Настоящее изобретение касается композиции катализатора без благородных металлов, которая применима, например, для окисления твердых частиц (ТЧ).
Уровень техники изобретения
Выхлопные газы дизельных двигателей содержат ТЧ, которые могут вызывать экологические проблемы. Для захвата ТЧ был разработан фильтр дизельных частиц (ФДЧ), в котором ТЧ отфильтровываются из выхлопного газа. Наиболее обычным типом ФДЧ является керамический стеннопроточный фильтр, изготовленный из SiС или кордиерита. Так как собранные ТЧ накапливаются в этом фильтре, противодавление увеличивается и мощность двигателя снижается. Поэтому стеннопроточный фильтр необходимо регенерировать непрерывно (пассивно) и/или периодически (активно), выжигая сажу (смотри, например, A.P. Walker et al, Controlling particulate emissions from Diesel vehicles, Topics in Catalysis Vol. 28, 2004, 165-170).
Например, в US 8114354 описано, что для пассивной регенерации на фильтр должна быть нанесена каталитическая композиция. Эта каталитическая композиция, содержащая элементы группы Аl, Се, Zr, Si, цеолиты, неблагородные и благородные металла, окисляет NО в NО2, который является лучшим окислителем для сажи, чем кислород. Активная некатализируемая регенерация работает около 650°С. Дополнительное впрыскивание топлива непосредственно перед дизельным катализатором окисления (ДКО) является самым обычным способом достижения этой температуры. Способ дополнительного впрыскивания имеет недостатки в увеличенном расходе топлива и разбавлении моторного масла.
Согласно презентации, сделанной S. Spiess (Umicore AG) на САРоС9 (август 2012), автомобили с прямым впрыскиванием бензина получают долю рынка, достигая новых пределов выбросов СО2. С помощью этой новой технологии потребление топлива и выброс СО2 могут быть снижены, но такие автомобили выделяют значительно больше частиц, чем традиционные бензиновые двигатели, и в 10 раз больше, чем новые дизельные двигатели. Это загрязнение можно предотвращать с помощью бензинового фильтра частиц (БФЧ), который может существенно снижать выбросы частиц.
Уже были сделаны усилия, чтобы обеспечить катализатор для каталитического окисления ТЧ кислородом при меньших температурах, чтобы уменьшить число циклов активной регенерации и время регенерации. Таким образом, расход топлива может быть уменьшен, а качество работы моторного масла может быть увеличено.
В DЕ 102 00 900 2182 раскрываются различные смешанные оксиды без благородных металлов для окисления сажи. Раскрываются оксиды на основе железа, хрома и кобальта.
ЕР 2 210 861 касается дизельного фильтра частиц, содержащего церийсодержащий смешанный оксид с Вi и Рr, в котором мольное отношение Се, Вi, Рr выражается в виде Се:Вi:Рr=(1-х-у):х:у, где 0<х≤0,3 и 0<у≤0,5.
В US 8071501 раскрывается катализатор очистки выхлопного газа, содержащий смешанный оксид и металл платиновой группы (МПГ, где МПГ включает Ru, Rh, Pd, Os, Ir и Pt), где данный смешанный оксид состоит из Се, Вi и лантанида за исключением Lа и Се. В ЕР 2 269 730 этот тип смешанных оксидов расширили с помощью дополнительного элемента, выбранного из группы 3, 4 и 13 периодической таблицы элементов.
В US 2009/0288401 раскрывается смешанный оксид для катализатора очистки выхлопного газа, содержащий Се, Вi и один или несколько элементов, выбранных из щелочноземельных металлов за исключением бериллия. Дополнительно, один или несколько добавочных элементов могут быть выбраны из Zr, Рr и Tb.
Смешанные оксиды перовскитного типа для окисления сажи заявляются в ЕР 1 829 609. Эти перовскитные композиции могут быть выражены структурной формулой RТО3, где R содержит один или несколько элементов, выбранных из группы, состоящей из Lа, Sr, Ва, Са и Li; а Т содержит один или несколько элементов, выбранных из группы, состоящей из Mn, Fe, Co, Cu, Zn, Ga, Zr, Mo, Mg, Al и Si.
В WО 2006/044822 (ЕР 1 817 096) раскрывается катализатор для окисления сажи, состоящий из щелочного металла, церия, кислорода и возможно металла платиновой группы и/или циркония. Раскрывается, что наиболее активные материалы являются комбинациями церия и калия или церия и цезия.
В Kripasindhu Sardar et al, "Nanocrystalline Cerium-Bismuth Oxides: Synthesis, Structural Characterization, and Redox Properties", Chemistry of Materials, vol. 22, no. 22, 23 November 2010 (2010-11-23), pages 6191-6201, ISSN: 0897-4756, doi: 10.1021/cm1025848 раскрывается смешанный оксид церия-висмута с фоновым уровнем натрия.
В ЕР 2 098289 раскрывается смешанный оксид для использования в катализаторе очистки выхлопного газа, содержащий Се, Вi и один или несколько элементов, выбранных из Mg, Ca, Sr и Ва. Раскрывается, что этот катализатор пригоден для сжигания ТЧ дизельного выхлопного газа при низких температурах, и он с трудом отравляется под действием оксида серы.
Задача и сущность настоящего изобретения
Композиции для каталитического окисления ТЧ, раскрытые в предшествующем уровне техники, не выполняют все требования по каталитической эффективности, сопротивлению соединениям серы и/или устойчивости к гидротермической обработке. Поэтому целью настоящего изобретения является обеспечить новую композицию для каталитического окисления ТЧ, которая имеет более высокую каталитическую активность по сравнению с материалами предшествующего уровня техники, демонстрирует более высокую гидротермическую устойчивость, и которая дружественна для окружающей среды.
В одном аспекте настоящее изобретение обеспечивает композицию с формулой
Се1-a-b-cNaMbDcOx I
в которой
М обозначает один или несколько элементов из группы щелочных металлов кроме натрия, предпочтительно калий,
N обозначает Вi и/или Sb, предпочтительно Вi,
D присутствует или отсутствует, и если присутствует выбирается из одного или нескольких элементов из
- Mg, Ca, Sr, Ba; предпочтительно Са, Sr, Ва; более предпочтительно Sr,
- Y, Lа, Рr, Nd, Sm, Gd, Еr; предпочтительно Y, Рr, Lа, Nd; более предпочтительно Рr,
- Fе, Zr, Nb, Аl; в одном аспекте предпочтительно Ае, в другом аспекте предпочтительно Аl,
а является числом в интервале 0<а≤0,9, например 0,01≤а≤0,9,
b является числом в интервале 0<b≤0,3, например 0,01≤b≤0,3, например 0,1≤b≤0,2,
с является числом в интервале 0≤с≤0,2; предпочтительно 0≤с≤0,1,
а плюс b плюс с равно <1,
и
х является числом в интервале 1,2≤х≤2.
В дополнительном аспекте в композиции с формулой I присутствует D.
В другом аспекте в композиции с формулой I D отсутствует, и в этом аспекте настоящее изобретение обеспечивает композицию, которая имеет формулу
Се1-a-bNaMbOx II
в которой
М обозначает один или несколько элементов из группы щелочных металлов кроме натрия,
N обозначает Вi и/или Sb,
а является числом в интервале 0<а≤0,9,
b является числом в интервале 0<b≤0,3,
а плюс b равно <1, и
х является числом в интервале 1,2≤х≤2.
В дополнительном аспекте настоящее изобретение обеспечивает композицию, которая выбрана из группы, состоящей из
Bi0,45Ce0,45K0,10O1,65-1,4,
Bi0,40Ce0,40K0,20O1,4-1,2,
Bi0,30Ce0,60K0,10O1,7-1,4,
Bi0,80Ce0,10K0,10O1,5-1,4,
Bi0,10Ce0,80K0,10O1,8-1,4,
Bi0,4Ce0,4K0,1Sr0,1O1,55-1,35,
Bi0,4Ce0,4K0,1Pr0,1O1,6-1,4, и
Bi0,4Ce0,4K0,1Fe0,1O1,6-1,4,
Композиция, обеспеченная настоящим изобретением, например с формулой I, также называется здесь "композиция (согласно) настоящего изобретения".
Для целей настоящего описания и формулы изобретения выражение "щелочной металл" означает щелочной металл или смесь щелочных металлов, например более чем один щелочной металл, кроме натрия. Согласно ЮПАК щелочной металл является элементом из группы 1 периодической таблицы элементов.
В другом аспекте настоящее изобретение обеспечивает способ приготовления композиции согласно настоящему изобретению, который отличается тем, что используют полимерный комплексный способ, в котором комплексные ионы металлов соединяют полимеризацией, предпочтительно полиэстерификацией.
Способ, обеспеченный настоящим изобретением, также называет здесь "способом (согласно) настоящего изобретения".
Полимерный комплексный способ, используемый для приготовления соединений настоящего изобретения, представляет собой способ, аналогичный способу Печини, например аналогичный способу, описанному в A. L. Quinelato et al, ʺSynthesis and sintering of ZrO2-CeO2 powder by use of polymeric precursor based on Pechini processʺ, Journal of Material Science Vol. 36, 2001, 3825-3830.
Более конкретно, способ настоящего изобретения содержит этапы
а) приготовления раствора соли висмута в смеси воды, неорганической кислоты, одного или нескольких предшественников полимера и возможно одного или нескольких комплексообразующих агентов, в частности путем растворения оксида висмута в азотной кислоте и разбавления данного раствора смесью воды, одного или нескольких предшественников полимера и возможно одного или нескольких комплексообразующих агентов,
b) приготовления раствора соли церия, щелочной соли и возможно одной или нескольких солей D, заданных в соединении с формулой I, в воде, одном или нескольких предшественниках полимера и возможно в одном или нескольких комплексообразующих агентах,
в частности, путем растворения соли церия, щелочной соли и возможно одной или нескольких солей D, заданных в соединении с формулой I, в смеси воды, одного или нескольких предшественников полимера и возможно одного или нескольких комплексообразующих агентов,
с) возможно добавления неорганической кислоты к раствору, полученному на этапе b),
d) смешивания растворов металлов, полученных в а) и b), или в а) и с), при перемешивании или закручивании, и
е) термической обработки раствора, полученного в d), на воздухе в температурном интервале от 300 до 1000°С в течение от 1 до 120 часов,
предпочтительно от 350°С до 600°С, наиболее предпочтительно 375-500°С, например 400°С,
предпочтительно от 1 до 50 часов, более предпочтительно от 4 до 10 часов, например 5 часов,
с предпочтительно одной поддерживаемой температурой в температурном интервале 70-120°С и более предпочтительно второй поддерживаемой температурой в температурном интервале 120-250°С.
В способе настоящего изобретения комплексообразующий агент может служить органическим растворителем.
В способе настоящего изобретения предшественник полимера может служить органическим растворителем.
В способе настоящего изобретения подходящий предшественник полимера может быть использован на этапах а) и b), предпочтительно один и тот же предшественник полимера может быть использован на этапах а) и b). Подходящие предшественники полимера содержат поликарбоновые кислоты, гидроксикарбоновые кислоты, многоатомные спирты и их смеси, предпочтительно многоатомные спирты и поликарбоновые кислоты и их смеси, более предпочтительно многоатомные спирты. Наиболее предпочтительно, этиленгликоль используют в качестве предшественника полимера.
В способе настоящего изобретения подходящий комплексообразующий агент может быть использован на этапе а) и b), предпочтительно один и тот же комплексообразующий агент может быть использован на этапах а) и b). Подходящие комплексообразующие агенты включают органические соединения, например органические кислоты, кетоны, альдегиды, спирты, амины и их смеси, предпочтительно поликарбоновые кислоты, более предпочтительно лимонную кислоту и щавелевую кислоту, и наиболее предпочтительно лимонную кислоту. Комплексообразующий агент может увеличивать растворимость солей металлов, а также сшивать связи в полимерной структуре и дополнительно увеличивать однородность распределения металла в полимерном геле.
В способе настоящего изобретения подходящая щелочная соль включает соли щелочных металлов кроме натрия, например нитраты, оксиды, гидроксиды, карбонаты, сульфаты, ацетаты, галогениды, предпочтительно нитраты и карбонаты, наиболее предпочтительно нитраты.
В способе настоящего изобретения неорганическая кислота на этапе а) и с) включает в себя подходящие неорганические кислоты, например азотную кислоту, серную кислоту, соляную кислоту и их смеси, более предпочтительно азотную кислоту, серную кислоту и их смеси, наиболее предпочтительно азотную кислоту.
В случае использования многоатомных спиртов в качестве предшественника полимера без комплексообразующего агента или другого предшественника полимера, окислительную неорганическую кислоту, такую как азотная кислота, предпочтительно используют в качестве неорганической кислоты, чтобы окислять часть многоатомных спиртов в поликарбоновые кислоты, которые подходят для полиэстерификации с неокислительными многоатомными кислотами. При использовании смеси многоатомных спиртов и поликарбоновых кислот, неорганическая кислота не обязана быть окислительной кислотой, чтобы начинать полимеризацию.
Неожиданно было обнаружено, что композиции настоящего изобретения показывают более высокую каталитическую активность в окислении ТЧ по сравнению с материалами предшествующего уровня техники.
Композиции настоящего изобретения в свежем состоянии (прокаленные при 400°С) демонстрируют прекрасную каталитическую активность (выраженную как меньшие Т50-величины), которая сильно превышает активность материалов предшествующего уровня техники (что видно из таблицы 2). Путем введения щелочного металла кроме натрия, предпочтительно калия, в систему СеВiОх каталитическая активность в окислении ТЧ может быть увеличена по сравнению с церий-висмут-содержащими материалами, описанными в предшествующем уровне техники. Т50-величины (температуры, при которых наблюдают потерю 50% массы между 200°С и конечной температурой) смешанных церий-висмут-щелочных оксидов настоящего изобретения до 110°С ниже относительно церий-висмут-содержащих соединений из сравнительных примеров, что ясно из примера 4 и сравнительного примера 2 в таблице 2.
Кроме того, неожиданно было обнаружено, что композиции согласно настоящему изобретению показывают высокую термическую устойчивость до 800°С. Термически выдержанные композиции настоящего изобретения показывают более высокую каталитическую активность в окислении сажи по сравнению с материалами предшествующего уровня техники, что опять видно из таблицы 2. Т50-величины композиций настоящего изобретения (смешанные церий-висмут-щелочные оксиды) до 115°С ниже по сравнению с церий-висмут-содержащими сравнительными композициями из сравнительных примеров, что ясно, например, из примера 2 и сравнительного примера 3 в таблице 2.
Неожиданно, материалы на основе калия показывают более высокую активность в свежем состоянии, а также в выдержанном состоянии, чем смешанные церий-висмут-щелочные оксиды на основе натрия. Т50-величины композиций настоящего изобретения (смешанные церий-висмут-щелочные оксиды) находятся в свежем состоянии до 71°С ниже по сравнению с церий-висмут-натрий сравнительным примером, что видно, например, из примера 4 и сравнительного примера 1 в таблице 2. Также после выдерживания Т50-величины композиций настоящего изобретения (смешанные церий-висмут-щелочные оксиды) до 65°С ниже по сравнению с церий-висмут-натрий сравнительным примером, что видно, например, из примера 2 и сравнительного примера 1 в таблице 2. Если D присутствует в композиции настоящего изобретения, ее активность в свежем состоянии может быть дополнительно увеличена.
Вследствие более высокой каталитической активности композиций настоящего изобретения температура может быть снижена до интервала, который уже может достигаться в обычном цикле движения. Поэтому величина последующего впрыскивания топлива для активной регенерации может быть снижена.
Так как выхлопные газы двигателей сгорания содержат определенное количество воды, дополнительным важным аспектом настоящего изобретения является гидротермическая устойчивость. Щелочные материалы, описанные в предшествующем уровне техники, например в ЕР 1 817 096, неустойчивы в отношении воды. Неожиданно было обнаружено согласно настоящему изобретению, что путем введения висмута в систему смешанных церий-щелочных оксидов данная система показывает улучшенную устойчивость в отношении гидротермической обработки.
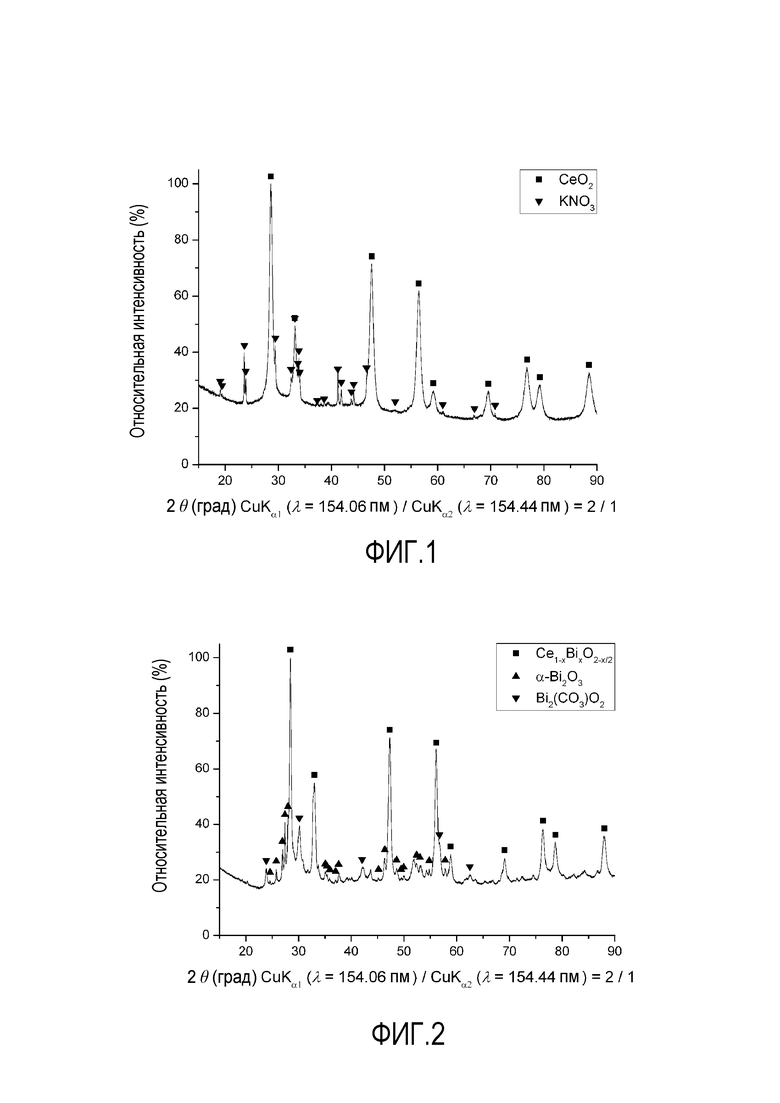
Кроме того, неожиданно было обнаружено, что композиции настоящего изобретения требуют меньших температур для окисления сажи после гидротермического выдерживания по сравнению материалами предшествующего уровня техники. Т50-величины композиции настоящего изобретения (смешанные церий-висмут-щелочные оксиды) после гидротермической обработки до 96°С ниже по сравнению с церий-калиевой системой (смотри пример 1 и сравнительный пример 5 в таблице 3). Считается, что этот аспект вызывается свободными калиевыми частицами в материале предшествующего уровня техники (смотри фиг.1). Напротив, никаких свободных калиевых частиц не наблюдается в композициях настоящего изобретения (смотри фиг.2).
Композиции настоящего изобретения представляют собой первые катализаторы на основе щелочных металлов для окисления ТЧ, которые показывают гидротермическую устойчивость.
Композиции настоящего изобретения представляют собой первые каталитические композиции, которые объединяют высокую каталитическую активность в окислении сажи материала на основе щелочных металлов и гидротермическую устойчивость материала без щелочных металлов.
Композиции настоящего изобретения применимы сами по себе или в комбинации с несущим материалом в покрытой или экструдированной форме для ФДЧ систем. В таком варианте осуществления композиции настоящего изобретения также применимы сами по себе или с носителем для фильтров частиц для бензиновых двигателей.
Композиции согласно настоящему изобретению могут быть использованы в применении для удаления сажи, в частности в системах постобработки выхлопного газа дизельных или бензиновых двигателей внутреннего сгорания, и дополнительно композиции настоящего изобретения также могут быть использованы в других применениях, например для удаления ТЧ в электростанциях, например в электростанциях на ископаемом топливе или электростанциях на биомассе.
В другом аспекте настоящее изобретение обеспечивает применение композиции настоящего изобретения для систем постобработки выхлопного газа, например, дизельных двигателей, бензиновых двигателей сгорания, двигателей на бедной смеси и электростанций.
Характеризация
Композиции настоящего изобретения частично характеризовали с помощью РФА.
Графики порошкового РФА (рентгеновская дифракция) получали, используя систему PANalytical X'Pert PRO с Ni-отфильтрованным Сu излучением (Сu-Кα1 и Сu-Кα2 дублет с длиной волны 1,5406 и 1,5444 Å). Прибор работал в геометрии Брегга-Брентано с PIXcel.
Для каталитического тестирования эффективности удаления ТЧ композиции подвергали условиям тестирования каталитического порошка, как описано ниже.
Условия для каталитического тестирования порошка
Приготовление образца
Синтезированные твердые образцы настоящего изобретения вручную перетирали в агатовой ступке. Порошкообразные образцы и углеродную сажу (CB, Printex 90, Evonik Degussa GmbH (способ A), или CB, Printex U, Evonik Carbon Black GmbH (способ B)) тщательно перемешивали шпателем в массовом отношении 4:1 до получения однородной смеси в режиме свободного контакта.
Измерение каталитической активности
Определение характерных температур сгорания сажи (Т50-величины, т.е. температуры, когда потерю 50% массы наблюдали между 200°С и конечной температурой) выполняли путем регистрации термогравиметрических данных с помощью двух разных способов.
Способ А
выполняли с помощью одновременного термического анализатора ТGА/DSС 1 (Mettler Toledo Corp.). Смесь 8% О2, 350 ч/млн СО, 250 ч/млн NО, 50 ч/млн пропана, 50 ч/млн SО2 и N2 в качестве баланса использовали в качестве модельного исходного газа. Полный поток газа был 50 мл/мин. Активности катализаторов в сгорании сажи измеряли в динамических условиях при скорости нагрева 10°С/мин в температурном интервале от 25°С до 700°С.
Способ В
выполняли с помощью NЕТSСН SТА 409 С/СD. Смесь 20% О2 в N2 использовали в качестве модельного исходного газа. Полный поток газа был 50 мл/мин. Активности катализаторов в сгорании сажи измеряли в динамических условиях при скорости нагрева 5°С/мин в температурном интервале от 25°С до 700°С.
Для тестирования термической устойчивости композиции подвергали следующим условиям в течение термического выдерживания:
Условия для термического выдерживания
Предварительную термическую обработку выполняли путем прокаливания порошкообразных образцов при 800°С в течение 2 часов в обычной муфельной печи.
Для тестирования гидротермической устойчивости каталитические композиции подвергали предварительной гидротермической обработке, описанной ниже.
Условия для предварительной гидротермической обработки
Предварительную гидротермическую обработку выполняли в изготовленном на заказ, 7-местном мультиклаве с тефлоновыми входами. Образцы (100-120 мг) суспензировали в исходном виде в 10 мл деионизованной воды (уровень наполнения: 33%). Автоклав нагревали до 150°С. Мультиклав вращали вокруг его цилиндрической оси в течение 60 минут в печи при 150°С и позволяли снова остывать до комнатной температуры. Гидротермически выдержанные образцы отделяли от жидкости фильтрованием, промывали деионизованной водой и сушили в вакуумном сушильном шкафу при 60°С и пониженном давлении (<10 мбар).
Более подробное описание изобретения
Настоящее изобретение будет теперь объяснено более подробно со ссылками на примеры и сравнительные примеры без ограничения ими. Температуры приводятся в градусах Цельсия (°С).
Синтез
Пример 1
Вi0,45Се0,45К0,10О1,65-1,4
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 50 мл деионизованной воды, 33,46 мл этиленгликоля (ЭГ) и 47,285 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (0,1048 г Вi2О3) растворяли в 0,1477 мл концентрированной азотной кислоты (69%) и добавляли 0,704 мл смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (0,1954 г Се(NО3)3*6Н2О) растворяли в 0,842 мл смеси Н2О/ЭГ/ЛК и добавляли 9 мкл концентрированной азотной кислоты (69%). Нитрат калия (0,0101 г КNО3) растворяли в 0,187 мл смеси Н2О/ЭГ/ЛК и добавляли 2 мкл концентрированной азотной кислоты (69%). Три полученных раствора смешивали и перемешивали в течение 60 минут с помощью орбитального миксера. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 90°С при скорости нагрева 10°С/час. После времени выдерживания 5 часов при 90°С смесь нагревали до 200°С при скорости нагрева 5°С/час. После поддерживания этой температуры в течение 5 часов образец нагревали до 400°С при скорости нагрева 10°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Примеры 2-5
Композиции согласно примерам 2-5 были такими, как описано в таблицах 1А и 1В ниже, и их готовили аналогично процедуре, описанной в примере 1, но используя соответствующий исходный материал и количества. Количества исходных материалов, использованных для приготовления согласно примерам 2-5, приведены в таблицах 1А и 1В. Смесь 50 мл деионизованной воды, 33,46 мл этиленгликоля (ЭГ) и 47,285 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Таблица 1A
[г]
[мл]
Таблица 1B
[mg]
[μl]
[мл]
*) для растворения Bi2O3
**) для растворения других солей металлов
Пример 6
Вi0,4Се0,4К0,1Sr0,1О1,55-1,35
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 200 мл деионизованной воды, 133,84 мл этиленгликоля (ЭГ) и 189,14 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (5,26 г Вi2О3) растворяли в 11,81 г концентрированной азотной кислоты (69%) и добавляли 40,77 г смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (9,81 г Се(NО3)3*6Н2О), нитрат калия (0,57 г КNО3) и карбонат стронция (0,83 г SrСО3) растворяли в 59,52 г смеси Н2О/ЭГ/ЛК и добавляли 0,85 г концентрированной азотной кислоты (69%). Два полученных раствора перемешивали в течение 60 минут с помощью магнитной мешалки. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 70°С при скорости нагрева 7,5°С/час. После времени выдерживания 24 часа при 70°С смесь нагревали до 200°С при скорости нагрева 26°С/час. После поддерживания этой температуры в течение 24 часов образец нагревали до 400°С при скорости нагрева 200°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Пример 7
Вi0,4Се0,4К0,1Рr0,1О1,6-1,4
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 200 мл деионизованной воды, 133,84 мл этиленгликоля (ЭГ) и 189,14 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (5,09 г Вi2О3) растворяли в 11,81 г концентрированной азотной кислоты (69%) и добавляли 39,12 г смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (9,48 г Се(NО3)3*6Н2О), нитрат калия (0,55 г КNО3) и гексагидрат нитрата празеодима (2,37 г Рr(NО3)3*6Н2О) растворяли в 59,52 г смеси Н2О/ЭГ/ЛК и добавляли 0,85 г концентрированной азотной кислоты (69%). Два полученных раствора перемешивали в течение 60 минут с помощью магнитной мешалки. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 70°С при скорости нагрева 7,5°С/час. После времени выдерживания 24 часа при 70°С смесь нагревали до 200°С при скорости нагрева 26°С/час. После поддерживания этой температуры в течение 24 часов образец нагревали до 400°С при скорости нагрева 200°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Пример 8
Вi0,4Се0,4К0,1Fе0,1О1,6-1,4
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 200 мл деионизованной воды, 133,84 мл этиленгликоля (ЭГ) и 189,14 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (5,33 г Вi2О3) растворяли в 11,81 г концентрированной азотной кислоты (69%) и добавляли 41,03 г смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (9,94 г Се(NО3)3*6Н2О), нитрат калия (0,58 г КNО3) и нонагидрат нитрата железа (III) (2,31 г Fе(NО3)3*9Н2О) растворяли в 59,52 г смеси Н2О/ЭГ/ЛК и добавляли 0,85 г концентрированной азотной кислоты (69%). Два полученных раствора перемешивали в течение 60 минут с помощью магнитной мешалки. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 70°С при скорости нагрева 7,5°С/час. После времени выдерживания 24 часа при 70°С смесь нагревали до 200°С при скорости нагрева 26°С/час. После поддерживания этой температуры в течение 24 часов образец нагревали до 400°С при скорости нагрева 200°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Сравнительный пример 1
Вi0,45Се0,45Nа0,1О1,65-1,4
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 50 мл деионизованной воды, 33,46 мл этиленгликоля (ЭГ) и 47,285 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (0,1048 г Вi2О3) растворяли в 0,1477 мл концентрированной азотной кислоты (69%) и добавляли 0,704 мл смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (0,1954 г Се(NО3)3*6Н2О) растворяли в 0,842 мл смеси Н2О/ЭГ/ЛК и добавляли 9 мкл концентрированной азотной кислоты (69%). Нитрат натрия (0,0085 г NаNО3) растворяли в 0,187 мл смеси Н2О/ЭГ/ЛК и добавляли 2 мкл концентрированной азотной кислоты (69%). Три полученных раствора смешивали и перемешивали в течение 60 минут с помощью орбитального миксера. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 70°С при скорости нагрева 7,5°С/час. После времени выдерживания 24 часа при 70°С смесь нагревали до 200°С при скорости нагрева 26°С/час. После поддерживания этой температуры в течение 24 часов образец нагревали до 400°С при скорости нагрева 200°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Сравнительный пример 2
Вi10Се80Sr10Ох (ЕР 2 438 984 А1, пример 2)
Нитратные соли металлов (0,5988 г Се(NО3)3*6Н2О, 0,0836 г Вi(NО3)3*5Н2О и 0,0365 г Sr(NО3)2) смешивали в мольном отношении Се/Вi/Sr=0,8/0,1/0,1 и добавляли 5 мл деионизованной воды. После растворения нитратных солей формировался белый осадок, и добавляли 3 мл концентрированной азотной кислоты (69%). Полученную смесь перемешивали до получения прозрачного раствора. К полученному раствору снова добавляли воду, так что полный объем конечного раствора был 50 мл. К полученному раствору медленно добавляли 40 мл осадителя (1 молярный вводный раствор карбоната аммония) при перемешивании. Полученную суспензию дополнительно перемешивали в течение 30 минут. Получали осадок, который отфильтровывали, промывали деионизованной водой и сушили при 125°С в течение 15 часов в воздушной атмосфере. Высушенное твердое вещество прокаливали при 400°С в течение 5 часов.
Сравнительный пример 3
Вi10Се50Рr40Ох (ЕР 2 210 861 В1, пример 1)
Сначала 0,6809 г оксида празеодима (Рr6О11, 99,9%, АВСR) растворяли в 4,5 мл концентрированной азотной кислоты (69%). Затем 2,1711 г гексагидрата нитрата церия (Се(NО3)3*6Н2О, 99,9%, ChemPur) и 0,485 пентагидрата нитрата висмута (Вi(NО3)3*5Н2О, ≥99,99%, Sigma-Aldrich) добавляли к азотнокислому раствору Рr в мольном отношении Се/Вi/Рr=0,5/0,1/0,4. К полученному раствору медленно добавляли 45 мл осадителя (1 молярный вводный раствор карбоната аммония) при перемешивании в течение 30 минут. Полученную суспензию отфильтровывали и промывали деионизованной водой, сушили при 125°С в течение 15 часов на воздухе. Полученное высушенное твердое вещество прокаливали при 400°С в течение 5 часов.
Сравнительный пример 4
Се50К50Ох (WО 2006/04482)
Сравнительный пример 4 готовили путем плавления соответствующих нитратных солей. Для этого сравнительного примера 1,0856 г Се(NО3)3*6Н2О и 0,2528 г КNО3 смешивали вручную. Полученную смесь нагревали от комнатной температуры до 350°С со скоростью нагрева 50°С/час. Температуру 350°С поддерживали постоянной в течение 12 часов и затем опять снижали до комнатной температуры со скоростью 120°С/час. Полученное твердое вещество прокаливали при 400°С в течение 5 часов.
Сравнительный пример 5
Се50К50Ох (WО 2006/044822 А1)
Синтезировали путем растворения 4,3422 г Се(NО3)3*6Н2О в 10 мл деионизованной воды и добавления 0,6910 г К2СО3 к водному Се раствору. Полученный раствор уменьшали в объеме путем выпаривания при 120°С в течение 24 часов на воздухе. Полученное твердое вещество прокаливали при 400°С в течение 5 часов.
Сравнительный пример 6
Се66,7К33,3Ох (WО 2006/044822 А1)
Готовили путем плавления соответствующих нитратных солей. Для этого сравнительного примера 1,7369 г Се(NО3)3*6Н2О и 0,2022 г КNО3 смешивали вручную. Полученную смесь нагревали от комнатной температуры до 350°С со скоростью нагрева 50°С/час. Температуру 350°С поддерживали постоянной в течение 12 часов и затем опять снижали до комнатной температуры со скоростью 120°С/час. Полученное твердое вещество прокаливали при 400°С в течение 5 часов.
Сравнительный пример 7
Се66,7К33,3Ох (WО 2006/044822 А1)
Синтезировали путем растворения 4,3422 г Се(NО3)3*6Н2О в 10 мл деионизованной воды и добавления 0,3455 г К2СО3 к водному Се раствору. Полученный раствор уменьшали в объеме путем выпаривания при 120°С в течение 24 часов на воздухе. Полученное твердое вещество прокаливали при 400°С в течение 5 часов.
Сравнительный пример 8
Вi0,45Се0,45Sr0,1Ох
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 200 мл деионизованной воды, 133,84 мл этиленгликоля (ЭГ) и 189,14 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (5,17 г Вi2О3) растворяли в 11,81 г концентрированной азотной кислоты (69%) и добавляли 39,77 г смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (9,64 г Се(NО3)3*6Н2О) и карбонат стронция (0,71 г SrСО3) растворяли в 59,52 г смеси Н2О/ЭГ/ЛК и добавляли 0,85 г концентрированной азотной кислоты (69%). Два полученных раствора перемешивали в течение 60 минут с помощью магнитной мешалки. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 70°С при скорости нагрева 7,5°С/час. После времени выдерживания 24 часа при 70°С смесь нагревали до 200°С при скорости нагрева 26°С/час. После поддерживания этой температуры в течение 24 часов образец нагревали до 400°С при скорости нагрева 200°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Сравнительный пример 9
Вi0,45Се0,45Рr0,1Ох
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 200 мл деионизованной воды, 133,84 мл этиленгликоля (ЭГ) и 189,14 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (5,23 г Вi2О3) растворяли в 11,81 г концентрированной азотной кислоты (69%) и добавляли 40,26 г смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (9,38 г Се(NО3)3*6Н2О) и гексагидрат нитрата празеодима (2,09 г Рr(NО3)3*6Н2О) растворяли в 59,52 г смеси Н2О/ЭГ/ЛК и добавляли 0,85 г концентрированной азотной кислоты (69%). Два полученных раствора перемешивали в течение 60 минут с помощью магнитной мешалки. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 70°С при скорости нагрева 7,5°С/час. После времени выдерживания 24 часа при 70°С смесь нагревали до 200°С при скорости нагрева 26°С/час. После поддерживания этой температуры в течение 24 часов образец нагревали до 400°С при скорости нагрева 200°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Сравнительный пример 10
Вi0,45Се0,45Fе0,1Ох
синтезировали с помощью золь-гель способа с полимерным комплексом.
Смесь 200 мл деионизованной воды, 133,84 мл этиленгликоля (ЭГ) и 189,14 г моногидрата лимонной кислоты (ЛК) использовали в качестве растворителя.
Стехиометрическое количество оксида висмута (III) (5,03 г Вi2О3) растворяли в 11,81 г концентрированной азотной кислоты (69%) и добавляли 38,7 г смеси Н2О/ЭГ/ЛК (после растворения Вi2О3 может возникать белый осадок, который растворяется после добавления смеси Н2О/ЭГ/ЛК). Гексагидрат нитрата церия (III) (9,38 г Се(NО3)3*6Н2О) и нонагидрат нитрата железа (III) (1,94 г Fе(NО3)3*9Н2О) растворяли в 59,52 г смеси Н2О/ЭГ/ЛК и добавляли 0,85 г концентрированной азотной кислоты (69%). Два полученных раствора перемешивали в течение 60 минут с помощью магнитной мешалки. Затем растворитель растворов испаряли и полученный остаток испарения прокаливали на воздухе. Чтобы испарить растворитель, растворы нагревали от комнатной температуры до 70°С при скорости нагрева 7,5°С/час. После времени выдерживания 24 часа при 70°С смесь нагревали до 200°С при скорости нагрева 26°С/час. После поддерживания этой температуры в течение 24 часов образец нагревали до 400°С при скорости нагрева 200°С/час. Образцы прокаливали при 400°С в течение 5 часов. Порошки прокаленного оксида охлаждали до комнатной температуры со скоростью 20°С/час (свежие образцы).
Результаты каталитического тестирования
Таблица 2 показывает эффективность удаления ТЧ, измеренную способом А, церий-висмут-щелочных композиций настоящего изобретения, приготовленных согласно примерам 1-6, а также для сравнительных примеров 1 и 2 в свежем состоянии (прокаленные при 400°С/2 часа) и после термического выдерживания порошков при 800°С/2 часа.
Таблица 2
Результаты каталитического тестирования показали, что все материалы из примеров 1-5 имеют меньшую Т50-величину после термического выдерживания и в свежем состоянии, чем материалы сравнительных примеров 1, 2 и 3.
Результаты каталитического тестирования после гидротермической обработки:
Таблица 3 ниже показывает эффективность удаления ТЧ, измеренную способом А, трех композиций настоящего изобретения и композиций сравнительных примеров 3, 4, 5 и 6, все в свежем состоянии, а также после гидротермической обработки. Композиции сравнительных примеров 3-6 показывают прекрасную каталитическую активность в свежем состоянии. В противоположность этому, однако, композиции сравнительных примеров теряют свою каталитическую активность после гидротермического выдерживания в противоположность примерам настоящего изобретения, которые все еще показывают каталитическую активность.
Таблица 3
Влияние добавок калия на различные Се-Вi-М-смешанные оксиды металлов:
Таблица 4 ниже показывает влияние легирования калием трех разных Се-Вi-М-смешанных оксидов металлов на эффективность удаления ТЧ, измеренную способом В. Все композиции, легированные калием, являются более каталитически активными в окислении сажи, чем не легированные композиции в свежем состоянии, а также после термической обработки.
Таблица 4
Краткое описание чертежей (фигуры 1-2)
Фиг.1 показывает график порошковой рентгеновской дифракции сравнительного примера 4, рефлексы относятся к СеО2 и КNО3.
Фиг.2 показывает график порошковой рентгеновской дифракции примера 1, рефлексы относятся к Се1-хВiхО2-х/2, α-Вi2О3 и Вi2(СО3)О2, не обнаружено кристаллических соединений калия.
Изобретение относится к композиции для систем постобработки выхлопного газа дизельных двигателей, бензиновых двигателей сгорания, двигателей на обедненной смеси и электростанций, имеющей формулу Се1-a-b-cNaMbDcOx (I), в которой М обозначает калий, N обозначает Вi и/или Sb, D присутствует или отсутствует, и если присутствует, то выбирается из одного или нескольких элементов из Mg, Ca, Sr, Ba; Y, Lа, Рr, Nd, Sm, Gd, Еr; Fе, Zr, Nb, Аl; а является числом в интервале 0<а≤0,9, b является числом в интервале 0<b≤0,3, с является числом в интервале 0≤с≤0,2; а плюс b плюс с равно <1, и х является числом в интервале 1,2≤х≤2. Изобретение также относится к применению заявленной композиции для систем постобработки выхлопного газа дизельных двигателей, бензиновых двигателей сгорания, двигателей на бедной смеси и электростанций. Технический результат заключается в получении новой композиции, обладающей более высокой каталитической активностью. 2 н. и 11 з.п. ф-лы, 2 ил., 4 табл., 18 пр.
1. Композиция для систем постобработки выхлопного газа дизельных двигателей, бензиновых двигателей сгорания, двигателей на обедненной смеси и электростанций, имеющая формулу
Се1-a-b-cNaMbDcOx (I),
в которой
М обозначает калий,
N обозначает Вi и/или Sb,
D присутствует или отсутствует, и если присутствует, то выбирается из одного или нескольких элементов из
- Mg, Ca, Sr, Ba;
- Y, Lа, Рr, Nd, Sm, Gd, Еr;
- Fе, Zr, Nb, Аl;
а является числом в интервале 0<а≤0,9,
b является числом в интервале 0<b≤0,3,
с является числом в интервале 0≤с≤0,2;
а плюс b плюс с равно <1, и
х является числом в интервале 1,2≤х≤2.
2. Композиция по п. 1, в которой D присутствует.
3. Композиция по любому из пп. 1 или 2, в которой D представляет собой Ca, Sr, Ba; особенно Sr.
4. Композиция по любому из пп. 1 или 2, в которой D представляет собой Y, Рr, Lа, Nd; особенно Рr.
5. Композиция по любому из пп. 1 или 2, в которой D представляет собой Fе, Zr, Nb, Аl; особенно Fе, Аl.
6. Композиция по любому из пп. 1-5, в которой с является числом в интервале 0≤с≤0,1.
7. Композиция по п. 1, которая имеет формулу
Се1-a-bNaMbOx (II),
в которой
М обозначает калий,
N обозначает Вi и/или Sb,
а является числом в интервале 0<а≤0,9,
b является числом в интервале 0<b≤0,3,
а плюс b равно <1, и
х является числом в интервале 1,2≤х≤2.
8. Композиция по любому из пп. 1-7, в которой N представляет собой Вi.
9. Композиция по любому из пп. 1-8, в которой а является числом в интервале 0,01≤а≤0,9.
10. Композиция по любому из пп. 1-9, в которой b является числом в интервале 0,01≤b≤0,3.
11. Композиция по п. 10, в которой b является числом в интервале 0,1≤b≤0,2.
12. Композиция по любому из пп. 1 и 7-11, которая выбрана из группы, состоящей из
Bi0,45Ce0,45K0,10O1,65-1,4,
Bi0,40Ce0,40K0,20O1,4-1,2,
Bi0,30Ce0,60K0,10O1,7-1,4,
Bi0,80Ce0,10K0,10O1,5-1,4,
Bi0,10Ce0,80K0,10O1,8-1,4,
Bi0,4Ce0,4K0,1Sr0,1O1,55-1,35,
Bi0,4Ce0,4K0,1Pr0,1O1,6-1,4 и
Bi0,4Ce0,4K0,1Fe0,1O1,6-1,4.
13. Применение композиции по любому из пп. 1-12 для систем постобработки выхлопного газа дизельных двигателей, бензиновых двигателей сгорания, двигателей на бедной смеси и электростанций.
| СПОСОБ ПРИГОТОВЛЕНИЯ КАТАЛИЗАТОРА ДЛЯ ОКИСЛЕНИЯ И АММОКСИДАЦИИ ОЛЕФИНОВ | 2003 |
|
RU2341327C2 |
| US 4460706 А1, 17.07.1984 | |||
| УСТРОЙСТВО ДЛЯ ПРИЕМА СИГНАЛОВ С ПСЕВДОСЛУЧАЙНОЙ ПЕРЕСТРОЙКОЙ РАБОЧЕЙ ЧАСТОТЫ | 2002 |
|
RU2210861C1 |
| WO 2006044822 А1, 27.04.2006. | |||

