Настоящее изобретение относится к способу получения оптически активных энантиомеров пирлиндола в форме свободного основания или в форме фармацевтически приемлемых солей.
Оптически активные энантиомеры пирлиндола по настоящему изобретению представляют собой (R)-пирлиндол и (S)-пирлиндол.
Продукты, полученные по настоящему изобретению, являются энантиомерно чистыми и применимы в медицине.
УРОВЕНЬ ТЕХНИКИ
Пирлиндол, 2,3,3a,4,5,6-гексагидро-1H-8-метилпиразин [3,2,1-j,k]карбазол, является тетрациклическим соединением формулы I
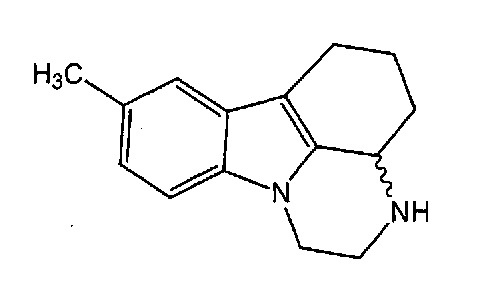
(I)
Пирлиндол является обратимым ингибитором моноаминоксидазы A, в настоящее время применяющимся в качестве лекарственного средства для лечения депрессии.
Пирлиндол содержит асимметрический атом углерода, и это означает, что существуют два энантиомера, (S)-пирлиндол и (R)-пирлиндол.
В современном уровне техники описаны разные методики разделения энантиомеров пирлиндола. Например, в публикации The Journal of Pharmaceutical and Biomedical Analysis, 18(1998) 605-614, "Enantiomeric separation of pirlindole by liquid chromatography using different types of chiral stationary phases", Ceccato et al. раскрыто разделение энантиомеров пирлиндола с помощью жидкостной хроматографии (LC) с использованием трех разных хиральных стационарных фаз.
Кроме того, в публикации The Journal of Pharmaceutical and Biomedical Analysis 27(2002) 447-455, "Automated determination of pirlindole enantiomers in plasma by on-line coupling of a pre-column packed with restricted access material to a chiral liquid chromatographic column", Chiap et al., раскрыто применение предколонки, содержащей материал с ограниченным доступом, для очистки образца, соединенной с колонкой, содержащей хиральную стационарную фазу на основе целлюлозы, предназначенную для разделения и количественного анализа энантиомеров.
По данным предшествующего уровня техники, Chirality 11:261-266 (1999) оказались неудачными все попытки получения энантиомеров пирлиндола путем селективной кристаллизации с оптически активными кислотами, и их удалось получить лишь в лабораторном масштабе (несколько граммов) в виде гидрохлоридной соли с помощью методики получения производных совместно с препаративной хроматографией.
Характеристики способа, раскрытого в предшествующем уровне техники, определенным образом ограничивают его применение в промышленном или полупромышленном масштабе вследствие необходимости проведения крупномасштабного разделения с помощью хроматографии, что затруднительно осуществить и характеризуется плохой воспроизводимостью.
В данной области техники необходим поиск новых способов, которые конкурентоспособны и могли бы легко использоваться в промышленности для получения энантиомеров пирлиндола в форме их свободного основания или в форме фармацевтически приемлемых солей.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
В настоящем изобретении (R)-пирлиндол и (S)-пирлиндол, в отличие от данных предшествующего уровня техники, можно получить в форме свободного основания или в форме фармацевтически приемлемой соли путем кристаллизации (рац)-пирлиндола в форме свободного основания с оптически активными кислотами, и это позволяет затем получить их в форме свободного основания или в форме фармацевтически приемлемых солей.
Поэтому объектом настоящего изобретения является способ получения оптически активных (R)- и (S)-энантиомеров пирлиндола в форме свободного основания или в форме фармацевтически приемлемой соли, отличающийся тем, что проводят разделение путем кристаллизации с оптически активными кислотами (рац)-пирлиндола в форме свободного основания.
Оптически активные энантиомеры пирлиндола представляют собой энантиомерно чистые (S)-пирлиндол или (R)-пирлиндол.
Поэтому другим объектом настоящего изобретения является способ, отличающийся тем, что он включает следующие стадии:
i) растворение гидрохлорида (рац)-пирлиндола в водном растворителе с последующей экстракцией с помощью хлорированного растворителя и полным удалением растворителя с получением (рац)-пирлиндола в форме свободного основания;
ii) растворение (рац)-пирлиндола, полученного на стадии i), в органическом растворителе с последующим добавлением оптически активной кислоты для разделения;
iii) перемешивание в течение от 15 мин до 2 ч суспензии, образованной на стадии ii), с осаждением диастереоизомерной соли;
iv) фильтрование полученной диастереоизомерной соли и ее очистка путем суспендирования в органическом растворителе с получением (S)-энантиомера пирлиндола или (R)-энантиомера пирлиндола в форме фармацевтически приемлемой соли, образованной с оптически активной кислотой; и необязательно,
v) получение энантиомерно чистого (S)-пирлиндола и/или (R)-пирлиндола в виде свободного основания путем растворения продукта, полученного на стадии iv), в водном растворителе, последующая экстракция с помощью хлорированного растворителя и полное удаление растворителя; и, кроме того, необязательно,
vi) получение (S)-пирлиндола или (R)-пирлиндола в форме фармацевтически приемлемых солей присоединения кислот путем образования соли энантиомерно чистого (S)-пирлиндола и/или (R)-пирлиндола в форме свободного основания, полученного на стадии v), с фармацевтически приемлемой кислотой с образованием фармацевтически приемлемой соли присоединения кислоты (S)-энантиомера пирлиндола или (R)-энантиомера пирлиндола.
Дополнительным объектом настоящего изобретения также является способ, определенный выше, в котором оптически активная кислота, используемая на стадии ii), выбрана из группы, сотоящей из: (R)-миндальной кислоты, (R)-(+)-α-метокси-α-трифторметилфенилуксусной кислоты, (1R,3S)-(+)-камфорной кислоты, D(+)-яблочной кислоты, (S)-миндальной кислоты, (S)-(-)-α-метокси-α-трифторметилфенилуксусной кислоты, (1S,3R)-(+)-камфорной кислоты или L(-)-яблочной кислоты.
Другим объектом настоящего изобретения также является способ, определенный выше, в котором органический растворитель, используемый на стадиях ii) и iv), выбран из группы, состоящей из: метанола, этанола, пропанола, 1-бутанола, 2-бутанола, трет-бутилового спирта, 2-бутанона, ацетона, метилэтилкетона, метилизобутилкетона, диметилсульфоксида, 1,2-дихлорэтана, диэтилового эфира, диметилового эфира, диметилформамида, метил-трет-бутилового эфира, 2-пропанола, пиридина, толуола, ксилола или их смеси в любом соотношении.
Кроме того, другим объектом настоящего изобретения является способ, определенный выше, отличающийся тем, что полученное соединение представляет собой энантиомерно чистый (S)-пирлиндол в виде соли (R)-манделата, (R)-пирлиндол в виде соли (S)-манделата, гидробромидную соль (S)-пирлиндола, гидробромидную соль (R)-пирлиндола, цитратную соль (S)-пирлиндола, цитратную соль (R)-пирлиндола, мезилатную соль (S)-пирлиндола, мезилатную соль (R)-пирлиндола, (R)-(+)-α-метокси-α-трифторметилфенилацетатную соль (R)-пирлиндола и (R)-(+)-α-метокси-α-трифторметилфенилацетатную соль (S)-пирлиндола.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении (R)-пирлиндол и (S)-пирлиндол, в отличие от данных предшествующего уровня техники, можно получить в форме свободного основания или в форме фармацевтически приемлемой соли путем кристаллизации (рац)-пирлиндола в форме свободного основания с оптически активными кислотами и это позволяет затем получить их в форме свободного основания или в форме фармацевтически приемлемых солей.
В соответствии с настоящим изобретением более предпочтительно, если (R)-пирлиндол и (S)-пирлиндол в форме свободного основания или в форме фармацевтически приемлемой соли можно получить путем кристаллизации (рац)-пирлиндола в форме свободного основания с оптически активными кислотами в органическом растворителе и необязательно с последующим получением соли с образованием фармацевтически приемлемых солей с фармацевтически приемлемыми кислотами.
Авторы настоящего изобретения установили, что при некоторых особых условиях проведения способа действительно можно провести разделение рацемического диастереоизомерного пирлиндола.
Существенные условия проведения указанного выше способа включают предварительное выделение рацемического пирлиндола, без которого неожиданно оказалось невозможным эффективное проведение разделения, период перемешивания (размешивания) после добавления оптически активной кислоты, что следует особенно тщательно контролировать для исключения рацемизации, и конкретный тип органического растворителя, использующийся на стадии добавления оптически активной кислоты и очистку.
Авторы настоящего изобретения установили, что в течение непродолжительных периодов перемешивания после добавления оптически активной кислоты к (рац)-пирлиндолу в форме свободного основания разделение неэффективно и при более продолжительных периодах перемешивания происходит рацемизация.
Как правило, перемешивание проводят в течение периода времени продолжительностью от 15 мин до 2 ч. Предпочтительно, перемешивание проводят в течение периода времени продолжительностью от 30 мин до 1 ч.
Авторы настоящего изобретения также установили, что выбор конкретного органического растворителя для стадии добавления оптически активной кислоты (разделения) и очистки весьма важен, поскольку он заметно влияет на эффективность и выход процедуры разделения.
Способ, предлагаемый в настоящем изобретении, в дополнение к тому, что он является способом, который легко применять в промышленном масштабе, в отличие от способов, известных в данной области техники, дает возможность впервые получить (R)-пирлиндол и (S)-пирлиндол в форме свободного основания или фармацевтически приемлемой соли в количествах, которые позволяют провести доклинические и клинические исследования.
Молекула пирлиндола содержит вторичную аминогруппу, которая обладает основным характером и поэтому может образовывать соли присоединения кислот, которые являются фармацевтически приемлемыми солями.
Способ, описанный в настоящем изобретении, позволяет получить (R)-пирлиндол и (S)-пирлиндол оба в форме их свободного основания и в форме фармацевтически приемлемых солей.
Для задач настоящего изобретения соединение считается энантиомерно чистым, если энантиомерная чистота, рассчитанная с помощью хиральной хроматографии, больше или равна 97%.
Способ, предлагаемый в настоящем изобретении, если не учитывать гидрохлорид (рац)-пирлиндола, включает следующие стадии:
i) растворение гидрохлорида (рац)-пирлиндола в водном растворителе с последующей экстракцией с помощью хлорированного растворителя и полным удалением растворителя с получением (рац)-пирлиндола в форме свободного основания;
ii) растворение (рац)-пирлиндола, полученного на стадии i), в органическом растворителе с последующим добавлением оптически активной кислоты для разделения;
iii) перемешивание в течение от 15 мин до 2 ч суспензии, образованной на стадии ii), с осаждением диастереоизомерной соли;
iv) фильтрование полученной диастереоизомерной соли и ее очистка путем суспендирования в органическом растворителе с получением (S)-энантиомера пирлиндола или (R)-энантиомера пирлиндола в форме фармацевтически приемлемой соли, образованной с оптически активной кислотой;
В дополнение к подробно описанным стадиям способа и если получаемыми продуктами являются (S)-энантиомер пирлиндола или (R)-энантиомер пирлиндола в виде свободного основания или в виде фармацевтически приемлемой соли присоединения подходящих органических и неорганических кислот, указанный способ необязательно включает по меньшей мере одну из следующих стадий:
v) получение энантиомерно чистого (S)-пирлиндола и/или (R)-пирлиндола в виде свободного основания путем растворения продукта, полученного на стадии iv), в водном растворителе, последующая экстракция с помощью хлорированного растворителя и полное удаление растворителя; и
vi) получение (S)-пирлиндола или (R)-пирлиндола в форме фармацевтически приемлемых солей присоединения кислот путем образования соли энантиомерно чистого (S)-пирлиндола и/или (R)-пирлиндола в форме свободного основания, полученного на стадии v), с фармацевтически приемлемой кислотой.
Типичные проводимые операции, получаемые выходы, отсутствие стадий, проводимых в жестких условиях (например, при повышенной температуре) и, в особенности, отсутствие необходимости проведения разделения с помощью хроматографии делают этот способ особенно подходящим для применения в промышленности и уникальным и отличающимся от предшествующего уровня техники.
Для задач настоящего изобретения термин "фармацевтически приемлемая соль" означает такие соли, которые в рамках тщательной медицинской оценки являются подходящими для применения при соприкосновении с тканями и органами людей и низших животных без проявления токсичности, раздражения, аллергической реакции и т. п. и соответствуют разумному отношению польза/риск. Фармацевтически приемлемые соли включают фармацевтически приемлемые соли присоединения кислот, образованные с органическими и неорганическими кислотами и фармацевтически приемлемые соли, образованные с оптически активными кислотами в соответствии с настоящим изобретением.
Типичные соли присоединения кислот включают, но не ограничиваются, ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, фумарат, глицерофосфат, гемисульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидройодид, 2-гидроксиэтансульфонат (изетионат), лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, тартрат, тиоцианат, фосфат, глутамат, бикарбонат, п-толуолсульфонат и ундеканоат.
Неограничивающие примеры кислот, которые можно использовать для образования фармацевтически приемлемых солей присоединения кислот с соединениями, предлагаемые в настоящем изобретении, включают неорганические кислоты, такие как хлористоводородную кислоту, бромистоводородную кислоту (HBr) серную кислоту и фосфорную кислоту, и органические кислоты, такие как лимонную кислоту, безводную лимонную кислоту, миндальную кислоту, янтарную кислоту и метансульфоновую кислоту.
Для задач настоящего изобретения "оптически активные кислоты" включают (S)-миндальную кислоту, (R)-миндальную кислоту, (R)-(+)-α-метокси-α-трифторметилфенилуксусную кислоту, (S)-(-)-α-метокси-α-трифторметилфенилуксусную кислоту, (1S,3R)-(-)-камфорную кислоту, (1R, 3S)-(+)-камфорную кислоту, L-(-)-яблочную кислоту, D-(+)-яблочную кислоту или аналогичные хорошо известные в данной области техники оптически активные кислоты.
При использовании в настоящем изобретении термин "водный растворитель" означает воду или смеси воды с другими органическими растворителями, в которых вода является основным компонентом, т. е. содержится в количестве, составляющем не менее 95% (об./об.).
При использовании в настоящем изобретении термин "органический растворитель" означает растворители, обычно использующиеся в органической химии, или их смеси в любых соотношениях.
Неограничивающие примеры органических растворителей, использующихся на стадиях ii) и iv), способа, предлагаемого в настоящем изобретении, выбраны из группы, включающей: метанол, этанол, пропанол, 1-бутанол, 2-бутанол, трет-бутиловый спирт, 2-бутанон, ацетон, метилэтилкетон, метилизобутилкетон, диметилсульфоксид, 1,2-дихлорэтан, диэтиловый эфир, диметиловый эфир, диметилформамид, метил-трет-бутиловый эфир, 2-пропанол, пиридин, толуол, ксилол и т. п. и их смеси в любом соотношении.
Предпочтительными являются следующие растворители: этанол, метанол, 1-бутанол, 2-бутанол, трет-бутиловый спирт, ацетон, метилэтилкетон и изопропанол, а также их смеси в любых соотношениях, такие как изопропанол/ацетон (1:1), этанол/ацетон (1:1), этанол/метилизобутилкетон (1:1) и этанол/1-бутанол (1:4).
При использовании в настоящем изобретении термин "хлорированный растворитель" означает хлороформ, дихлорметан, метиленхлорид, трихлорметан или тетрахлорид углерода, или их смеси в любых соотношениях.
Соединениями, полученными в соответствии с настоящим изобретением, являются:
(S)-манделат (S)-пирлиндола;
(R)-манделат (S)-пирлиндола;
(R)-(+)-α-метокси-α-трифторметилфенилацетат (R)-пирлиндола;
гидробромид (R)-пирлиндола;
мезилат (R)-пирлиндола;
цитрат (S)-пирлиндола;
цитрат (R)-пирлиндола;
(R)-пирлиндол (свободное основание);
(S)-пирлиндол (свободное основание);
Примерами других соединений, которые можно получить способом, предлагаемым в настоящем изобретении, являются:
гидробромид (S)-пирлиндола
мезилат (S)-пирлиндола
бензолсульфонат (S)-пирлиндола
п-толуолсульфонат (R)-пирлиндола
бисульфат (S)-пирлиндола
оксалат (R)-пирлиндола
малеат (R)-пирлиндола
ацетат (S)-пирлиндола
глутамат (S)-пирлиндола
лактат (S)-пирлиндола
адипат (R)-пирлиндола
бензоат (R)-пирлиндола
малат (S)-пирлиндола
ПРИМЕРЫ
Приведенные ниже примеры предназначены для иллюстрации настоящего изобретения и их не следует рассматривать в качестве ограничивающих настоящее изобретение.
ПРИМЕР 1
(S)-Манделат (R)-пирлиндола
100 г (0,38 моль) Гидрохлорида (R,S)-пирлиндола растворяли в 16 л деионизированной воды при комнатной температуре. К раствору добавляли 42,4 г (0,4 моль) безводного карбоната натрия и содержимое перемешивали в течение 1 ч.
Указанный выше раствор экстрагировали с помощью 3×4 л дихлорметана и объединенные органические фазы сушили над сульфатом натрия и выпаривали в вакууме досуха.
К концентрату добавляли 2 л ацетона.
К указанному выше раствору при перемешивании добавляли раствор 27,6 г (0,18 моль) (S)-миндальной кислоты в 150 мл ацетона.
Перемешивание продолжали в течение 45 мин.
Осадившийся продукт отфильтровывали, промывали с помощью 2×100 мл ацетона и сушили в вакууме при 35°C-45°C.
Указанный выше продукт суспендировали в этаноле (250 мл) и затем фильтровали и сушили в вакууме при 35°C-45°C и получали 48,5 г (0,13 моль) (S)-манделата (R)-пирлиндола (выход=68%). Хиральная ВЭЖХ (высокоэффективная жидкостная хроматография) (энантиомерная чистота=98,2%).
ПРИМЕР 2
(R)-Манделат (S)-пирлиндола
По такой же методике, как в примере 1 (с тем отличием, что длительность перемешивания после добавления хиральной кислоты равнялась 60 мин), с использованием в качестве исходного вещества 100 г (0,38 моль) гидрохлорида (R,S)-пирлиндола и с использованием 27,6 г (0,18 моль) (R)-миндальной кислоты получали 45,6 г (0,12 моль) (R)-манделата (S)-пирлиндола (выход=63%). Хиральная ВЭЖХ (энантиомерная чистота=98,7%).
ПРИМЕР 3
(R)-Манделат (S)-пирлиндола
По такой же методике, как в примере 1, с тем отличием, что в качестве органического растворителя использовали смесь изопропанол/ацетон (1:1) и длительность перемешивания после добавления хиральной кислоты равнялась 35 мин, с использованием в качестве исходного вещества 10 г (0,038 моль) гидрохлорида (R,S)-пирлиндола и с использованием 2,8 г (0,018 моль) (R)-миндальной кислоты получали 4,1 г (0,011 моль) (R)-манделата (S)-пирлиндола (выход=57,9%). Хиральная ВЭЖХ (энантиомерная чистота=98,1%).
ПРИМЕР 4
(R)-(+)-α-Метокси-α-трифторметилфенилацетат (R)-пирлиндола
По такой же методике, как в примере 1, с тем отличием, что в качестве органического растворителя использовали смесь этанол/ацетон (1:1), длительность перемешивания после добавления хиральной кислоты равнялась 55 мин и в качестве оптически активной кислоты использовали (R)-(+)-α-метокси-α-трифторметилфенилуксусную кислоту (8,3 г) (0,018 моль), с использованием в качестве исходного вещества 10 г (0,038 моль) гидрохлорида (R,S)-пирлиндола получали 4,8 г (0,010 моль) (R)-(+)-α-метокси-α-трифторметилфенилацетата (R)-пирлиндола (выход=52,6%). Хиральная ВЭЖХ (энантиомерная чистота=97,7%).
ПРИМЕР 5
Гидробромид (R)-пирлиндола
Продукт, полученный в примере 1 (10 г, 0,027 моль) растворяли в 550 мл деионизированной воды. Водную фазу экстрагировали с помощью 3×300 мл хлороформа. Объединенные органические фазы сушили над сульфатом натрия, выпаривали досуха в вакууме и добавляли 200 мл ацетона.
К указанному выше раствору при перемешивании добавляли раствор 6 мл HBr (48% водный раствор) (0,04 моль).
Высушенный осажденный продукт фильтровали, промывали с помощью 2×10 мл ацетона и сушили в вакууме при 35°C-45°C.
Указанный выше продукт суспендировали в смеси этанол/метилизобутилкетон (1:1) (250 мл) и затем фильтровали и сушили в вакууме при 35°C-45°C и получали 6,5 г (0,021 моль) гидробромида (R)-пирлиндола (выход=77,8%). Хиральная ВЭЖХ (энантиомерная чистота=97,9%).
ПРИМЕР 6
Цитрат (R)-пирлиндола
Продукт, полученный в примере 1 (10 г, 0,027 моль), растворяли в 550 мл деионизированной воды. Водную фазу экстрагировали с помощью 3×300 мл трихлорэтана. Объединенные органические фазы сушили над сульфатом натрия, выпаривали досуха в вакууме и добавляли 200 мл ацетона.
К указанному выше раствору при перемешивании добавляли 7,7 г безводной лимонной кислоты (0,04 моль).
Высушенный осажденный продукт отфильтровывали, промывали с помощью 2×10 мл ацетона и сушили в вакууме при 35°C-45°C.
Указанный выше продукт суспендировали в смеси этанол/1-бутанол (1:4) (250 мл) и затем отфильтровывали и сушили в вакууме при 35°C-45°C и получали 9,2 г (0,020 моль) цитрата (R)-пирлиндола (выход=74,1%). Хиральная ВЭЖХ (энантиомерная чистота=97,6%).
ПРИМЕР 7
Мезилат (R)-пирлиндола
С использованием в качестве исходного вещества 10 г (S)-манделата (R)-пирлиндола, полученного в примере 1, и по методике, описанной в примере 5, с использованием метансульфоновой кислоты в качестве фармацевтически приемлемой кислоты получали 7,4 г (0,023 моль) мезилата (R)-пирлиндола (выход=85,2%). Хиральная ВЭЖХ (энантиомерная чистота=98,0%).
ПРИМЕР 8
Гидробромид (S)-пирлиндола
С использованием в качестве исходного вещества 10 г (R)-манделата (S)-пирлиндола, полученного в примере 2, с использованием бромистоводородной кислоты в качестве фармацевтически приемлемой кислоты и по методике, описанной в примере 6, получали 7,4 г (0,024 моль) гидробромида (S)-пирлиндола (выход=88,9%). Хиральная ВЭЖХ (энантиомерная чистота=98,2%).
ПРИМЕР 9
Мезилат (S)-пирлиндола
С использованием в качестве исходного вещества 10 г (R)-манделата (S)-пирлиндола, полученного в примере 2, и по методике, описанной в примере 6, с использованием метансульфоновой кислоты в качестве фармацевтически приемлемой кислоты получали 6,8 г (0,021 моль) мезилата (S)-пирлиндола (выход=77,8%). Хиральная ВЭЖХ (энантиомерная чистота=98,0%).
ПРИМЕР 10
Цитрат (S)-пирлиндола
С использованием в качестве исходного вещества 10 г (R)-манделата (S)-пирлиндола, полученного в примере 2, и по методике, описанной в примере 6, с использованием лимонной кислоты в качестве фармацевтически приемлемой кислоты получали 9,5 г (0,021 моль) цитрата (R)-пирлиндола (выход=77,8%). Хиральная ВЭЖХ (энантиомерная чистота=98,5%).
ПРИМЕР 11
(R)-Пирлиндол (свободное основание)
Продукт, полученный в примере 1 (2 г, 0,005 моль) растворяли в 110 мл деионизированной воды. Водную фазу экстрагировали с помощью 3×75 мл дихлорметана. Объединенные органические фазы сушили над сульфатом натрия, выпаривали в вакууме до полного удаления растворителя и выдерживали при 0°C/5°C в течение ночи. Происходила кристаллизация. Получали 1,1 г (0,0049 моль) (R)-пирлиндола (свободное основание) (выход=98%). Хиральная ВЭЖХ (энантиомерная чистота=98,3%).
ПРИМЕР 12
(S)-Пирлиндол (свободное основание)
Продукт, полученный в примере 2 (2 г, 0,005 моль), растворяли в 110 мл деионизированной воды. Водную фазу экстрагировали с помощью 3×75 мл трихлорэтана. Объединенные органические фазы сушили над сульфатом натрия, выпаривали в вакууме до полного удаления растворителя и выдерживали при 0°C/5°C в течение ночи. Происходила кристаллизация. Получали 1,1 г (0,0049 моль) (S)-пирлиндола (свободное основание) (выход=98%). Хиральная ВЭЖХ (энантиомерная чистота=97,8%).
| название | год | авторы | номер документа |
|---|---|---|---|
| ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ЭНАНТИОМЕРОВ ПИРЛИНДОЛА ДЛЯ ПРИМЕНЕНИЯ В МЕДИЦИНЕ | 2014 |
|
RU2706688C2 |
| (S)-ПИРЛИНДОЛ И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ДЛЯ ПРИМЕНЕНИЯ В МЕДИЦИНЕ | 2014 |
|
RU2695647C2 |
| (R)-ПИРЛИНДОЛ И ЕГО ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫЕ СОЛИ ДЛЯ ПРИМЕНЕНИЯ В МЕДИЦИНЕ | 2014 |
|
RU2695607C2 |
| СЛОЖНЫЕ ЭФИРЫ АМИНОКИСЛОТ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ПРОДУКТЫ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1998 |
|
RU2197489C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРНО ЧИСТЫХ СОЕДИНЕНИЙ ИМИДАЗОЛИЛА, ЭНАНТИОМЕРНО ЧИСТАЯ КИСЛАЯ АДДИТИВНАЯ СОЛЬ ИМИДАЗОЛИЛА И D- ИЛИ L-ПИРОГЛУТАМИНОВОЙ КИСЛОТЫ, МОНОГИДРАТ ГИДРОХЛОРИДА ЭНАНТИОМЕРНО ЧИСТЫХ СОЕДИНЕНИЙ ИМИДАЗОЛИЛА | 1996 |
|
RU2162085C2 |
| СПОСОБ ПОЛУЧЕНИЯ ЭНАНТИОМЕРА АМЛОДИПИНА ВЫСОКОЙ ОПТИЧЕСКОЙ ЧИСТОТЫ | 2005 |
|
RU2376288C2 |
| СПОСОБ РАЦЕМИЗАЦИИ ОПТИЧЕСКИ ЧИСТОГО ИЛИ ОБОГАЩЕННОГО ПИПЕРАЗИН-2- ТРЕТ.БУТИЛКАРБОКСАМИДНОГО СУБСТРАТА И РАЦЕМИЧЕСКИЙ 2-ТРЕТ-БУТИЛКАРБОКСАМИД-4-(3- ПИКОЛИЛ)ПИПЕРАЗИН | 1995 |
|
RU2135482C1 |
| ОПТИЧЕСКИ ЧИСТЫЕ NA, MG, LI, K ИЛИ СА СОЛИ (-)-5-МЕТОКСИ-2[[(4-МЕТОКСИ-3,5-ДИМЕТИЛ-2-ПИРИДИНИЛ)МЕТИЛ] СУЛЬФИНИЛ]-1H-БЕНЗИМИДАЗОЛА, СПОСОБ ИХ ПОЛУЧЕНИЯ, ФАРМКОМПОЗИЦИЯ НА ИХ ОСНОВЕ И ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ | 1994 |
|
RU2137766C1 |
| ДИМЕТИЛБЕНЗОФУРАНЫ И ДИМЕТИЛБЕНЗОПИРАНЫ, ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ 5-НТ-АНТАГОНИСТОВ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ | 1993 |
|
RU2116072C1 |
| КОМПОЗИЦИИ И СПОСОБЫ ПРИМЕНЕНИЯ (R)-ПРАМИПЕКСОЛА | 2007 |
|
RU2491068C2 |
Изобретение относится к новому способу получения оптически активных энантиомеров пирлиндола, выбранных из группы, состоящей из энантиомерно чистых (S)-пирлиндола и (R)-пирлиндола, в форме свободного основания или в форме фармацевтически приемлемой соли, отличающийся тем, что проводят разделение путем кристаллизации с оптически активными кислотами (рац)-пирлиндола в форме свободного основания, где оптически активные кислоты выбраны из группы, состоящей из: (R)-миндальной кислоты, (R)-(+)-α-метокси-α-трифторметилфенилуксусной кислоты и (S)-миндальной кислоты. Технический результат: разработан новый способ получения оптически активных энантиомеров пирлиндола, который является обратимым ингибитором моноаминооксидазы А и может применяться для лечения депрессии. 13 з.п. ф-лы, 12 пр.
1. Способ получения оптически активных энантиомеров пирлиндола, выбранных из группы, состоящий из энантиомерно чистых (S)-пирлиндола и (R)-пирлиндола, в форме свободного основания или в форме фармацевтически приемлемой соли, отличающийся тем, что проводят разделение путем кристаллизации с оптически активными кислотами (рац)-пирлиндола в форме свободного основания, где оптически активные кислоты выбраны из группы, состоящей из: (R)-миндальной кислоты, (R)-(+)-α-метокси-α-трифторметилфенилуксусной кислоты и (S)-миндальной кислоты.
2. Способ по п. 1, отличающийся тем, что он включает следующие стадии:
i) растворение гидрохлорида (рац)-пирлиндола в водном растворителе с последующей экстракцией с помощью хлорированного растворителя и полным удалением растворителя с получением (рац)-пирлиндола в форме свободного основания;
ii) растворение (рац)-пирлиндола, полученного на стадии i), в органическом растворителе с последующим добавлением оптически активной кислоты для разделения;
iii) перемешивание в течение от 15 мин до 2 ч суспензии, образованной на стадии ii), с осаждением диастереоизомерной соли;
iv) фильтрование полученной диастереоизомерной соли и ее очистка путем суспендирования в органическом растворителе с получением (S)-энантиомера пирлиндола или (R)-энантиомера пирлиндола в форме фармацевтически приемлемой соли, образованной с оптически активной кислотой; и необязательно,
v) получение энантиомерно чистого (S)-пирлиндола и/или (R)-пирлиндола в виде свободного основания путем растворения продукта, полученного на стадии iv), в водном растворителе, последующей экстракции с помощью хлорированного растворителя и полного удаления растворителя; и необязательно,
vi) получение (S)-пирлиндола или (R)-пирлиндола в форме фармацевтически приемлемых солей присоединения с кислотами путем образования соли энантиомерно чистого (S)-пирлиндола и/или (R)-пирлиндола в форме свободного основания, полученного на стадии v), с фармацевтически приемлемой кислотой с образованием фармацевтически приемлемой соли присоединения с кислотой (S)-энантиомера пирлиндола или (R)-энантиомера пирлиндола.
3. Способ по любому из пп. 1, 2, в котором органический растворитель, используемый на стадиях ii) и iv), выбран из группы, состоящей из: этанола, изопропанола и ацетона.
4. Способ по любому из предыдущих пунктов, отличающийся тем, что полученное соединение представляет собой энантиомерно чистый (S)-пирлиндол в виде соли (R)-манделата.
5. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистый (R)-пирлиндол в виде соли (S)-манделата.
6. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистую гидробромидную соль (S)-пирлиндола.
7. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистую гидробромидную соль (R)-пирлиндола.
8. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистую цитратную соль (S)-пирлиндола.
9. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистую цитратную соль (R)-пирлиндола.
10. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистую мезилатную соль (S)-пирлиндола.
11. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистую мезилатную соль (R)-пирлиндола.
12. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистую (R)-(+)-α-метокси-α-трифторметилфенилацетатную соль (R)-пирлиндола.
13. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистый (R)-пирлиндол в форме свободного основания.
14. Способ по любому из пп. 1-3, отличающийся тем, что полученное соединение представляет собой энантиомерно чистый (S)-пирлиндол в форме свободного основания.
| RU 2004115690 A, 10.11.2005 | |||
| Pascal De Tullio et al., First Preparative Enantiomer Resolution of Pirlindole, a Potent Antidepressant Drug, Helvetica Chimica Acta, 81, 3-4, стр.: 539 - 547, 1998 | |||
| Chiap P et | |||
| al., Automated determination of pirlindole enantiomers in plasma by on-line coupling of a pre-column packed with restricted access material to a chiral liquid chromatographic column, Journal of Pharmaceutical and Biomedical Analysis, 27, 3-4, стр.: 447-455, 2002. |

