Эта заявка испрашивает приоритет китайской заявки на патент №201610802100,5 под названием «Новый ингибитор рецептора фактора роста фибробластов и его применение», поданной в Патентное ведомство Китая 1 сентября 2016 г., и китайской заявки на патент №201710351160,4 под названием «Ингибиторы рецептора фактора роста фибробластов и их применение», поданной в Патентное ведомство Китая 18 мая 2017 г., которые полностью включены в настоящее описание посредством ссылки.
Область техники
Настоящее изобретение относится к области медицинской технологии и касается необратимого ингибитора рецептора фактора роста фибробластов (FGFR) или его фармацевтически приемлемой соли, стереоизомера, и их применения.
Предшествующий уровень техники
Рецепторы тирозинкиназы играют важную роль в ангиогенезе, пролиферации, миграции и инфильтрации опухолевых клеток. Более 100 лекарств-ингибиторов тирозинкиназы были выпущены на рынок или последовательно вступали в фазы клинических испытаний. Эти низкомолекулярные ингибиторы тирозинкиназы (TKI) функционируют способом обратимого ингибирования, что имеет некоторые недостатки: 1) селективность не является достаточно хорошей, 2) эффективность не является достаточно сильной и продолжительной, 3) легко вызывается лекарственная устойчивость. Поэтому ученым рекомендуется сосредоточить свои исследования на разработке необратимых TKI.
Необратимые TKI, как правило, основаны на основной структуре обратимых TKI с электрофильной функциональной группой, присоединенной в соответствующем положении. Электрофильная функциональная группа может образовывать ковалентную связь посредством электрофильной реакции с остатком цистеина (обогащенной электронами нуклеофильной структурой) вблизи АТФ-связывающего домена тирозинкиназы, тем самым необратимо ингибируя киназную активность. По сравнению с обратимыми TKI необратимые TKI обладают многими уникальными преимуществами: 1) необратимые TKI функционируют в режиме постоянной инактивации, и этот способ ингибирования активности фермента делает их эффект более сильным и продолжительным, тем самым сохраняя эффективность, даже если молекула лекарства полностью удаляется из кровеносной системы; 2) развитие лекарственной устойчивости снижается или предотвращается из-за отсутствия конкуренции АТФ за связывание TKI с киназой, что снижает вероятность мутации киназы; и 3) селективность необратимых TKI очень высока из-за того, что электрофильная функциональная группа в молекулярной структуре TKI селективно реагирует с тиольной группой в остатке цистеина. Исходя из вышеизложенных характеристик, разработка необратимых TKI постепенно становится горячей точкой для исследований и разработок.
Рецептор фактора роста фибробластов (FGFR) является важным членом семейства рецепторов тирозинкиназы. FGFR включает четыре члена, а именно FGFR-1, FGFR-2, FGFR-3 и FGFR-4. В основном, это одноцепочечная молекула гликопротеина с массой молекулы в диапазоне от 110 кДа до 150 кДа, включающая внеклеточную область, трансмембранную область и внутриклеточную область. В нормальных физиологических условиях FGFR связывается со своим лигандом, фактором роста фибробластов (FGF), что приводит к его димеризации и фосфорилированию, тем самым активируя нисходящий сигнальный путь, такой как путь JAK/STAT, путь фосфолипазы С, путь фосфатидилинозитол-3-киназы PI3K и сигнальный путь MAPK (митоген-активируемой протеинкиназы), которые играют важную роль в росте опухоли и ангиогенезе. Аномально высокая экспрессия FGFR тесно связана с развитием различных опухолей, таких как рак легких, рак печени, глиома, рабдомиосаркома и меланома.
В настоящее время не существует лекарств, которые являются необратимыми ингибиторами FGFR, особенно необратимыми ингибиторами с высокой селективностью в отношении пан-FGFR.
В WO 2015061572 А1 раскрыты соединения в качестве ингибиторов FGFR-4, фармацевтические композиции, содержащие эти соединения, и применение этих соединений и фармацевтических композиций.
Сущность изобретения
Задачей настоящего изобретения является создание нового необратимого ингибитора пан-FGFR, обладающего высокой селективностью. Такое соединение обладает мощной ингибирующей активностью в отношении пан-FGFR и обеспечивает возможности для лечения заболеваний, патологически опосредованных пан-FGFR. Настоящее изобретение также предусматривает применение вышеуказанного ингибитора FGFR.
Технические решения, принятые настоящим изобретением, заключаются в следующем.
Решение 1: Необратимый ингибитор рецептора фактора роста фибробластов (FGFR), представленный общей формулой (I), или его фармацевтически приемлемая соль или стереоизомер:
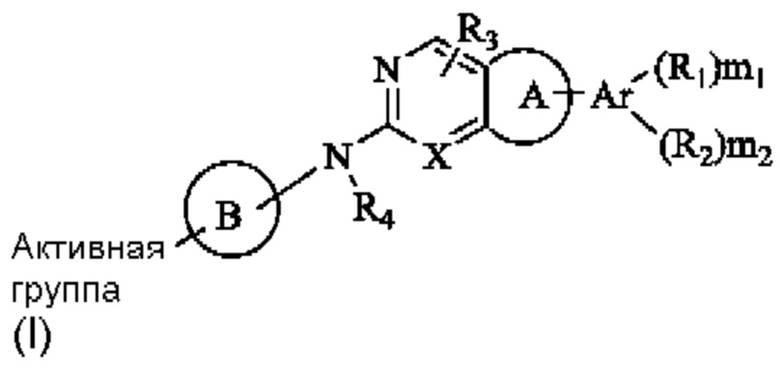
где,
R1 и R2 каждый независимо выбран из группы, состоящей из водорода, гидрокси, амино, циано, нитро, галогена, карбокси, С1-6 алкила C1-6 алкокси, С1-6 алкиламино, (C1-6 алкил)2амино, галоС1-6 алкила, галоС1-6 алкокси, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила, С1-6 алкилкарбониламино, C1-6 алкил-замещенного 3-8-членного циклоалкила, С1-6 алкил-замещенного 3-8-членного гетероциклила, альтернативно, R1 и R2 вместе с двумя атомами на ароматическом кольце или гетероароматическом кольце, к которому они присоединены соответственно, могут образовывать 3-8-членный циклоалкил, 3-8-членный гетероциклил, 6-14-членный арил или 5-10-членный гетероарил, и атом S в любом кольце может быть необязательно окислен до S(O) или S(O)2, и атом углерода в любом кольце может быть необязательно окислен до С(О);
R3 и R4 каждый независимо выбран из группы, состоящей из водорода, гидрокси, амино, циано, нитро, галогена, карбокси, С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галоС1-6 алкила, галоС1-6 алкокси, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила, С1-6 алкилкарбониламино, С1-6 алкиламинокарбонила, 3-8-членного циклоалкила, 3-8-членного гетероциклила, 6-14-членного арила или 5-10-членного гетероарила, С1-6 алкил-замещенного 3-8-членного циклоалкила, С1-6 алкил-замещенного 3-8-членного гетероциклила, С1-6 алкил-замещенного 6-14-членного арила или С1-6 алкил-замещенного 5-10-членного гетероарила;
Ar представляет собой 6-14-членное ароматическое кольцо или 5-10-членный гетероарил, возможно содержащий 0-3 атома О, S и/или N;
Кольцо А выбрано из группы, состоящей из 3-8-членного циклоалкила, 3-8-членного гетероциклила, 6-14-членного арила и 5-10-членного гетероарила, имеющего 0-3 атома О, S и/или N, возможно замещенного 1-3 группами R5, где атом S в любом кольце может быть необязательно окислен до S(O) или S(O)2, и атом углерода в любом кольце может быть необязательно окислен до С(О);
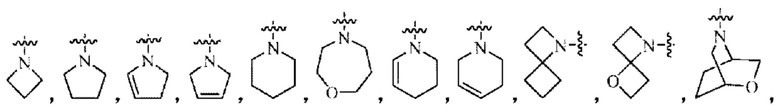
Кольцо В представляет собой 3-10-членный насыщенный или ненасыщенный гетероциклил, содержащий по меньшей мере один гетероатом N, или 5-6-членный N-содержащий гетероарил, возможно замещенный 1-3 группами R6, и атом N на кольце В непосредственно связан с Активной группой, где любой атом S в кольце В может быть необязательно окислен до S(O) или S(O)2, и любой атом углерода в кольце В может быть необязательно окислен до С(О);
X представляет собой R7 или N;
R5, R6, и R7 каждый независимо выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, амино, карбокси, циано, нитро или галогена,
(iii) С1-6 алкила, С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, галоС1-6 алкила, галоС1-6 алкокси, С2-8 алкенила, С2-8 алкинила, С1-6 алкилсульфонила или С1-6 алкилтио, возможно замещенного гидрокси, амино, карбокси, циано, нитро, галогеном, С1-6 алкилом, С1-6 алкокси, С1-6 алкокси С1-6 алкокси, С1-6 алкиламино, (С1-6 алкил)2 амино, С1-6 алкилкарбониламино, С1-6 алкилсульфониламино или 3-8-членным гетероциклилом, где 3-8-членный гетероциклил может быть необязательно замещен гидрокси, амино, карбокси, циано, нитро, галогеном, С1-6 алкилом, С1-6 алкокси, С1-6 алкиламино или (С1-6 алкил)2 амино,
(iv) 3-8-членного циклоалкила или 3-8-членного гетероциклил а, возможно замещенного гидрокси, амино, карбокси, циано, нитро, галогеном, С1-6 алкилом, С1-6 алкокси, С1-6 алкиламино или (С1-6 алкил)2 амино, и
(v) аминокарбонила, цианокарбонила, С1-6 алкилкарбонила, С1-6 алкиламинокарбонила, (С1-6 алкил)2 аминокарбонила, С1-6 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила;
m1 и m2 равны 1, 2 или 3, и сумма m1 и m2 меньше или равна 5; и
«Активная группа» относится к группе, которая способна образовывать ковалентную связь с нуклеофилом.
Решение 2: Соединение согласно Решению 1 или его фармацевтически приемлемая соль или стереоизомер,
где,
R1 независимо выбран из группы, состоящей из водорода, галогена и гидрокси;
R2 независимо выбран из группы, состоящей из водорода, галогена, гидрокси, C1-4 алкила, C1-4 алкокси, циано, галоС1-4 алкила, и галоС1-4 алкокси;
Ar представляет собой 6-14-членное ароматическое кольцо или 5-6-членный гетероарил, возможно содержащий 0-3 атома О, S и/или N;
m1 и m2 равны 1, 2 или 3, и сумма m1 и m2 меньше или равна 5.
Решение 3: Соединение согласно Решению 2 или его фармацевтически приемлемая соль или стереоизомер,
где,
R1 независимо выбран из группы, состоящей из водорода, галогена, и гидрокси;
R2 независимо выбран из группы, состоящей из водорода, галогена, гидрокси, C1-4 алкила, C1-4 алкокси, циано, галоС1-4 алкила и галоС1-4 алкокси;
R3 и R4 каждый независимо выбран из группы, состоящей из водорода, гидрокси, амино, циано, нитро, галогена, карбокси, C1-4 алкила, C1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, галоС1-4 алкила, галоС1-4 алкокси, С2-4 алкенила, С2-4 алкинила, C1-4 алкилсульфонила и С1-4 алкилкарбониламино;
Ar представляет собой фенил;
Кольцо А представляет собой фенил, возможно замещенный 1-3 группами R5; Кольцо В представляет собой 4-10-членный насыщенный или ненасыщенный гетероциклил, содержащий по меньшей мере один N гетероатом, возможно замещенный 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой;
X представляет собой CR7 или N;
R5 и R7 каждый независимо выбран из группы, состоящей из водорода, гидрокси, амино, карбокси, циано, нитро, галогена, С1-4 алкила и C1-4 алкокси;
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, амино, карбокси, циано, нитро или галогена,
(iii) C1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, галоС1-4 алкила, галоС1-4 алкокси, С2-4 алкенила, С2-4 алкинила, С1-4 алкилсульфонила или C1-4 алкилтио, возможно замещенного гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкокси С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино или 3-8-членным гетероциклилом, где 3-8-членный гетероциклил может быть необязательно замещен гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкиламино или (С1-4 алкил)2 амино,
(iv) 3-8-членного циклоалкила или 3-8-членного гетероциклил а, возможно замещенного гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкиламино или (С1-4 алкил)2 амино, и
(v) аминокарбонила, цианокарбонила, С1-4 алкилкарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2 аминокарбонила, С1-4 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила;
m1 и m2 равны 1, 2 или 3, и сумма m1 и m2 меньше или равна 5;
«Активная группа» относится к группе, которая способна образовывать ковалентную связь с нуклеофилом.
Решение 4: Соединение формулы (I) согласно любому из Решений 1-3 или его фармацевтически приемлемая соль или стереоизомер, имеющее структуру, показанную общей формулой (II):
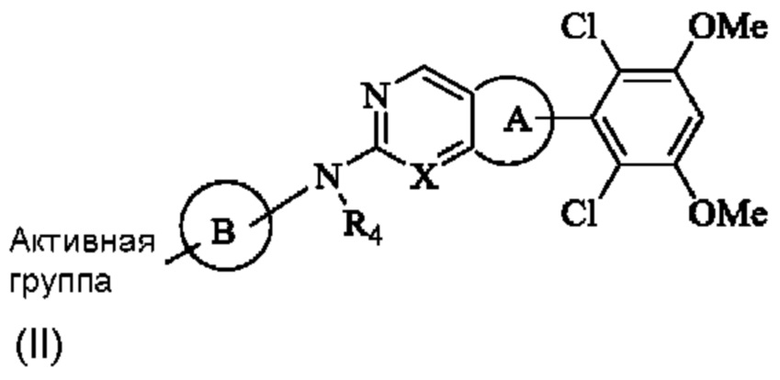
R4 представляет собой Н или С1-4 алкил;
Кольцо А представляет собой фенил;
Кольцо В выбрано из группы, состоящей из 4-6-членного насыщенного или ненасыщенного моногетероциклила или 6-10-членного насыщенного или ненасыщенного конденсированного гетероциклила, содержащего по меньшей мере один N гетероатом, возможно замещенного 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой;
X представляет собой N;
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, амино, карбокси, циано, нитро или галогена,
(iii) С1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, галоС1-4 алкила, галоС1-4 алкокси, С2-4 алкенила, С2-4 алкинила, С1-4 алкилсульфонила или С1-4 алкилтио, возможно замещенного гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкокси С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино или 3-8-членным гетероциклилом, где 3-8-членный гетероциклил может быть необязательно замещен гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкиламино или (С1-4 алкил)2 амино, и
(iv) аминокарбонила, цианокарбонила, С1-4 алкилкарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2 аминокарбонила, С1-4 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила;
Активная группа выбрана из группы, состоящей из
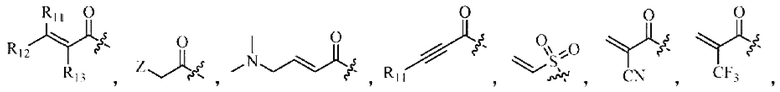
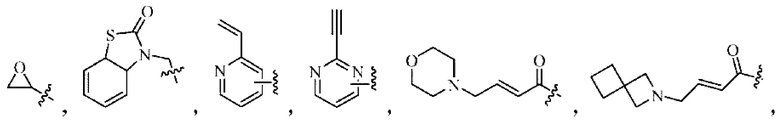
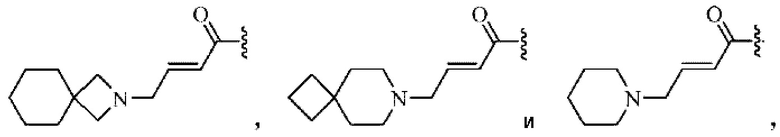
Z относится к уходящей группе или активированной гидроксильной группе,
R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода, галогена, циано, С1-4 алкила, галоС1-4 алкила, 3-8-членного циклоалкила, 3-8-членного гетероциклила, 5-8-членного арила и 5-10-членного гетероарила, где С1-4 алкил, галоС1-4 алкил, 3-8-членный циклоалкил, 3-8-членный гетероциклил, 5-8-членный арил или 5-10-членный гетероарил возможно замещен заместителем, где заместитель выбран из группы, состоящей из гидрокси, амино, карбокси, циано, нитро, галогена, С1-4 алкила, С1-4 алкокси, С1-4 алкокси С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино и 3-8-членного гетероциклила; и R11, R12 и R13 являются предпочтительно водородом.
Решение 5: Соединение согласно любому из Решений 1-4 или его фармацевтически приемлемая соль или стереоизомер, имеющее структуру, показанную общей формулой (II):
Активная группа непосредственно связана с атомом N в кольце В следующим образом:
где Кольцо В выбрано из группы, состоящей из:
Решение 6: Соединение согласно любому из Решений 1-5 или его фармацевтически приемлемая соль или стереоизомер, где
Активная группа выбрана из группы, состоящей из:
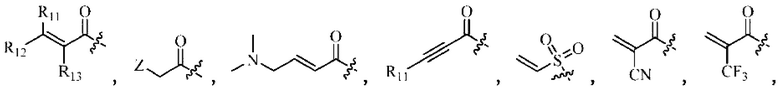
Z относится к уходящей группе или активированной гидроксильной группе, и
R11, R12 и R13 каждый независимо представляет собой Н или C1-4 алкил.
Решение 7: Соединение согласно настоящему изобретению, его фармацевтически приемлемая соль или стереоизомер представляет собой:
Решение 8: Соединение согласно Решению 4 или его фармацевтически приемлемая соль или стереоизомер, где
Кольцо В выбрано из 5-6-членного насыщенного моногетероциклила, содержащего по меньшей мере один N гетероатом, возможно замещенного 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой.
Решение 9: Соединение согласно решению 8 или его фармацевтически приемлемая соль или стереоизомер, имеющее структуру, показанную общей формулой (III):
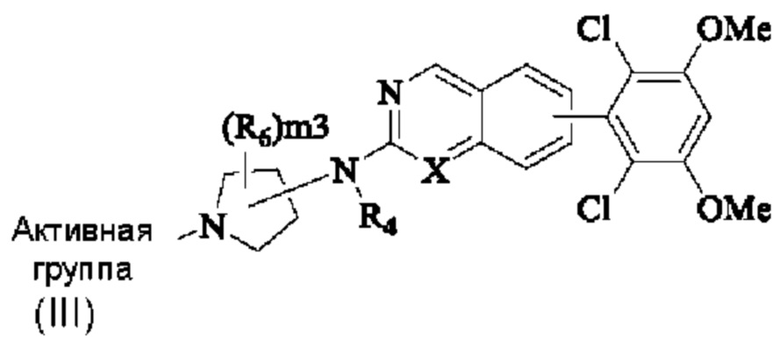
R4 представляет собой Н или C1-4 алкил;
X представляет собой N;
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, амино, карбокси, циано, нитро или галогена,
(iii) С1-4 алкила, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, галоС1-4 алкила, галоС1-4 алкокси, С2-4 алкенила, С2-4 алкинила, С1-4 алкилсульфонила или С1-4 алкилтио, возможно замещенного гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкокси С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино или 3-8-членным гетероциклилом, где 3-8-членный гетероциклил может быть необязательно замещен гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкиламино или (С1-4 алкил)2 амино, и
(iv) аминокарбонила, цианокарбонила, С1-4 алкилкарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2 аминокарбонила, С1-4 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила;
m является целым числом от 1 до 3;
Активная группа выбрана из группы, состоящей из
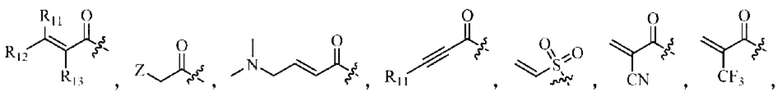
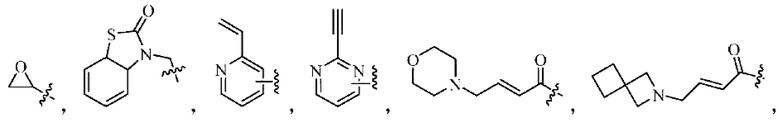
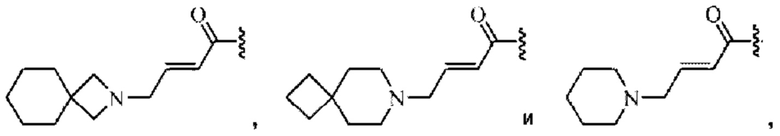
Z относится к уходящей группе или активированной гидроксильной группе;
R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода, галогена, циано, С1-4 алкила, галоС1-4 алкила, 3-8-членного циклоалкила, 3-8-членного гетероциклила, 5-8-членного арила и 5-10-членного гетероарила, причем С1-4 алкил, галоС1-4 алкил, 3-8-членный циклоалкил, 3-8-членный гетероциклил, 5-8-членный арил или 5-10-членный гетероарил возможно замещен заместителем, где заместитель выбран из группы, состоящей из гидрокси, амино, карбокси, циано, нитро, галогена, С1-4 алкила, С1-4 алкокси, С1-4 алкокси С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино и 3-8-членного гетероциклила; и R11, R12 и R13 являются предпочтительно водородом.
Решение 10: Соединение согласно Решению 9 или его фармацевтически приемлемая соль или стереоизомер, где
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, амино, карбокси, циано, нитро или галогена,
(iii) С1-4 алкила или С1-4 алкокси, возможно замещенного гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкокси С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2 амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино или 3-8-членным гетероциклилом, где 3-8-членный гетероциклил может быть необязательно замещен гидрокси, амино, карбокси, циано, нитро, галогеном, С1-4 алкилом, С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2амино, где 3-8-членный гетероциклил предпочтительно представляет собой 4-6-членный насыщенный гетероциклил, более предпочтительно - азетидинил, пирролидинил, пиперединил или морфолинил; и
(iv) аминокарбонила, цианокарбонила, С1-4 алкилкарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2 аминокарбонила, С1-4 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила;
Активная группа выбрана из группы, состоящей из
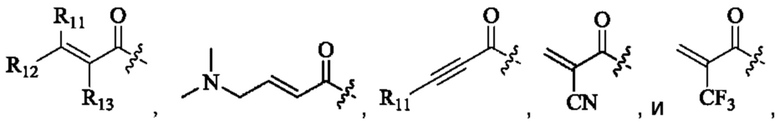
R11, R12, и R13 каждый независимо представляет собой Н или С1-4 алкил, и Активная группа представляет собой предпочтительно 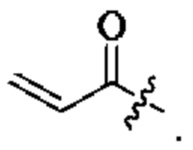
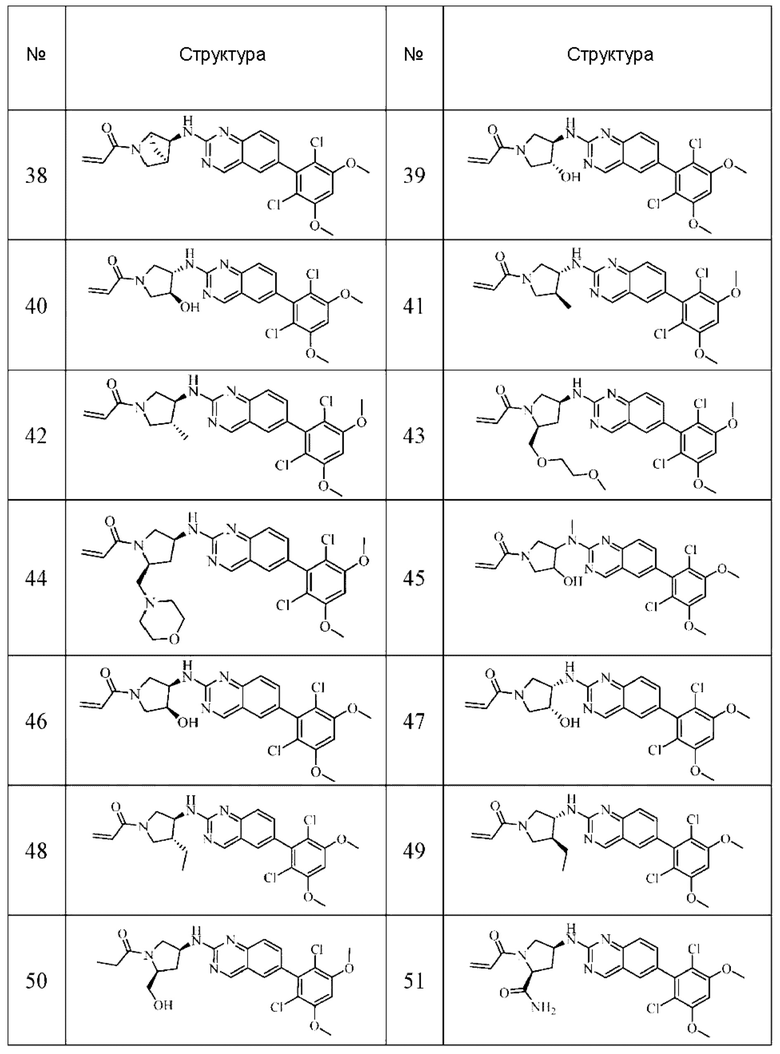
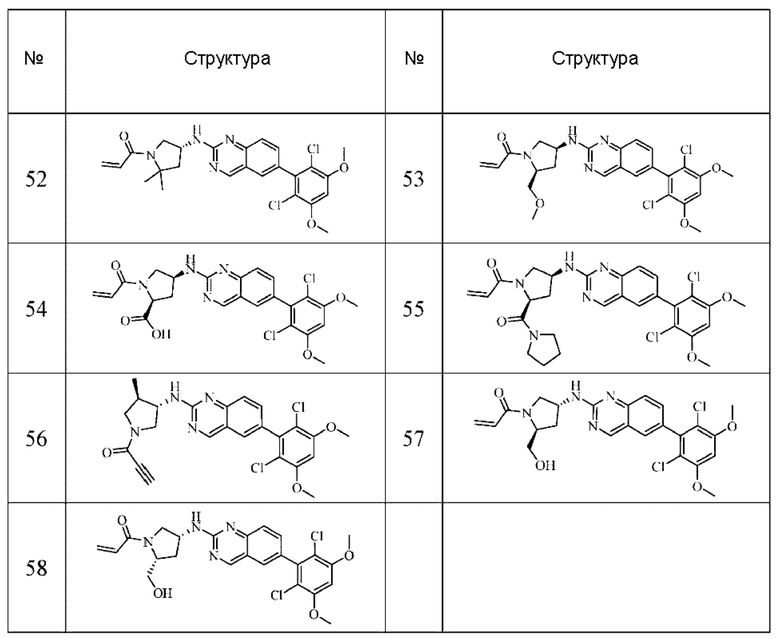
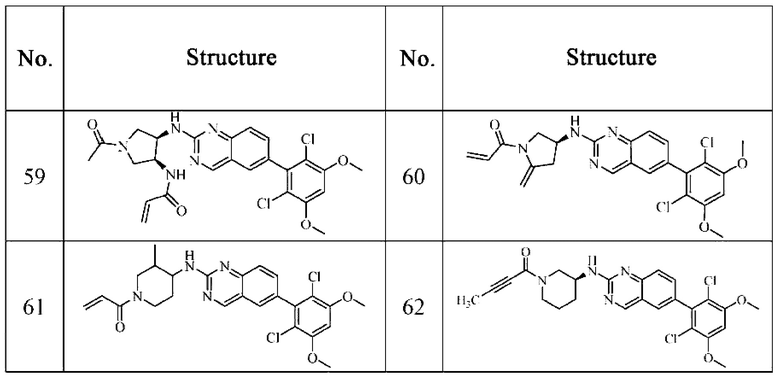
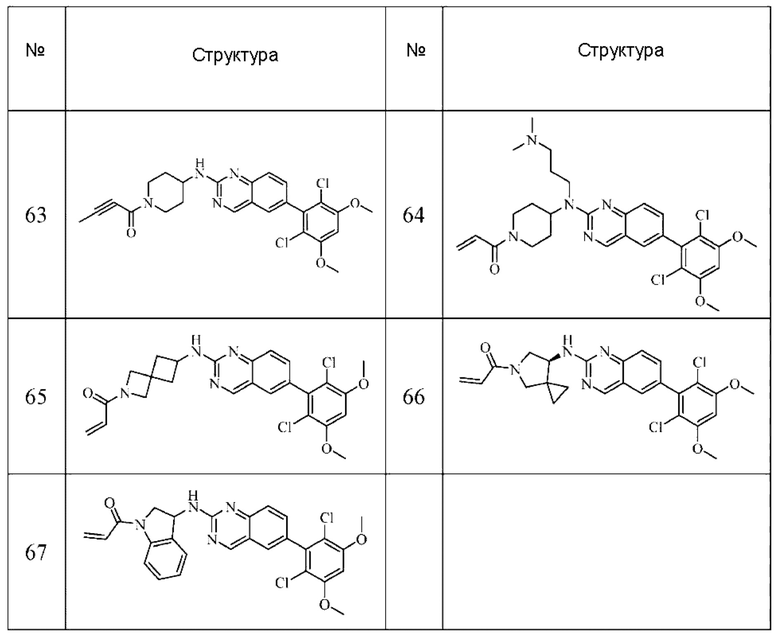
Решение 11: Соединение согласно Решению 10 или его фармацевтически приемлемая соль или стереоизомер, которое может иметь структуру, выбранную из группы, состоящей из:
Соединение согласно настоящему изобретению, его фармацевтически приемлемая соль или стереоизомер представляет собой:
Настоящее изобретение также относится к фармацевтической композиции любого из соединений или их фармацевтически приемлемых солей или стереоизомеров в соответствии с настоящим изобретением. Фармацевтическая композиция дополнительно содержит один или более фармацевтически приемлемых носителей.
Фармацевтические носители в соответствии с настоящим изобретением могут представлять собой один или более твердых или жидких наполнителей или гелевых материалов, подходящих для использования у человека. Предпочтительно фармацевтические носители имеют достаточную чистоту и достаточно низкую токсичность, которые совместимы с активным ингредиентом согласно настоящему изобретению, и существенно не снижают эффективность активного ингредиента. Например, фармацевтическими носителями могут быть наполнители, связующие, дезинтегранты, лубриканты, водные растворители или неводные растворители, и тому подобное.
Фармацевтическая композиция согласно настоящему изобретению может быть приготовлена в любой фармацевтически приемлемой лекарственной форме и может вводиться пациенту или субъекту, нуждающемуся в этом, любым подходящим способом введения, таким как пероральное, парентеральное, ректальное или внутрилегочное введение. Для перорального введения ее можно приготовить в виде таблеток, капсул, пилюль, гранул и тому подобного. Для парентерального введения ее можно приготовить в виде раствора для инъекций, стерильного порошка для инъекций и тому подобного.
Фармацевтическая композиция согласно настоящему изобретению дополнительно содержит один или более вторых терапевтически активных агентов. Вторыми терапевтически активными агентами являются антиметаболиты, ингибиторы факторов роста, митотические ингибиторы, противоопухолевые гормоны, алкилирующие агенты, металлы, ингибиторы топоизомеразы, гормональные лекарства, иммуномодуляторы, гены-супрессоры опухолей, противораковые вакцины или антитела и низкомолекулярные лекарства, относящиеся к иммунным контрольным точкам или иммунотерапии опухолей.
В настоящем изобретении также заявляется применение любого из соединений или их фармацевтически приемлемых солей или стереоизомеров в соответствии с настоящим изобретением, или фармацевтической композиции в соответствии с настоящим изобретением для изготовления лекарственного средства для лечения заболевания, опосредованного аномалией FGF/FGFR. Заболевание, опосредованное аномалией FGF/FGFR согласно настоящему изобретению, представляет собой рак, и рак включает в себя рак легкого, плоскоклеточный рак эпителиальных клеток, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, карциному протока молочной железы, рак головы и шеи, рак эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, рак почечной лоханки, рак пищевода, аденокарциному пищевода, глиому, рак простаты, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому, нейрофиброматоз, рак костей, рак кожи, рак головного мозга, рак толстой кишки, рак яичек, желудочно-кишечную стромальную опухоль, рак ротовой полости, рак глотки, множественную миелому, лейкемию, неходжкинскую лимфому, хориоаденому толстой кишки, меланому, цитому и саркому, и миелодиспластический синдром.
Настоящее изобретение также относится к способу лечения заболевания, опосредованного аномалией FGF/FGFR. Этот способ включает введение субъекту, нуждающемуся в этом, любого из соединений согласно настоящему изобретению или его фармацевтически приемлемых солей или стереоизомеров, или фармацевтической композиции согласно настоящему изобретению. Заболевание, опосредованное аномалией FGF/FGFR по настоящему изобретению, представляет собой рак, причем рак включает в себя рак легкого, плоскоклеточный рак эпителиальных клеток, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, карциному протока молочной железы, рак головы и шеи, рак эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, рак почечной лоханки, рак пищевода, аденокарциному пищевода, глиому, рак простаты, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому, нейрофиброматоз, рак костей, рак кожи, рак головного мозга, рак толстой кишки, рак яичек, желудочно-кишечную стромальную опухоль, рак ротовой полости, рак глотки, множественную миелому, лейкемию, неходжкинскую лимфому, хориоаденому толстой кишки, меланому, цитому и саркому, и миелодиспластический синдром.
Настоящее изобретение также относится к любому из соединений согласно настоящему изобретению, его фармацевтически приемлемым солям или стереоизомерам, или фармацевтической композиции по настоящему изобретению для применения в качестве лекарственного средства.
Подробное описание изобретения
«Галоген», используемый в настоящем изобретении, означает фтор, хлор, бром, йод или тому подобное, предпочтительно - фтор и хлор.
«Оксо», используемый в настоящем изобретении, означает, что любой С в заместителе может быть замещен «-С(О)-»; если содержится гетероатом, гетероатом может образовывать оксид, например, 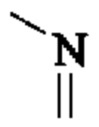 может быть замещен
может быть замещен 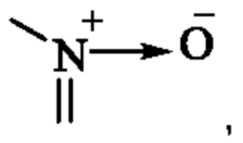 и S может быть окислен до S(O) или S(O)2.
и S может быть окислен до S(O) или S(O)2.
"Гало", используемый в настоящем изобретении, означает, что любой атом водорода в заместителе может быть замещен одним или более одинаковых или различных атомов галогена. "Галоген" является таким, как описано выше.
"С1-6 алкил", используемый в настоящем изобретении, означает линейную или разветвленную алкильную группу, полученную путем удаления одного атома водорода из углеводородного фрагмента, имеющего от 1 до 6 атомов углерода, такую как метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, 2-метилбутил, неопентил, 1-этилпропил, н-гексил, изогексил, 4-метилпентил, 3-метилпентил, 2-метилпентил, 1-метилпентил, 3,3-диметилбутил, 2,2-диметилбутил, 1,1-диметилбутил, 1,2-диметилбутил, 1,3-диметилбутил, 2,3-диметилбутил, 2-этилбутил и 1-метил-2-метилпропил, и т.п. "С1-4 алкил", используемый в настоящем изобретении, означает вышеперечисленные примеры, содержащие 1-4 атома углерода.
"С2-8 алкенил", используемый в настоящем изобретении, означает линейную или разветвленную или циклическую алкеновую группу, полученную путем удаления одного атома водорода из олефиновой группы, имеющую от 2 до 8 атомов углерода и содержащую углерод-углеродные двойные связи, такую как винил, 1-пропенил, 2-пропенил, 1-бутенил, 2-бутенил, 1,3-бутадиенил, 1-пентенил, 2-пентенил, 3-пентенил, 1,3-пентадиенил, 1,4-пентадиенил, 1-гексенил, 1,4-гексадиенил, циклобутенил, циклопентенил, циклогексенил, 1,4-циклогексадиенил, циклогептенил, 1,4-циклогептадиенил, циклооктенил, 1,5-циклооктадиенил, и т.п., и возможно образованную полицикпическую систему, например, спиро-циклоолефин, орто-конденсированный циклоолефин, мостиковый циклоолефин и т.д.
"С2-8 алкинил", используемый в настоящем изобретении, означает линейную или разветвленную алкиновую группу, полученную удалением одного атома водорода из алкиновой группы, имеющую от 2 до 8 атомов углерода и содержащую углерод-углеродные тройные связи, такую как этинил, пропинил, 2-бутинил, 2-пентинил, 3-пентинил, 4-метил-2-пентинил, 2-гексинил, 3-гексинил и т.п.
"С1-6 алкиламино", "(С1-6 алкил)2 амино", "С1-6 алкилкарбониламино", "С1-6 алкилсульфониламино", "С1-6 алкиламинокарбонил", "(С1-6 алкил)2 аминокарбонил", "С1-6 алкоксикарбонил", "С1-6 алкилсульфонил", "С1-6 алкилтио", "С1-6 алкилкарбонил", "3-8-членный циклоалкилкарбонил", "3-8-членный гетероциклилкарбонил", используемый в настоящем изобретении, означает С1-6 алкил-NH-, (С1-6 алкил)(С1-6 алкил)N-, С1-6 алкил-С(O)-NH-, С1-6 алкил-S(O)2-NH2-, С1-6 алкил-NH-С(O)-, (С1-6 алкил)(С1-6 алкил)N-С(O)-, С1-6 алкил-О-С(О)-, С1-6 алкил-S(O)2-, С1-6 алкил-S-, С1-6 алкил-С(О)-, 3-8-членный циклоалкил-С(О)-, 3-8-членный гетероциклил-С(О)-, соответственно; "С1-6 алкил" является таким, как описано выше, предпочтительно - "C1-4 алкилом".
"С1-6 алкокси", используемый в настоящем изобретении, означает группу, в которой "С1-6 алкил", как определено выше, связан с исходной молекулой через атом кислорода, то есть группу "С1-6 алкил-О-", такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси, трет-бутокси, н-пентилокси, неопентилокси, н-гексилокси и т.д. Термин "С1-4 алкокси", используемый в настоящем изобретении, означает приведенные выше примеры, содержащие от 1 до 4 атомов углерода, то есть группу "C1-4 алкил-О-".
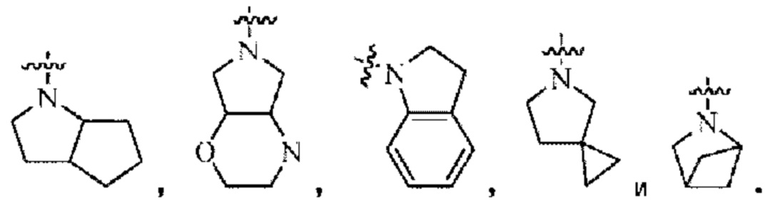
«Конденсированное кольцо», используемое в настоящем изобретении, означает полициклическую структуру, образованную двумя или более циклическими структурами, соединенными в виде орто-конденсированного кольца, спиро-кольца или мостикового кольца. Орто-конденсированное кольцо относится к конденсированной кольцевой структуре, образованной двумя или более циклическими структурами, имеющими два общих соседних кольцевых атома (то есть, имеющими одну общую связь). Мостиковое кольцо относится к структуре конденсированного кольца, образованной двумя или более циклическими структурами, соединенными двумя несмежными атомами кольца друг с другом. Спиро-кольцо относится к конденсированной кольцевой структуре, образованной двумя или более циклическими структурами, имеющими один общий кольцевой атом.
«Циклоалкил», используемый в настоящем изобретении, относится к моноциклической циклоалкильной, бициклической циклоалкильной или полициклической циклоалкильной системе (также известной как система конденсированных колец). Моноциклическая циклоалкильная система представляет собой циклоалкильную группу, содержащую от 3 до 8 атомов углерода. Примеры 3-8-членного циклоалкила включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил и т.п. Конденсированный циклоалкил включает орто-конденсированный циклоалкил, мостиковый циклоалкил, спиро-циклоалкил.
Орто-конденсированный циклоалкил может представлять собой 6-12-членный орто-конденсированный циклоалкил, 7-10-членный орто-конденсированный циклоалкил, и типичные примеры включают, но не ограничиваются ими, бициклический[3,1,1]гептан, бициклический[2,2,1]гептан, бициклический[2,2,2]октан, бициклический[3,2,2]нонан, бициклический[3,3,1]нонан и бициклический[4,2,1]нонан.
Спиро-циклоалкил может представлять собой 6-12-членную спиро-кольцевую группу и 7-11-членную спиро-кольцевую группу, и примеры включают, но не ограничиваются ими: 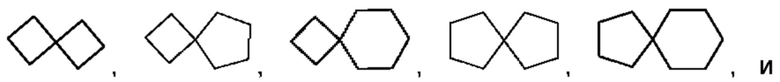
Мостиковый циклоалкил может представлять собой 6-12-членную мостиковую кольцевую группу и 7-11-членную мостиковую кольцевую группу, и примеры включают, но не ограничиваются ими: 
"Гетероциклил", используемый в настоящем изобретении, означает неароматическую циклическую группу, в которой по меньшей мере один атом углерода в кольце замещен гетероатомом, выбранным из О, S и N, предпочтительно замещенным 1-3 гетероатомами, и где а атом углерода, атом азота и атом серы могут быть окислены.
«Гетероциклил» означает моноциклическую гетероцикл ильную, бициклическую гетероцикл ильную или полициклическую гетероцикл ильную систему (также известную как система конденсированных колец), включая насыщенный и частично насыщенный гетероциклил, но исключая ароматические кольца. Моногетероциклил может представлять собой 3-8-членный гетероциклил, 3-8-членный насыщенный гетероциклил, 3-6-членный гетероциклил, 4-7-членный гетероциклил, 5-7-членный гетероциклил, 5-6-членный гетероциклил, 5-6-членный кислород-содержащий гетероциклил, 5-6-членный азот-содержащий гетероциклил, 5-6-членный насыщенный гетероциклил или тому подобное. Примеры «3-8-членного насыщенного гетероциклила» включают в себя, но не ограничиваются этим, азиридинил, оксиранил, тииранил, азетидинил, оксетанил, тиетанил, тетрагидрофуранил, тетрагидропирролил, тетрагидротиофенил, имидазолидинил, пиразолидинил, 1,2-оксазолидинил, 1,3-оксазолидинил, 1,2-тиазолидинил, 1,3-тиазолидинил, тетрагидро-2Н-пиранил, тетрагидро-2Н-тиопиранил, пиперидинил, пиперазинил, морфолинил, 1,4-диоксанил, 1,4-оксатианил. Примеры 3-8-членного частично насыщенного гетероциклила включают, но не ограничиваются ими, 4,5-дигидроизооксазолил, 4,5-дигидрооксазолил, 2,5-дигидрооксазолил, 2,3-дигидрооксазолил, 3,4-дигидро-2Н-пирролил, 2,3-дигидро-1Н-пирролил, 2,5-дигидро-1Н-имидазолил, 4,5-дигидро-1Н-имидазолил, 4,5-дигидро-1Н-пиразолил, 4,5-дигидро-3Н-пиразолил, 4,5-дигидротиазолил, 2,5-дигидротиазолил, 2Н-пиранил, 4Н-пиранил, 2Н-тиопиранил, 4Н-тиопиранил, 2,3,4,5-тетрагидропиридил, 1,2-изооксазинил, 1,4-изооксазинил или 6Н-1,3-оксазинил и тому подобное. Конденсированное гетероциклическое кольцо включает орто-конденсированный гетероциклил, спиро-гетероциклил, мостиковый гетероциклил, и может быть насыщенным, частично насыщенным или ненасыщенным, но не ароматическим. Конденсированный гетероциклил представляет собой 5-6-членное моноциклическое гетероциклическое кольцо, конденсированное с бензольным кольцом, 5-6-членным моноциклическим циклоалкилом, 5-6-членным моноциклическим циклоалкенилом, 5-6-членным моноциклическим гетеро цикл илом или 5-6-членным моноциклическим гетероарилом. Орто-конденсированный гетероциклил может быть 6-12-членным орто-конденсированным гетеро цикл ил ом, 7-10-членным орто-конденсированным гетероциклил ом, 6-10-членным орто-конденсированным гетеро цикл илом и 6-12-членным насыщенным орто-конденсированным гетероциклилом, и типичные примеры включают, но не ограничиваются ими, 3-азабицикло[3,1,0.]гексил, 3,6-диазабицикло[3,2,0]гептил, 3,8-диазабицикло[4,2,0]октил, 3,7-диазабицикло[4,2.0]октил, октагидропирроло[3,4-с]пирролил, октагидропирроло[3,4-b]пирролил, октагидропирроло[3,4-b][1,4]оксазинил, октагидро-1Н-пирроло[3,4-с]пиридинил, 2,3-дигидробензофуран-2-ил, 2,3-дигидробензофуранил-3-ил, индолин-1-ил, индолин-2-ил, индолин-3-ил, 2,3-дигидробензотиофен-2-ил, октагидро-1Н-индолил, октагидробензофуранил.
Спиро-гетероциклил может представлять собой 6-12-членный спиро-гетероциклил, 7-11-членный спиро-гетероциклил и 6-12-членный насыщенный спироциклил, и примеры включают, но не ограничиваются ими: 
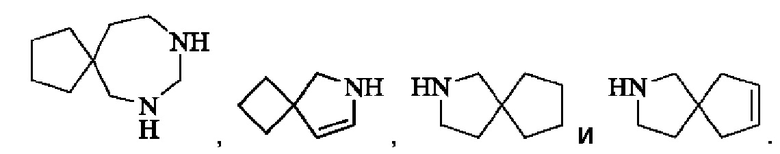
Мостиковый гетероциклил может представлять собой 6-12-членный мостиковый гетероциклил, 7-11-членный мостиковый гетероциклил, и 6-12-членный насыщенный мостиковый гетероциклил, и примеры включают, но не ограничиваются ими: 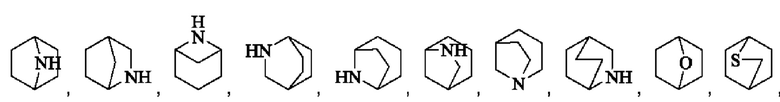
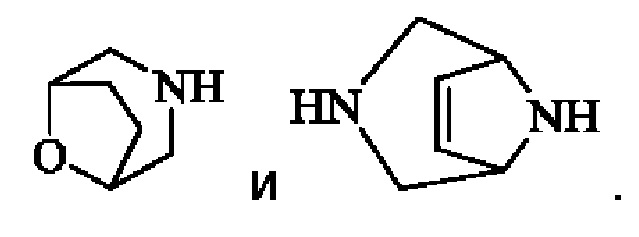
"6-14-членный арил", используемый в настоящем изобретении, означает циклическую ароматическую группу, имеющую от 6 до 14 атомов углерода, и включает «6-8-членный моноциклический арил», такой как фенил, «8-14-членное конденсированное арильное кольцо», такое как пенталенил, нафтил, фенантрил и тому подобное.
«Гетероарил», используемый в настоящем изобретении, может представлять собой 5-10-членную гетероарильную группу, и относится к ароматической циклической группе, в которой по меньшей мере один атом углерода в кольце замещен гетероатомом, выбранным из О, S и N, предпочтительно 1-3 гетероатомами, включая условие, что атом углерода или атом серы окислен, например, атом углерода замещен С(О), а атом серы замещен S(O) или S(O)2. Гетероарил включает в себя моноциклический гетероарил и конденсированный гетероарил. Моноциклический гетероарил может представлять собой 5-7-членный гетероарил или 5-6-членный гетероарил, и примеры моноцикпического гетероарила включают, но не ограничиваются этим, фуранил, имидазолил, изоксазолил, тиазолил, изотиазолил, оксадиазолил, оксазолил, пиридил, пиридазинил, пиримидинил, пиразинил, пиразолил, пирролил, тетразолил, тиадиазолил, тиенил, триазолил и триазинил. В некоторых вариантах воплощения изобретения конденсированный гетероарил представляет собой 5- или 6-членный моноциклический моноциклический циклоалкил, 5- или 6-членный моноциклический циклоалкенил, 5- или 6-членный моноциклический гетероциклил, или 5- или 6-членный моноциклический гетероарил, где конденсированный циклоалкил, конденсированный циклоалкенил и конденсированный гетероциклил возможно замещены одной или двумя группами в качестве независимой оксо или тио группы. Конденсированный гетероарил может представлять собой 8-12-членный орто-конденсированный гетероарил или 9-10-членный орто-конденсированный гетероарил, и примеры конденсированного гетероарила включают, но не ограничиваются ими, бензимидазолил, бензофуранил, бензотиенил, бензооксадиазолил, бензотиадиазолил, бензотиазолил, циннолинил, 5,6-дигидрохинолин-2-ил, 5,6-дигидроизохинолин-1-ил, фуропиридинил, индазолил, индолил, изохинолил, нафтиридинил, пуринил, хинолил, 5,6,7,8-тетрагидрохинол-2-ил, 5,6,7,8-тетрагидрохинолил, 5,6,7,8-тетрагидрохинол-4-ил, 5,6,7,8-тетрагидроизохинол-1-ил, тиенопиридинил, 4,5,6,7-тетрагидро[с][1,2,5]оксадиазолил и 6,7-дигидро[с][1,2,5]оксадиазол-4(5Н)кето.
«Фармацевтически приемлемые соли», используемые в данном описании, означают фармацевтически приемлемые аддитивные соли кислот и оснований, и сольваты. Такие фармацевтически приемлемые соли включают соли кислот, таких как соляная кислота, фосфорная кислота, бромистоводородная кислота, серная кислота, сернистая кислота, муравьиная кислота, толуолсульфоновая кислота, метансульфоновая кислота, азотная кислота, бензойная кислота, лимонная кислота, винная кислота, малеиновая кислота, йодистоводородная кислота, алкановая кислота (такая как уксусная кислота, НООС-(СН2)n-СООН (где n равно 0-4)) и тому подобное; и включают соли оснований, таких как натрий, калий, кальций, аммоний и тому подобное. Специалистам в данной области известно множество нетоксичных фармацевтически приемлемых аддитивных солей.
«Стереоизомер» соединения формулы (I) в соответствии с настоящим значением означает энантиомер в случае, когда соединение формулы (I), (II) или (III) имеет асимметричный атом углерода; цис-транс-изомер в том случае, если соединение имеет углерод-углеродную двойную связь или циклическую структуру; таутомеры в том случае, если в соединении присутствует кетон или оксим. Энантиомеры, диастереомеры, рацемические изомеры, цис-транс-изомеры, таутомеры, геометрические изомеры, эпимеры соединений формулы (I), (II) или (III) и их смеси включены в объем изобретения.
«Активная группа», используемая в настоящем изобретении, относится к группе, которая способна образовывать ковалентную связь с нукпеофилом. «Нуклеофил» относится к веществу, которое поставляет электронные пары электрофилу для образования химической связи в реакции. В некоторых вариантах нуклеофил может представлять собой кислородный нуклеофил, например, воду или гидрокси; азотный нуклеофил, например, амин; или нуклеофил серы, например, тиол, такой кактиол в боковой цепи остатка цистина.
«Активная группа», используемая в настоящем изобретении, относится к фрагменту в ингибиторе, который обратимо или необратимо участвует в реакции донора (например белка) с субстратом. Например, активная группа может образовывать ковалентную связь с белком, или может образовывать стабильное переходное состояние, или является обратимым или необратимым алкилирующим агентом. Например, активная группа может быть функциональной группой на ингибиторе, который участвует в реакции образования связи, где новая ковалентная связь образуется между частью активной группы и донором (например аминокислотным остатком белка). Активная группа представляет собой электрофил, а «донор» представляет собой нуклеофил, такой как боковая цепь остатка цистеина. Подходящая активная группа включает, но не ограничивается следующими структурами:
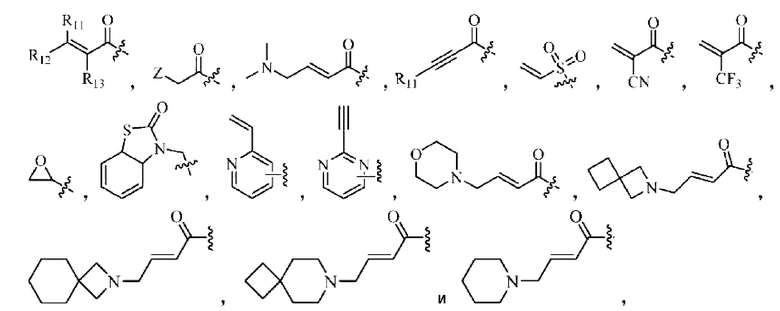
где,
Z относится к уходящей группе (такой как галоген) или активированной гидроксильной группе (такой как трифлат);
R11, R12 и R13 независимо выбраны из группы, состоящей из водорода, галогена, циано, С1-4 алкила, галоС1-4 алкила, 3-8-членного циклоалкила, 3-8-членного гетероциклила, 5-8-членного арила и 5-10-членного гетероарила, причем С1-4 алкил, галоС1-4 алкил, 3-8-членный циклоалкил, 3-8-членный гетероциклил, 5-8-членный арил или 5-10-членный гетероарил возможно замещен заместителем, где заместитель выбран из группы, состоящей из гидрокси, амино, карбокси, циано, нитро, галогена, С1-4 алкила, С1-4 алкокси, С1-4 алкокси-С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино, 3-8-членного гетероциклила; и R11, R12 и R13 являются предпочтительно водородом.
Аббревиатура «NMP», использованная в настоящем документе, означает N-метилпирролидон; «DIPEA» означает N,N-диизопропилэтиламин; «TLC» означает тонкослойную хроматографию; «РЕ : ЕА» означает петролейный эфир : этилацетат; «TFA» означает три фтор уксусную кислоту; «THF» означает тетрагидрофуран; «ЕА» означает этилацетат; «DCM : МеОН» означает дихлорметан : метанол; «DCM» означает дихлорметан; «МТВЕ» означает метил-трет-бутиловый эфир; и «TFAA» означает трифторуксусный ангидрид.
Варианты осуществления изобретения
ПРИМЕР 1: Синтез соединения 1
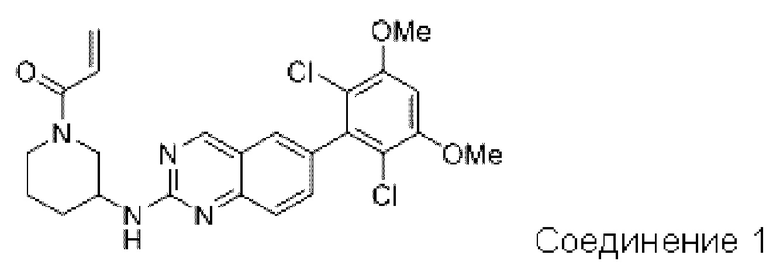
Путь синтеза является следующим:
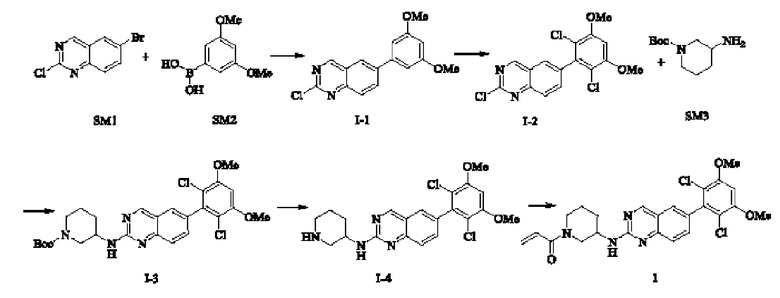
Синтез промежуточного соединения I-1
SM1 (5,00 г × 20,5 ммоль) и SM2 (3,73 г × 20,5 ммоль) растворяли в тетрагидрофуране (30 мл), добавляли раствор карбоната цезия (20,00 г × 61,5 ммоль) в воде (30 мл) и добавляли с каталитическое количество Pd(PPh3)Cl2. Полученную смесь кипятили с обратным холодильником в течение 4 ч в атмосфере азота.
Реакционный раствор концентрировали досуха и экстрагировали этилацетатом. Органическую фазу промывали один раз насыщенным хлоридом натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный сырой продукт подвергали колоночной хроматографии (силикагель 200-300 меш, петролейный эфир / этил ацетат = 10/1) с получением промежуточного соединения I-1 (3,80 г, выход 62%) в виде бледно-желтого твердого вещества.
Синтез промежуточного соединения I-2
I-1 (3,80 г, 12,6 ммоль) растворяли в тетрагидрофуране (100 мл), охлаждали до температуры от -20 до 30°С в атмосфере азота и по каплям добавляли сульфонилклорид (5,11 г, 37,9 ммоль). Полученная смесь реагировала в течение 2 ч при той же температуре.
Реакционный раствор медленно нагревали до температуры окружающей среды, добавляли ацетонитрил (100 мл) и перемешивали в течение 10 минут. Полученное твердое вещество собирали фильтрацией и сушили с получением промежуточного соединения 1-2 (2,80 г, выход 60%) в виде бледно-желтого твердого вещества.
Синтез промежуточного соединения I-3
SM3 (81 мг, 0,40 ммоль) и I-2 (100 мг, 0,35 ммоль) добавляли в N-метилпирролидон (3 мл) и добавляли N,N-диизопропилэтиламин (80 мг, 0,80 ммоль). Полученную смесь нагревали до 100°С и она реагировала в течение 4 часов.
Реакционный раствор выливали в ледяную воду и выпавшее в осадок твердое вещество собирали фильтрацией. Твердое вещество подвергали колоночной хроматографии (силикагель 200-300 меш, петролейный эфир / этилацетат = 3/1) с получением промежуточного соединения I-3 (100 мг, выход 54%) в виде бледно-желтого твердого вещества.
Синтез промежуточного соединения I-4
I-3 (100 мг, 0,19 ммоль) растворяли в дихлорметане (5 мл), добавляли трифторуксусную кислоту (1 мл) и полученную смесь подвергали реакции при температуре окружающей среды в течение 4 часов.
Реакционный раствор концентрировали досуха, добавляли толуол и затем концентрировали при пониженном давлении для удаления остаточной трифторуксусной кислоты. Полученный сырой продукт I-4 (81 мг, выход 100%) в виде желтого масла использовали непосредственно в следующей реакции.
Синтез соединения 1
I-4 (81 мг, 0,18 ммоль) растворяли в тетрагидрофуране (10 мл), добавляли триэтиламин (0,1 мл, 0,54 ммоль) и акрилоилхлорид (25 мг, 0,21 ммоль), и полученную смесь подвергали реакции при температуре окружающей среды в течение 4 ч. Небольшое количество метанола добавляли, чтобы погасить оставшийся акрилоилхлорид, и полученную смесь концентрировали при пониженном давлении. Полученный сырой продукт подвергали колоночной хроматографии (силикагель 200-300 меш, петролейный эфир / этил ацетат = 1/1) с получением соединения 1 (11 мг, выход 15%) в виде бледно-желтого твердого вещества.
1Н NMR (400 MHz, DMSO-d6) δ (ppm) 9.18 (s, 1H), 7.51-7.69 (m, 4H), 7.01 (s, 1H), 6.79-6.86 (in, 1H), 6.04-6.09 (m, 1H), 5.66-5.87 (m, 1H), 3.86-4.40 (m, 9H), 3.07-3.17 (m, 1H), 2.90-2.95 (m, 1H), 1.99-2.01 (m, 1H), 1.36-1.84 (m, 3H); LC-MS [M+H]=488.
Обозначения: 1Н NMR - 1Н ЯМР, MHz - МГц, DMSO - диметилсульфоксид, ppm - млн-1, LC-MS - ЖК-МС (жидкостная хроматография-масс-спектроскопия)
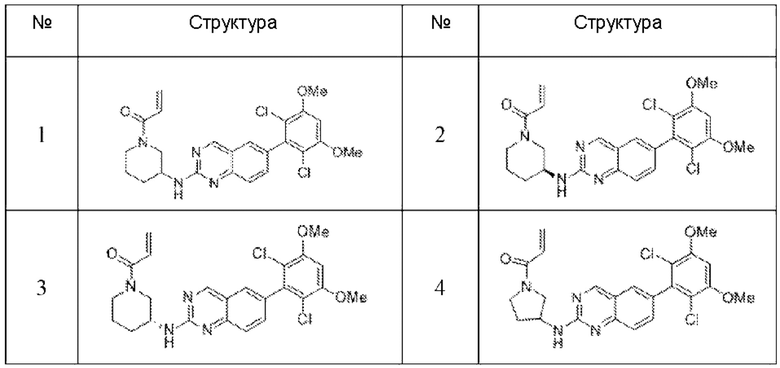
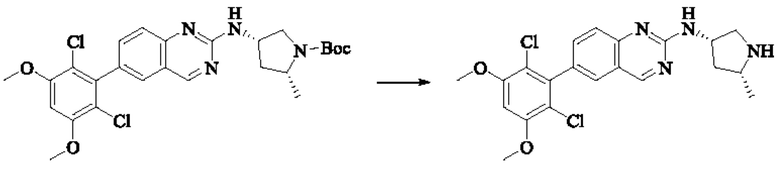
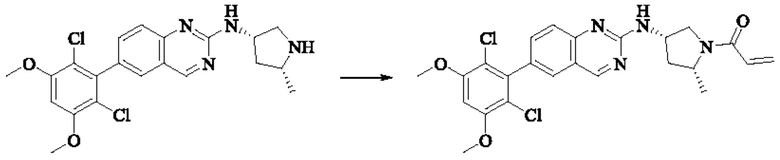
Пример 2: Синтез (S)-1-(3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)пропил-2-ен-1-она (соединение 5)
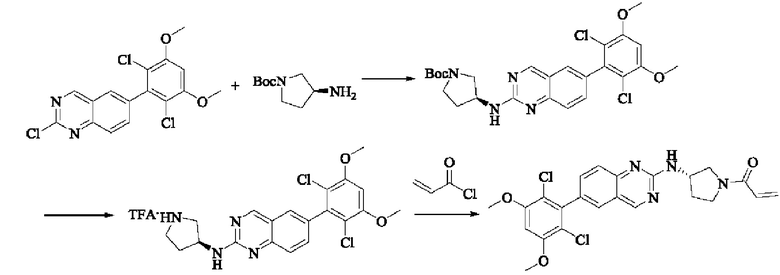
Стадия 1: Синтез трет-бутил(S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
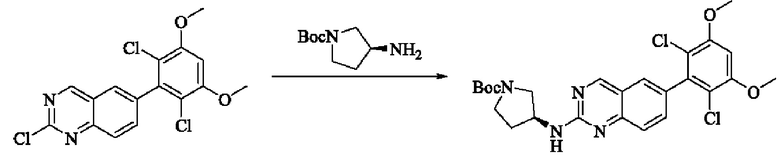
Вещество 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (100,0 мг, 0,27 моль, 1,0 экв.) растворяли в NMP (3 мл), добавляли трет-бутил(S)-3-аминопирролидин-1-карбоксилат (55,4 мг, 0,30 ммоль, 1,1 экв.) и DIPEA (104,9 мг). 0,81 моль, 3,0 экв.), нагревали до 100°С и проводили реакцию в течение 4 часов. Завершение реакции детектировали с помощью TLC и реакционный раствор охлаждали до комнатной температуры, выливали в ледяную воду и фильтровали с помощью отсасывания для сбора осадка на фильтре. Неочищенный продукт подвергали колоночной хроматографии на силикагеле (РЕ : ЕА = от 10:1 до 2:1) с получением трет-бутил(S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (78,7 мг, выход 47%) в виде желтой жидкости.
Стадия 2: Синтез (S)-6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пирролидин-3-ил)хиназолин-2-амина трифторацетата
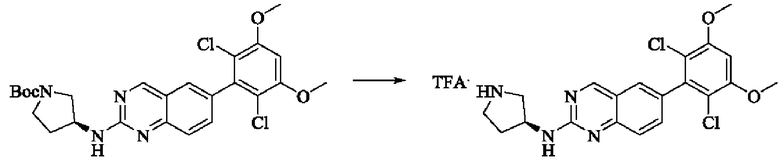
Трет-бутил(S)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (78,7 мг, 0,15 ммоль, 1,0 экв.) растворяли в дихлорметане (8 мл), добавляли TFA (1 мл) и проводили реакцию при комнатной температуре в течение 8 часов. Завершение реакции детектировали с помощью TLC. Путем концентрирования получали (S)-6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пирролидин-3-ил)хиназолин-2-амина трифторацетат (77,9 мг, выход: 100%) в виде желтой жидкости.
Стадия 3: Синтез (S)-1-(3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)пропил-2-ен-1-она
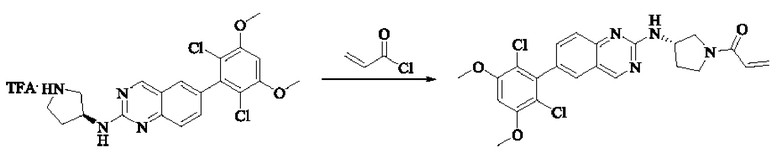
(S)-6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пирролидин-3-ил)хиназолин-2-амина трифторацетат (77,9 мг, 0,15 ммоль, 1,0 экв) растворяли в THF (10 мл), добавляли триэтиламин (45,5 мг, 0,45 ммоль, 2,0 экв), перемешивали для реакции в течение 30 мин, добавляли акрилоилхлорид (27,0 мг, 0,30 ммоль, 2,0 экв) и проводили реакцию в течение 8 ч. Завершение реакции детектировали с помощью TLC. Добавляли насыщенный раствор бикарбоната натрия (20 мл) с последующей экстракцией ЕА (3×20 мл). Органические фазы объединяли, сушили безводным сульфатом магния, фильтровали и концентрировали. Неочищенный продукт очищали колоночной хроматографией на си лика геле (силикагель 200-300 меш, DCM : МеОН = от 100:1 до 20:1) с получением (S)-1-(3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она (28,4 мг, выход: 40%).
1H NMR(400 MHz, DMSO-d6) δ (ppm): 9.19 (s, 1H), 7.84-7.89 (m, 1H), 7.69 (s, 1H), 7.50-7.59 (m, 2H), 7.01 (s, 1H), 6.52-6.63 (m, 1H), 6.10-6.17 (m, 1H), 5.61-5.70 (m, 1H), 4.40-4.60 (m, 1H), 3.93 (s, 6H), 3.49-3.78 (m, 4H), 2.20-2.35 (m, 3H).
Молекулярная формула: C23H22Cl2N4O3 Молекулярная масса: 473,35 LC-MS (m/z)=473,25[M+H+].
Пример 3: Синтез (R)-1-(3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она (соединение 6)
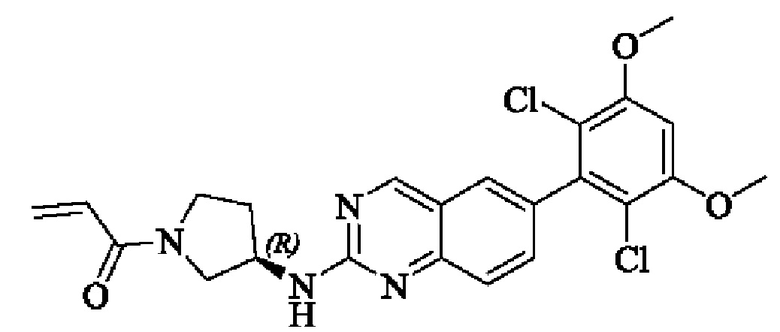
Стадия 1: Синтез трет-бутил(R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
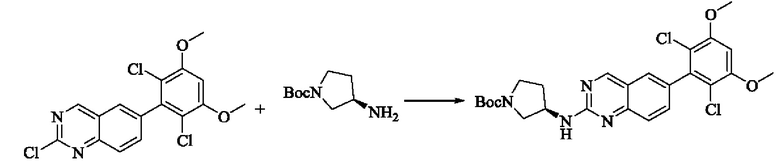
Вещество 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (100,0 мг, 0,27 моль, 1,0 экв) растворяли в NMP (3 мл), добавляли трет-бутил(R)-3-аминопирролидин-1-карбоксилат (57,4 мг, 0,31 ммоль, 1,5 экв) и DIPEA (80,2 мг, 0,62 моль, 2,3 экв), нагревали до 100°С и проводили реакцию в течение 4 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до комнатной температуры, выливали в ледяную воду и фильтровали с помощью отсасывания для сбора осадка на фильтре. Сырой продукт подвергали колоночной хроматографии на силикагеле (РЕ : ЕА = от 10:1 до 2:1) с получением трет-бутил(R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (88 мг, выход: 67%).
Стадия 2: Синтез (R)-6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пирролидин-3-ил)хиназолин-2-амина трифторацетата
Промежуточное вещество трет-бутил(R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (88,0 мг, 0,17 ммоль, 1,0 экв) растворяли в DCM (15 мл), добавляли TFA (2,5 мл) и проводили реакцию при комнатной температуре в течение 8 ч. Завершение реакции детектировали с помощью TLC и раствор концентрировали непосредственно до сухости с получением (R)-6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пирролидин-3-ил)хиназолин-2-амин трифторацетата (90,4 мг, выход: 100%).
Стадия 3: Синтез (R)-1-(3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она
Промежуточное вещество (R)-6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пирролидин-3-ил)хиназолин-2-амин трифторацетат (90,4 мг, 0,17 ммоль, 1,0 экв) растворяли в THF (8 мл), добавляли триэтиламин (68,6 мг, 0,68 ммоль, 4,0 экв), перемешивали для реакции в течение 30 минут и добавляли акрилоилхлорид (18,4 мг, 0,20 ммоль, 1,2 экв) для реакции в течение 8 часов. Завершение реакции детектировали с помощью TLC. Добавляли насыщенный раствор бикарбоната натрия (20 мл) с последующей экстракцией ЕА (3×15 мл). Органические фазы объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (DCM : МеОН = от 100:1 до 40:1) с получением (R)-1-(3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она (56,0 мг, выход: 70%).
1H NMR (400 MHz, DMSO-d6) δ (ppm): 9.19 (m, 1H), 7.82 (m, 2H), 7.51-7.69 (m, 2H), 7.02 (s, 1H), 6.53-6.63 (m, 1H), 6.11-6.17 (m, 1H), 5.62-5.69 (m, 1H), 4.53-5.61 (d, 1H), 3.97 (s, 6H), 3.52-3.78 (m, 3H), 2.07-2.29 (m, 3H).
Молекулярная формула: C23H22Cl2N4O3 Молекулярная масса: 473,35
Пример 4: Синтез 1-(4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она (соединение 10)
Стадия 1: Синтез трет-бутил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пиперидин-1-карбоксилата
Вещество 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (100,0 мг, 0,27 моль, 1,0 экв) растворяли в NMP (3 мл), добавляли трет-бутил-4-аминопиперидин-1-карбоксилат (61,7 мг, 0,31 ммоль, 1,2 экв) и DIPEA (80,0 мг, 0,62 моль, 2,3 экв), нагревали до 100°С и проводили реакцию в течение 4 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до комнатной температуры и выливали в ледяную воду, фильтровали с отсосом для сбора осадка на фильтре. Сырой продукт подвергали колоночной хроматографии на силикагеле (РЕ : ЕА = от 10:1 до 2:1) с получением трет-бутил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пиперидин-1-карбоксилата (62,6 мг, выход: 43%).
Стадия 2: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пиперидин-4-ил)хиназолин-2-амина трифторацетата
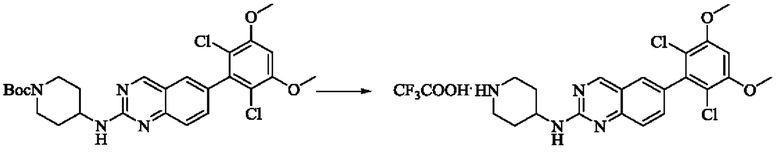
Промежуточное вещество трет-бутил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пиперидин-1-карбоксилат (62,6 мг, 0,12 ммоль, 1,0 экв) растворяли в этаноле (5 мл), добавляли TFA (2,5 мл), проводили реакцию при комнатной температуре в течение 8 ч. Завершение реакции детектировали с помощью TLC, и раствор концентрировали до сухого состояния с получением 6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пиперидин-4-ил)хиназолин-2-амина трифторацетата (64,2 мг, выход: 100%).
Стадия 3: Синтез 1-(4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она
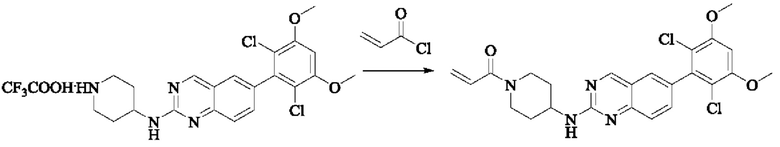
Промежуточное вещество 6-(2,6-дихлор-3,5-диметоксифенил)-Н-(пиперидин-4-ил)хиназолин-2-амина трифторацетат (64,2 мг, 0,12 ммоль, 1,0 экв) растворяли в THF (8 мл), добавляли триэтиламин (48,5 мг, 0,48 ммоль, 4,0 экв), перемешивали для реакции в течение 30 мин, добавляли акрилоилхлорид (12,7 мг, 0,14 ммоль, 1,2 экв) для реакции в течение 8 ч. Завершение реакции детектировали с помощью TLC. Добавляли насыщенный раствор бикарбоната натрия (20 мл) с последующей экстракцией ЕА (3×15 мл). Органические фазы объединяли, сушили над безводным сульфатом магния, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (DCM : МеОН = от 100:1 до 40:1) с получением 1-(4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пиперидин-1-ил)проп-2-ен-1-она (40,0 мг, выход: 70%).
1H NMR (400 MHz, DMSO) δ (ppm): 9.15 (m, 1H), 7.47-7.66 (m, 3H), 7.01 (s, 1H), 6.82-6.89 (m, 1H), 6.09-6.14 (m, 1H), 5.66-5.70 (m, 1H), 4.05-4.36 (m, 8H), 2,90-2.96 (m, 1H), 1.99 (m, 2H), 1.44-1.47 (m, 2H).
Молекулярная формула: C24H24Cl2N4O3 Молекулярная масса: 487,38 LC-MS (m/z)=487,28[M+H+].
Пример 5: Синтез 1-(транс-5-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азабицикпо[2,1,1]гексан-2-ил)проп-2-ен-1-она (соединение 11)
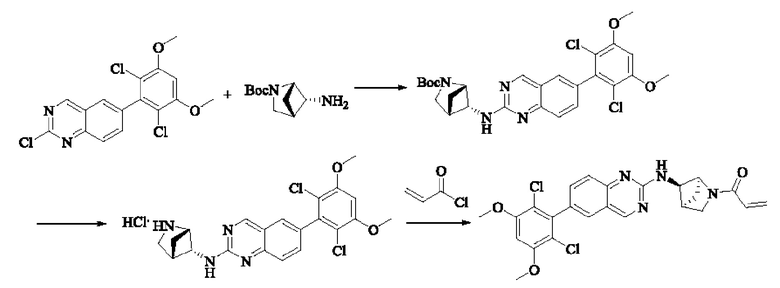
Стадия 1: Синтез трет-бутил-транс-5-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азабицикло[2,1,1]гексан-2-карбоксилата
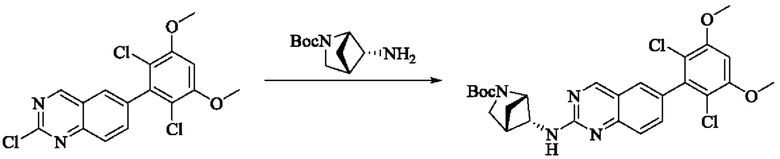
Вещество 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (500,0 мг, 1,35 моль, 1,0 экв) растворяли в NMP (5 мл), добавляли трет-бутил-транс-5-амино-2-азабицикло[2,1,1]гексан-2-карбоксилат (402,3 мг, 2,03 ммоль, 1,5 экв) и DIPEA (524,6 мг, 4,06 моль, 3,0 экв), нагревали до 100°С и проводили реакцию в течение 4 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до комнатной температуры и выливали в ледяную воду, фильтровали отсасыванием для сбора осадка на фильтре. Сырой продукт подвергали колоночной хроматографии на силикагеле (силикагель 200-300 меш, РЕ : ЕА = от 10:1 до 2:1) с получением трет-бутил-транс-5-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азабицикло[2,1,1]гексан-2-карбоксилата (520 мг, выход 72%) в виде бледно-желтого твердого вещества.
Стадия 2: Синтез транс-Н-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-2-азабицикпо[2,1,1]гексан-5-амина гидрохлорида
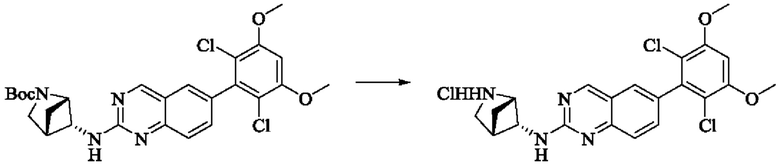
Трет-бутил-транс-5-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)2-азабицикло[2,1,1]гексан-2-карбоксилат (510 мг, 0,96 ммоль, 1,0 экв) растворяли в этаноле (10 мл), добавляли HCl-этанол (10 мл) и проводили реакцию при комнатной температуре в течение 8 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор концентрировали с получением транс-Н-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-2-азабицикло[2,1,1]гексан-5-амина гидрохлорида (480 мг, выход: 100%) в виде желтого твердого вещества.
Стадия 3: Синтез 1-(транс-5-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азабицикло[2,1,1]гексан-2-ил)проп-2-ен-1-она (соединение 11)
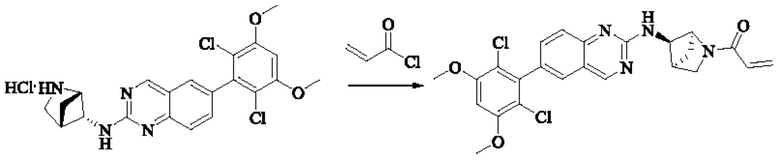
Транс-Н-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)-2-азабицикло-[2,1,1]гексан-5-амина гидрохлорид (480 мг, 1,03 ммоль, 1,0 экв) растворяли в THF (22 мл), добавляли триэтиламин (415,3 мг, 4,10 ммоль, 4,0 экв), перемешивали для реакции в течение 30 мин и добавляли акрилоилхлорид (138,6 мг, 1,54 ммоль, 1,5 экв) для реакции в течение 8 ч. Завершение реакции детектировали с помощью TLC. Добавляли насыщенный раствор бикарбоната натрия (50 мл) с последующей экстракцией ЕА (3×30 мл). Органические фазы объединяли, сушили безводным сульфатом магния, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (силикагель 200-300 меш, DCM : МеОН = от 100:1 до 40:1) с получением 1-(транс-5-((6-(2,6-дихлор-3,5-диметокси-фенил)хиназолин-2-ил)амино)-2-азабицикло[2,1,1]гексан-2-ил)проп-2-ен-1-она (350 мг, выход: 70%) в виде белого твердого вещества.
1H NMR(400 MHz, DMSO-d6) δ (ppm): 9.15-9.17 (d, 1H), 7.51-7.69 (m, 4H) 7.00 (s, 1H), 6.43-6.55 (m, 1H), 6.07-6.12 (m, 1H), 5.92-5.97 (m, 1H), 5.65-5.68 (m, 1H), 4.77-4.79 (m, 1H), 4.12 (m, 1H), 3.92 (s, 6H), 3.83-3.85 (m, 1H), 3.26-3.59 (m, 211), 2.96-3.01 (m, 1H), 1.85-1.87 (m, 1H). 1.21-1.23 (m, 1H).
Молекулярная формула: C24H22Cl2N4O3 Молекулярная масса: 485,37 LC-MS (m/z)=485,00[M+H+].
Пример 6: Синтез 1-((3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-ил)пропил-2-ен-1-она (соединение 20)
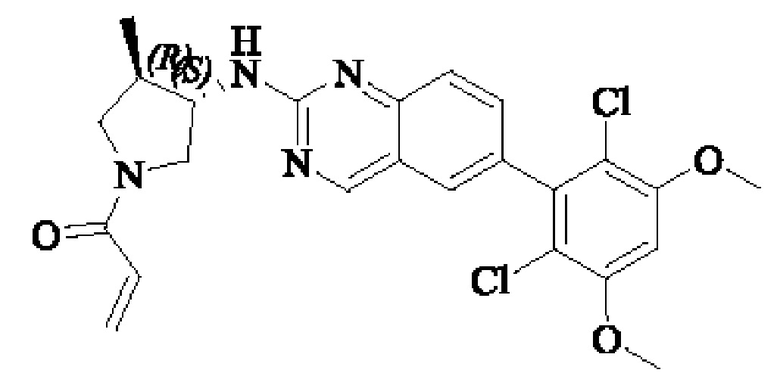
Стадия 1: Синтез трет-бутил(3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-карбоксилата
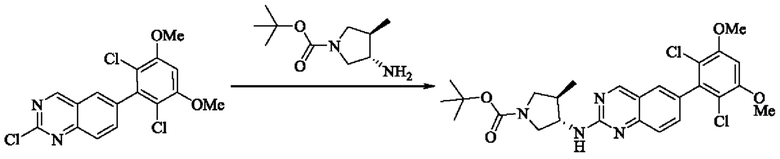
Вещество 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (400,0 мг, 1,1 ммоль, 1,0 экв) и трет-бутил-(3S,4R)-3-амино-4-метилпирролидин-1-карбоксилат (440,6 мг, 2,2 ммоль, 2,0 экв) растворяли в N-метилпирролидоне (3,0 мл), добавляли N,N-диизопропилэтиламин (568,7 мг, 4,4 ммоль, 4,0 экв) и постепенно нагревали до 110°С для реакции в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до комнатной температуры, добавляли 15 мл ледяной воды и фильтровали. Осадок на фильтре промывали небольшим количеством ледяной воды, растворяли в дихлорметане (5 мл), сушили над безводным сульфатом магния, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (силикагель 200-300 меш, РЕ : ЕА = от 10:1 до 3:1) с получением трет-бутил(3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-карбоксилата (346,0 мг, выход: 59,0%) в виде желтого твердого вещества.
Стадия 2: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,4R)-4-метилпирролидин-3-ил)хиназолин-2-амина гидрохлорида
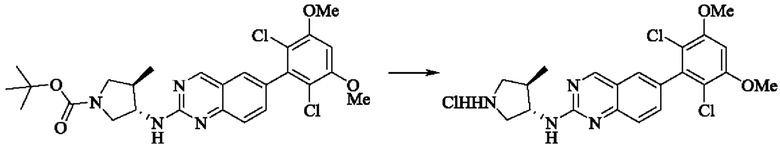
Трет-бутил(3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-карбоксилат (346,0 мг, 0,65 ммоль, 1,0 экв) растворяли в этаноле (5,0 мл), охлаждали до 0°С на ледяной бане, добавляли этанольный раствор хлористого водорода (5,0 мл) и постепенно нагревали до комнатной температуры для реакции в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор непосредственно концентрировали с получением 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,4R)-4-метилпирролидин-3-ил)хиназолин-2-амина гидрохлорида (313,0 мг сырого продукта, выход: 100%) в виде желтого твердого вещества.
Стадия 3: Синтез 1-((3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-ил)пропил-2-ен-1-она (соединение 20)
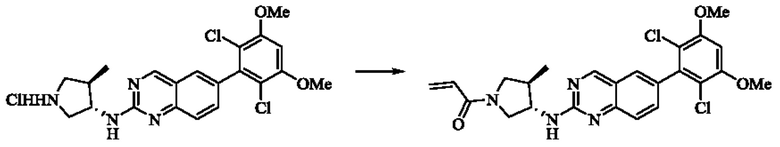
6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,4R)-4-метилпирролидин-3-ил)хиназолин-2-амина гидрохлорид (313,0 мг, 0,67 ммоль, 1,0 экв) растворяли в THF (10,0 мл), добавляли триэтиламин (203,0 мг, 2,0 ммоль, 3,0 экв), перемешивали при комнатной температуре в течение 30 мин, охлаждали до 0°С, медленно добавляли акрилоилхлорид (122,4 мг, 1,35 ммоль, 2,0 экв), нагревали до комнатной температуры постепенно для реакции в течение 1 ч. Завершение реакции детектировали с помощью TLC. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (20 мл) с последующим добавлением этилацетата (8,0 мл) для разделения раствора. Водную фазу экстрагировали этилацетатом (8,0 мл × 2). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (DCM : МеОН = от 125:1 до 80:1) с получением 1-((3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-ил)пропил-2-ен-1-она (160,5 мг, выход: 49,2%) в виде желтого твердого вещества.
1H NMR (400 MHz, DMSO) δ (ppm): 9.18 (s, 1Н), 7.82 (s, 1H), 7.79 (s, 1Н), 7.49-7.54 (m, 2Н), 7.02 (s, 1H), 6.60-6.71 (m, 1H), 6.16 (d, 1H), 5.67 (m, 1H), 4.12-4.20 (m, 2H). 4.03 (s, 6H), 3.97 (m, 1H), 3.24 (m, 1H), 3.05 (m, 1Н), 2.48 (m, 1H), 1.23 (s, 3H).
Молекулярная формула: C24H24Cl2N4O3 Молекулярная масса: 487,39 LC-MS (m/z)=489,07[M+H+].
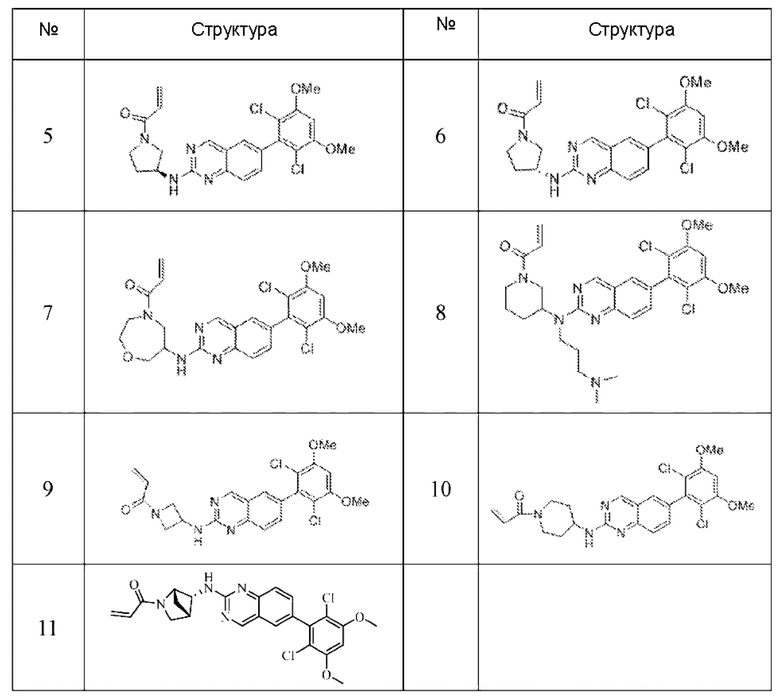
Пример 7: Синтез 1-((2R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-метилпирролидин-1-ил)пропил-2-ен-1-она (соединение 21)
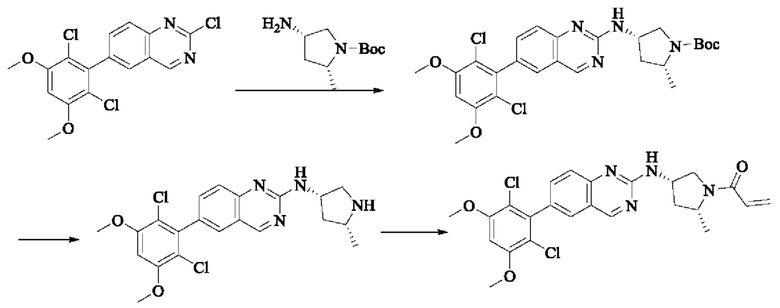
Стадия 1: Синтез трет-бутил(2R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-метилпирролидин-1-карбоксилата
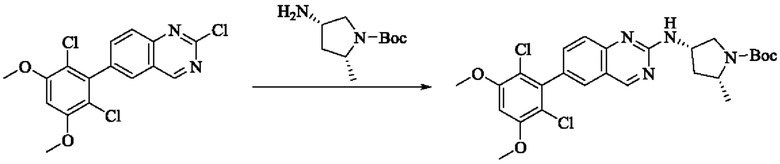
2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (200 мг, 0,514 ммоль, 1,0 экв), трет-бутил(2R,4S)-4-амино-2-метилпирролидин-1-карбоксилат (162,63 мг, 0,812 ммоль, 1,5 экв) и N,N-диизопропилэтиламин (139,85 мг, 1,082 ммоль, 2,0 экв) растворяли в N-метилпирролидоне (2 мл), нагревали до 120°С для реакции в течение ночи. Было обнаружено путем детектирования TLC, что все еще присутствует небольшое количество оставшегося вещества. Реакционный раствор выливали в холодную воду (20 мл) для осаждения коричневого твердого вещества и фильтровали с помощью отсасывания. Фильтрат экстрагировали с помощью ЕА (10 мл × 3) и коричневое твердое вещество растворяли в DCM. Органические фазы объединяли и промывали водой (20 мл × 3) с последующим разделением раствора. Полученную органическую фазу сушили безводным сульфатом натрия, фильтровали и осадок на фильтре промывали DCM. Маточную жидкость концентрировали при пониженном давлении. Сырой продукт подвергали колоночной хроматографии на силикагеле (РЕ : ЕА = от 5:1 до 3:1) с получением твердого трет-бутил(2R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-ил-метилпирролидин-1-карбоксилата (100 мг, выход: 35%).
Стадия 2: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,5R)-5-метилпирролидин-3-ил)хиназолин-2-амина
Трет-бутил(2R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-метилпирролидин-1-карбоксилат (100 мг, 0,187 ммоль, 1,0 экв) растворяли в HCl-этаноле (15 мл), нагревали до 45°С и проводили реакцию в течение 3 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор концентрировали. Сырой продукт растворяли в ТГФ и концентрировали, повторяя трижды. Сырой продукт использовали на следующей стадии без очистки.
Стадия 3: Синтез 1-((2R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-метилпирролидин-1-ил)проп-2-ен-1-она (соединение 21)
6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,5R)-5-метилпирролидин-3-ил)хиназолин-2-амин (88 мг, 0,203 ммоль, 1,0 экв) растворяли в THF (15 мл), добавляли триэтиламин (102,71 мг, 1,015 ммоль, 5,0 экв), охлаждали до 0°С и медленно добавляли акрилоилхлорид (18,37 мг, 0,203 ммоль, 1,0 экв)для реакции в течение ночи. Завершение реакции детектировали с помощью TLC. Для разделения раствора добавляли насыщенный раствор бикарбоната натрия (10 мл). Водную фазу экстрагировали ЕА (10 мл × 3) и отделяли. Органическую фазу сушили безводным сульфатом натрия и фильтровали. Осадок на фильтре промывали ЕА и маточный раствор концентрировали при пониженном давлении. Сырой продукт отделяли с помощью PTLC (DCM : МеОН = 15:1) с получением твердого 1-((2R,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-метилпирролидин-1-ил)проп-2-ен-1-она (21 мг, выход: 21%).
1H NMR (400 MHz, DMSO) δ (ppm): 9.18 (s, 1H), 7.82 (s, 1H), 7.68 (s, 1H), 7.49-7.57 (m, 2H), 7.00 (s, 1H), 6,54-6.58 (m, 1H), 6.11-6.18 (m, 1H), 5.63-5.67 (m, 1H), 4.45-4,47 (m, 3H), 4.11-4.13 (m, 6H), 3.40-3.44 (m, 1H).
Молекулярная формула: C24H24Cl2N4O3 Молекулярная масса: 487,38 LC-MS(m/z)=487,1[M+H+].
Пример 8: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-ил)проп-2-ен-1-она (соединение 22)
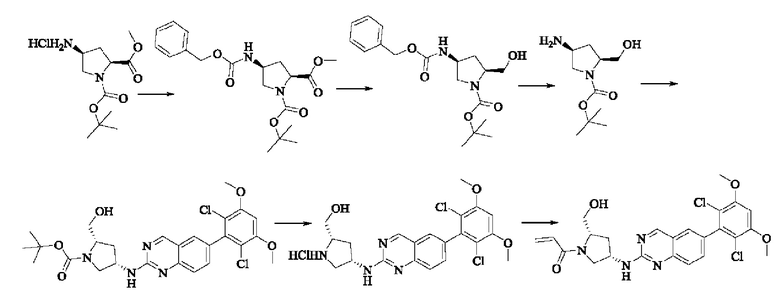
Стадия 1: Синтез 1-трет-бутил-2-метил(2S,4S)-4-(((бензилокси)карбонил)амино)пирролидин-1,2-дикарбоксилата
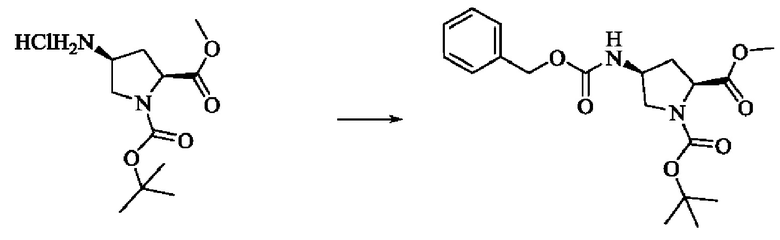
Вещество 1-(трет-бутил)-2-метил(2S,4S)-4-аминопирролидин-1,2-дикарбоксилата гидрохлорид (10,5 г, 36,8 ммоль, 1,0 экв) растворяли в DCM (100 мл), добавляли триэтиламин (11,7 г, 115,8 ммоль, 3,0 экв), перемешивали в течение 0,5 ч при комнатной температуре и охлаждали до 0°С в ледяной бане, затем медленно добавляли бензилхлорформиат (7,9 г, 46,3 ммоль, 1,2 экв) растворяли в DCM с помощью воронки с постоянным давлением, нагревали до комнатной температуры постепенно и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. Для разделения раствора добавляли насыщенный раствор бикарбоната натрия (100 мл). Водную фазу экстрагировали DCM (50 мл × 2) и органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (200-300 меш силикагель, РЕ : ЕА = от 10:1 до 3:1) с получением 1-трет-бутил-2-метил(2S,4S)-4-(((бензилокси)карбонил)амино)пирролидин-1,2-дикарбоксилата (3,6 г, выход: 24,7%) в виде бесцветного масла.
Стадия 2: Синтез трет-бутил-(2S,4S)-4-((((бензилокси)карбонил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилата
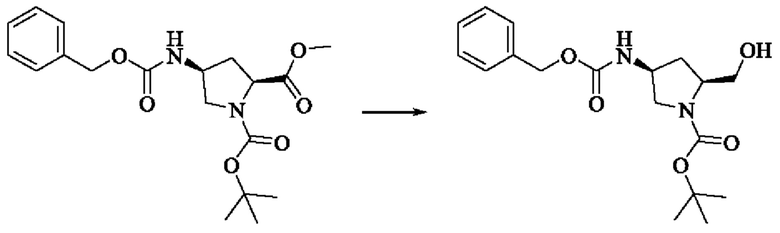
Литийалюминийгидрид (0,7 г, 19,0 ммоль, 2,0 экв) растворяли в безводном THF (20 мл) при 0°С, перемешивали в течение 0,5 ч, медленно добавляли 1-трет-бутил-2-метил-(2S,4S)-4-(((бензилокси)карбонил)амино)пирролидин-1,2-дикарбоксилат (3,6 г, 9,5 ммоль, 1,0 экв), растворенный в растворе THF, нагревали до комнатной температуры постепенно и проводили реакцию в течение 2 часов. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до 0°С, медленно добавляли воду (0,7 мл) и 10% раствор гидроксида натрия (0,7 мл), добавляли воду (2,1 мл) и перемешивали в течение 0,5 ч. Реакционный раствор фильтровали и фильтрат концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (200-300 меш силикагель, РЕ : ЕА = от 10:1 до 2:1) с получением трет-бутил(2S,4S)-4-((((бензилокси)карбонил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилата (1,05 г, выход: 30,0%) в виде бесцветного масла.
Стадия 3: Синтез трет-бутил-(2S,4S)-4-амино-2-(гидроксиметил)пирролидин-1-карбоксилата
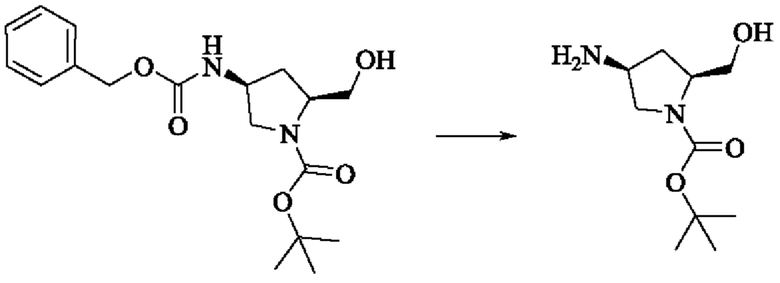
Трет-бутил(2S,4S)-4-((((бензилокси)карбонил)амино)-2-(гидроксиметил)-пирролидин-1-карбоксилат (250 мг, 0,71 ммоль, 1,0 экв) растворяли в метаноле (5,0 мл), добавляли палладий на угле (75,2 мг, 7,2 ммоль, 0,01 экв), добавляли водород 4 раза, перемешивали при комнатной температуре и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор фильтровали путем отсасывания через целит и фильтрат концентрировали с получением трет-бутил(2S,4S)-4-амино-2-(гидроксиметил)пирролидин-1-карбоксилата (123,0 мг, выход 80,2%) в виде бесцветного прозрачного масла.
Стадия 4: Синтез трет-бутил-(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилата
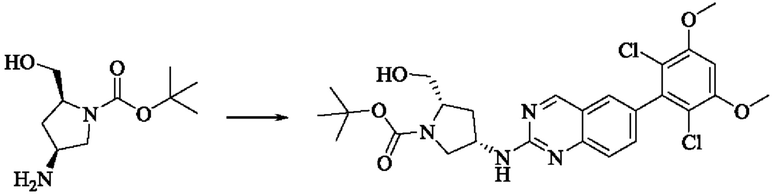
Вещества трет-бутил(2S,4S)-4-амино-2-(гидроксиметил)пирролидин-1-карбоксилат (123,0 мг, 0,57 ммоль, 1,0 экв) и 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (231,5 мг, 0,63 ммоль, 1,1 экв) растворяли в N-метилпирролидоне (4,0 мл), добавляли N,N-диизопропилэтиламин (147,3 мг, 1,14 ммоль, 2,0 экв), постепенно нагревали до 110°С, перемешивали и проводили реакцию в течение 5 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до комнатной температуры, добавляли 20 мл ледяной воды, перемешивали в течение 10 мин и фильтровали. Осадок на фильтре промывали небольшим количеством ледяной воды, затем растворяли в дихлорметане (10 мл), сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (200-300 меш силикагеля, DCM : МеОН = от 100:1 до 50:1) с получением трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилата (113,0 мг, выход: 36,1%) в виде кленового твердого вещества.
Стадия 5: Синтез ((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин)-2-амино)пирролидин)-2-метанола гидрохлорида
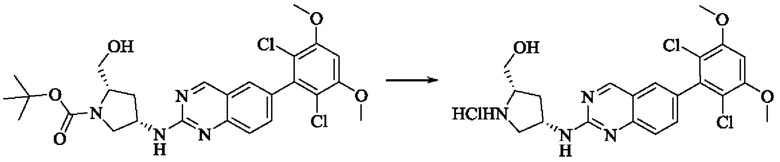
Вещество трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилат (113,0 мг, 0,21 ммоль, 1,0 экв) растворяли в этаноле (5,0 мл), охлаждали до 0°С в ледяной бане, добавляли этанольный раствор хлористого водорода (5,0 мл), нагревали постепенно до комнатной температуры и проводили реакцию в течение ночи.
Завершение реакции детектировали с помощью TLC. Реакционный раствор прямо концентрировали с получением ((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин)-2-амино)пирролидин)-2-метанола гидрохлорида (180,0 мг сырого продукта, выход: 100%) в виде желтого твердого вещества.
Стадия 6: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин)-1-пропил-2-ен-1-она (соединение 22)
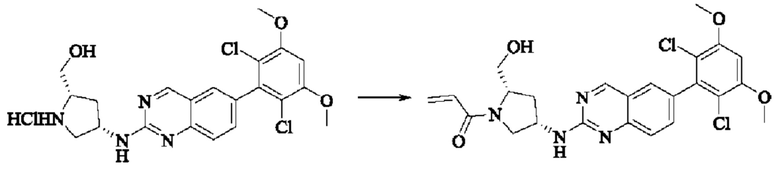
Вещество ((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин)-2-амино)пирролидин)-2-метанола гидрохлорид (160,0 мг, 0,35 ммоль, 1,0 экв) растворяли в THF (10,0 мл), добавляли триэтиламин (106,0 мг, 1,1 ммоль, 3,0 экв), перемешивали при комнатной температуре для реакции в течение 0,5 ч, охлаждали до 0°С, медленно добавляли акрилоилхлорид (38,0 мг, 0,42 ммоль, 1,2 экв), нагревали до комнатной температуры постепенно и проводили реакцию в течение 1 ч. Завершение реакции детектировали с помощью TLC. К реакционному раствору добавляли насыщенный раствор бикарбоната натрия (15 мл) с последующим добавлением этилацетата (8 мл) для разделения раствора. Водную фазу экстрагировали ЕА (8,0 мл × 2) и органические фазы объединяли, промывали насыщенным рассолом, сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (200-300 меш силикагель, DCM: МеОН= от 100:1 до 50:1) с получением 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-ил)проп-2-ен-1-она (75,0 мг, выход: 42,5%) в виде бледно-желтого твердого вещества.
1H NMR (400 MHz, DMSO) δ (ppm): 9.18 (s, 1H), 7.89 (s, 1H), 7.80 (s, 1H), 7.55 (d, 1H), 7.52 (d, 1H), 7.00 (s, 1Н), 6.60-6.71 (m, 1Н), 6,16 (d, 1H), 5.67 (d, 1H), 5.09 (s, 1H), 4.55 (m, 1H), 4.12-4.20 (m, 2H), 4.03 (s, 6H), 3.97 (m, 1H), 3.74 (m, 1H), 3.52 (m, 1H), 1.98 (m, 2H).
Молекулярная формула: C24H24Cl2N4O4 Молекулярная масса: 503,39 LC-MS (m/z)=505,40 [M+H+].
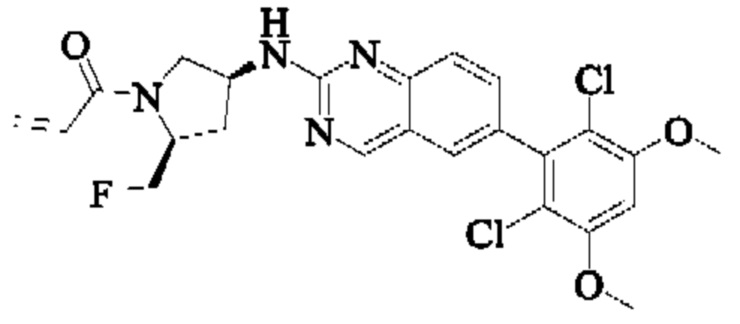
Пример 9: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(фторметил)пирролидин-1-ил)проп-2-ен-1-она (соединение 24)
Стадия 1: Синтез 1-(трет-бутил)-2-метил(2S,4R)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1,2-дикарбоксилата
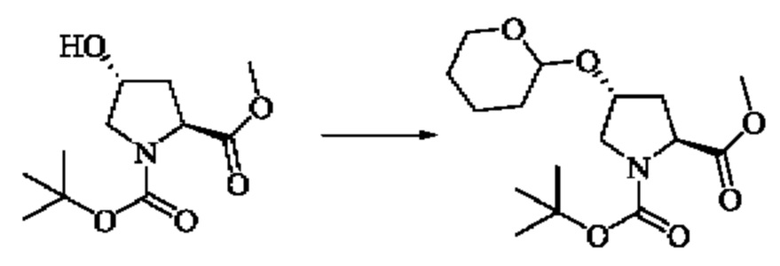
Вещество 1-(трет-бутил)-2-метил(2S,4R)-4-гидроксипирролидин-1,2-дикарбоксилат (10,0 г, 40,8 экв) растворяли в трет-бутилметиловом эфире (100 мл), добавляли пара-толуолсульфокислоту (234,2 мг, 1,36 ммоль), охлаждали до 0°С в ледяной бане, медленно добавляли 3,4-дигидро-2Н-пиран (10,3 г, 122,4 ммоль), медленно нагревали до комнатной температуры и проводили реакцию в течение 3 ч. Завершение реакции детектировали с помощью TLC. 1-(трет-бутил)2-метил(2S,4R)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1,2-дикарбоксилат (14 г) получали путем концентрирования при пониженном давлении.
Стадия 2: Синтез трет-бутил(2S,4R)-2-(гидроксиметил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилата
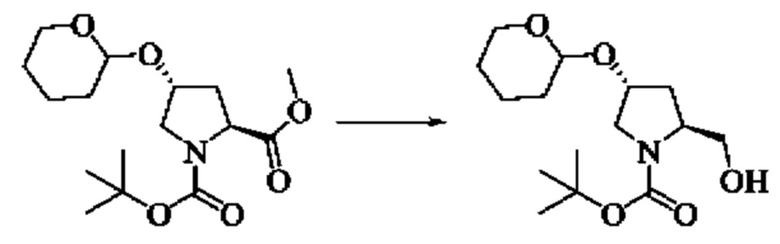
Алюминийгидрид лития (3,1 г, 81,6 ммоль) добавляли к тетрагидрофурану (100,0 мл), охлаждали до -20°C с использованием этанола, воды и сухого льда, по каплям добавляли 1-(трет-бутил)-2-метил(2S,4R)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1,2-дикарбоксилат (13,43 г, 40,8 ммоль) в растворе тетрагидрофурана (100 мл), перемешивали при -20°С в атмосфере азота и проводили реакцию в течение 3 ч. Завершение реакции детектировали с помощью TLC. Воду (3,1 мл), 10% NaOH (3,1 мл) и воду (9,3 мл) добавляли к реакционному раствору в ледяной бане, перемешивали в течение 10 мин, фильтровали с помощью отсасывания через целит. Фильтрат концентрировали непосредственно досуха с получением трет-бутил(2S,4R)-2-(гидроксиметил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилата (13,1 г сырого продукта, выход 100%).
Стадия 3: Синтез трет-бутил(2S,4R)-2-(((метилсульфонил)окси)метил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилата
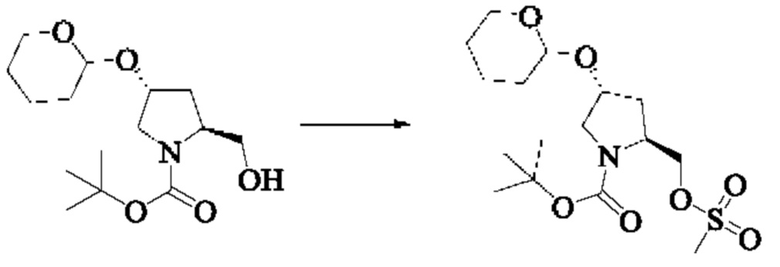
Вещество трет-бутил(2S,4R)-2-(гидроксиметил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилат (2,0 г, 6,65 ммоль) растворяли в дихлорметане (10,0 мл), охлаждали до 0°С в ледяной бане, добавляли триэтиламин (1,35 г, 13,3 ммоль), медленно добавляли метансульфонилхлорид (1,15 г, 9,975 ммоль) при 0°С, медленно нагревали до комнатной температуры и проводили реакцию в течение 1 ч. Завершение реакции детектировали с помощью TLC. Добавляли дихлорметан (10 мл), затем насыщенный хлорид аммония (10 мл) для разделения. Органические фазы объединяли, промывали водой (3×5 мл), затем промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, и подвергали колоночной хроматографии (РЕ: ЕА = 15:1, 10:1, DCM: МеОН = 100:1) с получением трет-бутил(2S,4R)-2-(((метилсульфонил)окси)метил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилата (2,0 г, выход 79,3%).
Стадия 4: Синтез трет-бутил(2S,4R)-2-(fluro метил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилата
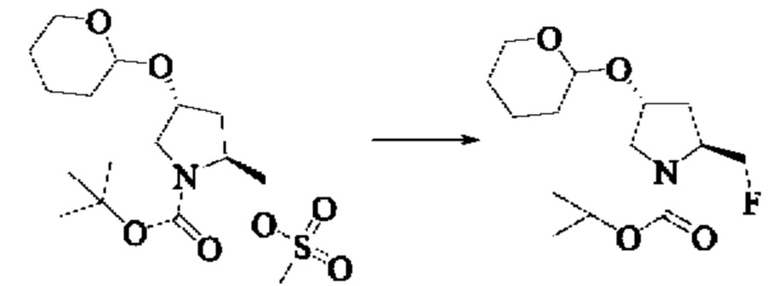
Вещество трет-бутил(2S,4R)-2-(((метилсульфонил)окси)метил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилат (7,5 г, 19,8 ммоль) растворяли в тетрагидрофуране (45,0 мл), добавляли тетраметиламмония фторид (1,5 г, 4,75 ммоль), нагревали до 80°С и кипятили с обратным холодильником. Завершение реакции детектировали с помощью TLC. Реакционный раствор сушили при пониженном давлении, добавляли этилацетат (10 мл), промывали водой (5×10 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия, и подвергали колоночной хроматографии (DCM: МеОН = 200:1, 150:1, 100:1) с получением трет-бутил(2S,4R)-2-(((метилсульфонил)окси)метил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилата (3,2 г, выход 53,3%).
Стадия 5: Синтез трет-бутил(2S,4R)-2-(фторметил)-4-гидроксипирролидин-1-карбоксилата
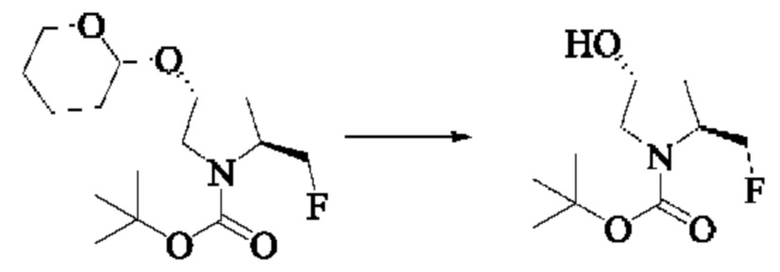
Вещество трет-бутил(2S,4R)-2-(фторметил)-4-((тетрагидро-2Н-пиран-2-ил)окси)пирролидин-1-карбоксилат (3,2 г, 10,55 ммоль) растворяли в смешанном растворителе из уксусной кислоты (33,0 мл), тетрагидрофурана (33,0 мл) и воды (33,0 мл), и кипятили с обратным холодильником. Завершение реакции детектировали с помощью TLC. рН доводили насыщенным бикарбонатом натрия (8-9) и удаляли тетрагидрофуран путем выпаривания. Водную фазу экстрагировали этилацетатом (3×50 мл). Органические фазы объединяли, промывали водой (1×10 мл), затем насыщенным солевым раствором, сушили над безводным сульфатом натрия, и подвергали колоночной хроматографии (DCM: МеОН = 200:1, 150:1, 100:1, 50:1) с получением трет-бутил(2S,4R)-2-(фторметил)-4-гидроксипирролидин-1-карбоксилата (2,0 г, выход 87%).
Стадия 6: Синтез трет-бутил(2S,4S)-4-азидо-2-(фторметил)пирролидин-1-карбоксилата
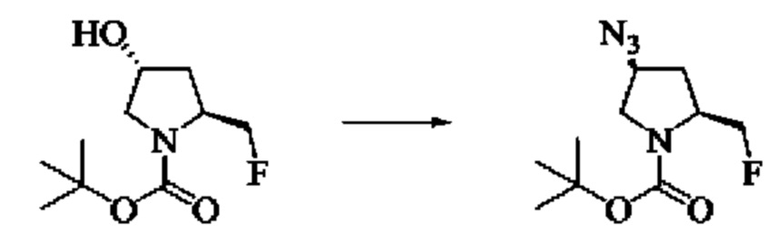
Вещество трет-бутил(2S,4R)-2-(фторметил)-4-гидроксипирролидин-1-карбоксилат (100,0 мг, 0,46 ммоль) растворяли в тетрагидрофуране (1,5 мл), охлаждали до 0°С в ледяной бане, добавляли трифенилфосфин (144,8 мг, 0,552 ммоль), затем добавляли диизопропилазодикарбоксилат (111,6 мг, 0,552 ммоль) при 0°С, перемешивали при 0°С в течение 20 мин, медленно добавляли DDPA (151,9 мг, 0,552 ммоль) при 0°С и медленно нагревали до комнатной температуры в течение ночи. Завершение реакции детектировали с помощью TLC и тетрагидрофуран удаляли путем выпаривания, затем проводили колоночную хроматографию (РЕ: ЕА = 5:1, 2:1) с получением трет-бутил(2S,4S)-4-азидо-2-(фторметил)пирролидин-1-карбоксилата (170,0 мг, выход 100%).
Стадия 7: Синтез трет-бутил(2S,4S)-4-амино-2-(фторметил)пирролидин-1-карбоксилата
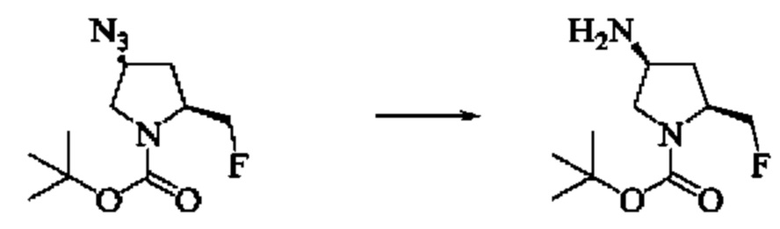
Вещество трет-бутил(2S,4S)-4-азидо-2-(фторметил)пирролидин-1-карбоксилат (112,3 мг, 0,46 ммоль) растворяли в метаноле (2 мл), добавляли палладий на угле (5,62 мг, 5%wt) и добавляли водород три раза, затем гидрировали в течение ночи. Завершение реакции детектировали с помощью TLC. Смесь фильтровали путем отсасывания через целит и фильтрат выпаривали с получением трет-бутил(2S,4S)-4-амино-2-(фторметил)пирролидин-1-карбоксилата (105,0 мг).
Стадия 8: Синтез трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(фторметил)пирролидин-1-карбоксилата
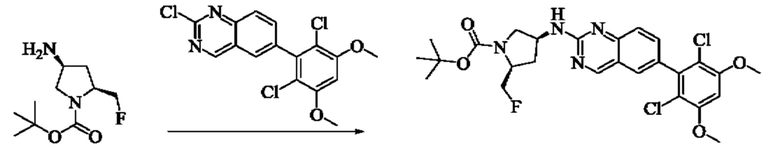
Вещество трет-бутил(2S,4S)-4-амино-2-(фторметил)пирролидин-1-карбоксилат (100,3 мг, 0,46 ммоль) растворяли в N-метилпирролидоне (2,0 мл), добавляли N,N-диизопропилэтиламин (178,21 мг, 1,38 ммоль) и 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (203,1 мг, 0,552 ммоль), постепенно нагревали до 110°С и нагревали с обратным холодильником в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до комнатной температуры, добавляли 10 мл воды, и фильтровали. Остаток на фильтре промывали малым количеством ледяной воды, растворяли в дихлорметане (10 мл), сушили над безводным сульфатом натрия, очищали на препаративной пластине силикагеля (DCM: МеОН = 20:1) с получением трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(фторметил)пирролидин-1-карбоксилата (50,0 мг, выход: 19,8%).
Стадия 9: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,5S)-5-(фторметил)пирролидин-3-ил)хиназолин-2-амина
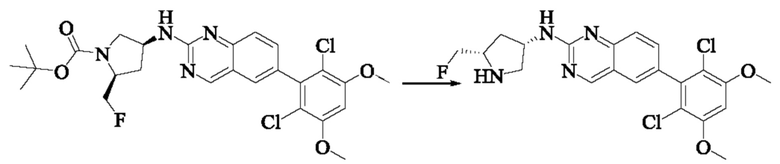
Вещество трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-2-(фторметил)пирролидин-1-карбоксилат (45,0 мг, 0,082 ммоль) растворяли в DCM (2,0 мл), охлаждали до 0°С в ледяной бане, добавляли этанольный раствор хлористого водорода (1,0 мл), нагревали постепенно до комнатной температуры и проводили реакцию в течение 2 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор концентрировали прямо досуха с получением 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,5S)-5-(фторметил)пирролидин-3-ил)хиназолин-2-амина (40,0 мг).
Стадия 10: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(фторметил)пирролидин-1-ил)проп-2-ен-1-она
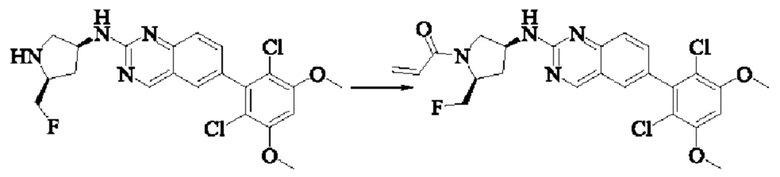
Вещество 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,5S)-5-(фторметил)пирролидин-3-ил)хиназолин-2-амин (37,0 мг, 0,082 ммоль) растворяли в THF (1,0 мл), охлаждали до 0°С в ледяной бане, добавляли триэтиламин (24,0 мг, 0,246 ммоль), затем добавляли акрилоилхлорид (11,13 мг, 0,123 ммоль) при 0°С, нагревали постепенно до комнатной температуры и проводили реакцию в течение 2 ч. Завершение реакции детектировали с помощью TLC. После добавления воды (5 мл), водную фазу экстрагировали этилацетатом (8,0 мл×3). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили безводным сульфатом натрия и подвергали колоночной хроматографии (DCM: МеОН = 250:1, 200:1, 150:1, 100:1) с получением 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(фторметил)пирролидин-1-ил)проп-2-ен-1-она (11,5 мг, выход: 27,7%).
1H NMR (400 MHz, DMSO-d6) δ (ppm): 11.93 (s, 1H), 9.19 (s, 1H), 7.85-7.83 (d, 1Н), 7.69 (s, 1H), 7.58-7.50 (m, 2H), 7.01 (s, 1H), 6.59 (s, 2H), 6.19-6.15 (d, 1H), 5.71-5.69 (d, 1H), 5.31 (s, 1H), 4.65-4.54 (d, 4H), 4.44 (s, 1H), 4.27-4.19 (d, 3H), 3.97 (s, 6H), 3.81-3.75 (d, 2H), 2.19-2.16 (t, 2H), 2.10-1.98 (m, 3H).
Молекулярная формула: C24H22Cl2N4O3 Молекулярная масса: 485,37 LC-MS (Pos, m/z)=485,43 [M+H+].
Пример 10: Синтез 2-((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)ацетонитрила (соединение 27)
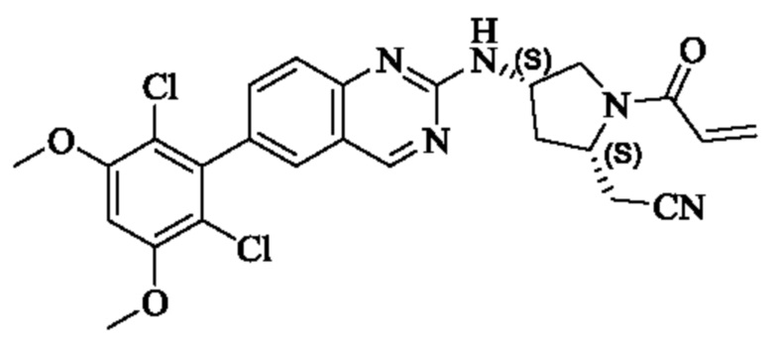
Стадия 1: Синтез трет-бутил(2S,4S)-2-(бромметил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
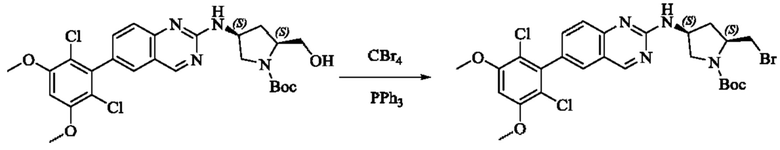
Вещество трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилат (1571 мг, 2,859 ммоль, 1,0 экв) растворяли в DCM (20 мл), перемешивали, затем добавляли тетрабромид углерода (1185 мг, 3,574 ммоль, 1,25 экв) и охлаждали до 0°С. Трифенилфосфин (2250 мг, 8,577 ммоль, 3 экв) добавляли порциями, реакционную смесь медленно нагревали до комнатной температуры в атмосфере азота и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. В реакционный раствор прямо добавляли силикагель и растворитель удаляли путем выпаривания, затем разделяли колоночной хроматографией (элюент, РЕ: ЕА = 3:1) с получением трет-бутил(2S,4S)-2-(бромметил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (800 мг, выход 45,7%).
Стадия 2: Синтез трет-бутил(2S,4S)-2-(цианометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
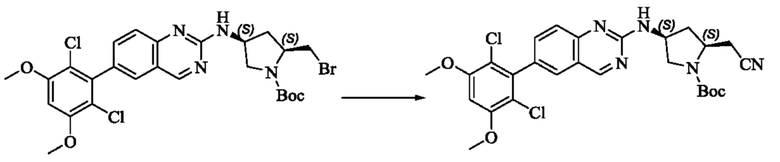
Промежуточное вещество трет-бутил(2S,4S)-2-(бромметил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (800 мг, 1,3064 ммоль, 1 экв) растворяли в ацетонитриле (10 мл), добавляли TMSCN (259 мг, 2,6128 ммоль, 2 экв) и необходимое количество тетраэтиламмония фторида при комнатной температуре, нагревали до 60°С и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали, добавляли 100 мл воды, экстрагировали DCM (200 мл×3) для разделения раствора. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали досуха при пониженном давлении, и separated колоночной хроматографией (элюент, РЕ: ЕА = 5:1-4:1) с получением трет-бутил(2S,4S)-2-(цианометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (215 мг, выход 29,5%).
Стадия 3: Синтез 2-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)ацетонитрила
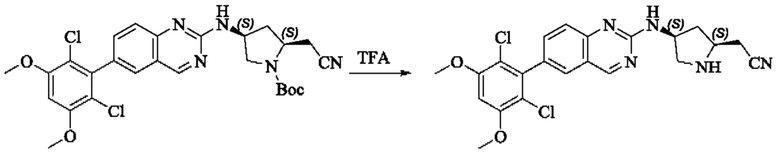
Промежуточное вещество трет-бутил(2S,4S)-2-(цианометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (215 мг) растворяли в DCM (4 мл), охлаждали до 0°С, и добавляли TFA (3 мл). после этого реакционную систему медленно нагревали до комнатной температуры и завершение реакции детектировали с помощью TLC. Смесь концентрировали досуха при пониженном давлении при комнатной температуре, добавляли DCM и концентрировали досуха при пониженном давлении. TFA удаляли из системы как можно более полно. 2-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)ацетонитрил (теоретический выход, 176 мг) получали и использовали непосредственно на следующей стадии.
Стадия 4: Синтез 2-((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)ацетонитрила
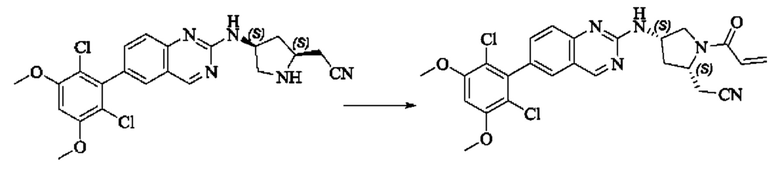
Промежуточное вещество 2-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)ацетонитрил (в теоретическом количестве 176 мг, 0,3840 ммоль) растворяли в THF (4 мл), добавляли TEA, и охлаждали до -10°С. Раствор акрилоилхлорида (41 мг, 0,4608 ммоль, 1,2 экв, растворенный в 1 мл DCM) медленно добавляли по каплям в реакционную систему. Завершение реакции детектировали с помощью TLC. Добавляли насыщенный раствор бикарбоната натрия при охлаждении с получением щелочной реакционной системы, затем экстрагировали DCM (100 мл× 3) для разделения раствора. Органические фазы объединяли, сушили над безводным сульфатом натрия, и фильтровали. Фильтрат концентрировали и подвергали колоночной хроматографии (DCM: МеОН = от 200:1 до 50:1) с получением 2-((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)ацетонитрила (138 мг, выход: 70,4%).
1H NMR (400 MHz, DMSO) δ (ppm): 9.21 (s, 1H), 7.98-8.00 (d, 1H), 7.70-7.71 (d, 1H), 7.52-7.6l (m, 2H), 7.02 (s, 1H), 6.60-6.67 (q, 1H), 6.18-6.23 (q, 1H), 5.72-5.76 (t, 1H), 4.57-4.59 (t, 1H), 4.51-4.53 (d, 1H), 4.24-4.25 (d, 2H), 3.98 (s, 3H), 3.48-3.50 (d, 3H), 3.42-3.43 (d, 2H), 3.13-3.22 (m, 1H), 3.01-3.10 (m, 1H), 1.98-2.05 (q, 1H).
Молекулярная формула: C25H23Cl2N5O3, Молекулярная масса: 512,39, LC-MS (Pos, m/z)=512,1 [M+H+].
Пример 11: Синтез (2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбонитрила (соединение 28)
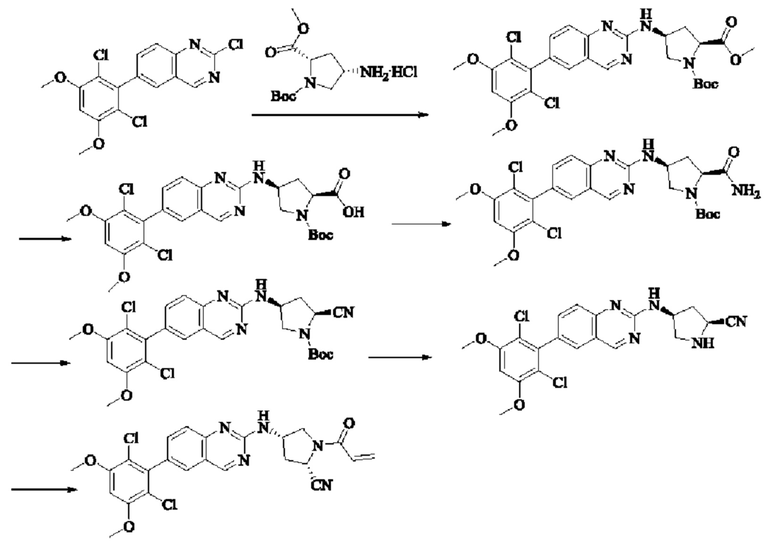
Стадия 1: Синтез 1-трет-бутил-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата
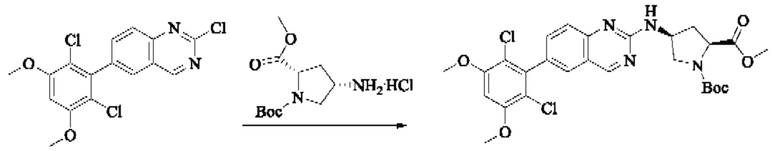
Вещества 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (500,0 мг, 1,353 ммоль, 1,0 экв), 1-трет-бутил-2-метил(2S,4S)-4-аминопирролидин-1,2-дикарбоксилата гидрохлорид (644 мг, 2,030 ммоль, 1,5 экв) и N,N-диизопропилэтиламин (874,4 мг, 6,765 ммоль, 5,0 экв) растворяли в N-метилпирролидоне (5 мл), нагревали до 120°С и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC на следующее утро. Реакционный раствор выливали в ледяную воду (20 мл), осаждали коричневое твердое вещество. Смесь фильтровали путем отсасывания, и фильтрат экстрагировали этилацетатом (10 мл×3). Коричневое твердое вещество растворяли в этилацетате, органические фазы объединяли, промывали водой (20 мл×3) для разделения раствора. Органическую фазу сушили над безводным сульфатом натрия, фильтровали и остаток на фильтре промывали этилацетатом. Маточный раствор концентрировали при пониженном давлении и сырой продукт подвергали колоночной хроматографии на силикагеле (РЕ: ЕА = 5:1) с получением твердого 1-трет-бутил-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата (384 мг, выход: 49%).
Стадия 2: Синтез (2S,4S)-1-трет-бутоксикарбонил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты
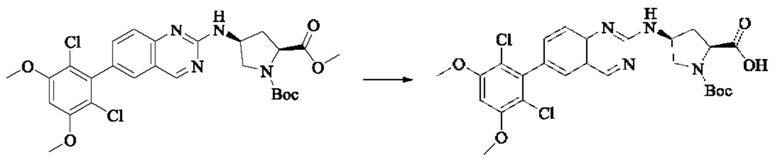
1-трет-бутил-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилат (300 мг, 0,5195 ммоль, 1,0 экв) растворяли в метаноле (5 мл), перемешивали и растворяли, охлаждали до 0°С и по каплям добавляют раствор моногидрата гидроксида лития (65 мг, 1,558 ммоль, 3,0 экв.) в воде (1 мл). После реакции в течение 3 часов TLC использовали для обнаружения оставшихся материалов. Добавляли раствор моногидрата гидроксида лития (65 мг, 1,558 ммоль, 3,0 экв.) в воде (1 мл). Метанол добавляли для получения осветленной системы и оставляли реагировать в течение ночи. Завершение реакции детектировали с помощью TLC и реакционную систему концентрировали при пониженном давлении, добавляли воду (10 мл) и метил-трет-бутиловый эфир (10 мл), перемешивали и разделяли. Этилацетат (20 мл) добавляли в водную фазу и охлаждали до 0°С. рН доводили до 5-6 лимонной кислотой и органическую фазу отделяли. Водную фазу экстрагировали этилацетатом (10 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Осадок на фильтре промывали этилацетатом. Маточный раствор концентрировали при пониженном давлении с получением (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты (260 мг, выход: 89%) в виде бледно-желтого твердого вещества.
Стадия 3: Синтез трет-бутил(2S,4S)-2-карбамоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
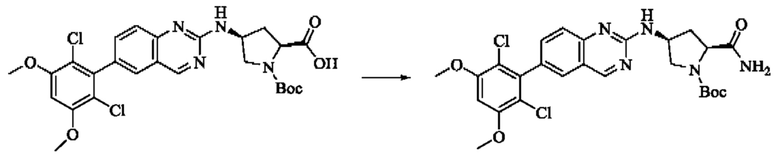
(2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-2-карбоновую кислоту (260 мг, 0,462 ммоль, 1,0 экв) растворяли в THF (10 мл), добавляли триэтиламин (140,1 мг, 1,384 ммоль, 3,0 экв), охлаждали до примерно -15°С, и медленно добавляли изобутилхлорформиат (75,64 мг, 0,554 ммоль, 1,2 экв). После реагирования в течение 2 ч завершение реакции детектировали с помощью TLC. Реакционный раствор добавляли по каплям к водному раствору аммиака, охлаждали до 0°С (5 мл). После реагирования в течение 3 ч завершение реакции детектировали с помощью TLC. Добавляли насыщенный раствор карбоната калия (10 мл), насыщенный солевой раствор (10 мл) и МТВЕ (20 мл), перемешивали и разделяли. Водную фазу экстрагировали МТВЕ (10 мл). Органические фазы объединяли, сушили и концентрировали с получением трет-бутил(2S,4S)-2-карбамоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-1-карбоксилата (350 мг сырого) в виде бледно-желтого твердого вещества.
Стадия 4: Синтез трет-бутил(2S,4S)-2-циано-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
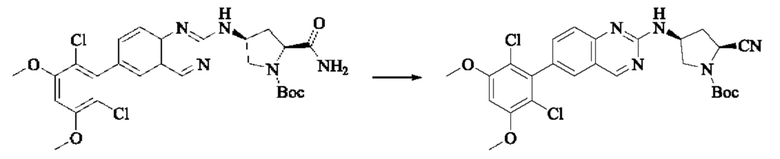
Трет-бутил(2S,4S)-2-карбамоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-1-карбоксилат (250 мг, 0,445 ммоль, 1,0 экв) растворяли в THF (10 мл), добавляли триэтиламин (112,6 мг, 1,113 ммоль, 2,5 экв), охлаждали до примерно 0°С, и медленно добавляли TFAA (186,9 мг, 0,89 ммоль, 2,0 экв). Через 0,5 ч температуру повышали до 40°С и проводили реакцию в течение ночи. Оставшийся материал детектировали TLC. TFAA (93,46 мг, 0,445 ммоль, 1,0 экв) добавляли и реакцию продолжали в течение 1 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали, добавляли DCM (20 мл) и насыщенный бикарбонат натрия (10 мл), перемешивали и разделяли. Органическую фазу отделяли, сушили над безводным сульфатом натрия и фильтровали. Остаток на фильтре промывали DCM и маточную жидкость концентрировали при пониженном давлении с получением твердого трет-бутил(2S,4S)-2-циано-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (350 мг сырого).
Стадия 5: Синтез (2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбонитрила
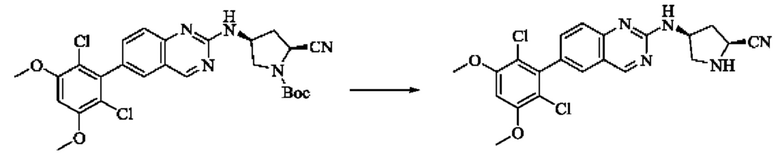
Промежуточное вещество трет-бутил(2S,4S)-2-циано-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (350 мг, 0,445 ммоль, 1,0 экв) растворяли в DCM (5 мл), добавляли трифторуксусную кислоту (5 мл), нагревали до 30°С и проводили реакцию в течение 4 ч. Завершение реакции детектировали с помощью TLC и реакционный раствор концентрировали. В сырой продукт добавляли ЕА и концентрировали, повторяли три раза. МТВЕ (20 мл) добавляли и осаждали твердое вещество, затем фильтровали путем отсасывания с получением твердого (2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбонитрила (143 мг, выход: 50%).
Стадия 6: Синтез (2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбонитрила (соединение 28)
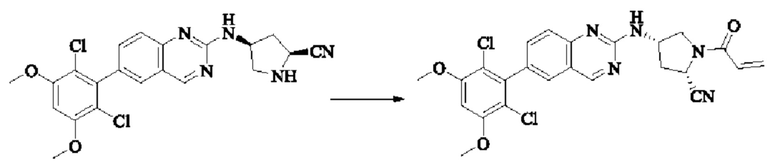
(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-пирролидин-2-карбонитрил (143 мг, 0,322 ммоль, 1,0 экв) растворяли в THF (10 мл), добавляли триэтиламин (162,81 мг, 1,61 ммоль, 5,0 экв), охлаждали до 0°С, медленно добавляли акрилоилхлорид (29,12 мг, 0,322 ммоль, 1,0 экв). Завершение реакции детектировали с помощью TLC После реагирования в течение 4 ч добавляли раствор насыщенного бикарбоната натрия (10 мл) для разделения раствора. Водную фазу экстрагировали ЕА (10 мл×3), и органические фазы объединяли и концентрировали. Сырой продукт разделяли на толстой препаративной пластине силикагеля (РЕ: ЕА = 2:1) с получением твердого (2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-пирролидин-2-карбонитрила (10 мг, выход: 6%).
1H NMR (400 MHz, DMSO) δ (ppm): 9.04 (s, 1H), 7.72 (m, 1H), 7.60-7.70 (m, 2H), 6.65 (s, 1H), 6.51-6.55 (m, 1H), 6.42-6.51 (m, 1H), 5.81-5.84 (m, 1H), 5.65 (s, 1H), 5.30-5.36 (m, 1H), 4.90 (s, 2H), 4.13-4.16 (m, 1H), 4.01 (s, 6H), 3.98 (m, 1H), 2.74 (m, 1H), 2.50 (m, 1H), 2.20 (m, 1H), 2.00 (s, 2H), 1.63 (m, 1H).
Молекулярная формула: C24H21Cl2N5O3 Молекулярная масса: 498,36 LC-MS(m/z)=498,1[M-H+].
Пример 12: Синтез (2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,H-диметилпирролидин-2-карбоксамида (соединение 30)
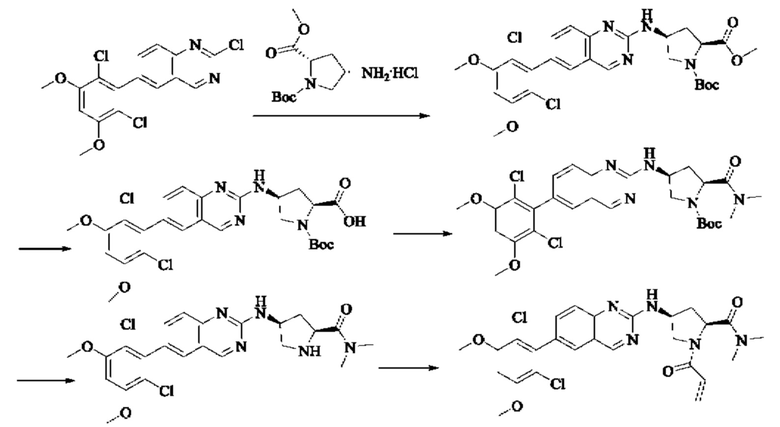
Стадия 1: Синтез 1-трет-бутил-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата
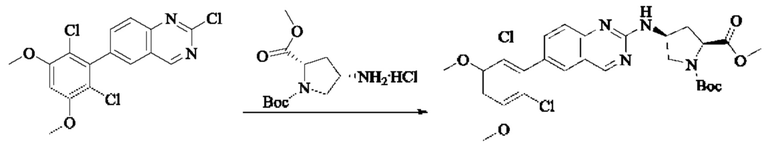
Вещества 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (500,0 мг, 1,353 ммоль, 1,0 экв), 1-трет-бутил-2-метил(2S,4S)-4-аминопирролидин-1,2-дикарбоксилата гидрохлорид (644 мг, 2,030 ммоль, 1,5 экв) и N,N-диизопропилэтиламин (874,4 мг, 6,765 ммоль, 5,0 экв) растворяли в N-метилпирролидоне (5,0 мл), нагревали до 120°С и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор выливали в ледяную воду (20 мл) и осаждали коричневое твердое вещество, затем фильтровали путем отсасывания. Фильтрат экстрагировали этилацетатом (10 мл×3). Коричневое твердое вещество растворяли в этилацетате, и органические фазы объединяли, промывали водой (20 мл×3), сушили над безводным сульфатом натрия и фильтровали. Остаток на фильтре промывали этилацетатом. Маточный раствор концентрировали при пониженном давлении и сырой продукт подвергали колоночной хроматографии на силикагеле (РЕ: ЕА = 5:1) с получением твердого 1-трет-бутил-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата (384 мг, выход: 49%).
Стадия 2: Синтез (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты
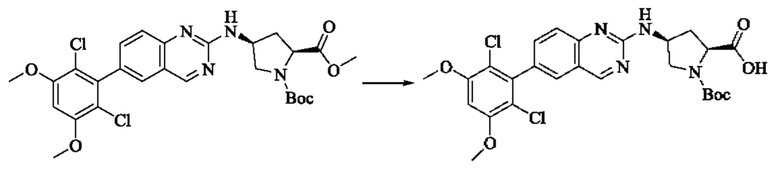
Промежуточное вещество 1-трет-бутил-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилат (300 мг, 0,520 ммоль, 1,0 экв) растворяли в метаноле (5 мл), перемешивали и растворяли, охлаждали до 0°С, и по каплям добавляли раствор лития хлорида моногидрата (65 мг, 1,558 ммоль, 3,0 экв) в воде (1 мл). После реагирования в течение 3 ч использовали TLC для детектирования оставшегося материала и добавляли раствор лития хлорида моногидрата (65 мг, 1,558 ммоль, 3,0 экв) в воде (1 мл). Метанол добавляли с получением очищенной системы и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC и реакционный раствор концентрировали при пониженном давлении, добавляли воду (10 мл) и метил-трет-бутиловый эфир (10 мл), перемешивали и разделяли. Этилацетат (20 мл) добавляли в водную фазу и охлаждали до 0°С. рН доводили до 5-6 лимонной кислотой и органическую фазу разделяли. Водную фазу экстрагировали этилацетатом (10 мл). Органические фазы объединяли и концентрировали при пониженном давлении с получением (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты (260 мг, выход: 89%) в виде бледно-желтого твердого вещества.
Стадия 3: Синтез трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(диметилкарбамоил)пирролидин-1-карбоксилата
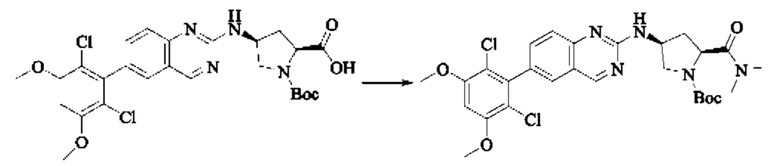
Промежуточное вещество (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновую кислоту (200 мг, 0,355 ммоль, 1,0 экв) растворяли в THF (15 мл), добавляли триметиламин (107,8 мг, 1,065 ммоль, 3,0 экв), охлаждали до примерно -15°С и медленно добавляли изобутилхлорформиат (48,49 мг, 0,355 ммоль, 1,0 экв), разбавленный THF. После реагирования в течение 1 ч завершение реакции детектировали с помощью TLC и реакционный раствор добавляли по каплям в раствор диметиламина и тетрагидрофурана (5 мл), охлажденный до 0°С. После реагирования в течение 4 ч завершение реакции детектировали с помощью TLC. Добавляли насыщенный раствор карбоната калия (10 мл), насыщенный солевой раствор (10 мл) и ЕА (20 мл), перемешивали и разделяли. Водную фазу экстрагировали ЕА (10 мл×2). Органические фазы объединяли, сушили и концентрировали. Сырой продукт использовали на следующей стадии без очистки.
Стадия 4: Синтез (2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,H-диметилпирролидин-2-карбоксамида
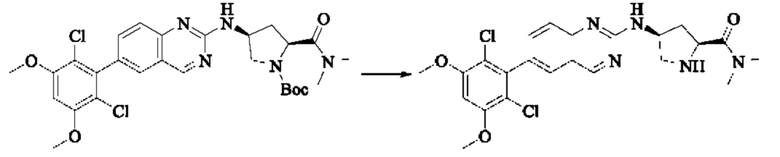
Промежуточное вещество трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(диметилкарбамоил)пирролидин-1-карбоксилат (сырой продукт, полученный на предыдущей стадии) растворяли в DCM (10 мл), добавляли трифторуксусную кислоту (10 мл), нагревали до 30°С и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC на следующее утро и реакционный раствор концентрировали при пониженном давлении. В сырой продукт добавляли этилацетат и концентрировали, повторяли три раза. Добавляли МТВЕ (10 мл), осаждали твердое вещество и фильтровали путем отсасывания с получением твердого (2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,H-диметилпирролидин-2-карбоксамида (60 мг, выход: 34%).
Стадия 5: Синтез (2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,H-диметилпирролидин-2-карбоксамида (соединение 30)
Промежуточное вещество (2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,H-диметилпирролидин-2-карбоксамид (60 мг, 0,122 ммоль, 1,0 экв) растворяли в THF (10 мл), добавляли TEA (61,73 мг, 0,61 ммоль, 5,0 экв), охлаждали до 0°С и медленно добавляли акрилоилхлорид (11,04 мг, 0,122 ммоль, 1,0 экв), разбавленный THF. Завершение реакции детектировали с помощью TLC после реагирования в течение 2 ч. Добавляли раствор насыщенного бикарбоната натрия (10 мл) для разделения раствора. Водную фазу экстрагировали ЕА (10 мл×3), органические фазы объединяли, концентрировали и разделяли на толстой препаративной пластине силикагеля (DCM: МеОН = 15:1) с получением твердого (2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-N,H-диметилпирролидин-2-карбоксамида (7 мг, выход: 10%).
1H NMR (400 MHz, DMSO) δ (ppm): 9.17 (s, 1H), 7.81 (s, 1H), 7.69 (s, 1H), 7.50-7.58 (m, 2H), 7.00 (s, 1H), 6.62-6.67 (m, 1H), 6.10-6.14 (m, 1H), 5.66-5.69 (m, 1H), 5.32 (s, 1H), 4.91 (m, 1H), 4.75 (m, 1H), 4.14-4.16 (s, 1H), 3.97 (s, 6H), 3.11 (s, 1H), 3.11-3.32 (m, 2H), 2.85 (s, 3H), 2.00 (m, 2H), 1.23 (s, 6H), 1.10 (s, 2H).
Молекулярная формула: C26H27Cl2N5O4 Молекулярная масса: 544,43 LC-MS(Neg, m/z)=544,2 [M-H+].
Пример 13: Синтез 1-((2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(2-гидроксипропил-2-ил)пирролидин-1-ил)проп-2-ен-1-она (соединение 31)
Стадия 1: Синтез 1-(трет-бутил)-2-метил(2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата
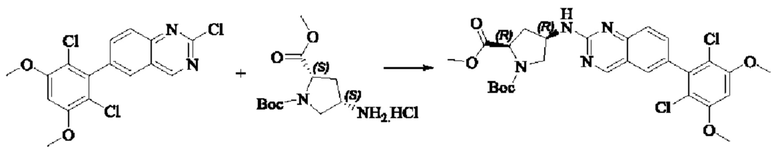
Вещества 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (500,0 мг, 1,353 ммоль, 1,0 экв), 1-(трет-бутил)-2-метил(2R,4R)-4-аминопирролидин-1,2-дикарбоксилата гидрохлорид (644 мг, 2,030 ммоль, 1,5 экв) и N,N-диизопропилэтиламин (874,4 мг, 6,765 ммоль, 5,0 экв) растворяли в N-метилпирролидоне (5,0 мл), нагревали до 120°С и проводили реакцию в течение 16 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор выливали в холодную воду (20 мл), осаждали твердое коричневое вещество и фильтровали путем отсасывания. Фильтрат экстрагировали этилацетатом (10 мл×3). Коричневое твердое вещество растворяли в этилацетате и органические фазы объединяли, промывали водой (20 мл×3), сушили над безводным сульфатом натрия, затем концентрировали при пониженном давлении и подвергали колоночной хроматографии (РЕ: ЕА = 5:1) с получением 1-(трет-бутил)-2-метил(2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата (384 мг, выход: 49%).
Стадия 2: Синтез трет-бутил(2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(2-гидроксипропил-2-ил)пирролидин-1-карбоксилата
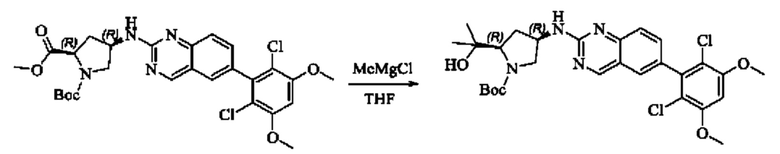
1-(трет-бутил)-2-метил(2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилат (300 мг, 0,5195 ммоль, 1,0 экв) растворяли в THF (5 мл), перемешивали с получением прозрачного раствора, охлаждали до 0°С и по каплям добавляли 3М раствор метилмагния хлорида (0,8 мл, 2,5975 ммоль, 5,0 экв) в THF. После реагирования в течение 1 ч при 0°С TLC не показала наличия исходного вещества. Добавляли насыщенный раствор хлорида аммония (5 мл), затем экстрагировали дихлорметаном (20 мл×3). После разделения водные фазы отбрасывали. Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Маточный раствор концентрировали при пониженном давлении с получением сырого продукта. Сырой продукт подвергали колоночной хроматографии, элюентом была смесь метанол: дихлорметан = 1:50. Желаемый компонент собирали и концентрировали при пониженном давлении с получением трет-бутил(2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(2-гидроксипропил-2-ил)пирролидин-1-карбоксилата (150 мг, выход: 50%).
Стадия 3: Синтез 2-((2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)пропан-2-ола
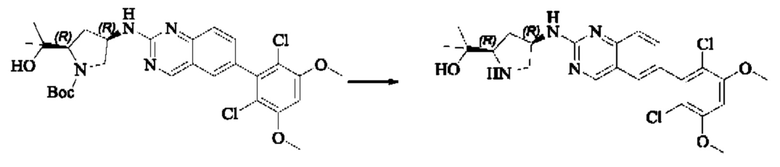
Трет-бутил(2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(2-гидроксипропил-2-ил)пирролидин-1-карбоксилат (150 мг, 0,2597 ммоль, 1,0 экв) растворяли в метаноле (5 мл), перемешивали с получением прозрачного раствора, охлаждали до 0°С, по каплям добавляли 35% этанольный раствор соляной кислоты (5 мл), перемешивали при комнатной температуре в течение 2 ч. TLC не показала наличия исходного вещества. Реакционный раствор концентрировали, растворяли путем добавления THF (50 мл) и концентрировали, повторяли 3 раза с получением 2-((2R,4R)-4-((6-(2,6-дихлор-3,5-диметокси-фенил)хиназолин-2-ил)амино)пирролидин-2-ил)пропан-2-ола. Следующую реакцию проводили сразу в соответствии с теоретическим количеством.
Стадия 4: Синтез 1-((2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(2-гидроксипропил-2-ил)пирролидин-1-ил)проп-2-ен-1-она
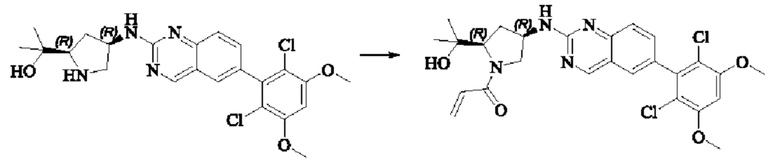
2-((2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-пирролидин-2-ил)пропан-2-ол (123 мг, 0,2597 ммоль, 1,0 экв) растворяли в THF (10 мл), добавляли TEA (131 мг, 1,2985 ммоль, 5,0 экв), охлаждали до 0°С и медленно добавляли акрилоилхлорид (15,7 мг, 0,2597 ммоль, 1,0 экв). Завершение реакции детектировали с помощью TLC после реагирования в течение 4 ч. Добавляли насыщенный раствор бикарбоната натрия (10 мл) для разделения раствора, водную фазу экстрагировали ЕА (10 мл×3). Органические фазы объединяли, концентрировали и разделяли на толстой препаративной пластине силикагеля (РЕ:ЕА=2:1) с получением 1-((2R,4R)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(2-гидроксипропил-2-ил)пирролидин-1-ил)проп-2-ен-1-она (8 мг, выход: 6%).
1H NMR (400 MHz, DMSO) δ (ppm): 9.18 (s, 1H), 7.69 (s, 1H), 7.50-7.57 (d, 3H), 7.01 (s, 1H), 6.21 (m, 1H), 6.17 (m, 1H), 5.76 (m, 1H), 4.44(s, 6H), 4.24 (m, 1H), 3.5l (m, 1H), 3.22-3.41 (m, 2H), 2.67-2.70 (m, 2H), 1.24 (s, 6H).
Молекулярная формула: C26H28Cl2N4O4, Молекулярная масса: 531,43, LC-MS (Pos, m/z)=531,1 [M-H+].
Пример 14: Синтез Н-(((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)ацетамида (соединение 32)
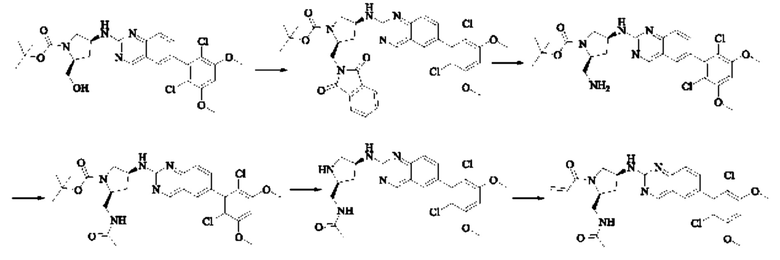
Стадия 1: Синтез трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-((1,3-диохоизоиндолин-2-ил)амино)метил)пирролидин-1-карбоксилата
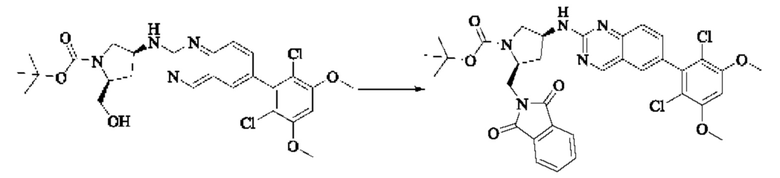
Трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилат (550,0 мг, 1,0 моль, 1,0 экв) растворяли в тетрагидрофуране (15 мл), добавляли трифенилфосфин (918,0 мг, 3,5 ммоль, 1,5 экв) и фтальимид (206,1 мг, 1,4 ммоль, 1,4 экв), охлаждали до 0°С в ледяной бане, добавляли диэтилазодикарбоксилат (523,0 мг, 3,0 ммоль) при 0°С и медленно нагревали до комнатной температуры в течение ночи. Завершение реакции детектировали с помощью TLC, реакцию гасили H2O (2 мл). Растворитель удаляли путем выпаривания. Добавляли H2O (5 мл) и этилацетат (20 мл). Органическую фазу разделяли и концентрировали. Сырой продукт очищали путем колоночной хроматографии на силикагеле (DCM: МеОН = от 100:1 до 40:1) с получением трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-((1,3-диохоизоиндолин-2-ил)амино)метил)пирролидин-1-карбоксилата (600,0 мг, выход: 88,6%) в виде желтого коллоидного твердого вещества.
Стадия 2: Синтез трет-бутил(2S,4S)-2-(аминометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
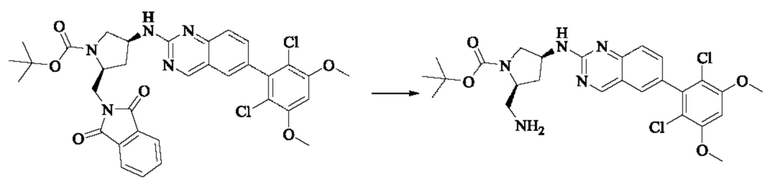
Трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-((1,3-диохоизоиндолин-2-ил)амино)метил)пирролидин-1-карбоксилат (600 мг, 0,88 ммоль, 1,0 экв) растворяли в этаноле (12 мл), добавляли гидразингидрат (6 мл) и проводили реакцию при комнатной температуре в течение 2 ч. Завершение реакции детектировали с помощью TLC. Добавляли H2O (6 мл) и концентрировали реакционный раствор. Водную фазу экстрагировали этилацетатом. Органические фазы объединяли и сушили над безводным сульфатом натрия. Сырой продукт очищали путем колоночной хроматографии на силикагеле (DCM: МеОН = 100:1 до 40:1) с получением трет-бутил(2S,4S)-2-(аминометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (500,0 мг, выход 100%) в виде желтого коллоидного твердого вещества.
Стадия 3: Синтез трет-бутил(2S,4S)-2-(ацетиламинометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
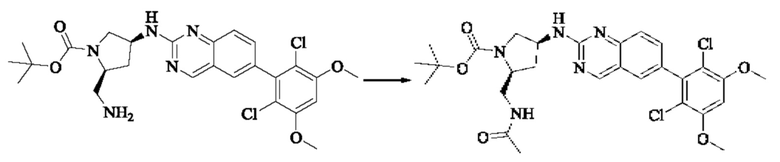
Трет-бутил(2S,4S)-2-(аминометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-1-карбоксилат (150,0 мг, 0,274 ммоль, 1,0 экв) растворяли в DCM (3 мл), охлаждали до 0°С в ледяной бане, добавляли триэтиламин (55,7 мг, 0,55 ммоль) и уксусный ангидрид (30,61 мг, 0,3 ммоль), медленно нагревали до комнатной температуры и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. Дихлорметан (5 мл) добавляли в реакционный раствор и органическую фазу промывали насыщенным хлоридом аммония, затем водой и насыщенным солевым раствором, сушили над безводным сульфатом магния, фильтровали и концентрировали с получением трет-бутил(S5,4S)-2-(ацетиламинометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-1-карбоксилата (100,0 мг, выход: 62,1%) в виде белого коллоидного твердого вещества.
Стадия 4: Синтез Н-(((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)ацетамида
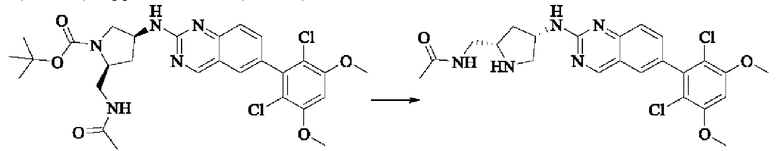
Трет-бутил(2S,4S)-2-(ацетиламинометил)-4-((6-(2,6-дихлор-3,5-диметокси-фенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (100,0 мг, 0,17 ммоль, 1,0 экв) растворяли в DCM (4 мл), охлаждали до 0°С в ледяной бане, добавляли трифторуксусную кислоту (2 мл) при 0°С, медленно нагревали до комнатной температуры в течение ночи и перемешивали в течение 2 ч. Завершение реакции детектировали с помощью TLC и реакционный раствор концентрировали досуха с получением Н-(((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)ацетамида(136,0 мг сырого) в виде желтого коллоидного твердого вещества.
Стадия 5: Синтез Н-(((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)ацетамида (соединение 32)
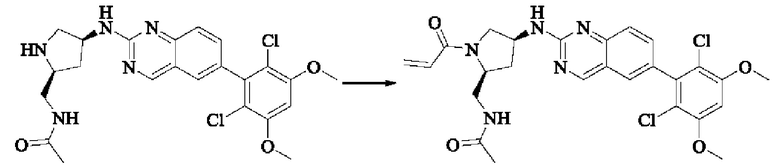
Н-(((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-пирролидин-2-ил)метил)ацетамид (134,0 мг сырого, 0,17 ммоль, 1,0 экв) растворяли в DCM (2 мл), охлаждали до 0°С в ледяной бане, добавляли триэтиламин (83,2 мг, 0,822 ммоль) и акрилоилхлорид (37,2 мг, 0,41 ммоль) при 0°С, медленно нагревали до комнатной температуры и перемешивали в течение 2 ч. Завершение реакции детектировали с помощью TLC и реакционный раствор концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (DCM: МеОН = от 200: 1 до 100: 1) с получением H-(((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)ацетамида (20,0 мг, выход 13,3%) в виде белого твердого вещества.
1HNMR (400MHz, CDCl3) δ (ppm): 9.19 (s, 1H), 8.14-7.91 (m, 2H), 7.89 (s, 1H), 7.70-7.51 (m, 2H), 7.01 (s, 1H), 6,80-6.55 (m, 2H), 6.23-6.17 (t, 1H), 5.72-5.66 (t, 1H), 4.48-4.45 (t, 1H), 4.18-4.10 (m, 2H), 4.08 (s, 6H), 3.53-3.49 (m, 1H), 3.21-3.11 (m, 1H), 2.33-3.32 (d, 1H), 2.01-1.99 (d, 1H), 1.85-1.81 (d, 3H).
Молекулярная формула: C26H27Cl2N5O4 Молекулярная масса: 544,43 LC-MS(Pos, m/z)=544,2[M+H+].
Пример 15: Синтез Н-(((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)-метансульфонамида (соединение 33)
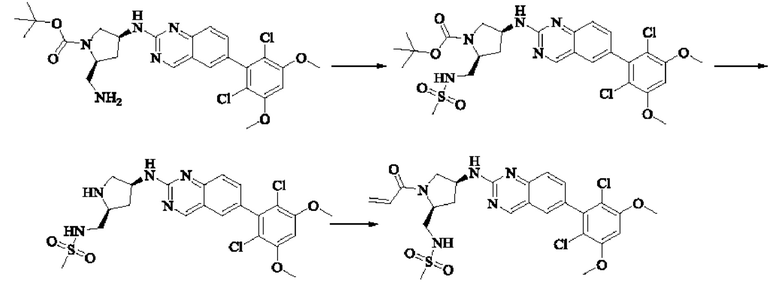
Стадия 1: Синтез трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метилсульфониламинометил)-пирролидин-1-карбоксилата
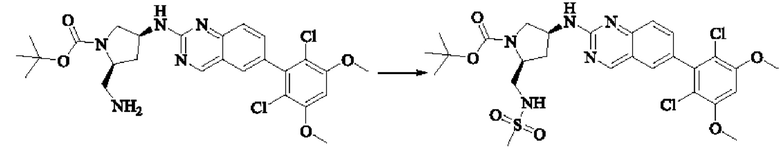
Промежуточное вещество трет-бутил(2S,4S)-2-(аминометил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (150,0 мг, 0,274 ммоль, 1,0 экв) растворяли в DCM (3 мл), охлаждали до 0°С в ледяной бане, добавляли триметиламин (55,7 мг, 0,55 ммоль), затем добавляли метансульфонилхлорид (36,9 мг, 0,324 ммоль) при 0°С, медленно нагревали до комнатной температуры и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. Дихлорметан (5 мл) добавляли в реакционный раствор и органическую фазу промывали насыщенным хлоридом аммония, водой и насыщенным солевым раствором последовательно, сушили над безводным сульфатом магния, фильтровали и концентрировали с получением трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метилсульфониламинометил)пирролидин-1-карбоксилата (90,0 мг, выход: 53,3%) в виде желтого коллоидного твердого вещества.
Стадия 2: Синтез Н-(((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)-метансульфонамида
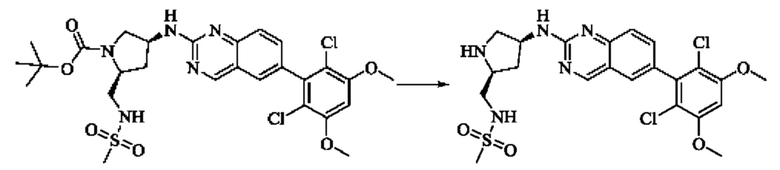
Трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метилсульфониламинометил)пирролидин-1-карбоксилат (90,0 мг, 0,27 ммоль, 1,0 экв) растворяли в DCM (4 мл), охлаждали до 0°С в ледяной бане, добавляли трифторуксусную кислоту (2 мл) и медленно нагревали до комнатной температуры в течение ночи и перемешивали в течение 2 ч. Завершение реакции детектировали с помощью TLC и реакционный раствор концентрировали в вакууме с получением Н-(((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)метансульфонамида (142,0 мг, выход:100%).
Стадия 3: Синтез Н-(((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)-метансульфонамида (соединение 33)
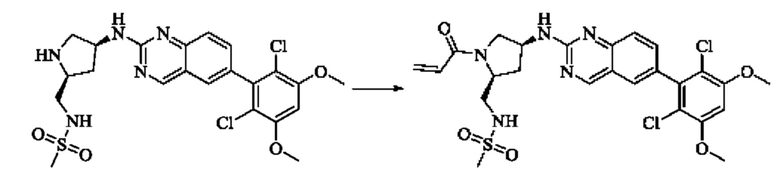
Н-(((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-пирролидин-2-ил)метил)метансульфонамид (141,8 мг, 0,27 ммоль, 1,0 экв) растворяли в THF (2 мл), охлаждали до 0°С в ледяной бане, добавляли триэтиламин (83,2 мг, 0,822 ммоль) и акрилоилхлорид (37,2 мг, 0,41 ммоль) последовательно, медленно нагревали до комнатной температуры и перемешивали в течение 2 ч. Завершение реакции детектировали с помощью TLC и реакционный раствор концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (DCM: МеОН=200: 1 до 100: 1) с получением Н-(((2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)метил)метансульфонамида (соединение 33) (21,0 мг, выход 13,3%) в виде белого твердого вещества.
1HNMR (400 MHz, CDCl3) δ (ppm): 9.19 (s,1H), 7.81-7.80 (d, 1H), 7.70 (s, 1H), 7.59-7.51 (m, 2H), 7.30-7.17 (m, 1H), 7.01 (s, 1H), 6.80-6.55 (m, 2H), 6.23-6.17 (t, 1H), 5.72-5.66 (t, 1H), 4.48-4.45 (t, 1H), 4.18-4.10 (m, 2H), 4.08 (s, 6H), 3.53-3.49 (m, 1H), 3.21-3.11 (m, 1H), 2.73 (s, 3H), 2.33-3.32 (d, 1H), 2.01-1.99 (d, 1H).
Молекулярная формула: C25H27CI2N5O5S Молекулярная масса: 580,48 LC-MS (Pos, m/z)=580,1 [M+H+].
Пример 16: Синтез 1-((2S,4S)-2-(азетидин-1-карбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она (соединение 35)
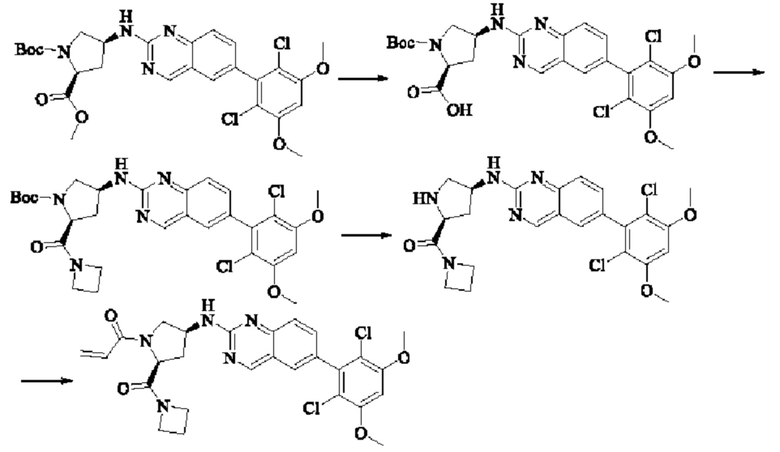
Стадия 1: Синтез (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты
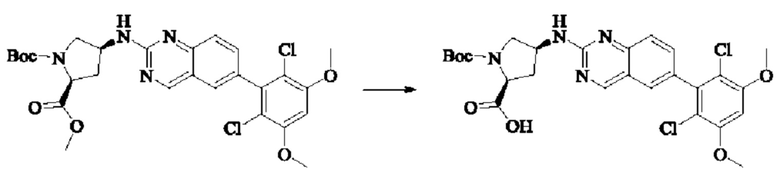
Вещество 1-трет-бутил-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметокси-фенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилат (400,7 мг, 0,69 ммоль, 1,0 экв) растворяли в метаноле (6 мл), охлаждали до 0°С, добавляли водный раствор (3 мл) лития хлорида моногидрата (87,34 мг, 2,08 ммоль, 3,0экв), затем медленно нагревали до комнатной температуры и перемешивали в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор концентрировали, растирали с метил-трет-бутиловым эфиром (5 мл) в течение 30 мин и затем фильтровали путем отсасывания. Остаток на фильтре растворяли в воде (10 мл). рН доводили до 5-6 водным насыщенным раствором бикарбоната натрия, затем экстрагировали DCM (20 мл×1). Органическую фазу сушили над безводным сульфатом натрия, фильтровали путем отсасывания и концентрировали с получением (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты (288,7 мг, выход: 73,8%).
Стадия 2: Синтез трет-бутил(2S,4S)-2-(азетидин-1-карбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата
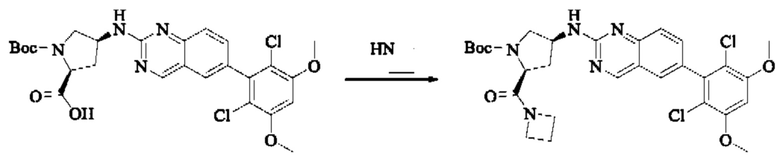
(2S, 4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-2-карбоновую кислоту (421,3 мг, 0,74 ммоль, 1,0 экв) растворяли в смешанном растворителе из тетрагидрофурана (6 мл) и ацетонитрила (1 мл), охлаждали до 0°С при перемешивании, медленно добавляли порциями 2-(7-O-бензотриазол)-N,N,N',N'-тетраметилурония гексафторфосфат (312,7 мг, 0,82 ммоль, 1,1 экв) и затем по каплям добавляли N,N-диизопропилэтиламин (193,1 мг, 1,49 ммоль, 2,0 экв). После добавления по каплям проводили реакцию при 0°С в течение 1 ч. Добавляли по каплям раствор азетидина (42,68 мг, 0,74 ммоль, 1,0 экв) в тетрагидрофуране (0,5 мл). После этого смесь нагревали до комнатной температуры и перемешивали в течение ночи в атмосфере азота. Завершение реакции детектировали с помощью TLC. Систему концентрировали, добавляли насыщенный водный хлорид натрия (30 мл) и экстрагировали этилацетатом (3×10 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали путем отсасывания и концентрировали. Сырой продукт очищали колоночной хроматографией (200-300 меш силикагель, дихлорметан: метанол = от 200: 1 до 100: 1) с получением трет-бутил(2S,4S)-2-(азетидин-1-карбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилата (194,3 мг, выход: 43,1%).
Стадия 3: Синтез азетидин-1-ил((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-амино)пирролидин-2-ил)метанона
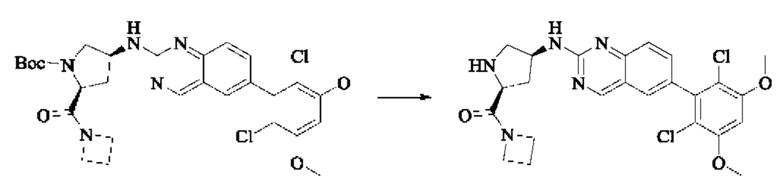
Трет-бутил(2S,4S)-2-(азетидин-1-карбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-карбоксилат (194,3 мг, 0,32 ммоль, 1,0 экв) растворяли в DCM (5 мл), охлаждали до 0°С и по каплям добавляли трифторуксусную кислоту (5 мл). После добавления смесь нагревали до комнатной температуры и проводили реакцию в течение 2,5 ч. Завершение реакции детектировали с помощью TLC и систему охлаждали до 0°С. рН системы доводили до 7-8 насыщенным водным раствором карбоната натрия, затем экстрагировали путем добавления дихлорметана (3×10 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали путем отсасывания и концентрировали с получением азетидин-1-ил((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-амино)пирролидин-2-ил)метанона (151,1 мг, выход: 93,2%).
Стадия 4: Синтез 1-((2S,4S)-2-(азетидин-1-карбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1-ил)проп-2-ен-1-она (соединение 35)
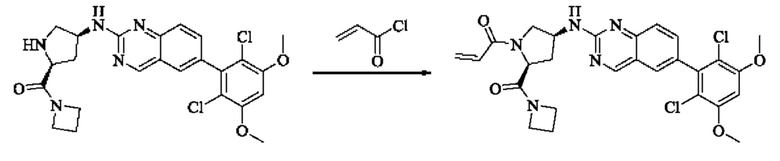
Азетидин-1-ил((2S,4S)-4-(6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-амино)пирролидин-2-ил)метанон (150 мг, 0,32 ммоль, 1,0 экв) растворяли в THF (3 мл), охлаждали до 0°С, по каплям добавляли триэтиламин (65,2 мг, 0,64 ммоль, 2,0 экв) и акрилоилхлорид (35,0 мг, 0,39 ммоль, 1,2 экв) последовательно. После добавления смесь постепенно нагревали до комнатной температуры и перемешивали в течение 4 ч. Завершение реакции детектировали с помощью TLC. Добавляли насыщенный водный раствор хлорида аммония, затем экстрагировали этилацетатом (3×10 мл). Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали путем отсасывания и концентрировали. Сырой продукт очищали колоночной хроматографией (200-300 меш силикагель, дихлорметан: метанол = 100: 1) с получением продукта 1-((2S,4S)-2-(азетидин-1-карбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-пирролидин-1-ил)проп-2-ен-1-она (41,0 мг, выход: 22,3%).
1H NMR (400MHz, DMSO) δ (ppm):9.18 (s, 1H), 7.95 (s, 1H), 7.51-7.70 (m, 3Н), 7.01 (s, 1H), 6.61-6.65 (m, 1H), 6.14-6.19 (m, 1H), 5.69-5.76 (m, 1H), 4.74 (m, 1H), 4.41-4.48 (m, 411), 1.11-1.12 (s, 6H), 3.84-3.92 (m, 1H), 3.50-3.54 (t, 1H), 1.90-1.91 (m, 1H), 2.55-2.89 (m, 1H).
Молекулярная формула: C17H27Cl12N5O4 Молекулярная масса: 555,14 LC-MS(Neg, m/z)=556,2[М+Н+].
Пример 17: Синтез этил(2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоксилата (соединение 36)
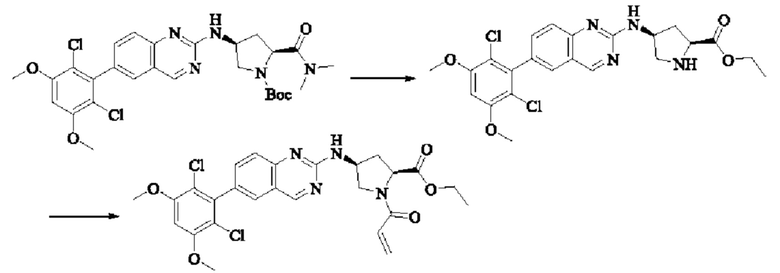
Стадия 1: Синтез этил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоксилата
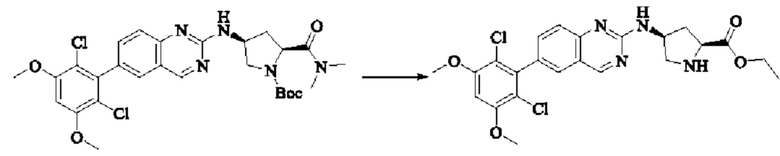
Промежуточное вещество трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(диметилкарбамоил)пирролидин-1-карбоксилат (183 мг, 0,310 ммоль, 1,0 экв) растворяли в HCl-этаноле (15 мл) и проводили реакцию в течение 3 ч при 45°С. Завершение реакции детектировали с помощью TLC. Реакционный раствор концентрировали. Сырой продукт растворяли в THF и концентрировали, повторяли 3 раза. Сырой продукт использовали на следующей стадии без очистки (теоретическое количество: 170 мг).
Стадия 2: Синтез этил(2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоксилата (соединение 36)
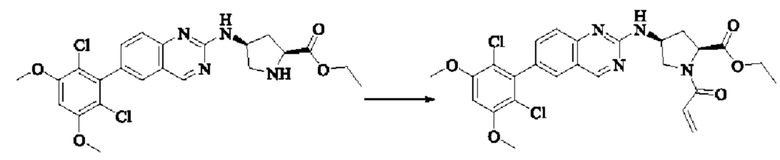
Промежуточное вещество этил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоксилат (170 мг, 0,347 ммоль, 1,0 экв) растворяли в THF (15 мл), добавляли триметиламин (175,6 мг, 1,735 ммоль, 5,0 экв), охлаждали до 0°С и медленно добавляли акрилоилхлорид (31,4 мг, 0,347 ммоль, 1,0 экв), разбавленный THF. После реагирования в течение 2 ч завершение реакции детектировали с помощью TLC и добавляли насыщенный раствор бикарбоната натрия (10 мл). После разделения водную фазу экстрагировали ЕА (10 мл × 3), органические фазы объединяли, концентрировали и разделяли тонкослойной хроматографией (DCM: МеОН=15: 1) с получением твердого этил(2S,4S)-1-акрилоил-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоксилата (21 мг, выход: 11%).
1HNMR (400MHz, DMSO) δ (ppm): 9.02 (s, 1H), 7.70 (m, 1Н),7.68 (s, 2H), 6.66 (s, 1H),6.4 5-6.47 (m, 2H), 6.11-6.13 (m, 1H), 5.73-5.76 (m, 1H), 5.00 (s, 1H), 4.66 (m, 1H), 4.28 (m, 2H), 4.12-4.14 (m, 1H), 3.99 (s, 6H), 3.77-3.80 (m, 1H), 2.69 (m, 1H), 2.15 (m, 1H), 1.23-1.25 (m, 2H), 1.25-1.27 (m, 3H).
Молекулярная формула: C26H26Cl2N4O5 Молекулярная масса: 545,42 LC-MS(Pos, m/z)=545,1[M-H+].
Пример 18: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метоксиметил)пирролидин-1-ил)проп-2-ен-1-она (соединение 53)
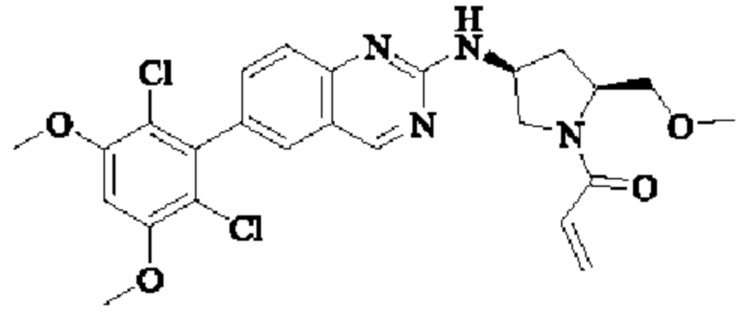
Стадия 1: Синтез трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метоксиметил)пирролидин-1-карбоксилата
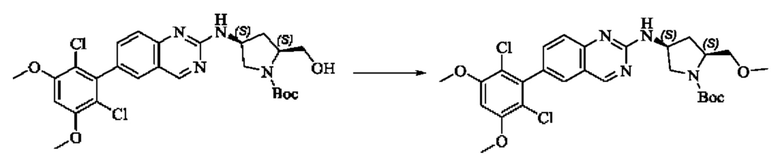
Вещества трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-2-(гидроксиметил)пирролидин-1-карбоксилат (210 мг, 0,382 ммоль, 1,0 экв) и трет-бутоксид натрия (91,78 мг, 0,955 ммоль, 2,5 экв) растворяли в THF (10 мл), охлаждали до 0°С и перемешивали в течение 1 ч. Затем добавляли иодометан (108,44 мг, 0,764 ммоль, 2,0 экв), разбавленный THF (1 мл), для реакции в течение ночи. TLC показала, что малое количество исходного вещества осталось на следующее утро. Добавляли насыщенный раствор хлорида аммония (10 мл) и DCM (20 мл) для разделения раствора. В водной фазе путем TLC не обнаружено продукта. Органическую фазу сушили, концентрировали и подвергали колоночной хроматографии (РЕ: ЕА=5: 1) с получением трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метоксиметил)пирролидин-1-карбоксилата (200 мг, выход: 93%).
Стадия 2: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,5S)-5-(метоксиметил)пирролидин-3-ил)хиназолин-2-амина
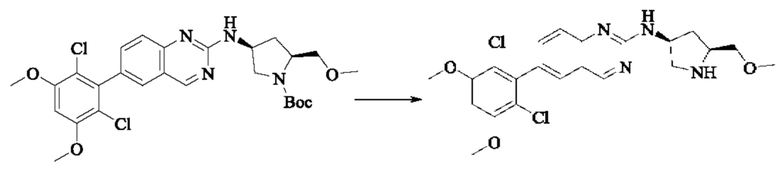
Промежуточное вещество трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метоксиметил)пирролидин-1-карбоксилат (200 мг, 0,355 ммоль, 1,0 экв) растворяли в DCM (5 мл), перемешивали с получением прозрачного раствора, охлаждали до 0°С и добавляли трифторуксусную кислоту (5 мл). После реагирования в течение 1,5 ч завершение реакции детектировали с помощью TLC. Реакционный раствор концентрировали, растворяли путем добавления THF (50 мл) и концентрировали, повторяли 3 раза. Сырой продукт использовали на следующей стадии.
Стадия 3: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-2-(метоксиметил)пирролидин-1-ил)проп-2-ен-1-она
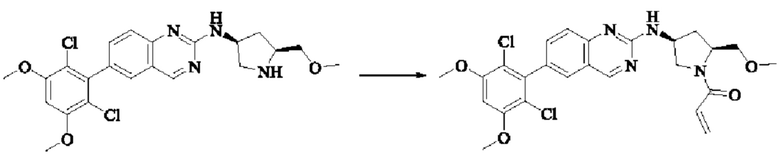
Промежуточное вещество 6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,5S)-5-(метоксиметил)пирролидин-3-ил)хиназолин-2-амин (160 мг, 0,345 ммоль, 1,0 экв) и триэтиламин (174,55 мг, 1,725 ммоль, 5,0 экв) растворяли в THF (10 мл), охлаждали до 0°С. Добавляли акрилоилхлорид (31,22 мг, 0,345 ммоль, 1,0 экв), разбавленный THF (1 мл), для реакции в течение ночи. Завершение реакции детектировали с помощью TLC на следующее утро. Добавляли насыщенный раствор бикарбоната натрия (10 мл) и ЕА (20 мл) для разделения раствора. Водную фазу экстрагировали ЕА. В водной фазе путем TLC не обнаружено продукта. Органические фазы объединяли, сушили, концентрировали и подвергали колоночной хроматографии (РЕ: ЕА=5: 1 до 3: 1) с получением 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(метоксиметил)пирролидин-1-ил)проп-2-ен-1-она (34 мг, выход: 19%).
1H NMR (400MHz, DMSO) δ (ppm): 9.19 (s, 1H), 7.80 (s, 1H), 7.69 (s, 1H), 7.51-7.59 (m, 2H), 7.02 (s, 1H), 6.70-6.75 (m, 1H), 6.57-6.70 (m, 1H), 6.18 (m, 1H), 5.67-5.76 (m, 1H), 4.44-4.52 (m, 2H), 4.22-4.36 (m, 2H), 3.98 (s, 6H), 3.61 (s, 1H), 3.28-3.32 (m, 1H), 3.22 (s, 3H), 2.02 (s, 1H), 1.23 (s, 1H).
Молекулярная формула: C25H26Cl2N4O4, Молекулярная масса: 517,41, LC-MS (Neg, m/z)=517,2[M-H+].
Пример 19: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(пирролидин-1-карбонил)пирролидин-1-ил)проп-2-ен-1-она (соединение 55)
Стадия 1: Синтез 1-(трет-бутил)-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата
Вещество 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (3,28 г, 8,87 ммоль, 1,0 экв) растворяли в N-метилпирролидоне (8 мл). Добавляли в систему N,N-диизопропилэтиламин (3,44 г, 26,6 ммоль, 3,0 экв) и 1-(трет-бутил)-2-метил(2S,4S)-4-аминопирролидин-1,2-дикарбоксилата гидрохлорид (3,73 г, 13,3 ммоль, 1,5 экв) при комнатной температуре. После добавления систему нагревали до 120°С с обратным холодильником и перемешивали в течение ночи. Завершение реакции детектировали с помощью TLC на следующий день. Затем реакционный раствор охлаждали, добавляли воду (32 мл) и осаждали твердое вещество. После перемешивания в течение 2 ч получали твердое вещество фильтрацией путем отсасывания и экстрагировали дихлорметаном (150×3 мл). Органическую фазу сушили над безводным сульфатом натрия и затем очищали колоночной хроматографией (РЕ: ЕА=5: 1) с получением 1-(трет-бутил)-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилата (1,91 г, 38,1%).
Стадия 2: Синтез (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты
Промежуточное вещество 1-(трет-бутил)-2-метил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-1,2-дикарбоксилат (1,05 г, 1,81 ммоль, 1,0 экв) растворяли в метаноле (14 мл), охлаждали до примерно 0°С при перемешивании при комнатной температуре и по каплям добавляли водный раствор (5,5 мл) лития хлорида моногидрата (229,5 мг, 5,46 ммоль, 3,0 экв). После добавления смесь медленно нагревали до комнатной температуры и перемешивали в течение ночи. Завершение реакции детектировали с помощью TLC на следующий день. Систему концентрировали досуха, добавляли метил-трет-бутиловый эфир (30 мл) и перемешивали в течение 0,5 ч, и затем фильтровали путем отсасывания. Остаток на фильтре растворяли в воде (20 мл) и рН доводили до 6-7, и затем добавляли дихлорметан (40 мл) для экстрагирования. Органическую фазу сушили над безводным сульфатом натрия и затем концентрировали с получением продукта (2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-карбоновой кислоты (900,8 мг, выход:89,8%).
Стадия 3: Синтез трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(пирролидин-1-карбонил)пирролидин-1-карбоксилата
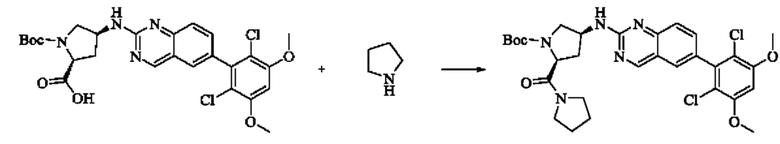
(2S,4S)-1-(трет-бутоксикарбонил)-4-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)пирролидин-2-карбоновую кислоту (287,3 мг, 0,50 ммоль, 1,0 экв) растворяли в смешанном растворе тетрагидрофурана (11 мл) и ацетонитрила (1,5 мл), охлаждали до примерно 0°С при перемешивании и по каплям добавляли 7-азобензотриазол (213,27 мг, 0,56 ммоль, 1,1 экв) и N,N-диизопропилэтиламин (131,7 мг, 1,01 ммоль, 2,0 экв). После добавления проводили реакцию при 0°С в течение 1 ч. После этого добавляли по каплям пирролидин при той же температуре для реакции в течение 3 ч. Затем завершение реакции детектировали с помощью TLC. Систему концентрировали досуха и затем промывали этилацетатом (30 мл) и насыщенным водным хлоридом натрия (15 мл) три раза. Органические фазы объединяли и сушили над безводным сульфатом натрия, затем концентрировали с получением (2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(пирролидин-1-карбонил)пирролидин-1-карбоксилата (471,8 мг, выход: 100%).
Стадия 4: Синтез ((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)(пирролидин-1-ил)метанона
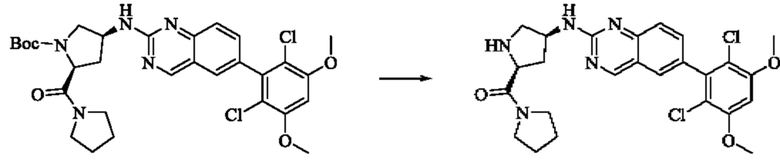
Трет-бутил(2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(пирролидин-1-карбонил)пирролидин-1-карбоксилат (374,1 мг, 0,60 ммоль, 1,0 экв) растворяли в DCM (6 мл), охлаждали до примерно 0°С при перемешивании и по каплям добавляли трифторуксусную кислоту (4 мл). После добавления систему медленно нагревали до комнатной температуры и перемешивали в течение 3 ч. После этого завершение реакции детектировали с помощью TLC. Систему охлаждали до примерно 0°С. рН системы доводили до 6-7 путем добавления по каплям насыщенного водного раствора бикарбоната натрия, затем добавляли дихлорметан (15 мл) для экстрагирования. Органическую фазу сушили над безводным сульфатом натрия и затем концентрировали с получением ((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)пирролидин-2-ил)(пирролидин-1-ил)метанона (226,0 мг, выход: 72,2%).
Стадия 5: Синтез 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(пирролидин-1-карбонил)пирролидин-1-ил)проп-2-ен-1-она
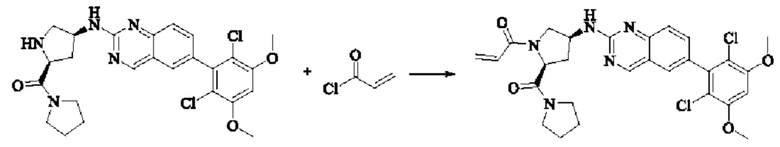
((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-пирролидин-2-ил)(пирролидин-1-ил)метанон растворяли в THF (5 мл), охлаждали до 0°С при перемешивании и по каплям добавляли триэтиламин (132,8 мг, 1,31 ммоль, 3,0 экв) и акрилоилхлорид (55,4 мг, 0,61 ммоль, 1,4 экв) последовательно. После добавления систему нагревали до комнатной температуры и перемешивали в течение ночи. Завершение реакции детектировали с помощью TLC на следующий день. В систему добавляли насыщенный водный раствор хлорида аммония (10 мл) и DCM (20 мл) для разделения. Органическую фазу сушили над безводным сульфатом натрия, концентрировали досуха, затем добавляли метил-трет-бутиловый эфир (10 мл), перемешивали в течение 2 ч и затем фильтровали путем отсасывания с получением продукта 1-((2S,4S)-4-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-(пирролидин-1-карбонил)пирролидин-1-ил)проп-2-ен-1-она (162,8 мг, выход: 66,1%).
1HNMR (400MHz, DMSO) δ (ppm): 9.18 (s, 1H), 7.95 (s 1H), 7 70 (s, 1H), 7.51-7,59 (m, 2H), 7.01 (s, 1H), 6.63-6.67 (m, 1H), 6.12-6.16 (m, 1H), 5.67-5.70 (m, 1H), 4.70-4.71 (m, 1H), 4.68 (s, 1H), 4.13 (s, 5H), 4.21 (m, 1H), 3.74-3.76 (m, 1H), 3.49-3.54 (m, 1H), 1.89-1.94 (m, 3H), 1.78-1.83 (m, 2H).
Молекулярная формула: C28H29C12N5O4 Молекулярная масса: 569,16 LC-MS (Neg, m/z)=570,2[М+Н+].
Пример 20: Синтез 1-((3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-ил)проп-2-ин-1-она (соединение 56)
Стадия 1: Синтез трет-бутил(3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-карбоксилата
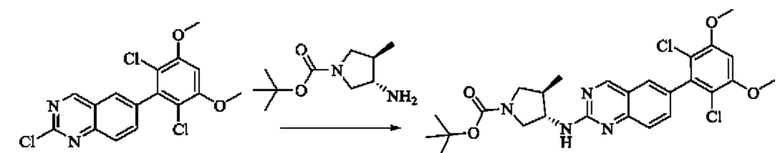
Вещества 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (400,0 мг, 1,1 ммоль, 1,0 экв) и трет-бутил(3S,4R)-3-амино-4-метилпирролидин-1-карбоксилат (440,6 мг, 2,2 ммоль, 2,0 экв) растворяли в N-метилпирролидоне (3,0 мл), добавляли N,N-диизопропилэтиламин (568,7 мг, 4,4 ммоль, 4,0 экв), постепенно нагревали до 110°С и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC. Реакционный раствор охлаждали до комнатной температуры, добавляли ледяную воду (15 мл) и фильтровали. Остаток на фильтре промывали малым количеством ледяной воды, растворяли в этилацетате (20 мл). Фазу этилацетата промывали насыщенным солевым раствором (10 мл) и сушили над безводным сульфатом натрия, фильтровали и концентрировали. Сырой продукт подвергали колоночной хроматографии на силикагеле (DCM: МеОН = от 200: 1 до 100:1) с получением трет-бутил(3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-4-метилпирролидин-1-карбоксилата (320,6 мг, выход: 54,7%) в виде желтого твердого вещества.
Стадия 2: Синтез (6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,4R)-4-метилпирролидин-3-ил)хиназолин-2-амина гидрохлорида
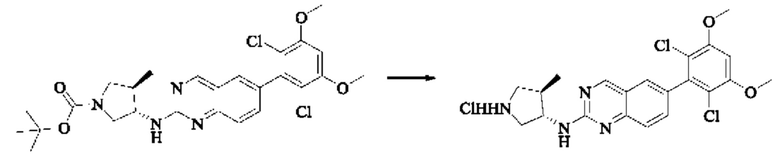
Вещество трет-бутил(3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-4-метилпирролидин-1-карбоксилат (320,6 мг, 1,0 экв) растворяли в этаноле (8,0 мл), охлаждали до 0°С в ледяной бане, добавляли этанольный раствор хлористого водорода (8,0 мл), перемешивали и проводили реакцию в течение 2 ч. Завершение реакции детектировали с помощью TLC. Реакционный раствор прямо концентрировали с получением (6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,4R)-4-метилпирролидин-3-ил)хиназолин-2-амина гидрохлорида (339,2 мг сырого, выход: 100%) в виде бледно-желтого твердого вещества.
Стадия 3: Синтез 1-((3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-ил)проп-2-ин-1-она
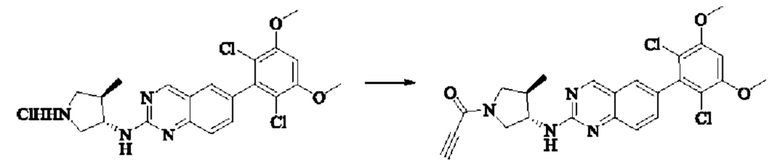
6-(2,6-дихлор-3,5-диметоксифенил)-Н-((3S,4R)-4-метилпирролидин-3-ил)хиназолин-2-амина гидрохлорид (339,2 мг) растворяли в THF (10,0 мл), добавляли триэтиламин (203,0 мг, 2,0 ммоль, 3,0 экв), обрабатывали ультразвуком в течение 5 мин, добавляли воду (15 мл) и этилацетат (10 мл), и разделяли. Водную фазу экстрагировали этил ацетатом (8,0 мл×2). Органические фазы объединяли, промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали с получением бледно-желтого твердого вещества (216,0 мг). Полученное твердое вещество (108,0 мг, 0,25 ммоль) растворяли в дихлорметане (2 мл), добавляли пропионовую кислоту (37,8 мг, 0,54 ммоль), 4-диметиламинопиридин (3,1 мг, 0,025 ммоль) и N,N'-дициклогексилкарбамид (56,5 мг, 0,27 ммоль), и проводили реакцию в микроволновой печи при 40°С в течение 30 мин. Завершение реакции детектировали с помощью TLC. Реакционный раствор промывали водой (5 мл×3), концентрировали досуха и разделяли препаративной тонкослойной хроматографией на пластине с получением 1-((3S,4R)-3-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-4-метилпирролидин-1-ил)проп-2-ин-1-она (24,2 мг, выход: 20%) в виде желтого твердого вещества.
1HNMR (400MHz, DMSO-d6) δ (ppm): 9.20-9.19 (d, 1H), 7.96 (s, 1H), 7.86-7.81 (t, 1H), 7.69 (s, 1H), 7.56-7.52 (m, 2H), 7.01 (s, 1Н), 4.49-4.42 (d, 1H), 4.25-4.15 (t, 3H), 3.97 (s, 6H), 4.03 (s, 6H), 3.77-3.72 (m, 1H), 3.45-3.41 (m, 1H), 3.20-3.15 (m, 1H), 3.06-3.01 (m, 1H), 2.89 (s, 2H),2.73 (s, 2H), 2.40-2.33 (d, 2H),1.24 (s, 2H), 1.10 (s, 3H).
Молекулярная формула: C24H22Cl2N4O3 Молекулярная масса: 485,37 LC-MS(Pos, m/z)=485,43[M+H+].
Пример 21: Синтез 1-(6-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азаспиро[3,3]гептан-2-ил)проп-2-ен-1-она (соединение 65)
Стадия 1: Синтез трет-бутил-6-((6-(2,6-дихлор-3,5-диметоксифенил)-хиназолин-2-ил)амино)-2-азаспиро[3,3]гептан-2-карбоксилата
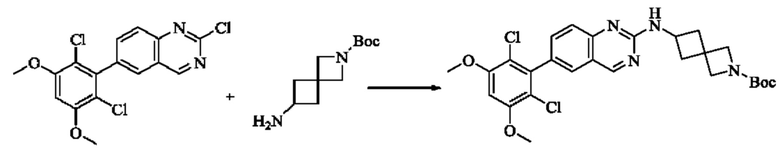
Вещества 2-хлор-6-(2,6-дихлор-3,5-диметоксифенил)хиназолин (400 мг, 1,082 ммоль, 1,0 экв) и трет-бутил-6-амино-2-азаспиро[3,3]гептан-2-карбоксилат (248,53 мг, 1,082 ммоль, 1,0 экв) растворяли в NMP (1,5 мл), добавляли DIPEA (699,24 мг, 5,41 ммоль, 5,0 экв), нагревали до 110°С и проводили реакцию в течение ночи. Завершение реакции детектировали с помощью TLC на следующее утро. После охлаждения реакционный раствор медленно выливали в воду (50 мл) и фильтровали путем отсасывания. Остаток на фильтре растворяли в DCM. Водную фазу экстрагировали ЕА (100 мл×2) и разделяли. В водной фазе путем TLC не обнаружено продукта. Органические фазы объединяли, сушили и концентрировали, и затем подвергали колоночной хроматографии (РЕ: ЕА=5: 1) с получением трет-бутил-6-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азаспиро-[3,3]гептан-2-карбоксилата (812 мг) в виде густой жидкости, которую использовали непосредственно в следующей реакции.
Стадия 2: Синтез 6-(2,6-дихлор-3,5-диметоксифенил)-Н-(2-азаспиро[3,3]гептан-6-ил)хиназолин-2-амина
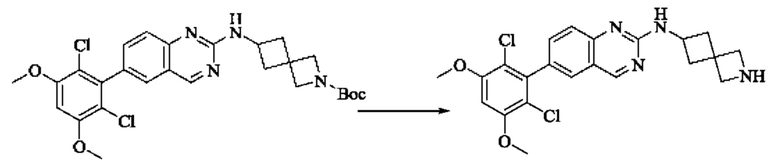
Промежуточное вещество трет-бутил-6-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азаспиро[3,3]гептан-2-карбоксилат (590,3 мг, 0,355 ммоль, 1,0 экв) растворяли в DCM (5 мл), перемешивали с получением прозрачного раствора, охлаждали до 0°С, добавляли трифторуксусную кислоту (5 мл) и проводили реакцию в течение 1 ч. Затем завершение реакции детектировали с помощью TLC. Реакционный раствор концентрировали, растворяли путем добавления THF (50 мл) и концентрировали, повторяли 3 раза. Сырой продукт использовали на следующей стадии.
Стадия 3: Синтез 1-(6-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азаспиро[3,3]гептан-2-ил)проп-2-ен-1-она
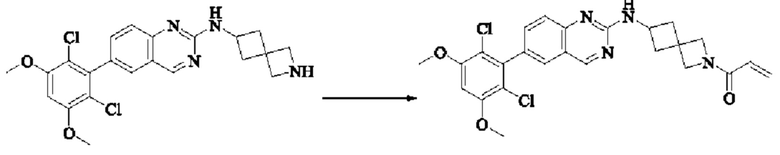
Промежуточное вещество 6-(2,6-дихлор-3,5-диметоксифенил)-Н-(2-азаспиро[3,3]гептан-6-ил)хиназолин-2-амин (480 мг, 1,08 ммоль, 1,0 экв) и триэтиламин (546,4 мг, 5,4 ммоль, 5,0 экв) растворяли в THF (10 мл). рН системы доводили до примерно 8 и затем охлаждали до 0°С. Добавляли акрилоилхлорид (97,74 мг, 1,08 ммоль, 1,0 экв), разбавленный THF (1 мл). TLC показала, что малое количество исходного вещества осталось через 1 час. Добавляли каплю акрилоилхлорида. TLC показала, что малое количество исходного вещества осталось через 0,5 ч. Добавляли насыщенный раствор бикарбоната натрия (10 мл) и ЕА (20 мл), перемешивали и разделяли. Водную фазу экстрагировали ЕА (10 мл×2). В водной фазе путем TLC не обнаружено продукта. Органические фазы объединяли, сушили, концентрировали и подвергали колоночной хроматографии (РЕ: ЕА = от 2: 1 до 1: 1) с получением 1-(6-((6-(2,6-дихлор-3,5-диметоксифенил)хиназолин-2-ил)амино)-2-азаспиро[3,3]гептан-2-ил)проп-2-ен-1-она (337 мг, выход: 62%).
1HNMR (400MHz, DMSO) δ (ppm): 9.14 (s, 1H), 7.81 (s, 1H), 7.66 (s, 1Н), 7.48-7.52 (m, 211), 7.01 (s, 1H), 6.31-6.34 (m, 1H), 6.07-6.11 (m, 1H), 5.64-5.68 (m, 1H), 4.54 (s, 1H), 4.40 (s, 1H), 4.26 (m, 1H), 4.20 (s, 1H), 3.98 (s, 6H), 3.80 (s, 1H), 2.62 (s, 2H), 2.26 (s, 2H).
Молекулярная формула: C25H24Cl2N4O3, Молекулярная масса: 499,39, LC-MS (Neg, m/z)=499,1[M-H+].
В соответствии с вышеописанным способом синтеза можно получить следующие соединения:
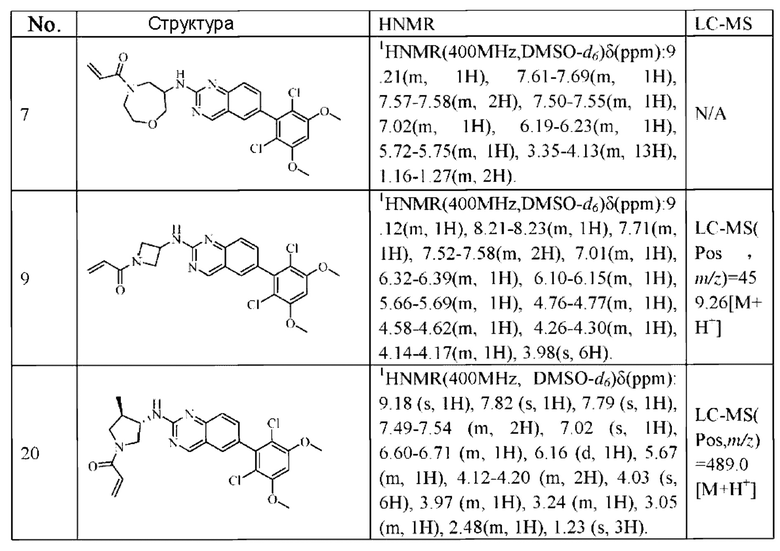
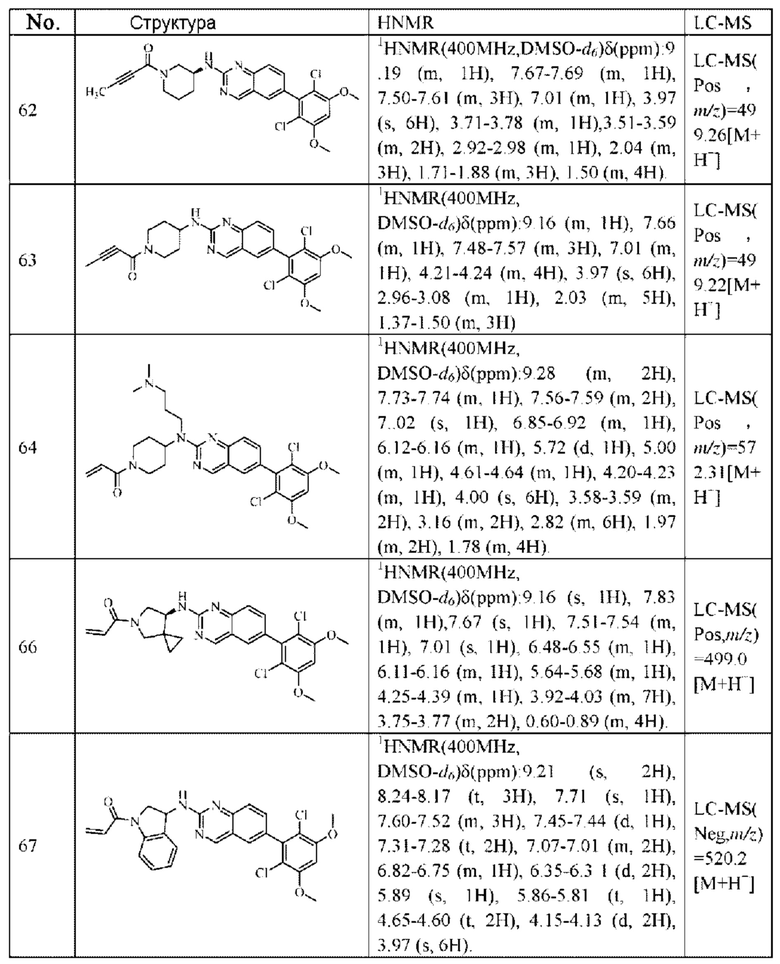
Настоящее изобретение может быть лучше понято из следующих экспериментальных примеров. Однако специалисты в данной области техники легко поймут, что описание экспериментальных примеров предназначено только для иллюстрации изобретения и не должно ограничивать изобретение, подробно описанное в формуле изобретения.
Экспериментальный Пример 1: Тест на ферментативную активность соединений согласно настоящему изобретению
Тестируемое вещество: Соединения согласно настоящему изобретению, структура которых показана выше.
Тестовый прибор: платформа для проверки лекарств LabChip EZ Reader II.
Метод испытания:
1. Подготовка планшетов для соединений
а) 96-луночные планшеты, 10 групп доз, 3-кратные серийные разведения, добавляли ДМСО в каждую лунку, максимальная концентрация 500 мкМ.
б) 384-луночные планшеты, разбавление 1Х киназным буфером (50 мМ HEPES, рН 7,5; 0,0015% Brij-35; 2 мМ DTT), каждая лунка содержит 5Х соединение, растворенное в 5 мкл 10% ДМСО. Лунка с отрицательным контролем - это 5 мкл 1Х киназного буфера, содержащего 10% ДМСО.
2. Методика эксперимента
Проводили реакцию FGFR1-4 (h) в 1Х киназном буфере с раствором фермента 2,5Х при комнатной температуре в течение 10 минут и затем добавляли меченный FAM полипептидный субстрат и АТФ для инициации реакции. После инкубации в течение 30 мин для прекращения реакции добавляли 25 мкл терминального раствора (100 мМ HEPES, рН 7,5; 0,015% Brij-35; 0,2% покрывающего реагента №3; 50 мМ EDTA) и окончательные данные считывали датчиком.
Результаты тестирования представлены в Таблице 1.
Таблица 1. Ингибирующая активность соединений согласно настоящему изобретению против FGFR (IC50)
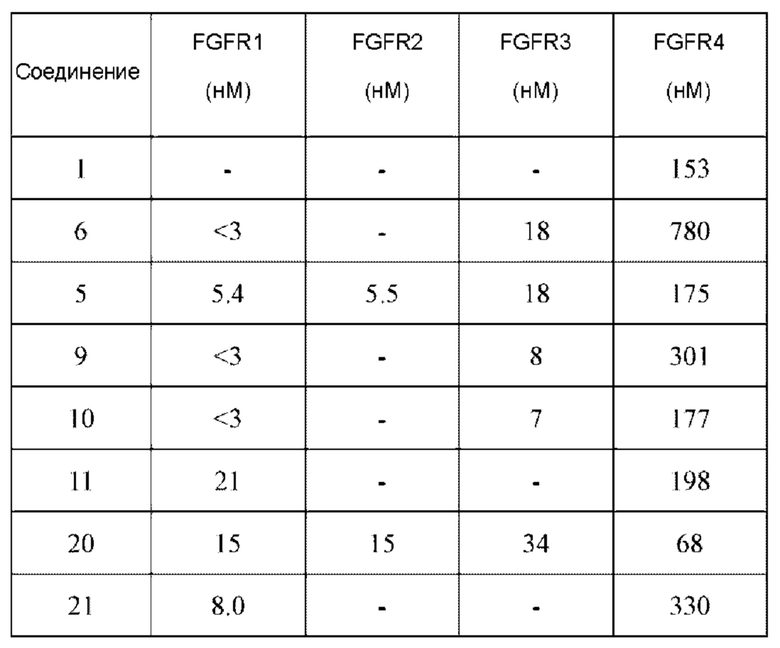
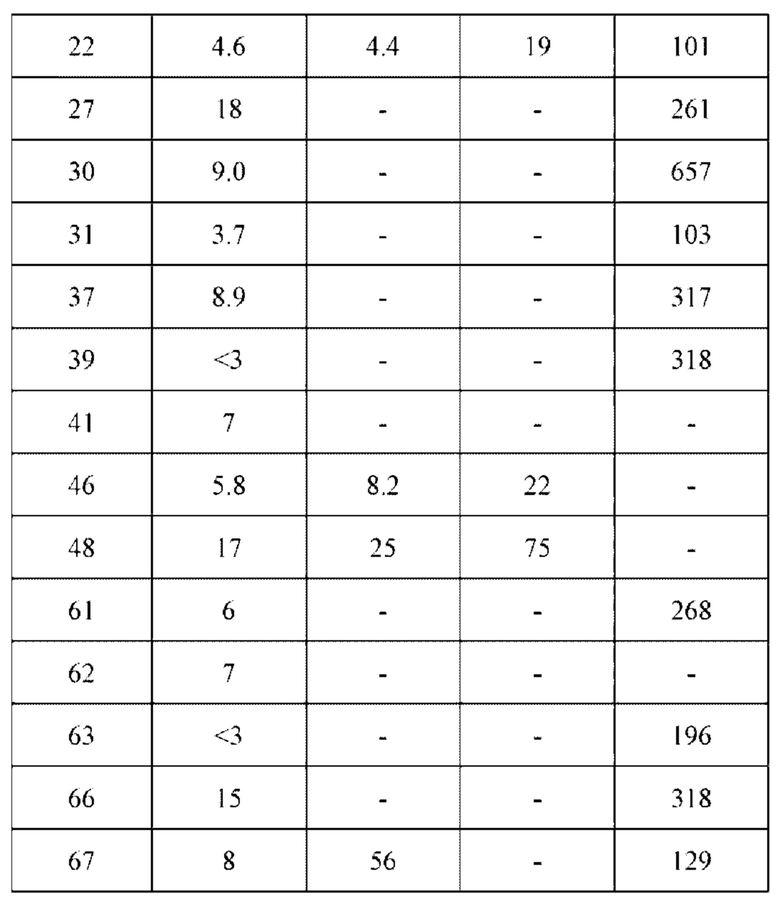
"-" означает «не проверяли».
Как видно из результатов эксперимента в Таблице 1, соединения согласно настоящему изобретению обладают хорошей ингибирующей активностью в отношении FGFR, что указывает на то, что соединения согласно настоящему изобретению имеют хороший клинический потенциал применения при лечении заболеваний, опосредованных аномалией FGFR.
Экспериментальный Пример 2: Тест на клеточную активность соединений согласно настоящему изобретению
Нер3В - это клетка с аномальным FGFR при гепатоцеллюлярной карциноме
RT112/84 - это клетка с аномальным FGFR при раке мочевого пузыря
DMS114 - это клетка с аномальным FGFR при мелкоклеточном раке легкого
AN3CA - это клетка с аномальным FGFR при раке эндометрия
SNU-16 - это клетка с аномальным FGFR при раке желудка
Тестируемое вещество: Соединения согласно настоящему изобретению, структуры которых показаны выше.
Тестовый прибор: многофункциональное считывающее устройство для микропланшет Espire.
Метод испытания:
Каждую клеточную линию высевали в 96-луночный планшет и прилипшие к субстрату культивировали в течение ночи. Затем добавляли соединения в различных концентрациях (12 групп доз, 3Х серийные разведения в ДМСО) до конечной концентрации 0,17-30000 нМ, с конечным содержанием ДМСО 5%. Лунка с отрицательным контролем была средней, содержащей 5% ДМСО. После инкубации при 37°С, 5% CO2, 95% влажности в течение 72 ч культуры испытывали. В каждую лунку добавляли 30 мкл реагента Cell titer-Glo и инкубировали в течение 30 мин при комнатной температуре. Окончательные данные считывали считывающим устройством микропланшетов Espire.
Результаты испытаний приведены в таблице 2.
Таблица 2. Ингибирующая активность соединений согласно настоящему изобретению против клеток (IC50)
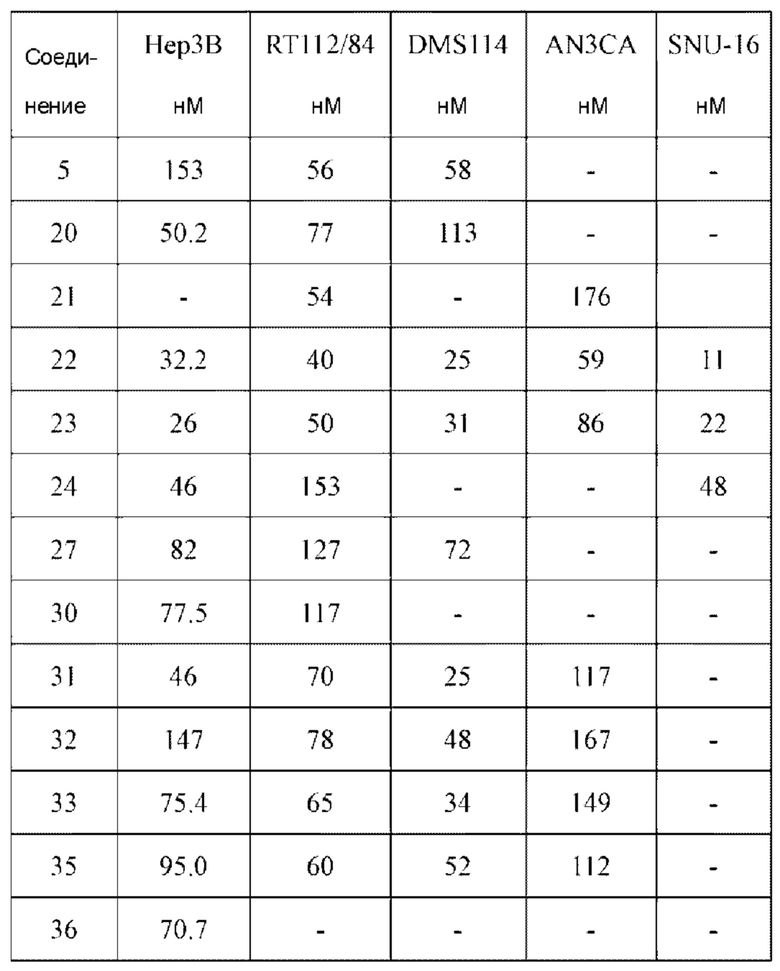
"-" означает «не проверяли».
Как видно из результатов эксперимента в Таблице 2, соединения согласно настоящему изобретению обладают хорошей ингибирующей активностью в отношении клеток с аномальным FGFR, таких как Нер3В, RT112/84, DMS114, AN3CA, SNU-16 и т.д., что указывает на то, что соединения согласно настоящему уровню могут использоваться для лечения рака, опосредованного аномалией FGF/FGFR, такого как рак печени, рак желудка, мелкоклеточный рак, рак мочевого пузыря, рак эндометрия, и имеют очень хорошую ценность клинического применения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2014 |
|
RU2704112C2 |
| МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СООТВЕТСТВУЮЩИЕ СПОСОБЫ ПРИМЕНЕНИЯ | 2019 |
|
RU2805511C2 |
| Производные хинолона как ингибиторы рецептора фактора роста фибробластов | 2015 |
|
RU2721723C2 |
| УЛУЧШЕННЫЕ АГОНИСТЫ АПЕЛИНОВОГО РЕЦЕПТОРА (APJ) И ИХ ИСПОЛЬЗОВАНИЕ | 2016 |
|
RU2766148C1 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| ИНГИБИТОРЫ РЕЦЕПТОРА ФАКТОРА РОСТА ФИБРОБЛАСТОВ | 2013 |
|
RU2679130C2 |
| ПИРАЗИНОВЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2019 |
|
RU2809631C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| СОЕДИНЕНИЯ С КОНДЕНСИРОВАННЫМИ КОЛЬЦАМИ | 2019 |
|
RU2783414C2 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
Изобретение относится к области органической химии, а именно к необратимому ингибитору рецептора фактора роста фибробластов (FGFR) общей формулы (I) или его фармацевтически приемлемой соли или стереоизомеру:
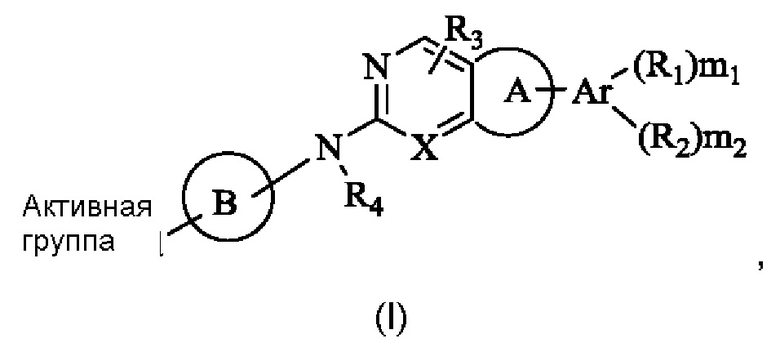
где R1 и R2 каждый независимо выбран из группы, состоящей из водорода, галогена и C1-6 алкокси; R3 и R4 каждый независимо выбран из группы, состоящей из водорода, С1-6 алкила и (С1-6 алкил)2-амино-С1-6 алкила; Ar представляет собой фенил; кольцо А представляет собой фенил; кольцо В представляет собой 3-10-членный насыщенный или ненасыщенный гетероциклил, содержащий по меньшей мере один гетероатом N, возможно замещенный 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой; X представляет собой N; R6 выбран из группы, состоящей из: (i) водорода, (ii) гидрокси, амино, карбокси, циано, атома галогена, С2-4 алкенилкарбониламино или =СН2, (iii) С1-6 алкила, C1-6 алкокси, С1-6 алкиламино, (C1-6 алкил)2-амино, галоС1-6 алкила, галоС1-6 алкокси или C1-6 алкилтио, возможно замещенных гидрокси, амино, циано, галогеном, C1-6 алкилом, C1-6 алкокси, C1-6 алкокси-С1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2-амино, C1-6 алкилкарбониламино, С1-6 алкилсульфониламино или 3-8-членным гетероциклилом, содержащим 1-3 гетероатома, выбранных из О, S и N, и (iv) аминокарбонила, С1-6 алкилкарбонила, C1-6 алкиламинокарбонила, (С1-6 алкил)2-аминокарбонила, С1-6 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила, содержащего 1-3 гетероатома, выбранных из О, S и N; m1 и m2 равны 1 или 2; «Активная группа» выбрана из группы, состоящей из
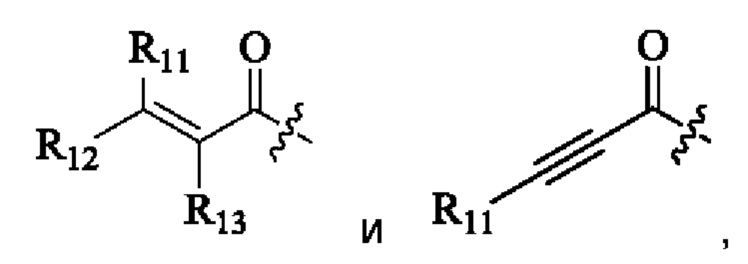 R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода и С1-4 алкила. Также предложена фармацевтическая композиция, применение и способ лечения заболевания. Технический результат: предложено новое соединение общей формулы (I), обладающее высокой эффективностью и высокой селективностью в отношении ингибирования рецептора фактора роста фибробластов и может применяться для лечения заболеваний, опосредованных аномалией FGF/FGFR. 4 н. и 13 з.п. ф-лы, 2 табл., 23 пр.
R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода и С1-4 алкила. Также предложена фармацевтическая композиция, применение и способ лечения заболевания. Технический результат: предложено новое соединение общей формулы (I), обладающее высокой эффективностью и высокой селективностью в отношении ингибирования рецептора фактора роста фибробластов и может применяться для лечения заболеваний, опосредованных аномалией FGF/FGFR. 4 н. и 13 з.п. ф-лы, 2 табл., 23 пр.
1. Необратимый ингибитор рецептора фактора роста фибробластов (FGFR) общей формулы (I) или его фармацевтически приемлемая соль или стереоизомер:
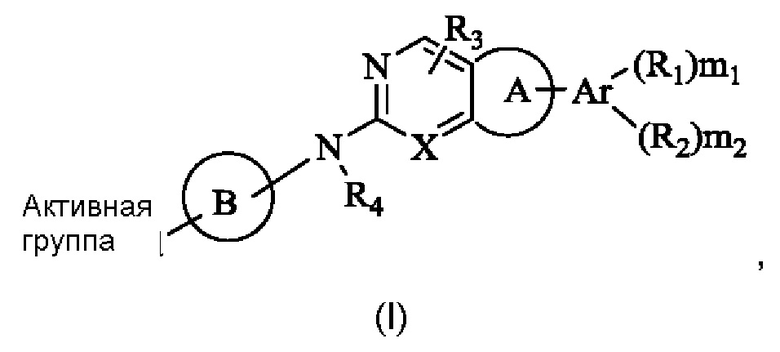
где
R1 и R2 каждый независимо выбран из группы, состоящей из водорода, галогена и C1-6 алкокси;
R3 и R4 каждый независимо выбран из группы, состоящей из водорода, С1-6 алкила и (С1-6 алкил)2-амино-С1-6 алкила;
Ar представляет собой фенил;
кольцо А представляет собой фенил;
кольцо В представляет собой 3-10-членный насыщенный или ненасыщенный гетероциклил, содержащий по меньшей мере один гетероатом N, возможно замещенный 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой;
X представляет собой N;
R6 выбран из группы, состоящей из:
(i) водорода,
(ii) гидрокси, амино, карбокси, циано, атома галогена, С2-4 алкенилкарбониламино или =СН2,
(iii) С1-6 алкила, C1-6 алкокси, С1-6 алкиламино, (C1-6 алкил)2-амино, галоС1-6 алкила, галоС1-6 алкокси или C1-6 алкилтио, возможно замещенных гидрокси, амино, циано, галогеном, C1-6 алкилом, C1-6 алкокси, C1-6 алкокси-С1-6 алкокси, C1-6 алкиламино, (C1-6 алкил)2-амино, C1-6 алкилкарбониламино, С1-6 алкилсульфониламино или 3-8-членным гетероциклилом, содержащим 1-3 гетероатома, выбранных из О, S и N, и
(iv) аминокарбонила, С1-6 алкилкарбонила, C1-6 алкиламинокарбонила, (С1-6 алкил)2-аминокарбонила, С1-6 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила, содержащего 1-3 гетероатома, выбранных из О, S и N;
m1 и m2 равны 1 или 2;
«Активная группа» выбрана из группы, состоящей из
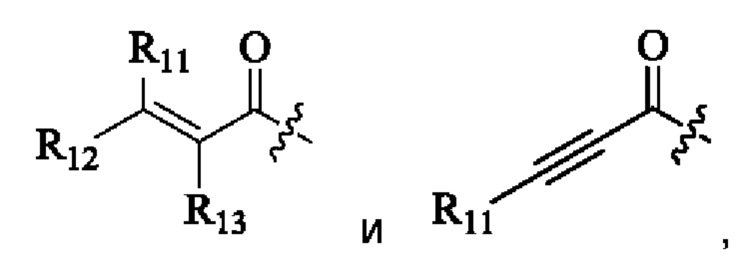
R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода и С1-4 алкила.
2. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по п. 1, где
R1 представляет собой галоген;
R2 представляет собой C1-4 алкокси;
Ar представляет собой фенил;
m1 и m2 равны 1 или 2.
3. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по п. 2, где
R1 представляет собой галоген;
R2 представляет собой C1-4 алкокси;
R3 и R4 каждый независимо выбран из группы, состоящей из водорода, С1-4 алкила и (С1-4 алкил)2-амино-С1-4 алкила;
Ar представляет собой фенил;
кольцо А представляет собой фенил,
кольцо В представляет собой 4-10-членный насыщенный или ненасыщенный гетероциклил, содержащий по меньшей мере один гетероатом N, возможно замещенный 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой;
X представляет собой N;
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, карбокси, циано или =СН2,
(iii) C1-4 алкила, возможно замещенного гидрокси, циано, галогеном, С1-4 алкокси, С1-4 алкокси-С1-4 алкокси, С1-4 алкиламино, (C1-4 алкил)2-амино, C1-4 алкилкарбониламино, С1-4 алкилсульфониламино или 3-8-членным гетероциклилом, содержащим 1-3 гетероатома, выбранных из О, S и N, и
(iv) аминокарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2-аминокарбонила, C1-4 алкоксикарбонила или 3-8-членного гетероциклилкарбонила, содержащего 1-3 гетероатома, выбранных из О, S и N;
m1 и m2 равны 1 или 2;
«Активная группа» выбрана из группы, состоящей из
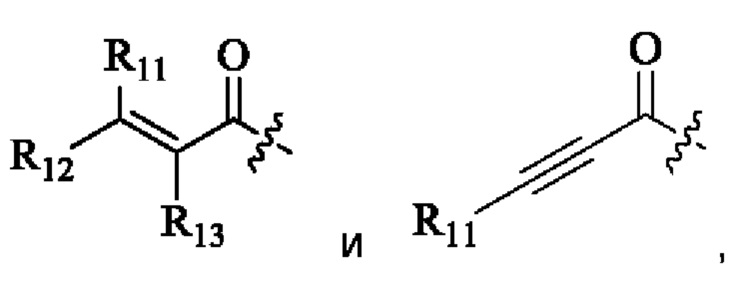
R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода и С1-4 алкила.
4. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по любому из пп. 1-3, имеющий структуру, показанную общей формулой (II):
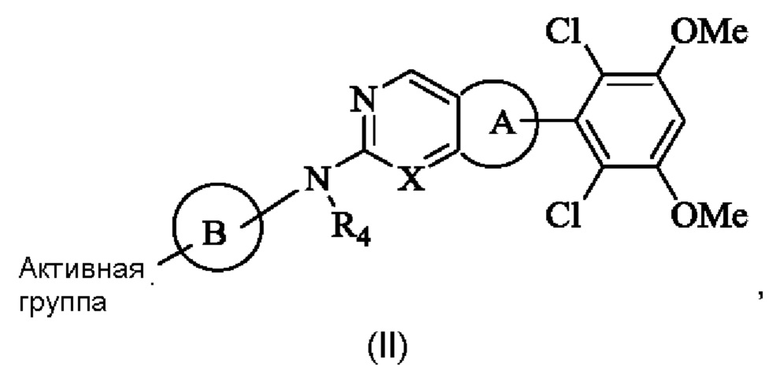
где
R4 выбран из группы, состоящей из водорода, С1-4 алкила и (С1-4 алкил)2-амино-С1-4 алкила;
кольцо А представляет собой фенил;
кольцо В выбрано из группы, состоящей из 4-6-членного насыщенного или ненасыщенного моногетероциклила и 6-10-членного насыщенного или ненасыщенного конденсированного гетероциклила, содержащего по меньшей мере один гетероатом N, возможно замещенного 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой;
X представляет собой N;
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, карбокси, циано или =СН2,
(iii) С1-4 алкила, возможно замещенного гидрокси, циано, галогеном, С1-4 алкокси, С1-4 алкокси-С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2-амино, C1-4 алкилкарбониламино, C1-4 алкилсульфониламино или 3-8-членным гетероциклилом, содержащим 1-3 гетероатома, выбранных из О, S и N, и
(iv) аминокарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2-аминокарбонила, С1-4 алкоксикарбонила или 3-8-членного гетероциклилкарбонила, содержащего 1-3 гетероатома, выбранных из О, S и N;
«Активная группа» выбрана из группы, состоящей из
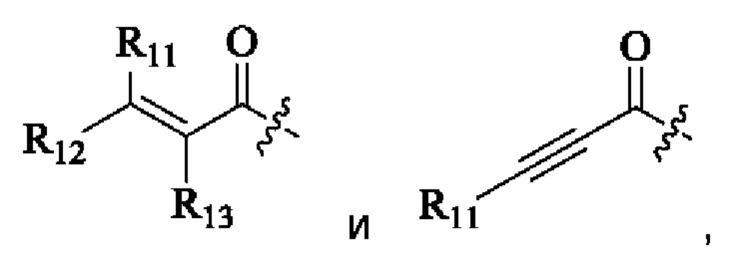
R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода и С1-4 алкила.
5. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по любому из пп. 1-4, имеющий структуру, показанную общей формулой (II):
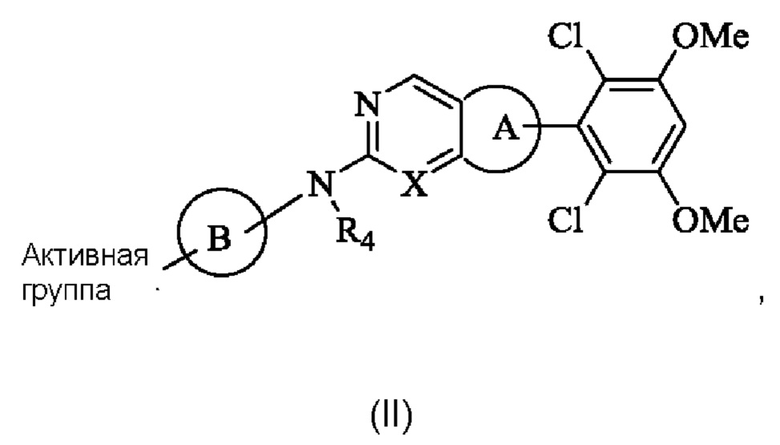
где Активная группа непосредственно связана с атомом N на кольце В следующим образом:
кольцо В выбрано из группы, состоящей из:
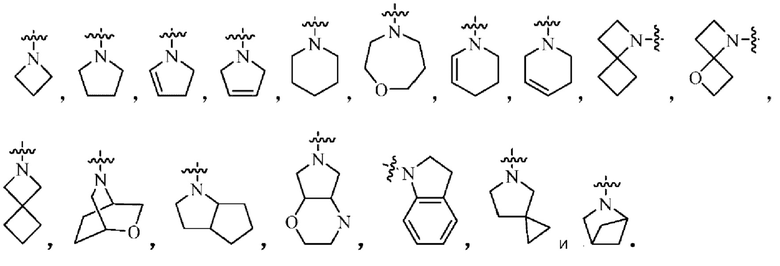
6. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по любому из пп. 1-5, где ингибитор имеет структуру, выбранную из группы, состоящей из:
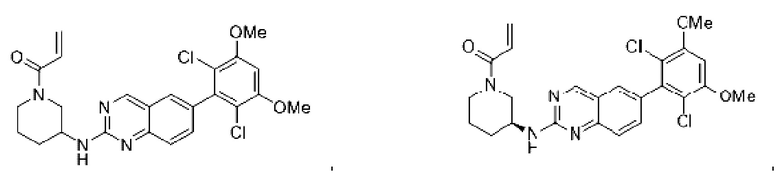
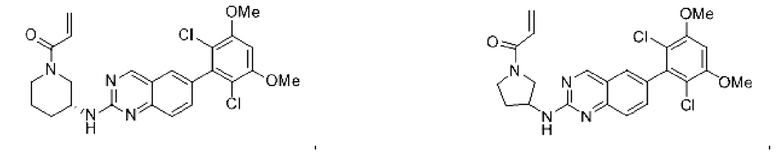
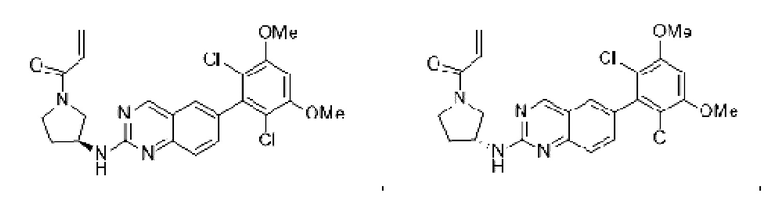
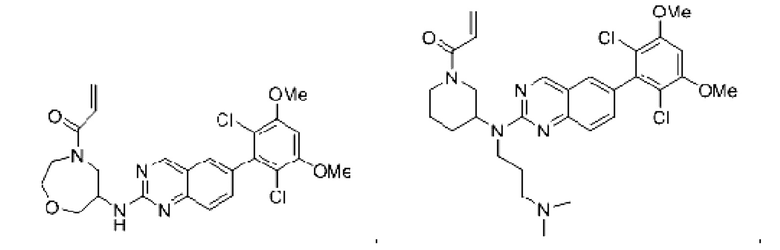
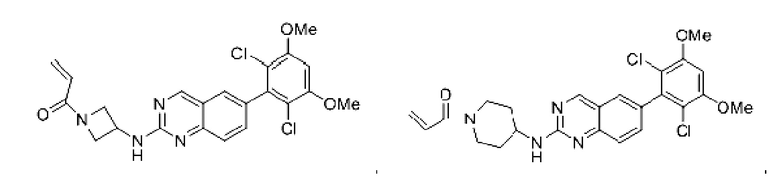
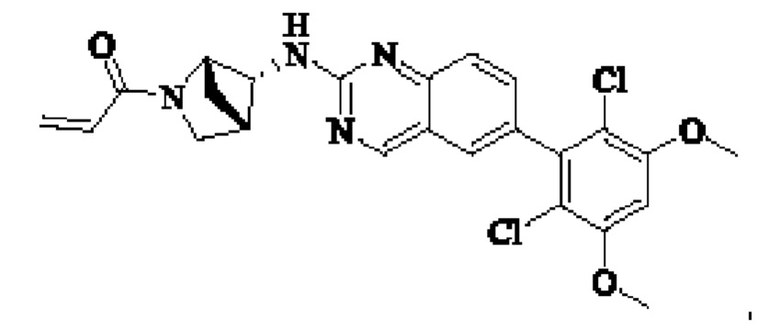
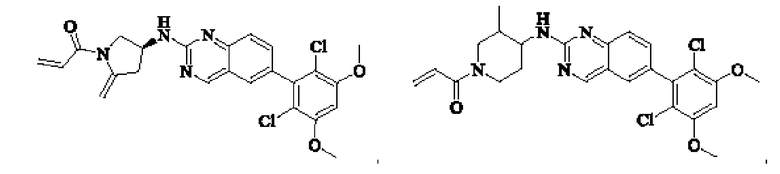
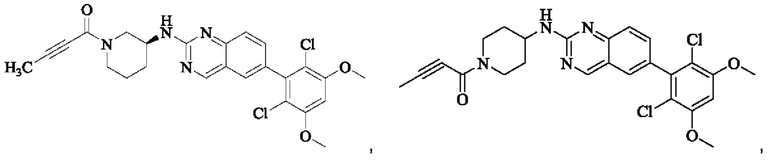
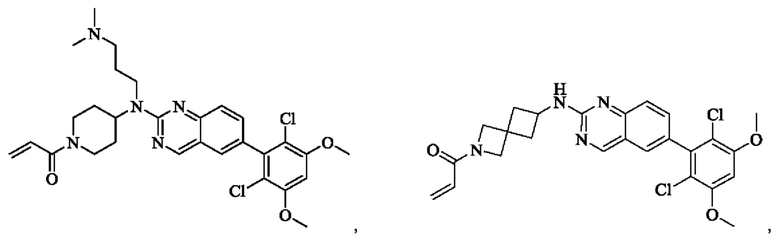
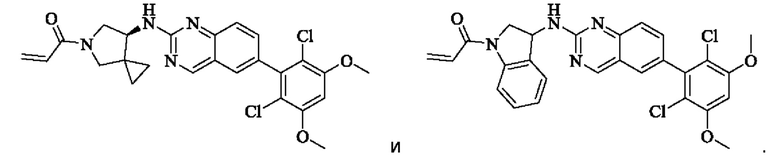
7. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по п. 4, где кольцо В выбрано из 5-6-членного насыщенного моногетероциклила, содержащего по меньшей мере один гетероатом N, возможно замещенного 1-3 группами R6, и атом N в кольце В непосредственно связан с Активной группой.
8. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по п. 1, имеющий структуру, показанную общей формулой (III):
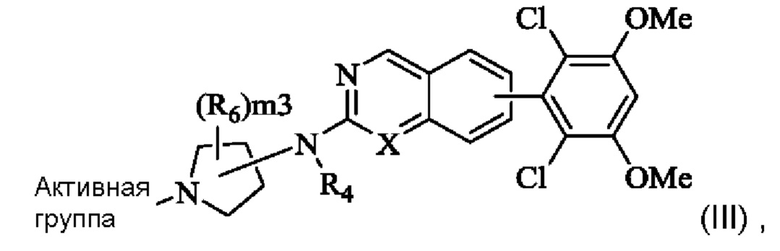
где
R4 представляет собой Н или С1-4 алкил;
X представляет собой N;
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, амино, карбокси, циано или галогена,
(iii) С1-4 алкила, С1-4 алкокси, возможно замещенных гидрокси, амино, циано, галогеном, С1-4 алкилом, С1-4 алкокси, C1-4 алкокси-С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2-амино, С1-4 алкилкарбониламино, С1-4 алкилсульфониламино или 3-8-членным гетероциклилом, содержащим 1-3 гетероатома, выбранных из О, S и N, и
(iv) аминокарбонила, С1-4 алкилкарбонила, C1-4 алкиламинокарбонила, (C1-4 алкил)2-аминокарбонила, C1-4 алкоксикарбонила, 3-8-членного циклоалкилкарбонила или 3-8-членного гетероциклилкарбонила, содержащего 1-3 гетероатома, выбранных из О, S и N;
m3 является целым числом от 1 до 3;
«Активная группа» выбрана из группы, состоящей из
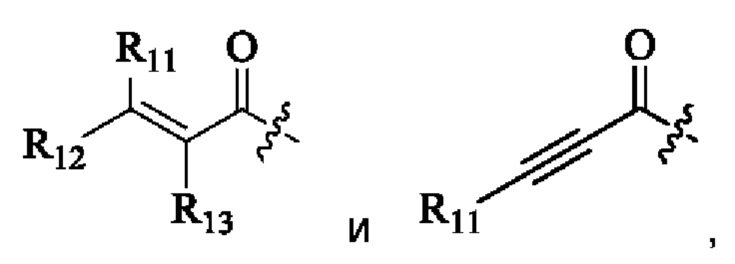
R11, R12 и R13 каждый независимо выбран из группы, состоящей из водорода и C1-4 алкила.
9. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по п. 8, где
R6 выбран из группы, состоящей из
(i) водорода,
(ii) гидрокси, карбокси или циано,
(iii) C1-4 алкила, возможно замещенного гидрокси, циано, галогеном, С1-4 алкокси, C1-4 алкокси-С1-4 алкокси, С1-4 алкиламино, (С1-4 алкил)2-амино, C1-4 алкилкарбониламино, C1-4 алкилсульфониламино или 3-8-членным гетероциклилом, содержащим 1-3 гетероатома, выбранных из О, S и N, и
(iv) аминокарбонила, С1-4 алкиламинокарбонила, (С1-4 алкил)2-аминокарбонила, C1-4 алкоксикарбонила или 3-8-членного гетероциклилкарбонила, содержащего 1-3 гетероатома, выбранных из О, S и N;
Активная группа представляет собой 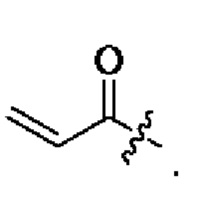
10. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по п. 1, имеющий структуру, выбранную из группы, состоящей из:
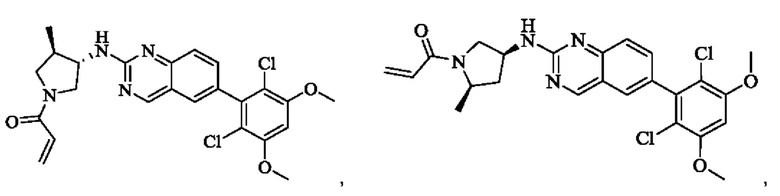
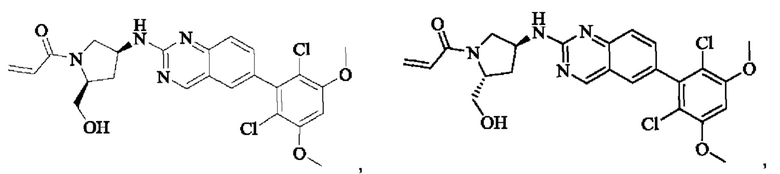
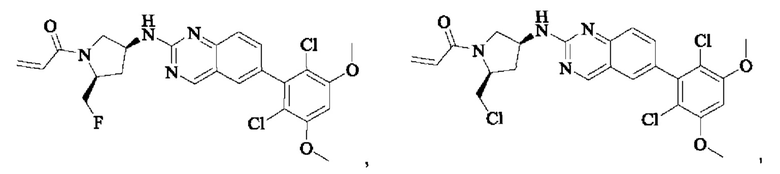
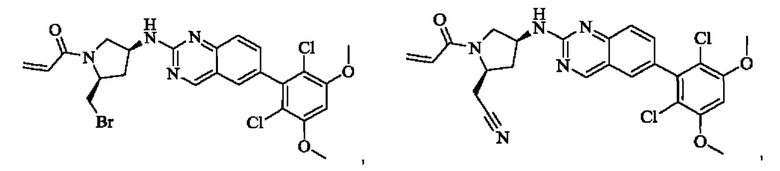
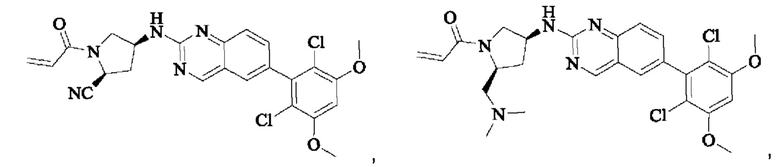
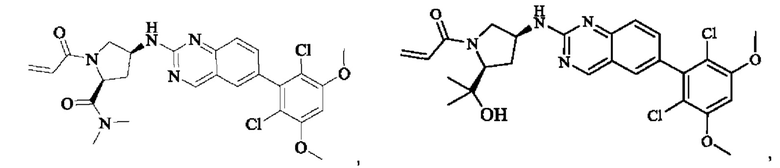
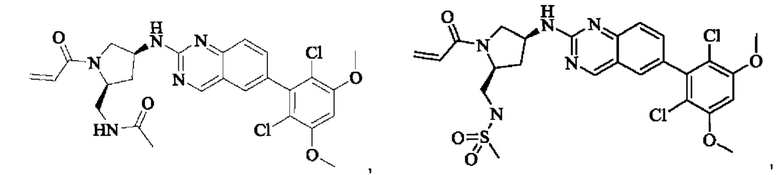
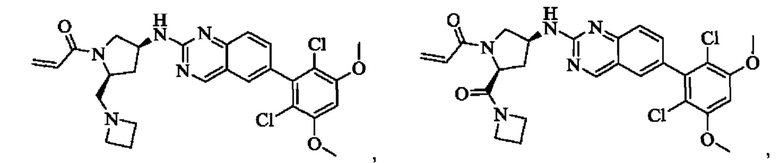
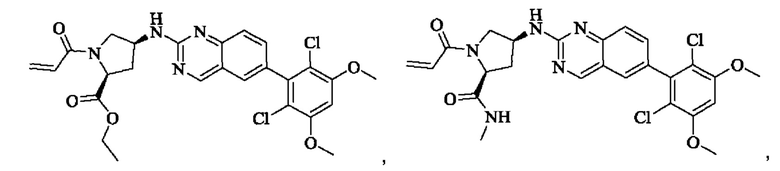
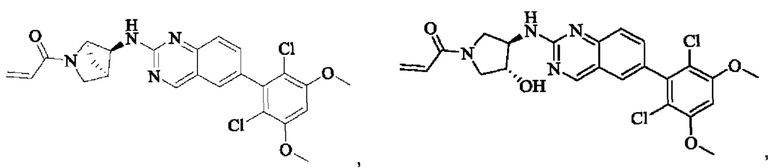
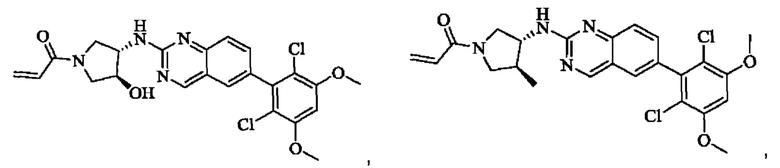
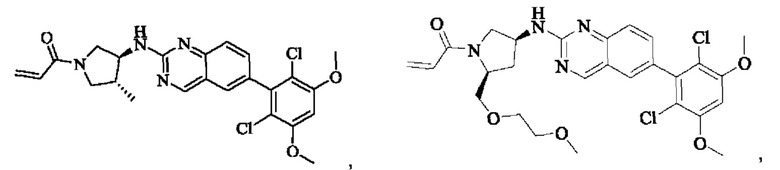
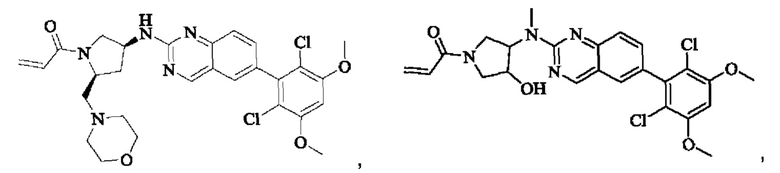
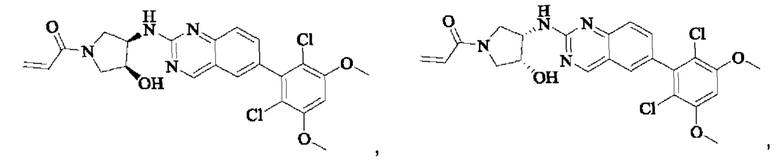
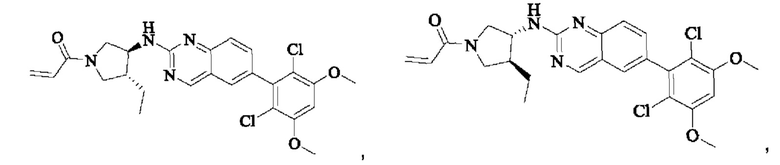
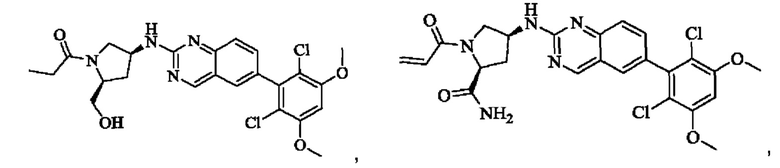
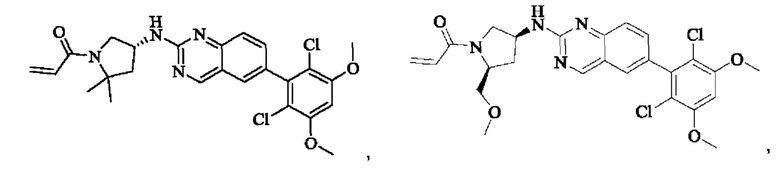
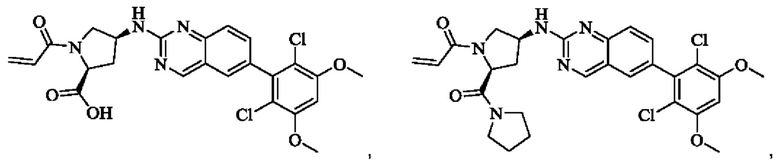
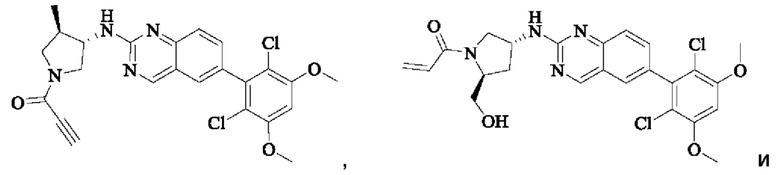
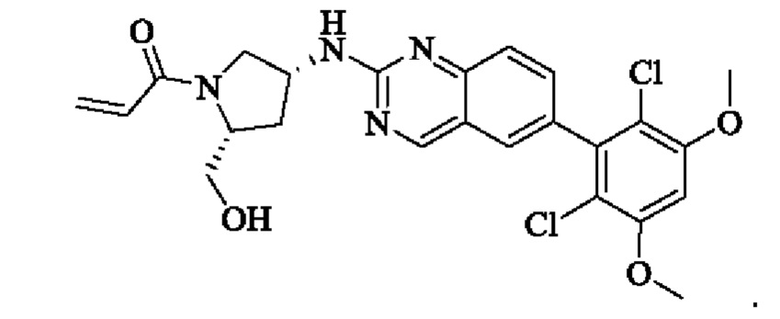
11. Фармацевтическая композиция, содержащая эффективное количество ингибитора или его фармацевтически приемлемой соли или стереоизомера по любому из пп. 1-10 и один или более фармацевтически приемлемых носителей.
12. Фармацевтическая композиция по п. 11, дополнительно содержащая один или более вторых терапевтически активных агентов, где вторыми терапевтически активными агентами являются антиметаболиты, ингибиторы факторов роста, митотические ингибиторы, противоопухолевые гормоны, алкилирующие агенты, металлы, ингибиторы топоизомеразы, гормональные лекарства, иммуномодуляторы, гены-супрессоры опухолей, противораковые вакцины или антитела и низкомолекулярные лекарства, относящиеся к иммунным контрольным точкам или иммунотерапии опухолей.
13. Применение ингибитора или его фармацевтически приемлемой соли или стереоизомера по любому из пп. 1-10 или фармацевтической композиции по любому из пп. 11, 12 для изготовления лекарственного средства для лечения заболевания, опосредованного аномалией FGF/FGFR.
14. Применение по п. 13, где заболевание, опосредованное аномалией FGF/FGFR, представляет собой рак, и рак включает в себя рак легкого, плоскоклеточный рак эпителиальных клеток, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, карциному протока молочной железы, рак головы и шеи, рак эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, рак почечной лоханки, рак пищевода, аденокарциному пищевода, глиому, рак простаты, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому, нейрофиброматоз, рак костей, рак кожи, рак головного мозга, рак толстой кишки, рак яичек, желудочно-кишечную стромальную опухоль, рак ротовой полости, рак глотки, множественную миелому, лейкемию, неходжкинскую лимфому, хориоаденому толстой кишки, меланому, цитому и саркому, и миелодиспластический синдром.
15. Способ лечения заболевания, опосредованного аномалией FGF/FGFR, включающий введение субъекту, нуждающемуся в этом, ингибитора или его фармацевтически приемлемой соли или стереоизомера по любому из пп. 1-10 или фармацевтической композиции по любому из пп. 11, 12, где заболевание, опосредованное аномалией FGF/FGFR, представляет собой рак, причем рак включает в себя рак легкого, плоскоклеточный рак эпителиальных клеток, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, карциному протока молочной железы, рак головы и шеи, рак эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, рак почечной лоханки, рак пищевода, аденокарциному пищевода, глиому, рак простаты, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому, нейрофиброматоз, рак костей, рак кожи, рак головного мозга, рак толстой кишки, рак яичек, желудочно-кишечную стромальную опухоль, рак ротовой полости, рак глотки, множественную миелому, лейкемию, неходжкинскую лимфому, хориоаденому толстой кишки, меланому, цитому и саркому, и миелодиспластический синдром.
16. Ингибитор или его фармацевтически приемлемая соль или стереоизомер по любому из пп. 1-10 для применения в качестве лекарственного средства для лечения заболевания, опосредованного аномалией FGF/FGFR, где заболевание, опосредованное аномалией FGF/FGFR, представляет собой рак, причем рак включает в себя рак легкого, плоскоклеточный рак эпителиальных клеток, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, карциному протока молочной железы, рак головы и шеи, рак эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, рак почечной лоханки, рак пищевода, аденокарциному пищевода, глиому, рак простаты, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому, нейрофиброматоз, рак костей, рак кожи, рак головного мозга, рак толстой кишки, рак яичек, желудочно-кишечную стромальную опухоль, рак ротовой полости, рак глотки, множественную миелому, лейкемию, неходжкинскую лимфому, хориоаденому толстой кишки, меланому, цитому и саркому, и миелодиспластический синдром.
17. Фармацевтическая композиция по любому из пп. 11, 12 для применения в качестве лекарственного средства для лечения заболевания, опосредованного аномалией FGF/FGFR, где заболевание, опосредованное аномалией FGF/FGFR, представляет собой рак, причем рак включает в себя рак легкого, плоскоклеточный рак эпителиальных клеток, рак мочевого пузыря, рак желудка, рак яичника, рак брюшины, рак молочной железы, карциному протока молочной железы, рак головы и шеи, рак эндометрия, рак тела матки, рак прямой кишки, рак печени, рак почки, рак почечной лоханки, рак пищевода, аденокарциному пищевода, глиому, рак простаты, рак щитовидной железы, рак женской репродуктивной системы, карциному in situ, лимфому, нейрофиброматоз, рак костей, рак кожи, рак головного мозга, рак толстой кишки, рак яичек, желудочно-кишечную стромальную опухоль, рак ротовой полости, рак глотки, множественную миелому, лейкемию, неходжкинскую лимфому, хориоаденому толстой кишки, меланому, цитому и саркому, и миелодиспластический синдром.
| WO 2015061572 A1, 30.04.2015 | |||
| EP 1878727 A1, 16.01.2008 | |||
| CN 104540809 A, 22.04.2015 | |||
| WO 2015108992 A1, 23.07.2015 | |||
| WO 2014174307 A1, 30.10.2014 | |||
| RU 2011122539 A, 10.12.2012. |

