Область техники
Изобретение относится к способу получения материала для хроматографии. Более конкретно, изобретение относится к способу получения материала для обращенно-фазной хроматографии (ОФХ) посредством модификации поверхности частиц для хроматографии.
Предпосылки создания изобретения
Адсорбционная хроматография зависит от химических взаимодействий между молекулами растворенного вещества и специально разработанными лигандами, привитыми на хроматографическую матрицу. За прошедшие годы множество различных типов лигандов были привиты на хроматографические носители для очистки биомолекул, используя с этой целью различные биохимические свойства от электронного заряда до биологического сродства. Важным дополнением к арсеналу адсорбционных методик для препаративной хроматографии биомолекул стала обращенно-фазная хроматография, в которой связывание вещества, растворенного в подвижной фазе, с н-алкил-углеводородным или ароматическим лигандом происходит через гидрофобное взаимодействие. Обращенно-фазная хроматография нашла применение как в аналитических, так и в препаративных целях в области биохимического разделения и очистки. Молекулы, которые проявляют некоторую степень гидрофобное™, такие как, например, белки, пептиды и нуклеиновые кислоты, можно разделить с помощью обращенно-фазной хроматографии с прекрасной степенью извлечения и разделения. Кроме того, применение ион-парных модификаторов в подвижной фазе позволяет применять обращенно-фазную хроматографию в случае растворенных веществ, имеющих заряд, таких как, например, полностью незащищенные олигонуклеотиды и гидрофильные пептиды. Препаративная обращенно-фазная хроматография нашла широкое применение от области микроочистки фрагментов белков при секвенировании до процесса очистки рекомбинантных белковых продуктов в промышленном масштабе.
Механизм разделения в обращенно-фазной хроматографии зависит от гидрофобного связывания между молекулами вещества, растворенного в подвижной фазе, и привитым гидрофобным лигандом, то есть неподвижной фазой. Сама по себе действительная природа гидрофобного связывания является предметом горячих дискуссий, но в свете традиционных представлений допускается, что связывание является результатом положительного энтропийного эффекта. Обязательным условием в отношении начальной подвижной фазы, используемой в обращенно-фазной хроматографии, в первую очередь, является ее водная основа, о чем свидетельствует высокая степень структурированной воды, окружающей как молекулу растворенного вещества, так и привитого лиганда. Поскольку растворенное вещество связывается с привитым гидрофобным лигандом, гидрофобная область, подверженная действию растворителя, минимизирована. Следовательно, степень структурированной воды уменьшается при соответствующем положительном росте энтропии системы. Таким образом, связывание является преимущественным с энергетической точки зрения для гидрофобных составляющих, то есть растворенного вещества и лиганда.
По экспериментальному воплощению обращенно-фазовая хроматография представляет собой адсорбционный процесс, основанный на механизме распределения для осуществления разделения. Молекулы растворенного вещества распределяются (то есть устанавливается равновесие) между подвижной фазой и неподвижной фазой. Распределение растворенного вещества между двумя фазами зависит от связывающих свойств среды, гидрофобности растворенного вещества и состава подвижной фазы. Первоначально условия эксперимента подбираются для предпочтительной адсорбции растворенного вещества из подвижной фазы на неподвижную фазу. После этого состав подвижной фазы модифицируется для предпочтительной десорбции растворенного вещества с неподвижной фазы обратно в подвижную фазу. В данном случае адсорбция считается крайним вариантом состояния равновесия, когда распределение молекул растворенного вещества в неподвижной фазе составляет по существу 100%. С другой стороны, десорбция представляет собой крайний вариант состояния равновесия, когда растворенное вещество по существу на 100% распределено в подвижной фазе.
При обращенно-фазной хроматографии биомолекул обычно используют градиентное, а не изократическое элюирование. Поскольку биомолекулы прочно адсорбируются поверхностью обращенно-фазной матрицы в условиях водной среды, их десорбция с матрицы проходит в очень узком диапазоне концентраций органических модификаторов. Наряду с такими высокомолекулярными биомолекулами, имеющими уникальные адсорбционные свойства, типичные биологические образцы содержат смесь разнообразных биомолекул с соответствующим разнообразным диапазоном адсорбционных свойств. Единственным практическим способом обращенно-фазного разделения сложных биологических образцов является, таким образом, градиентное элюирование.
Вкратце, разделение в методе обращенно-фазной хроматографии зависит от обратимой адсорбции/десорбции молекул растворенного вещества с различающейся степенью гидрофобности на гидрофобной неподвижной фазе.
Первая стадия в хроматографическом процессе состоит в уравновешивании набитой колонки, содержащей среду с обращенной фазой, при подходящих для начальной подвижной фазы условиях pH, ионной силы и полярности (гидрофобности подвижной фазы). Полярность подвижной фазы контролируют путем добавления органических модификаторов, таких как, например, ацетонитрил. Ион-парные агенты, такие как, например, трифторуксусная кислота, тоже могут подойти. Полярность начальной подвижной фазы (обычно называемой подвижной фазой А) должна быть достаточно низкой, чтобы растворить частично гидрофобное растворенное вещество, но достаточно высокой, чтобы обеспечить связывание растворенного вещества с обращенно-фазной хроматографической матрицей. На второй стадии вводят образец, содержащий растворенные вещества, подлежащие разделению. Идеально, если образец растворяют в той же подвижной фазе, которую используют для установления равновесия в хроматографическом слое. Образец вводят в колонку при скорости потока, при которой происходит оптимальное связывание. После того, как образец введен, хроматографический слой продолжают промывать подвижной фазой A для того, чтобы удалить любые несвязанные и нежелательные молекулы растворенных веществ.
Связанные растворенные вещества затем десорбируются из обращенно-фазной среды путем регулирования полярности подвижной фазы таким образом, что связанные молекулы растворенного вещества последовательно десорбируются и вымываются из колонки. В обращенно-фазном варианте хроматографии для этого обычно уменьшают полярность подвижной фазы путем увеличения процентного содержания органического модификатора в подвижной фазе. Это приводит к обеспечению высокой концентрации органического модификатора в конечной подвижной фазе (подвижная фаза В). Как правило, pH растворов начальной и конечной подвижной фаз остается постоянным. Градиентное уменьшение полярности подвижной фазы (увеличение гидрофобности подвижной фазы) достигается путем увеличения линейного градиента от 100% начальной фазы А, содержащей небольшое количество или не содержащей органического модификатора, до 100% (или менее) подвижной фазы В, содержащей более высокую концентрацию органического модификатора. Связанные растворенные вещества десорбируются со среды с обращенной фазой в соответствии с характерной для них гидрофобностью.
Четвертая стадия процесса включает извлечение веществ, не десорбировавшихся ранее. Обычно это осуществляют путем изменения подвижной фазы В на практически 100% органический модификатор для того, чтобы обеспечить полное удаление всех связанных веществ перед тем, как повторно использовать эту же колонку.
Пятая стадия представляет собой повторное уравновешивание хроматографической среды от состояния со 100% подвижной фазой В обратно к условиям с начальной подвижной фазы. Разделение в обращенно-фазной хроматографии происходит благодаря различным связывающим свойствам растворенных веществ, присутствующих в образце, являющимся результатом различий в их гидрофобных свойствах. Степень связывания молекулы растворенного вещества со средой с обращенной фазой можно регулировать путем варьирования гидрофобных свойств начальной подвижной фазы. Хотя гидрофобность молекулы растворенного вещества трудно оценить количественно, разделение растворенных веществ, гидрофобные свойства которых лишь немного различаются, легко осуществимо. Благодаря своей превосходной разрешающей способности, обращенно-фазная хроматография является незаменимым методом для высокоэффективного разделения сложных биомолекул.
Обычно обращенно-фазное разделение первоначально достигается с помощью градиента в широком диапазоне от 100% подвижной фазы A до 100% подвижной фазы В. Количество органического модификатора в начальной и конечной подвижных фазах тоже может сильно варьировать. Однако рабочее процентное содержание органического модификатора составляет 5% или менее в подвижной фазе А и 95% или более в подвижной фазе В.
Метод обращенно-фазной хроматографии допускает существенную гибкость условий разделения, так что исследователь может выбирать, связать представляющее интерес растворенное вещество, позволив примесям без задержки пройти через колонку, или связать примеси, позволив представляющему интерес растворенному веществу свободно пройти. Как правило, более рационально связать представляющее интерес растворенное вещество, поскольку десорбированное растворенное вещество вымывается с хроматографической среды в концентрированном виде. Кроме того, поскольку связывание завершается в условиях начальной подвижной фазы, начальная концентрация целевого растворенного вещества в растворе образца не является важной, позволяя вводить в колонку разбавленные образцы.
Хроматографическая среда с обращенной фазой состоит из гидрофобных лигандов, привитых на пористую нерастворимую послойную матрицу. Матрица должна быть химически и механически стабильной. Основная матрица для приемлемой в промышленных масштабах среды с обращенной фазой обычно состоит из силикагеля или синтетического органического полимера, такого как, например, полистирол.
Когда выбирают буферные условия для обращенно-фазного разделения, pH является одним из параметров, оказывающих существенное влияние на профиль разделения. Кроме того, стабильность целевой молекулы тоже должна быть принята во внимание. Следовательно, существует необходимость в хроматографической среде обращенной фазой, которую можно использовать в широком диапазоне pH, таком как, например, pH от 3 до 12, чтобы предоставить потребителю полную свободу в выборе наиболее оптимального pH.
Хотя ОФХ среды из силикагеля и полистирола удовлетворительно функционируют во многих случаях, их невозможно использовать в широком диапазоне pH. Ранее в US 7048858 В2 было показано, что прививание стирола на полимерную (например, сшитый полистирол) подложку, приводящее к изменению пористой структуры, дает некоторые улучшения при разделении инсулина. Полистирол - химически стабилен в широком диапазоне pH, но, в сравнении с силикагелем при многих значениях pH, обладает меньшей селективностью. С другой стороны, силикагель не стабилен при длительном использовании при pH более ~8.
Таким образом, по-прежнему существует необходимость в улучшенных ОФХ средах, которые проявляют хорошую селективность в широком диапазоне pH.
Краткое описание изобретения
Настоящее изобретение относится к способу получения ОФХ материала на основе частиц пористого углевода, который соответствует требованиям механической прочности и дает высокую селективность в широком диапазоне pH.
Первый аспект настоящего изобретения относится к способу получения материала для обращенно-фазной хроматографии (ОФХ), включающему следующие стадии: введение ненасыщенных групп на поверхность частиц пористого углевода и прививание стирольных мономеров на указанные частицы, содержащие ненасыщенную группу.
Частицы пористого углевода предпочтительно сделаны из полисахаридного материала, наиболее предпочтительно из агарозы.
Агарозу прежде удачно использовали для метода гидрофобной хроматографии (ГХ), и на рынке доступны многие продукты промышленного назначения, как, например, Butyl Sepharose Fast Flow (производства компании GE Healthcare). Продукты для ГХ должны быть лишь умеренно гидрофобными, поэтому агарозу не рассматривали для применения в методе обращенно-фазной хроматографии, где требуется высоко гидрофобная подложка, из-за присущей ей гидрофильности и сложности сделать ее в достаточной степени гидрофобной.
Авторы изобретения случайно обнаружили, что путем прививания стирола на поверхность частиц сшитой агарозы можно получить приемлемую гидрофобность в комбинации с хорошей селективностью во всем диапазоне pH, которую не проявляют ни силикагель, ни полистирол.
Предпочтительно, чтобы ненасыщенные группы в заявленном способе получения представляли собой аллильные группы.
В одном из воплощений способа аллилирование осуществляют с помощью аллилглицидилового эфира (АГЭ).
Стирольные мономеры могут быть выбраны, например, из стирола, трет-бутилстирола или пентафторстирола.
Предпочтительно, чтобы объемное содержание стирольного мономера в прививающем растворе составляло от 5 до 95% об./об., предпочтительно от 25 до 75%.
В предпочтительном воплощении аллилирование проводят с помощью АГЭ и стирольный мономер представляет собой стирол или трет-бутилстирол, присутствующий в прививающем растворе в количестве 50% об./об.
Второй аспект настоящего изобретения относится к ОФХ материалу, полученному в соответствии с указанным выше способом.
Третий аспект настоящего изобретения относится к использованию указанного выше полученного ОФХ материала для осуществления обращенно-фазной хроматографии.
Краткое описание графических материалов
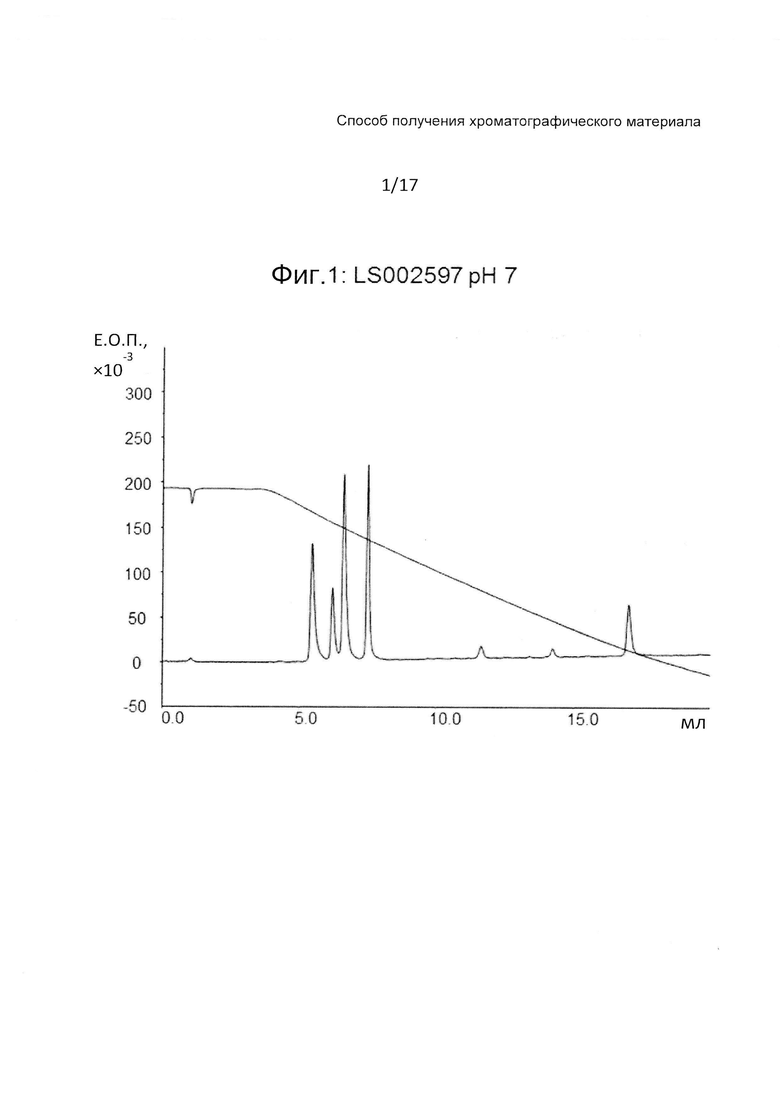
На Фиг. 1 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002597 (смотри ниже Таблицу 6) при pH 7.
На Фиг. 2 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002597 (смотри ниже Таблицу 6) при pH 3.
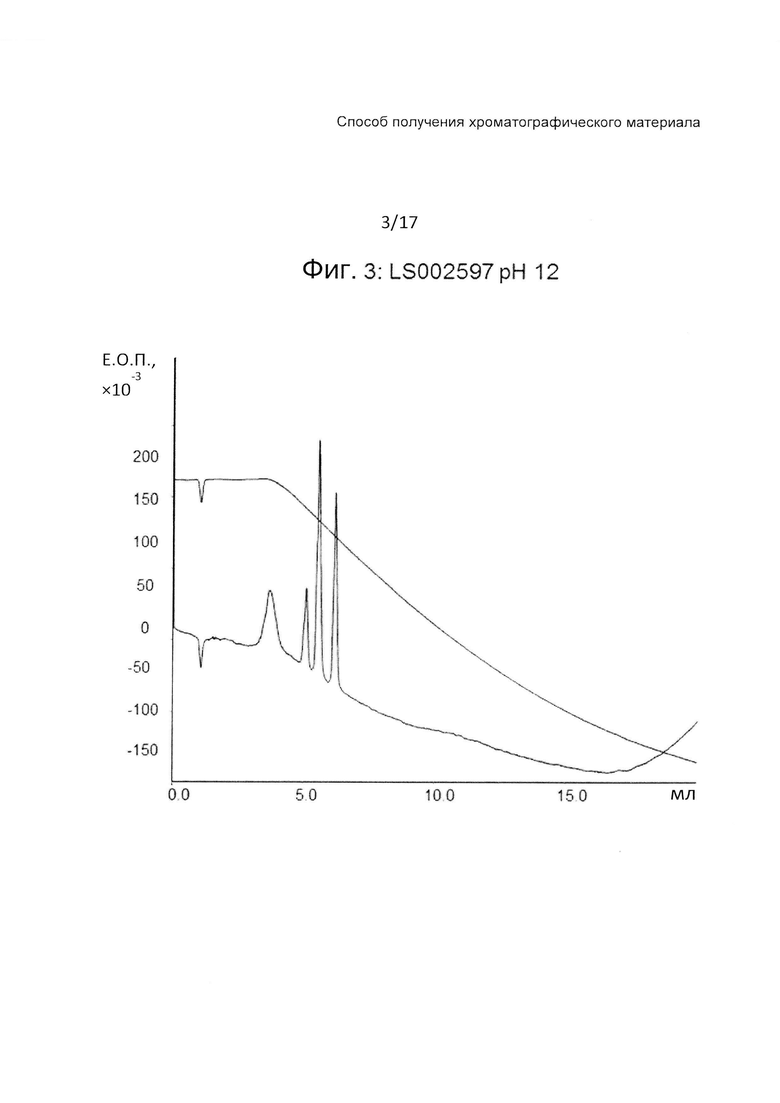
На Фиг. 3 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002597 (смотри ниже Таблицу 6) при pH 12.
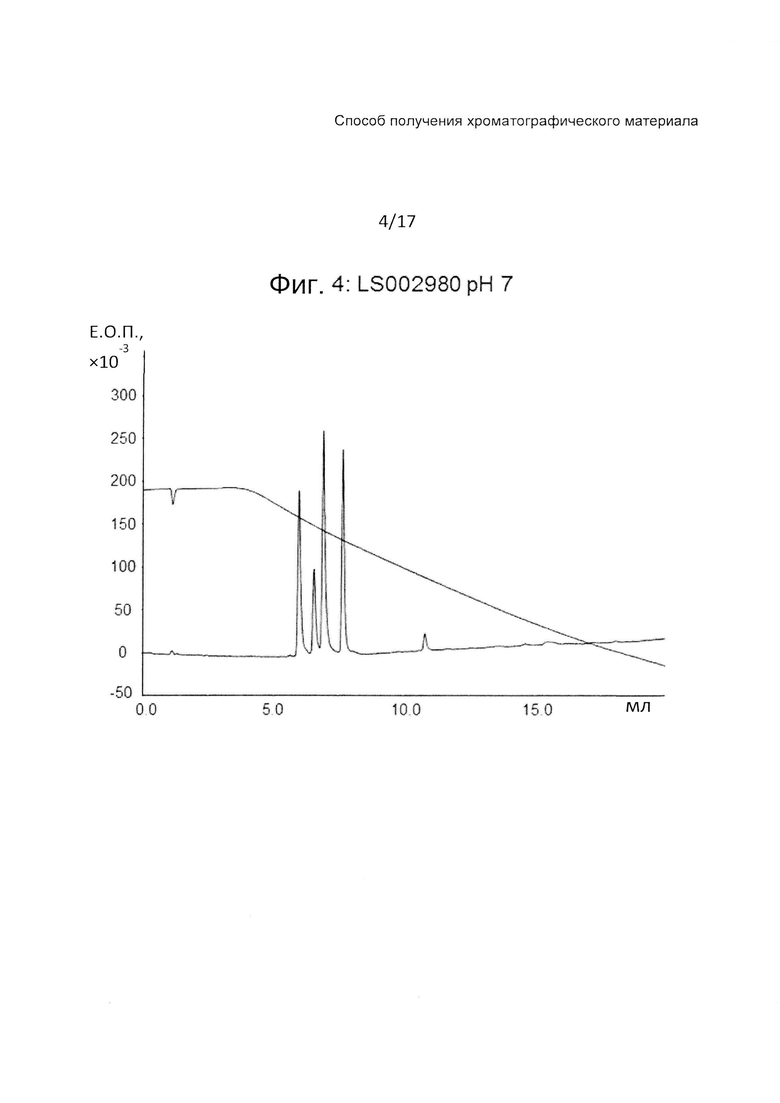
На Фиг. 4 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002980 (смотри ниже Таблицу 6) при pH 7.
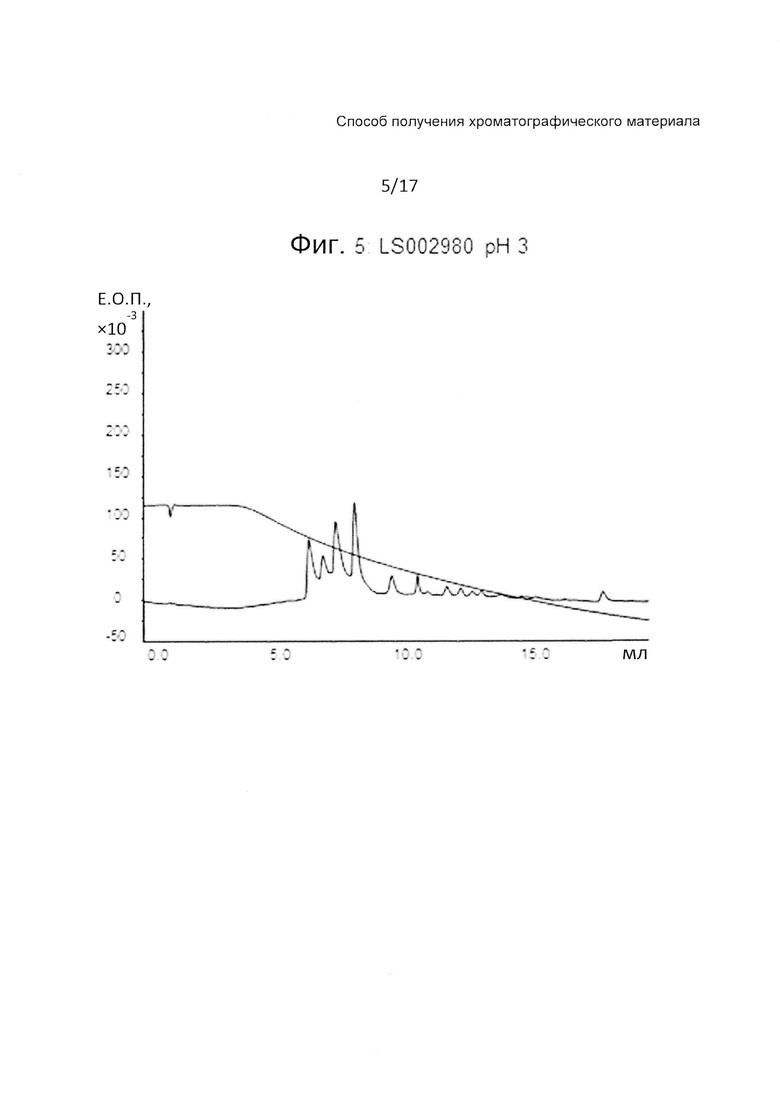
На Фиг. 5 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002980 (смотри ниже Таблицу 6) при pH 3.
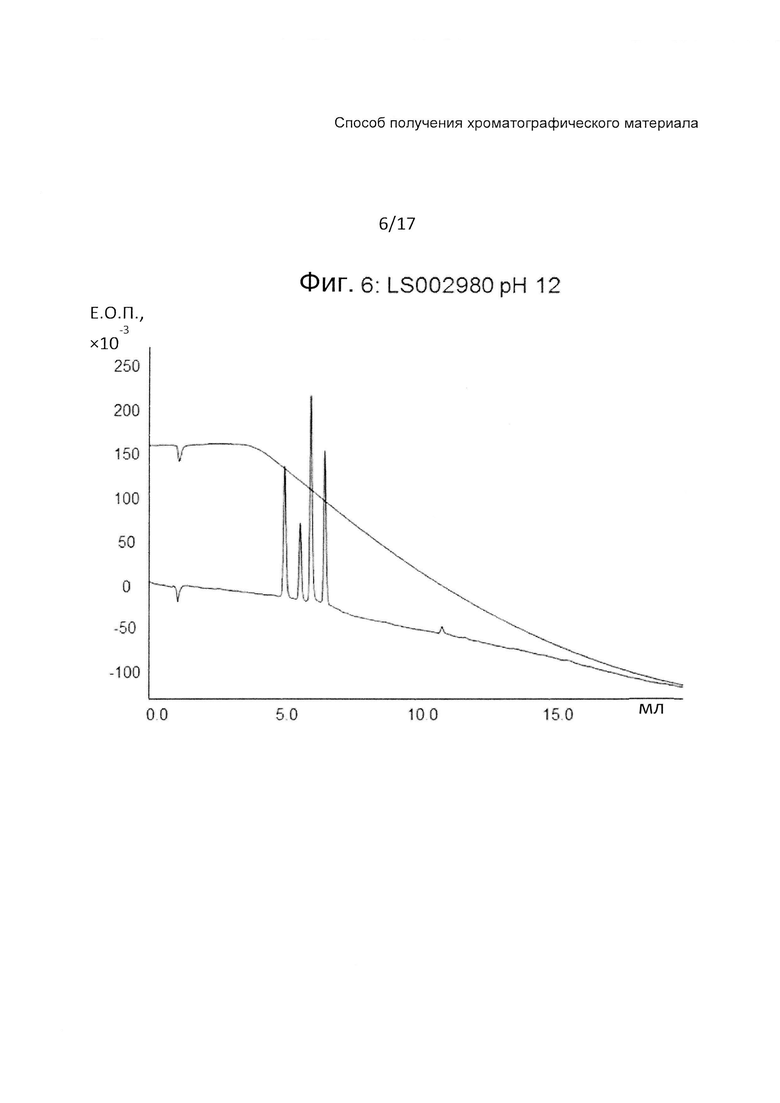
На Фиг. 6 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002980 (смотри ниже Таблицу 6) при pH 12.
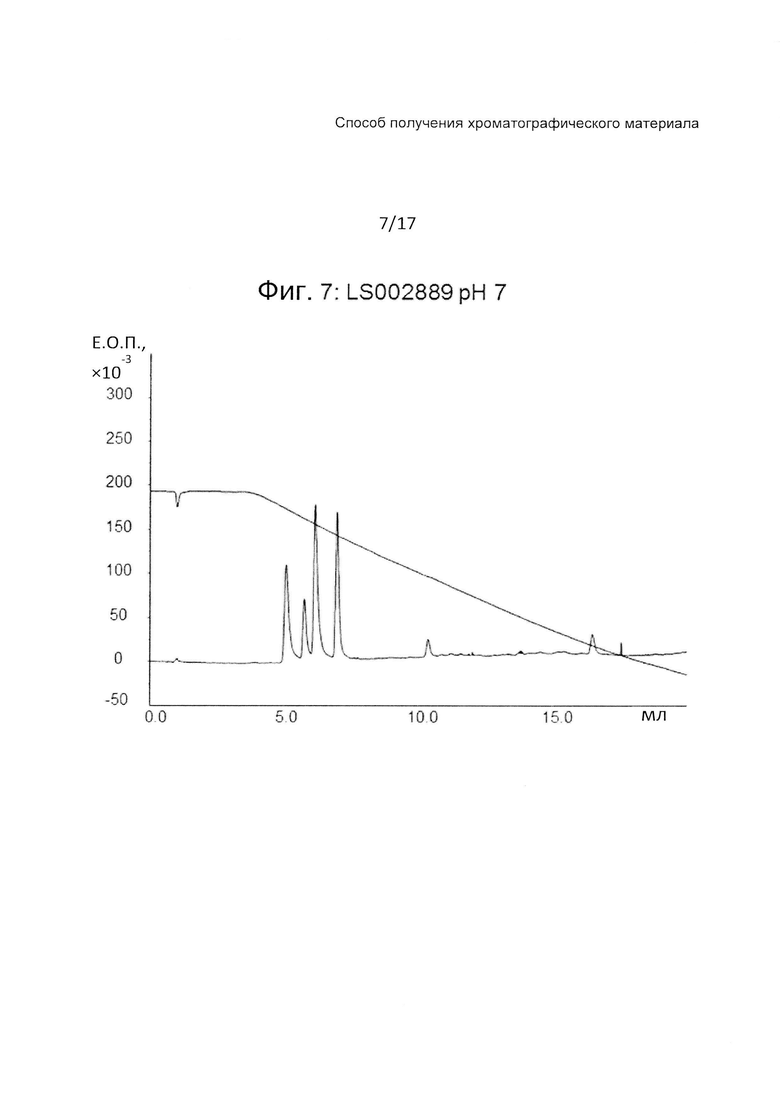
На Фиг. 7 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002889 (смотри ниже Таблицу 6) при pH 7.
На Фиг.8 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002889 (смотри ниже Таблицу 6) при pH 3.
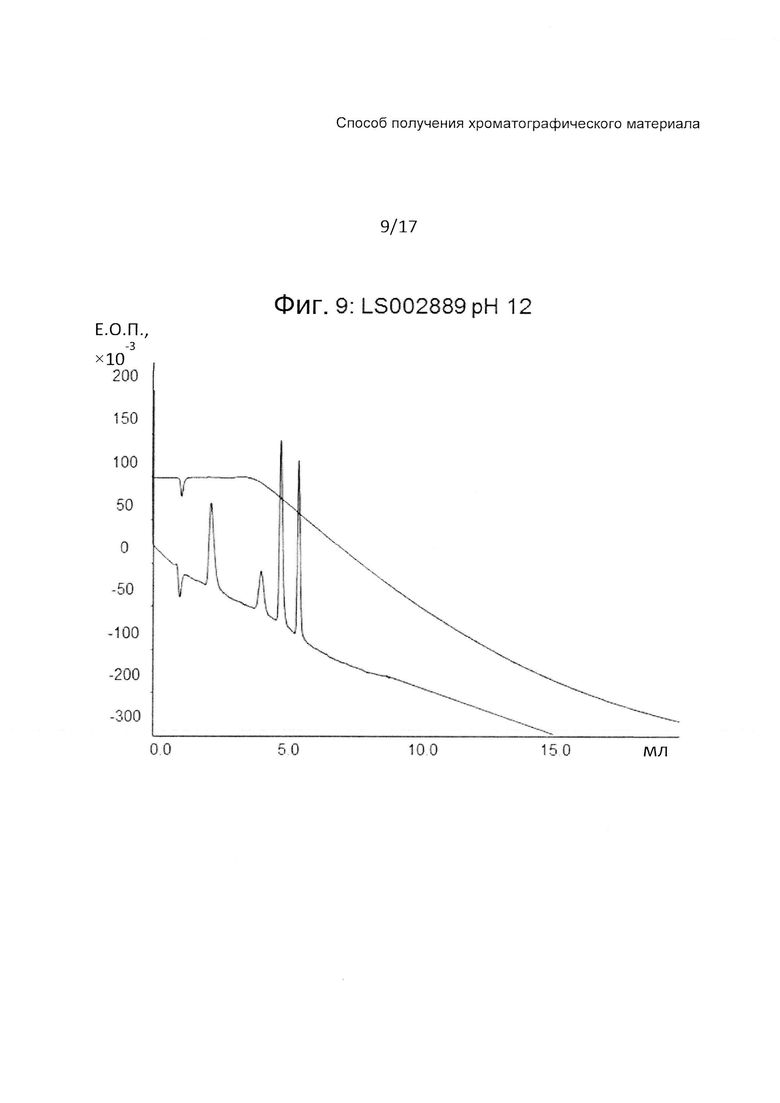
На Фиг. 9 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS002889 (смотри ниже Таблицу 6) при pH 12.
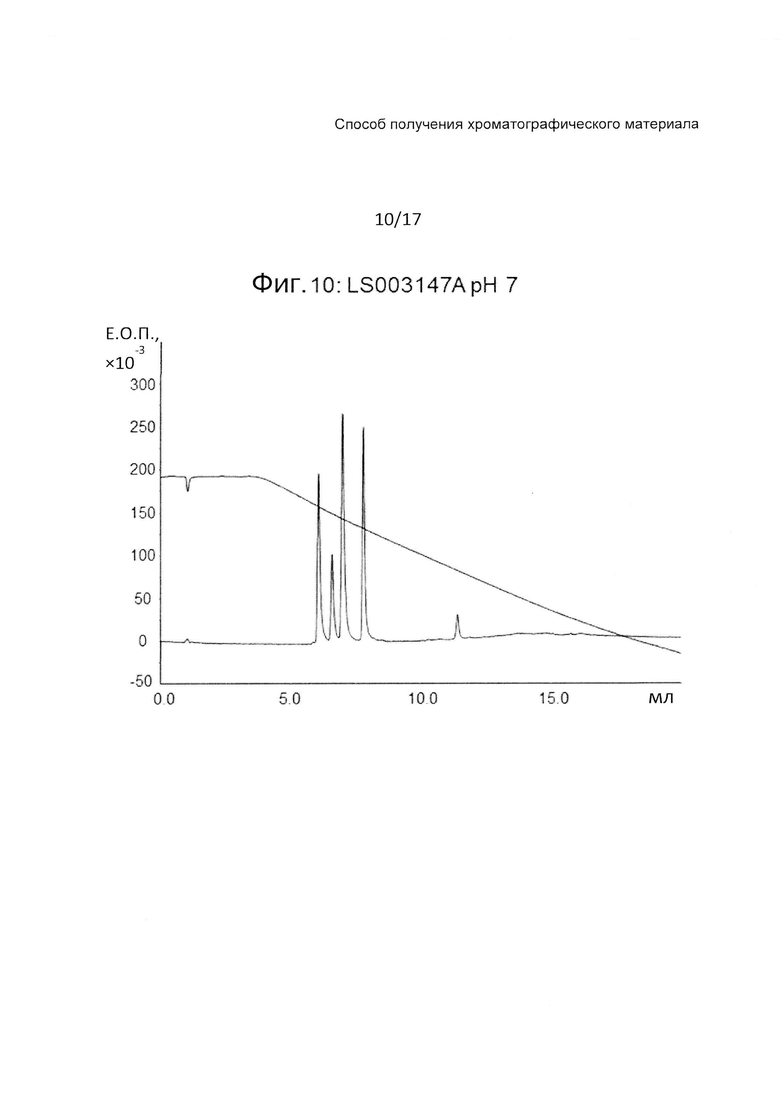
На Фиг. 10 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS003147A (смотри ниже Таблицу 6) при pH 7.
На Фиг. 11 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS003147A (смотри ниже Таблицу 6) при pH 3.
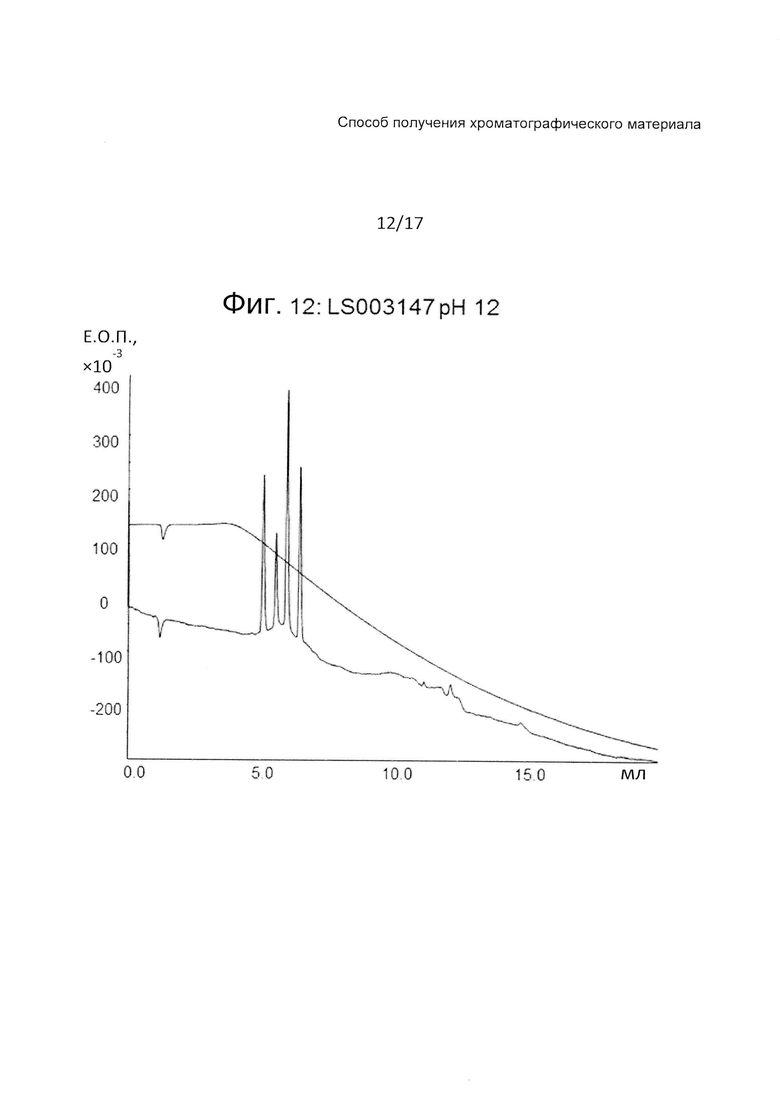
На Фиг. 12 представлена хроматограмма разделения четырех тестовых пептидов (смотри ниже Таблицу 3) на прототипе ОФХ LS003147A (смотри ниже Таблицу 6) при pH 12.
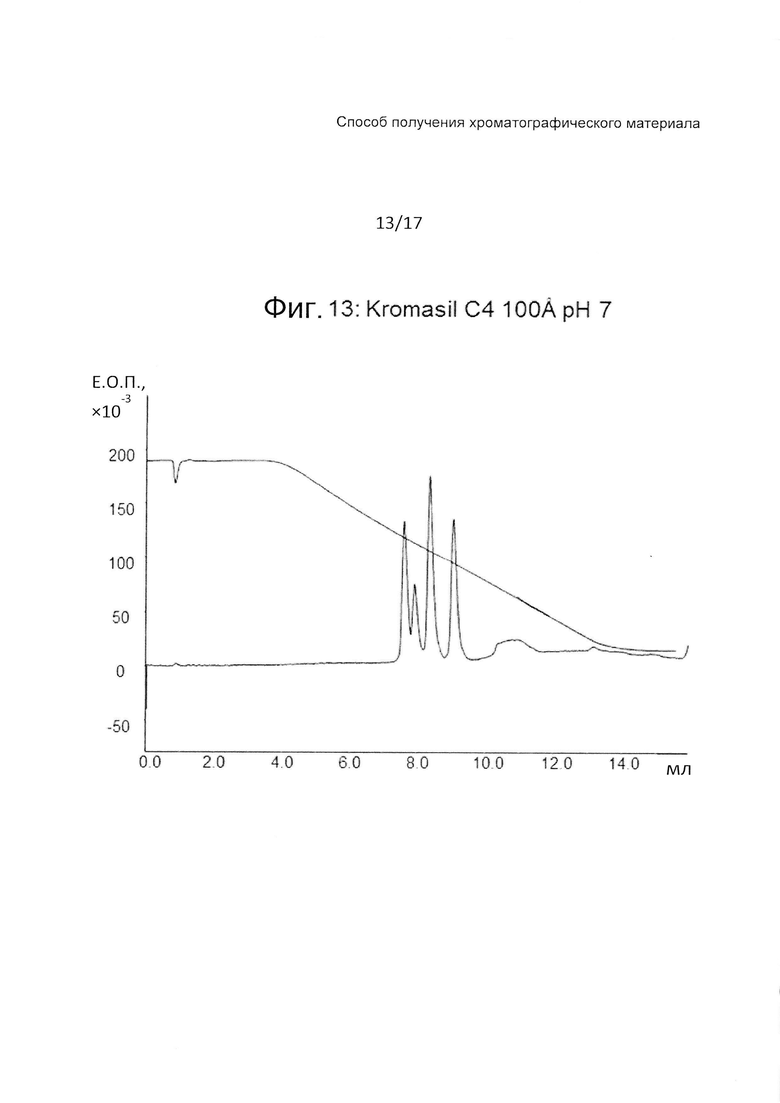
На Фиг. 13 представлена хроматограмма сравнительного испытания тех же четырех тестовых пептидов (Таблица 3) на колонке с силикагелем (уровень техники) при pH 7.
На Фиг. 14 представлена хроматограмма сравнительного испытания тех же четырех тестовых пептидов (Таблица 3) на колонке с силикагелем (уровень техники) при pH 3.
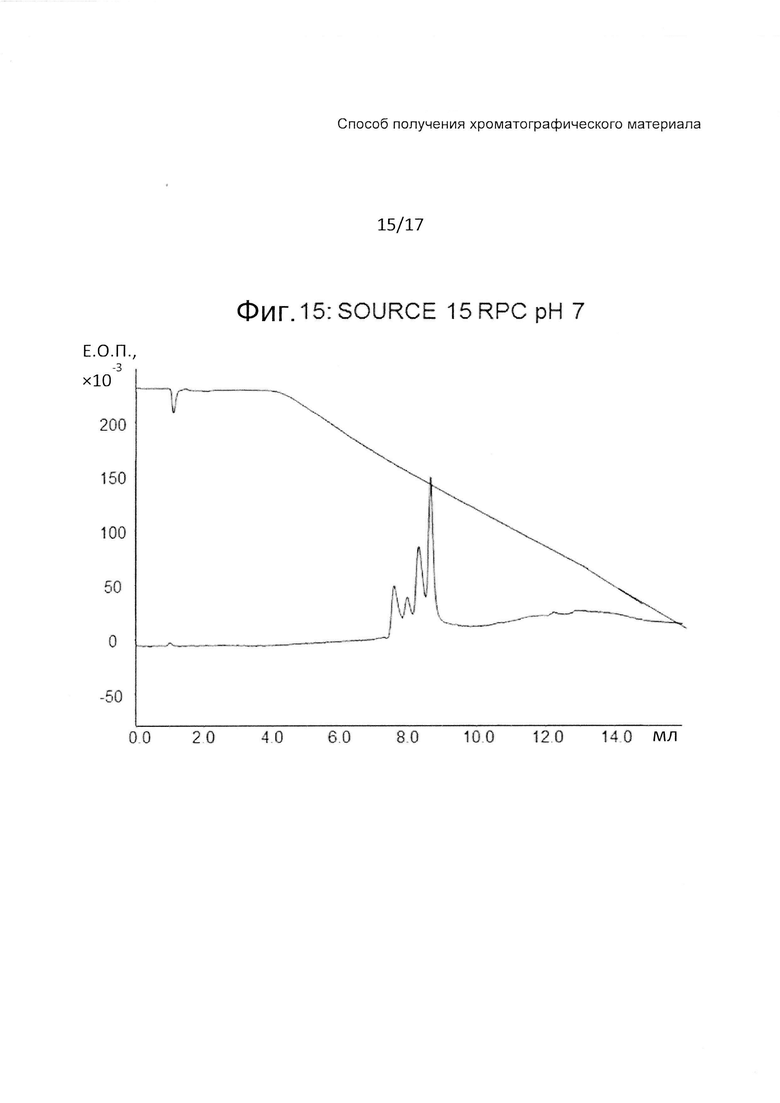
На Фиг. 15 представлена хроматограмма сравнительного испытания тех же четырех тестовых пептидов (Таблица 3) на колонке с полистиролом при pH 7.
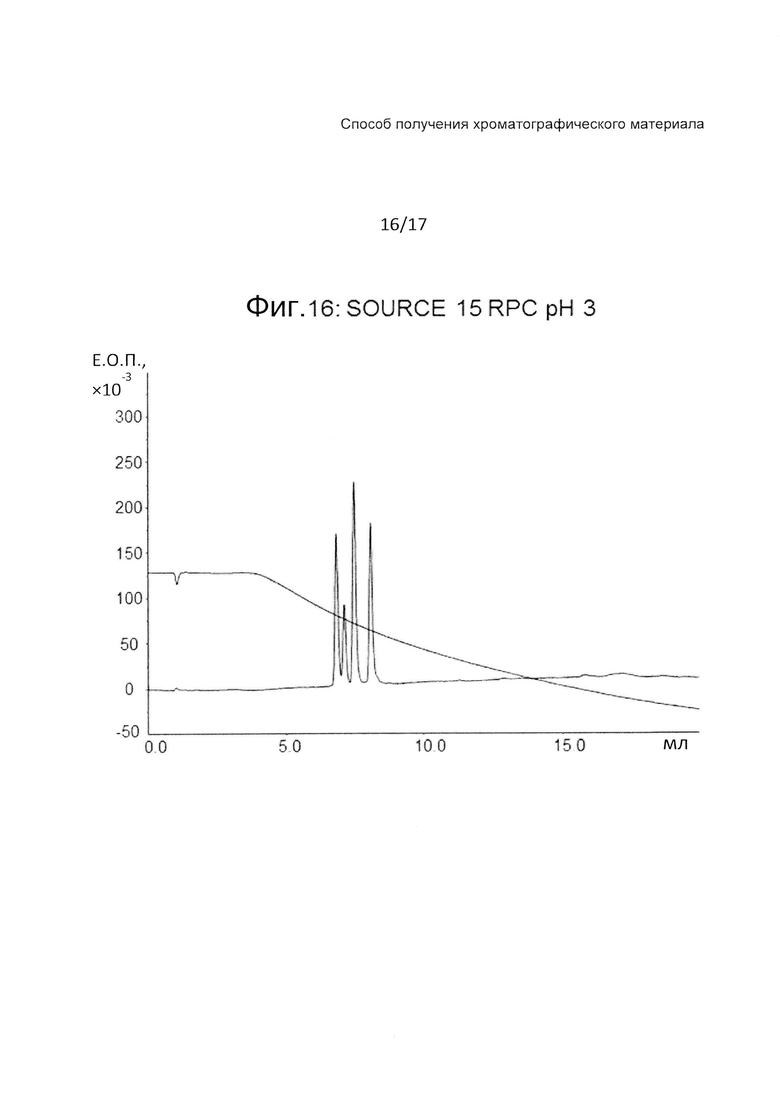
На Фиг. 16 представлена хроматограмма сравнительного испытания тех же четырех тестовых пептидов (Таблица 3) на колонке с полистиролом при pH 3.
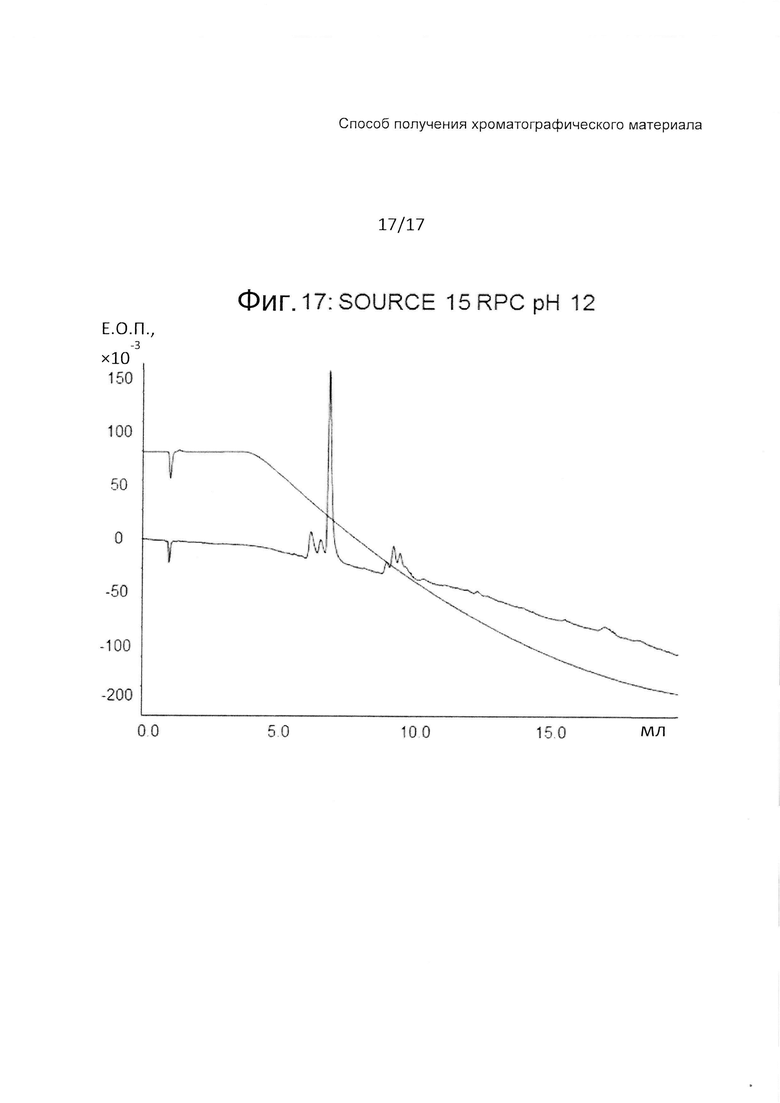
На Фиг. 17 представлена хроматограмма сравнительного испытания тех же четырех тестовых пептидов (Таблица 3) на колонке с полистиролом при pH 12.
Подробное описание изобретения
Далее настоящее изобретение будет описано более подробное со ссылкой на некоторые неограничивающие примеры и прилагаемые графические материалы.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы
Для всех экспериментов использовали частицы пористой сшитой агарозы со средним размером частиц 8,35 мкм.
Модифицирующие реагенты перечислены в Таблице 1.
ЭКСПЕРИМЕНТ 1: LS002597 Аллилирование и прививание полистирола на частицы агарозы
Аллилирование
50 мл частиц агарозы промывали на фильтре из пористого стекла 500 мл дистиллированной воды. Готовили раствор гидроксида натрия в дистиллированной воде с концентрацией 50% масс, и промывали частицы 300 мл 50%-ного раствора гидроксида натрия. Частицы высушивали над вакуумом и переносили в круглодонную колбу объемом 250 мл, снабженную механической мешалкой. Добавляли 40 мл 50%-ного раствора гидроксида натрия и повышали температуру до 50°C. Скорость перемешивания устанавливали 250 оборотов/мин. Когда устанавливалась температура, добавляли 50 мл аллилглицидилового эфира. Реакционную смесь выдерживали в течение ночи.
Суспензию, содержащую частицы, переносили на фильтр из пористого стекла и частицы промывали 500 мл дистиллированной воды, 500 мл этанола и 500 мл 20%-ного этанола.
Количество привитых аллильных групп определяли методом титрования и обнаружили, что оно составляет 625 мкмоль/мл частиц. Прививание полистирола
10 мл аллилированных частиц агарозы, полученных как описано выше, промывали на фильтре из пористого стекла 100 мл толуола. Частицы высушивали над вакуумом и переносили во флакон фирмы Falcon объемом 50 мл. Добавляли 15 мл толуола, 15 мл стирола и 270 мг ДАК (толуол и стирол образуют прививающий раствор). Газообразный азот пропускали через суспензию частиц в течение 5 минут. Флакон закрывали крышкой и размещали на нагреваемом вибростоле при 70°C. Реакционную смесь выдерживали в течение 18 часов.
Суспензию частиц переносили на фильтр из пористого стекла, и частицы промывали 300 мл толуола, 300 мл этанола и 100 мл 20%-ного этанола.
ЭКСПЕРИМЕНТ 2: LS002980 Привитая полимеризация частиц аллилированной агарозы с полистиролом (увеличенное количество стирола)
10 мл аллилированных частиц агарозы, полученных как описано в эксперименте 1, промывали на фильтре из пористого стекла 100 мл толуола. Частицы высушивали над вакуумом и переносили во флакон фирмы Falcon объемом 50 мл. Добавляли 10 мл толуола, 20 мл стирола и 360 мг ДАК (толуол и стирол образуют прививающий раствор). Газообразный азот пропускали через суспензию частиц в течение 5 минут. Флакон закрывали крышкой и размещали на нагреваемом концентрирующем вибростоле при 70°C. Реакционную смесь выдерживали в течение 18 часов.
Суспензию частиц переносили на фильтр из пористого стекла и частицы промывали 300 мл толуола, 300 мл этанола и 100 мл 20%-ного этанола.
ЭКСПЕРИМЕНТ 3: LS002597 Аллилирование и прививание поли(пентафторстирола) на частицы агарозы
Аллилирование
200 мл частиц агарозы промывали на фильтре из пористого стекла 2000 мл дистиллированной воды. Готовили раствор гидроксида натрия в дистиллированной воде с концентрацией 50% масс, и промывали частицы 1200 мл 50%-ного раствора гидроксида натрия. Частицы высушивали над вакуумом и переносили в круглодонную колбу объемом 1000 мл, снабженную механической мешалкой. Добавляли 160 мл 50%-ного раствора гидроксида натрия и 1,2 г боргидрида натрия, а температуру повышали до 50°C. Скорость перемешивания устанавливали 600 оборотов/мин. Когда устанавливалась температура, добавляли 200 мл аллилглицидилового эфира. Реакционную смесь выдерживали в течение ночи.
Суспензию, содержащую частицы, переносили на фильтр из пористого стекла и частицы промывали 500 мл дистиллированной воды, 500 мл этанола и 500 мл 20%-ного этанола.
Количество привитых аллильных групп определяли методом титрования и обнаружили, что оно составляет 501 мкмоль/мл частиц. Прививание поли(пентафторстирола)
10 мл аллилированных частиц агарозы промывали на фильтре из пористого стекла 100 мл толуола. Частицы высушивали над вакуумом и переносили во флакон фирмы Falcon объемом 50 мл. Добавляли 15 мл толуола, 15 мл пентафторстирола и 270 мг ДАК (толуол и стирол образуют прививающий раствор). Газообразный азот пропускали через суспензию частиц в течение 5 минут. Флакон закрывали крышкой и размещали на нагреваемом концентрирующем вибростоле при 70°C. Реакционную смесь выдерживали в течение 18 часов.
Суспензию частиц переносили на фильтр из пористого стекла и частицы промывали 300 мл ацетона, 300 мл этанола и 100 мл 20%-ного этанола.
ЭКСПЕРИМЕНТ 4: LS003147A Аллилирование и прививание поли(трет-бутилстирола) на частицы агарозы
Аллилирование
200 мл частиц агарозы аллилировали, как описано в Эксперименте 3. Количество привитых аллильных групп определяли методом титрования и обнаружили, что оно составляет 501 мкмоль/мл частиц. Прививание поли(трет-бутилстирола)
10 мл аллилированных частиц агарозы промывали на фильтре из пористого стекла 100 мл толуола. Частицы высушивали над вакуумом и переносили во флакон фирмы Falcon объемом 50 мл. Добавляли 15 мл толуола, 15 мл трет-бутилстирола и 270 мг ДАК (толуол и стирол образуют прививающий раствор). Газообразный азот пропускали через суспензию частиц в течение 5 минут.Флакон закрывали крышкой и размещали на нагреваемом концентрирующем вибростоле при 70°C. Реакционную смесь выдерживали в течение 18 часов.
Суспензию частиц переносили на фильтр из пористого стекла и частицы промывали 300 мл толуола, 300 мл этанола и 100 мл 20%-ного этанола.
ЭКСПЕРИМЕНТ 5: Разделение пептидов на прототипах и материалах сравнения
Четыре пептида с различными значениями pH использовали в качестве испытуемых пептидов для способа хроматографического анализа. Некоторые свойства пептидов представлены в Таблице.

Прототипы и колонки
Прототипы ОФХ материалов согласно настоящему изобретению (см. Эксперименты 1-4) набивали в колонки Tricorn 5/50 (производства GE Healthcare Bio-Sciences АВ) с объемом колонки (ОК) 0,98 мл. Кроме того, с целью сравнения SOURCE 15 RPC (производства GE Healthcare Bio-Sciences АВ) и Krornasil 100-13-С4 (производства Akzo Nobel) набивали в колонки Tricorn 5/50. Для осуществления метода разделения использовали систему АКТА (ТМ) Explorer 10S (производства GE Healthcare Bio-Sciences АВ).
Материалы, используемые в способе разделения, перечислены в Таблице 3.
Приготовление буферного раствора
15 мМ буферный раствор фосфата натрия с pH 3,0:
0,176 мл фосфорной кислоты и 1,71 г дигидрофосфата натрия моногидрата растворяли до конечного объема в 1 л в воде, подвергнутой очистке в системе Milli Q.
15 мМ буферный раствор фосфата натрия с pH 7,0:
1,032 г дигидрофосфата натрия моногидрата и 1,068 г гидрофосфата динатрия растворяли до конечного объема в 1 л.
10 мМ раствор гидроксида натрия использовали в качестве раствора с pH 12. Раствор готовили с помощью ампулы фирмы Titrisol, которую разбавляли водой, подвергнутой очистке в системе Milli Q, до конечного объема в 1 л.
Способ разделения пептидов
Испытуемые пептиды: Ангиотензин I, IIе7-Ангиотензин III, VaI4-Ангиотензин III и Ангиотензин III растворяли в воде, подвергнутой очистке в системе Milli Q, до конечной концентрации 0,125 мг/мл для каждого пептида.
Разделение проводили при pH 3,0, pH 7,0 и pH 12,0.
Буферный раствор фазы А представляет собой 15 мМ раствор фосфата натрия с pH 3,0 или pH 7,0 или 10 мМ раствор NaOH с pH 12. Буферный раствор фазы В представляет собой ацетонитрил.
Общее представление о способе приведено ниже.

УФ 215 нм использовали в качестве детектирующей длины волны.
В зависимости от pH пептиды будут иметь положительный заряд (pH 3), будут почти нейтральны (pH 7) или отрицательно заряжены (pH 12). Заряд пептидов может влиять на разделение. Если, например, в частицах присутствуют отрицательно заряженные группы, это может привести к уширению пиков при низких pH, поскольку тогда положительно заряженные пептиды будут удерживаться как за счет ионного, так и за счет гидрофобного взаимодействия.
На Фиг. 1-3 приведены хроматограммы разделения на прототипе LS002597 при pH 7, pH 3 и pH 12, соответственно.
LS002597 обладает очень хорошими рабочими характеристиками с острыми пиками при всех значениях pH. Один из пептидов не связывается при значении pH 12, когда пептиды сильно отрицательно заряжены.
На Фиг. 4-6 приведены хроматограммы разделения на прототипе LS002980 при pH 7, pH 3 и pH 12, соответственно. LS002980 обладает очень хорошими рабочими характеристиками и является одним из нескольких прототипов, обладающих приемлемой гидрофобностью для удержания всех четырех пептидов при pH 12, когда получают прекрасное разделение. Разделение при pH 3 дает несколько более широкие пики, чем, например, LS002597, разделение при pH 7 вполне сопоставимо с таковым на Kromasil С4 100 А.
На Фиг. 7-9 приведены хроматограммы разделения на прототипе LS002889 при pH 7, pH 3 и pH 12, соответственно.
Этот прототип, с привитым поли(пентафторстиролом), (LS002889) дает хорошее разделение при всех значениях pH, характер разделения похож на наблюдаемый с прототипом LS 002597.
На Фиг. 10-12 приведены хроматограммы разделения на прототипе LS003147A при pH 7, pH 3 и pH 12, соответственно. Трет-бутилстирол (LS003147A) дает очень хорошие рабочие характеристики.
Фиг. 13-14 являются фигурами сравнения, на которых представлены хроматограммы с Kromasil С4 100 А при pH 7 и pH 3, соответственно.
Колонка с Kromasil дает хорошее разделение при pH 7, но не может разделить пептиды при pH 3, при этом наблюдается только три пика. Времена удерживания для всех пептидов значительно более высокие, чем для прототипов на основе агарозы. Это означает, что в этом случае, чтобы выделить пептиды, придется использовать больше органических растворителей. Разделение на колонке с Kromasil при pH 12 не проводили, поскольку продукты на основе силикагеля не стабильны при pH выше ~8.
Фиг. 15-17 являются фигурами сравнения, на которых представлены хроматограммы с Source 15 RPC при pH 7, pH 3 и pH 12, соответственно. Колонка с SOURCE 15 RPC демонстрирует хорошее разделение при pH3, но дает плохое разделение и широкие пики как при pH 7, так и при 12.
| название | год | авторы | номер документа |
|---|---|---|---|
| ХРОМАТОГРАФИЧЕСКИЙ ЛИГАНД | 2005 |
|
RU2396246C2 |
| СПОСОБ ОЧИСТКИ АНТИТЕЛ | 2005 |
|
RU2389552C2 |
| ХРОМАТОГРАФИЧЕСКИЙ ЛИГАНД | 2005 |
|
RU2541429C2 |
| СПОСОБ ПОЛУЧЕНИЯ ХРОМАТОГРАФИЧЕСКОЙ МАТРИЦЫ | 2005 |
|
RU2367517C2 |
| СПОСОБ ИЗГОТОВЛЕНИЯ РАЗДЕЛЯЮЩЕЙ МАТРИЦЫ | 2006 |
|
RU2411081C2 |
| СПОСОБ ПОЛУЧЕНИЯ СОРБЕНТОВ ДЛЯ ХРОМАТОГРАФИИ | 1999 |
|
RU2163911C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ТРОКСЕРУТИНА, ДЕКСПАНТЕНОЛА, БЕНЗОКАИНА И МЕТИЛПАРАГИДРОКСИБЕНЗОАТА В ЛЕКАРСТВЕННОМ ПРЕПАРАТЕ МЕТОДОМ ВЭЖХ | 2011 |
|
RU2475733C1 |
| СПОСОБ СИНТЕЗА РЕКОМБИНАНТНОГО ПАРАТИРЕОИДНОГО ГОРМОНА ЧЕЛОВЕКА | 2007 |
|
RU2441019C2 |
| ОПТИМИЗИРОВАННЫЙ МЕТОД ЗАХВАТА АНТИТЕЛ ХРОМАТОГРАФИЕЙ СМЕШАННОГО ТИПА | 2011 |
|
RU2591523C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ СОДЕРЖАНИЯ ЭСТРОГЕНА И ДЕКСПАНТЕНОЛА В ДВУХКОМПОНЕНТНОМ ЛЕКАРСТВЕННОМ ПРЕПАРАТЕ МЕТОДОМ ВЭЖХ | 2011 |
|
RU2476873C1 |




Изобретение относится к способу получения материала для хроматографии. Предложен способ получения материала для хроматографии (ОФХ), включающий следующие стадии: аллилирование частиц пористого полисахаридного материала с помощью аллилглицидилового эфира и прививание стирольных мономеров, выбранных из стирола, трет-бутилстирола и пентафторстирола, на указанные частицы, содержащие аллильную группу. Материал обеспечивает улучшенное разделение пептидов. 4 н. и 3 з.п. ф-лы, 17 ил., 3 табл.
1. Способ получения материала для обращенно-фазной хроматографии, ОФХ, включающий следующие стадии: аллилирование частиц пористого полисахаридного материала с помощью аллилглицидилового эфира и прививание стирольных мономеров, выбранных из стирола, трет-бутилстирола и пентафторстирола, на указанные частицы, содержащие аллильную группу.
2. Способ по п. 1, в котором частицы пористого полисахаридного материала сделаны из агарозы.
3. Способ по п. 1, в котором объемное содержание стирольного мономера в прививающем растворе составляет от 5 до 95% об./об., предпочтительно от 25 до 75%.
4. Способ по одному из пп. 1-3, в котором стирольный мономер представляет собой стирол или трет-бутилстирол, объемное содержание которого в прививающем растворе составляет 50% об./об.
5. Материал для ОФХ, полученный в соответствии с одним из пп. 1-4.
6. Материал для ОФХ, полученный в соответствии с п. 4.
7. Применение материала для ОФХ по п. 5 или 6 для осуществления обращенно-фазной хроматографии.
| US 7048858 B2 23.05.2006 | |||
| Пресс для выдавливания из деревянных дисков заготовок для ниточных катушек | 1923 |
|
SU2007A1 |
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |
| US 6602990 B1 05.08.2003 | |||
| СПОСОБ ПОЛУЧЕНИЯ ХРОМАТОГРАФИЧЕСКОЙ МАТРИЦЫ | 2005 |
|
RU2367517C2 |

