Область изобретения
Настоящее изобретение относится к соединениям, способам их получения, композициям, содержащим их, их применению в лечении или предупреждении различных расстройств, в частности аллергических заболеваний и других воспалительных состояний, например аллергического ринита и астмы, инфекционных заболеваний и рака, а также в качестве адъювантов вакцин.
Предшествующий уровень техники
Позвоночные животные постоянно находятся под угрозой заражения микроорганизмами и имеют развитые механизмы иммунной защиты для элиминации инфекционных патогенов. У млекопитающих эта иммунная система включает две ветви: врожденный иммунитет и приобретенный иммунитет. В первую очередь иммунную защиту организма обеспечивает врожденная иммунная система, опосредованная макрофагами и дендритными клетками. Приобретенный иммунитет задействован в элиминации патогенов на поздней стадии инфицирования, а также способствует формированию иммунологической памяти. Приобретенный иммунитет является высокоспецифичным вследствие огромного репертуара лимфоцитов с антиген-специфическими рецепторами, которые подвергаются перестройке генов.
Центральное место в формировании эффективного врожденного иммунного ответа у млекопитающих занимают механизмы, которые вызывают индукцию интерферонов и других цитокинов, которые действуют на клетки, вызывая целый ряд эффектов. У человека интерфероны I типа представляют собой семейство родственных белков, кодируемых генами, расположенными на хромосоме 9 и кодирующими по меньшей мере 13 изоформ интерферона альфа (IFNα) и одну изоформу интерферона бета (IFNβ). Интерферон впервые был описан как вещество, которое может защищать клетки от вирусной инфекции (Isaacs & Lindemann, J. Virus Interference. Proc. R. Soc. Lon. Ser. B. Biol. Sci. 1957: 147, 258-267). Рекомбинантный IFNα был первым разрешенным биологическим терапевтическим средством и стал важным препаратом в терапии вирусных инфекций и рака. Помимо оказания прямого антивирусного действия на клетки, интерфероны, как известно, являются мощными модуляторами иммунного ответа, воздействуя на клетки иммунной системы (Gonzalez-Navajas J.M. et al Nature Reviews Immunology, 2012; 2, 125-35).
Толл-подобные рецепторы (TLR) представляют собой семейство из десяти паттерн-распознающих рецепторов, описанных у человека (Gay, N.J. et al, Annu. Rev. Biochem., 2007: 46, 141-165). TLR преимущественно экспрессируются клетками врожденной иммунной системы, где их функция заключается в контроле окружающей среды на предмет наличия признаков инфекции и, при их активации, мобилизации защитных механизмов, направленных на элиминацию проникающих патогенов. Ранние врожденные иммунные ответы, запускаемые TLR, ограничивают распространение инфекции, а провоспалительные цитокины и хемокины, индуцируемые ими, приводят к рекрутингу и активации антиген-представляющих клеток, В-клеток и Т-клеток. TLR могут модулировать природу приобретенных иммунных ответов, обеспечивая соответствующую защиту посредством активации дендритных клеток и высвобождения цитокинов (Akira S. et al, Nat. Immunol., 2001: 2, 675-680). Наблюдаемый профиль иммунного ответа, вызываемого различными агонистами TLR, зависит от типа активируемых клеток.
TLR7 является членом подгруппы TLR (TLR 3, 7, 8 и 9), локализованной в эндосомальном отделе клеток, которые становятся способными обнаруживать чужеродные нуклеиновые кислоты. TLR7 играет ключевую роль в противовирусной защите посредством распознавания оцРНК (одноцепочечная рибонуклеиновая кислота) (Diebold S.S. et al, Science, 2004: 303, 1529-1531; и Lund J. M. et al, PNAS, 2004: 101, 5598-5603). TLR7 имеет ограниченный профиль экспрессии у человека и экспрессируется преимущественно В-клетками и плазмоцитоидными дендритными клетками (pDC) и в меньшей степени моноцитами. Плазмоцитоидные DC представляют собой уникальную популяцию дендритных клеток лимфоидного происхождения (0,2-0,8% мононуклеарных клеток периферической крови (РВМС)), которые являются первичными продуцирующими интерферон I типа клетками, секретирующими высокие уровни интерферона альфа (IFNα) и интерферона бета (IFNβ) в ответ на вирусные инфекции (Liu Y-J, Annu. Rev. Immunol., 2005: 23, 275-306).
Введение имеющего небольшую молекулу соединения, которое может стимулировать врожденный иммунный ответ, включая активацию интерферонов I типа и других цитокинов через толл-подобные рецепторы, может стать важной стратегией лечения или предупреждения заболеваний человека. Были описаны имеющие небольшую молекулу агонисты TLR7, которые могут индуцировать интерферон альфа у животных и у человека (Takeda K. et al, Annu. Rev. Immunol., 2003: 21, 335-76). Агонисты TLR7 включают имидазохинолиновые соединения, такие как имиквимод и резиквимод, аналоги оксоаденина, а также нуклеозидные аналоги, такие как локсорибин и 7-тиа-8-оксогуанозин, которые, как уже давно известно, индуцируют интерферон альфа (Czarniecki. М., J. Med, Chem., 2008: 51, 6621-6626; Hedayat М. et al, Medicinal Research Reviews, 2012: 32, 294-325). При этом типе иммуномодуляторной стратегии имеется возможность идентифицировать соединения, которые могут быть полезны в лечении аллергических заболеваний (Moisan J. et al, Am. J. Physiol. Lung Cell Mol. Physiol., 2006: 290, L987-995), вирусных инфекций (Horcroft N.J. et al, J. Antimicrob. Chemther, 2012: 67, 789-801), рака (Krieg A., Curr. Oncol. Rep., 2004: 6(2), 88-95), других воспалительных состояний, таких как синдром раздраженного кишечника (Rakoff-Nahoum S., Cell., 2004, 23, 118(2): 229-41), и в качестве адъювантов вакцин (Persing etal. Trends Microbiol. 2002: 10(10 Suppl), S32-7).
Более конкретно, аллергические заболевания ассоциированы с Th2-смещенным иммунным ответом на аллергены. Th2-ответы ассоциированы с повышенными уровнями IgE (иммуноглобулин Е), который посредством воздействия на тучные клетки способствует гиперчувствительности к аллергенам, приводя к симптомам, наблюдаемым, например, при астме и аллергическом рините. У здоровых индивидуумов иммунный ответ на аллергены в большей степени сбалансирован со смешанным Th2/Th1 и регуляторным T-клеточным ответом. Было показано, что лиганды TLR7 снижают высвобождение цитокинов Th2 и повышают высвобождение цитокинов Th1 in vitro и ослабляют воспалительные ответы Th2-типа в моделях аллергического легкого in vivo (Duechs M.J., Pulmonary Pharmacology & Therapeutics, 2011: 24, 203-214; Fili L. et al, J. All Clin. Immunol., 2006: 118, 511-517; Tao et al, Chin. Med. J., 2006: 119, 640-648; Van LP. Eur. J. Immunol., 2011: 41, 1992-1999). Таким образом, лиганды TLR7 обладают потенциалом восстановления баланса иммунного ответа у индивидуумов, страдающих аллергией, и модифицирования заболевания. Недавние клинические исследования агониста TLR7 показали, что повторная интраназальная стимуляция TLR7 вызывает длительное снижение реактивности на аллерген у пациентов как с аллергическим ринитом, так и с аллергической астмой (Greiff L. Respiratory Research, 2012: 13, 53; Leaker B.R. et al, Am. J. Respir. Crit. Care Med., 2012: 185, A4184).
При поиске новых имеющих небольшую молекулу индукторов человеческого интерферона IFNα была разработана стратегия анализа для характеристики небольших молекул (вне зависимости от механизма), которая основана на стимуляции соединениями первичных человеческих донорских клеток или цельной крови и которая раскрыта здесь.
Краткое изложение сущности изобретения
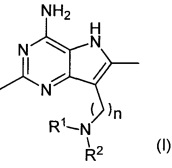
В первом аспекте настоящее изобретение относится к соединениям формулы (I) и их солям:
 ,
,
где R1, R2 и n являются такими, как описано ниже.
Некоторые соединения по изобретению, как было показано, являются индукторами человеческого интерферона и могут обладать требуемым подходящим для разработки профилем по сравнению с известными индукторами человеческого интерферона. В одном воплощении некоторые соединения по изобретению могут проявлять селективность к IFNα относительно TNFα (фактор некроза опухоли альфа). В дополнительном воплощении некоторые соединения по изобретению могут быть желательны для разработки, поскольку они могут быть менее сильнодействующие, чем другие индукторы человеческого интерферона.
Соединения, которые индуцируют человеческий интерферон, могут быть полезны в лечении или предупреждении различных расстройств, например в лечении или предупреждении аллергических заболеваний и других воспалительных состояний, например аллергического ринита и астмы, в лечении или предупреждении инфекционных заболеваний и рака. Поэтому, изобретение дополнительно относится к фармацевтическим композициям, содержащим соединение формулы (I) или его фармацевтически приемлемую соль. Настоящее изобретение дополнительно относится к способам лечения или предупреждения связанных с этим расстройств при использовании соединения формулы (I) или его фармацевтически приемлемой соли или фармацевтической композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль.
Соединения по изобретению также могут быть использованы в качестве адъювантов вакцин. Следовательно, настоящее изобретение дополнительно относится к вакцинной композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и антиген или антигенную композицию.
Некоторые соединения по изобретению могут быть сильнодействующими иммуномодуляторами и, соответственно, при обращении с ними следует проявлять осторожность.
Подробное описание изобретения
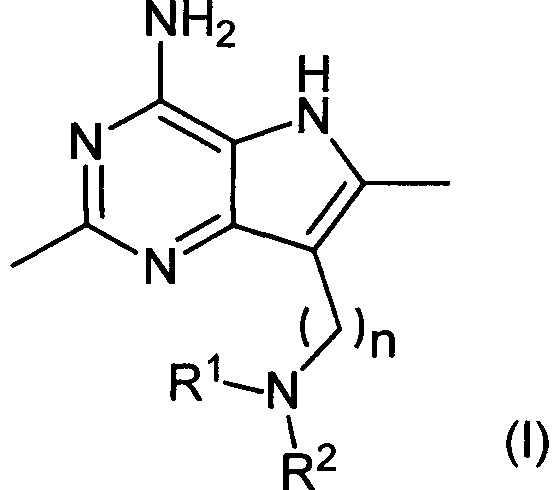
В первом аспекте настоящее изобретение относится к соединениям формулы (I) и их солям:
 ,
,
где:
R1 представляет собой водород, метил или -(СН2)2OR3,
R2 представляет собой метил или -(СН2)2OR4, или
R1 и R2 вместе с атомом азота, к которому они присоединены, связаны с образованием 5- или 6-членного гетероциклила, где 6-членный гетероциклил возможно замещен двумя заместителями гидрокси;
каждый из R3 и R4 независимо представляет собой водород или метил; и
n равен целому числу, имеющему значение 5 или 6.
В одном воплощении R1 представляет собой водород, метил или -(СН2)2OR3, и R2 представляет собой метил или -(CH2)2OR4. В другом воплощении R1 представляет собой водород, и R2 представляет собой -(CH2)2OR4, например -(СН2)2OH. В другом воплощении R1 и R2 оба представляют собой метил. В дополнительном воплощении R1 представляет собой -(CH2)2OR3, и R2 представляет собой -(CH2)2OR4.
В одном воплощении R1 и R2 вместе с атомом азота, к которому они присоединены, связаны с образованием 5- или 6-членного гетероциклила, где 6-членный гетероциклил возможно замещен двумя заместителями гидрокси. В другом воплощении R1 и R2 вместе с атомом азота, к которому они присоединены, связаны с образованием пирролидина. В другом воплощении R1 и R2 вместе с атомом азота, к которому они присоединены, связаны с образованием пиперидина, возможно замещенного двумя заместителями гидрокси. В дополнительном воплощении R1 и R2 вместе с атомом азота, к которому они присоединены, связаны с образованием пиперидин-3,5-диола.
В одном воплощении, когда R1 представляет собой -(CH2)2OR3, и R2 представляет собой -(СН2)2OR4, тогда R3 и R4 оба представляют собой водород. В другом воплощении, когда R1 представляет собой -(CH2)2OR3, и R2 представляет собой -(CH2)2OR4, тогда R3 представляет собой водород, и R4 представляет собой метил. В дополнительном воплощении, когда R1 представляет собой -(СН2)2OR3, и R2 представляет собой -(СН2)2OR4, тогда R3 и R4 оба представляют собой метил.
В одном воплощении n равен 5. В другом воплощении n равен 6.
Примеры соединений формулы (I) представлены в следующем перечне и составляют дополнительный аспект изобретения:
2,6-диметил-7-(6-(пиперидин-1-ил)гексил)-5Н-пирроло[3,2-d]пиримидин-4-амин;
2,6-диметил-7-(5-(пирролидин-1-ил)пентил)-5Н-пирроло[3,2-d]пиримидин-4-амин;
2,2'-((5-(4-амино-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-7-ил)пентил)азандиил)диэтанол;
2,2'-((6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)азандиил)диэтанол;
2-((6-(4-амино-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-7-ил)гексил)(2-метоксиэтил)амино)этанол;
7-(6-(бис(2-метоксиэтил)амино)гексил)-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амин;
2-((6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)амино)этанол;
(3R,5S)-1-(6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)пиперидин-3,5-диол;
(3R,5R)-1-(6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)пиперидин-3,5-диол; и
7-(6-(диметиламино)гексил)-2,6-диметил-5Н-пирроло[3,2-]пиримидин-4-амин;
и их соли.
Использованный здесь термин "гетероциклил" относится к моноциклическому насыщенному гетероциклическому кольцу, содержащему конкретное число атомов углерода и один гетероатом, который представляет собой азот. Такие гетероциклические кольца представляют собой пирролидин или пиперидин.
Следует понимать, что ссылки, данные здесь на соединения по изобретению, означают соединение формулы (I) в виде свободного основания или в виде соли, например фармацевтически приемлемой соли.
В одном аспекте изобретения соединение формулы (I) находится в форме свободного основания. В другом аспекте изобретения соединение формулы (I) находится в форме фармацевтически приемлемой соли.
Соли соединений формулы (I) включают фармацевтически приемлемые соли и соли, которые могут не быть фармацевтически приемлемыми, но могут быть полезны в получении соединений формулы (I) и их фармацевтически приемлемых солей. В одном аспекте изобретения соединение формулы (I) находится в форме фармацевтически приемлемой соли. Соли могут быть получены из некоторых неорганических или органических кислот.
Примерами солей являются фармацевтически приемлемые соли. Фармацевтически приемлемые соли включают соли присоединения кислот. Для обзора подходящих солей см. Berge et al., J. Pharm. Sci., 66:1-19 (1977).
Примеры фармацевтически приемлемых солей присоединения кислот соединения формулы (I) включают соли с неорганическими кислотами, такими как, например, соляная кислота, бромистоводородная кислота, ортофосфорная кислота, азотная кислота, фосфорная кислота или серная кислота, или с органическими кислотами, такими как, например, метансульфоновая кислота, этансульфоновая кислота, пара-толуолсульфоновая кислота, уксусная кислота, пропионовая кислота, молочная кислота, лимонная кислота, фумаровая кислота, яблочная кислота, янтарная кислота, салициловая кислота, малеиновая кислота, глицерофосфорная кислота, винная, бензойная, глутаминовая, аспарагиновая, бензолсульфоновая, нафталинсульфоновая, такая как 2-нафталинсульфоновая, капроновая кислота или ацетилсалициловая кислота.
В объем изобретения включены все возможные стехиометрические и нестехиометрические формы солей соединений формулы (I).
Соли могут быть образованы с использованием методик, хорошо известных в данной области техники, например, путем осаждения из раствора с последующей фильтрацией или путем выпаривания растворителя.
Обычно фармацевтически приемлемая соль присоединения кислоты может быть образована путем взаимодействия соединения формулы (I) с подходящей кислотой (такой как бромистоводородная, соляная, серная, малеиновая, пара-толуолсульфоновая, метансульфоновая, нафталинсульфоновая или янтарная кислоты), возможно в подходящем растворителе, таком как органический растворитель, с получением соли, которую обычно выделяют, например, путем кристаллизации и фильтрации.
Следует понимать, что многие органические соединения могут образовывать комплексы с растворителями, в которых они подвергаются взаимодействию или из которых их осаждают или кристаллизуют. Эти комплексы известны как "сольваты". Например, комплекс с водой известен как "гидрат". Растворители с высокими точками кипения и/или растворители с высокой способностью к образованию водородных связей, такие как вода, этанол, изопропиловый спирт и N-метил пиррол идинон, могут быть использованы для образования сольватов. Способы идентификации сольватов включают ЯМР (ядерный магнитный резонанс) и микроанализ, но не ограничиваются ими. Сольваты соединений формулы (I) включены в объем изобретения. Использованный здесь термин "сольват" включает сольваты как соединения в виде свободного основания, так и любой его соли.
Некоторые соединения по изобретению могут содержать хиральные атомы и, следовательно, могут существовать в одной или более чем одной стереоизомерной форме. Настоящим изобретением охватываются все стереоизомеры соединений по изобретению, включая оптические изомеры, как в виде индивидуальных стереоизомеров, так и в виде их смесей, включая рацемические модификации. Любой стереоизомер может содержать менее 10 мас. %, например менее 5 мас. % или менее 0,5 мас. %, любого другого стереоизомера. Например, любой оптический изомер может содержать менее 10 мас. %, например менее 5 мас. % или менее 0,5 мас. %, своего антипода.
Некоторые соединения по изобретению могут существовать в таутомерных формах. Следует понимать, что настоящим изобретением охватываются все таутомеры соединений по изобретению, как в виде индивидуальных таутомеров, так и в виде их смесей.
Соединения по изобретению могут находиться в кристаллической или аморфной форме. Более того, некоторые из кристаллических форм соединений по изобретению могут существовать в виде полиморфов, все из которых включены в объем настоящего изобретения. Особенный интерес представляет наиболее термодинамически стабильная полиморфная форма или формы соединений по изобретению.
Полиморфные формы соединений по изобретению могут быть охарактеризованы и дифференцированы с использованием ряда традиционных аналитических методик, включая дифракцию рентгеновских лучей на порошке (ДРЛП), инфракрасную спектроскопию (ИКС), рамановскую спектроскопию, дифференциальную сканирующую калориметрию (ДСК), термогравиметрический анализ (ТГА) и твердотельный ядерный магнитный резонанс (ттЯМР), но не ограничиваясь ими.
Настоящее изобретение также включает все подходящие изотопные варианты соединения формулы (I) или его фармацевтически приемлемой соли. Изотопный вариант соединения формулы (I) или его фармацевтически приемлемой соли определяют как вариант, в котором по меньшей мере один атом замещен атомом, имеющим такой же атомный номер, но атомная масса отличается от атомной массы, которая обычно обнаруживается в природе. Примеры изотопов, которые могут быть включены в соединения по изобретению, включают изотопы водорода, углерода, азота, кислорода, фтора и хлора, такие как 2Н, 3Н, 13С, 14С, 15N, 17O, 18O, 18F и 36Cl, соответственно. Некоторые изотопные варианты соединения формулы (I) или его соли или сольвата, например, варианты, в которые включен радиоактивный изотоп, такой как 3Н или 14С, полезны в исследованиях распределения лекарственных средств и/или субстратов в тканях. Тритий, то есть 3Н, и углерод-14, то есть 14С, являются особенно предпочтительными изотопами из-за простоты их получения и обнаружения. Кроме того, замещение изотопами, такими как дейтерий, то есть 2Н, может дать некоторые терапевтические преимущества, обусловленные более высокой метаболической стабильностью, например, увеличенный период полувыведения in vivo или потребность в более низких дозах и, следовательно, в некоторых случаях может быть предпочтительным. Изотопные варианты соединения формулы (I) или его фармацевтически приемлемой соли обычно могут быть получены традиционными методиками, такими как, например, иллюстративные способы или получения, описанные ниже в Примерах, с использованием соответствующих изотопных вариантов подходящих реагентов.
Из вышесказанного понятно, что сольваты, гидраты, изомеры и полиморфные формы соединений формулы (I) и их солей и сольватов включены в объем изобретения.
Получение соединений
Соединения формулы (I) и их соли могут быть получены с использованием описанной далее методологии, составляющей дополнительные аспекты этого изобретения.
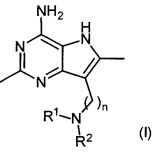
Соответственно, предложен способ получения соединения формулы (I) или его соли:
 ,
,
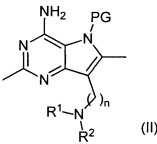
где R1, R2 и n являются такими, как определено выше, включающий снятие защиты с соединения формулы (II):
 ,
,
где R1, R2 и n являются такими, как определено выше для соединения формулы (I), a PG представляет собой защитную группу, такую как бензилоксиметил (ВОМ) или пара-толуолсульфонил, и после этого, при необходимости, получение соли соединения формулы (I).
Например, соединение формулы (II), где PG представляет собой ВОМ, растворяют в подходящем растворителе, например метаноле или этаноле, и пропускают через катализатор, например 10% палладий на угле, в присутствии водорода, при подходящей температуре, например 20-60°C, в подходящем проточном гидрирующем реакторе, таком как Thales H-cube™. Продукт (I) выделяют путем удаления растворителя и очистки, при необходимости.
Соединение формулы (II) может быть получено путем взаимодействия соединения формулы (III):
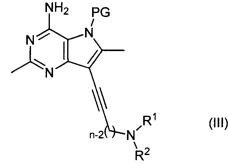 ,
,
где R1, R2 и n являются такими, как определено выше для соединения формулы (II), a PG представляет собой защитную группу, с водородом в присутствии катализатора.
Например, соединение формулы (III) растворяют в подходящем растворителе, например метиловом спирте или этиловом спирте, и пропускают через катализатор, например 10% палладий на угле, в присутствии водорода, при подходящей температуре, например 20-60°C, в подходящем проточном гидрирующем реакторе, таком как Thales H-Cube™. Продукт (II) выделяют путем удаления растворителя и очистки, при необходимости.
Когда защитная группа представляет собой бензилоксиметильную (ВОМ) группу, реакция восстановления алкина может привести к одновременному удалению защитной группы с получением непосредственно соединений формулы (I).
Соединение формулы (III) может быть получено путем взаимодействия соединения формулы (IV):
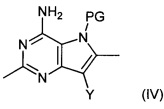 ,
,
где Y представляет собой уходящую группу, например галоген, такой как йод или бром, или алкилсульфонат, такой как трифторметансульфонат, a PG представляет собой защитную группу, с соединением формулы (V):
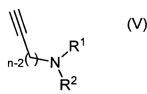
где R1, R2 и n являются такими, как определено для соединения формулы (I).
Например, соединение формулы (IV) и соединение формулы (V) растворяют в подходящем растворителе, например N,N-диметилформамиде, в присутствии йодида меди(I), подходящего катализатора, например дихлорида бис(трифенилфосфин)палладия(II), и подходящего основания, например триэтиламина, и нагревают при подходящей температуре, например 20-55°C, в течение подходящего периода времени, например 0,5-17 часов. Продукт (III) выделяют после водной обработки и очистки.
Соединение формулы (V) может быть получено путем взаимодействия соединения формулы (VI):
 ,
,
где n является таким, как определено для соединения формулы (I), и X представляет собой уходящую группу, такую как галоген, например хлор, бром или йод, или алкилсульфонат, например лара-толуолсульфонат, с соединением формулы (VII):
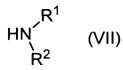 ,
,
где R1 и R2 являются такими, как определено для соединения формулы (I).
Например, соединение формулы (VI) и соединение формулы (VII) и подходящее основание, например гидрокарбонат натрия, растворяют в подходящем растворителе, например N,N-диметилформамиде, и нагревают при подходящей температуре, например 80-100°C, в течение подходящего периода времени, например 16-18 часов. Продукт (V) выделяют после водной обработки и очистки, например путем выделения подходящей кристаллической соли, например оксалата.
Соединения формулы (VI) и формулы (VII) либо имеются в продаже, либо могут быть получены способами, описанными в литературе.
Альтернативно, соединение формулы (III) может быть получено путем взаимодействия соединения формулы (VIII):
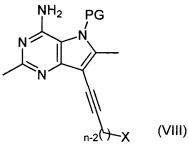 ,
,
где n является таким, как определено выше для соединения формулы (I), X представляет собой уходящую группу, как определено выше для соединений формулы (VI), и PG представляет собой защитную группу, с соединением формулы (VII).
Например, соединение формулы (VIII), соединение формулы (VII) и подходящее основание, например триэтиламин, растворяют в подходящем растворителе, например ацетонитриле, и нагревают при подходящей температуре, например 60-80°C, в течение подходящего периода времени, например 16-26 часов. Продукт (III) выделяют после водной обработки и очистки.
Соединения формулы (VIII) могут быть получены путем взаимодействия соединений формулы (IV) с соединениями формулы (VI). Например, соединение формулы (IV) и соединение формулы (VI) растворяют в подходящем растворителе, например N,N-диметилформамиде, в присутствии йодида меди(I), подходящего катализатора, например дихлорида бис(трифенилфосфин)палладия(II), и подходящего основания, например триэтиламина, и нагревают при подходящей температуре, например 65°C, в течение подходящего периода времени, например 18-20 часов. Продукт (VIII) выделяют после водной обработки и очистки.
Соединения формулы (IV) могут быть получены путем взаимодействия соединений формулы (IX):
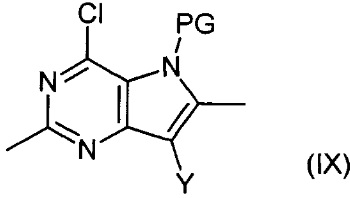 ,
,
где Y является таким, как определено для соединения формулы (IV), и PG представляет собой защитную группу, с раствором аммиака.
Например, водный раствор аммиака (0,88) добавляют в раствор соединения формулы (IX) в подходящем растворителе, например изопропиловом спирте. Полученную смесь затем нагревают в микроволновом нагревателе при подходящей температуре, например 120-150°С, в течение подходящего периода времени, например 1-2 часов. Продукт (IV) выделяют после водной обработки и очистки.
Соединения формулы (IX) могут быть получены путем взаимодействия соединений формулы (X):
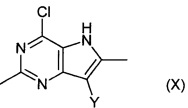 ,
,
где Y является таким, как определено для соединения формулы (IV), с соединением формулы (XI):
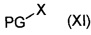 ,
,
где соединение формулы (XI) является подходящим предшественником защитной группы PG, например бензилхлорметиловым эфиром.
Например, соединение формулы (X) в подходящем растворителе, например N,N-диметилформамиде или тетра гидрофура не, подвергают взаимодействию с подходящим основанием, например суспензией гидрида натрия в масле. Добавляют соединение формулы (XI), например бензилхлорметиловый эфир, и реакционную смесь перемешивают при подходящей температуре, например 20°C, в течение подходящего периода времени, например 1-4 часов. Продукт (IX) выделяют после водной обработки и очистки.
Соединения формулы (X) могут быть получены путем взаимодействия соединений формулы (XII):
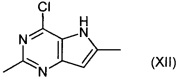 ,
,
с галогенирующим агентом, например N-йодсукцинимидом.
Например, соединение формулы (XII) растворяют в подходящем растворителе, например тетрагидрофуране, подвергают взаимодействию с N-йодсукцинимидом при подходящей температуре, например 20°C, в течение подходящего периода времени, например 1-2 часов. Продукт (X) выделяют после водной обработки и очистки.
Соединения формулы (XII) могут быть получены путем взаимодействия соединений формулы (XIII):
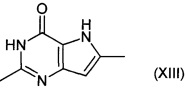
с хлорирующим агентом, например хлорангидридом фосфорной кислоты.
Например, соединение формулы (XIII) суспендируют в хлорангидриде фосфорной кислоты и нагревают при подходящей температуре, например 90-120°C, в течение подходящего периода времени, например 3-30 часов. Избыток хлорангидрида фосфорной кислоты может быть удален под вакуумом, затем остаток выливают на лед, и рН смеси доводят до 7-9. Продукт затем экстрагируют подходящим органическим растворителем, например этилацетатом. Продукт (XIII) выделяют путем удаления растворителя и очистки, при необходимости.
Соединения формулы (XIII) могут быть получены путем взаимодействия соединений формулы (XIV):
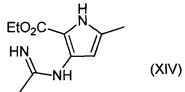
с подходящим основанием, например гидроксидом натрия.
Например, раствор соединений формулы (XIV) в подходящем растворителе, например этиловом спирте, обрабатывают водным раствором гидроксида натрия, и реакционную смесь перемешивают при подходящей температуре, например 80-100°C, в течение подходящего периода времени, например 2-18 часов. Продукт (XIII) выделяют после водной обработки и очистки.
Соединения формулы (XIV) могут быть получены путем взаимодействия соединений формулы (XV):
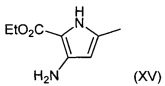
с ацетонитрилом.
Например, суспензию соединения формулы (XV) в ацетонитриле обрабатывают раствором хлороводорода в подходящем растворителе, например раствором хлороводорода в 1,4-диоксане, и нагревают при подходящей температуре 50-70°C в течение подходящего периода времени, например 16-96 часов. Продукт (XIV) выделяют после фильтрации.
Соединения формул (VI), (VII), (XI) и (XV) либо известны в литературе, либо имеются в продаже, например от Sigma-Aldrich, UK, или могут быть получены аналогично известным способам, например способам, раскрытым в стандартных руководствах по методике синтеза, таких как J. March, Advanced Organic Chemistry, 6th Edition (2007), WileyBlackwell или Comprehensive Organic Synthesis (Trost B.M. and Fleming I., (Eds.), Pergamon Press, 1991), каждое из которых включено здесь посредством ссылки как относящееся к таким способам.
Примеры других защитных групп, которые могут быть использованы в путях синтеза, описанных здесь, и способы их удаления можно найти в Т.W. Greene 'Protective Groups in Organic Synthesis', 4th Edition, J. Wiley and Sons, 2006, включенном здесь посредством ссылки как относящийся к таким способам.
Для любых описанных здесь выше взаимодействий или способов могут быть использованы общепринятые способы нагревания и охлаждения, например масляные бани с регулируемой температурой или нагревательные плитки с регулируемой температурой и бани со льдом и солью или бани с сухим льдом и ацетоном, соответственно. Могут быть использованы общепринятые способы выделения, например экстракция из водных или неводных растворителей или в водные или неводные растворители. Могут быть использованы общепринятые способы сушки органических растворителей, растворов или экстрактов, такие как встряхивание с безводным сульфатом магния или безводным сульфатом натрия, или пропускание через гидрофобную фритту. В случае необходимости могут быть использованы общепринятые способы очистки, например кристаллизация и хроматография, например хроматография на диоксиде кремния или обращенно-фазовая хроматография. Кристаллизация может быть осуществлена с использованием традиционных растворителей, таких как этилацетат, метанол, этанол или бутанол или их водные смеси. Следует понимать, что конкретное время и температуры реакций обычно могут быть определены методами мониторинга реакций, например посредством тонкослойной хроматографии и ЖХ-МС (жидкостная хроматография-масс-спектрометрия).
Когда это целесообразно, отдельные изомерные формы соединений по изобретению могут быть получены в виде индивидуальных изомеров с использованием общепринятых способов, таких как фракционная кристаллизация диастереоизомерных производных или хиральная высокоэффективная жидкостная хроматография (хиральная ВЭЖХ).
Абсолютная стереохимия соединений может быть определена с использованием общепринятых способов, таких как рентгеновская кристаллография.
Способы применения
Примеры болезненных состояний, при которых соединения формулы (I) и их фармацевтически приемлемые соли оказывают потенциально полезные эффекты, включают аллергические заболевания и другие воспалительные состояния, например аллергический ринит и астму, инфекционные заболевания и рак. Соединения формулы (I) и их фармацевтически приемлемые соли также являются потенциально полезными в качестве адъювантов вакцин.
В качестве модуляторов иммунного ответа соединения формулы (I) и их фармацевтически приемлемые соли также могут быть полезны в лечении и/или предупреждении иммуноопосредованных расстройств, включая воспалительные или аллергические заболевания, такие как астма, аллергический ринит и риноконъюнктивит, пищевая аллергия, состояния легочной гиперчувствительности, эозинофильный пневмонит, состояния гиперчувствительности замедленного типа, атеросклероз, панкреатит, гастрит, колит, остеоартрит, псориаз, саркоидоз, легочный фиброз, респираторный дистресс-синдром, бронхиолит, хроническая обструктивная болезнь легких, синусит, муковисцидоз, актинический кератоз, кожная дисплазия, хроническая крапивница, экзема и все типы дерматита, но не ограничиваются ими.
Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть полезны в лечении и/или предупреждении реакций на респираторные инфекции, включая обострения, вызванные вирусными инфекциями дыхательных путей, и тонзиллит, но не ограничиваются ими. Соединения также могут быть полезны в лечении и/или предупреждении аутоиммунных заболеваний, включая ревматоидный артрит, псориатический артрит, системную красную волчанку, болезнь Шегрена, анкилозирующий спондилит, склеродерму, дерматомиозит, диабет, отторжение трансплантата, включая заболевание "трансплантат против хозяина", воспалительные заболевания кишечника, включая, без ограничения, болезнь Крона и неспецифический язвенный колит, но не ограничиваются ими.
Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть полезны в лечении или предупреждении инфекционных заболеваний, включая заболевания, вызываемые вирусами гепатита (например вирусом гепатита В, вирусом гепатита С), вирусом иммунодефицита человека, папилломавирусами, вирусами герпеса, респираторными вирусами (например, вирусами гриппа, респираторно-синцитиальным вирусом, риновирусом, метапневмовирусом, вирусом парагриппа, SARS (тяжелый острый респираторный синдром)) и вирусом лихорадки Западного Нила, но не ограничиваются ими. Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть полезны в лечении или предупреждении микробных инфекций, вызываемых, например, бактериями, грибами или простейшими. Эти инфекции включают туберкулез, бактериальную пневмонию, аспергиллез, гистоплазмоз, кандидоз, пневмоцитоз, лепру, хламидии, криптококковое заболевание, криптоспоридиоз, токсоплазмоз, лейшманию, малярию и трипаносомоз, но не ограничиваются ими.
Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть полезны в лечении или предупреждении различных видов рака, в частности в лечении или предупреждении видов рака, которые, как известно, реагируют на иммунотерапию, включая почечно-клеточную карциному, рак легкого, рак молочной железы, колоректальный рак, рак мочевого пузыря, меланому, лейкоз, лимфомы и рак яичника, но не ограничиваясь ими.
Специалистам в данной области техники следует понимать, что в данном описании ссылки на лечение относятся к лечению установленных состояний. Однако, соединения формулы (I) и их фармацевтически приемлемые соли в зависимости от состояния также могут быть полезны в предупреждении некоторых заболеваний. Таким образом, в одном воплощении предложено лечение или предупреждение заболевания. В другом воплощении предложено лечение заболевания. В дополнительном воплощении предложено предупреждение заболевания.
Таким образом, в дополнительном аспекте изобретения предложено соединение формулы (I) или его фармацевтически приемлемая соль для применения в терапии.
Следует понимать, что, когда соединение формулы (I) или его фармацевтически приемлемую соль применяют в терапии, его(ее) применяют в качестве активного терапевтического агента.
Таким образом, предложено также соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении или предупреждении аллергических заболеваний и других воспалительных состояний, инфекционных заболеваний и рака.
Таким образом, предложено также соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении или предупреждении аллергического ринита.
Таким образом, предложено также соединение формулы (I) или его фармацевтически приемлемая соль для применения в лечении или предупреждении астмы.
Дополнительно предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или предупреждения аллергических заболеваний и других воспалительных состояний, инфекционных заболеваний и рака.
Дополнительно предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или предупреждения аллергического ринита.
Дополнительно предложено применение соединения формулы (I) или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или предупреждения астмы.
Дополнительно предложен способ лечения или предупреждения аллергических заболеваний и других воспалительных состояний, инфекционных заболеваний и рака, включающий введение субъекту-человеку, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Дополнительно предложен способ лечения или предупреждения аллергического ринита, включающий введение субъекту-человеку, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Дополнительно предложен способ лечения или предупреждения астмы, включающий введение субъекту-человеку, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его фармацевтически приемлемой соли.
Соединения формулы (I) и его фармацевтически приемлемые соли также потенциально полезны в качестве адъювантов вакцин.
Таким образом, в дополнительном аспекте изобретения предложена вакцинная композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и антиген или антигенную композицию, для применения в терапии.
Таким образом, в дополнительном аспекте изобретения предложено применение соединения формулы (I) или его фармацевтически приемлемой соли и антигена или антигенной композиции в изготовлении лекарственного средства для применения в терапии.
Дополнительно предложен способ лечения или предупреждения заболевания, включающий введение субъекту-человеку, страдающему заболеванием или восприимчивому к заболеванию, вакцинной композиции, содержащей соединение формулы (I) или его фармацевтически приемлемую соль и антиген или антигенную композицию.
Композиции
Соединения формулы (I) и их фармацевтически приемлемые соли будут, как правило, но не обязательно, приготовлены в виде фармацевтических композиций перед введением пациенту. Соответственно, в другом аспекте изобретения предложена фармацевтическая композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и один или более чем один фармацевтически приемлемый эксципиент.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть приготовлены в виде препаратов для введения любым подходящим путем. Соединения формулы (I) и их фармацевтически приемлемые соли могут быть приготовлены, например, в виде препаратов для перорального, местного, ингаляционного, интраназального, трансбуккального, парентерального (например внутривенного, подкожного, внутрикожного или внутримышечного) или ректального введения. В одном аспекте соединения формулы (I) и их фармацевтически приемлемые соли приготовлены в виде препарата для перорального введения. В дополнительном аспекте соединения формулы (I) и их фармацевтически приемлемые соли приготовлены в виде препарата для местного введения, например интраназального или ингаляционного введения.
Таблетки и капсулы для перорального введения могут содержать традиционные эксципиенты, такие как связывающие агенты, например сироп, аравийскую камедь, желатин, сорбит, трагакант, растительный клей из крахмала, целлюлозу или поливинилпирролидон; наполнители, например лактозу, микрокристаллическую целлюлозу, сахар, маисовый крахмал, фосфат кальция или сорбит; смазывающие вещества, например стеарат магния, стеариновую кислоту, тальк, полиэтиленгликоль или диоксид кремния; разрыхлители, например картофельный крахмал, кроскармеллозу натрия или крахмал-гликолят натрия; или смачивающие агенты, такие как лаурилсульфат натрия. Таблетки могут быть покрыты оболочкой с использованием методик, хорошо известных в данной области техники.
Пероральные жидкие препараты могут быть в форме, например, водных или масляных суспензий, растворов, эмульсий, сиропов или эликсиров, или могут быть представлены в виде сухого продукта для разведения водой или другим подходящим носителем перед применением. Такие жидкие препараты могут содержать традиционные добавки, такие как суспендирующие агенты, например сорбитовый сироп, метилцеллюлозу, глюкозо-сахарный сироп, желатин, гидроксиметилцеллюлозу, карбоксиметилцеллюлозу, гель стеарата алюминия или гидрогенизированные пищевые жиры; эмульгаторы, например лецитин, сорбитанмоноолеат или аравийскую камедь; неводные носители (которые могут включать пищевые масла), например миндальное масло, фракционированное кокосовое масло, масляные эфиры, пропиленгликоль или этиловый спирт; или консерванты, например метил- или пропил-пара-гидроксибензоаты или сорбиновую кислоту. Препараты могут также содержать буферные соли, ароматизаторы, красители и/или подсластители (например маннит), если это целесообразно.
Композиции для интраназального введения включают водные композиции, которые вводят в нос каплями или посредством нагнетательного насоса. Подходящие композиции содержат воду в качестве разбавителя или носителя для этой цели. Композиции для введения в легкое или нос могут содержать один или более чем один эксципиент, например один или более чем один суспендирующий агент, один или более чем один консервант, одно или более чем одно поверхностно-активное вещество, один или более чем один агент, регулирующий тоничность, один или более чем один сорастворитель, и могут включать компоненты для регулирования рН композиции, например, буферную систему. Дополнительно, композиции могут содержать другие эксципиенты, такие как антиоксиданты, например метабисульфит натрия, и маскирующие вкус агенты. Композиции также могут быть введены в нос или другие участки дыхательных путей посредством распыления.
Интраназальные композиции могут обеспечивать доставку соединения(й) формулы (I) или его(их) фармацевтически приемлемой(ых) соли(ей) во все участки носовых пазух (ткань-мишень), а также могут давать возможность соединению(ям) формулы (I) или его(их) фармацевтически приемлемой(ым) соли(ям) оставаться в контакте с тканью-мишенью в течение длительных периодов времени. Подходящим для пациента режимом дозирования интраназальной композиции может быть медленная ингаляция через нос после очистки носовой полости. Во время ингаляции композицию можно вводить в одну ноздрю, зажимая рукой другую. Эту процедуру затем можно повторить в отношении другой ноздри. Обычно по одному или двум впрыскиваниям в каждую ноздрю можно вводить вышеописанной методикой один, два или три раза в сутки, предпочтительно один раз в сутки. Особый интерес представляют интраназальные композиции, подходящие для введения один раз в сутки.
Суспендирующий(ие) агент(ы), если они включены в состав, обычно будут присутствовать в количестве от 0,1 до 5% (масс./масс.), например от 1,5% до 2,4% (масс./масс.), в расчете на общую массу композиции. Примеры фармацевтически приемлемых суспендирующих агентов включают Avicel® (микрокристаллическая целлюлоза и карбоксиметилцеллюлоза натрия), карбоксиметилцеллюлозу натрия, вигум, трагакант, бентонит, метилцеллюлозу, ксантановую камедь, карбопол и полиэтиленгликоли, но не ограничиваются ими.
Композиции для введения в легкое или нос могут содержать один или более чем один эксципиент и могут быть защищены от микробного или грибкового заражения или роста посредством включения в состав одного или более чем одного консерванта. Примеры фармацевтически приемлемых антимикробных агентов или консервантов включают четвертичные аммониевые соединения (например хлорид бензалкония, хлорид бензэтония, цетримид, хлорид цетилпиридиния, хлорид лауралкония и хлорид миристилпиколиния), ртутьсодержащие агенты (например нитрат фенилртути, ацетат фенилртути и тимеросал), спиртовые агенты (например хлорбутанол, фенилэтиловый спирт и бензиловый спирт), антибактериальные эфиры (например эфиры пара-гидроксибензойной кислоты), хелатирующие агенты, такие как динатриевая соль этилендиаминтетрауксусной кислоты (EDTA), и другие антимикробные агенты, такие как хлоргексидин, хлоркрезол, сорбиновая кислота и ее соли (такие как сорбат калия) и полимиксин, но не ограничиваются ими. Примеры фармацевтически приемлемых противогрибковых агентов или консервантов включают бензоат натрия, сорбиновую кислоту, пропионат натрия, метилпарабен, этилпарабен, пропилпарабен и бутилпарабен, но не ограничиваются ими. Консервант(ы), если он(и) включен(ы) в состав, может(гут) присутствовать в количестве от 0,001 до 1% (масс./масс.), например от 0,015% до 0,5% (масс./масс.), в расчете на общую массу композиции.
Композиции (например те, в которых по меньшей мере одно соединение находится в суспензии) могут включать одно или более чем одно поверхностно- активное вещество для облегчения растворения частиц лекарственного средства в водной фазе композиции. Например, используемое количество поверхностно-активного вещества представляет собой количество, которое не будет вызывать пенообразование во время смешивания. Примеры фармацевтически приемлемых поверхностно-активных веществ включают жирные спирты, сложные эфиры и простые эфиры, такие как моноолеат полиоксиэтилен (20) сорбитана (Polysorbate 80), простые эфиры макрогола и полоксамеры. Поверхностно-активное вещество может присутствовать в количестве от примерно 0,01 до 10% (масс./масс.), например от 0,01 до 0,75% (масс./масс.), например примерно 0,5% (масс./масс.), в расчете на общую массу композиции.
Один или более чем один агент, регулирующий тоничность, может быть включен в состав композиции для достижения тоничности с жидкостями организма, например жидкостями носовой полости, что приводит к снижению степени раздражения. Примеры фармацевтически приемлемых агентов, регулирующих тоничность, включают хлорид натрия, декстрозу, ксилит, хлорид кальция, глюкозу, глицерин и сорбит, но не ограничиваются ими. Агент, регулирующий тоничность, если он присутствует, может быть включен в количестве от 0,1 до 10% (масс./масс.), например от 4,5 до 5,5% (масс./масс.), например примерно 5,0% (масс./масс.), в расчете на общую массу композиции.
Композиции по изобретению могут быть забуферены посредством добавления подходящих буферных агентов, таких как цитрат натрия, лимонная кислота, трометамол, фосфаты, такие как динатрийфосфат (например додекагидрат, гептагидрат, дигидрат и безводные формы) или фосфат натрия и их смеси.
Буферный агент, если он присутствует, может быть включен в количестве от 0,1 до 5% (масс./масс.), например от 1 до 3% (масс./масс.), в расчете на общую массу композиции.
Примеры маскирующих вкус агентов включают сукралозу, сахарозу, сахарин или его соль, фруктозу, декстрозу, глицерин, кукурузный сироп, аспартам, ацесульфам-K, ксилит, сорбит, эритрит, глицирризинат аммония, тауматин, неотам, маннит, ментол, эвкалиптовое масло, камфору, натуральный ароматизатор, искусственный ароматизатор и их комбинации.
Один или более чем один сорастворитель может быть включен в состав композиции для повышения растворимости лекарственного(ых) соединения(ий) и/или других эксципиентов. Примеры фармацевтически приемлемых сорастворителей включают пропиленгликоль, дипропиленгликоль, этиленгликоль, глицерин, этанол, полиэтиленгликоли (например PEG300 или PEG400) и метанол, но не ограничиваются ими. В одном воплощении сорастворитель представляет собой пропиленгликоль.
Сорастворитель(и), если он(и) присутствует(ют), может(гут) быть включен(ы) в количестве от 0,05 до 30% (масс./масс.), например от 1 до 25% (масс./масс.), например от 1 до 10% (масс./масс.), в расчете на общую массу композиции.
Композиции для ингаляционного введения включают водные, органические или водно-органические смеси, порошковые или кристаллические композиции, которые вводят в дыхательные пути посредством нагнетательного насоса или ингалятора, например с помощью ингаляторов с резервуаром для сухого порошка, однодозовых ингаляторов сухого порошка, дозирующих многодозовых ингаляторов сухого порошка, назальных ингаляторов или аэрозольных ингаляторов под давлением, небулайзеров или инсуффляторов. Подходящие композиции содержат воду в качестве разбавителя или носителя для этой цели и могут быть приготовлены с традиционными эксципиентами, такими как буферные агенты, агенты, модифицирующие тоничность, и им подобные. Водные композиции также могут быть введены в нос и другие отделы дыхательных путей посредством распыления. Такие композиции могут представлять собой водные растворы или суспензии или аэрозоли, доставляемые из упаковок под давлением, таких как дозирующий ингалятор, с использованием подходящего сжиженного пропеллента.
Композиции для введения местно в нос (например, для лечения ринита) или в легкое включают находящиеся под давлением аэрозольные композиции и водные композиции, доставляемые в полости носа посредством нагнетательного насоса. Композиции, не находящиеся под давлением и подходящие для введения местно в носовую полость, представляют особый интерес. Подходящие композиции содержат воду в качестве разбавителя или носителя для этой цели. Водные композиции для введения в легкое или нос могут быть приготовлены с традиционными эксципиентами, такими буферные агенты, агенты, модифицирующие тоничность, и им подобные. Водные композиции можно также вводить в нос посредством распыления.
Для доставки жидкой композиции в полости носа обычно может быть использован жидкостный дозатор. Жидкая композиция может быть водной или неводной, но обычно водной. Соединение формулы (I) или его фармацевтически приемлемая соль может быть приготовлено в виде суспензии или раствора. Такой жидкостный дозатор может иметь дозирующую насадку или дозирующее сопло, через которое отмеренная доза жидкой композиции дозируется при нажатии пользователем на нагнетательный механизм жидкостного дозатора. Такие жидкостные дозаторы, как правило, снабжены резервуаром для многократных отмериваемых доз жидкой композиции, причем дозы дозируются при последовательных срабатываниях насоса. Альтернативно, жидкостный дозатор для доставки жидкой композиции в полости носа может быть выполнен с возможностью ограничения дозы, например одноразовый дозатор, содержащий однократную дозу. Дозирующая насадка или сопло могут иметь конфигурацию, подходящую для введения в ноздри пользователя для дозированного распыления жидкой композиции в полость носа. Жидкостный дозатор вышеупомянутого типа описан и проиллюстрирован в публикации международной заявки на патент WO 2005/044354 (Glaxo Group Limited). Дозатор имеет корпус, который содержит выпускающее жидкость устройство с нагнетательным насосом, установленным на контейнере, содержащем жидкую композицию. Корпус имеет по меньшей мере один приводимый в действие пальцами боковой рычаг, который двигается внутрь относительно корпуса, перемещая контейнер вверх в корпусе посредством эксцентрика, заставляя насос сжимать и нагнетать отмеренную дозу композиции из штока насоса через назальную насадку корпуса. В одном воплощении жидкостный дозатор представляет собой дозатор общего типа, проиллюстрированный на Фиг. 30-40 в WO 2005/044354.
Водные композиции, содержащие соединение формулы (I) или его фармацевтически приемлемую соль, также могут быть доставлены с помощью насоса, как раскрыто в публикации международной заявки на патент WO 2007/138084 (Glaxo Group Limited), например, как описано в этом документе со ссылкой на Фиг. 22-46, или как описано в заявке на патент Великобритании GB 0723418.0 (Glaxo Group Limited), например, как описано со ссылкой на Фиг. 7-32. Насос может быть приведен в действие с помощью пускателя, как изображено на Фиг. 1-6 в GB 0723418.0.
Сухие порошковые композиции для местной доставки в легкое посредством ингаляции могут быть представлены, например, в капсулах и картриджах, например из желатина, или в блистерах, например из ламинированной алюминиевой фольги, для использования в ингаляторе или инсуффляторе. Композиции в форме порошковых смесей обычно содержат порошковую смесь для ингаляции соединения формулы (I) или его фармацевтически приемлемой соли и подходящей порошковой основы (вещества, являющегося носителем/разбавителем/эксципиентом), такой как моно-, ди- или полисахариды (например лактоза или крахмал). Помимо лекарственного средства и носителя сухие порошковые композиции могут также содержать дополнительный эксципиент (например тройной агент, такой как сложный эфир сахара, например октаацетат целлобиозы, стеарат кальция или стеарат магния).
В одном воплощении композиция, подходящая для ингаляционного введения, может быть включена во множество герметичных дозовых контейнеров, которыми снабжена(ы) упаковка(и) лекарственного средства, вставленная(ые) внутрь подходящего ингаляционного устройства. Контейнеры могут быть разрываемыми, открываемыми путем снятия предохранительной пленки или иным образом вскрываемыми последовательно, и дозы сухой порошковой композиции могут быть введены посредством ингаляции из наконечника ингаляционного устройства, как известно в данной области техники. Упаковка лекарственного средства может принимать ряд различных форм, например форму диска или длинной полоски. Типичными ингаляционными устройствами являются устройства DISKHALER™ и DISKUS™, продаваемые фирмой GlaxoSmithKline.
Сухая порошковая ингалируемая композиция также может быть представлена в виде объемной массы в резервуаре в ингаляционном устройстве, которое к тому же снабжено дозирующим механизмом для отмеривания дозы композиции из резервуара в ингаляционный канал, откуда отмеренную дозу пациент может вдыхать через наконечник устройства. Примерами имеющихся в продаже устройств этого типа являются TURBUHALER™ (AstraZeneca), TWISTHALER™ (Schering) и CLICKHALER™ (Innovata).
Еще один способ доставки сухой порошковой ингалируемой композиции предназначен для отмеренных доз композиции, находящихся в капсулах (одна доза на капсулу), которые затем загружаются в ингаляционное устройство обычно самим пациентом, когда это требуется. Устройство имеет средства для разрывания, прокалывания или иного открытия капсулы, чтобы доза могла поступить в легкие пациента, когда он вдыхает через наконечник устройства. В качестве примеров таких устройств, имеющихся в продаже, могут быть упомянуты ROTAHALER™ (GlaxoSmithKline) и HANDIHALER™ (Boehringer Ingelheim).
Аэрозольные композиции под давлением, подходящие для ингаляции, могут представлять собой либо суспензию, либо раствор и могут содержать соединение формулы (I) или его фармацевтически приемлемую соль и подходящий пропеллент, такой как фторуглерод или водородсодержащий хлорфторуглерод или их смеси, в частности гидрофторалканы, особенно 1,1,1,2-тетрафторэтан, 1,1,1,2,3,3,3-гептафтор-н-пропан или их смесь. Аэрозольная композиция возможно может содержать дополнительные эксципиенты композиции, хорошо известные в данной области техники, такие как поверхностно-активные вещества, например олеиновую кислоту, лецитин или олигомолочную кислоту или их производные, описанные, например, в WO 94/21229 и WO 98/34596 (Minnesota Mining and Manufacturing Company), и сорастворители, например этанол. Композиции под давлением обычно находятся в закрытом баллончике (например алюминиевом баллончике) с клапаном (например дозирующим клапаном), встроенным в пускатель, оснащенный наконечником.
Мази, кремы и гели могут быть приготовлены, например, в виде препарата с водной или масляной основой с добавлением подходящего загустителя и/или желатинирующего агента и/или растворителей. Таким образом, такие основы могут включать, например, воду и/или масло, такое как вазелиновое масло или растительное масло, такое как арахисовое масло или касторовое масло, или растворитель, такой как полиэтиленгликоль. Загустители и желатинирующие агенты, которые могут быть использованы в соответствии с природой основы, включают мягкий парафин, стеарат алюминия, цетостеариловый спирт, полиэтиленгликоли, ланолин, пчелиный воск, карбоксиполиметилен и производные целлюлозы и/или глицерилмоностеарат и/или неионные эмульгаторы.
Лосьоны могут быть приготовлены в виде препарата с водной или масляной основой и будут, как правило, также содержать один или более чем один эмульгатор, стабилизатор, диспергирующий агент, суспендирующий агент или загуститель.
Порошки для наружного применения могут быть приготовлены с помощью любой подходящей порошковой основы, например талька, лактозы или крахмала. Капли могут быть приготовлены в виде препарата с водной или неводной основой, также содержащей один или более чем один диспергирующий агент, солюбилизатор, суспендирующий агент или консервант.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть, например, приготовлены для трансдермальной доставки путем введения в состав пластырей или других устройств (например, устройств со сжатым газом), которые доставляют активный компонент в кожу.
Для трансбуккального введения композиции могут быть приготовлены в форме таблеток или лепешек, изготовленных традиционным способом.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть также приготовлены в виде суппозиториев, содержащих, например, традиционные суппозиторные основы, такие как масло какао или другие глицериды.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть также приготовлены в виде препаратов для парентерального введения посредством болюсной инъекции или непрерывной инфузии и могут быть представлены в стандартной лекарственной форме, например в ампулах, флаконах, инфузионных системах небольшого объема или предварительно заполненных шприцах, или в многодозовых контейнерах с добавлением консерванта. Композиции могут быть приготовлены в таких формах, как растворы, суспензии или эмульсии в водных или неводных носителях, и могут содержать вспомогательные агенты, такие как антиоксиданты, буферные агенты, антимикробные агенты и/или агенты, регулирующие тоничность. Альтернативно, активный ингредиент может находиться в форме порошка для разведения подходящим носителем, например стерильной апирогенной водой, перед применением. Сухая твердая форма может быть приготовлена путем асептического заполнения индивидуальных стерильных контейнеров стерильным порошком или путем асептического заполнения каждого контейнера стерильным раствором и посредством сублимационной сушки.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть также включены в состав вакцин в качестве адъювантов для модулирования их активности. Такие композиции могут содержать антитело(а) или фрагмент(ы) антител(а) или антигенный компонент, включая белок, ДНК, живые или мертвые бактерии и/или вирусы или вирусоподобные частицы, но не ограничиваясь ими, вместе с одним или более чем одним компонентом с адъювантной активностью, включая соли алюминия, масляные и водные эмульсии, белки теплового шока, препараты и производные липида А, гликолипиды, другие агонисты TLR, такие как CpG ДНК или подобные агенты, цитокины, такие как GM-CSF (гранулоцитарно-макрофагальный колониестимулирующий фактор) или IL-12 (интерлейкин 12) или подобные агенты, но не ограничиваясь ими.
В дополнительном аспекте изобретения предложен адъювант вакцин, содержащий соединение формулы (I) или его фармацевтически приемлемую соль.
Дополнительно предложена вакцинная композиция, содержащая соединение формулы (I) или его фармацевтически приемлемую соль и антиген или антигенную композицию.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы отдельно или в комбинации с другими терапевтически активными агентами. В дополнительном аспекте согласно изобретению предложена комбинация, содержащая соединение формулы (I) или его фармацевтически приемлемую соль вместе с по меньшей мере одним другим терапевтически активным агентом.
Соединения формулы (I) и их фармацевтически приемлемые соли и другой терапевтически активный агент(ы) могут быть введены вместе или по отдельности, и при введении по отдельности введение можно проводить одновременно или последовательно в любом порядке. Количества соединения(ий) формулы (I) или его(их) фармацевтически приемлемой(ых) соли(ей) и другого(их) терапевтически активного(ых) агента(ов) и относительные моменты времени введения выбирают так, чтобы достичь желаемого комбинированного терапевтического эффекта. Введение комбинации соединения формулы (I) или его фармацевтически приемлемой соли с другими терапевтическими агентами может быть совместным в единой фармацевтической композиции, содержащей оба соединения, или в отдельных фармацевтических композициях, каждая из которых содержит одно из соединений. Альтернативно, комбинация может быть введена по отдельности последовательно, когда один терапевтический агент вводят первым, а другой вторым, или наоборот. Такое последовательное введение может быть близким по времени или отдаленным по времени.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в комбинации с одним или более чем одним агентом, полезным в предупреждении или лечении вирусных инфекций. Примеры таких агентов включают ингибиторы полимеразы, такие, как раскрыты в WO 2004/037818-А1, а также в WO 2004/037818 и WO 2006/045613; JTK-003, JTK-019, NM-283, HCV-796, R-803, R1728, R1626, а также раскрытые в WO 2006/018725, WO 2004/074270, WO 2003/095441, US 2005/0176701, WO 2006/020082, WO 2005/080388, WO 2004/064925, WO 2004/065367, WO 2003/007945, WO 02/04425, WO 2005/014543, WO 2003/000254, EP 1065213, WO 01/47883, WO 2002/057287, WO 2002/057245, и подобные агенты; ингибиторы репликации, такие как ацикловир, фамцикловир, ганцикловир, цидофовир, ламивудин и подобные агенты; ингибиторы протеаз, такие как ингибиторы протеазы HIV (вирус иммунодефицита человека): саквинавир, ритонавир, индинавир, нелфинавир, ампренавир, фосампренавир, бреканавир, атазанавир, типранавир, палинавир, лазинавир, и ингибиторы протеазы HCV (вирус гепатита С): BILN2061, VX-950, SCH503034, и подобные агенты; нуклеозидные и нуклеотидные ингибиторы обратной транскриптазы, такие как зидовудин, диданозин, ламивудин, залцитабин, абакавир, ставидин, адефовир, адефовир дипивоксил, фозивудин, тодоксил, эмтрицитабин, аловудин, амдоксовир, элвуцитабин и подобные агенты; ненуклеозидные ингибиторы обратной транскриптазы (включая агент, обладающий антиоксидантной активностью, такой как иммунокал, олтипраз и тому подобное), такие как невирапин, делавирдин, эфавиренц, ловирид, иммунокал, олтипраз, каправирин, ТМС-278, ТМС-125, этравирин и подобные агенты; ингибиторы проникновения, такие как энфувиртид (Т-20), Т-1249, PRO-542, PRO-140, TNX-355, BMS-806, 5-Helix и подобные агенты; ингибиторы интегразы, такие как L-870,180 и подобные агенты; ингибиторы почкования, такие как РА-344 и РА-457 и подобные агенты; ингибиторы хемокиновых рецепторов, такие как викривирок (Sch-C), Sch-D, TAK779, маравирок (UK-427,857), TAK449, а также те, которые раскрыты в WO 02/74769, WO 2004/054974, WO 2004/055012, WO 2004/055010, WO 2004/055016, WO 2004/055011 и WO 2004/054581, и подобные агенты; ингибиторы нейраминидазы, такие как CS-8958, занамивир, оселтамивир, перамивир и подобные агенты; блокаторы ионных каналов, такие как амантадин или римантадин и подобные агенты; и интерферирующая РНК и антисмысловые олигонуклеотиды и такие как ISIS-14803 и подобные агенты; противовирусные агенты с неустановленным механизмом действия, например, раскрытые в WO 2005/105761, WO 2003/085375, WO 2006/122011, рибавирин и подобные агенты, но не ограничиваются ими. Соединения формулы (I) и их фармацевтически приемлемые соли также могут быть использованы в комбинации с одним или более чем одним другим агентом, который может быть полезен в предупреждении или лечении вирусных инфекций, например при видах иммунной терапии (например, интерфероном или другими цитокинами/хемокинами, модуляторами рецепторов цитокинов/хемокинов, агонистами или антагонистами цитокинов и подобными агентами); и терапевтическими вакцинами, антифиброзными агентами, противовоспалительными агентами, такими как кортикостероиды или нестероидные противовоспалительные агенты (NSAID), и подобными агентами.
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в комбинации с одним или более чем одним другим агентом, который может быть полезен в предупреждении или лечении аллергического заболевания, воспалительного заболевания, аутоиммунного заболевания, например с антигенными иммунотерапевтическими средствами, антигистаминными средствами, стероидами, NSAID, бронходилататорами (например бета-2-агонистами, адренергическими агонистами, антихолинергическими агентами, теофиллином), метотрексатом, модуляторами лейкотриенов и подобными агентами; терапевтическими моноклональными антителами, такими как анти-IgE, анти-TNF, анти-IL-5, анти-IL-6, анти-IL-12, анти-IL-1 и подобными агентами; препаратами рецепторной терапии, например энтанерсептом и подобными агентами; препаратами антигенной неспецифической иммунотерапии (например интерфероном или другими цитокинами/хемокинами, модуляторами рецепторов цитокинов/хемокинов, агонистами или антагонистами цитокинов, агонистами TLR и подобными агентами).
Соединения формулы (I) и их фармацевтически приемлемые соли могут быть использованы в комбинации с одним или более чем одним другим агентом, который может быть полезным в предупреждении или лечении рака, например химиотерапевтическими средствами, такими как алкилирующие агенты, ингибиторы топоизомеразы, антиметаболиты, антимитотические агенты, ингибиторы киназ и подобные агенты; терапевтическими моноклональными антителами, такими как трастузумаб, гемтузумаб, и другие подобные агенты; и препаратами гормональной терапии, такими как тамоксифен, гозерелин, и подобные агенты.
Фармацевтические композиции по изобретению также могут быть использованы отдельно или в комбинации по меньшей мере с одним другим терапевтическим агентом, применяемым в других терапевтических областях, например при заболеваний желудочно-кишечного тракта. Композиции по изобретению также могут быть использованы в комбинации с генной заместительной терапией.
В дополнительном аспекте изобретение включает комбинацию, содержащую соединение формулы (I) или его фармацевтически приемлемую соль вместе с по меньшей мере одним другим терапевтически активным агентом.
Вышеупомянутые комбинации для удобства могут быть представлены для применения в форме фармацевтической композиции, и, соответственно, фармацевтические композиции, содержащие комбинацию, как она определена выше, вместе с по меньшей мере одним фармацевтически приемлемым разбавителем или носителем, составляют дополнительный аспект изобретения.
Терапевтически эффективное количество соединения формулы (I) или его фармацевтически приемлемой соли будет зависеть от целого ряда факторов. Например, вид, возраст и масса реципиента, точное состояние, требующее лечения, и его тяжесть, природа композиции и путь введения все являются факторами, которые нужно учитывать. В конечном счете терапевтически эффективное количество будет определяться на усмотрение лечащего врача. Тем не менее, эффективное количество соединения по настоящему изобретению для лечения людей в болезненном состоянии, как правило, должно находиться в диапазоне от 0,0001 до 100 мг/кг массы тела реципиента в сутки. Еще точнее эффективное количество должно находиться в диапазоне от 0,001 до 10 мг/кг массы тела в сутки. Таким образом, для взрослого человека с массой тела 70 кг фактическое количество в сутки будет обычно составлять от 7 до 700 мг. Для интраназального и ингаляционного путей введения типичные дозы для взрослого человека с массой тела 70 кг должны находиться в диапазоне от 0,1 мкг до 1 мг в сутки, например 1 мкг, 10 мкг или 100 мкг. Это количество может быть введено в однократной дозе в сутки или в многократных (например двух, трех, четырех, пяти или более) субдозах в сутки, так чтобы суммарная суточная доза была той же. Эффективное количество фармацевтически приемлемой соли соединения формулы (I) может быть определено пропорционально эффективному количеству соединения формулы (I) или его фармацевтически приемлемой соли как таковой. Подобные дозировки должны подходить для лечения других состояний, упомянутых здесь.
Соединения формулы (I) и их фармацевтически приемлемые соли также можно вводить с любой соответствующей частотой, например 1-7 раз в неделю. Точный режим дозировки будет конечно же зависеть от таких факторов, как терапевтическое показание, возраст и состояние пациента, а также выбранный конкретный путь введения. В одном аспекте изобретения соединение формулы (I) или его фармацевтически приемлемую соль можно вводить один раз в неделю в течение периода времени от 4 до 8 недель, например 4, 5, 6, 7 или 8 недель.
Фармацевтические композиции могут быть представлены в стандартных лекарственных формах, содержащих предварительно определенное количество активного ингредиента на стандартную дозу. Такие стандартные дозы могут содержать в качестве неограничивающего примера от 0,5 мг до 1 г соединения формулы (I) или его фармацевтически приемлемой соли в зависимости от состояния, подлежащего лечению, пути введения и возраста, массы и состояния пациента. Предпочтительными композициями в стандартной лекарственной форме являются композиции, содержащие суточную дозу или субдозу, как указано выше, или ее соответствующую долю активного ингредиента. Такие фармацевтические композиции могут быть получены любым из способов, известных в области фармации.
Предложен также способ изготовления такой фармацевтической композиции, включающий смешивание соединения формулы (I) или его фармацевтически приемлемой соли с одним или более чем одним фармацевтически приемлемым эксципиентом.
Аспекты изобретения проиллюстрированы посредством ссылки на следующие примеры, но никак ими не ограничены.
Примеры
Аналитические методики
1Н ЯМР
Спектры 1Н ЯМР регистрировали либо в CDCl3, либо в DMSO-d6 (диметилсульфоксид-d6) на спектрометре либо Bruker DPX 400, либо Bruker Avance DRX, либо Varian Unity 400, либо JEOL Delta, каждый из которых работает при 400 МГц. Используемым внутренним стандартом был либо тетраметилсилан, либо остаточный протонированный растворитель при 7,25 м.д. (миллионные доли) для CDCl3 или 2,50 м.д. для DMSO-d6.
ЖХ-МС
Система А
Колонка: 50 мм × 2,1 мм в.д. (внутренний диаметр), 1,7 мкм Acquity UPLC ВЕН C18
Скорость потока: 1 мл/мин
Температура: 40°C
Детектирование в УФ-диапазоне: от 210 до 350 нм
Масс-спектр: регистрировали на масс-спектрометре с использованием режима электрораспылительной ионизации с попеременной регистрацией положительных и отрицательных ионов
Растворители:
А: 0,1% об./об. муравьиная кислота в воде
В: 0,1% об./об. муравьиная кислота в ацетонитриле
Градиент:
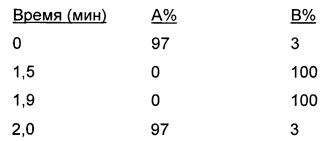
Система В
Колонка: 50 мм × 2,1 мм в.д., 1,7 мкм Acquity UPLC BEH C18
Скорость потока: 1 мл/мин
Температура: 40°C
Детектирование в УФ-диапазоне: от 210 до 350 нм
Масс-спектр: регистрировали на масс-спектрометре в режиме электрораспылительной ионизации с попеременной регистрацией положительных и отрицательных ионов
Растворители:
А: 10 мМ бикарбонат аммония в воде, доведенный до рН 10 раствором аммиака
В: ацетонитрил
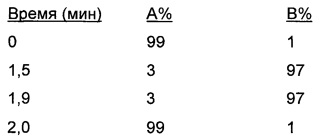
Градиент:
Масс-направленная автоматизированная препаративная ВЭЖХ (МНАП) Масс-направленную автоматизированную препаративную ВЭЖХ выполняли в условиях, приведенных ниже. Посредством УФ-детектирования регистрировали усредненный сигнал при длине волны от 210 до 350 нм и масс-спектры регистрировали на масс-спектрометре с использованием режима электрораспылительной ионизации с попеременной регистрацией положительных и отрицательных ионов.
Способ А
Способ А осуществляли на колонке Sunfire C18 (типично 150 мм × 30 мм в.д., диаметр упаковки 5 мкм) при температуре окружающей среды. Использовали растворители:
А: 0,1% об./об. раствор муравьиной кислоты в воде
В: 0,1% об./об. раствор муравьиной кислоты в ацетонитриле.
Способ В
Способ В осуществляли на колонке XBridge С18 (типично 100 мм × 30 мм в.д., диаметр упаковки 5 мкм) при температуре окружающей среды. Использовали растворители:
А: 10 мМ водный бикарбонат аммония, доведенный до рН 10 раствором аммиака
В: ацетонитрил.
Сокращения
В приведенном ниже перечне представлены определения некоторых использованных здесь сокращений. Следует понимать, что этот перечень не является исчерпывающим, но значение тех сокращений, которые не определены в нем, будет очевидно специалистам в данной области техники.
DCM - дихлорметан;
DMF - N,N-диметилформамид;
DMSO - диметилсульфоксид;
THF - тетрагидрофуран;
EtOAc - этилацетат;
МеОН - метанол;
EtOH - этанол;
MeCN - ацетонитрил;
HCl - соляная кислота;
ВЭЖХ - высокоэффективная жидкостная хроматография;
МНАП - масс-направленная автоматизированная препаративная ВЭЖХ;
ТФЭ - твердофазная экстракция;
МеОН - метанол;
ТВМЕ - трет-бутилметиловый эфир;
TFA - трифторуксусная кислота;
DIPEA - N,N-диизопропилэтиламин.
Получение промежуточных соединений
Промежуточное соединение 1: этил-3-ацетимидамидо-5-метил-1H-пиррол-2-карбоксилат
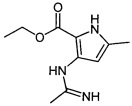
К суспензии этил-3-амино-5-метил-1H-пиррол-2-карбоксилата (10,876 г; 64,7 ммоль) в ацетонитриле (200 мл) добавляли 4 М HCl в диоксане (81 мл; 323 ммоль). Смесь нагревали при 60°C в атмосфере азота в течение 4 суток. Реакционную смесь фильтровали с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (12,738 г).
ЖХ-МС (система В): tRET (время удерживания): 0,45, 0,47 мин; МН+ 210.
Промежуточное соединение 2: 2,6-диметил-3Н-пирроло[3,2-d]пиримидин-4(5Н)-он
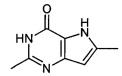
К суспензии гидрохлорида этил-3-ацетимидамидо-5-метил-1H-пиррол-2-карбоксилата (12,7 г; 51,7 ммоль) в этаноле (200 мл) добавляли 5 М NaOH (41,4 мл; 207 ммоль) одной порцией. Смесь помещали в атмосферу азота и нагревали до 90°C в течение 2 часов. Реакционную смесь концентрировали под вакуумом и добавляли в нее 5%-ный водный раствор лимонной кислоты (200 мл) до тех пор, пока рН не становился нейтральным. При рН 7 образовывалось твердое вещество, его отфильтровывали и промывали водой с получением указанного в заголовке соединения в виде бледно-коричневого твердого вещества (6,806 г).
ЖХ-МС (система В): tRET: 0,45 мин; МН+ 164.
Фильтрат концентрировали под вакуумом и фильтровали с получением дополнительной порции указанного в заголовке соединения (0,5 г).
ЖХ-МС (система В): tRET: 0,45 мин; МН+ 164.
Промежуточное соединение 3: 4-хлор-2,6-диметил-5H-пирроло[3,2-d]пиримидин
T i N
В круглодонную колбу вносили 2,6-диметил-3H-пирроло[3,2-d]пиримидин-4(5H)-он (6,15 г; 37,7 ммоль) и растворяли в POCl3 (70,3 мл; 754 ммоль). Реакционную смесь затем нагревали до 90°C в течение 30 часов. Реакционную смесь упаривали под вакуумом с получением коричневого масла. Его растворяли в толуоле и упаривали снова для удаления оставшегося в смеси POCl3. Остаток затем разбавляли водой (50 мл) и добавляли бикарбонат натрия до тех пор, пока рН не достигал значения 7-8. Появлялся белый осадок, и его отфильтровывали и затем промывали водой. Его сушили в течение 2 часов с получением указанного в заголовке соединения (5,617 г) в виде белого твердого вещества.
ЖХ-МС (система В): tRET: 0,59 мин; МН+ 182/184.
Фильтрат концентрировали под вакуумом, фильтровали, промывали и сушили с получением дополнительной порции указанного в заголовке соединения (0,22 г).
ЖХ-МС (система В): tRET: 0,59 мин; МН+ 182/184.
Промежуточное соединение 4: 4-хлор-7-йод-2,6-диметил-5H-пирроло[3,2-d]пиримидин
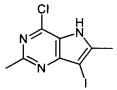
К перемешиваемому раствору 4-хлор-2,6-диметил-5Н-пирроло[3,2-d]пиримидина (6,68 г; 36,8 ммоль) в тетрагидрофуране (100 мл) в атмосфере азота при комнатной температуре порциями добавляли N-йодсукцинимид (9,52 г; 42,3 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. Реакционную смесь затем разбавляли ТВМЕ. Смесь промывали раствором тиосульфата натрия (150 мл) и рассолом. Органический слой затем пропускали через гидрофобную фритту и концентрировали под вакуумом. Образец предварительно абсорбировали на флоросиле и очищали на диоксиде кремния (750 г) с использованием градиента 0-100% TBME-циклогексан в количестве 11 объемов колонки. Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде желтого твердого вещества (9,854 г).
ЖХ-МС (система A): tRET: 0,75 мин; МН+ 308/310.
Промежуточное соединение 5: 5-((бензилокси)метил)-4-хлор-7-йод-2,6-диметил-5H-пирроло[3,2-d]пиримидин
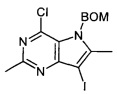
К раствору 4-хлор-7-йод-2,6-диметил-5H-пирроло[3,2-d]пиримидина (1,098 г; 3,57 ммоль) в N,N-диметилформамиде (25 мл), охлажденному на ледяной бане, порциями добавляли гидрид натрия (0,286 г; 7,14 ммоль) в течение 5 минут. Реакционную смесь перемешивали в течение 30 минут перед добавлением бензилхлорметилового эфира (0,891 мл; 6,43 ммоль). Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 1 часа. Реакционную смесь гасили водой (50 мл) и распределяли между этилацетатом (200 мл) и водой (200 мл). Органический слой промывали водой и рассолом (200 мл) и пропускали через гидрофобную фритту перед тем, как концентрировать под вакуумом. Образец растворяли в дихлорметане и очищали на картридже с диоксидом кремния (70 г) при использовании градиента 0-25% этилацетат-циклогексан в течение 40 минут. Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (1,4 г).
ЖХ-МС (система A): tRET: 1,18 мин; МН+ 428/430.
Промежуточное соединение 6: 5-((бензилокси)метил)-7-йод-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амин
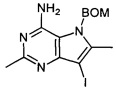
5-((Бензилокси)метил)-4-хлор-7-йод-2,6-диметил-5Н-пирроло[3,2-d]пиримидин (3,458 г) распределяли в три пробирки на 20 мл для микроволновой печи. В каждую пробирку для микроволновой печи добавляли 1,15 г исходного вещества и содержимое разбавляли IPA (10 мл) перед добавлением в каждую пробирку 0,88 аммиака (2,5 мл). Пробирки герметизировали и нагревали в микроволновой печи (Biotage) при 150°C (High Power) в течение 5 часов. Мониторинг с помощью ЖХ-МС показал, что реакция окончена в двух герметизированных пробирках для микроволновой печи из трех: в пробирку, содержащую не вступившее в реакцию исходное вещество, добавляли дополнительно 0,88 аммиака (2,5 мл) и смесь нагревали в микроволновой печи в течение еще 5 часов. Реакционную смесь из трех пробирок объединяли, а пробирки промывали IPA. Затем все концентрировали под вакуумом с получением оранжевого масла, которое растирали с ТВМЕ (15 мл) с получением указанного в заголовке соединения в виде желтого твердого вещества (2,352 г).
ЖХ-МС (система A): tRET: 0,69 мин; МН+ 409.
Фильтрат концентрировали под вакуумом и повторно растирали с ТВМЕ (10 мл) с получением второй порции указанного в заголовке соединения в виде желтого твердого вещества (267 мг).
ЖХ-МС (система A): tRET: 0,67 мин; МН+ 409.
Промежуточное соединение 7: 1-(гекс-5-ин-1-ил)пиперидин
Раствор 6-хлоргекс-1-ина (5 мл; 41,3 ммоль), пиперидина (4,08 мл; 41,3 ммоль) и гидрокарбоната натрия (4,16 г; 49,5 ммоль) в DMF (50 мл) нагревали с обратным холодильником в течение 16 часов. Реакционную смесь концентрировали под вакуумом и остаток распределяли между эфиром (150 мл) и водой (150 мл). Органический слой отделяли, а водный слой подвергали обратной экстракции диэтиловым эфиром (50 мл). Объединенные органические слои промывали рассолом (150 мл), сушили (MgSO4), фильтровали и концентрировали под вакуумом с получением неочищенного образца указанного в заголовке соединения (3,74 г). К неочищенному продукту добавляли щавелевую кислоту (2,161 г; 24 ммоль). Полученное твердое вещество перекристаллизовывали из этанола, собирали посредством фильтрации и сушили под вакуумом с получением оксалата 1-(гекс-5-ин-1- ил)пиперидина (4,66 г). Твердое вещество распределяли между диэтиловым эфиром (150 мл) и насыщенным водным бикарбонатом натрия (150 мл). Органический слой отделяли и сушили (MgSO4), фильтровали и концентрировали под вакуумом с получением указанного в заголовке соединения в виде желтого масла (1,93 г).
1Н ЯМР (400 МГц, CDCl3) δ м.д. 2.31-2.52 (m, 6Н) 2.18-2.26 (m, 2Н) 1.92-1.96 (m, 1Н) 1.40-1.72 (m, 10Н).
Промежуточное соединение 8: 5-((бензилокси)метил)-2,6-диметил-7-(6-(пиперидин-1-ил)гекс-1-ин-1-ил)-5Н-пирроло[3,2-d]пиримидин-4-амин
К перемешиваемой дегазированной суспензии 5-((бензилокси)метил)-7-йод-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина (407 мг; 0,997 ммоль), йодида меди(I) (27,5 мг; 0,145 ммоль) и дихлорида бис(трифенилфосфин)палладия(II) (49,0 мг; 0,070 ммоль) добавляли триэтиламин (0,221 мл; 1,595 ммоль) в безводном DMF (8 мл). Раствор оставляли для перемешивания в течение 30 минут, затем порциями добавляли раствор 1-(5-гексин-1-ил)пиперидина (247 мг; 1,495 ммоль) в DMF (2 мл) в течение 5 минут. Реакционную смесь перемешивали при 55°C в течение 2 часов с последующим перемешиванием при 60°C в течение 1 часа. Добавляли еще одну порцию 1-(5-гексин-1-ил)пиперидина (247 мг; 1,495 ммоль) и реакционную смесь оставляли для перемешивания в течение 1,5 часа при 60°C, затем оставляли для перемешивания при комнатной температуре в течение ночи. Реакционную смесь концентрировали под вакуумом с получением коричневого масла. Масло распределяли между DCM (100 мл) и водой (100 мл). Органическую фазу отделяли и водную фазу подвергали обратной экстракции с использованием DCM (100 мл). Объединенные органические экстракты сушили при использовании гидрофобной фритты и концентрировали под вакуумом с получением темно-оранжевого масла (902 мг). Неочищенное вещество растворяли в дихлорметане и очищали на картридже с диоксидом кремния (100 г) с использованием градиента 0-25% метанол-дихлорметан в течение 60 минут. Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде двух партий.
Партия 1: прозрачное масло (42 мг).
ЖХ-МС (система В): tRET: 1,08 мин; МН+ 446.
Партия 2: бледно-желтое твердое вещество (117 мг).
ЖХ-МС (система В): tRET: 1,11 мин; МН+ 446.
Промежуточное соединение 9: 5-((бензилокси)метил)-7-(5-хлорпент-1-ин-1-ил)-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амин
К дегазированному азотом раствору 5-((бензилокси)метил)-7-йод-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амина (300 мг; 0,735 ммоль) в безводном N,N-диметилформамиде (7 мл) в атмосфере азота при комнатной температуре добавляли йодид меди(I) (30,8 мг; 0,162 ммоль), дихлорид бис(трифенилфосфин)палладия(II) (61,9 мг; 0,088 ммоль) и в конце триэтиламин (0,205 мл; 1,470 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 10 минут и затем добавляли раствор 5-хлорпент-1-ина (151 мг; 1,470 ммоль) в безводном N,N-диметилформамиде (5 мл). Реакционную смесь перемешивали при 60°C в течение 2 часов. Реакционную смесь упаривали под вакуумом с получением коричневого масла. Масло распределяли между смесью вода/рассол (1:1) (100 мл) и DCM (100 мл). Органический слой отделяли, пропускали через гидрофобную фритту и упаривали под вакуумом с получением темно-оранжевого масла (538 мг). Вещество растворяли в смеси DMSO/MeOH 50:50 (6×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (132 мг).
ЖХ-МС (система В): tRET: 1,10 мин; МН+ 383.
Промежуточное соединение 10: 5-((Бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амин
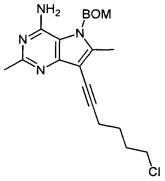
К дегазированному азотом раствору 5-((бензилокси)метил)-7-йод-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина (482,7 мг; 1,182 ммоль) в безводном N,N-диметилформамиде (7 мл) в атмосфере азота при комнатной температуре добавляли йодид меди(I) (49,5 мг; 0,260 ммоль), дихлорид бис(трифенилфосфин)палладия(II) (100 мг; 0,142 ммоль) и в конце триэтиламин (0,330 мл; 2,365 ммоль). Смесь перемешивали при комнатной температуре в атмосфере азота в течение 10 минут и затем добавляли раствор 6-хлоргекс-1-ина (276 мг; 2,365 ммоль) в безводном N,N-диметилформамиде (1 мл). Реакционную смесь перемешивали при 65°C в течение 15 часов, затем реакционную смесь концентрировали под вакуумом. Неочищенный продукт растворяли в DMSO и очищали посредством колоночной обращенно-фазовой хроматографии (колонка С18 120 г) при использовании градиента от 35 до 90% ацетонитрила плюс смесь 0,1% аммиака/10 мМ бикарбоната аммония в воде, доведенная до рН 10 с помощью раствора аммиака, в количестве 12 объемов колонки. Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества (215 мг).
ЖХ-МС (система В): tRET: 1,15 мин; МН+ 397.
Получение соединений по примерам
Пример 1: 2,6-диметил-7-(6-(пиперидин-1-ил)гексил)-5H-пирроло[3,2-d]пиримидин-4-амин
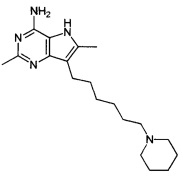
Фильтрованный раствор 5-((бензилокси)метил)-2,6-диметил-7-(6-(пиперидин-1-ил)гекс-1-ин-1-ил)-5H-пирроло[3,2-d]пиримидин-4-амина (159 мг; 0,357 ммоль) в этаноле (15 мл) и уксусной кислоте (1,5 мл) гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Раствор повторно гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и того же картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Раствор концентрировали под вакуумом. Твердое вещество растворяли в смеси DMSO/MeOH 50:50 (2×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде кремового твердого вещества (62 мг).
ЖХ-МС (система В): tRET: 0,80 мин; МН+ 330.
Пример 2: малеат 2,6-диметил-7-(5-(пирролидин-1-ил)пентил)-5Н-пирроло[3,2-d]пиримидин-4-амина
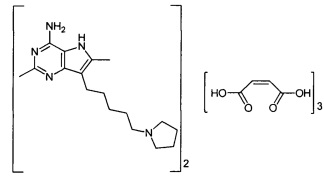
К раствору 5-((бензилокси)метил)-7-(5-хлорпент-1-ин-1-ил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина (251 мг; 0,656 ммоль) в безводном ацетонитриле (4 мл) добавляли пирролидин (0,164 мл; 1,967 ммоль) и триэтиламин (0,274 мл; 1,967 ммоль). Реакционную смесь перемешивали при 70°C в течение 22 часов. Добавляли дополнительную порцию пирролидина (1,5 эквивалента) и триэтиламина (1,5 эквивалента) и смесь оставляли для перемешивания в течение еще 24 часов. Реакционную смесь концентрировали под вакуумом с получением коричневого масла. Масло распределяли между EtOAc и водой. Органическую фазу отделяли и водную фазу подвергали обратной экстракции EtOAc. Объединенные органические экстракты сушили при использовании гидрофобной фритты и концентрировали под вакуумом с получением коричневого масла (315 мг). Неочищенный продукт растворяли в этаноле (40 мл) и гидрировали при использовании аппарата H-cube (параметры: 25°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Раствор концентрировали под вакуумом с получением бледно-желтого масла (275 мг). Вещество растворяли в дихлорметане и очищали на картридже с диоксидом кремния (20 г) при использовании градиента 0-50% метанол-дихлорметан в течение 40 минут. Фракции, содержащие требуемый продукт, объединяли и концентрировали под вакуумом с получением 174 мг желтого масла. Вещество повторно растворяли в EtOH (25 мл) и уксусной кислоте (2,5 мл) и пропускали через аппарат H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Раствор пропускали через аппарат H-cube до тех пор, пока реакция не завершалась и не наступало полное удаление ВОМ группы (3 раза при использовании тех же параметров). Этанольный раствор упаривали досуха с получением прозрачного масла (149 мг). Вещество растворяли в смеси DMSO/MeOH 50:50 (2×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением свободного основания указанного в заголовке соединения в виде белого твердого вещества (60 мг).
ЖХ-МС (система В): tRET: 0,65 мин; МН+ 302.
2,6-Диметил-7-(5-(пирролидин-1-ил)пентил)-5H-пирроло[3,2-d]пиримидин- 4-амин (11,3 мг; 0,037 ммоль) растворяли в смеси MeOH/DCM и добавляли малеиновую кислоту (6,53 мг; 0,056 ммоль). Раствор упаривали досуха с получением указанного в заголовке соединения в форме малеата в виде прозрачного масла (16,7 мг).
ЖХ-МС (система В): tRET: 0,66 мин; МН+ 302.
Пример 3: Формиат 2,2'-((5-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)пентил)азандиил)диэтанола
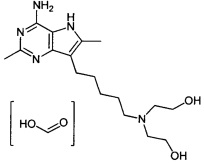
К перемешиваемому раствору 5-((бензилокси)метил)-7-(5-хлорпент-1-ин-1-ил)-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амина (132 мг; 0,345 ммоль) в N,N-диметилформамиде (3 мл) добавляли триэтиламин (0,144 мл; 1,034 ммоль) и диэтаноламин (109 мг; 1,034 ммоль). Полученную смесь нагревали при 70°C в течение 22 часов. В реакционную смесь добавляли дополнительное количество диэтаноламина (109 мг; 1,034 ммоль) и триэтиламина (0,144 мл; 1,034 ммоль) и продолжали нагревание при 70°C в течение 48 часов. Реакционную смесь концентрировали под вакуумом и остаток растворяли в MeOH (25 мл) и добавляли уксусную кислоту (2 мл). Раствор гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Метанольный раствор повторно пропускали через аппарат Н- cube при использовании тех же параметров, которые описаны выше. Метанольный раствор упаривали досуха и неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (6×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде белого твердого вещества (82 мг).
ЖХ-МС (система В): tRET: 0,59 мин; МН+ 336.
Пример 4: формиат 2,2'-((6-(4-амино-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-7-ил)гексил)азандиил)диэтанола
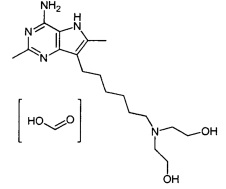
Получали аналогично соединению по примеру 3 из 5-((бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина и диэтаноламина.
ЖХ-МС (система В): tRET: 0,64 мин; МН+ 350.
Пример 5: 2-((6-(4-амино-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-7-ил)гексил)(2-метоксиэтил)амино)этанол
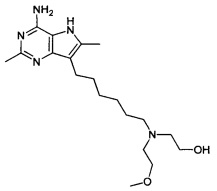
К перемешиваемому раствору 5-((бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина (215 мг; 0,542 ммоль) в N,N-диметилформамиде (4 мл) добавляли триэтиламин (0,226 мл; 1,625 ммоль) и 2-((2-метоксиэтил)амино)этанол (0,191 мл; 1,625 ммоль). Полученную смесь нагревали при 70°C в течение 40 часов. В реакционную смесь добавляли дополнительное количество 2-((2-метоксиэтил)амино)этанола (0,191 мл; 1,625 ммоль) и триэтиламина (0,226 мл; 1,625 ммоль) и продолжали нагревание при 75°C в течение 24 часов. Реакционную смесь концентрировали под вакуумом и остаток растворяли в МеОН (25 мл), затем добавляли уксусную кислоту (2 мл). Раствор гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора.
Раствор повторно пропускали через аппарат H-cube еще 3 раза при использовании тех же параметров, которые описаны выше. Растворитель выпаривали под вакуумом. Неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (4×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением бледно-желтого масла (93 мг). Масло повторно очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения (27 мг).
ЖХ-МС (система В): tRET: 0,70 мин; МН+ 364.
Пример 6: 7-(6-(бис(2-метоксиэтил)амино)гексил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амин
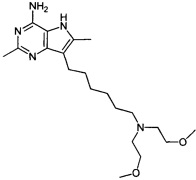
К перемешиваемому раствору 5-((бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина (315,6 мг; 0,795 ммоль) в N,N-диметилформамиде (5 мл) добавляли триэтиламин (0,332 мл; 2,385 ммоль) и бис(2-метоксиэтил)-амин (0,349 мл; 2,385 ммоль). Полученную смесь нагревали при 70°C в течение 20 часов. В реакционную смесь добавляли дополнительное количество бис(2-метоксиэтил)-амина (0,349 мл; 2,385 ммоль) и триэтиламина (0,332 мл; 2,385 ммоль) и продолжали нагревание при 70°C в течение 32 часов. Реакционную смесь концентрировали под вакуумом и остаток растворяли в МеОН (40 мл) и добавляли уксусную кислоту (4 мл). Раствор гидрировали при использовании аппарата H-cube (параметры: 65°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Раствор повторно пропускали через аппарат H-cube еще 3 раза при использовании тех же параметров, которые описаны выше, и замене картриджа CatCart при каждом пропускании. Метанол выпаривали под вакуумом. Неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (3×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения (84,5 мг). ЖХ-МС (система В): tRET: 0,82 мин; МН+ 378.
Пример 7: 2-((6-(4-амино-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-7-ил)гексил)амино)этанол
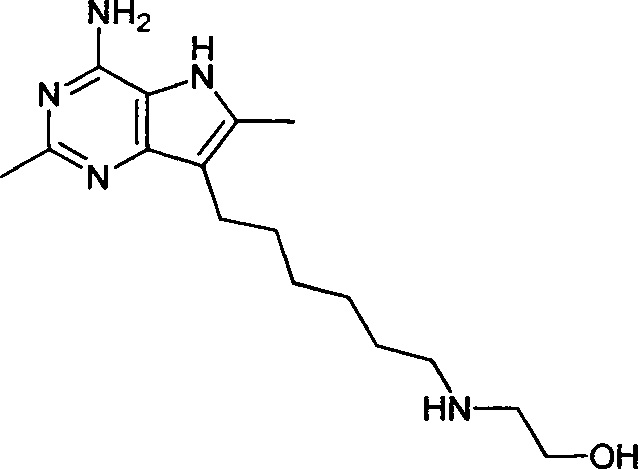
К перемешиваемому раствору 5-((бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина (220,6 мг; 0,556 ммоль) в ацетонитриле (4 мл) добавляли триэтиламин (0,232 мл; 1,667 ммоль) и 2-аминоэтанол (0,101 мл; 1,667 ммоль). Полученную смесь нагревали при 70°C в течение 21 часа. В реакционную смесь добавляли дополнительное количество 2-аминоэтанола (0,101 мл; 1,667 ммоль) и триэтиламина (0,232 мл; 1,667 ммоль) и продолжали нагревание при 70°C в течение 46 часов. Реакционную смесь концентрировали под вакуумом и остаток растворяли в МеОН (25 мл) и добавляли уксусную кислоту (2 мл). Раствор гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Метанольный раствор повторно пропускали через аппарат Н- cube еще 2 раза при использовании тех же параметров, которые описаны выше. Метанол выпаривали под вакуумом. Неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (5×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде прозрачного масла (43,7 мг).
ЖХ-МС (система В): tRET: 0,55 мин; МН+ 306.
Пример 8: (3R,5S)-1-(6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)пиперидин-3,5-диол
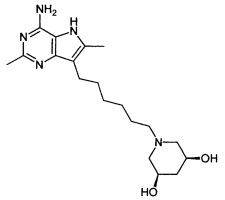
К перемешиваемому раствору 5-((бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амина (215 мг; 0,542 ммоль) в N,N-диметилформамиде (4 мл) добавляли триэтиламин (0,226 мл; 1,625 ммоль) и (3R,5S)-пиперидин-3,5-диол (250 мг; 1,625 ммоль). Полученную смесь нагревали при 70°C в течение 16 часов. В реакционную смесь добавляли дополнительное количество (3R,5S)-пиперидин-3,5-диола (250 мг; 1,625 ммоль) и триэтиламина (0,226 мл; 1,625 ммоль) и продолжали нагревание при 75°C в течение 72 часов. Реакционную смесь концентрировали под вакуумом и остаток растворяли в метаноле (45 мл) и добавляли уксусную кислоту (5 мл). Раствор гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Метанольный раствор повторно пропускали через аппарат H-cube еще 2 раза при использовании тех же параметров, которые описаны выше, затем упаривали под вакуумом. Неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (8×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде кремового твердого вещества (160,7 мг).
ЖХ-МС (система В): tRET: 0,60 мин; МН+ 362.
Пример 9: (3R,5R)-1-(6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)пиперидин-3,5-диол
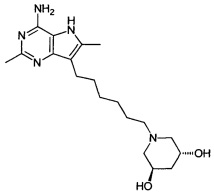
К перемешиваемому раствору 5-((бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амина (165 мг; 0,416 ммоль) в N,N-диметилформамиде (3 мл) добавляли триэтиламин (0,174 мл; 1,247 ммоль) и (3R,5R)-пиперидин-3,5-диол (192 мг; 1,247 ммоль) (Tetrahedron 67(7), 1485, 2011). Полученную смесь нагревали при 70°C в течение 32 часов. В реакционную смесь добавляли дополнительное количество (3R,5R)-пиперидин-3,5-диола (192 мг; 1,247 ммоль) и дополнительное количество триэтиламина (0,174 мл; 1,247 ммоль) и продолжали нагревание при 75°C в течение 5 часов. Реакционную смесь концентрировали под вакуумом и остаток растворяли в МеОН (30 мл) и добавляли уксусную кислоту (3 мл). Раствор гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Метанольный раствор повторно пропускали через аппарат H-cube еще 2 раза при использовании тех же параметров, которые описаны выше, затем упаривали под вакуумом. Неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (7×1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде прозрачного масла (95,2 мг).
ЖХ-МС (система В): tRET: 0,62 мин; МН+ 362.
Пример 10: 7-(6-(диметиламино)гексил)-2,6-диметил-5Н-пирроло[3,2-d]пиримидин-4-амин
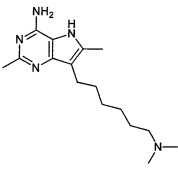
К перемешиваемому раствору 5-((бензилокси)метил)-7-(6-хлоргекс-1-ин-1-ил)-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амина (183,7 мг; 0,463 ммоль) в N,N-диметилформамиде (3 мл) добавляли триэтиламин (0,194 мл; 1,388 ммоль) и 2-аминоэтанол (0,084 мл; 1,388 ммоль). Полученную смесь нагревали при 70°C в течение 18 часов. Реакционную смесь концентрировали под вакуумом и неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (3×1 мл) и очищали посредством МНАП (способ В). Фракции, содержащие требуемый продукт, объединяли и концентрировали с получением бледно-желтого масла (73 мг). Масло растворяли в МеОН (20 мл) и гидрировали при использовании аппарата H-cube (параметры: 60°C, режим полного гидрирования Full Н2, скорость потока 1 мл/мин) и картриджа CatCart 30 с 10% Pd/C в качестве катализатора. Метанол выпаривали под вакуумом и неочищенное вещество растворяли в смеси DMSO/MeOH 50:50 (1 мл) и очищали посредством МНАП (способ В). Соответствующие фракции объединяли и упаривали под вакуумом с получением указанного в заголовке соединения в виде кремового твердого вещества (24,2 мг).
ЖХ-МС (система В): tRET: 0,66 мин; МН+ 290.
Оценка биологического действия
Соединения по изобретению тестировали на биологическую активность in vitro в соответствии со следующим анализом.
Анализ индуцирования интерферона-α и TNF-α с использованием свежей человеческой цельной крови (WB)
Приготовление соединений
Соединения готовили в DMSO до получения 100× требуемой концентрации в плоскодонных микротитрационных планшетах в объеме 1,5 мкл. Колонки 1-10 содержали 1/4 серийное разведение тестируемого соединения. Каждый планшет содержал серийное разведение агониста TLR7/8 резиквимода в качестве стандарта, и колонка 11 содержала 1,5 мкл 200 мкМ резиквимода (давая 2 мкМ конечную концентрацию, которую использовали для определения приблизительного максимального ответа на резиквимод). Каждое соединение анализировали в двух повторностях для каждого донора. Инкубация и анализы на интерферон-α и TNF-α
Образцы крови, взятые у трех доноров-людей, собирали в гепарин- натрий (10 Ед/мл). 150 мкл цельной крови вносили в колонки с 1 по 11 аналитического планшета, содержащие 1,5 мкл тестируемого соединения или стандарта в DMSO. Планшеты помещали в термостат на ночь (37°C, 95% воздуха, 5% СО2). После инкубирования в течение ночи планшеты извлекали из термостата и перемешивали на орбитальном шейкере в течение приблизительно 1 минуты. В каждую лунку добавляли 100 мкл 0,9% физиологического раствора и планшеты снова перемешивали на орбитальном шейкере. Планшеты затем центрифугировали (2500 об/мин, 10 минут), после чего образец извлекали при использовании Biomek FX и анализировали как на IFN-α, так и на TNF-α при использовании платформы электрохемилюминесцентного анализа MSD (Mesoscale Discovery). Анализ на IFN-α проводили аналогично анализу, описанному выше. Анализ на TNF-α проводили согласно инструкциям к набору (№ по каталогу K111BHB).
Высвобождаемый цитокин выражали в процентах от контрольного 2 мкМ резиквимода (колонка 11). Этот процент наносили на график зависимости от концентрации соединения и значение pEC50 для ответной реакции определяли путем аппроксимации нелинейной кривой методом наименьших квадратов. Для IFN-α ответов обычно выбирали 4-параметрическую логистическую модель. Для TNF-α ответов, если получали четкий максимальный ответ (то есть если наблюдали четко определенное плато в ответе), тогда обычно использовали 4-параметрическую модель. Если верхняя асимптота кривой не была четко определенной, тогда аппроксимацию кривой обычно ограничивали максимальным ответом 100% (то есть ответом на 2 мкМ резиквимод) или ответом самой высокой тестируемой концентрации, если этот ответ был больше, чем ответ на резиквимод. Некоторые кривые имели колоколообразную форму для одного или обоих цитокинов, и данные по цитокинам на наклоне кривой колоколообразного ответа (то есть концентрации, выше тех, которые дают максимальный ответ) обычно исключали из аппроксимации, обычно с исключением концентрации непосредственно выше максимального ответа. Аппроксимация кривой, таким образом, сосредоточена на восходящей кривой доза-ответ.
Результаты
Соединения по Примерам с 1 по 10 имели средние значения рЕС50 для IFN-α не более 5,7.
Соединения по Примерам с 1 по 10 имели средние значения рЕС50 для TNF-α не более 4,3.
Анализ индуцирования интерферона-α и TNF-α с использованием свежих человеческих мононуклеарных клеток периферической крови (РВМС)
Приготовление соединений
Соединения последовательно разводили в DMSO до получения 100× требуемой концентрации (1 мкл/лунка в плоскодонном 96-луночном планшете для культуры ткани). Каждое соединение анализировали в двух повторностях для каждого донора. Каждый планшет содержал серию разведений агониста TLR7/8 резиквимода в качестве стандарта, и колонка 11 содержала 1 мкл 200 мкМ резиквимода (давая 2 мкМ конечную концентрацию, которую использовали для определения приблизительного максимального ответа на резиквимод).
Получение РВМС
Образцы крови, взятые у трех доноров-людей, собирали в гепарин- натрий (10 Ед/мл). Объемы по 25 мл цельной крови наслаивали на 15 мл среды Histopaque в пробирках Leucosep, которые центрифугировали при 400g в течение 30 минут, и слой между плазмой и средой Histopaque осторожно извлекали в стерильные конические пробирки на 50 мл. Объем пробирки доводили до 50 мл стерильным DPBS (фосфатно-солевой буфер в модификации Дульбекко, -CaCl2/MgCl2) и центрифугировали при 300g в течение 10 мин. Осадок ресуспендировали в 20 мл среды (RPMI 1640 (низкий уровень эндотоксинов), дополненной 10% об./об. фетальной телячьей сыворотки (FCS, низкий уровень эндотоксинов), 100 Ед/мл пенициллина G, 100 мкг/мл стрептомицина, 10 мМ L-глутамина и 1× заменимых аминокислот, и клетки подсчитывали с использованием Nucleoview 3000 (Chemometec, Via-1 Cassette). Концентрацию PBMC доводили до получения конечной концентрации 2×106 /мл и 100 мкл этой клеточной суспензии добавляли в лунки, содержащие 1 мкл разведенного тестируемого соединения.
Инкубация и анализы на интерферон-α и TNF-α
Препараты клеток инкубировали в течение 24 часов (37°C, 95% воздуха, 5% CO2), после чего образец супернатанта извлекали с использованием Biomek FX и анализировали как на IFN-α, так и на TNF-α при использовании платформы электрохемилюминесцентного анализа MSD (Mesoscale Discovery). Анализ на IFN-α проводили аналогично анализу, описанному выше. Анализ на TNF-α проводили согласно инструкциям к набору (№ по каталогу K111BHB).
Высвобождаемый цитокин выражали в процентах от контрольного 2 мкМ резиквимода (колонка 11). Этот процент наносили на график зависимости от концентрации соединения и значение pEC50 для ответной реакции определяли путем аппроксимации нелинейной кривой методом наименьших квадратов. Для IFN-α ответов обычно выбирали 4-параметрическую логистическую модель. Для TNF-α ответов, если получали четкий максимальный ответ (то есть если наблюдали четко определенное плато в ответе), тогда обычно использовали 4-параметрическую модель. Если верхняя асимптота кривой не была четко определенной, тогда аппроксимацию кривой обычно ограничивали максимальным ответом 100% (то есть ответом на 2 мкМ резиквимод) или ответом самой высокой тестируемой концентрации, если этот ответ был больше, чем ответ на резиквимод. Некоторые кривые имели колоколообразную форму для одного или обоих цитокинов, и данные по цитокинам на наклоне кривой колоколообразного ответа (то есть концентрации, выше тех, которые дают максимальный ответ) обычно исключали из аппроксимации, обычно с исключением концентрации непосредственно выше максимального ответа. Аппроксимация кривой, таким образом, сосредоточена на восходящей кривой доза-ответ.
Результаты
Соединения по Примерам с 1 по 9 имели средние значения pEC50 Для IFN-α от 5,4 до 6,3.
Соединения по Примерам с 1 по 9 имели средние значения рЕС50 для TNF-α не более 4,3.
| название | год | авторы | номер документа |
|---|---|---|---|
| Новые соединения | 2013 |
|
RU2640200C2 |
| НОВЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2643371C2 |
| ТРИЦИКЛИЧЕСКИЕ ПИРРОЛО ПРОИЗВОДНЫЕ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2012 |
|
RU2591191C2 |
| НЕКОТОРЫЕ ИНГИБИТОРЫ ПРОТЕИНКИНАЗЫ | 2015 |
|
RU2671494C2 |
| СОЕДИНЕНИЯ, ГЕТЕРОБИЦИКЛО-ЗАМЕЩЕННЫЕ-[1,2,4]ТРИАЗОЛО[1,5c]ХИНАЗОЛИН-5-АМИНА, ОБЛАДАЮЩИЕ СВОЙСТВАМИ А2А АНТАГОНИСТОВ | 2013 |
|
RU2671628C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИМИДИНЫ, ИНГИБИРУЮЩИЕ HCV | 2005 |
|
RU2380101C2 |
| ИНГИБИТОРЫ HPK1 И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2839132C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДОНА ИЛИ ПИРРОЛОТРИАЗОНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ФАРМАЦЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2014 |
|
RU2674977C2 |
| СОЕДИНЕНИЯ ДИОКСИН- И ОКСАЗИН[2,3-D]ПИРИМИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ ФОСФОИНОЗИТИД-3-КИНАЗЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2013 |
|
RU2612251C2 |
| ПИРРОЛОПИРИМИДИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ ЗЛОКАЧЕСТВЕННОЙ ОПУХОЛИ | 2012 |
|
RU2631655C2 |
Изобретение относится к новому соединению формулы (I) и его фармацевтически приемлемой соли. Соединение обладает активностью индукторов интерферона альфа (IFN-α) и может найти применение для лечения или предупреждения аллергического заболевания, воспалительного состояния, инфекционного заболевания или рака. В формуле (I)
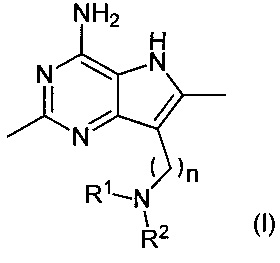 ,
,
R1 представляет собой водород, метил или -(CH2)2OR3, R2 представляет собой метил или -(CH2)2OR4, или R1 и R2 вместе с атомом азота, к которому они присоединены, связаны с образованием 5- или 6-членного гетероциклила, где 6-членный гетероциклил возможно замещен двумя заместителями гидрокси; каждый из R3 и R4 независимо представляет собой водород или метил; и n равен целому числу, имеющему значение 5 или 6. 3 н. и 8 з.п. ф-лы, 10 пр.
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
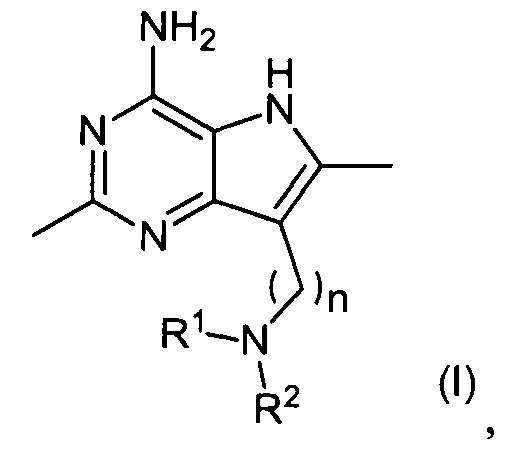
где:
R1 представляет собой водород, метил или -(CH2)2OR3,
R2 представляет собой метил или -(CH2)2OR4, или
R1 и R2 вместе с атомом азота, к которому они присоединены, связаны с образованием 5-членного или 6-членного гетероциклила, содержащего один атом азота, где 6-членный гетероциклил возможно замещен двумя заместителями гидрокси;
каждый из R3 и R4 независимо представляет собой водород или метил; и
n равен целому числу, имеющему значение 5 или 6.
2. Соединение по п. 1 или его соль, где R1 представляет собой водород, метил или -(CH2)2OR3, и R2 представляет собой метил или -(CH2)2OR4.
3. Соединение по п. 1, где n равен 5.
4. Соединение по п. 1, где n равен 6.
5. Соединение или его фармацевтически приемлемая соль, выбранные из:
2,6-диметил-7-(6-(пиперидин-1-ил)гексил)-5H-пирроло[3,2-d]пиримидин-4-амина;
2,6-диметил-7-(5-(пирролидин-1-ил)пентил)-5H-пирроло[3,2-d]пиримидин-4-амина;
2,2'-((5-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)пентил)азандиил)диэтанола;
2,2'-((6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)азандиил)диэтанола;
2-((6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)(2-метоксиэтил)амино)этанола;
7-(6-(бис(2-метоксиэтил)амино)гексил)-2,6-диметил-5H-пирроло[3,2-d]пиримидин-4-амина;
2-((6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)амино)этанола;
(3R,5S)-1-(6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)пиперидин-3,5-диола;
(3R,5R)-1-(6-(4-амино-2,6-диметил-5H-пирроло[3,2-d]пиримидин-7-ил)гексил)пиперидин-3,5-диола или
7-(6-(диметиламино)гексил)-2,6-диметил-5H-пирроло[3,2-]пиримидин-4-амина.
6. Соединение по любому из пп. 1-5, которое находится в форме фармацевтически приемлемой соли.
7. Соединение по любому из пп. 1-5, которое находится в форме свободного основания.
8. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, обладающие активностью индукторов интерферона альфа (IFN-α), для применения в терапии.
9. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, обладающие активностью индукторов интерферона альфа (IFN-α), для применения в лечении или предупреждении аллергического заболевания, воспалительного состояния, инфекционного заболевания или рака.
10. Применение соединения по любому из пп. 1-5 или его фармацевтически приемлемой соли в изготовлении лекарственного средства для лечения или предупреждения аллергического заболевания, воспалительного состояния, инфекционного заболевания или рака.
11. Способ лечения или предупреждения аллергического заболевания, воспалительного состояния, инфекционного заболевания или рака, включающий введение субъекту-человеку, нуждающемуся в этом, терапевтически эффективного количества соединения по любому из пп. 1-5 или его фармацевтически приемлемой соли, обладающих активностью индукторов интерферона альфа (IFN-α).
| WO 2014081645 A1, 30.05.2014 | |||
| Waleed M Hussein, et al.,Toll-like receptor agonists: a patent review (2011 2013), EXPERT OPINION on THERAPEUTICAL PATENTS, 24.01.2014, pp.1-18 | |||
| Michael Czarniecki, Small molecule Modulators of Toll-like Receptors, Journal of Medicinal Chemistry, 2008, vol.51, no.21, pp.6621-6626 | |||
| Новые соединения | 2013 |
|
RU2640200C2 |
| Прибор с двумя призмами | 1917 |
|
SU27A1 |
| Новые соединения | 2013 |
|
RU2640200C2 |
| Прибор с двумя призмами | 1917 |
|
SU27A1 |
| WO 2014081644 A1, 30.05.2014 & RU 2643371 01.02.2018 C2, приоритет 20.11.2012 | |||
| US 20090233948 A1, 17.09.2009 | |||
| Электролит латунирования | 1980 |
|
SU981458A1 |

