ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к соединениям из класса фторхинолонов, содержащим молекулу пиперазина, замещенную длинной алкильной цепью, а также к их фармацевтически приемлемым солям, сольватам и пролекарствам.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Огромное количество заболеваний и синдромов обусловлены бактериальными инфекциями, в частности, к ним относятся инфекционные заболевания мочеполовой системы, инфекционные заболевания кожи и мягких тканей, инфекции, передающиеся половым путем, столбняк, брюшной тиф, туберкулез, холера, сифилис, различные пневмонии и сальмонеллез. Несмотря на огромное количество и разнообразие антибактериальных агентов, бактериальные инфекции остаются основной причиной смерти людей по всему миру, в особенности в развивающихся странах. Более того, постоянное появление резистентных бактерий является большой проблемой, как в развитых, так и в развивающихся странах.
Избыточное назначение антибиотиков, по всей видимости, является одной из основных причин появления резистентных бактерий. Однако, такие факторы, как использование антибиотиков при разведении сельскохозяйственных животных, а также все более частое использование антибактериальных агентов в составе чистящих средств, также влияют на появление резистентных штаммов. Кроме того, даже без воздействия антибиотиков, мутации ДНК и приобретение экстрахромосомной ДНК происходят у бактерий естественным образом и потенциально могут участвовать в развитии резистентности.
В зависимости от степени устойчивости, резистентные бактерии классифицируются на три группы: бактерии с множественной лекарственной устойчивостью (MDR), бактерии с широкой лекарственной устойчивостью (XDR) и бактерии с полной лекарственной устойчивостью (PDR) (Magiorakos, А.Р. et al, Clinical Microbiology and Infection, 2012, pp. 268-281). Таким образом, существует необходимость в разработке антибиотиков, которые были бы активны в отношении как бактерий дикого типа, так и в отношении различных классов лекарственно-устойчивых бактерий. Кроме того, любая бактерия, выжившая после воздействия антибиотика, будет реплицироваться и продуцировать новое потомство резистентных бактерий, поэтому антибиотики должны обладать максимальной активностью для полной эрадикации бактерий.
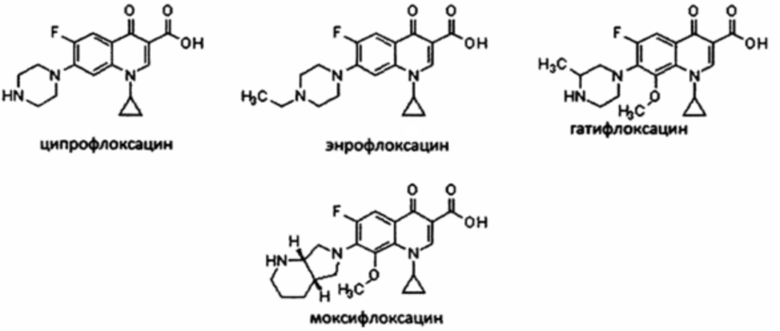
Хинолоны образуют большой класс антибиотиков, разработанных в 60-х годах и обладающих активностью в отношении широкого спектра бактерий. Добавление атома хлора к ароматическому кольцу привело к открытию в 70-х годах класса фторхинолонов. Указанные молекулы обладают улучшенными фармакокинетическими свойствами по сравнению с хинолонами, такими, как хорошая абсорбция при пероральном введении, хорошее проникновение в ткани и относительно длительный период активности. Фторхинолоны, такие как ципрофлоксацин (US 4,670,444, ЕР 0049355), энрофлоксацин (US 4,670,444, ЕР 0049355), гатифлоксацин (US 4,980,470) и моксифлоксацин (US 5,607,942) в настоящее время широко используются для лечения бактериальных инфекций, вызванных различными типами бактерий, в качестве терапии первой или второй линии.
Механизм действия фторхинолонов на бактерии заключается в ингибировании фермента ДНК гиразы, необходимого для репликации ДНК, таким образом, резистентность к фторхинолонам обусловлена главным образом появлением мутаций ДНК гиразы.
Таким образом, несмотря на то, что фторхинолоны высоко эффективны в отношении широкого спектра бактериальных инфекций и обладают прекрасными фармакокинетическими параметрами, даже к ним может развиваться высокая резистентность микроорганизмов.
Следовательно, существует необходимость в разработке новых соединений, активных как в отношении бактерий дикого типа, так и в отношении резистентных бактерий, таких, как MDA, XDR и PDR бактерии. Такие соединения должны преодолевать механизмы устойчивости, которые развиваются у бактерий против используемых в настоящее время антибиотиков, и обладать максимальной эрадикационной способностью одновременно с низкой токсичностью.
Настоящее изобретение относится к соединениям из класса фторхинолонов, содержащим молекулу пиперазина, замещенную длинной алкильной цепью. Соединения, заявленные в соответствии с настоящим изобретением, обладают повышенной антибактериальной активностью по сравнению с фторхинолонами, которые используются в настоящее время против бактерий дикого типа и против резистентных бактерий.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В соответствии с настоящим изобретением, заявленные соединения имеют общую
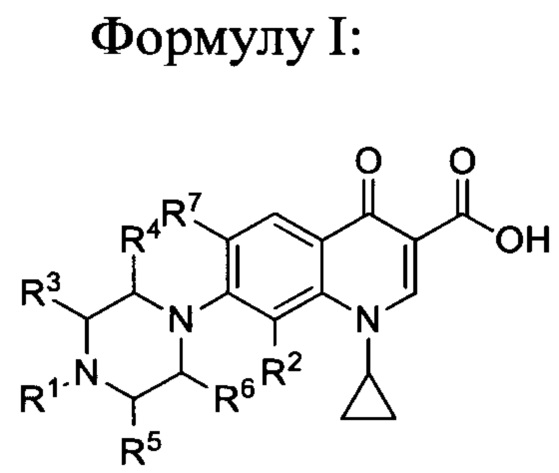
и также включают фармацевтически приемлемые соли, сольваты и пролекарства, при этом:
- R1 представляет собой насыщенную или ненасыщенную, замещенную или незамещенную, разветвленную или неразветвленную алкильную группу, включающую от 4 до 20 атомов углерода, предпочтительно, от 5 до 16, более предпочтительно, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или 16 атомов углерода, при этом алкильная группа является замещенной, а заместитель выбран из группы, включающей гало-, гидроксил-, оксо-, нитро-, амидо-, карбокси-, амино-, циано-, алкокси-, галоалкокси- или галоалкил.
- R2 представляет собой заместитель, выбранный из группы, включающей водород, алкил, алкен, алкин, циклоалкил, арил, гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси, или галоалкил, предпочтительно, R2 представляет собой заместитель, выбранный из группы, включающей водород, метил, метокси, этокси, хлор, фтор, более предпочтительно, R2 представляет собой водород или метокси группу, еще более предпочтительно, R2 представляет собой метокси группу.
- R3, R4, R5 и R6 могут быть одинаковыми или разными, при этом каждый представляет собой заместитель, выбранный из группы, включающей водород, алкил, алкен, алкин, циклоалкил, арил, гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси или галоалкил, предпочтительно, R3, R4, R5 и R6 являются одинаковыми и представляют собой водород.
- R7 представляет собой заместитель, выбранный из группы, включающей водород, алкил, алкен, алкин, циклоалкил, арил, гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси, или галоалкил, предпочтительно, R7 представляет собой водород, -NH2 или фтор,
при условии, что соединения, имеющие формулу I, не являются:
- 7-(4-бутилпиперазин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
- 1-циклопропил-6-фтор-7-(4-гептилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбьоксильной кислотой,
- 7-(4-бутил-3-метилпиперазин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
- 1-циклопропил-6-фтор-7-(4-гексил-3-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
- 1-циклопропил-6-фтор-7-(4-(4-гидроксибутил)пиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
при условии, что R2 представляет собой водород, a R7 представляет собой фтор, R3, R4, R5 и R6 не являются метильной группой.
Согласно другому варианту осуществления изобретения, заявленные соединения имеют общую формулу II, которая согласуется с общей формулой I, при этом R7 представляет собой фтор:
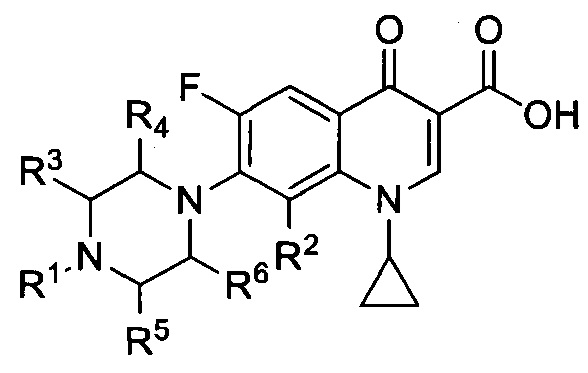
и также включают фармацевтически приемлемые соли, сольваты и пролекарства.
Согласно еще одному варианту осуществления изобретения, заявленные соединения имеют общую формулу III, которая согласуется с общей формулой II, при этом R3, R4, R5 и R6 являются одинаковыми и представляют собой атом водорода:
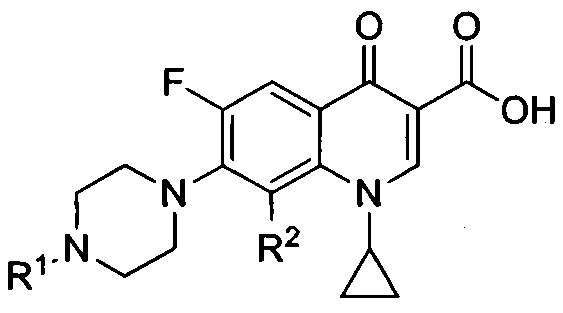
и также включают фармацевтически приемлемые соли, сольваты и пролекарства.
Согласно предпочтительному варианту осуществления изобретения, заявленные соединения выбраны из группы, включающей 1-циклопропил-6-фтор-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-7-(4-гексилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-7-(4-нонилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-4-оксо-7-(4-ундецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-7-(4-додецилпиперазин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-4-оксо-7-(4-тридецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карббоксильную кислоту, 1-циклопропил-6-фтор-4-оксо-7-(4-тетрадецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-4-оксо-7-(4-пентадецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-7-(4-гексилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-7-(4-гептилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-8-метокси-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-8-метокси-7-(4-нонилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-8-метокси-4-окси-7-(4-ундецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-7-(4-додецилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-тридецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-тетрадецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентадецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту, 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту, а также их фармацевтически приемлемые соли, сольваты и пролекарства.
Согласно еще одному варианту, настоящее изобретение относится к фармацевтической композиции, включающей, по крайней мере, одно из заявленных соединений, или его фармацевтически доступную соль, сольват или пролекарство, а также, по крайней мере, один фармацевтически приемлемый носитель, растворитель, эксципиент и/или адьювант.
Согласно еще одному варианту, настоящее изобретение также относится к лекарственному препарату, содержащему, по крайней мере, одно из заявленных соединений, или его фармацевтически доступную соль или сольват.
Согласно одному из вариантов осуществления изобретения, фармацевтически приемлемый носитель, растворитель, эксципиент и/или адьювант выбран из группы, включающей: Этанол 5%, Глицерин 15%, Полиэтиленгликоль 300 50%, Полиэтиленгликоль 400 9%, Полисорбат 80 0,4%, Пропиленгликоль 68%, 2-гидроксипропил-циклодекстрин 20%, Метилцеллюлозу 0,5% и кукурузное масло.
Согласно одному из вариантов осуществления изобретения, фармацевтически приемлемый носитель, растворитель, эксципиент и/или адьювант представляет собой Полисорбат 80 0,4% или кукурузное масло.
Согласно другому варианту осуществления изобретения, фармацевтическая композиция или лекарственный препарат, заявленный в соответствии с настоящим изобретением, дополнительно содержит другой терапевтический агент и/или активный ингредиент.
Согласно еще одному из вариантов осуществления изобретения, соединение, фармацевтическая композиция или лекарственный препарат, заявленные в соответствии с настоящим изобретением, подходят для лечения бактериальных инфекций.
Согласно другому варианту осуществления изобретения, соединение, фармацевтическая композиция или лекарственный препарат, заявленные в соответствии с настоящим изобретением, подходят для лечения бактериальных инфекций, при этом бактериальные инфекции вызваны бактериями следующих видов: Mycobacterium, в частности, tuberculosis или leprae; Грам-положительными бактериями, такими, как Streptococcus, Staphylococcus или Bacillus; enterobacteriaceae такими, как Escherichia, Klebsiella, Enterobacter, Proteus, Serratia, Shigella, Citrobacter, Salmonella или Yersinia, неферментирующими Грам-отрицательными бациллами, такими, как Bacteroides, Fusobacterium, Eubacterium, Propionibacterium, Peptococcus, Clostridium, Peptostreptococcus, или Veillonella; Helicobacter pylori или патогенами, вызывающими инфекции, передающиеся половым путем, такими, как Neisseria, Haemophilus, Chlamydia, или Mycoplasma.
ОПРЕДЕЛЕНИЯ
В описании настоящего изобретения представленные ниже термины имеют следующие значения:
- термин ''арил'' относится к полиненасыщенной, ароматической углеводородной группе, имеющей простое кольцо (например, фенил) или несколько ароматических колец, соединенных вместе (например, нафтил) или связанных ковалентно, включающих обычно от 5 до 12 атомов; предпочтительно, от до 10 атомов, при этом, по крайней мере, одно из колец является ароматическим.
- термин ''алифатическая группа'' относится к любой карбонизированной, ациклической или циклической, насыщенной или ненасыщенной, разветвленной или неразветвленной группе, необязательно, замещенной, исключая ароматические соединения. В соответствии с настоящим изобретением, алифатическая группа, предпочтительно, содержит от 4 до 20 атомов углерода, предпочтительно, от 5 до 16 атомов углерода, еще более предпочтительно, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или 16 атомов. Согласно одному предпочтительному варианту осуществления изобретения, разветвленные и неразветвленные алифатические группы включают алкиловые, алкенильные, алкинильные группы.
- термин ''алкил'' относится к любой насыщенной линейной или разветвленной углеводородной цепи, необязательно, замещенной, содержащей от 4 до 20 атомов углерода и, предпочтительно, от 5 до 16 атомов углерода; более предпочтительно, термин объединяет бутил, пентил, гексил, гептил, октил, нонил, децил, ундецил, додецил, тридецил, тетрадецил, пентадецил, гексадецил.
- термин ''циклоалкил'' относится к циклической или полициклической, необязательно разветвленной, замещенной или незамещенной алкильной группе; предпочтительно, к циклопропильной, циклопентильной или циклогексильной группе.
- термин ''карбокси'' относится к -СООН группе.
- термин ''алкенил'' относится к любой линейной или разветвленной, необязательно замещенной углеводородной цепи, имеющей, по крайней мере, одну двойную связь.
- термин ''алкинил'' относится к любой линейной или разветвленной, необязательно замещенной углеводородной цепи, имеющей, по крайней мере, одну тройную связь.
- термин ''алкокси'' относится к О-алкильной группе. Одной из самых предпочтительных групп в соответствии с настоящим изобретением является метокси группа.
- термин ''ароматическая группа'' относится к моно- или полициклической системе, включающей от 6 до 12 атомов углерода, с одним или несколькими ароматическими кольцами (при этом, когда имеется два кольца они обозначаются термином биарил), к которым относятся: фенильная группа, бифенильная группа, 1-нафтильная группа, 2-нафтильная группа, антраценильная группа, пиренильная группа, пиренильная группа, тетрагидронафтильная группа, инданильная группа и бинафтильная группа. Термин ароматическая группа также относится к любому ароматическому кольцу, содержащему, по крайней мере, один гетероатом, выбранный из группы, включающей кислород, азот или атом серы; к таким группам относятся хинолин, терпиридинил, бипиридинил, гуанин, фенантролин, гидроксихинолин. Ароматическая группа может включать от 1 до 3 заместителей, которые независимо друг от друга выбраны из группы, включающей соединения с 1, 2, 3, 4, 5 или 6 атомами углерода, в частности, метил, этил, пропил, бутил, алкокси группу или атом галогена, в частности, бром, хлор или йод. В том случае, когда ароматическая группа является замещенной, она может быть мета и/или пара и/или орто замещенной.
- термин ''гало'' относится к атому фтора, хлора, брома или йода. Одной из предпочтительных гало-групп в соответствии с настоящим изобретением является фтор.
- термин ''гидроксил'' обозначает -ОН группу.
- термин ''оксо'' относится к - C=O функциональной группе.
- термин ''нитро'' относится к группе - NO2;
- термин ''циано'' относится к группе -CN.
- термин ''амино'' относится к NH2 группе или любой группе, полученной из NH2 путем замещения одного или нескольких атомов водорода алифатической или ароматической, замещенной или незамещенной органической группой, при этом указанная алифатическая или ароматическая группа является замещенной одним или несколькими заместителями, выбранными из группы, включающей гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, галоалкокси, галоалкил-группы. - NH2 - производные предпочтительно представляют собой алкиламино группы, другими словами, N-алкил группы, включая моноалкиламино- и диалкиламино-группы.
- термин ''амидо'' относится к -NR-CO функциональной группе, при этом R представляет собой Н или алкил.
- термин ''лекарственная устойчивость или резистентность'' означает, что бактериальный штамм является устойчивым, по крайней мере, к одному лекарственному препарату; при этом такие штаммы могут иметь множественную устойчивость, широкую устойчивость или полную лекарственную устойчивость.
- термин ''множественная устойчивость'' означает, что бактериальный штамм является устойчивым более чем к одному противомикробному агенту, предпочтительно, штамм является устойчивым, по крайней мере, к одному агенту из трех или более групп противомикробных препаратов.
- термин ''широкая лекарственная устойчивость'' эквивалентен термину ''высокая лекарственная устойчивость'' и означает, что бактериальный штамм является устойчивым, по крайней мере, к одному агенту во всех, кроме максимум двух, группах антимикробных препаратов.
- термин ''полная лекарственная устойчивость'' означает, что бактериальный штамм является устойчивым ко всем агентам из всех групп антимикробных препаратов.
- термин ''пролекарство'' относится к фармацевтически приемлемым производным соединений по Формуле I, II или III, например таким как амиды и эфиры, которые in vivo подвергаются биотрансформации с образованием биологически активного лекарственного вещества. В целом пролекарства характеризуются повышенной биодоступностью, легко метаболизируются в биологически активные соединения in vivo.
- термин ''сольват'' относится к соединению, заявленному в соответствии с настоящим изобретением, которое содержит стехиометрическое или субстехиометрическое количество одного или более фармацевтически приемлемого растворителя, такого, как этанол или вода.
- термины ''лечить'' или ''лечение'' используемые в соответствии с настоящим изобретением, означают облегчение, уменьшение или полное подавление заболевания или патологического состояния и/или сопутствующих ему симптомов. Предпочтительно, термины ''лечить'' или ''лечение'' означают эрадикацию бактерий, вызвавших заболевание.
- термин ''фармацевтически приемлемый'' относится к ингредиентам фармацевтической композиции, которые являются совместимыми друг с другом и не опасны для пациента.
- термин ''фармацевтический носитель'' относится к носителю или инертной среде, которые используются в качестве растворителя или разбавителя для приготовления лекарственной композиции или введения фармацевтически активного агента. Примерами таких фармацевтически приемлемых носителей являются кремы, гели, лосьоны, растворы, липосомы.
ДЕТАЛЬНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям, имеющим общую формулу I
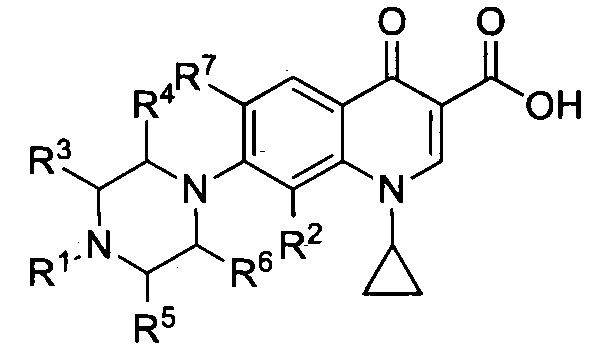
а также к их фармацевтически приемлемым солям, сольватам и пролекарствам, при этом:
- R1 представляет собой насыщенную или ненасыщенную, замещенную или незамещенную, разветвленную или неразветвленную алкильную группу, включающую от 4 до 20 атомов углерода, предпочтительно, от 5 до 16, более предпочтительно, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или 16 атомов углерода, при этом алкильная группа является замещенной, а заместитель выбран из группы, включающей гало-, гидроксил-, оксо-, нитро-, амидо-, карбокси-, амино-, циано-, алкокси-, галоалкокси- или галоалкил.
- R2 представляет собой заместитель, выбранный из группы, включающей водород, алкил, алкен, алкин, циклоалкил, арил, гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси, или галоалкил, предпочтительно, R2 представляет собой заместитель, выбранный из группы, включающей водород, метил, метокси, этокси, хлор, фтор, более предпочтительно, R2 представляет собой водород или метокси группу, еще более предпочтительно, R2 представляет собой метокси группу.
- R3, R4, R5 и R6 могут быть одинаковыми или разными, при этом каждый представляет собой заместитель, выбранный из группы, включающей водород, алкил, алкен, алкин, циклоалкил, арил, гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси или галоалкил, предпочтительно, R3, R4, R5 и R6 являются одинаковыми и представляют собой водород.
- R7 представляет собой заместитель, выбранный из группы, включающей водород, алкил, алкен, алкин, циклоалкил, арил, гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси, или галоалкил, предпочтительно, R7 представляет собой водород, -NH2 или фтор,
при условии, что соединения, имеющие формулу I, не являются:
- 7-(4-бутилпиперазин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
- 1-циклопропил-6-фтор-7-(4-гептилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбьоксильной кислотой,
- 7-(4-бутил-3-метилпиперазин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
- 1-циклопропил-6-фтор-7-(4-гексил-3-метилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
- 1-циклопропил-6-фтор-7-(4-(4-гидроксибутил)пиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислотой,
при условии, что R2 представляет собой водород, a R7 представляет собой фтор, R3, R4, R5 и R6 не являются метальной группой.
Согласно одному варианту, изобретение относится к соединениям, имеющим общую Формулу I, при этом R2, R3, R4, R5 и R6 представляют собой водород, a R7 представляют собой фтор, a R1 не является С1-С7 алкилом или R1 не является алкилом, замещенным оксо-группой. Согласно еще одному варианту, настоящее изобретение относится к соединениям, имеющим общую Формулу I, при этом R2, R3, R4, R5 и R6 представляют собой водород, a R7 представляет собой фтор, a R1 не является пентильной или гексильной группой, или R1 не является алкилом, замещенным оксо-группой.
Согласно еще одному варианту осуществления изобретения, когда R2, R3, R4, R5 и R6 представляют собой водород, a R7 представляет собой фтор, R1 не является пентильной или гексильной группой. Согласно еще одному варианту осуществления изобретения, предпочтительные соединения, имеющие общую Формулу I, не могут представлять собой 1-циклопропил-6-фтор-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту. Согласно еще одному варианту осуществления изобретения, предпочтительные соединения, имеющие общую Формулу I, не могут представлять собой 1-циклопропил-6-фтор-7-(4-гексилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту.
Согласно одному из вариантов осуществления изобретения, соединения, имеющие общую Формулу I, не включают соединения, в которых R1 представляет собой алкильную группу, замещенную, по крайней мере, одной оксо-функциональной группой; Согласно еще одному варианту осуществления изобретения, соединения, имеющие общую формулу I не включают соединения, в которых R1 представляет собой алкильную группу, замещенную только одной оксо-функциональной группой.
Согласно еще одному варианту осуществления изобретения, R2, R3, R4, R5 и R6 представляют собой водород, a R7 представляет собой фтор, R1 не является алкильной группой, замещенной оксо-функциональной группой. Согласно одному варианту осуществления изобретения, соединения, имеющие общую формулу I, не включают следующие соединения;
- 7-(4-бутурилпиперазин-1-ил)-1-циклопропил-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-пентаноилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-гексаноилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-гептаноилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-нонаноилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-деканоилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-додеканоилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-тетрадеканоилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-пальмитоилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-(3-оксобутил)пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту
Согласно одному из вариантов осуществления изобретения, R1 не является алкильной группой, замещенной более чем одним заместителем, выбранным из группы, включающей гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси или галоалкил. Согласно еще одному варианту осуществления изобретения, R1 не является алкильной группой, замещенной более чем одним заместителем, выбранным из группы, включающей гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси или галоалкил. Согласно еще одному варианту осуществления изобретения, R1 не является алкильной группой, замещенной как карбокси, так и оксо-группами. Согласно еще одному варианту осуществления изобретения, R1 не является алкильной группой, замещенной как оксо-, так и амино-группами.
Согласно одному из вариантов осуществления изобретения, предпочтительными соединениями, имеющими Формулу I, являются соединения, где:
- R1 представляет собой насыщенную или ненасыщенную, замещенную или незамащенную, разветвленную или неразветвленную алкильную группу, включающую от 4 до 20 атомов углерода, предпочтительно, от 5 до 16, более предпочтительно, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15 или 16 атомов углерода, при этом алкильная группа является насыщенной, а заместитель выбран из группы, включающей гало, гидроксил, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси или галоалкил, и
- R2, R3, R4, R5, R6, R7 определены выше.
Согласно изобретению, предпочтительные соединения, имеющие общую Формулу I, представляют собой соединения, где:
- R1 представляет собой насыщенную или ненасыщенную, замещенную или незамащенную, разветвленную или неразветвленную алкильную группу, включающую от 8 до 12 атомов углерода, предпочтительно, 8, 9, 10, 11, 12 атомов углерода, более предпочтительно, включающую 8, 9 или 10 атомов углерода, при этом алкильная группа является замещенной, а заместитель выбран из группы, включающей гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоакокси, или галоалкил, и
- R2, R3, R4, R5, R6, R7 определены выше.
Согласно одному из вариантов осуществления изобретения, предпочтительные соединения, имеющие общую Формулу I, представляют собой соединения, где R1 представляет собой октил-, нонил-, децил-, ундецил- или додецил-группу; предпочтительно, R1 представляет собой октил, нонил или децил-группу.
Согласно еще одному варианту осуществления изобретения, предпочтительные соединения, имеющие общую Формулу I, представляют собой соединения, где:
- R1 представляет собой насыщенную или ненасыщенную, замещенную или незамащенную, разветвленную или неразветвленную алкильную группу, включающую от 7 до 12 атомов углерода, предпочтительно, 7, 8, 9, 10, 11, 12 атомов углерода, более предпочтительно, включающую 7, 8, 9 или 10 атомов углерода, при этом алкильная группа является замещенной, а заместитель выбран из группы, включающей гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоакокси, или галоалкил, и
- R2, R3, R4, R5, R6, R7 определены выше.
Согласно одному из вариантов осуществления изобретения, предпочтительные соединения, имеющие общую Формулу I, представляют собой соединения, где R1 представляет собой гептил, октил, нонил, децил, ундецил или додецил-группу; предпочтительно, R1 представляет собой гептил, октил, нонил или децил-группу.
Согласно еще одному варианту, настоящее изобретение относится к соединениям, имеющим общую Формулу II:
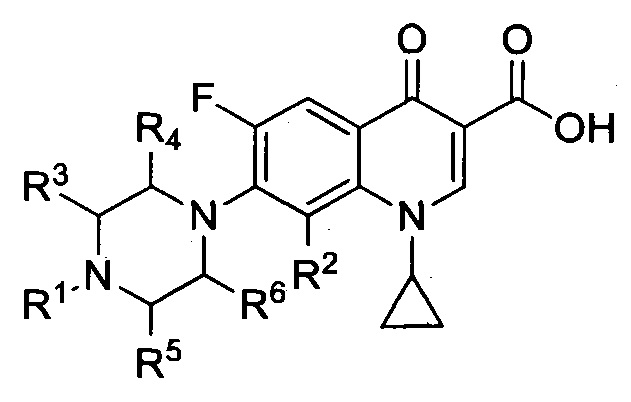
а также к их фармацевтически приемлемым солям, сольватам и пролекарствам; что согласуется с общей формулой I, при этом R7 представляет собой атом фтора.
Согласно другому варианту, настоящее изобретение относится к соединениям, имеющим общую Формулу III:
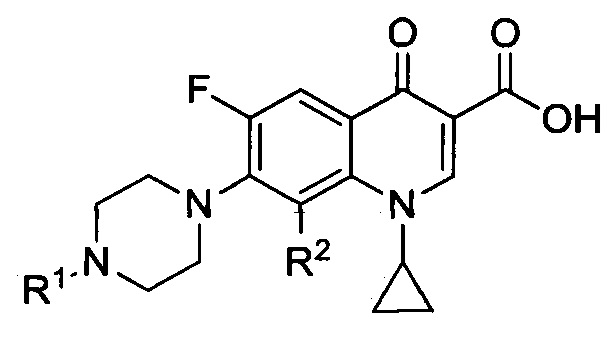
а также к их фармацевтически приемлемым солям, сольватам и пролекарствам, что согласуется с общей формулой II, при этом R3, R4, R5, R6 являются идентичными и представляют собой атом водорода.
Согласно предпочтительному варианту, настоящее изобретение относится к соединениям, имеющим общую Формулу III:
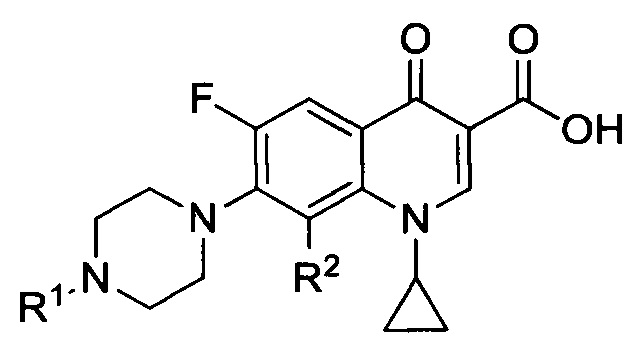
а также к их фармацевтически приемлемым солям, сольватам и пролекарствам, при этом R2 представляет собой атом водорода или метокси-группу, более предпочтительно, R2 представляет собой метокси группу, a R1 определен выше.
В соответствии с настоящим изобретением, предпочтительными соединениями, имеющими общую Формулу III, являются соединения, где:
- R2 представляет собой метокси-группу, и
- R1 представляет собой насыщенную или ненасыщенную, замещенную или незамещенную, разветвленную или неразветвленную алкильную группу, включающую от 8 до 12 атомов углерода, предпочтительно, 8, 9, 10, 11, 12 атомов углерода, более предпочтительно, 8, 9 или 10 атомов углерода, при этом алкильная группа является замещенной, а заместитель выбран из группы, включающей гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси или галоалкил.
Согласно предпочтительному варианту осуществления изобретения, предпочтительными соединениями, имеющими общую Формулу III, являются соединения, где R2 представляет собой метокси группу, a R1 представляет собой октил-, нонил-, децил-, ундецил- или додецил-группу; предпочтительно, R2 представляет собой метокси группу, R1 представляет собой октил, нонил или децил-группу.
В соответствии с одним из вариантов осуществления изобретения, предпочтительными соединениями, имеющими общую Формулу III, являются соединения, где:
- R2 представляет собой метокси-группу, и
- R1 представляет собой насыщенную или ненасыщенную, замещенную или незамещенную, разветвленную или неразветвленную алкильную группу, включающую от 7 до 12 атомов углерода, предпочтительно, 7, 8, 9, 10, 11, 12 атомов углерода, более предпочтительно, 7, 8, 9 или 10 атомов углерода, при этом алкильная группа является замещенной, а заместитель выбран из группы, включающей гало, гидроксил, оксо, нитро, амидо, карбокси, амино, циано, алкокси, галоалкокси или галоалкил.
Согласно предпочтительному варианту осуществления изобретения, предпочтительными соединениями, имеющими общую Формулу III, являются соединения, где R2 представляет собой метокси группу, a R1 представляет собой гептил-, октил-, нонил-, децил-, ундецил- или додецил-группу; предпочтительно, R2 представляет собой метокси группу, a R1 представляет собой гептил, октил, нонил или децил-группу.
Согласно предпочтительному варианту осуществления изобретения, заявленные соединения представляют собой соединения, перечисленные ниже в
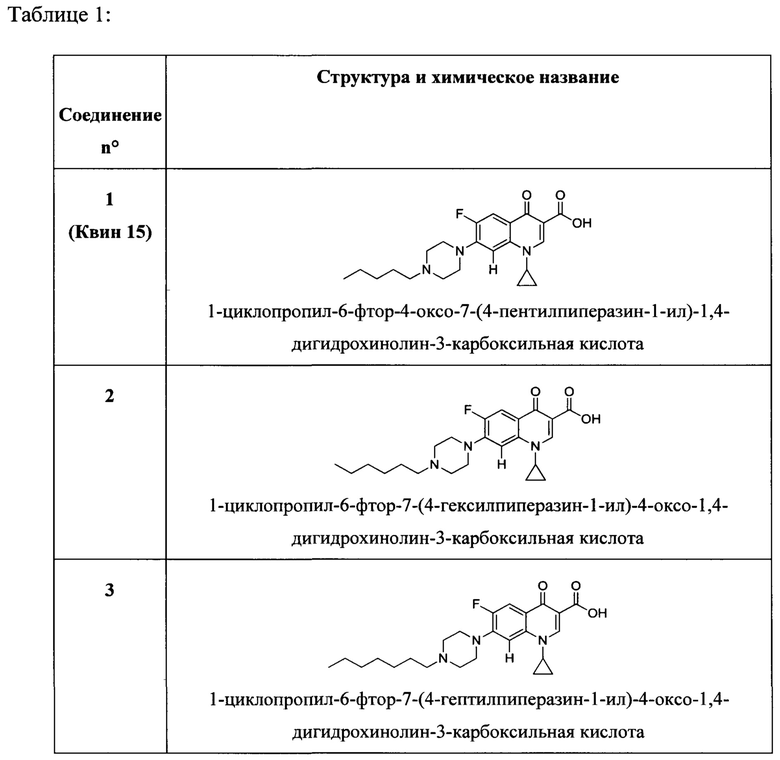
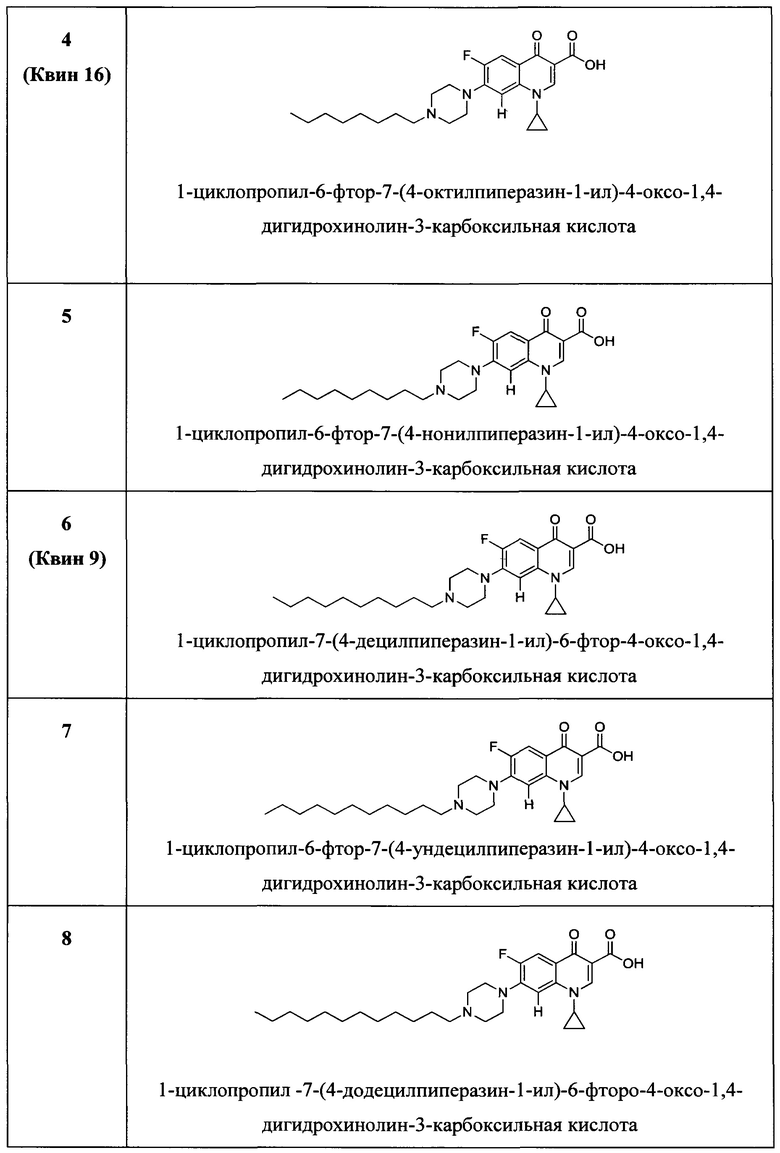
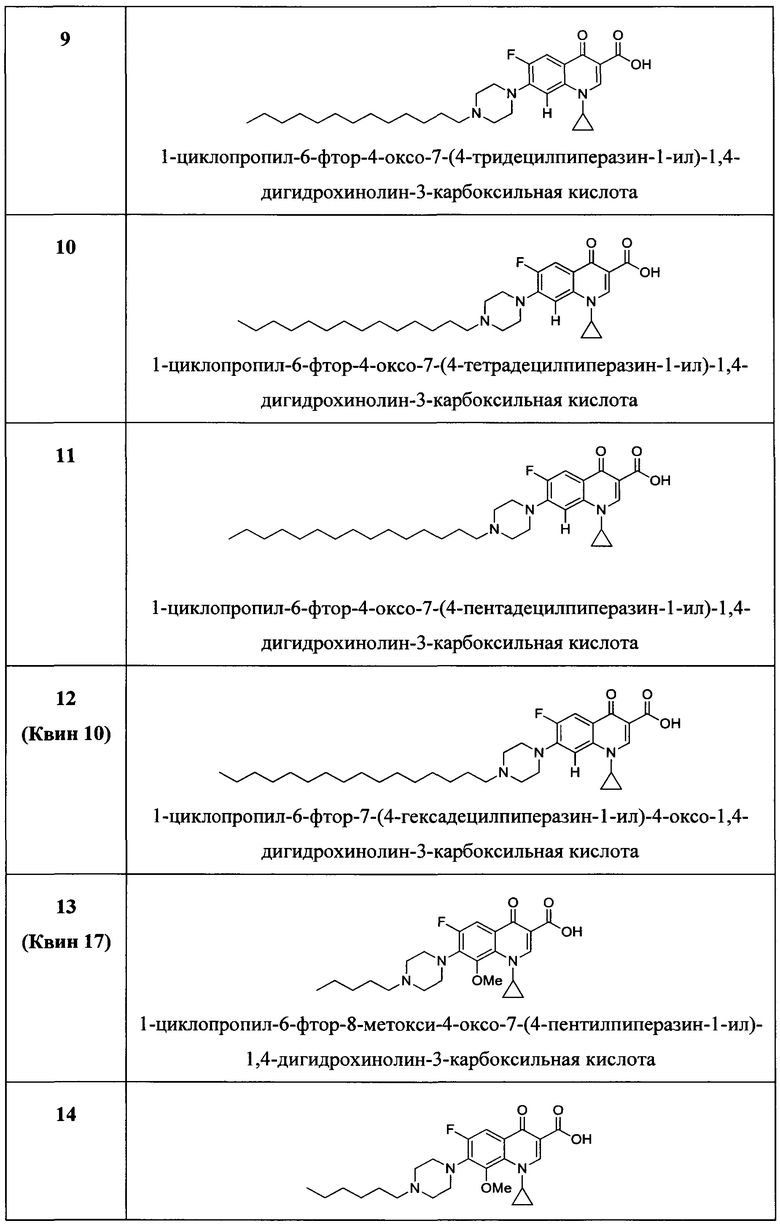
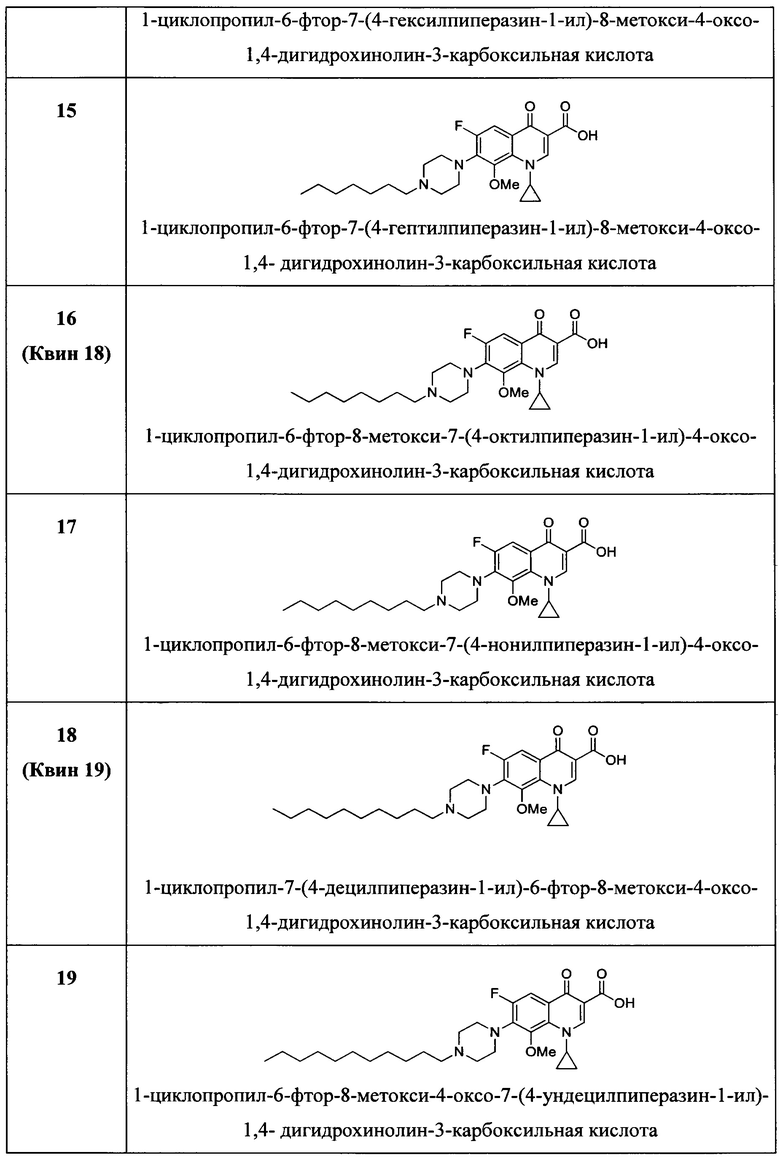
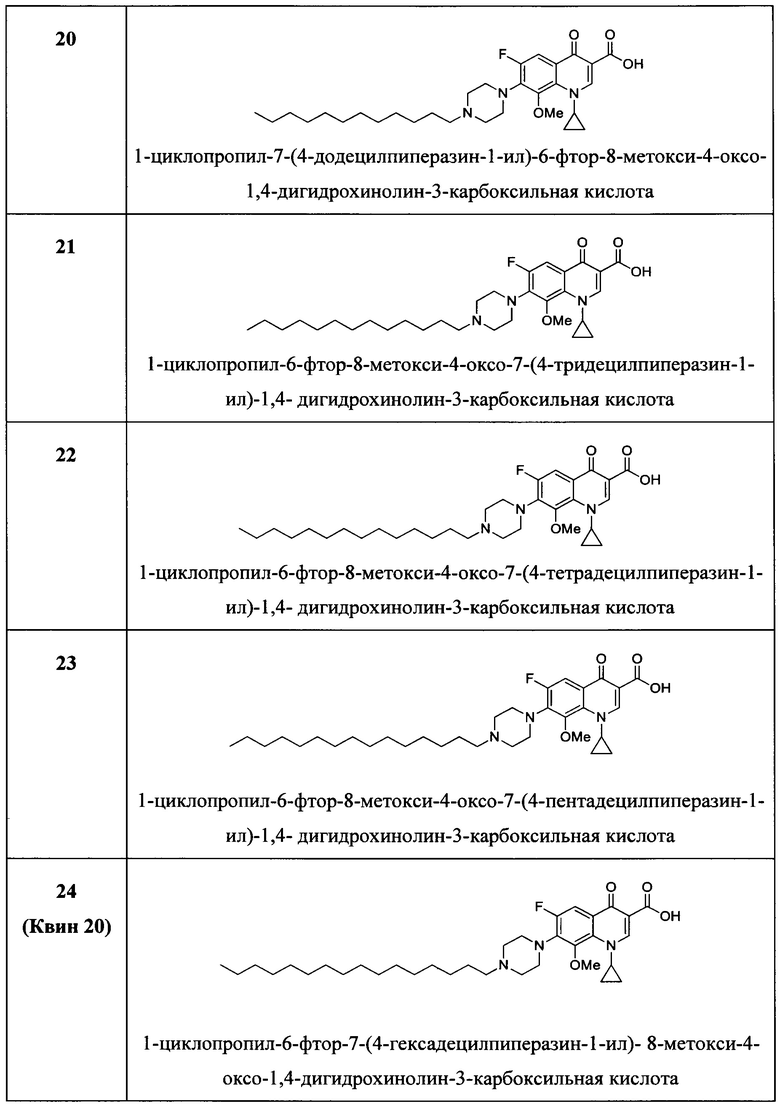
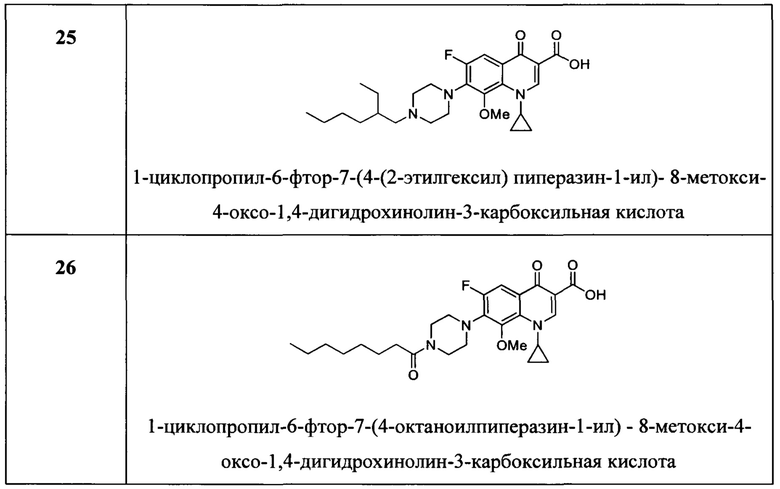
Согласно наиболее предпочтительному варианту осуществления изобретения, заявленные соединения выбраны из группы, включающей 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту (Хин 17), 1-циклопропил-6-фтор-8-метокси-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (Хин 18), 1-циклопропил-7-(децилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (Хин 19) и 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (Квин 20).
Согласно другому варианту осуществления, настоящее изобретение относится к фармацевтической композиции, включающей, по крайней мере, одно заявленное соединение, или его фармацевтически приемлемую соль, сольват или пролекарство; и, по крайней мере, один фармацевтически приемлемый носитель, растворитель, эксципиент и/или адьювант. Настоящее изобретение также относится к фармацевтической композиции которая содержит помимо, по крайней мере, одного заявленного соединения или его фармацевтически приемлемой соли, сольвата или пролекарства в качестве активного ингредиента, дополнительные лекарственные агенты и/или активные ингредиенты.
Согласно еще одному варианту осуществления, настоящее изобретение относится к лекарственному средству, содержащему, по крайней мере, одно заявленное соединение или его фармацевтически приемлемую соль, сольват или про лекарство.
Согласно другому варианту осуществления изобретения, заявленное лекарственное средство помимо, по крайней мере, одного заявленного соединения или его фармацевтически приемлемой соли, сольвата или пролекарства в качестве активного ингредиента, содержит дополнительные лекарственные агенты и/или активные ингредиенты.
Примерами дополнительных лекарственных агентов и/или активных ингредиентов являются, без ограничений указанными, фторхинолоны, такие, как, ципрофлоксацин, энрофлоксацин, гатифлоксацин, моксифлоксацин, офлоксацин, левофлоксацин и спарфлоксацин.
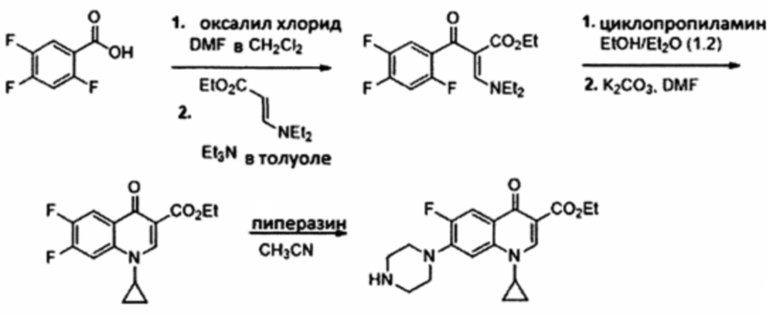
Согласно одному из вариантов осуществления изобретения, соединения, имеющие общую Формулу III, синтезируют согласно технологии, показанной на схеме 1, начиная с фторхинолона как предшественника, путем замещения атома фтора в положении 7 пиперазином. Добавление длинной алкильной цепи в положение 4 пиперазиновой группы приводит к получению конечных соединений, имеющих общую Формулу III:
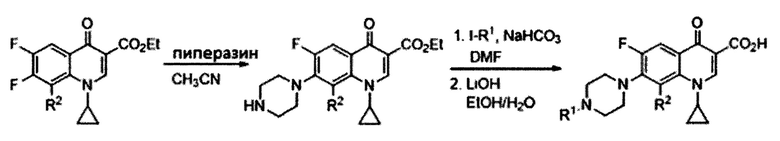
Схема 1: Синтез соединений, имеющих общую формулу III
Согласно одному из вариантов осуществления изобретения, соединения, имеющие общую Формулу III, где R2 представляет собой атом водорода, получают согласно следующей схеме, начиная с 2,4,5-трифторбензойной кислоты:
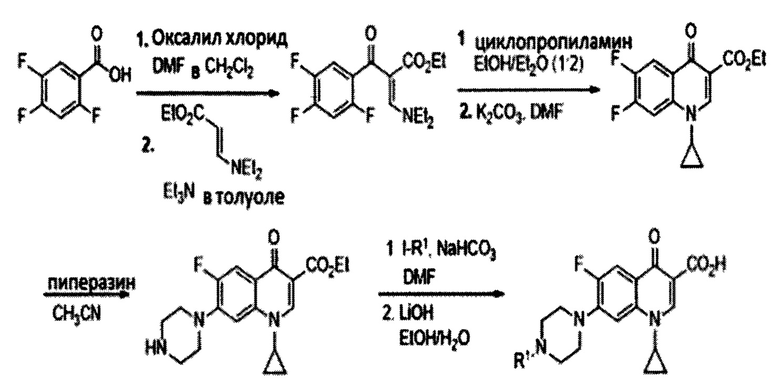
Схема 2: Синтез соединений, имеющих общую Формулу III, где R2=Н
Согласно другому варианту осуществления изобретения, соединения, имеющие общую Формулу III, где R2 представляет собой метокси группу, получают согласно следующей схеме, начиная с 2,4,5-трифтор-3-метокси-бензойной кислоты.
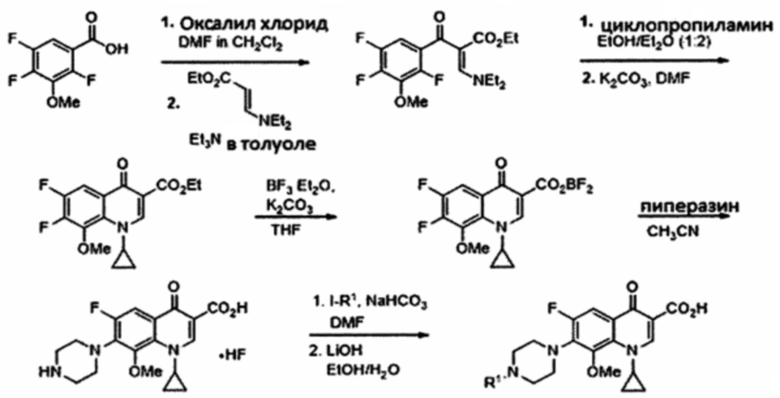
Схема 3: Синтез соединений, имеющих общую Формулу III, где R2=ОМе
Согласно одному из вариантов осуществления изобретения, заявленные соединения не имеют хирального центра. Следовательно, стоимость их получения должна быть ниже по сравнению со стоимостью синтеза уже известных хинолонов, таких, как DC-159а, левофлоксацина или моксифлоксацина. Кроме того, синтез является конвергентным и включает разнообразные превращения, что позволяет получить множество соединений.
Настоящее изобретение также относится к лечению бактериальных инфекций. Молекулы веществ из семейства хинолонов и фторхинолонов являются активными в отношении широкого спектра бактерий. Механизм действия хинолонов и фторхинолонов заключается в ингибировании топоизомераз II типа, то есть ДНК гираз (активных как комплекс GyrA и GyrB) и ДНК топоизомеразы IV. Следовательно, мутации топоизомераз II типа - это основной путь развития резистентности бактерий к хинолонам и фторхинолонам. В случае определенных бактерий, таких как Mycobacterium tuberculosis, ДНК гираза является единственным типом топоизомеразы II и, следовательно, является единственной мишенью для хинолонов и фторхинолонов. Штаммы Mycobacterium tuberculosis, которые являются резистентными к хинолонам и фторхинолонам, следовательно, имеют, по крайней мере, одну мутацию в GyrA и/или GyrB субъединицах ДНК гиразы. Интересно, что соединения, заявленные в соответствии с настоящим изобретением, ингибируют рост бактерий, хотя при этом они очень слабо ингибируют ДНК гиразу в низких концентрациях.
Авторы изобретения показывают, что заявленные соединения могут ингибировать рост бактерий посредством уникального механизма, который не затрагивает ДНК гиразу или механизма, который затрагивает ДНК гиразу, а также другие клеточные сигнальные пути и мишени.
Согласно одному из вариантов осуществления изобретения, соединения, фармацевтические композиции и лекарственные средства, заявленные в соответствии с настоящим изобретением, являются полезными для лечения бактериальных инфекций.
Согласно еще одному варианту осуществления изобретения, заявленные соединения являются полезными для лечения инфекций, вызванных, по крайней мере, одной Грам-негативной или Грам-позитивной бактерией.
Определенные бактерии, такие, как бактерии Salmonella, Legionella и Mycobacterium, в особенности Mycobacteria tuberculosis, могут сохранять жизнеспособность в макрофагах. Согласно одному из вариантов осуществления изобретения, заявленные соединения обладают способностью проникать в макрофаги и проявлять свою бактерицидную активность.
Согласно одному варианту осуществления изобретения, заявленные соединения являются полезными для лечения бактериальной инфекции, вызванной, по крайней мере, одной бактерией из рода, выбранного из группы, включающей (без ограничений указанными) Mycobacterium, такие, как tuberculosis или leprae; Грам-позитивные бактерии, такие, как Streptococcus, Staphylococcus или Bacillus; enterobacteriaceae такие как Escherichia, Klebsiella, Enterobacter, Proteus, Serratia, Shigella, Citrobacter, Salmonella или Yersinia, неферментирующие Грам-негативные бациллы, такие, как Pseudomonas, Alcaligenes, или Acitenobacter; анаэробы, такие как Bacteroides, Fusobacterium, Eubacterium, Propionibacterium, Peptococcus, Clostridium, Peptostreptococcus, или Veillonella; Helicobacter pylori и патогены, вызывающие инфекции, передающиеся половым путем, такие, как Neisseria, Haemophilus, Chlamydia, или Mycoplasma. С
Согласно еще одному варианту осуществления изобретения, заявленные соединения являются полезными для лечения бактериальной инфекции, вызванной, по крайней мере, одной бактерией, выбранной из группы, включающей (без ограничений указанными): Mycobacterium leprae, Mycobacterium tuberculosis комплекс, включая Mycobacterium tuberculosis и нетуберкулезные микобактерии, такие, как Mycobacterium chelonae, Mycobacterium avium, Mycobacterium abscessus, Mycobacterium fortuitum, Mycobacterium malmoense, Mycobacterium gordonae, Mycobacterium terrae, Mycobacterium nonchromogenicium, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium phlei, Mycobacterium xenopi, Mycobacterium marinum, или Mycobacterium ulcerans; Грам-позитивные бактерии, такие как Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis, Bacillus anthracis, Staphylococcus epidermidis, или Streptococcus pyogenes; энтеробактерии, такие как Escherichia coli, Klebsiella pneumonia, Enterobacter aerogenes, Enterobacter cloacae, Proteus vulgaris, Shigella flexneri, Serratia marcescens, Citrobacter freundii, Yersinia enterocolitica, или Salmonella enteritidis; неферментирующие Грам-негативные бациллы, такие как Pseudomonas aeruginosa, Acitenobacter baumannii, Burkholderia cepacia, or Stenotrophomonas maltophilia; анаэробы, такие, как Bacteroides fragilis, Bacteroides distasonis, Bacteroides thetaiotaomicron, Bacteroide vulgatus, Fusobacterium mortiferum, Fusobacterium necrophorum, Fusobacterium varium, Eubacterium lentum, Propionibacterium acens, Clostridium difficile, Clostridium perfringens, Clostridium ramosum, Peptostreptococcus anaerobius, Peptostreptococcus micros, или Veillonella parvula; Helicobacter pylori и патогены, вызывающие инфекции, передающиеся половым путем, такие, как Neisseria gonorrhoeae, Haemophulis ducreyi, Chlamydia trachomatis, or Mycoplasma genitallium.
Согласно предпочтительному варианту осуществления изобретения, заявленные соединения являются полезными для лечения бактериальных инфекций, вызванных Mycobacterium tuberculosis дикого типа, а также MDR, XDR штаммами, и PDR штаммами.
Согласно одному из вариантов осуществления изобретения, заявленные соединения являются полезными для лечения бактериальной инфекции, вызванной, по крайней мере, штаммами с множественной, широкой и полной лекарственной резистентностью, выбранных из группы, включающей, но не ограниченной Mycobacterium leprae, Mycobacterium tuberculosis комплекс, включая Mycobacterium tuberculosis и нетуберкулезные микобактерии, такие, как Mycobacterium chelonae, Mycobacterium avium и avium комплекс, Mycobacterium abscessus, Mycobacterium fortuitum, Mycobacterium malmoense, Mycobacterium gordonae, Mycobacterium terrae, Mycobacterium nonchromogenicium, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium phlei, Mycobacterium kansasii, Mycobacterium xenopi, Mycobacterium marinum, или Mycobacterium ulcerans; Грам-позитивные бактерии, такие, как Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis, Bacillus anthracis, Staphylococcus epidermidis, или Streptococcus pyogenes; энтеробактерии, такие, как Escherichia coli, Klebsiella pneumonia, Enterobacter aerogenes, Enterobacter cloacae, Proteus vulgaris, Shigella flexneri, Serratia marcescens, Citrobacter freundii, Yersinia enterocolitica, или Salmonella enteritidis; неферментирующие Грам-отрицательные бациллы, такие, как Pseudomonas aeruginosa, Acitenobacter baumannii, Burkholderia cepacia, или Stenotrophomonas maltophilia; анаэробы, такие, как Bacteroides fragilis, Bacteroides distasonis, Bacteroides thetaiotaomicron, Bacteroide vulgatus, Fusobacterium mortiferum, Fusobacterium necrophorum, Fusobacterium varium, Eubacterium lentum, Propionibacterium acens, Clostridium difficile, Clostridium perfringens, Clostridium ramosum, Peptostreptococcus anaerobius, Peptostreptococcus micros, или Veillonella parvula; Helicobacter pylori и патогены, вызывающие инфекции, передающиеся половым путем, такие, как Neisseria gonorrhoeae, Haemophulis ducreyi, Chlamydia trachomatis, or Mycoplasma genitallium.
Согласно еще одному варианту осуществления изобретения, заявленные соединения являются полезными для лечения инфекций, вызванных, по крайней мере, одним бактериальным штаммом с множественной, широкой или полной лекарственной устойчивостью, который выбран из группы, включающей (без ограничений указанными): Mycobacterium leprae, Mycobacterium tuberculosis комплекс, включая Mycobacterium tuberculosis и нетуберкулезные микобактерии, такие, как Mycobacterium chelonae, Mycobacterium avium и avium комплекс, Mycobacterium abscessus, Mycobacterium fortuitum, Mycobacterium malmoense, Mycobacterium gordonae, Mycobacterium terrae, Mycobacterium nonchromogenicium, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium phlei, Mycobacterium kansasii, Mycobacterium xenopi, Mycobacterium marinum, или Mycobacterium ulcerans; Грам-позитивные бактерии, такие, как Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis, Bacillus anthracis, Staphylococcus epidermidis, or Streptococcus pyogenes; энтеробактерии, такие, как Escherichia coli, Klebsiella pneumonia, Enterobacter aerogenes, Enterobacter cloacae, Proteus vulgaris, Shigella flexneri, Serratia marcescens, Citrobacter freundii, Yersinia enterocolitica, или Salmonella enteritidis; неферментирующие Грам-отрицательные бациллы, такие, как Pseudomonas aeruginosa, Acitenobacter baumannii, Burkholderia cepacia, или Stenotrophomonas maltophilia; анаэробы, такие, как Bacteroides fragilis, Bacteroides distasonis, Bacteroides thetaiotaomicron, Bacteroide vulgatus, Fusobacterium mortiferum, Fusobacterium necrophorum, Fusobacterium varium, Eubacterium lentum, Propionibacterium acens, Clostridium difficile, Clostridium perfringens, Clostridium ramosum, Peptostreptococcus anaerobius, Peptostreptococcus micros, или Veillonella parvula; Helicobacter pylori и патогены, вызывающие инфекции, передающиеся половым путем, такие, как Neisseria gonorrhoeae, Haemophulis ducreyi, Chlamydia trachomatis, or Mycoplasma genitallium.
Согласно еще одному варианту осуществления изобретения, заявленные соединения являются полезными для лечения инфекций, вызванных, по крайней мере, одним бактериальным штаммом с множественной, широкой или полной лекарственной устойчивостью, который выбран из группы, включающей (без ограничений указанными): Mycobacterium leprae, Mycobacterium tuberculosis комплекс, включая Mycobacterium tuberculosis и нетуберкулезные микобактерии, такие, как Mycobacterium chelonae, Mycobacterium avium и avium комплекс, Mycobacterium abscessus, Mycobacterium fortuitum, Mycobacterium malmoense, Mycobacterium gordonae, Mycobacterium terrae, Mycobacterium nonchromogenicium, Mycobacterium simiae, Mycobacterium scrofulaceum, Mycobacterium phlei, Mycobacterium kansasii, Mycobacterium xenopi, Mycobacterium marinum, или Mycobacterium ulcerans; Грам-позитивные бактерии, такие, как Staphylococcus aureus, Streptococcus pneumoniae, Enterococcus faecalis, Bacillus anthracis, Staphylococcus epidermidis, or Streptococcus pyogenes; энтеробактерии, такие, как Escherichia coli, Klebsiella pneumonia, Enterobacter aerogenes, Enterobacter cloacae, Proteus vulgaris, Shigella flexneri, Serratia marcescens, Citrobacter freundii, Yersinia enterocolitica, или Salmonella enteritidis; неферментирующие Грам-отрицательные бациллы, такие, как Pseudomonas aeruginosa, Acitenobacter baumannii, Burkholderia cepacia, или Stenotrophomonas maltophilia; анаэробы, такие, как Bacteroides fragilis, Bacteroides distasonis, Bacteroides thetaiotaomicron, Bacteroide vulgatus, Fusobacterium mortiferum, Fusobacterium necrophorum, Fusobacterium varium, Eubacterium lentum, Propionibacterium acens, Clostridium difficile, Clostridium perfringens, Clostridium ramosum, Peptostreptococcus anaerobius, Peptostreptococcus micros, или Veillonella parvula; Helicobacter pylori и патогены, вызывающие инфекции, передающиеся половым путем, такие, как Neisseria gonorrhoeae, Haemophulis ducreyi, Chlamydia trachomatis, or Mycoplasma genitallium.
Согласно еще одному варианту осуществления изобретения, заявленные соединения являются полезными для лечения инфекции, вызванной, по крайней мере, одним штаммом бактерий с множественной, широкой или полной лекарственной резистентностью, при этом указанный штамм является, предпочтительно, штаммом Mycobacterium tuberculosis.
Согласно другому варианту осуществления изобретения, бактериальный штамм является устойчивым к фторхинолонам главным образом по причине одной или нескольких(множественных) мутаций субъединицы GyrA фермента ДНК гиразы, или одной или нескольких (множественных) мутаций субъединицы GyrB фермента ДНК гиразы, или по причине множественных мутаций в субъединицах GyrA или GyrB фермента ДНК гиразы.
Согласно одному из вариантов осуществления изобретения, Mycobacterium tuberculosis являются устойчивыми к фторхинолонам по причине одной мутации в субъединице GyrA фермента ДНК гиразы, указанная мутация выбрана из группы, включающей (без ограничений указанными): D89N, D94A, D94N, D94G, D94H, D94F, D94Y, D94V, A74S, A90V, Т80А, G88A, G88C, S91A, и S91P (Maruri et al., J. Antimicrob. Chemother., 2012, p 1-13).
Согласно еще одному варианту осуществления изобретения, Mycobacterium tuberculosis являются устойчивыми к фторхинолонам по причине одной мутации в субъединице GyrB фермента ДНК гиразы, указанная мутация выбрана из группы, включающей (без ограничений указанными) N538D, N538T, D500A, D500N, D500H, Т539Р, E540D, и E540V (Maruri et al., J. Antimicrob. Chemother., 2012, p 1-13).
Согласно другому варианту осуществления изобретения, Mycobacterium tuberculosis являются устойчивыми к фторхинолонам по причине множественных мутаций в субъединице GyrA ДНК гиразы, указанные множественные мутации выбраны из группы, включающей (без ограничений указанными): A74S+D94G, Т80А+А90Е, T80A+A90G+D94G, G88A+A90V, G88A+D94T, A90V+G94A, A90V+P102H, A90V+S91P, A90V+D94N, A90V+D94G, S91P+D94G, S91P+D94G+G94A, D94A+D94Y, D94N+D94G, D94N+D94G+D94Y, и D94G+D94A (Maruri et al., J. Antimicrob. Chemother., 2012, p 1-13).
Согласно еще одному варианту осуществления изобретения, Mycobacterium tuberculosis являются устойчивыми к фторхинолонам по причине множественных мутаций в субъединицах GyrA и GyrB ДНК гиразы, указанные множественные мутации выбраны из группы, включающей (без ограничений указанными) A90V+T500P, D94A+D461N, D94G+N499K, D94G+N499T, и D94N+A504V (Maruri et al., J. Antimicrob. Chemother., 2012, p 1-13).
Согласно еще одному варианту осуществления изобретения, заявленные соединения могут использоваться в качестве терапии первой линии. Согласно другому варианту осуществления изобретения, заявленные соединения могут использоваться в качестве терапии второй линии.
Настоящее изобретение также относится к способу лечения бактериальной инфекции у субъекта, нуждающегося в подобном лечении, при этом указанный способ включает введение субъекту терапевтически эффективного количества, по крайней мере, одного соединения или композиции, заявленных в настоящем изобретении, как описано выше. Предпочтительно, субъектом является теплокровное животное, предпочтительно, человек
Согласно одному из вариантов осуществления изобретения, заявленные соединения могут вводиться в составе комбинированной терапии. Таким образом, в объем настоящего изобретения включены варианты, включающие одновременное введение композиций и лекарственных средств, которые помимо заявленного соединения или его фармацевтически приемлемого сольвата в качестве активного ингредиента, содержат дополнительные лекарственные агенты и/или активные ингредиенты.
В описанных выше комбинированных вариантах осуществления изобретения, заявленное соединение, его фармацевтически приемлемый сольват, и другие лекарственные агенты могут вводиться как в составе одной лекарственной формы, так по отдельности, а что касается времени введения, они могут вводиться как одновременно, так и последовательно. Таким образом, введение одного компонента/агента может осуществляться до, во время или после введения другого компонента (ов)/агента(ов).
В целом, для фармакологического применения, заявленные соединения могут находиться в составе фармацевтических лекарственных форм, содержащих, по крайней мере, одно заявленное соединение и, по крайней мере, один фармацевтически приемлемый носитель, растворитель, наполнитель и/или адьювант, и, необязательно, одно или более дополнительное фармакологически активное соединение.
Неограниченные примеры указанных лекарственных форм включают лекарственные формы, подходящие для перорального введения, парентерального введения (посредством внутривенных, внутримышечных или подкожных инъекций или внутривенных инфузий), местного введения (включая глазные композиции), для ингаляционного введения, для введения посредством кожных пластырей, посредством имплантации, посредством суппозиториев и т.д. Такие подходящие лекарственные формы могут быть твердыми, полутвердыми или жидкими, в зависимости от способа введения, а также от способов и носителей, растворителей и эксципиентов, которые используются при изготовлении лекарственных форм, что очевидно специалисту в данной области (Remington's Pharmaceutical Sciences).
Согласно одному из вариантов осуществления изобретения, заявленные соединения вводятся перорально (per os) или посредством внутривенных инъекций.
Некоторыми предпочтительными лекарственными формами являются (без ограничений указанными): таблетки, пилюли, порошки, таблетки для рассасывания, саше, капсулы, эликсиры, суспензии, эмульсии, растворы, сиропы, аэрозоли, мази, кремы, лосьоны, мягкие и твердые желатиновые капсулы, суппозитории, капли, стерильные инъекционные растворы и стерильные упакованные порошки (которые обычно растворяют перед использованием) для болюсного и/или непрерывного введения, при этом указанные лекарственные формы могут включать носители, эксципиенты и растворители, которые являются подходящими для указанных лекарственных форм, такие, как лактоза, декстроза, сахароза, сорбитол, маннитол, крахмалы, гуммиарабик, фосфат кальция, альгинаты, трагакант, желатин, силикат кальция, микрокристаллическая целлюлоза, поливинилпирролидон, полиэтиленгликоль, целлюлоза, (стерильная) вода, метилцеллюлоза, метил- и пропилгидроксибензоаты, тальк, стеарат магния, пищевые масла, овощные масла и минеральные масла, а также их подходящие смеси. Лекарственные формы также могут необязательно содержать другие вещества, которые обычно применяются в фармацевтических композициях, такие, как смягчающие компоненты, увлажняющие агенты, эмульгирующие и суспендирующие агенты, диспергирующие агенты, дезинтегранты, объемообразующие агенты, наполнители, консерванты, подсластители, ароматизаторы, регуляторы расхода, высвобождающие агенты и т.д. Указанные фармацевтические композиции также могут быть сформулированы таким образом, чтобы обеспечивать быстрое, замедленное или отсроченное высвобождение активного ингредиента(ов), который они содержат.
Фармацевтические композиции, заявленные в соответствии с настоящим изобретением, предпочтительно, находятся в составе одной лекарственной формы и соответствующим образом упакованы, например, в коробки, блистеры, флаконы, бутылки, саше, ампулы или в любые другие подходящие контейнеры или упаковки (которые должны быть соответствующим образом помечены) для единичных и множественных доз, которые также могут включать один или более вкладыш с информацией о продукте и/или инструкцией по применению.
Обычно, в зависимости от заболевания, для которого проводится профилактика или лечение при помощи заявленных соединений, а также в зависимости от способа введения, активного ингредиента, дозировка составляет от 0,001 до 200 мг на килограмм массы тела, предпочтительно, от 1 до 160 мг на килограмм массы тела, предпочтительно, около 25, 50, 100, 150 кг на килограмм массы тела пациента в день, которые могут вводиться как единичная доза или быть разделенными на несколько доз, или же вводиться непрерывно, например, посредством капельной инфузии.
Согласно одному из вариантов осуществления изобретения, заявленное соединение вводится в концентрации, составляющей от 0 до 4 мг/мл; предпочтительно, в концентрации, составляющей 10 или 20 мл/мг.
Согласно одному варианту осуществления изобретения, заявленное соединение вводится в объеме, составляющем более чем 0-100 мг/кг; предпочтительно, объем введения составляет 10 мл/кг.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
Фиг. 1 представляет собой графическое изображение изменений выживаемости мышей, инфицированных штаммом H37rv Mycobacterium tuberculosis дикого типа и получавших лечение посредством кормления через зонд питанием, содержавшим Quin (Квин)18 и Квин 19.
Фиг. 2 представляет собой графическое изображение изменений выживаемости мышей, инфицированных штаммом H37rv Mycobacterium tuberculosis дикого типа и получавших лечение Квин 18 посредством внутривенных инъекций.
Фиг. 3 представляет собой графическое изображение изменений массы тела мышей, получавших лечение с D0 по D14 Квин 18 или Квин 19 в различных дозировках.
ПРИМЕРЫ
Настоящее изобретение дополнительно иллюстрируется следующими примерами.
1. Синтез соединений, заявленных в настоящем изобретении
Материалы и Методы
Все материалы были получены у коммерческих поставщиков и использовались без дополнительной очистки. Тонкослойную хроматографию осуществляли на листах пластика TLC силикагель 60F254 (толщина слоя 0,2 мм) компании Merck. Хроматографическое очищение на колонке осуществляли с силикагелем 60 (70-230 ячеек, ASTM, Merck). Точки плавления определяли как при помощи ручного аппарата (Electrothermal IA 8103), при этом полученные данные являлись нескорректированными, так и при помощи оборудования Kofler WME (Wagner & Munz). IR, lH, 19F и 13C ЯМР спектры подтверждали структуру всех соединений. ИК спектры регистрировали на ИК спектрометре Perkin Elmer Spectrum 100 FT-IR, а ЯМР-спектр регистрировали при помощи CDCl3, CD3CN, D2O или DMSO-d6 в качестве растворителя, на спектрометре BRUKER АС 300 или 400 при 300 или 400 МГц для 1Н, 75 или 100 МГц для 13С и 282 или 377 МГц для 19F спектра. Химические сдвиги (δ) выражали в частях на миллион относительно сигнала косвенно (i) к СНСl3 (δ 7.27) для 1Н и (ii) к CDCl3 (δ 77.2) для 13С и напрямую (iii) к CFCl3 (внутренний стандарт) (δ 0) для 19F. Химические сдвиги представлены в ppm, а мультиплеты обозначены следующим образом: s, синглет; br s, широкий синглет; d, дублет; dd, дублет дублета; t, триплет; q, квадриплет; квинт, квинтиплет; m, мультиплет.
Масс-спектры высокого разрешения (HRMS) получали из сервиса ''Service Central d'analyse de Solaize'' (Centre Nationale de la Recherche Scientifique) и регистрировали на спектрометре Waters с использованием ионизации электроспреем-TOF (ESI-TOF).
Результаты
Пример 1: Синтез предшественника для последующего получения соединений, имющих общую Формулу III, где R2 представляет собой атом водорода.
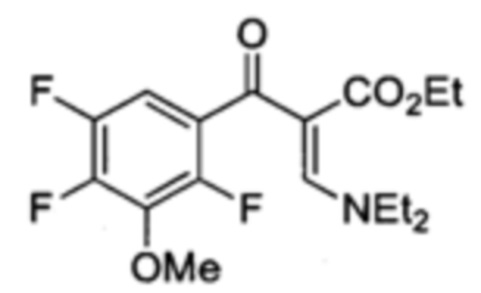
Этил α(Z)-[(диэтиламино)метилен]-2,4,5-трифтор-β-оксо-бензенпропаноат
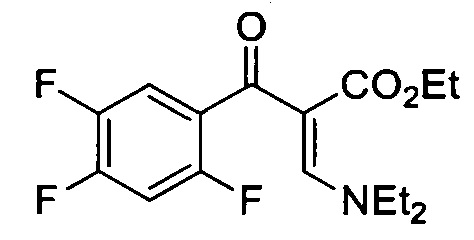
Безводный раствор CH2Cl2 (50 мл) 2,4,5-трифторбензойной кислоты (3.49 г, 19.81 ммоль), оксалил хлорид (2.18 мл, 25.75 ммоль) и пять капель DMF перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь затем подвергали концентрирующему выпариванию под пониженным давлением, растворяли в толуоле (30 мл) и по каплям добавляли к раствору толуола (20 мл) триэтиламин (8.26 мл, 59.43 ммоль) и этил-3-(диэтиламино)-2Е-пропеноат (4.40 г, 25.75 ммоль). Через 18 часов перемешивания при 90°С, охлажденную реакционную смесь отмывали водой. Органический слой высушивали над Na2SO4, фильтровали и выпаривали. Сырой осадок очищали посредством флэш-хроматографии на силикагеле (95:5 к 60:40 гексан/AcOEt) с получением Этил α(Z)-[(диэтиламино)метилен]-2,4,5-трифтор-β-оксо-бензенпропаноата (4.180 г, 12.7 ммоль, 64% в два этапа) в форме бесцветного масла.
1H ЯМР (400 МГц, CDCl3, δ): 0.97 (t, 3Н, ОСН2СН3, 3JH-H=7.1 Гц), 1.03 (br s, 3Н, NCH2CH3), 1.33 (br s, 3Н, NCH2CH3), 3.45 (br s, 4Н, NCH2CH3), 3.98 (q, 2Н, ОСН2СН3, 3J=7.1 Гц), 6.87 (Td, 1Н, H5, 3JH-F=9.7 Гц, 4JH-F=6.3 Гц), 7.45 (m, 1H, H8), 7.75 (s, 1Н);
19F ЯМР (376 МГц, CDCl3, δ): -115.6 (dd, F2, 5JF-F=15.5 Гц, 4JF-F=5.3 Гц), -130.2 (br s, F4); -142.8 (dd, F5, 3JF-F=21.5 Гц, 5JF-F=15.5 Гц).
Получение этилового эфира 1-циклопропил-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата
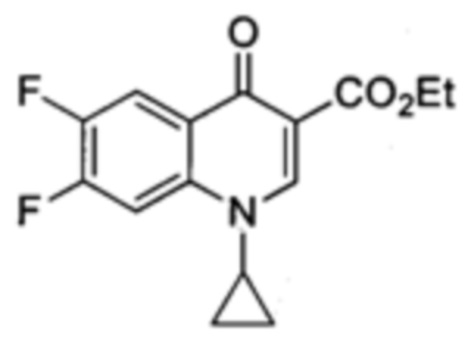
Этил α(Z)-[(диэтиламино)метилен]-2,4,5-трифтор-β-оксо-бензенпропаноата (1.977 г, 6.10 ммольl) в 1:2 EtOH/Et2O (50 мл) добавляли к циклопропиламину (0.98 мл, 10.40 ммоль). Через 3 ч перемешивания при комнатной температуре, реакционную смесь выпаривали под пониженным давлением. Маслянистый осадок растворяли в DMF (40 мл) и добавляли K2СО3 (3.386 г, 24.4 ммоль). Через 16 ч перемешивания при 100°С, добавляли холодную воду (20 мл). Желтый преципитат фильтровали и высушивали с получением этилового эфира 1-циклопропил-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата (1.336 г, 4.56 ммоль, 76%).
1Н ЯМР (400 МГц, CD3CN, δ): 1.07 [m, 2Н, CH2(cPr)], 1.26-1.30 [m, 2Н, CH2(cPr)], 1.32 [t, 3Н, ОСН2СН3, 3JН-H=7.1 Гц], 3.50 (tt, 1Н, СН(cPr), 3JH-H=6.9 Гц, 3JH-H=3.8 Гц), 4.26 (q, 2Н, ОСН2СН3, 3JH-H=7.1 Гц), 7.95 (dd, 1Н, Н5, 3JH-H=12.1 Гц, 4JH-f=6.6 Гц), 7.95 (dd, 1H, Н8, 3JH-F=10.8 Гц, 4JH-F=8.8 Гц), 8.53 (s, 1Н, Н2). 19F ЯМР (376 МГц, CD3CN, δ): -131.1 и -142.4 (2d, 2F, этиловый эфир F6 и F7, 3JF-F=21.7 Гц); MP=181-182°С.
Получение этилового эфира 1-циклопропил-6-фтор-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата
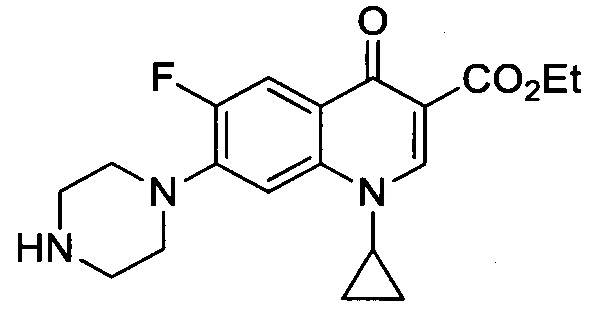
Раствор пиперазина (387 мг, 4.50 ммоль) и этиловый эфир 1-циклопропил-6,7-дифтор-4-оксо-1,4-дигидрохинолин-3-карбоксилата (600 мг, 2.05 ммоль) в безводном CH3CN (10 мл) дефлегмировали в течение одной недели. После выпаривания под пониженным давлением, сырой осадок разделяли в соотношении 1: CHCl3/H2O. Органический слой отмывали водой, высушивали над Na2SO4, фильтровали и выпаривали с получением этилового эфира 1-циклопропил-6-фтор-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (657 мг, 1.83 ммоль, 89%) в форме желтого порошка.
1H ЯМР (400 МГц, CD3CN, δ): 1.04-1.08 (m, 2Н, CH2(cPr)), 1.26-1.29 (m, 2Н, CH2(cPr)), 1.32 (t, 3Н, СН3, 3JH-H=7.1 Гц), 2.96 (dd, 4Н, H1', H4', 3Jн-н=3.9 Гц, 5.9 Гц), 3.19 (dd, 4Н, Н2', Н3', 3JH-H=3.9 Гц, 5.9 Гц), 3.50 (tt, 1H, CH(cPr), 3JH-H=3.7 Гц, 7.0 Гц), 4.25 (q, 2Н, СН2, 3JH-H=7.1 Гц), 7.42 (d, 1Н, Н8, 4JH-F=7.4 Гц), 7.83 (d, 1H, Н5, 3JH-F=13.7 Гц), 8.47 (s, 1Н, Н2);
19F ЯМР (376 МГц, CD3CN, δ):, -125.9 (s, 1F, F6).
Пример 2: Синтез предшественника для получения соединений, имеющих общую формулу III, где R2 представляет собой метокси-группу.
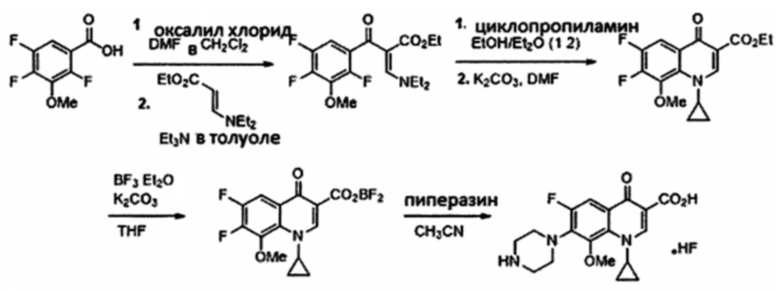
Получение Этил α(Z)-[(диэтиламино)метилен]-2,4,5-трифтор-3-метокси-β-оксо-бензенпропаноата
Безводный раствор CH2Cl2 (30 мл) 2,4,5-трифтор-3-метокси-бензойной кислоты (1.36 г, 6.60 ммоль), оксалил хлорид (0.80 мл, 9.17 ммоль) и пять капель DMF перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь затем подвергали концентрирующему выпариванию под пониженным давлением, растворяли в толуоле (15 мл) и по каплям добавляли к раствору толуола (15 мл) триэтиламин (3 мл, 16.5 ммоль) и этил-3-(диэтиламино)-2Е-пропеноат (1.29 г, 7.54 ммоль). Через 5 часов перемешивания при 90°С, охлажденную реакционную смесь отмывали водой. Органический слой высушивали над Na2SO4, фильтровали и выпаривали. Сырой осадок очищали посредством флэш-хроматографии на силикагеле (95:5 к 60:40 гексан/AcOEt) с получением этил α(Z)-[(диэтиламин6)метилен]-2,4,5-трифтор-3-метокси-β-оксо-бензенпропаноата (1.88 г, 5.22 ммоль, 79% в два этапа) в форме бесцветного порошка.
1H ЯМР (400 МГц, CDCl3, δ): 0.77 (t, 3Н, ОСН2СН3, 3J=7.1 Гц), 0.84 (br s, 3Н, NCH2CH3), 1.08 (br s, 3Н, NCH2CH3), 3.25 (br s, 4H, NCH2CH3), 3.76 (q, 2H, OCH2CH3, 3J=7.1 Гц), 3.78 (s, 3Н, OCH3), 6.87 (ddd, 1H, HAr, 3JH-F=10.1 Гц, 4JH-F=8.5 Гц, 4JH-F=6.0 Гц), 7.53 (s, 1H);
19F ЯМР (376 МГц, CDCl3, δ): -135.1 (dd, F2, 5JF-F=13.8 Гц, 4JF-F=7.6 Гц), -141.5 (dd, F5, 3JF-F=20.6 Гц, 5JF-F=13.7 Гц), -149.2 (br d, F4, 3JF-F=6.5 Гц);
13C ЯМР (100 МГц, CDCl3, δ): 10.8, 14.2, 13.4, 45.0, 53.8, 59.4, 61.5, 101.6, 109.5, 126.2, 137.1, 145.1, 146.6, 149.1, 154.4, 167.3, 184.5.
Получение этилового эфира 1-циклопропил-6,7-дифтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксилата
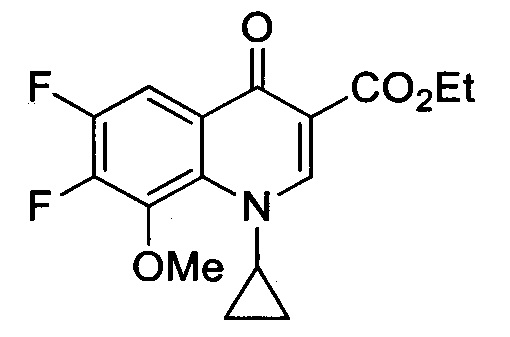
Этилα(Z)-[(диэтиламино)метилен]-2,4,5-трифтор-3-метокси-β-оксо-бензенпропаноат (850 мг, 2.37 ммоль) в 1:2 EtOH/Et2O (20 мл) добавляли к циклопропиламину (0.38 мл, 5.48 ммоль). Через 3 ч перемешивания при комнатной температуре реакционную смесь выпаривают под пониженным давлением. Маслянистый осадок растворяют в DMF (10 мл) и затем добавляют K2СО3 (1.32 г, 9.57 ммоль). Через 5 ч перемешивания при 100°С добавляют холодную воду (5 мл). Желтый преципитат фильтруют и высушивают с получением этилового эфира 1-циклопропил-6,7-дифтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксилата (627 мг, 1.94 ммоль, 82%).
1Н ЯМР (400 МГц, CDCl3, δ): 1.04 и 1.19 [2m, 4Н, СН2(cPr)], 1.37 (t, 3Н, ОСН2СН3, 3JH-H=7.1 Гц), 3.97 (tt, 1H, CH(cPr), 3JH-H=7.5 Гц, 3JH-H=3.7 Гц), 4.07 (d, 3Н, ОСН3, 5JH-H=1.9 Гц), 4.35 (q, 2Н, ОСН2СН3, 3JH-H=7.1 Гц), 7.97 (dd, 1Н, Н5, 3JH-F=10.0 Гц, 4JH-F=8.8 Гц), 8.56 (s, 1Н, Н2);
19F ЯМР (376 МГц, CDCl3, δ): -136.9 и -145.1 (2d, 2F, F6 и F7, 3JF-F=21.3 Гц);
13С ЯМР (100 МГц, CDCl3, δ): 9.2 [СН2(cPr)], 14.5 (ОСН2СН3), 39.8 [CH(cPr)], 61.1 (ОСН2СН3), 62.9 (d, ОСН3, 4Jc-f=7.7 Гц), 108.7 (dd, С5, 2Jc-f=18.7 Гц, 3JC-f=1.1 Гц), 110.1 (С3), 126.1 (dd, С10, 3JC-f=5.9 Гц, 4JC-F=1.8 Гц), 131.6 (dd, С9, 3JC-f=3.7 Гц, 4Jс-F=2.2 Гц), 140.4 (d, С8, 2JC-F=12.1 Гц), 148.2 (dd, С7, 1JC-F=253.7 Гц, 2JC-F=15.6 Гц), 149.2 (dd, С6, JC-F=251.4 Гц, 2JC-F=12.4 Гц), 150.7 (С2), 165.2 [С(O)O], 172.3 (С4). MP=183-184°С.
Получение 1-циклопропил-6,7-дифтор-1,4-дигидро-8-метокси-4-оксо-3-хинолин-карбоксилато-O3,O4)дифтор-бора
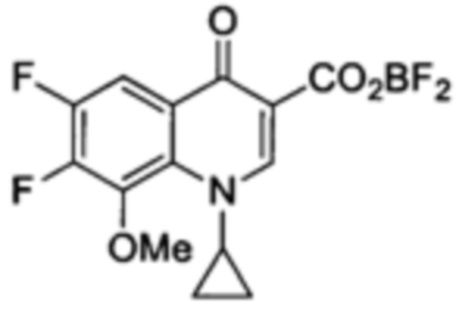
К раствору этилового эфира 1-циклопропил-6,7-дифтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксилата (621 мг, 1.92 ммоль) и K2СО3 (305 мг, 2.21 ммоль) в безводном THF (20 мл), по каплям в течение пяти минут добавляли BF3.Et2O (0.4 мл, 3.18 ммольl) После дефл*егмирования в течение 96 ч чистую реакционную смесь разводили, Et2O (40 мл), а полученную смесь фильтровали и отмывали Et2O. Полученное сырое белое твердое вещество растворяли в CH3CN и фильтровали. Сырое твердое вещество повторно растворяли в CH3CN и фильтровали. Фильтраты объединяли и выпаривали с получением 1-циклопропил-6,7-дифтор-1,4-дигидро-8-метокси-4-оксо-3-хинолин-карбоксилато-О3,O4)дифтор-бора в форме белого твердого вещества (514 мг, 1.50 ммоль, 78%).
1H ЯМР (400 МГц, CD3CN, δ): 1.25-1.37 [m, 4Н, 2СН2(cPr)], 4.19 (d, 3Н, ОСН3, 5JH-F=2.4 Гц), 4.48 (tt, 1Н, CH(cPr), 3JH-H=7.3 Гц, 3JH-H=3.8 Гц), 8.17 (dd, 1Н, Н5, 3JH-F=9.8 Гц, 4JH-F=8.1 Гц), 9.17 (s, 1Н, Н2);
19F ЯМР (376 МГц, CD3CN, δ): -131.7 и -139.0 (2d, 2F, F6 и F7, 3JF-F=19.9 Гц), -144.0 (s, 0.5F, 10BF2), -144.1 (s, 2.4F, 11BF2); MS (+ESI) m/z: [M+Na]+ расч для C14H10BF4NO4: 343.06; обнаружено: 344.2; Mp=221-223°С.
Получение 1-циклопропил-6-фтор-8-метокс-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты
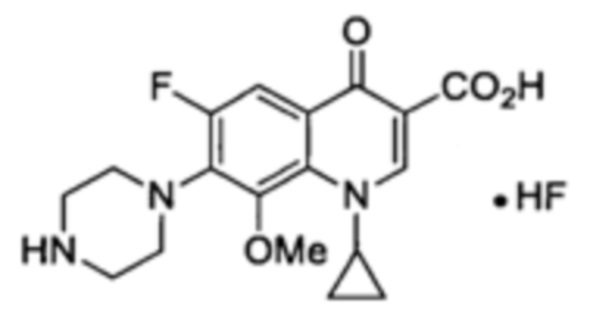
Раствор пиперазина (450 мг, 5.2 ммоль) и 1-циклопропил-6,7-дифтор-1,4-дигидро-8-метокси-4-оксо-3-хинолин-карбоксилато-O3,O4)дифтор-бора (650 мг, 1.89 ммоль) в безводном CH3CN (25 мл) дефлегмировали в течение 96 часов. Твердое вещество отмывали CH3CN и Et2O с получением 1-циклопропил-6-фтор-8-метокс-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (550 мг, 1.44 ммоль, 76%) в форме твердого вещества бежевого цвета.
1Н ЯМР (400 МГц, D2O/CD3CN:4/1, δ): 0.89 (m, 2Н, CH2(cPr)), 1.08 (m, 2Н, CH2(cPr)), 3.30 (m, 4Н, H1', Н4'), 3.52 (m, 4Н, H2', H3'), 3.74 (s, 3Н, ОСН3), 4.03 (m, 1Н, CH(cPr)), 7.68 (d, 1Н, Н5, 3JH-F=12.1 Гц), 8.59 (s, 1H, Н2);
19F ЯМР (376 МГц, CD3CN, δ): -121.6 (bs, 1F, F-), -122.2 (s, 1F, F6). MP=191-193°C.
Пример 3: Получение соединений, имеющих общую Формулу III, где R2=Н
Получение 1-циклопропил-6-фтор-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (15)
К раствору этилового эфира 1-циклопропил-6-фтор-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (63 мг, 0.18 ммоль) в сухом DMF (18 мл) добавляли 1-йодопентан (760 мг, 3.8 ммоль) и NaHCO3 (150 мг, 1.8 ммоль). Реакционную смесь перешивали при комнатной температуре в течение 20 часов и концентрировали при пониженном давлении. Полученный маслянистый осадок собирали в DCM (40 мл). Органический слой отмывали водой (20 мл), высушивали над безводным MgSO4 и концентрировали в вакууме с получением этилового этилового эфира 1-циклопропил-6-фтор-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (65 мг, 86%) в форме желтого порошка. Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
Этиловый эфир 1-циклопропил-6-фтор-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (65 мг, 0.15 ммоль) растворяли в ЕtOН/Н2O (5/2) смеси (21 мл) и затем добавляли LiOH (30 мг, 1.25 ммоль). Затем реакционную смесь перемешивали при комнатной температуре в течение 20 часов и закисляли до величины рН 1 при помощи HClaq 3N. EtOH удаляли при пониженном давлении и водный слой экстрагировали DCM (40 мл). Затем органический слой отмывали насыщенным водным раствором NaHCO3 (20 мл), высушивали над безводным MgSO4 и концентрировали в вакууме с получением 1-циклопропил-6-фтор-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (48 мг, 80%) в форме белого порошка.
1Н ЯМР (400 МГц, CDCl3, δ): 14.78 (широкий s, 1Н, СO2H), 8.73 (s, 1Н, Н2), 7.90 (d, 3JH-F=12.6 Гц, 1Н, Н5), 7.41 (d, 4JH-F=7.0 Гц, 1Н, Н8), 3.79 (широкий s, 4Н, Н2' и Н3'), 3.55 (m, 1Н, CH(cPr)), 3.54 (широкий s, 4H, H1' и H4'), 3.02 (m, 2Н, NCH2СН2СН2), 1.94 (широкий s, 2Н, NСН2СN2СН2), 1.47-1.37 (m, 6Н, CH2(cPr), NСН2СН2СH2СH2), 1.28-1.19 (m, 2Н, CH2(cPr)), 0.94 (t, 3JH-H=6.7 Гц, 3Н, СН3);
19F ЯМР (376 МГц, CDCl3, δ): -121.9 (s, F6);
13С ЯМР (100 МГц, CDCl3, δ): 177.1 (d, J=2.6 Гц, С4), 166.7 (s, СO2Н), 153.5 (d, J=250.6 Гц, С6), 147.9 (s, С2), 144.3 (d, J=10.4 Гц, С7), 139.0 (s, С9), 121.0 (d, J=8.0 Гц, С10), 112.6 (d, J=23.1 Гц, С5), 108.4 (s, С3), 106.1 (d, J=2.3 Гц, С8), 58.0 (s, NCH2CH2CH2), 51.9 (s, С1' и C4'), 46.9 (s, C2' и C3'), 35.6 (s, CH(cPr)), 29.0 (s, CH2), 23.6 (s, CH2), 22.3 (s, CH2), 13.9 (s, CH3), 8.5 (s, 2CH2(cPr));
IR (чистый): v = 3433, 2956, 2933, 2872, 1729, 1630, 1505, 1471, 1390, 1337, 1302, 1266, 1099, 979, 944, 892, 831, 805, 733 cm-1;
HRMS (+ESI) m/z: [M+Na]+ расч для C22H29FN3O3: 402.2194, обнаружено: 402.2192.
Контрольные соединения Квин 16, Квин 9 и Квин 10 получали аналогичным образом, что и соединение Квин 15 с использованием соответствующего исходного материала.
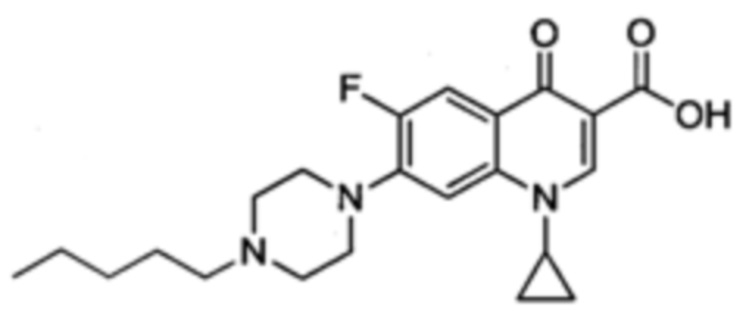
Получение 1-циклопропил-6-фтор-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (Квин 16)
Синтезировали этиловый эфир 1-циклопропил-6-фтор-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (70 мг, 87%, бледно-желтый порошок) как описано для Quin 15, начиная с этилового эфира 1-циклопропил-6-фтор-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (63 мг, 0.18 ммоль), 1-йодоктан (133 мг, 0.55 ммоль) и NaHCO3 (80 мг, 0.95 ммоль) в сухом DMF (15 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-кароксильную кислоту (45 мг, 68%, белый порошок) получали аналогично получению Квин 15, начиная с этилового эфира 1-циклопропил-6-фтор-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (70 мг, 0.15 ммоль) и LiOH (40 мг, 1.67 ммоль) в ЕtOН/Н2O (5/2) смесь (21 мл).
1H ЯМР (400 МГц, CDCl3, δ): 15.02 (широкий s, 1Н, СO2H), 8.71 (s, 1Н, Н2), 7.94 (d, 3JH-F=13.1 Гц, 1Н, Н5), 7.34 (d, 4JH-F=7.1 Гц, 1H, H8), 3.55 (широкий s, 1H, CH(cPr)), 3.36 (широкий s, 4Н, Н2' и Н3'), 2.67 (широкий s, 4Н, Н1' и Н4'), 2.42 (m, 2Н, NCH2СН2СН2), 1.58-1.48 (m, 2Н, NСН2СH2СН2), 1.42-1.21 (m, 12Н, CH2(cPr), 5СН2), 1.15-1.21 (m, 2Н, CH2(cPr)), 0.87 (t, 3JH-H=6.8 Гц, 3Н, CH3);
19F ЯМР (376 МГц, CDCl3, δ): -120.7 (s, F6);
13С ЯМР (100 МГц, CDCl3, δ): 177.2 (d, J=2.5 Гц, С4), 167.1 (s, СO2Н), 153.8 (d, J=251.6 Гц, С6), 147.4 (s, С2), 146.1 (d, J=10.3 Гц, С7), 139.2 (s, С9), 119.8 (d, J=7.9 Гц, С10), 112.4 (d, J=23.5 Гц, С5), 108.2 (s, С3), 104.9 (d, J=3.3 Гц, С8), 58.8 (s, NCH2CH2CH2), 53.0 (s, C1' и С4'), 50.0 (s, С2' и С3'), 35.4 (s, СН(сРr)), 31.9 (s, СН2), 29.6 (s, СН2), 29.4 (s, СН2), 27.6 (s, СН2), 26.9 (s, СН2), 22.8 (s, СН2), 14.2 (s, СН3), 8.3 (s, 2СН2(сРr));
IR (чистый): v=2926, 2854, 2816, 2778, 1725, 1626, 1611, 1544, 1494, 1464, 1452, 1378, 1344, 1299, 1254, 1223, 1185, 1143, 1128, 1109, 1094, 1043, 1027, 1009, 991, 945, 890, 859, 833, 805, 778, 747, 706 cm-1;
HRMS (+ESI) m/z. [М+Н]+ расч для C25H34FN3O3: 444.2663, обнаружено: 444.2668.
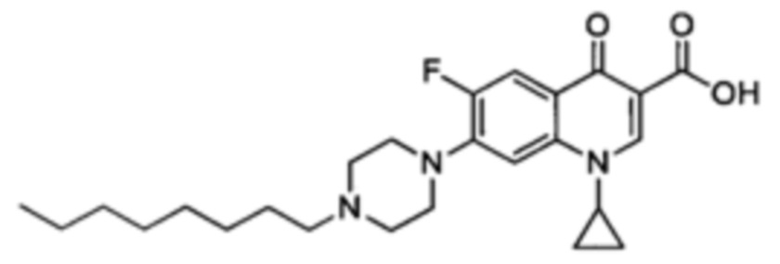
Получение 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (Квин 9)
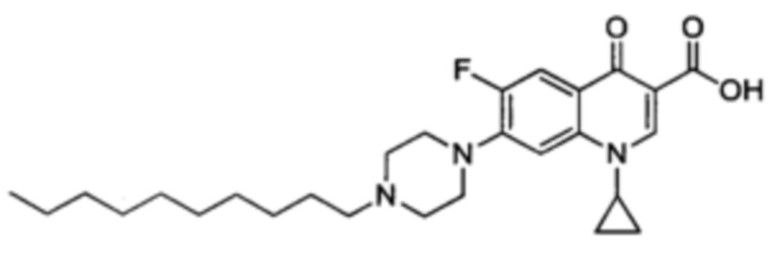
Синтезировали этиловый эфир 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-1,4-дигидрохинолин-3-карбоксилата (117 мг, 72%, бежевый порошок) как описано для Квин 15, начиная с этилового эфира 1-циклопропил-6-фтор-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (117 мг, 0.33 ммоль), 1-йододекан (0,10 мл, 0.49 ммоль) и NaHCO3 (82 мг, 0.98 ммоль). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-кароксильную кислоту (52 мг, 92%, белый порошок) получали аналогично получению Квин 15, начиная с этилового эфира 1-циклопропил-6-фтор-7-(4-децилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (60 мг, 0.12 ммоль) и LiOH.H2O (55 мг, 1.32 ммоль) в 39 мл о 4:1 МеОН/Н2O.
1Н ЯМР (400 МГц, CDCl3, δ): 15.03 (широкий s, 1H, СO2H), 8.78 (s, 1Н, Н2), 8.03 (d, 3JH-F=13.0 Гц, 1Н, Н5), 7.36 (d, 4JH-F=7.1 Гц, 1H, Н8), 3.53 (широкий s, 1H, CH(сРr)), 3.38 (широкий s, 4Н, H2' и Н3'), 2.71 (широкий s, 4Н, Н1' и Н4'), 2.45 (m, 2Н, NCH2CH2CH2), 1.60-1.49 (m, 2Н, NСН2СH2СН2), 1.42-1.16 (m, 18Н, 2СH2(сРr), 7СН2), 0.88 (t, 3JH-H=6.9 Гц, 3Н, СН3);
19F ЯМР (376 МГц, CDCl3, δ): s, 1F, F6);
HRMS (+ESI) m/z: расч для С27Н39FN3О3: 472.2975; обнаружено 472.2965 [М+Н]+;
IR (чистый): 2928 (ацид), 2853 (алкил), 1625 (цетон), 1488 (С=С), 1413 (С=С), 1306 (C-N, С-С, С-О), 1254 (C-N, С-С, С-О). MP=209°С.
Получение 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (Квин 10)
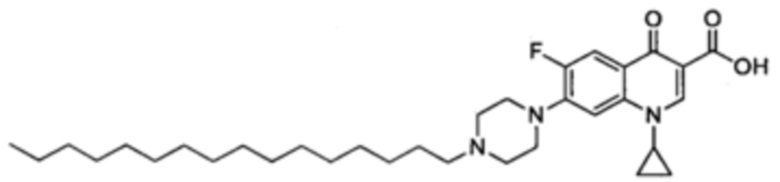
Синтезировали этиловый эфир 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (146 мг, 90%, белое твердое вещество) как описано для Quin 15, начиная с этилового эфира 1-цикл опропил-6-фтор-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (100 мг, 0.28 ммоль), 1-йодогексадекан (147 мг, 0.42 ммоль) и NaHCO3 (70 мг, 0.84 ммоль). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
Этиловый эфир 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (60 мг, 0.1 ммоль) и LiOH.H2O (47 мг, 1.10 ммоль) в 34 мл 4:1 МеОН/Н2O. Реакционную смесь перемешивали при комнатной температуре в течение 24 ч и оставляли перемешиваться в колбе с обратным холодильником еще на 22 ч. После закисления ледяной АсОН до величины рН 5-6, растворитель выпаривали до полного высыхания и добавляли небольшое количество воды. Суспензию фильтовали, а твердое вещество высушивали в вакууме с получением желаемого соединения в форме желтого порошка (35 мг, 0.06 ммоль, 61%).
1Н ЯМР (400 МГц, CDCl3, δ): 15.03 (широкий s, 1H, СO2H), 8.78 (s, 1Н, Н2), 8.03 (d, 3JH-F=13.0 Гц, 1H, Н5), 7.36 (d, 4JH-F=6.2 Гц, 1Н, H8), 3.54 (широкий s, 1H, CH(сРr)), 3.36 (широкий s, 4Н, Н2' и Н3'), 2.67 (широкий s, 4Н, Н1' и Н4'), 2.42 (dd, 3JH-H=7.8, 7.3 Гц, 2Н, NСH2СН2СН2), 1.60-1.49 (m, 2Н, NСН2СH2СН2), 1.42-1.15 (m, 30Н, 2CH2(сРr), 13СН2), 0.88 (t, 3JH-H=6.5 Гц, 3Н, СН3);
19F ЯМР (376 МГц, CDCl3, δ): δ -120.7 (s, 1F, F6);
13С ЯМР (100 МГц, CDCl3, δ): δ 8.3 (s, СН2(сРr)), 14.2 (s, СН3), 22.8 (s, СН2), 26.9 (s, NCH2CH2), 27.7, 29.5, 29.6, 29.7, 29.8, 32.0 (s, СН2), 49.9 (d, С2' и C3', 4JC-F=4.4 Гц), 50.9 (s, CH(cPr)), 52.9 (s, C1' и C4'), 58.7 (s, NCH2CH2), 104.8 (d, C8,3JC-F=2.9 Гц), 108.2 (s, C3), 112.4 (d, C5, 2JC-F=23.3 Гц), 119.8 (d, C10, 3JC-F=6.3 Гц), 139.2 (s, C9), 146.1 (d, C7, 2Jc-f=11.2 Гц), 147.5 (s, C2), 153.3 (d, C6, 1JC-F=249.7 Гц), 167.2 (s, C5'), 177.2 (s, C4);
HRMS (+ESI) m/z: расч для C33H51FN3O3: 556.3914; обнаружено: 556.3891 [M+H]+ IR (чистый): 2916 (кислый), 2850 (алкил), 1742 (ацид), 1626 (цетон), 1501 (С=С), 1464 (С=С), 1335 (C-N, С-С, С-О), 1254 (C-N, С-С, С-О), 1131 (C-N, С-С, С-О), 1029 (C-F). MP=150°С.
Пример 4: Получение соединений, имеющих общую Формулу III, где R2=ОМе
Получение 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (Квин 17)
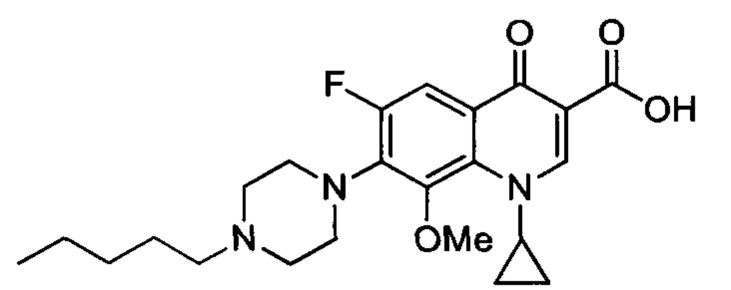
К суспензии 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (65 мг, 0.17 ммоль) в сухом DMF (18 мл), добавляли 1-йодопентан (152 мг, 0.77 ммоль) и NaHCO3 (200 мг, 2.4 ммоль). Реакционную смесь перемешивали при температуре 40°С в течение 40 часов и затем концентрировали при пониженном давлении. Осадок собирали в DCM (40 мл) и органический слой отмывали водой (30 мл) и высушивали над безводным MgSO4 с получением смеси 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты и 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилового пентилового эфира (150 мг).
Полученную смесь растворяли в смеси EtOH/H2O (5/2) (21 мл) и затем добавляли LiOH (40 мг, 1.67 ммоль). Реакционную смесь перемешивал в течение ночи при комнатной температуре и затем закисляли при помощи HClaq 1N до величины рН 3. Этанол удаляли при пониженном давлении и водный слой экстрагировали DCM (40 мл). Органический слой отмывали насыщенным водным раствором NaHCO3 (20 мл), высушивали над безводным MgSO4 и концентрировали в вакууме. Сырое твердое вещество отмывали Et2O (3×5 mL) с получением 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты в виде желтого порошка.
1H ЯМР (400 МГц, CD2Cl2, δ): 14.83 (широкий s, 1Н, СO2H), 8.78 (s, 1Н, Н2), 7.82 (d, 3JH-F=12.4 Гц, 1Н, Н5), 4.04 (m, 1H, CH(сРr)), 3.76 (s, 3Н, ОСН3), 3.43 (широкий s, 4Н, Н2' и Н3'), 2.58 (широкий 4Н, Н1' и Н4'), 2.39 (m, 2Н, NCH2СН2СН2), 1.52 (m, 2Н, NCH2CH2CH2), 1.42-1.24 (m, 4Н, NСН2СН2СH2СH2), 1.20 (q, J=6.8 Гц, 2Н, СH2(сРr)), 1.01-0.95 (m, 2Н, СH2(сРr)), 0.92 (t, 3JH-H=6.9 Гц, 3Н, СН3);
19F ЯМР (376 МГц, CD2Cl2, δ): -120.1 (s, F6);
13С ЯМР (75 МГц, CD2Cl2, δ): 177.5 (d, J=2.9 Гц, С4), 166.9 (s, CO2Н), 156.7 (d, J=250.9 Гц, С6), 150.3 (s, С2), 145.9 (d, J=5.8 Гц, С8), 140.1 (d, J=11.6 Гц, С7), 134.6 (s, С9), 121.9 (d, J=9.3 Гц, С10), 108.0 (s, С3), 107.9 (d, J=23.3 Гц, С5), 62.8 (s, ОСН3), 59.2 (s, NCH2CH2CH2), 54.3 (s, C1' и C4'), 51.2 (d, J=4.6 Гц, C2' и C3'), 41.0 (s, CH(cPr)), 30.1 (s, CH2), 26.9 (s, NCH2CH2CH2), 23.1 (s, CH2), 14.3 (s, CH3), 9.8 (s, 2CH2(cPr));
IR (чистый): v=3081, 2954, 2931, 2857, 2810, 2771, 1729, 1665, 1616, 1580, 1534, 1506, 1440, 1372, 1314, 1277, 1238, 1206, 1187, 1148, 1128, 1114, 1089, 1057, 1039, 1001, 958, 936, 888, 851, 821, 807, 776, 732, 709 см-1;
HRMS (+ESI) m/z: [M+H]+ расч для C23H30FN3O4: 432.2299, обнаружено: 432.2311.
Контрольные соединения Квин 18, Квин 19 и Квин 20,а также соединения 15, 17, 19, 20 и 22, получали аналогичным образом, что и контрольное соединение Quin 17, используя соответствующий исходный материал.
Получение 1-циклопропил-6-фтор-8-метокси-7-(4-гептилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (соединение 15)
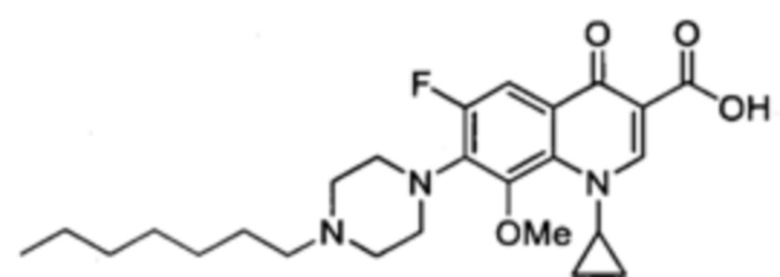
Смесь 1-циклопропил-6-фтор-8-метокси-7-(4-гептилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты и гептилового эфира 1-циклопропил-6-фтор-8-метокси-7-(4-гептилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата получали согласно схеме получения контрольного соединения Квин 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (2.370 г, 6.21 ммоль), 1-йодогептана (4.75 г, 21.2 ммоль) и NaHCO3 (3.69 г, 43.9 ммоль) в сухом DMF (450 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-8-метокси-7-(4-гептилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (1.03 г, 2.17 ммоль, 35% за два этапа, желтый порошок) получали согласно схеме получения контрольного соединения Quin 17, начиная с последней смеси и LiOH (580 мг, 24.2 ммоль) в смеси EtOH/H2O (5/2) (400 мл).
1Н ЯМР (400 МГц, CD2Cl2, δ): 14.77 (широкий s, 1H, СO2H), 8.77 (s, 1Н, Н2), 7.80 (d, 3JH-F=12.4 Гц, 1H, Н5), 4.04 (m, 1Н, СH(сРr)), 3.77 (s, 3Н, OCH3), 3.43 (широкий s, 4Н, Н2' и Н3'), 2.58 (широкий s, 4Н, Н1' и Н4'), 2.39 (m, 2Н, NCH2CH2CH2), 1.50 (m, 2Н, СН2СH2СН2), 1.42-1.24 (m, 8Н, СН2), 1.20 (q, J=6.9 Гц, 2Н, CH2(сРr)), 1.02-0.95 (m, 2Н, CH2(сРr)), 0.89 (t, 3JH-H=6.8 Гц, 3Н, СН3);
13С ЯМР (100 МГц, CD2Cl2, δ): 177.1 (d, J=3.1 Гц, С4), 166.9 (s, СO2Н), 157.8 (d, J=250.8 Гц, С6), 154.5 (s, С2), 145.4 (d, J=5.8 Гц, С8), 139.5 (d, J=11.7 Гц, С7), 134.1 (s, С9), 121.9 (d, J=9.2 Гц, С10), 108.4 (s, С3), 107.9 (d, J=23.3 Гц, С5), 62.7 (s, ОСН3), 59.1 (s, NCH2CH2CH2), 53.9 (s, С1' и С4'), 50.5 (d, J=4.6 Гц, С2' и С3'), 40.6 (s, СН(сРr)), 31.9 (s, СН2), 29.3 (s, СН2), 29.0 (s, СН2), 27.6 (s, СН2), 27.2 (s, СН2), 26.7 (s, СН2), 22.7 (s, СН2), 14.2 (s, СН3), 9.7 (s, 2СН2(сРr)). MP=157.2°С. Элементный анализ: С=64.62%, Н=7.52%, N=8.83%, расч С=65.14%, Н=7.47%, N=9.12%.
Получение 1-циклопропил-6-фтор-8-метокси-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (Квин 18)
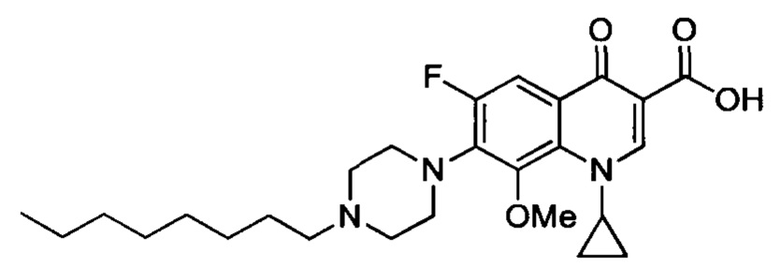
Смесь 1-циклопропил-6-фтор-8-метокси-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты и октилового эфира 1-циклопропил-6-фтор-8-метокси-7-(4-октиллпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (80 мг), получали согласно схеме получения контрольного соединения Quin 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (74 г, 0.20 ммоль), 1-йодоктана (133 мг, 0.55 ммоль) и NaHCO3 (100 мг, 1.20 ммоль) в сухом DMF (450 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-8-метокси-7-(4-гептилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (30 мг, 32% за два этапа, желтый порошок) получали согласно схеме получения контрольного соединения Quin 17, начиная с последней смеси (80 мг) и LiOH (40 мг, 1.67 ммоль) в смеси ЕtOН/Н2O (5/2) (21 мл).
1Н ЯМР (400 МГц, CD2Cl2, δ): 14.77 (широкий s, 1H, СO2H), 8.77 (s, 1H, Н2), 7.80 (d, 3JH-F=12.4 Гц, 1Н, Н5), 4.04 (m, 1Н, СH(сРr)), 3.77 (s, 3Н, ОСH3), 3.43 (широкий s, 4Н, Н2' и Н3'), 2.58 (широкий s, 4Н, Н1' и H4'), 2.39 (m, 2Н, NCH2CH2CH2), 1.50 (m, 2Н, NCH2CH2CH2), 1.42-1.24 (m, 10Н, СН2), 1.20 (q, J=6.9 Гц, 2Н, СH2(сРr)), 1.02-0.95 (m, 2Н, СH2(сРr)), 0.89 (t, 3JH-H=6.8 Гц, 3Н, СН3);
19F ЯМР (376 МГц, CD2Cl2, δ): -120.1 (s, F6);
13С ЯМР (100 МГц, CD2Cl2, δ): 177.5 (d, J=3.1 Гц, С4), 166.8 (s, СO2Н), 156.7 (d, J=250.8 Гц, С6), 150.3 (s, С2), 145.9 (d, J=5.8 Гц, С8), 140.1 (d, J=11.7 Гц, С7), 134.6 (s, С9), 121.9 (d, J=9.2 Гц, С10), 108.0 (s, С3), 107.9 (d, J=23.3 Гц, С5), 62.8 (s, ОСН3, 59.3 (s, NCH2CH2CH2), 54.3 (s, С1' и С4'), 51.2 (d, J=4.6 Гц, С2' и С3'), 41.0 (s, СН(сРr)), 32.3 (s, СН2), 30.0 (s, СН2), 29.7 (s, СН2), 27.9 (s, СН2), 27.2 (s, СН2), 23.1 (s, СН2), 14.3 (s, СН3), 9.8 (s, 2СН2(сРr));
IR (чистый): v=3084, 2926, 2853, 2809, 2770, 1728, 1617, 1601, 1554, 1539, 1505, 1436, 1383, 1376, 1312, 1280, 1238, 1204, 1187, 1144, 1128, 1115, 1091, 1055, 1040, 1008, 993, 957, 934, 887, 831, 821, 805, 730, 710 cm-1;
HRMS (+ESI) m/z: [M+H]+ расч для C26H36FN3O4: 474.2769, found: 474.2765. Элементный анализ: С=66.11%, Н=7.78%, N=8.82%, расч С=65.94%, Н=7.66%, N=8.87%.
Получение 1-циклопропил-6-фтор-8-метокси-7-(4-нонилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (соединение 17)
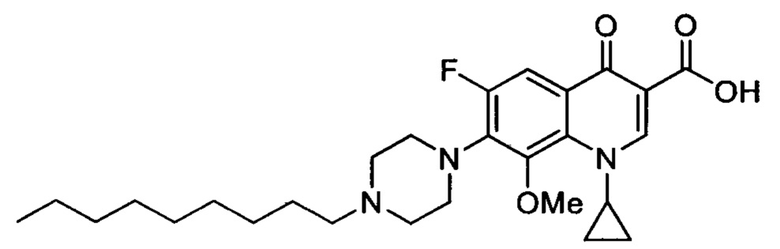
Смесь 1-циклопропил-6-фтор-8-метокси-7-(4-нонилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты и нонилового эфира 1-циклопропил-6-фтор-8-метокси-7-(4-нониллпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата (80 мг), получали согласно схеме получения контрольного соединения Квин 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (2,04 г, 5.35 ммоль), 1-йодоноан (4,45 мг, 17.5 ммоль) и NaHCO3 (3,04 г, 36.2 ммоль) в сухом DMF (450 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-8-метокси-7-(4-нонилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (1,02 г, 2,03 ммоль, 38% за два этапа, желтый порошок) получали согласно схеме получения контрольного соединения Quin 17, начиная с последней смеси и LiOH (580 мг, 24.2 ммоль) в смеси ЕtOН/Н2O (5/2) (400 мл).
1Н ЯМР (400 МГц, CD2Cl2, δ): 14.77 (широкий s, 1Н, СO2H), 8.77 (s, 1Н, Н2), 7.80 (d, 3JH-F=12.4 Гц, 1H, Н5), 4.04 (m, 1Н, СH(сРr)), 3.77 (s, 3Н, ОСH3), 3.43 (широкий s, 4Н, Н2' и Н3'), 2.58 (широкий s, 4Н, Н1' и Н4'), 2.39 (m, 2Н, NСH2СН2СН2), 1.50 (m, 2Н, NCH2CH2CH2), 1.42-1.24 (m, 12Н, СН2), 1.20 (q, J=6.9 Гц, 2Н, СH2(сРr)), 1.02-0.95 (m, 2Н, СH2(сРr)), 0.89 (t, 3JH-H=6.8 Гц, 3Н, СH3);
13С ЯМР (100 МГц, CD2Cl2) δ): 177.1 (d, J=3.1 Гц, С4), 166.9 (s, СO2Н), 157.9 (d, J=250.8 Гц, С6), 154.5 (s, С2), 145.3 (d, J=5.8 Гц, С8), 139.7 (d, J=11.7 Гц, С7), 134.0 (s, С9), 121.6 (d, J=9.2 Гц, С10), 108.2 (s, С3), 107.9 (d, J=23.3 Гц, C5), 62.6 (s, ОСН3), 59.1 (s, NCH2CH2CH2), 54.0 (s, C1' и C4'), 50.8 (d, J=4.6 Гц, C2' и С3'), 40.6 (s, CH(cPr)), 32.0 (s, CH2), 29.7 (s, CH2), 29.6 (s, CH2), 29.4 (s, CH2), 27.7 (s, CH2), 26.9 (s, CH2), 25.5 (s, CH2), 22.8 (s, CH2), 14.2 (s, CH3), 9.7 (s, 2CH2(cPr)). MP=144.4°C. Элементный анализ: С=66.36%, H=7.86%, N=8.50%, расч С=66.51%, Н=7.85%, N=8.62%.
Получение 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (Квин 19)
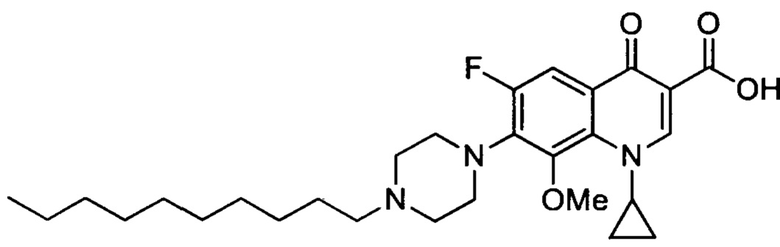
Смесь 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты и децилового эфира 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-децилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксилата (90 мг), получали согласно схеме получения контрольного соединения Quin 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (80 мг, 0.21 ммоль), 1-йододекана (163 мг, 0.61 ммоль) и NaHCO3 (120 мг, 1.40 ммоль) в сухом DMF (20 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (30 мг, 29% за два этапа, белое твердое вещество) получали согласно схеме получения контрольного соединения Quin 17, начиная с последней смеси (90 мг) и LiOH (40 мг, 1.67 ммоль) в смеси ЕtOН/Н2O (5/2) (21 мл).
1Н ЯМР (400 МГц, CD2Cl2, δ): 14.78 (широкий s, 1H, СO2H), 8.78 (s, 1Н, Н2), 7.82 (d, 3JH-H=12.4 Гц, 1Н, Н5), 4.04 (m, 1H, СH(сРr)), 3.76 (s, 3Н, OCH3, 3.43 (широкий s, 4Н, Н2' и Н3'), 2.57 (широкий s, 4Н, Н1' и H4'), 2.39 (m, 2Н, NCH2CH2CH2), 1.51 (m, 2H, NCH2CH2CH2), 1.37-1.24 (m, 14H, CH2), 1.20 (m, 2H, CH2(cPr)), 1.02-0.95 (m, 2H, CH2(cPr)), 0.89 (t, 3JH-H=6.7 Гц, 3Н, CH3);
19F ЯМР (376 МГц, CD2Cl2, δ): -120.1 (s, F6);
13C ЯМР (100 МГц, CD2Cl2) δ): 177.5 (d, J=3.1 Гц, C4), 166.9 (s, CO2H), 156.7 (d, J=250.9 Гц, C6), 150.3 (s, C2), 145.9 (d, J=5.8 Гц, C8), 140.1 (d, J=11.8 Гц, C7), 134.6 (s, C9), 121.9 (d, J=9.1 Гц, С10), 108.0 (s, C3), 107.9 (d, J=23.2 Гц, C5), 62.8 (s, OCH3), 59.3 (s, NCH2CH2CH2), 54.3 (s, C1' и C4'), 51.2 (d, J=4.7 Гц, С2' и C3'), 41.0 (s, CH(cPr)), 32.3 (s, CH2), 30.1 (s, CH2), 30.0 (s, 2CH2), 29.8 (s, CH2), 27.9 (s, CH2), 27.3 (s, CH2), 23.1 (s, CH2), 14.3 (s, CH3), 9.8 (s, 2CH2(cPr));
IR (чистый): v=3071; 2924, 2852, 2770, 1729, 1618, 1601, 1536, 1506, 1441, 1394, 1384, 1313, 1281, 1239, 1206, 1188, 1148, 1129, 1116, 1091, 1055, 1042, 1003, 959, 937, 888, 879, 831, 821, 805, 730, 710 cm-1;
HRMS (+ESI) m/z: [M+H]+ расч для C28H40FN3O4: 502.3082, found, 502.3077. Элементный анализ: С=67.28%, H=8.21%, N=8.31%, расч С=67.04%, Н=8.04%, N=8.38%.
Получение 1-циклопропил-6-фтор-8-метокси-7-(4-ундецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (соединение 19)
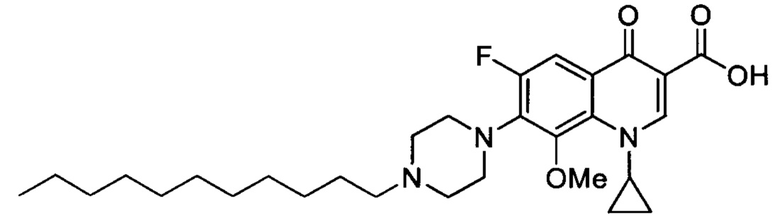
Смесь 1-циклопропил-6-фтор-8-метокси-7-(4-ундецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты и ундецилового эфира 1-циклопропил-6-фтор-8-метокси-7-(4-ундецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата получали согласно схеме получения контрольного соединения Квин 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (2,13 г, 5.60 ммоль), 1-йодоундекана (4,94 г, 17.5 ммоль) и NaHCO3 (3,04 г, 36,2 ммоль) в сухом DMF (450 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-8-метокси-7-(4-ундецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (1,01 г, 1,96 ммоль, 35% за два этапа, желтый порошок) получали согласно схеме получения контрольного соединения Quin 17, начиная с последней смеси и LiOH (580 мг, 24.2 ммоль) в смеси ЕtOH/Н2O (5/2) (400 мл).
1Н ЯМР (400 МГц, CD2Cl2, δ): 14.77 (широкий s, 1H, СO2Н), 8.77 (s, 1Н, Н2), 7.80 (d, 3JH-F=12.4 Гц, 1Н, Н5), 4.04 (m, 1Н, СH(сРr)), 3.77 (s, 3Н, ОСH3, 3.43 (широкий s, 4Н, Н2' и Н3'), 2.58 (широкий s, 4Н, Н1' и H4'), 2.39 (m, 2Н, NHCH2СН2СН2), 1.50 (m, 2Н, NCH2СH2СН2), 1.40-1.16 (m, 18Н, СН2, СH2(сРr)), 1.02-0.95 (m, 2Н, СH2(сРr)), 0.89 (t, 3JH-H=6.8 Гц, 3Н, СН3);
13С ЯМР (100 МГц, CD2Cl2, δ): 177.2 (d, J=3.1 Гц, С4), 166.9 (s, СO2Н), 157.9 (d, J=250.8 Гц, С6), 154.6 (s, С2), 145.3 (d, J=5.8 Гц, С8), 139.6 (d, J=11.7 Гц, С7), 134.1 (s, С9), 121.8 (d, J=9.2 Гц, С10), 108.2 (s, С3), 107.9 (d, J=23.3 Гц, С5), 62.6 (s, ОСН3), 59.1 (s, NCH2CH2CH2), 54.0 (s, С1' и С4'), 50.8 (d, J=4.6 Гц, С2' и С3'), 40.7 (s, СН(сРr)), 32.1 (s, СН2), 29.8 (s, СН2), 29.7 (s, СН2), 29.5 (s, СН2), 27.7 (s, СН2), 26.9 (s, СН2), 25.5 (s, СН2), 22.8 (s, СН2), 14.3 (s, СН3), 9.7 (s, 2CH2(cPr)). MP=136.3°С. Элементный анализ: С=67.27%, Н=8.14%, N=8.02%), расч С=67.55%, Н=8.21%, N=8.15%.
Получение 1-циклопропил-6-фтор-8-метокси-7-(4-додецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (соединение 20)
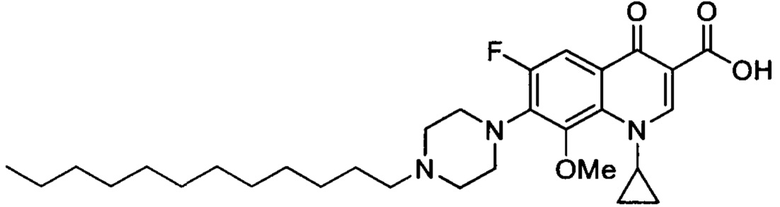
Смесь 1-циклопропил-6-фтор-8-метокси-7-(4-додецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты и додецилового эфира 1-циклопропил-6-фтор-8-метокси-7-(4-ундецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата получали согласно схеме получения контрольного соединения Квин 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (2,32 г, 6.08 ммоль), 1-йодододекана (4,94 г, 17.5 ммоль) и NaHCO3 (3,04 г, 36,2 ммоль) в сухом DMF (450 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-8-метокси-7-(4-додецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (1,03 г, 1,94 ммоль, 32% за два этапа, желтый порошок) получали согласно схеме получения контрольного соединения Квин 17, начиная с последней смеси и LiOH (580 мг, 24.2 ммоль) в смеси EtOH/H2O (5/2) (400 мл).
1Н ЯМР (400 МГц, CD2Cl2, δ): 14.77 (широкий s, 1Н, СO2H), 8.77 (s, 1H, Н2), 7.80 (d, 3JH-F=12.4 Гц, 1H, Н5), 4.04 (m, 1H, СH(сРr)), 3.77 (s, 3Н, ОСH3), 3.43 (широкий s, 4Н, Н2' и Н3'), 2.58 (широкий s, 4Н, Н1' и Н4'), 2.39 (m, 2Н, NCH2СН2СН2), 1.50 (m, 2Н, NCH2СH2СН2), 1.40-1.16 (m, 20Н, СН2, СH2(сРr)), 1.02-0.95 (m, 2Н, СH2(сРr)), 0.89 (t, 3JH-H=6.8 Гц, 3Н, СН3);
13С ЯМР (100 МГц, CD2Cl2, δ): 177.1 (d, J=3.1 Гц, С4), 166.9 (s, СO2Н), 157.8 (d, J=250.8 Гц, С6), 154.5 (s, С2), 145.4 (d, J=5.8 Гц, С8), 139.7 (d, J=11.7 Гц, С7), 134.0 (s, С9), 121.6 (d, J=9.2 Гц, С10), 108.2 (s, С3), 107.8 (d, J=23.3 Гц, С5), 62.6 (s, ОСН3), 59.1 (s, NCH2CH2CH2), 53.9 (s, С1' и С4'), 50.7 (d, J=4.6 Гц, C2' и С3'), 40.6 (s, CH(cPr)), 32.0 (s, CH2), 29.7 (s, CH2), 29.6 (s, CH2), 29.4 (s, CH2), 27.7 (s, CH2), 26.8 (s, CH2), 22.8 (s, CH2), 14.2 (s, CH3), 9.6 (s, 2CH2(cPr)). MP=134.6°C. Элементный анализ: С=67.89%, H=8.49%, N=7.82%, расч С=68.03%, Н=8.37%, N=7.93%.
Получение 1-циклопропил-6-фтор-8-метокси-7-(4-тетрадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (соединение 22)
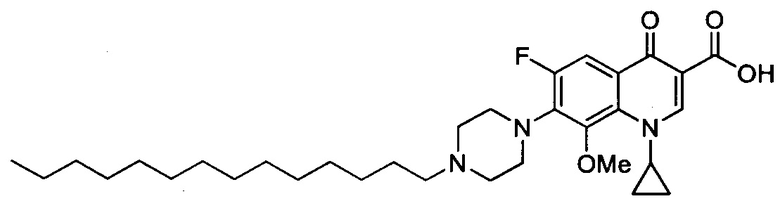
Смесь 1-циклопропил-6-фтор-8-метокси-7-(4-тетрадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты и тетрадецилового эфира 1-циклопропил-6-фтор-8-метокси-7-(4-ундецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксилата получали согласно схеме получения контрольного соединения Квин 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (2,18 г, 5.71 ммоль), 1-бромотетрадекана (4,714 г, 17.0 ммоль) и NaHCO3 (2,94 г, 35,0) ммоль) в сухом DMF (450 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-8-метокси-7-(4-тетрадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (1,02 г, 1,83 ммоль, 32% за два этапа, желтый порошок) получали согласно схеме получения контрольного соединения Квин 17, начиная с последней смеси и LiOH (580 мг, 24.2 ммоль) в смеси ЕtOН/Н2O (5/2) (400 мл).
1Н ЯМР (400 МГц, CD2Cl2, δ): 14.77 (широкий s, 1Н, СO2H), 8.77 (s, 1Н, Н2), 7.80 (d, 3JH-H=12.4 Гц, 1Н, Н5), 4.04 (m, 1Н, СH(сРr)), 3.77 (s, 3Н, ОСН3), 3.43 (широкий s, 4Н, Н2' и Н3'), 2.58 (широкий s, 4Н, Н1' и Н4'), 2.39 (m, 2Н, NCH2СН2СН2), 1.50 (m, 2Н, NСН2СH2СН2), 1.40-1.16 (m, 24Н, СН2, СH2(сРr)), 1.02-0.95 (m, 2Н, СH2(сРr)), 0.89 (t, 3JH-H=6.8 Гц, 3Н, СН3);
13С ЯМР (100 МГц, CD2Cl2, δ): 177.1 (d, J=3.1 Гц, С4), 166.8 (s, СO2Н), 157.8 (d, J=250.8 Гц, С6), 154.5 (s, С2), 145.5 (d, J=5.8 Гц, С8), 139.7 (d, J=11.7 Гц, С7), 134.0 (s, С9), 121.7 (d, J=9.2 Гц, С10), 107.9 (s, С3), 107.8 (d, J=23.3 Гц, С5), 62.5 (s, ОСН3), 59.1 (s, NCH2CH2CH2), 54.0 (s, C1' и C4'), 50.7 (d, J=4.6 Гц, C2' и C3'), 40.6 (s, CH(cPr)), 32.0 (s, CH2), 29.7 (s, CH2), 29.6 (s, CH2), 29.4 (s, CH2), 27.7 (s, CH2), 26.6 (s, CH2), 22.8 (s, CH2), 14.2 (s, CH3), 9.6 (s, 2CH2(cPr)). MP=131.1°C. Элементный анализ: С=69.10%, H=8.85%, N=7.44%, расч С=68.91%, Н=8.67%, N=7.53%.
Получение 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (Квин 20)
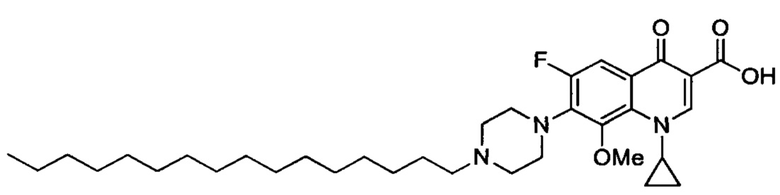
Гексадециловый эфир 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксилата (100 мг, 68%) получали как и контрольное соединение Квин 17, начиная с 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(пиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильной кислоты (70 мг, 0.18 ммоль), 1-йодогексадодекана (230 мг, 0.65 ммоль) и NaHCO3 (200 мг, 2,40 ммоль) в сухом DMF (18 мл). Полученный продукт был достаточно чистым для последующего использования в реакциях синтеза.
1-циклопропил-6-фтор-7-(4-тетрадецилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (50 мг, 47% за два этапа, белое твердое вещество) получали согласно схеме получения контрольного соединения Quin 17, начиная с соответствующего эфира (100 мг, 0,123 ммоль) и LiOH (40 мг, 1.67 ммоль) в (580 мг, 24.2 ммоль) в ЕtOН/Н2O (5/2) смеси (400 mL).
1Н ЯМР (400 MHz, CD2Cl2, δ): 14.79 (широкий s, 1Н, СO2H), 8.78 (s, 1Н, Н2), 7.82 (d, 3JH-F=12.4 Hz, 1Н, H5), 4.04 (m, 1H, СH(сРr)), 3.76 (s, 3Н, OCH3), 3.43 (широкий s, 4H, H2' и H3'), 2.58 (широкий s, 4H, H1' и H4'), 2.39 (m, 2H, NCH2СН2СН2), 1.50 (m, 2Н, NCH2СH2СН2), 1.40-1.16 (m, 28Н, СН2, СH2(сРr)), 1.01-0.95 (m, 2Н, СH2(сРr)), 0.88 (t, 3JH-H=6.8 Hz, 3Н, СH3);
19F ЯМР (376 MHz, CD2Cl2, δ): -120.1 (s, F6);
13C ЯМР (100 MHz, CD2Cl2, δ): 177.5 (d, J=3.0 Hz, C4), 166.9 (s, CO2H), 156.7 (d, J=250.8 Hz, C6), 150.3 (s, C2), 145.9 (d, J=5.8 Hz, C8), 140.1 (d, J=11.7 Hz, C7), 134.6 (s, C9), 121.9 (d, J=9.2 Hz, С10), 108.1 (s, C3), 108.0 (d, J=23.3 Hz, C5), 62.8 (s, OCH3), 59.3 (s, NCH2CH2CH2), 54.3 (s, C1' и C4'), 51.2 (d, J=4.6 Hz, С2' и C3'), 41.0 (s, CH(cPr)), 32.4 (s, CH2), 30.11 (s, 5CH2), 30.07 (s, 3CH2), 30.03 (s, CH2), 29.8 (s, CH2), 27.9 (s, CH2), 27.3 (s, CH2), 23.1 (s, CH2), 14.3 (s, CH3), 9.8 (s, 2CH2(cPr));
IR (чистый): v=3078, 3004, 2917, 2850, 2770, 1733, 1620, 1601, 1511, 1443, 1393, 1384, 1370, 1328, 1312, 1281, 1239, 1208, 1187, 1149, 1130, 1115, 1090, 1053, 993, 959, 937, 889, 878, 831, 822, 805, 730, 719 cm-1;
HRMS (-ESI) m/z: [M-H]- расч для C34H52FN3O4: 584.3863, обнаружено, 584.3843.
Получение 1-циклопропил-6-фтор-7-(4-(2-этилгексил)пиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (соединение 25)
К соединению 2 (1 экв.) в дихлорметане добавляли 2-этилгексанал (1.2 экв.) и уксусную кислоту (6 экв.). NaBH(OAc)3 (1.3 экв) добавляли порционно и смесь перемешивали при комнатной температуре в течение ночи. Добавляли воду (450 мл) и реакционную смесь фильтровали через целит. Осадок отмывали дихлорметаном и фазы разделяли. Органическую фазу отмывали водой, высушивали над сульфатом магнияя, фильтровали и концентрировали до высыхания. После очищения на силикагеле (DCM/Метанол 90/10, 0.5% АсОН) получали 1-циклопропил-6-фтор-7-(4-(2-этилгексил)пиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильую кислоту в форме белого твердого вещества (35%).
1Н ЯМР (400 MHz, CD2Cl2, δ): 8.85 (s, 1Н, Н2), 7.90 (d, 3JH-F=12.4 Hz, 1H, Н5), 4.06 (m, 1Н, СH(сРr)), 3.89 (s, 3Н, ОСH3), 3.56 (широкий s, 4Н, Н2' и Н3'), 2.59 (широкий s, 4Н, Н1' и Н4'), 2.28 (bs, 2Н, NCH2СН), 1.49-1.21 (m, 11Н, СН2, СН, СH2(сРr)), 1.06-1.00 (m, 2Н, СH2(сРr)), 0.89 (m, 6Н, 2СH3);
13С ЯМР (100 MHz, CD2Cl2, δ): 177.4 (d, J=3.1 Hz, C4), 167.3 (s, CO2H), 156.5 (d, J=250.8 Hz, C6), 150.2 (s, C2), 145.6 (d, J=5.8 Hz, C8), 140.1 (d, J=11.7 Hz, C7), 134.4 (s, C9), 121.8 (d, J=9.2 Hz, С10), 117.2 (s, C3), 108.4 (d, J=23.3 Hz, C5), 63.6 (s, OCH3), 62.7 (s, NCH2CH), 54.7 (s, C1' и C4'), 51.2 (d, J=4.6 Hz, C2' и C3'), 41.0 (s, CH(cPr)), 36.5 (s, CH), 31.9 (s, CH2), 29.4 (s, CH2), 25.0 (s, CH2), 23.6 (s, CH2), 14.6 и 11.2 (s, CH3), 9.8 (s, 2CH2(cPr)). Элементный анализ: С=65.66%, H=7.49%, N=8.57%, расч С=65.94%, Н=7.66%, N=8.87%.
Получение 1-циклопропил-6-фтор-7-(4-октаноилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты (соединение 26)
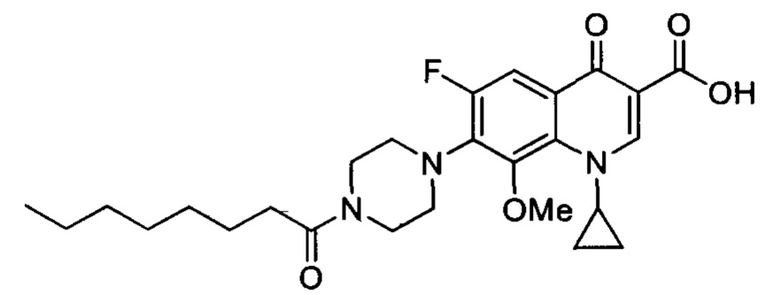
1-циклопропил-6-фтор-7-пиперазин-1-ил-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту (1 экв.) растворяли в дихлорметане. При 0°С добавляли триэтиламин (1.3 экв.) и ацилхлорид (1.5 экв.) и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли циклогексан и фильтровали смесь. Фильтрат выпаривали и полученное твердое вещество очищали при помощи хроматографии на колонке (силикагель, градиент DCM/Метанол 95/15) с получением 1-циклопропил-6-фтор-7-(4-октаноилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильной кислоты в форме твердого белого вещества (40%).
1Н ЯМР (400 MHz, CD2Cl2, δ): 8.87 (s, 1H, Н2), 7.95 (d, 3JH-F=-12.4 Hz, 1H, H5), 4.06 (m, 1H, СH(сРr)), 3.77 (s, 3H, OCH3), 3.71 (широкий s, 4H, H1' и H4'), 3.44 (широкий s, 4H, H2' и H3'), 2.42 (m, 2H, NCH2CH2CH2), 1.68 (m, 2H, NCH2CH2CH2), 1.42-1.23 (m, 12H, CH2), 1.02-0.95 (m, 2H, CH2(cPr)), 0.94 (t, 3JH-H=6.8 Hz, 3H, CH3);
13C ЯМР (100 MHz, CD2Cl2, δ): 177.4 (d, J=3.1 Hz, C4), 172.5 (s, CO), 167.0 (s, CO2H), 156.4 (d, J=250.8 Hz, C6), 150.5 (d, J=5.8 Hz, C8), 146.0 (d, J=11.7 Hz, C7), 139.4 (s, C9), 134.3 (s, C3), 122.9 (d, J=9.2 Hz, C10), 108.7 (d, J=23.3 Hz, C5), 63.0 (s, OCH3), 51.3 (bs, C1', C4', C2' и C3'), 40.9 (s, CH(cPr)), 33.9 (s, CH2), 32.1 (s, CH2), 29.9 (s, CH2), 29.5 (s, CH2), 25.8 (s, CH2), 23.0 (s, CH2), 14.5 (s, CH3), 10.0 (s, 2CH2(cPr)). Элементный анализ: С=63.31%, H=7.10%, N=8.08%, calcd С=64.05%, Н=7.03%, N=8.62%.
2. Биологические данные
2.1 Исследование противомикробной активности in vitro в отношении роста H37Rv М. tuberculosis, а также в отношении суперспирализации ДНК фермента ДНК гиразы
А) Для соединений от Квин 9 до Квин 20
Материалы и Методы
Реагенты
Следующие три хинолона были предоставлены их производителями: гатифлоксацин ( , Levallois-Perret, Франция); ципрофлоксацин и моксифлоксацин (Bayer Pharma, Puteaux, Франция).
, Levallois-Perret, Франция); ципрофлоксацин и моксифлоксацин (Bayer Pharma, Puteaux, Франция).
Противомикробная активность in vitro
H37Rv М. tuberculosis и мутантные штаммы, несущие мутации ДНК гиразы, которые обычно встречаются у штаммов, устойчивых к хинолонам, в клинической практике (GyrA A90V и GyrA D94G) выращивали на среде Левенштейна-Йенсена. Максимальные ингибирующие концентрации (МИК) определяли методом пропорций, как описано ранее (Guillemin, I.; Jarlier V.; Cambau Е. Antimicrob. Agents Chemother. 1998, 42, 2084). 103 и 105 КОЕ распыляли на 7H11 агар, обогащенный 10% комплексом олеиновая кислота+альбумин+декстроза+каталаза, и содержащий двухкратные разведения изучаемого соединения. Колонии подсчитывали через 21-30 дней после инкубации при температуре 37°С. МИК определяли как концентрацию лекарственного препарата при которой происходило уменьшение роста бактерий на 1% или менее по сравнению с ростом, который наблюдался для контрольной культуры на среде, не содержащей лекарственного препарата (Inderlied, С.В.; Nash K.A. In Antibiotics in laboratory medicine, 4th ed.; M. D. V. Lorian, Eds.; The Williams & Wilkins Co.; Baltimore, Md., 1996; pp 127-175).
Нарушение репликации M. tuberculosis внутри макрофагов регистрировали при помощи фенотипического клеточного исследования, в котором используется конфокальный флуоресцентный микроспок для высокоточного скрининга химических соединений (Christophe Т. et al. High Content Screening Identifies Decaprenyl-Phosphoribose 29 Epimerase as a Target for Intracellular Antimycobacterial Inhibitors, PloS pahogens, 2009 Oct; 5(10): e1000645).
Исследование суперспирализации ДНК
ДНК гиразу М. tuberculosis выделяли и очищали как описано ранее (Aubry, А.; Pan, X.-S.; Fisher, L.М.; Jarlier, V.; Cambau, E. Antimicrob. Agents Chemother. 2004, 48, 1281). Реакционная смесь (общий объем 30 мкл) содержала аналитический буфер для ДНК гиразы (40 mМ Трис-HCl [рН 7.5], 25 мМ KCl,6 мМ стеарат магния, 2 мМ спермидина, 4 мМ дитиотреитола, альбумин бычьей сыворотки [0.36 мкг/мл], 10 мМ глутамат натрия, 1 мМ АТФ [рН 8.0]) и ослабленную pBR322 ДНК (0.4 мкг) в качестве субстрата. Белки гиразы (300 нг GyrA и 250 нг GyrB) перемешивали в присутствии увеличивающихся концентраций хинолонов в течение 1 ч при 37°С для М. tuberculosis. Реакцию останавливали добавлением 50% глицерола, содержащего 0,25% бромфенолового голубого, и общую реакционную смесь подвергали электрофорезу в 1% агарозном геле в 0,5Х ТВЕ (Трис-борат-ЭДТА, рН 8.3) буфере. После электрофореза в течение 5,5 ч при 50 В, гель окрашивали бромидом этидия (0,7 мкг/мл). Ингибирующий эффект хинолонов на ДНК гиразу оценивали путем определения концентрации лекарственного вещества, которая необходима для полного ингибирования суперспирализующей активности фермента на 50% (IC50). Суперспирализующая активность оценивалась на основании яркости полос, соответствующих суперспирализованной pBR322 ДНК, при помощи программного обеспечения Molecular Analyst (Bio-Rad).
Результаты
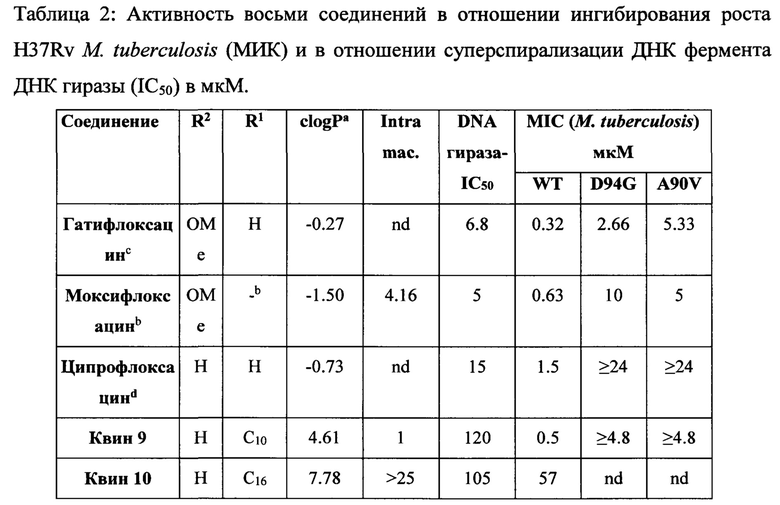
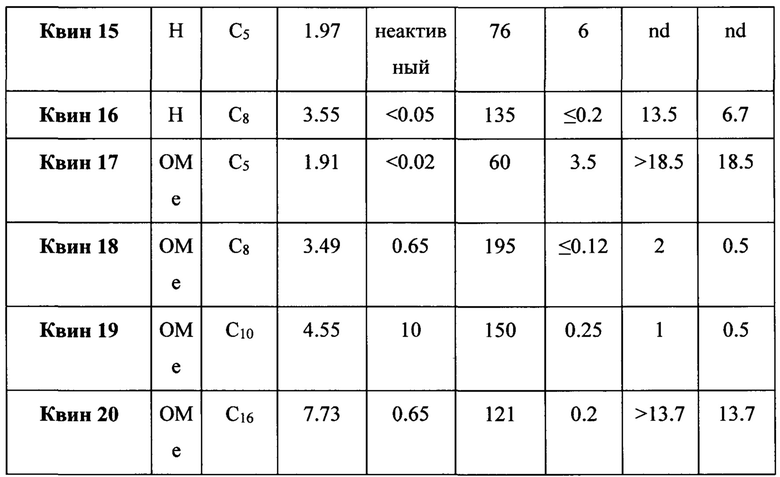
nd - не определялось
Intra mac. - активность внутри макрофагов
MIC - максимальная ингибирующая концентрация
aclogP - рассчитывали при помощи программы ChemdrawUltra 12.0
b - в моксифлоксацине присутствует другая замена в R7 и отсутствует алкильная цепь на терминальном атоме азота
с - представленные здесь данные относительно биологической активности гатифлоксацина и моксифлоксацина соответствуют данным, опубликованным ранее (Poissy et al., Antimicrob. Agents Chemother., 2010, p 4765-71)
d - представленные здесь данные относительно биологической активности ципрофлоксацина в отношении тех же штаммов, которые были описаны в ранее изданной публикации, опубликованы здесь впервые (Poissy, Antimicrob. Agents Chemother., 2010, p. 4765-71)
Все синтезированные фторхинолоны продемонстрировали антибактериальную активность в отношении М. tuberculosis дикого типа, в частности, соединения Квин 9, 16, 18, 19 и 20 (МИК<1 мкМ). Ни одно из указанных соединений не ингибировало (только в очень высоких концентрациях) ДНК гиразы М. tuberculosis дикого типа. Более того, два из представленных новых фторхинолонов (Квин 18 и Квин 19) представляют особый интерес. Они продемонстрировали высокую антибактериальную активность как в отношении бактерий дикого типа (H37Rv штамм), так и в отношении хинолон-устойчивых штаммов М. tuberculosis (D94V и A90V штаммы).
Все вместе полученные результаты позволяют предположить (i) другой (или дополнительный) механизм действия препаратов по сравнению с традиционными хинолонами, и (ii) высокую антибактериальную активность в отношении штаммов М. tuberculosis, включая штаммы XDR-TB (то есть штаммы, устойчивые к хинолонам).
В) Для соединений 15-20, Квин 18 и Квин 19
Материалы и Методы
Реагенты
Моксифлоксацин (МОХ), используемый в качестве позитивного контроля, был предоставлен его производителем (Bayer Pharma, Франция).
Противомикробная активность in vitro
М. tuberculosis H37Rv дикого типа и мутантные штаммы, несущие мутации ДНК гиразы, которые обычно встречаются у штаммов, устойчивых к хинолонам, в клинической практике (GyrA A90V и GyrA D94G и GyrB D500N), а также мультирезистентные клинические штаммы М. tuberculosis (MDR; что определяется как устойчивость к ключевым противомикробным лекарственным средствам - изониазиду и рифампицину; 2 из 3-х штаммов также являются устойчивыми к аминогликозидам и, следовательно, классифицируются как пре-XDR штаммы (с широкой лекарственной устойчивостью); GV1503014223, KC1503006247, ХСС1503082245) выращивали на среде Левенштейна-Йенсена. Максимальные ингибирующие концентрации (МИК) определяли методом пропорций как описано ранее (Guillemin, I.; Jarlier V.; Cambau Е. Antimicrob. Agents Chemother. 1998, 42, 2084). 103 и 105 КОЕ распыляли на 7H11 агар, обогащенный 10% комплексом олеиновая кислота+альбумин+декстроза+каталаза, и содержащий двухкратные разведения изучаемого соединения. Колонии подсчитывали через 3-30 дней после инкубации при температуре 37°С (в зависимости от вида микобактерий). МИК определяли как концентрацию лекарственного препарата при которой происходило уменьшение роста бактерий на 1% или менее по сравнению с ростом, который наблюдался для контрольной культуры на среде, не содержащей лекарственного препарата culture (Inderlied, С.В.; Nash K.A. In Antibiotics in laboratory medicine, 4th ed.; M. D. V. Lorian, Eds; The Williams & Wilkins Co.; Baltimore, Md., 1996; pp 127-175).
Результаты
Результаты представлены в Таблице 3.
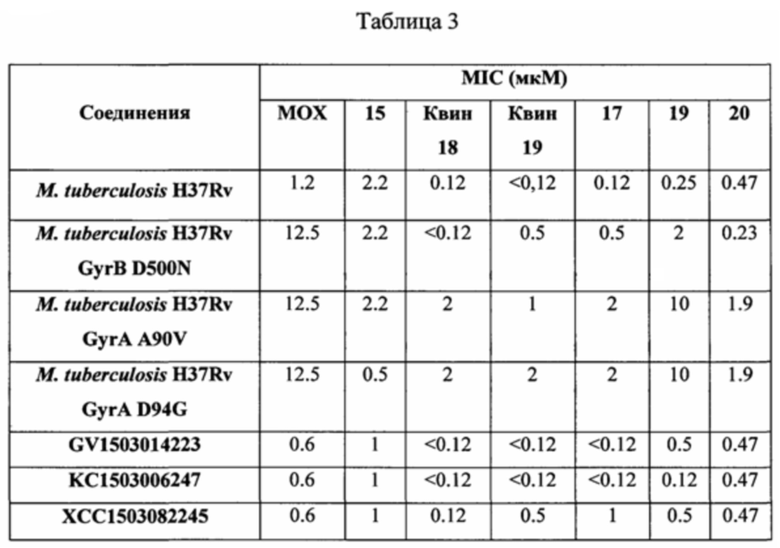
МИК - максимальная ингибирующая концентрация
Все новые синтезированные соединения продемонстрировали антибактериальную активность в отношении штаммов М. tuberculosis дикого типа. В частности, МИК Квин 18, Квин 19, соединений 17, 19 и 20 была ниже, чем МИК моксифлоксацина (1,2 мкМ). Интересно, что все указанные соединения демонстрировали высокую антибактериальную активность против хинолон-устойчивых штаммов М. tuberculosis (GyrB D500N, GyrA D94G и GyrA A90V штаммы). Необходимо отметить, что для Квин 18, Квин 19, соединений 17, 19 и 20 МИК для ФХ-устойчивых штаммов была такой же или даже ниже, в зависимости от мутации, но при этом оставалась на уровне ≤2 мкМ для Квин 18, Квин 19, соединений 17 и 20, то есть была такой же, как и МИК моксифлоксацина в отношении штаммов дикого типа. Очень интересно, что МИК большинства потенциально активных соединений была одинакова для MDR устойчивых штаммов и штаммов H37Rv М. tuberculosis дикого типа.
2.2 Определение минимальной эффективной дозы (МЭД) для Квин 18 и Квин 19 на мышиной ТБ модели
Материалы и методы
4-х недельных мышей женского пола линии Balb/C/J инфицировали штаммом H37Rv М. tuberculosis посредством внутривенного введения в дозе 106 КОЕ. Через день после инфицирования (Д1) 10 мышей умерщвляли для того, чтобы определить точные исходные величины, такие, как масса селезенки и количество КОЕ в легких. Оставшихся мышей распределяли на следующие группы: негативный контроль, не получавший лечения, для мониторинга выживаемости; позитивный контроль, получавший лечение в течение 1 месяца изониазидом (INH) в дозе 25 мг/кг в день, десять тестируемых групп, получавших лечение Квин 18 и Квин 19 в увеличивающихся дозировках (25 мг/кг/д; 50 мг/кг/д; 100 мг/кг/д; 150 мг/кг/д), которые вводили перорально через зонд, и две дополнительные тестируемые группы, которые получали лечение Квин 18 посредством внутривенного введения в дозе 25 мг/кг/д и 100 мг/кг/д. В каждой группе было 10 мышей, которые получали лечение с первого дня (Д1) по 28 день (Д28) 5 дней в неделю. Для оценки тяжести инфекции и эффективности лечения использовали такие параметры как выживаемость, масса селезенки, наличие крупных очагов поражения в легких и число КОЕ в легких.
Минимальная эффективная доза определяется как минимальная доза, способная предотвратить гибель животных, увеличение селезенки и появление обширных повреждений легких. Подсчет КОЕ считается более точным способом для определения предела эффективных доз для Квин 18 и Квин 19. Данные о выживаемости в различных группах представляли на кривой Каплана-Мейера и сравнивали с использованием лог-рангового критерия.
Результаты
Результаты представлены в Таблице 4 и на Фиг. 1-2.
Выживаемость среди мышей, инфицированных штаммом H37Rv М. tuberculosis и получавших лечение как Квин 18, так и Квин 19, независимо от дозы (25, 50, 100 и 150 мг/кг/д) и способа введения (через зонд, Фиг. 1, или внутривенно, Фиг. 2, для Квин 18) была статистически выше по сравнению с контрольной группой и аналогична выживаемости животных в группе, получавшей лечение контрольным препаратом, то есть изоциазидом в дозе 25 мг/кг/д (кривые можно наложить одну на другую).
Вес мышей был ниже в группах, не получавших лечения, по сравнению с весом животных в тестируемых группах, в которых вес животных был одинаковым как у животных, получавших лечение новыми соединениями, так и у животных, получавших лечение изониазидом. Массы селезенки, увеличение которой является косвенным признаком инфекции, была выше в группе, не получавшей лечения, по сравнению с группами, получавшими лечение как двумя изучаемыми соединениями через зонд, так и изониазидом, при этом масса селезенки мышей, получавших внутривенное лечение Квин 18, была аналогична массе селезенки животных из группы, не получавшей лечения.
Зависимость выживаемости от дозы представлена на Фиг. 1 и 2. Выживаемость была одинаковой у животных из тестируемых групп, при этом выживаемость существенно различалась от выживаемости животных из группы, не получавшей лечение, и была сравнима с выживаемостью животных из группы позитивного контроля, получавшей лечение изониазидом, даже в той же дозе, то есть 25 мг/кг/д.
Заключение
Полученные результаты показывают, что Квин 18 и Квин 19 при пероральном введении демонстрируют чрезвычайную эффективность на мышиных моделях туберкулеза. Создание нового лекарственного средства для перорального применения, в особенности для лечения туберкулеза, вызванного MDR и XDR штаммами, в настоящее время действительно необходимо, и у Квин 18 и Квин 19 имеются реальные перспективы в этом отношении.
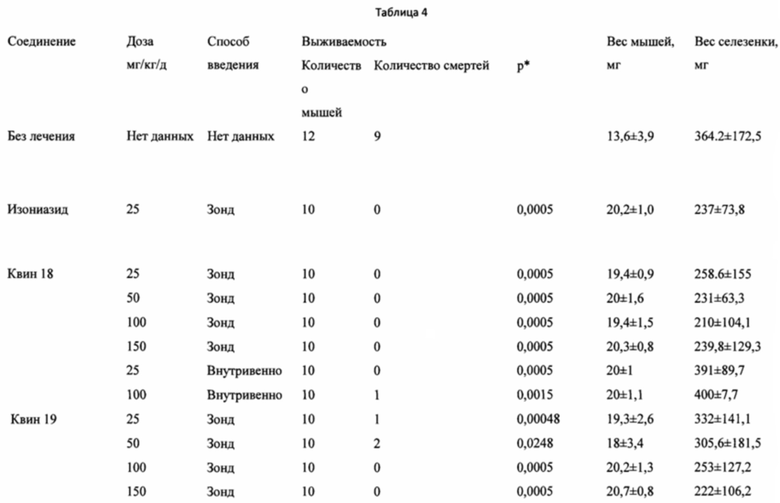
2.3. Оценка острой токсичности Квин 18 и Квин 19 на здоровых мышах женского пола линии CD-1 после однократного введения.
Цель исследования состояла в том, чтобы оценить острую токсичность двух новых соединений (Квин 18 и Квин 19) у здоровых мышей женского пола линии CD-1 после однократного перорального введения на День 0 (Д0).
Предварительно изучали растворимость обоих соединений, Квин 18 и Квин 19, в подходящих для перорального введения растворителях для того чтобы в последующем получить тонко дисперсную суспензию с максимальной концентрацией (МК) каждого соединения. Растворимость каждого соединения в каждом носителе оценивали путем наблюдения за потенциальным преципитатом. В исследовании изучали девять традиционных эксципиента Этанол 5%, Глицерин 15%, Полиэтиленгликоль 300 50%, Полиэтиленгликоль 400 9%, Полисорбат 80 0.4%), Пропиленгликоль 68%, 2-гидроксипропил-циклодекстрин 20%, Метилцеллюлоза 0.5% и кукурузное масло).
Наилучшие результаты были получены для полисорбата 80. 04% и кукурузного масла, именно эти эксципиенты обеспечивали необходимое растворение соединений Квин 18 и Квин 19, соответственно.
Первая доза (МК) представляла собой максимальную концентрацию соединения которая необходима для получения тонкодисперсной суспензии тестируемых соединений в адаптированном для использования в ветеринарии. В соответствии с полученными результатами для наибольших доз каждого вещества, для каждого из них концентрация может быть уменьшена или увеличена.
Токсичность лечения оценивалась путем мониторинга животных (общие проявления фармакологических и токсических эффектов, клинические проявления, смертность и очевидные признаки токсического действия, а также два раза в неделю контроль клинических симптомов и массы тела) до завершения эксперимента (День 14). В конце эксперимента животных умерщвляли и производили аутопсию.
Материалы и методы
Тестируемые соединения
Оба тестируемых соединения Квин 18 (MW: 472.13.6 г/моль, чистота >99%) и Квин 19 (MW: 501.70 г/моль, чистота >99%), были предоставлены компанией C.RIS Pharma и хранились при комнатной температуре.
Получение и содержание животных
Тридцать шесть (36) мышей женского пола линии CD-1 (RjOr1:SWISS), 7-ми недельного возраста получали в лаборатории Janvier (Le Genest-Saint-Isle, Франция). Животных в течение пяти дней содержали в привычных для них условиях до начала исследования. Содержание и использование животных контролировалось Министерством сельского хозяйства и научных исследований Франции (соглашение No. В 35 288-1). Все эксперименты над животными проводились в соответствии с этическими руководствами по использованию лабораторных животных.
Условия окружающей среды
Животных содержали в помещениях с контролируемыми параметрами, такими, как температура (22±3°С), влажность (50±20%), световой период (12 ч свет/12 ч темнота) и воздухообмен. Система кондиционирования воздуха запрограммирована на 14-кратный воздухообмен в течение часа, без рециркуляции. Свежий воздух извне проходит черещ фильтры перед точной подачей в каждое помещение. Весь персонал работающий в указанных принятых условиях следует специфическим руководствам, касающимся гигиены и рабочей одежды, особенно при входе в помещения, где содержатся животные, в соответствии со стандартным действующим протоколом No GEN-006.
Содержание животных
Животных содержали в макролоновых клетках (03120133, Genestil, Франция), оборудованных для поставки питания и воды. Стандартный размер клеток составлял 820 см2, при этом в одной клетке содержали максимум 10 животных, согласно стандартномй действующему протоколу N°. ТЕС-106. Подстилками для животных служили деревянные стружки (Toplit select fine, SAFE, Франция), которые меняли один раз в неделю.
Для того, чтобы улучшить условия быта животных использовали некоторые материалы, позволяющие улучшить состояние клеток, а именно полоски из древесины тополя (TOP WOODWOOL, Safe, Франция).
Кормление и питьевой режим
Еду для животных получали в компании SAFE Франция. Использовали контролируемые гранулы типа А04. Еду предоставляли не ограничено, помещали ее в металлическую крышечку на верхней части клетки. Воду также давали неограниченно, из бутылок, оснащенных резиновыми ограничителями и специальными поильниками. Бутылки с водой мыли и меняли один раз в неделю.
Идентификация животных в клетках
Мышей идентифицировали при помощи одной 8 мм ISO метки (Genestil, Франция), в соответствии со стандартным действующим протоколом No. ТЕС-167. Метки считывались регистратором GES (Rumitag, Испания). В случае поломки или не считывания метки в любое время, животному вводили новую. Каждую клетку помечали специфическим кодом, соответствующим номеру исследования и номеру группы.
Распределение животных по группам
После периода акклиматизации мышей взвешивали и распределяли по весу на 12 групп (по 3 животных в группе), в соответствии с действующим протоколом No. ТЕС-086. Средний вес животных в группе статистически не различался.
Подготовка изучаемых соединений и носителей
Носитель A (VA) представлял собой раствор полисорбата 80 0.4%. Его изготавливали путем взвешивания полисорбата 80 и разведения в NaCl 0.9%. Носитель В (VB) представлял собой готовое к использованию кукурузное масло.
Квин 18 заготавливали в концентрации 10 мг/мл или 20 мг/мл путем растворения нужного количества Квин 18 в полисорбате 80 0.4% с последующей обработкой ультразвуком или без нее. Квин 18 использовали чистым или разведенным в полисорбате 80 0.4% для получения рабочих концентраций. Квин 18 хранили при комнатной температуре в течение всего периода лечения, и вводили при комнатной температуре раствора.
Квин 19 заготавливали в концентрации 10 мг/мл или 20 мг/мл путем растворения нужного количества Квин 19 в кукурузном масле с последующей обработкой ультразвуком или без нее. Квин 19 использовали чистым или разведенным в кукурузном масле для получения рабочих концентраций. Квин 19 хранили при комнатной температуре в течение всего периода лечения, и вводили при комнатной температуре раствора.
Мыши из каждой группы получали соединения перорально (per os) в объеме 10 мл/кг в соответствии со стандартным действующим протоколом No. ТЕС-078. Рекомендуемый объем для введения per os (РО) для мышей составляет 10 мл/кг.
Ниже и в Таблице 5 представлены обозначения экспериментальных групп:
Тестируемое соединение Квин (Quin) 18:
Группа VA1 получала лечение полисорбатом 80 0.4% (носитель А) в объеме 10 мл/кг в соответствии с протоколом лечения Q1Dx1.
Группа A1 получала дозу 100 мг/кг Квин 18 в объеме 10 мг/мл (с обработкой ультразвуком) в соответствии с протоколом лечения Q1Dx1.
Группа А2 получала дозу 200 мг/кг Квин 18 в объеме 20 мг/мл (с обработкой ультразвуком) в соответствии с протоколом лечения Q1Dx1.
Группа A3 получала дозу 100 мг/кг Квин 18 в объеме 10 мг/мл (без обработки ультразвуком) в соответствии с протоколом лечения Q1Dx1.
Группа VА4 получала полисорбат 80 0.4% (носитель А) в дозе 10 мл/кг в соответствии с протоколом лечения 2Q1Dx1 (с 2-х часовым интервалом между двумя введениями).
Группа А4 получала дозу 200 мг/кг Квин 18 в объеме 10 мг/мл (без обработки ультразвуком) в соответствии с протоколом лечения 2Q1Dx1 (с 2-х часовым интервалом между двумя введениями).
Тестируемое соединение Квин (Quin) 19:
Группа VВ1 получала кукурузное масло (Носитель В) в объеме 10 мл/кг в соответствии с режимом лечения Q1Dx1.
Группа В1 получала дозу 100 мг/кг тестируемого соединения в оьъеме 10 мг/мл (с обработкой ультразвуком) в соответствии с режимом лечения Q1Dx1.
Группа В2 получала дозу 200 мг/кг тестируемого соединения в объеме 20 мг/мл (с обработкой ультразвуком) в соответствии с режимом лечения Q1Dx1.
Группа В3 получала дозу 200 мг/кг тестируемого соединения в объеме 20 мг/мл (без обработки ультразвуком) в соответствии с режимом лечения Q1Dx1.
Группа VВ4 получала кукурузное масло (Носитель В) в объеме 10 мл/кг в соответствии с режимом лечения 2Q1Dx1 (с 2-х часовым интервалом между двумя введениями).
Группа В4 получала дозу 400 мг/кг тестируемого соединения в объеме 20 мг/мл (без обработки ультразвуком) в соответствии с режимом лечения 2Q1Dx1 (с 2-х часовым интервалом между двумя введениями).

На Д0 мышам начинали лечение путем перорального введения тестируемых соединений.
Мониторинг животных
Заболеваемость, смертность и явные признаки токсичности у мышей оценивали ежедневно начиная с Д0 до окончания эксперимента (Д14 или Д15).
Детальное наблюдение за изменениями поведения и клиническими симптомами осуществляли два раза в неделю, начиная с Д0 и до окончания эксперимента (Д14 или Д15) в соответствии со стандартным действующим протоколом No. ТЕС-192.
Массу тела животных регистрировали на Д1, Д2 и затем два раза в неделю до окончания эксперимента (Д14 или Д15) в соответствии со стандартным действующим протоколом No. ТЕС-108. Потерю массы тела оценивали по сравнению с исходной массой каждого животного на день начала эксперимента Д0. В течение эксперимента животных умерщвляли если наблюдалось хотя бы одно из указанных явлений:
- Признаки страдания животного (кахексия, слабость, животному трудно передвигаться и принимать пищу),
- Токсическое действие препарата (съеживания, судороги),
- 25% потери массы тела на любой день эксперимента.
Умерщвление животных
На Д14 или Д15, животных умерщвляли при помощи ингаляции СО2 и производили вскрытие.
Результаты Анализ массы тела
Результаты, выраженные при помощи графиков, отражающих среднюю массу тела (СМТ), представлены на Фиг. 3.
У мышей из Группы VA1, получавших лечение носителем А, полисорбатом 80 0.4% (10 мл/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. Средняя масса тела (СМТ) с Д0 до Д14 составляла 31.6±1.23 г и 32.13±1.79 г, соответственно. Тестируемое соединение носитель А, полисорбат 80 0,4% (10 мл/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы А1, получавших лечение Квин 18/полисорбат 80 0.4% (10 мг/мл, с обработкой ультразвуком, 10 мл/кг, 100 мг/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д14 составляла 31.5±1.48 г и 31.77±0.76 г, соответственно. Тестируемое соединение Квин 18/полисорбат 80 0.4% (10 мг/мл, с обработкой ультразвуком, 10 мл/кг, 100 мг/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы А2, получавших лечение Квин 18/полисорбат 80 0.4% (20 мг/мл, с обработкой ультразвуком, 10 мл/кг, 200 мг/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д14 составляла 30.2±1.15 г и 32.6±1.35 г, соответственно. Тестируемое соединение Квин 18/полисорбат 80 0.4% (20 мг/мл, с обработкой ультразвуком, 10 мл/кг, 200 мг/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы A3, получавших лечение Квин 18/полисорбат 80 0.4% (10 мг/мл, без обработки ультразвуком, 10 мл/кг, 100 мг/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д15 составляла 29.9±1.25 г и 32.17±1.94 г, соответственно. Тестируемое соединение Квин 18/полисорбат 80 0.4% (10 мг/мл, с обработкой ультразвуком, 10 мл/кг, 100 мг/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы VA4, получавших лечение носителем А, полисорбатом 80 0.4% (10 мл/кг, РО, 2Q1Dx1, 2-х часовой интервал между двумя введениями) не наблюдалось какой либо потери массы тела. Средняя масса тела (СМТ) с Д0 до Д14 составляла 29.83±1.56 г и 32.17±2.91 г, соответственно. Тестируемое соединение носитель А, полисорбат 80 0,4% (10 мл/кг, РО, 2Q1Dx, 2-х часовой интервал между двумя введениями) переносилось хорошо.
У мышей из Группы А4, получавших лечение Квин 18/полисорбат 80 0.4% (20 мг/мл, с обработкой ультразвуком, 10 мл/кг, 200 мг/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д14 составляла 29.9±0.89 г и 31.33±1.63 г, соответственно. Тестируемое соединение Квин 18/полисорбат 80 0.4% (20 мг/мл, с обработкой ультразвуком, 10 мл/кг, 200 мг/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы A B1, получавших лечение носителем В, кукурузным маслом (10 мг/мл, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д14 составляла 30.57±1.32 г и 31.5±1.64 г, соответственно. Тестируемое соединение Носитель В, кукурузное масло (10 мг/мл, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы В1, получавших лечение Квин 19/кукурузное масло (10 мг/мл, с обработкой ультразвуком, 10 мл/кг, 100 мг/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д14 составляла 31.67±2.25 г и 31.4±0.44 г, соответственно. Тестируемое соединение Квин 19/кукурузное масло (10 мг/мл, с обработкой ультразвуком, 10 мл/кг, 100 мг/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы В2, получавших лечение Квин 19/кукурузное масло (20 мг/мл, с обработкой ультразвуком, 20 мл/кг, 200 мг/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д14 составляла 30.67±0.21 г и 31.9±1.15 г, соответственно. Тестируемое соединение Квин 19/кукурузное масло (20 мг/мл, с обработкой ультразвуком, 10 мл/кг, 200 мг/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы В3, получавших лечение Квин 19/кукурузное масло (20 мг/мл, без обработки ультразвуком, 20 мл/кг, 200 мг/кг, РО, Q1Dx1) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д15 составляла 33.6±2.11 г и 35.43±1.99 г, соответственно. Тестируемое соединение Квин 19/кукурузное масло (20 мг/мл, с обработкой ультразвуком, 10 мл/кг, 200 мг/кг, РО, Q1Dx1) переносилось хорошо.
У мышей из Группы VB4, получавших лечение Носителем В, кукурузным маслом (10 мг/мл, РО, 2Q1Dx1, 2-х часовой интервал между двумя введениями) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д14 составляла 31.23±1.23 г и 32±0.7, соответственно. Тестируемое соединение Носитель В, кукурузноемасло (10 мл/кг, РО, 2Q1Dx1, 2-х часовой интервал между двумя введениями) переносилось хорошо.
У мышей из Группы В4, получавших лечение Квин 19/кукурузное масло (20 мг/мл, без обработки ультразвуком, РО, 2Q1Dx1, 2-х часовой интервал между двумя введениями) не наблюдалось какой либо потери массы тела. СМТ с Д0 до Д15 составляла 31.03±1.86 г и 32.53±1, соответственно. Тестируемое соединение Квин 19/кукурузное масло (20 мг/мл, без обработки ультразвуком, РО, 2Q1Dx1, 2-х часовой интервал между двумя введениями) переносилось хорошо.
Мониторинг животных
Мониторинг мышей (оценку заболеваемости, смертности и явных признаков токсичности) проводили два раза в неделю, данные обобщены в Таблице 6. Никаких особенных симптомов выявлено не было.
Вскрытие
В Таблице 7 представлены причины смерти каждого животного, а также макроскопические наблюдения. Каких-либо особенных симптомов у животных, получавших лечение Квин 18 или Квин 19 и/или носителями выявлено не было.
*В группах VA4, у 2 мышей были обнаружены пузырьки воздуха в толстом кишечнике, а в группах VB1 и В1 у одного животного из каждой группы были обнаружены пузырьки воздуха в слепой кишке.
Заключение
Тестируемое соединение Квин 18/полисорбат 80 0,4% хорошо переносилось животными вплоть до объема 200 мг/кг (максимальная растворимость; не было отмечено каких-либо существенных изменений массы тела и других симптомов, а также значимых макроскопических проявлений у мышей женского пола).
Тестируемое соединение Квин 19/кукурузный крахмал хорошо переносилось животными вплоть до объема 400 мг/кг (максимальная растворимость; не было отмечено каких-либо существенных изменений массы тела и других симптомов, а также значимых макроскопических проявлений у мышей женского пола).
Таким образом, результаты исследования показывают, что Квин 18 и Квин 19 прекрасно переносятся мышами, без признаков токсичности в тестируемом режиме дозирования.
| название | год | авторы | номер документа |
|---|---|---|---|
| Антибактериальные средства на основе производных ципрофлоксацина | 2016 |
|
RU2636751C1 |
| ДЕГАЛОГЕНИРОВАННЫЕ СОЕДИНЕНИЯ, АНТИБАКТЕРИАЛЬНОЕ СРЕДСТВО И ТЕРАПЕВТИЧЕСКОЕ СРЕДСТВО НА ИХ ОСНОВЕ, СПОСОБ ПОЛУЧЕНИЯ АНТИБАКТЕРИАЛЬНОГО ИЛИ ЛЕКАРСТВЕННОГО СРЕДСТВА, ПРИМЕНЕНИЕ ДЕГАЛОГЕНИРОВАННЫХ СОЕДИНЕНИЙ ДЛЯ ПОЛУЧЕНИЯ АНТИБАКТЕРИАЛЬНОГО ИЛИ ЛЕКАРСТВЕННОГО СРЕДСТВА | 2001 |
|
RU2298006C2 |
| Соединения фторхинолонового ряда на основе производных пиридоксина, обладающие антибактериальными свойствами | 2019 |
|
RU2713932C1 |
| КОМПОЗИЦИИ ТЕТРАГИДРОХИНОЛИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ БРОМОДОМЕНА ВЕТ | 2014 |
|
RU2727169C2 |
| СРЕДСТВО ПРОТИВ КИСЛОТОУСТОЙЧИВЫХ БАКТЕРИЙ, СОДЕРЖАЩЕЕ ПИРИДОНКАРБОНОВЫЕ КИСЛОТЫ В КАЧЕСТВЕ АКТИВНОГО КОМПОНЕНТА | 2001 |
|
RU2299205C2 |
| ФАРМАЦЕВТИЧЕСКИЕ СОЕДИНЕНИЯ | 2020 |
|
RU2840815C2 |
| ХИНОЛИНИЛ-СОДЕРЖАЩЕЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЕ | 2020 |
|
RU2803116C2 |
| СРЕДСТВО ПРОТИВ КИСЛОТОУСТОЙЧИВЫХ БАКТЕРИЙ, СОДЕРЖАЩЕЕ ГЕТЕРОЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ БЕНЗОКСАЗИНА В КАЧЕСТВЕ АКТИВНОГО КОМПОНЕНТА | 2001 |
|
RU2297420C2 |
| СОЕДИНЕНИЕ ИЗОИНДОЛИН, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2813232C2 |
| ЗАМЕЩЕННОЕ ПРОПАНАМИДНОЕ ПРОИЗВОДНОЕ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СОДЕРЖАЩАЯ ТАКОЕ ПРОИЗВОДНОЕ | 2006 |
|
RU2394560C2 |
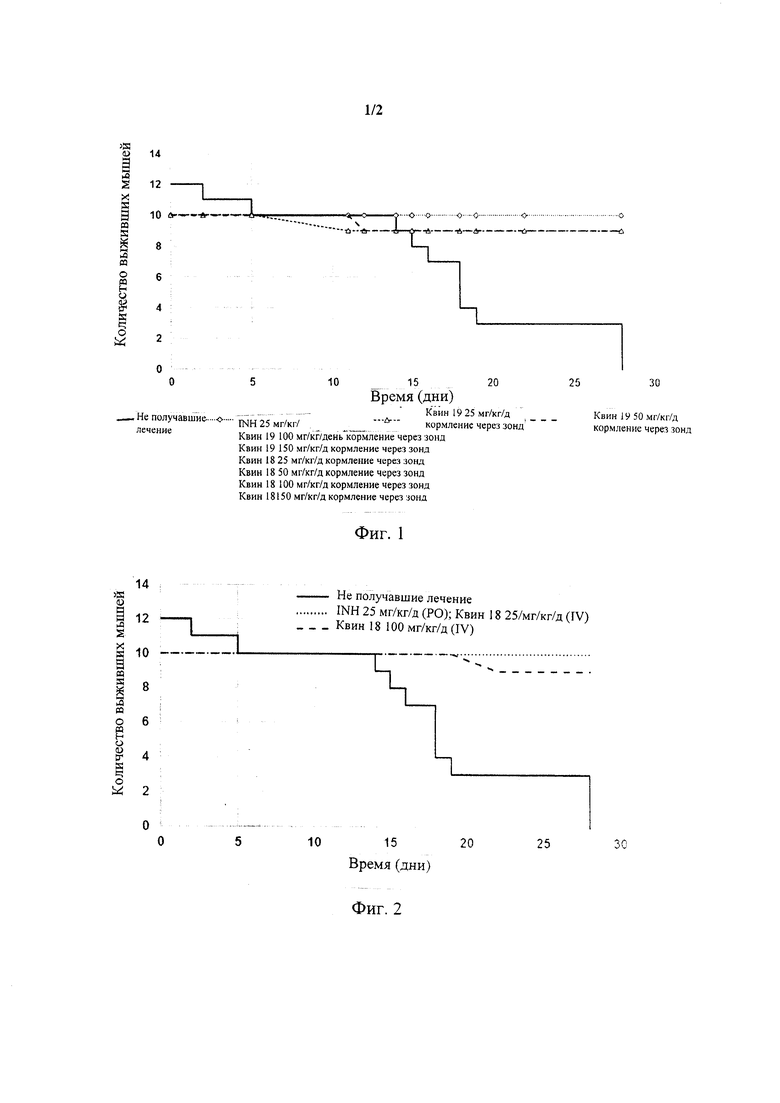
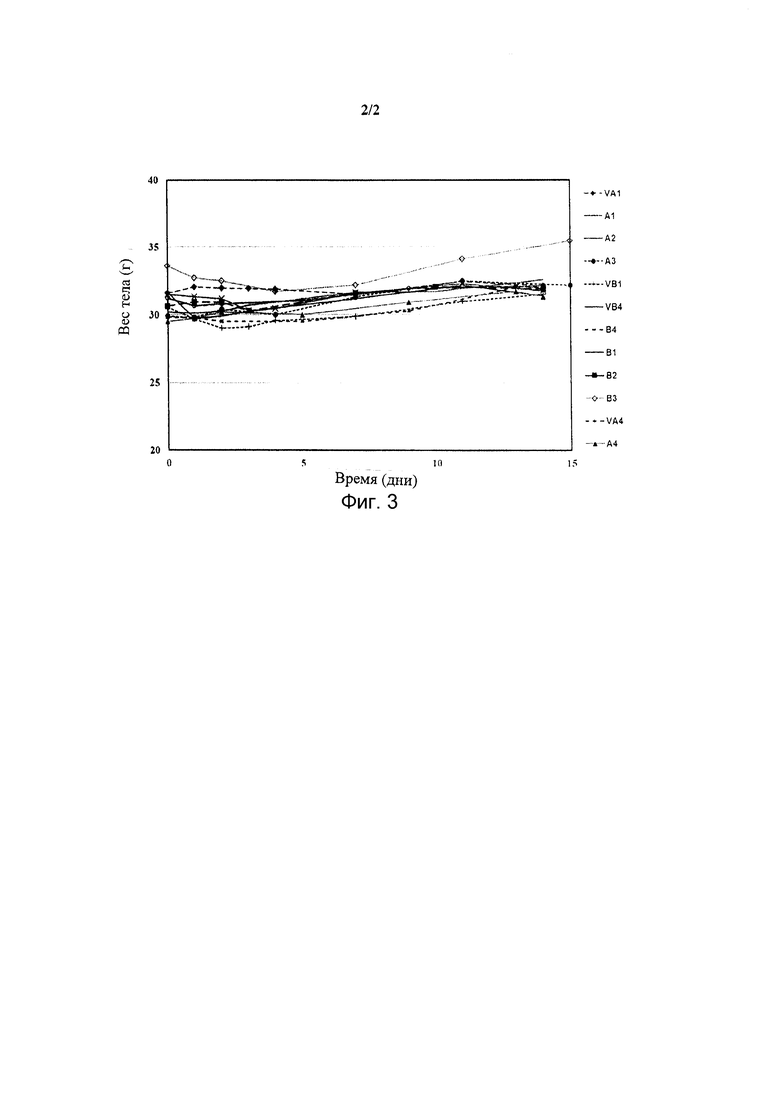
Изобретение относится к новым фторхинолонам, имеющим общую Формулу I:
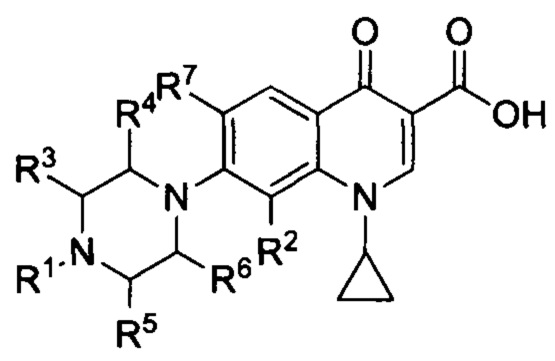 , фармацевтическим композициям или лекарственным средствам, содержащим указанные хинолоны, и их использованию для лечения бактериальной инфекции. Технический результат: получены новые соединения формулы I, пригодные для лечения бактериальной инфекции М. tuberculosis H37Rv. 3 н. и 4 з.п. ф-лы, 3 ил., 7 табл.
, фармацевтическим композициям или лекарственным средствам, содержащим указанные хинолоны, и их использованию для лечения бактериальной инфекции. Технический результат: получены новые соединения формулы I, пригодные для лечения бактериальной инфекции М. tuberculosis H37Rv. 3 н. и 4 з.п. ф-лы, 3 ил., 7 табл.
1. Соединение, имеющее общую Формулу I:
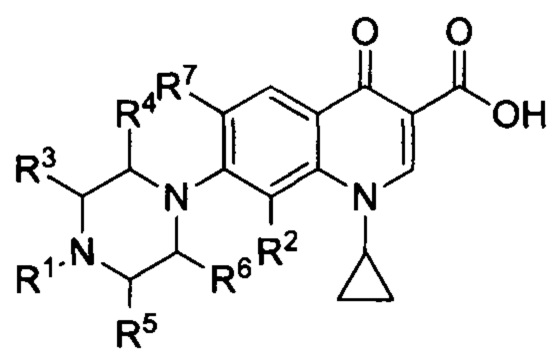
- а также его фармацевтически приемлемые соли, при этом:
- R1 представляет собой насыщенную, незамещенную, разветвленную или неразветвленную алкильную группу, включающую от 8 до 16 атомов углерода;
- R2 представляет собой заместитель, выбранный из группы, включающей водород, или метокси группу;
- R3, R4, R5 и R6 являются одинаковыми и представляют собой водород;
- R7 представляет собой гало, предпочтительно фтор.
2. Соединение по п. 1, имеющее общую формулу III:
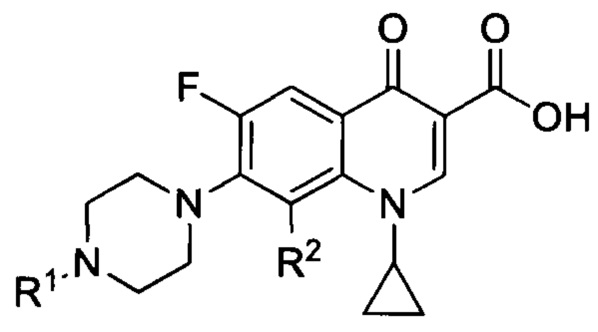
а также его фармацевтически приемлемые соли.
3. Соединение по любому из пп. 1-2, выбранное из группы, включающей:
- 1-циклопропил-6-фтор-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-нонилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-ундецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-7-(4-додецилпиперазин-1-ил)-6-фтор-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-тридецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-тетрадецилпиперазин-1-ил)1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-4-оксо-7-(4-пентадецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-8-метокси-7-(4-октилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-8-метокси-7-(4-нонилпиперазин-1-ил)-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-7-(4-децилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-ундецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-7-(4-додецилпиперазин-1-ил)-6-фтор-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-тридецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-тетрадецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-8-метокси-4-оксо-7-(4-пентадецилпиперазин-1-ил)-1,4-дигидрохинолин-3-карбоксильную кислоту;
- 1-циклопропил-6-фтор-7-(4-гексадецилпиперазин-1-ил)-8-метокси-4-оксо-1,4-дигидрохинолин-3-карбоксильную кислоту
или их фармацевтически приемлемые соли.
4. Фармацевтическая композиция, содержащая по крайней мере эффективное количество одного соединения по любому из пп. 1-3 или его фармацевтически приемлемую соль, а также по крайней мере один фармацевтически приемлемый носитель, растворитель, эксципиент и/или адъювант для лечения бактериальной инфекции М. tuberculosis H37Rv.
5. Лекарственное средство для лечения бактериальной инфекции М. tuberculosis H37Rv, включающее эффективное количество соединения по любому из пп. 1-3 или его фармацевтически приемлемую соль.
6. Соединение по любому из пп. 1-3, фармацевтическая композиция или лекарственное средство по любому из пп. 4-5, предназначенные для лечения бактериальной инфекции М. tuberculosis H37Rv.
7. Соединение, фармацевтическая композиция или лекарственное средство по любому из пп. 1-6, предназначенные для лечения бактериальной инфекции М. tuberculosis H37Rv.
| A | |||
| HAEMERS ET AL | |||
| "Influence of N substitution on antimycobacterial activity of ciprofloxacin.", ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, 34, 3, стр | |||
| 496 - 497, 1990 | |||
| M | |||
| BERMEJO ET AL | |||
| "Validation of a biophysical drug absorption model by the PATQSAR system", JOURNAL OF PHARMACEUTICAL SCIENCES, 88, 4, стр | |||
| Приспособление для выключения электрических цепей катодного генератора | 1922 |
|
SU398A1 |
| AZEMA J ET AL | |||

