Область техники, к которой относится изобретение
Настоящее изобретение предлагает новые 2-пиридоновые соединения, производящие эффект активации глюкокиназы, и лекарственное средство, содержащее такое соединение в качестве активного ингредиента. Кроме того, настоящее изобретение предлагает кристаллы этих соединений и способ их получения.
Уровень техники
Во всем мире увеличивается число пациентов, страдающих диабетом второго типа, причем прогноз изменения состояния пациентов и развития осложнений является неутешительным; в таких обстоятельствах весьма желательной является разработка нового профилактического или лекарственного средства. Считается, что риск развития диабета второго типа значительно повышается на фоне генетической предрасположенности и старения, предположительно связанного с его развитием, при обычном образе жизни в развитых странах, а именно в состоянии потребления избыточной энергии по сравнению с физическими нагрузками. Кроме того, метаболические расстройства, которые представляют собой предрасполагающие факторы, включают неудовлетворительное усвоение глюкозы в скелетных мышцах и жировых тканях, нарушение секреции инсулина бета-клетками поджелудочной железы и недостаточное регулирование высвобождения глюкозы из печени, и считается, что лекарственное средство, способное устранить эти расстройства, является полезным для профилактики и лечения диабета второго типа.
Для усвоения глюкозы в скелетных мышцах и жировых тканях считается эффективной лекарственная терапия с использованием повышающих чувствительность к инсулину препаратов, представляющих собой производные тиазолидина (например, пиоглитазон); однако было отмечено, что возникают обостряющееся ожирение, задержка жидкости в организме, повышенный риск сердечной недостаточности, повышенная вероятность рака мочевого пузыря и т.д., и, следовательно, требуется всестороннее обследование при использовании этих лекарственных средств. Кроме того, при нарушении секреции инсулина считаются эффективными лекарственные средства на основе сульфонилмочевины (например, глимепирид, глибенкламид, глипизид); однако часто возникает гипогликемия и/или избыточная масса, а также может возникать неудовлетворительное регулирование уровня глюкозы в крови (вторичная резистентность) вследствие снижения терапевтических эффектов при использовании в течение продолжительного периода времени, и, таким образом, остаются нерешенными вопросы безопасности и эффективности. В случае возникающей после приема пищи гипергликемии используются ингибиторы α-глюкозидазы (например, акарбоза, воглибоза и миглитол) или глинидные лекарственные средства (например, натеглинид, репаглинид и митиглинид), но они производят ограниченные терапевтические эффекты на диабет. Для регулирования высвобождения глюкозы из печени эффективными являются бигуанидные лекарственные средства (например, метформин), но гликемический контроль становится затруднительным по мере прогрессирования состояния, и, кроме того, в некоторых случаях использование лекарственного средства может быть ограниченным вследствие его неблагоприятного воздействия на пищеварительный тракт, риска лактоцидоза и т.д. В результате исследования вышеупомянутых основных лекарственных средств становится очевидным, что существующие лекарственные средства не всегда соответствуют медицинским требованиям. В частности, метформин представляет собой практически единственное лекарственное средство для непосредственного улучшения метаболизма глюкозы в печени, и в таких обстоятельствах становится чрезвычайно важной разработка лекарственного средства, способного улучшать метаболизм глюкозы в печени посредством нового механизма действия.
Глюкокиназа (далее сокращенно называется «GK») принадлежит к семейству гексокиназ и катализирует фосфорилирование глюкозы, содержащейся в клетках, таких как бета-клетки поджелудочной железы или клетки печени. Глюкокиназы, содержащиеся в клетках печени и бета-клетках поджелудочной железы, отличаются друг от друга в отношении последовательности 15 аминокислот с концевым атомом азота вследствие различного сплайсинга, но являются ферментативно идентичными. GK проявляет высокое сродство к глюкозе (S0,5 составляет приблизительно 10 мМ), и ее не ингибирует продукт, а именно глюкоза-6-фосфат. Таким образом, ее скорость реакции чутко реагирует на физиологические изменения уровня глюкозы в крови. GK в бета-клетках поджелудочной железы модулирует зависимую от глюкозы секрецию инсулина, в то время как GK в печени модулирует путь расщепления глюкозы или образование гликогена, таким образом, что уровень глюкозы в крови поддерживается и регулируется. Таким образом, считается, что GK функционирует как датчик глюкозы, поддерживая гомеостаз уровня глюкозы в крови (см. непатентный документ 1).
Генетически модифицированные мыши и генетические мутации, обнаруженные у человека, подтверждают гипотезу, что GK функционирует как датчик глюкозы в организме. Гомозиготные по GK мыши умирали от гипергликемии немедленно после рождения, а у гетерозиготных мышей наблюдалась гипергликемия и нарушенная переносимость глюкозы (см. непатентный документ 2). С другой стороны, у имеющих повышенную экспрессию GK мышей подтверждено наличие гипогликемии (см. непатентный документ 3). Кроме того, в случае юношеского инсулиннезависимого сахарного диабета второго типа (MODY2) человека, когда наблюдается мутация гена GK, диабет развивается в молодости (см. непатентный документ 4). Было подтверждено, что эти генные мутации снижают активность GK. С другой стороны, были описаны семьи, имеющие генные мутации, повышающие активность GK (см. непатентный документ 5). Согласно наблюдениям, эти генные мутации повышали сродство GK к глюкозе и вызывали симптомы гипогликемии натощак в сочетании с повышением концентрации инсулина в крови.
Таким образом, было показано, что GK функционирует как датчик глюкозы у млекопитающих, включая человека.
Повышающие активность GK вещества (далее называются «активирующие GK вещества») могут улучшать состояние при гипергликемии посредством усиления метаболизма глюкозы и образования гликогена в печени и зависимой от глюкозы секреции инсулина из бета-клеток поджелудочной железы. В частности, вещества, которые повышают активность GK преимущественно в печени, могут улучшать состояние при гипергликемии посредством усиления метаболизма глюкозы в печени независимым от инсулина путем. Можно также предполагать, что улучшение состояние при гипергликемии приведет к лечению и профилактике хронических диабетических осложнений, таких как ретинопатия, нефропатия, невроз, ишемическая болезнь сердца и артериосклероз, а также к лечению и профилактике связанных с диабетом заболеваний, такие как ожирение, гиперлипидемия, повышенное артериальное давление и метаболический синдром. Таким образом, предполагается, что соединения, усиливающие функцию GK, должны представлять собой эффективные средства для лечения диабета.
С другой стороны, была отмечена экспрессия GK не только в поджелудочной железе и печени, но также в пищевом центре, а также ее важная функция в противопищевом эффекте глюкозы (см. непатентный документ 6). Соответственно, активирующие GK вещества могут воздействовать на пищевой центр и производить противопищевой эффект, и можно предполагать, что они должны действовать не только как средства для лечения диабета, но также как лекарственные средства против ожирения.
В связи с этим отмечено, что некоторые соединения, содержащие 2-пиридон, представляют собой активирующие GK вещества, но значительно отличаются по структуре от соединений согласно настоящему изобретению (см. патентные документы 1 и 2). Описаны и другие 2-пиридоновые соединения, имеющие близкое структурное сходство (см. патентные документы 3 и 4), но соединения согласно настоящему изобретению ранее не были описаны определенным образом. Настоящее изобретение отличается от предшествующих сообщений тем, что в них не содержится описание в отношении медицинского применения, и тем, что они сосредоточены на способе синтеза 2-пиридоновых соединений (см. непатентный документ 7). Кроме того, некоторые ацилмочевинные соединения, которые могут иметь псевдоциклическую структуру, были описаны как активирующие GK вещества (см. патентные документы 5 и 6), но они представляют собой нециклические соединения и отличаются от соединений согласно настоящему изобретению.
Список цитируемой литературы
Непатентная литература
Непатентный документ 1: F. M. Matschinsky и M. A. Magnuson, Frontiers in Diabetes (Ограничения при диабете), 2004 г., с. 16
Непатентный документ 2: A. Grupe и др., Cell, 1995 г., т. 83, № 1, с. 69-78
Непатентный документ 3: T. Ferre и др., Proc. Natl. Acad. Sci., 1996 г., т. 93, № 14, с. 7225-7230
Непатентный документ 4: N. Vionnet и др., Nature, 1992 г., т. 356, № 6371, с. 721-722
Непатентный документ 5: B. Glaser и др., N. Engl. J. Med., 1998 г., т. 338, № 4, с. 226-230
Непатентный документ 6: L. Kang и др., Diabetes, 2006 г., т. 55, № 2, с. 412-420
Непатентный документ 7: R. Latif и др., J. Chem. Soc. C. Organic, 1968 г., т. 17, с. 2140-2144,
Патентная литература
Патентный документ 1: международная патентная заявка WO 2008/079787
Патентный документ 2: международная патентная заявка WO 2010/013161
Патентный документ 3: патент США US 4275069
Патентный документ 4: международная патентная заявка WO 2011/068211
Патентный документ 5: международная патентная заявка WO 2000/58293
Патентный документ 6: международная патентная заявка WO 2001/44216
Сущность изобретения
Техническая проблема
Задача настоящего изобретения заключается в том, чтобы предложить соединения, которые производят превосходный эффект активации GK и являются пригодными для использования в качестве лекарственных средств.
Решение проблемы
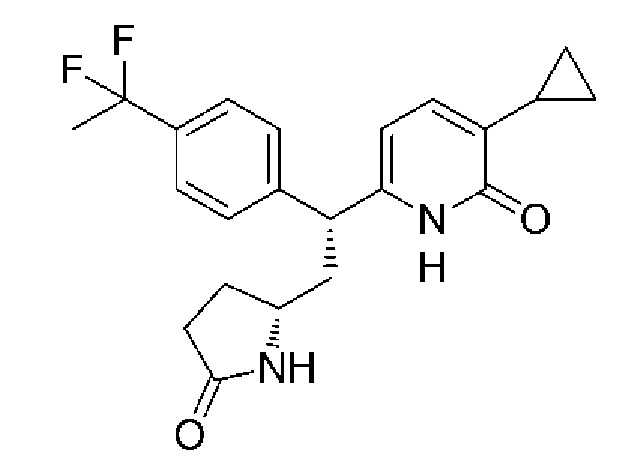
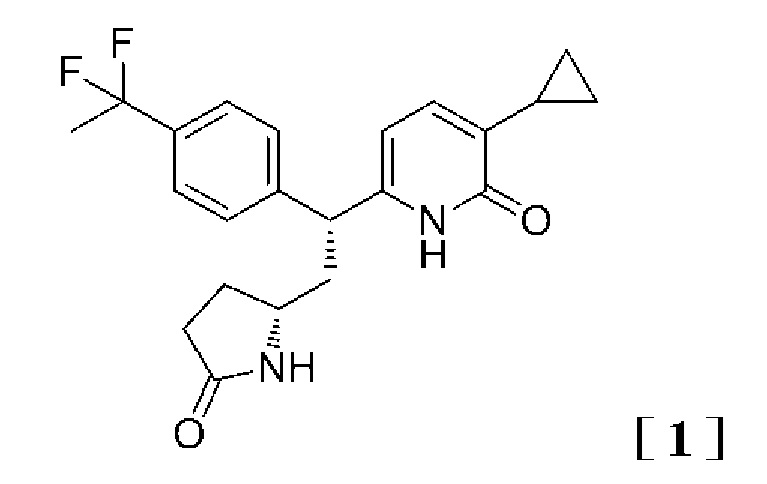
Принимая во внимание вышеупомянутые обстоятельства, авторы настоящего изобретения провели всесторонние исследования, чтобы найти соединения, производящие эффект активации GK, и в результате этого обнаружили, что данную задачу могут решить 2-пиридоновое соединение, представленное следующей формулой [1] (название соединения: 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он), таутомер данного соединения, его фармацевтически приемлемая соль (далее 2-пиридоновое соединение, таутомер данного соединение или его фармацевтически приемлемая соль называются термином «2-пиридоновое соединение или родственные соединения»), или сольват 2-пиридонового соединения или родственных соединений, и, таким образом, было выполнено настоящее изобретение.
(I) Вариант осуществления настоящего изобретения предлагает 2-пиридоновое соединение, представленное формулой [1]:
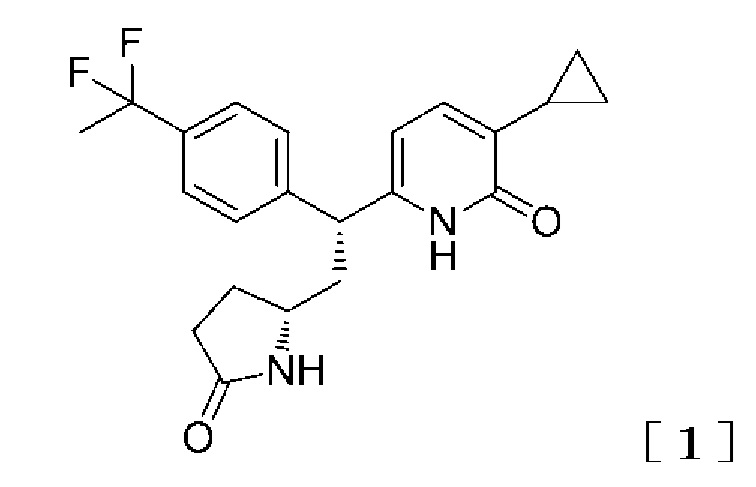 ,
,
таутомер данного соединение или его фармацевтически приемлемая соль, или сольват 2-пиридонового соединения или родственных соединений.
(II) Следующий вариант осуществления настоящего изобретения предлагает кристаллический 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он по пункту (I), представленный вышеупомянутой формулой [1] и имеющий следующее физическое свойство (a):
(a) рентгеновская порошковая дифрактограмма (Cu-Kα), проявляющая пики при дифракционных углах 2θ, составляющих 8,5, 13,4, 19,1 и 24,5°.
(III) Следующий вариант осуществления настоящего изобретения предлагает кристаллический 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он по пункту (I), представленный вышеупомянутой формулой [1] и имеющий следующие физические свойства (a)-(c):
(a) рентгеновская порошковая дифрактограмма (Cu-Kα), проявляющая пики при дифракционных углах 2θ, составляющих 8,5, 13,4, 19,1 и 24,5°;
(b) инфракрасный спектр поглощения, проявляющий характеристические полосы поглощения при 916, 1146, 1167, 1295, 1651, 1664, 2909, 2955, 3003 и 3146 см-1; и
(c) температура плавления, составляющая от 199 до 201°C.
(IV) Следующий вариант осуществления настоящего изобретения предлагает способ получения кристаллического 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она, имеющего следующие физические свойства (a)-(c):
(a) рентгеновская порошковая дифрактограмма (Cu-Kα), проявляющая пики при дифракционных углах 2θ, составляющих 8,5, 13,4, 19,1 и 24,5°;
(b) инфракрасный спектр поглощения, проявляющий характеристические полосы поглощения при 916, 1146, 1167, 1295, 1651, 1664, 2909, 2955, 3003 и 3146 см-1; и
(c) температура плавления, составляющая от 199 до 201°C,
включающий растворение 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она, представленного вышеупомянутой формулой [1], в спиртовом растворителе при нагревании для получения раствора; последующее добавление воды в качестве растворителя в раствор; охлаждение полученного в результате раствора до температуры 5°C или ниже для получения кристаллов; и высушивание полученных кристаллов при температуре 60°C или ниже.
(V) Следующий вариант осуществления настоящего изобретения предлагает лекарственное средство содержащее, в качестве активного ингредиента, 2-пиридоновое соединение, таутомер данного соединение или его фармацевтически приемлемую соль, или сольват 2-пиридонового соединения или родственных соединений по пункту (I).
(VI) Следующий вариант осуществления настоящего изобретения предлагает лекарственное средство по пункту (V), где данное лекарственное средство используется для профилактики или лечения заболевания или состояния, которое может улучшаться посредством эффекта активации глюкокиназы.
(VII) Следующий вариант осуществления настоящего изобретения предлагает лекарственное средство по пункту (V), которое представляет собой гипогликемическое средство.
(VIII) Следующий вариант осуществления настоящего изобретения предлагает лекарственное средство по пункту (V), где данное лекарственное средство представляет собой средство для профилактики или лечения диабета.
Полезные эффекты изобретения
Согласно настоящему изобретению, предлагаются 2-пиридоновые соединения, производящие превосходный эффект активации GK.
Кроме того, согласно настоящему изобретению, предлагается кристаллический 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он, который представляет собой новое кристаллическое соединение, пригодное для использования в качестве лекарственного средства. Данное соединение существует в устойчивой кристаллической форме в условиях комнатной температуры и проявляет хорошую устойчивость при хранении.
Кроме того, согласно настоящему изобретению, предлагается новый способ получения, позволяющий получать безопасным и устойчивым путем вышеупомянутое кристаллическое соединение, имеющее однородное качество.
Краткое описание чертежей
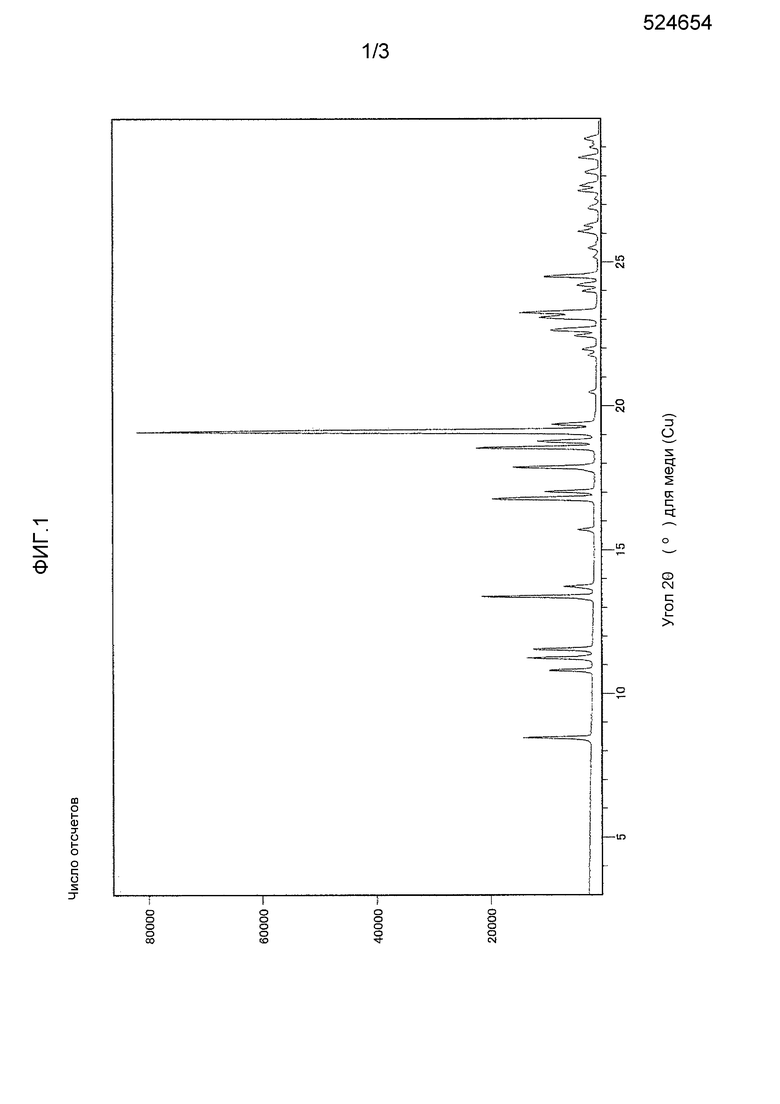
Фиг. 1 представляет рентгеновскую порошковую дифрактограмму кристаллического 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она согласно настоящему изобретению.
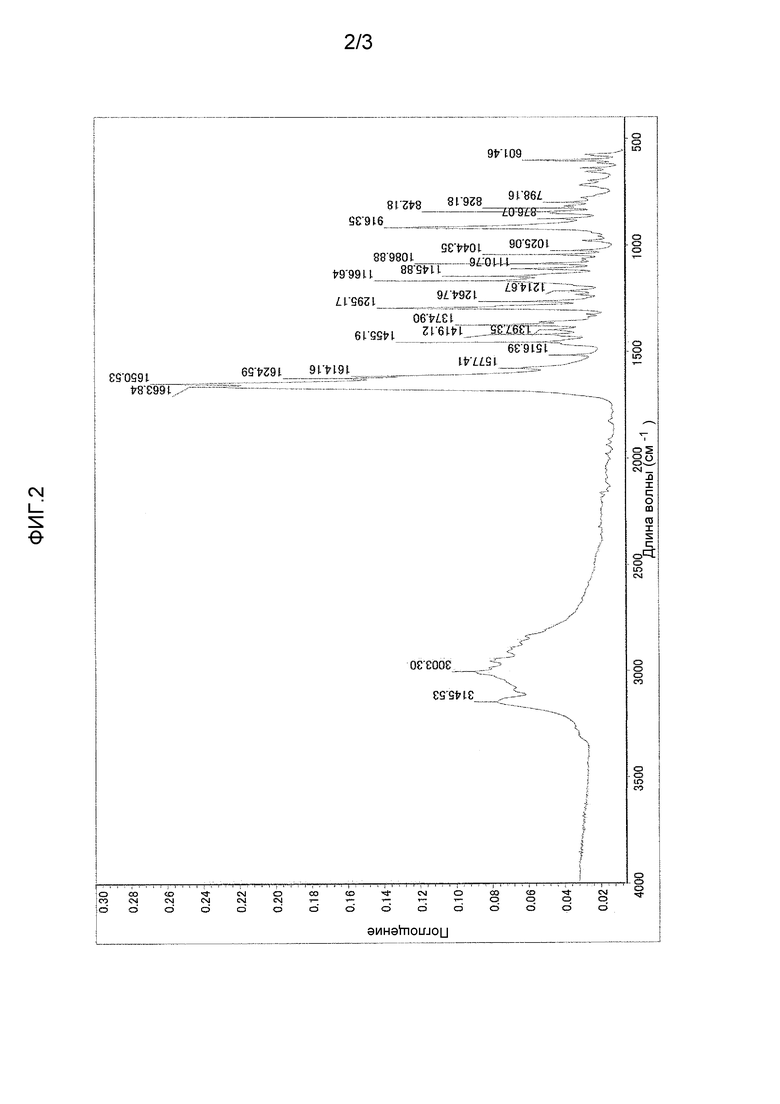
Фиг. 2 представляет инфракрасный спектр поглощения (метод нарушенного полного внутреннего отражения (НПВО), алмазный кристалл) кристаллического 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она согласно настоящему изобретению.
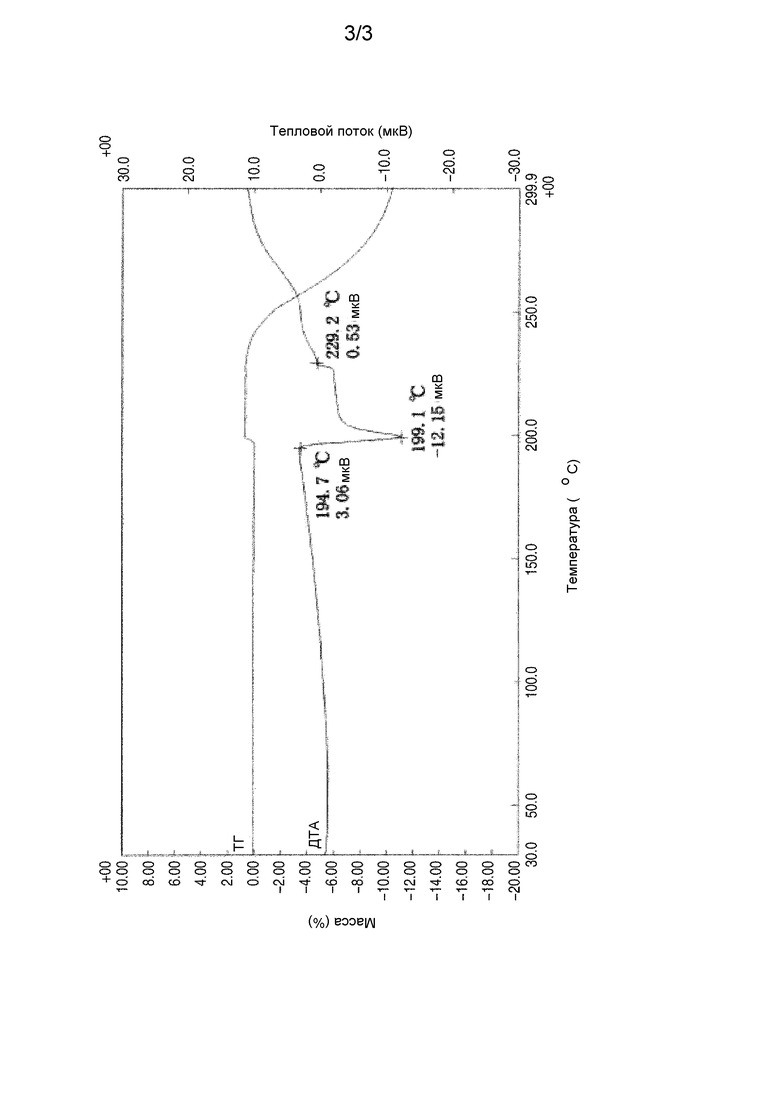
Фиг. 3 представляет кривые дифференциального термического анализа/термогравиметрического анализа кристаллического 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она согласно настоящему изобретению.
Описание вариантов осуществления
Далее настоящее изобретение будет описано подробно, но оно не ограничивается определенным образом примерными вариантами осуществления.
Согласно настоящему изобретению, «н» означает «нормальный», «и» означает «изо», «в» и «втор» означают «вторичный», «трет» означает «третичный», «ц» означает «цикло», «o» означает «орто», «м» означает «мета» и «п» означает «пара».
Сначала будут описаны соединения согласно настоящему изобретению.
Примеры фармацевтически приемлемых солей согласно настоящему изобретению включают соли минеральных кислот, такие как гидрохлориды, гидробромиды, гидройодиды, фосфаты, сульфаты и нитраты; сульфонаты, такие как метансульфонаты, этансульфонаты, бензолсульфонаты и п-толуолсульфонаты; карбоксилаты, такие как оксалаты, тартраты, цитраты, малеаты, сукцинаты, ацетаты, бензоаты, манделаты, аскорбаты, лактаты, глюконаты и малаты; соли аминокислот, такие как соли глицина, соли лизина, соли аргинина, соли орнитина, глутаматы и аспартаты; и минеральные соли, такие как соли лития, соли натрия, соли калия, соли кальция и соли магния, и соли органических оснований, такие как соли аммония, соли триэтиламина, соли диизопропиламина и соли циклогексиламина. Предпочтительные примеры включают гидрохлориды, гидробромиды, фосфаты, сульфаты, метансульфонаты, п-толуолсульфонаты, оксалаты, тартраты, цитраты, ацетаты, лактаты, глутаматы, аспартаты, соли натрия, соли калия, соли аммония и соли триэтиламина.
Сольваты согласно настоящему изобретению представляют собой фармацевтически приемлемые сольваты соединений или их соли согласно настоящему изобретению. Соединения и их соли согласно настоящему изобретению могут абсорбировать влагу, содержать адсорбированную воду или образовывать гидраты при воздействии воздуха, перекристаллизации и т.д. Соединения согласно настоящему изобретению также включают такие гидраты.
Соединения согласно настоящему изобретению имеют два центра асимметрии и представляют собой оптически активные соединения, причем оба центра асимметрии в данных соединениях имеют абсолютную конфигурацию (R). Соединения согласно настоящему изобретению можно получать путем разделения оптических изомеров из соответствующего рацемата или смеси диастереомеров. Приемлемые способы разделения оптических изомеров включают способы, хорошо известные специалисту в данной области техники, такие как способ фракционной кристаллизации или хиральная колоночная хроматография. В качестве альтернативы, оптически активные соединения согласно настоящему изобретению можно также получать хорошо известным способом, практически используемым в органической химии для этой цели. Кроме того, геометрические изомеры, такие как изомер (E) и изомер (Z), могут присутствовать как промежуточные соединения в синтезе для получения соединений согласно настоящему изобретению, и соотношение этих изомеров может представлять собой любую пропорцию.
Соединения согласно настоящему изобретению включают таутомеры. Таутомер в настоящем документе означает кето-енольный таутомер соединений, представленных вышеупомянутой формулой [1]. Соединения, представленные вышеупомянутой формулой [1], и соответствующий таутомер [1'] приведены ниже в качестве примера.
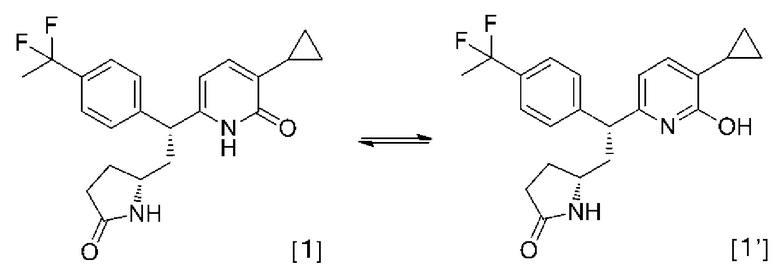
2-Пиридоновые соединения согласно настоящему изобретению могут представлять собой соответствующие фармацевтически приемлемые соли или сольваты 2-пиридонового соединения или родственных соединений. Далее 2-пиридоновые соединения, таутомеры соединений, их фармацевтически приемлемые соли или сольваты 2-пиридоновых соединений или родственных соединений в совокупности называются «соединения согласно настоящему изобретению».
Эти «соединения согласно настоящему изобретению» также включают соединения, обычно называемые термином «пролекарства», которые имеют химически или метаболически разлагающуюся группу и образуют фармакологически активные соединения согласно настоящему изобретению в результате сольволиза или в физиологических условиях живого организма.
Соединения согласно настоящему изобретению производят эффект активации GK. Таким образом, соединения согласно настоящему изобретению могут приводить к улучшению при гипергликемии посредством усиления метаболизма глюкозы, главным образом, в печени. Соответственно, данные соединения можно использовать как новые лекарственные средства, которые отличаются по механизму действия от существующих средств для лечения диабета. Диабет включает диабет первого типа, диабет второго типа и диабет других типов, вызванный определенными причинами. Соединения согласно настоящему изобретению также являются эффективными для лечения и профилактики диабетических осложнений, такие как кетоацидоз, микроангиопатия (ретинопатия или нефропатия), артериосклероз (такой как атеросклероз, инфаркт миокарда, церебральный инфаркт или окклюзионная болезнь периферических артерий), нейропатия (такая как сенсорная нейропатия, моторная нейропатия или автономная нейропатия), гангрена ступни и инфекции.
Данные соединения можно также использовать для лечения и профилактики связанных с диабетом заболеваний, таких как ожирение, гиперлипидемия, повышенное артериальное давление, метаболический синдром, отек, гиперурикемия и подагра.
Соединения согласно настоящему изобретению можно также использовать в сочетании с лекарственными средствами, имеющими механизм действия, который представляет собой эффект активации GK, такие как средства для лечения диабета, диабетических осложнений, гиперлипидемии, повышенного артериального давления и т.д. Посредством сочетания соединений согласно настоящему изобретению с такими другими лекарственными средствами можно предполагать аддитивный эффект в лечении вышеупомянутых заболеваний по сравнению с эффектом, достигаемым с помощью каждого из отдельно взятых соответствующих лекарственных средств.
Примеры средств для лечения диабета и средств для лечения диабетических осложнений, которые являются пригодными для использования в сочетании с соединениями согласно настоящему изобретению, включают препараты инсулина, повышающие чувствительность к инсулину вещества, такие как агонисты γ-рецепторов, активируемых пролифератором пероксисом (PPAR), агонисты α/γ-PPAR, агонисты δ-PPAR и агонисты α/γ/δ-PPAR (например, пиоглитазон, росиглитазон, алеглитазар, пнлиглитазар, AVE-0897 и MBX-8025), ингибиторы α-глюкозидазы (например, воглибоза, акарбоза и миглитол), бигуанидные лекарственные средства (например, метформин, буформин и фенформин), усилители секреции инсулина (например, глибенкламид, глимепирид, репаглинид, натеглинид и митиглинид), антагонисты глюкагоновых рецепторов, усилители киназы инсулиновых рецепторов, ингибиторы дипептидилпептидазы IV (например, вилдаглиптин, алоглиптин, ситаглиптин, линаглиптин, саксаглиптин, тенелиглиптин, анаглиптин), ингибиторы натрий-зависимых переносчиков глюкозы (SGLT) (например, дапаглифлозин, лузеоглифлозин, канаглифлозин, эмпаглифлозин, ипраглифлозин, тофоглифлозин), ингибиторы протеинтирозинфосфатазы 1b (PTP1b) (например, ванадат натрия), ингибиторы глюкоза-6-фосфатазы, ингибиторы гликогенфосфорилазы (например, PSN-357 и FR-258900), ингибиторы фруктоза-1,6-бисфосфатазы (FBP) (например, MB-07803), ингибиторы фосфоэнолпируваткарбоксиназы (PEPCK), ингибиторы киназы пируватдегидрогеназы, D-хиро-инозит, ингибиторы киназы гликогенсинтазы-3 (GSK3), агонисты глюкагоноподобного пептида-1 (GLP-1) (например, лираглютид и эксенатид), агонисты амилина (например, прамлинтид), антагонисты глюкокортикоидных рецепторов, ингибиторы 11-β-гидроксистероиддегидрогеназы типа 1 (11βHSD1) (например, INCB-13739, LY-2523199, Ro-5027838, Ro-5093151 и S-707106), ингибиторы протеинкиназы C (например, рубоксистаурин), агонисты адреналиновых рецепторов β3, ингибиторы фосфатидилинозиткиназы, ингибиторы фосфатидилинозитфосфатазы, ингибиторы ацетил-СоА-карбоксилазы (ACC), агонисты рецепторов свободных жирных кислот (GPR40) (например, TAK-875), агонисты протеинсвязанных рецепторов G119 (GPR119) (например, APD-597, PSN-821, MBX-2982 и DS-8500), агонисты протеинсвязанных рецепторов G120 (GPR120), агонисты рецепторов TGR5, активаторы 5'- аденозинмонофосфатпротеинкиназы (AMPK), ингибиторы альдозоредуктазы (например, эпалрестат, ранирестат, фидарестат) и ингибиторы конечных продуктов усиленного гликозилирования (AGE).
Кроме того, примеры средств для лечения связанных с диабетом заболеваний, которые являются пригодными для использования в сочетании с соединениями согласно настоящему изобретению, включают ингибиторы редуктазы 3-гидрокси-3-метилглутарил-СоА (HMG-CoA), ингибиторы синтазы сквалена, адсорбенты желчной кислоты, ингибиторы подвздошного переноса желчной кислоты (IBAT), ингибиторы белка переноса холестерилового эфира (CETP), ингибиторы карнитинпальмитоилтрансферазы (CPT), фибраты, ингибиторы ацил-CoA-холестеринацилтрансферазы (ACAT), ингибиторы моноацилглицеринацилтрансферазы (MGAT), ингибиторы диацилглицеринацилтрансферазы (DGAT), ингибиторы абсорбции холестерина, ингибиторы липазы поджелудочной железы, ингибиторы белка переноса микросомальных триглицеридов (MTP), производные никотиновой кислоты, агонисты печеночных рецепторов X (LXR), усилители рецепторов липопротеинов низкой плотности (LDL), ингибиторы превращающего ангиотензин фермента, антагонисты ангиотензина II, антагонисты рениновых рецепторов, антагонисты альдостерона, диуретики, антагонисты кальция, альфа-блокаторы, бета-блокаторы, ингибиторы превращающего эндотелин фермента, антагонисты эндотелиновых рецепторов, подавляющие аппетит вещества, ингибиторы синтеза мочевой кислоты и способствующие выведению мочевой кислоты средства.
Соединения согласно настоящему изобретению можно вводить в чистом виде или вместе с фармацевтически или фармакологически приемлемыми носителями или разбавителями. Соединения согласно настоящему изобретению, используемые в качестве активирующих GK веществ, можно вводить перорально или парентерально в чистом виде. Соединения согласно настоящему изобретению можно также вводить перорально или парентерально в виде средств, содержащих данные соединения в качестве активных ингредиентов. Примеры парентерального введения включают внутривенное введение, назальное введение, чрескожное введение, подкожное введение, внутримышечное введение и подъязычное введение.
Дозировка соединения согласно настоящему изобретению изменяется в зависимости от пациента, пути введения, рассматриваемого заболевания, симптома и т.д., причем обычно она составляет, например, приблизительно от 0,01 до 1000 мг и предпочтительно от 0,1 до 100 мг в однократной дозе при пероральном введении взрослому пациенту, страдающему диабетом; оказывается желательным введение данной дозы один, два или три раза в сутки.
Далее будет описан способ получения соединения согласно настоящему изобретению.
Соединения согласно настоящему изобретению можно синтезировать, осуществляя способы, представленные ниже. Следующие способы получения представляют общие примеры способов получения и не ограничивают способы получения.
Соединения согласно настоящему изобретению можно также синтезировать, используя способ, общеизвестный в области химии, или способ, включающий один или несколько способов, аналогичных этому способу. Примеры таких способов включают способы, описанные в книгах «Синтезы органических функциональных групп», второе издание, издательство Academic Press, Inc., 1986 г.; «Полное описание превращений органических веществ», издательство VCH Publishers Inc., 1989 г.; и «Основы и эксперименты синтеза пептидов», издательство Maruzen Co., Ltd., 1985 г.
Подходящие способы введения и удаления функциональных защитных групп, содержащихся в исходных материалах, промежуточных продуктах и т.д., при синтезе соединений согласно настоящему изобретению можно осуществлять, используя способы, хорошо известных специалисту в данной области техники, такие как способы, описанные в книге Greene «Защитные группы в органическом синтезе», издательство John Wiley & Sons, 2006 г.
Общие способы получения соединения согласно настоящему изобретению представлены на схемах 1 и 2. Способы получения не ограничиваются следующими способами получения. Соединения согласно настоящему изобретению можно также получать, используя способы, хорошо известные специалисту в данной области техники, например, изменяя последовательность осуществления стадий; используя защитную группу для гидроксильной группы и т.д., осуществляя реакцию и удаление защитной группы на последующей стадии; или добавляя новую стадию в ходе соответствующих стадий.
Схема 1: способ синтеза соединения [1] согласно настоящему изобретению из соединения (1-a).
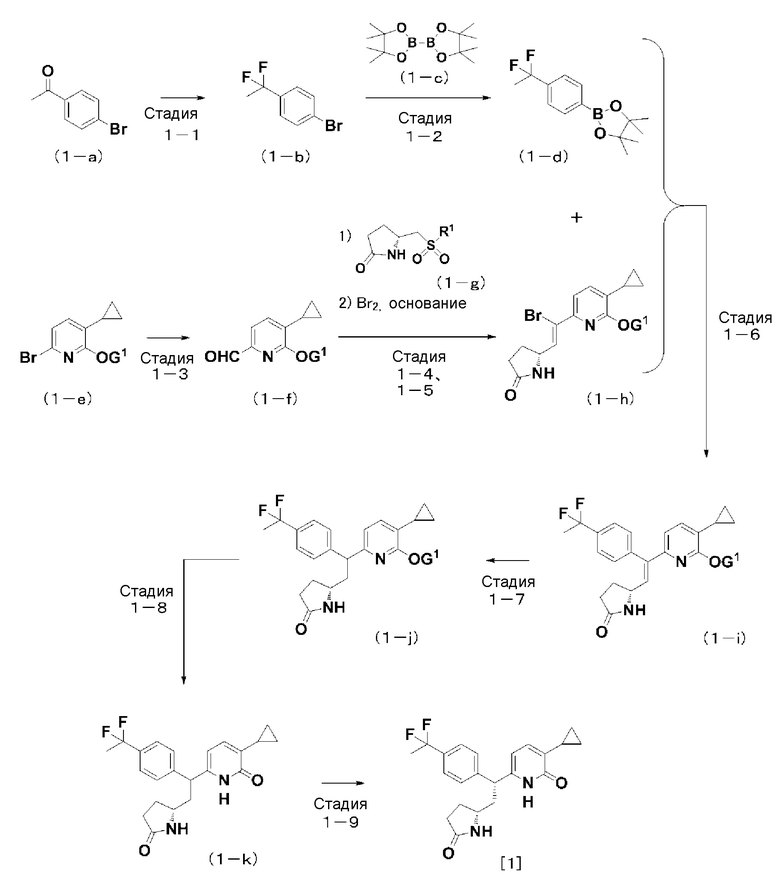
(На данной схеме G1 представляет собой защитную группу для гидроксильной группы в гидроксипиридильной группе, R1 представляет собой 2-бензотиазолильную группу или 1-фенил-1H-тетразол-5-ильную группу.)
Соединение (1-g), используемое на стадии (1-4), можно получать как имеющееся в продаже соединение, общеизвестное соединение или соединение, легко получаемое с использованием разнообразных методик органического синтеза, которые известны специалист в данной области техники.
Стадия (1-1):
Способ получения соединения (1-b): соединение (1-b) можно получить, осуществляя «фторирование» с использованием фторирующего реагента, такого как трифторид N,N-диметоксиэтиламиносеры (трифторид бис(2-метоксиэтил)аминосеры или Deoxo-Fluor (зарегистрированный товарный знак)).
Примеры фторирования включают способ, согласно которому фторирующий реагент, такой как Deoxo-Fluor (зарегистрированный товарный знак) реагирует с соединением (1-a) при отсутствии растворителя или в инертном растворителе при температуре, составляющей от 0°C до 100°C, и образуется соединение (1-b).
Стадия (1-2):
Способ получения соединения (1-d): соединение (1-d) можно получить, вводя в реакцию соединение (1-c) и соединение (1-b) в инертном растворителе в присутствии основания, такого как ацетат калия и палладиевый катализатор.
Стадия (1-3):
Способ получения соединения (1-f): соединение (1-f) можно получить, вводя в реакцию основание, такое как н-бутиллитий или хлорид н-бутилмагния, и соединение (1-e) в инертном растворителе, а затем вводя в реакцию с ними N,N-диметилформамид.
Соединение (1-e) как исходное вещество, можно получать способом, описанным в международной патентной заявке WO 2011/068211, или соответствующим ему способом.
Стадии (1-4, 1-5):
Способ получения соединения (1-h): соединение (1-h) можно получить, вводя в реакцию в инертном растворителе бром и (5R)-5-[2-(5-циклопропил-6-метоксипиридин-2-ил)этенил]пирролидин-2-он (смесь изомеров E и Z), который производит «реакция сочетания», где используются карбонильное соединение (1-f) и соединение (1-g), и после этого в реакцию с ними вводится основание, такое как 1,8-диазабицикло[5,4,0]ундец-7-ен (DBU).
Примерные «реакции сочетания» включают способ получения соединения (1-h) путем образования аниона, где используются соединение (1-g) в качестве матрицы и металлоорганический реагент, такой как н-бутиллитий, втор-бутиллитий или трет-бутиллитий, или основание, такое как гексаметилдисилазид лития или гексаметилдисилазид калия при температуре, составляющей от -78°C до 100°C, в инертном растворителе, и после этого реагируют анион и карбонильное соединение (1-f). Получаемое олефиновое соединение, как правило, получают в форме смеси изомеров E и Z, которые, однако, можно разделять соответствующим образом, используя колоночную хроматографию с силикагелем или высокоэффективную жидкостную хроматографию (ВЭЖХ).
Соединение (1-g), используемое для реакции сочетания, можно получать способом, описанным в международной патентной заявке WO 2011/068211, или соответствующим ему способом.
Стадия (1-6):
Способ получения соединения (1-i): соединение (1-i) можно получить, осуществляя «реакцию сочетания» с фенилборным соединением (1-d), где используется соединение (1-h) в качестве матрицы в присутствии палладиевого катализатора.
Примерные реакции сочетания включают способ, где реагируют соединение (1-h) и фенилборное соединение в инертном растворителе при температуре, составляющей от 20°C до 160°C, в присутствии палладиевого катализатора и основания. Реакцию можно также осуществлять, используя микроволновое излучение.
Примерные палладиевые катализаторы, используемые для реакции сочетания, включают палладиевые катализаторы, известные специалисту в данной области техники, такие как тетракистрифенилфосфинпалладий(0), бис(дибензилиденацетон)палладий(0), трис(дибензилиденацетон)дипалладий(0), дихлорид бис(трифенилфосфин)палладия(II), ацетат бис(трифенилфосфин)палладия(II) и комплекс дихлорида [1,1'-бис(дифенилфосфин)ферроцен]палладия(II) и дихлорметана (1:1). Кроме того, в присутствии основания для реакции можно использовать трис(дибензилиденацетон)дипалладий(0) и три(2-фурил)фосфин.
Стадия (1-7):
Способ получения соединения (1-j): соединение (1-j) можно получить, восстанавливая соединение (1-i) в качестве матрицы посредством реакции каталитического гидрирования, где используется в каталитическом количестве палладий на активированном угле, родий на активированном угле или платина на активированном угле в инертном растворителе в присутствии или при отсутствии кислоты при температуре, составляющей от 0°C до 80°C.
Стадия (1-8):
Способ получения соединения (1-k): соединение (1-k) можно получить, осуществляя «реакцию удаление защитной группы» в отношении защитной группы G1, которую содержит соединение (1-j).
Примерные реакции удаления защитной группы включают (i) реакции удаления защитной группы, где защитная группа G1 представляет собой алкильную группу или аллильную группу, такие как реакции гидролиза для удаления защитной группы в инертном растворителе в присутствии кислоты или сильной кислоты при температуре, составляющей от 0°C до 200°C, реакции с использованием триметилсилилйодида и т.д., и реакции, где используются хлорид алюминия и алкилтиол, и (ii) реакции удаления защитной группы, где в качестве защитной группы G1 присутствуют бензильная группа, 4-метоксибензильная группа, 2,4-диметоксибензильная группа, бензилоксикарбонильная группа, бензгидрильная (дифенилметильная) группа и т.д., такие как реакции удаления защитной группы посредством гидрогенолиза, где в каталитическом количестве используются палладий на активированном угле, родий на активированном угле и т.д., в инертном растворителе в присутствии или при отсутствии кислоты при температуре, составляющей от 0°C до 80°C, или реакции с использованием окислителя, такого как двойной нитрат аммония и церия(IV) или 2,3-дихлор-5,6-дициано-п-бензохинон.
Стадия (1-9):
Способ получения соединения [1] согласно настоящему изобретению: соединение [1] согласно настоящему изобретению можно получить, разделяя диастереомеры соединения (1-k) с использованием, например, высокоэффективной жидкостной хроматографии (ВЭЖХ).
Соединение (1-a) и соединение (1-c), используемые в качестве соединений исходного материала на приведенной выше схеме 1, можно получить, приобретая как товарные продукты или синтезируя известным способом.
Схема 2: способ синтеза соединения [1] согласно настоящему изобретению из соединения (2-a)
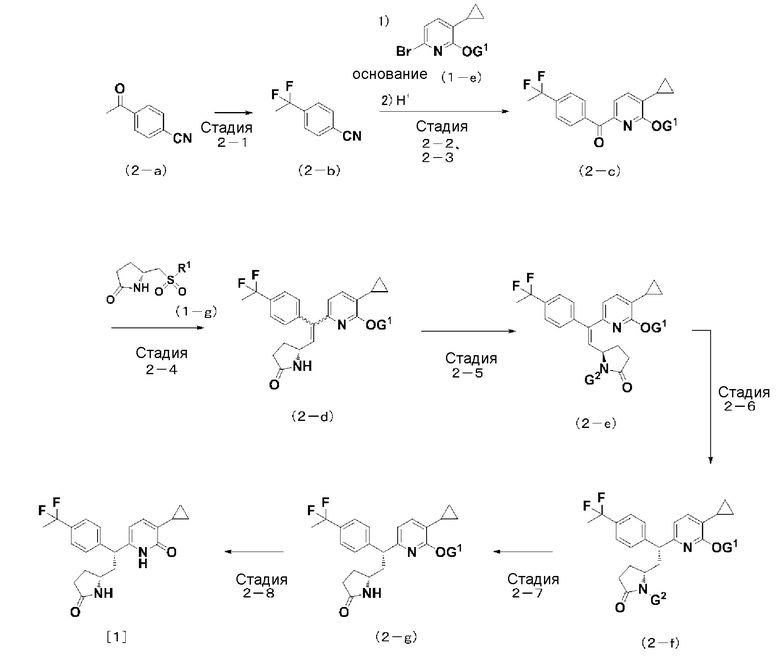
(На данной схеме G1 и R1 являются такими, как определено выше. G2 представляет собой защитную группу для атома азота в пирролидинильной группе, замещенной оксогруппой.)
Стадия (2-1):
Способ получения соединения (2-b): соединение (2-b) можно получить способом, описанным в международной патентной заявке WO 2008/103185, или соответствующим ему способом.
Стадии (2-2, 2-3):
Способ получения соединения (2-c): соединение (2-c) можно получить, осуществляя «реакцию присоединения» и используя соединение (2-b) и анион, такой как соединение лития реагент, например, гетероариллитий, реактив Гриньяра (Grignard), например, бромид гетероарилмагния, и обрабатывая полученное соединение кислотой, такой как хлористоводородная кислота.
Примерные «реакции присоединения» включают способ, где реагируют соединение (2-b) и анион, который получают, используя соединение (1-e) в качестве матрицы и металлоорганический реагент, такой как н-бутиллитий, втор-бутиллитий, трет-бутиллитий, или бромид изопропилмагния, металлический реагент, такой как магний, или основание, такое как гексаметилдисилазид лития или гексаметилдисилазид калия, в инертном растворителе при температуре, составляющей от -78°C до 100°C.
Стадия (2-4):
Способ получения соединения (2-d): соединение (2-d) можно получить, осуществляя «реакцию сочетания», где используются карбонильное соединение (2-c) и соединение (1-g).
Примерные «реакции сочетания» включают такие же реакции сочетания, как реакции, описанные выше для стадии (1-4).
Полученное таким способом соединение (2-d) может привести к соединению [1] согласно настоящему изобретению способом, осуществляемым на стадиях (1-7)-(1-9) описанной выше схемы 1.
В качестве альтернативы, соединение [1] согласно настоящему изобретению можно также получить следующим способом.
Стадия (2-5):
Способ получения соединения (2-e): соединение (2-e), содержащее защитную группу G2 можно получить, вводя в реакцию ди-трет-бутилдикарбонат и т.д. и соединение (2-d), содержащее пирролидинильную группу, замещенную оксогруппой.
Стадия (2-6):
Способ получения соединения (2-f): соединение (2-f) можно получить, восстанавливая соединение (2-e) в качестве матрицы в реакции каталитического гидрирования, где используются в каталитическом количестве палладий на активированном угле, родий на активированном угле, платина на активированном угле, и т.д., в инертном растворителе в присутствии или при отсутствии кислоты при температуре, составляющей от 0°C до 80°C.
Стадия (2-7):
Способ получения соединения (2-g): соединение (2-g) можно получить, осуществляя «реакцию удаления защитной группы» в отношении защитной группы G2, содержащейся в соединении (2-f).
Примерная реакция удаления защитной группы включает реакцию с использованием кислоты, такой как хлористоводородная кислота или трифторуксусная кислота.
Стадия (2-8):
Способ получения соединения [1] согласно настоящему изобретению: соединение [1] согласно настоящему изобретению можно получить, осуществляя «реакцию удаления защитной группы» в отношении защитной группы G1, содержащейся в соединении (2-g).
Примерные «реакция удаление защитной группы» включают такие же реакции удаление защитной группы, которые были описаны выше для стадии (1-8).
Соединение (2-a), используемое в качестве соединения исходного материала в представленной выше схеме 2, можно получить, приобретая как товарный продукт или синтезируя известным способом.
В заключение будут описаны кристаллический 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он согласно настоящему изобретению и способ его получения.
Кристаллический 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он согласно настоящему изобретению (далее иногда называется «кристалл согласно настоящему изобретению») имеет химическую структурную формулу, представленную вышеупомянутой формулой [1]. Кроме того, кристаллическое соединение согласно настоящему изобретению можно получить в форме монокристаллов, имеющих устойчивое качество, как описано выше, с хорошей воспроизводимостью, его можно устойчиво поставлять как кристаллическое лекарственное вещество, используемое для изготовления лекарственных средств, и оно имеет хорошую устойчивость при хранении.
Кристалл согласно настоящему изобретению имеет следующие физические свойства (a)-(c):
(a) рентгеновская порошковая дифрактограмма (Cu-Kα, метод измерения: метод пропускания), проявляющая пики при дифракционных углах 2θ, составляющих 8,5, 10,8, 11,2, 11,6, 13,4, 16,8, 17,0, 17,9, 18,5, 18,8, 19,1, 19,4, 22,6, 23,1, 23,2 и 24,5°, в частности, проявляющая характеристические пики при дифракционных углах 2θ, составляющих 8,5, 13,4, 19,1 и 24,5°;
(b) инфракрасный спектр поглощения (метод НПВО, алмазный кристалл), проявляющий характеристические полосы поглощения при 916, 1146, 1167, 1295, 1375, 1614, 1625, 1651, 1664, 2837, 2866, 2909, 2955, 2986, 3003, 3088 и 3146 см-1, в частности, проявляющий определенные характеристические полосы при 916, 1146, 1167, 1295, 1651, 1664, 2909, 2955, 3003 и 3146 см-1; и
(c) температура плавления, составляющая от 199°C до 201°C.
Однако характеристические пики на рентгеновской порошковой дифрактограмме могут смещаться в зависимости от условий измерения. По этой причине, пики на рентгеновской порошковой дифрактограмме соединений согласно настоящему изобретению могут иногда иметь различия или могут оказаться нечеткими.
Кристалл согласно настоящему изобретению характеризуют рентгеновская порошковая дифрактограмма, которая представлена на фиг. 1, инфракрасный спектр поглощения (метод НПВО, алмазный кристалл), который представлен на фиг. 2, и кривые дифференциального термического анализа/термогравиметрического анализа, которые представлены на фиг. 3.
Кристалл согласно настоящему изобретению можно получать, кристаллизуя 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он с использованием спиртового растворителя.
Используемый в качестве исходного материала 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он является аморфным или кристаллическим перед растворением в спиртовом растворителе.
Когда кристалл согласно настоящему изобретению получают путем кристаллизации или перекристаллизации, используя спиртовой растворитель, 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он растворяют в спиртовом растворителе и кристаллизуют из спиртового раствора, это можно осуществлять традиционным способом. Например, используется способ, согласно которому аморфный 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он растворяется в спиртовом растворителе при нагревании с последующим охлаждением.
Примерные растворители, совместимые со спиртовым растворителем, включают воду в качестве растворителя и углеводородные растворители, такие как гептан.
Соотношение при смешивании спиртового растворителя и растворителя, совместимого со спиртовым растворителем в смешанном растворителе можно изменять соответствующим образом.
Используемый спиртовой растворитель представляет собой предпочтительно спирт, содержащий от 1 до 4 атомов углерода, такой как метанол, этанол, 1-пропанол, изопропиловый спирт, трет-бутиловый спирт, 1-бутанол, 2-бутанол, 2-этоксиэтанол, 2-метоксиэтанол, трифторэтанол, этиленгликоль, и пропиленгликоль; предпочтительнее метанол, этанол, изопропиловый спирт или пропиленгликоль; и еще предпочтительнее метанол или этанол.
Растворенный 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он присутствует в концентрации, составляющей от 1 до 50 мас. % и предпочтительно от 17 до 25 мас. %. При использовании в настоящем документе термин «мас. %» означает массовую процентную долю, которую составляет 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он в растворе.
Кристаллизацию 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она можно осуществлять при температуре, составляющей от -78°C до температуры дефлегмации растворителя, но предпочтительный пример представляет собой способ, согласно которому 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он растворяется в спиртовом растворителе при нагревании раствора от 55°C до 75°C; после этого в некоторых случаях в раствор добавляется растворитель, совместимый со спиртовым растворителем, такой как вода; и при охлаждении раствора до температуры, составляющей 5°C или ниже, происходит осаждение кристаллов.
Продолжительность охлаждения не ограничивается определенным образом, при том условии, что она составляет 10 секунд или более, но, как правило, она составляет от 10 минут до 24 часов и предпочтительно от 30 минут до 5 часов. С точки зрения промышленного производства она составляет предпочтительно от 2 часов до 4 часов.
Осажденные кристаллы отделяют от раствора посредством фильтрования или центрифугирования суспензии, а затем высушивают при температуре 60°C или ниже.
Кроме того, для кристаллизации можно использовать затравочный кристалл. Затравочный кристалл можно заблаговременно получить способом, хорошо известным специалисту в данной области техники, таким как трение стенки контейнера, содержащего раствор для осаждения кристаллов, с использованием шпателя.
Температура реакции в обычных способах получения соединения согласно настоящему изобретению составляет от -78°C до 250°C и предпочтительно от -20°C до 80°C. Продолжительность реакции составляет от 5 минут до 3 суток и предпочтительно от 30 минут до 18 часов. Способы получения можно осуществлять, например, при нормальном давлении, при повышенном давлении или при микроволновом облучении.
Далее будут более подробно описаны основание, кислота и инертный растворитель, используемые в обычных способах получения соединения согласно настоящему изобретению, но они не ограничиваются следующими иллюстративными примерами. Кроме того, будут подробно описаны пригодные для использования способы выделения, но, аналогичным образом, они не ограничиваются следующими иллюстративными примерами.
Примерные «основания» включают неорганические основания, такие как гидриды щелочных металлов или щелочноземельных металлов (такие как гидрид лития, гидрид натрия, гидрид калия и гидрид кальция), амиды щелочных металлов или щелочноземельных металлов (такие как амид лития, амид натрия, диизопропиламид лития, дициклогексиламид лития, гексаметилдисилазид лития и гексаметилдисилазид калия), C1-C6-алкоксиды щелочных металлов или щелочноземельных металлов (такие как метоксид натрия, этоксид натрия и трет-бутоксид калия), гидроксиды щелочных металлов или щелочноземельных металлов (такие как гидроксид натрия, гидроксид калия, гидроксид лития и гидроксид бария), карбонаты щелочных металлов или щелочноземельных металлов (такие как карбонат натрия, карбонат калия, карбонат кальция и карбонат цезия), бикарбонаты щелочных металлов (такие как бикарбонат натрия и бикарбонат калия) и фосфаты щелочных металлов или щелочноземельных металлов (такие как трехзамещенный фосфат калия), амины (такие как триэтиламин, диизопропилэтиламин и N-метилморфолин) и основные гетероциклические соединения (такие как пиридин, 4-диметиламинопиридин, DBU (1,8-диазабицикло[5,4,0]ундец-7-ен), DBN (1,5-диазабицикло[4,3,0]нонан-5-ен), имидазол и 2,6-лутидин).
Примерные «кислоты» включают неорганические кислоты (такие как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота и фосфорная кислота), органические кислоты (такие как п-толуолсульфоновая кислота, метансульфоновая кислота, трифторуксусная кислота, муравьиная кислота, уксусная кислота и камфорсульфоновая кислота) и кислоты Льюиса (Lewis) (такие как трифторид бора, трибромид бора, хлорид алюминия, трифлат скандия и трифлат иттербия).
«Инертный растворитель» не ограничивается, при том условии, что он не ингибирует реакцию и растворяет исходный материал в некоторой степени, и соответствующие примеры включают нитрильные растворители, амидные растворители, галогеноуглеродные растворители, простоэфирные растворители, ароматические растворители, углеводородные растворители, сложноэфирные растворители, спиртовые растворители, сульфоксидные растворители и воду. Эти растворители можно использовать в форме смеси двух или более растворителей в соответствующей пропорции.
Примерные нитрильные растворители включают ацетонитрил и пропионитрил. Примерные амидные растворители включают N,N-диметилформамид (далее иногда обозначается сокращением «DMF»), N,N-диметилацетамид и N-метилпирролидон. Примерные галогеноуглеродные растворители включают дихлорметан, хлороформ, 1,2-дихлорэтан и четыреххлористый углерод. Примерные простоэфирные растворители включают диэтиловый эфир (далее иногда обозначается сокращением «эфир»), тетрагидрофуран (далее иногда обозначается сокращением «THF»), 1,4-диоксан и 1,2-диметоксиэтан. Примерные ароматические растворители включают бензол, толуол, ксилол и пиридин. Примерные углеводородные растворители включают гексан, пентан и циклогексан. Примерные сложноэфирные растворители включают этилацетат и этилформиат. Примерные спиртовые растворители включают метанол, этанол, изопропиловый спирт, трет-бутиловый спирт и этиленгликоль. Примерные сульфоксидные растворители включают диметилсульфоксид (далее иногда обозначается сокращением «DMSO»).
Соединения, полученные вышеупомянутыми способами получения, можно выделять и очищать известными средствами, такими как экстракция растворителем, изменение текучести, перенос, кристаллизация, перекристаллизация и разнообразные виды хроматографических методик.
Далее будут описаны защитные группы, которые можно использовать для соединений в общих способах получения соединений согласно настоящему изобретению, но они не ограничиваются данными иллюстративными примерами; можно также соответствующим образом выбирать и другие защитные группы.
Примеры защитной группы G2 включают C1-C6-ацильные группы (такие как формильная, ацетильная и пропионильная), C2-C15-алкоксикарбонильные группы (такие как метоксикарбонильная, этоксикарбонильная, трет-бутоксикарбонильная, бензилоксикарбонильная и 9-флуоренилметиленоксикарбонильная), арилкарбонильные группы (такие как бензоильная), тритильная группа, фталоильная группа, N,N-диметиламинометиленовая группа, замещенные силильные группы (такие как триметилсилильная, триэтилсилильная, диметилфенилсилильная, трет-бутилдиметилсилильная и трет-бутилдиэтилсилильная) и C2-C6-алкенильные группы (такие как 1-аллильная), каждая из которых обычно используется в синтезе пептидов. Эти группы могут быть замещенными и содержать один или несколько заместителей, в качестве которых выбираются атомы галогенов, C1-C6-алкоксильные группы (такие как метоксильная, этоксильная и пропоксильная) и нитрогруппа.
ПРИМЕРЫ
Далее настоящее изобретение будет описано более подробно посредством следующих примеров и примерных исследований. Эти примеры не ограничивают настоящее изобретение и могут изменяться в пределах объема настоящего изобретения.
В следующих примерах, колоночная хроматография с силикагелем NH означает хроматографическое разделение и очистку на колонке с использованием силикагеля типа NH2 (Chromatorex (зарегистрированный товарный знак) типа NH2, картридж Biotage (зарегистрированный товарный знак) SNAP KP-NH). Соотношение элюирующего растворителя выражается как объемное соотношение, если не определено другое условие.
Для колоночной хроматографии с силикагелем использовали Silica Gel 60 от компании Kanto Chemical Co., PSQ60B от компании Fuji Silysia или наполненную колонку (колонка YAMAZEN Hi-FlashTM, MORITEX Purif Pack или картридж Biotage (зарегистрированный товарный знак) SNAP KP-Sil).
В настоящем описании используются следующие сокращения.
с: синглет
д: дублет
т: триплет
к: квартет
дд: дублет дублетов
м: мультиплет
ш: широкий
J: константа спин-спинового взаимодействия
Гц: герц
CDCl3: дейтерированный хлороформ
Протонные спектры ядерного магнитного резонанса (ЯМР 1H) измеряли, используя следующие спектрометры ЯМР с преобразованием Фурье (Fourier):
300 МГц: JNM-ECP300 (JEOL), JNM-ECX300 (JEOL)
600 МГц: JNM-ECA600 (JEOL)
Для анализа использовали программное обеспечение ACD/SpecManager версии 12.01 (торговое наименование) и т.д.
Масс-спектры (МС) измеряли, используя следующие устройства:
Micromass ZQ (Waters)
LTQ XL (Thermo Fisher Scientific)
LCMS-2010EV (Shimadzu)
LCMS-IT-TOF (Shimadzu)
Agilent 6150 (Agilent)
LCQ Deca XP (Thermo Fisher Scientific)
В качестве метода ионизации использовали метод электрораспылительной ионизации (ESI) или метод двойной ионизации, сочетающий ESI и химическую ионизацию при атмосферном давлении (APCI).
Измерение оптического вращения осуществляли, используя поляриметр модели P-1020 от компании JASCO Corporation.
Измерение рентгеновской дифракции порошка осуществляли, используя дифрактометр X'Pert PRO MPD от компании PANalytical (источник излучения: Cu-Kα).
Измерение инфракрасного спектра поглощения осуществляли методом нарушенного полного внутреннего отражения (НПВО), используя спектрометр Nicolet iS5 от компании Thermo Fisher Scientific.
Измерение температуры плавления осуществляли, используя автоматическую систему определения температуры плавления MP90 от компании Mettler Toledo.
Для препаративной высокоэффективной жидкостной хроматографии (ВЭЖХ) использовали колонку CHIRALPAK IB 5 мкм (внутренний диаметр 20 мм, длина 250 мм) от компании Daicel Chemical Industries, Ltd., или аналогичную колонку.
Для аналитической ВЭЖХ использовали колонку CHIRALPAK IB 5 мкм (внутренний диаметр 4,6 мм, длина 250 мм) от компании Daicel Chemical Industries, Ltd., или аналогичную колонку.
Номенклатура химических соединений соответствовала программному обеспечению ACD/Name версии 12.01 (торговое наименование) или аналогичному.
ПРИМЕР 1
3-Циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он:
(1) 1-Бром-4-(1,1-дифторэтил)бензол:
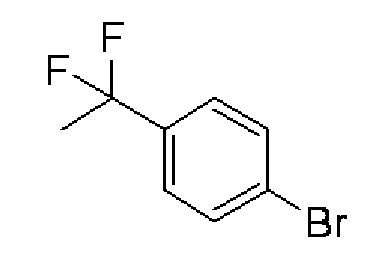
Deoxo-Fluor (зарегистрированный товарный знак) (22,2 г) добавляли в 1-(4-бромфенил)этанон (20,0 г) и смесь перемешивали при 85°C в течение 15 часов. При охлаждении льдом ледяную воду и водный раствор карбоната калия добавляли в реакционный раствор, после чего осуществляли экстракцию хлороформом. Растворитель испаряли при пониженном давлении, и полученный остаток очищали колоночной хроматографией с силикагелем (гексан), получая целевое соединение (13,0 г, выход 59%) в форме желтого масла.
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 1,91 (т, J=18,2 Гц, 3H), 7,50 (д, J=8,3 Гц, 2H), 7,86 (д, J=8,3 Гц, 2H).
(2) 2-[4-(1,1-Дифторэтил)фенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолан:
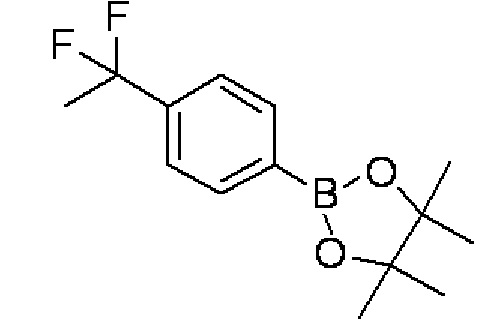
Бис(пинаколято)дибор (22,5 г), комплекс дихлорида 1,1'-бис(дифенилфосфин)ферроцен]палладия(II) и дихлорметана (904 мг) и ацетат калия (8,70 г) добавляли в раствор 1-бром-4-(1,1-дифторэтил)бензола (9,80 г), синтезированного в примере 1-(1), в 1,4-диоксане (60 мл) и перемешивали при 90°C в течение 10 часов. Реакционный раствор выливали в воду, и после этого осуществляли экстракцию хлороформом. Растворитель испаряли при пониженном давлении, и полученный остаток очищали колоночной хроматографией с силикагелем (гексан), получая целевое соединение (7,87 г, выход 66%) в форме бесцветного твердого вещества.
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 1,35 (с, 12H), 1,91 (т, J=18,2 Гц, 3H), 7,50 (д, J=7,8 Гц, 2H), 7,86 (д, J=7,8 Гц, 2H).
(3) 5-Циклопропил-6-метоксипиридин-2-карбальдегид:
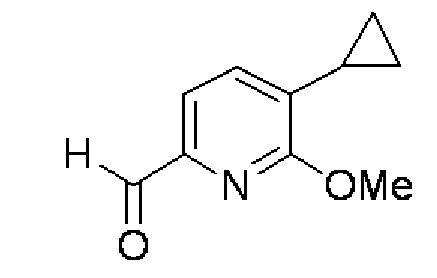
Раствор 2 М хлорида н-бутилмагния в тетрагидрофуране (74,5 мл) добавляли в смешанный растворитель, содержащий толуол (433 мл) и тетрагидрофуран (116 мл), в атмосфере аргона. Раствор 1,6 М н-бутиллития в тетрагидрофуране (186 мл) добавляли каплями при -12°C, перемешивали в течение 40 минут, и после этого в раствор добавляли 6-бром-3-циклопропил-2-метоксипиридин (34,0 г). После дополнительного перемешивания в течение 1 часа в раствор добавляли каплями N,N-диметилформамид (32,7 г). После дополнительного перемешивания в течение 1,5 часов реакционный раствор добавляли в 13% водный раствор лимонной кислоты и экстрагировали, и после этого органический слой промывали водой. Растворитель испаряли при пониженном давлении, и полученный остаток очищали колоночной хроматографией с силикагелем (гексан/этилацетат=95:5→90:10), получая целевое соединение (21,2 г, выход 80%) в форме желтого масла.
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 0,73-0,78 (м, 2H), 1,03-1,08 (м, 2H), 2,14-2,21 (м, 1H), 4,07 (с, 3H), 7,19 (д, J=7,4 Гц, 1H), 7,49 (д, J=7,4 Гц, 1H), 9,92 (с, 1H).
МС (+): 178[M+H]+.
(4), (5) (5R)-5-[(Z)-2-Бром-2-(5-циклопропил-6-метоксипиридин-2-ил)этенил]пирролидин-2-он:
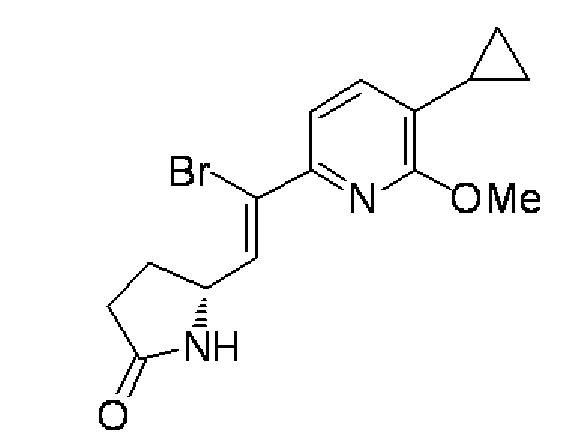
(4) Раствор 1 М гексаметилдисилазида калия в тетрагидрофуране (405 мл) добавляли каплями при -78°C в раствор, содержащий (5R)-5-[(1,3-бензотиазол-2-илсульфонил)метил]пирролидин-2-он (30,0 г) и хлорид лития (8,58 г) в тетрагидрофуране (1,2 л), и после этого перемешивали в течение 1 часа. Раствор 5-циклопропил-6-метоксипиридин-2-карбальдегида (17,9 г), синтезированного в примере 1-(3), в тетрагидрофуране (600 мл) добавляли каплями в раствор и дополнительно перемешивали в течение 0,5 часа. Насыщенный раствор хлорида аммония (500 мл) добавляли в реакционный раствор, и после этого осуществляли экстракцию этилацетатом. Органический слой промывали солевым раствором, высушивали над сульфатом магния, а затем отделяли осушающее вещество путем фильтрования, и растворитель испаряли при пониженном давлении. Полученный остаток дважды очищали колоночной хроматографией с силикагелем (гексан/этилацетат=100:0→70:30), получая (5R)-5-[(Z)-2-(5-циклопропил-6-метоксипиридин-2-ил)этенил]пирролидин-2-он (8,40 г, выход 34%) в форме желтого масла.
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 0,62-0,68 (м, 2H), 0,93-0,99 (м, 2H), 1,89-1,97 (м, 1H), 2,03-2,09 (м, 1H), 2,33-2,56 (м, 3H), 3,98 (с, 3H), 5,53-5,55 (м, 1H), 5,70 (дд, J=11,56, 7,84 Гц, 1H), 5,94-6,03 (ш. с, 1H), 6,34 (дд, J=11,56, 1,24 Гц, 1H), 6,70 (д, J=7,43 Гц, 1H), 7,07 (д, J=7,43 Гц, 1H).
МС (+): 259[M+H]+.
(5) Бром (1,33 мл) добавляли каплями в раствор, содержащий (5R)-5-[(Z)-2-(5-циклопропил-6-метоксипиридин-2-ил)этенил]пирролидин-2-он (8,40 г), синтезированный в примере 1-(4), в хлороформе (126 мл) при 0°C. После перемешивания в течение 1 часа, раствор, содержащий 1,8-диазабицикло[5,4,0]ундец-7-ен (9,7 мл) в хлороформе (42 мл), добавляли каплями в течение 30 минут и перемешивали в течение 15 минут. Раствор 1 М хлористоводородной кислоты (200 мл) добавляли в реакционный раствор, и после этого осуществляли экстракцию этилацетатом. Органический слой промывали солевым раствором, высушивали над сульфатом магния, а затем отделяли осушающее вещество путем фильтрования, и растворитель испаряли при пониженном давлении. Полученный остаток очищали колоночной хроматографией с силикагелем (гексан/этилацетат=50:50→0:100), получая целевое соединение (7,50 г, выход 68%) в форме желтого твердого вещества.
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 0,65-0,68 (м, 2H), 0,95-1,00 (м, 2H), 1,96-2,10 (м, 2H), 2,40-2,59 (м, 3H), 4,00 (с, 3H), 4,80 (к, J=7,4 Гц, 1H), 5,68-5,70 (ш. с, 1H), 7,10 (д, J=7,8 Гц, 1H), 7,15 (д, J=7,8 Гц, 1H), 7,24 (д, J=7,8 Гц, 1H).
МС (+): 337[M+H]+.
(6) (5R)-5-{(E)-2-(5-Циклопропил-6-метоксипиридин-2-ил)-2-[4-(1,1-дифторэтил)фенил]этенил}пирролидин-2-он:
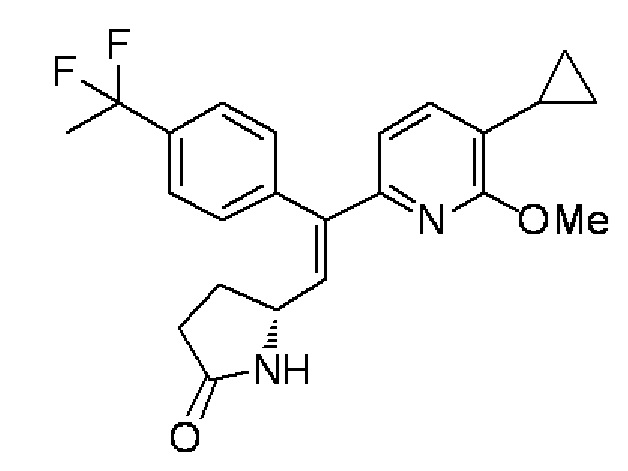
В атмосфере азота 2-[4-(1,1-дифторэтил)фенил]-4,4,5,5-тетраметил-1,3,2-диоксаборолан (1,59 г), синтезированный в примере 1-(2), карбонат цезия (1,92 г), трис(дибензилиденацетон)дипалладий (0) (271 мг), три(2-фурил)фосфин (412 мг) и дистиллированную воду (10 мл) добавляли в раствор, содержащий (5R)-5-[(Z)-2-бром-2-(5-циклопропил-6-метоксипиридин-2-ил)этенил]пирролидин-2-он (1,0 г), синтезированный в примере 1-(5), в 1,4-диоксане (50 мл), и смесь перемешивали при 90°C в течение 2 часов. Реакционный раствор выливали в воду, и после этого осуществляли экстракцию этилацетатом. Органический слой высушивали над безводным сульфатом магния, а затем отделяли осушающее вещество путем фильтрования, и растворитель испаряли при пониженном давлении. Полученный остаток очищали колоночной хроматографией с силикагелем (гексан/этилацетат=100:0→0:100) и дополнительно колоночная хроматография с силикагелем NH (гексан/этилацетат=100:0→0:100), получая целевое соединение (1,13 г, выход 96%) в форме бесцветного аморфного вещества.
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 0,56-0,64 (м, 2H), 0,90-0,97 (м, 2H), 1,93-2,09 (м, 5H), 2,20-2,34 (м, 2H), 2,37-2,45 (м, 1H), 4,04 (с, 3H), 4,07-4,16 (м, 1H), 5,73-5,75 (ш. с, 1H), 6,23 (д, J=7,4 Гц, 1H), 6,90 (д, J=9,9 Гц, 1H), 6,92 (д, J=7,8 Гц, 1H), 7,24 (д, J=8,3 Гц, 2H), 7,57 (д, J=7,8 Гц, 2H).
МС (+): 399[M+H]+.
(7) (5R)-5-{2-(5-Циклопропил-6-метоксипиридин-2-ил)-2-[4-(1,1-дифторэтил)фенил]этил}пирролидин-2-он:
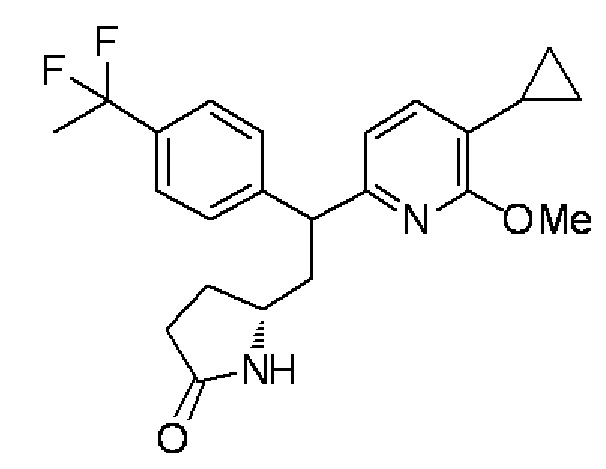
В атмосфере азота 10% палладия на активированном угле (110 мг) добавляли в раствор, содержащий (5R)-5-{(E)-2-(5-циклопропил-6-метоксипиридин-2-ил)-2-[4-(1,1-дифторэтил)фенил]этенил}пирролидин-2-он (1,1 г), синтезированный в примере 1-(6), в метаноле (44 мл), и смесь перемешивали при комнатной температуре в течение 1 часа в атмосфере водорода. После фильтрования реакционного раствора с использованием Celite (зарегистрированный товарный знак) растворитель испаряли при пониженном давлении, получая целевое соединение (1,10 г, выход 99%) в форме бесцветного аморфного вещества.
MS(+): 401[M+H]+.
(8) 3-Циклопропил-6-{1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он:
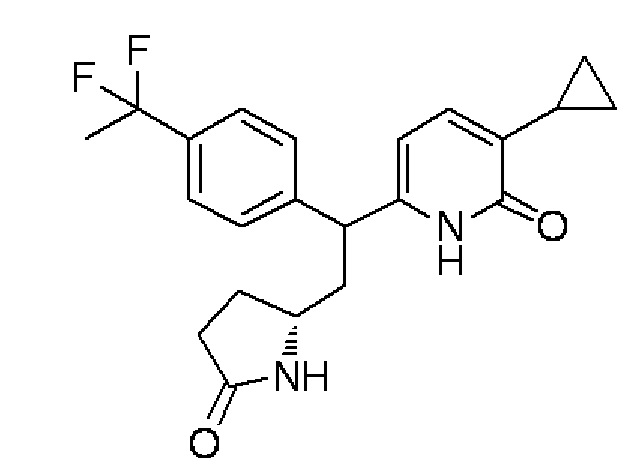
Хлортриметилсилан (707 мкл) и йодид калия (1,37 г) добавляли в раствор, содержащий (5R)-5-{2-(5-циклопропил-6-метоксипиридин-2-ил)-2-[4-(1,1-дифторэтил)фенил]этил}пирролидин-2-он (1,1 г), синтезированный в примере 1-(7), в ацетонитриле (30 мл), и смесь перемешивали при 60°C в течение 1 часа. Реакционный раствор выливали в воду, и после этого осуществляли экстракцию этилацетатом. Органический слой промывали солевым раствором, высушивали над безводным сульфатом магния, а затем отделяли осушающее вещество путем фильтрования и растворитель испаряли при пониженном давлении. Полученный остаток очищали колоночной хроматографией с силикагелем (хлороформ/метанол=100:0→80:20), получая целевое соединение (880 мг, выход 83%) в форме бесцветного аморфного вещества.
MS(+):387[M+H]+.
(9) 3-Циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он:
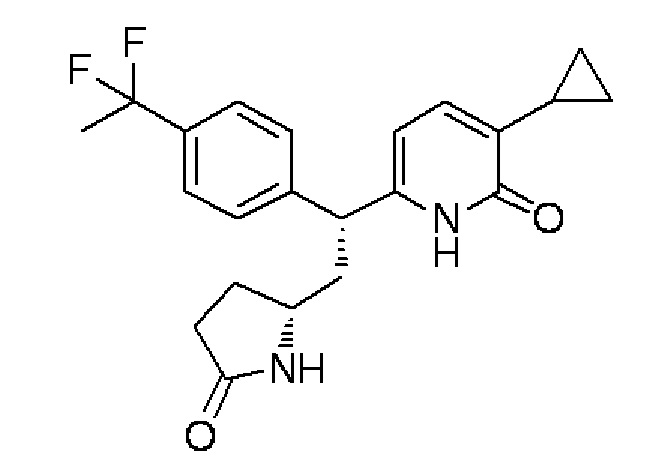
Смесь RS (180 мг) 3-циклопропил-6-{1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она, синтезированного в примере 1-(8), фракционировали, используя высокоэффективную жидкостную хроматографию (ВЭЖХ) с колонкой, содержащей хиральный наполнитель CHIRALPAK IB (гексан/этанол=70:30 по объему, 40°C, 12 мл/мин, 254 нм), получая целевое соединение (70 мг) в форме бесцветного аморфного вещества и диастереомер (67 мг) целевого соединения в форме бесцветного аморфного вещества.
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 0,54-0,67 (м, 2H), 0,90-0,98 (м, 2H), 1,68-1,75 (м, 1H), 1,88 (т, J=18,2 Гц, 3H), 2,07-2,14 (м, 1H), 2,14-2,40 (м, 5H), 3,43-3,52 (м, 1H), 4,07-4,12 (м, 1H), 6,00 (д, J=7,0 Гц, 1H), 6,92 (д, J=7,0 Гц, 1H), 7,41-7,47 (м, 4H), 7,60-7,68 (м, 1H), 12,28-12,49 (ш. с, 1H).
МС (+): 387[M+H]+.
CHIRALPAK IB 4,6×250 мм 5 мкм (DAICEL), гексан/этанол=70:30 по объему, 40°C, 1,0 мл/мин, 210 нм, время удерживания Rt=7,5 мин.
Диастереомер:
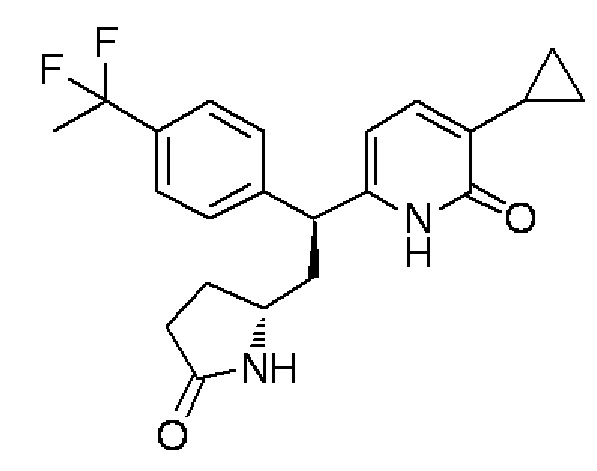
Спектр ЯМР 1H (600 МГц, CDCl3) δ м. д. 0,54-0,66 (м, 2H), 0,95-1,05 (м, 2H), 1,75-1,84 (м, 1H), 1,90 (т, J=18,0 Гц, 3H), 2,15-2,41 (м, 6H), 3,54-3,64 (м, 1H), 4,16 (дд, J=10,1, 5,57 Гц, 1H), 6,01 (с, 1H), 6,96 (д, J=7,0 Гц, 1H), 7,39 (д, J=8,26 Гц, 2H), 7,47 (с, 2H), 7,83-7,92 (м, 1H), 13,14-13,34 (ш. с, 1H).
МС (+): 387[M+H]+.
CHIRALPAK IB 4,6×250 мм 5 мкм (DAICEL), гексан/этанол=70:30 по объему, 40°C, 1,0 мл/мин, 210 нм, время удерживания Rt=18,9 мин.
ПРИМЕР 2
(5R)-5-{2-(5-Циклопропил-6-метоксипиридин-2-ил)-2-[4-(1,1-дифторэтил)фенил]этенил}пирролидин-2-он:
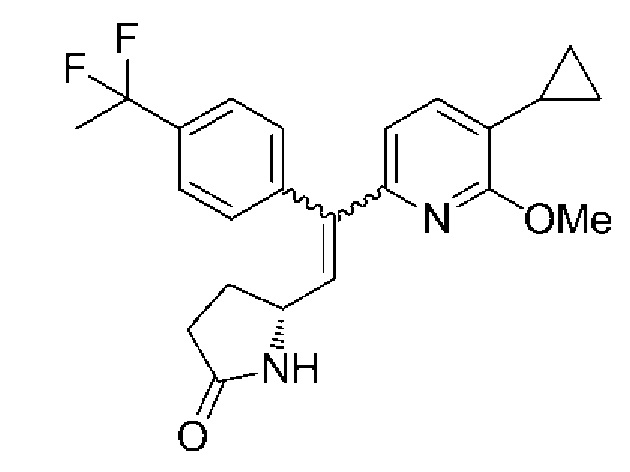
(1) (5-Циклопропил-6-метоксипиридин-2-ил)[4-(1,1-дифторэтил)фенил]метанон:
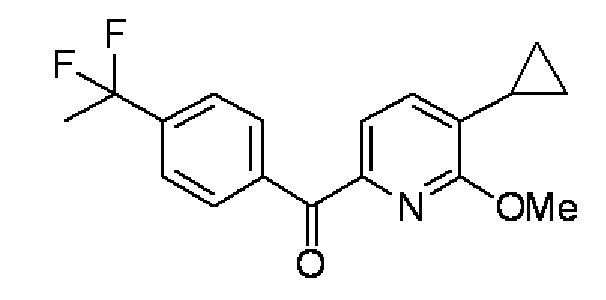
В атмосфере азота, 1,6 M раствор н-бутиллития в тетрагидрофуране (127 мл) добавляли каплями в раствор 6-бром-3-циклопропил-2-метоксипиридина (41,5 г) в тетрагидрофуране (273 мл) при -78°C в течение 50 минут, а затем осуществляли перемешивание при -78°C в течение 1 часа. После этого раствор 4-(1,1-дифторэтил)бензонитрила (24,3 г) в тетрагидрофуране (137 мл) добавляли каплями в реакционный раствор при поддержании температуры на уровне -78°C в течение 75 минут, а затем осуществляли дополнительное перемешивание в течение 1 часа. После повышения температуры реакционного раствора до 0°C, в раствор последовательно добавляли каплями раствор 1 М хлористоводородной кислоты (437 мл), тетрагидрофуран (365 мл) и раствор 1 М хлористоводородной кислоты (146 мл).
Реакционный раствор разделяли на органический слой и водный слой, а затем осуществляли экстракцию водного слоя этилацетатом (1000 мл). Объединенные органические слои высушивали над безводным сульфатом магния, осушающее вещество отделяли путем фильтрации, и после этого растворитель испаряли при пониженном давлении. Полученный остаток очищали колоночной хроматографией с силикагелем (гексан/этилацетат=100:0→95:5), получая целевое соединение (34,0 г, выход 74%) в форме бесцветного масла.
Спектр ЯМР 1H (300 МГц, CDCl3) δ м. д. 0,72-0,81 (м, 2H), 1,00-1,10 (м, 2H), 1,96 (т, J=18,2 Гц, 3H), 2,10-2,25 (м, 1H), 3,95 (с, 3H), 7,24 (д, J=6,9 Гц, 1H), 7,59 (д, J=9,0 Гц, 2H), 7,67 (д, J=7,8 Гц, 1H), 8,21 (д, J=8,6 Гц, 2H).
МС (+): 318[M+H]+.
(2) (5R)-5-{2-(5-Циклопропил-6-метоксипиридин-2-ил)-2-[4-(1,1-дифторэтил)фенил]этенил}пирролидин-2-он:
В атмосфере азота раствор 1,0 M гексаметилдисилазида лития в тетрагидрофуране (317 мл) добавляли каплями в раствор, содержащий (5-циклопропил-6-метоксипиридин-2-ил)[4-(1,1-дифторэтил)фенил]метанон (33,5 г), полученный в примере 2-(1), и (5R)-5-[(1,3-бензотиазол-2-илсульфонил)метил]пирролидин-2-он (37,5 г) в дихлорметане (1007 мл) в течение 50 минут при -78°C, и смесь перемешивали при -78°C в течение 4 часов 40 минут.
После того, как температуру реакционного раствора повышали до 0°C, насыщенный водный раствор хлорида аммония (335 г) добавляли каплями для завершения реакции. Реакционный раствор разделяли на органический слой и водный слой, а затем осуществляли экстракцию водного слоя хлороформом (339 мл) и объединенные органические слои промывали водой (502 г). Органический слой высушивали над безводным сульфатом магния, осушающее вещество отделяли путем фильтрации, и после этого растворитель испаряли при пониженном давлении. Полученный остаток очищали колоночной хроматографией с силикагелем (гексан/этилацетат=50:50→0:100), получая целевое соединение, которое представляло собой смесь изомеров E и Z, в форме светло-желтого аморфного вещества (42,7 г, E:Z=50:50).
Соотношение в смеси определяли по процентному соотношению площади пиков методом жидкостной хроматографии.
Жидкостную хроматографию осуществляли в следующих условиях.
Колонка для жидкостной хроматографии (L), содержащая октадецилсилан (ODS), CH3CN/ацетатный буферный раствор 0,01 М (водный раствор 0,01 M уксусной кислоты + водный раствор 0,01 M ацетата натрия в соотношении 8:1)=80:20 по объему, 1,0 мл/мин, 40°C, 254 нм, время удерживания Rt: изомер E 5,40 мин, изомер Z 5,08 мин.
МС(+): 399[M+H]+.
После этого целевое соединение можно превратить в соединение [1], осуществляя способы, описанные в примерах 1 (7)-(9) или соответствующие способы.
ПРИМЕР 3
Кристаллическое соединение [1]
Способ кристаллизации соединения [1]
Воду (29 г) добавляли каплями в раствор, содержащий 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он (14,4 г) в этаноле (72 г) при 75°C. При постепенном охлаждении от 75°C затравочный кристалл добавляли во время достижения внутренней температуры 40°C, а затем раствор охлаждали до комнатной температуры. После этого температуру дополнительно снижали до 0°C, и раствор перемешивали в течение ночи, получая суспензию.
Температуру возвращали на уровень комнатной температуры, и полученное твердое вещество собирали путем фильтрации, промывали водой и высушивали при 50°C в течение 6 часов), получая 8,7 г (выход 60%) бесцветных кристаллов.
(a) Рентгеновская порошковая дифрактограмма (Cu-Kα, метод измерения: метод пропускания) проявляет пики при дифракционных углах 2θ, составляющих 8,5, 10,8, 11,2, 11,6, 13,4, 16,8, 17,0, 17,9, 18,5, 18,8, 19,1, 19,4, 22,6, 23,1, 23,2 и 24,5°.
(b) Инфракрасный спектр поглощения (метод НПВО, алмазный кристалл) проявляет характеристические полосы поглощения при 916, 1146, 1167, 1295, 1375, 1614, 1625, 1651, 1664, 2837, 2866, 2909, 2955, 2986, 3003, 3088 и 3146 см-1.
(c) Температура плавления составляет от 199°C до 201°C.
(d) Удельное оптическое вращение составляет [α]D23=+36 (c0,1, MeOH).
Эффект активации GK, которые проявляют соединения согласно настоящему изобретению можно оценивать, осуществляя известные способы, такие как способ, описанный в примерных исследованиях.
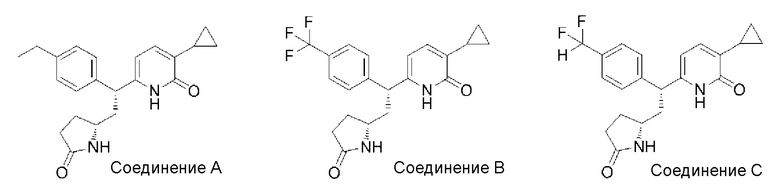
Эффекты активации GK, которые проявляют соединение [1] согласно настоящему изобретению, соединение A (пример 4-302), соединение B (пример 4-248) и соединение C (пример 4-340), описанные в международной патентной заявке WO 2011/068211, измеряли, используя способ, описанный в следующих примерных исследованиях.
Структуры соединения A, соединения B и соединения C, описанных в международной патентной заявке WO 2011/068211, представлены ниже.
Примерное исследование 1. Исследование активации GK
Исследование активации GK для исследуемых соединений осуществляли способом Van Schaftingen и др. (Eur. J. Biochem., 1989 г., т. 179, с. 179-184) с частичными изменениями. Активность GK измеряли по изменению поглощения на основании количества тио-NADH, который представляет собой восстановленную форму продукта, в который превращается тио-NAD+ (тионикотинамид-адениндинуклеотид), когда дегидрируется глюкоза 6-фосфат, произведенный GK с использованием глюкозы в качестве матрицы под действием G6PDH (глюкоза-6-фосфатдегидрогеназа).
Источник фермента, который использовали в данном исследовании, а именно GK печени человека, проявлял экспрессию в кишечной палочке (E. coli) как белок слияния с GST (глутатион-S-трансфераза), которая присоединялась к аминному концу цепи, и очистку осуществляли, используя глутатионсефарозу 4B от компании Amersham Biosciences.
Исследование осуществляли, используя микропланшеты, содержащие 96 плоскодонными лунками половинного объема от компании Corning. Раствор исследуемого соединения в диметилсульфоксиде (DMSO) при конечной концентрации 1% в DMSO и DMSO в качестве контрольного вещества добавляли в каждую лунку планшетов. Кроме того, раствор 25 мМ HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота)-KOH (pH=7,1), 25 мМ KCl, 2 мМ MgCl2, 2 мМ тио-NAD+, 4 мМ глюкозы, 1 мМ DTT (дитиотреитол), 0,01 ед/мкл G6PDH и 2 мкг/мл GK печени человека добавляли в конечных концентрациях, соответственно, в каждую из лунок. После этого в каждую из лунок добавляли ATP (аденозин-5'-трифосфат), получая конечную концентрацию 2 мМ, и реакция начиналась. Микропланшеты выдерживали при комнатной температуре. Через 15 минут после начала реакции поглощение при 405 нм измеряли, используя абсорбционный спектрометр для микропланшетов.
Активность GK, максимально активированной исследуемым соединением, принимали как максимальную способность активации, и концентрацию исследуемого соединения (нМ), требуемую для активации на уровне 50% от этой максимальной способности активации, выражали как EC50.
Результаты представлены ниже.
Примерное исследование 2. Исследование гипогликемии с использованием мышей линии C57BL6/J
Исследование для подтверждения гипогликемического эффекта исследуемых соединений осуществляли согласно обычно используемому способу, который представили Grimsby и др. (Science 2003 г., т. 301, с. 370-373).
Измеряли массу тела шести мышей линии C57BL6/J, которые получали неограниченное количество пищи. Исследуемое соединение суспендировали или растворяли в основании для введения (0,5% метилцеллюлозы) при концентрации от 0,06 до 20 мг/мл. Мыши перорально вводили 5 мл/кг раствора лекарственного средства (эквивалент от 0,3 до 100 мг/кг исследуемого соединения) или контрольного раствора (содержащего только основание). Приблизительно 60 мкл крови отбирали из хвостовой вены, используя капиллярную трубку, непосредственно перед введением исследуемого соединения и через 0,5, 1, 2, 4 и 6 часов после введение исследуемого соединения. Образцы крови центрифугировали и после этого измеряли концентрацию глюкозы в плазме. Площадь под кривой (AUC) вычисляли по изменению концентрации глюкозы в плазме с течением времени после введение исследуемого соединения, и определяли степень уменьшения (%) по отношению к AUC для контрольной группы. Дозу, при которой процентное уменьшение AUC составляло 20% (значение ED20; мг/кг), вычисляли по кривой зависимости от дозы, где степень уменьшения AUC показывала вертикальная ось, а дозу показывала горизонтальная ось.
Результаты представлены ниже.
Результаты описанного выше исследования подтвердили, что соединения согласно настоящему изобретению производят хорошие гипогликемические эффекты, начиная с интервала низких доз. Следовательно, соединения согласно настоящему изобретению являются пригодными для использования в качестве средства для профилактики/лечения диабета и связанных с ним заболеваний, причем их терапевтический интервал, очевидно, является шире, чем для других соединений.
Кроме того, оказалось, что по сравнению с тремя соединениями, описанными в международной патентной заявке WO 2011/068211, соединения согласно настоящему изобретению производят значительно более сильный гипогликемический эффект.
Кроме того, соединения согласно настоящему изобретению имеют свойства, желательные для лекарственных средств. Примеры таких свойств включают хорошие гипогликемические эффекты посредством проявления хороших физических свойств и фармакокинетики (например, устойчивость печеночного метаболизма).
ПРОМЫШЛЕННАЯ ПРИМЕНИМОСТЬ
Соединения согласно настоящему изобретению проявляют превосходный эффект активации GK и могут быть использованы как средства для лечения и профилактики не только диабета, но также связанных с диабетом заболеваний, таких как ожирение и гиперлипидемия, или хронических диабетических осложнений, таких как ретинопатия, нефропатия и артериосклероз.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ 2-ПИРИДОНА | 2010 |
|
RU2551847C2 |
| НОВОЕ ПРОИЗВОДНОЕ 3-ГИДРОКСИ-5-АРИЛИЗОТИАЗОЛА | 2010 |
|
RU2567755C2 |
| (ГЕТЕРО)АРИЛЦИКЛОПРОПИЛАМИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ LSD1 | 2012 |
|
RU2681211C2 |
| ПИРАЗОЛЬНЫЕ СОЕДИНЕНИЯ, ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ, И ИХ ПРИМЕНЕНИЕ В ФАРМАЦЕВТИКЕ | 2019 |
|
RU2805312C2 |
| ПРОИЗВОДНЫЕ 2-ОКСОХИНАЗОЛИНА КАК ИНГИБИТОРЫ МЕТИОНИН АДЕНОЗИЛТРАНСФЕРАЗЫ 2A | 2019 |
|
RU2830169C2 |
| ЦИКЛОАЛКИЛНИТРИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС-КИНАЗЫ | 2014 |
|
RU2655380C2 |
| АМИНОТЕТРАГИДРОПИРАНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ДИПЕПТИДИЛПЕПТИДАЗЫ-IV ДЛЯ ЛЕЧЕНИЯ ИЛИ ПРЕДУПРЕЖДЕНИЯ ДИАБЕТА | 2010 |
|
RU2550508C2 |
| КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛЬПАИНА | 2010 |
|
RU2540856C2 |
| ПРОИЗВОДНЫЕ ЦИКЛОПРОПИЛАМИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ H-ГИСТАМИНОВОГО РЕЦЕПТОРА | 2007 |
|
RU2449989C2 |
| ИНГИБИТОРЫ ЦИСТЕИНПРОТЕАЗ КАТЕПСИНОВ | 2014 |
|
RU2692799C2 |
Изобретение относится к 2-пиридоновому соединению, представленному формулой [1]:
 ,
,
таутомеру данного соединения, его фармацевтически приемлемой соли (далее называется 2-пиридоновое соединение, таутомер данного соединения или его фармацевтически приемлемая соль, называемая термином «2-пиридоновое соединение или родственные соединения»). Изобретение также относится к кристаллическому 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-ону, его способу получения, к лекарственному средству для профилактики или лечения заболевания или состояния, которое может улучшаться посредством эффекта активации глюкокиназы. Технический результат: получено новое 2-пиридоновое соединение, обладающее свойствами активатора глюкокиназы, которое может быть использовано в качестве лекарственного средства для профилактики или лечения диабета. 5 н. и 2 з.п. ф-лы, 3 ил., 2 табл., 3 пр.
1. 2-Пиридоновое соединение, представленное формулой [1]:
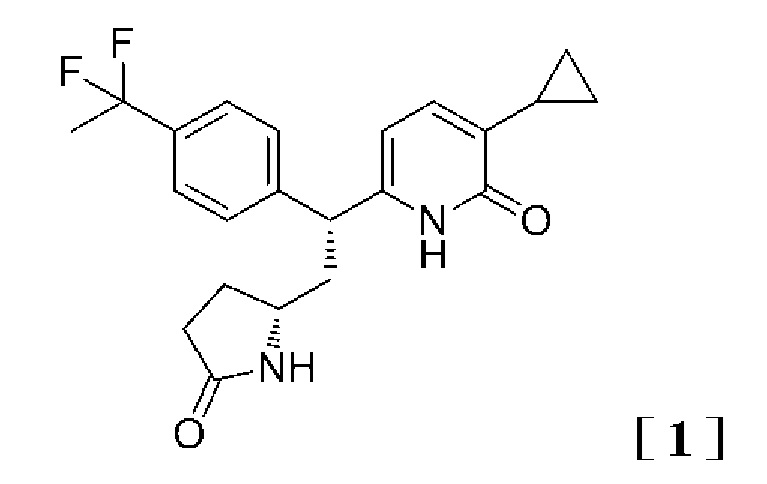 ,
,
таутомер данного соединения, его фармацевтически приемлемая соль (далее называется 2-пиридоновое соединение, таутомер данного соединения или его фармацевтически приемлемая соль, называемая термином «2-пиридоновое соединение или родственные соединения»).
2. Кристаллический 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он по п. 1, представленный вышеупомянутой формулой [1] и имеющий следующее физическое свойство (a):
(a) рентгеновская порошковая дифрактограмма (Cu-Kα), проявляющая пики при дифракционных углах 2θ, составляющих 8,5, 13,4, 19,1 и 24,5°.
3. Кристаллический 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-он по п. 1, представленный вышеупомянутой формулой [1] и имеющий следующие физические свойства (a)-(c):
(a) рентгеновская порошковая дифрактограмма (Cu-Kα), проявляющая пики при дифракционных углах 2θ, составляющих 8,5, 13,4, 19,1 и 24,5°;
(b) инфракрасный спектр поглощения, проявляющий характеристические полосы поглощения при 916, 1146, 1167, 1295, 1651, 1664, 2909, 2955, 3003 и 3146 см-1; и
(c) температура плавления, составляющая от 199 до 201°C.
4. Способ получения кристаллического 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она, имеющего следующие физические свойства (a)-(c):
(a) рентгеновская порошковая дифрактограмма (Cu-Kα), проявляющая пики при дифракционных углах 2θ, составляющих 8,5, 13,4, 19,1 и 24,5°;
(b) инфракрасный спектр поглощения, проявляющий характеристические полосы поглощения при 916, 1146, 1167, 1295, 1651, 1664, 2909, 2955, 3003 и 3146 см-1; и
(c) температура плавления, составляющая от 199 до 201°C,
включающий растворение 3-циклопропил-6-{(1R)-1-[4-(1,1-дифторэтил)фенил]-2-[(2R)-5-оксопирролидин-2-ил]этил}пиридин-2(1H)-она, представленного вышеупомянутой формулой [1], в спиртовом растворителе при нагревании для получения раствора; последующее добавление воды в качестве растворителя в раствор; охлаждение полученного в результате раствора до температуры 5°C или ниже для получения кристаллов и высушивание полученных кристаллов при температуре 60°C или ниже.
5. Лекарственное средство для профилактики или лечения заболевания или состояния, которое может улучшаться посредством эффекта активации глюкокиназы, содержащее в качестве активного ингредиента 2-пиридоновое соединение, таутомер данного соединения, его фармацевтически приемлемую соль или родственные соединения по п. 1.
6. Лекарственное средство по п. 5, которое представляет собой гипогликемическое средство.
7. Лекарственное средство по п. 5, где данное лекарственное средство представляет собой средство для профилактики или лечения диабета.
| УСТРОЙСТВО ДЛЯ СУШКИ ЗЕРНА | 2012 |
|
RU2508513C1 |
| УСТРОЙСТВО ДЛЯ СУШКИ ЗЕРНА | 2012 |
|
RU2508513C1 |
| Калорифер для подогревания воздуха в сушилках | 1929 |
|
SU16595A1 |

