Изобретение относится к области исследования химических и физических характеристик вещества и может быть использовано в области медицины, пищевой промышленности, при геологических, криминалистических, технологических и др. исследованиях, а также для контроля качества технологического производства и выпускаемой и продаваемой продукции.
Известен интегрально-сцинтилляционный способ исследования вещества с введением его в плазму, включающий переведение вещества в порошковое состояние, съемку с использованием интегрально-сцинтилляционного спектрометра спектров известных веществ и образцов сравнения, близких по химическому составу к исследуемому веществу, с виртуальным делением исследуемого вещества на большое число частей (микронавесок), путем осуществления периодической прерывистой синхронной кратковременной покадровой регистрации спектрального излучения плазмы источника возбуждения спектров с временем накопления спектральных сигналов в кадре меньшем удвоенной длительности сцинтилляционных импульсов отдельных частиц вещества, попадающих в плазму, калибровку («профилирование») шкалы спектрометра по спектрам известных веществ, нахождение в зарегистрированных спектрах веществ местоположения спектральных аналитических линии и фона вблизи них, покадровую сортировку аналитических сигналов, полученных при исследовании виртуальных микронавесок вещества с учетом фонового излучения плазмы, расчет по аналитическим сигналам, полученным после их покадровой сортировки, суммарной интенсивности аналитической спектральной линии определяемого химического элемента с учетом сигналов всех полученных спектров виртуальных микронавесок исследованного образца сравнения, построение градуировочных графиков химических элементов с использованием рассчитанных суммарных интенсивностей спектральных аналитических линий определяемых химических элементов в образцах сравнения, проведение с емки спектров исследуемого вещества, сортировку аналитических сигналов микронавесок исследуемого вещества с учетом фона, расчет суммарных интенсивностей спектральных линий определяемых химических элементов с учетом всех виртуальных микронавесок исследуемого вещества, определение по суммарным интенсивностям с использованием; построенных градуировочных графиков «условных содержаний» химических элементов, входящих в состав анализируемой навески вещества, а за тем расчет реальных содержаний химических элементов в исследуемом веществе по «соотношению условных содержаний», при котором предполагается, что сумма всех содержаний химических элементов в исследуемом веществе составляет 100%, расчет характеристик исследуемого вещества, осуществление фазовой диагностики вещества путем сравнения рассчитанных соотношений содержаний химических элементов в исследуемом веществе с соотношением содержаний этих же элементов в известных веществах и оценку качество исследуемого вещества, (см. Патент РФ №2172949, Способ спектрального анализа, Аполицкий В.Н. 1998 г., 13 стр., Патент РФ №2272277, Способ интегрально-сцинтилляционного спектрального анализа вещества, Аполицкий В.Н. 2004 г., 11 стр).
Наиболее близким по техническому решению и достигаемому эффекту к данному техническому решению является интегрально-сцинтилляционный способ исследования вещества с введением его в плазму, включающий переведение вещества в порошковое состояние, съемку с использованием интегрально-сцинтилляционного спектрометра спектров известных веществ и образцов сравнения, близких по химическому составу к исследуемому веществу, с виртуальным делением исследуемого вещества на большое число частей (микронавесок), путем осуществления периодической прерывистой синхронной кратковременной покадровой регистрации спектрального излучения плазмы источника возбуждения спектров с временем накопления спектральных сигналов в кадре; меньшем удвоенной длительности сцинтилляционных импульсов отдельных частиц вещества, попадающих в плазму, калибровку («профилирование») шкалы спектрометра по спектрам известных веществ, нахождение местоположения спектральных аналитических линии и фона вблизи этих линии в зарегистрированных спектрах веществ, покадровую сортировку аналитических сигналов, полученных при исследовании покадровых виртуальных микронавесок вещества с учетом фонового излучения плазмы, расчет по аналитическим сигналам, полученным после их покадровой сортировки, суммарной интенсивности аналитической спектральной линии определяемого химического элемента; с учетом сигналов всех полученных спектров виртуальных микронавесок исследованного образца сравнения, построение градуировочных графиков химических элементов с использованием рассчитанных суммарных интенсивностей спектральных аналитических линий определяемых химических элементов в образцах сравнения, проведение с емок спектров исследуемого вещества, сортировку аналитических сигналов микронавесок исследуемого вещества с учетом фона, расчет суммарных интенсивностей спектральных линий определяемых химических элементов с учетом всех виртуальных микронавесок исследуемого вещества, определение по суммарным интенсивностям с использованием построенных градуировочных графиков «условных содержаний» химических элементов, входящих в состав анализируемой навески вещества, расчет реальных содержаний химических элементов в исследуемом веществе по «соотношению условных содержаний», при котором предполагается, что сумма всех содержаний химических элементов в исследуемом веществе должна составлять 100%, расчет содержаний химический элементный, входящих в состав микронавесок вещества и отдельных инородных фазовых частиц, определение поэлементной и фазовой неоднородности вещества по содержаниям химических элементов в микронавесках вещества, итерационный расчет содержаний химических элементов в веществе, при котором исключаются аналитические сигналы инородных фаз и производят расчет характеристик вещества без инородных фаз, диагностику веществ с использованием сравнения; процентных соотношений содержаний химических элементов в изучаемом веществе с процентными соотношениями этих элементов в известных веществах, а также оценку качества исследуемого вещества (см. журнал «Заводская лаборатория. Диагностика материалов», Оценка неоднородности вещества с использованием интегрально-сцинтилляционного спектрального метода исследования, том 77, №5, Аполицкий В.Н., стр. 3-9, 2011 г., прототип).
Недостатками известных интегрально-сцинтилляционных способов исследования вещества с введением его в плазму (аналогов и прототипа) является невысокая точность определения содержаний химических элементов в веществах, определение небольшого числа фазовых характеристик веществ.
Целью данного предложения является повышение правильности и точности характеристик вещества и одновременное получение дополнительных его характеристик.
Поставленная цель достигается за счет того, что согласно интегрально-сцинтилляционному способу исследования вещества с введением его в плазму, включающему переведение вещества в порошковое состояние, съемку с использованием интегрально-сцинтилляционного спектрометра спектров известных веществ и образцов сравнения, близких по химическому составу к исследуемому веществу, с виртуальным делением исследуемого вещества на большое число частей (микронавесок), путем осуществления периодической прерывистой синхронной кратковременной покадровой регистрации спектрального излучения плазмы источника возбуждения спектров с временем накопления спектральных сигналов в кадре меньшем удвоенной длительности сцинтилляционных импульсов отдельных частиц вещества, попадающих в плазму, калибровку («профилирование») шкалы спектрометра по спектрам известных веществ, нахождение местоположения спектральных аналитических линии и фона вблизи этих линии в зарегистрированных спектрах веществ, покадровую сортировку аналитических сигналов, полученных при исследовании виртуальных микронавесок вещества с учетом фона, расчет по аналитическим сигналам, полученных после их покадровой сортировки, суммарной интенсивности аналитической спектральной линии определяемого химического элемента с учетом сигналов всех полученных спектров виртуальных микронавесок исследованного образца сравнения, построение градуировочных графиков химических элементов с использованием рассчитанных суммарных интенсивностей спектральных аналитических линий определяемых химических элементов в образцах сравнения, проведение съемки спектров исследуемого вещества, сортировку аналитических сигналов микронавесок исследуемого вещества с учетом фона, расчет суммарных интенсивностей спектральных линий определяемых химических элементов с учетом всех виртуальных микронавесок исследуемого вещества, определение по суммарным интенсивностям с использованием построенных градуировочных графиков «условных содержаний» химических элементов, входящих в состав анализируемой навески вещества, расчет реальных содержаний химических элементов в исследуемом веществе по «соотношению условных содержаний», при котором предполагается, что сумма всех содержаний химических элементов в исследуемом веществе составляет 100%, расчет содержаний химических элементов в отдельных микронавесок вещества и отдельных инородных фазовых частиц, определение поэлементной и фазовой неоднородность вещества по содержаниям химических элементов в микронавесках вещества, итерационный расчет содержаний химических элементов в веществе, при котором исключают аналитические сигналы инородных фаз и производят расчет характеристик вещества без инородных фаз, сравнение процентных соотношений содержаний химических элементов в изучаемом веществе с процентными соотношениями их в известных веществах, а также оценку качества исследуемого вещества, после калибровки (профилирования) измерительной шкалы интегрально-сцинтилляционного спектрометра осуществляют покадровую съемку спектров одинаковых аналитических навесок образцов сравнения с большим содержанием определяемых химических элементов, но не большим, чем содержания, при котором наблюдается реабсорбция спектральных линии определяемых элементов, проводят покадровую регистрацию аналитических фоновых сигналов электронной схемы спектрометра без горения плазмы и с плазмой без введения в нее вещества и с введением вещества, рассматривают покадровые зарегистрированные спектры, в которых присутствуют и отсутствуют аналитические линии определяемых элементов, с целью правильного отыскания местоположения фона вблизи аналитической спектральной линии определяемого элемента, выбирают покадровый участок спектра так, чтобы величина фона на этом участке спектра была близка к величине фона, находящегося под аналитической линией, при этом величину фона на выбранном участке спектра находят и рассчитывают так же, как величину аналитического сигнала спектральной линии определяемого химического элемента по максимальному значению аналитического сигнала на выбранном участке спектра, с использованием измерительной шкалы спектрометра определяют максимальную величину разброса флуктуаций фона («пороговый сигнал») путем наблюдения изменений величин покадровых фоновых аналитических сигналов, зарегистрированных в кадрах без горения плазмы, с введения вещества в плазму и без его введения, величину «порогового сигнала» выбирают больше максимального разброса флуктуаций фона, определяют величину аналитического сигнала на спектральной линии определяемого химического элемента по максимальному значению аналитического сигнала на выбранном участке спектра, из этой величины вычитают величину фона, расположенного рядом со спектральной линией определяемого элемента, и выбранный «пороговый сигнал», рассчитанную величину используют в качестве основного полезного сигнала, по которому осуществляют покадровую сортировку полезных аналитических спектральных сигналов образцов сравнения, и производят расчет суммарной интенсивности аналитической линии определяемых элементов с учетом всех микронавесок вещества, построение градуировочных графиков химических элементов осуществляют в простых координатах, по оси абсцисс откладывают содержания химического элемента в образцах сравнения, а по оси ординат величину суммарной интенсивности аналитического сигнала спектральной линии определяемого элемента в образце сравнения с учетом фона и его флуктуаций Iэл.ср.=Σ(Iэл.ф.-Iпр. сиг), градуировочные графики должны исходить из начала координат в виде прямой, проходящей через экспериментальные точки образцов сравнения, в которых имеются относительно большим содержания определяемых элементов, контроль правильности построенных градуировочных графиков осуществляют с использованием спектров холостых проб и контрольных образцов сравнения, зарегистрированных в разное время, после построения градуировочных графиков осуществляют съемку покадровых спектров исследуемого вещества, используя любые аналитические навески исследуемого вещества или частиц без их взвешивания, проводят покадровую сортировку аналитических сигналов исследуемых веществ с учетом фона и ранее полученного «порогового сигнала» подобно тому, как это делалось в случае сортировки сигналов образцов сравнения, по которым проводят расчет характеристик вещества. Определение тех или иных характеристик веществ осуществляют в зависимости от необходимости решения конкретных задач. Для определения правильности, точности исследований и предела обнаружения химических элементов используют нахождение полезных аналитических сигналов определяемых химических элементов, производя просмотр покадровых спектров виртуальных микронавесок исследуемого вещества на экране монитора, при котором оценивают величину аналитического сигнала, его форму и количество кадров, в которых наблюдается полезный сигнал. С целью определения дополнительных фазовых характеристик исследуемого вещества и более точного выявления инородных фаз при съемке спектров образцов сравнения берут одинаковые аналитические навески веществ массой 100 мг, а при построении в простых координатах градуировочных графиков по оси абсцисс откладывают величину массы в мг химического элемента, находящуюся в 100 мг навеске используемого образца сравнения, при осуществлении итерационного расчета исключают сигналы матричных элементов и элементов, находящихся в рассеянной фазовой форме, за счет осуществления покадровых вычетов из аналитических спектральных сигналов определяемых элементов, полученных с учетом фона и «порогового сигнала», рассчитанных до итерационного расчета, аналитических спектральных сигналов определяемых элементов, полученных с учетом фона и «порогового сигнала», полученных после итерационного расчета, при этом величины рассчитанных вычетов используют в качестве основных сигналов, по которым производят расчет процентных соотношения определяемых элементов в отдельных фазовых частицах, фазовую диагностику частиц, определение диаметров исследуемых частиц. Для повышения точности исследований длительность кадра делают такой, чтобы она была меньше удвоенной длительности спектрального сцинтилляционного импульса отдельной инородной частицы, попадающей в плазму, но не меньше длительности самого этого сцинтилляционного импульса. Предела обнаружения химических элементов в исследуемом веществе определяют с применением непараметрической статистики, используя пуассоновский закон распределения, по обнаружению двух частиц в исследуемой навеске вещества, при этом количественную оценку исследования оценивать в случае, если в исследуемом веществе находится более 25-30 частиц, содержащих определяемый элемент, с использованием параметрической статистике и градуировочных графиков, построенным в простых координатах, исходящих из начала координат. После получения характеристик вещества осуществляют исследование его с использованием других известных способов интегрально-сцинтилляционного исследования вещества с целью выявления правильности получения характеристик вещества. Способ применяют при интегрально-сцинтилляционном методе исследовании веществ в случае введения их в плазму способом испарения вещества из кратера электрода дуги в ее плазму.
Сущность предлагаемого способа.
Для совершенствования современного производства и дальнейшего развития науки необходимо создавать более точные, чувствительные, детальные и дешевые методы исследования разнообразных веществ. К таким методам исследования можно отнести прямой атомный интегрально-сцинтилляционный спектральный метод исследования веществ. Этот метод обладает более высокой точностью и чувствительностью определения содержаний химических элементов в различных веществах. Он позволяет кроме содержаний химических элементов в веществах определять и его фазовые характеристики. Изучение веществ с использования интегрально-сцинтилляционного метода исследования показали, что все вещества состоят из матричных фазовых частиц и инородных фазовых частиц, химический состав которых существенно отличается от состава частиц матрицы. Изучение форм нахождения химических элементов в веществах позволяет получать дополнительные, важные технологические характеристики вещества, которые дают возможность осуществлять надежную идентификацию, диагностику и оценку качества вещества, а также контроль производства.
Многочисленные исследования показывают, что точность и чувствительность прямых атомных эмиссионных спектральных методов анализа порошковых веществ существенно зависит от правильности определения величины интенсивности фона вблизи аналитических спектральных линий определяемых химических элементов и построения градуировочных графиков химических элементов по образцам сравнения. Обычно градуировочные графики представляют сложную кривую, построенную в логарифмических координатах по большому числу экспериментальных точек. Особенно труден процесс построения кривой в области определения низких содержаний химических элементов в веществе. В этом случае правильность построения кривой зависит от правильности нахождения местоположения фона в спектре, оценки величины сигнала самого фона и его флуктуаций, от случайных и неслучайных загрязнений вещества при подготовки его к исследованию и в процессе его исследования, от неточности паспортных значений содержаний химического элемента в образцах сравнения при малых содержаниях элементов в них, от неоднородности образцов. Использование нового интегрально-сцинтилляционного метода исследования вещества с делением навески исследуемого вещества на малые виртуальные части, за счет короткого покадрового накопления аналитических сигналов, позволило четко наблюдать молекулярную структуру фонового излучения плазмы и обнаружить существенные непредсказуемые флуктуации фона, связанные с шумами электронной схемы спектрометра. Это дает основание для нахождение фона вблизи спектральной линии и оценки его величины использовать структуру молекулярного фона с учетом не только величины фона, находящегося под аналитической линией, но и с учетом его флуктуаций. Это позволяет исключить искривление градуировочных графиков при малых содержаниях химических элементов в веществах.
Предлагается для более точного построения градуировочных графиков строить их в простых координатах (Iан.эл, Сер.эл). В этом случаи градуировочный график, построенный в простых координатах, должен проходить через начало координат. Что существенно упрощает и уточняет построение градуировочных графиков. График может быть построен в этом случае без использования неоднородных образцов сравнения, в которых имеются низкие содержания химических элементов. Он строится только по образцам сравнения, в которых содержания определяемых химических элементов относительно большое, когда влияние фона на правильность построения графика ничтожно. При этом точность определения низких содержаний химических элементов в исследуемых веществах в основном зависит не от точности построения калибровочного графика, а от правильного учета фона и его флуктуаций при расчете величин полезных аналитических спектральных сигналов исследуемых веществ. Особое значение при этом имеет выбор местонахождения и оценка величины самого фона, расположенного вблизи аналитической линии определяемого элемента. Исследования показали также, что результаты исследований искажаются за счет неправильного учета фона, расположенного под аналитической спектральной линией определяемого элемента. Это связано с тем, что аналитический сигнал на месте аналитической линии определяется по максимальной величине сигнала, а фон вблизи спектральной линии определяется по минимальной величине сигнала. Если в исследуемом веществе отсутствует определяемый элемент, то измеряемая величина фон на месте аналитической линии по максимальному сигналу будет всегда больше, чем величина фон, измеряемая на выбранном участке спектра, по минимальной величине сигнала. Для правильного учета фона необходимо измерять фон на выбранном участке спектра так же, как это делается при измерении аналитического сигнала спектральной аналитической линии определяемого элемента с поиском местоположения и измерении величины аналитического сигнала по максимальной амплитуде сигнала на выбранном спектральном участке. В этом случае градуировочный график проходит через начало координат. Это позволяет более точно исключать наложение молекулярного фона на спектральные аналитические линии определяемых элементов и найти максимальную величину разброса флуктуаций фона - «пороговый сигнал», путем наблюдения за флуктуациями фона в кадровых спектрах на экране дисплея, зарегистрированных при горящей плазме источника возбуждения спектров и без нее. В процессе покадровой сортировки сигналов из полученных аналитических спектральных сигналов химического элемента с учетом фона вычитают найденный «пороговый сигнал». Полученную разность используют для расчетах как содержаний определяемых химических элементов веществе, так и его виртуальных микронавесках при различных характеристик вещества.
Способы получения различных характеристик веществ с использованием нового интегрально-сцинтилляционного элементно-фазового метода исследования вещества изложены в прототипе и аналогах, в опубликованных статьях и патентах. Поэтому они детально не рассматриваются в случае предлагаемого способа.
Примеры конкретной реализации предлагаемого способа.
Пример 1. Необходимо оценить качество известных искусственных стандартных образцов, изготовляемых Бронницкой экспедицией ИМГРЭ, серия «Гранит», состоящей из 6 образцов, содержащих различные химические элементы.
При проведении исследований использовался интегрально-сцинтилляционный спектрометр «Спектроплазма-3» (см. аналоги и прототип) с подачей порошкового вещества с использованием «просыпки-вдувания» порошка вещества в плазму 3х-полусной дуги постоянного тока с двумя анодными и одним катодным электродом, который был создан на базе спектрографа СТЭ-1 с применением 9 ПЗС-линеек. Длительность накопления аналитических сигналов в кадре составляла менее 100 мс. Спектрометр позволяет определять более 50 химических элементов в широком диапазоне содержаний химических элементов в матрицы и инородных фазах. При проведении анализов веществ использовались образцы сравнения, имеющие различную матрицу (известные искусственные стандартные образцы типа «Гранит», «Ультраосновная порода» и др., изготовленные в Бронницкой экспедиции), и «холостые» пробы, в которых отсутствуют определяемые химические элементы.
Исследование веществ начинают с их подготовки к исследованию, с переведения вещества в порошковое состояние с крупностью частиц менее 500 мкм. Для этого используют измельчение твердых веществ или высушивания жидкостей и измельчение их сухих остатков. Далее осуществляют съемку спектров известного вещества с целью осуществления калибровки (профилирования) измерительной шкалы спектрометра. После калибровки измерительной шкалы интегрально-сцинтилляционного спектрометра проводят покадровую съемку спектров известных веществ, образцов сравнения и исследуемых веществ с виртуальным делением исследуемого вещества на большое число частей (микронавесок), путем осуществления периодической прерывистой синхронной кратковременной покадровой регистрации спектрального излучения плазмы источника возбуждения спектров с временем накопления спектральных сигналов в кадре меньшем удвоенной длительности сцинтилляционных импульсов отдельных частиц вещества, попадающих в плазму.
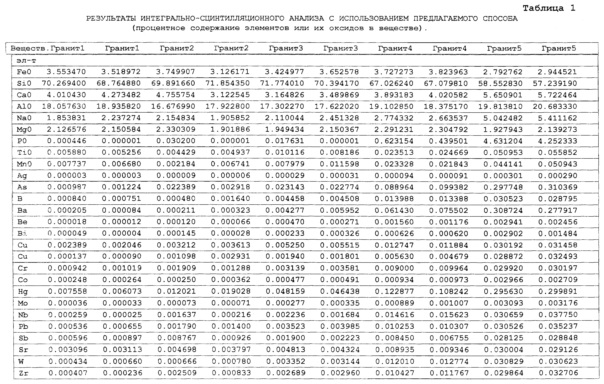
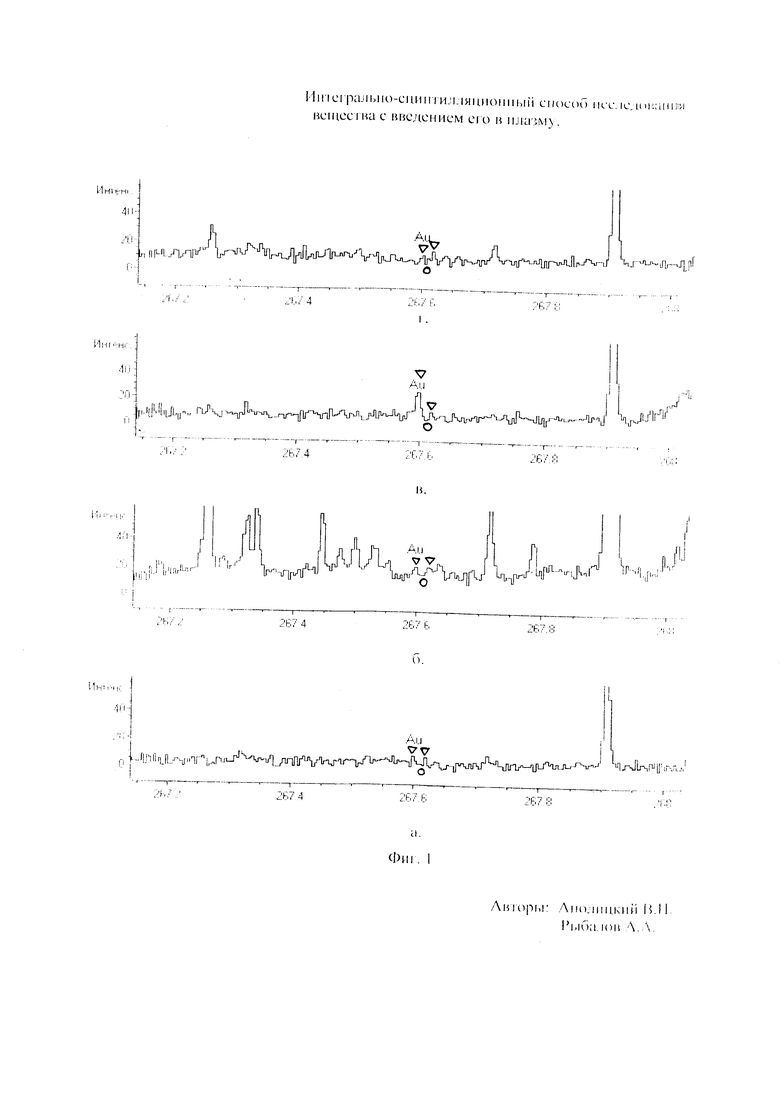
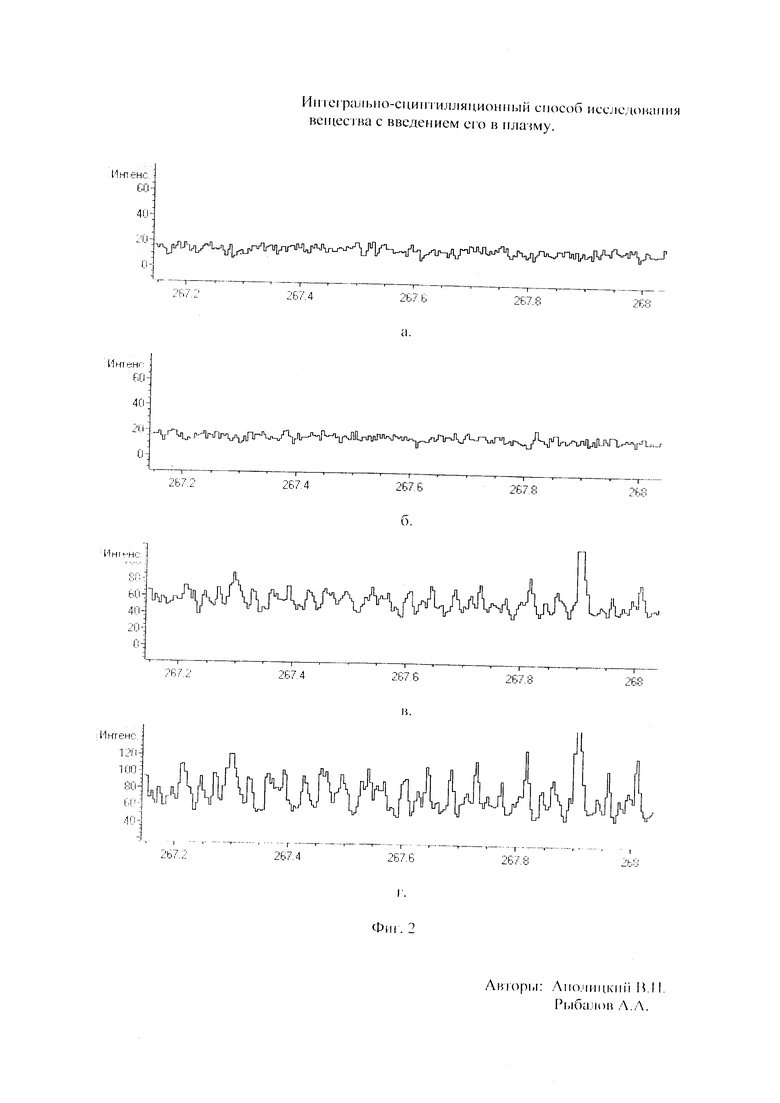
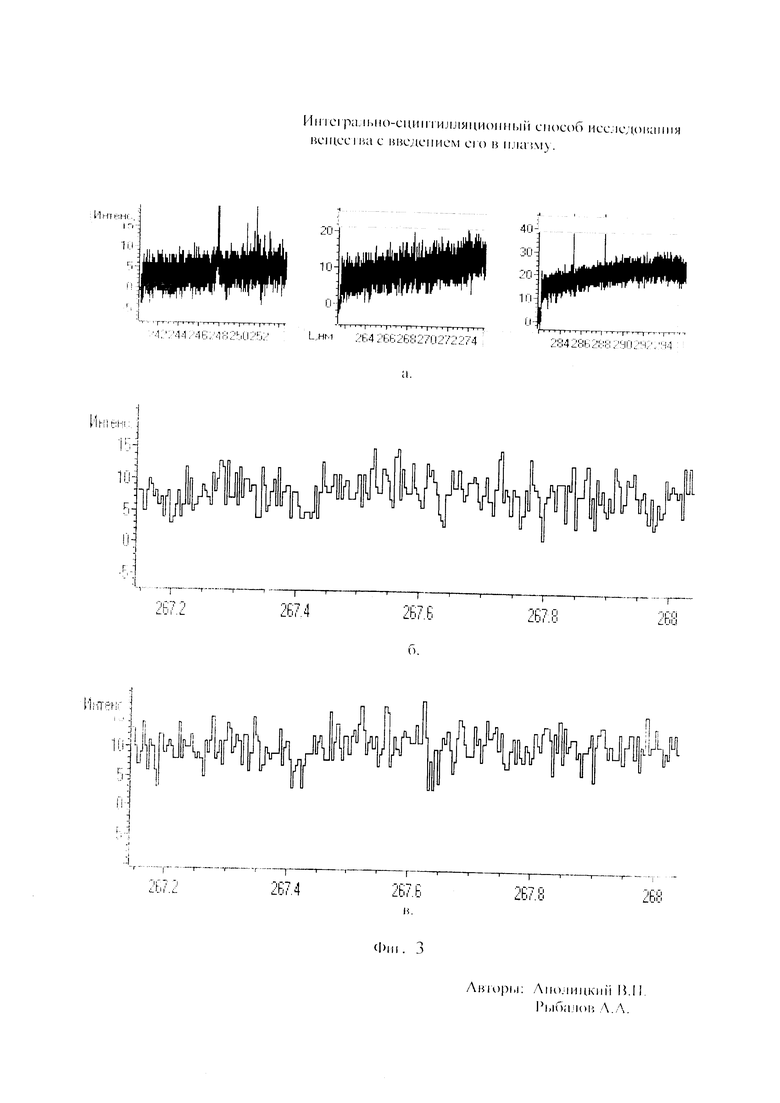
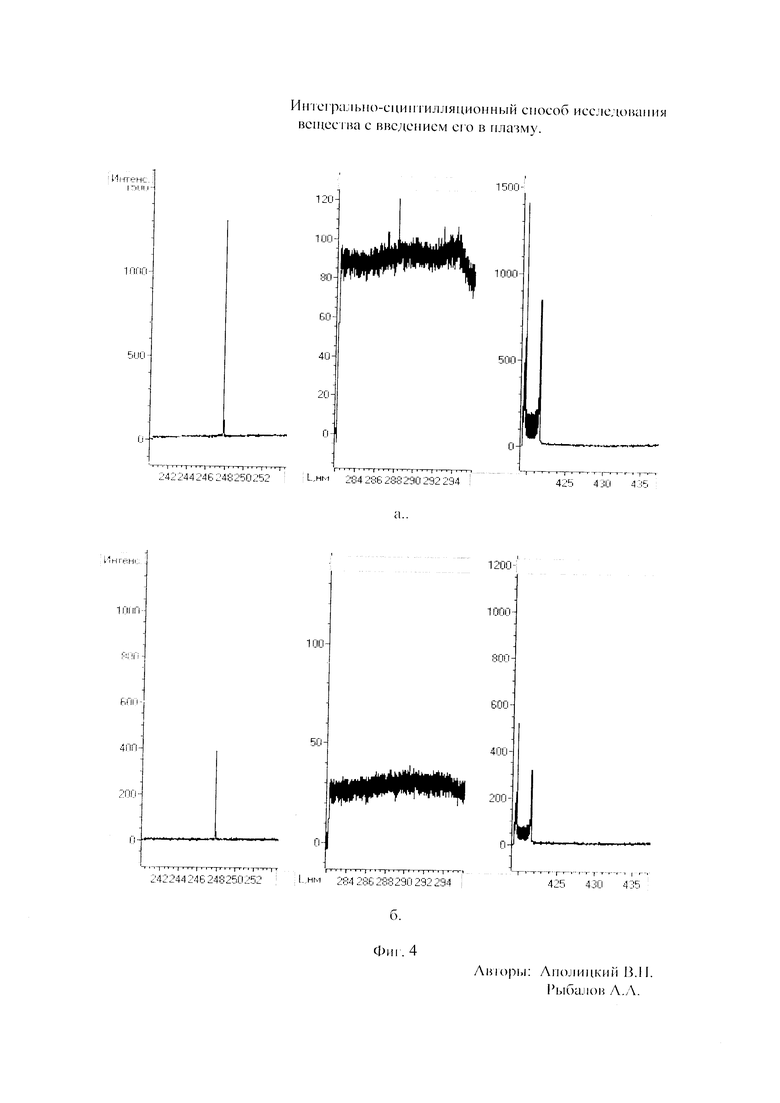
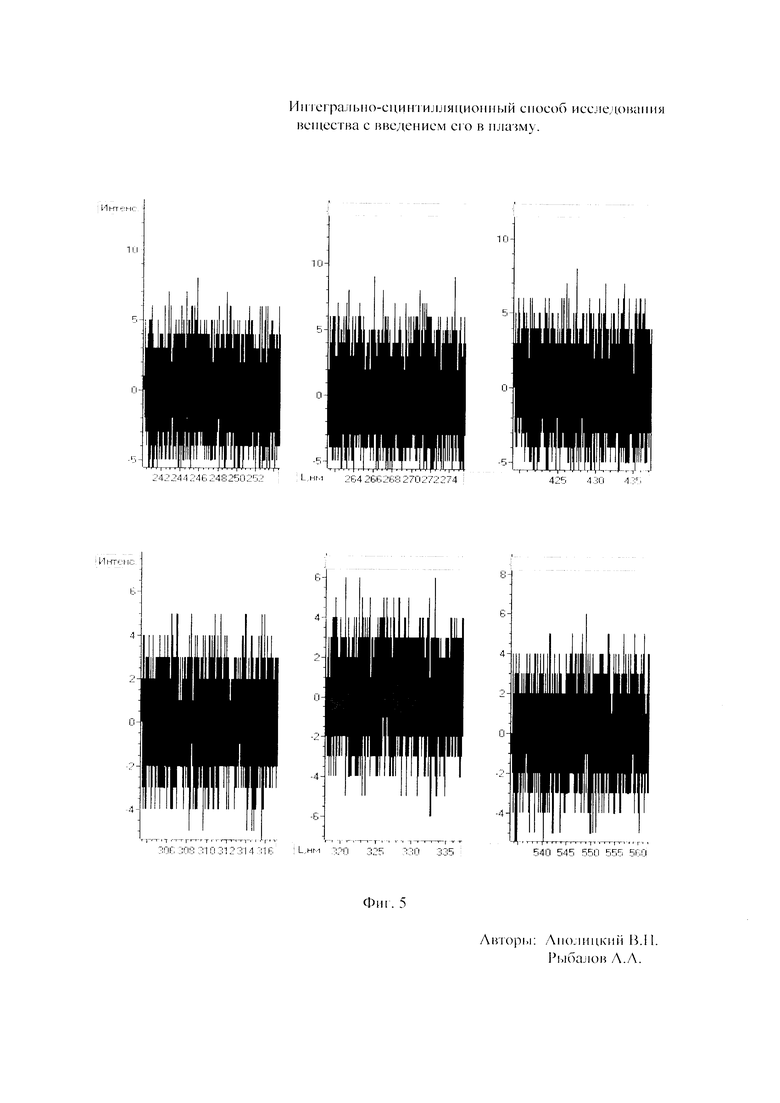
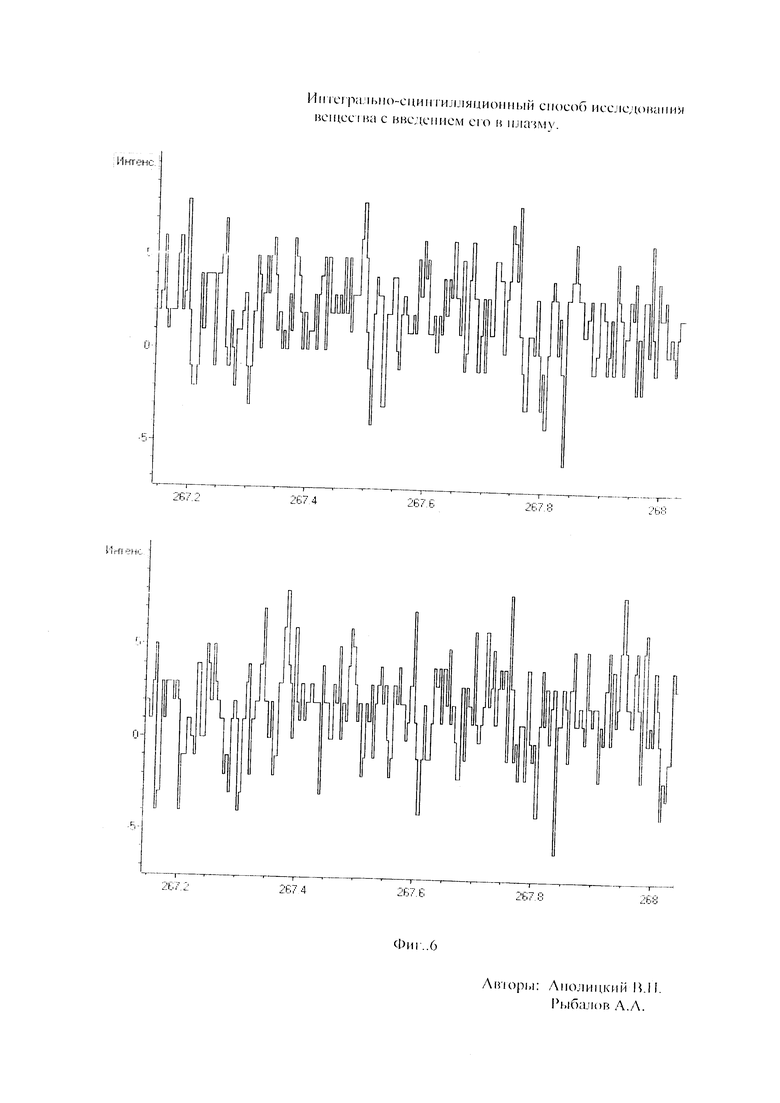
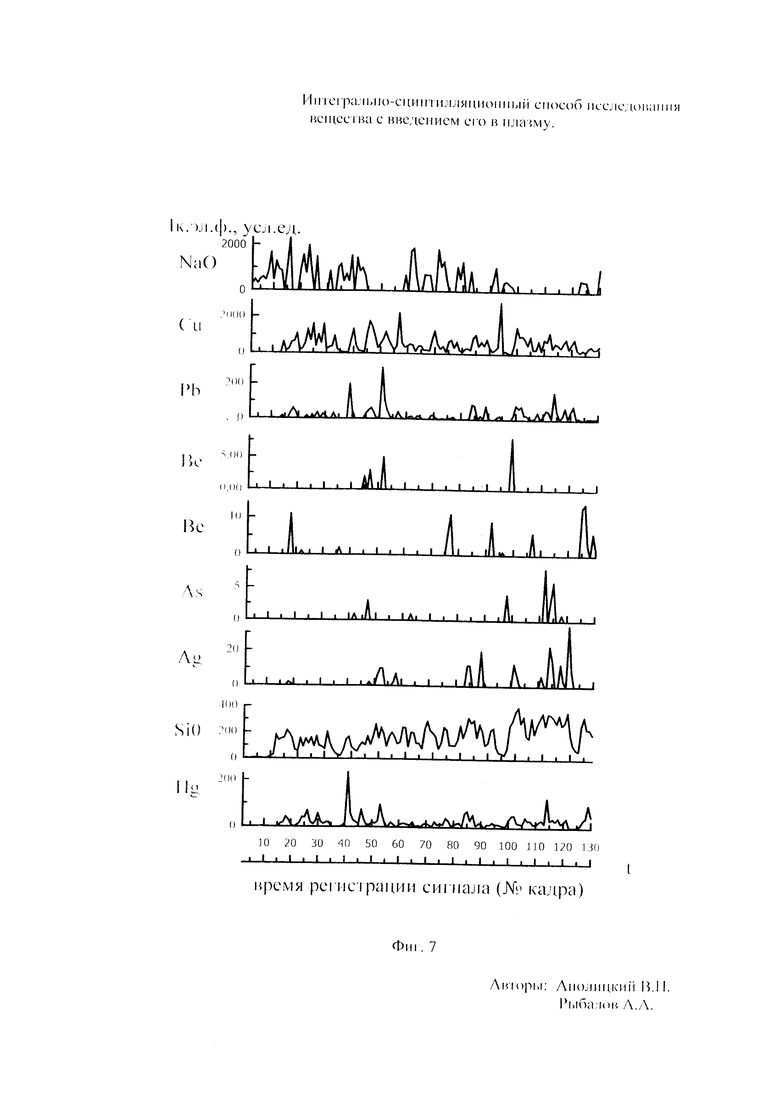
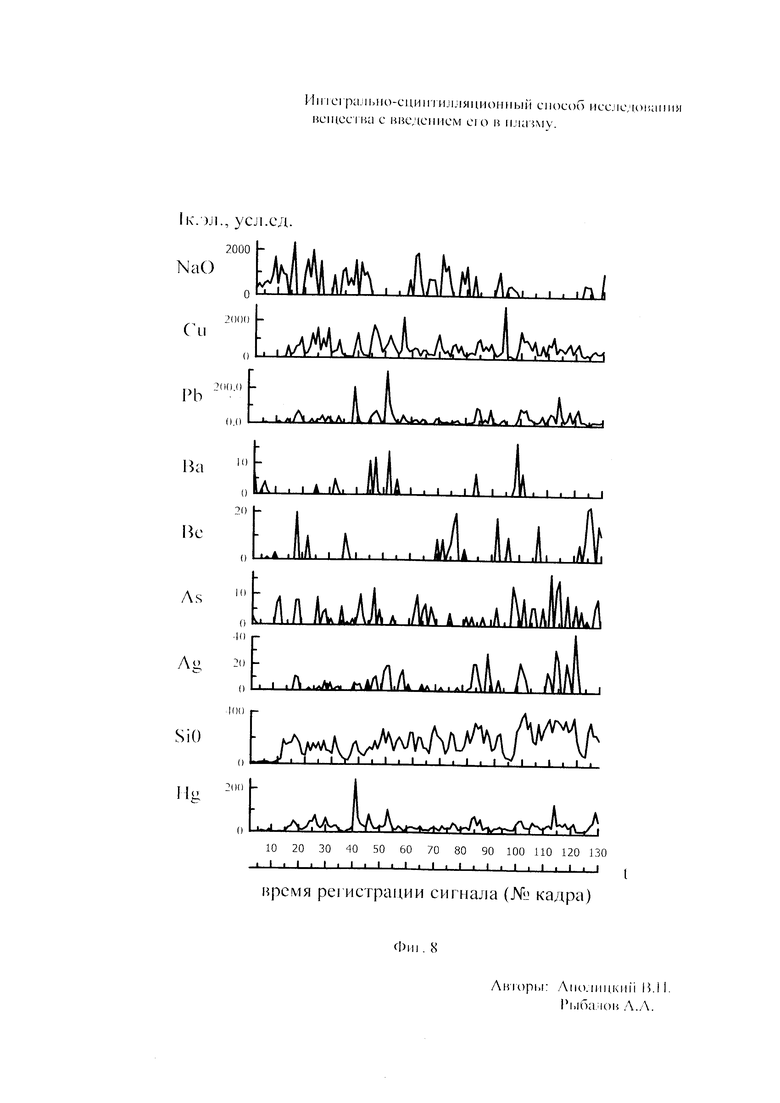
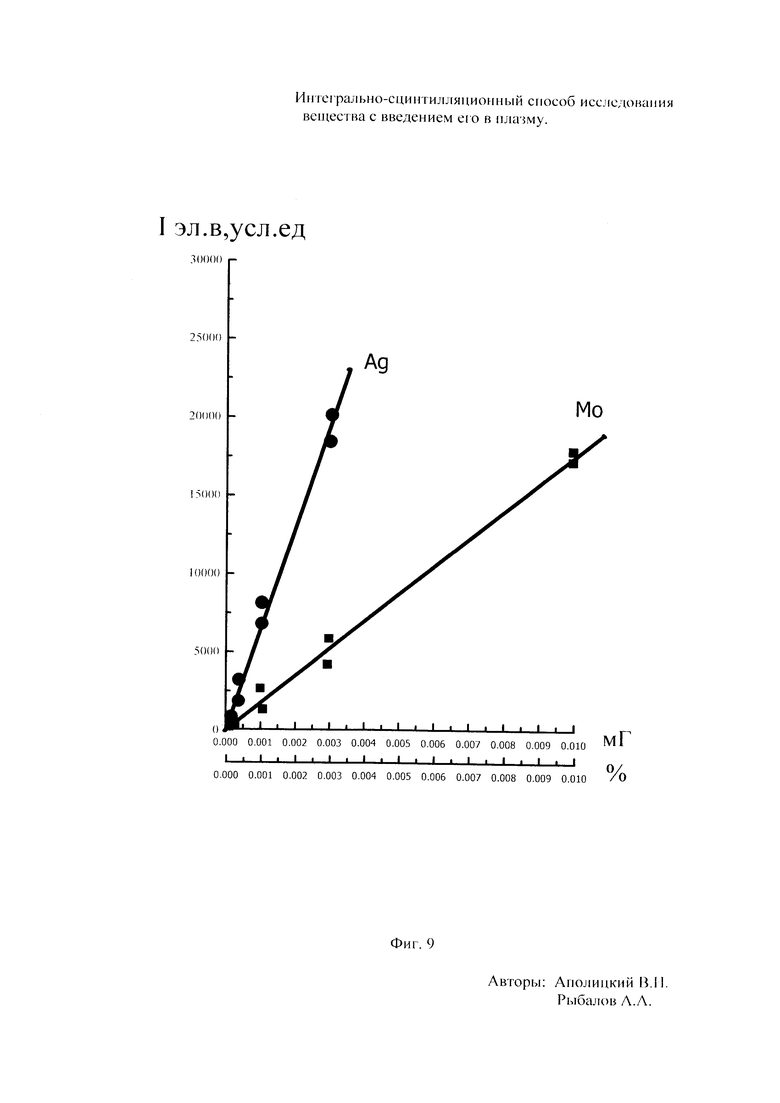
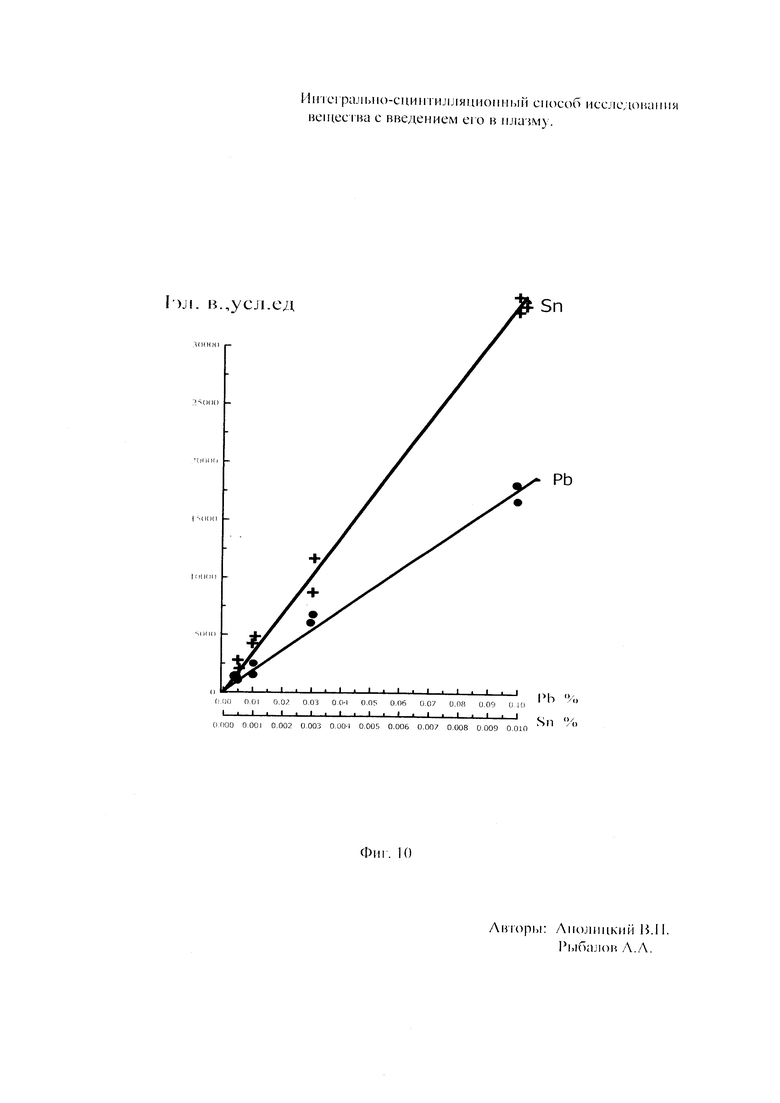
Как показали исследования точность и чувствительность интегрально-сцинтилляционного метода исследования существенно зависят от правильности нахождения в покадровых спектрах местоположения спектральной аналитической линии определяемого элемента и местоположение фона вблизи этой линии. На Фиг. 1 представлены 4 покадровых спектра виртуальных микронавесок (показана зависимость интенсивности излучения плазмы Iл, усл. ед. от длины волны в диапазоне длин волн от 267 до 268 нм) вещества, содержащего небольшие количество золота. Интегрально-сцинтилляционный метод исследования позволяет осуществить покадровую регистрацию спектров сотен виртуальных микронавесок вещества с небольшой длительностью кадров, меньшей 100 мс. Это позволяет уменьшить более чем в 100 раз фон в спектре кадра, не изменяя величину полезного сцинтилляционного сигнала, так как длительность его меньше времени длительности кадра. Это позволяет не только регистрировать отдельные сцинтилляционные сигналы инородных частиц вещества в отдельных кадрах, но и обнаружить молекулярное спектральное излучение плазмы и отдельные молекулярные полосы в спектрах микронавесок. На Фиг. 1 в спектрах видны сцинтилляционные сигналы различных химических элементов, в том числе и небольшой сцинтилляционный I аналитический сигнал спектральной линии золота (267,596 нм) (см. Фиг. 1в). При этом явно наблюдается наложение на эту линию фоновой молекулярной полосы излучаемой плазмой, искажающей сигнал золота. Обычно рекомендуется определять местонахождения фона по минимальному сигналу фона на выбранном участке спектра (см. на Фиг. 1, эти места отмечены снизу спектра кругляшками), а местоположение аналитической линии определять по максимальному сигналу на выбранном участке спектра (см. на Фиг. 1 - эти места в спектрах отмечены сверху треугольниками). Из-за различия способа нахождения сигнала фона Iф (по минимуму) и сигнала аналитической линии определяемого элемента Iэл.л (по максимуму) могут наблюдаться существенные искажения исследований при малых содержаний элементов в веществе. Для более правильного отыскания фона предлагается выбирать участок спектра не по минимальной величине сигнала на выбранном участке, а по максимальному сигналу фона на выбранном участке спектра так, чтобы величина фона была близка к величине фона, находящегося под аналитической линией. При этом величину фона на выбранном участке спектра находят и рассчитывают так же, как величину аналитического сигнала спектральной линии определяемого химического элемента по максимальному значению аналитического сигнала на выбранном участке спектра. Выполнение этих условий позволяет правильно учитывать молекулярный фон под аналитической линией элемента как в случае наложения молекулярной полосы на линию, так и без нее. При этом в рассматриваемом случае правильно в качестве фона выбрать рядом с аналитической линией элемента молекулярную полосу, подобную молекулярной полосе, находящейся под аналитической линией. Проведенные исследования показывают, что молекулярная структура фона в спектрах исследуемых веществ зависит от химического состава исследуемых веществ. На Фиг. 2 представлены покадровые спектры плазмы угольной 3-полюсной дуги без введения в нее вещества (см. фиг. 2а, б) и при введении в нее порошка кварца (см. в, г). В случае угольной дуги характерная структура молекулярного фона практически не наблюдается, а при введении кварца в плазму явно возрастает величина фона плазмы и проявляется характерная молекулярная структура фона в спектрах зарегистрированных кадров, при этом величина фона изменяется от кадра к кадру, т.е. зависит от номера кадра. Это требует покадрового учета фона, что повышает правильность и точность исследований. Подобная характерная структуре фона, наблюдающаяся и на Фиг. 1. Покадровый учет фона позволяет надежней обнаружить наложение молекулярных полос на аналитические спектральные линии и правильно выбрать местоположения фона и его величину. В некоторых случаях найти фон равный фону под аналитической линией сложно, в этом случае можно использовать экспериментальный коэффициент, на который умножают выбранный фон, чтобы он был более близким к фону расположенному под аналитической линией определяемого элемента (как это делается в случае при учете наложения спектральных линий различных элементов друг на друга). После нахождения величины фона и величины аналитического сигнала аналитической спектральной линии определяемого элемента проводят покадровый расчет величины интенсивности аналитического спектрального сигнала определяемого элемента с учетом фона 1к.эл.=Iэл,л-Iф. Исследования показали, что правильность и точность исследований существенно зависит не только от величины найденного фона в кадре, но и от флуктуации покадрового фонового излучения. Для обнаружения этих флуктуаций необходимо провести покадровую съемку спектров плазмы без введения в нее вещества, съемку спектров с введением в плазму «холостых проб» и съемку «спектров» фона электронной схемы спектрометра без горения плазмы. На Фиг. 3а представлен вид покадровых спектров фона плазмы без введения в нее вещества, зарегистрированных на трех ПЗС-линейках. В случае осуществления регистрации сигналов на различных линейках величина фоновых флуктуаций примерно одинакова и составляет величину меньшую 10 делений шкалы спектрометра. На Фиг. 3(б, в) показана более детальная структура этого фона в области длин волн от 267 нм до 268 нм в двух разных кадрах. Величина сигналов в этих спектрах изменяется хаотично (непредсказуемо), не наблюдается связь флуктуаций и с величиной самого молекулярного фона, при этом четко наблюдается разброс (флуктуации) сигналов фона с величиной меньшей 10 делений шкалы спектрометра. С целью понимания природы (происхождения) этих флуктуаций были проведены эксперименты, при которых световой поток, исходящий из плазмы, ослаблялся нейтральным ослабителем (сеткой). На Фиг. 4 приведены спектры плазмы, зарегистрированные с помощью 3 ПЗС-линеек, без ослабления потока (а) и при его ослаблении (б). В случае ослабления потока величина интенсивности молекулярного фона и спектральных линий элементов снижаются в три раза, а флуктуации фона остаются практически не изменяемыми (см. средний спектр - флуктуации), т.е. регистрируемые флуктуации не связаны с плазмой источника возбуждения спектров. На Фиг. 5 представлены спектры фоновых сигналов только электронной схемы спектрометра, которые были получены без горения плазмы, зарегистрированные с помощью 6 ПЗС-линеек в диапазоне длин волн от 240 нм до 560 нм. Наблюдается характерный вид фоновых спектральных сигналов, одинаковый для всех ПЗС-линеек, максимальная величина сигналов в спектрах изменяется от -5 до +5 делений шкалы спектрометра с максимальным разбросом сигналов в 10 дел. При этом изменения сигналов фона носит случайный характер. Сравнение детальной структуры флуктуаций покадровых сигналов, полученных при съемке спектра плазмы дуги без введения в нее вещества (см. Фиг. 3б, в), со структурой сигналов фона электронной схемы спектрометра, которая представлена на Фиг. 6, позволяет считать, что по природе происхождения и по характеру флуктуаций сигналов близки. Аналитические сигналы фона электронной схемы спектрометра имеют значительную величину и могут существенно изменять (искажать) результаты исследования и изменять вид градуировочных графиков при определении малых содержаний элементов в веществе. С целью повышения правильности, точности и чувствительности интегрально-сцинтилляционных исследований предлагается после расчета величины интенсивности сигнала аналитической линии определяемого элемента с учетом фона, определить по измерительной шкале спектрометра максимальную величину разброса флуктуаций фона («пороговый сигнал») Iпор.сиг. путем наблюдения изменений величин покадровых фоновых аналитических сигналов, зарегистрированных в кадрах без горения плазмы, с введения вещества в плазму и без его введения, при этом величину «порогового сигнала» выбирают больше максимального значения разброса флуктуаций фона по рассмотренным покадровым спектрам. Величина «порогового сигнала» фона Iпор.сиг зависит от используемой конструкции электронной схемы спектрометра. Поэтому для уменьшения влияния флуктуаций фона на результат исследования необходимо снижать шумы электронной схемы спектрометра, например, за счет выбора светочувствительных элементов и их охлаждения. Величина «порогового сигнала» Iпор.сиг зависит от используемой конструкции электронной схемы спектрометра. По этому для уменьшения влияния флуктуаций фона на результат исследования необходимо снижать шумы электронной схемы спектрометра, например, за счет выбора светочувствительных элементов и их охлаждения, В нашем случае, величина «порогового сигнала» оказалась Iпор.сиг.=10 усл. ед. шкалы спектрометра. Величина «порогового сигнала» практически не меняется при длительной работе спектрометра. После определения «порогового сигнала» осуществляют покадровую сортировку аналитических сигналов образцов сравнения, в процессе покадровой сортировки из величины аналитического сигнала определяемого химического элемента с учетом фона вычитают выбранную величину «порогового сигнала» Iк.эл.ф.=Iк.эл.-Iпор.сиг, далее полученную разность используют в качестве основного аналитического сигнала определяемого элемента. Результат процесса сортировки аналитических сигналов различных химических элементов при использовании предлагаемого способа и способа, используемого в прототипе, при исследовании искусственного стандартного образца «Гранит» №2, представлен на «временных покадровых развертках» (см. Фиг. 7 и 8). На этих фигурах приведены зависимости величин интенсивностей аналитических спектральных сигналов различных химических элементов (Iк.эл.ф. и Iк.эл) от времени попадания частиц, содержащих определяемый элемент, в плазму t (или от номера № зарегистрированного спектрального кадра). В случае предлагаемого способа (см. Фиг. 7) по сравнению с прототипом (см. Фиг. 8) величина полезных аналитических сигналов, за счет более правильного нахождения местоположения фона и учета флуктуаций фона, уменьшается, при этом число полезных аналитических сигналов (больших нуля) существенно снижается, сигналы меньшие нуля при расчетах не используется. После покадровой сортировки сигналов осуществляют расчет суммарных интенсивностей аналитических линии определяемых элементов с учетом всех микронавесок исследуемого образца сравнения.. После расчета суммарных интенсивностей спектральных линий элемента Iэл.об.ср=Σ(Iк.эл.ф.об.ср.) в исследуемых образцах сравнения осуществляют построение, градуировочных графиков химических элементов. В силу детального учета фона и его флуктуаций при расчете Iэл.об.ср оказалось возможным градуировочные графики строить в простых координатах, по оси абсцисс откладывают содержания химического элемента в образцах сравнения (Сэл.об.ср,), а по оси ординат суммарную величину аналитического сигнала спектральной линии определяемого элемента с учетом величины фона и его флуктуаций (Iэл.об.ср,) (см. Фиг. 9, 10). За счет тщательного, правильного учета фона и его флуктуаций, градуировочные графики химических элементов исходят из начала координат виде прямой линии, наклон которой зависит от экспериментально полученных аналитических сигналов образцов сравнения. Для повышения правильности, точности исследований рационально использовать при построении графиков образцы сравнения с большими содержаниями определяемых химических элементов, но не большим, чем содержания, при котором начинается реабсорбция спектральной линии определяемого элемента. Такой способ построения графиков позволяет повысить точность и чувствительность исследований. В качестве образца сравнения нами используется длительное время только один стандартный искусственные образец №5 стандарта серии «Гранит», при использовании которого реабсорбция спектральных линий обычно еще не наблюдается. Контроль правильности и корректировка построенных градуировочных графиков осуществляют с использованием спектров холостых проб и веществ с известным содержаниями элементов, полученных в разное время. Это позволяет использовать градуировочные графики длительное время. При съемке спектров образцов сравнения рационально использовать наиболее удобную аналитическую навеску образца сравнения равную 100 мг, которая позволяет определять по построенным градуировочным графикам (см. фиг. 9) как процентное содержания определяемых элементов, так и массу определяемого элемента в мг в виртуальных микронавесок вещества и отдельных инородных фазовых частицах. Это позволяет рассчитывать дополнительных фазовые характеристик вещества. После построения градуировочных графиков осуществляют покадровую съемку спектров исследуемого вещества при этом можно использовать любые аналитические навески исследуемого вещества или отдельных частиц без их взвешивания. После получения покадровых спектров исследуемого вещества, подобно тому, как это делалось при расчете суммарной интенсивности спектральной аналитической линии определяемого элемента в образцах сравнения с использованием покадровой сортировки сигналов с учетом фона и выбранного «порогового сигнала», осуществляют расчет суммарной покадровой интенсивности спектральной аналитической линии определяемого элемента исследуемого вещества Iэл.в=Σ(Iк.эл.ф.в) Далее с использованием имеющихся градуировочных графиков, построенных по образцам сравнения близких по составу к исследуемому веществу, рассчитывают «условные содержания» матричных и инородных химических элементов в исследуемом веществе, а за тем их реальные содержания по «соотношению условных содержаний» элементов веществе. Если полученный матричный химический состав исследуемого вещества близок к составу образцов сравнения, по которым строились выбранные градуировочные графики, то производят расчет содержаний химических элементов в виртуальных микронавесках вещества и отдельных фазовых частицах микронавесок вещества. В случае, если матричный состав вещества существенно отличается от состава образцов сравнения, с помощью которых строились использованные градуировочный график, то выбирают из имеющихся градуировочных графиков графики, который строились по образцам сравнения близким по составу к исследуемому веществу. При этом осуществляют повторный расчет всех химических элементов в исследуемом веществе, его виртуальных микронавесках и отдельных фазовых частицах. В таблице 1 приведены полученные предлагаемым интегрально-сцинтилляционным способом содержания химических элементов матрицы и элементов инородных фаз в 5 стандартных искусственных образцах «Гранит» №1-№5. В таблице приводятся результаты исследования двух параллельных навесок каждого из стандартных образцов. Паспортные содержания химических элементов и их оксидов в стандартных образцах «Гранита» приведены в таблице 2. Паспортные данные образцов «Гранита» в большинстве случаев в пределах допустимых погрешностей совпадают с полученными результатами (см. Таблица 1 и 2). Конечно, с уменьшением содержаний химических элементов точность результатов анализов снижается, но это в первую очередь связана с поэлементной неоднородностью исследуемых веществ.
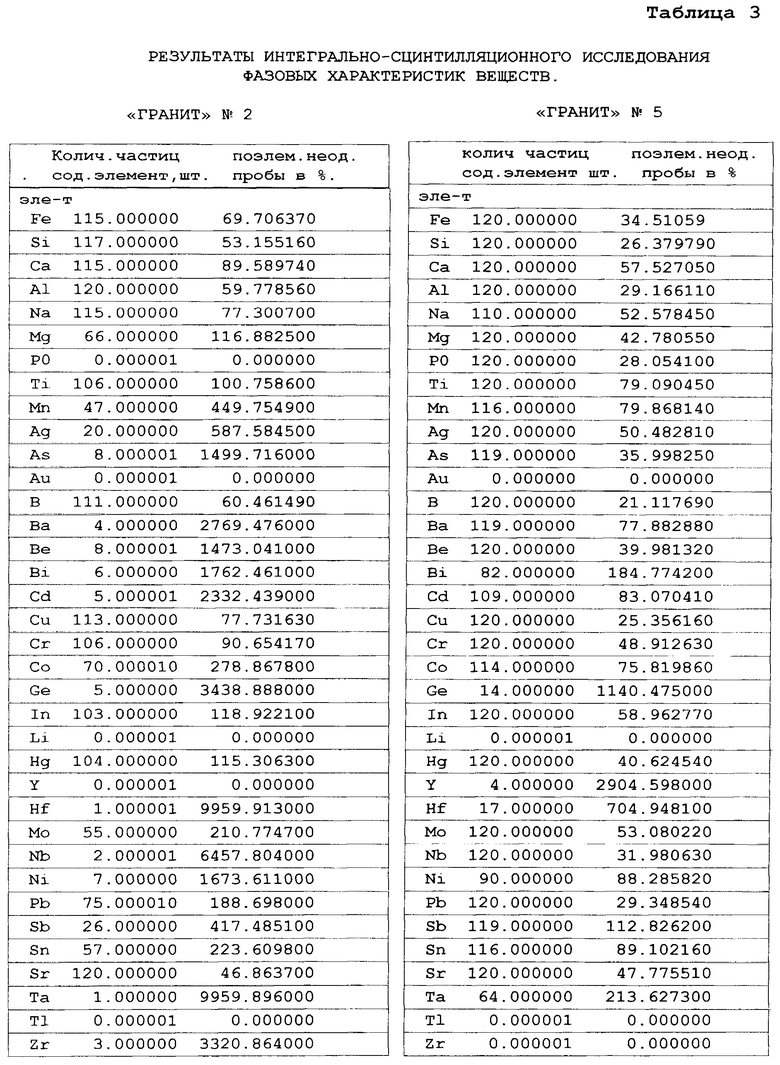
Далее, для оценки качества исследуемых вещества по полученным содержаниям определяемых химических элементов в виртуальных микронавесках вещества (они не приводятся из-за большого их количества, тонкости см. прототип) осуществляют оценку поэлементной неоднородности исследуемого вещества по разбросу полученных содержаний химических элементов виртуальных микронавесок вещества. Исследовалось более 120 микронавесок вещества. Поэлементная неоднородность вещества оценивалась по относительной среднеквадратической погрешности содержаний химических элементов в 120 микронавесках исследуемого вещества (см. таблица 3). Из таблицы 3 видно, что при определении содержаний элементов матрицы вещества поэлементная неоднородность составляет менее 50%. Если учесть то, что при расчете результатов исследования использовалось 120 параллельных исследований микронавесок, то в этом случае с учетом всех 120 измерений относительная среднеквадратическая погрешность исследований вещества составляет менее 5%. При содержаниях элементов меньших 0.001% из-за дискретного неравномерного нахождения элементов в веществе поэлементная неоднородность веществ составляет более 100-1000%. В этом случае точность исследований зависит от вероятности попадания отдельных частиц, содержащих определяемый химический элемент, в исследуемую навеску вещества (см. фиг. 1). Так как при этом наблюдается дискретный пуассоновский характер распределения частиц в веществе, то для оценки предела обнаружения химического элемента в веществе: необходимо использовать непараметрическую статистику, при которой учитывается число отдельных частиц, попавших в исследуемую аналитическую навеску вещества (подробней см. - Аполицкий В.Н. «Оценка предела обнаружения элементов при анализе порошковых; проб», Заводская лаборатория, Диагностика материалов. 1998, Т. 64, №8, С. 24-28). В процессе расчета содержаний химических элементов в микронавеске вещества несложно определять число виртуальных микронавесок в исследуемом веществе, в которых находятся определяемые химические элементы. В таблице 3, столбец 1 указано число частиц (микронавесок), в которых присутствует определяемый элемент. Если оценивать предел обнаружения по нахождению двух частиц в аналитической навеске вещества, то вероятность обнаружения определяемого химического элемента в веществе составляет 80%. Из таблицы 3 в «Граните» №2 видно, что количественные определения содержаний элементов большого числа элементов (As, Ba, Be, Bi, Cd, Ge, Ni, Zr) не возможно. Это содержания элементов на уровне 0.001% и ниже (см. Таблицу 2). Более точное количественное определение содержание элемента в веществе может осуществляться при нахождении в аналитической навеске вещества более 25-30 частиц, содержащих определяемый элемент. В этом случае пуассоновское распределение переходит в гауссовское (нормальное) распределение и появляется возможность применения параметрических статистических методов для оценки точности исследования. С целью подтверждения факта правильности обнаружения полезного сигнала в виртуальных микронавесках вещества необходимо осуществлять визуальное наблюдение на экране монитора (см. фиг. 1) аналитические спектральные линии определяемых элементов в покадровых спектрах виртуальных микронавесок вещества, оценивая величину аналитического сигнала, форму спектральной линии элемента и количеством частиц, зарегистрированных в кадрах исследуемого вещества.
Важной характеристикой любого вещества является форма нахождения химического элемента в веществе (элемент может находиться в собственной и рассеянной форме). Если химический элемент находится в отдельной частице (фазе) вещества и его содержание больше 10%, то считается, что он находит в собственной фазовой форме. Обычно это частицы инородных фаз. Если содержания химического элемента в частице меньше 1-10%, и элемент равномерно распределен (рассеян) по объему вещества, то этот элемент находится в рассеянной форме (обычно этот элемент входит в состав матрицы вещества). Для обнаружения фазовой формы нахождения элемента необходимо создать условия для того, чтобы фазовая частица, в которой содержится определяемый элемент, попало в отдельную виртуальную микронавеску исследуемого вещества, попадающую в плазму источника возбуждения. Это можно сделать, если содержание элемента в веществе менее 0.01%, (частицы должны быть пространственно разделены в веществе при введении вещества в плазму), при этом длительность накопления аналитических сигналов в кадре должна быть более удвоенной длительности сцинтилляционного сигнала, возникающего при попадании отдельной частицы в плазму, для полной регистрации ее сцинтилляционного излучения. При длительности кадра меньшей длительности сцинтилляционного импульса частицы происходит уменьшение полезного аналитического сигнала, неполная регистрация его в отдельном кадре. Регистрация сцинтилляционного излучения частицы в нескольких кадрах делает невозможным фазовую диагностику частиц. Фазовую собственную форму нахождения химического элемента в веществе можно определять по отсутствию определяемого химического элемента в более чем одной виртуальной микронавеске вещества. Присутствие определяемого химического элемента во всех виртуальных микронавесках вещества дает основание считать, что химический элемент находится в рассеянной фазовой форме. Определение присутствия и отсутствия химического элемента в виртуальных микронавесках вещества осуществляется в процессе расчета содержаний определяемых химических элементов во всех микронавесках исследуемого вещества (см. таблица 3, столбцы 1 и 3). В процессе исследования вещества регистрировалось 120 кадров (исследовалось 120 виртуальных микронавесок). Элементы матрицы в случае хорошего перемешивания искусственных стандартных образцов «Гранит» должны присутствовать во всех микронавесках стандартных образцов. Что подтверждается при исследовании стандартного образца «Гранит» - №5, в котором содержания химических элементов более 0,01%. Все определяемые элементы присутствуют во всех 120 микронавесках этого стандарта. Не смотря на то, что элементы, не входящие в состав матрицы искусственных стандартов «Гранит», находятся в собственной фазовой форме они присутствуют во всех микронавесках. Это связано с большим количеством этих частиц в микронавесках вещества, при содержаниях определяемых элементов в веществе больших 0,01%. Фазовая форма нахождения элемента может обнаруживаться только при содержаниях определяемых химических элементов в веществе меньшем 0,01%, В случае исследования вещества искусственных стандартных образцов «Гранита» с номером меньшим №3 (см. Таблица 3) можно видеть, что большинство определяемых химических элементов не присутствуют во всех микронавесках образца, что дает основание считать, что эти элементы находятся в собственной фазовой форме. Это подтверждается паспортными данными образцов - они вводились в образцах в виде частиц оксидов химических элементов. Количество частиц в аналитической навеске вещества меняется от 0 до 120 шт., что приводит к изменению поэлементной неоднородности вещества от 30 до величин больших 1000%, что дает основания считать качество стандартных образцов с номером меньшим №3 неудовлетворительным. Раздельное присутствие фазовых частиц в микронавесках вещества позволяет определять не только поэлементную неоднородность веществ и фазовую форму нахождения элемента, но и массу фазовой частицы, ее крупность и процентное содержание различных химических элементов в них (см. прототип и аналгии, в которых рассматриваются эти вопросы). Полученные характеристики вещества с использованием предлагаемого способа позволяют диагностировать вещества путем сравнения полученных характеристик с характеристиками известных веществ, как это делалось в прототипе.
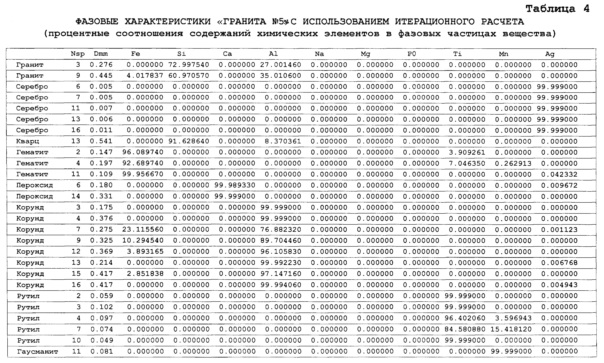
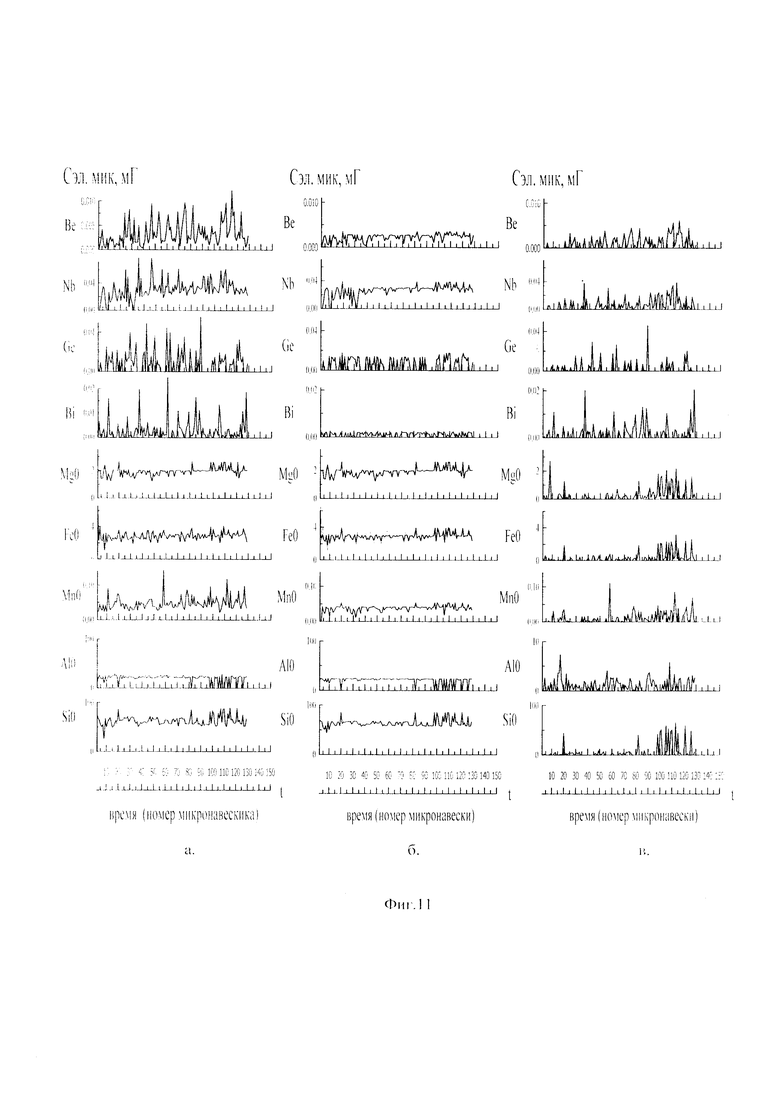
Для более правильного определения элементного и фазового состава матрицы исследуемого вещества и его отдельных инородных фазовых частиц рационально использовать итерационный расчет содержаний химических элементов и характеристик вещества. При пересчетах содержаний элементов в веществе осуществляют исключение аналитических спектральных сигналов инородных фаз, зарегистрированных в кадрах. Это позволяет осуществлять не только расчет процентных содержаний химических элементов без инородных фаз в матрице исследуемого вещества, но и выявлять фазовые формы нахождения химических элементов в инородных частицах, в которых содержатся даже матричные элементы. Для этого при съемке спектров образцов сравнения берут одинаковые аналитические навески веществ массой 100 мг, а при построении в простых координатах градуировочных графиков (см. фиг. 9) по оси абсцисс откладывают величину массы в мг химического элемента, находящуюся в 100 мг навеске используемого образца сравнения, при осуществлении итерационного расчета исключают сигналы матричных элементов и элементов, находящихся в рассеянной фазовой форме, путем осуществления покадровых вычетов из аналитических спектральных сигналов определяемых элементов, полученных с учетом фона и «порогового сигнала», рассчитанных до итерационного расчета, аналитических спектральных сигналов определяемых элементов с учетом фона и «порогового сигнала», полученных после итерационного расчета. При этом величины рассчитанных вычетов используют в качестве основных сигналов, по которым производят расчет процентных соотношения определяемых элементов в исследуемом веществе с учетом того, что сумма всех процентных содержаний химических элементов в исследуемом веществе должна быть равна 100%. Это позволяет осуществлять фазовую диагностику частиц, определять диаметр исследуемых частиц в веществе (подобно см. аналоги). На фиг. 11 показаны «временные покадровые развертки» содержаний химических элеметов в виртуальных микронавесках мг от временного номера микронавесок при исследовании стандартного образца «Гранит» №5, полученные до итерационного расчета (см. фиг. 11а), после итерационного расчета (см. фиг. 11б) и содержаний элементов мг в виртуальных микронавесках, полученных путем покадрового вычета из содержаний элементов, полученных до итерационного расчета, содержаний: химических элементов, полученных после итерационного расчета (см. фиг. 11в). Это позволяет после итерационного расчета по полученным покадровым содержаниям химических элементов производить расчет процентных соотношений содержаний химических элементов в инородных фазовых частицах, с предположением что сумма всех; процентных содержаний химических элементов в фазовых инородных частицах должна составлять 100%. Сравнивая полученные процентные соотношения содержаний химических элементов в исследованных инородных частицах с соотношением содержаний этих элементов в известных веществах позволяет осуществлять диагностику веществ (как это делается в прототипе). Результаты такого расчета содержаний определяемых химических элементов в стандартном образце «Гранит» №5 и диагностика частиц представлены в таблице 4 (приведена часть таблицы). В первом столбце таблицы 4 указаны результаты фазовой диагностики различных виртуальных микронавесок исследованного вещества, во втором столбце указан номер исследованного кадра (виртуальной микронавески), по которому несложно вывести спектр изучаемого кадр на экран дисплея для просмотра, в третьем столбце указан диаметр инородной частицы в мм, в последующих столбцах указано процентные содержания химических элементов в 1 микронавесках вещества (отдельных фазовых частицах) в зависимости от определяемого элемента и номера кадра (микронавески). Из таблицы 4 видно, что в исследуемом веществе присутствуют большое число минералов, в состав которых входят только отдельные химические элементы. Количество частиц (минералов), в которых присутствуют более одного определяемого элемента, мало. Это связано, по-видимому, со; случайными загрязнениями, со сросткам минералов и совместным попаданием фазовых частиц в виртуальную микронавеску. В целом полученные результаты хорошо согласуются со способом создания стандартных образцов Бронницкой экспедиции - стандартные образцы типа «Гранит» готовились с использованием смешивания основы (матрицы) вещества с частицами различных оксидов химических элементов.
Таким образом, за счет более правильного учета величины фона при сортировке аналитических сигналов (отыскания фона на заданном участке спектра вблизи аналитической линии определяемого элемента по максимальному аналитическому сигналу и осуществление учета «порогового сигнала») и предлагаемого способа построения градуировочных графиков в простых координатах, которые исходят из начало координат и проходят через экспериментальные точки образцов сравнения, имеющих большие содержания химических элементов. Это повышает точность исследования вещества, упрощает процесс построения градуировочных графиков химических элементов, существенно уменьшает число используемых образцов сравнения при построении градуировочных графиков, исключает использование малонадежных образцов сравнения с низкими содержаниями химических элементов. Использование одной и той же аналитической навески образцов сравнения в 100 мг позволяет уточнять и контролировать построение калибровочных графиков во времени, использовать градуировочные графики химических элементов для определения массы химических элементов в исследуемом веществе в мг и его виртуальных микронавесках, а так же определять содержания химических элементов в отдельных исследуемых частицах вещества с целью фазовой диагностики частиц, массу частицы и их крупность (размеры) без взвешивания исследуемого вещества. Предлагаемый способ позволяет исключить искажения результатов исследования за счет выполнения при расчетах содержаний химических элементов условия, что сумма вех процентных содержаний элементов в веществе должна быть ровна 100%, если эти искажения в одинаковой степени изменяют аналитические сигналы определяемых химических элементов в процессе исследования (прерывание подачи вещества в плазму, сдвиг струи подаваемого вещества в плазму, изменение положения концов электродов при с.емки спектров, изменение положения и загрязнение оптики, одинаковое изменение аналитических сигналов элементов во времени). Это позволяет использовать градуировочные графики длительное время. Предлагаемый способ исследования может успешно применяться при использовании интегрально-сцинтилляционного метода исследования в случае введения вещества в плазму различных источников возбуждения, в том числе и с испарением вещества в плазму из кратера электрода.
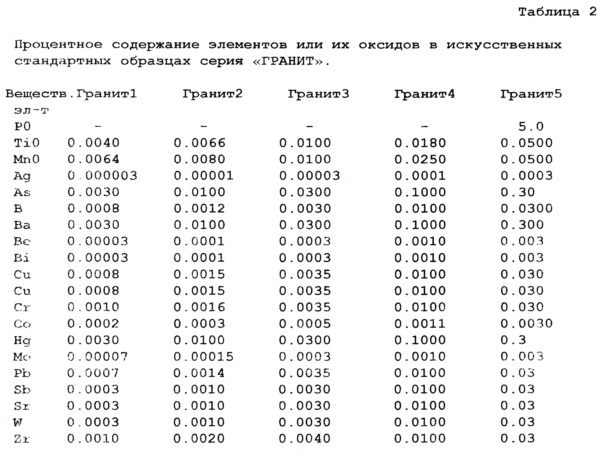
Содержание оксидов матричных элементов в стандартах «Гранит», в которых содержание фосфора менее 1%, составляет SiO2 - 65%, Al2O3 - 20%, CaO - 6.0%, Na2CO3 - 5.0%, Fe2O3 - 3.0%, MgO - 2.0%, а в «Граните» №5, в который специально ввели фосфор (Р2O5 6%), содержание оксидов матричных элементов в стандартах составляет SiO2 - 59%, Al2O3 - 19%, CaO - 5.6%, Na2CO3 - 4.7%, Fe2O3 - 2.8%, MgO - 1.9%.
Изобретение относится к исследованию химических и физических характеристик вещества. Интегрально-сцинтилляционный способ исследования вещества с введением его в плазму включает: переведение вещества в порошковое состояние, съемку покадровых спектров аналитических навесок исследуемых веществ с использованием интегрально-сцинтилляционного спектрометра с виртуальным делением исследуемого вещества на большое число частей путем осуществления периодической прерывистой синхронной кратковременной покадровой регистрации спектрального излучения плазмы источника возбуждения спектров, калибровку шкалы спектрометра, нахождение в зарегистрированных спектрах веществ местоположения спектральных аналитических линий, покадровую сортировку аналитических сигналов, расчет по аналитический сигналам суммарной интенсивности аналитической спектральной линии определяемого химического элемента, построение градуировочных графиков, сортировку аналитических сигналов микронавесок, расчет суммарных интенсивностей спектральных линий определяемых химических элементов, определение по суммарным интенсивностям спектральных линий, расчет реальных содержаний химических элементов в исследуемом веществе, определение поэлементной и фазовой неоднородности вещества и оценку качества исследуемого вещества. Причем после калибровки измерительной шкалы интегрально-сцинтилляционного спектрометра осуществляют покадровую регистрацию спектральных аналитических фоновых сигналов электронной схемы спектрометра без горения плазмы и с плазмой без введения и с введением в нее вещества. Проводят рассмотрение покадровых зарегистрированных спектров, выбирают покадровый участок спектра так, чтобы величина фона на этом участке спектра была близка к величине фона, находящегося под аналитической линией, после этого рассчитывают покадровую величину аналитического сигнала определяемого элемента с учетом фона, определяют по измерительной шкале спектрометра максимальную величину разброса флуктуаций фона, осуществляют исследования покадровых спектров снятых образцов сравнения, строят градуировочные графики химических элементов в простых координатах, контроль правильности построенных градуировочных графиков осуществляют с использованием спектров холостых проб и контрольных образцов сравнения, расчет суммарного аналитического сигнала спектральной линии осуществляют с учетом выбранного «порогового сигнала». Технический результат заключается в повышении точности и чувствительности интегрально-сцинтилляционного атомного эмиссионного спектрального метода. 7 з.п. ф-лы, 11 ил., 4 табл.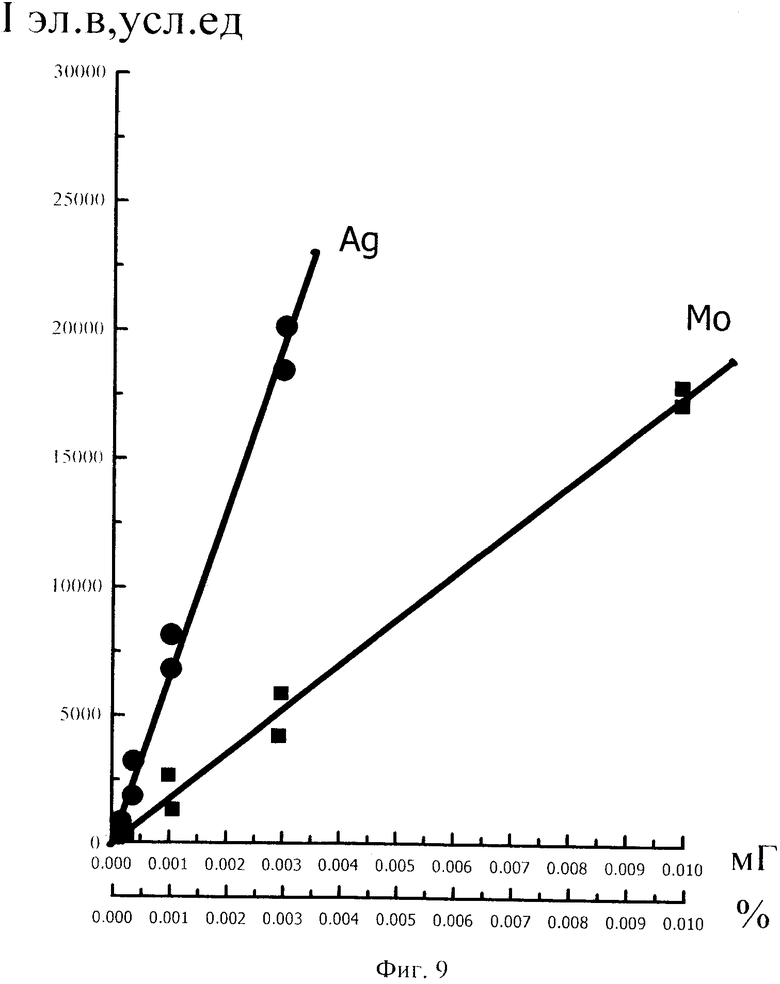
1. Интегрально-сцинтилляционный способ исследования вещества с введением его в плазму, включающий переведение вещества в порошковое состояние, съемку покадровых спектров аналитических навесок исследуемых веществ, образцов сравнения, близких по химическому составу к исследуемому веществу, и известных веществ с использованием интегрально-сцинтилляционного спектрометра с виртуальным делением исследуемого вещества на большое число частей (микронавесок) путем осуществления периодической прерывистой синхронной кратковременной покадровой регистрации спектрального излучения плазмы источника возбуждения спектров с временем накопления спектральных сигналов в кадре, меньшем удвоенной длительности сцинтилляционных, импульсных сигналов вещества, попадающего в плазму, калибровку («профилирование») шкалы спектрометра по спектрам известных веществ, нахождение в зарегистрированных спектрах веществ местоположения спектральных аналитических линий и фона вблизи них, покадровую сортировку аналитических сигналов, полученных виртуальных микронавесок вещества, с учетом фона излучения плазмы, расчет по аналитический сигналам, полученным после их покадровой сортировки, суммарной интенсивности аналитической спектральной линии определяемого химического элемента с учетом сигналов всех спектров виртуальных микронавесок образца сравнения, построение градуировочных графиков по суммарным интенсивностям спектральных аналитических линий определяемых химических элементов, сортировку аналитических сигналов микронавесок самого исследуемого вещества с учетом фона, расчет суммарных интенсивностей спектральных линий определяемых химических элементов с учетом всех виртуальных микронавесок исследуемого вещества, определение по суммарным интенсивностям спектральных линий с использованием построенных градуировочных графиков «условных содержаний» химических элементов, входящих в состав анализируемой навески вещества, а затем расчет реальных содержаний химических элементов в исследуемом веществе по «соотношению условных содержаний», при котором предполагается, что сумма всех содержаний химических элементов в исследуемом веществе составляет 100%, осуществление расчета содержаний химических элементов в отдельных микронавесках вещества и отдельных инородных фазовых частицах, определение поэлементной и фазовой неоднородности вещества по содержаниям химических элементов в микронавесках вещества, фазовые характеристики вещества с применением итерационного расчета содержаний химических элементов в веществе, при котором исключаются аналитические сигналы инородных фаз, и производят расчет характеристик вещества без инородных фаз, фазовую диагностику с использованием сравнений процентных соотношений содержаний химических элементов в изучаемой частице с процентными соотношениями их в известных веществах, а также оценку качества исследуемого вещества, отличающийся тем, что после калибровки (профилирования) измерительной шкалы интегрально-сцинтилляционного спектрометра осуществляют покадровую регистрацию спектральных аналитических фоновых сигналов электронной схемы спектрометра без горения плазмы и с плазмой, без введения и с введением в нее вещества, проводят рассмотрение покадровых зарегистрированных спектров, в которых присутствуют и отсутствуют аналитические линии определяемых элементов, с целью правильного отыскания местоположения фона вблизи аналитической спектральной линии определяемого элемента, выбирают покадровый участок спектра так, чтобы величина фона на этом участке спектра была близка к величине фона, находящегося под аналитической линией, при этом величину фона на выбранном участке спектра находят и рассчитывают так же, как величину аналитического сигнала спектральной линии определяемого химического элемента по максимальному значению аналитического сигнала на выбранном участке спектра, после этого рассчитывают покадровую величину аналитического сигнала определяемого элемента с учетом фона, определяют по измерительной шкале спектрометра максимальную величину разброса флуктуаций фона («пороговый сигнал») путем наблюдения изменений величин покадровых фоновых аналитических сигналов, зарегистрированных в кадрах без горения плазмы, с введением вещества в плазму и без его введения, при этом величину «порогового сигнала» выбирают больше максимального разброса флуктуаций фона в рассмотренных покадровых спектрах, после выбора «порогового сигнала» осуществляют исследования покадровых спектров снятых образцов сравнения, при котором производят покадровую сортировку аналитических сигналов образцов сравнения с учетом фона и его флуктуаций («порогового сигнала»), в процессе покадровой сортировки из величины аналитического сигнала определяемого химического элемента с учетом фона вычитают выбранную величину «порогового сигнала», далее полученную разность используют в качестве основного аналитического сигнала определяемого элемента, по которому осуществляют расчет суммарной интенсивности аналитической линии определяемых элементов с учетом всех микронавесок вещества, градуировочные графики химических элементов строят в простых координатах, по оси абсцисс откладывают содержания химического элемента в образцах сравнения, а по оси ординат величину суммарного аналитического сигнала спектральной линии определяемого элемента в образце сравнения с учетом фона и его флуктуаций («порогового сигнала»), для построения графиков используют одинаковые аналитические навески образцов сравнения с большим содержанием определяемых химических элементов, но не большим, чем содержание, при котором наблюдается реабсорбция спектральной линии, градуировочные графики исходят из начала координат в виде прямой, проходящей через экспериментальные точки, контроль правильности построенных градуировочных графиков осуществляют с использованием спектров холостых проб и контрольных образцов сравнения, зарегистрированных в разное время, расчет суммарного аналитического сигнала спектральной линии определяемых химических элементов в исследуемом веществе осуществляют с учетом выбранного «порогового сигнала», подобно тому, как это делалось в случае расчета суммарного сигнала образцов сравнения, при съемке покадровых спектров исследуемого вещества используют любые аналитические навески исследуемого вещества или частиц без их взвешивания.
2. Способ по п. 1, отличающийся тем, что определение тех или иных характеристик веществ осуществляют в зависимости от необходимости решения конкретных производственных задач.
3. Способ по п. 1, отличающийся тем, что для определения правильности, точности и предела обнаружения химических элементов проводимых исследований используют нахождение полезного аналитического сигнала определяемого химического элемента, производят просмотр покадровых спектров виртуальных микронавесок исследуемого вещества, оценивая величину сигнала, его форму и количество кадров, в которых наблюдается этот сигнал.
4. Способ по п. 1, отличающийся тем, что при съемке спектров образцов сравнения берут одинаковые аналитические навески веществ массой 100 мг, а при построении в простых координатах градуировочных графиков по оси абсцисс откладывают величину массы в мг химического элемента, находящуюся в 100-мг навеске используемого образца сравнения, при осуществлении итерационного расчета исключают сигналы матричных элементов и элементов, находящихся в рассеянной фазовой форме, за счет осуществления покадровых вычетов из аналитических спектральных сигналов определяемых элементов, полученных с учетом фона и «порогового сигнала», рассчитанных до итерационного расчета, аналитических спектральных сигналов определяемых элементов, полученных с учетом фона и «порогового сигнала», полученных после итерационного расчета, при этом величины рассчитанных вычетов используют в качестве основных сигналов, по которым производят расчет процентных соотношений определяемых элементов в отдельных фазовых частицах, фазовую диагностику частиц, определение диаметров исследуемых частиц.
5. Способ по п. 1, отличающийся тем, что длительность кадра делают такой, чтобы она была меньше удвоенной длительности спектрального сцинтилляционного импульса отдельной инородной частицы, попадающей в плазму, но не меньше длительности самого этого сцинтилляционного импульса.
6. Способ по п. 1, отличающийся тем, что предел обнаружения химических элементов в исследуемом веществе определяют с применением непараметрической статистики, используя пуассоновский закон распределения, по обнаружению двух частиц в исследуемой навеске вещества, при этом количественная оценка исследования может оцениваться в случае, если в исследуемом веществе находится более 25-30 частиц, содержащих определяемый элемент, с использованием параметрической статистики и градуировочных графиков, построенных в простых координатах, исходящих из начала координат.
7. Способ по п. 1, отличающийся тем, что после получения характеристик вещества осуществляют исследование его с использованием других известных способов исследования веществ с целью выявления правильности получения характеристик вещества.
8. Способ по п. 1, отличающийся тем, что его применяют при интегрально-сцинтилляционном методе исследования различных веществ в случае введения их в плазму способом испарения вещества из кратера электрода дуги в ее плазму.

