Область техники
Настоящее изобретение относится к антагонисту рецептора нейропептида нейрокинина-1 (NK1 или NK-1).
Предшествующий уровень техники
Тахикинин является пептидным лигандом нейрокининовых рецепторов. Рецепторы нейрокинина, такие как NK1, NK2 и NK3, участвуют в различных биологических процессах. Их можно найти в нервной и кровеносной системе и в окружающих тканях млекопитающих. Поэтому изучали модуляцию таких рецепторов для потенциального лечения или профилактики различных заболеваний у млекопитающих. Типичные антагонисты нейрокининовых рецепторов и их применение включают: US 5760018 (1998) (боль, воспаление, мигрень и рвота), US 5620989 (1997) (боль, ноцицепция и воспаление), WO 95/19344 (1995), WO 94/13639 (1994) и WO 94/10165 (1994). Другие типы антагонистов рецептора NK1 включают: Wu et al., Tetrahedron 56, 3043-3051(2000); Rombouts et al., Tetrahedron Letters 42, 7397-7399(2001); и Rogiers et al., Tetrahedron 57, 8971-8981(2001).
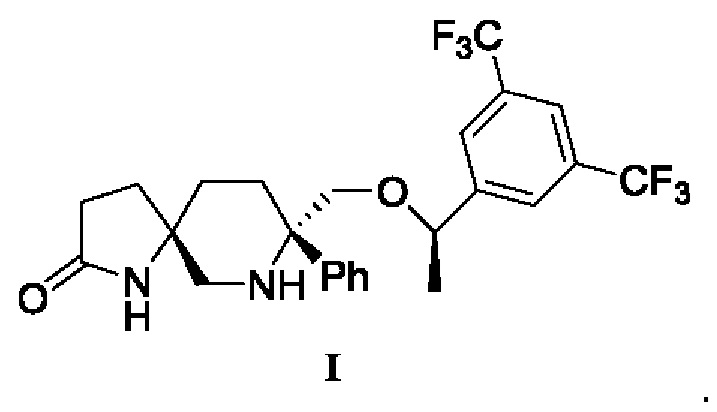
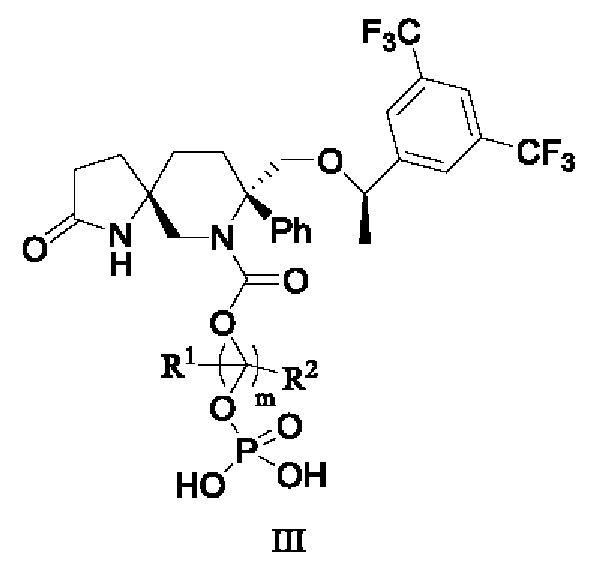
US 7049320 предлагает эффективный и селективный антагонист NK1, (5S,8S)-8-[{(1R)-1-(3,5-бис-(трифторметил)фенил)-этокси}-метил]-8-фенил-1,7-диазаспиро[4.5]дец-2-он (соединение формулы I) с полезными терапевтическими и фармакологическими свойствами и хорошей метаболической стабильностью, который может быть в форме свободного основания или фармацевтически приемлемой соли и подходит для преперата для парентерального введения,
US 9101615 предлагает пролекарство соединения формулы I, то есть пролекарство и его соль, в которых свободный амин (или два амина) соединения формулы I замещен группой, выбранной из -Y и -X, где Y выбран из группы, состоящей из -Р(O)(ОН)2, -S(O)n1R1, -C(O)(C1-6 алкил)Х, -C(O)(C1-6 алкил)(арил) и -C(O)OR4; X выбран из группы, состоящей из -NR2R3, -Р(O)(ОН)2 и -S(O)n1R1; R1 представляет собой Н или С1-6 алкил; R2 представляет собой Н или С1-6 алкил; R3 представляет собой Н или C1-6 алкил; R4 представляет собой Н или C1-6 алкил; n1 равно 0-4. Пролекарство можно использовать в подходящей жидкой композиции (включая или исключая парентеральный носитель для доставки) для лечения пациентов, нуждающихся в этом.
В другой стороны, после попадания лекарственного средства в организм человека возникает лекарственный гемолиз в результате массивного разрушения эритроцитов, вызванного иммунными факторами. Клинически проявляющимися симптомами гемолиза являются анемия, желтуха, моча, похожая на соевый соус, и тому подобное. Лекарственно-индуцированную гемолитическую анемию можно разделить на следующие три типа: (1) лекарственно-индуцированный иммунитет, приводящий к опосредованным антителами гемолитическим реакциям; (2) действие лекарства на эритроциты с дефицитом генетического фермента (например, с дефицитом G6PD); (3) лекарственная гемолитическая реакция на аномальный гемоглобин. Ключом к лечению таких заболеваний является прекращение использования соответствующих лекарств и контроль возникновения гемолиза с целью предупреждения возникновения осложнений. Чтобы решить проблему низкой растворимости соединения формулы I при физиологических значениях рН, исследователи использовали состав на основе сорастворителя, содержащий каптизол, пропиленгликоль и этанол, для значительного улучшения растворимости соединения 1, но состав на основе сорастворителя обладает выраженным гемолитическим эффектом при внутривенном введении. CN 102573475 раскрывает улучшенную формулу, содержащую 15-гидроксистеарат полиэтиленгликоля и триглицериды со средней длиной цепи. Однако, даже если соединение формулы I получают в виде пролекарства, содержащего фосфаты, гемолитический эффект фармацевтической композиции все же не устраняется полностью.
В настоящей заявке предложено новое пролекарственное соединение антагониста NK1, которое эффективно при лечении различных физиологических нарушений, состояний и заболеваний и имеет минимальные побочные эффекты.
Сущность изобретения
Настоящее изобретение предлагает соединение формулы II:
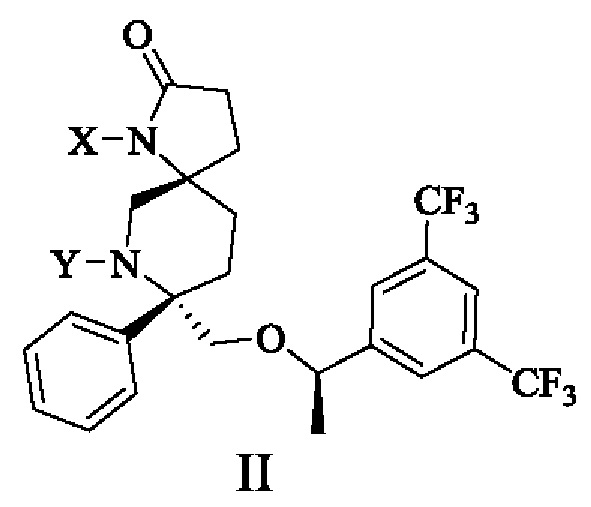
или его фармацевтически приемлемую соль, или его стереоизомер, ротамер или таутомер, или его дейтерид,
где X выбран из группы, состоящей из водорода, гетероциклила, арила, гетероарила, -C(O)OAmR3, -C(O)NR4AmR3, -Am[C(R1)(R2)]C(O)OAnR3, -AmOC(O)[C(R1)(R2)]AnR3, -AmC(O)NR4AnR3, -AmNR4C(O)AnR3 и -AmR5, где указанный гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
Y выбран из группы, состоящей из водорода, -C(O)OAmR3, -C(O)NR4AmR3, -Am[C(R1)(R2)C(O)OAnR3, -AmOC(O)[C(R1)(R2)]AnR3, -AmC(O)NR4R3, -AmNR4C(O)AnR3 и -AmR5;
А независимо выбран из группы, состоящей из -C(R1)(R2)(B)p- и -(B)qC(R1)(R2)-, каждый R1, R2 и R4 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где указанный алкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
R3 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила, гетероарила, поли(оксиэтиленокси)(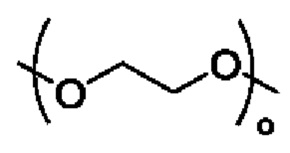 ), поли(этиленокси)(
), поли(этиленокси)(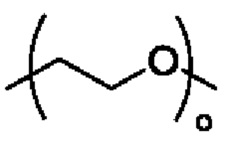 ), OPO(R6)2, PO(R6)2, OSO2(R6)2, SO2(R6)2, OC(O)R6 и C(O)OR6, где указанный алкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
), OPO(R6)2, PO(R6)2, OSO2(R6)2, SO2(R6)2, OC(O)R6 и C(O)OR6, где указанный алкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
R5 выбран из группы, состоящей из гетероциклила, гетероарила, OSO2R7, OC(O)R7, SR', 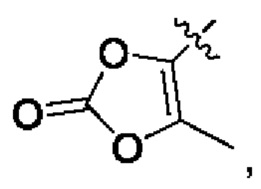 SO2R' и NR'(R");
SO2R' и NR'(R");
каждый R6 независимо выбран из группы, состоящей из водорода, гидроксила, алкила, циклоалкила, гетероциклила, арила, гетероарила, алкоксила, гидроксиалкила и NR'(R"), где указанный алкил, гидроксиалкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
каждый R7 независимо выбран из группы, состоящей из алкила, гидроксила, циклоалкила, гетероциклила, арила, гетероарила, гидроксиалкила и NR'(R"), где указанный алкил, гидроксиалкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
каждый R' и R" независимо выбран из группы, состоящей из водорода, гидроксила, алкила (предпочтительно выбран из группы, состоящей из C1-12 алкила, включая, но не ограничиваясь этим, метил, этил или изопропил), алкоксила (предпочтительно выбран из группы, состоящей из С1-12 алкоксила), алкенила и ацила;
каждый В независимо выбран из группы, состоящей из О, N и SC(O);
каждый m, n и о независимо выбран из группы, состоящей из 1~10, и может быть равен 1, 2, 3, 4, 5, 6, 7, 8, 9 или 10;
каждый р и q независимо выбран из группы, состоящей из 0 и 1; и
X и Y одновременно не являются водородом.
Соединение формулы II согласно настоящему изобретению обладает лучшей растворимостью, чем исходное лекарственное средство, соединение формулы I, и, таким образом, подходит для внутривенного введения. Кроме того, описанное выше соединение будет разлагаться и высвобождать исходное лекарство в физиологических условиях при попадании в организм человека после приготовления внутривенного препарата, что задерживает высвобождение лекарственного средства и продлевает период высвобождения лекарственного средства.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II X выбран из группы, состоящей из водорода, гетероциклила, арила, гетероарила, -C(O)O[C(R1)(R2)(O)p]mR3, -C(О)NR4[C(R1)(R2)(O)p]mR3, -[C(R1)(R2)(O)p]mC(O)[C(R1)(R2)(O)p]nR3, -[C(R1)(R2)(O)p]m[C(R1)(R2)]C(O)[(O)qC(R1)(R2)nR3, -[C(R1)(R2)(O)p]mC(O)NR4[C(R1)(R2)(O)p]nR3 и -[C(R1)(R2)(O)p]mR5, где указанный алкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила (предпочтительно выбраного из C1-12 алкила, включая, но не ограничиваясь этим, метил, этил или изопропил), циклоалкила (предпочтительно выбраного из C1-12 циклоалкила, такого как циклогексил и циклопентанил), алкоксила (предпочтительно выбраного из C1-12 алкоксила), гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
Y выбран из группы, состоящей из водорода, -C(O)O[C(R1)(R2)(O)p]mR3, -C(O)NR4[C(R1)(R2)(O)p]mR3, -[C(R1)(R2)(O)p]mC(O)[C(R1)(R2)(O)p]nR3, -[C(R1)(R2)(O)p]mC(O)[(O)qC(R1)(R2)]nR3, -[C(R1)(R2)(O)p]mC(O)NR4[C(R1)(R2)(O)p]nR3 и -[C(R1)(R2)(O)p]mR5, и X и Y одновременно не являются водородом.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из группы, состоящей из -C(O)O[C(R1)(R2)]mR3, -C(O)NR4[C(R1)(R2)]mR3, -[C(R1)(R2)O]mC(O)[C(R1)(R2)]nR3, -[C(R1)(R2)]mC(O) [OC(R1)(R2)]nR3, -[C(R1)(R2)N]mC(O)[C(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[OC(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[NC(R1)(R2)]nR3, -[C(R1)(R2)(O)p]mC(O)NR4[C(R1)(R2)(O)p]nR3 и -[C(R1)(R2)(O)p]nR5, X представляет собой водород или 3-6 членный гетероциклил.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из группы, состоящей из -C(O)O[C(R1)(R2)]mR3, -C(O)NR4[C(R1)(R2)]mR3, -[C(R1)(R2)O]mC(O)[C(R1)(R2)]nR3, -[C(R1)(R2)]mC(O)[OC(R1)(R2)]nR3, -[C(R1)(R2)N]mC(O)[C(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[OC(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[NC(R1)(R2)]nR3, -[C(R1)(R2)(O)p]mC(O)NR4 [C(R1)(R2)(O)p]nR3 и -[C(R1)(R2)(O)p]nR5, X представляет собой водород или 3-6 членный гетероциклил; каждый m, n и o независимо выбран из группы, состоящей из 1, 2, 3, 4, 5 и 6; каждый p и q независимо выбран из 0.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из группы, состоящей из -C(O)O[C(R1)(R2)]mR3, -C(O)NR4[C(R1)(R2)]mR3, -[C(R1)(R2)O]mC(O)[C(R1)(R2)]nR3, -[C(R1)(R2)]mC(O)[OC(R1)(R2)]nR3, -[C(R1)(R2)N]mC(O)[C(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[OC(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[NC(R1)(R2)]nR3, -[C(R1)(R2)(O)p]mC(O)NR4[C(R1)(R2)(O)p]nR3 и -[C(R1)(R2)(O)p]nR5, X представляет собой водород или 3-6 членный гетероциклил; каждый m, n и о независимо выбран из группы, состоящей из 1, 2, 3, 4, 5 и 6; каждый р и q независимо выбран из 1.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из группы, состоящей из -C(O)O[C(R1)(R2)]R3, -C(O)NR4[C(R1)(R2)]R3, -[C(R1)(R2)O]C(O)[C(R1)(R2)]R3, -[C(R1)(R2)]C(O)[OC(R1)(R2)]R3, -[C(R1)(R2)N]C(O)[C(R1)(R2)]R3, [C(R1)(R2)N]C(O)[OC(R1)(R2)]R3, [C(R1)(R2)N]C(O)[NC(R1)(R2)]R3, -[C(R1)(R2)(O)p]C(O)NR4[C(R1)(R2)(O)p]R3 и -[C(R1)(R2)(O)p]R5, X представляет собой водород или 3-6 членный гетероциклил.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из группы, состоящей из -C(O)O[C(R1)(R2)]2R3, -C(O)NR4[C(R1)(R2)]2R3, -[C(R1)(R2)O]2C(O)[C(R1)(R2)]2R3, -[C(R1)(R2)]2C(O)[OC(R1)(R2)]2R3, -[C(R1)(R2)N]2C(O)[C(R1)(R2)]2R3, [C(R1)(R2)N]2C(O)[OC(R1)(R2)]2R3, [C(R1)(R2)N]2C(O)[NC(R1)(R2)]2R3, -[C(R1)(R2)(O)p]2C(O)NR4[C(R1)(R2)(O)p]2R3 и -[C(R1)(R2)(O)p]2R5, X представляет собой водород или 3-6 членный гетероциклил.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II X выбран из группы, состоящей из -C(O)O[C(R1)(R2)]mR3, -C(O)NR4[C(R1)(R2)]mR3, -[C(R1)(R2)O]mC(O)[C(R1)(R2)]nR3, -[C(R1)(R2)]mC(O)[OC(R1)(R2)]nR3 и -[C(R1)(R2)]nR5, Y представляет собой водород.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II X выбран из группы, состоящей из -C(O)O[C(R1)(R2)]mR3, -C(O)NR4[C(R1)(R2)]mR3, -[C(R1)(R2)O]mC(O)[C(R1)(R2)]nR3, -[C(R1)(R2)]mC(O)[OC(R1)(R2)]nR3 и -[C(R1)(R2)]nR5; Y представляет собой водород; каждый m, n и o независимо выбран из группы, состоящей из 1, 2, 3, 4, 5 и 6; каждый p и q независимо выбран из 0.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II X выбран из группы, состоящей из -[C(R1)(R2)]C(O)[OC(R1)(R2)]R3, -C(O)O[C(R1)(R2)]R3, -C(O)NR4[C(R1)(R2)]R3, -[C(R1)(R2)O]C(O)[C(R1)(R2)]R3, -[C(R1)(R2)]C(O)NR4[C(R1)(R2)]R3 и -[C(R1)(R2)]R5, Y представляет собой водород.
Кроме того, в альтернативном варианте осуществления настоящего изобретения в соединении формулы II R3 выбран из группы, состоящей из водорода, поли(оксиэтиленокси)(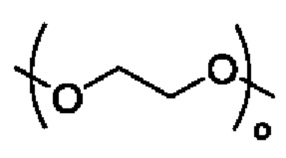 ), поли(этиленокси)(
), поли(этиленокси)(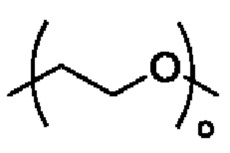 ), OPO(R6)2, PO(R6)2, OSO2(R6)2, SO2(R6)2, OC(O)R6 и C(O)OR6, R6 является таким, как определено в соединении формулы II.
), OPO(R6)2, PO(R6)2, OSO2(R6)2, SO2(R6)2, OC(O)R6 и C(O)OR6, R6 является таким, как определено в соединении формулы II.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из группы, состоящей из -C(O)O[C(R1)(R2)]mR3, -C(O)NR4[C(R1)(R2)]mR3, -[C(R1)(R2)O]mC(O)[C(R1)(R2)]nR3, -[C(R1)(R2)]mC(O)[OC(R1)(R2)]nR3, -[C(R1)(R2)N]mC(O)[C(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[OC(R1)(R2)]nR3, [C(R1)(R2)N]mC(O)[NC(R1)(R2)]nR3, -[C(R1)(R2)(O)p]mC(O)NR4[C(R1)(R2)(O)p]nR3 и -[C(R1)(R2)(O)p]nR5, X представляет собой водород или 3-6 членный гетероциклил; R3 выбран из водорода, поли(оксиэтиленокси)(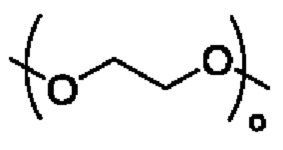 ), поли(этиленокси)(
), поли(этиленокси)(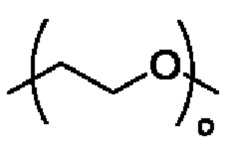 ), OPO(R6)2, PO(R6)2, OSO2(R6)2, SO2(R6)2, OC(O)R6 и C(O)OR6, R6 является таким, как определено в соединении формулы II.
), OPO(R6)2, PO(R6)2, OSO2(R6)2, SO2(R6)2, OC(O)R6 и C(O)OR6, R6 является таким, как определено в соединении формулы II.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из [C(R1)(R2)(O)p]nR5, R5 выбран из группы, состоящей из С6-10 гетероциклила, OPO(R6)2, OSO2R6, SR', SO2R', 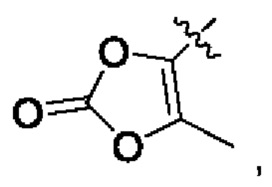 OC(O)R7 и NR'(R").
OC(O)R7 и NR'(R").
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из [C(R1)(R2)]nR5, R5 выбран из группы, состоящей из С6-10 гетероциклила, OPO(R6)2, OSO2R6, SR', SO2R', 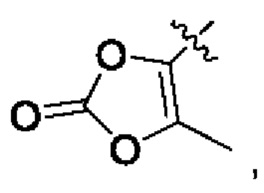 OC(O)R7 и NR'(R").
OC(O)R7 и NR'(R").
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II Y выбран из [C(R1)(R2)O]nR5, R5 выбран из группы, состоящей из С6-10 гетероциклила, OPO(R6)2, OSO2R6, SR', SO2R', 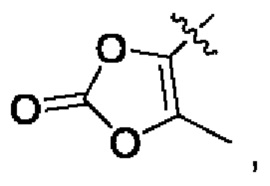 OC(O)R7 и NR'(R").
OC(O)R7 и NR'(R").
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II R5 выбран из группы, состоящей из С6-10 гетероциклила, OPO(R6)2, OSO2R6, SR', SO2R', 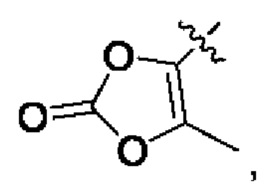 OC(O)R7 и NR'(R").
OC(O)R7 и NR'(R").
Кроме того, в соединении формулы II указанный R6 выбран из группы, состоящей из водорода, C1-6 алкила, С3-6 циклоалкила, 3-6 членного гетероциклила (такого как пиперидин), OR' и NR'(R"), R' и R" являются такими, как определено в соединении формулы II.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II R' и R" выбраны из группы, состоящей из водорода и алкила, где указанный алкил предпочтительно выбран из С1-10 алкила, более предпочтительно - выбран из С1-6 алкила, такого как метил, этил, пропил и изопропил.
В альтернативном варианте осуществления настоящего изобретения в соединении формулы II, где m=1, 2, 3 или 4, n=1, 2, 3 или 4, и о=1~8.
В некоторых вариантах осуществления изобретения в соединении формулы II каждый R6 и R7 независимо выбран из группы, состоящей из:
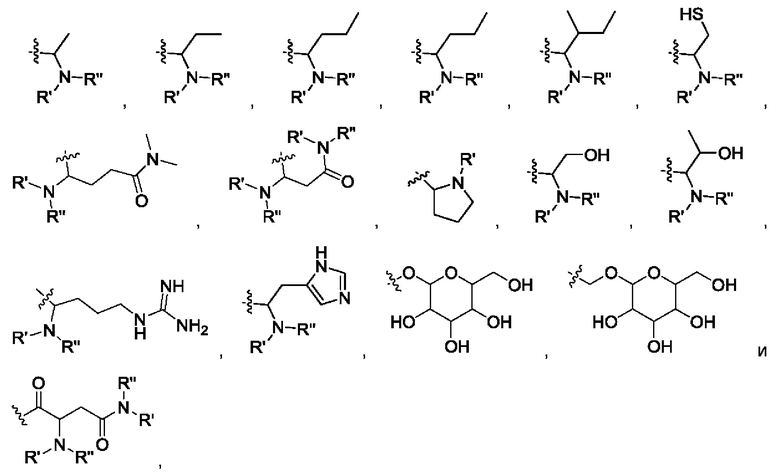
где R' и R" выбраны из группы, состоящей из водорода и алкила, где указанный алкил предпочтительно выбран из C1-10 алкила, более предпочтительно - из C1-6 алкила, такого как метил, этил, пропил и изопропил.
В некоторых вариантах осуществления изобретения предложено соединение II, где R3 выбран из OPO(R6)2, R6 выбран из группы, состоящей из гидроксила, C1-6 алкила, С3-7 циклоалкила, C1-6 алкоксила и 3-7 членного гетероциклила.
В некоторых других вариантах осуществления изобретения предложено соединение II, где m=1, 2, 3 или 4.
В некоторых других вариантах осуществления изобретения предложено соединение II, где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, C1-6 алкила и С3-7 циклоалкила.
В некоторых других вариантах осуществления изобретения соединение формулы II представляет собой
или его фармацевтически приемлемую соль, или его стереоизомер, ротамер или таутомер, или его дейтерид,
где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где указанный алкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
каждый R' и R" независимо выбран из группы, состоящей из водорода, гидроксила, алкила, алкоксила, алкенила и ацила;
m=1, 2, 3 или 4.
В некоторых других вариантах осуществления изобретения в соединении формулы II R6 выбран из группы, состоящей из:
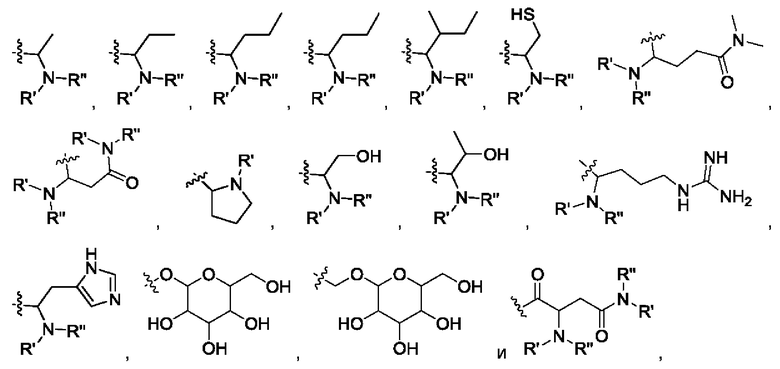
где R' и R" выбраны из группы, состоящей из водорода и алкила, где указанный алкил предпочтительно выбран из С1-10 алкила, более предпочтительно - из C1-6 алкила, такого как метил, этил, пропил и изопропил.
В некоторых других вариантах осуществления изобретения соединение формулы III представляет собой
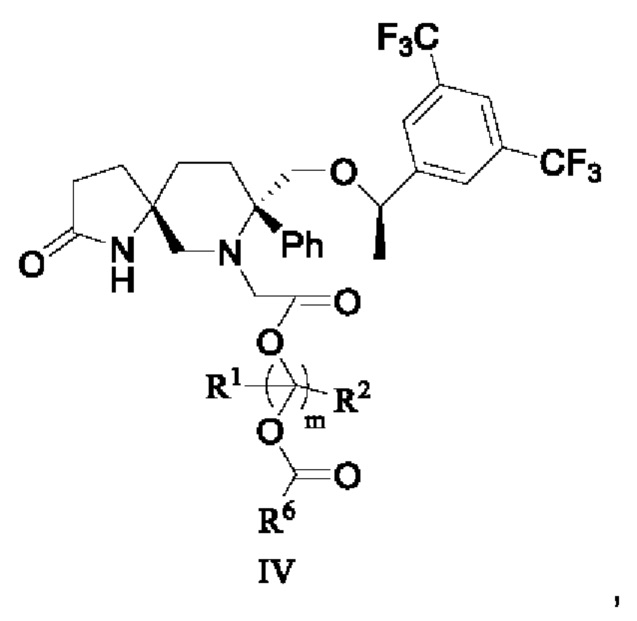
или его фармацевтически приемлемую соль, или его стереоизомер, ротамер или таутомер, или его дейтерид,
где каждый R1 и R2 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где указанный алкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
каждый R6 независимо выбран из группы, состоящей из водорода, гидроксила, алкила, циклоалкила, гетероциклила, арила, гетероарила, алкоксила, гидроксиалкила и NR'(R"), где указанный алкил, гидроксиалкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, циклоалкила, алкоксила, гидроксиалкила, алкенила, алкинила, арила, гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R");
каждый R' и R" независимо выбран из группы, состоящей из водорода, гидроксила, алкила, алкоксила, алкенила и ацила.
Кроме того, в альтернативном варианте в соединении формулы IV R6 выбран из группы, состоящей из С1-12 алкила (включая, но не ограничиваясь этим, метил, этил, пропил или изопропил), С3-12 циклоалкила (включая, но не ограничиваясь этим, циклопропил, циклопентил, циклогексил), 3-12 членного гетероциклила (включая, но не ограничиваясь этим, пирролил), С6-12 арила (включая, но не ограничиваясь этим, фенил, нафтил), 3-12 членного гетероарила (включая, но не ограничиваясь этим, пиридин, пиперидин), С1-12 алкоксила (включая, но не ограничиваясь этим, метоксил, этоксил, пропоксил или изопропоксил), С1-12 гидроксиалкила и NR'(R"), где указанный алкил, гидроксиалкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из C1-6 алкила, С3-6 циклоалкила, 3-12 членного гетероциклила, С1-12 алкоксила, C1-6 гидроксиалкила, С2-4 алкенила, С2-4 алкинила, С6-10 арила, 3-10 членного гетероарила, нитро, циано, гидроксила, галогена, SR', NR'(R"), COOR' и CONR'(R"); каждый R' и R" независимо выбран из группы, состоящей из водорода, C1-6 алкила, C1-6 алкоксила, С2-4 алкенила, С1-6 алканоила (такого как ацетил, формил), бензоил и п-толил.
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы II указанный R6 выбран из группы, состоящей из водорода, C1-6 алкила, С3-6 циклоалкила, 3-6 членного гетероциклила (такого как пиперидин), OR' и NR'(R").
В альтернативном варианте осуществления настоящего изобретения в соединении формулы IV R' и R" выбраны из группы, состоящей из водорода и алкила, где указанный алкил предпочтительно выбран из С1-10 алкила, более предпочтительно - из С1-6 алкила, такого как метил, этил, пропил и изопропил.
В некоторых вариантах осуществления изобретения в соединении формулы IV R6 выбран из группы, состоящей из:
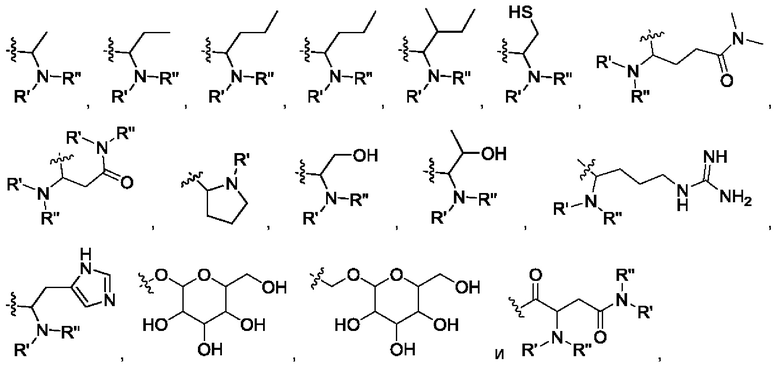
где R' и R" выбраны из группы, состоящей из водорода и алкила, где указанный алкил предпочтительно выбран из С1-10 алкила, более предпочтительно - из C1-6 алкила, такого как метил, этил, пропил и изопропил.
В предпочтительном варианте осуществления настоящего изобретения в соединении формулы II указанный R6 выбран из группы, состоящей из водорода, C1-6 алкила, С3-6 циклоалкила, 3-6 членного гетероциклила (такого как пиперидин), OR' и NR'(R").
В альтернативном варианте осуществления настоящего изобретения в соединении формулы IV R' и R" выбраны из группы, состоящей из водорода и алкила, где указанный алкил предпочтительно выбран из С1-10 алкила, более предпочтительно - из С1-6 алкила, такого как метил, этил, пропил и изопропил.
В некоторых других вариантах в соединении формулы IV R6 выбран из группы, состоящей из:
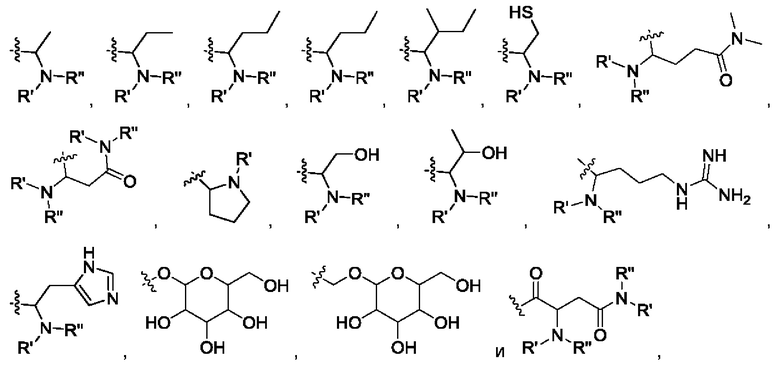
где R' и R" выбраны из группы, состоящей из водорода и алкила, где указанный алкил предпочтительно выбран из С1-10 алкила, более предпочтительно - из C1-6 алкила, такого как метил, этил, пропил и изопропил.
Типичные соединения формулы II включают, но не ограничиваются следующими:
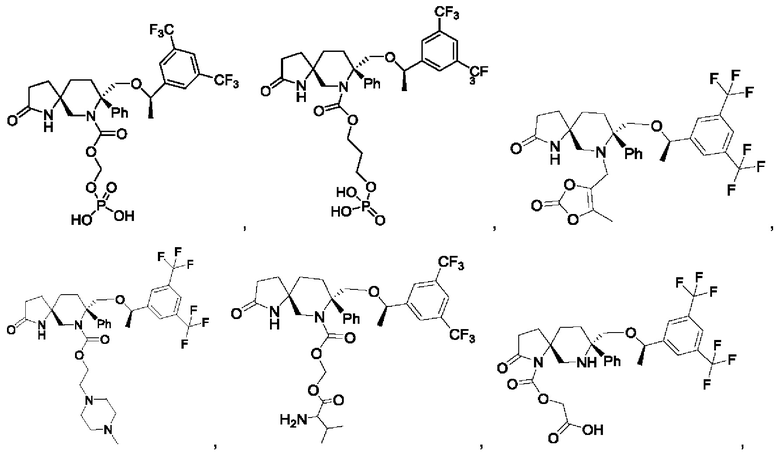
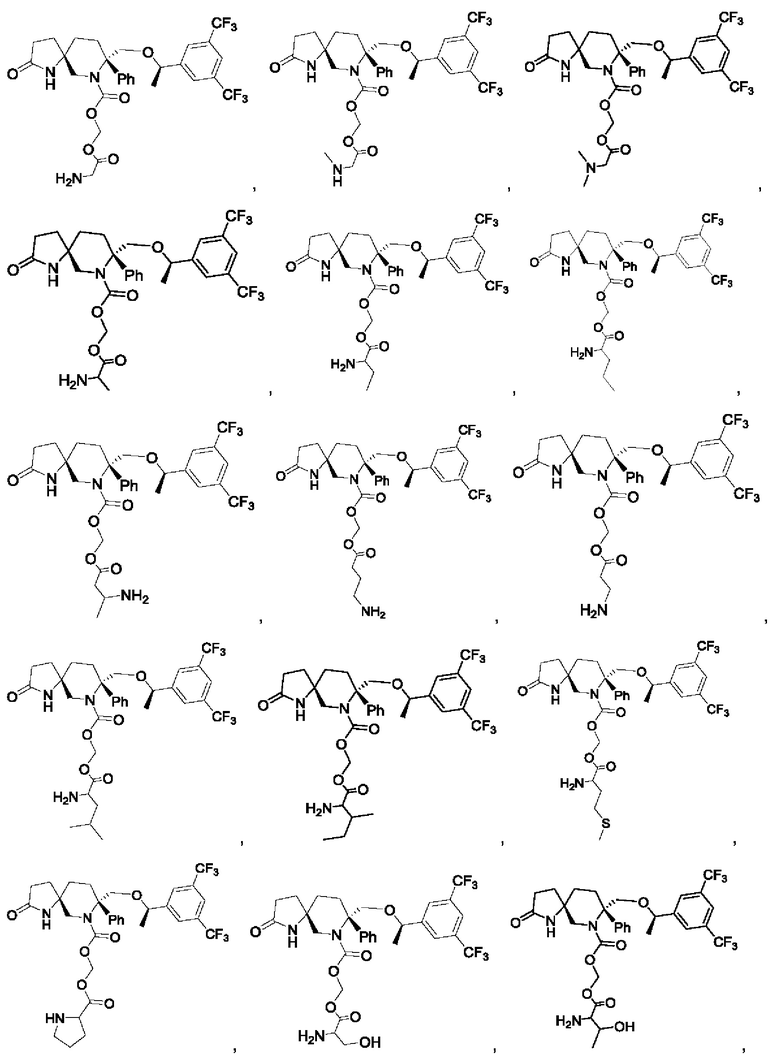
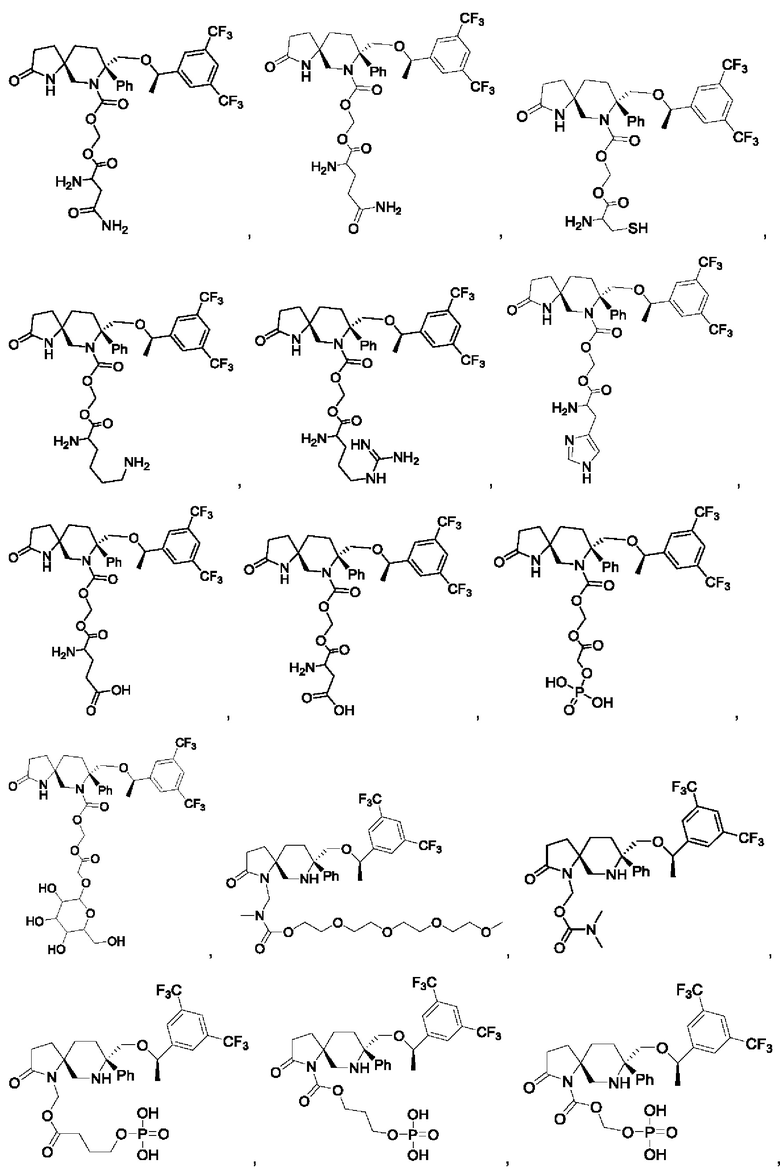
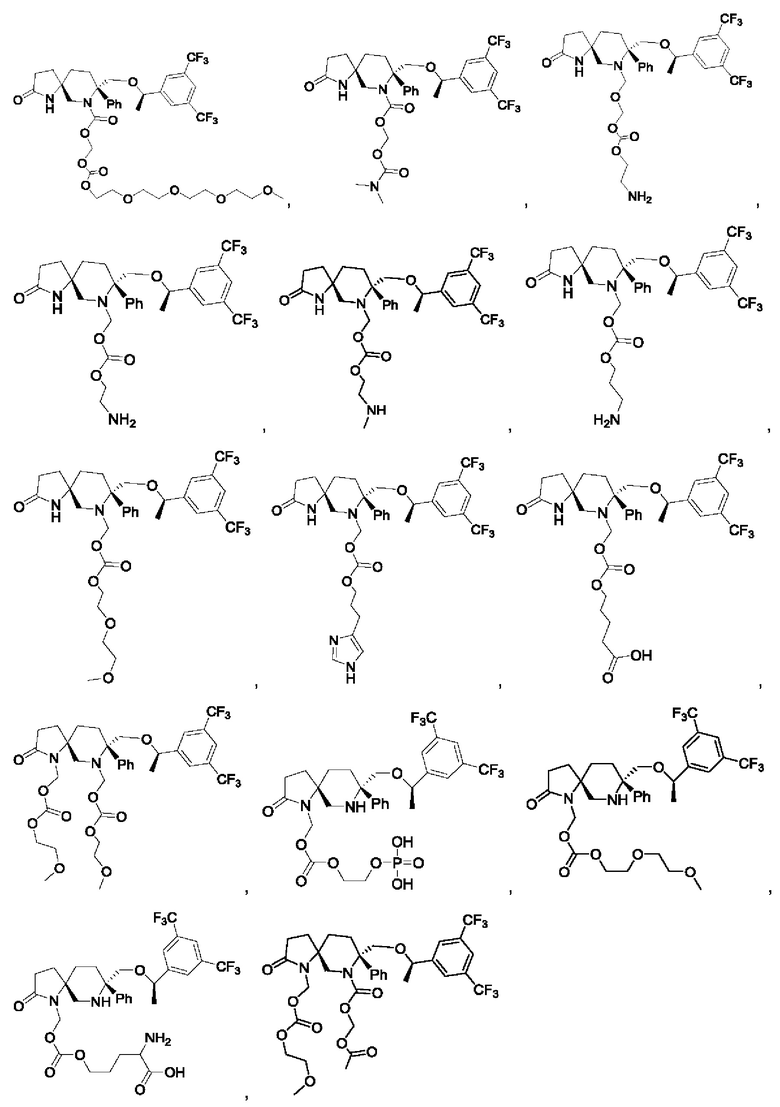
или их фармацевтически приемлемую соль, или их стереоизомер, ротамер или таутомер.
Кроме того, соединение формулы IA представляет собой:
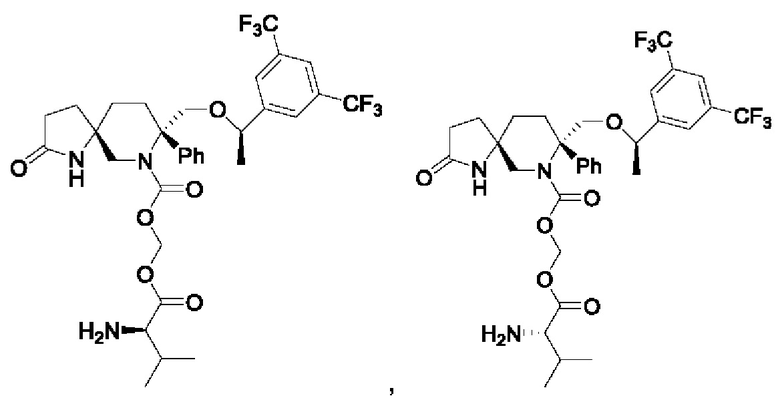
или его фармацевтически приемлемую соль.
В другом аспекте соединение согласно настоящему изобретению обладает более высокой растворимостью и лучшим превращением in vivo по сравнению с известными соединениями. В некоторых вариантах осуществления соединение согласно настоящему изобретению обладает низким гемолитическим эффектом и уменьшенными побочными эффектами после введения лекарственного средства, а также улучшает соблюдение пациентом режима приема лекарственного средства.
Настоящее изобретение также предлагает фармацевтическую композицию, содержащую терапевтически эффективное количество по меньшей мере одного из вышеупомянутых соединений или его фармацевтически приемлемой соли, а также фармацевтически приемлемый носитель, разбавитель или эксципиент.
В другом аспекте, водород в функциональной группе соединения согласно настоящему изобретению может быть дейтерирован с получением соответствующего дейтерированного соединения, которое сохраняет селективность и потенциал, сравнимые с водородным аналогом. Дейтериевая связь является более стабильной, что приводит к иным показателям «АРМЭ» (абсорбции, распределения, метаболизма, элиминации), т.е. «токсической фармакокинетики», тем самым обеспечивая клинически полезные эффекты.
Токсическая фармакокинетика относится к процессам абсорбции, распределения, метаболизма, элиминации из организма экзогенных химических веществ.
Настоящее изобретение также относится к применению соединения или его фармацевтически приемлемой соли или фармацевтической композиции в соответствии с вышеприведенными вариантами осуществления для изготовления лекарственных средств для лечения физиологических расстройств, состояний или заболеваний у пациента, где физиологическим расстройством, состоянием или заболеванием является респираторное заболевание, кашель, воспалительное заболевание, кожное расстройство, офтальмологическое расстройство, депрессия, тревожность, фобии, биполярное расстройство, алкогольная зависимость, злоупотребление веществами со значительным воздействием на нервную систему, эпилепсия, ноцицепция, психоз, шизофрения, болезнь Альцгеймера, деменция, связанная со СПИДом, болезнь Тауна, расстройство, связанное со стрессом, обсессивно-компульсивное расстройство, булемия, нервная анорексия, компульсивное переедание, мания, предменструальный синдром, желудочно-кишечная дисфункция, атеросклероз, фиброзное расстройство, ожирение, диабет II типа, головная боль, невропатическая боль, послеоперационная боль, хронический болевой синдром, расстройство мочевого пузыря, мочеполовое расстройство или рвота или тошнота, и дополнительно относится к применению для изготовления лекарственных средств для лечения астмы, рвоты, тошноты, депрессии, тревожности, кашля или мигрени.
В другом аспекте фармацевтически приемлемая соль соединения выбрана из группы, состоящей из неорганической соли и органической соли. Соединение согласно настоящему изобретению реагирует с кислотой, такой как трифторуксусная кислота, с образованием соответствующей соли. Указанную кислоту выбирают из группы, состоящей из, но не ограничиваясь этим, уксусной кислоты, соляной кислоты, салициловой кислоты, яблочной кислоты, аскорбиновой кислоты, фосфорной кислоты, лимонной кислоты, бензойной кислоты и фумаровой кислоты. Соединение согласно настоящему изобретению реагирует с основанием, таким как N-метил-D меглумин или дициклогексиламин, с образованием соответствующей соли. Указанное основание выбирают из группы, состоящей из, но не ограничиваясь ими, основания натрия, щелочноземельного металла и аминокислоты (такого как аргинин, лизин).
В другом аспекте настоящее изобретение также включает меченые изотопами соединения согласно настоящему изобретению, которые являются такими же, как соединения, описанные в настоящем описании, но с заменой одного или более атомов атомами, имеющими атомную массу или массовое число, которые отличаются от обычно встречаемых в природе. Примеры изотопов, которые могут быть включены в соединения согласно настоящему изобретению, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, такие как 2Н, 3Н, 11С, 13С, 14С, 13N, 15N, 15O, 17O, 18O, 31Р, 32Р, 5S, 18F, 123I, 125I и 36Cl, и т.п.
Соединения согласно настоящему изобретению могут содержать неприродную пропорцию атомных изотопов в одном или более атомах, составляющих соединение. Например, соединения могут быть помечены радиоизотопами, такими как тритий (3Н), йод-125 (125I) или С-14 (14С). Другой пример: водород можно заменить дейтерием с образованием дейтерида. Связь между дейтерием и углеродом сильнее, чем связь между обычным водородом и углеродом. По сравнению с недейтерированными соединениями, дейтерированные соединения обладают такими преимуществами, как снижение токсических и побочных эффектов, повышенная стабильность лекарственного средства, повышенная эффективность, увеличенный биологический период полураспада и т.п. Все изменения в изотопном составе соединений согласно настоящему изобретению, независимо от того, радиоактивны они или нет, входят в объем настоящей заявки.
Кроме того, замена более тяжелыми изотопами (такими как дейтерий (т.е. 2Н)) может обеспечить определенные терапевтические преимущества в результате более высокой метаболической стабильности (например, увеличение периода полувыведения in vivo или условия сниженной дозировки), и поэтому может быть более предпочтительной при определенных обстоятельствах, где замена на дейтерий может быть частичной или полной, причем под частичной заменой на дейтерий понимается замена по меньшей мере одного водорода на по меньшей мере один дейтерий.
Объяснение терминов
«Алкил» относится к насыщенной алифатической углеводородной группе, включая линейную или разветвленную группу, с 1-20 атомами углерода, предпочтительно к алкилу с 1-12 атомами углерода и более предпочтительно к алкилу с 1-6 атомами углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил и их различные разветвленные изомеры. Алкил может быть замещенным или незамещенным. При замещении замещающая группа (группы) может быть замещена в любой доступной точке соединения. Заместитель(и) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из арила, гетероарила и галогена.
«Алкенил» относится к разветвленному или прямому олефину с 2-12 атомами углерода или к олефину, содержащему алифатические углеводородные группы. Например, «С2-6 алкенил» означает алкенил с 2, 3, 4, 5 или 6 атомами углерода. Примеры алкенильных групп включают, но не ограничиваются ими, винил, аллил, 1-пропенил, 1-бутенил, 2-бутенил, 3-бутенил, 2-метилбут-2-енил, 3-метилбут-1-енил, 1-пентенил, 3-пентенил и 4-гексенил.
Термин «циклоалкил» относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю. Циклоалкильное кольцо содержит от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептантриенил, циклооктил и т.п. Полициклические циклоалкилы включают циклоалкилы, имеющие спирокольцо, конденсированное кольцо или мостиковое кольцо.
Указанное циклоалкильное кольцо может быть сконденсировано с арильным, гетероарильным или гетероциклильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и т.п. Циклоалкил может быть необязательно замещенным или незамещенным. При замещении заместитель предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкантио, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкоксила, гетероциклоалкоксила, циклоалкилатио, гетероциклилатио, оксо, карбоксила или карбоксилата.
Термин «гетероциклил» относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю, который содержит от 3 до 20 атомов в кольце, где один или более атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из азота, кислорода и S(O)m (где m представляет собой целое число от 0 до 2), но исключая кольцевую группу -ОО-, -OS- или -SS-, а остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил содержит от 3 до 12 кольцевых атомов, где от 1 до 4 атомов представляют собой гетероатомы; более предпочтительно содержит от 3 до 6 атомов в кольце. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидротиофенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.п., предпочтительно - пиперидинил и пирролидинил. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо.
Указанное гетероциклическое кольцо может быть сконденсировано с арильным, гетероарильным или циклоалкильным кольцом, где кольцо, присоединенное к исходной структуре, представляет собой гетероциклил. Неограничивающие примеры включают:
Гетероциклил может быть необязательно замещенным или незамещенным. При замещении группа(ы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилтиола, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкоксила, гетероциклоалкоксила, циклоалкилатио, гетероциклилатио, оксо, карбоксила или карбоксилата.
«Алкинил» включает разветвленный или прямой алкинил с 2-12 атомами углерода или олефин, содержащий алифатические углеводородные группы, или, если количество атомов углерода указано, это означает именно это конкретное количество, например, этинил, пропинил (например, 1-пропинил, 2-пропинил), 3-бутинил, пентинил, гексинил и 1-метилпент-2-инил.
Термин «арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо имеет общую соседнюю пару атомов углерода), имеющему сопряженную π-электронную систему, предпочтительно - к 6-12-членному, например, к фенилу или нафтилу. Указанный арильный цикл может быть сконденсирован с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, присоединенное к исходной структуре, представляет собой арильное кольцо. Неограничивающие примеры включают:
Арил может быть замещенным или незамещенным. При замещении группа(ы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилтио, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкоксила, гетероциклоалкоксила, циклоалкилатио, гетероциклилатио, карбоксила и карбоксилата, предпочтительно фенила.
Термин «гетероарил» относится к гетероароматической системе, включающей от 1 до 4 гетероатомов и от 5 до 14 кольцевых атомов, где гетероатом выбран из группы, состоящей из кислорода, серы и азота. Гетероарил предпочтительно представляет собой 6-12-членный гетероарил, более предпочтительно - 5- или 6-членный гетероарил, например, имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, тетразолил, пиридил, пиримидинил, тиадиазол, пиразинил и т.п.; предпочтительно - имидазолил, пиразолил, пиримидинил или тиазолил; более предпочтительно - пиразолил или тиазолил. Указанное кольцо гетероарила может быть сконденсировано с кольцом арила, гетероциклила или циклоалкала, где кольцо, присоединенное к исходной структуре, представляет собой кольцо гетероарила. Неограничивающие примеры включают:
Гетероарил может быть необязательно замещенным или незамещенным. При замещении группа(ы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилтиола, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкоксила, гетероциклоалкоксила, циклоалкилатио, гетероциклилатио, карбоксила и карбоксилата.
Термин «алкоксил» относится к группе -О-(алкил) или -O-(незамещенный циклоалкил), где алкил имеет значения, указанные выше. Неограничивающие примеры алкоксила включают метоксил, этоксил, пропоксил, бутоксил, циклопропоксил, циклобутоксил, циклопентилоксил, циклогексилоксил. Алкоксил может быть необязательно замещенным или незамещенным. При замещении группа(ы) заместителя предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилтиола, алкиламино, галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкоксила, гетероциклоалкокси, циклоалкилатио, гетероциклилатио, карбоксила или карбоксилата.
Термин «гидроксиалкил» относится к алкилу, замещенному гидроксилом(ами), где алкил является таким, как определено выше.
Термин «галоалкил» относится к алкилу, замещенному галогеном(галогенами), где алкил имеет значение, определенное выше.
Термин «дейтерированный алкил» относится к алкилу, замещенному атомом(атомами) дейтерия, где алкил является таким, как определено выше.
Термин «гидроксил» относится к группе -ОН.
Термин «галоген» относится к фтору, хлору, брому или йоду.
Термин "амино" относится к -NH2.
Термин «циано» относится к -CN.
Термин «нитро» относится к -NO2.
«Необязательный» или «необязательно» означает, что событие или обстоятельство, описываемое впоследствии, может, но не должно произойти, и описание включает ситуацию, в которой событие или обстоятельство происходит или не происходит. Например, «гетероциклил, необязательно замещенный алкилом» означает, что алкил может присутствовать, но не обязательно должен присутствовать, и описание включает ситуацию, когда гетероциклил замещен алкилом и когда гетероциклил не замещен алкилом.
«Замещенный» относится к ситуации, когда один или более атомов водорода в группе, предпочтительно до 5, более предпочтительно от 1 до 3 атомов, независимо замещены соответствующим количеством заместителей. Само собой разумеется, что заместители существуют только в их химически возможных положениях. Специалисты в данной области способны определить, возможно или невозможно замещение, с помощью экспериментов или теории без чрезмерных усилий. Например, связь амино или гидроксила, имеющего свободный водород, с атомом углерода, имеющим ненасыщенную связь (таким как олефин), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси, включающей одно или более соединений, описанных в настоящем документе, или их физиологически/фармацевтически приемлемую соль или пролекарство, и другие химические компоненты, а также другие компоненты, такие как физиологически/фармацевтически приемлемые носители и вспомогательные вещества. Целью фармацевтической композиции является облегчение введения соединения в организмы, что способствует абсорбции активного ингредиента для проявления биологической активности.
Известные исходные вещества согласно настоящему изобретению могут быть синтезированы или в соответствии со способами, известными в данной области техники, или могут быть в равной степени приобретены у Acros Organics или Aldrich Chemical Company и других компаний, или могут быть в равной степени получены способом, описанным в CN102775401A.
Структуры соединений идентифицируют методами ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Сдвиг ЯМР (δ) приведен в единицах 10-6 (млн-1). ЯМР определяют на ядерно-магнитном спектрометре BrukerAVANCE-400. Растворители для определения - дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), внутренний стандарт - тетраметилсилан (ТМС). Масс-спектрометр FINNIGANLCQAd (ESI) (производитель: Thermo, модель: FinniganLCQadvantageMAX) используют для определения ESI-MS. ЖХ-МС определяют на высокоэффективном жидкостном хроматографе (производитель: Agilent, модель: 1200) с градиентным элюированием, режимом сканирования положительными ионами и диапазоном сканирования качества 100-1500.
Краткое описание графических материалов
Фиг. 1: График тенденции превращения соединения примера 5 в плазме человека.
Подробное описание изобретения
Настоящее изобретение будет дополнительно описано со ссылкой на следующие примеры, но примеры не следует рассматривать как ограничивающие объем настоящего изобретения.
Методы экспериментов, в которых условия не описаны конкретно в примерах настоящего изобретения, в целом соответствуют обычным условиям или условиям, рекомендованным производителем сырья или продукта. Реагенты, для которых не указаны источники, представляют собой обычные реагенты, приобретаемые на рынке.
Пример 1:

Стадия 1:
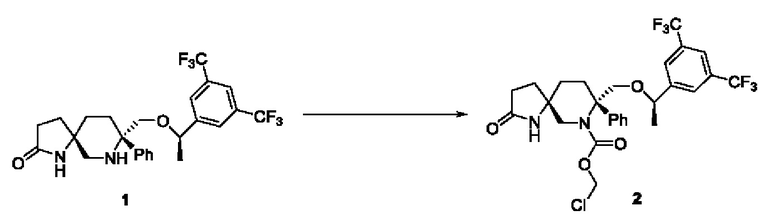
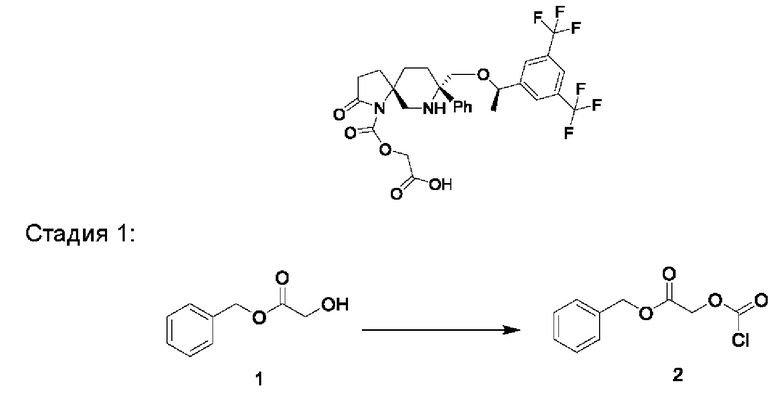
Соединение 1 (2,43 г, 4,86 ммоль, 1 экв.) взвешивали и растворяли в дихлорметане (36 мл) в трехгорлой колбе на 100 мл в атмосфере N2. Добавляли диизопропилэтиламин (5 г, 38,76 ммоль, 8 экв.) и смесь охлаждали до -30°С. Добавляли триметилхлорсилан (1,36 г, 12,52 ммоль, 2,6 экв.) и смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь охлаждали до -25°С. Добавляли по каплям раствор хлорметилхлорформиата (0,77 г, 6 ммоль, 1,23 экв.) в дихлорметане и смесь перемешивали при контролируемой температуре от -20°С до -5°С до завершения реакции. Реакционный раствор выливали в воду со льдом, подвергали разделению и экстрагировали дихлорметаном. Добавляли воду и 1 н. раствор соляной кислоты и подвергали разделению. Затем органический слой последовательно промывали солевым раствором, насыщенным водным раствором бикарбоната натрия и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 3,0 г желтого желе с выходом 104%.
Стадия 2:
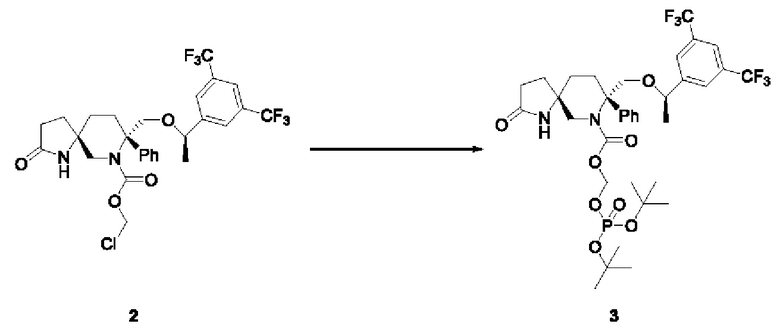
Соединение 2 (2,8 г, 4,53 ммоль, 1 экв.), йодид тетрабутиламмония (1,68 г, 4,55 ммоль, 1 экв.), ди-трет-бутилфосфатную калиевую соль (5,63 г, 22,67 ммоль, 5 экв.) и диоксан (84 мл) добавляли в трехгорлую колбу на 500 мл в атмосфере N2. Реакционную смесь нагревали до 55°С и перемешивали в течение 4 часов. Реакционный раствор охлаждали, выливали в этилацетат и воду, подвергали разделению и экстрагировали этилацетатом. Органический слой промывали водным раствором сульфита натрия, затем последовательно промывали водой и солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 3,73 г желтой пены с выходом 107%.
Стадия 3:
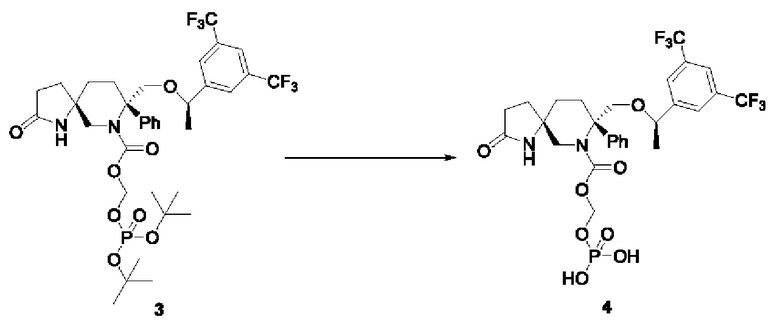
Соединение 3 (1,95 г, 2,543 ммоль, 1 экв.) добавляли в одногорлую колбу на 100 мл и растворяли в дихлорметане (40 мл) в атмосфере N2. Медленно добавляли трифторуксусную кислоту (1,45 мл, 19,52 ммоль, 8 экв.) при охлаждении ледяной водой. Реакционную смесь перемешивали до завершения реакции и затем концентрировали с получением 2,29 г масла, которое затем подвергали очистке с получением 1,39 г белого пенистого твердого вещества с выходом 83,5%.
1Н-ЯМР (400 МГц, CD3OD): δ (млн-1 (ppm)) 7.89 (s, 2Н), 7.86 (s, 1Н), 7.41-7.27 (m, 5Н), 5.66 (d, J=12 Гц, 1Н), 5.50-5.47 (m, 1Н), 4.60 (d, J=8 Гц, 1Н), 4.20-3.88 (m, 3Н), 2.51-2.10 (m, 5Н), 1.86-1.66 (m, 3Н), 1.44-1.31 (m, 4Н).
Стадия 4:
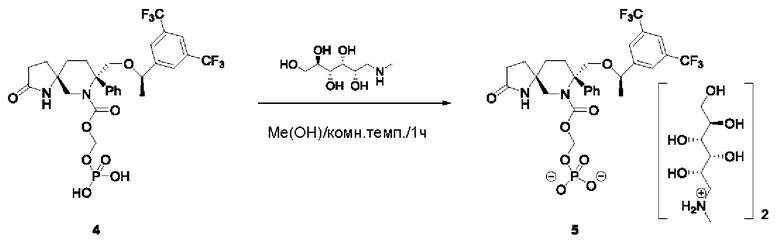
Соединение 4 (111 мг, 0,17 ммоль) и меглумин (59,6 мг, 0,305 ммоль) добавляли в одногорлую колбу на 50 мл и растворяли в метаноле (5 мл). Реакционную смесь перемешивали при комнатной температуре в течение 1,5 ч и затем концентрировали с получением 4 мг белой твердой соли.
Пример 2:
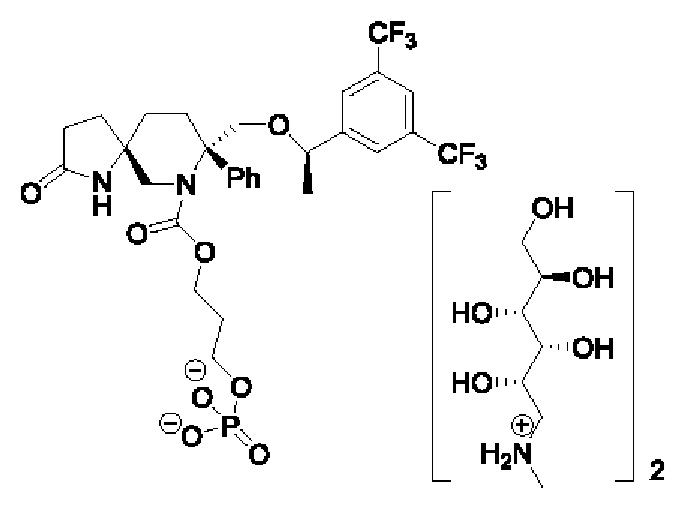
Стадия 1:
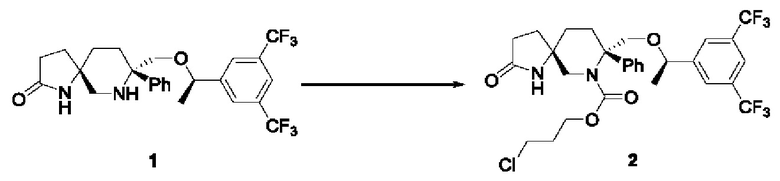
Соединение 1 (5 г, 10 ммоль, 1 экв.) помещали в трехгорлую колбу на 250 мл и добавляли 50 мл дихлорметана. Реакционную смесь продували N2. Добавляли диизопропилэтиламин (5,1 г, 40 ммоль, 4 экв.) и смесь охлаждали до 0°С. Медленно по каплям добавляли 3-хлорпропилхлорформиат (4,71 г, 30 ммоль, 3 экв.) и реакционную смесь перемешивали до завершения реакции. Реакционный раствор промывали водой (20 мл × 2), сушили над безводным сульфатом натрия и концентрировали. Неочищенный продукт суспендировали с 20 мл трет-бутилметилового эфира, фильтровали и сушили с получением 5,3 г продукта в виде белого твердого вещества с выходом 85,5% и чистотой по ВЭЖХ 95,2%. Стадия 2:
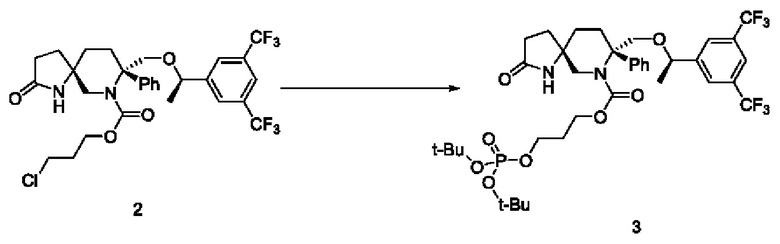
Соединение 2 (500 мг, 0,833 ммоль, 1 экв.) помещали в круглодонную колбу на 25 мл. Добавляли 5 мл диметилформамида, 5 мг йодида калия и ди-трет-бутилфосфат тетрабутилчетвертичного аммония (564 мг, 1,25 ммоль, 1,5 экв.). Температуру повышали до 100°С до завершения реакции. Реакционную смесь концентрировали и остаток очищали ВЭЖХ с получением 370 мг продукта с выходом 57,8% и чистотой по ВЭЖХ 97%.
Стадия 3:
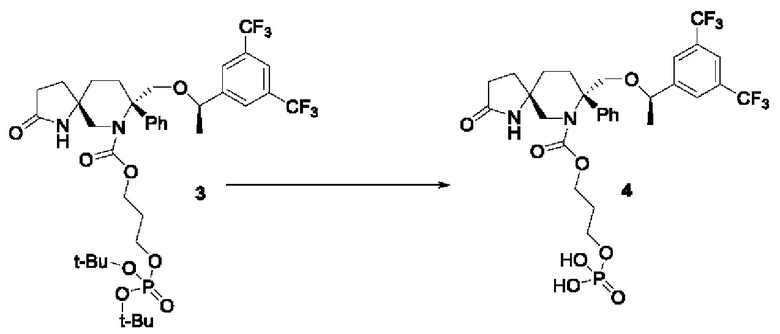
Соединение 3 (2 г, 2,52 ммоль) растворяли в растворе соляной кислоты в диоксане (25 мл, 4 М) и перемешивали при комнатной температуре в течение 30 минут. Реакционную смесь упаривали досуха при пониженном давлении с получением соединения 4 (1,4 г, 2,05 ммоль) с выходом 81%.
Стадия 4:
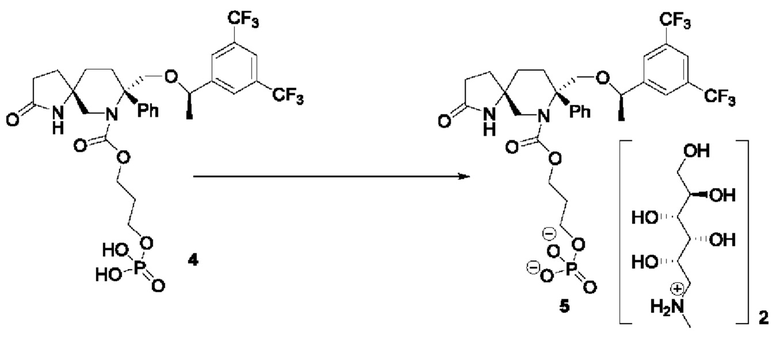
Соединение 4 (700 мг, 1,025 ммоль) и меглумин (310 мг, 2 ммоль) растворяли в метаноле (10 мл) при 25°С и перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении, получая неочищенный продукт соединения 5 (1,1 г), который затем суспендировали с метил-трет-бутиловым эфиром, фильтровали и сушили, получая чистый продукт соединения 5 (1 г, 0,932 ммоль) с выходом 91%.
1Н-ЯМР (400 МГц, CD3OD): δ7.90-7.84 (m, 3Н), 7.32-7.25 (m, 5Н), 4.14-3.61 (m, 25Н), 2.81 (m, 5Н), 2.47-2.29 (m, 12Н), 1.79-1.63 (m, 5H), 1.46-1.29 (m, 3Н), 1.21-1.12 (m, 6H).
Пример 3:
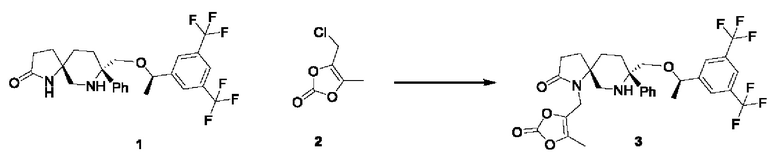
В колбу вместимостью 25 мл добавляли 2 мл ацетона и 100 мг соединения 1 и перемешивали. Карбонат калия (42 мг, 0,3 ммоль, 1,5 экв.) добавляли порциями и смесь перемешивали при комнатной температуре в течение получаса. В реакционную колбу добавляли 36 мг соединения 2. Реакционную смесь перемешивали при комнатной температуре в течение примерно 18 часов до завершения реакции и очищали колоночной хроматографией с получением 50 мг соединения 3 (выход 40,8%).
1Н-ЯМР (400 МГц, CDCl3): δ=7.79 (s, 1Н), 7.72 (s, 2Н), 7.42-7.40 (d, J=8 Гц, 2Н), 7.31-7.27 (m, 2Н), 7.27-7.21 (m, 1Н), 5.58 (s, 1Н), 4.55-4.53 (m, 1Н), 4.06-3.99 (m, 2Н), 3.69-3.67 (d, J=8 Гц, 1 Н), 3.53-3.49 (d, J=16 Гц, 1Н), 3.25-3.21 (d, J=16 Гц, 1 Н), 2.77-2.74 (d, J=12 Гц, 1H), 2.59-2.57 (d, J=8 Гц, 1H), 2.34-2.31 (m, 3Н), 1.97-1.71 (m, 7Н), 1.46-1.45 (d, J=4 Гц, 3Н).
Пример 4:
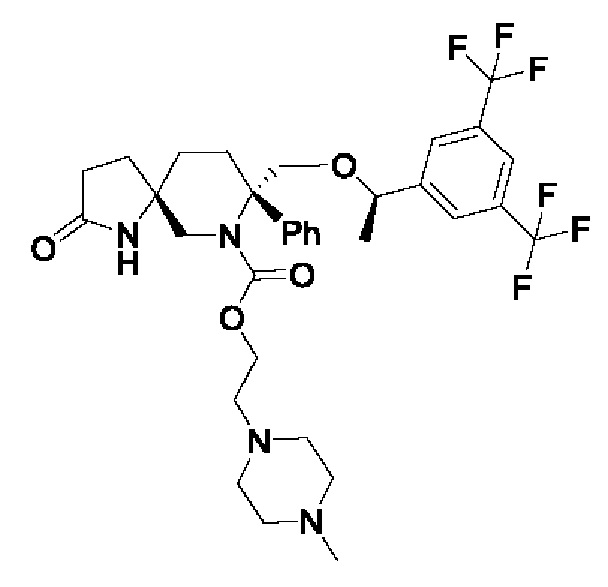
Стадия 1:
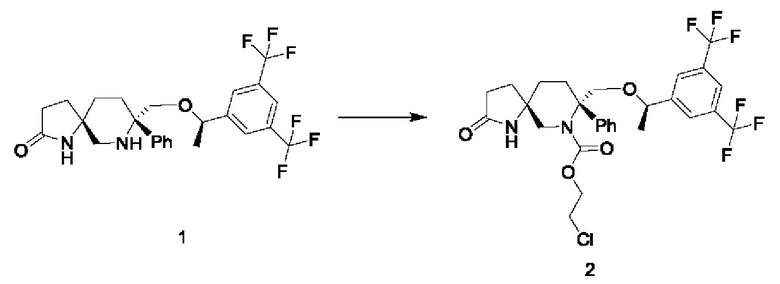
В трехгорлую колбу в атмосфере азота добавляли 750 мг соединения 1 и 2,1 мл диизопропилэтиламина. Затем добавляли 12 мл безводного дихлорметана. Смесь охлаждали до -40°С и по каплям добавляли 0,5 мл триметилхлорсилана. Смесь перемешивали при комнатной температуре в течение двух часов. Затем реакционную смесь охлаждали до температуры от -30°С до -20°С и по каплям добавляли 0,024 мл хлорэтилхлорформиата, растворенного в 3 мл безводного дихлорметана. Реакционную смесь перемешивали при температуре от -20°С до 5°С до завершения реакции. Реакцию гасили добавлением воды. Жидкость подвергали разделению и промывали последовательно разбавленной 1Н соляной кислотой, насыщенным раствором соли, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и сушили в вакууме с получением 1 г соединения 2 в виде белого твердого вещества.
Стадия 2:
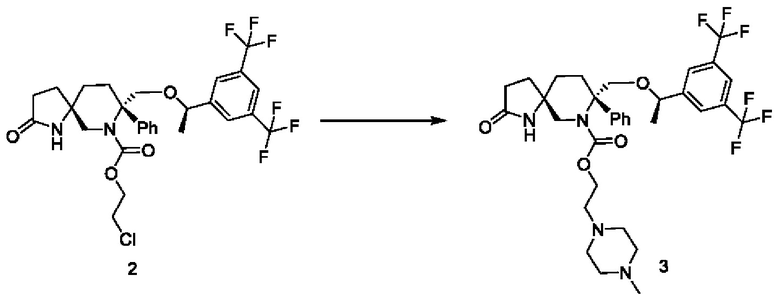
1 г соединения 2 и 0,99 г йодида натрия растворяли в 10 мл диметилформамида. Добавляли 1,15 мл диизопропилэтиламина и 0,75 мл метилпиперазина. Реакционную смесь нагревали до 90°С и перемешивали до завершения реакции. Реакционный раствор сразу концентрировали и остаток очищали с помощью ВЭЖХ с получением 430 мг соединения 3.
1Н-ЯМР (400 МГц, CDCl3): δ7.77 (s, 1Н), 7.73 (s, 2Н), 7.37-7.26 (m, 5Н), 6.56 (s, 1 Н), 4.44-4.40 (m, 1Н), 4.29-4.24 (m, 2Н), 4.10-4.07 (m, 1Н), 3.90-3.87 (d, J=12 Гц, 1Н), 3.79-3.76 (d, J=12 Гц, 1Н), 3.01-2.97 (d, J=16 Гц, 1Н), 2.52-2.32 (m, 15Н), 1.93-1.65 (m, 6Н), 1.29-1.28 (d, J=4 Гц, 3Н).
Пример 5:
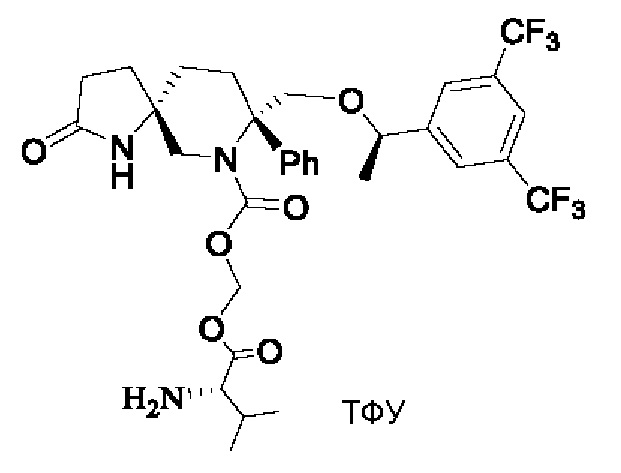
Стадия 1:
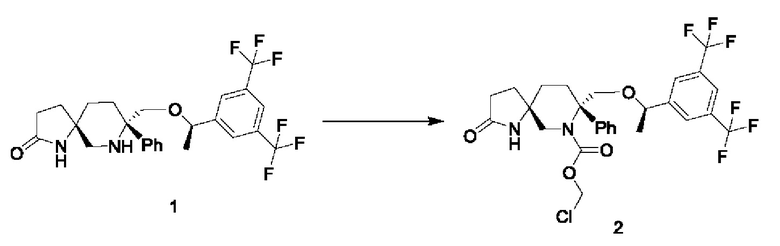
750 мг соединения 1 и 2,1 мл диизопропилэтиламина растворяли в 13 мл безводного дихлорметана в атмосфере азота. Реакционную смесь охлаждали до -10°С и по каплям добавляли 0,5 мл триметилхлорсилана. Затем смесь нагревали до комнатной температуры и перемешивали в течение трех часов. Смесь снова охлаждали до -10°С и добавляли 3 мл раствора хлорметилхлорформиата (0,288 г) в дихлорметане. Затем реакционную смесь подвергали реакции при -10°С до завершения реакции. Реакцию гасили водой. Жидкость подвергали разделению и промывали последовательно разбавленной соляной кислотой, насыщенным раствором соли, насыщенным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и очищали остаток, получая 400 мг соединения 2 в виде белого твердого вещества.
Стадия 2:
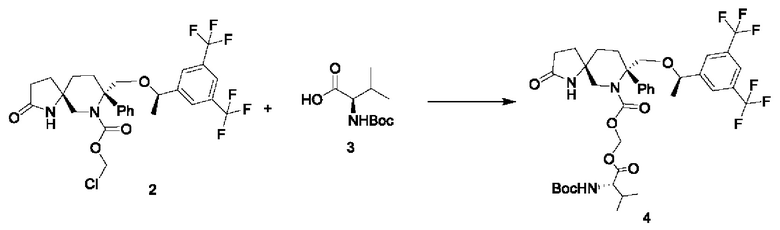
147 мг соединения 3 и 203 мг йодида натрия добавляли к 2 мл диметилформамида с последующим добавлением 136 мг бикарбоната калия. Реакционную смесь перемешивали при комнатной температуре в течение получаса, затем по каплям добавляли 400 мг соединения 2, растворенного в 10 мл диметилформамида. Смесь реагировала в течение ночи. Реакцию гасили добавлением воды. Реакционную смесь дважды экстрагировали этилацетатом. Органические фазы собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Концентрат очищали на колонке с силикагелем, получая 450 мг соединения 4 в виде масла.
Стадия 3:
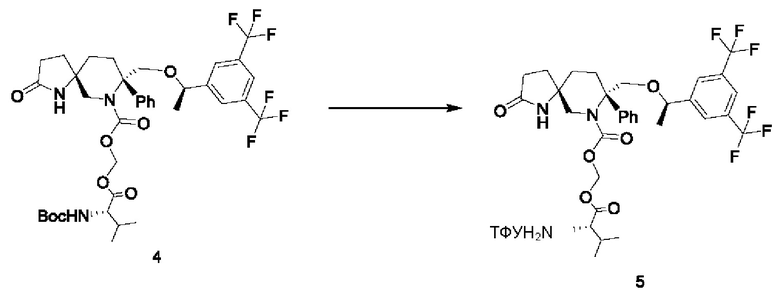
405 мг соединения 4 растворяли в 18 мл дихлорметана. Добавляли по каплям 4,5 мл трифторуксусной кислоты при охлаждении на бане со льдом. После добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение двух часов. Реакционную смесь концентрировали и остаток очищали колоночной хроматографией с получением 330 мг продукта, соединения 5, с выходом 80%.
1Н-ЯМР (400 МГц, CDCl3): δ 8.34 (s, 1 Н), 7.72 (s, 1 Н), 7.63 (s, 2Н), 7.40-7.28 (m, 5Н), 6.19 (s, 1 Н), 5.68-5.67 (d, J=4 Гц, 1 Н), 4.30-4.29 (d, J=4 Гц, 1Н), 4.20-4.17 (d, J=12 Гц, 1 Н), 3.99-3.91 (m, 2Н), 3.79 (s, 1 Н), 2.70-2.67 (d, J=12 Гц, 1Н), 2.49-2.21 (m, 8Н), 1.83-1.70 (m, 4Н), 1.29-1.28 (m, 3Н), 1.09-1.07 (m, 6Н).
Пример 6:
1,294 г трифосгена растворяли в 7,5 мл безводного тетрагидрофурана. Раствор охлаждали на бане со льдом и трижды продували азотом. Затем по каплям добавляли 0,33 мл пиридина. После добавления добавляли по каплям раствор 500 мг соединения 1, растворенного в 7,5 мл безводного тетрагидрофурана. После добавления реакционную смесь перемешивали при 5°С в течение 3 часов. Для разбавления добавляли 30 мл дихлорметана. Раствор промывали последовательно разбавленной соляной кислотой, водой и насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали, получая 700 мг неочищенного продукта.
Стадия 2:

128 мг соединения 3 добавляли к 2 мл безводного тетрагидрофурана. Реакционную смесь трижды продували азотом. Реакционную смесь охлаждали примерно до -65°С и по каплям добавляли 0,28 мл гексаметилдисилазида лития (1 моль/л, растворенного в н-гексане). Смесь перемешивали в течение получаса. При этом 60 мг неочищенного продукта соединения 2 с предыдущей стадии растворяли в 1 мл безводного тетрагидрофурана. Смесь охлаждали до -65°С после трехкратной продувки азотом. Литиевую соль соединения 3, полученную выше, переносили в реакционную колбу, содержащую ацилхлоридное соединение 2. После добавления реакцию завершали при -65°С и гасили добавлением насыщенного водного раствора хлорида аммония. Реакционную смесь экстрагировали этилацетатом. Органическую фазу концентрировали и очищали непосредственно колоночной хроматографией с получением 62 мг соединения 4.
Стадия 3:
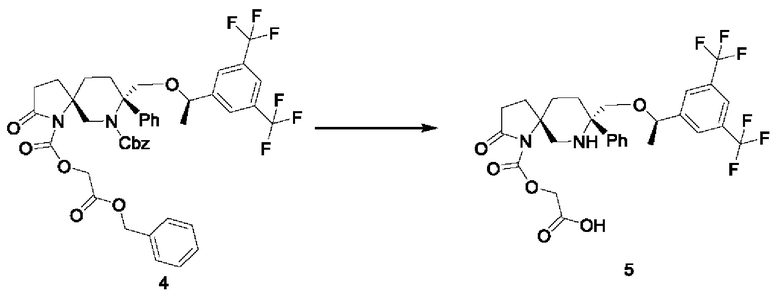
62 мг соединения 4 и 31 мг 20% влажного гидроксида палладия добавляли к 1,5 мл этилацетата и начинали перемешивание. Реакционную смесь трижды продували водородом, перемешивали при комнатной температуре в течение 5 часов и фильтровали. Осадок на фильтре промывали этилацетатом. Фильтрат концентрировали и очищали остаток, получая 41 мг соединения 5 в виде белого твердого вещества.
1Н-ЯМР (400 МГц, CDCl3) δ=7.72 (s, 1Н), 7.57 (s, 2Н), 7.50-7.41 (m, 5Н), 4.80-4.59 (m, 3Н), 4.17-4.13 (d, J=16 Гц, 1Н), 3.83-3.80 (d, J=12 Гц, 1Н), 3.62-3.59 (d, J=12 Гц, 1Н), 3.26-3.22 (d, J=16 Гц, 1Н), 2.60-2.47 (m, 4Н), 2.26-2.18 (m, 2Н), 1.85-1.80 (m, 2Н), 1.45-1.43 (d, J=8 Гц, 3Н), 0.89-0.83 (m, 1Н).
Пример 7:
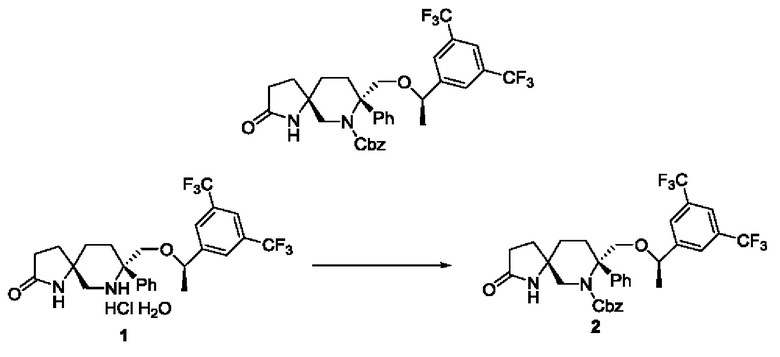
Карбонат калия (11,7 г, 84,66 ммоль, 8,47 экв.) растворяли в воде (40 мл) до использования. Соединение 1 (5,55 г, 10 ммоль, 1 экв.) суспендировали в этилацетате (80 мл) в атмосфере N2, к которому добавляли вышеупомянутый водный раствор карбоната калия при охлаждении на бане со льдом. Реакционный раствор постепенно становился прозрачным при перемешивании и затем по каплям добавляли Cbz-Cl (1,7 мл, 12 ммоль, 1,2 экв.). После добавления реакционную смесь перемешивали в течение 10 мин и перемешивали при комнатной температуре в течение ночи. Реакционный раствор подвергали разделению и экстрагировали этилацетатом. Органические фазы собирали, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Остаток очищали колоночной хроматографией с получением 4,3 г твердого вещества белого цвета с выходом 56%.
Пример 8:
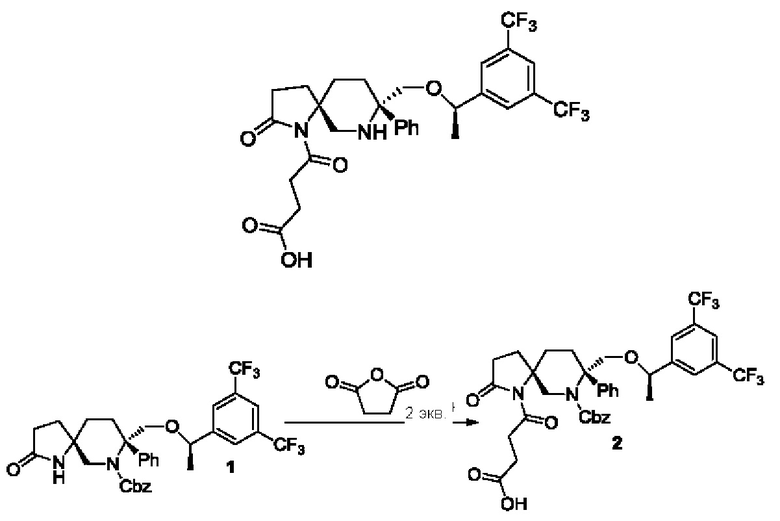
Соединение 1 (317 мг, 0,5 ммоль) и ТГФ (7,2 мл) добавляли в одногорлую колбу на 50 мл в атмосфере азота, затем перемешивали до растворения и охлаждали до -20°С. По каплям добавляли NaHMDS (2 М, 0,5 мл, 1 ммоль) и перемешивали реакционную смесь до завершения реакции. Реакцию гасили насыщенным раствором хлорида аммония. Реакционную смесь экстрагировали метил-трет-бутиловым эфиром. Органический слой промывали насыщенным раствором соли, сушили над безводным сульфатом натрия и концентрировали. Остатки очищали колоночной хроматографией с получением соединения 2 (250 мг) с выходом 68%.
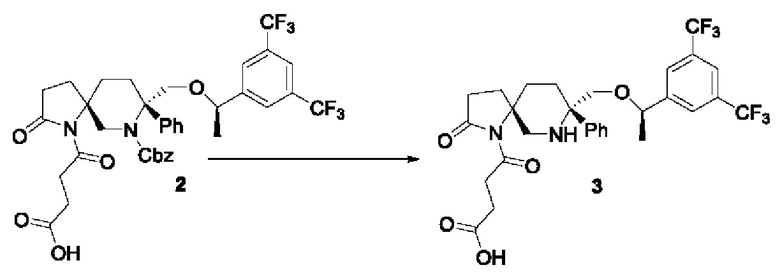
В одногорлую колбу на 50 мл добавляли соединение 2 (250 мг, 0,34 ммоль), метанол (10 мл) и палладий на угле (10%, 250 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода до завершения реакции. Реакционную смесь фильтровали и остаток концентрировали с получением соединения 3 (150 мг) с выходом 74%.
ЖХМС: 601 [М+1].
Пример 9:
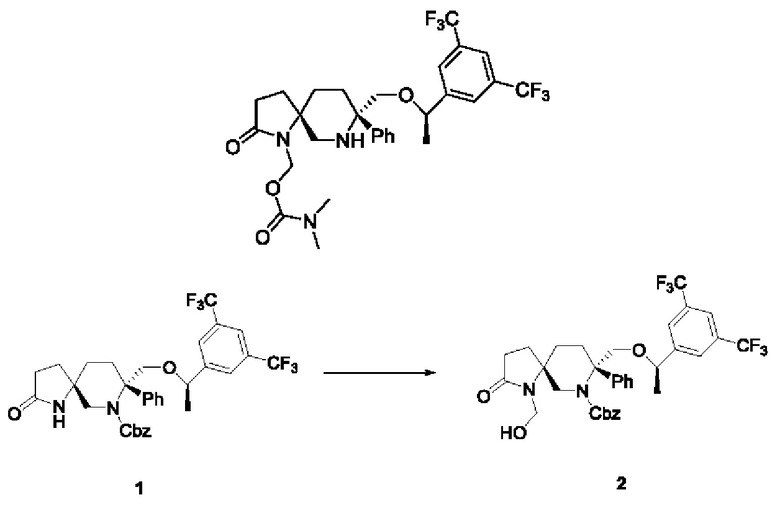
Соединение 1 (215 мг, 0,339 ммоль, 1 экв.), безводный карбонат калия (55 мг, 0,396 ммоль, 1,1 экв.), параформальдегид (37 мг, 1,23 ммоль, 3,3 экв.) и ТГФ (5 мл) добавляли в реакционную колбу на 50 мл в атмосфере N2. Реакционную смесь нагревали и перемешивали до завершения реакции. Реакционную смесь фильтровали и концентрировали, получая неочищенный продукт, который очищали колоночной хроматографией с получением 216 мг масла соединения 2 с выходом 95%.
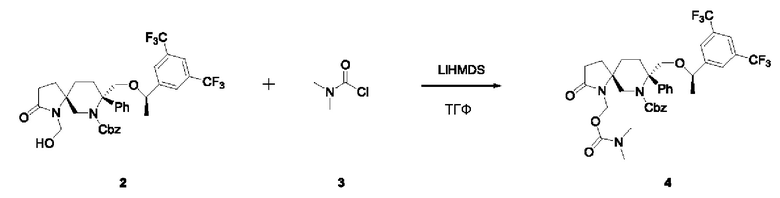
Соединение 2 (66 мг, 0,1 ммоль, 1 экв.) и ТГФ (5 мл) добавляли в реакционную колбу в атмосфере N2. Добавляли LiHMDS (1M в ТГФ, 0,2 мл, 0,2 ммоль, 2 экв.) затем по каплям соединение 3 (40 мг, 0,37 ммоль, 3,7 экв.) и реакционную смесь перемешивали до завершения реакции. Реакционную смесь экстрагировали этилацетатом и концентрировали с получением неочищенного продукта, который очищали колоночной хроматографией с получением 27 мг неочищенного продукта 4 в виде масла с выходом 36%.
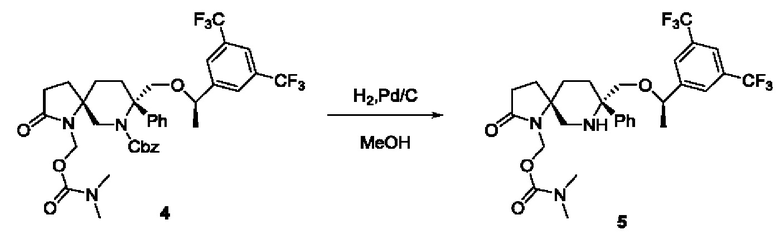
Соединение 4 (27 мг, 0,367 ммоль, 1 экв.), Pd/C (33 мг) и метанол (5 мл) добавляли в реакционную колбу при комнатной температуре и перемешивали в атмосфере водорода до завершения реакции. Реакционную смесь фильтровали и концентрировали, получая неочищенный продукт, который подвергали колоночной хроматографии с получением 13 мг масла соединения 5 с выходом 58,8%.
ЖХМС: 602 [М+1].
Пример 10:
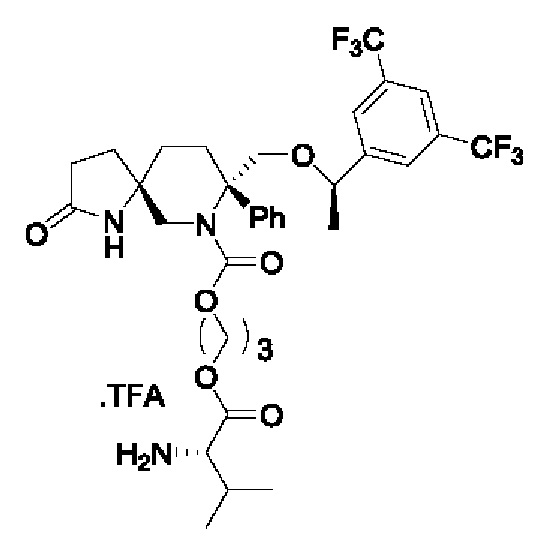
Целевое соединение синтезировали по методике Примера 5 с заменой хлорметилхлорформиата на хлорпропилхлорформиат. ЖХМС: 702 [М+1].
Пример 11:
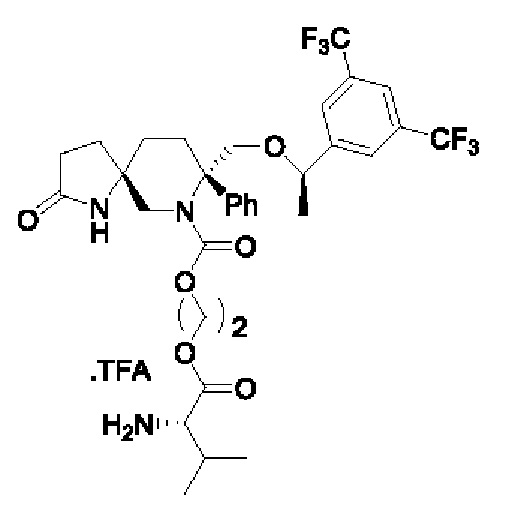
Целевое соединение синтезировали по методике Примера 5 с заменой хлорметилхлорформиата на хлорэтилхлорформиат. ЖХМС: 688 [М+1].
Пример 12:
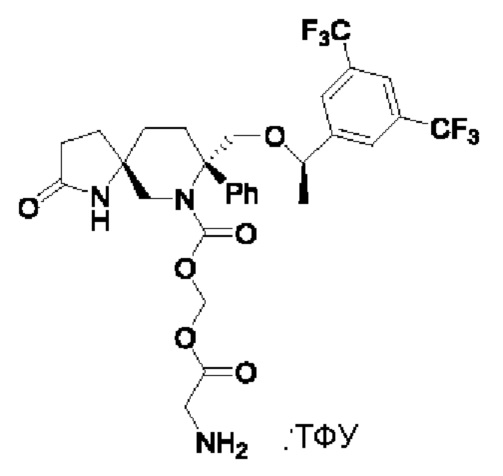
Целевое соединение синтезировали по методике Примера 5 с заменой Boc-L-валина на N-Boc-глицин. ЖХМС: 632 [М+1].
Пример 13:
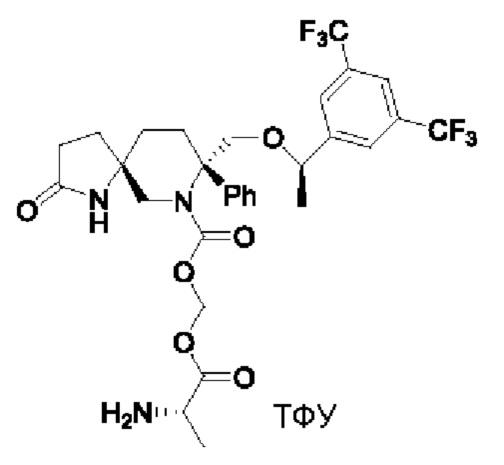
Целевое соединение синтезировали по методике Примера 5 с заменой Boc-L-валина на N-Boc-аланин. ЖХМС: 646 [М+1].
Пример 14:
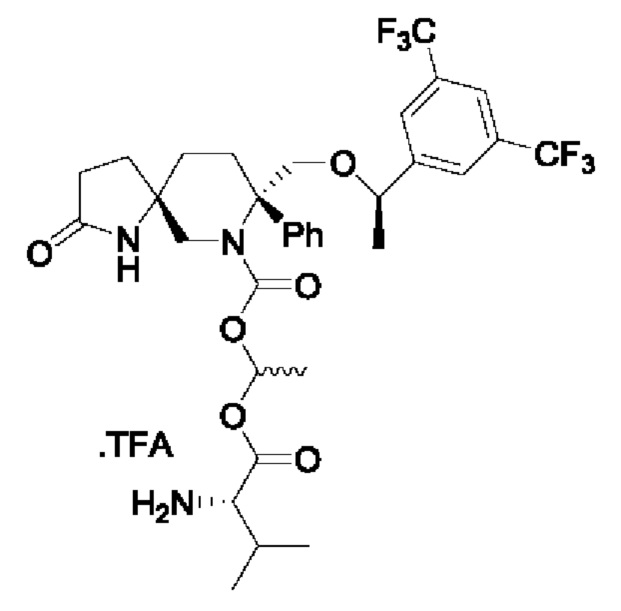
Целевое соединение (изомеры приблизительно 1/1) синтезировали по методике Примера 5 с заменой хлорметилхлорформиата на 1-хлорэтилхлорформиат. ЖХМС: 688 [М+1].
Пример 15:
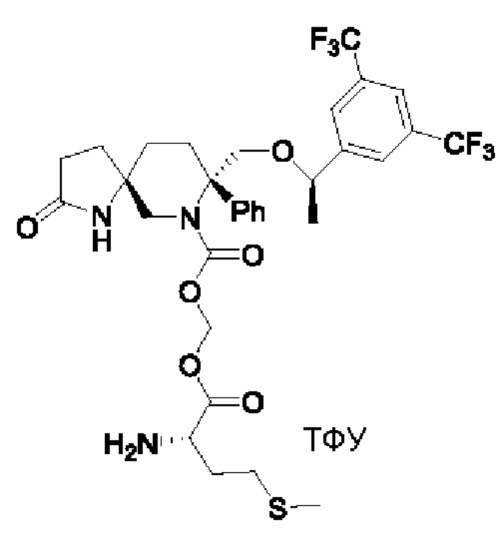
Целевое соединение синтезировали по методике Примера 5 с заменой Boc-L-валина на Boc-L-метионин. ЖХМС: 706 [М+1].
Пример 16:
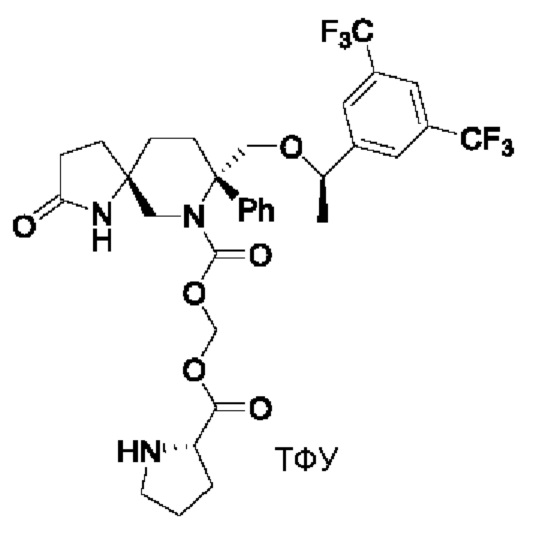
Целевое соединение синтезировали по методике Примера 5 с заменой Boc-L-валина на Boc-L-пролин. ЖХМС: 672 [М+1].
Пример 17:
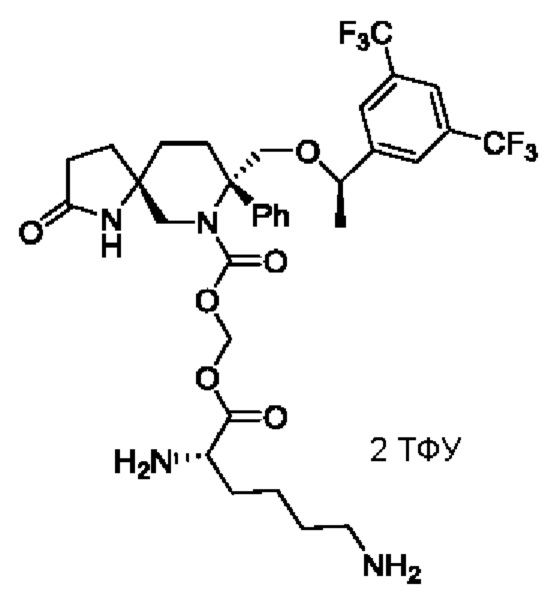
Целевое соединение синтезировали по методике Примера 5 с заменой Boc-L-валина на (S)-2,6-ди-трет-бутилкарбониламинокапроновую кислоту. ЖХМС: 703 [М+1].
Пример 18:
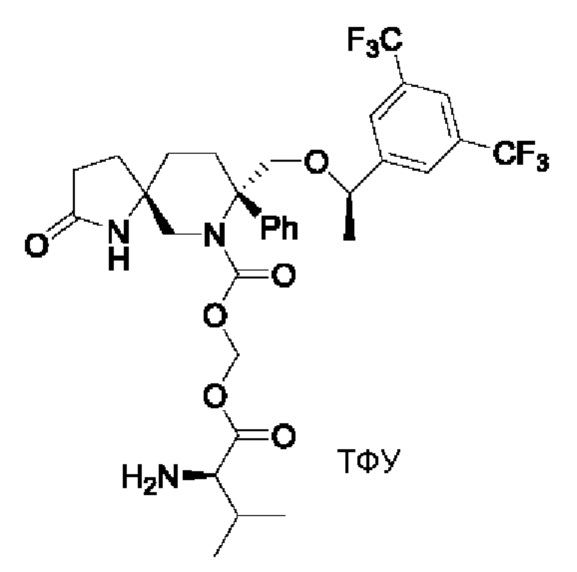
Целевое соединение синтезировали по методике Примера 5 с заменой Boc-L-валина на Boc-D-валин. ЖХМС: 674 [М+1].
Тестовый Пример 1: Данные растворимости в воде и химическая стабильность
1.1. Получение реагентов
Реагент: NaH2PO4⋅2H2O
1.2. Способ получения
Реагенты на 100 мл готовили следующим образом:
рН=3.0: фосфатный буферный раствор: 100 мл 20 ммоль/л NaH2PO4, 0.1 М H3PO4, доведение рН до 3.0.
рН=4.0: фосфатный буферный раствор: 100 мл 20 ммоль/л NaH2PO4, 0.1 М H3PO4, доведение рН до 4.0.
рН=7.0: сверхчистая вода
рН=9.0: фосфатный буферный раствор: 100 мл 20 ммоль/л Na2HPO4, раствор 0.1 М NaOH, доведение рН до 9.0.
1.3. Метод испытаний
Соответствующее количество тестируемого соединения точно взвешивали. Раствор добавляли каждый раз небольшими порциями несколько раз и перемешивали до растворения соединения и определяли содержание соединения в растворе. Данные представлены в таблице 1.
2.1 Испытание соединений на стабильность
Во флакон взвешивали около 1 мг образца, затем флакон помещали в вакуумный пакет и пакет вакуумировали. Затем пакет помещали в контейнер с изменяющим цвет силикагелем и запаивали. Готовили два параллельных образца. Готовили достаточное количество образцов в соответствии с моментом времени отбора проб и помещали при 4°С и комнатной температуре соответственно. Данные представлены в таблице 1.
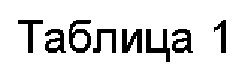
Примечание: Хорошая: чистота снизилась на <0,5% после выдерживания в течение 7 дней; Умеренная: чистота снизилась на 0,5%~2,0% после выдерживания в течение 7 дней; Плохая: чистота снизилась более чем на 2,0% после выдерживания в течение 7 дней.
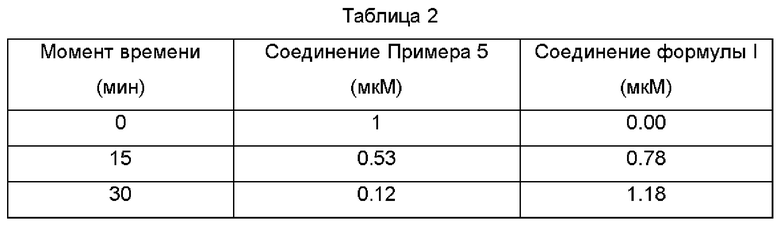
Тестовый Пример 2: Испытание на стабильность в плазме
Протокол испытаний
1.1 Препарат для испытаний
Соединение примера 5 и соединение формулы I.
1.2 Плазма для испытаний
Свежая человеческая плазма была сдана добровольцами с информированного согласия.
1.3 Приготовление раствора соединения
Взвешивали определенное количество соединения Примера 5 и добавляли ДМСО для приготовления исходного раствора с концентрацией 30 мМ. Некоторый объем исходного раствора разбавляли ДМСО, получая раствор I с концентрацией 1600 мкМ. Затем определенный объем 1600 мкМ раствора I разбавляли 45% метанолом для приготовления рабочего раствора II с концентрацией 16 мкМ. Исходный раствор 30 мМ и раствор II 1600 мкМ для соединения формулы I также готовили вышеописанным способом.
1.4 Инкубирование образцов
5 мкл 16 мкМ рабочего раствора соединения Примера 5 добавляли к 75 мкл плазмы с получением конечной концентрации 1 мкМ. Образцы инкубировали на водяной бане при 37°С в течение 0, 15, 30, 60, 90, 120 и 180 мин. По окончании инкубации в образцы добавляли 240 мкл внутреннего стандарта, содержащего ацетонитрил, затем встряхивали на шейкере при 800 об/мин в течение 10 мин и центрифугировали при 3700 об/мин при 4°С в течение 20 мин. Супернатант анализировали методом ЖХ-МС, объем инъекции составлял 2 мкл.
1.5 Получение стандартной кривой
Предварительно 1600 мкМ раствор I разбавляли ацетонитрилом для приготовления рабочего раствора для стандартной кривой с концентрациями 160, 400, 1600, 4000, 8000, 16000 и 32000 нг/мл. Концентрация рабочего раствора для контроля качества составляла 480, 1920 и 25600 нг/мл. 5 мкл рабочего раствора для стандартной кривой и рабочего раствора для контроля качества добавляли к 75 мкл плазмы для получения образцов стандартной кривой с конечными концентрациями 10, 25, 100, 250, 500, 1000 и 2000 нг/мл и образцов для контроля качества с конечными концентрациями 30, 120 и 1600 нг/мл. Затем в образцы быстро добавляли 240 мкл ацетоциано, содержащего внутренний стандарт, которые затем встряхивали на шейкере при 800 об/мин в течение 10 мин и центрифугировали при 3700 об/мин при 4°С в течение 20 мин. Супернатант собирали и анализировали методом ЖХ-МС, объем инъекции составлял 2 мкл.
Стандартную кривую соединения формулы I и образец для контроля качества получали вышеуказанным способом.
2. Результаты
Превращение соединения Примера 5 согласно настоящему изобретению в свежей человеческой плазме происходит следующим образом, данные представлены в Таблице 2:

Тестовый Пример 3: Испытание на стабильность в плазме
Протокол испытаний
1.1 Препарат для испытаний
Соединения примеров 4, 6, 10 и 11.
1.2 Плазма для испытаний
Свежая человеческая плазма была сдана добровольцами с информированного согласия.
1.3 Стадии эксперимента
1) Тестируемые соединения из Таблицы 3 соответственно готовили в виде 30 мМ исходных растворов с ДМСО для последующего использования.
2) Исходный раствор 30 мМ разбавляли раствором ДМСО до раствора I с концентрацией 1600 мкМ. Затем 1600 мкМ раствор I разбавляли ацетонитрилом (АЦН) до рабочего раствора II с концентрацией 16 мкМ.
3) В эксперименте задавали 7 моментов времени 0, 15, 30, 60, 90, 120 и 180 мин с двумя параллельными образцами для каждого момента времени. Для каждого соединения были установлены две группы образцов. В каждую группу добавляли 75 мкл плазмы и 5 мкл приготовленного выше рабочего раствора II с концентрацией 16 мкМ. Реакционную систему инкубировали при 37°С до заданного времени, после чего реакцию останавливали добавлением 300 мкл раствора АЦН, содержащего внутренний стандарт. Реакционную смесь центрифугировали при 3700 об/мин в течение 10 мин и отбирали супернатант для анализа.
4) Построение стандартной кривой: предварительно разбавленный 1600 мкМ раствор I разбавляли ацетонитрилом до 1,5 мкМ/мл раствора III для стандартной кривой для дальнейшего использования. Концентрацию стандартной кривой устанавливали на 0,32, 0,8, 1,6, 4,0, 8, 12, 16 и 42 мкМ. После разбавления каждой концентрации стандартной кривой к 5 мкл каждой точки концентрации добавляли 75 мкл плазмы при конечной концентрации 0,02, 0,05, 0,1, 0,25, 0,75, 1,0 и 1,5 мкМ соответственно. Затем к образцам быстро добавляли 300 мкл стоп-раствора, которые затем центрифугировали при 3700 об/мин в течение 10 мин, и супернатант собирали для анализа ЖХ-МСМС. Данные представлены в таблице 3.
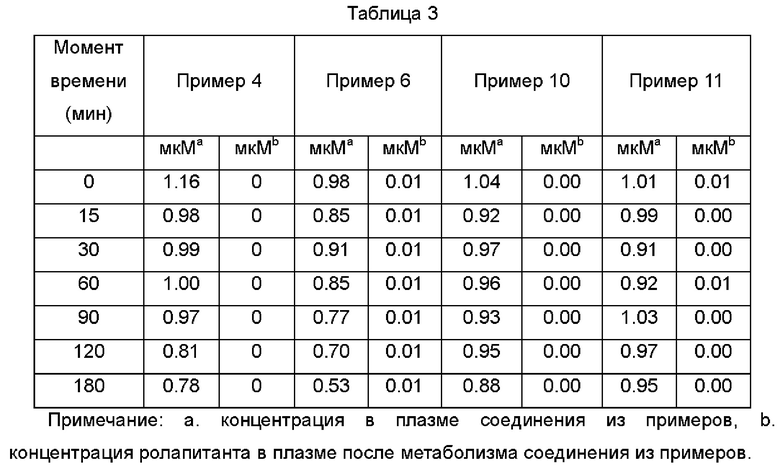
Вывод. Соединения Примера 4, Примера 10 и Примера 11 относительно стабильны в плазме с относительно более длительным периодом полужизни в плазме, но только небольшая часть этих трех соединений метаболизируется в плазме до ролапитанта. Соединение Примера 6 может метаболизироваться до ролапитанта в плазме, но из приведенных выше данных видно, что общий объем метаболизма в плазме относительно невелик.
Тестовый Пример 4:
Метаболизм соединений Примера 1, Примера 2 и Примера 8 в плазме мыши, крысы и человека определяли с помощью метода испытаний, описанного в Тестовом Примере 2. Данные представлены в Таблице 4.
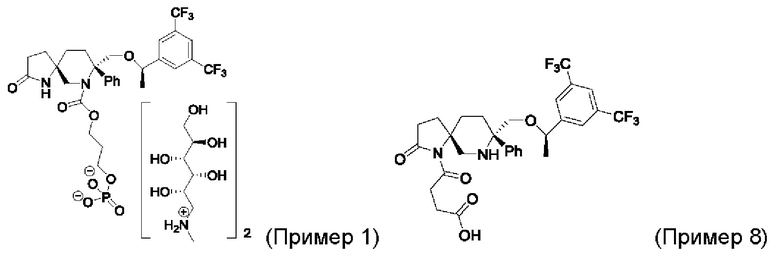
Выводы. Соединение Примера 8 может превращаться в ролапитант в плазме мыши, крысы и человека, причем степень превращения в плазме человека составляет около 46%. Между тем, соединения Примера 1 и Примера 2 практически не претерпевают превращения в ролапитант в плазме человека или претерпевают лишь незначительное превращение.
Тестовый Пример 5: Фармакокинетическое испытание in vivo на крысах В качестве подопытных животных использовали крыс. Концентрацию лекарственного средства в плазме в разные моменты времени после введения соединений Примера 1 и Примера 2 путем инъекции определяли с использованием метода ЖХ/МС/МС. Фармакокинетику соединений in vivo изучали на крысах и оценивали фармакокинетические характеристики. Приготовление лекарства
Взвешивали определенное количество соединений Примера 1 и Примера 2 и готовили в виде раствора с рН=4,0 с использованием 20 ммоль/л дигидрофосфата натрия для последующего использования.
1.1 Введение лекарства
Лекарство вводили путем внутривенной болюсной инъекции со временем введения около 5 мин, дозой введения 2 мг/кг, концентрацией введения 0,4 мг/мл и объемом введения 5 мл/кг.
1.2 Проведение опыта
Кровь отбирали из глазничной вены до введения и через 5 мин, 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч, 24 ч и 48 ч после введения. Около 0,6 мл отбирали для каждого образца, который подвергали антикоагуляции с использованием гепарина натрия и помещали на лед сразу после отбора. Образцы крови после отбора помещали в маркированные центрифужные пробирки, а плазму отделяли центрифугированием (условия центрифугирования: центробежная сила 2200 g, центрифугирование при 2-8°С в течение 10 мин).
1.3 Результаты фармакокинетических параметров
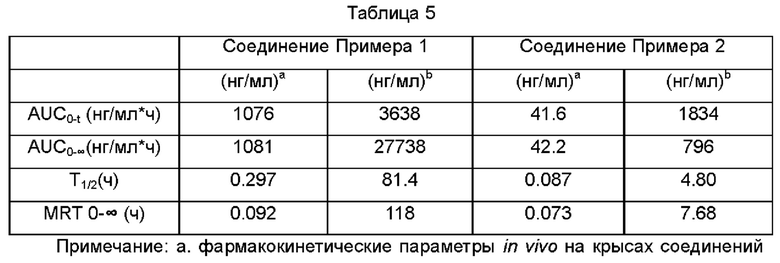
Примеров, b. фармакокинетические параметры ролапитанта in vivo после метаболизма соединений Примеров у крыс.
Выводы: хотя соединение Примера 1 практически не метаболизируется in vitro, особенно в плазме человека, до активного вещества ролапитанта, оно демонстрирует отличные фармакокинетические данные ролапитанта у крыс, что указывает на то, что соединение Примера 1 метаболизируется до ролапитанта in vivo. Более того, по данным AUC0-∞, AUC0-t и Т1/2 метаболический цикл in vivo соединения 1 после введения более продолжительный, а уровень абсорбции и воздействия соединения 1 сравним с таковыми для ролапитанта.
Тестовый Пример 6: Фармакокинетические испытания соединений на яванских макаках
В качестве подопытных животных использовали яванских макак. Концентрацию лекарственного средства в плазме в различные моменты времени после внутривенного введения соединения Примера 5 путем инъекции определяли с использованием метода ЖХ/МС/МС. У яванских макак изучали фармакокинетику соединения согласно настоящему изобретению in vivo и оценивали фармакокинетические характеристики.
Приготовление лекарства
Взвешивали определенное количество соединения Примера 5 и готовили в виде раствора с рН=4,0 с использованием 20 ммоль/л дигидрофосфата натрия для последующего использования.
1.1 Введение лекарства
Лекарство вводили внутривенно со временем введения около 30 мин, дозой введения 2 мг/кг, концентрацией введения 0,4 мг/мл и объемом введения 5 мл/кг.
1.2 Проведение опыта
Кровь отбирали из бедренной вены до введения и через 5 мин, 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч, 24 ч и 48 ч после введения. Около 0,6 мл отбирали для каждого образца, который подвергали антикоагуляции с использованием гепарина натрия и помещали на лед сразу после отбора. Образцы крови после отбора помещали в маркированные центрифужные пробирки, а плазму отделяли центрифугированием (условия центрифугирования: центробежная сила 2200 g, центрифугирование при 2-8°С в течение 10 мин).
Содержание соединения Примера 5 и ролапитанта в образцах плазмы определяли методом ЖХ/МС/МС.
1.3 Результаты фармакокинетических параметров
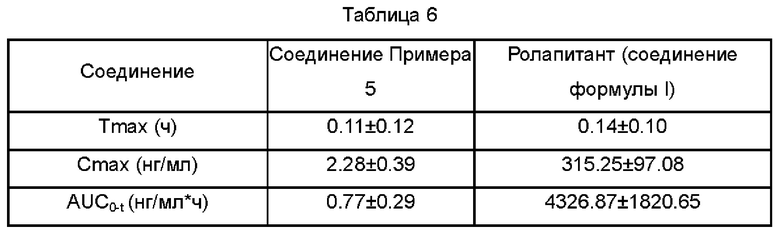
Выводы. При фармакокинетическом исследовании соединения Примера 5 у яванских макак большая часть соединения быстро превращалась в активный метаболит ролапитант, обладающий хорошими фармакокинетическими свойствами.
Пример 19:
Соединение 3 получали в соответствии со стадиями 1-2 Примера 1. Затем соединение 3 (6,65 г, 8,67 ммоль, 1 экв.) растворяли в дихлорметане (200 мл) в одногорлой колбе на 500 мл в атмосфере азота и медленно добавляли трифторуксусную кислоту (9,89 г, 86,7 ммоль, 10,0 экв.) при охлаждении в ледяной воде. Реакционную смесь перемешивали до завершения реакции. Реакционную смесь концентрировали с получением 2,29 г масла, которое очищали на колонке с силикагелем (С18) с обращенной фазой (раствор А: 20 ммоль водный раствор NH4HCO3, раствор В: ацетонитрил) и затем доводили до рН=1~2 с помощью 1 М фосфорной кислоты, экстрагировали дихлорметаном, промывали насыщенным раствором соли, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением 2,7 г целевого продукта.
Тестовый Пример 7: Растворимость
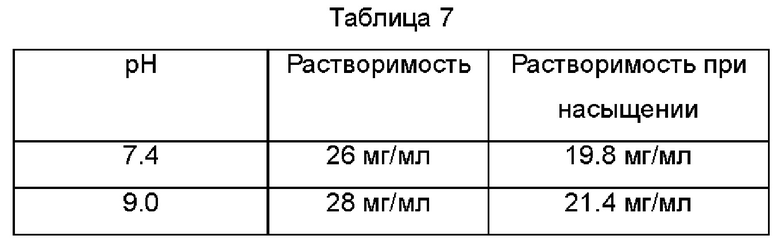
Измеряли растворимость соединения Примера 19 при различных значениях рН с использованием метода определения растворимости из Тестового Примера 1. Получены следующие данные:
Тестовый Пример 8: Гемолитический эффект
Красные кровяные клетки (эритроциты) собирали из яремной вены или центральной ушной артерии кроликов (10 мл цельной крови с ЭДТА). Кровь помещали в коническую колбу со стеклянными шариками и встряхивали в течение 10 мин для удаления фибриногена, в результате чего кровь дефибринировалась. К дефибринированной крови добавляли примерно 10-кратное количество раствора хлорида натрия, после чего хорошо встряхивали и центрифугировали при 1500 об/мин в течение 10 мин. Супернатант удаляли, а выпавшие в осадок эритроциты промывали 3 раза введением хлорида натрия по описанной выше методике до тех пор, пока супернатант не становился бесцветным. Полученные эритроциты готовили в виде 2% (об./об.) суспензии с введением хлорида натрия для последующего использования.
Исследуемые образцы (соединение Примера 5 и соединение Примера 19) соответственно растворяли в фосфатно-солевом буферном растворе (PBS) (рН 7,4 или рН 5) и фильтровали для получения 0,4 мг/мл, 0,8 мг/мл, 1,2 мг/мл, 1,6 мг/мл и 2 мг/мл растворов для последующего использования.
Некоторое количество раствора испытуемого образца добавляли к вышеуказанному гемоглобину для тестирования в супернатанте.
Если раствор в пробирке является прозрачным и красным, а оставшихся клеток нет или есть небольшое количество оставшихся эритроцитов на дне пробирки, это указывает на то, что произошел гемолиз. Если все эритроциты опускаются, а надосадочная жидкость бесцветная и прозрачная, это свидетельствует об отсутствии гемолиза. Если в растворе имеются буро-красные или красно-коричневые хлопьевидные осадки, которые после 3-5-кратного осторожного переворачивания все еще не диспергируются, это свидетельствует о возможной коагуляции эритроцитов. Образец следует дополнительно исследовать под микроскопом, и, если эритроциты видны как агрегированные, значит, произошла коагуляция. С использованием этого метода определяли гемолитический эффект соединений согласно настоящему изобретению.
Вывод: соединение Примера 19 не оказывает гемолитического действия в концентрации до 2 мг/мл, а соединение Примера 5 оказывает гемолитическое действие в концентрации 0,04 мг/мл и выше.
Тестовый Пример 9: Гемолитический эффект эмульсии ролапитанта
Эмульсию ролапитанта готовили способом, описанным в CN 102573475 (состав: 4,4% полиэтиленгликоль-15-гидроксистеарата, 1,1% триглицерида со средней длиной цепи и 0,66% соевого масла), и получали концентрации 0,18 мг/мл, 0,09 мг/мл, 0,045 мг/мл, 0,023 мг/мл, 0,011 мг/мл, 0,056 мг/мл и 0,028 мг/мл с PBS для последующего использования.
Гемолитический эффект определяли способом, описанным в Тестовом примере 8.
Вывод: эмульсии ролапитанта при всех концентрациях проявляли гемолитический эффект.
Тестовый Пример 10: Фармакокинетические испытания на яванских макаках
В качестве подопытных животных использовали яванских макак. Концентрацию лекарственного средства в плазме в различные моменты времени после внутривенного введения соединения Примера 19 путем инъекции определяли с использованием метода ЖХ/МС/МС.У яванских макак изучали фармакокинетику соединений согласно настоящему изобретению in vivo и оценивали фармакокинетические характеристики.
Приготовление лекарства
Взвешивали определенное количество тестируемого соединения и готовили в виде раствора с рН=4,0 с использованием 20 ммоль/л дигидрофосфата натрия для последующего применения.
1.1 Введение лекарства
Лекарство вводили внутривенно со временем введения около 30 мин, дозой введения 3,54 мг/кг, концентрацией введения 2 мг/мл и объемом введения 5 мл/кг.
1.2 Проведение опыта
Кровь отбирали из бедренной вены до введения и через 5 мин, 0,25 ч, 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч после введения. Отбирали около 0,6 мл каждого образца, который подвергали антикоагуляции с использованием гепарина натрия и помещали на лед сразу после отбора. Образцы крови после отбора помещали в маркированные центрифужные пробирки, а плазму отделяли центрифугированием (условия центрифугирования: центробежная сила 2200 g, центрифугирование при 2-8°С в течение 10 мин).
Содержание соединения Примера 24 и ролапитанта в образцах плазмы определяли методом ЖХ/МС/МС.
1.3 Результаты фармакокинетических параметров
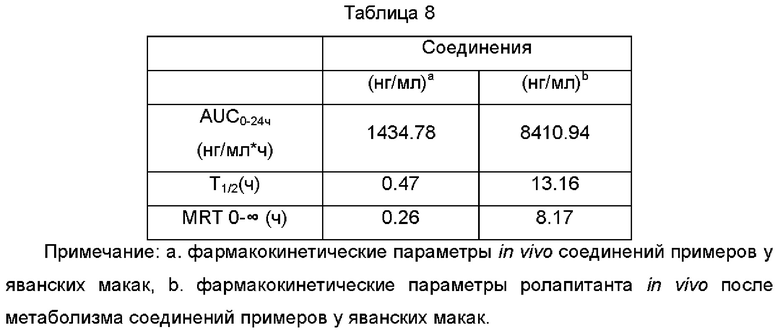
Вывод: при исследовании фармакокинетики соединения in vivo на яванских макаках большая часть соединения быстро превращается в активный метаболит ролапитант у яванских макак, и соединение обладает хорошими фармакокинеическими свойствами. Кроме того, по сравнению с соединением Примера 5 это соединение имеет более высокую биодоступность in vivo у яванских макак.

Изобретение относится к соединению формулы III или его фармацевтически приемлемой соли, где каждый R1, R2 независимо выбран из группы, состоящей из водорода и С1-6 алкила; m=1, 2, 3 или 4. Соединение формулы III является антагонистом рецептора нейрокинина-1, может быть использовано для лечения заболеваний, связанных с рецептором нейрокинина-1, например для лечения рвоты, тошноты, депрессии или тревожности у пациента, а также позволяет избежать гемолитических эффектов лекарственных средств и уменьшить побочные эффекты введения лекарственных средств. 3 н. и 1 з.п. ф-лы, 1 ил., 8 табл., 18 пр.
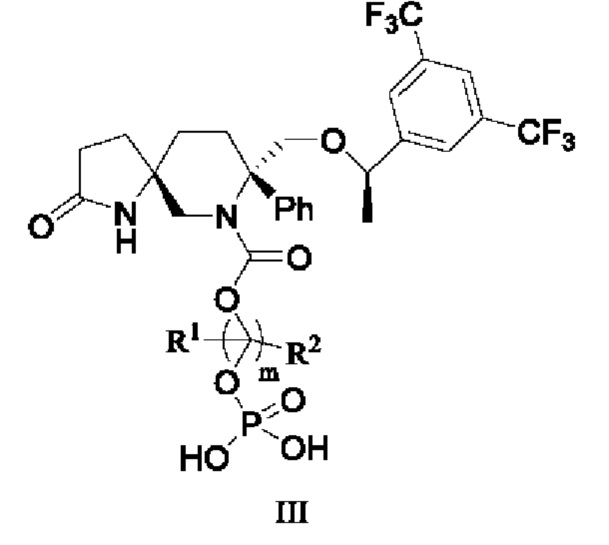
1. Соединение формулы III
или его фармацевтически приемлемая соль, где
каждый R1, R2 независимо выбран из группы, состоящей из водорода и С1-6 алкила;
m=1, 2, 3 или 4.
2. Соединение по п. 1, где соединение формулы (III) выбрано из группы, состоящей из:
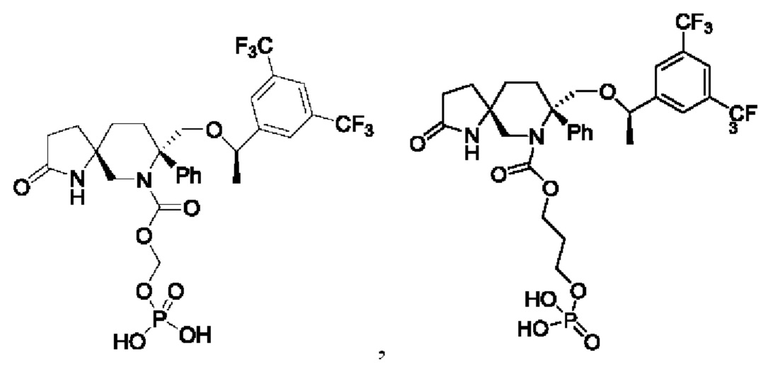
или их фармацевтически приемлемой соли.
3. Фармацевтическая композиция для лечения рвоты, тошноты, депрессии или тревожности у пациента, содержащая терапевтически эффективное количество по меньшей мере одного из соединений по любому из пп. 1, 2, а также фармацевтически приемлемый носитель, разбавитель или эксципиент.
4. Применение соединения по любому из пп. 1, 2 или фармацевтической композиции по п. 3 для получения лекарственных средств для лечения физиологических расстройств, состояний или заболеваний у пациента, где физиологическое расстройство, состояние или заболевание выбрано из группы, состоящей из рвоты, тошноты, депрессии и тревожности.
| RU 2012109405 A, 20.09.2013 | |||
| WO 2005063243 A1, 14.07.2005 | |||
| CN 107383008 A, 24.11.2017. |

