ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет, согласно 35 U.S.C. 119(e), по заявке США с серийным номером № 61/222918, поданной 2 июля 2009 года, которая включена в настоящий документ в качестве ссылки в полном объеме для всех целей.
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Предусмотрены соединения пиразолопиримидина формулы I, которые являются ингибиторами одной или нескольких Janus-киназ, а также композиции, содержащие эти соединения, и способы применения, включая, но не ограничиваясь ими, диагностику in vitro, in situ и in vivo на клетках млекопитающих или лечение.
УРОВЕНЬ ТЕХНИКИ
Каскады цитокинов опосредуют широкий диапазон биологических функций, включая многие аспекты воспаления и иммунитета. Janus-киназы (JAK), включающие JAK1, JAK2, JAK3 и TYK2, представляют собой цитоплазматические протеинкиназы, которые связываются с рецепторами цитокинов типа I и типа II и регулируют передачу сигнала цитокинов. Встреча цитокина с его собственными рецепторами запускает активацию ассоциированных с рецептором JAK и это приводит к опосредуемому JAK фосфорилированию тирозина белков трансдукторов сигнала и активаторов транскрипции (STAT) и в конечном итоге к транскрипционной активации определенных групп генов (Schindler et al., 2007, J Biol. Chem. 282: 20059-63). JAK1, JAK2 и TYK2 проявляют широкие паттерны экспрессии генов, в то время как экспрессия JAK3 ограничивается лейкоцитами. Рецепторы цитокинов, как правило, являются функциональными в качестве гетеродимеров, и в результате с комплексами цитокин-рецептор обычно ассоциировано более одного типа JAK-киназы. Конкретные JAK, ассоциированные с различными комплексами цитокин-рецептор, были определены во многих случаях с помощью генетических исследований и подтверждены другими экспериментальными данными.
JAK1 первоначально была идентифицирована в скрининге новых киназ (Wilks A.F., 1989, Proc. Natl. Acad. Sci. U.S.A. 86:1603-1607). Генетические и биохимические исследования показали, что JAK1 функционально и физически ассоциирована с комплексами рецептор-цитокин интерферона типа I (например, IFN-альфа), интерферона типа II (например, IFN-гамма), IL-2 и IL-6 (Kisseleva et al., 2002, gene 285:1-24; Levy et al., 2005, Nat. Rev. Mol. Cell Biol. 3:651-662; O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). Мыши с нокаутом JAK1 погибают перинатально вследствие дефектов передачи сигнала рецептора LIF (Kisseleva et al., 2002, gene 285:1-24; O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). Охарактеризация тканей, полученных из мышей с нокаутом JAK1, продемонстрировала критические роли для этой киназы в каскадах IFN, IL-10, IL-2/IL-4 и IL-6. Недавно Европейская комиссия по лечению ревматоидного артрита от умеренного до тяжелого одобрила гуманизированное моноклональное антитело, нацеленное на каскад IL-6 (тоцилизумаб) (Scheinecker et al., 2009, Nat. Rev. Drug Discov. 8:273-274).
Биохимические и генетические исследования показали ассоциацию между JAK2 и семействами одноцепочечных рецепторов цитокинов (например, EPO), IL-3 и интерферона-гамма (Kisseleva et al., 2002, gene 285:1-24; Levy et al., 2005, Nat. Rev. Mol. Cell Biol. 3:651-662; O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). В соответствии с этим, мыши с нокаутом JAK2 погибают от анемии (O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). Активирующие киназу мутации в JAK2 (например, JAK2 V617F) ассоциированы с миелопролиферативными нарушениями (MPD) у человека.
JAK3 связывается исключительно с гамма-цепью общего рецептора цитокинов, который присутствует в комплексах рецептор-цитокин IL-2, IL-4, IL-7, IL-9, IL-15 и IL-21. JAK3 важна для развития и пролиферации лимфоидных клеток и мутации в JAK3 приводят к тяжелому комбинированному иммунодефициту (SCID) (O'Shea et al., 2002, Cell, 109 (suppl.): S121-S131). Исходя из их роли в регуляции лимфоцитов, проводили нацеливание на опосредуемые JAK3 и JAK3 каскады для иммунодепрессивных показаний (например, отторжение трансплантата и ревматоидный артрит) (Baslund et al., 2005, Arthritis & Rheumatism 52:2686-2692; Changelian et al., 2003, Science 302:875-878).
TYK2 связывается с комплексами рецептор-цитокин интерферона типа I (например, IFNальфа), IL-6, IL-10, IL-12 и IL-23 (Kisseleva et al., 2002, gene 285:1-24; Watford, W.T. & O'Shea, J.J., 2006, Immunity 25:695-697). В соответствии с этим, первичные клетки, происходящие из человека с дефицитом TYK2, являются дефектными по передаче сигнала интерферона типа I, IL-6, IL-10, IL-12 и IL-23. Недавно полностью человеческое моноклональное антитело, нацеленное на общую субъединицу p40 цитокинов IL-12 и IL-23 (устекинумаб), было одобрено Европейской комиссией по лечению бляшковидного псориаза от умеренного до тяжелого (Krueger et al., 2007, N. Engl. J. Med. 356:580-92; Reich et al., 2009, Nat. Rev. Drug Discov. 8:355-356). Кроме того, проведены клинические испытания антитела, нацеленного на каскады IL-12 и IL-23, для лечения болезни Крона (Mannon et al., 2004, N. Engl. J. Med. 351:2069-79).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Один из вариантов осуществления относится к соединению формулы I:

его энантиомерам, диастереомерам, таутомерам или фармацевтически приемлемым солям, где R1, R2 и R3 являются такими, как определено в настоящем документе.
Другой вариант осуществления относится к фармацевтической композиции, которая включает соединение формулы I и фармацевтически приемлемый носитель, адъювант или наполнитель.
Другой вариант осуществления включает способ лечения или снижения тяжести заболевания или состояния, отвечающего на ингибирование активности одной или нескольких Janus-киназ, выбранных из JAK1, JAK2, JAK3 и TYK2, у пациента. Способ включает введение пациенту терапевтически эффективного количества соединения формулы I.
Другой вариант осуществления относится к применению соединения формулы I для терапии.
Другой вариант осуществления относится к применению соединения формулы I для профилактики, лечения или снижения тяжести заболевания. В одном варианте осуществления заболевание представляет собой аутоиммунное заболевание.
Другой вариант осуществления относится к применению соединения формулы I для изготовления лекарственного средства для профилактики, лечения или снижения тяжести заболевания. В одном варианте осуществления заболевание представляет собой аутоиммунное заболевание.
Другой вариант осуществления относится к набору для лечения заболевания или нарушения, отвечающего на ингибирование Janus-киназы. Набор включает первую фармацевтическую композицию, содержащую соединение формулы I, и инструкции по применению.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Далее приводится подробное описание определенных вариантов осуществления, примеры которых проиллюстрированы прилагаемыми структурами и формулами. Хотя изобретение описано применительно к названным вариантам осуществления, подразумевают, что изобретение охватывает все альтернативы, модификации и эквиваленты, которые могут входить в объем настоящего изобретения, определяемый формулой изобретения. Специалисту в данной области известны способы и материалы, сходные или эквивалентные способам и материалам, описанным в настоящем документе, которые можно использовать при осуществлении на практике этого изобретения.
ОПРЕДЕЛЕНИЯ
Термин "алкил" относится к насыщенному линейному или разветвленному одновалентному углеводородному радикалу, где алкильный радикал необязательно может быть независимо замещен одним или несколькими заместителями, описанными в настоящем документе. В одном примере, алкильный радикал имеет от одного до восемнадцати атомов углерода (C1-C18). В других примерах, алкильный радикал представляет собой C0-C6, C0-C5, C0-C3, C1-C12, C1-C10, C1-C8, C1-C6, C1-C5, C1-C4 или C1-C3. C0алкил относится к связи. Примеры алкильных групп включают метил (Me, -CH3), этил (Et, -CH2CH3), 1-пропил (n-Pr, н-пропил, -CH2CH2CH3), 2-пропил (i-Pr, изопропил, -CH(CH3)2), 1-бутил (n-Bu, н-бутил, -CH2CH2CH2CH3), 2-метил-1-пропил (i-Bu, изобутил, -CH2CH(CH3)2), 2-бутил (s-Bu, втор-бутил, -CH(CH3)CH2CH3), 2-метил-2-пропил (t-Bu, трет-бутил, -C(CH3)3), 1-пентил (н-пентил, -CH2CH2CH2CH2CH3), 2-пентил (-CH(CH3)CH2CH2CH3), 3-пентил (-CH(CH2CH3)2), 2-метил-2-бутил (-C(CH3)2CH2CH3), 3-метил-2-бутил (-CH(CH3)CH(CH3)2), 3-метил-1-бутил (-CH2CH2CH(CH3)2), 2-метил-1-бутил (-CH2CH(CH3)CH2CH3), 1-гексил (-CH2CH2CH2CH2CH2CH3), 2-гексил (-CH(CH3)CH2CH2CH2CH3), 3-гексил (-CH(CH2CH3)(CH2CH2CH3)), 2-метил-2-пентил (-C(CH3)2CH2CH2CH3), 3-метил-2-пентил (-CH(CH3)CH(CH3)CH2CH3), 4-метил-2-пентил (-CH(CH3)CH2CH(CH3)2), 3-метил-3-пентил (-C(CH3)(CH2CH3)2), 2-метил-3-пентил (-CH(CH2CH3)CH(CH3)2), 2,3-диметил-2-бутил (-C(CH3)2CH(CH3)2), 3,3-диметил-2-бутил (-CH(CH3)C(CH3)3, 1-гептил и 1-октил.
Группы типа (C0-Cnалкил)R включают алкильные группы, замещенные группой R в любом из атомов в группе, доступных для замещения (в иллюстративном варианте осуществления n представляет собой число, равное 1-6, и R представляет собой -OH, -OCH3, -NH2, -N(CH3)2, -CN, галоген, C3-C6циклоалкил, фенил или 3-9-членный гетероциклил). Например, группа (C0-C3алкил)CN включает группы -CN, -CH2CN, -CH2CH2CN, -CH(CN)CH3, -CH2CH2CH2CN, -CH(CN)CH2CH3, -CH2CH(CN)CH3, -C(CH3)2CN, -C(CH2CN)CH3. Например, группа (C0-C2алкил)C3циклоалкил включает группы: 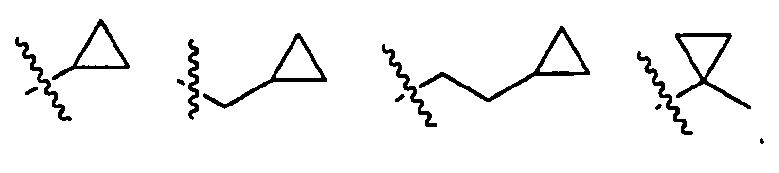 .
.
Термин "алкенил" относится к линейному или разветвленному одновалентному углеводородному радикалу по меньшей мере с одним участком ненасыщенности, т.е. углерод-углеродной двойной связью, где алкенильный радикал может быть необязательно независимо замещен одним или несколькими заместителями, описанными в настоящем документе, и включает радикалы, имеющие ориентацию "цис" и "транс", или альтернативно ориентацию "E" и "Z". В одном примере алкенильный радикал имеет от двух до восемнадцати атомов углерода (C2-C18). В других примерах алкенильный радикал представляет собой C2-C12, C2-C10, C2-C8, C2-C6 или C2-C3. Примеры включают, но не ограничиваются ими, этенил или винил (-CH2=CH2), проп-1-енил (-CH=CHCH3), проп-2-енил (-CH2CH=CH2), 2-метилпроп-1-енил, бут-1-енил, бeт-2-енил, бут-3-енил, бута-1,3-диенил, 2-метилбута-1,3-диен, гекс-1-енил, гекс-2-енил, гекс-3-енил, гекс-4-енил и гекса-1,3-диенил.
Термин "алкинил" относится к линейному или разветвленному одновалентному углеводородному радикалу по меньшей мере с одним участком ненасыщенности, т.е. углерод-углеродной тройной связью, где алкинильный радикал необязательно может быть независимо замещен одним или несколькими заместителями, описанными в настоящем документе. В одном примере алкинильный радикал имеет от двух до восемнадцати атомов углерода (C2-C18). В других примерах алкинильный радикал представляет собой C2-C12, C2-C10, C2-C8, C2-C6 или C2-C3. Примеры включают, но не ограничиваются ими, этинил (-C≡CH), проп-1-инил (-C≡CCH3), проп-2-инил (пропаргил, -CH2C≡CH), бут-1-инил, бут-2-инил и бут-3-инил.
"Циклоалкил" относится к неароматической, насыщенной или частично ненасыщенной группе углеводородного кольца, где циклоалкильная группа может быть необязательно независимо замещена одним или несколькими заместителями, описанными в настоящем документе. В одном примере циклоалкильная группа имеет от 3 до 12 атомов углерода (C3-C12). В других примерах циклоалкил представляет собой C3-C8, C3-C10 или C5-C10. В других примерах циклоалкильная группа, в качестве моноцикла, представляет собой C3-C8, C3-C6 или C5-C6. В другом примере циклоалкильная группа, в качестве бицикла, представляет собой C7-C12. Примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, 1-циклопент-1-енил, 1-циклопент-2-енил, 1-циклопент-3-енил, циклогексил, 1-циклогекс-1-енил, 1-циклогекс-2-енил, 1-циклогекс-3-енил, циклогексадиенил, циклогептил, циклооктил, циклононил, циклодецил, циклоундецил и циклододецил. Иллюстративные структуры бициклических циклоалкилов, имеющих от 7 до 12 атомов в кольце, включают, но не ограничиваются ими, [4,4], [4,5], [5,5], [5,6] или [6,6] кольцевые системы. Иллюстративные соединенные мостиковой связью бициклические циклоалкилы включают, но не ограничиваются ими, бицикло[2.2.1]гептан, бицикло[2.2.2]октан и бицикло[3.2.2]нонан.
"Арил" относится к циклической ароматической углеводородной группе, необязательно независимо замещенной одним или несколькими заместителями, описанными в настоящем документе. В одном примере арильная группа имеет 6-20 атомов углерода (C6-C20). В другом примере арильная группа представляет собой C6-C9. В другом примере арильная группа представляет собой C6арильную группу. Арил включает бициклические группы, содержащие ароматическое кольцо, с конденсированным неароматическим или частично насыщенным кольцом. Примеры арильных групп включают, но не ограничиваются ими, фенил, нафталенил, антраценил, инденил, инданил, 1,2-дигидронафталенил и 1,2,3,4-тетрагидронафтил. В одном примере арил включает фенил.
"Галоген" или "галоген" относятся к F, Cl, Br или I.
Термины "гетероцикл", "гетероциклил" и "гетероциклическое кольцо" используют в настоящем документе взаимозаменяемо и они относятся к: (i) насыщенной или частично ненасыщенной циклической группе (т.е. имеющей одну или несколько двойных и/или тройных связей в кольце) ("гетероциклоалкил"), или (ii) ароматическую циклическую группу ("гетероарил"), и, в каждом случае, в которых по меньшей мере один атома кольца представляет собой гетероатом, независимо выбранный из азота, кислорода, фосфора и серы, а остальные атомы кольца представляют собой углерод. Гетероциклильная группа необязательно может быть замещена одним или несколькими заместителями, описанными ниже. В одном варианте осуществления гетероциклил включает моноциклы или бициклы, имеющие от 1 до 9 углеродных членов кольца (C1-C9), а остальные атомы кольца представляют собой гетероатомы, выбранные из N, O, S и P. В других примерах гетероциклил включает моноциклы или бициклы, имеющие C1-C5, C3-C5 или C4-C5, где остальные атомы кольца представляют собой гетероатомы, выбранные из N, O, S и P. В другом варианте осуществления гетероциклил включает 3-7-членные кольца или 3-6-членные кольца, содержащие один или несколько гетероатомов, независимо выбранных из N, O, S и P. В других примерах гетероциклил включает моноциклические 3-, 4-, 5-, 6- или 7-членные кольца, содержащие один или несколько гетероатомов, независимо выбранных из N, O, S и P. В другом варианте осуществления гетероциклил включает би- или полициклические или соединенные мостиковой связью 4-, 5-, 6-, 7-, 8- и 9-членные кольцевые системы, содержащие один или несколько гетероатомов, независимо выбранных из N, O, S и P. Примеры бициклических систем включают, но не ограничиваются ими, [3,5], [4,5], [5,5], [3,6], [4,6], [5,6] или [6,6] системы. Примеры соединенных мостиковой связью систем включают, но не ограничиваются ими, [2.2.1], [2.2.2], [3.2.2] и [4.1.0] структуры, имеющие от 1 до 3 гетероатомов, выбранных из N, O, S и P. В другом варианте осуществления гетероциклил включает спирогруппы, имеющие от 1 до 4 гетероатомов, выбранных из N, O, S и P. Гетероциклильная группа может представлять собой присоединенную через углерод группу или присоединенную через гетероатом группу. "Гетероциклил" включает гетероциклильную группу, конденсированную с циклоалкильной группой.
Иллюстративные гетероциклильные группы включают, но не ограничиваются ими, оксиранил, азиридинил, тиоранил, азетидинил, оксетанил, тиэтанил, 1,2-дитиэтанил, 1,3-дитиэтанил, пирролидинил, пиперидинил, морфолинил, тиоморфолинил, тиоксанил, пиперазинил, гомопиперазинил, гомопиперидинил, оксепанил, тиэпанил, оксазепинил, оксазепанил, диазепанил, 1,4-диазепанил, диазепинил, тиазепинил, тиазепанил, дигидротиенил, дигидропиранил, дигидрофуранил, тетрагидрофуранил, тетрагидротиенил, тетрагидропиранил, тетрагидротиопиранил, 1-пирролинил, 2-пирролинил, 3-пирролинил, индолинил, 2H-пиранил, 4H-пиранил, диоксанил, 1,3-диоксоланил, пиразолинил, пиразолидинил, дитианил, дитиоланил, пиразолидинилимидазолинил, имидазолидинил, 3-азабицикло[3.1.0]гексанил, 3,6-диазабицикло[3.1.1]гептанил, 6-азабицикло[3.1.1]гептанил, 3-азабицикло[3.1.1]гептанил, 3-азабицикло[4.1.0]гептанил и азабицикло[2.2.2]гексанил. Примерами гетероциклильной группы, где атом кольца замещен оксо (=O), являются дигидропиридинонил, пиридинонил, пиперидинонил, пирролидинонил, пиримидинонил, дигидропиримидинонил, пиперазинонил, пиразинонил, пиридазинонил, дигидропиридазинонил, дигидропирролонил, пирролонил, оксазолидинонил, тиазолидинонил, имидазолидинонил, 1-оксотиенил, 1,1-диоксотиенил, 1-оксотетрагидротиенил, 1,1-диоксотетрагидротиенил и 1,1-диоксотиоморфолинил. Гетероциклильные группы, представленные в настоящем документе, необязательно независимо замещены одним или несколькими заместителями, описанными в настоящем документе. Гетероциклы описаны в Paquette, Leo A.; "Principles of Modern Heterocyclic Chemistry" (W.A. Benjamin, New York, 1968), в частности, главы 1, 3, 4, 6, 7 и 9; "The Chemistry of Heterocyclic Compounds, A series of Monographs" (John Wiley & Sons, New York, с 1950 по настоящее время), в частности, тома 13, 14, 16, 19 и 28; и J. Am. Chem. Soc. (1960) 82:5566.
Термин "гетероарил" относится к ароматическому карбоциклическому радикалу, в котором по меньшей мере один атом кольца представляет собой гетероатом, независимо выбранный из азота, кислорода и серы, а остальные атомы кольца представляют собой углерод. Гетероарильные группы необязательно могут быть замещены одним или несколькими заместителями, описанными в настоящем документе. В одном примере гетероарильная группа содержит от 1 до 9 углеродных атомов кольца (C1-C9). В других примерах гетероарильная группа представляет собой C1-C5, C3-C5 или C4-C5. В одном варианте осуществления иллюстративные гетероарильные группы включают моноциклические ароматические 5-, 6- и 7-членные кольца, содержащие один или несколько гетероатомов, независимо выбранных из азота, кислорода и серы. В другом варианте осуществления иллюстративные гетероарильные группы включают конденсированные кольцевые системы из вплоть до 9 атомов углерода, где по меньшей мере одно ароматическое кольцо содержит один или несколько гетероатомов, независимо выбранных из азота, кислорода и серы. Конденсированные системы могут быть конденсированными в одной или нескольких точках на кольцах. "Гетероарил" включает гетероарильные группы, конденсированные с арилом, циклоалкилом или другой гетероциклильной группой. Примеры гетероарильных групп включают, но не ограничиваются ими, пиридинил, имидазолил, имидазопиридинил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, хинолинил, изохинолинил, индолил, бензимидазолил, бензофуранил, циннолинил, индазолил, индолизинил, фталазинил, пиридазинил, триазинил, изоиндолил, птеридинил, пуринил, оксадиазолил, триазолил, тиадиазолил, фуразанил, бензофуразанил, бензотиофенил, бензотиазолил, бензоксазолил, хиназолинил, хиноксалинил, нафтиридинил и фуропиридинил.
В определенных вариантах осуществления гетероциклильная или гетероарильная группа присоединена через C. В качестве неограничивающего примера, связанные через углерод гетероциклилы включают расположение связей в положении 2, 3, 4, 5 или 6 пиридина, в положении 3, 4, 5 или 6 пиридазина, в положении 2, 4, 5 или 6 пиримидина, в положении 2, 3, 5 или 6 пиразина, в положении 2, 3, 4 или 5 фурана, тетрагидрофурана, тиофурана, тиофена, пиррола или тетрагидропиррола, в положении 2, 4 или 5 оксазола, имидазола или тиазола, в положении 3, 4 или 5 изоксазола, пиразола или изотиазола, в положении 2 или 3 азиридина, в положении 2, 3 или 4 азетидина, в положении 2, 3, 4, 5, 6, 7 или 8 хинолина или в положении 1, 3, 4, 5, 6, 7 или 8 изохинолина (2-пиридил, 3-пиридил, 4-пиридил, 5-пиридил, 6-пиридил).
В определенных вариантах осуществления гетероциклильная или гетероарильная группа присоединена через N. В качестве неограничивающего примера, связанная через азот гетероциклильная или гетероарильная группа включает расположение связи в положении 1 азиридина, азетидина, пиррола, пирролидина, 2-пирролина, 3-пирролина, имидазола, имидазолидина, 2-имидазолина, 3-имидазолина, пиразола, пиразолина, 2-пиразолина, 3-пиразолина, пиперидина, пиперазина, индола, индолина, 1H-индазола, положении 2 изоиндола или изоиндолина, положении 4 морфолина и положении 9 карбазола или β-карболина.
"Лечить" или "лечение" включает как терапевтические, так и профилактические или превентивные меры, где задачей является предотвращение или замедление (уменьшение) нежелательного физиологического изменения или нарушения, такого как развитие или распространение злокачественной опухоли. Для целей этого изобретения, благоприятные или желательные клинические результаты включают, но не ограничиваются ими, смягчение симптомов, уменьшение степени заболевания, стабилизацию (т.е. не ухудшение) состояния заболевания, задержку или замедление прогрессирования заболевания, смягчение или облегчение болезненного состояния и ремиссию (как частичную, так и общую), как поддающиеся выявлению, так и не поддающиеся выявлению. "Лечение" также может означать продление выживания по сравнению с ожидаемым выживанием без проведения лечения. Нуждающиеся в лечении индивидуумы включают индивидуумов, уже имеющих состояние или нарушение, а также индивидуумов, предрасположенных к состоянию или нарушению (например, вследствие генетической мутации), или индивидуумов, у которых состояние или нарушение подлежит профилактике.
Выражение "терапевтически эффективное количество" означает количество соединения по настоящему изобретению, которое (i) лечит или предупреждает конкретное заболевание, состояние или нарушение, (ii) ослабляет, смягчает или устраняет один или несколько симптомов конкретного заболевания, состояния или нарушения, или (iii) предупреждает или замедляет возникновение одного или нескольких симптомов конкретного заболевания, состояния или нарушения, описанного в настоящем документе. В случае злокачественной опухоли, терапевтически эффективное количество лекарственного средства может уменьшить количество злокачественных клеток; уменьшить размер опухоли; ингибировать (т.е. замедлять в некоторой степени и предпочтительно останавливать) инфильтрацию злокачественных клеток в периферические органы; ингибировать (т.е. замедлять в некоторой степени и предпочтительно останавливать) метастазирование опухоли; ингибировать, в некоторой степени, рост опухоли; и/или смягчать в некоторой степени один или несколько симптомов, ассоциированных со злокачественной опухолью. По степени, с которой лекарственное средство может предупреждать рост и/или уничтожать существующие злокачественные клетки, оно может быть цитостатическим и/или цитотоксическим. Для терапии злокачественной опухоли эффективность можно определять, например, путем оценки времени до прогрессирования заболевания (TTP) и/или определения показателя ответа (RR).
Термин "биодоступность" относится к системной доступности (т.е. уровням в крови/плазме) данного количества лекарственного средства, введенного пациенту. Биодоступность представляет собой абсолютный показатель для введенной дозированной формы, который указывает на меру как времени (скорости), так и общего количества (степени) лекарственного средства, которое достигает общего кровотока.
"Воспалительное нарушение", как используют в настоящем документе, может относиться к любому заболеванию, нарушению или синдрому, при котором чрезмерный или нерегулируемый воспалительный ответ приводит к чрезмерным воспалительным симптомам, повреждению тканей хозяина или потере функции ткани. "Воспалительное нарушение" также относится к патологическому состоянию, опосредуемому притоком лейкоцитов и/или хемотаксисом нейтрофилов.
"Воспаление", как используют в настоящем документе, относится к локализованному защитному ответу, вызываемому повреждением или разрушением тканей, который служит для разрушения, ослабления или отгораживания (изолирования) как повреждающего агента, так и поврежденной ткани. Воспаление в значительной мере ассоциировано с притоком лейкоцитов и/или хемотаксисом нейтрофилов. Воспаление может быть результатом инфекции патогенными организмами и вирусами и неинфекционных путей, таких как травма или реперфузия после инфаркта миокарда или инсульта, иммунный ответ на чужеродный антиген и аутоиммунные ответы. Таким образом, воспалительные нарушения, поддающиеся лечению соединениями формулы I, охватывают нарушения, ассоциированные с реакциями специфической защитной системы, а также с реакциями неспецифической защитной системы.
"Специфическая защитная система" относится к компоненту иммунной системы, который реагирует на присутствие определенных антигенов. Примеры воспаления вследствие ответа специфической защитной системы включают классический ответ на чужеродные антигены, аутоиммунные заболевания и ответ в виде гиперчувствительности замедленного типа, опосредуемый T-клетками. Другими примерами воспалительных реакций специфической защитной системы являются хронические воспалительные заболевания, отторжение трансплантированных солидных тканей и органов, например, трансплантатов почки и костного мозга и реакция "трансплантат против хозяина" (GVHD).
Термин "неспецифическая защитная система", как используют в настоящем документе, относится к воспалительным нарушениям, которые опосредуются лейкоцитами, которые неспособны к иммунологической памяти (например, гранулоциты и макрофаги). Примеры воспаления, которое является следствием, по меньшей мере частично, реакции неспецифической защитной системы, включают воспаление, ассоциированное с состояниями, такими как взрослый (острый) респираторный дистресс-синдром (ARDS) или синдромы полиорганного повреждения; реперфузионное повреждение; острый гломерулонефрит; реактивный артрит; дерматоз с острыми воспалительными компонентами; острый гнойный менингит или другие воспалительные нарушения центральной нервной системы, такие как инсульт; термическое повреждение; воспалительное заболевание кишечника; ассоциированные с трансфузией гранулоцитов синдромы и индуцируемую цитокинами токсичность.
"Аутоиммунное заболевание", как используют в настоящем документе, относится к любой группе нарушений, в которых повреждение ткани ассоциировано с гуморальными или клеточно-опосредуемыми ответами на собственные компоненты организма.
"Аллергическое заболевание", как используют в настоящем документе, относится к любым симптомам, повреждению ткани или утрате функции ткани вследствие аллергии. "Артритическое заболевание", как используют в настоящем документе, относится к любому заболеванию, которое характеризуется воспалительными очагами повреждения в суставах, приписываемыми различной этиологии. "Дерматит", как используют в настоящем документе, относится к любому из широкого семейства заболеваний кожи, которые характеризуются воспалением кожи, приписываемым различной этиологии. "Отторжение трансплантата", как используют в настоящем документе, относится к любой иммунной реакции, направленной против трансплантированной ткани, такой как органы или клетки (например, костный мозг), характеризующейся утратой функции трансплантированных и окружающих тканей, болью, припухлостью, лейкоцитозом и тромбоцитопенией. Терапевтические способы по настоящему изобретению включают способы лечения нарушений, ассоциированных с активацией воспалительных клеток.
"Активация воспалительных клеток" относится к индукции стимулом (включая, но не ограничиваясь ими, цитокины, антигены или аутоантитела) пролиферативного клеточного ответа, продукции растворимых медиаторов (включая, но не ограничиваясь ими, цитокины, радикалы кислорода, ферменты, простаноиды или вазоактивные амины) или экспрессии на клеточной поверхности новых медиаторов или увеличенных количеств медиаторов (включая, но не ограничиваясь ими, антигены главного комплекса гистосовместимости или молекулы клеточной адгезии) в воспалительных клетках (включая, но не ограничиваясь ими, моноциты, макрофаги, T-лимфоциты, B-лимфоциты, гранулоциты (т.е. полиморфноядерные лейкоциты, такие как нейтрофилы, базофилы и эозинофилы), тучные клетки, дендритные клетки, клетки Лангерганса и эндотелиальные клетки). Специалистам в данной области будет понятно, что активация одного или комбинации этих фенотипов в этих клетках может приводить к инициации, сохранению или обострению воспалительного нарушения.
Термин "NSAID" является сокращенным обозначением для "нестероидного противовоспалительного средства" и представляет собой лекарственное средство с болеутоляющими, жаропонижающими (снижение повышенной температуры тела и смягчение боли без нарушения сознания) и, при более высоких дозах, противовоспалительными эффектами (снижение воспаления). Термин "нестероидный" используют, чтобы отличить эти лекарственные средства от стероидов, которые (среди широкого диапазона других эффектов) имеют сходное подавляющее эйкозаноиды противовоспалительное действие. В качестве анальгетиков NSAID являются необычными в том, что они являются ненаркотическими. NSAID включают аспирин, ибупрофен и напроксен. NSAID обычно показаны для лечения острых или хронических состояний, где присутствуют боль и воспаление. NSAID, как правило, показаны для симптоматического смягчения следующих состояний: ревматоидный артрит, остеоартрит, воспалительные артропатии (например, анкилозирующий спондилит, псориатический артрит, синдром Рейтера, острая подагра), дисменорея, метастатическая боль в костях, головная боль и мигрень, послеоперационная боль, боль от мягкой до умеренной вследствие воспаления и повреждения тканей, гипертермия, непроходимость кишечника и почечная колика. Большинство NSAID действуют в качестве неселективных ингибиторов фермента циклооксигеназы, ингибирующих изоферменты как циклооксигеназу-1 (COX-1), так и циклооксигеназу-2 (COX-2). Циклооксигеназа катализирует образование простагландинов и тромбоксана из арахидоновой кислоты (которая сама образуется из клеточного фосфолипидного бислоя с помощью фосфолипазы A2). Простагландины действуют (помимо прочего) в качестве молекул-посредников в процессе воспаления. Ингибиторы COX-2 включают целекоксиб, эторикоксиб, люмиракоксиб, парекоксиб, рофекоксиб, рофекоксиб и вальдекоксиб.
Термины "злокачественная опухоль" и "злокачественный" относятся к или описывают физиологическое состояние у млекопитающих, которое, как правило, характеризуется нерегулируемым ростом клеток. "Опухоль" включает одну или несколько злокачественных клеток. Примеры злокачественной опухоли включают, но не ограничиваются ими, карциному, лимфому, бластому, саркому и лейкоз или лимфоидные злокачественные опухоли. Более конкретные примеры таких злокачественных опухолей включают плоскоклеточный рак (например, эпителиальный плоскоклеточный рак), рак легкого, включая мелкоклеточный рак легкого, немелкоклеточный рак легкого ("NSCLC"), аденокарциному легкого и плоскоклеточную карциному легкого, злокачественную опухоль брюшины, печеночноклеточный рак, гастральный рак или рак желудка, включая желудочно-кишечный рак, рак поджелудочной железы, глиобластому, рак шейки матки, рак яичника, рак печени, рак мочевого пузыря, гепатому, рак молочной железы, рак толстого кишечника, рак прямой кишки, рак ободочной и прямой кишки, карциному эндометрия или матки, карциному слюнной железы, рак почки или ренальный рак, рак предстательной железы, рак женских наружных половых органов, рак щитовидной железы, карциному печени, анальную карциному, карциному полового члена, а также рак головы и шеи.
"Химиотерапевтическое средство" представляет собой химическое соединение, пригодное для лечения злокачественной опухоли. Примеры химиотерапевтических средств включают эрлотиниб (TARCEVA®, Genentech, Inc./OSI Pharm.), трастузумаб (HERCEPTIN®, Genentech, Inc.); бевацизумаб (AVASTIN®, Genentech, Inc.); ритуксимаб (RITUXAN®, Genentech, Inc./Biogen Idec, Inc.), бортезомиб (VELCADE®, Millennium Pharm.), фулвестрант (FASLODEX®, AstraZeneca), сутент (SU11248, Pfizer), летрозол (FEMARA®, Novartis), иматиниба мезилат (GLEEVEC®, Novartis), PTK787/ZK 222584 (Novartis), оксалиплатин (Eloxatin®, Sanofi), 5-FU (5-фторурацил), лейковорин, рапамицин (сиролимус, RAPAMUNE®, Wyeth), лапатиниб (GSK572016, Glaxo Smith Kline), лонафарниб (SCH 66336), сорафениб (BAY43-9006, Bayer Labs) и гефитиниб (IRESSA®, AstraZeneca), AG1478, AG1571 (SU 5271; Sugen), алкилирующие средства, такие как тиотепа и циклофосфамид CYTOXAN®; алкилсульфонаты, такие как бусульфан, импросульфан и пипосульфан; азиридины, такие как бензодопа, карбоквон, метуредопа и уредопа; этиленимины и метиламеламины, включая алтретамин, триэтиленмеламин, триэтиленфосфорамид, триэтилентиофосфорамид и триметилoмеламин; ацетогенины (особенно буллатацин и буллатацинон); камптотецин (включая синтетический аналог топотекан); бриостатин; каллистатин; CC-1065 (включая его синтетические аналоги адозелезин, карзелезин и бизелезин); криптофицины (в частности криптофицин 1 и криптофицин 8); доластатин; дуокармицин (включая синтетические аналоги, KW-2189 и CB1-TM1); элеутеробин; панкратистатин; саркодиктиин; спонгистатин; азотистые иприты, такие как хлорамбуцил, хлорнафазин, хлофосфамид, эстрамустин, ифосфамид, мехлорэтамин, мехлорэтамина оксида гидрохлорид, мелфалан, новэмбихин, фенестерин, преднимустин, трофосфамид, иприт урацила; нитрозомочевину, такую как кармустин, хлорзотоцин, фотемустин, ломустин, нимустин и ранимустин; антибиотики, такие как энедииновые антибиотики (например, калихеамицин, особенно калихеамицин гамма1I и калихеамицин омегаI1 (Angew Chem. Intl. Ed. Engl. (1994) 33:183-186); динемицин, включая динемицин A; бисфосфонаты, такие как клодронат; эсперамицин; а также хромофор на основе неокарциностатина и сходные хромопротеиновые хромофоры на основе энедииновых антибиотиков), аклациномизины, актиномицин, аутрамицин, азасерин, блеомицины, кактиномицин, карабицин, карминомицин, карцинофилин, хромомицины, дактиномицин, даунорубицин, деторубицин, 6-диазо-5-оксо-L-норлейцин, ADRIAMYCIN® (доксорубицин), морфолинодоксорубицин, цианоморфолинодоксорубицин, 2-пирролинодоксорубицин и дезоксидоксорубицин), эпирубицин, эзорубицин, идарубицин, марселломицин, митомицины, такие как митомицин C, микофеноловую кислоту, ногаламицин, оливомицины, пепломицин, потфиромицин, пуромицин, квеламицин, родорубицин, стрептонигрин, стрептозоцин, туберцидин, убенимекс, зиностатин, зорубицин; антиметаболиты, такие как метотрексат и 5-фторурацил (5-FU); аналоги фолиевой кислоты, такие как деноптерин, метотрексат, птероптерин, триметрексат; аналоги пуринов, такие как флударабин, 6-меркаптопурин, тиамиприн, тиогуанин; аналоги пиримидинов, такие как анцитабин, азацитидин, 6-азауридин, кармофур, цитарабин, дидезоксиуридин, доксифлуридин, эноцитабин, флоксуридин; андрогены, такие как калустерон, дромостанолона пропионат, эпитиостанол, мепитиостан, тестолактон; антиадренальные средства, такие как аминоглутетимид, митотан, трилостан; средства для восполнения фолиевой кислоты, такие как фролиновая кислота; ацеглатон; альдофосфамид гликозид; аминолевулиновую кислоту; энилурацил; амсакрин; бестрабуцил; бисантрен; эдатраксат; дефофамин; демекольцин; диазиквон; эльфорнитин; эллиптиний ацетат; эпотилон; этоглуцид; нитрат галлия; гидроксимочевину; лентинан; лонидаинин; майтанзиноиды, такие как майтанзин и ансамитоцины; митогуазон; митоксантрон; мопиданмол; нитраэрин; пентостатин; фенамет; пирарубицин; лозоксантрон; подофиллиновую кислоту; 2-этилгидразид; прокарбазин; полисахаридный комплекс PSK® (JHS Natural Products, Eugene, OR); разоксан; ризоксин; сизофиран; спирогерманий; тенуазоновую кислоту; триазиквон; 2,2',2''-трихлортриэтиламин; трихотецены (особенно токсин T-2, верракурин A, роридин A и ангуидин); уретан; виндезин; дакарбазин; манномустин; митобронитол; митолактол; пипоброман; гацитозин; арабинозид ("Ara-C"); циклофосфамид; тиотепу; таксоиды, например, TAXOL® (паклитаксел; Bristol-Myers Squibb Oncology, Princeton, N.J.), ABRAXANE® (без кремофора), составы сконструированного с альбумином паклитаксела в форме наночастиц (American Pharmaceutical Partners, Schaumberg, Illinois), и TAXOTER® (доксетаксел; Rhône-Poulenc Rorer, Antony, Франция); хлоранбуцил; GEMZAR® (гемцитабин); 6-тиогуанин; меркаптопурин; метотрексат; аналоги платины, такие как цисплатин и карбоплатин; винбластин; этопозид (VP-16); ифосфамид; митоксантрон; винкристин; NAVELBINE® (винорелбин); новантрон; тенипозид; эдатрексат; дауномицин; аминоптерин; кселоду; ибандронат; CPT-11; ингибитор топоизомеразы RFS 2000; дифторметилорнитин (DMFO); ретиноиды, такие как ретиноевая кислота; капецитабин; и фармацевтически приемлемые соли, кислоты и производные любого из указанных выше.
Также в определение "химиотерапевтическое средство" включены: (i) антигормональные средства, которые действуют, регулируя или ингибируя действие гормона на опухоли, такие как антиэстрогены и селективные модуляторы рецепторов эстрогена (SERM), включая, например, тамоксифен (в том числе NOLVADEX®; тамоксифена цитрат), ралоксифен, дролоксифен, 4-гидрокситамоксифен, триоксифен, кеоксифен, LY117018, онапристон и FARESTON® (торемифина цитрат); (ii) ингибиторы ароматазы, которые ингибируют фермент ароматазу, который регулирует продукцию эстрогена в надпочечниках, например, такие как 4(5)-имидазолы, аминоглутетимид, MEGASE® (мегестрола ацетат), AROMASIN® (экземестан; Pfizer), форместан, фадрозол, RIVISOR® (ворозол), FEMARA® (летрозол; Novartis) и ARINIDEX® (анастрозол; AstraZeneca); (iii) антиандрогены, такие как флутамид, нилутамид, бикалутамид, леупролид и гозерелин; а также троксацитабин (1,3-диоксолановый нуклеозидный аналог цитозина); (iv) ингибиторы протеинкиназ; (v) ингибиторы киназ липидов; (vi) антисмысловые олигонуклеотиды, в частности олигонуклеотиды, которые ингибируют экспрессии генов каскадов передачи сигнала, вовлеченных в измененную пролиферацию клеток, например, PKC-альфа, Ralf и H-Ras; (vii) рибозимы, такие как ингибиторы экспрессии VEGF (например, ANGIOZYME®); и (viii) вакцины, такие как вакцины для генной терапии, например, ALLOVECTIN®, LEUVECTIN® и VAXID®; PROLEUKIN® rIL-2; ингибиторы топоизомеразы 1, такие как LURTOTECAN®; ABARELIX® rmRH; (ix) антиангиогенные средства; и (х) фармацевтически приемлемые соли, кислоты и производные любого из указанных выше.
Гуманизированные моноклональные антитела с терапевтическим потенциалом в качестве средств в комбинации с ингибиторами Janus-киназы по изобретению включают: адалимумаб, этанерцепт, инфликсимаб, алемтузумаб, аполизумаб, азелизумаб, атлизумаб, бапинеузумаб, бевацизумаб, биватузумаб, мертансин, кантузумаб мертансин, цеделизумаб, цертолизумаб пегол, цидфузитузумаб, цидтузумаб, даклизумаб, экулизумаб, эфализумаб, эпратузумаб, эрлизумаб, фелвизумаб, фонтолизумаб, гемтузумаб озогамицин, инотузумаб озогамицин, ипилимумаб, лабетузумаб, линтузумаб, матузумаб, меполизумаб, мотавизумаб, мотовизумаб, натализумаб, нимотузумаб, ноловизумаб, нумавизумаб, окрелизумаб, омализумаб, паливизумаб, пасколизумаб, пекфузитузумаб, пектузумаб, пертузумаб, пекселизумаб, раливизумаб, ранибизумаб, ресливизумаб, реслизумаб, резивизумаб, ровелизумаб, руплизумаб, сибротузумаб, сиплизумаб, сонтузумаб, такатузумаб тетраксетан, тадоцизумаб, тализумаб, тефибазумаб, тоцилизумаб, торализумаб, трастузумаб, тукотузумаб целмолейкин, тукузитузумаб, умавизумаб, уртоксазумаб, устекинумаб, визилизумаб и антитело против интерлейкина-12 (ABT-874/J695, Wyeth Research и Abbott Laboratories), которое представляет собой рекомбинантную исключительно человеческую последовательность, полноразмерное антитело IgG1 λ, генетически модифицированное для распознавания белка p40 интерлейкина-12.
Термин "пролекарство", как используют в настоящей заявке, относится к форме предшественника или производного соединения по изобретению, которая является менее эффективной для пациента или менее цитотоксической для злокачественных клеток по сравнению с исходным лекарственном средством и может быть ферментативно или гидролитически активирована или превращена в более активную исходную форму. См., например, Wilman, "Prodrugs in Cancer Chemotherapy" Biochemical Society Transactions, 14, pp. 375-382, 615th Meeting Belfast (1986) и Stella et al., "Prodrugs: A Chemical Approach to Targeted Drug Delivery", Directed Drug Delivery, Borchardt et al., (ed.), pp. 247-267, Humana Press (1985). Пролекарства по этому изобретению включают, но не ограничиваются ими, содержащие фосфат пролекарства, содержащие тиофосфат пролекарства, содержащие сульфат пролекарства, содержащие пептид пролекарства, пролекарства с модифицированными D-аминокислотами, гликозилированные пролекарства, содержащие β-лактам пролекарства, содержащие необязательно замещенный феноксиацетамид пролекарства, содержащие необязательно замещенный фенилацетамид пролекарства, пролекарства с 5-фторцитозином и другие пролекарства с 5-фторуридином, которые могут превращаться в более активное цитотоксическое свободное лекарственное средство. Примеры цитотоксических лекарственных средств, которые могут быть преобразованы в форму пролекарства для применения в этом изобретении, включают, но не ограничиваются ими, химиотерапевтические средства, описанные выше.
"Липосома" относится к везикуле, состоящей из одного или нескольких липидов, фосфолипидов и/или поверхностно-активных веществ, которые пригодны для доставки лекарственного средства (такого как соединение формулы I и, необязательно, химиотерапевтическое средство) млекопитающему. Компоненты липосомы обычно образуют двухслойную структуру, сходную с размещением липидов биологических мембран.
Термин "вкладыш в упаковку" используют для обозначения инструкций, обычно включаемых в коммерческие упаковки терапевтических продуктов, которые содержат информацию о показаниях, применении, дозировке, введении, противопоказаниях, и/или предупреждения, касающиеся применения таких терапевтических продуктов.
Термин "хиральный" относится к молекулам, которые обладают свойством несовпадения с аналогами, являющимися их зеркальным отображением, а термин "ахиральный" относится к молекулам, которые совпадают с их аналогами, являющимися их зеркальными отображениями.
Термин "стереоизомеры" относится к соединениям, которые обладают идентичным химическим строением, но отличаются расположением атомов или групп в пространстве.
"Диастереомер" относится к стереоизомеру с двумя или более центрами хиральности, молекулы которого не являются зеркальными отображениями друг друга. Диастереомеры обладают различными физическими свойствами, например, температурой плавления, температурой кипения, спектральными свойствами и реакционной способностью. Смеси диастереомеров можно разделять аналитическими способами высокого разрешения, такими как электрофорез и хроматография.
"Энантиомеры" относятся к двум стереоизомерам соединения, которые не являются совпадающими зеркальными отображениями друг друга.
Стереохимические определения и обозначения, используемые в настоящем документе, главным образом, соответствуют S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. and Wilen, S., Stereochemistry of Organic Compounds (1994) John Wiley & Sons, Inc., New York. Многие органические соединения существуют в оптически активных формах, т.е. они обладают способностью к вращению плоскости плоскополяризованного света. При описании оптически активного соединения приставки D и L, или R и S, используют для обозначения абсолютной конфигурации молекулы в области ее хирального центра(ов). Приставки d и l или (+) и (-) используют для обозначения признака вращения плоскополяризованного света соединением, причем (-) или l означает, что соединение является левовращающим. Соединение с приставкой (+) или d является правовращающим. Для данной химической структуры эти стереоизомеры являются идентичными, за исключением того, что они являются зеркальными отображениями друг друга. Конкретный стереоизомер также может быть обозначен как энантиомер, а смесь таких изомеров часто называют энантиомерной смесью. Смесь энантиомеров 50:50 называют рацемической смесью или рацематом, которая может встречаться, только когда отсутствует стереоселекция или стереоспецифичность в химической реакции или процессе. Термины "рацемическая смесь" и "рацемат" относятся к эквимолярной смеси двух энантиомерных соединений, лишенной оптической активности.
Выражение "фармацевтически приемлемая соль", как используют в настоящем документе, относится к фармацевтически приемлемым органическим или неорганическим солям соединения формулы I. Иллюстративные соли включают, но не ограничиваются ими, сульфаты, цитраты, ацетаты, оксалаты, хлориды, бромиды, йодиды, нитраты, бисульфаты, фосфаты, кислые фосфаты, изоникотинаты, лактаты, салицилаты, кислые цитраты, тартраты, олеаты, таннаты, пантотенаты, битартраты, аскорбаты, сукцинаты, малеаты, гентизинаты, фумараты, глюконаты, глюкуронаты, сахараты, формиаты, бензоаты, глутаматы, метансульфонаты, этансульфонаты, бензолсульфонаты, п-толуолсульфонаты и памоаты (т.е. 1,1'-метилен-бис(2-гидрокси-3-нафтоат)). Фармацевтически приемлемая соль может включать другую молекулу, такую как ион ацетата, ион сукцината или другой противоион. Противоион может представлять собой любую органическую или неорганическую группу, которая стабилизирует заряд на исходном соединении. Более того, фармацевтически приемлемая соль может иметь более одного заряженного атома в ее структуре. В случаях, когда множество заряженных атомов являются частью фармацевтически приемлемой соли, может существовать множество противоионов. Таким образом, фармацевтически приемлемая соль может иметь один или несколько заряженных атомов и/или один или несколько противоионов.
"Сольват" относится к ассоциации или комплексу одной или нескольких молекул растворителя и соединения формулы I. Примеры растворителей, которые образуют сольваты, включают, но не ограничиваются ими, воду, изопропанол, этанол, метанол, DMSO, этилацетат, уксусную кислоту и этаноламин. Термин "гидрат" относится к комплексу, где молекулой растворителя является вода.
Термин "защитная группа" или "Pg" относится к заместителю, который обычно используют для блокирования или защиты конкретной функциональной группы в ходе реакции других функциональных групп на соединении. Например, "амино-защитная группа" представляет собой заместитель, связанный с аминогруппой, который блокирует или защищает функциональную аминогруппу в соединении. Пригодные амино-защитные группы включают ацетил, трифторацетил, фталимидо, трет-бутоксикарбонил (BOC), бензилоксикарбонил (CBZ) и 9-флуоренилметиленоксикарбонил (Fmoc). Аналогично, "гидрокси-защитная группа" относится к заместителю гидроксигруппы, который блокирует или защищает функциональную гидроксигруппу. Пригодные гидрокси-защитные группы включают ацетил, триалкилсилил, диалкилфенилсилил, бензоил, бензил, бензилоксиметил, метил, метоксиметил, триарилметил и тетрагидропиранил. "Карбокси-защитная группа" относится к заместителю карбоксигруппы, который блокирует или защищает функциональную группу карбокси. Распространенные карбокси-защитные группы включают -CH2CH2SO2Ph, цианоэтил, 2-(триметилсилил)этил, 2-(триметилсилил)этоксиметил, 2-(п-толуолсульфонил)этил, 2-(п-нитрофенилсульфенил)этил, 2-(дифенилфосфино)этил, нитроэтил и т.п. Для общего описания защитных групп и их применения см. T.W. Greene, Protective Groups in Organic Synthesis, Third Ed., John Wiley & Sons, New York, 1999; и P. Kocienski, Protecting Groups, Third Ed., Verlag, 2003.
Термин "пациент" включает пациентов-людей и пациентов-животных. Термин "животное" включает животных-компаньонов (например, собак, кошек и лошадей), животных, являющихся источниками пищи, животных зоопарков, морских животных, птиц и другие сходные виды животных.
Выражение "фармацевтически приемлемый" указывает на то, что вещество или композиция должно быть совместимо химически и/или токсикологически с другими ингредиентами, составляющими состав, и/или млекопитающим, подвергаемым лечению ими.
Термины "JAK-киназа" и "Janus-киназа" относятся к протеинкиназам JAK1, JAK2, JAK3 и TYK2.
Термины "соединение по изобретению" и "соединения по настоящему изобретению" и "соединения формулы I", если нет иных указаний, включают соединения формулы I, формул 1a-1n и их стереоизомеры, таутомеры, сольваты, метаболиты, соли (например, фармацевтически приемлемые соли) и пролекарства. Если нет иных указаний, структуры, изображенные в настоящем документе, также предполагают включение соединений, которые отличаются только присутствием одного или нескольких изотопно обогащенных атомов. Например, соединения формулы I и формул 1a-1n, где один или несколько атомов водорода заменены дейтерием или тритием, или один или несколько атомов углерода заменены 13C- или 14C-обогащенным углеродом, входят в объем этого изобретения.
ИНГИБИРУЮЩИЕ JANUS-КИНАЗУ СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА
В одном варианте осуществления предусмотрено соединение формулы I и его фармацевтические составы, которые пригодны для лечения заболеваний, состояний и/или нарушений, отвечающих на ингибирование одной или нескольких Janus-киназ.
Другой вариант осуществления относится к соединениям формулы I

I
их энантиомерам, диастереомерам или фармацевтически приемлемым солям, где
R1 представляет собой водород, C1-C6алкил, -OR6, -NR6R7 или галоген;
R2 представляет собой 5- или 6-членный гетероарил, где R2 необязательно замещен 1-3 R4;
R3 представляет собой фенил, 5-6-членный гетероарил, C3-C6циклоалкил или 3-10-членный гетероциклил, где R3 необязательно замещен 1-5 R5;
R4 независимо представляет собой C1-C6алкил, C2-C6алкенил, C2-C6алкинил, галоген, -(C0-C6алкил)CN, -(C0-C6алкил)OR6, -(C0-C6алкил)SR6, -(C0-C6алкил)NR6R7, -(C0-C6алкил)CF3, -(C0-C6алкил)C(O)R6, -(C0-C6алкил)C(O)OR6, -(C0-C6алкил)C(O)NR6R7, -(C0-C6алкил)NR6C(O)R7, -(C0-C6алкил)C(O)3-6-членный гетероциклил, -(C0-C6алкил)(C3-C6циклоалкил), -(C0-C6алкил)фенил, -(C0-C6алкил)5-6-членный гетероарил или -(C0-C6алкил)(3-6-членный гетероциклил), где R4 независимо необязательно замещен R15;
R5 независимо представляет собой C1-C6алкил, C2-C6алкенил, C2-C6алкинил, оксо, галоген, -(C0-C3алкил)CN, -(C0-C3алкил)OR11, -(C0-C3алкил)SR11, -(C0-C3алкил)NR11R12, -(C0-C3алкил)OCF3, -(C0-C3алкил)CF3, -(C0-C3алкил)NO2, -(C0-C3алкил)C(O)R11, -(C0-C3алкил)C(O)OR11, -(C0-C3алкил)C(O)NR11R12, -(C0-C3алкил)NR11C(O)R12, -(C0-C3алкил)S(O)1-2R11, -(C0-C3алкил)NR11S(O)1-2R12, -(C0-C3алкил)S(O)1-2NR11R12, -(C0-C3алкил)(C3-C6циклоалкил), -(C0-C3алкил)(3-6-членный гетероциклил), -(C0-C3алкил)C(O)(3-6-членный гетероциклил), -(C0-C3алкил)(5-6-членный гетероарил) или -(C0-C3алкил)фенил, где R5 независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -CF3, -(C0-C3алкил)OR13 или -(C0-C3алкил)NR13R14; или
два R5, взятые вместе, образуют -O(CH2)1-3O-;
R6 и R7 независимо представляют собой водород, C1-C6алкил, C2-C6алкенил, C2-C6алкинил, -CN, -OR8, -NR8R9, -C(O)R8, -C(O)OR8, -C(O)NR8R9, -NR8C(O)R9, -NR8C(O)OR9, -OC(O)NR8, -S(O)1-2R8, -NR8S(O)1-2R9, -S(O)1-2NR8R9, C3-C6циклоалкил, фенил, 3-6-членный гетероциклил или 5-6-членный гетероарил, где указанные R6 и R7 независимо необязательно замещены R20; или
R6 и R7, взятые вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом;
R8 и R9 независимо представляют собой водород или C1-C3алкил; или
R8 и R9, взятые вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом;
R11 независимо представляет собой водород, C1-C6алкил, C3-C6циклоалкил, 3-6-членный гетероциклил, -C(O)R13, -C(O)OR13, -C(O)NR13R14, -NR13C(O)R14, -S(O)1-2R13, -NR13S(O)1-2R14 или -S(O)1-2NR13R14, где указанные алкил, циклоалкил и гетероциклил независимо необязательно замещены оксо, C1-C3алкилом, OR13, NR13R14 или галогеном;
R12 независимо представляет собой водород или C1-C3алкил, где указанный алкил независимо необязательно замещен галогеном или оксо; или
R11 и R12, взятые вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом;
R13 и R14 независимо представляют собой водород или C1-C3алкил, необязательно замещенный галогеном или оксо; или
R13 и R14, взятые вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом;
R15 представляет собой C1-C6алкил, C2-C6алкенил, C2-C6алкинил, оксо, галоген, -CN, -OR16, -SR16, -NR16R17, -OCF3, -CF3, -C(O)R16, -C(O)OR16, -C(O)NR16R17, -NR16C(O)R17, -NR16C(O)OR17, -OC(O)NR16, C3-C6циклоалкил, 3-6-членный гетероциклил, -C(O)(3-6-членный гетероциклил, 5-6-членный гетероарил или фенил), где R15 независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -CN, -CF3, -OR18, -NR18R19;
R16 и R17 независимо представляют собой водород или C1-C6алкил, необязательно замещенный оксо или галогеном; или
R16 и R17, взятые вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил, необязательно замещенный оксо, галогеном или C1-C3алкилом;
R18 и R19 независимо представляют собой водород или C1-C6алкил, необязательно замещенный оксо или галогеном; или
R18 и R19, взятые вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил, необязательно замещенный оксо, галогеном или C1-C3алкилом;
R20 представляет собой C1-C6алкил, оксо, галоген, -OR21, -NR21R22, -CN, C3-C6циклоалкил, фенил, 3-6-членный гетероциклил или 5-6-членный гетероарил, где R20 необязательно замещен оксо, галогеном или C1-C3алкилом; и
R20 и R21 независимо представляют собой водород или C1-C6алкил, необязательно замещенный оксо или галогеном; или
R20 и R21, взятые вместе с атомом, к которому они присоединены, образуют 3-6-членный гетероциклил, необязательно замещенный оксо, галогеном или C1-C3алкилом.
Другой вариант осуществления относится к соединениям формулы I

I
их энантиомерам, диастереомерам или фармацевтически приемлемым солям, где
R1 представляет собой водород, C1-C6алкил, -OR6, -NR6R7 или галоген;
R2 представляет собой 5- или 6-членный гетероарил, где R2 необязательно замещен 1-3 R4;
R3 представляет собой фенил, 5- или 6-членный гетероарил, где R3 необязательно замещен 1-5 R5;
R4 независимо представляет собой C1-C6алкил, C2-C6алкенил, C2-C66алкинил, галоген, -(C0-C6алкил)OR6, -(C0-C6алкил)SR6, -(C0-C6алкил)NR6R7, -(C0-C6алкил)CF3, -(C0-C6алкил)C(O)R6, -(C0-C6алкил)C(O)OR6, -(C0-C6алкил)C(O)NR6R7, -(C0-C6алкил)(C3-C6циклоалкил) или -(C0-C6алкил)(3-6-членный гетероциклил), где R4 независимо необязательно замещен C1-C3алкилом, оксо, галогеном, -CF3, -OR8 или -NR8R9;
R5 независимо представляет собой C1-C6алкил, C2-C6алкенил, C2-C6алкинил, галоген, -(C0-C3алкил)CN, -(C0-C3алкил)OR11, -(C0-C3алкил)SR11, -(C0-C3алкил)NR11R12, -(C0-C3алкил)OCF3, -(C0-C3алкил)CF3, -(C0-C3алкил)NO2, -(C0-C3алкил)C(O)R11, -(C0-C3алкил)C(O)OR11, -(C0-C3алкил)C(O)NR11R12, -(C0-C3алкил)NR11C(O)R12, -(C0-C3алкил)S(O)1-2R11, -(C0-C3алкил)NR11S(O)1-2R12, -(C0-C3алкил)S(O)1-2NR11R12, -(C0-C3алкил)(C3-C6циклоалкил), -(C0-C3алкил)(3-6-членный гетероциклил), -(C0-C3алкил)C(O)(3-6-членный гетероциклил), -(C0-C3алкил)(5-6-членный гетероарил) или -(C0-C3алкил)фенил, где R5 независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -CF3, -(C0-C3алкил)OR13 или -(C0-C3алкил)NR13R14; или
два R5, взятые вместе, образуют -O(CH2)1-3O-;
R6 независимо представляет собой водород, C1-C3алкил, -C(O)R8, -C(O)OR8, -C(O)NR8R9, -NR8C(O)R9, -S(O)1-2R8, -NR8S(O)1-2R9 или -S(O)1-2NR8R9, где указанный алкил независимо необязательно замещен оксо, OH или галогеном;
R7 независимо представляет собой водород или C1-C3алкил, где указанный алкил независимо необязательно замещен галогеном; или
R6 и R7, взятые вместе с атомом, к которому они присоединены, образуют 5- или 6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом;
R8 и R9 независимо представляют собой водород или C1-C3алкил; или
R8 и R9, взятые вместе с атомом, к которому они присоединены, образуют 5- или 6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом;
R11 независимо представляет собой водород, C1-C3алкил, -C(O)R13, -C(O)OR13, -C(O)NR13R14, -NR13C(O)R14, -S(O)1-2R13, -NR13S(O)1-2R14 или -S(O)1-2NR13R14, где указанный алкил независимо необязательно замещен оксо, OH или галогеном;
R12 независимо представляет собой водород или C1-C3алкил, где указанный алкил независимо необязательно замещен галогеном; или
R11 и R12, взятые вместе с атомом, к которому они присоединены, образуют 5- или 6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом; и
R13 и R14 независимо представляют собой водород или C1-C3алкил; или
R13 и R14, взятые вместе с атомом, к которому они присоединены, образуют 5- или 6-членный гетероциклил, необязательно замещенный галогеном, оксо, -CF3 или C1-C3алкилом.
Другой вариант осуществления включает соединения формулы I, отличные от:
N-(5-метил-4-(4-пропилфенил)тиазол-2-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида,
N-(4-(4-хлорфенил)тиазол-2-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида или
N-(3-метил-1-фенил-1H-пиразол-5-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида.
В определенных вариантах осуществления R2 выбран из пиридинила, имидазолила, пиримидинила, пиразолила, триазолила, пиразинила, тетразолила, фурила, тиенила, изоксазолила, тиазолила, оксазолила, изотиазолила, пирролила, пиридазинила, триазинила, оксадиазолила, триазолила, тиадиазолила или фуразанила, и где R2 необязательно замещен 1-3 R4. В одном варианте осуществления R2 выбран из пиридинила или пиразолила, необязательно замещенного 1-3 R4.
В определенных вариантах осуществления R2 выбран из тиазолила, пиридинила или пиразолила, необязательно замещенного 1-3 R4.
В определенных вариантах осуществления, R4 независимо представляет собой C1-C6алкил, -(C0-C6алкил)OR6, -(C0-C6алкил)SR6, -(C0-C6алкил)NR6R7, -(C0-C6алкил)CF3, -(C0-C6алкил)C(O)R6, -(C0-C6алкил)C(O)OR6, -(C0-C6алкил)C(O)NR6R7, -(C0-C6алкил)(C3-C6циклоалкил) или -(C0-C6алкил)(3-6-членный гетероциклил), где R4 независимо необязательно замещен C1-C3алкилом, оксо, галогеном, -CF3, -OR8 или -NR8R9.
В определенных вариантах осуществления R4 представляет собой C1-C6алкил, галоген, -(C0-C6алкил)CN, -(C0-C6алкил)OR6, -(C0-C6алкил)NR6R7, -(C0-C6алкил)CF3, -(C0-C6алкил)C(O)R6, -(C0-C6алкил)C(O)OR6, -(C0-C6алкил)C(O)NR6R7, -(C0-C6алкил)NR6C(O)R7, -(C0-C6алкил)C(O)3-6-членный гетероциклил, -(C0-C6алкил)(C3-C6циклоалкил), -(C0-C6алкил)фенил, -(C0-C6алкил)5-6-членный гетероарил или -(C0-C6алкил)(3-6-членный гетероциклил), где R4 независимо необязательно замещен R15.
В определенных вариантах осуществления R3 представляет собой фенил, пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, пиридазинил, триазинил, оксадиазолил, триазолил, тиадиазолил или фуразанил, и где R3 необязательно замещен 1-5 R5. В одном варианте осуществления R3 представляет собой фенил, необязательно замещенный 1-3 R5.
В определенных вариантах осуществления R3 представляет собой фенил, пиридинил, имидазолил, пиримидинил, пиразолил, триазолил, пиразинил, тетразолил, фурил, тиенил, изоксазолил, тиазолил, оксазолил, изотиазолил, пирролил, пиридазинил, триазинил, оксадиазолил, триазолил, тиадиазолил, циклогексенил, дигидробензофуранил, пиперидинил, пиридинонил, пирролидинил или фуразанил, и где R3 необязательно замещен 1-5 R5.
В определенных вариантах осуществления R5 независимо представляет собой C1-C6алкил, галоген, -CN, -(C0-C3алкил)OR11, -(C0-C3алкил)SR11, -(C0-C3алкил)NR11R12, -(C0-C3алкил)OCF3 или -CF3, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -CF3, -(C0-C3алкил)OR13 или -(C0-C3алкил)NR13R14.
В определенных вариантах осуществления R3 представляет собой фенил, необязательно замещенный 1-3 R5; и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -(C0-C3алкил)OR11, -(C0-C3алкил)SR11, -(C0-C3алкил)NR11R12, -(C0-C3алкил)OCF3 или -CF3, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -CF3, -(C0-C3алкил)OR13 или -(C0-C3алкил)NR13R14.
В определенных вариантах осуществления R3 представляет собой фенил, пиридинил, дигидробензофуранил, пиперидинил, пирролидинил, пиридинонил, имидазолил или изоксазолил, где R3 необязательно замещен 1-3 R5; и R5 независимо представляет собой оксо, C1-C6алкил, C2-C6алкенил, C2-C6алкинил, C3-C6циклоалкил, галоген, -CN, -O(C1-C6алкил), -S(C1-C6алкил), -O(C3-C6циклоалкил), -S(C3-C6циклоалкил), -(C0-C3алкил)NR11R12, -OCF3, -OCHF2 или -CF3, где указанный алкил, алкенил, алкинил и циклоалкил независимо необязательно замещены галогеном, C1-C3алкилом, OH, OCH3, NH2, NMe2, оксо или -CF3.
В определенных вариантах осуществления R1 представляет собой водород, OR8 или -NR6R7. В одном варианте осуществления R1 представляет собой водород. В одном варианте осуществления R1 представляет собой -NH2.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)OR6 или -(C0-C6алкил)SR6, и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9. В одном варианте осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)OR6 или -(C0-C3алкил)SR6, и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)CF3, и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9. В одном варианте осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)CF3, и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)NR6R7 или -(C0-C6алкил)(3-6-членный гетероциклил), и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9. В одном варианте осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)NR6R7 или -(C0-C6алкил)(3-6-членный гетероциклил), и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)(C3-C6циклоалкил), и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9. В одном варианте осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)(C3-C6циклоалкил), и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)C(O)OR6 или -(C0-C6алкил)C(O)NR6NR7, и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9. В одном варианте осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)C(O)OR6, и где R4 независимо необязательно замещен C1-C3алкилом, -OR8 или -NR8R9, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой C0-C6алкил, и где R4 независимо необязательно замещен галогеном, -OR8 или -NR8R9. В одном варианте осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой C0-C6алкил, и где R4 независимо необязательно замещен галогеном, -OR8 или -NR8R9, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)CN, и где R4 независимо необязательно замещен галогеном, -OR8 или -NR8R9. В одном варианте осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -(C0-C6алкил)CN, и где R4 независимо необязательно замещен галогеном, -OR8 или -NR8R9, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой C1-C6алкил, необязательно замещенный оксо, -OR8, -NR8R9, -CN, галогеном, C3-C6циклоалкилом или 5-6-членным гетероциклилом, 5-6-членный гетероциклил, необязательно замещенный -OR8, -NR8R9, -CN, галогеном или оксо, -CH2C(O)NR6NR7, необязательно замещенный -OR8, -NR8R9, -CN, галогеном или C3-C6циклоалкилом, или -CH2(5-6-членный гетероциклил, необязательно замещенный оксо, -OR8, -NR8R9, -CN, галогеном или C1-C3алкилом), и R3 представляет собой фенил, необязательно замещенный 1-3 R5.
В определенных вариантах осуществления R2 представляет собой пиразолил, необязательно замещенный R4, где R4 представляет собой -CH2C(OH)(C1-C3алкил, необязательно замещенный галогеном), -CH2C(O)NR6NR7 или -CH2C(O)(4-6-членный гетероциклил), где R4 необязательно замещен оксо, -OR8, -NR8R9, -CN, галогеном, C1-C6алкилом или C3-C6циклоалкилом, и R3 представляет собой фенил, необязательно замещенный 1-3 R5.
В определенных вариантах осуществления R2 представляет собой пиридинил, необязательно замещенный C1-C6алкилом, и указанный алкил необязательно замещен галогеном. В одном варианте осуществления R2 представляет собой пиридинил, необязательно замещенный C1-C6алкилом, и указанный алкил необязательно замещен галогеном, R3 представляет собой фенил, необязательно замещенный 1-3 R5, и R5 независимо представляет собой C1-C6алкил, галоген, -CN, -OR11, -SR11 или -CF3, где указанный алкил независимо необязательно замещен галогеном, -CF3, -OR13 или -NR13R14.
В определенных вариантах осуществления R4 представляет собой -(C0-C3алкил)OR6 или -(C0-C6алкил)SR6, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:

 ,
,
где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)CF3, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:
 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)NR6R7, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:
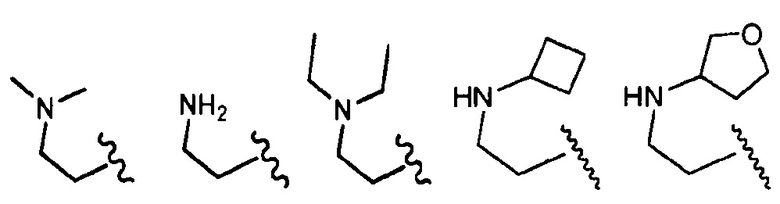 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)(3-6-членный гетероциклил), где указанные алкил и гетероциклил независимо необязательно замещены галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления указанный 3-6-членный гетероциклил представляет собой оксетанил, морфолинил, пиперидинил, пиперазинил, пирролидинил, пирролидинонил, тетрагидрофуранил, оксазолил, изоксазолил и тетрагидропиранил, необязательно замещенный галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:
 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)C(O)(3-6-членный гетероциклил), где указанные алкил и гетероциклил независимо необязательно замещены галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления указанный 3-6-членный гетероциклил представляет собой оксетанил, морфолинил, пиперидинил, пиперазинил, пирролидинил, тетрагидрофуранил, оксазолил, изоксазолил, дигидропирролил и тетрагидропиранил, необязательно замещенный галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:


 ,
,
где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)(C3-C6циклоалкил), где указанные алкил и циклоалкил независимо необязательно замещены галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)(C3-C6циклоалкил), где указанный циклоалкил представляет собой циклопентил или циклогексил, необязательно замещенный C1-C3алкилом, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:
 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)C(O)OR6, -(C0-C6алкил)C(O)NR6NR7, -(C0-C6алкил)OC(O)NR6, -(C0-C6алкил)NR6C(O)OR7, -(C0-C6алкил)NR6C(O)NR7 или -(C0-C6алкил)NR6C(O)R7, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:


 ,
,
где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой C1-C6алкил, C2-C6алкенил или C2-C6алкинил, необязательно замещенный галогеном, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 выбран из:
 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R4 представляет собой -(C0-C6алкил)CN, где указанный алкил необязательно замещен галогеном, оксо, -OR8 или -NR8R9. В одном варианте осуществления R4 представляет собой -CH2CN, -CH2CH2CN или -CH(CH3)CN.
В определенных вариантах осуществления R4 представляет собой галоген. В одном варианте осуществления R4 представляет собой F, Cl, Br или I. В одном варианте осуществления R4 представляет собой F или Cl.
В определенных вариантах осуществления R3 представляет собой:
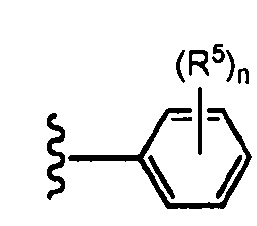 , где n равно 0, 1, 2 или 3 и волнистая линия соответствует точке присоединения к R2. В одном варианте осуществления n равно 2. В одном варианте осуществления R3 выбран из:
, где n равно 0, 1, 2 или 3 и волнистая линия соответствует точке присоединения к R2. В одном варианте осуществления n равно 2. В одном варианте осуществления R3 выбран из:
 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R3 представляет собой фенил или пиридинил, необязательно замещенный C1-C6алкилом, галогеном, -CN, -(C0-C3алкил)OR6, -(C0-C3алкил)SR6, -(C0-C3алкил)NR6R7, -(C0-C3алкил)OCF3 или -CF3, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -CF3, -(C0-C3алкил)OR8 или -(C0-C3алкил)NR8R9. В одном варианте осуществления R3 выбран из:


 , где волнистая линия соответствует точке присоединения к R2. В одном варианте осуществления R3 выбран из:
, где волнистая линия соответствует точке присоединения к R2. В одном варианте осуществления R3 выбран из:
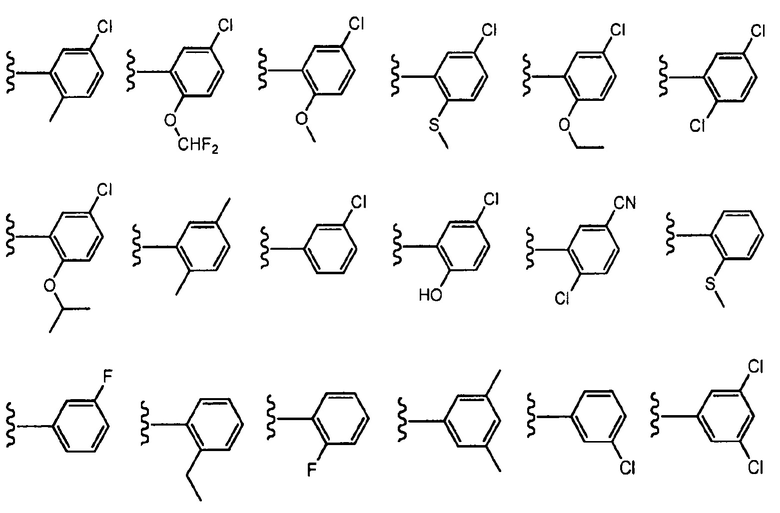
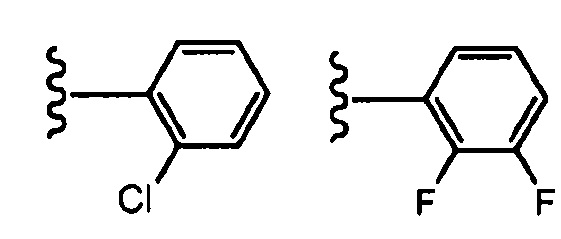 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R3 представляет собой 4-6-членный гетероциклил, необязательно замещенный C1-C6алкилом, галогеном, -CN, -(C0-C3алкил)OR6, -(C0-C3алкил)SR6, -(C0-C3алкил)NR6R7, -(C0-C3алкил)OCF3 или -CF3, где указанный алкил независимо необязательно замещен галогеном, C1-C3алкилом, оксо, -CF3, -(C0-C3алкил)OR8 или -(C0-C3алкил)NR8R9. В определенных вариантах осуществления указанный гетероциклил выбран из азетидинила, пирролидинила, пиперидинила, пиперазинила, морфолинила, тетрагидропиридинила и пиридинонила. В определенных вариантах осуществления R3 выбран из:
 , где волнистая линия соответствует точке присоединения к R2.
, где волнистая линия соответствует точке присоединения к R2.
В определенных вариантах осуществления R2 выбран из:
 , где R10 независимо выбран из водорода или R4 и волнистая линия соответствует точке присоединения к формуле I.
, где R10 независимо выбран из водорода или R4 и волнистая линия соответствует точке присоединения к формуле I.
В определенных вариантах осуществления R2 выбран из:
 , где R10 независимо выбран из водорода или R4 и волнистая линия соответствует точке присоединения к формуле I.
, где R10 независимо выбран из водорода или R4 и волнистая линия соответствует точке присоединения к формуле I.
В определенных вариантах осуществления R2 выбран из:
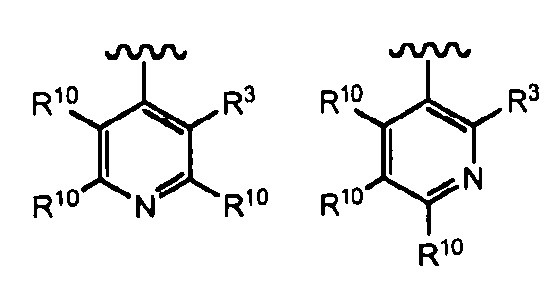 , где R10 независимо выбран из водорода или R4 и волнистая линия соответствует точке присоединения к формуле I.
, где R10 независимо выбран из водорода или R4 и волнистая линия соответствует точке присоединения к формуле I.
В определенных вариантах осуществления R2 выбран из:
 , R3 представляет собой
, R3 представляет собой  или пиридинил, R10 представляет собой водород или R4, и волнистая линия соответствует точке присоединения к формуле I и R2, соответственно.
или пиридинил, R10 представляет собой водород или R4, и волнистая линия соответствует точке присоединения к формуле I и R2, соответственно.
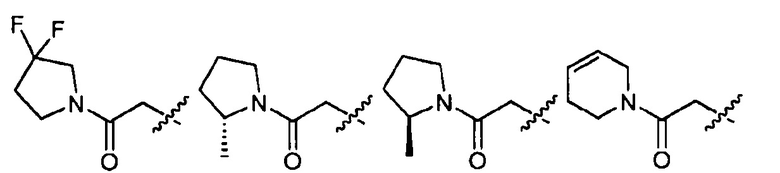
В определенных вариантах осуществления -R2-R3 в формуле I представляет собой  , где R10 представляет собой водород или R4 и волнистая линия соответствует точке присоединения к формуле I.
, где R10 представляет собой водород или R4 и волнистая линия соответствует точке присоединения к формуле I.
В определенных вариантах осуществления -R2-R3 в формуле I представляет собой  , где n равно 0, 1, 2 или 3, R10 представляет собой водород или R4 и волнистая линия соответствует точке присоединения R2 к формуле I.
, где n равно 0, 1, 2 или 3, R10 представляет собой водород или R4 и волнистая линия соответствует точке присоединения R2 к формуле I.
Другой вариант осуществления относится к соединению формулы I, которое имеет Ki и/или EC50, которые по меньшей мере в 15 раз, альтернативно в 10 раз или в 5 раз или более являются более селективными в отношении ингибирования одного вида активности Janus-киназы относительно ингибирования одного или нескольких других видов активности Janus-киназы.
Соединения формулы I могут содержать асимметричные или хиральные центры, и, таким образом, существуют в различных стереоизомерных формах. Подразумевают, что все стереоизомерные формы соединений формулы I, включая, но не ограничиваясь ими, диастереомеры, энантиомеры и атропизомеры, а также их смеси, такие как рацемические смеси, формируют часть настоящего изобретения. Кроме того, настоящее изобретение охватывает все геометрические и позиционные изомеры. Например, если соединение формулы I включает двойную связь или конденсированные кольца, объем изобретения охватывает как цис-, так и транс-формы, а также их смеси. Также в объем настоящего изобретения входят как единичные позиционные изомеры, так и смеси позиционных изомеров, например, вследствие N-окисления пиримидинильного и пиррозолильного колец, или E- и Z-формы соединений формулы I (например, группы оксимов).
В структурах, представленных в настоящем документе, когда стереохимия какого-либо конкретного хирального атома не указана, все стереоизомеры предусматриваются и включены в качестве соединений по изобретению. Когда стереохимия указана закрашенным клином или пунктирной линией, соответствующими конкретной конфигурации, тогда таким путем указывают и определяют этот стереоизомер.
Соединения по настоящему изобретению могут существовать в несольватированной, а также в сольватированной формах с фармацевтически приемлемыми растворителями, такими как вода, этанол и т.п., и подразумевают, что изобретение, как определено в формуле изобретения, охватывает как сольватированные, так и несольватированные формы.
В одном варианте осуществления соединения формулы I могут существовать в различных таутомерных формах, и все такие формы охватываются объемом изобретения, как определено в формуле изобретения. Термин "таутомер" или "таутомерная форма" относится к структурным изомерам с различной энергией, которые являются взаимопревращающимися с низким энергетическим барьером. Например, протонные таутомеры (также известные как прототропные таутомеры) включают взаимопревращения посредством миграции протона, такие как кето-енольная и имин-энаминовая изомеризации. Валентные таутомеры включают взаимопревращения путем реорганизации некоторых электронов связи.
Настоящее изобретение также охватывает изотопно меченные соединения формулы I, которые идентичны соединениям, приведенным в настоящем документе, за исключением того факта, что один или несколько атомов заменены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или атомного числа, обычно встречающихся в природе. Объем изобретения предусматривает все изотопы какого-либо конкретного указанного атома или элемента. Иллюстративные изотопы, которые могут быть включены в соединения формулы I, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, хлора и йода, такие как 2H, 3H, 11C, 13C, 14C, 13N, 15N, 15O, 17O, 18O, 32P, 33P, 35S, 18F, 36Cl, 123I и 125I, соответственно. Определенные изотопно меченные соединения формулы I (например, соединения, меченные 3H и 14C) пригодны в анализах распределения соединения и/или субстрата в тканях. Тритированные изотопы (т.е. 3H) и изотопы углерода 14 (т.е. 14C) пригодны вследствие их простоты получения и возможности выявления. Кроме того, замена более тяжелыми изотопами, такими как дейтерий (т.е. 2H), может обеспечить определенные терапевтические преимущества вследствие более высокой метаболической стабильности (например, увеличенное время полужизни in vivo или сниженные потребности в дозировках) и, таким образом, могут быть предпочтительными в некоторых случаях. Позитронно-активные изотопы, такие как 15O, 13N, 11C и 18F, пригодны для исследований способом позитронно-эмиссионной томографии (PET) для исследования способности рецептора быть занятым субстратом. Изотопно меченные соединения формулы I, главным образом, можно получать способами, аналогичными способам, описанным на схемах и/или в примерах настоящего документа ниже, путем замещения изотопно меченным реагентом не меченного изотопно реагента.
СИНТЕЗ ИНГИБИРУЮЩИХ JAK СОЕДИНЕНИЙ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА
Соединения формулы I можно синтезировать синтетическими способами, описанными в настоящем документе. В определенных вариантах осуществления можно использовать способы, хорошо известные в области химии, в дополнение к описанию или с учетом описания, содержащегося в настоящем документе. Исходные материалы, главным образом, доступны из коммерческих источников, таких как Aldrich Chemicals (Milwaukee, Wis.), или их легко получать с использованием способов, хорошо известных специалистам в данной области (например, их можно получать способами, главным образом, описанными в Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, N.Y. (1967-1999 ed.), Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer-Verlag, Berlin, включая дополнения (также доступные через интерактивную базу данных Beilstein)), или Comprehensive Heterocyclic Chemistry, Editors Katrizky and Rees, Pergamon Press, 1984.
Соединения формулы I можно получать отдельно или в качестве библиотек соединений, содержащих по меньшей мере 2, например, от 5 до 1000 соединений, или от 10 до 100 соединений формулы I. Библиотеки соединений формулы I можно получать путем комбинаторного подхода "разделения и смешивания" или путем множественного параллельного синтеза с использованием либо жидкофазной, либо твердофазной химии, способами, известными специалистам в данной области. Таким образом, согласно следующему аспекту изобретения, предусмотрена библиотека соединений, содержащая по меньшей мере 2 соединения формулы I, их энантиомеров, диастереомеров, таутомеров или фармацевтически приемлемых солей.
Для целей иллюстрации, на схемах реакции 1-13, показанных ниже, предоставлены пути синтеза соединений по настоящему изобретению, а также ключевые промежуточные соединения. Для более подробного описания отдельных стадий реакции см. раздел "Примеры" ниже. Специалистам в данной области понятно, что для синтеза соединений по изобретению можно использовать другие способы синтеза. Хотя конкретные исходные материалы и реагенты представлены на схемах и обсуждаются ниже, их можно легко заменять другими исходными материалами и реагентами для получения различных производных и/или условий реакции. Кроме того, многие соединения, полученные способами, описанными ниже, можно далее модифицировать с учетом этого описания с использованием общепринятых химических реакций, хорошо известных специалистам в данной области.
При получении соединений по настоящему изобретению может быть необходима защита отдельных функциональных групп (например, первичного или вторичного амина) промежуточных соединений. Необходимость в такой защите может варьировать, в зависимости от природы отдельной функциональной группы и условий способов получения. Пригодные амино-защитные группы (NH-Pg) включают ацетил, трифторацетил, трет-бутоксикарбонил (BOC), бензилоксикарбонил (CBz) и 9-флуоренилметиленоксикарбонил (Fmoc). Необходимость в такой защите может легко определить специалист в данной области. Для общего описания защитных групп и их применения см. T.W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
Соединения по изобретению можно получать из хорошо доступных исходных материалов с использованием общих способов, проиллюстрированных на схемах реакции 1-21 ниже.
Схема реакции 1

Соединения формулы I можно синтезировать, как показано на схеме реакции 1. Например, коммерчески доступную бензойную кислоту можно подвергать реакции с 3-этокси-3-оксопропаноатом калия в присутствии карбонилдиимидазола (CDI) и хлорида магния с получением β-кето-сложного эфира 2. Соединение 2 можно нагревать с 1,1-диметокси-N,N-диметилметанамином (DMFDMA) с получением соединения 3. Циклизация соединения 3 гидразином в этаноле дает соединение пиразола 4. Метилирование соединения 4 с йодметаном в присутствии основания, такого как карбонат цезия, дает смесь региоизомеров 5a и 5b. Гидролиз этилового сложного эфира, с последующей перегруппировкой Курциуса с использованием дифенилфосфонового азида (dppa) и трет-бутанола дает защищенный трет-бутилкарбаматом амино-пиразол, защитную группу из которого удаляют с помощью HCl с получением амино-пиразольных соединений 6a и 6b. Региоизомеры 6a и 6b можно разделять на этой стадии с использованием хроматографии с диоксидом кремния. Реакция сочетания каждого региоизомера с коммерчески доступной пиразолo[1,5-a]пиримидин-3-карбоновой кислотой в присутствии гексафторфосфата 7-азабензотриазол-1-илокси-трис(пирролидинo)фосфония (PyAOP), диизопропилэтиламина (DIEA) и 4-диметиламинопиридина (DMAP) дает соединения формулы 1a и 1b.
Схема реакции 2
Альтернативный способ синтеза соединений формулы I проиллюстрирован на схеме реакции 2. Алкилирование ди-трет-бутилиминодикарбоната гидридом натрия и различными α-бромкетонами 7 дает соединение 8. Соединение 8 можно нагревать с DMFDMA с получением соединения 9. Циклизация соединения 9 с гидразином в этаноле дает пиразольное соединение 10. Реакция сочетания соединения 10 с пиразоло[1,5-a]пиримидин-3-карбоновой кислотой в присутствии PyAOP, DIEA и DMAP дает соединения формулы 1c. Соединения формулы 1c можно подвергать алкилированию йодметаном в присутствии карбоната цезия с получением соединений формулы 1a и 1b.
Схема реакции 3

На схеме реакции 3 проиллюстрирован синтез соединений формулы 1d. Проведение диазотизации коммерчески доступных анилинов и опосредуемого хлоридом олова восстановления дает соединение 11. Конденсация соединения 11 с 3-аминокротонитрилом в хлористоводородной кислоте в этаноле дает аминопиразольное соединение 12. Реакция сочетания соединения 12 с пиразоло[1,5-a]пиримидин-3-карбоновой кислотой в присутствии PyAOP, DIEA и DMAP дает соединения формулы 1d.
Схема реакции 4

На схеме реакции 4 проиллюстрирован синтез соединений формулы 1e. 5-Бром-2-метилпиридин можно окислять м-хлорпероксибензойной кислотой с получением соединения 13. Нитрирование соединения 13 с получением соединения 14, а затем восстановление хлоридом олова дает аминопиридин 15. Пиразоло[1,5-a]пиримидин-3-карбоновую кислоту можно превращать в соответствующий хлорангидрид с помощью оксалилхлорида, а затем подвергать реакции с соединением 15 или другими коммерчески доступными аминопиридинами с образованием соединения 16. Эти соединения можно подвергать условиям перекрестного сочетания Сузуки с арилбороновыми кислотами с получением соединений формулы 1e.
Схема реакции 5

Альтернативный способ синтеза соединений формулы 1 описан на схеме реакции 5. Алкилирование фталимида калия α-бромкетонами 7 дает соединение 17. Конденсация с DMFDMA дает соединения 18. Соединения формулы 18 можно подвергать циклизации с N-метилгидразином с получением поддающейся разделению смеси региоизомеров 6a и 6b. Реакция сочетания каждого региоизомера отдельно согласно способам образования амидов с использованием гексафторфосфата о-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония (HATU) с коммерчески доступной пиразоло[1,5-a]пиримидин-3-карбоновой кислотой дает соединения формулы 1a и 1b.
Схема реакции 6

На схеме реакции 6 проиллюстрирован способ синтеза соединений формулы 1g и 1f. Соединение 18 можно подвергать циклизации с гидразином с получением соединения 10, которое затем можно подвергать реакции сочетания с пиразоло[1,5-a]пиримидин-3-карбоновой кислотой в условиях образования амида с использованием HATU с получением соединений формулы 1c. Алкилирование соединения 1c алкилгалогенидами в присутствии карбоната цезия дает соединения формулы 1f. Реакция соединения 1c с 2,2-диметилоксираном в присутствии карбоната цезия дает соединения формулы 1g.
Схема реакции 7
На схеме реакции 7 проиллюстрирован синтез соединений формулы 1h. Образование амида с использованием HATU с 4-бром-1-метил-1H-пиразол-3-амином и пиразоло[1,5-a]пиримидин-3-карбоновой кислотой дает соединение 19. Катализируемая палладием реакция сочетания Сузуки соединения 19 с различными бороновыми кислотами дает соединения формулы 1h.
Схема реакции 8
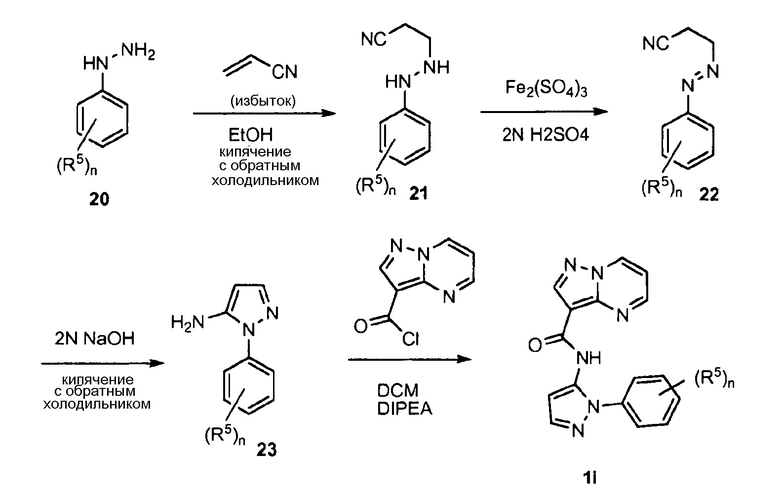
На схеме реакции 8 иллюстрируется синтез соединений формулы 1i. Раствор надлежащим образом замещенного фенилгидразина можно нагревать при кипячении с обратным холодильником с избытком акрилонитрила в растворителе, таком как этанол, с получением соединений формулы 21. Гидразины 21 можно окислять с использованием сульфата железа(III) в разбавленной серной кислоте с получением соединений формулы 22. Соединения 22 можно подвергать циклизации в разбавленном растворе гидроксида натрия с получением соединений формулы 23. Реакция 22 с ранее полученным пиразоло[1,5-a]пиримидин-3-карбонилхлоридом дает конечные соединения формулы 1i.
Схема реакции 9

Альтернативный способ синтеза соединений формулы 1a и 1b представлен на схеме реакции 9. Коммерчески доступный 4-нитро-1H-пиразол можно защищать с помощью [β-(триметилсилил)этокси]метильной (SEM) группы путем обработки гидридом натрия и (2-(хлорметокси)этил)триметилсиланом. Полученное соединение 24 можно арилировать с помощью арилбромидов или йодидов в катализируемых палладием условиях с получением 4-нитро-5-арилпиразолов формулы 25. Нитрогруппу соединений 25 можно восстанавливать в присутствии хлорида железа и аммония с получением аминопиразолов 26. Реакция сочетания по амидной связи с коммерчески доступной пиразоло[1,5-a]пиримидин-3-карбоновой кислотой в присутствии PyAOP, DIEA и DMAP дает соединения 27. Удаление защитной группы SEM с помощью водного раствора HCl в этаноле дает соединения 1c, которые можно алкилировать алкилгалогенидами в присутствии пригодного основания, такого как карбонат цезия, или акцепторами Михаэля с получением соединений формулы 1a и 1b.
Схема реакции 10
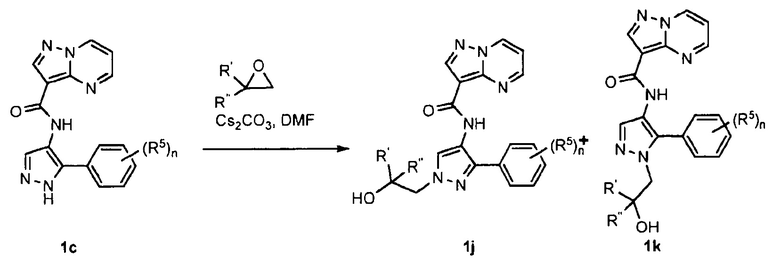
На схеме реакции 10 иллюстрируется синтез соединений формулы 1j и 1k. Пиразольные соединения 1c (полученные, как описано либо на схеме реакции 2, либо на схеме реакции 9) можно алкилировать замещенными эпоксидами в присутствии карбоната цезия с получением региоизомерных соединений формулы 1j и 1k.
Схема реакции 11
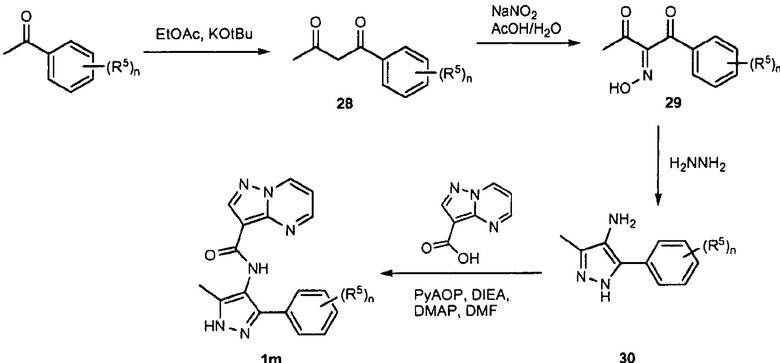
Соединения формулы 1m можно синтезировать, как показано на схеме реакции 11. Коммерчески доступные ацетофеноны можно ацилировать трет-бутоксидом калия и этилацетатом с получением дикетоновых соединений формулы 28. Соединения 28 можно обрабатывать нитритом натрия в присутствии уксусной кислоты и воды с получением гидроксииминосоединений 29, которые можно затем подвергать циклизации с гидразином с получением аминопиразольных соединений 30. Реакция сочетания по амидной связи с коммерчески доступной пиразоло[1,5-a]пиримидин-3-карбоновой кислотой в присутствии PyAOP, DIEA и DMAP дает соединения формулы 1m.
Схема реакции 12

Альтернативный способ синтеза соединений формулы 1a и 1b представлен на схеме реакции 12. Коммерчески доступный 4-нитро-1H-пиразол можно подвергать реакции с алкилбромидами в присутствии карбоната цезия при 55°C в течение 12 часов с получением соединения 31. Соединение 31 можно подвергать реакции с арилбромидами в N,N-диметилацетамиде в присутствии ацетата палладия(II), ди-(1-адамантил)-н-бутилфосфина, карбоната калия и триметилуксусной кислоты с получением соединений 32a и 32b. Соотношение продуктов 32a:32b варьирует, в зависимости от заместителя R1, однако реакция, как правило, идет в пользу образования продукта 32b. Соединения 32a и 32b можно восстанавливать до соединений 33a и 33b в присутствии хлорида железа и аммония в этаноле и воде. Реакция сочетания соединений 33a и 33b с пиразоло[1,5-a]пиримидин-3-карбоновой кислотой в присутствии PyAOP, DIEA и DMAP может дать соединения формулы 1a и 1b.
Схема реакции 13

Соединения формулы 1n можно синтезировать, как показано в схеме реакции 13. Трихлорацетонитрил можно подвергать реакции с этиловым эфиром цианоуксусной кислоты с получением соединения 34. Соединение 34 можно конденсировать с гидразином с получением соединения 35, которое затем можно конденсировать с 1,1,3,3-тетраметоксипропаном с получением соединения 36. Амин 36 можно дважды защищать с помощью Boc с получением соединения 37, которое затем можно гидролизовать гидроксидом лития с получением карбоновой кислоты 38. Затем карбоновую кислоту 38 можно подвергать реакции сочетания с различными аминами в присутствии PyAOP, DIEA и DMAP с получением соединений формулы 1n.
Схема реакции 14
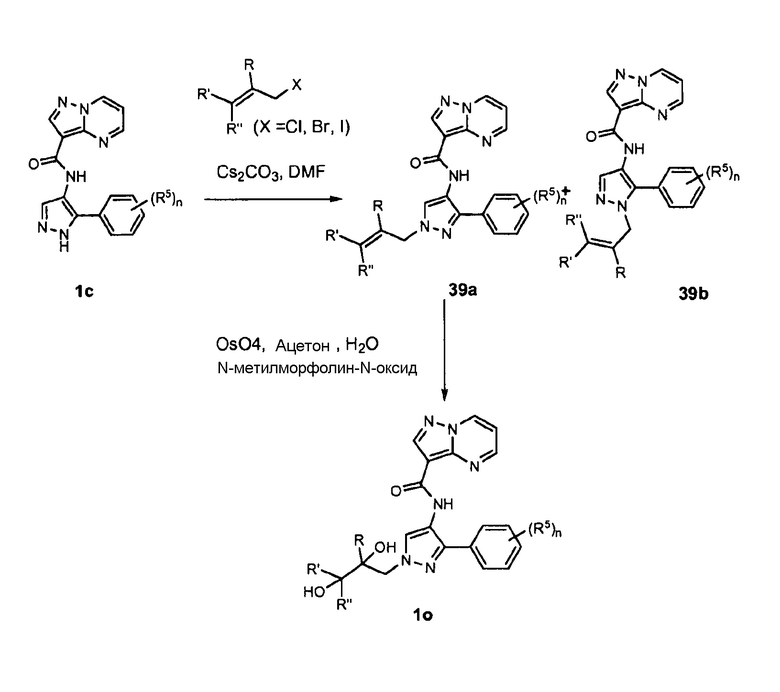
Соединения формулы 1o также можно синтезировать, как показано на схеме реакции 14. Пиразольные соединения 1c (получаемые, как описано либо на схеме реакции 2, либо на схеме реакции 9) можно алкилировать аллилгалогенидами в присутствии карбоната цезия с получением региоизомерных соединений формулы 39a и 39b. Региоизомерные соединения можно разделять хроматографией на силикагеле и соответствующий изомер можно подвергать реакции с тетроксидом осмия в присутствии N-метилморфолин-N-оксида с получением соединений формулы 1o.
Схема реакции 15

Соединения формулы 1p и 1q можно синтезировать, как показано на схеме реакции 15. Пиразольные соединения 1c (получаемые, как описано либо на схеме реакции 2, либо на схеме реакции 9) можно алкилировать 2-(хлорметил)оксираном в присутствии карбоната цезия, а затем обрабатывать аминами с получением региоизомерных соединений формулы 1p и 1q.
Схема реакции 16
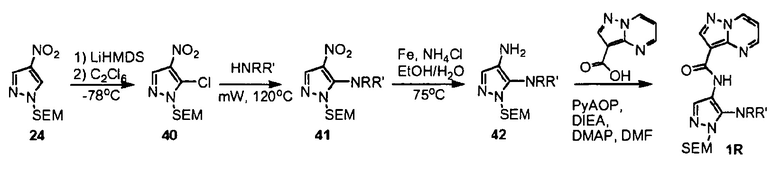
На схеме реакции 16 проиллюстрирован синтез соединений формулы 1R. Нитро-SEM-пиразольное соединение 24, полученное как на схеме реакции 9, можно региоселективно депротонировать гексаметилдисилазидом лития при низкой температуре и гасить гексахлорэтаном с получением 40. При нагревании при микроволновом излучении (mW) с амином HNRR', где RR', взятые вместе с азотом, к которому они присоединены, образуют R3 (например, 3-10-членную гетероциклильную группу), это соединение можно превращать в 41. Нитрогруппу промежуточного соединения 41 можно восстанавливать в присутствии хлорида железа и аммония с получением аминопиразолов 42. Реакция сочетания по амидной связи с коммерчески доступной пиразоло[1,5-a]пиримидин-3-карбоновой кислотой в присутствии PyAOP, DIEA и DMAP дает 1R. 1R можно далее обрабатывать удалением группы SEM и функционализацией, как на схемах реакции 9, 10, 14, 15, 18 или 19.
Схема реакции 17

На схеме реакции 17 проиллюстрирован альтернативный синтез соединений формулы 10. Нитро-SEM-пиразольное соединение 24, полученное, как на схеме реакции 9, можно региоселективно депротонировать гексаметилдисилазидом лития при низкой температуре и гасить йодом с получением 43. Нитрогруппу соединения 43 можно восстанавливать в присутствии хлорида железа и аммония, с последующей защитой с помощью Boc с получением соединения 44. Соединение 44 можно подвергать реакции сочетания в условиях Сузуки с арилбороновыми кислотами или арилборонатами с получением соединений 45. После отщепления группы Boc тетрахлоридом олова получают соединения формулы 10.
Схема реакции 18

Соединения формулы 1s можно синтезировать, как показано на схеме реакции 18. Пиразольные соединения 1c (получаемые, как описано либо на схеме реакции 2, либо на схеме реакции 9) можно алкилировать трет-бутилбромацетатом в присутствии карбоната цезия с получением промежуточного соединения 46. Соединение 46 можно обрабатывать трифторуксусной кислотой с получением кислот 47, которые затем можно подвергать реакции с первичными или вторичными аминами в присутствии реагента реакции сочетания, такого как гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (HATU), с получением соединений формулы 1s.
Схема реакции 19

Соединения формулы 1t можно синтезировать, как показано на схеме реакции 19. Пиразольные соединения 1c (получаемые, как описано либо на схеме реакции 2, либо на схеме реакции 9) можно алкилировать 2-хлорэтил-пара-толуолсульфонатом в присутствии карбоната цезия с получением алкилхлоридов 48. Затем 48 можно подвергать реакции с первичными или вторичными аминами в присутствии соответствующего основания, такого как N,N-диизопропилэтиламин, с получением соединений формулы 1t.
Схема реакции 20
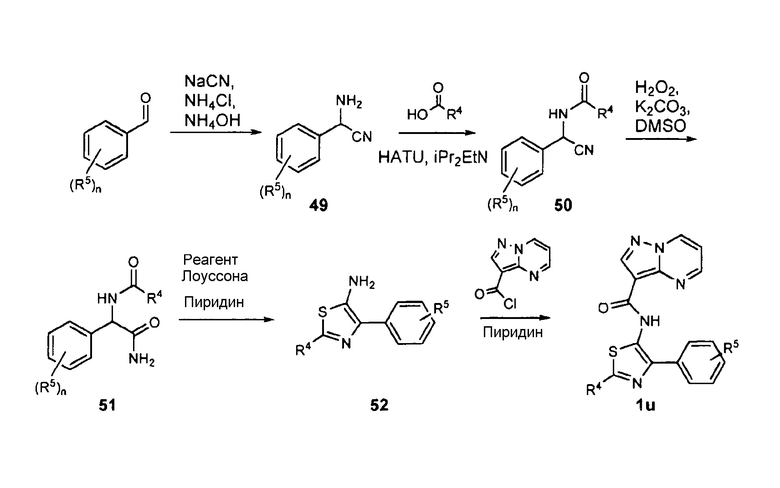
Соединения формулы 1u можно синтезировать, как представлено на схеме реакции 20. Например, коммерчески доступные замещенные бензальдегиды можно превращать в соединения 49 обработкой источником цианида, таким как цианид натрия, в присутствии хлорида аммония и гидроксида аммония. Соединения 49 можно подвергать реакции сочетания с карбоновыми кислотами с получением соединений 50 с использованием амидного реагента реакции сочетания, такого как HATU, и основания, такого как диизопропилэтиламин. Соединения 50 можно обрабатывать пероксидом водорода и карбонатом калия с получением соединений 51. Циклизация диамидосоединений 51 с использованием реагента Лоуссона и пиридина дает аминотиазолосоединения 52. Соединения формулы 1u можно получать путем обработки соединений 52 ранее полученным пиразоло[1,5-a]пиримидин-3-карбонилхлоридом в пиридине.
Схема реакции 21

Альтернативно, соединения формулы 1u можно синтезировать, как показано на схеме реакции 21. Например, соединения 53 можно получать путем обработки коммерчески доступных замещенных ацетофенонов диэтилкарбонатом и последующего бромирования с использованием, например, брома в диоксане. Обработка соединений 53 соответствующим образом замещенным тиоамидом или тиомочевиной дает тиазольные соединения 54. Соединения 54 можно гидролизовать с использованием водного раствора основания, такого как гидроксид калия, в совместимом растворителе, таком как THF, с получением кислотных соединений 55. Соединения 56 можно получать путем обработки соединений 55 дифенилфосфорилазидом (DPPA) в трет-бутаноле. Удаление защитной группы из соединений 56 в кислотных условиях дает аминосоединения 52. Соединения формулы 1u можно получать путем обработки соединений 52 ранее полученным пиразоло[1,5-a]пиримидин-3-карбонилхлоридом в пиридине.
Будет понятно, что, когда существуют подходящие функциональные группы, соединения различных формул или любые промежуточные соединения, используемые при их получении, можно далее преобразовывать в производные одним или несколькими стандартными способами синтеза с использованием реакций конденсации, замещения, окисления, восстановления или расщепления. Конкретные подходы для замещения включают общепринятые способы алкилирования, арилирования, гетероарилирования, ацилирования, сульфонилирования, галогенирования, нитрирования, формилирования и сочетания.
В следующем примере группы первичных или вторичных аминов можно превращать в амидные группы (-NHCOR' или -NRCOR') ацилированием. Ацилирование можно проводить путем реакции с соответствующим хлорангидридом в присутствии основания, такого как триэтиламин, в пригодном растворителе, таком как дихлорметан, или путем реакции с соответствующей карбоновой кислотой в присутствии пригодного реагента реакции сочетания, такого как HATU (гексафторфосфат O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония), в пригодном растворителе, таком как дихлорметан. Аналогично, аминогруппы можно превращать в сульфонамидные группы (-NHSO2R' или -NR''SO2R') путем реакции с соответствующим сульфонилхлоридом в присутствии пригодного основания, такого как триэтиламин, в пригодном растворителе, таком как дихлорметан. Первичные или вторичные аминогруппы можно превращать в группы мочевины (-NHCONR'R'' или -NRCONR'R'') путем реакции с соответствующим изоцианатом в присутствии пригодного основания, такого как триэтиламин, в пригодном растворителе, таком как дихлорметан.
Амин (-NH2) можно получать путем восстановления нитрогруппы (-NO2), например, путем каталитической гидрогенизации, с использованием, например, водорода в присутствии металлического катализатора, например, палладия на подложке, такой как уголь, в растворителе, таком как этилацетат или спирт, например, метанол. Альтернативно, превращение можно проводить путем химического восстановления с использованием, например, металла, например, олова или железа, в присутствии кислоты, такой как хлористоводородная кислота.
В следующем примере аминогруппы (-CH2NH2) можно получать путем восстановления нитрилов (-CN), например, каталитической гидрогенизацией, с использованием, например, водорода в присутствии металлического катализатора, например, палладия на подложке, такой как уголь, или никеля Ренея, в растворителе, таком как простой эфир, например, циклический простой эфир, такой как тетрагидрофуран, при подходящей температуре, например от приблизительно -78°C до температуры кипячения растворителя с обратным холодильником.
В следующем примере аминогруппы (-NH2) можно получать из групп карбоновых кислот (-CO2H) путем превращения соответствующего ацилазида (-CON3), перегруппировки Курциуса и гидролиза полученного изоцианата (-N=C=O).
Альдегидные группы (-CHO) можно превращать в аминогруппы (-CH2NR'R'') восстановительным аминированием с использованием амина и боргидрида, например, триацетоксиборгидрида натрия или цианоборгидрида натрия, в растворителе, таком как галогенированный углеводород, например, дихлорметан, или спирт, такой как этанол, где необходимо, в присутствии кислоты, такой как уксусная кислота, приблизительно при температуре окружающей среды.
В следующем примере альдегидные группы можно превращать в алкенильные группы (-CH=CHR') с использованием реакции Виттига или Вадсворта-Эммонса с использованием соответствующего фосфорана или фосфоната в стандартных условиях, известных специалистам в данной области.
Альдегидные группы можно получать восстановлением сложноэфирных групп (таких как -CO2Et) или нитрилов (-CN) с использованием гидрида диизобутилалюминия в пригодном растворителе, таком как толуол. Альтернативно, альдегидные группы можно получать окислением спиртовых групп с использованием любого пригодного окислителя, известного специалистам в данной области.
Сложноэфирные группы (-CO2R') можно превращать в соответствующую кислотную группу (-CO2H) с помощью катализируемого кислотой или основанием гидролиза, в зависимости от природы R. Если R представляет собой трет-бутил, катализируемый кислотой гидролиз можно проводить, например, путем обработки органической кислотой, такой как трифторуксусная кислота, в водном растворителе, или путем обработки неорганической кислотой, такой как хлористоводородная кислота, в водном растворителе.
Группы карбоновых кислот (-CO2H) можно превращать в амиды (CONHR' или -CONR'R'') реакцией с соответствующим амином в присутствии пригодного реагента реакции сочетания, такого как HATU, в пригодном растворителе, таком как дихлорметан.
В следующем примере карбоновые кислоты можно превращать в гомологи, отличающиеся на один атом углерода (т.е. с -CO2H в -CH2CO2H), путем превращения в соответствующий хлорангидрид (-COCl) с последующим синтезом Арндта-Айстерта.
В следующем примере -OH-группы можно получать из соответствующего сложного эфира (например, -CO2R') или альдегида (-CHO) путем восстановления с использованием, например, комплексного гидрида металла, такого как алюмогидрид лития, в диэтиловом эфире или тетрагидрофуране, или боргидрид натрия в растворителе, таком как метанол. Альтернативно, спирт можно получать восстановлением соответствующей кислоты (-CO2H), с использованием, например, алюмогидрида лития в растворителе, таком как тетрагидрофуран, или с использованием борана в растворителе, таком как тетрагидрофуран.
Спиртовые группы можно превращать в уходящие группы, такие как атомы галогенов или сульфонилоксигруппы, такие как алкилсульфонилокси, например, трифторметилсульфонилокси или арилсульфонилокси, например, п-толуолсульфонилоксигруппа, с использованием условий, известных специалистам в данной области. Например, спирт можно подвергать реакции с тиоилхлоридом в галогенированном углеводороде (например, дихлорметане) с получением соответствующего хлорида. Также в реакции можно использовать основание (например, триэтиламин).
В другом примере спиртовую, фенольную или амидную группы можно алкилировать реакцией сочетания фенола или амида со спиртом в растворителе, таком как тетрагидрофуран, в присутствии фосфина, например, трифенилфосфина, и активатора, такого как диэтил-, диизопропил или диметилазодикарбоксилат. Альтернативно, алкилирование можно проводить путем депротонирования с использованием пригодного основания, например, гидрида натрия, с последующим добавлением алкилирующего агента, такого как алкилгалогенид.
Галогеновые заместители ароматической группы в соединениях можно подвергать обмену галоген-металл путем обработки основанием, например, литиевым основанием, таким как н-бутил- или трет-бутиллитий, необязательно при низкой температуре, например приблизительно -78°C, в растворителе, таком как тетрагидрофуран, а затем гасить электрофилом для внесения желаемого заместителя. Таким образом, например, формильную группу можно вносить с использованием N,N-диметилформамида в качестве электрофила. Галогеновые заместители ароматических групп альтернативно можно подвергать катализируемым металлами (например, палладием или медью) реакциям для внесения, например, кислотного, сложноэфирного, циано, амидного, арильного, гетероарильного, алкенильного, алкинильного, тио- или аминозаместителей. Пригодные способы, которые можно использовать, включают способы, описанные Хеком, Сузуки, Стилле, Бухвальдом или Хартвигом.
Галогеновые заместители ароматических групп также можно подвергать нуклеофильному вытеснению после реакции с соответствующим нуклеофилом, таким как амин или спирт. Преимущественно, такую реакцию можно проводить при повышенной температуре в присутствии микроволнового излучения.
СПОСОБЫ РАЗДЕЛЕНИЯ
На каждой из иллюстративных схем может быть преимущественным отделение продуктов реакции друг от друга и/или от исходных материалов. Желаемые продукты каждой стадии или серии стадий отделяют и/или очищают (в дальнейшем, в настоящем документе, отделяют) до желаемой степени гомогенности способами, известными в данной области. Как правило, такое разделение вовлекает многофазную экстракцию, кристаллизацию из растворителя или смеси растворителей, дистилляцию, сублимацию или хроматографию. Хроматография может вовлекать любое количество способов, включая, например: обращенно-фазовую и нормально-фазовую, эксклюзионную, ионообменную хроматографии; способы и устройство для жидкостной хроматографии высокого, среднего и низкого давления; мелкомасштабную аналитическую хроматографию; хроматографию с псевдодвижущимся слоем (SMB) и препаративную тонкослойную или толстослойную хроматографию, а также способы мелкомасштабной тонкослойной или флэш-хроматографии.
Другой класс способов разделения вовлекает обработку смесей реагентом, выбранным для связывания с желаемым продуктом, не вступившим в реакцию исходным материалом, побочным продуктом реакции или сходными с ними, или иным образом обеспечивающим их отделение. Такие реагенты включают адсорбенты или абсорбенты, такие как активированный уголь, молекулярные сита, ионообменный носитель или сходные с ними. Альтернативно, реагенты могут представлять собой кислоты в случае основного материала, основания в случае кислотного материала, связывающие реагенты, такие как антитела, связывающие белки, селективные хелаторы, такие как краун-эфиры, реагенты для жидкостно/жидкостной ионной экстракции реагенты (LIX), или сходные с ними.
Выбор соответствующих способов разделения зависит от природы вовлеченных материалов, например, от температуры кипения и молекулярной массы при дистилляции и сублимации, присутствия или отсутствия полярных функциональных групп при хроматографии, стабильности материалов в кислотных и основных средах при многофазной экстракции и т.п. Специалист в данной области может применить способы для наиболее вероятного достижения желаемого разделения.
Диастереомерные смеси можно разделять на их отдельные диастереоизомеры на основе их физико-химических отличий способами, хорошо известными специалистам в данной области, такими как хроматография и/или фракционная кристаллизация. Энантиомеры можно разделять превращением энантиомерной смеси в диастереомерную смесь путем реакции с соответствующим активным соединением (например, хиральным вспомогательным веществом, таким как хиральный спирт или хлорангидрид Мошера), разделения диастереоизомеров и превращения (например, гидролизом) отдельных диастереоизомеров в соответствующие чистые энантиомеры. Также некоторые из соединений по настоящему изобретению могут представлять собой атропизомеры (например, замещенные биарилы), и они считаются частью этого изобретения. Энантиомеры также можно разделять с использованием хиральной колонки для ВЭЖХ.
Отдельный стереоизомер, например, энантиомер, по существу свободный от его стереоизомера, можно получать разделением рацемической смеси с использованием способа, такого как образование диастереомеров с использованием оптически активных разделяющих агентов (Eliel, E. and Wilen, S., Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York, 1994; Lochmuller, C.H., J. Chromatogr., 113(3):283-302 (1975)). Рацемические смеси хиральных соединений по изобретению можно разделять и выделять любым пригодным способом, включая: (1) образование ионных диастереомерных солей с хиральными соединениями и разделение фракционной кристаллизацией или другими способами, (2) образование диастереомерных соединений с помощью хиральных преобразующих в производное реагентов, разделение диастереомеров и превращение в чистые стереоизомеры, и (3) разделение по существу чистых или обогащенных стереоизомеров непосредственно в хиральных условиях. См.: Drug Stereochemistry, Analytical Methods and Pharmacology, Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
Диастереомерные соли можно получать реакцией энантиомерно чистых хиральных оснований, таких как бруцин, хинин, эфедрин, стрихнин, α-метил-β-фенилэтиламин (амфетамин) и т.п., с асимметричными соединениями, несущими кислотную функциональную группу, такую как карбоновая кислота и сульфоновая кислота. Разделение диастереомерных солей можно индуцировать фракционной кристаллизацией или ионной хроматографией. Для разделения оптических изомеров аминосоединений, добавление хиральных карбоновых или сульфоновых кислот, таких как камфорсульфоновая кислота, виннокаменная кислота, миндальная кислота или молочная кислота, может приводить к образованию диастереомерных солей.
Альтернативно, субстрат, подлежащий разделению, подвергают реакции с энантиомером хирального соединения с образованием диастереомерной пары (Eliel, E. and Wilen, S., Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York, 1994, p. 322). Диастереомерные соединения можно получать реакцией асимметричных соединений с энантиомерно чистыми хиральными преобразующими в производное реагентами, такими как производные ментила, с последующим разделением диастереомеров и гидролизом с получением чистого или обогащенного энантиомера. Способ определения оптической чистоты вовлекает получение хиральных сложных эфиров, таких как ментиловый сложный эфир, например, (-)ментилхлорформиат в присутствии основания, или сложный эфир Мошера, α-метокси-α-(трифторметил)фенилацетат (Jacob, J. Org. Chem. 47:4165 (1982)), рацемической смеси и анализ ЯМР-спектра в отношении присутствия двух атропизомерных энантиомеров или диастереомеров. Стабильные диастереомеры атропизомерных соединений можно разделять и выделять нормальной и обращенно-фазовой хроматографией согласно способам разделения атропизомерных нафтилизохинолинов (WO 96/15111). Способом (3) можно разделять рацемическую смесь двух энантиомеров посредством хроматографии с использованием хиральной стационарной фазы (Chiral Liquid Chromatography, W.J. Lough, Ed., Chapman и Hall, New York, (1989); Okamoto, J. of Chromatogr. 513:375-378 (1990)). Обогащенные или очищенные энантиомеры можно различать способами, используемыми для различения других хиральных молекул с асимметричными атомами углерода, такими как оптическое вращение и круговой дихроизм.
Позиционные изомеры, например, E- и Z-формы соединений формулы I и промежуточных соединений для их синтеза, можно выявлять способами охарактеризации, такими как ЯМР и аналитическая ВЭЖХ. Для определенных соединений, где энергетический барьер для взаимопревращения является достаточно высоким, E- и Z-изомеры можно разделять, например, препаративной ВЭЖХ.
БИОЛОГИЧЕСКАЯ ОЦЕНКА
В предшествующих исследованиях было показано, что выделенные киназные домены JAK1, JAK2, JAK3 или TYK2 человека фосфорилируют пептидные субстраты в анализах киназ in vitro (Saltzman et al., Biochem. Biophys. Res. Commun. 246:627-633 (2004)). Каталитически активный киназный домен JAK1, JAK2, JAK3 или TYK2 человека очищали из экстрактов клеток насекомых SF9, инфицированных рекомбинантным бакуловирусным экспрессирующим вектором, кодирующим киназные домены JAK1, JAK2, JAK3 или TYK2 человека (аминокислотные остатки JAK1 N852-D1154 согласно нумерации последовательности GenBank с регистрационным номером P23458, аминокислотные остатки JAK2 D812-G1132 согласно нумерации последовательности GenBank с регистрационным номером NP_004963.1; аминокислотные остатки JAK3 S783-S1124 согласно нумерации последовательности GenBank с регистрационным номером P52333, и аминокислотные остатки TYK2 N873-C1187 согласно нумерации последовательности GenBank с регистрационным номером P29597). Активность киназных доменов JAK1, JAK2, JAK3 или TYK2 можно измерять рядом прямых и непрямых способов, включая количественное определение фосфорилирования пептидных субстратов, происходящих из белка JAK3 человека (Saltzman et al., Biochem. Biophys. Res. Commun. 246:627-633 (2004)). Активность киназных доменов JAK1, JAK2, JAK3 или TYK2 измеряли in vitro путем мониторинга фосфорилирования происходящих из JAK3 пептидов с использованием технологии Caliper LabChip (см. примеры).
Соединения по настоящему изобретению тестируют в отношении их способности ингибировать активность и активацию Janus-киназы (первичные анализы) и в отношении их биологических эффектов на растущие клетки (вторичные анализы), как описано в настоящем документе. Соединения, имеющие Ki менее 10 мкМ (предпочтительно менее 5 мкМ, более предпочтительно менее 1 мкМ, наиболее предпочтительно менее 0,5 мкМ) в соответствующем анализе активности и активации Janus-киназы (см. примеры A и B) и EC50 менее 20 мкМ (предпочтительно менее 10 мкМ, более предпочтительно менее 5 мкМ, наиболее предпочтительно менее 1 мкМ) в соответствующих клеточных анализах (см. пример C), пригодны в качестве ингибиторов Janus-киназы.
ВВЕДЕНИЕ СОЕДИНЕНИЙ, ИНГИБИРУЮЩИХ JANUS-КИНАЗУ
Другой вариант осуществления относится к способу лечения или снижения тяжести заболевания или состояния, отвечающего на ингибирование активности Janus-киназы у пациента. Способ включает стадию введения пациенту терапевтически эффективного количества соединения формулы I. В одном варианте осуществления заболевание представляет собой аутоиммунное заболевание.
Другой вариант осуществления включает применение соединения формулы I для терапии.
Другой вариант осуществления относится к применению соединения формулы I для предупреждения, лечения или снижения тяжести заболевания. В одном варианте осуществления заболевание представляет собой аутоиммунное заболевание.
Другой вариант осуществления включает применение соединения формулы I для изготовления лекарственного средства для предупреждения, лечения или снижения тяжести заболевания. В одном варианте осуществления заболевание представляет собой аутоиммунное заболевание.
В одном варианте осуществления соединение формулы I вводят пациенту в терапевтически эффективном количестве для лечения или снижения тяжести заболевания или состояния, отвечающего на ингибирование активности Janus-киназы, и указанное соединение является по меньшей мере в 15 раз, альтернативно в 10 раз, альтернативно в 5 раз или более селективным в отношении ингибирования активности Janus-киназы относительно ингибирования одного или нескольких других видов активности Janus-киназы.
В одном варианте осуществления заболевание или состояние представляет собой злокачественную опухоль, инсульт, диабет, гепатомегалию, сердечно-сосудистое заболевание, рассеянный склероз, болезнь Альцгеймера, кистозный фиброз, вирусное заболевание, аутоиммунные заболевания, атеросклероз, рестеноз, псориаз, аллергические нарушения, воспаление, неврологические нарушения, связанное с гормонами заболевание, состояние, ассоциированное с трансплантацией органов, иммунодефицитные нарушения, деструктивные нарушения костей, пролиферативные нарушения, инфекционные заболевания, состояния, ассоциированные с гибелью клеток, индуцируемую тромбином агрегацию тромбоцитов, заболевание печени, патологические иммунные состояния, вовлекающие активацию T-клеток, нарушения ЦНС или миклопролиферативное нарушение.
В одном варианте осуществления заболевание или состояние представляет собой злокачественную опухоль.
В одном варианте осуществления заболевание представляет собой миелопролиферативное нарушение.
В одном варианте осуществления миелопролиферативное нарушение представляет собой истинную полицитемию, идиопатический тромбоцитоз, миелофиброз или хронический миелогенный лейкоз (CML).
В одном варианте осуществления злокачественная опухоль представляет собой злокачественную опухоль молочной железы, яичника, шейки матки, предстательной железы, яичка, полового члена, мочеполовых путей, семиному, злокачественную опухоль пищевода, глотки, гастральную злокачественную опухоль, злокачественную опухоль желудка, желудочно-кишечную злокачественную опухоль, злокачественную опухоль кожи, кератоакантому, фолликулярную карциному, меланому, мелкоклеточную карциному легкого, немелкоклеточную карциному легких (NSCLC), аденокарциному легкого, плоскоклеточную карциному легкого, злокачественную опухоль толстого кишечника, поджелудочной железы, щитовидной железы, папиллярный рак, злокачественную опухоль мочевого пузыря, печени, желчных путей, почки, кости, миелоидные нарушения, лимфоидные нарушения, волосатоклеточный рак, рак щечной поверхности и глотки (полости рта), губ, языка, рта, слюнных желез, глотки, тонкого кишечника, толстого кишечника, прямой кишки, ануса, почки, предстательной железы, наружных женских половых органов, щитовидной железы, толстого кишечника, эндометрия, матки, головного мозга, центральной нервной системы, злокачественную опухоль брюшины, печеночноклеточный рак, рак головы, рак шеи, лимфому Ходжкина или лейкоз.
В одном варианте осуществления сердечно-сосудистое заболевание представляет собой рестеноз, кардиомегалию, атеросклероз, инфаркт миокарда или застойную сердечную недостаточность.
В одном варианте осуществления нейродегенеративное заболевание представляет собой болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, болезнь Хантингтона и ишемию головного мозга и нейродегенеративное заболевание, вызываемое травматическим повреждением, глутаматную нейротоксичность или гипоксию.
В одном варианте осуществления воспалительное заболевание представляет собой воспалительное заболевание кишечника, ревматоидный артрит, псориаз, контактный дерматит или замедленную реакцию гиперчувствительности.
В одном варианте осуществления аутоиммунное заболевание представляет собой волчанку или рассеянный склероз.
В одном варианте осуществления аутоиммунное заболевание представляет собой болезнь Крона, язвенный колит, коллагенозный колит, лимфоцитарный колит, ишемический колит, воспаление в отключенной кишке, синдром Бехчета, инфекционный колит и колит неустановленной этиологии.
Оценку индуцируемой лекарственным средством иммуносупрессии соединениями по изобретению можно проводить с использованием функциональных тестов in vivo, таких как модели на грызунах индуцируемого артрита и терапевтическое или профилактическое лечение для оценки показателя заболевания, зависимого от T-клеток антительного ответа (TDAR) и гиперчувствительности замедленного типа (DTH). Можно рассматривать другие системы in vivo, включающие модели на мышах для защиты хозяина против инфекций или устойчивости к опухоли (Burleson GR, Dean JH, and Munson AE. Methods in Immunotoxicology, Vol. 1. Wiley-Liss, New York, 1995) для установления природы или механизмов наблюдаемой иммуносупрессии. Тест-системы in vivo можно дополнять общепризнанными функциональными анализами in vitro или ex vivo для оценки иммунной компетентности. Эти анализы могут включать пролиферацию B- или T-клеток в ответ на митогены или конкретные антигены, определение передачи сигнала через один или несколько каскадов Janus-киназы в B- или T-клетках или иммортализованных B- или T-клеточных линиях, измерение маркеров клеточной поверхности в ответ на B-клеточную или T-клеточную передачу сигнала, активность естественных киллерных клеток (NK), активность тучных клеток, дегрануляцию тучных клеток, фагоцитоз или активность уничтожения макрофагов, и нейтрофильный окислительный взрыв и/или хемотаксис. В каждый из этих тестов может быть включено определение продукции цитокинов конкретными эффекторными клетками (например, лимфоцитами, NK, моноцитами/макрофагами, нейтрофилами). Анализы in vitro и ex vivo можно применять как в доклиническом, так и в клиническом тестировании с использованием лимфоидных тканей и/или периферической крови (House RV. "Theory and practice of cytokine assessment in immunotoxicology" (1999) Methods 19:17-27; Hubbard AK. "Effects of xenobiotics on macrophage function: evaluation in vitro" (1999) Methods; 19:8-16; Lebrec H, et al. (2001) Toxicology 158:25-29).
6-Недельное детальное исследование индуцированного коллагеном артрита (CIA) с использованием аутоиммунного механизма для имитации артрита человека; модели на крысах и мышах (пример 68). Индуцированный коллагеном артрит (CIA) представляет собой одну из наиболее широко используемых моделей на животных для ревматоидного артрита (RA) человека. Воспаление суставов, которое развивается у животных с CIA, в высокой степени сходно с воспалением, наблюдаемым у пациентов с RA. Блокирование фактора некроза опухоли (TNF) является эффективным способом лечения CIA, также как оно является высокоэффективной терапией при лечении пациентов с RA. CIA опосредуется как T-клетками, так и антителами (B-клетки). Полагают, что макрофаги играют важную роль в опосредовании повреждения тканей в ходе развития заболевания. CIA индуцируют иммунизацией животных коллагеном, эмульгированным в полном адъюванте Фрейнда (CFA). Наиболее часто его индуцируют в линии мышей DBA/1, однако заболевание также можно индуцировать у крыс Lewis.
Существуют убедительные доказательства того, что B-клетки играют ключевую роль в патогенезе аутоиммунного и/или воспалительного заболевания. Лекарственные средства на основе белка, которые истощают B-клетки, такие как ритуксан, являются эффективными против запускаемых аутоантителами воспалительных заболеваний, таких как ревматоидный артрит (Rastetter et al. (2004) Annu Rev Med 55:477). CD69 является ранним маркером активации в лейкоцитах, включая T-клетки, тимоциты, B-клетки, NK-клетки, нейтрофилы и эозинофилы. Анализ CD69 в цельной крови человека (пример 69) определяет способность соединений ингибировать продукцию CD69 B-лимфоцитами в цельной крови человека, активированными связыванием поверхностного IgM F(ab')2 козы против IgM человека.
Зависимый от T-клеток антительный ответ (TDAR) представляет собой прогнозирующий анализ для тестирования иммунной функции, когда необходимо исследовать потенциальные иммунотоксические эффекты соединений. Анализ IgM-бляшкообразующих клеток (PFC), с использованием эритроцитов овцы (SRBC) в качестве антигена, в настоящее время является широко признанным и проверенным стандартным тестом. Было показано, что TDAR является в высокой степени прогнозирующим анализом для детекции иммунотоксичности при воздействии на взрослых мышей, на основе базы данных US National Toxicology Program (NTP) (M.I. Luster et al. (1992) Fundam. Appl. Toxicol. 18:200-210). Применимость этого анализа основана на том факте, что он представляет собой целостное измерение, вовлекающее несколько важных компонентов иммунного ответа. TDAR зависит от функций следующих групп клеток: (1) антигенпредставляющие клетки, такие как макрофаги или дендритные клетки; (2) T-хелперные клетки, которые являются критическими участниками в формировании ответа, а также в переключении изотипов; и (3) B-клетки, которые являются конечными эффекторными клетками и ответственны за продукцию антител. Химически индуцируемые изменения в любой из групп могут вызывать существенные изменения в общем TDAR (M.P. Holsapple In: G.R. Burleson, J.H. Dean и A.E. Munson, Editors, Modern Methods in Immunotoxicology, Volume 1, Wiley-Liss Publishers, New York, NY (1995), pp. 71-108). Обычно этот анализ проводят либо в качестве ELISA для измерения растворимых антител (R.J. Smialowizc et al. (2001) Toxicol. Sci. 61:164-175), либо в качестве анализа бляшкообразующих (или антителообразующих) клеток (L. Guo et al. (2002) Toxicol. Appl. Pharmacol. 181:219-227) для детекции плазматических клеток, секретирующих специфические к антигену антитела. Предпочтительным для выбора антигеном являются либо цельные клетки (например, эритроциты овцы), либо растворимые белковые антигены (T. Miller et al. (1998) Toxicol. Sci. 42:129-135).
Соединение формулы I можно вводить любым путем, пригодным для заболевания или состояния, подвергаемого лечению. Пригодные пути включают пероральный, парентеральный (включая подкожный, внутримышечный, внутривенный, внутриартериальный, внутрикожный, интратекальный и эпидуральный), трансдермальный, ректальный, назальный, местный (включая буккальный и сублингвальный), вагинальный, внутрибрюшинный, внутрилегочный и интраназальный. Для местного иммуносупрессивного лечения соединения можно вводить путем введения в очаг повреждения, включая перфузию или иное контактирование трансплантата с ингибитором перед трансплантацией. Будет понятно, что предпочтительный путь может варьировать, в зависимости, например, от состояния реципиента. Когда соединение формулы I вводят перорально, его можно изготавливать в качестве пилюли, капсулы, таблетки и т.д. с фармацевтически приемлемым носителем или эксципиентом. Когда соединение формулы I вводят парентерально, его можно изготавливать с фармацевтически приемлемым парентеральным носителем и единичной дозированной инъецируемой формой, как подробно описано ниже.
Доза для лечения пациентов-людей может находиться в диапазоне от приблизительно 10 мг до приблизительно 1000 мг соединения формулы I. Типичная доза может составлять от приблизительно 100 мг до приблизительно 300 мг соединения формулы I. Дозу можно вводить один раз в сутки (QD), два раза в сутки (BID) или более часто, в зависимости от фармакокинетических и фармакодинамических свойств, включая абсорбцию, распределение, метаболизм и экскрецию конкретного соединения. Кроме того, на дозировку и режим введения могут влиять токсические факторы. При пероральном введении пилюлю, капсулу или таблетку можно принимать внутрь каждые сутки или менее часто в течение определенного периода времени. Режим можно повторять в течение ряда циклов терапии.
ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ СОЕДИНЕНИЙ, ИНГИБИРУЮЩИХ JANUS-КИНАЗУ
Другой вариант осуществления относится к фармацевтической композиции, которая содержит соединение формулы I и фармацевтически приемлемый носитель, адъювант или наполнитель.
В одном варианте осуществления фармацевтическая композиция также содержит дополнительное лекарственное средство, выбранное из антипролиферативного средства, противовоспалительного средства, иммуномодулирующего средства, нейротрофического фактора, средства для лечения сердечно-сосудистого заболевания, средства для лечения заболевания печени, противовирусного средства, средства для лечения нарушений крови, средства для лечения диабета или средства для лечения иммунодефицитных нарушений.
В одном варианте осуществления соединение формулы I присутствует в фармацевтическом составе в количестве, требуемом для поддающегося детекции ингибирования активности Janus-киназы, вместе с фармацевтически приемлемым носителем, адъювантом или наполнителем.
В одном варианте осуществления соединение формулы I присутствует в фармацевтическом составе в количестве, требуемом для поддающегося детекции ингибирования активности Janus-киназы, и является по меньшей мере в 15 раз, альтернативно в 10 раз или в 5 раз или более, более селективным в отношении ингибирования одного вида активности Janus-киназы относительно ингибирования одного или нескольких других видов активности JAK1, JAK2, JAK3 и/или Tyk-2.
Типичный состав получают смешиванием соединения по настоящему изобретению и носителя, разбавителя или эксципиента. Пригодные носители, разбавители и эксципиенты хорошо известны специалистам в данной области и включают материалы, такие как углеводы, воски, растворимые и/или набухающие в воде полимеры, гидрофильные или гидрофобные материалы, желатин, масла, растворители, воду и т.п. Конкретный используемый носитель, разбавитель или эксципиент может зависеть от способов и назначения, для которых применяют соединение по настоящему изобретению. Растворители обычно выбирают на основе растворителей, признанных специалистами в данной области в качестве безопасных (GRAS) для введения млекопитающему. Как правило, безопасные растворители представляют собой нетоксичные водные растворители, такие как вода и другие нетоксичные растворители, которые являются растворимыми в воде или смешивающимися с ней. Пригодные водные растворители включают воду, этанол, пропиленгликоль, полиэтиленгликоли (например, PEG400, PEG300) и т.д. и их смеси. Составы также могут включать один или несколько буферов, стабилизаторов, поверхностно-активных веществ, смачивающих веществ, смазывающих веществ, эмульгаторов, суспендирующих веществ, консервантов, антиоксидантов, замутнителей, веществ, способствующих скольжению, технологических добавок, красителей, подсластителей, отдушек, вкусовых добавок и других известных добавок для обеспечения хорошего представления лекарственного средства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или облегчения изготовления фармацевтического продукта (т.е. лекарственного средства).
Составы можно получать с использованием общепринятых способов растворения и смешивания. Например, нерасфасованное лекарственное вещество (т.е. соединение по настоящему изобретению или стабилизированная форма соединения, такая как комплекс с производным циклодекстрина или другим комплексообразующим агентом) растворяют в пригодном растворителе в присутствии одного или нескольких из эксципиентов, описанных выше. Соединение по настоящему изобретению, как правило, изготавливают в фармацевтических дозированных формах для обеспечения легко контролируемой дозировки лекарственного средства и для обеспечения соблюдения пациентом назначенного режима.
Фармацевтическую композицию (или состав) для применения можно упаковывать различными способами, в зависимости от способа, используемого для введения лекарственного средства. Как правило, изделие для распределения включает контейнер, в котором находится фармацевтический состав в надлежащей форме. Пригодные контейнеры хорошо известны специалистам в данной области и включают материалы, такие как бутыли (пластмассовые и стеклянные), саше, ампулы, пластмассовые мешки, металлические цилиндры и т.п. Контейнер также может включать безопасную в отношении проникновения систему для предупреждения непредусмотренного доступа к содержимому упаковки. Кроме того, на контейнере расположен ярлык, на котором описано содержимое контейнера. Ярлык также может включать соответствующие предупреждения.
Фармацевтические составы соединения формулы I можно получать для различных путей и типов введения. Соединение формулы I, имеющее желаемую степень чистоты, необязательно смешивают с фармацевтически приемлемыми разбавителями, носителями, эксципиентами или стабилизаторами (Remington's Pharmaceutical Sciences (1980) 16th edition, Osol, A. Ed.), в форме лиофилизированного состава, измельченного порошка или водного раствора. Состав можно получать смешиванием при температуре окружающей среды и надлежащем pH, и при желаемой степени чистоты, с физиологически приемлемыми носителями, т.е. носителями, которые являются нетоксичными для реципиентов в используемых дозировках и концентрации. pH состава зависит, главным образом, от конкретного применения и концентрации соединения, но может находиться в диапазоне от приблизительно 3 до приблизительно 8. Состав в ацетатном буфере при pH 5 является приемлемым вариантом осуществления.
В одном варианте осуществления соединение формулы I для применения в фармацевтической композиции является по существу стерильным. Соединение исходно можно хранить в качестве твердой композиции, хотя лиофилизированные составы или водные растворы являются приемлемыми.
Фармацевтические композиции по изобретению можно изготавливать, дозировать и вводить таким образом, т.е. в количествах, концентрациях, с расписаниями, с курсом, носителем и путем введения, которые согласуются с Good medical practice. Факторы для рассмотрения в этом контексте включают конкретное нарушение, подвергаемое лечению, конкретное млекопитающее подвергаемое лечению, клиническое состояние отдельного пациента, причину нарушения, область доставки средства, способ введения, расписание введения и другие факторы, известные практикующим врачам. "Терапевтически эффективное количество" соединения, подлежащего введению, определяется такими факторами, и оно представляет собой минимальное количество, требуемое для предупреждения, смягчения или лечения нарушения. Такое количество предпочтительно находится в количестве ниже количества, которое является токсичным для хозяина.
В качестве общего предложения, начальное фармацевтически эффективное количество ингибитора, вводимого парентерально на дозу, может находиться в диапазоне приблизительно 0,01-100 мг/кг, а именно приблизительно от 0,1 до 20 мг/кг массы тела пациента в сутки, причем типичный используемый начальный диапазон соединения составляет от 0,3 до 15 мг/кг/сутки.
Приемлемые разбавители, носители, эксципиенты и стабилизаторы являются нетоксичными для реципиентов в используемых дозировках и концентрациях, и включают буферы, такие как фосфат, цитрат и другие органические кислоты; антиоксиданты, включая аскорбиновую кислоту и метионин; консерванты (такие как хлорид октадецилдиметилбензиламмония; хлорид гексаметэтония; хлорид бензалкония, хлорид бензэтония; фенол, бутил или бензиловый спирт; алкилпарабены, такие как метил- или пропилпарабен; катехол; резорцинол; циклогексанол; 3-пентанол; и м-крезол); низкомолекулярные (менее 10 остатков) полипептиды; белки, такие как сывороточный альбумин, желатин или иммуноглобулины; гидрофильные полимеры, такие как поливинилпирролидон; аминокислоты, такие как глицин, глутамин, аспарагин, гистидин, аргинин или лизин; моносахариды, дисахариды и другие углеводы, включая глюкозу, маннозу или декстрины; хелатирующие агенты, такие как EDTA; сахара, такие как сахароза, маннит, трегалоза или сорбит; солеобразующие противоионы, такие как натрий; комплексы металлов (например, комплексы Zn-белок); и/или неионные поверхностно-активные вещества, такие как TWEEN®, PLURONICS® или полиэтиленгликоль (PEG). Активные фармацевтические ингредиенты также могут быть заключены в микрокапсулы, полученные, например, способами коацервации или межфазной полимеризации, например, микрокапсулы на основе гидроксиметилцеллюлозы или желатина и микрокапсулы на основе полиметилметакрилата, соответственно, в коллоидные системы для доставки лекарственного средства (например, липосомы, микросферы на основе альбумина, микроэмульсии, наночастицы и нанокапсулы), или в микроэмульсии. Такие способы описаны в Remington's Pharmaceutical Sciences 16th edition, Osol, A. Ed. (1980).
Можно получать препараты с замедленным высвобождением. Пригодные примеры препаратов с замедленным высвобождением включают полупроницаемые матрицы твердых гидрофобных полимеров, содержащие антитело, которые представлены в виде имеющих форму изделий, например, пленок или микрокапсул. Примеры матриц с замедленным высвобождением включают сложные полиэфиры, гидрогели (например, поли(2-гидроксиэтилметакрилат) или поли(виниловый спирт)), полилактиды, сополимеры L-глутаминовой кислоты и γ-этил-L-глутамата, недеградируемый этиленвинилацетат, деградируемые сополимеры молочная кислота-гликолевая кислота, такие как LUPRON DEPOT® (инъецируемые микросферы, состоящие из сополимера молочной кислоты и гликолевой кислоты и ацетата леупролида), и поли-D-(-)-3-гидроксимасляную кислоту.
Составы для применения для введения in vivo должны быть стерильными. Этого легко достигать фильтрацией через стерильные фильтрующие мембраны.
Составы включают составы, пригодные для путей введения, подробно описанных в настоящем документе. Составы могут быть традиционно представлены в единичной дозированной форме, и их можно получать любым из способов, хорошо известных в области фармацевтики. Способы и составы, главным образом, находятся в Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA). Такие способы включают стадию объединения активного ингредиента с носителем, который представляет собой один или несколько вспомогательных ингредиентов. Как правило, составы получают путем гомогенного и однородного объединения активного ингредиента с жидкими носителями или тонко измельченными твердыми носителями или обоими из них, а затем, если необходимо, придания продукту формы.
Составы соединения формулы I, пригодные для перорального введения, можно получать в качестве отдельных единиц, таких как пилюли, капсулы, крахмальные капсулы или таблетки, каждая из которых содержит заданное количество соединения формулы I.
Прессованные таблетки можно получать прессованием в пригодном устройстве активного ингредиента в свободнотекущей форме, такой как порошок или гранулы, необязательно в смеси со связующими веществом, смазывающим веществом, инертным разбавителем, консервантом, поверхностно-активным веществом или диспергирующим веществом. Прессованные таблетки можно получать формованием в пригодном устройстве смеси порошкового активного ингредиента, увлажненного инертным жидким разбавителем. На таблетки необязательно можно наносить покрытие и борозды и необязательно их изготавливают так, чтобы обеспечить медленное или контролируемое высвобождение из них активного ингредиента.
Для перорального применения можно изготавливать таблетки, лепешки, пастилки, водные или масляные суспензии, диспергируемые порошки или гранулы, эмульсии, твердые или мягкие капсулы, например, желатиновые капсулы, сиропы или эликсиры. Составы соединения формулы I, предназначенные для перорального применения, можно получать любым способом изготовления фармацевтических композиций, известным в данной области, и такие композиции могут содержать одно или несколько средств, включая подсластители, вкусовые добавки, красители и консерванты, для обеспечения препарата с приятным вкусом. Являются приемлемыми таблетки, содержащие активный ингредиент в смеси с нетоксичным фармацевтически приемлемым эксципиентом, который пригоден для изготовления таблеток. Эти эксципиенты могут представлять собой, например, инертные разбавители, такие как карбонат кальция или натрия, лактоза, фосфат кальция или натрия; гранулирующие и дезинтегрирующие вещества, такие как кукурузный крахмал или альгиновая кислота; связующие вещества, такие как крахмал, желатин или гуммиарабик; и смазывающие вещества, такие как стеарат магния, стеариновая кислота или тальк. Таблетки могут быть непокрытыми или они могут быть покрыты известными способами, включающими микроинкапсуляцию для замедления дезинтеграции и всасывания в желудочно-кишечном тракте, и, тем самым, обеспечения непрерывного действия в течение более длительного периода. Например, можно использовать задерживающий материал, такой как глицерилмоностеарат или глицерилдистеарат отдельно или с воском.
Для инфекций глаза или других наружных тканей, например, полости рта и кожи, составы предпочтительно наносят в качестве мази или крема для местного применения, содержащих активный ингредиент(ы) в количестве, например, от 0,075 до 20% масс./масс. При изготовлении в виде мази активные ингредиенты можно использовать либо с парафиновой, либо со смешивающейся с водой основой мази. Альтернативно, активные ингредиенты можно изготавливать в виде крема с основой крема типа "масло-в-воде".
Если желательно, водная фаза основы крема может включать многоатомный спирт, т.е. спирт, имеющий две или более гидроксильных групп, такой как пропиленгликоль, бутан-1,3-диол, маннит, сорбит, глицерин и полиэтиленгликоль (включая PEG 400) и их смеси. Составы для местного применения, желательно, могут включать соединение, которое усиливает всасывание или проникновение активного ингредиента через кожу или другие пораженные области. Примеры таких усилителей проникновения через кожу включают диметилсульфоксид и сходные аналоги.
Масляная фаза эмульсий по этому изобретению может быть изготовлена из известных ингредиентов известным способом. Хотя фаза может содержать только эмульгатор (также известный как эмульгирующее вещество), она желательно содержит смесь по меньшей мере одного эмульгатора с жиром или маслом, или как с жиром, так и с маслом. Предпочтительно, гидрофильный эмульгатор включают вместе с липофильным эмульгатором, который действует в качестве стабилизатора. Также предпочтительно включать как масло, так и жир. Взятые вместе, эмульгатор(ы) с или без стабилизатора(ов) составляют так называемый эмульгирующий воск, и воск вместе маслом и жиром составляют так называемую эмульгирующую основу мазь, которая формирует маслянистую дисперсную фазу составов кремов. Эмульгирующие вещества и стабилизаторы эмульсии, пригодные для применения в составе по изобретению, включают Tween® 60, Span® 80, цетостеариловый спирт, бензиловый спирт, миристиловый спирт, глицерилмоностеарат и лаурилсульфат натрия.
Водные суспензии по изобретению содержат активные материалы в смеси с эксципиентами, пригодными для изготовления водных суспензий. Такие эксципиенты включают суспендирующее вещество, такое как натрий карбоксиметилцеллюлоза, кроскармеллоза, повидон, метилцеллюлоза, гидроксипропилметилцеллюлоза, альгинат натрия, поливинилпирролидон, трагакантовая камедь и гуммиарабик, и диспергирующие или смачивающие вещества, такие как встречающийся в природе фосфатид (например, лецитин), продукт конденсации алкилоксида (например, оксид этилена, оксид пропилена) с жирной кислотой (например, стеарат полиоксиэтилена), продукт конденсации оксида этилена с алифатическим спиртом длинной цепи (например, гептадекаэтиленоксицетанол), продукт конденсации оксида этилена с неполным эфиром, образованным из жирной кислоты и гекситного ангидрида (например, моноолеат полиоксиэтилена сорбитана). Водная суспензия также может содержать один или несколько консервантов, таких как этил- или н-пропил-п-гидроксибензоат, один или несколько красителей, одну или несколько вкусовых добавок или один или несколько подсластителей, таких как сахароза или сахарин.
Фармацевтическая композиция соединения формулы I может быть в форме стерильного инъецируемого препарата, такого как стерильная инъецируемая водная или масляная суспензия. Эта суспензия может быть изготовлена известными способами с использованием пригодных диспергирующих или смачивающих веществ и суспендирующих веществ, которые упомянуты выше. Стерильный инъецируемый препарат также может представлять собой стерильный инъецируемый раствор или суспензию в нетоксичном парентерально приемлемом разбавителе или растворителе, таком как раствор в 1,3-бутандиоле, или он может быть получен в качестве лиофилизированного порошка. Среди приемлемых носителей и растворителей, которые можно использовать, находятся вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, в качестве растворителя или суспендирующей среды обычно можно использовать стерильные жирные масла. Для этой цели можно использовать любое легкое жирное масло, включая синтетические моно- или диглицериды. Кроме того, аналогично для получения инъецируемых препаратов можно использовать жирные кислоты, такие как олеиновая кислота.
Количество активного ингредиента, которое можно комбинировать с материалом носителя для получения единичной дозированной формы, варьирует, в зависимости от подвергаемого лечению хозяина и конкретного способа введения. Например, состав с замедленным высвобождением, предназначенный для перорального введения человеку, может содержать приблизительно от 1 до 1000 мг активного материала, изготовленного с соответствующим и удобным количеством материала носителя, которое может варьировать от приблизительно 5 до приблизительно 95% от всей композиции (масс.:масс.). Фармацевтическую композицию можно получать для обеспечения легко определяемых количеств для введения. Например, водный раствор, предназначенный для внутривенной инфузии, может содержать приблизительно от 3 до 500 мкг активного ингредиента на миллилитр раствора, для того чтобы можно было проводить инфузию пригодного объема со скоростью приблизительно 30 мл/ч.
Составы, пригодные для парентерального введения, включают водные и неводные стерильные растворы для инъекции, которые могут содержать антиоксиданты, буферы, бактериостатические средства и растворенные вещества, которые делают состав изотоничным с кровью предполагаемого реципиента; и водные и неводные стерильные суспензии, которые могут включать суспендирующие вещества и загустители.
Составы, пригодные для местного введения в глаз, также включают капли для глаз, где активный ингредиент растворен или суспендирован в пригодном носителе, особенно в водном растворителе для активного ингредиента. Активный ингредиент предпочтительно присутствует в таких составах в концентрации от 0,5 до 20%, преимущественно от 0,5 до 10%, в частности, приблизительно 1,5% масс./масс.
Составы, пригодные для местного введения в полость рта, включают пастилки, содержащие активный ингредиент во вкусовой основе, обычно сахарозе и гуммиарабике или трагаканте; пастилки, содержащие активный ингредиент в инертной основе, такой как желатин и глицерин, или сахароза и гуммиарабик; и жидкости для полоскания рта, содержащие активный ингредиент в пригодном жидком носителе.
Составы для ректального введения могут присутствовать в качестве суппозитория с пригодной основой, содержащей, например, масло какао или салицилат.
Составы, пригодные для внутрилегочного или назального введения, имеют размер частиц, например, в диапазоне от 0,1 до 500 микрометров (включая размер частиц в диапазоне от 0,1 до 500 микрометров с шагом в микрометрах, таким как 0,5, 1, 30 микрометров, 35 микрометров и т.д.), которые вводят путем быстрой ингаляции через носовой ход или путем ингаляции через рот, так чтобы достигнуть альвеолярных мешков. Пригодные составы включают водные и масляные растворы активного ингредиента. Составы, пригодные для введения аэрозоля или сухого порошка, можно получать общепринятыми способами и можно доставлять с другими лекарственными средствами, такими как соединения, до настоящего времени используемые для лечения или профилактики ВИЧ-инфекции, как описано ниже.
Составы, пригодные для вагинального введения, могут быть предоставлены в качестве пессариев, тампонов, кремов, гелей, паст, пен или составов спреев, содержащих в дополнение к активному ингредиенту носители, которые известны в данной области в качестве приемлемых.
Составы могут быть упакованы в контейнеры с единичной дозой или с многократными дозами, например, запаянные ампулы и флаконы, и их можно хранить в сублимированном (лиофилизированном) состоянии, требующем только добавления стерильного жидкого носителя, например воды, для инъекции непосредственно перед применением. Инъекционные растворы и суспензии для немедленного введения получают из стерильных порошков, гранул и таблеток ранее описанного типа. Предпочтительные единичные дозированные составы представляют собой составы, содержащие суточную дозу или единичную суточную субдозу активного ингредиента, как описано в настоящем документе выше, или ее соответствующую фракцию.
Кроме того, изобретение относится к ветеринарным композициям, содержащим по меньшей мере один активный ингредиент, как определено выше, вместе с ветеринарным носителем для него. Ветеринарные носители представляют собой материалы, пригодные для цели введения композиции, и они могут представлять собой твердые, жидкие или газообразные материалы, которые в любом случае являются инертными или приемлемыми в ветеринарной области и совместимыми с активным ингредиентом. Эти ветеринарные композиции можно вводить парентерально, перорально или любым другим желаемым путем.
КОМБИНИРОВАННАЯ ТЕРАПИЯ
Соединения формулы I можно использовать отдельно или в комбинации с другими лекарственными средствами для лечения заболевания или нарушения, описанного в настоящем документе, такого как иммунологическое нарушение (например, псориаз или воспаление) или гиперпролиферативное нарушение (например, злокачественная опухоль). В определенных вариантах осуществления соединение формулы I комбинируют в фармацевтическом комбинированном составе или в режиме дозирования в качестве комбинированной терапии со вторым терапевтическим соединением, которое обладает противовоспалительными или антипролиферативными свойствами и которое пригодно для лечения воспаления, нарушения иммунного ответа или гиперпролиферативного нарушения (например, злокачественной опухоли). Второе лекарственное средство может представлять собой NSAID или другое противовоспалительное средство. Второе лекарственное средство может представлять собой химиотерапевтическое средство. Второе лекарственное средство фармацевтического комбинированного состава или режима дозирования предпочтительно имеет дополняющую активность для активности соединения формулы I, так что они не оказывают неблагоприятного действия друг на друга. Такие соединения соответственно присутствуют в комбинации в количествах, которые являются эффективными для предполагаемого назначения. В одном варианте осуществления композиция по этому изобретению содержит соединение формулы I или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемую соль или пролекарство, в комбинации с лекарственным средством, таким как NSAID.
Таким образом, другой вариант осуществления относится к способу лечения или снижения тяжести заболевания или состояния, отвечающего на ингибирование Janus-киназы у пациента, включающему введение указанному пациенту терапевтически эффективного количества соединения формулы I, и, кроме того, включающему введение второго лекарственного средства.
Комбинированную терапию можно проводить в качестве одновременного или последовательного режима. При введении последовательно комбинацию можно вводить за два или более введений. Комбинированное введение включает совместное введение с использованием отдельных составов или единого состава, и последовательное введение в любом порядке, где предпочтительно существует период времени, когда оба (или все) активных средства одновременно проявляют их биологическую активность.
Пригодные дозировки любого из описанных выше совместно вводимых средств представляют собой дозировки, используемые в настоящее время, и их можно снижать вследствие комбинированного действия (синергии) вновь идентифицированного средства и других химиотерапевтических средств или способов лечения.
Комбинированная терапия может обеспечить "синергию" и оказаться "синергической", т.е. эффект, достигаемый, когда активные ингредиенты используют вместе, превышает сумму эффектов вследствие применения соединений по отдельности. Синергического эффекта можно достигать, когда активные ингредиенты: (1) совместно включают в состав и вводят или доставляют одновременно в комбинированном единичном дозированном составе; (2) доставляют поочередно или параллельно в качестве отдельных составов; или (3) с помощью любого другого режима. При доставке в чередующейся терапии, синергического эффекта можно достигать, когда соединения вводят или доставляют последовательно, например, путем различных инъекций в отдельных шприцах. Как правило, в ходе поочередной терапии эффективную дозировку каждого активного ингредиента вводят последовательно, т.е. по порядку, в то время как при комбинированной терапии эффективные дозировки двух или более активных ингредиентов вводят вместе.
В конкретном варианте осуществления терапии соединение формулы I или его стереоизомер, геометрический изомер, таутомер, сольват, метаболит или фармацевтически приемлемую соль или пролекарство можно комбинировать с другими терапевтическими, гормональными средствами или средствами на основе антител, такими как средства, описанные в настоящем документе, а также комбинировать с хирургическим лечением и лучевой терапией. Комбинированная терапия в соответствии с настоящим изобретением, таким образом, включает введение по меньшей мере одного соединения формулы I или его стереоизомера, геометрического изомера, таутомера, сольвата, метаболита или фармацевтически приемлемой соли или пролекарства, и использование по меньшей мере одного другого способа лечения злокачественной опухоли или способа лечения иммунологического нарушения. Количество соединения(ий) формулы I и другого фармакологически активного иммунологического или химиотерапевтического средства (средств) и соотносительное расписание введения можно выбирать для достижения желаемого комбинированного терапевтического эффекта.
МЕТАБОЛИТЫ СОЕДИНЕНИЙ, ИНГИБИРУЮЩИХ JANUS-КИНАЗУ
Другой вариант осуществления относится к метаболическим продуктам in vivo вводимого соединения формулы I. Такие продукты могут быть результатом, например, окисления, восстановления, гидролиза, амидирования, дезамидирования, этерификации, деэтерификации, ферментативного расщепления и т.п. вводимого соединения.
Метаболические продукты, как правило, идентифицируют получением радиоактивно меченного (например, 14C или 3H) изотопа соединения по изобретению, введением его парентерально в поддающейся детекции дозе (например, превышающей 0,5 мг/кг) животному, такому как крыса, мышь, морская свинка, обезьяна, или человеку, обеспечивая достаточно времени для протекания метаболизма (как правило, приблизительно от 30 секунд до 30 часов) и выделяя его продукты превращения из мочи, крови или других биологических образцов. Эти продукты легко выделять, поскольку они являются мечеными (другие продукты выделяют с использованием антител, способных связывать эпитопы, сохраняющиеся в метаболите). Структуры метаболитов определяют общепринятым способом, например, посредством анализа MS, LC/MS или ЯМР. Как правило, анализ метаболитов проводят аналогично тому, как в общепринятых исследованиях метаболизма лекарственных средств, хорошо известных специалистам в данной области. Продукты превращения, при условии, что они не встречаются в ином случае in vivo, пригодны в диагностических анализах для терапевтического дозирования соединения формулы I.
ИЗДЕЛИЯ
Другой вариант осуществления относится к способу изготовления соединения формулы I. Способ включает реакцию соединения формулы (i)
 ,
,
где R1 определен в формуле I, и X представляет собой уходящую группу, например, галоген, -OH, -O(C1-C12алкил); с соединением формулы (iia-c)
 ,
,
где R5 определен в формуле I, и R10 представляет собой водород или R4, и R4 определен в формуле I; в условиях, достаточных для образования соединения формулы I.
В одном варианте осуществления указанные условия включают основные условия, например, проведение реакции в присутствии основания, такого как аминовое основание, например, пиридин или алкилированный амин, такой как диалкил- или триалкиламин (например триметиламин, триэтиламин, диизопропиламин или диэтиламин).
В одном варианте осуществления указанные условия включают условия реакции сочетания, например, проведение реакции в присутствии PyAOP, DMAP и алкилированного амина, например, диизопропиламина.
Другой вариант осуществления относится к набору для лечения заболевания или нарушения, отвечающего на ингибирование Janus-киназы. Набор включает:
первую фармацевтическую композицию, содержащую соединение формулы I; и
инструкции по применению.
В другом варианте осуществления набор, кроме того, включает:
вторую фармацевтическую композицию, которая включает химиотерапевтическое средство.
В одном варианте осуществления инструкции включают инструкции для одновременного, последовательного или раздельного введения указанных первой и второй фармацевтических композиций пациенту, нуждающемуся в них.
В одном варианте осуществления первая и вторая композиции находятся в отдельных контейнерах.
В одном варианте осуществления первая и вторая композиции находятся в одном контейнере.
Контейнеры для применения включают, например, бутыли, флаконы, шприцы, блистерную упаковку и т.д. Контейнеры могут быть образованы из различных материалов, таких как стекло или пластмасса. Контейнер включает соединение формулы I или его состав, которые являются эффективными для лечения состояния и могут иметь стерильное отверстие для доступа (например, контейнер может представлять собой мешок для внутривенного раствора или флакон, имеющий пробку, проницаемую иглой для подкожной инъекции). Контейнер включает композицию, содержащую по меньшей мере одно соединение формулы I. На ярлыке или вкладыше в упаковку указано, что композицию используют для лечения определенного состояния, такого как злокачественная опухоль. В одном варианте осуществления на ярлыке или вкладыше в упаковку указано, что композицию, содержащую соединение формулы I, можно применять для лечения нарушения. Кроме того, на ярлыке или вкладыше в упаковку может быть указано, что пациент, подвергаемый лечению, представляет собой пациента, имеющего нарушение, характеризующегося избыточной или неправильной киназной активностью. На ярлыке или вкладыше в упаковку также может быть указано, что композицию можно применять для лечения других нарушений.
Изделие может содержать (a) первый контейнер с соединением формулы I, содержащимся в нем; и (b) второй контейнер со вторым фармацевтическим составом, содержащимся в нем, где второй фармацевтический состав содержит химиотерапевтическое средство. Изделие в этом варианте осуществления изобретения может, кроме того, содержать вкладыш в упаковку, на котором указано, что первое и второе соединения можно применять для лечения пациентов, имеющих риск инсульта, тромба или тромботического нарушения. Альтернативно или дополнительно, изделие может, кроме того, содержать второй (или третий) контейнер, содержащий фармацевтически приемлемый буфер, такой как бактериостатическая вода для инъекции (BWFI), фосфатно-солевой буфер, раствор Рингера и раствор декстрозы. Кроме того, он может включать другие материалы, желаемые с коммерческой и пользовательской точки зрения, включая другие буферы, разбавители, фильтры, иглы и шприцы.
Для иллюстрации изобретения включены следующие примеры. Однако следует понимать, что эти примеры не ограничивают изобретение и представлены только для предложения способа осуществления изобретения на практике. Специалистам в данной области будет понятно, что описанные химические реакции можно легко адаптировать для получения других соединений формулы I, и альтернативные способы получения соединений формулы I находятся в объеме этого изобретения. Например, синтез непроиллюстрированных соединений согласно изобретению можно успешно проводить с помощью модификаций, очевидных специалистам в данной области, например, путем надлежащей защиты мешающих групп, путем применения других пригодных реагентов, известных в данной области, отличных от описанных реагентов, и/или путем проведения общепринятых модификаций условий реакции. Альтернативно, другие реакции, описанные в настоящем документе, или известные в данной области, могут быть признаны в качестве применимых для получения других соединений по изобретению.
БИОЛОГИЧЕСКИЕ ПРИМЕРЫ
Соединения формулы I можно анализировать в отношении способности модулировать активность протеинкиназ Janus, тирозинкиназ, дополнительных серин/треониновых киназ и/или киназ с двойной специфичностью in vitro и in vivo. Анализы in vitro включают биохимические и клеточные анализы, которые определяют ингибирование активности киназы. Альтернативно, анализы in vitro количественно определяют способность соединения формулы I связываться с киназами, и ее можно измерять либо путем радиоактивного мечения соединения формулы I перед связыванием, выделения комплекса соединение формулы I/киназа и определения количества связанной радиоактивной метки, или путем проведения конкурентного эксперимента, где соединение формулы I инкубируют с известными меченными радиоактивной меткой лигандами. Эти и другие пригодные анализы in vitro хорошо известны специалистам в данной области.
В одном варианте осуществления соединения формулы I можно применять для контроля, модулирования или ингибирования активности тирозинкиназы, например, активности протеинкиназы Janus, дополнительной серин/треониновой киназы и/или киназы с двойной специфичностью. Таким образом, они пригодны в качестве фармакологических стандартов для применения в разработке новых биологических тестов и анализов, и в поиске новых фармакологических средств.
Пример A
Протокол анализа ингибирования JAK1, JAK2 и TYK2
Активность выделенного киназного домена JAK1, JAK2 или TYK2 измеряли путем мониторинга фосфорилирования пептида, происходящего из JAK3 (Val-Ala-Leu-Val-Asp-Gly-Tyr-Phe-Arg-Leu-Thr-Thr), меченного флуоресцентной меткой на N-конце 5-карбоксифлуоресцеином с использованием технологии Caliper LabChip (Caliper Life Sciences, Hopkinton, MA). Для определения констант ингибирования (Ki) соединений примеров 1-508, соединения серийно разбавляли в DMSO и добавляли в 50-мкл реакционные смеси киназы, содержащие 1,5 нМ JAK1, 0,2 нМ очищенный фермент JAK2 или 1 нМ очищенный фермент TYK2, 100 мМ Hepes pH7,2, 0,015% Brij-35, 1,5 мкМ пептидный субстрат, 25 мкМ ATP, 10 мМ MgCl2, 4 мМ DTT в конечной концентрации DMSO 2%. Реакционные смеси инкубировали при 22°С в 384-луночном полипропиленовом микропланшете для титрования в течение 30 минут, а затем реакцию останавливали добавлением 25 мкл содержащего ЭДТА раствора (100 мМ Hepes pH 7,2, 0,015% Brij-35, 150 мМ ЭДТА), с получением конечной концентрации ЭДТА 50 мМ. После завершения киназной реакции долю фосфорилированного продукта определяли в качестве фракции общего пептидного субстрата с использованием Caliper LabChip 3000 согласно спецификациями изготовителя. Затем определяли величины Ki с использованием модели прочного связывания Morrison. Morrison, J.F., Biochim. Biophys. Acta. 185:269-296 (1969); William, J.W. and Morrison, J.F., Meth. Enzymol., 63:437-467 (1979).
Пример B
Протокол анализа ингибирования JAK3
Активность выделенного киназного домена JAK3 измеряли путем мониторинга фосфорилирования пептида, происходящего из JAK3 (Leu-Pro-Leu-Asp-Lys-Asp-Tyr-Tyr-Val-Val-Arg), меченного флуоресцентной меткой на N-конце 5-карбоксифлуоресцеином с использованием технологии Caliper LabChip (Caliper Life Sciences, Hopkinton, MA). Для определения констант ингибирования (Ki) соединений примеров 1-508, соединения серийно разбавляли в DMSO и добавляли в 50-мкл реакционные смеси киназы, содержащие 5 нМ очищенный фермент JAK3, 100 мМ Hepes pH 7,2, 0,015% Brij-35, 1,5 мкМ пептидный субстрат, 5 мкМ ATP, 10 мМ MgCl2, 4 мМ DTT в конечной концентрации DMSO 2%. Реакционные смеси инкубировали при 22°C в 384-луночных полипропиленовых микропланшетах для титрования в течение 30 минут, а затем реакцию останавливали добавлением 25 мкл содержащего ЭДТА раствора (100 мМ Hepes pH 7,2, 0,015% Brij-35, 150 мМ ЭДТА), с получением конечной концентрации ЭДТА 50 мМ. После завершения киназной реакции долю фосфорилированного продукта определяли в качестве фракции общего пептидного субстрата с использованием Caliper LabChip 3000 согласно спецификациями изготовителя. Затем определяли величины Ki с использованием модели прочного связывания Morrison. Morrison, J.F., Biochim. Biophys. Acta. 185:269-296 (1969); William, J.W. and Morrison, J.F., Meth. Enzymol., 63:437-467 (1979).
Пример C
Клеточные анализы фармакологии
Активность соединений примеров 1-508 определяли в клеточных анализах, которые были предназначены для изменения зависимой от Janus-киназы передачи сигнала. Соединения серийно разбавляли в DMSO и инкубировали с клетками Set-2 (German Collection of Microorganisms and Cell Cultures (DSMZ); Braunschweig, Германия), которые экспрессируют мутантный белок JAK2V617F, в 96-луночных микропланшетах для титрования в течение 1 ч при 37°C в среде RPMI с конечной плотностью клеток 105 клеток на лунку и в конечной концентрации DMSO 0,57%. Затем в лизатах инкубированных клеток измеряли опосредуемые соединением эффекты на фосфорилирование STAT5 с использованием технологии Meso Scale Discovery (MSD) (Gaithersburg, Maryland) согласно протоколу изготовителя и определяли величины EC50. Альтернативно, серийно разбавленные соединения добавляли к клеткам NK92 (American Type Culture Collection (ATCC); Manassas, VA) в 96-луночные микропланшеты для титрования в среде RPMI с конечной плотностью клеток 105 клеток на лунку и в конечной концентрации DMSO 0,57%. Затем добавляли рекомбинантный IL-2 или IL-12 человека (R&D systems; Minneapolis, MN) в конечной концентрации 10 нг/мл или 30 нг/мл, соответственно, в микропланшеты для титрования, содержащие клетки NK92 и соединение, и планшеты инкубировали в течение 1 ч при 37°C. Затем в лизатах инкубированных клеток измеряли опосредуемые соединением эффекты на фосфорилирование STAT5 (IL-2-опосредемое) или STAT4 (IL-12-опосредуемое) с использованием технологии Meso Scale Discovery (MSD) (Gaithersburg, Maryland) согласно протоколу изготовителя и определяли величины EC50.
ПРИМЕРЫ ПОЛУЧЕНИЯ
Сокращения
CD3OD дейтерированный метанол
CDCl3 дейтерированный хлороформ
DAST трифторид диэтиламиносеры
DCM дихлорметан
DIPEA диизопропилэтиламин
DMAP 4-диметиламинопиридин
DMF диметилформамид
DMF-DMA диметилацеталь N,N-диметилформамида
DMSO диметилсульфоксид
DMSO-d6 дейтерированный DMSO
DME 1,2-диметоксиэтан
DMF диметилформамид
DPPA дифенилфосфорилазид
EtOAc этилацетат
EtOH этанол
HOAc уксусная кислота
г грамм
HATU (O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат)
HCl хлористоводородная кислота
Hex гексан
HM-N Isolute® HM-N представляет собой модифицированную форму диатомитовой земли
IMS промышленные метилированные спирты
л литр
МeOH метанол
мг миллиграмм
мл миллилитр
POCl3 оксихлорид фосфора
NaH гидрид натрия
Na2SO4 сульфат натрия
NaHCO3 бикарбонат натрия
NaOH гидроксид натрия
Pd(PPh3)4 Тетракис(трифенилфосфин)палладий(0)
PyAOP гексафторфосфат (7-азабензотриазол-1-илокси)трипирролидинофосфония
Реагент Лоуссона 2,4-бис(4-метоксифенил)-1,3-дитиа-2,4-дифосфэтан-2,4-дисульфид
NEt3 триэтиламин
Pd2dba3 трис(дибензилиденацетон)дипалладий(0)
Si-SPE предварительно упакованная кассета для флэш-хроматографии с диоксидом кремния Isolute®
Si-ISCO предварительно упакованная кассета для флэш-хроматографии с диоксидом кремния ISCO®
THF тетрагидрофуран
SEM 2-(триметилсилил)этоксиметил
SEMCl 2-(триметилсилил)этоксиметилхлорид
TEA триэтиламин
TFA трифторуксусная кислота
Общие экспериментальные условия
Спектры 1H-ЯМР регистрировали при температуре окружающей среды с использованием спектрометра Varian Unity Inova (400 МГц) с 5-мм зондом тройного резонанса, спектрометра Bruker AVIII (400 МГц) с использованием 5-мм зонда BBI Broad Band Inverse, спектрометра Bruker Avance DRX400 (400 МГц) с 5-мм зондом тройного резонанса или спектрометра Bruker AVIII (500 МГц) с использованием 5-мм зонда QNP (Quad Nucleus detect). Химические сдвиги выражают в м.д. относительно тертаметилсилана. Используются следующие сокращения: уш. = широкий сигнал, с = синглет, д = дублет, дд = двойной дублет, т = триплет, кв.=квартет, м = мультиплет.
Эксперименты с высокоэффективной жидкостной хроматографией - масс-спектрометрией (LCMS) для определения времени удерживания (RT) и ассоциированных масс ионов проводили с использованием одного из следующих способов.
Способ A: Эксперименты проводили на квадрупольном масс-спектрометре Waters Micromass ZQ, соединенном с системой Hewlett Packard HP1100 LC с детектором с диодной матрицей. В этой системе используется 5-микронная колонка Higgins Clipeus C18 100×3,0 мм и скорость потока 1 мл/минута. Первоначальная система растворителей представляла собой 95% воду, содержащую 0,1% муравьиную кислоту (растворитель A) и 5% ацетонитрил, содержащий 0,1% муравьиную кислоту (растворитель B) в течение первой минуты, а затем градиент из вплоть до 5% растворителя A и 95% растворителя B в течение последующих 14 минут. Конечную систему растворителей держали постоянной в течение дальнейших 5 минут.
Способ B: Эксперименты проводили на квадрупольном масс-спектрометре Waters Platform LC, соединенном с системой Hewlett Packard HP1100 LC с детектором с диодной матрицей и 100-позиционным автоматическим пробоотборником с использованием колонки Phenomenex Luna C18(2) 30×4,6 мм и скорости потока 2 мл/минута. Система растворителей представляла собой 95% воду, содержащую 0,1% муравьиную кислоту (растворитель A) и 5% ацетонитрил, содержащий 0,1% муравьиную кислоту (растворитель B) в течение первых 0,50 минут, с последующим градиентом из вплоть до 5% растворителя A и 95% растворителя B в течение последующих 4 минут. Конечную систему растворителей держали постоянной в течение дальнейших 0,50 минут.
Для очистки определенных соединений использовали препаративную высокоэффективную жидкостную хроматографию (ВЭЖХ). Использованная система представляла собой Varian LC, оборудованную автоматизированным УФ-запускаемым коллектором фракций и колонкой Gemini NX (3×10 см). Подвижная фаза представляла собой градиент из 5-85% ацетонитрила в воде, содержавший 0,1% муравьиную кислоту или гидроксид аммония в течение 10 минут.
Хиральную сверхкритическую жидкостную хроматографию (SFC) использовали для разделения некоторых рацемических соединений на их составляющие энантиомеры. Использованная система представляла собой Berger Pronto SFC, оборудованную автоматизированным УФ-запускаемым коллектором фракций. Примерами использованных колонок являются Chiral Technologies AD, OD, OJ или AS (21,2×250 мм). Длина пробега варьировала от 5 до 10 минут, и использовали изократическую подвижную фазу, состоящую из смеси 5-50% MeOH:диоксид углерода.
Эксперименты с микроволновым излучением проводили с использованием Biotage Initiator 60™, в котором используется одномодовый резонатор и регулировка нестационарного поля. Можно достигать температуры 40-250°C и давления вплоть до 30 бар.
ПРИМЕР 1
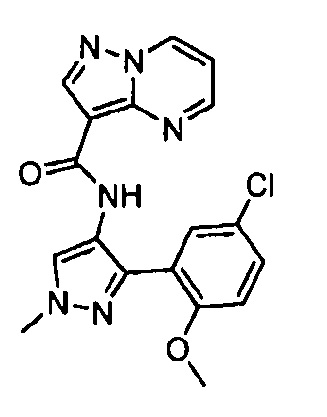
N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
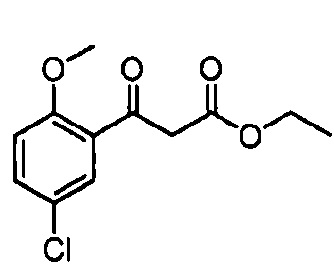
этил-3-(5-хлор-2-метоксифенил)-3-оксопропаноат
К перемешиваемому раствору 5-хлор-2-метоксибензойной кислоты (1,87 г, 10,0 ммоль, 1 экв.) в 20 мл тетрагидрофурана добавляли N,N-карбонилдиимидазол (1,64 г, 10,1 ммоль, 1,01 экв.), и перемешивание продолжали в течение двадцати минут, получая ацилимидазол. Отдельно, этилмалонат калия (4,08 г, 24,0 ммоль, 2,39 экв.) и хлорид магния (1,15 г, 12,1 ммоль, 1,20 экв.) суспендировали в 20 мл тетрагидрофуране. К смеси с хлоридом магния добавляли раствор ацилимидазола. Перемешивание продолжали при 50°C в течение 1,5 часа. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть сушили над сульфатом магния, фильтровали через слой целита и концентрировали с получением этил-3-(5-хлор-2-метоксифенил)-3-оксопропаноата, который использовали без дальнейшей очистки. LCMS (ESI) m+H=257,2.

этил-2-(5-хлор-2-метоксибензоил)-3-(диметиламино)акрилат
Перемешиваемую смесь этил-3-(5-хлор-2-метоксифенил)-3-оксопропаноата (10,02 ммоль, 1 экв.) и 1,1-диметокси-N,N-диметилметанамина (3,0 мл, 22 ммоль, 2,2 экв.) нагревали при 90°C в течение 2 часов. После выпаривания избытка 1,1-диметокси-N,N-диметилметанамина неочищенный продукт очищали флэш-хроматографией на силикагеле (0-80% этилацетат в дихлорметане) с получением 2,493 г (80%) этил-2-(5-хлор-2-метоксибензоил)-3-(диметиламино)акрилата. LCMS (ESI) m+H=312,2.

этил-5-(5-хлор-2-метоксифенил)-1H-пиразол-4-карбоксилат
Раствор этил-2-(5-хлор-2-метоксибензоил)-3-(диметиламино)акрилата (2,49 г, 8,00 ммоль, 1 экв.) и гидразина (0,40 мл, 13 ммоль, 1,6 экв.) в 20 мл этанола нагревали при 70°C в течение 2 часов. Затем растворитель и избыток гидразина выпаривали с получением этил-5-(5-хлор-2-метоксифенил)-1H-пиразол-4-карбоксилата, который использовали без дальнейшей очистки. LCMS (ESI) m+H=281,3.

этил-3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-карбоксилат
К раствору этил-5-(5-хлор-2-метоксифенил)-1H-пиразол-4-карбоксилата (2,24 г, 8,00 ммоль, 1 экв.) в 20 мл N,N-диметилформамида добавляли карбонат цезия (3,417 г, 10,49 ммоль, 1,3 экв.) и йодметан (0,60 мл, 9,6 ммоль, 1,2 экв.). Реакционную смесь перемешивали при 40°C в течение 4 часов, затем добавляли дополнительный йодметан (0,20 мл, 3,2 ммоль, 0,4 экв.). После дополнительных 2,5 часов реакционную смесь распределяли между этилацетатом и водой. Органическую часть сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-35% этилацетат в дихлорметане) с получением 2,18 г (92%) смеси 1:1 региоизомерных продуктов: этил-3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-карбоксилата и этил-5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-карбоксилата. LCMS (ESI) m+H=295,1.
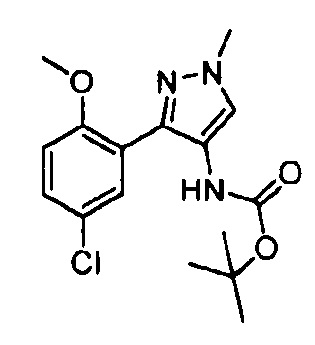
трет-бутил-3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамат
Раствор этил-3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-карбоксилата и этил-5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-карбоксилата (смесь региоизомеров 1:1, 2,18 г, 7,40 ммоль, 1 экв.) в 15 мл этанола обрабатывали 1,0 М водным раствором гидроксида натрия (12 мл, 0,020 ммоль, 4,0 экв.). Реакционную смесь нагревали при 50°C в течение 14 часов. После выпаривания этанола осадок разбавляли в воде и доводили до pH 2 1,0 M водным раствором фосфорной кислоты. Этот раствор экстрагировали два раза дихлорметаном. Объединенные экстракты сушили над MgSO4 и концентрировали с получением 1,79 г (91%) соответствующих карбоновых кислот, которые немедленно использовали далее. LCMS (ESI) m+H=267,2. К раствору этого материала в 15 мл диоксана добавляли триэтиламин (2,0 мл, 14 ммоль, 4,3 экв.) и дифенилфосфоновый азид (1,6 мл, 7,4 ммоль, 2,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа, и в это время реакционную смесь нагревали до 90°C и добавляли 15 мл трет-бутилового спирта. После перемешивания при 90°C в течение 2,5 часов растворитель выпаривали и осадок распределяли между этилацетатом и водой. Органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-50% этилацетат в дихлорметане), разделяя два стериоизомера с получением: 543,2 мг (48%) трет-бутил-3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамата; LCMS (ESI) m+H=338,3; 1H ЯМР (400 МГц, CDCl3) δ: 7,84 (с, 1H), 7,39 (д, 1H), 7,23 (с, 1H), 6,96 (д, 1H), 5,92 (с, 1H), 3,89 (перекрывающийся с и с, 6H), 1,48 (с, 9H); и 773,7 мг (68%) трет-бутил-5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамата. LCMS (ESI) m+H=338,3; 1H ЯМР (400 MГц, CDCl3) δ: 7,84 (с, 1H), 7,59 (с, 1H), 7,29 (д, 1H), 6,93 (д, 1H), 3,89 (перекрывающийся с и с, 6H), 1,48 (с, 9H).

3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-амин
К перемешиваемому раствору трет-бутил-3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамата (543,2 мг, 1,608 ммоль, 1 экв.) добавляли хлористоводородную кислоту (5,0 мл 4,0 M раствора в 1,4-диоксане, 0,020 моль, 12 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 9 часов, а затем упаривали до сухого состояния. Твердый осадок распределяли между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Водную часть экстрагировали еще раз дихлорметаном, и объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-амина, который использовали далее без очистки. LCMS (ESI) m+H=238,2; 1H ЯМР (400 MГц, CDCl3) δ: 7,53 (с, 1H), 7,28 (д, 1H), 7,02 (с, 1H), 6,91 (д, 1H), 3,91 (д, 2H), 3,87 (с, 3H), 3,84 (с, 3H).

N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-амина (258 мг, 0,869 ммоль, 1,00 экв.), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (157 мг, 0,961 ммоль, 1,11 экв.), гексафторфосфата 7-азабензотриазол-1-илокси-трис(пирролидинo)фосфония (541 мг, 1,04 ммоль, 1,20 экв.), N,N-диизопропилэтиламина (0,40 мл, 2,3 ммоль, 2,6 экв.) и 4-диметиламинопиридина (30,7 мг, 0,251 ммоль, 0,29 экв.) в 8,0 мл N,N-диметилформамида перемешивали при 50°C в течение 3 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали с получением 232,3 мг (70%) неочищенного продукта. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=383,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,66 (с, 1H), 9,32 (д, 1H), 8,76 (д, 1H), 8,65 (с, 1H), 8,26 (с, 1H), 7,48 (д, 1H), 7,40 (с, 1H), 7,28 (м, 2H), 3,91 (с, 3H), 3,84 (с, 3H).
ПРИМЕР 2

N-(3-(5-хлор-2-гидроксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (145 мг, 0,379 ммоль, 1 экв.) в 3,0 мл дихлорметана добавляли трибромид бора (1,1 мл 1,0 M раствора в дихлорметане, 1,1 ммоль, 2,9 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем реакционную смесь гасили метанолом, разбавляли в этилацетате и промывали насыщенным водным раствором бикарбоната натрия и рассолом, сушили над сульфатом магния и концентрировали. Полученный твердый материал растирали с 2 мл DMSO, и твердые вещества собирали и сушили в вакууме с получением 47,8 мг (34%) N-(3-(5-хлор-2-гидроксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=369,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 10,45 (с, 1H), 9,99 (с, 1H), 9,33 (д, 1H), 8,76 (д, 1H), 8,65 (с, 1H), 8,27 (с, 1H), 7,45 (с, 1H), 7,30 (м, 2H), 7,08 (д, 1H), 3,92 (с, 3H).
ПРИМЕР 3

N-(5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-амин
К раствору трет-бутил-5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамата (774 мг, 2,29 ммоль, 1 экв.) добавляли хлористоводородную кислоту (6,0 мл 4,0 M раствор в 1,4-диоксане, 24 ммоль, 10 экв.). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, а затем выпаривали до сухого состояния. Твердый осадок распределяли между дихлорметаном и насыщенным водным раствором бикарбоната натрия. Водную часть экстрагировали еще один раз дихлорметаном, и объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-амина, который использовали далее без очистки. LCMS (ESI) m+H=238,3; 1H ЯМР (400 МГц, CDCl3) δ: 7,35 (д, 1H), 7,22 (перекрывающийся с и с, 2H), 6,94 (д, 1H), 3,83 (с, 3H), 3,70 (д, 2H), 3,66 (с, 3H).

N-(5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-амина (0,100 г, 0,421 ммоль, 1 экв.), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (77,2 мг, 0,473 ммоль, 1,12 экв.), гексафторфосфата 7-азабензотриазол-1-илокси-трис-(пирролидинo)фосфония (269 мг, 0,519 ммоль, 1,23 экв.), N,N-диизопропилэтиламина (0,15 мл, 0,86 ммоль, 2,0 экв.) и 4-диметиламинопиридина (20,1 мг, 0,164 ммоль, 0,39 экв.) в 5,0 мл N,N-диметилформамиде перемешивали при 50°C в течение 2 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 82,2 мг (51%) N-(5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=383,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,51 (с, 1H), 9,31 (д, 1H), 8,67 (д, 1H), 8,63 (с, 1H), 7,98 (с, 1H), 7,58 (д, 1H), 7,54 (с, 1H), 7,31 (д, 1H), 7,28 (дд, 1H), 3,85 (с, 3H), 3,70 (с, 3H).
ПРИМЕР 4 И ПРИМЕР 5



ди-трет-бутил-2-(2,5-диметилфенил)-2-оксоэтилиминодикарбонат
В высушенной в сушильном шкафу колбе ди-трет-бутил-иминодикарбоксилат (2,566 г, 11,81 ммоль, 1,10 экв.) объединяли с гидридом натрия (60% на минеральном масле, 0,586 г, 14,6 ммоль, 1,37 экв.) и 30 мл N,N-диметилформамидом. Реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов, а затем добавляли 2-бром-1-(2,5-диметилфенил)этанон (2,432 г, 10,71 ммоль, 1 экв.). Реакционную смесь перемешивали при комнатной температуре в течение дополнительных 1,5 часов, а затем распределяли между этилацетатом и водой. Органическую часть промывали водой и рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-40% этилацетат в гептане) с получением 3,008 г (77%) ди-трет-бутил-2-(2,5-диметилфенил)-2-оксоэтилиминодикарбоната. LCMS (ESI) m+Na=386,2.
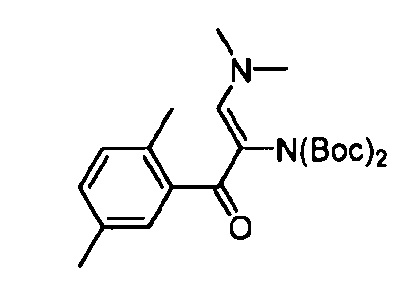
ди-трет-бутил-1-(диметиламино)-3-(2,5-диметилфенил)-3-оксопроп-1-ен-2-илиминодикарбонат
Перемешиваемую смесь ди-трет-бутил-2-(2,5-диметилфенил)-2-оксоэтилиминодикарбоната (3,008 г, 8,275 ммоль, 1 экв.) и 1,1-диметокси-N,N-диметилметанамина (6,0 мл, 45 ммоль, 5,4 экв.) нагревали при 70°C в течение 17 часов, а затем при 100°C в течение 24 часов. После выпаривания избытка 1,1-диметокси-N,N-диметилметанамина неочищенный продукт очищали флэш-хроматографией на силикагеле (0-50% этилацетат в гептане) с получением 1,305 г (38%) ди-трет-бутил-1-(диметиламино)-3-(2,5-диметилфенил)-3-оксопроп-1-ен-2-илиминодикарбоната. LCMS (ESI) m+H=419,3.

3-(2,5-диметилфенил)-1H-пиразол-4-амин
Ди-трет-бутил-1-(диметиламино)-3-(2,5-диметилфенил)-3-оксопроп-1-ен-2-илиминодикарбонат (1,305 г, 3,118 ммоль, 1 экв.) и гидразин (0,20 мл, 6,4 ммоль, 2,0 экв.) растворяли вместе в 15 мл этанола. Реакционную смесь перемешивали при 70°C в течение 1 часа, а затем упаривали до сухого состояния в вакууме. Твердый осадок растворяли в 8 мл дихлорметана и хлористоводородной кислоте (8,0 мл 4,0 M раствор в 1,4-диоксане, 0,10 моль, 40 экв.) и перемешивали при комнатной температуре в течение 3,5 часов. Растворитель и избыток хлористоводородной кислоты выпаривали, и неочищенный продукт распределяли между насыщенным водным раствором бикарбоната натрия и дихлорметаном. Водный слой экстрагировали еще один раз дихлорметаном, и объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 3-(2,5-диметилфенил)-1H-пиразол-4-амина, который использовали далее без очистки. LCMS (ESI) m+H=188,3.

N-(3-(2,5-диметилфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 3-(2,5-диметилфенил)-1H-пиразол-4-амина (0,300 г, 1,60 ммоль, 1 экв.), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (289,2 мг, 1,773 ммоль, 1,11 экв.), гексафторфосфата 7-азабензотриазол-1-илокси-трис-(пирролидинo)фосфония (1,087 г, 2,096 ммоль, 1,31 экв.), N,N-диизопропилэтиламина (1,0 мл, 5,7 ммоль, 3,6 экв.) и 4-диметиламинопиридина (42,3 мг, 0,346 ммоль, 0,22 экв.) в 8,0 мл N,N-диметилформамида перемешивали при 50°C в течение 3 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (40-100% этилацетат в дихлорметане) с получением 299,8 мг (56%) N-(3-(2,5-диметилфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=333,3; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,67 (с, 0,7H), 9,55 (с, 0,3H), 9,32 (д, 1H), 8,64 (с, 1H), 8,49 (д, 1H), 8,27 (с, 1H), 7,29 (д, 1H), 7,22 (м, 3H), 2,34 (с, 3H), 2,24 (с, 3H).

N-(3-(2,5-диметилфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

N-(5-(2,5-диметилфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(2,5-диметилфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (0,255 г, 0,767 ммоль, 1 экв.) в 10 мл N,N-диметилформамида добавляли йодметан (60,0 мкл, 0,964 ммоль, 1,26 экв.) и карбонат цезия (0,562 г, 1,72 ммоль, 2,25 экв.). Реакционную смесь перемешивали при 40°C в течение 2,5 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Смесь региоизомерных продуктов разделяли и очищали флэш-хроматографией на силикагеле (20-90% этилацетат в дихлорметане) с получением: 84,3 мг (32%) N-(3-(2,5-диметилфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида; LCMS (ESI) m+H=347,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,65 (с, 1H), 9,30 (д, 1H), 8,65 (с, 1H), 8,47 (д, 1H), 8,27 (с, 1H), 7,27 (д, 1H), 7,23-7,19 (м, 3H), 3,91 (с, 3H), 2,33 (с, 3H), 2,25 (с, 3H); и 81,8 мг (31%) N-(5-(2,5-диметилфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида; LCMS (ESI) m+H=347,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,42 (с, 1H), 9,30 (д, 1H), 8,61 (с, 1H), 8,43 (д, 1H), 8,01 (с, 1H), 7,36 (д, 1H), 7,30 (д, 1H) 7,20 (м, 2H), 3,62 (с, 3H), 2,35 (с, 3H), 2,12 (с, 3H).
ПРИМЕР 6
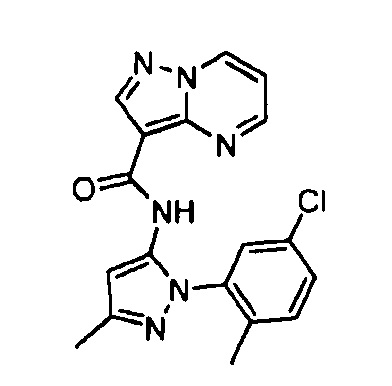
N-(1-(5-хлор-2-метилфенил)-3-метил-1H-пиразол-5-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

гидрохлорид (5-хлор-2-метилфенил)гидразина
К перемешиваемой суспензии 5-хлор-2-метиланилина (1,436 г, 10,14 ммоль, 1 экв.) в 10 мл воды при 0°C добавляли 10 мл концентрированной хлористоводородной кислоты. К этой реакционной смеси капельно добавляли раствор нитрита натрия (0,791 г, 11,5 ммоль, 1,13 экв.) в 5 мл воды. Реакционную смесь перемешивали при 0°C в течение 2 часов при одновременном медленном добавлении дигидрата хлорида олова (5,8826 г, 25,839 ммоль, 2,55 экв.), растворенного в 8 мл концентрированной хлористоводородной кислоты. При необходимости добавляли воду для обеспечения перемешивания во время образования твердых веществ. Реакционную смесь поддерживали при 0°C в течение 45 минут. Белые твердые вещества фильтровали и промывали двумя порциями по 50 мл диэтилового эфира, затем сушили в вакууме с получением 1,49 г (76%) гидрохлорида (5-хлор-2-метилфенил)гидразина. 1H ЯМР (400 МГц, DMSO-d6) δ: 10,08 (с, 3H), 7,98 (с, 1H), 7,13 (д, 1H), 6,97 (с, 1H), 6,91 (д, 1H), 2,14 (с, 3H).

1-(5-хлор-2-метилфенил)-3-метил-1H-пиразол-5-амин
К раствору гидрохлорида (5-хлор-2-метилфенил)гидразина (1,49 г, 7,72 ммоль, 1 экв.) в 8 мл этанола добавляли хлористоводородную кислоту (4,0 мл 5 M водного раствора, 20 ммоль, 2 экв.) и 3-аминокротонитрил (0,664 г, 8,09 ммоль, 1,05 экв.). Реакционную смесь перемешивали при 80°C в течение 16 часов, а затем доводили до нейтрального значения pH насыщенным водным раствором бикарбоната натрия. Полученный раствор дважды экстрагировали дихлорметаном, и объединенные экстракты сушили над сульфатом магния и концентрировали с получением 1-(5-хлор-2-метилфенил)-3-метил-1H-пиразол-5-амина, который использовали далее без дальнейшей очистки. LCMS (ESI) m+H=222,2; 1H ЯМР (400 МГц, DMSO-d6) δ: 7,39 (д, 1H), 7,37 (д, 1H), 7,25 (с, 1H), 5,22 (с, 1H), 5,00 (с, 2H), 2,04 (перекрывающийся с и с, 6H).

N-(1-(5-хлор-2-метилфенил)-3-метил-1H-пиразол-5-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 1-(5-хлор-2-метилфенил)-3-метил-1H-пиразол-5-амина (95,3 мг, 0,430 ммоль, 1 экв.), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (79,5 мг, 0,487 ммоль, 1,13 экв.), гексафторфосфатаа 7-азабензотриазол-1-илокси-трис-(пирролидинo)фосфония (0,290 г, 0,558 ммоль, 1,30 экв.), N,N-диизопропилэтиламина (0,2 мл, 1,1 ммоль, 2,7 экв.) и 4-диметиламинопиридина (11,0 мг, 0,09 ммоль, 0,21 экв.) в 4,0 мл N,N-диметилформамида перемешивали при 75°C в течение 3 суток. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния, фильтровали через слой силикагеля и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 50,0 мг (30%) N-(1-(5-хлор-2-метилфенил)-3-метил-1H-пиразол-5-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=367,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 10,03 (с, 1H), 9,32 (д, 1H), 8,68 (с, 1H), 8,36 (д, 1H), 7,60 (д, 1H), 7,58 (с, 1H), 7,55 (д, 1H), 7,27 (дд, 1H), 6,53 (с, 1H), 2,24 (с, 3H), 2,05 (с, 3H).
ПРИМЕР 7
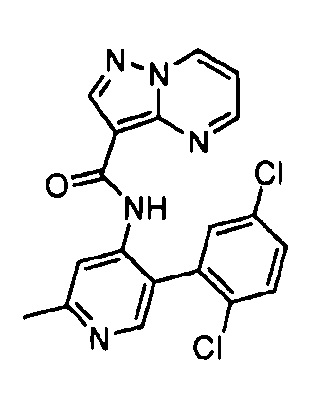
N-(5-(2,5-дихлорфенил)-2-метилпиридин-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
1-оксид 5-бром-2-метилпиридина
Раствор 5-бром-2-метилпиридина (2,0 г, 12 ммоль, 1,0 экв.) и м-хлорпероксибензойной кислоты (70%, 2,89 г, 12,9 ммоль, 1,1 экв.) в 30 мл хлороформа перемешивали при комнатной температуре в течение 4 часов. Затем реакционную смесь распределяли между дихлорметаном и 2 M водным раствором карбоната натрия. Водный слой экстрагировали еще один раз дихлорметаном, и объединенные органические части сушили над сульфатом магния и концентрировали с получением 1-оксида 5-бром-2-метилпиридина. LCMS (ESI) m+H=189,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 8,57 (с, 1H), 7,50 (д, 1H), 7,45 (д, 1H), 2,30 (с, 3H).

1-оксид 5-бром-2-метил-4-нитропиридина
1-Оксид 5-бром-2-метилпиридина (2,269 г, 12,07 ммоль, 1 экв.) растворяли в серной кислоте (4 мл, 80 ммоль, 6 экв.) и охлаждали при 0°C. Капельно добавляли дымящую азотную кислоту (3 мл, 60 ммоль, 5 экв.). После завершения добавления азотной кислоты, реакционную смесь сначала нагревали до комнатной температуры, а затем нагревали до 90°C. Через 2 часа реакционную смесь охлаждали на ледяной бане и медленно доводили до pH=10 2 M водным раствором карбоната натрия. Этот раствор экстрагировали два раза дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 2,54 г (90%) 1-оксида 5-бром-2-метил-4-нитропиридина. LMCS (ESI) m+H=233,0.

5-бром-2-метилпиридин-4-амин
К раствору 1-оксида 5-бром-2-метил-4-нитропиридина (2,54 г, 10,9 ммоль, 1 экв.) в 10 мл концентрированной хлористоводородной кислоты добавляли дигидрат хлорида олова (9,96 г, 43,8 ммоль, 4,01 экв.). Реакционную смесь перемешивали при 90°C в течение 24 часов, а затем добавляли дополнительный дигидрат хлорида олова (3,15 г, 13,8 ммоль, 1,27 экв.) и 5 мл концентрированной хлористоводородной кислоты. Реакционную смесь поддерживали при 90°C в течение дополнительных 24 часов, а затем охлаждали до комнатной температуры и доводили до нейтральных значений pH 2 M водным раствором карбоната натрия. Раствор экстрагировали три раза дихлорметаном, и объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 5-бром-2-метилпиридин-4-амина. LCMS (ESI) m+H=187,2; 1H ЯМР (400 МГц, DMSO-d6) δ: 8,07 (с, 1H), 6,51 (с, 1H), 6,13 (с, 2H), 2,22 (с, 3H).

N-(5-бром-2-метилпиридин-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К суспензии пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (165,0 мг, 1,011 ммоль, 1,2 экв.) в 6 мл дихлорметана добавляли оксалилхлорид (0,90 мл 2,0 M раствора в дихлорметане, 1,8 ммоль, 2,1 экв.) и три капли N,N-диметилформамида. После перемешивания при комнатной температуре в течение 1 часа, реакционную смесь концентрировали и сушили в вакууме с получением ацилхлорида. Твердый осадок перерастворяли в 6 мл дихлорметана, и к этому раствору добавляли 5-бром-2-метилпиридин-4-амин (157,6 мг, 0,8426 ммоль, 1,0 экв.) и триэтиламин (0,50 мл, 3,6 ммоль, 4,2 экв.). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, а затем концентрировали на диоксиде кремния. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-100% этилацетат в дихлорметане) с получением 197,5 мг (71%) N-(5-бром-2-метилпиридин-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=332,2.
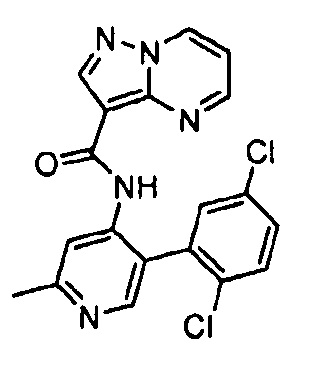
N-(5-(2,5-дихлорфенил)-2-метилпиридин-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
N-(5-Бром-2-метилпиридин-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (82,2 мг, 0,247 ммоль, 1 экв.), 2,5-дихлорфенилбороновую кислоту (122,1 мг, 0,6399 ммоль, 2,59 экв.) и хлорид бис(трифенилфосфин)палладия(II) (15,4 мг, 0,0219 ммоль, 0,09 экв.) объединяли в емкости для обработки микроволновым излучением. К этим твердым веществам добавляли карбонат натрия (1,0 мл 1,0 M водного раствора, 1,0 ммоль, 4,0 экв.) и 3,0 мл ацетонитрила. Емкость закрывали и подвергали воздействию микроволнового излучения (120°C, 30 Вт) в течение 30 минут. Реакционную смесь распределяли между этилацетатом и водой, и органический слой сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 45,3 мг (46%) N-(5-(2,5-дихлорфенил)-2-метилпиридин-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=398,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,75 (с, 1H), 9,32 (д, 1H), 8,71 (с, 1H), 8,48 (с, 1H), 8,26 (с, 1H), 8,21 (д, 1H), 7,78 (д, 1H), 7,75 (д, 1H), 7,69 (с, 1H), 7,26 (дд, 1H), 2,55 (с, 3H).
ПРИМЕР 8

N-(3-(3-хлорфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

2-(2-(3-хлорфенил)-2-оксоэтил)изоиндолин-1,3-дион
Смесь 2-бром-3'-хлорацетофенона (0,927 г, 3,97 ммоль, 1 экв.) и фталимида калия (0,813 г, 4,39 ммоль, 1,1 экв.) в 15 мл N,N-диметилформамида перемешивали при 50°C в течение одного часа. Растворитель удаляли в вакууме. Полученные твердые вещества растирали с этилацетатом и фильтровали. Собранные твердые вещества сушили в вакууме с получением 2-(2-(3-хлорфенил)-2-оксоэтил)изоиндолин-1,3-диона, который использовали далее без очистки. 1H ЯМР (400 МГц, DMSO-d6) δ: 8,13 (с, 1H), 8,05 (д, 1H), 7,96 (м, 2H), 7,93 (м, 2H), 7,83 (д, 1H), 7,65 (т, 1H), 5,29 (с, 2H).

2-(3-(3-хлорфенил)-1-(диметиламино)-3-оксопроп-1-ен-2-ил)изоиндолин-1,3-дион
Перемешиваемую смесь 2-(2-(3-хлорфенил)-2-оксоэтил)изоиндолин-1,3-диона (782,2 мг, 2,610 ммоль, 1 экв.) и 1,1-диметокси-N,N-диметилметанамина (1,5 мл, 11 ммоль, 4,3 экв.) нагревали при 100°C в течение 18 часов. Избыток 1,1-диметокси-N,N-диметилметанамина удаляли в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (50-100% этилацетат в гептане) с получением 740 мг (80%) 2-(3-(3-хлорфенил)-1-(диметиламино)-3-оксопроп-1-ен-2-ил)изоиндолин-1,3-диона. LCMS (ESI) m+H=355,2; 1H ЯМР (400 МГц, CDCl3) δ: 7,90 (дд, 2H), 7,77 (дд, 2H), 7,57 (с, 1H), 7,44 (д, 1H), 7,37 (перекрывающийся д и с, 2H), 7,31 (т, 1H), 3,00 (с, 6H).
3-(3-хлорфенил)-1-метил-1H-пиразол-4-амин
К раствору 2-(3-(3-хлорфенил)-1-(диметиламино)-3-оксопроп-1-ен-2-ил)изоиндолин-1,3-диона (2,30 г, 6,48 ммоль, 1 экв.) в 50 мл этанола добавляли N-метилгидразин (1,4 мл, 26 ммоль, 4,0 экв.). Реакционную смесь перемешивали при 80°C в течение 2 часов, а затем концентрировали на силикагеле. Неочищенную смесь региоизомеров разделяли и очищали флэш-хроматографией на силикагеле (0-80% этилацетат в дихлорметане) с получением: 715 мг (53%) 3-(3-хлорфенил)-1-метил-1H-пиразол-4-амина; LCMS (ESI) m+H=208,2; 1H ЯМР (400 МГц, CDCl3) δ: 7,78 (с, 1H), 7,63 (д, 1H), 7,33 (т, 1H), 7,25 (перекрывающийся CDCl3, 1H), 7,04 (с, 1H), 3,84 (с, 3H); и 274,6 мг (20%) 5-(3-хлорфенил)-1-метил-1H-пиразол-4-амина; LCMS (ESI) m+H=208,2; 1H ЯМР (400 МГц, CDCl3) δ: 7,43 (т, 1H), 7,38 (перекрывающийся д и с, 2H), 7,27 (д, 1H), 7,23 (с, 1H), 3,76 (с, 3H).

N-(3-(3-хлорфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 3-(2,5-диметилфенил)-1H-пиразол-4-амина (400,0 мг, 1,926 ммоль, 1 экв.), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (342,9 мг, 2,102 ммоль, 1,09 экв.), гексафторфосфата 7-азабензотриазол-1-илокси-трис-(пирролидинo)фосфония (1,205 г, 2,324 ммоль, 1,21 экв.), N,N-диизопропилэтиламина (0,80 мл, 4,6 ммоль, 2,4 экв.) и 4-диметиламинопиридина (43,4 мг, 0,355 ммоль, 0,18 экв.) в 15,0 мл N,N-диметилформамида перемешивали при 50°C в течение 15 часов. Реакционную смесь распределяли между дихлорметаном и водой, и водный слой экстрагировали еще один раз дихлорметаном. Объединенные органические части сушили над сульфатом магния и концентрировали на силикагеле. Неочищенный продукт разделяли флэш-хроматографией на силикагеле (0-70% этилацетат (содержащий 2% метанол) в дихлорметане). Полученный твердый материал растирали с этилацетатом. После обработки ультразвуком твердые вещества собирали фильтрацией и сушили в вакууме с получением 0,502 г (74%) N-(3-(3-хлорфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=353,0; 1H ЯМР (400 МГц, CDCl3) δ: 10,00 (с, 1H), 9,38 (д, 1H), 8,85 (д, 1H), 8,70 (с, 1H), 8,33 (с, 1H), 7,84 (с, 1H), 7,76 (д, 1H), 7,57 (т, 1H), 7,49 (д, 1H), 7,34 (дд, 1H), 3,93 (с, 3H).
ПРИМЕР 9

N-(3-(3-хлорфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

3-(3-хлорфенил)-1H-пиразол-4-амин
Смесь 2-(3-(3-хлорфенил)-1-(диметиламино)-3-оксопроп-1-ен-2-ил)изоиндолин-1,3-диона (255 мг, 0,719 ммоль, 1 экв.) и гидразина (0,15 мл, 4,8 ммоль, 6,6 экв.) в 10 мл этанола перемешивали при кипячении с обратным холодильником в течение 2 часов. Этанол и избыток гидразина удаляли в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (50-100% этилацетат в гептане) с получением 111,4 мг (80%) 3-(3-хлорфенил)-1H-пиразол-4-амина. LCMS (ESI) m+H=194,0; 1H ЯМР (400 МГц, CD3OD) δ: 7,75 (уш., 1H), 7,65 (уш., 1H), 7,41 (т, 1H), 7,32 и 7,30 (перекрывающийся с и с, 2H).

N-(3-(3-хлорфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 3-(3-хлорфенил)-1H-пиразол-4-амина (95,3 мг, 0,492 ммоль, 1 экв.), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (90,6 мг, 0,555 ммоль, 1,13 экв.), гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (247,3 мг, 0,6504 ммоль, 1,32 экв.), N,N-диизопропилэтиламина (0,20 мл, 1,1 ммоль, 2,3 экв.) и 4-диметиламинопиридина (24,1 мг, 0,197 ммоль, 0,40 экв.) в 5,0 мл N,N-диметилформамида перемешивали при 50°C в течение 3 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 33,8 мг (20%) N-(3-(3-хлорфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=339,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 13,05 (с, 1H), 9,99 (с, 1H), 9,38 (д, 1H), 8,84 (д, 1H), 8,70 (с, 1H), 8,31 (с, 1H), 7,86 (с, 1H), 7,78 (д, 1H), 7,58 (т, 1H), 7,50 (д, 1H), 7,34 (дд, 1H).
ПРИМЕР 10

N-(3-(3-хлорфенил)-1-изопропил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
N-(3-(3-Хлорфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (68,9 мг, 0,203 ммоль, 1 экв.) растворяли в 4 мл N,N-диметилформамида. К этому раствору добавляли карбонат цезия (148 мг, 0,454 ммоль, 2,23 экв.) и изопропилйодид (23,0 мкл, 0,230 ммоль, 1,13 экв.). Реакционную смесь перемешивали при 50°C в течение 2 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 47,2 мг (61%) N-(3-(3-хлорфенил)-1-изопропил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=381,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,99 (с, 1H), 9,37 (д, 1H), 8,85 (д, 1H), 8,70 (с, 1H), 8,36 (с, 1H), 7,85 (с, 1H), 7,77 (д, 1H), 7,57 (т, 1H), 7,48 (д, 1H), 7,34 (дд, 1H), 4,59 (дкв., 1H), 1,48 (д, 6H).
ПРИМЕР 11

N-(3-(3-хлорфенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
N-(3-(3-Хлорфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (58,9 мг, 0,174 ммоль, 1 экв.) растворяли в 5 мл N,N-диметилформамида. К этому раствору добавляли изобутиленоксид (0,5 мл, 6 ммоль, 30 экв.) и карбонат цезия (56,4 мг, 0,173 ммоль, 1,0 экв.). Реакционную смесь перемешивали при 50°C в течение 7,5 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 33,1 мг (46%) N-(3-(3-хлорфенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=411,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 10,02 (с, 1H), 9,36 (д, 1H), 8,83 (д, 1H), 8,69 (с, 1H), 8,35 (с, 1H), 7,84 (с, 1H), 7,76 (д, 1H), 7,58 (т, 1H), 7,49 (д, 1H), 7,33 (дд, 1H), 4,73 (с, 1H), 4,09 (с, 2H), 1,13 (с, 6H).
ПРИМЕР 12
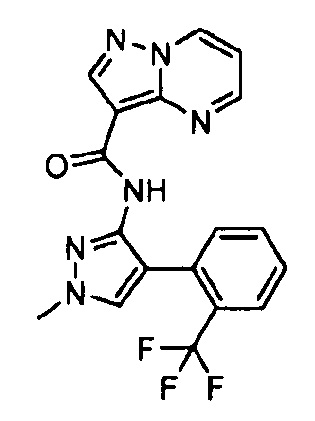
N-(1-метил-4-(2-(трифторметил)фенил)-1H-пиразол-3-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

N-(4-бром-1-метил-1H-пиразол-3-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Раствор пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (165,5 мг, 1,014 ммоль, 1,01 экв.), 4-бром-1-метил-1H-пиразол-3-амина (177,4 мг, 1,008 ммоль, 1,0 экв.), гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (497 мг, 1,31 ммоль, 1,30 экв.), N,N-диизопропилэтиламина (0,25 мл, 1,4 ммоль, 1,4 экв.) и 4-диметиламинопиридина (33,8 мг, 0,277 ммоль, 0,27 экв.) в 5 мл N,N-диметилформамида нагревали при 50°C в течение 4 суток. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (20-80% этилацетат в дихлорметане (содержащий 5% метанол)) с получением 203,6 мг (63%) N-(4-бром-1-метил-1H-пиразол-3-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=321,1; 1H ЯМР (400 МГц, CD3OD) δ: 9,16 (д, 1H), 8,84 (д, 1H), 8,68 (с, 1H), 7,76 (с, 1H), 7,28 (дд, 1H), 3,88 (с, 3H).

N-(1-метил-4-(2-(трифторметил)фенил)-1H-пиразол-3-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
В колбу для обработки микроволновым излучением, оборудованную мешалкой, добавляли: N-(4-бром-1-метил-1H-пиразол-3-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (101 мг, 0,313 ммоль, 1 экв.), 2-трифторметилфенилбороновую кислоту (126,6 мг, 0,6666 ммоль, 2,13 экв.), хлорид бис(трифенилфосфин)палладия(II) (26,8 мг, 0,0382 ммоль, 0,12 экв.), карбонат натрия (1,0 мл 2,0 M водного раствора, 2 ммоль, 6 экв.) и 3 мл ацетонитрила. Реакционную смесь подвергали воздействию микроволнового излучения при 130°C в течение 30 минут. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 4,7 мг (4%) N-(1-метил-4-(2-(трифторметил)фенил)-1H-пиразол-3-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=387,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,58 (с, 1H), 9,32 (д, 1H), 8,74 (д, 1H), 8,57 (с, 1H), 7,78 (с, 1H), 7,74 (д, 1H), 7,60 (т, 1H), 7,49 (м, 2H), 7,28 (дд, 1H), 3,87 (с, 3H).
ПРИМЕР 13

N-(1-(2,5-дифторфенил)-1H-пиразол-5-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

3-(2-(2,5-дифторфенил)гидразинил)пропаннитрил
Раствор акрилoнитрила (10 мл), этанола (20 мл) и 2,5-дифторфенилгидразина (790 мг) нагревали до температуры кипячения с обратным холодильником в течение 2 суток. Реакционную смесь концентрировали в вакууме, и продукт очищали флэш-хроматографией на силикагеле с метиленхлоридом с получением 790 мг (73%) 3-(2-(2,5-дифторфенил)гидразинил)пропаннитрила. LCMS (ESI) m+H=198,2; 1H ЯМР (400 МГц, CDCl3) δ 6,99 (ддд, 1H), 6,89 (ддд, 1H), 6,38 (тт, 1H), 5,45 (с, 1H), 3,91 (с, 1H), 3,19 (тд, 2H), 2,61 (т, 2H).

(E)-3-((2,5-дифторфенил)диазенил)пропаннитрил
К раствору 3-(2-(2,5-дифторфенил)гидразинил)пропаннитрила (790 мг) в 15 мл 2 Н серной кислоты добавляли 2,0 г гидрата сульфата железа(III). Реакционную смесь перемешивали при температуре окружающей среды в течение 30 мин по мере того, как сульфат железа медленно растворялся и выпадало в осадок желтое масло. Реакционную смесь экстрагировали простым эфиром, и экстракты промывали водой, рассолом, сушили над сульфатом натрия, фильтровали через слой силикагеля и концентрировали с получением 710 мг (91%) (E)-3-((2,5-дифторфенил)диазенил)пропаннитрила. LCMS (ESI) m+H=196,2. 1H ЯМР (400 МГц, CDCl3) δ 7,34-7,08 (м, 3H), 4,54-4,31 (м, 2H), 2,99 (т, 2H).
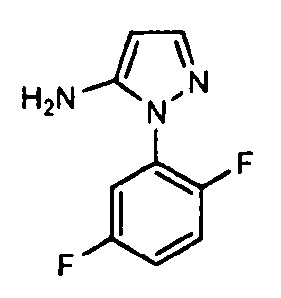
1-(2,5-дифторфенил)-1H-пиразол-5-амин
Смесь (E)-3-((2,5-дифторфенил)диазенил)пропаннитрила (710 мг) и 20 мл 1 Н NaOH нагревали до температуры кипячения с обратным холодильником при перемешивании в течение 30 мин. Реакционную смесь охлаждали до температуры окружающей среды и экстрагировали простым эфиром. Фазу простого эфира промывали водой, рассолом, сушили над сульфатом натрия и фильтровали через слой силикагеля. Концентрирование в вакууме дало 630 мг (88%) 1-(2,5-дифторфенил)-1H-пиразол-5-амина. LCMS (ESI) m+H=196,0; 1H ЯМР (400 МГц, CDCl3) δ 7,54-7,43 (м, 1H), 7,35-7,00 (м, 3H), 5,63 (д, 1H), 3,96-3,71 (м, 2H).

N-(1-(2,5-дифторфенил)-1H-пиразол-5-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Пиразолo[1,5-a]пиримидин-3-карбоновую кислоту (0,020 г, 0,12 ммоль), 3 мл фосфорилхлорида и 32 мкл N,N-диизопропилэтиламина нагревали до 120°C в течение 2 ч и упаривали до сухого состояния. Осадок отбирали в дихлорметан и добавляли к смеси 1-(2,5-дифторфенил)-1H-пиразол-5-амина (18 мг), N,N-диизопропилэтиламина (32 мкл) и дихлорметана (1 мл) и перемешивали в течение ночи. Реакционную смесь концентрировали, и продукт очищали флэш-хроматографией на силикагеле (95/5 дихлорметан/7 M NH3 в MeOH) с получением 8,0 мг (20%) N-(1-(2,5-дифторфенил)-1H-пиразол-5-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=341,2; 1H ЯМР (500 МГц, CDCl3) δ 10,14 (с, 1H), 8,80 (дд, 1H), 8,72 (д, 1H), 8,38 (дд, 1H), 7,72 (т, 1H), 7,38 (ддд, 1H), 7,33-7,27 (м, 1H), 7,22 (ддд, 1H), 7,03-6,99 (м, 1H), 6,88 (д, 1H).


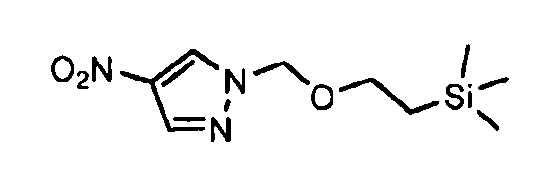
4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол
В высушенной в сушильном шкафу колбе, оборудованной мешалкой, 4-нитро-1H-пиразол (6,598 г, 58,35 ммоль) растворяли в 50 мл THF. Добавляли гидрид натрия (4,83 г 60% дисперсии с минеральным маслом, 121 ммоль) при охлаждении на ледяной бане, и затем реакционную смесь перемешивали при комнатной температуре в течение 10 минут. Затем добавляли [β-(триметилсилил)этокси]метилхлорид (12,0 мл, 67,8 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение 1,5 часов. Реакционную смесь гасили 50 мл воды и экстрагировали этилацетатом. Органический экстракт сушили над сульфатом магния и концентрировали. Полученный неочищенный материал очищали флэш-хроматографией на силикагеле (0-30% этилацетат в гептане) с получением 14,1 г (99%) 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола. LCMS (ESI) m+H=244,2; 1H ЯМР (400 МГц, CDCl3) δ: 8,28 (с, 1H), 8,08 (с, 1H), 5,43 (с, 2H), 3,61 (т, 2H), 0,92 (т, 2H).
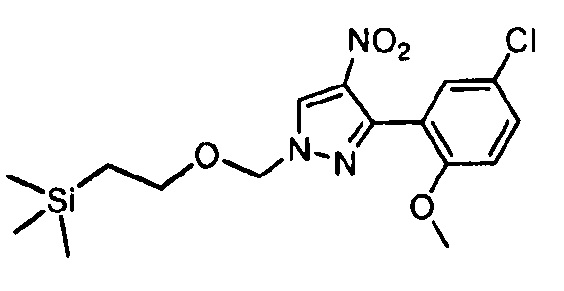
3-(5-хлор-2-метоксифенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол
К раствору 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (4,26 г, 17,5 ммоль) в 40 мл N,N-диметилацетамида добавляли 2-бром-4-хлоранизол (3,35 мл, 24,6 ммоль), ацетат палладия(II) (197,2 мг, 0,878 ммоль), ди(1-адамантил)-н-бутилфосфин (469,5 мг, 1,309 ммоль), карбонат калия (7,27 г, 52,6 ммоль) и триметилуксусную кислоту (0,452 г, 4,43 ммоль). При перемешивании при комнатной температуре через реакционную смесь барботировали газообразный азот в течение 10 минут, а затем реакционную смесь нагревали при 120°C в течение 6 часов. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и промывали водой и рассолом, сушили над сульфатом магния и концентрировали. Неочищенный материал очищали флэш-хроматографией на силикагеле (0-25% этилацетат в гептане) с получением 6,719 г (89%) 3-(5-хлор-2-метоксифенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола. LCMS (ESI) m+H=384,2; 1H ЯМР (400 МГц, CDCl3) δ: 8,22 (с, 1H), 7,46 (дд, 1H), 7,36 (д, 1H), 6,95 (д, 1H), 5,23 (м, 2H), 3,56 (м, 2H), 0,87 (м, 2H).

3-(5-хлор-2-метоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-амин
К раствору 3-(5-хлор-2-метоксифенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (5,97 г, 15,6 ммоль) в 25 мл этанола добавляли 50 мл воды, хлорид аммония (3,37 г, 62,9 ммоль) и железный порошок (4,367 г, 78,2 ммоль). Реакционную смесь перемешивали при 75°C в течение 1,5 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном и фильтровали через слой целита, промывая дополнительным дихлорметаном. Фильтрат добавляли к 150 мл насыщенного водного раствора бикарбоната натрия и экстрагировали два раза дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 5,50 г (100%) 3-(5-хлор-2-метоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-амина, который использовали далее без очистки. LCMS (ESI) m+H=354,3; 1H ЯМР (400 МГц, CDCl3) δ: 7,44 (д, 1H), 7,34 (дд, 1H), 7,28 (с, 1H), 6,92 (д, 1H), 5,24 (с, 2H), 3,52 (т, 2H), 0,85 (т, 2H), -0,04 (с, 9H).

N-(5-(5-хлор-2-метоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 3-(5-хлор-2-метоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-амина (179,6 мг, 0,5075 ммоль), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (91 мг, 0,560 ммоль), гексафторфосфата 7-азабензотриазол-1-илокси-трис-(пирролидинo)фосфония (326 мг, 0,630 ммоль), N,N-диизопропилэтиламина (0,25 мл, 1,4 ммоль) и 4-диметиламинопиридина (11,5 мг, 0,094 ммоль) в 5,0 мл N,N-диметилформамида перемешивали при 40°C в течение 1,5 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния, фильтровали через слой силикагеля и концентрировали с получением 0,212 г (84%) N-(5-(5-хлор-2-метоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида, который использовали далее без очистки. LCMS (ESI) m+H=499,2; 1H ЯМР (400 МГц, CDCl3) δ: 9,56 (с, 1H), 8,76 (д, J=7,0, 1H), 8,72 (с, 1H), 8,50 (д, J=4,1, 1H), 8,39 (с, 1H), 7,54 (д, J=2,6, 1H), 7,44 (дд, J=8,8, 2,6, 1H), 7,04-6,92 (м, 2H), 5,35 (д, 2H), 3,82 (с, 3H), 3,57 (м, 2H), 0,86 (м, 2H), -0,04 (с, 9H).

N-(5-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(5-хлор-2-метоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (2,75 г, 5,51 ммоль) в 105 мл этанола добавляли HCl (8,0 мл 5 M раствора в воде, 40 ммоль). Затем реакционную смесь перемешивали при 70°C в течение 2 часов. После охлаждения до комнатной температуры продукт фильтровали в виде светло-желтого твердого вещества, промывая метанолом и диэтиловым эфиром. Объем фильтрата уменьшали и фильтровали дополнительный твердый продукт. Объединенные собранные твердые вещества сушили в вакууме с получением 1,81 г (89%) N-(5-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=369,2; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,66 (с, 1H), 9,33 (дд, J=7,0, 1,6, 1H), 8,78 (дд, J=4,2, 1,6, 1H), 8,65 (с, 1H), 8,20 (с, 1H), 7,50 (дд, J=8,8, 2,7, 1H), 7,44 (д, J=2,7, 1H), 7,29 (дд, J=7,8, 4,8, 2H), 3,86 (с, 3H).

N-(3-(5-хлор-2-метоксифенил)-1-(2-морфолиноэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

N-(5-(5-хлор-2-метоксифенил)-1-(2-морфолиноэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (39,1 мг, 0,106 ммоль) в 3 мл DMF добавляли карбонат цезия (109,7 мг, 0,3367 ммоль) и 4-(2-хлорэтил)морфолин HCl. Реакционную смесь перемешивали при 50°C в течение 5 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Смесь региоизомерных продуктов разделяли и очищали обращенно-фазовой ВЭЖХ, и лиофилизировали с получением 19,4 мг (38%) N-(3-(5-хлор-2-метоксифенил)-1-(2-морфолиноэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида; LCMS (ESI) m+H=482,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,50 (с, 1H), 9,31 (дд, 1H), 8,66 (д, 1H), 8,63 (с, 1H), 8,03 (с, 1H), 7,58 (дд, 1H), 7,54 (д, 1H), 7,32 (д, 1H), 7,27 (дд, 1H), 4,05 (м, 2H), 3,85 (с, 3H), 3,44 (м, 2H), 2,71 (м, 2H), 2,60 (м, 2H), 2,23 (м, 4H); и 9,3 мг (18%) N-(5-(5-хлор-2-метоксифенил)-1-(2-морфолиноэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида; LCMS (ESI) m+H=482,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,67 (с, 1H), 9,31 (дд, 1H), 8,76 (д, 1H), 8,65 (с, 1H), 8,34 (с, 1H), 7,48 (дд, 1H), 7,39 (д, 1H), 7,30 (д, 1H), 7,38 (т, 1H), 4,29 (т, 2H), 3,84 (с, 3H), 3,58 (т, 2H), 2,77 (т, 2H), 2,46 (м, 4H).
ПРИМЕР 16

N-(3-(5-хлор-2-метоксифенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида
К раствору N-(5-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (102,7 мг, 0,279 ммоль) в 3 мл DMF добавляли изобутиленоксид (0,20 мл, 2,2 ммоль) и карбонат цезия (180,0 мг, 0,5524 ммоль). Реакционную смесь перемешивали при 40°C в течение 15 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 49,3 мг (40%) N-(3-(5-хлор-2-метоксифенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=441,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,68 (с, 1H), 9,32 (дд, 1H), 8,76 (д, 1H), 8,64 (с, 1H), 8,30 (с, 1H), 7,48 (дд, 1H), 7,39 (д, 1H), 7,31 (с, 1H), 7,28 (т, 1H), 4,72 (с, 1H), 4,07 (с, 2H), 3,84 (с, 3H), 1,12 (с, 6H).
ПРИМЕР 17

N-(3-(5-хлор-2-этоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

N-(3-(5-хлор-2-гидроксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (синтезированному способами, описанными для примера 14) (193,9 мг, 0,5065 ммоль) в 8,0 мл дихлорметана добавляли трибромид бора (1,50 мл 1,0 M раствора в дихлорметане, 1,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь гасили 1 мл метанола, разбавляли этилацетатом и промывали насыщенным водным раствором бикарбоната натрия и рассолом, сушили над сульфатом магния и концентрировали с получением 191,5 мг (100%) N-(3-(5-хлор-2-гидроксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида, который использовали далее без очистки. LCMS (ESI) m+H=369,1.

N-(3-(5-хлор-2-этоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(5-хлор-2-гидроксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (61,1 мг, 0,166 ммоль) в 3,0 мл ацетона добавляли йодэтан (26,0 мкл, 0,325 ммоль) и карбонат калия (70,6 мг, 0,511 ммоль). Реакционную смесь перемешивали при 45°C в течение 3 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 34,8 мг (54%) N-(3-(5-хлор-2-этоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=397,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,68 (с, 1H), 9,33 (дд, J=7,0, 1,6, 1H), 8,73 (дд, J=4,2, 1,6, 1H), 8,66 (с, 1H), 8,28 (с, 1H), 7,46 (дд, J=8,8, 2,7, 1H), 7,38 (д, J=2,7, 1H), 7,32-7,23 (м, 2H), 4,12 (кв., J=6,9, 2H), 3,91 (с, 3H), 1,04 (т, J=6,9, 3H).
ПРИМЕР 18

N-(3-(5-хлор-2-(дифторметокси)фенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

2-бром-4-хлор-1-(дифторметокси)бензол
К раствору 2-бром-4-хлорфенола (4,98 г, 24,0 ммоль) в 25 мл DMF добавляли хлордифторацетат натрия (8,42 г, 55,2 ммоль), карбонат цезия (10,97 г, 33,67 ммоль) и 2,5 мл воды. Реакционную смесь перемешивали при 100°C в течение 16 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-20% этилацетат в гептане) с получением 2,98 г (48%) 2-бром-4-хлор-1-(дифторметокси)бензола в виде прозрачного бесцветного масла. LCMS (ESI): нет сигнала m/z; 1H ЯМР (400 МГц, DMSO-d6) δ: 7,90 (д, 1H), 7,54 (дд, 1H), 7,38 (д, 1H), 7,28 (т, 1H).

N-(3-(5-хлор-2-(дифторметокси)фенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
С использованием 2-бром-4-хлор-1-(дифторметокси)бензола указанное в заголовке соединение синтезировали способами синтеза, описанными для примера 14. LCMS (ESI) m+H=419,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,71 (с, 1H), 9,34 (дд, J=7,0, 1,6, 1H), 8,67 (дд, J=4,7, 2,0, 1H), 8,66 (с, 1H), 8,30 (с, 1H), 7,62 (дд, J=8,8, 2,7, 1H), 7,59 (д, J=2,6, 1H), 7,45 (д, J=8,7, 1H), 7,29 (дд, J=7,0, 4,2, 1H), 7,23 (т, 1H), 3,93 (с, 3H).
ПРИМЕР 19

N-(3-(5-хлор-2-(метилтио)фенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

(4-хлор-2-йодфенил)(метил)сульфан
К раствору при 0°C концентрированной серной кислоты (0,3 мл, 5,0 ммоль) в 5,0 мл воды и 5,0 мл ацетонитрила добавляли 5-хлор-2-(метилтио)анилин (472 мг, 2,72 ммоль), а затем медленно добавляли нитрит натрия (210 мг, 3,0 ммоль) в виде раствора в 1 мл воды. Реакционную смесь перемешивали при 0°C в течение 30 минут. Затем эту смесь медленно добавляли к раствору при 0°C йодида калия (691,7 мг, 4,167 ммоль) в 5 мл воды. Реакционную смесь перемешивали в течение 1 часа, позволяя ледяной бане нагреться до комнатной температуры. Затем реакционную смесь распределяли между водой и этилацетатом. Органический слой сушили рассолом и сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-30% этилацетат в дихлорметане) с получением 598,4 мг (77%) (4-хлор-2-йодфенил)(метил)сульфана. LCMS (ESI): нет сигнала m/z; 1H ЯМР (400 МГц, DMSO-d6) δ: 7,87 (д, 1H), 7,47 (дд, 1H), 7,20 (д, 1H), 2,47 (с, 3H).

N-(3-(5-хлор-2-(метилтио)фенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
С использованием (4-хлор-2-йодфенил)(метил)сульфана указанное в заголовке соединение получали способами синтеза, описанными для примера 14. LCMS (ESI) m+H=399,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,69 (с, 1H), 9,32 (дд, J=7,0, 1,6, 1H), 8,65 (с, 1H), 8,57 (дд, J=4,2, 1,6, 1H), 8,27 (с, 1H), 7,55 (дд, J=8,6, 2,4, 1H), 7,45 (д, J=8,6, 1H), 7,40 (д, J=2,4, 1H), 7,25 (дд, J=7,0, 4,2, 1H), 3,91 (с, 3H), 2,38 (с, 3H).
ПРИМЕР 20

N-(3-(5-хлор-2-этоксифенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
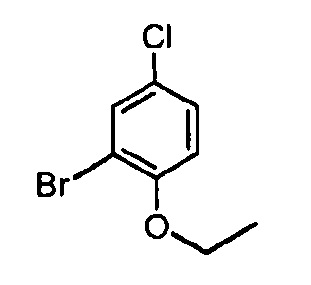
2-бром-4-хлор-1-этоксибензол
К раствору 2-бром-4-хлорфенола (2,12 г, 10,2 ммоль) в 25 мл ацетона добавляли йодэтан (0,850 мл, 10,6 ммоль) и карбонат цезия (4,08 г, 12,5 ммоль). Реакционную смесь перемешивали при 50°C в течение 2 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали с получением 2,37 г (98%) 2-бром-4-хлор-1-этоксибензола в виде желтого масла, которое использовали без дальнейшей очистки. LCMS (ESI): нет сигнала m/z; 1H ЯМР (400 МГц, DMSO-d6) δ: 7,67 (д, J=2,6, 1H), 7,39 (дд, J=8,8, 2,6, 1H), 7,12 (д, J=8,9, 1H), 4,11 (кв., J=7,0, 2H), 1,35 (т, J=7,0, 3H).

N-(3-(5-хлор-2-этоксифенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
С использованием 2-бром-4-хлор-1-этоксибензола указанное в заголовке соединение получали с использованием способов синтеза, описанных для примеров 14 и 16. LCMS (ESI) m+H=455,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,71 (с, 1H), 9,33 (дд, J=7,0, 1,6, 1H), 8,73 (дд, J=4,2, 1,6, 1H), 8,66 (с, 1H), 8,33 (с, 1H), 7,47 (дд, J=8,8, 2,7, 1H), 7,38 (д, J=2,7, 1H), 7,28 (м, 2H), 4,73 (с, 1H), 4,12 (кв., J=6,9, 2H), 4,07 (с, 2H), 1,13 (с, 6H), 1,03 (т, J=6,9, 3H).
ПРИМЕР 21

N-(1-(2-аминоэтил)-3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(5-хлор-2-метоксифенил)-1-(2-(1,3-диоксоизоиндолин-2-ил)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (полученного способами синтеза, описанными для примера 14) (104,1 мг, 0,192 ммоль) в 8 мл этанола добавляли гидразин (78 мкл, 2,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, а затем концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 17,7 мг (15,4%) N-(1-(2-аминоэтил)-3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=412,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,68 (с, 1H), 9,33 (дд, J=7,0, 1,6, 1H), 8,77 (дд, J=4,2, 1,6, 1H), 8,65 (с, 1H), 8,30 (с, 1H), 7,49 (дд, J=8,8, 2,7, 1H), 7,42 (д, J=2,7, 1H), 7,29 (м, 2H), 4,14 (т, J=6,2, 2H), 3,85 (с, 3H), 2,99 (т, J=6,2, 2H).


Рацемический N-(3-(5-хлор-2-метоксифенил)-1-(2-гидрокси-3-метилбутил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (полученный, как описано для примера 16) подвергали хиральной хроматографии SFC с получением указанных в заголовке соединений. LCMS (ESI) m+H=455,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,67 (с, 1H), 9,33 (дд, J=7,0, 1,6, 1H), 8,77 (дд, J=4,2, 1,6, 1H), 8,65 (с, 1H), 8,30 (с, 1H), 7,49 (дд, J=8,8, 2,8, 1H), 7,40 (д, J=2,7, 1H), 7,34-7,24 (м, 2H), 4,86 (д, J=5,8, 1H), 4,19 (дд, J=13,8, 3,7, 1H), 4,04 (дд, J=13,8, 8,1, 1H), 3,84 (с, 3H), 3,64 (м, 1H), 1,63 (м, 1H), 0,93 (т, J=7,2, 6H).
ПРИМЕР 24

N-(3-(5-хлор-2-метоксифенил)-1-((1-гидроксициклопентил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

1-(йодметил)циклопентанол
В 100-мл колбу помещали порошковый самарий (номер 40, 3,07 г, 2,02 ммоль) и эту колбу охлаждали в атмосфере азота на ледяной бане. В капельную воронку помещали раствор циклопентанона (0,90 мл, 10,1 ммоль) и дийодметан (2,40 мл, 29,8 ммоль) в 50 мл тетрагидрофурана, и этот раствор капельно добавляли к перемешиваемому порошковому самарию в течение 1 часа. После завершения добавления реакционную смесь перемешивали в течение одного дополнительного часа при 0°C. реакционную смесь обрабатывали 40 мл 1 Н водного раствора HCl и экстрагировали 100 мл диэтилового эфира. Слой простого эфира промывали 4% водным раствором Na2S2O3 и рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-30% этилацетат в гептане) с получением 1,17 г (51%) 1-(йодметил)циклопентанола в виде желтого масла. 1H ЯМР (400 МГц, CDCl3) δ: 3,47 (с, 2H), 1,70-1,92 (м, 8H).

N-(3-(5-хлор-2-метоксифенил)-1-((1-гидроксициклопентил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (полученного, как описано для примера 14, 115,4 мг, 0,313 ммоль) в 3 мл DMF добавляли 1-(йодметил)циклопентанол (195,4 мг, 2,762 ммоль) и карбонат цезия (339,2 мг, 3,327 ммоль). Реакционную смесь перемешивали в закрытой емкости 140°C в течение 18 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт отделяли от другого региоизомерного продукта и очищали обращенно-фазовой ВЭЖХ, и лиофилизировали с получением 32,1 мг (22%) N-(3-(5-хлор-2-метоксифенил)-1-((1-гидроксициклопентил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=467,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,68 (с, 1H), 9,33 (дд, J=7,0, 1,6, 1H), 8,77 (дд, J=4,2, 1,6, 1H), 8,64 (с, 1H), 8,33 (с, 1H), 7,49 (дд, J=8,9, 2,7, 1H), 7,39 (д, J=2,7, 1H), 7,32-7,25 (м, 2H), 4,67 (с, 1H), 4,19 (с, 2H), 3,84 (с, 3H), 1,61 (м, 8H).
ПРИМЕР 25

N-(3-(3-хлорфенил)-5-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

1-(3-хлорфенил)бутан-1,3-дион
К раствору 3-хлорацетофенона (1,30 мл, 10,0 ммоль) в 20 мл THF добавляли трет-бутоксид калия (11,0 мл 1,0 M раствора в THF, 11,0 ммоль), а затем безводный этилацетат (1,05 мл, 10,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем при 50°C в течение 15 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-50% этилацетат в гептане) с получением 0,75 г (38%) 1-(3-хлорфенил)бутан-1,3-диона. LCMS (ESI) m+H=197,2.

1-(3-хлорфенил)-2-(гидроксиимино)бутан-1,3-дион
К раствору 1-(3-хлорфенил)бутан-1,3-диона (0,75 г, 3,8 ммоль) при 0°C в 15 мл уксусной кислоты медленно добавляли нитрит натрия (0,560 г, 8,11 ммоль) в виде раствора в 1,5 мл воды. Реакционную смесь перемешивали при 0°C в течение 30 минут, а затем нагревали до комнатной температуры. После дополнительных 4 часов реакционную смесь выливали в насыщенный водный раствор бикарбоната натрия и экстрагировали три раза дихлорметаном. Объединенные экстракты сушили над сульфатом магния и концентрировали с получением 765,7 мг (89%) 1-(3-хлорфенил)-2-(гидроксиимино)бутан-1,3-диона, который использовали далее без дальнейшей очистки. LCMS (ESI) m+H=226; 1H ЯМР (400 МГц, DMSO-d6) δ: 7,81 (д, 1H), 7,80 (с, 1H), 7,75 (д, 1H), 7,62 (т, 1H), 2,60 (с, 1H), 2,48 (с, 3H).

5-(3-хлорфенил)-3-метил-1H-пиразол-4-амин
К раствору 1-(3-хлорфенил)-2-(гидроксиимино)бутан-1,3-диона (224,8 мг, 0,996 ммоль) в 5 мл этанола при 0°C капельно добавляли гидразин (0,30 мл, 9,6 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 15 часов. Неочищенную реакционную смесь концентрировали и очищали флэш-хроматографией на силикагеле (20-100% этилацетат в дихлорметане) с получением 113,3 мг (55%) 5-(3-хлорфенил)-3-метил-1H-пиразол-4-амина. LCMS (ESI) m+H=208,2; 1H ЯМР (400 МГц, DMSO-d6) δ: 12,20 (с, 1H), 7,94-7,57 (м, 2H), 7,40 (с, 1H), 7,27 (д, J=9,1, 1H), 2,12 (с, 3H).

N-(3-(3-хлорфенил)-5-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 5-(3-хлорфенил)-3-метил-1H-пиразол-4-амина (113,3 мг, 0,546 ммоль), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (109,5 мг, 0,6712 ммоль), гексафторфосфата 7-азабензотриазол-1-илокси-трис-(пирролидинo)фосфония (338,2 мг, 0,652 ммоль), N,N-диизопропилэтиламина (0,30 мл, 1,7 ммоль) и 4-диметиламинопиридина (19,1 мг, 0,156 ммоль) в 8,0 мл N,N-диметилформамида перемешивали при 50°C в течение 15 часов. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 86,9 мг (45%) N-(3-(3-хлорфенил)-5-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=353,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 12,90 (с, 1H), 9,37 (д, J=7,0, 1H), 9,30 (с, 1H), 8,86 (дд, J=4,1, 1,3, 1H), 8,69 (с, 1H), 7,80 (д, J=12,1, 1H), 7,72 (д, J=7,6, 1H), 7,37 (т, J=7,8, 1H), 7,32 (дд, J=6,9, 4,4, 2H), 2,18 (с, 3H).
ПРИМЕР 26
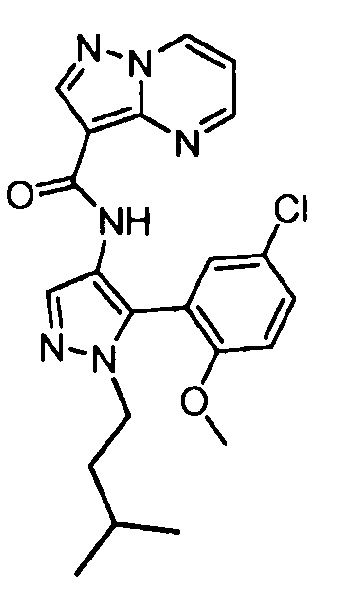
N-(5-(5-хлор-2-метоксифенил)-1-изопентил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

1-изопентил-4-нитро-1H-пиразол
Смесь 4-нитро-1H-пиразола (234 мг, 2,06 ммоль, 1,0 экв.), 1-бром-3-метилбутана (0,30 мл, 2,48 ммоль, 1,2 экв.) и карбоната цезия (1,01 г, 3,10 ммоль, 1,5 экв.) в 5,0 мл 1,2-диметоксиэтана перемешивали при 55°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли 25 мл этилацетата и фильтровали. Затем фильтрат концентрировали, и осадок растворяли в 5 мл дихлорметана и очищали колоночной флэш-хроматографией (диоксид кремния, 0-80% этилацетат в гептане в течение 30 минут) с получением 357,6 мг (94,32%) изопентил-4-нитро-1H-пиразола в виде белого твердого вещества. LCMS (ESI) m+H=184,1; 1H ЯМР (400 МГц, CDCl3) δ 8,11 (с, 1H), 8,06 (с, 1H), 4,20-4,14 (м, 2H), 1,80 (дд, J=14,8, 7,1, 2H), 1,60 (dp, J=13,4, 6,7, 1H), 0,97 (д, J=6,6, 6H).
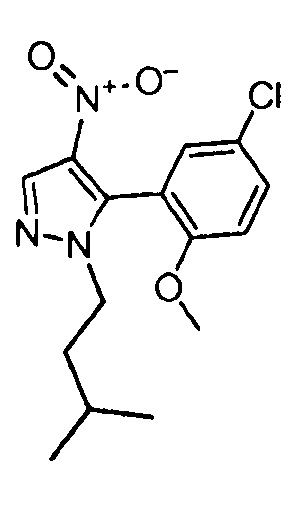
N-(5-(5-хлор-2-метоксифенил)-1-изопентил-4-нитро-1H-пиразол
Смесь 1-изопентил-4-нитро-1H-пиразола (356,9 мг, 1,95 ммоль, 1,00 экв.), 2-бром-4-хлоранизола (0,37 мл, 2,72 ммоль, 1,40 экв.), ацетата палладия(II) (88 мг, 0,39 ммоль, 0,20 экв.), ди(1-адамантил)-н-бутилфосфина (209 мг, 0,58 ммоль, 0,30 экв.), карбоната калия (807 мг, 5,84 ммоль, 3,00 экв.) и триметилуксусной кислоты (52 мг, 0,50 ммоль, 0,26 экв.) в 5,0 мл N,N-диметилацетамида перемешивали при 120°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры и разбавляли 20 мл этилацетата и фильтровали. Затем фильтрат концентрировали и использовали как есть для следующей стадии. LCMS (ESI) m+H=324,3.
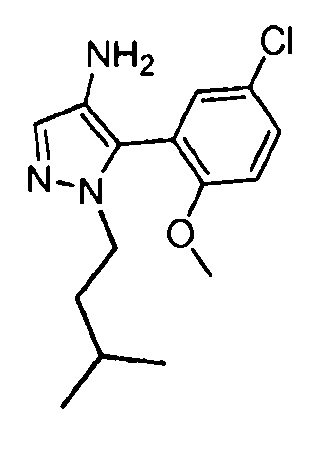
N-(5-(5-хлор-2-метоксифенил)-1-изопентил-1H-пиразол-4-амин
Смесь 5-(5-хлор-2-метоксифенил)-1-изопентил-4-нитро-1H-пиразола (632 мг, 1,95 ммоль, 1,00 экв.), железа (642,5 мг, 11,50 ммоль, 5,90 экв.) и хлорида аммония (500,7 мг, 9,36 ммоль, 4,80 экв.) в 5,0 мл этанола и 10 мл воды перемешивали при 75°C в течение 2 часов. Реакционную смесь концентрировали, добавляли 10 мл насыщенного раствора бикарбоната, и водный слой экстрагировали дихлорметаном (20 мл×3). Объединенные слои дихлорметана сушили сульфатом магния, фильтровали и концентрировали. Красновато-желтое масло использовали как есть для следующей стадии. LCMS (ESI) m+H=293,8.

N-(5-(5-хлор-2-метоксифенил)-1-изопентил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Смесь 5-(5-хлор-2-метоксифенил)-1-изопентил-1H-пиразол-4-амина (573,0 мг, 1,95 ммоль, 1,00 экв.), пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (349,9 мг, 2,145 ммоль, 1,10 экв.), гексафторфосфата 7-азабензотриазол-1-илокси-трис-(пирролидинo)фосфония (1,25 г, 2,42 ммоль, 1,24 экв.) и N,N-диизопропилэтиламина (0,95 мл, 5,5 ммоль, 2,8 экв.) в 5,0 мл N,N-диметилформамида перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 105,2 мг (12,3%) N-(5-(5-хлор-2-метоксифенил)-1-изопентил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=439,1; 1H ЯМР (400 МГц, DMSO) δ: 9,50 (с, 1H), 9,32 (дд, J=7,0, 1,6, 1H), 8,73-8,57 (м, 2H), 8,01 (с, 1H), 7,60 (дд, J=8,9, 2,7, 1H), 7,48 (д, J=2,7, 1H), 7,33 (д, J=9,0, 1H), 7,27 (дд, J=7,0, 4,2, 1H), 3,97 (дкв., J=16,6, 6,8, 2H), 3,84 (с, 3H), 1,54 (кв., J=7,1, 2H), 1,45-1,31 (м, 1H), 0,75 (дд, J=12,0, 6,6, 6H).
ПРИМЕР 27

2-амино-N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

(Z)-этил 3-амино-4,4,4-трихлор-2-цианобут-2-еноат
К смеси трихлорацетонитрила (38 мл, 0,38 моль) и этилового эфира цианоуксусной кислоты (20 мл, 0,2 моль) в этаноле (63 мл) добавляли триэтиламин (1 мл, 7 ммоль). Реакционная смесь начинала краснеть и через ~1 минуту достигала экзотермического эффекта. Реакционную смесь охлаждали до 0°C, затем перемешивали в течение двух часов при одновременном медленном нагревании до комнатной температуры. Реакционную смесь концентрировали в вакууме до красного масла, которое отбирали в DCM, фильтровали через слой силикагеля и концентрировали в вакууме с получением 44,95 г (90%) (Z)-этил 3-амино-4,4,4-трихлор-2-цианобут-2-еноата в виде бесцветного масла, которое медленно застывало до белого твердого вещества. 1H ЯМР (400 МГц, CDCl3) δ 10,19 (с, 1H), 6,85 (с, 1H), 4,31 (кв., J=7,1, 1H), 1,37 (т, J=7,1, 3H).
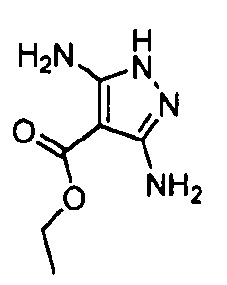
этил-3,5-диамино-1H-пиразол-4-карбоксилат
К (Z)-этил 3-амино-4,4,4-трихлор-2-цианобут-2-еноату (15,0 г, 58 ммоль) в DMF (50 мл) добавляли гидразин (2,19 мл, 70 ммоль). Реакционную смесь нагревали до 100°C в течение 1 ч, а затем охлаждали до комнатной температуры. DMF удаляли в вакууме, затем осадок суспендировали в смеси 95:5 DCM:2 M раствор аммиака в метаноле. Полученный осадок отфильтровывали, промывали смесью 95:5 DCM:MeOH и сушили в вакууме с получением 5,72 г (58%) этил-3,5-диамино-1H-пиразол-4-карбоксилата в виде белого твердого вещества. 1H ЯМР (400 МГц, DMSO) δ 10,53 (с, 1H), 5,28 (уш., 4H), 4,14 (кв., J=7,1, 2H), 1,33-1,15 (т, J=7,1, 3H).

этил-2-аминопиразолo[1,5-a]пиримидин-3-карбоксилат
Смесь этил-3,5-диамино-1H-пиразол-4-карбоксилата (1,0 г, 5,9 ммоль), 1,1,3,3-тетраметоксипропана (2,9 мл, 18 ммоль), триэтиламина (2 мл, 10 ммоль) и DMF (15 мл) нагревали при 100°C в течение 14 ч, затем добавляли дополнительные 2 мл 1,1,3,3-тетраметоксипропана. После добавления дополнительного 1,1,3,3-тетраметоксипропана отмечали существенное количество побочного продукта и нагревание немедленно прекращали. Реакционную смесь охлаждали до комнатной температуры и DMF удаляли в вакууме. Осадок распределяли между DCM и водой, затем органический слой концентрировали и осадок очищали хроматографией на диоксиде кремния, элюируя смесью 95:5 DCM: 2 M раствор аммиака в метаноле с получением 420 мг (35%) этил 2-аминопиразолo[1,5-a]пиримидин-3-карбоксилата. 1H ЯМР (500 МГц, CDCl3) δ 8,57 (дд, J=4,3, 1,6, 1H), 8,43 (дд, J=6,7, 1,6, 1H), 6,84 (дд, J=6,7, 4,4, 1H), 5,52 (с, 2H), 4,48 (кв., J=7,1, 2H), 1,45 (т, J=7,1, 3H).

этил-2-(бис(трет-бутоксикарбонил)амино)пиразолo[1,5-a]пиримидин-3-карбоксилат
К раствору этил-2-аминопиразолo[1,5-a]пиримидин-3-карбоксилата (810 мг, 3,9 ммоль), 4-диметиламинопиридина (96 мг, 0,78 ммоль) и N,N-диизопропилэтиламина (1,4 мл, 7,8 ммоль) в ацетонитриле (100 мл) добавляли ди-трет-бутилдикарбонат (1,30 г, 5,9 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, а затем концентрировали в вакууме. Осадок распределяли между EtOAc и водой, затем слои разделяли, и органический слой промывали рассолом, затем сушили над Na2SO4, фильтровали и концентрировали. Осадок очищали хроматографией с диоксидом кремния, элюируя смесью 97:3 DCM: 2 M раствор аммиака в метаноле с получением 370 мг (31%) этил-2-(бис(трет-бутоксикарбонил)амино)пиразолo[1,5-a]пиримидин-3-карбоксилата. 1H ЯМР (500 МГц, CDCl3) δ 8,79 (дд, J=4,2, 1,8, 1H), 8,69 (дд, J=7,0, 1,8, 1H), 7,05 (дд, J=7,0, 4,2, 1H), 4,40 (кв., J=7,1, 2H), 1,43 (с, 18H), 1,38 (т, J=7,1, 3H).
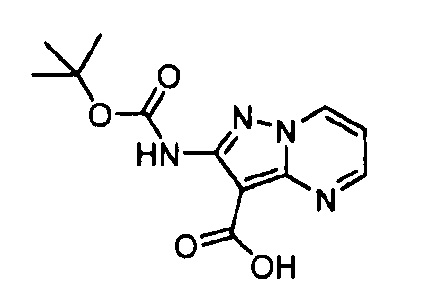
2-(трет-бутоксикарбониламино)пиразолo[1,5-a]пиримидин-3-карбоновая кислота
К раствору 2-(бис(трет-бутоксикарбонил)амино)пиразолo[1,5-a]пиримидин-3-карбоксилата (220 мг, 0,54 ммоль) в этаноле (15 мл) добавляли 4 мл 10% водного раствора гидроксида лития. Реакционную смесь нагревали до 70°C в течение 18 ч, затем охлаждали до комнатной температуры. Добавляли 15 мл 10% водного раствора лимонной кислоты и реакционную смесь концентрировали в вакууме. Осадок распределяли между EtOAc и насыщенным водным раствором лимонной кислоты, затем органический слой промывали водой и рассолом, затем сушили над Na2SO4, фильтровали и концентрировали в вакууме с получением 150 мг 2-(трет-бутоксикарбониламино)пиразолo[1,5-a]пиримидин-3-карбоновой кислоты. 1H ЯМР (400 МГц, DMSO) δ 9,34 (с, 1H), 9,12 (дд, J=6,9, 1,7, 1H), 8,71 (дд, J=4,3, 1,7, 1H), 7,20 (дд, J=6,9, 4,3, 1H), 1,49 (с, 9H).

трет-бутил-3-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамоил)пиразолo[1,5-a]пиримидин-2-илкарбамат
К раствору 2-(трет-бутоксикарбониламино)пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (88 мг, 0,32 ммоль), 5-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-амина (75 мг, 0,32 ммоль), 4-диметиламинопиридина (7,7 мг, 0,063 ммоль) и N,N-диизопропилэтиламина (0,16 мл, 0,95 ммоль) в DMF (3 мл) добавляли PyAOP (200 мг, 0,38 ммоль). Реакционную смесь перемешивали в течение 14 ч при 50°C, а затем разбавляли EtOAc. Органический слой промывали два раза водой и один раз рассолом, затем сушили над Na2SO4, фильтровали и концентрировали. Осадок очищали хроматографией на силикагеле, элюируя EtOAc, с получением 98 мг (62%) трет-бутил-3-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамоил)пиразолo[1,5-a]пиримидин-2-илкарбамата. 1H ЯМР (400 МГц, CDCl3) δ 9,85 (с, 1H), 9,65 (с, 1H), 8,76 (дд, J=6,8, 1,7, 1H), 8,44 (дд, J=4,3, 1,7, 1H), 8,20 (с, 1H), 7,56 (д, J=2,7, 1H), 7,36 (дд, J=8,8, 2,7, 1H), 6,97 (д, J=8,9, 1H), 6,90 (дд, J=6,9, 4,3, 1H), 3,97 (с, 3H), 3,87 (с, 3H), 1,56 (с, 9H).
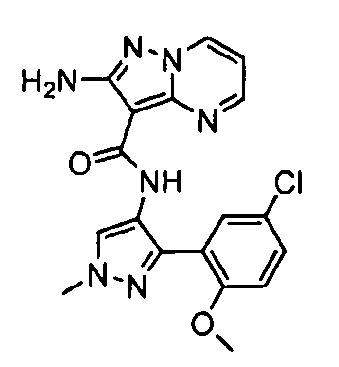
2-амино-N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору трет-бутил-3-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-илкарбамоил)пиразолo[1,5-a]пиримидин-2-илкарбамата (80 мг, 0,2 ммоль) в DCM (10 мл) добавляли TFA (0,5 мл). Реакционную смесь перемешивали в течение 3 ч при комнатной температуре, а затем концентрировали в вакууме. Осадок очищали хроматографией на силикагеле с получением 55 мг (90%) 2-амино-N-(3-(5-хлор-2-метоксифенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида в виде белого твердого вещества. LCMS (ESI) m+H=398,1; 1H ЯМР (400 МГц, DMSO) δ 9,46 (с, 1H), 8,91 (дд, J=6,7, 1,6, 1H), 8,44 (дд, J=4,5, 1,6, 1H), 8,21 (с, 1H), 7,47 (дд, J=8,9, 2,7, 1H), 7,37 (д, J=2,7, 1H), 7,27 (д, J=8,9, 1H), 6,99 (дд, J=6,7, 4,5, 1H), 6,56 (с, 2H), 3,89 (с, 3H), 3,83 (с, 3H).
Соединения примеров 28-131, представленные в таблице 1, получали согласно описанным выше примерам. Для каждого соединения, представленного в таблице 1, в столбце "способ" приведен номер примера, которому следовали.
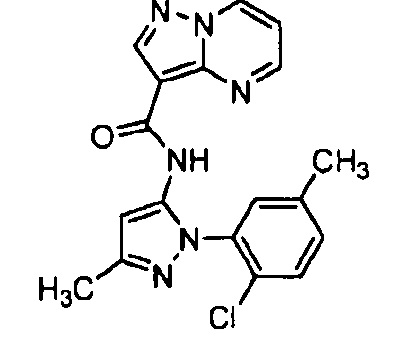

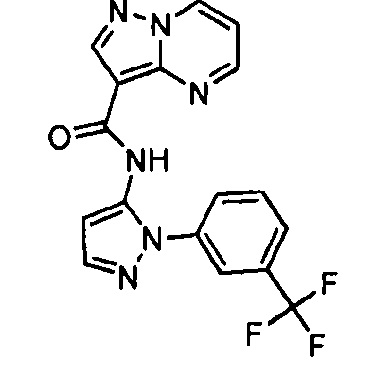

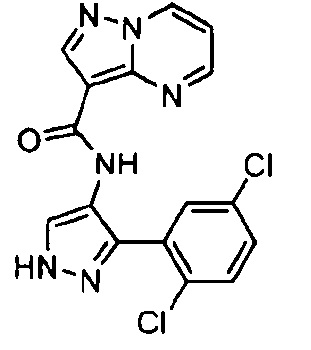
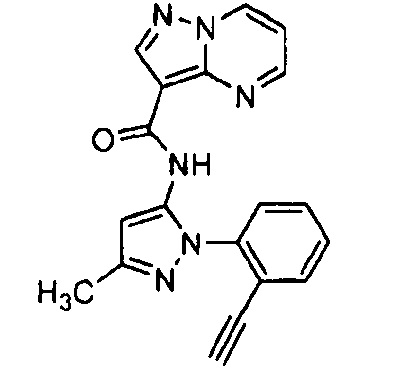
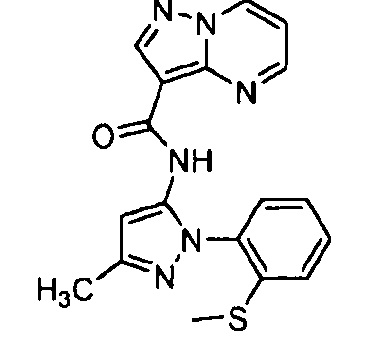


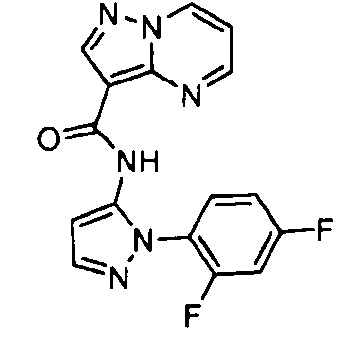





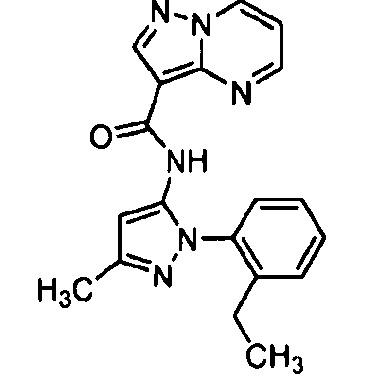










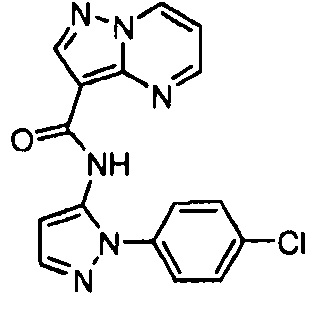




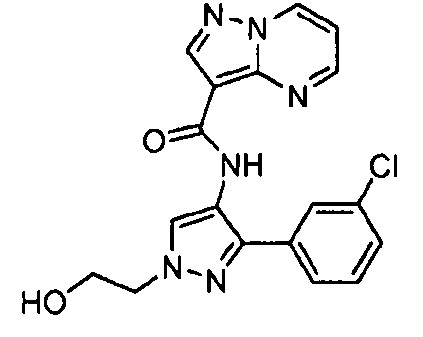


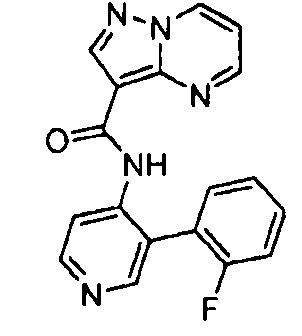

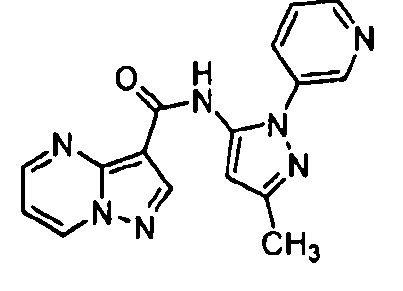





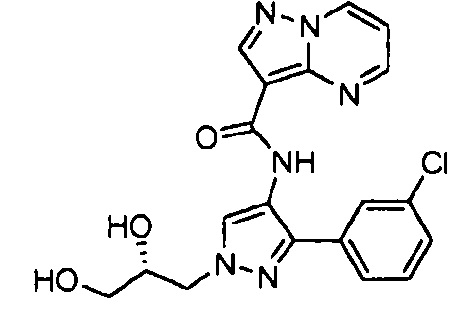


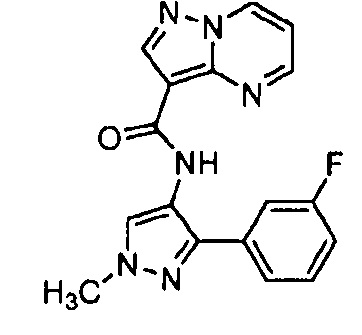



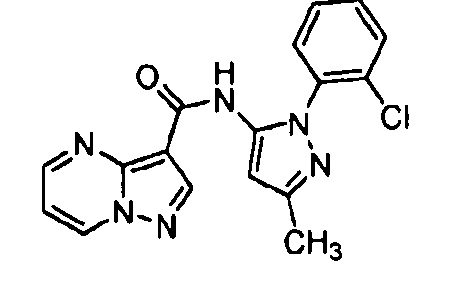



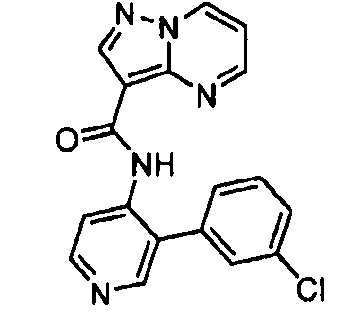










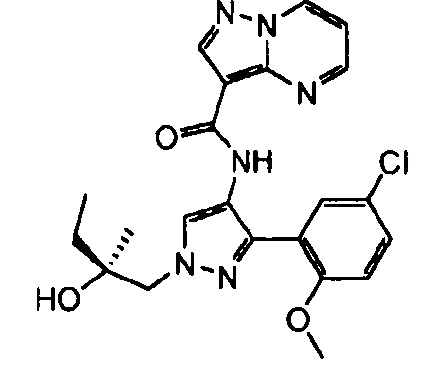



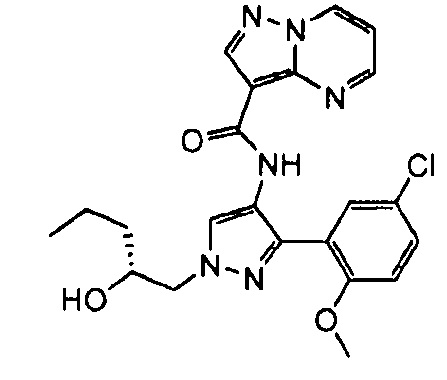
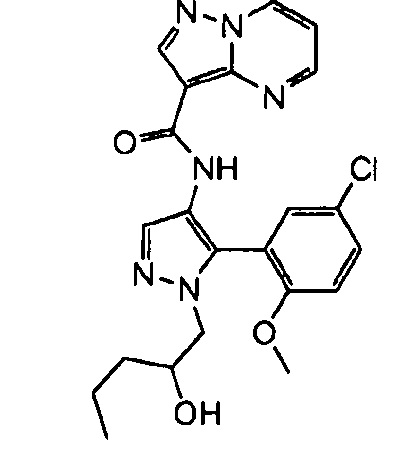




и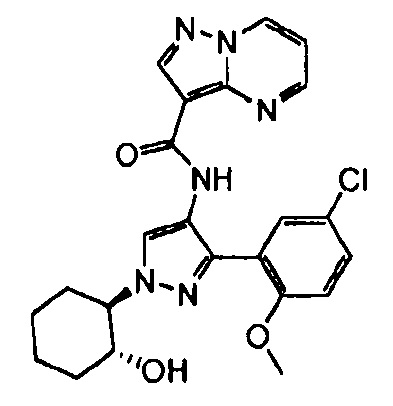
и
N-(3-(5-хлор-2-метоксифенил)-1-((1R,2R)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиридимин-3-карбоксамид




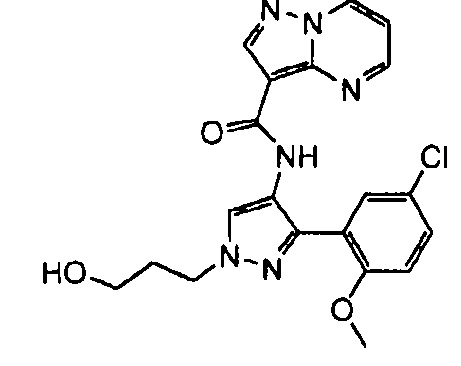










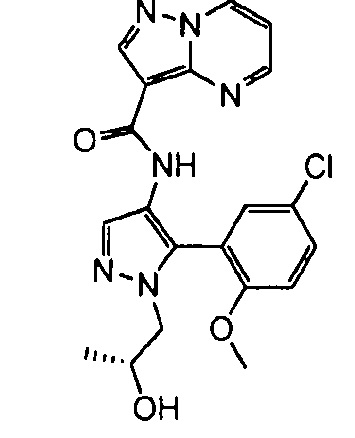






18


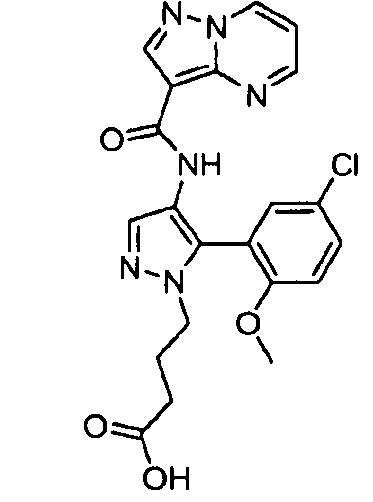
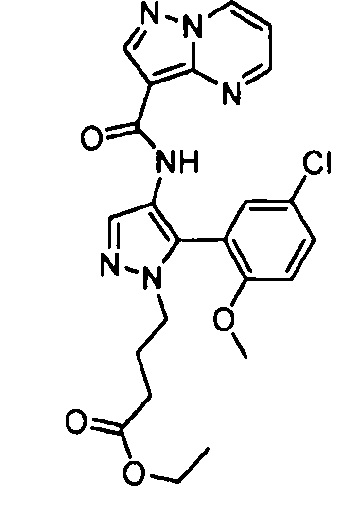


ПРИМЕР 132

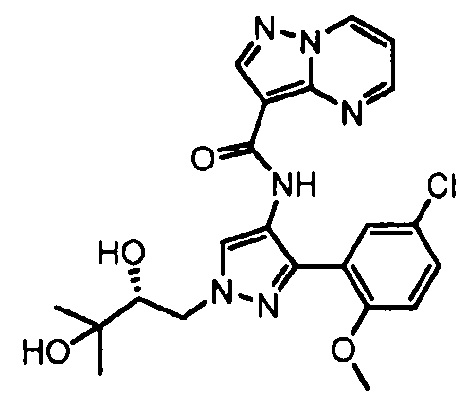
К суспензии N-(5-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (114,0 мг, 0,31 ммоль) и карбоната цезия (201,4 мг, 0,62 ммоль) в N,N-диметилформамиде (2 мл) добавляли 4-бром-2-метил-2-бутен (53 мкл, 0,46 ммоль). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре, затем добавляли EtOAc. Органический слой промывали 1x каждый раз водой и насыщенным рассолом. Органический слой отделяли, затем сушили над Na2SO4 и концентрировали в вакууме. Осадок очищали хроматографией на силикагеле (20-100% EtOAc:Hex) с получением N-(3-(5-хлор-2-метоксифенил)-1-(3-метилбут-2-енил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида в виде светло-желтой пены (67 мг, 0,15 ммоль). Затем добавляли ацетон и воду (по 1 мл каждого), а затем тетраоксид осмия (1,89 мг, 0,0074 ммоль) и N-метилморфолин-N-оксид в воде (1:1, N-метилморфолин-N-оксид:вода, 37 мг). Реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Добавляли целит и смесь концентрировали в вакууме. Осадок очищали хроматографией на силикагеле (50-100 % EtOAc*:Hex, *EtOAc также содержал 10% MeOH), а затем хиральной SFC для разделения энантиомеров с получением 11,2 мг и 15,8 мг указанных в заголовке соединений в виде белых твердых веществ. LCMS (ESI) m+H=471,1; 1H ЯМР (500 МГц, DMSO) δ 9,69 (с, 1H), 9,34 (д, J=6,9, 1H), 8,78 (д, J=4,0, 1H), 8,66 (с, 1H), 8,30 (с, 1H), 7,49 (дд, J=8,8, 2,6, 1H), 7,42 (д, J=2,6, 1H), 7,29 (дд, J=10,0, 4,6, 2H), 5,05 (д, J=6,3, 1H), 4,53 (с, 1H), 4,43 (д, J=13,5, 1H), 3,95 (дд, J=13,5, 10,1, 1H), 3,85 (с, 3H), 3,60 (с, 1H), 1,16 (с, 3H), 1,10 (с, 3H).
ПРИМЕР 133
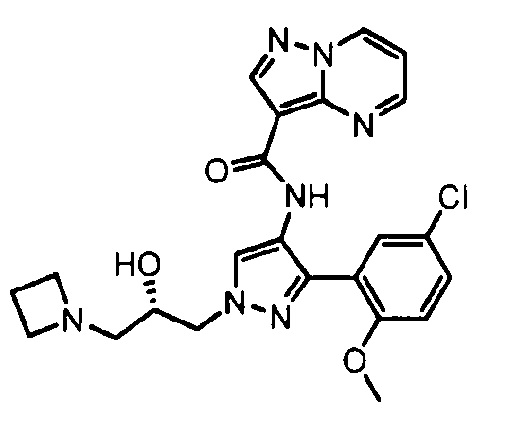
(S)-N-(1-(3-(азетидин-1-ил)-2-гидроксипропил)-3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К суспензии N-(5-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (60 мг, 0,16 ммоль) и карбоната цезия (212 мг, 0,65 ммоль) в N,N-диметилформамиде (1 мл) добавляли (S)-2-(хлорметил)оксиран (26,3 мг, 0,28 ммоль). Реакционную смесь перемешивали в течение 8 часов при комнатной температуре, и после этого анализ LCMS показал полное потребление исходного материала. Добавляли азетидин (44 мкл, 0,65 ммоль) и реакционную смесь перемешивали в течение дополнительных 16 часов при комнатной температуре. Реакционную смесь фильтровали и очищали обращенно-фазовой ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (7,8 мг). LCMS (ESI) m+H=482,2; 1H ЯМР (400 МГц, DMSO) δ 9,69 (с, 1H), 9,34 (дд, J=7,0, 1,6, 1H), 8,78 (дд, J=4,2, 1,6, 1H), 8,66 (с, 1H), 8,27 (с, 1H), 7,50 (дд, J=8,9, 2,7, 1H), 7,40 (д, J=2,7, 1H), 7,33-7,26 (м, 2H), 4,94 (д, J=5,4, 1H), 4,21 (дд, J=13,7, 3,8, 1H), 3,99 (дд, J=13,8, 7,8, 1H), 3,84 (с, 3H), 3,77 (с, 1H), 3,17 (т, J=6,9, 4H), 2,39 (кв.д, J=12,0, 5,9, 2H), 2,02-1,93 (м, 2H).
ПРИМЕР 134

N-(3-(5-хлор-2-метоксифенил)-1-(2-(3,3-диметилпирролидин-1-ил)-2-оксоэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

трет-бутил-2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)ацетат
N-(3-(5-Хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (2,0 г, 5,4 ммоль), трет-бутилбромацетат (0,88 мл, 6,0 ммоль) и карбонат цезия (2,1 г, 6,5 ммоль) объединяли и перемешивали при 30°C в течение ночи. Смесь нагревали до 65°C, после чего добавляли дополнительный карбонат и трет-бутилбромацетат и перемешивали в течение 8 ч. Смесь охлаждали до температуры окружающей среды и перемешивали в течение ночи, затем распределяли между EtOAc/водой. Органическую фазу отделяли, промывали рассолом, сушили (Na2SO4), фильтровали через силикагель и концентрировали до твердого вещества. Твердое вещество промывали смесью 1:1 EtOAc/гексан с получением 1,85 г (71%) трет-бутил-2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)ацетата в виде желтых кристаллов. LCMS (ESI) m+H=483,0. 1H ЯМР (400 МГц, CDCl3) δ 9,74 (с, 1H), 8,77 (м, 1H), 8,73 (с, 1H), 8,51 (м, 1H), 8,38 (с, 1H), 8,02 (с, 1H), 7,58 (с, 1H), 7,31 (м, 1H), 6,97 (м, 2H), 4,85 (с, 2H), 3,83 (с, 3H), 1,49 (с, 9H).

2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)уксусная кислота
К трет-бутил-2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)ацетату (1,85 г, 3,83 ммоль) в 50 мл дихлорметана добавляли 30 мл TFA. Смесь перемешивали в течение 2 ч при температуре окружающей среды, затем концентрировали и перекристаллизовывали из EtOAc с получением 1,4 г (86%) of 2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)уксусной кислоты в виде бесцветного твердого вещества. LCMS (ESI) m+H=427,1. 1H ЯМР (400 МГц, CD3OD) δ 9,95 (с, 1H), 9,08 (м, 1H), 8,69 (м, 1H), 8,63 (с, 1H), 8,34 (с, 1H), 7,47 (м, 2H), 7,21 (м, 2H), 5,03 (с, 2H), 3,86 (с, 3H).
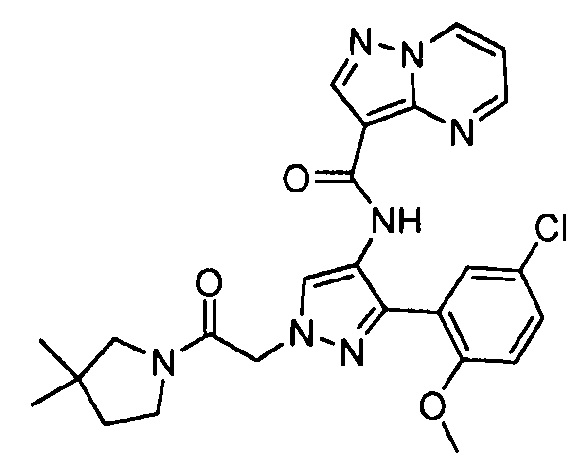
N-(3-(5-хлор-2-метоксифенил)-1-(2-(3,3-диметилпирролидин-1-ил)-2-оксоэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К 2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)уксусной кислоте (31,9 мг, 0,075 ммоль) в 1 мл DMF добавляли 3,3-диметилпирролидин HCl (15 мг, 0,11 ммоль), затем гексафторфосфат N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (43 мг, 0,11 ммоль), а затем триэтиламин (42 мкл, 0,30 ммоль) и смесь целиком перемешивали в течение 30 мин. Неочищенную смесь очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 28,9 мг (76%) в виде бесцветного твердого вещества. LCMS (ESI) m+H=508,1. 1H ЯМР (400 МГц, DMSO) δ 9,72 (с, 1H), 9,34 (д, J=6,9 Гц, 1H), 8,77 (с, 1H), 8,67 (с, 1H), 8,28 (с, 1H), 7,50 (д, J=8,9 Гц, 1H), 7,37 (с, 1H), 7,30 (т, J=9,3 Гц, 2H), 5,09 (д, J=17,8 Гц, 2H), 3,85 (с, 3H), 3,62 (т, J=6,7 Гц, 1H), 3,43 (т, J=7,1 Гц, 1H), 3,29 (с, 1H), 3,12 (с, 1H), 1,74 (т, J=7,0 Гц, 1H), 1,63 (т, J=6,9 Гц, 1H), 1,08 (д, J=9,9 Гц, 6H).
ПРИМЕР 135

N-(3-(5-хлор-2-метоксифенил)-1-(2-(циклопропиламино)-1-фтор-2-оксоэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
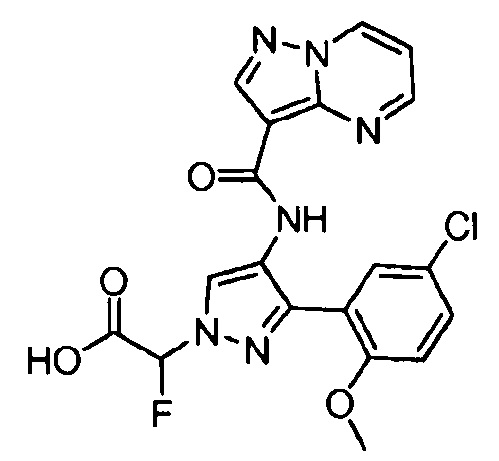
2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-2-фторуксусная кислота
К N-(3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамиду (37,1 мг, 0,10 ммоль) в 1,5 DMF добавляли гидрид натрия (10 мг, 0,40 ммоль) и смесь перемешивали в течение 5 мин, после чего добавляли этилбромфторацетат (37 мг, 0,20 ммоль). Смесь перемешивали в течение ночи, затем очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 13,3 мг (30%) 2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-2-фторуксусной кислоты в виде бесцветного твердого вещества. LCMS (ESI) m+H=445,1. 1H ЯМР (400 МГц, DMSO) δ 9,71 (с, 1H), 9,33 (д, J=7,0 Гц, 1H), 8,76-8,71 (м, 1H), 8,66 (д, J=8,3 Гц, 1H), 8,38 (с, 1H), 7,58-7,20 (м, 5H), 5,98 (д, J=56,5 Гц, 1H), 3,84 (с, 3H).

N-(3-(5-хлор-2-метоксифенил)-1-(2-(циклопропиламино)-1-фтор-2-оксоэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К 2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-2-фторуксусной кислоте (39,1 мг, 0,088 ммоль) и циклопропиламину (10 мг, 0,18 ммоль) в 1,0 мл DMF добавляли гексафторфосфат (7-азабензотриазол-1-илокси)трипирролидинофосфония (91 мг, 0,18 ммоль) и смесь перемешивали в течение 1 ч. Неочищенную смесь очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 9,1 мг (21%) N-(3-(5-хлор-2-метоксифенил)-1-(2-(циклопропиламино)-1-фтор-2-оксоэтил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида в виде бесцветного твердого вещества. LCMS (ESI) m+H=484,1. 1H ЯМР (400 МГц, DMSO) δ 9,77 (с, 1H), 9,34 (дд, J=7,0, 1,4 Гц, 1H), 8,83 (д, J=4,4 Гц, 1H), 8,76 (дд, J=4,2, 1,5 Гц, 1H), 8,68 (с, 1H), 8,55 (с, 1H), 7,58 (дд, J=8,9, 2,7 Гц, 1H), 7,37 (т, J=5,7 Гц, 2H), 7,29 (дд, J=7,0, 4,2 Гц, 1H), 6,80 (д, J=50,7 Гц, 1H), 3,85 (с, 3H), 2,82 (дд, J=7,3, 3,3 Гц, 1H), 0,70 (т, J=7,0 Гц, 2H), 0,64-0,57 (м, 2H).
ПРИМЕР 136
 и
и 
N-(3-(5-хлор-2-метоксифенил)-1-((1S,2R)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
и
N-(3-(5-хлор-2-метоксифенил)-1-((1R,2S)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К перемешиваемому раствору N-(3-(5-хлор-2-метоксифенил)-1-((1S,2S)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида и N-(3-(5-хлор-2-метоксифенил)-1-((1R,2R)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (смесь транс-энантиомеров; 107 мг, 0,229 ммоль), 4-нитробензойной кислоты (50,6 мг, 0,303 ммоль) и трифенилфосфина (81,8 мг, 0,312 ммоль) в 3,0 мл тетрагидрофурана капельно добавляли диэтилазодикарбоксилат (47,0 мкл, 0,298 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение одного часа, а затем нагревали при 50°C в течение 2,5 часов. Добавляли 4-нитробензойную кислоту (51 мг), трифенилфосфин (86 мг) и диэтилазодикарбоксилат (53 мкл) и реакционную смесь нагревали при 50°C в течение ночи. Реакционную смесь распределяли между этилацетатом и водой, и органическую часть промывали рассолом, сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт подвергали флэш-хроматографии на силикагеле (0-100% этилацетат в дихлорметане) с получением смеси энантиомеров (1R,2S)- и (1S,2R)-2-(3-(5-хлор-2-метоксифенил)-4-(пиразолo[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)циклогексил-4-нитробензоата в качестве смеси с трифенилфосфиноксидом. Этот материал использовали далее без дальнейшей очистки.
Неочищенный материал с предыдущей стадии растворяли в 3 мл тетрагидрофурана с 5,0 M гидроксидом натрия в воде (1,0 мл). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь выливали в этилацетат и промывали два раза 2 M водным раствором гидроксида натрия. Органический слой сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 8,5 мг смеси энантиомеров N-(3-(5-хлор-2-метоксифенил)-1-((1S,2R)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида и N-(3-(5-хлор-2-метоксифенил)-1-((1R,2S)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=467,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,68 (с, 1H), 9,33 (дд, J=7,0, 1,6 Гц, 1H), 8,78 (дд, J=4,2, 1,6 Гц, 1H), 8,65 (с, 1H), 8,33 (с, 1H), 7,49 (дд, J=8,8, 2,7 Гц, 1H), 7,42 (д, J=2,7 Гц, 1H), 7,33-7,25 (м, 2H), 4,84 (д, J=4,3 Гц, 1H), 4,24 (д, J=12,1 Гц, 1H), 4,11 (с, 1H), 3,84 (с, 3H), 2,16 (тд, J=12,4, 8,6 Гц, 1H), 1,81 (м, 3H), 1,62 (дт, J=26,0, 13,0 Гц, 2H), 1,43 (д, J=6,1 Гц, 2H).
ПРИМЕР 137
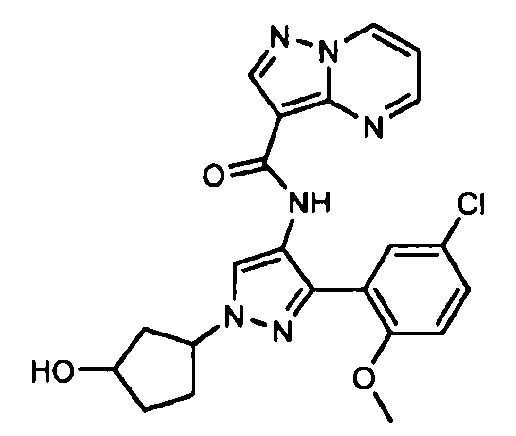
N-(3-(5-хлор-2-метоксифенил)-1-(3-гидроксициклопентил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(5-хлор-2-метоксифенил)-1-(3-гидроксициклопентил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (171,1 мг, 0,3795 ммоль) в 5,0 мл тетрагидрофурана при -78°C добавляли 1,0 M три-трет-бутоксиалюмогидрид лития в тетрагидрофуране (0,6 мл, 0,6 ммоль). Реакционную смесь поддерживали при -60°C в течение 4 часов, а затем добавляли 1,0 M три-трет-бутоксиалюмогидрид лития в тетрагидрофуране (0,6 мл, 0,6 ммоль). Реакционную смесь поддерживали при -25°C в течение 8 часов. Добавляли 1,0 M три-трет-бутоксиалюмогидрид лития в тетрагидрофуране (0,9 мл, 0,9 ммоль) и реакционную смесь поддерживали при -25°C в течение дополнительных 6 часов до тех пор, пока с помощью LCMS не определяли полное восстановление кетона. Затем реакционную смесь охлаждали при -40°C и гасили 3 мл насыщенного водного раствора хлорида аммония. После нагревания до комнатной температуры эту смесь экстрагировали дихлорметаном, и органический экстракт сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 34 мг (20%) N-(3-(5-хлор-2-метоксифенил)-1-(3-гидроксициклопентил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=453,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,67 (с, 1H), 9,34 (дд, J=7,0, 1,6 Гц, 1H), 8,78 (дд, J=4,2, 1,6 Гц, 1H), 8,64 (с, 1H), 8,38 (с, 1H), 7,50 (дд, J=8,8, 2,7 Гц, 1H), 7,40 (д, J=2,7 Гц, 1H), 7,34-7,25 (м, 2H), 4,90 (д, J=4,6 Гц, 1H), 4,83-4,69 (м, 1H), 4,21 (дд, J=10,2, 5,5 Гц, 1H), 3,85 (с, 3H), 2,42 (ддд, J=14,6, 8,7, 6,2 Гц, 1H), 2,21-2,04 (м, 2H), 2,00-1,87 (м, 1H), 1,86-1,70 (м, 2H).
ПРИМЕР 138

N-(3-(5-хлор-2-циклопропилфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

4-хлор-2-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенол
К раствору 5-(5-хлор-2-(4-метоксибензилокси)фенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (полученного способом, описанным для примера 14) (1,193 г, 2,434 ммоль) в 17 мл дихлорметана добавляли 3 мл воды, а затем дихлордицианохинон (1,2208 г). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов, а затем добавляли дополнительный дихлордицианохинон (0,4946 г). После дополнительных 24 часов реакционную смесь выливали в насыщенный водный раствор бикарбоната натрия и экстрагировали дихлорметаном. Органическую часть сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-35% этилацетат в гептане) с получением 745,2 мг (83%) 4-хлор-2-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенола. LCMS (ESI) m+H=270,2; 1H ЯМР (400 МГц, DMSO-d6) δ: 10,47 (с, 1H), 8,44 (с, 1H), 7,47 (д, J=2,6, 1H), 7,43 (дд, J=8,8, 2,7, 1H), 7,00 (д, J=8,8, 1H), 5,30 (дд, J=65,1, 10,8, 2H), 3,42 (т, J=8,1, 2H), 0,74 (т, 2H), -0,08 (с, 9H).

4-хлор-2-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенилтрифторметансульфонат
К раствору 4-хлор-2-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенола (190,4 мг, 0,5148 ммоль) в 8 мл дихлорметана при -40°C добавляли триэтиламин (0,30 мл, 2,2 ммоль), а затем трифторметансульфоновый ангидрид (0,15 мл, 0,89 ммоль). После 30 минут при этой температуре реакционную смесь нагревали до комнатной температуры. Через 3 часа реакционную смесь выливали в воду и экстрагировали два раза дихлорметаном. Объединенные органические части сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-30% этилацетат в гептане) с получением 239,8 мг (93%) 4-хлор-2-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенилтрифторметансульфоната. LCMS (ESI) m+Na=524,0.

5-(5-хлор-2-циклопропилфенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол
К смеси 4-хлор-2-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)фенилтрифторметансульфоната (72,4 мг, 0,144 ммоль), циклопропилбороновой кислоты (55,0 мг, 0,640 ммоль), тетракис(трифенилфосфин)палладия(0) (79,2 мг, 0,685 ммоль), фосфата калия (152,7 мг, 0,7194 ммоль) и бромида натрия (149,2 мг, 1,450 ммоль) добавляли воду (13,0 мкл , 0,722 ммоль) и толуол (3,0 мл). Реакционную смесь нагревали при 90°C в течение 72 часов. Реакционную смесь распределяли между этилацетатом и водой, и органический слой промывали рассолом, сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-30% этилацетат в гептане) с получением 30,1 мг (53%) 5-(5-хлор-2-циклопропилфенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола. LCMS (ESI) m+Na=416,2.

N-(3-(5-хлор-2-циклопропилфенил)-1-метил-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Указанное в заголовке соединение синтезировали из 5-(5-хлор-2-циклопропилфенил)-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола способами, описанными для примера 14. LCMS (ESI) m+H=393,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,66 (с, 1H), 9,31 (дд, J=7,0, 1,5 Гц, 1H), 8,65 (с, 1H), 8,54-8,50 (м, 1H), 8,29 (с, 1H), 7,43 (дд, J=8,5, 2,3 Гц, 1H), 7,35 (д, J=2,3 Гц, 1H), 7,24 (дд, J=7,0, 4,2 Гц, 1H), 7,00 (д, J=8,6 Гц, 1H), 3,92 (с, 3H), 1,98 (с, 1H), 0,79 (дт, J=6,2, 4,3 Гц, 2H), 0,66-0,59 (м, 2H).
ПРИМЕР 139
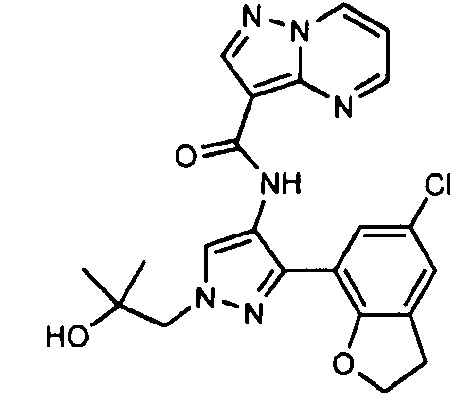
N-(3-(5-хлор-2,3-дигидробензофуран-7-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
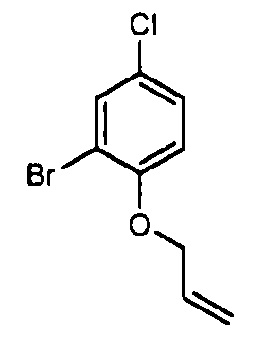
1-(аллилокси)-2-бром-4-хлорбензол
К раствору 2-бром-4-хлорфенола (7,7897 г, 37,549 ммоль) в 20 мл DMF добавляли карбонат калия (5,784 г, 41,85 ммоль) и аллилбромид (3,30 мл, 38,1 ммоль). Реакционную смесь перемешивали при 50°C в течение 15 часов. Реакционную смесь распределяли между этилацетатом и водой, и органический слой промывали рассолом, сушили над сульфатом магния и упаривали в вакууме с получением 9,4 г (100%) 1-(аллилокси)-2-бром-4-хлорбензола, который использовали далее без дальнейшей очистки. 1H ЯМР (400 МГц, CDCl3) δ: 7,54 (д, J=2,5, 1H), 7,21 (дд, J=8,8, 2,5, 1H), 6,81 (д, J=8,8, 1H), 6,10-5,98 (м, 1H), 5,46 (дд, J=17,3, 1,4, 1H), 5,31 (дд, J=10,6, 1,3, 1H), 4,59 (д, J=5,0, 2H).

2-аллил-6-бром-4-хлорфенол
Раствор 1-(аллилокси)-2-бром-4-хлорбензола (4,122 г, 16,65 ммоль) в N,N-диэтиланилине (20 мл, 100 ммоль) нагревали при 200°C в течение 15 часов. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом и 1 M водным раствором HCl, и органический слой промывали дополнительной частью 1 M водного раствора HCl, а затем рассолом. Органический слой сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-20% этилацетат в гептане) с получением 3,1761 г (77%) 2-аллил-6-бром-4-хлорфенола в виде прозрачного бесцветного масла. 1H ЯМР (400 МГц, CDCl3) δ: 7,33 (д, J=2,4, 1H), 7,07 (д, J=2,3, 1H), 5,94 (ддт, J=16,8, 10,3, 6,6, 1H), 5,53 (с, 1H), 5,18-5,06 (м, 2H), 3,40 (д, J=6,6, 2H).

2-бром-4-хлор-6-(2-гидроксиэтил)фенол
Раствор 2-аллил-6-бром-4-хлорфенола (1,378 г, 5,567 ммоль) в 20 мл дихлорметана охлаждали при -78°C. При перемешивании при этой температуре, через реакционный раствор барботировали озон в течение 6,5 часов. После продувания реакционной емкости кислородом, все еще при -78°C, реакционную смесь гасили тетрагидроборатом натрия (1,064 г, 28,12 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Реакционную смесь распределяли между этилацетатом и водой, и органический слой промывали рассолом, сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (10-60% этилацетат в гептане) с получением 0,5911 г (42%) 2-бром-4-хлор-6-(2-гидроксиэтил)фенола. 1H ЯМР (400 МГц, CDCl3) δ: 7,38 (д, J=2,5, 1H), 7,27 (с, 1H), 7,06 (д, J=2,4, 1H), 3,96 (уш. с, 2H), 2,91 (т, J=5,7, 2H), 1,98 (с, 1H).
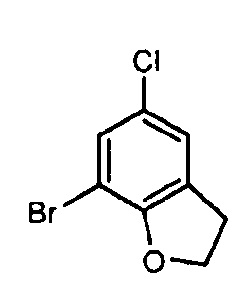
7-бром-5-хлор-2,3-дигидробензофуран
К смеси 2-бром-4-хлор-6-(2-гидроксиэтил)фенола (99,8 мг, 0,397 ммоль), триэтиламина (0,40 мл, 2,9 ммоль) и дихлорметана (4 мл) при 0°C добавляли метансульфонилхлорид (56,0 мкл, 0,724 ммоль). Реакционную смесь перемешивали при 0°C в течение 1,5 часов, после чего добавляли дополнительный метансульфонилхлорид (10 мкл) и реакционную смесь нагревали до комнатной температуры. После перемешивания в течение ночи реакционную смесь повторно охлаждали до 0°C и добавляли триэтиламин (0,2 мл) и метансульфонилхлорид (15 мкл). Через два часа реакционную смесь распределяли между этилацетатом и водой, и органический слой промывали рассолом, сушили над сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-30% этилацетат в гептане) с получением 40,0 мг (40%) 7-бром-5-хлор-2,3-дигидробензофурана. 1H ЯМР (400 МГц, CDCl3) δ: 7,26 (д, 1H), 7,09 (д, 1H), 4,67 (т, J=8,8, 2H), 3,30 (т, J=8,8, 2H).

N-(3-(5-хлор-2,3-дигидробензофуран-7-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Указанное в заголовке соединение получали с использованием 7-бром-5-хлор-2,3-дигидробензофурана и способами, описанными для примеров 14 и 16. LCMS (ESI) m+H=453,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,72 (с, 1H), 9,33 (д, J=7,0, 1H), 8,82 (дд, J=4,2, 1,5, 1H), 8,65 (с, 1H), 8,35 (с, 1H), 7,36 (с, 1H), 7,33-7,23 (м, 2H), 4,70 (с, 1H), 4,62 (т, J=8,8, 2H), 4,07 (с, 2H), 3,37 (т, J=8,8, 2H), 1,12 (с, 6H).
ПРИМЕР 140

N-(1-метил-3-(3-метилпиперидин-1-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

5-хлор-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол
В высушенной в сушильном шкафу колбе 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол (412,4 мг, 1,695 ммоль) растворяли в 5 мл THF и охлаждали при -78°C. К этому раствору медленно добавляли 1,0 M гексаметилдисилазид лития в тетрагидрофуране (2,0 мл, 2,0 ммоль). После перемешивания в течение 30 минут при -78°C, медленно добавляли раствор гексахлорэтана (455,2 мг, 1,923 ммоль) в 3 мл THF. Реакционную смесь поддерживали при -78°C в течение дополнительного часа, а затем гасили насыщенным водным раствором хлорида аммония и нагревали до комнатной температуры. Реакционную смесь распределяли между этилацетатом и водой, и органический слой сушили над сульфатом магния и упаривали в вакууме с получением 0,4592 г (98%) 5-хлор-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола, который использовали далее без дальнейшей очистки. LCMS (ESI) m+H=220,2; 1H ЯМР (400 МГц, DMSO-d6) δ: 8,52 (с, 1H), 5,56 (с, 2H), 3,67-3,57 (м, 2H), 0,91-0,83 (м, 2H), -0,04 (с, 9H).

3-метил-1-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)пиперидин
К раствору 5-хлор-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (348,3 мг, 1,254 ммоль) в 1 мл н-бутанола добавляли 3-метилпиперидин (0,20 мл, 1,7 ммоль). Реакционную смесь подвергали облучению микроволновым излучением при температуре 120°C в течение 30 минут. Растворитель выпаривали в вакууме, и неочищенный продукт очищали флэш-хроматографией на силикагеле (0-40% этилацетат в гептане) с получением 486,5 мг 3-метил-1-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)пиперидина. LCMS (ESI) m+H=341,4; 1H ЯМР (400 МГц, DMSO-d6) δ: 8,04 (с, 1H), 5,36 (с, 2H), 3,71-3,62 (м, 2H), 3,20 (м, 3H), 2,96-2,85 (м, 1H), 1,84-1,76 (м, 4H), 1,15 (м, 1H), 0,97-0,87 (м, 5H), 0,00 (с, 9H).
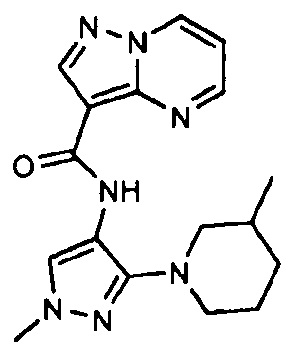
N-(1-метил-3-(3-метилпиперидин-1-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Указанное в заголовке соединение получали с использованием 3-метил-1-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)пиперидина и способами, описанными для примера 14. LCMS (ESI) m+H=340,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,59 (с, 1H), 9,36 (дд, J=7,0, 1,5, 1H), 8,83 (дд, J=4,2, 1,6, 1H), 8,66 (с, 1H), 7,98 (с, 1H), 7,32 (дд, J=7,0, 4,2, 1H), 3,71 (с, 3H), 3,25 (с, 1H), 2,64 (с, 1H), 2,41-2,29 (м, 1H), 1,96-1,70 (м, 4H), 1,04 (с, 1H), 0,91 (д, J=6,7, 3H).
ПРИМЕР 141
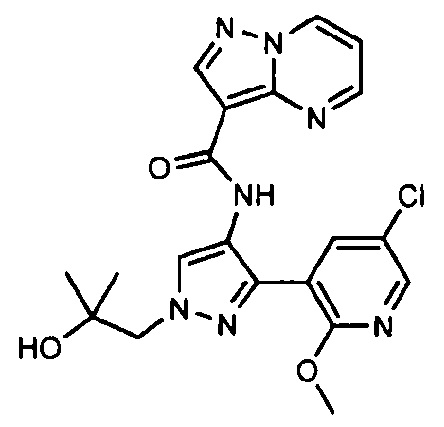
N-(3-(5-хлор-2-метоксипиридин-3-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

5-йод-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол
В высушенной в сушильном шкафу колбе 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол (1,5192 г, 6,2432 ммоль) растворяли в 20 мл THF и охлаждали при -78°C. К этому раствору медленно добавляли 1,0 M гексаметилдисилазид лития в тетрагидрофуране (7,5 мл, 7,5 ммоль). После перемешивания в течение 40 минут при -78°C, медленно добавляли раствор йода (1,7602 г, 6,9351 ммоль) в 8 мл THF. Реакционную смесь поддерживали при -78°C в течение дополнительных 1,5 часов, а затем гасили насыщенным водным раствором хлорида аммония и нагревали до комнатной температуры. Реакционную смесь распределяли между этилацетатом и полунасыщенным водным раствором Na2S2O3. Органический слой сушили сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-15% этилацетат в гептане) с получением 2,2349 г (97%) 5-йод-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола. 1H ЯМР (400 МГц, DMSO-d6) δ: 8,48 (с, 1H), 5,59 (с, 2H), 3,61 (т, J=8,0, 2H), 0,86 (т, J=8,0, 2H), -0,04 (с, 9H).
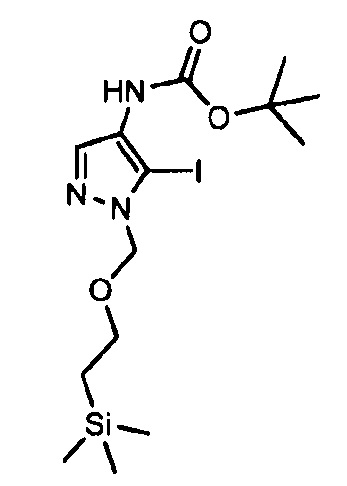
трет-бутил-5-йод-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамат
К раствору 5-йод-4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (2,234 г, 6,050 ммоль) в этаноле (20 мл) добавляли хлорид аммония (1,303 г, 24,36 ммоль), железный порошок (1,695 г, 30,35 ммоль) и воду (30 мл). Затем реакционную смесь перемешивали при 70°C в течение 45 минут, а затем охлаждали до комнатной температуры, разбавляли дихлорметаном и фильтровали через целит, промывая дополнительным дихлорметаном. К фильтрату добавляли насыщенный водный раствор бикарбоната натрия и слои разделяли. Водный слой экстрагировали еще раз дихлорметантом, а затем объединенные органические части сушили над сульфатом магния, фильтровали и упаривали в вакууме. К полученному осадку добавляли диоксан (20 мл), триэтиламин (2,0 мл, 14 ммоль) и ди-трет-бутилдикарбонат (1,513 г, 6,932 ммоль). Эту смесь перемешивали при 60°C в течение 4 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-40% этилацетат в гептане) с получением 1,4353 г (54%) трет-бутил-5-йод-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамата. 1H ЯМР (400 МГц, DMSO-d6) δ: 8,44 (с, 1H), 7,56 (с, 1H), 5,39 (с, 2H), 3,53 (т, J=8,0, 2H), 1,43 (с, 9H), 0,83 (т, J=8,0, 2H), -0,04 (с, 9H).

трет-бутил-5-(5-хлор-2-метоксипиридин-3-ил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамат
Смесь трет-бутил-5-йод-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамата (390,7 мг, 0,8892 ммоль), пинаколового эфира 5-хлор-2-метоксипиридин-3-бороновой кислоты (364,0 мг, 1,350 ммоль), аддукта трис(дибензилиденацетон)дипалладий(0)-хлороформ (48,5 мг, 0,0468 ммоль), S-Phos (74,2 мг, 0,181 ммоль), фосфата калия (597,1 мг, 2,813 ммоль) и 1-бутанола (10 мл) дегазировали азотом, а затем перемешивали при 80°C в течение 15 часов. Реакционную смесь разбавляли этилацетатом, промывали водой и рассолом, сушили над сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-50% этилацетат в гептане) с получением 212,3 мг (52%) трет-бутил-5-(5-хлор-2-метоксипиридин-3-ил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамата. LCMS (ESI) m+H=455,2; 1H ЯМР (400 МГц, DMSO-d6) δ: 8,53 (с, 1H), 8,33 (с, 1H), 7,79 (д, J=2,6, 1H), 5,22 (с, 2H), 3,85 (с, 3H), 3,35 (т, 2H), 1,38 (с, 9H), 0,70 (т, J=8,1, 2H), -0,10 (с, 9H).
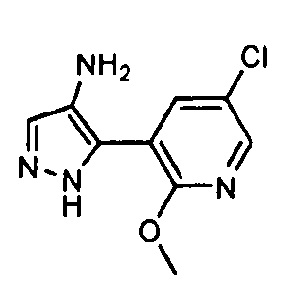
5-(5-хлор-2-метоксипиридин-3-ил)-1H-пиразол-4-амин
К раствору трет-бутил-5-(5-хлор-2-метоксипиридин-3-ил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамата (211,7 мг, 0,4652 ммоль) в 5 мл этилацетата добавляли тетрахлорид олова (0,52 мл, 4,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, а затем упаривали в вакууме. Остаточное масло распределяли между этилацетатом и насыщенным водным раствором бикарбоната натрия, и водный слой экстрагировали еще два раза этилацетатом. Объединенные органические слои сушили над сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-20% метанол в дихлорметане) с получением 32,4 мг (31%) 5-(5-хлор-2-метоксипиридин-3-ил)-1H-пиразол-4-амина. LCMS (ESI) m+H=225,1.
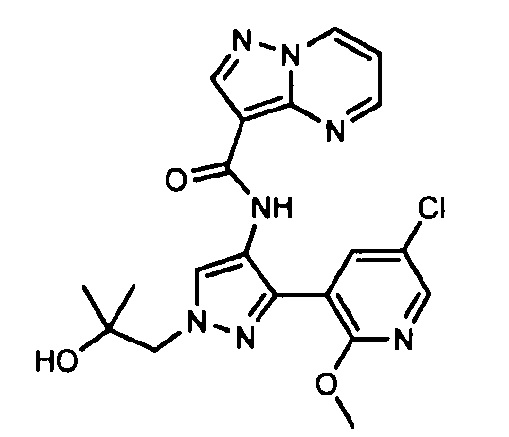
N-(3-(5-хлор-2-метоксипиридин-3-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Указанное в заголовке соединение получали с использованием 5-(5-хлор-2-метоксипиридин-3-ил)-1H-пиразол-4-амина и способами, описанными для примеров 4 и 16. LCMS (ESI) m+H=442,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,72 (с, 1H), 9,34 (д, J=6,8 Гц, 1H), 8,73 (с, 1H), 8,66 (с, 1H), 8,35 (с, 2H), 7,88 (с, 1H), 7,28 (с, 1H), 4,75 (с, 1H), 4,09 (с, 2H), 3,95 (с, 3H), 1,13 (с, 6H).
ПРИМЕР 142

N-(1-(2-гидрокси-2-метилпропил)-3-(1-метил-2-оксо-5-(трифторметил)-1,2-дигидропиридин-3-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

3-бром-1-метил-5-(трифторметил)пиридин-2(1H)-он
К раствору 3-бром-2-гидрокси-5-(трифторметил)пиридина (1,0097 г, 4,1724 ммоль) в хлороформе (20 мл) добавляли карбонат серебра (1,1964 г, 4,3388 ммоль) и метилйодид (0,40 мл, 6,4 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение двух часов, а затем при 40°C в течение 24 часов. Добавляли дополнительный метилйодид (0,40 мл, 6,4 ммоль) и реакционную смесь поддерживали при 40°C в течение дополнительных 15 часов. Затем реакционную смесь разбавляли дихлорметаном, фильтровали через целит и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-80% этилацетат в гептане) с получением 0,5297 г (50%) 3-бром-1-метил-5-(трифторметил)пиридин-2(1H)-она. 1H ЯМР (400 МГц, DMSO-d6) δ: 8,47 (д, J=0,9, 1H), 8,22 (д, J=2,5, 1H), 3,56 (с, 3H).

N-(1-(2-гидрокси-2-метилпропил)-3-(1-метил-2-оксо-5-(трифторметил)-1,2-дигидропиридин-3-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Указанное в заголовке соединение получали с использованием 3-бром-1-метил-5-(трифторметил)пиридин-2(1H)-она и способами, описанными для примеров 14 и 16. LCMS (ESI) m+H=476,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 10,23 (с, 1H), 9,31 (дд, J=7,0, 1,5, 1H), 8,77 (дд, J=4,1, 1,6, 1H), 8,64 (с, 1H), 8,58 (с, 1H), 8,39 (с, 1H), 7,88 (д, J=2,7, 1H), 7,27 (дд, J=7,0, 4,2, 1H), 4,74 (с, 1H), 4,08 (с, 2H), 3,71 (с, 3H), 1,11 (с, 6H).
ПРИМЕР 143

N-(1-(2-гидрокси-2-метилпропил)-3-(5-метокси-2-(трифторметил)пиридин-4-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

4-йод-5-метокси-2-(трифторметил)пиридин
В высушенной в сушильном шкафу колбе 5-метокси-2-(трифторметил)пиридин растворяли в THF (20 мл). Эту смесь охлаждали при -78°C, а затем добавляли 2,5 M н-бутиллитий в гексане (2,60 мл, 6,5 ммоль). После перемешивания при той же температуре в течение 40 минут добавляли 1-хлор-2-йодэтан (0,60 мл, 6,6 ммоль). Реакционную смесь поддерживали при -78°C в течение дополнительных 30 минут, а затем гасили насыщенным водным раствором хлорида аммония. Смесь нагревали до комнатной температуры, распределяли между этилацетатом и водой, и органический слой сушили сульфатом магния и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (0-40% этилацетат в гептане) с получением 0,3128 г (16%) 4-йод-5-метокси-2-(трифторметил)пиридина. 1H ЯМР (400 МГц, DMSO-d6) δ: 8,38 (с, 1H), 8,29 (с, 1H), 4,05 (с, 3H).

N-(1-(2-гидрокси-2-метилпропил)-3-(5-метокси-2-(трифторметил)пиридин-4-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Указанное в заголовке соединение получали с использованием 4-йод-5-метокси-2-(трифторметил)пиридина и способами, описанными для примеров 14 и 16. LCMS (ESI) m+H=476,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,71 (с, 1H), 9,36 (д, J=6,9, 1H), 8,88 (д, J=3,8, 1H), 8,82 (с, 1H), 8,67 (с, 1H), 8,41 (с, 1H), 7,85 (с, 1H), 7,36-7,27 (м, 1H), 4,77 (с, 1H), 4,12 (с, 2H), 4,09 (с, 3H), 1,13 (с, 6H).
ПРИМЕР 144




К раствору 2-бром-6-йод-3-метоксипиридин (627,4 мг, 1,999 ммоль) в N-метилпирролидиноне (10 мл) добавляли цианид меди (202,6 мг, 2,262 ммоль). Реакционную смесь перемешивали при 130°C в течение 3,5 часов, а затем охлаждали до комнатной температуры. Неочищенную реакционную смесь распределяли между этилацетатом и водой, органический слой промывали рассолом, сушили сульфатом магния, фильтровали и упаривали в вакууме. Неочищенный продукт очищали флэш-хроматографией на силикагеле (20-100% этилацетат в гептане) с получением 157,8 мг (37%) смеси двух региоизомерных продуктов: 6-бром-5-метоксипиколинонитрила и 6-йод-3-метоксипиколинонитрила. 1H ЯМР (400 МГц, DMSO) δ: 8,12 (д, J=8,3 Гц, 0,75H), 7,98 (д, J=9,0 Гц, 1H), 7,80 (д, J=9,1 Гц, 1H), 7,69 (д, J=8,5 Гц, 0,75H), 3,98 (перекрывающийся с и с, 6H).

N-(3-(6-циано-3-метоксипиридин-2-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

N-(3-(6-циано-5-метоксипиридин-2-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Указанные в заголовке соединения получали с использованием смеси 6-бром-5-метоксипиколинонитрила и 6-йод-3-метоксипиколинонитрила и способами, описанными для примеров 14 и 16, разделяя два региоизомерных конечных продукта посредством обращенно-фазовой ВЭЖХ с получением: N-(3-(6-циано-3-метоксипиридин-2-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида; LCMS (ESI) m+H=433,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 10,55 (с, 2H), 9,35 (д, J=6,9 Гц, 2H), 8,93 (с, 2H), 8,68 (с, 2H), 8,48 (с, 2H), 8,10 (д, J=8,6 Гц, 2H), 7,80 (д, J=8,4 Гц, 2H), 7,42-7,26 (м, 2H), 6,54 (с, 1H), 4,80 (с, 2H), 4,12 (с, 4H), 3,94 (с, 6H), 1,12 (с, 12H); и N-(3-(6-циано-5-метоксипиридин-2-ил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида; LCMS (ESI) m+H=433,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 10,97 (с, 1H), 9,36 (д, J=6,8 Гц, 1H), 9,09 (с, 1H), 8,70 (с, 1H), 8,57 (с, 1H), 8,26 (д, J=9,4 Гц, 1H), 7,94 (д, J=9,1 Гц, 1H), 7,42-7,32 (м, 1H), 4,78 (с, 1H), 4,11 (с, 2H), 4,04 (с, 3H), 1,13 (с, 6H).
ПРИМЕР 145

N-(5-(3-бромфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

5-(3-бромфенил)-4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол
К раствору 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (4 г, 16,4 ммоль) в 40 мл N,N-диметилацетамида добавляли 1,3-дибромбензол (4,6 г, 19,7 ммоль), ацетат палладия(II) (242 мг, 1,08 ммоль), ди-(1-адамантил)-н-бутилфосфин (565 мг, 1,58 ммоль), карбонат калия (8,28 г, 60 ммоль) и триметилуксусную кислоту (552 мг, 5,2 ммоль). При перемешивании при комнатной температуре через реакционную смесь барботировали газообразный азот в течение 10 минут, а затем реакционную смесь нагревали при 120°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и промывали водой и рассолом, сушили над сульфатом магния и концентрировали. Неочищенный материал очищали флэш-хроматографией на силикагеле (0-25% этилацетат в гексане) с получением 800 мг (12%) 5-(3-бромфенил)-4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразола. LCMS (ESI) m+H=398,0; 1H ЯМР (400 МГц, CDCl3) δ: 8,24 (д, 1H), 7,70 (м, 1H), 7,53 (с, 1H), 7,46 (д, 1 H), 7,42 (д, 1H), 7,41 (д, 1H), 5,27 (с,1H), 3,72 (м, 2H), 0,95 (м, 2H), 0,00 (с,9H).

5-(3-бромфенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амин
К раствору 5-(3-бромфенил)-4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразола (800 мг, 2,4 ммоль) в 25 мл этанола добавляли 50 мл воды, хлорид аммония (636 мг, 12 ммоль) и железный порошок (806 мг, 14 ммоль). Реакционную смесь перемешивали при 75°C в течение 6 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном и фильтровали через слой целита, промывая дополнительным дихлорметаном. Фильтрат добавляли к 150 мл насыщенного водного бикарбоната натрия и экстрагировали два раза дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 530 мг (71%) 5-(3-бромфенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амина, который использовали далее без очистки. LCMS (ESI) m+H=368,0.
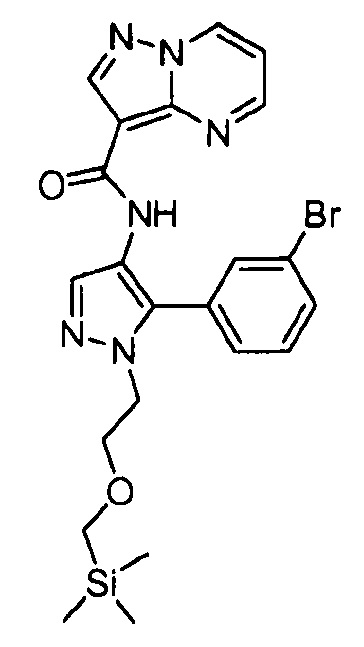
N-(5-(3-бромфенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К смеси 5-(3-бромфенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амина (533 мг, 1,45 ммоль) в тетрагидрофуране (5 мл) добавляли пиразолo[1,5-a]пиримидин-3-карбонилхлорид (262 мг, 1,45 ммоль) в тетрагидрофуране (5 мл) при 0°C. После добавления смесь нагревали до комнатной температуры, а затем перемешивали в течение ночи при этой температуре. Смесь концентрировали с получением 742 мг (99%) N-(5-(3-бромфенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида, который использовали далее без очистки. LCMS (ESI) m+H=513,1.
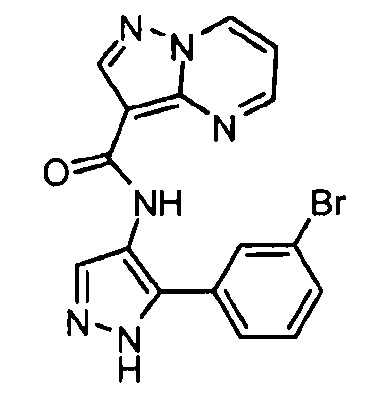
N-(5-(3-бромфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(3-бромфенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (742 мг, 1,45 ммоль) в 20 мл этанола добавляли HCl (1,0 мл 6 M раствора в воде, 6 ммоль). Затем реакционную смесь перемешивали при 70°C в течение 6 часов. После охлаждения до комнатной температуры образовывался светло-желтый осадок, который выделяли фильтрацией и промывали метанолом и диэтиловым эфиром. Объем фильтрата уменьшали и фильтровали дополнительный твердый продукт. Объединенные собранные твердые вещества сушили в вакууме с получением 320 мг (58%) N-(5-(3-бромфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=383,0; 1H ЯМР (400 МГц, DMSO-d6) δ: 13,01 (с, 1H), 9,93 (с, 1H), 9,33 (дд, 1H), 8,83 (дд, 1H), 8,66 (д, 1H), 8,26 (с, 1H), 7,57 (д, 1H), 7,47 (т, 1H), 7,28 (дд, 1H).
ПРИМЕР 146
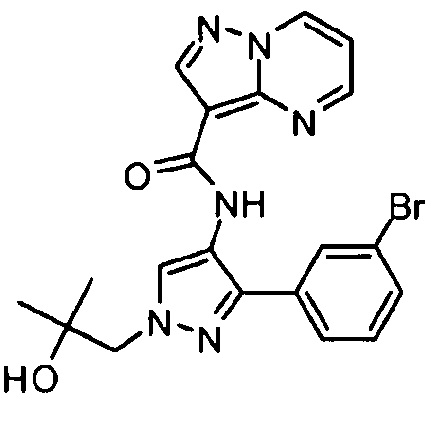
К раствору N-(5-(3-бромфенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (200 мг, 0,523 ммоль) в 20 мл DMF добавляли изобутиленоксид (0,20 мл, 2,2 ммоль) и карбонат цезия (340 мг, 1,04 ммоль). Реакционную смесь перемешивали при 80°C в течение 6 часов, а затем охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали. Фильтрат промывали рассолом, сушили над сульфатом магния и концентрировали. Осадок очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 14,2 мг (6%) требуемого соединения. LCMS (ESI) m+H=456,8. 1H ЯМР (CDCl3, 400 МГц): δ 10,16 (д, J=1,2 Гц, 1H), 8,83-8,77 (м, 2H), 8,76 (т, J=5,2 Гц, 1H), 8,42 (с, 1H), 8,03 (с, 1H), 7,76 (д, J=8,4 Гц, 1H), 7,55 (д, J=8,4 Гц, 1H), 7,37 (т, J=8,0 Гц, 1H), 7,06 (дд, J=3,6, 6,8 Гц, 1H), 4,12 (с, 2H), 1,30 (с, 6H).
ПРИМЕР 147

N-(5-(5-циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

4-метокси-3-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)бензонитрил
К раствору 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (2 г, 8,2 ммоль) в 50 мл N,N-диметилацетамида добавляли 3-бром-4-метоксибензонитрил (2,1 г, 9,8 ммоль), ацетат палладия(II) (120 мг, 0,54 ммоль), ди-(1-адамантил)-н-бутилфосфин (250 мг, 0,7 ммоль), карбонат калия (6,0 г, 43,3 ммоль) и триметилуксусную кислоту (200 мг, 1,95 ммоль). При перемешивании при комнатной температуре, через реакционную смесь барботировали газообразный азот в течение 10 минут, а затем реакционную смесь нагревали при 120°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и промывали водой и рассолом, сушили над сульфатом магния и концентрировали. Неочищенный материал очищали флэш-хроматографией на силикагеле (0-25% этилацетат в гептане) с получением 730 мг (12%) 4-метокси-3-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)бензонитрила. LCMS (ESI) m+H=375,1; 1H ЯМР (400 МГц, CDCl3) δ: 8,24 (д, 1H), 7,86 (м, 1H), 7,72 (с, 1H), 7,12 (д, 1 H), 5,27 (кв., 2H), 3,86 (с, 3H), 3,68 (м, 2H), 0,89 (м, 2H), 0,00 (с, 9H).

3-(4-амино-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)-4-метоксибензонитрил
К раствору 4-метокси-3-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)бензонитрила (900 мг, 2,4 ммоль) в 25 мл этанола добавляли 50 мл воды, хлорид аммония (636 мг, 12 ммоль) и железный порошок (806 мг, 14 ммоль). Реакционную смесь перемешивали при 75°C в течение 6 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном и фильтровали через слой целита, промывая дополнительным дихлорметаном. Фильтрат добавляли к 150 мл насыщенного водного раствора бикарбоната натрия и экстрагировали два раза дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 717 мг (84%) 3-(4-амино-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)-4-метоксибензонитрила, который использовали далее без очистки. LCMS (ESI) m+H=375,1.

N-(5-(5-циано-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
3-(4-Амино-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)-4-метоксибензонитрил (717 мг, 2,08 ммоль) в тетрагидрофуране (20 мл) добавляли к пиразолo[1,5-a]пиримидин-3-карбонилхлориду (262 мг, 1,45 ммоль) в тетрагидрофуране (5 мл) при 0°C. После добавления смесь нагревали до комнатной температуры, а затем перемешивали в течение ночи при этой температуре. Смесь концентрировали с получением 1,0 г (98%) N-(5-(5-циано-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида, который использовали далее без очистки. LCMS (ESI) m+H=490,1.
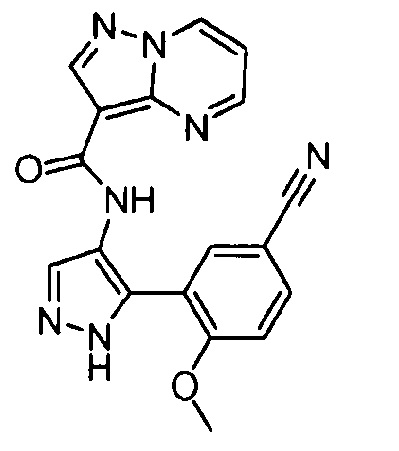
N-(5-(5-циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(5-циано-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (1,0 г, 2,04 ммоль) в 20 мл этанола добавляли HCl (2,0 мл 6 M раствора в воде, 12 ммоль). Затем реакционную смесь перемешивали при 70°C в течение 4 часов. После охлаждения до комнатной температуры образовывался светло-желтый осадок, который отфильтровывали и промывали метанолом и диэтиловым эфиром. Объем фильтрата уменьшали для осаждения дополнительного твердого продукта, который отфильтровывали. Объединенные собранные твердые вещества сушили в вакууме с получением 530 мг (72%) N-(5-(5-циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=360,1; 1H ЯМР (400 МГц, DMSO-d6) δ 12,98 (с, 1H), 9,59 (с, 1H), 9,30 (дд, 1H), 8,73 (д, 1H), 8,61 (с, 1H), 8,23 (с, 1H), 8,22 (с, 1H), 7,78 (с, 1H), 7,43 (д, 1H), 7,25 (с, 1H), 3,9 (с, 3H).
ПРИМЕР 148

N-(3-(5-циано-2-метоксифенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(5-циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (200 мг, 0,557 ммоль) в 20 мл DMF добавляли изобутиленоксид (0,20 мл, 2,2 ммоль) и карбонат цезия (363 мг, 1,11 ммоль). Реакционную смесь перемешивали при 80°C в течение 6 часов, затем разбавляли этилацетатом и фильтровали, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 27,2 мг (11%) требуемого продукта. LCMS (ESI) m+H=431,9. 1H ЯМР (CDCl3, 400 МГц): δ 9,68 (с, 1H), 8,78 (дд, J=2,0, 6,8 Гц, 1H), 8,71 (с, 1H), 8,56 (кв., J=1,6 Гц, 1H), 8,37 (с, 1H), 7,87 (д, J=2,4 Гц, 1H), 7,72 (дд, J=2,0, 8,4 Гц, 1H), 7,1 (д, J=8,8 Гц, 1H), 7,01 (дд, J=4,4, 7,2 Гц, 1H), 4,11 (с, 2H), 3,91 (с, 3H), 1,23 (с, 6H).
ПРИМЕР 149

N-(5-(5-фтор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

5-(5-фтор-2-метоксифенил)-4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол
К раствору 4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразола (2,98 г, 12,28 ммоль) в 25 мл N,N-диметилацетамида добавляли 2-бром-4-фтор-1-метоксибензол (3,54 г, 17,35 ммоль), ацетат палладия(II) (144 мг, 0,62 ммоль), ди-(1-адамантил)-н-бутилфосфин (330 мг, 0,93 ммоль), карбонат калия (5,1 г, 37,1 ммоль) и триметилуксусную кислоту (330 мг, 2,68 ммоль). При перемешивании при комнатной температуре через реакционную смесь барботировали газообразный азот в течение 10 минут, а затем реакционную смесь нагревали при 120°C в течение 12 часов. Затем реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом и промывали водой и рассолом, сушили над сульфатом магния и концентрировали. Неочищенный материал очищали флэш-хроматографией на силикагеле (0-25% этилацетат в гептане) с получением 1,0 г (22%) 5-(5-фтор-2-метоксифенил)-4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразола. LCMS (ESI) m+H=368,1; 1H ЯМР (400 МГц, CDCl3) δ: 8,25 (с, 1H), 7,29 (м, 1 H), 7,18 (дд, 1H), 7,01 (дд, 1H), 5,31(д, 2H), 3,77 (с, 3H), 3,65 (т, 2H), 2,03 (т, 2H), 0,92 (кв., 2H), 0 (с, 9H).
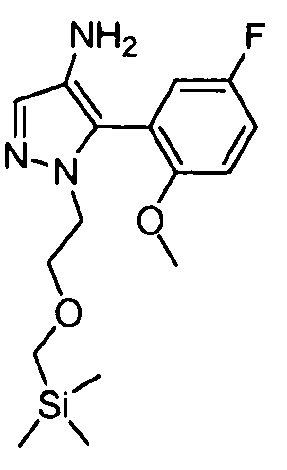
5-(5-фтор-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амин
К раствору 5-(5-фтор-2-метоксифенил)-4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразола (1,0 г, 2,7 ммоль) в 8 мл этанола добавляли 16 мл воды, хлорид аммония (570 мг, 10,8 ммоль) и железный порошок (760 мг, 13,5 ммоль). Реакционную смесь перемешивали при 75°C в течение 6 часов. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном и фильтровали через слой целита, промывая дополнительным дихлорметаном. Фильтрат добавляли к 150 мл насыщенного водного раствора бикарбоната натрия и экстрагировали два раза дихлорметаном. Объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 640 мг (70%) 5-(5-фтор-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амина, который использовали далее без очистки. LCMS (ESI) m+H=338,1. 1H ЯМР (400 МГц, CDCl3) δ: 7,37 (м, 2H), 7,20 (т, 1H), 7,04 (дд, 1H), 5,42 (т, 2H), 3,90 (д, 3H), 3,65 (т, 2H), 0,96 (т, 2H), 0 (с, 9H).

N-(5-(5-фтор-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К 5-(5-фтор-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амину (580 мг, 1,72 ммоль) в тетрагидрофуране (40 мл) добавляли пиразолo[1,5-a]пиримидин-3-карбонилхлорид (320 мг, 1,72 ммоль) в THF (5 мл) при 0°C. После добавления смесь нагревали до комнатной температуры, а затем перемешивали в течение ночи при этой температуре. Смесь концентрировали с получением 330 мг (40%) N-(5-(5-фтор-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида, который использовали далее без очистки. LCMS (ESI) m+H=483,1. 1H ЯМР (400 МГц, CDCl3) δ: 9,63 (с, 1H), 8,82 (м, 1H), 8,76 (с, 1H), 8,52 (м, 1H), 8,42 (с, 1H), 7,38 (дд, 1H), 7,31 (д, 1H), 7,22 (м, 1H), 7,06 (м, 2H), 5,42 (д, 2H), 3,86(с, 3H), 3,72 (м, 2H), 0,92 (кв., 2H), 0 (с, 9H).

N-(5-(5-фтор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(5-фтор-2-метоксифенил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (330 мг, 0,68 ммоль) в 14 мл этанола добавляли HCl (1,0 мл 6 M раствора в воде, 6,0 ммоль). Затем реакционную смесь перемешивали при 70°C в течение 6 часов. После охлаждения до комнатной температуры образовывался светло-желтый осадок, который отфильтровывали и промывали метанолом и диэтиловым эфиром. Объем фильтрата уменьшали для осаждения дополнительного продукта, который отфильтровывали. Объединенные собранные твердые вещества сушили в вакууме с получением 220 мг (92%) N-(5-(5-фтор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=353,1; 1H ЯМР (400 МГц, DMSO-d6) δ: 9,68 (с, 1H), 9,32 (дд, 1H), 8,77 (дд, 1H), 8,63 (с, 1H), 8,19 (с, 1H), 7,30 (м, 4H), 3,82 (д, 3H).
ПРИМЕР 150

N-(3-(5-фтор-2-метоксифенил)-1-(2-гидрокси-2-метилпропил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору соединения 8 (200 мг, 0,57 ммоль) в 10 мл DMF добавляли изобутиленоксид (0,41 г, 5,7 ммоль) и карбонат цезия (560 мг, 1,71 ммоль). Реакционную смесь перемешивали при 80°C в течение 6 часов, затем разбавляли этилацетатом и фильтровали, и органическую часть промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 69,5 мг (29%) требуемого соединения. LCMS (ESI) m+Na=447,1. 1H ЯМР (CDCl3, 400 МГц) δ: 9,83 (с, 1H), 8,80-8,78 (дд, J=2,0, 6,8 Гц, 1H), 8,73 (с, 1H), 8,56-8,54 (дд, J=1,6, 4,0 Гц, 1H), 8,36 (с, 1H), 7,33 (дд, J=3,2, 8,8 Гц, 1H), 7,11-7,08 (м, 1H), 7,02-6,97 (м, 2H), 4,12 (с, 1H), 3,85 (с, 3H), 1,24 (с, 6H).
ПРИМЕР 151

N-(3-(5-хлор-2-циклопропоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

метил-5-хлор-2-(2-хлорэтокси)бензоат
К раствору метил-5-хлор-2-гидроксибензоата (54,06 г, 0,29 моль) в 300 мл безводного DMF добавляли 2-хлорэтил-4-метилбензолсульфонат (81,6 г, 0,35 моль) и Cs2CO3 (142 г, 0,44 моль). Смесь перемешивали при 60-70°C в течение ночи, затем добавляли воду. Из раствора осаждалось белое твердое вещество, которое собирали фильтрацией и совместно упаривали с толуолом с получением 76,1 г (88%) метил-5-хлор-2-(2-хлорэтокси)бензоата. 1H ЯМР (400 МГц, CDCl3) δ: 7,76 (д, 1H), 7,40 (дд, 1H), 6,91 (д, 1H), 4,27 (т, 2H), 3,87 (с, 3H), 3,83 (т, 2H).

метил-5-хлор-2-(винилокси)бензоат
К раствору 5-хлор-2-(2-хлорэтокси)бензоата (76 г, 0,3 моль) в 600 мл THF добавляли порционно трет-бутоксид калия (40,4 г, 0,36 моль) при 0°C. После перемешивания при этой температуре в течение 1 часа, TLC продемонстрировало полное потребление исходного материала, и смесь выливали в ледяную воду. Водный слой экстрагировали два раза EtOAc (200 мл), и объединенные органические слои упаривали до сухого состояния. Осадок очищали колоночной хроматографией на силикагеле (гексан/EtOAc=50:1~30:1) с получением 22 г (34%) метил-5-хлор-2-(винилокси)бензоата. 1H ЯМР (400 МГц, CDCl3) δ: 7,76 (д, 1H), 7,38 (дд, 1H), 7,00 (д, 1H), 6,52 (дд, 1H), 4,67 (дд, 1H), 4,44 (дд, 1H), 3,83 (с, 3H).
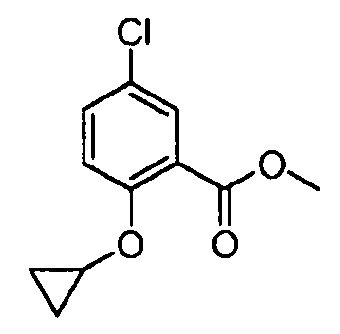
метил-5-хлор-2-циклопропоксибензоат
К раствору метил-5-хлор-2-(винилокси)бензоата в 100 мл дихлорметана добавляли диэтилцинк (1 M раствор в гексане) (200 мл, 0,2 моль) в атмосфере N2. Раствор охлаждали на ледяной бане и в смесь очень медленно капельно добавляли раствор трифторуксусной кислоты (16 мл) в дихлорметане (100 мл). После перемешивания в течение 20 минут капельно добавляли раствор CH2I2 (16,4 мл, 0,2 моль) в дихлорметане (100 мл). После перемешивания в течение дополнительных 20 минут, добавляли раствор метил-5-хлор-2-(винилокси)бензоата (21,3 г, 0,1 моль) в дихлорметане (100 мл) и ледяную баню удаляли. Через 8 часов смесь гасили насыщенным раствором NH4Cl и экстрагировали два раза дихлорметаном. Объединенные органические слои сушили над Na2SO4, фильтровали и упаривали до сухого состояния с получением 21 г (92%) метил-5-хлор-2-циклопропоксибензоата. 1H ЯМР (400 МГц, CDCl3) δ: 7,75 (д, 1H), 7,43 (кв., 1H), 3,86 (с, 3H), 3,80 (м, 1H), 0,85 (м, 4H).

5-хлор-2-циклопропоксибензойная кислота
Метил-5-хлор-2-циклопропоксибензоат растворяли в смеси THF/H2O (1:1, 400 мл), затем добавляли гидроксид натрия (16 г, 0,4 моль). Смесь нагревали до 60°C и перемешивали в течение 1 часа. Реакционную смесь охлаждали до комнатной температуры, затем pH доводили до 4 с использованием 4 Н водного раствора HCl, в результате чего продукт выпадал в осадок. Осадок собирали фильтрацией и подвергали азеотропной перегонке с толуолом с получением 19 г (96%) 5-хлор-2-циклопропоксибензойной кислоты. 1H ЯМР (400 МГц, CDCl3) δ: 12,96 (уш., 1H), 7,58 (м, 2H), 7,43 (кв., 1H), 3,91 (м, 1H), 0,81 (м, 4H).
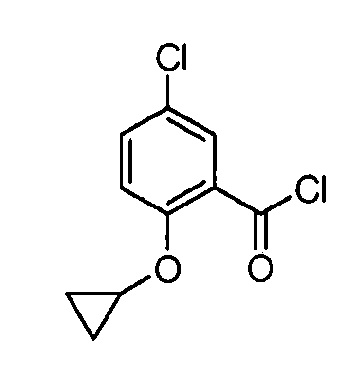
5-хлор-2-циклопропоксибензоилхлорид
5-Хлор-2-циклопропоксибензойную кислоту (18,5 г, 87 ммоль) растворяли в SOCl2 и раствор кипятили с обратным холодильником в течение 4 часов. Реакционную смесь концентрировали в вакууме, затем совместно упаривали с толуолом с получением желаемого хлорангидрида в виде бесцветного масла, который непосредственно использовали на следующей стадии.
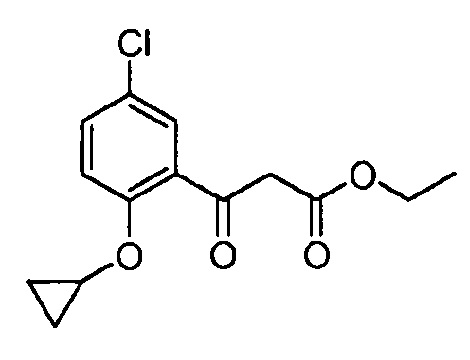
этил-3-(5-хлор-2-циклопропоксифенил)-3-оксопропаноат
В 1000-мл колбе трехгорлой колбе к 3-этокси-3-оксопропаноату калия (31,1 г, 182,7 ммоль) добавляли ацетонитрил (200 мл) в атмосфере N2 при перемешивании. Реакционную смесь охлаждали до 0°C, затем добавляли триэтиламин (38,8 мл, 278,4 ммоль), а затем MgCl2 (20,71 г, 217,5 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение дополнительных 2,5 ч. Полученную суспензию охлаждали до 0°C и капельно добавляли 5-хлор-2-циклопропоксибензоилхлорид (18,5 г, 87 ммоль), а затем добавляли дополнительный триэтиламин (3,9 мл, 28 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, затем концентрировали в вакууме. Добавляли 1 л толуола, смесь охлаждали до 0°C, затем добавляли 125 мл HCl (13% водный раствор). Ледяную баню удаляли, смесь перемешивали в течение 30 минут, а затем слои разделяли, и органические слои промывали водой и упаривали до сухого состояния. Осадок очищали хроматографией на силикагеле (гексан/EtOAc=3:1) с получением 21,8 г этил-3-(5-хлор-2-циклопропоксифенил)-3-оксопропаноата. LCMS (ESI) m+H=283,0; 1H ЯМР (400 МГц, CDCl3) δ: 7,81 (т, 1H), 7,44 (дд, 1H), 7,29 (д, 1H), 4,24 (м, 2H), 3,87 (с, 2H), 3,78 (м, 1H), 1,23 (т, 2H), 0,85 (м, 4H).

этил-2-(5-хлор-2-циклопропоксибензоил)-3-(диметиламино)акрилат
Этил-3-(5-хлор-2-циклопропоксифенил)-3-оксопропаноат (21,8 г, 77 ммоль) растворяли в 150 мл DMF-DMA. Смесь нагревали до температуры кипячения с обратным холодильником в течение 2 часов. Упаривание дало желтое твердое вещество, которое использовали на следующей стадии без дальнейшей очистки. 1H ЯМР (CDCl3, 400 МГц) δ: 7,69 (с, 1H), 7,39 (с, 1H), 7,29 (дд, J=2,4, 8,8 Гц, 1H), 7,15 (д, J=8,8 Гц, 1H), 3,92 (кв., J=6,8 Гц, 2H), 3,69-3,65 (м, 1H), 3,10 (уш., 6H), 0,88 (т, J=6,8 Гц, 3H), 0,77-0,71 (м, 4H).

этил-3-(5-хлор-2-циклопропоксифенил)-1H-пиразол-4-карбоксилат
Этил-2-(5-хлор-2-циклопропоксибензоил)-3-(диметиламино)акрилат (24 г, 71 ммоль) растворяли в 150 мл HOAc. Реакционную смесь охлаждали до 0°C, затем капельно добавляли 85% гидразин в воде (25 мл). Смесь нагревали до комнатной температуры и перемешивали в течение 6 часов, затем концентрировали в вакууме. Осадок очищали смесью EtOAc/гексан=1:2 с получением 24 г указанного в заголовке соединения в виде сиропа. 1H ЯМР (DMSO-d6, 400 МГц) δ: 13,41 (с, 1H), 8,26 (с, 0,5H), 7,88 (с, 0,5H), 7,49-7,24 (м, 3H), 4,07-3,99 (м, 2H), 3,82-3,74 (м, 1H), 1,17-1,10 (м, 3H), 0,73-0,46 (м, 4H).
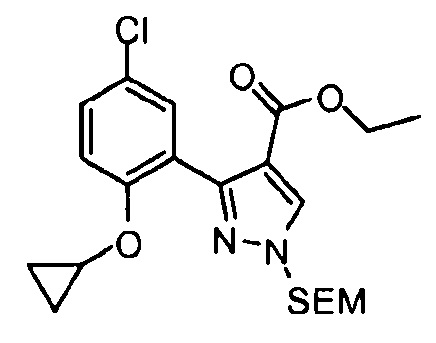

К раствору этил-3-(5-хлор-2-циклопропоксифенил)-1H-пиразол-4-карбоксилата (12 г, 39 ммоль) в THF (200 мл) добавляли 60% гидрид натрия в минеральном масле (1,72 г, 43 ммоль) и реакционную смесь перемешивали в течение 10 мин. Добавляли SEMCl (7,2 г, 43 ммоль) и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь гасили ледяной водой и экстрагировали EtOAc (300 мл×2). Объединенные органические слои сушили над сульфатом натрия и концентрировали в вакууме с получением 16 г указанных в заголовке соединений.

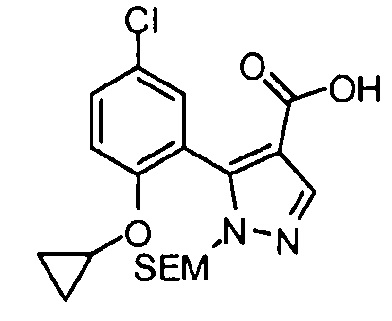
Полученный неочищенный сложный эфир с последней стадии растворяли в 200 мл воды. К раствору добавляли NaOH (7,32 г, 0,18 ммоль) и смесь кипятили с обратным холодильником в течение 6 часов, затем охлаждали до комнатной температуры и нейтрализовывали 4 Н HCl до pH~7. Полученный осадок собирали фильтрацией, затем растворяли в MeOH. Оставшиеся твердые вещества отфильтровывали и удаляли, и фильтрат упаривали до сухого состояния с получением указанных в заголовке соединений (11,9 г) в виде желтого твердого вещества.

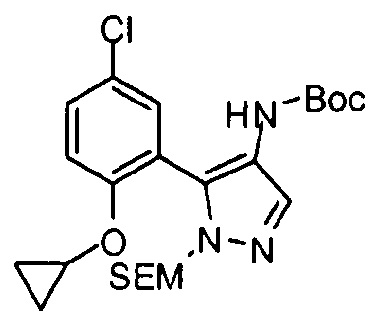
Продукт с предыдущей стадии (8,5 г, 20,8 ммоль) растворяли в толуоле. Добавляли DPPA (5,4 мл, 25 ммоль) и TEA (3,5 мл, 25 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли трет-бутиловый спирт (5,3 мл, 50 ммоль). Смесь нагревали до 90°C и перемешивали в течение ночи. Реакционную смесь концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле, элюируя смесью гексан/EtOAc=10:1 с получением 2,5 г указанных в заголовке соединений (не разделенных), которые использовали непосредственно на следующей стадии.

3-(5-хлор-2-циклопропоксифенил)-1H-пиразол-4-амин
Трет-бутил-3-(5-хлор-2-циклопропоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамат и трет-бутил-5-(5-хлор-2-циклопропоксифенил)-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-4-илкарбамат растворяли в 50 мл MeOH. Капельно добавляли 50 мл 4 M HCl/MeOH. Смесь перемешивали при комнатной температуре в течение ночи. Смесь упаривали до сухого состояния и очищали хроматографией на силикагеле, элюируя EtOAc с получением 0,25 г 3-(5-хлор-2-циклопропоксифенил)-1H-пиразол-4-амина. LCMS (ESI) m+H=249,8. 1H ЯМР (DMSO-d6, 400 МГц) δ: 12,32-12,10 (уш., 1H), 7,41-7,34 (м, 3H), 7,09 (с, 1H), 4,12 (кв., J=5,2 Гц, 2H), 2,94-3,88 (м, 3H), 0,79-0,74 (м, 4H).
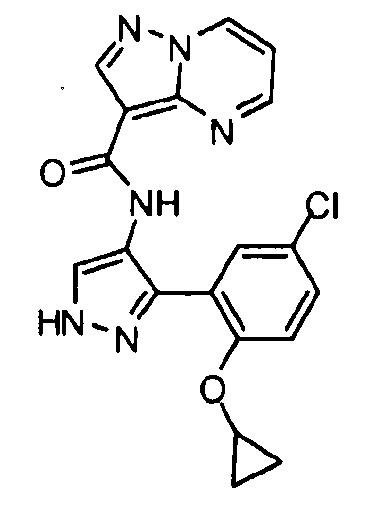
N-(3-(5-хлор-2-циклопропоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
3-(5-Хлор-2-циклопропоксифенил)-1H-пиразол-4-амин (0,2 г, 0,8 ммоль) растворяли в 15 мл безводного THF. Добавляли пиразолo[1,5-a]пиримидин-3-карбонилхлорид (0,17 г, 0,96 ммоль), а затем DIPEA (0,2 г, 1,6 ммоль). Смесь перемешивали при комнатной температуре в течение 2 ч, затем упаривали до сухого состояния. Осадок очищали хроматографией на силикагеле (EtOAc в качестве элюента) с получением 0,3 г указанного в заголовке соединения. Выход: 95%. 1H ЯМР (DMSO-d6, 400 МГц) δ: 12,93 (д, 1H), 9,56 (д, J=5,2 Гц, 1H), 9,34 (д, J=7,2 Гц, 1H), 8,75-8,74 (м, 1H), 8,65 (с, 1H), 8,22 (с, 0,5H), 8,05 (с, 0,5H), 7,55-7,25 (м, 4H), 3,97-3,94 (м, 1H), 0,70-0,44 (м, 4H).
ПРИМЕР 152
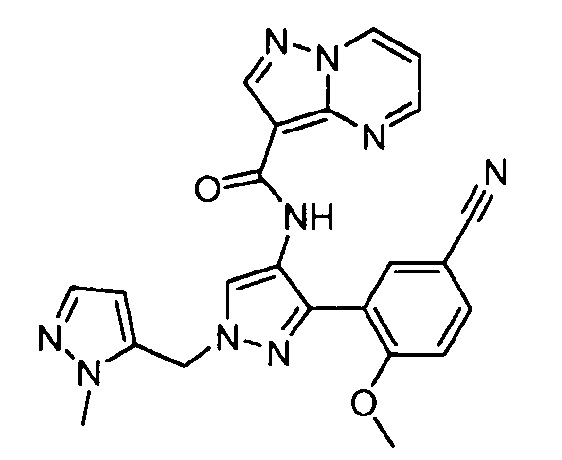
N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-пиразол-5-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
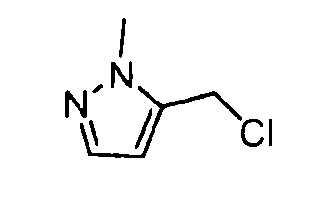
5-(хлорметил)-1-метил-1H-пиразол
К перемешиваемому раствору (1-метил-1H-пиразол-5-ил)метанола (200 мг, 1,8 ммоль) в 20 мл DCM добавляли SOCl2 (262 мг, 2,2 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 2 часов, затем растворитель выпаривали, и осадок совместно упаривали с DCM. Осадок распределяли между DCM и водой. Органическую фазу отделяли и промывали насыщенным водным раствором NaHCO3, водой и рассолом, затем сушили над сульфатом магния, фильтровали и концентрировали с получением 93 мг 5-(хлорметил)-1-метил-1H-пиразола. 1H ЯМР (400 МГц, CDCl3) δ: 7,40 (с, 1H), 6,27 (с, 1H), 4,60 (с, 2H), 3,92 (с, 3H).
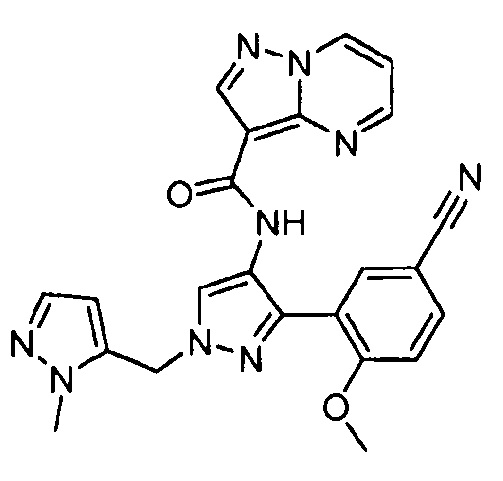
N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-пиразол-5-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(5-циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (144 мг, 0,40 ммоль) в 3 мл DMF добавляли 5-(хлорметил)-1-метил-1H-пиразол (78 мг, 0,6 ммоль) и карбонат цезия (391 мг, 1,20 ммоль). Реакционную смесь перемешивали при 60°C в течение 6 часов, затем охлаждали до комнатной температуры, разбавляли этилацетатом и фильтровали. Фильтрат промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 58,3 мг (32%) N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-пиразол-5-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=454,1; 1H ЯМР (400 МГц, CDCl3) δ: 9,58 (с, 1H), 8,74 (дд, 1H), 8,64 (с, 1H), 8,48 (дд, 1H), 8,21 (с, 1H), 7,81 (с, 1H), 7,69 (дд, 1H), 7,39 (с, 1H), 7,07 (д, 1H), 6,96 (дд, 1H), 6,31 (с, 2H), 5,33 (с, 2H), 3,86 (д, 6H).
ПРИМЕР 153
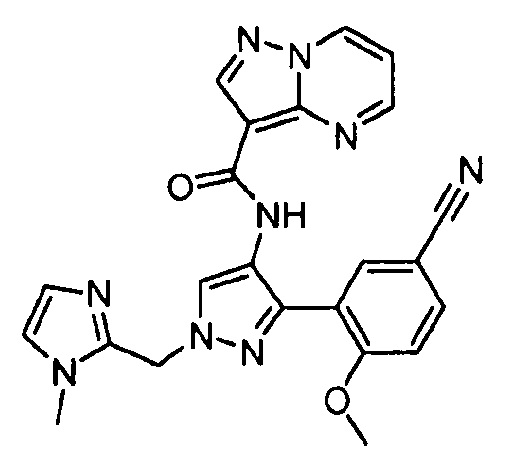
N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-имидазол-2-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(3-(5-циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (200 мг, 0,56 ммоль) в 3 мл DMF добавляли 2-(хлорметил)-1-метил-1H-имидазол (109 мг, 0,84 ммоль) и карбонат цезия (547 мг, 1,68 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем разбавляли этилацетатом и фильтровали. Фильтрат промывали рассолом, сушили над сульфатом магния и концентрировали. Неочищенный продукт очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 82,2 мг (32%) N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-имидазол-2-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=454,1; 1H ЯМР (400 МГц, CDCl3) δ: 9,53 (с, 1H), 8,73 (дд, 1H), 8,64 (с, 1H), 8,48 (дд, 1H), 8,26 (с, 1H), 7,80 (д, 1H), 7,68 (дд, 1H), 7,06 (д, 1H), 6,97 (м, 2H), 6,83 (с, 1H), 5,42 (с, 2H), 3,85 (с, 3H), 3,63 (с, 3H).
ПРИМЕР 154

N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
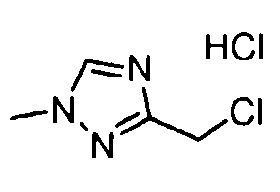
гидрохлорид 3-(хлорметил)-1-метил-1H-1,2,4-триазола
(1-Метил-1H-1,2,4-триазол-3-ил)метанол (0,25 г, 2,2 ммоль) растворяли в 10 мл SOCl2 и кипятили с обратным холодильником в течение 2 часов. Смесь упаривали до сухого состояния и совместно упаривали с толуолом. Полученное белое твердое вещество (0,2 г) использовали на следующей стадии без дальнейшей очистки.

N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
N-(3-(5-Циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (0,15 г, 0,42 ммоль) растворяли в 10 мл DMF, затем добавляли гидрохлорид 3-(хлорметил)-1-метил-1H-1,2,4-триазола (82 мг, 0,63 ммоль) и Cs2CO3 (0,41 г, 1,26 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов, затем фильтровали через целит и очищали обращенно-фазовой ВЭЖХ с получением 44,8 мг (23%) N-(3-(5-циано-2-метоксифенил)-1-((1-метил-1H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=455,1; 1H ЯМР (400 МГц, CDCl3) δ: 9,64 (с, 1H), 9,34 (д, 1H), 8,75 (д, 1H), 8,64 (с, 1H), 8,44 (с, 1H), 8,35 (с, 1H), 7,95 (д, 1H), 7,73 (д, 1H), 7,47 (д, 1H), 7,28 (дд, 1H), 5,39 (с, 2H), 3,89 (с, 3H), 3,84 (с, 3H).
ПРИМЕР 155

N-(3-(5-циано-2-метоксифенил)-1-((2-метил-2H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

гидрохлорид 5-(хлорметил)-1-метил-1H-1,2,4-триазола
(2-Метил-2H-1,2,4-триазол-3-ил)метанол (0,1 г, 0,88 ммоль) растворяли в 10 мл SOCl2 и кипятили с обратным холодильником в течение 2 часов. Смесь упаривали до сухого состояния и совместно упаривали с толуолом. Полученное белое твердое вещество (приблизительно 0,1 г) использовали на следующей стадии без дальнейшей очистки.

N-(3-(5-циано-2-метоксифенил)-1-((2-метил-2H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
N-(3-(5-Циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (0,21 г, 0,58 ммоль) растворяли в 10 мл DMF. Добавляли гидрохлорид 5-(хлорметил)-1-метил-1H-1,2,4-триазола (приблизительно 0,1 г, 0,88 ммоль) и Cs2CO3 (0,57 г, 1,75 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали через целит и очищали обращенно-фазовой ВЭЖХ с получением 60,8 мг (23%) N-(3-(5-циано-2-метоксифенил)-1-((2-метил-2H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пирамидин-3-карбоксамида. LCMS (ESI) m+H=455,1; 1H ЯМР (400 МГц, CDCl3) δ: 9,64 (с, 1H), 9,64 (с, 1H), 9,33 (дд, 1H), 8,74 (дд, 1H), 8,64 (с, 1H), 8,42 (с, 1H), 7,95 (дд, 1H), 7,89 (с, 1H), 7,74 (д, 1H), 7,46 (д, 1H), 7,28 (дд, 1H), 5,65 (с, 2H), 3,90 (с, 3H), 3,88 (с, 3H).
ПРИМЕР 156
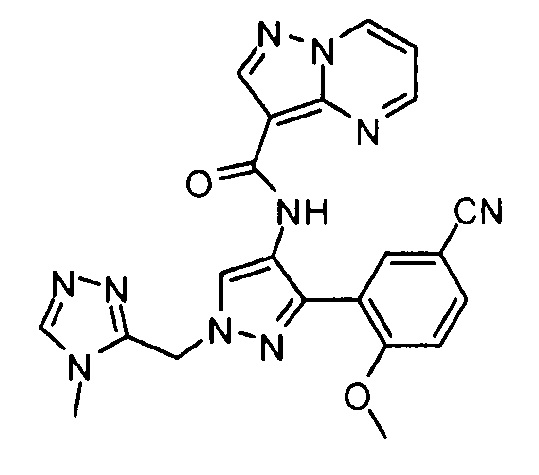
N-(3-(5-циано-2-метоксифенил)-1-((4-метил-4H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

2-гидроксиацетогидразид
К раствору моногидрата гидразина (10,4 г, 0,1 моль) в EtOH (50 мл) капельно добавляли этил-2-гидроксиацетат (6 г, 0,12 моль) при 0°C. Смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь концентрировали в вакууме с получением требуемого продукта, который использовали на следующей стадии без очистки. Выход: 97%. 1H ЯМР (DMSO-d6, 400 МГц) δ: 8,82 (с, 1H), 5,32-5,13 (м, 1H), 4,38-4,09 (м, 2H), 3,80 (с, 2H).

2-(2-гидроксиацетил)-N-метилгидразинкарбимидотиовая кислота
К раствору 2-гидроксиацетогидразида (4,5 г, 50 ммоль) в EtOH капельно добавляли метилизотиоцианат (3,7 г, 50 ммоль) при охлаждении льдом. После завершения добавления реакционную смесь доводили до комнатной температуры, а затем перемешивали в течение 24 ч при 60°C. Затем добавляли ледяную воду и перемешивание продолжали в течение 15 мин. Реакционную смесь концентрировали в вакууме с получением указанного в заголовке соединения. Выход: 100%. LCMS (ESI) m+H=164,0.

(5-меркапто-4-метил-4H-1,2,4-триазол-3-ил)метанол
К раствору 2-(2-гидроксиацетил)-N-метилгидразинкарбимидотиовой кислоты (8,15 г, 50 ммоль) в EtOH добавляли 5 Н NaOH (50 ммоль), а затем смесь перемешивали в течение 4 ч при 60°C. Смесь охлаждали на ледяной бане и pH доводили до ~5-6 концентрированной HCl. Выпавшее в осадок твердое вещество фильтровали, промывали EtOH и сушили в вакууме с получением указанного в заголовке соединения. Выход: 83%. 1H ЯМР (DMSO-d6, 400 МГц) δ: 5,64 (с, 1H), 4,46 (с, 2H), 3,44 (с, 3H).

(4-метил-4H-1,2,4-триазол-3-ил)метанол
NaNO2 (70 мг, 1 ммоль) добавляли к 5 Н HNO3 (10 мл) при комнатной температуре. Реакционную смесь охлаждали на ледяной бане, затем добавляли (5-меркапто-4-метил-4H-1,2,4-триазол-3-ил)метанол (360 мг, 2,5 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение одного часа. Добавляли воду (30 мл), затем полученную смесь нейтрализовывали с использованием твердого K2CO3, пока значение pH не становилось ~7-8. Реакционную смесь концентрировали в вакууме, и осадок растворяли в DCM/MeOH (5:1), фильтровали и концентрировали в вакууме с получением указанного в заголовке соединения. Выход: 92%. 1H ЯМР (DMSO-d6, 400 МГц) δ: 8,38 (с, 1H), 5,57 (с, 1H), 4,56 (д, J=10,8 Гц, 2H), 3,65 (с, 3H).
3-(хлорметил)-4-метил-4H-1,2,4-триазол
Раствор (4-метил-4H-1,2,4-триазол-3-ил)метанола (260 мг, 2,3 ммоль) в SOCl2 (10 мл) кипятили с обратным холодильником в течение 1 часа, а затем концентрировали в вакууме с получением требуемого соединения. Выход: 100%. LCMS (ESI) m+H=132,1.

N-(3-(5-циано-2-метоксифенил)-1-((4-метил-4H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
N-(3-(5-Циано-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид (120 мг, 0,334 ммоль), 3-(хлорметил)-4-метил-4H-1,2,4-триазол (65 мг, 0,5 ммоль) и Cs2CO3 (325 мг, 1 ммоль) суспендировали в DMF. Смесь перемешивали в течение ночи при комнатной температуре, затем фильтровали и концентрировали в вакууме. Осадок очищали препаративной ВЭЖХ с получением N-(3-(5-циано-2-метоксифенил)-1-((4-метил-4H-1,2,4-триазол-3-ил)метил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (23,2 мг, выход: 15%). 1H ЯМР (CDCl3, 400 МГц) δ: 9,59 (с, 1H), 8,77 (дд, J=2,0, 7,2 Гц, 1H), 8,70 (с, 1H), 8,51 (дд, J=2,0, 4,4 Гц, 1H), 8,39 (с, 1H), 8,11 (с, 1H), 7,82 (д, J=2,0 Гц, 1H), 7,73 (дд, J=2,0, 8,4 Гц, 1H), 7,10 (д, J=8,4 Гц, 1H), 6,99 (кв., J=4,4, 6,8 Гц, 1H), 3,91 (с, 3H), 3,72 (с, 3H).
ПРИМЕР 157

N-(3-(3,3-дифторпиперидин-1-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

1-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)пиперидин-3-он
К раствору оксалилхлорида (0,82 г, 6,44 ммоль) и DMSO (1,07 г, 13,75 ммоль) капельно добавляли 1-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)пиперидин-3-ол (2,0 г, 5,85 ммоль) при -78°C. Реакционную смесь перемешивали при этой температуре в течение 15 мин, затем добавляли Et3N (4,1 мл, 29,25 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение дополнительных 90 минут. Реакционную смесь концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (гексан/EtOAc=50:1) с получением 1,9 г (95%) 1-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)пиперидин-3-она. 1H ЯМР (400 МГц, CDCl3) δ: 8,09 (с, 1H), 5,30 (с, 2H), 3,68 (м, 2H), 3,69 (м, 2H), 3,48 (м, 2H), 2,63 (м, 2H), 2,14 (м, 2H), 0,93 (м, 2H), 0 (с, 9H).

3,3-дифтор-1-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)пиперидин
К раствору 1-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)пиперидин-3-она (1,8 г, 5,28 ммоль) в 20 мл этанола добавляли DAST (1,3 г, 7,92 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 часов, затем к смеси добавляли 100 мл воды. Смесь экстрагировали два раза дихлорметаном, и объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 1,4 г (73%) 3,3-дифтор-1-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)пиперидина. 1H ЯМР (400 МГц, CDCl3) δ: 8,08 (с, 1H), 5,41 (м, 2H), 3,70 (м, 6H), 2,11 (м, 2H), 1,92 (с, 2H), 0,94 (м, 2H), 0 (с, 9H).

5-(3,3-дифторпиперидин-1-ил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амин
К раствору 3,3-дифтор-1-(4-нитро-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-5-ил)пиперидина (1,4 г, 3,86 ммоль) в 20 мл этанола добавляли 40 мл воды, хлорид аммония (0,82 мг, 15,44 ммоль) и железный порошок (1,08 г, 19,3 ммоль). Реакционную смесь перемешивали при 70°C в течение 1 часа. После охлаждения до комнатной температуры реакционную смесь разбавляли дихлорметаном и фильтровали через слой целита, промывая дополнительным дихлорметаном. Фильтрат экстрагировали два раза дихлорметаном, затем объединенные органические экстракты сушили над сульфатом магния и концентрировали с получением 1,2 г (93%) 5-(3,3-дифторпиперидин-1-ил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амина. LCMS (ESI) m+H=333,1; 1H ЯМР (400 МГц, CDCl3) δ: 7,09 (с, 1H), 5,38 (м, 2H), 3,74 (м, 6H), 2,01 (с, 1H), 1,96 (с, 2H), 1,52 (м, 2H), 0,92 (м, 2H), 0 (с, 9H).

N-(5-(3,3-дифторпиперидин-1-ил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору 5-(3,3-дифторпиперидин-1-ил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-амина (1,2 г, 3,61 ммоль) в 30 мл THF добавляли DIPEA (1,3 мл, 7,22 ммоль) и пиразолo[1,5-a]пиримидин-3-карбонилхлорид (720 мг, 3,97 ммоль) в THF (50 мл) при 0°C. После завершения добавления смесь нагревали до комнатной температуры, а затем перемешивали в течение ночи. Смесь концентрировали в вакууме, и осадок очищали колоночной хроматографией на силикагеле (гексан:EtOAc=от 3:1 до 1:1) с получением 1,0 г (60%) N-(5-(3,3-дифторпиперидин-1-ил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=478,2; 1H ЯМР (400 МГц, CDCl3) δ: 9,49 (с, 1H), 8,87 (дд, 1H), 8,75 (с, 1H), 8,71 (дд, 1H), 7,94 (с, 1H), 7,10 (дд, 1H), 5,40 (с, 2H), 3,67 (м, 6H), 2,06 (с, 2H), 1,93 (м, 2H), 0,95 (м, 2H), 0 (с, 9H).
N-(3-(3,3-дифторпиперидин-1-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К раствору N-(5-(3,3-дифторпиперидин-1-ил)-1-(2-((триметилсилил)метокси)этил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (1,0 г, 2,1 ммоль) в 50 мл этанола добавляли HCl (1,75 мл 6 M раствора в воде, 10,5 ммоль). Реакционную смесь перемешивали при 70°C в течение 1 часа. После охлаждения до комнатной температуры образовывался светло-желтый осадок, который отфильтровывали и промывали метанолом и диэтиловым эфиром. Затем объем фильтрата уменьшали, и дополнительный твердый продукт выделяли фильтрацией. Объединенные собранные твердые вещества сушили в вакууме с получением 0,77 г (94%) N-(3-(3,3-дифторпиперидин-1-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=348,1; 1H ЯМР (400 МГц, CDCl3) δ: 9,59 (с, 1H), 9,37 (дд, 1H), 8,84 (м, 2H), 8,01 (с, 1H), 7,32 (дд, 1H), 3,74 (с, 1H), 3,33 (м, 2H), 3,12 (м, 2H), 2,10 (м, 2H), 2,00 (м, 2H).
ПРИМЕР 158

N-(3-(5-циано-2-метоксипиридин-3-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
6-гидрокси-5-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)никотинoнитрил
Указанное в заголовке соединение получали способами, описанными для N-(5-(5-хлор-2-метоксифенил)-1-изопентил-4-нитро-1H-пиразола.

6-метокси-5-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)никотинoнитрил
К охлажденному льдом раствору 6-метокси-5-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)никотинoнитрила (57 мг, 0,16 ммоль, 1 экв.) в 1:1 растворе метанол/толуол (3 мл) добавляли триметилсилилдиазометан (0,8 мл, 1,6 ммоль, 2,0 M раствор в диэтиловом эфире). Через 50 мин реакционную смесь нагревали до 24°C и концентрировали в вакууме. Очистка колоночной флэш-хроматографией (1:1 гептан/этилацетат) дала продукт (3,3 мг, 5,6%). 1H ЯМР (400 МГц, CDCl3) δ: 8,63 (д, J=2,2 Гц, 1H), 8,21 (с, 1 H), 7,96 (д, J=2,2 Гц, 1 H), 5,27 (м, 2H), 3,97 (с, 3H), 3,63 (м, 2H), 0,86 (м, 2H), -0,02 (с, 9H).

5-(4-амино-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)-6-метоксиникотинoнитрил
Раствор 6-метокси-5-(4-нитро-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)никотинoнитрила (11 мг, 0,029 ммоль, 1 экв.) в метаноле (4 мл) циркулировали через реактор для гидрогенизации с непрерывным потоком H-Cube® (ThalesNano), оборудованный кассетой с катализатором в виде палладия на углероде при 30°C. Полученный раствор концентрировали в вакууме с получением продукта (7,1 мг, выход неочищенного вещества 70%). 1H ЯМР (400 МГц, CDCl3) δ: 8,52 (д, J=2,2 Гц, 1H), 8,09 (д, J=2,2 Гц, 1H), 5,21 (с, 2H), 4,06 (с, 3H), 3,58 (м, 2H), 0,85 (м, 2H), -0,04 (с, 9H).

N-(3-(5-циано-2-метоксипиридин-3-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
К суспензии пиразолo[1,5-a]пиримидин-3-карбоновой кислоты (22,0 мг, 0,135 ммоль, 6,56 экв.), 5-(4-амино-1-((2-(триметилсилил)этокси)метил)-1H-пиразол-5-ил)-6-метоксиникотинoнитрила (7,1 мг, 0,020 ммоль, 1 экв.) и 2-хлор-2,4-диметокси-1,3,5-триазина (23,7 мг, 0,135 экв., 6,56 экв.) в ацетонитриле (2 мл) добавляли 4-метилморфолин (23 мкл, 0,21 ммоль, 10 экв.) при 24°C. Через 2 суток реакционную смесь распределяли между насыщенным водным раствором бикарбоната натрия (2 мл), насыщенным водным раствором хлорида натрия (2 мл) и этилацетатом (5 мл). Органический слой отделяли, и водный слой экстрагировали этилацетатом (2×5 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный материал растворяли в этаноле (3 мл) и 6 Н водном растворе хлористоводородной кислоты (1 мл) и нагревали до 50°C. Через 5 ч реакционную смесь концентрировали в вакууме и очищали препаративной ВЭЖХ с получением белого твердого вещества (1,1 мг, выход 15%). 1H ЯМР (400 МГц, CDCl3) δ: 9,72 (с, 1H), 8,80 (дд, J=7,0, 1,6 Гц, 1H), 8,74 (с, 1H), 8,66 (м, 1H), 8,54 (д, J=2,2 Гц, 1H), 8,47 (с, 1H), 8,22 (д, J=1,9 Гц, 1H), 7,04 (дд, J=7,0, 4,2 Гц, 1H), 4,11 (с, 3H). LCMS (ESI): M+H=361,1.
ПРИМЕР 159

N-(1-(азетидин-3-ил)-3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид
Суспензию N-(3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамида (50,0 мг, 0,136 ммоль, 1 экв.), трет-бутил-3-йодазетидин-1-карбоксилата (230,3 мг, 0,813 ммоль, 6,00 экв.) и карбоната цезия (177 мг, 0,542 ммоль, 4,00 экв.) в N,N-диметилформамиде (1 мл) нагревали при 50°C. Через 5 ч реакционную смесь распределяли между насыщенным водным раствором хлорида натрия (5 мл) и этилацетатом (5 мл). Водный слой экстрагировали этилацетатом (2×5 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Неочищенный осадок растворяли в дихлорметане (2 мл) и трифторуксусной кислоте (2 мл) при 24°C. Через 5 ч реакционную смесь концентрировали в вакууме и очищали препаративной ВЭЖХ с получением продукта (12,7 мг, выход 22%). 1H ЯМР (400 МГц, DMSO-d6) δ: 9,75 (с, 1H), 9,36 (м, 1H), 8,79 (м, 1H), 8,67 (с, 1H), 8,46 (с, 1H), 7,53-7,56 (м, 2H), 7,29-7,34 (м, 2H), 5,45 (м, 1H), 4,33 (м, 2H), 4,23 (м, 2H), 3,86 (с, 3H). LCMS (ESI): M+H=424,1.
ПРИМЕР 160

N-(3-(5-хлор-2-метоксифенил)-1-(1-(оксетан-3-иламино)-1-оксопропан-2-ил)-1H-пиразол-4-ил)пиразолo[1,5-a]пиримидин-3-карбоксамид

трет-бутил-2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)пропаноат
Суспензию N-(3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (1,0 г, 2,7 ммоль, 1 экв.), трет-бутил-2-бромпропаноата (0,54 мл, 3,2 ммоль, 1,2 экв.) и карбоната цезия (1,1 г, 3,2 ммоль, 1,2 экв.) в N,N-диметилформамиде (15 мл) нагревали при 75°C в течение 3 ч. Реакционную смесь концентрировали, и полученный осадок распределяли между полунасыщенным водным раствором хлорида натрия (50 мл) и этилацетатом (50 мл). Водный слой экстрагировали этилацетатом (2×50 мл). Собранные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очистка колоночной флэш-хроматографией (90% этилацетат в гептане) дала продукт (0,945 г, 70%). LCMS (ESI): M+H=497,2.
2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)пропионовая кислота
К раствору трет-бутил-2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)пропаноата (0,945 г, 1,90 ммоль, 1 экв.) в дихлорметане (10 мл) капельно добавляли трифторуксусную кислоту (10 мл). Через 2 ч реакционную смесь концентрировали в вакууме с получением неочищенного продукта (количественный), который использовали без дальнейшей очистки. LCMS (ESI): M+H=441,1.

N-(3-(5-хлор-2-метоксифенил)-1-(1-(оксетан-3-иламино)-1-оксопропан-2-ил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Раствор 2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)пропионовой кислоты (66,0 мг, 0,150 ммоль, 1 экв.), гидрохлорида оксетан-3-амина (41,0 мг, 0,374 ммоль, 2,50 экв.), гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (114 мг, 0,299 ммоль, 2,00 экв.) и N,N-диизопропилэтиламина (522 мкл, 2,99 ммоль, 20,0 экв.) в N,N-диметилформамиде (1,0 мл) нагревали при 50°C. Через 6 ч реакционную смесь концентрировали, и полученный осадок распределяли между насыщенным водным раствором бикарбоната натрия (10 мл) и этилацетатом (10 мл). Водный слой экстрагировали этилацетатом (2×5 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очистка препаративной ВЭЖХ дала продукт (39 мг, выход 51%). 1H ЯМР (400 МГц, DMSO-d6) δ: 9,70 (с, 1H), 9,33 (м, 1H), 9,02 (д, J=6,5 Гц, 1H), 8,77 (дд, J=4,2, 1,5 Гц, 1H), 8,65 (с, 1H), 8,36 (с, 1H), 7,50 (дд, J=8,9, 2,7 Гц, 1H), 7,38 (д, J=2,6 Гц, 1H), 7,27-7,31 (м, 2H), 5,06 (кв., J=7,1 Гц, 1H), 4,80 (м, 1H), 4,73 (м, 2H), 4,44 (м, 2H), 3,84 (с, 3H), 1,66 (д, J=7,1 Гц, 3H). LCMS (ESI): M+H=496,1.
ПРИМЕР 161

N-(1-(1-(азетидин-1-ил)-2-метил-1-оксопропан-2-ил)-3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид

этил-2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-2-метилпропаноат
Суспензию N-(3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (1,0 г, 2,7 ммоль, 1 экв.), этил-2-бромизобутирата (0,597 мл, 4,07 ммоль, 1,50 экв.) и карбоната цезия (1,77 г, 5,42 ммоль, 2,00 экв.) в N,N-диметилформамиде (15 мл) нагревали при 50°C. Через 5 ч реакционную смесь концентрировали в вакууме, и полученный осадок распределяли между насыщенным водным раствором хлорида натрия (30 мл) и этилацетатом (20 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенного продукта (1,13 г, выход 84%). LCMS (ESI): M+H=483,2.

2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-2-метилпропионовая кислота
К раствору этил-2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-2-метилпропаноата (0,918 г, 1,90 ммоль, 1 экв.) добавляли 2 Н водный раствор гидроксида натрия (4 мл). Через 18 ч добавляли дополнительный 2 Н водный раствор гидроксида натрия (4 мл). Через 5 ч реакционную смесь концентрировали в вакууме, и полученный осадок растворяли в воде (15 мл). Водный раствор подкисляли 6 Н водным раствором хлористоводородной кислоты до pH=2. Полученный водный раствор экстрагировали этилацетатом (3×30 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением неочищенной кислоты (количественный). LCMS (ESI): M+H=455,1.

N-(1-(1-(азетидин-1-ил)-2-метил-1-оксопропан-2-ил)-3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Раствор 2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-2-метилпропионовой кислоты (0,050 г, 0,11 ммоль, 1 экв.), азетидина (18,5 мкл, 0,275 ммоль, 2,50 экв.), гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (84 мг, 0,20 ммоль, 2,00 экв.) и N,N-диизопропилэтиламина (380 мкл, 2,2 ммоль, 20 экв.) в N,N-диметилформамиде (1 мл) нагревали при 50°C. Через 6 ч реакционную смесь концентрировали, и полученный осадок распределяли между насыщенным водным раствором бикарбоната натрия (5 мл) и этилацетатом (5 мл). Водный слой экстрагировали этилацетатом (2×5 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очистка препаративной ВЭЖХ дала продукт (32 мг, выход 60%). 1H ЯМР (400 МГц, DMSO-d6) δ: 9,70 (с, 1H), 9,34 (м, 1H), 8,77 (дд, J=4,2, 1,5 Гц, 1H), 8,64 (с, 1H), 8,36 (с, 1H), 7,52 (дд, J=8,9, 2,7 Гц, 1H), 7,41 (д, J=2,6 Гц, 1H), 7,28-7,32 (м, 2H), 3,88 (с, 2H), 3,86 (с, 3H), 3,41 (м, 2H), 2,04 (м, 2H), 1,72 (с, 6H). LCMS (ESI): M+H=494,1.
ПРИМЕР 162

N-(3-(5-хлор-2-метоксифенил)-1-(2-(тетрагидрофуран-3-иламино)этил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид

N-(3-(5-хлор-2-метоксифенил)-1-(2-хлорэтил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Суспензию N-(3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (1,0 г, 2,7 ммоль, 1 экв.), 2-хлорэтил-пара-толуолсульфоната (745 мкл, 4,11 ммоль, 1,5 экв.) и карбоната цезия (2,16 г, 6,62 ммоль, 2,4 экв.) в N,N-диметилформамиде (10 мл) нагревали при 50°C. Через 3 ч реакционную смесь концентрировали в вакууме, и полученный осадок распределяли между этилацетатом (30 мл) и полунасыщенным водным раствором хлорида натрия (30 мл). Водный слой экстрагировали этилацетатом (2×20 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очистка колоночной флэш-хроматографией (этилацетат) дала продукт (950 мг, выход 77%). LCMS (ESI): M+H=432,1.

N-(3-(5-хлор-2-метоксифенил)-1-(2-(тетрагидрофуран-3-иламино)этил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Раствор N-(3-(5-хлор-2-метоксифенил)-1-(2-хлорэтил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (0,040 г, 0,090 ммоль, 1 экв.), гидрохлорида 3-аминотетрагидрофурана (29 мг, 0,23 ммоль, 2,50 экв.) и N,N-диизопропилэтиламина (73 мкл, 0,42 ммоль, 4,5 экв.) в N-метилпирролидиноне (1 мл) нагревали при 100°C. Через 18 ч реакционную смесь распределяли между этилацетатом (10 мл) и полунасыщенным водным раствором хлорида натрия (10 мл). Водный слой экстрагировали этилацетатом (2×10 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очистка препаративной ВЭЖХ дала продукт (13,3 мг, выход 30%). 1H ЯМР (400 МГц, DMSO-d6) δ: 9,67 (с, 1H), 9,32 (дд, J=7,0, 1,5 Гц, 1H), 8,77 (дд, J=4,2, 1,6 Гц, 1H), 8,65 (с, 1H), 8,31 (с, 1H), 7,49 (дд, J=8,9, 2,7 Гц, 1H), 7,40 (д, J=2,7 Гц, 1H), 7,27-7,30 (м, 2H), 4,21 (т, J=6,4 Гц, 2H), 3,85 (с, 3H), 3,60-3,76 (м, 4H), 3,38 (м, 2H), 2,96 (м, 2H), 1,96 (м, 1H), 1,62 (м, H). LCMS (ESI): M+H=482,2.
ПРИМЕР 163

N-(3-(5-хлор-2-метоксифенил)-1-(2-(циклопропанкарбоксамидо)этил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
К раствору N-(1-(2-аминоэтил)-3-(5-хлор-2-метоксифенил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (0,030 г, 0,073 ммоль, 1 экв.) и N,N-диизопропилэтиламина (51 мкл, 0,29 ммоль, 4,0 экв.) в дихлорметане (1 мл) капельно добавляли циклопропанкарбонилхлорид (0,020 мл, 0,22 ммоль, 3,0 экв.) при 24°C. Через 1 ч реакционную смесь концентрировали в вакууме. Очистка препаративной ВЭЖХ дала продукт (16,5 мг, выход 47%). 1H ЯМР (400 МГц, DMSO-d6) δ: 9,69 (с, 1H), 9,33 (дд, J=7,0, 1,6 Гц, 1H), 8,78 (дд, J=4,1, 1,5 Гц, 1H), 8,66 (с, 1H), 8,28 (с, 1H), 8,22 (т, J=5,5 Гц, 1H), 7,50 (дд, J=8,8, 2,8 Гц, 1H), 7,44 (д, J=2,7 Гц, 1H), 7,27-7,31 (м, 2H), 4,22 (т, J=6,1 Гц, 2H), 3,85 (с, 3H), 3,52 (кв., J=6,0 Гц, 2H), 1,55 (м, 1H), 0,62-0,71 (м, 4 H). LCMS (ESI): M+H=480,2.
ПРИМЕР 164

(S)-N-(3-(5-хлор-2-метоксифенил)-1-(2-(2-гидроксипропиламино)-2-оксоэтил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Раствор 2-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)уксусной кислоты (51 мг, 0,12 ммоль, 1 экв.), (2S)-1-аминопропан-2-ола (24 мкл, 0,30 ммоль, 2,5 экв.), триэтиламина (20 мкл) и гексафторфосфата N,N,N',N'-тетраметил-O-(7-азабензотриазол-1-ил)урония (91 мг, 0,24 ммоль, 2,0 экв.) в N,N-диметилформамиде (1,2 мл) нагревали при 80°C. Через 6 ч реакционную смесь концентрировали в вакууме, и полученный осадок распределяли между насыщенным водным раствором бикарбоната натрия (5 мл) и этилацетатом (5 мл). Водный слой экстрагировали этилацетатом (2×5 мл). Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали. Очистка препаративной ВЭЖХ дала продукт (17 мг, выход 29%). 1H ЯМР (400 МГц, DMSO-d6) δ: 9,70 (с, 1H), 9,35 (дд, J=7,0, 1,5 Гц, 1H), 8,70 (дд, J=4,2, 1,5 Гц, 1H), 8,66 (с, 1H), 8,32 (с, 1H), 8,11 (т, J=5,8 Гц, 1H), 7,50 (дд, J=8,9, 2,7 Гц, 1H), 7,38 (д, J=2,7 Гц, 1H), 7,27-7,32 (м, 2H), 4,88 (с, 2H), 4,74 (д, J=4,7 Гц, 1H), 3,85 (с, 3H), 3,68 (м, 1H), 3,06 (м, 2H), 1,04 (д, J=6,2 Гц, 3H). LCMS (ESI): M+H=484,2.
ПРИМЕР 165
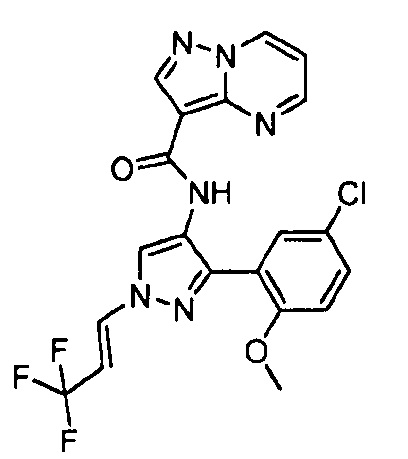
(E)-N-(3-(5-хлор-2-метоксифенил)-1-(3,3,3-трифторпроп-1-енил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
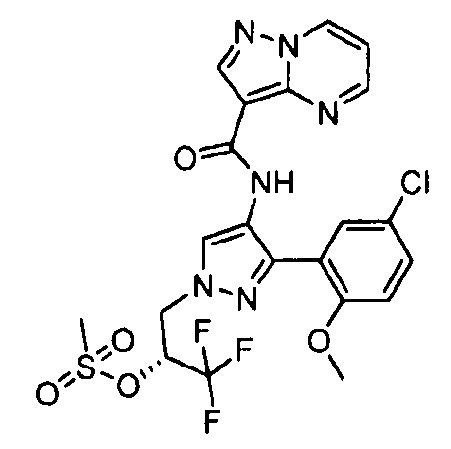
(R)-3-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-1,1,1-трифторпропан-2-ил метансульфонат
В атмосфере азота к раствору (R)-N-(3-(5-хлор-2-метоксифенил)-1-(3,3,3-трифтор-2-гидроксипропил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (188,7 мг, 0,3724 ммоль, смесь 88:12 региоизомеров пиразола при определении с помощью 1H ЯМР) в безводном дихлорметане (3,5 мл, 0,1 M) добавляли мезилхлорид (61 мкл, 0,78 ммоль), а затем дегазированный триэтиламин (164 мкл, 1,18 ммоль). После перемешивания в течение 16,5 ч реакционную смесь разбавляли дихлорметаном и промывали насыщенным водным раствором хлорида аммония. Органический слой сушили над сульфатом магния. Концентрирование в вакууме дало осадок, очистка которого колоночной флэш-хроматографией (дихлорметан/метанол, 100:0-96:4) дала указанное в заголовке соединение в виде желтого твердого вещества (198,1 мг, 90%); RF=0,28 (CH2Cl2:MeOH, 95:5); Основной региоизомер: 1H ЯМР (400 МГц, CDCl3) δ 9,75 (с, 1H), 8,79 (дд, J=7,1, 1,3 Гц, 1H), 8,72 (с, 1H), 8,54 (дд, J=4,0, 1,4 Гц, 1H), 8,41 (с, 1H), 7,55 (д, J=2,6 Гц, 1H), 7,41 (дд, J=8,7, 2,6 Гц, 1H), 7,03-6,98 (м, 2H), 5,56 (м, 1H), 4,67 (дд, J=14,7, 2,4 Гц, 1H), 4,43 (дд, J=14,6, 10,2 Гц, 1H), 3,84 (с, 3H), 2,76 (с, 3H).
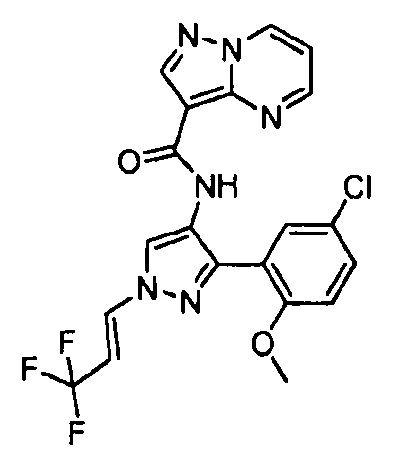
(E)-N-(3-(5-хлор-2-метоксифенил)-1-(3,3,3-трифторпроп-1-енил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Смесь (R)-3-(3-(5-хлор-2-метоксифенил)-4-(пиразоло[1,5-a]пиримидин-3-карбоксамидо)-1H-пиразол-1-ил)-1,1,1-трифторпропан-2-илметансульфоната (35,4 мг, 0,0633 ммоль), карбоната цезия (310 мг, 0,950 ммоль) и гидрохлорида диметиламина (155 мг, 1,90 ммоль) в N,N-диметилформамиде (1 мл, 0,06 M) подвергали облучению микроволновым излучением (180°C) в течение 45 мин. После разбавления дихлорметаном и фильтрации твердых веществ органические вещества концентрировали до сухого состояния. Очистка колоночной флэш-хроматографией (дихлорметан:метанол, 100:0-95:5) дала заданное соединение в виде желтого твердого вещества (18,8 мг, 58%). Нежелательный региоизомер удаляли с помощью ОФ-ВЭЖХ, после чего оставалось 8,0 мг продукта; RF=0,36 (CH2Cl2:iPrOH, 90:10); LCMS (ESI) m+H=463,1; 1H ЯМР (400 МГц, CDCl3) δ 9,82 (с, 1H), 8,80 (дд, J=7,0, 1,6 Гц, 1H), 8,74 (с, 1H), 8,56 (с, 1H), 8,52 (дд, J=4,1, 1,6 Гц, 1H), 7,58 (д, J=2,6 Гц, 1H), 7,52 (дд, J=14,0, 1,9 Гц, 1H), 7,45 (дд, J=8,8, 2,6 Гц, 1H), 7,07-6,95 (м, 2H), 6,24 (м, 1H), 3,85 (с, 3H).
ПРИМЕР 166

N-(4-(5-хлор-2-метоксифенил)-2-(2-гидрокси-2-метилпропил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
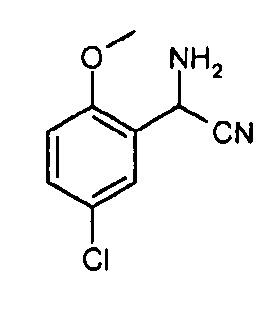
2-амино-2-(5-хлор-2-метоксифенил)ацетонитрил
К перемешиваемому раствору цианида натрия (0,90 г) и хлорида аммония (1,50 г) при 0°C в водном растворе гидроксида аммония (33% раствор, 30 мл) добавляли раствор 5-хлор-2-метокси-бензальдегида (полученный согласно патенту US 4602035) (2,04 г, 12,0 ммоль) в метаноле (80 мл), а затем нагревали до комнатной температуры в течение 18 часов. Смесь упаривали до сухого состояния, осадок распределяли между DCM и водой, органические слои отделяли, промывали рассолом и упаривали в вакууме с получением 2,21 г (94%) 2-амино-2-(5-хлор-2-метоксифенил)ацетонитрила в виде оранжевого масла. LCMS (ESI) m+H=197,2.

3-(трет-бутилдиметилсилилокси)-N-((5-хлор-2-метоксифенил)(циано)метил)-3-метилбутанамид
К раствору 3-(трет-бутилдиметилсиланилокси)-3-метилмасляной кислоты (полученному согласно патенту EP2025667) (590 мг, 2,54 ммоль), диизопропилэтиламина (0,87 мл, 5,08 ммоль) и HATU (966 мг, 2,54 ммоль) в DMF (5 мл) добавляли 2-амино-2-(5-хлор-2-метоксифенил)ацетонитрил (500 мг, 2,54 ммоль) в DMF (2 мл) и перемешивали в течение 18 часов. Добавляли этилацетат, органические слои промывали гидрокарбонатом натрия (насыщенный водный раствор) и рассолом, а затем упаривали до сухого состояния. Осадок очищали флэш-хроматографией на силикагеле (0-5% этилацетат в дихлорметане) с получением 740 мг (73%) 3-(трет-бутилдиметилсилилокси)-N-((5-хлор-2-метоксифенил)(циано)метил)-3-метилбутанамида. LCMS (ESI) m+H=411,4.

N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-(трет-бутилдиметилсилилокси)-3-метилбутанамид
К раствору 3-(трет-бутилдиметилсилилокси)-N-((5-хлор-2-метоксифенил)(циано)метил)-3-метилбутанамида (740 мг, 1,80 ммоль) и карбоната калия (609 мг, 4,41 ммоль) в DMSO (4 мл) добавляли пероксид водорода (50% водн., 413 мкл) и перемешивали в течение 18 часов. Добавляли этилацетат, органические слои промывали водой и рассолом, а затем упаривали до сухого состояния с получением 667 мг (86%) N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-(трет-бутилдиметилсилилокси)-3-метилбутанамида в виде белого твердого вещества. LCMS (ESI) m+H=429,3.

2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-амин
Смесь N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-(трет-бутилдиметилсилилокси)-3-метилбутанамида (327 мг, 0,76 ммоль), реагента Лоуссона (308 мг, 0,76 ммоль) и пиридина (2,5 мл) нагревали до 95°C в течение 18 часов. Добавляли DCM, органические слои промывали гидрокарбонатом натрия (насыщенный водный раствор) и рассолом, а затем упаривали до сухого состояния. Осадок очищали флэш-хроматографией на силикагеле (DCM) с получением 107 мг (33%) 2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-амина. LCMS (ESI) m+H=427,3.

N-(2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Раствор пиразоло[1,5-a]пиримидин-3-карбонилхлорида (52,0 мг, 0,29 ммоль), 2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-амина (103 мг, 0,24 ммоль) и пиридина (2 мл) перемешивали при 60°C в течение 2 часов, а затем при комнатной температуре в течение дополнительных 72 часов. Добавляли DCM, органические слои промывали водой, гидрокарбонатом натрия (насыщенный водный раствор) и рассолом, а затем упаривали до сухого состояния. Осадок очищали флэш-хроматографией на силикагеле (0-20% этилацетат в дихлорметане) с получением 90,6 мг (66%) N-(2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида в виде желтого твердого вещества. LCMS (ESI) m+H=572,1; 1H ЯМР (400 МГц, DMSO-d6): δ 10,45 (с, уш., 1H); 9,38 (дд, 1H); 8,76 (с, 1H); 8,73-8,72 (м, 1H); 7,51 (дд, 1H); 7,45 (д, 1H); 7,35 (д, 1H); 7,31 (д, 1H); 3,79 (с, 3H); 3,06 (с, 2H); 1,32 (с, 6H); 0,90 (с, 9H); 0,11 (с, 6H).
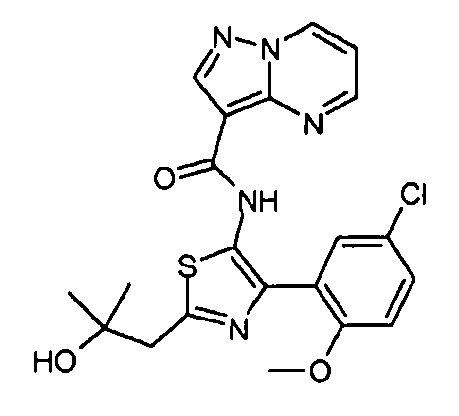
N-(4-(5-хлор-2-метоксифенил)-2-(2-гидрокси-2-метилпропил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
Раствор N-(2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида (20 мг, 35,0 мкмоль) в трифторуксусной кислоте (3 мл) перемешивали в течение 7 суток при комнатной температуре. Реакционную смесь упаривали в вакууме, и неочищенный продукт очищали обращенно-фазовой ВЭЖХ, а затем лиофилизировали с получением 11,3 мг (71%) N-(4-(5-хлор-2-метоксифенил)-2-(2-гидрокси-2-метилпропил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида в виде белого твердого вещества. LCMS (ESI) m+H=458,0; 1H ЯМР (400 МГц, DMSO-d6): δ 10,43 (с, уш., 1H); 9,38 (дд, 1H); 8,74 (дд, 1H); 8,73 (с, 1H); 7,51 (дд, 1H); 7,46 (д, 1H); 7,35 (с, 1H); 7,32-7,31 (м, 1H); 4,76 (с, 1H); 3,79 (с, 3H); 3,01 (с, 2H); 1,19 (с, 6H).
ПРИМЕР 167
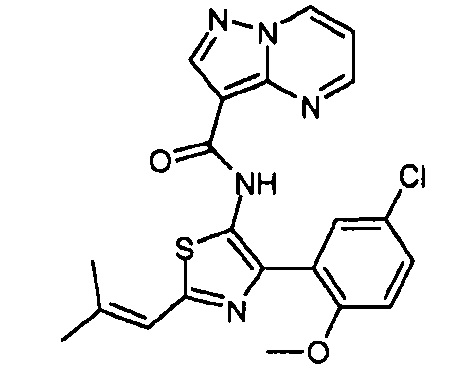
N-(4-(5-хлор-2-метоксифенил)-2-(2-метилпроп-1-енил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид

N-((5-хлор-2-метоксифенил)(циано)метил)-3-гидрокси-3-метилбутанамид
С использованием амино-(5-хлор-2-метокси-фенил)ацетонитрила и 3-гидрокси-3-метилмасляной кислоты указанное в заголовке соединение синтезировали согласно способам синтеза, описанным для 3-(трет-бутилдиметилсилилокси)-N-((5-хлор-2-метоксифенил)(циано)метил)-3-метилбутанамида, с получением N-((5-хлор-2-метоксифенил)(циано)метил)-3-гидрокси-3-метилбутанамида. LCMS (ESI) m+H=297,1.

N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-гидрокси-3-метилбутанамид
С использованием N-((5-хлор-2-метоксифенил)(циано)метил)-3-гидрокси-3-метилбутанамида указанное в заголовке соединение синтезировали способами синтеза, описанными для N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-(трет-бутилдиметилсилилокси)-3-метилбутанамида, с получением N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-гидрокси-3-метилбутанамида в виде желтой камеди. LCMS (ESI) m+Na=337,4.
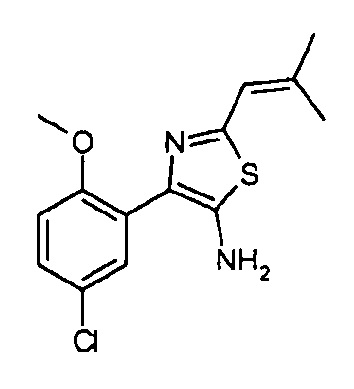
4-(5-хлор-2-метоксифенил)-2-(2-метилпроп-1-енил)тиазол-5-амин
Смесь N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-гидрокси-3-метилбутанамида (240 мг, 0,762 ммоль), реагента Лоуссона (308 мг, 0,76 ммоль) и пиридина (2,5 мл) нагревали до 100°C в течение 18 часов. После охлаждения добавляли DCM и смесь промывали водой, гидрокарбонатом натрия (насыщенный водный раствор) и рассолом и концентрировали до сухого состояния. 4-(5-Хлор-2-метоксифенил)-2-(2-метилпроп-1-енил)тиазол-5-амин (51,0 мг) выделяли в виде светло-желтого твердого вещества. LCMS (ESI) m+H=295,3; 1H ЯМР (400 МГц, DMSO-d6): δ 7,38 (д, 1H); 7,31 (дд, 1H); 7,09 (д, 1H); 6,24 (т, 1H); 5,50 (с, уш., 2H); 3,83 (с, 3H); 2,02 (д, 3H); 1,89 (с, 3H).

N-(4-(5-хлор-2-метоксифенил)-2-(2-метилпроп-1-енил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
С использованием 4-(5-хлор-2-метоксифенил)-2-(2-метилпроп-1-енил)тиазол-5-амина и пиразоло[1,5-a]пиримидин-3-карбонилхлорида указанное в заголовке соединение синтезировали способами синтеза, описанными для N-(2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида, с дальнейшей очисткой обращенно-фазовой ВЭЖХ и лиофилизировали с получением N-(4-(5-хлор-2-метоксифенил)-2-(2-метилпроп-1-енил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=440,0; 1H ЯМР (400 МГц, DMSO-d6): δ 10,50 (с, уш., 1H); 9,39 (дд, 1H); 8,74 (д, 1H); 8,73 (с, 1H); 7,54 (дд, 1H); 7,49 (д, 1H); 7,37 (д, 1H); 7,34 (дд, 1H); 6,47 (т, 1H); 3,80 (с, 3H); 2,16 (с, 3H); 1,97 (с, 3H).
ПРИМЕР 168

N-(2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид

этил-3-(5-хлор-2-метоксифенил)-3-оксопропаноат
К перемешиваемому раствору 1-(5-хлор-2-метоксифенил)этанона (10,0 г, 54,2 ммоль) в THF (100 мл) при 0°C порционно добавляли гидрид натрия (60% дисперсия в минеральном масле, 2,17 г, 54,2 ммоль). Затем смесь перемешивали в течение 10 минут, после чего добавляли диэтилкарбонат (7,68 г, 65,0 ммоль), а затем в течение дополнительного 1 часа. Смесь нагревали до комнатной температуры в течение 2 часов, а затем нагревали до 65°C в течение 2 часов. Добавляли диэтиловый эфир, органические слои промывали водой и рассолом, а затем упаривали до сухого состояния. Осадок очищали флэш-хроматографией на силикагеле (50-100% дихлорметан в циклогексане) с получением 3,41 г этил-3-(5-хлор-2-метоксифенил)-3-оксопропаноата. LCMS (ESI) m+H=257,2; 1H ЯМР (400 МГц, CDCl3): δ 7,59 (д, 1H); 7,38 (дд, 1H); 6,89 (д, 1H); 4,18 (кв., 2H); 3,95 (с, 2H); 3,88 (с, 3H); 1,24 (т, 3H).

этил-2-бром-3-(5-хлор-2-метоксифенил)-3-оксопропаноат
К раствору этил-3-(5-хлор-2-метоксифенил)-3-оксопропаноата (3,39 г, 13,2 ммоль) в диоксане (25 мл) добавляли бром (0,70 мл, 13,6 ммоль) и перемешивали в течение 1 часа. Реакционную смесь выливали в ледяную воду, экстрагировали этилацетатом, органические слои промывали водой и рассолом и упаривали до сухого состояния с получением этил-2-бром-3-(5-хлор-2-метоксифенил)-3-оксопропаноата. LCMS (ESI) m+H=337,2.

этил-2-амино-4-(5-хлор-2-метоксифенил)тиазол-5-карбоксилат
Смесь этил-2-бром-3-(5-хлор-2-метоксифенил)-3-оксопропаноата (согласно расчетам 13,2 ммоль) и тиомочевины (1,01 г, 13,3 ммоль) в этаноле (25 мл) нагревали до температуры кипячения с обратным холодильником в течение 3 часов, затем охлаждали до комнатной температуры в течение 18 часов. Полученное твердое вещество удаляли фильтрацией, и фильтрат упаривали в вакууме. К осадку добавляли DCM, органические слои промывали гидрокарбонатом натрия (насыщенный водный раствор), водой и рассолом и упаривали до сухого состояния. Осадок растирали (DCM) с получением 1,30 г (31%) этил-2-амино-4-(5-хлор-2-метоксифенил)тиазол-5-карбоксилата в виде желтого твердого вещества. LCMS (ESI) m+H=313,2. 1H ЯМР (400 МГц, DMSO-d6): δ 7,77 (с, уш., 2H); 7,39 (дд, 1H); 7,22 (д, 1H); 7,05 (д, 1H); 4,00 (кв., 2H); 3,70 (с, 3H); 1,04 (т, 3H).

этил-2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-карбоксилат
Бромид меди (1,07 г, 4,79 ммоль) в ацетонитриле (20 мл) дегазировали азотом и охлаждали до 0°C, а затем добавляли трет-бутилнитрит (0,80 мл, 6,00 ммоль), а затем суспензию этил-2-амино-4-(5-хлор-2-метоксифенил)тиазол-5-карбоксилата (1,25 г, 3,99 ммоль) в ацетонитриле (20 мл) и перемешивали при комнатной температуре в течение 18 часов. Реакционную смесь концентрировали в вакууме, добавляли этилацетат, органические вещества промывали гидрокарбонатом натрия (насыщенный водный раствор) и рассолом, затем упаривали до сухого состояния с получением 1,40 г (93%) этил-2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-карбоксилата. LCMS (ESI) m+H=378,1. 1H ЯМР (400 МГц, DMSO-d6): δ 7,50 (дд, 1H); 7,42 (д, 1H); 7,14 (д, 1H); 4,16 (кв., 2H); 3,73 (с, 3H); 1,12 (т, 3H).
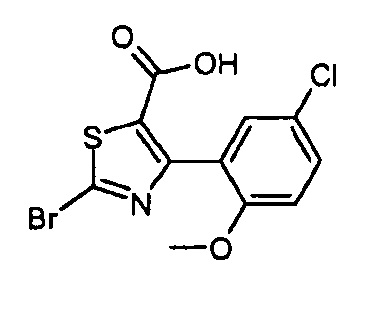
2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-карбоновая кислота
Смесь этил-2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-карбоксилата (1,40 г, 3,72 ммоль), гидроксида калия (278 мг) в THF (40 мл) и воды (10 мл) перемешивали в течение 20 часов при температуре окружающей среды. Смесь обрабатывали 1 M водным раствором HCl (приблизительно 8 мл, 2 экв.), добавляли DCM, и органические вещества отделяли и упаривали до сухого состояния с получением 1,23 г (95%) 2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-карбоновой кислоты в виде желтого твердого вещества. LCMS (ESI) m+H=350,1. 1H ЯМР (400 МГц, DMSO-d6): δ 7,47 (дд, 1H); 7,39 (д, 1H); 7,13 (д, 1H); 3,73 (с, 3H).

трет-бутил-2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-илкарбамат
2-Бром-4-(5-хлор-2-метоксифенил)тиазол-5-карбоновую кислоту (1,22 г, 3,50 ммоль), дифенилфосфорилазид (963 мг, 3,50 ммоль) и триэтиламин (354 мг, 3,50 ммоль) в трет-бутаноле (30 мл) перемешивали при 85°C в течение 4 часов. После охлаждения реакционную смесь распределяли между этилацетатом и водой, органические вещества отделяли, затем промывали рассолом и упаривали до сухого состояния. Полученный осадок очищали флэш-хроматографией на силикагеле (50-100% дихлорметан в циклогексане) с получением 970 мг (66%) трет-бутил-2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-илкарбамата. LCMS (ESI) m+H=421,2; 1H ЯМР (400 МГц, DMSO-d6): δ 7,43 (дд, 1H), 7,30 (д, 1H); 7,11 (д, 1H); 3,77 (с, 3H); 1,45 (с, 9H).

2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-амин
К раствору трет-бутил-2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-илкарбамата (360 мг, 0,86 ммоль) в DCM (10 мл) и воде (3 капли) добавляли TFA (4,0 мл). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре, а затем упаривали до сухого состояния. Осадок отбирали в DCM и промывали гидрокарбонатом натрия (насыщенный водный раствор), водой и рассолом и концентрировали в вакууме с получением 2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-амина в виде оранжевого осадка. LCMS (ESI) m+H=321,3. 1H ЯМР (400 МГц, DMSO-d6): δ 7,36 (д, 1H); 7,34-7,32 (м, 1H); 7,10 (д, 1H); 3,83 (с, 3H).

N-(2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
С использованием 2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-амина и пиразоло[1,5-a]пиримидин-3-карбонилхлорида указанное в заголовке соединение получали способами синтеза, описанными для N-(2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида, с дальнейшей очисткой флэш-хроматографией на силикагеле (0-40% этилацетат в DCM) с получением N-(2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=465,8. 1H ЯМР (400 МГц, DMSO-d6): δ 10,68 (с, уш., 1H); 9,41 (дд, 1H); 8,78 (с, 1H); 8,76 (дд, 1H); 7,57 (дд, 1H); 7,50 (д, 1H); 7,37-7,34 (м, 2H); 3,81 (с, 3H).
ПРИМЕР 169
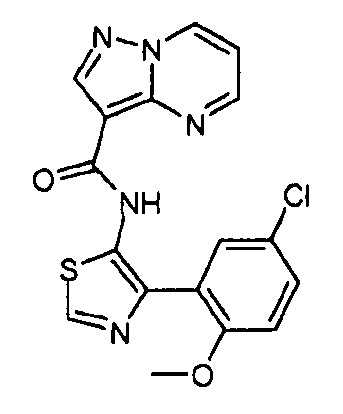
N-(4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
N-(2-Бром-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид (50,0 мг, 0,11 ммоль), Pd(PPh3)4 (6,20 мг, 5,35 мкмоль), формиат натрия (10,9 мг, 0,16 ммоль) и DMF (0,5 мл) герметизировали в атмосфере азота в емкости для обработки микроволновым излучением и нагревали до 130°C в течение 10 минут с использованием микроволнового излучения. Реакционную смесь охлаждали, затем добавляли DCM, и органические слои промывали гидрокарбонатом натрия (насыщенный водный раствор), водой и рассолом, сушили и упаривали до сухого состояния. Осадок очищали флэш-хроматографией на силикагеле (0-40% этилацетат в DCM), а затем далее очищали обращенно-фазовой ВЭЖХ и лиофилизировали с получением 11,1 мг (27%) N-(4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида в виде бежевого твердого вещества. LCMS (ESI) m+H=386,0. 1H ЯМР (400 МГц, DMSO-d6): δ 10,56 (с, 1H); 9,39 (дд, 1H); 8,74-8,73 (м, 3H); 7,55 (дд, 1H); 7,49 (д, 1H); 7,37 (д, 1H); 7,34 (дд, 1H); 3,80 (с, 3H).
ПРИМЕР 170

N-(4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид

этил-4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-карбоксилат
С использованием этилового эфира 2-бром-3-(5-хлор-2-метоксифенил)-3-оксопропионовой кислоты и тиоацетамида, указанное в заголовке соединение получали способами синтеза, описанными для этил-2-амино-4-(5-хлор-2-метоксифенил)тиазол-5-карбоксилата, с дополнительной очисткой флэш-хроматографией на силикагеле (50-100% DCM в циклогексане) с получением этил-4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-карбоксилата. LCMS (ESI) m+H=321,4; 1H ЯМР (400 МГц, CDCl3): δ 7,38 (д, 1H); 7,33 (дд, 1H); 6,87 (д, 1H); 4,20 (кв., 2H); 3,75 (с, 3H); 2,76 (с, 3H); 1,20 (т, 3H).

4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-карбоновая кислота
Смесь этил-4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-карбоксилата (520 мг, 1,67 ммоль) и гидроксида калия (125 мг) в THF (18 мл) и воде (4,5 мл) перемешивали в течение 20 часов при комнатной температуре, а затем при 75°C в течение дополнительных 8 часов. Раствор обрабатывали 1 М водным раствором HCl (pH 2) и экстрагировали DCM. Органические слои упаривали до сухого состояния с получением 450 мг (95%) 4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-карбоновой кислоты в виде белого твердого вещества. LCMS (ESI) m+H=284,3; 1H ЯМР (400 МГц, DMSO-d6): δ 7,43 (дд, 1H); 7,31 (д, 1H); 7,10 (д, 1H); 3,71 (с, 3H); 2,69 (с, 3H).

трет-бутил-4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-илкарбамат
С использованием 4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-карбоновой кислоты указанное в заголовке соединение получали способами синтеза, описанными для трет-бутил-2-бром-4-(5-хлор-2-метоксифенил)тиазол-5-илкарбамата, с получением трет-бутил-4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-илкарбамата. LCMS (ESI) m+H=355,3; 1H ЯМР (400 МГц, DMSO-d6): δ 7,40 (дд, 1H); 7,32 (д, 1H); 7,11 (д, 1H); 3,77 (с, 3H); 2,55 (с, 3H); 1,42 (с, 9H).
4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-амин
К раствору трет-бутил-4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-илкарбамата (315 мг, 0,89 ммоль) в DCM (10 мл) и воде (3 капли) добавляли TFA (4,0 мл). Реакционную смесь перемешивали в течение 1,5 часов при комнатной температуре, затем упаривали до сухого состояния с получением 215 мг (95%) 4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-амина в виде желтого твердого вещества. LCMS (ESI) m+H=255,2.

N-(4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
С использованием 4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-амина и пиразоло[1,5-a]пиримидин-3-карбонилхлорида указанное в заголовке соединение синтезировали способами синтеза, описанными для N-(2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида, с дальнейшей очисткой флэш-хроматографией на силикагеле (0-100% этилацетат в DCM) с получением N-(4-(5-хлор-2-метоксифенил)-2-метилтиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида в виде оранжевого твердого вещества. LCMS (ESI) m+H=400,0; 1H ЯМР (400 МГц, DMSO-d6): δ 10,43 (с, уш., 1H); 9,38 (дд, 1H); 8,75-8,73 (м, 1H); 8,72 (с, 1H); 7,52 (дд, 1H); 7,47 (д, 1H); 7,34-7,32 (м, 2H); 3,79 (с, 3H); 2,62 (с, 3H).
ПРИМЕР 171

N-(4-(5-хлор-2-метоксифенил)-2-(тетрагидро-2H-пиран-4-ил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид

N-((5-хлор-2-метоксифенил)(циано)метил)тетрагидро-2H-пиран-4-карбоксамид
С использованием амино-(5-хлор-2-метоксифенил)ацетонитрила и тетрагидро-2H-пиран-4-карбоновой кислоты, указанное в заголовке соединение получали способами синтеза, описанными для 3-(трет-бутилдиметилсилилокси)-N-((5-хлор-2-метоксифенил)(циано)метил)-3-метилбутанамида, с получением N-((5-хлор-2-метоксифенил)(циано)метил)тетрагидро-2H-пиран-4-карбоксамида в виде белого твердого вещества. LCMS (ESI) m+H=309,3.

N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)тетрагидро-2H-пиран-4-карбоксамид
С использованием N-((5-хлор-2-метоксифенил)(циано)метил)тетрагидро-2H-пиран-4-карбоксамида, указанное в заголовке соединение получали способами синтеза, описанными для N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)-3-(трет-бутилдиметилсилилокси)-3-метилбутанамида, с получением N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)тетрагидро-2H-пиран-4-карбоксамида в виде белого твердого вещества. LCMS (ESI) m+H=327,3; 1H ЯМР (400 МГц, DMSO-d6): δ 8,30 (д, уш., 1H); 7,31-7,31 (м, 2H); 7,24 (с, уш., 1H); 7,13 (с, уш., 1H); 7,05-7,01 (д, 1H); 5,62 (д, 1H); 3,91-3,81 (м, 2H); 3,79 (с, 3H); 3,30-3,24 (м, 2H); 2,55-2,54 (м, 1H); 1,56-1,55 (м, 4H).

4-(5-хлор-2-метоксифенил)-2-(тетрагидро-2H-пиран-4-ил)тиазол-5-амин
С использованием N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)тетрагидро-2H-пиран-4-карбоксамида, указанное в заголовке соединение получали способами синтеза, описанными для 2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-амина, с получением 4-(5-хлор-2-метоксифенил)-2-(тетрагидро-2H-пиран-4-ил)тиазол-5-амина в виде белой камеди. LCMS (ESI) m+H=325,2; 1H ЯМР (400 МГц, DMSO-d6): δ 7,38 (д, 1H); 7,31 (дд, 1H); 7,09 (д, 1H); 5,31 (с, уш., 2H); 3,90 (д, 2H); 3,84 (с, 3H); 3,43 (м, 2H); 3,04-2,99 (м, 1H); 1,90 (д, 2H); 1,66-1,66 (м, 2H).

N-(4-(5-хлор-2-метоксифенил)-2-(тетрагидро-2H-пиран-4-ил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
С использованием N-(2-амино-1-(5-хлор-2-метоксифенил)-2-оксоэтил)тетрагидро-2H-пиран-4-карбоксамида указанное в заголовке соединение получали способами синтеза, описанными для N-(2-(2-(трет-бутилдиметилсилилокси)-2-метилпропил)-4-(5-хлор-2-метоксифенил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида, с дополнительной очисткой путем растирания в диэтиловом эфире с получением N-(4-(5-хлор-2-метоксифенил)-2-(тетрагидро-2H-пиран-4-ил)тиазол-5-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида. LCMS (ESI) m+H=470,0; 1H ЯМР (400 МГц, DMSO-d6): δ 10,44 (с, уш., 1H); 9,38 (дд, 1H); 8,75 (дд, 1H); 8,73 (с, 1H); 7,52 (дд, 1H); 7,46 (д, 1H); 7,35 (д, 1H); 7,32-7,31 (м, 1H); 3,95-3,94 (м, 2H); 3,80 (с, 3H); 3,47 (м, 2H); 3,23 (м, 1H); 2,00 (дд, 2H); 1,77 (м, 2H).
Соединения примеров 172-508, представленные в таблице 2, получали, главным образом, согласно описанным выше примерам. Для каждого соединения, представленного в таблице 2, в столбце "Способ" приведен номер примера, которому следовали.







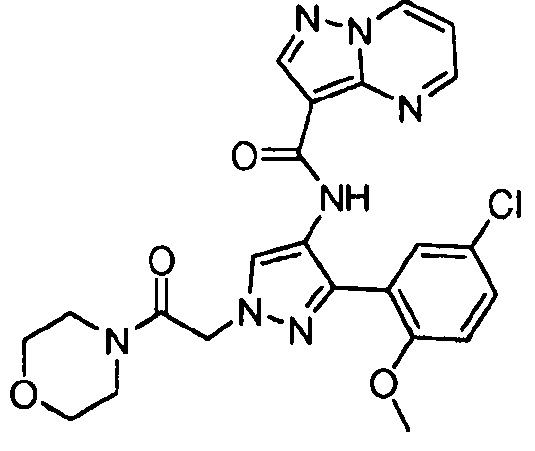
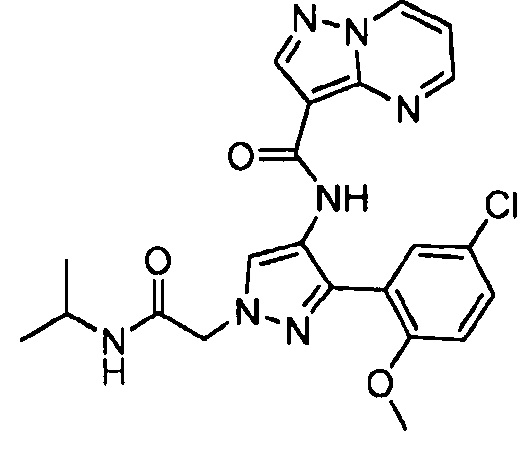
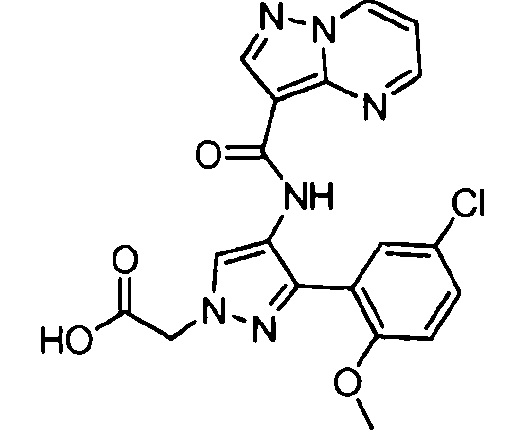
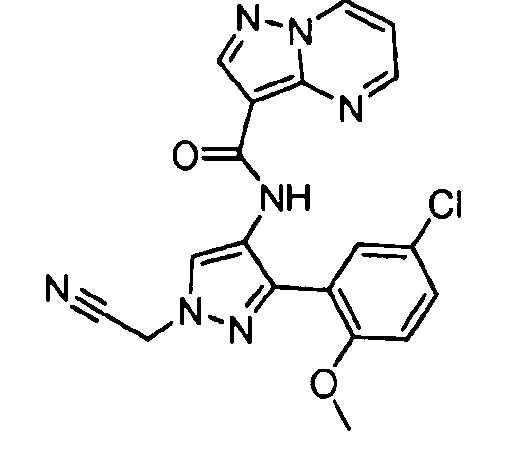





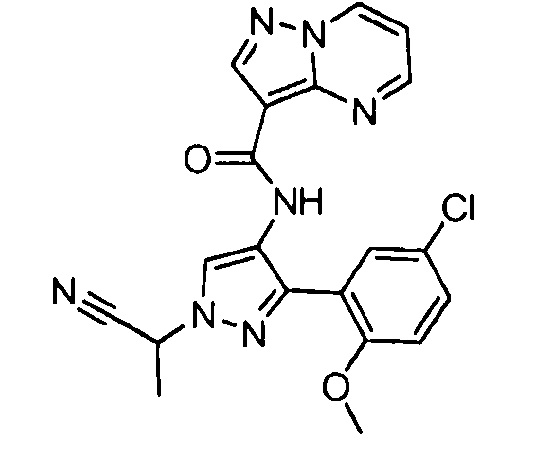


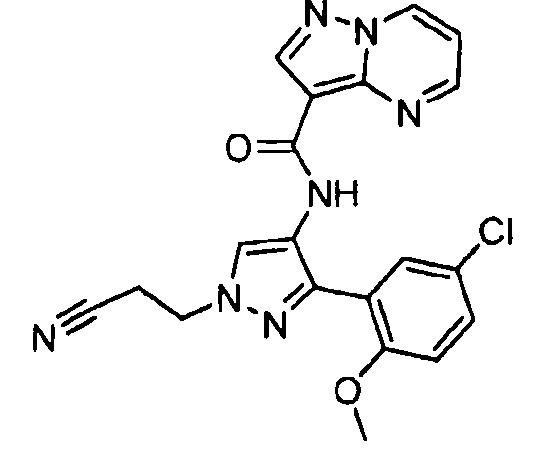
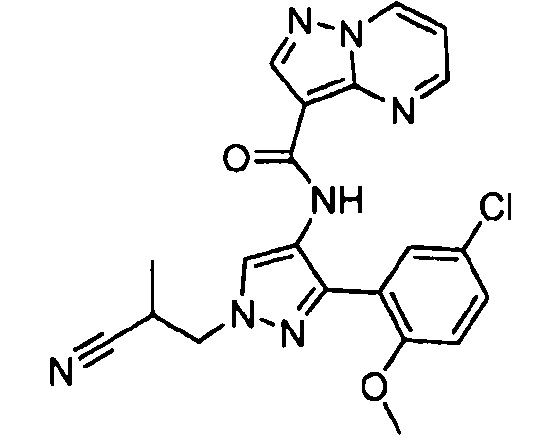




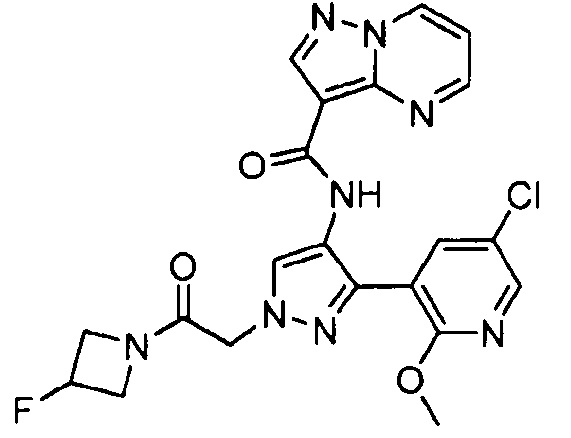

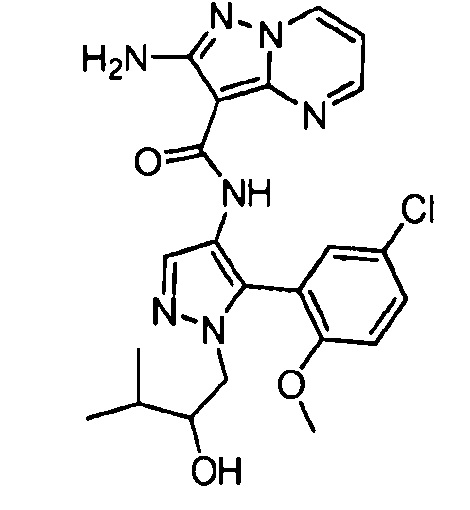









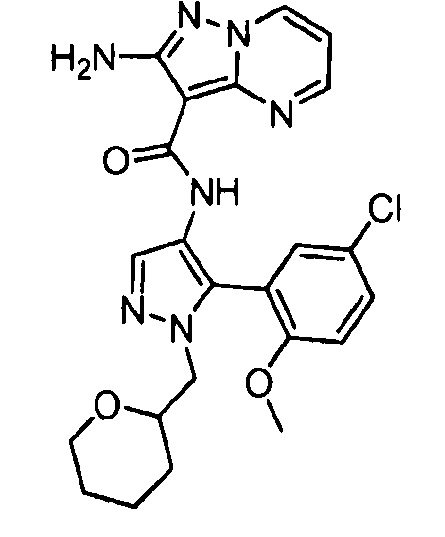
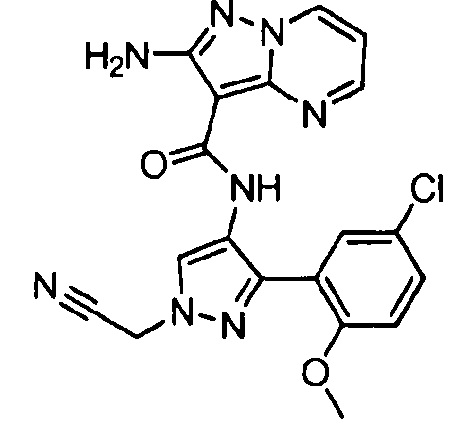
















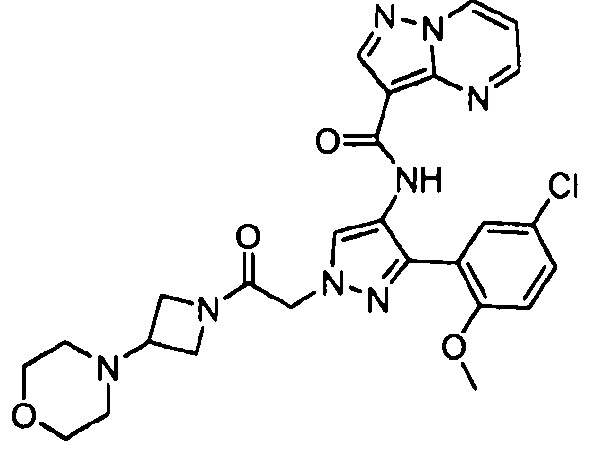

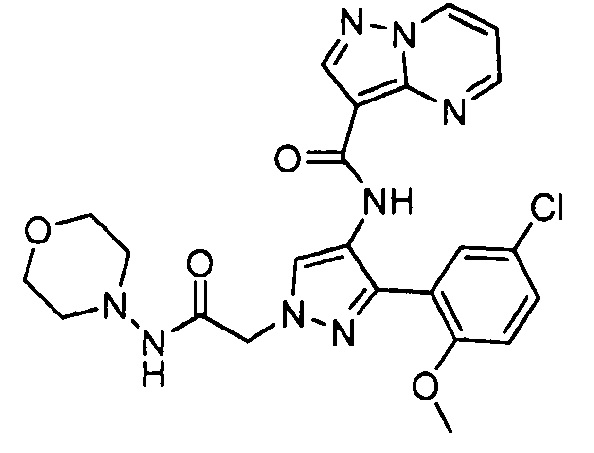


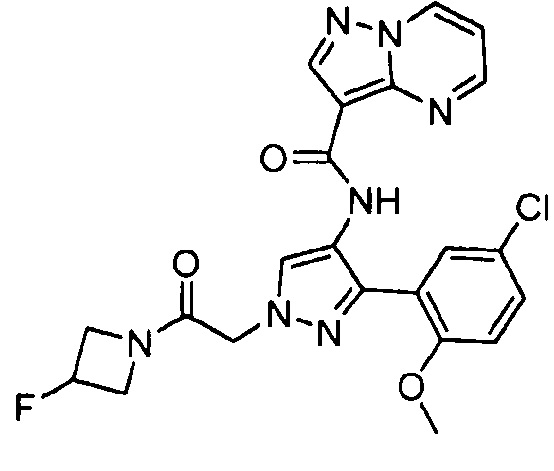

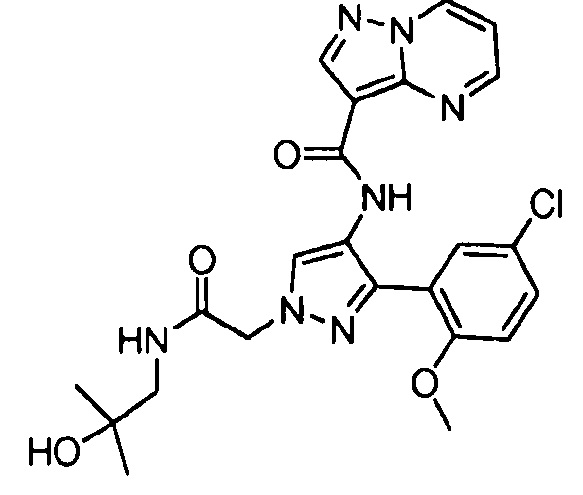
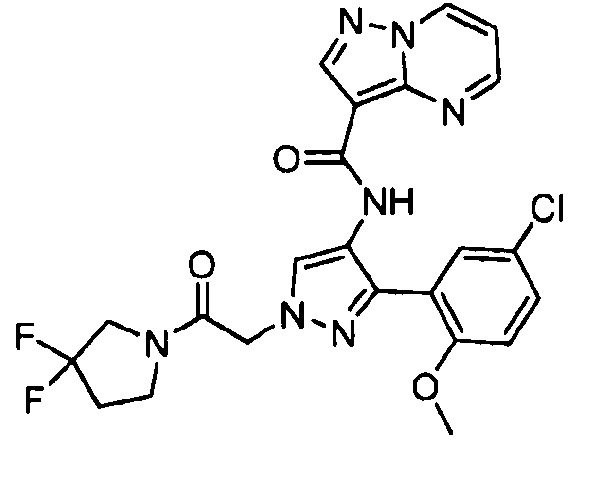



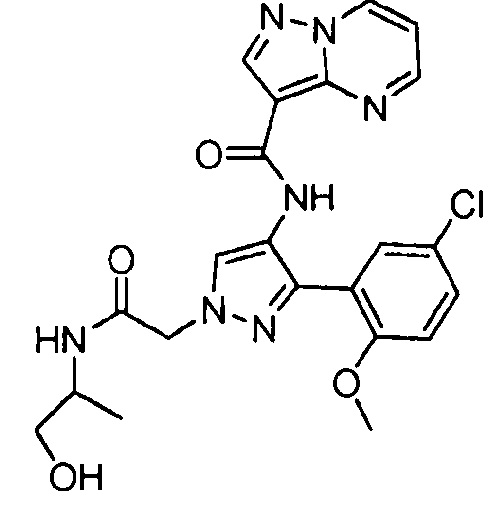
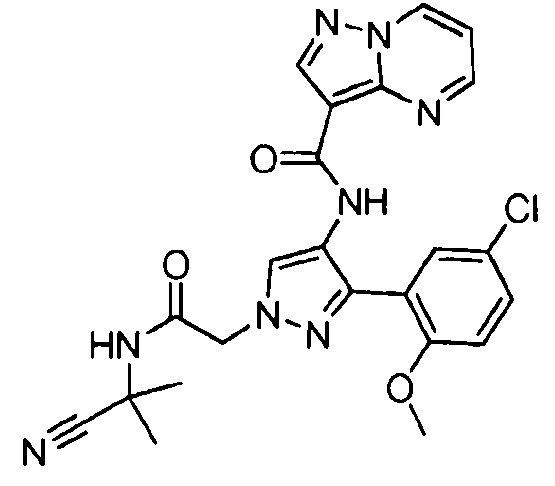

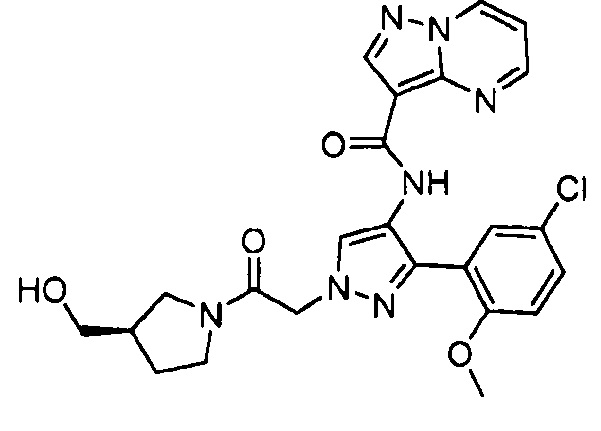
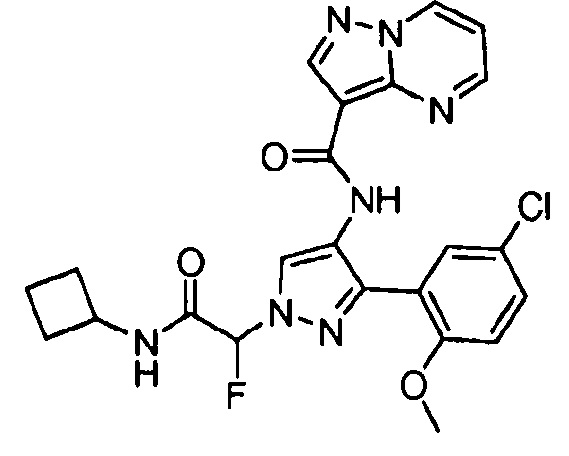





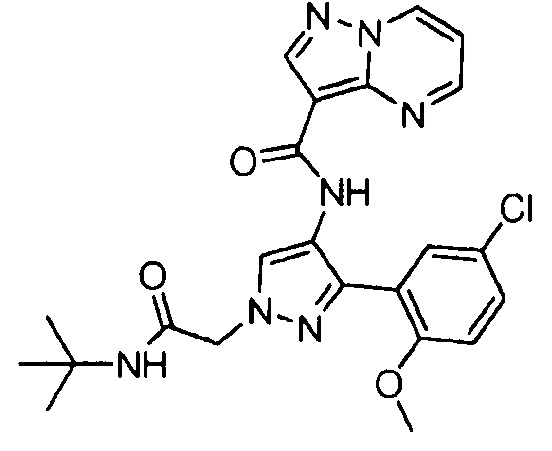


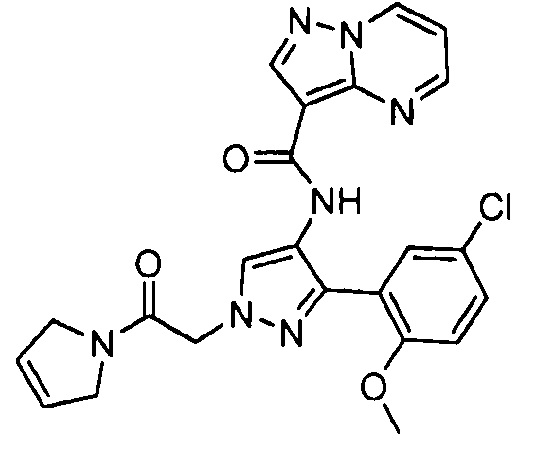
















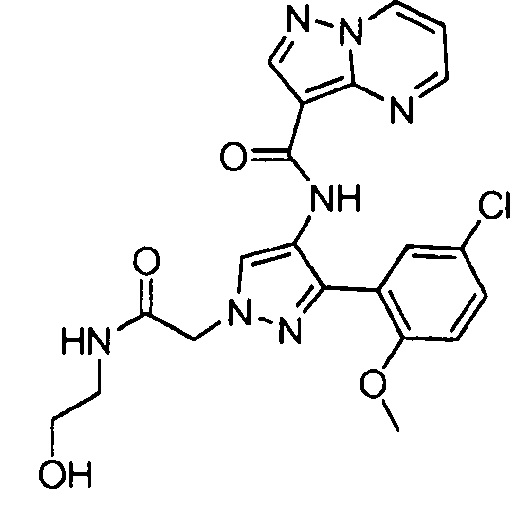

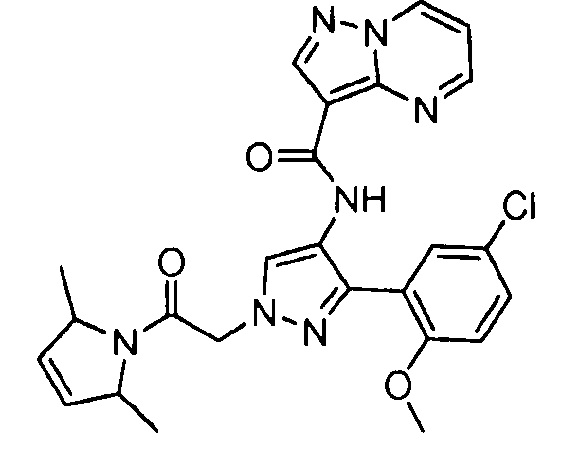
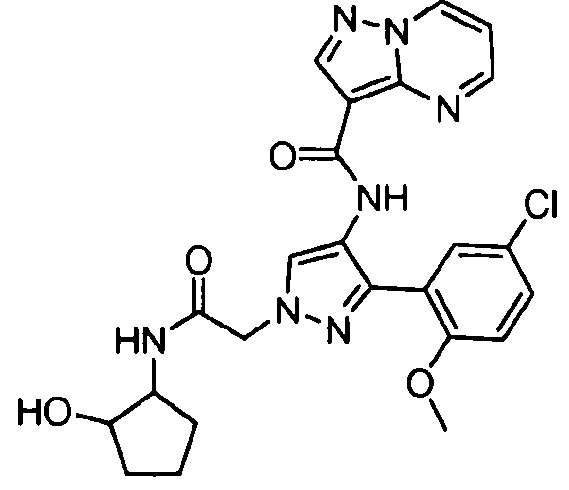





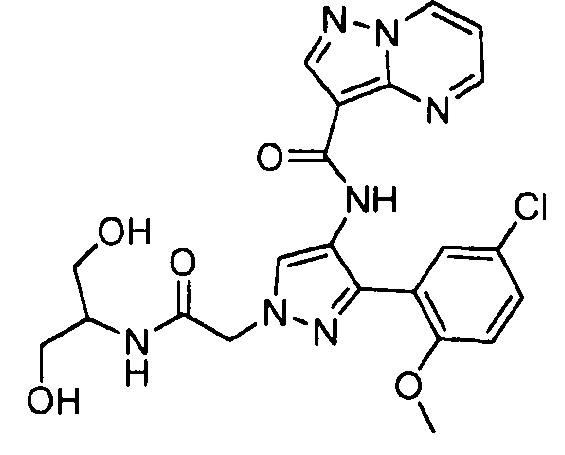

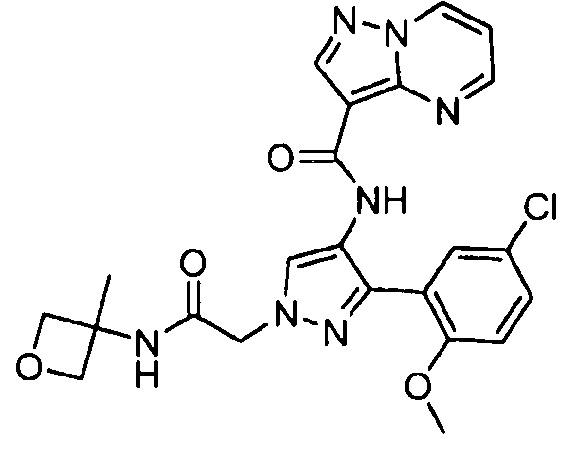












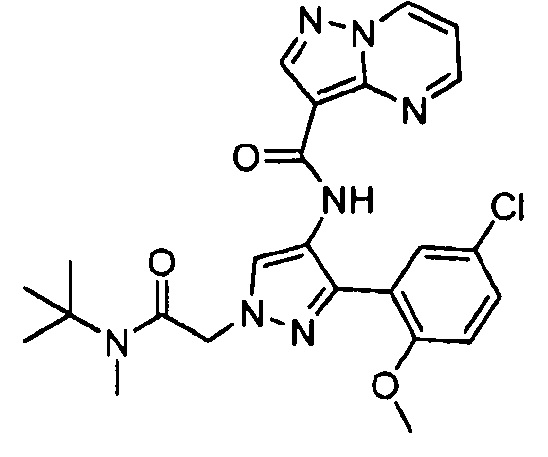


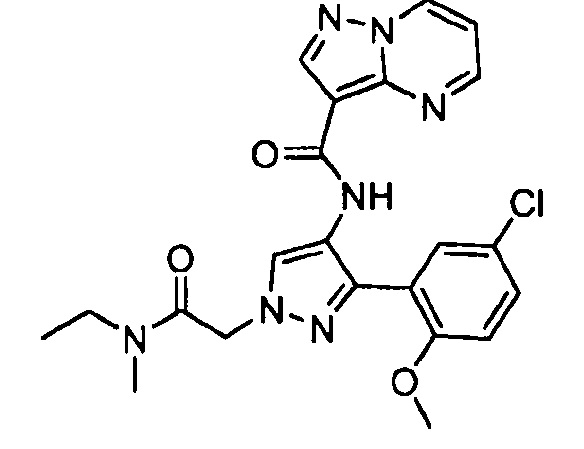


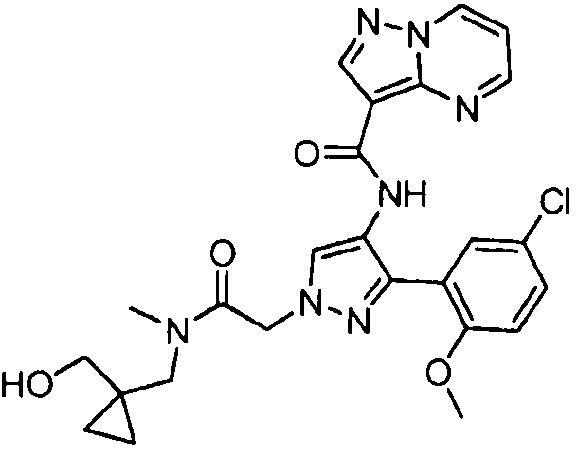




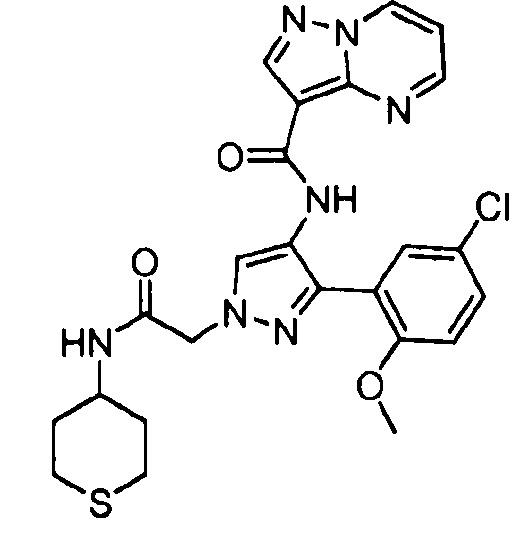
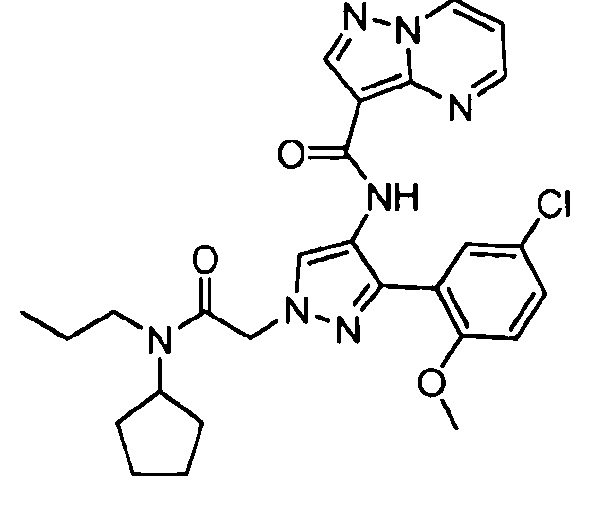





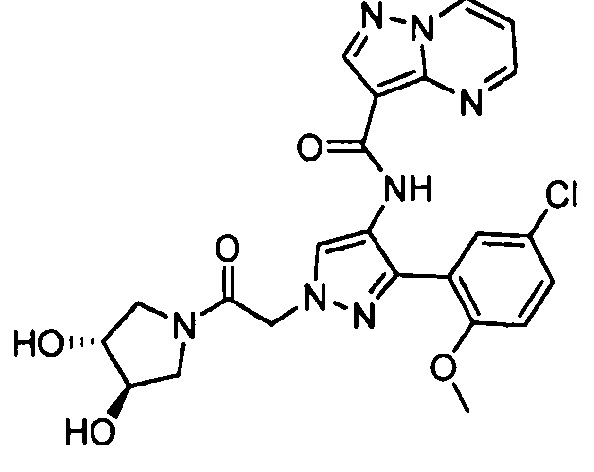






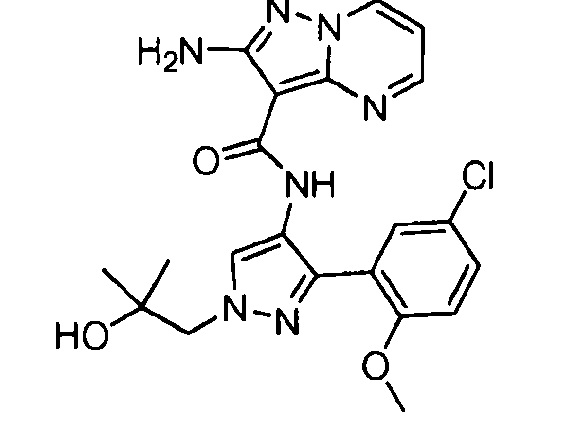
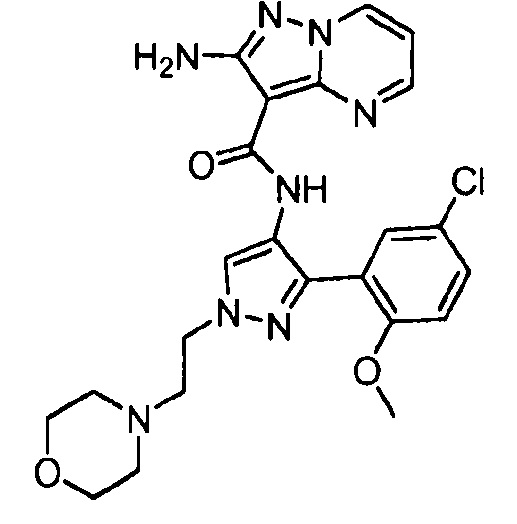

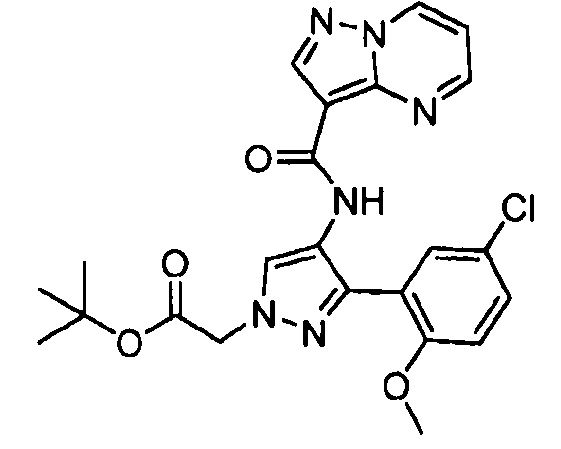

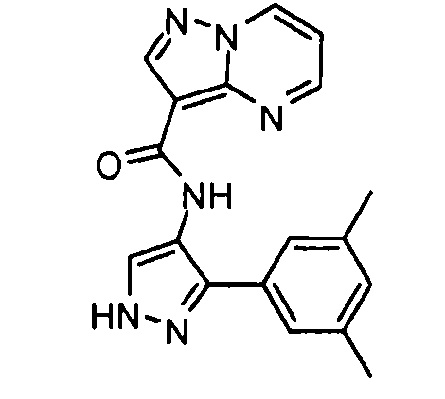


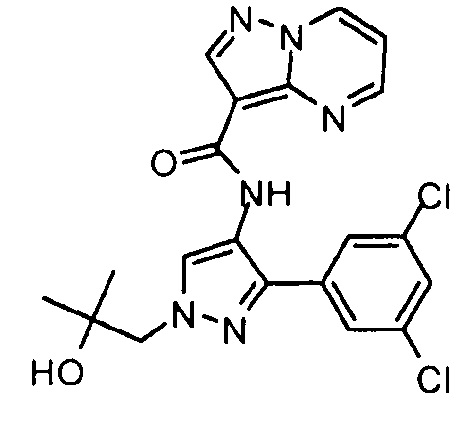


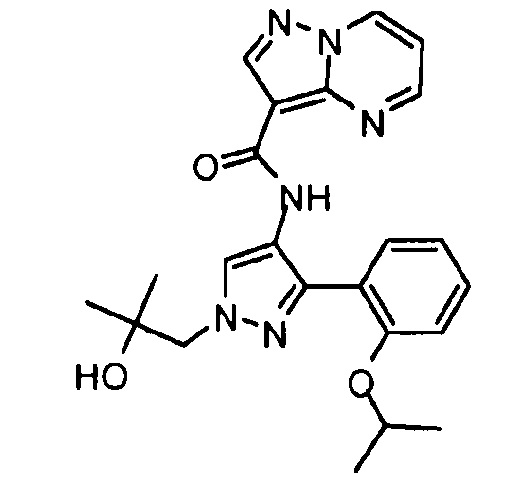






















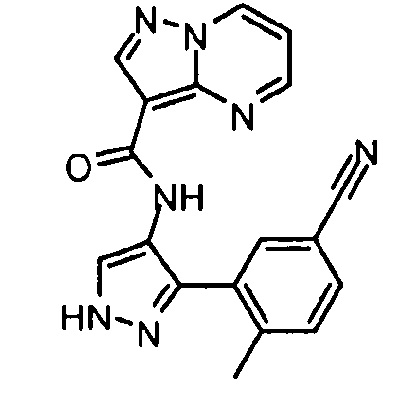
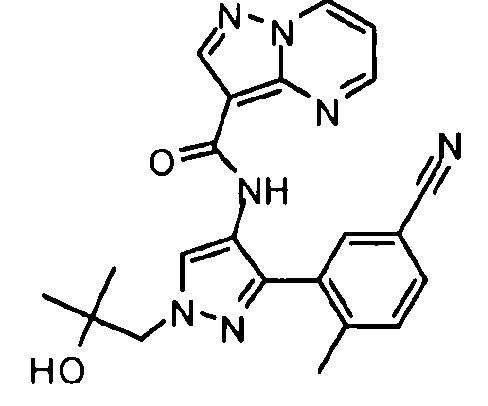
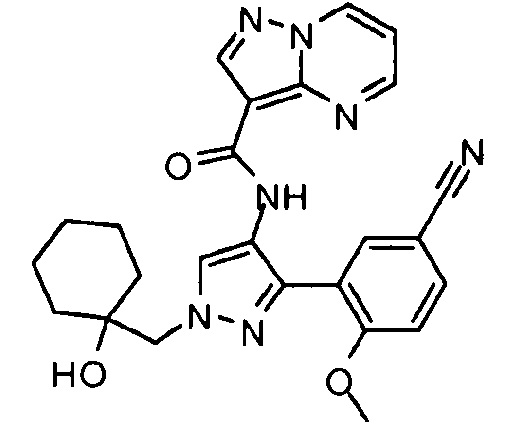





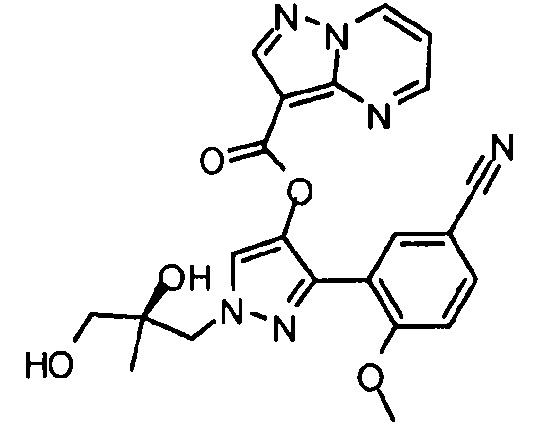
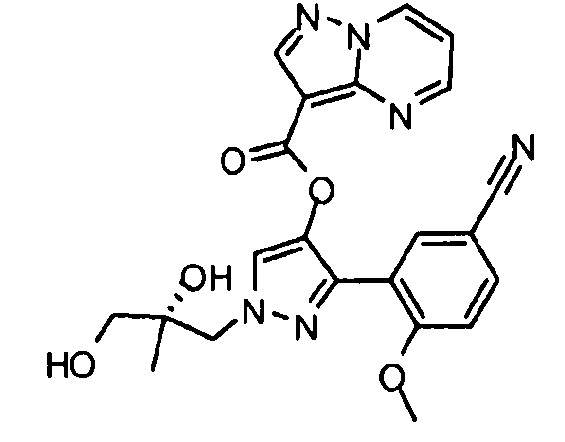


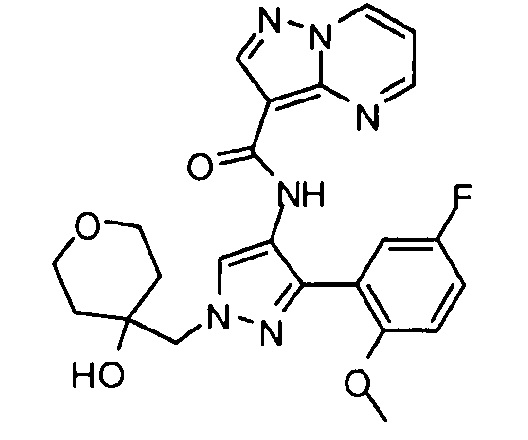



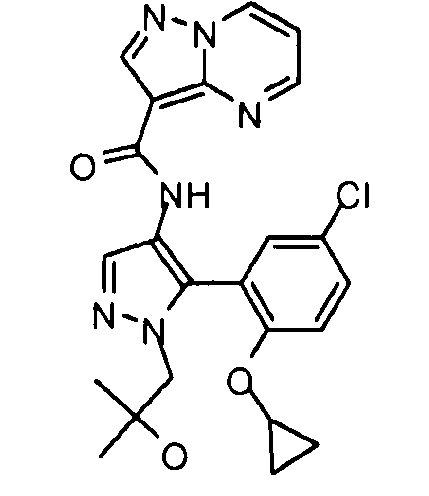


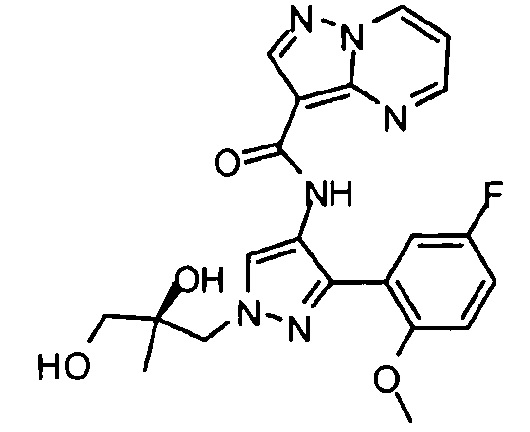


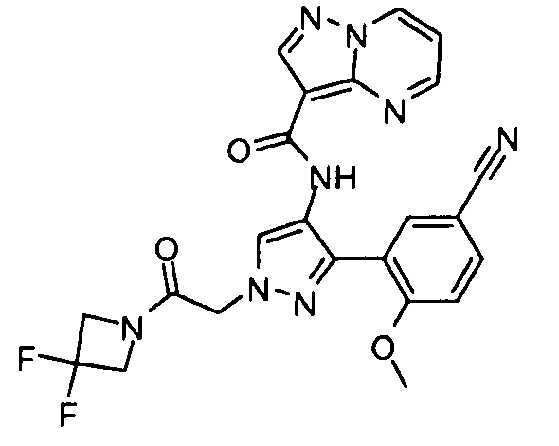



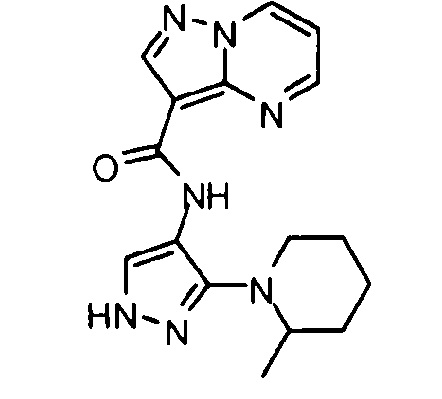

















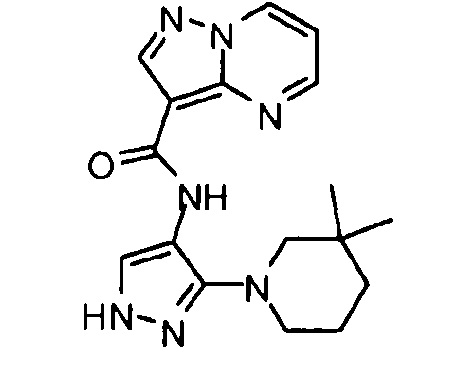




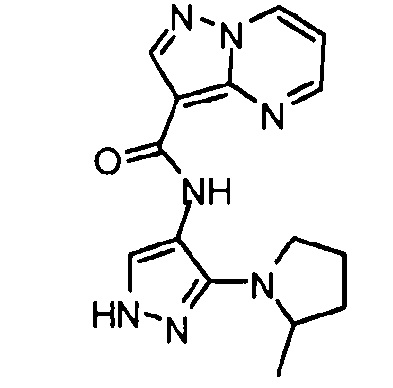










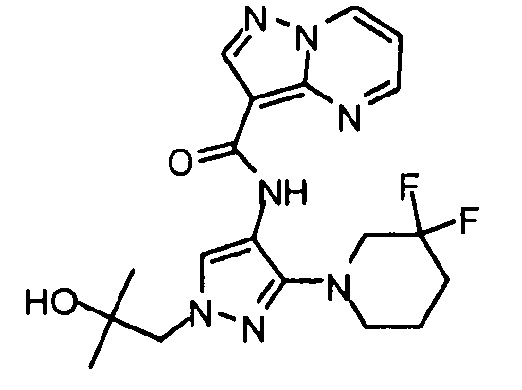
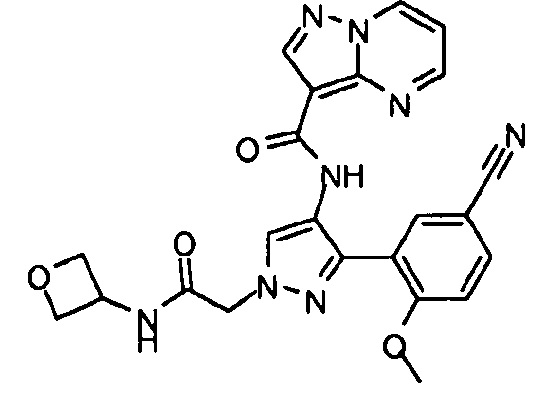
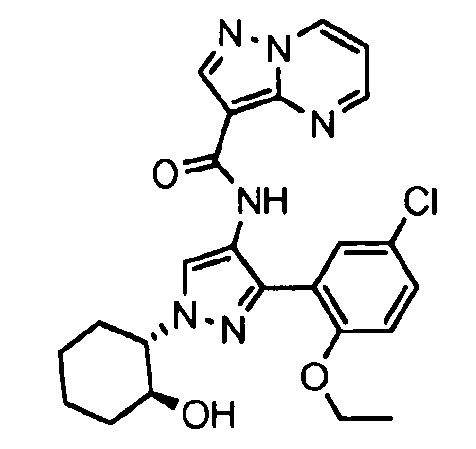
и
и
N-(3-(5-хлор-2-этоксифенил)-1-((1R,2R)-2-гидроксициклогексил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид
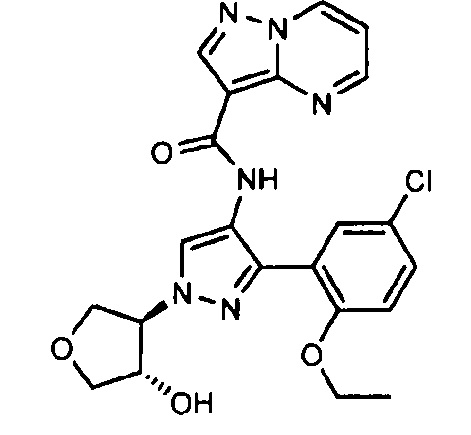
и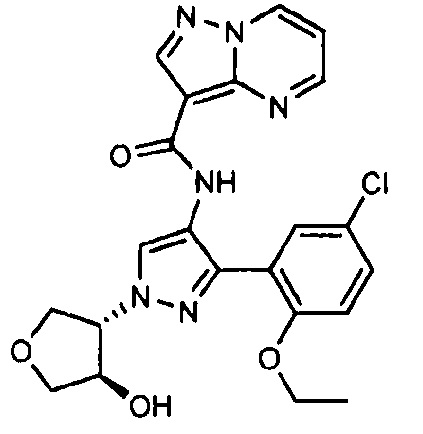
и
N-(3-(5-хлор-2-этоксифенил)-1-((3S,4R)-4-гидрокситетрагидро-фуран-3-ил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид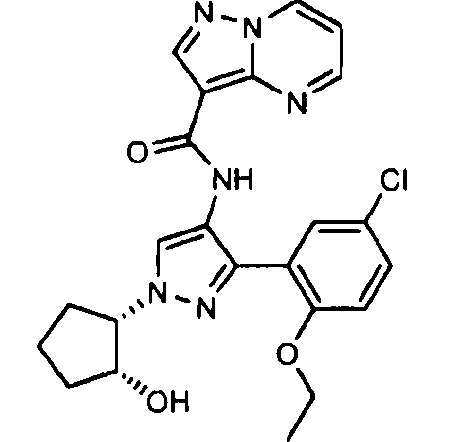
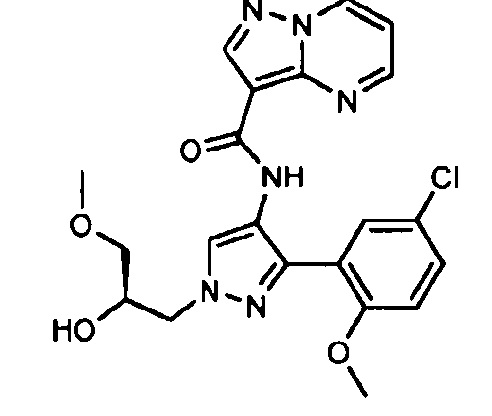
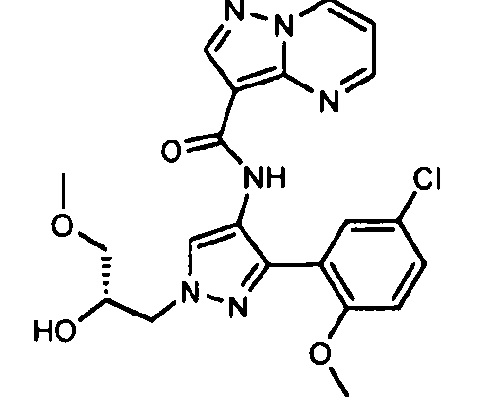






и
и
N-(3-(5-хлор-2-метоксифенил)-1-((3S,4R)-4-гидрокситетрагидро-фуран-3-ил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-
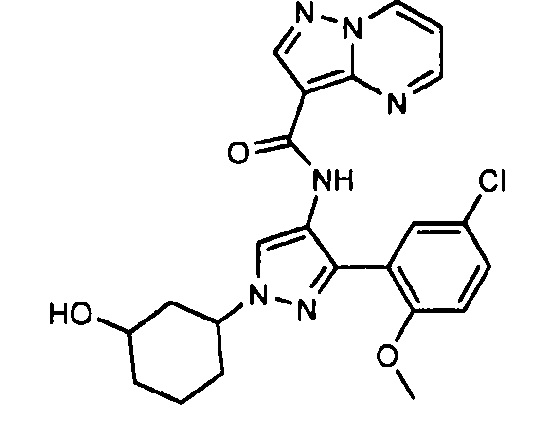

и
и
N-(3-(5-хлор-2-метоксифенил)-1-((1R,2R)-2-гидроксициклопентил)-1H-пиразол-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамид


и
136
и
16

16




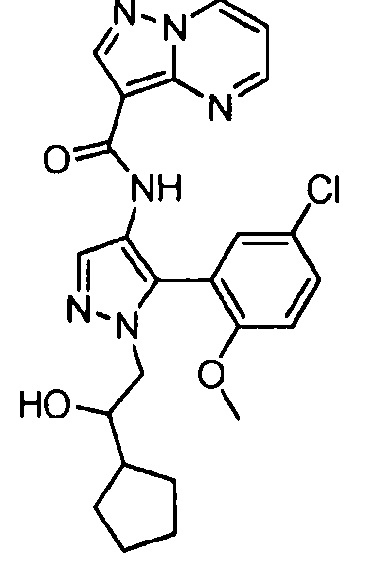






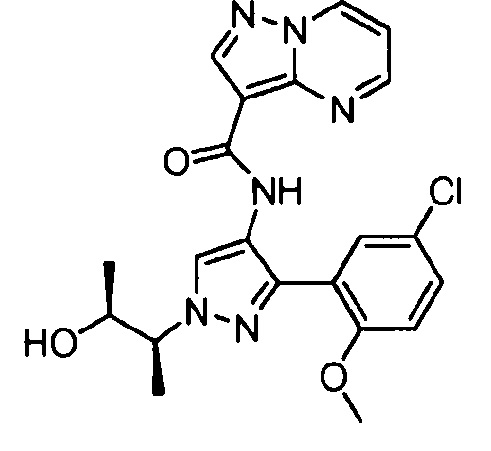


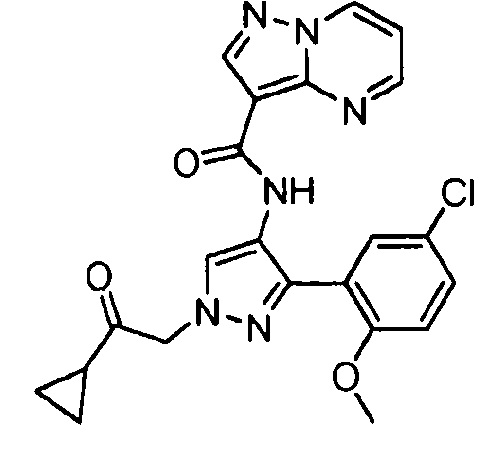

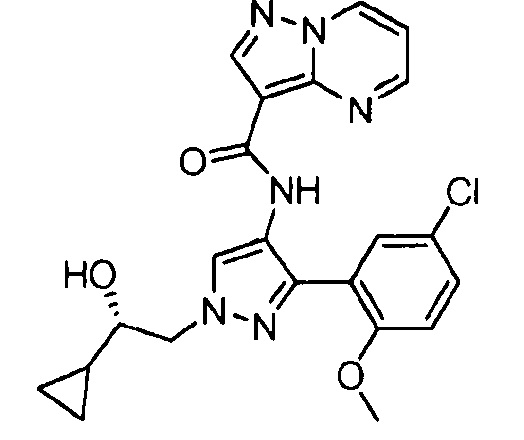
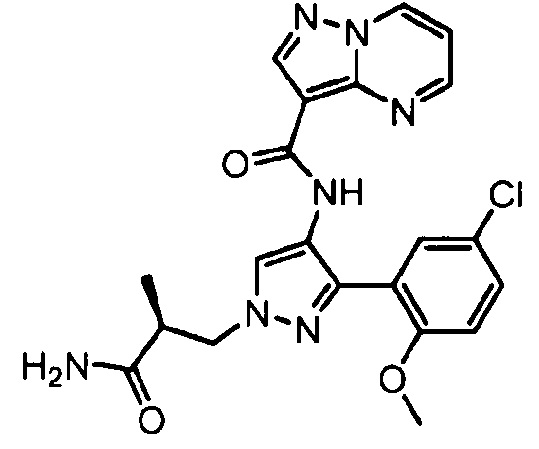

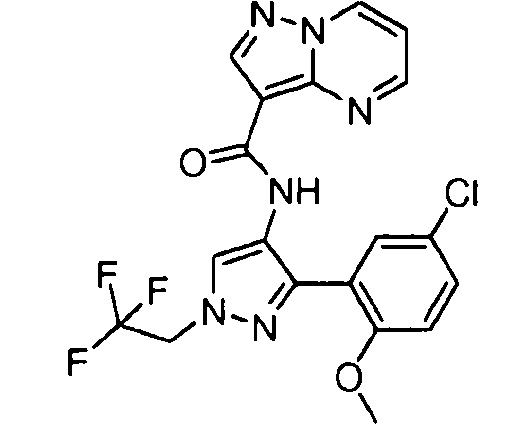




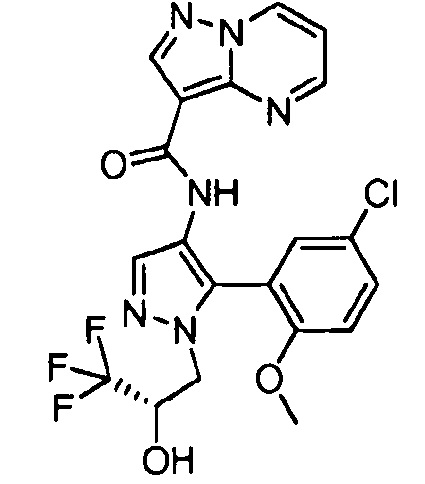


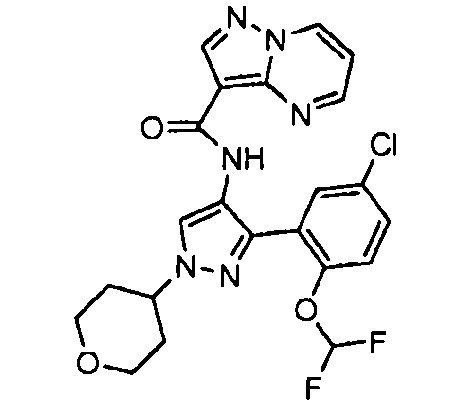

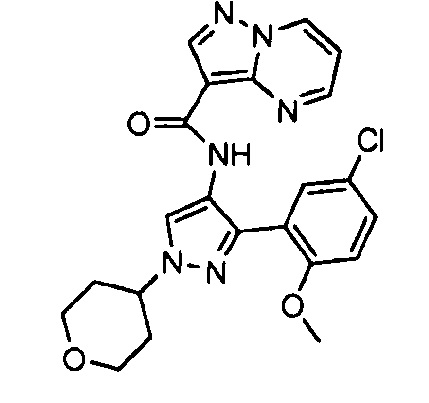


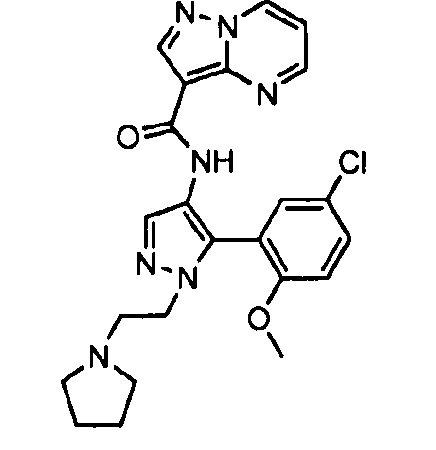


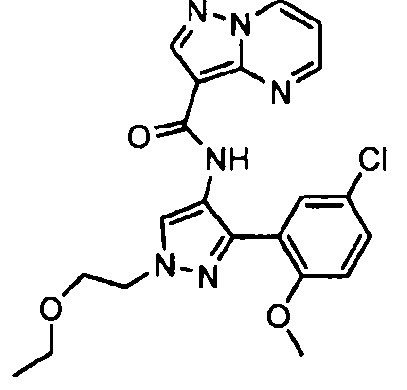





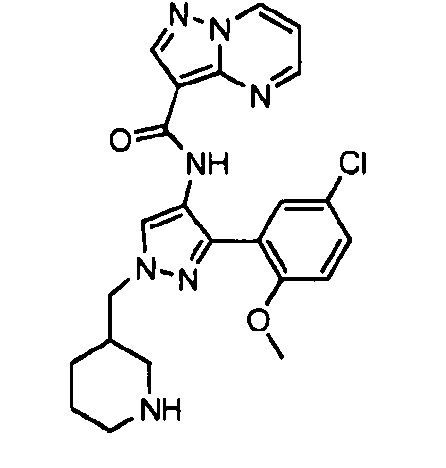
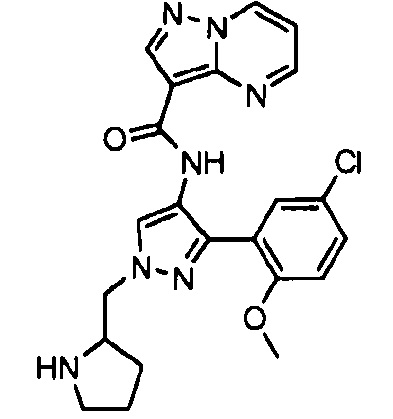




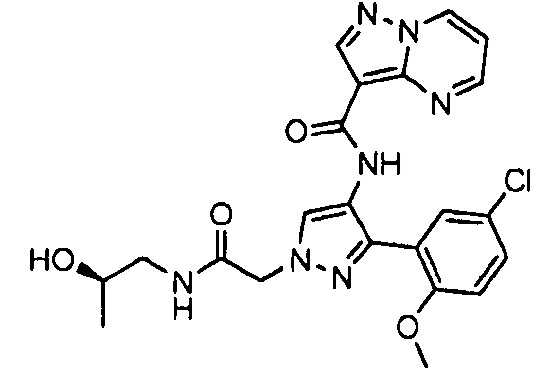
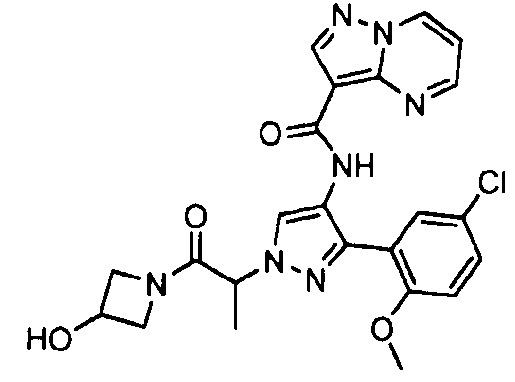
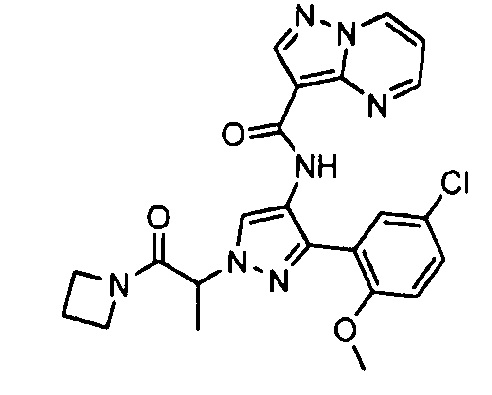




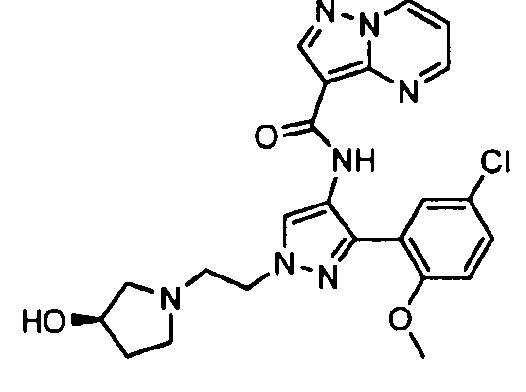
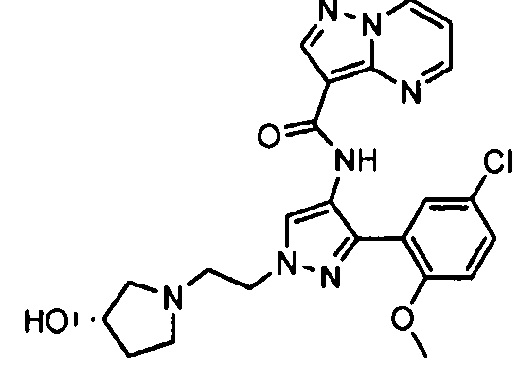


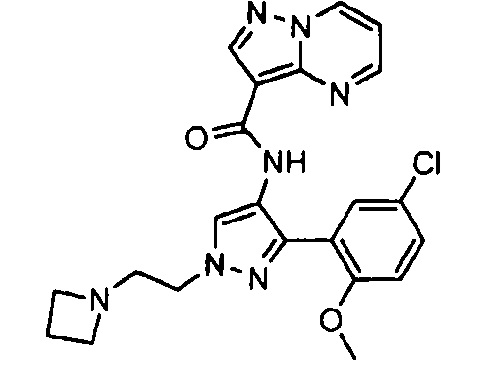




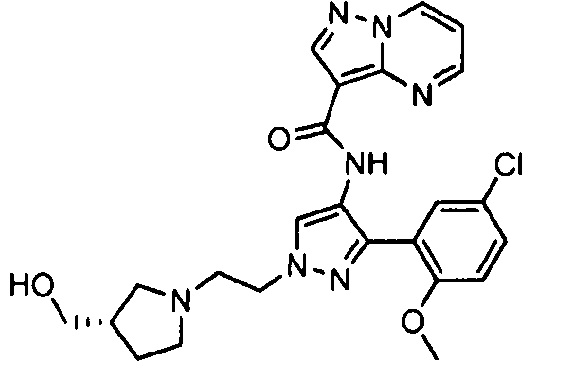








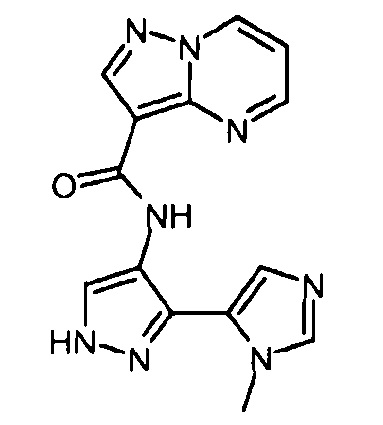

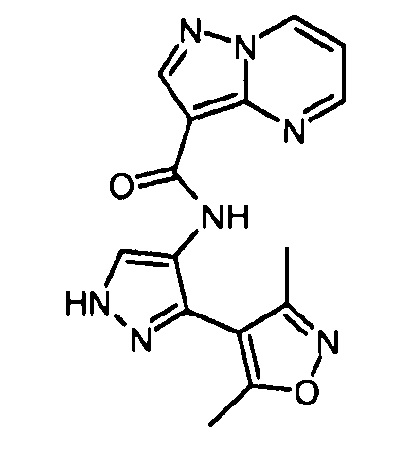

Соединения примеров 1-508 тестировали в описанных выше анализах (примеры A-C) и было выявлено, что они имеют величины Ki менее чем приблизительно 1 мкМ для ингибирования одной или обеих из киназ JAK1 и JAK2. Соединения примеров 1-132 были протестированы в описанных выше анализах (примеры A-C) и было выявлено, что они имеют величины Ki менее чем приблизительно 1 мкМ для ингибирования киназы JAK2. Соединения примеров 132-508 тестировали в описанных выше анализах (примеры A-C) и было выявлено, что они имеют величины Ki менее чем приблизительно 1 мкМ для ингибирования киназы JAK1.
В таблице 3 ниже представлены данные о ферментативной активности (Ki, мкМ) для определенных соединений по настоящему изобретению, исследованных в описанных выше анализах (примеры A-C).
Приводится отсылка на предварительную заявку США с серийным номером № 61/222918, поданную 2 июля 2009 года, которая включена в настоящий документ в качестве ссылки в полном объеме для любых целей.
Хотя изобретение описано и проиллюстрировано с определенной степенью подробности, понятно, что настоящее описание представлено только в качестве примера, и что специалисты в данной области могут прибегнуть к множеству изменений в комбинации и порядке частей без отклонения от сущности и объема изобретения, определенного формулой изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИРУЮЩИЕ JAK СОЕДИНЕНИЯ НА ОСНОВЕ ПИРАЗОЛОПИРИМИДИНА И СПОСОБЫ | 2010 |
|
RU2567238C2 |
| ПИРАЗОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ-ИНГИБИТОРЫ JAK И СПОСОБЫ | 2009 |
|
RU2539568C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА В КАЧЕСТВЕ ИНГИБИТОРОВ КАЛЛИКРЕИНА | 2017 |
|
RU2739447C2 |
| АНТАГОНИСТЫ ЕР4 | 2016 |
|
RU2761341C2 |
| Соединения 1-циано-пирролидинов в качестве ингибиторов USP30 | 2016 |
|
RU2717238C2 |
| ПИРАЗОЛО[1,5-A]ПИРИМИДИНИЛКАРБОКСАМИДЫ И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ПАТОЛОГИЧЕСКИХ СОСТОЯНИЙ | 2017 |
|
RU2799332C2 |
| ПУРИНОВЫЕ ИНГИБИТОРЫ ЧЕЛОВЕЧЕСКОЙ ФОСФАТИДИЛИНОЗИТ 3-КИНАЗЫ ДЕЛЬТА | 2013 |
|
RU2658006C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОРЫ MELK | 2011 |
|
RU2582610C2 |
| N-((ГЕТ)АРИЛМЕТИЛ)-ГЕТЕРОАРИЛ-КАРБОКСАМИДНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ПЛАЗМЕННОГО КАЛЛИКРЕИНА | 2015 |
|
RU2707870C2 |

Изобретение относится к соединению формулы I или к его фармацевтически приемлемой соли. В формуле I R1 представляет собой водород; R2 представляет собой пиридинил, где R2 необязательно замещен 1-3 R4; R3 представляет собой фенил, где R3 необязательно замещен 1-5 R5; R4 независимо представляет собой C1-C6алкил; R5 независимо представляет собой C1-C6алкил, галоген или -(C0-C3алкил)CF3. Заявленные соединения пригодны в качестве ингибиторов одной или нескольких Janus-киназ. Изобретение также относится к фармацевтической композиции, которая включает соединение формулы I и фармацевтически приемлемый носитель, адъювант или наполнитель, способу лечения или снижения тяжести заболевания или состояния, отвечающего на ингибирование активности Janus-киназы у пациента, и применению соединения формулы I. 4 н. и 2 з.п. ф-лы, 1 ил., 3 табл., 508 пр.
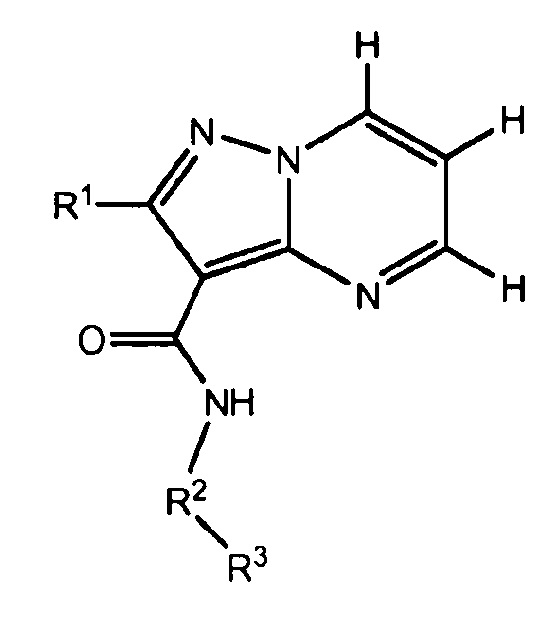
I
1. Соединение формулы I
 ,
,
I
или его фармацевтически приемлемые соли, где
R1 представляет собой водород;
R2 представляет собой пиридинил, где R2 необязательно замещен 1-3 R4;
R3 представляет собой фенил, где R3 необязательно замещен 1-5 R5;
R4 независимо представляет собой C1-C6алкил;
R5 независимо представляет собой C1-C6алкил, галоген или -(C0-C3алкил)CF3.
2. Соединение по п.1, выбранное из:
N-(3-(2-фторфенил)пиридин-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида,
N-(3-(3-фторфенил)пиридин-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида и
N-(3-(3-хлорфенил)пиридин-4-ил)пиразоло[1,5-a]пиримидин-3-карбоксамида.
3. Фармацевтическая композиция, ингибирующая активность Janus-киназы, содержащая соединение по п.1 и фармацевтически приемлемый носитель, адъювант или наполнитель.
4. Способ профилактики, лечения или снижения тяжести заболевания или состояния, отвечающего на ингибирование активности Janus-киназы у пациента, включающий введение указанному пациенту терапевтически эффективного количества соединения по п.1.
5. Способ по п.4, где указанное заболевание или состояние представляет собой злокачественную опухоль, истинную полицитемию, идиопатический тромбоцитоз, миелофиброз, хронический миелогенный лейкоз (CML), ревматоидный артрит, синдром воспаленного кишечника, болезнь Крона, псориаз, контактный дерматит или реакцию гиперчувствительности замедленного типа.
6. Применение соединения по п.1 для лечения злокачественной опухоли, истинной полицитемии, идиопатического тромбоцитоза, миелофиброза, хронического миелогенного лейкоза (CML), ревматоидного артрита, синдрома воспаленного кишечника, болезни Крона, псориаза, контактного дерматита или реакции гиперчувствительности замедленного типа.
| Колосоуборка | 1923 |
|
SU2009A1 |
| US 20070083044 A1, 12.04.2007 | |||
| ВИСЯЧАЯ НАРУЖНАЯ СТЕНА ДЛЯ ЖИЛЫХ СТРОЕНИЙ | 1927 |
|
SU6227A1 |

