Настоящее изобретение относится к применению бициклических пиримидинов в качестве ингибиторов репликации HCV, а также их применению в фармацевтических композициях, предназначенных для лечения или борьбы с инфекциями HCV. Кроме того, настоящее изобретение относится к способам получения таких фармацевтических композиций. Настоящее изобретение также охватывает комбинации настоящих бициклических пиримидинов с другими противо-HCV агентами.
После открытия в 1989 г. в качестве агента, вовлеченного в большинство вирусных гепатитов, не относящихся к типу А и типу В (Choo et al. Science 244, 359-362, 1989), вирус гепатита С (HCV) стал центром значительного числа медицинских исследований (Lauer, G.M and Walker, B.D., New Eng. J Med. 345, 41-52, 2001). HCV является членом семейства вирусов Flaviviridae рода hepacivirus и близко связан с родом flavivirus, который включает ряд вирусов, вовлеченных в заболевания человека, таких как вирус денге и вирус желтой лихорадки, и с семейством pestivirus животных, которое включает вирус бычьей вирусной диареи (BVDV). HCV представляет собой положительно-смысловой одноцепочечный РНК вирус с геномом около 9600 пар нуклеотидов. Геном включает как 5' так и 3' нетранслируемые области, которые принимают вторичные структуры РНК и центральную открытую рамку считывания, которая кодирует единственный полипротеин, состоящий приблизительно из 3010-3030 аминокислот. Полипротеин кодирует десять генных продуктов, которые генерируются из предшественника полипротеина посредством серии сочетающихся со- и пост-трансляционных эндопротеолитических расщеплений, опосредованных с помощью протеаз хозяина и вируса. Структурные белки вируса включают ядерный нуклеокапсидный белок и два обрамляющих гликопротеина Е1 и Е2. Неструктурные (NS) белки кодируют некоторые неотъемлемые вирусные ферментативные функции (геликаза, полимераза, протеаза), а также белки с неизвестными функциями. Репликация вирусного генома опосредована РНК-зависимой полимеразой РНК, кодируемой неструктурным белком 5b (NS5B). В дополнение к полимеразе, на модели инфекции шимпанзе было показано, что функции как вирусной геликазы, так и протеазы, кодируемые в бифункциональном белке NS3, являются существенными для репликации РНК HCV (Kolykhalov, A.A., Mihalik, K., Feinstone, S.M., and Rice, C.M. J Virol. 74, 2046-2051, 2000). Помимо серинпротеазы NS3 HCV также кодирует металлопротеиназу в области NS2.
Репликация HCV предпочтительно протекает в гепатоцитах, но не является непосредственно цитопатической, приводя к стабильной инфекции. В частности, оказалось, что недостаток сильной ответной реакции Т-лимфоцитов и высокая склонность вируса к мутации промотируют высокую скорость хронической инфекции. Существует 6 основных генотипов HCV и более 50 подтипов, которые географически распределены различным образом. HCV типа 1 является доминирующим генотипом в США и Европе. Например, HCV типа 1 насчитывает от 70 до 75 процентов всех HCV инфекций в США. Большая генетическая гетерогенность HCV имеет важные диагностические и клинические последствия, возможно объясняя трудности в разработке вакцин и недостаточную ответную реакцию на лечение. Как оценивается, 170 миллионов человек во всем мире инфицировано вирусом гепатита С (HCV). После первоначальной острой инфекции у большинства инфицированных индивидуумов развивается хронический гепатит, который может прогрессировать в фиброз печени, приводящий к циррозу, конечной стадии заболевания печени и HCC (гепатоклеточная карцинома) (National Institutes of Health Consensus Development Conference Statement: Management of Hepatitis C. Hepatology, 36, 5 Suppl. S3-S20, 2002). Цирроз печени, возникший вследствие инфекции HCV является ответственным за примерно 10000 смертей в год только в США и является основной причиной для трансплантации печени. Передача HCV может происходить за счет контакта с зараженной кровью и продуктами крови, например, после переливания крови или внутривенного применения наркотиков. Введение диагностических тестов, используемых при скрининге крови, привело к нисходящей тенденции случаев HCV после переливания крови. Однако, принимая во внимание медленное развитие до конечной стадии заболевания печени, существующая инфекция будет продолжать оставаться серьезной медицинской и экономической проблемой на протяжении десятилетий (Kim, W.R. Hepatology, 36, 5 Suppl. S30-S34, 2002).
Лечение данного хронического заболевания является неудовлетворенной клинической потребностью, поскольку существующая в настоящее время терапия является эффективной только частично и ограничена нежелательными побочными действиями.
Имеющаяся в настоящее время терапия HCV основана на (пэгилированном) интерфероне-альфа (IFN-α) в сочетании с рибавирином. Такая комбинированная терапия проводит к продолжительному вирологическому ответу более чем у 40% пациентов, инфицированных вирусами генотипа 1 и примерно у 80% пациентов, инфицированных генотипами 2 и 3. В сравнении с ограниченной эффективностью на HCV типа 1 комбинированная терапия имеет существенные побочные действия и плохо переносится большинством пациентов. Например, в зарегистрированных исследованиях пэгилированного интерферона и рибавирина заметные побочные действия приводили к приостановке лечения у приблизительно от 10 до 14 процентов пациентов. Основные побочные действия при комбинированной терапии включают гриппоподобные симптомы, гематологические аномалии и нейропсихиатрические симптомы. Разработка более эффективного, удобного и легче переносимого лечения является главной задачей общественного здравоохранения. Таким образом, существует повышенная медицинская необходимость в низкомолекулярных соединениях, которые приводили бы к ингибированию репликации HCV. За счет ингибирования репликации HCV достигается быстрое снижение вирусной нагрузки у пациентов, инфицированных вирусом гепатита С. Снижение вирусной нагрузки является доказательством принципа клинической антивирусной активности ингибиторов HCV. При поддержании вирусной нагрузки у пациентов от низкого до необнаруживаемого уровня замедляется или даже останавливается прогрессирование заболевания, исключая таким образом развитие клинического гепатита, фиброза печени, цирроза, конечной стадии заболевания печени и HCC (гепатоклеточной карциномы).
Резистентность вирусов и, в частности, вируса HCV в отношении ингибиторов также является причиной неудачного лечения. Многие пациенты, получающие противо-HCV терапию, полностью не поддаются лечению, главным образом вследствие резистентности вируса к одному или нескольким использованным лекарственным средствам. Более того, было показано, что резистентный вирус переносится на вновь инфицированных индивидуумов, приводя к весьма ограниченным возможностям терапии для данных не получавших каких-либо лекарственных средств пациентов. Следовательно, в данной области существует необходимость в новых соединениях для противовирусной терапии, более конкретно, для терапии гепатита. Особенно острой в этой области является необходимость в соединениях, которые являются активными не только в отношении вируса HCV дикого типа, но также в большей степени в отношении более обычных резистентных вирусов HCV.
Соединения, использованные в настоящем изобретении, представляют собой производные пиримидина или триазина. В PCT публикации WO01/47921 описаны соединения пиримидина и триазина, которые представляют собой ингибиторы активности киназ, связанной с различными воспалительными состояниями. Кроме того, в PCT публикациях WO00/12497 и WO02/076976 описаны производные хиназолина, которые представляют собой ингибиторы киназы рецептора TGFβR и TGF-β-опосредованной передачи сигнала.
WO04/087056 относится к бициклическим пиримидиновым ингибиторам TGF-β, в которых пиримидиновое кольцо связано мостиком, соединяющим положения (5) и (6), и дополнительно замещенных в положениях (2) и (4) заместителями, включающими ароматические фрагменты. Данные соединения можно использовать для лечения субъектов, состояние которых улучшается при ингибирования активности TGFβ.
WO03/097615 касается способов лечения фибропролиферативных нарушений, связанных с TGF-β-опосредованной передачей сигнала, путем введения непептидных небольших молекул ингибиторов TGF-β, специфически связывающихся с рецептором TGF-β типа I (TGF-β-R1). Данные ингибиторы представляют собой производные хиназолина.
WO04/065392 относится к конденсированным пиридинам и пиримидинам и их применению в качестве лигандов рецепторов ALK-5. Данные соединения являются терапевтически активными, в частности при лечении и профилактике нарушений, характеризуемых сверхэкспрессией трансформирующего фактора роста β (TGF-β). Описаны фармацевтические композиции для применения при такой терапии.
В WO03/078426 описаны азолиламиноазины в качестве ингибиторов протеинкиназ, фармацевтически приемлемые композиции, включающие указанные соединения и способы применения композиций для лечения различных заболеваний, состояний или нарушений.
В WO03/078427 описаны азолиламиноазины в качестве ингибиторов протеинкиназ, фармацевтически приемлемые композиции, включающие указанные соединения и способы применения композиций для лечения различных заболеваний, состояний или нарушений.
В WO2003077921 описаны азиниламиноазолы в качестве ингибиторов протеинкиназ, фармацевтически приемлемые композиции, включающие указанные соединения и способы применения композиций для лечения различных заболеваний, состояний или нарушений.
В WO03/078423 описаны соединения, полезные в качестве ингибиторов протеинкиназ, фармацевтически приемлемые композиции, включающие указанные соединения и способы применения композиций для лечения различных заболеваний, состояний или нарушений.
В WO02/22601 описаны композиции пиразола, включающие фармацевтически приемлемый носитель, и соединения, которые, которые могут использоваться в качестве ингибиторов протеинкиназ, в особенности ингибиторов аврора-2 и GSK-3, для лечения таких заболеваний, как рак, диабет и болезнь Альцгеймера.
Описание изобретения
Соединения по настоящему изобретению представляют собой производные пиримидина, которые содержат конденсированное кольцо, связывающее в виде мостика положения 5 и 6. Соединения имеют формулу
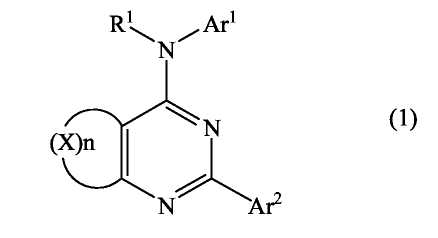
или являются ее фармацевтически приемлемой солью, где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, является насыщенным, ненасыщенным или ароматическим. Конденсированное кольцо является необязательно замещенным кольцом и содержит 4-7 члена, где каждый член независимо представляет собой C, N, O или S. Однако если указанное конденсированное кольцо содержит 6 членов, оно не является ароматическим.
Каждый из Ar1 и Ar2 независимо представляет собой необязательно замещенный ароматический фрагмент или необязательно замещенный гетероароматический фрагмент, содержащий один или несколько O, S и/или N; обычно данные фрагменты содержат 5-12 членов.
R1 представляет собой H или необязательно замещенный алкил (1-10C), алкенил (2-10C) или алкинил (2-10C).
Соединения, используемые в данном изобретении, представляют собой производные пиримидина, содержащие мостик в положениях 5 и 6, и обязательные заместители в положениях, соответствующими 2- и 4-положениям пиримидина. Дополнительно также могут быть включены не мешающие заместители.
Настоящее изобретение относится к применению соединения формулы

или его фармацевтически приемлемой соли;
где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, является необязательно замещенным насыщенным, ненасыщенным или ароматическим кольцом, содержащим 4-7 членов, где Х представляет собой атом, выбранный из N, O или S;
n равно 0, 1, 2 или 3;
каждый из Ar1 и Ar2 независимо представляет собой необязательно замещенный ароматический фрагмент или необязательно замещенный гетероароматический фрагмент, где указанный гетероароматический фрагмент содержит один или несколько O, S и/или N; и данные ароматический и гетероароматический фрагменты содержат 5-12 членов;
R1 представляет собой H или необязательно замещенный алкил (1-10C), алкенил (2-10C) или алкинил (2-10C);
при условии, что если указанное конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, содержит 6 членов, то оно не является ароматическим кольцом;
для получения лекарственного средства, которое может быть использовано для ингибирования активности HCV, профилактики или лечения состояний, связанных с HCV, у млекопитающих, инфицированных HCV.
В одном варианте осуществления настоящего изобретения разработано применение соединения формулы (1) для получения лекарственного средства, используемого для предотвращения прогрессирования заболевания, ведущего к хроническому гепатиту, фиброзу печени, циррозу, заболеванию печени последней стадии, HCC (гепатоклеточной карциноме) и тому подобному у млекопитающих, инфицированных HCV.
В одном варианте осуществления настоящего изобретения разработано применение соединения формулы (1) для получения лекарственного средства, используемого для лечения хронического гепатита, фиброза печени, цирроза, заболевания печени последней стадии, HCC (гепатоклеточной карциномы) и тому подобному у млекопитающих, инфицированных HCV.
Соединения, используемые в данном изобретении, представляют собой производные пиримидина, содержащие мостик между положениями 5 и 6, и обязательные заместители в положениях, соответствующих 2- и 4-положениям пиримидина. Дополнительно также могут быть включены не мешающие заместители.
В одном варианте осуществления настоящего изобретения каждый из Ar1 и Ar2 независимо представляет собой необязательно замещенный ароматический фрагмент или необязательно замещенный гетероароматический фрагмент, где указанный гетероароматический фрагмент содержит один или два O, S и/или N; и данные ароматический и гетероароматический фрагменты содержат 5-12 членов.
В одном варианте осуществления настоящее изобретение относится к применению соединения формулы

или его фармацевтически приемлемой соли,
где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, является необязательно замещенным насыщенным, ненасыщенным или ароматическим кольцом, содержащим 5 или 6 членов, где Х представляет собой атом, выбранный из N или О;
n равно 0, 1 или 2;
каждый из Ar1 и Ar2 независимо представляет собой необязательно замещенный ароматический фрагмент или необязательно замещенный гетероароматический фрагмент, где указанный гетероароматический фрагмент содержит один или два N; и данные ароматический и гетероароматический фрагменты содержат 5-7 членов;
R1 представляет собой H или необязательно замещенный алкил (1-10C);
при условии, что если указанное конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, содержит 6 членов, то оно не является ароматическим кольцом;
для получения лекарственного средства, которое может быть использовано для ингибирования активности HCV, профилактики или лечения состояний, связанных с HCV, у млекопитающих, инфицированных HCV.
В одном варианте осуществления настоящее изобретение относится к применению соединения формулы

или его фармацевтически приемлемой соли,
где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, является необязательно замещенным насыщенным, ненасыщенным или ароматическим кольцом, содержащим 5 или 6 членов, где Х представляет собой атом, выбранный из N или O;
n равно 0, 1 или 2;
каждый из Ar1 и Ar2 независимо представляет собой необязательно замещенный фенил или необязательно замещенный пиридил;
R1 представляет собой H или необязательно замещенный алкил (1-6C),
при условии, что если указанное конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, содержит 6 членов, то оно не является ароматическим кольцом,
для получения лекарственного средства, которое может быть использовано для ингибирования активности HCV, профилактики или лечения состояний, связанных с HCV, у млекопитающих, инфицированных HCV.
В одном варианте осуществления настоящее изобретение относится к применению соединения формулы

или его фармацевтически приемлемой соли,
где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, является необязательно замещенным насыщенным, ненасыщенным или ароматическим кольцом, содержащим 5 или 6 членов, где Х представляет собой атом, выбранный из N или O;
n равно 0, 1 или 2;
R1 представляет собой H или необязательно замещенный алкил (1-6C);
каждый R3 независимо представляет собой водород, галоген, циано, нитро, алкил(1-6C), полигалогеналкил(1-6C), -COR, -CONR2, -COOR, -OR, -OCOR, -NR2 или -NRCOR; каждый R4 независимо представляет собой водород, галоген, циано, нитро, полигалогеналкил(1-6C) или алкил(1-6C);
где каждый R независимо представляет собой водород, гидрокси, амино, моно- или диалкил(1-6C)амино, циклоалкил(3-7C), Het или алкил(1-6C), необязательно замещенный одним или двумя заместителями, выбранными из гидрокси, циклоалкила(3-7C), амино, моно- или диалкил(1-6C)амино и Het;
Het представляет собой 5 или 6-членное насыщенное, частично ненасыщенное или полностью ненасыщенное гетероциклическое кольцо, содержащее 1-2 гетероатома, каждый из которых независимо выбирают из азота, кислода и серы, и где Het как целое может быть необязательно замещенным одним, двумя или тремя заместителями, каждый из которых независимо выбирают из группы, состоящей из галогена, алкила(1-6С), гидрокси и оксо;
при условии, что если указанное конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, содержит 6 членов, то оно не является ароматическим кольцом;
для получения лекарственного средства, которое может быть использовано для ингибирования активности HCV, профилактики или лечения состояний, связанных с HCV, у млекопитающих, инфицированных HCV.
В одном варианте осуществления настоящее изобретение относится к применению соединения формулы

или его фармацевтически приемлемой соли,
где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, вместе с пиримидиновым кольцом образует группу, выбранную из
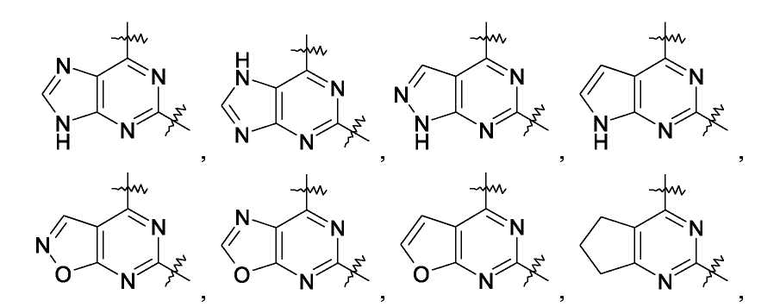
где любые из данных групп необязательно могут быть замещены одним или двумя заместителями, выбранными из алкила(1-6C), фенила и бензила;
R3 представляет собой водород, галоген, алкил(1-6C), -CF3, -COR, -CONR2 или -COOR;
каждый из R4a R4b независимо представляет собой водород или галоген,
где каждый R независимо представляет собой водород, гидрокси, амино, моно- или диалкил(1-6C)амино, циклоалкил(3-7C), Het или алкил(1-6C), необязательно замещенный одним или двумя заместителями, выбранными из гидрокси, циклоалкила(3-7C), амино, моно- или диалкил(1-6C)амино и Het;
Het представляет собой группу, выбранную из

где группа Het необязательно может быть замещена одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из алкила(1-6C) и оксо;
для получения лекарственного средства, которое может быть использовано для ингибирования активности HCV, профилактики или лечения состояний, связанных с HCV, у млекопитающих, инфицированных HCV.
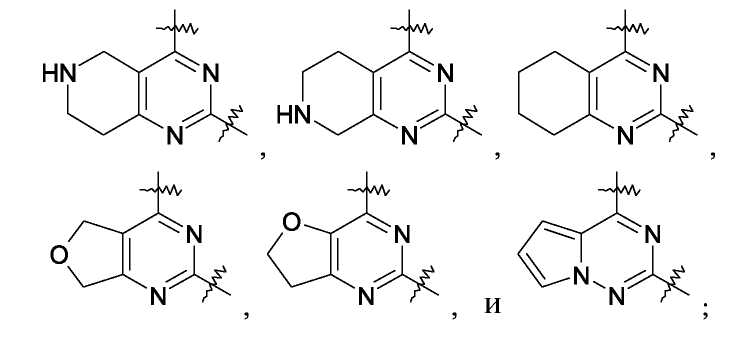
В одном варианте осуществления настоящее изобретение относится к применению соединения формулы

или его фармацевтически приемлемой соли,
где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, вместе с пиримидиновым кольцом образует группу, выбранную из


где любая из данных групп необязательна может быть замещена одним или двумя алкилами(1-6C);
R3 представляет собой водород, алкил(1-6C) или -CONR2;
каждый R4a R4b независимо представляет собой водород или галоген,
где каждый R независимо представляет собой водород, амино, моно- или диалкил(1-6C)амино или алкил(1-6C), необязательно замещенный одним гидрокси;
для получения лекарственного средства, которое может быть использовано для ингибирования активности HCV, профилактики или лечения состояний, связанных с HCV, у млекопитающих, инфицированных HCV.
Как использовано в данном описании, «не мешающий заместитель» представляет собой заместитель, который качественно не затрагивает способность соединения формулы (1) ингибировать активность HCV. Таким образом, заместитель может изменять степень ингибирования, но до тех пор, пока соединение формулы (1) сохраняет способность ингибировать активность, заместитель будет классифицироваться как «не мешающий».
Как использовано в данном описании, термин "алкил," "алкенил" и "алкинил" включают линейные, разветвленные и циклические одновалентные заместители, содержащие только С+Н, когда они являются не замещенными. Примеры включают метил, этил, изобутил, циклогексил, циклопентилэтил, 2-пропенил, 3-бутинил и тому подобные. Обычно алкильные, алкенильные и алкинильные заместители содержат 1-10С (алкил) или 2-10С (алкенил или алкинил). Предпочтительно они содержат 1-6C (низший алкил) или 2-6C (низший алкенил или низший алкинил).
Гетероалкил, гетероалкенил и гетероалкинил определены аналогично, но могут содержать 1 или более O, S или N гетероатомов и их комбинаций в остатке основной цепи.
Как использовано в данном описании, термин "ацил" охватывает определения алкила, алкенила, алкинила, а гетероацил включает соответствующие гетероатомы, которые связаны с дополнительным остатком через карбонильную группу.
Термин "ароматический" фрагмент или "арильный" фрагмент относится к моноциклическому или конденсированному бициклическому фрагменту, такому как фенил или нафтил; термин «гетероатоматический» также относится к моноциклической или конденсированной бициклической кольцевой системе, содержащей одни или несколько гетероатомов, выбранных из O, S и N. Включение гетероатома допускает включение 5-членного кольца, так же как и 6-членных колец. Таким образом, типичные ароматические/гетероароматические системы включают пиридил, пиримидил, индолил, бензимидазолил, бензотриазолил, изохинолил, хинолил, бензотиазолил, бензофуранил, тиенил, фурил, пирролил, тиазолил, оксазолил, имидазолил и тому подобные. Поскольку теоретически возможны таутомеры, фталимидо также рассматривается как ароматический. Любая моноциклическая или конденсированная кольцевая бициклическая система, которая имеет признаки ароматичности с точки зрения электронного распределения по кольцевой системе, включена в данное определение. Обычно кольцевые системы содержат 5-12 кольцевых атомов.
Аналогично, термины "арилалкил" и "гетероарилалкил" относятся к ароматическим и гетероароматическим системам, которые связаны с другими остатком через углеродную цепь, включая замещенные и незамещенные, насыщенные и ненасыщенные углеродные цепи, обычно 1-8C, и их гетероформы. Данные углеродные цепи также могут включать карбонильную группу, что дает им возможность обеспечивать такие заместители, как ацильный и гетероацильный фрагменты.
Термин "полигалогеналкил(1-6С)" в качестве группы или части группы определен как моно- или полигалогензамещенный алкил(1-6С), в частности алкил(1-6С), замещенный одним, двумя, тремя, четырьмя, пятью, шестью или более атомов галогена, такой как метил или этил с одним или несколькими атомами фтора, например дифторметил, трифторметил, трифторэтил. Предпочтительным является трифторметил. Определение также включает группы перфторалкил(1-6С), которые представляют собой группы алкил(1-6С), где все атомы водорода заменены на атомы фтора, например пентафторэтил. В том случае, если в рамках определения полигалогеналкила(1-6С) более одного атома галогена присоединено к алкильной группе, атомы галогена могут быть одинаковыми или разными.
Обычно любая алкильная, алкенильная, алкинильная, ацильная или арильная группа, содержащаяся в заместителе, сама необязательно может быть замещена дополнительными заместителями. Природа данных заместителей аналогична указанному в отношении самих основных заместителей. Таким образом, когда вариантом осуществления заместителя является алкил, данный алкил необязательно может быть замещен остальными заместителями, перечисленными в качестве заместителей, когда это имеет химический смысл и когда это не затрагивает предельный размер самого алкила; например, алкил, замещенный алкилом или алкенилом, будет просто превышать верхний предел числа атомов углерода для данных вариантов осуществления. Однако сюда включен алкил, замещенный арилом, амино, алкокси и тому подобными. Отличительные особенности соединений по изобретению определены формулами (1), (2), и (3), и природа заместителей менее важна до тех пор, пока заместители не служат помехой указанной биологической активности данной основной структуры.
Термин «галоген» является общим для фтора, хлора, брома и иода.
Как использовано в данном описании, термин «оксо» или (=O) относится к карбонильному фрагменту, который присоединен к атому углерода. Когда кольцо или кольцевая система замещена оксогруппой, атом углерода, с которым связана оксогруппа, является насыщенным атомом углерода.
Радикалы, использованные в определениях переменных, включают все возможные изомеры, если не указано другого. Например, пиридил включает 1-пиридил, 2-пиридил, 3-пиридил и 4-пиридил; пентил включает 1-пентил, 2-пентил и 3-пентил.
Когда любая переменная возникает более одного раза в любом компоненте, каждое определение является независимым.
Не мешающие заместители в Ar1 или Ar2 включают, но не ограничиваются указанным, алкил, алкенил, алкинил, галоген, -OR, -NR2, -SR, -SOR, -SO2R, -OCOR, -NRCOR, -NRCONR2, -NRCOOR, -OCONR2, -RCO, -COOR, -NRSOR, -NRSO2R, -SO3R, -CONR2, SO2NR2, где каждый R независимо представляет собой H или алкил(1-8C), -CN, -CF3 и NO2, и тому подобные заместители. Не мешающие заместители в Ar1 или Ar2 включают, но не ограничиваются указанным, алкил, алкенил, алкинил, галоген, -OR, -NR2, -SR, -SOR, -SO2R, -OCOR, -NRCOR, -NRCONR2, -NRCOOR, -OCONR2, -RCO, -COR, -COOR, -NRSOR, -NRSO2R, -SO3R, -CONR2, SO2NR2, где каждый R независимо представляет собой H или алкил(1-8C), -CN, -CF3 и NO2, и тому подобные заместители. Предпочтительным вариантом осуществления для R являются H, алкил (1-10C) или его гетероатомсодержащие формы, каждая их которых необязательно является замещенной, в особенности, (1-4C)алкилом; алкокси (1-8C), ациламидо, арилокси, арилалкилокси, в особенности, когда арильная группа представляет собой фталимидогруппу, и алкил или арилалкиламином.
Предпочтительно Ar1 и Ar2 представляют собой необязательно замещенный фенил, 2-, 3- или 4-пиридил, индолил, 2- или 4-пиримидил, пиридазинил, бензотриазолил или бензимидазолил. Более предпочтительно Ar1 и Ar2 представляют собой фенил, пиридил или пиримидил. Предпочтительно Ar1 представляет собой пиридил или пиримидил, и Ar2 представляет собой фенил. Каждый из данных вариантов осуществления необязательно может быть замещен такой группой как алкил, алкенил, алкинил, арил, -O-арил, -O-алкиларил, -O-ароил, -NR-арил, -N-алкиларил, -NR-ароил, галоген, -OR, -NR2, -SR, -OOCR, -NROCR, -RCO, -COOR, -CONR2 и/или SO2NR2, где каждый R независимо представляет собой H или алкил (1-8C), и/или такой группой как -CN, -CF3 и/или NO2. Каждый из данных вариантов осуществления необязательно может быть замещен одной или двумя группами, такими как алкил, алкенил, алкинил, арил, -O-арил, -O-алкиларил, -O-ароил, -NR-арил, -N-алкиларил, -NR-ароил, галоген, -OR, -NR2, -SR, -OOCR, -OCOR, -NROCR, -NRCOR, -RCO, -COR, -COOR, -CONR2 и/или SO2NR2, где каждый R независимо представляет собой H или алкил (1-8C), и/или такими группами, как -CN, -CF3 и/или NO2. Алкильные, алкенильные, алкинильные и арильные части данных заместителей могут быть дополнительно замещены аналогичными заместителями. Предпочтительные заместители в Ar1 или Ar2 включают алкил, алкенил, алкинил, галоген, -OR, -SR, -NR2, где R представляет собой H или алкил (1-4C); и/или ариламино, арилалкиламино, включают алкиламино, который замещен более чем одним арилом. Как указано выше, любая арильная или алкильная группа, включенная в заместитель, сама может быть замещена аналогичным образом. Данные заместители могут занимать все возможные положения кольца, предпочтительно 1-2 положения и наиболее предпочтительно только одно положение.
Любой из арильных фрагментов, включая указанные в формулах (1), (2), и (3), в особенности фенильные фрагменты, могут также включать два заместителя, которые, взятые вместе, образуют 5-7-членное карбоциклическое или гетероциклическое алифатическое кольцо. Мостик между положениями 5 и 6 пиримидинового кольца образует конденсированную кольцевую систему, где, если конденсированное кольцо содержит 6 членов, то оно не является ароматическим. Однако мостик также может содержать пи-связи, и может содержать один или несколько гетероатомов, которые выбраны из N, O и S. Предпочтительные варианты осуществления включают те, в которых мостик приводит к образованию 5-членного кольца, необязательно содержащего один или два азота, азот и кислород, кислород и дополнительную двойную связь, насыщенный мостик, или 6-членное кольцо, образованное мостиком, которое является насыщенным. В одном варианте осуществления 6-членное насыщенное кольцо, образованное за счет мостика, содержит один или два азота. Кольцо, образованное мостиком, само может быть замещенным. Заместители, описанные выше для Ar1 и Ar2, также могут присутствовать в качестве конденсированных кольцевых систем.
Всякий раз при последующем использовании подразумевается, что термин «соединения формулы (1)», «соединения формулы (2)», «соединения формулы (3)» или «настоящие соединения» или аналогичные термины включают соединения формул (1), (2) и (3), их пролекарства, N-оксиды, аддитивные соли, четвертичные амины, комплексы металлов и стереохимически изомерные формы. Один вариант осуществления включает соединения формулы (1) или любую подгруппу определенных в данном описании соединений формулы (1), включая соединения формулы (2) и (3), а также N-оксиды, соли в виде их возможных стереоизомерных форм. Другой вариант осуществления включает соединения формулы (1) или любую подгруппу определенных в данном описании соединений формулы (1), а также их соли в виде их возможных стереоизомерных форм.
Для терапевтического применения соли соединений формулы (1) представляют собой те, в которых противоион является фармацевтически приемлемым. Однако соли кислот и оснований, которые не являются фармацевтически приемлемыми, также могут находить применение, например, при получении или очистке фармацевтически приемлемого соединения. Все соли, являются ли они фармацевтически приемлемыми или нет, включены в объем настоящего изобретения.
Подразумевается, что фармацевтически приемлемые кислотные и основные аддитивные соли, как отмечено выше, включают терапевтически активные нетоксичные формы аддитивных солей кислот и оснований, которые способны образовывать соединения формулы (1). Фармацевтически приемлемые кислотно-аддитивные соли могут быть удобным образом получены путем обработки основной формы такой подходящей кислотой. Подходящие кислоты включают, например, неорганические кислоты, такие как галогенводородные кислоты, например хлористоводородная и бромистоводородная кислота, серная, азотная, фосфорная и тому подобные кислоты; или органические кислоты, например, такие как уксусная, пропановая, гидроксиуксусная, молочная, пировиноградная, щавелевая (т.е. этандионовая), малоновая, янтарная (т.е. бутандионовая кислота), бензойная, малеиновая, фумаровая, яблочная (т.е. гидроксибутандионовая кислота), винная, лимонная, метансульфоновая, этансульфоновая, бензолсульфоновая, п-толуолсульфоновая, цикламовая, салициловая, п-аминосалициловая, памовая и тому подобные кислоты.
Наоборот, указанные солевые формы могут быть преобразованы при обработке подходящим основанием в форму свободного основания.
Соединения формулы (1), содержащие кислый протон, также могут быть преобразованы в их нетоксичные аддитивные формы солей металлов или аминов путем обработки подходящими органическими и неорганическими основаниями. Таким образом, если в соединении формулы (1) присутствует карбоксильный фрагмент, соединение также может быть получено в виде соли с фармацевтически приемлемым катионом. Подходящие солевые формы оснований включают, например, соли аммония, соли щелочных и щелочноземельных металлов, например, лития, натрия, калия, магния, соли кальция и тому подобные, соли с органическими основаниями, например бензатином, N-метил-D-глюкамином, соли гидрабамина и соли с аминокислотами, такими как, например, аргинин, лизин и тому подобные.
Термин «аддитивная соль» как он использован выше, также включает сольваты, которые способны образовывать как соединения формулы (1), так и их соли. Такие сольваты представляют собой, например, гидраты, алкоголяты и тому подобные.
Соединения формулы (1) также могут быть представлены в виде «пролекарства», которое разработано для высвобождения соединения формулы (1) при введении субъекту. Термин «пролекарство», как он использован в данном описании, означает фармакологически приемлемые производные, такие как сложные эфиры, амиды и фосфаты, так, что образующийся продукт биотрансформации производного in vivo представляет собой активное лекарственное средство, как определено для соединений формулы (1). В общем виде в описание включена ссылка на Goodman Gilman (The Pharmacological Basis of Therapeutics, 8th ed, McGraw-Hill, Int. Ed. 1992, "Biotransformation of Drugs", p 13-15), описывающая пролекарства. Пролекарства предпочтительно обладают превосходной растворимостью в воде, повышенной биодоступностью и легко метаболизируют в активные ингибиторы in vivo. Пролекарства соединения по настоящему изобретению могут быть получены путем модификации функциональных групп, присутствующих в соединениях, таким образом, что модифицированные фрагменты отщепляются или путем обычных манипуляций, или in vivo с образованием исходного соединения.
Образующие пролекарства структуры хорошо известны в данной области и зависят от заместителей, содержащихся в соединениях формулы (1). Например, заместитель, содержащий сульфгидрильную группу, может быть сконденсирован с носителем, который придает соединению биологическую неактивность до его удаления эндогенными ферментами, или, например, ферментами, нацеленными на конкретный рецептор или местоположение в организме субъекта.
Предпочтительными являются фармацевтически приемлемые сложноэфирные пролекарства, которые являются гидролизуемыми in vivo и являются производными тех соединений формулы (1), которые имеют гидроксильную или карбоксильную группу. Гидролизуемый in vivo сложный эфир представляет собой сложный эфир, который гидролизуется в организме человека или животного с образованием исходной кислоты или спирта. Подходящие фармацевтически приемлемые сложные эфиры карбоксильных групп включают алкоксиметиловые сложные эфиры, например метоксиметиловый, алканоилоксиметиловые сложные эфиры, например пивалоилоксиметиловый, фталидиловые сложные эфиры, циклоалкоксикарбонилоксиалкиловые сложные эфиры, например 1-циклогексилкарбонилоксиэтиловый; 1,3-диоксолен-2-онилметиловые сложные эфиры, например 5-метил-1,3-диоксолен-2-онилметиловый; и алкоксикарбонилоксиэтиловые эфиры, например 1-метоксикарбонилоксиэтиловый, которые могут быть образованы по любой карбоксильной группе в соединениях по изобретению.
Гидролизуемые in vivo сложные эфиры соединений формулы (1), содержащих гидроксильную группу, включают неорганические сложные эфиры, такие как фосфатные эфиры и α-ацилоксиалкиловые простые эфиры и родственные соединения, которые в результате гидролиза in vivo, протекающего с расщеплением сложного эфира, приводят к исходной гидроксильной группе. Примеры α-ацилоксиалкиловых простых эфиров включают ацетоксиметокси и 2,2-диметилпропионилоксиметокси. Выбор гидролизуемых in vivo образующих сложный эфир групп для гидроксильной группы включает алканоил, бензоил, фенилацетил и земещенный бензоил и фенилацетил, алкоксикарбонил (с получением алкилкарбонатных сложных эфиров), диалкилкарбамоил и N-(диалкиламиноэтил)-N-алкилкарбамоил (с получением карбаматов), диалкиламиноацетил и карбоксиацетил. Примеры заместителей у бензоила включают морфолино и пиперазиногруппы, с образованием связи от кольцевого атома азота через метиленовую группу к 3- или 4-положению бензоильного кольца.
Термин «четвертичный амин», как он использован ранее в данном описании, относится к определению солей четвертичного аммония, которые способны образовывать соединения формулы (1) при реакции между основным азотом в соединении формулы (1) и подходящим кватернизующим агентом, например, таким как необязательно замещенный алкилгалогенид, арилгалогенид и арилалкилгалогенид, например метилиодид или бензилиодид. Также можно использовать другие реагенты с хорошими уходящими группами, такие как алкилтрифторметансульфонаты, алкилметансульфонаты и алкил-п-толуолсульфонаты. Четвертичный амин содержит положительно заряженный азот. Фармацевтически приемлемые противоионы включают хлор, бром, иод, трифторацетат и ацетат. Выбранный противоион может быть введен с использованием ионообменных смол.
Подразумевается, что N-оксидные формы настоящих соединений включают соединения формулы (1), где один или несколько атомов азота окислены в так называемые N-оксиды.
Следует понимать, что соединения формулы (1) могут обладать металлсвязывающими, хелатирующими, комплексообразующими свойствами и, следовательно, могут существовать как комплексы металлов и хелаты металлов. Предполагается, что такие металлированные производные соединений формулы (1) включены в объем настоящего изобретения.
Некоторые соединения формулы (1) также могут существовать в их таутомерных формах. Подразумевается, что такие формы, хотя и не указаны подробно в вышеуказанной формуле, включены в объем настоящего изобретения.
Соединения формулы (1) имеют несколько центров хиральности и существуют в виде стереохимически изомерных форм. Термин «стереохимически изомерные формы», как он использован в данном описании, определяет все возможные соединения, полученные из тех же атомов, связанных той же последовательностью связей, но имеющие другие трехмерные структуры, которые не являются взаимозаменяемыми, которыми могут обладать соединения формулы (1).
При ссылке на случаи, когда обозначения (R) или (S) используются для обозначения абсолютной конфигурации хирального атома в заместителе, обозначение приведено, принимая во внимание соединение целиком, а не отдельный заместитель.
В том случае, когда любые заместители формулы (1) содержат хиральные центры, как это действительно имеет место для некоторых из них, соединения формулы (1) включают все стереоизомерные формы, как и все выделенные стереоизомеры и смеси таких стереоизомерных форм. Указанные смеси могут содержать все диастереомеры и/или энантиомеры основной молекулярной структуры указанного соединения. Подразумевается, что объем настоящего изобретения охватывает все стереохимические изомерные формы соединений настоящего изобретения как в чистом виде, так и в смеси друг с другом.
Чистые стереоизомерные формы соединений и промежуточных соединений, как отмечено в данном описании, определены как изомеры, по существу не содержащие других энантиомерных или диастереомерных форм той же основной молекулярной структуры указанных соединений или промежуточных соединений. В частности, термин «стереохимически чистый» относится к соединениям или промежуточных соединениям, имеющим стереоизомерный избыток по крайней мере 80% (т.е. минимум 90% одного изомера и максимум 10% других возможных изомеров), вплоть до стереоизомерного избытка в 100% (т.е. 100% одного изомера при отсутствие всех других), более конкретно, к соединениям или промежуточных соединениям, имеющим стереоизомерный избыток от 90 до 100%, еще более конкретно, имеющим стереоизомерный избыток от 94 до 100%, и наиболее конкретно, имеющим стереоизомерный избыток от 97 до 100%. Термины «энантиомерно чистый» и «диастереомерно чистый» следует понимать аналогичным образом, но имеющим отношение соответственно к энантиомерному избытку и дистереомерному избытку в рассматриваемых смесях.
Чистые стереоизомерные формы соединений и промежуточных соединений по данному изобретению могут быть получены с использованием известных в данной области способов. Например, энантиомеры могут быть отделены друг от друга путем селективной кристаллизации их диастереомерных солей с оптически активными кислотами или основаниями. Примерами являются винная кислота, дибензоилвинная кислота, дитолуилвинная кислота и камфорсульфоновая кислота. Альтернативно, энантиомеры могут быть разделены хроматографическими методами с использованием хиральных стационарных фаз. Указанные стереохимически чистые изомерные формы могут быть получены из соответствующих стереохимически чистых изомерных форм подходящих исходных веществ при условии, что реакция протекает стереоспецифически. Предпочтительно, если желателен определенный стереоизомер, указанное соединение синтезировать стереоспецифическими способами получения. В данных способах преимущественно будут использовать энантиомерно чистые исходные вещества.
Диастереомерные рацематы соединений формулы (1) могут быть получены отдельно удобными способами. Подходящие физические способы разделения, которые преимущественно можно использовать, представляют собой, например, селективную кристаллизацию и хроматографию, например колоночную хроматографию.
Для некоторых соединений формулы (1), их пролекарств, N-оксидов, солей, сольватов, четвертичных аминов, или комплексов металлов и промежуточных соединений, использованных для их получения, абсолютная стереохимическая конфигурация не была определена экспериментально. Специалист в данной области способен определить абсолютную конфигурацию таких соединений с использованием способов, известных в данной области, например таких, как рентгенодифракционное исследование.
Настоящее изобретение предназначено для охвата всех изотопов атомов, существующих в настоящих соединениях. Изотопы включают те атомы, которые имеют тот же самый атомный номер, но различные массовые числа. В качестве общего примера и без ограничения изотопы водорода включают тритий и дейтерий. Изотопы углерода включают C-13 и C-14.
Синтез соединений изобретения
Для получения соединений по изобретению можно использовать ряд синтетических путей. Обычно их можно синтезировать с использованием реакций, известных в данной области. Можно использовать любой известный метод синтеза. Однако следующие пути синтеза являются удобными для получения соединений по изобретению. Типичные соединения по изобретению показаны ниже.

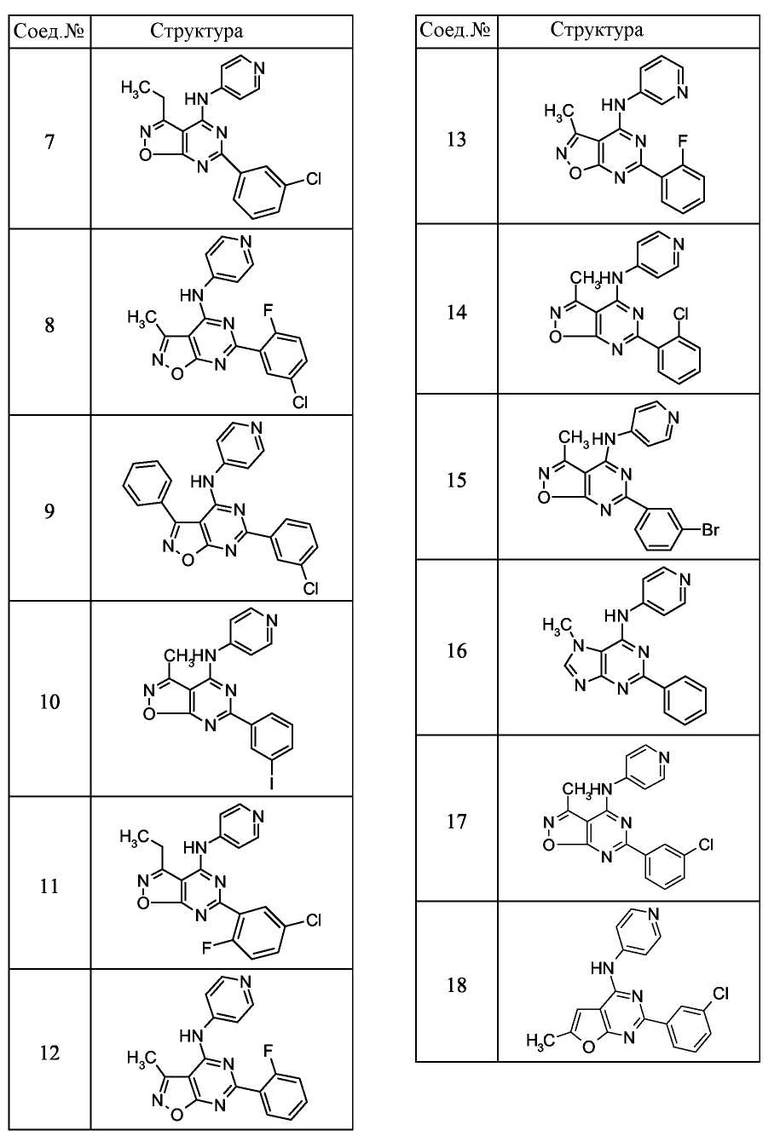







Схема A (Синтез соединений 1 и 26):
Данная общая схема была использована для получения соединений 1 и 16.

Получение соединения формулы (I):
2,53 г 4-Амино-5-имидазолкарбоксамида растворяли в 30 мл хлороформа и 30 мл диметилформамида. К этому раствору добавляли при 0°C 3,02 мл 3-хлорбензоилхлорида с последующим добавлением 5,4 мл диизопропилэтиламина. Реакционную смесь оставляли нагреваться до комнатной температуры и выдерживали при комнатной температуре в течение ночи. Реакционную смесь разбавляли хлороформом и промывали водой, 10% водным раствором карбоната натрия и насыщенным раствором соли. Органический слой сушили над безводным сульфатом натрия и фильтровали. Неочищенный остаток, полученный после концентрирования, помещали в минимальное количество хлороформа и хроматографировали на силикагеле с использованием этилацетата, 5% метанола, получая 4,81 г соединения формулы (I).
Примечание: для синтеза соединения 16 использовали метил-4-амино-5-имидазолкарбоксамид
Получение соединения формулы (II):
2,74 г Соединения формулы (I) суспендировали в 75 мл этанола, добавляли 5 мл 10 н. гидроксида натрия к реакционной смеси и реакционную смесь нагревали при кипении с обратным холодильником в течение четырех часов. После охлаждения до комнатной температуры реакционную смесь концентрировали для удаления этанола и затем разбавляли водой. Раствор затем подкисляли, добавляя 1 н. соляной кислоты при 0°C, до pH 6,5. Образовавшийся белый осадок собирали фильтрованием, промывали водой и простым эфиром и сушили в высоком вакууме, получая 0,84 г соединения формулы (II).
Получение соединения формулы (III):
0,84 г Соединения формулы (II) суспендировали в 60 мл хлороформа, к этой суспензии добавляли 1,1 мл хлористого тионила и 2 мл диметилформамида. Полученную смесь нагревали при кипении с обратным холодильником в токе азота в течение трех часов. Реакционную смесь охлаждали до комнатной температуры и концентрировали, получая желтый остаток. Этот остаток растворяли в хлороформе и к реакционной смеси добавляли лед. Холодный раствор промывали 5%-ным водным раствором бикарбоната натрия и насыщенным раствором соли. Органический слой сушили над безводным сульфатом натрия и фильтровали. После концентрирования полученный остаток обрабатывали холодным этилацетатом. Отделяется белое твердое вещество. Это твердое вещество отфильтровывали и промывали этилацетатом, получая 0,46 г соединения формулы (III).
Получение соединения формулы (1):
0,46 г Соединения формулы (III) растворяли в 10 мл сухого диметилформамида, к раствору добавляли 0,67 мл диизопропилэтиламина. Реакционную смесь нагревали до 60°C и добавляли к ней раствор 0,21 г 4-аминопиридина. Реакционную смесь нагревали при кипении с обратным холодильником в течение одного часа. После охлаждения до комнатной температуры реакционную смесь концентрировали до минимального объема, и продукт очищали препаративной ВЭЖХ с обращенной фазой с использованием колонки C18 Vydac® и градиента воды, ацетонитрила (оба из которых содержат 0,1% трифторуксусной кислоты). После лиофилизации фракций, содержащих целевой продукт, получали 15 мг соединения формулы (1). Анализ: 1H ЯМР, d6 ДМСО, ЖХМС, M+ 323.
Получение соединения формулы (26):
Соединение 26 получали в соответствии со способом, представленным на схеме А, с использованием N-1-фенил-2-аминоимидазол-3-карбоксамида и хлористого бензоила.
Схема B (Синтез соединений 3, 5-15 и 17):
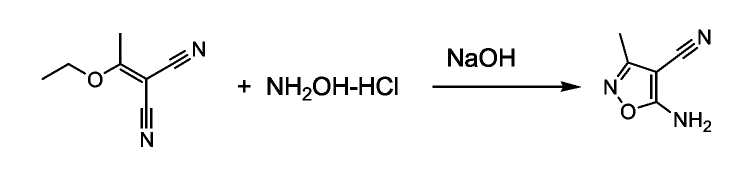
Получение 3-амино-4-циано-5-метилизоксазола:
Гидрохлорид гидроксиламина (12,78 г, 0,184 моль) растворяли в 40 мл воды, и обрабатывали гидроксидом натрия (7,36 г, 0,184 моль). Добавляли 60 мл этанола (безводный) и при перемешивании осторожно добавляли (1-этоксиэтилиден)малонитрил (25 г, 0,184 моль). Реакционную смесь нагревали при 50°C в течение 30 минут, затем перемешивали в течение ночи при комнатной температуре. Этанол удаляли в вакууме, отфильтрованный твердый продукт промывали водой, сушили в вакууме, получая 21,93 г (96,8% выход).
Примечание: Для синтеза 7 и 11 использовали соответствующий этилгидроксиламин, а для синтеза 9 использовали соответствующий фенилгидроксиламин.

Получение N-(3-хлорбензоил)-4-циано-3-метил-5-карбоксамида:
3-амино-4-циано-5-метилизоксазол (6,0 г, 0,0487 моль) суспендировали в смеси ацетонитрил/тетрагидрофуран (30 мл/10 мл). Добавляли диизопропилэтиламин (8,26 мл, 0,0487 моль) с последующим добавлением по каплям 3-хлорбензоилхлорида. Смесь перемешивали при комнатной температуре в течение ночи. Выпавший в осадок продукт выделяли фильтрованием, промывали хлороформом. Получали 1,31 г продукта (11% выход).
Примечание: Для синтеза соединений 3, 5, 6, 8, 10, 12, 13, 14, 15 и 17 использовали соответствующие хлорангидриды кислот.

Получение 3-метил-6-(3-хлорфенил)изоксазол[5,4d]пиримидона:
N-(3-хлорбензоил)-4-циано-3-метил-5-карбоксамид (1,0 г, 15 ммоль) суспендировали в 20 мл 1 M раствора гидроксида натрия и обрабатывали 8 мл 30% перекиси водорода. Смесь нагревали при кипении с обратным холодильником в течение ночи. Охлажденную реакционную смесь помещали на ледяную баню и обрабатывали 1 M соляной кислотой до pH 6. Фильтровали продукт в виде белого осадка и сушили в вакууме, получая 537 мг (53% выход).

Получение 3-метил-4-хлор-6-(3-хлорфенил)изоксазол[5,4d]пиримидина:
3-метил-6-(3-хлорфенил)изоксазол[5,4d]пиримидон (535 мг, 2,04 ммоль) суспендировали в оксихлориде фосфора (6 мл) и нагревали при кипении с обратным холодильником в течение 4 часов. Избыток оксихлорида фосфора удаляли, добавляли лед и хлороформ (10 мл), подщелачивали насыщенным раствором бикарбоната натрия, продукт экстрагировали хлороформом и экстракты сушили над сульфатом натрия (безв.). Продукт очищали хроматографией на силикагеле, элюируя хлороформом. Получали 200 мг продукта.

Получение 3-метил-4-(4-аминопиридил)-6-(3-хлорфенил)изоксазол[5,4d]пиримидина (17):
4-аминопиридин (80,6 мг, 0,859 ммоль) растворяли в N-метилпирролидоне, добавляли диизопропилэтиламин (149 микролитров) с последующим добавлением 3-метил-4-хлор-6-(3-хлорфенил)изоксазол[5,4d]пиримидина (120 мг, 0,428 ммоль). Смесь нагревали при 50°C при перемешивании в течение 1 часа. Продукт очищали препаративной ВЭЖХ на колонке С18.
Схема C (Синтез соединений 2 и 4):

Получение 2-фенил-пирроло[2,3d]пиримидинона:
Гидрохлорид бензамидина (4,0 г, 0,25 моль) растворяли в 64 мл этанола. К этому раствору добавляли 8,0 мл 25 вес.% раствора метоксида натрия. Реакционную смесь затем перемешивали при комнатной температуре в течение 5 часов и фильтровали. Фильтрат затем добавляли к этил-2-циано-4,4-диэтоксибутирату (4,80 г, 0,21 моль). Этот раствор нагревали при кипении с обратным холодильником в течение 5 часов. Половину растворителя удаляли при пониженном давлении, затем добавляли 80 мл ледяной воды и pH доводили до 7 с помощью уксусной кислоты. Вещество затем охлаждали в течение 6 часов и продукт выделяли фильтрованием в вакууме.
Примечание: для синтеза соединения 4 использовали 3-хлорбензамидин.

Получение 4-хлор-2-фенил-пирроло[2,3d]пиримидина:
2-фенил-пирроло[2,3d]пиримидинон (1,0 г 4,73 ммоль) обрабатывали оксихлоридом фосфора (7 мл, 27,7 ммоль) и нагревали при кипении с обратным холодильником в течение 5 часов. Избыток оксихлорида фосфора удаляли при пониженном давлении и затем экстрагировали хлороформом, промывали раствором бикарбоната натрия. Органический слой сушили над сульфатом натрия (безв.), затем концентрировали досуха, получая продукт.

Получение 4-(4-аминопиридил)-2-фенил-пирроло[2,3d]пиримидина (2):
4-хлор-2-фенил-пирроло[2,3d]пиримидин (0,12 г, 1,27 ммоль) растворяли в 4 мл NMP. Добавляли N,N'-диизопропилэтиламин (0,229 мл) с последующим добавлением 4-аминопиридина (0,15 г, 0,635 ммоль). Реакционную смесь нагревали при кипении с обратным холодильником в течение 2 часов, охлаждали и очищали препаративной ВЭЖХ.
Схема D (Синтез соединения 18):

Получение ацетилметилдиметилмалоната:
Диметилмалонат (5 г, 0,189 моль) обрабатывали карбонатом калия (34,78 г, 0,25 моль), иодидом натрия (1,00 г, 0,0067 моль) и затем нагревали, добавляя при этом быстро порциями хлорацетон (23,1 г, 0,25 моль). Реакционную смесь нагревали при 100°C в течение 20 минут. К охлажденной реакционной смеси добавляли 50 мл этанола и отфильтрованное твердое вещество промывали этанолом. Этанол из фильтрата удаляли в вакууме. Продукт выделяли перегонкой в вакууме. Получали 11,26 г продукта (32% выход).

Получение соединения формулы (IV):
Этиленгликоль (3,90 г, 0,0628 моль), ацетилметилдиметилмалонат (11,26 г, 0,06 моль), и п-толуолсульфоновую кислоту (0,21 г, 0,0011 моль) объединяли в 25 мл бензола. Реакционную смесь нагревали при кипении с обратным холодильником, собирая воду в насадке Дина-Старка в течение ночи. Реакционную смесь промывали 10% раствором бикарбоната натрия (2×10 мл) и бензол сушили над сульфатом натрия. Растворитель удаляли, получая продукт в виде масла, 14,1 г.
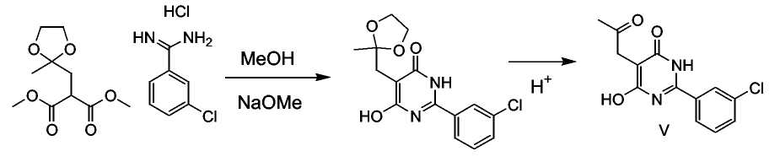
Получение соединения формулы (V):
Защищенное производное диметилмалоната (5,0 г, 0,0215 моль) растворяли в метаноле (20 мл), добавляли гидрохлорид 3-хлорбензамидина с последующим добавлением 25% раствора метоксида натрия (16 мл, 0,0646 моль). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней. Смесь разбавляли водой (50 мл), добавляли 60 мл 1 M HCl и перемешивали в течение 1 часа при комнатной температуре, затем добавляли 4 мл концентрированной HCl и перемешивали в течение ночи, получая конечный продукт. Метанол удаляли в вакууме, и продукт получали фильтрованием и высушиванием в вакууме. Получали 5 г продукта.

Получение 6-метил-2-хлорфенил-фурано[3,2d]пиримидона:
6-гидрокси-5-ацетилметил-2-(3-хлорфенил)пиримидон (5,0 г) обрабатывали концентрированной серной кислотой (80 мл). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов, затем нейтрализовали карбонатом натрия экстрагировали хлороформом, хлороформенный экстракт промывали водой, сушили над сульфатом натрия (безв.) и растворитель удаляли, получая продукт (1,10 г).

Получение 6-метил-4-хлор-2-хлорфенил-фурано[3,2d]пиримидина:
6-метил-2-хлорфенил-фурано[3,2-d]пиримидон (480 мг, 1,84 ммоль) суспендировали в дихлорметане (4 мл). Добавляли хлористый тионил (1,6 мл, 22,5 ммоль) и диметилформамид (0,5 мл) и нагревали при кипении с обратным холодильником в течение 3 часов. Избыток растворителя удаляли, остаток обрабатывали льдом, экстрагировали хлороформом, промывали 10% раствором бикарбоната натрия и водой, сушили над сульфатом натрия (безв.) и растворитель удаляли, получая 480 мг продукта.

Получение 6-метил-4-(4-аминопиридил)-2-хлорфенилфурано[3,2d]пиримидина (18):
6-метил-4-хлор-2-хлорфенил-фурано[3,2d]пиримидин (480 мг, 1,72 ммоль, 1 экв.), BINAP (8 мг, 0,013 ммоль, 0,0075 экв.), Pd2(dba)3 (3,9 мг, 0,0043 ммоль, 0,0025 экв.), трет-бутоксид натрия (231 мг, 2,4 ммоль, 1,4 экв.), 4-аминопиридин (194 мг, 2,06 ммоль, 1,2 экв.) объединяли в 5 мл диоксана и нагревали при 50°C в течение 5 часов. Продукт выделяли препаративной ВЭЖХ на колонке С18.
Схема E (Синтез соединения 19)

Получение этил-2-метил-5-этокси-4-оксазол-4-карбоксилата:
Диэтилацетамидомалонат (15,0 г, 69,1 ммоль) растворяли в 60 мл хлороформа, затем обрабатывали 60 г пентаоксида фосфора. Реакционную смесь нагревали при кипении в течение 6 часов, затем охлаждали до комнатной температуры. Этот раствор обрабатывали гидроксидом натрия (1 M) для нейтрализации реакционной смеси. Органический слой промывали водой и сушили над сульфатом натрия (безв.). Неочищенный продукт перегоняли в вакууме для выделения продукта. Получали 8,26 г продукта (60% выход).
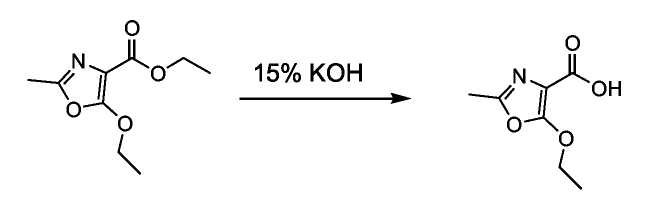
Получение 2-метил-5-этокси-4-оксазол-4-карбоновой кислоты:
Этил-2-метил-5-этокси-4-оксазол-4-карбоксилат (8,26 г, 41,5 ммоль) обрабатывали 74 мл 15%-ного раствора KOH. Эту смесь нагревали при кипении с обратным холодильником в течение 15 минут, затем охлаждали и подкисляли с использованием 10%-ного раствора HCl. Продукт собирали фильтрованием в вакууме.

Получение 2-метил-5-этокси-4-оксазол-4-карбонилхлорида:
2-метил-5-этокси-4-оксазол-4-карбоновую кислоту (2,56 г, 14,9 ммоль) растворяли в дихлорметане. Добавляли две капли диметилформамида и реакционную смесь охлаждали на ледяной бане. Добавляли по каплям оксалилхлорид (12 мл, 22,3 ммоль). Ледяную баню удаляли и реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Растворитель удаляли при пониженном давлении.

Получение 4-(3-хлорбензамидинамид)-2-метил-5-этокси-4-оксазол-4-карбонилхлорида:
3-хлорбензамидин (2,29 г, 14,8 ммоль) растворяли в 40 мл ДХМ и доводили температуру до 0°C. Затем добавляли 15 мл 2,0 M раствора гидроксида натрия. К реакционной смеси добавляли по каплям 2-метил-5-этокси-4-оксазол-4-карбонилхлорид (2,8 г, 14,8 ммоль), растворенный в 30 мл ДХМ, и перемешивали при комнатной температуре в течение 3 часов. Органический растворитель затем промывали водой, затем бикарбонатом натрия, затем сушили сульфатом натрия и концентрировали.

Получение соединения формулы (VI):
4-(3-хлорбензамидинамид)-2-метил-5-этокси-4-оксазол-4-карбонилхлорид (2,0 г, 6,5 ммоль) растворяли в 30 мл толуола и нагревали при кипении с обратным холодильником в течение 1,5 часов. Растворитель затем удаляли при пониженном давлении.

Получение 7-метил-2-(3-хлорфенил)оксазоло[2,3d]пиримидона:
1,86 г, 5,93 ммоль сложного эфира оксазола обрабатывали 0,86 г, 15,4 ммоль KOH в 20 л этанола. Эту смесь перемешивали при комнатной температуре в течение ночи. Органический растворитель упаривали при пониженном давлении и соединение растворяли в воде и подкисляли с использованием 15%-ного раствора HCl. Твердое вещество собирали фильтрованием в вакууме.

Получение 7-метил-4-хлор-2-(3-хлорфенил)оксазоло[2,3d]пиримидина:
7-метил-2-(3-хлорфенил)оксазоло[2,3d]пиримидон (1,32 г, 5,06 ммоль) обрабатывали оксихлоридом фосфора (13,2 мл, 141,7 ммоль) и нагревали при кипении с обратным холодильником в течение трех часов. Реакционную смесь охлаждали, растворитель удаляли при пониженном давлении и остаток помещали в хлороформ. К органическому растворителю добавляли лед, затем органический растворитель промывали бикарбонатом натрия, сушили над сульфатом натрия и затем концентрировали. Неочищенный продукт очищали колоночной флэш-хроматографией.
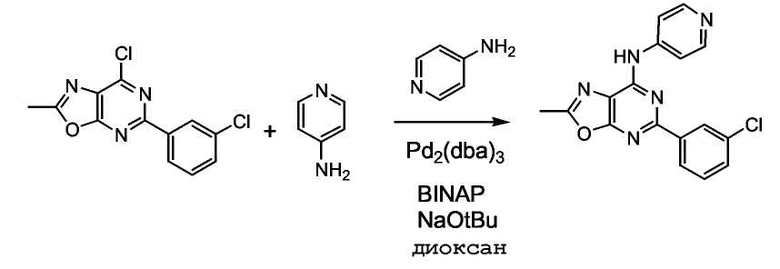
Получение 7-метил-4-(4-аминопиридил)-2-(3-хлорфенил)оксазоло[2,3d]пиримидина (19):
7-метил-4-хлор-2-(3-хлорфенил)оксазоло[2,3d]пиримидин (0,100 г, 0,358 ммоль), 4-аминопиридин (0,040 г, 0,430 ммоль), трет-бутоксид натрия (0,048 г, 0,501 ммоль), бис(дифенилфосфино)-1,1'-бинафтил (0,0009 г, 0,0014 ммоль) и Pd2(dba)3 (0,0004 г, 0,0043 ммоль) объединяли и растворяли в 2 мл безводного диоксана и нагревали при кипении с обратным холодильником в течение 3,5 часов. Реакционную смесь охлаждали, затем фильтровали через целит® и затем очищали с помощью ВЭЖХ.
Схема F
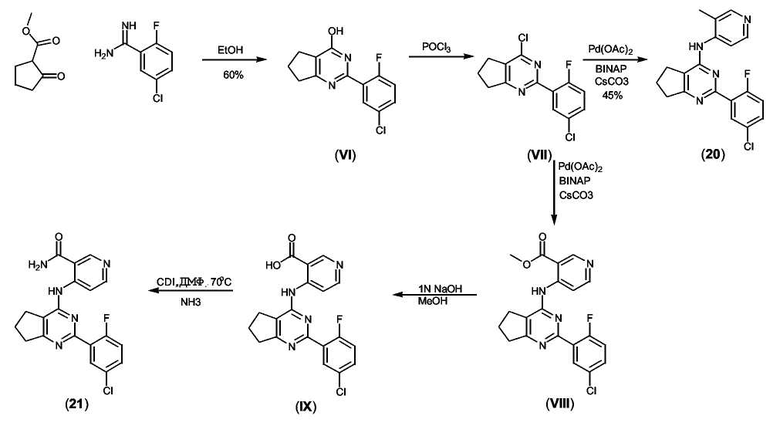
Получение соединения формулы (VI):
К раствору метил-2-оксоциклопентанкарбоксилата (4,10 г, 28,9 ммоль, 1 экв.) в безводном этаноле (20 мл) добавляли раствор 2-фтор-5-хлорбензамидина (5,0 г, 28,9 ммоль, 1 экв.) в этаноле (20 мл) и реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры (к.т.) и белый осадок отфильтровывали и промывали холодным этилацетатом (2×20 мл). Неочищенный остаток распределяли между хлороформом и водой. Водный слой подкисляли до pH 4, и продукт экстрагировали хлороформом (3×50 мл). Органические слои объединяли, промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая неочищенное белое твердое вещество (VI) (4,5 г, 60%), которое не подвергали дополнительной очистке.
Получение соединения формулы (VII):
Суспензию соединения формулы (VI) (200 мг, 0,757 ммоль) в POCl3 (5 мл) перемешивали при кипении с обратным холодильником в течение 1 часа. Раствор затем охлаждали до комнатной температуры и концентрировали при пониженном давлении, получая белое твердое вещество, которое растворяли в сухом хлористом метилене. Раствор охлаждали до 0°C и добавляли лед, а затем насыщенный раствор NaHCO3. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая неочищенное твердое белое вещество, которое дополнительно не очищали.
Получение соединения формулы (20): (Общая методика реакции Бухвальда):
Неочищенное иминохлоридное соединение формулы (VII) (210 мг, 0,76 ммоль, 1 экв.) растворяли в диоксане (5 мл) и добавляли к раствору Pd(OAc)2 (9 мг, 0,04 ммоль, 0,05 экв.), затем BINAP (35 мг, 0,056 ммоль, 0,075 экв.), 4-амино-3-пиколин (82 мг, 0,760 ммоль, 1 экв.) и Cs2CO3 (370 мг, 1,13 ммоль, 1,5 экв.). Реакционную смесь нагревали при 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит® и неочищенное вещество очищали колоночной флэш-хроматографией (3:2/этилацетат:гексан), получая соединение формулы (20) (110 мг, 41%).
Получение соединения формулы (IX):
К суспензии соединения формулы (VIII) (100 мг, 0,25 ммоль, 1 экв.) в MeOH (5 мл) добавляли 1н. NaOH(водн.) раствор (500 мкл, 0,50 ммоль, 2 экв.) и реакционную смесь нагревали при кипении с обратным холодильником в течение 2 часов. Смесь охлаждали до комнатной температуры и концентрировали в вакууме. К неочищенному веществу добавляли воду (10 мл) и водный слой подкисляли до pH 4. Твердое вещество отфильтровывали, промывали водой (2×5 мл) и сушили в течение ночи, получая соединение формулы (IX) (50 мг, 52%) в виде твердого вещества кремового цвета.
Получение соединения формулы (21):
К суспензии соединения формулы (IX) (50 мг, 0,13 ммоль, 1 экв.) в безводном ДМФ (2 мл) добавляли 1-1'-карбонилдиимидазол (42 мг, 0,26 ммоль, 2 экв.) и реакционную смесь нагревали при 70°C в течение 2 часов. Смесь охлаждали до комнатной температуры и барботировали через нее NH3 (газ) в течение 10 минут. Реакционную смесь перемешивали при комнатной температуре дополнительно в течение 1 часа. Реакционную смесь концентрировали в вакууме. К неочищенному веществу добавляли воду (10 мл) и твердое вещество отфильтровывали, промывали водой (2×5 мл) и сушили в течение ночи, получая соединение формулы (21) (30 мг, 60%) в виде твердого вещества кремового цвета.
Дополнительные соединения, полученные согласно схеме F
Соединения формулы (32) были получены в соответствии со способом, показанным на схеме F для получения соединения формулы (VIII), с использованием этилового эфира 4-аминопиридин-3-карбоновой кислоты. Соединения формулы (36) получали способом, описанным для синтеза соединения формулы (20), с использованием 4-амино-3-трифторметил-пиколина вместо 4-амино-3-пиколина. Соединения формулы (35) получали способом, описанным для синтеза соединения формулы (21), с использованием метиламина вместо аммиака. Соединения формулы (37) получали способом, описанным для синтеза соединения формулы (21), с использованием пирролидина вместо аммиака. Соединения формулы (41) получали способом, описанным для синтеза соединения формулы (21), с использованием циклопропиламина вместо аммиака. Соединения формулы (42) получали способом, описанным для синтеза соединения формулы (21), с использованием циклопропилметиламина вместо аммиака. Соединения формулы (51) получали способом, описанным для синтеза соединения формулы (21), с использованием 2-амино-этанола вместо аммиака. Соединения формулы (52) получали способом, описанным для синтеза соединения формулы (21), с использованием 1-аминопропан-2-(S)-ола вместо аммиака. Соединения формулы (53) получали способом, описанным для синтеза соединения формулы (21), с использованием 3-аминопропана-1,2(S)-диола вместо аммиака. Соединения формулы (54) получали способом, описанным для синтеза соединения формулы (21), с использованием HO-NH2 вместо аммиака. Соединения формулы (55) получали способом, описанным для синтеза соединения формулы (21), с использованием 1-амино-пропан-2-(R)-ола вместо аммиака. Соединения формулы (56) получали способом, описанным для синтеза соединения формулы (21), с использованием N-метилэтилендиамина вместо аммиака. Соединения формулы (58) получали способом, описанным для синтеза соединения формулы (21), с использованием гидразина вместо аммиака. Соединения формулы (67) получали способом, описанным для синтеза соединения формулы (21), с использованием бензамидина. Соединения формулы (68) получали способом, описанным для синтеза соединения формулы (21), с использованием метиламина вместо аммиака и бензамидина вместо 2-F,3-Cl-бензамидина. Соединения формулы (69) получали способом, описанным для синтеза соединения формулы (21), с использованием N,N'-диметил-1,3-пропандиамина вместо аммиака. Соединения формулы (70) получали способом, описанным для синтеза соединения формулы (21), с использованием 4-(3-аминопропил)морфолина вместо аммиака. Соединения формулы (71) получали способом, описанным для синтеза соединения формулы (21), с использованием 1-(3-аминопропил)имидазола вместо аммиака. Соединения формулы (72) получали способом, описанным для синтеза соединения формулы (21), с использованием 1-(3-аминопропил)-2-пирролидинона вместо аммиака. Соединения формулы (73) получали способом, описанным для синтеза соединения формулы (21), с использованием 2-(2-аминоэтил)-1-метилпирролидина вместо аммиака. Соединения формулы (74) получали способом, описанным для синтеза соединения формулы (21), с использованием 1-(3-аминопропил)-2-пипеколина вместо аммиака. Соединения формулы (75) получали способом, описанным для синтеза соединения формулы (21), с использованием 1-(2-аминоэтил)пирролидина вместо аммиака. Соединения формулы (78) получали способом, описанным для синтеза соединения формулы (21), с использованием 1-(2-аминоэтил)пиперидина вместо аммиака. Соединения формулы (79) получали способом, описанным для синтеза соединения формулы (21), с использованием N,N-диэтилэтендиамин вместо аммиака.
Схема G (Синтез cоединений формул (22), (24), (25) и (30))
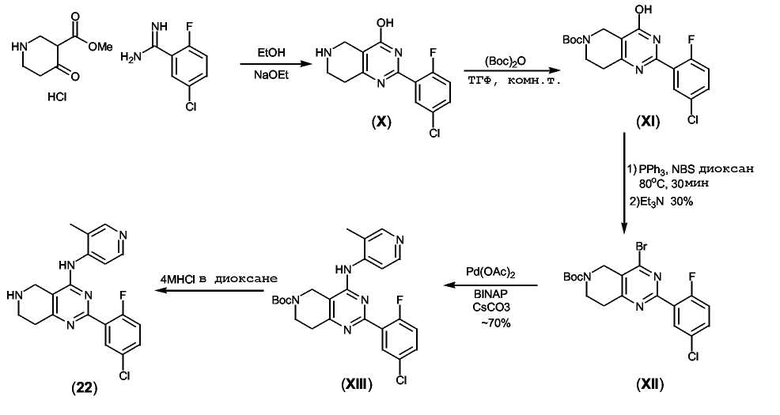
Получение соединения формулы (X):
К раствору 2-фтор-5-хлорбензамидина (1,79 г, 10,4 ммоль, 1 экв.) в EtOH (10 мл) добавляли твердый NaOEt (705 мг, 10,4 ммоль, 1 экв.) с последующим добавлением метил-4-оксо-3-пиперидинкарбоксилата•HCl (2,0 г, 10,4 ммоль, 1 экв.). Реакционную смесь нагревали при 70°C в течение 2 часов, затем охлаждали до комнатной температуры. Осадок отфильтровывали и промывали этилацетатом (2×20 мл), получая белое твердое вещество (2,2 г, 76%), которое не подвергали дополнительной очистке.
Получение соединения формулы (XI):
К суспензии соединения формулы (X) (300 мг, 1,08 ммоль, 1 экв.) в безводном ТГФ (10 мл) добавляли раствор Boс2O (258 мг, 1,18 ммоль, 1,1 экв.) в безводном ТГФ (10 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов и затем раствор концентрировали в вакууме, получая неочищенный остаток, который очищали колоночной флэш-хроматографией, получая соединение формулы (XI) (320 мг, 79%) в виде белого твердого вещества.
Получение соединения формулы (XII):
К раствору PPh3 (813 мг, 3,03 ммоль, 5 экв.) в безводном диоксане (20 мл) добавляли NBS (540 мг, 3,03 ммоль, 5 экв.) в виде одной порции и суспензию перемешивали при комнатной температуре в течение 30 минут. Добавляли раствор соединения формулы (XI) (230 мг, 0,61 ммоль, 1 экв.) в безводном диоксане (5 мл) и реакционную смесь нагревали при 80°C в течение 45 минут. Реакционную смесь охлаждали до комнатной температуры и добавляли Et3N (160 мкл, 1,21 ммоль, 2 экв.). Смесь концентрировали в вакууме и неочищенный остаток очищали колоночной флэш-хроматографией (1:9 этилацетат:гексан), получая соединение формулы (XII) (72 мг, 30%).
Получение соединения формулы (XIII):
К раствору соединения формулы (XII) (72 мг, 0,16 ммоль, 1 экв.) в безводном диоксане (2 мл) добавляли Pd(OAc)2 (2 мг, 0,008 ммоль, 0,05 экв.) с последующим добавлением BINAP (8 мг, 0,001 ммоль, 0,075 экв.), 4-амино-3-пиколина (18 мг, 0,16 ммоль, 1 экв.) и Cs2CO3 (80 мг, 0,24 ммоль, 1,5 экв.). Реакционную смесь нагревали при 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит® и неочищенное вещество очищали колоночной флэш-хроматографией (7:3/этилацетат:гексан), получая соединение формулы (XIII) (65 мг, 85%).
Получение соединения формулы (22):
К раствору соединения формулы (XIII) (65 мг, 0,14 ммоль, 1 экв.) в безводном диоксане (2 мл) добавляли 4 M раствор HCl в диоксане (1 мл). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Осадок отфильтровывали и промывали хлороформом (1×5 мл), этилацетатом (1×5 мл) и холодным метанолом (1×2 мл), получая соединение формулы (22) (35 мг, 68%) в виде белого твердого вещества.
Получение соединения формулы (24):
Соединения формулы (24) получали в соответствии со способом, показанным на схеме G, с использованием этил 1-бензил-4-оксопиперидин-3-карбоксилата и бензамидина.
Получение соединения формулы (25):
Соединения формулы (25) получали в соответствии со способом, показанным на схеме G, с использованием бензамидина.
Получение соединения формулы (30):
Соединения формулы (30) получали в соответствии со способом, показанным на схеме G, с использованием этил 1-бензил-4-оксопиперидин-3-карбоксилата.
Схема H (Синтез соединения формулы (23))

Получение соединения формулы (XIV):
К раствору этил-N-бензил-3-оксо-4-пиперидинкарбоксилата•HCl (2 г, 6,73 ммоль, 1 экв.) в этаноле (60 мл) добавляли 10% Pd/C. Воздух откачивали и заменяли на водород из баллона. Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 4 часов. Реакционную смесь фильтровали через короткий слой целита®, получая соединение формулы (XIV), которое дополнительно не очищали.
Получение соединения формулы (XV):
К раствору 2-фтор-5-хлорбензамидина (1,16 г, 6,71 ммоль, 1 экв.) в EtOH (10 мл) добавляли твердый NaOEt (457 мг, 6,71 ммоль, 1 экв.) с последующим добавлением соединения формулы (XIV) (1,39 г, 6,71 ммоль, 1 экв.). Реакционную смесь нагревали при 70°C в течение 2 часов и затем охлаждали до комнатной температуры. Осадок отфильтровывали и промывали этилацетатом (2×20 мл), получая соединение формулы (XV) (1,12 г, 60%) в виде белого твердого вещества, которое дополнительно не очищали.
Получение соединения формулы (XVI):
К суспензии неочищенного соединения формулы (XV) (1,12 мг, 4,01 ммоль, 1 экв.) в безводном ТГФ (10 мл) добавляли раствор Boс2O (960 мг, 4,42 ммоль, 1,1 экв.) в безводном ТГФ (10 мл) при комнатной температуре. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов, и затем раствор концентрировали в вакууме, получая неочищенный остаток, который очищали колоночной флэш-хроматографией, получая соединение формулы (XVI) (750 мг, 50%) в виде белого твердого вещества.
Получение соединения формулы (XVII):
К раствору PPh3 (2,28 г, 8,70 ммоль, 5 экв.) в безводном диоксане (20 мл) добавляли NBS (1,55 мг, 8,71 ммоль, 5 экв.) в виде одной порции и суспензию перемешивали при комнатной температуре в течение 30 минут. Добавляли раствор соединения формулы (XVI) (660 мг, 1,74 ммоль, 1 экв.) в безводном диоксане (5 мл) и реакционную смесь нагревали при 80°C в течение 45 минут. Реакционную смесь охлаждали до комнатной температуры и добавляли 2 эквивалента Et3N. Смесь концентрировали в вакууме и неочищенный остаток очищали колоночной флэш-хроматографией (1:9 этилацетат:гексан), получая соединение формулы (XVII) (230 мг, 30%).
Получение соединения формулы (XVIII):
К раствору соединения формулы (XVII) (230 мг, 0,52 ммоль, 1 экв.) в безводном диоксане (5 мл) добавляли Pd(OAc)2 (6 мг, 0,03 ммоль, 0,05 экв.) с последующим добавлением BINAP (8 мг, 0,004 ммоль, 0,075 экв.), 4-амино-3-пиколина (67 мг, 0,62 ммоль, 1,2 экв.) и Cs2CO3 (271 мг, 0,83 ммоль, 1,5 экв.). Реакционную смесь нагревали 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит® и неочищенное вещество очищали колоночной флэш-хроматографией (9:1/этилацетат:гексан), получая соединение формулы (XVIII) (38 мг, 16%).
Получение соединения формулы (23):
К раствору соединения формулы (XVIII) (38 мг, 0,08 ммоль, 1 экв.) в безводном диоксане (2 мл) добавляли 4 M раствор HCl в диоксане (1 мл). Полученную суспензию перемешивали при комнатной температуре в течение 2 часов. Осадок отфильтровывали и промывали хлороформом (1×3 мл), этилацетатом (1×3 мл) и холодным метанолом (1×1 мл), получая соединение формулы (23) (32 мг, 95%) в виде белого твердого вещества.
Схема I (Синтез соединения формулы (27))
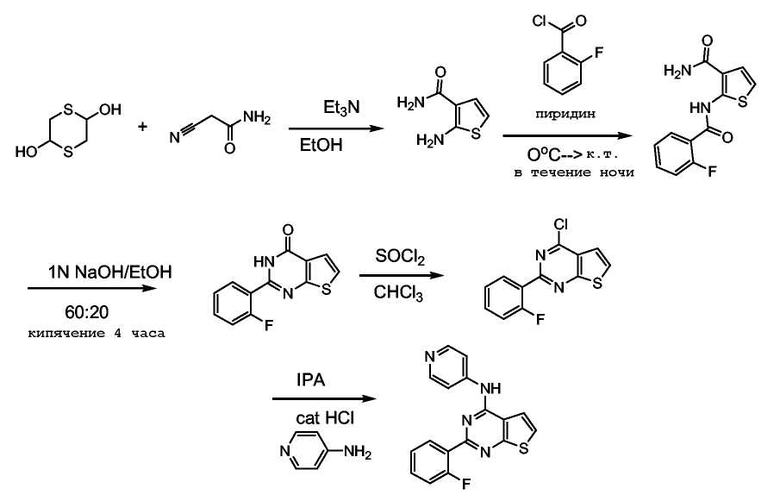
Амид 2-аминотиофен-3-карбоновой кислоты:
1,4-дитиан-2,5-диол (4,56 г, 30 ммоль) и 2-цианоацетамид (2,52 г, 30 ммоль) объединяли в этаноле (50 мл). Добавляли триэтиламин (6 мл) и нагревали при 70°C в течение 1 часа. Объем растворителя уменьшали в вакууме и продукт выделяли фильтрованием. Продукт перекристаллизовывали из этанола, получая 2,71 г продукта (выход 64%).
Амид 2-(2-фторбензоиламино)тиофен-3-карбоновой кислоты:
Амид 2-амино-тиофен-3-карбоновой кислоты (8,73 г, 61,4 ммоль) растворяли в пиридине (100 мл), охлаждали до 0°C и добавляли по каплям в течение 20 минут 2-фторбензоилхлорид, затем реакционную смесь оставляли нагреваться до комнатной температуры при перемешивании в течение ночи. Пиридин удаляли в вакууме, добавляли дихлорметан и воду. Продукт выпадал в осадок в виде серого твердого вещества, и его промывали разбавленной соляной кислотой, водой и сушили на воздухе. Слой дихлорметана отделяли разбавленной соляной кислотой и водой, сушили над сульфатом натрия (безв.) и растворитель удаляли, получая всего 12,45 г продукта (77% выход).
2-(2-Фторфенил)-3H-тиено[2,3-d]пиримидин-4-он:
Амид 2-(2-фтор-бензоиламино)тиофен-3-карбоновой кислоты (8,56 г, 32,4 ммоль) растворяли в смеси 20 мл 1 M раствора гидроксида натрия и 60 мл этанола. Смесь кипятили с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали и выливали на лед. Раствор подкисляли разбавленной соляной кислотой и продукт выделяли фильтрованием. После сушки в вакууме получали 5,42 г продукта (выход: 68%).
4-Хлор-2-(2-фторфенил)тиено[2,3-d]пиримидин:
2-(2-Фтор-фенил)-3H-тиено[2,3-d]пиримидин-4-он (900 мг, 3,65 ммоль) растворяли в хлороформе и добавляли к смеси хлористый тионил (0,532 мл, 7,30 ммоль) с последующим добавлением 1 мл диметилформамида. Реакционную смесь нагревали при кипении с обратным холодильником в течение 2,5 часов, охлажденную смесь промывали 10% раствором карбоната натрия, раствор в хлороформе сушили над сульфатом натрия (безв.) и растворитель удаляли. Неочищенный продукт хроматографировали на силикагеле, элюируя хлороформом. После удаления растворителя получали 438 мг продукта (выход: 45%).
[2-(2-Фторфенил)тиено[2,3-d]пиримидин-4-ил]пиридин-4-ил-амин:
4-Хлор-2-(2-фтор-фенил)тиено[2,3-d]пиримидин (110 мг, 0,41 ммоль) и 4-аминопиридин (78 мг, 0,830 ммоль) объединяли в изопропаноле (3 мл), затем добавляли 4 капли 4 M HCl/диоксане и реакционную смесь нагревали при 80°C в течение 7 часов. Реакционную смесь охлаждали и продукт выделяли фильтрованием, промывали минимальным количеством холодного метанола и сушили в вакууме, получая 116 мг продукта (выход: 86%).
Схема J (Синтез соединения формулы (28))

5-Амино-1,3-диметил-1H-пиразол-4-карбонитрил:
Метилгидразин (5 г, 108,5 ммоль) добавляли по каплям к раствору (1-этокси-этилиден)малононитрила (14,7 г, 108,5 ммоль) в 250 мл этанола. Смесь затем нагревали при кипении с обратным холодильником в течение 2,5 часов. Реакционную смесь оставляли охлаждаться, объем уменьшали примерно до 70 мл и продукт выделяли фильтрованием, промывали холодным этанолом и сушили, получая 13,5 г продукта (выход: 91%).
N-(4-Циано-2,5-диметил-2H-пиразол-3-ил)-2-фторбензамид:
5-Амино-1,3-диметил-1H-пиразол-4-карбонитрил (10 г, 73,4 ммоль) суспендировали в пиридине (90 мл) и добавляли по каплям 2-фторбензоилхлорид при охлаждении реакционной смеси на ледяной бане. Реакционную смесь перемешивали в течение ночи при комнатной температуре. Большую часть пиридина удаляли и для осаждения продукта добавляли 100 мл холодной воды. Продукт выделяли фильтрованием, промывали холодной водой и небольшим количеством холодного этанола и сушили в вакууме, получая 9,15 г продукта (выход: 50%).
6-(2-Фторфенил)-1,3-диметил-1,5-дигидро-пиразолo[3,4-d]пиримидин-4-он:
N-(4-Циано-2,5-диметил-2H-пиразол-3-ил)-2-фтор-бензамид (5,0 г, 19,36 ммоль) суспендировали в 38 мл 1 M раствора гидроксида натрия и нагревали при 90°C в течение 3 часов с последующим добавлением 30%-ной перекиси водорода (10 мл) и дополнительным нагреванием в течение ночи. Реакционную смесь охлаждали до комнатной температуры и подкисляли разбавленной соляной кислотой. Продукт выделяли фильтрованием, промывали водой и сушили в течение ночи в вакууме, получая 3,39 г продукта. (Выход 67%).
4-Хлор-6-(2-фторфенил)-1,3-диметил-1H-пиразолo[3,4-dl]пиримидин:
6-(2-Фтор-фенил)-1,3-диметил-1,5-дигидро-пиразолo[3,4-d]пиримидин-4-он (2,0 г, 7,74 ммоль) обрабатывали 40 мл оксихлорида фосфора и нагревали при кипении с обратным холодильником в течение ночи. Избыток оксихлорида фосфора удаляли в вакууме и к остатку добавляли ледяную воду. Продукт экстрагировали этилацетатом, промывали 10%-ным карбонатом натрия, водой, сушили над сульфатом натрия (безв.) и растворитель удаляли, получая неочищенный продукт. Неочищенный продукт хроматографировали на колонке с силикагелем, элюируя хлороформ и получая 1,10 г чистого продукта (выход: 51%).
[6-(2-Фторфенил)-1,3-диметил-1H-пиразолo[3,4-d]пиримидин-4-ил]пиридин-4-иламин:
4-Хлор-6-(2-фтор-фенил)-1,3-диметил-1H-пиразолo[3,4-d]пиримидин (84 мг, 0,304 ммоль) и 4-аминопиридин (57 мг, 0,608 ммоль) объединяли в 4 мл изопропапнола, добавляли 3 капли 4M HCl/диоксане и реакционную смесь нагревали при 80°C в течение 5 часов. Реакционную смесь охлаждали и продукт отфильтровывали, получая 83 мг после сушки в вакууме (выход: 81%).
Схема K (Синтез соединения формулы (29))

5-Амино-1-бутил-3-метил-1H-пиразол-4-карбонитрил:
Бутилгидразиноксалат (14,25 г, 80 ммоль) и (1-этоксиэтилиден)малонитрил (10,82 г, 80 ммоль) объединяли в этаноле (200 мл) и смесь обрабатывали диизопропилэтиламином (10,39 г, 80 ммоль). Смесь нагревали при кипении с обратным холодильником в течение 2 часов, затем перемешивали при комнатной температуре в течение ночи. Большую часть растворителя удаляли в вакууме и остаток растирали с хлороформом, некоторое количество твердого вещества отфильтровывали и хлороформенный фильтрат сохраняли. Хлороформенный фильтрат промывали водой, сушили над сульфатом натрия (безв.) и растворитель удаляли, получая продукт в виде твердого вещества, 13,13 г (выход: 74%).
N-(2-Бутил-4-циано-5-метил-2H-пиразол-3-ил)-2-фторбензамид:
5-Амино-1-бутил-3-метил-1H-пиразол-4-карбонитрил (10,0 г, 56 ммоль) растворяли в смеси дихлорметан/пиридин (45 мл/15 мл) и охлаждали до 0°C. Добавляли по каплям 2-фторбензоилхлорид (8,87 г, 56 ммоль) и реакционную смесь перемешивали в течение 1 часа при 0°C и при комнатной температуре в течение ночи. Растворитель удаляли в вакууме. Остаток помещали в этилацетат, промывали разбавленной соляной кислотой, 0,1 M раствором гидроксида натрия и водой и сушили над сульфатом натрия (безв.). Растворитель удаляли и твердое вещество растирали в смеси 30% этилацетат/гексаны. Продукт отфильтровывали, получая 4,16 г продукта (выход: 24%).
1-Бутил-6-(2-фторфенил)-3-метил-1,5-дигидропиразолo[3,4-d]пиримидин-4-он:
N-(2-Бутил-4-циано-5-метил-2H-пиразол-3-ил)-2-фторбензамид (4,0 г, 13,3 ммоль) суспендировали в 26 мл 1M раствора гидроксида натрия и добавляли 30% перекись водорода (10 мл) и этанол (5 мл). Реакционную смесь нагревали при кипении с обратным холодильником в течение 4 часов, затем добавляли дополнительное количество 30% перекиси водорода (10 мл) добавляли и нагревали при кипении с обратным холодильником в течение ночи. Реакционную смесь охлаждали и подкисляли разбавленной соляной кислотой до pH 6,0. Продукт собирали фильтрованием и сушили в вакууме, получая 1,29 г продукта (выход: 32%).
1-Бутил-4-хлор-6-(2-фторфенил)-3-метил-1H-пиразолo[3,4-d]пиримидин:
1-Бутил-6-(2-фторфенил)-3-метил-1,5-дигидропиразолo[3,4-d]пиримидин-4-он (1,23 г, 4,1 ммоль) растворяли в оксихлориде фосфора (15 мл) и нагревали при кипении с обратным холодильником в течение ночи. Избыток оксихлорида фосфора удаляли в вакууме, остаток обрабатывали ледяной водой, продукт экстрагировали этилацетатом, промывали водой, насыщенным раствором хлорида натрия, затем сушили над сульфатом натрия (безв.) и растворитель удаляли, получая неочищенный продукт. Продукт хроматографировали на силикагеле элюируя хлороформом, получая 723 мг очищенного продукта (выход: 55%).
[1-Бутил-6-(2-фторфенил)-3-метил-1H-пиразолo[3,4-d]пиримидин-4-ил]пиридин-4-иламин:
1-Бутил-4-хлор-6-(2-фторфенил)-3-метил-1H-пиразолo[3,4-d]пиримидин (100 мг, 0,31 ммоль) и 4-аминопиридин (58 мг, 0,626 ммоль) объединяли в диметиловом эфире этиленгликоля и нагревали при кипении с обратным холодильником в течение 4 часов. Реакционную смесь охлаждали и продукт выделяли фильтрованием, промывали минимальным количеством холодного растворителя и сушили, получая 132 мг продукта. 50 мг этого вещества подвергали ВЭЖХ очистке с обращенной фазой на колонке С18, элюируя с повышающимся градиентом смесью вода/ацетонитрил/0,1% трифторуксусная кислота.
Схема L (Синтез соединения формулы (9))

2-(Этоксифенилметилен)малононитрил:
Триэтилортобензоат (25 г, 0,112 моль), малонитрил (9,07 г, 0,137 моль) и уксусный ангидрид (50 мл) нагревали при кипении с обратным холодильником в течение ночи. Избыток уксусного ангидрида удаляли в вакууме и продукт хроматографировали на силикагеле, элюируя смесью 30% этилацетат/гексаны, получая 21,7 г продукта (выход: 97%).
5-Амино-3-фенил-изоксазол-4-карбонитрил:
Гидрохлорид гидроксиламина суспендировали в воде (30 мл) и добавляли гидроксид натрия (4,4 г, 0,11 моль) и затем этанол (40 мл) с последующим порционным добавлением 2-(этокси-фенилметилен)малононитрила (21,7 г, 0,11 моль). Реакционную смесь нагревали при 50°C в течение 2 часов. Этанол удаляли в вакууме и осадок отфильтровывали. Осадок повторно растворяли в смеси 50% этилацетат/гексаны и хроматографировали на силикагеле, получая 8,2 г продукта после удаления растворителя (выход: 40%). Остальную последовательность реакций проводили по аналогии с заменой метила в фенильном аналоге.
Схема M (Синтез соединения формулы (80))

1H-Пиррол-2-карбонитрил:
Пиррол-2-карбоксальдегид (3,00 г, 0,0316 моль) объединяли с гидроксиламин-O-сульфоновой кислотой (12,5 г, 0,11 моль) в 100 мл воды и перемешивали при комнатной температуре в течение ночи. Реакционную смесь охлаждали до 0°C и добавляли по каплям в течение 1 часа раствор гидркосида калия (12,06 г, 0,603 моль) в 80 мл воды. Реакционную смесь перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь экстрагировали дихлорметаном (3 × 100 мл), экстракт сушили над сульфатом натрия (безв.). Растворитель удаляли, получая 3,36 г продукта в виде жидкости (выход: 100%).
1-Амино-1Н-пиррол-2-карбонитрил:
1Н-Пиррол-2-карбонитрил (3,36 г, 36,5 ммоль) растворяли в 100 мл диметилформамида, к смеси добавляли карбонат калия (7,51 г, 54,75 ммоль) с последующим добавлением O-(4-нитро-2-трифторметилфенил)гидроксиламина (12,15 г, 54,73 ммоль). Смесь перемешивали при комнатной температуре в течение ночи. Добавляли 80 мл воды и осадок отфильтровывали. pH фильтрата доводили до 10 и экстрагировали этилацетатом (3×100 мл). Экстракт промывали водой, насыщенным раствором хлорида натрия и сушили над сульфатом натрия. Растворитель удаляли в вакууме, получая 6,31 г продукта, содержащего остаточное количество диметилформамида. Выход был оценен по данным ЯМР как 64%.
Амид 1-амино-1Н-пиррол-2-карбоновой кислоты:
1-Амино-1Н-пиррол-2-карбонитрил (2,52 г, 23,5 ммоль) суспендировали в 75 мл воды, обрабатывали гидроксидом калия (32 г, 0,57 моль), 30% перекисью водорода (2 мл) и перемешивали в течение ночи при комнатной температуре. Реакционную смесь охлаждали до 0°C в течение 30 минут и продукт выделяли фильтрованием, промывали холодной водой и сушили в вакууме, получая 2,55 г продукта (выход: 87%).
Амид 1-(5-хлор-2-фтор-бензоиламино)-1Н-пиррол-2-карбоновой кислоты:
Амид 1-амино-1Н-пиррол-2-карбоновой кислоты (1,25 г, 10 ммоль) частично растворяли в 45 мл ацетонитрила, добавляли триэтиламин (1,39 мл, 10 ммоль) с последующим добавлением по каплям 5-хлор-2-фторбензоилхлорида (1,93 г, 10 ммоль) в 3 мл хлороформа. Реакционную смесь перемешивали при комнатной температуре в течение ночи, растворитель удаляли в вакууме и остаток помещали в хлороформ, промывали 10% раствором бикарбоната натрия, водой и сушили над сульфатом натрия (безв.). Твердое вещество кристаллизовалось из раствора при перемешивании. Дополнительное количество продукта получали путем хроматографирования фильтрата на силикагеле при элюировании смесью 3% метанол/хлороформ. Получали 600 мг продукта (выход: 21%).
2-(5-Хлор-2-фторфенил)-3H-пирроло[2,1-f][1,2,4]триазин-4-он:
Амид 1-(5-хлор-2-фтор-бензоиламино)-1Н-пиррол-2-карбоновой кислоты (200 мг, 0,71 ммоль) растворяли в 5 мл 28% гидроксида аммония в запаянной трубке и нагревали при 80°C в течение ночи. Раствор продували азотом для удаления избытка аммиака и подкисляли 1 M соляной кислотой до pH 2. Продукт выделяли фильтрованием, промывали водой и сушили в вакууме, получая 90 мг продукта (выход: 48%).
4-Хлор-2-(5-хлор-2-фторфенил)пирроло[2,1-f][1,2,4]триазин:
2-(5-Хлор-2-фторфенил)-3H-пирроло[2,1-f][1,2,4]триазин-4-он (60 мг, 0,228 ммоль) добавляли к оксихлориду фосфора (1 мл). Добавляли 57 микролитров N,N-диметиланилина (катализатор) и нагревали при 110°C в течение ночи. Избыток оксихлорида фосфора удаляли в вакууме, остаток обрабатывали льдом и продукт экстрагировали хлороформом. Хлороформенный экстракт сушили над сульфатом натрия (безв.) и растворитель удаляли, получая неочищенный продукт. Продукт очищали хроматографией на силикагеле, элюируя хлороформом, получая 36 мг продукта (выход: 56%).
[2-(5-Хлор-2-фторфенил)пирроло[2,1-f][1,2,4]триазин-4-ил]-(3-метил-пиридин-4-ил)амин:
4-Хлор-2-(5-хлор-2-фторфенил)пирроло[2,1-f][1,2,4]триазин (30 мг, 0,106 ммоль), карбонат цезия (48,5 мг, 0,149 ммоль), ацетат палладия(II) (1,19 мг, 0,0053 ммоль), BINAP (4,96 мг, 0,0080 ммоль) и 4-амино-пиколин (13,8 мг, 0,128 ммоль) объединяли в 4 мл диоксана (безв.) и нагревали при 90°C при перемешивании в течение ночи. Реакционную смесь фильтровали для удаления твердого вещества, фильтрат упаривали досуха, остаток растворяли в хлороформе (8 мл), промывали 0,5 M раствором гидроксида натрия (1 мл), сушили над сульфатом натрия (безв.), упаривали досуха и затем остаток растворяли в диметилформамиде и очищали ВЭЖХ с обращенной фазой. Выделяли продукт.
Схема N (Синтез соединений формулы (81) и (82))
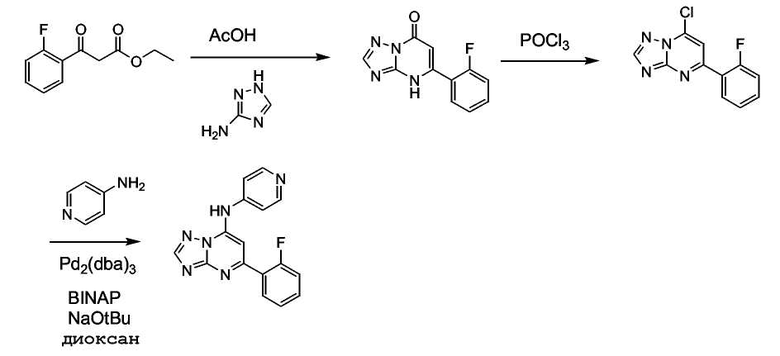
5-(2-Фтор-фенил)-4H-[1,2,4]триазоло[1,5-a]пиримидин-7-он:
3-амино-1,2,4-триазол (3,64 г, 43,25 ммоль) и этил 2-фторбензоилацетат (10 г, 47,57 ммоль) объединяли в уксусной кислоте (45 мл) и нагревали при кипении с обратным холодильником в течение ночи. Реакционную смесь охлаждали и продукт отфильтровывали, промывали диэтиловым эфиром, получая 3,47 г (выход: 35%).
7-Хлор-5-(2-фтор-фенил)-[1,2,4]триазоло[1,5-a]пиримидин:
5-(2-Фтор-фенил)-4H-[1,2,4]триазоло[1,5-a]пиримидин-7-он (840 мг, 3,64 ммоль) суспендировали в оксихлориде фосфора (5 мл) и нагревали при кипении с обратным холодильником в течение 45 минут. Избыток оксихлорида фосфора удаляли в вакууме, остаток обрабатывали льдом, продукт экстрагировали хлороформом, затем хлороформ промывали 10% раствором бикарбоната натрия, сушили над сульфатом натрия и растворитель удаляли в вакууме, получая 420 мг продукта (выход 46%).
[5-(2-Фтор-фенил)-[1,2,4]триазоло[1,5-a]пиримидин-7-ил]пиридин-4-иламин:
7-Хлор-5-(2-фтор-фенил)-[1,2,4]триазоло[1,5-a]пиримидин (124 мг, 0,5 ммоль) суспендировали в диоксане (5 мл), добавляли 4-аминопиридин (56,4 мг, 0,6 ммоль), трет-бутоксид натрия (67 мг, 0,7 ммоль), BINAP (2,3 мг, 0,00375 ммоль) и Pd2(dba)3 (1,14 мг, 0,00125 моль) и нагревали при 90°C в течение ночи. Диоксан удаляли в вакууме, остаток помещали в метанол, фильтровали и очищали ВЭЖХ с обращенной фазой и фракции лиофилизовали, получая продукт в виде трифторацетатной соли.
Соединение формулы (81) получали с использованием способа, аналогичного описанному выше, с использованием этил 5-хлор-2-фторбензоилацетата. Соединение формулы (83) получали с использованием способа, аналогичного описанному выше, с использованием 4-аминопиримидина.
Схема O (Синтез соединения формулы (60) и (61))

Получение 4,4-диметилциклогексанона:
Ссылка: Tetrahedron Lett, 1992, 33(35), 5009.
Раствор 4,4-диметилциклогексен-2-она (10 г, 78,11 ммоль) и триэтиламина (10,89 мл, 78,11 ммоль) в 100% этаноле (30 мл) подвергали гидрированию при давлении 30 фунт/кв.дюйм в аппарате Парра при комнатной температуре в течение ночи. Фильтрование содержимого через целит® и упаривание фильтрата давали чистый продукт в виде бесцветного масла (10,08 г, 99% выход).
Получение 3,3-диметилгександионовой кислоты
Ссылка: J. Med. Chem. 1970, 13(3), 531.
К раствору 4,4-диметилциклогексанона (2 г, 15,85 ммоль) в ледяной AcOH (100 мл) добавляли раствор CrO3 (4,75 г, 47,54 ммоль) в ледяной AcOH (20 мл) и воде (20 мл). Смесь перемешивали при 60°C в течение ночи, затем охлаждали и разбавляли 40%-ным водным раствором NaOH до pH 14. Смесь промывали диэтиловым эфиром (4×100 мл) и водный слой подкисляли концентрированной HCl (водн.) до pH 1. Раствор экстрагировали диэтиловым эфиром (4×100 мл); органические экстракты сушили (насыщенный раствор соли и MgSO4) и упаривали, получая неочищенную дикислоту, которую непосредственно этерифицировали.
Получение диметилового эфира 3,3-диметилгександионовой кислоты:
Неочищенную 3,3-диметилгександионовую кислоту растворяли в метаноле (50 мл), добавляли хлористый тионил (1 мл) и раствор нагревали при 60°C в течение 6 часов, затем смесь охлаждали и упаривали, получая неочищенный диэфир, который очищали с помощью хроматографии (1:1 гексан/этилацетат), получая чистый продукт в виде бесцветного масла (2,90 г, 91% выход по 2 стадиям).
Получение метилового эфира 4,4-диметил-2-оксоциклопентанкарбоновой кислоты:
Ссылка: J. Chem. Soc. Perkin Trans. 1, 1984, 799.
К раствору диметилового эфира 3,3-диметилгександионовой кислоты (2,90 г, 14,36 ммоль) и метанола (100 мкл) в сухом толуоле (10 мл) добавляли металлический натрий (0,66 г, 28,72 ммоль). Смесь нагревали при кипении с обратным холодильником в течение ночи, затем охлаждали и упаривали. Остаток очищали с помощью хроматографии (1:1 гексан/этилацетат), получая целевой продукт циклизации в виде бесцветного масла (2,08 г, 85% выход).
Получение 2-(5-хлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-ола:
Раствор метилового эфира 4,4-диметил-2-оксоциклопентанкарбоновой кислоты (2,08 г, 12,21 ммоль) и 5-хлор-2-фтор-бензамидина (2,32 г, 13,44 ммоль) в 100% этаноле (40 мл) нагревали при кипении с обратным холодильником в течение ночи, затем охлаждали и упаривали. Остаток растворяли в 1н. (водн.) NaOH (50 мл) и промывали хлористым метиленом (2 x 50 мл). Водный слой затем подкисляли ледяной уксусной кислотой до pH 4 и экстрагировали хлористым метиленом (2 x 100 мл). Органические экстракты сушили (насыщенный раствор соли и MgSO4) и упаривали, получая неочищенный продукт, который очищали хроматографией (CH2Cl2, 0-10% MeOH), получая целевой продукт в виде кремового твердого вещества (2,10 г, 59% выход).
Получение 2-(5-хлор-2-фтор-фенил)-4-иод-6,6-диметил-6,7-дигидро-5H-циклопентапиримидина:
Суспензию 2-(5-хлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-ола (2,10 г, 7,16 ммоль) в оксихлориде фосфора (40 мл) нагревали при кипении с обратным холодильником в течение 2 часов, затем охлаждали и упаривали. Остаток растворяли в хлористом метилене и раствор фильтровали через короткий слой силикагеля. Фильтрат упаривали, получая остаток, который суспендировали в иодистоводородной кислоте (10 мл) и нагревали при 90°C с иодидом натрия (5,37 г, 35,82 ммоль) в течение 3 часов. Смесь охлаждали и разбавляли водой (50 мл). Добавляли водный раствор тиосульфата натрия (50 мл) и смесь встряхивали с хлористым метиленом (3 × 100 мл). Органические экстракты сушили над MgSO4 и упаривали, получая неочищенный продукт, который очищали флэш-хроматографией (CH2Cl2), получая целевой иодсодержащий продукт в виде кремового твердого вещества (1,75 г, 66% выход).
Получение [2-(5-хлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-ил]-(3 -метил-пиридин-4-ил)амина:
К раствору 2-(5-хлор-2-фтор-фенил)-4-иод-6,6-диметил-6,7-дигидро-5H-циклопентапиримидина (100 мг, 0,25 ммоль), 3-метил-пиридин-4-иламина (30 мг, 0,27 ммоль), Pd(OAc)2 (3 мг, 12,42 мкмоль) и рац-BINAP (12 мг, 18,63 мкмоль) в сухом диоксане (3 мл) добавляли Cs2CO3 (121 мг, 0,37 ммоль). Смесь нагревали в течение 48 часов при 85°C, охлаждали и упаривали. С помощью ВЭЖХ очистки получали после лиофилизации целевой продукт формулы (60) в виде соли ТФУК, представлявшей собой белое твердое вещество (6,4 мг). Тот же способ использовали для получения соединения формулы (61).
Схема P (Синтез соединений формул (62) - (66))
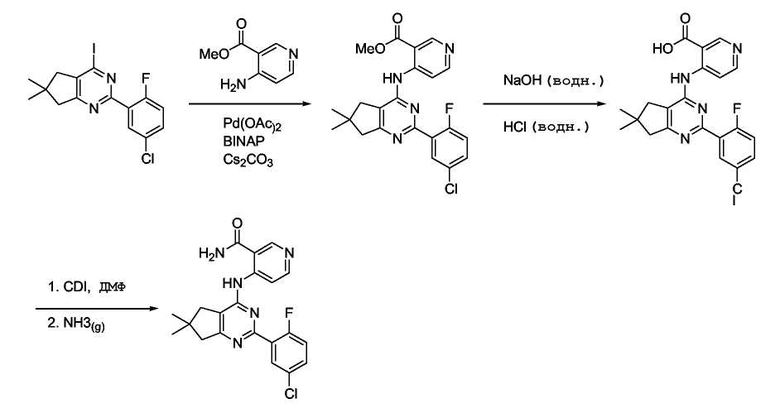
Получение метилового эфира 4-[2-(5-хлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты.
К раствору 2-(5-хлор-2-фтор-фенил)-4-иод-6,6-диметил-6,7-дигидро-5H-циклопентапиримидина (500 мг, 1,24 ммоль), метилового эфира 4-аминоникотиновой кислоты (208 мг, 1,37 ммоль), Pd(OAc)2 (14 мг, 62,09 мкмоль) и рац-BINAP (60 мг, 93,14 мкмоль) в безводном диоксане (10 мл) добавляли Cs2CO3 (607 мг, 1,86 ммоль). Смесь нагревали в течение 12 часов при 85°C, охлаждали и упаривали. Остаток очищали с помощью хроматографии (CH2Cl2, 0-10% MeOH), получая целевой продукт (178 мг, 34% выход).
Получение 4-[2-(5-хлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты:
К раствору метилового эфира 4-[2-(5-хлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты (178 мг, 0,42 ммоль) в диоксане (10 мл) добавляли NaOH (водн.) (451 мкл, 0,44 ммоль, 0,97н. раствор). Смесь нагревали при 60°C в течение 1 часа, затем охлаждали и добавляли HCl (водн.) (425 мкл, 0,44 ммоль, 1,03 н. раствор). После прибавления кислоту, выпавшую в осадок из раствора, отфильтровывали и сушили в вакууме, получая 137 мг продукта.
Получение 4-[2-(5-хлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотинамида:
Суспензию 4-[2-(5хХлор-2-фтор-фенил)-6,6-диметил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты (25 мг, 60,56 мкмоль) и карбонилдиимидазола (20 мг, 121,11 мкмоль) в сухом ДМФ (3 мл) нагревали при 70°C в течение 1 часа и затем охлаждали до комнатной температуры. Через раствор в течение 30 минут пропускали газообразный NH3, что приводило к беспримесному преобразованию в продукт амида. Упаривание раствора с последующей очисткой методом ВЭЖХ приводило после лиофилизации к чистому амиду формулы (62) в виде соли ТФУК (20 мг).
Соединения формулы (63)-(66) были получены с использованием того же способа.
Схема Q (Синтез соединений формулы (76) и (77))

Метиловый эфир 4-[6-(5-хлор-2-фтор-фенил)-3-метил-изоксазоло[5,4-d]пиримидин-4-иламино]никотиновой кислоты:
4-Хлор-6-(5-хлор-2-фтор-фенил)-3-метил-изоксазоло[5,4-d]пиримидин (получен способом, использованным для получения соединения формулы (43)) (298 мг, 1 ммоль) растворяли в диоксане (4 мл) и к смеси добавляли BINAP (4,67 мг, 0,0075 ммоль), метиловый эфир 4-амино-никотиновой киислоты (182 мг, 1,2 ммоль), карбонат цезия (456 мг, 1,4 ммоль) и Pd2(dba)3 (2,29 мг, 0,0025 ммоль) и нагревали при 90°C в течение ночи. Диоксан удаляли в вакууме и остаток растирали с этилацетатом (5 мл) и продукт выделяли фильтрованием. Получали 525 мг твердого вещества, которое содержало также карбонат цезия в качестве побочного продукта.
4-[6-(5-Хлор-2-фтор-фенил)-3-метил-изоксазоло[5,4-d]пиримидин-4-иламино]никотиновая кислота:
Метиловый эфир 4-[6-(5-хлор-2-фтор-фенил)-3-метил-изоксазоло[5,4-d]пиримидин-4-иламино]никотиновой кислоты (525 мг) суспендировали в метаноле (4 мл), добавляли 1 M раствор гидркосида натрия (4 мл) и затем нагревали при 70°C в течение 30 минут. Метанол удаляли в вакууме и раствор подкисляли до pH 4 с использованием 6 M соляной кислоты. Твердое вещество отфильтровывали, промывали водой и сушили в печи, получая 137 мг продукта.
4-[6-(5-Хлор-2-фтор-фенил)-3-метил-изоксазоло[5,4-d]пиримидин-4-иламино]-N-циклопропилникотинамид:
4-[6-(5-Хлор-2-фтор-фенил)-3-метил-изоксазоло[5,4-d]пиримидин-4-иламино]никотиновую кислоту (130 мг, 0,325 ммоль) объединяли с карбонилдиимидазолом (105 мг, 0,650 ммоль) в 2 мл диметилформамида и нагревали при 70°C в течение 1 часа. Смесь охлаждали до комнатной температуры, добавляли циклопропиламин (74 мг, 1,3 ммоль) и перемешивали в течение 1 часа при комнатной температуре. Раствор фильтровали и фильтрат подвергали очистке с помощью ВЭЖХ с обращенной фазой. После лиофилизации фракций, содержавших чистый продукт, получали 13,7 мг чистого продукта.
Соединения формулы (77) получали с использованиеим вышеописанного способа с использованием метиламина.
Схема R (Синтез соединения формулы (33))
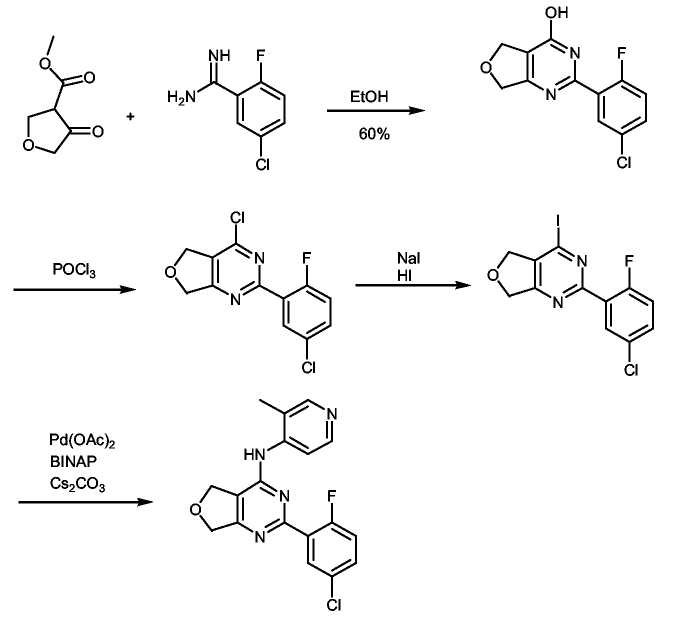
2-(5-Хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-ол:
К суспензии метилового эфира 4-оксо-тетрагидрофуран-3-карбоновой кислоты (получен согласно Dowd, P.; Choi, S-C. Tetrahedron, 1991, 47, 4847-4860; 800 мг, 5,55 ммоль, 1 экв.) в этаноле (20 мл) добавляли раствор 2-фтор-5-хлорбензамидина (961 мг, 5,55 ммоль, 1 экв.) в EtOH (10 мл). Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры, и белый осадок отфильтровывали и промывали холодным этилацетатом (2×20 мл). Неочищенный остаток распределяли между хлороформом и водой. Водный слой подкисляли до pH 4 и продукт экстрагировали хлороформом (3 × 50 мл). Органические слои объединяли, промывали насыщенным раствором соли, сушили над MgSO4, фильтровали и концентрировали в вакууме, получая неочищенное твердое вещество, которое очищали колоночной флэш-хроматографией (5% MeOH в EtOAc), получая белое твердое вещество 2-(5-хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-ол (440 мг, 30%).
4-Хлор-2-(5-хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин:
Суспензию 2-(5-хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-ола (100 мг, 0,36 ммоль, 1 экв.) в POCl3 (5 мл) перемешивали при кипении в течение 1 часа. Раствор затем охлаждали до комнатной температуры и концентрировали при пониженном давлении, получая белое твердое вещество, которое растворяли в безводном хлористом метилене. Раствор охлаждали до 0°C и добавляли лед с последующим добавлением насыщенного раствора NaHCO3. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая неочищенное белок твердое вещество, которое очищали колоночной флэш-хроматографией (1:9 EtOAc:гексан), получая 4-хлор-2-(5-хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин (78 мг, 73%) в виде белого твердого вещества.
2-(5-Хлор-2-фторфенил)-4-иод-5,7-дигидрофуро[3,4-d]пиримидин:
К суспензии 4-хлор-2-(5-хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидина (78 мг, 0,275 ммоль, 1 экв.) в 57% HI (водн.) (2 мл) при комнатной температуре добавляли NaI (206 мг, 1,37 ммоль, 5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выливали на лед. Продукт экстрагировали хлороформом и водный слой нейтрализовали NaHCO3 и дополнительно экстрагировали хлороформом. Органические слои объединяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая неочищенный остаток 2-(5-хлор-2-фторфенил)-4-иод-5,7-дигидрофуро[3,4-d]пиримидина, который не подвергали дополнительной очистке.
[2-(5-Хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-ил]-(3-метил-пиридин-4-ил)амин, соединение формулы (33):
К раствору 2-(5-хлор-2-фторфенил)-4-иод-5,7-дигидрофуро[3,4-d]пиримидина (80 мг, 0,21 ммоль, 1 экв.) в безводном диоксане (5 мл) добавляли Pd(OAc)2 (2 мг, 0,01 ммоль, 0,05 экв.) с последующим добавлением BINAP (10 мг, 0,02 ммоль, 0,075 экв.), 4-амино-3-пиколина (25 мг, 0,23 ммоль, 1,2 экв.) и Cs2CO3 (100 мг, 0,32 ммоль, 1,5 экв.). Реакционную смесь нагревали при 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит® и неочищенное вещество очищали колоночной флэш-хроматографией (9:1/этилацетат:гексан), получая [2-(5-хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-ил]-(3-метил-пиридин-4-ил)амин, соединение формулы (33), (20 мг, 26%) в виде белого твердого вещества.
Схема S (Синтез соединения формулы (34))

Этиловый эфир 4-[2-(5-хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотиновой кислоты:
В соответствии с общим способом реакции, использованным для синтеза [2-(5-хлор-2-фторфенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-ил]-(3-метил-пиридин-4-ил)амина, т.е. соединения формулы (33), этиловый эфир 4-[2-(5-хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотиновой кислоты выделяли с выходом 73%.
4-[2-(5-Хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотиновая кислота:
К суспензии этилового эфира 4-[2-(5-хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотиновой кислоты (60 мг, 0,14 ммоль, 1 экв.) в MeOH (5 мл) добавляли 1н. раствор NaOH(водн.) (300 мкл, 0,30 ммоль, 2 экв.) и реакционную смесь нагревали при кипении с обратным холодильникком в течение 2 часов. Раствор охлаждали до комнатной температуры и концентрировали в вакууме. К неочищенному веществу добавляли воду (20 мл) и водный слой подкисляли до pH 4. Твердое вещество отфильтровывали, промывали водой (2×5 мл) и сушили в течение ночи, получая 4-[2-(5-хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотиновую кислоту (50 мг, 90%).
4-[2-(5-Хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотинамид, соединение формулы (34):
К суспензии 4-[2-(5-хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотиновой кислоты (50 мг, 0,13 ммоль, 1 экв.) в ДМФ (2 мл) добавляли 1-1'-карбонилдиимидазол (50 мг, 0,31 ммоль, 2,4 экв.) и реакционную смесь нагревали при 70°C в течение 2 часов. Смесь охлаждали до комнатной температуры и NH3 (газ) барботировали через нее в течение 10 минут. Реакционную смесь перемешивали при комнатной температуре дополнительно в течение 1 часа. Реакционную смесь концентрировали в вакууме и остаток растирали с водой (2 × 5 мл). К неочищенному остатку добавляли 1н. NaOH (5 мл) и суспензию нагревали при 100°C в течение 2 часов. Реакционную смесь охлаждали до комнатной температуры и нейтрализовали 1н. HCl и твердое вещество отфильтровывали, получая 4-[2-(5-хлор-2-фтор-фенил)-5,7-дигидрофуро[3,4-d]пиримидин-4-иламино]никотинамид, соединение формулы (34), (25 мг, 50%) в виде белого твердого вещества.
Схема T (Синтез соединения формулы (38))

2-(5-Хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-ол:
К раствору метил-2-оскоциклопентанкарбоксилата (2 г, 11,8 ммоль, 1 экв.) в безводном этаноле (20 мл) добавляли раствор 2-фтор-5-хлорбензамидина (2,04 г, 11,8 ммоль, 1 экв.) в этаноле (20 мл) и реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в вакууме, получая неочищенный остаток, который очищали перекристаллизацией из горячего этилацетата, получая 2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-ол (2,56 г, 78%) в виде белого твердого вещества.
4-Хлор-2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин:
Суспензию 2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-ола (500 мг, 1,89 ммоль) в POCl3 (6 мл) перемешивали при кипении с обратным холодильником в течение 1 часа. Раствор затем охлаждали до комнатной температуры и концентрировали при пониженном давлении, получая белое твердое вещество, которое растворяли в хлористом метилене. Раствор охлаждали до 0°C и добавляли лед с последующим добавлением насыщенного раствора NaHCO3, Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая неочищенное белое вещество 4-хлор-2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолина, который дополнительно не очищали.
2-(5-Хлор-2-фторфенил)-4-иод-5,6,7,8-тетрагидрохиназолин:
К суспензии 4-хлор-2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолина (534 мг, 1,89 ммоль, 1 экв.) в 57%-ном растворе HI в воде (10 мл) при комнатной температуре добавляли NaI (1,42 г, 9,47 ммоль, 5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выливали на лед. Продукт экстрагировали хлороформом и водный слой нейтрализовали NaHCO3 и дополнительно экстрагировали хлороформом. Органические слои объединяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая неочищенное белое твердое вещество 2-(5-хлор-2-фторфенил)-4-иод-5,6,7,8-тетрагидрохиназолина, которое дополнительно не очищали.
[2-(5-Хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-ил]-(3-метилпиридин-4-ил]амин, соединение формулы (38):
Неочищенный 2-(5-хлор-2-фторфенил)-4-иод-5,6,7,8-тетрагидрохиназолин (130 мг, 0,35 ммоль, 1 экв.) растворяли в диоксане (5 мл) и добавляли к раствору Pd(OAc)2 (4 мг, 0,02 ммоль, 0,05 экв.) с последующим добавлением BINAP (16 мг, 0,03 ммоль, 0,075 экв.), 4-амино-3-пиколина (49 мг, 0,45 ммоль, 1,3 экв.) и Cs2CO3 (170 мг, 0,52 ммоль, 1,5 экв.). Реакционную смесь нагревали при 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит® и неочищенное вещество очищали колоночной флэш-хроматографией (1:1/этилацетат:гексан), получая [2-(5-хлор-2-фтор-фенил)-5,6,7,8-тетрагидрохиназолин-4-ил]-(3-метилпиридин-4-ил)амин, соединение формулы (38), (110 мг, 86%).
Схема U (Синтез соединений формулы (50), (40), (45), (57), (59))

Этиловый эфир 4-[2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]никотиновой кислоты:
В соответствии с общим способом проведения реакции для синтеза [2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-ил]-(3-метилпиридин-4-ил)амина, т.е. соединения формулы (38), этиловый эфир 4-[2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]никотиновой кислоты выделяли с 67% выходом.
4-[2-(5-Хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]никотиновая кислота 50:
К суспензии этилового эфира 4-[2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]никотиновой кислоты (150 мг, 0,35 ммоль, 1 экв.) в MeOH (5 мл) добавляли 1н. раствор NaOH (водн.) (423 мкл, 0,42 ммоль, 1,2 экв.) и реакционную смесь нагревали при кипении с обратным холодильником в течение 1 часа. Раствор охлаждали до комнатной температуры и концентрировали в вакууме. Добавляли воду (20 мл) к неочищенному веществу и водный слой подкисляли до pH 4. Твердое вещество отфильтровывали, промывали водой (2×5 мл) и сушили в течение ночи, получая 4-[2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]никотиновую кислоту, соединения формулы (50) (132 мг, 94%), в виде твердого вещества кремового цвета
4-[2-(5-Хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]-N-циклопропилникотинамид, соединение формулы (45):
К суспензии 4-[2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]никотиновой кислоты (40 мг, 0,10 ммоль, 1 экв.) в сухом ДМФ (1 мл) добавляли триэтиламин (15 мкл, 0,11 ммоль, 1,1 экв.) с последующим добавлением циклопропиламина (70 мкл, 0,10 ммоль, 10 экв.). К суспензии добавляли по каплям раствор PyBrOP (56 мг, 0,21 ммоль, 1,2 экв.) в ДМФ (500 мкл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали в вакууме, и остаток растирали с простым эфиром (2×20 мл). Неочищенный остаток очищали колоночной флэш-хроматографией (0-5% MeOH в CHCl3), получая 4-[2-(5-хлор-2-фторфенил)-5,6,7,8-тетрагидрохиназолин-4-иламино]-N-циклопропилникотинамид, соединение формулы (45), (15 мг, 34%) в виде белого твердого вещества.
Соединение формулы (40) получали способом, описанным для синтеза соединения формулы (45), с использованием метиламина вместо циклопропиламина. Соединение формулы (57) получали способом, описанным для синтеза соединения формулы (45), с использованием 1-аминопропан-2-(S)-ола вместо циклопропиламина. Соединение формулы (59) получали способом, описанным для синтеза соединения формулы (45), с использованием N,N-диэтилэтендиамина вместо циклопропиламина.
Схема V (Синтез соединения формулы (39))
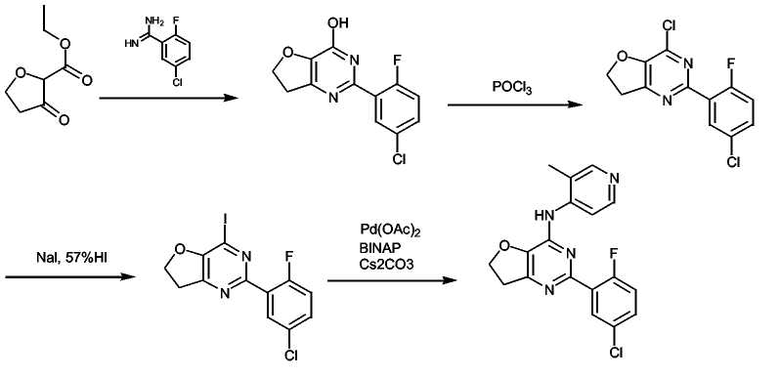
2-(5-Хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-ол:
К раствору 2-фтор-5-хлорбензамидина (1,05 г, 6,08 ммоль, 1,2 экв.) в EtOH (20 мл) добавляли этиловый эфир 3-оксотетрагидрофуран-2-карбоновой кислоты (получен согласно Moyer, M.P; Feldman, P.L.; Rapoport, H. J.Org. Chem, 1985, 50, 5223-5230; 800 мг, 5,06 ммоль, 1 экв.) в этаноле (5 мл). Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и неочищенный остаток очищали колоночной флэш-хроматографией (5% MeOH в CHCl3), получая белое твердое вещество 2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-ола (650 мг, 53%).
4-Хлор-2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин:
Суспензию 2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-ола (100 мг, 0,36 ммоль, 1 экв.) в POCl3 (5 мл) перемешивали при кипении с обратным холодильником в течение 1 часа. Раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении, получая белое твердое вещество, которое растворяли в хлористом метилене. Раствор охлаждали до 0°C и добавляли лед с последующим добавлением насыщенного раствора NaHCO3. Органический слой отделяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая неочищенное белое твердое вещество, которое очищали колоночной флэш-хроматографией (1:9 EtOAc:гексан), получая 4-хлор-2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин (78 мг, 73%) в виде белого твердого вещества.
2-(5-Хлор-2-фторфенил)-4-иод-6,7-дигидрофуро[3,2-d]пиримидин:
К суспензии 4-хлор-2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидина (80 мг, 0,28 ммоль, 1 экв.) в 57%-ном растворе HI в воде (2 мл) при комнатной температуре добавляли NaI (206 мг, 1,41 ммоль, 5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выливали на лед. Продукт экстрагировали хлороформом и водный слой нейтрализовали NaHCO3 и дополнительно экстрагировали хлороформом. Органические слои объединяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая неочищенный остаток 2-(5-хлор-2-фторфенил)-4-иод-6,7-дигидрофуро[3,2-d]пиримидина, который не подвергали дополнительной очистке.
[2-(5-Хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-ил]-(3-метилпиридин-4-ил)амин, соединение формулы (39):
К раствору 2-(5-хлор-2-фторфенил)-4-иод-6,7-дигидрофуро[3,2-d]пиримидина (106 мг, 0,28 ммоль, 1 экв.) в диоксане (5 мл) добавляли Pd(OAc)2 (3 мг, 0,01 ммоль, 0,05 экв.) с последующим добавлением BINAP (13 мг, 0,02 ммоль, 0,075 экв.), 4-амино-3-пиколина (40 мг, 0,37 ммоль, 1,3 экв.) и Cs2CO3 (138 мг, 0,42 ммоль, 1,5 экв.). Реакционную смесь нагревали при 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит® и неочищенное вещество очищали колоночной флэш-хроматографией (4:1/этилацетат:гексан-100%этилацетат), получая [2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-ил]-(3-метил-пиридин-4-ил)амин, т.е. соединение формулы (39) (30 мг, 30%) в виде белого твердого вещества.
Схема W (Синтез соединения формулы (44))

Этиловый эфир 4-[2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]никотиновой кислоты:
К раствору 4-хлор-2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидина (320 мг, 1,13 ммоль, 1 экв.) в диоксане (5 мл) добавляли Pd(OAc)2 (13 мг, 0,06 ммоль, 0,05 экв.) с последующим добавлением BINAP (53 мг, 0,08 ммоль, 0,075 экв.), этилового эфира 4-аминоникотиновой кислоты (206 мг, 1,24 ммоль, 1,1 экв.) и Cs2CO3 (478 мг, 1,46 ммоль, 1,5 экв.). Реакционную смесь нагревали при 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит®, и неочищенное вещество очищали колоночной флэш-хроматографией (0-5% MeOH в CHCl3), получая этиловый эфир 4-[2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]никотиновой кислоты (100 мг, 21%) в виде белого твердого вещества.
4-[2-(5-Хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]никотиновая кислота:
К суспензии этилового эфира 4-[2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]никотиновой кислоты (100 мг, 0,24 ммоль, 1 экв.) в MeOH (5 мл) добавляли 1н раствор NaOH (водн.) (290 мкл, 0,29 ммоль, 1,2 экв.) и реакционную смесь нагревали при кипении с обратным холодильником в течение 5 часов. Раствор охлаждали до комнатной температуры и концентрировали в вакууме. К неочищенному веществу добавляли воду (20 мл) и водный слой подкисляли до pH 4. Твердое вещество отфильтровывали, промывали водой (2 × 5 мл) и сушили в течение ночи, получая 4-[2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]никотиновую кислоту (88 мг, 94%) в виде твердого вещества кремового цвета.
4-[2-(5-Хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]-N-метилникотинамид, соединение формулы (44):
К суспензии 4-[2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]никотиновой кислоты (75 мг, 0,19 ммоль, 1 экв.) в ДМФ (2 мл) добавляли триэтиламин (30 мкл, 0,21 ммоль, 1,1 экв.) с последующим добавлением метиламина (1,17 мл, 3,89 ммоль, 2 M раствор в ТГФ, 20 экв.). К суспензии добавляли по каплям раствор PyBrOP (100 мг, 0,21 ммоль, 1,2 экв.) в ДМФ (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали в вакууме и остаток растирали с простым эфиром (2 × 20 мл). Неочищенный остаток очищали колоночной флэш-хроматографией (5% MeOH в CHCl3), получая 4-[2-(5-хлор-2-фторфенил)-6,7-дигидрофуро[3,2-d]пиримидин-4-иламино]-N-метилникотинамид, т.е. соединения формулы (44) (20 мг, 26%) в виде белого твердого вещества.
Схема X (Синтез соединения формулы (43))

2-(5-Хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-ол:
К раствору 2-фтор-5-хлорбензамидина (1,95 г, 11,27 ммоль, 1,1 экв.) в EtOH (20 мл) добавляли раствор этилового эфира 2-метил-5-оксоциклопентанкарбоновой кислоты (получен согласно Wang, C; Gu, X.; Yu, M.S.; Curran, D.P.: Tetrahedron, 1998, 29, 8355-8370; 1,60 г, 10,26 ммоль, 1 экв.) в этаноле (5 мл). Реакционную смесь нагревали при 80°C в течение ночи. Реакционную смесь охлаждали до комнатной температуры и растворитель удаляли в вакууме, получая неочищенный остаток, который очищали перекристаллизацией из горячего этилацетата, получая 2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-ол (1,0 г, 35%) в виде белого кристаллического твердого вещества, а фильтрат дополнительно очищали колоночной флэш-хроматографией, получая дополнительно 850 мг 2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-ола.
2-(5-Хлор-2-фторфенил)-4-иод-5-метил-6,7-дигидро-5H-циклопентапиримидин:
Суспензию 2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-ола (200 мг, 0,72 ммоль, 1 экв.) в POCl3 (5 мл) перемешивали при кипении с обратным холодильником в течение 1 часа. Раствор затем охлаждали до комнатной температуры и концентрировали при пониженном давлении, получая белое твердое вещество. Остаток POCl3 удаляли путем азеотропной отгонки с хлороформом. К суспензии неочищенного иминохлорида в 57%-ном растворе HI в воде (5 мл) при комнатной температуре добавляли NaI (540 мг, 3,60 ммоль, 5 экв.). Реакционную смесь перемешивали при комнатной температуре в течение ночи и затем выливали на лед. Продукт экстрагировали хлороформом, и водный слой нейтрализовали NaHCO3 и дополнительно экстрагировали хлороформом. Органические слои объединяли, промывали насыщенным раствором соли, сушили (MgSO4), фильтровали и упаривали в вакууме, получая в неочищенном остатке 2-(5-хлор-2-фторфенил)-4-иод-5-метил-6,7-дигидро-5H-циклопентапиримидин, который дополнительно не очищали.
[2-(5-Хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-ил]-(3-метилпиридин-4-ил)амин, соединение формулы (43):
К раствору 2-(5-хлор-2-фторфенил)-4-иод-5-метил-6,7-дигидро-5H-циклопентапиримидина (275 мг, 0,71 ммоль, 1 экв.) в диоксане (5 мл) добавляли Pd(OAc)2 (8 мг, 0,04 ммоль, 0,05 экв.) с последующим добавлением BINAP (33 мг, 0,05 ммоль, 0,075 экв.), 4-амино-3-пиколина (84 мг, 0,78 ммоль, 1,1 экв.) и Cs2CO3 (347 мг, 1,06 ммоль, 1,5 экв.). Реакционную смесь нагревали при 80°C в течение 15 часов. Реакционную смесь охлаждали до комнатной температуры и фильтровали через целит®, и неочищенное вещество очищали колоночной флэш-хроматографией (1:1/этилацетат:гексан-100% этилацетат), получая [2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-ил]-(3-метил-пиридин-4-ил)амин, соединение формулы (43), (250 мг, 96%).
Схема Y (Синтез соединений формулы (48), (49), (47), (46))
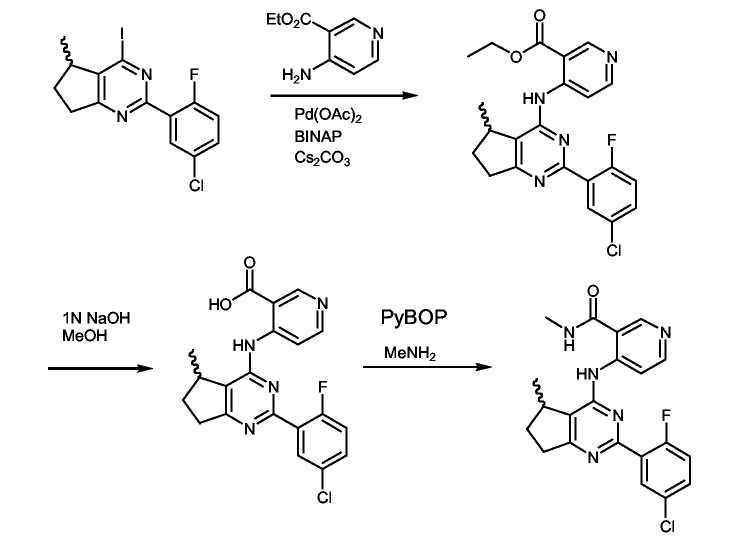
Этиловый эфир 4-[2-(5-хлор-2-фтор-фенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты, соединение формулы (48):
В соответствии с общим способом проведения реакции синтеза [2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-ил]-(3-метил-пиридин-4-ил)-амина, т.e. соединения формулы (43), этиловый эфир 4-[2-(5-хлор-2-фтор-фенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты, соединение формулы (48), выделяли с 68% выходом.
4-[2-(5-Хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновая кислота, соединение формулы (49):
К суспензии этилового эфира 4-[2-(5-хлор-2-фтор-фенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты (180 мг, 0,42 ммоль, 1 экв.) в MeOH (5 мл) добавляли 1н. раствор NaOH (водн.) (634 мкл, 0,63 ммоль, 1,5 экв.) и реакционную смесь нагревали при кипении с обратным холодильником в течение 1 часа. Раствор охлаждали до комнатной температуры и концентрировали в вакууме. К неочищенному веществу добавляли воду (20 мл) и водный слой подкисляли до pH 4. Твердое вещество отфильтровывали, промывали водой (2 × 5 мл) и сушили в течение ночи, получая 4-[2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновую кислоту, соединения формулы (49) (160 мг, 95%) в виде белого твердого вещества.
4-[2-(5-Хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопента пиримидин-4-иламино]-N-метилникотинамид, соединение формулы (47):
К суспензии 4-[2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]никотиновой кислоты (20 мг, 0,05 ммоль, 1 экв.) в ДМФ (2 мл) добавляли триэтиламин (8 мкл, 0,05 ммоль, 1,1 экв.) с последующим добавлением метиламина (250 мкл, 3,89 ммоль, 2 M раствор в ТГФ, 10 экв.). К суспензии добавляли по каплям раствор PyBOP (40 мг, 0,08 ммоль, 1,5 экв.) в ДМФ (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь концентрировали в вакууме и остаток растирали с простым эфиром (2×20 мл). Неочищенный остаток очищали колоночной флэш-хроматографией (100% этилацетат), получая 4-[2-(5-хлор-2-фторфенил)-5-метил-6,7-дигидро-5H-циклопентапиримидин-4-иламино]-N-метилникотинамид, т.е. соединение формулы (47), (12 мг, 58%) в виде белого твердого вещества.
Соединение формулы (46) получали способом, описанным для синтеза соединения формулы (47), с использованием аммиака вместо метиламина.
Соединения формулы (1) могут быть преобразованы друг в друга в соответствии с известными в данной области реакциями преобразования функциональных групп, включая те, которые описаны ниже. Ряд промежуточных соединений, использованных для получения соединений формулы (1), представляют собой известные соединения или аналоги известных соединений, которые могут быть получены в соответствии с модификациями известных в данной области методологий, легко доступных специалистам в данной области.
Соединения формулы (1) могут быть преобразованы в соответствующие N-оксидные формы в соответствии с известными в данной области способами преобразования трехвалентного азота в его N-оксидную форму. Указанную реакцию N-окисления обычно можно проводить посредством взаимодействия исходного вещества формулы (1) с подходящим органическим или неорганическим пероксидом. Подходящие неорганические пероксиды включают, например, перекись водорода, пероксиды щелочных металлов или щелочноземельных металлов, например пероксид натрия, пероксид калия; подходящие органические пероксиды могут включать пероксокислоты, например, такие как бензолкарбопероксокислота или галогензамещенная бензолкарбопероксокислота, например 3-хлорбензолкарбопероксокислота, пероксоалкановые кислоты, например пероксиуксусная кислота, алкилгидропероксиды, например трет-бутилгидропероксид. Подходящими растворителями являются, например, вода, низшие спирты, например этанол и тому подобные, углеводороды, например толуол, кетоны, например 2-бутанон, галогенированные углеводороды, например, дихлорметан и смеси таких растворителей.
Стереохимически чистые изомерные формы соединений формулы (1) могут быть получены с применением известных в данной области способов. Диастереомеры могут быть разделены физическими методами, такими как селективная кристаллизация и хроматографические методы, например противоточное распределение, жидкостная хроматография и тому подобные.
Соединения формулы (1) могут быть получены в виде рацемических смесей энантиомеров, которые могут быть отделены друг от друга в соответствии со способами разделения, известными в данной области. Рацемические соединения формулы (1), которые являются достаточно основными или кислыми, могут быть преобразованы в соответствующие диастереомерные солевые формы с помощью реакции с подходящей хиральной кислотой, соответственно с хиральным основанием. Указанные диастереомерные солевые формы впоследствии разделяют, например, путем селективной или дробной кристаллизации и энантиомеры высвобождают из них под действием щелочи или кислоты. Альтернативный способ разделения энантиомерных форм соединений формулы (1) включает жидкостную хроматографию, в частности жидкостную хроматографию с использованием хиральной стационарной фазы. Указанные стереохимически чистые изомерные формы также могут быть получены из соответствующих стереохимически чистых изомерных форм подходящих исходных веществ при условии, что реакция протекает стереоспецифически. Предпочтительно, если желателен определенный стереоизомер, указанное соединение может быть синтезировано стереоспецифическими способами получения. В данных способах преимущественно используются энантиомерно чистые исходные вещества.
Способ введения и получение препаратов соединений, использованных в изобретении и родственных им соединений, будет зависеть от природы болезненного состояния, тяжести болезненного состояния, конкретного субъекта, подвергаемого лечению и точки зрения лечащего врача; тип препарата будет зависеть от способа введения. С целью введения соединения по настоящему изобретению или любая их подгруппа могут быть введены в состав различных фармацевтических форм. В качестве подходящих композиций здесь можно упомянуть все композиции, обычно используемые для системного введения лекарственных средств.
Для получения фармацевтических композиций по данному изобретению терапевтически эффективное количество конкретного соединения, необязательно в виде аддитивной соли или комплекса металла, в качестве активного ингредиента объединяют в виде однородной смеси с фармацевтически приемлемым носителем, где носитель может принимать множество форм в зависимости от препарата, желательного для введения. Терапевтически эффективное количество в данном контексте представляет собой количество, достаточное для профилактического действия против вирусной инфекции, стабилизации или снижения вирусной инфекции и в частности вирусной инфекции HCV или для предотвращения прогрессирования заболевания, приводящего к хроническому гепатиту, фиброзу печени, циррозу, конечной стадии заболевания печени, HCC (гепатоклеточная карцинома) и тому подобного у инфицированных субъектов или у субъектов, подверженных риску инфицирования.
Желательно, чтобы фармацевтические композиции представляли собой единичные препаративные лекарственные формы, подходящие, в частности, для перорального, ректального, подкожного введения или введения с помощью парентеральной инъекции. При получении композиций в виде пероральной препаративной лекарственной формы можно использовать любую из обычных фармацевтических сред, например такую как вода, гликоли, масла, спирты и тому подобные в случае пероральных жидких препаратов, таких как суспензии, сиропы, эликсиры, эмульсии и растворы; или твердые носители, такие как крахмалы, сахара, каолин, смазочные вещества, связующие агенты, дезинтегрирующие агенты и тому подобные в случае порошков, пилюль, капсул и таблеток. Благодаря легкости их введения, таблетки и капсулы представляют наиболее выигрышные пероральные препаративные единичные лекарственные формы, в таком случае с очевидностью используют твердые носители. Для парентеральных композиций носитель обычно будет включать стерильную воду, по крайней мере большей частью, хотя другие ингредиенты, например те, которые способствуют растворимости, могут быть включены в состав. Например, могут быть получены растворы для инъекций, в которых носитель включает физиологический раствор, раствор глюкозы или смесь физиологического раствора и раствора глюкозы. Также могут быть получены суспензии для инъекций, в таком случае можно использовать подходящие жидкие носители, суспендирующие агенты и тому подобное. К ним относятся также препараты в твердом виде, которые предназначены для преобразования, незадолго до применения в жидкие препараты. В композициях, подходящих для подкожного введения, носитель необязательно включает агент, усиливающий проницаемость и/или подходящий смачивающий агент, необязательно в сочетании с подходящими вспомогательными добавками любой природы в небольших долях, указанные добавки не оказывают существенного опасного действия на кожу.
Поскольку соединения по изобретению представляют собой небольшие молекулы, их удобно вводить посредством перорального введения путем их компаундирования с подходящими фармацевтическими эксципиентами таким образом, чтобы получать таблетки, капсулы, сиропы и тому подобное. Подходящие препараты для перорального введения также могут включать минорные компоненты, такие как буферы, ароматизаторы и тому подобные. Обычно количество активного ингредиента в препарате будет колебаться в диапазоне 5%-95% от общей композиции, но допускается широкое варьирование в зависимости от носителя. Подходящие носители включают сахарозу, пектин, стеарат магния, лактозу, арахисовое масло, оливковое масло, воду и тому подобное.
Композиции, используемые в изобретении, также можно вводить с помощью суппозиториев или других чрезслизистых носителей. Обычно такие препараты будут включать эксципиенты, которые облегчают прохождение соединения через слизистую, такие как фармацевтически приемлемые детергенты.
Соединения также можно вводить наружно в случае локально проявляющихся болезненных состояний, таких как псориаз, или в препаратах, предназначенных для проникновения через кожу. Они включают лосьоны, кремы, мази и тому подобные, которые могут быть получены известными способами.
Композиции также можно вводить посредством инъекции, включая внутривенную, внутримышечную, подкожную и внутрибрюшинную инъекцию. Типичными препаратами для такого применения являются жидкие препараты в изотонических носителях, таких как раствор Хэнка или раствор Рингенра.
Соединения по настоящему изобретению также можно вводить посредством ингаляции в ротовую полость или вдувания с помощью способов и препаратов, используемых в данной области для введения таким образом. Таким образом, обычно соединения по настоящему изобретению можно вводить в легкие в виде раствора, суспензии или сухого порошка, при этом раствор является предпочтительным. Любая система, разработанная для доставки растворов, суспензий или сухих порошков посредством ингаляции в ротовую полость или вдувания, является подходящей для введения настоящих соединений.
Таким образом настоящее изобретение также относится к фармацевтическим композициям, приспособленным для введения путем ингаляции или вдувания через рот, включающим соединение формулы (1) и фармацевтически приемлемый носитель. Предпочтительно соединения по настоящему изобретению вводят путем ингаляции раствора в дозах из небулайзера или аэрозоля.
Альтернативные препараты включают назальные спреи, липосомальные препараты, препараты замедленного высвобождения и тому подобные, как это известно в данной области.
Можно использовать любой подходящий препарат. Краткое руководство по известным в данной области препаратам можно найти в последнем издании Remington's Pharmaceutical Sciences, Mack Publishing Company, Easton, PA. Ссылка на данный справочник является обычной в данной области.
Особенно выгодно получать вышеуказанные фармацевтические композиции в виде единичных препаративных лекарственных форм для легкости введения и единообразия дозировки. Термин «единичная препаративная лекарственная форма», как он используется в данном описании, относится к физически дискретным единицам, подходящим в качестве стандартных лекарственных доз, при этом каждая стандартная форма содержит определенное количество активного ингредиента, рассчитанное для оказания желаемого терапевтического действия, в сочетании с требуемым фармацевтическим носителем. Примерами таких стандартных препаративных лекарственных форм являются таблетки (включая таблетки с насечками и покрытые оболочкой), капсулы, пилюли, суппозитории, пакетики с порошками, облатки, растворы или суспензии для инъекций и тому подобное и определенные их множества.
Дозировки соединений по изобретению будут зависеть от ряда факторов, которые будут изменяться от пациента к пациенту. Однако предполагается, что обычно дневная пероральная дозировка будет составлять 0,001-100 мг/кг общего веса тела, предпочтительно от 0,01-50 мг/кг и более предпочтительно примерно 0,01 - 10 мг/кг. Режим дозировки будет варьироваться, однако, в зависимости от состояния, подвергаемого лечению, и точки зрения лечащего врача.
В общем подразумевается, что противовирусное эффективное дневное количество будет составлять от 0,01 до 500 мг/кг веса тела, более предпочтительно от 0,1 до 50 мг/кг веса тела. Может оказаться удобным вводить требуемую дозу в виде двух, трех, четырех или более поддоз с подходящими интервалами в течение дня. Указанные поддозы могут быть получены в виде стандартных лекарственных форм, например, содержащих от 1 до 1000 мг и, в частности, от 5 до 200 мг активного ингредиента на стандартную лекарственную форму.
Точная дозировка и частота введения зависят от конкретного используемого соединения формулы (1), конкретного состояния, подвергаемого лечению, тяжести состояния, подвергаемого лечению, возраста, веса, пола, степени нарушения и общего физического состояния конкретного пациента, а также других лекарственных средств, которые может принимать пациент, что хорошо известно специалистам в данной области. Кроме того, очевидно, что указанное эффективное дневное количество может быть снижено или повышено в зависимости от ответной реакции субъекта, подвергаемого лечению, и в зависимости от оценки врача, предписавшего соединения по настоящему изобретению. Отмеченные выше диапазоны эффективного дневного количества, следовательно, являются только руководящими указаниями.
Благодаря своим благоприятным противовирусным свойствам, как будет очевидно из примеров, соединения по настоящему изобретению можно использовать для лечения индивидуумов, инфицированных HCV, и для профилактики данных индивидуумов. Обычно соединения по настоящему изобретению можно использовать для лечения теплокровных животных, инфицированных флавивирусами. Состояния, которые могут быть предотвращены или которые можно лечить с помощью соединений по настоящему изобретению, представляют собой в частности состояния, связанные с HCV и другими патогенными флавивирусами, такие как желтая лихорадка, лихорадка денге (типы 1-4), энцефалит Ст.Луиса, японский энцефалит, энцефалит долины Муррея, вирус Западного Нила и вирус Кунджин. Состояния, связанные с HCV, включают прогрессирующий фиброз печени, воспаление и некроз, приводящие к циррозу, конечную стадию заболевания печени и НСС, а для других патогенных флавивирусов состояния влючают желтую лихорадку, лихорадку денге, геморрагическую лихорадку и энцефалит.
Следовательно, соединения по настоящему изобретению или любая их подгруппа могут использоваться в качестве лекарственных средств против вышеуказанных состояний. Указанное применение в качестве лекарственного средства или способ лечения включают системное введение HCV-инфицированным субъектам количества, эффективного для борьбы с состояниями, связанными с HCV и другими патогенными флавивирусами. Следовательно, соединения настоящего изобретения можно использовать для изготовления лекарственного средства, используемого для лечения состояний, связанных с HCV и другими патогенными флавивирусами.
В варианте осуществления изобретение относится к применению соединения формулы (1) или любой его подгруппы, как определено в данном описании, для получения лекарственного средства для лечения или борьбы с инфекцией или заболеванием, связанным с HCV инфекцией у млекопитающих Изобретение также относится к способу лечения флавивирусной инфекции или заболевания, связанного с флавивирусной инфекцией, включающему введение нуждающемуся в этом млекопитающему эффективного количества соединения формулы (1) или его подгруппы, как определено в данном описании.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы (1) или любой его подгруппы, как определено в данном описании, для получения лекарственного средства, которое можно использовать для ингибирования активности HCV у млекопитающего, инфицированного флавивирусом, в частности HCV.
В другом варианте осуществления настоящее изобретение относится к применению соединения формулы (1) или любой его подгруппы, как определено в данном описании, для получения лекарственного средства, которое можно использовать для ингибирования активности HCV у млекопитающего, инфицированного флавивирусом, где указанный HCV ингибируется на стадии его репликации.
Следует отметить, что соединения формулы (1) можно вводить в виде индивидуальных активных ингредиентов или в виде смесей нескольких вариантов осуществления данной формулы. Соединения по изобретению можно использовать в качестве единственных терапевтических агентов или в сочетании с другими терапевтическими агентами.
По существу, комбинация ранее известного противо-HCV соединения, такого как, например, интерферон-α (IFN-α), пэгилированный интерферон-α и/или рибавирин, и соединения по настоящему изобретению может использоваться в качестве лекарственного средства в комбинированной терапии. Термин «комбинированная терапия» относится к продукту, содержащему обязательно (а) соединение по настоящему изобретению и (b) необязательно другое противо-HCV соединение, в качестве комбинированного препарата для совместного, отдельного или последовательного применения для лечения инфекций HCV, в частности для лечения инфекций HCV типа 1. Таким образом для борьбы или лечения инфекций HCV соединения по данному изобретению могут быть введены совместно в сочетании, например, с интерфероном-α (IFN-α), пэгилированным интерфероном-α и/или рибавирином, а также с лекарственными препаратами, основанными на антителах, нацеленных против эпитопов HCV, небольших мешающих РНК (Si РНК), рибозимах, ДНКзимах, антисмысловых РНК, небольшими молекулами, являющимися антагонистами, например протеазы NS3, геликазы NS3 и полимеразы NS5B.
Соответственно настоящее изобретение относится к применению соединения формулы (1) или любой его подгруппы, как определено в данном описании, для получения лекарственного средства, которое можно использовать для ингибирования активности HCV у млекопитающего, инфицированного вирусом HCV, где указанное лекарственное средство используется в комбинированной терапии, при этом указанная комбинированная терапия предпочтительно включает соединение формулы (1) и (пэгилированный) интерферон-α и/или рибавирин и, возможно, противо-HCV соединение.
Специалисту в данной области следует понимать, что соединения формулы (1) могут быть протестированы в клеточных системах репликона HCV на основании данных Lohmann et al. (1999) Science 285:110-113, с дополнительными модификациями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624 (включенными в данное описание путем ссылки), что дополнительно проиллюстрировано в разделе примеров. Данная модель, хотя и не является полной моделью инфекции HCV, широко принята в качестве наиболее надежной и эффективной модели автономной репликации РНК HCV, доступной в настоящее время. Соединения, проявляющие противо-HCV активность в данной клеточной модели, рассматриваются как кандидаты для дальнейшей разработки для лечения инфекций HCV у млекопитающих. Следует понимать, что важно делать различия между соединениями, которые специфически мешают функциям HCV, и теми, которые обладают цитотоксическим или цитостатическим действием в модели репликона HCV и в качестве следствия вызывают снижение РНК HCV или концентрации связанного репортерного фермента. В данной области для оценки клеточной цитотоксичности известны анализы, основанные, например, на активности митохондриальных ферментов с использованием флуорогенных восстановительно-окислительных красителей, таких как резазурин. Кроме того, для оценки неселективного ингибирования активности связанного репортерного гена существуют клеточные контрзаслоны, такие как люцифераза светлячка. Подходящие типы клеток могут быть снабжены стабильной трансфекцией с репортерным геном люциферазы, чья экспрессия зависит от конститутивно активного генного промотора и такие клетки можно использовать в качестве контрзаслона для исключения неселективных ингибиторов.
Все патенты, патентные заявки и статьи, упоминаемые выше и ниже, включены в данное описание путем ссылки.
Примеры.
Следующие примеры предназначены для иллюстрации настоящего изобретения, а не для его ограничения.
Пример 1: активность соединений формулы 91) в анализах репликона HCV
Репортерные анализы стабильного клеточного репликона
Соединения настоящего изобретения исследовали на активность в отношении ингибирования репликации РНК HCV в клеточном анализе. Данный анализ показал, что настоящие соединения проявляют активность против репликонов HCV, функциональных в клеточной культуре. Клеточный анализ основан на бицистронной экспрессионной конструкции, как описано Lohmann et al. (1999) Science vol. 285 pp. 110-113, с модификациями, описанными Krieger et al. (2001) Journal of Virology 75: 4614-4624, при многоцелевой стратегии скрининга. В сущности, метод представлял собой следующее.
В анализе использовали стабильно трансфектированную клеточную линию Huh-7 luc/neo (далее упоминаемую как Huh-Luc). Данная клеточная линия дает прибежище РНК, кодирующей бицистронную конструкцию экспрессии, включающую области NS3-NS5B дикого типа HCV типа Ib, транслируемые из внутреннего сайта входа рибосом (IRES) вируса энцефаломиокардита (EMCV), с предшествующей репортерной частью (FfL-люцифераза) и частью селектируемого маркера (neo®, неомицин фосфотрансфераза). Конструкция была ограничена 5' и 3' NTRs (нетранслируемые области) HCV типа Ib. Постоянная культура клеток репликона в присутствии G418 (neo®) была зависимой от репликации РНК HCV. Для скрининга противовирусных соединений использовали стабильно трансфектированные клетки репликона, которые экспрессировали РНК HCV, которая реплицировала автономно и до высоких уровней кодировала, помимо прочего, люциферазу.
Экспериментальный метод клеточного анализа.
Клетки репликона размещали в 384-луночных планшетах в присутствии тестируемых и контрольных соединений, которые добавляли в различных концентрациях. После инкубирования в течение трех дней измеряли репликацию HCV путем анализирования активности люциферазы (с использованием стандартных субстратов и реагентов анализа люциферазы и микропланшетного визуализатора Perkin Elmer ViewLuxTm ultraHTS). Клетки репликона в контрольных культурах имели высокую экспрессию люциферазы в отсутствие какого-либо ингибитора. Ингибирующую активность соединения на активность люциферазы контролировали на клетках Huh-Luc, получая кривую доза-ответная реакция для каждого тестируемого соединения. Затем рассчитывали значения ЕС50, где значение представляло собой количество соединения, требуемое для снижения на 50% уровня обнаруживаемой активности люциферазы или, более конкретно, способности генетически связанной РНК репликона HCV к репликации.
В следующей таблице в колонке 1 приведен идентификационный номер, в колонке 2 показаны результаты вышеуказанного анализа Huh-7Luc.
HCV (в мкМ)
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕТЕРОБИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ВИРУСА ГЕПАТИТА С (HCV) | 2006 |
|
RU2405783C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ АММОНИЯ, СПОСОБ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ИХ | 2016 |
|
RU2743098C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ | 2006 |
|
RU2441010C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ИНГИБИТОРЫ ТИРОЗИНКИНАЗЫ БРУТОНА | 2013 |
|
RU2619465C2 |
| БИЦИКЛИЧЕСКИЕ ПИРИДИНЫ И ПИРИМИДИНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ Р38 | 2003 |
|
RU2301233C2 |
| АНТАГОНИСТЫ CXCR7 | 2013 |
|
RU2649004C2 |
| Соединения триазоло-пиримидина и их применение | 2019 |
|
RU2802866C2 |
| ПРОИЗВОДНЫЕ ПИПЕРИДИНОВ | 2011 |
|
RU2554353C2 |
| СОЕДИНЕНИЯ-ИНГИБИТОРЫ ФОСФОИНОЗИТИД 3-КИНАЗЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2007 |
|
RU2470936C2 |
Настоящее изобретение относится к области фармацевтики и медицины и касается применения бициклических пиримидинов для получения лекарственных средств, которые могут быть использованы для ингибирования HCV, профилактики или лечения состояний, связанных с HCV. Изобретение расширяет число средств, воздействующих на живой организм, а также предоставляет соединения, которые активны как в отношении вируса HCV дикого типа, так и в отношении обычных резистентных вирусов HCV. 10 з.п. ф-лы, 1 табл.
1. Применение соединения формулы
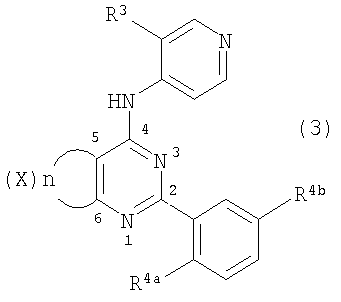
или его фармацевтически приемлемой соли;
где конденсированное кольцо, связывающее в виде мостика положения 5 и 6 пиримидинового кольца, вместе с пиримидиновым кольцом образует группу, выбранную из
 ,
,  ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,  ,
,
 ,
,  ,
,  ,
,
 ,
, 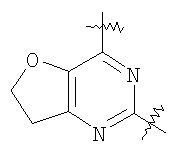 , и
, и  ;
;
где любые из данных групп необязательно могут быть замещены одним или двумя заместителями, выбранными из алкила(1-6С), фенила и бензила;
R3 представляет собой водород, галоген, алкил(1-6С), -CF3, -COR, -CONR2 или -COOR;
каждый из R4a R4b независимо представляет собой водород или галоген;
где каждый R независимо представляет собой водород, гидрокси, амино, моно- или диалкил(1-6С)амино, циклоалкил(3-7С), Het или алкил(1-6С), необязательно замещенный одним или двумя заместителями, выбранными из гидрокси, циклоалкила(3-7С), амино, моно- или диалкил(1-6С)амино и Het;
Het представляет собой группу, выбранную из
 ,
,  ,
,  ,
,  , и
, и  ;
;
где группа Het необязательно может быть замещена одним или двумя заместителями, каждый из которых независимо выбран из группы, состоящей из. алкила(1-6С) и оксо;
для получения лекарственного средства, которое может быть использовано для ингибирования активности HCV, профилактики или лечения состояний, связанных с HCV, у млекопитающих, инфицированных HCV.
2. Применение по п.1, где соединение представляет собой
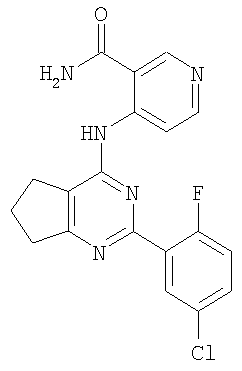
3. Применение по п.1, где соединение представляет собой

4. Применение по п.1, где соединение представляет собой

5. Применение по п.1, где соединение представляет собой
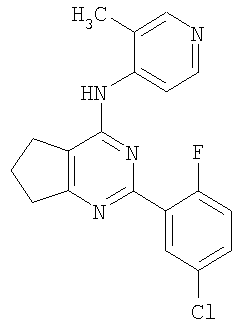
6. Применение по п.1, где соединение представляет собой

7. Применение по п.1, где соединение представляет собой

8. Применение по п.1, где соединение представляет собой

9. Применение по п.1, где соединение представляет собой

10. Применение по п.1, где лекарственное средство используют для профилактики прогрессирования заболевания, приводящего к хроническому гепатиту, фиброзу печени, циррозу, последней стадии заболевания печени, НСС (гепатоклеточная карцинома) и тому подобному у млекопитающего, инфицированного HCV.
11. Применение по п.1, где лекарственное средство используют для лечения хронического гепатита, фиброза печени, цирроза, последней стадии заболевания печени, НСС (гепатоклеточная карцинома) и тому подобного у млекопитающего, инфицированного HCV.
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Способ приготовления мыла | 1923 |
|
SU2004A1 |
| ЛИПОСОМАЛЬНОЕ ПРОТИВОВИРУСНОЕ ЛЕКАРСТВЕННОЕ СРЕДСТВО ДЛЯ ПЕРОРАЛЬНОГО ПРИМЕНЕНИЯ | 1996 |
|
RU2123328C1 |
| Соринсон О.Н., «Вирусные гепатиты», СПб., ТЕЗА, 1996, стр.314. | |||

