использования в лечении заболеваний или состояний, для которых показан ингибитор бромодомена.
16. Применение соединения формулы (I) или его фармацевтически приемлемой соли по п. 12, в изготовлении лекарственного средства для лечения заболеваний или состояний, для которых показан ингибитор бромодомена.
17. Способ лечения заболеваний или состояний, для которых показан ингибитор бромодомена, у субъекта, нуждающегося в этом, включающий введение терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли по п. 12.
18. Способ лечения по п. 17, где субъект представляет собой человека.
| WO 2011054553 A1, 12.05.2011 | |||
| WO 2013027168 A1 | |||
| D.L.J | |||
| Clive et al | |||
| "Model Studies and First Synthesis of Antifungal and Antibacterial Agent Cladobotryal", Jounal of Organic Chemistry, 2004, v.16, no.12, 1872-1879 | |||
| Chun-Wa Chung et al | |||
| "Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains", Journal of Medicinal Chemistry, 2011, v.54, no.11, p.3827-3838 | |||
| Железобетонное гнездо для телеграфных и т.п. деревянных столбов | 1925 |
|
SU2527A1 |

