Область техники
Настоящее изобретение относится к терапевтическому и/или профилактическому средству против боли, содержащему производное 1-индансульфамида, его соль или его пролекарство.
Предпосылки изобретения
В соответствии с классификацией Международной ассоциации по изучению боли (IASP), "боль" определяется как "неприятный сенсорный и эмоциональный опыт, связанный с текущим или потенциальным повреждением ткани или описанный с точки зрения такого повреждения" (непатентный литературный источник 1).
Как правило, боль классифицируют как острую или хроническую. Острая боль представляет собой боль, которая присутствовала не более трех месяцев. Острая боль начинается внезапно и в большинстве случаев является резкой по своим свойствам. Острая боль может быть вызвана многими явлениями или обстоятельствами, такими как хирургическое вмешательство, переломы костей, стоматологическое лечение, ожоги или порез и т.д. Хроническая боль представляет собой боль, которая длится не менее трех месяцев. Обычная хроническая боль включает головную боль, боль в пояснице, боль, связанную с раковым заболеванием, боль при артрите, невропатическую боль, психогенную боль (боль, возникающую в отсутствие физической причины боли, например, после перенесенного заболевания или травмы). Невропатическая боль, которая также была переведена как нейрогенная боль, представляет собой рефрактерную боль в результате повреждений или заболеваний периферической или центральной соматической сенсорной нервной системы, в том числе диабетической невропатии, невралгии тройничного нерва и постгерпетической невралгии (непатентные литературные источники 2 и 3).
Боль является общей медицинской проблемой, и ослабление боли представляет собой важную терапевтическую цель. Наиболее часто боль лечат с помощью обезболивающих средств. Обезболивающие средства условно разделяют на три категории: (1) опиоидные обезболивающие средства; (2) неопиоидные обезболивающие средства, такие как, противовоспалительные стероиды, ацетаминофен и дипирон; и (3) “вспомогательные обезболивающие средства” (неоднородная группа лекарственных средств, известных как “лекарственные средства, которые не обладают обезболивающим действием в качестве первичного фармакологического действия, но могут усиливать обезболивающий эффект при применении в комбинации с обезболивающими средствами и демонстрируют обезболивающий эффект в выбранных обстоятельствах”).
Поскольку опиоидные обезболивающие средства обеспечивают сильный обезболивающий эффект, воздействуя на опиоидный рецептор в центральной нервной системе, их применение ограничено из-за серьезных побочных реакций и зависимости от лекарственного средства. Хотя неопиоидные обезболивающие средства обладают обезболивающим эффектом, данный эффект является слабым, и могут быть вызваны различные побочные реакции на лекарственное средство. Кроме того, ни одно терапевтическое лекарственное средство, эффективное при хронической боли, такой как невропатическая боль, связанная с диабетической невропатией, невропатией тройничного нерва и опоясывающем герпесе, не было обнаружено, и является необходимой разработка лекарственного средства, эффективного для широкого спектра боли, в том числе острой боли и хронической боли.
Хотя в настоящее время доступны различные обезболивающие средства для лечения боли, все еще существует значительная нереализованная медицинская потребность в лечении боли. Оценки последних отчетов свидетельствуют о том, что достаточный обезболивающий эффект относительно острой боли осуществляется только у одного из четырех пациентов, подвергаемых хирургическому лечению (непатентные литературные источники 4 и 5). Кроме того, недостаточное лечение острой боли может привести к различным симптомам, в том числе тревоге, депрессии, бессоннице, утомлению, сниженному аппетиту, тошноте и рвоте. Кроме того, неослабленная острая боль может прогрессировать в хроническую боль.
С другой стороны, относительно хронической боли ВОЗ приводит оценки, что 20% населения мира имеет некоторую степень хронической боли. Хроническая боль имеет значительное воздействие как на непосредственные затраты на здравоохранение, так и связанные с этим непрямые затраты (например, выплаты по инвалидности, снижение производительности труда). Поскольку достаточное ослабление не может быть достигнуто у примерно 40% пациентов с хронической болью, хроническая боль в настоящее время считается серьезной проблемой общественного здравоохранения (непатентный литературный источник 6).
Эффективное лечение острой боли и хронической боли все еще остается нереализованной медицинской потребностью для многих пациентов. Вследствие этого, чрезвычайно желательной является разработка терапевтического средства, эффективного против острой боли и хронической боли.
В качестве животных моделей острой боли известны модели для оценки непостоянной боли, например, испытание с отдергиванием хвоста, испытание с вздрагиванием/отпрыгиванием, испытание с горячей пластинкой, испытание с защемлением. В качестве модели для острой постоянной боли применяют испытание с формалином (непатентный литературный источник 7).
С другой стороны, в качестве моделей хронической боли, известны модель хронического компрессионного повреждения нерва крысы (модель CCI) и подобные. Классифицированная по причине боли, модель CCI считается моделью заболевания, соответствующей невропатической боли (непатентный литературный источник 8).
В качестве производных сульфамида, обладающих обезболивающим эффектом, известны низкомолекулярные соединения, раскрытые в патентных литературных источниках 1 и 2. Однако производные 1-индансульфамида с обезболивающим эффектом не были известны.
Список использованной литературы
Патентная литература
[0008]
Патентный литературный источник 1: WO2007/095615.
Патентный литературный источник 2: WO2007/075752.
Непатентная литература
[0009]
Непатентный литературный источник 1: International Association for the Study of Pain (1979), “Pain Definitions", Pain 6(3): 247-248.
Непатентный литературный источник 2: “Japanese translation of Neuropathic Pain - A report of Terminology Committee, the Japan Society of Pain Clinicians", Journal of Japan Society of Pain Clinicians (2009), 16(4) 509-514.
Непатентный литературный источник 3: Treede, R.D., Jensen, T.S., Campbell, J. N., et al., (2008), “Neuropathic pain: Redefinition and a grading system for clinical and research purposes", Neurology 70: 1630-1635.
Непатентный литературный источник 4: Phillip, D.M. (2000), “JCAHO: Pain management standards are unveiled", JAMA 284(4): 428-429.
Непатентный литературный источник 5: Wu, C.L. and Raja, S.N. (2011), “Treatment of acute postoperative pain", Lancet 377(9784): 2215-2225.
Непатентный литературный источник 6: Breivik, H., Collett, B., Ventafridda, V., Cohen, R. and Gallacher, D. (2006), “Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment", European Journal of Pain 10 (4): 287-333.
Непатентный литературный источник 7: Dubuisson, D. and Stephen, G. (1977), “The formalin test: A quantitative study of the analgesic effects of morphine, meperidine, and brain stem stimulation in rats and cats", Pain 4(2): 161-174.
Непатентный литературный источник 8: Wang, L.X. and Wang, Z. (2003), “Animal and cellular models of chronic pain" Advanced Drug Delivery Reviews 55(8): 949-965.
Сущность изобретения
Техническая проблема
Целью настоящего изобретения является обеспечение терапевтического и/или профилактического средства против боли, который проявляет обезболивающий эффект в различных животных моделях и может быть применимо при различных типах боли.
Решение проблемы
Авторы настоящего изобретения проводили исследования с использованием испытания с горячей пластинкой на мышах для подтверждения обезболивающего эффекта при острой боли и с использованием модели хронического компрессионного повреждения нерва крысы (CCI) для подтверждения обезболивающего эффекта при хронической боли, соответственно.
В качестве результатов исследования с использованием испытания с горячей пластинкой на мышах авторы настоящего изобретения обнаружили, что производные 1-индансульфамида обладают ингибирующим эффектом при острой боли, вызванной ноцицептивным раздражителем. Кроме того, в качестве результатов исследования с использованием модели хронического компрессионного повреждения нерва крысы (CCI) авторы настоящего изобретения обнаружили, что производные 1-индансульфамида обладают ингибирующим эффектом при хронической боли, вызванной лигированием нерва. Таким образом авторы настоящего изобретения выполняли настоящее изобретение.
В частности, настоящее изобретение относится к:
[1] терапевтическому и/или профилактическому средству против боли, содержащему соединение, выбранное из следующей группы:
(1) N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамид,
(2) (-)-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамид,
(3) N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамид и
(4) N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамид,
или его фармацевтически приемлемую соль;
[2] терапевтическому и/или профилактическому средству в соответствии с [1], где боль представляет собой острую боль или хроническую боль;
[3] терапевтическому и/или профилактическому средству против боли в соответствии с [1], где боль представляет собой невропатическую боль;
[4] терапевтическому и/или профилактическому средству против боли в соответствии с [1], где боль представляет собой диабетическую невропатию, невропатию тройничного нерва или постгерпетическую невралгию;
[5] терапевтическому и/или профилактическому средству против боли в соответствии с любым из [1]-[4], где средство вводят перорально, сублингвально, интраназально, ректально, посредством введения через десны, внутривенно, внутримышечно, интраартикулярно, подкожно, ингаляционно, чрескожно или эпидурально; и
[6] терапевтическому и/или профилактическому средству против боли в соответствии с любым из [1]-[4], где средство вводят перорально, сублингвально, внутривенно, внутримышечно, интраартикулярно, подкожно, чрескожно или эпидурально.
Полезные эффекты изобретения
Средство в соответствии с настоящим изобретением обладает ингибирующим эффектом при острой боли, вызванной ноцицептивными раздражителями в испытании с горячей пластинкой на мышах и при хронической боли, вызванной лигированием нерва в крысиной модели CCI. Кроме того, средство в соответствии с настоящим изобретением проявляет обезболивающий эффект в испытании с формалином на мышах, которое представляет собой животную модель острой постоянной боли. Таким образом, средство по настоящему изобретению можно применять в качестве терапевтического средства и/или профилактического средства при острой боли и хронической боли.
Краткое описание графических материалов
Фиг. 1 представляет собой график, демонстрирующий результаты примера испытания 1, в котором вводят испытуемые соединения 1, 2, 3 и 4.
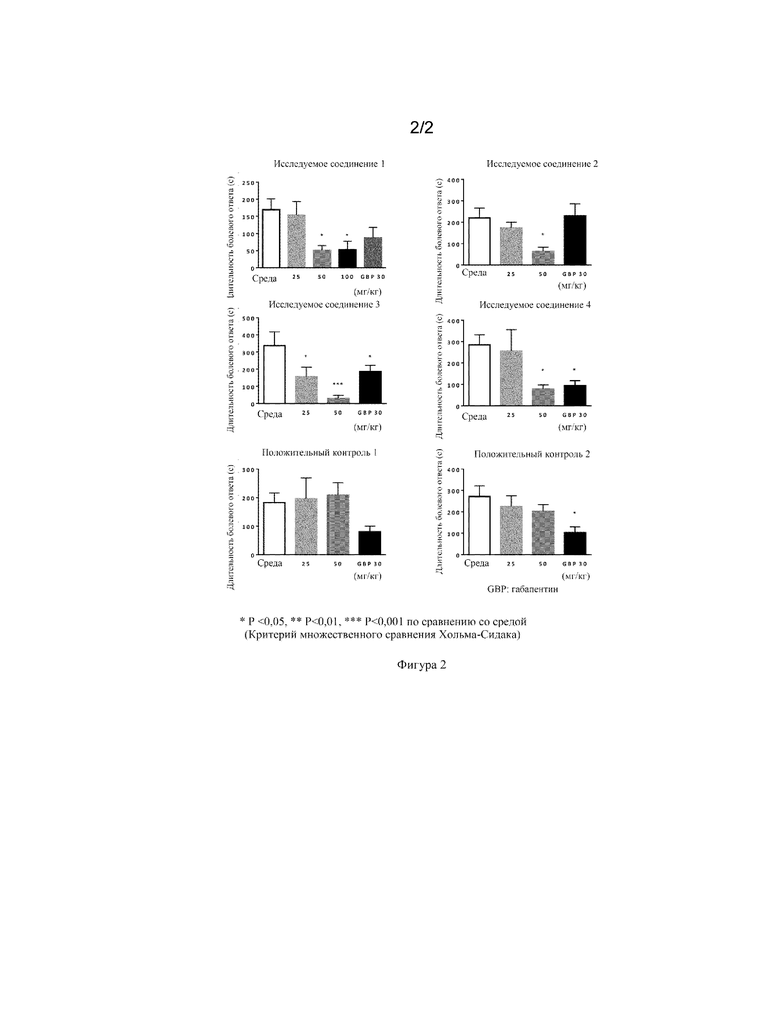
Фиг. 2 представляет собой график, демонстрирующий результаты примера испытания 3, в котором вводят испытуемые соединения 1, 2, 3 и 4.
Описание вариантов осуществления
Настоящее изобретение подробно описано ниже.
Хотя соединение 1-индансульфамида, применяемое в настоящем изобретении, может иметь кристаллические полиморфы, соединение не ограничено каким-либо из полиморфов и может присутствовать в виде единичной кристаллической формы или смеси единичных кристаллических форм. И аморфная форма также включена.
Кроме того, соединение может образовывать фармацевтически приемлемые соли и различные сольваты.
В дальнейшем в данном документе объясняются значения терминов, символов и т. п., описанных в настоящем описании.
Термин “фармацевтически приемлемая соль” в настоящем описании конкретно не ограничен при условии, что она образует соль с соединением и является фармацевтически приемлемой.
"Сольват" означает состояние, в котором растворитель, применяемый в реакции или кристаллизации, вводят в кристалл без образования ковалентной связи с молекулой или ионом соединения. Примерами сольвата являются гидрат, алкоголят (этанолят) и т. п.
Соединения исходного материала, промежуточные соединения и различные реагенты при получении соединения, применяемого в настоящем изобретении, могут образовывать соли или сольваты, при этом все они варьируют в зависимости от исходного материала, применяемого растворителя или т. п., и конкретно не ограничены при условии, что они не ингибируют реакцию. Кроме того, само собой разумеется, что применяемый растворитель варьирует в зависимости от исходного материала, реагента или т. п. и конкретно не ограничен при условии, что он не ингибирует реакцию и растворяет исходный материал до определенной степени. Если соединения получены в виде свободных форм, их можно преобразовать в приемлемые соли или сольваты с помощью обычных способов.
Различные изомеры соединений или промежуточных соединений по настоящему изобретению (например, геометрические изомеры, оптические изомеры, ротамеры, стереоизомеры, таутомеры и т. п.) можно очищать и выделять с использованием общих способов разделения, например, перекристаллизации, образования диастереомерной соли, ферментативного расщепления и различных хроматографических способов (например, тонкослойная хроматография, колоночная хроматография и газовая хроматография).
Соединения или их фармацевтически приемлемые соли, применяемые в настоящем изобретении, могут быть составлены с помощью обычных способов, и примеры дозированных форм включают составы для перорального применения (например, таблетки, гранулы, порошки, капсулы и сиропы), сублингвальные таблетки, инъекционные растворы (для внутривенного введения, внутримышечного введения, подкожного введения, внутрибрюшинного введения, интраартикулярного введения или эпидурального введения) и препараты для наружного применения (например, составы для всасывания через кожу (например, мази и пластыри), назальные препараты, суппозитории и т. п.).
Твердые составы, такие как таблетки, капсулы, гранулы и порошки, как правило, могут содержать от 0,001 до 99,5 вес. %, предпочтительно от 0,01 до 90 вес. % или т. п. соединений или их фармацевтически приемлемых солей, применяемых в настоящем изобретении.
При изготовлении твердых составов для перорального применения таблетки, гранулы, порошки и капсулы могут быть получены путем добавления разбавителей, связующих средств, средств для улучшения распадания, смазывающих средств, красителей и т. п. к соединениям или их фармацевтически приемлемым солям, применяемым в настоящем изобретении, при необходимости, и обработки с помощью обычных способов. Данные составы при необходимости также могут быть покрыты пленкой.
Примеры разбавителей включают лактозу, кукурузный крахмал и микрокристаллическую целлюлозу, примеры связующих средств включают гидроксипропилцеллюлозу и гидроксипропилметилцеллюлозу, и примеры средств для улучшения распадания включают карбоксиметилцеллюлозу кальция и кроскармеллозу натрия.
Примеры смазывающих средств включают стеарат магния и стеарат кальция, и примеры красителей включают оксид титана.
Примеры пленкообразователей включают гидроксипропилцеллюлозу, гидроксипропилметилцеллюлозу и метилцеллюлозу.
Разумеется, не существует ограничения для вспомогательных средств, упомянутых выше.
Для получения инъекционного раствора (для внутривенного введения, внутримышечного введения, подкожного введения, внутрибрюшинного введения, интраартикулярного введения или эпидурального введения) к соединению могут быть добавлены регулятор pH, буферное средство, суспендирующее средство, эмульгаторы, солюбилизирующие средства, антиоксидант, консервант (антисептическое средство), изотоническое средство или т. п., или их фармацевтически приемлемые соли в случае необходимости, и получение проводят с помощью обычного способа. Он также может быть лиофилизирован в виде лиофилизированного препарата для растворения во время применения. Такие инъекционные растворы можно вводить, например, в вену, под кожу или в мышцу.
Примеры регуляторов pH и буферных средств включают органические кислоты или неорганические кислоты и/или их соли, примеры суспендирующих средств включают метилцеллюлозу, полисорбат 80 и карбоксиметилцеллюлозу натрия, примеры эмульгаторов включают полиоксиэтиленовое касторовое масло, гидроксипропилцеллюлозу и лецитин, примеры солюбилизирующих средств включают полисорбат 80 и полиоксиэтилен сорбитан монолаурат, примеры антиоксидантов включают α-токоферол, примеры консервантов включают метилпарагидроксибензоат и этилпарагидроксибензоат, и примеры изотонических средств включают глюкозу, хлорид натрия и маннит, естественно, без их особого ограничения.
Такие инъекционные растворы могут содержать, как правило, от 0,00001 до 99,5 вес. %, предпочтительно от 0,0001 до 90 вес. % соединений или их фармацевтически приемлемых солей, применяемых в настоящем изобретении.
Для получения препарата для наружного применения к соединению по настоящему изобретению может быть добавлен основной исходный материал или его фармацевтически приемлемая соль, и, при необходимости, любой из приведенных выше эмульгаторов, консервантов, регуляторов pH или красителей с помощью обычного способа добавляют для получения, например, препарата для всасывания через кожу (мази, медицинского пластыря или т. п.), капель в нос или суппозитория.
Традиционно применяемое различное сырье для фармацевтических препаратов, препаратов общего воздействия, косметических препаратов и т. п. могут быть применены в качестве основных материалов, и примеры включают сырье, такое как масла животного и растительного происхождения, минеральные масла, сложноэфирные синтетические масла, воски, высшие спирты и очищенная вода.
Такие препараты для наружного применения могут содержать, как правило, от 0,00001 до 99,5 вес. %, предпочтительно от 0,0001 до 90 вес. % соединений или их фармацевтически приемлемых солей, применяемых в настоящем изобретении.
Дозировка лекарственного препарата в соответствии с настоящим изобретением, как правило, варьирует в зависимости от симптома, возраста, пола, веса или т. п., но является приемлемой, если она представляет собой дозу, достаточную для проявления необходимого эффекта. Например, для взрослого применяют дозировку приблизительно от 0,1 до 5000 мг (предпочтительно от 0,5 до 1000 мг, более предпочтительно от 1 до 600 мг) в день в одной дозе в течение одного или нескольких дней или в 2-6 отдельных дозах в течение одного дня.
Примеры
Соединения, применяемые в настоящем изобретении, могут быть получены с помощью способов, описанных, например, в примерах получения ниже, и эффекты соединений могут быть подтверждены с помощью способов, описанных в примерах испытаний ниже. Однако данные способы являются иллюстративными и могут быть изменены без отступления от сущности и объема настоящего изобретения, и настоящее изобретение никоим образом не ограничено следующими конкретными примерами.
Соединения, к которым привязаны названия публикаций или т. п., получали в соответствии с публикациями или т. п.
Все сокращения, применяемые в данном описании, представляют собой общепринятые сокращения, известные специалисту в данной области. Следующие сокращения применены в следующих примерах.
BAST: бис(2-метоксиэтил)аминосеры трифторид
Bn: бензил
Boc: трет-бутоксикарбонил
DCM: дихлорметан
DMF: N,N-диметилформамид
DMSO: диметилсульфоксид
1H-ЯМР: спектрометрия на основе протонного ядерного магнитного резонанса
HPLC: высоэффективная жидкостная хроматография
I.D.: внутренний диаметр
LC-MS: жидкостная хроматография-масс-спектрометрия
m-: мета-
n-: нормальный-
NBS: N-бромсукцинимид
o-: орто-
p-: пара-
PPTS: пиридиния п-толуолсульфонат
Selectfluor™: N-фтор-N’-хлорметил-триэтилендиамин-бис(тетрафторборат)
t-: третичный-
TBS: трет-бутилдиметилсилил
TEA: триэтиламин
THF: тетрагидрофуран
THP: тетрагидропиран
Z(Cbz): бензилоксикарбонил
“Комнатная температура” в следующих примерах получения, как правило, относится к температуре от приблизительно 10°C до приблизительно 35°C. “%” указывает вес. %, если не указано иное. Соотношение растворителей в хроматографии на силикагеле, однако, демонстрирует объемное соотношение растворителей, подлежащих смешиванию.
Химические сдвиги спектров протонного ядерного магнитного резонанса регистрируются в единицах измерения δ (ppm) относительно тетраметилсилана, а константы взаимодействия регистрируются в герцах (Гц). Обозначения мультиплетности сигналов являются следующими: s, синглет; d, дублет; t, триплет; q, квартет; m, мультиплет; brs, широкий синглет.
Оптическое разделение соединений осуществляли посредством системы GILSON HPLC (насос; ведущий насос модели 305, ведомый насос модели 306, головка насоса 50SC, динамический смеситель модели 811D/A, манометрический модуль модели 806, УФ-детектор; детектор УФ/видимой части спектра модели 155, инжектор, коллектор фракций; модель 215, колонка, выбранная из DAICEL CHIRALPAK® AD-H, IA, IB, IC, ID, IE, IF, DAICEL CHIRALCEL®, OD-H, OJ-H, 20 мм I.D.×250 мм). После обнаружения фракций посредством УФ-детектора измеряли оптическое вращение (+/−) с использованием детектора оптического вращения (OR-2090, JASCO, ртутно-ксеноновая (Hg—Xe) лампа, 150 Вт).
Относительно хроматографии, если описана колоночная хроматография на силикагеле, применяли YAMAZEN Parallel Prep (колонка: колонка YAMAZEN Hi-Flash™ (Silicagel), размер: S (16×60 мм), M (20×75 мм), L (26×100 мм), 2L (26×150 мм) или 3L (46×130 мм)), сферический силикагель для хроматографии PSQ 60B™ FUJI SILYSIA CHEMICAL CO., LTD., силикагель для хроматографии BW-300™ Fuji Silysia Chemical Co., Ltd., Wakogel® C-200 (Wako Pure Chemical Industries, Ltd.) или силикагель 60 (70-230 меш) Merck Ltd., Япония.
Кроме того, если присутствует описание с NH колоночной хроматографией на силикагеле, применяли YAMAZEN Parallel Prep (колонка: колонка YAMAZEN Hi-Flash™ (амино), размер: S (16×60 мм), M (20×75 мм), L (26×100 мм), 2L (26×150 мм) или 3L (46×130 мм)), или NH SILICA GEL (200-350 меш) FUJI SILYSIA CHEMICAL CO., LTD.
В номенклатуре соединений в настоящем описании (+)-, (−)-, (R) и (S) обозначают конфигурации энантиомеров (+), (−), (R) и (S), соответственно. И “*” в стерической конфигурации обозначает относительную конфигурацию и, если конкретно не указано, означает определенный энантиомер.
Пример получения 1
Синтез N -[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида
(1) Синтез 2,5,7-трифтор-2,3-дигидро-1H-инден-1-она
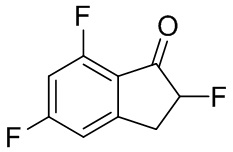
В раствор 5,7-дифтор-1-инданона (№ в реестре CAS 84315-25-3, 500 мг, 2,97 ммоль) в MeOH (20 мл) добавляли SelectfluorTM (1,16 г, 3,27 ммоль) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 2 часов и охлаждали до комнатной температуры. Затем растворитель отгоняли при пониженном давлении. Остаток обрабатывали DCM и нерастворимые вещества отфильтровывали. Затем растворитель отгоняли при пониженном давлении. Остаток растворяли в MeCN (10 мл) и 5 N HCl (5 мл). Раствор перемешивали при комнатной температуре в течение 1 часа и затем концентрировали под вакуумом. Остаток разделяли между AcOEt и H2O. Органический слой промывали солевым раствором, высушивали над MgSO4 и фильтровали. Растворитель концентрировали под вакуумом с получением титульного соединения (547 мг, 2,94 ммоль).
1H- ЯМР (400 МГц, CDCl3): δ ppm 3,11-3,36 (m, 1H) 3,49-3,77 (m, 1H) 5,10-5,40 (m, 1H) 6,82 (td, J=9,0, 1,9 Гц, 1H) 6,90-7,04 (m, 1H).
(2) Синтез 2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-она
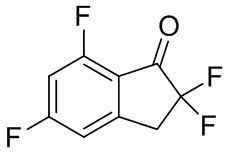
т-Бутилдиметилсилил трифторметансульфонат (1,00 мл, 4,35 ммоль) добавляли в раствор продукта, полученного в примере получения 1-(1) (540 мг, 2,90 ммоль), и TEA (1,21 мл, 8,70 ммоль) в DCM (20 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 5 часов. Затем в реакционную смесь добавляли диэтиловый эфир и насыщенный водный Na2CO3 и слои разделяли. Органический слой последовательно промывали 1N HCl, насыщенным водным Na2CO3 и солевым раствором и высушивали над Na2SO4. Растворитель выпаривали под вакуумом и остаток высушивали при пониженном давлении.
Остаток растворяли в MeCN (20 мл) и добавляли Selectfluor™ (1,13 г, 3,19 ммоль) при комнатной температуре. После перемешивания смесь при той же температуре в течение 11 часов растворитель отгоняли при пониженном давлении. Остаток растворяли в DCM и нерастворимые вещества отфильтровывали. Фильтрат концентрировали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии (колонка Yamazen HI-FLASH™ Silicagel размера L, 20 мл/мин., градиент 10%-50% AcOEt в н-гептане) с получением титульного соединения в виде белого твердого вещества (532 мг, 2,61 ммоль).
1H- ЯМР (400 МГц, CDCl3): δ (ppm) 3,57 (t, J=12,4 Гц, 2H) 6,74-6,94 (m, 1H) 6,95-7,08 (m, 1H).
(3) Синтез 2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-амина
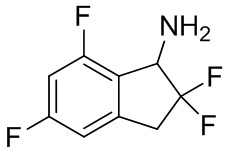
Ацетат аммония (4,27 г, 55,4 ммоль) добавляли в раствор продукта, полученного в примере получения 1-(2) (377 мг, 1,85 ммоль) в изопропаноле (16 мл) при комнатной температуре, и смесь нагревали с обратным холодильником в течение 30 мин. В реакционную смесь добавляли цианоборгидрид натрия (348 мг, 5,54 ммоль) и перемешивали с обратным холодильником в течение 7 часов. После охлаждения до комнатной температуры в реакционную смесь добавляли AcOEt и 2N NaOH и слои разделяли. Органический слой концентрировали под вакуумом. К остатку добавляли воду и разделяли между AcOEt и 1N HCl. Водный слой подщелачивали 2N NaOH и экстрагировали AcOEt. Органический слой высушивали над Na2SO4, выпаривали и высушивали с получением титульного соединения (210 мг, 1,02 ммоль).
ESI-MS; масса/заряд 206 [M+H] +.
1H ЯМР (400 МГц, CDCl3): δ (ppm) 3,26-3,55 (m, 2H) 4,59 (dd, J=13,3, 5,3 Гц, 1H) 6,61-6,86 (m, 2H).
(4) Синтез бензил-N-(2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
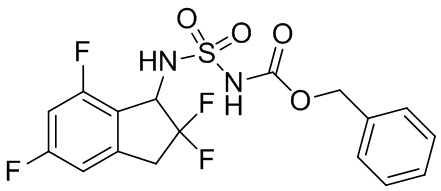
К раствору DCM (10 мл) продукта, полученного в примере получения 1-(3) (200 мг, 0,975 ммоль), добавляли [(бензилокси)карбонил]{[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (№ в реестре CAS 1037211-09-8, 654 мг, 1,95 ммоль, полученный в соответствии со способом, описанным в WO2008083248) и TEA (0,55 мл, 3,90 ммоль) при комнатной температуре. Полученный в результате раствор перемешивали в течение 24 часов с обратным холодильником. После охлаждения до комнатной температуры в реакционную смесь добавляли AcOEt и 1N HCl. Слои разделяли и органический слой высушивали над MgSO4 и выпаривали под вакуумом. Остаток очищали с помощью колоночной хроматографии (Silicagel, 30% AcOEt в н-гептане) с получением титульного соединения в виде белого твердого вещества (316 мг, 0,755 ммоль).
ESI-MS; масса/заряд 441 [M+Na]+.
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 3,25-3,54 (m, 2H) 5,14-5,38 (m, 3H) 5,72 (brs, 1H) 6,72 (t, J=9,4Гц, 1H) 6,79 (d, J=7,8Гц, 1H) 7,30-7,46 (m, 5H) 7,51 (brs, 1H).
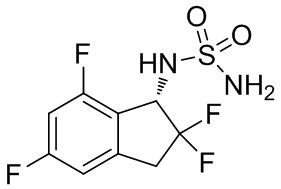
(5) Синтез N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида
В раствор продукта, полученного в примере получения 1-(4) (310 мг, 0,741 ммоль), в MeOH (5 мл) и AcOEt (5 мл) при комнатной температуре добавляли палладированный уголь (10 вес/вес %, 30 мг, 0,028 ммоль). Полученный в результате раствор перемешивали в течение 30 мин. при комнатной температуре в атмосфере H2. Р реакционную смесь добавляли AcOEt и фильтровали через Celite® для удаления палладированного углерода. Фильтрат концентрировали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии (колонка Yamazen HI-FLASH™ Silicagel размера M, 10 мл/мин., градиент 30%-70% AcOEt в н-гептане) с получением титульного соединения в виде рацемата (181 мг, 0,637 ммоль). Оптическое разделение полученного рацемата (180 мг, 0,633 ммоль) проводили с помощью HPLC (CHIRALPAK™ IA, 20 мм I.D.×250 мм, 10 мл/мин., 15% EtOH в гексане) с получением S-формы титульного соединения в виде белого твердого вещества (76 мг, 0,267 ммоль, 98% энантиомерного избытка), который элюировали вторым со временем удержания 44 мин. среди 2 изомеров.
ESI-MS; масса/заряд307 [M+Na] +.
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 3,32-3,60 (m, 2H), 4,70 (brs, 2H), 4,93 (d, J=9,3Гц, 1H), 5,30 (q, J=9,3Гц, 1H), 6,70-6,86 (m, 2H).
Пример получения 2
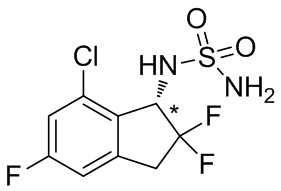
Синтез (−)- N -(7-хлор-2,2,5-трифтор-2,3-дигидро-1 H -инден-1-ил)сульфамида
(1) Синтез 7-хлор-2,5-дифтор-2,3-дигидро-1H-инден-1-она
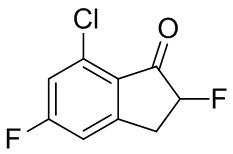
В раствор 7-хлор-5-фтор-1-инданона (№ в реестре CAS 1260008-48-7, 1,08 г, 5,85 ммоль) в MeOH (30 мл) при комнатной температуре добавляли Selectfluor™ (2,49 г, 7,02 ммоль). Смесь нагревали с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении. К остатку добавляли DCM и нерастворимые вещества отфильтровывали. Фильтрат концентрировали под вакуумом. Остаток растворяли в MeCN (20 мл) и 5 N HCl (10 мл) и раствор перемешивали при комнатной температуре в течение 1 часа. После концентрации раствора под вакуумом остаток разделяли между AcOEt и H2O. Органический слой промывали солевым раствором, высушивали над MgSO4 и фильтровали. Фильтрат концентрировали под вакуумом с получением титульного соединения (1,13 г, 5,58 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ(ppm): 3,13 - 3,33 (m, 1H) 3,47-3,71 (m, 1H) 5,25 (ddd,J=51,0, 8,0, 4,5Гц, 1H) 7,07 (dt,J=7,6, 2,0Гц, 1H) 7,14 (dd,J=8,8, 2,0Гц, 1H).
(2) Синтез 7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-она
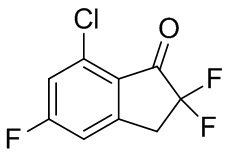
В раствор продукта, полученного в примере получения 2-(1) (1,13 г, 5,58 ммоль), и TEA (3,11 мл, 22,3 ммоль) в DCM (30 мл) при 0°C добавляли т-бутилдиметилсилил трифторметансульфонат (2,56 мл, 11,2 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь разводили диэтиловым эфиром и насыщенным водным Na2CO3 и слои разделяли. Органический слой последовательно промывали 1N HCl, насыщенным водным Na2CO3 и солевым раствором и высушивали над Na2SO4. После фильтрации растворитель выпаривали под вакуумом. Остаток растворяли в MeCN (30 мл) и при комнатной температуре добавляли Selectfluor™ (2,17 г, 6,11 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов и затем полученную в результате смесь выпаривали при пониженном давлении. К остатку добавляли DCM и нерастворимые вещества отфильтровывали. Растворитель выпаривали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии (колонка Yamazen HI-FLASH™ Silicagel размер L, 20 мл/мин., градиент 0%-30% AcOEt в н-гептане) с получением титульного соединения (2) (1,11 г, 5,03 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ(ppm): 3,47-3,63 (m, 2H) 7,06-7,13 (m, 1H) 7,17-7,23 (m, 1H).
(3) Синтез 7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-амина
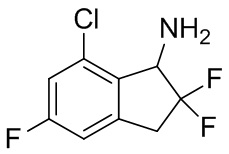
Ацетат аммония (11,5 г, 150 ммоль) добавляли в раствор продукта, полученного в примере получения 2-(2) (1,10 г, 4,98 ммоль), в изопропаноле (40 мл) при комнатной температуре. Смесь нагревали с обратным холодильником в течение 30 мин. В реакционную смесь добавляли цианоборгидрид натрия (940 мг, 15,0 ммоль) и смесь нагревали с обратным холодильником в течение 12 часов. После охлаждения до комнатной температуры реакционную смесь разводили AcOEt и добавляли 2N NaOH. Слои разделяли и органический слой концентрировали под вакуумом. К остатку добавляли воду, AcOEt и 1N HCl и слои разделяли. Водный слой подщелачивали 2N NaOH и экстрагировали AcOEt. Органический слой высушивали над Na2SO4. После фильтрации растворитель выпаривали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии (колонка Yamazen HI-FLASH™ Silicagel размер L, 20 мл/мин., градиент 10%-50% AcOEt в н-гептане) с получением титульного соединения (3) (699 мг, 3,15 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 3,24-3,41 (m, 1H) 3,47-3,65 (m, 1H) 4,50 (d,J=14,6Гц, 1H) 6,85-6,93 (m, 1H) 7,02 (dd,J=9,0, 2,2Гц, 1H).
(4) Синтез т-бутил-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
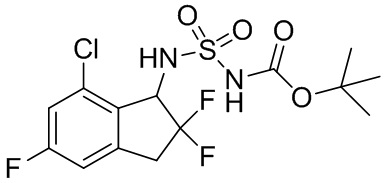
[(т-Бутокси)карбонил] {[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (№ в реестре CAS 872496-91-8, 1,90 г, 6,31 ммоль, полученный в соответствии со способом, описанным в Organic Letters, 3, 2241 (2001)) и TEA (1,76 мл, 12,6 ммоль) добавляли в раствор продукта, полученного в примере получения 2-(3) (699 мг, 3,15 ммоль), в DCM (20 мл) при комнатной температуре. Полученную в результате смесь нагревали в течение 12 часов с обратным холодильником. После охлаждения до комнатной температуры к реакционной смеси добавляли AcOEt и 1N HCl и слои разделяли. Органический слой высушивали над MgSO4. После фильтрации растворитель выпаривали под вакуумом. Остаток очищали с помощью колоночной хроматографии на силикагеле (Silicagel, 30 % AcOEt в н-гептане) с получением титульного соединения (4) (1,08 г, 2,69 ммоль).
1H-ЯМР (400 МГц, CDCl3)
δ (ppm): 1,49 (s, 9H) 3,28-3,55 (m, 2H) 5,07-5,36 (m, 1H) 5,51-5,70 (m, 1H) 6,89 (d, J=7,8Гц, 1H) 7,07 (d, J=9,2Гц, 1H) 7,29 (brs, 1H).
(5) Синтез (-)- N -(7-хлор-2,2,5-трифтор-2,3-дигидро-1 H -инден-1-ил)сульфамида
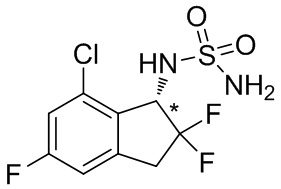
К раствору продукта, полученного в примере получения 2-(4) (1,08 г, 2,69 ммоль), в AcOEt (25 мл) добавляли 4N HCl в AcOEt (26,9 мл, 108 ммоль) и смесь перемешивали при комнатной температуре в течение 5 часов. Растворитель выпаривали под вакуумом и остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ размер L, 20 мл/мин., градиент 30%-70% AcOEt в н-гептане) с получением титульного соединения в виде рацемата (627 мг, 2,09 ммоль). Оптическое разделение полученного рацемата (200 мг, 0,665 ммоль) проводили с помощью HPLC (CHIRALPAK™ IB, 20 мм I.D. x 250 мм, 10 мл/мин., 10% EtOH в н-гексане) с получением титульного соединения (-)-формы (83 мг, 0,276 ммоль, 96% энантиомерного избытка), которое элюировали вторым со временем удержания 49 мин. среди 2 оптических изомеров.
ESI-MS; масса/заряд: 323[M+Na]+
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 3,35-3,64(m,2H), 4,74(brs,2H), 4,86(d, J=8,6Гц,1H), 5,07-5,28(m,1H), 6,83-6,95(m,1H), 7,09(dd, J=8,7,2,3Гц,1H).
Пример получения 3
N -[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1 H -инден-1-ил]сульфамид
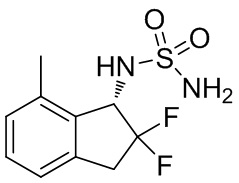
(1) Синтез 2-фтор-7-метил-2,3-дигидро-1H-инден-1-она
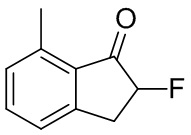
К раствору 7-метил-1-инданона (№ в реестре CAS 39627-61-7, 513 мг, 3,51 ммоль) в MeOH (18 мл) добавляли Selectfluor™ (1,49 г, 4,21 ммоль) при комнатной температуре. Реакционную смесь нагревали в течение 2 часов с обратным холодильником. После охлаждения до комнатной температуры растворитель выпаривали при пониженном давлении. Остаток обрабатывали DCM и нерастворимые вещества отфильтровывали. Фильтрат концентрировали под вакуумом. Остаток растворяли в MeCN (10 мл) и 5 N HCl (5 мл). Раствор перемешивали при комнатной температуре в течение 30 мин. После концентрации раствора под вакуумом остаток разделяли между AcOEt и H2O. Водный слой экстрагировали AcOEt дважды. Объединенные органические слои промывали солевым раствором, высушивали над MgSO4, фильтровали и концентрировали под вакуумом с получением титульного соединения (555 мг, 3,38 ммоль).
1H ЯМР (400 МГц, CDCl3) δ ppm 2,64 (s, 3H), 3,18 (ddd, J= 23,4, 16,8, 4,3 Гц, 1H), 3,57 (ddd, J= 16,8, 7,8, 7,5 Гц, 1H), 5,21 (ddd, J=51,2, 7,8, 4,3 Гц, 1H), 7,17 (d, J =7,4 Гц, 1H), 7,26 (d, J= 7,4 Гц, 1H), 7,51 (t, J= 7,4 Гц, 1H).
(2) Синтез 2,2-дифтор-7-метил-2,3-дигидро-1 H -инден-1-она
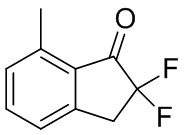
Трет-бутилдиметилсилил трифторметансульфонат (1,55 мл, 6,74 ммоль) добавляли в раствор продукта, полученного в примере получения 3-(1) (555 мг, 3,38 ммоль), и TEA (1,88 мл, 13,49 ммоль) в DCM (30 мл) при 0°C. Смесь перемешивали при комнатной температуре в течение 1,5 часа. Реакцию гасили насыщ. NaHCO3 и слои разделяли. Водный слой экстрагировали DCM. Объединенные органические слои промывали солевым раствором и высушивали над MgSO4. Нерастворимые вещества отфильтровывали и фильтрат концентрировали под вакуумом. Остаток растворяли в MeCN (20 мл) и при комнатной температуре добавляли Selectfluor™ (1,32 г, 3,73 ммоль). После перемешивания реакционной смеси в течение 1 ч. при комнатной температуре растворитель выпаривали при пониженном давлении. Остаток растворяли в DCM и нерастворимые вещества отфильтровывали. Фильтрат концентрировали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ размера L, 20 мл/мин., градиент 15%-20% AcOEt в н-гептане) с получением титульного соединения (563 мг, 3,09 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ ppm 2,66 (s, 3H), 3,51 (t, J= 13,1Гц, 1H), 7,23 (d, J=7,8Гц, 1H), 7,28 (d, J=7,8 Гц, 1H), 7,57 (t, J=7,8 Гц, 1H).
(3) Синтез 2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ола
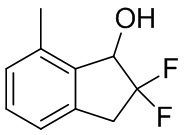
К раствору продукта, полученного с помощью способа, описанного в примере получения 3-(2) (1,09 г, 5,99 ммоль), в MeOH (20 мл) добавляли боргидрид натрия (453 мг, 12,0 ммоль) при 0°C. После перемешивания в течение 45 минут при той же температуре в реакционную смесь добавляли воду и AcOEt и слои разделяли. Отделенный водный слой экстрагировали AcOEt дважды. Объединенный органический слой промывали солевым раствором и высушивали над MgSO4. После фильтрации фильтрат концентрировали и высушивали под вакуумом с получением титульного соединения (1,05 г, 5,72 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,23 (br. s, 1H), 2,43 (s, 3H), 3,26-3,39 (m, 1H), 3,44-3,58 (m, 1H), 5,08-5,15 (m, 1H), 7,07 (d,J= 7,8Гц, 1H), 7,10 (d,J= 7,8Гц, 1H), 7,23-7,26(m, 1H).
(4) Синтез 1-азидо-2,2-дифтор-7-метил-2,3-дигидро-1H-индена
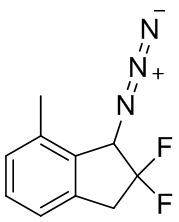
TEA (3,59 мл, 25,8 ммоль) и хлорметансульфонил хлорид (1,02 мл, 11,4 ммоль) добавляли к раствору продукта, полученного в примере получения 3-(3) (1,05 г, 5,72 ммоль), в DCM (25 мл) при 0°C. После перемешивания в течение 2 часов при комнатной температуре реакционную смесь разводили диэтиловым эфиром и гасили насыщ. NaHCO3. Водный слой экстрагировали диэтиловым эфиром в 3 раза. Объединенный органический слой промывали солевым раствором и высушивали над MgSO4. Экстракт фильтровали и концентрировали под вакуумом. Остаток растворяли в DMF (50 мл) и к раствору добавляли азид натрия (753 мг, 11,6 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 2 часов при 70°C. После охлаждения смеси до комнатной температуры добавляли воду и диэтиловый эфир. Слои разделяли и водный слой экстрагировали диэтиловым эфиром 3 раза. Объединенный органический слой промывали водой и солевым раствором и высушивали над MgSO4. Экстракт фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ размера L, 20 мл/мин., 20% AcOEt в н-гептане) с получением титульного соединения (641 мг, 3,06 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ ppm: 2,41 (s, 3H), 3,30-3,43 (m, 1H), 3,51 (ddd,J= 20,3, 16,8, 10,9Гц, 1H), 4,77 (d,J= 13,3Гц, 1H), 7,09 (d,J= 7,8Гц, 1H), 7,14 (d,J= 7,8Гц, 1H), 7,26-7,31 (m, 1H).
(5) Синтез N-(2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамида
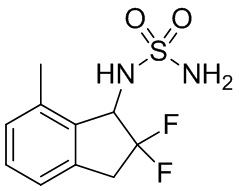
К раствору продукта, полученного в примере получения 3-(4) (641 мг, 3,06 ммоль), в воде (4 мл) и тетрагидрофуране (16 мл) добавляли трифенилфосфин (1,21 г, 4,61 ммоль) при комнатной температуре. Реакционную смесь перемешивали в течение 1 часа при 80°C. После охлаждения до комнатной температуры добавляли AcOEt (20 мл) и 1N HCl (20 мл). Отделенный органический слой экстрагировали 10 мл 1N HCl дважды. Водный слой объединяли и подщелачивали 20 мл 2N NaOH. Слой экстрагировали AcOEt 3 раза и объединенный органический слой промывали солевым раствором и высушивали над MgSO4. Экстракт фильтровали и концентрировали под вакуумом. К раствору остатка и TEA (1,1 мл, 7,89 ммоль) в DCM (26 мл) небольшими порциями при комнатной температуре добавляли сульфамоилхлорид (№ в реестре CAS 7778-42-9, 915 мг, 7,92 ммоль, полученный в соответствии со способом, описанным в US2008/96903). Реакционную смесь затем перемешивали в течение 1 часа при комнатной температуре. К смеси добавляли 20 мл 1N HCl и водный слой экстрагировали DCM дважды. Объединенный органический слой высушивали над MgSO4, фильтровали и концентрировали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ размера L, 20 мл/мин., градиент 50%-65% AcOEt в н-гептане) с получением титульного соединения (348 мг, 1,33 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ ppm: 2,45 (s, 3 H), 3,32-3,56 (m, 2H), 4,70-4,80 (m, 3H), 5,17-5,26 (m, 1H), 7,06 (d,J= 7,4Гц, 1H), 7,12 (d,J= 7,4Гц, 1H), 7,23-7,29 (m, 1H).
(6) Синтез N -[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1 H -инден-1-ил]сульфамида
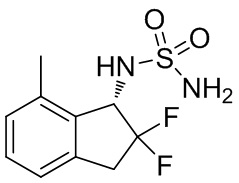
Оптическое разделение рацемата, полученного в примере получения 3-(5) (348 мг, 1,33 ммоль), проводили с помощью HPLC (CHIRALPAK™ IA, I.D. 20 мм x 250 мм, 10 мл/мин., 15% EtOH в н-гексане) с получением титульного соединения (1S)-формы (107 мг, 0,409 ммоль; >99% энантиомерного избытка) в виде белого твердого вещества, которое элюировали вторым со временем удержания 25 мин. среди 2 оптических изомеров.
ESI-MS масса/заряд: 285[M+Na]+
1H-ЯМР (400 МГц, CDCl3) δ ppm: 2,45 (s, 3 H), 3,32-3,56 (m, 2H), 4,70-4,80 (m, 3H), 5,17-5,26 (m, 1H), 7,06 (d, J=7,4Гц, 1H), 7,12 (d, J=7,4Гц, 1H), 7,23-7,29(m, 1H).
Пример получения 4
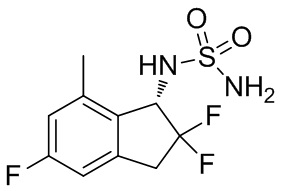
Синтез N -[(1 S )-2,2,5-трифтор-7-метил-2,3-дигидро-1 H -инден-1-ил]сульфамида
(1) Синтез 7-бром-2,2,5-трифтор-2,3-дигидро-1H-инден-1-она
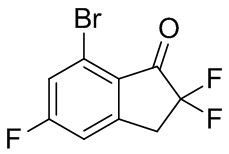
Титульное соединение (5,10 г, 19,2 ммоль) получали из 7-бром-5-фтор-1-инданона (№ в реестре CAS 1260016-95-2, 4,55 г, 19,9 ммоль) с помощью подобного способа, как описано в примере получения 1-(1) и 1-(2).
1H ЯМР (400 МГц, CDCl3) δ (ppm): 3,53(t, J=12,5Гц, 2H), 7,14(d, J=7,6Гц, 1H), 7,41(d, J=8,4Гц, 1H).
(2) Синтез 7-бром-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ола
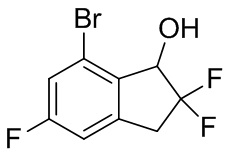
Титульное соединение (4,78 г, 17,9 ммоль) получали из продукта, полученного в примере получения 4-(1) (5,10 г, 19,2 ммоль), с помощью подобного способа, как описано в примере получения 3-(3).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,50(s,1H), 3,38(td,J=17,0,2,7Гц,1H), 3,50-3,69(m,1H), 5,06(dd, J=12,5,4,3Гц,1H), 6,95(dd, J=8,0,1,0Гц,1H), 7,22(dd, J=8,6,2,3Гц,1H).
(3) Синтез 2-[(7-бром-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)окси]тетрагидро-2H-пирана
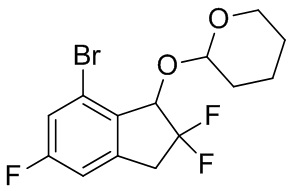
К раствору продукта, полученного в примере получения 4-(2) (2,78 г, 10,4 ммоль), и 3,4-дигидро-2H-пирану (2,18 мл, 23,9 ммоль) в DCM (40 мл) при комнатной температуре добавляли PPTS (52 мг, 0,208 ммоль). И реакционную смесь перемешивали в течение 86 часов при комнатной температуре. Растворитель выпаривали под вакуумом и остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ размер M, 10 мл/мин., градиент 10%-25% AcOEt в н-гептане) с получением титульного соединения (3,42 г, 9,74 ммоль) в виде смеси приблизительно 1:1 рацемических диастеремеров.
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,51-1,84(m,6H), 3,26-3,52(m,1H), 3,52-3,68(m,2H), 4,05-4,19(m,1H), 5,00-5,21 (m,2H), 6,92(d,J=8,2Гц,1H), 7,21(dt,J=8,2,2,6Гц,1H).
(4) Синтез 2-[(2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил)окси]тетрагидро-2H-пирана
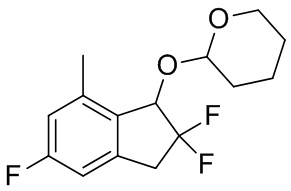
К раствору продукта, полученного в примере получения 4-(3) (1,70 г, 4,84 ммоль), в 1,4-диоксане (10 мл) добавляли по капле 2M раствор диметилцинка в н-гексане (4,84 мл, 9,68 ммоль). Затем после добавления [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладия (II) (177 мг, 0,242 ммоль) реакционную смесь перемешивали в течение 3 часов при 100°C в атмосфере азота. После охлаждения до комнатной температуры добавляли воду и смесь экстрагировали AcOEt. Органический слой промывали солевым раствором и высушивали над безводным Na2SO4. Нерастворимые вещества отфильтровывали и фильтрат выпаривали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ Silicagel размера M, 10 мл/мин., градиент 0%-25% AcOEt в н-гептане) с получением титульного соединения в виде смеси приблизительно 1:1 рацемических диастереомеров (1,06 г, 3,70 ммоль).
ESI-MS; масса/заряд: 309[M+Na]+
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 1,51-1,90(m,6H), 2,35(s,1,5H), 2,43(s,1,5H), 3,19-3,29(m,1H), 3,45-3,64(m,2H), 3,98-4,11(m,1H), 4,88(t,J=3,4Гц,0,5H), 4,95(d,J=5,1Гц,0,5H), 5,01(dd, J=11,6,2,8Гц,0,5H), 5,16(d,J=11,7Гц,0,5H), 6,74-6,81 (m,2H)
(5) Синтез 2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ола
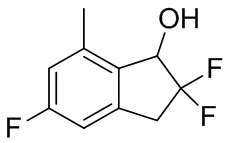
К раствору продукта, полученного в примере получения 4-(4) (1,06 г, 3,70 ммоль), в MeOH (10 мл) добавляли PPTS (46 мг, 0,185 ммоль). И реакционную смесь перемешивали в течение 1 часа при 60°C. После охлаждения до комнатной температуры к реакционной смеси добавляли насыщенный NaHCO3 и смесь экстрагировали AcOEt. Органический слой промывали солевым раствором и высушивали над безводным Na2SO4. Нерастворимые вещества отфильтровывали и фильтрат выпаривали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ размера M, 10 мл/мин., градиент 5%-25% AcOEt в н-гептане) с получением титульного соединения (692 мг, 3,42 ммоль).
1H-ЯМР (400 МГц, CDCl3) δ (ppm): 2,23(dd, J=5,7,2,5Гц,1H), 2,42(s,3H), 3,30(td, J=16,8,5,2Гц,1H), 3,50(td, J=16,8,11,6Гц,1H), 5,05(dd, J=12,1,5,1Гц,1H), 6,77(d, J=8,2Гц,1H), 6,82(d, J=10,2Гц,1H).
(6) Синтез 1-азидо-2,2,5-трифтор-7-метил-2,3-дигидро-1H-индена
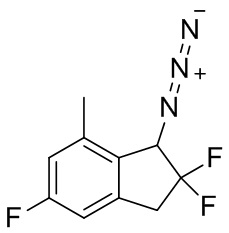
К раствору продукта, полученного в примере получения 4-(5) (692 мг, 3,42 ммоль), и TEA (1,43 мл, 10,3 ммоль) в DCM (10 мл) добавляли хлорметансульфонил хлорид (765 мг, 5,13 ммоль) при 0°C. И реакционную смесь перемешивали в течение 2 часов при комнатной температуре. К реакционной смеси добавляли насыщенный NaHCO3 и смесь экстрагировали диэтиловым эфиром. Органический слой последовательно промывали 1N HCl и солевым раствором, затем высушивали над безводным Na2SO4. После фильтрации фильтрат выпаривали под вакуумом. К раствору остатка в DMF (10 мл) добавляли азид натрия (442 мг, 6,80 ммоль) при комнатной температуре и смесь перемешивали в течение 2 часов при 70°C. После охлаждения до комнатной температуры смесь разделяли между диэтиловым эфиром и H2O. Водный слой экстрагировали диэтиловым эфиром. Объединенный органический слой промывали солевым раствором и высушивали над безводным Na2SO4. После фильтрации фильтрат выпаривали под вакуумом. Остаток очищали с помощью колоночной флеш-хроматографии на силикагеле (колонка Yamazen HI-FLASH™ размера M, 10 мл/мин., градиент 10%-30% AcOEt в н-гептане) с получением титульного соединения (320 мг, 1,41 ммоль).
1H-ЯМР (400 МГц, CDCl3)
δ (ppm): 2,41(s,3H), 3,30-3,56(m,2H), 4,74(d, J=13,3Гц,1H), 6,81(d, J=7,8Гц,1H), 6,86(d, J=9,4Гц,1H).
(7) Синтез 2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-амина
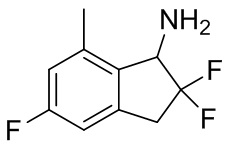
К раствору продукта, полученного в примере получения 4-(6) (320 мг, 1,41 ммоль), в воде (1 мл) и THF (5 мл) добавляли трифенилфосфин (554 мг, 2,11 ммоль) при комнатной температуре и смесь перемешивали в течение 2 часов при 80°C. После охлаждения до комнатной температуры смесь разделяли между AcOEt и 1N HCl. Полученный водный слой подщелачивали 5N NaOH. Водный слой экстрагировали AcOEt 3 раза и объединенный экстракт высушивали над безводным Na2SO4. После фильтрации фильтрат концентрировали под вакуумом с получением титульного соединения (180 мг, 0,895 ммоль).
ESI-MS; масса/заряд: 202 [M+H]+
(8) Синтез трет-бутил-N-(2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил)сульфамоилкарбамата
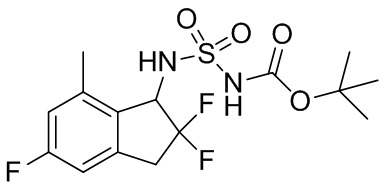
К раствору продукта примера получения 4-(7) (180 мг, 0,895 ммоль) в DCM (10 мл) добавляли [(трет-бутокси)карбонил]{[4-(диметилимино)пиридин-1(4H)-ил]сульфонил}амид (297 мг, 0,984 ммоль) и TEA (0,374 мл, 2,68 ммоль) при комнатной температуре. Полученную в результате смесь нагревали с обратным холодильником в течение 65,5 часов. После охлаждения до комнатной температуры смесь разделяли между AcOEt и 1N HCl. Органический слой высушивали над безводным Na2SO4, фильтровали и выпаривали под вакуумом с получением титульного соединения (257 мг, 0,676 ммоль).
ESI-MS; масса/заряд: 403 [M+Na]+
(9) Синтез N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
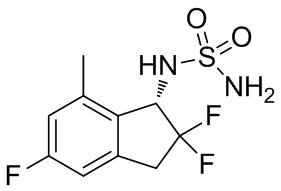
К раствору продукта примера получения 4-(8) (257 мг, 0,676 ммоль) в MeOH (4 мл) добавляли 4N HCl в AcOEt (3,38 мл, 13,5 ммоль) при комнатной температуре и перемешивали в течение 14 часов. Растворитель выпаривали под вакуумом и остаток очищали с помощью колоночной хроматографии на силикагеле (AcOEt) с получением титульного соединения (162 мг, 0,578 ммоль) в виде рацемата. Оптическое разделение полученного рацемата (162 мг, 0,578 ммоль) осуществляли с помощью HPLC (CHIRALPAK™ IF, I.D. 20 мм x 250 мм, 10 мл/мин., 10% EtOH в н-гексане) с получением титульного соединения (S)-изомера (71 мг, 0,253 ммоль; 98% энантиомерный избыток) в виде белого твердого вещества, который элюировали вторым со временем удержания 30 мин. среди 2 оптических изомеров.
ESI-MS; масса/заряд: 303[M+Na]+
1H-ЯМР (400 МГц, DMSO-d6) δ (ppm): 2,37(s,3H), 3,20-3,31(m,1H), 3,38-3,64(m,1H), 4,79(dd, J=14,3,8,8Гц,1H), 6,77(s,2H), 6,90-7,03(m,2H), 7,51(d, J=9,0Гц,1H).
(Справочный пример 1)
Кристаллографический анализ с помощью рентгеновских лучей N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамида
Белое твердое вещество, полученное в примере получения 1-(5), растворяли в MeOH и толуоле и перекристаллизовывали с помощью способа выпаривания растворителя. Рентгеноструктурный анализ проводили с использованием полученного одного кристалла. Результаты сбора данных и кристаллографического анализа кратко изложены в таблице 1, и данные атомных координат приведены в таблице 2. Абсолютную конфигурацию титульного соединения определяли исходя из таких результатов.
[Таблица 1]
число отдельных отражений
[Таблица 2]
(Справочный пример 2)
Кристаллографический анализ с помощью рентгеновских лучей N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
Белые твердые вещества, полученные в примере получения 3-(6), растворяли в EtOH и н-гексане и перекристаллизовывали с помощью температурного градиента с получением микрокристаллов. Микрокристаллы растворяли в Et2O и далее перекристаллизовывали с помощью сопособа выпаривания растворителя. Рентгеноструктурный анализ проводили с использованием полученного одного кристалла. Результаты сбора данных и кристаллографического анализа кратко изложены в таблице 3, и данные атомных координат приведены в таблице 4. Абсолютную конфигурацию титульного соединения определяли исходя из таких результатов.
[Таблица 3]
число отдельных отражений
[Таблица 4]
(Справочный пример 3)
Кристаллографический анализ с помощью рентгеновских лучей N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамида
Белые твердые вещества, полученные в примере получения 4-(9), растворяли в EtOH и перекристаллизовывали посредством способа диффузии пара с использованием толуола в качестве резервуарного раствора. Рентгеноструктурный анализ проводили с использованием одного кристалла, полученного выше.
Результаты сбора данных и кристаллографического анализа кратко изложены в таблице 5, и данные атомных координат приведены в таблице 6. Абсолютную конфигурацию титульного соединения определяли исходя из таких результатов.
[Таблица 5]
число отдельных отражений
[Таблица 6]
Примеры фармакологических испытаний
Авторы настоящего изобретения проводили исследования с использованием испытания с горячей пластинкой на мышах для подтверждения обезболивающего эффекта при острой боли и с использованием модель хронического компрессионного повреждения нерва крысы (CCI) для подтверждения обезболивающего эффекта при хронической боли, соответственно. Кроме того, авторы настоящего изобретения проводили исследование с использованием испытания с формалином на мышах для подтверждения обезболивающего эффекта при острой постоянной боли.
Соединение, раскрытое в примере 1 патентного литературного источника 1, (N-(бензо[b]тиофен-3-илметил)сульфамид), и соединение, раскрытое в примере 7 патентного литературного источника 2, ((2S)-(-)-N-(6-хлор-2,3-дигидро-бензо[1,4]диоксин-2-илметил)-сульфамид), получали в соответствии с патентными источниками 1 и 2 и применяли в качестве эталонных соединений (1 и 2), соответственно.
(Пример испытания 1)
Испытание с горячей пластинкой на мышах
Испытание с горячей пластинкой на мышах проводили для определения эффективности при острой боли. В данной модели скрытое состояние ответа на первую боль записывали как меру чувствительности к боли (Malmberg, A. and Yaksh, T., Pain. 1995, 60: 83-90).
<Способы>
В данном эксперименте применяли самцов мышей C57BL/6NCrlCrlj (Charles River, Япония) в возрасте 6 недель (n = 4-10, для каждой обработки). Горячую пластину (модель MK 350, Muromachi Kikai Co. Ltd.) устанавливали на 53°C. Для перорального введения испытуемое соединение суспендировали в водном растворе, содержащем 0,45% метилцеллюлозы/4,5% кремофора/10% диметилсульфоксида, с получением суспензии с объемом дозирования 10 мл/кг и суспензию вводили перорально за 60 минут до испытания с горячей пластинкой. В качестве положительного контроля применяли морфин и в качестве отрицательного контроля применяли отдельный смешанный растворитель (среду), не содержащий испытуемое соединение.
Каждую мышь помещали на горячую пластину и немедленно начинали измерение с секундомером. Измеряли время скрытого состояния до первого болевого ответа (облизывание лапы или прыжок для побега). Время окончания устанавливали на 30 секунд во избежание излишнего повреждения тканей и мышь немедленно удаляли с горячей пластины после измерения. Все данные выражены как среднее±SEM. Все статистические анализы проводили с использованием критерия множественного сравнения Хольма-Сидака. p-Значения менее 0,05 оценивали как статистически значимые в данном эксперименте.
<Результаты>
Результаты испытания с горячей пластинкой на мышах приведены на фигуре 1. Значения времени скрытого состояния до первого болевого ответа были значительно увеличенными при предварительном введении испытуемых соединений 1, 2, 3 и 4. Данные результаты демонстрируют обезболивающий эффект испытуемых соединений при острой боли.
(Пример испытания 2)
Модель хронического компрессионного повреждения нерва крысы (CCI)
Модель хронического компрессионного повреждения нерва крысы (CCI) применяли для определения эффективности при хронической боли. В данной модели тактильную аллодинию, характерный симптом у пациентов с хронической болью, можно оценивать с использованием порога ответа на механическую стимуляцию нитями фон Фрея в виде индекса (Bennett, G. J. и Xie, Y. K., (Pain 1988; 33: p87-107).
<Способы>
В данном эксперименте использовали самцов крыс SD (Charles River, Япония) в возрасте 6 недель (n=4, каждую обработку проводили в общей сложности дважды). Модель хронического компрессионного повреждения нерва крысы (CCI) получали в соответствии со способом Bennett и Xie, выше.
Общий седалищный нерв подвергали отслаиванию на уровне середины бедра через двуглавую мышцу бедра под анестезией изофлураном. Четыре лигатуры (4-0 шелк) неплотно накладывали вокруг седалищного нерва на расстоянии приблизительно 1 мм. Двуглавую мышцу бедра и кожу зашивали.
На 14 день после хирургического вмешательства оценивали эффективность соединений при тактильной аллодинии. Животных помещали в клетки с дном из проволочной сетки на 30 минут перед началом эксперимента для акклиматизации. Испытуемое соединение суспендировали в водном растворе, содержащем 0,45% метилцеллюлозы/4,5% кремофора/10% диметилсульфоксида, с получением суспензии с объемом дозирования 10 мл/кг и суспензию вводили перорально. Отдельный указанный выше смешанный растворитель (среду), не содержащий испытуемое соединение, применяли в качестве отрицательного контроля. Порог ответа на механическую стимуляцию измеряли посредством применения нитей фон Фрея при силах изгибания (0,4, 0,6, 1, 2, 4, 6, 8, и 15 г) к подошвенной поверхности задней лапы в соответствии с Chaplan et al. (J Neuroscience Methods 1994; 53(1): p.55-63). Нити фон Фрея подносили к лапе в течение 6 секунд для оценки реакции избегания. 50% порог ответа определяли в соответствии со способом "вверх и вниз" Dixon (Annual Review of Pharmacology and Toxicology 1980; 20: p.441-462). Измерение проводили перед введением и в моменты времени 30, 90 и 180 минут после введения. Все данные выражены как среднее±SEM. Все статистические анализы проводили с использованием двусторонных повторных измерений ANOVA с последующим тестом Даннета. p-Значения менее 0,05 оценивали как статистически значимые в данном эксперименте.
<Результаты>
Результаты тактильной аллодинии в модели CCI приведены в таблицах 7, 8, 9 и 10. Испытуемые соединения 1 и 2 проявляли статистически значимый обезболивающий эффект при 50 мг/кг.
[Таблица 7]
50% порог ответа в испытании с нитями фон Фрея
10 мг/кг
50 мг/кг
**P < 0,01 по сравнению со средой (тест Даннета)
[Таблица 8]
50% порог ответа в испытании с нитями фон Фрея
10 мг/кг
50 мг/кг
** P < 0,01 по сравнению со средой (тест Даннета)
[Таблица 9]
50% порог ответа в испытании с нитями фон Фрея
10 мг/кг
50 мг/кг
10 мг/кг
50 мг/кг
[Таблица 10]
50% порог ответа в испытании с нитями фон Фрея
10 мг/кг
50 мг/кг
10 мг/кг
50 мг/кг
* P < 0,05, **P < 0,01 по сравнению со средой (тест Даннета)
Как показано в таблицах 7 и 8, порог ответа на механическую стимуляцию увеличивался примерно дозозависимым способом при предварительном введении испытуемых соединений 1 и 2.
Данные результаты демонстрируют обезболивающий эффект испытуемых соединений на животной модели хронической боли.
(Пример испытания 3)
Испытание с формалином
Соединения исследовали во второй фазе испытания с формалином для определения обезболивающего эффекта при острой постоянной боли. В данной модели продолжительность ответов, вызванных болью (такое поведение, как облизывание или кусание лапы с инъецированным формалином) применяли в качестве индекса оценки (Dubuisson and Dennis (Pain, 4: p.161-174 (1977)).
<Способы>
В данном эксперименте применяли самцов мышей CD1 (ICR) (Charles River, Япония) в возрасте от 5 до 6 недель (n = 6-16, для каждой обработки). Ответы, вызванные болью, измеряли с помощью автоматического устройства для анализа поведения (MicroAct (Neuroscience, Inc.)) в соответствии с определением изменений магнитного поля. За день до оценки болевого ответа небольшой магнит для определения изменений в магнитном поле помещали в тыл стопы задней конечности под анестезией изофлераном.
Для перорального введения испытуемое соединение суспендировали в растворе, содержащем 0,45% метилцеллюлозы/4,5% кремофора/10% диметилсульфоксида, с получением суспензии с объемом дозирования 10 мл/кг и суспензию вводили перорально. Отдельный смешанный растворитель (среду), как указано выше, не содержащий испытуемое соединение, применяли в качестве отрицательного контроля. Габапентин вводили внутрибрюшинно в качестве положительного контроля. 2,5% раствор формалина готовили путем разведения раствора формальдегида (Wako Pure Chemical Industries) физиологическим раствором (Otsuka Pharmaceutical). Через промежуток времени от 40 минут до часа после введения испытуемого соединения или положительного контроля 10 мкл 2,5% раствора формалина вводили подкожно в левую заднюю подушечку стопы. Мышь помещали в камеру для наблюдения сразу после введения для измерения.
Для оценки обезболивающего эффекта во второй фазе измеряли длительность показателей поведения, таких как облизывание или кусание лапы, с помощью автоматического устройства для анализа поведения через 15-45 минут после введения 2,5% раствора формалина.
Все данные выражены как среднее±SEM. Все статистические анализы проводили с использованием критерия множественного сравнения Хольма-Сидака. p-Значения менее 0,05 оценивали как статистически значимые в данном эксперименте.
<Результаты>
Результаты испытания с формалином приведены на фигуре 2. Как показано на фигуре 2, длительность болевого ответа была значительно сниженной с точки зрения статистики при предварительном введении испытуемых соединений 1, 2, 3 и 4. Данные результаты демонстрируют обезболивающий эффект соединений в животной модели острой постоянной боли.
| название | год | авторы | номер документа |
|---|---|---|---|
| НОВОЕ ИНДАНСУЛЬФАМИДНОЕ ПРОИЗВОДНОЕ | 2013 |
|
RU2619937C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2007 |
|
RU2456278C2 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
| СП0СОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ ДИГИДРОИНДЕНАМИДА, ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИИ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ | 2009 |
|
RU2528408C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОАРИЛОМ ПИРИДИНЫ И СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2756743C2 |
| АМИДЫ КОНДЕНСИРОВАННОГО ПИПЕРИДИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ ИОННЫХ КАНАЛОВ | 2014 |
|
RU2741810C2 |
| СОЕДИНЕНИЯ, КОТОРЫЕ УСИЛИВАЮТ РЕЦЕПТОР ГЛУТАМАТА, И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2005 |
|
RU2403242C2 |
| ПИРАНОДИПИРИДИНОВОЕ СОЕДИНЕНИЕ | 2016 |
|
RU2743998C2 |
| ТРИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ИНГИБИТОР JAK | 2015 |
|
RU2674262C2 |
| ПИРИМИДИНОВОЕ СОЕДИНЕНИЕ И СПОСОБ ЕГО ПОЛУЧЕНИЯ | 2020 |
|
RU2790017C1 |

Изобретение относится к медицине, в частности к терапевтическому и/или профилактическому средству против острой и/или хронической боли. Терапевтическое и/или профилактическое средство содержит в качестве активного агента соединение на основе 1-индансульфамидов, выбранное из группы: N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамид, (-)-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамид, N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамид N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамид, или его фармацевтически приемлемую соль. Осуществление изобретения обеспечивает обезболивающий эффект в различных животных моделях и может быть применимо при различных типах боли. 4 з.п. ф-лы, 10 табл., 9 пр., 2 ил.
1.Терапевтическое и/или профилактическое средство против острой и/или хронической боли, содержащее соединение, выбранное из следующей группы:
(1) N-[(1S)-2,2,5,7-тетрафтор-2,3-дигидро-1H-инден-1-ил]сульфамид,
(2) (-)-N-(7-хлор-2,2,5-трифтор-2,3-дигидро-1H-инден-1-ил)сульфамид,
(3) N-[(1S)-2,2-дифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамид и
(4) N-[(1S)-2,2,5-трифтор-7-метил-2,3-дигидро-1H-инден-1-ил]сульфамид,
или его фармацевтически приемлемую соль.
2.Терапевтическое и/или профилактическое средство против боли по п. 1, где боль представляет собой невропатическую боль.
3.Терапевтическое и/или профилактическое средство против боли по п. 1, где боль представляет собой боль в результате диабетической невропатии, невропатии тройничного нерва или постгерпетической невралгии.
4.Терапевтическое и/или профилактическое средство против боли по любому из пп. 1-3, где средство вводят перорально, сублингвально, интраназально, ректально, посредством введения в десны, внутривенно, внутримышечно, интраартикулярно, подкожно, ингаляционно, чрескожно или эпидурально.
5.Терапевтическое и/или профилактическое средство против боли по любому из пп. 1-3, где средство вводят перорально, сублингвально, внутривенно, внутримышечно, интраартикулярно, подкожно, чрескожно или эпидурально.
| US 2008096903 A1, 24.04.2008 | |||
| US 2007191452 A1, 16.08.2007 | |||
| WO 2007075752 A1, 05.07.2007 | |||
| RU 2012110908 A, 27.09.2013. |

