ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к области медицины, в частности, к новому соединению сальвианоловой кислоты, способу его получения и применения.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Radix Salviae Miltiorrhizae представляет собой корень растений рода Salvia семейства Labiatae, горький на вкус и имеющий слегка охлаждающий эффект, действующий на протоки сердца и печени, и обладающий функциями снятия боли путем устранения застоя, активации циркуляции крови и облегчения напряженного состояния путем очищения сердца. Современные фармакологические исследования показали, что Radix Salviae Miltiorrhizae обладает эффектами дилатации коронарной артерии, улучшая микроциркуляцию и защищая сердце, а также способностью ингибировать и устранять агрегацию тромбоцитов, увеличивая способность организма в отношении устойчивости к гипоксии и активности против гепатита, опухолей и вирусов и т.д. В 2001 году Институт фармакологии Китайской академии наук и Пекинский объединенный медицинский колледж сообщили о наличии 13 соединений фенолокислот водорастворимых действующих компонентов в Radix Salviae Miltiorrhizae и растениях того же рода, в том числе сальвианоловой кислоты А, В, С, D, Е, F, G, Н, I, J, литоспермовой кислоты, розмариновой кислоты и изосальвианоловой кислоты С и т.д. (Lianniang, Li et al. Bulletin of Medical Research. 2001. Vol. 30(7)), а также было раскрыто фармакологическое действие этих 13 фенолокислот. В 2002 году. Rena. Kasimu et al. описали химическую структуру сальвианоловой кислоты K (Rena. Kasimu et al., Journal of Xinjiang Medical University. 2002. Vol. 25(3)). Водорастворимые действующие компоненты Radix Salviae Miltiorrhizae также были изучены зарубежными исследователями. В 1999 году Университет Джорджа Вашингтона подал заявку и в итоге получил патент США касательно действия химических структур 13 типов сальвианоловой кислоты против интегразы ВИЧ и других вирусов, и в котором говорится о том, что Radix Salviae Miltiorrhizae представляет собой источник лекарственного растительного сырья, который имеет большой потенциал и представляет ценность для разработки.
Указанная сальвианоловая кислота Т по настоящему изобретению представляет собой новое соединение, которое было найдено в Radix Salviae Miltiorrhizae в процессе обширного скрининга. До сих пор не сообщалось о структуре и фармакологическом действии, относящихся к данному соединению.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
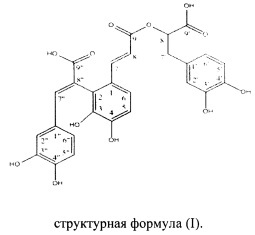
Целью настоящего изобретения является получение соединения сальвианоловой кислоты Т структурной формулы (I), его фармацевтически приемлемых солей, сольватов и гидролизуемых сложных эфиров,
Другой целью настоящего изобретения является обеспечение способа получения сальвианоловой кислоты Т.
Дополнительной целью настоящего изобретения является получение фармацевтической композиции, антиоксиданта, ловушки свободных радикалов, содержащих сальвианоловую кислоту Т.
Другой целью настоящего изобретения является обеспечение применения сальвианоловой кислоты Т в получении лекарственных средств для лечения острого инфаркта миокарда и острой ишемии миокарда.
Другой целью настоящего изобретения является обеспечение применения сальвианоловой кислоты Т в получении лекарственных средств для лечения фиброзных заболеваний легких.
Другой целью настоящего изобретения является обеспечение применения сальвианоловой кислоты Т в получении антиокислителей.
Другой целью настоящего изобретения является применение сальвианоловой кислоты Т для лечения острого инфаркта миокарда, острой ишемии миокарда или фиброзных заболеваний легких.
Другой целью настоящего изобретения является применение сальвианоловой кислоты Т для замедления старения.
Другой целью настоящего изобретения является применение сальвианоловой кислоты Т для антиокислительного действия.
В частности, настоящее изобретение относится к следующим (1)-(37) терминам по настоящему изобретению:
[1] сальвианоловой кислоте Т, представленной структурной формулой (I), ее фармацевтически приемлемым солям, хиральным изомерам, сольватам и гидролизуемым сложным эфирам,

[2] Способ получения сальвианоловой кислоты Т описан по [1], при этом способ включает следующие стадии:
(1а) экстракцию: экстрагирование лекарственного сырья из Radix Salviae Miltiorrhizae или смеси Radix Salviae Miltiorrhizae и другого лекарственного сырья водой, концентрирование фильтрата с получением водного экстракта, последующее добавление спирта к осадку и получение надосадочной жидкости, концентрирование надосадочной жидкости с получением спиртового экстракта;
(1b) разделение: разбавление спиртового экстракта из стадии (1а) водой, перенос на макропористую адсорбционную смолу, промывание смолы кислым водным раствором с целью удаления примесей и последующее элюирование смолы этанолом с получением этанольного элюата, концентрирование этанольного элюата с получением экстракта;
или замещение вышеуказанных стадий (1а) и (1b) следующей стадией (1):
(1) синтеза: растворение сальвианоловой кислоты В в воде, нагревание;
(2) очистки: доведение рН реакционной жидкости, полученной на стадии (1), до кислого значения или очистка экстракта, полученного на стадии (1b), с помощью препаративной жидкостной хроматографии высокого давления, с применением обращенно-фазовой колонки с силикагелем С18 в качестве наполнителя для хроматографии, ацетонитрила-воды-муравьиной кислоты в качестве элюента, выполнение изократического элюирования или градиентного элюирования, с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего сальвианоловую кислоту Т; концентрирование с получением сальвианоловой кислоты Т.
[3] Способ получения хиральных изомеров сальвианоловой кислоты Т, описанной как [1], при этом способ включает следующие стадии:
(1а) экстракцию: экстрагирование лекарственного сырья из Radix Salviae Miltiorrhizae или смеси Radix Salviae Miltiorrhizae и другого лекарственного сырья водой, концентрирование фильтрата с получением водного экстракта, последующее добавление спирта к осадку и получение надосадочной жидкости, концентрирование надосадочной жидкости с получением спиртового экстракта;
(1b) разделение: разбавление полученного на стадии (1а) спиртового экстракта водой, перенос на макропористую адсорбционную смолу, промывание смолы кислым водным раствором с целью удаления примесей и последующее элюирование смолы этанолом с получением этанольного элюата, концентрирование этанольного элюата с получением экстракта;
или замещение вышеуказанных стадий (1а) и (1b) следующей стадией (1):
(1) синтеза: растворение сальвианоловой кислоты В в воде, нагревание;
(2) очистки: доведение рН полученной на стадии (1) реакционной жидкости до кислого значения или очистка экстракта, полученного на стадии (1b) путем препаративной жидкостной хроматографии высокого давления, с применением обращенно-фазовой колонки с силикагелем С18 в качестве наполнителя для хроматографии, ацетонитрила-воды-муравьиной кислоты в качестве элюента, выполнение изократического элюирования, с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, объединение элюата, содержащего сальвианоловую кислоту Т; концентрирование с получением сальвианоловой кислоты Т.
(3) получения хиральных изомеров: отделение хиральных изомеров сальвианоловой кислоты Т, полученной на стадии (2), с помощью препаративной жидкостной хроматографии с применением обращенно-фазовой хиральной колонки в качестве хроматографической колонки, ацетонитрила-воды-муравьиной кислоты в качестве элюента, выполнение изократического элюирования или градиентного элюирования, с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего отдельно (S)-сальвианоловую кислоту Т и (R)-сальвианоловую кислоту Т, лиофильная сушка с получением чистых продуктов в виде (S)-сальвианоловой кислоты Т и (R)-сальвианоловой кислоты Т.
[4] Способ получения, как описано в [2] или [3], где на стадии (1а) лекарственное сырье из Radix Salviae Miltiorrhizae или смесь из Radix Salviae Miltiorrhizae и другого лекарственного сырья представляют собой отваренные кусочки, измельченные частички или порошки, при этом указанное другое лекарственное сырье представляет собой Radix Notoginseng или Radix Astragali, или комбинацию из двух, которые являются совместимыми с Radix Salviae Miltiorrhizae.
[5] Способ получения, как описано в [2] или [3], где на стадии (1а) указанная водная экстракция состоит в следующем: вываривание лекарственного сырья в воде, объем которой в 4-8 раз превышает объем лекарственного сырья, в течение 1,5-4 ч.; фильтрование; концентрирование фильтрата с получением водного экстракта с относительной плотностью 1,10-1,30 (80°С).
[6] Способ получения, как описано в [2] или [3], где на стадии (1а) указанная водная экстракция состоит в следующем: вываривание лекарственного сырья в воде, объем которой в 6 раз превышает объем лекарственного сырья, в течение 3 ч.; фильтрование; концентрирование фильтрата с получением водного экстракта с относительной плотностью 1,22 (80°С).
[7] Способ получения, как описано в [2] или [3], где на стадии (1а) для указанного этапа водной экстракции применяют щелочной водный раствор, при этом указанный щелочной раствор представляет собой по меньшей мере один выбранный из группы, включающей раствор бикарбоната натрия, водный раствор карбоната натрия, раствор гидрокарбоната калия, раствор карбоната калия, водный раствор гидроксида натрия, водный раствор гидроксида калия.
[8] Способ получения, как описано в [7], где указанный щелочной водный раствор представляет собой водный раствор бикарбоната натрия в концентрации 0,3%-0,45% (масса/объем).
[9] Способ получения, как описано в [2] или [3], где на стадии (1а) указанное осаждение спиртом состоит в следующем: добавление 95% (объем/объем) этанола в водный экстракт для осаждения до тех пор, пока содержание этанола не будет составлять 50%-70% (объем/объем) (25°С), и отстаивание в течение 8-36 ч.; получение надосадочной жидкости, извлечение этанола в условиях пониженного давления, концентрирование с получением спиртового экстракта с относительной плотностью 1,25-1,5(60°С).
[10] Способ получения, как описано в [2] или [3], где на стадии (1а) указанное осаждение спиртом состоит в следующем: добавление 95% (объем/объем) этанола в водный экстракт до образования осадка до тех пор, пока содержание этанола не будет составлять 60% (объем/объем) (25°С), отстаивание в течение 24 ч.; получение надосадочной жидкости, извлечение этанола в условиях пониженного давления, концентрирование с получением спиртового экстракта с относительной плотностью 1,32 (60°С).
[11] Способ получения, как описано в [2] или [3]. где на стадии (1b) указанная макропористая адсорбционная смола может представлять собой неполярную или слабополярную макропористую адсорбционную смолу.
[12] Способ получения, как описано в [11], где неполярная или слабополярная макропористая адсорбционная смола представляет собой макропористую адсорбционную смолу АВ-8 типа, HPD450 типа, D101 типа или Х5 типа.
[13] Способ получения, как описано в [12], где неполярная или слабополярная макропористая адсорбционная смола относится к АВ-8 типу.
[14] Способ получения, как описано в [2] или [3], где на стадии (1а) весовое отношение лекарственного сырья к макропористой адсорбционной смоле составляет 5:1-1:1.
[15] Способ получения, как описано в [14], где на стадии (1а) весовое отношение лекарственного сырья к макропористой адсорбционной смоле составляет 3:1.
[16] Способ получения, как описано в [2] или [3], где на стадии (1b) указанный кислый водный раствор представляет собой по меньшей мере один выбранный из группы, включающей водный раствор соляной кислоты, водный раствор серной кислоты, водный раствор азотной кислоты и водный раствор уксусной кислоты или их комбинацию; рН раствора доводят до 1,0-5,0, с промыванием кислым водным раствором, пока элюат не станет почти бесцветным.
[17] Способ получения, как описано в [16], где указанный кислый водный раствор представляет собой водный раствор соляной кислоты; рН раствора доводят до 3,0.
[18] Способ получения, как описано в [2] или [3], где на стадии (1b) для промывания колонки 4-10 раз применяют 50%-95% (объем/объем) этанол, затем элюат концентрируют с получением экстракта без запаха спирта.
[19] Способ получения, как описано в [18], где для промывания колонки 5 раз применяют 95% (объем/объем) этанол.
[20] Способ получения, как описано в [2] или [3], где на стадии (1) сырьевым материалом для реакции является сальвианоловая кислота В или ее соли.
[21] Способ получения, как описано в [2] или [3], где на стадии (1) массовое отношение указанной сальвианоловой кислоты В к указанному водному раствору составляет 1:0,1-1:100000, температура реакции составляет 10-150°С, время реакции составляет от 10 мин. до 24 ч.
[22] Способ получения, как описано в [2] или [3], где на стадии (1) массовое отношение указанной сальвианоловой кислоты В к указанному водному раствору составляет 1:200, температура реакции составляет 90°С, время реакции составляет 1 ч.
[23] Способ получения, как описано в [2] или [3], где на стадии (1) указанный водный раствор представляет собой кислый водный раствор, нейтральный водный раствор или щелочной водный раствор.
[24] Способ получения, как описано в [2] или [3], где на стадии (1) указанный водный раствор представляет собой щелочной водный раствор, при этом указанный щелочной водный раствор выбран по меньшей мере из следующих водных растворов: раствор бикарбоната натрия, водный раствор карбоната натрия, раствор гидрокарбоната калия, раствор карбоната калия, водный раствор гидроксида натрия и водный раствор гидроксида калия.
[25] Способ получения, как описано в [8], где указанный щелочной водный раствор представляет собой раствор бикарбоната натрия в концентрации 0,05%-0,45% (масса/объем).
[26] Способ получения, как описано в [2] или [3], где на стадии (2) для доведения рН реакционной жидкости до 1,0-6,0, применяют любой раствор или комбинацию водного раствора соляной кислоты, водного раствора серной кислоты, водного раствора азотной кислоты и водного раствора уксусной кислоты.
[27] Способ получения, как описано в [26] где для доведения реакционной жидкости до 3,0, применяют водный раствор соляной кислоты.
[28] Способ получения, как описано в [2] или [3], где на стадии (2) указанный жидкостный хроматограф высокого давления представляет собой жидкостный хроматограф высокого давления с колонкой динамического аксиального сжатия, насадкой для хроматографии является обращенно-фазовая колонка с силикагелем С18, растворение реакционной жидкости, рН которой доводят на стадии (1) или экстракта, полученного на указанном этапе (1b), подвижной фазой, при этом указанная подвижная фаза представляет собой ацетонитрил : воду : муравьиную кислоту (объемное отношение) = (10:90:1)-(90:10:1); элюент применяют при соотношении, указанном выше для подвижной фазы, элюирование представляет собой изократическое элюирование или градиентное элюирование; скорость потока составляет 300 мл/мин.; длина волны детектирования составляет 280 нм; для мониторинга процесса элюирования применяют высокоэффективную жидкостную хроматографию, сбор компонентов, время удерживания которых составляет 21,2-24,0 мин., концентрирование досуха, получение образца сальвианоловой кислоты Т.
[29] Способ получения, как описано в [28], где указанная подвижная фаза представляет собой ацетонитрил : воду : муравьиную кислоту (объемное отношение) = (10:90:1)-(50:50:1).
[30] Способ получения, как описано в [28], где указанная подвижная фаза представляет собой ацетонитрил : воду : муравьиную кислоту (объемное отношение) = 15:85:1.
[31] Способ получения, как описано в [28], где для выполнения изократического элюирования применяют указанное элюирование с подвижной фазой ацетонитрил : вода : муравьиная кислота (объемное отношение) = 15:85:1.
[32] Способ получения, как описано в [3], где на стадии (3) для выполнения разделения хиральных изомеров применяют препаративную жидкостную хроматографию, хроматографическая колонка представляет собой обращенно-фазовую колонку, растворение образца сальвианоловой кислоты Т, полученной на стадии (2) подвижной фазой, при этом указанная подвижная фаза представляет собой ацетонитрил : воду : муравьиную кислоту (объемное отношение) = (90:10:1)-(10:90:1); элюент применяют при соотношении, указанном выше для подвижной фазы, элюирование представляет собой изократическое элюирование или градиентное элюирование; скорость потока составляет 25 мл/мин.; длина волны детектирования составляет 280 нм; для мониторинга процесса элюирования применяют высокоэффективную жидкостную хроматографию, сбор отдельно компонента в виде (S)-сальвианоловой кислоты Т со временем удерживания 19,5-21,1 мин., компонента в виде (R)-сальвианоловой кислоты Т со временем удерживания 23.9-25,3 мин., лиофильная сушка после концентрирования при низкой температуре, получение чистого продукта в виде (S)-сальвианоловой кислоты Т и чистого продукта в виде (R)-сальвианоловой кислоты Т.
[33] Способ получения, как описано в [32], где указанная подвижная фаза представляет собой ацетонитрил : воду : муравьиную кислоту (объемное отношение) = 17:83:1.
[34] Способ получения, как описано в [32], где для выполнения изократического элюирования применяют указанное элюирование с подвижной фазой. ацетонитрил : вода : муравьиная кислота (объемное отношение) = 17:83:1.
[35] Способ получения, как описано в [32], где указанная низкая температура составляет 10-40°С.
[36] Способ получения, как описано в [32], где указанная низкая температура составляет 30°С.
[37] Фармацевтическая композиция, содержащая указанную сальвианоловую кислоту Т, ее фармацевтически приемлемые соли, хиральные изомеры, сольваты и гидролизуемые сложные эфиры, описанные в [1].
[38] Антиоксидант, содержащий сальвианоловую кислоту Т, ее фармацевтически приемлемые соли, хиральные изомеры, сольваты и гидролизуемые сложные эфиры, описанные в [1].
[39] Ловушка свободных радикалов, содержащая сальвианоловую кислоту Т, ее фармацевтически приемлемые соли, хиральные изомеры, сольваты и гидролизуемые сложные эфиры, описанные в [1].
[40] Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров, описанных в [1], в получении лекарственных средств для лечения острого инфаркта миокарда и острой ишемии миокарда.
[41] Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров, описанных в [1], в получении лекарственных средств для лечения фиброзного заболевания легких.
[42] Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров, описанных в [1], в получении антиоксидантов.
[43] Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров, описанных в [1], для лечения острого инфаркта миокарда, острой ишемии миокарда или фиброзного заболевания легких.
[44] Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров, описанных в [1], для замедления старения.
[45] Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров, описанных в [1], для антиокислительного действия.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На фиг. 1 представлена масс-спектрограмма сальвианоловой кислоты Т высокого разрешения, А: (R)-сальвианоловая кислота Т; В: (S)-сальвианоловая кислота Т.
На фиг. 2 представлена диаграмма 1Н-ЯМР сальвианоловой кислоты Т при 500 МГц, с применением DMSO, А: (R)-сальвианоловая кислота Т; В: (S)-сальвианоловая кислота Т.
На фиг. 3 представлена диаграмма 13С-ЯМР сальвианоловой кислоты Т при 125 МГц, с применением DMSO, А: (R)-сальвианоловая кислота Т; В: (S)-сальвианоловая кислота Т.
На фиг. 4 представлен DEPT спектр сальвианоловой кислоты Т, A: (R)-сальвианоловая кислота Т; В: (S)-сальвианоловая кислота Т.
На фиг. 5 представлен COSY спектр сальвианоловой кислоты Т, A: (R)-сальвианоловая кислота Т; В:(S)-сальвианоловая кислота Т.
На фиг. 6 представлен ROESY спектр сальвианоловой кислоты Т, A: (R)-сальвианоловая кислота Т; В: (S)-сальвианоловая кислота Т.
На фиг. 7 представлен HSQC спектр сальвианоловой кислоты Т, A: (R)-сальвианоловая кислота Т; В: (S)-сальвианоловая кислота Т.
На фиг. 8 представлен НМВС спектр сальвианоловой кислоты Т, A: (R)-сальвианоловая кислота Т; В:(S)-сальвианоловая кислота Т.
На фиг. 9 представлен CD спектр сальвианоловой кислоты Т, A: (R)-сальвианоловая кислота Т; В:(S)-сальвианоловая кислота Т.
На фиг. 10 представлено сравнение CD спектра и моделируемый спектр ECD сальвианоловой кислоты Т, А: (R)-сальвианоловая кислота Т; В: (S)-сальвианоловая кислота Т.
На фиг. 11 представлены диаграммы результатов биопсии сердца для каждой группы в исследовании эффекта (S)-сальвианоловой кислоты Т в отношении острого инфаркта миокарда.
На фиг. 12 показан ингибиторный эффект (R)-сальвианоловой кислоты Т и (S)-сальвианоловой кислоты Т в отношении индуцированной TGF-β1 пролиферации клеток L929.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Целью настоящего изобретения является получение соединения сальвианоловой кислоты Т структурной формулы (I), его фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров,

Согласно настоящему изобретению структура нового соединения фенолокислоты была идентифицирована по физико-химическим свойствам, с помощью масс-спектрометрии высокого разрешения (QFT-ESI), масс-спектрометрии с ионизацией электрораспылением (ESI-MS), 1Н-ЯМР, 13С-ЯМР, DEPT, COSY, НМВС, HMQC и CD (фиг. 1-фиг. 10).
1Н-ЯМР (спектр водорода) демонстрирует 1 сигнал метенильного протона, присоединенного к кислороду, при δ 4,93 (1Н, dd, 8,0, 4,5 Гц); 11 сигналов ароматического протона при δ 6,85 (1Н, d, 8,5 Гц), δ 7,31 (1Н, d, 8,5 Гц), δ 7,41 (1Н, d, 15,5 Гц), δ 6,27 (1Н, d, 15,5 Гц), δ 6,62 (1Н, s), δ 6,63 (1Н, d, 8,0 Гц), δ 6,47 (1Н, d, 8,0 Гц), δ 6,44 (1H, d, 2,0 Гц), δ 6,55 (1H, d, 8,5 Гц), δ 6,43 (1H, dd, 8,5, 2,0 Гц), δ 7,69 (1H, s); 2 сигнала алифатического протона при δ 2,89 (2Н, ddd, 14,0, 8,0, 4,5 Гц).
13С-ЯМР (спектр углерода) демонстрирует 27 сигналов углерода, в том числе 1 алифатического углерода при δ 36,0, 1 метенильного протона, присоединенного к кислороду при δ 72,8, 3 сигнала карбонильного углерода при δ 166,0, δ 170,6, δ 168,4, и 22 сигнала углерода с двойной связью при δ 123,7, δ 126,4, δ 142,9, δ 147,7, δ 115,0, δ 118,4, δ 143,7, δ 113,9, δ 127,1, δ 116,5, δ 143,9, δ 144,8, δ 115,5, δ 120,0, δ 126,0, δ 117,3, δ 144,8, δ 147,2, δ 115,3, δ 122,9, δ 141,1, δ 123,4.
Удельное вращение 2 изомеров соединения по настоящему изобретению составляет -157,5°, 196,6°, соответственно. Молекулярное строение соединения, абсолютная конфигурация С-8' которого определена как оптимизированная конфигурация S/R соответственно, затем для вычисления при базовых наборах 6-31++G (2d, р) применяли способ BPV86 с TD-SCF, считывали результаты вычислений и сравнивали со спектрами CD соединения по настоящему изобретению, с помощью конечных результатов было обнаружено, что результаты вычисления по соединениям в 2 конфигурациях в основном накладываются с экспериментальными диаграммами CD спектра соединений по настоящему изобретению, из чего сделан вывод что абсолютные конфигурации С-8' 2 изомеров соединений по настоящему изобретению представляют собой S конфигурацию и R конфигурацию соответственно (ссылаясь на фиг. 10). Основная корреляция НМВС соединения по настоящему изобретению состоит в следующем:

Соединение по настоящему изобретению представляет собой новое соединение сальвианоловой кислоты, названное "сальвианоловой кислотой Т".
В связи с изменениями в конфигурации и конформации, которые произошли в пределах соединения по настоящему изобретению в процессе экстрагирования, следовательно, изменения могут наблюдаться в его спектральных данных. Но различные виды изомеров, образованных в результате конфигурационных и конформационных изменений, будут входить в объем правовой охраны настоящего изобретения.
Сальвианоловую кислоту Т по настоящему изобретению, в соответствии с общеизвестными техническими знаниями и уровнем техники, можно применять в форме ее фармацевтически приемлемых солей или сольватов. Фармацевтически приемлемые соли сальвианоловой кислоты Т согласно настоящему изобретению включают известные и фармацевтически приемлемые соли, полученные из неорганического или органического основания, и которые получают известным способом образования солей. Пригодные примеры солей включают натриевую соль, калиевую соль, литиевую соль, магниевую соль, алюминиевую соль, кальциевую соль, цинковую соль и т.д., или соли, образованные при взаимодействии с N,N'-дибензилэтилендиамином, хлорпрокаином, холином, диэтаноламином, этилендиамином, N-метилглюкозимином, прокаином и берберином. До описания второй цели настоящего изобретения, описанная ниже сальвианоловая кислота Т включает сальвианоловою кислоту Т, представленную формулой (I) и ее фармацевтически приемлемые соли, хиральные изомеры, сольваты и гидролизуемые сложные эфиры.
Сальвианоловую кислоту Т по настоящему изобретению соответствующим образом вводят в форме фармацевтической композиции, которую можно применять обычным образом в смеси с одним или более видами фармацевтически приемлемых носителей или наполнителей. По возможности, сальвианоловую кислоту Т по настоящему изобретению можно вводить с терапевтической целью в виде сырья для лекарственного средства, предпочтительно активные компоненты выбирают для непосредственного применения в виде фармацевтического препарата. С точки зрения совместимости с другими компонентами и безопасности для пациентов, носители должны быть фармацевтически приемлемыми.
Таким образом, настоящее изобретение предусматривает фармацевтические препараты сальвианоловой кислоты Т, которые содержат сальвианоловую кислоту Т по настоящему изобретению и один или более видов фармацевтически приемлемых носителей, с другими или без других терапевтических и/или профилактических компонентов. Эти препараты можно вводить перорально, парентерально (в том числе подкожно, например, в виде инъекции или таблеток резервуарного типа, внутрикожно, интратекально, внутримышечно, например, резервуарного типа и внутривенно), ректально и местно (например, сублингвально). Тем не менее, наиболее подходящий путь введения зависит от заболевания пациентов. Указанные фармацевтические препараты могут представлять собой препарат единичной дозы и могут быть получены любым хорошо известным в области фармацевтики способом. Все эти способы включают этап объединения сальвианоловой кислоты Т по настоящему изобретению с носителем, представляющим собой один или более видов вспомогательных компонентов. В целом, указанные препараты по настоящему изобретению получают следующим образом: путем равномерного и плотного объединения сальвианоловой кислоты Т по настоящему изобретению с жидкой средой или тонкоизмельченными твердыми носителями или смесью двух вариантов, с последующим, по необходимости, формированием продукта в требуемый препарат.
Как правило, для получения фармацевтической композиции по настоящему изобретению с использованием сальвианоловой кислоты Т и фармацевтических носителей можно применять ряд стандартных фармацевтических технологий. Технологии включают смешивание, гранулирование и прессование. Как известно специалисту в данной области техники, характеристики и формы фармацевтически приемлемых носителей или разбавителей зависят от количества смешиваемых активных компонентов, пути введения и других известных факторов.
Для данного применения указанные фармацевтически приемлемые носители относятся ко всем типам органических и неорганических носителей, которые можно вводить совместно с композицией, например, наполнитель, скользящее вещество, связующее средство, средство для улучшения распадаемости таблеток и средство для образования оболочки, применяемые для твердого препарата; или фармацевтическим добавкам, таким как краситель и подсластитель. Указанные фармацевтические носители выбраны из группы, включающей сахароспирт, такой как маннитол, сорбитол, ксилитол; аминокислоту, такую как гидрохлорид цистеина, метионин, глицин; витамин С; двунатриевый ЭДТА, ЭДТА кальция-натрия, пиросульфит натрия; неорганические соли, такие как карбонаты, ацетаты, фосфаты одновалентных щелочных металлов или их водные растворы; натрий хлорид, калий хлорид; метабисульфит натрия, бисульфит натрия, тиосульфат натрия; карбонат кальция, бикарбонат кальция; стеарат, такой как стеарат кальция, стеарат магния; неорганическая кислота, такая как соляная кислота, уксусная кислота, серная кислота, фосфорная кислота; соли органических кислот, такие как лактат натрия; олигосахарид, полисахарид, целлюлоза и их производные, такие как мальтоза, глюкоза, фруктоза, декстран, сахароза, лактоза, циклодекстрин (такой как β-циклодекстрин), крахмал; производные силикона; альгинат; желатин; поливинилпирролидон; глицерин; агар; поверхностно-активное вещество, такое как Tween-80; полиэтиленгликоль; фосфолипиды; каолин; тальковая пудра и т.д.
Форма фармацевтических препаратов может представлять собой любую фармацевтически приемлемую лекарственную форму, в том числе таблетки, такие как таблетки, покрытые сахарной оболочкой, таблетки, покрытые пленочной оболочкой и таблетки, покрытые кишечнорастворимой оболочкой; капсулы, такие как твердые капсулы и мягкие капсулы; раствор для перорального применения; буккальные таблетки; гранулы; гранулы, принимаемые после растворения в кипящей воде; пилюли; порошки; пасты; пеллеты; суспензии; пудра; растворы; инъекции; суппозитории; пасты, такие как мази и пластичные пасты; крема; спреи; капли и пластыри. Предпочтительно, препараты представлены лекарственной формой для перорального применения, такой как капсулы, таблетки, растворы, гранулы, пилюли, порошки, пеллеты и пасты; и в форме инъекций, такие как инъекционные грануляты, инъекции и внутривенные вливания и т.д. Наиболее предпочтительно, чтобы препараты были в форме таблеток.
Указанные препараты для перорального применения могут содержать широко используемый наполнитель, связующее средство, объемообразующее средство, разбавитель, средство для прессования таблеток, скользящее вещество, средство для улучшения распадаемости таблеток, красители, ароматизатор и увлажняющее средство, а также, по необходимости, таблетки могут быть покрыты оболочкой.
Предпочтительные примеры указанного наполнителя включают лактозу, D-маннитол, D-сорбитол, крахмал, такой как α-крахмал, декстрин, кристаллическую целлюлозу, гидроксипропилцеллюлозу с низкой степенью замещения, карбоксиметилцеллюлозу натрия, гуммиарабик, амилопектин, легкую безводную кремниевую кислоту, синтетический силикат алюминия или алюмосиликат магния и т.д.
Предпочтительные примеры указанного скользящего вещества включают стеарат магния, стеарат кальция, тальковую пудру и силикагель и т.д.
Предпочтительные примеры указанного связующего средства включают α-крахмал, сахарозу, желатин, гуммиарабик, метилцеллюлозу, карбоксиметилцеллюлозу, карбоксиметилцеллюлозу натрия, кристаллическую целлюлозу, сахар, D-маннитол, трегалозу, декстрин, амилопектин, гидроксипропилцеллюлозу, гидроксипропил метилцеллюлозу, пирролидон и т.д.
Предпочтительные примеры указанного средства для улучшения распадаемости таблеток включают лактозу, сахар, крахмал, карбоксиметилцеллюлозу, кальциевую соль карбоксиметилцеллюлозы, аминоалкил натрия, натриевую соль карбоксиметилкрахмала, легкую безводную кремниевую кислоту, гидроксипропилцеллюлозу с низкой степенью замещения и т.д.
Предпочтительные примеры указанного средства для образования оболочки включают гидроксипропил метилцеллюлозу, гидроксипропилцеллюлозу, этилцеллюлозу, карбоксиметилцеллюлозу, поливиниловый спирт и т.д.
Предпочтительные примеры указанного красителя включают водорастворимый пищевой краситель тартразин (пищевой краситель, такой как пищевой красный №2 и №3, пищевой желтый №4 и №5, пищевой синий №1 и №2); нерастворимые в воде красочные «лаки» (такие как алюминиевая соль вышеупомянутого водорастворимого пищевого красителя тартразина) и природный краситель (такой как β-каротин, хлорофилл и крокус) и т.д.
Предпочтительные примеры указанного подсластителя включают сахарин натрия, глицирретиновую кислоту, аспартам и стевиозид и т.д.
Известный способ получения таблеток включает объединение сальвианоловой кислоты Т по настоящему изобретению с одним или более видами фармацевтически приемлемого наполнителя и последующее прессование, и формование.
Сальвианоловую кислоту Т по настоящему изобретению также можно получать в виде жидких препаратов для перорального применения, к примеру, водорастворимых или растворимых в масле суспензий, растворов, эмульсий, сиропов и т.д. Сальвианоловую кислоту Т по настоящему изобретению можно получать в виде сухого продукта, повторно смешиваемого с водой или другими пригодными, ранее используемыми носителями. Такой тип жидких препаратов может содержать известные добавки, в том числе суспендирующее средство, такое как сорбитный сироп, метилцеллюлоза, глюкоза/сироп, желатин, гидроксиэтилцеллюлоза, карбоксиметилцеллюлоза, гель стеарата алюминия или гидрогенизованный пищевой жир; эмульгирующее средство, такое как лецитин, сорбитмоноолеат или гуммиарабик; безводный носитель (в том числе пищевое масло), такой как миндальное масло, фракционированное кокосовое масло, маслянистый сложный эфир, пропиленгликоль или этанол; а также консервант, такой как метилпарабен, нипазол или сорбиновая кислота.
Вводимые парентеральным путем, препараты включают водные и неводные стерильные инъекции, где данные препараты могут содержать антиоксидант, буферное средство, бактериостатическое средство, изотоническое средство и т.д.; и водные и неводные стерильные суспензии, где данные препараты могут содержать суспендирующее средство и средство для сгущения. Препараты могут храниться в сосуде для единичной дозы или многократной дозы, таком как запаянные ампулы и флаконы, которые можно сохранять в условиях лиофильной сушки (лиофилизации) и повторно растворять перед применением в стерильном жидком носителе, таком как вода для инъекции.
Вводимые ректальным путем, препараты могут представлять собой суппозитории, содержащие известную суппозиторную основу, такую как кокосовое масло, стеариновую кислоту или другие глицериды, или этиленгликоль.
Препараты, вводимые местно в полость рта, например, препараты, вводимые буккально или сублингвально, включают пастилки, где активный компонент заключен в ароматизированную основу, такую как сахароза и гуммиарабик; а также ароматизированные таблетки, где активный компонент заключен в основу, такую как желатин и глицерин, или сахарозу и гуммиарабик.
Сальвианоловую кислоту Т по настоящему изобретению также можно получать в виде препаратов резервуарного типа, таких как препарат с замедленным высвобождением, который можно вводить путем имплантации (как например, подкожной или внутримышечной имплантации) или внутримышечной инъекции. Таким образом, сальвианоловую кислоту Т по настоящему изобретению можно получать с пригодными полимерами или гидрофобными материалами (такими как эмульсия в приемлемом масле), или ионообменными смолами, или можно получать в виде мало растворимых производных, как например, малорастворимой соли.
В соответствии с общеизвестными техническими знаниями и уровнем техники, лечение, относящееся к настоящему изобретению, включает предупреждение и лечение определенных заболеваний или симптомов. Кроме того, терапевтически эффективное количество сальвианоловой кислоты Т по настоящему изобретению зависит от характера заболеваний и индивидуального состояния пациентов, или соответствует рекомендации лечащего врача. Как правило, терапевтически эффективное количество для взрослого человека находится в диапазоне 0,02-5000 мг в день, предпочтительно 1-1500 мг в день. Количество может быть представлено единичной дозой или многократной дозой, которую пациенты будут принимать через определенные интервалы, например, дважды в день, три раза в день, четыре раза в день или более. Указанный препарат по настоящему изобретению содержит 0,1-99% по массе активных компонентов, предпочтительно 30-95%) по массе в случае таблеток и капсул; и предпочтительно 3-50% по массе в случае жидких препаратов.
Относительно второй цели, настоящее изобретение относится к способу получения сальвианоловой кислоты Т, при этом указанный способ включает следующие стадии:
(1a) экстракцию: экстрагирование лекарственного сырья из Radix Salviae Miltiorrhizae или смеси Radix Salviae Miltiorrhizae и другого лекарственного сырья водой, концентрирование фильтрата с получением водного экстракта, последующее добавление спирта к осадку с получением надосадочной жидкости, концентрирование надосадочной жидкости с получением спиртового экстракта;
(1b) разделение: разбавление спиртового экстракта из стадии (1а) водой, перенос на макропористую адсорбционную смолу, промывание смолы кислым водным раствором с целью удаления примесей и последующее элюирование смолы этанолом с получением этанольного элюата, концентрирование этанольного элюата с получением экстракта;
или замещение вышеуказанных стадий (1а) и (1b) следующей стадией (1):
(1) синтеза: растворение сальвианоловой кислоты В в воде, нагревание;
(2) очистки: доведение рН реакционной жидкости, полученной на стадии (1), до кислого значения или очистка экстракта, полученного на этапе (1b), путем препаративной жидкостной хроматографии высокого давления, с применением обращенно-фазовой колонки с силикагелем С18 в качестве наполнителя для хроматографии, ацетонитрила:воды:муравьиной кислоты в качестве элюента, выполнение изократического элюирования или градиентного элюирования, с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего сальвианоловую кислоту Т; концентрирование с получением сальвианоловой кислоты Т.
Дополнительно, настоящее изобретение относится к способу получения хиральных изомеров сальвианоловой кислоты Т, при этом способ включает следующие стадии:
(1а) экстракцию: экстрагирование лекарственного сырья из Radix Salviae Miltiorrhizae или смеси Radix Salviae Miltiorrhizae и другого лекарственного сырья водой, концентрирование фильтрата с получением водного экстракта, последующее добавление спирта к осадку с получением надосадочной жидкости, концентрирование надосадочной жидкости с получением спиртового экстракта;
(1b) разделение: разбавление полученного на этапе (1а) спиртового экстракта водой, перенос на макропористую адсорбционную смолу, промывание смолы кислым водным раствором с целью удаления примесей и последующее элюирование смолы этанолом с получением этанольного элюата, концентрирование этанольного элюата с получением экстракта;
или замещение вышеуказанных стадий (1а) и (1b) следующей стадией (1):
(1) синтеза: растворение сальвианоловой кислоты В в воде, нагревание;
(2) очистки: доведение рН реакционной жидкости, полученной на стадии (1), до кислого значения или очистка экстракта, полученного на этапе (1b) путем препаративной жидкостной хроматографии высокого давления, с применением обращенно-фазовой колонки с силикагелем С18 в качестве наполнителя для хроматографии, ацетонитрила : воды : муравьиной кислоты в качестве элюента, изократическое элюирование или градиентное элюирование, с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего сальвианоловую кислоту Т; концентрирование с получением сальвианоловой кислоты Т.
(3) получения хиральных изомеров: отделение хиральных изомеров от сальвианоловой кислоты Т, полученной на этапе (2), с помощью препаративной жидкостной хроматографии с применением хиральной колонки для обращенно-фазовой хроматографии в качестве хроматографической колонки, ацетонитрила:воды:муравьиной кислоты в качестве элюента, выполнение изократического элюирования или градиентного элюирования, с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего (S)-сальвианоловую кислоту Т и (R)-сальвианоловую кислоту Т раздельно, лиофильная сушка с получением чистых продуктов (S)-сальвианоловой кислоты Т и (R)-сальвианоловой кислоты Т.
На указанном этапе (1а), лекарственное сырье Radix Salviae Miltiorrhizae или смесь Radix Salviae Miltiorrhizae и другого лекарственного сырья представляет собой отваренные кусочки, измельченные частички или порошки, предпочтительно отваренные кусочки; указанное другое лекарственное сырье может представлять собой хорошо известное специалисту в данной области техники китайское лекарственное сырье, совместимое с Radix Salviae Miltiorrhizae, предпочтительно Radix Notoginseng или Radix Astragali, или комбинацию из двух.
На указанном этапе (1а), указанная водная экстракция состоит в следующем: вываривание лекарственного сырья в воде, объем которой в 4-8 раз превышает объем лекарственного сырья, в течение 1,5-4 ч., предпочтительно в течение 3 ч.; фильтрование; концентрирование фильтрата с получением водного экстракта с относительной плотностью 1,10-1,30 (80°С), предпочтительно 1,22 (80°С). С целью улучшения эффективности экстракции и образования солей, веществ фенокислоты, на указанном этапе водной экстракции применяют щелочной водный раствор, при этом указанная щелочь представляет собой по меньшей мере одну выбранную из группы, включающей раствор бикарбоната натрия, водный раствор карбоната натрия, раствор гидрокарбоната калия, раствор карбоната калия, водный раствор гидроксида натрия, водный раствор гидроксида калия, предпочтительно водный раствор бикарбоната натрия в концентрации 0,3%-0,45% (масса/объем).
На этапе (1а), указанное осаждение спиртом состоит в следующем: добавление 95% (объем/объем) этанола к водному экстракту до образования осадка до тех пор, пока содержание этанола не будет составлять 50%-70% (объем/объем) (25°С), предпочтительно 60% (объем/объем), отстаивание в течение 8-36 ч., предпочтительно 24 ч.; получение надосадочной жидкости, извлечение этанола в условиях пониженного давления, концентрирование с получением спиртового экстракта с относительной плотностью 1,25-1,5 (60°С), предпочтительно спиртового экстракта с относительной плотностью 1,32 (60°С).
На этапе (1b), указанная макропористая адсорбционная смола представляет собой неполярную или слабополярную макропористую адсорбционную смолу, которая может быть выбрана из макропористой адсорбционной смолы АВ-8 типа, HPD450 типа, D101 типа или Х5 типа, предпочтительно АВ-8 типа; весовое отношение лекарственного сырья к макропористой адсорбционной смоле на этапе (1) составляет 5:1-1:1, предпочтительно 3:1; указанный кислый водный раствор представляет собой по меньшей мере один выбранный из группы, включающей водный раствор соляной кислоты, водный раствор серной кислоты, водный раствор азотной кислоты и водный раствор уксусной кислоты или их комбинацию, предпочтительно водный раствор соляной кислоты; рН раствора доводят до 1,0-5,0, предпочтительно 3,0; промывание кислым водным раствором, пока элюат не станет почти бесцветным. Затем, для промывания колонки 4-10 раз применяют 50%-95% (объем/объем) этанол, предпочтительно 5 раз 95% (объем/объем) этанол; элюат концентрируют с получением экстракта без запаха спирта.
На указанном этапе (1) указанным сырьевым материалом для реакции является сальвианоловая кислота В или ее соли.
На указанном этапе (1) массовое отношение указанной сальвианоловой кислоты В к указанному водному раствору составляет 1:0.1-1:100000, предпочтительно 1:200; температура реакции составляет 10-150°С, предпочтительно 90°С; время реакции составляет от 10 мин. до 24 ч., предпочтительно 1 ч.
На указанном этапе (1) указанный водный раствор может представлять собой кислый водный раствор, нейтральный водный раствор или щелочной водный раствор, предпочтительно щелочной водный раствор, указанный водный раствор представляет собой щелочной водный раствор, указанный щелочной водный раствор выбран по меньшей мере из следующих водных растворов: раствора бикарбоната натрия, водного раствора карбоната натрия, раствора гидрокарбоната калия, раствора карбоната калия, водного раствора гидроксида натрия и водного раствора гидроксида калия; более предпочтительно, указанный щелочной водный раствор представляет собой раствор бикарбоната натрия в концентрации 0,05%-0,45% (масса/объем).
На этапе (2) для доведения рН до 1,0-6,0 можно применять любой из водного раствора соляной кислоты, водного раствора серной кислоты, водного раствора азотной кислоты и водного раствора уксусной кислоты, предпочтительно, для доведения реакционной жидкости до 3,0 применяют водный раствор соляной кислоты.
На этапе (2) указанный жидкостный хроматограф высокого давления может представлять собой жидкостный хроматограф высокого давления с колонкой динамического аксиального сжатия, такой как NOVASEP LC80-600 от французского производителя, предпочтительно насадкой для хроматографии является обращенно-фазовая колонка с силикагелем С18 (10 мкм, YMC Company), растворение экстракта, полученного на указанном этапе (1b) подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = (10:90:1)-(90:10:1)), предпочтительно, ацетонитрил : вода : муравьиная кислота (объемное отношение) = (10:90:1)-(50:50:1), более предпочтительно, ацетонитрил:вода:муравьиная кислота (объемное отношение)=15:85:1; элюент применяют при соотношении, указанном выше для подвижной фазы, элюирование представляет собой изократическое элюирование или градиентное элюирование, предпочтительно изократическое элюирование ацетонитрилом : водой : муравьиной кислотой (объемное отношение) = 15:85:1; скорость потока составляет 300 мл/мин.; длина волны детектирования составляет 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор компонентов, при котором время удерживания составляет 21,2-24,0 мин., концентрирование досуха, получение образца сальвианоловой кислоты Т.
На этапе (3) для выполнения разделения хиральных изомеров применяют препаративный жидкостный хроматограф Waters Prep 400, хроматографическая колонка представляет собой обращенно-фазовую хроматографическую колонку CHIRALCEL® OD-RH (250×20 мм, 5 мкм), растворение образца сальвианоловой кислоты Т, полученного на этапе (2) подвижной фазой (ацетонитрил : вода : муравьиная кислота) (объемное отношение) = (10:90:1)-(90:10:1)), предпочтительно ацетонитрил : вода : муравьиная кислота (объемное отношение) = 15:85:1; элюент применяют при соотношении, указанном выше для подвижной фазы, элюирование представляет собой изократическое элюирование или градиентное элюирование, предпочтительно изократическое элюирование ацетонитрилом : водой : муравьиной кислотой (объемное отношение) = 15:85:1; скорость потока 25 мл/мин.; длина волны детектирования составляет 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор компонента в виде (S)-сальвианоловой кислоты Т со временем удерживания 19,5-21,1 мин., компонента в виде (R)-сальвианоловой кислоты Т со временем удерживания 23,9-25,3 мин. раздельно, лиофильная сушка после концентрирования при низкой температуре (10-40°С, предпочтительно 30°С), получение чистого продукта в виде (S)-сальвианоловой кислоты Т и чистого продукта в виде (R)-сальвианоловой кислоты Т.
Результаты фармакодинамического теста в настоящем изобретении продемонстрировали, что сальвианоловая кислота Т по настоящему изобретению обладает активностью в отношении предупреждения острого инфаркта миокарда и острой ишемии миокарда, характеризуется высоким уровнем захвата свободных радикалов и восстановительной способностью, а также активностью в отношении лечения фиброза легких.
Таким образом, настоящее изобретение также относится к следующему:
антиоксиданту, ловушке свободных радикалов, содержащим сальвианоловую кислоту Т, ее фармацевтически приемлемые соли, хиральные изомеры, сольваты и гидролизуемые сложные эфиры.
Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров в получении лекарственных средств для лечения острого инфаркта миокарда и острой ишемии миокарда.
Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров в получении лекарственных средств для лечения фиброзного заболевания легких.
Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров в получении антиоксидантов.
Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров для лечения острого инфаркта миокарда, острой ишемии миокарда или фиброзного заболевания легких.
Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров для замедления старения.
Применение сальвианоловой кислоты Т, ее фармацевтически приемлемых солей, хиральных изомеров, сольватов и гидролизуемых сложных эфиров, описанных в [1], для антиокислительного действия.
Примеры
Технические предложения по настоящему изобретению дополнительно представлены следующими примерами получения и экспериментальными примерами. Но следует понимать, что объем правой защиты настоящего изобретения не ограничивается данными примерами получения и экспериментальными примерами.
Пример получения 1. Получение сальвианоловой кислоты Т, (S)-сальвианоловой кислоты Т, (R)-сальвианоловой кислоты Т.
Отваренные кусочки Salvia miltiorrhiza помещали в аппарат китайской медицины для вываривания, добавляли воду с содержанием 0,3% (масса/объем) бикарбоната натрия, в 6 раз превышающую количество отваренных кусочков Salvia miltiorrhiza, вываривали в течение 2 ч., фильтровали, фильтрат концентрировали с получением водного экстракта с относительной плотностью 1,22 (80°С).
К вышеупомянутому экстракту добавляли 95% (объем/объем) этанол, для осуществления осаждения пока конечное содержание этанола не будет составлять 60% (объем/объем) (25°С), отстаивали в течение 24 ч.; получали надосадочную жидкость и концентрировали в условиях пониженного давления с получением этанольного экстракта с относительной плотностью 1,37 (60°С).
Вышеупомянутый этанольный экстракт растворяли водой, а затем переносили в колонку с макропористой адсорбционной смолой АВ-8, для промывания колонки применяли кислый водный раствор со значением рН 3,0, пока элюат не становился почти бесцветным, затем для элюирования применяли 95% (объем/объем) этанол в объеме, 5 раз превышающем объем колонки, элюат концентрировали с получением экстракта без запаха спирта.
Экстракт, полученный на предыдущем этапе, растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 15:85:1), для очистки применяли жидкостный хроматограф высокого давления с колонкой динамического аксиального сжатия NOVASEP LC80-600 от французского производителя, насадкой для хроматографии являлась обращенно-фазовая колонка с силикагелем С18 (10 мкм, YMC Company), для изократического элюирования применяли ацетонитрил : воду : муравьиную кислоту (объемное отношение) = 15:85:1; скорость потока была 300 мл/мин.; длина волны детектирования была 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонентов со временем удерживания 21,2-24,0 мин., концентрирование досуха с помощью ротационного испарителя, с получением образца сальвианоловой кислоты Т.
Вышеупомянутый образец сальвианоловой кислоты Т растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 17:83:1), для разделения хиральных изомеров применяли препаративный жидкостный хроматограф, хроматографическая колонка представляла собой обращенно-фазовую колонку CHIRALCEL® OD-RH (250×20 мм, 5 мкм), для изократического элюирования применяли ацетонитрил : воду : муравьиную кислоту (объемное отношение) = 17:83:1; скорость потока была 25 мл/мин.; длина волны детектирования была 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонентов, (S)-сальвианоловой кислоты Т со временем удерживания 19,5-21,1 мин., компонент, (R)-сальвианоловой кислоты Т со временем удерживания 23,9-25,3 мин., концентрирование элюата с помощью ротационного испарителя при 30°С, затем осуществляли лиофильную сушку с получением чистого продукта в виде (S)-сальвианоловой кислоты Т и чистого продукта (R)-сальвианоловой кислоты Т.
Пик квазимолекулярного иона (S)-сальвианоловой кислоты Т. полученный с помощью масс-спектрометрии высокого разрешения имел значения m/z=537,1033; пик квазимолекулярного иона (R)-сальвианоловой кислоты Т имел значения [М-Н]- m/z 537,1034.
Интерпретация спектральных данных ядерного магнитного резонанса по (S)-сальвианоловой кислоте Т и (R)-сальвианоловой кислоте Т продемонстрирована в следующей таблице:


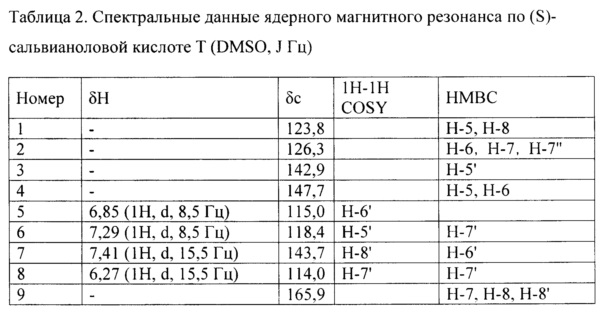
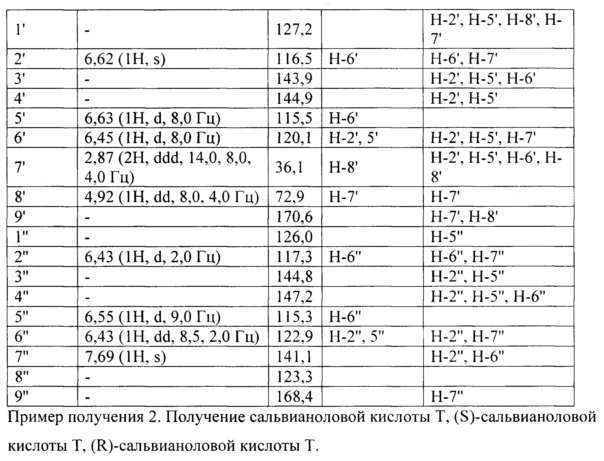
Отваренные кусочки Salvia miltiorrhiza и Radix Notoginseng помещали в аппарат китайской медицины для вываривания, добавляли воду с содержанием 0,45% (масса/объем) бикарбоната натрия, в 4 раза превышающей по объему отваренные кусочки Salvia miltiorrhiza и Radix Notoginseng, вываривали в течение 2 ч., фильтровали, фильтрат концентрировали с получением водного экстракта с относительной плотностью 1,25 (80°С).
К вышеупомянутому водному экстракту добавляли 95% (объем/объем) этанол, для осуществления осаждения пока конечное содержание этанола не будет составлять 65% (объем/объем) (25°С), отстаивали в течение 12 ч.; получали надосадочную жидкость и концентрировали в условиях пониженного давления с получением этанольного экстракта с относительной плотностью 1,28 (60°С).
Вышеупомянутый этанольный экстракт растворяли водой, а затем переносили в колонку с макропористой адсорбционной смолой АВ-8, для промывания колонки применяли кислый водный раствор со значением рН 2.5, пока элюат не становился почти бесцветным, затем для элюирования применяли 95% (объем/объем) этанол в объеме, 4 раз превышающем объем колонки, элюат концентрировали с получением экстракта без запаха спирта.
Полученный на предыдущем этапе экстракт растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 15:85:1), для очистки применяли жидкостный хроматограф высокого давления с колонкой динамического аксиального сжатия NOVASEP LC80-600 от французского производителя, насадкой для хроматографии являлась обращенно-фазовая колонка с силикагелем С18 (10 мкм, YMC Company), для линейного градиента применяли следующие условия: ацетонитрил : вода : муравьиная кислота (объемное отношение) заменяли от 15:85:1 до 20:80:1, от 0 мин до 60 мин; скорость потока: 300 мл/мин.; длина волны детектирования: 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонентов со временем удерживания 29,5-32,1 мин., концентрирование досуха с помощью ротационного испарителя, с получением образца сальвианоловой кислоты Т.
Вышеупомянутый образец сальвианоловой кислоты Т растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 17:83:1), для разделения хиральных изомеров применяли препаративный жидкостный хроматограф Waters Prep 400, хроматографическая колонка представляла собой обращенно-фазовую колонку CHIRALCEL® OD-RH (250×20 мм, 5 мкм), для линейного градиента применяли следующие условия: ацетонитрил : вода : муравьиная кислота (объемное отношение) от 17:83:1 до 22:78:1, от 0 мин до 45 мин; скорость потока: 20 мл/мин.; длина волны детектирования: 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонента в виде (S)-сальвианоловой кислоты Т со временем удерживания 25,2-27,1 мин., компонента в виде (R)-сальвианоловой кислоты Т со временем удерживания 32,4-34,2 мин., концентрирование элюата с помощью ротационного испарителя при 30°С, затем осуществляли лиофильную сушку с получением чистого продукта в виде (S)-сальвианоловой кислоты Т и чистого продукта в виде (R)-сальвианоловой кислоты Т.
Пик квазимолекулярного иона (S)-сальвианоловой кислоты Т, полученный с помощью масс-спектрометрии высокого разрешения имел значения m/z=537,1035; пик квазимолекулярного иона (R)-сальвианоловая кислота Т имел значения [М-Н]- m/z 537,1034.
Интерпретация спектральных данных ядерного магнитного резонанса по (S)-сальвианоловой кислоте Т и (R)-сальвианоловой кислоте Т продемонстрирована в следующей таблице:

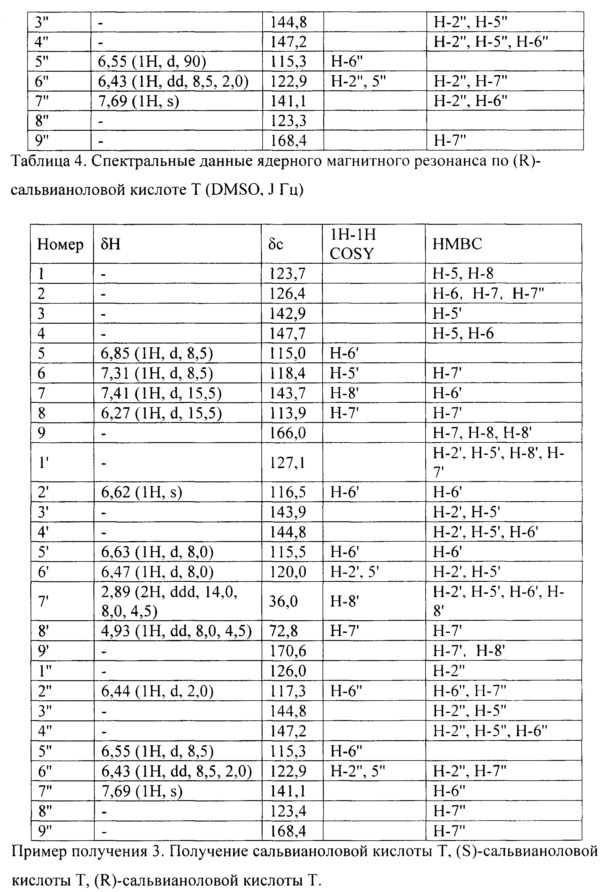
Получали сальвианоловую кислоту В и растворяли в воде с содержанием 0,3% (масса/объем) бикарбоната натрия, в 200 раз (массовое отношение) превышающей сальвианоловую кислоту В, помещали в круглодонную колбу, нагревали с обратным холодильником в течение 1 ч. при 90°С.
После реакции для доведения рН до 3,0 применяли водный раствор соляной кислоты в концентрации 0,1 моль/л, затем растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 15:85:1), для очистки применяли жидкостный хроматограф высокого давления с колонкой динамического аксиального сжатия NOVASEP LC80-600 от французского производителя, насадкой для хроматографии являлась обращенно-фазовая колонка с силикагелем С18 (10 мкм, YMC Company), для изократического элюирования применяли ацетонитрил : воду : муравьиную кислоту (объемное отношение) = 15:85:1; ацетонитрил : вода : муравьиная кислота (объемное отношение) меняли от 15:85:1 до 20:80:1, от 0 мин. до 60 мин.; скорость потока была 300 мл/мин.; длина волны детектирования была 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонентов со временем удерживания 21,2-24,0 мин., концентрирование досуха с помощью ротационного испарителя, с получением образца сальвианоловой кислоты Т.
Вышеупомянутый образец сальвианоловой кислоты Т растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 17:83:1), для разделения хиральных изомеров применяли препаративный жидкостный хроматограф Waters Prep 400, хроматографическая колонка представляла собой обращенно-фазовую колонку CHIRALCEL® OD-RH (250×20 мм. 5 мкм), для изократического элюирования применяли ацетонитрил : воду : муравьиную кислоту (объемное отношение) = 17:83:1; скорость потока была 25 мл/мин.; длина волны детектирования была 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонента в виде (S)-сальвианоловой кислоты Т со временем удерживания 19,5-21,1 мин., компонента в виде (R)-сальвианоловой кислоты Т со временем удерживания 23,9-25,3 мин., концентрирование элюата с помощью ротационного испарителя при 30°С, затем осуществляли лиофильную сушку с получением чистого продукта в виде (S)-сальвианоловой кислоты Т и чистого продукта в виде (R)-сальвианоловой кислоты Т.
Пример получения 4. Получение сальвианоловой кислоты Т, (S)-сальвианоловой кислоты Т, (R)-сальвианоловой кислоты Т.
Получали магниевую соль сальвианоловой кислоты В и растворяли в воде с содержанием 0,05% бикарбоната натрия, в 300 раз (массовое отношение) превышающей магниевую соль сальвианоловой кислоты В, помещали в круглодонную колбу, нагревали с обратным холодильником в течение 2 ч. при 90°С.
После реакции для доведения рН до 3,0 применяли водный раствор соляной кислоты в концентрации 0,1 моль/л, затем растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 15:85:1), для очистки применяли жидкостный хроматограф высокого давления с колонкой динамического аксиального сжатия NOVASEP LC80-600 от французского производителя, насадкой для хроматографии являлась обращенно-фазовая колонка с силикагелем С18 (10 мкм, YMC Company), для линейного градиента применяли следующие условия: ацетонитрил : вода : муравьиная кислота (объемное отношение) заменили от 15:85:1 до 20:80:1, от 0 мин до 60 мин; скорость потока: 250 мл/мин.; длина волны детектирования: 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонентов со временем удерживания 29,5-32,1 мин., концентрирование досуха с помощью ротационного испарителя, с получением образца сальвианоловой кислоты Т.
Вышеупомянутый образец сальвианоловой кислоты Т растворяли подвижной фазой (ацетонитрил : вода : муравьиная кислота (объемное отношение) = 17:83:1), для разделения хиральных изомеров применяли препаративный жидкостный хроматограф Waters Prep 400, хроматографическая колонка представляла собой обращенно-фазовую колонку CHIRALCEL® OD-RH (250×20 мм, 5 мкм), для линейного градиента применяли следующие условия: ацетонитрил : вода : муравьиная кислота (объемное отношение) меняли от 17:83:1 до 22:78:1, от 0 мин. до 45 мин.; скорость потока: 20 мл/мин.; длина волны детектирования: 280 нм. Мониторинг процесса элюирования осуществляли с помощью высокоэффективной жидкостной хроматографии, сбор компонента в виде (S)-сальвианоловой кислоты Т со временем удерживания 25,2-27,1 мин., компонента в виде (R)-сальвианоловой кислоты Т со временем удерживания 32,4-34,2 мин., концентрирование элюата с помощью ротационного испарителя при 30°С, затем осуществляли лиофильную сушку с получением чистого продукта в виде (S)-сальвианоловой кислоты Т и чистого продукта в виде (R)-сальвианоловой кислоты Т.
Пример составления 1. Получение таблеток сальвианоловой кислоты Т, (S)-сальвианоловой кислоты Т, (R)-сальвианоловой кислоты Т.

Способ получения:
1. Грануляция
Сальвианоловую кислоту Т, (S)-сальвианоловую кислоту Т, (R)-сальвианоловая кислоту Т и другие вспомогательные вещества, перечисленные в составе, просеивали через сито 100 меш в указанном порядке. Согласно дозировке состава, сальвианоловая кислота Т, авицел, крахмал и натрия карбоксиметилкрахмал хорошо перемешивали в соответствии со способом постепенного возрастания эквивалентов, для получения мягких материалов применяли необходимое количество 5% (масса/объем) PVP в безводном этаноле, гранулировали с помощью сита 14 меш и сушили при 50-60°С в течение 1 ч. Стеарат магния, согласно дозировке состава, добавляли для просеивания гранул с помощью сита 14 меш.
2. Прессование таблеток
Полученные гранулы прессовали с помощью пуансона для получения таблеток.
Пример составления 2. Получение капсул сальвианоловой кислоты Т, (S)-сальвианоловой кислоты Т, (R)-сальвианоловой кислоты Т.

Способ получения:
1. Грануляция
Сальвианоловую кислоту Т, (S)-сальвианоловую кислоту Т, (R)-сальвианоловая кислоту Т и другие вспомогательные вещества, перечисленные в составе, просеивали через сито 100 меш в указанном порядке. Согласно дозировке состава, сальвианоловую кислоту Т, крахмал и натрий карбоксиметилкрахмал хорошо перемешивали в соответствии со способом постепенного возрастания эквивалентов, для получения мягких материалов применяли необходимое количество 5% (масса/объем) PVP в безводном этаноле, гранулировали с помощью сита 14 меш и сушили при 50-60°С в течение 1 ч. Стеарат магния, согласно дозировке состава, добавляли для просеивания гранул с помощью сита 14 меш.
2. Инкапсуляция
Полученными гранулами заполняли капсулы.
Пример составления 3. Получение инъекционных форм сальвианоловой кислоты Т, (S)-сальвианоловой кислоты Т, (R)-сальвианоловой кислоты Т.

Способ получения:
Сальвианоловую кислоту Т, (S)-сальвианоловую кислоту Т, (R)-сальвианоловую кислоту Т брали в соответствии с дозировкой состава, растворяли в 1000 мл воды для инъекций и равномерно перемешивали; маннитол брали в соответствии с. дозировкой состава, растворяли в 500 мл воды для инъекций и добавляли к вышеуказанному раствору, равномерно перемешивали, в раствор добавляли 0,5 г активированного угля при перемешивании при постоянной температуре в течение 30 мин. и фильтровали, рН фильтрата доводили до 4,5-5,0, разбавляли водой для инъекций до 2500 мл, фильтровали в стерильных условиях, наполняли по отдельности с получением продукта.
Пример составления 4. Получение лиофилизированного порошка сальвианоловой кислоты Т, (S)-сальвианоловой кислоты Т, (R)-сальвианоловой кислоты Т.

Способ получения:
Сальвианоловую кислоту Т, (S)-сальвианоловую кислоту Т, (R)-сальвианоловую кислоту Т и маннитол взвешивали в соответствии с дозировкой состава и растворяли в 1500 мл воды для инъекций путем перемешивания, в раствор добавляли 0,5 г активированного угля для обесцвечивания при перемешивании в течении 20 мин., раствор фильтровали через микропористую фильтровальную мембрану (0,45 мкм) с целью удаления угля и разбавляли водой для инъекций до 2000 мл. Полученный раствор фильтровали в стерильных условиях, упаковывали по отдельности и лиофилизировали с получением продукта.
Пример фармакодинамического теста 1. Эффект (S)-сальвианоловой кислоты Т в отношении предупреждения острого инфаркта миокарда при перевязке коронарной артерии
Материалы
1. Материалы для исследования и реагенты
(S)-сальвианоловая кислота Т, № партии: 120301, предоставлена Tasly Holding Group Academy,
Покрытые кишечнорастворимой оболочкой таблетки аспирина: описание: 100 мг/штуку, Bayer healthcare Co., Ltd., № партии: BJ07160.
Инъекционный хлорид натрия в количестве 0,9% (масса/объем), Nanjing Xiaoying Pharmaceutical Group Co. Ltd., № партии: 2012051205.
хлоралгидрат: AR, Sinopharm Chemical Reagent Co., Ltd., № партии: 20100111.
Тетразолий красный (TTC), Sinopharm Chemical Reagent Co., Ltd., № партии: F20040308.
Набор реактивов для определения креатинкиназы (СК), № партии: 20120917; набор реактивов для определения молочной кислоты (LD), № партии: 20120919; набор реактивов для определения малонового диальдегида (MDA), № партии: 20120919; набор реактивов для определения супероксиддисмутазы (SOD), № партии: 20120918; набор реактивов для определения изофермента креатинкиназы (СК-МВ), № партии: 20120922; набор реактивов для определения фермента АТФ, № партии: 20120921. Все предоставлены Институтом биоинженерии Nanjing Jiancheng.
2. Основные приборы:
респиратор НХ-300: Chengdu Taimeng Science and Technology Co., Ltd.
электрокардиограф ECG-6511: Shanghai Photoelectric Medical Electronic Instrument Co., Ltd.
Баня-термостат с цифровым экраном НН-2: Guohua Electric Appliance Co., Ltd.
Электронные весы типа BS 224s: Beijing Sartorius Instrument System Engineering Co., Ltd.
Электронные весы типа BS 110s: Beijing Sartorius Instrument System Engineering Co., Ltd.
3. Экспериментальные животные:
крысы SD, масса тела 210~230 г, самцы, предоставлены Beijing Vital River Laboratory Animal Technology Co., Ltd.; № сертификата SCXK (Su) 2009-0001.
Экспериментальные способы и результаты
1. Схема введения дозы
Доза (S)-сальвианоловой кислоты Т в виде лиофилизированного порошка составляла 20 мг/кг массы тела, 10 мг/кг массы тела. Аспирин: 30 мг/кг массы тела.
2. Экспериментальные способы
Брали самцов крыс чистой линии SD и рандомизировано распределяли в группы в соответствии с массой тела, включая группу имитации операции (дистиллированная вода), модельную группу (дистиллированная вода), группу аспирина, группу низкой дозы (S)-сальвианоловой кислоты Т, группу высокой дозы (S)-сальвианоловой кислоты Т, и в каждой группе было 10 крыс. Для каждой группы крыс осуществляли введение путем введения в желудок один раз в день, непрерывно в течение 10 дней. Вводимый объем составлял 1 мл/100 г массы тела. Через 1 ч. после окончания времени введения, делали инъекцию хлоралгидрата в концентрации 300 мг/кг массы тела путем внутрибрюшинной инъекции, наркотизировали, фиксировали в положении на спине, дезинфицировали операционные поля йодом и спиртом последовательно, затем вскрывали грудную клетку в области третьего и четвертого ребер, и обеспечивали искусственную вентиляцию легких, освобождали сердце, перевязывали левую переднюю нисходящую ветвь коронарной артерии с помощью медицинской атравматической хирургической иглы 5/0, быстро закрывали полость грудной клетки и дезинфицировали обычным образом, время операционной процедуры составляло менее 30 с, искусственная вентиляция легких продолжалась в течение 1~2 мин. после операции, для предотвращения инфекции делали внутримышечную инъекцию (i. m.) 200 тысяч единиц пенициллина. Запись ЭКГ во II стандартном отведении за 5 минут до операции, через 0, 1 мин., 5 мин., 15 мин., 1 ч., 4 ч. после перевязки с целью исследования изменений J точки ЭКГ.
Извлечение сердца непосредственно после окончания эксперимента, промывание от крови физиологическим раствором, отрезание предсердия и нижней части кровеносных сосудов, взвешивание массы желудочка, разрезание желудочка на 5 кусков посередине, параллельно предсердно-желудочковой борозде, и помещение в 1% (масса/объем) раствор ТТС, окрашивание на водяной бане при постоянной температуре 37°С в течение 5 мин., получение цифровых фото сразу по извлечении, затем отделение неокрашенных частей (то есть частей с инфарктом) и взвешивание для вычисления процента их массы от массы всего желудочка (процентный показатель инфаркта миокарда), и проведение t-теста для модельной группы ишемии. Формула для вычисления процентного показателя инфаркта представляет собой следующее:
Процентный показатель инфаркта (%) = (масса светлых участков/масса желудочка) × 100%
После этого кровь осаждали центрифугированием при 2000 об./мин. в течение 25 минут, отделение сыворотки, определение содержания или активности в сыворотке креатинкиназы (СК), лактатдегидрогеназы (LD), изофермента креатинкиназы (СК-МВ), малональдегида (MDA), супероксиддисмутазы (SOD), АТФазы. Результаты приведены в таблице 5, 6, 7 и фиг. 11.
Как показано в таблице 5, J точки ЭКГ у крыс каждой группы после операции по перевязке коронарной артерии были очевидно выше, чем такие до операции (Р<0,01), что указывало на успешность моделирования. По сравнению с модельной группой, через 5 минут после операции в группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось значительное замедление повышения J точек (Р<0,05); через 15 минут после операции в группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось значительное замедление повышения J точек (Р<0,05); через 1 ч. и 4 ч. после операции, и в группе аспирина, и в группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось замедление повышения J точек (Р<0,05).
Как показано в таблице 6, степень выраженности инфаркта миокарда в модельной группе и в каждой группе, которой вводили лекарственный препарат, была очевидно выше, чем в группе имитации операции (Р<0.01), что указывало на успешность моделирования. По сравнению с модельной группой, и в группе аспирина, и в группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось значительное уменьшение степени выраженности инфаркта миокарда (Р<0,05).
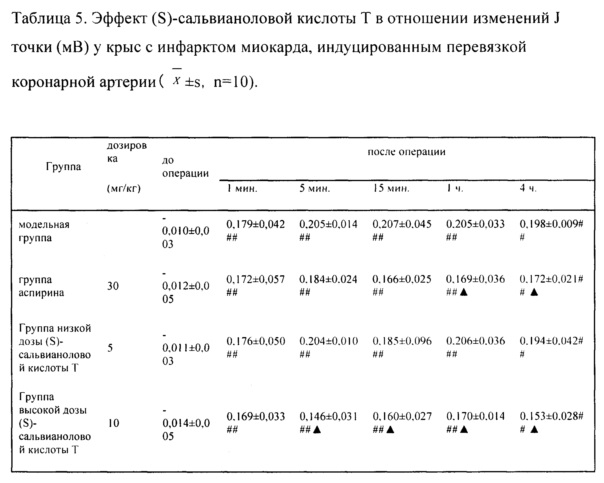
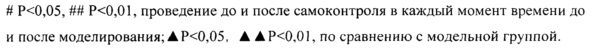


Как показано в таблице 7, уровни MDA, LD, СК, СК-МВ сыворотки в модельной группе были очевидно выше чем в группе имитации операции (Р<0,01), в то время как уровни SOD, Na+-К+-АТФазы были очевидно ниже чем в группе имитации операции (Р<0,01), что указывало на успешность моделирования. По сравнению с модельной группой, в группе аспирина, группе низкой дозы (S)-сальвианоловой кислоты Т, группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось очевидное снижение содержания в сыворотке MDA и LD у крыс с ишемией миокарда (Р<0,05); в группе аспирина, группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось очевидное повышение активности SOD сыворотки у крыс с ишемией миокарда (Р<0,05, Р<0,01 соответственно); в группе аспирина, группе низкой дозы (S)-сальвианоловой кислоты Т, группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалась очевидное снижение активности СК в сыворотке у крыс с ишемией миокарда (Р<0,05, Р<0,01); в группе аспирина, группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось очевидное снижение активности СК-МВ (Р<0,01); в группе аспирина, группе низкой дозы (S)-сальвианоловой кислоты Т, группе высокой дозы (S)-сальвианоловой кислоты Т наблюдалось очевидное повышение активности Na+-К+-АТФазы.
Как показано на фигуре 11, в группе высокой дозы (S)-сальвианоловой кислоты Т и группе аспирина наблюдали одинаковый эффект в отношении предупреждения острого инфаркта миокарда, индуцированного перевязкой коронарной артерии. Дополнительный тест продемонстрировал, что аналогично (S)-сальвианоловой кислоте Т, в группе высокой дозы (R)-сальвианоловой кислоты Т и группе аспирина также наблюдали одинаковый эффект в отношении предупреждения острого инфаркта миокарда, индуцированного перевязкой коронарной артерии.
Пример фармакодинамического теста 2. Защитный эффект (R)-сальвианоловой кислоты Т в отношении экспериментальной острой ишемии миокарда у крыс.
Материалы для эксперимента
1. Материалы для исследования и реагенты: инъекционный питуитрин (Pit) производства Nanjing Xinbai Pharmaceutical Co., Ltd., с № партии 070302. Физиологический раствор производства Tianjin Tian'an Pharmaceutical Co., Ltd., с № партии 201009231, описание: 500 мл/флакон. (R)-сальвианоловая кислота Т с более чем 95% чистотой была предоставлена от института TASLY HOLDING GROUP.CO.LTD.
2. Основные приборы: 8-канальный регистратор биофизиологических параметров MedLab производства Nanjing Medease Science and Technology Co., Ltd.
3. Животные: крысы SD, поровну самцов и самок с массой тела 220-230 г, предоставлены Beijing Vital River Laboratory Animal Technology Co., Ltd.; с №сертификата SCXK (Jing) 2007-0001. Всех крыс кормили в соответствии со специальным рационом для крыс (производства Beijing Keaoxieli Diet Co., Ltd.) и поили водопроводной водой в комнате для кормления животных при комнатной температуре 20-25□, освещаемой в течение 12 ч.
Экспериментальные способы
1. Схема введения дозы
В группе высокой дозы вводимая доза (R)-сальвианоловой кислоты Т в виде лиофилизированного порошка составляла 10 мг/кг массы тела; в группе низкой дозы 5,0 мг/кг массы тела.
2. Группирование:
2.1 Скрининг животных: перед официальным экспериментом крысам делали инъекцию питуитрина в хвостовую вену (Pit) (1 ед./кг). Нормальную ЭКГ и ЭКГ через 5 мин. после инъекции записывали с целью наблюдения подъема J точки и отклонения от нормы зубца Т. Животные с аномальной ЭКГ до инъекции, или которые были нечувствительны к Pit, были отклонены.
2.2 Группирование животных: необходимых крыс делили на 3 группы, называли соответственно: □ модельная контрольная группа, □ группа низкой дозы (R)-сальвианоловой кислоты Т в виде лиофилизированного порошка; □ группа низкой дозы (R)-сальвианоловой кислоты Т в виде лиофилизированного порошка.
3. Экспериментальные способы: крыс SD с массой тела 220-250 г, поровну самцов и самок, рандомизированно делили на группы, 10 животных на каждую группу. Крысам в группах лечения каждый день перорально вводили водные суспензии разных образцов, в то время как крысам модельной контрольной группы перорально вводили такой же объем физиологического раствора. Осуществляли введение всем животным последовательно в течение 7 дней. Через 40 мин. после последнего введения крыс наркотизировали и подсоединяли к приборам для записи нормальной ЭКГ во II отведении. Делали инъекцию питуитрина (Pit) при постоянной скорости в дозе 1 ед./кг массы тела в хвостовую вену в течение приблизительно 10 с. Изменения ECG записывали на 0 с, 5 с, 10 с, 15 с, 30 с, 45 с, 1 мин., 2 мин., 3 мин., 4 мин., 5 мин., 10 мин. и 15 мин. после введения. Различия между случаями до инъекции и после инъекции Pit для каждой группы, а также между группой лечения и модельной контрольной группой, сравнивали с целью анализа изменений J точки и зубца Т, и анализировали данные с помощью t-теста.
Экспериментальные результаты
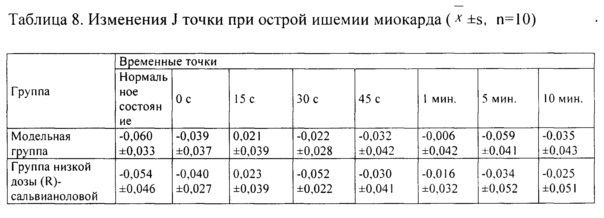
1. Эффект в отношении J точки
Как показывают результаты таблицы 8, по сравнению с модельной контрольной группой, повышение зоны J точки на ЭКГ в группе высокой дозы (R)-сальвианоловой кислоты Т составляло меньше на 15 с, 30 с и 45 с при острой ишемии миокарда, вызванной питуитрином, и разница характеризовалась статистической значимостью в данных условиях эксперимента (Р<0,05).

Как показывают результаты таблицы 9, по сравнению с модельной контрольной группой, повышение зоны Т зубца на ЭКГ в группе высокой дозы (R)-сальвианоловой кислоты Т составляло меньше на 15 с и 30 с, и разница характеризовалась статистической значимостью в данных условиях эксперимента (Р<0,05).
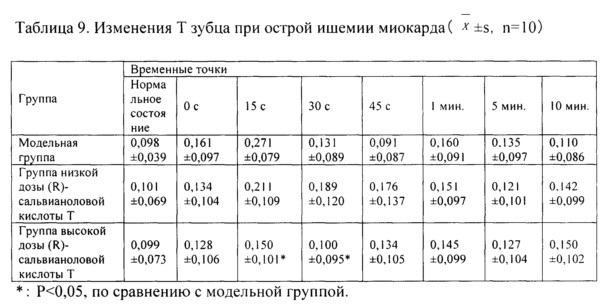
Выводы
По сравнению с модельной контрольной группой, повышение зоны J точки на ЭКГ и Т зубца в группе высокой дозы (Я)-сальвианоловой кислоты Т составляло меньше на 15 с и 30 с, и разница характеризовалась статистической значимостью (Р<0,05). Как показали результаты, в данном исследовании (R)-сальвианоловая кислота Т (10,0 мг/кг) обладала эффектом в отношении острой ишемии миокарда. Дополнительный эксперимент показал, что аналогично (R)-сальвианоловой кислоте Т, (S)-сальвианоловая кислота обладала аналогичным эффектом в отношении острой ишемии миокарда.
Пример фармакодинамического теста 3. Реакция захвата свободных радикалов (S)/(R)-сальвианоловой кислоты Т.
В связи с их прямым или опосредованным эффектом окисления, было показано, что свободные радикалы повсеместно участвуют в физиологических и патологических процессах. При наличии избыточного количества свободных радикалов, посредством окисления они постоянно повреждают макромолекулы организма. Соединения сальвианоловой кислоты являются донорами фенольной гидроксильной группы, характеризуясь структурным основанием для их антиоксидантной активности. В данном исследовании для наблюдения активности захвата свободных радикалов (S)/(R)-сальвианоловой кислотой Т применяли модельную реакцию захвата свободных радикалов 1,1-дифенил-2-пикрил-гидразила (DPPH).
1. Реагенты и приборы
(S)/(R)-сальвианоловая кислота Т с более чем 95% чистотой была предоставлена Tasly Group Academy. Витамин С и DPPH были приобретены от SIGMA Inc. Ультрафиолетовый спектрофотометр (UV-1800) был приобретен от Beijing Rayleigh Analytical Instrument Co., Ltd.
2. Экспериментальные способы
Общий объем реакции составлял 2 мл. Добавляли 1 мл растворов образцов в разных концентрациях в 80% метаноле к 100 мкМ метанольному раствору DPPH, равномерно перемешивали, и давали раствору прореагировать в течение 20 мин. при 25°С, в темноте. Поглощение реакционного раствора измеряли при 517 нм. В данном исследовании витамин С считали положительным контролем. Скорость захвата свободных радикалов рассчитывали в соответствии со следующим уравнением:
Скорость захвата свободных радикалов
(%) = [1-А образец/А контроль)/А контроль] × 100%
Где А образец означает поглощение исследуемых образцов, а А контроль означает поглощение холостой пробы.
Экспериментальные результаты
(S)/(R)-сальвианоловая кислота Т обладает более высокой степенью захвата свободных радикалов, чем витамин С, но между двумя изомерами значительная разница в захвате свободных радикалов не наблюдалась (Р<0,05).

Пример фармакодинамического теста 4. Определение восстановительной способности (S)/(R)-сальвианоловой кислоты Т.
В определенной степени, потенциал профилактического антиокислительного действия представлен восстановительной способностью лекарственного средства. Проводили исследование касательно восстановительной способности (S)/(R)-сальвианоловой кислоты Т по настоящему изобретению.
1. Реагенты и приборы
(S)/(R)-сальвианоловая кислота Т с более чем 95% чистотой была предоставлена Tasly group Academy. Химически чистый ферроцианид калия был приобретен от Tianjin No. 1 Chemical Reagent Factory. Химически чистая трихлоруксусная кислота была приобретена от Sinopharm Chemical Reagent Co., Ltd. Химически чистый хлорид железа был приобретен от Tianjin Fengchuan Chemical Reagent Science and Technology Co., Ltd. Витамин С был приобретен от SIGMA Inc. Ультрафиолетовый спектрофотометр (UV-1800) был приобретен от Beijing Rayleigh Analytical Instrument Co., Ltd. Центрифуга с охлаждением (Z323K) была приобретена от HEMMLE, Германия.
2. Экспериментальные способы
Соответствующим образом отбирали 0,5 мл 200 мМ фосфатного буфера (рН 6,8) с содержанием (S)/(R)-сальвианоловой кислоты Т в разных концентрациях и 0,5 мл 1,0% (масса/объем) раствора ферроцианида калия и охлаждали на ледяной бане после нагревания на водяной бане (50□) в течение 20 мин. Добавляли 0,5 мл 10% раствора трихлоруксусной кислоты и осаждали центрифугированием при 1000 g/мин. в течение 10 мин. Отбирали 1,0 мл полученной надосадочной жидкости, добавляли в нее 1,0 мл дистиллированной воды и 0,2 мл 0,1% (масса/объем) раствора хлорида железа, отстаивали в течение 10 мин. и измеряли поглощение при 700 нм. В то же время проводили контрольный эксперимент. Витамин С представляет собой вещество с сильной восстановительной способностью, в данном исследовании служащее в качестве положительного контроля. Восстановительная способность образца представлена в виде вычитанного от поглощения исследуемого образца поглощения холостой пробы. Таким образом, это означает, что чем выше поглощение, тем сильнее восстановительная способность.
3. Экспериментальные результаты
В каждом случае восстановительная способность (S)/(R)-сальвианоловой кислоты Т была намного сильнее, чем такая витамина С, но между двумя значительная разница восстановительной способности не наблюдалась (Р<0,05).

Пример фармакодинамического теста 5. Эксперимент с (8)/(Я)-сальвианоловой кислотой Т в отношении ее действия против фиброза легких у мышей.
Фиброз легких является типичной реакцией и осложнением при поражении легких, патологические изменения при котором представляют собой воспаление альвеол, образование фибробластических очагов и повторяющееся восстановление, и избыточное накопление внеклеточного матрикса. Фиброз легких, как правило, заканчивается окончательной потерей дыхательной функции, и характеризуется отсутствием эффективных средств предупреждения и лечения. Мыши с фиброзом легких, индуцируемым блеомицином, являются общепринятой моделью, применяемой для изучения фиброза легких у людей, в данной модели, свободный кислородный радикал, который прямо или опосредованно образуется под действием блеомицина, является одним из механизмов, который вызывает фиброз легких.
1. Реагенты и приборы
(S)/(R)-сальвианоловая кислота Т с более чем 95% чистотой была предоставлена Tasly group Academy. Набор для определения супероксиддисмутазы (SOD), набор для определения каталазы (CAT), набор для определения пероксидазы (POD), набор для определения малонового диальдегида (MDA) были приобретены от Института биоинженерии Nanjing Jiancheng. Блеомицин был приобретен от SIGMA.
2. Животные
Мыши Kunming, все самки, с массой тела 22-25 г, были предоставлены Beijing Vital River Laboratory Animal Technology Co., Ltd., кормление осуществляли при температуре 23±2°С, при относительной влажности 65%-75%, и световом периоде 12 ч.:12 ч.
3. Экспериментальные способы
Делили 40 мышей на 4 группы: контрольная группа с нормальным состоянием, которой давали физиологический раствор путем закапывания носа, перорально вводили физиологический раствор; модельная контрольная группа: давали блеомицин путем закапывания носа, перорально вводили физиологический раствор; группа (S)-сальвианоловой кислоты Т (16 мг/кг): давали блеомицин путем закапывания носа, перорально вводили раствор (S)-сальвианоловой кислоты Т; группа (R)-сальвианоловой кислоты Т (16 мг/кг): давали блеомицин путем закапывания носа, перорально вводили раствор (R)-сальвианоловой кислоты Т; 10 мышей в каждой группе.
На первый день эксперимента мышей наркотизировали хлоралгидратом, каждой мыши давали 50 мкг блеомицина путем однократного закапывания носа с целью создания модели фиброза легких. На второй день эксперимента двум группам лечения перорально вводили (S)/(R)-сальвианоловую кислоту Т, модельной группе и группе с нормальным состоянием перорально вводили такой же объем физиологического раствора, один раз каждый день, 3 недели подряд. На 21-ый день эксперимента от крови, собранной из глазной вены, отделяли сыворотку с целью выявления TGF-β1, мышей умерщвляли для забора легких, которые измельчали с дважды дистиллированной водой (соотношение легких и дважды дистиллированной воды составляло 1:5 (масса/объем)) с образованием тканевого гомогената, осаждали центрифугированием для выявления в тканях MDA, супероксиддисмутазы (SOD), каталазы (CAT), пероксидазы (POD).
Статистический способ: Все результаты были продемонстрированы как x±s, анализ проводили с помощью однофакторного дисперсионного анализа между группами, с применением программного обеспечения для статистики, парное сравнение выполняли с двумя группами в t-тесте с независимыми выборками.
4. Экспериментальные результаты
Эффект в отношении индуцированного блеомицином фиброза легких у мышей: по сравнению с мышами с нормальным состоянием, уровень TGF-β1 в легких мышей с индуцированным блеомицином фиброзом легких был повышен в 9,96 раз, MDA был повышен в 1,83 раза, уровень антиоксидазы SOD, POD, CAT снижен в 1,5 раза, и TGF-β1 сыворотки также повышен в 4,44 раза, и модель была создана успешно. (S)/(R)-сальвианоловая кислота Т могла как ингибировать индуцированное блеомицином повышение TGF-β1 в сыворотке мышей (таблица 12), так и предупреждать индуцированное блеомицином снижение SOD, POD, CAT в легких мышей (таблица 13).


Пример фармакодинамического теста 6. Ингибиторный эффект (S)/(R)-сальвианоловой кислоты Т в отношении фибробластов, индуцированных TGF-β.
Чрезмерная пролиферация фибробластов, индуцированная TGF-β и дифференциация фибробластов, активных в отношении образования миофибробластов, играет важную роль в возникновении фиброза легких, и ингибирование передачи сигнала TGF-β, которое могло бы предупредить пролиферацию и активацию фибробластов легких, представляет собой важное средство эффективного предупреждения и лечения фиброза легких.
1. Реагенты и приборы
(S)/(R)-сальвианоловая кислота Т с более чем 95% чистотой была предоставлена Tasly group Academy. Культуральная среда DMEM, МТТ, рекомбинантный TGF-β1 были приобретены у компании Sigma. Пенициллин и стрептомицин производства CSPC фармацевтический Group Limited. Фетальная бычья сыворотка производства Hangzhou Sijiqing института материалов для биологической инженерии. Набор реактивов для определения коллагена производства компании Biocolor. Набор Laminin(LN) R1A был приобретен от Beijing Beifang биотехнологического института.
Планшет-ридер EL-800X типа был приобретен от компании BIO-ТЕК; CO2-инкубатор был приобретен от компании Thermo; проточный цитометр был приобретен от компании FACS.
2. Клеточный штамм
Клетки L929 были приобретены от Института изучения клетки Военной академии медицинских наук.
3. Экспериментальные способы
Клетки L929 в клеточной популяции 5×107/л, которым добавляли культуральный раствор DMEM с содержанием 10% фетальной бычьей сыворотки, высеивали на 96-луночный планшет, культивировали в течение 24 ч. Проводили дополнение такой же культуральной средой с содержанием 2 мкг/л TGF-β1 и (S)/(R)-сальвианоловой кислоты Т в разных концентрациях. В каждой группе было 6 разных лунок, конечная установленная концентрация (S)/(R)-сальвианоловой кислоты Т была 0, 1, 3, 10, 20, 40, 80, 150 мкМ/л. Удаление среды после культивирования в течение 72 ч., анализ клеточной активности способом МТТ. 4. Экспериментальные результаты
(S)-сальвианоловая кислота Т и (R)-сальвианоловая кислота Т характеризовались ингибиторным эффектом в отношении индуцированной TGF-β1 пролиферации клеток L929 (Фиг. 12), значение IC50 (S)-сальвианоловой кислоты Т составляло 26,1 мкмоль/л, значение IC50 (R)-сальвианоловой кислоты Т составляло 26,9 мкмоль/л, между указанными двумя значительная разница не наблюдалась.
Результаты фармакодинамического теста в настоящем изобретении демонстрируют, что сальвианоловая кислота Т по настоящему изобретению обладает активностью в отношении предупреждения острого инфаркта миокарда и острой ишемии миокарда, характеризуется высоким уровнем захвата свободных радикалов и восстановительной способностью, а также активностью в отношении лечения фиброза легких.






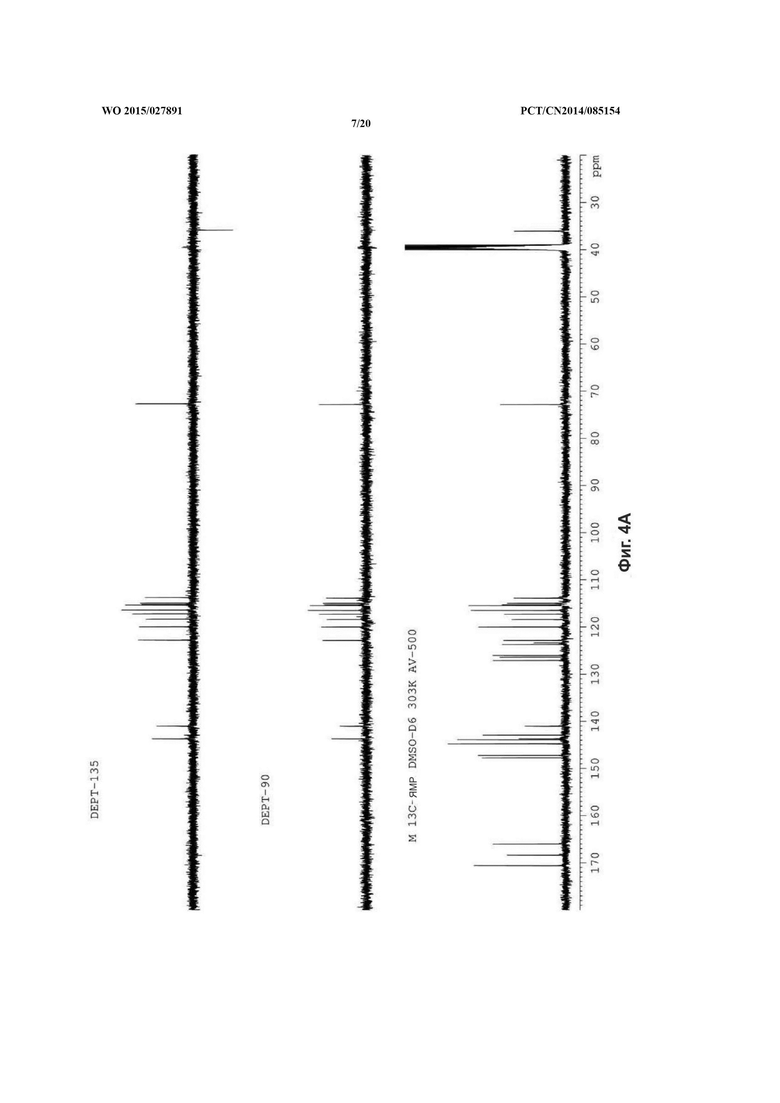





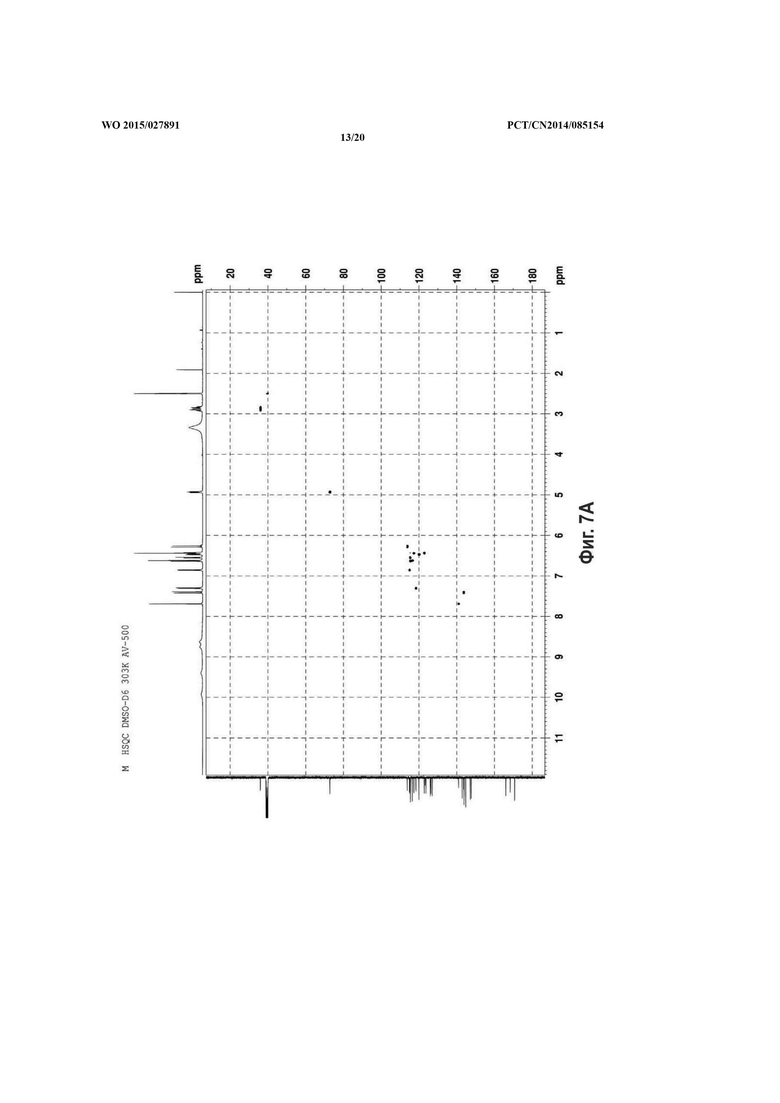

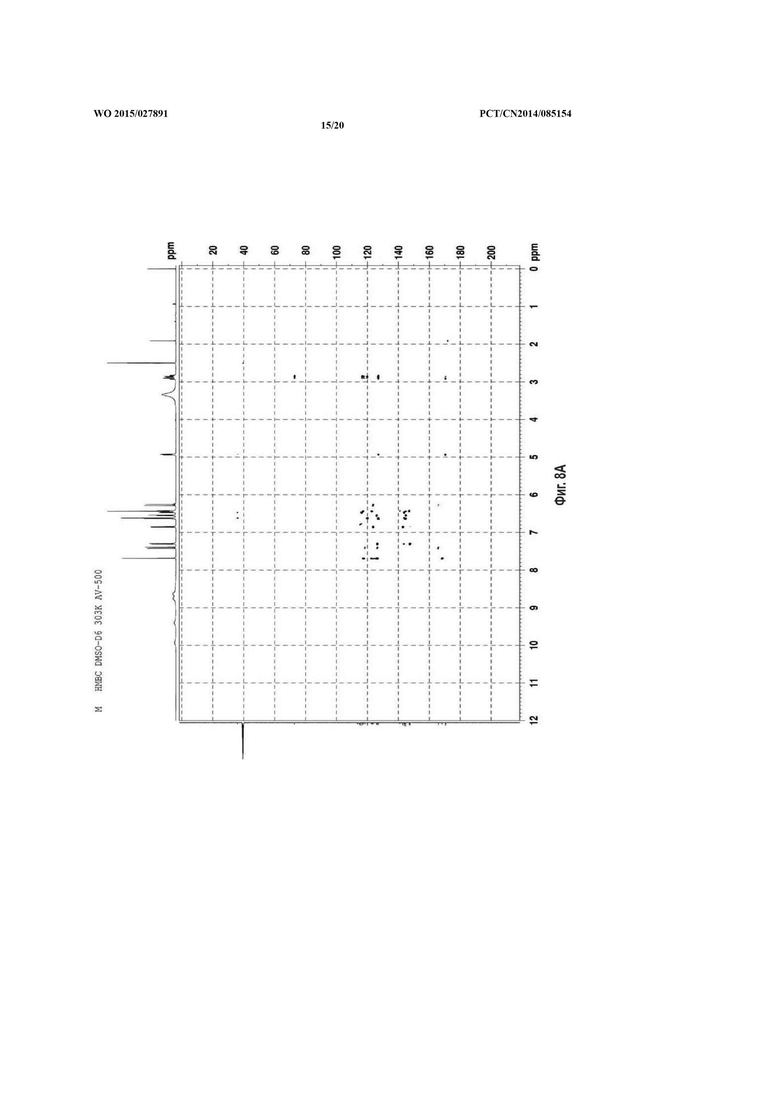

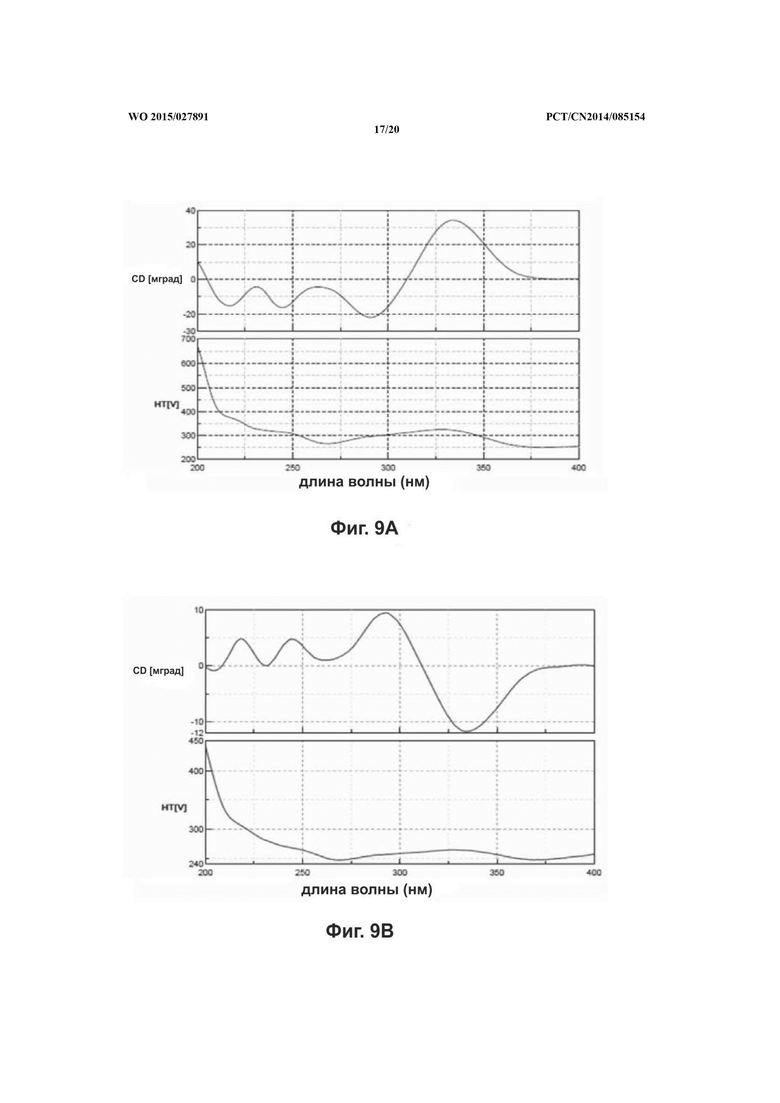



Изобретение относится к области медицины, в частности к сальвианоловой кислоте T, описанной структурной формулой (I), или ее фармацевтически приемлемой соли, или ее R- или S-изомеру. Изобретение также относится к применению соединения сальвианоловой кислоты Т или ее фармацевтически приемлемой соли или ее R- или S-изомера для антиокислительного действия, для замедления старения или для лечения острого инфаркта миокарда, острой ишемии миокарда или фиброзного заболевания легких. 9 н. и 16 з.п. ф-лы, 22 ил., 13 табл., 4 пр.

1. Соединение сальвианоловой кислоты Т, представленной структурной формулой (I), или ее фармацевтически приемлемой соли, или ее R- или S-изомера,

2. Способ получения сальвианоловой кислоты Т по п. 1, где способ предусматривает следующие стадии:
(1а) экстракции: экстрагирование лекарственного сырья из Radix Salviae Miltiorrhizae или смеси Radix Salviae Miltiorrhizae и другого лекарственного сырья водой, концентрирование фильтрата с получением водного экстракта, затем добавление спирта к осадку и получение надосадочной жидкости, концентрирование надосадочной жидкости с получением спиртового экстракта;
(1b) разделения: разбавление спиртового экстракта из стадии (1а) в воде, перенос на макропористую адсорбционную смолу, промывание смолы кислым водным раствором с целью удаления примесей и затем элюирование смолы этанолом с получением этанольного элюата, концентрирование этанольного элюата с получением экстракта;
(2) очистки: доведение рН реакционной жидкости, полученной на стадии (1), до кислого значения или очистка экстракта, полученного на стадии (1b), с помощью препаративного жидкостного хроматографа высокого давления с применением обращенно-фазовой колонки с силикагелем С18 в качестве наполнителя для хроматографии, ацетонитрилом-водой-муравьиной кислотой в качестве элюента, причем осуществляют изократическое элюирование или градиентное элюирование с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего сальвианоловую кислоту Т; концентрирование с получением сальвианоловой кислоты Т.
3. Способ получения сальвианоловой кислоты Т по п. 1, где способ предусматривает следующие стадии:
(1) синтеза: растворение сальвианоловой кислоты В в воде, нагревание;
(2) очистки: доведение рН реакционной жидкости, полученной на стадии (1), до кислого значения или очистка экстракта, полученного на стадии (1b), с помощью препаративного жидкостного хроматографа высокого давления с применением обращенно-фазовой колонки с силикагелем С18 в качестве наполнителя для хроматографии, ацетонитрилом-водой-муравьиной кислотой в качестве элюента, причем осуществляют изократическое элюирование или градиентное элюирование с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего сальвианоловую кислоту Т; концентрирование с получением сальвианоловой кислоты Т.
4. Способ получения по п. 2, где на стадии (1а) указанное лекарственное сырье из Radix Salviae Miltiorrhizae или смесь Radix Salviae Miltiorrhizae и другого лекарственного сырья представляют собой отваренные кусочки, измельченные частицы или порошки, при этом указанное другое лекарственное сырье представляет собой Radix Notoginseng, или Radix Astragali, или комбинацию из двух, которые совместимы с Radix Salviae Miltiorrhizae, при этом указанная водная экстракция заключается в следующем: отваривание лекарственного сырья в воде, объем которой в 4-8 раз превышает объем лекарственного сырья, в течение 1,5-4 ч; фильтрование; концентрирование фильтрата с получением водного экстракта с относительной плотностью 1,10-1,30 (80°С), предпочтительно отваривание лекарственного сырья в воде, объем которой в 6 раз превышает объем лекарственного сырья, в течение 3 ч; фильтрование; концентрирование фильтрата с получением водного экстракта с относительной плотностью 1,22 (80°С).
5. Способ получения по п. 2, где на стадии (1а) щелочной водный раствор применяют на указанной стадии водной экстракции, при этом указанная щелочь представляет собой по меньшей мере одну выбранную из группы, включающей раствор бикарбоната натрия, водный раствор карбоната натрия, раствор гидрокарбоната калия, раствор карбоната калия, водный раствор гидроксида натрия, водный раствор гидроксида калия, предпочтительно представляет собой водный раствор бикарбоната натрия в концентрации 0,3-0,45% (масса/объем).
6. Способ получения по п. 2, где на стадии (1а) указанное осаждение спиртом заключается в следующем: добавление 95% (объем/объем) этанола в водный экстракт для осаждения до тех пор, пока содержание этанола не будет составлять 50-70% (объем/объем) (25°С), и отстаивание в течение 8-36 ч; получение надосадочной жидкости, извлечение этанола в условиях пониженного давления, концентрирование с получением спиртового экстракта с относительной плотностью 1,25-1,5 (60°С), предпочтительно добавление 95% (объем/объем) этанола в водный экстракт для осаждения до тех пор, пока содержание этанола не будет составлять 60% (объем/объем) (25°С), и отстаивание в течение 24 ч; получение надосадочной жидкости, извлечение этанола в условиях пониженного давления, концентрирование с получением спиртового экстракта с относительной плотностью 1,32 (60°С).
7. Способ получения по п. 2, где на стадии (1b) указанная макропористая адсорбционная смола может представлять собой неполярную или слабополярную макропористую адсорбционную смолу.
8. Способ получения по п. 2, где на стадии (1b) весовое отношение применяемого на стадии (1а) лекарственного сырья к макропористой адсорбционной смоле составляет 5:1-1:1, предпочтительно 3:1, при этом указанный кислый водный раствор представляет собой по меньшей мере один выбранный из группы, включающей водный раствор соляной кислоты, водный раствор серной кислоты, водный раствор азотной кислоты и водный раствор уксусной кислоты или их комбинацию; причем рН раствора доводят до 1,0-5,0, предпочтительно 3,0, промывание кислым водным раствором осуществляют до тех пор, пока элюат не станет почти бесцветным.
9. Способ получения по п. 2, где на стадии (1b) для промывания колонки 4-10 раз, предпочтительно 5 раз, применяют 50-95% (объем/объем) этанол, затем элюат концентрируют с получением экстракта без запаха спирта.
10. Способ получения по п. 3, где на стадии (1) сырьевым материалом для реакции является сальвианоловая кислота В или ее соли, массовое отношение указанной сальвианоловой кислоты В к указанному водному раствору составляет от 1:0,1 до 1:100000, предпочтительно 1:200, температура реакции составляет 10-150°С, предпочтительно 90°С, время реакции составляет от 10 мин до 24 ч, предпочтительно 1 ч.
11. Способ получения по п. 3, где на стадии (1) указанный водный раствор представляет собой кислый водный раствор, нейтральный водный раствор или щелочной водный раствор.
12. Способ получения по п. 3, где на стадии (1) указанный водный раствор представляет собой щелочной водный раствор, при этом указанный щелочной водный раствор выбран по меньшей мере из следующих водных растворов: раствора бикарбоната натрия, водного раствора карбоната натрия, раствора гидрокарбоната калия, раствора карбоната калия, водного раствора гидроксида натрия и водного раствора гидроксида калия, предпочтительно представляет собой раствор бикарбоната натрия в концентрации 0,05-0,45% (масса/объем).
13. Способ получения по п. 2 или 3, где на стадии (2) для доведения рН реакционной жидкости до 1,0-6,0, предпочтительно до 3,0, применяют любой из водного раствора соляной кислоты, водного раствора серной кислоты, водного раствора азотной кислоты и водного раствора уксусной кислоты или их комбинацию.
14. Способ получения по п. 2 или 3, где на стадии (2) указанный жидкостный хроматограф высокого давления представляет собой жидкостный хроматограф высокого давления с колонкой динамического аксиального сжатия, наполнителем для хроматографии на обращенно-фазовой колонке является силикагель С18, причем осуществляют растворение реакционной жидкости, рН которой доводят на стадии (1), или экстракта, полученного на указанной стадии (1b), в подвижной фазе, при этом указанная подвижная фаза представляет собой ацетонитрил:воду:муравьиную кислоту (объемное соотношение) = (10:90:1)-(90:10:1); элюент применяют при соотношении, указанном выше для подвижной фазы, элюирование представляет собой изократическое элюирование или градиентное элюирование; скорость потока составляет 300 мл/мин; длина волны детектирования составляет 280 нм; для мониторинга процесса элюирования применяют высокоэффективную жидкостную хроматографию, причем осуществляют сбор компонентов, время удерживания которых составляет 21,2-24,0 мин, концентрирование досуха, получение образца сальвианоловой кислоты Т.
15. Способ получения по п. 14, где указанная подвижная фаза представляет собой ацетонитрил:воду:муравьиную кислоту (объемное соотношение) = (10:90:1)-(50:50:1), предпочтительно 15:85:1.
16. Способ получения по п. 14, где для указанного элюирования применяют подвижную фазу ацетонитрил:вода:муравьиная кислота (объемное соотношение) = 15:85:1 с осуществлением изократического элюирования.
17. Способ получения хиральных изомеров сальвианоловой кислоты Т по п. 1, где способ предусматривает следующие стадии:
(1a) экстракции: экстрагирование лекарственного сырья из Radix Salviae Miltiorrhizae или смеси Radix Salviae Miltiorrhizae и другого лекарственного сырья водой, концентрирование фильтрата с получением водного экстракта, затем добавление спирта к осадку и получение надосадочной жидкости, концентрирование надосадочной жидкости с получением спиртового экстракта;
(1b) разделения: разбавление полученного на стадии (1а) спиртового экстракта водой, перенос на макропористую адсорбционную смолу, промывание смолы кислым водным раствором с целью удаления примесей и последующее элюирование смолы этанолом с получением этанольного элюата, концентрирование этанольного элюата с получением экстракта;
или замещение вышеуказанных стадий (1а) и (1b) следующей стадией (1):
(1) синтеза: растворение сальвианоловой кислоты В в воде, нагревание;
(2) очистки: доведение рН реакционной жидкости, полученной на стадии (1), до кислого значения или очистка экстракта, полученного на стадии (1b), с помощью препаративного жидкостного хроматографа высокого давления с применением обращенно-фазовой колонки с силикагелем С18 в качестве наполнителя для хроматографии, ацетонитрилом-водой-муравьиной кислотой в качестве элюента, причем осуществляют изократическое элюирование с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, объединение элюата, содержащего сальвианоловую кислоту Т; концентрирование с получением сальвианоловой кислоты Т;
(3) получения хиральных изомеров: разделение хиральных изомеров сальвианоловой кислоты Т, полученной на стадии (2), с помощью препаративного жидкостного хроматографа с применением обращенно-фазовой хиральной колонки в качестве хроматографической колонки, ацетонитрила-воды-муравьиной кислоты в качестве элюента, причем осуществляют изократическое элюирование или градиентное элюирование с длиной волны детектирования 280 нм; мониторинг процесса элюирования с помощью высокоэффективной жидкостной хроматографии, сбор элюата, содержащего отдельно (S)-сальвианоловую кислоту Т и (R)-сальвианоловую кислоту Т, лиофильная сушка с получением чистых продуктов в виде (S)-сальвианоловой кислоты Т и (R)-сальвианоловой кислоты Т.
18. Способ получения по п. 17, где на стадии (3) для осуществления разделения хиральных изомеров применяют препаративную жидкостную хроматографию, хроматографическая колонка представляет собой обращенно-фазовую колонку, причем осуществляют растворение образца сальвианоловой кислоты Т, полученной на стадии (2), в подвижной фазе, при этом указанная подвижная фаза представляет собой ацетонитрил:воду:муравьиную кислоту (объемное соотношение) = (90:10:1)-(10:90:1), предпочтительно 17:83:1, элюент применяют при соотношении, указанном выше для подвижной фазы, элюирование представляет собой изократическое элюирование или градиентное элюирование; скорость потока составляет 25 мл/мин; длина волны детектирования составляет 280 нм; для мониторинга процесса элюирования применяют высокоэффективную жидкостную хроматографию, причем осуществляют сбор отдельно компонента в виде (S)-сальвианоловой кислоты Т со временем удерживания 19,5-21,1 мин, компонента в виде (R)-сальвианоловой кислоты Т со временем удерживания 23,9-25,3 мин, лиофильную сушку после концентрирования при низкой температуре, получение чистого продукта в виде (S)-сальвианоловой кислоты Т и чистого продукта в виде (R)-сальвианоловой кислоты Т.
19. Способ получения по п. 17, где для указанного элюирования применяют подвижную фазу ацетонитрил:вода:муравьиная кислота (объемное соотношение) = 17:83:1 с осуществлением изократического элюирования.
20. Способ получения по п. 17, где указанная низкая температура составляет 10-40°С, предпочтительно 30°С.
21. Фармацевтическая композиция для лечения острого инфаркта миокарда, острой ишемии миокарда или фиброзных заболеваний легких, содержащая терапевтически эффективное количество соединения сальвианоловой кислоты Т по п. 1, или ее фармацевтически приемлемой соли, или ее R- или S-изомера и фармацевтически приемлемый носитель.
22. Антиоксидант, содержащий указанное соединение сальвианоловой кислоты Т по п. 1, или ее фармацевтически приемлемой соли, или ее R- или S-изомера.
23. Ловушка свободных радикалов, содержащая указанное соединение сальвианоловой кислоты Т по п. 1, или ее фармацевтически приемлемой соли, или ее R- или S-изомера.
24. Применение соединения сальвианоловой кислоты Т по п. 1, или ее фармацевтически приемлемой соли, или ее R- или S-изомера в получении антиокислителей или лекарственных средств для лечения острого инфаркта миокарда и острой ишемии миокарда или фиброзного заболевания легких.
25. Применение соединения сальвианоловой кислоты Т по п. 1, или ее фармацевтически приемлемой соли, или ее R- или S-изомера для антиокислительного действия, для замедления старения или для лечения острого инфаркта миокарда, острой ишемии миокарда или фиброзного заболевания легких.
| WO 2010111935 A1, 07.10.2010 | |||
| СN 1927858 A, 14.03.2007 | |||
| Пневматический краскораспылитель | 1989 |
|
SU1669573A1 |
| КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ СЕРДЕЧНЫХ ЗАБОЛЕВАНИЙ, СПОСОБ ПРИГОТОВЛЕНИЯ УКАЗАННОЙ КОМПОЗИЦИИ И ЕЕ ПРИМЕНЕНИЕ | 2003 |
|
RU2328300C2 |
| СПОСОБ ПОЛУЧЕНИЯ ОБЩЕГО КОЛИЧЕСТВА ФЕНОЛЬНОЙ КИСЛОТЫ ИЗ ШАЛФЕЯ МНОГОКОРНЕВОГО (ДАНЬШЕНЯ) И ЕЕ ПРИМЕНЕНИЕ | 2003 |
|
RU2317973C2 |

