Область техники
[0001] Настоящее изобретение относится к производным карбоксиметилпиперидина, полезным в качестве лекарственных средств.
[0002] Более конкретно, настоящее изобретение относится к производным карбоксиметилпиперидина или их фармацевтически приемлемым солям, которые обладают антагонистической активностью в отношении рецептора вещество P/нейрокинин 1 (NK1), и которые полезны в качестве средств для предупреждения или лечения индуцированной противораковой химиотерапией тошноты и рвоты (CINV), и так далее.
Уровень техники
[0003] CINV происходит, когда рвотный центр, расположенный в боковой ретикулярной формации продолговатого мозга, получает стимул. Площадь самого заднего поля и одиночное ядро продолговатого мозга содержат рецепторы NK1, и рецепторы NK1, как полагают, должны активно вызывать рвоту.
Введение противоопухолевого средства способствует секреции серотонина из энтерохромаффинных клеток (EC) в пищеварительном тракте, и серотонин непосредственно стимулирует рвотный центр через рецепторы 5-гидрокситриптамина 3 (5-HT3) в ЖКТ. Кроме того, когда серотонин стимулирует рвотный центр через хеморецепторную триггерную зону (CTZ), расположенную в области самого заднего поля четвертого желудочка, возникают тошнота и рвота. Вещество P, подобно серотонину, обнаруживается в клетках EC в пищеварительном тракте и способствует их секреции при введении противоопухолевого средства. Недавно было обнаружено, что вещество P вызывает рвоту через рецепторы NK1 в CTZ или путем связывания с рецепторами NK1 в центральной нервной системе, и поэтому рецепторы NK1 привлекают внимание в качестве мишени для разработки противорвотных средств (непатентная литература 1).
[0004] Апрепитант является первым селективным антагонистом рецепторов NK1 в мире, который был утвержден в качестве превентивного средства против тошноты и рвоты, связанных с введением противоопухолевых средств. Что касается механизма действия апрепитанта, полагают, что апрепитант избирательно ингибирует связывание вещества Р и рецепторов NK1 в центральной нервной системе, которая является одним из путей, вызывающих CINV, и таким образом предотвращает CINV. Апрепитант был выпущен на рынок в качестве превентивного средства против CINV (непатентная литература 2).
[0005] Известно, что апрепитант метаболизируется цитохромом P450 (CYP) 3A4. Кроме того, апрепитант известен, как обладающий дозозависимым эффектом на CYP3A4, вызывающим CYP3A4 эффект и вызывающим CYP2C9 эффект. Соответственно, апрепитант может вызвать межлекарственное взаимодействие с лекарственными средствами, которые ингибируют или индуцируют CYP3A4, или с лекарственными средствами, которые метаболизируются CYP3A4 или CYP2C9. Например, сообщается, что ингибирующее действие апрепитанта на CYP3A4 иногда тормозит метаболизм дексаметазона, и, таким образом, необходимо корректировать дозу дексаметазона в сочетании с апрепитантом (непатентная литература 3).
Поэтому, когда используется апрепитант, следует с особым вниманием контролировать межлекарственные взаимодействия, основанные на ингибирующем воздействии апрепитанта на CYP3A4.
[0006] По указанным выше причинам, требуется новый антагонист рецепторов NK1 с наименьшими межлекарственными взаимодействиями для профилактики или лечения CINV.
[0007] Известны соединения с антагонистической активностью в отношении рецепторов NK1, такие как касопитант, нетупитант, эзлопитант, ролапитант, вестипитант, вофопитант и т.д.
Однако сообщается, что касопитант обладает ингибирующим воздействием на CYP3A4 и вызывает межлекарственные взаимодействия под этим воздействием (непатентная литература 4). Были проведены клинические испытания касопитанта в США и Европе в качестве превентивного средства против тошноты и рвоты, индуцированной противораковой химиотерапией; однако его внедрение было прекращено после применения. Нетупитант в настоящее время находится в стадии разработки в качестве превентивного средства против тошноты и рвоты, индуцированной противораковой химиотерапией; однако сообщается, что нетупитант обладает ингибирующим воздействием на CYP3A4 и вызывает межлекарственные взаимодействия за счет данного воздействия (непатентная литература 5). В США были проведены клинические испытания эзлопитанта в качестве превентивного средства против тошноты и рвоты, индуцированной противораковой химиотерапией; однако, его внедрение было прекращено. В Европе были проведены клинические испытания вофопитанта в качестве превентивного средства против тошноты и рвоты, индуцированной противораковой химиотерапией; однако, его внедрение было прекращено.
Разработка многих из указанных выше соединений в результате была приостановлена.
[0008] Производные пиридина, обладающие антагонистической активностью в отношении рецептора NK1, описаны в патентной литературе 1-16. И пролекарства производных пиридина описаны в патентной литературе 17 и 18.
Однако производные карбоксиметилпиперидина настоящего изобретения не описаны в приведенной выше литературе.
Перечень ссылочных материалов
Патентная литература
[0009] Патентная литература 1: патент США 6479483
Патентная литература 2: патент США 6770637
Патентная литература 3: патент США 7939533
Патентная литература 4: европейский патент 1103545
Патентная литература 5: патент США 7211579
Патентная литература 6: публикация патентной заявки США № 2006/0030600
Патентная литература 7: патент США 6576762
Патентная литература 8: патент США 6225316
Патентная литература 9: патент США 7683056
Патентная литература 10: патент США 8344005
Патентная литература 11: публикация международной патентной заявки № WO2011/054773
Патентная литература 12: публикация патентной заявки США № 2007/0071813
Патентная литература 13: публикация патентной заявки США № 2003/0083345
Патентная литература 14: публикация патентной заявки США № 2003/0004157
Патентная литература 15: патент США 6849624
Патентная литература 16: патент США 6297375
Патентная литература 17: патент США 6593472
Патентная литература 18: патент США 8426450
Непатентная литература
[0010] Непатентная литература 1: P. J. Hesketh et al., European Journal of Cancer, 2003, Vol. 39, pp. 1074-1080
Непатентная литература 2: Toni M. Dando et al., Drugs, 2004, Vol. 64, No. 7, pp. 777-794
Непатентная литература 3: Jacqueline B. McCrea et al., CLINICAL PHARMACOLOGY & THERAPEUTICS, 2003, Vol. 74, No. 1, pp. 17-24
Непатентная литература 4: Stefano Zamuner et al., British Journal of Clinical Pharmacology, 2010, Vol. 70, No. 4, pp. 537-546
Непатентная литература 5: Corinna Lanzarotti et al., Support Care Cancer, 2013, Vol. 21, No. 10, pp. 2783-2791
Сущность изобретения
Задачи, решаемые настоящим изобретением
[0011] Задачей настоящего изобретения является предоставление нового соединения, обладающего антагонистической активностью в отношении рецептора NK1, чья CYP3A4 ингибирующая активность ослаблена, по сравнению с апрепитантом, и которое полезно для предупреждения или лечения индуцированной противораковой химиотерапией тошноты или рвоты. Предпочтительной задачей настоящего изобретения является предоставление указанного выше соединения, чье свойство транспортировки в центральную нервную систему и лечебный эффект пролонгированного действия являются превосходными.
Средства для решения задачи
[0012] Настоящее изобретение относится к соединению, представленному следующей формулой изобретения (I), или его фармацевтически приемлемой соли.
[0013] То есть, настоящее изобретение относится к следующим вариантам осуществления [1]-[12] и тому подобное.
[1] Соединение, представленное формулой (I):
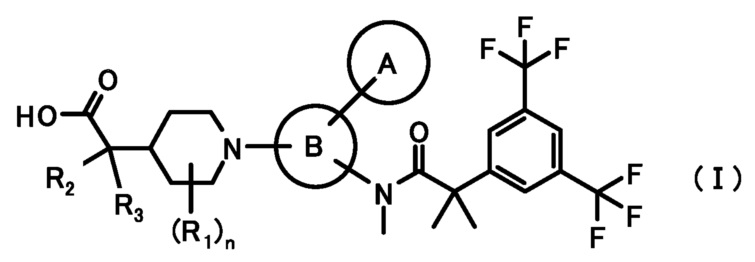
где
кольцо A является группой, представленной формулой:
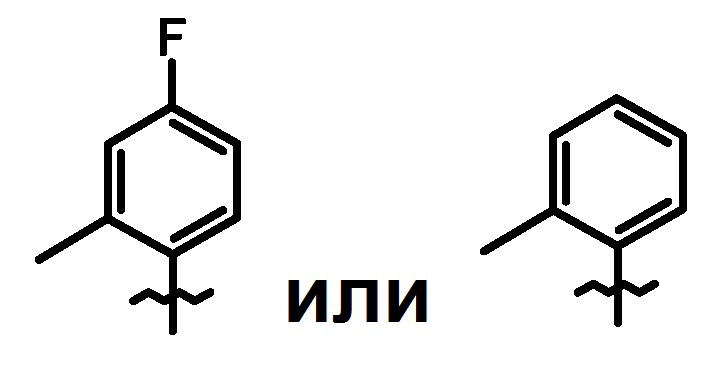 ;
;
кольцо B является группой, представленной формулой:
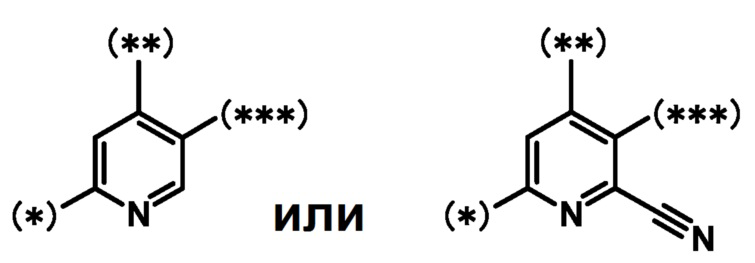 ;
;
при условии, что связи, обозначенные (*), являются местом связывания с формулой:
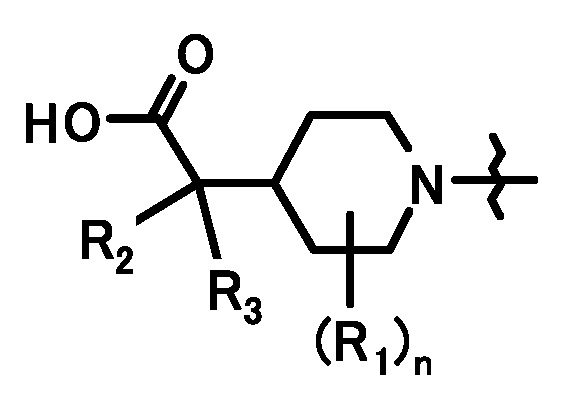 ;
;
связи, обозначенные (**), являются местом связывания с кольцом A;
связи, обозначенные (***), являются местом связывания с формулой:
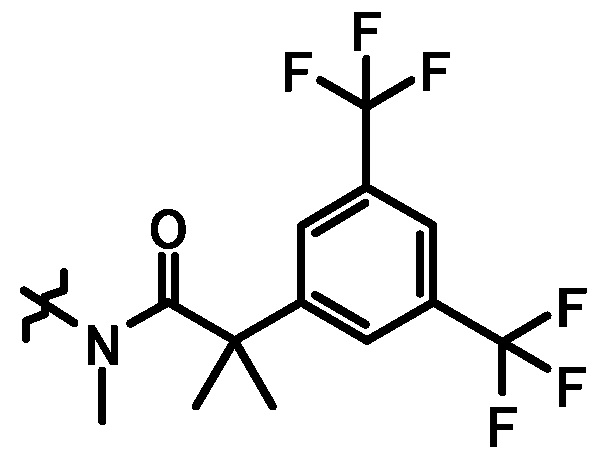 ;
;
R1 представляет собой C1-6алкил или C1-6алкокси;
R2 и R3, каждый независимо, представляет собой атом водорода или метил;
n равен 0, 1, 2, 3, 4 или 5;
или его фармацевтически приемлемая соль.
[2] Соединение, представленное формулой (Ia), по приведенному выше пункту [1]:
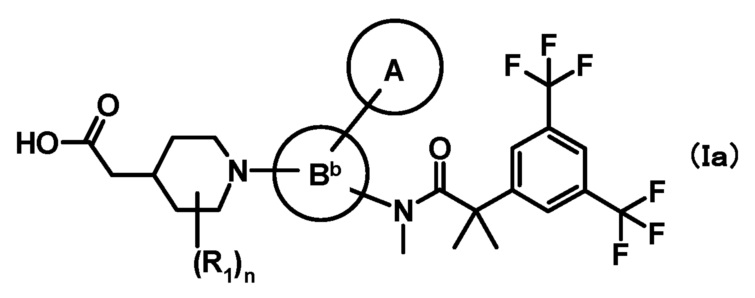
где
кольцо A, R1 и n имеют такие же значения, как описано в пункте [1];
кольцо Bb является группой, представленной формулой:
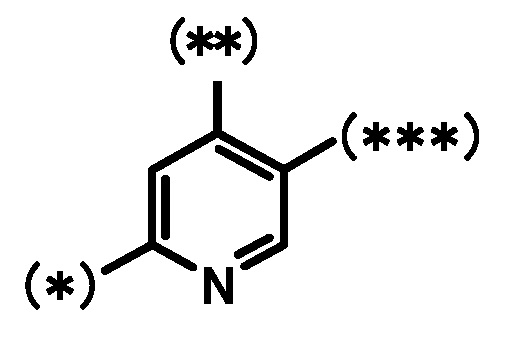 ;
;
где связь, обозначенная (*), является местом связывания с формулой:
 ;
;
(**) и (***) имеют такие же значения, как описано в пункте [1];
или его фармацевтически приемлемая соль.
[3] Соединение, представленное формулой (Ib), по приведенному выше пункту [2]:
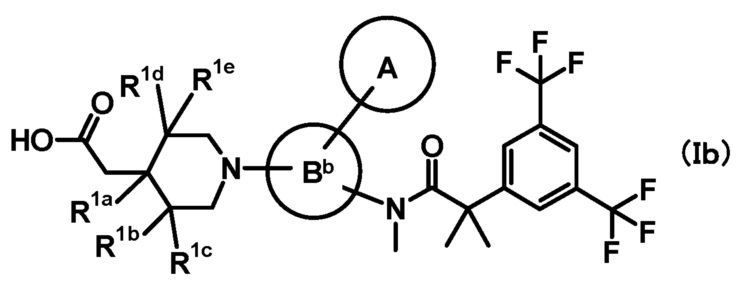
где
кольцо A и кольцо Bb имеют такие же значения, как описано в пункте [2];
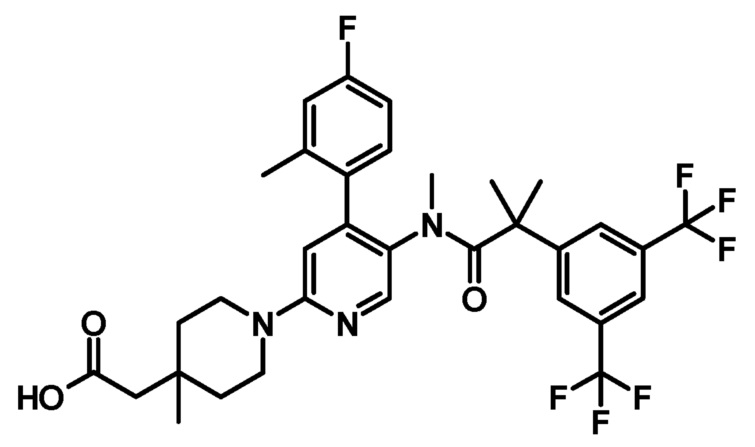
при условии, что связь, обозначенная (*), является местом связывания с формулой:
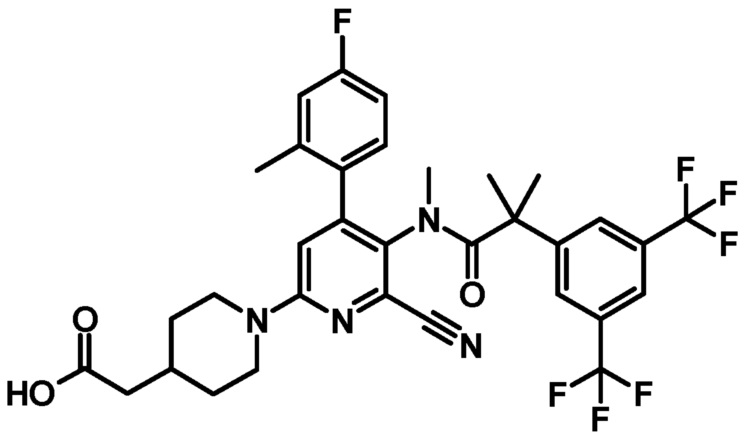
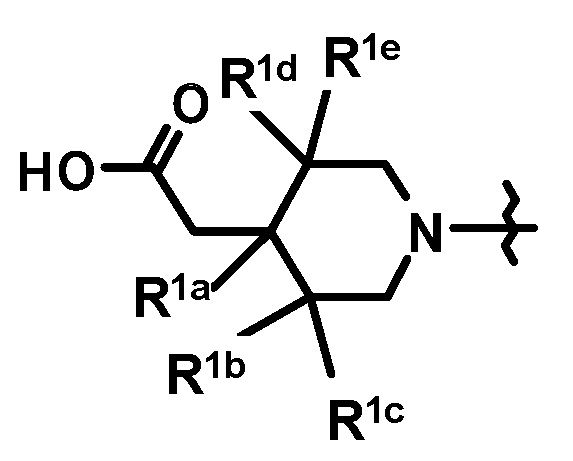 ;
;
(**) и (***) имеют такие же значения, как описано в пункте [2];
R1a, R1b, R1c, R1d и R1e, каждый независимо, представляет собой любой из атома водорода, метила или метокси;
или его фармацевтически приемлемая соль.
[4] Соединение по приведенному выше пункту [1], где кольцо B представлено следующей формулой:
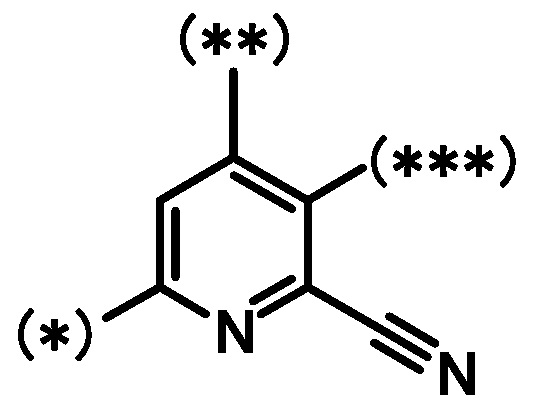
где (*), (**) и (***) имеют такие же значения, как описано в пункте [1];
или его фармацевтически приемлемая соль.
[5] Соединение по приведенному выше пункту [4], где R1 представляет собой метил, и n равен 0 или 1, или его фармацевтически приемлемая соль.
[6] Соединение по приведенному выше пункту [2], представленное следующей формулой:
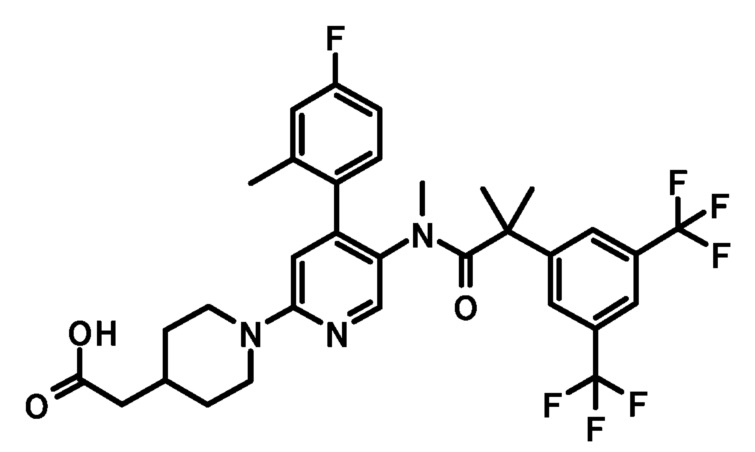
или его фармацевтически приемлемая соль.
[7] Соединение по приведенному выше пункту [2], представленное следующей формулой:
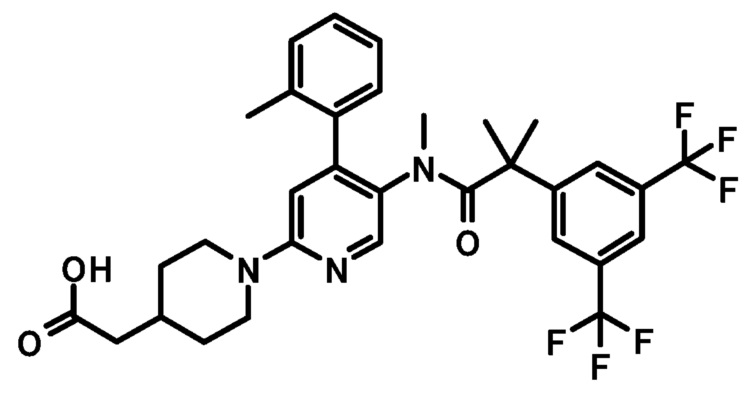
или его фармацевтически приемлемая соль.
[8] Соединение по приведенному выше пункту [2], представленное следующей формулой:
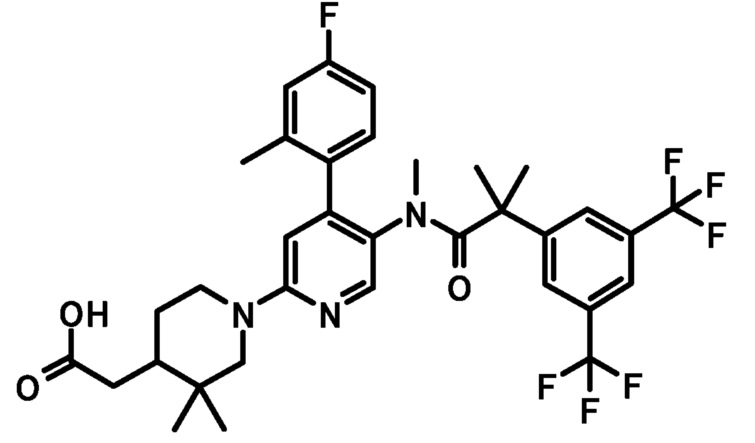
или его фармацевтически приемлемая соль.
[9] Соединение по приведенному выше пункту [2], представленное следующей формулой:
или его фармацевтически приемлемая соль.
[10] Соединение по приведенному выше пункту [1], представленное следующей формулой:
или его фармацевтически приемлемая соль.
[11] Фармацевтическая композиция, содержащая в качестве активного ингредиента соединение по любому из пунктов [1]-[10] или его фармацевтически приемлемую соль.
[12] Фармацевтическая композиция по приведенному выше пункту [11] для применения при предупреждении или лечении индуцированной противораковой химиотерапией тошноты или рвоты.
Эффект настоящего изобретения
[0014] Соединения по настоящему изобретению обладают великолепной антагонистической активностью в отношении рецептора NK1. И ингибирующее действие на CYP3A4 соединений по настоящему изобретению ослаблено по сравнению с апрепитантом. Кроме того, предпочтительные соединения настоящего изобретения превосходны в способности транспортировки в центральную нервную систему и длительности лечебного эффекта.
Поэтому, соединения по настоящему изобретению или их фармацевтически приемлемые соли полезны в качестве средства для предупреждения или лечения индуцированной противораковой химиотерапией тошноты или рвоты.
Краткое описание фигур
[0015] [Фигура 1] На фиг.1 показано воздействие на индуцированный цисплатином острый и задержанный рвотный ответ. На фигуре каждая линейная диаграмма показывает значения для контрольной группы (контроль), группы внутривенного ведения 3 мг/кг соединения примера 1 (пример № 1, 3 мг/кг, вв), и группы перорального введения 1 мг/кг соединения примера 1 (пример № 1, 1 мг/кг, по) в фазе обострения (острый), и значения для контрольной группы, внутривенного ведения 3 мг/кг соединения примера 1 (пример № 1 3 мг/кг, вв) и группы перорального введения 1 мг/кг соединения примера 1 (пример № 1, 1 мг/кг, по) в стадии задержки слева, соответственно. На вертикальных осях показано число рвотных позывов и рвоты (позыв к рвоте+рвота) (среднее значение+стандартная ошибка для 5 примеров контрольной группы, среднее значение+стандартная ошибка для 5 примеров группы внутривенного введения и среднее значение+стандартная ошибка для 5 примеров группы перорального введения).
Способ осуществления настоящего изобретения
[0016] В настоящем изобретении каждый термин имеет соответствующее значение, если специально не указано иное.
[0017] Термин “C1-6алкил” означает алкильную группу с прямой или разветвленной цепью, имеющую 1-6 атомов углерода, например, проиллюстрированную как метил, этил, пропил, изопропил, бутил, изобутил, втор-бутил, трет-бутил.
Термин “C1-6алкокси” означает алкоксигруппу с прямой или разветвленной цепью, имеющую 1-6 атомов углерода, например, проиллюстрированную как метокси, этокси, пропокси, изопропокси.
[0018] В соединении, представленном формулой (I) настоящего изобретения, символ "R1" обозначает заместитель пиперидинового кольца.
[0019] Дополнительно настоящее изобретение проиллюстрировано ниже.
[0020] В случае, когда соединения, представленные формулой (I) настоящего изобретения, содержат один или более асимметрических атомов углерода, все стереоизомеры в R- или S-конфигурации на каждом из асимметричных атомов углерода и их смеси включены в настоящее изобретение. В таких случаях, рацемические соединения, рацемические смеси, индивидуальные энантиомеры и смеси диастереоизомеров включены в объем настоящего изобретения. В случае, когда соединения, представленные формулой (I) настоящего изобретения, имеют цис-транс изомеры, все цис-транс изомеры включены в настоящее изобретение.
[0021] В настоящем изобретении определение стереохимии может быть также осуществлено согласно известным в данной области методам. Например, см. также "Tokuron NMR rittai kagaku", Kodansha, 2012, p. 59.
[0022] Соединение, представленное формулой (I), настоящего изобретения может быть преобразовано в его фармацевтически приемлемые соли согласно общему методу. В качестве таких солей могут быть указаны кислотно-аддитивные соли и соли присоединения оснований.
[0023] В качестве кислотно-аддитивных солей могут быть проиллюстрированы соли с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, йодистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и подобные, соли с органическими кислотами, такими как муравьиная кислота, уксусная кислота, трифторуксусная кислота, метансульфоновая кислота, бензолсульфоновая кислота, п-толуолсульфоновая кислота, пропионовая кислота, лимонная кислота, янтарная кислота, винная кислота, фумаровая кислота, масляная кислота, щавелевая кислота, малоновая кислота, малеиновая кислота, молочная кислота, яблочная кислота, угольная кислота, бензойная кислота, глутаминовая кислота, аспарагиновая кислота и подобные.
[0024] В качестве солей с основаниями могут быть соли, образованные с неорганическим основанием, такие как соль лития, соль натрия, соль калия, соль кальция, соль магния и подобные, и соли, образованные с органическим основанием, такие как N-метил-D-глутамин, N,N'-дибензилэтилендиамин, триэтиламин, пиперидин, морфолин, пирролидин, аргинин, лизин, холин и подобные.
[0025] В настоящем изобретении фармацевтически приемлемая соль также включает ее сольват с фармацевтически приемлемым растворителем, таким как вода, этанол или подобные.
[0026] В варианте осуществления соединения, представленного формулой (I) настоящего изобретения, n равен 0, 1 или 2.
[0027] Соединение, представленное формулой (I) настоящего изобретения, также может быть получено, например, способом, описанным ниже, или аналогичным ему способом, или способом, описанным в литературе, или подобным ему способом.
Схема 1
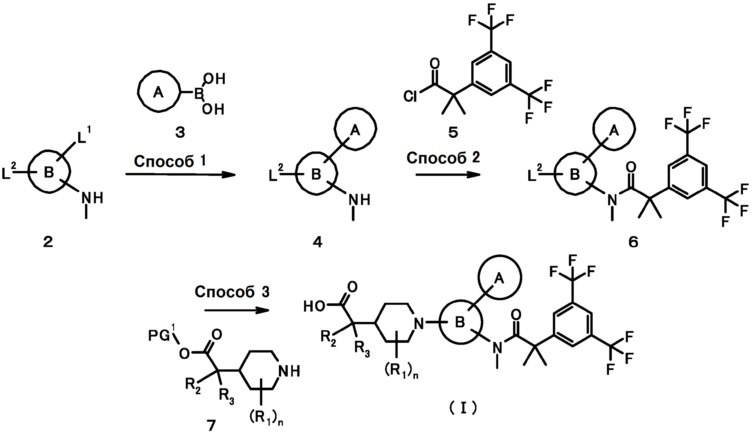
В данных формулах L1 и L2, каждый независимо, представляет собой уходящую группу, такую как атом хлора, атом брома, атом йода, трифторметансульфонилоксигруппу или подобную, PG1 представляет собой защитную группу, и кольцо A, кольцо B, R1, R2, R3 и n имеют такие же значения, как определено выше.
[0028] Способ 1
Соединение (4) может быть получено путем проведения реакции сочетания соединения (2) с соединением (3) в инертном растворителе в присутствии основания и палладиевого катализатора.
[0029] Способ 2
Соединение (6) также может быть получено путем проведения реакции конденсации соединения (4) с соединением (10) в инертном растворителе в присутствии основания.
[0030] Способ 3
Соединение (I) также может быть получено путем подвергания соединения (6) взаимодействию с соединением (7) и проведения удаления защиты в инертном растворителе в присутствии или отсутствии основания.
В качестве инертного растворителя, например, может быть указан N,N-диметилформамид, N-метилпирролидон, диметилсульфоксид, простой диэтиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол и их смешанные растворители. В качестве основания может быть указан карбонат калия, карбонат натрия, карбонат цезия, гидроксид натрия, гидроксид калия, гидроксид лития, фторид калия, фторид цезия, триэтиламин, пиридин, N,N-диизопропилэтиламин, 2,6-лютидин и 1,8-диазабицикло[5,4,0]-7-ундецен. Температура реакции обычно составляет от 0°C до температуры кипячения с обратным холодильником. Время реакции обычно составляет от 30 минут до 7 дней, варьируемое в зависимости от используемого исходного вещества, растворителя и температуры реакции, и тому подобное. Описанная выше реакция может быть проведена с использованием микроволнового реактора (Biotage). Когда используется микроволновый реактор, данную реакцию проводят при давлении в интервале: 1-30 бар, мощности в диапазоне: 1-400 В, реакционной температуре: от комнатной температуры до 300°C, и времени реакции: от минуты до 1 дня, варьируемого в зависимости от используемого исходного вещества, растворителя и методики.
[0031]
Схема 2
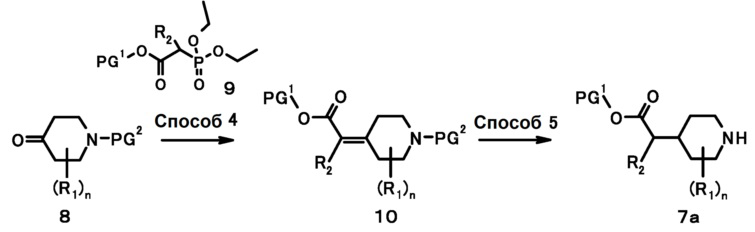
В данных формулах PG1 и PG2 представляют собой защитную группу, и R1, R2 и n имеют такие же значения, как определено выше.
[0032] Способ 4
Соединение (10) также может быть получено путем проведения реакции олефинирования соединения (9) с соединением (8) в инертном растворителе в присутствии основания. В качестве инертного растворителя, например, может быть указан N,N-диметилформамид, N-метилпирролидон, диметилсульфоксид, простой диэтиловый эфир, тетрагидрофуран, 1,4-диоксан, 1,2-диметоксиэтан, бензол, толуол, ксилол и их смешанные растворители. В качестве основания может быть указан гидрид натрия, метоксид натрия, карбонат калия, карбонат цезия, трет-бутоксид калия, триэтиламин, N,N-диизопропилэтиламин и 1,8-диазабицикло[5,4,0]-7-ундецен. Температура реакции обычно составляет от -78°C до температуры кипячения с обратным холодильником. Время реакции обычно составляет от 30 минут до 7 дней, варьируемое в зависимости от используемого исходного вещества, растворителя и реакционной температуры или подобного. Данная реакция может быть проведена с использованием микроволнового реактора (Biotage).
[0033] Способ 5
Соединение (7a) также может быть получено путем проведения восстановления олефинов, такого как метод каталитического восстановления соединения (10) и т.д., и проведения удаления защиты. Метод каталитического восстановления может быть проведен, например, путем подвергания соединения (10) взаимодействию с использованием катализатора в атмосфере водорода в растворителе. В качестве растворителя, например, может быть указан метанол, этанол, этилацетат, тетрагидрофуран, уксусная кислота и тому подобное. В качестве катализатора, например, может быть указан палладий на порошкообразном углероде, родий на порошкообразном углероде, платина на порошкообразном углероде, платина на порошкообразном углероде с нанесенным ванадием. Температура реакции обычно составляет от комнатной температуры до температуры кипячения с обратным холодильником. Время реакции обычно составляет от 30 минут до 7 дней, варьируемое в зависимости от используемого исходного вещества, растворителя и температуры реакции или подобного.
[0034] Приведенные выше схемы являются иллюстративными для получения соединений, представленных формулой (I), настоящего изобретения и синтеза их промежуточных продуктов. Приведенные выше схемы могут быть изменены и модифицированы в схемы, которые могут быть легко истолкованы специалистом в данной области техники.
[0035] На приведенных выше схемах, когда защитная группа является необходимой на основе изменения функциональной группы, операции введения и удаления защиты также могут проводиться при необходимости в комбинации, согласно общему способу.
[0036] Соединения, представленные формулой (I), настоящего изобретения и их промежуточные соединения также могут быть выделены и очищены, если требуется, согласно общепринятым методам выделения и очистки, хорошо известным специалистам в соответствующей области техники, таким как экстракция растворителем, кристаллизация, перекристаллизация, хроматография, препаративная высокоэффективная жидкостная хроматография или тому подобное.
[0037] Соединения по настоящему изобретению обладают превосходной антагонистической активностью в отношении рецептора NK1 и, таким образом, могут быть использованы в качестве средства для предупреждения или лечения различных заболеваний, опосредованных рецептором NK1. Например, соединения по настоящему изобретению полезны в качестве антиэметика, особенно полезны в качестве превентивного средства против желудочно-кишечного симптома, индуцированного противораковой химиотерапией (например, цисплатином) (например, тошноты и рвоты). Предпочтительно, соединения настоящего изобретения не только полезны при острой, вызванной противораковой химиотерапией тошноте и рвоте, но также при задержанной, вызванной противораковой химиотерапией тошноте и рвоте.
[0038] В одном из вариантов осуществления соединения по настоящему изобретению обладают превосходной антагонистической активностью в отношении рецептора NK1 и, таким образом, также могут использоваться в качестве средства для предупреждения послеоперационной тошноты и рвоты (PONV), тошноты и рвоты, связанных с лучевой терапией, морфин-индуцированной рвоты или при укачивании, и при лечении шизофрении, социальной фобии, тревоги и депрессии, алкоголизма, синдрома раздраженного кишечника, язвенного колита, кашля, астмы, атопического дерматита, псориаза, зуда, боли, мигрени, шума в ушах, доброкачественной гиперплазии предстательной железы, гиперактивного мочевого пузыря или недержания мочи.
[0039] Фармацевтические композиции настоящего изобретения могут быть введены в различных дозированных формах в зависимости от их применения. Такими дозированными формами, например, являются порошки, гранулы, тонкие гранулы, сухие сиропы, таблетки, капсулы, инъекции, жидкости, мази, суппозитории и припарки, которые вводятся перорально или парентерально.
[0040] Фармацевтические композиции настоящего изобретения могут быть получены с использованием соединения, представленного формулой (I), или его фармацевтически приемлемой соли и по меньшей мере одной фармацевтической добавки. Такие фармацевтические композиции могут быть сформулированы смешиванием, разведением или растворением с подходящими фармацевтическими добавками, такими как эксципиенты, дезинтегранты, связующие, лубриканты, разбавители, буферы, тонизирующие агенты, консерванты, смачивание агенты, эмульгирующие агенты, диспергирующие агенты, стабилизирующие агенты, солюбилизирующие агенты и тому подобное, согласно общепринятой методике составления композиций, в зависимости от их дозированных форм.
[0041] Когда фармацевтическая композиция настоящего изобретения используется для предупреждения или лечения, доза соединения, представленного формулой (I), или его фармацевтически приемлемой соли в качестве активного ингредиента соответствующим образом зависит от возраста, пола, массы тела, сложности заболевания и лечения каждого пациента, и тому подобное. Дозировка для взрослых может составлять диапазон, например, 1-1000 мг в день, 0,1-500 мг в день, 0,1-100 мг в день или 0,1-50 мг в день в случае перорального введения, и ежедневная доза может быть разделена на одноразовое, двухразовое, трехразовое или четырехразовое введение в сутки.
[0042] Когда фармацевтическая композиция настоящего изобретения используется для предупреждения индуцированной противораковой химиотерапией тошноты или рвоты, такая фармацевтическая композиция может быть также введена перед введением противоопухолевых средств. Например, данная фармацевтическая композиция может быть введена непосредственно за полтора часа до проведения химиотерапии и спустя два дня, фармацевтическая композиция также может вводиться в первой половине дня.
[0043] В одном из вариантов осуществления соединение, представленное формулой (I) настоящего изобретения, или его фармацевтически приемлемая соль также может быть использовано в комбинации с другим лекарственным средством, отличным от антагониста рецептора NK1. В качестве таких других лекарственных средств, используемых в комбинации, можно указать, например, кортикостероид и противорвотное средство-антагонист рецептора 5-HT3.
[0044] Когда соединение, представленное формулой (I) настоящего изобретения, или его фармацевтически приемлемая соль используется в комбинации с другим лекарственным средством, оно может быть введено в виде состава, сформулированного совместно с другими активными ингредиентами, или в виде составов, в каждом из которых отдельно сформулирован каждый из активных ингредиентов. В случае отдельно сформулированных составов, эти составы могут быть введены раздельно или одновременно.
Кроме того, доза соединения, представленного формулой (I) настоящего изобретения, может быть снижена в зависимости от дозы другого лекарственного средства, используемого в комбинации.
ПРИМЕРЫ
[0045] Настоящее изобретение далее проиллюстрировано более подробно ссылочными примерами, примерами и примерами тестирования. Однако настоящее изобретение не ограничивается ими.
[0046] Ссылочный пример 1
Этиловый эфир (3-метилпиперидин-4-ил)уксусной кислоты
К суспензии гидрида натрия (60%, 0,17 г) в тетрагидрофуране (5 мл) добавляли триэтилфосфоноацетат (1,04 г) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли раствор трет-бутилового эфира 3-метил-4-оксопиперидин-1-карбоновой кислоты (0,50 г) в тетрагидрофуране (5 мл) при комнатной температуре, и полученную смесь перемешивали при 50°C в течение 2,5 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный остаток очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат), с получением трет-бутилового эфира 4-этоксикарбонилметилен-3-метилпиперидин-1-карбоновой кислоты (0,65 г). К раствору полученного соединения (0,65 г) в этаноле (12 мл) добавляли 10% палладий на углероде (250 мг, влажный) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в атмосфере газообразного водорода в течение 18 часов. Реакционную смесь разбавляли этилацетатом, и полученную в результате смесь перемешивали при комнатной температуре в течение 15 минут. Реакционную смесь фильтровали через рыхлый слой целита, и фильтрат концентрировали при пониженном давлении, с получением трет-бутилового эфира 4-этоксикарбонилметил-3-метилпиперидин-1-карбоновой кислоты (0,64 г). К раствору полученного соединения (0,64 г) в этилацетате (10 мл) добавляли 4 моль/л хлористоводородную кислоту (этилацетатный раствор, 10 мл) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 39 часов. Полученную реакционную смесь концентрировали при пониженном давлении, и к полученному остатку добавляли насыщенный водный раствор гидрокарбоната натрия. Полученную в результате смесь дважды экстрагировали смешанным растворителем дихлорметан-изопропиловый спирт (дихлорметан/изопропиловый спирт=3/1). Объединенный органический слой сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,39 г).
[0047] Ссылочные примеры 2 и 3
Соединения ссылочных примеров 2-3 получали по методике, аналогично описанной в ссылочном примере 1, с использованием соответствующих исходных веществ.
[0048] Ссылочный пример 4
трет-Бутиловый эфир (6-хлор-4-йодпиридин-3-ил)карбаминовой кислоты
К раствору трет-бутилового эфира (6-хлорпиридин-3-ил)карбаминовой кислоты (5,0 г) и N,N,N',N'-тетраметилэтан-1,2-диамина (7,7 г) в простом диэтиловом эфире (120 мл) по каплям добавляли н-бутиллитий (2,65 моль/л н-гексановый раствор, 25 мл) при -78°C в атмосфере аргона. После перемешивания смеси при -10°C в течение 2 часов, по каплям добавляли раствор йодина (11,4 г) в простом диэтиловом эфире (40 мл) при -78°C. Полученную в результате смесь перемешивали при комнатной температуре в течение 1 дня. К реакционной смеси добавляли насыщенный водный раствор хлористого аммония, и полученную в результате смесь экстрагировали диэтиловым эфиром. Органический слой промывали 10% водным пиросульфитом натрия и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат), с получением указанного в заголовке соединения (2,59 г).
[0049] Ссылочный пример 5
трет-Бутиловый эфир (6-хлор-4-йодпиридин-3-ил)метилкарбаминовой кислоты
К раствору трет-бутилового эфира (6-хлор-4-йодпиридин-3-ил)карбаминовой кислоты (2,59 г) в N,N-диметилформамиде (30 мл) добавляли гидрид натрия (60%, 0,32 г) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К данной смеси добавляли йодметан (2,60 г) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат), с получением указанного в заголовке соединения (2,66 г).
[0050] Ссылочный пример 6
(6-Хлор-4-йодпиридин-3-ил)метиламин
К раствору трет-бутилового эфира (6-хлор-4-йодпиридин-3-ил)метилкарбаминовой кислоты (2,66 г) в дихлорметане (10 мл) добавляли трифторуксусную кислоту (8,23 г) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Полученную реакционную смесь концентрировали при пониженном давлении, и к полученному остатку добавляли насыщенный водный раствор карбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (1,89 г).
[0051] Ссылочный пример 7
[6-Хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]метиламин
К смешанному раствору (6-хлор-4-йодпиридин-3-ил)метиламина (1,89 г) и 4-фтор-2-метилфенилбороновой кислоты (1,30 г) в смеси 1,2-диметоксиэтан (20 мл)-вода (20 мл) добавляли ацетат палладия(II) (0,16 г), трифенилфосфин (0,37 г) и карбонат натрия (3,73 г) при комнатной температуре, и полученную смесь перемешивали при 90°C в течение ночи. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат), с получением указанного в заголовке соединения (1,56 г).
[0052] Ссылочный пример 8
(6-Хлор-4-орто-толилпиридин-3-ил)метиламин
К смешанному раствору (6-хлор-4-йодпиридин-3-ил)метиламина (0,70 г) и 2-метилфенилбороновой кислоты (0,42 г) в смеси 1,2-диметоксиэтан (10 мл)-вода (10 мл) добавляли ацетат палладия(II) (0,058 г), трифенилфосфин (0,14 г) и карбонат натрия (1,38 г) при комнатной температуре, и полученную смесь перемешивали при 90°C в течение ночи. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат), с получением указанного в заголовке соединения (0,54 г).
[0053] Ссылочный пример 9
2-(3,5-Бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламид
К раствору 2-(3,5-бистрифторметилфенил)-2-метилпропионовой кислоты (0,66 г) в дихлорметане (10 мл) добавляли оксалилхлорид (0,56 г) и N,N-диметилформамид (2 капли) при комнатной температуре, и полученную смесь перемешивали при этой же температуре в течение 1 часа. Полученную реакционную смесь концентрировали при пониженном давлении до получения остатка. В атмосфере аргона, к раствору [6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]метиламина (0,50 г) в тетрагидрофуране (10 мл) добавляли бис(триметилсилил)амид калия (0,500 моль/л толуольный раствор, 5,0 мл) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси по каплям добавляли раствор полученного выше остатка в тетрагидрофуране (5 мл) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. К реакционной смеси добавляли 1,0 моль/л водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат), с получением указанного в заголовке соединения (1,03 г).
[0054] Ссылочный пример 10
2-(3,5-Бистрифторметилфенил)-N-(6-хлор-4-орто-толилпиридин-3-ил)-N-метилизобутиламид
К раствору 2-(3,5-бистрифторметилфенил)-2-метилпропионовой кислоты (0,77 г) в дихлорметане (12 мл) добавляли оксалилхлорид (0,65 г) и N,N-диметилформамид (2 капли) при комнатной температуре, и полученную смесь перемешивали при этой же температуре в течение 1 часа. Полученную реакционную смесь концентрировали при пониженном давлении до получения остатка. В атмосфере аргона к раствору (6-хлор-4-орто-толилпиридин-3-ил)метиламина (0,54 г) в тетрагидрофуране (12 мл) добавляли бис(триметилсилил)амид калия (0,500 моль/л толуольный раствор, 6,0 мл) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси по каплям добавляли раствор полученного выше остатка в тетрагидрофуране (6 мл) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли 1,0 моль/л водный раствор гидрокарбоната натрия, и полученную смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат), с получением указанного в заголовке соединения (1,0 г).
[0055] Ссылочный пример 11
Этиловый эфир [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты
Суспензию 2-(3,5-бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,79 г), этилового эфира (пиперидин-4-ил)уксусной кислоты (0,38 г) и карбоната калия (0,41 г) в диметилсульфоксиде (4,5 мл) перемешивали при 180°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат=100/0-60/40). Полученное вещество очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-60/40), с получением указанного в заголовке соединения (0,38 г).
[0056] Ссылочный пример 12
Этиловый эфир (5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-орто-толил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил)уксусной кислоты
Суспензию 2-(3,5-бистрифторметилфенил)-N-(6-хлор-4-орто-толилпиридин-3-ил)-N-метилизобутиламида (0,50 г), этилового эфира (пиперидин-4-ил)уксусной кислоты (0,25 г) и карбоната калия (0,27 г) в диметилсульфоксиде (3,0 мл) перемешивали при 180°C при микроволновом облучении в течение 2 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-50/50), с получением указанного в заголовке соединения (0,35 г).
[0057] Ссылочный пример 13
Этиловый эфир 5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,3-диметил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты
Раствор 2-(3,5-бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,05 г) и этилового эфира (3,3-диметилпиперидин-4-ил)уксусной кислоты (0,094 г) в 1-метил-2-пирролидоне (0,5 мл) перемешивали при 180°C при микроволновом облучении в течение 3 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-50/50), с получением указанного в заголовке соединения (0,043 г).
[0058] Ссылочный пример 14
Этиловый эфир 5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты
Раствор 2-(3,5-бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,05 г) и этилового эфира (3-метилпиперидин-4-ил)уксусной кислоты (0,087 г) в 1-метил-2-пирролидоне (0,5 мл) перемешивали при 180°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-50/50), с получением указанного в заголовке соединения (0,040 г).
[0059] Ссылочный пример 15
Этиловый эфир [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3-метоксил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты
Раствор 2-(3,5-бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,05 г) и этилового эфира (3-метоксипиперидин-4-ил)уксусной кислоты (0,094 г) в 1-метил-2-пирролидоне (0,5 мл) перемешивали при 180°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-60/40), с получением указанного в заголовке соединения (0,025 г).
[0060] Ссылочный пример 16
Этиловый эфир 2-метил-2-пиперидин-4-илпропионовой кислоты
К раствору диизопропиламида лития (1,09 моль/л тетрагидрофуран/н-гексановый раствор, 30,0 мл) в тетрагидрофуране (40 мл) по каплям добавляли раствор этилизобутилата в тетрагидрофуране (20 мл) при -78°C в атмосфере аргона, и полученную смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси добавляли раствор бензилового эфира 4-оксопиперидин-1-карбоновой кислоты (5,83 г) в тетрагидрофуране (50 мл) при -78°C, и полученную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор хлористого аммония, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=90/10-10/90), с получением бензилового эфира 4-(1-этоксикарбонил-1-метилэтил)-4-гидроксипиперидин-1-карбоновой кислоты (6,13 г). К раствору полученного соединения (6,13 г) в толуоле (100 мл) добавляли внутреннюю соль (метоксикарбонилсульфамоил)триэтиламмонийгидроксида (5,00 г) при комнатной температуре, и полученную смесь перемешивали при 90°C в течение 2 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия, водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-50/50), с получением бензилового эфира 4-(1-этоксикарбонил-1-метилэтил)-3,6-дигидро-2H-пиридин-1-карбоновой кислоты (5,85 г). В атмосфере газообразного водорода, смесь полученного соединения (5,85 г) и катализатора Перлмана (0,600 г) в метаноле (65 мл) и этилацетате (65 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через рыхлый слой целита, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения (3,18 г).
[0061] Ссылочный пример 17
Этиловый эфир 2-пиперидин-4-илпропионовой кислоты
К суспензии гидрида натрия (60%, 0,78 г) в N,N-диметилформамиде (20 мл) добавляли этиловый эфир 2-(диэтоксифосфорил)пропионовой кислоты (4.44 г) при охлаждении льдом, и полученную смесь перемешивали при этой же температуре в течение 30 минут. К реакционной смеси добавляли раствор бензилового эфира 4-оксопиперидин-1-карбоновой кислоты (3,50 г) в N,N-диметилформамиде (10 мл) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 1 часа. К реакционной смеси добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-50/50), с получением бензилового эфира 4-(1-этоксикарбонилэтилиден)пиперидинe-1-карбоновой кислоты (4,60 г). В атмосфере газообразного водорода, смесь полученного соединения (4,60 г) и 10% палладия на углероде (500 мг, влажный) в метаноле (50 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь фильтровали через рыхлый слой целита, и фильтрат концентрировали при пониженном давлении, с получением указанного в заголовке соединения (2,85 г).
[0062] Ссылочный пример 18
Этиловый эфир (4-метилпиперидин-4-ил)уксусной кислоты
К раствору трет-бутилового эфира 4-гидроксиметил-4-метилпиперидин-1-карбоновой кислоты (0,67 г) и триэтиламина (0,44 г) в дихлорметане (15 мл) добавляли метансульфонилхлорид (0,40 г) при комнатной температуре, и полученную смесь перемешивали при этой же температуре в течение 3 часов. Реакционную смесь промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, с получением трет-бутилового эфира 4-метансульфонилоксиметил-4-метилпиперидин-1-карбоновой кислоты (0,85 г). К раствору полученного соединения (0,85 г) в N,N-диметилформамиде (6 мл) добавляли цианид натрия (0,27 г) при комнатной температуре, и полученную смесь перемешивали при 50°C в течение 5 часов и при 80°C в течение 13 часов. К реакционной смеси добавляли цианид натрия (0,22 г) и йодид натрия (0,02 г), и полученную смесь перемешивали при 120°C в течение 8 часов. К реакционной смеси добавляли цианид натрия (0,72 г), и полученную смесь перемешивали при 140°C в течение 17 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали смешанным растворителем из н-гексана и этилацетата (смесь н-гексан/этилацетат=1/4). Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=80/20-50/50), с получением трет-бутилового эфира 4-цианометил-4-метилпиперидин-1-карбоновой кислоты (0,53 г). Смесь полученного соединения (0,53 г) и концентрированной хлористоводородной кислоты (12 мл) перемешивали при 110°C в течение 47 часов. Полученную реакционную смесь охлаждали до комнатной температуры, и к данной смеси добавляли воду (24 мл), водный раствор гидроксида натрия (2,0 моль/л, 45 мл) и ди-трет-бутилдикарбонат (0,51 г), и полученную в результате смесь перемешивали при комнатной температуре в течение 21 часа. Растворитель удаляли при пониженном давлении, и к полученному остатку добавляли воду и хлористоводородную кислоту (2,0 моль/л, 3 мл), и полученную в результате смесь экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении, с получением трет-бутилового эфира 4-карбоксиметил-4-метилпиперидин-1-карбоновой кислоты (0,34 г). К раствору полученного соединения (0,34 г) в N,N-диметилформамиде (5 мл) добавляли карбонат калия (0,27 г) и этилйодид (0,41 г) при комнатной температуре, и полученную в результате смесь перемешивали при этой же температуре в течение 22 часов. К реакционной смеси добавляли воду, и полученную в результате смесь экстрагировали диэтиловым эфиром. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-60/40), с получением трет-бутилового эфира 4-этоксикарбонилметил-4-метилпиперидин-1-карбоновой кислоты (0,28 г). К раствору полученного соединения (0,28 г) в 1,4-диоксан (5 мл) добавляли хлористый водород (4,0 моль/л 1,4-диоксаноый раствор, 5,0 мл) при комнатной температуре, и полученную смесь перемешивали при этой же температуре в течение 26 часов. Растворитель удаляли при пониженном давлении, и к полученному остатку добавляли насыщенный водный гидрокарбонат натрия, и полученную в результате смесь дважды экстрагировали смешанным растворителем дихлорметан/изопропиловый спирт (дихлорметан/изопропиловый спирт=3/1). Объединенный органический слой сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,17 г).
[0063] Ссылочный пример 19
6-Хлор-3-нитропиридин-2-карбонитрил
К раствору 2,6-дихлор-3-нитропиридина (2,50 г) в N-метилпирролидоне (25 мл) добавляли цианид меди(I) (2,32 г) при комнатной температуре, и полученную смесь перемешивали при 180°C в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры, и к данной смеси добавляли этилацетат и воду. Полученную смесь фильтровали через рыхлый слой целита. Фильтрат промывали насыщенным раствором соли, и отделенный водный слой повторно экстрагировали этилацетатом. Объединенный органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=90/10-70/30), с получением указанного в заголовке соединения (0,90 г).
[0064] Ссылочный пример 20
3-Амино-6-хлорпиридин-2-карбонитрил
К раствору 6-хлор-3-нитропиридин-2-карбонитрила (0,32 г) и концентрированной хлористоводородной кислоты (1,2 мл) в этаноле (3,6 мл) добавляли порошковое железо (0,34 г) при комнатной температуре, и полученную смесь нагревали при кипячении с обратным холодильником в течение 30 минут. Полученную реакционную смесь охлаждали до комнатной температуры, и подщелачивали добавлением насыщенного водного раствора гидрокарбоната натрия. К данной смеси добавляли этилацетат, и полученную в результате смесь фильтровали через рыхлый слой целита, и фильтрат экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, сушили над безводным сульфатом магния. Растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,24 г).
[0065] Ссылочный пример 21
3-Амино-4-бром-6-хлорпиридин-2-карбонитрил
К раствору 3-амино-6-хлорпиридин-2-карбонитрила (0,24 г) в N,N-диметилформамиде (8 мл) добавляли N-бромсукцинимид (0,37 г) при комнатной температуре, и полученную смесь перемешивали при этой же температуре в течение ночи. К реакционной смеси добавляли насыщенный водный раствор тиосульфата натрия, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат=75/25-50/50), с получением указанного в заголовке соединения (0,30 г).
[0066] Ссылочный пример 22
3-Амино-6-хлор-4-(4-фтор-2-метилфенил)пиридин-2-карбонитрил
Смесь 3-амино-4-бром-6-хлорпиридин-2-карбонитрила (0,15 г), 4-фтор-2-метилфенилбороновой кислоты (0,08 г), тетракис(трифенилфосфин)палладия(0) (0,07 г), карбоната натрия (0,20 г), 1,2-диметоксиэтана (3,2 мл) и воды (0,8 мл) перемешивали при 100°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным водным раствором гидрокарбоната натрия и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат=50/50-0/100), с получением указанного в заголовке соединения (0,14 г).
[0067] Ссылочный пример 23
2-(3,5-Бистрифторметилфенил)-N-[6-хлор-2-циано-4-(4-фтор-2-метилфенил)пиридин-3-ил]изобутиламид
К раствору 2-(3,5-бистрифторметилфенил)-2-метилпропионовой кислоты (0,31 г) в дихлорметане (2,6 мл) добавляли оксалилхлорид (0,26 г) и N,N-диметилформамид (2 капли) при комнатной температуре, и полученную смесь перемешивали при этой же температуре в течение 1 часа. Полученную реакционную смесь концентрировали при пониженном давлении до получения остатка. К раствору 3-амино-6-хлор-4-(4-фтор-2-метилфенил)пиридин-2-карбонитрила (0,14 г) в тетрагидрофуране (5 мл) добавляли бис(триметилсилил)амид натрия (1,0 моль/л тетрагидрофурановый раствор, 1,1 мл) при охлаждении льдом, и полученную смесь перемешивали при этой же температуре в течение 30 минут. К реакционной смеси по каплям добавляли раствор полученного выше остатка в тетрагидрофуране (2,0 мл) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение 30 минут. К реакционной смеси добавляли насыщенный водный раствор гидрокарбоната натрия, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат=85/15-40/60), с получением указанного в заголовке соединения (0,21 г).
[0068] Ссылочный пример 24
2-(3,5-Бистрифторметилфенил)-N-[6-хлор-2-циано-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламид
К раствору 2-(3,5-бистрифторметилфенил)-N-[6-хлор-2-циано-4-(4-фтор-2-метилфенил)пиридин-3-ил]изобутиламида (0,21 г) в N,N-диметилформамиде (2,4 мл) добавляли гидрид натрия (60%, 0,018 г) при охлаждении льдом, и полученную смесь перемешивали при этой же температуре в течение 5 минут. К реакционной смеси добавляли йодметан (0,11 г) при охлаждении льдом, и полученную смесь перемешивали при комнатной температуре в течение ночи. К реакционной смеси добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=90/10-50/50), с получением указанного в заголовке соединения (0,09 г).
[0069] Ссылочный пример 25
Этиловый эфир [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты
Суспензию 2-(3,5-бистрифторметилфенил)-N-[6-хлор-2-циано-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,03 г), этилового эфира пиперидин-4-илуксусной кислоты (0,05 г) и карбоната калия (0,02 г) в диметилсульфоксиде (1,0 мл) перемешивали при 100°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат=80/20-20/80), с получением указанного в заголовке соединения (0,02 г).
[0070] Ссылочный пример 26
Этиловый эфир 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]-2-метилпропионовой кислоты
Смесь 2-(3,5-бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,15 г), этилового эфира 2-метил-2-пиперидин-4-илпропионовой кислоты (0,28 г) и N-метилпирролидона (1,5 мл) перемешивали при 190°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-60/40), с получением указанного в заголовке соединения (0,14 г).
[0071] Ссылочный пример 27
Этиловый эфир 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовой кислоты
Суспензию 2-(3,5-бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,48 г), этилового эфира 2-пиперидин-4-илпропионовой кислоты (0,83 г) и карбоната калия (0,25 г) в N-метилпирролидоне (3,6 мл) перемешивали при 190°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-50/50), с получением указанного в заголовке соединения (0,51 г).
[0072] Ссылочные примеры 28 и 29
N-[4-[(S)-2-((S)-4-Бензил-2-оксооксазолидин-3-ил)-1-метил-2-оксоэтил]-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-5'-ил]-2-(3,5-бистрифторметилфенил)-N-метилизобутиламид (ссылочный пример 28) и
N-[4-[(R)-2-((S)-4-бензил-2-оксооксазолидин-3-ил)-1-метил-2-оксоэтил]-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-5'-ил]-2-(3,5-бистрифторметилфенил)-N-метилизобутиламид (ссылочный пример 29)
Смесь этилового эфира 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовой кислоты (0,50 г), водного раствора гидроксида натрия (1,0 моль/л, 2,0 мл), тетрагидрофурана (2 мл) и метанола (6 мл) перемешивали при 140°C при микроволновом облучении в течение 2 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли хлористоводородную кислоту (1,0 моль/л, 3,0 мл). Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=50/50-0/100), с получением 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовой кислоты (0,42 г). К раствору (S)-4-бензилоксазолидин-2-она (0,06 г) в тетрагидрофуране (4 мл) по каплям добавляли н-бутиллитий (2,65 моль/л в н-гексановом растворе, 0,12 мл) при -78°C в атмосфере аргона, и полученную смесь перемешивали при этой же температуре в течение 30 минут, с получением литиевого раствора (S)-4-бензилоксазолидин-2-она. В атмосфере аргона к раствору 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовой кислоты (0,20 г) и триэтиламина (0,04 г) в простом диэтиловом эфире (4 мл) добавляли пивалоилхлорид (0,04 г) при охлаждении льдом, и полученную смесь перемешивали при этой же температуре в течение 1 часа. К реакционной смеси по каплям добавляли полученный выше литиевый раствор при -78°C, и полученную смесь перемешивали при охлаждении льдом в течение 1 часа. К реакционной смеси добавляли насыщенный водный раствор хлористого аммония, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=90/10-10/90). Затем полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=100/0-50/50). Затем полученный неочищенный продукт очищали препаративной тонкослойной хроматографией (толщина силикагеля: 0,5 мм, элюент: смесь н-гексан/этилацетат=2/1), с получением соединения ссылочного примера 28 (0,09 г) и ссылочного примера 29 (0,09 г). По данным указанной выше хроматографии, соединение ссылочного примера 28 находилось на стороне высокой полярности, и соединение ссылочного примера 29 находилось на стороне низкой полярности.
[0073] Ссылочный пример 30
Этиловый эфир 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовой кислоты
Суспензию 2-(3,5-бистрифторметилфенил)-N-[6-хлор-2-циано-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,06 г), этилового эфира 2-пиперидин-4-илпропионовой кислоты (0,10 г) и карбоната калия (0,03 г) в диметилсульфоксиде (1,0 мл) перемешивали при 180°C при микроволновом облучении в течение 2 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат=90/10-65/35), с получением указанного в заголовке соединения (0,05 г).
[0074] Ссылочный пример 31
Этиловый эфир 5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-4-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты
Смесь 2-(3,5-бистрифторметилфенил)-N-[6-хлор-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,05 г), этилового эфира (4-метилпиперидин-4-ил)уксусной кислоты (0,09 г) и N-метилпирролидона (0,5 мл) перемешивали при 190°C при микроволновом облучении в течение 30 минут. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=90/10-60/40), с получением указанного в заголовке соединения (0,03 г).
[0075] Ссылочный пример 32
Этиловый эфир [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-4-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты
Суспензию 2-(3,5-бистрифторметилфенил)-N-[6-хлор-2-циано-4-(4-фтор-2-метилфенил)пиридин-3-ил]-N-метилизобутиламида (0,06 г), этилового эфира (4-метилпиперидин-4-ил)уксусной кислоты (0,09 г) и карбоната калия (0,03 г) в диметилсульфоксиде (1,0 мл) перемешивали при 180°C при микроволновом облучении в течение 1 часа. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли воду. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на аминопропилированном силикагеле (элюент: смесь н-гексан/этилацетат=90/10-65/35), с получением указанного в заголовке соединения (0,06 г).
[0076] Пример 1
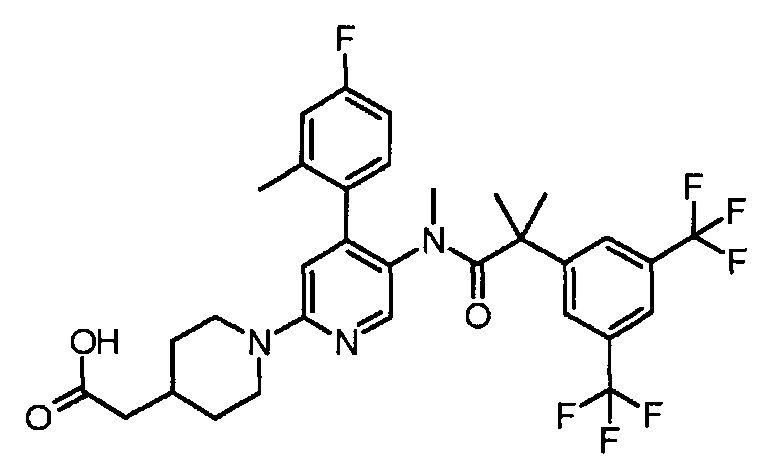
[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,38 г) в тетрагидрофуране (6 мл), метанола (6 мл) и воды (2 мл) добавляли моногидрат гидроксида лития (0,12 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь нейтрализовали добавлением 2 моль/л хлористоводородной кислоты (1,5 мл), и полученную в результате смесь концентрировали при пониженном давлении. К полученному остатку добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,37 г).
[0077] Пример 2
(5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-орто-толил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил)уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-орто-толил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,30 г) в тетрагидрофуране (4 мл), метанола (2 мл) и воды (2 мл) добавляли моногидрат гидроксида лития (0,084 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь нейтрализовали добавлением 2 моль/л хлористоводородной кислоты (1,1 мл), и полученную в результате смесь концентрировали при пониженном давлении. К полученному остатку добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,29 г).
[0078] Пример 3
5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,3-диметил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,3-диметил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,043 г) в тетрагидрофуране (1 мл), метанола (0,5 мл) и воды (0,5 мл) добавляли моногидрат гидроксида лития (0,012 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь нейтрализовали добавлением 2 моль/л хлористоводородной кислоты (0,14 мл). К полученной смеси добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,033 г).
[0079] Пример 4
5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,040 г) в тетрагидрофуране (1 мл), этанола (0,5 мл) и воды (0,5 мл) добавляли моногидрат гидроксида лития (0,011 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь нейтрализовали добавлением 2 моль/л хлористоводородной кислоты (0,13 мл). К полученной смеси добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,035 г).
[0080] Пример 5
[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3-метоксил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3-метокси-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,025 г) в тетрагидрофуране (1 мл), этанола (0,5 мл) и воды (0,5 мл) добавляли моногидрат гидроксида лития (0,006 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь нейтрализовали добавлением 2 моль/л хлористоводородной кислоты (0,075 мл). К полученной смеси добавляли воду, и полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,021 г).
[0081] Пример 6
[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,02 г) в тетрагидрофуране (0,30 мл), метанола (0,15 мл) и воды (0,15 мл) добавляли моногидрат гидроксида лития (0,007 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь нейтрализовали добавлением уксусной кислоты. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,02 г).
[0082] Пример 7
2-[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]-2-метилпропионовая кислота
Смесь этилового эфира 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]-2-метилпропионовой кислоты (0,14 г), водного раствора гидроксида натрия (1,0 моль/л, 0,60 мл), тетрагидрофурана (0,6 мл) и метанола (1,8 мл) перемешивали при 140°C при микроволновом облучении в течение 4,5 часов. К реакционной смеси добавляли водный раствор гидроксида натрия (2,0 моль/л, 0,50 мл), и полученную смесь перемешивали при 140°C при микроволновом облучении в течение 2 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли хлористоводородную кислоту (1,0 моль/л, 2,0 мл). Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,11 г).
[0083] Пример 8
2-[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовая кислота
К смеси этилового эфира 2-[5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовой кислоты (0,03 г) в тетрагидрофуране (0,4 мл), метанола (0,2 мл) и воды (0,2 мл) добавляли моногидрат гидроксида лития (0,008 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь нейтрализовали добавлением уксусной кислоты. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат/метанол=50/50/0-0/100/0-0/90/10), с получением указанного в заголовке соединения (0,02 г).
[0084] Пример 9
5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-4-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-4-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,03 г), тетрагидрофурана (0,375 мл), метанола (0,375 мл) и воды (0,15 мл) добавляли моногидрат гидроксида лития (0,02 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 3 часов и затем при 50°C в течение 4 часов. Полученную реакционную смесь охлаждали до комнатной температуры и добавляли хлористоводородную кислоту (2,0 моль/л, 0,2 мл). Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали водой и насыщенным раствором соли, и сушили над безводным сульфатом натрия, и растворитель удаляли при пониженном давлении, с получением указанного в заголовке соединения (0,025 г).
[0085] Пример 10
[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-4-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусная кислота
К смеси этилового эфира [5'-{[2-(3,5-бистрифторметилфенил)-2-метилпропионил]метиламино}-6'-циано-4'-(4-фтор-2-метилфенил)-4-метил-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]уксусной кислоты (0,06 г), тетрагидрофурана (0,50 мл), метанола (0,25 мл) и воды (0,25 мл) добавляли моногидрат гидроксида лития (0,02 г) при комнатной температуре, и полученную смесь перемешивали при комнатной температуре в течение 1 часа, и затем перемешивали при 50°C в течение 3 часов. Полученную реакционную смесь охлаждали до комнатной температуры и нейтрализовали добавлением уксусной кислоты. Полученную в результате смесь экстрагировали этилацетатом. Органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат/метанол=20/80/0-0/100/0-0/90/10), с получением указанного в заголовке соединения (0,03 г).
[0086] Пример 11
(S)-2-[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовая кислота
К смеси N-[4-[(S)-2-((S)-4-бензил-2-оксооксазолидин-3-ил)-1-метил-2-оксоэтил]-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-5'-ил]-2-(3,5-бистрифторметилфенил)-N-метилизобутиламида (0,08 г) в тетрагидрофуране (1,5 мл) и воды (0,5 мл) добавляли моногидрат гидроксида лития (0,008 г) и раствор пероксида водорода (30%, 0,06 мл) при охлаждении льдом, и полученную смесь перемешивали при этой же температуре в течение ночи. К реакционной смеси добавляли водный раствор сульфита натрия (10%, 0,75 мл), и полученную в результате смесь перемешивали в течение 30 минут. Растворитель удаляли при пониженном давлении. Полученный остаток подкисляли хлористоводородной кислотой, и полученную в результате смесь экстрагировали этилацетатом. Водный слой повторно экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=90/10-10/90), с получением указанного в заголовке соединения (0,014 г).
[0087] Пример 12
(R)-2-[5'-{[2-(3,5-Бистрифторметилфенил)-2-метилпропионил]метиламино}-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-4-ил]пропионовая кислота
К смеси N-[4-[(R)-2-((S)-4-бензил-2-оксооксазолидин-3-ил)-1-метил-2-оксоэтил]-4'-(4-фтор-2-метилфенил)-3,4,5,6-тетрагидро-2H-[1,2']бипиридинил-5'-ил]-2-(3,5-бистрифторметилфенил)-N-метилизобутиламида (0,04 г) в тетрагидрофуране (0,75 мл) и воды (0,25 мл) добавляли моногидрат гидроксида лития (0,004 г) и раствор пероксида водорода (30%, 0,03 мл) при охлаждении льдом, и полученную смесь перемешивали при этой же температуре в течение ночи. К реакционной смеси добавляли водный раствор сульфита натрия (10%, 0,375 мл), и полученную в результате смесь перемешивали в течение 30 минут. Растворитель удаляли при пониженном давлении. Полученный остаток подкисляли хлористоводородной кислотой, и полученную в результате смесь экстрагировали этилацетатом. Водный слой повторно экстрагировали этилацетатом. Объединенный органический слой промывали насыщенным раствором соли и сушили над безводным сульфатом магния, и растворитель удаляли при пониженном давлении. Полученный неочищенный продукт очищали колоночной хроматографией на силикагеле (элюент: смесь н-гексан/этилацетат=90/10-10/90), с получением указанного в заголовке соединения (0,004 г).
[0088] В таблицах 1-7 показаны химические структуры полученных выше соединений ссылочных примеров 1-32, и химические структуры и физические свойства полученных выше соединений примеров 1-12. Аббревиатуры в этих таблицах: “ссыл. №”, “пример №”, “структура”, “физические данные”, “1H-ЯМР”, “ДМСО-d6” и “CDCl3” обозначают номер ссылочного примера, номер примера, химическую структуру, физические свойства, спектр водородного ядерно-магнитного резонанса, диметилсульфоксид-d6 и хлороформ-d1, соответственно. И “МС” и “ESI_APCI” обозначают масс-спектрометрию и измерение химической ионизации с электрораспылением при ионизации при атмосферном давлении, соответственно.
[0089]
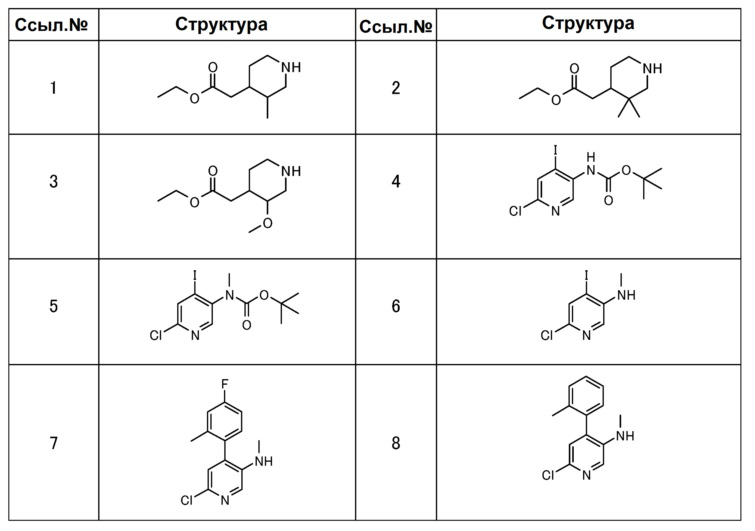
[0090]
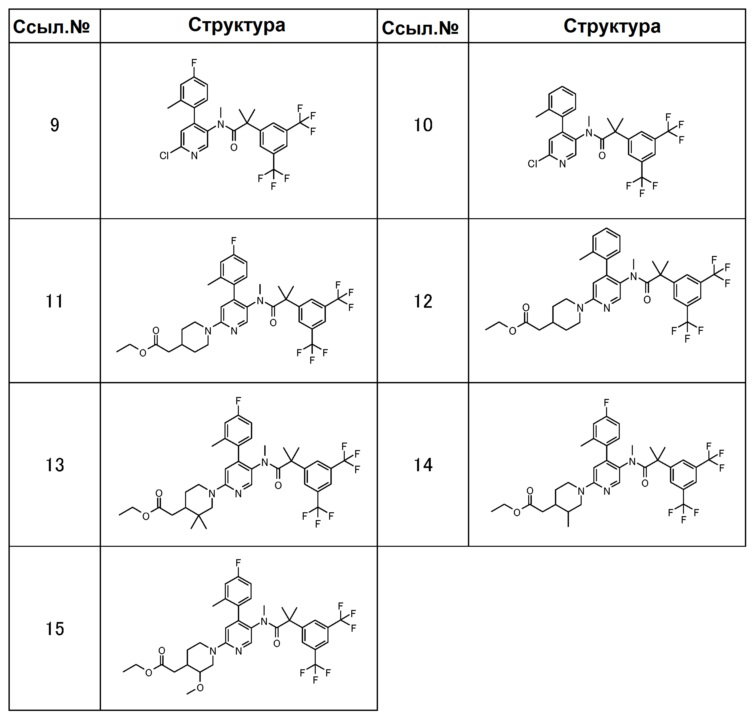
[0091]
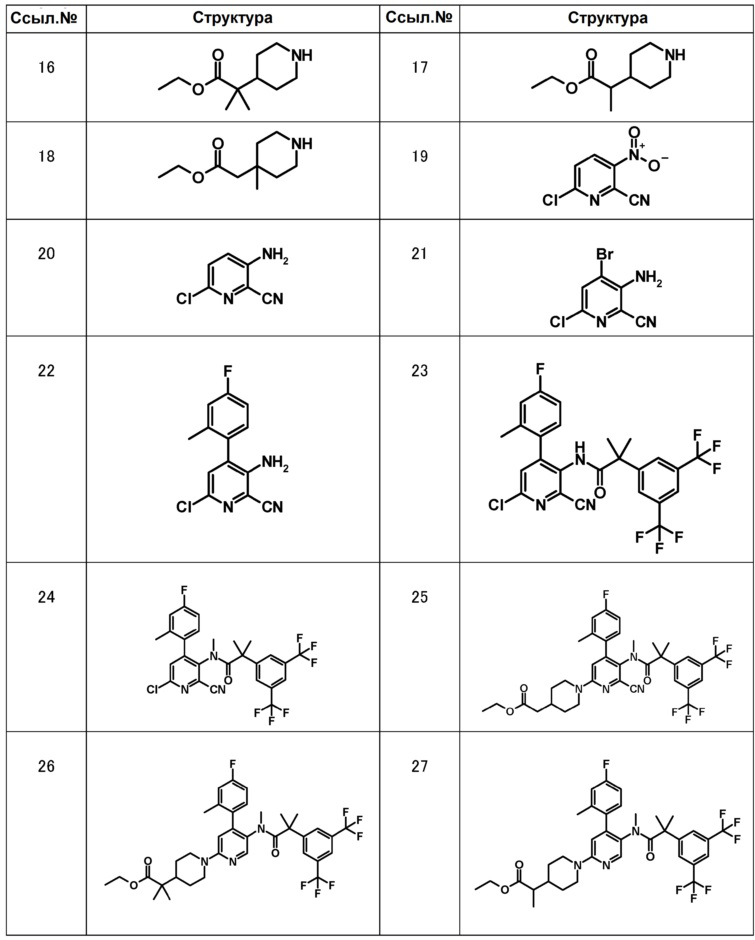
[0092]
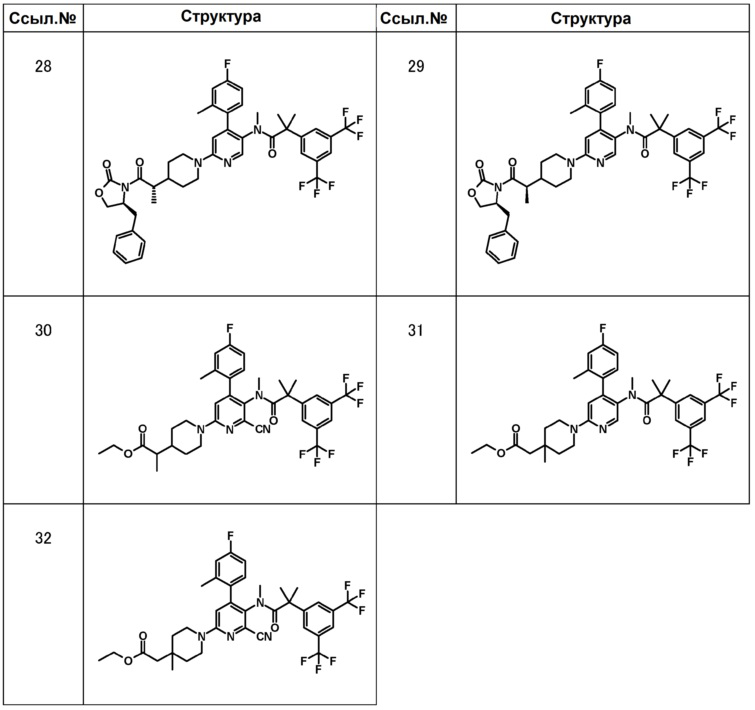
[0093]
МС (ESLAPCI, m/z):640 (M+H)+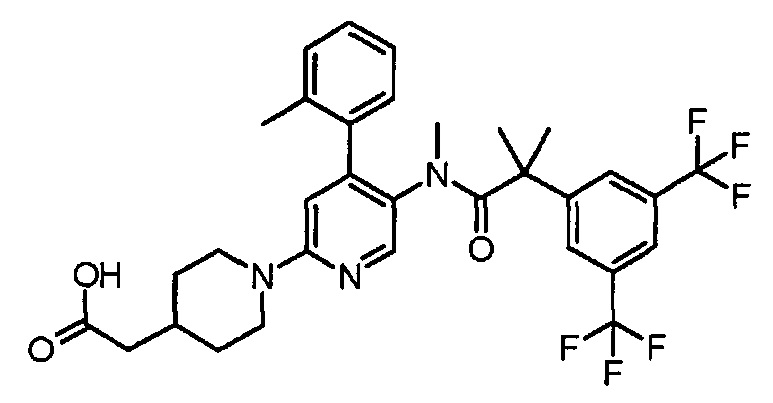
МС (ESLAPCI, m/z):622 (M+H)+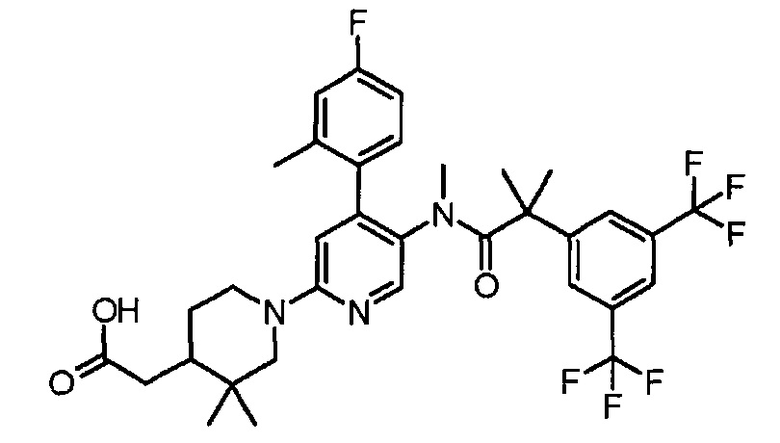
МС (ESLAPCI, m/z):668 (M+H)+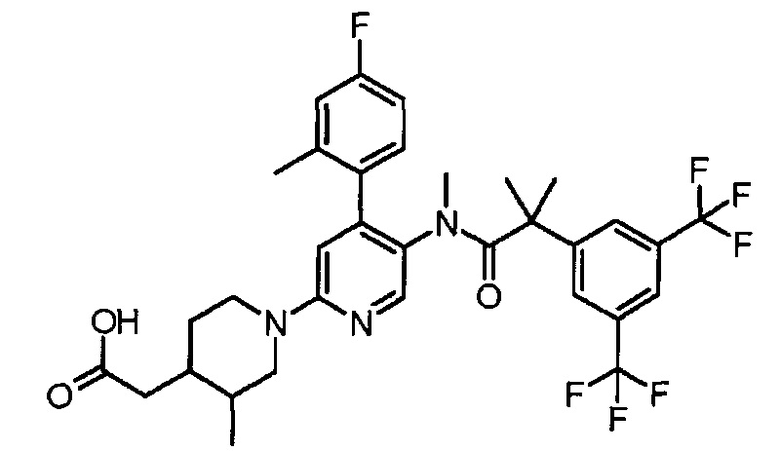
МС (ESLAPCI, m/z):654 (M+H)+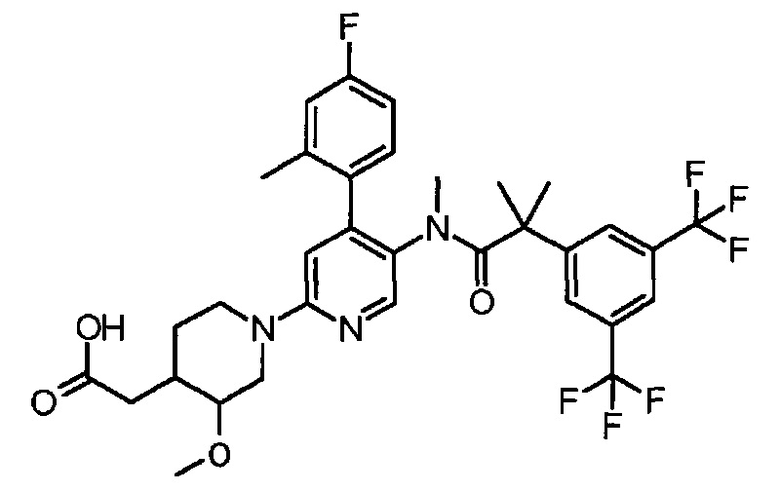
МС (ESLAPCI, m/z):670 (M+H)+
[0094]
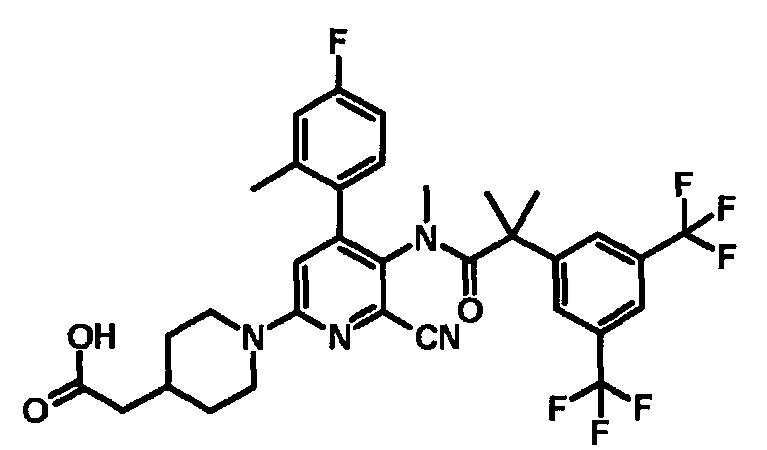
МС (ESLAPCI, m/z): 665 (M+H)+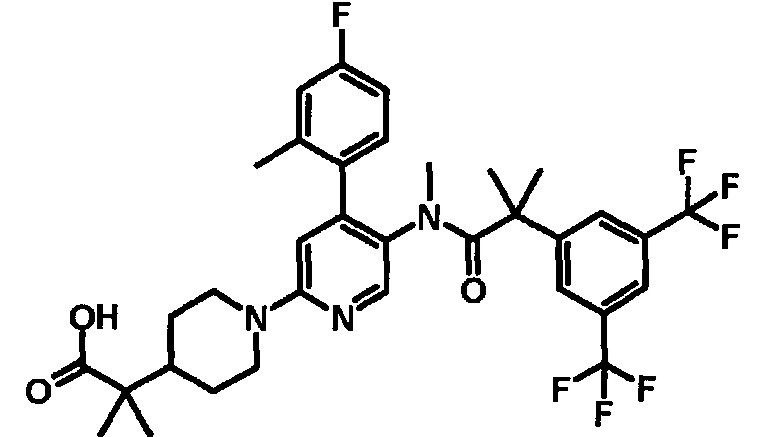
МС (ESLAPCI, m/z): 668 (M+H)+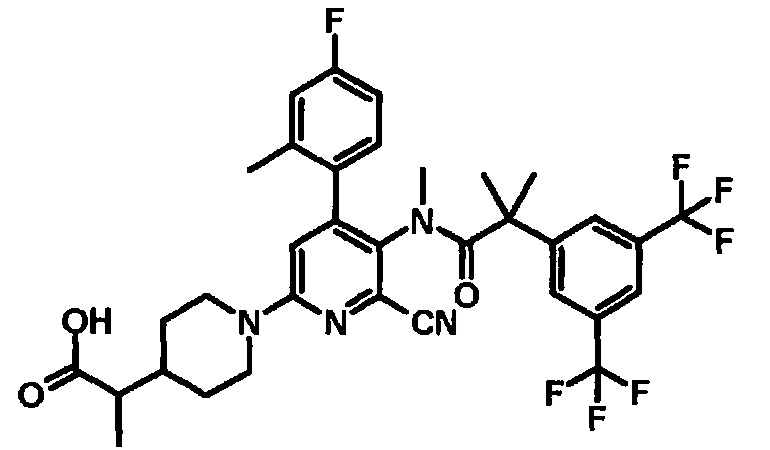
МС (ESLAPCI, m/z): 679 (M+H)+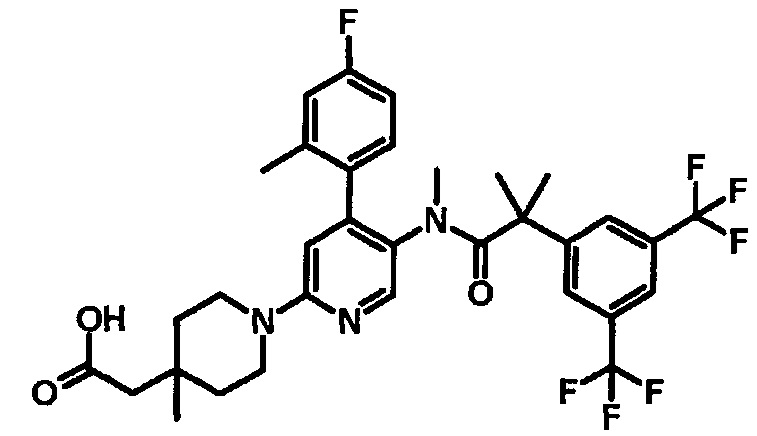
МС (ESLAPCI, m/z): 654 (M+H)+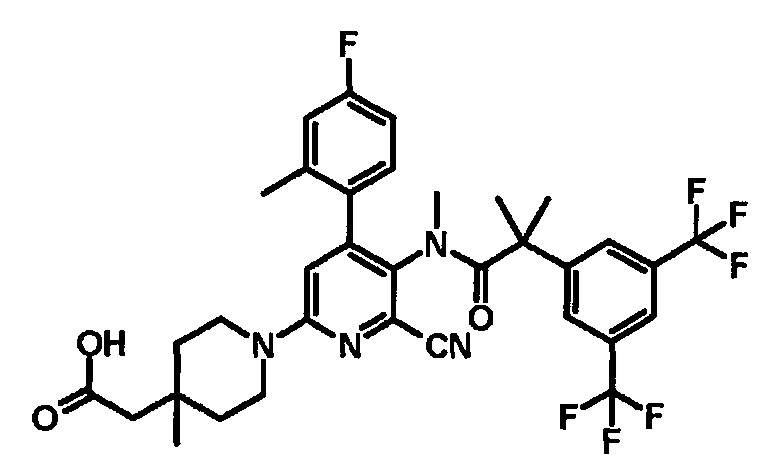
МС (ESLAPCI, m/z): 679 (M+H)+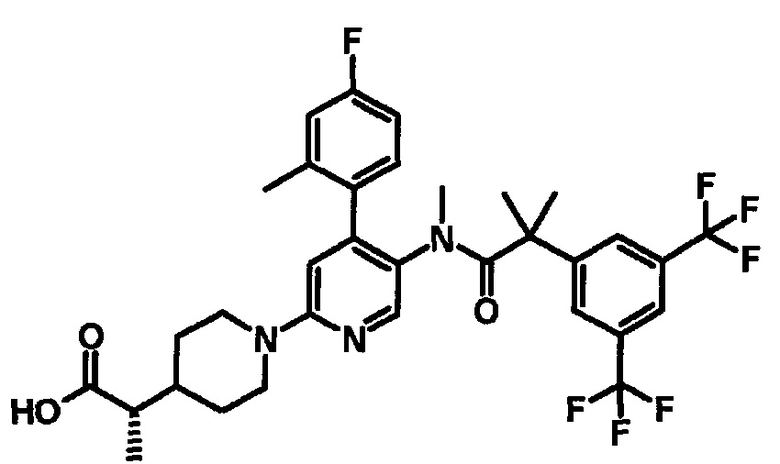
МС (ESLAPCI, m/z): 654 (M+H)+
[0095]
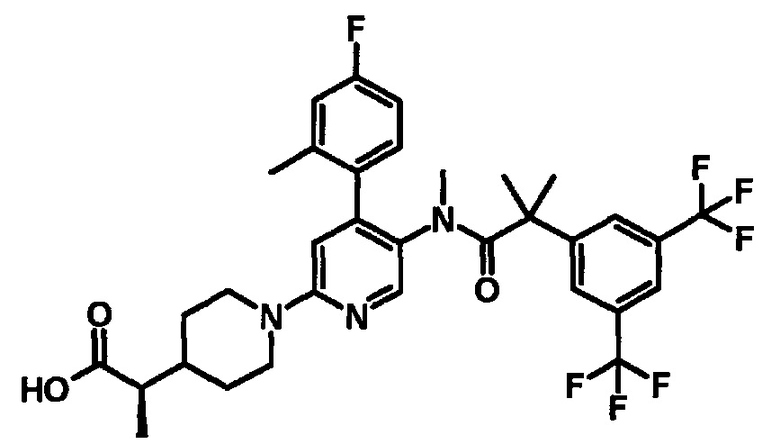
МС (ESLAPCI, m/z): 654 (M+H)+
[0096] Пример тестирования 1
Аффинность в отношении рецептора NK1 человека
(1) Получение вектора экспрессии рецептора NK1 человека
ПЦР проводили с использованием кДНК нормальных тканей взрослого человека, полученных из мозга (BioChain), в качестве матрицы, с прямым праймером SEQ ID No:1 и обратным праймером SEQ ID NO:2, с использованием фермента ПЦР, ДНК-полимеразы PrimeSTAR Макс или PrimeSTAR GXL ДНК полимеразы (зарегистрированная торговая марка, Takara Bio). Амплифицированный продукт вставляли в плазмиду (ПЦР-BluntII-TOPO (зарегистрированная торговая марка), Life Technologies) с использованием набора для клонирования Zero Blunt PCR (зарегистрированная торговая марка, Life Technologies). По общей методике Escherichia coli (One Shot TOP10 компетентные клетки, Life Technologies) трансформировали плазмидой, в которую амплифицированный продукт был вставлен. Клетки Escherichia coli культивировали на LB агарозной среде, содержащей 50 мкг/мл канамицина, в течение суток. После культивирования, колонию разделяли и культивировали в LB среде, содержащей 50 мкг/мл канамицина. После культивирования, плазмиду очищали с использованием набора Quantum Prep Plasmid Miniprep (Bio-Rad). Данную плазмиду дважды расщепляли в течение около двух часов с использованием рестрикционных ферментов, XhoI и HindIII (New England Biolabs). Затем осуществляли электрофорез с использованием 1% агарозного геля, и фрагменты, которые были расщеплены, собирали и очищали с использованием TaKaRa RICOCHIP (Takara Bio). Отдельно, плазмиду также очищали от Escherichia coli, которая была трансформирована вектором (pкДНК3.1(-) (pcDNA3.1(-)) (зарегистрированная торговая марка), Life Technologies), плазмиду дважды расщепляли в течение около двух часов с использованием рестрикционных ферментов, XhoI и HindIII (New England Biolabs). Затем осуществляли электрофорез с использованием 1% агарозного геля, и вектор, который был расщеплен, собирали и очищали с использованием TaKaRa RICOCHIP (Takara Bio). Фрагмент, вырезанный из ПЦР-Blunt-II, и вектор pкДНК3.1(-), обработанный рестрикционными ферментами, лигировали с использованием набора для лигирования ДНК <Mighty Mix> (Takara Bio). По общей методике, Escherichia coli (One Shot TOP10 компетентные клетки, Life Technologies) трансформировали плазмидой, полученной при лигировании. Клетки Escherichia coli культивировали на LB агарозной среде, содержащей 50 мкг/мл ампициллина, в течение суток. После культивирования, колонию разделяли и культивировали в LB среде, содержащей 50 мкг/мл ампициллина, и затем плазмиду очищали с использованием набора Quantum Prep Plasmid Miniprep (Bio-Rad). Белок, кодирующий нуклеотидную последовательность (SEQ ID NO:3) полученной плазмиды, был полностью идентичен нуклеотидной последовательности (NM_001058.3) рецептора 1 тахикинина человека (TACR1, NK1R), зарегистрированной в известной базе данных (NCBI). Таким образом, было подтверждено, что последовательностью клонированного гена была нуклеотидная последовательность рецептора NK1 человека, и что аминокислотной последовательностью, которая может быть переведена, была последовательность рецептора NK1 человека. pкДНК3.1(-) (зарегистрированная торговая марка), в которую нуклеотидная последовательность SEQ ID NO:3 была вставлена, использовали в качестве экспрессирующей плазмиды рецептора NK1 человека.
[0097] (2) Получение клеток, экспрессирующих рецептор NK1 человека
(2-1) Культура клеток 293T
При использовании жидкой среды D-MEM (модифицированная по Дульбекко среда Игла) (низкое содержание глюкозы, содержащей L-глутамин, Wako Pure Chemical Industries), дополненной раствором антибиотика пенициллин/стрептомицин (Life Technologies, конечная концентрация пенициллина 100 Ед./мл и конечная концентрация стрептомицина 100 мкг/мл) и фетальной телячьей сывороткой (конечная концентрация 10%), клетки 293T (RIKEN) культивировали в инкубаторе в условиях газообразного 5% CO2 при 37°C.
(2-2) Субкультура клеток 293T
Почти слившиеся клетки промывали PBS (забуференный фосфатом физиологический раствор, Wako Pure Chemical Industries), разъединяли с использованием 0,05% трипсин-EDTA (Life Technologies) и суспендировали в жидкой среде. Суспезию клеток разводили указанной выше жидкой средой таким образом, чтобы коэффициент распространения становился 1:10, и затем клетки культивировали.
(2-3) Получение клеток, экспрессирующих рецептор NK1 человека
Слившиеся клетки промывали PBS, разъединяли с использованием 0,05% трипсин-EDTA (Life Technologies) и суспендировали в жидкой среде D-MEM (пониженное содержание глюкозы, содержащей L-глутамин, Wako Pure Chemical Industries), дополненной фетальной телячьей сывороткой (конечная концентрация 10%). Суспензию клеток разводили жидкой средой, и клетки высевали в лунки 96-луночного микропланшета, покрытого поли-D-лизином (BD Biocoat (зарегистрированная торговая марка), Nippon Becton Dickinson) при плотности 5×104 клеток/лунка и объеме жидкой среды 100 мкл/лунка. После высевания, клетки культивировали в инкубаторе в условиях газообразного 5% CO2 при 37°C в течение от около четырех до пяти часов, и клетки трансфицировали с полученной, таким образом, экспрессирующей плазмидой рецептора NK1 человека.
(2-4) Трансфекция экспрессирующей плазмиды рецептора NK1 человека в клетки 293T
Для трансфекции экспрессирующей плазмиды рецептора NK1 человека использовали липофектамин 2000 (зарегистрированная торговая марка, Life Technologies). Экспрессирующую плазмиду рецептора NK1 человека разводили в редуцированной сывороточной среде I Opti-MEM (зарегистрированная торговая марка) (Life Technologies) до конечной концентрации в 0,2 мкг/25 мкл/лунка. В то же самое время, липофектамин 2000 (зарегистрированная торговая марка, Life Technologies) разводили в редуцированной сывороточной среде I Opti-MEM (зарегистрированная торговая марка) (Life Technologies) до конечной концентрации в 0,4 мкл/25 мкл/лунка и инкубировали при комнатной температуре в течение пяти минут. Спустя пять минут, для образования комплекса экспрессионная плазмида рецептора NK1 человека/липофектамин 2000, разведенную экспрессионную плазмиду рецептора NK1 человека и разведенный липофектамин 2000 смешивали и инкубировали при комнатной температуре в течение 20-25 минут. После инкубации, 50 мкл/лунка раствора полученного комплекса добавляли к клеткам для трансфекции с экспрессирующей плазмидой рецептора NK1 человека, и клетки культивировали в инкубаторе в условиях газообразного 5% CO2 при 37°C в течение от около 48 часов. Клетки, которые культивировали в течение 48 часов, использовали для анализа в качестве клеток, экспрессирующих рецептор NK1 человека.
[0098] (3) Измерение аффинности связывания рецептора NK1 человека
(3-1) Получение мембранной фракции из клеток, экспрессирующих рецептор NK1 человека
Клетки, экспрессирующие рецептор NK1 человека, получали в 175 см2 флаконе для культивирования (Nippon Becton Dickinson). Формирование комплекса экспрессирующей плазмиды рецептора NK1 человека и липофектамина 2000 осуществляли путем расчета отношения площади культуры и увеличения масштаба метода, описанного выше в 2-4. Клетки, экспрессирующие рецептор NK1 человека, собирали в буферный раствор для получения мембранной фракции (50 мМ Tris (Wako Pure Chemical), 120 мМ хлорид натрия (Wako Pure Chemical Industries), 5 мМ хлорид калия (Wako Pure Chemical Industries), 1 мМ этилендиаминтетрауксусная кислота (Sigma), 0,002 мг/мл цитостатин (Peptide Institute), 0,04 мг/мл бацитрацин (Wako Pure Chemical Industries), 0,005 мг/мл фосфорамидон (Peptide Institute) и 0,5 мМ фенилметилсульфонилфторид (Wako Pure Chemical Industries), pH 7,4) и центрифугировали при 1880 g в течение 10 минут, и осажденные клетки суспендировали в буферном растворе для получения мембранной фракции. После замораживания и оттаивания клеток один раз, клетки гомогенизировали с использованием гомогенизатора типа Dounce (охлажденные на льду, 1000 об./мин., 20 раз). Суспензию гомогенизированных клеток центрифугировали при 20000 об./мин. в течение 10 минут, и супернатант удаляли, с получением осажденных клеток. Осажденные клетки снова суспендировали в буферном растворе для получения мембранной фракции и гомогенизировали с использованием гомогенизатора типа Dounce (охлаждение на льду, 1000 об./мин., 30 раз). Суспензию клеток центрифугировали при 20000 об./мин, в течение 10 минут, и супернатант удаляли, с получением осажденных клеток. Ту же самую гомогенизацию и центрифугирование снова повторяли и получали конечные осажденные клетки. Конечные осажденные клетки суспендировали в буферном растворе для теста на связывание рецептора (50 мМ Tris (Wako Pure Chemical Industries), 3 мМ хлорид магния (Wako Pure Chemical Industries), 0,002 мг/мл цитостатин (Peptide Institute), 0,04 мг/мл бацитрацин (Wako Pure Chemical Industries) и 0,02% бычий сывороточный альбумин (Sigma), pH 7,4), и измеряли концентрацию белка с использованием набора для анализа белка BCA Protein Assay (Pierce).
(3-2) Тест на связывание рецептора
Буферный раствор для теста на связывание рецептора распределяли в лунки 96-луночного планшета для анализов (Greiner) при 22,5 мкл/лунка. ДМСО растворы тестируемого соединения, которые получали при 80-кратно высокой концентрации с использованием 100% диметилсульфоксида (ДМСО), добавляли в лунки при 2,5 мкл/лунка (конечные концентрации от 1 нМ до 100 нМ), и растворы смешивали. В качестве радиомеченного лиганда использовали 125I-вещество P (вещество P, [125I]Tyr8-, PerkinElmer). 125I-вещество P разводили буферным раствором для тестирования на связывание рецептора до конечной концентрации в 125 пмоль/25 мкл/лунка и добавляли в 96-луночный планшет для анализов, и растворы смешивали. Мембранные фракции, полученные из клеток, экспрессирующих рецептор NK1 человека, разводили буферным раствором для тестирования на связывание рецептора до конечной концентрации в 8-10 мкг/лунка, суспендировали до тех пор, пока суспензия не приходила в такое гомогенное состояние, при котором суспензия могла плавно протекать через иглу для инъекций 27G и затем добавляли в 96-луночный планшет для анализов при 150 мкл/лунка. Затем планшет инкубировали при комнатной температуре в течение 60 минут при встряхивании. Реакционные растворы фильтровали при всасывании через мультискрининговый фильтр для 96-луночных планшетов (Millipore), который был предварительно обработан 0,3% полиэтиленимином, и реакцию прекращали промывкой раствором для промывки (50 мМ Tris и 0,02% бычий сывороточный альбумин, pH 7,4) четыре раза. Нижнюю часть микропланшета сушили при 60°C, и затем 100 мкл/лунка MicroScint 20 (PerkinElmer) распределяли в лунки. Верхнюю часть планшета герметизировали TopSeal A (PerkinElmer), и планшет встряхивали в течение 5-10 минут. Затем измеряли радиоактивность с помощью счетчика TopCount NXT (зарегистрированная торговая марка) (PerkinElmer). Радиоактивность каждой лунки вычисляли путем вычитания радиоактивности лунки, в которую добавляли 10 мкм апрепитанта (неспецифическое связывание). Рассчитывали скорость связывания по уравнению:
Скорость связывания (%) 125I-вещества P=(радиоактивность группы, к которой добавлено тестируемое соединение)/(радиоактивность группы, к которой добавлен наполнитель)×100.
С использованием программного обеспечения для анализов GraphPad Prism (GraphPad Software), скорость связывания (%) определяли нанесением на график зависимости от концентрации тестируемого соединения и линейного приближения, и рассчитывали концентрацию, требуемую для 50% ингибирования, IC50. Эти результаты показаны в таблице 8. В данной таблице пример № обозначает номер примера, и IC50 (нМ) представляет собой концентрацию, требуемую для 50% ингибирования.
[0099] (4) Результаты
Как показано в таблице 8, продемонстрировано, что соединения по настоящему изобретению проявляют высокую аффинность связывания в отношении рецептора NK1 человека.
[0100] Пример тестирования 2
Ингибирующий эффект на рецептор NK1 человека
(1) Получение клеток, экспрессирующих рецептор NK1 человека
Клетки, экспрессирующие рецептор NK1 человека, получали методами, аналогично описанным в 2-3 и 2-4 примера тестирования 1.
[0101] (2) Исследование влияния ингибирующего эффекта на увеличение концентрации внутриклеточного кальция
Клетки, экспрессирующие рецептор NK1 человека, промывали 300 мкл/лунка раствора для промывки (20 мМ HEPES/сбалансированный солевой раствор Хэнка (HBSS) pH 7,3). Флуоресцентный индикатор кальция (набор Fluo-4 Direct Calcium Assay, Life Technologies, содержащий 0,42 мМ пробенецид и 0,1% бычий сывороточный альбумин, полученный согласно протоколу для продукта) добавляли в лунки при 150 мкл/лунка, и планшет инкубировали при 37°C в течение 30 минут в инкубаторе. Затем ДМСО растворы тестируемого соединения, которые были получены при 80-кратно повешенной концентрации с использованием 100% диметилсульфоксида (ДМСО), добавляли в лунки при 2,5 мкл/лунка (конечные концентрации 0,1, 1 и 10 мкМ), и растворы смешивали. Затем планшет дополнительно инкубировали при 37°C в течение 30 минут в инкубаторе. Спустя 30 минут, немедленно измеряли концентрации внутриклеточного кальция.
Каждую из концентраций внутриклеточного кальция измеряли в виде флуоресцентного сигнала с использованием FDSS (зарегистрированная торговая марка) 7000 (Hamamatsu Photonics). Раствор вещества P (Peptide Institute, Inc.), который был получен при 0,4 мкМ или 4 мкМ с использованием буфера для анализа (20 мМ HEPES/сбалансированный солевой раствор Хэнка (HBSS) pH 7,3, содержащий 0,1% бычий сывороточный альбумин) автоматически добавляли в каждую лунку при 50 мкл/лунка (конечная концентрация 0,1 или 1 мкМ) 10 секунд после начала считывания, и флуоресцентный сигнал измеряли вплоть до 120 секунд.
Концентрации внутриклеточного кальция (%) клеток, к которым добавляли тестируемое соединение, рассчитывали по уравнению ниже, где флуоресцентный сигнал группы, к которой добавляли наполнитель (ДМСО), принимали за 100%, и флуоресцентный сигнал перед добавлением вещества P принимали за 0%.
Концентрация внутриклеточного кальция (%)=(Флуоресцентный сигнал группы добавленного тестируемого соединения)/(Флуоресцентный сигнал группы добавленного наполнителя)×100
Рассчитанную концентрацию внутриклеточного кальция (%) принимали за остаточную агонистическую активность вещества P (вещество P-остаточный ответ: SPRR). Эти результаты показаны в таблице 9. В данной таблице пример № обозначает номер примера. SPRR (%) представляет собой значение, полученное, когда концентрация вещества P составляла 1 мкМ, и концентрация соединения составляла 0,1 мкМ.
[0102] (3) Результаты
Как показано в таблице 9, продемонстрировано, что соединения по настоящему изобретению проявляют антагонистическую активность в отношении рецептора NK1 человека.
[0103] Пример тестирования 3
Ингибирующий эффект на CYP3A4
Получали диметилсульфоксидный (ДМСО) раствор тестируемого соединения с концентрацией в 1000 раз выше, чем оцениваемая концентрация, и реакционный раствор получали разведением указанного раствора. Ферментативную реакцию осуществляли путем инкубирования в забуференном фосфатом калия растворе (pH 7,4), содержащем от 1 нМ до 20 мкМ тестируемого соединения, 3,2 мМ хлорид магния, 0,2 пмоль CYP3A4 человека (BD Biosciences), 0,5 мМ ослабленный никотинамидадениндинуклеотидфосфат (NADPH) и 3 мкМ люциферин-IPA (Promega) при 37°C в течение 10 минут. Объем реакционного раствора составлял 50 мкл/лунка. Группу, подвергнутую 30-минутной предварительной инкубации, инкубировали при 37°C в течение 30 минут, затем добавляли субстрат, раствор люциферин-IP (12,5 мкл/лунка). В конце ферментативной реакции в лунки добавляли 50 мкл/лунка реагента обнаружения люциферина (Promega), и планшет выдерживали при комнатной температуре в течение 20 минут. Затем измеряли интенсивность эмиссии с использованием Infinite M1000 (TECAN). Вычисляли значение ферментативной активности (%) относительно группы, к которой тестируемое соединение не добавляли. Наносили кривую доза-ответ с использованием программного обеспечения для анализов GraphPad Prism (GraphPad Software), и рассчитывали концентрацию каждого из соединений, которые проявляли 50% ингибирование, IC50. В качестве сравнительного примера, по аналогичной методике тестировали апрепитант, который является антагонистом рецептора NK1.
[0104] Измеряли значения IC50 для группы, подвергнутой 30-минутной предварительной инкубации, с использованием тестируемых соединений, описанным выше способом измерения, и результаты показаны в таблице 10. В данной таблице пример № обозначает номер примера, и IC50 (мкМ) является концентрацией, требуемой для 50% ингибирования.
Продемонстрировано, что CYP3A4-ингибирующая активность соединений по настоящему изобретению сокращена по сравнению с апрепитантом. Поэтому ожидается, что соединения по настоящему изобретению имеют меньшее межлекарственное взаимодействие на основе ингибирующего эффекта на CYP3A4, чем апрепитант.
[0105] Пример тестирования 4
Воздействие на постукивание лапой
(1) Воздействие на постукивание лапой
Самцов песчанки (Япония SLC) анестезировали изофлюраном, и вводили 0,3 мг/кг тестируемого соединения в яремную вену. Спустя четыре часа, вводили GR73632 (5 пмоль/5 мкл), который является агонистом рецептора NK1, в мозговой желудочек по 1 мМ в боковую часть и 4,5 мМ нижнюю часть брегмы головы, под анестезией изофлюраном. После введения, песчанок переносили в клетку наблюдения, и измеряли число постукиваний лапой в течение периода пяти минут после восстановления восстанавливающего рефлекса. Ингибирование скорости постукивания лапой (%) рассчитывали для каждого тестируемого соединения по следующему уравнению.
Ингибирование скорости постукивания лапой (%)={1-(Период постукивания лапой, когда вводится тестируемое соединение)/(Период постукивания лапой, когда вводится растворитель)}×100
[0106] (2) Измерение концентраций лекарственного средства
После окончания постукивания лапой, лапаротомию осуществляли непосредственно под эфирным наркозом, и отбирали образец крови из верхней полой вены живота. В то же самое время экстрагировали мозг. Через количественный анализ с использованием жидкостной хроматографии-масс-спектрометрии (ЖХ/МС), измеряли концентрации тестируемого соединения в плазме и мозге.
[0107] (3) Результаты
Измеряли воздействия на постукивание лапой описанным выше методом тестирования, и результаты показаны в таблице 11. В данной таблице пример № обозначает номер примера. Ингибирование (%) представляет собой ингибирование скорости постукивания лапой, и концентрация (нМ) обозначает концентрацию лекарственного средства в мозге.
Соединения по настоящему изобретению проникали в центральную нервную систему, а также проявляли великолепную антагонистическую активность в отношении рецептора NK1 in vivo.
[0108] Пример тестирования 5
Тест на выявление фармакокинетики (на хорьках)
(1) Методы
Раствор тестируемого соединения получали растворением тестируемого соединения в наполнителе (50% N,N-диметилформамид (Wako Pure Chemical Industries), 30% пропиленгликоль (Wako Pure Chemical Industries) и 4% 2-гидроксипропил-β-циклодекстрин (Wako Pure Chemical Industries)). Под анестезией изофлюраном, самцам хорька (Marshall BioResources Japan) внутривенно вводили 0,3 мг/кг тестируемого соединения через яремную вену. В случае перорального введения, 1 мг/кг тестируемого соединения перорально вводили бодрствующим животным. После введения тестируемого соединения, последовательно отбирали образцы крови из вены плечевого предлежания, и измеряли концентрации тестируемого соединения в плазме количественным анализом с использованием жидкостной хроматографии-масс-спектрометрии (ЖХ/МС).
[0109] (2) Результаты
Тест на выявление фармакокинетики на хорьках осуществляли описанным выше методом тестирования, и результаты показаны в таблице 12 и таблице 13. В данных таблицах пример № обозначает номер примера. CLtot и Vss обозначают общий клиренс организма и стационарный объем распределения, основанный на концентрации в плазме в случае внутривенного введения, соответственно. Cmax, AUC и BA обозначают максимальную концентрацию тестируемого соединения в плазме, площадь под кривой концентрация тестируемого соединения в плазме от времени, и биодоступность, в случае перорального введения, соответственно.
Как показано в таблице 12 и таблице 13, соединение настоящего изобретения проявляет великолепную фармакокинетику.
[0110] Пример тестирования 6
Воздействие на индуцированный цисплатином острый и замедленный рвотный ответ
(1) Методы
(1-1) Тест при внутривенном введении
Раствор тестируемого соединения получали растворением тестируемого соединения в наполнителе (смесь 50% N,N-диметилформамида (Wako Pure Chemical Industries), 30% пропиленгликоля (Wako Pure Chemical Industries) и 4% 2-гидроксипропил-β-циклодекстрина (Wako Pure Chemical Industries)). Контрольной группе вводили только наполнитель.
Под анестезией изофлюраном, 3 мг/кг тестируемого соединения последовательно внутривенно вводили самцам хорьков (Marshall BioResources Japan) через яремную вену. Цисплатин, который был нагрет до 40-50°C, внутрибрюшинно вводили при 5 мг/кг через один час после введения лекарственного средства. Хорьков наблюдали в течение 72 часов сразу же после введения цисплатина, и подсчитывали число рвотных позывов (периодические сжатия брюшной полости без рвоты содержимого желудка) и рвоты.
(1-2) Тест при пероральном введении
Раствор тестируемого соединения получали растворением тестируемого соединения в наполнителе (1% N-метил-2-пирролидон, 0,5% Tween 80, 20% полиэтиленгликоль 400 и 78,5% физиологический солевой раствор). Контрольной группе вводили только наполнитель.
Тестируемое соединение перорально вводили самцам хорьков (Marshall BioResources Japan). Пероральное введение осуществляли каждые 24 часа при 1 мг/кг, в общем три раза. Цисплатин, который был нагрет до 40-50°C, внутрибрюшинно вводили при 5 мг/кг через один час после перорального введения тестируемого соединения. Хорьков наблюдали в течение 72 часов сразу же после введения цисплатина, и подсчитывали число рвотных позывов (периодические сжатия брюшной полости без рвоты содержимого желудка) и рвоты.
[0111] (2) Результаты
Результаты показаны на фиг.1. В контрольной группе увеличение числа рвотных позывов и рвоты наблюдалось в острой фазе (вплоть до 24 часов после введения цисплатина) и в замедленной фазе (от 24 часов до 72 часов после введения цисплатина). В группе, которой внутривенно вводили соединение примера 1, и в группе, которой перорально вводили данное соединение, ингибирование числа рвотных позывов и рвоты наблюдалось в острой фазе и в замедленной фазе.
Продемонстрировано, что соединение настоящего изобретения обладает лечебным эффектом длительного действия и ингибирующим эффектом на индуцированные цисплатином острые и замедленные рвотные ответы.
[0112] Пример тестирования 7
Оценка распространения hERG
(1) Метод
Получали диметилсульфоксидный (ДМСО) раствор тестируемого соединения с концентрацией в 1000 раз выше, чем оцениваемая концентрация (10 мкМ), и раствор с конечной применяемой концентрацией получали разведением указанного раствора. Распространение hERG измеряли методом цельных клеток с использованием системы "открытия-закрытия" (пэтч-кламп), где покрывное стекло, на котором были высеяны 293 клетки hERG канала, экспрессирующие человеческие эмбриональные клетки почек, помещали на перфузионную баню и вызывали течение перфузионного раствора. Измеряли изменение распространения hERG каналов, вызванное протоколом пульсации (выдерживаемый потенциал -80 МВ, пульс деполяризации +20 МВ в течение 1,9 секунд, пульс реполяризации -50 МВ в течение 2 секунд, интервал стимуляции в 15 секунд) приобретение данных/анализ данных программным обеспечением, pCLAMP9 (Axon Instruments, Inc.). Условиями измерения была скорость потока около 1,5 мл/мин и температура около 33°C. Анализировали две формы волн непосредственно перед применением тестируемого соединения, и две формы волн сразу же после его применения в течение 10 минут, и осуществляли статистический анализ. Значение перед применением тестируемого соединения принимали за 100%, и определяли значение изменения скорости на основании этого значения.
(2) Результаты
Воздействие тестируемого соединения на распространение hERG оценивали описанным выше способом (результат показан в таблице 14). В данной таблице пример № обозначает номер примера, и значение представлено как среднее±стандартная ошибка.
Как показано в таблице 14, соединение настоящего изобретения не вызывает каких-либо изменений в распространении hERG со статистической значимостью по сравнению с контролем (0,1% ДМСО).
Промышленная применимость
[0113] Соединения по настоящему изобретению или их фармацевтически приемлемые соли обладают превосходной антагонистической активностью в отношении рецептора NK1 и, таким образом, полезны в качестве средства для предупреждения или лечения индуцированной противораковой химиотерапией тошноты или рвоты.
Свободный перечень последовательностей
[0114] <Перечень последовательности 1>
SEQ ID NO:1 представляет собой последовательность прямого праймера, который использован для амплификации ДНК SEQ ID NO:3.
<Перечень последовательности 2>
SEQ ID NO:2 представляет собой последовательность обратного праймера, который использован для амплификации ДНК SEQ ID NO:3.
<Перечень последовательности 3>
SEQ ID NO:3 представляет собой последовательность ДНК белка сайта экспрессии, который амплифицирован с использованием праймеров SEQ ID NO:1 и 2 для экспрессии рецептора 1 тахикинина человека.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ЦИКЛОГЕКСИЛПИРИДИНА | 2015 |
|
RU2681316C2 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛПИРИДИНА И ЛЕКАРСТВЕННОЕ СРЕДСТВО НА ИХ ОСНОВЕ | 2001 |
|
RU2277087C2 |
| ПИРАНОДИПИРИДИНОВОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2743998C2 |
| ПРОИЗВОДНЫЕ ДИФЕНИЛА И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2000 |
|
RU2238266C2 |
| ПРОИЗВОДНЫЕ 3-ФЕНИЛПИРИДИНА И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2000 |
|
RU2236402C2 |
| ПРОИЗВОДНЫЕ 4-ФЕНИЛПИРИДИНА И ЛЕКАРСТВЕННОЕ СРЕДСТВО | 2001 |
|
RU2276139C2 |
| ПРОИЗВОДНЫЕ (3-МЕТИЛПИРРОЛИДИН-3-ИЛ)-МЕТИЛ ПИРИДИНИЛОВОГО ЭФИРА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ РЕЦЕПТОРА NK-3 | 2011 |
|
RU2625798C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ, ИХ ПРИМЕНЕНИЕ И СПОСОБ ЛЕЧЕНИЯ | 2003 |
|
RU2294927C2 |
| 3-АЗЕТИДИНИЛАЛКИЛПИПЕРИДИНЫ ИЛИ ПИРРОЛИДИНЫ В КАЧЕСТВЕ АНТАГОНИСТОВ ТАХИКИНИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ И СПОСОБ ЛЕЧЕНИЯ | 1996 |
|
RU2158264C2 |
| ДВОЙНЫЕ АНТАГОНИСТЫ NK1/NK3 ДЛЯ ЛЕЧЕНИЯ ШИЗОФРЕНИИ | 2005 |
|
RU2374229C2 |
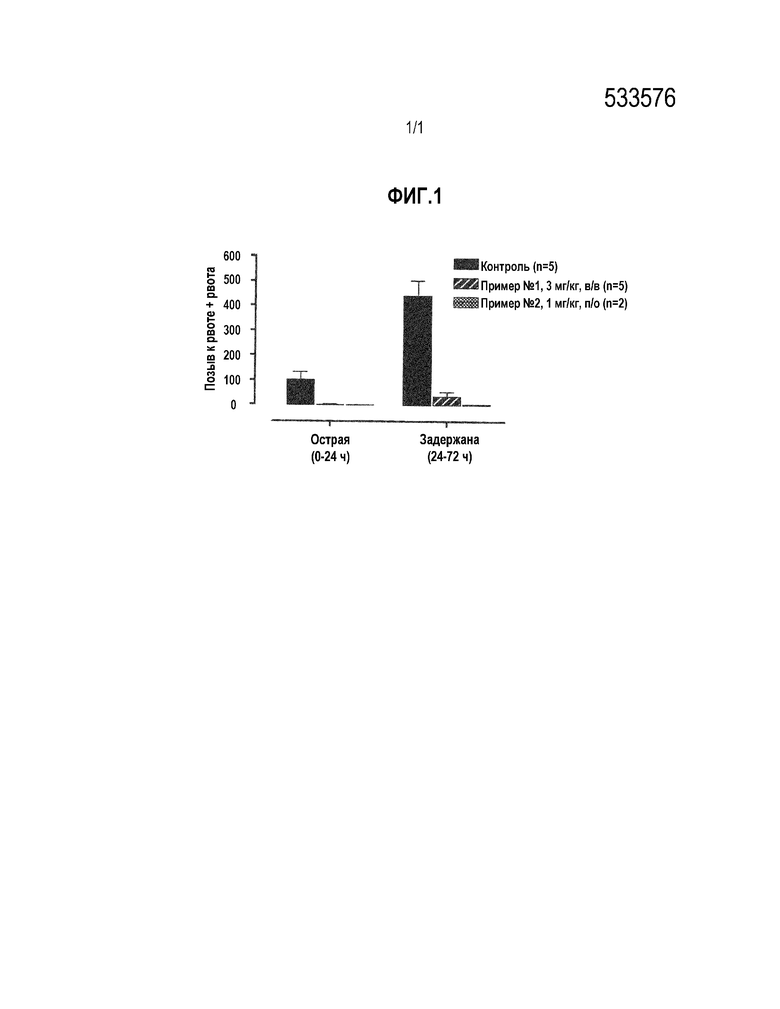
Настоящее изобретение относится к производным карбоксиметилпиперидина, представленным следующей формулой (I):
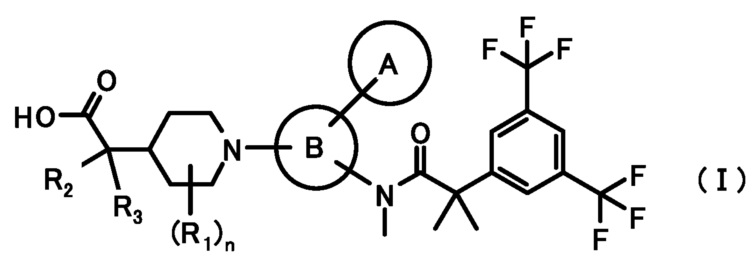
где кольцо A является группой, представленной формулой:
 ;
;
кольцо B является группой, представленной формулой:
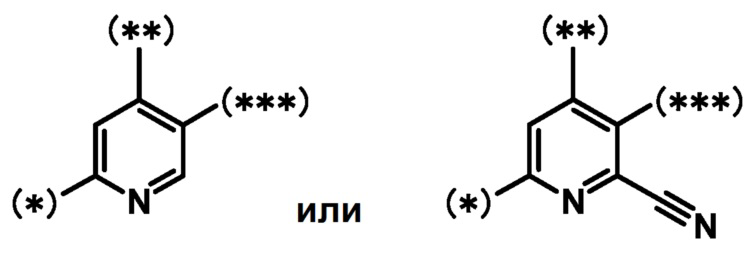 ,
,
при условии, что связи, обозначенные (*), являются местом связывания с формулой:
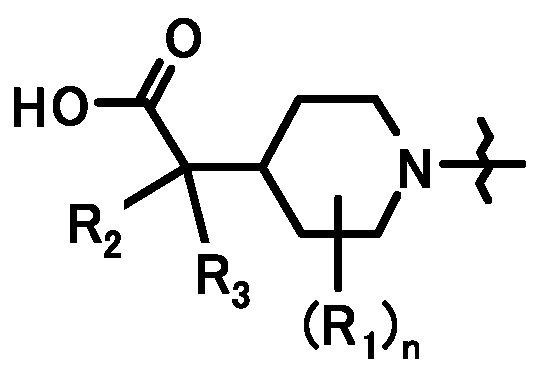 ;
;
связи, обозначенные (**), являются местом связывания с кольцом A; связи, обозначенные (***), являются местом связывания с формулой:
 ;
;
R1 представляет собой C1-6алкил или C1-6алкокси; R2 и R3, каждый независимо, представляют собой атом водорода или метил; n равен 0, 1, 2, 3, 4 или 5; или его фармацевтически приемлемым солям, которые обладают антагонистической активностью в отношении рецептора NK1 и которые полезны для предупреждения или лечения индуцированной противораковой химиотерапией тошноты или рвоты. 7 н. и 5 з.п. ф-лы, 1 ил., 14 табл., 12 пр.
1. Соединение, представленное формулой (I):
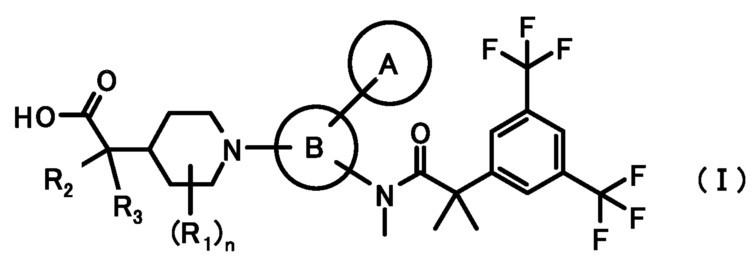
где кольцо A является группой, представленной формулой:
 ;
;
кольцо B является группой, представленной формулой:
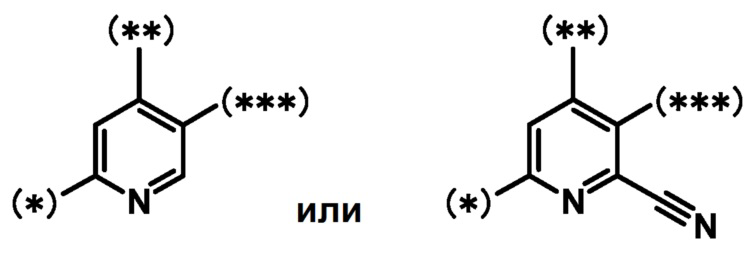 ,
,
при условии, что связи, обозначенные (*), являются местом связывания с формулой:
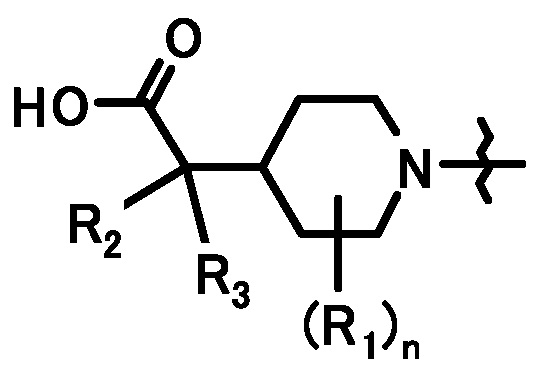 ;
;
связи, обозначенные (**), являются местом связывания с кольцом A;
связи, обозначенные (***), являются местом связывания с формулой:
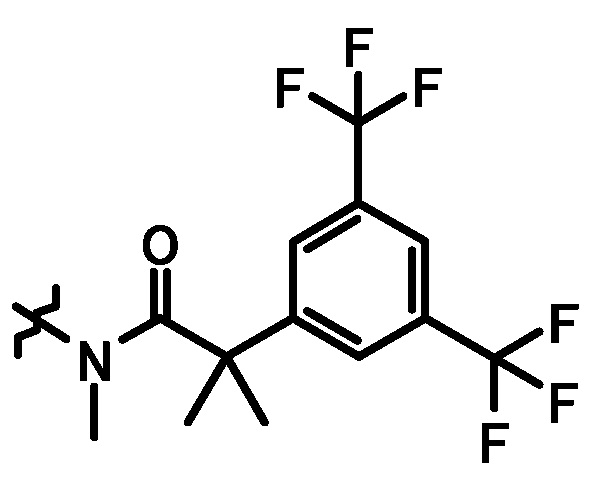 ;
;
R1 представляет собой C1-6алкил или C1-6алкокси;
R2 и R3, каждый независимо, представляют собой атом водорода или метил;
n равен 0, 1, 2, 3, 4 или 5;
или его фармацевтически приемлемая соль.
2. Соединение по п.1, представленное формулой (Ia):
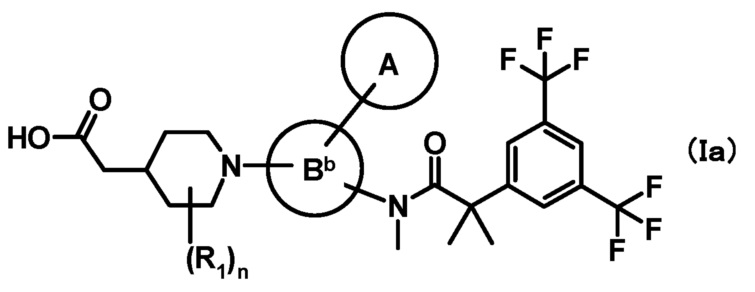 ,
,
где кольцо A, R1 и n имеют такие же значения, как описано в п.1;
кольцо Bb является группой, представленной формулой:
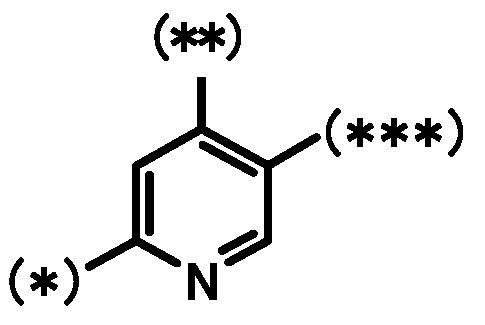 ,
,
где связь, обозначенная (*), является местом связывания с формулой:
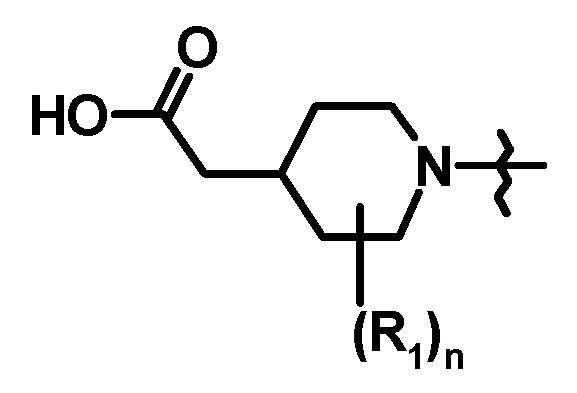 ;
;
(**) и (***) имеют такие же значения, как описано в п.1;
или его фармацевтически приемлемая соль.
3. Соединение по п.2, представленное формулой (Ib):
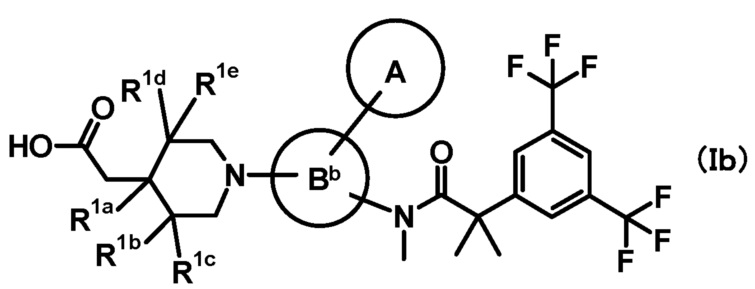
где кольцо A и кольцо Bb имеют такие же значения, как описано в п.2,
при условии, что связь, обозначенная (*), является местом связывания с формулой:
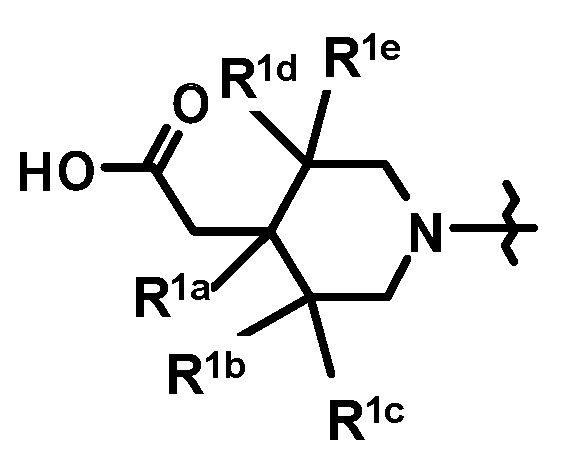 ;
;
(**) и (***) имеют такие же значения, как описано в п.2;
R1a, R1b, R1c, R1d и R1e, каждый независимо, представляют собой любой из атома водорода, метила или метокси;
или его фармацевтически приемлемая соль.
4. Соединение по п.1, где кольцо B представлено следующей формулой:
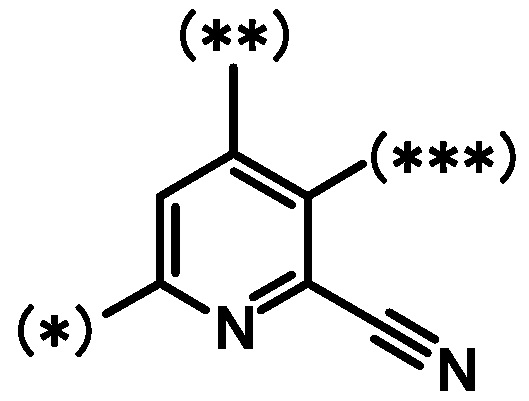 ,
,
где (*), (**) и (***) имеют такие же значения, как описано в п.1;
или его фармацевтически приемлемая соль.
5. Соединение по п.4, где R1 представляет собой метил и n равен 0 или 1, или его фармацевтически приемлемая соль.
6. Соединение, представленное следующей формулой:
 ,
,
или его фармацевтически приемлемая соль.
7. Соединение, представленное следующей формулой:
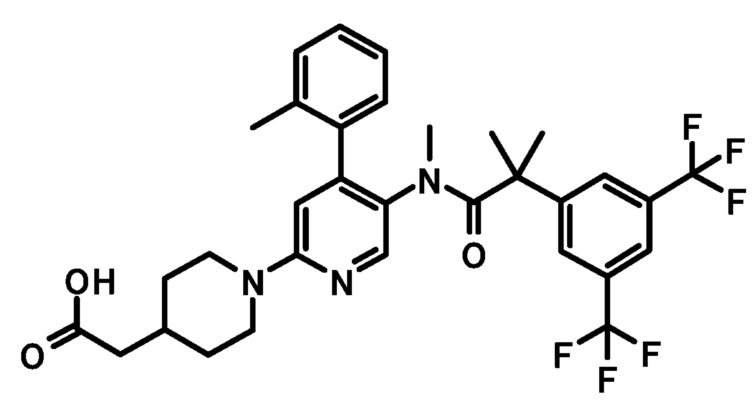 ,
,
или его фармацевтически приемлемая соль.
8. Соединение, представленное следующей формулой:
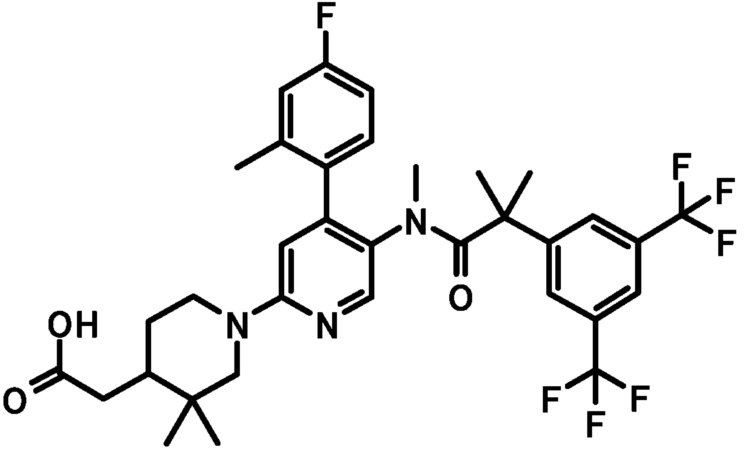 ,
,
или его фармацевтически приемлемая соль.
9. Соединение, представленное следующей формулой:
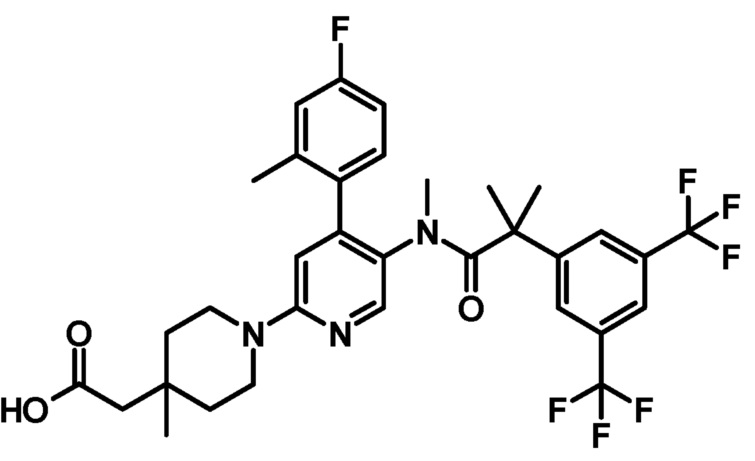 ,
,
или его фармацевтически приемлемая соль.
10. Соединение, представленное следующей формулой:
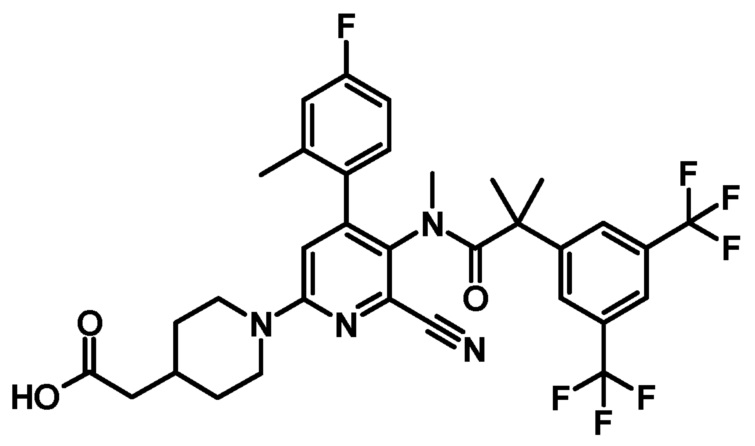 ,
,
или его фармацевтически приемлемая соль.
11. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении рецептора NK1, содержащая в качестве активного ингредиента соединение по любому из пп.1-10 или его фармацевтически приемлемую соль.
12. Фармацевтическая композиция по п.11 для применения при предупреждении индуцированной противораковой химиотерапией тошноты или рвоты.
| СПОСОБ ВЫРАБОТКИ КОНСЕРВОВ "СИЧЕНИКИ РЫБНЫЕ УКРАИНСКИЕ" | 2007 |
|
RU2347422C1 |
| WO 2005002577 A1, 13.01.2005 | |||
| US 6770637 B2, 03.08.2004 | |||
| 2,6-ЗАМЕЩЕННЫЕ-4-МОНОЗАМЕЩЕННЫЕ АМИНОПИРИМИДИНЫ КАК АНТАГОНИСТЫ РЕЦЕПТОРОВ ПРОСТАГЛАНДИНА D2 | 2007 |
|
RU2431631C2 |
| 2-АМИНОБЕНЗОКСАЗОЛКАРБОКСАМИДЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ 5-НТ3 | 2007 |
|
RU2448105C2 |

