Родственные заявки
В настоящей заявке испрашивается приоритет в связи с предварительной заявкой US 61/274051, поданной 12 августа 2009 г, содержание которой включено в полном объеме в настоящее описание в качестве ссылки.
Область изобретения
В настоящем изобретении предлагается кристаллическая форма (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида, его солей и гидратов. В настоящем изобретении предлагается также твердый пероральный состав, включающий (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамид, его фармацевтически приемлемые соли, сольваты (включая гидраты), а также способы лечения с их использованием.
Предпосылки создания настоящего изобретения
(S)-N-((S)-1-Циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамид характеризуется формулой (I):
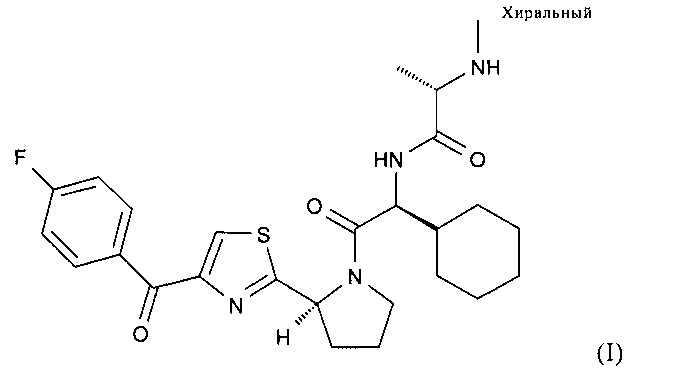
и является ингибитором белка апоптоза, который защищает раковые клетки от гибели в результате апоптоза.
Соединение формулы (I) («соединение (I)») в общем виде и/или более подробно описано в WO 05/097791 и WO 08/016893, содержание которых включено в полном объеме в настоящее описание в качестве ссылок.
Краткое описание сущности изобретения
В настоящем изобретении предлагаются пероральные составы, включающие (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамид, включая его соль(соли) и/или сольват(сольваты). В предпочтительных вариантах осуществления настоящего изобретения предлагаются составы таблеток, включающих (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамид, характеризующихся высоким содержанием лекарственного вещества и профилем быстрого высвобождения. Соединение (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида характеризуется формулой (I):
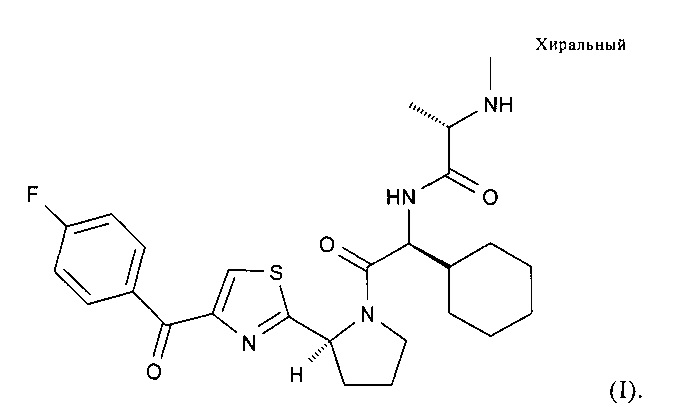
В настоящем изобретении предлагаются также кристаллические формы (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида, включая его соль(соли) и/или сольват(сольваты). В первом варианте осуществления настоящего изобретения предлагается кристаллическая форма HA, которая представляет собой полугидрат свободной формы соединения формулы (I). Во втором, третьем, четвертом и/или пятом вариантах предлагаются соответственно кристаллические формы A, B, C и/или D, каждая из которых представляет собой безводную свободную форму соединения формулы (I).
Краткое описание фигур
Настоящее изобретение проиллюстрировано прилагаемыми фигурами, описанными ниже.
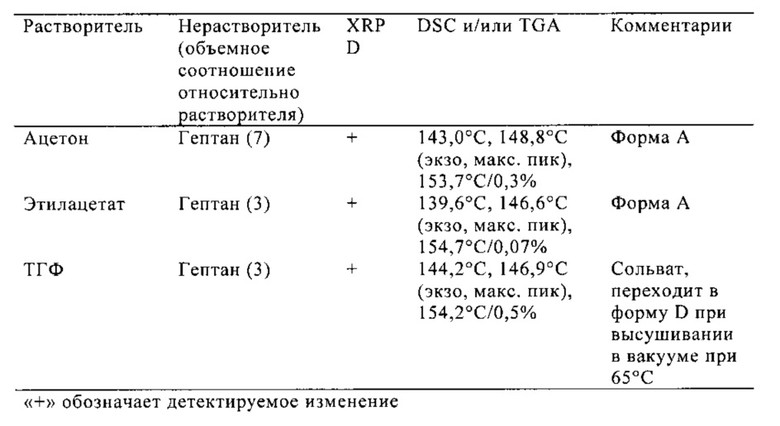
На фиг. 1 показаны порошковые рентгеновские дифрактограммы (XRPD) для форм HA, А, В, С и D соединения формулы (I).
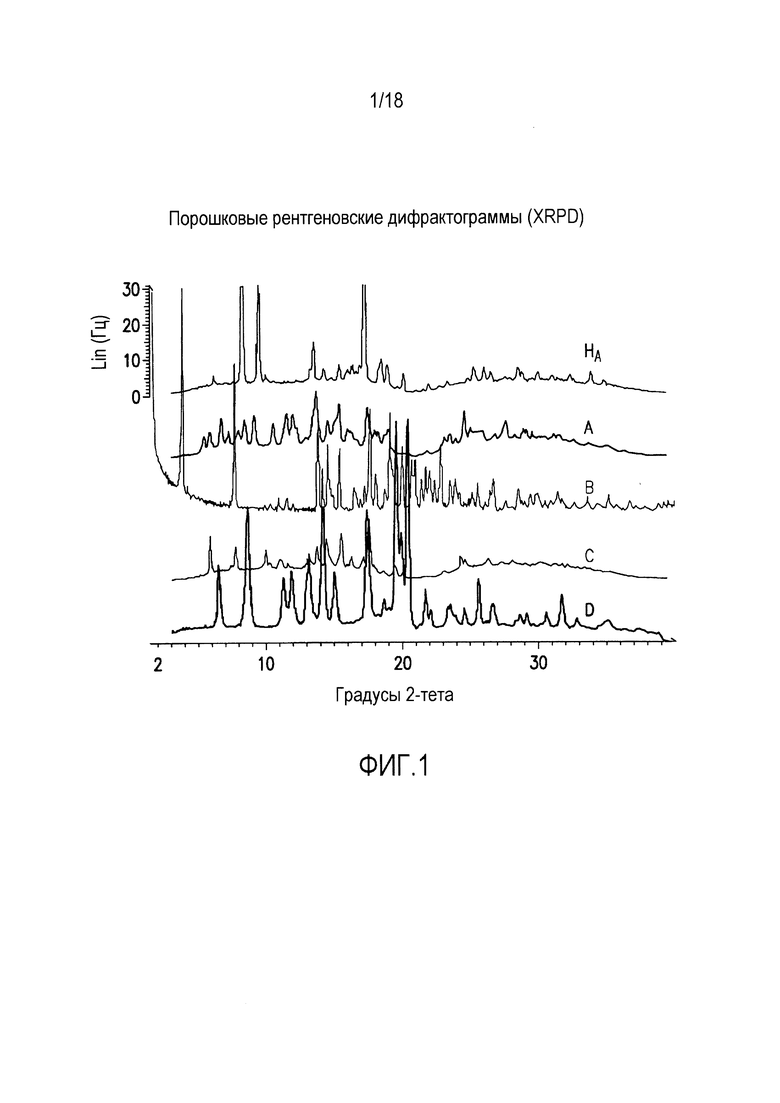
На фиг. 2 представлены кривые дифференциальной сканирующей калориметрии (DSC) и термогравиметрического анализа (TGA) для формы HA соединения формулы (I).
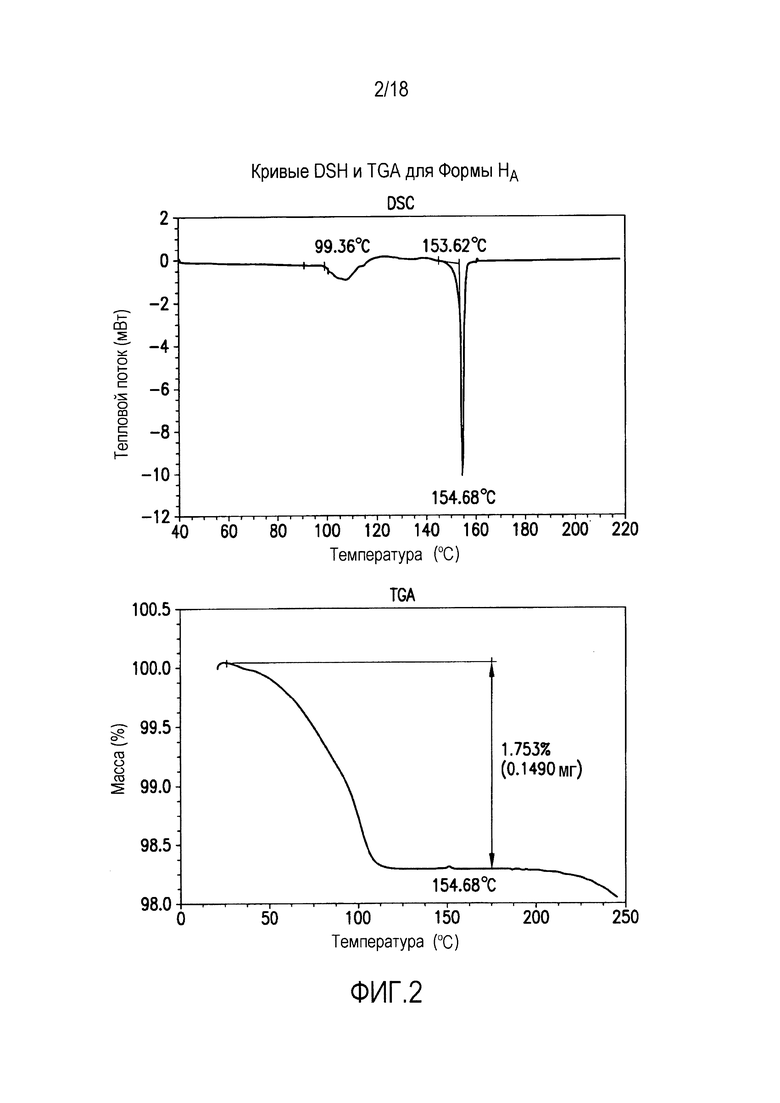
На фиг. 3 представлены кривые дифференциальной сканирующей калориметрии (DSC) и термогравиметрического анализа (TGA) для формы A соединения формулы (I).
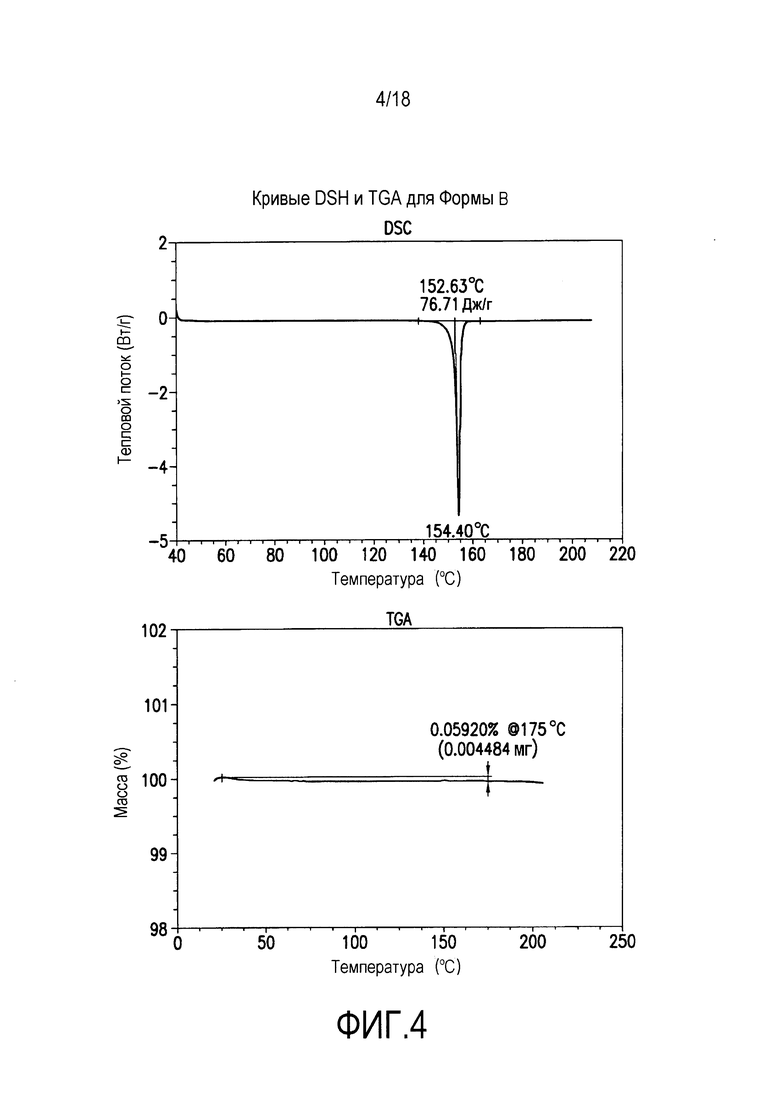
На фиг. 4 представлены кривые дифференциальной сканирующей калориметрии (DSC) и термогравиметрического анализа (TGA) для формы B соединения формулы (I).
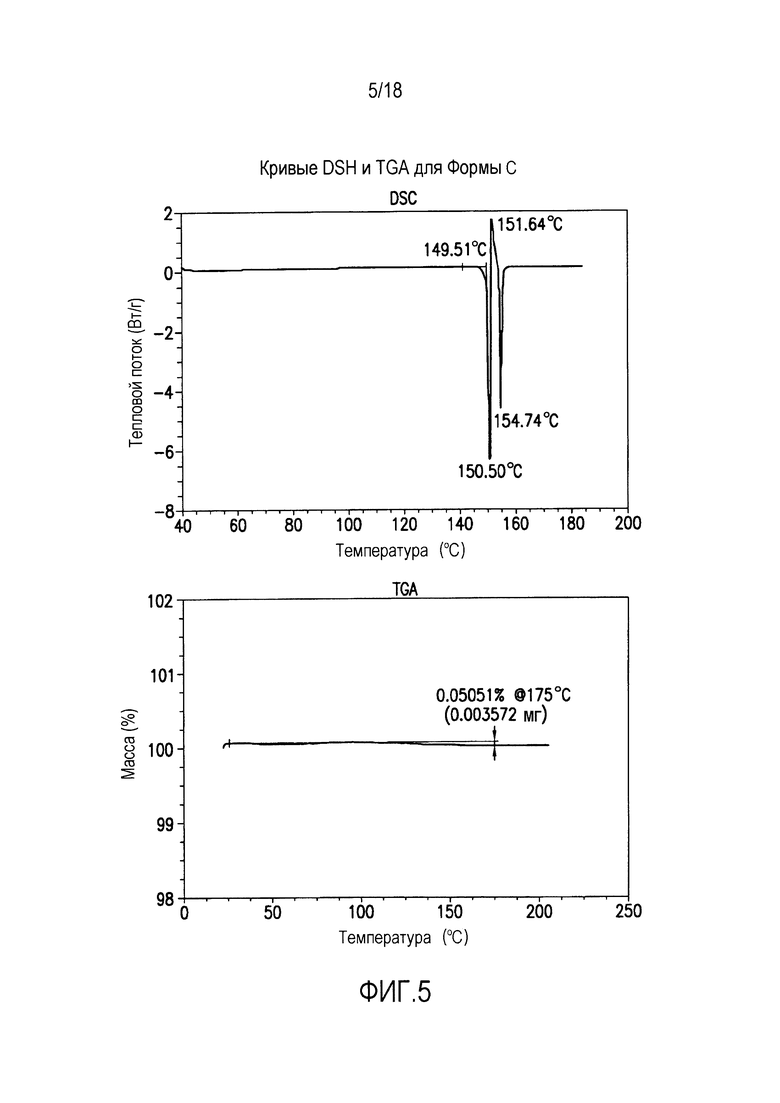
На фиг. 5 представлены кривые дифференциальной сканирующей калориметрии (DSC) и термогравиметрического анализа (TGA) для формы C соединения формулы (I).
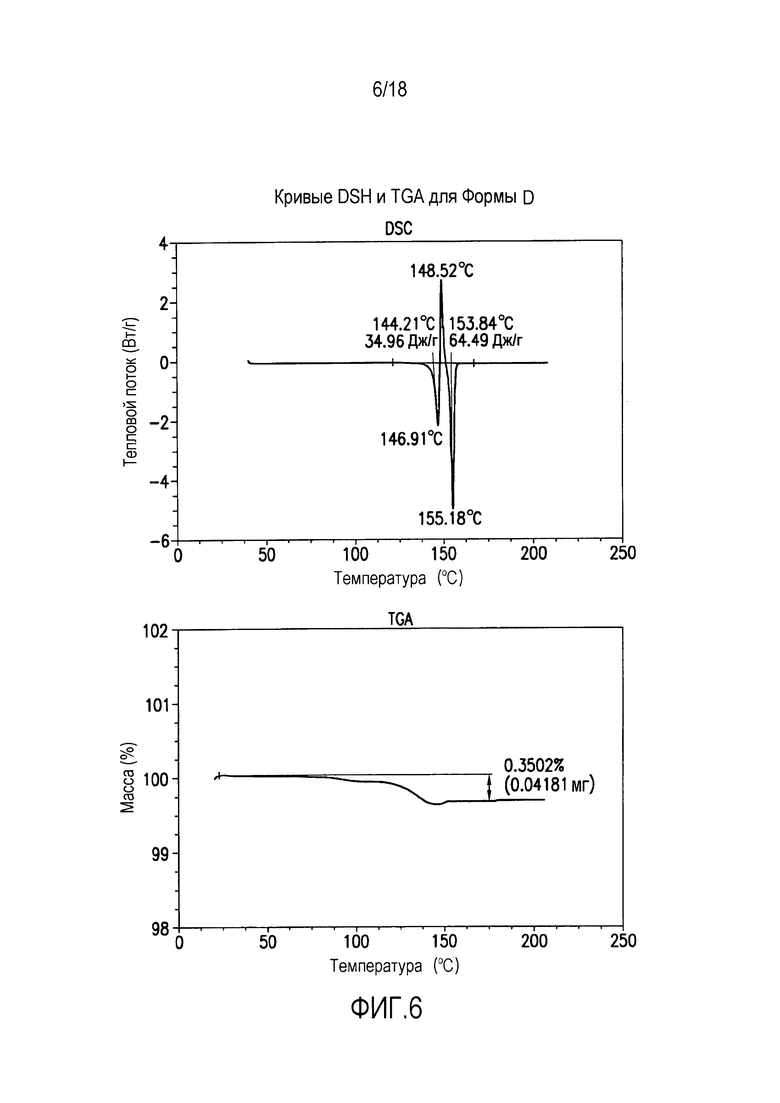
На фиг. 6 представлены кривые дифференциальной сканирующей калориметрии (DSC) и термогравиметрического анализа (TGA) для формы D соединения формулы (I).
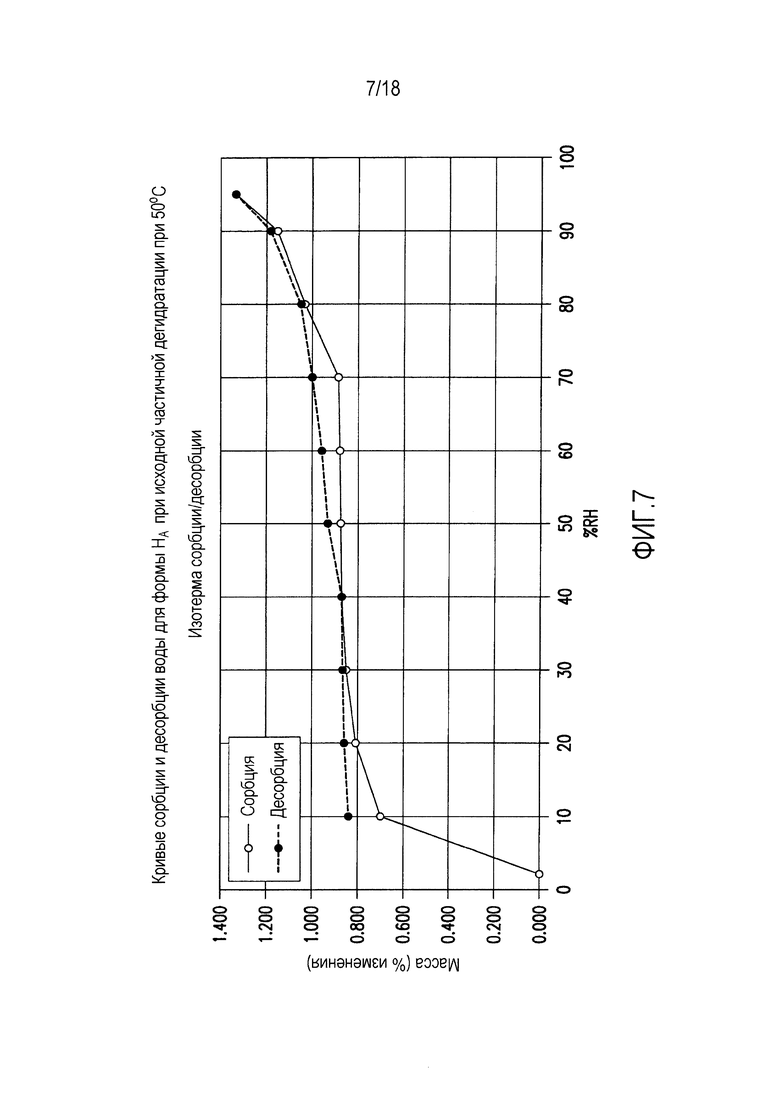
На фиг. 7 представлены кривые сорбции и десорбции воды для формы HA соединения формулы (I) при исходной частичной дегидратации при 50°C.
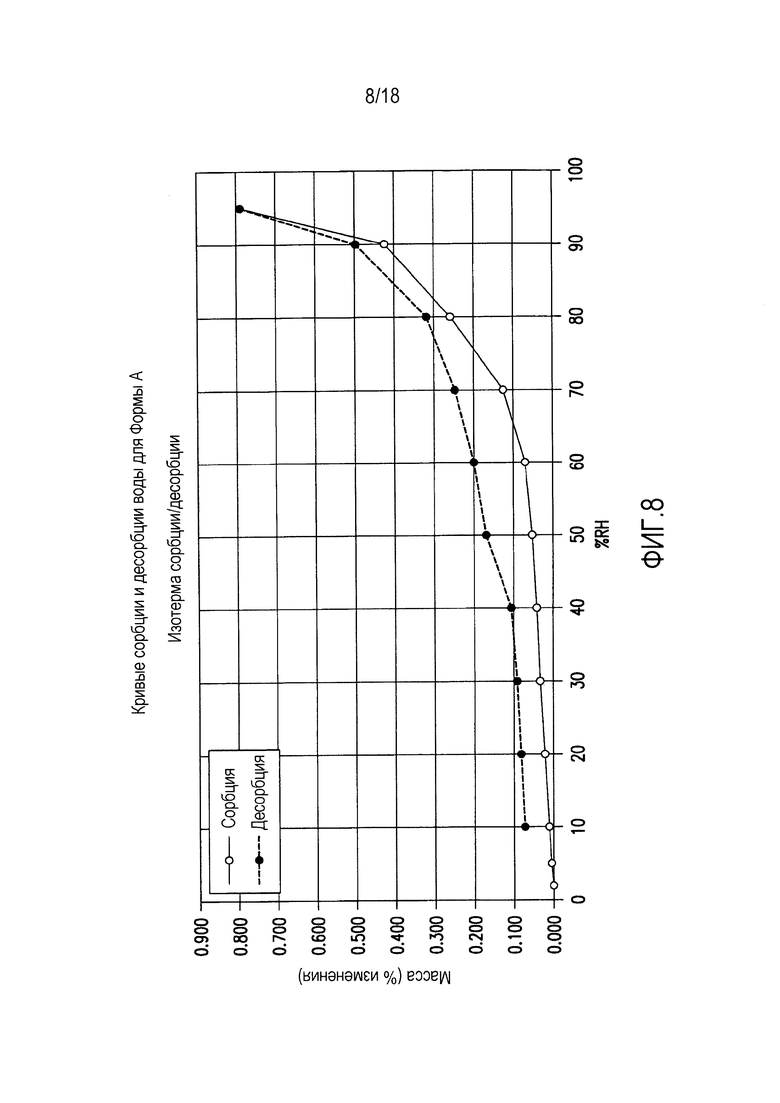
На фиг. 8 представлены кривые сорбции и десорбции воды для формы A соединения формулы (I).
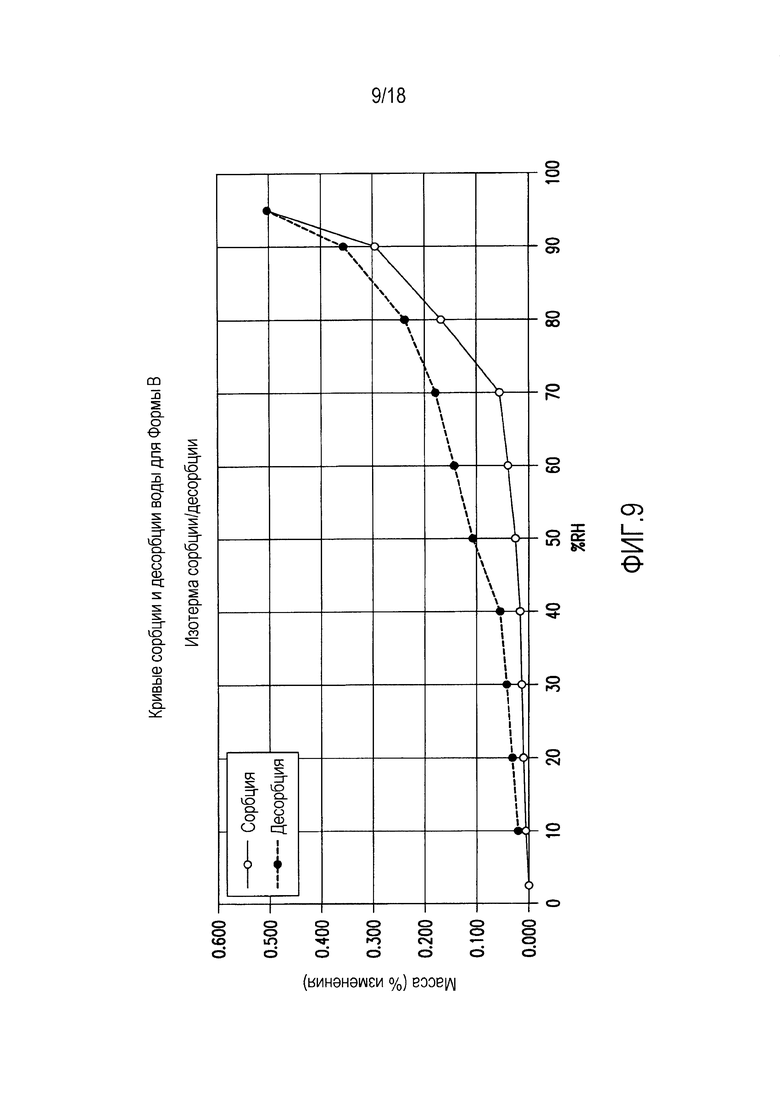
На фиг. 9 представлены кривые сорбции и десорбции воды для формы B соединения формулы (I).
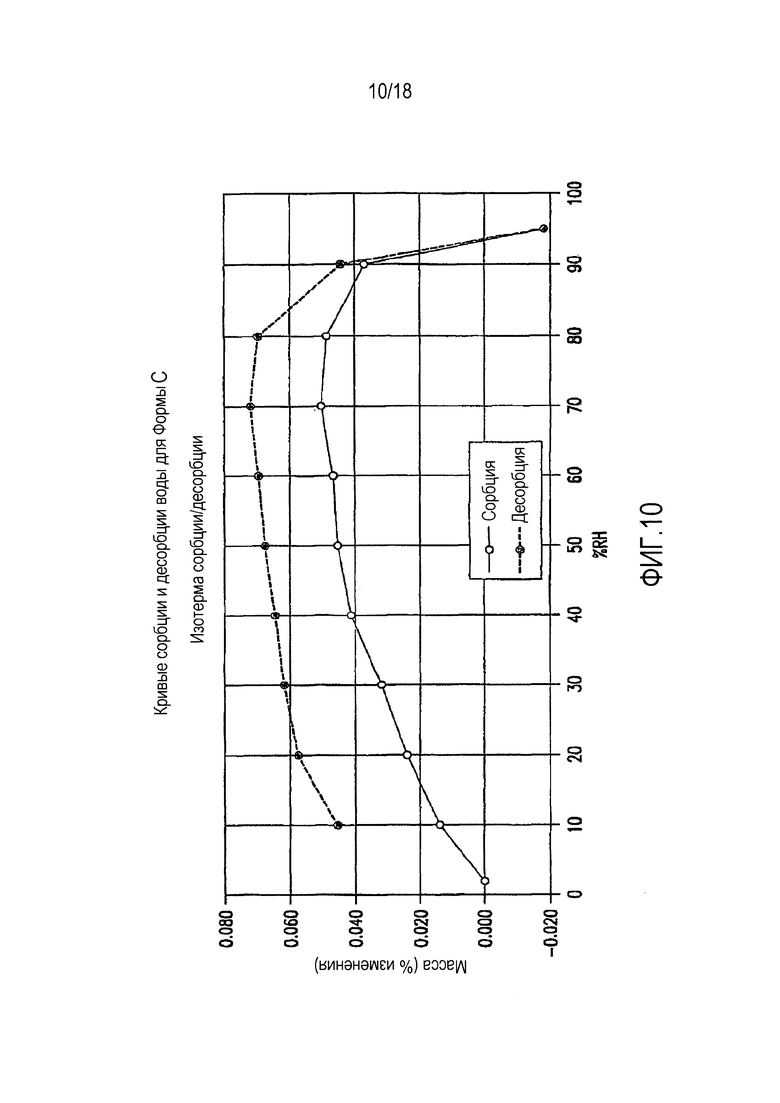
На фиг. 10 представлены кривые сорбции и десорбции воды для формы C соединения формулы (I).
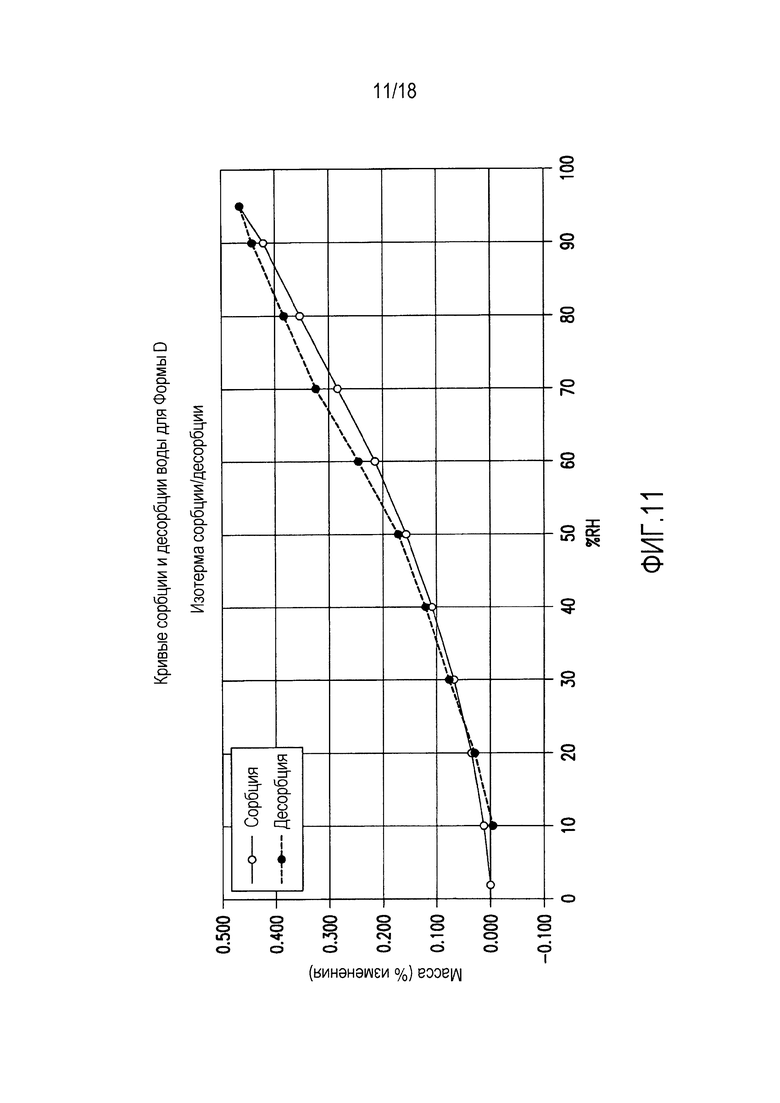
На фиг. 11 представлены кривые сорбции и десорбции воды для формы D соединения формулы (I).
На фиг. 12 показана микрофотография формы HA соединения формулы (I).
На фиг. 13 показана микрофотография формы A соединения формулы (I).
На фиг. 14 показана микрофотография формы B соединения формулы (I).
На фиг. 15 показана микрофотография формы C соединения формулы (I).
На фиг. 16 показана микрофотография формы D соединения формулы (I).
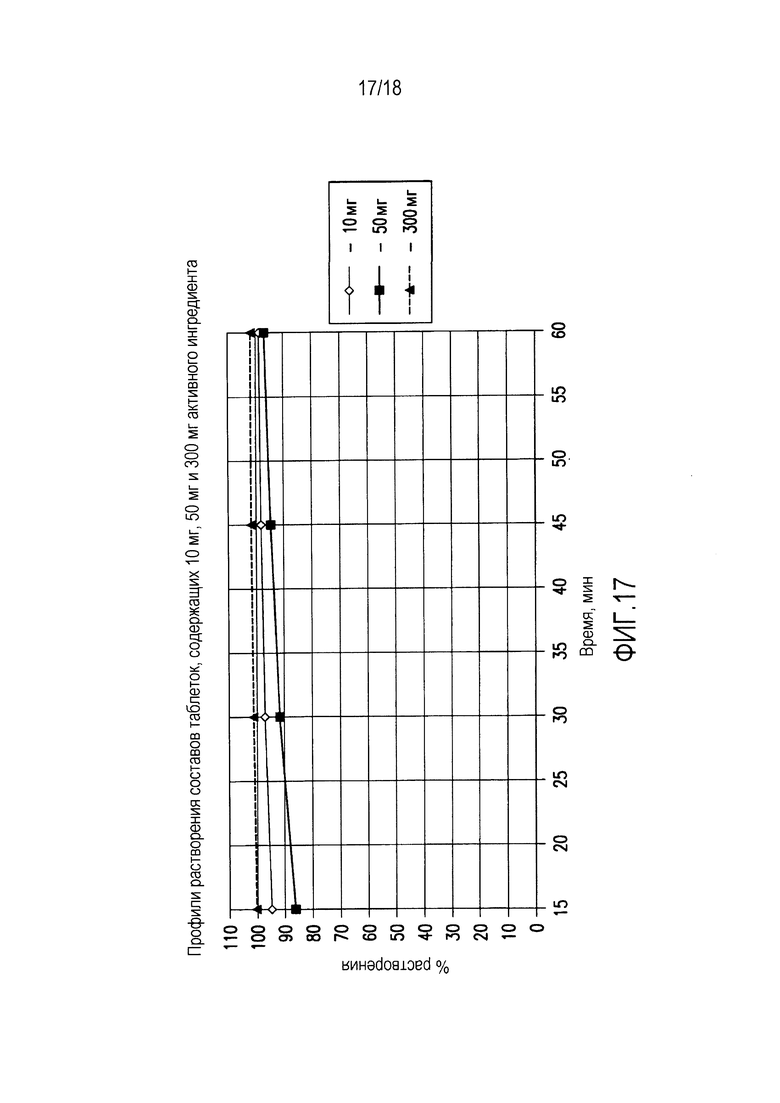
На фиг. 17 показаны профили растворения составов таблеток, содержащих 10 мг, 50 мг и 300 мг активного ингредиента, полученных как описано в примерах 1-3.
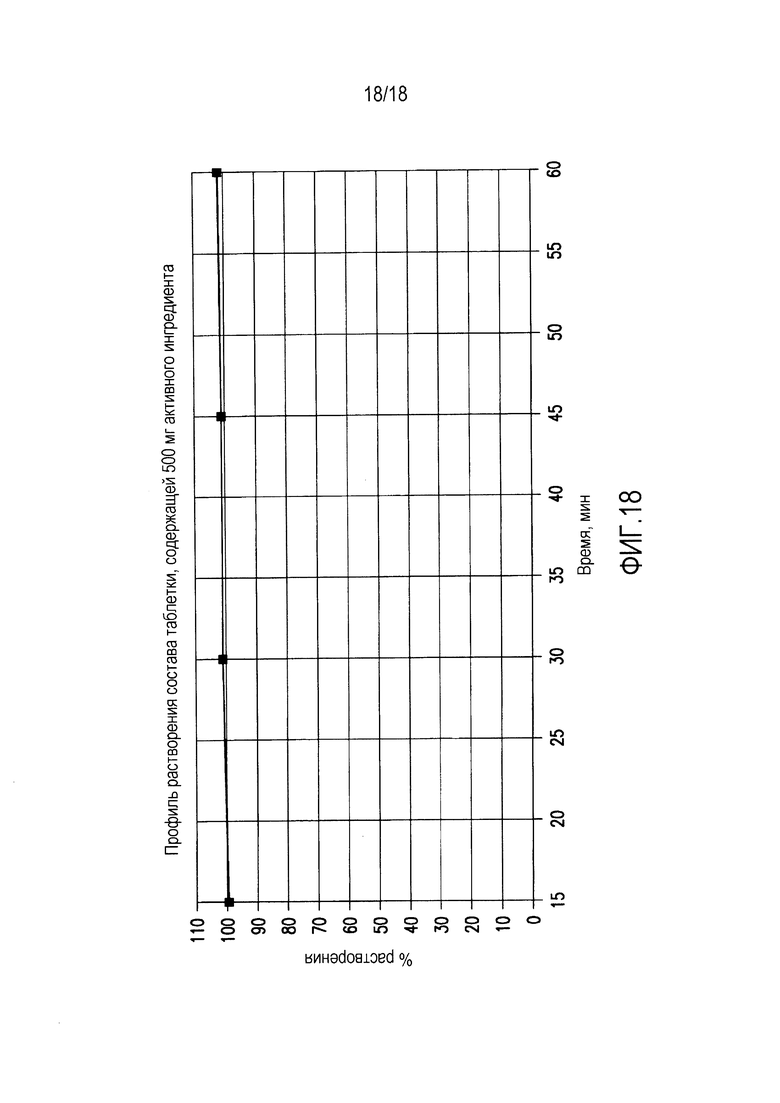
На фиг. 18 показан профиль растворения состава таблетки, содержащей 500 мг активного ингредиента, полученной как описано в примере 4.
Подробное описание изобретения
Соединение (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида (структура формулы (I)) и его гидрат(гидраты) существуют в различных формах. Настоящее описание относится к кристаллическим формам HA, А, B, C и D соединения формулы (I).
Кристаллические формы соединения (I), его солей и сольватов можно характеризовать рядом методов, включая, но не ограничиваясь только ими, порошковую рентгеновскую дифракцию (PXRD), моделированные порошковые рентгеновские дифрактограммы (Yin. S., Scaringe R. P., DiMarco J., Galella M. и Gougoutas J. Z., American Pharmaceutical Review, 6, 2, c. 80 (2003)), анализ методом дифференциальной сканирующей калориметрии (DSC), метод твердофазного ЯМР С-13 (Earl W.L. и VanderHart D.L., J. Magn. Reson., 48, стр. 35-54 (1982)), спектроскопию комбинационного (рамановского) рассеяния света, инфракрасную спектроскопию, изотермы поглощения влаги (изотермы в переменном температурном режиме), а также высокотемпературные методики.
Формы можно характеризовать и различать с использованием анализа, который основан на рентгеноструктурном анализе монокристалла конкретной формы при фиксированной температуре и на расчете параметров элементарной ячейки. Подробное описание элементарных ячеек приведено в руководстве Stout и Jensen, X-Ray Structure Determination: A Practical Guide, Macmillan Co., Нью-Йорк, глава 3 (1968), которая включена в настоящее описание в качестве ссылки. В другом варианте, уникальное пространственное расположение атомов в кристаллической решетке можно характеризовать в соответствии с наблюдаемыми относительными атомными координатами. Другим способом характеризации кристаллической структуры является анализ методом порошковой рентгеновской дифракции, в ходе которого дифракционный профиль сравнивают с моделированным профилем, полученным для чистого порошкообразного материала, при этом оба профиля получают при одной и той же температуре анализа и результаты измерений для анализируемой формы представляют в виде серии значений угла 2θ.
Специалисту в данной области техники представляется очевидным, что ошибка измерения при получении рентгеновской дифрактограммы зависит от используемых условий измерения. Прежде всего, общеизвестно, что в зависимости от используемых условий измерения изменяются значения интенсивности на рентгеновской дифрактограмме. Кроме того, следует понимать, что относительные значения интенсивности изменяются также в зависимости от экспериментальных условий, и соответственно, точное значение интенсивности не учитывают. Кроме того, ошибка измерения угла дифракции на стандартной рентгеновской дифрактограмме обычно составляет приблизительно 5% или менее, и следует учитывать, что указанная величина ошибки измерения также относится к величинам упомянутых выше дифракционных углов. В связи с этим, следует также понимать, что кристаллические формы по настоящему изобретению не ограничиваются кристаллическими формами, которые характеризуются рентгеновскими дифрактограммами, полностью идентичными рентгеновским дифрактограммам, указанных на прилагаемых здесь фигурах. В объем настоящего изобретения включены любые кристаллические формы, которые характеризуются рентгеновскими дифрактограммами, в значительной степени идентичными рентгеновским дифрактограммам, приведенным на представленных в настоящем изобретении фигурах. Степень идентичности рентгеновских дифрактограмм определяет специалист в данной области техники.
Аналогичным образом, следует понимать, что в объем настоящего изобретения включены любые кристаллические формы, которые характеризуются кривыми дифференциальной сканирующей калориметрии (DSC), термогравиметрического анализа (TGA) и/или изотермами поглощения влаги, которые в основном идентичны указанным на прилагаемых фигурах. Степень идентичности указанных кривых определяет специалист в данной области техники.
Форма HA
Форму HA получают, как показано на схеме А. Исходные соединения B1 и В3 являются коммерческими продуктами.
Форма HA представляет собой кристаллический полугидрат, содержание воды в котором составляет ~1,7%. Указанная форма является незначительно гигроскопичной. Содержание воды в указанной форме составляет ~1,7% при относительной влажности (ОВ) от 10% до 70%, и указанная форма поглощает дополнительные ~0,4% влаги при ОВ от 70% до 95%. При нагревании выше 100°C форма HA теряет воду и переходит в форму В.
Форма HA характеризуется порошковой рентгеновской дифрактограммой, содержащей три или более значения 2θ, выбранных из группы, включающей 8,3±0,2, 9,5±0,2, 13,5±0,2, 17,3±0,2, 18,5±0,2 и 18,9±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма HA характеризуется порошковой рентгеновской дифрактограммой, содержащей четыре или более значения 2θ, выбранных из группы, включающей 8,3±0,2, 9,5±0,2, 13,5±0,2, 17,3±0,2, 18,5±0,2 и 18,9±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма HA характеризуется порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ, выбранных из группы, включающей 8,3±0,2, 9,5±0,2, 13,5±0,2, 17,3±0,2, 18,5±0,2 и 18,9±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма HA характеризуется порошковой рентгеновской дифрактограммой при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C), в основном как указано на фиг. 1.
Форма HA характеризуется термограммой дифференциальной сканирующей калориметрии (DSC), в основном как указано на фиг. 2.
Форма HA характеризуется диаграммой термогравиметрического анализа (TGA), в основном как указано на фиг. 2.
Схема A
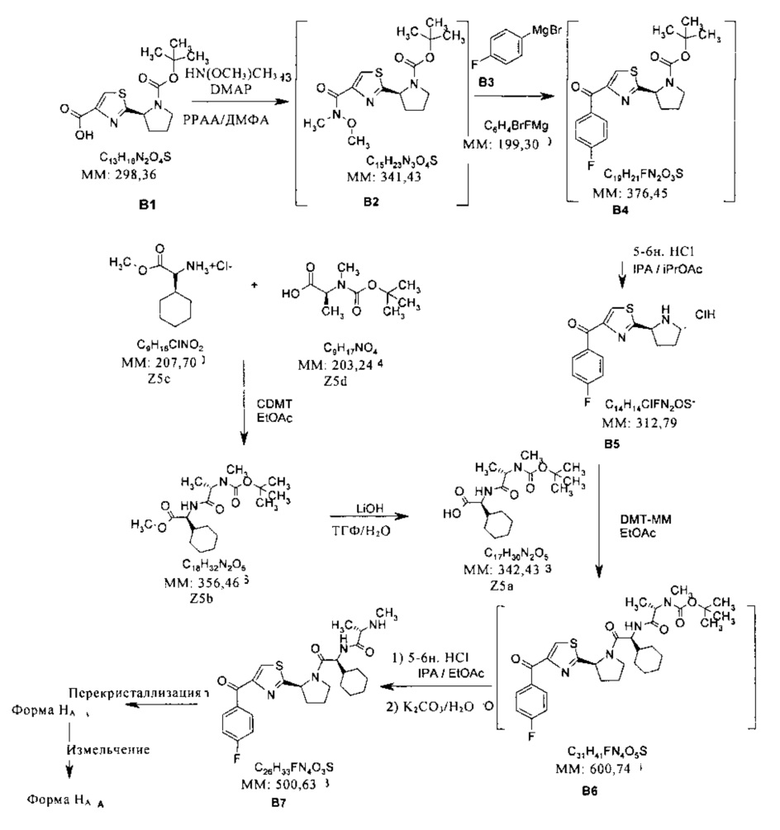
Форма A
Форму A получают при уравновешивании полугидрата формы HA в ряде органических растворителей, например в ацетоне, ацетонитриле, этаноле и т.п.
Форма A представляет собой безводное кристаллическое соединение. Форма A незначительно гигроскопична. Максимальное поглощение воды при 25°C и ОВ вплоть до 95% составляет приблизительно 0,8%. Начало плавления формы A регистрируется методом дифференциальной сканирующей калориметрии (DSC) при ~149°C, затем при дальнейшем нагревании до ~155°C происходит перекристаллизация в форму В. На микроскопическом изображении формы A видно, что она представляет собой агрегаты иголок небольшого размера.
Форма A характеризуется порошковой рентгеновской дифрактограммой, содержащей три или более значения 2θ, выбранных из группы, включающей 5,3±0,2, 6,7±0,2, 9,1±0,2, 13,4±0,2, 13,6±0,2, 15,0±0,2, 15,3±0,2, 17,4±0,2, 18,2±0,2, 18,7±0,2, 18,9±0,2, 20,2±0,2, 21,3±0,2, 21,8±0,2, 23,0±0,2, 23,5±0,2, 24,6±0,2 и 27,6±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма A характеризуется порошковой рентгеновской дифрактограммой, содержащей четыре или более значения 2θ, выбранных из группы, включающей 5,3±0,2, 6,7±0,2, 9,1±0,2, 13,4±0,2, 13,6±0,2, 15,0±0,2, 15,3±0,2, 17,4±0,2, 18,2±0,2, 18,7±0,2, 18,9±0,2, 20,2±0,2, 21,3±0,2, 21,8±0,2, 23,0±0,2, 23,5±0,2, 24,6±0,2 и 27,6±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма A характеризуется порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ, выбранных из группы, включающей 5,3±0,2, 6,7±0,2, 9,1±0,2, 13,4±0,2, 13,6±0,2, 15,0±0,2, 15,3±0,2, 17,4±0,2, 18,2±0,2, 18,7±0,2, 18,9±0,2, 20,2±0,2, 21,3±0,2, 21,8±0,2, 23,0±0,2, 23,5±0,2, 24,6±0,2 и 27,6±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма A характеризуется порошковой рентгеновской дифрактограммой, содержащей шесть или более значений 2θ, выбранных из группы, включающей 5,3±0,2, 6,7±0,2, 9,1±0,2, 13,4±0,2, 13,6±0,2, 15,0±0,2, 15,3±0,2, 17,4±0,2, 18,2±0,2, 18,7±0,2, 18,9±0,2, 20,2±0,2, 21,3±0,2, 21,8±0,2, 23,0±0,2, 23,5±0,2, 24,6±0,2 и 27,6±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма A характеризуется порошковой рентгеновской дифрактограммой при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C), в основном как указано на фиг. 1.
Форма A характеризуется термограммой дифференциальной сканирующей калориметрии (DSC), в основном как указано на фиг. 3.
Форма A характеризуется диаграммой термогравиметрического анализа (TGA), в основном как указано на фиг. 3.
Форма B
Форму B получают при нагревании полугидрата формы HA выше 100°C в течение периода времени, достаточного для полного удаления воды, при уравновешивании полугидрата формы HA в гептане при 50°C, или кристаллизацией при охлаждении из метилизобутилкетона.
Форма B представляет собой безводное кристаллическое соединение. Форма B незначительно гигроскопична. Максимальное поглощение воды при 25°C и ОВ вплоть до 95% составляет приблизительно 0,5%. Начало плавления формы B регистрируется методом DSC при ~153°C. Форма B представляет собой длинные палочкообразные кристаллы.
Форма B характеризуется порошковой рентгеновской дифрактограммой, содержащей три или более значения 2θ, выбранных из группы, включающей 3,8±0,2, 7,7±0,2, 13,8±0,2, 14,6±0,2, 15,4±0,2, 17,6±0,2, 19,1±0,2, 19,2±0,2, 19,4±0,2, 20,0±0,2, 20,7±0,2, 20,9±0,2 и 22,8±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма B характеризуется порошковой рентгеновской дифрактограммой, содержащей четыре или более значения 2θ, выбранных из группы, включающей 3,8±0,2, 7,7±0,2, 13,8±0,2, 14,6±0,2, 15,4±0,2, 17,6±0,2, 19,1±0,2, 19,2±0,2, 19,4±0,2, 20,0±0,2, 20,7±0,2, 20,9±0,2 и 22,8±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма B характеризуется порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ, выбранных из группы, включающей 3,8±0,2, 7,7±0,2, 13,8±0,2, 14,6±0,2, 15,4±0,2, 17,6±0,2, 19,1±0,2, 19,2±0,2, 19,4±0,2, 20,0±0,2, 20,7±0,2, 20,9±0,2 и 22,8±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма B характеризуется порошковой рентгеновской дифрактограммой, содержащей шесть или более значений 2θ, выбранных из группы, включающей 3,8±0,2, 7,7±0,2, 13,8±0,2, 14,6±0,2, 15,4±0,2, 17,6±0,2, 19,1±0,2, 19,2±0,2, 19,4±0,2, 20,0±0,2, 20,7±0,2, 20,9±0,2 и 22,8±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма B характеризуется порошковой рентгеновской дифрактограммой при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C), в основном как указано на фиг. 1.
Форма B характеризуется термограммой дифференциальной сканирующей калориметрии (DSC), в основном как указано на фиг. 4.
Форма B характеризуется диаграммой термогравиметрического анализа (TGA), в основном как указано на фиг. 4.
Форма C
Форму C получают кристаллизацией при охлаждении из ацетонитрила, а затем после фильтрования растворитель упаривают.
Форма C представляет собой безводное кристаллическое соединение. Форма B не гигроскопична. Максимальное поглощение воды при 25°C и ОВ вплоть до 95% составляет менее 0,2%. Начало плавления формы C регистрируется методом DSC при ~150°C, затем при дальнейшем нагревании до ~155°C наблюдается перекристаллизация указанной формы в форму В. Форма C представляет собой длинные палочкообразные кристаллы.
Форма C характеризуется порошковой рентгеновской дифрактограммой, содержащей три или более значения 2θ, выбранных из группы, включающей 5,8±0,2, 7,7±0,2, 9,9±0,2, 13,0±0,2, 14,3±0,2, 15,5±0,2, 17,5±0,2, 19,4±0,2, 20,0±0,2, 22,9±0,2 и 24,3±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма C характеризуется порошковой рентгеновской дифрактограммой, содержащей четыре или более значения 2θ, выбранных из группы, включающей 5,8±0,2, 7,7±0,2, 9,9±0,2, 13,0±0,2, 14,3±0,2, 15,5±0,2, 17,5±0,2, 19,4±0,2, 20,0±0,2, 22,9±0,2 и 24,3±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма C характеризуется порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ, выбранных из группы, включающей 5,8±0,2, 7,7±0,2, 9,9±0,2, 13,0±0,2, 14,3±0,2, 15,5±0,2, 17,5±0,2, 19,4±0,2, 20,0±0,2, 22,9±0,2 и 24,3±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма C характеризуется порошковой рентгеновской дифрактограммой, содержащей шесть или более значений 2θ, выбранных из группы, включающей 5,8±0,2, 7,7±0,2, 9,9±0,2, 13,0±0,2, 14,3±0,2, 15,5±0,2, 17,5±0,2, 19,4±0,2, 20,0±0,2, 22,9±0,2 и 24,3±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма C характеризуется порошковой рентгеновской дифрактограммой при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C), в основном как указано на фиг. 1.
Форма C характеризуется термограммой дифференциальной сканирующей калориметрии (DSC), в основном как указано на фиг. 5.
Форма C характеризуется диаграммой термогравиметрического анализа (TGA), в основном как указано на фиг. 5.
Форма D
Форму D получают кристаллизацией при охлаждении из ацетона и затем после фильтрования растворитель упаривают.
Форма D представляет собой безводное кристаллическое соединение. Форма D незначительно гигроскопична. Максимальное поглощение воды при 25°C и ОВ вплоть до 95% составляет менее 0,5%. Начало плавления формы D регистрируется методом DSC при ~144°C, затем при дальнейшем нагревании до ~155°C происходит перекристаллизация в форму В. Форма D представляет собой пучки палочкообразных кристаллов.
Форма D характеризуется порошковой рентгеновской дифрактограммой, содержащей три или более значения 2θ, выбранных из группы, включающей 6,5±0,2, 8,6±0,2, 11,3±0,2, 11,9±0,2, 13,1±0,2, 14,2±0,2, 15,1±0,2, 17,4±0,2, 19,6±0,2, 19,9±0,2, 20,4±0,2, 21,7±0,2, 25,6±0,2 и 31,7±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма D характеризуется порошковой рентгеновской дифрактограммой, содержащей четыре или более значения 2θ, выбранных из группы, включающей 6,5±0,2, 8,6±0,2, 11,3±0,2, 11,9±0,2, 13,1±0,2, 14,2±0,2, 15,1±0,2, 17,4±0,2, 19,6±0,2, 19,9±0,2, 20,4±0,2, 21,7±0,2, 25,6±0,2 и 31,7±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма D характеризуется порошковой рентгеновской дифрактограммой, содержащей пять или более значений 2θ, выбранных из группы, включающей 6,5±0,2, 8,6±0,2, 11,3±0,2, 11,9±0,2, 13,1±0,2, 14,2±0,2, 15,1±0,2, 17,4±0,2, 19,6±0,2, 19,9±0,2, 20,4±0,2, 21,7±0,2, 25,6±0,2 и 31,7±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма D характеризуется порошковой рентгеновской дифрактограммой, содержащей шесть или более значений 2θ, выбранных из группы, включающей 6,5±0,2, 8,6±0,2, 11,3±0,2, 11,9±0,2, 13,1±0,2, 14,2±0,2, 15,1±0,2, 17,4±0,2, 19,6±0,2, 19,9±0,2, 20,4±0,2, 21,7±0,2, 25,6±0,2 и 31,7±0,2 при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C).
Форма D характеризуется порошковой рентгеновской дифрактограммой при комнатной температуре (т.е. температуре от приблизительно 20°C до 25°C), в основном как указано на фиг. 1.
Форма D характеризуется термограммой дифференциальной сканирующей калориметрии (DSC), в основном как указано на фиг. 6.
Форма D характеризуется диаграммой термогравиметрического анализа (TGA), в основном как указано на фиг. 6.
Получение составов, содержащих соединение формулы (I)
Соединение (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида характеризуется низкой насыпной плотностью и низкой текучестью. Разработка перорального фармацевтического состава, прежде всего с высоким содержанием активного агента, т.е. содержащей более 100 мг лекарственного вещества (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида (соединения (I)), представляет значительные трудности. Как правило, высокое содержание лекарственного вещества в составе составляет приблизительно 100 мг, 125 мг, 200 мг, 250 мг, 300 мг, 400 мг, 500 мг или 600 мг.
В каждой пероральной дозированной лекарственной форме количество (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида, его соли(солей) и сольватов (включая гидраты) изменяется в диапазоне 5-600 мг. В одном варианте указанное количество составляет 10-100 мг. В другом варианте указанное количество составляет от 100 мг до 600 мг. В еще одном варианте указанное количество составляет 200-600 мг. В другом варианте указанное количество составляет 250-500 мг. Особенно предпочтительно, количество составляет 10 мг, 20 мг, 50 мг, 100 мг, 125 мг, 150 мг, 200 мг, 250 мг, 300 мг, 400 мг, 500 мг и 600 мг. Еще более предпочтительно, количество составляет более 125 мг и более 200 мг.
Способ получения в случае низкой дозы, включая от 10 мг вплоть до 50 мг, заключается во взвешивании эксципиентов и лекарственного вещества. Затем лекарственное вещество смешивают с эксципиентами, такими как микрокристаллическая целлюлоза, маннит, дикальций фосфат, высушенная распылением лактоза, поливинилпирролидон XL, крахмал, коллоидный диоксид кремния и стеарат магния, предпочтительно с дикальций фосфатом, микрокристаллической целлюлозой, поливинилпирролидоном XL и коллоидным диоксидом кремния, при этом получают предварительную смесь. В предварительную смесь добавляют стеарат магния в качестве смазывающего вещества, и прессуют с получением ядер, на которые наносят пленочное покрытие. Содержание лекарственного вещества изменяется от 7% вплоть до 36%, но предпочтительно составляет от приблизительно 10% до приблизительно 18%.
В способе получения таблеток с более высоким содержанием лекарственного вещества, включая 250 мг и более, предпочтительно 300 мг или более, 400 мг или более, 500 мг или более, сначала взвешивают эксципиенты и лекарственное вещество. Как только все эксципиенты и лекарственное вещество взвешены, лекарственное вещество смешивают в сухом состоянии с микрокристаллической целлюлозой, особенно с продуктом под торговым названием Avicel РН101, в смесителе с высоким сдвигом. Смешанный материал смачивают, предпочтительно ПВП-К30, в водном растворе. Влажную массу перемешивают с получением гранулята. Гранулят высушивают, предпочтительно в сушилке с псевдоожиженным слоем, затем просеивают. В просеянный гранулят добавляют смазывающее вещество с получением конечной смеси, которую прессуют, получают при этом ядра, на которые наносят пленочное покрытие.
Соединение (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида в виде небольших частиц и крупных частиц получают в кристаллической или аморфной форме, а также в формах гидратов или их смесей. Соли (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида включают соли соляной кислоты (HCl), пара-толуолсульфокислоты, метансульфоновой кислоты, бензолсульфоновой кислоты, щавелевой кислоты, этансульфоновой кислоты, аспарагиновой кислоты, малеиновой кислоты и серной кислоты (H2SO4).
Использованный здесь термин «фармацевтически приемлемые соли» обозначает нетоксичные соли кислот или щелочноземельных металлов и (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида по настоящему изобретению. Указанные соли получают in situ в ходе конечного выделения и очистки (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида, или отдельно при взаимодействии основных или кислотных функциональных групп с пригодной органической или неорганической кислотой или основанием, соответственно. Примеры солей включают, но не ограничиваются только ими, следующие соли: ацетат, адипат, альгинат, цитрат, аспартат, бензоат, бензолсульфонат, соль желчной кислоты, бисульфат, бутират, камфорат, камфорсульфонат, диглюконат, циклопентанпропионат, додецилсульфат, этансульфонат, глюкогептаноат, глицерофосфат, полусульфат, гептаноат, гексаноат, фумарат, гидрохлорид, гидробромид, гидроиодид, 2-гидроксиэтансульфонат, лактат, малеат, метансульфонат, никотинат, 2-нафталинсульфонат, оксалат, памоат, пектинат, персульфат, 3-фенилпропионат, пикрат, пивалат, пропионат, сукцинат, сульфат, тартрат, тиоцианат, пара-толуолсульфанат и ундеканоат. Из основных азотсодержащих групп можно также получать группы четвертичного азота в присутствии таких агентов, как алкилгалогениды, такие как метил-, этил-, пропил- и бутилхлориды, -бромиды и -иодиды, диалкилсульфаты, такие как диметил-, диэтил-, дибутил- и диамилсульфаты, длинноцепочечные галогениды, такие как децил-, лаурил-, миристил- и стеарилхлориды, -бромиды и -иодиды, аралкилгалогениды, такие как бензил- и фенилбромиды, и другие. Таким образом, получают водо- или маслорастворимые или диспергируемые продукты.
Примеры кислот, которые можно использовать для получения кислотно-аддитивных фармацевтически приемлемых солей, включают неорганические кислоты, такие как соляная кислота, серная кислота и фосфорная кислота, а также органические кислоты, такие как щавелевая кислота, малеиновая кислота, метансульфоновая кислота, янтарная кислота и лимонная кислота. Основно-аддитивные соли получают in situ в ходе конечного выделения и очистки (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида, или отдельно при взаимодействии остатков карбоновых кислот с пригодным основанием, таким как гидроксид, карбонат или гидрокарбонат фармацевтически приемлемого катиона металла или аммония, или органического первичного, вторичного или третичного амина. Фармацевтически приемлемые соли включают, но не ограничиваясь только ими, катионы щелочных и щелочноземельных металлов, такие как соли натрия, лития, калия, кальция, магния, алюминия и т.п., а также нетоксичные катионы аммония, четвертичного аммония и аминов, включая, но не ограничиваясь только ими, аммоний, тетраметиламмоний, тетраэтиламмоний, метиламин, диметиламин, триметиламин, триэтиламин, этиламин и т.п. Другие примеры органических аминов, пригодных для получения основно-аддитивных солей, включают диэтиламин, этилендиамин, этаноламин, диэтаноламин, пиперазин и т.п.
Состав по настоящему изобретению может содержать фармацевтически приемлемые эксципиенты, обычно используемые в фармацевтических составах, прежде всего предназначенных для перорального введения.
В одном варианте осуществления настоящего изобретения композицию получают в форме составы твердой пероральной дозированной лекарственной формы, включающей (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамид или его соль, необязательно в смеси с одним или более дополнительных эксципиентов. Примеры дополнительных эксципиентов включают дезинтегрирующий агент или супер-дезинтегрирующий агент, наполнитель, скользящее вещество или смазывающее вещество. Соединение (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамид получают в форме частиц небольшого размера со средним размером частицы от приблизительно 10 нм до приблизительно 40 мкм.
Состав по настоящему изобретению необязательно может включать поверхностно-активное вещество. Поверхностно-активные вещества, пригодные для целей настоящего изобретения, включают витамин E TPGS, полисорбат 80, полисорбат 20, лаурилсульфат натрия, анионные поверхностно-активные вещества типа алкилсульфатов, например н-додецилсульфат, н-тетрадецилсульфат, н-гексадецилсульфат или н-октадецилсульфат натрия, калия или магния, типа сульфатов простых алкиловых эфиров, например н-додецилоксиэтилсульфат, н-тетрадецилоксиэтилсульфат, н-гексадецилоксиэтилсульфат или н-октадецилоксиэтилсульфат натрия, калия или магния, или типа алкансульфонатов, например н-додекансульфонат, н-тетрадекансульфонат, н-гексадекансульфонат или н-октадекансульфонат натрия, калия или магния, или неионогенные поверхностно-активные вещества типа эфиров жирных кислот и полигидроксиспиртов, такие как монолаурат, моноолеат, моностеарат или монопальмитат сорбита, тристеарат или триолеат сорбита, аддукты полиоксиэтилена и эфиров жирных кислот и полигидроксиспиртов, такие как монолаурат, моноолеат, моностеарат, монопальмитат, тристеарат или триолеат сорбита и полиоксиэтилена, эфиры жирных кислот и полиэтиленгликоля, такие как полиоксиэтилстеарат, полиэтиленгликоль-400-стеарат, полиэтиленгликоль-2000-стеарат, прежде всего блок-сополимеры этиленоксида и пропиленоксида типа плюроников (BWC) или синпероника (ICI).
Витамин E TPGS (сукцинат d-α-токоферилполиэтиленгликоля 1000) при комнатной температуре обычно представляет собой воскообразное вещество, практически не поддающееся переработке, однако его можно получить в форме частиц при замораживании и последующим измельчении, что позволяет напрямую смешивать витамин E TPGS с ингредиентами. Способ прямого смешивания включает сухую переработку эксципиента, такого как витамин Е TPGS, и активного ингредиента, в данном случае (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. Сухая переработка обозначает, что эксципиенты перерабатывают в сухом состоянии, а не в расплавленном состоянии, и кроме того не получают твердый раствор или твердую дисперсию. Витамин E TPGS, полученный замораживанием и измельчением, можно смешивать напрямую и перерабатывать более простым способом, и его содержание в составы составляет вплоть до приблизительно 20 мас. %, приблизительно 25 мас. %, или приблизительно 35 мас. %, или приблизительно 40 мас. %, или менее 50 мас. %. Витамин Е TPGS после сухой переработки по настоящему изобретению получают в порошкообразной форме или в форме частиц.
Согласно настоящему изобретению количество поверхностно-активного вещества в составе составляет от приблизительно 0,5 мас. % до приблизительно 95 мас. %, от приблизительно 1 мас. % до приблизительно 85 мас. % и от приблизительно 5 мас. % до приблизительно 75 мас. % в расчете на общую массу составы. Кроме того предлагаются составы, содержащие приблизительно 5 мас. %, приблизительно 10 мас. %, приблизительно 15 мас. %, приблизительно 20 мас. %, приблизительно 25 мас. %, приблизительно 30 мас. %, приблизительно 35 мас. % и приблизительно 45 мас. % ПАВ.
Состав по настоящему изобретению необязательно может включать кислоты. Кислоты, пригодные для применения по настоящему изобретению, включают любую фармацевтически приемлемую кислоту, включая органические кислоты, такие как янтарная кислота, винная кислота, лимонная кислота, уксусная кислота, пропионовая кислота, малеиновая кислота, яблочная кислота, фталевая кислота, метансульфоновая кислота, толуолсульфоновая кислота, нафталинсульфоновая кислота, камфорсульфоновая кислота, бензолсульфоновая кислота, молочная кислота, масляная кислота, гидроксималеиновая кислота, малоновая кислота, сорбиновая кислота, гликолевая кислота, глюкуроновая кислота, фумаровая кислота, муциновая кислота, глюконовая кислота, бензойная кислота, щавелевая кислота, фенилуксусная кислота, салициловая кислота, сульфаниловая кислота, аспарагиновая кислота, глутаминовая кислота, этилендиаминтетрауксусная кислота, стеариновая кислота, пальмитиновая кислота, олеиновая кислота, лауриновая кислота, пантотеновая кислота, дубильная кислота, валериановая кислота или аскорбиновая кислота, и полимерную кислоту, такую как сополимер метакриловой кислоты, продукты с торговым названием EUDRAGIT E РО, EUDRAGIT L100-55, EUDRAGIT L-30 D-55, EUDRAGIT FS 30 D, EUDRAGIT NE 30 D, EUDRAGIT LI00, EUDRAGIT SI00, полиаминокислота (например, полиглутаминовая кислота, полиаспарагиновая кислота и их комбинации), полинуклеиновые кислоты, полиакриловая кислота, полигалактуроновая кислота, а также поливинилсульфат или анионную аминокислоту, такую как полиглутаминовая кислота или полиаспарагиновая кислота. В объеме настоящего изобретения следует понимать, что органические кислоты включают полимерные кислоты. Кислоты включают также неорганические кислоты, такие как соляная кислота, фосфорная кислота, фосфоновая кислота, фосфиновая кислота, бороновая кислота, бромистоводородная кислота, серная кислота, сульфаминовая кислота, азотная кислота или сульфоновая кислота. Кислота присутствует в качестве буферного вещества.
Согласно настоящему изобретению количество кислот в составы составляет от приблизительно 2 мас. % до приблизительно 80 мас. %, от приблизительно 2 мас. % до приблизительно 60 мас. % и от приблизительно 5 мас. % до приблизительно 40 мас. % в расчете на общую массу составы. Кроме того предлагаются составы, содержащие приблизительно 10 мас. %, приблизительно 20 мас. %, приблизительно 25 мас. %, приблизительно 35 мас. %, приблизительно 40 мас. % и приблизительно 45 мас. % кислоты.
Состав по настоящему изобретению необязательно может содержать антиоксидант. Примеры антиоксидантов включают, но не ограничиваются только ими, сульфит натрия, бисульфит натрия, метабисульфит натрия, аскорбиновую кислоту, тиоглицерин, тиосорбит, тиокарбамид, тиосульфат натрия, тиоуксусную кислоту, цистеин, метионин, бутилированный гидрокситолуол (БГТ), бутилированный гидроксианизол (БГА), аскорбилпальмитат, гидрохинон, пропилгаллат, нордигидрогуайретовую кислоту, витамин Е (α-токоферол) и лецитин. Предпочтительными антиоксидантами являются микронизированный пропилгаллат, микронизированный БГА, микронизированный БГТ, витамин E, аскорбиновая кислота, тиосульфат натрия и цистеин. Концентрация антиоксиданта составляет 1-3 мас. %.
Дезинтегрирующие агенты, предназначенные для применения по настоящему изобретению, включают стандартные дезинтегрирующие агенты, такие как крахмал, альгиновая кислота или смолы типа Амберлит, а также включены супер-дезинтегрирующие агенты, такие как кросповидон, натриевая соль гликолята крахмала, натриевая соль кроскармеллозы и полисахарид сои. Термин «супер-дезинтегрирующий агент» является общеизвестным в данной области и обозначает дезинтегрирующий агент, которые является эффективным при низких концентрациях по сравнению с крахмалом, обычно при концентрациях от 2 мас. % до 4 мас. %.
Скользящие вещества, предназначенные для использования по настоящему изобретению, включают диоксид кремния, такой как коллоидный диоксид кремния (высокодисперсный кремнезем) и тальк.
В одном варианте способом влажной грануляции получают состав по настоящему изобретению, содержащий соединение (I) и следующие эксципиенты:
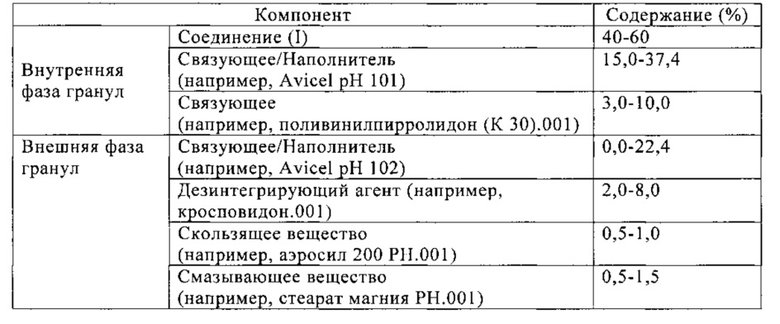
В другом варианте способом влажной грануляции получают состав по настоящему изобретению, содержащий соединение (I) и следующие эксципиенты:

В еще одном варианте способом влажной грануляции получают состав по настоящему изобретению, содержащий соединение (I) и следующие эксципиенты:


Наполнители: МКЦ, включая продукты с торговым названием Avicel pH101, 102, 105, 201 и т.п., Celous, сахара, такие как лактоза, маннит, декстроза, крахмал и т.п., или другие неорганические наполнители, такие как дикальций гидрофосфат, трикальций фосфат, сульфат кальция и т.п.
В способе влажной грануляции можно использовать различные растворители. Примеры растворителей включают, но не ограничиваясь только ими, воду, спирты (например, этиловый спирт, изопропанол) или их смеси, прежде всего смеси воды и спирта (спиртов).
В одном варианте способом сухой грануляции получают состав по настоящему изобретению, содержащий соединение (I) и следующие эксципиенты:

В другом варианте способом сухой грануляции получают состав по настоящему изобретению, содержащий соединение (I) и следующие эксципиенты:


В еще одном варианте способом сухой грануляции получают состав по настоящему изобретению, содержащий соединение (I) и следующие эксципиенты:

Наполнители: МКЦ, включая продукты с торговым названием Avicel pH101, 102, 105, 201 и т.п., Celous®, сахара, такие как лактоза, маннит, декстроза, крахмал и т.п., или другие неорганические наполнители, такие как дикальций гидрофосфат, трикальций фосфат, сульфат кальция и т.п.
Для получения составов по настоящему изобретению можно использовать любые кристаллические формы соединения (I), его солей или сольватов (включая гидраты), включая, но не ограничиваясь только ими, формы HA, А, В, С и D, или их смеси. При получении составов, содержащих соединение (I), может происходить или не происходить изменение его формы.
Примером смазывающего вещества, которое можно использовать в настоящем изобретении, является стеарат магния, стеариновая кислота, стеарат кальция, тальк, гидрированное растительное масло, глицерилбегенат, стеарилфумарат натрия, полиэтиленгликоль (ПЭГ) 4000/6000, лаурилсульфат натрия, изолейцин, бензоат натрия или высоко дисперсный кремнезем.
В настоящем изобретении используют наполнители: микрокристаллическая целлюлоза (МКЦ), например типа AVICEL (фирмы FMC Corp.), например типа AVICEL РН101, 102, 105, RC581 или RC 591, типа EMCOCEL (фирмы Mendell Corp.) или типа ELCEMA (фирмы Degussa), совместно осажденную МКЦ, такую как силилированная МКЦ (фирмы Prosolv-JRS pharma), переработанную смесь, такую как Ludipress (фирмы BASF), которая состоит из лактозы, а также коллидона 30, и коллидон CL, углеводороды, такие как сахара, альдиты, крахмалы или производные крахмалов, например, лактоза, декстроза, сахароза, глюкоза, сорбит, маннит, ксилит, картофельный крахмал, маисовый крахмал, рисовый крахмал, пшеничный крахмал или амилопектин, трикальций фосфат, гидрофосфат кальция, сульфат кальция, дикальций фосфаты, оксид магния или трисиликат магния.
Пригодные связующие, которые можно использовать по настоящему изобретению, включают желатин, трагакант, агар, альгиновую кислоту, альгинат натрия, аравийскую камедь, простые эфиры целлюлозы, например метилцеллюлозу, карбоксиметилцеллюлозу или гидроксипропилметил-целлюлозу, гидроксипропилцеллюлозу, полиэтиленгликоли или гомополимеры этиленоксида, прежде всего характеризующиеся степенью полимеризации приблизительно от 2,0×103 до 1,0×105 и средней молекулярной массой приблизительно от 1,0×105 до 5,0×106, например, эксципиенты, известные под торговыми названием POLYOX (фирмы Union Carbide), поливинилпирролидон или повидоны, прежде всего характеризующиеся средней молекулярной массой приблизительно 1000 и степенью полимеризации приблизительно от 500 до 2500, а также агар или желатин.
Пригодными полимерами, которые можно использовать для нанесения пленочных покрытий, являются гидроксипропилметилцеллюлоза, фталат гидроксипропилметилцеллюлозы, этилцеллюлоза, метилцеллюлоза, производные поливинилового спирта, производные поливинилацетата, или акрилатные полимеры, такие как Eudragit ЕРО, Eudragit RL и RS3D, Eudragit L30D (фирмы Evonik).
Состав по настоящему изобретению можно получить стандартным способом, таким как прямое смешивание, прямое прессование, грануляция, грануляция из растворителя, влажная грануляция, грануляция в псевдоожиженном слое, грануляция (высокотемпературная) из расплава, сухая грануляция, ротационное прессование, комкование, таблетирование с лиофильным высушиванием, влажное или сухое агрегирование, а также экструзия и сферонизация.
В одном варианте осуществления настоящего изобретения состав представляет собой капсулу, такую как твердая желатиновая капсула или мягкая эластичная капсула. В другом варианте состав представляет собой таблетку или пилюлю. В таких твердых пероральных составах, количество (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида составляет величину в диапазонах 1-900 мг, 2,5-600 мг, 2,5-300 мг или 2,5-100 мг, при этом предпочтительные примеры включают 10 мг, 50 мг, 100 мг, 200 мг, 250 мг, 300 мг, 400 мг, 500 мг и 600 мг.
Твердые пероральные составы по настоящему изобретению можно вводить для лечения заболеваний, связанных с ингибированием белка апоптоза. Белок апоптоза защищает раковые клетки от гибели в результате апоптоза.
Точную дозировку S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида в составах по настоящему изобретению определяет специалист в данной области в зависимости от состояния и требований пациента. Например, состав по настоящему изобретению можно вводить ежедневно, через день или раз в неделю.
Настоящее изобретение кроме того описано в разделе «Примеры». Следующие примеры, не ограничивающие объем настоящего изобретения, иллюстрируют настоящее изобретение, и не ограничивают объем прилагаемых пунктов формулы изобретения.
Пример 1
В Таблице 1 ниже приведен состав таблетки, включающей 10 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида.
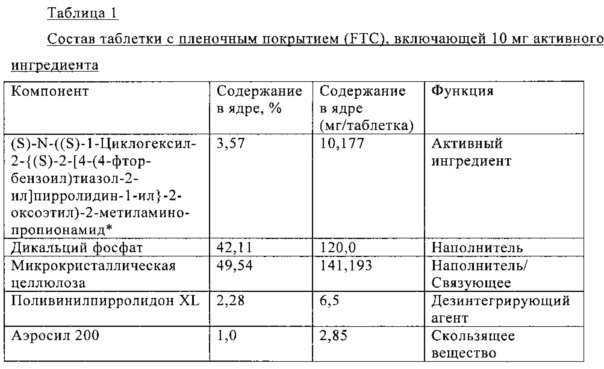
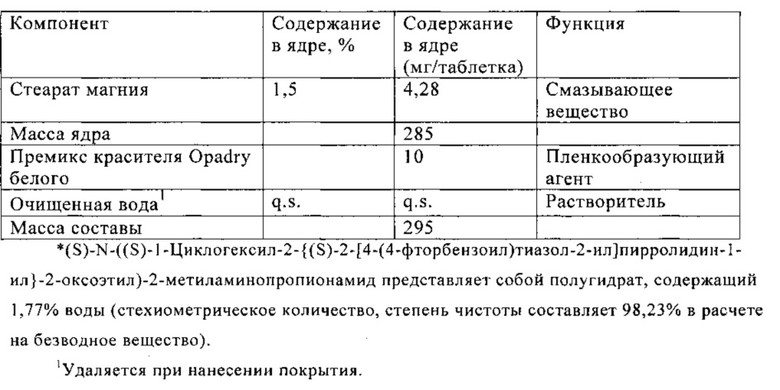
Средний профиль растворения показан на фиг. 17.
Способ прямого прессования применяют для получения таблеток, содержащих 10 мг активного ингредиента, с использованием пригодных для прямого прессования эксципиентов, таких как микрокристаллическая целлюлоза, маннит, дикальций фосфат и высушенная распылением лактоза, в комбинации с дезинтегрирующими агентами (такими как поливинилпирролидон XL, крахмал), смазывающим веществом (стеарат магния) и скользящим веществом (коллоидный диоксид кремния). Содержание лекарственного средства изменяется от 7% вплоть до 36%.
При переработке составов, содержащих маннит, наблюдается высокая сила выталкивания. Указанную проблему можно исключить при замене маннита на дикальций фосфат или лактозу и снижении содержания лекарственного вещества. В некоторых случаях наблюдается слипание и значительно изменение силы прессования, что обычно связано с недостаточным смазыванием и низкой текучестью. Указанные недостатки можно исключить при снижении содержания лекарственного вещества.
Пример 2
В Таблице 2 ниже приведен состав таблетки, включающей 50 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида.
Таблица 2
Состав FTC, включающей 50 мг активного ингредиента
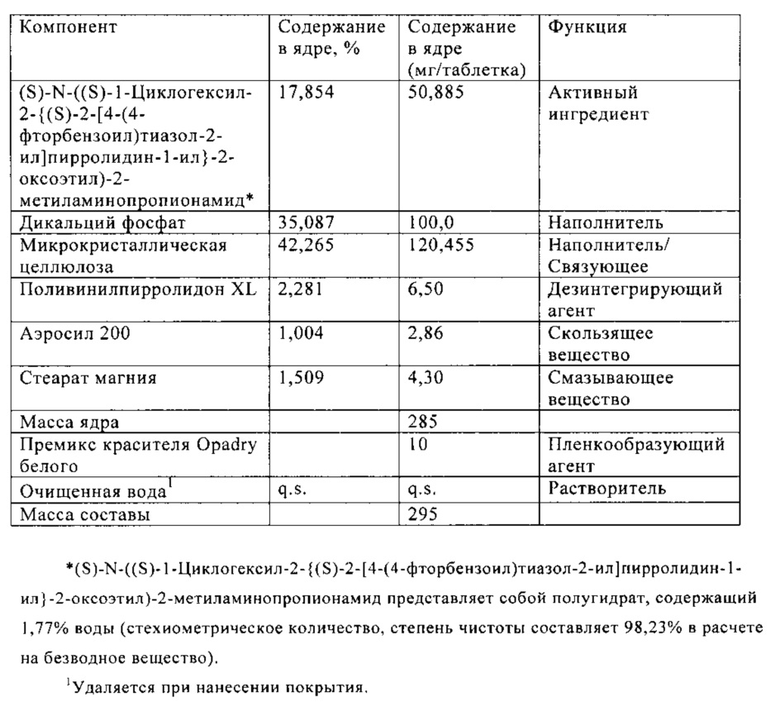
Способ прямого прессования применяют для получения таблеток, содержащих 10 мг активного ингредиента, с использованием пригодных для прямого прессования эксципиентов, таких как МКЦ, маннит, дикальций фосфат и высушенная распылением лактоза, в комбинации с дезинтегрирующими агентами (таким как поливинилпирролидон XL, крахмал), смазывающим веществом (стеарат магния) и скользящим веществом (коллоидный диоксид кремния). Содержание лекарственного средства изменяется от 7% вплоть до 36%.
Для составов, содержащих маннит, наблюдается высокая сила выталкивания. Указанную проблему можно исключить при замене маннита на дикальций фосфат или лактозу и снижении содержания лекарственного вещества. В некоторых случаях наблюдается слипание и значительно изменение силы прессования, что обычно связано с недостаточным смазыванием и низкой текучестью. Указанный недостаток можно исключить при снижении содержания лекарственного вещества.
Средний профиль растворения показан на фиг. 17.
Пример 3
В Таблице 3 ниже приведен состав таблетки, включающей 300 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида.
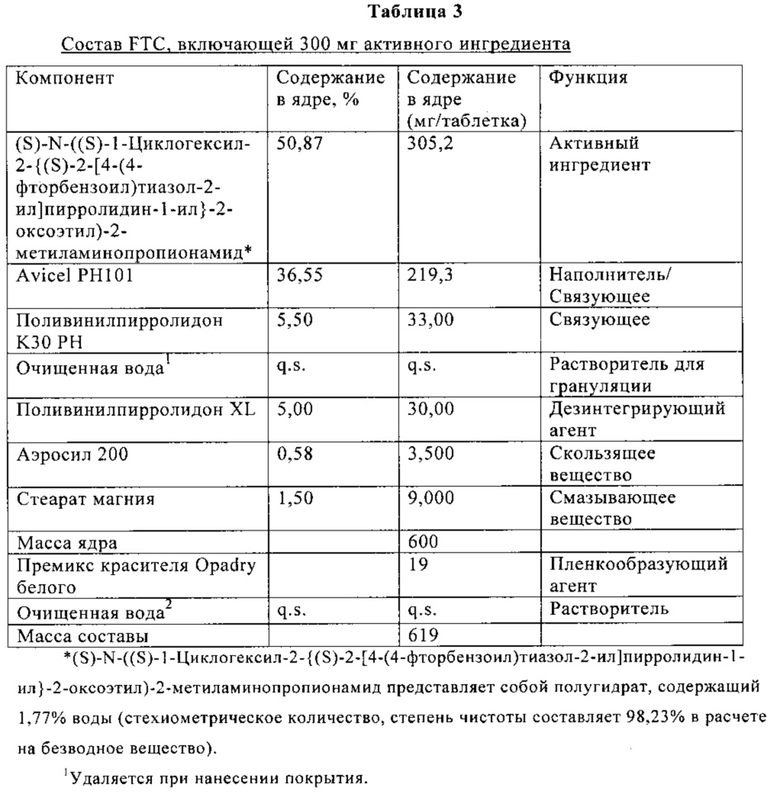
Средний профиль растворения показан на фиг. 17.
На основании результатов, полученных при разработке композиций, включающих 10 мг и 50 мг активного ингредиента, был проведен ряд испытаний по моделированию способа ротационного прессования в прессе для таблетирования с одним пуансоном с целью разработки композиций с более высоким содержанием активного ингредиента (например, 250 мг). Для оценки технологических характеристик таких составов проводили ряд испытаний с использованием комбинации эксципиентов, таких как микрокристаллическая целлюлоза, предварительно желатинизированный крахмал, дикальций фосфат и маннит в качестве наполнителей, а также гидроксипропилцеллюлозы, коллидона VA64 в качестве связующих. Ряд проблем, таких как низкая текучесть, низкая прессуемость вследствие слипания, наблюдали даже при содержании лекарственного вещества приблизительно 30%. Указанные проблемы нельзя исключить за счет качественных или количественных изменений эксципиентов. Предполагали, что измельченное лекарственное вещество, характеризующееся большей площадью поверхности (следовательно, большей площадью связывания), обеспечит более плотно спрессованные/гранулированные гранулы при измельчении, которые можно перерабатывать, однако значительный успех не наблюдается. Указанные результаты моделирования прессования были неожиданными. Дальнейшее снижение содержания лекарственного вещества ниже 30% не проводили, т.к. при этом происходит значительное увеличение размера таблетки, что снижает согласие субъекта с курсом лечения, прежде всего, если в ходе клинического испытания необходим прием нескольких таблеток.
Технологические проблемы были успешно решены при использовании способа влажной грануляции и/или сухой грануляции, которые обеспечивают получение лекарственного средства с более высоким содержанием активного агента (более 40 мас. %, более 50 мас. %, более 60 мас. %, более 70 мас. % или более 80 мас. %). В способе влажной грануляции высокая дозировка с высоким содержанием лекарственного вещества (например, 50 мас. %) достигается за счет специального выбора стандартных эксципиентов и растворителя для грануляции и подбора их количеств.
Пример 4
В Таблице 4 ниже приведен состав таблетки, включающей 500 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. На таблетку можно наносить пленочное покрытие.

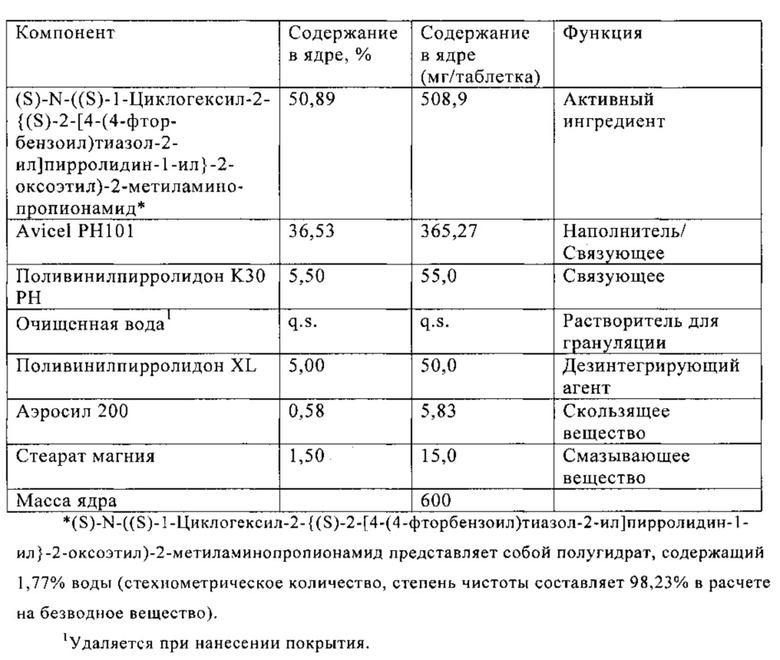
Средний профиль растворения показан на фиг. 18.
Пример 5
В Таблице 5 ниже приведен состав таблетки, включающей 300 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. На таблетку необязательно наносят пленочное покрытие. Таблетку получают влажной грануляцией, как показано на схеме Б.
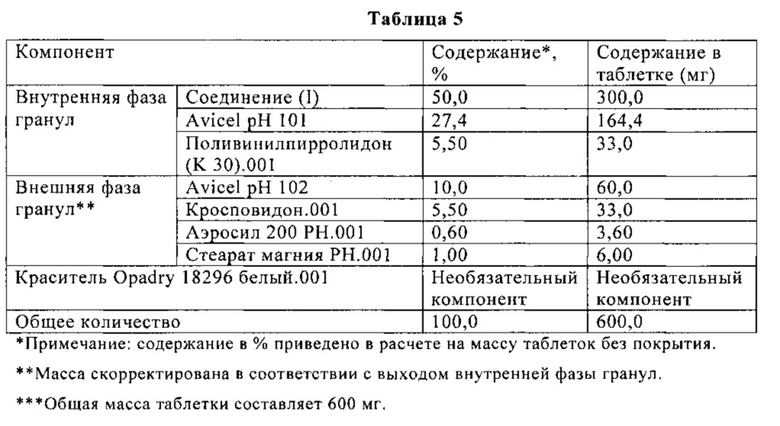
Пример 6
В Таблице 6 ниже приведен состав таблетки, включающей 400 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. На таблетку необязательно наносят пленочное покрытие. Таблетку получают влажной грануляцией, как показано на схеме Б.
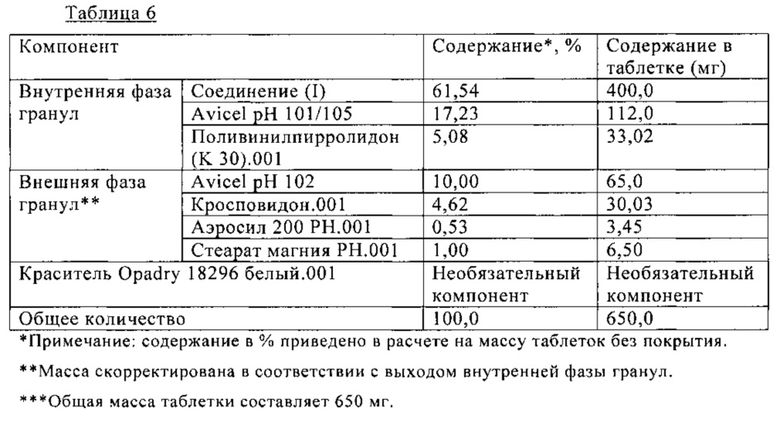
Пример 7
В Таблице 7 ниже приведен состав таблетки, включающей 500 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. На таблетку необязательно наносят пленочное покрытие. Таблетку получают влажной грануляцией, как показано на схеме Б.
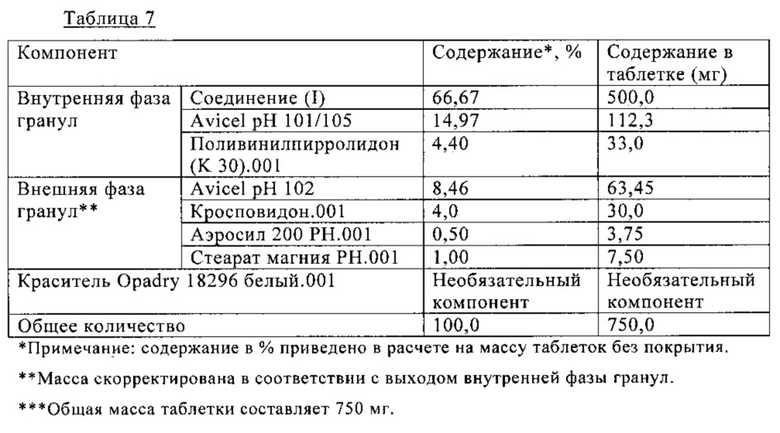
Схема Б
Получение таблеток, содержащих 300 мг, 400 мг и 500 мг активного ингредиента, способом влажной грануляции
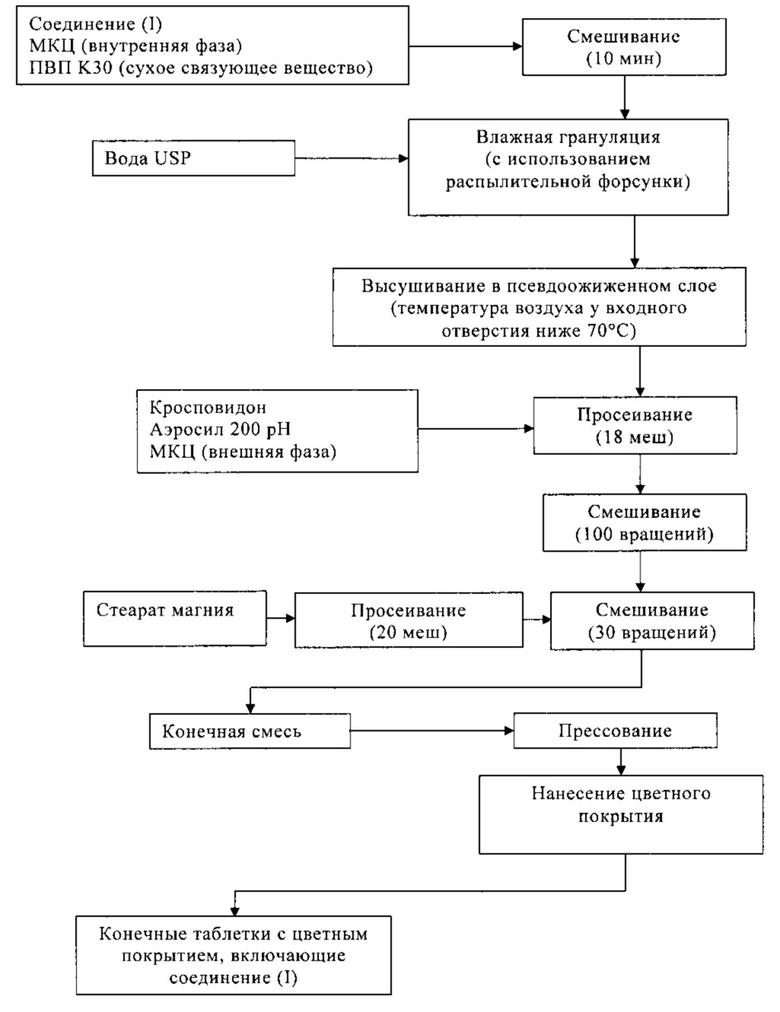
Пример 8
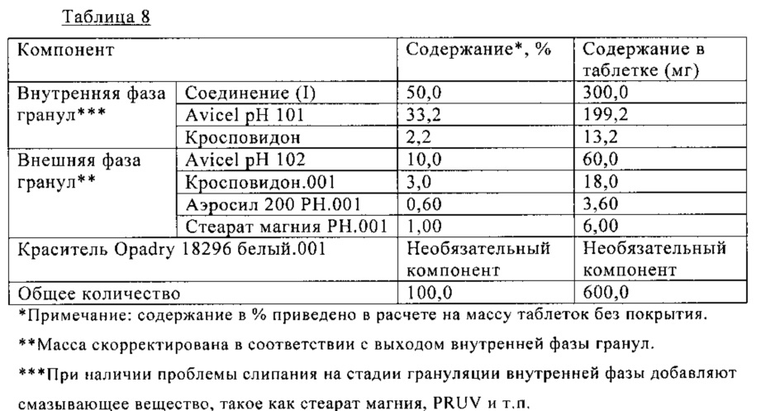
В Таблице 8 ниже приведен состав таблетки, включающей 300 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. На таблетку необязательно наносят пленочное покрытие. Таблетку получают сухой грануляцией, как показано на схеме В.
Пример 9
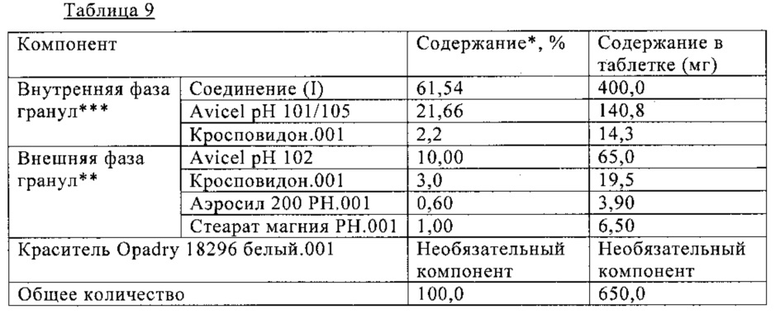
В Таблице 9 ниже приведен состав таблетки, включающей 400 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. На таблетку необязательно наносят пленочное покрытие. Таблетку получают сухой грануляцией, как показано на схеме В.

Пример 10
В Таблице 10 ниже приведен состав таблетки, включающей 500 мг (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида. На таблетку необязательно наносят пленочное покрытие. Таблетку получают сухой грануляцией, как показано на схеме В.
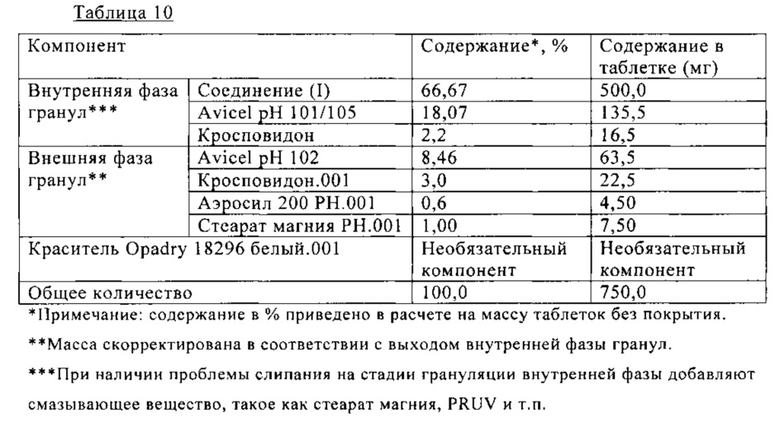
Схема В
Получение таблеток, содержащих 300 мг, 400 мг и 500 мг активного ингредиента, способом сухой грануляции
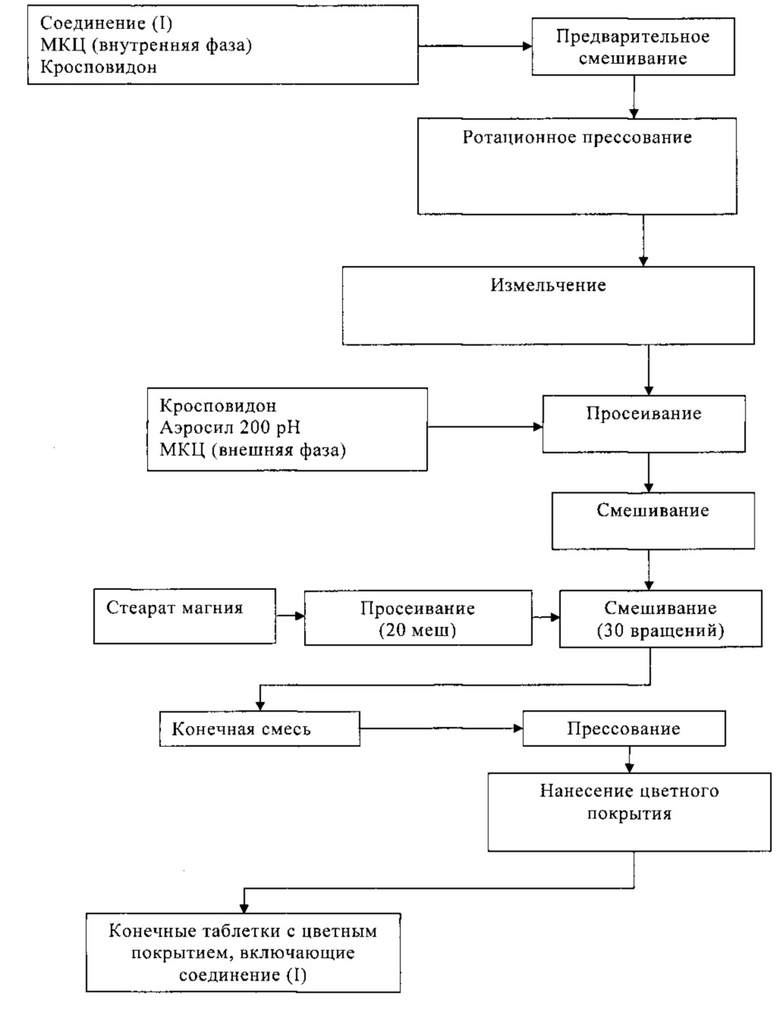
Пример 11
Соединение (I) и форму HA соединения (I) получали, как указано на общей схеме A и подробно описано ниже. На схеме A указаны также другие соединения.
Получение соединения В4 из соединения В2
2-(S)-1-(Трет-бутоксикарбонилпирролидин-2-ил)тиазол-4-карбоновую кислоту (В1, 27 г), гидрохлорид N,N-диметилгидроксиламина (9,7 г) и N,N-диметилформамид (157 г) загружали в реактор Argonaut (объемом 1 л). Суспензию нагревали при 24±3°C в течение 20 мин, при этом получали гомогенный раствор. Содержимое охлаждали до 20±3°C в течение 15 мин, затем в реактор добавляли триэтиламин (35 г) при 20±3°C в течение 15 мин, при этом получали суспензию светло-коричневого цвета. В реактор добавляли раствор циклического ангидрида 1-пропилфосфорной кислоты в этилацетате (50 мас. %, 69 г) при 20±3°C в течение 30 мин. Суспензию перемешивали при 20±3°C в течение 30 мин. После контроля управления процессом (КУП #1) в реактор медленно добавляли воду (200 г) при 20±3°C в течение 20 мин, при этом получали гомогенный раствор. В реактор добавляли толуол (360 г) и смесь перемешивали при 20±3°C в течение 15 мин. Нижний водный слой и слой диспергированного осадка отбрасывали. Верхний органический слой промывали раствором бикарбоната натрия (1 г) в воде (100 г). Нижний водный слой отбрасывали. Верхний органический слой промывали дважды водой (общее количество 200 г). Экстракт толуола концентрировали при 60±3°C (10 мбар), при этом получали вязкое масло (~36 г). Остаток промывали дважды толуолом (общее количество 66 г) при 60±3°C (10 мбар), получая при этом трет-бутиловый эфир (S)-2-[4-(метоксиметилкарбамоил)тиазол-2-ил]пирролидин-1-карбоновой кислоты (В2, 33,5 г) в виде вязкого масла желто-коричневого цвета. В реактор добавляли толуол (90 г). Толуол (~11,5 г) отгоняли из содержимого при 60±5°C (10 мбар), при этом получали раствор В2 в толуоле (-25 мас. %, 112 г). После КУП #2 и анализа на содержание воды (методом Карла Фишера, Н2O <0,1%) раствор В2 в толуоле (~25 мас. %) затем использовали на стадии получения соединения В4.
Получение соединения В4 из соединения В2
Получение раствора уксусной кислоты
Воду (156,9 г) и ледяную уксусную кислоту (39,2 г) загружали в круглодонную колбу (объемом 500 мл) в атмосфере азота. Раствор перемешивали в течение 5 мин и хранили до применения.
Взаимодействие соединения В2 с соединением В3
Предварительно полученный раствор соединения В2 (109,8 г) в толуоле (329,3 г) загружали в 4-горлую колбу (объемом 0,5 л), снабженную устройством для продувки азотом, охлаждающей баней, закрепленной сверху мешалкой и внутренним датчиком температуры. Раствор охлаждали до -10°C±5°C, добавляли раствор соединения ВЗ (386 г, 1,0 М раствор в тетрагидрофуране (ТГФ)) в течение 2,0 ч, поддерживая температуру -10±5°C. Содержимое колбы перемешивали в течение 1,0 ч при -10°C±5°C, добавляли водный раствор уксусной кислоты (19,6 г, 20 мас. %) в течение 0,5 ч. Затем добавляли еще одну порцию водного раствора уксусной кислоты (176 г, 20 мас. %) в течение 1,5 ч, поддерживая температуру -10±5°C, добавляли воду (200 г) в течение 0,5 ч, поддерживая температуру -10±5°C. Фазы перемешивали в течение 1 ч. Массу нагревали до 25±3°C в течение 0,5 ч. Перемешивание прекращали и фазы выдерживали для разделения. Нижний водный слой удаляли. Добавляли воду (200 г). Фазы перемешивали в течение 5 мин. Перемешивание прекращали и фазы выдерживали для разделения. Нижний водный слой удаляли. Добавляли воду (200 г). Фазы перемешивали в течение 5 мин. Перемешивание прекращали и фазы выдерживали для разделения. Нижний водный слой удаляли. Органический слой концентрировали до общего объема 500 мл, добавляли изопропилацетат (435 г). Органический слой концентрировали до общего объема 500 мл. Добавляли изопропилацетат (435 г). Органический слой концентрировали до общего объема 500 мл. Полученный раствор использовали на следующей стадии без дополнительной очистки.
Получение соединения В5 из соединения В4
В круглодонную колбу (объемом 0,5 л), продуваемую азотом и снабженную мешалкой и ледяной баней, загружали изопропанол (192,0 г). Массу охлаждали до 10°C±3°C и добавляли газообразный HCl (48,4 г, массу определяли по разнице массы цилиндра) с использованием вакуума. Раствор перемешивали в течение 15 мин при 10°C±3°C и массу нагревали до 20°C±3°C. При наличии вакуума раствор продували азотом, или направляли в скруббер, если давление повышалось выше атмосферного.
Получение соединения В5
Предварительно полученный раствор соединения В4 (55,0 г) в толуоле и изопропилацетате (231,0 г) помещали в отдельную 4-горлую колбу (объемом 0,5 л), снабженную устройством для продувки азотом, охлаждающей баней, закрепленной сверху мешалкой и внутренним датчиком температуры, и повышали внутреннюю температуру до 27°C±3°C. Добавляли предварительно полученный раствор HCl (168 г, 5,5 М) в изопропаноле в течение 10 мин, поддерживая температуру 27°C±3°C. Содержимое колбы перемешивали в течение 5,5 ч при 27°C±3°C. Реакционную смесь охлаждали до 20°C±5°C, и смесь концентрировали до общего объема 250 мл при давлении 100-120 мбар (температура в рубашке 35-45°C). Добавляли изопропилацетат (217,0 г). Органический слой концентрировали до общего объема 250 мл (давление 100-120 мбар, температура в рубашке 35-45°C). Добавляли изопропилацетат (217,0 г). Полученное твердое вещество отделяли фильтрованием и промывали изопропилацетатом (130,0 г). Твердое вещество высушивали в сушильном шкафу при температуре 80°C±3°C в течение 8 ч, при этом получали соединение В5 (40,1 г).
Получение соединения В6 из соединения В5
Соединение В5 (67,98 г, 200 ммолей), содержащее толуол и iPrOAc (общее содержание 8,67 мас. %), Z5a (75,70 г, 210 ммолей), содержащее воду (5,01%), DMT-MM (60,9 г, 220 ммолей) и этилацетат (700 мл, 631,4 г) загружали в реактор Argonaut (объемом 2 л). Суспензию перемешивали и охлаждали до -1±3°C, медленно добавляли N-метилморфолин (50,6 г, 0,5 моля), поддерживая температуру при -1±3°C в течение 40 мин, смесь перемешивали и выдерживали при температуре -1±3°C в течение 30 мин, затем нагревали до 19±3°C и выдерживали при указанной температуре в течение 3,5 ч. Отбирали образец для контроля управления процессом (КУП). После КУП медленно добавляли воду (300 г) и этилацетат (300 мл, 270,6 г), поддерживая температуру при 20±3°C. Перемешивали при 20±3°C в течение 30 мин, затем два слоя разделяли. Верхний слой сохраняли, т.к. указанная органическая фаза содержала соединение В6. Органический слой промывали 1н. раствором NaOH (250 мл, 260 г). Нижний (водный) слой отделяли. В верхний слой добавляли 2н. раствор HCl (250 мл, 254,6 г) и перемешивали в течение 15 мин. Нижний слой отделяли. В верхний слой добавляли солевой раствор (250 мл, 286,6 г) и перемешивали в течение 15 мин. Нижний слой отделяли и органический слой упаривали до объема 200 мл, выдерживали в колбе в вакууме при температуре 30°C и давлении 735 мм. Добавляли этилацетат (300 мл, 270,6 г) и органический слой упаривали в вакууме при 30°C и 735 мм, при этом в колбе получали остаток объемом 400~500 мл, который использовали на следующей стадии без дополнительной очистки.
Получение соединения В7 (соединения (I)) из соединения В6
Раствор неочищенного соединения В6 (120 г, 20 ммолей) в этилацетате (360,8 г, 400 мл) загружали в реактор Argonaut (объемом 2 л). Раствор нагревали до 45±3°C, медленно добавляли раствор HCl (5-6н.) в изопропиловом спирте (109,1 г, 120 мл), поддерживая температуру при 45±3°C в течение 30 мин. Смесь перемешивали и выдерживали при 45±3°C в течение 2 ч, отбирали образец для контроля управления процессом (КУП). После КУП реакционную смесь охлаждали до 18±3°C. Указанный раствор медленно добавляли в реактор Argonaut (объемом 2 л), содержащий раствор карбоната калия (82,9 г) в воде (500 г), поддерживая температуру при 5±3°C, смесь перемешивали при 5±3°C в течение 30 мин, добавляли этилацетат (451 г, 500 мл). Раствор нагревали до 20±3°C и выдерживали при указанной температуре в течение 1 ч. Два слоя разделяли. Верхний слой сохраняли, т.к. указанная органическая фаза содержала соединение В7. Органический слой промывали солевым раствором (286,6 г, 250 мл). Нижний (водный) слой отделяли. Верхний органический слой концентрировали до объема 500 мл в вакууме при 30°C, медленно добавляли гептан (1368 г, 2 л), поддерживая температуру при 30±3°C. Суспензию охлаждали до 18±3°C и выдерживали при 18±3°C в течение 1 ч. Твердое вещество отделяли фильтрованием и промывали гептаном (136 г, 200 мл). Твердое вещество высушивали в сушильном шкафу при 45°C в течение 16 ч, при этом получали соединение В7 (80 г, выход 80%).
Получение формы HA соединения (I) из соединения В7
Соединение В7 (78,0 г) и этанол (616,2 г, 780 мл) этанола (крепость 200) загружали в реактор Argonaut (объемом 2 л). Раствор нагревали до 30±3°C и добавляли воду (100 г). Раствор фильтровали, затем медленно добавляли воду (1750 г), поддерживая температуру при 30±3°C в течение 40 мин. Суспензию охлаждали до 18±3°C и выдерживали при указанной температуре в течение 2 ч. Твердое вещество отделяли фильтрованием и промывали 20% раствором этанола в воде (60 мл). Твердое вещество высушивали в сушильном шкафу при 45°C и 25 мбар в течение 16 ч, при этом получали форму HA соединения (I) (72 г, выход 90%).
Пример 12
Уравновешивание при комнатной температуре
Анализ проводили с использованием ряда различных растворителей. Форму HA соединения (I) (50-60 мг), полученную, как описано в примере 11, уравновешивали в каждом растворителе (1 мл) при комнатной температуре в течение по крайней мере 24 ч. Если твердое вещество растворялось, добавляли дополнительное количество формы HA соединения (I) до образования суспензии. Уравновешенные суспензии фильтровали, и твердое вещество высушивали на открытом воздухе в течение 10 мин. Форму A получали из определенных растворителей, как подробно описано ниже.

Пример 13
Уравновешивание при 50°C
Анализ проводили с использованием ряда различных растворителей. Форму HA соединения (I) (50-60 мг), полученную, как описано в примере 11, уравновешивали в каждом растворителе (1 мл) при 50°C в течение по крайней мере 24 ч. Если твердое вещество растворялось, добавляли дополнительное количество формы HA соединения (I) до образования суспензии. Уравновешенные суспензии фильтровали, и твердое вещество высушивали на открытом воздухе в течение 10 мин. Формы A и B получали из определенных растворителей, как подробно описано
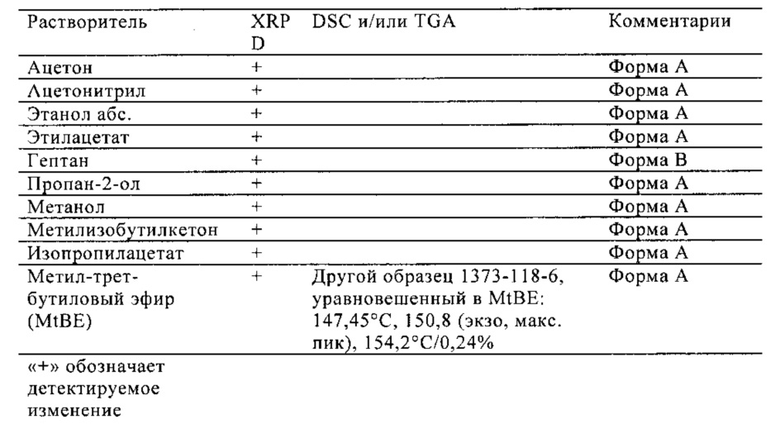
Пример 14
Кристаллизация при упаривании при комнатной температуре Уравновешенные суспензии, описанные в примере 12, фильтровали, и отфильтрованные прозрачные растворы выдерживали при комнатной температуре для упаривания растворителей. Форма A образуется из этилацетата.
Пример 15
Кристаллизация из горячих насыщенных растворов
Концентрированные (>50 мг/мл) или насыщенные растворы фильтровали при 60°C и затем охлаждали до 4°C. Осадки отделяли фильтрованием, высушивали на воздухе и анализировали. Формы B, С или D, которые образуются в определенных растворителях, подробно описаны ниже.
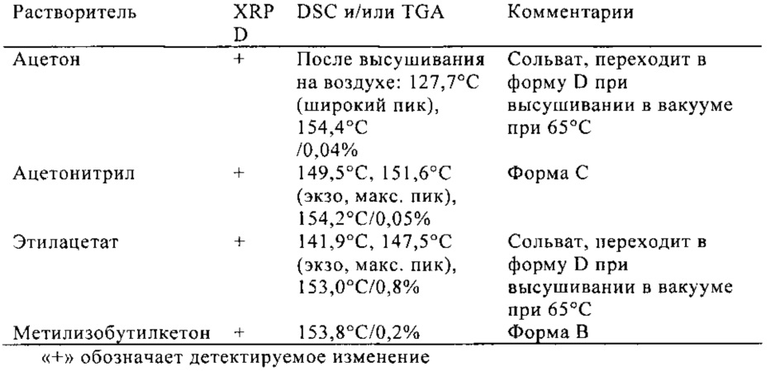
Пример 16
Осаждение при добавлении антирастворителя
Анализировали различные комбинации растворителей. Форму HA соединения (I) растворяли в среде, в которой указанное форма характеризуется высокой растворимостью, и добавляли растворитель, в котором соединение (I) характеризуется чрезвычайно низкой растворимостью. Каждый осадок отделяли фильтрованием, и твердое вещество высушивали на открытом воздухе в течение 10 мин. Формы A или D образуются в определенных комбинациях растворителей, как подробно описано ниже.
Пример 17
Кристаллические формы HA, А, В, С и D, полученные как описано в примерах 11-16, анализировали различными стандартными методами: XRPD, DSC, TGA, микроскопией. Определяли также сорбцию и десорбцию воды. Результаты представлены на фиг. 1-16.





Настоящее изобретение относится к фармацевтическому составу, ингибирующему белок апоптоза, включающему в качестве активного ингредиента кристаллическую полугидратную форму HA (S)-N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида, характеризующегося формулой (I), в количестве более 100 мг и фармацевтически приемлемые эксципиенты. Кристаллическая полугидратная форма HA соединения (I) характеризуется порошковой рентгеновской дифрактограммой, содержащей три или более значения 2θ, выбранных из группы, включающей 8,3±0,2, 9,5±0,2, 13,5±0,2, 17,3±0,2, 18,5±0,2 и 18,9 ±0,2 при комнатной температуре. Состав по изобретению дополнительно включает поверхностно-активное вещество, кислоту, антиоксидант. Технический результат – пероральный фармацевтический состав с высоким содержанием активного агента. 23 з.п. ф-лы, 18 ил., 17 пр., 10 табл.
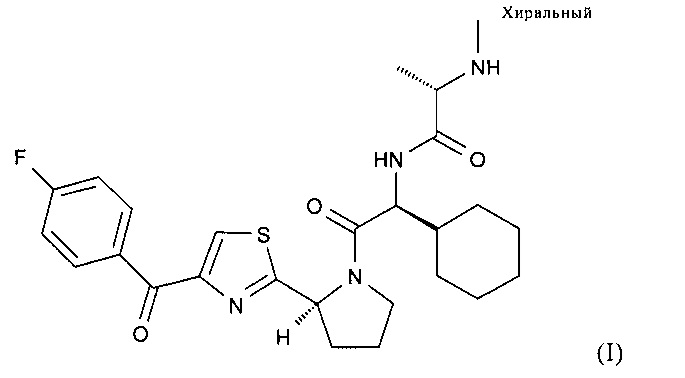
1. Фармацевтический состав, ингибирующий белок апоптоза, включающий в качестве активного ингредиента кристаллическую полугидратную форму HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида, характеризующуюся формулой (I):
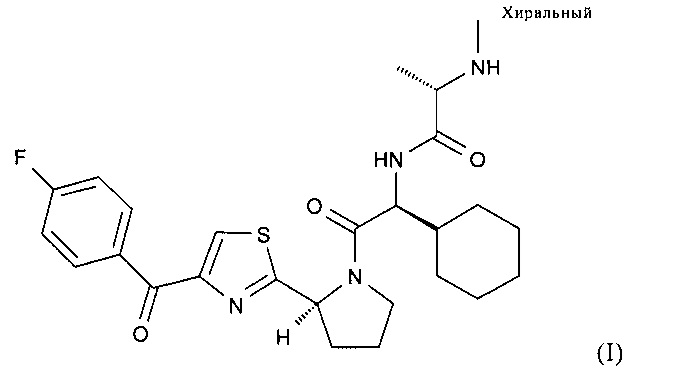
в количестве более 100 мг и фармацевтически приемлемые эксципиенты.
2. Состав по п.1, где кристаллическая полугидратная форма HA соединения (I) характеризуется порошковой рентгеновской дифрактограммой, содержащей три или более значения 2θ, выбранных из группы, включающей 8,3±0,2, 9,5±0,2, 13,5±0,2, 17,3±0,2, 18,5±0,2 и 18,9 ±0,2 при комнатной температуре.
3. Состав по п.1, где кристаллическая полугидратная форма HA соединения (I) характеризуется при комнатной температуре порошковой рентгеновской дифрактограммой, в основном как указано на фиг. 1.
4. Состав по п.1, где кристаллическая полугидратная форма HA соединения (I) характеризуется термограммой дифференциальной сканирующей калориметрии (DSC), в основном как указано фиг. 2.
5. Состав по п.1, где кристаллическая полугидратная форма HA соединения (I) характеризуется диаграммой термогравиметрического анализа (TGA), в основном как указано на фиг. 2.
6. Состав по любому из пп.1-5, где количество кристаллической полугидратной формы HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида составляет более 125 мг.
7. Состав по любому из пп.1-5, где количество кристаллической полугидратной формы HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида составляет более 200 мг.
8. Состав по любому из пп.1-5, где количество кристаллической полугидратной формы HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида составляет более 250 мг.
9. Состав по любому из пп.1-5, где количество кристаллической полугидратной формы HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида составляет более 300 мг.
10. Состав по любому из пп.1-5, где количество кристаллической полугидратной формы HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида составляет более 400 мг.
11. Состав по любому из пп.1-5, где количество кристаллической полугидратной формы HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида составляет более 500 мг.
12. Состав по любому из пп.1-5, полученный влажной грануляцией.
13. Состав по любому из пп.1-5, полученный сухой грануляцией.
14. Состав по любому из пп.1-5, дополнительно включающий поверхностно-активное вещество.
15. Состав по любому из пп.1-5, дополнительно включающий кислоту.
16. Состав по любому из пп.1-5, дополнительно включающий антиоксидант.
17. Состав по любому из пп.1-5, которой представляет собой твердую пероральную дозированную лекарственную форму, а кристаллическая полугидратная форма HA (S)N-((S)-1-циклогексил-2-{(S)-2-[4-(4-фторбензоил)тиазол-2-ил]пирролидин-1-ил}-2-оксоэтил)-2-метиламинопропионамида может присутствовать в форме частиц небольшого размера со средним размером от приблизительно 10 нм до приблизительно 40 мкм.
18. Фармацевтический состав по п. 17, дополнительно включающий дезинтегрирующий агент или супердезинтегрирующий агент.
19. Фармацевтический состав по п. 17, дополнительно включающий скользящее вещество, смазывающее вещество или оба этих вещества - скользящее вещество и смазывающее вещество.
20. Фармацевтический состав по п. 17, где содержание активного ингредиента составляет более 40 мас.% в расчете на общую массу состава.
21. Фармацевтический состав по п. 17, где содержание активного ингредиента составляет более 50 мас.% в расчете на общую массу состава.
22. Фармацевтический состав по п. 17, где содержание активного ингредиента составляет более 60 мас.% в расчете на общую массу состава.
23. Фармацевтический состав по п. 17, где содержание активного ингредиента составляет более 70 мас.% в расчете на общую массу состава.
24. Фармацевтический состав по п. 17, где содержание активного ингредиента составляет более 80 мас.% в расчете на общую массу состава.
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| Способ обработки целлюлозных материалов, с целью тонкого измельчения или переведения в коллоидальный раствор | 1923 |
|
SU2005A1 |
| ЕА 200701467 А1, 28.12.2007 | |||
| CAIRA M.R | |||
| et al.: "CRYSTALLINE POLYMORPHISM OF ORGANIC COMPOUNDS", TOPICS IN CURRENT, 1998, vol.198, p.163-208. | |||

