Изобретение относится к химической технологии неорганических веществ и может быть использовано в атомной промышленности для получения тетрафторида урана из диоксида урана сухим методом. Тетрафторид урана, полученный по предложенному способу, может применяться в производстве гексафторида урана или металлического урана.
Способы получения тетрафторида урана делят на три большие группы - сухие, полусухие и мокрые, в зависимости от используемых фторирующих реагентов. Сухие способы предполагают взаимодействие оксидов урана с газообразными реагентами (фтористым водородом, бифторидом аммония, фреонами). При реализации сухих способов получения тетрафторида урана используется оснастка из никеля или сплавов на его основе: монель-металл (Ni -Cu - Fe - Mn); хастелой (Ni - Mo - Fe); ИНОР-8 (Ni -Mo -Cr- Fe).
При получении тетрафторида урана сухим способом вследствие коррозии аппаратуры в нем могут содержаться эти элементы (Ni, Fe, Cr), что особо нежелательно при использовании такого тетрафторида для получения металлического урана. Эти элементы либо растворяются в уране, либо образуют интерметаллические соединения и эвтектики с ним, что делает невозможным удаление этих примесей при рафинировочной плавке чернового урана.
В основе полусухого способа получения тетрафторида урана лежит реакция диоксида урана с водными растворами плавиковой кислоты. В основе мокрых способов лежат химические реакции, протекающие в жидкой фазе и приводящие к осаждению UF4. Основная проблема полусухого и мокрого способов заключается в обезвоживании полученного гидрата тетрафторида урана (nUF4⋅H2O). Обезвоживание UF4 является сложной задачей и в этом заключается существенный недостаток полусухого и мокрого способов.
Известен способ получения тетрафторида урана (патент GB №2222824, МПК C01G 43/06, опубл. 06.09.1989), по которому тетрафторид урана получают осаждением фтористоводородной кислотой при 95°С из раствора урана в концентрированной соляной кислоте. Недостатком этого способа является использование избытка фтористоводородной кислоты при 95°С, являющейся опасным и коррозионно-активным веществом.
Также известен способ (патент RU №2257351, МПК C01G 43/06, опубл. 27.02.2005), по которому осаждение тетрафторида урана проводят из хлоридного неводного раствора урана фторидом щелочного металла или бифторидом аммония. Недостатками этого способа являются использование агрессивных хлорсодержащих растворов и трибутилфосфата, а также необходимость проведения операций промывки, фильтрации и сушки тетрафторида урана и утилизации (переработки) промывных вод.
Известен сухой способ получения тетрафторида урана, по которому смесь диоксида урана и бифторида аммония нагревают до температуры выше точки плавления бифторида (125°С), выдерживают в течение 8 часов, а полученную двойную соль урана разлагают в вакуумной печи (Н.С. Тураев, И.И. Жерин «Химия и технология урана», Москва, Цнииатоминформ, 2005 г., стр. 372). К недостаткам этого способа можно отнести большую длительность и многостадийность процесса и сложность аппаратурного оформления.
Известен способ получения тетрафторида урана, по которому для сокращения продолжительности процесса в смесь оксида урана и бифторида аммония добавляют карбамид и процесс синтеза двойной соли проводят при температуре выше точки кипения карбамида, но ниже температуры кипения бифторида аммония (RU №2601477, МПК C01G 43/06 (2006.1), опубл. 10.11.2016). Предложенный способ позволяетсократить продолжительность процесса. Однако по этому способу избыток бифторида аммония достигает 80% от стехиометрического количества, а разложение двойной соли проводят в вакууме или в инертной атмосфере. Кроме того, использование карбамида снижает объем полезной загрузки шихты более чем на 30%.
Наиболее близким по технической сущности к заявляемому техническому решению является способ получения тетрафторида урана (патент РФ №2625871, МПК C01G 43/06 (2006.01), опубл. 19.07.2017), по которому смесь порошков диоксида урана и бифторида аммония размещают в замкнутой емкости с ограниченным доступом воздуха, устанавливают замкнутую емкость в другую емкость с зазором, который заполняют засыпкой из углеграфитового материала в виде гранул таким образом, чтобы гранулы полностью укрывали упомянутую замкнутую емкость. Термообработку емкостей проводят в воздушной атмосфере в две стадии: на первой стадии (на стадии образования двойной соли урана) - при температуре выше точки плавления бифторида аммония, но ниже точки его кипения и на второй стадии (на стадии разложения полученной соли до тетрафторида урана) - при температуре выше начала окисления углеграфитового материала, но ниже температуры плавления тетрафторида урана. Изобретение позволяет значительно упростить аппаратурное оформление процесса, так как процесс синтеза и разложения двойной соли до тетрафторида урана осуществляют в воздушной атмосфере с использованием стандартного простого оборудования.
К недостаткам этого способа можно отнести повышенное содержание примесей в получаемом тетрафториде урана, обусловленное коррозией материала оснастки, в которой происходит взаимодействие бифторида аммония и оксида урана. При этом содержание такого элемента как никель (материал оснастки) в тетрафториде, в зависимости от продолжительности использования оснастки составляет 0,05-0,1 мас. %. Черновой уран, полученный при металлотермическом восстановлении такого тетрафторида, концентрирует в себе примесь никеля, удаление которого при рафинировочной плавке затруднительно из-за растворения никеля в уране. Кроме того, большой избыток бифторида аммония, используемого для получения тетрафторида урана (избыток 80-100% от стехиометрически необходимого количества), усугубляет процесс коррозии оснастки и усложняет процесс улавливания и утилизации непрореагировавшего реагента и продуктов его разложения (HF, NH3).
Задачей изобретения является снижение содержания примесей в получаемом тетрафториде урана, уменьшение коррозии используемой оснастки и увеличение продолжительности ее использования.
Для решения поставленной задачи и достижения при использовании изобретения технического результата в способе получения тетрафторида урана, включающем смешивание диоксида урана с бифторидом аммония, загрузку смеси порошков диоксида урана и бифторида аммония в замкнутую емкость с ограниченным доступом воздуха, помещение этой емкости в другую емкость с зазором, заполнение зазора засыпкой из углеграфитового материала, термообработку полученной смеси на стадии синтеза двойной соли урана и термообработку двойной соли на стадии ее разложения до тетрафторида урана, согласно изобретению перед загрузкой смеси порошков диоксида урана и бифторида аммония в первую емкость на ее внутренней поверхности размещают углеграфитовую ткань, на которую наносят слой оксида или фторида щелочноземельного металла, а термообработку на стадии синтеза двойной соли урана вначале проводят при температуре выше точки плавления бифторида аммония, но ниже точки его кипения, а затем - при температуре, превышающей точку кипения бифторида аммония на 10-20°С.
В частных случаях осуществления изобретения термообработку на стадии синтеза двойной соли урана проводят вначале при 200-220°С, а затем при температуре 250-260°С.
В качестве оксида или фторида щелочноземельного металла используют оксид или фторид кальция.
В качестве оксида или фторида щелочноземельного металла используют оксид или фторид магния.
В качестве углеграфитовой ткани используют углеграфитовый войлок, толщина которого может составлять 1,0-2,0 мм.
Толщину слоя оксида или фторида щелочноземельного металла выбирают 0,3-0,5 мм.
Бифторид аммония берут в избытке 30-40 мас. % от стехиометрически необходимого количества.
Слой оксида или фторида щелочноземельного металла наносят на углеграфитовую ткань путем ее побелки водной суспензией компонентов.
Проведение первой половины процесса синтеза двойной соли урана при температуре ниже точки кипения бифторида аммония, т.е. ниже (238-240°С) продиктовано необходимостью более полного использования реагента - бифторида аммония. Проведение второй половины процесса синтеза двойной соли при температуре, превышающей точку кипения бифторида аммония на 10-20°С, приводит к «объемному» вскипанию бифторида на развитой поверхности углеродных волокон. Это приводит к перемешиванию жидкого бифторида и увеличению скорости реакции, что позволяет снизить избыток бифторида аммония в шихте с 80-100% до 30-40% от стехиометрического количества. Это уменьшение концентрации агрессивного компонента в шихте также способствует снижению коррозии аппаратуры и увеличению ресурса ее использования. Увеличение температуры второй половины синтеза двойной соли урана более чем на 20°С выше точки кипения бифторида аммония нецелесообразно из-за увеличения упругости паров бифторида аммония, «холостого» его проскока и обусловленного этим необходимостью увеличения его избытка в шихте. Проведение всего процесса синтеза двойной соли при температуре выше точки кипения бифторида аммония нецелесообразно, так как сопряжено с необходимостью еще большего его избытка в шихте.
Скорость реакции расплавленного бифторида аммония с оксидом урана мало зависит от температуры (125-238°С) и в большей мере определяется величиной дисперсности (удельной поверхностью) порошка оксида урана. По мере увеличения толщины слоя на поверхности частиц оксида урана образовавшейся двойной соли, скорость реакции замедляется и определяется диффузией реагента через образовавшийся слой. В зависимости от величины дисперсности оксида урана продолжительность реакции расплава бифторида аммония с оксидом урана с образованием двойной соли может составлять 8-20 ч.
Продолжительность процесса термообработки в предлагаемом способе на каждом этапе стадии синтеза двойной соли урана (200-220°С и 250-260°С) может составлять от 0,75 ч до 1,5 ч и зависит от дисперсности диоксида урана, толщины слоя шихты и толщины углеграфитового материала. Кроме того, на стадии разложения двойной соли урана до тетрафторида урана, которую проводят при температуре выше начала окисления углеграфитового материала (≥600°С), но ниже температуры плавления тетрафторида урана (≤950°С), углерод ткани начинает взаимодействовать с кислородом воздуха, находящегося в ткани и порах шихты с образованием монооксида углерода, являющегося хорошим восстановителем. Образование восстановительной среды в реакционном пространстве на стадии разложения двойной соли NH4UF5 до тетрафторида урана приводит к существенному снижению коррозии оснастки.
В качестве углеграфитовой ткани могут использоваться ткани типа «Урал», «Десна», ТГН, ТГН-2М и т.п. Также интерес представляет углеродная вуаль (вейл, углеродная бумага) толщиной 0,52 мм и поверхностной плотностью 40 г/м2. Использование в качестве углеграфитовой ткани углеграфитового войлока является предпочтительным из-за его большой удельной поверхности (более 100 м2 на 1 г войлока).
Слой оксида или фторида щелочноземельного металла, нанесенный на углеграфитовую ткань, экранирует ее поверхность от контакта с образующимся тетрафторидом урана. Целесообразность использования в качестве оксидов и фторидов щелочноземельных металлов оксидов и фторидов кальция или магния обусловлена тем, что кальций и магний применяют для металлотермического получения урана из его тетрафторида, при этом они легко удаляются при рафинировочной плавке чернового урана.
Осуществление способа.
В соответствии с заявляемым способом порошок диоксида урана смешивали с порошком бифторида аммония, взятым в избытке 30-40 мас. % от стехиометрически необходимого количества. Перед загрузкой полученной смеси порошков в никелевый контейнер с закрывающейся крышкой его внутреннюю поверхность покрывали углеграфитовым войлоком типа «Карбопон» толщиной 1,0-2,0 мм, на который методом побелки наносили водную суспензию гидроксида или фторида щелочноземельного металла, например - кальция или магния. Нанесенное покрытие водной суспензии сушили при 100-200°С. Этот контейнер со смесью порошков указанных ингредиентов размещали с зазором 1,0-2,0 см в другом контейнере из жаропрочной стали, заполняли гранулами углеграфитового материала до верхнего уровня таким образом, чтобы он полностью закрывал крышку первого контейнера. Систему емкостей с углеграфитовой засыпкой (гранулы графита ГМЗ размером 0,5-3,0 мм) нагревали в воздушной атмосфере.
Режимы термообработки и соотношение ингредиентов шихты приведены в таблице 1.
Стадия синтеза двойной соли фторида урана происходила по реакции: 2UO2+5NH4HF2=2NH4UF5+3NH3+4H2O
При нагревании контейнеров до температуры 750°С происходило разложение двойной соли до тетрафторида урана. Соотношение ингредиентов и результаты химического и рентгенофазового анализов полученного UF4 приведены в таблицах 1 и 2.
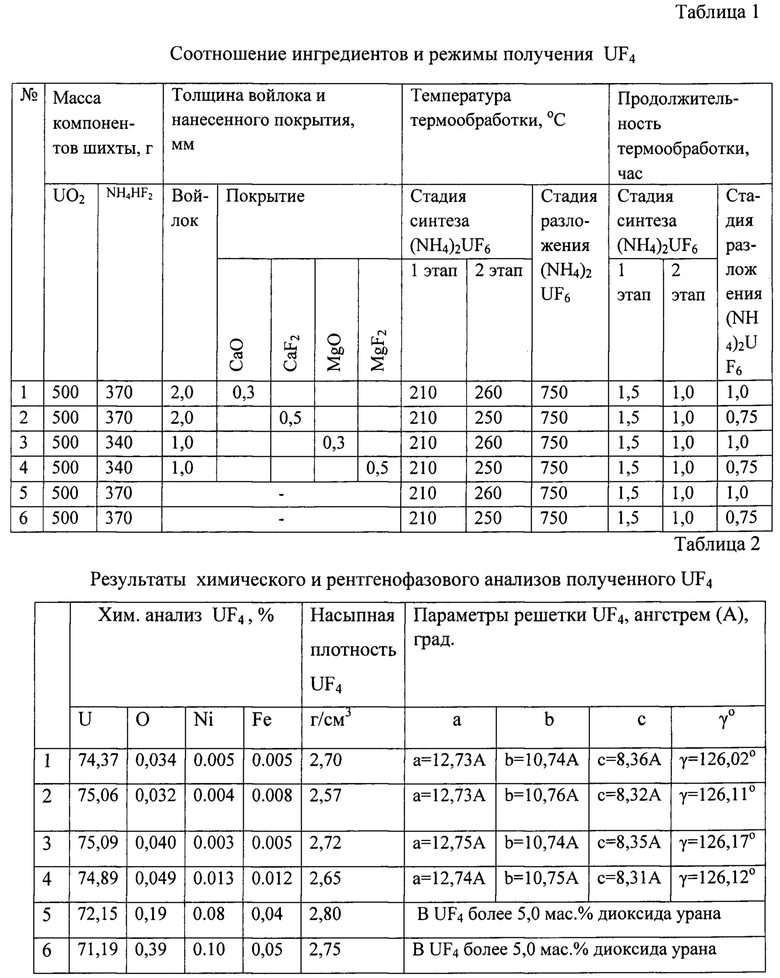
Примечание: в опытах №№1, 2, 3, 5, 6 - оснастка из никеля, в №4 - оснастка из монеля.
Из данных таблицы 2 видно, что в опытах №№1, 2, 3, 4 был получен UF4 хорошего качества с низким содержанием никеля и железа. Насыпная плотность полученного тетрафторида урана составляла 2,57-2,80 г/см3. В опытах №5 и №6, проведенных без углеграфитового войлока, отмечено повышенное содержание никеля, железа и кислорода.
Таким образом, предложенный способ позволяет сократить содержание примеси металла оснастки (никеля) в тетрафториде урана с 0,1-0,08 до 0,003 мас. %. Кроме того, в 2-3 раза повышается ресурс использования оснастки для получения тетрафторида урана в контейнерах, изготовленных из никеля или сплавов на его основе (монель, инконель). Предложенный способ по сравнению с прототипом позволяет уменьшить избыток используемого бифторида аммония с 80-100% от стехиометрически необходимого количества до 30-40%, что, в свою очередь, упрощает процесс улавливания и утилизации непрореагировавшего избыточного агрессивного реагента, а также приводит к уменьшению коррозии аппаратуры и увеличению ресурса ее использования.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРИДА УРАНА | 2016 |
|
RU2625871C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРИДА УРАНА | 2015 |
|
RU2601477C1 |
| СПОСОБ ПЕРЕРАБОТКИ ЦИРКОНИЕВОГО КОНЦЕНТРАТА | 1993 |
|
RU2048559C1 |
| Способ переработки полифторида аммония и способ получения трифторида азота, применяемый в нем | 2024 |
|
RU2829882C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТРИФТОРИДА АЗОТА | 2006 |
|
RU2317251C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРИДА КРЕМНИЯ | 2001 |
|
RU2182558C1 |
| СПОСОБ ОЧИСТКИ ТЕТРАФТОРИДА УРАНА | 2013 |
|
RU2542286C1 |
| СПОСОБ ПЕРЕРАБОТКИ МОНАЦИТОВОГО СЫРЬЯ | 2017 |
|
RU2667932C1 |
| Способ получения фторида водорода из смеси дифторида кальция и диоксида кремния | 2020 |
|
RU2757017C1 |
| СПОСОБ СИНТЕЗА НЕОРГАНИЧЕСКИХ ФТОРСОДЕРЖАЩИХ СОЕДИНЕНИЙ | 2004 |
|
RU2278073C1 |
Изобретение относится к химической технологии неорганических веществ, а именно к способу получения тетрафторида урана сухим методом, который может применяться в производстве гексафторида урана или металлического урана. Способ включает смешивание порошков диоксида урана с бифторидом аммония, загрузку смеси порошков в замкнутую емкость с ограниченным доступом воздуха, на внутренней поверхности которой предварительно размещают углеграфитовую ткань с нанесенным слоем оксида или фторида щелочноземельного металла, помещение этой емкости в другую емкость с зазором, заполнение зазора засыпкой из углеграфитового материала и термообработку емкостей в воздушной атмосфере на стадии синтеза двойной соли урана, осуществляемую вначале при температуре выше точки плавления бифторида аммония, но ниже точки его кипения, а затем при температуре, превышающей точку кипения бифторида аммония на 10-20°С, и на стадии разложения двойной соли урана до тетрафторида урана, осуществляемую при температуре выше начала окисления углеграфитового материала, но ниже температуры плавления тетрафторида урана. Изобретение обеспечивает снижение содержания примесей в получаемом тетрафториде урана, уменьшение коррозии используемой оснастки и увеличение продолжительности ее использования. 8 з.п. ф-лы, 2 табл.
1. Способ получения тетрафторида урана, включающий смешивание диоксида урана с бифторидом аммония, загрузку смеси порошков диоксида урана и бифторида аммония в замкнутую емкость с ограниченным доступом воздуха, помещение этой емкости в другую емкость с зазором, заполнение зазора засыпкой из углеграфитового материала, термообработку емкостей в воздушной атмосфере на стадии синтеза двойной соли урана и на стадии разложения двойной соли урана до тетрафторида урана, которую проводят при температуре выше начала окисления углеграфитового материала, но ниже температуры плавления тетрафторида урана, отличающийся тем, что перед загрузкой смеси порошков диоксида урана и бифторида аммония в первую емкость на ее внутренней поверхности размещают углеграфитовую ткань, на которую наносят слой оксида или фторида щелочноземельного металла, а термообработку на стадии синтеза двойной соли урана вначале проводят при температуре выше точки плавления бифторида аммония, но ниже точки его кипения, а затем - при температуре, превышающей точку кипения бифторида аммония на 10-20°С.
2. Способ по п. 1, отличающийся тем, что термообработку на стадии синтеза двойной соли урана проводят вначале при 200-220°С, а затем - при температуре 250-260°С.
3. Способ по п. 1, отличающийся тем, что в качестве оксида или фторида щелочноземельного металла используют оксид или фторид кальция.
4. Способ по п. 1, отличающийся тем, что в качестве оксида или фторида щелочноземельного металла используют оксид или фторид магния.
5. Способ по п. 1, отличающийся тем, что в качестве углеграфитовой ткани используют углеграфитовый войлок.
6. Способ по п. 5, отличающийся тем, что толщину углеграфитового войлока выбирают равной 1,0-2,0 мм.
7. Способ по п. 1, отличающийся тем, что толщину слоя оксида или фторида щелочноземельного металла выбирают 0,3-0,5 мм.
8. Способ по п. 1, отличающийся тем, что бифторид аммония берут в избытке 30-40 мас.% от стехиометрически необходимого количества.
9. Способ по п. 1, отличающийся тем, что слой оксида или фторида щелочноземельного металла наносят на углеграфитовую ткань путем ее побелки водной суспензией компонентов.
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРИДА УРАНА | 2016 |
|
RU2625871C1 |
| СПОСОБ ПОЛУЧЕНИЯ ТЕТРАФТОРИДА УРАНА | 2015 |
|
RU2601477C1 |
| СПОСОБ ПОЛУЧЕНИЯ ФТОРИДОВ УРАНА | 2003 |
|
RU2257351C2 |
| ИТЕРАЦИОННЫЙ СПОСОБ ВЫДЕЛЕНИЯ ТРЕНДА И УСТРОЙСТВО ДЛЯ ЕГО ОСУЩЕСТВЛЕНИЯ | 2001 |
|
RU2222824C2 |

