Настоящее изобретение относится к новым производным морфолинилантрациклина, к способам их получения, к содержащим их фармацевтическим композициям и их применению при лечении заболеваний с аномальной клеточной пролиферацией. В качестве примера, соединения настоящего изобретения могут быть использованы при лечении опухолей.
Антрациклины являются соединениями, относящимися к антибиотикам, которые проявляют цитотоксическую активность. Некоторые исследования показали, что антрациклины могут воздействовать на уничтожение клеток рядом различных механизмов, включая: 1) интеркаляцию клеток ДНК, таким образом ингибируя синтез ДНК-зависимых нуклеиновых кислот; 2) продукцию свободных радикалов, которые затем реагируют с клеточными макромолекулами, вызывая повреждение клетки, или 3) взаимодействие с клеточной мембраной [см., например, C. Peterson et al., ”Transport and storage of Anthracycline in experimental systems and human leukemia” in Anthracycline Antibiotics in Cancer Therapy (1982), pp.132-146; и N.R. Bachur, “Free Radical Damage” id, pp.97-102]. Из-за их цитотоксической активности, антрациклины используют при лечении многочисленных видов рака, таких как лейкоз, карцинома груди, карцинома легких, аденокарцинома яичников и саркомы [см., например, P.H- Wiernik, in Anthracycline: Current Status and New Developments (1980), p 11]. Часто используемые антрациклины включают доксорубицин, эпирубицин, идарубицин и дауномицин.
В последние годы было синтезировано много новых производных высоко цитотоксических антрациклинов.
Производные антрациклина, содержащие замещенное морфолинoвое кольцо, связанное в положении C-3' сахарной части молекулы, показали многообещающую противоопухолевую активность в экспериментах на мышиных опухолях [см.: J. W. Lown, Bioactive Molecules, vol 6, (1988), pp.55-101] и в клинических испытаниях при лечении гепатоцеллюлярной карциномы [см.: C. Sessa, O. Valota, C. Geroni, Cardiovascular Toxicology, vol. 7(2), (2007), pp. 75-79].
Новые производные морфолинилантрациклина, в которых морфолинoвое кольцо связано мостиковой связью с атомом кислорода в положении C-4’ сахарного остатка, раскрыты в качестве противоопухолевых средств в международной патентной заявке WO9802446 на имя Pharmacia & Upjohn SPA.
4-Амино и 4-фтор производные антрациклина также раскрыты в качестве противоопухолевых средств в патентных заявках EP 288268 и EP 381989 на имя Farmitalia Carlo Erba Srl.
Несмотря на усилия в противораковых исследованиях, рак остается занимающей центральное место и недостижимой целью, поэтому по-прежнему существует потребность в новых противоопухолевых средствах.
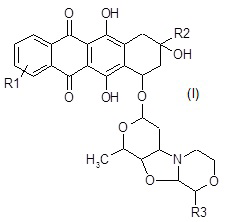
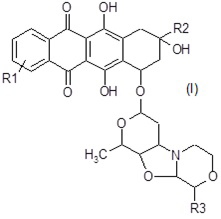
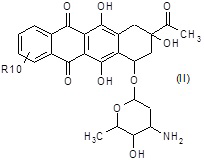
Настоящее изобретение относится к производным морфолинилантрациклина формулы (I):
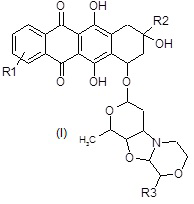
где:
R1 представляет собой галоген или NR4R5;
R2 представляет собой необязательно замещенную группу, выбранную их прямого или разветвленного C2-C6 алкила, NR7R8-C2-C6 алкила, R6O-C2-C6 алкила и COR9;
R3 представляет собой водород или прямой или разветвленный C1-C4 алкокси;
R4 и R5 независимо представляют собой водород, монозамещенный бензил, дизамещенный бензил или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, NR7R8-C1-C6 алкила, R6O-C1-C 6алкила, R7R8N-C1-C6 алкилкарбонила, R6O-C1-C6 алкилкарбонила, R7R8N-C1-C6 алкиламинокарбонила, R6O-C1-C6 алкиламинокарбонила, R7R8N-C1-C6 алкилсульфонила, R6O-C1-C6 алкилсульфонила, R7R8N-C1-C6 алкоксикарбонила и R6O-C1-C6 алкоксикарбонила; или
R4 и R5, взятые вместе с атомом N, к которому они присоединены, образуют гетероциклил, замещенный R4;
R6, R7 и R8 независимо представляют собой водород или необязательно замещенный прямой или разветвленный C1-C6 алкил;
R9 представляет собой OR6, NR7R8 или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C4 алкила, NR7R8-C1-C4 алкила и R6O-C1-C4 алкила,
или их фармацевтически приемлемым солям.
Настоящее изобретение также предоставляет способы синтеза производных морфолинилантрациклина (I), полученных посредством способов, состоящих из стандартных синтетических преобразований, и их изомеров, таутомеров, гидратов, сольватов, комплексов, метаболитов, пролекарств, носителей, N-оксидов.
Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, и по меньшей мере один фармацевтически приемлемый эксципиент, носитель или разбавитель.
Настоящее изобретение дополнительно предоставляет фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы (I) и одно или более химиотерапевтических средств.
Настоящее изобретение также обеспечивает фармацевтическую композицию, содержащую терапевтически эффективное количество соединения формулы (I) в комбинации с известным противораковым лечением, таким как лучевая терапия или химиотерапия, в комбинации с цитостатическими или цитотоксическими средствами, средствами типа антибиотиков, алкилирующими средствами, антиметаболитами, гормональными средствами, иммунологическими средствами, средствами типа интерферона, ингибиторами циклооксигеназы (например, ингибиторами COX-2), ингибиторами матриксметаллопротеазы, ингибиторами теломеразы, ингибиторами тирозинкиназы, средствами против рецептора фактора роста, средствами против HER2, средствами против EGFR, средствами против ангиогенеза (например, ингибиторами ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами пути передачи сигнала ras-raf, ингибиторами клеточного цикла, ингибиторами других cdk, тубулинсвязывающими средствами, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и подобными.
Дополнительно настоящее изобретение предоставляет продукт, содержащий соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, и одно или более химиотерапевтических средств, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в противораковой терапии.
В еще одном аспекте настоящее изобретение предоставляет соединение формулы (I) или его фармацевтически приемлемую соль, как определено выше, для применения в качестве лекарственного средства.
Настоящее изобретение также предоставляет соединение формулы (I), как определено выше, для применения в способе лечения рака, расстройства клеточной пролиферации и вирусных инфекций.
Предпочтительно, соединение формулы (I), как определено выше, предназначено для применения в способе лечения специфических типов рака, таких как, но, не ограничиваясь ими: карциномы, в том числе мочевого пузыря, груди, толстой кишки, почки, печени, легких, включая мелкоклеточный рак легких, пищевода, желчного пузыря, яичников, поджелудочной железы, желудка, шейки матки, щитовидной железы, предстательной железы, и карциномы кожи, включая плоскоклеточный рак; гемопоэтические опухоли лимфоидной клеточной линии, включая лейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, В-клеточную лимфому, Т-клеточную лимфому, лимфому Ходжкина, неходжкинскую лимфому, лимфому волосатых клеток и лимфому Беркитта; гемопоэтические опухоли миелоидной клеточной линии, включая острую и хроническую миелоцитарную лейкемию, миелодиспластический синдром и промиелоцитарную лейкемию; опухоли мезенхимального происхождения, включая фибросаркому и рабдомиосаркому; опухоли центральной и периферической нервной системы, включая астроцитому, нейробластому, глиому и шванному; и другие опухоли, включая меланому, семиному, тератокарциному, остеосаркому, пигментную ксеродерму, кератоксантому, фолликулярный рак щитовидной железы, саркому Капоши и мезотелиому.
Кроме того, соединение формулы (I), как определено выше, предназначено для применения в способе лечения специфических расстройств клеточной пролиферации, таких как, например, доброкачественная гиперплазия предстательной железы, семейный аденоматозный полипоз (FAP), нейрофиброматоз, псориаз, гладкая сосудистая пролиферация, связанная с атеросклерозом, легочный фиброз, артрит, гломерулонефрит и послеоперационный стеноз и рестеноз.
В дополнение, соединение формулы (I), как определено выше, предназначено для применения в способе ингибирования ангиогенеза и метастазирования опухоли, а также в способе лечения отторжения трансплантата органа и лечения реакции трансплантат против хозяина.
Настоящее изобретение также предоставляет способ лечения рака, включающий введение млекопитающему, нуждающемуся в этом, эффективного количества соединения формулы (I), как определено выше. Млекопитающим, нуждающимся в этом, может быть, например, человек.
Кроме того, настоящее изобретение предоставляет применение соединения формулы (I) или его фармацевтически приемлемой соли, как определено выше, в производстве лекарственного средства для лечения рака.
Если не указано иное, следующие термины и фразы, использованные в данном описании, имеют следующие значения.
Под термином “прямой или разветвленный C1-C6 алкил” подразумеваются любые группы, такие как, например, метил, этил, н-пропил, н-бутил, н-пентил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил.
Под термином “прямой или разветвленный C1-C4 алкокси” подразумеваются любые группы, такие как, например, метокси, этокси, пропокси и т.д.
Под термином “галоген” подразумеваются фтор, хлор, бром или йод.
Под термином “монозамещенный бензил” подразумеваются любые группы, такие как 4-метоксибензил, 4-метилбензил, 4-фторбензил, 3-метоксибензил, 3-метилбензил, 3-фторбензил, 2-метоксибензил, 2-метилбензил, 2-фторбензил и т.д.
Под термином “дизамещенный бензил” подразумеваются любые группы, такие как 2,4-диметоксибензил, 2,4-диметилбензил, 2,4-дифторбензил, 2,3-диметоксибензил, 2,3-диметилбензил, 2,3-дифторбензил, 2,5-диметоксибензил, 2,5-диметилбензил, 2,5-дифторбензил, 2-фтор-4-метоксибензил, 2-фтор-4-метилбензил и т.д.
Под термином “прямой или разветвленный C1-C6 алкилкарбонил” подразумеваются любые группы, такие как, например, метилкарбонил, этилкарбонил, н-бутилкарбонил, изопропилкарбонил и т.д.
Под термином “прямой или разветвленный C1-C6 алкиламинокарбонил” подразумеваются любые группы, такие как, например, метиламинокарбонил, этиламинокарбонил, н-бутиламинокарбонил, изопропиламинокарбонил и т.д.
Под термином “прямой или разветвленный C1-C6 алкилсульфонил” подразумеваются любые группы, такие как, например, метилсульфонил, этилсульфонил, н-бутилсульфонил, изопропилсульфонил и т.д.
Под термином “прямой или разветвленный C1-C6 алкоксикарбонил” подразумеваются любые группы, такие как, например, этоксикарбонил, н-бутоксикарбонил, изопропоксикарбонил, н-пропоксикарбонил и т.д.
Термин "гетероциклил", как используется в данном описании, относится к насыщенному или ненасыщенному неароматическому 5-7-членному карбоциклическому кольцу, где от 1 до 3 атомов углерода заменены гетероатомами, такими как азот, кислород и сера, где указанные гетероатомы могут быть непосредственно связаны друг с другом, азот и сера могут быть необязательно окисленными, азот может быть необязательно кватернизирован или иметь заместитель R4. Неограничивающими примерами гетероциклильных групп являются, например, пиперидинил, пиперазинил, оксазолидинил, 4-метилпиперазинил, 4-этилпиперазинил и т.д.
Термин "арил", как используется в данном описании, относится к карбоциклическим углеводородам, имеющим от 1 до 2 кольцевых фрагментов, или конденсированных или связанных друг с другом простыми связями, где по меньшей мере одно из колец является ароматическим. Примерами арильных групп, согласно настоящему изобретению, являются, например, фенил, бифенил, α- или β-нафтил, дигидронафтил и подобные.
Термин “уходящая группа” относится к группе, которая может быть замещена другой группой в реакции замещения. Такие уходящие группы хорошо известны в данной области техники, и примеры включают, но, не ограничиваясь ими, галогениды (фторид, хлорид, бромид и йодид), азиды, сульфонаты (например, необязательно замещенный C1-C6 алкансульфонат, такой как метансульфонат и трифторметансульфонат, или необязательно замещенный C7-C12 алкилбензолсульфонат, такой как п-толуолсульфонат), сукцинимид-N-оксид, п-нитрофеноксид, пентафторфеноксид, тетрафторфеноксид, карбоксилаты, аминокарбоксилаты (карбаматы) и алкоксикарбоксилаты (карбонаты). Для замещения в насыщенном углероде, галогениды и сульфонаты являются предпочтительными уходящими группами. Для замещения в карбонильном углероде, в качестве уходящей группы могут быть использованы, например, галогенид, сукцинимид-N-оксид, п-нитрофеноксид, пентафторфеноксид, тетрафторфеноксид, карбоксилат или алкоксикарбоксилат (карбонат). Термин "уходящая группа" также относится к группе, которая удаляется вследствии реакции элиминирования, например, реакции электронного каскада или реакции спироциклизации. В этом случае, в качестве уходящей группы могут быть использованы, например, галогенид, сульфонат, азид, аминокарбоксилат (карбамат) или алкоксикарбоксилат (карбонат).
Термин “азот-защитная группа” относится к группе, которая с атомом азота образует карбаматы, амиды, циклические имиды, N-алкил- и N-ариламины. Такие защитные группы хорошо известны в данной области техники (см., например, Green T. W., Wuts P. G. M.; “Protecting groups in organic synthesis”). Неограничивающими примерами карбамат-защитных групп являются, например, метил- и этилкарбамат, 9-флуоренилметилкарбамат (Fmoc), 2,2,2-трихлорэтилкарбамат (Troc), трет-бутилкарбамат (BOC), винилкарбамат (Voc), аллилкарбамат (Alloc), бензилкарбамат (Cbz), п-нитробензил и подобные. Неограничивающими примерами амидов являются, например N-трихлорацетамид, N-трифторацетамид (TFA) и подобные. Неограничивающими примерами циклических амидных защитных групп являются, например, N-фталимид, N-дитиасукциноилимид (dts) и подобные. Неограничивающими примерами N-алкил и N-арил-защитных групп являются, например, N-аллиламин, N-бензиламин и подобные.
Термин “гидроксил-защитная группа” относится к группе, которая с атомом кислорода образует простые эфиры, сложные эфиры, циклические ацетали или кетали. Такие защитные группы хорошо известны в данной области техники (см., например, Green T. W., Wuts P. G. M.; “Protecting groups in organic synthesis”). Неограничивающими примерами простых эфирных защитных групп являются, например, простые алкиловые и бензиловые эфиры, такие как простой метоксиметиловый эфир (MOM-OR), простой тетрагидропираниловый эфир (THP-OR), простой аллиловый эфир (Аллил-OR), простой бензиловый эфир (Bn-OR), простой трифенилметиловый эфир (Tr-OR) и подобные, или простые силиловые эфиры, такие как простой триметилсилиловый эфир (TMS-OR), простой трет-бутилдиметилсилиловый эфир (TBS-OR или TBDMS-OR), простой трет-бутилдифенилсилиловый эфир (TBDPS-OR), простой дифенилметилсилиловый эфир (DPMS-OR) и подобные. Неограничивающими примерами сложноэфирных защитных групп являются, например, трифторацетат, бензоат (Bz-OR) и карбонаты, такие как этилкарбонат и подобные. Неограничивающими примерами циклических ацетальных или кетальных защитных групп являются, например, метиленацеталь, этилиденацеталь, метоксиметиленацеталь и подобные.
Термин "активный сложный эфир" относится к функциональной группе, в которой алкоксигруппа сложноэфирного фрагмента представляет собой хорошо уходящую группу. Примеры таких алкоксигрупп включают, но, не ограничиваясь ими, сукцинимид-N-оксид, п-нитрофеноксид, пентафторфеноксид, тетрафторфеноксид, 1-гидроксибензотриазол и 1-гидрокси-7-азабензотриазол, и группы с сопоставимой способностью к удалению. Незамещенные на основе алкила алкоксигруппы, такие как метокси, этокси, изопропокси и трет-бутокси не квалифицируются как хорошо уходящие группы, и поэтому сложные метиловые, этиловые, изопропиловые и трет-бутиловые эфиры не считаются активными сложными эфирами.
Под терминами “соединения формулы (Ia)" или “соединения формулы (Ib)" имеются в виду соответственно соединения, изображенные ниже:
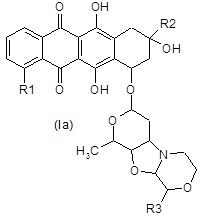
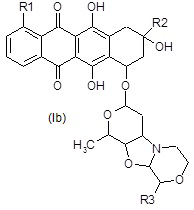
где R1, R2 и R3 являются такими, как определено выше.
Фармацевтически приемлемые соли соединения формулы (I) также включают соли с неорганическими или органическими основаниями, например, щелочными или щелочноземельными металлами, особенно натрием, калием, кальцием, гидроксидами аммония или магния, карбонатами или бикарбонатами, ациклическими или циклическими аминами.
Если в соединении настоящего изобретения присутствует стереогенный центр или другая форма изомерного центра, все формы такого изомера или изомеров, включая энантиомеры и диастереомеры, предназначены для включения в настоящее изобретение. Соединения, содержащие стереогенный центр могут быть использованы в виде рацемической смеси, энантиомерно обогащенной смеси, или рацемическая смесь может быть разделена с использованием хорошо известной технологии, и индивидуальные энантиомеры могут быть использованы по отдельности. В случаях, в которых соединения имеют ненасыщенные углерод-углеродные двойные связи, как цис (Z), так и транс (E) изомеры входят в объем настоящего изобретения.
В случаях, когда соединения могут существовать в таутомерных формах, каждая форма рассматривается как включенная в настоящее изобретение, существует ли она в равновесии или в преимущественно в одной форме.
Предпочтительными соединениями формулы (I) являются соединения формулы (Ia) или (Ib):
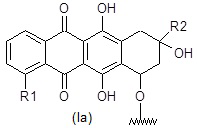

где R1 представляет собой фтор или NR4R5, где один из R4 или R5 представляет собой водород, и другой представляет собой водород или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, NR7R8-C1-C6 алкила, R6O-C1-C6 алкила, R7R8N-C1-C6 алкилкарбонила, R6O-C1-C6 алкилкарбонила, R7R8N-C1-C6 алкиламинокарбонила, R6O-C1-C6 алкиламинокарбонила, R7R8N-C1-C6 алкоксикарбонила и R6O-C1-C6 алкоксикарбонила.
Более предпочтительными соединениями формулы (I) являются соединения формулы (Ia):

где R1 представляет собой фтор или NR4R5, где R4 и R5 являются такими, как определено выше, и
R2 представляет собой COR9, где R9 является таким, как определено выше.
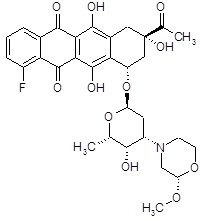
Конкретными, но не ограничивающими, предпочтительными соединениями (соединение) настоящего изобретения, необязательно в форме фармацевтически приемлемой соли, являеются следующие:
1) (8S,10S)-8-ацетил-1-фтор-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион,
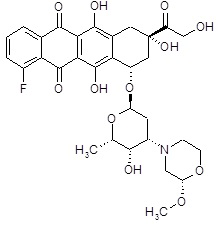
2) (8S,10S)-1-фтор-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион,
3) (8S,10S)-1-амино-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион,
4) (8S,10S)-8-ацетил-1-амино-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион,
5) (8S,10S)-8-ацетил-6,8,11-тригидрокси-1-[(2-гидроксиэтил)амино]-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион,
6) (8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-[(2-гидроксиэтил)амино]-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион,
7) (8S,10S)-8-ацетил-1-[(2-аминоэтил)амино]-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион и
8) (8S,10S)-1-[(2-аминоэтил)амино]-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион.
В качестве справочной информации по любому конкретному соединению формулы (I) настоящего изобретения, необязательно в форме фармацевтически приемлемой соли, см. экспериментальный раздел и формулу изобретения.
Настоящее изобретение также предоставляет способ получения соединения формулы (I), как определено выше, с применением реакционных путей и схем синтеза, описанных ниже, при использовании методик, доступных в данной области техники, и легко доступных исходных веществ. Получение по некоторым вариантам осуществления настоящего изобретения описано в примерах, которые следуют ниже, но специалисту в данной области техники будет понятно, что описанные получения могут быть легко адаптировны для подготовки других вариантов осуществления настоящего изобретения. Например, синтез непроиллюстрированного соединения согласно настоящему изобретению может быть осуществлен проведением модификаций, очевидных для специалиста в данной области техники, например, защитой надлежащим образом интерферирующих групп, заменой другими подходящими реагентами, известными в данной области техники, или проведением рутинных модификаций реакционных условий. Альтернативно, другие реакции, на которые ссылаются в данном описании или известные в данной области техники, будут пониматься как имеющие адаптируемость для получения других соединений настоящего изобретения.
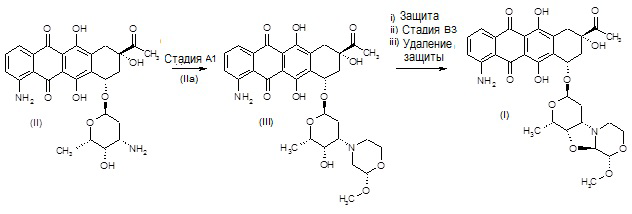
Соединение формулы (I) получают согласно любому из пяти альтернативных путей A-E, суммированных на схеме 1 ниже; также суммированным на схеме 1 является получение промежуточного соединения формулы (V), согласно пути F, и получение исходного соединения формулы (II), согласно пути G.
Схема 1
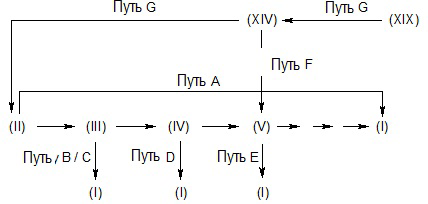
Путь A
Соединение формулы (I), где R1 и R3 являются такими, как определено выше, и R2 представляет собой COR9, где R9 представляет собой OR6 или NR7R8, где R6, R7 и R8 являются такими, как определено выше, получают, как суммировано на схеме 2 ниже.
Схема 2
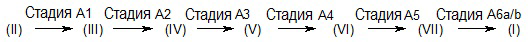
Соответственно, способ настоящего изобретения включает следующие стадии:
A1) взаимодействие соединения формулы (II):
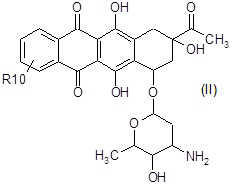
где R10 представляет собой R1 или группу формулы NR19R20, где R19 и R20 независимо представляют собой подходящую азот-защитную группу, или один из R19 или R20 представляет собой водород, и другой представляет собой подходящую азот-защитную группу, и R1 является таким, как определено выше,
с соединением формулы (IIa):
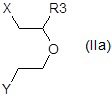
где R3 является таким, как определено выше, и X и Y представляют собой одинаковые или различные уходящие группы, предпочтительно галоген;
A2) взаимодействие полученного в результате соединения формулы (III):
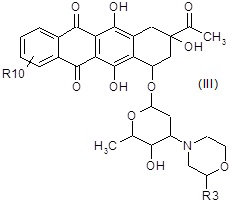
где R3 и R10 являются такими, как определено выше,
с этилортоформиатом и бромом, затем добавление HBr;
A3) взаимодействие полученного в результате соединения формулы (IV):
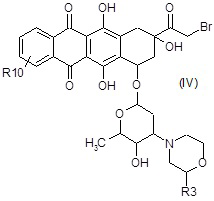
где R3 и R10 являются такими, как определено выше,
с формилирующим агентом;
A4) окисление полученного в результате соединения формулы (V):
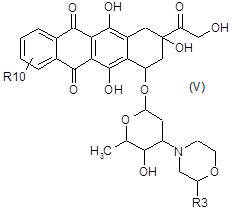
где R10 и R3 являются такими, как определено выше;
A5) взаимодействие полученного в результате соединения формулы (VI):
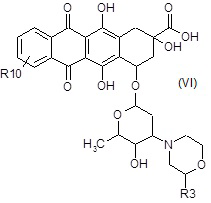
где R3 и R10 являются такими, как определено выше,
с соединением формулы (VIa) или (VIb):
R6-OH (VIa); R7R8NH (VIb),
где R6, R7 и R8 являются такими, как определено выше;
A6a) взаимодействие полученного в результате соединения формулы (VII):
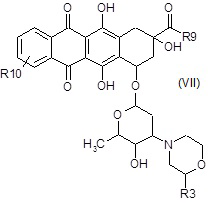
где R3, R10 и R9 являются такими, как определено выше,
сначала с DMDO;
A6b) затем взаимодействие полученного в результате соединения формулы (XX):

где R3 и R10 являются такими, как определено выше, и R2 представляет собой COR9, где R9 является таким, как определено выше, с хлорангидридом циануровой кислоты или с солью железа (II), и, наконец, если требуется, удаление азот и/или гидрокси-защитной группы/групп, с получением соединения формулы (I):
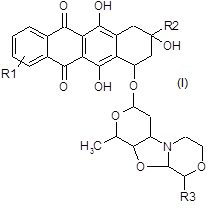
где R1 и R3 являются такими, как определено выше, и R2 представляет собой COR9, где R9 является таким, как определено выше;
необязательно преобразование первого соединения формулы (I) во второе соединение формулы (I) известными химическими реакциями; и/или, если требуется, преобразование такого соединения формулы (I) в его фармацевтически приемлемую соль или преобразование соли в свободное соединение формулы (I).
Путь B
Соединение формулы (I), где R1 и R3 являются такими, как определено выше, и R2 представляет собой этил или COCH3, получают, как суммировано на схеме 3 ниже.
Схема 3
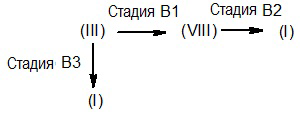
Соответственно, способ настоящего изобретения включает следующие стадии:
B1) взаимодействие соединения формулы (III), как определено выше, с производным гидразина формулы (IIIa):
R11-NH-NH2 (IIIa)
где R11 представляет собой арил, предпочтительно фенил, 4-метилфенил или 4-галогенфенил, и затем восстановление гидразида;
B2) взаимодействие полученного в результате соединения формулы (VIII):
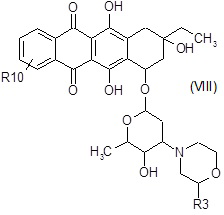
где R3 и R10 являются такими, как определено выше, в таких же условиях, как описано выше для стадий A6a) и A6b),
с получением соединения формулы (I):
где R1 и R3 являются такими, как определено выше, и R2 представляет собой этил,
или
B3) взаимодействие соединения формулы (III), как определено выше, в таких же условиях, как описано выше для стадий A6a) и A6b),
с получением соединения формулы (I):
где R1 и R3 являются такими, как определено выше, и R2 представляет собой COCH3;
необязательно преобразование первого соединения формулы (I) во второе соединение формулы (I) известными химическими реакциями; и/или, если требуется, преобразование такого соединения формулы (I) в его фармацевтически приемлемую соль или преобразование соли в свободное соединение формулы (I).
Путь C
Соединение формулы (I), где R1 и R3 являются такими, как определено выше, и R2 выбран из прямого или разветвленного C3-C6 алкила, NR7R8-C3-C6 алкила, R6O-C3-C6 алкила и COR9, где R9 представляет собой прямой или разветвленный C2-C4 алкил, NR7R8-C2-C4 алкил или R6O-C2-C4 алкил, где R6, R7 и R8 являются такими, как определено выше, получают, как суммировано на схеме 4 ниже.
Схема 4
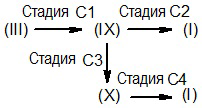
Соответственно, способ настоящего изобретения включает следующие стадии:
C1) взаимодействие соединения формулы (III), как определено выше, с соединением формулы (IIIb):
R12-X (IIIb)
где R12 представляет собой группу, выбранную из прямого или разветвленного C1-C4 алкила, NR7R8-C1-C4 алкила и R6O-C1-C4 алкила, и X представляет собой уходящую группу, предпочтительно галоген;
C2) взаимодействие полученного в результате соединения формулы (IX):
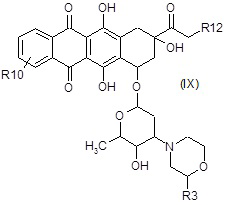
где R3, R10 и R12 являются такими, как определено выше, в таких же условиях, как описано выше для стадий A6a) и A6b),
с получением соединения формулы (I):
где R1 и R3 являются такими, как определено выше, и R2 представляет собой COR9, где R9 представляет собой прямой или разветвленный C2-C4 алкил, NR7R8-C2-C4 алкил или R6O-C2-C4 алкил, где R6, R7 и R8 являются такими, как определено выше;
или, альтернативно,
C3) взаимодействие соединения формулы (IX), как определено выше, в таких же условиях, как описано выше для стадии B1;
C4) взаимодействие полученного в результате соединения формулы (X):
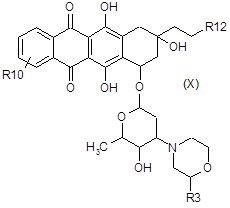
где R3, R10 и R12 являются такими, как определено выше, в таких же условиях, как описано выше для стадий A6a) и A6b),
с получением соединения формулы (I):
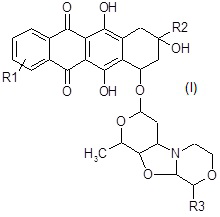
где R1 и R3 являются такими, как определено выше, и R2 представляет собой группу, выбранную из прямого или разветвленного C3-C6 алкила, NR7R8-C3-C6 алкила и R6O-C3-C6 алкила, где R6, R7 и R8 являются такими, как определено выше;
необязательно преобразование первого соединения формулы (I) во второе соединение формулы (I) известными химическими реакциями; и/или, если требуется, преобразование такого соединения формулы (I) в его фармацевтически приемлемую соль или преобразование соли в свободное соединение формулы (I).
Путь D
Соединение формулы (I), где R1 и R3 являются такими, как определено выше, и R2 представляет собой CH2-CH2NR7R8, CH2-CH2OR6 или COR9, где R9 представляет собой -CH2NR7R8 или -CH2OR6, где R6, R7 и R8 являются такими, как определено выше, получают, как суммировано на схеме 5 ниже.
Схема 5
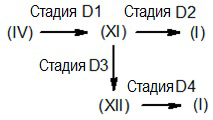
Соответственно, способ настоящего изобретения включает следующие стадии:
D1) взаимодействие соединения формулы (IV), как определено выше, где функция карбонила может быть необязательно активирована в виде производного фенилгидразона, с соединением формулы (IVa) или (IVb):
HN-R7R8 (IVa), HOR6 (IVb)
где R6, R7 и R8 являются такими, как определено выше, и где группа OH может быть необязательно активирована в виде, например, производного тозила или мезила, и затем, если присутствует, удаление функции гидразона гидролизом;
D2) взаимодействие полученного в результате соединения формулы (XI) или (XIa):
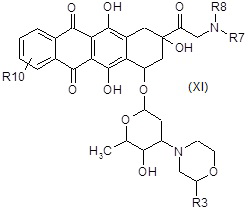 ,
, 
где R3, R6, R7, R8 и R10 являются такими, как определено выше, в таких же условиях, как описано выше для стадий A6a) и A6b),
с получением соединения формулы (I):
где R1 и R3 являются такими, как определено выше, и R2 представляет собой COR9, где R9 представляет собой -CH2-NR7R8 или -CH2-OR6, где R6, R7 и R8 являются такими, как определено выше;
или, альтернативно,
D3) взаимодействие полученного в результате соединения формулы (XI) или (XIa), как определено выше, в таких же условиях, как описано выше для стадии B1;
D4) взаимодействие полученного в результате соединения формулы (XII) или (XIIa):
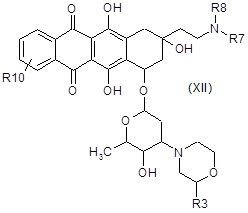 ,
, 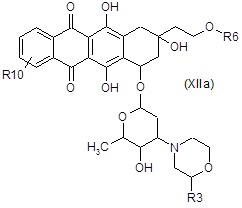
где R3, R6, R7, R8 и R10 являются такими, как определено выше, в таких же условиях, как описано выше для стадий A6a) и A6b),
с получением соединения формулы (I):
где R1 и R3 являются такими, как определено выше, и R2 представляет собой -CH2-CH2-NR7R8 или -CH2-CH2-OR6, где R6, R7 и R8 являются такими, как определено выше;
необязательно преобразование первого соединения формулы (I) во второе соединение формулы (I) известными химическими реакциями; и/или, если требуется, преобразование такого соединения формулы (I) в его фармацевтически приемлемую соль или преобразование соли в свободное соединение формулы (I).
Путь E
Соединение формулы (I), где R1 и R3 являются такими, как определено выше, и R2 представляет собой CH2OH или COR9, где R9 представляет собой CH2OH, получают, как суммировано на схеме 6 ниже.
Схема 6
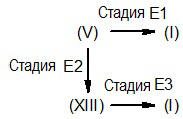
Соответственно, способ настоящего изобретения включает следующие стадии:
E1) взаимодействие соединения формулы (V), как определено выше, в таких же условиях, как описано выше для стадий А6a) и A6b),
с получением соединения формулы (I):
где R1 и R3 являются такими, как определено выше, и R2 представляет собой COR9, где R9 представляет собой CH2OH;
или, альтернативно,
E2) взаимодействие соединения формулы (V), как определено выше, в таких же условиях, как описано выше для стадии B1;
E3) взаимодействие полученного в результате соединения формулы (XIII):
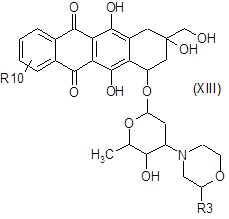
где R10 и R3 являются такими, как определено выше, в таких же условиях, как описано выше для стадий A6a) и A6b),
с получением соединения формулы (I):
где R1, R3 являются такими, как определено выше, и R2 представляет собой CH2OH;
необязательно преобразование первого соединения формулы (I) во второе соединение формулы (I) известными химическими реакциями; и/или, если требуется, преобразование такого соединения формулы (I) в его фармацевтически приемлемую соль или преобразование соли в свободное соединение формулы (I).
Согласно стадии A1), взаимодействие соединения формулы (II) с соединением формулы (IIa) осуществляют в органическом растворителе, предпочтительно ДМФА, при комнатной температуре,, следуя хорошо известным методикам, описанным в данной области техники (см., например WO91/09046).
Согласно стадии A2), взаимодействие соединения формулы (III) с этилортоформиатом и бромом, и затем с HBr, осуществляют в две стадии, следуя хорошо известным методикам, описанным в данной области техники (см., например Doxorubicin Anticancer Antibiotics Vol. 17, 1981, p.168; F. Arcamone et al. J. Med. Chem. 1974, 17, p. 335).
Согласно стадии A3), взаимодействие для получения соединения формулы (V) осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например Doxorubicin Anticancer Antibiotics Vol. 17, 1981, p.168; US3803124). Пример, который не предназначен для ограничения данного способа, представляет собой взаимодействие соединения формулы (IV) с формиатом натрия. Данное взаимодействие осуществляют в CH3CN или ацетоне или их смеси, при температуре в интервале примерно от 20°C до температуры кипения с обратным холодильником и в течение времени примерно от 30 минут до примерно 24 часов.
Согласно стадии A4), окисление соединения формулы (V) осуществляют с реагентом окисления, предпочтительно NaIO4. Данное взаимодействие осуществляют в MeOH или воде или их смеси, при температуре в интервале примерно от 20°C до температуры кипения с обратным холодильником и в течение времени примерно от 30 минут до примерно 24 часов.
Согласно стадии A5), реакцию сочетания между соединением формулы (VI) и соединением формулы (VIa) или (VIb) осуществляют, следуя хорошо известным методикам, описанным в данной области техники (как правило, реагенты сочетания см., например, в Amino Acids, Peptides and Proteins in Organic Chemistry: Building Blocks, Catalysis and Coupling Chemistry, Volume 3; Andrew B. Hughes, Ayman El- Faham, Fernando Albericio, 2010). Пример, который не предназначен для ограничения данного способа, представляет собой взаимодействие соединения формулы (VI) с соединением формулы (VIa) в присутствии конденсирующего агента, такого как, например, DCC или EDC. Данное взаимодействие осуществляют в органическом растворителе, предпочтительно ДМФА, при температуре в интервале примерно от 20°C до температуры кипения с обратным холодильником и в течение времени примерно от 30 минут до примерно 24 часов.
Согласно стадиям A6a) и 6Ab), взаимодействие соединения формулы (VII) сначала с DMDO и затем взаимодействие полученного в результате соединения формулы (XX) с хлорангидридом циануровой кислоты или с солью железа (II) осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например, GB2296495A; WO2012073217; WO9802446).
Удаление азот и/или гидроксил-защитных групп, если необходимо, осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например, Protective Groups in Organic Synthesis; Theodora W. Greeen, Peter G. M. Wuts 4th edition).
Согласно стадии B1), взаимодействие соединения формулы (III) с соединением формулы (IIIa) осуществляют в органическом растворителе, предпочтительно MeOH, при температуре в интервале примерно от -10°C до примерно 50°C и в течение времени примерно от 30 минут до примерно 96 часов. Последующие восстановление производного гидразида для получения соединения формулы (VIII) осуществляют с NaBH4 или NaBH3CN в присутствии камфорсульфоновой кислоты. Данное взаимодействие осуществляют в органическом растворителе, предпочтительно MeOH, при температуре в интервале примерно от 20°C до температуры кипения с обратным холодильником и в течение времени примерно от 30 минут до примерно 5 часов (см., также Doxorubicin Anticancer Antibiotics Vol. 17, 1981, p. 165).
Согласно стадиям B2 и B3), данное взаимодействие соответственно осуществляют, как описано выше для стадий A6a) и A6b).
Согласно стадии C1), взаимодействие соединения формулы (III) с соединением формулы (IIIb) осуществляют в органическом растворителе, предпочтительно ДМФА, следуя хорошо известным методикам, описанным в данной области техники (см., например, Smith T. H., Fujiwara A.N., Henry D. W.; J. Med. Chem.1979, 22, p. 40).
Согласно стадии C2), взаимодействие соединения формулы (IX) осуществляют, как описано выше для стадий A6a) и A6b).
Согласно стадии C3), взаимодействие соединения формулы (IX) осуществляют, как описано выше для стадии B1.
Согласно стадии C4), взаимодействие соединения формулы (X) осуществляют, как описано выше для стадий A6a) и A6b).
Согласно стадии D1), взаимодействие соединения формулы (IV) с соединением формулы (IVa) или (IVb) необязательно проводят в присутствии основания, предпочтительно диэтиламина. Данное взаимодействие осуществляют в органическом растворителе, предпочтительно ацетоне, при температуре в интервале примерно от 20°C до температуры кипения с обратным холодильником и в течение времени примерно от 30 минут до примерно 24 часов (см., US4133877).
Удаление функции гидразона может быть осуществлено в гидролитических условиях, как сообщалось Baker, T. S.; Exley, D.; Steroids 1977, 29, p. 429; Sugimoto, K.; Sunakawa, N.; Ohki, S.; Chem Pharm Bull 1966, 14, p.147.
Согласно стадии D2), взаимодействие соединения формулы (XI) или (XIa) осуществляют, как описано выше для стадий A6a) и A6b).
Согласно стадии D3), взаимодействие соединения формулы (XI) или (XIa) осуществляют, как описано выше для стадии B1.
Согласно стадии D4), взаимодействие соединения формулы (XII) или (XIIa) осуществляют, как описано выше для стадий A6a) и A6b).
Согласно стадии E1), взаимодействие соединения формулы (V) осуществляют, как описано выше для стадий A6a) и A6b).
Согласно стадии E2), взаимодействие соединения формулы (V) осуществляют, как описано выше для стадии B1.
Согласно стадии E3), взаимодействие соединения формулы (XIII) осуществляют, как описано выше для стадий A6a) и A6b).
Путь F
Промежуточные соединения формулы (V), как определено выше, альтернативно получают согласно схеме 7 ниже.
Схема 7
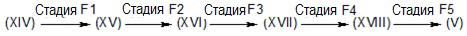
Соответственно, способ настоящего изобретения включает следующие стадии:
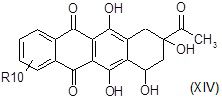
F1) взаимодействие соединения формулы (XIV):

где R10 является таким, как определено выше, с бромом и ацетатом калия;
F2) взаимодействие полученного в результате соединения формулы (XV):
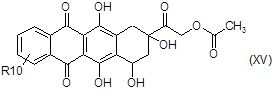
где R10 является таким, как определено выше, с сахаром формулы (XVa):
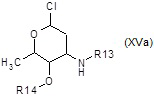
где R13 и R14 независимо представляют собой водород или подходящую азот и/или гидрокси-защитную группу, как например, трифторацетил или бензил;
F3) взаимодействие полученного в результате соединения формулы (XVI):

где R10, R13 и R14 являются такими, как определено выше, с этилортоформиатом и п-толуолсульфонатом пиридиния (PPTS);
F4) взаимодействие полученного в результате соединения формулы (XVII):
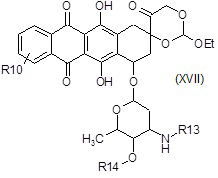
где R13 представляет собой водород, и R10 и R14 являются такими, как определено выше, с соединением формулы (IIa), как определено выше;
F5) удаление защиты с полученного в результате соединения формулы (XVIII):
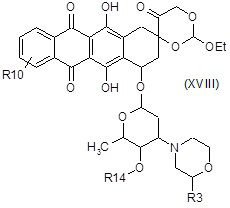
где R3, R10 и R14 являются такими, как определено выше, с получением соединения формулы (V), как определено выше.
Согласно стадии F1), данное взаимодействие осуществляют в органическом растворителе, предпочтительно ацетоне или диоксане, при температуре в интервале от 20°C до температуры кипения с обратным холодильником и в течение времени примерно от 30 минут до примерно 24 часов.
Согласно стадии F2), реакцию гликозидирования соединения формулы (XV) осуществляют в присутствии трифторметансульфоната серебра, следуя хорошо известным методикам, описанным в данной области техники (см., например GB2225781; GB2215332A).
Согласно стадии F3), для получения соединения формулы (XVII) осуществляют взаимодействие соединения формулы (XVI) с этилортоформиатом и PPTS. Данное взаимодействие осуществляют в органическом растворителе, предпочтительно DCM, при температуре в интервале от 0°C до температуры кипения с обратным холодильником и в течение времени примерно от 30 минут до примерно 24 часов.
Согласно стадии F4), данное взаимодействие осуществляют, как описано выше для стадии A1.
Согласно стадии F5), удаление гидрокси-защитной группы осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например, Protective Groups in Organic Synthesis; Theodora W. Greeen, Peter G. M. Wuts 4th edition).
Путь G
Соединение формулы (II), как определено выше, где R10 является таким, как определено выше, за исключением NH2 и галогена, получают согласно схеме 8, описанной ниже.
Схема 8

Соответственно, способ настоящего изобретения включает следующие стадии:
либо
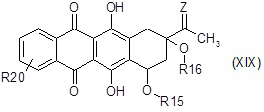
G1) взаимодействие соединения формулы (XIX):
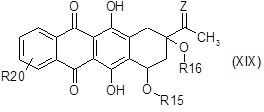
где R15 и R16 независимо представляют собой водород или подходящую гидрокси-защитную группу, такую как, например, трифторацетил, 9-флуоренилметил, ди-трет-бутилметилсилил, трет-бутилдифенилсилил или дифенилметилсилил, R20 представляет собой R10, где R10 представляет собой NH-R19, где R19 независимо представляет собой водород или подходящую азот-защитную группу, и Z является кислородом или подходящей карбонильной защитной группой, такой как ацеталь или кеталь, предпочтительно 1-3-диоксан или 1-3-диоксолан,
либо
i) с соединением формулы (XIXa), (XIXb), (XIXc) или (XIXd):
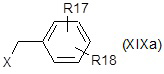 ;
;  ;
;  ;
; 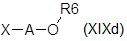
где X представляет собой уходящую группу, предпочтительно галоген; R17 и R18, одинаковые или различные, независимо представляют собой водород, галоген, прямой или разветвленный C1-C3 алкил или C1-C3 алкокси; A представляет собой прямой или разветвленный C1-C6 алкил; и R6, R7 и R8 являются такими, как определено выше, с получением после удаления защитных групп, если присутствуют, соответствующего соединения формулы (XIV), как определено выше, где R10 представляет собой группу R1 формулы NR4R5, где R4 и R5 независимо представляют собой водород, монозамещенный бензил, дизамещенный бензил или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, NR7R8-C1-C6 алкила и R6O-C1-C6 алкила, но оба не являются водородом;
или
ii) с соединением формулы (XIXe) или (XIXf):
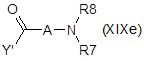 ;
; 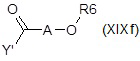
где Y’ представляет собой OH или уходящую группу, предпочтительно хлор, и A, R6, R7 и R8 являются такими, как определено выше, с получением после удаления защитных групп, если присутствуют, соответствующего соединения формулы (XIV), как определено выше, где R10 представляет собой группу R1 формулы NR4R5, где один из R4 или R5 представляет собой водород, и другой представляет собой группу R7R8N-C1-C6 алкилкарбонила или R6O-C1-C6 алкилкарбонила;
или
iii) с соединением формулы (XIXg) или (XIXh):
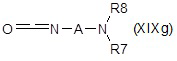 ;
; 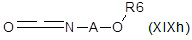
где A, R6, R7 и R8 являются такими, как определено выше, с получением после удаления защитных групп, если присутствуют, соответствующего соединения формулы (XIV), как определено выше, где R10 представляет собой группу R1 формулы NR4R5, где один из R4 или R5 представляет собой водород, и другой представляет собой группу R7R8N-C1-C6 алкиламинокарбонила или R6O-C1-C6 алкиламинокарбонила;
или
iv) с соединением формулы (XIXi) или (XIXm):
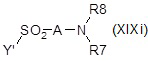 ;
; 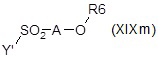
где Y представляет собой OH или уходящую группу, предпочтительно хлор, и A, R6, R7 и R8 являются такими, как определено выше, с получением после удаления защитных групп, если присутствуют, соответствующего соединения формулы (XIV), как определено выше, где R10 представляет собой группу R1 формулы NR4R5, где один из R4 или R5 представляет собой водород, и другой представляет собой группу R7R8N-C1-C6 алкилсульфонила или R6O-C1-C6 алкилсульфонила;
или
v) с соединением формулы (XIXn) или (XIXo):
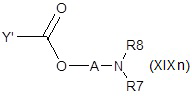 ;
; 
где Y’, A, R6, R7 и R8 являются такими, как определено выше, с получением после удаления защитных групп, если присутствуют, соответствующего соединения формулы (XIV), как определено выше, где R10 представляет собой группу R1 формулы NR4R5, где один из R4 или R5 представляет собой водород, и другой представляет собой группу R7R8N-C1-C6 алкоксикарбонила или R6O-C1-C6 алкоксикарбонила;
или
vi) с соединением формулы (XIXp):
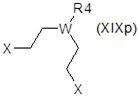
где W представляет собой CH или N, и R4 и X являются такими, как определено выше, с получением после удаления защитных групп, если присутствуют, соединения формулы (XIV), как определено выше, где R10 представляет собой группу R1 формулы NR4R5, где R4 и R5 взятые вместе с атомом N, с которым они связаны, образуют 6-членный гетероциклил, замещенный R4;
или
G2) взаимодействие соединения формулы (XIX):
где R15, R16 и Z являются такими, как определено выше, и R20 представляет собой подходящую уходящую группу, такую как, например, мезилат, тозилат или 4-фторбензолсульфонат, с соединением формулы (XIXq):
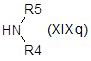
где R4 и R5 независимо представляют собой водород, монозамещенный бензил, дизамещенный бензил или необязательно замещенную группу, выбранную из прямого или разветвленного C1-C6 алкила, NR7R8-C1-C6 алкила и R6O-C1-C6 алкила; или R4 и R5, взятые вместе с атомом N, с которым они связаны, образуют замещенный гетероциклил;
с получением после удаления защитных групп, если присутствуют, соответствующего соединения формулы (XIV), как определено выше, где R10 представляет собой группу R1 формулы NR4R5, где R4 или R5 являются такими, как определено выше;
G3) взаимодействие полученного в результате соединения формулы (XIV):
где R10 являются такими, как определено на стадиях G1 или G2,
с соединением формулы (XVa), как определено выше, в таких же условиях, как описано выше для стадии F2, с получением соединения формулы (II), где R10 является таким, как определено выше.
Согласно стадии G1i), данное взаимодействие осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например Ngu, K.; Patel, D. V. Tetrahedron Lett 1997, 38 (6), pp. 973-976). В качестве примера, который не предназначен для ограничения данного способа, данное взаимодействие осуществляют в DCM, при температуре в интервале от 20°C до температуры кипения с обратным холодильником и в течение времени в интервале от 30 минут до примерно 24 часов.
Согласно стадии G1ii), реакцию сочетания между соединением формулы (XIX) и соединением формулы (XIXe) или (XIXf) осуществляют, следуя хорошо известным методикам, описанным в данной области техники (как правило, для реагентов сочетания см., например, Amino Acids, Peptides and Proteins in Organic Chemistry: Building Blocks, Catalysis and Coupling Chemistry, Volume 3; Andrew B. Hughes, Ayman El- Faham, Fernando Albericio, 2010). Примером, который не предназначен для ограничения данного способа, является взаимодействие в присутствии конденсирующего агента, такого как, например, DCC, EDC формиат натрия. Данное взаимодействие осуществляют в органическом растворителе, предпочтительно ДМФА, при температуре в интервале от 20°C до температуры кипения с обратным холодильником и в течение времени в интервале от 30 минут до примерно 24 часов.
Согласно стадии G1iii), данное взаимодействие осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например Gopalsamy A.; et al. Bioorg Med Chem Lett 2005, 15 (6), pp. 1591-1594; Lee Y. S. et al. Bioorg Med Chem Lett 2004, 3 14, (13), pp. 3379-3384). В качестве примера, который не предназначен для ограничения данного способа, данное взаимодействие осуществляют в пиридине, DCM, при температуре в интервале от 20°C до температуры кипения с обратным холодильником и в течение времени в интервале от 30 минут до примерно 24 часов.
Согласно стадии G1iv), данное взаимодействие осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например Filimonov S. J Heterocycl Chem 2006, 43, pp. 663-671; Rockway, T. W.; et al.; Bioorg Med Chem Lett 2006, 16, p. 3833).
Согласно стадии G1v), данное взаимодействие осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например Fukuoka S. et al. J. Chem. Soc. Chem. Commun. 1984, 6, p. 399).
Согласно стадии G1vi), данное взаимодействие осуществляют, следуя хорошо известным методикам, описанным в данной области техники (см., например Ismailov, V. et al.; Russ J Org Chem, 2004, 40 (2), pp. 284-285; Mewshaw R. E.; et al.; Bioorg Med Chem Lett 1998, 8 (19), pp. 2675-2680; Mishani E. et al.; Tetrahedron Lett 1996, 37 (3), pp. 319-322). В качестве примера, который не предназначен для ограничения данного способа, данное взаимодействие осуществляют в ДМСО, DCM, MeOH или их смеси, необязательно в присутствии основания или кислоты Льюиса (например, Al2Cl3) при температуре в интервале от 20°C до температуры кипения с обратным холодильником и в течение времени в интервале от 30 минут до примерно 24 часов.
Согласно стадии G2), данное взаимодействие осуществляют, как описано в патентной заявке GB2215322. В качестве примера, который не предназначен для ограничения данного способа, данное взаимодействие осуществляют в CH3CN, ТГФ или ДМФА, необязательно в присутствии основания, при температуре в интервале от 20°C до температуры кипения с обратным холодильником и в течение времени в интервале от 1 до 72 часов. Согласно стадии G3), данное взаимодействие осуществляют, как описано выше для стадии F2.
Соединения формулы (II), где R10 представляет собой NH2, могут быть получены, как описано в патентной заявке EP288268.
Соединения формулы (II), где R10 представляет собой водород, могут быть получены, как описано в патентной заявке WO9802446; и в Gary W et al. J.O.C 1987, 52, p. 713.
Соединения формулы (XIX) могут быть получены, как описано в патентной заявке EP288268.
Соединения формул (IIa), (IIIa), (IIIb), (IVa), (IVb), (XVa), (XIXa)-(XIXp) или являются коммерчески доступными или могут быть получены известными способами.
Из изложенного выше, специалисту в данной области техники будет очевидно, что когда получение соединений формулы (I) согласно любому из приведенных выше вариантов способа, необязательные функциональные группы в исходных веществах или их промежуточных соединениях, которые могут вызвать нежелательные побочные реакции, нуждаются в надлежащей защите, согласно общепринятым технологиям. Аналогичным образом, преобразование последних в свободные незащищенные соединения может быть осуществлено согласно известным методикам.
Как это будет легко оценено, если соединения формулы (I), полученные согласно способам, описанным выше, получены в виде смеси изомеров, их разделение с использованием общепринятых методов на отдельные изомеры формулы (I) входит в объем настоящего изобретения.
Фармакология
Новые производные морфолинилантрациклина настоящего изобретения полезны в качестве противоопухолевых средств.
Млекопитающее, например, человек или животное, может быть подвергнуто лечению способом, включающим введение фармацевтически эффективного количества производного морфолинилантрациклина формулы (I).
Состояние человека или животного может стать здоровым или улучшено таким образом.
Оценку цитотоксичности соединения формулы (I) проводили, как описано ниже.
In vitro анализ на клеточную пролиферацию
Линии клеток рака человека высевали в белые 384-луночные планшеты (1250 клеток/лунка) в полной среде (RPMI1640 или E-MEM плюс 10% фетальной телячьей сыворотки) и обрабатывали соединения растворением в 0,1% ДМСО в течение 24 часов после высевания. Полученные клетки инкубировали при 37°C и 5% CO2, и спустя 72 час планшеты подвергали обработке с использованием CellTiter-Glo анализа (Promega), следуя инструкциям производителя.
CellTiter-Glo представляет собой гомогенетический метод на основе количественной оценки присутствия АТФ, индикатора метаболически активных клеток. АТФ является количественным определителем с использованием системы на основе люциферазы и D-люциферина в результате легкой генерации. Кратко, 25 мл/лунка раствора реагента добавляли в каждую лунку, и после 5 минут встряхивания микропланшеты считывали люминометром. Люминесцентный сигнал пропорционален числу живых клеток, присутствующих в культуре.
Кривые доза-ответ получали путем интерполяции сигмовидной функции 8 точек концентрации, и антипролиферативную активность соединений представляли как половину максимальной ингибирующей концентрации (IC50).
Репрезентативные соединения настоящего изобретения формулы (I) тестировали в специфическом анализе in vitro на клеточную пролиферацию, описанном выше.
Как может быть оценено специалистом, все репрезентативные соединения, таким образом, являются особенно выгодными при противоопухолевой терапии.
Соединения настоящего изобретения могут быть введены либо в виде отдельных средств или, альтернативно, в комбинации с известными противораковыми лечениями, такими как лучевая терапия или химиотерапия, в комбинации с цитостатическими или цитотоксическими средствами, средствами типа антибиотиков, алкилирующими средствами, антиметаболитами, гормональными средствами, иммунологическими средствами, средствами типа интерферона, ингибиторами циклооксигеназы (например, ингибиторами COX-2), ингибиторами матриксметаллопротеазы, ингибиторами теломеразы, ингибиторами тирозинкиназы, средствами против рецептора фактора роста, средствами против HER, средствами против EGFR, средствами против ангиогенеза (например, ингибиторами ангиогенеза), ингибиторами фарнезилтрансферазы, ингибиторами пути передачи сигнала ras-raf, ингибиторами клеточного цикла, ингибиторами других cdk, тубулинсвязывающими средствами, ингибиторами топоизомеразы I, ингибиторами топоизомеразы II и подобными.
В такой комбинации продуктов, если она сформулирована в виде фиксированной дозы, используются соединения настоящего изобретения в диапазоне доз, описанных ниже, и другой фармацевтически активный ингредиент в утвержденном к применению диапазоне доз.
Соединения формулы (I) могут применяться последовательно с известными противораковыми средствами, когда составление комбинации является неуместным.
Соединения формулы (I) настоящего изобретения, подходящие для введения млекопитающим, например, людям, могут быть введены обычными путями, и уровень дозирования зависит от возраста, массы, состояния здоровья пациента и пути введения.
Например, подходящая доза, адаптированная для перорального введения соединения формулы (I), может составлять диапазон примерно от 1 до примерно 300 мг на дозу, от 1 до 5 раз в сутки. Соединения настоящего изобретения могут быть введены в различных дозированных формах, например, перорально, в форме таблеток, капсул, покрытых сахаром или пленкой таблеток, жидких растворов или суспензий; ректально в форме суппозиториев; парентерально, например, подкожно, внутримышечно или через внутривенное введение, и/или интратекальной и/или интраспинальной инъекцией или инфузией.
Настоящее изобретение также включает фармацевтические композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль в сочетании с фармацевтически приемлемым эксципиентом, которым может быть носитель или разбавитель.
Фармацевтические композиции, содержащие соединения настоящего изобретения, обычно получают, следуя общепринятым способам, и вводят в подходящей фармацевтической форме. Например, твердые формы для перорального введения могут содержать, вместе с активным соединением, разбавители, например, лактозу, декстрозу, сахарозу, целлюлозу, кукурузный крахмал или картофельный крахмал; лубриканты, например, оксид кремния, тальк, стеариновую кислоту, стеарат магния или кальция и/или полиэтиленгликоли; связующие агенты, например, крахмалы, гуммиарабик, желатинизированную метилцеллюлозу, карбоксиметилцеллюлозу или поливинилпирролидон; дезинтегрирующие агенты, например, крахмал, альгиновую кислоту, альгинаты или гликолят натрийкрахмала; шипучие смеси; красители; подсластители; смачивающие агенты, такие как лецитин, полисорбаты, лаурилсульфаты; и, как правило, нетоксичные и фармакологически неактивные вещества, используемые при составлении фармацевтических композиций. Такие фармацевтические препараты могут быть изготовлены известными способами, например, путем смешивания, гранулирования, таблетирования, способами покрытия сахаром или покрытия пленкой.
Жидкими дисперсиями для перорального введения могут быть, например, сиропы, эмульсии и суспензии. В качестве примера, сиропы могут содержать в качестве носителя сахарозу или сахарозу с глицерином и/или маннитом и сорбитом.
Суспензии и эмульсии могут содержать, в качестве примеров носителей, природную камедь, агар, альгинат натрия, пектин, метилцеллюлозу, карбксиметилцеллюлозу или поливиниловый спирт. Суспензии или растворы для внутримышечных инъекций могут содержать, вместе с активным соединением, фармацевтически приемлемый носитель, например, стерильную воду, оливковое масло, этилолеат, гликоли, например, пропиленгликоль, и, если требуется, подходящее количество гидрохлорида лидокаина. Растворы для внутривенных инъекций или инфузий могут содержать в качестве носителя стерильную воду или предпочтительно они могут быть в форме стерильных, водных, изотоничных, физиологических растворов, или они могут содержать пропиленгликоль в качестве носителя. Суппозитории могут содержать, вместе с активным соединением, фармацевтически приемлемый носитель, например, масло какао, полиэтиленгликоль, сложный эфир полиоксиэтиленсорбитана и жирной кислоты в качестве поверхностно-активного вещества или лецитин.
С целью лучшей иллюстрации настоящего изобретения, но без какого-либо его ограничения, приводятся следующие примеры.
ПРИМЕРЫ
Синтетическое получение некоторых соединений формулы (I) настоящего изобретения описано в следующих примерах. Соединения настоящего изобретения, полученные согласно следующим примерам, также характеризовали 1H-ЯМР и/или точными данными масс ESI(+).
1H-ЯМР спекты записывали при постоянной температуре 28°C на спектрометре Varian INOVA 400, работающем при 400,50 МГц и оборудованном 5 мм z-axis PFG Indirect Detection Probe (1H{15N-31P}).
Химические сдвиги выражали в соответствии с остаточными сигналами растворителя (ДМСО-d6: 2,50 ч./млн. для 1H, если не указано иное). Данные предоставлены следующим образом: химический сдвиг (δ), мультиплетность (с=синглет, д=дублет, т=триплет, кв=квартет, ушир.с.=широкий синглет, тд=триплет дублетов, дд=дублет дублетов, ддд=дублет дублетов дублетов, м=мультиплет, спт=септет), константы связи (J, Гц) и число протонов.
Точные данные масс ESI(+) получали на масс-спектрометре Waters Q-Tof Ultima, напрямую подключенном к Agilent 1100 ВЭЖХ микро-системе, как описано ранее (M. Colombo, F. Riccardi-Sirtori, V. Rizzo, Rapid Commun. Mass Spectrom. 2004, 18, 511-517).
В примерах ниже, а также по всему описанию, следующие аббревиатуры имеют следующие значения. Если нет указаний, то термины имеют их обычно принятые значения.
Пример 1
Стадия A1, стадия B3 (согласно A6a и A6b)
(8S,10S)-8-ацетил-1-фтор-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион [(I)] (соединение 1)
[R1=F, R2=CH3CO-, R3=CH3O-]
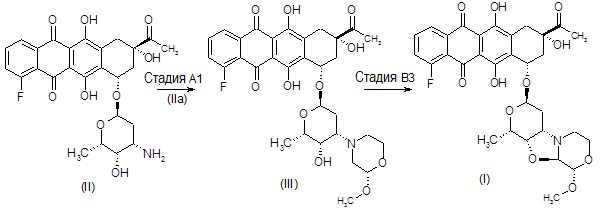
Стадия A1
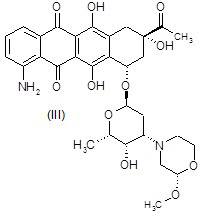
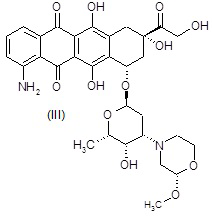
(1S,3S)-3-ацетил-10-фтор-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозид [(III)]
(1S,3S)-3-ацетил-10-фтор-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-3-амино-2,3,6-тридезокси-α-L-ликсогексопиранозид (70,0 мг, 0,136 ммоль) [полученный, как описано WO90/09392] растворяли в сухом ДМФА (3 мл); добавляли раствор диизопропилэтиламина (106 мг, 0,82 ммоль) в сухом ДМФА (2 мл) и раствор (1S)-2-йод-1-(2-йодэтокси)-1-метоксиэтана (IIa) (965 мг, 2,71 ммоль) в сухом ДМФА (10 мл). Реакционную смесь перемешивали при комнатной температуре в темноте в течение 48 часов, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Реакционную смесь разбавляли DCM и промывали водой. Органическую фазу сушили над безводным Na2SO4, растворитель выпаривали в вакууме, и остаток очищали флэш-хроматографией (элюент: EtOH/DCM; 0,2/9,8) на силикагеле (230-400 меш), с получением требуемого продукта (35 мг, красный воск).
ESI МС: m/z 616(MH+)
1H ЯМР (500 МГц, CHCl3-d) δ ч./млн. 1,39 (д, J=6,71 Гц, 3H), 1.78-1,85 (м, 2H), 2,09-2,14 (м, 1H), 2,46-2,56 (м, 3H), 2,61 (дд, J=11,41, 3,97 Гц, 1H), 3,03 (д, J=19,04 Гц, 1H), 3,27 (дд, J=19,10, 1,77 Гц, 1H), 3,40 (с, 3H), 3,57 (ддд, J=11,57, 5,34, 3,11 Гц, 1H), 3,70 (с, 1H), 3,92-3,99 (м, 1H), 4,04 (кв, J=6,47 Гц, 1H), 4,48-4,52 (м, 1H), 4,67 (с, 1H), 5,28-5,30 (м, 1H), 5,56 (ушир.с, 1H), 7,54 (дд, J=10,44, 8,48 Гц, 1H), 7,83 (тд, J=7,97, 4,58 Гц, 1H), 8,25 (д, J=7,69 Гц, 1H), 13,31 (с, 1H), 13,72 (с, 1H).
По аналогичной методике получали следующее соединение:
(1S,3S)-10-фтор-3,5,12-тригидрокси-3-(гидроксиацетил)-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозид [(III)]
ESI МС: m/z 632(MH+)
Стадия B3 (A6a)
(1S,3S)-3-ацетил-10-фтор-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-(3ξ)-2,3,6-тридезокси-3-[(2S)-2-метокси-4-оксидоморфолин-4-ил]-α-L-треогексопиранозид [(XX)]
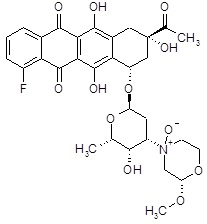
(1S,3S)-3-ацетил-10-фтор-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозид (28 мг, 0,045 ммоль) [полученный, как описано на стадии A1] растворяли в DCM (3,0 мл). Полученный раствор обрабатывали 0,1M раствором DMDO в ацетоне (0,8 мл) при комнатной температуре в течение 30 минут, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Реакционную смесь затем концентрировали досуха в вакууме, с получением требуемого промежуточного продукта (красный воск, 24,1 мг).
ESI МС: m/z 632(MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,23 (д, J=6,7 Гц, 3H), 1,96-2,00 (м, 1H), 2,10 (м, 1H), 2,35 (с, 3H), 2,33-2,38 (м, 1H), 2,56-2,64 (м, 2H), 2,94-3,00 (м, 1H), 3,07-3,12 (м, 1H), 3,13 -3,16 (м, 1H), 3,23-3,29 (м, 1H), 3,37 (с, 3H), 3,38-3,46 (м, 2H), 3,86-3,95 (м, 1H), 3,99 (кв, J=6,7 Гц, 1H), 4,14 (с, 1H), 4,22-4,29 (м, 1H), 4,32 (ушир.с, 1H), 4,91 (дд, J=8,1, 2,3 Гц, 1H), 5,20 (дд, J=4,6, 1,9 Гц, 1H), 5,60 (д, J=3,9 Гц, 1H), 7,62 (дд, JHH=8,3, JHF=10,8 Гц, 1H), 7,91 (м, 1H), 8,20 (д, JHH=7,7 Гц, 1H), 13,26 (ушир.с, 1H), 13,69 (ушир.с, 1H).
По аналогичной методике получали следующее соединение:
(1S,3S)-10-фтор-3,5,12-тригидрокси-3-(гидроксиацетил)-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-(3ξ)-2,3,6-тридезокси-3-[(2S)-2-метокси-4-оксидоморфолин-4-ил]-α-L-треогексопиранозид [(XX)]
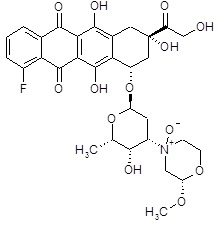
ESI МС: m/z 648(MH+)
Стадия B3 (A6b)
Указанное в заголовке соединение (соединение 1)
К раствору соединения (1S,3S)-3-ацетил-10-фтор-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-(3ξ)-2,3,6-тридезокси-3-[(2S)-2-метокси-4-оксидоморфолин-4-ил]-α-L-треогексопиранозида [(XX)] (20 мг, 0,032 ммоль) в 5,0 мл сухого CH3CN добавляли K2CO3 (13,2 мг, 0,096 ммоль) и хлорангидрид циануровой кислоты (11,8 мг, 0,064 ммоль). Реакционную смесь энергично перемешивали в темноте при комнатной температуре в течение 20 минут, до тех пор, пока исходные вещества прекращали детектироваться. Затем в реакционную смесь добавляли раствор 3-амино-1,2-пропандиола (17,5 мг, 0,192 ммоль) в воде (0,84 мл), и водную фазу экстрагировали DCM (4×10 мл). Объединенные органические фазы сушили над безводным Na2SO4, фильтровали и упаривали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией (AcOEt/толуол; 4/6) на силикагеле (230-400 меш), с получением 7,0 мг указанного в заголовке соединения в виде красного твердого вещества.
ESI МС: m/z 614(MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,29 (д, J=6,6 Гц, 3H), 1,68-1,73 (м, 1H), 1,86-1,91 (м, 1H), 2,05 (дд, J=14,8, 4,3 Гц, 1H), 2,34 (с, 3H), 2,42-2,47 (м, 1H), 2,67-2,81 (м, 2H), 2,93-2,98 (м, 1H), 3,05-3,11 (м, 1H), 3,37 (с, 3H), 3,42-3,47 (м, 1H), 3,52-3,58 (м, 1H), 3,71-3,76 (м, 1H), 4,03 (дд, J=7,1, 1,8 Гц, 1H), 4,06-4,12 (м, 1H), 4,26 (д, J=2,8 Гц, 1H), 4,53 (д, J=2,8 Гц, 1H), 4,54 (с, 1H), 5,20 (дд, J=4,3, 2,1 Гц, 1H), 5,35 (т, J=5,5 Гц, 1H), 7,60 (дд, JHH=8,3, JHF=11,6 Гц, 1H), 7,89 (м, 1H), 8,19 (дд, JHH=7,7, JHF=0,8 Гц, 1H), 13,25 (ушир.с, 1H), 13,61 (ушир.с, 1H).
Аналогично, с использованием подходящего исходного вещества, получали следующие соединения:
(8S,10S)-1-фтор-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион [(I)] (соединение 2) [R1=F, R2=HOCH2CO-, R3=CH3O-]
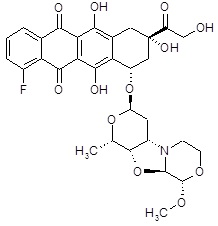
ESI МС: m/z 630(MH+)
(8S,10S)-8-ацетил-6,8,11-тригидрокси-1-[(2-гидроксиэтил)амино]-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион [(I)] (соединение 5) [R1=HO(CH2)2NH-, R2=CH3CO-, R3=CH3O-]
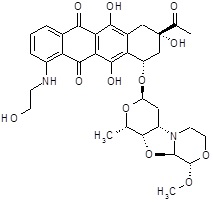
ESI МС: m/z 655(MH+)
(8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-[(2-гидроксиэтил)амино]-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион [(I)] (соединение 6) [R1=HO(CH2)2NH-, R2=HOCH2CO-, R3=CH3O-]
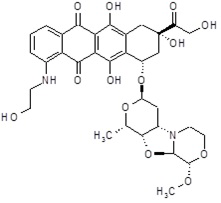
ESI МС: m/z 671(MH+)
(8S,10S)-8-ацетил-1-[(2-аминоэтил)амино]-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион [(I)] (соединение 7) [R1=H2N(CH2)2NH-, R2=CH3CO-, R3=CH3O-]
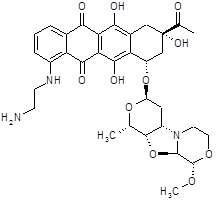
ESI МС: m/z 654(MH+)
(8S,10S)-1-[(2-аминоэтил)амино]-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион [(I)] (соединение 8) [R1=H2N(CH2)2NH-, R2=HOCH2CO-, R3=CH3O-]
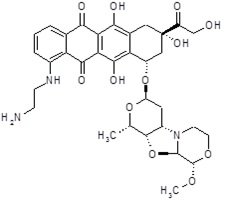
ESI МС: m/z 670(MH+)
Пример 2
Стадия A1, стадия B3 (согласно A6a и A6b)
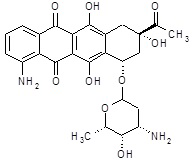
(8S,10S)-8-ацетил-1-амино-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион [(I)] (соединение 4)
[R1=NH2-, R2=CH3CO-, R3=CH3O-]
Стадия A1
(1S,3S)-3-ацетил-10-амино-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозид [(III)]
(1S,3S)-3-ацетил-10-амино-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-3-амино-2,3,6-тридезокси-α-L-ликсогексопиранозид (165,0 мг, 0,322 ммоль) [полученный, как описано ниже в примере 3] растворяли в сухом ДМФА (3,0 мл); добавляли раствор диизопропилэтиламина (221 мг, 1,71 ммоль) в сухом ДМФА (3 мл) и раствор (1S)-2-йод-1-(2-йодэтокси)-1-метоксиэтана (IIa) (2,0 г, 5,64 ммоль) в сухом ДМФА (10 мл). Реакционную смесь перемешивали при комнатной температуре в темноте в течение 48 часов, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Реакционную смесь разбавляли DCM и промывали водой. Органическую фазу сушили над безводным Na2SO4, растворитель выпаривали в вакууме, и остаток очищали флэш-хроматографией (элюент: EtOH/DCM; 0,2/9,8) на силикагеле (230-400 меш), с получением требуемого продукта (105,0 мг, красное твердое вещество).
ESI МС: m/z 613(MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,22-1,28 (м, 3H), 1,65-1,83 (м, 2H), 2,30-2,36 (м, 4H), 2,40 (дд, J=11,22, 4,94 Гц, 3H), 2,46-2,54 (м, 2H), 2,87-2,96 (м, 1H), 3,04-3,11 (м, 1H), 3,32 (с, 3H), 3,50 (ддд, J=11,34, 6,51, 2,83 Гц, 1H), 3,65 (ушир.с, 1H), 3,77-3,89 (м, 1H), 4,04 (д, J=6,51 Гц, 1H), 4,44 (дд, J=4,63, 2,41 Гц, 1H), 5,16 (д, J=1,99 Гц, 1H), 5,43-5,47 (м, 1H), 7,12-7,16 (м, 1H), 7,24 (ушир.с, 1H), 7,50-7,55 (м, 1H), 7,58-7,61 (м, 1H).
По аналогичной методике получали следующие соединения:
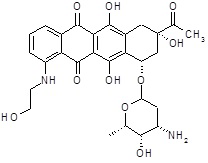
(1S,3S)-10-амино-3,5,12-тригидрокси-3-(гидроксиацетил)-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозид
ESI МС: m/z 629(MH+)
Аналогично, с использованием подходящего исходного вещества, получали следующие соединения:
(1S,3S)-3-ацетил-3,5,12-тригидрокси-10-[(2-гидроксиэтил)амино]-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-3-амино-2,3,6-тридезокси-L-ликсогексопиранозид
ESI МС: m/z 557(MH+)
(1S,3S)-3-ацетил-10-[(2-аминоэтил)амино]-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-3-амино-2,3,6-тридезокси-L-ликсогексопиранозид
ESI МС: m/z 556(MH+)
Защита
N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-5,12-диоксо-10-({2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозил}окси)-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамид
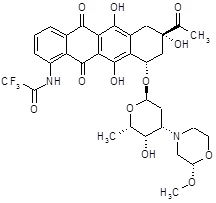
(1S,3S)-3-ацетил-10-амино-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозид [(III)] (80,0 мг, 0,130 ммоль) растворяли в сухом DCM (11 мл) и добаваляли трифторуксусный ангидрид (273,0 мг, 1,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в темноте в течение 30 минут, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Реакционную смесь разбавляли DCM и промывали насыщенным водным раствором NaHCO3 (3×10 мл) и затем водой (1×10 мл). Органическую фазу сушили над безводным Na2SO4, растворитель выпаривали в вакууме, и полученный таким образом остаток растирали в MeOH (10 мл) при комнатной температуре в течение 15 минут и затем упаривали в вакууме, с получением требуемого продукта (77,0 мг, красный воск).
ESI МС: m/z 709 (MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,25 (д, J=6,59 Гц, 3H), 1,76 (дд, J=8,68, 2,61 Гц, 2H), 2,27-2,45 (м, 8H), 2,52 (т, J=10,99 Гц, 2H), 2,96-3,03 (м, 1H), 3,08-3,15 (м, 1H), 3,30-3,33 (м, 3H), 3,50 (ддд, J=11,27, 6,57, 2,61 Гц, 1H), 3,66 (ушир.с, 1H), 3,79-3,87 (м, 1H), 4,05 (кв, J=6,62 Гц, 1H), 4,44 (дд, J=4,70, 2,35 Гц, 1H), 5,17 (д, J=2,27 Гц, 1H), 5,45 (с, 1H), 7,97 (т, J=8,15 Гц, 1H), 8,24 (д, J=7,50 Гц, 1H), 8,99 (д, J=8,18 Гц, 1H).
Стадия B3 (A6a)
N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-5,12-диоксо-10-({(3ξ)-2,3,6-тридезокси-3-[(2S)-2-метокси-4-оксидоморфолин-4-ил]-α-L-треогексопиранозил}окси)-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамид [(XX)]
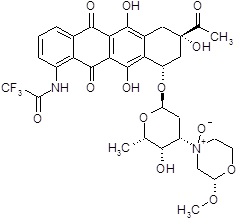
N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-5,12-диоксо-10-({2,3,6-тридезокси-3-[(2S)-2-метоксиморфолин-4-ил]-α-L-ликсогексопиранозил}окси)-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамид (72,0 мг, 0,102 ммоль) растворяли в DCM (6,4 мл). Полученный раствор обрабатывали 0,1M раствором DMDO в ацетоне (1,7 мл) при комнатной температуре в течение 30 минут, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Реакционную смесь затем концентрировали досуха в вакууме, с получением требуемого промежуточного продукта (красный воск, 73,0 мг).
ESI МС: m/z 725 (MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,23 (д, J=6,51 Гц, 3H), 2,32-2,39 (м, 4H), 2,57 (д, J=4,54 Гц, 1H), 2,74 (д, J=11,65 Гц, 1H), 2,96-3,02 (м, 1H), 3,08-3,15 (м, 1H), 3,25-3,45 (м, 7H), 3,57 (ушир.с, 1H), 3,92 (д, J=12,79 Гц, 1H), 4,04 (д, J=6,88 Гц, 1H), 4,18 (с, 1H), 4,21-4,28 (м, 1H), 4,92 (дд, J=8,21, 2,08 Гц, 1H), 5,19 (д, J=2,19 Гц, 1H), 5,61 (д, J=3,71 Гц, 1H), 7,97 (т, J=8,10 Гц, 1H), 8,23 (д, J=7,64 Гц, 1H), 8,98 (д, J=8,32 Гц, 1H).
По аналогичной методике может быть получено следующее соединение:
(1S,3S)-10-амино-3,5,12-тригидрокси-3-(гидроксиацетил)-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-(3ξ)-2,3,6-тридезокси-3-[(2S)-2-метокси-4-оксидоморфолин-4-ил]-α-L-треогексопиранозид [(XX)]
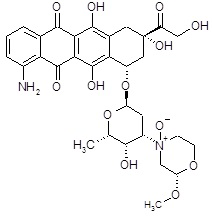
ESI МС: m/z 645(MH+)
Стадия B3 (A6b)
N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-5,12-диоксо-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамид [(I)]
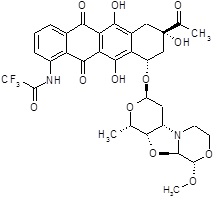
К раствору соединения N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-5,12-диоксо-10-({(3ξ)-2,3,6-тридезокси-3-[(2S)-2-метокси-4-оксидоморфолин-4-ил]-α-L-треогексопиранозил}окси)-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамида [(XX)] (60,0 мг, 0,083 ммоль) в 13 мл сухого CH3CN добавляли K2CO3 (34,4 мг, 0,249 ммоль) и хлорангидрид циануровой кислоты (30,6 мг, 0,166 ммоль). Реакционную смесь энергично перемешивали в темноте при комнатной температуре в течение 15 минут, до тех пор, пока исходные вещества прекращали детектироваться. Затем в реакционную смесь добавляли раствор 3-амино-1,2-пропандиола (45,3 мг, 0,5 ммоль) в воде (0,22 мл), и водную фазу экстрагировали DCM (4×10 мл). Объединенные органические фазы сушили над безводным Na2SO4, фильтровали и упаривали в вакууме. Неочищенный продукт очищали колоночной флэш-хроматографией (AcOEt/толуол; 4/6) на силикагеле (230-400 меш), с получением 12,0 мг указанного в заголовке соединения в виде красного твердого вещества.
ESI МС: m/z 707 (MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,29 (д, J=6,58 Гц, 4H), 1,70 (д, J=15,21 Гц, 1H), 1,90 (д, J=15,59 Гц, 2H), 2,04-2,08 (м, 2H), 2,45 (д, J=14,98 Гц, 1H), 2,69-2,76 (м, 1H), 2,77-2,83 (м, 1H), 2,97 (с, 1H), 3,08-3,14 (м, 2H), 3,38 (с, 4H), 3,45 (д, J=6,88 Гц, 2H), 3,56 (д, J=5,22 Гц, 2H), 3,74 (с, 1H), 4,04 (д, J=1,89 Гц, 2H), 4,09 (д, J=6,88 Гц, 1H), 4,26 (д, J=2,72 Гц, 1H), 4,52-4,54 (м, 2H), 5,22 (ушир.с, 1H), 5,36 (т, J=5,60 Гц, 1H), 7,98 (т, J=8,06 Гц, 1H), 8,26 (д, J=7,87 Гц, 1H), 9,00 (д, J=8,10 Гц, 1H).
Удаление защиты
Указанное в заголовке соединение (соединение 4)

Промежуточный N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-5,12-диоксо-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамид (4,8 мг, 0,00679 ммоль) охлаждали до 0°C и добавляли водный раствор 0,1Н NaOH (0,5 мл). Реакционную смесь перемешивали в темноте при 0°C в течение 15 минут, до тех пор, пока исходные вещества прекращали детектироваться. Реакционную смесь разбавляли H2O и экстрагировали DCM (4×5 мл). Объединенные органические фазы промывали насыщенным водным раствором NaCl (1×10 мл), сушили над безводным Na2SO4, фильтровали и упаривали в вакууме, с получением 4,0 мг указанного в заголовке соединения в виде красного твердого вещества.
ESI МС: m/z 611 (MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,70 (дт, J=15,06, 5,82 Гц, 2H), 1,87 (дт, J=15,17, 5,54 Гц, 1H), 2,34 (с, 3H), 2,43 (д, J=14,41 Гц, 1H), 2,68-2,81 (м, 2H), 2,91-2,96 (м, 1H), 3,07 (д, J=18,66 Гц, 1H), 3,37 (с, 3H), 3,44 (кв, J=5,87 Гц, 1H), 3,51-3,61 (м, 2H), 3,74 (ддд, J=11,63, 8,25, 2,96 Гц, 1H), 4,01-4,04 (м, 1H), 4,06-4,13 (м, 1H), 4,26 (д, J=2,66 Гц, 1H), 4,54 (д, J=2,58 Гц, 1H), 5,21 (ушир.с, 1H), 5,37 (т, J=5,61 Гц, 1H), 7,16 (д, J=8,57 Гц, 1H), 7,54 (т, J=7,89 Гц, 1H), 7,62 (д, J=7,06 Гц, 1H).
Аналогично, с использованием подходящих исходных веществ получали следующее соединение:
(8S,10S)-1-амино-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-дион (соединение 3)
[R1=NH2-, R2=HOCH2CO-, R3=CH3O-]
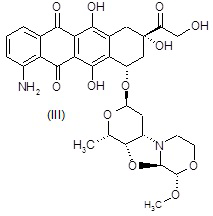
ESI МС: m/z 627(MH+)
Пример 3
Получение промежуточных продуктов формулы (II):
Стадия G2, удаление защиты, защита, стадия G3, удаление защиты
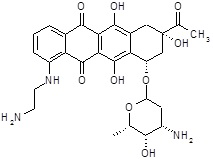
Синтез (1S,3S)-3-ацетил-10-амино-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-3-амино-2,3,6-тридезокси-L-ликсогексопиранозида
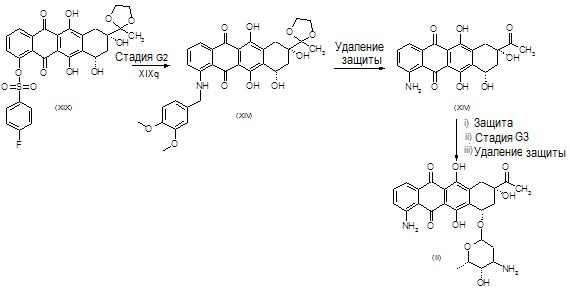
Стадия G2
Синтез промежуточного (8S,10S)-1-[(3,4-диметоксибензил)амино]-6,8,10,11-тетрагидрокси-8-(2-метил-1,3-диоксолан-2-ил)-7,8,9,10-тетрагидротетрацен-5,12-диона (XIV)
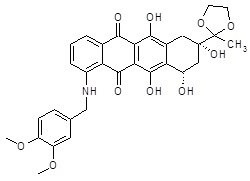
К раствору (8S,10S)-6,8,10,11-тетрагидрокси-8-(2-метил-1,3-диоксолан-2-ил)-5,12-диоксо-5,7,8,9,10,12-гексагидротетрацен-1-ил-4-фторбензолсульфоната (400 мг, 0,682 ммоль) [полученного, как описано в GB2215332] в ТГФ (10 мл) добавляли 3,4-диметоксибензиламин (0,532 мг, 3,1 ммоль). Полученный раствор нагревали до 60°C и перемешивали в течение 24 часов в темноте. Затем растворитель частично удаляли в вакууме, темно-фиолетовый осадок собирали фильтрованием, промывали ТГФ (3 мл) и зитем Et2O (10 мл). Полученное твердое вещество окончательно сушили в вакуумной печи при 30°C, с получением указанного в заголовке промежуточного соединения (188 мг, выход=48%).
Аналогично, с использованием подходящих аминов получали следующие соединения:
(8S,10S)-6,8,10,11-тетрагидрокси-1-[(2-гидроксиэтил)амино]-8-(2-метил-1,3-диоксолан-2-ил)-7,8,9,10-тетрагидротетрацен-5,12-дион
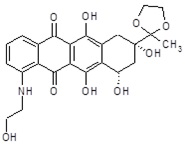
ESI МС: m/z 472(MH+)
1H ЯМР (499,75 МГц, ДМСО-d6) δ ч./млн. 1,33 (с, 3H), 1,82 (дд, J=14,3, 4,3 Гц, 1H), 2,20 (д, J=14,3 Гц, 1H), 2,67 (д, J=18,7 Гц, 1H), 3,10 (д, J=18,7 Гц, 1H), 3,45-3,49 (м, 2H), 3,68-3,72 (м, 2H), 3,92-4,01 (м, 4H), 5,00 (т, J=5,1 Гц, 1H), 5,05-5,11 (м, 1H), 5,35 (д, J=7,6 Гц, 1H), 5,44 (с, 1H), 7,35 (д, J=8,7 Гц, 1H), 7,56 (д, J=6,8 Гц, 1H), 7,70 (дд, J=8,7, 6,8 Гц, 1H), 9,61 (т, J=5,1 Гц, 1H), 13,52 (ушир.с, 1H), 13,74 (ушир.с, 1H).
(8S,10S)-1-[(2-аминоэтил)амино]-6,8,10,11-тетрагидрокси-8-(2-метил-1,3-диоксолан-2-ил)-7,8,9,10-тетрагидротетрацен-5,12-дион
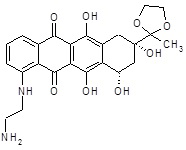
ESI МС: m/z 471(MH+)
Удаление защиты
Синтез промежуточного (8S,10S)-8-ацетил-1-амино-6,8,10,11-тетрагидрокси-7,8,9,10-тетрагидротетрацен-5,12-диона (XIV)
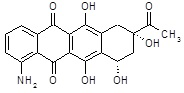
К охлажденной трифторуксусной кислоте (2 мл) добавляли (8S,10S)-1-[(3,4-диметоксибензил)амино]-6,8,10,11-тетрагидрокси-8-(2-метил-1,3-диоксолан-2-ил)-7,8,9,10-тетрагидротетрацен-5,12-дион (133 мг, 0,230 ммоль) и 2 капли анизола. Полученный раствор перемешивали при 5°C в течение 20 минут и затем при комнатной температуре в течение 2 часов до тех пор, пока исходные вещества прекращали детектироваться. Реакционную смесь разбавляли водой (5 мл), нейтрализовали раствором NaHCO3, затем водную фазу экстрагировали DCM (3×50 мл). Объединенные органические фазы сушили над безводным Na2SO4, фильтровали, растворитель выпаривали в вакууме, и неочищенный продукт обрабатывали Et2O (10 мл). Темно-фиолетовый осадок собирали фильтрованием и сушили в вакуумной печи при 30°C, с получением требуемого промежуточного продукта (82 мг, выход=93%).
ESI МС: m/z 384(MH+)
1H ЯМР (400,5 МГц, ДМСО-d6) δ ч./млн. 1,99 (дд, J=14,4, 4,6 Гц, 1H), 2,13-2,19 (м, 1H), 2,30 (с, 3H), 2,88-2,95 (м, 1H), 2,98-3,05 (м, 1H), 5,07 (м, 1H), 5,29 (ушир.с, 1H), 6,07 (с, 1H), 7,24 (дд, J=8,3, 1,1 Гц, 1H), 7,51 (дд, J=7,3, 1,1 Гц, , 1H), 7,55-7,60 (м, 1H), 8,05 (ушир.с, 2H), 13,49 (ушир.с, 1H), 13,85 (ушир.с, 1H).
Аналогично, с использованием подходящего исходного соединения могут быть получены следующие соединения:
(8S,10S)-8-ацетил-6,8,10,11-тетрагидрокси-1-[(2-гидроксиэтил)амино]-7,8,9,10-тетрагидротетрацен-5,12-дион
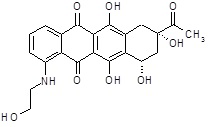
ESI МС: m/z 428(MH+)
1H ЯМР (500 МГц, ДМСО-d6) δ ч./млн. 1,98 (дд, J=14,2, 4,6 Гц, 1H), 2,16 (д, J=14,2 Гц, 1H), 2,31 (с, 3H), 2,88-2,94 (м, 1H), 2,98-3,04 (м, 1H), 3,46-3,49 (м, 2H), 3,68-3,72 (м, 2H), 5,01 (т, J=5,1 Гц, 1H), 5,05-5,10 (м, 1H), 5,30 (д, J=6,7 Гц, 1H), 6,10 (с, 1H), 7,35 (д, J=8,7 Гц, 1H), 7,56 (д, J=7,1 Гц, 1H), 7,70 (дд, J=8,7, 7,1 Гц, 1H), 9,62 (т, J=5,1 Гц, 1H), 13,47 (ушир.с, 1H), 13,76 (ушир.с, 1H)
(8S,10S)-8-ацетил-1-[(2-аминоэтил)амино]-6,8,10,11-тетрагидрокси-7,8,9,10-тетрагидротетрацен-5,12-дион
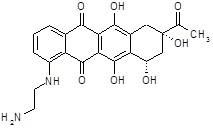
ESI МС: m/z 427(MH+)
1H ЯМР (500 МГц, ДМСО-d6) δ ч./млн. 1,97 (дд, J=14,2, 4,6 Гц, 1H), 2,14 (д, J=14,2 Гц, 1H), 2,31 (с, 3H), 2,88-2,94 (м, 1H), 2,98-3,04 (м, 1H), 3,05-3,22 (м, 4H), 5,05-5,10 (м, 1H), 5,30 (д, J=6,7 Гц, 1H), 6,10 (с, 1H), 7,35 (д, J=8,7 Гц, 1H), 7,54 (д, J=7,1 Гц, 1H), 7,70 (дд, J=8,7, 7,1 Гц, 1H), 9,60 (т, J=5,1 Гц, 1H), 13,46 (ушир.с, 1H), 13,76 (ушир.с, 1H).
Защита
Синтез промежуточного N-[(8S,10S)-8-ацетил-6,8,10,11-тетрагидрокси-5,12-диоксо-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамида
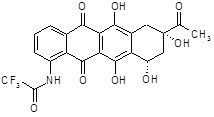
Промежуточный (8S,10S)-8-ацетил-1-амино-6,8,10,11-тетрагидрокси-7,8,9,10-тетрагидротетрацен-5,12-дион (600,0 мг, 1,56 ммоль) растворяли в сухом DCM (120 мл) и добавляли трифторуксусный ангидрид (1,2 мл). Реакционную смесь перемешивали при комнатной температуре в темноте в течение 5 минут, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Реакционную смесь разбавляли DCM (100 мл) и промывали насыщенным водным раствором NaHCO3 (3×100 мл) и затем водой (1×100 мл). Органическую фазу сушили над безводным Na2SO4, и растворитель удаляли в вакууме. Полученный таким образом остаток очищали флэш-хроматографией (элюент: CH3COCH3/DCM; 0,3/9,7) на силикагеле (230-400 меш), с получением требуемого продукта (494,1 мг, красное твердое вещество).
ESI МС: m/z 480(MH+)
1H ЯМР (500 МГц, CHCl3-d) δ ч./млн. 2,22 (дд, J=14,5, 4,9 Гц, 1H), 2,36-2,41 (м, 1H), 2,45 (с, 3H), 3,01 (д, J=18,7 Гц, 1H), 3,23 (дд, J=18,7, 2,2 Гц, 1H), 3,81 (д, J=5,2 Гц, 1H), 4,54 (с, 1H), 5,35 (м, 1H), 7,93 (дд, J=8,4, 7,7 Гц, 1H), 8,29 (дд, J=7,7, 1,1 Гц, 1H), 9,12 (дд, J=8,4, 1,1 Гц, 1H), 13,29 (ушир.с., 1H), 13,29 (с, 1H), 13,46 (с, 1H).
Аналогично, с использованием подходящих исходных веществ получали следующее соединение:
N-(2-{[(8S,10S)-8-ацетил-6,8,10,11-тетрагидрокси-5,12-диоксо-5,7,8,9,10,12-гексагидротетрацен-1-ил]амино}этил)-2,2,2-трифторацетамид
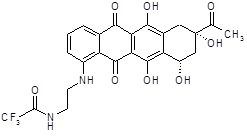
ESI МС: m/z 523(MH+)
Стадия G3
Синтез N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-5,12-диоксо-10-({2,3,6-тридезокси-3-[(трифторацетил)амино]-L-ликсогексопиранозил}окси)-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамида
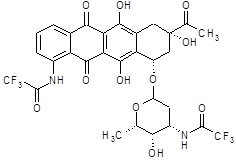
В высушенной трехгорлой круглодонной колбе в атмосфере аргона промежуточный N-[(8S,10S)-8-ацетил-6,8,10,11-тетрагидрокси-5,12-диоксо-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамид (480,0 мг, 1,0 ммоль) растворяли в сухом DCM (110 мл) и добавляли порошкообразные молекулярные сита (4Å, 20,0 мг). Реакционную смесь охлаждали до 10°C; затем добавляли раствор трифторметансульфоната серебра (334,0 мг, 1,3 ммоль) в сухом Et2O (15 мл) и раствор 2,3,6-тридезокси-4-O-(трифторацетил)-3-[(трифторацетил)амино]-δ-L-ликсогексопиранозилхлорида (511,4 мг, 1,43 ммоль) в сухом DCM (15 мл). Реакционную смесь перемешивали при 10°C в темноте в течение 45 минут, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Добаляли насыщенный водный раствор NaHCO3 (50 мл), и реакционную смесь перемешивали при комнатной температуре в течение 30 минут, затем фильтровали через рыхлый слой целита. Органическую фазу отделяли, промывали водой и сушили над безводным Na2SO4. Растворитель удаляли в вакууме, и полученный таким образом остаток охлаждали до 0°C и обрабатывали MeOH (20 мл) и твердым NaHCO3 в течение 15 минут. Растворитель выпаривали в вакууме, и остаток очищали флэш-хроматографией (элюент: CH3COCH3/DCM; 0,5/9,5) на силикагеле (230-400 меш), с получением требуемого продукта (320,0 мг, красное твердое вещество).
ESI МС: m/z 705(MH+)
1H ЯМР (500 МГц, CH3CN-d3) δ ч./млн. 1,22 (д, J=6,47 Гц, 3H), 1,77 (дд, J=13,18, 4,64 Гц, 1H), 2,00 (тд, J=13,15, 3,97 Гц, 1H), 2,10 (д, J=10,13 Гц, 1H), 2,33-2,35 (м, 1H), 2,87-3,00 (м, 1H), 3,01-3,13 (м, 1H), 3,22 (ушир.с, 1H), 3,61 (ушир.с, 1H), 4,09-4,16 (м, 1H), 4,22 (кв, J=6,47 Гц, 1H), 4,29 (с, 1H), 5,10 (ушир.с, 1H), 5,40 (д, J=3,54 Гц, 1H), 7,32 (д, J=7,81 Гц, 1H), 7,89 (т, J=7,63 Гц, 1H), 8,11 (д, J=7,08 Гц, 1H), 8,89 (д, J=8,30 Гц, 1H), 13,08 (ушир.с, 2H).
Аналогично, с использованием подходящего исходного вещества, получали следующие соединения:
N-(2-{[(8S,10S)-8-ацетил-6,8,11-тригидрокси-5,12-диоксо-10-({2,3,6-тридезокси-3-[(трифторацетил)амино]-L-ликсогексопиранозил}окси)-5,7,8,9,10,12-гексагидротетрацен-1-ил]амино}этил)-2,2,2-трифторацетамид
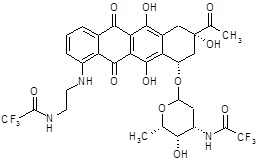
ESI МС: m/z 748(MH+)
(1S,3S)-3-ацетил-3,5,12-тригидрокси-10-[(2-гидроксиэтил)амино]-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-2,3,6-тридезокси-3-[(трифторацетил)амино]-L-ликсогексопиранозид
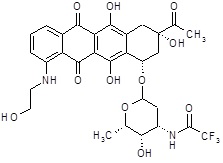
ESI МС: m/z 653(MH+)
Удаление защиты
Указанное в заголовке соединение (II)
Промежуточный N-[(8S,10S)-8-ацетил-6,8,11-тригидрокси-5,12-диоксо-10-({2,3,6-тридезокси-3-[(трифторацетил)амино]-α-L-ликсогексопиранозил}окси)-5,7,8,9,10,12-гексагидротетрацен-1-ил]-2,2,2-трифторацетамид (340,2 мг, 0,432 ммоль) охлаждали до 0°C в атмосфере аргона и обрабатывали 0,1Н водным раствором NaOH (12 мл). Реакционную смесь перемешивали в темноте при 0°C течение 1 часа, до тех пор, пока исходные вещества прекращали детектироваться (анализы ВЭЖХ). Реакционную смесь разбавляли DCM (50 мл), промывали насыщенным водным раствором NaHCO3 (3×30 мл), затем водой (1×30 мл) и окончательно насыщенным раствором NaCl (1×30 мл). Органическую фазу сушили над безводным Na2SO4, растворитель удаляли в вакууме, с получением требуемого продукта (180,0 мг, красное твердое вещество).
ESI МС: m/z 513(MH+)
1H ЯМР (500 МГц, ДМСО-d6) δ ч./млн. 1,14 (д, J=6,52 Гц, 2H), 1,47 (дд, J=12,61, 4,33 Гц, 1H), 1,60 (д, J=3,30 Гц, 1H), 2,06-2,21 (м, 2H), 2,24-2,27 (м, 3H), 2,86 (д, J=12,58 Гц, 1H), 2,88-3,01 (м, 2H), 3,28 (ушир.с, 1H), 4,09 (д, J=6,29 Гц, 1H), 4,45 (ушир.с, 1H), 4,94 (т, J=4,22 Гц, 1H), 5,19 (д, J=3,53 Гц, 1H), 5,44 (с, 1H), 7,24 (д, J=8,28 Гц, 1H), 7,50 (д, J=7,13 Гц, 1H), 7,51-7,52 (м, 0H), 7,55-7,59 (м, 1H), 8,06 (ушир.с, 2H).
Аналогично, с использованием подходящего исходного вещества, получали следующие соединения:
(1S,3S)-3-ацетил-10-[(2-аминоэтил)амино]-3,5,12-тригидрокси-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-3-амино-2,3,6-тридезокси-L-ликсогексопиранозид
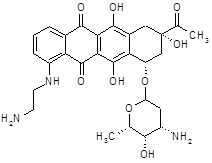
ESI МС: m/z 556(MH+)
(1S,3S)-3-ацетил-3,5,12-тригидрокси-10-[(2-гидроксиэтил)амино]-6,11-диоксо-1,2,3,4,6,11-гексагидротетрацен-1-ил-3-амино-2,3,6-тридезокси-L-ликсогексопиранозид
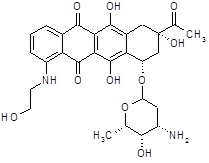
ESI МС: m/z 557(MH+).
| название | год | авторы | номер документа |
|---|---|---|---|
| ФУНКЦИОНАЛИЗИРОВАННЫЕ ПРОИЗВОДНЫЕ МОРФОЛИНИЛАНТРАЦИКЛИНА | 2015 |
|
RU2715902C2 |
| КОНЪЮГАТЫ ПРОИЗВОДНОГО АНТРАЦИКЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ПРОТИВООПУХОЛЕВЫХ СОЕДИНЕНИЙ | 2009 |
|
RU2523419C2 |
| N-ПИПЕРОНИЛЬНЫЕ ПРОИЗВОДНЫЕ ДАУНОРУБИЦИНА, ОБЛАДАЮЩИЕ АНТИПРОЛИФЕРАТИВНЫМИ СВОЙСТВАМИ | 2017 |
|
RU2642068C1 |
| РЕГУЛЯТОРЫ-ПРОИЗВОДНЫЕ СТЕРОИДОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2797408C2 |
| ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, ОБЛАДАЮЩАЯ УЛУЧШЕННЫМ ПРОТИВООПУХОЛЕВЫМ ДЕЙСТВИЕМ И/ИЛИ СНИЖЕННЫМИ ПОБОЧНЫМИ ЭФФЕКТАМИ, СОДЕРЖАЩАЯ ПРОТИВООПУХОЛЕВЫЙ АГЕНТ И ПРОИЗВОДНОЕ ГИДРОКСАМОВОЙ КИСЛОТЫ | 1998 |
|
RU2254129C2 |
| ВОДОРАСТВОРИМЫЕ АНАЛОГИ СС-1065 И ИХ КОНЪЮГАТЫ | 2007 |
|
RU2489423C2 |
| РЕАКЦИЯ ПРИНСА И ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ПРИМЕНИМЫЕ ПРИ СИНТЕЗЕ МАКРОЛИДОВ ГАЛИХОНДРИНОВОГО РЯДА И ИХ АНАЛОГОВ | 2017 |
|
RU2777913C2 |
| МОДУЛЯТОРЫ SHIP1 И ОТНОСЯЩИЕСЯ К НИМ СПОСОБЫ | 2014 |
|
RU2679805C2 |
| ПРОИЗВОДНОЕ ХИНАЗОЛИНОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И ПРИМЕНЕНИЯ | 2017 |
|
RU2730500C2 |
| ПРОИЗВОДНОЕ ВИТАМИНА D, СОДЕРЖАЩЕЕ ЦИКЛИЧЕСКИЙ АМИН В БОКОВОЙ ЦЕПИ | 2021 |
|
RU2819389C1 |
Изобретение относится к соединениям формулы (I)
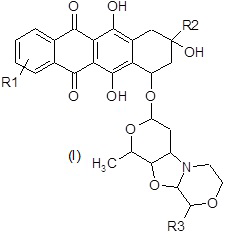 , а также к фармацевтическим композициям на их основе, способу их получения и применению таких соединений или содержащих их фармацевтических композиций при лечении заболеваний, таких как рак. Технический результат: получены новые соединения, обладающие цитотоксической активностью и пригодные для применения в лечении расстройств клеточной пролиферации. 5 н. и 4 з.п. ф-лы, 3 пр.
, а также к фармацевтическим композициям на их основе, способу их получения и применению таких соединений или содержащих их фармацевтических композиций при лечении заболеваний, таких как рак. Технический результат: получены новые соединения, обладающие цитотоксической активностью и пригодные для применения в лечении расстройств клеточной пролиферации. 5 н. и 4 з.п. ф-лы, 3 пр.
1. Соединение формулы (I)
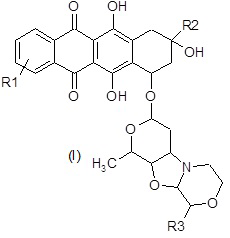
где
R1 представляет собой NR4R5;
R2 представляет собой группу COR9;
R3 представляет собой C1-C2 алкокси;
R4 и R5 независимо представляют собой водород, NR7R8-C1-C3 алкил, R6O-C1-C3 алкил, R7R8N-C1-C3 алкилкарбонил или R6O-C1-C3 алкилкарбонил;
R6, R7 и R8 независимо представляют собой водород или C1-C3 алкил;
R9 представляет собой OR6 или прямой C1-C3 алкил,
или его фармацевтически приемлемая соль.
2. Соединение формулы (Ia) по п.1
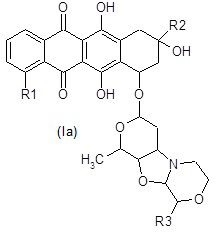
где R1 представляет собой фтор или NR4R5, где R4 представляет собой водород, а R5 представляет собой водород или NR7R8-C1-C3 алкила, R6O-C1-C3 алкила, R7R8N-C1-C3 алкилкарбонила или R6O-C1-C3 алкилкарбонила.
3. Соединение формулы (Ia) по п. 1 или 2, которое выбрано из группы, состоящей из:
(8S,10S)-1-амино-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона,
(8S,10S)-8-ацетил-1-амино-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона,
(8S,10S)-8-ацетил-6,8,11-тригидрокси-1-[(2-гидроксиэтил)амино]-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона,
(8S,10S)-6,8,11-тригидрокси-8-(гидроксиацетил)-1-[(2-гидроксиэтил)амино]-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона,
(8S,10S)-8-ацетил-1-[(2-аминоэтил)амино]-6,8,11-тригидрокси-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона и
(8S,10S)-1-[(2-аминоэтил)амино]-6,8,11-тригидрокси-8-(гидроксиацетил)-10-{[(1S,3R,4aS,9S,9aR,10aS)-9-метокси-1-метилоктагидро-1H-пирано[4',3':4,5][1,3]оксазоло[2,3-c][1,4]оксазин-3-ил]окси}-7,8,9,10-тетрагидротетрацен-5,12-диона.
4. Фармацевтическая композиция, содержащая терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли по п.1 и по меньшей мере один фармацевтически приемлемый эксципиент, носитель или разбавитель, пригодная для использования в лечении рака.
5. Продукт, содержащий соединение формулы (I) или его фармацевтически приемлемую соль по п.1 и одно или более химиотерапевтических средств, в виде комбинированного препарата для одновременного, раздельного или последовательного применения в противораковой терапии.
6. Соединение формулы (I) по п.1 для применения в качестве лекарственного средства для лечения рака.
7. Применение соединения формулы (I) по п.1 в производстве лекарственного средства для лечения рака.
8. Применение по п.7, где рак выбран из группы, состоящей из карцином, в том числе груди, толстой кишки, печени, легких, яичников, шейки матки, острого лимфоцитарного лейкоза, лимфомы Ходжкина, неходжкинской лимфомы, острой и хронической миелоцитарной лейкемии и рабдомиосаркому.
9. Способ получения соединения формулы (I) по п.1, включающий:
сначала взаимодействие
соединения формулы (VII)
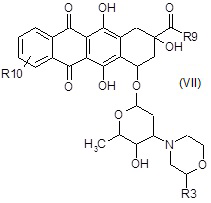
где R3 и R10 определены в п.1, и R9 представляет собой OR6 или NR7R8, где R6, R7 и R8 определены в п.1,
или
соединения формулы (VIII)
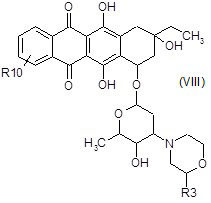
где R3 и R10 являются такими, как определено выше,
или
соединения формулы (III)
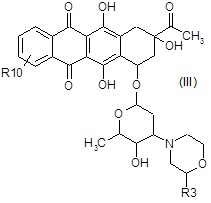
где R3 и R10 являются такими, как определено выше,
или
соединения формулы (IX)
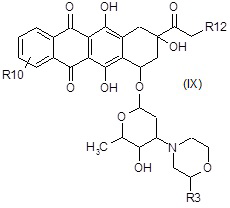
где R3 и R10 являются такими, как определено выше, и R12 представляет собой группу, выбранную из прямого или разветвленного C1-C4 алкила, NR7R8-C1-C4 алкила и R6O-C1-C4 алкила,
или
соединения формулы (X)
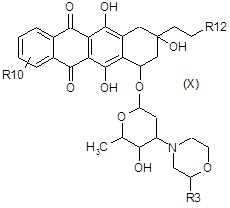
где R3, R10 и R12 являются такими, как определено выше,
или
соединения формулы (XI) или (XIa):
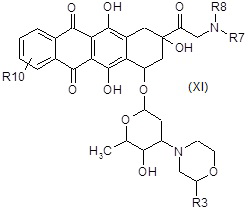 ,
, 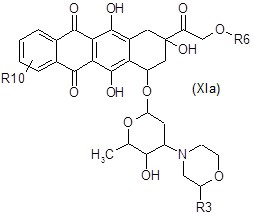
где R3, R6, R7, R8 и R10 являются такими, как определено выше,
или
соединения формулы (XII) или (XIIa)
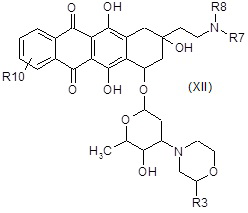 ,
, 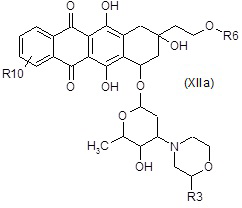
где R3, R6, R7, R8 и R10 являются такими, как определено выше,
или
соединения формулы (V)
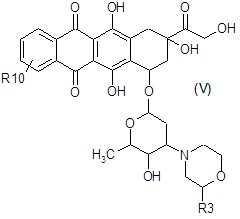
где R3 и R10 являются такими, как определено выше,
или
соединения формулы (XIII)
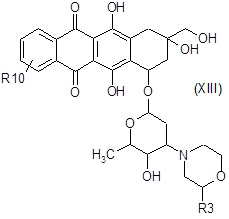
где R3 и R10 являются такими, как определено выше,
с DMDO;
затем
взаимодействие полученного в результате соединения формулы (XX):
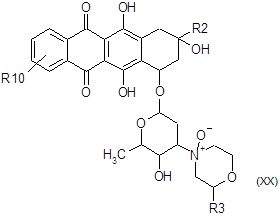
где R3, R10 и R2 определены в п.1, с хлорангидридом циануровой кислоты или с солью железа (II), и, наконец, если требуется, удаление защитной группы или групп с получением соединения формулы (I), как определено выше,
необязательно, преобразование такого соединения формулы (I) в его фармацевтически приемлемую соль или преобразование соли в свободное соединение формулы (I).
| WO 9802446 A1, 22.01.1998 | |||
| АВТОМАТИЧЕСКАЯ БЕССТУПЕНЧАТАЯ МЕХАНИЧЕСКАЯ ПЕРЕДАЧА | 2003 |
|
RU2247885C1 |
| АНТРАЦИКЛИН ГЛИКОЗИД И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 1992 |
|
RU2081878C1 |
| EA 200400729 A1, 30.12.2004. | |||

