Область техники
Настоящее изобретение относится к способу извлечения аполипопротеина A-I (Apo A-I) из белковой фракции, содержащей Apo A-I.
Уровень техники
Человеческий аполипопротеин A-I представляет собой основной белковый компонент (HDL) и является важным липопротеином крови. Он синтезируется в печени и кишечнике и отвечает за физиологическую функцию HDL в крови; удаление холестерина из периферических тканей, возвращение его либо в печень, либо к другим липопротеинам посредством механизма, известного как "обратный транспорт холестерина" (RCT).
Четкая корреляция между повышенным уровнем холестерина в сыворотке и развитием ишемической болезни сердца неоднократно подтверждалась на основе эпидемиологических и долгосрочных исследований.
Следовательно, Apo A-I, входящий в состав HDL, предположительно оказывает противовоспалительное действие и препятствует возникновению и развитию CHD. Кроме того, показано, что Apo A-I уменьшает уровень липопротеинов низкой плотности (LDL) в крови и, как известно, связывается с эндотоксинами, играя, таким образом, важную роль в антиэндотоксической функции HDL.
"Защитная" роль HDL и Apo A-I, как основного компонента HDL, была подтверждена в ряде исследований, что делает Apo A-I перспективным кандидатом (в частности, в составе восстановленных HDL) для применения в способах лечения атеросклероза, острого коронарного синдрома (ACS), воспаления, в качестве антитоксического средства, в производстве лекарственных средств, направленных на печень, и т.д.
В настоящее время плазму крови человека собирают в больших количествах и обрабатывают с получением отдельных фракций, некоторые из которых содержат аполипопротеин A-I. Фракционирование можно проводить с использованием этанола по способу, изначально разработанному в Соединенных Штатах и известному как метод Кона или метод Кона-Онкли [E. J. Cohn et al., J. Am. Chem. Soc. 68, 459-475, 1946; J. L. Qncley et al., J. Am. Chem. Soc. 71, 541-550, 1949]. Фракции плазмы, содержащие аполипопротеины, также можно получить с помощью модификации данного метода по Кистлеру-Нитчману [H. Nitschmann et al., Helv. Chim. Acta 37, 866-873, 1954; P. Kistler and H. Nitschmann, Vox Sang 7, 414-424, 1962]. Оба метода основаны на дифференциальном осаждении спиртом.
В литературе описаны разные методы извлечения аполипопротеина A-I из фракций, полученных осаждением этанолом:
Например, в US 5089602 описан способ получения аполипопротеинов из фракций плазмы или сыворотки человеческой крови путем ресуспендирования фракций в водном буферном растворе в диапазоне рН от 3 до 5 или от 6 до 9. Нежелательные примеси осаждают путем добавления короткоцепочечного алифатического спирта. Для осаждения примесей используют буферы с высоким содержанием этанола (68-96% этанола). Возможную агрегацию липопротеинов подавляют путем применения повышенной температуры, слабощелочных значений рН, или путем добавления хаотропных средств или поверхностно-активных веществ, которые затем удаляют методом гель-фильтрации. Для связывания примесей используют стадию анионообменной хроматографии, при этом аполипопротеины проходят через сорбент.
Kim et al., Manufacturing and Shelf Stability of Reconstituted High-density Lipoprotein for Infusion Therapy, Biotechnology and Bioprocess Engineering 16, 785-792 (2011), описывают применение буферов для экстракции, содержащих мочевину в концентрации 6 М.
В WO 98/07751 описано применение сочетания двух вышеуказанных методов для извлечения аполипопротеина A (Apo A-I) или аполипопротеина Е (ApoE) из человеческой плазмы. Указанный документ раскрывает способ, включающий в себя, по меньшей мере, одну стадию предварительной очистки путем применения буфера для экстракции, содержащего 8 М мочевину, и дополнительную стадию очистки с использованием анионообменной хроматографии. В WO 98/07751 описано извлечение 1% Apo A-I в экспериментах с использованием буфера для экстракции, содержащего 20% этанола. В WO2009/025754 описаны способы отделения Apo A-I от альфа-1 антитрипсина, в которых для осаждения Apo A-I используют более низкие концентрации этанола (от 8 до 14%).
Для извлечения Apo A-I также используют и другие методы, такие как ультрацентрифугирование и высокоэффективная жидкостная хроматография (ВЭЖХ). Однако указанные способы получения Apo A-I являются чрезвычайно трудоемкими и позволяют получать лишь небольшие количества Apo A-I. Следовательно, такие способы не подходят для промышленного получения Apo A-I.
Таким образом, остается потребность в способе экстрагирования и извлечения Apo A-I из белковых фракций, содержащих Apo A-I, который подходит для крупномасштабного производства и который позволил бы получать Apo A-I с высоким выходом.
Сущность изобретения
Изобретение предлагает способы получения Apo A-I из белковой фракции, содержащей Apo A-I (A), включающие в себя a) суспендирование белковой фракции, содержащей Apo A-I, в буферном растворе, содержащем от 15 до 30% линейного или разветвленного C1-С4 спирта (масс/масс), с получением суспензии, где суспендированная белковая фракция, содержащая Apo A-I (А), имеет рН в диапазоне от 6,4 до 10,0, (b) удаление примесей из суспензии при поддержании белков Apo A-I в солюбилизированном состоянии, (c) осаждение Apo A-I из суспензии и (d) сбор осадка Apo A-I.
В некоторых вариантах осуществления буферный раствор содержит 20% линейного или разветвленного C1-С4 спирта (масс/масс). В некоторых вариантах осуществления рН суспендированной белковой фракции, содержащей Apo A-I (А), находится в диапазоне от 8,1 до 8,5. В некоторых вариантах осуществления рН суспендированной белковой фракции, содержащей Apo A-I (А), находится в диапазоне от 7,0 до 7,6. В некоторых вариантах осуществления буферный раствор содержит от 5 мМ до 35 мМ NaHCO3. В некоторых вариантах осуществления линейный или разветвленный C1-С4 спирт представляет собой этанол.
В некоторых вариантах осуществления объемное соотношение белковой фракции Apo A-I и буферного раствора на стадии (а) составляет от 1:1 до 1:5.
В некоторых вариантах осуществления суспензия, полученная на стадии (a), имеет проводимость менее 5 мСм/см.
В некоторых вариантах осуществления примеси удаляют на стадии (b) путем осаждения в результате добавлении линейного или разветвленного C1-С4 спирта до концентрации спирта от 45 до 65% (масс/масс). В некоторых вариантах осуществления линейный или разветвленный C1-С4 спирт, добавляемый на стадии (b) имеет температуру от -5°С до 15°C. В некоторых вариантах осуществления примеси, осажденные на стадии (b), удаляют из суспензии путем фильтрования, центрифугирования, декантации, ультрафильтрации и/или седиментации.
В некоторых вариантах осуществления на стадии (с) рН суспензии доводят до значения, находящегося в диапазоне от 4,6 до 5,6. В некоторых вариантах осуществления на стадии (с) осаждение Apo A-I проводят при температуре суспензии от -2 до 20°С.
В соответствии с любыми вариантами осуществления способ может дополнительно включать в себя стадию (е), на которой собранный осадок Apo A-I делипидируют, например, путем добавления спирта, такого как этанол, концентрация которого может находиться в диапазоне примерно от 40% до 96% (масс/масс). В конкретных вариантах осуществления собранный осадок Apo A-I делипидируют путем добавления этанола в концентрации примерно 40%, или примерно 50%. Этанол может иметь любую степень чистоты фармацевтической категории. В конкретных вариантах осуществления изобретения этанол представляет собой 95% этанол фармацевтической категории (3A, содержит 5% метанола).
В соответствии с некоторыми вариантами осуществления выход очищенного Apo A-I в данном способе составляет, по меньшей мере, 0,50 г/л плазмы.
В другом аспекте настоящее изобретение предлагает очищенный Apo A-I, содержащий: (а) менее 0,3 мг IgA на грамм Apo A-I.
Изобретение также предлагает Аро А-I, полученный с помощью любого описанного здесь способа, фармацевтические композиции, содержащие такой Apo A-I и фармацевтически приемлемый носитель или разбавитель, и/или способы получения таких композиций.
Кроме того, изобретение предлагает восстановленные HDL, полученные из Apo A-I, полученного с помощью любого описанного здесь способа, фармацевтические композиции, содержащие такие HDL и фармацевтически приемлемый носитель или разбавитель, и/или способы получения восстановленных HDL и/или содержащих их композиций.
Краткое описание чертежей
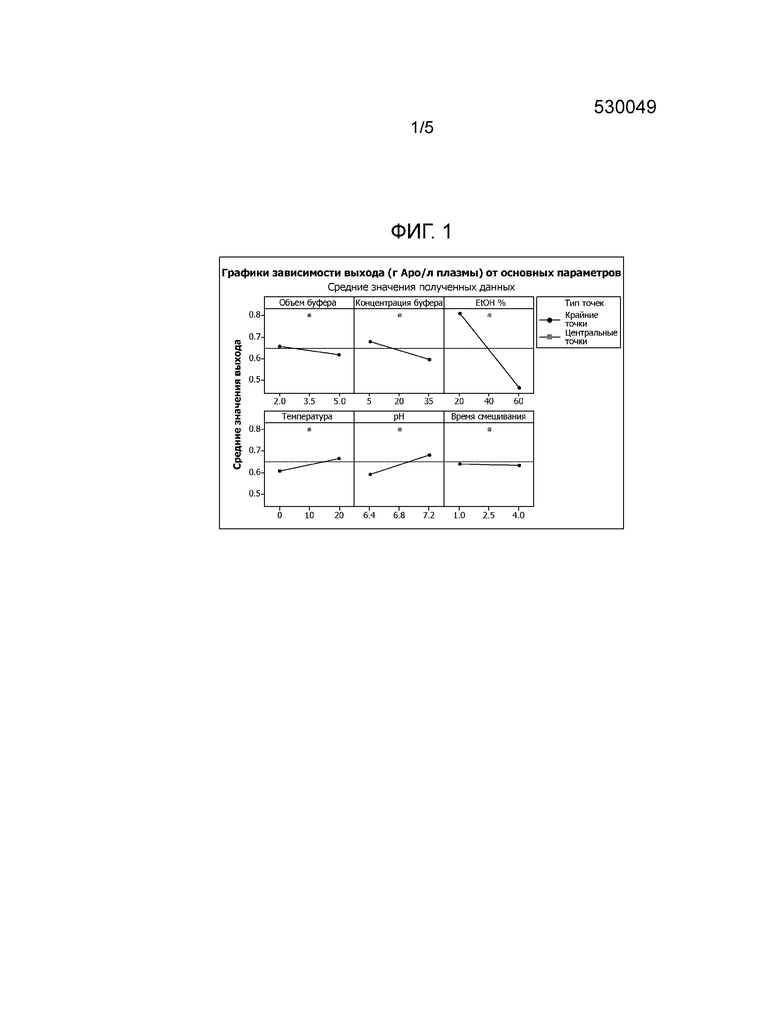
Фиг. 1: Влияние объема буфера, концентрации буфера, концентрации этанола, температуры, рН и времени смешивания на выход Apo A-I. На фиг. 1 показано, что выход Apo A-I, полученного из плазмы, достигает максимума при использовании буферного раствора (B), содержащего более низкие концентрации этанола.
Фиг. 2: Влияние концентрации этанола и рН на выход Apo A-I. На фиг. 2 показано, что выход Apo A-I, полученного из плазмы, достигает максимума при использовании буферного раствора (B), содержащего этанол в концентрации от 15 до 30%.
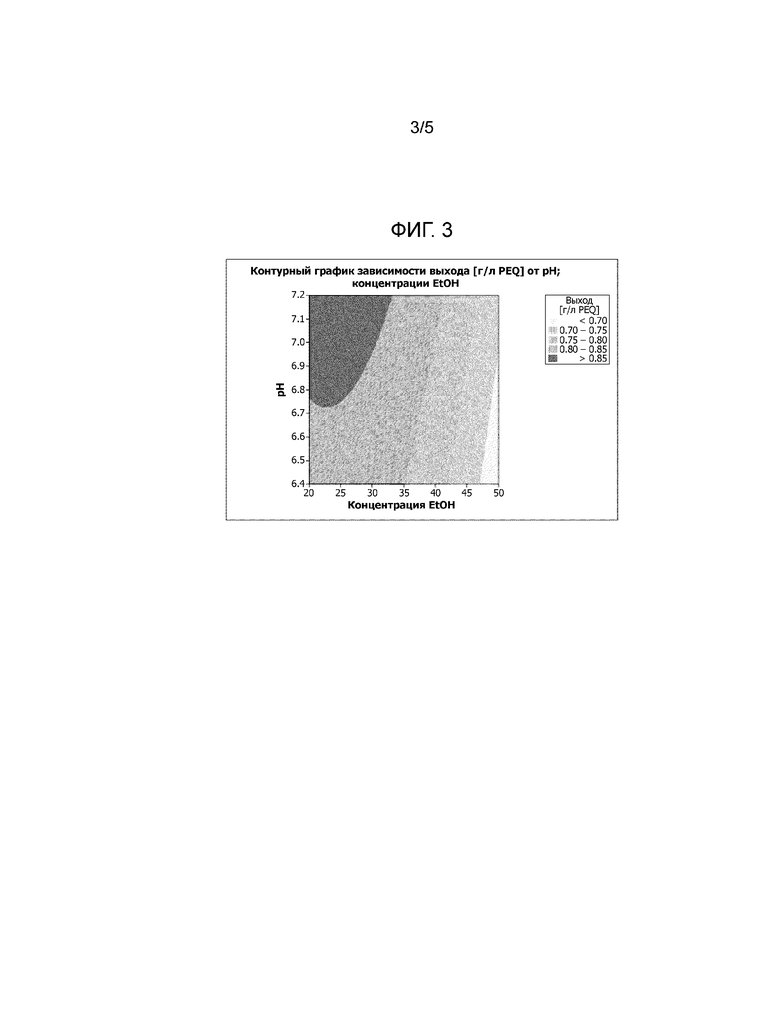
Фиг. 3: Контурный график, демонстрирующий влияние концентрации этанола и рН на выход Apo A-I. На фиг. 3 также показано влияние рН на выход Apo A-I при использовании концентрации этанола в диапазоне от 20 до 50%. Выход достигает максимума, если Apo A-I суспендируют в буферном растворе (В), содержащем этанол в концентрации от 15 до 30%, а рН суспензии находится в диапазоне от 6,4 до 10,0.
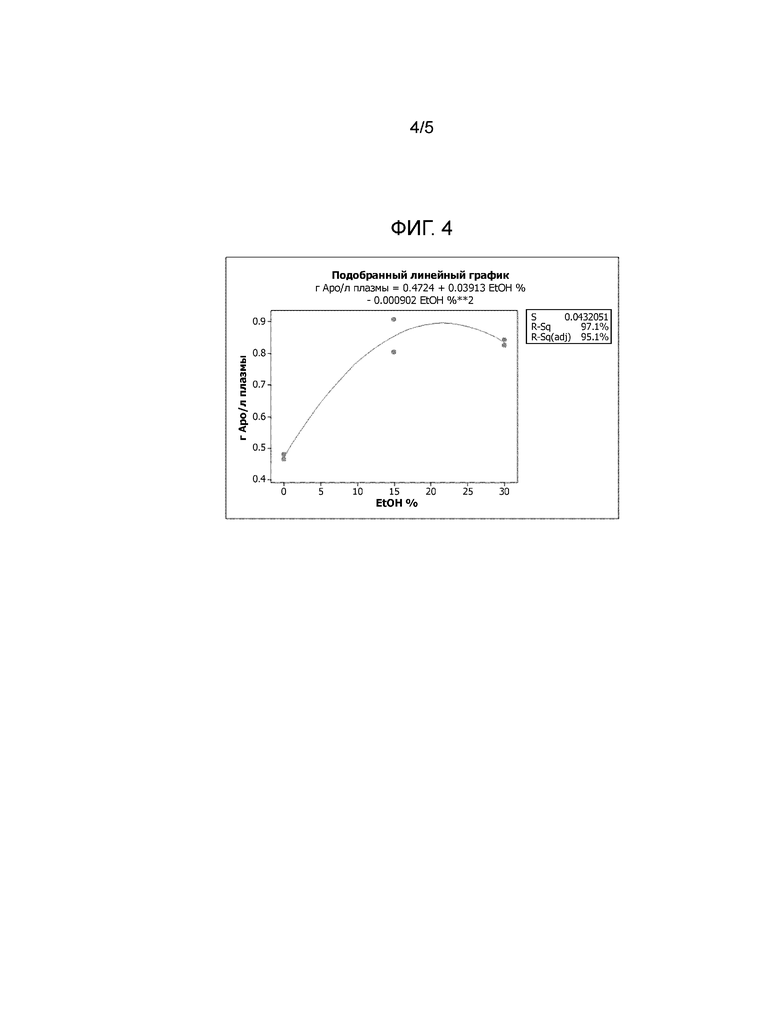
Фиг. 4: Подобранный линейный график, демонстрирующий влияние концентрации этанола на выход Apo A-I. На фиг. 4 показано, что выход Apo A-I, полученного из плазмы, достигает максимума при использовании буферного раствора (B), содержащего этанол в концентрации от 15 до 30%.
Фиг. 5: Протеолитическая активность (A) и неактивированное частичное тромбопластиновое время (NaPTT), время свертывания (B), в осадке Apo A-I (стадия d), очищенном по способу, описанному в примере 6. На фиг. 5 показано, что минимизация времени охлаждения в процессе ресолюбилизации вещества фракции IV уменьшает протеолитическую активность и активацию фактора свертывания в очищенных препаратах Apo A-I.
Подробное описание
В данном документе описаны способы получения Apo A-I из белковой фракции, содержащей Apo A-I (A), включающие в себя стадии a) суспендирования белковой фракции, содержащей Apo A-I (A), в буферном растворе (B), содержащем от 15 до 30% линейного или разветвленного C1-С4 спирта (масс/масс), где суспендированная белковая фракция, содержащая Apo A-I (А), имеет рН в диапазоне от 6,4 до 10,0, и (b) удаление примесей из суспензии при поддержании белков Apo A-I в солюбилизированном состоянии. В конкретных вариантах осуществления после стадии (b) проводят (с) осаждение Apo A-I из суспензии и, необязательно, (d) сбор осадка Apo A-I.
В контексте описанных здесь способов термин "суспендирование белковой фракции, содержащей Apo A-I", относится к солюбилизации белковой фракции, содержащей Apo A-I, с переводом в раствор, по меньшей мере, большей части белковой фракции.
Неожиданно было обнаружено, что, если белковую фракцию, содержащую Apo A-I (A), солюбилизировать в объеме буферного раствора (B), содержащего от 15 до 30% линейного или разветвленного C1-С4 спирта (масс/масс), где суспендированная белковая фракция, содержащая Apo A-I (А), имеет рН в диапазоне от 6,4 до 10,0, в том числе от 6,4 до 7,5 или от 8,1 до 8,5, в заданном объеме буфера можно суспендировать значительно большее количество белков Apo A-I, чем с помощью ранее известных способов, даже без добавления хаотропных веществ, таких как мочевина. Следовательно, можно значительно увеличить количество Apo A-I, полученного из исходной фракции (А), по сравнению с ранее известными способами.
Указанные факты заслуживают особого внимания, поскольку, согласно существующему уровню техники, концентрация спирта в диапазоне от 15 до 30% (масс/масс) является недостаточной для солюбилизации белков Apo A-I и позволяет достичь лишь очень низких выходов.
Вопреки ожиданиям, в некоторых вариантах осуществления уникальное сочетание рН и концентрации спирта, используемое в соответствии с описанными здесь способами, позволяет получить очищенный Apo A-I с выходом, составляющим, по меньшей мере, 0,50 грамм/литр плазмы (г/л плазмы). В некоторых вариантах осуществления с помощью описанных здесь способов Apo A-I получают с выходом, составляющим, по меньшей мере, 0,60 г/л плазмы (на стадии b). И наоборот, если количество этанола превышает 30% масс/масс, то количество извлекаемого Apo A-I снижается. Например, при концентрации 45% масс/масс выход Apo A-I составляет примерно 0,12 г/л плазмы, указывая на то, что описанные здесь способы позволяют увеличить выход Apo A-I, по меньшей мере, на 300%.
Описанные здесь процессы составляют способ солюбилизации Apo A-I, который является более простым и более экономичным, чем способы, существующие на предшествующем уровне техники. Кроме того, пониженная опасность взрыва благодаря применению более низких концентраций линейного или разветвленного C1-С4 спирта, является преимуществом, поскольку позволяет снизить затраты, связанные с конструированием производственного оборудования для стадии осаждения.
Кроме того, используемый в некоторых вариантах осуществления диапазон рН суспендированной белковой фракции, содержащей Apo A-I (А), составляющий от 6,4 до 9,0, предположительно позволяет предотвратить деамидирование белков, что приводит к уменьшению риска иммуногенности полученных биофармацевтических препаратов. В некоторых вариантах осуществления рН суспендированной белковой фракции, содержащей Apo A-I (А), находится в диапазоне от 6,4 до 9,0, в том числе от 7,2 до 8,8, или от 8,0 до 8,6, или от 8,1 до 8,5, или от 8,2 до 8,4, или от 6,4 до 7,4.
Описанные здесь способы являются быстрыми, надежными, специфичными и безопасными, и позволяют повысить выход и чистоту представляющего интерес продукта в процессе обработки. Следовательно, описанные здесь способы особенно хорошо подходят для крупномасштабной очистки Apo A-I. Термин "крупномасштабная очистка" в контексте данного описания означает, что исходное вещество, такое как фракция человеческой плазмы, используется в количестве, составляющем как минимум десятки килограмм, например, 50 кг или более, например, 500 кг исходной фракции, содержащей белок Apo A-I.
В данном описании термин "плазма" относится к жидким компонентам крови и включает в себя производные плазмы и плазму-содержащие композиции.
Способы настоящего изобретения тем самым способствуют достижению улучшенного и приемлемого баланса между выходом продукта и экономическими затратами по сравнению с традиционно используемыми способами.
Далее способы иллюстрируются применительно к конкретным вариантам осуществления. Из приведенного ниже описания следует понимать, что изобретение включает в себя все изменения и сочетания разных вариантов осуществления описанных ниже стадий, компонентов и параметров.
В описанных здесь способах можно использовать любое исходное вещество, содержащее Apo A-I, такое как любое исходное вещество, содержащее значительное количество Apo A-I, например, любые неочищенные смеси белков, содержащие значительные количества Apo A-I.
Термин "Apo A-I" относится к белкам Apo A-I и их фрагментам. Как правило, аполипопротеин A-I представляет собой либо полученный из плазмы, либо рекомбинантный аполипопротеин A-I, такой как Apo A-I, про-apo-A1, или его варианты, такие как Apo A-I Milano или так называемые устойчивые к окислению формы, такие как 4WF. В некоторых вариантах осуществления аполипопротеин A-I представляет собой полученный из плазмы Apo A-I. Изобретение также охватывает биологически активные фрагменты аполипопротеина A-I. Фрагменты могут представлять собой встречающиеся в природе, полученные путем химического синтеза или рекомбинантные фрагменты.
В соответствии с конкретными вариантами осуществления, исходное вещество представляет собой белковую фракцию, содержащую Apo A-I (А), такую как фракция человеческой плазмы, например, фракция плазмы IV-I или IV (иногда также называемая осадок IV, и т.п.), например, полученная путем фракционирования человеческой плазмы холодным этанолом по способу фракционирования Кона или по способу Кистлера и Нитчмана, или с помощью других подобных способов. Другие примеры могут включать в себя осадки, полученные с использованием сульфата аммония, которые содержат Apo A-I. При использовании фракции плазмы IV в качестве исходного вещества, преимущество описанных здесь способов заключается в том, что они позволяют полностью использовать ресурсы плазмы. В конкретных вариантах осуществления исходное вещество представляет собой фракцию IV или фракцию Кона IV1, или фракцию Кона IV4, или подобную фракцию. В других конкретных вариантах осуществления исходное вещество получают из осадка фракции IV. Другие Apo A-I-содержащие белковые фракции (A) могут включать в себя фракции, из которых предварительно удален альфа-1-антитрипсин (ААТ) с целью отдельного промышленного получения терапевтического продукта на основе ААТ, такого как Zemaira™. Примерами таких Apo A-I-содержащих белковых фракций описаны в WO 2009/025754. ААТ можно отделить от Apo A-I путем i) обработки исходной фракции человеческой плазмы, содержащей Apo A-I и AAT, с разделением Apo A-I-содержащей фракции и ААТ-содержащей фракции, где обработка включает в себя a) необязательно обработку исходной фракции человеческой плазмы, используемой в качестве исходного вещества, посредством которой и Apo A-I, и ААТ приводятся в солюбилизированное состояние; b) осаждение солюбилизированного Apo A-I добавлением этанола в концентрации 8-14% об/об и доведение рН до значения в диапазоне примерно от 5 до 6, чтобы Apo A-I осаждался, а ААТ оставался в растворе; и в) отделение осажденного Аро А-I от раствора, содержащего AAT. В конкретных вариантах осуществления настоящего изобретения указанный осажденный Apo A-I представляет собой белковую фракцию, содержащую Apo A-I (А). Исходная фракция человеческой плазмы, содержащая ААT и Apo A-I, может быть выбрана из одной или нескольких из фракций IV-I или IV (иногда также называемой осадок IV, и т.п.), полученных путем фракционирования человеческой плазмы холодным этанолом по способу фракционирования Кона или по способу Кистлера и Нитчмана, или с помощью других подобных способов. В некоторых случаях указанные фракции могут называться фракции Кона IV, осадки, полученные из супернатантов А и А+1 по методу Кистлера-Нитчмана (например, осадок А + IV). Другие примеры могут включать в себя осадки, полученные с использованием сульфата аммония, которые содержат AAT и Apo A-I. В некоторых вариантах осуществления фракция, содержащая белок Apo A-I, содержит тонкоизмельченный диоксид кремния, такой как Aerosil™.
Apo A-I-содержащую белковую фракцию (А) суспендируют в буферном растворе (В), содержащем от 15 до 30% линейного или разветвленного C1-С4 спирта (масс/масс), причем рН суспендированной Apo A-I-содержащей белковой фракции (А) находится в диапазоне от 6,4 до 10,0. В некоторых вариантах осуществления рН суспендированной Apo A-I-содержащей белковой фракции (А) доводят до значения в диапазоне от 6,4 до 10,0 после суспендирования Apo A-I-содержащей белковой фракции (А) в буферном растворе (В).
В некоторых вариантах осуществления буферный раствор (В) для солюбилизации белковой фракции Apo A-I (А) содержит от 16 до 28%, или от 17 до 26%, или от 18 до 24%, или от 19 до 22% линейного или разветвленного C1-C4 спирта (масс/масс).
В некоторых вариантах осуществления буферный раствор (В) для солюбилизации белковой фракции Apo A-I (А) содержит 20% линейного или разветвленного C1-C4 спирта (масс/масс). Данная низкая концентрация спирта, в отличие от концентраций, используемых в способах предыдущего уровня техники, не только является выгодной с экономической точки зрения, но также позволяет солюбилизировать повышенное количество белковой фракции Apo A-I (A), увеличивая общий выход очищенного Apo A-I из определенного количества исходного вещества.
В некоторых вариантах осуществления буферный раствор (В) имеет рН в диапазоне от 6,4 до 10,0, в том числе от 6,4 до 9,0, или от 7,2 до 8,8, или от 8,0 до 8,6, или от 6,4 до 7,4. В более конкретных вариантах осуществления рН буферного раствора (B) находится в диапазоне от 8,1 до 8,5, например, от может быть равен 8,3. Указанные диапазоны рН предположительно минимизируют дезаминирование и денатурацию белков Apo A-I.
В других более конкретных вариантах осуществления рН буферного раствора (B) находится в диапазоне от 7,0 до 7,4, например, он может составлять 7,3. Указанный диапазон рН предположительно минимизирует дезаминирование и денатурацию белков Apo A-I.
Для получения буферного раствора (B) можно использовать любую подходящую соль. Одной из солей, подходящих для получения буферного раствора (В), является NaHCO3. NaHCO3 представляет собой дешевую и коммерчески доступную в больших количествах соль, и, следовательно, хорошо подходит для применения в промышленном масштабе. В некоторых вариантах осуществления буферный раствор (В) содержит от 5 мМ до 35 мМ NaHCO3, например, 15 мМ или 25 мМ NaHCO3. Показано, что такие буферные условия являются особенно эффективными для солюбилизации белковой фракции Apo A-I (A). Кроме того, особым преимуществом NaHCO3 является ее низкая токсичность и простота применения. Однако можно использовать и другие подходящие забуферивающие средства. При выборе забуферивающего средства следует учитывать, что конкретное средство должно быть биологически совместимым и хорошо растворимым в водных/спиртовых растворах.
По соображениям стоимости, токсичности и применимости в промышленности в качестве линейного или разветвленного C1-C4 спирта, как правило, используют этанол. Дополнительное преимущество этанола заключается в том, что он прост в обработке и подходит для промышленного применения. Однако в описанных здесь способах помимо этанола можно использовать и другие спирты, такие как метанол, н-пропанол или изопропанол, н-бутанол, втор-бутанол, изобутанол или трет-бутанол.
В случае применения этанола, он, как правило, имеет любую фармацевтическую степень чистоты. В конкретных вариантах осуществления изобретения этанол представляет собой 96% этанол фармацевтической категории (например, 94% этанола и 2% метилэтилкетона (МЕК)). В конкретных вариантах осуществления изобретения этанол представляет собой 95% этанол фармацевтической категории (3A, содержащий 5% метанола).
Чтобы минимизировать активацию фактора коагуляции и протеазы, температуру суспензии Apo A-I-содержащей белковой фракции (A) (такой как, например, фракция IV) в буфере (B) понижают до 1,0±0,5°С в течение примерно 90 минут после приведения буфера (B) в контакт с Apo A-I-содержащей белковой фракцией (A). В конкретных вариантах осуществления суспензию охлаждают до 1,0±0,5°С в течение примерно 60 минут после приведения буфера В в контакт с Apo A-I-содержащей белковой фракцией (A) (такой как фракция IV).
В некоторых вариантах осуществления после суспендирования Apo A-I-содержащей фракции (А) в буфере (В) рН суспензии при необходимости доводят до значения, находящегося в диапазоне от 6,4 до 10,0, например, в диапазоне от 7,0 до 7,4, такого как 7,3, или до значения, находящегося в диапазоне от 8,1 до 8,5, такого как 8,3.
Чтобы облегчить суспендирование Apo A-I в буфере (В), температура буфера (В) во время приведения его в контакт с Apo A-I-содержащей белковой фракцией (A) должна составлять примерно от 10°С до 26°С, в том числе примерно от 10°С до 16°С, например, примерно 13°С.
В некоторых вариантах осуществления объемное отношение белковой фракции (A) к буферному раствору (В) составляет от 1:1 до 1:5, например, 1:2. При производстве в промышленном масштабе применение низких объемных отношений фракции, содержащей Apo A-I (A), к буферному раствору (B) имеет особые преимущества, связанные со снижением затрат на материалы и хранением буферного раствора.
В некоторых вариантах осуществления, проводимость суспензии составляет менее 5 мСм/см, например, она может находиться в диапазоне от 0,5 до 1,0 мСм/см.
Как указано выше, после солюбилизации Apo A-I-содержащей белковой фракции (А) в буферном растворе (B), из суспензии удаляют примеси, такие как другие нежелательные белки, или другие компоненты, присутствующие в белковой фракции, содержащей Apo A-I (A), при сохранении белков Apo A-I в растворенном состоянии (стадия (b)). В некоторых вариантах осуществления примеси осаждают путем добавления линейного или разветвленного С1-C4 спирта до концентрации в диапазоне от 45 до 65% (масс/масс), такой как 50% (масс/масс) или 60% (масс/масс). Полагают, что применение указанного диапазона позволяет эффективно солюбилизировать многие белковые компоненты, но не Apo A-I. Следовательно, данная стадия очистки представляет собой высокоспецифичный и эффективный способ отделения Apo A-I от других нежелательных компонентов.
Чтобы обеспечить осаждение примесей, линейный или разветвленный С1-C4 спирт добавляют к суспензии при температуре примерно от -5°С до 30°С, например, примерно при 5°С.
В некоторых вариантах осуществления линейный или разветвленный С1-C4 спирт представляет собой этанол. В конкретных вариантах осуществления концентрация этанола находится в диапазоне от 55% до 65% (масс/масс).
pН поддерживают в диапазоне от 6,4 до 10. В некоторых вариантах осуществления рН находится в диапазоне от 6,4 до 9,0, в том числе от 7,2 до 8,8, или от 3,0 до 8,6, или от 8,1 до 8,5, или от 8,2 до 8,4, или от 6,4 до 7,4.
В некоторых вариантах осуществления осажденные примеси удаляют из суспензии любым известным способом, таким как фильтрование, центрифугирование, декантация, ультрафильтрация и/или седиментация. В некоторых вариантах осуществления примеси удаляют фильтрацией. В соответствии с указанными вариантами осуществления можно специально выбрать фильтры, удерживающие осажденные примеси, но не целевой продукт, например, не целевые белки Apo A-I. Фильтры, используемые в таких способах, включают в себя полипропиленовые фильтровальные листы и CH9 целлюлозные фильтровальные листы.
В некоторых вариантах осуществления после удаления примесей из суспензии осаждают Apo A-I (стадия (c)). Осаждение Apo A-I можно проводить путем доведения рН суспензии до значения в диапазоне от 4,6 до 5,6, такого как 5,4. Полагают, что в указанном диапазоне рН осаждение Apo A-I происходит весьма избирательно с оставлением других нежелательных белков в растворе.
В некоторых вариантах осуществления, чтобы осадить Apo A-I, температуру суспензии доводят (при необходимости) до значения в диапазоне от -2 до 20°C.
В некоторых вариантах осуществления собирают осадок Apo A-I (стадия (d)). В соответствии с данными вариантами осуществления, после осаждения Apo A-I к суспензии можно добавить вспомогательное фильтрующее средство, которое облегчает прохождение жидкости через фильтр. Вспомогательные фильтрующие средства включают в себя неорганические минеральные порошки или органические волокнистые вещества, которые используют в сочетании с оборудованием для фильтрации для повышения эффективности фильтрации. В качестве вспомогательных фильтрующих средств обычно используют диатомит, перлит и целлюлозу. Подходящие вспомогательные фильтрующие средства известны в данной области и включают в себя Celite™ 574.
В некоторых вариантах осуществления осадок Apo A-I собирают любым подходящим способом, включающим в себя указанные выше способы, предлагаемые для фильтрации примесей. В некоторых вариантах осуществления осадок Apo A-I собирают фильтрацией. В соответствии с данными вариантами осуществления фильтр можно выбрать так, чтобы целевые белки Аро А-I удерживались, а другие более мелкие компоненты проходили через поры фильтра.
В конкретных вариантах осуществления способ дополнительно включает в себя удаление примесей из осадка Apo A-I с использованием раствора для промывания. В качестве раствора для промывания можно использовать воду или подходящий буфер для промывания. Если присутствует стадия промывания, ее проводят таким образом, чтобы белки Apo A-I оставались на фильтре, а примеси растворялись и вымывались.
В соответствии с другими конкретными вариантами осуществления способ включает в себя дополнительную стадию (e), на которой собранный осадок Apo A-I делипидируют, например, путем добавления спирта, такого как этанол, причем концентрация этанола может находиться в диапазоне примерно от 40% до 96% (масс/масс). Термин "делипидирование" в контексте описанных здесь способов относится к удалению липидов, и, следовательно, к уменьшению содержания липидов в продукте Apo A-I.
Делипидированный осадок Apo A-I (очищенный Apo A-I) можно хранить при -20°C или ниже в течение периода до одного года без потери функциональных свойств белков.
Apo A-I, полученный с помощью способов настоящего изобретения, можно дополнительно подвергнуть стадиям специфического уменьшения титра вирусов (включающим в себя инактивацию и/или удаление вирусов), чтобы обеспечить удаление вирусных патогенов. Традиционные способы инактивации вирусов включают в себя физические методы, такие как классическая процедура пастеризации (нагревание при 60°C в течение 10 часов), коротковолновое ультрафиолетовое облучение или гамма-облучение, и химические методы, такие как обработка растворитель/детергент или инкубация при низких значениях рН. Технологии удаления вирусов включают в себя методы, основанные на исключении по размеру, такие как фильтрация вирусов, также часто называемая нанофильтрацией. Показано, что указанные методы фильтрации вирусов позволяют эффективно удалять вирусы из белковых растворов. В некоторых вариантах осуществления очищенный Apo A-I по представленному изобретению подвергают процедуре уменьшения титра вирусов, включающую инактивацию нагреванием и/или фильтрацию вирусов, как описано в EP13179755 7.
Настоящее изобретение предлагает очищенный Apo A-I, имеющий следующие характеристики:
(a) менее 0,3 мг IgA на грамм Apo A-I;
(b) менее 0,7 мг IgG на грамм Apo A-I;
(c) менее 0,05 мг IgM на грамм Apo A-I;
(d) менее 4,9 мг гаптоглобина на грамм Apo A-I;
(e) менее 2,7 мг гемопексина на грамм Apo A-I;
(f) менее 6,4 мг фибриногена на грамм Apo A-I;
(g) менее 0,9 мг церулоплазмина на грамм Apo A-I;
(h) менее 14,6 мг альбумина на грамм Apo A-I;
(i) менее 2,3 мг альфа-2-макроглобулина на грамм Apo A-I;
(j) менее 12 мг альфа-1-антитрипсина на грамм Apo A-I; и
(k) менее 3,9 мг трансферина на грамм Apo A-I.
Содержание белков можно определить методом нефелометрии. Содержание Apo A-I также можно определить методом высокоэффективного капиллярного электрофореза.
В другом аспекте настоящее изобретение предлагает очищенный Apo A-I, содержащий: (а) менее 0,3 мг IgA на грамм Apo A-I. В конкретных вариантах осуществления препарат Apo A-I, содержит менее 0,2 мг IgA на грамм Аро А-I, или менее 0,1 мг IgA на грамм Apo A-I; или менее 0,075 мг IgA на грамм Apo A-I; или менее 0,05 мг IgA на грамм Apo A-I; или менее 0,02 мг IgA на грамм Apo A-I. Предпочтительно иметь низкое содержание IgA, поскольку он может влиять на профиль безопасности фармацевтических препаратов, содержащих Apo A-I, причем желательно достичь более низких уровней, чтобы свести к минимуму риск образования антител против IgA у пациентов с дефицитом IgA. Данное состояние является наиболее распространенным случаем дефицита первичного гуморального ответа, поражающим примерно 1 человека из 133, и, поскольку данное состояние является относительно мягким, его часто не диагностируют у пораженных им людей.
Описанный выше очищенный Apo A-I можно ввести в состав фармацевтических композиций, таких как восстановленные HDL для терапевтического применения. Такие фармацевтические композиции могут содержать фармацевтически приемлемый носитель или разбавитель. Неограничивающие примеры фармацевтически приемлемых носителей или разбавителей включают в себя воду, эмульгаторы, связующие средства, наполнители, поверхностно-активные вещества, буферы, стабилизаторы, соли, спирты и полиолы, детергенты, белки и пептиды, липиды, смолы, сахара и другие углеводы.
Восстановленные HDL, помимо Apo-AI, могут содержать, без ограничения, один или несколько из липида, детергента и стабилизатора. Неограничивающие примеры липидов включают в себя фосфолипиды, холестерин, сложные эфиры холестерина, жирные кислоты и/или триглицериды. Предпочтительно, липид представляет собой фосфолипид. Неограничивающие примеры фосфолипидов включают в себя фосфатидилхолин (PC) (лецитин), фосфатидную кислоту, фосфатидилэтаноламин (PE) (цефалин), фосфатидилглицерин (PG), фосфатидилсерин (PS), фосфатидилинозитол (PI) и сфингомиелин (SM), а также их природные или синтетические производные. В качестве стабилизатора можно использовать, без ограничения, углевод, такой как сахар (например, сахароза), или сахарный спирт (например, маннит или сорбит). В случае присутствия, детергент может представлять собой любой ионный (например, катионный, анионный, цвиттерионный) детергент, или неионный детергент, включающий в себя желчные кислоты и их соли, такие как холат натрия.
Терапевтическое применение Apo A-I и/или композиций восстановленных HDL может включать в себя лечение или профилактику сердечно-сосудистых заболеваний (таких как острый коронарный синдром (ACS), атеросклероз и инфаркт миокарда), или таких заболеваний, расстройств или состояний, как диабет, инсульт или инфаркт миокарда, которые свидетельствуют о предрасположенности к ACS, гиперхолестеринемия (например, повышенный уровень холестерина в сыворотке, или повышенный уровень холестерина в LDL) и гипохолестеринемия, возникающая в результате снижения уровня липопротеинов высокой плотности (HDL), например, являющаяся симптомом болезни Танжера.
Примеры
Как указано выше, в описанных здесь способах очистки в качестве исходного вещества можно использовать любую неочищенную смесь белков, содержащую значительное количество Apo A-I.
В нижеследующих примерах Apo A-I экстрагируют из человеческой плазмы путем фракционирования ее холодным этанолом в соответствии с методами фракционирования Кона или Кистлера и Нитчмана, с получением фракций плазмы IV-I или IV, которые используют в качестве исходного вещества. На стадии (а) фракцию IV суспендируют в буферном растворе (В) и затем доводят рН перед воздействием повышенной концентрации этанола, чтобы осадить разные примеси на стадии (b). На стадии (с) примеси удаляют путем глубинной фильтрации в присутствии вспомогательного фильтрующего средства, после чего рН доводят до значения, близкого к изоэлектрической точке Apo A-I, чтобы осадить Apo A-I. На стадии (d) добавляют вспомогательное фильтрующее средство и осадок Apo A-I собирают фильрацией. Необязательно проводят стадию (e), на которой осадок Apo A-I повторно суспендируют и добавляют этанол (например, 40-96% масс/масс), чтобы удалить оставшиеся липиды.
Содержание Apo A-I измеряют в разные выборочные моменты методом капиллярного электрофореза.
Чистоту, измеренную методом капиллярного электрофореза, а также визуально определенную на гелях SDS-PAGE, выражают в процентах.
Пример 1: Получение осадка фракции IV
Человеческую плазму охлаждают примерно до 0°С и ее рН доводят примерно до 7,2. Добавляют холодный этанол до концентрации примерно 8% (масс/масс), после чего температуру понижают примерно до -2°С. Образовавшийся осадок (фракция I) удаляют центрифугированием или фильтрацией. рН полученного фильтрата или супернатанта доводят примерно до 6,9 и добавляют холодный этанол до концентрации примерно 20% (масс/масс). Затем температуру понижают до -5°С и смесь снова подвергают центрифугированию или фильтрации. Образовавшийся осадок (фракция II + III) откладывают для других целей.
рН полученного в предыдущей процедуре фильтрата или супернатанта доводят примерно до 5, а концентрацию этанола доводят примерно до 20% (масс/масс). Температуру доводят примерно до -5°С. Образовавшийся осадок (фракция IV) удаляют центрифугированием или фильтрацией и хранят до применения в виде пасты. Указанная паста фракции IV содержит Apo A-I, а также примесные белки и липиды.
Пример 2: Факторы, влияющие на извлечение Apo A-I из осадка IV
В первоначальных экспериментах с использованием методологии планирования эксперимента (DOE) основное внимание уделяется растворению осадка IV (например, содержащего фракцию IV) в натрий-бикарбонатном буфере с целью солюбилизации большего количества Apo A-I. В таблице 1 приведены значения шести параметров процесса, используемых для схемы Плакетта-Бурмана с применением центральных точек. Исследуемые параметры включают в себя объем буфера, концентрацию этанола, температуру, рН и время смешивания. Начальный скрининг стадии солюбилизации осадка IV включает в себя тринадцать циклов.
Аликвоту пасты осадка IV растворяют в объеме буфера, указанном в схеме DOE. Значение рН доводят в соответствии со схемой DOE, после чего суспензию перемешивают при температуре, назначенной для данного эксперимента. Образец суспензии отбирают и центрифугируют в течение пятнадцати минут при 4500 об/мин. Полученный супернатант предоставляют для анализа. Результаты тестирования нормализуют и выражают в г Apo A-I/л плазмы.
Схема скрининга Плакетта-Бурмана для растворения осадка IV
В таблице 2 приведены результаты начального DOE-скрининга методом Плакетта-Бурмана, где нормализованный выход Apo A-I выражают в г Apo A-I/л плазмы.
Результаты анализа Minitab16 DOE позволяют сделать вывод, что только концентрация этанола оказывает влияние на выход с p<0,05, причем, чем меньше концентрация этанола в буфере для ресуспендирования, тем больше Apo A-I переходит в раствор (фигура 1).
Результаты скрининга по схеме планирования Плакетта-Бурмана для растворения осадка IV
Пример 3: Влияние концентрации этанола и рН на извлечение Apo A-I из осадка IV
Исследование включает в себя 10 экспериментов, в которых концентрация этанола варьирует от 20 до 50%, а рН - от 6 до 7,2. Исследование проводят по факторному полному плану DOE с двойными повторами 22, используя центральные точки (таблица 3). Данный метод включает в себя растворение аликвоты пасты осадка IV в 2-х частях буфера, содержащего 15 мМ NaHCO3 и этанол в концентрации, соответствующей DOE. Затем доводят рН в соответствии со схемой DOE, после чего температуру суспензии доводят до 0±1°С и перемешивают в течение одного часа. Образец суспензии центрифугируют в течение пятнадцати минут при 4500 оборотов в минуту. Полученный супернатант используют для анализа. Результаты тестирования нормализуют и выражают в г АроА-I/л плазмы.
Схема факторного полного плана DOE с оптимизацией 22 для растворения осадка IV
Анализ Minitab16 DOE позволяет сделать вывод, что единственным фактором, оказывающим влияние на выход с p<0,05, является концентрация этанола (таблица 4). Результаты показывают, что при уменьшении концентрации этанола в буфере для ресуспендирования большее количество Apo A-I переходит в раствор, причем максимальный выход наблюдается при концентрации этанола от 20 до 30% (фигура 2). Значение рН раствора, полученного после ресуспендирования, оказывает незначительное влияние при повышении рН от 6,4 до 7,2, приводящем к увеличению извлекаемого количества Apo A-I. Однако данный результат не является значимым (p=0,113).
Результаты анализа растворения осадка IV, проводимого по схеме факторного полного плана с оптимизацией 22
На фиг. 3 также показано влияние рН на выход Apo A-I при использовании концентрации этанола в диапазоне от 20 до 50%. Выход достигает максимального значения, если Apo A-I суспендируют в описанном здесь буферном растворе (В), содержащем этанол в концентрации от 15 до 30%, и имеющем рН в диапазоне от 6,4 до 7,2.
Пример 4: Влияние концентрации этанола на извлечение Apo A-I из осадка IV
Проводят линейный эксперимент, чтобы определить, при какой концентрации этанола выход Аро А-I на стадии солюбилизации достигает максимального значения. Концентрации этанола 0, 15 и 30% тестируют с двойными повторами, как указано выше. В таблице 5 приведены результаты линейного эксперимента по исследованию влияния концентрации этанола, где выход Apo A-I нормализуют и выражают в г Apo A-I/л плазмы, а чистоту выражают в процентах.
Результаты анализа растворения осадка IV, проводимого по схеме линейного эксперимента по исследованию влияния концентрации этанола
Как показано на фиг. 4, выход полученного из плазмы Apo A-I достигает максимального значения при использовании буферного раствора (B), содержащего этанол в концентрации от 15 до 30%.
Пример 5: Влияние концентрации этанола и рН на извлечение Apo A-I из осадка IV
Осадок IV растворяют в двух частях буфера для суспендирования, содержащего 15 мМ HaHCO3, 0,5 мМ ЭДТА и этанол в разных концентрациях в соответствии с таблицей 6, после чего рН доводят с использованием 0,5 М НСl или 0,5 М NaOH до значения, соответствующего приведенному в таблице 6. После достижения нужного значения рН образец суспензии центрифугируют в течение пятнадцати минут при 4500 об/мин. Полученный супернатант используют для анализа. Результаты нормализуют и выражают в граммах Apo A-I/л плазмы.
Предыдущие исследования демонстрируют, что на стадии ресуспендирования осадка IV выход Apo A-I достигает максимума, если концентрация этанола в буфере для суспендирования составляет 15%-30%. Показано, что pH суспензии, полученной после ресуспендирования, оказывает лишь незначительное влияние, однако исследуемый диапазон был узким, рН 6,4-рН 7,2. Чтобы определить, оказывает ли более широкий диапазон рН влияние на выход Apo A-I, проводят эксперименты в соответствии со схемой, приведенной в таблице 6, и рассчитывают выход Apo A-I.
Буфер для ресуспендирования и значения рН
Результаты данных экспериментов приведены в таблице 7. При рН 5,0 выход Apo A-I не детектируется независимо от концентрации этанола. Растворение в условиях 20% этанола, рН 7,4 и рН 10,0 в присутствии 15% или 30% этанола приводит к подобным уровням извлечения Apo A-I (стадия b) (таблица 7).
Результаты эксперимента по влиянию концентрации этанола и рН на растворение осадка IV
Пример 6: Протеазная активность и активированные факторы свертывания в извлеченном Apo A-I
Очистка Apo A-I включает в себя осаждение Apo A-I и определение протеолитической активности и наличия активированных факторов свертывания крови в осадке Apo A-I, полученном на стадии (d).
Аликвоту пасты осадка IV растворяют в 2-х частях буфера для суспендирования (B) (буфер, содержащий 15 мМ NaHCO3/20% этанол). Температуру суспензии доводят до 0±1°С. Охлаждающие устройства программируют так, чтобы температура суспензии достигала заданного значения 1°С в течение 10, 65 или 120 минут. Затем суспензию перемешивают при указанной температуре (примерно от 0±1°C) в течение одного часа. После перемешивания рН суспензии доводят до значения примерно 7,3, используя либо 0,5 М раствор KaOH, либо 0,5 М раствор НСl.
Этанол, предварительно охлажденный до -4°С, добавляют до достижения конечной концентрации 60%, поддерживая температуру суспензии в диапазоне от -1 до 2°C. После добавления этанола суспензию перемешивают в течение примерно 30 минут. После инкубации к суспензии добавляют вспомогательное фильтрующее средство. Растворимое вещество Apo A-I (фильтрат Apo A-I) отделяют от нерастворимых примесей путем фильтрации через глубинный фильтр, предварительно промытый смесью 15 мМ NaHCO3/60% этанол. Осадок на фильтре промывают смесью 15 мМ NaHCO3/60% этанол.
К фильтрату Apo A-I добавляют некоторое количество 0,5 М раствора НСl с получением суспензии, рН которой составляет 5,4±0,1. Температуру суспензии доводят примерно до 0°С и указанные значения рН/температуры поддерживают в течение примерно 2 часов.
Затем к суспензии добавляют вспомогательное фильтрующее средство и осадок Apo A-I собирают методом глубинной фильтрации. Глубинные фильтры предварительно промывают 60% этанолом. После фильтрации фильтры вначале промывают 96% этанолом и затем 20% этанолом.
Очищенный осадок АроА-I анализируют на протеолитическую активность и измеряют неактивированное частичное тромбопластиновое время (NaPTT), время свертывания. Указанные анализы предоставляют информацию о количестве остаточной протеазы в осадке Apo A-I, полученном на стадии (d).
Чтобы подготовить осадок Apo A-I, полученный на стадии (d), для анализа, его растворяют путем добавления одной части осадка к трем частям буфера, содержащего 2% SDS/100 мМ трис, рН 8,5. Полученную суспензию, содержащую Apo A-I, перемешивают в течение периода от 45 минут до 1 часа. Затем суспензию центрифугируют и супернатант используют для определения протеолитической активности и NaPTT.
Анализ протеолитической активности проводят с помощью колориметрического метода, в котором супернатант, содержащий Apo A-I, смешивают с буферным раствором (рН 8,0) в соотношении 1: 3. Затем полученный раствор (200 мкл) вместе с 20 мкл pNA-содержащего субстрата добавляют в лунки титрационного микропланшета. Планшет инкубируют при 37°С в течение 10 минут, после чего прочитывают первый раз при 405 нм. Инкубацию продолжают в тех же условиях с последующим прочтением (405 нм) через 30, 60 и 90 минут. В конце инкубации протеолитическую активность (нкат/л) рассчитывают путем измерения количества pNA, образовавшегося в период от 0 до 90 минут.
Наличие активированных факторов свертывания крови в партии осадка Apo A-I (полученного на стадии (d)) определяют с помощью традиционного анализа неактивированного частичного тромбопластинового времени (NaPTT) в соответствии со способом, описанным в Ph. Eur. monograph 2.6.22.
Результаты показывают, что уровни активности протеазы и активированных факторов свертывания крови в осадке Аро A-I
(полученном на стадии (d)) можно свести к минимуму путем сокращения времени, затрачиваемого на снижение температуры до 1°С в процессе ресолюбилизации фракции IV, полученной на стадии (а)
(фиг. 5).
Пример 7: Крупномасштабное извлечение Аро A-I
Партии более крупного масштаба (n=17) получают промышленным способом путем растворения пасты осадка IV (500-550 кг) в 2 частях буфера, содержащего 15 мМ NaHCO3/20% этанола. Затем температуру доводят до 0±1°С и суспензию перемешивают в течение примерно одного часа. После перемешивания рН доводят до 7,3, используя либо 0,5 М NaOH, либо 0,5 М НС1.
Добавляют этанол до конечной концентрации 60%, и суспензию перемешивают в течение примерно 15 минут. Затем суспензию фильтруют через фильтр-пресс, предварительно промытый смесью, содержащей 15 мМ NaHCO3/60% этанола. Полученный осадок на фильтре также промывают смесью, содержащей 15 мМ NaHCO3/60% этанола.
Результаты тестирования нормализуют и выражают в г Аро A-I на л плазмы. Среднее значение выхода от 17 партий составляет 0,6 г/л (на стадии (b)).
Пример 8: Выделение Аро A-I из фракции IV Кистлера-Нитчмана - влияние рН в процессе ресуспендирования
Влияние рН при ресуспендировании Аро A-I из осадка IV на общий выход очищенного Аро A-I определяют путем изменения рН в диапазоне 7,2 - 12,0.
Способ получения очищенного Аро A-I включает в себя растворение осадка IV в двух частях буфера для растворения осадка IV, содержащего 20% этанола и 15 мМ карбонат натрия (22±2°С). После добавления буфера к осадку IV температуру
суспензии снижают до 2°С (2±1°С) в течение примерно 15 минут. Суспензию перемешивают в течение примерно 15 минут, затем рН доводят до нужного значения в диапазоне от 7,2 до 12,0 и добавляют 96% этанол до достижения конечной концентрации 60%.
Полученную суспензию фильтруют в присутствии целита 574. Затем рН фильтрата снижают примерно до 5,3 и суспензию инкубируют в течение 2 ч при температуре примерно 0°С. После добавления вспомогательного фильтрующего средства суспензию перемешивают в течение примерно 30 минут, фильтруют и собирают осадок Apo A-I. Затем осадок Apo A-I подвергают делипидированию с помощью 96% этанола.
Чтобы определить содержание Apo A-I и другие характеристики делипидированного осадка Apo A-I, осадок растворяют при комнатной температуре в течение 45 минут в 3 частях буфера, содержащего 100 мМ Трис и 2% SDS, рН 8,5. Перед анализом образцы центрифугируют.
Исследуют влияние рН на стадии ресуспендирования осадка IV на общий выход Apo A-I в делипидированном осадке Apo A-I, где полученные результаты демонстрируют, что более высокого уровня извлечения можно достичь, используя при экстракции более высокие значения рН (таблица 8). Количество Apo A-I определяют методом высокоэффективного капиллярного электрофореза (Hewlett Packard 3D CE, Agilent Technology). Определение проводят в денатурирующих условиях, то есть в присутствии SDS. Содержание Apo A-I можно определить как в водных, так и в липид-содержащих образцах (таких как восстановленные HDL). Коротко говоря, получают образцы (150 мкл), содержащие примерно 2-3 мг/мл Apo A-I (при необходимости образцы разбавляют водой), 16% SDS (25 мкл) и фенилаланин (25 мкл, 2 или 4 нг/мл). Образцы инкубируют в бане с кипящей водой в течение 3 минут, разбавляют в соотношении 1:2,5 буфером для электрофореза (50 мМ борат натрия, 0,2% SDS, рН 9,1, 300 мкл) и фильтруют (0,45 мкм). Затем образцы загружают на капиллярную колонку из плавленого кварца (56 см, вн.д. 50 мкм, Agilent G1600-61232). Электрофорез проводят при 25 кВ. Строят стандартную кривую в диапазоне от 20 до 950 мкг/мл, используя в качестве стандарта Международный стандарт Apo A-I (BCR-393). Количество определяют по относительным площадям пиков при сравнении с внутренним стандартом (фенилаланин).
Выход очищенного Apo A-I (грамм на литр плазмы) в зависимости от значений рН, используемых для извлечения Apo A-I из фракции IV
Уровни других белков плазмы в очищенном препарате Apo A-I (мг белка плазмы на грамм Apo A-I) определяют методом нефелометрии (BN ProSpec, Siemens), используя полученные от Siemens стандарты, контроль и антисыворотку. Содержание IgA определяют с помощью набора N latex IgA, Siemens, в соответствии с инструкциями производителя.
Уровни других белков плазмы в очищенных препаратах Apo A-I остаются стабильными до достижения рН 8,5 на стадии экстракции фракции IV. При использовании в процессе экстракции более высоких значений рН, таких как 8,7, уровни белков плазмы повышаются (таблица 9).
Уровни примесей в очищенном Apo A-I в зависимости от значений рН, используемых для извлечения Apo A-I из фракции IV


Изобретение относится к биотехнологии, в частности к способу получения аполипопротеина A-I (Apo A-I) из белковой фракции. Осуществляют суспендирование белковой фракции, содержащей Apo A-I (A), имеющей рН в диапазоне от 6,4 до 10,0, в буферном растворе (B), содержащем от 15 до 30% линейного или разветвленного C1-С4 спирта (мас./мас.). Удаляют примеси из суспензии при поддержании белков Apo A-I в солюбилизированном состоянии путем осаждения добавлением линейного или разветвленного C1-С4 спирта до концентрации 55-65% (мас./мас.). Осаждают Apo A-I из суспензии, причем рН суспензии доводят до значения в диапазоне от 4,6 до 5,6, а температуру суспензии доводят до значения в диапазоне от -2 до 20°С. Собирают осадок Apo A-I. Предложенный способ подходит для крупномасштабного производства и позволяет получать Apo A-I с высоким выходом за счет суспендирования значительно большего количества Apo A-I без добавления хаотропных веществ. 14 з.п. ф-лы, 9 табл., 8 пр., 5 ил.
1. Способ получения аполипопротеина A-I (Apo A-I) из белковой фракции, содержащей Apo A-I (A), включающий:
a) суспендирование белковой фракции, содержащей Apo A-I (A), в буферном растворе (B), содержащем от 15 до 30% линейного или разветвленного C1-С4 спирта (мас./мас.), где суспендированная белковая фракция, содержащая Apo A-I (А), имеет рН в диапазоне от 6,4 до 10,0,
(b) удаление примесей из суспензии при поддержании белков Apo A-I в солюбилизированном состоянии,
(с) осаждение Apo A-I из суспензии и
(d) сбор осадка Apo A-I.
2. Способ по п.1, где буферный раствор (B) содержит 20% линейного или разветвленного C1-С4 спирта (мас./мас.).
3. Способ по п.1 или 2, где суспендированная Apo A-I-содержащая белковая фракция (А) имеет рН в диапазоне от 7,2 до 8,8.
4. Способ по п.1, где суспендированная Apo A-I-содержащая белковая фракция (А) имеет рН в диапазоне от 8,1 до 8,5.
5. Способ по п.1 или 2, где суспендированная Apo A-I-содержащая белковая фракция (А) имеет рН в диапазоне от 7,0 до 7,6.
6. Способ по п.1, где буферный раствор (В) содержит от 5 до 35 мМ NaHCO3.
7. Способ по п.1, где линейный или разветвленный C1-С4 спирт представляет собой этанол.
8. Способ по п.1, в котором на стадии (а) объемное отношение Apo A-I-содержащей белковой фракции (А) к буферному раствору (B) составляет от 1:1 до 1:5.
9. Способ по п.1, где проводимость суспензии, полученной на стадии (a), составляет менее 5 мСм/см.
10. Способ по п.1, где примеси удаляют на стадии (b) путем осаждения добавлением линейного или разветвленного C1-С4 спирта до концентрации 55-65% (мас./мас.).
11. Способ по п.1, где температура линейного или разветвленного C1-С4 спирта, добавляемого к суспензии на стадии (b), находится в диапазоне примерно от -5°С до 15°С.
12. Способ по п.1, где примеси удаляют на стадии (b) путем осаждения с последующими фильтрованием, центрифугированием, декантацией, ультрафильтрацией и/или седиментацией.
13. Способ по п.1, в котором на стадии (c) рН суспензии доводят до значения в диапазоне от 4,6 до 5,6.
14. Способ по п.1, в котором на стадии (с) температуру суспензии доводят до значения в диапазоне от -2 до 20°С, чтобы осадить Apo A-I.
15. Способ по п.1, дополнительно включающий в себя стадию (е), на которой проводят делипидирование собранного осадка Apo A-I путем добавления этанола в диапазоне концентраций от 40 до 96% (мас./мас.).
| US 5089602 A, 18.02.1992 | |||
| WO 2009025754 A2, 26.02.2009 | |||
| LERCH P | |||
| G | |||
| et al | |||
| "Production and Characterization of a Reconstituted High Density Lipoprotein for Therapeutic Applications", Vox Sanguinis, 1996, v | |||
| Контрольный стрелочный замок | 1920 |
|
SU71A1 |
| US 20080138394 A1, 12.06.2008 | |||
| RU 2011100801 A, 20.07.2012. | |||

