Данное изобретение относится к новым 6-метокси-1Н-бензотриазол- 5-карбоксамидным производным, характеризующимся тем, что они одновременно обладают превосходной противорвотной активностью и активностью, усиливающей сократительную способность желудочно-кишечного тракта и меньшей супрессорной активностью в отношении центральной нервной системы (CNS). Более конкретно, изобретение относится к 6-метокси-1Н-бензотиазол-5-карбоксамидным производным, у которых атом азота в амидной группе (-CONH-) замещен 1-замещенной-азациклоалкан-3-ил-группой 7-, 8- или 9-членного цикла, способам их получения, содержащим их фармацевтическим композициям и новым промежуточным соединениям.
Известный уровень техники
В JP-A 104572/1990 описано, что соединения, представленные приведенной ниже формулой [A] , обладают активностью, усиливающей подвижность желудочно-кишечного тракта, и полезны как противорвотные средства или средства, усиливающие подвижность желудочно-кишечного тракта: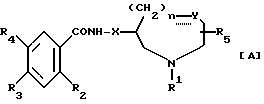
[где R1 обозначает низший алкил или необязательно замещенный арил(низший)алкил,
R2 обозначает гидрокси, алкокси, алкенилокси, циклоалкилокси или замещенный алкокси (заместителями являются галоген, гидрокси или оксо),
R3 обозначает амино, дизамещенный амино или ациламино,
R4 обозначает галоген или R3 и R4 вместе образуют - NH-N=N-,
R5 обозначает водород или низший алкил,
X обозначает простую связь или низший алкилен,
Y обозначает простую связь или группу, выражаемую формулой:
-CH2-, -O-, -S-, -SO-, -SO2 или -NR6-,
где R6 обозначает низший алкил или необязательно замещенный арил (низший) алкил; или может образовывать вместе с R1 этилен,
n равно 0 или 1, и
пунктирная линия обозначает двойную связь, которая может присутствовать, когда Y обозначает -CH2- и n равно 0, при условии, что:
i) когда Y обозначает -NR6- или простую связь, n равно 0;
ii) когда Y обозначает -O-, n равно 1;
iii) когда Y обозначает простую связь или -CH2- и n равно 0, P1 обозначает необязательно замещенный арил (низший) алкил;
и
iv) когда n равно 0, X обозначает низший алкилен].
Однако, названный JP-A-104572/1990 не содержит конкретного описания соединений по данному изобретению, представленных приведенной ниже формулой (I), одновременно обладающих 1H-бензотриазольным скелетом и азот-содержащим 7-, 8- или 9-членным алифатическим циклом, в частности, их оптически активных соединений и фармакологических активностей оптически активных соединений.
JP-A-83737/1977 описывает, что соединения, представленные приведенной ниже формулой [B], обладают высокой активностью по ослаблению условного рефлекса избегания поведения апоморфин-индуцируемой стереотипной реакции и метамфетамин- индуцируемого стереотипного взаимодействия, и поэтому полезны в качестве CNS-депрессантов, в частности, в качестве антипсихотических средств: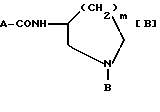
(где A-CO обозначает 4-амино-5-хлор-2-метоксибензоил, 5- этилсульфонил-2-метоксибензоил или 2-метокси-4,5- азимидобензоил;
В обозначает аллил или необязательно замещенную бензильную группу и m равно 1 или 2).
Кроме того, JP-A-100473/1977 описывает соединения, представленные приведенной ниже формулой [C]: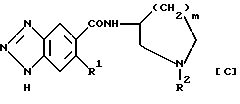
(в которой P1 обозначает низший алкокси, R2 обозначает необязательно замещенный бензил и m равно 1 или 2).
Однако в соединениях, представленных выше формулами [B] или [C], присоединенный к амидной составляющей (-CONH-) цикл является 5- или 6-членным и входящий в цикл атом азота замещен аллильной или бензильной группой и с этой точки зрения они отличаются от структуры соединений, представленных приведенной ниже формулой (I) по данному изобретению. Кроме того, их фармакологическая активность, опять же, отлична от активности соединений по изобретению.
С другой стороны известно, что 4-амино-5-хлор-N-[2 (диэтиламино)этил]-2-метоксибензамид [общее название: метоклопрамид; Cf. Merck Index, Ilth ed. 6063 (1989)] обладает противорвотной активностью и вместе с тем - активностью, усиливающей подвижность желудочно-кишечного тракта, и используется людьми преклонного возраста для лечения или профилактики функциональных нарушений желудочно-кишечного тракта, связанных с различными заболеваниями и приемом терапевтических средств, в качестве средства, усиливающего подвижность (сократительную способность) желудочно-кишечного тракта. Однако, метоклопрамид обладает CNS-депрессантной активностью, вытекающей из его допамин - D2-рецепторной антагонистической активности, что является препятствием для его клинического применения. В связи с возрастающей сложностью жизни в человеческом обществе и возрастающим количеством людей преклонного возраста, возрастает также количество пациентов, страдающих симптомами, связанными с дисфункцией желудочно- кишечного тракта, и необходимость лечения приводит к возрастанию спроса на соединение или соединения, обладающие низкой CNS- депрессантной активностью и вместе с тем обладающие высокой противорвотной активностью и активностью, усиливающей сокращения желудочно-кишечного тракта.
Описание изобретения
Проведены обширные исследования и установлено, что 6-метокси-1H-бензотриазол-5-карбоксамидные производные, в которых атом азота в амидном радикале (-CONH-) замещен 1-замещенной-азациклоалкан-3-ил-группой 7-, 8- или 9-членного кольца, в частности, (R)-6-метокси-1Н-бензотриазол-5-карбоксамидные производные конфигурации R, обладают одновременно превосходной противорвотной активностью и активностью, усиливающей сокращения желудочно-кишечного тракта и тем не менее обладают удивительно низкой CNS-депрессантной активностью. Таким образом данное изобретение является завершенной научной работой.
Цель данного изобретения состоит в получении новых 6-метокси-1H- бензотриазол-5-карбоксамидных производных, в частности, (R)-6- метокси-1H-бензотриазол-5-карбоксамидных производных, имеющих R- конфигурацию, обладающих одновременно прекрасной противорвотной активностью и активностью, усиливающей сокращения желудочно- кишечного тракта. Другая цель данного изобретения состоит в разработке способов получения названных соединений. Дальнейшая цель изобретения состоит в получении фармацевтических композиций, содержащих названные соединения. Кроме того, целью изобретения является также получение новых промежуточных соединений, полезных для получения соединений по изобретению. Эти и другие цели и преимущества по изобретению очевидны для специалистов, читающих данное описание и следующих ему.
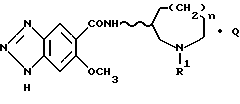
По изобретению предлагаются 6-метокси-1H- бензотриазол-5-карбоксамидные производные, представленные следующей формулой (I), их фармацевтически приемлемые соли кислотного присоединения и содержащие их фармацевтические композиции: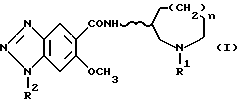
[где R1 обозначает этильную или циклопропилметильную группу,
R2 обозначает атом водорода, метильную или этильную группу,
n равно 1, 2 или 3, и
волнистая линия  обозначает, что конфигурация заместителей на углеродном атоме, связанном с атомом N в амидном радикале, является рацемической (RS) или оптический активной (R или S)].
обозначает, что конфигурация заместителей на углеродном атоме, связанном с атомом N в амидном радикале, является рацемической (RS) или оптический активной (R или S)].
Изобретение дает также соединения, представленные следующей формулой (II) и их соли кислотного присоединения, используемые в качестве промежуточных продуктов для получения соединений формулы (I), в которой R2 обозначает атом водорода;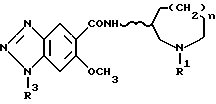 (II)
(II)
[где R3 обозначает амино-защитную группу и R1, n и волнистая линия имеют те же значения, что приняты выше для формулы (I)]. Кроме того, изобретение дает промежуточные соединения приведенной ниже формулы (IV) и их соли кислотного присоединения, используемые для получения соединений формулы (I) по данному изобретению: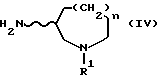
[где R1, n и волнистая линия имеют обозначения, принятые для формулы (I)] , в частности, соединения следующей формулы (IVa) и соли их кислотного присоединения, используемые в качестве промежуточных соединений для получения соединений формулы (I), имеющих R - конфигурацию: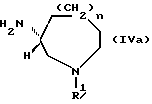
[где R1 и n имеют принятые для формулы (I) значения].
Фармацевтически приемлемые соли кислотного присоединения для соединений формулы (I) включают, например, соли неорганических кислот, безвредные для человеческого организма, такие как гидрохлорид, гидробромид, гидроиодид, сульфат, фосфат и т.д.; и соли органических кислот, безвредные для человеческого организма, такие как оксалат, малеат, фумарат, лактат, малат, цитрат, тартрат, бензоат, метансульфонат и т.д. Соединения формулы (I) и их соли кислотного присоединения могут быть получены в форме гидрата или сольвата, которые также входят в рамки объема данного изобретения и приложенных пунктов. Более конкретно, могут быть, например, названы: 1/4 гидрат, 1/2 гидрат, моногидрат, 3/2 фумарат•1/4 гидрат, 3/2 фумарат•1/2гидрат, дифумарат •1/2 гидрат и т.д.
Соли кислотного присоединения для промежуточных продуктов, представленных формулами (II) и (IV) или (IVa), включают, например, те же фармацевтически приемлемые соли кислотного соединения, что названы выше. Соединения формул (II) и (IV) или (IVa) и их соли кислотного присоединения могут быть получены в форме гидрата или сольвата, которые также входят в рамки объема данного изобретения и приложенных пунктов.
При получении соединений формулы (I) и их солей кислотного присоединения в кристаллической форме могут присутствовать различные виды полиморфизма, которые также входят в рамки объема изобретения и приложенных пунктов.
Следует учитывать, что соединения формулы (I), в которых R2 обозначает водород, присутствуют в форме таутомеров, в которых 6-метокси-1Н-бензотриазольный радикал может быть представлен формулой (I') или (I''), приведенные ниже: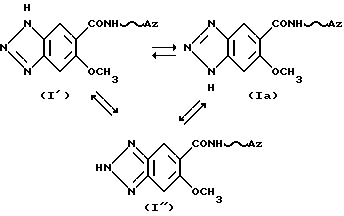
[в которых Az обозначает группу формулы [D], приведенной ниже: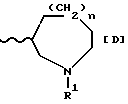
(в которой R1, n и волнистая линия имеют приведенные выше значения)].
Эти таутомеры также входят в рамки объема изобретения и приложенных пунктов.
Структура соединений по данному изобретению, в которых R2 в формуле (I) обозначает атом водорода, представлена формулой (Ia), и их химические названия также основаны на названной структуре.
Те соединения, в которых R2 в формуле (I) обозначает метильную или этильную группу, не обладают вышеупомянутым таутомеризмом.
Термин "галоген", как он используется в данном описании, обозначает фтор, хлор, бром или иод. Конкретные примеры "алкильных групп" включают метил, этил, пропил и изопропил. Конкретные примеры "алкокси-групп" включают метокси, этокси, пропокси и изопропокси. Конкретные примеры "низших алканоильных групп" включают ацетил и пропионил и соответствующие примеры "низших алкоксикарбонильных групп" включают метоксикарбонил и этоксикарбонил. "Необязательно замещенной бензильной группой" служат те бензильные группы, фенильная составляющая в которых необязательно замещена одним или двумя вышеупомянутыми атомами галогена, C1-C3 алкильными группами и, предпочтительно, C1-C3алкоксигруппами, конкретные примеры включают 2-, 3- или 4-хлорбензил, 3-бромбензил, 4- фторбензил, 2,4- или 3,4-дихлорбензил, 4-метилбензил, 2-,3- или 4- метоксибензил и т.д. Термин "необязательно замещенный бензилоксикарбонил" относится к тем бензилоксикарбонилам, у которых фенильный радикал необязательно замещен одним или двумя вышеупомянутыми атомами галогенов, C1-C3-алкильными группами, C1-C3 алкокси-группами, нитро группами и т. д., специфические примеры включают бензилоксикарбонил, 4-хлорбензилоксикарбонил, 4-бромбензилоксикарбонил, 2,4-дихлорбензилоксикарбонил и 4-метоксибензилоксикарбонил. Термин "амино-защитные группы" обозначает такие защитные группы, которые могут быть удалены гидролизом или гидрогенолизом, их примеры включают определенные ранее группы: низший алканоил, трифторацетил, низший алкоксикарбонил, необязательно замещенный бензил и необязательно замещенный бензилоксикарбонил, особенно предпочтительны бензил и ацетил.
В качестве предпочтительных примеров соединений по данному изобретению, представленных формулой (I), могут быть приведены следующие соединения и их фармацевтически приемлемые соли кислотного присоединения:
N-(1-этил-1H-гексагидроазепин-3-ил)-6-метокси-1H-бензотриазол-5-карбоксамид,
(R) - N-(1-этил-1Н-гексагидроазепин-3-ил)-б-метокси-1Н-бензотриазол- 5-карбоксамид,
N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил- 1Н-бензотриазол-5-карбоксамид,
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид,
(R)-1-этил-N-(1-этил-1H-гексагидроазепин-3-ил)-6- метокси-1H-бензотриазол-5-карбоксамид,
N-(1-этил-1Н-гептагидроазоцин-3-ил)-6-метокси-1Н-бензотриазол- 5-карбоксамид,
N-(1-этил-1H-гептагидроазоцин-3-ил)-6-метокси-1-метил-1H- бензотриазол-5-карбоксамид,
N-(1-этил-1Н-октагидроазонин-3-ил)-6-метокси-1Н-бензо-триазол- 5-карбоксамид,
(R)-N-(1-циклопропилметил-1Н-гексагидроазепин-3-ил)-6-метокси- 1Н-бензотриазол-5-карбоксамид и
(R)-N-(1-циклопропилметил-1Н-гексагидроазепин-3-ил)-6-метокси- 1-метил-1H-бензотриазол-5-карбоксамид.
В частности, соединения формулы (I), в которых R1 обозначает этил и R2 обозначает водород или метил, являются наиболее предпочтительными.
Предпочтительными в отношении конфигурации являются те соединения формулы (I), в которых конфигурация заместителей на углеродном атоме, связанном с N-атомом амидного радикала, является рацемической (RS) или оптически активной (R), в частности, последняя - более предпочтительна.
Что касается азациклоалканового цикла, то пригодным является 7-, 8- или 9-членный цикл, хотя 7-членный цикл особенно предпочтителен, т.е. соединения формулы (I), в которой n равно 1 - предпочтительны.
Из вышеупомянутых соединений особенно предпочтительны перечисленные далее соединения и их фармацевтически приемлемые соли кислотного присоединения:
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н-бензотриазол- 5-карбоксамид (далее обозначенный как соединение 7) и
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид (далее обозначенный как соединение 2).
В дополнение к перечисленным ранее соединениям, следующие соединения и их фармацевтически приемлемые соли кислотного присоединения могут быть приведены в качестве конкретных примеров других предпочтительных соединений, входящих в данное изобретение:
(R)-N-(1-этил-1Н-гептагидроазоцин-3-ил)-6-метокси-1Н-бензотриазол- 5-карбоксамид,
(R)-N-(1-этил-1Н-октагидроазонин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамид,
(R)-N-(1-этил-1Н-гептагидроазоцин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид и
(R)-N-(1-этил-1Н-октагидроазонин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид.
Соединения по данному изобретению могут быть получены, например, следующими способами.
Способ (a)
Соединения формулы (I) могут быть получены взаимодействием соединений, представленных формулой (III) ниже: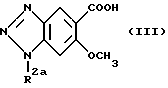
(в которой R2a обозначает водород, метил, этил или аминозащитную группу)
или их реакционноспособных производных с соединениями, представленными формулой (IV) ниже: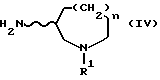
[в которой R1, n и волнистая линия имеют значения, принятые для формулы (I)].
В этом случае, когда R2a в формуле (III) обозначает амино-защитную группу, реакционный продукт должен быть затем подвергнут гидролизу или гидрогенолизу для превращения R2a в атом водорода с целью получения соединения формулы (I). Взаимодействие соединений формулы (III) с соединениями формулы (IV) может быть выполнено по хорошо известной реакции амидирования.
Примеры реакционноспособных производных соединений формулы (III) включают сложные низшие алкиловые эфиры (в том числе, метиловый эфир), активные сложные эфиры, ангидриды кислот и галоидангидриды кислот (в том числе, хлорангидрид кислоты) [когда используют соединение формулы (III), в котором R2a обозначает атом водорода, ангидриды кислот и галоидангидриды кислот исключаются] . Конкретные примеры активных сложных эфиров включают п-нитрофениловый эфир, пентахлорфениловый эфир, сложный эфир N-гидроксисукцинимида, сложный эфир N-гидроксифталимида, сложный эфир 1-гидроксибензотиазола, 8-гидроксихинолиновый эфир, 2-гидроксифениловый эфир и 2-гидрокси- 4,5-дихлорфениловый эфир. В качестве ангидрида кислоты используют симметричный ангидрид кислоты или смешанный ангидрид кислоты, конкретные примеры последнего включают ангидриды, смешанные с алкилхлорформиатами, такими как этилхлорформиат или изобутилхлорформиат; ангидриды, смешанные с аралкилхлорформиатами, такими как бензилхлорформиат; ангидриды, смешанные с арилхлорформиатами, такими как фенилхлорформиат, и ангидриды, смешанные с алкановыми кислотами, такими как изовалериановая кислота и пиваловая кислота.
В качестве амино-защитных групп, которые могут служить R2a, могут быть использованы такие защитные группы, которые можно удалить гидролизом или гидрогенолизом, их примеры включают: низший алканоил, трифторацетил, низший алкоксикарбонил, необязательно замещенный бензил и необязательно замещенный бензилоксикарбонил, особенно предпочтителен ацетил.
Когда используют сами по себе соединения формулы (III), реакция может быть проведена в присутствии конденсирующего агента, такого как N,N'-дициклогексилкарбодиимид, 1-этил-3-(3-диметиламинопропил) карбодиимид-гидрохлорид, N,N'-карбонилдиимидазол, N,N'-карбонилдисукцинимид, 1-этоксикарбонил-2-этокси-1,2-дигидрохинолин, дифенилфосфорилазид и пропанфосфоновый ангидрид. Когда в качестве конденсирующего средства используют N,N'-дициклогексилкарбодиимид или 1-этил-3-(3-диметиламинопропил)карбодиимид-гидрохлорид, то к реакционной смеси может быть добавлен N-гидроксисукцинимид, 1- гидроксибензотриазол, 3-гидрокси-1,2,3-бензотриазин-4-(3H)-он, N- гидрокси-5-норборнен-2,3-дикарбоксиимид или тому подобные.
Взаимодействие таких соединений формулы (III) или их реакционноспособных производных с соединениями формулы (IV) осуществляют либо в растворителе, либо в отсутствие растворителя. Используемые растворители должны быть подходящим образом выбраны в соответствии с видом исходного соединения и так далее, они включают: ароматические углеводороды, такие как бензол, толуол и ксилол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран и диоксан; галоидированные углеводороды, такие как метиленхлорид и хлороформ; спирты, такие как этанол и изопропиловый спирт; этилацетат, ацетон, ацетонитрил, диметилформамид, диметилсульфоксид, этиленгликоль и воду. Эти растворители могут быть использованы как отдельно, так и в виде смеси растворителей нескольких видов. При желании эту реакцию проводят в присутствии основания, конкретные примеры основания включают: гидроокись щелочного металла, такую как гидроокись натрия и гидроокись калия; карбонаты щелочных металлов, такие как карбонат натрия и карбонат калия; бикарбонат щелочного металла, такой как бикарбонат натрия и бикарбонат калия; и органические основания, такие как триэтиламин, трибутиламин, диизопропилэтиламин и N-метилморфолин. Избыточное количество соединения формулы (IV) может также действовать как основание. Реакционная температура меняется в зависимости от вида исходных соединений и т.д., но обычно находится в интервале приблизительно от -30oC до 200oC, предпочтительно, приблизительно от -10oC до 150oC.
Когда соединения формулы (III), в которых R2a обозначает аминозащитную группу, например, низший алканоил, трифторацетил, низший алкоксикарбонил или необязательно замещенный бензилоксикарбонил, взаимодействуют с соединениями формулы (IV), образуя соединения формулы (I), в которых R2a обозначает соответствующую защитную группу, продукты могут быть гидролизованы для превращения в соединения формулы (I), в которой R2 обозначает водород.
Реакция гидролиза может быть проведена по существу известными способами, например, взаимодействием продукта с водой в подходящем растворителе в кислой или основной среде. В качестве растворителей могут быть использованы, например, спирты, такие как метанол, этанол, изопропиловый спирт или тому подобные, диоксан, вода или их жидкие смеси. Конкретные примеры кислоты, используемой для создания кислой среды, включают неорганические кислоты, такие как соляная, бромистоводородная и серная кислоты; органические кислоты, такие как муравьиная, уксусная, пропионовая и щавелевая кислоты; и силикагель. Когда используют соединение формулы (III), в котором R2a обозначает ацетильную группу, использование силикагеля легко отщепляет ацетильную группу, превращая R2 в атом водорода. Конкретные примеры оснований для создания щелочной среды включают гидроокись щелочного металла, такую как гидроокись натрия или калия; и карбонат щелочного металла, такой как карбонат натрия или калия. Температура реакции обычно находится в интервале приблизительно от 20oC до 100oC.
Когда соединение формулы (III), в которой R2a обозначает, наряду с названными примерами амино-защитных групп, необязательно замещенную бензильную или бензилоксикарбонильную группу, реагирует с соединением формулы (IV) с образованием соединения формулы (I), в котором R2a обозначает соответствующую защитную группу, гидрогенолиз продукта может превращать такой R2 в водород. Гидрогенолиз может быть проведен в основном известным способом, например, взаимодействием продукта с водородом в подходящем растворителе в присутствии катализатора, такого как палладий-на-углероде, никель Ренея и т. д. В качестве используемых растворителей могут быть спирты, такие как метанол или этанол, уксусная кислота, диоксан, тетрагидрофуран, вода или их жидкие смеси. Реакционная температура обычно используется в интервале приблизительно от 0oC до 80oC. Реакцию проводят при нормальном или повышенном давлении.
Соединения формулы (III), в которых R2a обозначает водород или амино-защитную группу (низшую алканоильную группу, трифторацетильную группу, низшую алкоксикарбонильную группу или необязательно замещенную бензилоксикарбонильную группу) и их реакционноспособные производные могут быть получены по способу, описанному, например, в JP-A-80858/1976 (патент США 4.039.672) или аналогичными этому способами.
Соединения формулы (III), в которых R2a обозначает метил, этил или амино-защитную группу (необязательно замещенную бензильной группой), могут быть получены по способу, использующему 4-хлор-2-метокси-5-нитробензойную кислоту в качестве исходного вещества, включающему превращение этого соединения в соответствующий, адекватной структуры амид использованием подходящего амина, такого как пропиламин по приведенной ниже стадии 1 в схеме 4, введение метильной, этильной или необязательно замещенной бензильной группы в R2 положение и восстановление полученного продукта по приведенным ниже стадиям 2 и 3 в схеме 4, получение из этого соединения соответствующего 6- метокси-1Н-бензотриазол-5-карбоксамидного производного по способу (b), описанному ниже, и затем, гидролиз продукта по существу известным способом.
Конкретные примеры способов получения соединений формулы (IV) приведены ниже.
Те соединения формулы (IV), в которых n=1, могут быть получены, например, способом, иллюстрируемым ниже схемой 1.
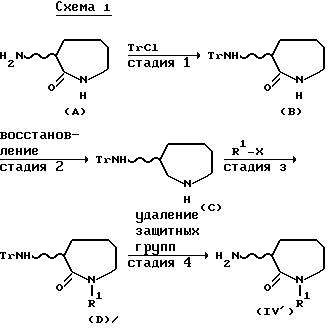
[В приведенной выше схеме I Tr обозначает трифенилметильную группу, X обозначает реакционноспособный сложноэфирный остаток спирта, R1 имеет определенное ранее значение и волнистая линия обозначает рацемическую или оптически активную конфигурацию, как определено ранее].
Стадия 1:
Реакцию между соединением формулы (A) и хлортрифенилметаном обычно проводят в подходящем растворителе в присутствии основания. Используемые растворитель и основание - те же, что названы для приведенного выше способа (а). Температура реакции обычно находится в интервалах приблизительно от -10oC до 150oC, предпочтительно от 0oC до 100oC. В качестве R и S изомеров формулы (A), являющихся исходными соединениями, могут быть использованы промышленные оптически активные соединения или промышленное рацемическое соединение может быть оптически разделено, например, способом, описанным в J. Org. Chem., 44, 4841-4847 (1979) или получено из оптически активного лизина, например, способом, описанным в Synthesis, 1978, 614-616. Этот способ оптического разрешения или синтеза оптически активного соединения сам по себе хорошо известен.
Стадия 2:
Соединения формулы (C) могут быть получены восстановлением соединений формулы (B) с использованием гидрида металла, такого как диизобутилалюмогидрид, литийалюмогидрид, натрий бис(2-метоксиэтокси)алюмогидрид или тому подобного. Конкретные примеры используемых растворителей включают, например, простые эфиры, такие как диэтиловый эфир, тетрагидрофуран, и т.д.; ароматические углеводороды, такие как бензол, толуол и т.д.; и галоидированные углеводороды, такие как метиленхлорид, хлороформ и т.д. Реакционная температура изменяется в зависимости от вида используемого гидрида металла, хотя обычно она остается в пределах приблизительно от -10oC до 100oC, предпочтительно, приблизительно от 0oC до 50oC.
Стадия 3:
Реакцию соединения формулы (C) с R1 - вводящим агентом, представленным формулой R1-X, обычно проводят в подходящем растворителе в присутствии основания. В качестве реакционноспособного сложноэфирного остатка спирта, обозначаемого X, могут быть названы, например, атомы галогена, такие как хлор, бром и иод; низшие алкилсульфонилоксигруппы, такие как метансульфонилокси; и арилсульфонилоксигруппы, такие как бензолсульфонилокси. Конкретные примеры растворителя включают: ароматические углеводороды, такие как бензол, толуол; кетоны, такие как ацетон, метилэтилкетон; простые эфиры, такие как тетрагидрофуран, диоксан; спирты, такие как этанол, изопропиловый спирт; ацетонитрил, хлороформ, этилацетат, диметилформамид, диметилсульфоксид, и их жидкие смеси. Конкретными примерами используемого основания являются те же, что перечислены выше для способа (a). Когда X в R1 - вводящем агенте (R1-X) обозначает хлор или бром, для спокойного протекания реакции добавляют иодид щелочного металла, такого как иодид натрия или иодид калия. Реакционная температура варьируется в зависимости от вида используемого R1 - вводящего агента, хотя обычно она находится в интервале приблизительно от 0oC до 200oC, предпочтительно приблизительно от 80oC до 150oC.
Стадия 4:
Реакцию на этой стадии обычно проводят в подходящем растворителе в присутствии неорганической кислоты, такой как разбавленная соляная кислота, разбавленная серная кислота или тому подобные. Конкретные примеры используемого растворителя включают спирты, такие как метанол, этанол; простые эфиры, такие как диэтиловый эфир, тетрагидрофуран; ацетон, ацетонитрил, этиленгликоль и смеси перечисленных выше жидкостей. Реакционную температуру варьируют в зависимости от вида используемого исходного соединения, хотя обычно она находится в интервале приблизительно от 0oC до 100oC.
Соединения формулы (IV') могут также быть получены, когда меняется местами порядок стадии 2 - реакции восстановления, и стадии 3 - реакции введения R1, в приведенной выше схеме 1. Это означает, что введение R1 в соединение формулы (B) (стадия 2') и последующее восстановление (стадия 3') могут приводить к соответствующему соединению формулы (D). В реакции такой стадии 2' предпочтительно использовать сильное основание, такое как гидрид натрия, вместо основания, описанного для способа (a). Кроме того, в восстановительной реакции 3' желательно использовать, например, бис (2-метоксиэтокси)-алюмогидрид натрия.
Соединения формулы (IV), в которой n равно 2 или 3, получают, например, по способу, иллюстрируемому приведенной ниже схемой 2: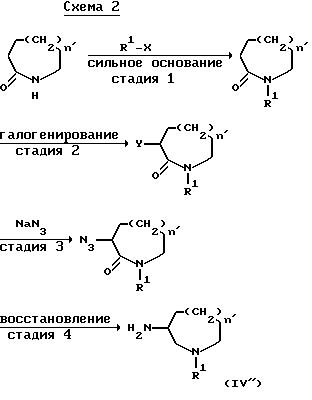
(в которой Y обозначает атом галогена, n' равно 2 или 3 и R1 и X имеют принятые ранее значения).
Стадию 1 в приведенной выше схеме 2 можно выполнять способом, аналогичным стадии 2', модифицированным способом подхода, принятого в иллюстрирующей схеме 1, используя в качестве исходного вещества, например, промышленно выпускаемые 2-азациклооктанон или 2-азациклононанон. Галоидирование стадии 2 может быть выполнено, например, согласно способу, описанному в J.Am. Chem. Soc. 80, 6233-6237 (1958). Стадии 3 и 4 можно выполнять, например, следуя способу, описанному в Helv.Chim.Acta, 41, 181-188 (1958).
Соединения формулы (IV) могут также быть получены способом, иллюстрируемым приведенной ниже схемой 3: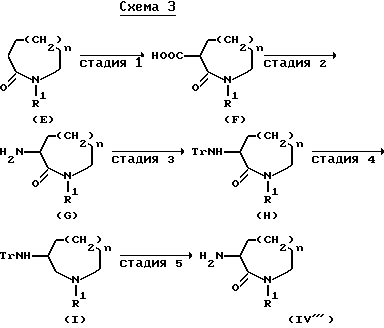
(в которой R1, Tr и n - имеют принятые выше значения).
Стадию 1 приведенной выше схемы 3 осуществляют в подходящем растворителе, приводя соединение формулы (E) к анионной форме под воздействием сильного основания и затем подвергая взаимодействию его с сухим льдом.
Превращение карбоксильной группы в амино-группу на стадии 2 может быть выполнено взаимодействием соединения формулы (F) с этилхлорформиатом и азидом натрия в подходящем растворителе, последующим нагреванием полученного ацилазида и воздействием кислоты на изоцианатный продукт. Трифенилметилирование на стадии 3, взаимодействие на стадии 4 и снятие трифенилметильной группы на стадии 5 могут быть выполнены способом, аналогичным, соответственно, стадиям 1, 2 и 4 схемы 1.
Исходные соединения формулы (E) могут быть получены способом стадии 1 схемы 2, использованием в качестве исходного соединения ∈-капролактама, 2-азациклооктанона или 2-азациклононанона.
По способу, иллюстрируемому схемой 1, конфигурация исходного соединения (A) превращается в конечный продукт формулы (IV'). Тогда как, конечные продукты (IV'') или (IV'''), полученные по способу схемы 2 или схемы 3, являются рацемическими. Рацемические соединения формулы (IV) могут быть разделены на два оптических изомера известными по существу способами. Например, такое соединение формулы (IV) обрабатывают оптически активной кислотой до образования солей или амидов диастереомеров, которые разделяют фракционированной перекристаллизацией или колоночной хроматографией, и затем превращают в свободные основания.
Соединения приведенной выше формулы (IV) являются новыми.
Соединения, представленные следующей формулой (IVa) и имеющие R - конфигурацию: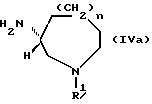
(в которой R1 и n имеют приведенные выше значения), используются в качестве новых промежуточных соединений формулы (I), имеющих R - конфигурацию, соединения, представленные ниже формулой (IVb):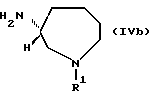
(в которой R1 имеет приведенные выше значения) являются особенно предпочтительными.
Способ (b)
Соединения формулы (I) могут быть получены диазотированием соединений формулы (V), приведенной ниже: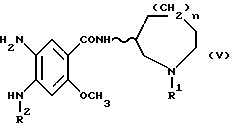
(в которой R1, R2, n и волнистая линия имеют приведенные выше значения) для образования бензотриазольного цикла.
Реакция замыкания цикла (реакция образования бензотриазольного цикла), приводящая к образованию соединений формулы (I) диазотированием соединений формулы (V), выполняется в условиях диазотирования, обычно используемых для ароматических аминов. В качестве диазотирующих агентов могут быть использованы, например, алкиловые эфиры азотистой кислоты, такие как нитрит натрия, трет- бутил нитрит и изоамилнитрит. Закрытие цикла с использованием азотистой кислоты обычно выполняют, добавляя сначала избыточное количество неорганической кислоты (например, соляной кислоты) или органической кислоты (например, уксусной кислоты) к водному раствору соединения формулы (V) или его соли кислотного присоединения, и затем добавляя водный раствор нитрита натрия. Реакционную температуру выдерживают обычно в интервале приблизительно от -20 до 60oC, предпочтительно приблизительно от 0oC до 25oC. Реакцию закрытия цикла с использованием алкилового эфира азотистой кислоты обычно проводят в подходящем растворителе путем взаимодействия соединения формулы (V) или его соли кислотного присоединения (например, гидрохлорида, ацетата) с алкиловым эфиром азотистой кислоты. Примеры подходящего растворителя включают метанол, уксусную кислоту, уксусную кислоту-диоксан, 1,2-диметоксиэтан, тетрагидрофуран, ацетон и метиленхлорид. Реакционную температуру обычно выдерживают в интервале приблизительно от 0oC до 100oC, предпочтительно приблизительно от 30oC до 80oC.
Исходные соединения, представленные формулой (V), могут быть получены, например, способом, который иллюстрируется приведенной ниже схемой 4.
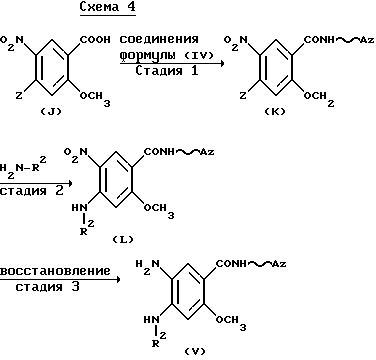
(в которой Z обозначает атом галогена и R2, Az и волнистая линия имеют приведенные выше значения).
Стадия 1:
Взаимодействие соединений формулы (J) или их реакционноспособных производных с соединениями формулы (IV) выполняют способом, аналогичным способу (a). Исходные соединения
формулы (J) могут быть получены, например, способом, описанным в Helv. Chim.Acta. 40, 369-372 (1957).
Стадия 2:
Взаимодействие соединений формулы (K) с соединениями, представленными формулой; H2N-R2, выполняют без использования растворителя или в адекватном растворителе.
Примеры используемого растворителя включают спирты, такие как метанол и этанол; диметилформамид, диметилсульфоксид и воду. Реакционную температуру обычно используют в интервале приблизительно от 0oC до 150oC.
Стадия 3:
Восстановление соединений формулы (L) осуществляют общепринятым способом. Например, соединение формулы (L) может быть обработано в подходящем растворителе восстанавливающим агентом. Конкретные примеры используемых восстанавливающих средств включают комбинации металлов (например, олова, цинка, железа) или солей металлов (например, хлористого олова) с кислотами (например, соляной кислотой, уксусной кислотой), хотя хлорид железа или олова сам по себе может быть использован в качестве восстанавливающего агента. Восстановление можно также выполнять гидрированием соединений формулы (L) в подходящем растворителе в присутствии катализатора. Конкретные примеры катализатора включают палладий- на-углероде, никель Ренея и окись платины. Растворитель следует выбирать согласно природе используемого восстанавливающего агента или вида используемого способа восстановления. В качестве растворителей обычно используют спирты, такие как метанол или этанол; этилацетат, ацетон, уксусная кислота, диоксан, вода или их жидкие смеси. Температуру реакции также варьируют в зависимости от восстанавливающего средства или способов восстановления, используемых в каждом случае, хотя обычно она находится в интервале приблизительно от 10oC до 100oC, а в случаях каталитического восстановления предпочтительный интервал составляет приблизительно от 10oC до 50oC.
Полученные таким образом соединения формулы (V) могут быть использованы в качестве исходного соединения для способа получения (b) без выделения и очистки.
Способ (c):
Соединения формулы (I), в которых R2 обозначает атом водорода, могут также быть получены гидрогенолизом соединений, представленных формулой (IIa) ниже: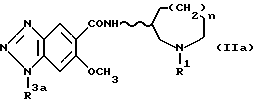
[в которой R3a обозначает амино-защитную группу (например, необязательно замещенный бензил или необязательно замещенный бензилоксикарбонил), и R1, n и волнистая линия имеют приведенные выше значения].
Гидрогенолиз может быть выполнен общепринятым способом, например, взаимодействием соединений либо с водородом в подходящем растворителе и в присутствии катализатора, такого как палладий-на-углероде, никель Ренея или тому подобного, либо с донором водорода (например, формиатом аммония, цикло-гексеном) в присутствии катализатора, такого как палладий-на-углероде. В качестве растворителя, например, используют спирты, такие как этанол или метанол; воду, уксусную кислоту, диоксан или тетрагидрофуран. Реакционную температуру обычно используют в интервалах приблизительно от 0oC до 80oC. Реакцию проводят при нормальном или повышенном давлении.
Соединения формулы (IIa), в которых R3a обозначает необязательно замещенную бензильную группу, могут быть получены способом (b) с использованием в качестве исходных соединений в схеме 4 тех соединений формулы (V), в которых R2 обозначает, например, необязательно замещенную бензильную группу. Исходные соединения (V) могут быть получены способом, иллюстрируемым схемой 4. В этом случае, восстановительную реакцию стадии 3 предпочтительно проводят, используя комбинацию металла или соли металла с кислотой, или железа, или хлористого олова.
Соединения формулы (IIa), в которых R3a обозначает необязательно замещенную бензилоксикарбонильную группу, могут быть получены по способу (a), использованием в качестве исходных соединений тех соединений формулы (III), в которых R2a обозначает необязательно замещенную бензилоксикарбонильную группу, которая в свою очередь может быть получена, например, по способу, описанному в JP-A-80858/1976 (патент США 4.039.672) или способами, аналогичными этому.
Способ (d)
Соединения формулы (I), в которой R2 обозначает атом водорода, могут также быть получены гидролизом соединений формулы (IIb), приведенной ниже: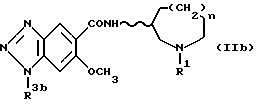
[в которой R3b обозначает амино-защитную группу (например, низший алканоил, трифторацетил, низший алкоксикарбонил или необязательно замещенный бензилоксикарбонил), и R1, n и волнистая линия имеют приведенные выше значения].
Реакция гидролиза может быть проведена по существу известным способом, например, путем контакта исходного соединения (IIb) с водой в подходящем растворителе, в кислой или щелочной среде. В качестве растворителя могут быть, например, использованы спирты, такие как метанол, этанол, изопропиловый спирт и тому подобные; диоксан, вода или их смеси. Конкретные примеры кислоты, используемой для создания кислотной среды, включают минеральные кислоты, такие как соляная, бромистоводородная и серная кислоты; органические кислоты, такие как муравьиная, уксусная, пропионовая и щавелевая кислота; и силикагель. Когда соединение формулы (IIb) содержит ацетильную группу в качестве R3b, использование силикагеля позволяет легко отщепить ацетильную группу и превратить это соединение в соединение формулы (I), в котором R2 обозначает атом водорода. Конкретные примеры основания, используемого для создания щелочной среды, включают гидроокись щелочного металла, такую как гидроокись натрия или калия; и карбонат щелочного металла, такой как карбонат натрия, карбонат калия и т.д. Температуру реакции обычно поддерживают в интервале приблизительно от 20oC до 100oC.
Соединения формулы (IIb), являющиеся промежуточными продуктами, по данному изобретению могут быть получены описанным ранее способом (a) с использованием в качестве исходных соединений тех соединений формулы (III), в которых R2a обозначает низший алканоил, трифторацетил, низший алкоксикарбонил или необязательно замещенный бензилоксикарбонил, который в свою очередь может быть получен, например, способом, описанным в JP-A-80858/1976 (патент США 4.039.672), или аналогичными ему способами.
По способам (a), (b), (c) и (d) конфигурация исходных соединений формул (IV), (V), (IIa) и (IIb) сохраняется при образовании соединений формулы (I). Поэтому, предпочтительно для получения соединений формулы (I), имеющих заданную конфигурацию, использовать исходные соединения, имеющие соответствующую конфигурацию. К тому же, возможно применять рацемическое исходное соединение для получения рацемического соединения формулы (I), которое затем может быть оптически разделено общепринятыми способами.
Соединения, полученные приведенными выше способами, могут быть выделены и очищены общепринятыми способами, такими как хроматография, перекристаллизация, переосаждение и т.д.
Соединения формулы (I) и соединения формул (IIa) и (IIb) получают в форме свободного основания или соли кислотного присоединения, что зависит от вида исходных соединений, условий реакции и обработки. Соли кислотного присоединения могут быть превращены в свободное основание обработкой их таким основанием, как карбонат щелочного металла или гидроокись щелочного металла. Тогда как свободные основания могут быть переведены в форму соли кислотного присоединения путем обработки их различными кислотами согласно общепринятым способам.
Далее представлены результаты испытаний характерных соединений по данному изобретению и моногидрата гидрохлорида метоклопрамида (metoclopramide) (Соединения A), промышленного средства для улучшения сократительной способности желудочно-кишечного тракта и обсуждается характеристика фармакологического действия соединений по изобретению.
Сначала приводится список соединений по данному изобретению, используемых в фармакологических испытаниях, с указанием их структуры и номеров соединений.
Соединение примера 1 (соединение 1)
N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид.
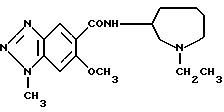
Соединение примера 2 (соединение 2)
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид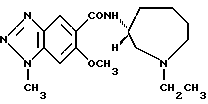
Соединение примера 4 (соединение 4)
1-этил-N-(1-этил-1H-гeкcaгидpoaзeпин-3-ил)-6-мeтoкcи 1Н- бензотриазол-5-карбоксамид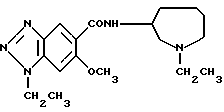
Соединение примера 5 (соединение 5)
(R)-1-этил-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамид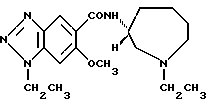
Соединение примера 6 (соединение 6) N-(1-этил-1Н- гексагидроазепин-3-ил)-6-метокси-1Н-бензотриазол-5-карбоксамид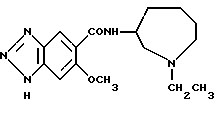
Соединение примера 7 (соединение 7a) N-(1-этил-1Н- гексагидроазепин-3-ил)-6-метокси-1H-бензотриазол-5-карбоксамид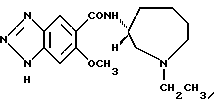
(соединение 7b)
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н-бензотриазол- 5-карбоксамид•3/2 фумарат
Соединение примера 9 (соединение 9)
N-(1-этил-1Н-гептагидроазоцин-3-ил)-6-метокси-1H-бензотриазол- 5-карбоксамид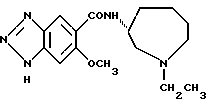
Соединение примера 10 (соединение 10)
N-(1-этил-1Н-октагидроазонин-3-ил)-6-метокси-1H-бензотриазол- 5-карбоксамид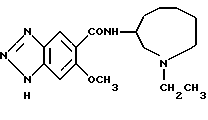
Соединение примера 11 (соединение 11)
N-(1-циклопропилметил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамид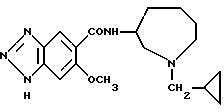
Соединение примера 12 (соединение 12)
(R)-N-(1-циклопропилметил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамид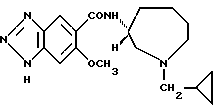
Соединение примера 13 (соединение 13)
(R)-N-(1-циклопропилметил-1Н-гексагидроазепин-3-ил)-6-метокси-1- метил-1Н-бензотриазол-5-карбоксамид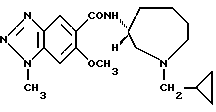
Соединение примера 14 (соединение 14)
N-(1-этил-1Н-гептагидроазоцин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид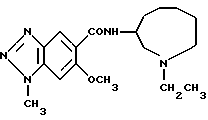
Соединение примера 15 (соединение 15)
N-(1-этил-1Н-октагидроазонин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид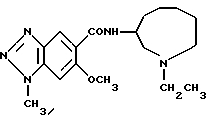
Соединение примера 3 (соединение 3)
(S)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил-1Н- бензотриазол-5-карбоксамид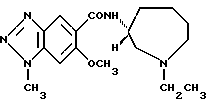
Соединение примера 8b (соединение 8b)
(S)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н-бензотриазол- 5-карбоксамид•3/2 фумарат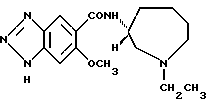
Соединение A
Моногидрат гидрохлорида 4-амино-5-хлор-N-[2-(диэтиламино) этил]-2-метоксибензамида [общее название: метоклопрамид; см., например, Merck Index, 11th ed. 6063 (1989)]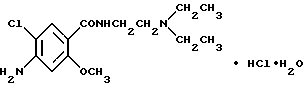
Эксперимент 1
Ингибирующее воздействие на апоморфин-индуцируемую рвоту
Группу из 3-4 собак (Гончая, 8-15 кг) используют для исследования ингибирующих воздействий испытуемых соединений на апоморфин-индуцируемую рвоту, согласно способу Chen and Ensor [cf.J.Pharmacol. Exp. Ther., 98 245-250 (1950)].
Испытуемые соединения, растворенные или суспендированные в 0,5% растворе трагаканта, вводят перорально за два часа до подкожного введения гидрохлорида апоморфина (0,3 мг/кг). Затем в течение одного часа подсчитывают количество приступов рвоты. Количество приступов рвоты в обработанной соединением группе сравнивают с количеством приступов в соответствующей контрольной (не получившей дозу) группе и рассчитывают процент ингибирования. Результаты приведены в таблице 1.
Из таблицы 1 очевидно, что почти все испытуемые соединения по данному изобретению показывают равную или большую чем метоклопрамид - гидрохлорид-моногидрат (соединение A) активность в ингибировании апоморфин-индуцируемой рвоты.
Эксперимент 2: Активность, усиливающая сократительную способность желудочно-кишечного тракта
Испытания проводят по способу Scarpignato et al. [cf. Arch.Int. Pharmacodyn., 246, 286-294(1980)].
Самцов крыс, весящих 130-150 г, выдерживают голодными в течение 18 часов перед началом эксперимента и дают перорально 1,5 мл пробного завтрака (фенол красный 0,05% в 1,5% водном растворе метилцеллюлозы). Через пятнадцать минут после введения завтрака удаляют желудок и измеряют количество фенола красного, оставшегося в желудке. Испытуемые соединения, растворенные или суспендированные в 0,5% растворе трагаканта, дают перорально за 60 минут до пробного завтрака. Степень сократительной способности желудочно-кишечного тракта рассчитывают по количеству фенола красного, оставшегося в желудке, и активность испытуемых соединений выражают через усиление степени сократительной способности по сравнению с контролем. Количество испытуемых животных составляет 4 для испытуемых соединений по данному изобретению и 5 для соединения A, используемого для сравнения. Результаты приведены в таблице 2.
Как следует из таблицы 2, каждое испытуемое соединение по данному изобретению показывает равную или большую чем моногидрат гидрохлорида метоклопрамида (соединение A) активность в усилении сократительной способности желудочно-кишечного тракта.
Эксперимент 3: Воздействие на сократительную способность желудочно-кишечного тракта, задержанную холецистокинином или морфином
Испытание проводят по способу Scarpignato et al.[cf. Arch.Int.Pharmacod yn., 246, 286-294(1980)].
Самцов Wistar крыс, весящих 130-150 г, выдерживают голодными в течение 18 часов перед началом эксперимента и дают перорально 1,5 мл пробного завтрака (фенол красный 0,05% в 1,5% водном растворе метилцеллюлозы). Через пятнадцать минут после введения завтрака удаляют желудок и измеряют количество фенола красного, оставшегося в желудке.
Испытуемые соединения, 10, 30 или 100 мг/кг каждого, растворенные или суспендированные в 0,5% растворе трагаканта, дают перорально за 60 минут до пробного завтрака. Сократительную способность желудочно-кишечного тракта замедляют подкожным введением холецистокинина 3 мг/кг или морфина 3 мг/кг за 5 минут до введения фенола красного. Количество используемых животных составляло 5-10. Результаты приведены в таблице 3.
Испытуемое соединение 2 (соединение примера 2) по данному изобретению при дозах 300-100 мг/кг значительно улучшает сократительную способность желудочно-кишечного тракта, задержанную холецистокинином.
Испытуемое соединение 7b (соединение примера 7b) по данному изобретению при дозах 10, 30 или 100 мг/кг значительно улучшает сократительную способность желудочно-кишечного тракта, замедленную холецистокинином или морфином, и проявляет превосходную активность, усиливающую сократительную способность желудочно-кишечного тракта (гастропрокинетическую активность) по сравнению с опорожнением кишечника, задержанным холецистокинином и морфином при каждой дозе. С другой стороны, моногидрат гидрохлорида метоклопрамида (соединение A) при дозах 10, 30 или 100 мг/кг не показывает улучшения активности по отношению к задержке опорожнения кишечника, вызванной холецистокинином или морфином.
Эксперимент 4
Воздействие на сократительную способность желудочно-кишечного тракта у находящихся в сознании собак.
Четыре здоровых гончих собаки на группу, обоего пола, весящие 10-12 кг, подвержены анестезии путем внутривенной инъекции пентобарбитала натрия (Nembutal, 30 мг/кг массы тела), и брюшная полость раскрыта в асептических условиях. По способу измерения сокращения круглой мышцы экстралуминарные силовые датчики вшивают в серозномышечный слой желудочной полости, 3 см - проксимально к кольцу привратника, двенадцатиперстной кишке, тощей кишке, средней кишке и окончанию подвздошной кишки, согласно методике Ito et al. [cf. Gastrointerol. Japan. 12, 275-283 (1977)].
Для внутрижелудочного (i.g.) введения лекарственных средств силастиковую трубку (Fr, размер 6,5) вводят в полость страдающего гастритом органа и трубку фиксируют на прилегающей серозной оболочке. Свинцовые проволоки этих датчиков и трубку для исследования желудка выводят из абдоминальной полости и затем проводят через кожный надрез, сделанный между лопаток. После операции на собаку надевают защитный фартук, чтобы предохранить свинцовые проволоки и силастиковую трубку. Собак помещают в изолированные экспериментальные клетки и дают пищу до 10 а.м. (до полудня), воду дают неограниченно.
Испытуемые соединения по 3 и 10 мг/кг суспендируют в 0,5% растворе трагаканта и вводят i.g. через находящуюся внутри силастиковую трубку.
Соединения 2 и 7b по данному изобретению при дозах 3 и 10 мг/кг вызывают сокращения, подобные внутренним пищеварительным перемещениям в состоянии кормления у находящихся в сознании собак. Таким образом, найдено, что испытуемые соединения проявляют значительную активность, усиливающую сократительную способность желудочно-кишечного тракта (гастропрокинетическую активность). Напротив, соединение A совсем не вызывает сокращения пищеварительного перемещения.
Эксперимент 5 Эффект ингибирования эксплоративной активности.
Используют группу из 5 самцов-мышей (Std-ddy strain 20-25 г). Через два часа после перорального введения испытуемых соединений в форме 0,5% трагакантного раствора или суспензии, мышей помещают отдельно в бокс для испытаний (23 х 35 х 30 см) на счетчик активности Animex'a (Farad Co.). После этого сразу же начинают счет активности и продолжают в течение трех минут. Среднее подсчитанное значение для обработанной соединением группы сравнивают с аналогичным значением для соответствующей контрольной (не получившей дозу) группы, и % ингибирования и 50% ингибиторную дозу (ID50) рассчитывают по способу.
Результаты приведены в таблице 4.
Как следует из таблицы 4, каждое испытуемое соединение по данному изобретению показывает намного меньший эффект ингибирования эксплоративной способности, чем метоклопрамид - гидрохлорид-моногидрат (соединение A). Этот результат приводит к выводу, что рассмотренные соединения по данному изобретению оказывают значительно меньшее подавление центральной нервной системы в сравнении с воздействием соединения A.
Эксперимент 6: Острая токсичность.
Используют группу из 5 мышей-самцов (Std-ddy strain 25-30 г). Испытуемое соединение вводят перорально испытуемому животному в форме 0,5% трагакантового раствора или суспензии и через 7 дней после введения испытуемых соединений наблюдают гибель животных и определяют 50% летальную дозу (LD50).
Результаты приведены в табл. 5.
Как видно из приведенных выше результатов испытаний, соединения формулы (I) по данному изобретению и их фармацевтически приемлемые соли кислотного присоединения обладают как значительным ингибирующим действием против рвоты, так и активностью, усиливающей сократительную способность желудочно-кишечного тракта (гастропрокинетической активностью) наряду с низкой CNS- депрессантной активностью и низкой токсичностью, и, следовательно, полезны в качестве агентов, усиливающих сократительную способность желудочно-кишечного тракта (гастропрокинетических средств) для лечения и профилактики различных желудочно-кишечных функциональных нарушений, связанных с различными болезнями и медикаментозными обработками: например, таких заболеваний, как: анорексия, тошнота, рвота, ощущение полноты желудка, дискомфорт верхней части желудочно-кишечного тракта, боль живота, изжога и отрыжка, которые наблюдаются при остром и хроническом гастрите, эзолафагеальный рефлюкс, язва желудка и двенадцатиперстной кишки, гастроневроз, гастроптоз, паралитическая непроходимость кишечника после операции, старческая непроходимость кишечника, демпинг-синдром, склеродермия, диабет, нарушение пищевода и желчных протоков, пуэрильная периодическая рвота, инфекции верхних дыхательных путей: например, слизистый колит, запор, понос у младенцев: и к примеру, тошнота и рвота, сопровождающие прием противораковых лекарственных средств или препаратов леводона, или после рентгеновского облучения.
Из соединений по данному изобретению, соединения 2, 7a и 7b показывают особенно высокую активность по усилению сокращения желудочно-кишечного тракта.
Фармацевтические препараты могут быть введены перорально, парентерально или прямокишечно. Клиническая доза варьируется в зависимости от видов соединений, путей введения, тяжести заболевания, возраста пациентов и тому подобного, но обычно используется в пределах от 0,01 до 10 мг/кг/день, предпочтительно от 0,1 до 3 мг/кг/день.
При использовании соединений формулы (I) или их фармацевтически приемлемых солей кислотного присоединения для описанного выше медицинского применения, их обычно вводят пациенту в форме фармацевтических препаративных составов, смешивая с носителями, обычно используемыми для получения медицинских составов и не реагирующими с соединениями по данному изобретению. Более конкретно, например, могут быть названы такие носители, как лактоза, инозит, глюкоза, маннитол, декстран, сорбитол, циклодекстрин, крахмал, частично преджелатинизированный крахмал, сахароза, магний алюмометасиликат, синтетический алюмосиликат, микрокристаллическая целлюлоза, натрий-карбоксиметилцеллюлоза, гидроксипропилкрахмал, кальций-карбоксиметилцеллюлоза, ионообменная смола, метилцеллюлоза, желатин, сок акации, пуллулан, гидроксипропилцеллюлоза, мало замещенная гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, поливинилпирролидон, поливиниловый спирт, альгиновая кислота, альгинат натрия, маловязкая безводная кремневая кислота, стеарат магния, тальк, трагакант, бентонит, веегам, карбоксивиниловый полимер, двуокись титана, сорбитановый эфир жирной кислоты, лаурилсульфат натрия, глицерин, глицериды насыщенных жирных кислот, безводный ланолин, глицерожелатин, полисорбат, макрогол, растительные масла, парафин, вода, пропиленгликоль, этанол, хлорид натрия, гидроокись натрия, соляная кислота, лимонная кислота, бензиловый спирт, глутаминовая кислота, глицин, метилпарагидроксибензоат, пропилпарагидроксибензоат и тому подобные.
Фармацевтические композиции могут быть взяты в любой препаративной форме, такой как таблетки, капсулы, гранулы, порошки, сиропы, суспензии, инъекции, припарки, суппозитории и тому подобной, получаемой общепринятыми способами. Жидкие препараты могут быть взяты в таких формах, которые перед использованием растворяют или суспендируют в воде или другой подходящей среде. Таблетки и гранулы могут быть покрыты оболочками по способам, которые сами по себе хорошо известны.
Соединения по данному изобретению, представленные формулой (I), в которой R2 обозначает водород, проявляют хорошую растворимость в воде и поэтому особенно удобны для жидких препаратов.
Эти препараты могут содержать, по крайней мере, 0,01% соединения формулы (I) по данному изобретению или его фармацевтически приемлемой соли кислотного присоединения, предпочтительно в соотношениях между 0,1 и 70%. Препараты могут также содержать другие терапевтически приемлемые компоненты.
Наилучший вариант воплощения изобретения
Далее изобретение раскрывается более конкретно путем стандартных примеров и экспериментальных примеров, понятно, что они не ограничивают рамки объема изобретения. Идентификация полученных соединений проведена на основании таких данных, как элементарный анализ, масс-спектр, УФ-спектр, ИК-спектр, ЯМР-спектр и т.д.
В приведенных далее стандартных и экспериментальных примерах иногда для простоты записи используются следующие обозначения:
[Растворитель для перекристаллизации]
A: этанол
E: диэтиловый эфир
[Заместитель]
Me: метил
Et: этил
Δ: циклопропил
Ph: фенил
ЯМР
J: постоянная связи
с: синглет
д: дублет
дд: двойной дублет
т: триплет
кв: квартет
м: мультиплет
ушир. с: уширенный синглет
[Другие обозначения]
ее: энантиомерный избыток
Пример 1 [реакция по способу (b)]
Получение N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил- 1Н-бензотриазол-5-карбоксамида (соединение 1):
К 40 мл водного раствора, содержащего около 3,0 г 5-амино-N -(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси-4-метиламинобензамида, полученного в приведенном ниже стандартном примере 4, добавляют 5 мл уксусной кислоты. Затем раствор охлаждают до 5oC, добавляют 10 мл водного раствора, содержащего 0,8 г нитрита натрия, после чего перемешивают в течение часа при той же температуре. Реакционную смесь подщелачивают водной гидроокисью натрия и экстрагируют хлороформом. Экстракт промывают водой, сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении, получая маслянистый остаток. Масло подвергают колоночной хроматографии на силикагеле, после чего элюируют и чистят смесью хлороформ-метанол (9:1). Полученный твердый продукт перекристаллизовывают из толуол- н-гексана, что дает 2,3 г указанного в заглавии соединения, т.пл. 103-104oC.
Пример 2 [реакция по способу (b)]
Получение (R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1-метил- 1Н-бензотриазол-5-карбоксамида (соединение 2):
Водный раствор уксусной кислоты, содержащий (R)-5- амино-N-(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси-4- метиламинобензамида, полученного ниже в стандартном примере 5, охлаждают до 5oC и добавляют по каплям 50 мл водного раствора, содержащего 6,6 г нитрита натрия. Смесь перемешивают один час при этой температуре и затем два часа при комнатной температуре. Реакционную смесь подщелачивают водным раствором гидроокиси натрия и экстрагируют этилацетатом. Экстракт промывают водой, сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении, получая маслянистый остаток. Масло подвергают колоночной хроматографии на силикагеле и затем элюируют и чистят смесью хлороформ-метанол (9:1). Образовавшийся твердый продукт перекристаллизовывают из толуол-н-гексана, получая 26,7 г указанного в заглавии соединения, т.пл. 118-120oC.
[α]
Пример 3 [реакция по способу (b)]
Получение (S)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1- метил-1Н-бензотриазол-5-карбоксамида (соединение 3):
Реакцию и обработку осуществляют способом, аналогичным примеру 2, используя водный раствор (S)-5-амино-N-(1-этил-1H-гексагидроазепин- 3-ил)-2-метокси-4-метиламинобензамид, полученный ниже в стандартном примере 6. Полученный продукт перекристаллизовывают из смеси толуол-н-гексан, получая указанное в заглавии соединение, т.пл. 119-120oC.
Пример 4 [реакция по способу (b)]
Получение 1-этил-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамида (соединение 4):
Реакцию и обработку проводят по способу примера 1, используя водный раствор 5-амино-N-(1-этил-1Н-гексагидро-азепин-3-ил) -4-этиламино-2-метоксибензамида, полученного ниже в стандартном примере 7. Образовавшийся продукт перекристаллизовывают из толуол-н-гексана, получая указанное в заглавии соединение, т.пл. 84-85oC.
Пример 5 [реакция по способу (b)]
Получение (R)-1-этил-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси- 1Н-бензотриазол-5-карбоксамида (соединение 5);
Реакцию и обработку выполняют по способу примера 1, используя водный раствор (R)-5-амино-N-(1-этил-1Н-гексагидроазепин-3-ил) -4-этиламино-2-метоксибензамида, полученного по стандартному примеру 8. Продукт получают в виде масла: масс-спектр (m/z): 346 (МН+).
Пример 6 [реакция по способу (а)]
Получение N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамида (соединение 6):
К 10 мл диметилформамидного раствора, содержащего 0,85 г 6-метокси-1Н-бензотриазол-5-карбоновой кислоты, добавляют 0,78 г N,N'-карбонилдиимидазола и перемешивают 6 часов при комнатной температуре. К реакционной смеси добавляют 0,75 г 3-амино-1-этил-1Н- гексагидроазепина и затем перемешивают в течение 14 часов при комнатной температуре. После прекращения реакции, растворитель упаривают при пониженном давлении и остаток подвергают колоночной хроматографии на силикагеле, используя элюирование и очистку с помощью хлороформ-метанола (10:1). Полученный продукт перекристаллизовывают из этанол-диэтилового эфира, получая 1,3 г указанного в заглавии соединения, т.пл. 156-158oC.
1H-ЯМР-спектр (CDCl3, δ м.д.): 1.09 (3H, т, J=7 Гц), 1.5-2.1 (6H, м), 2.5-3.1 (6H, м), 3.83 (3H, с), 4.4 (1H, м), 6.4 (1H, ушир.с), 7.07 (1H, с), 8.05 (1H, д, J=8 Гц), 8.78 (1H, с).
Пример 7 [реакция по способу (a)] и [(реакция по способу (d)]
Получение (R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6- метокси-1Н-бензотриазол-5-карбоксамид (соединение 7a):
a) [реакция по способу (a)]
К 150 мл диметилформамидного раствора, содержащего 10 г 6-метокси-1Н-бензотриазол-5-карбоновой кислоты, добавляют 9.0 г N,N'-кapбoнилдиимидaзoлa и перемешивают в течение 6 часов при комнатной температуре. К реакционной смеси добавляют 8.8 г (R)-3-амино-1-этил-1Н-гексагидроазепина и перемешивают 14 часов при комнатной температуре. После прекращения реакции, растворитель упаривают при пониженном давлении и остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая хлороформ-метанолом (10:1). Полученный продукт перекристаллизовывают из смеси диэтиловый эфир-н-гексан, получая 12 г указанного в заглавии соединения (соединение 7a), т.пл. 127-128oC.
(a') Указанное в заглавии соединение еще один раз перекристаллизовывают из этилацетат-н-гексана, получая указанное в заглавии соединение, т.пл. 142-144oC.
[α]
Этому соединению при жидкостной хроматографии высокого разрешения (ЖХВР) соответствует время удерживания 37,2 мин в приведенных ниже условиях и оно имеет оптическую чистоту 99% ее или выше.
Условия ЖХВР
ЖХВР-колонка: SUMICHIRAL ОА-4900; 4.6 мм х 250 мм, изготовлена Sumitomo Chemical Analysis Center
Подвижная фаза: н-гексан-метиленхлорид-этанол-трифторуксусная кислота (400:100:100:0.6)
Скорость истечения: 1.0 мл/мин
температура: 25oC
детектирование: 230 нм.
Из исходного вещества, имеющего R-конфигурацию, указанное выше в заглавии соединение с оптической чистотой 99% или выше получают методом ЖХВР, рацемизации не наблюдается. Кроме того, как показано ниже в примере 8, соединение S-конфигурации с оптической чистотой 99% ее или выше получают из исходного соединения S-конфигурации, без рацемизации. Эти факты ясно показывают, что конфигурация указанного в заглавии соединения, полученного в этом примере, R.
(b) Полученный выше на стадии (a) продукт обрабатывают фумаровой кислотой для превращения его в соответствующий фумарат, который перекристаллизовывают из изопропилового спирта-метанола, получая 3/2 фумарат указанного соединения (соединение 7b), т.пл. 131-133oC.
(b') Продукт, полученный выше на стадии (a), обрабатывают фумаровой кислотой для превращения его в соответствующий фумарат, который перекристаллизовывают из изопропилового спирта, получая 3/2 фумарат указанного в заглавии соединения, т.пл. 162-163oC.
(b'') Продукт, полученный выше на стадии (a), обрабатывают фумаровой кислотой и превращают в соответствующий фумарат, который перекристаллизовывают из этанол-изопропилового спирта, получая 3/2 фумарат•1/4 гидрат указанного в заглавии соединения, т.пл. 166-168oC.
(с) Реакция с использованием соединения формулы (III), в которой R2 обозначает ацетильную группу (аминозащитную группу)
[реакция по способу (а)]:
К 31,5 г 1-ацетил-6-метокси-1Н-бензотриазол-5-карбоновой кислоты добавляют 20,3 г триэтиламина и 400 мл этилацетата и к смеси добавляют по каплям при -7oC - -10oC 17,5 г этилхлорформиата. После последующего 2-часового перемешивания при -5oC - -7oC, к смеси добавляют по каплям 80 мл этил-ацетатного раствора, содержащего 22,8 г (R)-3-амино-1-этил-1H-гексагидроазепина и смесь перемешивают один час при этой температуре и затем 16 часов при комнатной температуре. Реакционную смесь промывают водой и насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом натрия, растворитель упаривают при пониженном давлении. Остаток растворяют в 1000 мл смеси хлороформ-метанол (8: 1) и затем добавляют к раствору 180 г силикагеля и впоследствии перемешивают 16 час при комнатной температуре.
Силикагель удаляют фильтрацией и остаток промывают 1000 мл смеси хлороформ-метанол (5: 1). Растворитель упаривают при пониженном давлении, получая 32 г сырого целевого продукта.
Двадцать четыре (24) г полученного выше сырого продукта обрабатывают 2,5-3-кратным объемом (22,5 г) фумаровой кислоты и образовавшийся фумарат перекристаллизовывают из метанол- изопропилового спирта, получая 25 г 3/2 фумарата указанного в заглавии соединения, т.пл. 131-133oC.
(d) [реакция по способу (d)]
К 1,85 г (R)-1-ацетил-N-(1-этил-1Н-гексагидроазепин-3-ил)- 6-метокси-1Н-бензотриазол-5-карбоксамиду, полученному по приведенному ниже примеру 17, добавляют 55 мл смеси хлороформ- метанол (8:1). Затем добавляют 18,5 г силикагеля и смесь перемешивают 16 часов при комнатной температуре. Силикагель удаляют фильтрацией и остаток промывают смесью хлороформ-метанол (9:1), содержащей 1% водный аммиак. Растворитель упаривают при пониженном давлении, получая 1,78 г сырого целевого продукта.
Пример 8 [реакция по способу (а)]
Получение (S)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси- 1Н-бензотриазол-5-карбоксамида (соединение 8):
a) Заменяя (R)-3-амино-1-этил-1Н-гексагидроазепин примера 7 на (S)-3-амино-1-этил-1Н-гексагидроазепин, который подвергают взаимодействию и обрабатывают по способу примера 7, получают указанное в заглавии соединение.
(b) Полученный выше продукт обрабатывают фумаровой кислотой и превращают в соответствующий фумарат. Перекристаллизация из изопропилового спирта дает 3/2 фумарат указанного в заглавии соединения, т.пл. 156-158oC.
Указанное в заглавии соединение показывает время удерживания 44,0 минут в ЖХВР в условиях анализа по примеру 7 и имеет оптическую чистоту 99% или выше.
(b') Полученный выше на стадии (a) продукт обрабатывают фумаровой кислотой аналогично приведенной выше стадии (b) и образующийся фумарат перекристаллизовывают из этанол-изопропилового спирта. Получают дифумарат•1/2 гидрат указанного в заглавии соединения, т.пл. 148-151oC.
Примеры 9-12 - реакция по способу (a)
Повторяют пример 6 за тем исключением, что 3-амино-1-этил-1Н- гексагидроазепин заменяют на соответствующие 3-амино-1-замещенный-1-гексагидроазепины.
Таким способом получают соединения, представленные в таблице 6.
Пример 13 [реакция по способу (b)]
Получение (R)-N-(1-циклопропилметил-1H-гексаридроазепин-3-ил)- 6-метокси-1-метил-1Н-бензотриазол-5-карбоксамида (соединение 13):
Используя соответствующее исходное соединение, водный раствор (R)-5-амино-N-(1-циклопропилметил-1Н-гексагидроазепин- 3-ил)-2-метокси-4-метиламинобензамид получают, используя реакцию и способ обработки, аналогичные приведенным ниже в примере 18 и стандартных примерах 1 и 4. Этот раствор обрабатывают и перерабатывают способом, аналогичным примеру 1, и полученный продукт перекристаллизовывают из толуола, что дает указанное в заглавии соединение, т.пл. 127-128oC.
Пример 14 [реакция по способу (a)]
Получение N-(1-этил-1Н-гептагидроазоцин-3-ил)-6-метокси-1-метил-1H-бензотриазол-5-карбоксамид (соединение 14).
Повторяют пример 6, за тем исключением, что 6-метокси-1Н- бензотриазол-5-карбоновую кислоту и 3-амино-1-этил-1Н- гексагидроазепин, используемые в примере 6, заменяют на 6-метокси-1-метил-1Н-бензотриазол-5-карбоновую кислоту и 3-амино- 1-этил-1Н-гептагидроазоцин, соответственно. Образовавшийся продукт перекристаллизовывают из этанол-диэтиловой эфир-смеси, получая указанное в заглавии соединение, т.пл. 116-118oC.
Пример 15 [реакция по способу (a)]
Получение N-(1-этил-1H-октагидроазонин-3-ил)-6-метокси-1-метил- 1Н-бензотриазол-5-карбоксамид (соединение 15):
Повторяют пример 6, за тем исключением, что 6-метокси-1Н-бензотриазол-5-карбоновую кислоту и 3-амино-1-этил-1Н-гексагидроазепин, используемые в примере 6, заменяют на 6-метокси-1-метил-1Н-бензотриазол-5-карбоновую кислоту и 3-амино-1-этил-1Н-октагидроазонин, соответственно. Полученный продукт перекристаллизовывают из этилацетата, получая указанное в заглавии соединение, т.пл. 155-156oC.
В приведенных далее примерах 16-24 описаны способы синтеза промежуточных продуктов, используемых для получения соединений по данному изобретению.
Пример 16 [реакция по способу (b)]
Получение 1-бензил-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н-бензотриазол-5-карбоксамида [соединение формулы (IIa), в которой R3a обозначает бензильную группу]:
К 6 мл водного раствора соляной кислоты, содержащей около 1,5 г 5-амино-4-бензиламино-N-(1-этил-1Н-гексагидроазепин-3-ил)- 2-метоксибензамида, полученного по стандартному примеру 9, добавляют 30 мл 5н соляной кислоты и 70 мл воды. Затем к раствору добавляют при охлаждении 1 мл водного раствора, содержащего 0,29 г нитрита натрия, и затем перемешивают 30 минут в тех же условиях и один час при комнатной температуре. Реакционную смесь подщелачивают 48% водным раствором гидроокиси натрия и экстрагируют хлороформом. Экстракт промывают насыщенным водным раствором хлористого натрия, сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении, получая маслянистый остаток. Остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая смесью хлороформ-метанол (20:1). Полученный твердый продукт перекристаллизовывают из диэтилового эфира, что дает 1,1 г указанного в заглавии соединения, т.пл. 136-137oC.
Пример 17 [реакция по способу (a)]
Получение 1-ацетил-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси- 1Н-бензотриазол-5-карбоксамида [соединение формулы (IIb), в которой R3b обозначает ацетильную группу]:
К 31,5 г 1-ацетил-6-метокси-1Н-бензотриазол-5-карбоновой кислоты добавляют 20,3 г триэтиламина и 400 мл этилацетата. Затем к раствору добавляют по каплям 17,5 г этилхлорформиата при -7oC - -10oC, с последующим перемешиванием в течение двух часов при -5oC - -7oC. К смеси добавляют по каплям восемьдесят (80 мл) этилацетатного раствора, содержащего 22,8 г (R)-3-амино-1-этил- 1Н-гексагидроазепина и перемешивают ее в течение одного часа, а затем в течение 16 часов при комнатной температуре. Реакционную смесь промывают последовательно водой и затем насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом натрия. Растворитель упаривают при пониженном давлении, получая 36 г указанного в заглавии соединения в виде твердого продукта, т.пл. 134-135oC (перекристаллизованный из этилацетата)
1H-ЯМР-спектр (CDCl3, δ м.д.): 1.04 (3H, т, J=7,0 Гц), 1,5-2,1 (6H, м), 2,45-2,95 (6H, м), 3,00 (3H, с), 4,09 (3H, с), 4,35 (1H, ушир.с), 7.76 (1H, с), 8.60 (1H, д, J=9,0 Гц), 8,95 (1H, с).
Пример 18 (реакция по схеме 1)
Получение (R)-3-амино-1-этил-1Н-гексагидроазепина;
(1) Стадия 1:
К перемешиваемой суспензии 173 г (R)-α-амино-∈-капролактам-гидрохлорида, 266 г триэтиламина и 1,700 мл хлороформа добавляют порциями при охлаждении льдом 293 г хлортрифенилметана. полученную смесь перемешивают один час в тех же условиях и затем два часа при комнатной температуре. Реакционную смесь промывают водой и сушат над безводным сульфатом магния. Растворитель упаривают при пониженном давлении и к полученному маслянистому остатку добавляют при нагревании и перемешивании 600 мл смеси н-гексан - этилацетат (2:1). Полученный кристаллический осадок собирают фильтрацией, промывают 1,000 мл смеси н-гексан- этилацетат (10:1) и сушат, что дает 290 г (R)-α-трифенилметиламино-∈-капролактама, т.пл. 189oC.
(2) Стадия 2':
К смеси 300 г полученного выше продукта, 193 г этан иодида и 1,500 мл тетрагидрофурана постепенно добавляют при перемешивании и комнатной температуре 42 г 60% гидрида натрия, после чего 1,5 часа перемешивают в тех же условиях. Затем сосуд охлаждают ледяной водой и к смеси медленно добавляют воду до растворения нерастворимых соединений. Реакционную смесь концентрируют при пониженном давлении и к остатку добавляют 1,000 мл этилацетата. Раствор промывают водой, органический слой сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении. К полученному маслянистому остатку добавляют 350 мл смеси гексан-этилацетат (50:1) и образовавшийся кристаллический осадок собирают фильтрацией и сушат, получая 287 г (R)-1-этил-3-трифенилметиламино-1Н-гексагидроазепин-2-она, т.пл. 127oC.
(3) Стадия 3':
К 615 г 70% толуольного раствора натрий бис(2-метокси- этокси)алюмогидрида добавляют 1400 мл толуола и к смеси добавляют 280 г полученного выше продукта при перемешивании и охлаждении льдом. Раствор перемешивают 1,5 часа в тех же условиях. К полученной реакционной смеси добавляют при охлаждении льдом 1,000 мл 15%-водного раствора гидроокиси натрия и отделяют органический слой, затем водный слой экстрагируют 1,500 мл толуола. Объединенный органический слой промывают водой и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении. Полученный таким образом маслянистый остаток кристаллизуют из этанола. Полученные кристаллы собирают фильтрацией и сушат, получая 247 г (R)-1-этил-3-трифенилметиламино-1Н-гексагидроазепина, т.пл. 83- 84oC.
(4) Стадия 4;
К смеси 177 г приведенного выше продукта и 50 мл тетрагидрофурана добавляют 700 мл 10% соляной кислоты, после чего 2 часа перемешивают при комнатной температуре. Реакционную смесь промывают диэтиловым эфиром. Затем к водному слою добавляют избыточное количество карбоната калия, после чего экстрагируют хлороформом. Жидкий экстракт сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении. Таким образом, получают 65 г указанного в заглавии соединения в виде масла.
1H-ЯМР-спектр (CDCl3, δ м.д.): 1.04 (3H, т, J=7.5 Гц), 1,3-1,9 (8H, м), 2,42 (1H, дд, J=13,5 Гц, 6,9 Гц), 2,5-2,6 (4H, м), 2.70 (1H, дд, J=13,5, J= 3,5 Гц), 2,98 (1H, м).
Пример 19 (реакция по схеме 1)
Получение (R)-3-амино-1-циклопропилметил-1Н-гексагидроазепина:
(1) Стадия 2;
К смеси 370 мл толуола и 37 г (R)-α-трифенилметиламино-∈-капролактама добавляют по каплям и при комнатной температуре 1,000 мл 1М толуольного раствора диизобутилалюмогидрида, с последующим 16-часовым перемешиванием. После прекращения реакции к смеси добавляют по каплям воду для разложения избытка диизобутилалюмогидрида. Осажденные соли удаляют фильтрацией и фильтрат промывают водным раствором хлористого натрия и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении. Таким образом получают 34 г (R)-3-трифенилметиламино-1Н-гексагидроазепина в виде масла.
(2) Стадия 3:
К смеси 10 г полученного продукта и 100 мл метилэтилкетона добавляют 10,5 г карбоната калия и 5,1 г циклопропилметилбромида и смесь нагревают 5 часов при температуре кипения с обратным холодильником. После прекращения реакции нерастворимые вещества удаляют фильтрацией и фильтрат концентрируют при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая хлороформ-метанолом (10: 1), что дает 10 г (R)-1-циклопропилметил-3- трифенилметиламино-1Н-гексагидроазепина в виде масла.
(3) Стадия 4:
К смеси 9.0 г полученного выше продукта и 10 мл тетрагидрофурана добавляют 100 мл 10% соляной кислоты и впоследствии перемешивают 5 часов при комнатной температуре. Полученную реакционную смесь промывают диэтиловым эфиром и водный слой насыщают карбонатом калия и экстрагируют хлороформом. Экстракт сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении, получая 4,0 г указанного в заглавии соединения в виде масла.
Пример 20 (реакция по схеме 1)
Получение 3-амино-1-этил-1H-гексагидроазепина:
(1) Стадия 1:
К перемешиваемой суспензии 125 г α-амино-∈-капролактама, 118 г триэтиламина и 600 мл хлороформа добавляют при охлаждении льдом 288 г хлортрифенилметана. Смесь дополнительно перемешивают один час при тех же условиях и затем перемешивают 2 часа при комнатной температуре. Полученный таким образом осадок собирают фильтрованием, тщательно промывают ацетоном и сушат, получая 330 г α-трифенилметиламино-∈-капролактама, т.пл. 240-241oC.
(2) Стадия 2':
К раствору 100 г полученного выше продукта и 65 г этан-иодида в 500 мл диметилформамида добавляют постепенно при перемешивании и комнатной температуре 12 г 60% гидрида натрия, с последующим перемешиванием в течение 4 часов при тех же условиях. Затем все выливают в ледяную воду и экстрагируют диэтиловым эфиром. Экстракт промывают водой и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении. Полученные кристаллы собирают фильтрацией и сушат, получая 88 г 1-этил-3-трифенилметиламино-1Н- гексагидроазепин-2-она, т.пл. 120-121oC.
(3) Стадия 3':
К 180 г 70% толуольного раствора, содержащего бис(2-метоксиэтокси) алюмогидрид натрия, добавляют 800 мл толуола. Далее при перемешивании и охлаждении льдом добавляют 83 г полученного выше продукта, после чего перемешивают один час при тех же условиях и 2 часа при комнатной температуре. К реакционной смеси добавляют при охлаждении льдом воду и 48% водный раствор гидроокиси натрия, органический слой отделяют. Органический слой промывают водой и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении. Полученный таким образом маслянистый остаток кристаллизуют из этанола. Кристаллы собирают фильтрацией и сушат, получая 65 г 1-этил-3- трифенилметиламино-1Н-гексагидроазепина, т.пл. 85-86oC.
(4) Стадия 4:
К смеси 134 г полученного выше продукта и 30 мл тетрагидрофурана добавляют 500 мл 10% соляной кислоты с последующим перемешиванием в течение 2 часов при комнатной температуре. Реакционную смесь промывают диэтиловым эфиром. Затем добавляют избыток карбоната калия к водному слою и экстрагируют хлороформом. Экстракт сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении, что дает 48 г указанного в заглавии соединения в виде масла.
1H-ЯМР-спектр (CDCl3, δ м.д.): 1,04 (3H, т. J=7,5 Гц), 1,3-1,9 (8H, м), 2,42 (1H, дд J=13,5 Гц, 6,9 Гц), 2,5-2,6 (4H, м), 2,70 (1H, дд, J=13,5, J= 3,5 Гц), 2,98 (1H, м).
Пример 21 (реакция по схеме 1)
Получение (S)-3-амино-1-этил-1Н-гексагидроазепина:
Повторяют пример 20, за тем исключением, что α-амино-∈-капролактам, используемый на стадии 1, здесь заменен на (S)-α-амино-∈-капролактам. Указанное в заглавии соединение получают в виде масла.
Пример 22 (реакция по схеме 1)
Получение 3-амино-1-циклопропилметил-1Н-гексагидроазепина:
Повторяют пример 18, за тем исключением, что (R)-α-трифенилметиламино-∈-капролактам, используемый на стадии 1, в данном случае заменяют на α -трифенилметиламино- ∈ -капролактам. Указанное в заглавии соединение получают в виде масла.
Пример 23 (реакция по схеме 2)
Получение 3-амино-1-этил-1Н-гептагидроазоцина:
(1) Стадия 1:
К раствору 27 г 2-азациклооктанона и 50 г этан иодида в 250 мл тетрагидрофурана постепенно добавляют при перемешивании и охлаждении льдом 10 г 60% гидрида натрия. Реакционную смесь перемешивают 4 часа при комнатной температуре и выливают в ледяную воду, с последующей экстракцией диэтиловым эфиром. Экстракт сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая хлороформ-метанолом (100:1), получая 36 г 1-этил-1Н- гептагидроазоцин-2-она в виде масла.
1H-ЯМР-спектр (CDCl3, δ м.д.): 1,15 (3H, т, J=7 Гц), 1,4-1,7 (6H, м), 1,82 (2H, м), 2,48 (2H, м), 3,38 (2H, кв. J=7 Гц), 3,47 (2H, м).
(2) Стадия 2:
К раствору 25 г полученного выше продукта в 250 мл хлороформа добавляют порциями при перемешивании и охлаждении льдом 34 г пентахлорида фосфора, с последующим дополнительным 30-минутным перемешиванием в тех же условиях. К полученной смеси добавляют при перемешивании и охлаждении льдом 0,4 г иода и затем медленно добавляют по каплям и в тех же условиях 25 г брома, с последующим нагреванием до температуры кипения с обратным холодильником в течение 2 часов. Растворитель упаривают при пониженном давлении. Остаток растворяют в этилацетате, промывают последовательно водой и водным раствором тиосульфата натрия и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении.
Остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая смесью н-гексан-этилацетат (4:1). Полученные таким образом кристаллы перекристаллизовывают из н-гексана, получая 10 г смеси 3-бром-1-этил-1Н-гептагидроазоцин-2-она и 3-хлор-1-этил-1Н-гептагидроазоцин-2-она.
(3) Стадия 3:
Смесь 10 г полученной выше смеси, 12 г азида натрия, 2,0 г иодида натрия и 100 мл диметилформамида перемешивают в течение ночи при 80oC, выливают в ледяную воду и экстрагируют диэтиловым эфиром. Экстракт сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении. Полученный остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая н-гексан-этилацетатом (4: 1), что дает 4,8 г 3-азид-1-этил- 1Н-гептагидроазоцин-2-она в виде масла.
1H-ЯМР-спектр (CDCl3, δ м.д.): 1,18 (3H, т, J=7 Гц), 1,5-1,8 (6H, м), 2,2 (2H, м), 3,08 (1H, м), 3,28 (1H, м), 3,53 (1H, м), 3,76 (1H, м), 4,0 (1H, дд, J=10,5 Гц, J=5,6 Гц).
(4) Стадия 4:
К 36,4 г 70% толуольного раствора бис (2- метоксиэтокси)-алюмогидрида натрия добавляют 100 мл толуола и к полученному раствору постепенно добавляют при перемешивании и охлаждении льдом дополнительно 4,8 г полученного выше продукта, с последующим перемешиванием при комнатной температуре в течение 2 часов. К реакционной смеси последовательно добавляют при перемешивании и охлаждении льдом воду и 48% водный раствор гидроокиси натрия и смесь экстрагируют диэтиловым эфиром. Экстракт сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении, получая 3,8 г указанного в заглавии соединения в виде масла.
Пример 24 (реакция по схеме 3)
Получение 3-амино-1-этил-1Н-октагидроазонина:
(1) К раствору 17 г 2-азациклононанона и 29 г иодида этана в 200 мл 1,2-диметоксиэтана постепенно добавляют при перемешивании и комнатной температуре 6,0 г 60% гидрида натрия, с последующим перемешиванием в этих условиях в течение 4 часов. Затем к смеси добавляют воду с последующей экстракцией хлороформом. Экстракт промывают водой и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении, получая 20 г 1-этил-1Н-октагидроазонин-2-она в виде масла.
(2) Стадия 1:
К 200 мл тетрагидрофуранового раствора, содержащего 20 г полученного выше продукта, добавляют по каплям и при охлаждении льдом 78 мл 2М-диизопропиламида лития в тетрагидрофуране, с последующим перемешиванием в течение одного часа. Реакционную смесь выливают на сухой лед и полученную смесь разбавляют водой и промывают этилацетатом. Водный слой подкисливают концентрированной соляной кислотой, экстрагируют хлороформом, промывают насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом магния. Растворитель упаривают при пониженном давлении, получая 14 г 3-карбокси-1-этил- 1Н-октагидроазонин-2-она, т.пл. 109-110oC (перекристаллизация из диэтилового эфир-н-гексана).
(3) Стадия (2):
К 100 мл ацетонового раствора, содержащего 12 г полученного выше продукта, добавляют 12 мл воды и 7,0 мл триэтиламина. К полученной смеси добавляют по каплям и при охлаждении льдом 30 мл ацетонового раствора, содержащего 8,0 г этилхлорформиата, с последующим 30-минутным перемешиванием. Затем, 30 мл водного раствора, содержащего 6,1 г азида натрия, добавляют по каплям к реакционной смеси, которую перемешивают потом 1,5 часа. Реакционную смесь выливают в ледяную воду и экстрагируют диэтиловым эфиром. Экстракт промывают насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом магния. Растворитель упаривают при пониженном давлении, к полученному остатку добавляют 200 мл толуола. Раствор перемешивают при нагревании до 70oC до прекращения вспенивания и затем температуру поднимают до 100oC, с последующим 2-часовым перемешиванием. После прекращения реакции, растворитель упаривают при пониженном давлении. К полученному остатку добавляют при перемешивании и охлаждении льдом 120 мл 20% соляной кислоты с последующим нагреванием до температуры кипения с обратным холодильником в течение 1,5 часов. Реакционную смесь промывают этилацетатом и водный слой подщелачивают избытком карбоната калия и экстрагируют хлороформом. Экстракт промывают насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом магния. Растворитель упаривают при пониженном давлении, получая 8.5 г 3-амино-1-этил-1Н-октагидроазонин-2-она в виде масла.
(4) Стадия 3:
К раствору 8,5 г полученного выше продукта в хлороформе добавляют 7,0 мл триэтиламина и затем добавляют порциями при охлаждении льдом 14 г хлортрифениламина, с последующим 3-часовым перемешиванием при комнатной температуре. Реакционную смесь промывают насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении. Остаток подвергают колоночной хроматографии, при которой элюируют и чистят этилацетатом-н-гексаном (1:10), получая 14 г 3-трифенилметиламино-1-этил-1Н-октагидроазонин-2-она в виде твердого вещества, т. пл. 160-162oC (перекристаллизация из н-гексан-этилацетата).
(5) Стадия 4:
К 30 г 70% толуольного раствора бис (2- метоксиэтокси)алюмогидрида натрия добавляют 100 мл толуола, к которому затем добавляют постепенно при перемешивании и охлаждении льдом 14 г вышеполученного продукта. Смесь перемешивают в течение ночи при комнатной температуре. После охлаждения реакционной смеси к ней добавляют по каплям 2н водный раствор гидроокиси натрия и затем добавляют 48% раствор гидроокиси натрия. Раствор экстрагируют этилацетатом. Экстракт промывают последовательно водой и насыщенным водным раствором хлористого натрия, сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая этил-ацетат-н-гексаном (1:10), что дает 13 г 3-трифенилметиламино-1-этил-1Н-октагидроазонин.
(6) Стадия 5:
К смеси 13 г полученного выше продукта и 3 мл тетрагидрофурана добавляют 45 мл 10% соляной кислоты с последующим перемешиванием в течение 5 часов при комнатной температуре. Реакционную смесь промывают диэтиловым эфиром. Водный слой подщелачивают избыточным количеством карбоната калия и экстрагируют хлороформом. Экстракт сушат над безводным сульфатом магния и растворитель упаривают при пониженном давлении, получая 5.0 г указанного в заглавии соединения в виде масла.
Стандартный пример 1 (реакция по стадии 1 схемы 4)
Получение 4-хлор-N-(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси-5-нитробензамида
К суспензии 14,7 г 4-хлор-2-метокси-5-нитробензойной кислоты, 300 мл хлороформа и 1 мл диметилформамида добавляют 22,7 г тионилхлорида и смесь нагревают в течение одного часа до температуры кипения с обратным холодильником. После прекращения реакции растворитель упаривают при пониженном давлении и остаток растворяют в 200 мл метиленхлорида, к смеси добавляют при охлаждении льдом 12,9 г триэтиламина и 9,0 г 3-амино- 1-этил-1Н-гексагидроазепина, с последующим перемешиванием при комнатной температуре в течение 15 мин. Реакционную смесь промывают последовательно водой и насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом магния, растворитель упаривают при пониженном давлении. Остаток подвергают колоночной хроматографии, элюируя и очищая хлороформ-метанолом (9:1), что дает 9,8 г указанного в заглавии соединения в виде твердого продукта.
Стандартный пример 2 (реакция по стадии 1 схемы 4)
Получение (R)-4-хлор-N-(1-этил-1Н-гексагидроазепин-3-ил) -2-метокси-5-нитробензамида:
Повторяют стандартный пример 1, за тем исключением, что используют (R)-3-амино-1-этил-1Н-гексагидроазепин. Указанное в заглавии соединение получают в виде твердого вещества.
Стандартный пример 3 (реакция по стадии 1 схемы 4)
Получение (S)-4-хлор-N-(1-этил-1Н-гексагидроазепин-3-ил) -2-метокси-5-нитробензамида:
Повторяют стандартный пример 1, за тем исключением, что используют (S)-3-амино-1-этил-1Н-гексагидроазепин. Указанное в заглавии соединение получают в виде твердого продукта.
Стандартный пример 4 (реакция по стадии 2 и 3 схемы 4)
Получение 5-амино-N-(1-этил-1Н-гексагидроазепин-3-ил)-2- метокси-4-метиламинобензамид:
(1) Стадия 2:
К 100 мл этанола, содержащего 4,9 г 4-хлор-N -(1-этил-1H-гексагидроазепин-3-ил)-2-метокси-5-нитробензамида добавляют 50 мл 30% этанольного раствора метиламина, с последующим нагреванием до температуры кипения с обратным холодильником в течение 1,5 часа. Растворитель упаривают при пониженном давлении и к образовавшемуся остатку добавляют воду. Кристаллы собирают фильтрацией, промывают водой и сушат, что дает 3,6 г N-(1-этил-1Н-гексагидроазепин-3- ил)-2-метокси-4-метиламино-5-нитробензамид.
(2) Стадия 3:
3,3 г полученного выше продукта растворяют в 200 мл 20%-водного метанола. К раствору добавляют 10%-палладий-на- углероде и смесь гидрируют при атмосферном давлении и комнатной температуре. После того, как абсорбируется теоретическое количество водорода, палладий-на-углероде удаляют фильтрацией и метанол из фильтрата упаривают при пониженном давлении. Таким образом получают водный раствор указанного в заглавии соединения.
Стандартный пример 5 (реакция по стадиям 2 и 3 схемы 4)
Получение (R)-5-амино-N-(1-этил-1Н-гексагидроазепин-3-ил)-2- метокси-4-метиламинобензамида:
(2) Стадия 2
К 600 мл этанольного раствора, содержащего 56,8 г (R) -4-хлор-N-(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси- 5-нитробензамида, добавляют 300 мл 40% водного раствора метиламина, с последующим нагреванием до температуры кипения с обратным холодильником в течение 2 часов. Растворитель упаривают при пониженном давлении и образовавшийся кристаллический осадок собирают фильтрацией. Осадок промывают водой и сушат, получая 32 г (R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси-4-метиламино- 5-нитробензамида.
(2) Стадия 3:
К 33,2 г полученного выше продукта добавляют 200 мл метанола, 400 мл воды, 80 мл уксусной кислоты и 2,0 г 10% - палладия-на-углероде и смесь гидрируют при атмосферном давлении и комнатной температуре. После того, как абсорбируется теоретическое количество водорода, палладий-на-углероде удаляют фильтрацией и метанол из фильтрата упаривают при пониженном давлении. Таким образом получают водный раствор уксусной кислоты, содержащий указанное в заглавии соединение.
Стандартные примеры 6-8 (реакция по стадиям 2 и 3 схемы 4)
Получение 4-замещенного амино-5-амино-N-(1-этил-1Н- гексагидроазепин-3-ил)-2-метоксибензамида или его оптических изомеров:
Используя 4-хлор-N-(1-этил-1Н-гексагидроазепин-3-ил)- 2-метокси-5-нитробензамиды, имеющие ту же конфигурацию, что и конечные продукты и соответствующие амины, взаимодействие и обработку проводят по способу стандартного примера 5, получая водные растворы соединений, перечисленных в таблице 7.
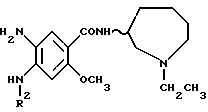
Стандартный пример 9 (реакция по стадиям 2 и 3 схемы 4)
Получение 5-амино-4-бензиламино-N-(1-этил-1Н- гексагидроазепин- 3-ил)-2-метоксибензамид:
(1) Стадия 2:
К 50 мл этанольного раствора, содержащего 2,0 г 4-хлор-N -(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси-5-нитробензамида добавляют 6,0 г бензиламина и нагревают до температуры кипения с обратным холодильником 22 часа. Растворитель упаривают при пониженном давлении. К остатку добавляют воду и затем экстрагируют хлороформом. Экстракт промывают насыщенным водным раствором хлористого натрия и сушат над безводным сульфатом натрия; растворитель упаривают при пониженном давлении. Остаток подвергают колоночной хроматографии на силикагеле, элюируя и очищая смесью хлороформ-метанол (12:1), что дает 2,4 г 4-бензиламино-N -(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси-5-нитробензамида в виде твердого вещества.
(2) Стадия 3:
К 1,6 г полученного выше продукта добавляют 6 мл конц. соляной кислоты и 3 мл этанола. Затем дополнительно добавляют раствор 2,6 г хлористого олова•дигидрат в 3 мл этанола, с последующим перемешиванием в течение 1 часа при 80oC. После прекращения реакции этанол упаривают при пониженном давлении, получая водный раствор соляной кислоты, содержащий указанное в заглавии соединение.
Стандартный пример 10
Получение 6-метокси-1-метил-1Н-бензотриазол-5-карбоновой кислоты [соединение формулы (III), R2a в которой обозначает метильную группу]:
1) Повторяют стандартный пример 1, за тем исключением, что 3-амино-1-этил-1Н-гексагидроазепин заменяют на пропиламин. Таким образом, получают 4-хлор-2-метокси- 5-нитpo-N-пропилбензамид в виде твердого продукта.
(2) 4-хлор-N-(1-этил-1Н-гексагидроазепин-3-ил)-2-метокси-5- нитробензамид, используемый в стандартном примере 4 (1), заменяют на приведенный выше продукт, и взаимодействие и обработку осуществляют способом, аналогичным стандартному примеру 4. Таким образом, получают 5-амино-2-метокси-4-метиламино-N-(1-пропил)бензамид в виде твердого вещества.
(3) 5-амино-N-(1-этил-1Н-гексагидроазепин-3-ил)-2- метокси-4-метиламинобензамид, используемый в примере 1, заменяют на полученный выше продукт. Взаимодействие и обработку проводят способом примера 1, что дает 6-метокси-1-метил-N-(1-пропил)-1Н- бензотриазол-5-карбоксамида в виде твердого соединения.
(4) Смесь 6.9 г полученного выше продукта и 100 мл конц. соляной кислоты нагревают до температуры кипения с обратным холодильником в течение 5,5 часов. Реакционную смесь охлаждают и концентрируют при пониженном давлении. Полученный кристаллический осадок собирают фильтрацией, промывают водой и сушат, получая 3,2 г указанного в заглавии соединения.
Пример состава 1
Получение таблеток (на 1.000 таблетки):
(R)-N-(1-этил-1Н- гексагидроазепин-3-ил)-6-метокси-1-метил-1Н-бензотриазол-5-карбоксамид (соединение 2) - 5 г
Лактоза - 80 г
Кукурузный крахмал - 30 г
Микрокристаллическая целлюлоза - 25 г
Гидроксипропилцеллюлоза - 3 г
Маловязкий кремниевый ангидрид - 0,7 г
Стеарат магния - 1,3 г
Перечисленные выше компоненты смешивают и гранулируют обычным способом, таблетированием получают 1.000 таблеток (145 мг/таблетка).
Пример состава 2
Получение капсул (на 1.000 капсул):
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н-бензотриазол- 5-карбоксамид•3/2 фумарат (соединение 7b) - 10 г
Лактоза - 160 г
Кукурузный крахмал - 22 г
Гидроксипропилцеллюлоза - 3,5 г
Маловязкий кремниевый ангидрид - 1,8 г
Стеарат магния - 2,7 г
Приведенные выше соединения смешивают, гранулируют общепринятым способом и заполняют в 1.000 капсул.
Пример состава 3
Получение порошка:
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамид•3/2 фумарат (соединение 7b) - 10 г
Лактоза - 960 г
Гидроксипропилцеллюлоза - 25 г
Маловязкий кремниевый ангидрид - 5 г
Приведенные компоненты смешивают и составляют в порошкообразный препарат общепринятым способом.
Пример состава 4
Препарат для инъекций (на 1.000 ампул):
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1Н- бензотриазол-5-карбоксамид•3/2 фумарат (соединение 7b) - 10 г
Сорбитол - 100 г
Вода для инъекций - g.s. - Всего 2.000 мл
(R)-N-(1-этил-1Н-гексагидроазепин-3-ил)-6-метокси-1H- бензотриазол-5-карбоксамид•3/2 фумарат и сорбитол растворяют в части воды для инъекций. Оставшуюся часть воды для инъекций добавляют к общему количеству. Полученный раствор фильтруют через мембранный фильтр (0,22 мкм). Фильтрат заполняют в 2 мл ампулы и стерилизуют при 121oC 20 минут.
Промышленная применимость
Как уже отмечалось раньше, соединения по данному изобретению, представленные формулой (I), и их фармацевтически приемлемые соли кислотного присоединения обладают одновременно превосходной противорвотной активностью и активностью, усиливающей сократительную способность желудочно-кишечного тракта, проявляя при этом меньшую супрессорную активность в отношении CNS и, поэтому, полезны в качестве средств, усиливающих сократительную способность желудочно-кишечного тракта для лечения или профилактики различных функциональных нарушений желудочно-кишечного тракта, связанных с различными заболеваниями и их терапией. Промежуточные соединения по данному изобретению полезны в качестве синтетических промежуточных веществ для соединений формулы (I), в которой R2 обозначает атом водорода. Кроме того, промежуточные соединения по данному изобретению, представленные формулой (IVa), полезны в качестве промежуточных веществ для соединений формулы (I), имеющих R-конфигурацию.
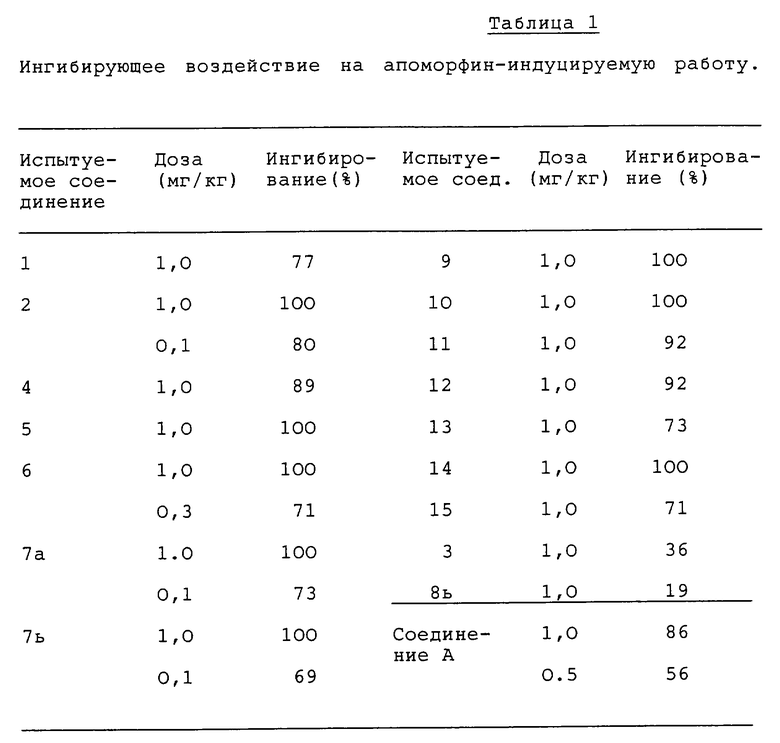
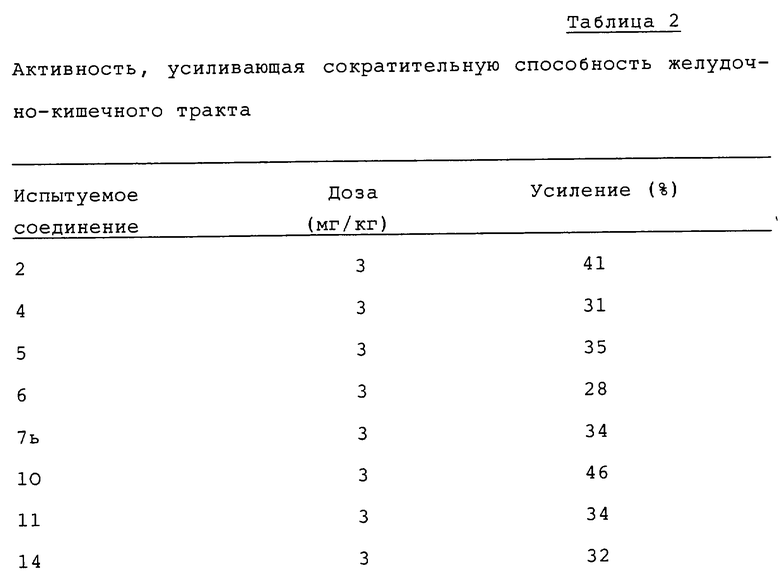



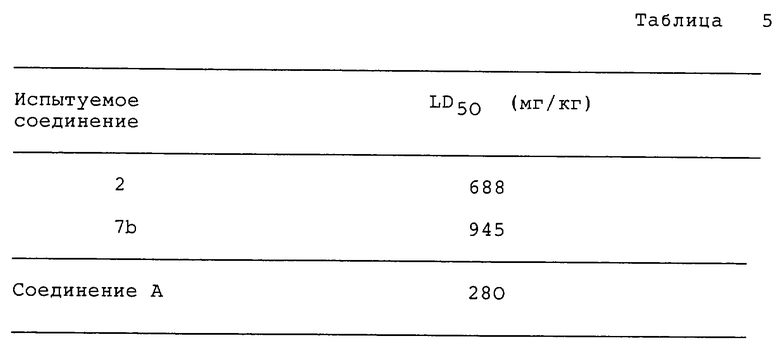

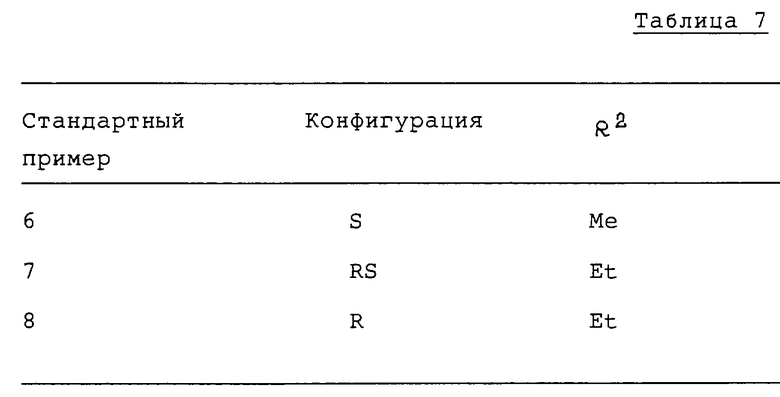
Описываются новые производные 6-метокси-1Н-бензотриазол- 5-карбоксамида формулы I, в которой R1 обозначает этил или циклопропилметил, R2 обозначает атом водорода, метил или этил, n равно 1, 2 или 3 и волнистая линия  означает, что конфигурация заместителей на углеродном атоме, связанном с N-атомом в амидной группе, является RS, R или S, или фармацевтически приемлемые кислые аддитивные кислые соли. Описываются также способы их получения; фармацевтические композиции, содержащие соединение формулы I или его фармацевтически приемлемую кислую аддитивную соль, и новые промежуточные соединения. Соединения обладают одновременно высокой противорвотной активностью и активностью, усиливающей сократительную способность желудочно-кишечного тракта. Кроме того, они обладают низкой депрессантной активностью относительно центральной нервной системы (CNS) и поэтому полезны для лечения и профилактики функциональных нарушений желудочно-кишечного тракта, связанных с различными болезнями и терапевтическими обработками, в качестве средства, усиливающего сократительную способность желудочно-кишечного тракта. 6 с. и 5 з.п.ф-лы., 7 табл.
означает, что конфигурация заместителей на углеродном атоме, связанном с N-атомом в амидной группе, является RS, R или S, или фармацевтически приемлемые кислые аддитивные кислые соли. Описываются также способы их получения; фармацевтические композиции, содержащие соединение формулы I или его фармацевтически приемлемую кислую аддитивную соль, и новые промежуточные соединения. Соединения обладают одновременно высокой противорвотной активностью и активностью, усиливающей сократительную способность желудочно-кишечного тракта. Кроме того, они обладают низкой депрессантной активностью относительно центральной нервной системы (CNS) и поэтому полезны для лечения и профилактики функциональных нарушений желудочно-кишечного тракта, связанных с различными болезнями и терапевтическими обработками, в качестве средства, усиливающего сократительную способность желудочно-кишечного тракта. 6 с. и 5 з.п.ф-лы., 7 табл.
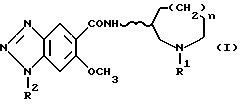
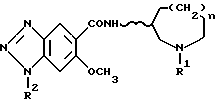
в которой R1 обозначает этил или циклопропилметил;
R2 обозначает атом водорода, метил или этил;
n равно 1,2 или 3;
волнистая линия  обозначает, что конфигурация заместителей на атоме углерода, связанном с атомом азота в амидной группе, является RS, R или S,
обозначает, что конфигурация заместителей на атоме углерода, связанном с атомом азота в амидной группе, является RS, R или S,
или его фармацевтически приемлемые аддитивные кислые соли.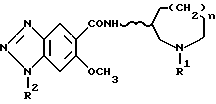
в которой R1 обозначает этил или циклопропилметил;
R2 обозначает атом водорода, метил или этил;
n обозначает 1,2 или 3;
волнистая линия  означает, что конфигурация заместителей на углеродном атоме, связанном с N-атомом в амидной группе, является RS, R или S,
означает, что конфигурация заместителей на углеродном атоме, связанном с N-атомом в амидной группе, является RS, R или S,
или его фармацевтически приемлемой аддитивной кислой соли, и фармацевтически приемлемый носитель.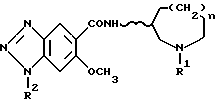
в которой R1 обозначает этил или циклопропилметил;
R2 обозначает атом водорода, метил или этил;
n равно 1, 2 или 3;
волнистая линия  означает, что конфигурация заместителей на углеродном атоме, связанном с атомом азота в амидном радикале, является RS, R или S,
означает, что конфигурация заместителей на углеродном атоме, связанном с атомом азота в амидном радикале, является RS, R или S,
или его фармацевтически приемлемой кислой аддитивной соли, включающий взаимодействие соединения, представленного формулой III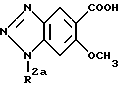
в которой R2а обозначает водород, метил, этил или аминозащитную группу,
или его реакционноспособного производного с соединением, представленным формулой IV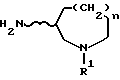
в которой R1 обозначает этил или циклопропилметил;
n обозначает 1, 2 или 3;
волнистая линия  означает, что конфигурация заместителей на атоме углерода, связанном с N-атомом амидной группы, является RS, R или S,
означает, что конфигурация заместителей на атоме углерода, связанном с N-атомом амидной группы, является RS, R или S,
и когда в качестве одного из реагентов используют соединение формулы III, в котором R2а обозначает аминозащитную группу, превращают R2а в соответствующем продукте в атом водорода путем гидролиза или гидрогенолиза названного продукта и, при необходимости, превращают полученный продукт в его фармацевтически приемлемую кислую аддитивную соль.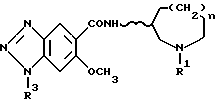
в которой R1 обозначает этил или циклопропилметил;
R3 обозначает аминозащитную группу;
n обозначает 1, 2 или 3;
волнистая линия  означает, что конфигурация заместителей на углеродном атоме, связанном с N-атомом в амидном радикале, является RS, R или S,
означает, что конфигурация заместителей на углеродном атоме, связанном с N-атомом в амидном радикале, является RS, R или S,
или его кислая аддитивная соль.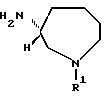
в которой R1 обозначает этил или циклопропилметил,
или его кислая аддитивная соль.
| Способ получения -(1"-аллилгирролидинил-2"-метил-2-метокси4,5)-азимидобензамида | 1975 |
|
SU577990A3 |
| Устройство для устранения мешающего действия зажигательной электрической системы двигателей внутреннего сгорания на радиоприем | 1922 |
|
SU52A1 |
| Способ получения производных бензтриазола | 1982 |
|
SU1179927A3 |
| SU 1227111 A, 1986. | |||

