Область техники, к которой относится изобретение
Данное изобретение относится к области лекарственных средств, в частности к способу получения производного филлигенина и глюкуроновой кислоты и к противовирусному эффекту такого производного филлигенина и глюкуроновой кислоты.
Уровень техники
Глюкурониды филлигенина - это активные вещества, выделяемые из высушенных листьев форсайтии пониклой (форзиции свисающей) Forsythia suspensa (Thunb.) Vahl. из семейства маслиновых (Oleaceae). Это растение используется в традиционной китайской медицине на протяжении двух тысячелетий; впервые оно описано в классическом фармакологическом сочинении о травах и лекарственных средствах «Shennong's Herbal Classic» («Трактат Шэнь-нуна о корнях и травах») следующим образом: «форсайтию применяют в основном для лечения озноба и жара, карбункулов, злокачественных язв, стойкой лихорадки и золотухи»
С тех пор, как Murakami впервые выделил из плодов форсайтии олеаноловую кислоту, сообщалось о более чем шестидесяти компонентах этого и родственных ему растений. В их число входят, главным образом, терпены, фенэтиловый спирт и его терпеновые гликозиды, лигнаны, флавоноиды и некоторые спирты, простые и сложные эфиры, кетоны и другие химические соединения.
Лигнаны в форсайтии представлены в основном лигнанолидами и бисэпоксилигнанами. В 1978 г. Nishibe с коллегами выделили из форсайтии арктигенин, матарезинол, арктиин и матайрезинозид. В 1985 г. Tsukamoto с коллегами выделили из плодов форсайтии филлигенин, (+)-пинорезинол, филлирин, (+)-пинорезинол-β-D-глюкозид и (+)-эпинорезинол-4-O-глюкозид. В 1977 г. Liu Donglei с коллегами выделили из плодов форсайтии (+)-пинорезинолмонометиловый эфир-β-D-глюкозид. Недавние исследования биосинтеза показали, что предшественником лигнанов в форсайтии является конифериловый спирт. Что касается лигнанов и лигнановых гликозидов форсайтии, то возможны полностью замещенные в положениях 3,3' метокси или полностью замещенные в положениях 4,4' гидроксилпроизводные, замещенные метокси или же образующие гликозиды с сахаридами.
Три глюкуронида филлигенина были впервые выделены из листьев форсайтии. Производные глюкуроновой кислоты обладают важными биологическими активностями, например производные глюкуроновой кислоты и артемизинина (Efficient Preparations of the β-Glucuronides of Dihydroartemisinin and Structural Confirmation of the Human Glucuronide Metabolite. Paul M. O'Neill, Feodor Scheinmann, Andrew V. Stachulski, James L. Maggs, и B. Kevin Park. J. Med. Chem., 2001, 44 (9), pp 1467-1470); производные глюкуроновой кислоты и эдаравона (Synthesis of the metabolites of a free radical scavenger edaravone (MCI-186, Radicut™). Kazutoshi Watanabe, Masao Taniguchi, Masaki Shinoda. Redox Report, Vol. 8, No. 3, 2003, 157-161), производные глюкуроновой кислоты и комбретастатина A-1 (Regio- and Stereospecific Synthesis of Mono-β-d-Glucuronic Acid Derivatives of Combretastatin A-1.Rajendra P. Tanpure, Tracy E. Strecker, David J. Chaplin, Bronwyn G. Siim, Mary Lynn Trawick и Kevin G. Pinney.J. Nat. Prod., 2010, 73 (6), pp 1093-1101); производные глюкуроновой кислоты и ресвератрола (WANG LAIXI; Heredia, A.; Song, HJ; ZHANG ZHAOJUN; YU BIAO; Davis, C.; Redfield, R. Resveratrol glucuronides Characterization, synthesis, and anti-HIV activity. J. Pharm. Sci. 2004, 93(10), 2448-2457); производные глюкуроновой кислоты и куркумина (K.S.Psrvathy, M.Sc. University of Mysore. 2009) и др. В связи с изложенным, изучаются способы получения трех глюкуронидов филлигенина и проводятся соответствующие фармакологические исследования.
Раскрытие изобретения
Цель данного изобретения - получить источник новых противовирусных лекарственных средств - производных филлигенина и глюкуроновой кислоты, способы получения производных филлигенина и глюкуроновой кислоты, и применение этих производных филлигенина и глюкуроновой кислоты в качестве противовирусного агента. Производные филлигенина и глюкуроновой кислоты, описываемые в данном изобретении, обладают противовирусным действием, и их можно использовать при получении лекарственных или иных медицинских продуктов, предназначенных для лечения и предотвращения инфекций вирусами гриппа. Способы получения производных глюкуроновой кислоты и филлигенина отличаются технологической простотой и удобством осуществления, а также пригодны для промышленного производства в больших масштабах.
Для достижения указанной цели данного изобретения предлагаются, с одной стороны, производное глюкуроновой кислоты и филлигенина общей молекулярной формулы (I):
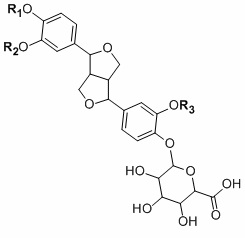
(I)
где R1=H, R2=CnH2n+1, R3=CnH2n+1 или R1=CnH2n+1, R2=CnH2n+1, R3=H или R1-R2=-CH2-, R3=CnH2n+1; n=1-30,
причем n в группе-заместителе равно 1.
С другой стороны, в данном изобретении описывается способ получения производного глюкуроновой кислоты и филлигенина. Этот способ включает последовательно следующие этапы:
1) смешивание листьев форсайтии с экстрагирующим растворителем, представляющим собой воду, нагревание, отваривание (кипячение) и экстракцию 2-3 раза, сбор и объединение порций раствора после экстракции, в результате чего получается водный экстракт форсайтии;
2) разделение водного экстракта форсайтии при помощи колонки с макропористой смолой, сбор и объединение порций элюата, в результате чего получается выделенный на колонке со смолой элюат из форсайтии;
3) высушивание полученного на колонке со смолой элюата из форсайтии, проведение хроматографии на колонке с силикагелем, фракционный сбор элюата и высушивание элюата, в результате чего получается желаемый продукт.
На этапе 1) нагревание и экстракцию предпочтительно осуществляют два раза.
В частном случае, при каждой процедуре отваривания (кипячения) и экстракции массовое соотношение листьев форсайтии и экстрагирующего растворителя, представляющего собой воду, составляет 1:(6-10), предпочтительно 1:(8-10).
Более конкретно, при первом отваривании (кипячении) массовое соотношение листьев форсайтии и экстрагирующего растворителя, представляющего собой воду, составляет 1:(9-10), при втором отваривании (кипячении) массовое соотношение листьев форсайтии и экстрагирующего растворителя (воды) составляет 1:8.
В частном варианте, способ получения дополнительно включает следующие этапы: концентрирование водного экстракта форсайтии, полученного на этапе 1); получение концентрированного раствора из форсайтии и осуществление разделения на колонке с макропористой смолой.
Говоря более конкретно, отношение объема концентрированного раствора из форсайтии, полученного путем концентрирования, к массе листьев форсайтии составляет (1-5):1, предпочтительно (2-2,5):1, причем процедура проведения колоночной хроматографии с использованием макропористой смолы на этапе 2) последовательно включает следующие подэтапы: инжектирование водного экстракта форсайтии в колонку с макропористой смолой и элюирование водой в качестве элюирующего растворителя; элюирование раствором этилового спирта с массовой концентрацией 3-50% в качестве элюирующего растворителя; и, наконец, элюирование абсолютным этиловым спиртом в качестве элюирующего растворителя; в результате получается элюат из форсайтии после пропускания через макропористую смолу.
В частном варианте, при процедуре колоночной хроматографии с использованием макропористой смолы отношение массы листьев форсайтии, взятых для приготовления водного экстракта, к объему макропористой смолы составляет 1:(0,8-2,5), предпочтительно 1:1,
Более конкретно, при разделении на колонке с макропористой смолой отношение диаметра колонки к высоте столба смолы в ней составляет 1:(5-10), предпочтительно 1:(5-7), более предпочтительно 1:(5,5-5,9), причем на этапе 2) в качестве макропористой смолы берут один из следующих продуктов: X-5, AB-8, NK-2, NKA-2, NK-9, D3520, D101 и WLD; предпочтительно используется X-5 или AB-8.
В частном варианте при элюировании с использованием воды в качестве элюирующего растворителя отношение используемого количества воды к объему колонки с макропористой смолой составляет (2-4):1, предпочтительно 4:1; при элюировании с использованием раствора этилового спирта с массовой концентрацией 3-50% в качестве элюирующего растворителя отношение используемого количества раствора этилового спирта с массовой концентрацией 3-50% к объему колонки с макропористой смолой составляет (2-8):1, предпочтительно (4-8):1, более предпочтительно 8:1; при элюировании с использованием абсолютного этилового спирта в качестве элюирующего растворителя отношение используемого количества абсолютного этилового спирта к объему колонки с макропористой смолой составляет (2-8):1, предпочтительно (4-8):1, более предпочтительно 8:1,
при этом проведение колоночной хроматографии с использованием силикагеля на этапе 3) последовательно включает следующие подэтапы:
A) концентрирование элюата из форсайтии, полученного в результате пропускания через колонку со смолой, и высушивание полученного концентрата, в результате чего получается неочищенный продукт из форсайтии;
B) растворение неочищенного продукта из форсайтии в воде, нанесение полученного раствора на колонку с силикагелем, проведение колоночной хроматографии с использованием силикагеля, фракционный сбор элюата и его высушивание, в результате чего получается желаемый продукт.
В частном варианте, на этапе В) колонку с силикагелем выбирают из колонок для хроматографии с обращенной фазой, причем наполнение обращенно-фазовой колонки выбирают из силикагеля С18 для обращенно-фазовой хроматографии; размер частиц в колонке с силикагелем составляет 5-10 мкм.
В частном варианте, внутренний диаметр колонки с силикагелем для обращенно-фазовой хроматографии составляет 10-100 мм, а длина 100-300 мм, предпочтительно внутренний диаметр колонки составляет 22,2 мм, длина - 250 мм, причем для проведения колоночной хроматографии с использованием силикагеля выбирают колонку для высокоэффективной жидкостной хроматографии.
В частном варианте, в качестве подвижной фазы при колоночной хроматографии с использованием силикагеля выбирают смешанный раствор - метиловый спирт (А) с водой (В), которые берут в объемном соотношении метиловый спирт (А) : вода (В) от 8:2 до 10:1.
В частном варианте, при проведении колоночной хроматографии с использованием силикагеля температура колонки составляет 20-35оС, а скорость потока 4-30 мл/мин.
В частном варианте, при проведении колоночной хроматографии с использованием силикагеля элюирование образцов подвижной фазой изократическое или градиентное, причем градиент элюирования составляет [0 мин (30% A) → 25 мин (50% A) → 50 мин (50% A)]; скорость потока составляет 4,0 мл/мин; температура колонки составляет 20°C; детекцию осуществляют на длине волны 273 нм.
В частном варианте, при проведении колоночной хроматографии с использованием силикагеля соблюдались следующие условия: наполняющим материалом служил силикагель С18 для обращенно-фазовой хроматографии; диаметр столба наполняющего материала 22,2 мм, высота 250 нм, размер частиц 10 мкм; в качестве подвижной фазы служила смесь метилового спирта (А) с водой (B); градиентное элюирование 0-25 мин при концентрации метилового спирта 30%-50%; градиентное элюирование 25-50 мин при концентрации метилового спирта 50%-50%; скорость потока 4,0 мл/мин; температура колонки 20°C; детекция в ультрафиолетовом диапазоне на длине волны 273 нм.
Производные филлигенина и глюкуроновой кислоты, полученные способом по данному изобретению, являются соединениями I, II и III. Их структура определена и подтверждена следующими аналитическими данными:
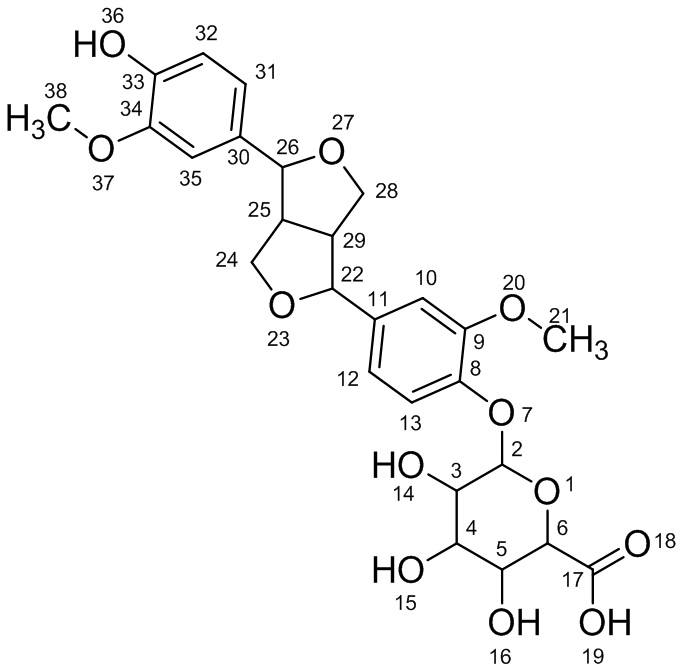
Соединение I: 33-гидроксифиллигенин-8-O-β-D-глюкуронид
Масс-спектрометрия с ионизацией распылением в электрическом поле (ESI-MS): m/z - 533,1658 [M-H]-; молекулярная масса - 534; химическая формула - C26H30O12.
Ядерный магнитный резонанс (1H-ЯМР) (400 MHz; d6-ДМСО): δ(ppm):12,0(1H; s; COOH); 7,119- 7,099(1H; d; J=8,0Hz; Ar-H); 6,530-6,943(2H; d; J=4,0Hz; Ar-H); 6,872(3H; s; Ar-H); 5,39(2H; s; J=4,8Hz); 5,23(1H; d; J=4,8Hz); 5,1(1H; d; J=4,8Hz); 4,800(1H; d; J=4,8Hz); 4,374-4,388(1H; d; J=9,6Hz); 4,105-4,085(1H; d; J=8,0Hz); 4,005-3,982(1H; d; J=9,2Hz); 3,75(8H; d; J=8,4Hz); 3,422(1H; t; J=8,7Hz); 3,08(1H; t; J=8,1Hz); 2,85(1H; d; J=7,2Hz).
Ядерный магнитный резонанс (13С-ЯМР) (100MHz; d6-ДМСО): δ(ppm):172,75(C-17); 149,51(C-9); 148,95(C-34); 148,09(C-33); 145,74(C-8); 136,26(C-11); 131,67(C-30); 118,55(C-12); 118,05(C-31); 115,72(C-13); 112,03(C-32); 111,07(C-10); 109,92(C-35); 100,21(C-2); 87,11(C-26); 81,74(C-22); 76,26(C-6); 75,70(C-3); 73,41(C-5); 71,91(C-4); 70,81(C-28); 69,46(C-24); 56,15(C-21); 55,99(C-38); 54,47(C-29); 49,79(C-25).
Соединение II: 9-гидроксифиллигенин-8-O-β-D-глюкуронид
ESI-MS: m/z - 533,1641 [M-H]-; молекулярная масса - 534; химическая формула - C26H30O12.
1H-ЯМР (400 MHz, d6-ДМСО): δ(ppm):12,0(1H; s; COOH); 7,119-7,099(1H; d; J=8,0Hz; Ar-H); 6,530-6,943(2H; d; J=4,0Hz; Ar-H); 6,872(3H; s; Ar-H); 5,39(2H; s; J=4,8Hz); 5,23(1H; d; J=4,8Hz); 5,1(1H; d; J=4,8Hz); 4,800(1H; d; J=4,8Hz); 4,374-4,388(1H; d; J=9,6Hz); 4,105-4,085(1H; d; J=8,0Hz); 4,005-3,982(1H; d; J=9,2Hz); 3,75(8H; d; J=8,4Hz); 3,422(1H; t; J=8,7Hz); 3,08(1H; t; J=8,1Hz); 2,85(1H; d; J=7,2Hz).
13С-ЯМР (100MHz; d6-ДМСО): δ(ppm):173,72(C-17); 149,51(C-33); 148,95(C-34); 148,09(C-9); 144,74(C-8); 136,26(C-11); 131,67(C-30); 121,45(C-12); 119,72(C-31); 118,05(C-13); 115,07(C-10); 113,03(C-32); 109,92(C-35); 100,21(C-2); 87,11(C-26); 81,74(C-22); 76,26(C-6); 75,70(C-3); 73,41(C-5); 71,91(C-4); 70,81(C-28); 69,46(C-24); 56,15(C-21); 55,99(C-38); 54,47(C-29); 50,16(C-25).

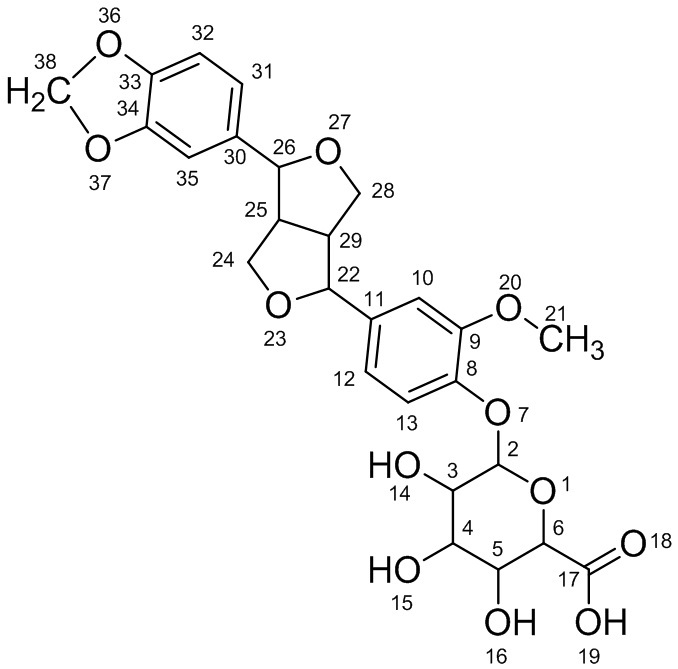
Соединение III: 33,34-метилендиоксифиллигенин-8-O-β-D-глюкуронид
ESI-MS: m/z - 531,4933 [M-H]-, молекулярная масса - 532; химическая формула - C26H28O12.
1Н-ЯМР (400 MHz; d6-ДМСО): δ(ppm):12,0(1H; s; COOH); 7,119-7,099(1H; d; J=8,0Hz; Ar-H); 6,530-6,943(2H; d; J=4,0Hz; Ar-H); 6,872(3H; s; Ar-H); 6,12(2H; s); 5,39(2H; s; J=4,8Hz); 5,23(1H; d; J=4,8Hz); 5,1(1H; d; J=4,8Hz); 4,800(1H; d; J=4,8Hz); 4,374-4,388(1H; d; J=9,6Hz); 4,105-4,085(1H; d; J=8,0Hz); 4,005-3,982(1H; d; J=9,2Hz); 3,75(8H; d; J=8,4Hz); 3,422(1H; t; J=8,7Hz); 3,08(1H; t; J=8,1Hz); 2,85(1H; d; J=7,2Hz);
13С-ЯМР (100MHz; d6-ДМСО): δ(ppm):169,75(C-17); 149,51(C-9); 148,95(C-34); 148,09(C-33); 145,74(C-8); 136,26(C-11); 131,67(C-30); 118,55(C-12); 118,05(C-13); 115,72(C-31); 112,03(C-32); 111,07(C-10); 109,92(C-35); 101,21(C-2); 100,29(C-38); 87,11(C-26); 81,74(C-22); 76,26(C-6); 75,70(C-3); 73,41(C-16); 71,91(C-4); 70,81(C-28); 69,46(C-24); 56,15(C-21); 54,47(C-29); 49,79(C-25).
В способе по изобретению большинство загрязнений удаляются путем очистки на колонке с макропористой смолой, которую можно использовать повторно, что снижает стоимость производства, и срок службы полупрепаративной хроматографической колонки на следующем этапе продлевается. Сама процедура несложная в исполнении, эффективность экстракции высока, загрязнение небольшое, так что получаемый продукт отличается высокой степенью чистоты и его получение легко может быть осуществлено в промышленных условиях.
В другом аспекте данного изобретения описывается противовирусное применение производных филлигенина и глюкуроновой кислоты.
В другом аспекте данного изобретения описывается назначение производных филлигенина и глюкуроновой кислоты, а именно их применение при получении лекарственных средств для предотвращения и/или лечения инфекций вирусами гриппа.
В данном изобретении описывается композиция лекарственного или иного медицинского продукта, обладающая противовирусным эффектом и содержащая производное филлигенина и глюкуроновой кислоты.
В частности, предлагаемая фармацевтическая композиция содержит производные филлигенина и глюкуроновой кислоты по данному изобретению, а также фармацевтически приемлемые эксципиенты.
Способ получения глюкуронидов филлигенина и их предназначенность для лекарственных средств, применяемых при предотвращении и лечении инфекций вирусами гриппа, не ограничиваются описанными в настоящем документе соединениями. Глюкурониды филлигенина также включают производные, полученные путем синтеза, ферментации и другими методами с использованием описанных в настоящем документе соединений в качестве исходных.
В данном изобретении фармацевтически приемлемые эксципиенты означают нетоксичные твердые, полужидкие или жидкие наполнители, разбавители, носители/несущие среды, агенты, влияющие на рН, агенты, влияющие на ионную силу, материалы для получения препаратов с замедленным высвобождением активных ингредиентов, покрывающие агенты или иные эксципиенты, используемые при изготовлении лекарственных препаратов. Носители/несущие среды выбирают в зависимости от лекарственной формы, используемой для введения в организм пациента. Известные специалистам в данной области техники эксципиенты могут входить в состав препаратов для инъекций, порошков, полученных путем лиофилизации (для инъекций), распыляемых/разбрызгиваемых препаратов (спреев), растворов и суспензий для перорального применения, таблеток, капсул, таблеток с кишечно-растворимой оболочкой, пилюль, порошков, гранул, препаратов с замедленным или задержанным высвобождением и др. В первом аспекте данного изобретения 4-деметилированный глюкуронид филлигенина предпочтительно вводят путем инъекций или через пищеварительный тракт. Соответственно, фармацевтическая композиция по данному изобретению предпочтительно представлена формами для инъекций или для введения через пищеварительный тракт, то есть эксципиенты по данному изобретению предпочтительно должны быть пригодны для включения в препараты, вводимые путем инъекций иди через пищеварительный тракт, причем по данному изобретению термин «введение через пищеварительный тракт» относится к способам введения лекарственных препаратов через пищеварительный тракт пациента, включая пероральное введение, интрагастральное введение, введение с помощью клизмы и др., предпочтительно пероральное введение. Например, известные специалистам в данной области техники эксципиенты по данному изобретению могут входить в состав растворов и суспензий для перорального применения, таблеток, капсул, таблеток с кишечно-растворимой оболочкой, пилюль, порошков, гранул, препаратов с замедленным или задержанным высвобождением и др., причем термин «препараты, вводимые путем инъекций» в настоящем документе относится в основном к растворам для инъекций и к порошкам для приготовления растворов, вводимых путем инъекций.
Новые соединения по данному изобретению, а именно производные филлигенина и глюкуроновой кислоты, обладают противовирусным действием и могут быть включены в состав противовирусных, жаропонижающих, болеутоляющих и противовоспалительных лекарственных или иных медицинских продуктов. В способе получения производных филлигенина и глюкуроновой кислоты по изобретению нетрудно регулировать рабочие условия и контролировать качество продукта; этот способ отличается высоким выходом, низкой энергоемкостью, он экологически безопасен и пригоден для крупномасштабного промышленного производства.
Осуществление изобретения
Далее данное изобретение описывается на примере его конкретных воплощений. Однако эти воплощения представлены в настоящем документе только в иллюстративных целях и их не следует считать ограничивающими объем изобретения.
Воплощение 1
1. Тепловая обработка для получения отвара
1-1) Листья форсайтии измельчают и пропускают через сито 20 меш, в результате чего получается порошок, к которому прибавляют воду из расчета 10 кг воды на 1 кг листьев. Порошок из листьев с водой перемешивают до однородности и нагревают, и проводят первую обработку для получения отвара, в рамках которой смесь лиственного порошка и воды в соотношении 1:10 нагревают и кипятят, отваривают и экстрагируют в течение 2 часов. Полученный отвар фильтруют, в результате чего получается первый раствор - продукт экстракции и первый остаток после экстракции.
1-2) В первый остаток после экстракции прибавляют 8 кг воды, нагревают и кипятят. При этой второй обработке для получения отвара соотношение добавленной воды и листьев форсайтии составляет 8:1, продолжительность отваривания и экстракции составляет 1 час. Полученный отвар фильтруют, в результате чего получается второй раствор - продукт экстракции и второй остаток после экстракции (его отбрасывают).
1-3) Объединяют первый и второй растворы, полученные в результате экстракции; этот объединенный раствор - водный экстракт форсайтии;
1-4) Проводят вакуумное концентрирование водного экстракта форсайтии с помощью роторного вакуумного испарителя, в результате чего извлекают растворитель и получают 2л концентрированного раствора форсайтии для последующего использования; отношение массы листьев форсайтии к объему полученного концентрированного раствора составляет 1:2.
2. Хроматография на колонке с макропористой смолой
2-1) В хроматографическую колонку с макропористой смолой наносят концентрированный раствор - экстракт листьев форсайтии и проводят разделение. При этом макропористую смолу выбирают из продуктов типа AB-8; объем макропористой смолы в колонке составляет 1л, диаметр колонки 60 мм, высота - 500 мм, высота столба смолы в колонке 354 мм); отношение объема смолы в колонке к сухой массе листьев форсайтии 1:1 (например, если сухая масса листьев форсайтии составляла 1 кг, то объем макропористой смолы в хроматографической колонке 1 л; если сухая масса лекарственного сырья была 1 г, то объем смолы 1 мл). Через колонку пропускают четырехкратный (по отношению к объему смолы в колонке) объем воды (4 л); после того, как концентрированный раствор - экстракт форсайтии, нанесенный на колонку, полностью уйдет в смолу, элюат удаляют. Затем проводят элюирование 8-кратным объемом раствора этилового спирта с массовой концентрацией 3% (то есть 8 л); элюат удаляют. Затем проводят элюирование 8-кратным объемом абсолютного этилового спирта (то есть 8 л) и собирают элюат.
2-2) Проводят вакуумное концентрирование элюата, полученного на этапе 2-1), с помощью роторного вакуумного испарителя. Концентрат высушивают, получая в результате 62 г неочищенного продукта из форсайтии.
3. Хроматография на колонке с силикагелем
3-1) Отвешивают 0,5 г неочищенного продукта из форсайтии, прибавляют соответствующее количество воды (2,5 мл), перемешивают и растворяют для последующего использования.
3-2) Проводят хроматографическую обработку неочищенного продукта из форсайтии путем высокоэффективной жидкостной хроматографии (HPLC), используя полупрепаративный жидкостной хроматограф Shimadzu с контроллером SCL-10AVP, насосом LC-8A и спектрофотометрическим детектором SPD-20A, для разделения и очистки неочищенного продукта из форсайтии. Растворенный в воде неочищенный продукт из форсайтии вводят в хроматограф и проводят градиентное элюирование смесью метиловый спирт-вода; при этом размеры хроматографической колонки 22,5х250 мм, наполнитель - силикагель С18 для обращенно-фазовой хроматографии, размер частиц 10 нм, количество введенного материала 500 мг, подвижная фаза - смесь метилового спирта с водой. Условия градиентного элюирования: 0-25 мин, концентрация метилового спирта 30%-50%; 25-50 мин, концентрация метилового спирта 50%-50%; скорость потока 4 мл/мин; температура колонки 20°C. Детекцию проводят в ультрафиолетовой области спектра на длине волны 273 нм. Собирают фракции с временем удерживания 25,5-27,5 мин, 30,5-32,5 мин и 35,5-37,5мин.
3-3) Проводят вакуумное концентрирование и высушивание трех собранных фракций, получая в результате соединение I (70,5 мг), соединение II (53,2 мг) и соединение III (46,6 мг).
Состав полученных образцов соединений I, II и III определяли путем HPLC при следующих условиях детекции: насос хроматографической системы модели Waters 515, детектор 2487, колонка Kromasil RP-C18; подвижная фаза - ацетонитрил/0,1%-ный раствор фосфорной кислоты в соотношении 13:87, длина волны 230 нм, скорость потока 1,0 мл/мин.
По данным HPLC чистота полученного соединения I составляет 99,6%, соединения II - 98,1% и соединения III - 98,3%.
Соединение I представляет собой твердое вещество белого цвета с температурой плавления 111°С, растворимое в воде и этиловом спирте. При тонкослойной хроматографии (элюирование раствором хлороформ/метиловый спирт 3:1, Rf = 0,25) с визуализацией реагентом 10%H2SO4-этиловый спирт соединение I на пластине выглядит пурпурно-красным.
ESI-MS: m/z - 533,1658 [M-H]-, молекулярная масса - 534.
1Н-ЯМР (400 MHz, d6-ДМСО): δ(ppm):12,0(1H; s; COOH); 7,119- 7,099(1H; d; J=8,0Hz; Ar-H); 6,530-6,943(2H; d; J=4,0Hz; Ar-H); 6,872(3H; s; Ar-H); 5,39(2H; s; J=4,8Hz); 5,23(1H; d; J=4,8Hz); 5,1(1H; d; J=4,8Hz); 4,800(1H; d; J=4,8Hz); 4,374-4,388(1H; d; J=9,6Hz); 4,105-4,085(1H; d; J=8,0Hz); 4,005-3,982(1H; d; J=9,2Hz); 3,75(8H; d; J=8,4Hz); 3,422(1H; t; J=8,7Hz); 3,08(1H; t; J=8,1Hz); 2,85(1H; d; J=7,2Hz).
13С-ЯМР (100MHz; d6-ДМСО): δ(ppm):172,75(C-17); 149,51(C-9); 148,95(C-34); 148,09(C-33); 145,74(C-8); 136,26(C-11); 131,67(C-30); 118,55(C-12); 118,05(C-31); 115,72(C-13); 112,03(C-32); 111,07(C-10); 109,92(C-35); 100,21(C-2); 87,11(C-26); 81,74(C-22); 76,26(C-6); 75,70(C-3); 73,41(C-5); 71,91(C-4); 70,81(C-28); 69,46(C-24); 56,15(C-21); 55,99(C-38); 54,47(C-29); 49,79(C-25).
На основании данных ESI-MS, 1Н-ЯМР и 13С-ЯМР соединение I решено назвать «33-гидроксифиллигенин-8-O-β-D-глюкуронид». Структурная формула соединения I имеет следующий вид:
Соединение II представляет собой твердое вещество белого цвета с температурой плавления 113°С, растворимое в воде и этиловом спирте. При тонкослойной хроматографии (элюирование раствором хлороформ/метиловый спирт 3:1, Rf = 0,32) с визуализацией реагентом 10%H2SO4-этиловый спирт соединение II на пластине выглядит пурпурно-красным.
ESI-MS: m/z - 533,1641 [M-H]-, молекулярная масса - 534.
1Н-ЯМР (400 MHz; d6-ДМСО): δ(ppm):12,0(1H; s; COOH); 7,119-7,099(1H; d; J=8,0Hz; Ar-H); 6,530-6,943(2H; d; J=4,0Hz; Ar-H); 6,872(3H; s; Ar-H); 5,39(2H; s; J=4,8Hz); 5,23(1H; d; J=4,8Hz); 5,1(1H; d; J=4,8Hz); 4,800(1H; d; J=4,8Hz); 4,374-4,388(1H; d; J=9,6Hz); 4,105-4,085(1H; d; J=8,0Hz); 4,005-3,982(1H; d; J=9,2Hz); 3,75(8H; d; J=8,4Hz); 3,422(1H; t; J=8,7Hz); 3,08(1H; t; J=8,1Hz); 2,85(1H; d; J=7,2Hz).
13С-ЯМР (100MHz; d6-ДМСО): δ(ppm):173,72(C-17); 149,51(C-33); 148,95(C-34); 148,09(C-9); 144,74(C-8); 136,26(C-11); 131,67(C-30); 121,45(C-12); 119,72(C-31); 118,05(C-13); 115,07(C-10); 113,03(C-32); 109,92(C-35); 100,21(C-2); 87,11(C-26); 81,74(C-22); 76,26(C-6); 75,70(C-3); 73,41(C-5); 71,91(C-4); 70,81(C-28); 69,46(C-24); 56,15(C-21); 55,99(C-38); 54,47(C-29); 50,16(C-25).
На основании данных ESI-MS, 1Н-ЯМР и 13С-ЯМР соединение II решено назвать «9-гидроксифиллигенин-8-O-β-D-глюкуронид». Структурная формула соединения II имеет следующий вид:
Соединение III представляет собой твердое вещество белого цвета с температурой плавления 119°С, растворимое в воде и этиловом спирте. При тонкослойной хроматографии (элюирование раствором хлороформ/метиловый спирт 3:1, Rf = 0,36) с визуализацией реагентом 10%H2SO4-этиловый спирт соединение III на пластине выглядит пурпурно-красным.
ESI-MS: m/z - 531,4933 [M-H]-, молекулярная масса - 532.
1Н-ЯМР (400 MHz; d6-ДМСО): δ(ppm):12,0(1H; s; COOH); 7,119-7,099(1H; d; J=8,0Hz; Ar-H); 6,530-6,943(2H; d; J=4,0Hz; Ar-H); 6,872(3H; s; Ar-H); 6,12(2H; s); 5,39(2H; s; J=4,8Hz); 5,23(1H; d; J=4,8Hz); 5,1(1H; d; J=4,8Hz); 4,800(1H; d; J=4,8Hz); 4,374-4,388(1H; d; J=9,6Hz); 4,105-4,085(1H; d; J=8,0Hz); 4,005-3,982(1H; d; J=9,2Hz); 3,75(8H; d; J=8,4Hz); 3,422(1H; t; J=8,7Hz); 3,08(1H; t; J=8,1Hz); 2,85(1H; d; J=7,2Hz).
13С-ЯМР (100MHz; d6-ДМСО): δ(ppm):169,75(C-17); 149,51(C-9); 148,95(C-34); 148,09(C-33); 145,74(C-8); 136,26(C-11); 131,67(C-30); 118,55(C-12); 118,05(C-13); 115,72(C-31); 112,03(C-32); 111,07(C-10); 109,92(C-35); 101,21(C-2); 100,29(C-38); 87,11(C-26); 81,74(C-22); 76,26(C-6); 75,70(C-3); 73,41(C-16); 71,91(C-4); 70,81(C-28); 69,46(C-24); 56,15(C-21); 54,47(C-29); 49,79(C-25).
На основании данных ESI-MS, 1Н-ЯМР и 13С-ЯМР соединение III решено назвать «33,34-метилендиоксифиллигенин-8-O-β-D-глюкуронид». Структурная формула соединения III имеет следующий вид:
Воплощение 2
1. Тепловая обработка для получения отвара
1-1) Листья форсайтии измельчают и пропускают через сито 20 меш, в результате чего получается порошок, к которому прибавляют воду из расчета 9 кг воды на 1 кг листьев. Порошок из листьев с водой перемешивают до однородности и нагревают, и проводят первую обработку для получения отвара, в рамках которой смесь лиственного порошка и воды (массовое отношение вода:листья = 9:1) нагревают и кипятят, отваривают и экстрагируют в течение 2,5 часов. Полученный отвар фильтруют, в результате чего получается первый раствор - продукт экстракции и первый остаток после экстракции.
1-2) В первый остаток после экстракции прибавляют 8 кг воды, нагревают и кипятят. При этой второй обработке для получения отвара соотношение добавленной воды и листьев форсайтии составляет 8:1, а продолжительность отваривания и экстракции - 1 час. Полученный отвар фильтруют, в результате чего получается второй раствор - продукт экстракции и второй остаток после экстракции (его отбрасывают).
1-3) Объединяют первый и второй растворы, полученные в результате экстракции; этот объединенный раствор представляет собой водный экстракт форсайтии.
1-4) Проводят вакуумное концентрирование водного экстракта форсайтии с помощью роторного вакуумного испарителя, в результате чего извлекают растворитель и получают 2,5 л концентрированного раствора для последующего использования; Отношение массы листьев форсайтии к объему полученного концентрированного раствора составляет 1:2,5.
2. Хроматография на колонке с макропористой смолой
2-1) В хроматографическую колонку с макропористой смолой наносят концентрированный экстракт листьев форсайтии и проводят разделение. Для этого макропористую смолу выбирают из продуктов типа Х-5; объем макропористой смолы в колонке составляет 1 л, диаметр колонки 60 мм, высота - 500 мм, высота столба смолы в колонке 354 мм); отношение объема смолы в колонке к сухой массе листьев форсайтии 1:1 (например, если сухая масса листьев форсайтии составляла 1 кг, то объем макропористой смолы в хроматографической колонке 1 л; если сухая масса лекарственного сырья была 1 г, то объем смолы 1 мл). Через колонку пропускают восьмикратный (по отношению к объему смолы) объем деионизованной воды (т.е. 8 л); после того, как концентрированный экстракт форсайтии, нанесенный на колонку, полностью уйдет в смолу, элюат удаляют. Затем проводят элюирование 4-кратным объемом 3%-ного (масса/масса) раствора этилового спирта (то есть 4 л); элюат удаляют. Затем проводят элюирование 8-кратным объемом абсолютного этилового спирта (то есть 8 л) и собирают элюат.
2-2) Проводят вакуумное концентрирование элюата, полученного на этапе 2-1), с помощью роторного вакуумного испарителя. Концентрат высушивают, получая в результате 58 г неочищенного продукта из форсайтии.
3. Хроматография на колонке с силикагелем
3-1) Отвешивают 0,8 г неочищенного продукта из форсайтии, прибавляют соответствующее количество воды (1,6 мл), перемешивают и растворяют продукт для последующего использования.
3-2) Проводят хроматографическую обработку неочищенного продукта из форсайтии путем высокоэффективной жидкостной хроматографии (HPLC), используя полупрепаративный жидкостной хроматограф Shimadzu с контроллером SCL-10AVP, насосом LC-8A и спектрофотометрическим детектором SPD-20A, разделение и очистку неочищенного продукта из форсайтии. Растворенный в воде неочищенный продукт из форсайтии вводят в хроматограф и проводят градиентное элюирование смесью метиловый спирт-вода; при этом размеры хроматографической колонки 22,5х250 мм, наполнитель - силикагель С18 для обращенно-фазовой хроматографии, размер частиц 10 нм, количество введенного материала 800 мг, подвижная фаза - смесь метилового спирта с водой. Условия градиентного элюирования: 0-25 мин, концентрация метилового спирта 30%-50%; 25-50 мин, концентрация метилового спирта 50%-50%; скорость потока 4 мл/мин; температура колонки 20°C. Детекцию проводят в ультрафиолетовой области спектра на длине волны 273 нм. Собирают фракции с временем удерживания 25,5-27,5 мин, 30,5-32,5 мин и 35,5-37,5мин.
3-3) Проводят вакуумное концентрирование и высушивание трех собранных фракций, получая в результате соединение А (104,2 мг), соединение В (74,3 мг) и соединение С (58,1 мг).
Состав полученных образцов соединений А, В и С определяли путем HPLC при следующих условиях детекции: насос хроматографической системы модели Waters 515, детектор 2487, колонка Kromasil RP-C18; подвижная фаза - ацетонитрил/0,1-ный раствор фосфорной кислоты в соотношении 13:87, длина волны 230 нм, скорость потока 1,0 мл/мин.
По данным HPLC чистота полученного соединения А составляет 99,3%, соединения В - 98,4% и соединения С - 98,5%.
Физико-химические свойства, результаты анализа методами масс-спектрометрии и ядерного магнитного резонанса соединений A, B и C, полученных в воплощении 2, такие же, как соединений I, II и III, полученных в воплощении 1.
Испытание продуктов 1. Определение противовирусного эффекта in vitro
1.1 Материалы
(1) Лекарственные средства
I. Испытываемые агенты
Глюкурониды филлигенина (то есть соединения I, II и III, полученные в воплощении 1 данного изобретения.
II. Агенты положительного контроля
Рибавирин для инъекций - бесцветная прозрачная жидкость; производство Henan Runhong Pharmaceutical Co., Ltd., серийный номер продукта 1206261, номер официального утверждения препарата Государственным управлением по контролю качества медикаментов и продуктов питания Китая (SFDA) H19993553, концентрация 100 мг/мл. Используется в качестве положительного контроля.
Оселтамивира фосфат - предоставлен Государственными институтами по контролю качества медикаментов и продуктов питания Китая (National Institutes for Food and Drug Control), серийный номер продукта 101096-200901, 100 мг/шт. Используется в качестве положительного контроля.
Филлигенин - белый порошок; производство Dalian Fusheng Natural Drug Development Co., Ltd.; чистота 99,2% (по данным высокоэффективной жидкостной хроматографии с детекцией 1) по поглощению в ультрафиолетовой области и 2) с помощью испарительного нефелометрического детектора с применением метода нормализации площадей).
Каждый из указанных агентов растворяли в очищенной воде, фильтровали и расфасовывали для последующего использования при температуре 4°C в качестве детектируемых агентов в данном испытании.
(2) Линии клеток
Линия Vero (почечные клетки африканской зеленой мартышки); коллекция Школы фундаментальной медицины Цзилиньского университета (Китай)
(3) Штаммы вирусов
I. Штаммы вирусов гриппа, парагриппа и респираторно-синцитиальных вирусов (RSV), предоставленные Институтом вирусологии Китайской академии профилактической медицины. II. Штаммы вирусов Коксаки B3 (CVB3), предоставленные Уханьским институтом вирусологии Китайской академии наук. III. Штаммы вирусов Коксаки A16 (CoxA16), штаммы энтеровирусов EV71, предоставленные Сендайской государственной больницей (Япония). IV. аденовирус (AdV), предоставленный Институтом педиатрии Первой больницы Медицинского колледжа им. Нормана Бетьюна Цзилиньского университета (ранее - бывший Медицинский университет им. Нормана Бетьюна). V. вирус простого герпеса типа I (HSV-1), предоставленный Государственными институтами по контролю качества медикаментов и продуктов питания Китая.
(4) Основное оборудование и реагенты
Боксы биологической безопасности BHC-1300ⅡA/B3, AIRTECH; CO2-инкубаторы MCO-18AIC, SANYO; инвертационные микроскопы CKX41, OLYMPUS; весы аналитические электронные AR1140/C, ОHAUS; культуральные среды DMEM, HyClone; эмбриональная телячья сыворотка HyClone; трипсин производства Gibco; тиазоловый синий (MTT) производства Sigma; диметилсульфоксид (ДМСО) производства Tianjin Beilian Fine Chemicals Development Co., Ltd.
1.2. Методика анализа
(1) Подготовка клеток
Проводили субкультивирование клеток Vero в течение 1-2 суток, чтобы они образовали пласт и имели четкие границы; проводили обработку трипсином под контролем с помощью микроскопа (при большой глубине изображаемого пространства и высокой оптической силе); когда на поверхности клеток возникают ямки, как от укола иглой, гидролизующий раствор полностью отсасывают; клетки смывают несколькими миллилитрами культуральной среды, подсчитывают, разводят до концентрации 5×107 клеток в 1 л средой DMEM, содержащей 10% эмбриональной телячьей сыворотки, вносят в лунки 96-луночного культурального планшета и оставляют расти, чтобы образовался один клеточный слой.
(2) Определение токсичности испытываемых агентов
Тест на цитотоксичность. Испытываемый агент растворяли в поддерживающей культуральной среде (среда DMEM, содержащая 2% эмбриональной телячьей сыворотки) в концентрациях, указанных в таблице 1-1, и определяли цитотоксичность полученных образцов
Таблица 1-1. Концентрации (г/л) испытываемых агентов при определении цитотоксичности
ции
Агент
Каждый из испытываемых агентов в той или иной концентрации из указанных в таблице 1-1 наносили на слой клеток Vero так, чтобы в каждую лунку попало 0,2 мл испытываемого образца (данной концентрации) и каждый образец был нанесен в 6 лунок; кроме того, в шести лунках был «нормальный» контроль (без испытываемых агентов) и в шести лунках - «пустая» проба (культуральная среда). Культивировали в инкубаторе в атмосфере, содержавшей 5% CO2, при температуре 37°C. Каждый день с помощью инвертационного микроскопа оценивали цитопатический эффект (в экспериментах in vitro имеющиеся в клетках вирусы гибнут в процессе культивирования и пересева; наблюдаемые под микроскопом в течение некоторого времени округление клеток, их отслаивание клеток от стенок емкости и гибель называются цитопатическим эффектом (CPE). Цитопатический эффект - проявление дегенерации клеток в культуре, зараженной вирусом; этот феномен можно использовать для оценки количества вирусных частиц). Величину СРЕ регистрировали. Через 72 часа прибавляли в лунки планшета краситель тиазоловый синий (3-(4,5-диметил-2-тиазолил)-2,5-дифенил-2-H-тетразолия бромид; МТТ) в количестве по 20 мкл (концентрация 5 мг/мл) и инкубировали непрерывно в течение 4 часов. Затем отсасывали из лунок культуральную среду, прибавляли в них по 100 мкл диметилсульфоксида (ДМСО), перемешивали, покачивая планшет, в течение 5 минут, после чего измеряли оптическую плотность (OD) на длине волны 492 нм. Определяли жизнеспособность клеток, используя статистическую модель пробит-регрессии, с помощью программы SPSS 18,0; рассчитывали максимальную переносимую концентрацию (TC0) и концентрацию, при которой гибнет половина клеток (TC50).
(3) Определение дозы инфекционного агента, при которой поражается 50% клеточной культуры (TCID50 - 50%-ная тканевая цитопатическая доза) для различных вирусов.
Готовили серию 10-кратных разведений взятых для анализа вирусов, а именно 10-1, 10-2, 10-3, 10-4, 10-5 и 10-6, и последовательно наносили полученные разведения вирусов на монослойную культуру клеток Vero в лунках 96-луночного планшета в количестве 100 мкл на лунку; каждое разведение каждого вируса вносили в 6 лунок. Так же, но без вирусов, обрабатывали контрольные клетки. Инкубировали в атмосфере, содержащей 5% CO2 при температуре 37°C в течение 2 часов. После этого удаляли раствор вируса, прибавляли в каждую лунку по 100 мкл поддерживающей среды и культивировали в атмосфере, содержащей 5% CO2 при температуре 37°C. Начиная с третьих суток, отслеживали цитопатический эффект с помощью микроскопа; на 7-8-е сутки производили оценку CPE и отмечали наибольшее разведение, при котором в 50% лунок с инфицированными клетками имели место явные признаки гибели клеток (конечная точка); рассчитывали титр вируса, применяя метод Карбера, по следующей формуле:
LogTCID50=XM+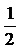

TCID50 - доза инфекционного агента, при которой поражается 50% клеточной культуры (50%-ная тканевая цитопатическая доза);
XM - логарифм наибольшего разведения вируса
d - логарифм коэффициента разведения;
Σpi - сумма долей (в процентах) пораженных клеток при разных степенях разведения;
(4) Влияние испытываемых агентов на обусловленный вирусами цитопатический эффект
Из лунок планшета с выращенной монослойной культурой клеток отсасывали культуральную среду и вносили тот или иной вирус в количестве, соответствующем 100TCID50. Держали планшет в инкубаторе в атмосфере, содержащей 5% CO2, при температуре 37°C в течение 2 часов, после чего прибавляли раствор испытываемого агента в определенной концентрации (в области максимальной переносимой концентрации) по 200 мкл в лунку, отводя на каждую концентрацию по 6 лунок. В качестве положительного контроля вместо соединений по данному изобретению брали рибавирин (препарат для инъекций) и оселтамивира фосфат; параллельно делали «нормальный» контроль (лунки без вирусов и без испытываемых агентов) и контроль «на вирус» (в лунки вносили вирус, но не добавляли испытываемых агентов). Наблюдали, как влияют испытываемые противовирусные агенты на обусловленный вирусами цитопатический эффект. Измеряли оптическую плотность (OD) на длине волны 492 нм, применяя колориметрический метод с использованием МТТ. Рассчитывали эффективность противовирусного действия (ER%) испытываемых агентов и по этим данным сравнивали различные противовирусные агенты методом дисперсионного анализа (ANOVA), используя программу SPSS 18,0.
ER%=(OD среднее значение для группы лунок, обработанных данным агентом - OD среднее значение для группы лунок с контролем «на вирус»)/( OD среднее значение для лунок с «нормальным» контролем - OD среднее значение для группы лунок с контролем «на вирус»)) ×100%
1.3 Результаты
(1)TCID50 для различных вирусов


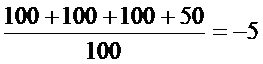
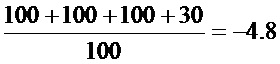
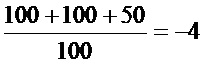
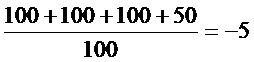
(2) Результаты определения токсичности испытываемых агентов
1) Цитотоксичность испытываемых агентов
Значения максимальной переносимой концентрации (TC0) и концентрации, при которой гибнет половина клеток (TC50), для различных агентов в опытах с клетками Vero представлены в таблице 1-2.
Таблица 1-2. Результаты определения цитотоксичности (г/л)
Концентр.
2) Результаты изучения защиты испытываемыми агентами от обусловленного вирусами цитопатического эффекта
Данные по эффективности защиты испытываемыми агентами от различных вирусов и результаты однофакторного дисперсионного анализа (ANOVA) см. таблицу 1-3.
Taблица 1-3. Данные по эффективности противовирусной защиты (ER%) испытываемыми агентами
Вирус
I
II
III
Примечание: при сравнении с контролем «на вирус» *P <0,05, **P <0,01; при сравнении с филлигенином #P <0,05, ##P <0,01.
Результаты, представленные в таблице 1-3, показывают, что 33-гидроксифиллигенин-8-O-β-D-глюкуронид (соединение I), 9-гидроксифиллигенин-8-O-β-D-глюкуронид (соединение II) и 33,34-метилендиоксифиллигенин-8-O-β-D-глюкуронид (соединение III) характеризуются степенью подавления цитопатического эффекта и эффективностью защиты от вируса гриппа, вируса парагриппа, вируса простого герпеса (HSV-I) и энтеровируса EV71, превышающими 90% и явно отличающимися от контроля «на вирус», причем эти данные статистически значимы; 33-гидроксифиллигенин-8-O-β-D-глюкуронид, 9- гидроксифиллигенин-8-O-β-D-глюкуронид и
33,34-метилендиоксифиллигенин-8-O-β-D-глюкуронид обладают лечебным эффектом в отношении многих вирусов, превышающим таковой филлигенина, рибаверина и оселтамивира фосфата.
Испытание продуктов 2. Определение противовирусного эффекта in vivo
2.1 Материалы
(1) Лабораторные животные
Мыши линии Куньмин (Kunming) с массой тела 18-22 г, половина особей самцы, половина - самки; предоставлены Центром лабораторных животных Даляньского медицинского университета (Китай); номер сертификата качества SCXK (13)2012-0003.
(2) Препараты
1) Соединение I, полученное в воплощении 1 данного изобретения (33- гидроксифиллигенин-8-O-β-D-глюкуронид).
2) Рибавирин для инъекций - бесцветная прозрачная жидкость; производство Henan Runhong Pharmaceutical Co., Ltd., серийный номер продукта 1206261, номер официального утверждения препарата Государственным управлением по контролю качества медикаментов и продуктов питания Китая (SFDA) H19993553, концентрация 100 мг/мл. Используется в качестве положительного контроля.
3) Оселтамивира фосфат - предоставлен Государственными институтами по контролю качества медикаментов и продуктов питания Китая (National Institutes for Food and Drug Control), серийный номер продукта 101096-200901, 100 мг/шт. Используется в качестве положительного контроля.
4) Филлигенин - белый порошок; производство Dalian Fusheng Natural Drug Development Co., Ltd.; чистота 99,2% (по данным высокоэффективной жидкостной хроматографии с детекцией 1) по поглощению в ультрафиолетовой области и 2) с помощью испарительного нефелометрического детектора с применением метода нормализации площадей).
Каждый из указанных агентов растворяли в очищенной воде, фильтровали и расфасовывали для последующего использования при температуре 4°C в качестве детектируемых агентов в данном испытании.
(2) Оборудование для детекции и реагенты
2.2 Методика анализа
(1) Определение полулетальной дозы вирусов гриппа и парагриппа для мышей
Готовили 10-кратные разведения вирусов гриппа и парагриппа в буферном растворе для лизиса клеток, получая растворы с концентрацией вирионов 10-1, 10-2, 10-3, 10-4 и 10-5. В опыт брали 120 мышей линии Куньмин (Kunming), из них 60 особей заражали вирусом гриппа и 60 особей - вирусом парагриппа. Для этого животных разделили случайным образом на 6 групп и каждой мыши под анестезией этиловым эфиром ввели ингаляционным путем интраназально раствор одного или другого вируса той или иной степени разведения в количестве 0,03 мл; параллельно делали контрольную обработку: вместо суспензии вирусных частиц брали физиологический раствор («пустой» контроль). Подопытных мышей наблюдали ежедневно в течение 14 суток после заражения, отмечая случаи смерти и количество выживших особей; при этом в расчет не включали животных, которые умерли от неспецифических причин в первые 24 часа после заражения. Для каждого раствора вирусных частиц рассчитывали полулетальную дозу (LD50) по методу Карбера, используя формулу LogLD50=XM+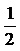

(2) Изучение эффекта 33-гидроксифиллигенин-8-O-β-D-глюкуронида при пневмонии, вызванной инфекцией вирусом гриппа и вирусом парагриппа
1) Лабораторные животные и экспериментальные группы
Для двух испытаний были взяты 960 мышей линии Куньмин в возрасте 4 недель. Для каждого из этих двух испытаний брали 480 особей и разделяли их случайным образом на 48 групп по 10 животных в каждой. В первом испытании определяли влияние 33-гидроксифиллигенин-8-O-β-D-глюкуронида на легочный показатель и на степень его снижения у мышей, зараженных вирусом гриппа; каждый опыт повторяли три раза, при этом всякий раз брали 80 особей. Во втором испытании определяли влияние 33-гидроксифиллигенин-8-O-β-D-глюкуронида на титр вируса в суспензии легочной ткани по реакции агглютинации; каждый опыт повторяли три раза и всякий раз брали 80 особей.
2) Метод заражения
В химический стакан на 200-300 мл помещали тампон из гигроскопической ваты, наливали такое количество этилового эфира, которое достаточно для смачивания тампона, переворачивали стакан вверх дном и сажали под него мышь. Когда животное после некоторого периода возбуждения явно ослабевало, его переворачивали на спину и в носовую полость вводили ингаляционным путем раствор вируса (гриппа или парагриппа) в количестве по 0,03 мл в каждую ноздрю; параллельно делали контрольную обработку, для которой брали вместо суспензии вирусных частиц физиологический раствор («нормальный» контроль).
3) Метод введения и дозировка испытываемых агентов
Подопытным животным перед вирусным заражением 1 раз в сутки на протяжении 5 суток (последний раз - за сутки до заражения) вводили интрагастрально 33-гидроксифиллигенин-8-O-β-D-глюкуронид; в качестве положительного контроля так же вводили рибавирин и оселтамивира фосфат. Концентрацию 33-гидроксифиллигенин-8-O-β-D-глюкуронида для введения брали высокую (10,0 мг на 1 кг массы тела), среднюю (5,0 мг/кг) и низкую (2,5 мг/кг); рибавирина - 58,5 мг/кг; оселтамивира фосфата - 19,5 мг/кг; филлигенина - 13,0 мг/кг. Контрольным животным (контроль «на вирус») вводили таким же образом и в таком же количестве физиологический раствор.
4) Наблюдаемые показатели
I. Определение легочного показателя
Подопытным мышам на пятые сутки после введения испытываемых агентов не давали есть и пить в течение 8 часов, после чего взвешивали, извлекали глаза, выпускали кровь и умерщвляли. Вскрывали грудную клетку, извлекали легкие целиком, промывали два раза физиологическим раствором, обсушивали поверхность органа фильтровальной бумагой, взвешивали легкие на электронных весах и рассчитывали легочный показатель и степень его снижения по следующим формулам:
Легочный показатель = (масса легких/масса тела) х 100%;
Степень снижения легочного показателя = (среднее значение легочного показателя у особей с выраженной инфекцией - среднее значение легочного показателя у особей, получавших испытываемый агент)/ среднее значение легочного показателя у особей с выраженной инфекцией х 100%
II. Определение титра вируса в суспензии легочной ткани по реакции гемагглютинации
На пятые сутки после описанной выше обработки у мышей различных групп извлекали легкие, измельчали в гомогенизаторе при низкой температуре, разбавляли гомогенизат легочной ткани физиологическим раствором до состояния суспензии концентрацией 10% и центрифугировали ее. Из полученного супернатанта готовили серию двукратных разведений, которые вносили по 0,2 мл в лунки титровального планшета, прибавляли в каждую лунку 0,2 мл 1%-ной суспензии куриных эритроцитов, перемешивали до однородности и оставляли при комнатной температуре на 30 минут, после чего наблюдали гемагглютинацию и определяли титр, приняв за конечную точку время агглютинации эритроцитов (++) и за титр - степень разведения суспензии,
2.3. Результаты и их анализ
(1) Результаты определения полулетальной дозы вирусов гриппа и парагриппа у мышей
Мышам линии Куньмин вводили ингаляционным путем в носовую полость по 30 мкл разведений вируса гриппа и вируса парагриппа различных концентраций. На третьи сутки после заражения в трех группах подопытных животных (концентрация вируса 10-1, 10-2 и 10-3) наблюдались следующие симптомы заболевания (в той или иной степени выраженности): пиломоторный рефлекс (шерсть «дыбом»), дрожь, пониженный аппетит и др.; на пятые сутки мыши шатались при ходьбе; на шестые сутки в группе особей, получивших разведение с наибольшей концентрацией вируса, начались случаи смерти, а в других группах животные стали умирать начиная с седьмого дня после заражения, Наблюдение вели 14 суток, после чего подсчитали, сколько мышей погибло в каждой группе; результаты представлены в таблицах 1-4 и 1-5, Для вируса гриппа полулетальная доза (LD50) составила разведение 10-2,9, для вируса парагриппа - 10-2,5.
Таблица 1-4. Результаты определения полулетальной дозы вируса гриппа
(разведение вируса гриппа)
(обработка без вирусов)
LD50 рассчитывали по методу Карбера. Для вируса гриппа расчет следующий:
LogLD50=XM+
Taблица 1-5. Результаты определения полулетальной дозы вируса парагриппа
без вирусов)
LD50 рассчитывали по методу Карбера. Для вируса парагриппа расчет следующий:
LogLD50=XM+
(2) Результаты влияния 33-гидроксифиллигенин-8-O-β-D-глюкуронида на развитие пневмонии, обусловленной инфекцией вируса гриппа и парагриппа,
I. Определение легочного показателя
После заражения мышей вирусами гриппа и парагриппа определяли средний легочный показатель, и это продемонстрировало, что по сравнению с группами особей, в которых этим инфекциям давали развиваться беспрепятственно, у животных, получавших 33-гидроксифиллигенин-8-O-β-D-глюкуронид в суточной дозе 2,25-10,0 мг на 1 кг массы тела, легочный показатель был явно ниже; лечебный эффект в группах мышей с инфекцией гриппа или парагриппа, получавших 33-гидроксифиллигенин-8-O-β-D-глюкуронид в высокой дозе, превышал таковой в группе особей, получавших филлигенин (P<0,05). Полученные результаты представлены в таблицах 1-6 и 1-7.
Taблица 1-6. Влияние соединения I на легочный показатель и на степень его снижения у мышей, зараженных вирусом гриппа (n=3)
(мг/кг/сут)
(
«на вирус»
При сравнении с контролем «на вирус» *P<0,05, **P0,01; при сравнении с группой мышей, получавших филлигенин, #P<0,05, ##P <0,01,
Taблица 1-7. Влияние соединения I на легочный показатель и на степень его снижения у мышей, зараженных вирусом парагриппа (n=3)
(мг/кг/сут)
(
«на вирус»
При сравнении с контролем «на вирус» *P<0,05, **P0,01; при сравнении с группой мышей, получавших филлигенин, #P<0,05, ##P <0,01.
II. Определение титра вируса в суспензии легочной ткани по реакции гемагглютинации
В группах мышей, у которых развивались инфекции вирусами гриппа и парагриппа, титр вируса в легочной ткани по реакции гемагглютинации (InX) после заражения составлял 31,64 и 32,06 соответственно. После того, как животным на протяжении 5 суток давали 33-гидроксифиллигенин-8-O-β-D-глюкуронид в различных концентрациях, титр вируса в легочной ткани по реакции гемагглютинации несколько снижался; по сравнению с группой особей, в которой вирусная инфекция развивалась беспрепятственно, разница явная (P<0,01). Титры вируса гриппа и вируса парагриппа в группах мышей, получавших 33-гидроксифиллигенин-8-O-β-D-глюкуронид в средней и высокой дозах явно ниже, чем в группе животных, у которых инфекция развивалась беспрепятственно, причем степень снижения титра явно выше, чем в группе мышей, получавших филлигенин (P<0,05; p<0,01). Эти результаты представлены в таблицах 1-8 и 1-9.
Taблица 1-8. Влияние соединения I на титр вируса в суспензии легочной ткани по реакции гемагглютинации у мышей, зараженных вирусом гриппа (n=3)
(мг/кг/сут)
«на вирус»
При сравнении с контролем «на вирус» *P<0,05, **P<0,01; при сравнении с группой мышей, получавших филлигенин, #P<0,05, ##P <0,01.
Taблица 1-9. Влияние соединения I на титр вируса в суспензии легочной ткани по реакции гемагглютинации у мышей, зараженных вирусом парагриппа (n=3)
(мг/кг/сут)
«на вирус»
При сравнении с контролем «на вирус» *P<0,05, **P<0,01; при сравнении с группой мышей, получавших филлигенин, #P<0,05, ##P <0,01.
2.4 Заключение
Результаты экспериментов in vivo, показали, что 33-гидроксифиллигенин-8-O-β-D-глюкуронид явно подавляет вирусы гриппа и парагриппа и развитие вирусной пневмонии у мышей, вызванной введением вирусов в дозировке от 2,25 мг на 1 кг массы тела в сутки до 10 мг/кг/сут. При этом наблюдается выраженный эффект снижения легочного показателя и титров этих вирусов по реакции гемагглютинации, а также ослабления патологических изменений в легких по сравнению с контролем - мышами, у которых указанные инфекции развивались беспрепятственно. Лечебный эффект в группах мышей, получавших 33-гидроксифиллигенин-8-O-β-D-глюкуронид в средней и высокой дозах, явно превышал таковой филлигенина (*P<0,05 или **P<0,01), и в меньшей степени - рибавирина и оселтамивира фосфата.
Соединения II и III так же, как соединение I, обладали выраженным подавляющим эффектом в отношении вирусов гриппа и парагриппа и вызываемой ими пневмонии у мышей, снижая легочный показатель и титр вируса (по реакции гемагглютинации), а также ослабляя патологические изменения в легких; при сравнении с контролем - мышами, у которых вирусная инфекция развивалась без воздействия указанными соединениями, разница очевидна.
Изобретение также относится к противовирусному производному общей формулы (I), фармацевтическим композициям на его основе, способу его получения, которые могут быть использованы в фармацевтической и медицинской отрасли:
 ,
,
где R1=CnH2n+1, R2=CnH2n+1, R3=H или R1-R2=-CH2-, R3=CnH2n+1; n=1-30. Предложенный способ включает смешивание листьев форсайтии с водой, нагревание с кипячением и экстракцией с получением водного экстракта форсайтии; разделение водного экстракта форсайтии на колонке с макропористой смолой; проведение хроматографии элюата из форсайтии, полученного на колонке с макропористой смолой, на колонке с силикагелем, сбор элюата, его высушивание. Предложены новое производное филлигенина и глюкуроновой кислоты, новый эффективный способ его получения, фармацевтические композиции на его основе для приготовления лекарств, эффективных для предотвращения и лечения гриппа. 4 н. и 6 з.п. ф-лы, 3 пр., 9 табл.
1. Производное филлигенина и глюкуроновой кислоты, характеризующееся молекулярной структурой общей формулы (I)

где R1=CnH2n+1, R2=CnH2n+1, R3=H или R1-R2=-CH2-, R3=CnH2n+1; n=1-30.
2. Производное по п. 1, в котором n равно 1.
3. Способ получения производного филлигенина и глюкуроновой кислоты, характеризующийся тем, что он включает последовательно следующие стадии:
1) смешивание листьев форсайтии с экстрагирующим растворителем, представляющим собой воду, 2-3-кратное нагревание с кипячением и экстракцией, сбор и объединение экстрагирующих растворов, в результате чего получают водный экстракт форсайтии;
2) разделение водного экстракта форсайтии на колонке с макропористой смолой и элюирование водой в качестве элюирующего растворителя; элюирование раствором этилового спирта с массовой концентрацией 3-50% в качестве элюирующего растворителя и, наконец, элюирование абсолютным этиловым спиртом в качестве элюирующего растворителя; сбор абсолютного этилового спирта, использованного в качестве элюирующего растворителя, в результате чего получают элюат из форсайтии на колонке с макропористой смолой; причем макропористая смола выбрана из следующих продуктов: X-5, AB-8, NK-2, NKA-2, NK-9, D3520, D101 и WLD;
3) проведение хроматографии элюата из форсайтии, полученного на колонке с макропористой смолой, на колонке с силикагелем, фракционный сбор элюата и его высушивание, в результате чего получают целевой продукт;
где производное филлигенина и глюкуроновой кислоты характеризуется молекулярной структурой общей формулы (I)
где R1=CnH2n+1, R2=CnH2n+1, R3=H или R1-R2=-CH2-, R3=CnH2n+1; n=1.
4. Способ по п. 3, отличающийся тем, что на этапе 1) при каждом нагревании с кипячением массовое соотношение листьев форсайтии и экстрагирующего растворителя, представляющего собой воду, составляет 1:(6-10).
5. Способ по п. 3 или 4, отличающийся тем, что способ дополнительно включает этапы концентрирования водного экстракта форсайтии на этапе 1), получения концентрированного раствора из форсайтии и проведения разделения на колонке с макропористой смолой.
6. Способ по п. 5, отличающийся тем, что отношение объема концентрированного раствора из форсайтии, полученного путем концентрирования, к массе листьев форсайтии составляет (1-5):1.
7. Способ по п. 3 или 4, отличающийся тем, что на этапе 3) наполнителем колонки для хроматографии с использованием силикагеля служит силикагель C18 для хроматографии на обращенной фазе; хроматографическая колонка имеет следующие параметры: внутренний диаметр 10-100 нм, длина 10-300 мм; размер частиц наполнителя 5-10 мкм; элюирование изократическое или градиентное.
8. Способ по п. 7, отличающийся тем, что при хроматографии на колонке с силикагелем подвижной фазой является смешанный раствор – метиловый спирт с водой, в котором объемное отношение метилового спирта и воды составляет 8:2-10:1.
9. Противовирусное средство, содержащее производное филлигенина и глюкуроновой кислоты по п. 1 в качестве активного ингредиента.
10. Применение производного филлигенина и глюкуроновой кислоты по п. 1 в приготовлении лекарственных средств для предотвращения и/или лечения инфекции вирусами гриппа.
| Soler A | |||
| et al | |||
| Food Chemistry, 2010, 119, N2, 703-714 | |||
| FAN Hongyu et al | |||
| Liaoning chemical industry, 2014, vol | |||
| Зубчатое колесо со сменным зубчатым ободом | 1922 |
|
SU43A1 |
| Одноколейная подвесная к козлам дорога | 1919 |
|
SU241A1 |
| Duan Linjian et al | |||
| Chinese General Practice, 2012, vol | |||
| Прибор для нагревания перетягиваемых бандажей подвижного состава | 1917 |
|
SU15A1 |
| Складная рамочная антенна | 1925 |
|
SU2082A1 |
| ЛЕКАРСТВЕННАЯ КОМПОЗИЦИЯ ДЛЯ ЛЕЧЕНИЯ БРОНХИТА И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 2009 |
|
RU2520745C2 |

