ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение принадлежит к области медицины и относится к пиперазингетероарильному производному, способу его получения и применению в медицине. В частности, настоящее изобретение относится к пиперазингетероарильному производному формулы (I), способу его получения, содержащей его фармацевтической композиции и их применению в качестве ингибитора капсидного белка, в частности, для предупреждения и/или лечения таких заболеваний, как гепатит В, грипп, герпес и СПИД (англ. AIDS, Acquired ImmunoDeficiency Syndrome - синдром приобретенного иммунодефицита).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Хроническая инфекция, вызываемая вирусом гепатита В (англ. HBV - Hepatitis В Virus), является уже глобальной проблемой здравоохранения. По данным Всемирной организации здравоохранения, около 2 миллиардов человек по всему миру заражены вирусом гепатита В (HBV), из них 240 миллионов являются хронически инфицированными HBV. Около 650000 человек умирают ежегодно от печеночной недостаточности, цирроза печени и гепато-целлюлярной карциномы (англ. НСС), вызванных HBV-инфекцией. Причиной цирроза печени у 30% пациентов и НСС у 45% во всем мире является HBV-инфекция. И несмотря на возможность использования профилактических вакцин против HBV, хроническая HBV-инфекция стала общемировой медицинской проблемой из-за отсутствия эффективных лекарственных средств.
В настоящее время существует два основных класса лекарственных средств для лечения хронической HBV-инфекции: препараты альфа-интерферона (такие как пегилированный альфа-интерферон) и аналоги нуклеозидов, ингибирующие ДНК-полимеразу HBV (такие как ламивудин, адефовир и др.). Однако препараты интерферона обладают серьезными побочными эффектами и плохой переносимостью, при этом лишь небольшая часть пациентов может иметь устойчивый клинический ответ на терапию интерферонами (Lancet. 2005 Jan 8-14; 365(9454): 123-9.; N. Engl. J. Med. 2005 Jun 30; 352(26):2682-95.; Hepatology. 2009 May; 49(5 Suppl): S103-11). Являясь конкурентными ингибиторами обратной транскриптазы, лекарственные средства на основе аналогов нуклеозидов оказывают противовирусное действие, блокируя синтез цепей ДНК HBV. Однако в случае существующих лекарственных средств на основе нуклеозидных аналогов возникают такие проблемы, как индуцирование обратной транскриптазы с появлением резистентных к лекарственным средствам мутаций и низкая эффективность в отношении штаммов, резистентных к лекарственным средствам. Кроме того, с помощью таких лекарственных средств зачастую сложно полностью устранить HBV-инфекцию даже при их приеме в течение длительного времени; после прекращения приема препарата может возникнуть серьезный синдром рикошета, поэтому часто требуется пожизненная медикаментозная терапия (Hepatology. 2009 May; 49(5 Suppl):S112-21). Таким образом, существует насущная потребность в разработке нового, безопасного и эффективного лекарственного средства для лечения хронического гепатита В.
Низкий уровень излечения хронической HBV-инфекции тесно связан с характеристиками вируса гепатита В (HBV). HBV представляет собой оболочечный, частично двухцепочечный ДНК-вирус (dsDNA) семейства Hepadnaviridae. Наружный слой зрелых вирусных частиц HBV представляет собой оболочечный белок, который инкапсулирует нуклеокапсид HBV. Нуклеокапсид, также называемый коровой частицей, состоит из капсидного белка, расслабленной кольцевой ДНК HBV (rcDNA) и обратной транскриптазы HBV, присоединенной к 5'-концу отрицательной цепи rcDNA. После инфицирования rcDNA превращается в ядре клетки-хозяина в ковалентно замкнутую кольцевую ДНК (cccDNA), выступающую в качестве матрицы для репликации HBV. Важным моментом процесса репликации HBV является капсидирование. Для завершения этапа сборки прегеномная РНК (pgRNA), транскрибируемая с cccDNA, должна быть инкапсулирована в капсидный белок вместе с обратной транскриптазой HBV, инициируя тем самым последующую обратную транскрипцию. Перед обратной транскрипцией обратная транскриптаза HBV и pgRNA должны быть надлежащим образом инкапсулированы капсидным белком. Следовательно, блокируя сборку капсидного белка или ускоряя разрушение капсидного белка, можно блокировать процесс сборки капсида и тем самым воздействовать на репликацию вируса. Кроме того, N-концевые 149 аминокислотных остатков (Ср149), составляющие мотив димеризации корового белка и домен сборки, не имеют гомологичных последовательностей человеческого белка. Вследствие этого ингибитор сборки капсидного белка можно рассматривать в качестве нового объекта для разработки лекарственных средств против гепатита В. Благодаря механизму действия, отличающемуся от традиционных противовирусных лекарственных средств, ингибитор капсидного белка может быть скомбинирован с ингибитором ДНК-пол имеразы для синергического ингибирования репликации HBV и предотвращения возникновения лекарственной устойчивости, обеспечивая при этом более безопасное и более эффективное лечение хронической инфекции гепатита В.
В настоящее время существует два основных класса ингибиторов капсидного белка: гетероарилдигидропиримидины (НАР) и фенилакриламиды, такие как GLS-4, NVR-3778 и тому подобное. К соответствующим заявкам на патент относятся WO 2001068642, WO 2014029193, WO 2015011281, WO 2016016196, WO 2017076791, WO 2016113273 и др. Однако большая часть соединений, направленных на эту мишень, находятся в стадии клинических исследований, а зарегистрированные для продажи лекарственные средства отсутствуют. Следовательно, сохраняется необходимость в постоянном усовершенствовании ингибиторов капсидного белка с целью повышения безопасности и эффективности лекарственных средств и решения проблемы хронической HBV-инфекции на ранней стадии.
Ингибитор капсидного белка препятствует нормальной сборке капсидного белка, связываясь с доменом сборки мотива димеризации корового белка, и тем самым влияет на репликацию HBV. Следовательно, хорошая фармакокинетическая абсорбция и высокая биодоступность (которые могут привести к повышению концентрации соединения в организме) позволят более эффективно блокировать репликацию HBV.
Настоящее изобретение предлагает новую структуру ингибитора капсидного белка формулы (I), где заместитель в гетероарильном фрагменте представляет собой ациламиногруппу, при этом аминогруппа, входящая в состав ациламиногруппы, предпочтительно является вторичной аминогруппой. В настоящем изобретении предусмотрен сравнительный пример (Пример 59), соответствующий соединению формулы (I), где в группе NR1R2 заместители R1 и R2 вместе с атомом азота образуют кольцо, содержащее третичную аминогруппу. Сравнительный пример показывает, что в случае, когда аминогруппа, входящая в состав ациламиногруппы в гетероарильном фрагменте, является вторичной аминогруппой, соединение проявляет значительно более высокую биологическую активность, оказывает заметное ингибирующее действие на нормальную сборку капсидного белка HPV и имеет хорошую фармакокинетическую абсорбцию и высокую биодоступность. В то же время, новая структура соединения формулы (I) не влияет или мало влияет на ингибирование пролиферации клеток HepG2 in vitro и демонстрирует высокую безопасность.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Цель настоящего изобретения - предложить соединение формулы (I):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую сол,
где:
кольцо А является арилом или гетероарилом;
Y представляет собой N или CR5;
Q представляет собой N или СН;
R1 выбран из группы, состоящей из алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила независимо друг от друга необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6;
R2 выбран из группы, состоящей из водорода, алкила, галогеналкила, гид роксиал кила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила независимо друг от друга необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6;
каждый из R4 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6;
R5 выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R6 выбран из группы, состоящей из водорода, алкила, галогеналкила, амино, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
n равно 0, 1, 2 или 3;
m равно 0, 1 или 2; и
s равно 0, 1, 2, 3 или 4.
Согласно предпочтительному варианту осуществления настоящего изобретения, соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (II):

где:
кольцо A, Y, Q, R1, R3, R4, s и n являются такими, как определены для формулы (I).
Согласно другому предпочтительному варианту осуществления настоящего изобретения, соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (III), формулы (IV), формулы (V) или формулы (VI):

где:
кольцо A, R1, R3, R4, s и n являются такими, как определены для формулы (I).
Согласно еще одному предпочтительному варианту осуществления настоящего изобретения, в соединении формулы (I) по настоящему изобретению кольцо А является фенилом или пиридилом.
Согласно другому предпочтительному варианту осуществления настоящего изобретения, соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (VII), формулы (VIII), формулы (IX) или формулы (X):

где:
G представляет собой С или N; а
R1, R3, R4, s и n являются такими, как определены для формулы (I).
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в соединении формулы (I) по настоящему изобретению R1 выбран из группы, состоящей из алкила, галогеналкила, циклоалкила, гетероциклила и арила, где алкил, циклоалкил, гетероциклил и арил необязательно дополнительно замещены одним или более заместителями, выбранными из группы, состоящей из галогена, алкила, алкокси и гидрокси.
Согласно другому предпочтительному варианту осуществления настоящего изобретения, соединение формулы (I) по настоящему изобретению представляет собой соединение формулы (VII-А), формулы (VIII-A), формулы (IX-A) или формулы (Х-А):

где:
G представляет собой С или N;
R8 представляет собой алкил, предпочтительно, метил;
R9 представляет собой алкил, где алкил необязательно дополнительно замещен одним или более атомами галогена; а
R3, R4, s и n являются такими, как определены для формулы (I).
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в соединении формулы (I) по настоящему изобретению R3 является водородом или алкилом.
Согласно другому предпочтительному варианту осуществления настоящего изобретения, в соединении формулы (I) по настоящему изобретению R4 выбран из группы, состоящей из водорода, галогена, галогеналкила и циано.
Типичные соединения формулы (I) включают, не ограничиваясь перечнем:








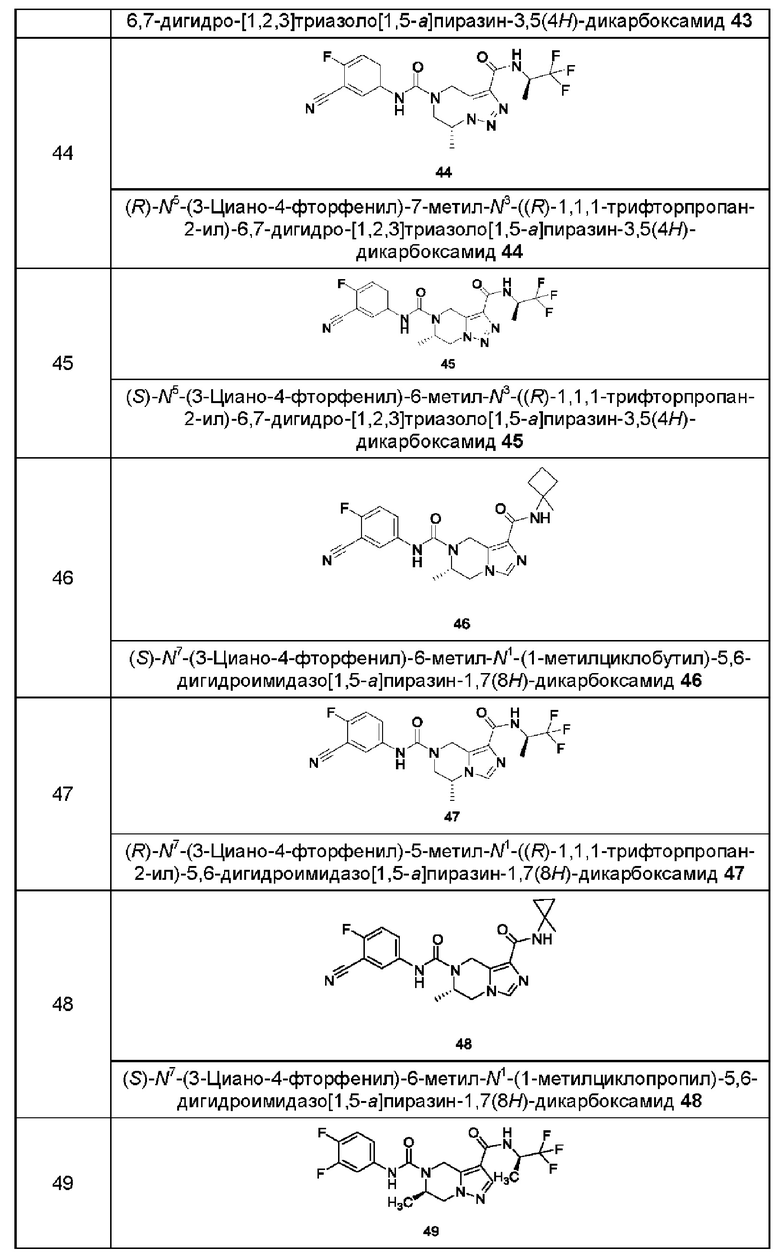


или их таутомер, мезомер, рацемат, энантиомер, диастереомер или их смеси, или их фармацевтически приемлемую соль.
Согласно другому аспекту, настоящее изобретение предлагает соединение формулы (IA):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
Ra представляет собой водород или алкил;
кольцо А является арилом или гетероарилом;
Y представляет собой N или CR5;
Q представляет собой N или СН;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6 и, предпочтительно, водорода или алкила;
каждый из R4 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6;
R5 выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R6 выбран из группы, состоящей из водорода, алкила, галогеналкила, амино, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
n равно 0, 1, 2 или 3;
m равно 0, 1 или 2; и
s равно 0, 1, 2, 3 или 4.
Согласно другому аспекту, настоящее изобретение предлагает соединение формулы (IC):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
кольцо А является арилом или гетероарилом;
Y представляет собой N или CR5;
Q представляет собой N или СН;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6 и, предпочтительно, водорода или алкила;
каждый из R4 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6;
R5 выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R6 выбран из группы, состоящей из водорода, алкила, галогеналкила, амино, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
n равно 0,1,2 или 3;
m равно 0, 1 или 2; и
s равно 0, 1, 2, 3 или 4.
Эти соединения являются промежуточными продуктами для получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли.
Типичные соединения формулы (IA) включают, не ограничиваясь перечнем:

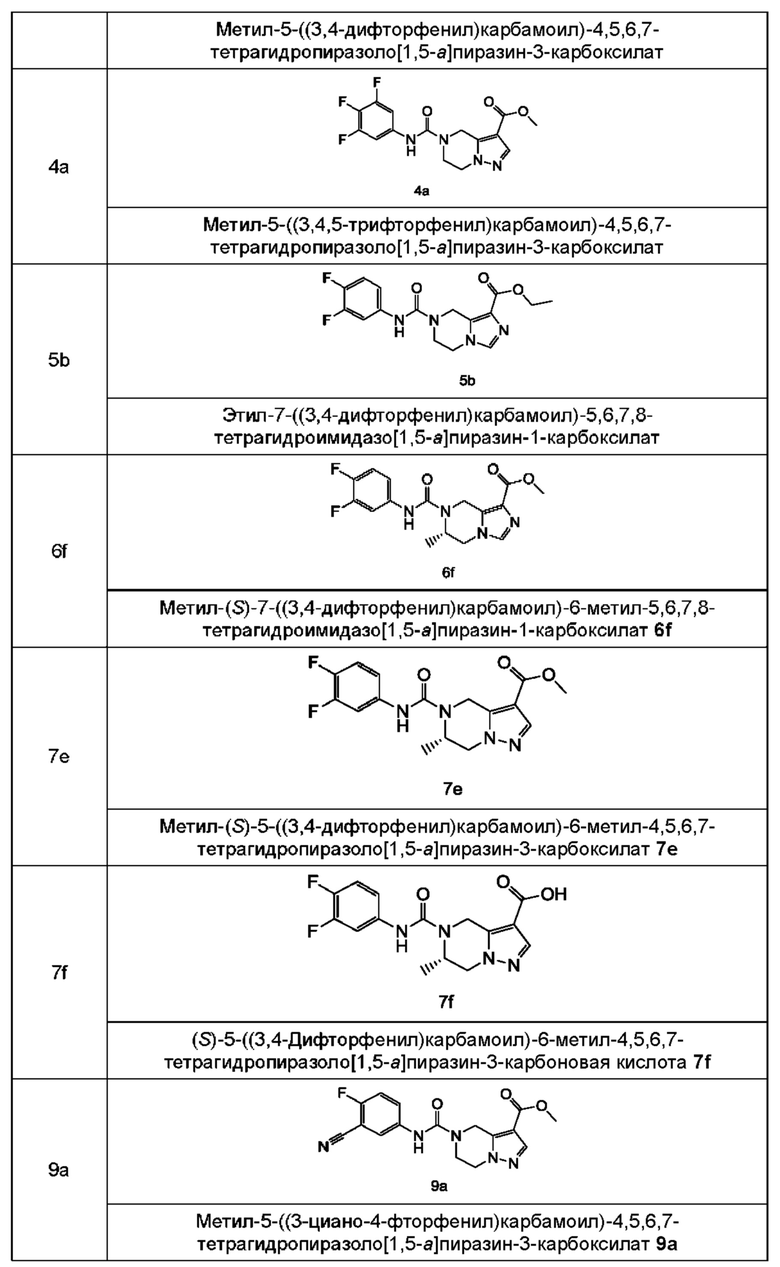


Согласно другому аспекту, настоящее изобретение предлагает соединение формулы (IIIA):

или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемую соль,
где:
М является трифторуксусной кислотой или соляной кислотой;
R1 выбран из группы, состоящей из алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый из алкила, галогеналкила, циклоалкила, гетероциклила, арила и гетероарила независимо друг от друга необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6 и, предпочтительно, водорода или алкила;
t равно 0 или 1; а
n равно 0, 1, 2 или 3.
Это соединение является промежуточным соединением для получения соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли.
Типичные промежуточные соединения включают, не ограничиваясь перечнем:
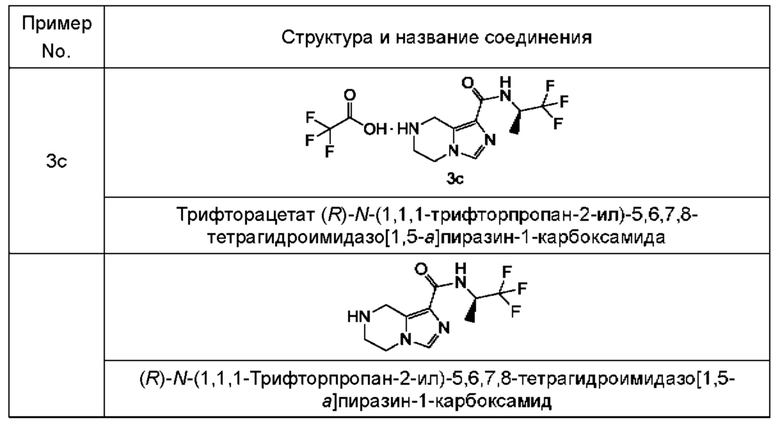

Согласно другому аспекту, настоящее изобретение предлагает способ получения соединения формулы (I) в соответствии с настоящим изобретением, включающий в себя стадию:
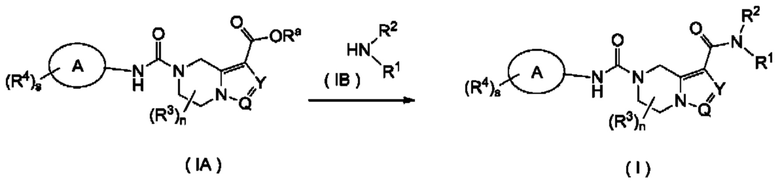
взаимодействия соединения формулы (IA) с соединением формулы (IB) или его солью с получением соединения формулы (I),
где:
Ra представляет собой водород или алкил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6 и, предпочтительно, водорода или алкила; и
кольцо A, Y, Q, R1, R2, R4, s и п являются такими, как определены для формулы (I).
Согласно другому аспекту, настоящее изобретение предлагает способ получения соединения формулы (I) в соответствии с настоящим изобретением, включающий в себя стадию:

взаимодействия соединения формулы (IC) с соединением формулы (IB) или его солью с получением соединения формулы (I),
где:
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6 и, предпочтительно, водорода или алкила; и
кольцо A, Y, Q, R1, R2, R4, s и n являются такими, как определены для формулы (I).
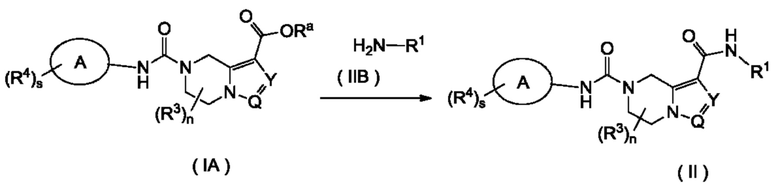
Согласно другому аспекту, настоящее изобретение предлагает способ получения соединения формулы (II) в соответствии с настоящим изобретением, включающий в себя стадию:

взаимодействия соединения формулы (IA) с соединением формулы (IIB) или его солью с получением соединения формулы (II),
где:
Ra представляет собой водород или алкил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; и
кольцо A, Y, Q, R1, R4, s и n являются такими, как определены для формулы (I).
Согласно другому аспекту, настоящее изобретение предлагает способ получения соединения формулы (II) в соответствии с настоящим изобретением, включающий в себя стадию:

взаимодействия соединения формулы (IC) с соединением формулы (IIB) или его солью с получением соединения формулы (II),
где:
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; и
кольцо A, Y, Q, R1, R4, s и n являются такими, как определены для формулы (II).
Согласно другому аспекту, настоящее изобретение предлагает способ получения соединения формулы (III), формулы (IV), формулы (V) или формулы (VI) в соответствии с настоящим изобретением, включающий в себя стадию:

взаимодействия соединения формулы (III-а) с соединением формулы (IIB) или его солью с получением соединения формулы (III) или

взаимодействия соединения формулы (IV-a) с соединением формулы (IIB) или его солью с получением соединения формулы (IV),
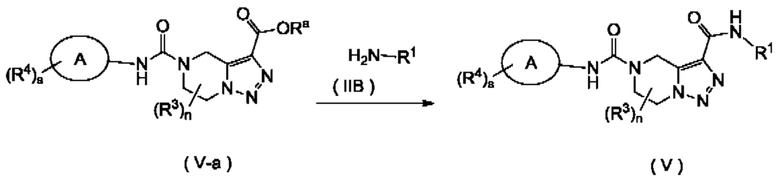
взаимодействия соединения формулы (V-a) с соединением формулы (IIB) или его солью с получением соединения формулы (V) или

взаимодействия соединения формулы (VI-a) с соединением формулы (IIB) или его солью с получением соединения формулы (VI),
где:
Ra представляет собой водород или алкил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
кольцо A, R1, R4, s и n являются такими, как определены для формулы (I).
Согласно другому аспекту, настоящее изобретение предлагает способ получения соединения формулы (III), включающий в себя стадию:

взаимодействия соединения формулы (IIIA) или его соли с соединением формулы (IIB') или его солью и бис(трихлорметил)карбонатом с получением соединения формулы (III),
где:
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; и
М является трифторуксусной кислотой или соляной кислотой;
t равно 0 или 1;
кольцо A, R1, R4, s и n являются такими, как определены для формулы (III).
Согласно другому аспекту, настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли и один или более фармацевтически приемлемых носителя (носителей), разбавителя (разбавителей) или вспомогательного вещества (веществ). Настоящее изобретение также относится к способу получения указанной выше композиции, включающему в себя стадию смешивания соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли с фармацевтически приемлемым носителем (носителями), разбавителем (разбавителями) или вспомогательным веществом (вспомогательными веществами).
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения ингибитора капсидного белка.
Настоящее изобретение также относится к применению соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для получения лекарственного средства для предупреждения и/или лечения вирусного инфекционного заболевания. Вирус может быть вирусом гепатита В, вирусом гриппа, вирусом герпеса и вирусом СПИД, а заболевание может быть гепатитом В, гриппом, герпесом и СПИД.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли для применения в качестве лекарственного препарата. Лекарственный препарат предпочтительно представляет собой лекарственный препарат для предупреждения и/или лечения вирусного инфекционного заболевания. Вирус может быть вирусом гепатита В, вирусом гриппа, вирусом герпеса и вирусом СПИД, а заболевание может быть гепатитом В, гриппом, герпесом и СПИД.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции для применения в качестве ингибитора капсидного белка.
Настоящее изобретение также относится к способу предупреждения и/или лечения вирусного инфекционного заболевания, включающему в себя стадию введения пациенту, нуждающемуся в этом, терапевтически эффективной дозы соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или содержащей их фармацевтической композиции в качестве ингибитора капсидного белка. Вирус может быть вирусом гепатита В, вирусом гриппа, вирусом герпеса и вирусом СПИД, а заболевание может быть гепатитом В, гриппом, герпесом и СПИД.
Фармацевтическая композиция, содержащая активный ингредиент, может иметь форму, подходящую для перорального введения, например, это может быть таблетка, лепешка, пастилка, водная или масляная суспензия, диспергируемый порошок или гранулы, эмульсия, твердая или мягкая капсула, сироп или эликсир. Композиция для перорального применения может быть приготовлена при помощи любого известного в данной области техники способа приготовления фармацевтической композиции. Для получения привлекательной и приятной на вкус рецептуры такая композиция может содержать один или более ингредиентов, выбранных из группы, состоящей из подсластителей, вкусо-ароматических добавок, красителей и консервантов. Таблетка содержит активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми вспомогательными веществами, подходящими для изготовления таблеток. Такими вспомогательными веществами могут быть инертные вспомогательные вещества, гранулирующие агенты, разрыхлители, связующие и смазывающие вещества. Таблетка может быть без покрытия или может иметь покрытие, нанесенное с помощью известных способов, для маскировки вкуса лекарственного средства или замедления распада и абсорбции активного ингредиента в желудочно-кишечном тракте, что обеспечивает замедленное высвобождение в течение длительного времени.
Состав для перорального применения также может быть представлен в виде мягких желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, либо активный ингредиент смешан с водорастворимым носителем или масляной средой.
Водная суспензия содержит активный ингредиент в смеси с вспомогательными веществами, подходящими для приготовления водной суспензии. Такими вспомогательными веществами являются суспендирующие агенты, диспергирующие вещества или увлажняющие агенты. Водная суспензия может также содержать один или более консервантов, один или более красителей, одну или более вкусо-ароматических добавок и один или более подсластителей.
Масляная суспензия может быть приготовлена путем суспендирования активного ингредиента в растительном или минеральном масле. Масляная суспензия может содержать загуститель. Для получения приятного на вкус состава могут быть добавлены упоминавшиеся выше подсластители и вкусо-ароматические добавки. Для сохранения таких композиций в них может быть добавлен антиокислитель.
Фармацевтическая композиция по настоящему изобретению также может иметь форму эмульсии типа "масло в воде". Масляная фаза может быть растительным маслом или минеральным маслом, или их смесью. Приемлемыми эмульгирующими агентами могут быть фосфолипиды природного происхождения. Кроме того, эмульсия может содержать подсластитель, вкусо-ароматическую добавку, консервирующую добавку и антиокислитель. Такой состав также может содержать средство, уменьшающее раздражение, консервирующую добавку, краситель и антиокислитель.
Фармацевтическая композиция по настоящему изобретению может иметь форму стерильного водного раствора для инъекций. В качестве подходящих носителей или растворителей можно использовать воду, раствор Рингера или изотонический раствор хлорида натрия. Стерильный состав для инъекций может представлять собой стерильную микроэмульсию типа "масло в воде" для инъекций, в которой активный ингредиент растворен в масляной фазе. Раствор или микроэмульсию для инъекций можно вводить в кровоток пациента при помощи локальной болюсной инъекции. В качестве альтернативы, раствор и микроэмульсию предпочтительно вводят таким образом, чтобы поддерживать постоянную циркулирующую концентрацию соединения по настоящему изобретению. Для поддержания этой постоянной концентрации можно использовать устройство для непрерывной внутривенной доставки. Примером такого устройства является насос для внутривенных инъекций Deltec CADD-PLUS. ТМ. 5400.
Фармацевтическая композиция по настоящему изобретению может иметь форму стерильной водной или масляной суспензии для инъекций, подходящей для внутримышечного и подкожного введения. Такая суспензия может быть приготовлена с использованием подходящих диспергирующих веществ или увлажняющих агентов и суспендирующих агентов, как описаны выше, с помощью известных способов. Стерильный состав для инъекций также может представлять собой стерильный раствор или суспензию для инъекций, приготовленные в нетоксичном, подходящем для парентерального введения разбавителе или растворителе. Кроме того, в качестве растворителя или суспендирующей среды можно использовать стерильные нелетучие масла. Для этой цели может быть использовано любое смешанное нелетучее масло. Кроме того, для приготовления инъекций также можно использовать жирные кислоты.
Соединение по настоящему изобретению можно вводить в форме суппозитория для ректального введения. Такие фармацевтические композиции могут быть приготовлены путем смешивания лекарственного средства с подходящим не вызывающим раздражения вспомогательным веществом, которое является твердым при обычных температурах, но становится жидким в прямой кишке и, следовательно, расплавляется в прямой кишке с высвобождением лекарственного средства.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от множества факторов, включая, но не ограничиваясь перечнем, следующие: активность конкретного соединения, возраст пациента, вес пациента, общее состояние здоровья пациента, образ жизни пациента, питание пациента, время введения, способ введения, скорость экскреции, комбинация лекарственных средств и тому подобное. Кроме того, оптимальное лечение, а именно, режим лечения, суточная доза соединения по настоящему изобретению или тип его фармацевтически приемлемой соли, может быть проверено с помощью традиционных схем лечения.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
Термин "алкил" относится к насыщенной алифатической углеводородной группе, представляющей собой группу с прямой или разветвленной цепью, содержащую от 1 до 20 атомов углерода, предпочтительно, алкильную группу, содержащую от 1 до 12 атомов углерода, и, более предпочтительно, алкильную группу, содержащую от 1 до 6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно, алкильная группа представляет собой низший алкил, содержащий от 1 до 6 атомов углерода, при этом неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметил пропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и тому подобное. Алкильная группа может быть замещенной или незамещенной. При наличии замещения заместитель (заместители) может находиться в любом доступном для присоединения месте. Заместитель (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетеро цикл о алкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси, алкокси карбон ил a, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6.
Термин "алкокси" относится к группе -О-(алкил) или -O-(незамещенный циклоалкил), где алкил и циклоалкил такие, как определены выше. Неограничивающие примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Алкоксильная группа может быть необязательно замещенной или незамещенной. При наличии замещения заместитель (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиол, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси, алкокси карбон ил a, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6.
Термин "циклоалкил" относится к насыщенному или частично ненасыщенному моноциклическому или полициклическому углеводородному заместителю, содержащему от 3 до 20 атомов углерода, предпочтительно, от 3 до 12 атомов углерода и, более предпочтительно, от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и тому подобное. Полициклический циклоалкил включает циклоалкил, содержащий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Термин "спироциклоалкил" относится к от 5 до 20-членной полициклической группе с отдельными кольцами, соединенными через один общий атом углерода (называемый спироатомом), где кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Спироциклоалкил предпочтительно представляет собой от 6 до 14-членный спироциклоалкил и, более предпочтительно, от 7 до 10-членный спироциклоалкил. В зависимости от числа спироатомов, общих для колец, спироциклоалкил можно подразделить на моноспироциклоалкил, диспироциклоалкил или полиспироциклоалкил, при этом спироциклоалкил предпочтительно представляет собой моноспироциклоалкил или диспироциклоалкил и, более предпочтительно, 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироциклоалкил. Неограничивающие примеры спироциклоалкила включают:

Термин "конденсированный циклоалкил" относится к от 5 до 20-членной полностью углеродной полициклической группе, где каждое кольцо в системе имеет общую смежную пару атомов углерода с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Конденсированный циклоалкил предпочтительно представляет собой от 6 до 14-членный конденсированный циклоалкил и, более предпочтительно, от 7 до 10-членный конденсированный циклоалкил. В зависимости от числа членных колец конденсированный циклоалкил можно подразделить на бицикпический, трициклический, тетра циклический или полициклический конденсированный циклоалкил, при этом конденсированный циклоалкил предпочтительно представляет собой бицикпический или трициклический конденсированный циклоалкил и, более предпочтительно, 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:

Термин "мостиковый циклоалкил" относится к от 5 до 20-членной полностью углеродной полициклической группе, где каждые два кольца в системе имеют два несвязанных атома углерода, где кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Мостиковый циклоалкил предпочтительно представляет собой от 6 до 14-членный мостиковый циклоалкил и, более предпочтительно, от 7 до 10-членный мостиковый циклоалкил. В зависимости от числа членных колец мостиковый циклоалкил можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, при этом мостиковый циклоалкил предпочтительно представляет собой бицикпический, трициклический или тетрациклический мостиковый циклоалкил и, более предпочтительно, бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостикового циклоалкила включают:

Циклоалкильное кольцо может быть сконденсировано с арильным, гетероарильным или гетероциклильным кольцом, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и тому подобное. Циклоалкил может быть необязательно замещенным или незамещенным. В случае замещения заместитель (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиол, гидрокси, нитро, циано, циклоалкила, гетеро цикл ил а, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси, алкокси карбон ил a, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6.
Термин "гетероциклил" относится к от 3 до 20-членной насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной группе, где один или более атомов кольца являются гетеро атомами, выбранными из группы, состоящей из N, О и S(O)m (где m - целое число от 0 до 2), но без -О-О-, -O-S- или -S-S- в кольце, а остальные атомы кольца представляют собой атомы углерода. Предпочтительно, гетероциклил содержит от 3 до 12 атомов в кольце, где от 1 до 4 атомов являются гетероатомами; более предпочтительно, от 3 до 8 атомов в кольце, где от 1 до 3 атомов являются гетероатомами; и, наиболее предпочтительно, от 3 до 6 атомов в кольце, где от 1 до 2 атомов являются гетероатомами. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидротиенил, дигидроимидазолил, дигидрофуранил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил, пиранил и тому подобное и, предпочтительно, пиперидинил, пиперазинил или морфолинил. Полициклический гетероциклил включает гетероциклил, содержащий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
Термин "спирогетероциклил" относится к от 5 до 20-членной полициклической гетероциклильной группе с отдельными кольцами, соединенными через один общий атом (называемый спироатом), где один или более атомов кольца являются гетероатомами, выбранными из группы, состоящей из N, О и S(O)m (где m - целое число от 0 до 2), а остальные атомы кольца представляют собой атомы углерода, при этом кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы. Спирогетероциклил предпочтительно представляет собой от 6 до 14-членный спирогетероциклил и, более предпочтительно, от 7 до 10-членный спирогетероциклил. В зависимости от числа спироатомов, общих для колец, спирогетероциклил можно подразделить на моноспирогетероциклил, диспирогетероциклил или полиспирогетероциклил, при этом спирогетероциклил предпочтительно представляет собой моноспирогетероциклил или диспирогетероциклил и, более предпочтительно, 3-членный/6-членный, 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспирогетероциклил. Неограничивающие примеры спирогетероциклила включают:

Термин "конденсированный гетероциклил" относится к от 5 до 20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую смежную пару атомов с другим кольцом, где одно или более колец может содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной π-электронной системы, и где один или более атомов кольца являются гетероатомами, выбранными из группы, состоящей из N, О и S(O)m (где m - целое число от 0 до 2), а остальные атомы кольца представляют собой атомы углерода. Конденсированный гетероциклил предпочтительно представляет собой от 6 до 14-членный конденсированный гетероциклил и, более предпочтительно, от 7 до 10-членный конденсированный гетероциклил. В зависимости от числа членных колец конденсированный гетероциклил можно подразделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, при этом конденсированный гетероциклил предпочтительно представляет собой бициклический или трициклический конденсированный гетероциклил и, более предпочтительно, 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:

Термин "мостиковый гетероциклил" относится к от 5 до 14-членной полициклической гетеро цикл ильной группе, где каждые два кольца в системе имеют два несвязанных атома, где кольца могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью сопряженной п-электронной системы, и где один или более атомов кольца являются гетероатомами, выбранными из группы, состоящей из N, О и S(O)m (где m - целое число от 0 до 2), а остальные атомы кольца представляют собой атомы углерода. Мостиковый гетероциклил предпочтительно представляет собой от 6 до 14-членный мостиковый гетероциклил и, более предпочтительно, от 7 до 10-членный мостиковый гетероциклил. В зависимости от числа членных колец мостиковый гетероциклил можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, при этом мостиковый гетероциклил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый гетероциклил и, более предпочтительно, бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостикового гетероциклила включают:


Гетероциклильное кольцо может быть сконденсировано с арильным, гетероарильным или циклоалкильным кольцом, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры этого включают:

Гетероциклил может быть необязательно замещенным или незамещенным. При наличии замещения заместитель (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси, алкокси карбонила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6.
Термин "арил" относится к от 6 до 14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (то есть каждое кольцо в системе имеет общую смежную пару атомов углерода с другим кольцом в системе), имеющему сопряженную п-электронную систему, предпочтительно, к от 6 до 10-членному арилу, например, фенилу и нафтилу. Более предпочтительно, арил является фенилом. Арильное кольцо может быть сконденсировано с гетероарильным, гетеро цикл ильным или циклоалкильным кольцом, где кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Неограничивающие примеры этого включают:


Арил может быть замещенным или незамещенным. При наличии замещения заместитель (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин "гетероарил" относится к от 5 до 14-членной гетероароматической системе, содержащей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N. Гетероарил предпочтительно представляет собой от 5 до 10-членный гетероарил, содержащий от 1 до 3 гетероатомов, более предпочтительно, 5 или 6-членный гетероарил, содержащий от 1 до 2 гетероатомов; предпочтительно, например, имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, тетразолил, пиридил, пиримидинил, тиадиазолил, пиразинил и тому подобное, предпочтительно, имидазолил, тетразолил, пиридил, тиенил, пиразолил, пиримидинил, тиазолил и, более предпочтительно, пиридил. Гетероарильное кольцо может быть сконденсировано с арильным, гетероциклильным или циклоалкильным кольцом, где кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Неограничивающие примеры этого включают:

Гетероарил может быть необязательно замещенным или незамещенным. При наличии замещения заместитель (заместители) предпочтительно представляет собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетеро циклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси, алкокси карбонила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6.
Термин "защитная группа для аминогруппы" относится к группе, которая препятствует реакциям аминогруппы в условиях, когда другие части молекулы подвергаются реакции, и которая впоследствии может быть легко удалена. Неограничивающие примеры включают трет-бутоксикарбонил, ацетил, бензил, аллил, п-метоксибензил и тому подобное. Эти группы могут быть необязательно замещены от 1 до 3 заместителями, выбранными из группы, состоящей из галогена, алкокси и нитро. Защитная группа для аминогруппы предпочтительно представляет собой п-метоксибензил.
Термин "галогеналкил" относится к алкильной группе, замещенной одним или более галогенами, где алкил такой, как определен выше.
Термин "галогеналкокси" относится к алкоксильной группе, замещенной одним или более галогенами, где алкоксильная группа такая, как определена выше.
Термин "гидроксиалкил" относится к алкильной группе, замещенной гидроксилом (гидроксилами), где алкил такой, как определен выше.
Термин "гидрокси" относится к группе -ОН.
Термин "галоген" относится к фтору, хлору, брому или йоду.
Термин "амино" относится к группе -NH2.
Термин "циано" относится к группе -CN.
Термин "нитро" относится к группе -NO2.
Термин "оксо" относится к группе=O.
Термин "карбонил" относится к группе С=O.
Термин "карбокси" относится к группе -С(O)ОН.
Термин "алкоксикарбонил" относится к группе -С(O)O(алкил) или -С(O)O(циклоалкил), где алкил и циклоалкил такие, как определены выше.
Термин "ацилгалогенид" относится к соединению, содержащему группу -С(O)-галоген.
"Необязательный" или "необязательно" означает, что описанное далее событие или обстоятельство может, но не обязательно должно, происходить, и такое описание включает ситуацию, в которой событие или обстоятельство происходит или не происходит. Например, "гетероциклил, необязательно замещенный алкилом" означает, что алкильная группа может, но не обязательно должна, присутствовать, и такое описание включает ситуации, когда гетероциклил замещен алкилом и когда гетероциклил не замещен алкилом.
"Замещенный" относится к одному или более атомам водорода в группе, предпочтительно, до 5 и, более предпочтительно, от 1 до 3 атомов водорода, независимо замещенным соответствующим числом заместителей. Само собой разумеется, что заместители присутствуют только в возможном для них с химической точки зрения положении. Специалист в данной области техники без чрезмерных усилий сможет экспериментально или теоретически определить, возможно или нет такое замещение. Например, комбинация аминогруппы или гидроксильной группы, имеющих свободный атом водорода, и атомов углерода с ненасыщенными (например, олефиновыми) связями может быть нестабильной.
"Фармацевтическая композиция" относится к смеси одного или более соединений, описанных в данной работе, или его физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и вспомогательные вещества. Назначение фармацевтической композиции заключается в облегчении введения соединения в организм, что способствует абсорбции активного ингредиента и тем самым проявлению его биологической активности.
"Фармацевтически приемлемая соль" относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной для млекопитающих и обладает требуемой биологической активностью.
R6 и m - такие, как определены для формулы (I).
Способ синтеза соединения по настоящему изобретению
Для достижения цели по настоящему изобретению в настоящем изобретении использованы следующие технические решения.
Схема I
Способ получения соединения формулы (I) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:
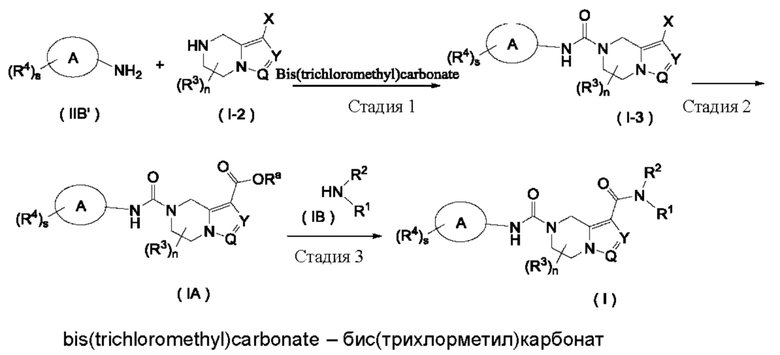
На стадии 1 соединение формулы (IIB') взаимодействует с соединением формулы (I-2) и бис(трихлорметил)карбонатом в щелочных условиях с получением соединения формулы (I-3);
на стадии 2 соединение формулы (I-3) взаимодействует с окисью углерода в присутствии катализатора в щелочных условиях с получением соединения формулы (IA);
на стадии 3 соединение формулы (IA) взаимодействует с соединением формулы (IB) или его солью с получением соединения формулы (I).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Катализатор включает, не ограничиваясь перечнем, Pd/C, Ni Ренея, диоксид платины, тетра(трифенилфосфин)палладий, дихлорид палладия, ацетат палладия, 2-дициклогексилфосфино-2,4,6-триизопропилбифенил, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, дихлорид 1,1'-бис(дибензилфосфорил)ферроценпалладия, трис(дибензилиденацетон)дипалладий и 2-дициклогексилфосфино-2',6'-диметоксибифенил и, предпочтительно, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий и 2-дициклогексилфосфино-2',6'-диметоксибифенил.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой водород или алкил и, предпочтительно, является водородом, метилом или этилом;
X представляет собой галоген и, предпочтительно, является бромом;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
кольцо A, Y, Q, R1, R2, R4, s и n являются такими, как определены для формулы (I).
Схема II
Способ получения соединения формулы (I) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:

На стадии 1 соединение формулы (IIB') взаимодействует с соединением формулы (I-4) и бис(трихлорметил)карбонатом в щелочных условиях с получением соединения формулы (IA);
на стадии 2 соединение формулы (IA) подвергают реакции гидролиза в щелочных условиях с получением соединения формулы (IC);
на стадии 3 соединение формулы (IC) и соединение формулы (IB) или его соль подвергают реакции конденсации с получением соединения формулы (I).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой алкил и, предпочтительно, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; и
кольцо A, Y, Q, R1, R2, R4, s и n являются такими, как определены для формулы (I).
Схема III
Способ получения соединения формулы (II) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, включает в себя следующую стадию:
Соединение формулы (IA) взаимодействует с соединением формулы (IIB) или его солью в щелочных условиях с получением соединения формулы (II).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой водород или алкил и, предпочтительно, водород, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6 и, предпочтительно, водорода или алкила; а
кольцо A, Q, Y, R1, R4, s и n являются такими, как определены для формулы (II).
Схема IV
Способ получения соединения формулы (II) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, включает в себя следующую стадию:

Соединение формулы (1С) взаимодействует с соединением формулы (ИВ) или его солью в щелочных условиях с получением соединения формулы (II).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6 и, предпочтительно, водорода или алкила; а
кольцо A, Q, Y, R1, R4, s и n являются такими, как определены для формулы (II).
Схема V
Способ получения соединения формулы (III) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:

На стадии 1 соединение формулы (IIB') взаимодействует с соединением формулы (III-1) и бис(трихлорметил)карбонатом в щелочных условиях с получением соединения формулы (III-а);
на стадии 2 соединение формулы (III-а) подвергают реакции гидролиза в щелочных условиях с получением соединения формулы (III-b);
на стадии 3 соединение формулы (III-b) взаимодействует с соединением формулы (IIB) или его солью с получением соединения формулы (III).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой алкил и, предпочтительно, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
кольцо A, R1, R4, s и n являются такими, как определены для формулы (III).
Схема VI
Способ получения соединения формулы (IV) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:

На стадии 1 соединение формулы (IIB') взаимодействует с соединением формулы (IV-1) и бис(трихлорметил)карбонатом в щелочных условиях с получением соединения формулы (IV-a);
на стадии 2 соединение формулы (IV-a) подвергают реакции гидролиза в щелочных условиях с получением соединения формулы (IV-b);
на стадии 3 соединение формулы (IV-b) взаимодействует с соединением формулы (IIB) или его солью с получением соединения формулы (IV).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Катализатор включает, не ограничиваясь перечнем, Pd/C, Ni Ренея, диоксид платины, тетра(трифенилфосфин)палладий, дихлорид палладия, ацетат палладия, 2-дициклогексилфосфино-2,4,6-триизопропилбифенил, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, дихлорид 1,1'-бис(дибензилфосфорил)ферроценпалладия, трис(дибензилиденацетон)дипалладий и 2-дициклогексилфосфино-2',6'-диметоксибифенил и, предпочтительно, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий и 2-дициклогексилфосфино-2',6'-диметоксибифенил.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой алкил и, предпочтительно, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
кольцо A, R1, R4, s и n являются такими, как определены для формулы (I).
Схема VII
Способ получения соединения формулы (III) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:
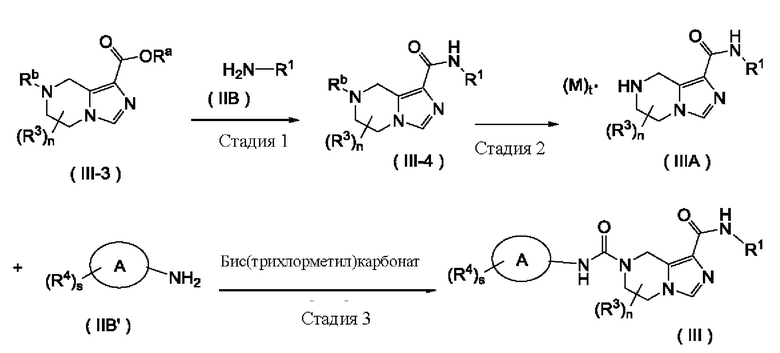
На стадии 1 соединение формулы (III-3) взаимодействует с соединением формулы (IIB) или его солью в присутствии конденсирующего агента в щелочных условиях с получением соединения формулы (III-4);
на стадии 2 с соединения формулы (III-4) удаляют защитные группы в кислых условиях с получением соединения формулы (IIIA) или его соли;
на стадии 3 соединение формулы (IIIA) или его соль взаимодействует с соединением формулы (IIB') или его солью и бис(трихлорметил)карбонатом с получением соединения формулы (III).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Реагент, создающий кислые условия, включает, не ограничиваясь перечнем, хлороводород, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту, метансульфокислоту, азотную кислоту, фосфорную кислоту, п-толуолсульфоновую кислоту, Me3SiCl и TMSOTf (англ. Trimethylsilyl trifluoromethanesulfonate - триметилсилилтрифторметансульфонат).
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
М является трифторуксусной кислотой или соляной кислотой; Ra представляет собой водород или алкил и, предпочтительно, водород, метил или этил;
Rb представляет собой защитную группу для аминогруппы и, предпочтительно, трет-бутоксикарбонил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
t равно 0 или 1; кольцо A, R1, R4, s и n являются такими, как определены для формулы (III).
Схема VIII
Способ получения соединения формулы (V) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:

На стадии 1 соединение формулы (IIB') взаимодействует с соединением формулы (V-1) в щелочных условиях с получением соединения формулы (V-2);
на стадии 2 соединение формулы (V-2) подвергают реакции восстановления в присутствии восстановителя с получением соединения формулы (V-3);
на стадии 3 соединение формулы (V-3) и окислитель подвергают реакции окисления с получением соединения формулы (V-4);
на стадии 4 соединение формулы (V-4) подвергают реакции окисления в присутствии окислителя с получением соединения формулы (V-a);
на стадии 5 соединение формулы (V-a) и соединение формулы (ИВ) или его соль подвергают реакции конденсации с получением соединения формулы (V).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Восстановитель включает, не ограничиваясь перечнем, алюмогидрид лития, NaBH4, NaBH4-ZnCl2 и гидрид диизобутилалюминия (англ. DIBAL-H).
Окислитель включает, не ограничиваясь перечнем, хлорхромат пиридиния (РСС), реактив Джонса, реактив Коллинза, дихромат пиридиния (PDC), оксалилхлорид (окисление по Сверну), карбодиимид, хлорит натрия и перманганат калия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N-дициклогексилкарбодиимид, N,N'-диизопропил карбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
кольцо A, R1, R4, s и n являются такими, как определены для формулы (V).
Схема IX
Способ получения соединения формулы (VI) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:

На стадии 1 соединение формулы (ИВ') взаимодействует с соединением формулы (VI-1) и бис(трихлорметил)карбонатом в щелочных условиях с получением соединения формулы (VI-2);
на стадии 2 соединение формулы (VI-а) взаимодействует с соединением формулы (ИВ) или его солью с получением соединения формулы (VI).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат O-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой водород или алкил и, предпочтительно, водород, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; и
кольцо A, R1, R4, s и n являются такими, как определены для формулы (I).
Схема X
Способ получения соединения формулы (VII) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:

На стадии 1 соединение формулы (VII-1) взаимодействует с соединением формулы (III-1) и бис(трихлорметил)карбонатом в щелочных условиях с получением соединения формулы (VII-a);
на стадии 2 соединение формулы (IV-a) подвергают реакции гидролиза в щелочных условиях с получением соединения формулы (VII-b);
на стадии 3 соединение формулы (VII-b) и соединение формулы (IIB) или его соль подвергают реакции конденсации с получением соединения формулы (VII).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат О-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой алкил и, предпочтительно, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетеро цикл ила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
R1, R4, s и n являются такими, как определены для формулы (I).
Схема XI
Способ получения соединения формулы (VIII) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли включает в себя следующие стадии:

На стадии 1 соединение формулы (VII-1) взаимодействует с соединением формулы (IV-1) и бис(трихлорметил)карбонатом в щелочных условиях с получением соединения формулы (VIII-a);
на стадии 2 соединение формулы (VIII-a) подвергают реакции гидролиза в щелочных условиях с получением соединения формулы (VIII-b);
на стадии 3 соединение формулы (VIII-b) и соединение формулы (IIB) или его соль подвергают реакции конденсации с получением соединения формулы (VIII).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат О-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
Ra представляет собой алкил и, предпочтительно, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетеро цикл ила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила; а
R1, R4, s и n являются такими, как определены для формулы (I).
Схема XII
Способ получения соединения формулы (VII-A) или формулы (VIII-А) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, включает в себя следующую стадию:

взаимодействия соединения формулы (VII-a) с соединением формулы (VII-В) или его солью с получением соединения формулы (VII-A); или взаимодействия соединения формулы (VIII-a) с соединением формулы (VII-В) или его солью с получением соединения формулы (VIII-A).
Реагент, создающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, не ограничиваясь перечнем, триэтиламин, 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия. Неорганические основания включают, не ограничиваясь перечнем, гидрид натрия, гидроксид натрия, фосфат калия, карбонат натрия, бикарбонат натрия, карбонат калия и карбонат цезия.
Конденсирующий агент включает, не ограничиваясь перечнем, гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида, N,N'-дициклогексилкарбодиимид, N,N'-диизопропилкарбодиимид, тетрафторборат O-бензотриазол-N,N,N',N'-тетраметилурония, 1-гидроксибензотриазол, 1-гидрокси-7-азобензотриазол, гексафторфосфат О-бензотриазол-N,N,N',N'-тетраметилурония, гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония, гексафторфосфат бензотриазол-1-илокситрис(диметиламино)фосфония и гексафторфосфат бензотриазол-1-ил-окситрипирролидинилфосфония и, предпочтительно, гексафторфосфат 2-(7-оксобензотриазол)-N,N,N',N'-тетраметилурония.
Указанную выше реакцию предпочтительно проводят в растворителе. Используемый растворитель включает, не ограничиваясь перечнем, уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N,N-диметилформамид и их смеси.
Где:
G представляет собой С или N;
Ra представляет собой водород или алкил и, предпочтительно, водород, метил или этил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, алкила, галогеналкила, алкокси, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетеро цикл ила, арила, гетероарила, -OR6, -C(O)R6, -C(O)OR6 и -S(O)mR6, и, предпочтительно, водорода или алкила;
R8 представляет собой алкил, предпочтительно, метил;
R9 представляет собой алкил, где алкил необязательно дополнительно замещен одним или более атомами галогена; а
R4 и s являются такими, как определены для формулы (I).
ОПИСАНИЕ ПРЕДПОЧТИТЕЛЬНЫХ ВАРИАНТОВ ОСУЩЕСТВЛЕНИЯ
ИЗОБРЕТЕНИЯ
Настоящее изобретение будет дополнительно описано со ссылкой на следующие примеры, однако эти примеры не следует рассматривать, как ограничивающие объем настоящего изобретения.
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Структуру соединений идентифицировали методом ядерно-магнитного резонанса (ЯМР, англ. NMR) и/или масс-спектрометрии (МС, англ. MS). Сдвиги ЯМР (δ) приведены в 10-6 (м.д.). Спектры ЯМР снимали на приборе Bruker AVANCE-400. В качестве растворителей для снятия спектров использовали дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), в качестве внутреннего стандарта -тетраметилсилан (ТМС, англ. TMS).
МС снимали при помощи масс-спектрометра FINNIGAN LCQAd (ESI) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Анализ методом высокоэффективной жидкостной хроматографии (ВЭЖХ, англ. HPLC) выполняли на жидкостных хроматографах высокого давления Agilent HPLC 1200DAD, Agilent HPLC 1200VWD и Waters HPLC e2695-2489.
Анализ методом хиральной ВЭЖХ выполняли при помощи жидкостного хроматографа высокого давления Agilent 1260 DAD.
Препаративную высокоэффективную жидкостную хроматографию выполняли на препаративных хроматографах Waters 2767, Waters 2767-SQ Detecor2, Shimadzu LC-20AP и Gilson-281.
Для проведения препаративной хиральной хроматографии использовали препаративный хроматограф Shimadzu LC-20AP.
Для быстрой хроматографии на системе CombiFlash использовали Combiflash Rf200 (TELEDYNE ISCO). Тонкослойную хроматографию на силикагеле (ТСХ, англ. TLC) выполняли с использованием силикагелевой пластины Yantai Huanghai HSGF254 или Qingdao GF254. Толщина слоя силикагеля пластины, используемой для ТСХ, составляла от 0,15 мм до 0,2 мм, а толщина слоя силикагеля пластины, используемой для очистки продукта - от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии на силикагеле использовали силикагель Yantai Huanghai 200 - 300 меш.
Средние скорости ингибирования киназы и величины IC50 определяли при помощи NovoStar ELISA(BMG Со, Germany).
Известные исходные вещества по настоящему изобретению могут быть синтезированы известными способами или приобретены у компаний ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc, Dari chemical Company и др.
Если не указано иное, реакции проводили в атмосфере аргона или атмосфере азота.
"Атмосфера аргона" или "атмосфера азота" означает, что реакционная колба соединена с баллоном, содержащим аргон или азот (емкостью приблизительно 1 л).
"Атмосфера водорода" означает, что реакционная колба соединена с баллоном, содержащим водород (емкостью приблизительно 1 л).
Для проведения реакций гидрирования под давлением использовали установку для гидрирования Parr 3916EKX и генератор водорода Qinglan QL-500 или установку для гидрирования HC2-SS.
При реакциях гидрирования реакционную систему обычно вакуумировали и заполняли водородом, указанную операцию повторяли трижды.
Для проведения микроволновых реакций использовали микроволновый реактор типа СЕМ Discover-S 908860.
Если не указано иное, раствор относится к водному раствору.
Если не указано иное, температура реакции соответствует комнатной температуре от 20°С до 30°С.
Ход реакции в примерах контролировали методом тонкослойной хроматографии (ТСХ). Проявляющий растворитель, используемый в реакциях, элюирующая система для колоночной хроматографии и проявляющая система растворителей в тонкослойной хроматографии для очистки соединений включали: А: систему дихлорметан/метанол, В: систему н-гексан/этилацетат и С: систему петролейный эфир/этилацетат. Объемное соотношение растворителей регулировали в зависимости от полярности соединений, при необходимости для регулирования может быть добавлено небольшое количество щелочного реагента, такого как триэтиламин, или кислого реагента, такого как уксусная кислота.
Пример 1
(R)-N7-(3,4,5-Трифторфенил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид

Стадия 1
1-Бром-N-(3,4,5-трифторфенил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8H)-карбоксамид 1с
3,4,5-Трифторанилин 1а (0,365 г, 2,48 ммоль, полученный в соответствии с известным способом, раскрытым в работе "Tempahedron Letters, 51(17), 2010, 2265-2268") и 1-бром-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин 1b (501,36 мг, 2,48 ммоль, Shanghai Shuya Pharmaceutical Technology Co, Ltd.) растворяли в 30 мл дихлорметана, затем добавляли триэтиламин (753,26 мг, 7,44 ммоль) и бис(трихлорметил)карбонат (294,53 мг, 992,54 мкмоль). После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 1с (300 мг, выход: 32,2%).
МС m/z (ESI): 376,1 [М+1].
Стадия 2
Метил-7-((3,4,5-трифторфенил)карбамоил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 1d
Октакарбонил дикобальта (525,03 мг, 1,54 ммоль) и карбонат калия (1,06 г, 7,68 ммоль) растворяли в 20 мл метанола в атмосфере окиси углерода. Раствор перемешивали при температуре 60°С в течение 15 минут, затем добавляли соединение 1с (300 мг, 767,71 мкмоль) и метил-2-хлорацетат (499,88 мг, 4,61 ммоль). После перемешивания в течение 8 часов реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 1d (70 мг, выход: 21,8%).
МС m/z (ESI): 355,3 [М+1].
Стадия 3
(R)-N7-(3,4,5-Трифторфенил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид
Соединение 1d (70 мг, 197,58 мкмоль) и гидрохлорид (2R)-1,1,1-трифторпропан-2-амина 1е (59,09 мг, 395,16 мкмоль, полученный в соответствии со способом, раскрытым в заявке на патент CN 102875270 A) растворяли в 10 мл тетрагидрофурана, после чего по каплям прибавляли 0,988 мкл 1М раствора бис(триметилсилил)амида лития в тетрагидрофуране при температуре 0°С. По окончании добавления реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 6 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток разбавляли 15 мл воды и экстрагировали этилацетатом (15 мл × 3). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 1 (15 мг, выход: 17,4%).
MC m/z (ESI): 436,2 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 9,31 (с, 1Н), 8,30 (д, 1Н), 7,70 (с, 1Н), 7,25-7,31 (м, 2Н), 5,05 (с, 2Н), 4,78-4,86 (м, 1Н), 4,21-4,24 (м, 2Н), 3,94-3,96 (м, 2Н), 1,31-1,44 (дд. 3Н).
Пример 2
(R)-N5-(3,4-Дифторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид
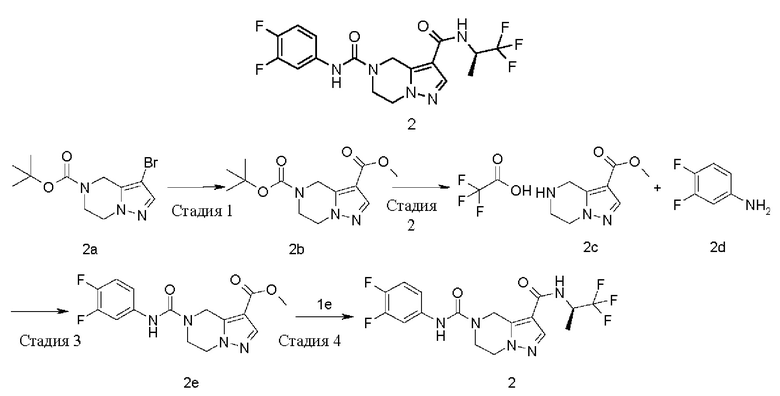
Стадия 1
5-трет-бутил-3-метил-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксилат 2b
Октакарбонил дикобальта (2,26 г, 6,62 ммоль) и карбонат калия (4,57 г, 33,09 ммоль) растворяли в 20 мл метанола в атмосфере окиси углерода. Раствор перемешивали при температуре 60°С в течение 15 минут, затем добавляли трет-бутил-3-бром-6,7-дигидропиразоло[1,5-а]пиразин-5(4H)-карбоксилат 2а (1 г, 3,31 ммоль, полученный в соответствии с известным способом, раскрытым в работе "ACS Medicinal Chemistry Letters, 2015, 6(1), 37-41") и метил-2-хлорацетат (2,15 г, 19,86 ммоль). После перемешивания в течение 16 часов реакционный раствор концентрировали при пониженном давлении, разбавляли 100 мл этилацетата, фильтровали и промывали 100 мл этилацетата. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 2b (0,5 г, выход: 53%).
МС m/z(ESI): 282,1 [М+1].
Стадия 2
Трифторацетат метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилата 2с
Соединение 2b (600 мг, 2,13 ммоль) растворяли в 10 мл дихлорметана, затем добавляли трифторуксусную кислоту (2,43 г, 21,33 ммоль). По окончании добавления реакционный раствор перемешивали в течение 12 часов и концентрировали при пониженном давлении с получением неочищенного, указанного в заголовке соединения 2 с (250 мг), которое использовали непосредственно на следующей стадии без очистки.
MC m/z (ESI): 182,0 [М+1].
Стадия 3
Метил-5-((3,4-дифторфенил)карбамоил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилат 2е
Неочищенное соединение 2с (200 мг, 1,10 ммоль), 3,4-дифторанилин 2d (142,51 мг, 1,10 ммоль) и триэтиламин (335,08 мг, 3,31 ммоль) растворяли в 10 мл тетрагидрофурана, после чего добавляли бис(трихлорметил)карбонат (114,64 мг, 386,33 мкмоль) при температуре 0°С. Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 2е (100 мг, выход: 26,9%).
MC m/z (ESI): 337,4 [М+1].
Стадия 4
(R)-N5-(3,4-Дифторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 2
Соединение 2е (100 мг, 297,36 мкмоль) и соединение 1е (133,40 мг, 892,08 мкмоль) растворяли в 20 мл тетрагидрофурана, после чего по каплям прибавляли 4,46 мл 1М раствора бис(триметилсилил)амида лития в тетрагидрофуране при температуре 0°С. По окончании прибавления реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. В реакционный раствор добавляли 2 мл насыщенного раствора хлорида аммония и 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 2 (15 мг, выход: 12,1%).
МС m/z (ESI): 418,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,07 (с, 1Н), 7,51-7,46 (м, 1Н), 7,22-7,16 (м, 2Н), 5,05 (с, 2Н), 4,88-4,82 (м, 1Н), 4,29-4,26 (м, 2Н), 4,05-4,02 (м, 2Н), 1,42-1,40 (м, 3Н).
Пример 3
(R)-N7-(3-Циано-4-фторфенил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8Н)-дикарбоксамид 3

Стадия 1
трет-бутил-(R)-1-((1,1,1-трифторпропан-2-ил)карбамоил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8H)-карбоксилат 3b
7-трет-Бутил-1-метил-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксилат 3а (469,78 мг, 1,67 ммоль, полученный в соответствии со способом, раскрытым в заявке на патент US 20110034443 A1) и соединение 1е (249,74 мг, 1,67 ммоль) растворяли в 20 мл тетрагидрофурана в атмосфере аргона, после чего по каплям прибавляли 3,3 мл 1М раствора бис(триметилсилил)амида лития в тетрагидрофуране при охлаждении на ледяной бане. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 3b (130 мг, выход: 21,5%).
MC m/z (ESI): 363,2 [М+1].
Стадия 2
Трифторацетат (R)-N-(1,1,1-трифторпропан-2-ил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксамида 3с
Соединение 3b (130 мг, 358,77 мкмоль) и трифторуксусную кислоту (204,54 мг, 1,79 ммоль) последовательно растворяли в 2 мл дихлорметана. Реакционный раствор перемешивали в течение 2 часов, концентрировали при пониженном давлении с получением неочищенного, указанного в заголовке соединения 3с (135 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
(R)-N7-(3-Циано-4-фторфенил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 3
Неочищенное соединение 3с (50 мг, 190,67 мкмоль), 5-амино-2-фторбензонитрил 3d (25,96 мг, 190,67 мкмоль, полученный в соответствии с известным способом, раскрытым в работе "Bioorganic & Medicinal Chemistry Letters, 2006, 16(19), 5176-5182") и триэтиламин (28,94 мг, 286,01 мкмоль) растворяли в 10 мл тетрагидрофурана, затем при охлаждении на ледяной бане добавляли бис(трихлорметил)карбонат (28,29 мг, 95,34 мкмоль). После перемешивания в течение 1 часа реакционный раствор концентрировали при пониженном давлении, остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 3 (2 мг, выход: 2,5%).
МС m/z (ESI): 425,1 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 9,22 (с, 1Н), 8,32 (д, 1Н), 7,95 (д, 1Н), 7,81-7,77 (м, 2Н), 7,47-7,43 (м, 1Н), 4,96 (с, 2Н), 4,81-4,74 (м, 1Н), 4,17-4,10 (м, 2Н), 3,89-3,86 (м, 2Н), 1,39-1,35 (дд. 3Н).
Пример 4
(R)-N5-(3,4,5-Трифторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 4

Стадия 1
Метил-5-((3,4,5-трифторфенил)карбамоил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилат 4а
Соединение 2с (100 мг, 338,74 мкмоль), соединение 1а (49,83 мг, 338,74 мкмоль) и трифторуксусную кислоту (342,77 мг, 3,39 ммоль) растворяли в 10 мл тетрагидрофурана, затем при температуре 0°С добавляли бис(трихлорметил)карбонат (35,18 мг, 118,56 3 мкмоль). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 4а (50 мг, выход: 25,0%).
МС m/z (ESI): 355,1 [М+1].
Стадия 2
(R)-N5-(3,4,5-Трифторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 4
Соединение 4а (50 мг, 141,13 мкмоль) и соединение 1е (63,31 мг, 423,39 мкмоль) растворяли в 20 мл тетрагидрофурана, после чего по каплям прибавляли 2,12 мл 1М раствора бис(триметилсилил)амида лития в тетрагидрофуране при температуре 0°С. По окончании прибавления реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. В реакционный раствор добавляли 2 мл насыщенного раствора хлорида аммония и 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 4 (5 мг, выход: 4%).
MC m/z (ESI): 436,0 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,07 (с, 1Н), 7,30-7,26 (м, 2Н), 5,04 (с, 2Н), 4,88-4,82 (м,1 Н), 4,28-4,26 (м, 2Н), 4,04-4,01 (м, 2Н), 1,42-1,40 (м, 3Н).
Пример 5
(R)-N7-(3,4-Дифторфенил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8Н)-дикарбоксамид 5

Стадия 1
Этил-7-((3,4-дифторфенил)карбамоил)-5,6,7,8-тетрагидроимидазо[1,5-а1пиразин-1-карбоксилат 5b
Этил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 5а (100 мг, 512,25 мкмоль, полученный в соответствии со способом, раскрытым в заявке на патент CN 102464661 А) и соединение 2d (79,36 мг, 614,70 мкмоль) добавляли в 20 мл тетрагидрофурана, после чего при охлаждении на ледяной бане добавляли бис(трихлорметил)карбонат (76,01 мг, 256,13 мкмоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 5b (110 мг, выход: 61,3%).
МС m/z (ESI): 351,1 [М+1].
Стадия 2
(R)-N7-(3,4-Дифторфенил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 5
Соединение 5b (75 мг, 214,09 мкмоль) и соединение 1е (48,02 мг, 321,14 мкмоль) добавляли в 20 мл тетрагидрофурана, затем при охлаждении на ледяной бане добавляли бис(триметилсилил)амид лития (107,47 мг, 642,27 мкмоль). После перемешивания в течение 1 часа реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 5 (20 мг, выход: 30,4%).
MC m/z (ESI): 418,2 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 9,09 (с, 1Н), 8,31 (д, 1Н), 7,76 (с, 1Н), 7,64-7,60 (м, 1Н), 7,34-7,23 (м, 2Н), 4,94 (с, 2Н), 4,79-4,77 (м, 1Н), 4,16-4,14 (м, 2Н), 3,88-3,85 (м,2Н), 1,37-1,35 (дд, 3Н).
Пример 6
(S)-N7-(3,4-Дифторфенил)-6-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8Н)-дикарбоксамид 6
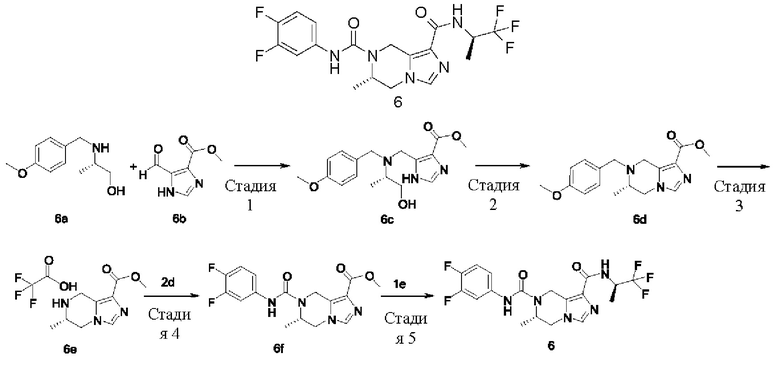
Стадия 1
Метил-(S)-5-(((1-гидроксипропан-2-ил)(4-метоксибензил)амино)метил)-1Н-имидазол-4-карбоксилат 6с
(S)-2-((4-Метоксибензил)амино)пропан-1-ол 6а (34,92 г, 179,08 ммоль, полученный в соответствии с известным способом, раскрытым в работе "Bioorganic & Medicinal Chemistry Letters, 2015, 25(5), 1086-1091") растворяли в 600 мл тетрагидрофурана, добавляли метил-5-формилимидазол-4-карбоксилат 6b (23 г, 149,23 ммоль, полученный в соответствии со способом, раскрытым в заявке на патент US 2008/318935), после чего порциями добавляли триацетоксиборгидрид натрия (47,44 г, 223,85 ммоль). После перемешивания в течение 12 часов реакционный раствор фильтровали, фильтрат концентрировали при пониженном давлении. Полученный остаток разбавляли 600 мл этилацетата, промывали водой (200 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного, указанного в заголовке соединения 6с (40 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ESI): 334,2 [М+1].
Стадия 2
Метил-(S)-7-(4-метоксибензил)-6-метил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 6d
Неочищенное соединение 6с (11 г, 33,03 ммоль) и трифенилфосфин (12,98 г, 49,49 ммоль) (Sinopharm Chemical Reagent Со, Ltd.) растворяли в 400 мл тетрагидрофурана, затем медленно добавляли диизопропилазодикарбоксилат (10 г, 49,45 ммоль, Shanghai Accela ChemBio Со, Ltd.) при охлаждении на ледяной бане. Реакционный раствор медленно нагревали до комнатной температуры, перемешивали в течение 12 часов и концентрировали при пониженном давлении. Полученный остаток разбавляли 400 мл этилацетата, промывали водой (100 мл × 2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле с использованием элюирующей системы А, в результате получали указанное в заголовке соединение 6d (4,5 г, выход: 43,2%).
MC m/z (ESI): 315,9 [М+1].
Стадия 3
Трифторацетат метил-(S)-6-метил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 6е
Соединение 6d (0,6 г, 2,33 ммоль) растворяли в 2 мл трифторуксусной кислоты. Реакционный раствор нагревали под воздействием микроволнового излучения до температуры 100°С в течение 5 минут, охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением неочищенного, указанного в заголовке соединения 6е (0,6 г), которое использовали непосредственно на следующей стадии без очистки.
MCm/z(ESI): 196,1 [М+1].
Стадия 4
Метил-(S)-7-((3,4-дифторфенил)карбамоил)-6-метил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 6f
Неочищенное соединение 6е (240 мг, 776,09 мкмоль) растворяли в 15 мл тетрагидрофурана, добавляли соединение 2d (150,3 мг, 1,16 ммоль) и триэтиламин (314,13 мг, 3,10 ммоль), после чего при охлаждении на ледяной бане добавляли бис(трихлорметил) карбонат (92,12 мг, 310,44 мкмоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 6f (0,19 г, выход: 69,9%).
MCm/z(ESI): 351,1 [М+1].
Стадия 5
(S)-N7-(3,4-Дифторфенил)-6-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 6
Соединение 6f (190 мг, 542,36 мкмоль) растворяли в 20 мл тетрагидрофурана, добавляли соединение 1е (121,66 мг, 813,54 мкмоль), после чего по каплям прибавляли 4 мл 1М раствора бис(триметилсилил)амида лития в тетрагидрофуране при охлаждении на ледяной бане. По окончании прибавления реакционный раствор перемешивали в течение 2 часов. Реакционный раствор разбавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении. Полученный остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 6 (25 мг, выход: 10,6%).
MCm/z(ESI): 432,1 [М+1].
1H ЯМР (400 МГц, ДМСО-d6): δ 9,28 (с, 1Н), 8,25 (д, 1Н), 7,73 (с, 1Н), 7,54-7,45 (м, 1Н), 7,21-7,15 (м, 2Н), 5,27 (с, 1Н), 4,95-4,92 (м, 1Н), 4,86-4,80 (м, 2Н), 4,28-4,22 (м, 2Н), 1,50-1,45 (м, 3Н), 1,20 (д, 3Н).
Пример 7
(S)-N5-(3,4-Дифторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 7

Стадия 1
5-трет-Бутил-3-метил-(S)-6-метил-6,7-дигидропиразоло[1.5-а]пиразин-3,5(4Н)-дикарбоксилат 7b
трет-Бутил-(S)-3-йод-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-5(4Н)-карбоксилат 7а (8,3 г, 22,85 ммоль, полученный в соответствии со способом, раскрытым в заявке на патент WO 2016113273 A1), ацетат палладия (1,03 г, 4,57 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (2,53 г, 4,57 ммоль) и триэтиламин (9,64 мл, 68,56 ммоль) растворяли в 150 мл метанола в атмосфере окиси углерода. После перемешивания при температуре 55°С в течение 15 часов реакционный раствор фильтровали через целит. Фильтрат концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле с использованием элюирующей системы С, в результате получали указанное в заголовке соединение 7b (6,3 г, выход: 93,34%).
Стадия 2
Трифторацетат метил-(S)-6-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилата 7с
Соединение 7b (6,3 г, 21,33 ммоль) растворяли в 20 мл дихлорметана, затем добавляли трифторуксусную кислоту (14,1 мл, 190,91 ммоль). Реакционный раствор перемешивали в течение 2 часов, концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 7 с (14 г), которое использовали непосредственно на следующей стадии без очистки.
MC m/z(ESI): 196,1 [М+1].
Стадия 3
Метил-(S)-5-((3,4-дифторфенил)карбамоил)-6-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилат 7е
Неочищенное соединение 7с (2 г, 3,23 ммоль) и триэтиламин (1,64 г, 16,17 ммоль) растворяли в 20 мл дихлорметана. После перемешивания в течение 10 минут в реакционный раствор добавляли 1,2-дифтор-4-изоцианатобензол (601,87 мг, 3,88 ммоль, полученный в соответствии с известным способом, раскрытым в работе "European Journal of Medicinal Chemistry, 2016, 115, 1-13") при охлаждении на ледяной бане, и проводили реакцию в течение 20 минут.Реакционный раствор наагревали до комнатной температуры и перемешивали в течение 20 минут. Реакционный раствор очищали колоночной хроматографией на силикагеле, используя последовательно элюирующие системы С и А, в результате получали указанное в заголовке соединение 7е (1 г, выход: 88,27%).
MC m/z(ESI): 351,1 [М+1].
Стадия 4
(S)-5-((3,4-Дифторфенил)карбамоил)-6-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоновая кислота 7f
Соединение 7е (500 мг, 1,43 ммоль) и гидроксид натрия (285,45 мг, 7,14 ммоль) растворяли в 5 мл смешанного растворителя, состоящего из метанола, тетрагидрофурана и воды (V:V:V=2:2:1). Реакционный раствор перемешивали сначала при температуре 40°С в течение 1 часа, затем - при температуре 35°С в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении, разбавляли 10 мл воды, добавляли по каплям 6 М соляную кислоту до получения рН 2 и фильтровали. Отфильтрованный осадок собирали и получали неочищенное указанное в заголовке соединение 7f (450 мг), которое использовали непосредственно на следующей стадии без очистки.
MCm/z(ESI): 337,4 [М+1].
Стадия 5
(S)-N5-(3,4-Дифторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 7
Неочищенное соединение 7f (80 мг, 237,89 мкмоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (67,16 мг, 285,47 мкмоль) и N,N-диизопропилэтиламин (122,98 мг, 951,55 мкмоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли соединение 1е (53,36 мг, 356,83 мкмоль) и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С.Полученный неочищенний продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 7 (70 мг, выход: 68,22%).
МС m/z (ESI): 432,1 [М+1].
1H ЯМР (400 МГц, CD3OD): δ 8,09 (м, 1Н), 7,49-7,47 (м, 1Н), 7,16-7,13 (м, 2Н), 5,32 (д, 1Н), 5,01-4,99 (м, 1Н), 4,84-4,80 (м, 1Н), 4,66 (д, 1Н), 4,32-4,29 (м, 1Н), 4,17 (д, 1Н), 1,40 (д, 3Н), 1,21 (д, 3Н).
Пример 8
(S)-N3-(трет-Бутил)-N5-(3,4-дифторфенил)-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 8
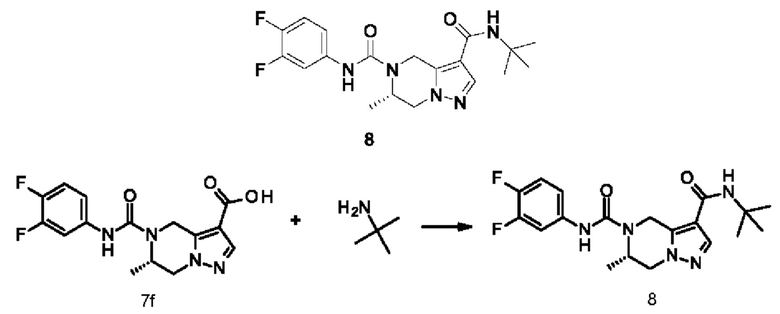
Неочищенное соединение 7f (150 мг, 446,04 мкмоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (157,41 мг, 669,06 мкмоль) и N,N-диизопропилэтиламин (230,58 мг, 1,78 ммоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли трет-бутиламин (48,93 мг, 669,06 мкмоль) и перемешивали в течение 3 часов. Реакционный раствор разбавляли 30 мл воды и экстрагировали этилацетатом (30 мл×2). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С.Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 8 (30 мг, выход: 17,18%).
MC m/z (ESI): 392,2 [М+1].
1H ЯМР(400 МГц, CD3OD): δ 8,02 (с, 1Н), 7,53-7,50 (м, 1Н), 7,20-7,17 (м, 2Н), 5,31 (д, 1Н), 5,01-4,98 (м, 1Н), 4,68 (д, 1Н), 4,33-4,29 (м, 1Н), 4,18 (д, 1Н), 1,46 (с, 9Н), 1,23 (д, 3Н).
Пример 9
(R)-N5-(3-Циано-4-фторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 9

Стадия 1
Метил-5-((3-циано-4-фторфенил)карбамоил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилат 9а
Соединение 2с (50 мг, 275,95 мкмоль), соединение 3d (38,56 мг, 275,95 мкмоль) и триэтиламин (279,23 мг, 2,76 ммоль) растворяли в 10 мл тетрагидрофурана, затем при температуре 0°С добавляли бис(трихлорметил)карбонат (32,76 мг, 110,38 мкмоль). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 9а (20 мг, выход: 21,1%).
MCm/z(ESI): 344,2 [М+1].
Стадия 2
5-((3-Циано-4-фторфенил)карбамоил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоновая кислота 9b
Соединение 9а (100 мг, 291,28 мкмоль) растворяли в 10 мл метанола, затем добавляли гидроксид лития (139,52 мг, 5,83 ммоль) и 2 мл воды. После перемешивания в течение 3 часов реакционный раствор концентрировали при пониженном давлении, разбавляли 10 мл воды и добавляли по каплям 6 М соляную кислоту до получения рН 2. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 9b (95,91 мг), которое использовали непосредственно на следующей стадии без очистки.
MCm/z(ESI): 330,1 [М+1].
Стадия 3
(R)-N5-(3-Циано-4-фторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 9
Неочищенное соединение 9b (95,91 мг, 291,28 мкмоль) растворяли в 10 мл N,N-диметилформамида, добавляли соединение 1е (34,34 мг, 303,69 мкмоль) и триэтиламин (92,19 мг, 911,06 мкмоль), затем добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (85,74 мг, 364,43 мкмоль) при температуре 0°С. После перемешивания в течение 12 часов реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 9 (5 мг, выход: 3,88%).
MCm/z (ESI): 425,1 [М+1].
1H ЯМР (400 МГц, CD3OD): δ 8,07 (с, 1Н), 7,87-7,85 (м, 2Н), 7,32-7,27 (м, 1Н), 5,06 (с, 2Н), 4,86-4,84 (м, 1Н), 4,29-4,27 (м, 2Н), 4,06-4,03 (м, 2Н), 1,42 (д, 3Н).
Пример 10
(S)-N3-(трет-Бутил)-6-метил-N5-(3,4,5-трифторфенил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 10

Стадия 1
Метил-(S)-6-метил-5-((3,4,5-трифторфенил)карбамоил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилат 10а
Соединение 1а (1,17 г, 7,92 ммоль) и соединение 7 с (1,63 г, 5,28 ммоль) растворяли в 30 мл дихлорметана, затем добавляли триэтиламин (2,14 г, 21,22 ммоль) и бис(трихлорметил)карбонат (626,8 мг, 2,11 ммоль). Реакционный раствор перемешивали в течение 2 часов, концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 10а (730 мг, выход: 37,5%).
МС m/z (ESI): 369,1 [М+1].
Стадия 2
(S)-6-Метил-5-((3,4,5-трифторфенил)карбамоил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоновая кислота 10b
Соединение 10а (730 мг, 1,98 ммоль) и гидроксид натрия (396,45 мг, 9,91 ммоль) растворяли в 10 мл смешанного растворителя, состоящего из метанола, тетрагидрофурана и воды (V:V:V=2:2:1). Реакционный раствор перемешивали сначала при температуре 40°С в течение 1 часа, затем - при температуре 35°С в течение 15 часов. Реакционный раствор концентрировали при пониженном давлении, разбавляли 10 мл воды, добавляли по каплям 6 М соляную кислоту до получения рН 2 и фильтровали. Отфильтрованный осадок собирали и получали неочищенное указанное в заголовке соединение 10b (702 мг, 1,98 ммоль), которое использовали непосредственно на следующей стадии без очистки.
MC m/z(ESI): 355,4 [М+1].
Стадия 3
(S)-N3-(трет-Бутил)-6-метил-N5-(3,4,5-трифторфенил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 10
Неочищенное соединение 10b (85 мг, 239,92 мкмоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (67,74 мг, 287,91 мкмоль), N,N-диизопропилэтиламин (129,21 мг, 719,76 мкмоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли трет-бутиламин (35,09 мг, 479,84 мкмоль) и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С.Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 10 (50 мг, выход: 50,90%).
MC m/z(ESI): 410,2 [M+1].
1H ЯМР (400 МГц, CD3OD): δ 8,00 (с, 1Н), 7,30-7,28 (м, 2Н), 5,31 (д, 1Н), 4,98-4,96 (м, 1Н), 4,69 (д, 1Н), 4,31-4,27 (м, 1Н), 4,17 (д, 1Н), 1,44 (с, 9Н), 1,22 (д, 3Н).
Пример 11
(S)-N7-(3-Циано-4-фторфенил)-6-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7-(8H)-дикарбоксамид 11
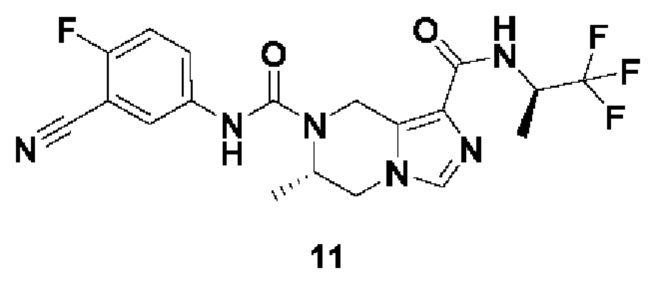

Стадия 1
Метил-(S)-7-((3-циано-4-фторфенил)карбамоил)-6-метил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 11а
Неочищенное соединение 6е (9,2 г, 47,13 ммоль) растворяли в 50 мл тетрагидрофурана, добавляли соединение 3d (6,5 г, 47,13 ммоль) и триэтиламин (5,82 г, 56,55 ммоль), затем при температуре 0°С добавляли бис(трихлорметил)карбонат (4,9 г, 16,49 ммоль). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор фильтровали, фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения На (16,84 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ESI): 358,1 [М+1].
Стадия 2
(S)-7-((3-Циано-4-фторфенил)карбамоил)-6-метил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновая кислота 11b
Неочищенное соединение 11а (16,84 г, 47,13 ммоль) растворяли в 50 мл метанола, после чего при температуре 0°С по каплям прибавляли приготовленный заранее раствор гидроксида натрия (полученный растворением гидроксида натрия (12 г, 282,76 ммоль) в 60 мл воды). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 4 часов. Реакционный раствор концентрировали при пониженном давлении для удаления органического растворителя. Полученный остаток промывали дихлорметаном, величину рН водной фракции доводили до 1-2 с помощью 6 М соляной кислоты. Раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 11b (16,18 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ESI): 344,1 [М+1].
Стадия 3
(S)-N7-(3-Циано-4-фторфенил)-6-метил-N1-((R))-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7-(8H)-дикарбоксамид 11
Неочищенное соединение 11b (13 г, 37,87 ммоль), соединение 1е (7,4 г, 49,23 ммоль) и триэтиламин (11,6 г, 113,6 ммоль) растворяли в 200 мл N,N-диметилформамида. После охлаждения до температуры 0°С в реакционный раствор добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (29 г, 75,73 ммоль). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор разбавляли 300 мл этилацетата и промывали водой (100 мл×3). Органическую фракцию концентрировали при пониженном давлении, полученный остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 11 (1,8 г, выход: 10,8%).
MCm/z (ESI): 439,0 [М+1].
1H ЯМР (400 МГц, CD3OD): δ 7,87-7,85 (м, 1Н), 7,73-7,70 (м, 2Н), 7,69-7,27 (м, 1Н), 5,30-5,26 (д, 1Н), 4,83-4,82 (м, 1Н), 4,85-4,72 (м, 2Н), 4,23-4,21 (м, 2Н), 1,43 (д, 3Н), 1,20 (д, 3Н).
Пример 12
(S)-N1-(трет-Бутил)-N7-(3,4-дифторфенил)-6-метил-5,6-дигидроимидазо[1.5-а]пиразин-1,7-(8H)-дикарбоксамид 12

Соединение 6f (2,11 г, 6,27 ммоль) растворяли в 15 мл тетрагидрофурана, добавляли трет-бутиламин (600 мг, 8,16 ммоль), после чего при температуре 0°С по каплям прибавляли 1М раствор бис(триметилсилил)амида лития в тетрагидрофуране (5 г, 31,37 ммоль). Реакционный раствор перемешивали в течение 2 часов, разбавляли 100 мл воды и экстрагировали этилацетатом (100 мл×3). Органические фракции объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовкесоединение 12 (120 мг, выход: 4,9%).
МС m/z (ESI): 392,1 [М+1].
1H ЯМР (400 МГц, CD3OD): δ 7,65 (с, 1Н), 7,50-7,49 (м, 1Н), 7,19-7,17 (м, 2Н), 5,25-5,21 (д, 1Н), 4,92-4,90 (м, 1Н), 4,77-4,73 (м, 1Н), 4,20-4,18 (м, 2Н), 1,46 (с, 9Н), 1,19-1,17 (д, 3Н).
Пример 13
(S)-6-Метил-N'-(3-метилоксетан-3-ил)-N7-(3,4,5-трифторфенил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 13

Стадия 1
Метил-(S)-6-метил-7-((3,4,5-трифторфенил)карбамоил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 13а
Неочищенное соединение 6е (140,00 мг, 452,72 мкмоль) растворяли в 15 мл тетрагидрофурана, добавляли соединение 1а (99,89 мг, 679,08 мкмоль) и триэтиламин (183,24 мг, 1,81 ммоль), затем при температуре 0°С добавляли бис(трихлорметил)карбонат (53,74 мг, 181,09 мкмоль). Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 13а (88 мг, выход: 52,8%).
МС m/z (ESI): 369,1 [М+1].
Стадия 2
(S)-6-Метил-7-((3,4,5-трифторфенил)карбамоил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновая кислота 13b
Соединение 13а (75 мг, 203,63 мкмоль) растворяли в 10 мл метанола, затем добавляли гидроксид натрия (81,45 мг, 2,04 ммоль) и 2 мл воды. После перемешивания в течение 3 часов реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 13b (75 мг), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ESI): 355,0 [М+1].
Стадия 3
(S)-6-Метил-N1-(3-метилоксетан-3-ил)-N7-(3,4,5-трифторфенил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8Н)-дикарбоксамид 13
Неочищенное соединение 13b (75 мг, 211,69 мкмоль) растворяли в 10 мл N,N-диметилформамида, добавляли 3-метил-3-амино-окситан 13 с (27,66 мг, 317,53 мкмоль, Shanghai Shuya Pharmaceutical Technology Co. Ltd.) и триэтиламин (64,26 мг, 635,07 мкмоль), затем при температуре 0°С добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (99,61 мг, 423,38 мкмоль). После перемешивания при комнатной температуре в течение 12 часов реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 13 (16 мг, выход: 17,9%).
МС m/z (ESI): 424,0 [М+1].
1H ЯМР (400 МГц, ДМСО-d6): δ 8,87 (с, 1Н), 8,36 (с, 1Н), 7,68 (с, 1Н), 7,33-7,27 (м, 2Н), 5,27-5,20 (м, 1Н), 4,92 (д, 2Н), 4,73 (д, 2Н), 4,78-4,73 (м, 1Н), 4,52 (д, 2Н), 4,26-4,20 (м, 2Н), 1,73 (с, 3Н), 1,20 (д, 3Н).
Пример 14
(S)-6-Метил-N3-(3-метилоксетан-3-ил)-N5-(3,4,5-трифторфенил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 14

Неочищенное соединение 10b (100 мг, 282,26 мкмоль), соединение 13 с (36,89 мг, 423,39 мкмоль) и N,N-диизопропилэтиламин (42,84 мг, 423,39 мкмоль) растворяли в 10 мл N,N-диметилформамида, затем добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (79,69 мг, 338,71 мкмоль). После перемешивания в течение 1 часа реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 14 (80 мг, выход: 66,94%).
MC m/z (ESI): 424,5 [М+1].
1H ЯМР (400 МГц, ДМСО-d6): δ 8,89 (с, 1Н), 8,37 (с, 1Н), 7,99 (с, 1Н), 7,33-7,17 (м, 2Н), 5,33 (д, 1Н), 5,00-4,98 (м, 1Н), 4,88 (д, 2Н), 4,71 (д, 1Н), 4,52 (д, 2Н), 4,34-4,31 (м, 1Н), 4,21-4,18 (м, 1Н), 1,72 (с, 3Н), 1,25 (д, 3Н).
Пример 15
(S)-N5-(3-Циано-4-фторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 15


Стадия 1
(S)-5-(трет-Бутоксикарбонил)-6-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоновая кислота 15а
Соединение 7b (525 мг, 1,78 ммоль) и моногидрат гидроксида лития (373 мг, 8,9 ммоль) растворяли в 10 мл смешанного растворителя, состоящего из метанола, тетрагидрофурана и воды (V:V:V=2:2:1). После перемешивания в течение 16 часов реакционный раствор концентрировали при пониженном давлении, разбавляли 10 мл воды, добавляли по каплям 6 М соляную кислоту до получения рН 2 и экстрагировали этилацетатом (20 мл×2). Органические фракции объединяли и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 15а (500 мг), которое использовали непосредственно на следующей стадии без очистки.
MC m/z(ESI): 282,2 [М+1].
Стадия 2
трет-Бутил-(S)-6-метил-3-(((R)-1,1,1-трифторпропан-2-ил)карбамоил)-6,7-дигидропиразоло[1,5-а]пиразин-5(4H)-карбоксилат 15b
Неочищенное соединение 15а (500 мг, 1,78 ммоль), соединение 1е (479 мг, 3,2 ммоль) и N,N-диизопропилэтиламин (360 мг, 3,56 ммоль) растворяли в 20 мл N,N-диметилформамида, затем добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (628 мг, 2,67 ммоль). Реакционный раствор перемешивали в течение 2 часов, концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали неочищенное соединение 15b (600 мг, выход: 89,56%).
MC m/z(ESI): 377,2 [М+1].
Стадия 3
Трифторацетат (S)-6-Метил-N-((R)-1,1,1-трифторпропан-2-ил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксамида 15с
Соединение 15b (500 мг, 1,33 ммоль) растворяли в 20 мл дихлорметана, затем добавляли трифторуксусную кислоту (758 мг, 6,65 ммоль). Реакционный раствор перемешивали в течение 2 часов, концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 15 с (500 мг), которое использовали непосредственно на следующей стадии без очистки.
MC m/z(ESI): 277,2 [М+1].
Стадия 4
(S)-N5-(3-Циано-4-фторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 15
Неочищенное соединение 15с (50 мг, 128 мкмоль) растворяли в 10 мл тетрагидрофурана, добавляли соединение 3d (26 мг, 192 мкмоль) и триэтиламин (26 мг, 256 мкмоль), затем при температуре 0°С добавляли бис(трихлорметил)карбонат (13 мг, 45 мкмоль). Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 15 (20 мг, выход: 35,61%).
MC m/z (ESI): 439,2 [М+1].
1H ЯМР (400 МГц, CDCl3): δ 7,87-7,86 (м, 1Н), 7,83 (с, 1Н), 7,70-7,68 (м, 1Н), 7,42 (с, 1Н), 7,20-7,16 (м, 1Н), 6,01 (д, 1Н), 5,34-5,23 (м, 2Н), 4,92-4,89 (м, 1Н), 4,84 (д, 1Н), 4,38-4,34 (м, 1Н), 4,25 (д, 1Н), 1,49 (д, 3Н), 1,27 (д, 3Н).
Пример 16
(S)-N5-(2,6-Дифторпиридин-4-ил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 16

Стадия 1
2,6-Дифтор-4-изоцианатопиридин 16b
2,6-Дифтор-4-аминопиридин 16а (500 мг, 3,84 ммоль) и N,N-диизопропилэтиламин (583 мг, 5,76 ммоль) растворяли в 25 мл толуола, затем добавляли бис(трихлорметил)карбонат (297 мг, 4,61 ммоль). После перемешивания при температуре 110°С в течение 4 часов реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 16b (1 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(S)-N5-(2,6-Дифторпиридин-4-ил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 16
Неочищенное соединение 15с (100 мг, 256 мкмоль) растворяли в 10 мл дихлорметана, затем добавляли неочищенное соединение 16b (80 мг) и триэтиламин (78 мг, 769 мкмоль). Реакционный раствор перемешивали в течение 2 часов, концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 16 (20 мг, выход: 18,05%).
MC m/z (ESI): 433,5 [М+1].
1H ЯМР (400 МГц, CDCl3): δ 8,19 (с, 1Н), 7,84 (с, 1Н), 7,31 (с, 1Н), 7,10 (с, 1Н), 6,05 (д, 1Н), 5,34 (д, 1Н), 5,27-5,25 (м, 1Н), 4,90-4,88 (м, 1Н), 4,84 (д, 1Н), 4,38-4,34 (м, 1Н), 4,28 (д, 1Н), 1,48 (д, 3Н), 1,29 (д, 3Н).
Пример 17
(R)-N5-(3,4,5-Трифторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 17

Стадия 1
(5-((3,4,5-Трифторфенил)карбамоил)-4,5,6,7-тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-ил)метилацетат 17b
(4,5,6,7-Тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-ил)метилацетат 17а (350 мг, 2 ммоль, полученный в соответствии с известным способом, раскрытым в работе "Journal of Medicine Chemistry, 2014, 57(9), 3687-3706") и диизопропилэтиламин (790 мг, 6 ммоль) растворяли в 20 мл дихлорметана. После перемешивания в течение 10 минут в реакционный раствор при охлаждении на ледяной бане добавляли 1,2,3-трифтор-5-изоцианатобензол 17f (400 мг, 2 ммоль, J&K Scientific Co. Ltd.) и проводили реакцию в течение 20 минут, после чего нагревали до комнатной температуры и перемешивали в течение 20 минут. Реакционный раствор концентрировали при пониженном давлении и очищали колоночной хроматографией на силикагеле с использованием элюирующих систем С и А, в результате получали указанное в заголовке соединение 17b (190 мг, выход: 25,24%).
MC m/z(ESI): 370,1 [М+1].
Стадия 2
3-(Гидроксиметил)-N-(3,4,5-трифторфенил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-5(4H)-карбоксамид 17с
Соединение 17b (190 мг, 0,51 ммоль) растворяли в 5 мл смешанного растворителя, состоящего из метанола и воды (V:V=4:1), затем добавляли гидроксид лития (108 мг, 2,57 ммоль). Реакционный раствор перемешивали в течение 2 часов, разбавляли 20 мл воды и экстрагировали этилацетатом (15 мл × 3). Органические фракции объединяли и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 17 с (65 мг), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ESI): 328,1 [М+1].
Стадия 3
3-Формил-N-(3,4,5-трифторфенил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-5(4Н)-карбоксамид 17d
Неочищенное соединение 17с (65 мг, 0,2 ммоль) растворяли в 5 мл дихлорметана, затем добавляли хлорхромат пиридиния (138 мг, 0,64 ммоль) и силикагель (140 мг, 100-200 меш). После перемешивания в течение 3 часов реакционный раствор фильтровали и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 17d (30 мг, выход: 43,12%).
МС m/z (ESI): 326,1 [М+1].
Стадия 4
5-((3,4,5-Трифторфенил)карбамоил)-4,5,6,7-тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-карбоновая кислота 17е
Соединение 17d (30 мг, 0,1 ммоль) растворяли в 5 мл смешанного растворителя, состоящего из ацетонитрила и воды (V:V=3:2), затем добавляли аминосульфоновую кислоту (20 мг, 0,2 ммоль). После перемешивания в течение 10 минут в раствор добавляли хлорит натрия (20 мг, 0,2 ммоль) и перемешивали в течение 2 часов. В реакционный раствор последовательно добавляли 0,5 мл насыщенного раствора сульфита натрия и 10 мл воды и экстрагировали этилацетатом (10 мл × 3). Органические фракции объединяли и концентрировали при пониженном давлении с получением неочищенного соединения 17е (25 мг), которое использовали непосредственно на следующей стадии без очистки.
MC m/z (ESI): 342,1 [М+1].
Стадия 5
(R)-N5-(3,4,5-Трифторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 17
Неочищенное соединение 17е (25 мг, 73,2 мкмоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (35 мг, 146,5 мкмоль) и N,N-диизопропилэтиламин (47 мг, 366,2 мкмоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли соединение 1е (33 мг, 220 мкмоль) и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С, в результате получали указанное в заголовке соединение 17 (5 мг, выход: 15,6%).
МС m/z (ESI): 437,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,29-7,27 (м, 2Н), 4,60 (ушир., 1Н), 4,54 (т, 2Н), 4,04 (т, 2Н), 2,80 (с, 2Н), 1,45 (д, 3Н).
Пример 18
(S)-6-Метил-N7-(3,4,5-трифторфенил)-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 18

В схеме синтеза Примера 6 исходное соединение 2d на стадии 4 заменяли исходным соединением 1а, соответственно, и получали указанное в заголовке соединение 18 (2,8 мг).
МС m/z (ESI): 450,2 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,86 (с, 1Н), 8,33 (д, 1Н), 7,73 (с, 1Н), 7,33-7,28 (м, 2Н), 5,29 (д, 1Н), 4,92 (д, 1Н), 4,85-4,76 (м, 2Н), 4,28-4,22 (м, 2Н), 1,44 (д, 3Н), 1,34 (д, 3Н).
Пример 19
N3-(3-Метилоксетан-3-ил)-N5-(3,4,5-триФторфенил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 19

Стадия 1
5-((3,4,5-Трифторфенил)карбамоил)-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоновая кислота 19а
Соединение 4а (3 г, 8,74 ммоль) растворяли в 20 мл метанола и 20 мл тетрагидрофурана, затем добавляли гидроксид натрия (3,39 г, 84,67 ммоль) и 10 мл воды. После перемешивания в течение 16 часов реакционный раствор концентрировали при пониженном давлении, разбавляли 20 мл воды и добавляли по каплям 6 М соляную кислоту до получения рН 2. Реакционный раствор концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 19а (100 мг), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ESI): 341,1 [М+1].
Стадия 2
N3-(3-Метилоксетан-3-ил)-N5-(3,4,5-трифторфенил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 19
Неочищенное соединение 19а (100 мг, 293,90 мкмоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (69,14 мг, 293,90 мкмоль) и триэтиламин (29,74 мг, 293,90 мкмоль) растворяли в 5 мл N,N-диметилформамида и проводили реакцию в течение 10 минут.В реакционный раствор добавляли соединение 13 с (108,96 мг, 881,69 мкмоль) и перемешивали в течение 3 часов. Реакционный раствор разбавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 19 (20 мг, выход: 16,62%).
МС m/z (ESI): 410,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,95 (с, 1Н), 7,30-7,26 (м, 2Н), 5,01 (с, 2Н), 4,88 (д, 2Н), 4,50 (д, 2Н), 4,28-4,25 (м, 2Н), 4,04-4,01 (м, 2Н), 1,71 (с, 3Н).
Пример 20
(S)-N7-(2,6-ДиФторпиридин-4-ил)-6-метил-N1-((R)-1,1,1-триФторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7-(8H)-дикарбоксамид 20

В схеме синтеза Примера 6 исходное соединение 2d на стадии 4 заменяли исходным соединением 2,6-дифтор-4-пиридиламин (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 20 (10 мг).
МС m/z (ESI):433,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,69 (с, 1Н), 7,11-7,09 (д, 2Н), 5,33-5,29 (м, 1Н), 4,97-4,77 (м, 1Н), 4,56-4,41 (м, 1Н), 4,27-4,21 (м, 1Н), 3,80-3,45 (м, 2Н), 1,38 (д, 3Н), 1,15(д, 3Н).
Пример 21
(R)-N3-(втор-Бутил)-N5-(3,4,5-трифторфенил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 21

Неочищенное соединение 19а (96 мг, 282,26 мкмоль) растворяли в 10 мл N,N-диметилформамида, добавляли (R)-бут-2-амин (20,64 мг, 282,26 мкмоль, TCI (Shanghai) Development Со, Ltd.) и триэтиламин (85,69 мг, 846,78 мкмоль), затем при температуре 0°С добавляли гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (79,69 мг, 338,71 мкмоль). Реакционный раствор нагревали до комнатной температуры и проводили реакцию в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 21 (10 мг, выход: 8,96%).
МС m/z (ESI): 396,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,00 (с, 1Н), 7,30-7,26 (м, 2Н), 5,04 (с, 2Н), 4,27-4,25 (м, 2Н), 4,04-4,00 (м, 3Н), 1,60-1,56 (м, 2Н), 1,22 (д, 3Н), 0,96 (т, 3Н).
Пример 22
(S)-N3-((R)-втор-Бутил)-N5-(3,4-диФторфенил)-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 22

В схеме синтеза Примера 7 исходное соединение 1е заменяли исходным соединением (R)-бут-2-амин, соответственно, и получали указанное в заголовке соединение 22 (20 мг).
MC m/z (ESI): 392,1 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,69 (с, 1Н), 7,53-7,50 (м, 1Н), 7,23 (с, 1Н), 7,06-7,02 (м, 2Н), 5,62 (д, 1Н), 5,23 (д, 2Н), 4,82 (д, 1Н), 4,31-4,28 (м, 1Н), 4,28 (д, 1Н), 4,07 (д, 1 Н), 1,60-1,57 (м, 2Н), 1,22 (д, 6Н), 0,99 (т, 3Н).
Пример 23
(S)-N5-(3,4-Дифторфенил)-N3-изопропил-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 23

В схеме синтеза Примера 7 исходное соединение 1е на стадии 5 заменяли исходным соединением изопропиламин, соответственно, и получали указанное в заголовке соединение 23 (25 мг).
МС m/z (ESI): 378,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,00 (с, 1Н), 7,50-7,45 (м, 1Н), 7,18-7,14 (м, 2Н), 5,35-5,30 (м, 1Н), 4,99-4,95 (м, 1Н), 4,69-4,62 (м, 1Н), 4,32-4,22 (м, 1Н), 4,22-4,12 (м, 2Н), 1,23-1,16 (м, 9Н).
Пример 24
(S)-N5-(3,4-Дифторфенил)-N3-(1-метокси-2-метилпропан-2-ил)-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 24

В схеме синтеза Примера 7 исходное соединение 1е на стадии 5 заменяли исходным соединением 1-метокси-2-метилпропан-2-амин (Sinopharm Chemical Reagent Со, Ltd. (Shanghai)), соответственно, и получали указанное в заголовке соединение 24 (15 мг).
MC m/z(ESI): 422,1 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,69 (с, 1Н), 7,56-7,51 (м, 1Н), 7,15-7,09 (м, 3Н), 6,13 (с, 1Н), 5,26-5,22 (м, 1Н), 5,20 (д, 1Н), 4,85 (д, 1Н), 4,36-4,32 (м, 1Н), 4,22 (д, 1Н), 3,47 (с, 3Н), 3,44 (с, 2Н), 1,50 (с, 6Н), 1,25 (д, 3Н).
Пример 25
(S)-N5-(3-Хлор-2-фторпиридин-4-ил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 25
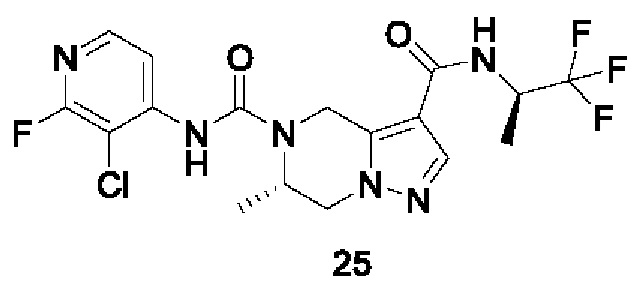
В схеме синтеза Примера 3 исходное соединение За на стадии 1 заменяли исходным соединением 7b, исходное соединение 3d на стадии 3 заменяли исходным соединением 2-хлор-3-фторпиридин-4-амин (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 25 (20 мг).
МС m/z (ESI): 449,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,09 (с, 1Н), 8,04 (д, 1Н), 7,87 (т, 1Н), 5,42 (д, 1Н), 5,02 (т, 1Н), 4,84 (т, 1Н), 4,77 (д, 1Н), 4,38-4,35 (м, 1Н), 4,21 (д, 1 Н), 1,41 (д, 3Н), 1,29 (д, 3Н).
Пример 26
(R)-N1-(втор-Бутил)-N7-(3,4,5-трифторфенил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 26

В схеме синтеза Примера 1 исходное соединение 1е на стадии 3 заменяли исходным соединением (R)-бут-2-амин, соответственно, и получали указанное в заголовке соединение 26 (4 мг).
MC m/z (ESI): 396,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,68 (с, 1Н), 7,31-7,29 (м, 2Н), 5,05 (с, 2Н), 4,23-4,21 (м, 2Н), 4,01-3,95 (м, 3Н), 1,61-1,58 (м, 2Н), 1,24-1,23 (м, 3Н), 0,96 (т, 3Н).
Пример 27
(S)-N3-(Тетрагидрофуран-3-ил)-N5-(3,4,5-трифторфенил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 27
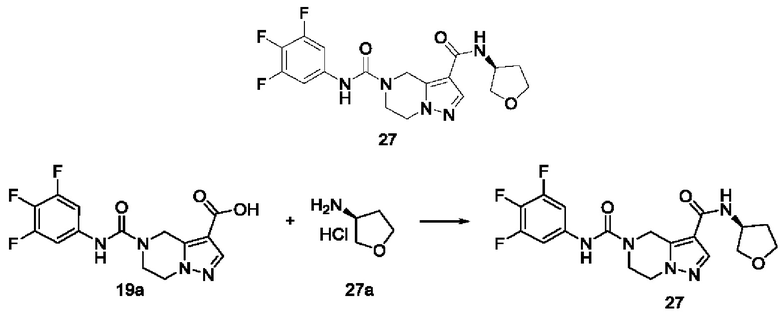
Неочищенное соединение 19а (100 мг, 293,90 мкмоль), гексафторфосфат 0-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (138,29 мг, 587,79 мкмоль) и триэтиламин (148,70 мг, 1,47 ммоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли гидрохлорид (S)-тетрагидрофуран-3-амина 27а (72,64 мг, 587,79 мкмоль, Shanghai Bide Pharmatech Ltd.) и перемешивали в течение 3 часов. Реакционный раствор разбавляли 10 мл воды и экстрагировали этилацетатом (20 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 27 (20 мг, выход: 16,62%).
МС m/z (ESI): 410,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,02 (с, 1Н), 7,30-7,26 (м, 2Н), 5,03 (с, 2Н), 4,56-4,55 (м, 1Н), 4,27-4,25 (м, 2Н), 4,04-3,95 (м, 4Н), 3,85-3,84 (м, 1Н), 3,72-3,71 (м, 1Н), 2,32-2,27 (м, 1Н), 2,02-1,96 (м, 1Н).
Пример 28
(S)-N5-(3,4-Дифторфенил)-N3-(1-гидрокси-2-метилпропан-2-ил)-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 28

В схеме синтеза Примера 7 исходное соединение 1е на стадии 5 заменяли исходным соединением 2-амино-2-метилпропан-1-ол (Accela ChemBio Inc), соответственно, и получали указанное в заголовке соединение 28 (10 мг).
МС m/z(ESI): 408,1 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,70 (с, 1Н), 7,57-7,52 (м, 1Н), 7,16-7,10 (м, 3Н), 5,94 (с, 1Н), 5,25-5,23 (м, 1Н), 5,22 (д, 1Н), 4,83 (д, 1Н), 4,59-4,58 (м, 1Н), 4,37-4,32 (м, 1Н), 4,21 (д, 1 Н), 3,77-3,69 (м, 2Н), 1,45 (с, 6Н), 1,25 (д, 3Н).
Пример 29
(R)-N5-(3,4-Дифторфенил)-2-метил-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 29
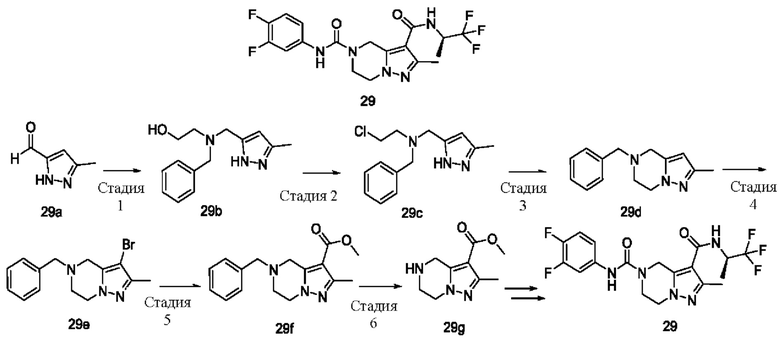
Стадия 1
2-(Бензил((3-метил-1Н-пиразол-5-ил)метил)амино)этан-1-ол 29b
3-Метил-1Н-пиразол-5-карбальдегид 29а (5,1 г, 45,41 ммоль, Shanghai Bide Pharmatech Ltd.) растворяли в 100 мл дихлорметана, затем добавляли 2-(бензиламино)этанол (6,87 г, 45,41 ммоль). После перемешивания в течение 1 часа в реакционный раствор добавляли ацетоксиборгидрид натрия (9,62 г, 45,41 ммоль) и перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле с использованием элюирующих систем С и А, в результате получали указанное в заголовке соединение 29b (11 г, выход: 98,75%).
МС m/z (ESI): 246,1 [М+1].
Стадия 2
N-Бензил-2-хлор-N-((3-метил-1Н-пиразол-5-ил)метил)этан-1-амин 29с
Соединение 29b (11 г, 44,84 ммоль) растворяли в 200 мл дихлорметана. После охлаждения реакционного раствора до температуры 0°С в него по каплям прибавляли дихлорсульфоксид (16,00 г, 134,52 ммоль) и проводили реакцию при температуре 40°С в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 29 с (11,8 г), которое использовали непосредственно на следующей стадии без очистки.
MCm/z(ESI): 264,1 [М+1].
Стадия 3
5-Бензил-2-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин 29d
Неочищенное соединение 29 с (11,8 г, 44,74 ммоль) растворяли в 200 мл ацетонитрила. После охлаждения до температуры 0°С в реакционный раствор добавляли триэтиламин (46 г, 447,37 ммоль) и проводили реакцию при температуре 80°С в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Остаток разбавляли 100 мл воды и экстрагировали этилацетатом (200 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 29d (10,17 г), которое использовали непосредственно на следующей стадии без очистки.
МС m/z (ESI): 228,1 [М+1].
Стадия 4
5-Бензил-3-бром-2-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин 29е
Неочищенное соединение 29d (228 мг, 1,0 ммоль) растворяли в 10 мл ацетонитрила, после чего добавляли N-бромсукцинимид (180 мг, 1,0 ммоль) при температуре 0°С. Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 29е (185 мг, выход: 60,6%).
MC m/z(ESI):306,1 [М+1].
Стадия 5
Метил-5-бензил-2-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилат 29f
Октакарбонил дикобальта (226 мг, 662 мкмоль) и карбонат калия (457 мг, 3,31 ммоль) добавляли в 10 мл метанола в атмосфере окиси углерода. После перемешивания при температуре 60°С в течение 45 минут в реакционный раствор добавляли соединение 29е (100 мг, 327 мкмоль) и метил-2-хлорацетат (215 мг, 1,98 ммоль) и перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, разбавляли 100 мл этилацетата, фильтровали и промывали 100 мл этилацетата. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 29f (60 мг, выход: 64,39%).
MCm/z (ESI):286,2 [М+1].
Стадия 6
Метил-2-метил-4,5,6,7-тетрагидропиразоло[1,5-а]пиразин-3-карбоксилат 29д
Соединение 29f (60 мг, 210,28 мкмоль) растворяли в 10 мл метанола в атмосфере водорода, затем добавляли катализатор гидрирования палладий на угле (влажный) (22,38 мг, 210,28 мкмоль). После перемешивания в течение 16 часов реакционный раствор фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 29g (30 мг), которое использовали непосредственно на следующей стадии без очистки.
MC m/z (ESI):196,2 [М+1].
В схеме синтеза Примера 2 исходное соединение 2с на стадии 3 заменяли неочищенным исходным соединением 29g, соответственно, и получали указанное в заголовке соединение 29 (10 мг).
MCm/z (ESI): 432,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,49-7,44 (м, 1Н), 7,19-7,14 (м, 2Н), 4,96 (с, 2Н), 4,87-4,83 (м, 1Н), 4,20-4,17 (м, 2Н), 4,01-3,96 (м, 2Н), 2,40(с, 3Н), 1,42-1,39 (м, 3Н).
Пример 30
N1-(3-Метилоксетан-3-ил)-N7-(3,4,5-трифторфенил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 30

В схеме синтеза Примера 1 исходное соединение 1е на стадии 5 заменяли исходным соединением 13 с, соответственно, и получали указанное в заголовке соединение 30 (5 мг).
MC m/z (ESI): 410,0 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,39 (с, 1Н), 7,80 (с, 1Н), 7,67 (с, 1Н), 7,31-7,26 (м, 2Н), 5,02 (с, 2Н), 4,54-4,51 (д, 2Н), 4,23-4,20 (м, 2Н), 3,96-3,93 (м, 2Н), 3,57-3,53 (м, 2Н), 1,72 (с, 3Н).
Пример 31
(R)-N5-(3,4-Дифторфенил)-N3-(1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 31

В схеме синтеза Примера 17 исходное соединение 17f на стадии 1 заменяли исходным соединением 7d, соответственно, и получали указанное в заголовке соединение 31 (20 мг).
МС m/z (ESI): 419,1 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 8,11 (с, 1Н), 8,09 (с, 1Н), 7,56-7,52 (м, 1Н), 7,13-7,08 (м, 1Н), 7,06 (ушир., 1Н), 5,35-5,32 (м, 1Н), 4,99 (с, 1Н), 4,94 (с, 1Н), 4,68 (с, 2Н), 2,26 (т, 2Н), 1,42 (д, 3Н).
Пример 32
(R)-N7-(2,6-Дифторпиридин-4-ил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 32

В схеме синтеза Примера 16 исходное соединение 15с на стадии 2 заменяли исходным соединением 3с, соответственно, и получали указанное в заголовке соединение 32 (25 мг).
MC m/z(ESI): 419,1 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,79 (с, 1Н), 8,33 (с, 1Н), 7,70 (с, 1Н), 7,10 (с, 2Н), 5,08 (с, 2Н), 4,84-4,80 (м, 1Н), 4,26-4,23 (м, 2Н), 3,99-3,96 (м, 2Н), 1,44 (д, 3Н).
Пример 33
(S)-6-Метил-N5-(3,4,5-триФторфенил)-N3-((R)-1,1,1-триФторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикар6оксамид 33

В схеме синтеза Примера 7 исходное соединение 7d на стадии 3 заменяли соединением 17f, соответственно, и получали указанное в заголовке соединение 33 (20 мг).
MC m/z (ESI): 450,0 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,09 (с, 1Н), 7,30-7,27 (м, 2Н), 5,36 (д, 1Н), 4,98-4,95 (м, 1Н), 4,83-4,80 (м, 1Н), 4,70 (д, 1Н), 4,31-4,27 (м, 1Н), 4,20 (д, 1Н), 1,41 (д, 3Н), 1,22 (д, 3Н).
Пример 34
(S)-N5-(3,4-Дифторфенил)-6-метил-N3-((S)-тетрагидрофуран-3-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 34

Неочищенное соединение 7f (150 мг, 446,04 мкмоль), гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (157,41 мг, 669,06 мкмоль) и N,N-диизопропилэтиламин (230,58 мг, 1,78 ммоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли соединение 27а (82,3 мг, 669,06 мкмоль) и перемешивали в течение 3 часов. Реакционный раствор разбавляли 30 мл воды и экстрагировали этилацетатом (30 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С. Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 34 (30 мг, выход: 16,54%).
MC m/z (ESI): 406,2 [М+1].
1H ЯМР (400 МГц, CD3OD): , 8,05 (с, 1Н), 7,52-7,47 (м, 1Н), 7,20-7,17 (м, 2Н), 5,33 (д, 1Н), 5,00-4,99 (м, 1Н), 4,70 (д, 1Н), 4,56-4,54 (м, 1Н), 4,34-4,32 (м, 1Н), 4,21-4,20 (м, 1Н), 4,04-3,95 (м, 2Н), 3,85-3,83 (м, 1Н), 3,73-3,72 (м, 1Н), 2,32-2,27 (м, 1Н), 2,01-1,94 (м, 3Н), 1,24 (д, 1Н).
Пример 35
N1-(3-Метилтетрагидрофуран-3-ил)-N7-(3,4,5-трифторфенил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7-(8H)-дикарбоксамид 35

В схеме синтеза Примера 1 исходное соединение 1е на стадии 5 заменяли исходным соединением 3-метилтетрагидрофуран-3-амин (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 35 (16 мг).
MC m/z(ESI): 424,1 [М+1].
1Н ЯМР (400 МГц, ДМСО-d6): δ 8,87 (с, 1Н), 8,35 (с, 1Н), 7,67 (с, 1Н), 7,31-7,27 (м, 2Н), 5,03 (с, 2Н), 4,21 (т, 2Н), 4,10 (д, 1Н), 3,96-3,92 (м, 4Н), 3,75 (д, 1Н), 2,46-2,41 (м, 1Н), 2,09-2,01 (м, 1Н), 1,59 (с, 3Н).
Пример 36
(R)-N2-(3,4-Дифторфенил)-N8-(1,1,1-трифторпропан-2-ил)-3,4-дигидропирроло[1,2-а]пиразин-2,8(1Н)-дикарбоксамид 36

Метилпирроло[1,2-а]пиразин-8-карбоксилат 36a (500 мг, 2,84 ммоль, полученный в соответствии со способом, раскрытым в заявке на патент US 2015/51189 A1) и палладий на угле (698,38 мг, 2,84 ммоль) растворяли в 10 мл метанола в атмосфере водорода. После перемешивания в течение 16 часов реакционный раствор фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта метил-1,2,3,4-тетрагидропирроло[1,2-а]пиразин-8-карбоксилата 36b (511 мг, выход: 100%).
MC m/z (ESI): 181,2 [М+1].
В схеме синтеза Примера 2 исходное соединение 2 с на стадии 3 заменяли неочищенным исходным соединением 36b, соответственно, и получали указанное в заголовке соединение 36 (10 мг).
MC m/z (ESI): 417,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,51-7,46 (м, 1Н), 7,19-7,15 (м, 2Н), 6,71-6,70 (м, 2Н), 5,00 (с, 2Н), 4,85-4,82 (м, 1Н), 4,11-4,09 (м, 2Н), 3,94-3,91 (м, 2Н), 1,39 (д, 3Н).
Пример 37
(R)-N7-(2-Дифторметил)пиридин-4-ил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 37
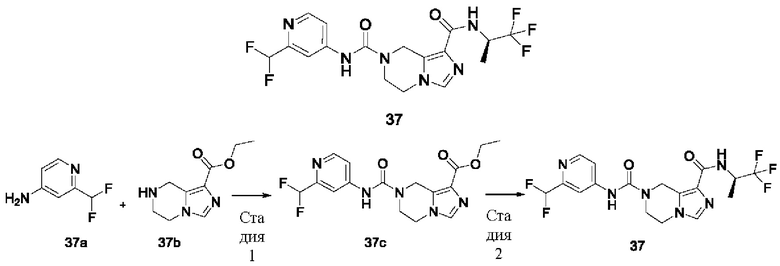
Стадия 1
Этил-7-((2-(дифторметил)пиридин-4-ил)карбамоил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 37с
2-(Дифторметил)пиридин-4-амин 37а (73,83 мг, 512,25 мкмоль, полученный в соответствии со способом, раскрытым в заявке на патент WO 2015/118057 А1), этил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилат 37b (100 мг, 512,25 мкмоль, Shanghai Bide Pharmatech Ltd.) и триэтиламин (103,67 мг, 1,02 ммоль) растворяли в 10 мл тетрагидрофурана, затем добавляли бис(трихлорметил)карбонат (60,80 мг, 204,09 мкмоль) при температуре 0°С. Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 37 с (120 мг, выход: 64,2%).
MC m/z (ESI): 366,2 [М+1].
Стадия 2
(R)-N7-(2-Дифторметил)пиридин-4-ил)-N1-(1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 37
Соединение 37с (100 мг, 273,72 мкмоль) и соединение 1е (30,95 мг, 273,72 мкмоль) растворяли в 10 мл тетра гидрофура на, после чего добавляли 1 мл 1М раствора бис(триметилсилил)амида лития в тетрагидрофуране при температуре 0°С. По окончании добавления раствор медленно нагревали до комнатной температуры и перемешивали в течение 6 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters 2767-SQ Detecor2, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 37 (20 мг, выход: 16,9%).
MC m/z (ESI): 433,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,42 (д, 1Н), 7,88-7,87 (м, 1Н), 7,70-7,66 (м, 2Н), 6,80-6,53 (м, 1Н), 5,10 (с, 2Н), 4,83-4,81 (м, 1Н), 4,26-4,24 (м, 2Н), 4,00-3,98 (м, 2Н), 1,44 (д, 3Н).
Пример 38
(S)-N3,N5-Бис(3,4-дифторфенил)-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 38

Неочищенное соединение 7f (80 мг, 237,89 мкмоль), гексафторфосфат О-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (83,95 мг, 356,83 мкмоль) и N,N-диизопропилэтиламин (92,23 мг, 713,66 мкмоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли соединение 2d (46,07 мг, 356,83 мкмоль) и перемешивали в течение 16 часов. Реакционный раствор разбавляли 10 мл воды и экстрагировали этилацетатом (40 мл × 2). Органические фракции объединяли и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С.Полученный неочищенный продукт очищали при помощи препаративной высокоэффективной жидкостной хроматографии (Waters-2767, элюирующая система: бикарбонат аммония, вода, ацетонитрил), в результате получали указанное в заголовке соединение 38 (10 мг, выход: 9,4%).
MC m/z (ESI): 448,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,18 (с, 1Н), 7,83-7,81 (м, 1Н), 7,52-7,50 (м, 1Н), 7,41-7,39 (м, 1Н), 7,24-7,17 (м, 3Н), 5,39 (д, 1Н), 5,04-5,01 (м, 1Н), 4,75 (д, 1Н), 4,38-4,36 (м, 1Н), 4,22 (д, 1Н), 1,26 (д, 3Н).
Пример 39
(S)-N5-(3,4-Дифторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 39

Стадия 1
(S)-4-((1-Гидроксипропан-2-ил)(4-метоксибензил)амино)бут-2-ин-1-илацетат 39b
Соединение 6а (3,00 г, 15,00 ммоль) растворяли в 60 мл диоксана, затем добавляли 4-хлорбут-2-ин-1-илацетат 39а (5,73 г, 39,00 ммоль, полученный в соответствии с известным способом, раскрытым в работе "Journal of Medicine Chemistry, 2014, 57(9),3687-3706") и триэтиламин (4,7 г, 46,00 ммоль). После перемешивания в течение 12 часов при температуре 60°С реакционный раствор фильтровали. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле с использованием элюирующей системы А, в результате получали указанное в заголовке соединение 39b (2,20 г, выход: 42,2%).
MC m/z (ESI): 306,2 [М+1].
Стадия 2
(S)-4-((1-Хлорпропан-2-ил)(4-метоксибензил)амино)бут-2-ин-1-илацетат 39с
Соединение 39b (2,20 г, 7,20 ммоль) и пиридин (854 мг, 10,08 ммоль) растворяли в 30 мл дихлорметана, после чего медленно по каплям прибавляй тионилхлорид (1,50 г, 12,60 ммоль) при охлаждении на ледяной бане. Реакционный раствор медленно нагревали до комнатной температуры и перемешивали в течение 2 часов. Реакционный раствор разбавляли 100 мл дихлорметана, промывали водой (50 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, в результате получали указанное в заголовке соединение 39 с (2,20 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
(S)-(5-(4-Метоксибензил)-6-метил-4,5,6,7-тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-ил)метилацетат 39d
Неочищенное соединение 39 с (2,20 г, 6,79 ммоль) растворяли в 20 мл N,N-диметилформамида, затем добавляли азид натрия (574 мг, 8,83 ммоль). После перемешивания при температуре 80°С в течение 12 часов реакционный раствор охлаждали до комнатной температуры, разбавляли 50 мл этилацетата, промывали водой (20 мл×2), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, полученный остаток очищали колоночной хроматографией на силикагеле с использованием элюирующей системы А, в результате получали указанное в заголовке соединение 39d (1,30 г, выход: 57,9%).
МС m/z (ESI): 331,1 [М+1].
Стадия 4
Трифторацетат (S)-(6-Метил-4,5,6,7-тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-ил)метилацетата 39е
Соединение 39d (1,30 г, 2,33 ммоль) растворяли в 5 мл трифторуксусной кислоты. Реакционный раствор нагревали под воздействием микроволнового излучения до температуры 100°С в течение 5 минут, охлаждали до комнатной температуры и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 39е (1,28 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 5
(S)-(5-((3,4-Дифторфенил)карбамоил)-6-метил-4,5,6,7-тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-ил)метилацетат 39f
Неочищенное соединение 39е (200 мг, 0,62 ммоль), соединение 2d (228 мг, 1,90 ммоль) и триэтиламин (290 мг, 2,86 ммоль) растворяли в 10 мл тетрагидрофурана, затем при охлаждении на ледяной бане добавляли бис(трихлорметил)карбонат (87 мг, 0,3 ммоль). После перемешивания в течение 3 часов реакционный раствор концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле с использованием элюирующей системы С, в результате получали указанное в заголовке соединение 39f (90 мг, выход: 40,1%).
МС m/z (ESI): 366,1 [М+1].
В схеме синтеза Примера 17 исходное соединение 17b на стадии 2 заменяли соединением 39f, соответственно, и получали указанное в заголовке соединение 39 (20 мг).
МС m/z (ESI): 433,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,50-7,48 (м, 1 Н), 7,18-7,16 (м, 2Н), 5,43 (д, 1 Н), 5,08-5,06 (м, 1 Н), 4,89-4,87 (м, 1Н), 4,74 (д, 1Н), 4,60 (д, 1 Н), 4,49-4,46 (м, 1 Н), 1,49 (д, 3Н), 1,21 (д, 3Н).
Пример 40
(S)-N5-(4-Фтор-3-метилфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 40

В схеме синтеза Примера 39 исходное соединение 2d на стадии 5 заменяли 3-метил-4-фторанилином 40а, соответственно, и получали указанное в заголовкесоединение 40 (25 мг).
МС m/z (ESI): 429,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,29-7,26 (м, 1Н), 7,21-7,20 (м, 1Н), 6,99-6,94 (м, 1Н), 5,45-5,41 (д, 1Н), 5,09-5,08 (м, 2Н), 4,76-4,71 (д, 1Н), 4,63-4,59 (м, 1Н), 4,52-4,51 (д, 1 Н), 2,27-2,26 (д, 3Н), 1,48-1,46 (д, 3Н), 1,23-1,22 (д, 3Н).
Пример 41
(S)-N7-(3,4-Дифторфенил)-6-метил-N1-((S)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 41

В схеме синтеза Примера 6 исходное соединение 1е заменяли исходным соединением гидрохлорид (2S)-1,1,1-трифторпропил-2-амина (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 41 (110 мг).
МС m/z (ESI): 432,1 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,54-7,51 (м, 1Н), 7,50 (с, 1Н), 7,21-7,02 (м, 3H), 6,83 (с, 1Н), 5,26-5,16 (м, 1Н), 5,15-5,04 (м, 1Н), 4,95-4,92 (м, 1Н), 4,86-4,80 (м, 1Н), 4,28-4,22 (м, 1Н), 4,01-4,05 (м, 1 Н), 1,48 (д, 3H), 1,20 (д, 3H).
Пример 42
(R)-N5-(3,4-Дифторфенил)-7-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 42

В схеме синтеза Примера 39 исходное соединение 6а на стадии 1 заменяли соединением (2R)-1-((4-метоксибензил)амино)пропан-1-ол (полученным в соответствии с известным способом, раскрытым в работе "Organic and Biomolecular Chemistry, 2014, 12, 16, 2584-2591"), соответственно, и получали указанное в заголовке соединение 42 (20 мг).
МС m/z (ESI): 433,1 [М+1].
1Н ЯМР(400 МГц, CD3OD): δ 7,50-7,48 (м, 1Н), 7,15-7,12 (м, 2Н), 5,16 (д, 1Н), 5,02 (д, 1Н), 4,77-4,75 (м, 1Н), 4,13-4,10 (м, 1Н), 3,77 (м, 1 Н), 3,06-3,03 (м, 1Н), 1,67 (д, 3H), 1,44 (д, 3H).
Пример 43
(S)-N5-(3,4-Дифторфенил)-6-метил-N3-((S)-1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 43

В схеме синтеза Примера 17 исходное соединение 17b на стадии 2 заменяли соединением 39f, а исходное соединение 1е заменяли исходным соединением гидрохлорид (2S)-1,1,1-трифторпропил-2-амина, соответственно, и получали указанное в заголовке соединение 43 (30 мг).
MCm/z (ESI): 433,3 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,53-7,50 (м, 1Н), 7,30 (д, 1Н), 7,15 (дд, 1Н), 7,06-7,04 (м, 1Н), 6,84 (с, 1Н), 5,37-5,32 (м, 1Н), 5,27 (д, 1Н), 4,89-4,85 (м, 2Н), 4,61 (д, 1 Н), 4,52-4,48 (м, 1 Н), 1,50 (д, 3H), 1,26 (д, 3H).
Пример 44
(R)-N5-(3-Циано-4-фторфенил)-7-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 44

В схеме синтеза Примера 39 исходное соединение 6а на стадии 1 заменяли соединением (2R)-1-((4-метоксибензил)амино)пропан-1-ол, а исходное соединение 2d на стадии 5 заменяли соединением 3d, соответственно, и получали указанное в заголовке соединение 44 (15 мг).
MC m/z (ESI): 440,3 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,86-7,84 (м, 1Н), 7,73-7,70 (м, 1 Н), 7,29 (т, 1Н), 5,19 (д, 1 Н), 5,05 (д, 1 Н), 4,85-4,82 (м, 1Н), 4,12 (д, 1 Н), 3,77-3,73 (м, 1Н), 3,64-3,62 (м, 1Н), 1,69 (д, 3H), 1,46 (д, 3H).
Пример 45
(S)-N5-(3-Циано-4-фторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 45
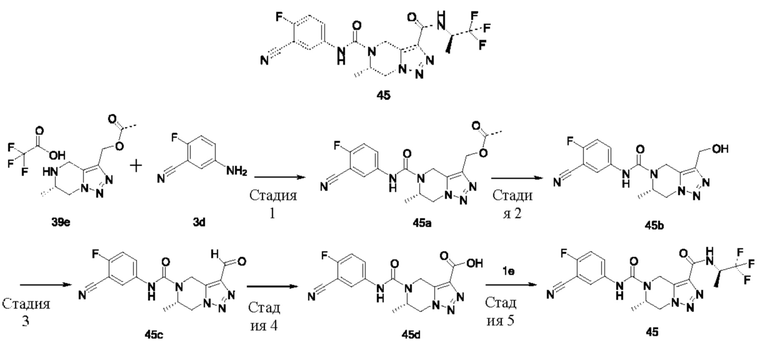
Стадия 1
(S)-(5-((3-Циано-4-фторфенил)карбамоил)-6-метил-4,5,6,7-тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-ил)метилацетат 45а
Неочищенное соединение 39е (400 мг, 1,24 ммоль), соединение 3d (450 мг, 3,80 ммоль) и триэтиламин (580 мг, 5,80 ммоль) растворяли в 20 мл тетрагидрофурана, затем при охлаждении на ледяной бане добавляли бис(трихлорметил)карбонат (180 мг, 0,6 ммоль). После перемешивания в течение 3 часов реакционный раствор концентрировали при пониженном давлении, остаток очищали колоночной хроматографией на силикагеле с использованием элюирующей системы С, в результате получали указанное в заголовке соединение 45а (195 мг, выход: 40,1%).
МС m/z (ESI): 373,1 [М+1].
Стадия 2
(S)-N-(3-Циано-4-фторфенил)-3-(гидроксиметил)-6-метил-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-5(4H)-карбоксамид 45b
Соединение 45а (190 мг, 0,51 ммоль) растворяли в 5 мл смешанного растворителя, состоящего из метанола и воды (V:V=4:1), после чего добавляли гидроксид лития (108 мг, 2,57 ммоль). Реакционный раствор перемешивали в течение 2 часов, разбавляли 20 мл воды и экстрагировали этилацетатом (15 мл×3). Органические фракции объединяли и концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 45b (120 мг), которое использовали непосредственно на следующей стадии без очистки.
MC m/z (ESI): 331,2 [М+1].
Стадия 3
(S)-N-(3-Циано-4-фторфенил)-3-формил-6-метил-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-5(4H)-карбоксамид 45с
Неочищенное соединение 45b (100 мг, 0,3 ммоль) растворяли в 5 мл дихлорметана, затем добавляли хлорхромат пиридиния (138 мг, 0,64 ммоль) и силикагель (140 мг, 100-200 меш). После перемешивания в течение 3 часов реакционный раствор фильтровали и концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей А, в результате получали указанное в заголовке соединение 45 с (60 мг, выход: 43,1%).
MC m/z (ESI): 329,5 [М+1].
Стадия 4
(S)-5-((3-Циано-4-фторфенил)карбамоил)-6-метил-4,5,6,7-тетрагидро-[1,2,3]триазоло[1,5-а]пиразин-3-карбоновая кислота 45d
Соединение 45 с (30 мг, 0,1 ммоль) растворяли в 5 мл смешанного растворителя, состоящего из ацетонитрила и воды (V:V=3:2), затем добавляли аминосульфоновую кислоту (20 мг, 0,2 ммоль). После перемешивания в течение 10 минут в раствор добавляли хлорит натрия (20 мг, 0,2 ммоль) и перемешивали в течение 2 часов. Затем в реакционный раствор последовательно добавляли 0,5 мл насыщенного раствора сульфита натрия и 10 мл воды и экстрагировали этилацетатом (10 мл×3). Органические фракции объединяли и концентрировали при пониженном давлении с получением неочищенного соединения 45d (25 мг), которое использовали непосредственно на следующей стадии без очистки.
MC m/z (ESI): 345,2 [М+1].
Стадия 5
(S)-N5-(3-Циано-4-фторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4H)-дикарбоксамид 45
Неочищенное соединение 45d (20 мг, 58,0 мкмоль), гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (105 мг, 146,5 мкмоль) и N,N-диизопропилэтиламин (150 мг, 366,2 мкмоль) растворяли в 3 мл N,N-диметилформамида и проводили реакцию в течение 10 минут. В реакционный раствор добавляли соединение 1е (50 мг, 110 мкмоль) и перемешивали в течение 3 часов. Реакционный раствор концентрировали при пониженном давлении, полученный остаток очищали при помощи тонкослойной хроматографии с использованием проявляющей системы растворителей С, в результате получали указанное в заголовке соединение 45 (8 мг, выход: 31,3%).
MC m/z (ESI): 440,3 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,87-7,84 (м, 1Н), 7,73-7,70 (м, 1 Н), 7,29 (т, 1Н), 5,45 (т, 1Н), 5,08-5,06 (м, 1 Н), 4,92-4,89 (м, 1Н), 4,77 (д, 1Н), 4,62 (д, 1Н), 4,50 (д, 1Н), 1,46 (д, 3H), 1,21 (д, 3H).
Пример 46
(S)-N7-(3-Циано-4-фторфенил)-6-метил-N1-(1-метилциклобутил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 46

В схеме синтеза Примера 11 исходное соединение 1е на стадии 3 заменяли соединением 1-метилциклобутил-1-амин (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 46 (20 мг).
MC m/z (ESI): 411,4 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,86-7,84 (м, 1Н), 7,72-7,70 (м, 1Н), 7,65 (с, 1 Н) 7,28 (т, 1Н), 5,26 (д, 1Н), 4,92-4,89 (м, 1Н), 4,78 (д, 1Н), 4,20-4,17 (м, 2Н), 2,45-2,43 (м, 2Н), 2,10-2,07 (м, 2Н), 1,92-1,90 (м, 2Н), 1,55 (с, 3H), 1,20 (д, 3H).
Пример 47
(R)-N7-(3-Циано-4-фторфенил)-5-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 47
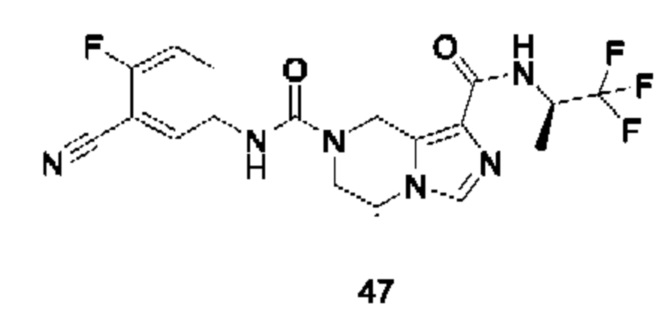
В схеме синтеза Примера 6 исходное соединение 6а на стадии 1 заменяли исходным соединением (2S)-1-((4-метоксибензил)амино)пропан-1-ол, а исходное соединение 2d на стадии 4 заменяли исходным соединением 3d, соответственно, и получали указанное в заголовке соединение 47 (12 мг).
MC m/z (ESI): 439,2 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,88 (дд, 1Н), 7,62-7,61 (м, 1Н), 7,57 (с, 1Н), 7,22-7,17 (м,3H), 5,20 (д, 1Н), 5,03 (д, 1Н), 4,84-4,80 (м, 1Н), 4,42-4,40 (м, 1Н),4,25-4,21 (дд, 1 Н), 3,58-3,53 (дд, 1Н), 1,64 (д, 3H), 1,47 (д, 3H).
Пример 48
(S)-N7-(3-Циано-4-фторфенил)-6-метил-N1-(1-метилциклопропил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 48

В схеме синтеза Примера 11 исходное соединение 1е на стадии 3 заменяли соединением 1-метилциклопропил-1-амин (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 48 (20 мг).
MC m/z (ESI): 397,3 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,86-7,84 (м, 1Н), 7,72-7,70 (м, 1Н), 7,63 (с, 1Н), 7,29 (т, 1Н), 5,28 (д, 1Н), 4,91-4,89 (м, 1Н), 4,80 (д, 1Н), 4,20-4,17 (м, 2Н), 1,44 (с, 3H), 1,20 (д, 3H), 0,71-0,68 (м, 2Н), 0,68-0,66 (м, 2Н).
Пример 49
(R)-N6-(3,4-Дифторфенил)-6-метил-N3-((R)-1,1,1-трифторпропан-2-ил)-6,7-дигидропиразоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 49

В схеме синтеза Примера 7 исходное соединение 7а на стадии 1 заменяли исходным соединением трет-бутил-(R)-3-йод-6-метил-6,7-дигидропиразоло[1,5-а]пиразин-5(4H)-карбоксилат (полученным в соответствии со способом, раскрытым в заявке на патент WO 2017198744A1), соответственно, и получали указанное в заголовке соединение 49 (20 мг).
MC m/z (ESI): 432,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8,11 (м, 1Н), 7,52-7,47 (м, 1 Н), 7,21-7,16 (м, 2Н), 5,38-5,33 (д, 2Н), 5,02-4,99 (м, 1Н), 4,74-4,69 (д, 1 Н), 4,36-4,33 (м, 1Н), 4,23-4,19 (д, 1Н), 1,43-1,42 (д, 3H), 1,27-1,25 (д, 3H).
Пример 50
(S)-N1-(трет-Бутил)-N7-(3-циано-4-фторфенил)-6-метил-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8Н)-дикарбоксамид 50

В схеме синтеза Примера 11 исходное соединение 1е на стадии 3 заменяли исходным соединением трет-бутиламин (Sinopharm Chemical Reagent Со, Ltd.), соответственно, и получали указанное в заголовке соединение 50 (180 мг).
MC m/z (ESI): 399,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,90-7,88 (м, 1 Н), 7,75-7,67 (м, 1 Н), 7,33 (с, 1 Н), 7,31-7,29 (м, 1Н), 5,28-5,24 (д, 1Н), 4,94 (м, 1Н), 4,81-4,77 (м, 1Н), 4,26-4,20 (м, 2Н), 1,48 (д, 9Н), 1,22-1,20 (д, 3H).
Пример 51
(S)-N7-(3-Циано-4-фторфенил)-N1-((R)-1-фторпропан-2-ил)-6-метил-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 51

В схеме синтеза Примера 11 исходное соединение 1е на стадии 3 заменяли исходным соединением гидрохлорид (S)-2-фтор-1-метилэтиламина (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 51 (20 мг).
MC m/z (ESI): 403,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,89-7,87 (м, 1 Н), 7,77-7,74 (м, 1 Н), 7,73 (с, 1 Н), 7,31-7,29 (м, 1Н), 5,31-5,27 (д, 1Н), 4,94-4,92 (м, 1Н), 4,83-4,78 (д, 1Н), 4,52-4,50 (д, 1Н), 4,40-4,39 (м, 2Н), 4,25-4,22 (м, 2Н), 1,32-1,30 (д, 3H), 1,22-1,20 (д, 3H).
Пример 52
(S)-N7-(3-Циано-4-фторфенил)-6-метил-N1-((S)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8Н)-дикарбоксамид 52

В схеме синтеза Примера 11 исходное соединение 1е на стадии 3 заменяли исходным соединением гидрохлорид (2S)-1,1,1-трифторпропил-2-амина, соответственно, и получали указанное в заголовке соединение 52 (20 мг).
MC m/z (ESI): 439,2 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,91 (дд, 1Н), 7,63-7,62 (м, 1Н), 7,50 (с, 1Н), 7,20-7,14 (м, 3H), 5,22-5,16 (м, 2Н), 4,95 (д, 1 Н), 4,85-4,83 (м, 1 Н), 4,26-4,23 (м, 1 Н), 4,11-4,08 (дд, 1Н), 1,48 (д, 3H), 1,26 (д, 3H).
Пример 53
(R)-N7-(3,4-Дифторфенил)-6-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 53

В схеме синтеза Примера 6 исходное соединение 6а на стадии 1 заменяли исходным соединением (R)-2-((4-метоксибензил)амино)пропан-1-ол (полученным в соответствии с известным способом, раскрытым в работе "Bioorganic & Medicinal Chemistry Letters, 2015, 25(5), 1086-1091"), соответственно, и получали указанное в заголовке соединение 53 (90 мг).
MC m/z (ESI): 432,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,71 (с, 1 Н), 7,52-7,47 (м, 1Н), 7,21-7,17 (м, 2Н), 5,31-5,27 (д, 1Н), 4,94-4,92 (м, 1Н), 4,76-4,84 (м, 2Н), 4,24-4,22 (м, 2Н), 1,45-1,43 (д, 3H), 1,23-1,21 (д, 3H).
Пример 54
(R)-N7-(3-Циано-4-фторфенил)-6-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 54

В схеме синтеза Примера 6 исходное соединение 6а на стадии 1 заменяли исходным соединением (S)-2-((4-метоксибензил)амино)пропан-1-ол, а исходное соединение 2d на стадии 4 заменяли соединением 3d, соответственно, и получали указанное в заголовке соединение 54 (88 мг).
MC m/z (ESI): 439,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,89-7,87 (м, 1Н), 7,75-7,73 (м, 1 Н), 7,71 (с, 1 Н), 7,33-7,29 (м, 1Н), 5,32-5,28 (д, 1Н), 4,95-4,93 (м, 1Н), 4,85-4,82 (м, 2Н), 4,25-4,22 (м, 2Н), 1,45-1,43 (д, 3H), 1,23-1,22 (д, 3H).
Пример 55
(R)-N7-(3-Циано-4-фторфенил)-6-метил-N1-((S)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 55

В схеме синтеза Примера 6 исходное соединение 6а на стадии 1 заменяли исходным соединением (R)-2-((4-метоксибензил)амино)пропан-1-ол, исходное соединение 2d на стадии 4 заменяли соединением 3d, а исходное соединение 1е на стадии 5 заменяли исходным соединением гидрохлорид (2S)-1,1,1-трифторпропил-2-амина, соответственно, и получали указанное в заголовке соединение 55 (20 мг).
MC m/z (ESI):439,2 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,86 (дд, 1 Н), 7,70-7,65 (м, 2Н), 7,30-7,26 (м, 3H), 5,30 (д, 1Н), 4,94-4,93 (м, 1Н), 4,81-4,76 (м, 2Н), 4,26-4,17 (м, 2Н), 1,43 (д, 3H), 1,20 (д, 3H).
Пример 56
(R)-N7-(3-Циано-4-фторфенил)-5-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 56

В схеме синтеза Примера 6 исходное соединение 6а на стадии 1 заменяли исходным соединением (S)-1-((4-метоксибензил)амино)пропан-2-ол (полученным в соответствии с известным способом, раскрытым в работе "Organic and Biomolecular Chemistry, 2014, 12, 16, 2584-2591"), а исходное соединение 2d на стадии 4 заменяли исходным соединением 3d, соответственно, и получали указанное в заголовке соединение 56 (10 мг).
MC m/z (ESI): 439,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,88-7,86 (м, 1Н), 7,83 (с, 1 Н), 7,76-7,72 (м, 1Н), 7,33-7,28 (м, 1Н), 5,16-5,11 (д, 1Н), 5,03-4,98 (м, 1Н), 4,85-4,81 (м, 2Н), 4,04-4,00 (м, 1Н), 3,72-3,66 (м, 1Н), 1,60-1,56 (д, 3H), 1,44-1,42 (д, 3H).
Пример 57
N5-(3-Циано-4-фторфенил)-7-метил-N3-((R)-1,1,1-триФторпропан-2-ил)-6,7-дигидро-[1,2,3]триазоло[1,5-а]пиразин-3,5(4Н)-дикарбоксамид 57

В схеме синтеза Примера 39 исходное соединение 6а на стадии 1 заменяли исходным соединением 1-((4-метоксибензил)амино)пропан-2-ол (полученным в соответствии с известным способом, раскрытым в работе "Organic and Biomolecular Chemistry, 2014, 12, 16, 2584-2591"), а исходное соединение 2d на стадии 5 заменяли исходным соединением 3d, соответственно, и получали указанное в заголовке соединение 57 (220 мг).
MC m/z (ESI): 440,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,85-7,87 (м, 1Н), 7,72-7,75 (м, 1Н), 7,28-7,33 (м, 1Н), 5,01-5,06 (д, 1Н), 5,16-5,21 (д, 1Н), 4,80-4,81 (м, 2Н), 4,13-4,17 (м, 1Н), 3,75-3,80 (м, 1Н), 1,69-1,70 (д, 3H), 1,18-1,21 (д, 3H).
Пример 58
(S)-N7-(4-Фтор-3-метилфенил)-6-метил-N1-((R)-1,1,1-трифторпропан-2-ил)-5,6-дигидроимидазо[1,5-а]пиразин-1,7(8H)-дикарбоксамид 58

В схеме синтеза Примера 6 исходное соединение 2d на стадии 4 заменяли исходным соединением 4-фтор-3-метиланилин (Shanghai Bide Pharmatech Ltd.), соответственно, и получали указанное в заголовке соединение 58 (14 мг).
MC m/z (ESI): 427,9 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 7,71 (с, 1Н), 7,29-7,27 (м, 1Н), 7,22-7,20 (м, 1Н), 6,99-6,94 (м, 1Н), 5,30-5,25 (д, 1Н), 4,94-4,92 (м, 1Н), 4,81-4,75 (м, 2Н), 4,24-4,21 (м, 2Н), 2,27-2,26 (д, 3H), 1,45-1,43 (д, 3H), 1,20-1,19 (д, 3H).
Пример 59
(S)-N-(3,4-Дифторфенил)-6-метил-1-(пирролидин-1-карбонил)-5,6-дигидроимидазо[1,5-а]пиразин-7(8Н)-карбоксамид 59

В схеме синтеза Примера 11 исходное соединение 3d на стадии 1 заменяли соединением 2d, исходное соединение 1е на стадии 3 заменяли исходным соединением пирролидин (Sinopharm Chemical Reagent Со, Ltd.), соответственно, и получали указанное в заголовке соединение 59 (10 мг).
MC m/z (ESI): 389,8 [М+1].
1Н ЯМР (400 МГц, CDCl3): δ 7,69 (с, 1Н), 7,52-7,47 (м, 1Н), 7,20-7,16 (м, 3H), 5,26 (д, 1 Н), 4,93-4,90 (м, 1Н), 4,77 (д, 1 Н), 4,23-4,21 (м, 2Н), 4,04-4,01 (м, 2Н), 3,63-3,60 (м, 2Н), 2,01-1,93 (м, 4Н), 1,22 (д, 3H).
Биологический анализ
Тестовый пример 1
Тест на анти-HBV активность in vitro соединения по настоящему изобретению (количественный анализ внутриклеточной ДНК HBV)
I. Экспериментальные материалы и приборы
1. Набор для выделения ДНК "QIAamp 96 DNAQIAcube НТ Kit" (Qiagen)
2. Лабораторный пластик QIAcube НТ plasticware (Qiagen)
3. Набор для количественного определения нуклеиновых кислот вируса гепатита В (Triplex International Biosciences Со, Ltd.)
4. Устройство для выделения ДНК (QIAcube) (Qiagen)
5. Система QuantStudio 6 Fiex (ABI, ThermFisher)
6. Считывающее устройство для микропланшетов (BMG)
7. Клетки HepG2.2.15 (Shanghai Ruilu Biotechnology Co, Ltd.)
II. Экспериментальная часть
Клетка HepG2.2.15 представляет собой стабильно экспрессирующую клеточную линию, несущую встроенный геном HBV. Вирусные частицы, содержащие ДНК HBV, могут синтезироваться в клетках путем репликации, транскрипции и капсидирования, а затем секретироваться. В данном исследовании количественный анализ ДНК HBV, продуцируемой при in vitro пролиферации HepG2.2.15, выполняли методом количественной ПЦР (англ. PCR, polymerase chain reaction - полимеразная цепная реакция), определяя таким образом активность соединения по настоящему изобретению в отношении ингибирования репликации ДНК HBV путем ингибирования сборки капсидного белка HPV.
Клетки HepG2.2.15 культивировали в среде DMEM (англ. Dulbecco modified Eagle's medium - модифицированная по способу Дульбекко среда Игла) с высоким содержанием глюкозы (10% FBS (англ. Fetal Bovine Serum - фетальная бычья сыворотка), 400 мкг/мл G418) и пассировали каждые три дня. В день проведения эксперимента готовили суспензию клеток со свежей средой для культивирования клеток и инкубировали в 96-луночном планшете (Corning, #3599) по 40000 клеток/лунку при 5% диоксида углерода, 37°С. На 2-ой день соединение растворяли в чистом ДМСО в концентрации 20 мМ, затем готовили первую концентрацию 2 мМ в ДМСО и последовательно разбавляли в 4 раза до 8 концентраций. В контрольную лунку добавляли 90 мкл ДМСО. Раствор соединения 200-кратно разбавляли средой DMEM с высоким содержанием глюкозы. Извлекали планшет для культивирования клеток, инокулированный в 1-й день. С помощью отсасывающего устройства с отрицательным давлением удаляли среду в лунках, и в каждую лунку соответственно добавляли по 200 мкл/лунку приготовленной среды, содержащей соединение при каждой концентрации. Планшет инкубировали при температуре 37°С в течение 72 часов. На 5-й день среду для культивирования клеток заменяли свежей средой, содержащей то же самое соединение, таким же образом, как во 2-й день, после чего планшет инкубировали при температуре 37°С в течение 72 часов. На 8-й день планшет для культивирования клеток извлекали, центрифугировали при 300 g в течение 3 минут и собирали супернатант клеточной культуры по 200 мкл/лунку. Выделяли ДНК HBV из супернатанта клеточной культуры с помощью устройства Qiagen для автоматического выделения ДНК и конкретного метода, описанного в инструкциях к реагентам и приборам. И, наконец, выделенную ДНК элюировали буфером для элюирования ДНК по 100 мкл/лунку. Выделенную ДНК подвергали количественному ПЦР-анализу на ДНК HBV с использованием набора для количественного определения нуклеиновых кислот вируса гепатита В (Triplex International Biosciences Со, Ltd.) и конкретного метода, описанного в инструкции к набору. Стандартную калибровочную кривую для количественного анализа получали с помощью стандартного образца, представленного в наборе, опыты проводили параллельно. Выполняли количественное преобразование каждого образца в соответствии со стандартной кривой. И, наконец, с помощью программного обеспечения GraphPad Prism рассчитывали величину ЕС50 соединения для каждой концентрации соединения и соответствующего значения ДНК. Emax - величина эффекта соединения с точки зрения максимального ингибирования репликации ДНК HBV.
С помощью описанного выше эксперимента определяли активность in vitro соединения по настоящему изобретению в отношении ингибирования репликации ДНК HBV путем ингибирования сборки капсидного белка HPV. Измеренные значения ЕС50 приведены в Таблице1.


Заключение: Соединение по настоящему изобретению оказывает значительное ингибирующее действие на репликацию ДНК HBV и имеет существенное преимущество по сравнению со Сравнительным примером 59. Основное отличие структуры соединения Сравнительного примера 59 от соединения по настоящей заявке состоит в том, что его аминогруппа, входящая в состав ациламиногруппы, представляет собой третичный амин, это указывает на то, что вторичная аминогруппа, присутствующая в ациламиногруппе соединения по настоящей заявке, значительно улучшает биологическую активность.
Тестовый пример 2
Влияние соединения по настоящему изобретению на пролиферацию клеток HepG2 in vitro
I. Экспериментальные материалы и приборы
1. Клетки HepG2 (АТСС)
2. Набор для детекции пролиферации клеток CellTiter-GloTM (Promega)
3. Автоматическая станция пипетирования (Bravo): Agilent Technologies Co.
4. Считывающее устройство для микропланшетов (VICTOR 3): PerkinElmer Co.
5. CO2-инкубатор (Fisher Scientific)
6. Центрифуга (Fisher Scientific)
II. Экспериментальная часть
Отбирали клетки HepG2, находящиеся в логарифмической фазе роста, и расщепляли трипсином для приготовления клеточной суспензии, которую инкубировали в 96-луночном планшете (96-луночный планшет белого цвета с прозрачным дном, PerkinElmer) при плотности 6000 клеток/лунку, 5% диоксида углерода, 37°С, в течение от 16 до 20 часов. На 2-ой день соединение растворяли в чистом ДМСО в концентрации 20 мМ. Соединение 3-кратно разбавляли с помощью автоматической станции пипетирования (Bravo), по 8 концентраций на каждое соединение, в качестве контрольной лунки использовали лунку с ДМСО. Соединение, приготовленное в ДМСО при каждой концентрации, 200-кратно разбавляли с помощью среды ЕМЕМ (содержащей 10% FBS). Извлекали культуральный планшет, инокулированный в 1-й день. С помощью отсасывающего устройства с отрицательным давлением удаляли среду в лунках, и в каждую лунку соответственно добавляли по 100 мкл/лунку приготовленной среды, содержащей соединение при каждой концентрации. Планшет инкубировали при температуре 37°С в течение 72 часов. На 5-й день 96-луночный культуральный планшет вынимали, и в каждую лунку добавляли свежеприготовленный CellTiter Glo по 100 мкл/лунку. 96-луночный планшет выстаивали в течение 5-10 минут, затем дно изолировали белой герметизирующей пленкой (PerkinElmer). Планшет помещали в считывающее устройство для микропланшетов для измерения сигнала люминесценции. Величину CC50 соединения рассчитывали с помощью программного обеспечения GraphPad Prism для каждой концентрации соединения и соответствующего значения сигнала ингибирования пролиферации.
С помощью указанного выше эксперимента определяли ингибирующее действие соединения по настоящему изобретению на пролиферацию клеток HepG2 in vitro. Измеренные значения CC50 приведены в Таблице 2.

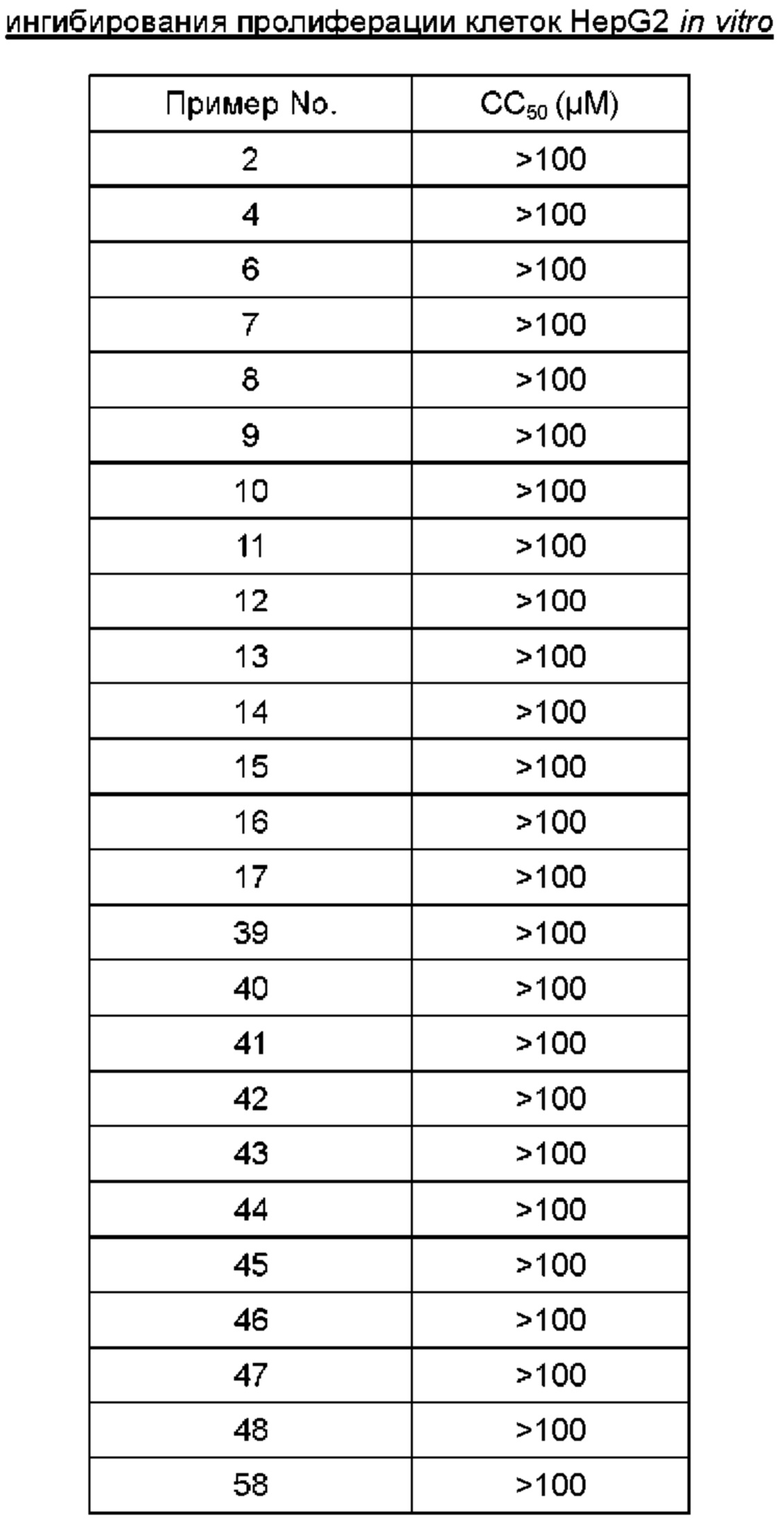
Заключение: Соединение по настоящему изобретению не влияет либо влияет незначительно на ингибирование пролиферации клеток HepG2 in vitro и демонстрирует высокую безопасность.
Оценка Фармакокинетических характеристик
Тестовый пример 3
Фармакокинетический анализ соединения по настоящему изобретению
1. Краткое описание
В качестве подопытных животных использовали крыс. Концентрацию лекарственного средства в плазме крыс в различные моменты времени определяли методом ЖХ/МС/МС после внутрижелудочного введения крысам соединения Примера 1, соединения Примера 2, соединения Примера 4, соединения Примера 6, соединения Примера 7, соединения Примера 11, соединения Примера 39, соединения Примера 42, соединения Примера 44, соединения Примера 45 и соединения Примера 47. Фармакокинетическое поведение и свойства соединения по настоящему изобретению изучали и оценивали на крысах.
2. Протокол испытаний
2.1. Испытываемое соединение
Соединение Примера 1, соединение Примера 2, соединение Примера 4, соединение Примера 6, соединение Примера 7, соединение Примера 11, соединение Примера 39, соединение Примера 42, соединение Примера 44, соединение Примера 45 и соединение Примера 47.
2.2. Подопытные животные
44 взрослые здоровые крысы линии SD (половина из них самцы и половина - самки, по 4 крысы/группу) были приобретены в компании Shanghai Jiesijie Laboratory Animal Co, LTD, Сертификат No.: SCXK (Шанхай) 2013-0006.
2.3. Приготовление испытываемого соединения
Взвешивали определенное количество испытываемого соединения, добавляли 5 об. % ДМСО, 5 об. % Твин 80 и 90 об. % физиологического раствора, в результате получали бесцветный чистый и прозрачный раствор концентрацией 0,2 мг/мл.
2.4. Введение
Испытываемое соединение вводили SD крысам внутрижелудочно утром натощак при дозе введения 2,0 мг/кг и объеме введения 10,0 мл/кг.
3. Способ
Крысам внутрижелудочно вводили соединение Примера 1, соединение Примера 2, соединение Примера 4, соединение Примера 6, соединение Примера 7, соединение Примера 11, соединение Примера 39, соединение Примера 42, соединение Примера 44, соединение Примера 45 и соединение Примера 47. Отбирали по 0,1 мл крови из глазницы перед введением и через 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 11,0 и 24,0 часа после введения. Образцы хранили в гепаринизированных пробирках и центрифугировалии в течение 10 минут при 3500 об/мин для отделения плазмы крови. Образцы плазмы хранили при температуре -20°С. Крыс кормили через 2 часа после введения.
Определяли содержание испытываемого соединения в плазме крыс после его внутрижелудочного введения в различных концентрациях: каждый раз после введения отбирали по 25 мкл плазмы крыс, добавляли 40 мкл внутреннего стандарта раствора камптотецина (100 нг/мл) и 200 мкл ацетонитрила, перемешивали на вортексе в течение 5 минут и центрифугировали в течение 10 минут (4000 об/мин). Из образцов плазмы отбирали по 0,2 мкл супернатанта для анализа методом ЖХ/МС/МС.
4. Результаты определения фармакокинетических параметров
Фармакокинетические параметры соединения по настоящему изобретению представлены ниже:


Заключение: Соединение по настоящему изобретению хорошо абсорбируется и обладает высокой биодоступностью и фармакокинетическим преимуществом.
Настоящее изобретение относится к гетероарильному производному пиперазина формулы I, где кольцо A представляет собой фенил или пиридил; Y представляет собой N или CR5; Q представляет собой N или CH; R1 выбран из группы, состоящей из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила, где каждый из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила независимо друг от друга необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-6-алкила, С1-6-алкокси и гидрокси; R2 выбран из группы, состоящей из водорода и С1-6-алкила, где С1-6-алкил необязательно дополнительно замещен одним или более С1-6-алкокси; каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода и С1-6-алкила; каждый из R4 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, С1-6-алкила, галоген-С1-6-алкила и циано; R5 выбран из группы, состоящей из водорода и С1-6-алкила; n равно 0, 1, 2 или 3 и s равно 0, 1, 2, 3 или 4. Также изобретение относится к соединению формулы IА, где Ra представляет собой водород или С1-6-алкил; кольцо A представляет собой фенил или пиридил; Y представляет собой N или CR5; Q представляет собой N или CH; каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода или С1-6-алкила; каждый из R4 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, С1-6-алкила, галоген-С1-6-алкила и циано; R5 выбран из группы, состоящей из водорода, галогена и С1-6-алкила; n равно 0, 1, 2 или 3 и s равно 0, 1, 2, 3 или 4 , и к соединению формулы IIIА, где M представляет собой трифторуксусную кислоту; R1 выбран из группы, состоящей из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила, где каждый из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила независимо друг от друга необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-6-алкила, С1-6-алкокси и гидрокси; каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода и С1-6-алкила; t равно 0 или 1; а n равно 0, 1, 2 или 3. Кроме того, изобретение относится к способу получения соединения формулы I, к фармацевтической композиции для ингибирования активности капсидного белка на основе соединения формулы I и к применению соединения формулы I для получения ингибитора капсидного белка. Технический результат: описано и получено новое соединение, которое может быть применимо в качестве ингибитора капсидного белка, в частности, для предупреждения и/или лечения таких заболеваний, как гепатит В, грипп, герпес, СПИД . 8 н. и 7 з.п. ф-лы, 6 табл., 59 пр.

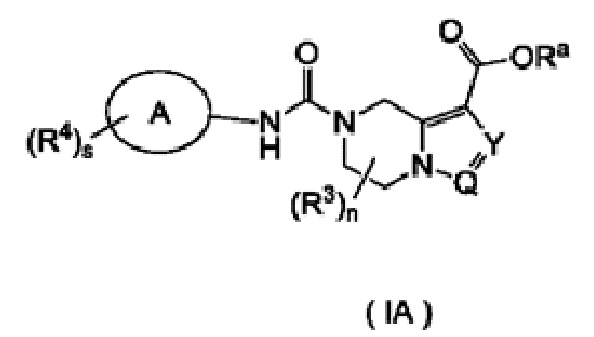
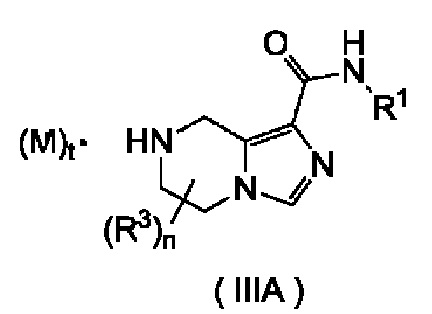
1. Соединение формулы (I):

или его фармацевтически приемлемая соль,
где:
кольцо A представляет собой фенил или пиридил;
Y представляет собой N или CR5;
Q представляет собой N или CH;
R1 выбран из группы, состоящей из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила, где каждый из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила независимо друг от друга необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-6-алкила, С1-6-алкокси и гидрокси;
R2 выбран из группы, состоящей из водорода и С1-6-алкила, где С1-6-алкил необязательно дополнительно замещен одним или более С1-6-алкокси;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода и С1-6-алкила;
каждый из R4 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, С1-6-алкила, галоген-С1-6-алкила и циано;
R5 выбран из группы, состоящей из водорода и С1-6-алкила;
n равно 0, 1, 2 или 3; и
s равно 0, 1, 2, 3 или 4 .
2. Соединение формулы (I) по п. 1, представляющее собой соединение формулы (II):

где:
кольцо A, Y, Q, R1, R3, R4, s и n являются такими, как определено в п. 1.
3. Соединение формулы (I) по п. 1 или 2, представляющее собой соединение формулы (III), формулы (IV), формулы (V) или формулы (VI):


где:
кольцо A, R1, R3, R4, s и n являются такими, как определено в п. 1.
4. Соединение формулы (I) по п. 1, представляющее собой соединение формулы (VII), формулы (VIII), формулы (IX) или формулы (X):


где:
G представляет собой С или N; и
R1, R3, R4, s и n являются такими, как определено в п. 1.
5. Соединение формулы (I) по п. 1, представляющее собой соединение формулы (VII-А), формулы (VIII-A), формулы (IX-А) или формулы (Х-А):
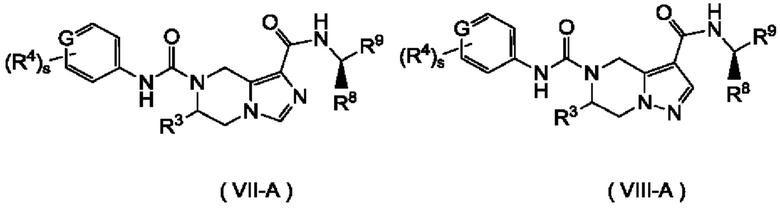

где:
G представляет собой С или N;
R8 представляет собой С1-6-алкил;
R9 представляет собой С1-6-алкил, где С1-6-алкил необязательно дополнительно замещен одним или более атомами галогена; и
R3, R4, s и n являются такими, как определено в п. 1.
6. Соединение формулы (I) по п. 5, где R8 представляет собой метил.
7. Соединение формулы (I) по п. 1, выбранное из группы, состоящей из:











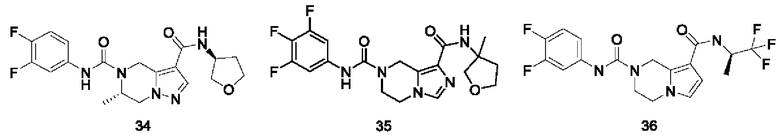

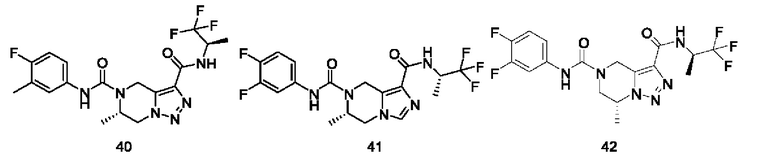
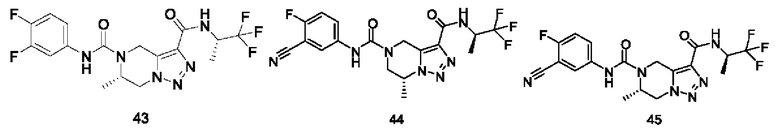





8. Соединение формулы (IA):

или его фармацевтически приемлемая соль,
где:
Ra представляет собой водород или С1-6-алкил;
кольцо A представляет собой фенил или пиридил;
Y представляет собой N или CR5;
Q представляет собой N или CH;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода или С1-6-алкила;
каждый из R4 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода, галогена, С1-6-алкила, галоген-С1-6-алкила и циано;
R5 выбран из группы, состоящей из водорода, галогена и С1-6-алкила;
n равно 0, 1, 2 или 3; и
s равно 0, 1, 2, 3 или 4 .
9. Соединение формулы (IA) по п. 8, выбранное из группы, состоящей из:
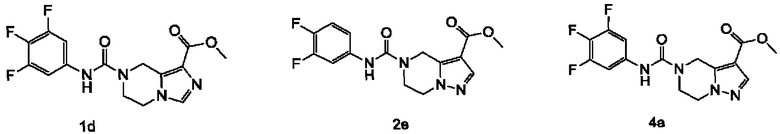

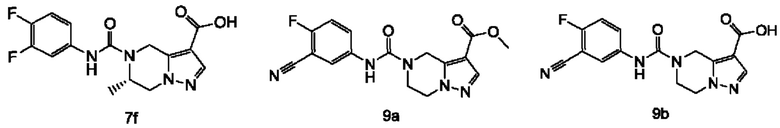

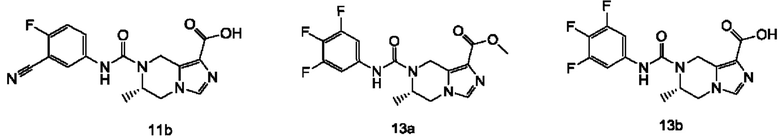


10. Соединение формулы (IIIA):

или его фармацевтически приемлемая соль,
где:
M представляет собой трифторуксусную кислоту;
R1 выбран из группы, состоящей из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила, где каждый из С1-6-алкила, галоген-С1-6-алкила, 3-6-членного циклоалкила, 3-6-членного гетероциклила и фенила независимо друг от друга необязательно дополнительно замещен одним или более заместителями, выбранными из группы, состоящей из галогена, С1-6-алкила, С1-6-алкокси и гидрокси;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода и С1-6-алкила;
t равно 0 или 1; а
n равно 0, 1, 2 или 3.
11. Соединение, выбранное из группы, состоящей из:


12. Способ получения соединения формулы (I) по п. 1, включающий стадию:

взаимодействия соединения формулы (IA) с соединением формулы (IB) или его солью с получением соединения формулы (I),
где:
Ra представляет собой водород или С1-6-алкил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода и С1-6-алкила; и
кольцо A, Y, Q, R1, R4, s и n являются такими, как определено в п. 1.
13. Способ получения соединения формулы (II) по п. 2, включающий стадию:

взаимодействия соединения формулы (IA) с соединением формулы (IIB) или его солью с получением соединения формулы (II),
где:
Ra представляет собой водород или С1-6-алкил;
каждый из R3 является одинаковым или различным, при этом каждый независимо выбран из группы, состоящей из водорода и С1-6-алкила; и
кольцо A, Y, Q, R1, R4, s и n являются такими, как определено в п. 1.
14. Фармацевтическая композиция для ингибирования активности капсидного белка, содержащая терапевтически эффективное количество соединения формулы (I) по любому из пп. 1-7 и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ.
15. Применение соединения формулы (I) по любому из пп. 1-7 или фармацевтической композиции по п. 14 для получения ингибитора капсидного белка, который используется для лечения вирусного инфекционного заболевания, где вирус предпочтительно выбран из группы, состоящей из вируса гепатита B, вируса гриппа, вируса герпеса и вируса СПИДа, и заболевание предпочтительно выбрано из группы, состоящей из гепатита B, гриппа, герпеса и СПИДа.
| WO2016113273 А1, 21.07.2016 | |||
| CN105916855 A, 31.08.2016 | |||
| CN101686989 А, 31.03.2010 | |||
| ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ЗАМЕЩЕННЫХ ДИГИДРО- И ТЕТРАГИДРООКСАЗОЛОПИРИМИДИНОНОВ | 2008 |
|
RU2470937C2 |

