Заявление приоритета
Настоящая заявка заявляет приоритет согласно патенту США с серийным номером 61/892086, поданному 17 октября 2013 года, патенту США с серийным номером 61/931204, поданному 24 января 2014 года, и патенту США с серийным номером 61/975229, поданному 4 апреля 2014 года, полное содержание каждого из которых включено в настоящий документ посредством ссылки в полном объеме.
Уровень техники
Настоящее изобретение относится к соединениям и композициям, пригодным для лечения расстройств, связанных с KIT и PDGFR.
Фермент KIT (также называемый CD117) представляет собой рецепторную тирозинкиназу, экспрессируемую во многих типах клеток. Молекула KIT содержит длинный внеклеточный домен, трансмембранный сегмент и внутриклеточную часть. Лиганд к KIT представляет собой фактор стволовых клеток (SCF), связывание которого с внеклеточным доменом KIT вызывает димеризацию рецептора и активацию последующих путей передачи сигналов. Мутации KIT встречаются, главным образом, в ДНК, кодирующей околомембранный домен (экзон 11). Реже они могут возникать также в экзонах 7, 8, 9, 13, 14, 17 и 18. Мутации приводят к независимости функции KIT от активации SCF, что обусловливает высокую скорость деления клеток и потенциальную геномную нестабильность. Мутантная KIT участвует в патогенезе некоторых расстройств и патологических состояний, включая системный мастоцитоз, GIST (желудочно-кишечные стромальные опухоли), AML (острый миелоидный лейкоз), меланому и семиному. Следовательно, существует потребность в терапевтических агентах, ингибирующих KIT, и особенно в агентах, ингибирующих мутантную KIT.
Рецепторы тромбоцитарного фактора роста (PDGF-R) представляет собой рецепторы к тирозинкиназе клеточной поверхности для членов семейства тромбоцитарного фактора роста (PDGF). Субъединицы PDGF-A и -B являются важными факторами, регулирующими пролиферацию клеток, клеточную дифференцировку, рост клеток, развитие и многие заболевания, включая рак. Мутация PDGFRA D842V обнаружена в отдельном подмножестве GIST, главным образом, желудка. Известно, что мутация D842V связана с резистентностью к ингибитору тирозинкиназы. Следовательно, существует потребность в агентах, нацеленных на данную мутацию.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В настоящем изобретении представлены соединения и композиции для лечения или предупреждения патологических состояний, таких как мастоцитоз и болезни тучных клеток, посредством модулирования активности KIT, указанные соединения имеют структурную Формулу I:
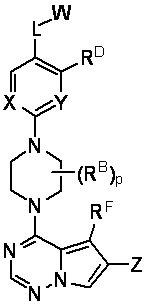
I
или их фармацевтически приемлемые соли, где:
W выбран из водорода или 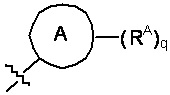 , где кольцо A выбрано из моноциклического или бициклического арила, моноциклического или бициклического гетероарила, циклоалкила или гетероциклила;
, где кольцо A выбрано из моноциклического или бициклического арила, моноциклического или бициклического гетероарила, циклоалкила или гетероциклила;
каждый X и Y независимо выбран из CR1 или N;
Z представляет собой C1-C6 алкил, циклоалкил, моноциклический или бициклический арил, моноциклический или бициклический аралкил, моноциклический или бициклический гетероарил, моноциклический или бициклический гетероциклил, моноциклический или бициклический гетероциклилалкил; где каждый C1-C6 алкил, циклоалкил, моноциклический или бициклический арил, моноциклический или бициклический аралкил, моноциклический или бициклический гетероарил, моноциклический или бициклический гетероциклил, моноциклический или бициклический гетероциклилалкил независимо замещен 0-5 группами RC;
L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкинилен)-, -(C2-C6 алкенилен)-, -(C1-C6 галогеналкилен)-, -(C1-C6 гетероалкилен)-, -(C1-C6 гидроксиалкилен)-, -C(O)-, -O-, -S-, -S(O), -SO2-, -N(R2)-, -O-(C1-C6 алкилен)-, -(C1-C6 алкилен)-O-, -N(R2)-CO-, -CO-N(R2)-, -(C1-C6 алкилен)-N(R2)-, -N(R2)-(C1-C6 алкилен)-, -N(R2)-CO-(C1-C6 алкилен)-, -CO-N(R2)-(C1-C6 алкилен)-, -N(R2)-SO2-, - SO2-N(R2)-, -N(R2)-SO2-(C1-C6 алкилен)- или -SO2-N(R2)-(C1-C6 алкилен)-;
каждый RA и RB независимо выбран из C1-C6 алкила, C1-C6 циклоалкила, C1-C6 гетероциклила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 гетероалкила, моноциклического или бициклического аралкила, -N(R2)(R2), циано, -OR2;
каждый RC независимо выбран из C1-C6 алкила, C1-C6 алкинила, галогена, C1-C6 гетероалкила, C1-C6 галогеналкила, C1-C6 галогеналкокси, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического арила, моноциклического или бициклического арилокси, моноциклического или бициклического аралкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, нитро, циано, -C(O)R2, -OC(O)R2, -C(O)OR2, -SR2, -S(O)2R2, -S(O)2-N(R2)(R2), -(C1-C6 алкилен)-S(O)2-N(R2)(R2), -N(R2)(R2), -C(O)-N(R2)(R2), -N(R2)(R2)-C(O)R2, -(C1-C6 алкилен)-N(R2)-C(O)R2, -NR2S(O)2R2, -P(O)(R2)(R2) и –OR2; где каждый гетероалкил, гелогеналкил, галогеналкокси, алкил, алкинил, циклоалкил, арил, арилокси, аралкил, гетероциклил, гетероциклилалкил независимо замещен 0-5 группами Ra; или 2 RC вместе с атомом(-ами) углерода, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо, замещенное 0-5 группами Ra;
каждый RD и RF независимо представляет собой водород, C1-C6 алкил, C1-C6 циклоалкил, гидроксил, галоген, C1-C6 алкокси, C1-C6 галогеналкил, -N(R2)(R2) или циано;
каждый R1 независимо выбран из водорода, C1-C6 алкила, моноциклического аралкила, C1-C6 гидроксиалкила, галогена, C1-C6 галогеналкила, -N(R2)(R2), -OR2;
каждый R2 независимо выбран из водорода, гидроксила, галогена, тиола, C1-C6 тиоалкила, -NR”R”, C1-C6 алкила, C1-C6 алкокси, C1-C6 галогеналкила, C1-C6 гидроксиалкила, циклоалкила, циклоалкилалкила, гетероциклила, гетероциклилалкила, где каждый C1-C6 алкил, циклоалкил и гетероциклил независимо замещен 0-5 группами Rb, или 2 R2 вместе с атомом углерода или азота, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо;
каждый Ra и Rb независимо представляет собой водород, галоген, циано, гидроксил, C1-C6 алкоксил, -C(O)R’, C(O)OR’, C1-C6 алкил, C1-C6 галогеналкил, C1-C6 гетероалкил, C1-C6 гидроксиалкил, -NR’R’ или циклоалкил, где циклоалкил замещен 0-5 группами R’;
каждый R’ представляет собой водород, гидроксил или C1-C6 алкил;
каждый R” представляет собой водород, C1-C6 алкил, -C(O)-C1-C6 алкил, -C(O)-NR’R’; -C(S)-NR’R’; и
m, p и q, каждый независимо представляют собой 0, 1, 2, 3 или 4.
Любые соединения, описанные в настоящем документе, могут быть использованы для лечения любых заболеваний, описанных в настоящем документе.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1 представляет собой линейный график кривых роста опухоли в различных экспериментальных группах: носитель (●), дазатиниб, 25 мг/кг, два раза в сутки*10 дней (■), Соединение 46, 3 мг/кг, перорально, один раз в сутки*10 дней (▲),Соединение 46, 10 мг/кг, перорально, один раз в сутки*10 дней (▼), Соединение 46, 30 мг/кг, перорально, один раз в сутки*10 дней (♦), и Соединение 46, 100 мг/кг, перорально, один раз в сутки*10 дней (●).
Фиг. 2 представляет собой линейный график, демонстрирующий результаты изменения массы тела мышей-опухоленосителей в различных экспериментальных группах: носитель (●), дазатиниб, 25 мг/кг, перорально, два раза в сутки*10 дней (■), Соединение 46, 3 мг/кг, перорально, один раз в сутки*10 дней (▲), Соединение 46, 10 мг/кг, перорально, один раз в сутки*10 дней (▼), Соединение 46, 30 мг/кг, перорально, один раз в сутки*10 дней (♦), и Соединение 46, 100 мг/кг, перорально, один раз в сутки*10 дней (●).
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
«Алифатическая группа» означает линейную, разветвленную или циклическую углеводородную группу и включает насыщенные и ненасыщенные группы, такие как алкильная группа, алкенильная группа и алкинильная группа.
«Алкилен» относится к двухвалентному радикалу алкильной группы, например, -CH2-, -CH2CH2- и CH2CH2CH2-.
«Алкенил» означает алифатическую группу, содержащую по меньшей мере одну двойную связь.
«Алкоксил» или «алкокси» означает алкильную группу, имеющую кислородный радикал, присоединенный к ней. Иллюстративные алкоксильные группы включают метокси, этокси, пропилокси, трет-бутокси и т.п. Термин «галогеналкокси» относится к алкокси, в котором один или более атомов водорода заменены на галоген, и включает алкокси-фрагменты, в которых все водороды заменены на галоген (например, перфторалкокси).
«Алкил» относится к одновалентному радикалу насыщенного прямого или разветвленного углеводорода, такому как прямая или разветвленная группа из 1-12, 1-10 или 1-6 атомов углерода, называемая в настоящем документе C1-C12 алкил, C1-C10 алкил и C1-C6 алкил, соответственно. Иллюстративные алкильные группы включают, но не ограничиваются ими, метил, этил, пропил, изопропил, 2-метил-1-пропил, 2-метил-2-пропил, 2-метил-1-бутил, 3-метил-1-бутил, 2-метил-3-бутил, 2,2-диметил-1-пропил, 2-метил-1-пентил, 3-метил-1-пентил, 4-метил-1-пентил, 2-метил-2-пентил, 3-метил-2-пентил, 4-метил-2-пентил, 2,2-диметил-1-бутил, 3,3-диметил-1-бутил, 2-этил-1-бутил, бутил, изобутил, трет-бутил, пентил, изопентил, неопентил, гексил, гептил, октил и т.д.
«Алкенилен» относится к алкенильной группе, имеющей две точки присоединения. Например, «этенилен» представляет собой группу -CH=CH-. Алкениленовые группы также могут быть в незамещенной форме или в замещенной форме с одним или более заместителями.
«Алкинил» относится к прямой или разветвленной углеводородной цепи, содержащей 2-12 атомов углерода и характеризующейся наличием одной или более тройных связей. Примеры алкинильных групп включают, но не ограничиваются ими, этинил, пропаргил и 3-гексинил. Один из атомов углерода у тройной связи может необязательно представлять собой точку присоединения алкинильного заместителя.
«Алкинилен» относится к алкинилу, имеющему две точки присоединения. Например, «этинилен» представляет собой группу -C≡C-. Алкиниленовые группы также могут быть в незамещенной форме или в замещенной форме с одним или более заместителями.
«Гидроксиалкилен» или «гидроксиалкил» относится к алкиленовому или алкильному фрагменту, в котором атом водорода алкилена или алкила заменен на гидроксильную группу. Гидроксиалкилен или гидроксиалкил включает группы, в которых более одного атома водорода заменены гидроксильной группой.
«Ароматическая кольцевая система» представляет собой известный в данной области техники термин и относится к моноциклической, бициклической или полициклической углеводородной кольцевой системе, в которой по меньшей мере одно кольцо является ароматическим.
«Арил» относится к одновалентному радикалу ароматической кольцевой системы. Иллюстративные арильные группы включают полностью ароматические кольцевые системы, такие как фенил, нафтил и антраценил, а также кольцевые системы, в которых ароматическое углеродное кольцо конденсировано с одним или более неароматическими углеродными кольцами, такие как инданил, фталимидил, нафтимидил или тетрагидронафтил и т.п.
«Арилалкил» или «аралкил» относится к алкильному фрагменту, в котором атом водорода алкила заменен на арильную группу. Аралкил включает группы, в которых более одного атома водорода заменены арильной группой. Примеры «арилалкила» или «аралкила» включают бензильную, 2-фенилэтильную, 3-фенилпропильную, 9-флуоренильную, бензгидрильную и тритильную группы.
«Арилокси» относится к -O-(арил), где гетероарильный фрагмент является таким, как описано выше.
«Галоген» относится к радикалу любого галогена, например, -F, -Cl, -Br или -I.
«Галогеналкил» и «галогеналкокси» относится к алкильным и алкокси-структурам, замещенным одной иле более группами галогена, или их комбинациями. Например, термины «фторалкил» и «фторалкокси» включают галогеналкильные и галогеналкокси-группы, соответственно, в которых галоген представляет собой фтор. «Галогеналкилен» относится к двухвалентному алкилу, например, -CH2-, -CH2CH2- и -CH2CH2CH2-, в котором один или более атомов водорода заменены галогеном, и включает алкильные фрагменты, в которых все атомы водорода заменены галогеном.
«Гетероалкил» относится к необязательно замещенному алкилу, который имеет один или более атомов в скелетной цепи, выбранных из атома, отличного от углерода, например, кислорода, азота, серы, фосфора или их комбинаций. Может быть указан числовой диапазон, например, C1-C6 гетероалкил, что относится к количеству атомов углерода в цепи, которое в данном примере включает от 1 до 6 атомов углерода. Например, радикал –CH2OCH2CH3 называют «C3» гетероалкилом. Связь с остальной частью молекулы может осуществляться через гетероатом или через атом углерода гетероалкильной цепи. «Гетероалкилен» относится к двухвалентному необязательно замещенному алкилу, который имеет один или более атомов в скелетной цепи, выбранных из атома, отличного от углерода, например, кислорода, азота, серы, фосфора или их комбинаций.
«Карбоциклическая кольцевая система» относится к моноциклической, бициклической или полициклической углеводородной кольцевой системе, в которой каждое кольцо является либо полностью насыщенным, либо содержит одну или более единиц ненасыщенности, но ни одно из колец не является ароматическим.
«Карбоциклил» относится к одновалентному радикалу карбоциклической кольцевой системы. Иллюстративные карбоциклильные группы включают циклоалкильные группы (например, циклопентил, циклобутил, циклопентил, циклогексил и т.п.) и циклоалкенильные группы (например, циклопентенил, циклогексенил, циклопентадиенил и т.п.).
«Циклоалкил» относится к циклическим, бициклическим, трициклическим или полициклическим неароматическим углеводородным группам, имеющим от 3 до 12 атомов углерода. Любой замещаемый кольцевой атом может быть замещен (например, одним или более заместителями). Циклоалкильные группы могут содержать конденсированные или спирокольца. Конденсированные кольца представляют собой кольца, которые имеют один общий атом углерода. Примеры циклоалкильных фрагментов включают, но не ограничиваются ими, циклопропил, циклогексил, метилциклогексил, адамантил и норборнил.
«Циклоалкилалкил» относится к радикалу –(циклоалкил)-алкил, где циклоалкил и алкил являются такими, как описано в настоящем документе. «Циклоалкилалкил» связан с основной молекулярной структурой через циклоалкильную группу.
«Гетероароматическая кольцевая система» представляет собой известный в данной области техники термин и относится к моноциклической, бициклической или полициклической кольцевой системе, в которой по меньшей мере одно кольцо одновременно является ароматическим и содержит по меньшей мере один гетероатом (например, N, O или S); и где другие ни одно другое кольцо не представляет собой гетероциклил (как описано ниже). В некоторых случаях кольцо, которое является ароматическим и содержит гетероатом, содержит 1, 2, 3 или 4 кольцевых гетероатома в таком кольце.
«Гетероарил» относится к одновалентному радикалу гетероароматической кольцевой системы. Иллюстративные гетероарильные группы включают кольцевые системы, в которых (i) каждое кольцо содержит гетероатом и является ароматическим, например, имидазолил, оксазолил, тиазолил, триазолил, пирролил, фуранил, тиофенил, пиразолил, пиридинил, пиразинил, пиридазинил, пиримидинил, индолизинил, пуринил, нафтиридинил и птеридинил; (ii) каждое кольцо является ароматическим или карбоциклическим, по меньшей мере одно ароматическое кольцо содержит гетероатом, и по меньшей мере одно другое кольцо представляет собой углеводородное кольцо, например, индолил, изоиндолил, бензотиенил, бензофуранил, дибензофуранил, индазолил, бензимидазолил, бензтиазолил, хинолил, изохинолил, циннолинил, фталазинил, хиназолинил, хиноксалинил, карбазолил, акридинил, феназинил, фенотиазинил, феноксазинил, пиридо[2,3-b]-1,4-оксазин-3-(4H)-он, 5,6,7,8-тетрагидрофинолинил и 5,6,7,-тетрагидроизохинолинил; и (iii) каждое кольцо является ароматическим или карбоциклическим, и по меньшей мере одно ароматическое кольцо имеет общий гетероатом головы мостика с другим ароматическим кольцом, например, 4H-хинолизинил.
«Гетероциклическая кольцевая система» относится к моноциклическим, бициклическим и полициклическим кольцевым системам, в которых по меньшей мере одно кольцо является насыщенным или частично ненасыщенным (но не ароматическим) и содержит по меньшей мере один гетероатом. Гетероциклическая кольцевая система может быть присоединена к своей боковой группе у любого гетероатома или атома углерода с образованием устойчивой структуры, и любой из кольцевых атомов может быть необязательно замещен.
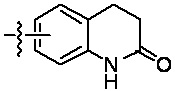
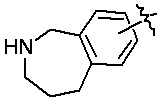
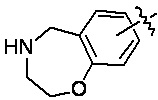
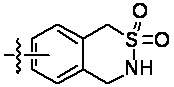
«Гетероциклил» относится к одновалентному радикалу гетероциклической кольцевой системы. Иллюстративные гетероциклилы включают кольцевые системы, в которых (i) каждое кольцо является неароматическим, и по меньшей мере одно кольцо содержит гетероатом, например, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, пирролидинил, пирролидонил, пиперидинил, пирролинил, декагидрохинолинил, оксазолидинил, пиперазинил, диоксанил, диоксоланил, диазепинил, оксазепинил, тиазепинил, морфолинил и хинуклидинил; (ii) по меньшей мере одно кольцо является неароматическим и содержит гетероатом, и по меньшей мере одно другое кольцо представляет собой ароматическое углеродное кольцо, например, 1,2,3,4-тетрагидрохинолинил, 1,2,3,4-тетрагидроизохинолинил; и (iii) по меньшей мере одно кольцо является неароматическим и содержит гетероатом, и по меньшей мере одно другое кольцо является ароматическим и содержит гетероатом, например, 3,4-дигидро-1H-пирано[4,3-c]пиридин и 1,2,3,4-тетрагидро-2,6-нафтиридин. В некоторых вариантах реализации изобретения гетероциклил может включать:
 ,
,  ,
, 
 ,
,  ,
,  и
и 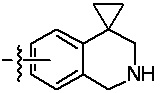 .
.
«Гетероциклилалкил» относится к алкильной группе, замещенной гетероциклильной группой.
«Циано» относится к радикалу –CN.
«Нитро» относится к –NO2.
«Гидрокси» или «гидроксил» относится к –OH.
«Гидроксиалкилен» относится к двухвалентному алкилу, например, -CH2-, -CH2CH2- и -CH2CH2CH2-, в котором один или более атомов водорода заменены гидрокси-группой, и включает алкильные фрагменты, в которых все атомы водорода заменены гидрокси-группой.
«Замещенный» с предшествующим термином «необязательно» или без него означает, что один или более атомов водорода в указанном фрагменте заменены на подходящий заместитель. Если не указано иное, то «необязательно замещенная» группа может иметь подходящий заместитель в каждом замещаемом положении этой группы, и если в любой данной структуре может быть замещено более одного положения более чем одним заместителем, выбранным из определенной группы, то этот заместитель может быть одинаковым или различным в каждом положении. Комбинации заместителей, предполагаемых настоящим изобретением, предпочтительно являются такими, которые приводят к образованию устойчивых или химически возможных соединений. Термин «устойчивый» в контексте настоящего документа относится к соединениям, которые существенно не изменяются при воздействии условий их получения, обнаружения и, в некоторых вариантах реализации, их выделения, очистки и применения для одной или более целей, описанных в настоящем документе.
В контексте настоящего документа определение каждого выражения, например, алкила, m, n и т.д., встречающегося в любой структуре более одного раза, следует понимать независимо от его определения в других местах той же структуры.
Некоторые соединения согласно настоящему изобретению могут существовать в определенных геометрических или стереоизомерных формах. Настоящее изобретение охватывает все такие соединения, включая цис- и транс-изомеры, R- и S-энантиомеры, диастереомеры, (D)-изомеры, (L)-изомеры, их рацемические смеси и других их смеси, входящие в границы объема настоящего изобретения. Дополнительные асимметричные атомы углерода могут присутствовать в заместителях, таких как алкильная группа. Все такие изомеры, а также их смеси, подразумеваются входящими в настоящее изобретение.
Если, например, желателен определенный энантиомер соединения согласно настоящему изобретению, он может быть получен посредством асимметричного синтеза или путем преобразования со вспомогательным хиральным агентом, при этом полученную диастеромерную смесь разделяют, и вспомогательную группу отщепляют с получением требуемых чистых энантиомеров. В альтернативном варианте, если молекула содержит основную функциональную группу, такую как аминогруппа, или кислотную функциональную группу, такую как карбоксил, то диастереомерные соли могут быть образованы с соответствующей оптически активной кислотой или основанием, с последующим разделением образовавшихся диастереомеров фракционной кристаллизацией или хроматографическими способами, известными в данной области, и последующим выделением чистых энантиомеров.
Если не указано иное, то соединение, имеющее один или более хиральных центров, названное или изображенное некоторой структурой без указания его стереохимии, следует понимать как представляющее все возможные стереоизомеры соединения, а также его энантиомерные смеси.
«Энантиомерный избыток» или «% энантиомерный избыток» композиции может быть рассчитан по уравнению, представленному ниже. В представленном ниже примере композиция содержит 90 % одного энантиомера, например, S энантиомера, и 10 % другого энантиомера, т.е. R энантиомера.
э.и. = (90-10)/100 = 80 %.
Следовательно, композиция, содержащая 90 % одного энантиомера и 10 % другого энантиомера, имеет энантиомерный избыток 80 %.
Соединения или композиции, описанные в настоящем документе, могут содержать энантиомерный избыток по меньшей мере 50 %, 75 %, 90 %, 95 % ил 99 % одной формы соединения, например, S-энантиомера. Другими словами, такие соединения или композиции содержат энантиомерный избыток S энантиомера относительно R энантиомера.
Соединения, описанные в настоящем документе, также могут содержать неприродные пропорции атомных изотопов у одного или более атомов, образующих указанные соединения. Например, соединения могут быть помечены радиоактивными изотопами, такими как, например, дейтерий (2H), тритий (3H), углерод-13 (13C) или углерод -14 (14C). Все изотопные варианты соединений, описанных в настоящем документе, радиоактивные или нерадиоактивные, подразумевают входящими в границы объема настоящего изобретения. Кроме того, в границы настоящего изобретения входят все таутомерные формы соединений, описанных в настоящем документе.
Соединение может быть пригодным в форме свободного основания или в форме соли. Иллюстративные соли включают гидробромидные, гидрохлоридные, сульфатные, бисульфатные, фосфатные, нитратные, ацетатные, валератные, олеатные, пальмитатные, стеаратные, лауратные, бензоатные, лактатные, фосфатные, тозилатные, цитратные, малеатные, фумаратные, сукцинатные, тартратные, нафтилатные, мезилатные, глюкогептонатные, лактобионатные и лаурилсульфонатные соли и т.п. (см., например, Berge et al. (1977) “Pharmaceutical Salts”, J. Pharm. Sci. 66:1-19.)
Некоторые соединения, описанные в настоящем документе, могут существовать в несольватированных формах, а также в сольватированных формах, включая гидратированные формы. Термин «гидрат» или «гидратированное» в контексте настоящего документа относится к соединению, образованному в результате объединения воды с исходным соединением.
В общем случае, сольватированные формы эквивалентны несольватированным формам и входят в границы объема настоящего изобретения. Некоторые соединения, описанные в настоящем документе, могут существовать в различных кристаллических или аморфных формах. В общем случае, все физические формы эквивалентны для применений, предполагаемых настоящим изобретением, и их подразумевают входящими в границы объема настоящего изобретения.
В контексте настоящего документа термин «пациент» относится к организмам, подлежащим лечению способами согласно настоящему изобретению. Такие организмы предпочтительно включают, но не ограничиваются ими, млекопитающих (например, мышей, обезьян, лошадей, быков, свиней, псовых, кошачьих и т.п.), и наиболее предпочтительно включают людей.
В контексте настоящего документа термин «эффективное количество» относится к количеству соединения (например, соединения согласно настоящему изобретению), эффективному для достижения благотворных или требуемых результатов. Эффективное количество может быть введено за один или более приемов, применений или доз, и не подразумевает ограничения до конкретного состава или способа введения. В контексте настоящего документа термин «лечение» включает любой эффект, например, ослабление, уменьшение, модулирование, облегчение или исключение, который приводит к облегчению патологического состояния, заболевания, расстройства и т.п., или к облегчению его симптома.
Соединения
В одном из вариантов реализации настоящего изобретения представлено соединение, имеющее структурную Формулу I, или его фармацевтически приемлемая соль, где:
I
W выбран из водорода и , где кольцо A выбрано из моноциклического или бициклического арила, моноциклического или бициклического гетероарила, циклоалкила и гетероциклила;
каждый X и Y независимо выбран из CR1 и N;
Z представляет собой C1-C6 алкил, циклоалкил, моноциклический или бициклический арил, моноциклический или бициклический аралкил, моноциклический или бициклический гетероарил, моноциклический или бициклический гетероциклил, моноциклический или бициклический гетероциклилалкил; где каждый C1-C6 алкил, циклоалкил, моноциклический или бициклический арил, моноциклический или бициклический аралкил, моноциклический или бициклический гетероарил, моноциклический или бициклический гетероциклил, моноциклический или бициклический гетероциклилалкил независимо замещен 0-5 группами RC;
L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкинилен)-, -(C2-C6 алкенилен)-, -(C1-C6 галогеналкилен)-, -(C1-C6 гетероалкилен)-, -(C1-C6 гидроксиалкилен)-, -C(O)-, -O-, -S-, -S(O), -SO2-, -N(R2)-, -O-(C1-C6 алкилен)-, -(C1-C6 алкилен)-O-, -N(R2)-CO-, -CO-N(R2)-, -(C1-C6 алкилен)-N(R2)-, -N(R2)-(C1-C6 алкилен)-, -N(R2)-CO-(C1-C6 алкилен)-, -CO-N(R2)-(C1-C6 алкилен)-, -N(R2)-SO2-, - SO2-N(R2)-, -N(R2)-SO2-(C1-C6 алкилен)- и -SO2-N(R2)-(C1-C6 алкилен)-;
каждый RA и RB независимо выбран из C1-C6 алкила, C1-C6 циклоалкила, C1-C6 гетероциклила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 гетероалкила, моноциклического или бициклического аралкила, -N(R2)(R2), циано и -OR2;
каждый RC независимо выбран из C1-C6 алкила, C1-C6 алкинила, галогена, C1-C6 гетероалкила, C1-C6 галогеналкила, C1-C6 галогеналкокси, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического арила, моноциклического или бициклического арилокси, моноциклического или бициклического аралкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, нитро, циано, -C(O)R2, -OC(O)R2, -C(O)OR2, -SR2, -S(O)2R2, -S(O)2-N(R2)(R2), -(C1-C6 алкилен)-S(O)2-N(R2)(R2), -N(R2)(R2), -C(O)-N(R2)(R2), -N(R2)(R2)-C(O)R2, -(C1-C6 алкилен)-N(R2)-C(O)R2, -NR2S(O)2R2, -P(O)(R2)(R2) и –OR2; где каждый гетероалкил, гелогеналкил, галогеналкокси, алкил, алкинил, циклоалкил, арил, арилокси, аралкил, гетероциклил, гетероциклилалкил независимо замещен 0-5 группами Ra; или 2 RC вместе с атомом(-ами) углерода, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо, замещенное 0-5 группами Ra;
каждый RD и RF независимо выбран из водорода, C1-C6 алкила, C1-C6 циклоалкила, гидроксила, галогена, C1-C6 алкокси, C1-C6 галогеналкила, -N(R2)(R2) и циано;
каждый R1 независимо выбран из водорода, C1-C6 алкила, моноциклического аралкила, C1-C6 гидроксиалкила, галогена, C1-C6 галогеналкила, -N(R2)(R2) и -OR2;
каждый R2 независимо выбран из водорода, гидроксила, галогена, тиола, C1-C6 тиоалкила, -NR”R”, C1-C6 алкила, C1-C6 алкокси, C1-C6 галогеналкила, C1-C6 гидроксиалкила, циклоалкила, циклоалкилалкила, гетероциклила и гетероциклилалкила, где каждый C1-C6 алкил, циклоалкил и гетероциклил независимо замещен 0-5 группами Rb, или 2 R2 вместе с атомом углерода или азота, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо;
каждый Ra и Rb независимо выбран из водорода, галогена, циано, гидроксила, C1-C6 алкоксила, -C(O)R’, C(O)OR’, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гетероалкила, C1-C6 гидроксиалкила, -NR’R’ и циклоалкила, где циклоалкил замещен 0-5 группами R’;
каждый R’ представляет собой водород, гидроксил или C1-C6 алкил;
каждый R” представляет собой водород, C1-C6 алкил, -C(O)-C1-C6 алкил, -C(O)-NR’R’;или -C(S)-NR’R’; и
m, p и q, каждый независимо, представляют собой 0, 1, 2, 3 или 4.
В некоторых вариантах реализации изобретения W представляет собой H. В некоторых вариантах реализации W представляет собой . В некоторых вариантах реализации кольцо A представляет собой моноциклический или бициклический арил, замещенный 0, 1, 2 или 3 RA. В некоторых вариантах реализации кольцо A представляет собой фенил. В некоторых вариантах реализации кольцо A представляет собой фенил, замещенный галогеном. В некоторых вариантах реализации кольцо A представляет собой фенил, замещенный фтором или хлором. В некоторых вариантах реализации кольцо A представляет собой 4-фторфенил. В некоторых вариантах реализации кольцо A представляет собой 2,4-дифторфенил. В некоторых вариантах реализации кольцо A представляет собой 2,4,6-трифторфенил. В некоторых вариантах реализации кольцо A представляет собой 4-хлорфенил.
В некоторых вариантах реализации изобретения каждый RA независимо выбран из C1-C6 алкила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, -N(R2)(R2), циано и -OR2. В некоторых вариантах реализации RA независимо выбран из C1-C6 алкила и галогена. В некоторых вариантах реализации RA независимо выбран из фтора, хлора и метила. В некоторых вариантах реализации RA независимо выбран из фтора и хлора. В некоторых вариантах реализации RA представляет собой метил. В некоторых вариантах реализации RA представляет собой фтор, и q равен 1, 2 или 3. В некоторых вариантах реализации RA представляет собой хлор и фтор, и q равен 2. В некоторых вариантах реализации RA представляет собой метил и фтор, и q равен 2.
В некоторых вариантах реализации изобретения каждый RB независимо выбран из C1-C6 алкила, C1-C6 гидроксиалкила, C1-C6 гетероалкила, -N(R2)(R2), циано и -OR2. В некоторых вариантах реализации RB представляет собой C1-C6 алкил или C1-C6 гидроксиалкил. В некоторых вариантах реализации RB представляет собой метил, этил или гидроксиметил. В некоторых вариантах реализации p равен 0 или 1. в некоторых вариантах реализации p равен 0. В некоторых вариантах реализации p равен 1.
В некоторых вариантах реализации изобретения по меньшей мере один из X и Y представляет собой N. В некоторых вариантах реализации X и Y представляют собой N. В некоторых вариантах реализации X и Y представляют собой CR1. В некоторых вариантах реализации X и Y представляют собой CH.
В некоторых вариантах реализации изобретения Z представляет собой моноциклический или бициклический арил. В некоторых вариантах реализации Z представляет собой моноциклический или бициклический гетероарил. В некоторых вариантах реализации Z представляет собой моноциклический или бициклический гетероциклил. В некоторых вариантах реализации Z представляет собой моноциклический гетероарил. В некоторых вариантах реализации Z выбран из пиразолила, изоксазолила, тиофенила, тиазолила и пиридила. В некоторых вариантах реализации Z замещен 0, 1 или 2 группами RC. В некоторых вариантах реализации Z замещен 0 или 1 группой RC.
В некоторых вариантах реализации изобретения RC независимо выбран из циано, C1-C6 алкила, C1-C6 алкинила, галогена, C1-C6 гетероалкила, C1-C6 галогеналкила, C1-C6 галогеналкокси, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, -C(O)R2, -OC(O)R2, -C(O)OR2, -N(R2)(R2), -C(O)-N(R2)(R2) и –OR2. В некоторых вариантах реализации RC независимо выбран из циано, C1-C6 алкила, галогена, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, -C(O)R2, -C(O)OR2, -N(R2)(R2), -C(O)-N(R2)(R2) и –OR2. В некоторых вариантах реализации каждый RC независимо выбран из C1-C6 алкила, галогена, моноциклического и бициклического гетероциклила.
В некоторых вариантах реализации изобретения RD представляет собой водород, C1-C6 алкил, C1-C6 алкокси, C1-C6 галогеналкил, -N(R2)(R2) или циано. В некоторых вариантах реализации RD представляет собой водород или -N(R2)(R2). В некоторых вариантах реализации RD представляет собой водород или -NH2.
В некоторых вариантах реализации изобретения RF представляет собой водород или галоген, например, хлор или фтор. В некоторых вариантах реализации RF представляет собой водород. В некоторых вариантах реализации RF представляет собой хлор или фтор.
В некоторых вариантах реализации изобретения L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкенилен)-, -(C1-C6 галогеналкилен)-, -(C1-C6 гидроксиалкилен)-, -S-, -S(O), -SO2- и -N(R2)-. В некоторых вариантах реализации L выбран из связи, -(C(R2)(R2))m-, -S- и -SO2-. В некоторых вариантах реализации L представляет собой -(C(R2)(R2))m-. В некоторых вариантах реализации L представляет собой связь или CH2. В некоторых вариантах реализации L представляет собой -(C(R2)(R2))m-, где каждый R2 независимо выбран из водорода, гидроксила, -NR”R”, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила и циклоалкила; и m равен 1.
В некоторых вариантах реализации изобретения каждый R2 независимо выбран из водорода, гидроксила, галогена, -NR”R”, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила и циклоалкила, где каждый C1-C6 алкил и циклоалкил независимо замещен 0-5 группами Rb, или 2 R2 вместе с атомом углерода или азота, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо. В некоторых вариантах реализации каждый R2 независимо выбран из галогена, водорода, гидроксила, -NR”R” и C1-C6 алкила, где C1-C6 алкил независимо замещен 0-5 группами Rb. В некоторых вариантах реализации Rb независимо представляет собой водород, галоген или гидроксил. В некоторых вариантах реализации L представляет собой -NR”R”. В некоторых вариантах реализации R” представляет собой водород или C1-C6 алкил. В некоторых вариантах реализации R” представляет собой водород. В некоторых вариантах реализации L представляет собой -S-. В некоторых вариантах реализации L представляет собой -CH2-.
В некоторых вариантах реализации изобретения m равен 0, 1 или 2. В некоторых вариантах реализации m равен 1. В некоторых вариантах реализации m равен 2.
В некоторых вариантах реализации изобретения p равен 0 или 1.
В некоторых вариантах реализации изобретения q равен 0, 1, 2 или 3. В некоторых вариантах реализации q равен 0. В некоторых вариантах реализации q равен 1. В некоторых вариантах реализации q равен 2. В некоторых вариантах реализации q равен 3.
В другом варианте реализации настоящего изобретения представлено соединение Формулы II или его фармацевтически приемлемая соль, где:
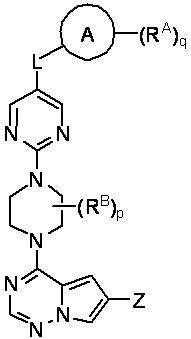
II
Кольцо A выбрано из моноциклического или бициклического арила, моноциклического или бициклического гетероарила, циклоалкила и гетероциклила;
Z выбран из C1-C6 алкила, циклоалкила, моноциклического или бициклического арила, моноциклического или бициклического аралкила, моноциклического или бициклического гетероарила, моноциклического или бициклического гетероциклила и моноциклического или бициклического гетероциклилалкила; где каждый C1-C6 алкил, циклоалкил, моноциклический или бициклический арил, моноциклический или бициклический аралкил, моноциклический или бициклический гетероарил, моноциклический или бициклический гетероциклил, моноциклический или бициклический гетероциклилалкил независимо замещен 0-5 группами RC;
L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкинилен)-, -(C2-C6 алкенилен)-, -(C1-C6 галогеналкилен)-, -(C1-C6 гетероалкилен)-, -(C1-C6 гидроксиалкилен)-, -C(O)-, -O-, -S-, -S(O), -SO2-, -N(R2)-, -O-(C1-C6 алкилен)-, -(C1-C6 алкилен)-O-, -N(R2)-CO-, -CO-N(R2)-, -(C1-C6 алкилен)-N(R2)-, -N(R2)-(C1-C6 алкилен)-, -N(R2)-CO-(C1-C6 алкилен)-, -CO-N(R2)-(C1-C6 алкилен)-, -N(R2)-SO2-, - SO2-N(R2)-, -N(R2)-SO2-(C1-C6 алкилен)- и -SO2-N(R2)-(C1-C6 алкилен)-;
каждый RA и RB независимо выбран из C1-C6 алкила, C1-C6 циклоалкила, C1-C6 гетероциклила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 гетероалкила, моноциклического или бициклического аралкила, -N(R2)(R2), циано и -OR2;
каждый RC независимо выбран из C1-C6 алкила, C1-C6 алкинила, галогена, C1-C6 гетероалкила, C1-C6 галогеналкила, C1-C6 галогеналкокси, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического арила, моноциклического или бициклического арилокси, моноциклического или бициклического аралкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, нитро, циано, -C(O)R2, -OC(O)R2, -C(O)OR2, -SR2, -S(O)2R2, -S(O)2-N(R2)(R2), -(C1-C6 алкилен)-S(O)2-N(R2)(R2), -N(R2)(R2), -C(O)-N(R2)(R2), -N(R2)(R2)-C(O)R2, -(C1-C6 алкилен)-N(R2)-C(O)R2, -NR2S(O)2R2, -P(O)(R2)(R2) и –OR2; где каждый гетероалкил, гелогеналкил, галогеналкокси, алкил, алкинил, циклоалкил, арил, арилокси, аралкил, гетероциклил, гетероциклилалкил независимо замещен 0-5 группами Ra; или 2 RC вместе с атомом(-ами) углерода, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо, замещенное 0-5 группами Ra;
каждый R2 независимо выбран из водорода, гидроксила, галогена, тиола, C1-C6 тиоалкила, -NR”R”, C1-C6 алкила, C1-C6 алкокси, C1-C6 галогеналкила, C1-C6 гидроксиалкила, циклоалкила, циклоалкилалкила, гетероциклила и гетероциклилалкила, где каждый C1-C6 алкил, циклоалкил и гетероциклил независимо замещен 0-5 группами Rb, или 2 R2 вместе с атомом углерода или азота, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо;
каждый Ra и Rb независимо выбран из водорода, галогена, циано, гидроксила, C1-C6 алкоксила, -C(O)R’, C(O)OR’, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гетероалкила, C1-C6 гидроксиалкила, -NR’R’ и циклоалкила, где циклоалкил замещен 0-5 группами R’;
каждый R’ представляет собой водород, гидроксил или C1-C6 алкил;
каждый R” представляет собой водород, C1-C6 алкил, -C(O)-C1-C6 алкил, -C(O)-NR’R’; -C(S)-NR’R’; и
m, p и q, каждый независимо, представляют собой 0, 1, 2, 3 или 4.
В некоторых вариантах реализации изобретения A представляет собой моноциклический или бициклический арил. В некоторых вариантах реализации кольцо A представляет собой моноциклический или бициклический арил, замещенный 0, 1, 2 или 3 RA. В некоторых вариантах реализации кольцо A представляет собой фенил. В некоторых вариантах реализации кольцо A представляет собой фенил, замещенный галогеном. В некоторых вариантах реализации кольцо A представляет собой фенил, замещенный фтором или хлором. В некоторых вариантах реализации кольцо A представляет собой 4-фторфенил. В некоторых вариантах реализации кольцо A представляет собой 2,4-дифторфенил. В некоторых вариантах реализации кольцо A представляет собой 2,4,6-трифторфенил. В некоторых вариантах реализации кольцо A представляет собой 4-хлорфенил.
В некоторых вариантах реализации изобретения каждый RA независимо выбран из C1-C6 алкила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, -N(R2)(R2), циано и -OR2. В некоторых вариантах реализации RA независимо выбран из C1-C6 алкила и галогена. В некоторых вариантах реализации RA независимо выбран из фтора, хлора и метила. В некоторых вариантах реализации RA независимо выбран из фтора и хлора. В некоторых вариантах реализации RA представляет собой метил. В некоторых вариантах реализации RA представляет собой фтор, и q равен 1, 2 или 3. В некоторых вариантах реализации RA представляет собой хлор и фтор, и q равен 2. В некоторых вариантах реализации RA представляет собой метил и фтор, и q равен 2.
В некоторых вариантах реализации изобретения каждый RB независимо выбран из C1-C6 алкила, C1-C6 гидроксиалкила, C1-C6 гетероалкила, -N(R2)(R2), циано и -OR2. В некоторых вариантах реализации RB представляет собой C1-C6 алкил или C1-C6 гидроксиалкил. В некоторых вариантах реализации RB представляет собой метил, этил или гидроксиметил. В некоторых вариантах реализации p равен 0 или 1. в некоторых вариантах реализации p равен 0. В некоторых вариантах реализации p равен 1.
В некоторых вариантах реализации изобретения Z представляет собой моноциклический или бициклический арил. В некоторых вариантах реализации Z представляет собой моноциклический или бициклический гетероарил. В некоторых вариантах реализации Z представляет собой моноциклический или бициклический гетероциклил. В некоторых вариантах реализации Z представляет собой моноциклический гетероарил. В некоторых вариантах реализации Z выбран из пиразолила, изоксазолила, тиофенила, тиазолила и пиридила. В некоторых вариантах реализации Z замещен 0, 1 или 2 группами RC. В некоторых вариантах реализации Z замещен 0 или 1 группой RC.
В некоторых вариантах реализации изобретения RC независимо выбран из циано, C1-C6 алкила, C1-C6 алкинила, галогена, C1-C6 гетероалкила, C1-C6 галогеналкила, C1-C6 галогеналкокси, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, -C(O)R2, -OC(O)R2, -C(O)OR2, -N(R2)(R2), -C(O)-N(R2)(R2) и –OR2. В некоторых вариантах реализации RC независимо выбран из циано, C1-C6 алкила, галогена, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, -C(O)R2, -C(O)OR2, -N(R2)(R2), -C(O)-N(R2)(R2) и –OR2. В некоторых вариантах реализации каждый RC независимо выбран из C1-C6 алкила, галогена, моноциклического или бициклического гетероциклила.
В некоторых вариантах реализации изобретения RD представляет собой водород, C1-C6 алкил, C1-C6 алкокси, C1-C6 галогеналкил, -N(R2)(R2) или циано. В некоторых вариантах реализации RD представляет собой водород или -N(R2)(R2). В некоторых вариантах реализации RD представляет собой водород или -NH2.
В некоторых вариантах реализации изобретения RF представляет собой водород или галоген, например, хлор или фтор. В некоторых вариантах реализации RF представляет собой водород. В некоторых вариантах реализации RF представляет собой хлор или фтор.
В некоторых вариантах реализации изобретения L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкенилен)-, -(C1-C6 галогеналкилен)-, -(C1-C6 гидроксиалкилен)-, -S-, -S(O), -SO2- и -N(R2)-. В некоторых вариантах реализации L выбран из связи, -(C(R2)(R2))m-, -S- и -SO2-. В некоторых вариантах реализации L представляет собой -(C(R2)(R2))m-. В некоторых вариантах реализации L представляет собой связь или CH2. В некоторых вариантах реализации L представляет собой -(C(R2)(R2))m-, где каждый R2 независимо выбран из водорода, гидроксила, -NR”R”, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила и циклоалкила; и m равен 1.
В некоторых вариантах реализации изобретения каждый R2 независимо выбран из водорода, гидроксила, галогена, -NR”R”, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила, циклоалкила, где каждый C1-C6 алкил и циклоалкил независимо замещен 0-5 группами Rb, или 2 R2 вместе с атомом углерода или азота, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо. В некоторых вариантах реализации каждый R2 независимо выбран из галогена, водорода, гидроксила, -NR”R”, C1-C6 алкила, где C1-C6 алкил независимо замещен 0-5 группами Rb. В некоторых вариантах реализации Rb независимо представляет собой водород, галоген или гидроксил. В некоторых вариантах реализации L представляет собой -NR”R”. В некоторых вариантах реализации R” представляет собой водород или C1-C6 алкил. В некоторых вариантах реализации R” представляет собой водород. В некоторых вариантах реализации L представляет собой -S-. В некоторых вариантах реализации L представляет собой -CH2-.
В некоторых вариантах реализации изобретения m равен 0, 1 или 2. В некоторых вариантах реализации m равен 1. В некоторых вариантах реализации m равен 2.
В некоторых вариантах реализации изобретения p равен 0 или 1.
В некоторых вариантах реализации изобретения q равен 0, 1, 2 или 3. В некоторых вариантах реализации q равен 0. В некоторых вариантах реализации q равен 1. В некоторых вариантах реализации q равен 2. В некоторых вариантах реализации q равен 3.
В другом варианте реализации настоящего изобретения представлено соединение Формулы III или его фармацевтически приемлемая соль, где:
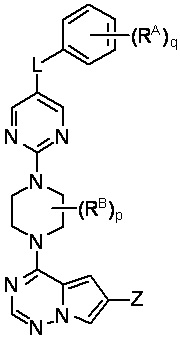
III
Z выбран из C1-C6 алкила, циклоалкила, моноциклического или бициклического арила, моноциклического или бициклического аралкила, моноциклического или бициклического гетероарила, моноциклического или бициклического гетероциклила и моноциклического или бициклического гетероциклилалкила; где каждый C1-C6 алкил, циклоалкил, моноциклический или бициклический арил, моноциклический или бициклический аралкил, моноциклический или бициклический гетероарил, моноциклический или бициклический гетероциклил, моноциклический или бициклический гетероциклилалкил независимо замещен 0-5 группами RC;
L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкинилен)-, -(C2-C6 алкенилен)-, -(C1-C6 галогеналкилен)-, -(C1-C6 гетероалкилен)-, -(C1-C6 гидроксиалкилен)-, -C(O)-, -O-, -S-, -S(O), -SO2-, -N(R2)-, -O-(C1-C6 алкилен)-, -(C1-C6 алкилен)-O-, -N(R2)-CO-, -CO-N(R2)-, -(C1-C6 алкилен)-N(R2)-, -N(R2)-(C1-C6 алкилен)-, -N(R2)-CO-(C1-C6 алкилен)-, -CO-N(R2)-(C1-C6 алкилен)-, -N(R2)-SO2-, - SO2-N(R2)-, -N(R2)-SO2-(C1-C6 алкилен)- и -SO2-N(R2)-(C1-C6 алкилен)-;
каждый RA и RB независимо выбран из C1-C6 алкила, C1-C6 циклоалкила, C1-C6 гетероциклила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, C1-C6 гетероалкила, моноциклического или бициклического аралкила, -N(R2)(R2), циано и -OR2;
каждый RC независимо выбран из C1-C6 алкила, C1-C6 алкинила, галогена, C1-C6 гетероалкила, C1-C6 галогеналкила, C1-C6 галогеналкокси, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического арила, моноциклического или бициклического арилокси, моноциклического или бициклического аралкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, нитро, циано, -C(O)R2, -OC(O)R2, -C(O)OR2, -SR2, -S(O)2R2, -S(O)2-N(R2)(R2), -(C1-C6 алкилен)-S(O)2-N(R2)(R2), -N(R2)(R2), -C(O)-N(R2)(R2), -N(R2)(R2)-C(O)R2, -(C1-C6 алкилен)-N(R2)-C(O)R2, -NR2S(O)2R2, -P(O)(R2)(R2) и –OR2; где каждый гетероалкил, гелогеналкил, галогеналкокси, алкил, алкинил, циклоалкил, арил, арилокси, аралкил, гетероциклил, гетероциклилалкил независимо замещен 0-5 группами Ra; или 2 RC вместе с атомом(-ами) углерода, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо, замещенное 0-5 группами Ra;
каждый R2 независимо выбран из водорода, гидроксила, галогена, тиола, C1-C6 тиоалкила, -NR”R”, C1-C6 алкила, C1-C6 алкокси, C1-C6 галогеналкила, C1-C6 гидроксиалкила, циклоалкила, циклоалкилалкила, гетероциклила и гетероциклилалкила, где каждый C1-C6 алкил, циклоалкил и гетероциклил независимо замещен 0-5 группами Rb, или 2 R2 вместе с атомом углерода или азота, к которому они присоединены, образуют циклоалкильное или гетероциклильное кольцо;
каждый Ra и Rb независимо выбран из водорода, галогена, циано, гидроксила, C1-C6 алкоксила, -C(O)R’, C(O)OR’, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гетероалкила, C1-C6 гидроксиалкила, -NR’R’ и циклоалкила, где циклоалкил замещен 0-5 группами R’;
каждый R’ представляет собой водород, гидроксил или C1-C6 алкил;
каждый R” представляет собой водород, C1-C6 алкил, -C(O)-C1-C6 алкил, -C(O)-NR’R’; -C(S)-NR’R’; и
m, p и q, каждый независимо, представляют собой 0, 1, 2, 3 или 4.
В некоторых вариантах реализации изобретения каждый RA независимо выбран из C1-C6 алкила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, -N(R2)(R2), циано и -OR2. В некоторых вариантах реализации изобретения каждый RA независимо выбран из C1-C6 алкила, галогена, C1-C6 галогеналкила, C1-C6 гидроксиалкила, -N(R2)(R2), циано, -OR2. В некоторых вариантах реализации RA независимо выбран из C1-C6 алкила и галогена. В некоторых вариантах реализации RA независимо выбран из фтора, хлора и метила. В некоторых вариантах реализации RA представляет собой галоген. В некоторых вариантах реализации RA независимо выбран из фтора и хлора. В некоторых вариантах реализации RA представляет собой метил. В некоторых вариантах реализации RA представляет собой фтор, и q равен 1, 2 или 3. В некоторых вариантах реализации RA представляет собой хлор и фтор, и q равен 2. В некоторых вариантах реализации RA представляет собой метил и фтор, и q равен 2.
В некоторых вариантах реализации изобретения Z представляет собой моноциклический или бициклический арил. В некоторых вариантах реализации Z представляет собой моноциклический или бициклический гетероарил. В некоторых вариантах реализации Z представляет собой моноциклический или бициклический гетероциклил. В некоторых вариантах реализации Z представляет собой моноциклический гетероарил. В некоторых вариантах реализации Z выбран из пиразолила, изоксазолила, тиофенила, тиазолила и пиридила. В некоторых вариантах реализации Z замещен 0, 1 или 2 группами RC. В некоторых вариантах реализации Z замещен 0 или 1 группой RC.
В некоторых вариантах реализации изобретения каждый RB независимо выбран из C1-C6 алкила, C1-C6 гидроксиалкила, C1-C6 гетероалкила, -N(R2)(R2), циано и -OR2. В некоторых вариантах реализации RB представляет собой C1-C6 алкил или C1-C6 гидроксиалкил. В некоторых вариантах реализации RB представляет собой метил, этил или гидроксиметил. В некоторых вариантах реализации p равен 0 или 1. в некоторых вариантах реализации p равен 0. В некоторых вариантах реализации p равен 1.
В некоторых вариантах реализации изобретения RC независимо выбран из циано, C1-C6 алкила, C1-C6 алкинила, галогена, C1-C6 гетероалкила, C1-C6 галогеналкила, C1-C6 галогеналкокси, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, -C(O)R2, -OC(O)R2, -C(O)OR2, -N(R2)(R2), -C(O)-N(R2)(R2) и –OR2. В некоторых вариантах реализации RC независимо выбран из циано, C1-C6 алкила, галогена, C1-C6 гидроксиалкила, циклоалкила, моноциклического или бициклического гетероциклила, моноциклического или бициклического гетероциклилалкила, -C(O)R2, -C(O)OR2, -N(R2)(R2), -C(O)-N(R2)(R2) и –OR2. В некоторых вариантах реализации каждый RC независимо выбран из C1-C6 алкила, галогена, моноциклического или бициклического гетероциклила.
В некоторых вариантах реализации изобретения L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкенилен)-, -(C1-C6 галогеналкилен)-, -(C1-C6 гидроксиалкилен)-, -S-, -S(O), -SO2- и -N(R2)-. В некоторых вариантах реализации L выбран из связи, -(C(R2)(R2))m-, -S- и -SO2-. В некоторых вариантах реализации L представляет собой -(C(R2)(R2))m-. В некоторых вариантах реализации L представляет собой связь или CH2. В некоторых вариантах реализации L представляет собой -(C(R2)(R2))m-, где каждый R2 независимо выбран из водорода, гидроксила, -NR”R”, C1-C6 алкила, C1-C6 галогеналкила, C1-C6 гидроксиалкила и циклоалкила; и m равен 1.
В некоторых вариантах реализации изобретения q равен 0, 1, 2 или 3. В некоторых вариантах реализации q равен 1, 2 или 3.
В настоящем изобретении представлены также фармацевтические композиции, содержащие фармацевтически приемлемый носитель и любое соединение Формул I-III.
В представленной ниже таблице изображены структуры соединений, описанных в настоящем документе.
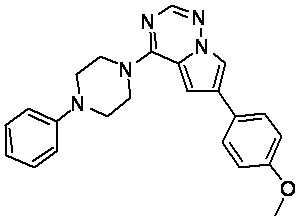
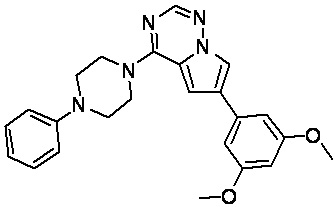
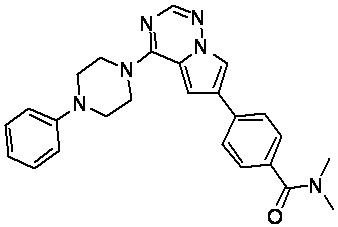
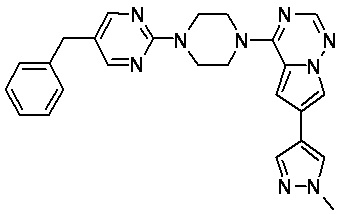
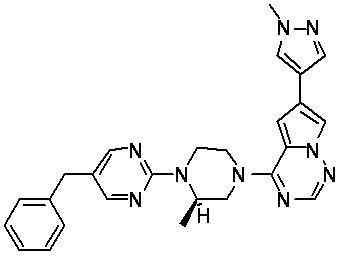
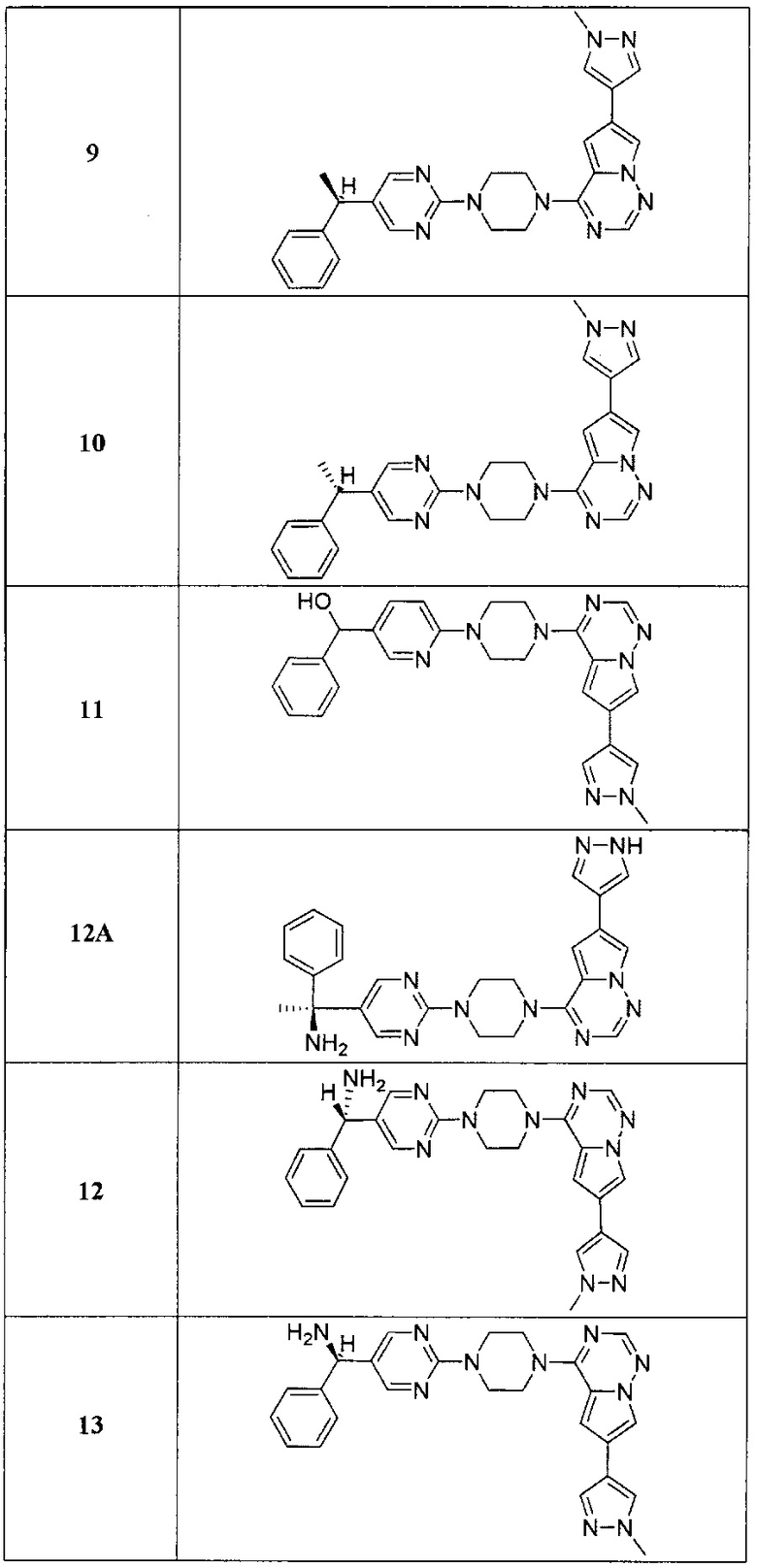
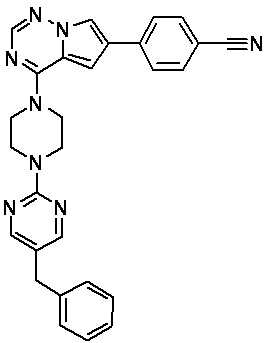
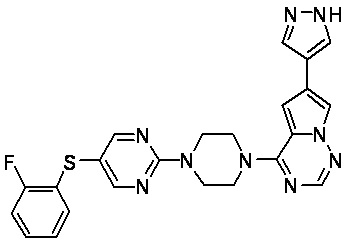
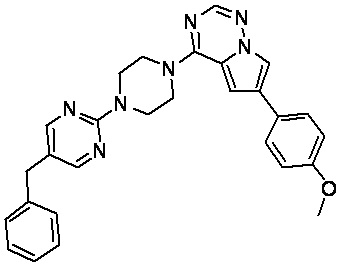
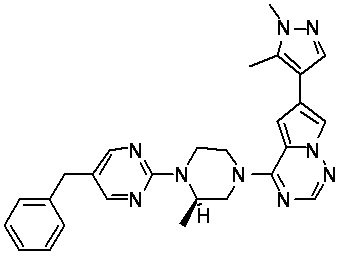
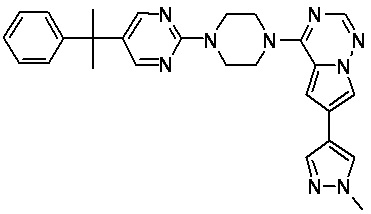
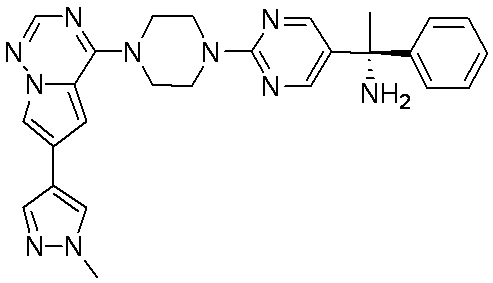
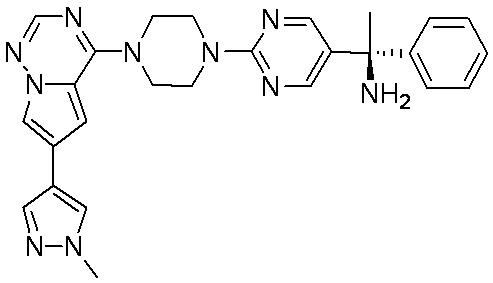
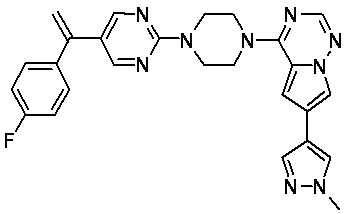
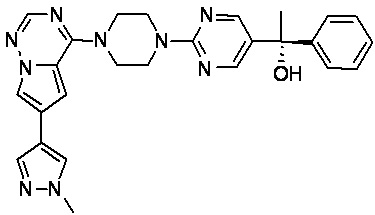
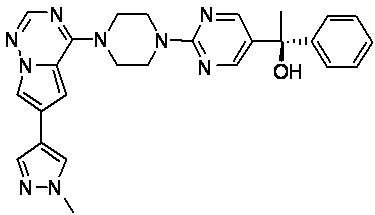
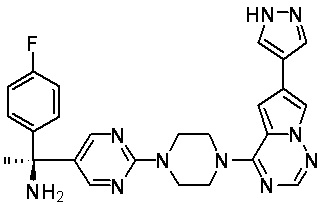
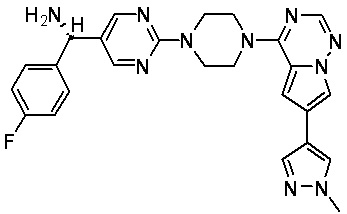
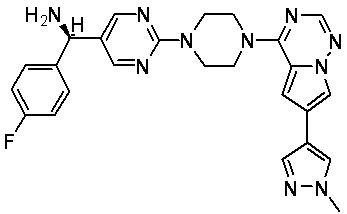
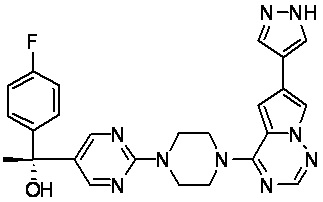
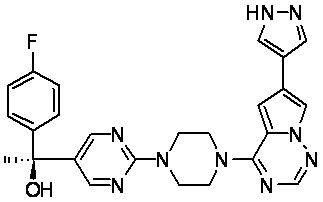
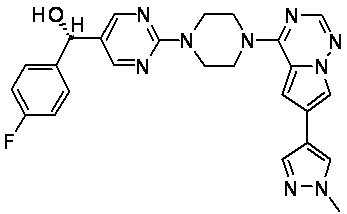
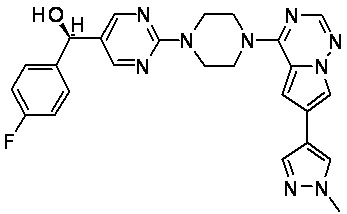
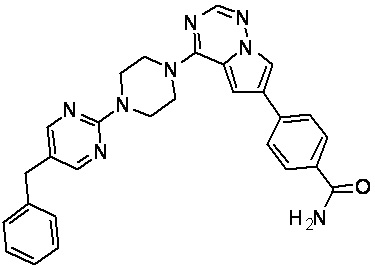
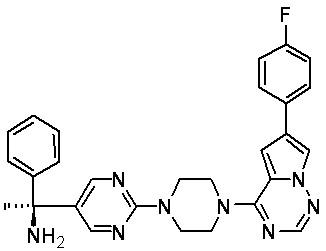
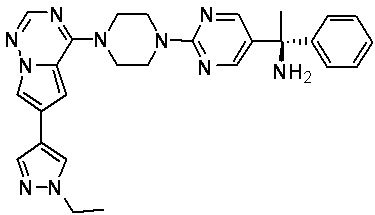
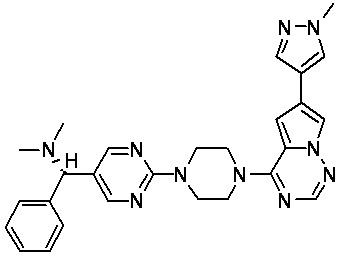
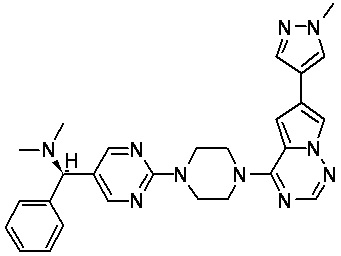
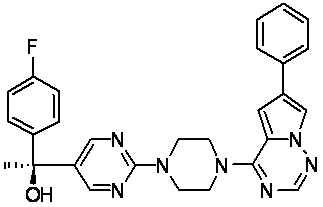
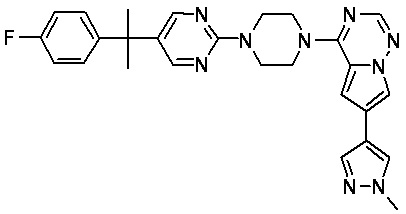
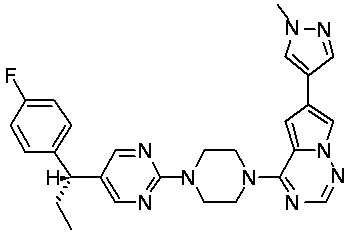
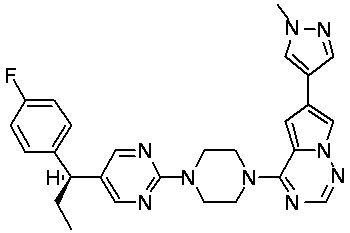
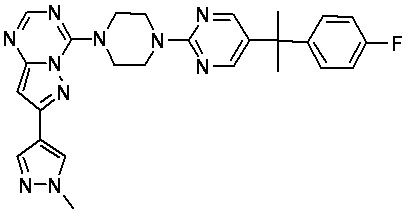
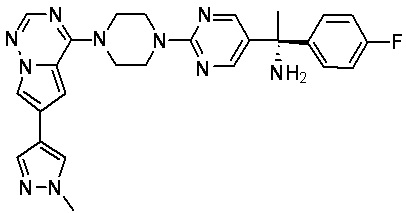
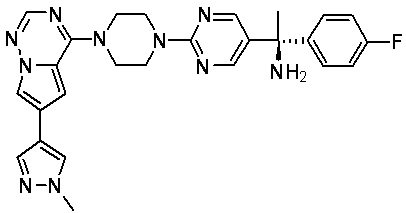
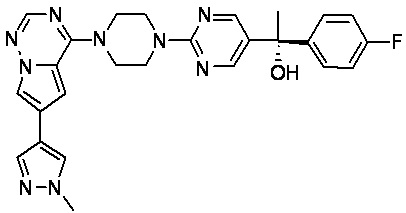
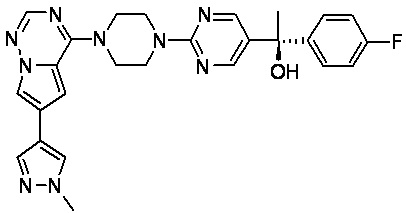
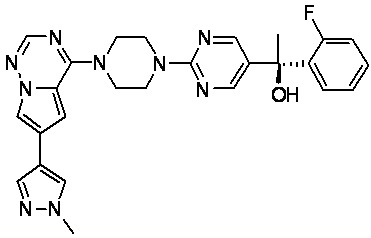
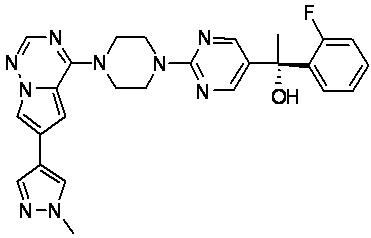
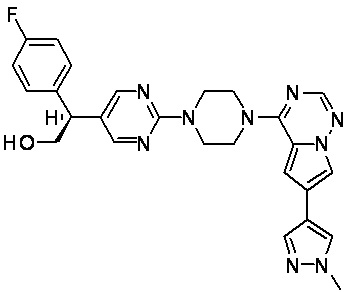
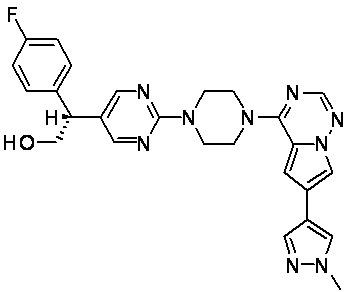
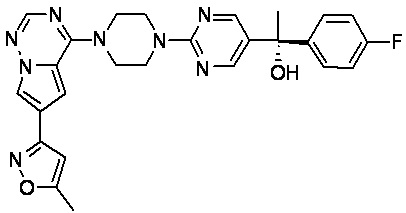
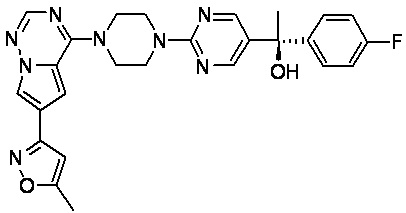
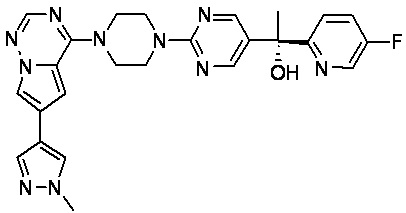
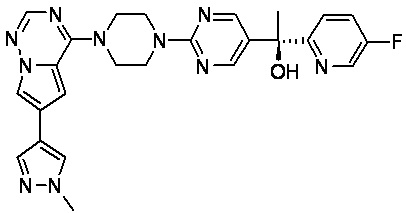
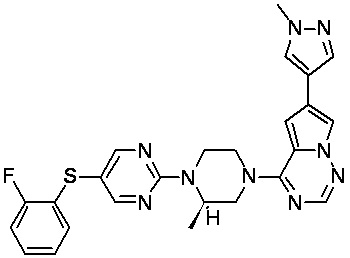
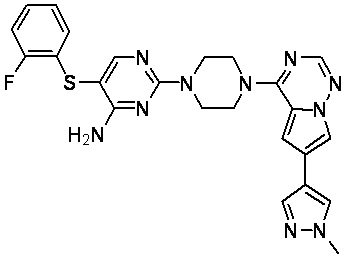
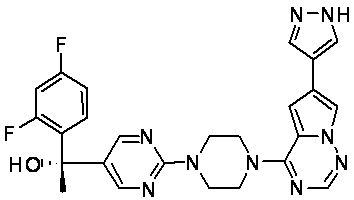
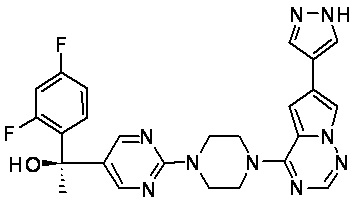
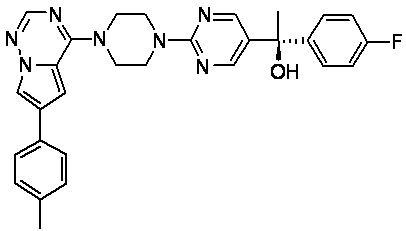
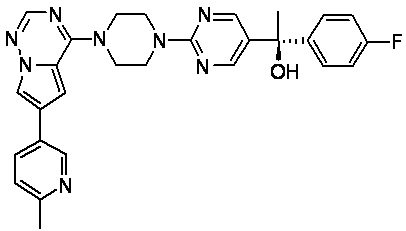
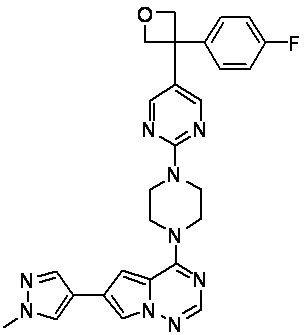
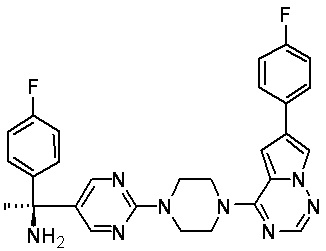
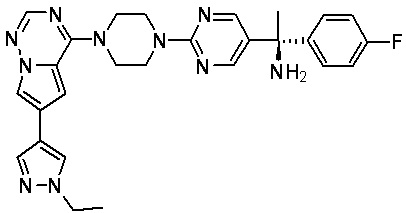
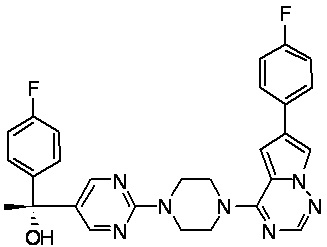
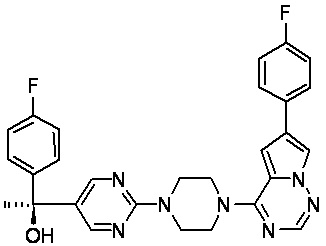
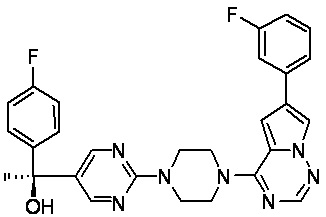
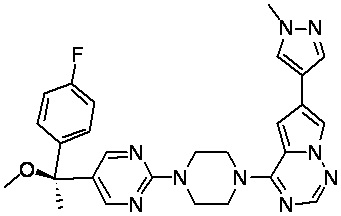
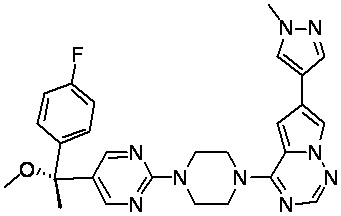
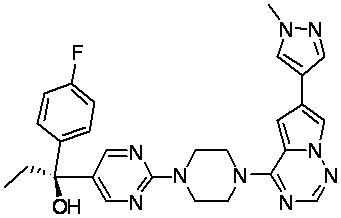
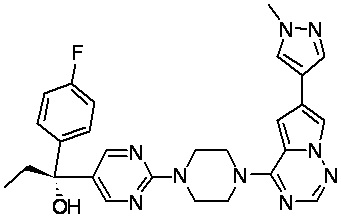
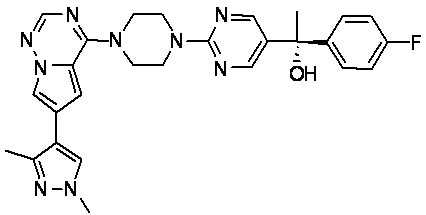
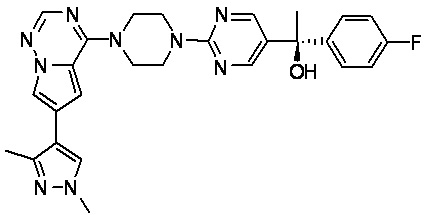
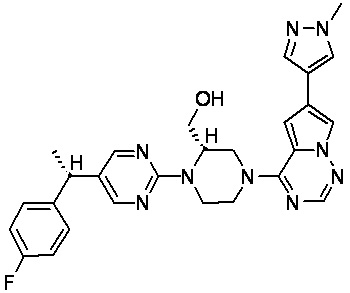
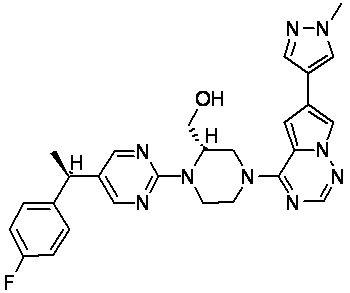
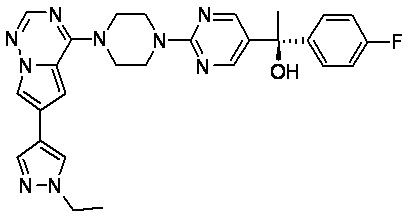
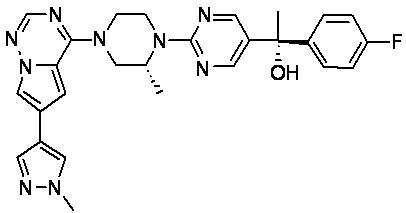
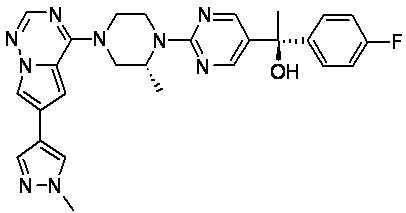
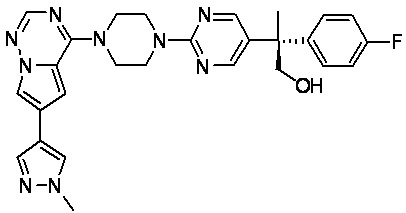
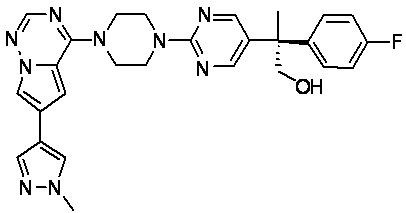
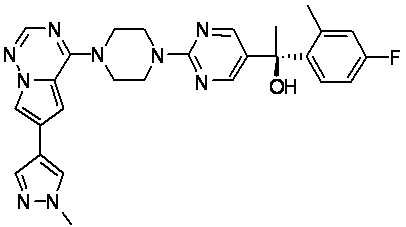
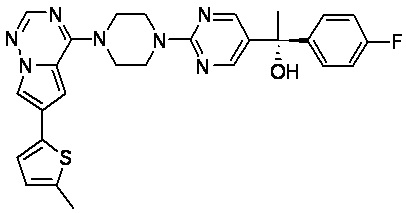
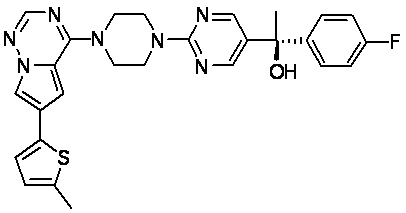
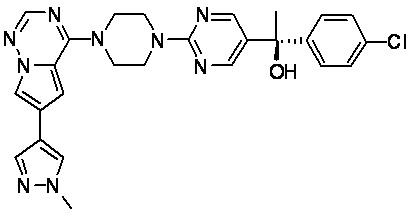
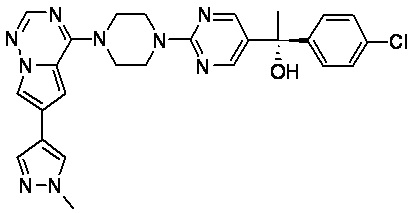
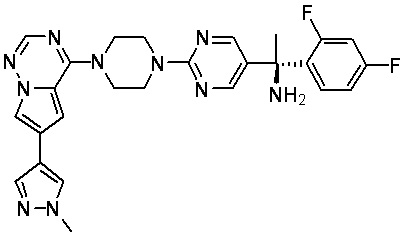
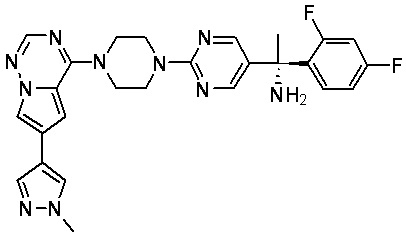
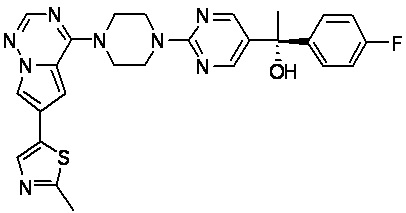
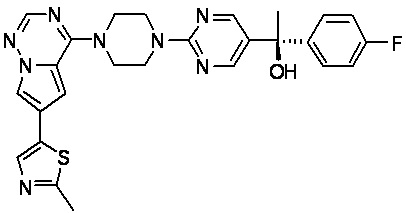
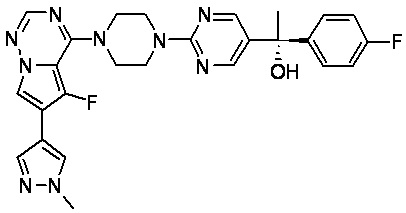
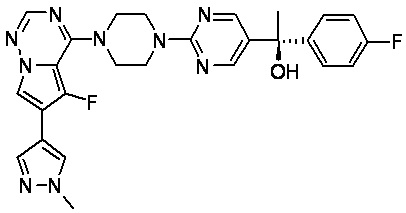
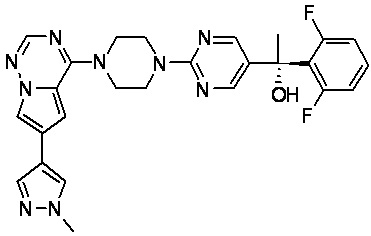
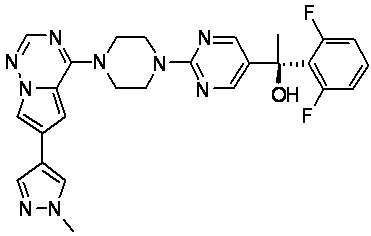
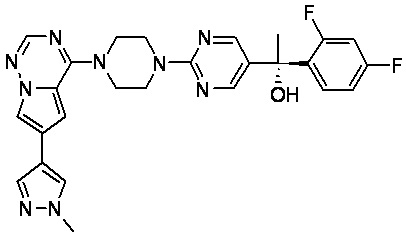
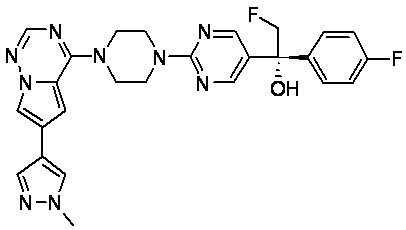
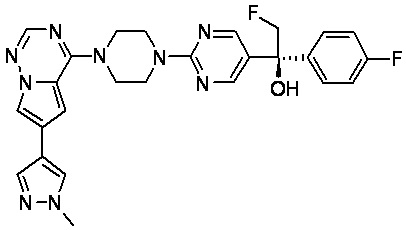
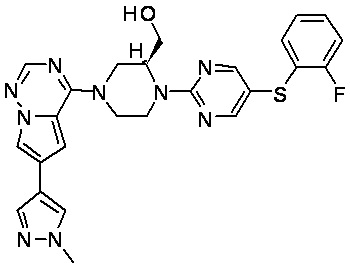
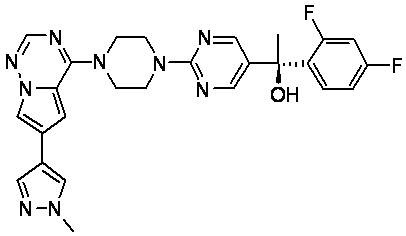
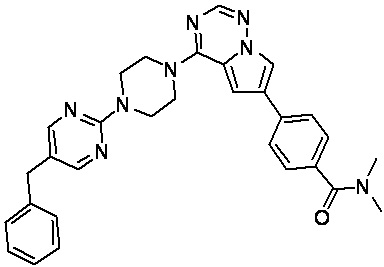
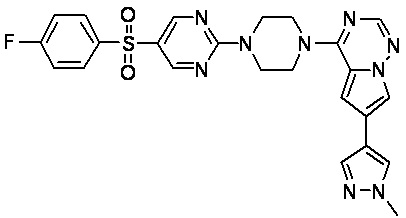
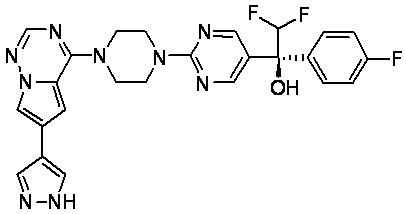
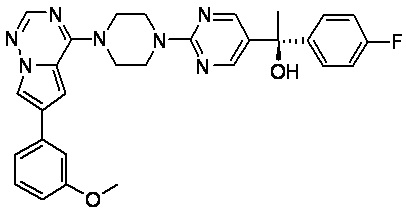
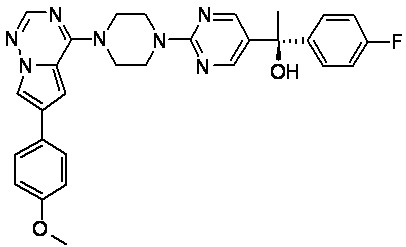
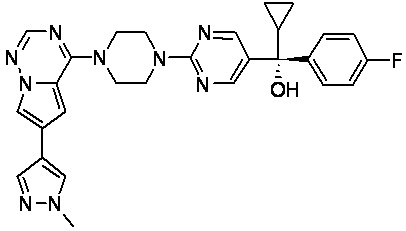
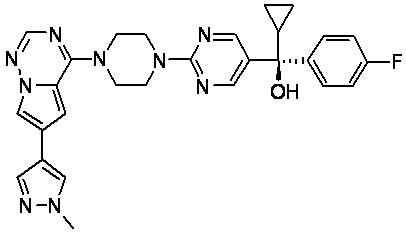
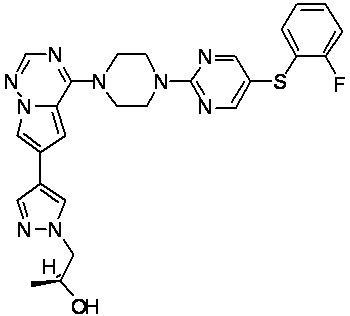
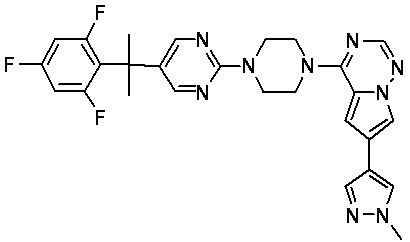
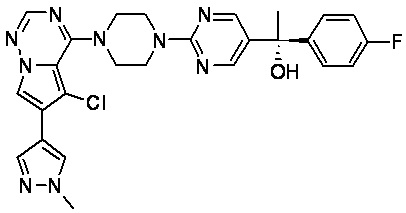
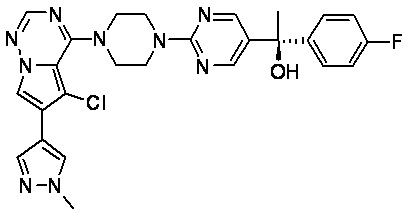
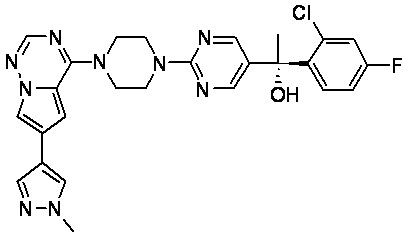
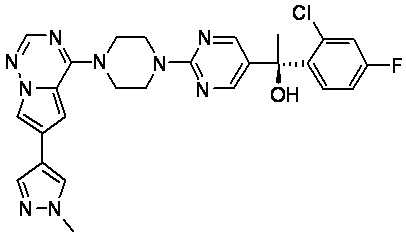
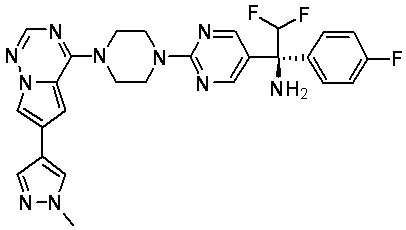
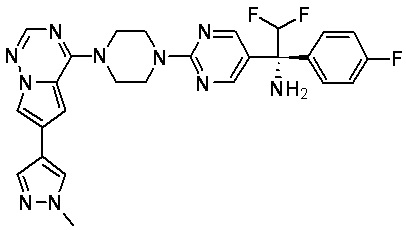
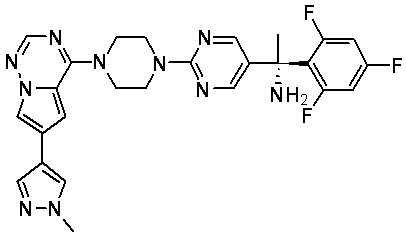
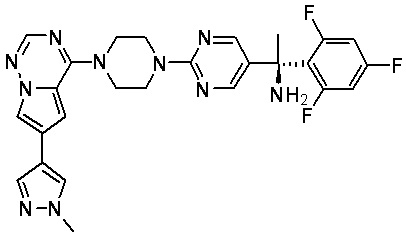
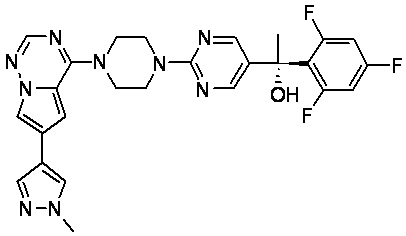
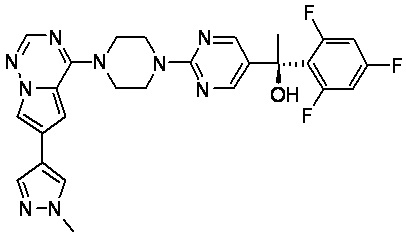
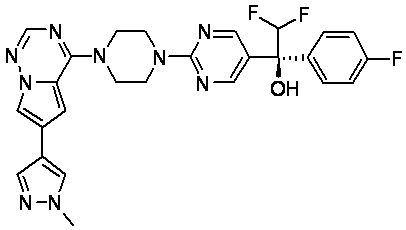
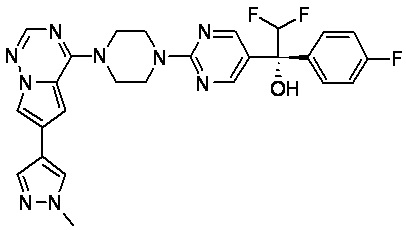
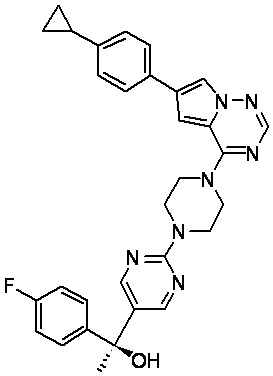
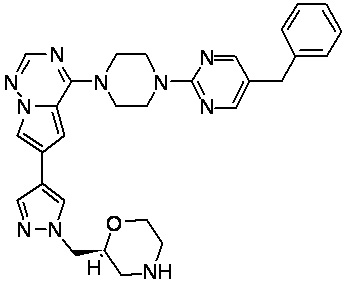
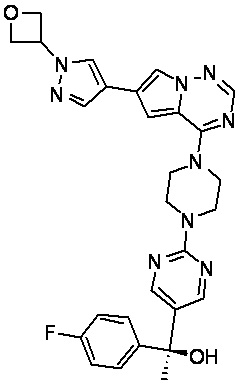
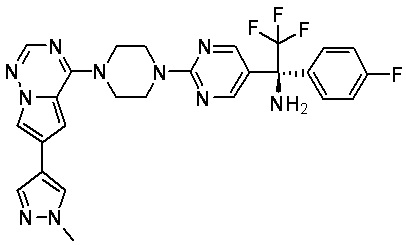
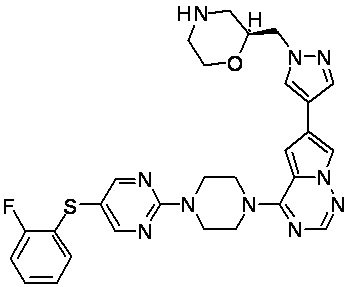
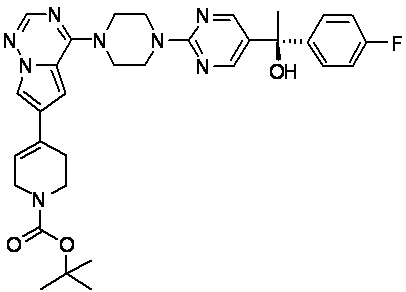
Синтез
Соединения согласно настоящему изобретению, включая их соли и N-оксиды, могут быть получены по известным методикам синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза, таких как пути, показанные на представленных Схемах. Реакции получения соединений согласно настоящему изобретению могут быть проведены в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящими растворителями могут быть растворители, в основном не взаимодействующие с исходными материалами (реагентами), промежуточными соединениями или продуктами при температурах проведения реакции, например, при температурах, которые могут находиться в диапазоне от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть проведена в одном растворителе или в смеси более чем одного растворителя. В зависимости от конкретной стадии реакции, специалистом в данной области техники могут быть выбраны подходящие растворители для конкретной стадии реакции.
Получение соединений согласно настоящему изобретению может включать защиту и снятие защиты с различных химических групп. Необходимость защиты или снятия защиты, а также выбор соответствующих защитных групп, может быть легко установлена специалистом в данной области техники. Химия защитных групп представлена, например, в публикации Wuts and Greene, Protective Groups in Organic Synthesis, 4ое изд., John Wiley & Sons: Нью-Джерси, (2006), которая включена в настоящую заявку посредством ссылки в полном объеме.
Реакции можно контролировать в соответствии с любым подходящим способом, известным в данной области техники. Например, образование продукта можно контролировать спектроскопическими способами, такими как спектроскопия ядерного магнитного резонанса (ЯМР) (например, 1H или 13C), инфракрасная (ИК) спектроскопия, спектрофотометрия (например, УФ-видимая), масс-спектрометрия (МС), или хроматографическими способами, такими как высокоэффективная жидкостная хроматография (ВЭЖХ) или тонкослойная хроматография (ТСХ).
Показания
Соединения, описанные в настоящем документе, могут быть пригодны для лечения патологических состояний, связанных с патологической активностью KIT, у людей или животных. Активирующие мутации в KIT встречаются при многих показаниях, включая системный мастоцитоз, GIST (желудочно-кишечные стромальные опухоли), AML (острый миелоидный лейкоз), меланому, семиному, внутричерепные герминогенные опухоли и средостенную B-клеточную лимфому.
Мастоцитоз относится к группе расстройств, характеризующихся избыточным накоплением тучных клеток в одной ткани или в нескольких тканях. Мастоцитоз подразделяют на две группы расстройств: (1) кожный мастоцитоз (CM) описывает формы, которые ограничены кожей; и (2) системный мастоцитоз (SM) описывает формы, в которых тучные клетки проникают во внекожные органы, затрагивая или не затрагивая кожу. SM дополнительно подразделяют на пять форм: индолентная (ISM), вялотекущая (SSM), агрессивная (ASM), SM с сопутствующим гематологическим заболеванием, не связанным с тучными клетками (SM-AHNMD), и тучноклеточный лейкоз (MCL).
Диагноз системного мастоцитоза частично основан на гистологических и цитологических исследованиях костного мозга, демонстрирующих инфильтрацию тучных клеток зачастую атипичной морфологии, которые часто патологически экспрессируют маркеры клеток, не являющихся тучными (CD25 и/или CD2). Диагноз SM подтверждают, если инфильтрация тучных клеток в костный мозг происходит в контексте одного из следующих: (1) патологическая морфология тучных клеток (веретенообразные клетки); (2) повышенный уровень триптазы в сыворотке, выше 20 нг/мл; или (3) наличие активирующей мутации KIT D816V.
Активирующие мутации в положении D816 встречаются в подавляющем большинстве случаев мастоцитоза (90-98 %), при этом наиболее распространенные мутации представляют собой D816V и D816H, и D816Y. Мутация D816V встречается в активационной петле домена киназы и приводит к конститутивной активации киназы KIT.
Соединения, описанные в настоящем документе, также могут быть пригодны для лечения GIST. Полная хирургическая резекция остается доминирующим предпочтительным способом лечения пациентов с первичными GIST. Хирургия эффективна примерно у 50 % пациентов с GIST; среди остальных пациентов часто встречается рецидив. Показано также, что первичное лечение ингибитором KIT, таким как иматиниб, является достаточным для первоначального лечения. Однако в течение нескольких месяцев развивается резистентность к иматинибу в результате соматической мутации. Такие вторичные мутации, резистентные к иматинибу, наиболее часто расположены в 11, 13, 14, 17 или 18 экзоне. Сунитиниб представляет собой стандарт второочередного лечения большинства резистентных к иматинибу опухолей и эффективен в отношении опухолей, содержащих мутации в 11, 13 и 14 экзонах. Однако вторичные мутации KIT в 17 и 18 экзонах являются резистентными к лечению сунитинибом и, более того, опухоли, содержащие третичные мутации резистентности в 17 и 18 экзоне, возникают спустя несколько месяцев после лечения сунитинибом. Регорафениб демонстрирует обнадеживающие результаты на 3 фазе клинических испытаний в отношении GIST, резистентных к иматинибу, сунитинибу, с активностью против нескольких, но не всех мутаций 17 и 18 экзона, одной из которых является D816. Следовательно, существует потребность в терапевтических агентах для лечения пациентов с GIST с мутациями в 17 экзоне, против которых неэффективен регорафениб.
Помимо применения соединений, описанных в настоящем документе, в качестве самостоятельных агентов при лечении рецидивирующих GIST, применение комбинаций иматиниба, сунитиниба и/или регорафениба с соединениями, описанными в настоящем документе, может обеспечивать возможность предотвращения возникновения резистентности к мутациям в 17 экзоне.
Существует подмножество пациентов с GIST с мутацией D842V в PDGFRα; указанная группа пациентов с GIST может быть разделена путем идентификации данной мутации. Указанное подмножество пациентов является невосприимчивым ко всем ингибиторам тирозинкиназы, доступным в настоящее время. Соединения, описанные в настоящем документе, благодаря их активности против D842V PDGFRα, могут быть пригодны для лечения указанных пациентов.
Соединения, описанные в настоящем документе, также могут быть пригодны для лечения AML. Пациенты с AML также имеют мутации KIT, и большинство указанных мутаций находятся в положении D816.
Кроме того, мутации в KIT связаны с саркомой Юинга, DLBCL (диффузной B-крупноклеточной лимфомой), дисгерминомой, MDS (миелодиспластическим синдромо), NKTCL (назальной NK/T-клеточной лимфомой), CMML (хроническим миеломоноцитарным лейкозом) и раком головного мозга.
Соединения, описанные в настоящем документе, могут быть использованы для лечения патологических состояний, связанных с мутациями KIT в 9 экзоне, 11 экзоне, 13 экзоне, 14 экзоне, 17 экзоне и/или 18 экзоне. Они также могут быть использованы для лечения патологических состояний, связанных с KIT дикого типа. Соединения, описанные в настоящем документе, могут быть использованы в качестве самостоятельных агентов для лечения патологических состояний, описанных в настоящем документе, или они могут быть использованы в комбинации с другими терапевтическими агентами, включая, без ограничения, иматиниб, сунитиниб и регорафениб. Другие агенты включают соединения, описанные в WO 2014/039714 и WO 2014/100620.
Соединения, описанные в настоящем документе, могут быть активны против одной или более мутаций KIT в 17 экзоне (например, D816V, D816Y, D816F, D816K, D816H, D816A, D816G, D820A, D820E, D820G, N822K, N822H, Y823D и A829P), и гораздо менее активным против KIT дикого типа. Указанные соединения могут быть введены в комбинации с агентом, который является (a) активным против других активирующих мутаций KIT, таких как мутации в экзоне 9 и 11, но (b) неактивными против мутаций в экзоне 17. Такие агенты включают иматиниб, сунитиниб и регорафениб. Следовательно, комбинация указанного соединения и указанного агента будет ингибировать мутантную в 17 экзоне KIT, а также ингибировать мутантную в 9/11 экзоне KIT. Указанное соединение и агент могут быть введены совместно или введены в чередующемся режиме. То есть в течение некоторого периода времени может быть отдельно введен ингибитор мутантной в 17 экзоне KIT; затем в течение некоторого последующего периода времени может быть отдельно введен ингибитор мутантной в 9/11 экзоне KIT. Затем указанный цикл может быть повторен. Полагают, что такой режим может замедлять развитие устойчивости к ингибитору мутантной в 17 экзоне KIT и/или к ингибитору мутантной в 9/11 экзоне KIT.
Кроме того, соединения, описанные в настоящем документе, которые могут быть селективными для мутаций KIT в 17 экзоне, могут быть введены с агентами, которые активны против мутаций в экзоне 9/11, в комбинации с третьим агентом, который охватывает мутации, пропущенные двухфакторной композицией. Комбинация трех агентов может ингибировать целый спектр мутаций KIT, а также в некоторых случаях KIT дикого типа. Агенты могут быть введены одновременно или в чередующемся режиме. Они могут быть введены одновременно, или два агента могут быть введены вместе в течение некоторого периода времени; затем может быть отдельно введен третий агент в течение некоторого последующего периода времени. Полагают, что такой режим может замедлять развитие устойчивости к ингибиторам мутантной KIT.
Фармацевтические композиции
Несмотря на то, что соединение, описанное в настоящем документе, может быть введено отдельно, предпочтительно вводить указанное соединение в виде фармацевтической композиции, в которой соединение комбинировано с одним или более фармацевтически приемлемыми вспомогательными веществами или носителями. Соединения, описанные в настоящем документе, могут быть составлены для введения любым удобным способом для применения в медицине или ветеринарии. В некоторых вариантах реализации изобретения соединение, включенное в фармацевтический препарат, может быть активно само по себе или может представлять собой пролекарство, например, с возможностью превращения в активное соединение в физиологических условиях.
Выражение «фармацевтически приемлемый» в настоящем документе использовано для обозначения таких соединений, материалов, композиций и/или лекарственных форм, которые, по результатам тщательной медицинской клинической оценки, пригодны для использования в контакте с тканями организма человека и животных, без избыточной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерно с соотношением приемлемой пользы и риска.
Примеры фармацевтически приемлемых носителей включают: (1) сахара, такие как лактоза, глюкоза и сахароза; (2) крахмалы, такие как кукурузный крахмал и картофельный крахмал; (3) целлюлозу и ее производные, такие как натрия карбоксиметилцеллюлоза, этилцеллюлоза и ацетат целлюлозы; (4) порошкообразный трагакант; (5) солод; (6) желатин; (7) тальк; (8) вспомогательные вещества, такие как масло какао и воски для суппозиториев; (9) масла, такие как арахисовое масло, хлопковое масло, сафлоровое масло, кунжутное масло, оливковое масло, кукурузное масло и соевое масло; (10) гликоли, такие как пропиленгликоль; (11) многоатомные спирты, такие как глицерин, сорбит, маннит и полиэтиленгликоль; (12) сложные эфиры, такие как этилолеат и этиллаурат; (13) агар; (14) буферные агенты, такие как гидроксид магния и гидроксид алюминия; (15) альгиновую кислоту; (16) апирогенную воду; (17) изотонический солевой раствор; (18) раствор Рингера; (19) этиловый спирт; (20) фосфатные буферные растворы; (21) циклодекстрины, такие как Captisol®; и (22) другие нетоксичные совместимые субстанции, используемые в фармацевтических композициях.
Примеры фармацевтически приемлемых антиоксидантов включают: (1) водорастворимые антиоксиданты, такие как аскорбиновая кислота, гидрохлорид цистеина, бисульфат натрия, метабисульфит натрия, сульфит натрия и т.п.; (2) маслорастворимые антиоксиданты, такие как аскорбилпальмитат, бутилированный гидроксианизол (BHA), бутилированный гидрокситолуол (BHT), лецитин, пропилгаллат, альфа-токоферол и т.п.; и (3) металлохелатирующие агенты, такие как лимонная кислота, этилендиаминтетрауксусная кислота (ЭДТК), сорбит, винная кислота, фосфорная кислота и т.п.
Твердые лекарственные формы (например, капсулы, таблетки, пилюли, драже, порошки, гранулы и т.п.) могут содержать один или более фармацевтически приемлемых носителей, таких как цитрат натрия или фосфат дикальция, и/или любой из следующих: (1) наполнители или сухие разбавители, такие как крахмалы, лактоза, сахароза, глюкоза, маннит и/или кремниевая кислота; (2) связующие агенты, такие как, например, карбоксиметилцеллюлоза, альгинаты, желатин, поливинилпирролидон, сахароза и/или гуммиарабик; (3) увлажнители, такие как глицерин; (4) средства для улучшения распадаемости таблеток, такие как агар-агар, карбонат кальция, картофельный или тапиоковый крахмал, альгиновая кислота, некоторые силикаты и карбонат натрия; (5) ингибиторы растворения, такие как парафин; (6) ускорители абсорбции, такие как четвертичные аммониевые соединения; (7) смачивающие агенты, такие как, например, цетиловый спирт и глицерина моностеарат; (8) абсорбенты, такие как каолин и бентонитовая глина; (9) смазывающие вещества, такие как тальк, стеарат кальция, стеарат магния, твердые полиэтиленгликоли, лаурилсульфат натрия и их смеси; и (10) красящие агенты.
Жидкие лекарственные формы могут включать фармацевтически приемлемые эмульсии, микроэмульсии, растворы, суспензии, сиропы и эликсиры. Помимо активного ингредиента, жидкие лекарственные формы могут содержать инертные разбавители, обычно используемые в данной области техники, такие как, например, вода или другие растворители, солюбилизирующие агенты и эмульгаторы, такие как этиловый спирт, изопропиловый спирт, этилкарбонат, этилацетат, бензиловый спирт, бензилбензоат, пропиленгликоль, 1,3-бутиленгликоль, масла (в частности, хлопковое масло, арахисовое масло, кукурузное масло, масло зародышей, оливковое масло, касторовое масло и кунжутное масло), глицерин, тетрагидрофурфуриловый спирт, полиэтиленгликоли и сложные эфиры жирных кислот сорбита, а также их смеси.
Суспензии, помимо активных соединений, могут содержать суспендирующие агенты, как например, этоксилированные изостеариловые спирты, полиоксиэтиленсорбит и сложные эфиры сорбита, микрокристаллическую целлюлозу, метагидроксид алюминия, бентонит, агар-агар и трагакант, и их смеси.
Мази, пасты, кремы и гели могут содержать, помимо активного соединения, вспомогательные вещества, такие как животные и растительные жиры, масла, воски, парафины, крахмал, трагакант, производные целлюлозы, полиэтиленгликоли, силиконы, бентониты, кремниевую кислоту, тальк и оксид цинка, или их смеси.
Порошки и спреи могут содержать, помимо активного соединения, вспомогательные вещества, такие как лактоза, тальк, кремниевая кислота, гидроксид алюминия, силикаты кальция и порошкообразный полиамид, или смеси этих веществ. Спреи могут дополнительно содержать обычные пропелленты, такие как хлорфторуглеводороды и летучие незамещенные углеводороды, такие как бутан и пропан.
Композиции могут быть удобно представлены в единичных лекарственных формах и могут быть получены любым из способов, хорошо известных в области фармации. Количество активного ингредиента, который может быть комбинирован с материалом носителя с получением единичной лекарственной формы, варьируется в зависимости от пациента, подлежащего лечению, конкретного способа введения. Количество активного ингредиента, которое может быть комбинировано с материалом носителя с получением единичной лекарственной формы, обычно является таким количеством соединения, которое вызывает терапевтический эффект.
Лекарственные формы для местного или трансдермального введения соединения согласно настоящему изобретению включают порошки, спреи, мази, пасты, кремы, лосьоны, гели, растворы, пластыри и средства для ингаляции. Активное соединение может быть смешано в стерильных условиях с фармацевтически приемлемым носителем и с любыми консервантами, буферами или пропеллентами, которые могут быть необходимы.
При введении соединений, описанных в настоящем документе, в виде фармацевтических средств людям и животным, они могут быть введены в чистом виде или в виде фармацевтической композиции, содержащей, например, от 0,1 % до 99,5 % (более предпочтительно, от 0,5 % до 90 %) активного ингредиента в комбинации с фармацевтически приемлемым носителем.
Композиции могут быть введены локально, перорально, трансдермально, ректально, вагинально, парентерально, интраназально, внутрилегочно, интраокулярно, внутривенно, внутримышечно, внутриартериально, интратекально, интракапсулярно, внутрикожно, внутрибрюшинно, подкожно, под кутикулу или ингаляцией.
Дозы
Фактические уровни доз активных ингредиентов в фармацевтических композициях согласно настоящему изобретению могут варьироваться для получения такого количества активного ингредиента, которое является эффективным для достижения требуемого терапевтического ответа для конкретного пациента, композиции и способа введения, и не является токсичным для пациента.
Выбранный уровень дозы зависит от множества факторов, включая активность конкретного используемого соединения, описанного в настоящем документе, или его сложного эфира, соли или амида, способа введения, времени введения, скорости экскреции конкретного используемого соединения, продолжительности лечения, других лекарств, соединений и/или материалов, применяемых в комбинации с конкретным используемым соединением, возраст, пол, массу, патологическое состояние, общее состояние здоровья и историю болезни пациента, подлежащего лечению, и подобные факторы, хорошо известные в области медицины.
Опытный врач или ветеринар может легко определить и прописать эффективное количество необходимой фармацевтической композиции. Например, врач или ветеринар может начать с более низких доз соединений согласно настоящему изобретению, используемых в фармацевтической композиции, чем необходимая доза для достижения требуемого терапевтического эффекта, с последующим постепенным повышением дозы до достижения требуемого эффекта.
В общем случае, подходящая суточная доза соединения согласно настоящему изобретению представляет собой такое количество соединения, которое представляет собой минимальную дозу, эффективную для обеспечения терапевтического эффекта. Такая эффективная доза, в общем случае, зависит от факторов, описанных выше. В общем случае, внутривенные, интрацеребровентрикулярные и подкожные дозы соединений согласно настоящему изобретению для пациента составляют от около 0,0001 до около 100 мг на килограмм массы тела в сутки. При необходимости эффективная суточная доза активного соединения может быть введена в виде двух, трех, четырех, пяти, шести или более субдоз, вводимых по отдельности с соответствующими интервалами в течение суток, необязательно в виде единичных лекарственных форм. В некоторых вариантах реализации изобретения доза для людей составляет 100-400 мг или 200-300 мг, которую вводят дважды в сутки; или 400-700 мг, или 500-600 мг, которую вводят один раз в сутки.
ПРИМЕРЫ
Следующие примеры представлены для иллюстрации, и их никоим образом не следует понимать как ограничение.
Представленные ниже Схемы предназначены для обеспечения общего руководства в отношении получения соединений согласно настоящему изобретению. Специалистам в данной области техники понятно, что способы получения, представленные на Схемах, могут быть модифицированы или оптимизированы с применением общих знаний в области органической химии для получения различных соединений согласно настоящему изобретению.
Протокол синтеза 1
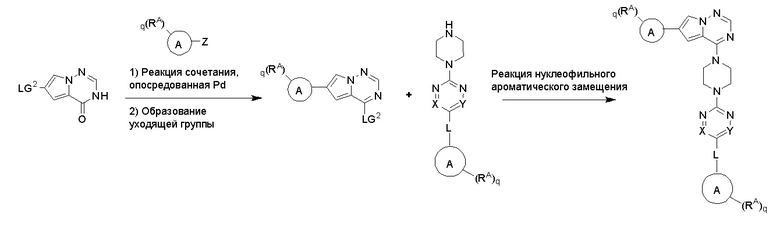
Пирролотриазинон может быть связан (LG2 может представлять собой, например, Cl, Br или I) с арильным, гетероарильным, алкенильным, алкильным реагентом бора, олова или цинка посредством реакции сочетания, опосредованной палладием, например, сочетания Сузуки, Стилла, Негиши, с получением промежуточного соединения с новой углерод-углеродной связью, образованной после последующего образования уходящей группы (с помощью POCl3 или других аналогичных реагентов). Полученный пирролотриазин может быть замещен амином в условиях реакции нуклеофильного ароматического замещения с применением основания, такого как диизопропилэтиламин (DIPEA) или триэтиламин (TEA) в полярном растворителе, таком как диоксан, с получением пиперазин-замещенного пирролотриазина. Как показано ниже, Соединения 9, 10 и 107 были получены по Протоколу синтеза 1.
Пример 1
Синтез (R)-6-(1-метил-1H-пиразол-4-ил)-4-(4-(5-(1-фенилэтил)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазина и (S)-6-(1-метил-1H-пиразол-4-ил)-4-(4-(5-(1-фенилэтил)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазина (Соединения 9 и 10)
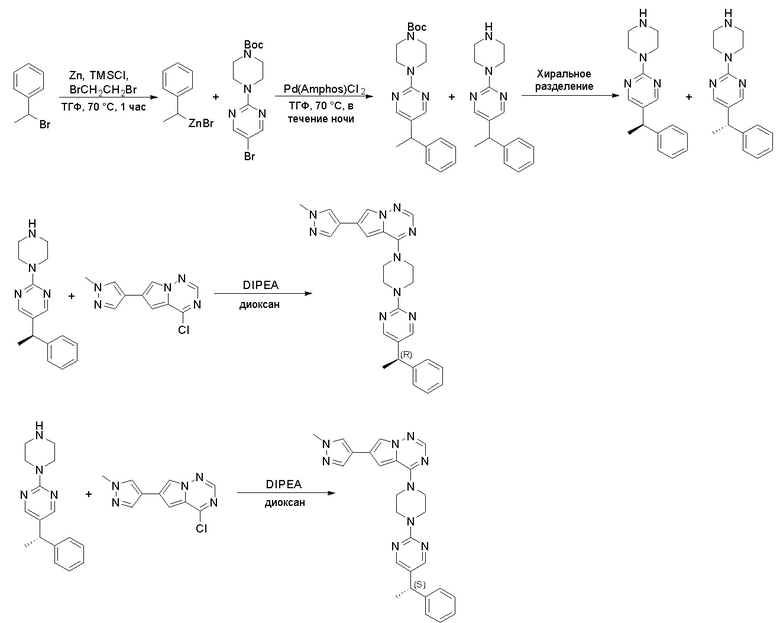
Стадия 1: Синтез бромида (1-фенилэтил)цинка (II):
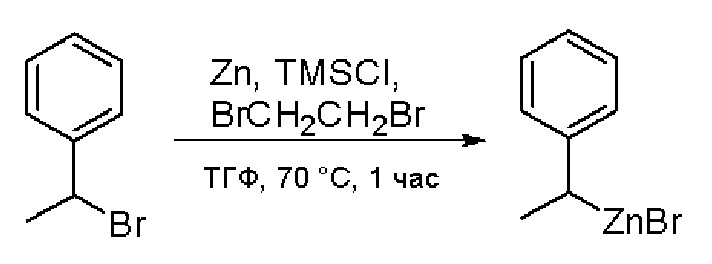
К суспензионной смеси порошкообразного цинка (активный, 5,1 г, 80,0 ммоль) в сухом ТГФ (20 мл) по каплям добавили 1,2-дибромэтан (0,28 мл, 5,7 ммоль) при 70 oC под атмосферой азота, затем добавили хлортриметилсилан (1,2 мл, 10,6 ммоль). Затем по каплям добавили (1-бромэтил)бензол (3,7 г, 20 ммоль). Полученную суспензию перемешивали при 70 °С еще 1 час. Реакционную смесь охладили до комнатной температуры и напрямую использовали на следующей стадии.
Стадия 2: Синтез 5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидина:
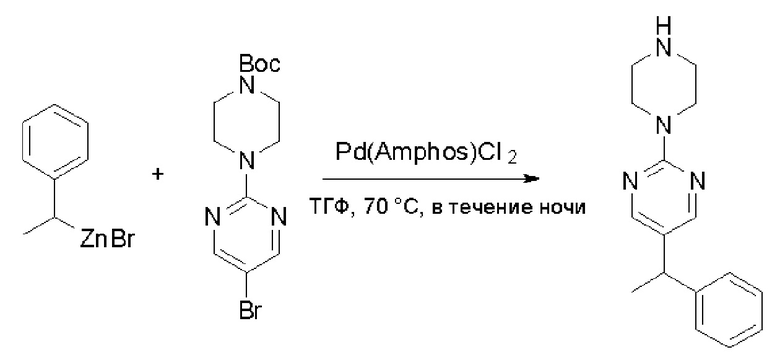
К раствору трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилата (4,1 г, 12,0 ммоль) и тетракис(трифенилфосфин)палладия (708 мг, 1,0 ммоль) в ТГФ (80 мл, сухой) по каплям добавили раствор бромида (1-фенилэтил)цинка (II) в ТГФ (20 мл, 1 M, 20 ммоль) под атмосферой азота и перемешивали смесь при 70 °С в течение ночи. Реакционную смесь охладили до комнатной температуры и отфильтровали через слой целита. Фильтрат концентрировали и очистили хроматографией на силикагеле с получением трет-бутил-4-(5-(1-фенилэтил)пиримидин-2-ил)пиперазин-1-карбоксилата (1,0 г, выход 23 %) в виде белого твердого вещества (этилацетат/петролейный эфир = 1/5 в качестве элюента) и 5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидина (2,4 г, 75 %) в виде желтого маслянистого вещества (метанол/дихлорметан = 1/20 в качестве элюента). МС (ЭС+) C16H20N4 рассчитано: 268, найдено: 269 [M + H ]+.
Стадия 3: Хиральное разделение (R)-5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидина и (S)-5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидина:
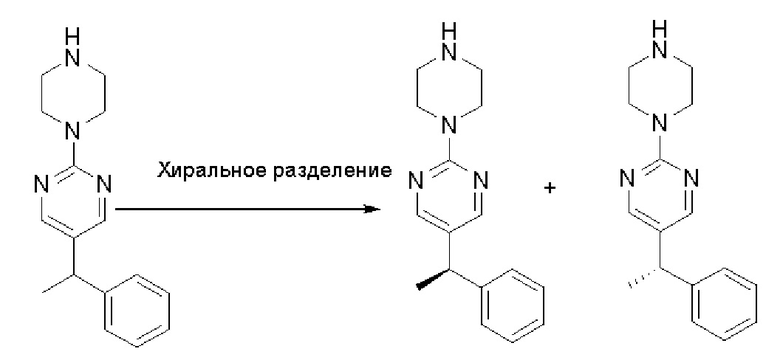
Рацемическое соединение 5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидин (900 мг) разделили с помощью хиральной ВЭЖХ в следующих условиях:
Хиральная колонка: AD-3 (150 * 4,6 мм, 3 мкм)
Подвижная фаза: гексан (0,1 % DEA)/EtOH (0,1 % DEA)
Получили (R)-5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидин (400 мг, 44 %) в виде желтого маслянистого вещества и (S)-5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидин (350 мг, 39 %) в виде желтого маслянистого вещества. Абсолютную стереохимию назначили произвольно. МС (ЭС+) C16H20N4 рассчитано: 268, найдено: 269 [M + H ]+.
Синтез 4-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина
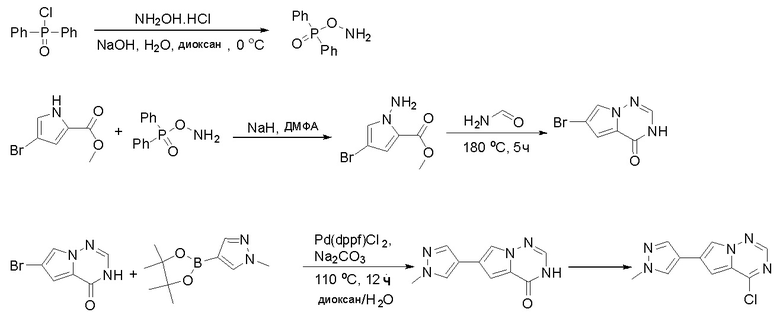
Стадия 4: Синтез O-(дифенилфосфорил)гидроксиламина:
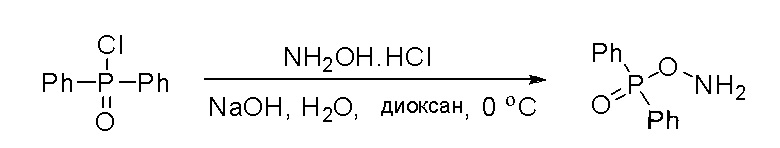
К раствору гидроксиламина гидрохлорида (7,3 г, 106 ммоль, 2,5 экв.) в воде (12 мл) и диоксане (12 мл) добавили раствор NaOH (4,07 г, 102 ммоль, 2,4 экв.) в воде (12 мл) и охладили смесь до -5 °С на бане изо льда/соли. К полученному выше раствору быстро добавили раствор дифенилфосфинхлорида (10 г, 42 ммоль, 1 экв.) в диоксане (12 мл), предварительно охлажденный до температуры ниже 10 °C, на бане изо льда/соли при энергичном перемешивании. По завершении добавления смесь перемешивали еще 5 минут на бане изо льда/соли, затем разбавили ледяной водой (150 мл) и отфильтровали. Осадок на фильтре промыли ледяной водой и лиофилизировали с получением о-(дифенилфосфорил)гидроксиламина (6,0 г, выход 61 %) в виде белого твердого вещества. МС (ЭС+) рассчитано: 233, найдено 234 [M+H]+; чистота: 75 %.
Стадия 5: Синтез метилового эфира 1-амино-4-бром-1H-пиррол-2-карбоновой кислоты:
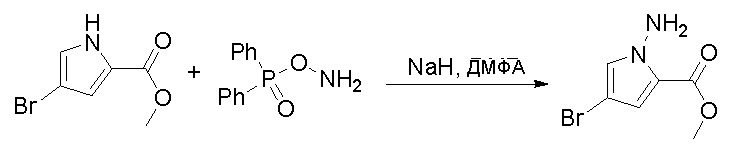
К раствору метилового эфира 4-бром-1H-пиррол-2-карбоновой кислоты (3,5 г, 17,2 ммоль, 1 экв.) в ДМФА (120 мл) добавили NaH (0,82 г, 20,6 ммоль, 1,2 экв.) при 0 °С и перемешивали смесь при 0 °С в течение 1 часа, после чего добавили о-(дифенилфосфинил)гидроксиламин (6 г, 25,8 ммоль). Реакционную смесь перемешивали еще 1 час, затем нейтрализовали 20 % раствором NH4Cl и экстрагировали EA. Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили над сульфатом натрия, отфильтровали и концентрировали выпариванием. Остаток очистили колоночной хроматографией на силикагеле (PE/EA = 4:1) с получением метилового эфира 1-амино-4-бром-1H-пиррол-2-карбоновой кислоты (2,9 г, выход 77 %) в виде светло-желтого твердого вещества. МС (ЭС+) рассчитано: 218, 220, найдено 219, 221 [M+H]+; чистота: 97 %.
Стадия 6: Синтез 6-бром-3H-пирроло[2,1-f][1,2,4]триазин-4-она:
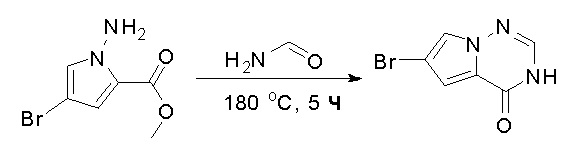
Раствор метилового эфира 1-амино-4-бром-1H-пиррол-2-карбоновой кислоты (2,9 г, 13,2 ммоль) в формамиде (12 мл) нагревали при 180 °С в течение 5 часов. Смесь разбавили этилацетатом (300 мл) и затем промыли водой (100 мл * 2), насыщенным солевым раствором (100 мл * 3). Органический слой высушили над сульфатом натрия, отфильтровали и концентрировали под пониженным давлением. Полученное твердое вещество промыли PE/EA (4:1, 50 мл) с получением 6-бром-3H-пирроло[2,1-f][1,2,4]триазин-4-она (1,4 г, выход 50 %) в виде желтого твердого вещества. МС (ЭС+) рассчитано: 213, 215, найдено 214, 216 [M+H]+; чистота: 92 %.
Стадия 7: Синтез 6-(1-метил-1H-пиразол-4-ил) пирроло[1,2-f][1,2,4] триазин-4(3H)-она:
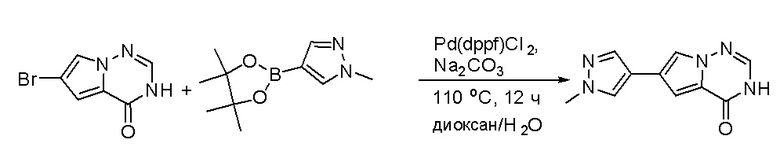
Смесь 6-бром-3H-пирроло[2,1-f][1,2,4]триазин-4-она (2,15 г, 10 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (4,2 г, 20 ммоль), Cs2CO3 (9,8 г, 30 ммоль), PdCl2dppf (814 мг, 1 ммоль), воды (15 мл), этанола (15 мл) и диоксана (70 мл) в колбе объемом 250 мл дегазировали N2 в течение 10 минут и затем нагревали при 120 °С под атмосферой N2 в течение ночи. Смесь охладили до комнатной температуры, затем добавили силикагель (~50 г). Остаток нанесли на силикагелевую колонку и элюировали ДХМ:MeOH (20:0 - 20:1) с получением 6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазин-4(3H)-она (600 мг, выход 28 %) в виде желтого твердого вещества. МС (ЭС+) рассчитано: 215, найдено 216,1 [M+H]+; чистота: 90 %.
Стадия 8: Синтез 4-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина:
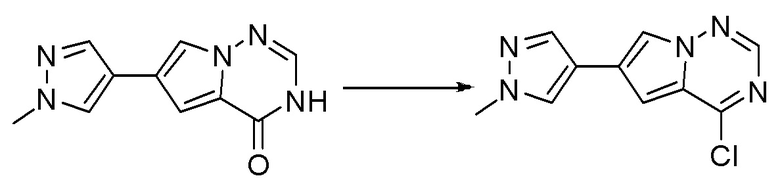
6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазин-4(3H)-он (600 мг, 2,8 ммоль) обрабатывали оксихлоридом фосфора (20 мл) при кипячении с обратным холодильником в течение 3 часов. Смесь охладили до комнатной температуры, концентрировали под пониженным давлением и разбавили остаток ледяной водой (100 мл). Смесь экстрагировали дихлорметаном (50 мл * 4), и объединенные органические слои высушили с помощью MgSO4, отфильтровали, концентрировали с получением 4-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина (600 мг, выход 92 %) в виде коричневого твердого вещества. МС (ЭС+) рассчитано: 233, 235, найдено 234, 236 [M+H]+; чистота: 90 %.
Стадия 9: Синтез (R)-6-(1-метил-1H-пиразол-4-ил)-4-(4-(5-(1-фенилэтил) пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазина:
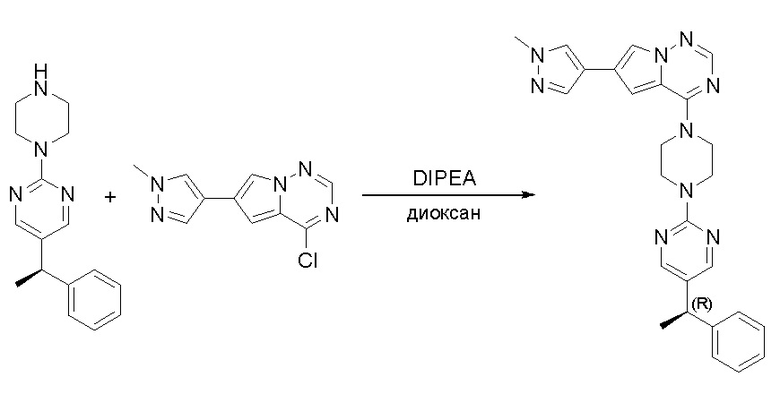
Смесь 4-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина (56 мг, 0,21 ммоль), (R)-5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидина (49 мг, 0,21 ммоль) и диизопропилэтиламина (97 мг, 0,84 ммоль) в диоксане (10 мл) перемешивали при комнатной температуре в течение ночи. Завершение реакции контролировали с помощью ЖХМС. Реакционную смесь концентрировали с получением остатка, который очистили препаративной ВЭЖХ с получением указанного в заголовке соединения (45 мг, 46 %) в виде белого твердого вещества. МС (ЭС+) C26H27N9 рассчитано: 465 найдено: 466 [M + H] +.
Стадия 10: Синтез (S)-6-(1-метил-1H-пиразол-4-ил)-4-(4-(5-(1-фенилэтил)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазина:
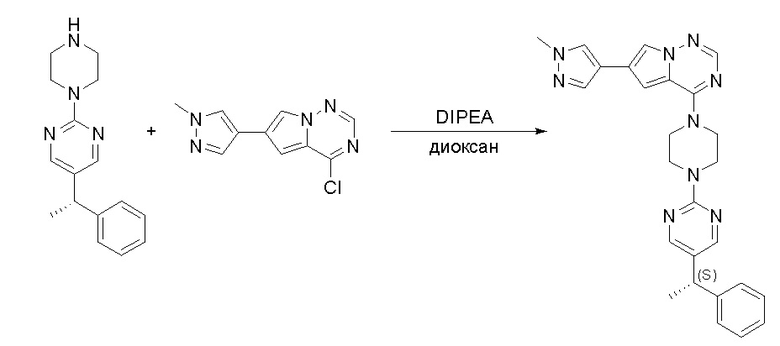
Смесь 4-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина (56 мг, 0,21 ммоль), (S)-5-(1-фенилэтил)-2-(пиперазин-1-ил)пиримидина (49 мг, 0,21 ммоль) и диизопропилэтиламина (97 мг, 0,84 ммоль) в диоксане (10 мл) перемешивали при комнатной температуре в течение ночи. Завершение реакции контролировали с помощью ЖХМС. Реакционную смесь концентрировали с получением остатка, который очистили препаративной ВЭЖХ с получением указанного в заголовке соединения (43 мг, 44 %) в виде белого твердого вещества. МС (ЭС+) C26H27N9 рассчитано: 465 найдено: 466 [M + H] +.
Пример 2: Синтез (S)-1-(2-(4-(5-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)-1-(4-фторфенил)этанола (Соединение 107)
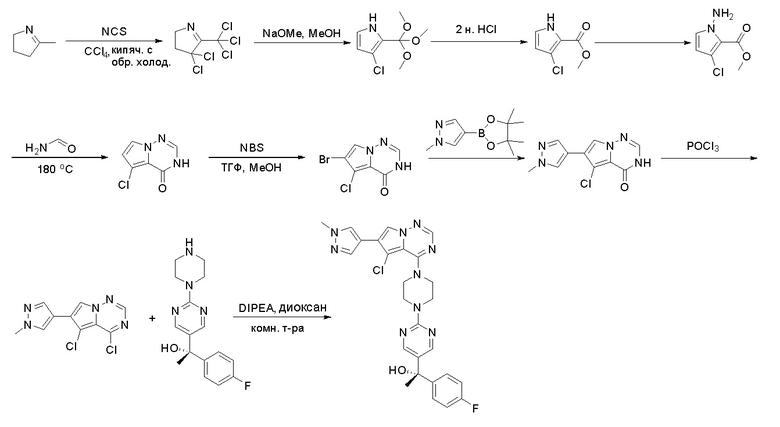
Стадия 1: Синтез метил-3-хлор-1H-пиррол-2-карбоксилата:
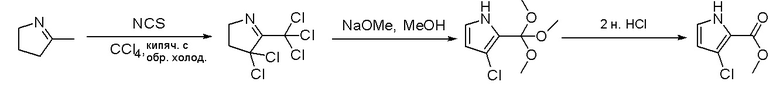
К раствору 5-метил-3,4-дигидро-2H-пиррола (2,50 г, 30,0 ммоль) в CCl4 (100 мл) добавили N-хлорсукцинимид (32,00 г, 240 ммоль) и затем нагревали смесь с обратным холодильником в течение 72 часов. Реакционную смесь охладили до 0 oC. Образовавшийся осадок отфильтровали и концентрировали фильтрат под пониженным давлением. Остаток растворили в метаноле (100 мл), затем добавили метоксид натрия (9,80 г, 180 ммоль). Полученную суспензию нагревали с обратным холодильником и перемешивали в течение 1,5 часов. Растворитель выпарили, а остаток суспендировали в эфире. Твердое вещество отфильтровали и концентрировали фильтрат под пониженным давлением. Остаток растворили в ДХМ (100 мл) и 2 M HCI (100 мл). Двухфазный раствор перемешивали в течение 10 минут. Органический слой отделили, высушили над MgSO4, отфильтровали и выпарили. Неочищенное маслянистое вещество очистили хроматографией на силикагеле, элюируя EtOAc и гексанами, с получением указанного в заголовке соединения (2,5 г, 52 %) в виде оранжевого твердого вещества. МС (ЭС+) C6H6ClNO2 рассчитано: 159, найдено: 160 [M+H]+.
Стадия 2: Синтез метил-1-амино-3-хлор-1H-пиррол-2-карбоксилата:
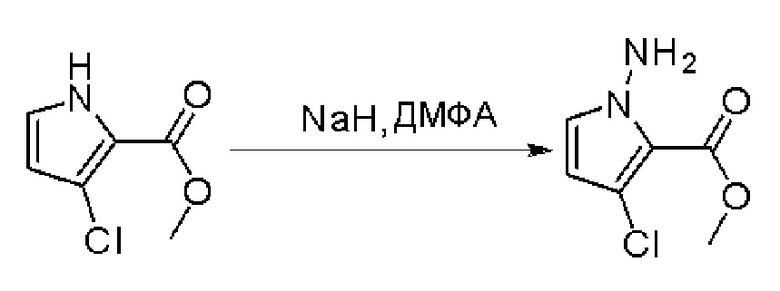
К суспензии гидрида натрия (60 процентов, 1,5 г, 37,5 ммоль) в ДМФА (250 мл) добавили метил-3-хлор-1H-пиррол-2-карбоксилат (5,0 г, 31,3 ммоль) при 0 °C и перемешивали смесь в течение 25 минут, затем добавили O-(дифенилфосфорил)гидроксиламин (10,0 г, 43,75 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 4 часов и погасили водным раствором Na2SO3, После перемешивания в течение еще 5 минут смесь экстрагировали EtOAc (3 x 300 мл). Объединенные органические слои промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали с получением неочищенного продукта в виде коричневого маслянистого вещества, которое очистили хроматографией на силикагеле (PE:EA = 4:1) с получением указанного в заголовке соединения (5,00 г, 91 %) в виде белого твердого вещества. МС (ЭС+) C6H7ClN2O2 рассчитано: 174, 176, найдено: 175, 177 [M+H]+.
Стадия 3: Синтез 5-хлорпирроло[1,2-f][1,2,4]триазин-4(3H)-она:
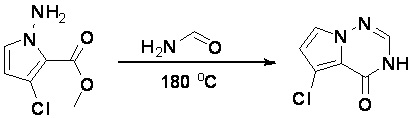
Смесь метил-1-амино-3-хлор-1H-пиррол-2-карбоксилата (4,00 г, 23 ммоль) и формамида (15 мл) нагревали до 180 oC в течение 3 часов. После охлаждения до комнатной температуры выпавшее в осадок твердое вещество собрали фильтрацией и промыли CH2CH2 с получением указанного в заголовке соединения (2,50 г, 64 %) в виде желтого твердого вещества. МС (ЭС+) C6H4ClN3O рассчитано: 169, найдено: 170 [M+H]+.
Стадия 4: Синтез 6-бром-5-хлорпирроло[1,2-f][1,2,4]триазин-4(3H)-она:
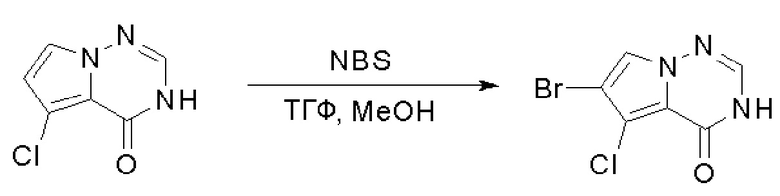
К смеси 5-хлорпирроло[1,2-f][1,2,4]триазин-4(3H)-она (2,50 г, 14,7 ммоль) в ТГФ (100 мл) и MeOH (50 мл) добавили N-бромсукцинимид (2,6 г, 14,7 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. Реакцию погасили водой и экстрагировали EA. Органический слой высушили (Na2SO4), отфильтровали и концентрировали. Остаток очистили хроматографией на силикагеле (EA:MeOH = 10:1) с получением указанного в заголовке соединения (2,00 г, 55 %) в виде желтого твердого вещества. МС (ЭС+) C6H3BrClN3O рассчитано: 246,9, найдено: 247,9 [M+H]+.
Стадия 5: Синтез 5-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазин-4(3H)-она:
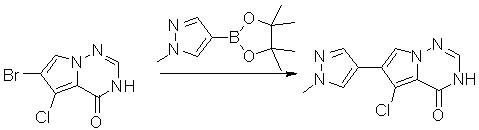
Смесь 6-бром-5-хлорпирроло[1,2-f][1,2,4]триазин-4(3H)-она (2,00 г, 8,1 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (2,5 г, 12,1 ммоль), K3PO4 (3,4 г, 16,1 ммоль) и Pd(dppf)Cl2 (589 мг, 0,81 ммоль) в 1,4-диоксане (30 мл) и воде (3 мл) продули N2 и затем нагревали до 90 °С в течение 15 часов. Реакционную смесь охладили до комнатной температуры и концентрировали. Остаток пропустили через колонку (силикагель, EA:ДХМ:MeOH = 10:10:1) с получением указанного в заголовке соединения (600 мг, 30 %) в виде желтого твердого вещества. МС (ЭС+) C10H8ClN5O рассчитано: 249, 251, найдено: 250, 252 [M+H]+.
Стадия 6: Синтез 4,5-дихлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина:
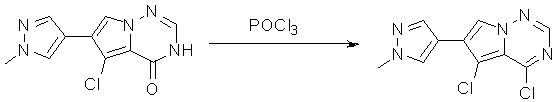
Смесь 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (600 мг, 2,4 ммоль) в POCl3 (4 мл) нагревали с обратным холодильником в течение 12 часов. Реакционную смесь охладили до комнатной температуры и концентрировали под пониженным давлением. Остаток промыли смесью ТГФ (20 мл) и 1,4-диоксана (10 мл) с получением указанного в заголовке соединения (450 мг, 69 %) в виде желтого твердого вещества. МС (ЭС+) C10H8ClN5O рассчитано: 267, 269, найдено: 268, 270 [M + H]+.
Стадия 7: Синтез (S)-1-(2-(4-(5-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)-1-(4-фторфенил)этанола
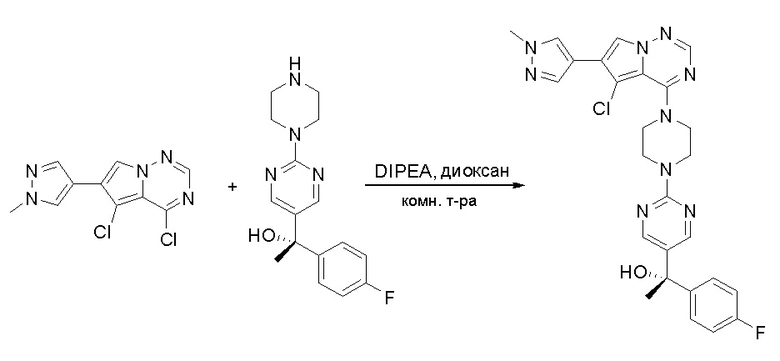
К смеси 4,5-дихлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина (150 мг, 0,56 ммоль) и (S)-1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола (135 мг, 0,45 ммоль) в 1,4-диоксане (10 мл) добавили DIPEA (361 мг, 2,8 ммоль). После перемешивания при комнатной температуре в течение 15 часов смесь концентрировали под пониженным давлением и очистили препаративной ВЭЖХ с получением указанного в заголовке соединения (40,9 мг, 17 %) в виде белого твердого вещества. МС (ЭС+) C26H25ClFN9O рассчитано: 533, найдено: 534 [M+H]+. 1H-ЯМР (400 МГц, ДМСО-d6) δ м.д. 8,41 (с, 1H), 8,39 (с, 2H), 8,13 (с, 1H), 8,09 (с, 1H), 7,48-7,44 (м, 2H), 7,22 (с, 1H), 7,14-7,10 (м, 2H), 5,91 (с, 1H), 3,95-3,89 (м, 7H), 3,71-3,68 (м, 4H), 1,82 (с, 3H).
Протокол синтеза 2
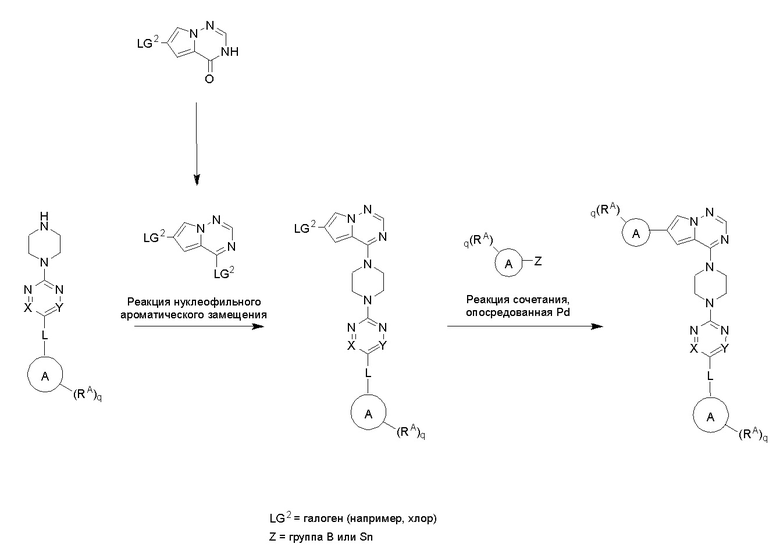
Пирролотриазинон может быть преобразован в пирролотриазин посредством обработки POCl3 или другими аналогичными реагентами. Пирролотриазин может быть замещен амином в условиях реакции нуклеофильного ароматического замещения с использованием аминного основания, такого как диизопропилэтиламин (DIPEA) или триэтиламин (TEA), в полярном растворителе, таком как диоксан, с получением пиперазин-замещенного пирролотриазина. Пирролотриазинон может быть связан (LG2 может представлять собой, например, Cl, Br или I) с арильным, гетероарильным, алкенильным или алкильным реагентом бора, олова или цинка посредством реакции сочетания, опосредованной палладием, например, сочетания Сузуки, Стилла, Негиши, с получением продукта. Как показано ниже, Соединение 123 было получено по Протоколу синтеза 2.
Пример 3: Синтез (S)-2-((4-(4-(4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)метил)морфолина (Соединение 123)
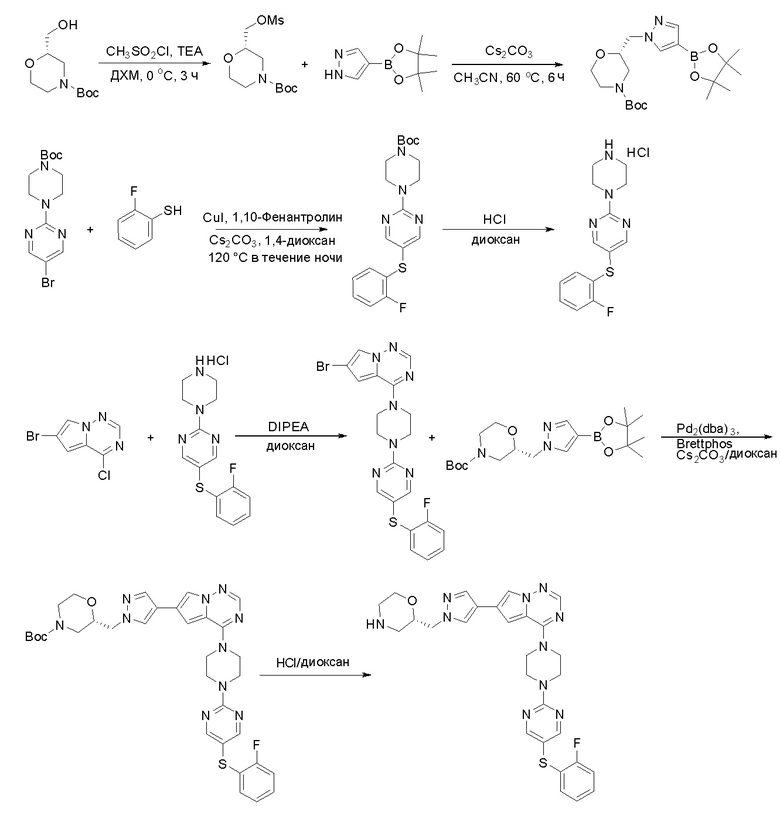
Стадия 1: Синтез (S)-трет-бутил-2-((метилсульфонилокси)метил) морфолин-4-карбоксилата:
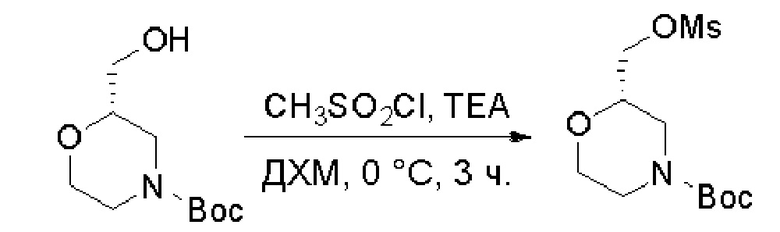
К смеси (S)-трет-бутил-2-(гидроксиметил)морфолин-4-карбоксилата (400 мг, 1,84 ммоль) в 10 мл дихлорметана по каплям добавили триэтиламин (372 мг, 3,68 ммоль) и метансульфонилхлорид (316 мг, 2,76 ммоль) при 0 oC. Реакционную смесь перемешивали при 0 °С в течение 3 часов, и ЖХМС показала, что реакция завершена. Реакционный раствор разбавили 20 мл дихлорметана и промыли насыщенным водным раствором NaHCO3 (30 мл× 3) и насыщенным солевым раствором. Органический слой отделили, высушили над сульфатом натрия, отфильтровали и концентрировали. Неочищенный продукт очистили хроматографией на силикагеле (петролейный эфир:этилацетат = 5:1) с получением указанного в заголовке продукта (430 мг, 79 %) в виде белого твердого вещества. МС (ЭС+) C11H21NO6S рассчитано: 295, найдено: 296 [M + H]+.
Стадия 2: Синтез (S)-трет-бутил-2-((4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)метил)морфолин-4-карбоксилата:
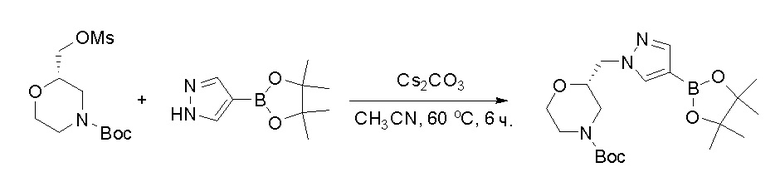
К смеси (S)-трет-бутил-2-((метилсульфонилокси)метил)морфолин-4-карбоксилата (430 мг, 1,46 ммоль) и 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (283 мг, 1,46 ммоль) в 50 мл ацетонитрила добавили карбонат церия (1,43 г, 4,37 ммоль) и перемешивали смесь при 60 °С в течение 3 часов. ТСХ и ЖХМС показали, что реакция завершена. После удаления растворителей под пониженным давлением остаток разбавили 50 мл этилацетата и промыли водой (50 мл × 3) и насыщенным солевым раствором. Органический слой отделили, высушили над сульфатом натрия, отфильтровали и концентрировали. Неочищенный продукт очистили хроматографией на силикагеле (петролейный эфир:этилацетат = 5:1) с получением указанного в заголовке продукта (300 мг, 52 %) в виде бесцветного маслянистого вещества. МС (ЭС+) C19H32BN3O5 рассчитано: 393, найдено: 394 [M + H]+.
Стадия 3: Синтез трет-бутил-4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-карбоксилата:
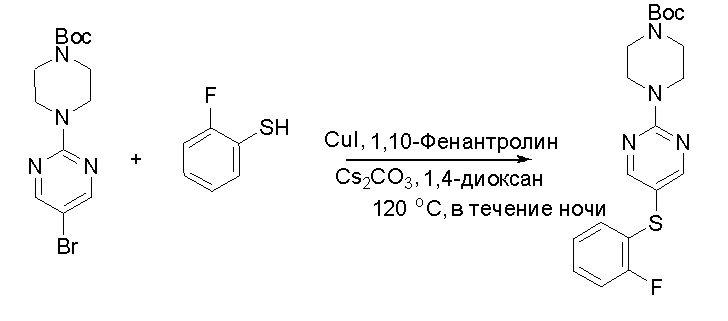
Смесь трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилата (5,0 г, 14,6 ммоль), 2-фторбензолтиола (9,3 г, 73 ммоль), 1,10-фенантролина (7,9 г, 43,8 ммоль), йодида меди (13,9 г, 73 ммоль) и карбоната церия (28,6 г, 87,6 ммоль) в диоксане (100 мл) кипятили с обратным холодильником в течение 3 дней. Реакционную смесь охладили до комнатной температуры и концентрировали. Остаток напрямую очистили хроматографией на силикагеле с получением указанного в заголовке соединения. МС (ЭС+) C19H23FN4O2S рассчитано: 390, найдено: 391 [M + H]+.
Стадия 4: Синтез 5-(2-фторфенилтио)-2-(пиперазин-1-ил)пиримидина HCl соли:
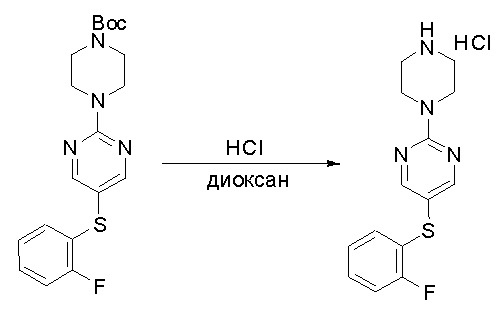
К раствору трет-бутил-4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-карбоксилата (5 г, 12,8 ммоль) в диоксане (150 мл) добавили HCl в диоксане (4 M, около 30 мл) и перемешивали смесь при 40 °С в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь концентрировали с получением 5-(2-фторфенилтио)-2-(пиперазин-1-ил)пиримидина HCl соли (3,6 г, 88 %) в виде твердого вещества. МС (ЭС+) C14H15FN4S рассчитано: 290, найдено: 291 [M + H]+.
Стадия 5: Синтез 6-бром-4-(4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазина:
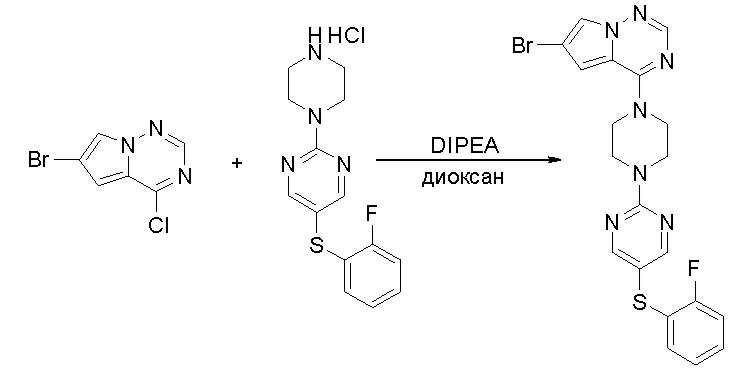
Смесь 6-бром-4-хлорпирроло[1,2-f][1,2,4]триазина (100 мг, 0,43 ммоль), 5-(2-фторфенилтио)-2-(пиперазин-1-ил)пиримидина HCl соли (126 мг, 0,43 ммоль) и диизопропилэтиламина (280 мг, 2,15 ммоль) в диоксане (5 мл) перемешивали при комнатной температуре в течение ночи. Реакционную смесь концентрировали и очистили хроматографией на силикагеле (петролейный эфир:этилацетат = 2:1) с получением указанного в заголовке соединения (100 мг, 49 %) в виде желтого твердого вещества. МС (ЭС+) C20H17BrFN7S рассчитано: 485, 487, найдено: 486, 488 [M + H]+.
Стадия 6: Синтез (S)-трет-бутил-2-((4-(4-(4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)метил)морфолин-4-карбоксилата:
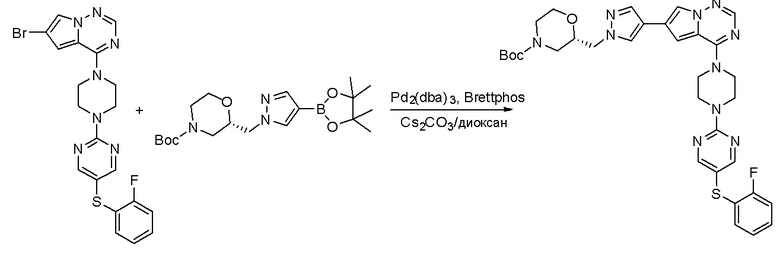
Смесь 6-бром-4-(4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазина (100 мг, 0,2 ммоль), (S)-трет-бутил-2-((4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)метил)морфолин-4-карбоксилата (80 мг, 0,2 ммоль), Pd2(dba)3 (34 мг, 0,02 ммоль), Brettphos (40 мг, 0,04 ммоль) и карбоната церия (260 мг, 0,4 ммоль) в диоксане (5 мл) дегазировали азотом три раза, а затем нагревали при 120 °С в течение ночи. Реакционную смесь охладили до комнатной температуры и концентрировали с получением остатка, который очистили хроматографией на силикагеле (дихлорметан:метанол = 15:1) с получением указанного в заголовке соединения (40 мг, 30 %) в виде белого твердого вещества. МС (ЭС+) C33H37FN10O3S рассчитано: 672, найдено: 617 [M-56+H]+.
Стадия 7: Синтез (S)-2-((4-(4-(4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)метил)морфолина:
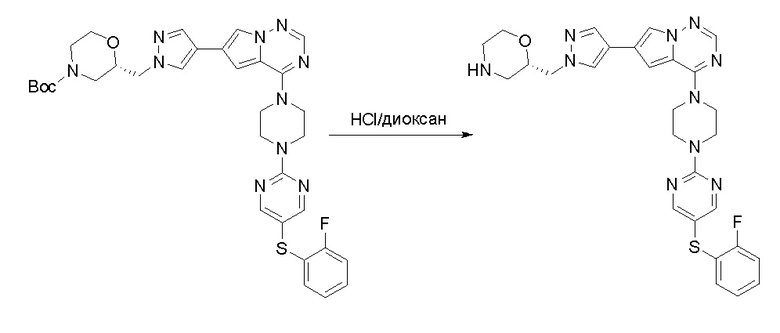
Смесь (S)-трет-бутил-2-((4-(4-(4-(5-(2-фторфенилтио)пиримидин-2-ил)пиперазин-1-ил)пирроло[1,2-f][1,2,4]триазин-6-ил)-1H-пиразол-1-ил)метил)морфолин-4-карбоксилата (40 мг, 0,06 ммоль) в HCl/диоксане (5 мл) перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали с получением остатка, который очистили препаративной ВЭЖХ с получением указанного в заголовке соединения (8,9 мг, 26 %) в виде желтого твердого вещества. МС (ЭС+) C28H29FN10OS рассчитано: 572, найдено: 573 [M + H]+.
Протокол синтеза 3
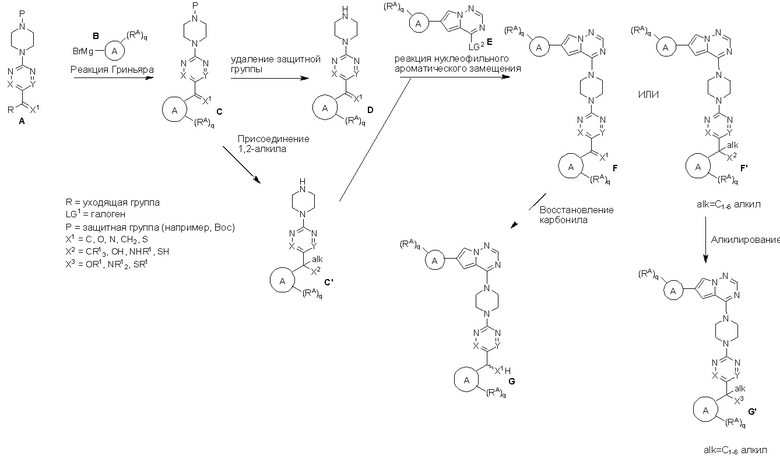
Карбонильное производное пиперазина, например, карбамоил (A, каждый X и Y представляют собой –CH-) может быть связано с бромидом Гриньяра (B, кольцо A представляет собой арил) с получением защищенного дизамещенного карбонила (C, X1 представляет собой CH2, S, NH или O). Если X1 представляет собой O, т.е. при получении карбонила, карбонил может быть дополнительно подвергнут взаимодействию с органометаллическим реагентом, таким как реагенты Гриньяра, лития, цинка и триалкилалюминий, например, триметилалюминий, что также может приводить к снятию защиты с атома азота пиперазина с получением дополнительно замещенного соединения (C’). Удаление защитной группы (P) из пиперазинового кольца (C) может быть проведено с помощью сильных кислот, таких как 4 М хлористоводородная кислота (HCl) в диоксане, или трифторуксусная кислота (ТФК) в полярном растворителе, таком как метанол или дихлорметан (ДХМ), с получением амина (D). Пирролотриазин (E) может быть замещен амином (C’) или (D) в условиях реакции нуклеофильного ароматического замещения с использованием амина, такого как диизопропилэтиламин (DIPEA) или триэтиламин (TEA), в полярном растворителе, таком как диоксан, с получением пиперазин-замещенного пирролотриазина (F) или (F’). Восстановление -C(=X1)-, где X1 представляет собой CH2, S, NH или O, например, карбонила в (F) может быть проведено с помощью восстанавливающего агента, такого как борогидрид натрия, с получением -C-(XH)-, например, спирта (G). В альтернативном варианте, алкилирование X2 может быть проведено с помощью алкилгалогенидов (альтернативные уходящие группы) с получением X3-содержащих аналогов (G’). Энантиомерно обогащенные продукты могут быть получены посредством каталитического асимметричного синтеза, синтеза с использованием вспомогательных хиральных реагентов и разделения рацемата. Как показано ниже, Соединения 40 и 41 были получены по Протоколу синтеза 3.
Пример 4: Синтез 4-(4-(5-(1-(4-фторфенил)пропил)пиримидин-2-ил)пиперазин-1-ил)-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина (Соединения 40 и 41)
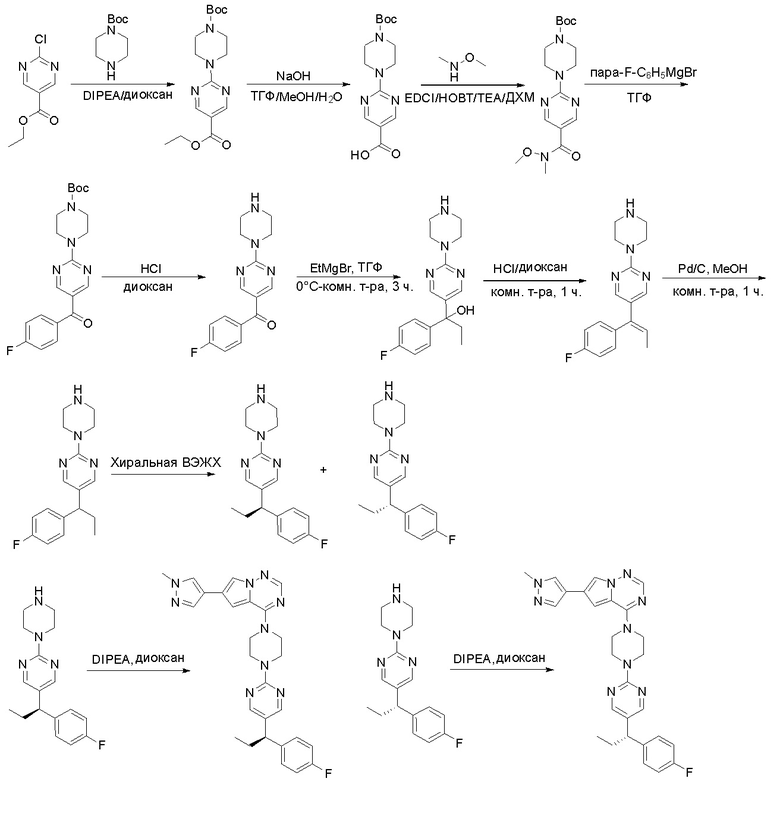
Стадия 1: Синтез этил-2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоксилата:
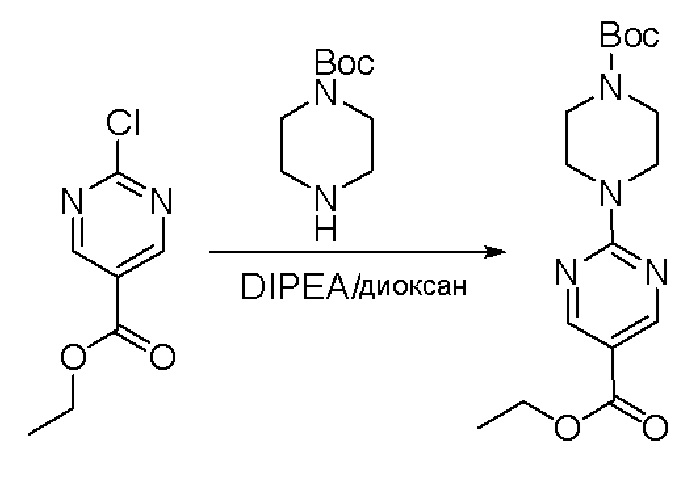
К раствору трет-бутил-пиперазин-1-карбоксилата (10,0 г, 53,7 ммоль) и диизопропилэтиламин (23,4 мл, 134,25 ммоль) в диоксане (80 мл) добавили этил-2-хлорпиримидин-5-карбоксилат (10 г, 53,7 ммоль) и перемешивали реакционную смесь при комнатной температуре в течение 3 часов. ЖХМС показала, что реакция завершена. Реакционную смесь концентрировали с получением указанного в заголовке соединения (17 г, неочищенное), которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C16H24N4O4 рассчитано: 336, найдено: 237, 281 [M -56+H]+.
Стадия 2: Синтез 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоновой кислоты:
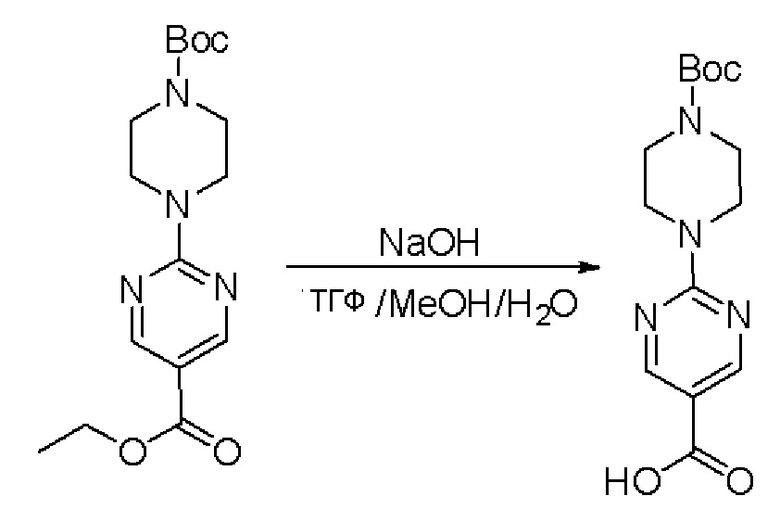
К раствору этил-2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоксилата (17 г, неочищенный) в ТГФ/MeOH/воде (300 мл) добавили гидроксид натрия (4,3 г, 107,5 ммоль) и перемешивали реакционную смесь при 70 oC в течение 2 часов. ЖХМС показала, что реакция завершена. Реакционную смесь охладили до комнатной температуры, подкислили до pH ≈ 5-6 с помощью 1 M HCl и отфильтровали. Твердое вещество собрали и высушили с получением указанного в заголовке соединения (16 г, 96 %) в виде белого твердого вещества, которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C14H20N4O4 рассчитано: 308, найдено: 253 [M -56+ H]+.
Стадия 3: Синтез трет-бутил-4-(5-(метокси(метил)карбамоил)пиримидин-2-ил)пиперазин-1-карбоксилата:
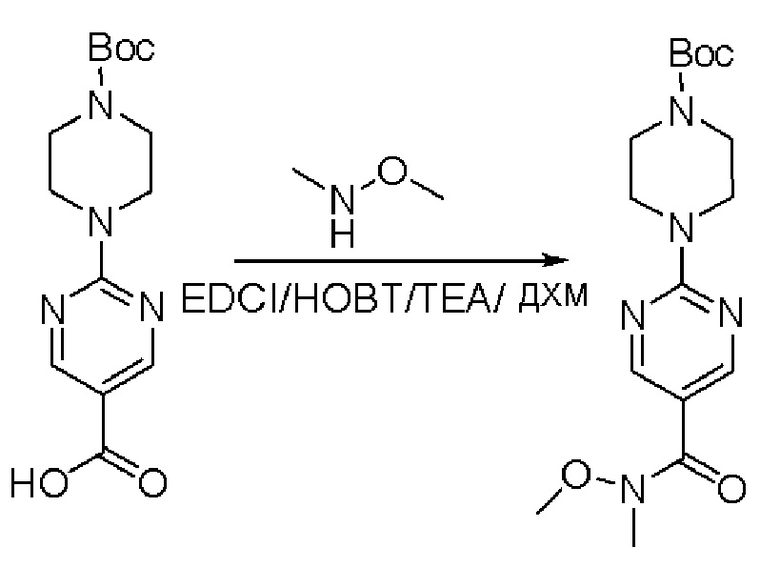
К суспензии 2-(4-(трет-бутоксикарбонил)пиперазин-1-ил)пиримидин-5-карбоновой кислоты (13,8 г, 44,8 ммоль), EDCI (12,8 г, 67,2 ммоль) и HOBT (7,2 г, 53,7 ммоль) в дихлорметане (200 мл) добавили триэтиламин (25 мл, 179,2 ммоль) и перемешивали смесь при комнатной температуре в течение 1 часа, затем добавили N,O-диметилгидроксиламин (5 г, 53,7 ммоль). Реакционную смесь перемешивали еще 3 часа. ЖХМС показала, что реакция завершена. Реакционную смесь промыли водой (100 мл), и органический слой высушили, отфильтровали и концентрировали. Остаток очистили хроматографией на силикагеле (петролейный эфир:этилацетат = 1:1) с получением указанного в заголовке соединения (11,2 г, 67 %) в виде белого твердого вещества. МС (ЭС+) C16H25N5O4 рассчитано: 351, найдено: 296 [M -56+ H]+.
Стадия 4: Синтез трет-бутил-4-(5-(4-фторбензоил)пиримидин-2-ил)пиперазин-1-карбоксилата:
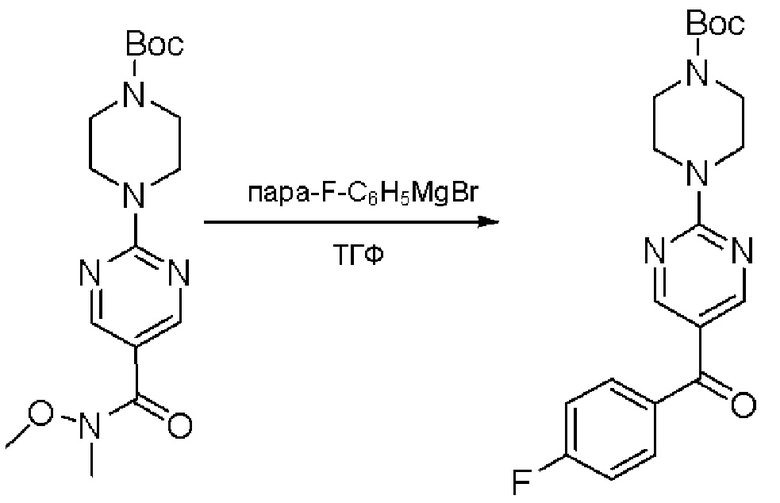
К раствору трет-бутил-4-(5-(метокси(метил)карбамоил)пиримидин-2-ил)пиперазин-1-карбоксилата (7,8 г, 22,22 ммоль) в сухом ТГФ (50 мл) добавили C6H5MgFBr (1 M в ТГФ, 50 мл) при 0 oC под азотом и перемешивали смесь при комнатной температуре в течение 3 часов. ЖХМС показала, что реакция завершена. Реакцию погасили 1 M раствором HCl и экстрагировали этилацетатом. Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили над сульфатом натрия, отфильтровали и концентрировали. Остаток очистили хроматографией на силикагеле (петролейный эфир:этилацетат = 5:1) с получением указанного в заголовке соединения (7.2г, 84 %) в виде желтого твердого вещества. МС (ЭС+) C20H23FN4O3 рассчитано: 386, найдено: 331 [M- 56 + H]+.
Стадия 5: Синтез (4-фторфенил)(2-(пиперазин-1-ил)пиримидин-5-ил)метанона:
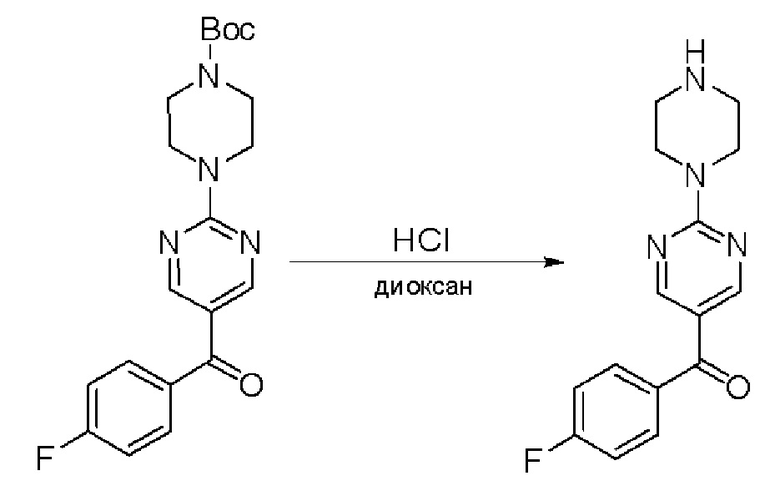
К раствору трет-бутил-4-(5-(4-фторбензоил)пиримидин-2-ил)пиперазин-1-карбоксилата (8,2 г, 21,24 ммоль) в диоксане (50 мл) добавили HCl в диоксане (4 M, 20 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Смесь концентрировали с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (5,5 г, 90 %). МС (ЭС+) C15H15FN4O рассчитано: 286, найдено: 287 [M + H]+.
Стадия 6: Синтез 1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)пропан-1-олм:
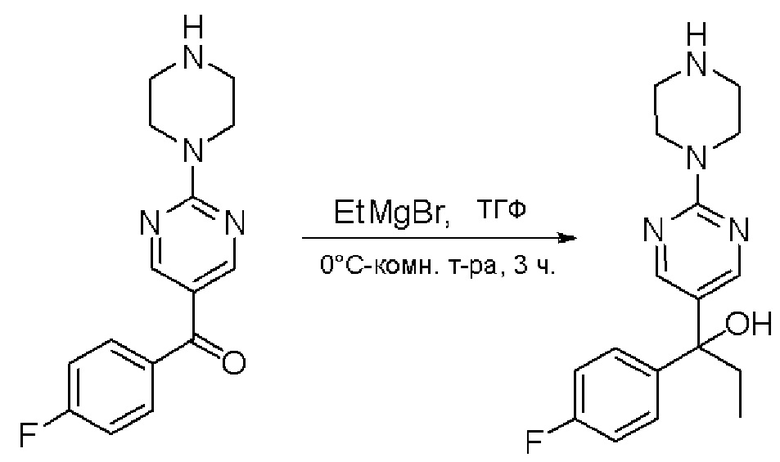
К раствору (4-фторфенил)(2-(пиперазин-1-ил)пиримидин-5-ил)метанона (4,0 г, 21,84 ммоль) в сухом ТГФ (150 мл) добавили EtMgBr (1 M в ТГФ, 150 мл) при 0 oC под азотом. Смесь перемешивали при комнатной температуре в течение 3 часов. Затем погасили раствором NH4Cl и экстрагировали этилацетатом (200 * 3 мл). Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили над сульфатом натрия, отфильтровали и концентрировали. Остаток очистили на колонке Combiflash с дихлорметаном:метанолом = 10:1 с получением указанного в заголовке соединения (440 мг, 10 %) в виде желтого твердого вещества. МС (ЭС+) C17H21FN4O рассчитано: 316, найдено: 317 [M + H]+.
Стадия 7: Синтез (E)-5-(1-(4-фторфенил)проп-1-енил)-2-(пиперазин-1-ил)пиримидина:
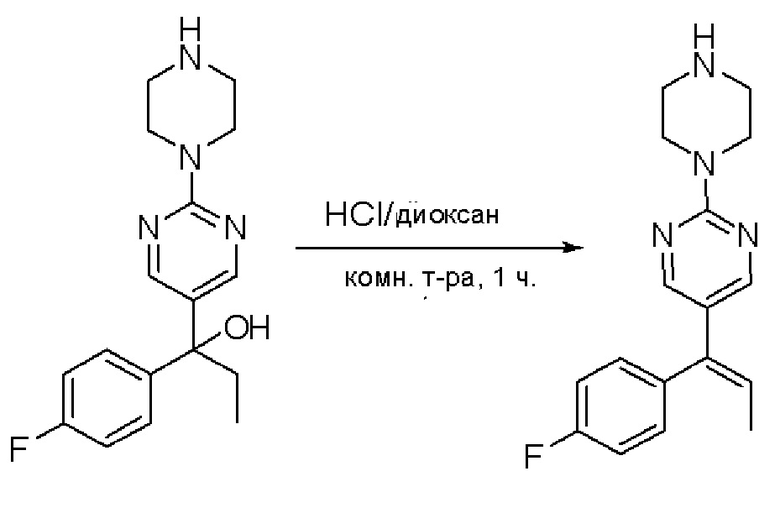
К раствору 1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)пропан-1-ола (200 мг, 0,6 ммоль) в диоксане (10 мл) добавили HCl в диоксане (4 M, 10 мл) и перемешивали реакционную смесь при комнатной температуре в течение 1 часа. ЖХМС показала, что реакция завершена. Смесь концентрировали до маслянистого вещества, которое очистили на колонке Combiflash с дихлорметаном:метанолом = 20:1 с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (185 мг, 98 %). МС (ЭС+) C17H19FN4 рассчитано: 298, найдено: 299 [M + H]+.
Стадия 8: Синтез 5-(1-(4-фторфенил)пропил)-2-(пиперазин-1-ил)пиримидина
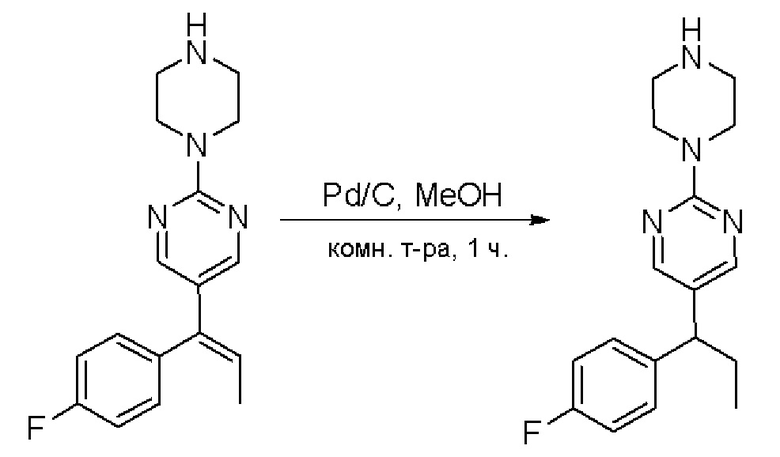
К раствору (E)-5-(1-(4-фторфенил)проп-1-енил)-2-(пиперазин-1-ил)пиримидина (170 мг, 0,57 ммоль) в метаноле (10 мл) добавили Pd/C (30 мг). Смесь обрабатывали водородом под давлением 1 атм. (из баллона) и перемешивали при комнатной температуре в течение 1 часа. Смесь отфильтровали, а фильтрат концентрировали до маслянистого вещества, которое очистили на колонке Combiflash с дихлорметаном:метанолом = 50:1 с получением указанного в заголовке соединения (рацемат, 90 мг, 53 %) в виде желтого маслянистого вещества.
Стадия 9: Хиральное разделение (R)-5-(1-(4-фторфенил)пропил)-2-(пиперазин-1-ил)пиримидина и (S)-5-(1-(4-фторфенил)пропил)-2-(пиперазин-1-ил)пиримидина:
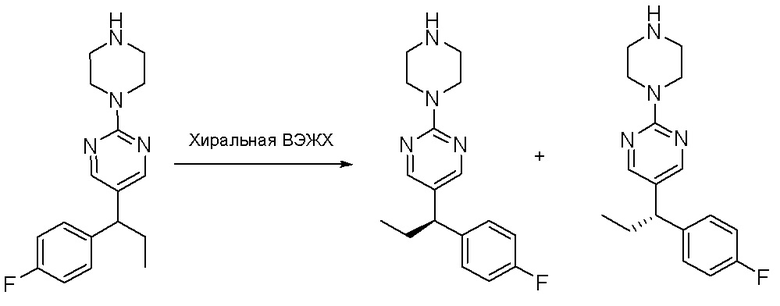
Полученное выше рацемическое соединение (90 мг) разделили с помощью хиральной ВЭЖХ с получением энантиомеров (35 мг). МС (ЭС+) C17H21FN4 рассчитано: 300, найдено: 301 [M + H]+. Абсолютную стереохимию назначили произвольно.
Условия хирального разделения: Хиральная колонка: OJ-H (250*4,6 мм, 5 мкм)
Подвижная фаза: н-гексан (0,1 % DEA):EtOH(0,1 % DEA)=95:5
Стадия 10a: Синтез (R)-4-(4-(5-(1-(4-фторфенил)пропил)пиримидин-2-ил)пиперазин-1-ил)-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина:
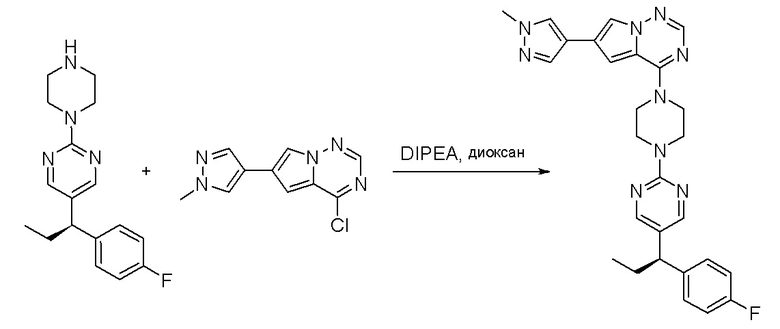
Раствор (R)-5-(1-(4-фторфенил)пропил)-2-(пиперазин-1-ил)пиримидина (36 мг, 0,12 ммоль), 4-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина (31 мг, 0,132 ммоль) и диизопропилэтиламина (47 мг, 0,36 ммоль) в 1,4-диоксане (5 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали, а остаток очистили препаративной ВЭЖХ с получением указанного в заголовке соединения (21,1 мг, 35 %) в виде белого твердого вещества. МС (ЭС+) C26H26FN9O рассчитано: 497, найдено: 498 [M + H]+.
Стадия 10b: Синтез (S)-4-(4-(5-(1-(4-фторфенил)пропил)пиримидин-2-ил)пиперазин-1-ил)-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина:
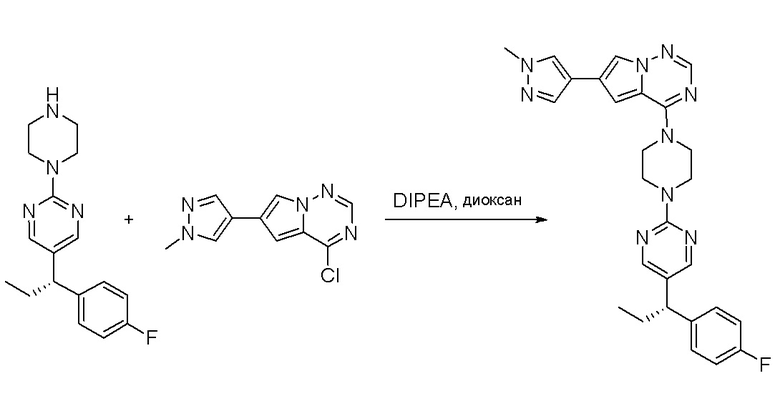
Смесь (S)-5-(1-(4-фторфенил)пропил)-2-(пиперазин-1-ил)пиримидина (35 мг, 0,12 ммоль), 4-хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[1,2-f][1,2,4]триазина (30 мг, 0,132 ммоль) и диизопропилэтиламина (47 мг, 0,36 ммоль) в 1,4-диоксане (5 мл) перемешивали при комнатной температуре в течение 3 часов. Реакционную смесь концентрировали, а остаток очистили препаративной ВЭЖХ с получением указанного в заголовке соединения (24,4 мг, 35 %) в виде белого твердого вещества. МС (ЭС+) C26H26FN9O рассчитано: 497, найдено: 498 [M + H]+.
Протокол синтеза 4
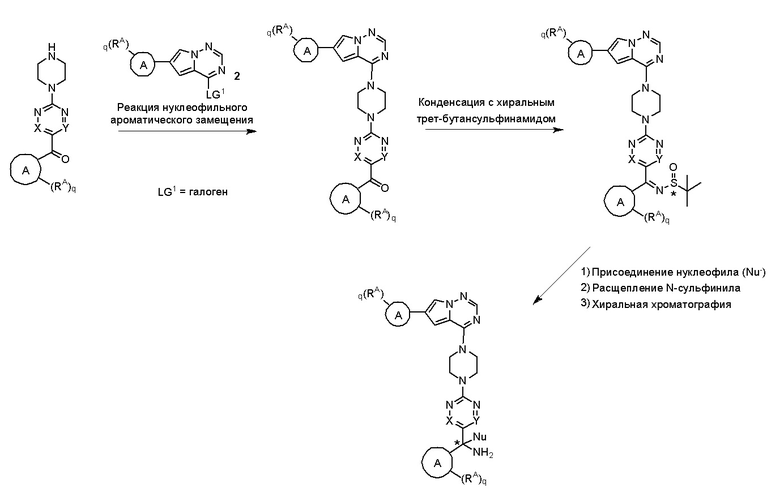
Пиперазин, изображенный выше, может быть получен по таким же методикам синтеза, как показаны в Протоколе синтеза 3. Пирролотриазин может быть замещен амином пиперазина в условиях реакции нуклеофильного ароматического замещения с использованием аминного основания, такого как диизопропилэтиламин (DIPEA) или триэтиламин (TEA), в полярном растворителе, таком как диоксан, с получением пиперазин-замещенного пирролотриазина. Непосредственная конденсация хирального трет-бутансульфинамида с кетоном пиперазин-замещенного пирролотриазина может приводить к получению хирального N-сульфинилимина. 1,2-Присоединение нуклеофила, такого как органометаллический реагент, например, алкил Гриньяра, или, например, енолята к N-сульфинилимину с последующим расщеплением N-сульфинильной группы, например, в кислотных условиях, может приводить к получению хирально обогащенного амина. Хирально чистый амин может быть получен хиральной хроматографией, например, СЖХ или ВЭЖХ. Соединения, полученные по Протоколу синтеза 4, разделяли хиральной СЖХ, используя следующие условия разделения:
Колонка: ChiralPak AS-H 20 x 250 мм
Подвижная фаза: 45 % этанол, содержащий 0,25 % DEA в CO2
Скорость потока: 70 мл/мин.
Образец: 93,7 мг рацемической смеси растворяли в 15 мл растворителя, состоящего из метанола/этанола = 1/1, содержащего 150 мкл диэтиламина
Объем вводимой пробы: 2 мл на одно хроматографирование
Обнаружение: 254 нм
Как показано ниже, Соединения 12, 13, 36, 43 и 44 были получены по Протоколу синтеза 4.
Пример 5: Синтез (S)-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанамина и (R)-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанамина (Соединения 12 и 13)
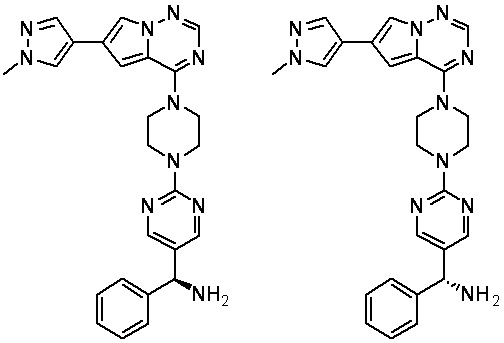
Стадия 1: Синтез (2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанимина:
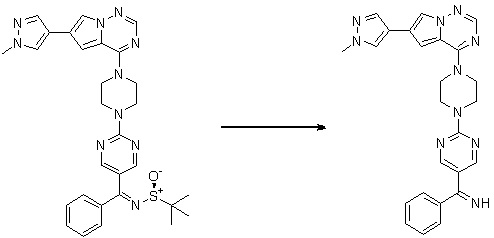
(S,Z)-2-Метил-N-((2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метилен)пропан-2-сульфинамид (490 мг, 0,862 ммоль) перемешивали в 4 M HCl в 1,4-диоксане (2 мл)/MeOH (2 мл) при комнатной температуре в течение 1 часа. Растворитель удалили in vacuo и растерли остаток в EtOAc с получением (2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанимина, HCl (490 мг, 0,861 ммоль, выход 100 % yield) в виде бледно-желтого твердого вещества, 88 % по массе.
МС (ЭС+) C25H24N10 рассчитано: 464, найдено: 465 [M + H]+.
Стадия 2: Синтез rac-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанамина:
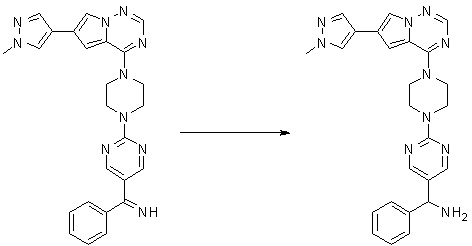
(2-(4-(6-(1-Метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанимин, HCl (410 мг, 0,818 ммоль) суспендировали в MeOH (8 мл). Одной порцией добавили борогидрид натрия (40 мг, 1,057 ммоль), в результате чего выделилось тепло, и образовался прозрачный раствор. Одной порцией добавили дополнительное количество борогидрида натрия (40 мг, 1,057 ммоль), в результате чего выделилось тепло, и образовалась суспензия. MeOH удалили in vacuo и разделили остаток между EtOAc-NaHCO3. Водную фазу второй раз экстрагировали EtOAc. Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo. Остаток перекристаллизовали из EtOH с получением (2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метнамина (182 мг, 0,390 ммоль, выход 47,7 %) в виде грязновато-белого твердого вещества.
МС (ЭС+) C25H26N10 рассчитано: 466, найдено: 467 [M + H]+.
Стадия 3: Разделение энантиомеров
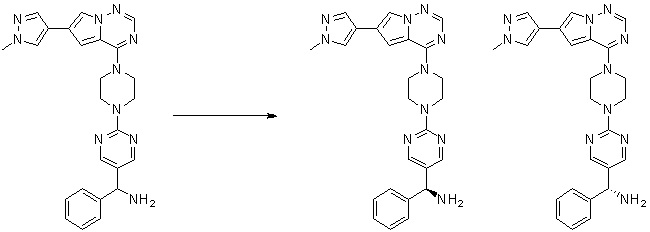
Энантиомеры рацемического (2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанамина (185 мг, 0,397 ммоль) разделили хиральной СЖХ с получением (S)-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанамина (74 мг, 0,159 ммоль, выход 80,0 %) и (R)-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанамина (94 мг, 0,201 ммоль, выход 100 %). Абсолютную стереохимию назначили произвольно.
МС (ЭС+) C25H26N10 рассчитано: 466, найдено: 467 [M + H]+.
Пример 6: Синтез (S)-N,N-диметил-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)-1-фенилметанамина (Соединение 36):
(S)-(2-(4-(6-(1-Метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)(фенил)метанамин (72 мг, 0,154 ммоль) и формальдегид (125 мг, 1,543 ммоль) растворили в MeCN (1,5 мл). Добавили цианоборгидрид натрия (25 мг, 0,398 ммоль), затем уксусную кислоту (0,02 мл, 0,349 ммоль) и перемешивали полученную смесь при комнатной температуре в течение 3 часов. Добавили насыщенный раствор NaHCO3 и экстрагировали продукты в ДХМ (x2). Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo. В результате очистки остатка с помощью ЖХСД (0-10 % MeOH-ДХМ) с последующей ЖХСД (0-10 % MeOH-EtOAc), с последующей ЖХСД (0-8 % MeOH-EtOAc) получили (S)-N,N-диметил-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)-1-фенилметанамин (15 мг, 0,030 ммоль, выход 19,65 %).
МС (ЭС+) C27H30N10 рассчитано: 494, найдено: 495 [M + H]+.
Пример 7: Синтез (R)-1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина и (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (Соединения 43 и 44)
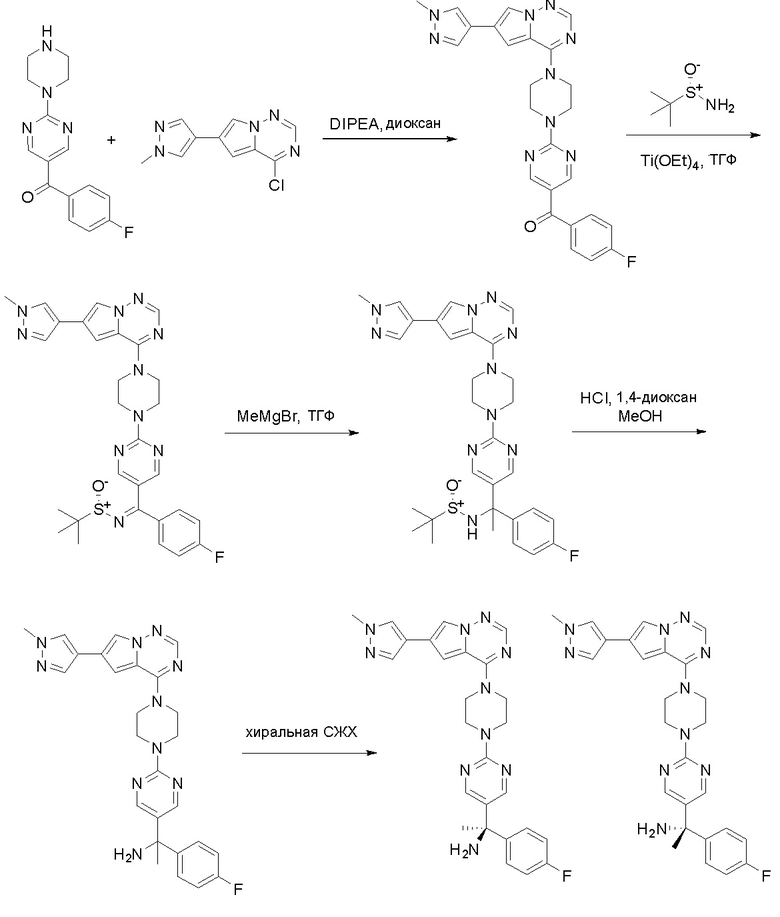
Стадия 1: Синтез (4-фторфенил)(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)метанона:
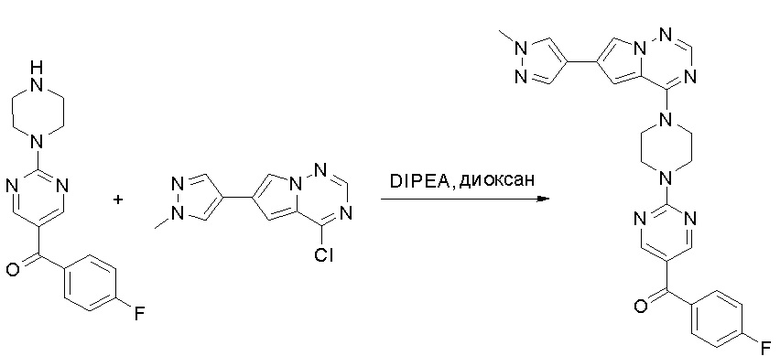
4-Хлор-6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин (180 мг, 0,770 ммоль), (4-фторфенил)(2-(пиперазин-1-ил)пиримидин-5-ил)метанон, HCl (265 мг, 0,821 ммоль) и DIPEA (0,40 мл, 2,290 ммоль) перемешивали в 1,4-диоксане (4 мл) при комнатной температуре в течение 18 часов. Добавили насыщенный раствор хлорида аммония и экстрагировали продукты в ДХМ (x2). Объединенные органические экстракты высушили над Na2SO4, отфильтровали через целит, элюируя ДХМ, и концентрировали фильтрат in vacuo. В результате очистки остатка с помощью ЖХСД (25-100 % EtOAc-ДХМ) получили (4-фторфенил)(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)метанон (160 мг, 0,331 ммоль, выход 43 %) в виде грязновато-белого твердого вещества. МС (ЭС+) C25H22FN9O рассчитано: 483, найдено: 484 [M + H]+.
Стадия 2: Синтез (S,Z)-N-((4-фторфенил)(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)метилен)-2-метилпропан-2-сульфинамида:
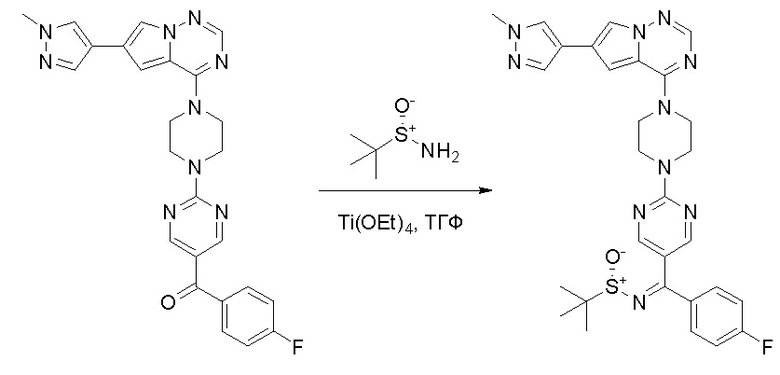
(S)-2-Метилпропан-2-сульфинамид (110 мг, 0,908 ммоль), (4-фторфенил)(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)метанон (158 мг, 0,327 ммоль) и этилортотитанат (0,15 мл, 0,715 ммоль) перемешивали в ТГФ (3,2 мл) при 70 °C в течение 18 часов. Охладили до комнатной температуры, добавили воду и экстрагировали продукты в EtOAc (x2). Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo на целите. В результате очистки остатка с помощью ЖХСД (0-10 % MeOH-EtOAc) получили (S,Z)-N-((4-фторфенил)(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)метилен)-2-метилпропан-2-сульфинамид (192 мг, 0,327 ммоль, выход 100 %) в виде оранжевого твердого вещества. МС (ЭС+) C29H31FN10OS рассчитано: 586, найдено: 587 [M + H]+.
Стадия 3: Синтез (S)-N-(1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этил)-2-метилпропан-2-сульфинамида:
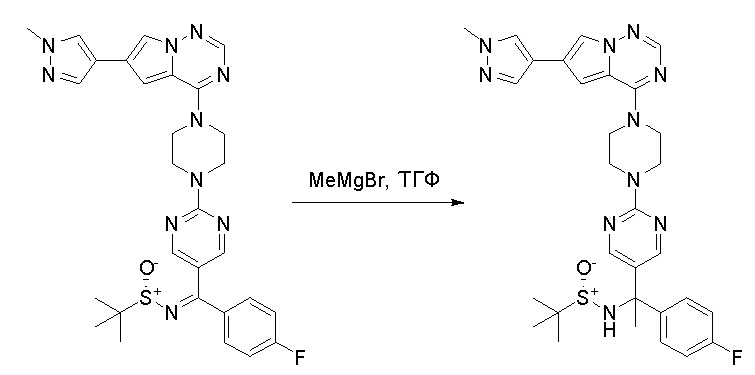
(S,Z)-N-((4-Фторфенил)(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)метилен)-2-метилпропан-2-сульфинамид (190 мг, 0,324 ммоль) растворили в ТГФ (3 мл) и охладили до 0 °C. Добавили метилмагнийбромид (3 M раствор в диэтиловом эфире, 0,50 мл, 1,500 ммоль) и перемешивали полученную смесь при 0 °С в течение 45 минут. Добавили дополнительное количество метилмагнийбромида (3 M раствор в диэтиловом эфире, 0,10 мл, 0,300 ммоль) и продолжали перемешивание при 0 °С в течение 20 минут. Добавили насыщенный раствор хлорида аммония и экстрагировали продукты в EtOAc (x2). Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo на целите. В результате очистки остатка с помощью ЖХСД (0-10 % MeOH-EtOAc) получили (S)-N-(1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этил)-2-метилпропан-2-сульфинамид (120 мг, 0,199 ммоль, выход 61,5 %) в виде желтого твердого вещества (смесь диастереоизомеров). МС (ЭС+) C30H35FN10OS рассчитано: 602, найдено: 603 [M + H]+.
Стадия 4: Синтез 1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина:
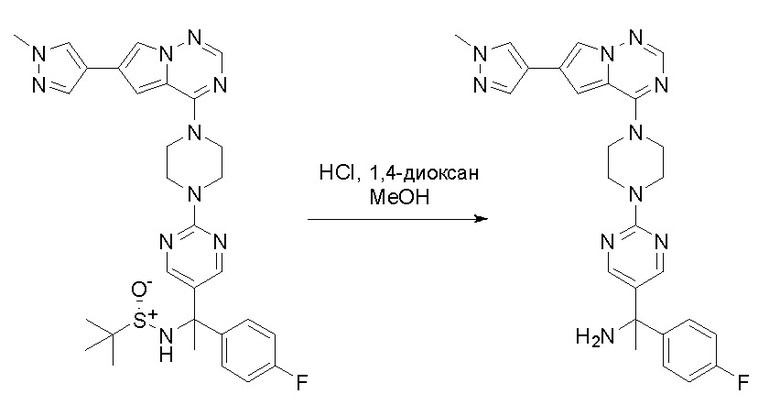
(S)-N-(1-(4-Фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этил)-2-метилпропан-2-сульфинамид (120 мг, 0,199 ммоль) перемешивали в 4 M HCl в 1,4-диоксане (1,5 мл)/MeOH (1,5 мл) при комнатной температуре в течение 1 часа. Растворитель удалили in vacuo, а остаток растерли в EtOAc с получением 1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина, HCl (110 мг, 0,206 ммоль, выход 103 %) в виде бледно-желтого твердого вещества. МС (ЭС+) C26H27FN10 рассчитано: 498, найдено: 482 [M-17 + H]+, 499 [M + H]+.
Стадия 5: Хиральное разделение (R)-1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина и (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина:
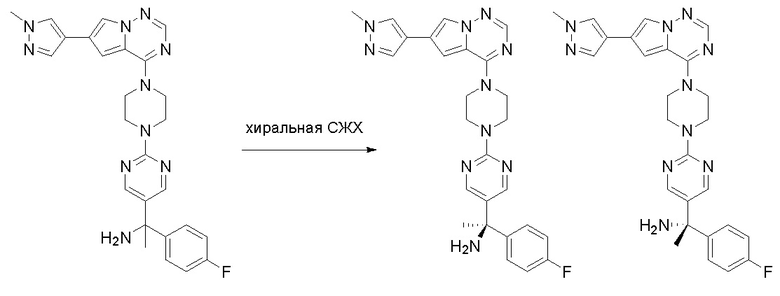
Энантиомеры рацемического 1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (94 мг, 0,189 ммоль) разделили хиральной СЖХ с получением (R)-1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (34,4 мг, 0,069 ммоль, выход 73,2 %) и (S)-1-(4-фторфенил)-1-(2-(4-(6-(1-метил-1H-пиразол-4-ил)пирроло[2,1-f][1,2,4]триазин-4-ил)пиперазин-1-ил)пиримидин-5-ил)этанамина (32,1 мг, 0,064 ммоль, выход 68,3 %). Абсолютную стереохимию назначили произвольно. МС (ЭС+) C26H27FN10 рассчитано: 498, найдено: 499 [M + H]+.
Получение общих промежуточных соединений
Синтез 5-(2-фенилпропан-2-ил)-2-(пиперазин-1-ил)пиримидина:
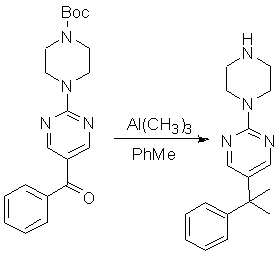
В герметичной пробирке перемешивали смесь трет-бутил-4-(5-бензоилпиримидин-2-ил)пиперазин-1-карбоксилата (500 мг, 1,36 ммоль) и триметилалюминия (2 M в толуоле, 2,7 мл) в сухом толуоле (10 мл) при 100 0°С в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь охладили до комнатной температуры, погасили ледяной водой и экстрагировали этилацетатом. Органический слой промыли водой и насыщенным солевым раствором, высушили над сульфатом натрия, отфильтровали и концентрировали. Остаток очистили препаративной ВЭЖХ с получением 5-(2-фенилпропан-2-ил)-2-(пиперазин-1-ил)пиримидина (40 мг, 7 %) в виде желтоватого твердого вещества. МС (ЭС+) C17H22N4 рассчитано: 282, найдено: 283 [M + H]+.
Синтез трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилата
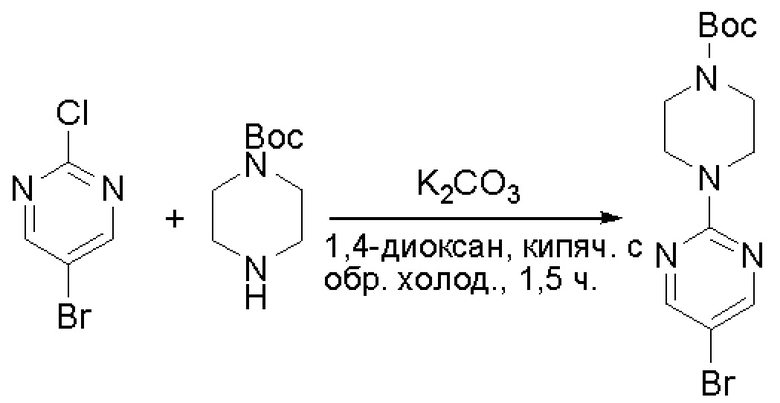
К раствору 5-бром-2-хлорпиримидина (50,0 г, 258 ммоль) и 1-трет-бутоксикарбонилпиперазина (72,2 г, 387 ммоль) в 1,4-диоксане (500 мл) добавили карбонат калия (67,8 г, 491 ммоль) и перемешивали смесь при кипячении с обратным холодильником в течение 1,5 часов. Реакционную смесь охладили до комнатной температуры, погасили водой (500 мл) и экстрагировали диэтиловым эфиром (1000 мл * 2). Объединенные органические слои высушили над сульфатом натрия, отфильтровали и концентрировали. Остаток очистили колоночной хроматографией на силикагеле (петролейный эфир:этилацетат = 8:1-4:1) с получением указанного в заголовке соединения (70,5 г, 80 %) в виде белого твердого вещества. МС (ЭС+) C13H19BrN4O2 рассчитано: 342, найдено: 243 [M + H - 100]+.
Синтез трет-бутил-4-(5-ацетилпиримидин-2-ил)пиперазин-1-карбоксилата
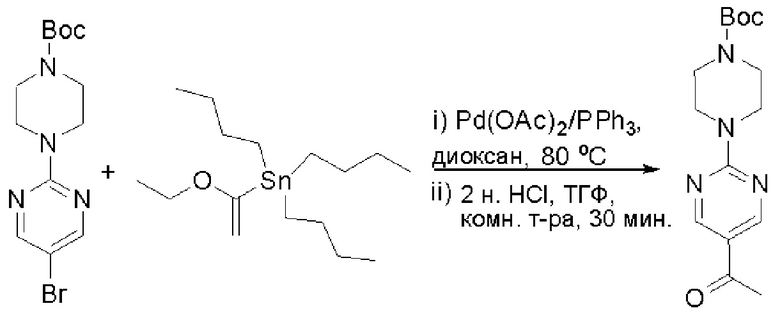
Смесь трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилата (5,0 г, 14,6 ммоль), диацетата палладия (240 мг, 1,46 ммоль), трифенилфосфина (376 мг, 2,92 ммоль) и трибутил(1-этоксивинил)станнана (5,3 мл, 16,1 мл) в диоксане (100 мл) дегазировали азотом три раза и перемешивали реакционную смесь при 80 °С в течение ночи. Реакционную смесь охладили до комнатной температуры и разбавили ТГФ (100 мл), затем добавили 2 н. HCl (100 мл). Смесь перемешивали при комнатной температуре в течение 30 минут, и ЖХМС показала, что реакция завершена. Реакционную смесь разбавили этилацетатом (200 мл). Органическую фазу отделили, промыли водой (3 × 100 мл), высушили над сульфатом натрия, отфильтровали и концентрировали. Остаток очистили хроматографией на силикагеле с получением указанного в заголовке соединения (3,0 г, 67 %). МС (ЭС+) C15H22N4O3 рассчитано: 306, найдено: 251 [M – 56 + H]+.
Синтез 1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанона
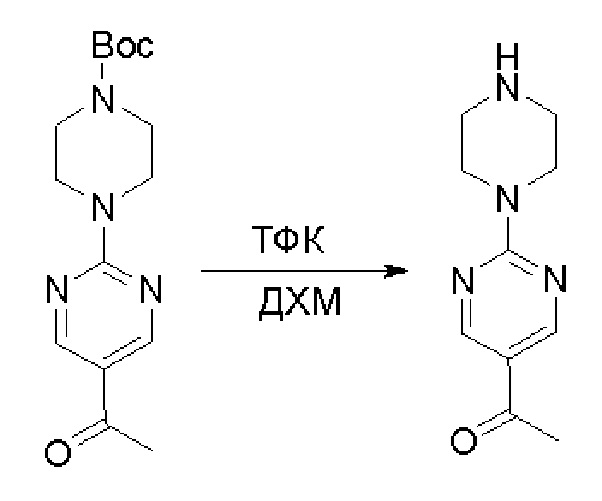
К раствору трет-бутил-4-(5-ацетилпиримидин-2-ил)пиперазин-1-карбоксилата (3 г, 9,8 ммоль) в дихлорметане (30 мл) добавили трифторэтилацетат (15 мл) и перемешивали смесь при комнатной температуре в течение 30 минут. ЖХМС показала, что реакция завершена. Реакционную смесь нейтрализовали раствором карбоната натрия и экстрагировали дихлорметаном. Органический слой высушили над сульфатом натрия, отфильтровали и концентрировали с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (2 г, 100 %), которое напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C10H14N4O рассчитано: 206, найдено: 207 [M + H]+.
Синтез 1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола
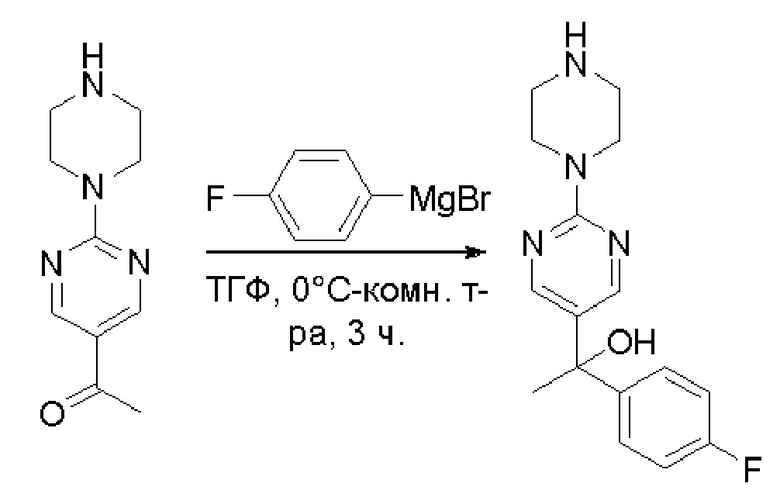
К раствору 1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанона (1,8 г, 8,73 ммоль) в сухом ТГФ (100 мл) добавили (4-фторфенил)магнийбромид (1 M в ТГФ, 87,3 мл) при 0 oC под N2. Смесь перемешивали при комнатной температуре в течение 3 часов. Затем погасили раствором хлорида аммония и экстрагировали дихлорметаном (300 мл). Органический слой высушили над сульфатом натрия, отфильтровали и концентрировали. Остаток очистили на колонке Combi-flash (дихлорметан:метанол = 10:1) с получением указанного в заголовке соединения (1,02 г, 38 %) в виде желтого твердого вещества.
Хиральное разделение 1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола:
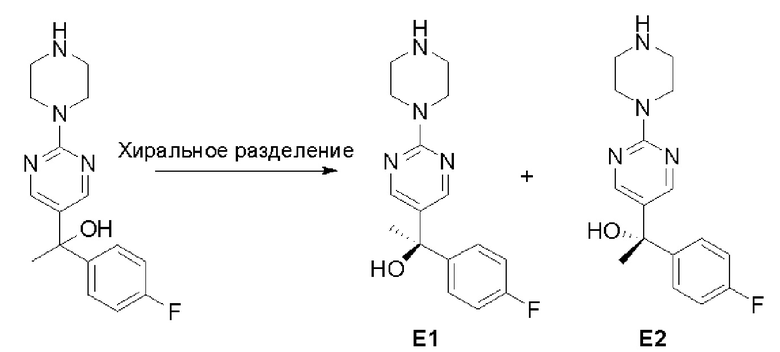
Рацемическое соединение (1,02 г) разделили хиральной ВЭЖХ с получением энантиомера 1 (E1, 320 мг) и энантиомера 2 (E2, 220 мг). МС (ЭС+) C16H19FN4O рассчитано: 302, найдено: 303 [M + H]+. Абсолютную конфигурацию назначили произвольно.
Условия хирального разделения: Хиральная колонка: OZ-H (4,6*250 мм, 5 мкм); Подвижная фаза: сорастворитель EtOH (0,1 % DEA)
Синтез (S)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола и (R)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола:
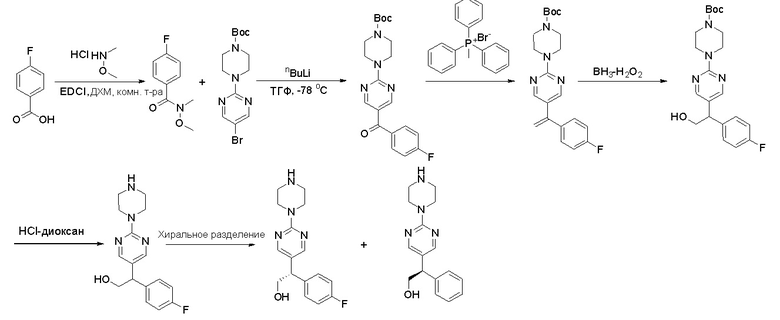
Стадия 1: Синтез 4-фтор-N-метокси-N-метилбензамида:
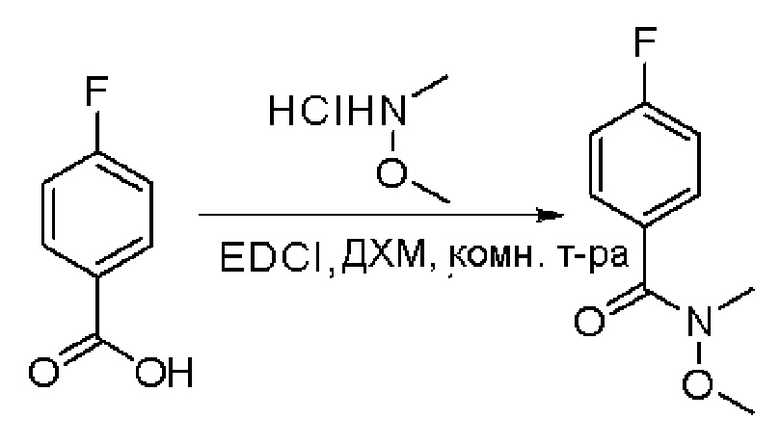
К раствору 4-фторбензойной кислоты (200 г, 1,43 моль), N,O-диметилгидроксиламина гидрохлорида (207 г, 2,14 моль) и EDCI (407 г, 2,14 моль) в дихлорметане (2 л) добавили диизопропилэтиламин (553 г, 4,28 моль) при 0 oC и перемешивали смесь при комнатной температуре в течение ночи. Затем реакционную смесь последовательно промыли водным раствором HCl (1 н., 1 л*4), водой (1 л) и насыщенным солевым раствором (1 л). Органический слой высушили над MgSO4, отфильтровали и концентрировали in vacuo с получением указанного в заголовке соединения (150 г, выход 57 %). МС (ЭС+) C9H10FNO2 рассчитано: 183, найдено 184 [M+H]+; чистота: 90 % (УФ254).
Стадия 2: Синтез трет-бутил-4-(5-(4-фторбензоил)пиримидин-2-ил)пиперазин-1-карбоксилата:
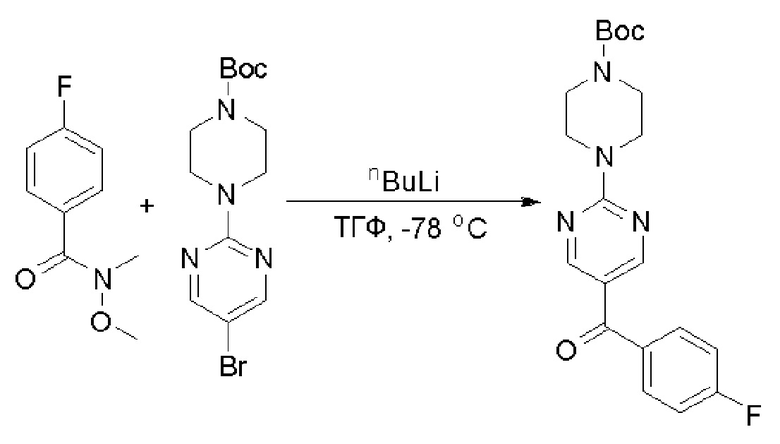
К раствору трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилата (50 г, 146,2 ммоль) в безводном ТГФ (700 мл) по каплям добавили n-BuLi (2,5 M в гексане, 70 мл, 175 ммоль) при -78 °C под азотом. Смесь перемешивали при -78 °C в течение 1 часа, затем добавили раствор 4-фтор-N-метокси-N-метилбензамида (30 г, 163,9 ммоль) в безводном ТГФ (100 мл). После перемешивания при -78 °С в течение еще 2 часов реакционную смесь погасили насыщенным водным раствором NH4Cl (250 мл) и экстрагировали этилацетатом (200 мл*3). Объединенные органические слои высушили над сульфатом натрия, отфильтровали и концентрировали in vacuo. Остаток разбавили пропан-2-олом (150 мл) и перемешивали при комнатной температуре в течение 30 минут. Твердые вещества собрали фильтрацией, промыли пропан-2-олом (100 мл) и петролейным эфиром (300 мл) и высушили под вакуумом с получением указанного в заголовке соединения (26 г, выход 46 %) в виде желтого твердого вещества. МС (ЭС+) C20H23FN4O3 рассчитано: 386, найдено 331 [M-56+H]+; чистота: 100 % (УФ214).
Стадия 3: Синтез трет-бутил-4-(5-(1-(4-фторфенил)винил)пиримидин-2-ил)пиперазин-1-карбоксилата:
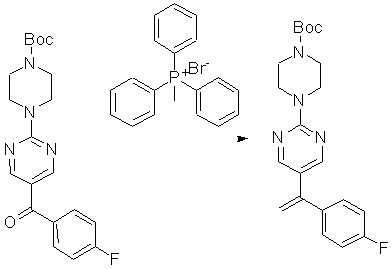
К раствору метилтрифенилфосфония бромида (6,0 г, 16,84 ммоль) в ТГФ (40 мл) при -78 oC по каплям добавили n-BuLi (2,4 M, 7,2 мл, 17,19 ммоль). После перемешивания при -78 oC в течение 1 часа добавили трет-бутил-4-(5-(4-фторбензоил)пиримидин-2-ил)пиперазин-1-карбоксилат (1,3 г, 3,37 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Реакцию погасили водным раствором NH4Cl и экстрагировали EA (2 × 50 мл). Органические фазы промыли H2O (3x30 мл) и насыщенным солевым раствором (50 мл), высушили над сульфатом натрия, отфильтровали, концентрировали и очистили хроматографией на силикагеле (PE:EA = 10:1) с получением указанного в заголовке соединения в виде белого твердого вещества (1,2 г, 93 %). МС (ЭС+) C21H25FN4O2 рассчитано: 384, найдено: 329 [M-56+1]+.
Стадия 4: Синтез трет-бутил-4-(5-(1-(4-фторфенил)-2-гидроксиэтил)пиримидин-2-ил)пиперазин-1-карбоксилата:
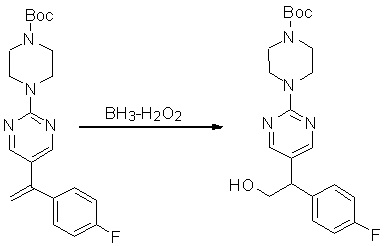
Трет-бутил-4-(5-(1-(4-фторфенил)винил)пиримидин-2-ил)пиперазин-1-карбоксилат (1,2 г, 3,12 ммоль) растворили в ТГФ (30 мл) и затем охладили до 0 oC, затем по каплям добавили BH3.ТГФ (6,24 мл, 6,24 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов. К смеси последовательно добавили H2O в ТГФ (10 %, 8 мл), раствор NaOH (1,25 г) в 30 мл H2O и H2O2 (35 %, 18 г) при 0 oC. Смесь перемешивали при комнатной температуре в течение ночи. Смесь подкислили 1 н. HCl и экстрагировали EA (3×50 мл). Органические фазы промыли водным раствором NaHCO3 (50 мл), насыщенным солевым раствором (50 мл), высушили над сульфатом натрия, отфильтровали, концентрировали и очистили хроматографией на силикагеле (ДХМ:CH3OH = 30:1) с получением указанного в заголовке соединения в виде белого твердого вещества (0,3 г, 24 %). МС (ЭС+) C21H27FN4O3 рассчитано: 402, найдено: 403 [M+H]+.
Стадия 5: Синтез 2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола:
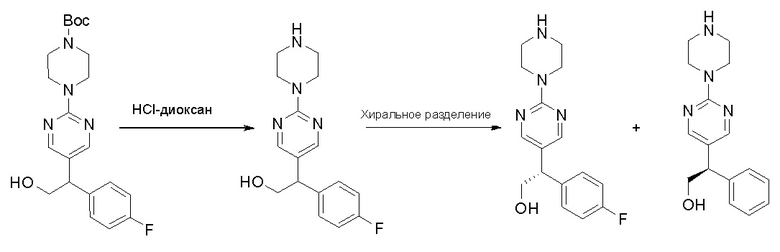
К раствору трет-бутил-4-(5-(1-(4-фторфенил)-2-гидроксиэтил)пиримидин-2-ил)пиперазин-1-карбоксилата (450 мг, 1,08 ммоль) в диоксане (5 мл) добавили HCl/диоксан (5 мл). Смесь перемешивали при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Раствор концентрировали и очистили хроматографией на силикагеле (ДХМ:CH3OH = 10:1) с получением указанного в заголовке соединения в виде желтого твердого вещества (0,2 г, 53 %). МС (ЭС+) C16H19FN4O рассчитано: 302, найдено: 303 [M+H]+.
Полученный выше пример (200 мг, 0,66 ммоль) разделили с помощью хиральной ВЭЖХ с получением обозначенного (S)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола (1ый пик, 50 мг, 25 %) и обозначенного (R)-2-фенил-2-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола (2ой пик, 50 мг, 25 %).
Синтез трет-бутил-4-(5-(4-фторфенокси)пиримидин-2-ил)пиперазин-1-карбоксилата:
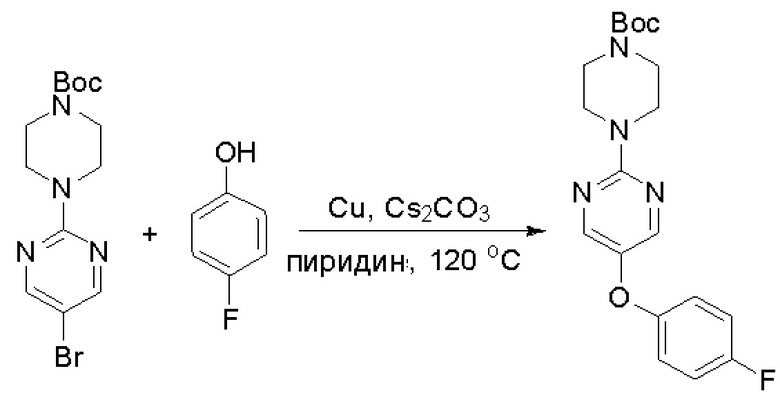
Смесь трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилата (684 мг, 2,0 ммоль), 4-фторфенола (1,1 г, 5,0 ммоль), меди (650 мг, 10,0 ммоль) и Cs2CO3 (6,5 г, 20,0 ммоль) в пиридине (15 мл) нагревали при 120 °С в течение 12 часов. Смесь охладили до комнатной температуры, разбавили этилацетатом (200 мл) и отфильтровали. Фильтрат концентрировали под пониженным давлением, а остаток очистили колоночной хроматографией на силикагеле (петролейный эфир/этилацетат = 20/1) с получением указанного в заголовке соединения (200 мг, 27 %) в виде коричневого твердого вещества. МС (ЭС+) C19H23FN4O3 рассчитано: 374, найдено: 319 [M-56+1]+.
Синтез 5-(4-фторфенилсульфонил)-2-(пиперазин-1-ил)пиримидина HCl соли:
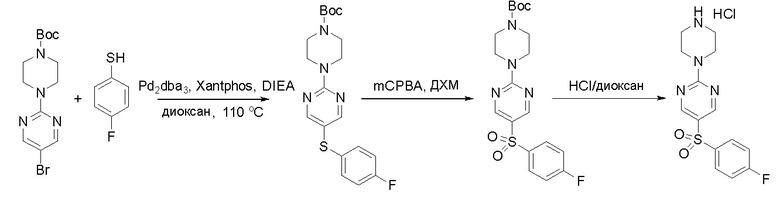
Стадия 1: Синтез трет-бутил-4-(5-(4-фторфенилтио)пиримидин-2-ил)пиперазин-1-карбоксилата:
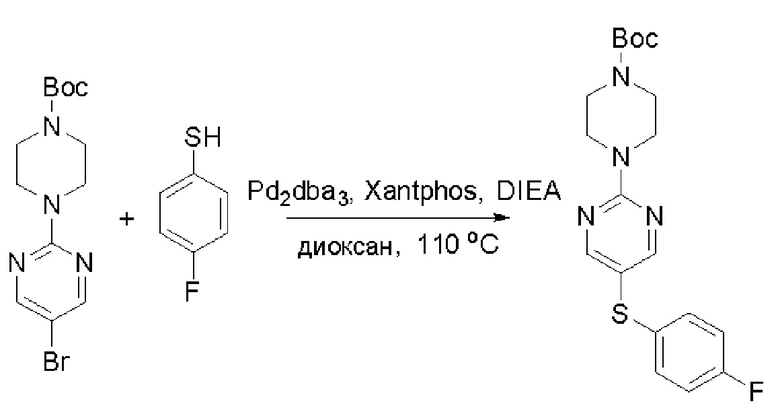
Перемешиваемый раствор трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилата (1 г, 2,924 ммоль), 4-фторбензолтиола (561 мг, 4,386 ммоль), Pd2(dba)3 (267 мг, 0,292 ммоль), Xantphos (169 мг, 0,292 ммоль) и DIPEA (754 мг, 5,848 ммоль) в диоксане (50 мл) дегазировали азотом три раза, а затем нагревали при 110 °С в течение 16 часов. Реакционную смесь охладили до комнатной температуры и концентрировали под пониженным давлением с получением остатка, который очистили флэш-хроматографией (силикагель, 0-20 % EtOAc/PE) с получением указанного в заголовке соединения в виде белого твердого вещества. (500 мг, выход 44 %, чистота: 99 %). МС (ЭС+) C19H23FN4O2S рассчитано: 390, найдено 391 [M+H]+.
Стадия 2: Синтез трет-бутил-4-(5-(4-фторфенилсульфонил)пиримидин-2-ил)пиперазин-1-карбоксилата:
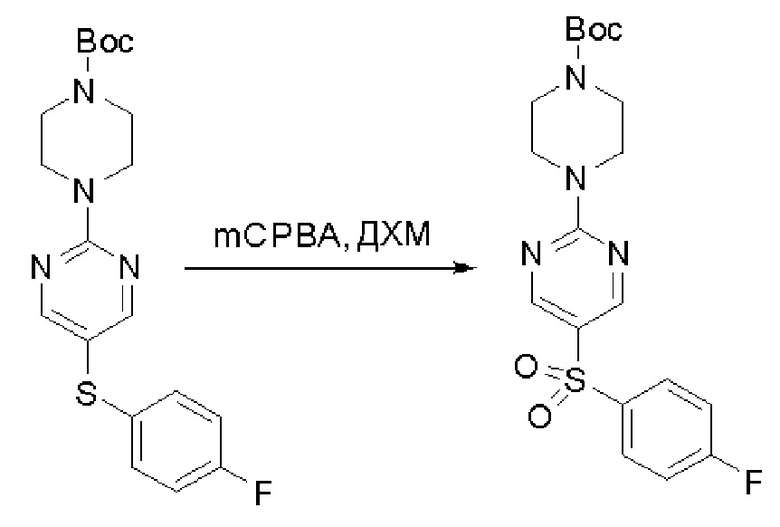
Раствор трет-бутил-4-(5-(4-фторфенилтио)пиримидин-2-ил)пиперазин-1-карбоксилата (700 мг, 1,799 ммоль) и mCPBA (619 мг, 3,599 ммоль) в ДХМ (20 мл) перемешивали при комнатной температуре в течение 16 часов. Реакционную смесь последовательно промыли насыщенным раствором карбоната калия, водой и насыщенным солевым раствором. Органический слой высушили над безводным сульфатом натрия, отфильтровали и концентрировали под вакуумом. Остаток очистили флэш-хроматографией (силикагель, 0-20 % EtOAc/PE) с получением указанного в заголовке соединения в виде белого твердого вещества. (600 мг, выход 83 %, чистота: 100 %). МС (ЭС+) C19H23FN4O4S рассчитано: 422, найдено 423 [M+H]+.
Стадия 3: Синтез 5-(4-фторфенилсульфонил)-2-(пиперазин-1-ил)пиримидина HCl соли:
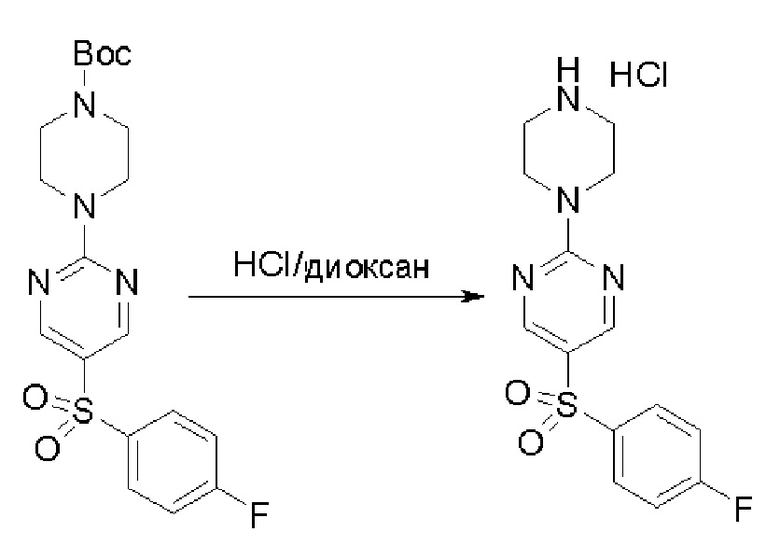
К раствору трет-бутил-4-(5-(4-фторфенилсульфонил)пиримидин-2-ил)пиперазин-1-карбоксилата (100 мг, 0,237 ммоль) в диоксане добавили 4 M HCl/диоксан (6 мл). Смесь перемешивали при комнатной температуре в течение 3 часов и концентрировали с получением указанного в заголовке соединения в виде желтого маслянистого вещества, которое использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C14H15FN4O2S рассчитано: 322, найдено 323 [M+H]+.
Синтез (R)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)пропан-1-ола и (S)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)пропан-1-ола:
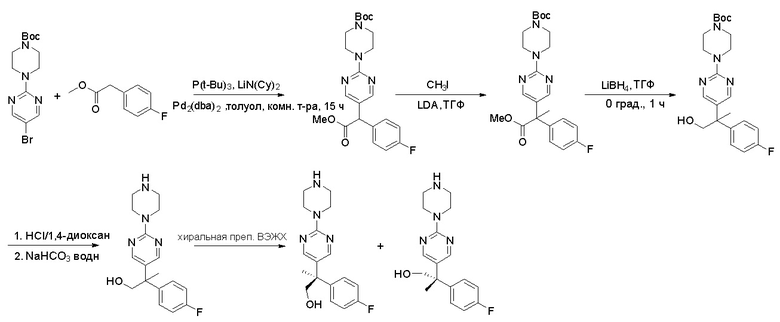
Стадия 1: Синтез трет-бутил-4-(5-(1-(4-фторфенил)-2-метокси-2-оксоэтил)пиримидин-2-ил)пиперазин-1-карбоксилата:
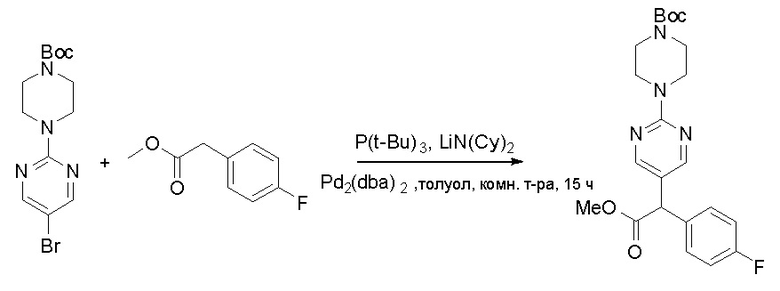
К раствору дициклогексиламина (3,43 г, 18,94 ммоль) в ТГФ (60 мл) при -78 oC по каплям добавили n-BuLi (2,5 M, 7,9 мл, 18,94 ммоль). Смесь перемешивали при комнатной температуре в течение 10 минут, затее мдобавили метил-2-(4-фторфенил)ацетат (2,69 г, 16,02 ммоль) в толуоле (60 мл). После перемешивания при комнатной температуре еще 10 минут последовательно добавили трет-бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилат (5,0 г, 14,57 ммоль), Pd2(dba)3 (667 мг, 0,728 ммоль) и P(t-Bu)3 (10 %, 1,47 г, 0,728 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 15 часов, погасили водой (150 мл) и экстрагировали EA. Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали. Остаток пропустили через колонку (силикагель, PE:EA = 6:1) с получением указанного в заголовке соединения (0,5 г, 8 %) в виде оранжевого твердого вещества. МС (ЭС+) C22H27FN4O4 рассчитано: 430, найдено: 431 [M+H]+.
Стадия 2: Синтез трет-бутил-4-(5-(2-(4-фторфенил)-1-метокси-1-оксопропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата:
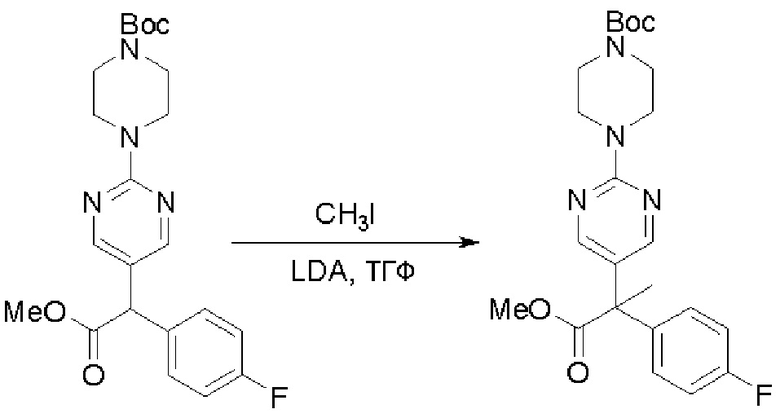
К смеси трет-бутил-4-(5-(1-(4-фторфенил)-2-метокси-2-оксоэтил)пиримидин-2-ил)пиперазин-1-карбоксилата (1,0 г, 2,33 ммоль) в ТГФ (20 мл) при -78 oC по каплям добавили LDA (2 M, 2,33 мл, 4,65 ммоль). После перемешивания при -78 °С в течение 30 минут добавили CH3I (0,66 г, 4,65 ммоль). После перемешивания при комнатной температуре в течение еще 1 часа реакцию погасили водой и экстрагировали EA. Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали. Остаток пропустили через колонку (силикагель, PE:EA = 6:1) с получением указанного в заголовке соединения (0,8 г, 77 %) в виде желтого твердого вещества. МС (ЭС+) C23H29FN4O4 рассчитано: 444, найдено: 445 [M+H]+.
Стадия 3: Синтез трет-бутил-4-(5-(2-(4-фторфенил)-1-гидроксипропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата:
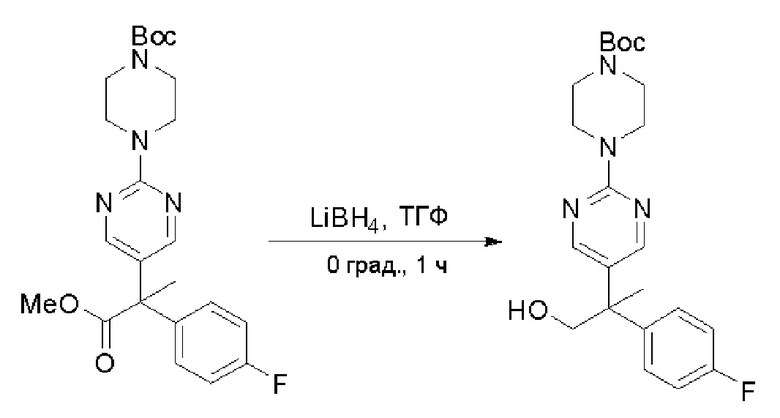
К смеси трет-бутил-4-(5-(2-(4-фторфенил)-1-метокси-1-оксопропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата (0,4 г, 0,9 ммоль) в ТГФ (10 мл) добавили LiBH4 (40 мг, 1,8 ммоль). После перемешивания при комнатной температуре в течение 2 часов реакцию погасили водным раствором NH4Cl и экстрагировали EA. Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали. Остаток пропустили через колонку (силикагель, PE:EA = 1:1) с получением указанного в заголовке соединения (225 мг, 60 %) в виде желтого твердого вещества. МС (ЭС+) C22H29FN4O3 рассчитано: 416, найдено: 417 [M+H]+.
Стадия 4: Синтез (R)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)пропан-1-ола:
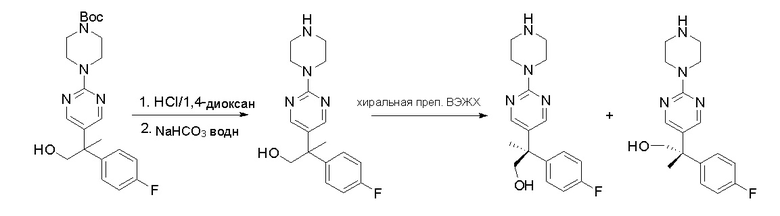
К смеси трет-бутил-4-(5-(2-(4-фторфенил)-1-гидроксипропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата (450 мг, 1,08 ммоль) в ДХМ (20 мл) добавили 4 M HCl/1,4-диоксан (3 мл). После перемешивания при комнатной температуре в течение 15 часов реакционную смесь концентрировали, остаток разбавили водным раствором NaHCO3 (20 мл) и экстрагировали ДХМ. Объединенные органические слои промыли водой и насыщенным солевым раствором, высушили (Na2SO4), отфильтровали и концентрировали. Остаток разделили хиральной препаративной ВЭЖХ с получением (R)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)пропан-1-ола (100 мг, 29 %) в виде белого твердого вещества. МС (ЭС+) C17H21FN4O рассчитано: 316, найдено: 317 [M+H]+. Хиральная ВЭЖХ, колонка: IC 4,6*150 мм, 5 мкм, сорастворитель: EtOH:гексан = 1:1 (0,1 % DEA), время удерживания = 4,22 мин.;
(S)-2-(4-фторфенил)-2-(2-(пиперазин-1-ил)пиримидин-5-ил)пропан-1-ола (100 мг, 29 %) в виде белого твердого вещества. МС (ЭС+) C17H21FN4O рассчитано: 316, найдено: 317 [M+H]+. Хиральная ВЭЖХ, колонка: IC 4,6*150 мм, 5 мкм, сорастворитель: EtOH:гексан = 1:1 (0,1 % DEA), время удерживания = 5,43 мин.
Синтез 5-(3-(4-фторфенил)оксетан-3-ил)-2-(пиперазин-1-ил)пиримидина
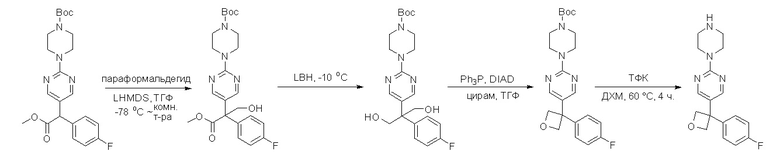
Стадия 1: Синтез трет-бутил-4-(5-(2-(4-фторфенил)-3-гидрокси-1-метокси-1-оксопропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата:
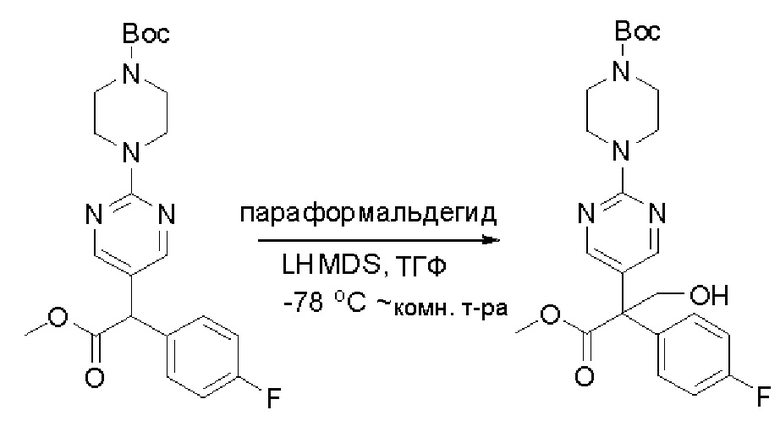
Раствор трет-бутил-4-(5-(1-(4-фторфенил)-2-метокси-2-оксоэтил)пиримидин-2-ил)пиперазин-1-карбоксилата (650 мг, 1,5 ммоль) в ТГФ (20 мл, сухой) охладили до -78 °C и закрыли газообразным N2. К полученному выше, охлажденному раствору за 5 минут добавили другой раствор n-BuLi (1,5 M, 3 мл, ~4,5 ммоль) в ТГФ. Реакционную смесь перемешивали при -78 °С в течение 1 часа, затем одной порцией добавили параформальдегид (405 мг, 15,0 ммоль) при -78 oC. Полученный раствор перемешивали при комнатной температуре в течение ночи. Реакцию погасили насыщенным водным раствором NH4Cl (50 мл) и водой (50 мл) и экстрагировали EtOAc (50 мл*4). Объединенные органические слои промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали под пониженным давлением. Остаток очистили на силикагелевой колонке, элюируя PE:EA (4:1), с получением указанного в заголовке соединения в виде желтого густого маслянистого вещества (300 мг, выход 44 %). МС (ЭС+) C23H29FN4O5 рассчитано: 460, найдено 461 [M+H]+; чистота: 93 % (УФ214).
Стадия 2: Синтез трет-бутил-4-(5-(2-(4-фторфенил)-1,3-дигидроксипропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата:
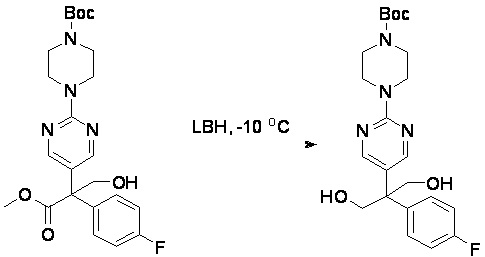
Раствор 4-(5-(2-(4-фторфенил)-3-гидрокси-1-метокси-1-oxoпропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата (700 мг, 1,5 ммоль) в 20 мл ТГФ охладили до -10 oC, затем медленно добавили LiBH4 (180 мг, 7,5 ммоль). Полученную смесь оставили нагреваться до комнатной температуры и перемешивали в течение ночи. Реакцию погасили MeOH (3 мл) и затем концентрировали in vacuo. Остаток очистили на силикагелевой колонке (ДХМ:MeOH, 10:1) с получением требуемого продукта (300 мг, выход 47 %) в виде желтого пенистого вещества. МС (ЭС+) C22H29FN4O4 рассчитано: 432, найдено 433 [M+H]+; чистота: 67 % (УФ254).
Стадия 3: Синтез трет-бутил-4-(5-(3-(4-фторфенил)оксетан-3-ил)пиримидин-2-ил)пиперазин-1-карбоксилата:
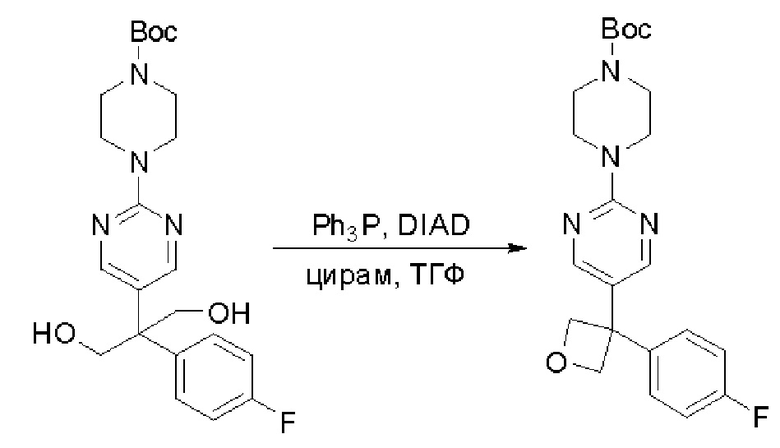
Смесь трет-бутил-4-(5-(2-(4-фторфенил)-1,3-дигидроксипропан-2-ил)пиримидин-2-ил)пиперазин-1-карбоксилата (650 мг, 1,5 ммоль), трифенилфосфина (470 мг, 1,8 ммоль), диизопропилазодикарбоксилата (360 мг, 1,8 ммоль) и цирама (500 мг, 1,8 ммоль) в ТГФ (50 мл) нагревали при 40 oC в течение ночи под N2 и концентрировали. Остаток разбавили EtOAc (40 мл), промыли водой (50 мл*2) и насыщенным солевым раствором, высушили над Na2SO4 и концентрировали. Остаток очистили препаративной ВЭЖХ с получением требуемого продукта (35 мг, выход 6 %) в виде белого твердого вещества. МС (ЭС+) C22H27FN4O3 рассчитано: 414, найдено 359 [M +H- 56]+; чистота: 93 % (УФ214).
Стадия 4: Синтез 5-(3-(4-фторфенил)оксетан-3-ил)-2-(пиперазин-1-ил)пиримидина:
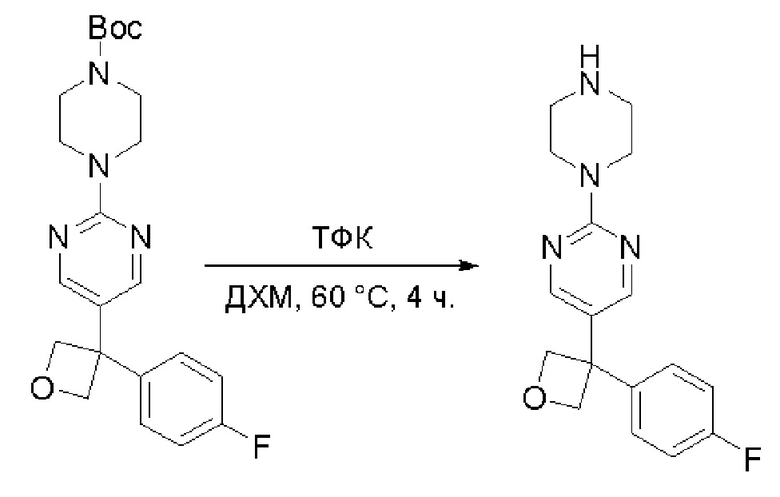
Раствор трет-бутил-4-(5-(3-(4-фторфенил)оксетан-3-ил)пиримидин-2-ил)пиперазин-1-карбоксилата (22 мг, 0,05 ммоль) в ДХМ (1 мл) обрабатывали ТФК (0,5 мл) при 60 oC в течение 3 часов, а затем концентрировали под пониженным давлением. Остаток (30 мг, неочищенный, твердое желтое вещество, выход 100 %) напрямую использовали на следующей стадии без дополнительной очистки. МС (ЭС+) C17H19FN4O рассчитано: 314, найдено 315 [M+H]+; чистота: 87 % (УФ254).
Синтез (S)-1-(5-фторпиридин-2-ил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола и (R)-1-(5-фторпиридин-2-ил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола:
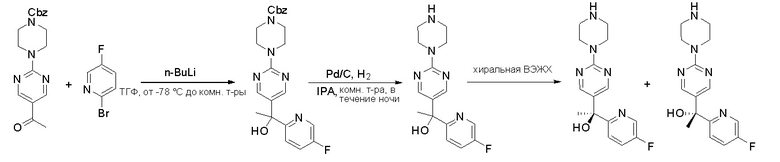
Стадия 1: Синтез бензил-4-(5-(1-(5-фторпиридин-2-ил)-1-гидроксиэтил)пиримидин-2-ил)пиперазин-1-карбоксилата:
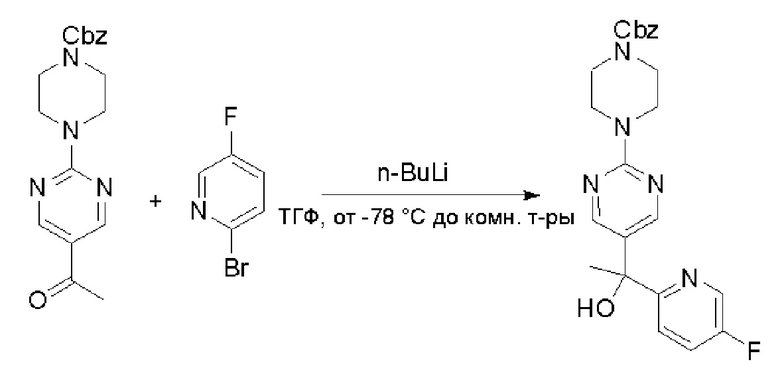
К раствору 2-бром-5-фторпиридина (1,3 г, 7,50 ммоль) в безводном ТГФ (30 мл) по каплям добавили n-BuLi (2,76 мл, 6,62 ммоль) при -78 oC и перемешивали смесь при -78 °С в течение 2 часов, затем добавили бензил-4-(5-ацетилпиримидин-2-ил)пиперазин-1-карбоксилат (1,5 г, 4,41 ммоль). Реакционную смесь оставили нагреваться до комнатной температуры и перемешивали в течение ночи. ЖХМС показала, что реакция завершена. Раствор погасили водным раствором NH4Cl (50 мл) и экстрагировали EtOAc (3 × 100 мл). Объединенные органические слои промыли водой (2 × 50 мл) и насыщенным солевым раствором (50 мл), высушили над сульфатом натрия, отфильтровали и концентрировали. Остаток очистили препаративной ВЭЖХ с получением указанного в заголовке соединения (0,4 г, 20 %) в виде белого твердого вещества. МС (ЭС+) C23H24FN5O3 рассчитано: 437, найдено: 438 [M + H]+.
Стадия 2: Синтез (S)-1-(5-фторпиридин-2-ил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола и (R)-1-(5-фторпиридин-2-ил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола:
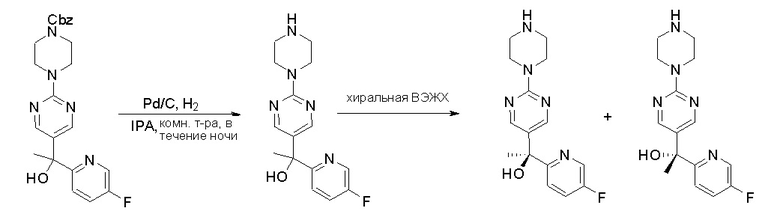
Суспензию бензил-4-(5-(1-(5-фторпиридин-2-ил)-1-гидроксиэтил)пиримидин-2-ил)пиперазин-1-карбоксилата (420,0 мг, 0,96 ммоль) и Pd/C (200,0 мг) в i-PrOH обрабатывали H2 под давлением 1 атм. (из баллона с H2) и перемешивали при комнатной температуре в течение ночи. ЖХМС показала, что реакция завершена. Реакционную смесь отфильтровали через слой целита. Фильтрат концентрировали и очистили хроматографией на силикагеле (ДХМ:CH3OH = 10:1) с получением указанного в заголовке соединения в виде белого твердого вещества (153 мг, 53 %). МС (ЭС+) C15H18FN5O рассчитано: 303, найдено: 304 [M+H]+.
Рацемическое соединение (290 мг, 0.96 ммоль) разделили с помощью хиральной ВЭЖХ с получением (S)-1-(5-фторпиридин-2-ил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола (пик 1, 80 мг, 28 %) и (R)-1-(5-фторпиридин-2-ил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанола (пик 2, 80 мг, 28 %).
Синтез (S)-трет-бутил-4-(5-(1-(4-фторфенил)винил)пиримидин-2-ил)-3-(гидроксиметил)пиперазин-1-карбоксилата:
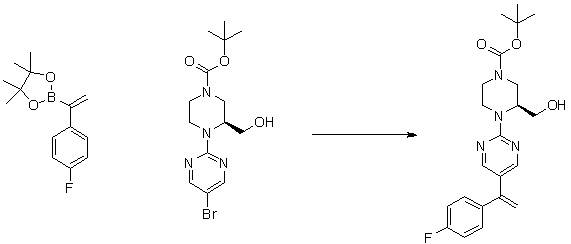
2-(1-(4-Фторфенил)винил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан (138 мг, 0,556 ммоль), (S)-трет-бутил-4-(5-бромпиримидин-2-ил)-3-(гидроксиметил)пиперазин-1-карбоксилат (103 мг, 0,276 ммоль), 2 M карбонат натрия (0,35 мл, 0,700 ммоль) и аддукт PdCl2(dppf)-ДХМ (17,3 мг, 0,021 ммоль) растворили в 1,4-диоксане (2 мл) в пробирке объемом 2-5 мл для работы в микроволновом реакторе. Реакционную смесь перемешивали при 100 °С в течение 18 часов. Охладили до комнатной температуры и отфильтровали реакционную смесь через слой целита, элюируя MeOH, и концентрировали фильтрат in vacuo на целите. Остаток очистили с помощью ЖХСД (0-100 % EtOAc-гексаны) с получением (S)-трет-бутил-4-(5-(1-(4-фторфенил)винил)пиримидин-2-ил)-3-(гидроксиметил)пиперазин-1-карбоксилата (107 мг, 0,258 ммоль, выход 94 %) в виде оранжевого смолистого вещества.
МС (ЭС+) C22H27FN4O3 рассчитано: 414, найдено: 415 [M + H]+.
Синтез (S)-2,2,2-трифтор-1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанамина:
Стадия 1: Синтез (R,Z)-2-метил-N-(2,2,2-трифтор-1-(4-фторфенил)этилиден)пропан-2-сульфинамида:
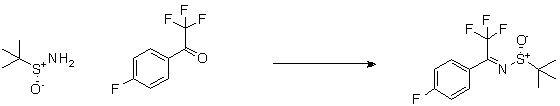
(R)-2-Метилпропан-2-сульфинамид (0,64 г, 5,28 ммоль), 2,2,2-трифтор-1-(4-фторфенил)этанон (0,53 г, 2,76 ммоль) и изопропоксид титана (IV) (1,5 мл, 5,12 ммоль) перемешивали в ТГФ (13 мл) при 70 °C в течение 18 часов. Охладили до комнатной температуры, добавили насыщенный раствор NaCl и EtOAc и перемешивали полученную двухфазную суспензию в течение 5 минут. Суспензию отфильтровали через целит для удаления остатков титана и отделили органическую фазу. Водную фазу второй раз экстрагировали EtOAc. Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo на целите. В результате очистки остатка с помощью ЖХСД (0-20 % EtOAc-гексаны) получили (R,Z)-2-метил-N-(2,2,2-трифтор-1-(4-фторфенил)этилиден)пропан-2-сульфинамид (0,23 г, 0,779 ммоль, выход 28,2 %) в виде желтого маслянистого вещества.
Стадия 2: Синтез трет-бутил-4-(5-((S)-1-(((R)-трет-бутилсульфинил)амино)-2,2,2-трифтор-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-карбоксилата:
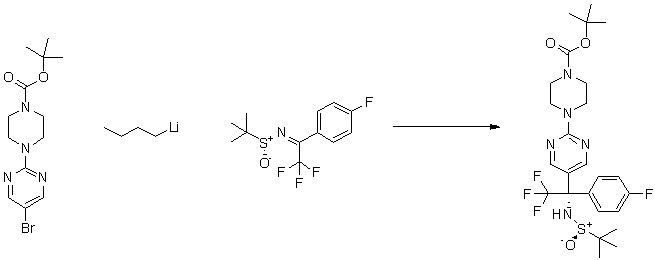
трет-Бутил-4-(5-бромпиримидин-2-ил)пиперазин-1-карбоксилат (0,242 г, 0,705 ммоль) растворили в ТГФ (3,5 мл) и охладили до -78 °C. По каплям, через шприц быстро добавили раствор nBuLi, 2,5 M в гексанах (0,31 мл, 0,775 ммоль). Полученную смесь перемешивали при -78 °С в течение 15 минут. По каплям, через шприц быстро добавили раствор (R,Z)-2-метил-N-(2,2,2-трифтор-1-(4-фторфенил)этилиден)пропан-2-сульфинамида (0,225 г, 0,762 ммоль) в ТГФ (0,5 мл). Реакционную смесь перемешивали при -78 °С в течение 5 минут, затем нагрели до комнатной температуре. Через 45 минут добавили насыщенный раствор NH4Cl и экстрагировали продукты в EtOAc (x2). Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo. В результате очистки остатка с помощью ЖХСД (0-80 % EtOAc-гексаны) получили трет-бутил-4-(5-((S)-1-(((R)-трет-бутилсульфинил)амино)-2,2,2-трифтор-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-карбоксилат (304 мг, 0,543 ммоль, выход 77 %) в виде бледно-желтого смолистого вещества. Абсолютную стереохимию назначили произвольно.
МС (ЭС+) C25H33F4N5O3S рассчитано: 559, найдено: 560 [M + H]+.
Стадия 3: Синтез (S)-2,2,2-трифтор-1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанамина:
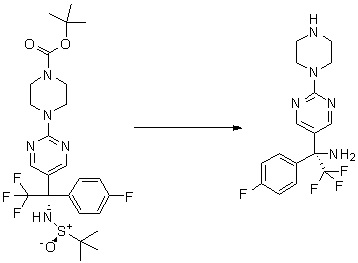
трет-Бутил-4-(5-((S)-1-(((R)-трет-бутилсульфинил)амино)-2,2,2-трифтор-1-(4-фторфенил)этил)пиримидин-2-ил)пиперазин-1-карбоксилат (302 мг, 0,540 ммоль) перемешивали в 4 M HCl в 1,4-диоксане (3 мл)/MeOH (3 мл) при комнатной температуре в течение 1 часа. Растворитель удалили in vacuo и разделили остаток между насыщенным раствором NaHCO3 и EtOAc. Органическую фазу второй раз экстрагировали EtOAc. Объединенные органические экстракты промыли насыщенным солевым раствором, высушили над Na2SO4, отфильтровали и концентрировали in vacuo с получением (S)-2,2,2-трифтор-1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанамина (190 мг, 0,535 ммоль, выход 99 %) э.и. ~88 %. В результате дополнительной очистки хиральной СЖХ получили (S)-2,2,2-трифтор-1-(4-фторфенил)-1-(2-(пиперазин-1-ил)пиримидин-5-ил)этанамин (123,7 мг, 0,348 ммоль, выход 71,2 %) с э.и. ~99 %.
МС (ЭС+) C16H17F4N5 рассчитано: 355, найдено: 356 [M + H]+.
Протоколы синтеза, которые могут быть использованы для получения соединений, описанных в настоящем документе, указаны ниже. Данные ЯМР и ЖХ МС, полученные для соединений, описанных в настоящем документе, также представлены ниже.
(M+1)
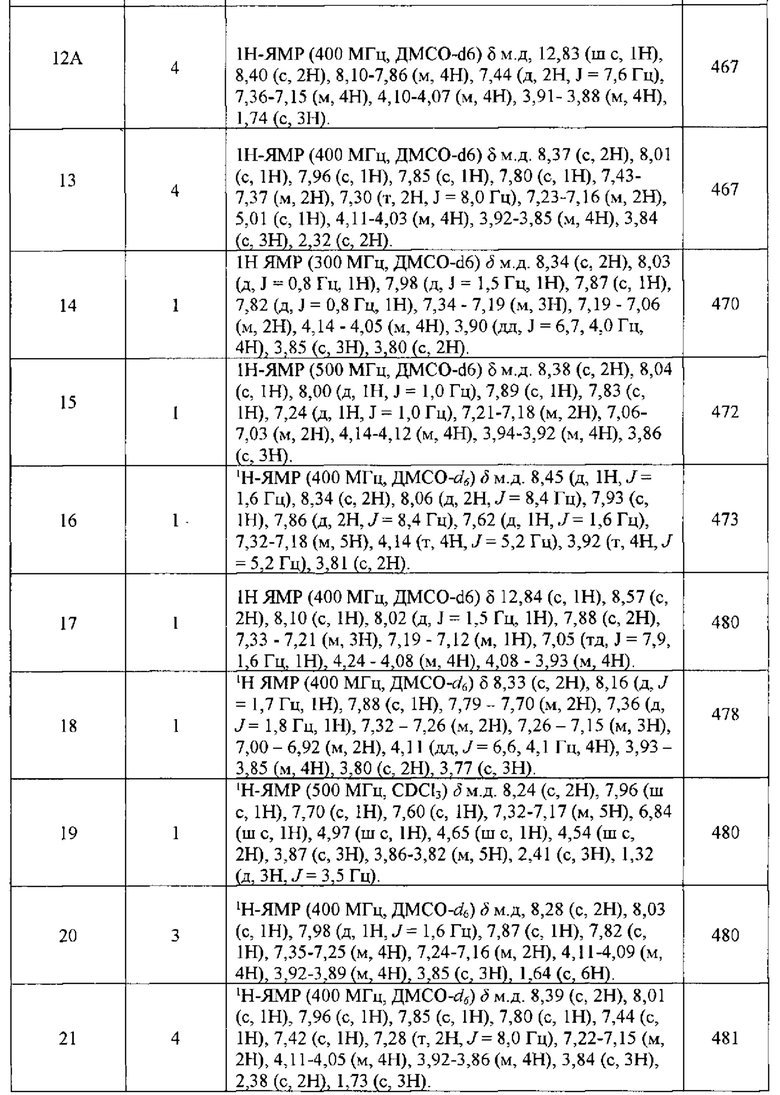
Биохимическая активность соединений
Для оценки активности химических соединений против рассматриваемой релевантной киназы использовали технологическую платформу изменения электрофоретической подвижности Caliper LifeSciences. Пептидный субстрат с флуоресцентной меткой инкубировали в присутствии киназы и АТФ для обеспечения фосфорилирования отражающей части пептида. По окончании реакции смесь фосфорилированного (продукт) и нефосфорилированного (субстрат) пептидов пропускали через микрожидкостную систему планшетридера Caliper EZ Reader 2 при приложенной разности потенциалов. Наличие фосфатной группе в пептиде-продукте обеспечивает разность массы и заряда относительно массы и заряда пептида-субстрата, что приводит к разделению пулов субстрата и продукта в образце. При прохождении пулов через LEDS, установленные в приборе, указанные пулы обнаруживали и анализировали как отдельные пики. Следовательно, соотношение между указанными пиками отражает активность химического вещества при данной концентрации в данной лунке и в данных условиях. Анализ D816V KIT при Km: В каждой лунке 384-луночного планшета инкубировали 0,04 нг/мкл (0,5 нМ) KIT D816V (Carna Bioscience 08-156) в общем объеме 12,5 мкл буфера (100 мМ HEPES с pH 7,5, 0,015 % Brij 35, 10 мМ MgCl2, 1 мМ DTT) с 1 мкМ Srctide (5-FAM-GEEPLYWSFPAKKK-NH2) и 15 мкМ АТФ при 25 С в течение 90 минут в присутствии или в отсутствие дозированных серий концентрации соединения (конечная концентрация ДМСО 1 %). Реакцию останавливали добавлением 70 мкл останавливающего буфера (100 мМ HEPES с pH 7,5, 0,015 % Brij 35, 35 мМ ЭДТК и 0,2 % реагента для покрытия 3 (Caliper Lifesciences)). Затем планшет считывали на планшетридере Caliper EZ Reader 2 (настройки протокола: -1,9 фунт/кв. дюйм, верхнее напряжение -700, нижнее напряжение -3000, время после впитывания образца 35 с). Данные нормализовали к контрольному ингибированию 0 % и 100 % и рассчитывали IC50 или EC50, используя 4-параметрический набор с помощью GraphPad Prism.
Анализ D842V PDGFRA при Km: В каждой лунке 384-луночного планшета инкубировали 0,7 нг/мкл (8 нМ) D842V PDGFRA (ProQinase 0761-0000-1) в общем объеме 12,5 мкл буфера (100 мМ HEPES с pH 7,5, 0,015 % Brij 35, 10 мМ MgCl2, 1 мМ DTT) с 1 мкМ CSKtide (5-FAM- KKKKEEIYFFF-NH2) и 15 мкМ АТФ при 25 С в течение 90 минут в присутствии или в отсутствие дозированных серий концентрации соединения (конечная концентрация ДМСО 1 %). Реакцию останавливали добавлением 70 мкл останавливающего буфера (100 мМ HEPES с pH 7,5, 0,015 % Brij 35, 35 мМ ЭДТК и 0,2 % реагента для покрытия 3 (Caliper Lifesciences)). Затем планшет считывали на планшетридере Caliper EZ Reader 2 (настройки протокола: -1,9 фунт/кв. дюйм, верхнее напряжение -500, нижнее напряжение -3000, время после впитывания образца 38 с). Данные нормализовали к контрольному ингибированию 0 % и 100 % и рассчитывали IC50 или EC50, используя 4-параметрический набор с помощью GraphPad Prism.
Клеточная активность
Анализ аутофосфорилирования HMC1.2: 10000 клеток HMC1.2 инкубировали в 22 мкл культуральной среды (IMDM без фенолового красного, без сыворотки) в каждой лунке 384-луночного планшета без сыворотки в течение ночи в инкубаторе для тканевых культур (5 % CO2, 37 oC). Затем к клеткам добавляли 10-точечные серии дозированных концентраций соединения (25 мкМ-95,4 пМ) в объеме 3,1 мкл в каждую лунку (конечная концентрация ДМСО 0,25 %). Через 90 минут в каждую лунку добавляли 6 мкл 5X лизирующего буфера AlphaLISA (Perkin Elmer) с добавлением коктейля ингибитора протеазы и фосфатазы (Cell Signaling Technologies) и встряхивали при 450 об./мин. В течение 15 минут при 4 °С. В каждую лунку добавляли 10 мкл c-KIT фосфо-Y719 и общих антител к c-KIT (конечная концентрация 15 нМ, Cell Signaling Technologies) и 50 мкг/мл акцепторных шариков кроликов AlphaLISA (Perkin Elmer) и встряхивали при 300 об./мин. при комнатной температуре в течение 2 часов. В каждую лунку добавляли 10 мкл стрептавидиновых донорных шариков (Perkin Elmer), закрывали от света алюминиевым адгезивом и встряхивали при 300 об./мин. при комнатной температуре в течение 2 часов. Сигнал флуоресценции получали на приборе Envision (Perkin Elmer) по 384-луночному протоколу AlphaScreen HTS. Данные нормализовали к контрольному ингибированию 0 % и 100 % и рассчитывали IC50 посредством построения четырехпараметрической логистической кривой IC50.
В представленной ниже таблице показана активность соединений в клеточной линии тучноклеточного лейкоза, HMC 1.2. Указанная клеточная линия содержит KIT, мутированную в положениях V560G и D816V, что приводит к конститутивной активации киназы. Следующие соединения испытывали в данном анализе для измерения прямого ингибирования активности киназы KIT D816V посредством измерения аутофосфорилирования KIT на тирозине 719 белка KIT.
В представленной ниже таблице в отношении биохимической активности D816V и D842V использованы следующие обозначения: < 1,00 нМ = A; 1,01-10,0 нМ = B; 10,01-100,0 нМ = C; >100 нМ = D; и н.о. = не определяли. Для клеточной активности в клеточной линии HMC1.2 использованы следующие обозначения: A означает < 50 нМ; B означает ≥50 и <100 нМ; C означает ≥100 и <1000 нМ; D означает ≥1000 и менее 10000нМ; E означает ≥10000 нМ; и н.о. = не определяли.
Эффективность в модели in vivo
Соединение 46 и дазатиниб анализировали в модели ксенотрансплантата мастоцитомы P815. Опухолевые клетки P815 (ATCC, Манассас, штат Вирджиния, кат. № ATCC® TIB-64) выдерживали in vitro в виде суспензии и однослойной культуры в среде RPMI1640 с добавлением 10 % эмбриональной телячьей сыворотки при 37 °С в атмосфере 5 % CO2 в воздухе. Опухолевые клетки пересевали два раза в неделю посредством обработки трипсин-ЭДТК. Клетки, растущие в экспоненциальной фазе роста, собирали и подсчитывали для инокуляции опухоли.
Для эксперимента использовали самок «голых» мышей BALB/c. Каждой мыши в правый бок подкожно инокулировали опухолевые клетки P815 (1 x 106) в 0,1 мл PBS для роста опухоли. Лечение начинали на 6 день после инокуляции опухоли по достижении среднего размера опухоли около 89 мм3. Исследуемое соединение и носитель вводили мышам в соответствии с режимом, показанным ниже.
Примечание: * QD = один раз в сутки, BID = два раза в сутки.
Размер опухоли измеряли один раз в два дня в двух направлениях, используя штангенциркуль, и объем опухоли выражали в мм3, рассчитывая по формуле: V = 0,5 a x b2, где a и b представляют собой наибольший и наименьший диаметры опухоли, соответственно. Затем размер опухоли использовали для расчетов значений T-C и T/C. T-C рассчитывали, используя T как среднее время (в сутках), необходимое для достижения предварительно определенного размера (например, 1000 мм3) опухоли в экспериментальной группе, и C как среднее время (в сутках), необходимое для достижения такого же размера опухоли в контрольной группе. Значение T/C (в процентах) является показателем противоопухолевой эффективности; T и C представляют собой средние объемы опухоли в экспериментальной и контрольной группах, соответственно, в данный день.
Для каждой группы рассчитывали TGI, используя формулу: TGI (%) = [1-(Ti-T0)/ (Vi-V0)] ×100; Ti представляет собой средний объем опухоли в экспериментальной группе в данный день, T0 представляет собой средний объем опухоли в экспериментальной группе в день начала лечения, Vi представляет собой средний объем опухоли в контрольной группе с носителем в тот же день, что и Ti, и V0 представляет собой средний объем опухоли в группе с носителем в день начала лечения. В конечной точке измеряли массу опухоли.
Статистический анализ разности объема опухоли и массы опухоли в группах проводили на основании данных, полученных в наилучший терапевтический момент времени после введения последней дозы (на 8 день после начала лечения). Выполнили однофакторный дисперсионный анализ для сравнения объема опухоли и массы опухоли в группах. Все данные анализировали с помощью программы Prism 5.0. p < 0,05 считали статистически значимым.
Результаты. Кривые роста опухоли в различных экспериментальных группах представлены на фиг. 1. Точки данных представляют средний объем опухоли в группе, величины ошибки представляют стандартную погрешность среднего значения (SEM). Как показано на фиг. 1, Соединение 46 было эффективным при ингибировании роста опухоли. Увеличение дозы Соединения 46 усиливает эффективность ингибирования опухоли.
Результаты изменения массы тела у опухоленесущих мышей представлены на фиг. 2. Точки данных представляют среднее изменение массы тела в группе. Величины ошибки представляют стандартную погрешность среднего значения (SEM). Как показано на фиг. 2, изменение массы тела составило менее 5 % даже при более высоких дозах Соединения 46. Напротив, животные, которым вводили носитель или дазатиниб, потеряли более 5 % массы тела.
Следовательно, Соединение 46, используемое в качестве самостоятельного агента, обеспечивало заметную противоопухолевую активность в модели ксенотрансплантата рака мастоцитомы P815 у мышей в данном исследовании. Кроме того, указанное соединение хорошо переносилось опухоленесущими животными, что продемонстрировано отсутствием потери массы.
Включение посредством ссылки
Все публикации и патенты, упомянутые в настоящем документе, включены посредством ссылки, как если бы каждая отдельная публикация или патент были бы указаны конкретно и отдельно для включения посредством ссылки.
Эквиваленты
Специалисты в данной области понимают или могут установить с помощью не более чем стандартных экспериментов множество эквивалентов конкретных вариантов реализации настоящего изобретения, описанных в данном документе. Подразумевается, что такие эквиваленты входят в границы объема следующей формулы изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЗАМЕЩЕННЫЕ 1,2-ДИГИДРО-3Н-ПИРАЗОЛО[3,4-D]ПИРИМИДИН-3-ОНЫ | 2019 |
|
RU2812726C2 |
| БЕНЗАМИДНЫЕ СОЕДИНЕНИЯ | 2019 |
|
RU2801647C2 |
| КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ KIT- И PDGFRA-ОПОСРЕДОВАННЫХ ЗАБОЛЕВАНИЙ | 2020 |
|
RU2817354C2 |
| ПРОИЗВОДНЫЕ ПИРИМИДИНА | 2002 |
|
RU2320658C2 |
| ИНГИБИТОРЫ КИНАЗ, ПРИМЕНИМЫЕ ДЛЯ ЛЕЧЕНИЯ МИЕЛОПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ И ДРУГИХ ПРОЛИФЕРАТИВНЫХ ЗАБОЛЕВАНИЙ | 2007 |
|
RU2482112C2 |
| ИНГИБИТОРЫ JAK2 И ALK2 И СПОСОБЫ ИХ ИСПОЛЬЗОВАНИЯ | 2014 |
|
RU2693480C2 |
| НОВЫЕ СОЕДИНЕНИЯ, КОТОРЫЕ ЯВЛЯЮТСЯ ИНГИБИТОРАМИ ERK | 2013 |
|
RU2660429C2 |
| ОКСАЗОЛИДИНОНОВЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ПРОТИВОБАКТЕРИАЛЬНЫХ СРЕДСТВ | 2016 |
|
RU2794494C2 |
| БИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ДИГИДРОПИРИМИДИНКАРБОКСАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ RHO-КИНАЗЫ | 2017 |
|
RU2778478C2 |
| ИНГИБИТОРЫ ПРОТЕИНКИНАЗ, ИХ СПОСОБ ПОЛУЧЕНИЯ И МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ | 2016 |
|
RU2749437C2 |
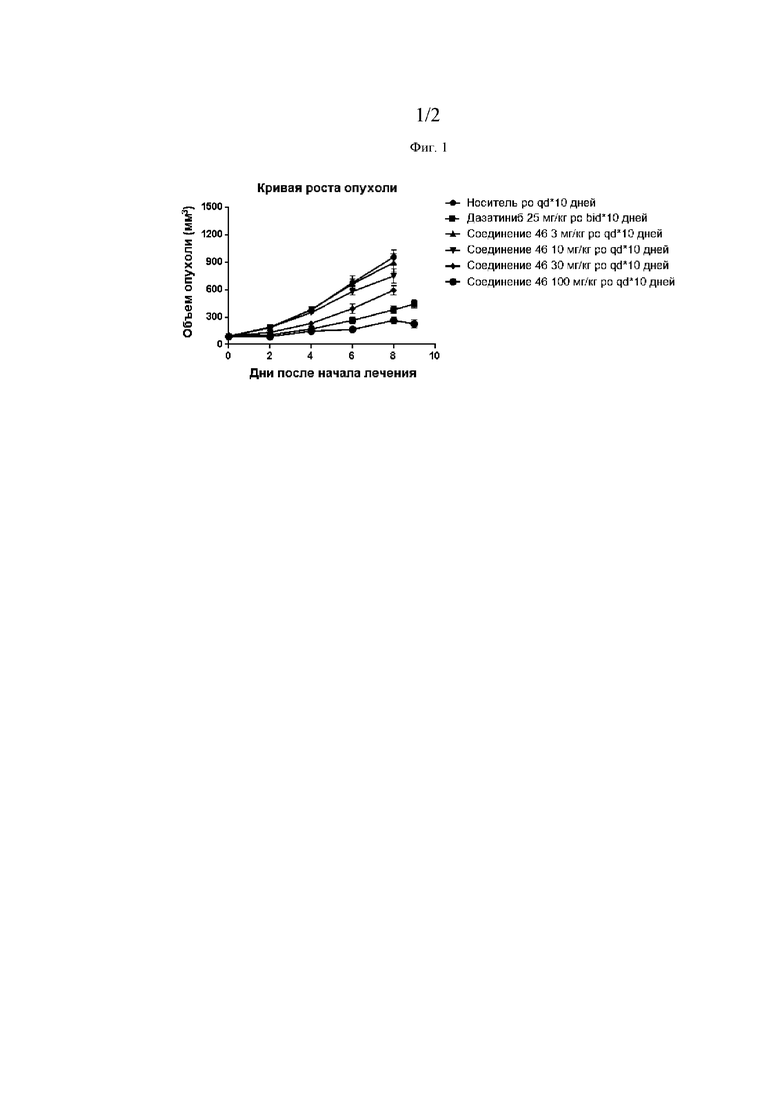
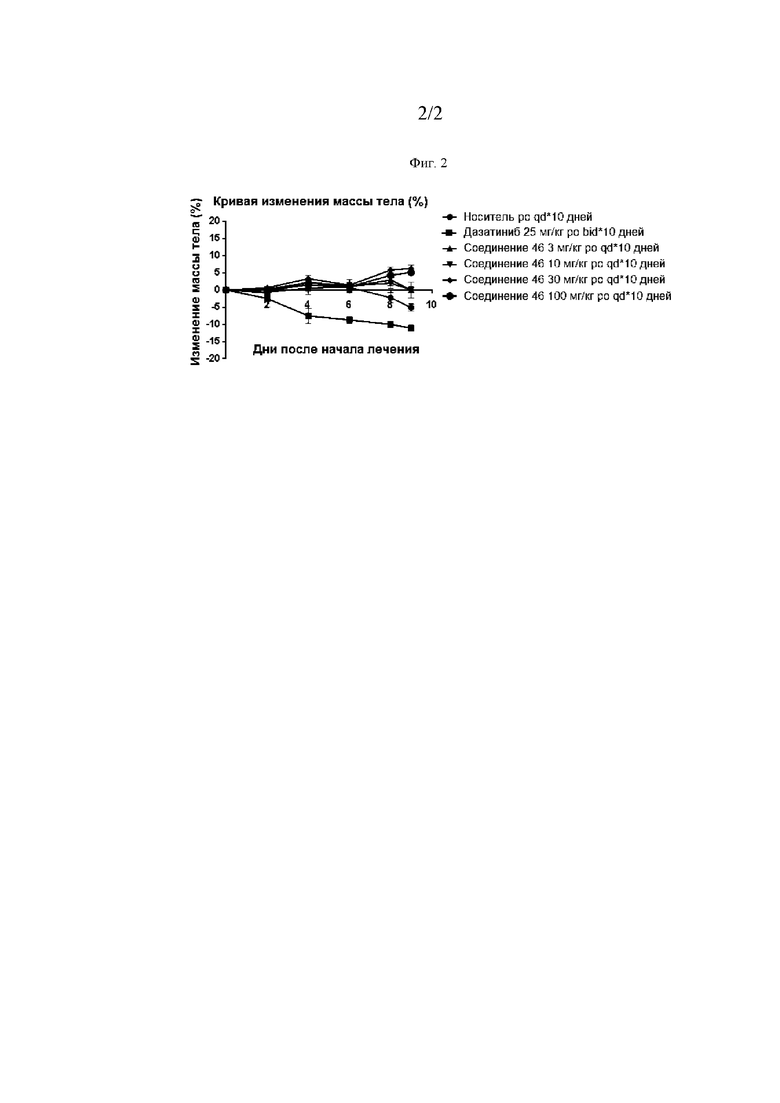
Изобретение относится к соединению Формулы I или к его фармацевтически приемлемой соли. В формуле I W выбран из водорода и 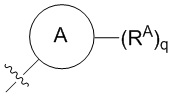 , где кольцо A выбрано из фенила или 6-членного моноциклического гетероарила с одним кольцевым гетероатомом, выбранным из N; каждый X и Y независимо выбран из CR1 и N; Z выбран из фенила, 5- или 6-членного моноциклического гетероарила с 1-2 кольцевыми гетероатомами, независимо выбранными из N, O и S, 6-членного моноциклического частично ненасыщенного гетероциклила с одним кольцевым гетероатомом, выбранным из N; где каждый из фенила, моноциклического гетероарила и моноциклического гетероциклила независимо замещен 0-2 группами RC; L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкинилен)-, -O-, -S- и -S(O)2-; каждый RA и RB независимо выбран из галогена, C1-C6 алкила и C1-C6 гидроксиалкила; каждый RC независимо выбран из C1-C6 алкила, C1-C6 алкокси, галогена, C1-C6 гидроксиалкила, С3-С7 циклоалкила, 4-членного гетероциклила с одним кольцевым гетероатомом, выбранным из О, (6-членного гетероциклила с 2 кольцевыми гетероатомами, выбранными из N, O)-C1-C6 алкила, циано, -C(O)OR2 и -C(O)-N(R2)(R2); каждый RD и RF независимо выбран из водорода и -N(R2)(R2); каждый R1 представляет собой водород; каждый R2 независимо выбран из водорода, гидроксила, -NR”R”, C1-C6 алкила, C1-C6 алкокси, C1-C6 галогеналкила, C1-C6 гидроксиалкила, С3-С7 циклоалкила, или 2 R2 вместе с атомами, к которым они присоединены, образуют 4-членное гетероциклильное кольцо с 1 кольцевым атомом, выбранным из О; каждый R” представляет собой водород или C1-C6 алкил; и m, p и q - каждый независимо представляет собой 0, 1, 2, 3 или 4. Изобретение также относится к индивидуальным соединениям, к фармацевтической композиции, к способам лечения. Технический результат: получены новые соединения формулы I, обладающие модулирующей активностью в отношении KIT и/или PDGFRα. 14 н. и 11 з.п. ф-лы, 2 ил., 3 табл., 7 пр.
, где кольцо A выбрано из фенила или 6-членного моноциклического гетероарила с одним кольцевым гетероатомом, выбранным из N; каждый X и Y независимо выбран из CR1 и N; Z выбран из фенила, 5- или 6-членного моноциклического гетероарила с 1-2 кольцевыми гетероатомами, независимо выбранными из N, O и S, 6-членного моноциклического частично ненасыщенного гетероциклила с одним кольцевым гетероатомом, выбранным из N; где каждый из фенила, моноциклического гетероарила и моноциклического гетероциклила независимо замещен 0-2 группами RC; L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкинилен)-, -O-, -S- и -S(O)2-; каждый RA и RB независимо выбран из галогена, C1-C6 алкила и C1-C6 гидроксиалкила; каждый RC независимо выбран из C1-C6 алкила, C1-C6 алкокси, галогена, C1-C6 гидроксиалкила, С3-С7 циклоалкила, 4-членного гетероциклила с одним кольцевым гетероатомом, выбранным из О, (6-членного гетероциклила с 2 кольцевыми гетероатомами, выбранными из N, O)-C1-C6 алкила, циано, -C(O)OR2 и -C(O)-N(R2)(R2); каждый RD и RF независимо выбран из водорода и -N(R2)(R2); каждый R1 представляет собой водород; каждый R2 независимо выбран из водорода, гидроксила, -NR”R”, C1-C6 алкила, C1-C6 алкокси, C1-C6 галогеналкила, C1-C6 гидроксиалкила, С3-С7 циклоалкила, или 2 R2 вместе с атомами, к которым они присоединены, образуют 4-членное гетероциклильное кольцо с 1 кольцевым атомом, выбранным из О; каждый R” представляет собой водород или C1-C6 алкил; и m, p и q - каждый независимо представляет собой 0, 1, 2, 3 или 4. Изобретение также относится к индивидуальным соединениям, к фармацевтической композиции, к способам лечения. Технический результат: получены новые соединения формулы I, обладающие модулирующей активностью в отношении KIT и/или PDGFRα. 14 н. и 11 з.п. ф-лы, 2 ил., 3 табл., 7 пр.
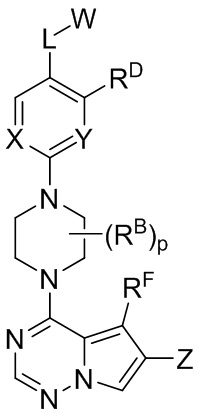 I
I
1. Соединение Формулы I:
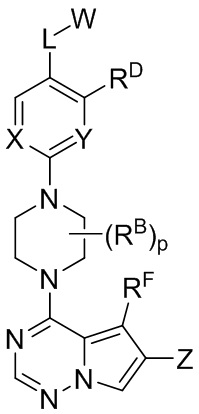
I
или его фармацевтически приемлемая соль, где:
W выбран из водорода и  , где кольцо A выбрано из фенила или 6-членного моноциклического гетероарила с одним кольцевым гетероатомом, выбранным из N;
, где кольцо A выбрано из фенила или 6-членного моноциклического гетероарила с одним кольцевым гетероатомом, выбранным из N;
каждый X и Y независимо выбран из CR1 и N;
Z выбран из фенила, 5- или 6-членного моноциклического гетероарила с 1-2 кольцевыми гетероатомами, независимо выбранными из N, O и S, 6-членного моноциклического частично ненасыщенного гетероциклила с одним кольцевым гетероатомом, выбранным из N; где каждый из фенила, моноциклического гетероарила и моноциклического гетероциклила независимо замещен 0-2 группами RC;
L выбран из связи, -(C(R2)(R2))m-, -(C2-C6 алкинилен)-, -O-, -S- и -S(O)2-;
каждый RA и RB независимо выбран из галогена, C1-C6 алкила и C1-C6 гидроксиалкила;
каждый RC независимо выбран из C1-C6 алкила, C1-C6 алкокси, галогена, C1-C6 гидроксиалкила, С3-С7 циклоалкила, 4-членного гетероциклила с одним кольцевым гетероатомом, выбранным из О, (6-членного гетероциклила с 2 кольцевыми гетероатомами, выбранными из N, O)-C1-C6алкила, циано, -C(O)OR2 и -C(O)-N(R2)(R2); каждый RD и RF независимо выбран из водорода и -N(R2)(R2);
каждый R1 представляет собой водород;
каждый R2 независимо выбран из водорода, гидроксила, -NR”R”, C1-C6 алкила, C1-C6 алкокси, C1-C6 галогеналкила, C1-C6 гидроксиалкила, С3-С7 циклоалкила или 2 R2 вместе с атомами, к которым они присоединены, образуют 4-членное гетероциклильное кольцо с 1 кольцевым атомом, выбранным из О;
каждый R” представляет собой водород или C1-C6 алкил; и
m, p и q - каждый независимо представляет собой 0, 1, 2, 3 или 4.
2. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что соединение представляет собой соединение Формулы II:
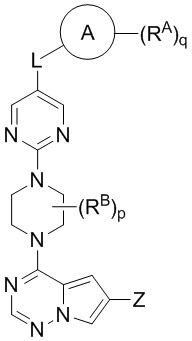
II.
3. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что соединение представляет собой соединение Формулы III:
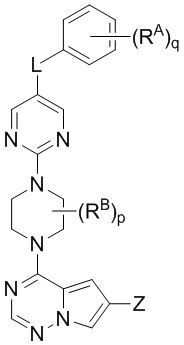
III.
4. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что по меньшей мере один из X и Y представляет собой N.
5. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что оба X и Y представляют собой N.
6. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что L представляет собой -(C(R2)(R2))m-.
7. Соединение по любому из пп.1, 2 или 4-6 или его фармацевтически приемлемая соль, отличающееся тем, что кольцо A представляет собой фенил.
8. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что Z представляет собой 5- или 6-членный гетероарил с 1-2 кольцевыми гетероатомами, независимо выбранными из N, O и S.
9. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что Z выбран из пиразолила, изоксазолила, тиофенила, тиазолила и пиридила.
10. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что Z представляет собой фенил.
11. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что Z представляет собой 6-членный моноциклический гетероциклил с одним кольцевым гетероатомом, выбранным из N.
12. Соединение по п.1 или его фармацевтически приемлемая соль, отличающееся тем, что RA представляет собой фтор и q равен 1.
13. Соединение, выбранное из представленных ниже соединений, или их фармацевтически приемлемых солей:
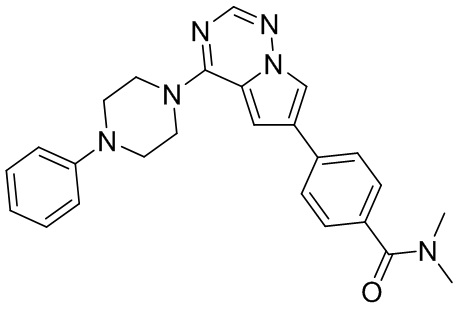
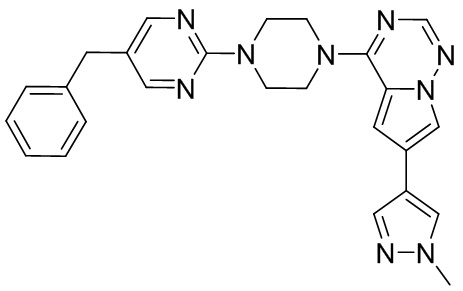
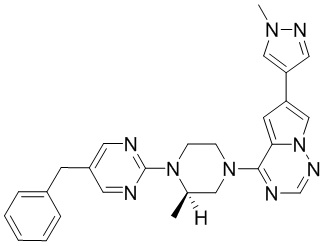
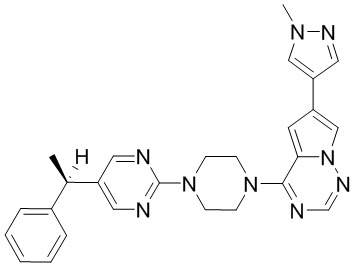
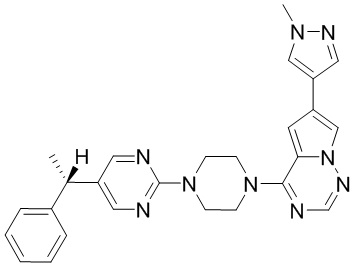
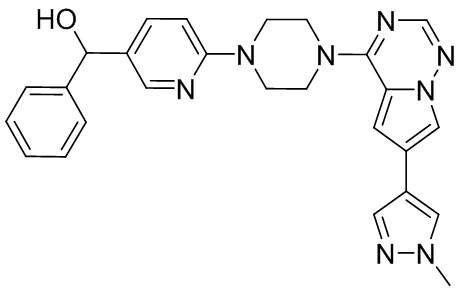
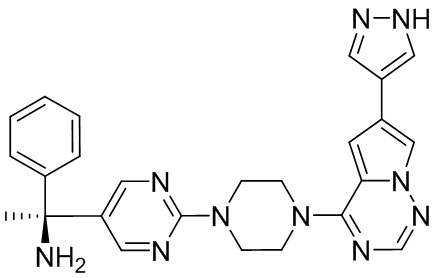
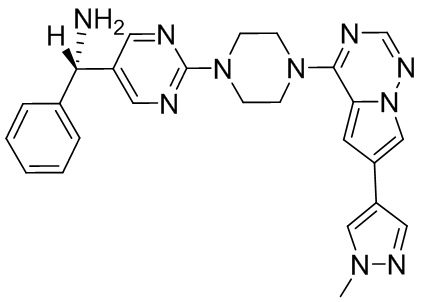
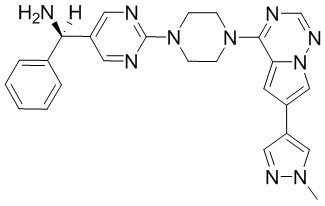
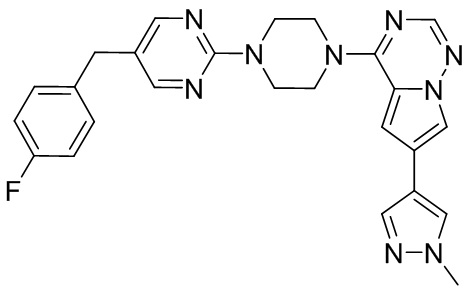
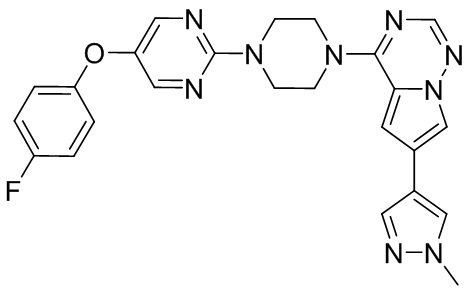
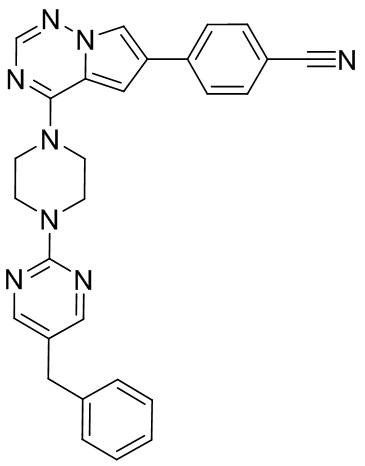
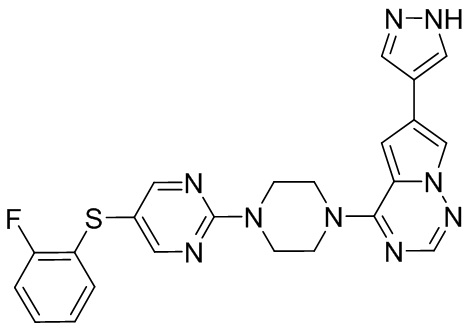
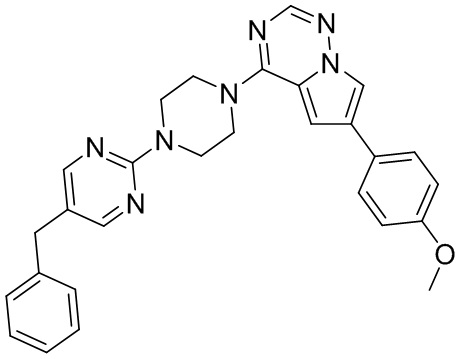
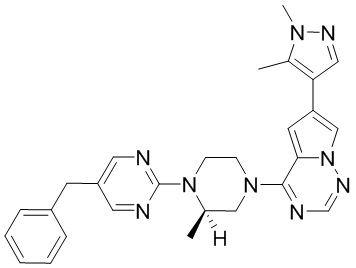
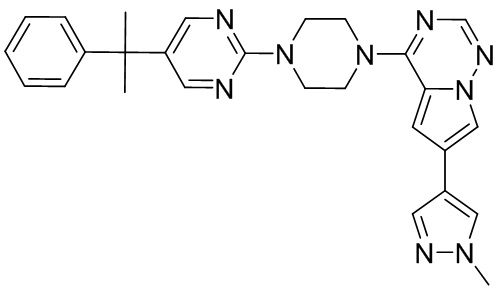
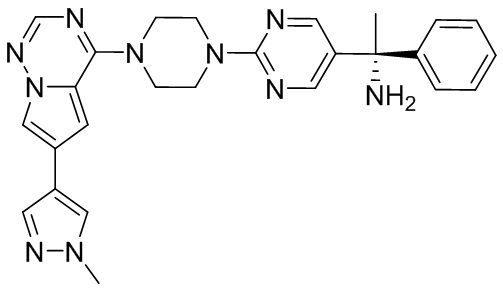
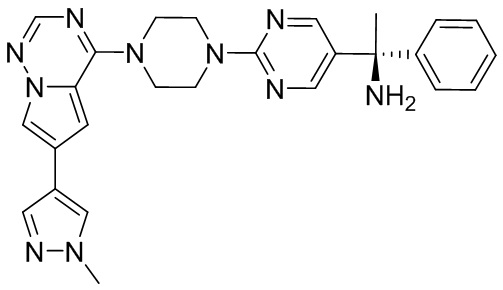
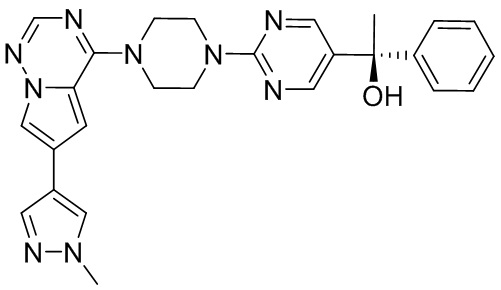
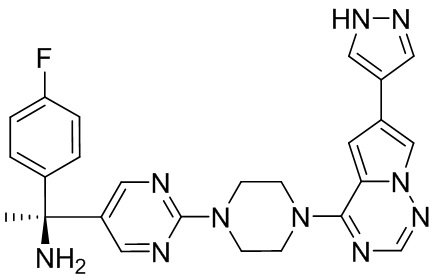
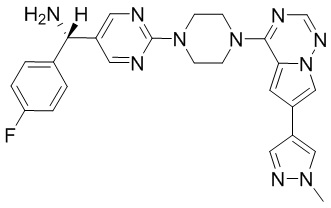
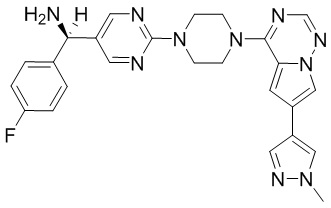
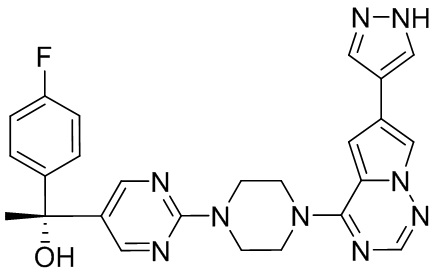
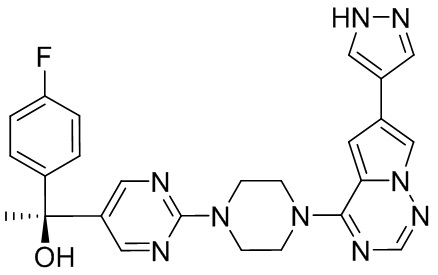
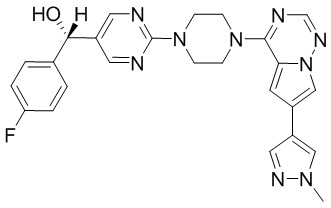
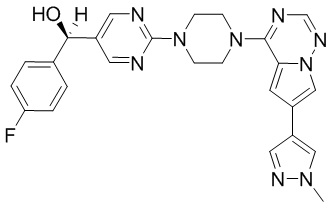
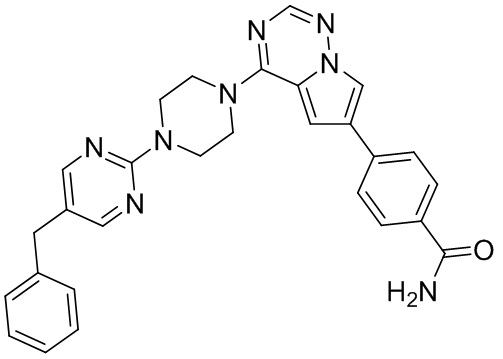
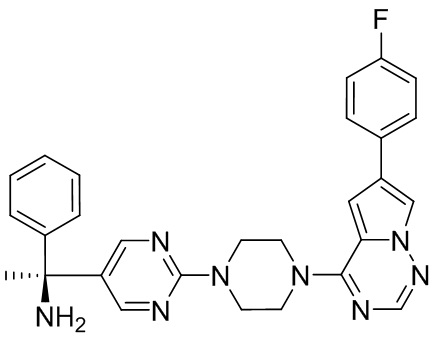
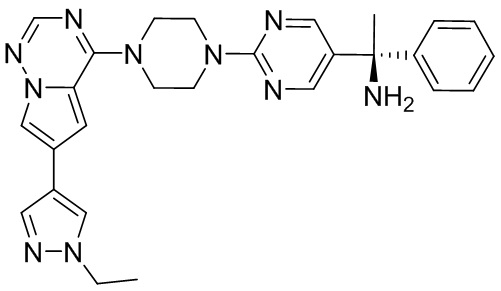
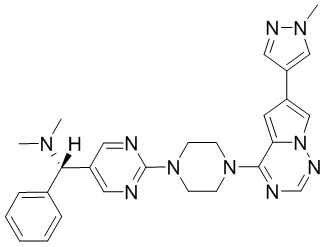
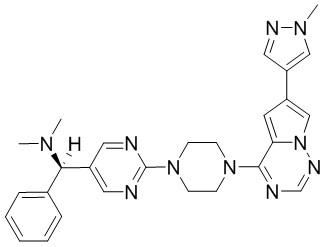
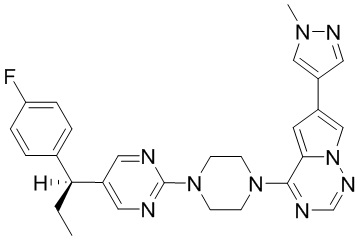
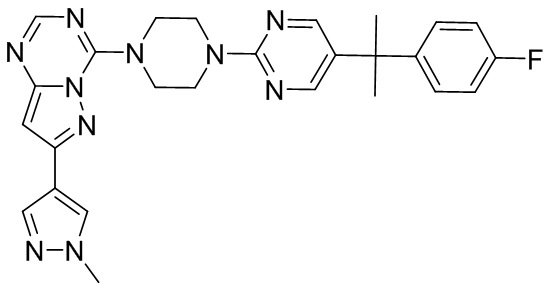
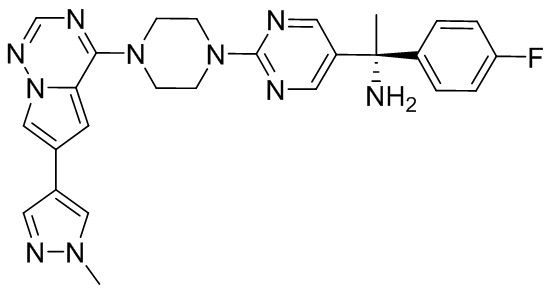
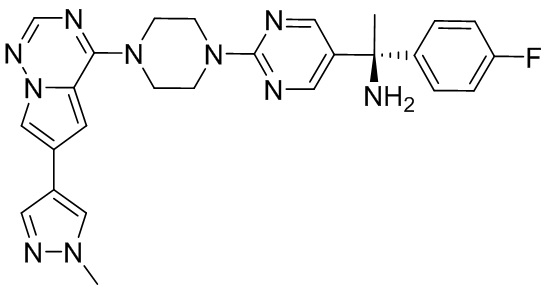
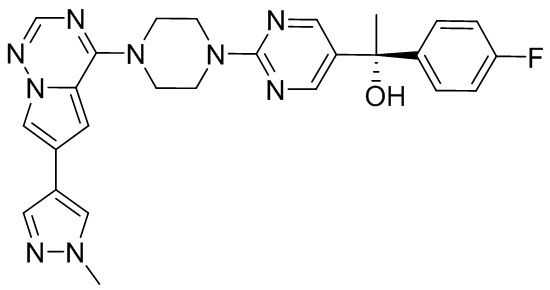
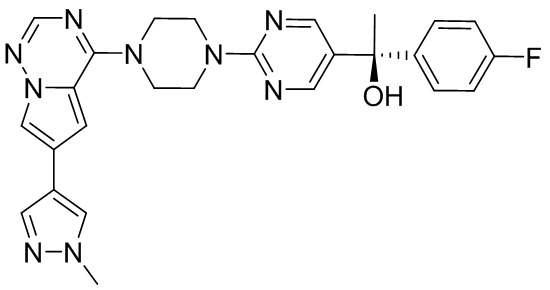
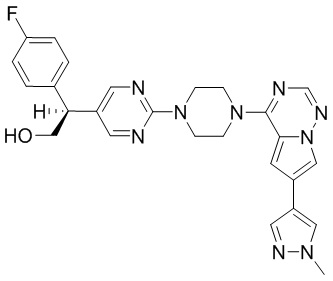
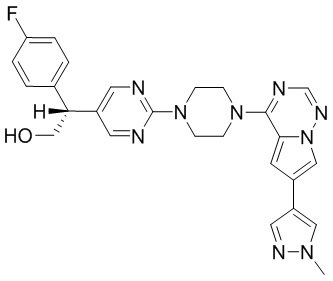
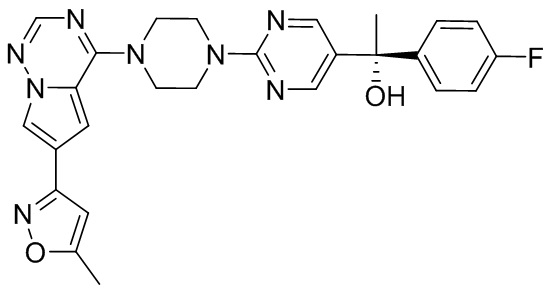
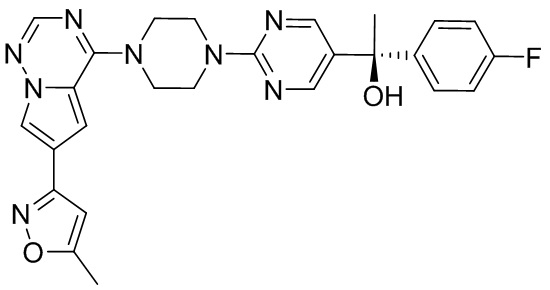
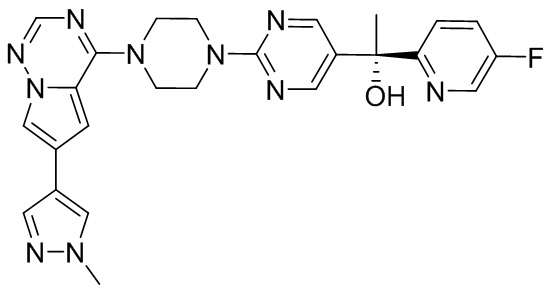
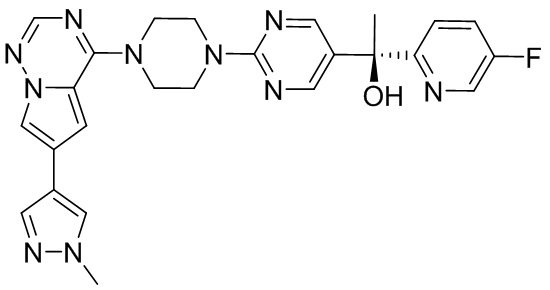
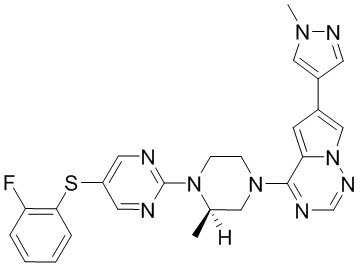
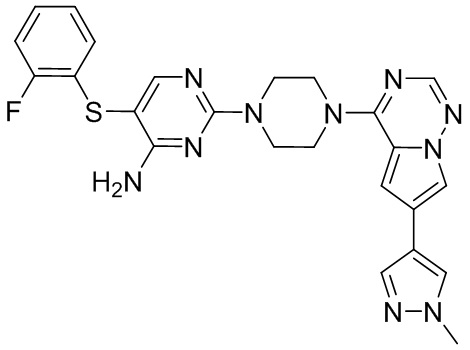
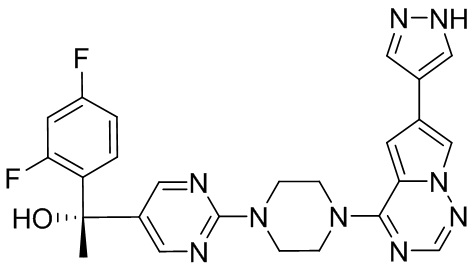
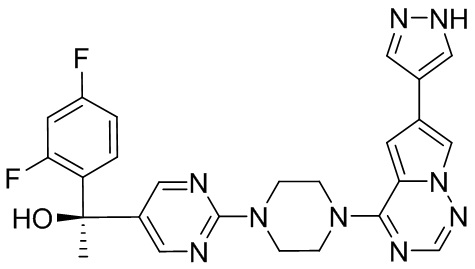
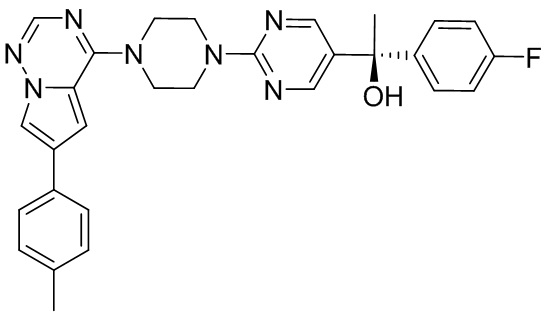
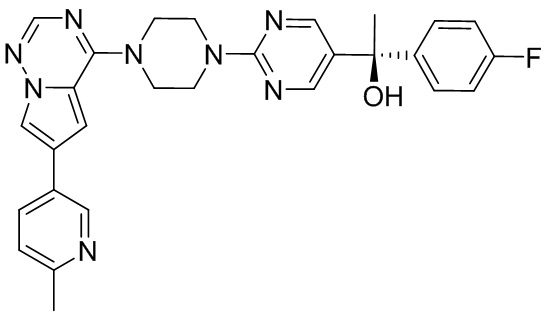
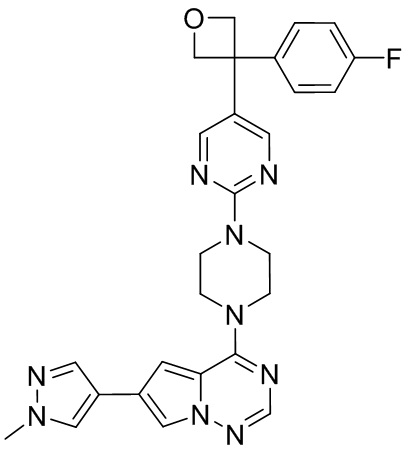
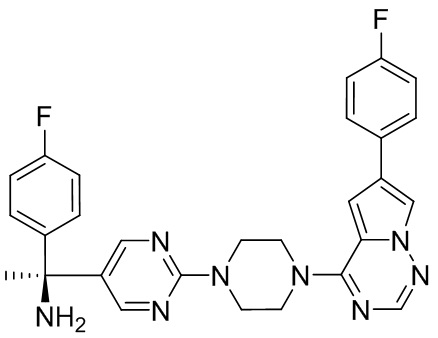
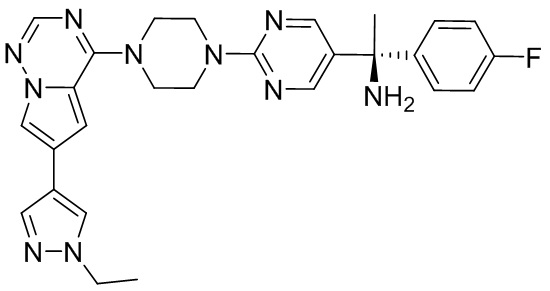
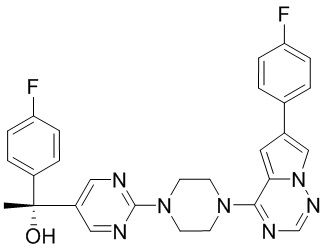
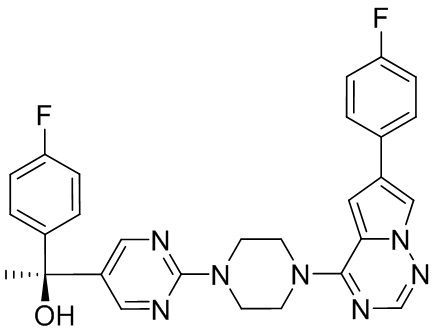
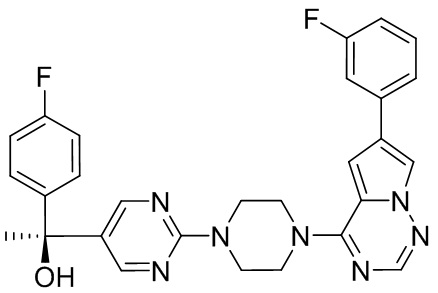
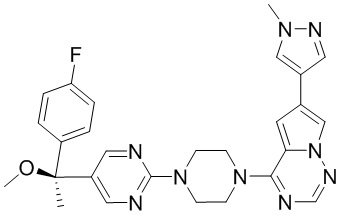
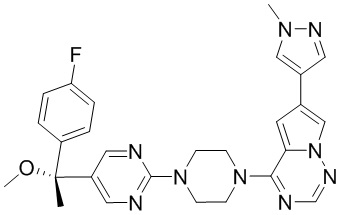
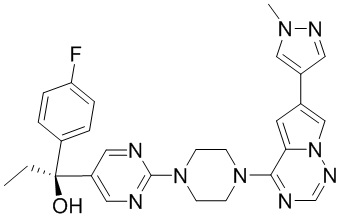
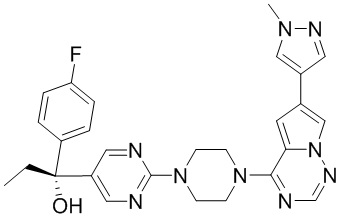
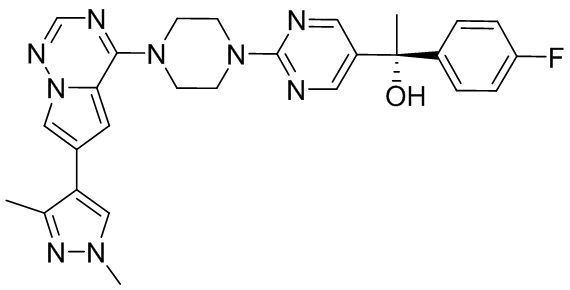
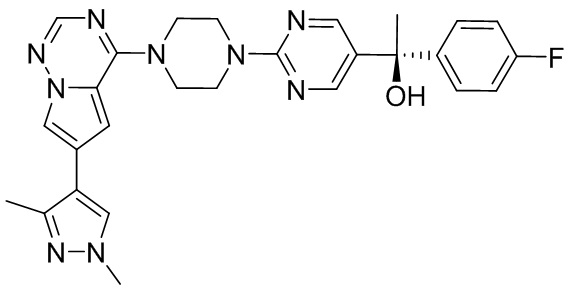
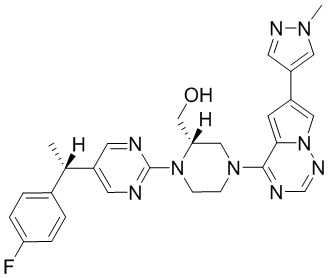
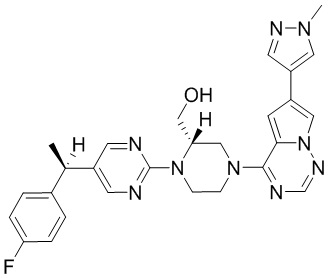
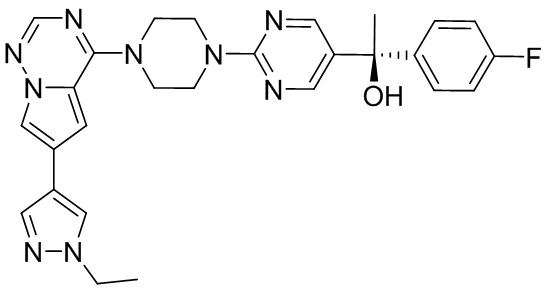
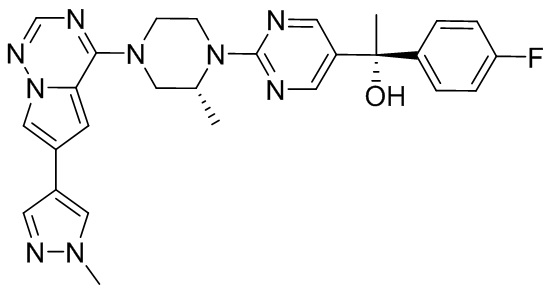
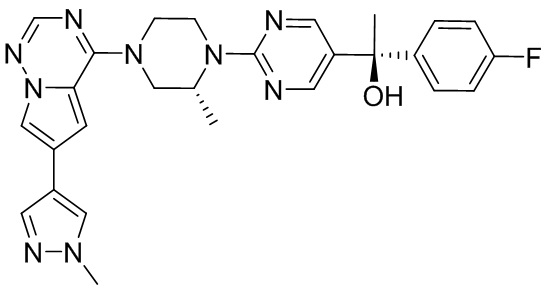
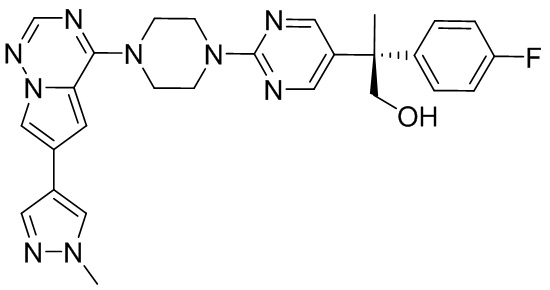
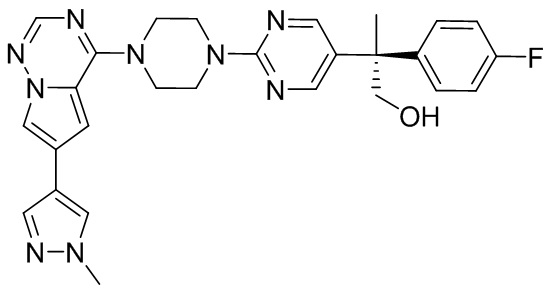
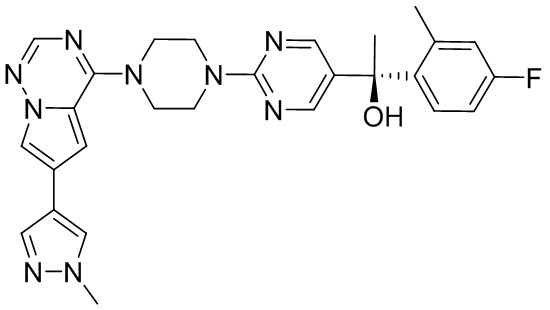
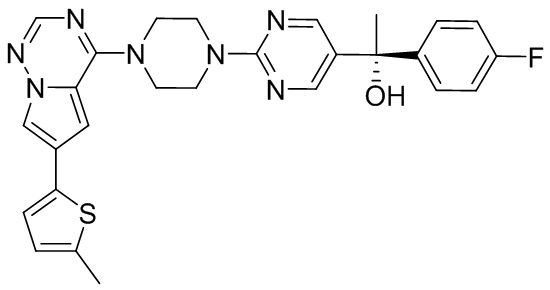
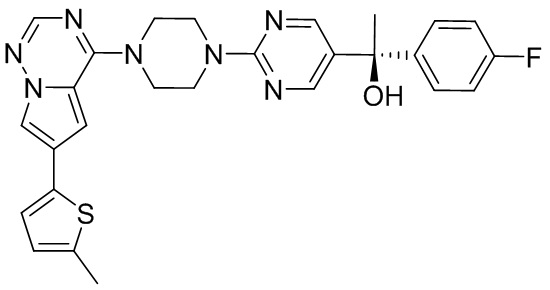
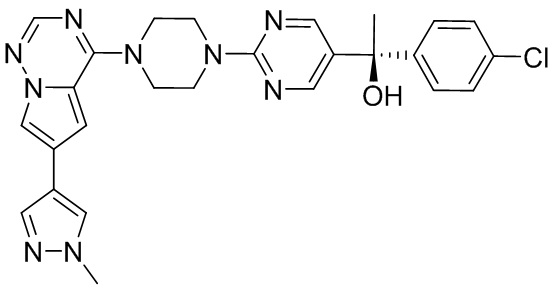
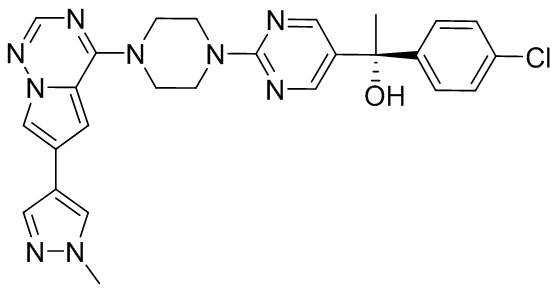
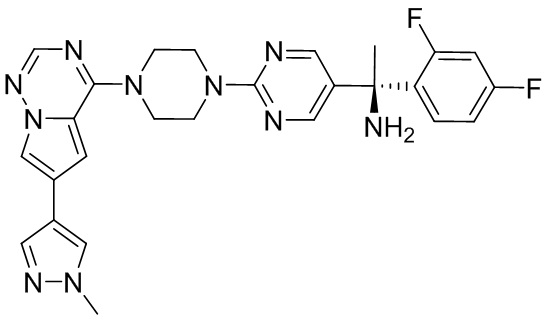
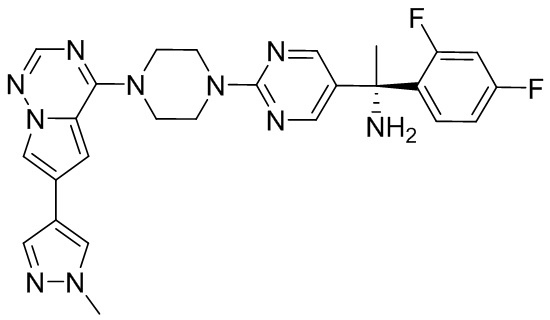
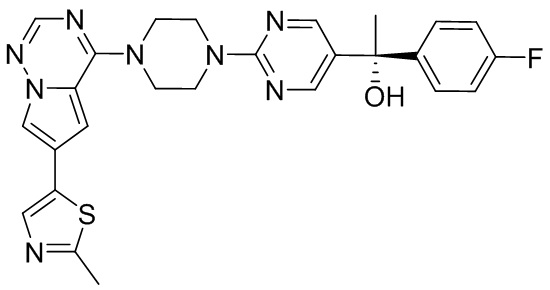
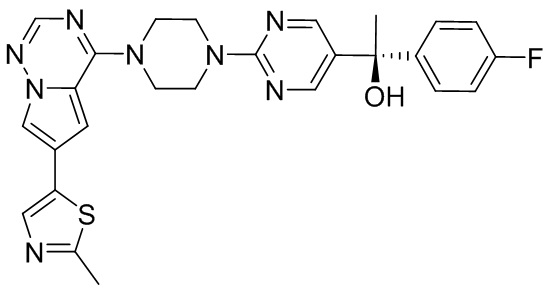
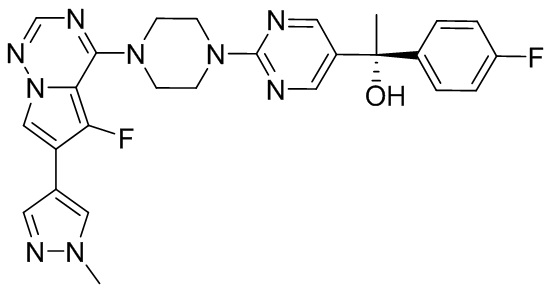
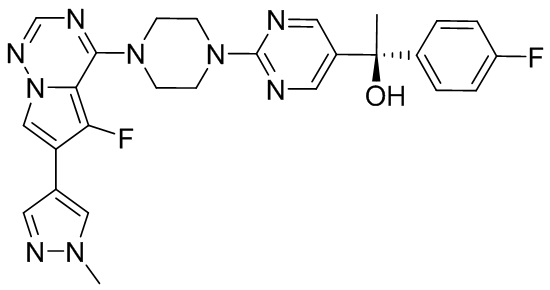
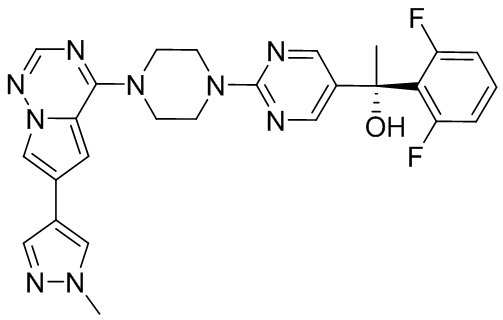
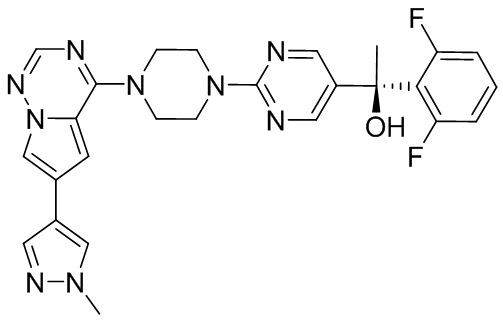
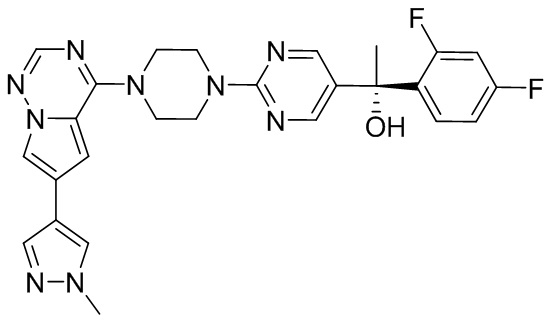
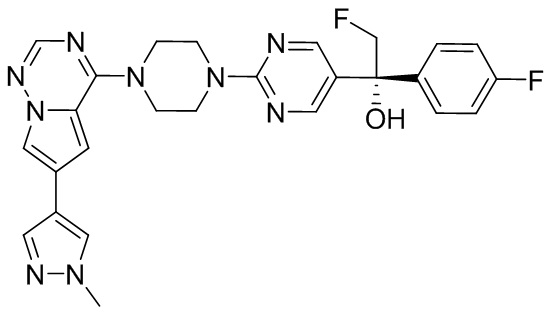
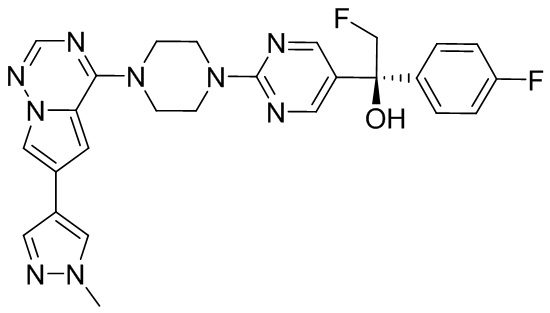
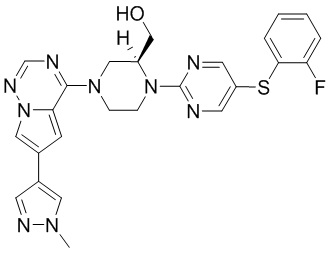
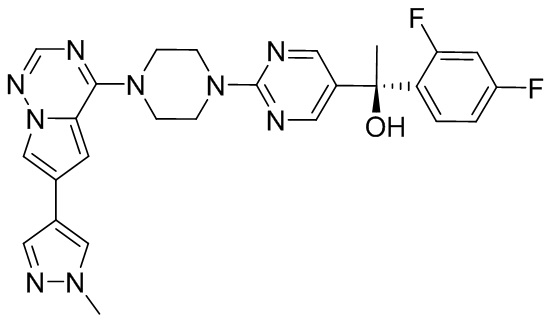
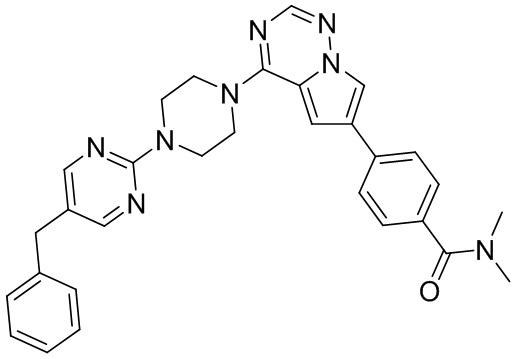
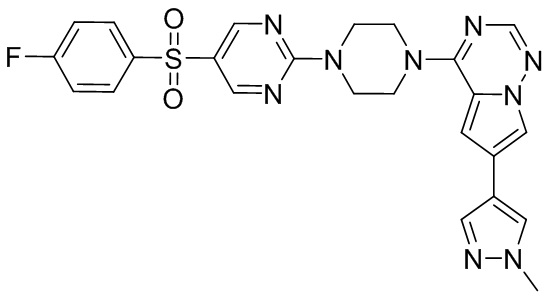
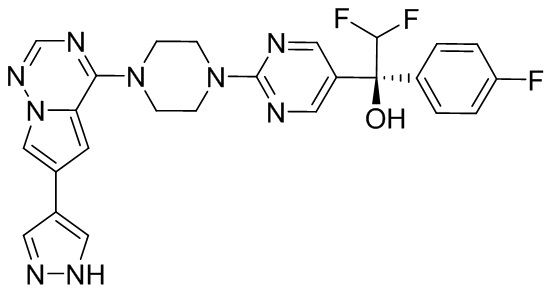
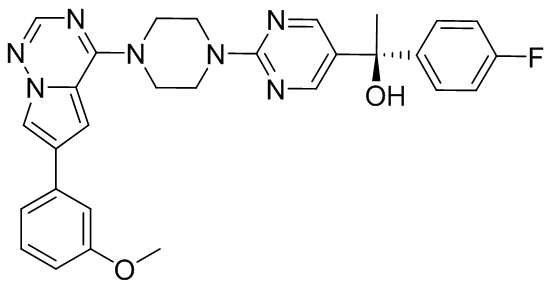
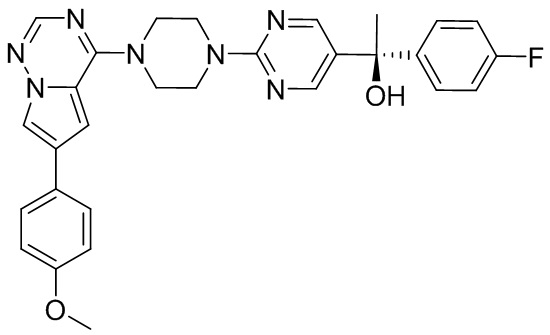
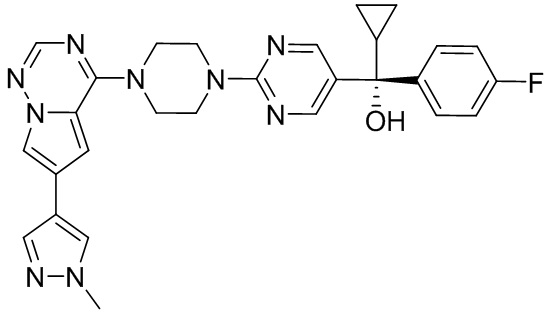
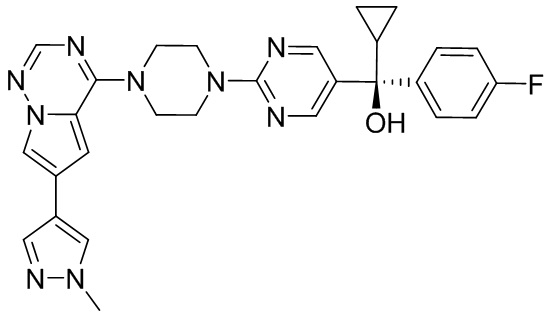
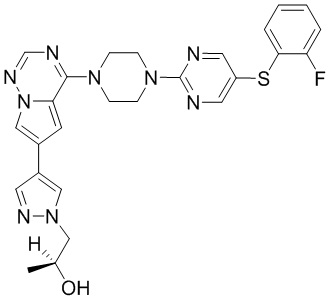
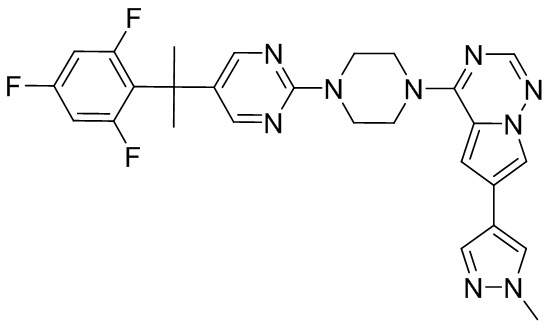
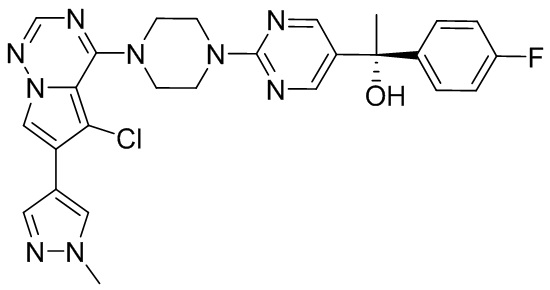
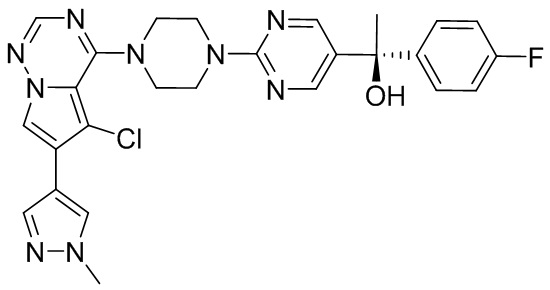
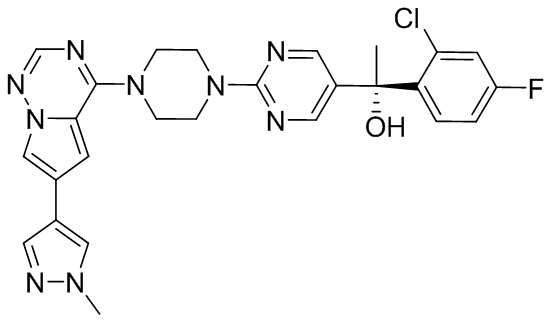
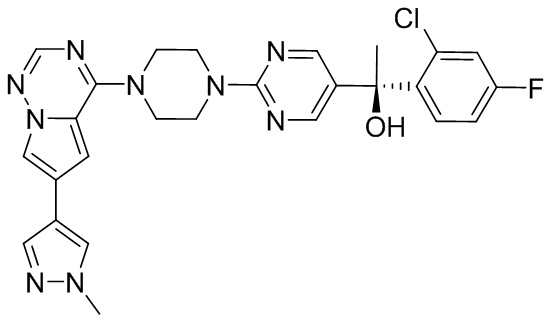
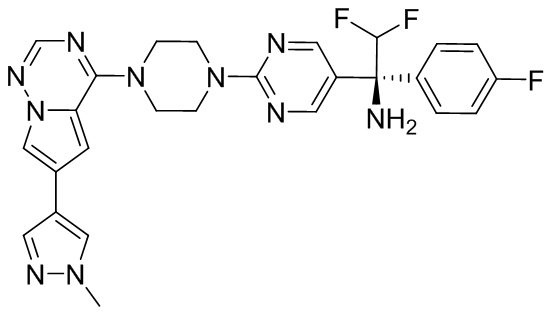
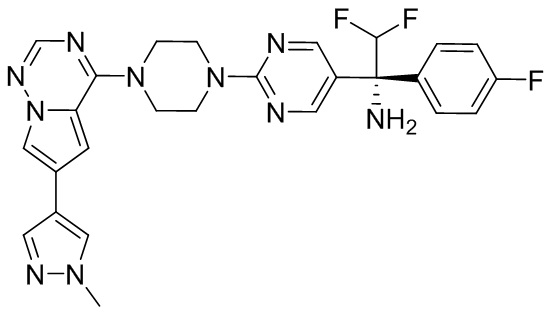
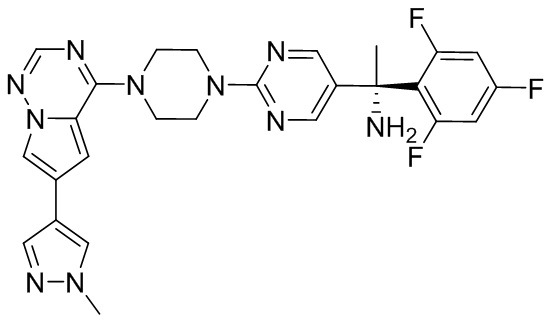
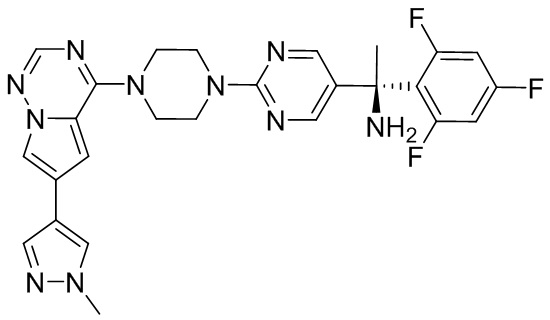
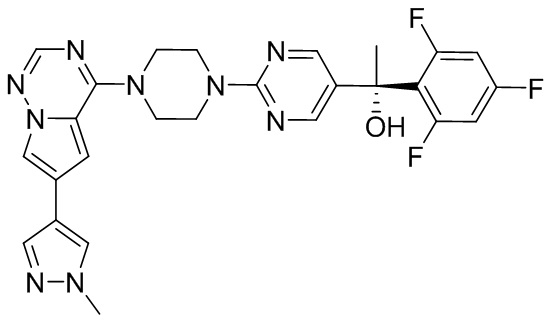
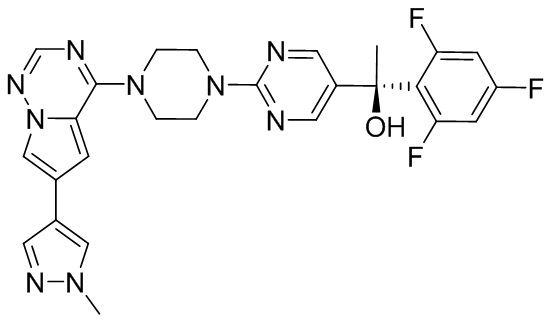
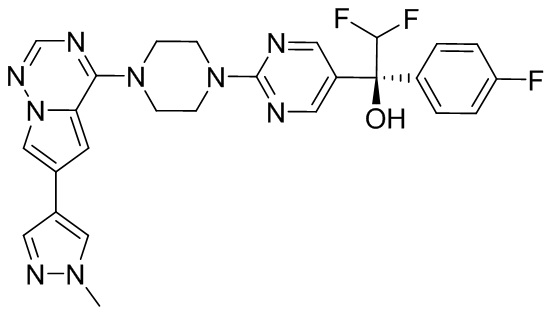
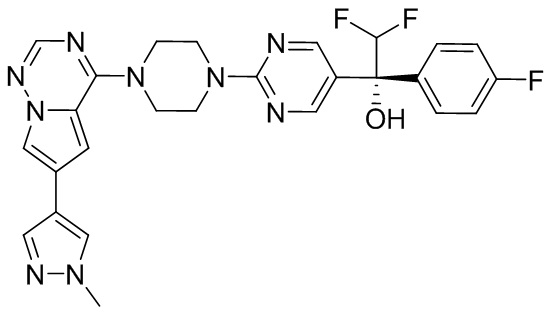
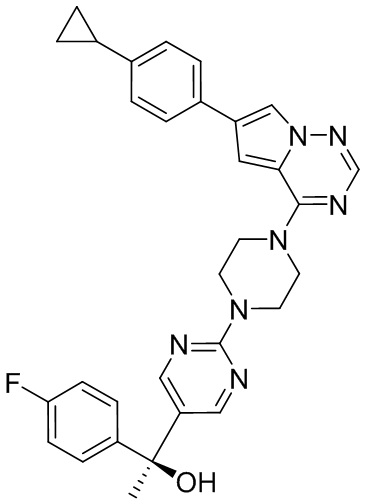
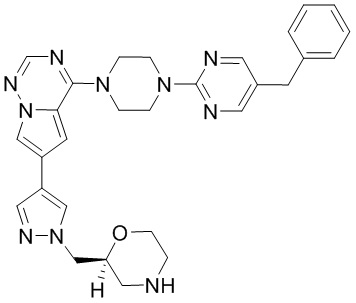
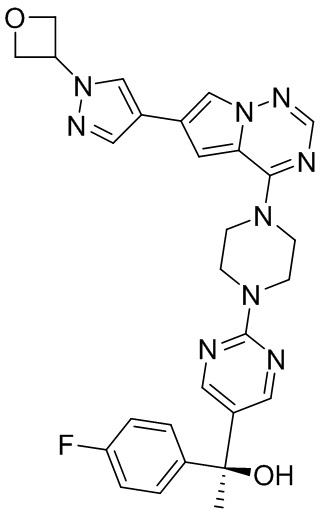
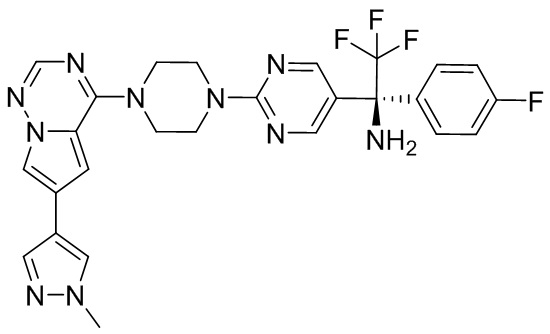
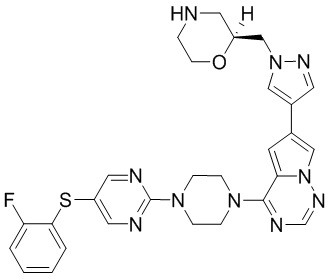
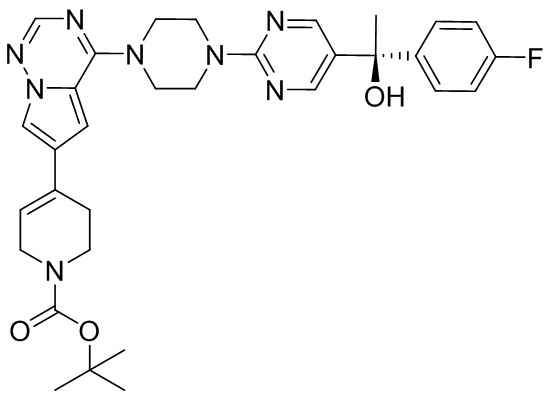
14. Соединение или его фармацевтически приемлемая соль, где соединение представляет собой
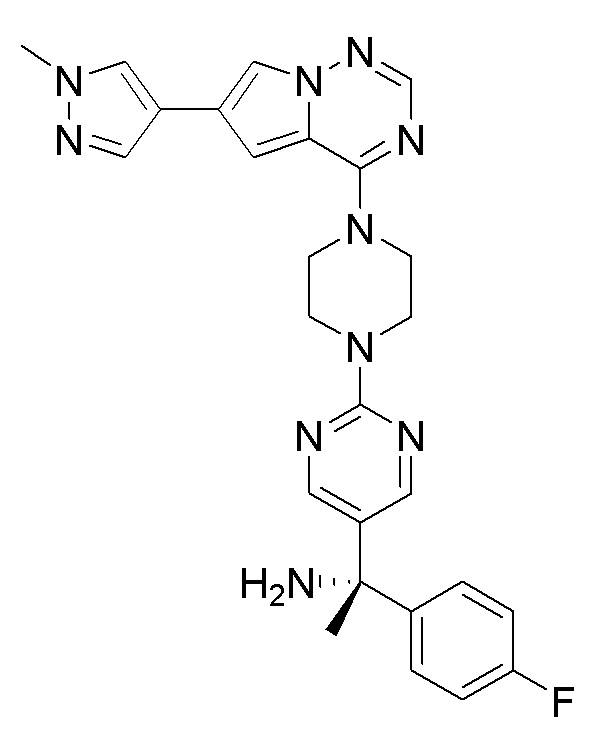
15. Соединение, представляющее собой
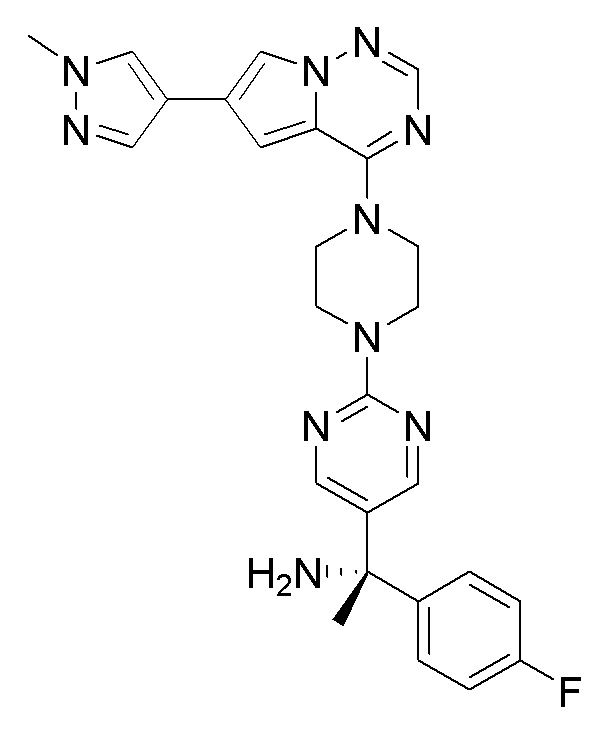
16. Фармацевтическая композиция, обладающая модулирующей активностью в отношении KIT и/или PDGFRα, содержащая фармацевтически приемлемый носитель и терапевтически эффективное количество соединения по любому из пп.1-15 или его фармацевтически приемлемой соли.
17. Способ лечения мастоцитоза, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
18. Способ лечения желудочно-кишечной стромальной опухоли, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
19. Способ лечения острого миелоидного лейкоза, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
20. Способ лечения патологического состояния, опосредованного мутантной KIT, причем указанная KIT имеет мутацию в 9 экзоне, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
21. Способ лечения патологического состояния, опосредованного мутантной KIT, причем указанная KIT имеет мутацию в 11 экзоне, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
22. Способ лечения патологического состояния, опосредованного мутантной KIT, причем указанная KIT имеет мутацию в 17 экзоне, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
23. Способ лечения патологического состояния, опосредованного мутантной KIT, причем указанная KIT мутирована в 816 остатке, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
24. Способ лечения патологического состояния, опосредованного мутантным PDGFRα, причем указанный PDGFRα имеет мутацию в 18 экзоне, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
25. Способ лечения патологического состояния, опосредованного KIT, включающий введение пациенту терапевтически эффективного количества соединения по любому из пп.1-15 или его фармацевтически приемлемой соли или фармацевтической композиции по п.16.
| WO 2005117909 A2, 15.12.2005 | |||
| WO 2012027495 A1, 01.03.2012 | |||
| ПИРРОЛТРИАЗИНОВЫЕ ИНГИБИТОРЫ КИНАЗ | 2000 |
|
RU2331640C2 |
| СПОСОБ СТАБИЛИЗАЦИИ НЕНАСЫЩЕННЫХ КАУЧУКОВ И РЕЗИН НА ИХ ОСНОВЕ | 0 |
|
SU309012A1 |
| WO 2007065100 A1, 07.06.2007. | |||

