Область техники, к которой относится изобретение
Настоящее изобретение относится к способу применения генетически модифицированных микроорганизмов для получения ксилита и способу получения генетически модифицированного микроорганизма, который способен превращать за одну стадию легкодоступные источники углерода, как, например, D-глюкозу, в ксилит.
Уровень техники
Ксилит представляет собой многоатомный спирт или сахарный спирт (альдит) формулы (CHOH)3(CH2OH)2, который применяют при гигиенических процедурах и в нутрицевтических составах и продуктах.
Ксилит используют в качестве подсластителя для диабетиков, который примерно настолько же сладкий насколько и сахароза, и при этом характеризуется на 33% меньшим количеством калорий. В отличие от других натуральных или синтетических подсластителей ксилит активно проявляет свое благоприятное воздействие в отношении здоровья зубов путем уменьшения степени проявления кариеса на треть при регулярном использовании и полезен при реминерализации.
Ксилит встречается в природе в низких концентрациях в волокнах многих фруктов и овощей, и его можно получать из разных ягод, овса и грибов, а также волокнистого материала, такого как кукурузная шелуха, и тростниково-сахарная багасса, и древесина березы.
Однако промышленное получение начинается с ксилана (гемицеллюлозы), полученного из древесины твердых пород или стержней кукурузных початков, который гидролизуют в ксилозу и каталитически гидрогенизируют в ксилит.
Очистка ксилозы, а также ксилита, тем самым, представляет существенную проблему. Известен ряд способов такого типа. Патенты США 4075406 и 4008285 могут быть упомянуты как примеры.
Восстановления D-ксилозы в ксилит также можно достичь при помощи микробиологического процесса с использованием либо штаммов дрожжей, полученных в чистом виде из природы (штаммов дикого типа), либо генетически сконструированных штаммов.
Однако, получение субстрата, D-ксилозы, в форме, пригодной для дрожжевого брожения, является проблемой, в силу того, что недорогостоящие источники ксилозы, такие как сульфитный щелок, полученный в результате способов обработки древесной массы и бумаги, содержат примеси, которые подавляют рост дрожжей.
Приемлемым альтернативным способом получения ксилита является получение его при помощи ферментации дешевого и легкодоступного субстрата, такого как D-глюкоза.
В уровне техники описаны несколько рекомбинантных микроорганизмов, способных продуцировать ксилит в определенных количествах в ходе одностадийной ферментации любых обычных источников углерода, отличных от D-ксилозы и D-ксилулозы.
Эти рекомбинантные микроорганизмы, в частности осмофильные дрожжи, представляют собой, например, Zygosaccharomyces rouxii, Candida polymorpha и Torulopsis candida, изначально известные как продуценты значительных количеств близкородственного ксилиту пентита, который представляет собой D-арабит, из D-глюкозы (Lewis D.H. & Smith D.C., 1967, New Phytol. 66:143-184).
Таким образом, в международной заявке на патент WO 94/10325 представлены способы конструирования таких рекомбинантных хозяев, способных продуцировать ксилит при росте на источниках углерода, отличных от D-ксилулозы или D-ксилозы и отличных от полимеров или олигомеров или их смесей.
В настоящей заявке на выдачу патента эту цель достигают посредством модификации метаболизма необходимого микроорганизма, предпочтительно встречающегося в природе дрожжевого микроорганизма, путем введения и экспрессирования необходимых гетерологичных генов.
Эту цель также достигают путем дополнительной модификации метаболизма такого необходимого микроорганизма так, чтобы сверхэкспрессировать и/или инактивировать активность или экспрессию определенных генов, гомологичных таковым микроорганизма в его нативном состоянии.
В предусмотренном в данной заявке на выдачу патента способе получения ксилита используют измененный путь биосинтеза D-арабита, и такой путь является в значительной мере измененным путем продления существующего пути для D-арабита при помощи введения генов, кодирующих образующую D-ксилулозу D-арабитдегидрогеназу (EC 1.1.1.11) и ксилитдегидрогеназу (EC 1.1.1.9), в продуцирующий D-арабит микроорганизм и их сверхэкспрессии в нем.
Однако, выход ксилита в испытаниях, описанных в WO 94/10325, составил только примерно 7,7 г/л через 48 часов культивирования в среде с дрожжевым экстрактом.
В попытке оптимизировать этот первый результат было дополнительно предложено в WO 94/10325 при помощи мутагенеза или разрушения генов инактивировать гены, кодирующие транскетолазу (EC 2.2.1.1), и/или ген, кодирующий D-ксилулокиназу (EC 2.7.1.17), а также сверхэкспрессировать гены, кодирующие ферменты окислительной ветки пентозофосфатного пути, в том числе ген D-глюкозо-6-фосфатдегидрогеназы (EC 1.1.1.49), и/или 6-фосфо-D-глюконатдегидрогеназы (EC 1.1.1.44), и/или D-рибулозо-5-фосфат-эпимеразы (EC 5.1.3.1), в таких микроорганизмах.
Но, независимо от применяемой генетической комбинации титр ксилита никогда не составлял более 9 г/л.
В силу этого все еще существует неудовлетворенная потребность в лучшей генетической манипуляции с продуцирующими ксилит штаммами для того, чтобы оптимизировать его продуцирование и, таким образом, сделать его коммерчески рентабельным.
Краткое описание изобретения
Настоящее изобретение относится к рекомбинантной клетке-хозяину, способной продуцировать ксилит, где указанная клетка-хозяин содержит:
последовательность гетерологичной нуклеиновой кислоты, кодирующую NAD+-специфическую D-арабит-4-оксидоредуктазу (EC 1.1.1.11), использующую D-арабит в качестве субстрата и дающую D-ксилулозу в качестве продукта; и
последовательность гетерологичной нуклеиновой кислоты, кодирующую NADPH-специфическую ксилитдегидрогеназу, использующую D-ксилулозу в качестве субстрата и дающую ксилит в качестве продукта.
Предпочтительно, клетка-хозяин не использует D-арабит в качестве единственного источника углерода. Более предпочтительно, клетку-хозяина выбирают из бактерий, грибов и дрожжей. Согласно предпочтительному варианту осуществления клетка-хозяин представляет собой клетку осмофильных или осмоустойчивых дрожжей, в частности Pichia ohmeri.
Предпочтительно, NAD+-специфическая D-арабит-4-оксидоредуктаза (EC 1.1.1.11) получена из E. coli или Ralstonia solanacearum. Более предпочтительно, NAD+-специфическая D-арабит-4-оксидоредуктаза (EC 1.1.1.11) содержит последовательность с SEQ ID NO 2 или 43 или последовательность с 1-3 добавлениями, заменами или делециями аминокислот или образована ею. Согласно предпочтительному варианту осуществления последовательность, кодирующая NAD+-специфическую D-арабит-4-оксидоредуктазу (EC 1.1.1.11), содержит последовательность с SEQ ID NO 3 или 42 или образована ею.
Предпочтительно, NADPH-специфическая ксилитдегидрогеназа является ксилитдегидрогеназой из Pichia stipitis или Gluconobacter oxydans, подвергнутых мутации в отношении изменения специфичности в отношении кофактара с NADH на NADPH. Более предпочтительно, NADPH-специфическая ксилитдегидрогеназа содержит последовательность с SEQ ID NO 5 или 8 или последовательность с 1-3 добавлениями, заменами или делециями аминокислот или образована ею. Согласно предпочтительному варианту осуществления последовательность, кодирующая NADPH-специфическую ксилитдегидрогеназу, содержит последовательность с SEQ ID NO 6 или 9 или образована ею.
Предпочтительно, клетка-хозяин способна продуцировать титр ксилита, составляющий по меньшей мере 15 г/л, в надосадочной жидкости через 48 ч. культивирования.
Предпочтительно, клетка-хозяин представляет собой клетку штамма, выбранного из штаммов I-4982, I-4960 и I-4981, депонированных в CNCM.
Предпочтительно, клетка-хозяин содержит несколько копий последовательности, кодирующей NAD+-специфическую D-арабит-4-оксидоредуктазу, и/или несколько копий последовательности, кодирующей NADPH-специфическую ксилитдегидрогеназу.
Настоящее изобретение также относится к способу получения ксилита, включающему культивирование рекомбинантной клетки-хозяина по любому из пунктов 1-13 и извлечение ксилита.
Оно дополнительно относится к нуклеиновой кислоте, которая содержит последовательность нуклеиновой кислоты, выбранную из группы, состоящей из SEQ ID NO 1, 3, 7 и 9, или образована ею, кассете экспрессии или вектору, содержащим указанную нуклеиновую кислоту.
И наконец, настоящее изобретение относится к применению рекомбинантных клеток-хозяев согласно настоящему изобретению для получения ксилита.
Подробное описание изобретения
Определения
Как используется в данном документе, под "источником углерода, отличным от D-ксилозы и D-ксилулозы" понимают углеродный субстрат для получения ксилита, отличный от D-ксилозы и D-ксилулозы или полимеров или олигомеров или их смесей (как, например, ксилана и гемицеллюлозы). Источник углерода, предпочтительно, включает D-глюкозу, и разные содержащие D-глюкозу патоки, и смеси D-глюкозы с другими сахарами.
Как используется в данном документе, под “геном” понимают последовательность нуклеиновой кислоты, которая может кодировать белок, в частности последовательность ДНК.
Как используется в данном документе, под “вектором” понимают плазмиду или любую другую последовательность ДНК, которая способна переносить генетическую информацию, в особенности ДНК, в клетку-хозяина. Вектор может дополнительно содержать маркер или репортер, пригодный для использования при идентификации клеток, трансформированных вектором, и точки начала репликации, которые обеспечивают поддержание и репликацию вектора в одном или нескольких прокариотических или эукариотических хозяевах. "Плазмида" представляет собой вектор, как правило, кольцевую ДНК, которую поддерживают и которая реплицируется самостоятельно по меньшей мере в одной клетке-хозяине.
Как используется в данном документе, под “вектором экспрессии” понимают вектор, схожий с вектором, но который содействует экспрессии гена или кодирующей нуклеиновой кислоты, которые клонировали в него, после трансформации им хозяина. Клонированный ген или кодирующую нуклеиновую кислоту обычно размещают под контролем (т. е. функционально связанными с определенными контрольными последовательностями) определенных контрольных последовательностей, таких как промоторные последовательности, которые могут обеспечиваться вектором или рекомбинантным конструированием клонированного гена. Контрольные последовательности экспрессии будут изменяться в зависимости от того сконструирован ли вектор для экспрессии функционально связанного гена в прокариотическом или эукариотическом хозяине и могут дополнительно содержать транскрипционные элементы, такие как энхансерные элементы (активирующие последовательности) и терминирующие последовательности, и/или сайты начала и завершения трансляции.
Как используется в данном документе, под “хозяином” понимают клетку, прокариотическую или эукариотическую, которую используют в качестве реципиента и носителя рекомбинантного материала.
Как используется в данном документе, под “окислительной веткой пентозофосфатного пути” понимают, что она включает часть пентозо-фосфатного шунта, который катализирует окислительные реакции, как, например, реакции, катализируемые D-глюкозо-6-фосфатдегидрогеназой (EC 1.1.1.49), глюконолактоназой (EC 3.1.1.17) и 6-фосфо-D-глюконатдегидрогеназой (EC 1.1.1.44), и при котором используются гексозные субстраты для образования пентозофосфатов. "Неокислительная" часть пентозофосфатного пути (которая также катализирует фактическое образование рибозы из D-глюкозы) характеризуется неокислительными изомеризациями, как, например, реакциями, катализируемыми транскетолазой (EC 2.2.1.1), рибозо-5-фосфатизомеразой (EC 5.3.1.6), D-рибулозо-5-фосфат-3-эпимеразой (EC 5.1.3.1) и трансальдолазой (EC 2.2.1.2). См. Biological Chemistry, H.R. Mahler & E.H.Cordes, Harper & Row, publishers, New York, 1966, pp. 448-454.
Как используется в данном документе, под "кодирующей нуклеиновой последовательностью" понимают молекулу нуклеиновой кислоты (предпочтительно, ДНК). Кодирующая нуклеиновая кислота способна кодировать белок и может быть получена из разнообразных источников. Эти источники включают геномную ДНК, кДНК, синтетическую ДНК и их комбинации.
Понимается, что под “гетерологичным”, используемым в данном документе, подразумевают, что ген или кодирующая последовательность были введены в клетку при помощи генной инженерии. Они могут присутствовать в эписомальной или хромосомной форме. Ген или кодирующая последовательность могут происходить из источника, отличного от клетки-хозяина, в которую их вводят. Однако, они также могут происходить из того же вида, что и клетка-хозяин, в которую их вводят, но они считаются гетерологичными из-за их окружающей среды, которая не является природной. Например, ген или кодирующую последовательность называют гетерологичными потому, что они находятся под контролем промотора, который не является их природным промотором, их вводят в местоположения, которые отличаются от их природных местоположений. Клетка-хозяин может содержать эндогенную копию гена перед введением гетерологичного гена, или она может не содержать эндогенную копию.
Объект изобретения
Согласно настоящему изобретению нативные метаболические пути специфического микробного хозяина подвергают манипуляции, чтобы сократить или исключить использование углерода в процессах, отличных от продуцирования ксилита.
Такой генетически модифицированный штамм-хозяин, таким образом, способен продуцировать ксилит за одну стадию ферментации с высоким выходом. К примеру, титр ксилита через 48 ч. культивирования в надосадочной жидкости составляет более 15 г/л, предпочтительно более 25 г/л, еще более предпочтительно более 50, 60, 70, 80, 90 или 100 г/л.
При практической реализации настоящего изобретения генетически модифицированный хозяин по настоящему изобретению также характеризуется его способностью синтезировать ксилит из структурно неродственных источников углерода, таких как D-глюкоза, а не только из D-ксилозы и/или D-ксилулозы.
Предпочтительно, генетически модифицированный хозяин по настоящему изобретению также способен секретировать синтезированный ксилит в среду.
Конкретно, согласно примерным и предпочтительным вариантам осуществления генетически модифицированный хозяин по настоящему изобретению характеризуется путем, при котором арабит является промежуточным соединением при образовании ксилита.
Соответственно, рекомбинантный штамм-хозяин по настоящему изобретению характеризуется следующими генетическими изменениями:
(1) гетерологичную нуклеиновую кислоту, кодирующую белок, обладающий активностью NAD+-специфической D-арабит-4-оксидоредуктазы (для образования D-ксилулозы), вводили в клетку-хозяина с обеспечением, таким образом, превращения D-арабита в D-ксилулозу и
(2) гетерологичную нуклеиновую кислоту, кодирующую белок, обладающий активностью NADPH-специфической ксилитдегидрогеназы, вводили в клетку-хозяина с обеспечением, таким образом, превращения D-ксилулозы в ксилит.
Выбор микроорганизма
Микроорганизмы или штаммы-хозяева, пригодные для настоящего изобретения, способны продуцировать D-арабит из глюкозы. Более конкретно, они способны продуцировать значительные количества D-арабита из глюкозы в условиях среды с высоким осмотическим давлением.
Под “средой с высоким осмотическим давлением” подразумевается в данном документе обозначение среды, содержащей 10-60% D-глюкозы, предпочтительно приблизительно 25% D-глюкозы.
Под “значительными количествами D-арабита” подразумевается по меньшей мере 100 г/л D-арабита. В частности, микроорганизм или штамм-хозяин считаются такими, которые продуцируют значительные количества D-арабита, когда микроорганизм или штамм-хозяин продуцирует 100 г/л D-арабита в среде, содержащей 25% D-глюкозы, в статических условиях.
Примеры штаммов-хозяев, способных продуцировать значительные количества D-арабита из глюкозы, включают осмофильные или осмоустойчивые дрожжи, в частности те, которые принадлежат видам Pichia, Kodamaea, Candida, Zygoaccharomyces, Debaromyces, Metschnikowia и Hansenula; или продуцирующие D-арабит грибы, в частности те, которые принадлежат видам Dendryphiella и Schizophyllum, в частности Dendryphiella salina и Schizophyllum commune.
Примеры микроорганизмов рода Pichia включают Pichia ohmeri, Pichia stipitis, Pichia farinosa, Pichia haplophila. Примеры микроорганизмов рода Candida включают Candida polymorpha и Candida tropicalis. Примеры микроорганизмов рода Zygoaccharomyces включают Zygoaccharomyces rouxii. Другие примеры включают Torulopsis candida и Torulaspora hansenii. Примеры микроорганизмов рода Metschnikowia включают Metschnikowia pulcherrima, Metschnikowia reukaufii, Metschnikowia bicuspidata, Metschnikowia lunata и Metschnikowia zobellii. В качестве конкретных штаммов могут быть упомянуты Metschnikowia pulcherrima ATCC 18406, Metschnikowia reukaufii ATCC 18407, Metschnikowia bicuspidata ATCC 24179, Metschnikowia lunata ATCC 22033, Metschnikowia zobellii ATCC 22302 и Metschnikowia pulcherrima FERM BP-7161. Эти штаммы можно получать из Американской коллекции типовых культур, расположенной по адресу: 12301 Parklawn Drive, Rockville, Мэриленд 20852, Соединенные Штаты Америки. Metschnikowia pulcherrima FERN BP-7161 сначала депонировали в Национальном институте бионауки и технологии человека (National Institute of Bioscience and Human-Technology), Агентства промышленной науки и технологии (Agency of Industrial Science and Technology), Министерства внешней торговли и промышленности (Ministry of International Trade and Industry) (почтовый индекс: 305-8566, 1-3 Higashi 1-Chome, Tsukuba-shi, Ibaraki-ken, Япония) 16 января 1998 года под номером депонирования FERM P-16592, и переносили из учреждения первоначального депонирования в международное учреждение для депонирования согласно Будапештскому договору 15 мая 2000 года, и депонировали с номером депонирования FERM BP-7161. Согласно конкретному аспекту микроорганизм имеет номер доступа FERM BP-7161. Для получения дополнительной информации обратитесь к EP1065276.
Микроорганизм может быть генетически сконструирован для улучшения его способности продуцировать D-арабит и/или снижения его способности использовать D-арабит для цели, отличной от продуцирования ксилита.
Для настоящего изобретения штамм-хозяина, преимущественно, выбирают по его конкретным метаболическим качествам:
- он может быть продуцентом значительных количеств D-арабита из глюкозы, как подробно описано выше, в частности, в условиях среды с высоким осмотическим давлением, например, среды, содержащей 10-60% D-глюкозы, и предпочтительно 25% D-глюкозы ("нормальная" среда, как правило, содержит только 2-3% глюкозы);
- он может не использовать D-арабит в качестве единственного источника углерода;
- его окислительно-восстановительное равновесие предусматривает образование кофакторов, необходимых для соответствующего превращения при помощи спирта кетопентоза/пентоза.
Согласно одному варианту осуществления настоящего изобретения осмофильные дрожжи Pichia ohmeri (и их мутировавшие производные) применяли в качестве модели и в качестве предпочтительного хозяина. Pichia ohmeri первоначально выделяли из огуречного рассола и повсеместно использовали в пищевой промышленности для ферментации солений, коры и фруктов.
Специалистам в данной области известно, что виды дрожжей, такие как Pichia, Zygosaccharomyces, Debaromyces и Hansenula, способны расти в окружающей среде с низкой активностью воды, в отличие от Saccharomyces cerevisiae. Эти осмоустойчивые или осмофильные дрожжи накапливают совместимый осмолит, подобный глицерину, D-арабиту, эритриту и манниту, который защищает и стабилизирует ферменты, обеспечивая, тем самым, клеточные функции осмотическими условиями для роста. Продуцируемые полиолы также играют роль в окислительно-восстановительном уравновешивании.
Согласно предпочтительному аспекту микроорганизм представляет собой Pichia ohmeri. В самом деле, главной характерной чертой штамма-хозяина Pichia ohmeri является продуцирование только D-арабита в качестве совместимого осмолита, в отличие от Zygosaccharomyces rouxii, продуцирующего глицерин и D-арабит. Кроме того, метаболический путь от глюкозы до D-арабита хорошо известен у Pichia ohmeri.
Как описано у Zygoaccharomyces rouxii (.M.INGRAM and W.A.WOOD, 1965, Journal of Bacteriology, Vol.89, N°5, 1186-1194), поток углерода у Pichia ohmeri проходит через окислительную часть пентозофосфатного пути (PPP) для превращения D-глюкозы в D-рибулозо-5-P с сопутствующим продуцированием двух молекул NADPH. D-рибулозо-5-P дефосфорилируют до D-рибулозы и затем восстанавливают до D-арабита. У штамма-хозяина Pichia ohmeri пентозофосфатный путь (PPP) очень активен и, как было определено, более чем на 50%.
Согласно предпочтительному варианту осуществления клетка-хозяин является клеткой мутантного Pichia ohmeri, депонированного 7 марта 2012 года в Collection Nationale de Cultures de Microorganismes [Национальной коллекции культур микроорганизмов] Института Пастера (CNCM), 25 rue du Docteur Roux, 75724 PARIS Cedex 15, под номером I-4605.
Окислительно-восстановительные реакции и ферменты
Кофакторы NADH и NADPH необходимы для множества биологических функций, действуя в так называемых окислительно-восстановительных реакциях в качестве переносчиков электронов от одной реакции к другой. Клеткам необходимо поддерживать метаболическое равновесие двух окислительно-восстановительных пар NADH/NAD+ и NADPH/NADP+, при этом известно, что пара NADPH/NADP+ поддерживается в более восстановленном состоянии, чем пара NADH/NAD+ для обеспечения термодинамической движущей силы. NADH, который главным образом встречается в окисленной форме NAD+, является главным кофактором в катаболических реакциях, где он вовлечен в окислительное высвобождение энергии из питательных веществ. В отличие от NADH, NADPH повторно окисляется исключительно в анаболических реакциях или в случаях оксидативного стресса.
Любая стратегия конструирования метаболизма, которая включает окислительно-восстановительные реакции, должна выполняться в рамках этих ограничений в клетке. Это было сделано в генетически модифицированном штамме, который является объектом настоящего изобретения.
Как было обнаружено изобретателем, в частности описано в докторской диссертации под названием “Contribution а l'йtude du metabolisme des pentilos chez Pichia ohmeri” (Sophie Huchette, Лилльский университет науки и техники, 1992), было показано, что реакции, вовлеченные в окисление-восстановление кетопентоз катализируется двумя различными ферментами.
Таким образом, штамм-хозяин имеет фермент, определенный как NADPH-специфическая D-кетопентозо-оксидоредуктаза, образующая D-арабит из D-рибулозы и образующая ксилит из D-ксилулозы. Штамм-хозяин также имеет NADH-специфическую D-кетопентозо-оксидоредуктазу, образующую рибит и ксилит соответственно из D-рибулозы и D-ксилулозы. Этот фермент близок хорошо известной NAD+-специфической ксилитдегидрогеназе E.C 1.1.1.9 из Pichia stipitis (XYL2). Поскольку доступна только внутриклеточная D-рибулоза в отличие от D-ксилулозу, то штамм-хозяин уравновешивает окислительно-восстановительную пару NADPH/NADP+ непосредственно путем повторного окисления NADPH посредством образования в цитозоле D-арабита из D-рибулозы. Затем, D-арабит секретируется в бульон посредством пассивной диффузии.
Изобретатели обнаружили, что недостаток внутриклеточной D-ксилулозы будет главной причиной для отсутствия продукции ксилита штаммом-хозяином, даже если Pichia ohmeri обладает всеми ферментативными инструментами для продуцирования этого полиола с помощью NADH- или NADPH-специфической-D-кетопентозо-оксидоредуктазы.
Действительно, для клонирования в штамм-хозяина дикого типа Pichia ohmeri выбирали ген, кодирующий белок, обладающий активностью NAD+-специфической D-арабит-4-оксидоредуктазы (для образования D-ксилулозы) (E.C.1.1.1.11), что обеспечивало возможность превращать цитозольный D-арабит в D-ксилулозу и NADH.
Итак, внутриклеточная D-ксилулоза становится доступной в генетически модифицированном штамме и может быть восстановлена при помощи внутренней NADH- и NADPH-специфической D-кетопентозо-оксидоредуктазы. Однако, штамм лишен эндогенных ферментов, способных эффективно преобразовывать D-ксилулозу в ксилит. Вследствие этого, необходимо генетически конструировать штамм для введения гетерологичной ксилитдегидрогеназы.
В патенте WO 94/10325 для клонирования выбирали NAD+-специфическую ксилитдегидрогеназу (E.C 1.1.1.9) из Pichia stipitis (XYL2), которая обеспечивала возможность продуцирования ксилита и уравновешивала окислительно-восстановительную пару NADH/NAD+ путем окисления NADH, продуцируемого на предыдущей стадии метаболизма. Но, как упоминалось ранее, результаты не совсем впечатляющие.
Изобретатели обнаружили, что при клонировании гена, кодирующего подвергнутый мутации белок, обладающий активностью NADPH-специфической ксилитдегидрогеназы, D-ксилулоза превращается в ксилит для уравновешивания окислительно-восстановительной пары NADPH/NADP+, как, например, делается при внутренней продукцией D-арабита из D-рибулозы.
Из-за низкой аффинности NADPH-специфической D-кетопентозо-оксидоредуктазы в отношении D-арабита штамм-хозяин дикого типа Pichia ohmeri не использует внеклеточный D-арабит.
Вследствие введения активности NAD+-специфической D-арабит-4-оксидоредуктазы (для образования D-ксилулозы) в генетически модифицированный штамм, D-арабит, продуцируемый в бульон, может удачно использоваться модифицированным штаммом таким же путем, что и цитолитический D-арабит.
Следовательно, ксилит продуцируется в одно и то же время из внутриклеточного и внеклеточного D-арабита.
Его продуцирование можно улучшить путем повышения эффективности продления пути продуцирования ксилита для полного избегания выведения промежуточного D-арабита.
Таким образом только ксилит будет продуцироваться из D-глюкозы с тем же физиологическим эффектом, что и D-арабит. Это улучшение может быть результатом генетических модификаций, но также приспособлением к условиям культивирования.
Выбор двух ферментативных активностей, подлежащих клонированию в штамм-хозяина
Выбору этих двух ферментативных активностей содействуют их специфичности в отношении кофактора, которые описаны выше.
Первый фермент окисляет D-арабит в D-ксилулозу.
Известны два типа D-арабитдегидрогеназ: образующая D-ксилулозу (EC 1.1.1.11) (D-арабинит-NAD+-4-оксидоредуктаза) и образующая D-рибулозу (EC 1.1.1.250). Если не указано иное, то образующая D-ксилулозу арабитдегидрогеназа является такой, которая предусматривается в данном документе и которую называют в данном документе как арабитдегидрогеназа. Образующие D-рибулозу дегидрогеназы встречаются в дрожжах и грибах дикого типа.
Образующие D-ксилулозу арабитдегидрогеназы в основном известны у бактерий. К примеру, их идентифицировали у Enterobacteriaceae, в частности E. coli, Klebsiella aerogenes и штамма PRL-R3 Aerobacter aerogenes, у Gluconobacter oxydans и также дополнительно у Pichia stipitis. В частности, несколько ферментов приведены в базе данных UniprotKB, как, например, Klebsiella pneumoniae (№ O52720), Ralstonia solanacearum (№ P58708), Yersinia pestis (№ P58709), Aerobacter aerogenes (№ L8BEF0), E. coli (№ K3EX35, I2ZSJ5, W1BYD6, W1H8N7, E7U4R7).
Для целей настоящего изобретения Escherichia coli является предпочтительным источником гена NAD+-специфической D-арабит-4-оксидоредуктазы (образующей D-ксилулозу). Более конкретно, ее аминокислотная последовательность раскрыта под SEQ ID NO 2. В частности, под SEQ ID NO 1 и 3 раскрывают нуклеиновые кислоты, кодирующие NAD+-специфическую D-арабит-4-оксидоредуктазу Escherichia coli. Кодирующая последовательность была оптимизирована для Pichia ohmeri с учетом ее специфичности кодонов.
Кроме того, Ralstonia solanacearum также является предпочтительным источником гена NAD+-специфической D-арабит-4-оксидоредуктазы (образующей D-ксилулозу). Более конкретно, ее аминокислотная последовательность раскрыта под SEQ ID NO 43. В частности, под SEQ ID NO 42 раскрывают нуклеиновые кислоты, кодирующие NAD+-специфическую D-арабит-4-оксидоредуктазу Ralstonia solanacearum. Кодирующая последовательность была оптимизирована для Pichia ohmeri с учетом ее специфичности кодонов.
Второй фермент превращает D-ксилулозу в ксилит.
Несмотря на то, что большинство дрожжей и грибов имеют эндогенный ген ксилитдегидрогеназы (EC 1.1.1.9), изменение их специфичностей в отношении кофактора с NADH на NADPH необходимо для реализации настоящего изобретения. Действительно, ключевым аспектом настоящего изобретения является использование NADPH-специфической ксилитдегидрогеназы. Кроме того, этот фермент предпочтительно сверхэкспрессируется в хозяине.
Известны многочисленные ксилитдегидрогеназы и известны несколько научных статей, в которых есть рекомендации по изменению специфичности в отношении кофактора с NADH на NADPH. В Watanabe et al (J; Biol. Chem., 2005, 280, 10340-10345) раскрывают подвергнутую мутации ксилитдегидрогеназу Pichia stipitis с модифицированной специфичностью в отношении кофактора, в особенности тройной мутант (D207A/I208R/F209S) и четверной мутант (D207A/I208R/F209S/N211R). Аминокислотная последовательность четверного мутанта раскрыта под SEQ ID NO 5. Двойной мутант ксилитдегидрогеназы Gluconobacter oxydans (D38S/M39R) со специфичностью в отношении кофактора NADPH раскрыт в Ehrensberger et al (2006, Structure, 14, 567-575). Аминокислотная последовательность двойного мутанта раскрыта под SEQ ID NO 8.
Мутация и клонирование последовательности нуклеиновой кислоты Pichia stipitis XYL2, кодирующей NADPH-специфическую ксилитдегидрогеназу, были осуществлены изобретателями. В частности, под SEQ ID NO 4 и 6 раскрывают нуклеиновые кислоты, кодирующие специфическую к NADPH ксилитдегидрогеназу Pichia stipitis.
Альтернативно, изобретатели также выполняли мутацию и клонирование последовательности нуклеиновой кислоты Gluconobacter oxydans, кодирующей NADPH-специфическую ксилитдегидрогеназу. В частности, под SEQ ID NO 7 и 9 раскрывают нуклеиновые кислоты, кодирующие NADPH-специфическую ксилитдегидрогеназу Gluconobacter oxydans. Кодирующая последовательность была оптимизирована для Pichia ohmeri с учетом ее специфичности кодонов.
Кассета экспрессии, вектор и рекомбинантная клетка-хозяин
Согласно конкретному аспекту настоящее изобретение относится к нуклеиновой кислоте, содержащей кодирующую последовательность, оптимизированную для Pichia ohmeri, выбранную из группы, состоящей из SEQ ID NO 1, 3, 7, 9 и 42.
Оно также относится к кассете экспрессии, содержащей нуклеиновую кислоту, содержащую кодирующую последовательность, оптимизированную для Pichia ohmeri, выбранную из группы, состоящей из SEQ ID NO 1, 3, 7, 9 и 42.
Оно также относится к конструкции нуклеиновой кислоты с SEQ ID NO 4 и нуклеиновой кислоте, содержащей указанную конструкцию нуклеиновой кислоты.
К тому же, оно относится к рекомбинантному вектору, в частности вектору экспрессии, содержащему указанную нуклеиновую кислоту или кассету экспрессии. Как правило, кассета экспрессии содержит все элементы, необходимые для транскрипции и трансляции гена в белок. В частности, она содержит промотор, необязательно энхансер, терминатор транскрипции и элементы для трансляции. Более конкретно, промотор, используемый для контроля экспрессии NADPH-специфической ксилитдегидрогеназы, выбран для управления сильной экспрессией. В действительности, этот фермент предпочтительно сверхэкспрессируется в клетке-хозяине. Такие промоторы хорошо известны из уровня техники. К примеру, промотор может быть промотором рибулозоредуктазы P. ohmeri (poRR) или фосфоглицераткиназы P. ohmeri (poPGK1).
Оно относится к рекомбинантному вектору, в частности вектору экспрессии, содержащему нуклеиновую кислоту, кодирующую NAD+-специфическую D-арабит-4-оксидоредуктазу, и нуклеиновую кислоту, кодирующую NADPH-специфическую ксилитдегидрогеназу. Оно также относится к набору, содержащему рекомбинантный вектор, в частности вектор экспрессии, содержащий нуклеиновую кислоту, кодирующую NAD+-специфическую D-арабит-4-оксидоредуктазу, и рекомбинантный вектор, в частности вектор экспрессии, содержащий нуклеиновую кислоту, кодирующую NADPH-специфическую ксилитдегидрогеназу.
Предпочтительно, указанные NAD+-специфическая D-арабит-4-оксидоредуктаза и NADPH-специфическая ксилитдегидрогеназа выбраны из числа ферментов, раскрытых выше. В частности, указанная NAD+-специфическая D-арабит-4-оксидоредуктаза содержит аминокислотную последовательность с SEQ ID NO 2 или 42 или последовательность с 1-3 добавлениями, заменами или делециями аминокислот или состоит из нее. В частности, указанная NADPH-специфическая ксилитдегидрогеназа содержит аминокислотную последовательность с SEQ ID NO 5 или 8 или последовательность с 1-3 добавлениями, заменами или делециями аминокислот или состоит из нее.
Предпочтительный вектор представляет собой плазмиду. Пригодные плазмиды хорошо известны специалисту в данной области и могут быть, к примеру, выбраны из числа тех, которые конкретно раскрыты в примерах.
Генетически модифицированного хозяина по настоящему изобретению сперва получают путем клонирования генов, кодирующих NAD+-специфическую D-арабит-4-оксидоредуктазу и NADPH-специфическую ксилитдегидрогеназу, под контролем соответствующих промоторов в рекомбинантный вектор и вводят в клетки-хозяева продуцирующего D-арабит организма при помощи трансформации.
Настоящее изобретение относится к рекомбинантной или генетически сконструированной клетке-хозяину, содержащей последовательность гетерологичной нуклеиновой кислоты, кодирующей NAD+-специфическую D-арабит-4-оксидоредуктазу (EC 1.1.1.11), и последовательность гетерологичной нуклеиновой кислоты, кодирующей NADPH-специфическую ксилитдегидрогеназу. NAD+-специфическая D-арабит-4-оксидоредуктаза использует D-арабит в качестве субстрата и дает D-ксилулозу в качестве продукта. NADPH-специфическая ксилитдегидрогеназа использует D-ксилулозу в качестве субстрата и дает ксилит. Последовательность, кодирующая NADPH-специфическую ксилитдегидрогеназу и NAD+-специфическую D-арабит-4-оксидоредуктазу, может быть эписомальной или может быть интегрирована в хромосому клетки-хозяина. В действительности, генетически стабильные трансформанты предпочтительно конструируют при помощи систем трансформации с использованием вектора, в результате чего необходимая ДНК интегрируется в хромосому хозяина. Такая интеграция происходит de novo в клетке или ей может содействовать трансформация вектором, который функционально сам вставляется в хромосому хозяина, ДНК-элементами, которые способствуют интеграции последовательностей ДНК в хромосомы.
Рекомбинантная или генетически сконструированная клетка-хозяин может содержать несколько копий последовательности, кодирующей NAD+-специфическую D-арабит-4-оксидоредуктазу, и/или несколько копий последовательности, кодирующей NADPH-специфическую ксилитдегидрогеназу, предпочтительно интегрированную в хромосому клетки-хозяина. В частности, рекомбинантная или генетически сконструированная клетка-хозяин может содержать две, три или четыре последовательности, кодирующие NAD+-специфическую D-арабит-4-оксидоредуктазу, и/или две, три или четыре последовательности, кодирующие NADPH-специфическую ксилитдегидрогеназу. К примеру, клетка-хозяин может содержать две или три NAD+-специфические D-арабит-4-оксидоредуктазы из E. coli и/или одну или две NAD+-специфические D-арабит-4-оксидоредуктазы из R. solanacearum, более конкретно две или три NAD+-специфические D-арабит-4-оксидоредуктазы из E. coli и/или одну NAD+-специфическую D-арабит-4-оксидоредуктазу из R. solanacearum. NAD+-специфические D-арабит-4-оксидоредуктазы могут происходить из одного и того же организма или из различных организмов. NADPH-специфические ксилитдегидрогеназы могут происходить из одного и того же организма или из различных организмов. К примеру, клетка-хозяин может содержать одну, две или три NADPH-специфические ксилитдегидрогеназы из P. stipitis и/или одну, две или три NADPH-специфические ксилитдегидрогеназы из G. oxydans, более конкретно одну NADPH-специфическую ксилитдегидрогеназу из P. stipitis и/или три NADPH-специфические ксилитдегидрогеназы из G. oxydans.
В конкретном аспекте настоящего изобретения рекомбинантная или генетически сконструированная клетка-хозяин является клеткой штамма Pichia ohmeri, содержащего:
- две NAD+-специфические D-арабит-4-оксидоредуктазы и две NADPH-специфические ксилитдегидрогеназы; или
- две NAD+-специфические D-арабит-4-оксидоредуктазы из E. coli и две NADPH-специфические ксилитдегидрогеназы, одна из P. stipitis и другая из G. oxydans; или
- две NAD+-специфические D-арабит-4-оксидоредуктазы и три NADPH-специфические ксилитдегидрогеназы; или
- две NAD+-специфические D-арабит-4-оксидоредуктазы из E. coli и три NADPH-специфические ксилитдегидрогеназы, одна из P. stipitis и две из G. oxydans; или
- три NAD+-специфические D-арабит-4-оксидоредуктазы и три NADPH-специфические ксилитдегидрогеназы; или
- три NAD+-специфические D-арабит-4-оксидоредуктазы, две из E. coli и одна из R. solanacearum, и три NADPH-специфические ксилитдегидрогеназы, одна из P. stipitis и две из G. oxydans; или
- четыре NAD+-специфические D-арабит-4-оксидоредуктазы и четыре NADPH-специфические ксилитдегидрогеназы; или
- четыре NAD+-специфические D-арабит-4-оксидоредуктазы, три из E. coli и одна из R. solanacearum, и четыре NADPH-специфические ксилитдегидрогеназы, одна из P. stipitis и три из G. oxydans.
Клетку-хозяина выбирают из числа микроорганизмов, подробно рассмотренных выше. Согласно предпочтительному варианту осуществления клетка-хозяин представляет собой клетку Pichia ohmeri. Начальная клетка-хозяин предпочтительно является клеткой мутантного Pichia ohmeri, депонированного в CNCM под номером I-4605.
Согласно конкретному аспекту настоящего изобретения клетка-хозяин является клеткой штамма, выбранного из штаммов I-4982, I-4960 и I-4981, депонированных в CNCM.
Настоящее изобретение относится к способу получения ксилита, включающему культивирование рекомбинантной или генетически сконструированной клетки-хозяина в культуральной среде и извлечение полученного ксилита. Предпочтительно, культуральная среда обеспечивает микроорганизм пригодным источником углерода. Источник углерода, предпочтительно, включает D-глюкозу, и разные содержащие D-глюкозу патоки, и смеси D-глюкозы с другими сахарами. Способ может дополнительно включать стадию очистки ксилита.
Настоящее изобретение относится к применению рекомбинантной или генетически сконструированной клетки-хозяина, которая раскрыта в данном документе, для получения ксилита.
Ксилит, продуцируемый такими генетически модифицированными штаммами, можно очищать от среды хозяев по настоящему изобретению согласно любой известной из уровня техники методике. Например, в US 5081026, включенном в данный документ при помощи ссылки, описано хроматографическое отделение ксилита от культур дрожжей. Таким образом, полученный на стадии ферментации ксилит может быть очищен от культуральной среды с использованием хроматографических стадий, которые описаны в US 5081026, с последующей кристаллизацией.
Другие характерные особенности и преимущества настоящего изобретения будут очевидны при прочтении изложенных ниже примеров. Однако, они даны в данном документе только в качестве иллюстраций, а не ограничений.
Фигуры и последовательности
Фигура 1. 12 ABYWMP, рестрикционная карта синтезированной NAD+-специфической D-арабит-4-оксидоредуктазы из E. Coli, фланкированной сайтами рестрикции для AscI и SphI.
Фигура 2a. lig7.78, рестрикционная карта NADH-специфической ксилитдегидрогеназы из Pichia stipitis.
Фигура 2b. 12AALQTP, рестрикционная карта синтезированной NADPH-специфической ксилитдегидрогеназы из Pichia stipitis, фланкированной сайтами рестрикции для HindIII и SacII.
Фигура 3. 13AAYSYP, рестрикционная карта синтезированной NADPH-специфической ксилитдегидрогеназы из Gluconobacter oxydans, фланкированной сайтами рестрикции для AscI и SphI.
Фигура 4. Конструирование кассеты экспрессии, состоящая из открытой рамки считывания, фланкированной промотором и терминатором poRR с помощью перекрывающейся ПЦР.
Фигура 5. 12 AAMCJP, рестрикционная карта синтезированной тагатозо-3-эпимеразы Pseudomonas cichorii, фланкированной сайтами рестрикции для HindIII и SacII.
Фигура 6. Конструирование "челночных" векторов P. ohmeri с маркерами отбора poLEU2 и poURA3.
Фигура 7. pEVE2523, рестрикционная карта вектора экспрессии pEVE2523 P. ohmeri с poURA3 с клонированной кассетой экспрессии, содержащей открытую рамку считывания тагатозо-3-эпимеразы Pseudomonas cichorii, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR).
Фигура 8. pEVE2560, рестрикционная карта вектора экспрессии pEVE2560 P. ohmeri с poLEU2 с клонированной кассетой экспрессии, содержащей открытую рамку считывания тагатозо-3-эпимеразы Pseudomonas cichorii, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR).
Фигура 9. Конструирование вектора P. ohmeri для сверхэкспрессии NADPH-специфической ксилитдегидрогеназы Gluconobacter oxydans.
Фигура 10. pEVE3284, рестрикционная карта вектора экспрессии pEVE3284 P. ohmeri с клонированной кассетой экспрессии, содержащей NADPH-специфическую ксилитдегидрогеназу Gluconobacter oxydans, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR).
Фигура 11. Конструирование векторов P. ohmeri для сверхэкспрессии NADPH-специфической ксилитдегидрогеназы Pichia stipitis.
Фигура 12. pEVE2562/pEVE2564, рестрикционная карта векторов экспрессии pEVE2562/pEVE2564 P. ohmeri с клонированной кассетой экспрессии, содержащей NADPH-специфическую ксилитдегидрогеназу Pichia stipitis, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR), соответственно с маркером отбора либо poURA3, либо poLEU2.
Фигура 13. Конструирование вектора P. ohmeri для сверхэкспрессии NADH-специфической ксилитдегидрогеназы Pichia stipitis.
Фигура 14. pEVE2563, рестрикционная карта вектора экспрессии pEVE2563 P. ohmeri с клонированной кассетой экспрессии, содержащей NADH-специфическую ксилитдегидрогеназу Pichia stipitis, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR).
Фигура 15. Конструирование вектора P. ohmeri для сверхэкспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора и терминатора рибулозоредуктазы P. ohmeri (poRR) с использованием маркера отбора poURA3.
Фигура 16. pEVE2839, рестрикционная карта вектора экспрессии pEVE2839 P. ohmeri с клонированной кассетой экспрессии, содержащей NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR).
Фигура 17. Конструирование вектора P. ohmeri для сверхэкспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы (poPGK1) и терминатора транскетолазы (poTKL) P. ohmeri с использованием маркера отбора poURA3.
Фигура 18. pEVE3102, рестрикционная карта вектора экспрессии pEVE3102 P. ohmeri с клонированной кассетой экспрессии, содержащей NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором фосфоглицераткиназы P. ohmeri (poPGK1) и терминатором рибулозоредуктазы (poRR).
Фигура 19. pEVE3123, рестрикционная карта вектора экспрессии pEVE3123 P. ohmeri с клонированной кассетой экспрессии, содержащей NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором фосфоглицераткиназы P. ohmeri (poPGK1), и терминатором транскетолазы (poTKL), и маркером отбора poURA3.
Фигура 20. Конструирование вектора P. ohmeri для сверхэкспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы (poPGK1) и терминатора транскетолазы (poTKL) P. ohmeri с использованием маркера отбора poLEU2.
Фигура 21. pEVE3157, рестрикционная карта вектора экспрессии pEVE3157 P. ohmeri с клонированной кассетой экспрессии, содержащей NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором фосфоглицераткиназы P. ohmeri (poPGK1), и терминатором транскетолазы (poTKL), и маркером отбора poLEU2.
Фигура 22. Конструирование вектора с loxP P. ohmeri с маркером отбора poLEU2.
Фигура 23. pEVE2787, рестрикционная карта вектора интеграции pEVE2787 P. ohmeri с клонированным маркером отбора LEU2 P. ohmeri под контролем эндогенного промотора и терминатора, фланкированным двумя сайтами loxP.
Фигура 24. 12ABTV4P, рестрикционная карта синтезированного гена nat1 из Streptomyces noursei, фланкированного сайтами рестрикции для AscI и SphI.
Фигура 25. pEVE2798, рестрикционная карта вектора экспрессии pEVE2798 P. ohmeri с клонированным маркером nat1 под контролем промотора рибулозоредуктазы (poRR) и терминатора оротидин-5'-фосфатдекарбоксилазы (poURA3).
Фигура 26. Конструирование вектора с loxP P. ohmeri с маркером отбора nat1.
Фигура 27. pEVE2852, рестрикционная карта вектора интеграции pEVE2852 P. ohmeri с клонированным маркером nat1 под контролем промотора рибулозоредуктазы (poRR) и терминатора оротидин-5'-фосфатдекарбоксилазы (poURA3), фланкированного двумя сайтами loxP.
Фигура 28. pEVE2855, рестрикционная карта вектора интеграции pEVE2855 P. ohmeri с клонированным фрагментом, гомологичным 5’-области, расположенной выше открытой рамки считывания LEU2, и маркером отбора nat1, фланкированным двумя сайтами loxP.
Фигура 29. Конструирование вектора с loxP P. ohmeri для делеции открытой рамки считывания LEU2.
Фигура 30. pEVE2864, рестрикционная карта вектора интеграции pEVE2864 P. ohmeri с клонированным фрагментом, гомологичным 5’-области, расположенной выше открытой рамки считывания LEU2, и фрагментом, гомологичным 3’-области, расположенной ниже открытой рамки считывания LEU2, и маркером отбора nat1, фланкированным двумя сайтами loxP.
Фигура 31. Конструирование двойных плазмид для экспрессии, содержащих NADPH-специфическую ксилитдегидрогеназу P. Stipitis и NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli.
Фигура 32. pEVE3318, рестрикционная карта вектора экспрессии pEVE3318 P. ohmeri, содержащего двойную конструкцию для экспрессии NADPH-специфической ксилитдегидрогеназы P. stipitis и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli.
Фигура 33. pEVE2862, рестрикционная карта вектора экспрессии pEVE2862 P. ohmeri, содержащего маркер LEU2 P. ohmeri, фланкированный промотором рибулозоредуктазы P. ohmeri (poRR) и терминатором оротидин-5'-фосфатдекарбоксилазы (poURA3).
Фигура 34. Конструирование интеграционного вектора для геномной экспрессии гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и гена NADPH-специфической ксилитдегидрогеназы P. Stipitis в P. ohmeri.
Фигура 35. pEVE2865, рестрикционная карта вектора интеграции pEVE2865 P. ohmeri, содержащего маркер LEU2 P. ohmeri, фланкированный двумя сайтами loxP.
Фигура 36. pEVE3387, рестрикционная карта вектора интеграции pEVE3387 P. ohmeri, содержащего двойную конструкцию для экспрессии гена NADPH-специфической ксилитдегидрогеназы P. stipitis и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli с маркером отбора LEU2 P. ohmeri, фланкированным двумя сайтами loxP.
Фигура 37. Конструирование двойной/тройной плазмид для экспрессии, содержащих NADPH-специфическую ксилитдегидрогеназу G. oxydans и NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli.
Фигура 38. pEVE3322/pEVE3324, рестрикционная карта векторов экспрессии pEVE3322/pEVE3324 P. ohmeri, содержащих либо двойную конструкцию для экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli, либо тройную конструкцию для экспрессии двух генов NADPH-специфической ксилитдегидрогеназы G. oxydans и одной NAD+-специфической D-арабит-4-оксидоредуктазы E. coli.
Фигура 39. Конструирование интеграционного вектора для геномной экспрессии гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и гена NADPH-специфической ксилитдегидрогеназы G. oxydans в P. ohmeri.
Фигура 40. pEVE3390/pEVE3392, рестрикционная карта векторов интеграции pEVE3390/pEVE3392 P. ohmeri, содержащих либо двойную конструкцию для экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli, либо тройную конструкцию для экспрессии двух генов NADPH-специфической ксилитдегидрогеназы G. oxydans и одной NAD+-специфической D-арабит-4-оксидоредуктазы E. coli, с маркером отбора LEU2 P. ohmeri, фланкированным двумя сайтами loxP.
Фигура 41. Конструирование интеграционного вектора для геномной экспрессии гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и гена NADPH-специфической ксилитдегидрогеназы G. oxydans в P. ohmeri.
Фигура 42. pEVE4390, рестрикционная карта вектора экспрессии pEVE4390 P. ohmeri, содержащего двойную конструкцию для экспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и гена NADPH-специфической ксилитдегидрогеназы G. oxydans с маркером отбора LEU2 P. ohmeri, фланкированным двумя сайтами loxP.
Фигура 43. 13AB2EGF, рестрикционная карта синтезированной NAD+-специфической D-арабит-4-оксидоредуктазы из R. solanacearum, фланкированной сайтами рестрикции для AscI и SphI.
Фигура 44. Конструирование интеграционного вектора для геномной экспрессии гена NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum и гена NADPH-специфической ксилитдегидрогеназы G. oxydans в P. ohmeri.
Фигура 45. pEVE3898, рестрикционная карта вектора экспрессии pEVE3898 P. ohmeri с клонированной кассетой экспрессии, содержащей NAD+-специфическую D-арабит-4-оксидоредуктазу Ralstonia solanacearum, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR).
Фигура 46. pEVE4077, рестрикционная карта вектора экспрессии pEVE4077 P. ohmeri с двойной конструкцией для экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum.
Фигура 47. pEVE4377, рестрикционная карта вектора интеграции pEVE4377 P. ohmeri с двойной конструкцией для экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum и маркером отбора poLEU2, фланкированным двумя сайтами loxP.
Перечень последовательностей
Примеры
Пример 1. Выбор штамма Pichia ohmeri в качестве предпочтительного хозяина для генетического конструирования
Выбранный штамм-хозяин Pichia ohmeri:
- является продуцентом значительных количеств арабита из глюкозы в условиях среды с высоким осмотическим давлением, например, среды, содержащей 10-60% D-глюкозы и предпочтительно 25% D-глюкозы ("нормальная" среда, как правило, содержит только 2-3% глюкозы).
- характеризуется окислительно-восстановительным балансом, который обеспечивает получение необходимых кофакторов.
Для иллюстрации указанных характеристик в следующих таблицах отображены ферментативные активности, вовлеченные в метаболический путь арабита у Pichia ohmeri (Sophie HUCHETTE Thesis, 1992).
Гексозомонофосфатный путь: от глюкозо-6-P до D-рибулозо-5-P и D-ксилулозо-5-P
Окислительная часть PPP, также называемого гексозомонофосфатным путем (HMP), представляет собой NADPH-продуцирующую часть пути. Два NADP+-зависимых фермента, представляющие собой глюкозо-6-P-дегидрогеназу (E.C.1.1.1.49) и 6-P-глюконатдегидрогеназу, (E.C.1.1.1.44) участвуют в окислении 1 моля глюкозо-6-P в 1 моль D-рибулозо-5-P и образовании 2 молей NADPH.
Таблица 1. Гексозомонофосфатный путь у P. ohmeri ATCC 20209
Одну единицу ферментативной активности определяли как потребление 1 мкмоля NAD(P)H или NAD(P)+ в 1 минуту на мл неочищенного экстракта. Одну единицу специфической активности определяли как одну единицу ферментативной активности на мг белков неочищенного экстракта.
Определяли кинетические параметры следующих ферментов: D-рибулозо-5-P-3-эпимераза (E.C 5.1.3.1), D-рибозо-5-P-кето-изомераза (E.C.5.3.1.6), транскетолаза (E.C.2.2.1.1) и кислые фосфатазы (E.C. 3.1.3.2).
Таблица 2. Кинетические параметры ферментов, использующих D-рибулозо-5-P в качестве субстрата у P. ohmeri ATCC 20209
Одну единицу ферментативной активности определяли как потребление 1 мкмоля NAD(P)H или NAD(P)+ в 1 минуту на мл неочищенного экстракта. Одну единицу специфической активности определяли как одну единицу ферментативной активности на мг белков неочищенного экстракта.
Таблица 3. Кинетические параметры ферментов, использующих D-ксилулозо-5-P в качестве субстрата у P. ohmeri ATCC 20209
Одну единицу ферментативной активности определяли как потребление 1 мкмоля NAD(P)H или NAD(P)+ в 1 минуту на мл неочищенного экстракта. Одну единицу специфической активности определяли как одну единицу ферментативной активности на мг белков неочищенного экстракта.
В условиях in vivo D-ксилулозо-5-P, синтезированный посредством эпимеризации D-рибулозо-5-P, эффективно участвует в неокислительной части PPP посредством реакции с транскетолазой. Следовательно, D-ксилулозо-5-P не доступен для его дефосфорилирования в D-ксилулозу.
NADH-и NADPH-специфические D-кетопентозо-оксидоредуктазы
D-рибулоза и D-ксилулоза образуются путем дефосфорилирования D-рибулозо-5-P и D-ксилулозо-5-P.
Константы Михаэлиса-Ментен отображают аффинности NADH- и NADPH-D-кетопентозо-оксидоредуктаз для каждого субстрата и соответствующих скоростей.
Таблица 4. Кинетические параметры NADH-специфической D-кетопентозо-оксидоредуктазы у P. ohmeri ATCC 20209
Одну единицу ферментативной активности определяли как потребление 1 мкмоля NAD(P)H или NAD(P)+ в 1 минуту на мл неочищенного экстракта. Одну единицу специфической активности определяли как одну единицу ферментативной активности на мг белков неочищенного экстракта.
NADH-специфическая D-кетопентозо-оксидоредуктаза, участвуя в образовании рибита и ксилита соответственно из D-рибулозы и D-ксилулоза, проявляет большую аффинность в отношении D-ксилулозы, чем D-рибулозы. Обратная реакция показывает хорошую аффинность в отношении ксилита и рибита, объясняющую хороший рост штамма-хозяина на этих двух полиолах.
Таблица 5. Кинетические параметры NADPH-специфической D-кетопентозо-оксидоредуктазы у P. ohmeri ATCC 20209
Одну единицу ферментативной активности определяли как потребление 1 мкмоля NAD(P)H или NAD(P)+ в 1 минуту на мл неочищенного экстракта. Одну единицу специфической активности определяли как одну единицу ферментативной активности на мг белков неочищенного экстракта.
NADPH-специфическая D-кетопентозо-оксидоредуктаза, участвуя в образовании D-арабита из D-рибулозы и участвуя в образовании ксилита из D-ксилулозы, проявляет большую аффинность в отношении D-рибулозы, чем D-ксилулозы. Обратная реакция показывает очень низкую аффинность в отношении D-арабита, объясняющую отсутствие роста штамма-хозяина на этом полиоле.
Две указанные кетопентозо-оксидоредуктазы из штамма-хозяина характеризовали как отличающиеся от предыдущих ферментов, описанных для Saccharomyces rouxii у Ingram и Wood, 1965 (Journal of Bacteriology, vol.89, n°5, 1186-1194). Действительно, у Saccharomyces rouxii не обнаруживали прямой реакции D-рибулозы и NADH и обратной реакции D-арабита с NADPH.
Кинетические модели поведения ферментов in vivo определяются отношением Холдейна.
Таблица 6. Определение констант Холдейна: NADH-специфическая D-кетопентозо-оксидоредуктаза
Определение констант Холдейна: NADPH-специфическая D-кетопентозо-оксидоредуктаза
Два указанных фермента способствуют прямой реакции (окисление D-кетопентозы), в отличие от обратной реакции (восстановление пентита).
PPP у штамма-хозяина является чрезвычайно эффективным, поскольку в результате потребления 1 моля глюкозы образуются 2 моля NADPH. Следовательно NADPH будет доступен в избытке как для анаболических реакций, так и для поддерживающих реакций. Штамм-хозяин должен продуцировать D-арабит из D-рибулозы или ксилит из D-ксилулозы для поддержания равновесия окислительно-восстановительной пары NADPH/NADP+.
Ингибирующий эффект NADP+ в отношении NADPH-специфической D-кетопентозо-оксидоредуктазы определяли в условиях in vitro. Активность составляет менее 80% при добавлении NADP+в избытке. Даже если данная концентрация не сопоставима с внутриклеточной концентрацией NADP+, такой результат дает некоторое представление о роли NADPH-специфической D-кетопентозо-оксидоредуктазы в равновесии окислительно-восстановительной пары NADPH/NADP+.
Штамм-хозяин продуцирует только D-арабит из D-рибулозы, поскольку D-ксилулоза недоступна из-за участия D-ксилулозо-5-P в неокислительной части PPP.
Связь между продукцией D-арабита и окислительно-восстановительного равновесия NADPH/NADP+ продемонстрирована у штамма-хозяина путем оценки влияния сверхэкспрессии глюкозо-6-P-дегидрогеназы на продуцирование D-арабита. Таким образом, полученный штамм характеризуется активностью G6PDH в 1,5 раза выше и продуцирует на 10% больше D-арабита, по сравнению со штаммом-хозяином (FR2772788).
Пример 2. Частота использования кодона Pichia ohmeri
Частоту использования кодона P. ohmeri определяли из имеющейся ДНК и соответствующей аминокислотной последовательности пяти генов P. ohmeri: транскетолазы, глюкозо-6-фосфатдегидрогеназы (FR 2772788), рибулозоредуктазы, бета-изопропилмалатдегидрогеназы - LEU2 (Piredda и Gaillardin, Yeast, vol.10:1601-1612 (1994) и оротидин-5'-фосфатдекарбоксилазы - URA3 (Piredda и Gaillardin, 1994, ранее).
Каждый отдельный ген разделяли на нуклеотидные триплеты, кодирующие одну аминокислоту. Пять генов составляли в общей сложности 2091 кодон.
Для каждой аминокислоты подсчитывали количество каждого кодона, присутствующее в пяти генах, делили на 2091 и умножали на 1000. Таким образом оценивали частоту конкретного кодона на 1000 кодонов.
Предварительная частота использования кодона P. ohmeri отображена в таблице 7.
Все гетерологичные гены, экспрессируемые P. ohmeri, за исключением гена ксилитдегидрогеназы из P. stipitis, были кодон-оптимизированы при помощи этой таблицы и программы Optimizer, полученной из http://genomes.urv.es/OPTIMIZER/.
Полученную последовательность отправляли на генный синтез после ручного добавления сайтов узнавания для ферментов рестрикции в соответствующие 5'- и 3'-концы последовательности, кодирующей фермент.

Пример 3. Клонирование гена бактериальной NAD+-специфической D-арабит-4-оксидоредуктазы (D-ксилулозо-образующей) E. coli
Фрагмент ДНК, кодирующий altD NAD+-специфической D-арабит-4-оксидоредуктазы из E. coli, синтезировали химическим путем (синтез генов при помощи GeneArt® Gene Synthesis, Life Technologies, Регенсбург, Германия) в соответствии с предоставленной последовательностью SEQ ID NO: 1.
Нуклеотиды 1441-2808 последовательности AF378082.1 (полученной из http://www.ncbi.nlm.nih.gov/nuccore/AF378082), кодирующие ген altD, использовали в качестве матрицы и подвергали оптимизации кодонов для применения у P. ohmeri ATCC 20209 согласно таблице 7 в примере 2, при помощи программы Optimizer, полученной из http://genomes.urv.es/OPTIMIZER/.
Для облегчения последующего клонирования к 5’- и 3’-концам результирующей последовательности добавляли нуклеотиды, кодирующие сайты распознавания ретрикционными ферментами, AscI (GGCGCGCC) и SphI (GCATGC) соответственно.
Дополнительно, включали аденозиновый триплет спереди от стартового ATG для обеспечения аденозина в положении -3 в Kozak-подобной последовательности у дрожжей.
Конечную последовательность (SEQ ID NO: 1) затем предоставляли для синтеза (GeneArt, Регенсбург, Германия).
Синтезированный фрагмент ДНК, кодирующий NAD+-специфическую D-арабит-4-оксидоредуктазу из E. coli, доставляли в виде 5 мкг лиофилизированной плазмидной ДНК в векторе на основе pMK-RQ (12ABYWMP, фигура 1).
Для дальнейшего субклонирования ген выделяли посредством рестрикционного вырезания при помощи ферментов AscI и SphI (New England Biolabs, Ипсуич, Массачусетс).
Пример 4. Мутагенез и клонирование NADH- и NADPH-специфической ксилитдегидрогеназы Pichia stipitis
Клонирование гена NADH-специфической ксилитдегидрогеназы Pichia stipitis
Известную нуклеотидную последовательность гена XYL2 дрожжей (Pichia stipitis), кодирующего ксилитдегидрогеназу (Kötter et al., Curr. Genet. 18:493-500 (1990)) клонировали в плазмидный вектор lig 7.78, следуя идее FR 2 765 589 (см. Пример 4 и фигуру 7 в данном патенте). Рестрикционная карта вектора представлена на фигуре 2a.
Мутагенез и клонирование гена NADPH-специфической ксилитдегидрогеназы Pichia stipitis
Фрагмент ДНК, кодирующий XYL2 NADPH-специфической ксилитдегидрогеназы из Pichia stipitis, синтезировали химическим путем (синтез генов при помощи GeneArt® Gene Synthesis, Life Technologies, Регенсбург, Германия) в соответствии с последовательностью SEQ ID NO: 4.
Нуклеотиды 319-1410 последовательности X55392.1 (полученной из http://www.ncbi.nlm.nih.gov/nuccore/X55392.1), кодирующей ген XYL2, были использованы в качестве матрицы.
Согласно документу Watanabe et al. (J; Biol. Chem., 2005, 280, 10340-10345), предпочтение в отношении кофактора для ксилитдегидрогеназы может быть изменено с NADH на NADPH посредством введения четырех опубликованных аминокислотных мутаций: D207A/I208R/F209S/N211R (нумерация основана на последовательности белка P22144, полученной из http://www.uniprot.org/uniprot/P22144).
В связи с этим, кодоны, кодирующие D207, I208, F209 и N211 вручную заменяли на GCT, AGA, ТСА и AGA в соответствующей последовательности, в указанном порядке.
Кроме того, для облегчения последующего клонирования, нуклеотиды, кодирующие сайты распознавания ретрикционными ферментами HindIII (AAGCTT) и SacII (CCGCGG), вручную добавляли к соответствующим 5’- и 3’-концам.
Кроме того, включали аденозиновый триплет спереди от стартового ATG для обеспечения аденозина в положении -3 в Kozak-подобной последовательности у дрожжей. Конечную последовательность (SEQ ID NO: 4) предоставляли для синтеза (GeneArt, Регенсбург, Германия).
Синтезированный фрагмент ДНК, кодирующий NADPH-специфическую ксилитдегидрогеназу из P. stipitis, доставляли в виде 5 мкг лиофилизированной плазмидной ДНК в векторе на основе pMA-T (12AALQTP, фигура 2b).
Пример 5. Мутагенез и клонирование гена NADPH-специфической ксилитдегидрогеназы Gluconobacter oxydans
Фрагмент ДНК, кодирующий Xdh NADPH-специфической ксилитдегидрогеназы из Gluconobacter oxydans, синтезировали химическим путем (синтез генов при помощи GeneArt® Gene Synthesis, Life Technologies, Регенсбург, Германия) в соответствии с предоставленной последовательностью SEQ ID NO: 7.
Нуклеотиды 1063-1851 последовательности AB091690.1 (полученной из http://www.ncbi.nlm.nih.gov/nuccore/AB091690.1), кодирующие ген Xdh, использовали в качестве матрицы и подвергали оптимизации кодонов для применения у P. ohmeri ATCC 20209 согласно таблице 7 (пример 2), при помощи программы Optimizer, полученной из http://genomes.urv.es/OPTIMIZER/.
Согласно публикации Ehrensberger et al. (Structure, 2006, 14, 567-575), специфичность в отношении кофактора фермента может быть изменена с NADH на NADPH посредством введения четырех опубликованных аминокислотных мутаций: D38S/M39R (нумерация основана на последовательности белка Q8GR61, полученной из http://www.uniprot.org/uniprot/Q8GR61).
Таким образом, кодоны, кодирующие D38 и M39 вручную заменяли на TCT и AGA в соответствующей последовательности, в указанном порядке. Кроме того, для облегчения последующего клонирования, нуклеотиды, кодирующие сайты распознавания ретрикционными ферментами AscI (GGCGCGCC) и SphI (GCATGC), вручную добавляли к соответствующим 5’- и 3’-концам.
Кроме того, включали аденозиновый триплет спереди от стартового ATG для обеспечения аденозина в положении -3 в Kozak-подобной последовательности у дрожжей. Конечную последовательность (SEQ ID NO: 7), предоставляли для синтеза (GeneArt, Регенсбург, Германия).
Синтезированный фрагмент ДНК, кодирующий NADPH-специфическую ксилитдегидрогеназу из Gluconobacter oxydans доставляли в виде 5 мкг лиофилизированной плазмидной ДНК в векторе на основе pMA-T (13AAYSYP, фигура 3). Для дальнейшего субклонирования ген выделяли посредством рестрикционного вырезания при помощи ферментов AscI и SphI (New England Biolabs, Ипсуич, Массачусетс).
Пример 6. Конструирование вектора P. ohmeri для экспрессии гетерологичного гена с применением маркера отбора poURA3
Клонирование вектора с замещаемыми:
- промотором,
- открытой рамкой считывания и
- элементами терминации
выполняли посредством двух последовательных перекрывающихся ПЦР трех отдельных фрагментов (фигура 4).
Первоначально вектор выступал в качестве модели экспрессии для проверки клонирования и сверхэкспрессии гена тагатозо-3-эпимеразы у рекомбинантного штамма Pichia ohmeri.
Как будет описано ниже, ген тагатозо-3-эпимеразы клонировали в специфическую в отношении AscI – SphI кассету рестрикционных сайтов, обеспечивая клонирование любого гена, представляющего интерес, при помощи таких же сайтов вставки.
Клонирование было организовано следующим образом.
В ходе первой ПЦР (PCR1) фрагмент промотора рибулозоредуктазы P. Ohmeri длиной 490 п.о., фланкированный при помощи сайтов SpeI и AscI (подчеркнуты в последовательности праймера), амплифицировали с применением:
- праймера EV2960:
GAACTAGTGGATCCGTAGAAATCTTG (SEQ ID No 12)
и
- праймера EV2961:
CTTTGTTCATTTTGGCGCGCCTTTTAGTTTAATAAGGGTCCGTG (SEQ ID No 13)
Кроме того, к 5’-концу обратного праймера EV2961 добавляли фрагмент длиной 13 нуклеотидов, представляющий собой 5’-конец гена тагатозо-3-эпимеразы.
Данный фрагмент совместно с 8 нуклеотидами сайта AscI и 10 последующими нуклеотидами 3’-конца промотора рибулозоредуктазы были необходимы в качестве перекрывающихся для слияния фрагмента PCR1 с фрагментом PCR2, описанных выше. Геномную ДНК P. ohmeri ATCC 20209 использовали в качестве матрицы.
Для данной цели свежеснятую способом штрихования колонию P. ohmeri ресуспендировали в 30 мкл 0,2% SDS и нагревали в течение 4 мин. при 95°C. После центрифугирования с максимальным числом оборотов, 0,5 мкл надосадочной жидкости применяли для ПЦР.
Матрицу амплифицировали в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 25 циклами в течение 10 сек при 98°C/20 сек при 50°C/15 сек при 72°C и конечной стадией удлинения в течение 10 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
В ходе второй ПЦР (PCR2) фрагмент тагатозо-3-эпимеразы Pseudomonas cichorii ST24 длиной 911 п.о., фланкированный при помощи сайтов AscI и SphI (подчеркнуты в последовательности праймера), амплифицировали с применением:
- праймера EV2962:
AAACTAAAAGGCGCGCCAAAATGAACAAAGTTGGCATG (SEQ ID No 14)
и
- праймера EV2963:
TTCTCTTCGAGAGCATGCTCAGGCCAGCTTGTCACG (SEQ ID No 15).
5-конец праймера EV2962 содержит фрагмент длиной 9 нуклеотидов, представляющий 3’-конец промотора рибулозоредуктазы.
Данный фрагмент совместно с 8 нуклеотидами сайта AscI и 12 последующими нуклеотидами открытой рамки считывания тагатозо-3-эпимеразы применяли для перекрывающейся ПЦР для слияния продукта PCR2 с описанным ранее продуктом PCR1.
Кроме того, 5’-конец обратного праймера EV2963 содержит фрагмент длиной 12 нуклеотидов, представляющий 5’-конец терминатора рибулозоредуктазы P. ohmeri.
Данный фрагмент совместно с 6 нуклеотидами сайта SphI и 12 последующими нуклеотидами 3’-конца открытой рамки считывания тагатозо-3-эпимеразы были необходимы в качестве перекрывающихся для слияния фрагмента PCR2 с фрагментом PCR из PCR3, описанных ранее.
В качестве матрицы применяли 25 нг вектора 12AAMCJP (фигура 5) (GeneArt, Регенсбург, Германия), содержащего синтезированную копию гена тагатозо-3-эпимеразы Pseudomonas cichorii ST24 (нуклеотиды 719-1591 AB000361.1, из http://www.ncbi.nlm.nih.gov/nuccore/AB000361) – SEQ ID No: 11.
Матрицу амплифицировали в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 25 циклами в течение 10 сек при 98°C/20 сек при 48°C/30 сек при 72°C и конечной стадией удлинения в течение 10 минут при 72°C.
В ходе третей ПЦР (PCR3), фрагмент терминатора рибулозоредуктазы P. ohmeri длиной 380 длиной п.о., фланкированный при помощи сайтов SphI и SacII (подчеркнуты в последовательности праймера), амплифицировали с применением:
- праймера EV2964
AAGCTGGCCTGAGCATGCTCTCGAAGAGAATCTAG (SEQ ID No 16)
и
- праймера EV2965
GTTCCGCGGAGAATGACACGGCCGAC (SEQ ID No 17)
5’-конец праймера EV2964 содержит фрагмент 3’-конца открытой рамки считывания тагатозо-3-эпимеразы длиной 12 нуклеотидов, который совместно с 6 нуклеотидами сайта SphI и 12 последующими нуклеотидами терминатора рибулозоредуктазы P. Ohmeri, применяли для слияния PCR3 с описанным ранее PCR2.
Геномную ДНК P. ohmeri ATCC 20209 использовали в качестве матрицы. После центрифугирования с максимальным числом оборотов, 0,5 мкл надосадочной жидкости применяли для ПЦР. Для данной цели свежеснятую способом штрихования колонию P. ohmeri ресуспендировали в 30 мкл 0,2% SDS и нагревали в течение 4 мин. при 95°C.
Матрицу амплифицировали в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера, а также 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 25 циклами в течение 10 сек при 98°C/20 сек при 50°C/15 сек при 72°C и конечной стадией удлинения в течение 10 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Слияние трех отдельных фрагментов ПЦР осуществляли следующим образом: 50 нг каждого продукта PCR1 и PCR2, очищенного от геля, использовали в качестве матрицы для реакции ПЦР с EV2960 и EV2963.
Гомологичный сегмент длиной 30 нуклеотидов в двух фрагментах, полученный в результате конструирования праймера, описанного выше, использовали в качестве перекрывающегося в реакции слияния.
Таким образом, фрагмент длиной 1,4 т.п.о., состоящий из промотора рибулозоредуктазы P. Ohmeri, фланкированного при помощи сайтов SpeI и AscI, сливали с открытой рамкой считывания тагатозо-3-эпимеразы Pseudomonas cichorii ST24.
Матрицы амплифицировали в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера, а также 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/20 сек при 62°C/45 сек при 72°C и конечной стадией удлинения в течение 10 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Очищенный фрагмент сливали во второй перекрывающейся ПЦР с продуктом PCR3. 40 нг каждого фрагмента использовали в качестве матрицы и амплифицировали с EV2960 и EV2965.
Гомологичный сегмент длиной 30 нуклеотидов в двух фрагментах, полученный в результате конструирования праймера, описанного выше, использовали в качестве перекрывающегося в слиянии.
Таким образом, фрагмент длиной 1,8 т.п.о., состоящий из промотора рибулозоредуктазы P. ohmeri, фланкированного при помощи SpeI и AscI, и открытой рамки считывания тагатозо-3-эпимеразы Pseudomonas cichorii ST24, фланкированной при помощи сайтов AscI и SphI, сливали с терминатором рибулозоредуктазы P. ohmeri.
Матрицы амплифицировали в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера, а также 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/20 сек при 65°C/55 сек при 72°C и конечной стадией удлинения в течение 10 минут при 72°C. Продукт ПЦР отделяли на агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Конечный продукт ПЦР, состоящий из фрагмента тагатозо-3-эпимеразы Pseudomonas cichorii ST24 длиной 1,7 т.п.о., фланкированный промотором рибулозоредуктазы и терминатором, расщепляли при помощи рестрикционных ферментов SpeI и SacII (New England Biolabs, Ипсуич, Массачусетс), очищали от геля и лигировали в течение ночи при 16°C с изолированным фрагментом SpeI/SacII длиной 9,8 т.п.о. остова вектора lig7.78, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 6).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния). Очищенную плазмидную ДНК использовали для дополнительного исследования посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Вновь клонированная плазмида для экспрессии pEVE2523 (фигура 7) представляет собой бифункциональный вектор E. coli - P. ohmeri, состоящий из бактериальной (E. coli) точки начала репликации и гена устойчивости к ампициллину, автономной последовательности репликации дрожжей (P. Ohmeri), а также гена poURA3 (P. ohmeri) для селекции у дрожжей.
Более того, она содержит взаимозаменяемый промоторный элемент рибулозоредуктазы P. ohmeri (посредством рестрикции SpeI и AscI ) и терминаторный элемент (посредством SphI и SacII), фланкирующие открытую рамку считывания тагатозо-3-эпимеразы Pseudomonas cichorii (взаимозаменяемая посредством рестрикции AscI и SphI).
Пример 7. Конструирование вектора P. ohmeri для экспрессии гетерологичного гена с применением маркера отбора poLEU2
Для конструирования второго вектора экспрессии P. ohmeri, кассету экспрессии pEVE2523 (фигура 7), описанную ранее в примере 6 клонировали в вектор, содержащий маркер отбора P. ohmeri poLEU2 (фигура 6).
Фрагмент «с затупленными концами» длиной 1,7 т.п.о. вектора pEVE2523 (фигура 7), вырезанный с помощью SpeI и SacII (New England Biolabs, Ипсуич, Массачусетс), использовали в качестве вставки. «Затупление концов» выполняли с помощью смеси ферментов для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре, с последующей термоинактивацией ферментов в течение 10 мин. при 70°C.
Остов вектора получали из вектора poARS (plig3 – FR 2772788), линеаризованного с помощью SalI (New England Biolabs, Ипсуич, Массачусетс), с «затуплением концов» и дефосфорилированием в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс). Вставку и вектор, очищенные от геля с помощью набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния), лигировали в течение 1 ч. при RT, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и использовали для дополнительного исследования посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Вновь клонированная плазмида для экспрессии pEVE2560 (фигура 8) представляет собой бифункциональный вектор E. coli - P. ohmeri, состоящий из бактериальной (E. coli) точки начала репликации и гена устойчивости к ампициллину, автономной последовательности репликации дрожжей (P. ohmeri), а также гена poLEU2 (P. ohmeri) для селекции у дрожжей.
Более того, открытая рамка считывания тагатозо-3-эпимеразы Pseudomonas cichorii, фланкированная при помощи промотора и терминатора рибулозоредуктазы P. ohmeri, является взаимозаменяемой посредством рестрикции AscI и SphI.
Пример 8. Конструирование вектора P. ohmeri для сверхэкспрессии NADPH-специфической ксилитдегидрогеназы Gluconobacter oxydans
Конструировали вектор P. ohmeri для сверхэкспрессии NADPH-специфической ксилитдегидрогеназы Gluconobacter oxydans.
Для клонирования в вектор экспрессии фрагмент ДНК, кодирующий NADPH-специфическую ксилитдегидрогеназу Gluconobacter oxydans, выделяли из вектора 13AAYSYP (фигура 3) посредством вырезания при помощи рестрикционных ферментов AscI и SphI (New England Biolabs, Ипсуич, Массачусетс).
Фрагмент длиной 803 п.о. очищали с помощью набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2523 длиной 9,8 т.п.о., расщепленным посредством AscI/SphI и очищенным от геля (фигура 7), с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 9).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE3284 (фигура 10) содержит кодон-оптимизированную NADPH-специфическую ксилитдегидрогеназу Gluconobacter oxydans, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri, и маркер отбора poURA3.
Пример 9. Конструирование вектора P. ohmeri для сверхэкспрессии NADPH-специфической ксилитдегидрогеназы Pichia stipitis
Для субклонирования в вектор экспрессии фрагмент ДНК, кодирующий NADPH-специфическую ксилитдегидрогеназу из Pichia stipitis, должен быть фланкированным рестрикционными сайтами AscI и SphI.
Для этой цели использовали:
- праймер EV3101
AAGGCGCGCCAAA ATGACTGCTAACCCTTCC (SEQ ID No 18), содержащий сайт AscI (подчеркнут), и
- праймер EV3102
GAGCATGCTTACTCAGGGCCGTCAATG (SEQ ID No 19), содержащий SphI (подчеркнут),
в реакции ПЦР с 30 нг вектора 12AALQTP (фигура 2b) в качестве матрицы.
Матрицу амплифицировали в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 25 циклами в течение 10 сек при 98°C/20 сек при 55°C/30 сек при 72°C и конечной стадией удлинения в течение 10 минут при 72°C.
Продукт ПЦР длиной 1,1 т.п.о. отделяли на 1% агарозном геле, экстрагировали, очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и расщепляли посредством рестрикции с помощью AscI и SphI (New England Biolabs, Ипсуич, Массачусетс). После очистки в колонке при помощи набора для очистки и концентрирования ДНК (DNA Clean & ConcentratorTM-5 Kit) (Zymo Research Corporation, Ирвайн, Калифорния), его лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2523 (фигура 7) длиной 10,6 т.п.о., расщепленным посредством AscI/SphI и очищенным от геля, и с остовом вектора pEVE2560 (фигура 8) длиной 11,8 т.п.о., расщепленным посредством AscI/SphI и очищенным от геля, соответственно, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 11).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученные в результате плазмиды pEVE2562 и pEVE2564 (фигура 12) содержат кодон-оптимизированную NADPH-специфическую ксилитдегидрогеназу Pichia stipitis, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri, и маркеры отбора либо poURA3, либо poLEU2 соответственно.
Пример 10. Конструирование вектора P. ohmeri для сверхэкспрессии NADH-специфической ксилитдегидрогеназы Pichia stipitis
Для субклонирования в вектор экспрессии фрагмент ДНК, кодирующий NADH-специфическую ксилитдегидрогеназу из Pichia stipitis, должен быть фланкированным рестрикционными сайтами AscI и SphI.
Для этой цели использовали:
- EV3101 (AAGGCGCGCCAAA ATGACTGCTAACCCTTCC) (SEQ ID No 18), содержащий сайт AscI (подчеркнут), и
- EV3102 (GAGCATGCTTACTCAGGGCCGTCAATG) (SEQ ID No 19), содержащий сайт SphI (подчеркнут)
в реакции ПЦР с 30 нг вектора lig7.78 (фигура 2a) в качестве матрицы.
Матрицу амплифицировали в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 25 циклами в течение 10 сек при 98°C/20 сек при 55°C/30 сек при 72°C и конечной стадией удлинения в течение 10 минут при 72°C.
Продукт ПЦР длиной 1,1 т.п.о. отделяли на 1% агарозном геле, экстрагировали, очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и расщепляли посредством рестрикции с помощью AscI и SphI (New England Biolabs, Ипсуич, Массачусетс).
После очистки в колонке при помощи набора для очистки и концентрирования ДНК (DNA Clean & ConcentratorTM-5 Kit) (Zymo Research Corporation, Ирвайн, Калифорния), его лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2560 (фигура 8) длиной 10,5 т.п.о., расщепленным посредством AscI/SphI и очищенным от геля, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 13).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2563 (фигура 14) содержит кодон-оптимизированную NADH-специфическую ксилитдегидрогеназу Pichia stipitis, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri, и маркер отбора poLEU2.
Пример 11. Конструирование векторов P. ohmeri для сверхэкспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli
Конструировали вектор P. ohmeri для сверхэкспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli.
Для клонирования в вектор экспрессии фрагмент ДНК, кодирующий кодон-оптимизированную NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, выделяли из вектора 12ABYWMP (фигура 1) посредством вырезания при помощи рестрикционных ферментов AscI и SphI (New England Biolabs, Ипсуич, Массачусетс).
Фрагмент длиной 1,4 т.п.о. очищали с помощью набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2523 длиной 9,8 т.п.о., расщепленным посредством AscI/SphI и очищенным от геля (фигура 7), с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 15).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2839 (фигура 16) содержит кодон-оптимизированную NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri, и маркер отбора poURA3.
Помимо промотора рибулозоредуктазы P. ohmeri, NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli также клонировали под контролем промотора фосфоглицераткиназы P. ohmeri (poPGK1) и терминатора транскетолазы (poTKL).
Клонирование осуществляли в ходе двух последовательных стадий, сначала заменив промотор рибулозоредуктазы на промотор poPGK1, с последующей заменой терминатора рибулозоредуктазы на терминатор poTKL.
Фрагмент промотора poPGK1 P. ohmeri длиной 611 п.о. амплифицировали из геномной ДНК P. ohmeri с использованием:
- праймера EV3177 (GAAGACTAGTTCACGTGATCTC) (SEQ ID No 20), содержащего сайт SpeI (подчеркнут), и
- праймера EV3178 (CACTGGCGCGCCTTTTGTGTGGTGGTGTCC) (SEQ ID No 21), содержащего сайт AscI (подчеркнут).
Матрицу геномной ДНК получали при помощи ресуспендирования свежеснятой способом штрихования колонии P. ohmeri в 30 мкл 0,2% SDS и нагревания в течение 4 мин. при 95°C. После центрифугирования с максимальным числом оборотов, 0,5 мкл надосадочной жидкости применяли для ПЦР.
Амплификацию осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР выполняли с начальной стадией денатурации в течение 2 мин. при 96°C, с последующими 25 циклами в течение 10 сек при 96°C/10 сек при 58°C/30 сек при 72°C и конечной стадией удлинения в течение 2 минут при 72°C.
Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Амплифицированный фрагмент промотора poPGK1 длиной 610 п.о. расщепляли посредством рестрикции с помощью SpeI и AscI (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2839 (фигура 16) длиной 11,5 т.п.о., расщепленным посредством SpeI/AscI и очищенным от геля, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 17).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE3102 (фигура 18) содержит кодон-оптимизированную NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором фосфоглицераткиназы (poPGK1) и терминатором рибулозоредуктазы P. ohmeri, и маркер отбора poURA3.
На следующей стадии терминатор рибулозоредуктазы pEVE3102 замещали на терминатор транскетолазы (poTKL) P. ohmeri.
Фрагмент терминатора poTKL P. ohmeri длиной 213 п.о. амплифицировали из геномной ДНК P. ohmeri с использованием:
- праймера EV3817 (TAGCAGCATGCATAGGTTAGTGAATGAGGTATG) (SEQ ID No 22), содержащего сайт SphI (подчеркнут), и
- праймера EV3818 (TAGGTCCGCGGGAGCTTCGTTAAAGGGC) (SEQ ID No 23), содержащего сайт SacII (подчеркнут).
Матрицу геномной ДНК получали как описано выше.
Амплификацию осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X.
ПЦР выполняли с начальной стадией денатурации в течение 2 мин. при 96°C, с последующими 25 циклами в течение 10 сек при 96°C/10 сек при 57°C/30 сек при 72°C и конечной стадией удлинения в течение 2 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Амплифицированный фрагмент терминатора poTKL длиной 213 п.о. расщепляли посредством рестрикции с помощью SphI и SacII (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE3102 (фигура 18) длиной 11,5 т.п.о., расщепленным посредством SphI/SacII и очищенным от геля, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 17).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE3123 (фигура 19) содержит кодон-оптимизированную NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором фосфоглицераткиназы (poPGK1) и терминатором транскетолазы (poTKL)P. ohmeri, и маркер отбора poURA3.
Для того, чтобы сделать возможной экспрессию NAD+-специфической D-арабит-4-оксидоредуктазы E. coli из плазмиды с использованием другого отбора, маркер poURA3 pEVE3123 заменяли на маркер poLEU2.
Для этой цели маркер poURA3 выделяли из вектора pEVE3123 (фигура 19) посредством рестрикционного расщепления при помощи PsiI и AfeI (New England Biolabs, Ипсуич, Массачусетс).
Остов вектора длиной 9,1 т.п.о. очищали от геля с помощью набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния), «затупляли концы» с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре, с последующей термоинактивацией ферментов в течение 10 мин. при 70°C и дефосфорилировали в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс).
В качестве вставки применяли очищенный от геля фрагмент маркера poLEU2 с «затупленными концами», длиной 3 т.п.о., выделенный из вектора pEVE2560 (фигура 8) посредством рестрикционного расщепления при помощи AseI и AfeI. Лигирование фрагментов осуществляли в течение 2 ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 20).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE3157 (фигура 21) содержит кодон-оптимизированную NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, фланкированную промотором фосфоглицераткиназы (poPGK1) и терминатором транскетолазы (poTKL)P. ohmeri, и маркер отбора poLEU2.
Пример 12. Экспрессия плазмидного гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и плазмидного гена NADPH-специфической ксилитдегидрогеназы Pichia stipitis у штамма ATCC 20209 Pichia ohmeri
Для биосинтетической конверсии арабита в ксилит необходима одновременная экспрессия NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и NADP-специфической ксилитдегидрогеназы P. stipitis.
Первый фермент приводит к образованию ксилулозы, а второй фермент преобразует ксилулозу в ксилит.
Штамм SRLU P. ohmeri (MATh- leu2 ura3), полученный из ATCC 20209 и ауксотрофный в отношении лейцина и урацила (Piredda и Gaillardin, 1994, ранее), использовали в качестве хозяина для конструирования штаммов дрожжей, секретирующих ксилит, посредством трансформации с помощью плазмид:
- pEVE2839 (NAD+-специфическая D-арабит-4-оксидоредуктаза E. coli) и
- pEVE2564 (NADPH-специфическая ксилитдегидрогеназа P. stipitis),
с получением штамма EYS2755.
Кроме того, в качестве контроля (следуя идее WO 94/10325) штамм, экспрессирующий NADH-специфическую ксилитдегидрогеназу дикого типа P. stipitis, конструировали посредством трансформации при помощи плазмид:
- pEVE2839 (NAD+-специфическая D-арабит-4-оксидоредуктаза E. coli) и
- pEVE2563 (NADH-специфическая ксилитдегидрогеназа P. stipitis)
в хозяин SRLU, с получением штамма EYS2962.
В качестве контроля также получали штаммы, трансформированные одной из плазмид:
- pEVE2839 (NAD+-специфическая D-арабит-4-оксидоредуктаза E. coli),
- pEVE2563 (NADH-специфическая ксилитдегидрогеназа P. stipitis) и
- pEVE2564 (NADPH-специфическая ксилитдегидрогеназа P. stipitis),
с получением EYS2943, EYS2696 и EYS2697 соответственно.
Трансформацию дрожжей проводили в основном с помощью способа с образованием сферопластов по Green et al. (Green E.D., Hieter, P., and Spencer F. A., chapter 5 in Genome Analysis: A Laboratory Manual, Vol. 3, Cloning Systems, Birren et al. (eds.), Cold Spring Harbor Press, New York, 1999) со следующими модификациями: Для получения сферопластов вместо литиказы использовали зимолиазу 100T и инкубацию с ферментом осуществляли при 37°C до тех пор, пока OD клеточной суспензии не вырастала на 20-30%, по сравнению с первоначальной OD до обработки зимолиазой.
Вкратце, клетки P. ohmeri выращивали в течение ночи при 30°C в среде YPD (дрожжевой экстракт 1% (вес/объем), пептон 2% (вес/объем), декстроза 2% (вес/объем)) до конечной OD600 3-5.
200 OD600 единиц собирали при помощи центрифугирования, промывали единоразово водой и 1 M сорбитом и ресуспендировали в буфере SCE (1 M сорбитол, 100 мM двуводной натриевой соли лимонной кислоты, 10 мM EDTA) до конечной концентрации 70 OD/мл.
DTT и зимолиазу (LuBio Science, Люцерн, Швейцария) добавляли до конечной концентрации 10 мM и 0,5 ЕД/OD, соответственно и смесь инкубировали при 37°C с медленным встряхиванием.
Выполняли расщепление клеточной стенки с последующим измерением оптической плотности раствора, разведенного водой. Когда это значение достигало 80% от первоначального, расщепление заканчивали осторожным центрифугированием и промыванием 1 M сорбитом и буфером STC (0,98 M сорбит, 10 мM Tris pH 7,5, 10 мM CaCl2).
Сферопласты осторожно ресуспендировали в буфере STC, содержащем 50 мкг/мл ДНК из зобной железы телёнка (Calbiochem/VWR, Дитикон, Швейцария), до конечной концентрации 200 OD/мл. Аликвоты 100 мкл перемешивали с 100-200 нг плазмидной ДНК и инкубировали в течение 10 мин. при комнатной температуре.
1 мл раствора PEG (19,6% PEG 8000 вес/объем, 10 мM Tris pH 7,5, 10 мM CaCl2) добавляли к суспензии, инкубировали в течение 10 минут и гранулировали. Сферопласты регенерировали при 30°C в течение 1-2 часов в 1 мл 1 M раствора сорбита, содержащего 25% YPD и 7 мM CaCl2.
К регенерированным клеткам добавляли 7 мл 50°C теплого агара для верхнего слоя (0,67% основы азотного агара для дрожжей без аминокислот, 0,13% порошка для отсеивания без лейцина/урацила/гистидина/триптофана/метионина, 0,086‰ требуемой недостающей аминокислоты, 2% глюкозы, 1 M сорбитола, pH 5,8 и 2,5% нобль-агара) и смесь равномерно выливали на предварительно нагретые планшеты для отбора с сорбитом (0,67% основы азотного агара для дрожжей без аминокислот, 0,13% порошка для отсеивания без лейцина/урацила/гистидина/триптофана/метионина, 0,086‰ требуемой недостающей аминокислоты, 2% глюкозы, 1 M сорбита, pH 5,8).
Планшеты инкубировали в течение 3-5 дней при 30°C. Трансформанты повторно отбирали в соответствующие планшеты для отбора.
Каждый полученный штамм трижды исследовали в отношении продукции арабита, ксилита и рибита.
Для этой цели клоны сначала выращивали при 30°C в течение ночи на среде для посева (0,67% основы азотного агара для дрожжей; 0,13% порошка для отсеивания без лейцина/урацила/гистидина/триптофана/метионина; 0,086‰ требуемой недостающей аминокислоты; 5% глюкозы; pH 5,7).
С помощью данной ночной культуры инокулировали главную культуру в среде для продукции (0,67% основы азотного агара для дрожжей; 0,13% порошка для отсеивания без лейцина/урацила/гистидина/триптофана/метионина; 0,086‰ требуемой недостающей аминокислоты; 15% глюкозы; pH 5,7) при начальной OD600 0,2.
Данную культуру выращивали при 37°C в течение 48 часов и концентрации арабита, ксилита и рибита концентрации в надосадочной жидкости определяли при помощи HPLC/MS с использованием колонки Aminex® HPX-87 (Bio-Rad, Геркулес, Калифорния) и Waters® TQ-Detector (Acquity® UPLC связан с тройным квардрупольным прибором для обнаружения, Waters, Милфорд, Массачусетс) и изократичных условий с 100% воды в качестве мобильной фазы.
Титры полиолов всех исследуемых штаммов отображены в таблице 8.
Таблица 8. Продукция полиолов штаммами SRLU P. Ohmeri, трансформированными в отношении NADH- и NADPH-специфической ксилитдегидрогеназы P. stipitis и/или в отношении NAD+-специфической D-арабит-4-оксидоредуктазы E. coli (среднее из трех параллелей)
н.о.– не определено
Применение NADPH-специфической ксилитдегидрогеназы P. stipitis приводит к значительному повышению титров ксилита, по сравнению с таковым у NADH-специфического фермента дикого типа.
Пример 13. Экспрессия плазмидного гена NADPH-специфической ксилитдегидрогеназы Gluconobacter oxydans у Pichia ohmeri
Помимо штамма, продуцирующего ксилит при помощи NADP-специфической ксилитдегидрогеназы P. stipitis, конструировали второй штамм, экспрессирующий NADP-специфическую ксилитдегидрогеназу G. oxydans.
Штамм SRLU P. ohmeri (MATh- leu2 ura3), полученный из ATCC 20209 и ауксотрофный в отношении лейцина и урацила (Piredda и Gaillardin, 1994, ранее), использовали в качестве хозяина для конструирования штаммов дрожжей, секретирующих ксилит, посредством трансформации с помощью плазмид pEVE3157 (NAD+-специфическая D-араби-4-оксидоредуктаза E. coli) и pEVE3284 (NADPH-специфическая ксилитдегидрогеназа G. oxydans), с получением штамма EYS3324.
В качестве контроля также получали штаммы, трансформированные одной из плазмид:
- pEVE3157 (NAD+-специфическая D-арабит-4-оксидоредуктаза E. coli) и
- pEVE3284 (NADH-специфическая ксилитдегидрогеназа G. oxydans), получением EYS3067 и EYS3323 соответственно.
D-арабит-4-оксидоредуктаза E. сoli, используемая для конструирования выше указанных штаммов, подвергается регуляции со стороны промотора poPGK1, в отличие от промотора poRR, используемого у штаммов, экспрессирующих ксилитдегидрогеназу P. stipitis.
Вместе с тем, для того, чтобы исключить влияние промотора и, таким образом, иметь возможность сравнивать уровни полиола у штаммов, экспрессирующих ксилитдегидрогеназу из G. oxydans, с таковыми, экспрессирующими соответствующий фермент из P. stipitis, был получен дополнительный штамм.
Данный штамм EYS2963 получали посредством трансформации хозяина SRLU при помощи
- pEVE3123 (NAD+-специфическая D-арабит-4-оксидоредуктаза E. coli) и
- pEVE2564 (NADPH-специфическая ксилитдегидрогеназа P. stipitis).
Трансформацию дрожжей проводили как описано в примере 12. Каждый полученный штамм трижды исследовали в отношении продукции арабита, ксилита и рибита как описано в примере 12.
Титры полиолов всех исследуемых штаммов отображены в таблице 9.
Таблица 9. Продукция полиолов штаммами SRLU P. ohmeri, трансформированными в отношении NADH- и NADPH-специфической ксилитдегидрогеназы G. oxydans и/или в отношении NAD+-специфической D-арабит-4-оксидоредуктазы E. coli (среднее из трех параллелей)
н.о.– не определено
Титры ксилита у штаммов, экспрессирующих NADPH-специфическую ксилитдегидрогеназу G. oxydans (EYS3324), аналогичны таковым у штаммов, экспрессирующих соответствующий фермент P. stipitis (EYS2963). Однако, фермент G. oxydans дает гораздо более низкие титры рибита, проявляя, таким образом, большую субстратную специфичность к ксилулозе.
Пример 14. Получение мутантного штамма P. ohmeri с повышенной секрецией арабита
Мутант-продуцент повышенных уровней арабита выбирали из UV-облученной суспензии P. ohmeri ATCC 20209.
Систему UV-облучения (Vilber Lourmat, Франция) оснащали радиометром с микропроцессорным управлением RMX-3 W. P.ohmeri выращивали на агаре YPD (декстроза 20 г/л) при 37°C в течение ночи.
Суспензию готовили до достижения 106 кое/мл (OD620=0,4) и 5 мл помещали в стерильную чашку Петри. Суспензию облучали после снятия крышки с чашки. Длина волны UV-излучения составляла 254 нм и энергия облучения составляла 1,8 10-2 Дж/см2. У дрожжевых клеток наблюдали 90% смертность. После прекращения облучения и замены крышки на чашке, суспензию переносили в стерильную тубу, помещенную на ледяную ванну.
20 мл жидкой среды YPD инокулировали мутированной суспензией и инкубировали в течение 12 часов при 37°C, 250 об/мин.
После инкубации мутированную культуру разбавляли стерильным 40% глицерином (объем/объем). Аликвоты распределяли в пробирки объемом 5 мл и замораживали при -80°C.
Отбор проводили на основе осмофильного свойства Pichia ohmeri, которые способны расти при очень высоких концентрациях декстрозы (выше 600 г/л).
Целью авторов настоящего изобретения был отбор мутантов, способных расти быстрее, чем материнский штамм на агаре YPD, содержащем декстрозу 600 г/л или 700 г/л.
Размороженные аликвоты распределяли на YPD600 и YPD700 и первые появившиеся колонии отбирали и исследовали в отношении продукции арабита во встряхиваемых колбах.
Среды для субкультивирования и продуцирования готовили с содержанием глюкозы 50 г/л или 100 г/л соответственно, дрожжевого экстракта 3 г/л, MgSO4 1 г/л и KH2PO4 2 г/л, pH 5,7. Субкультуру (10 мл в 100 мл колбах) инкубировали в течение 24 часов при 37°C, 250 об/мин. Продукцию (40 мл в 500 мл колбах) инокулировали 5 мл субкультуры и инкубировали в течение 64 часов при 37°C, 250 об/мин.
Мутантный штамм P. ohmeri выбирали по быстрому потреблению глюкозы и высокой продуктивности арабита и депонировали во Франции 7 марта 2012 года в Collection Nationale de Cultures de Microorganismes [Национальной коллекции культур микроорганизмов] Института Пастера (CNCM), 25 rue du Docteur Roux, 75724 PARIS Cedex 15, под номером I-4605.
Пример 15. Конструирование плазмиды для делеции LEU2
Для возможности применения недавно полученного штамма CNCM I-4605 для отбора плазмиды и интеграции генов, конструировали плазмиду для делеции открытой рамки считывания LEU2.
На первой стадии общий вектор интеграции, который может быть использован у P. ohmeri, был взят из системы CRE/loxP S. cerevisiae. Остов вектора выделяли из pUG73 (Gueldener et al., 2002, Nucleic Acid Res, 30, e23) посредством рестрикционного вырезания при помощи ферментов PstI и EcoRV (New England Biolabs, Ипсуич, Массачусетс).
В качестве вставки служил фрагмент ПЦР, содержащий маркер отбора LEU2 P. Ohmeri, фланкированный сайтами loxP, полученный с помощью пары праймеров:
- EV3043 (CACTGGCGCGCCCACTGCATGCGTCGACAACCCTTAATATAACTTCGTATAATGTATGCTATACGAAGTTATTAGGTCTAGACACATCGTGGATCCAAGCTATCAACGAGAGAGTC) (SEQ ID No 24) и
- EV3044 (AGTGGCTAGCAGTGCCATGGCCTAATAACTTCGTATAGCATACATTATACGAAGTTATATTAAGGGTTCTCGAGACGCGTCATCTAGCATCTCATCTACCAACTC) (SEQ ID No 25) и
- poARS (plig3 - FR 2772788 - см. фигуру 6) в качестве матрицы.
Прямой праймер EV3043 содержит сайт AscI (подчеркнут), предшествующий сайту SphI (подчеркнут), за которым следует фрагмент loxP длиной 48 п.о. (выделен жирным шрифтом) и сайт DraIII (подчеркнут). 3’-конец EV3043 содержит дополнительный фрагмент длиной 25 п.о. для амплификации гена LEU2 P. ohmeri. Обратный праймер EV3044, с другой стороны, содержит сайт NheI (подчеркнут), предшествующий сайту NcoI (подчеркнут), за которым следует loxP фрагмент длиной 48 п.о. (выделен жирным шрифтом) и сайт MluI (подчеркнут). 3’-конец EV3044 содержит дополнительный фрагмент длиной 25 п.о. для амплификации гена LEU2 P. ohmeri. Матрицу амплифицировали в реакционной смеси, состоящей из 200мкM каждого dNTP и 0,5мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. ПЦР проводили с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/10 сек при 65°C/50 сек при 72°C и конечной стадией удлинения в течение 7 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Амплифицированный фрагмент фланкировали с помощью сайтов PstI и EcoRV во второй реакции ПЦР для дальнейшего субклонирования. Амплификацию выполняли при помощи:
- праймера EV3056 (CACTCTGCAGCACTGGCGCGCCCACTGCAT) (SEQ ID No 26), содержащего сайт PstI (подчеркнут), и
- праймера EV3057 (CACTGATATCAGTGGCTAGCAGTGCCATGG) (SEQ ID No 27), содержащего сайт EcoRV (подчеркнут),
в реакционной смеси, состоящей из 200мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. ПЦР выполняли с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/45 сек при 72°C и конечной стадией удлинения в течение 7 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Амплифицированный маркер LEU2 длиной 2,5 т.п.н расщепляли посредством рестрикции с помощью ферментов PstI и EcoRV (New England Biolabs, Ипсуич, Массачусетс), очищали от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и лигировали в течение 2 ч. при комнатной температуре с PstI/EcoRV длиной 2,4 т.п.о. (New England Biolabs, Ипсуич, Массачусетс), очищенным от геля (при помощи набора для извлечения ДНК из геля ZymocleanTM - Zymo Research Corporation, Ирвайн, Калифорния) остовом вектора pUG73 (Gueldener et al., 2002 Nucleic Acid Res, 30, e23) с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) – (фигура 22). После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2787 (фигура 23) содержит маркер отбора LEU2 P. ohmeri под контролем эндогенного промотора и терминатора, фланкированный двумя сайтами loxP. Кроме того, сайты AscI и SphI вводили выше первого сайта loxP и сайты NheI и NcoI вводили ниже второго сайта loxP, для того, чтобы облегчить клонирование областей, гомологичных сайтам интеграции в геноме.
Поскольку делеция открытой рамки считывания эндогенного LEU2 служила в качестве цели, на второй стадии клонирования маркер LEU2 вектора интеграции затем заменяли на ген устойчивости nat1 Streptomyces noursei.
Фрагмент ДНК, кодирующий ген nat1 Streptomyces noursei, синтезировали химическим путем посредством синтеза генов при помощи GeneArt® Gene Synthesis (Life Technologies, Регенсбург, Германия) в соответствии с предоставленной последовательностью SEQ ID NO 28.
Нуклеотиды 204-776 последовательности S60706.1 (полученной из http://www.ncbi.nlm.nih.gov/nuccore/S60706.1), кодирующие ген nat1, использовали в качестве матрицы и подвергали оптимизации кодонов для применения у P. ohmeri ATCC 20209 согласно таблице 7 (выше), при помощи программы Optimizer, полученной из http://genomes.urv.es/OPTIMIZER/.
Для облегчения последующего клонирования к 5’- и 3’-концам результирующей последовательности вручную в текстовом файле добавляли нуклеотиды, кодирующие сайты распознавания ретрикционными ферментами AscI (GGCGCGCC) и SphI (GCATGC) соответственно. Дополнительно, включали аденозиновый триплет спереди от стартового ATG для обеспечения аденозина в положении -3 в Kozak-подобной последовательности у дрожжей.
Конечную последовательность (SEQ ID NO 28) затем предоставляли для синтеза с помощью GeneArt (Регенсбург, Германия). Синтезированный фрагмент ДНК, кодирующий ген nat1 доставляли в виде 5 мкг лиофилизированной плазмидной ДНК в векторе на основе pMA-T (12ABTV4P, фигура 24).
Для клонирования гена nat1 использовали вектор, содержащий промотор и терминатор рибулозоредуктазы (poRR). Терминатор заменяли на терминатор оротидин-5'-фосфатдекарбоксилазы (poURA3) и ген nat1 вводили между промоторной и терминаторной последовательностями.
Для этой цели терминатор оротидин-5'-фосфатдекарбоксилазы (poURA3) получали посредством ПЦР с помощью:
- праймера EV3393 (CAAGCATGCGGGAATGATAAGAGACTTTG) (SEQ ID No 29), содержащего сайт SphI (подчеркнут), и
- праймера EV3394 (GGACCGCGGAAAGGTGAGGAAGTATATGAAC) (SEQ ID No 30), содержащего сайт SacII (подчеркнут), и
- pEVE2523 (фигура 7) в качестве матрицы.
Амплификацию осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. ПЦР выполняли с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/10 сек при 59°C/10 сек при 72°C и конечной стадией удлинения в течение 5 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Терминатор poURA3 длиной 239 п.о. расщепляли посредством рестрикции с помощью ферментов SphI и SacII (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2681 длиной 11 т.п.о., линеаризованным при помощи рестрикционных ферментов SphI и SacII (New England Biolabs, Ипсуич, Массачусетс) и очищенным от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс). После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
На второй стадии клонирования ген nat1 выделяли из 12ABTV4P (фигура 24) посредством рестрикционного вырезания при помощи ферментов SphI и AscI (New England Biolabs, Ипсуич, Массачусетс). Кроме того, «затупление концов» сайта SphI с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) в течение15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10 мин. при 70°C проводили в промежутке между расщеплением SphI и AscI. Очищенный от геля фрагмент длиной 587 п.о. (при помощи набора для извлечения ДНК из геля ZymocleanTM - Zymo Research Corporation, Ирвайн, Калифорния) затем лигировали с очищенным от геля остовом вектора длиной 10,5 т.п.о. вектора, описанного выше, вырезанным при помощи рестрикционных ферментов SphI и AscI (New England Biolabs, Ипсуич, Массачусетс).
Также проводили «затупление концов» сайта SphI вектора в течение 15 мин. при комнатной температуре с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) с последующей стадией термоинактивации в течение 10 мин. при 70°C до расщепления с помощью AscI. Кроме того, вектор дефосфорилировали в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс). Лигирование осуществляли в течение 2 ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2798 (фигура 25) содержит маркер устойчивости к лекарственному средству nat1, фланкированный промотором рибулозоредуктазы P. ohmeri (poRR) и терминатором оротидин-5'-фосфатдекарбоксилазы (poURA3).
Кассету экспрессии nat1 использовали для замены маркера отбора LEU2 P. ohmeri в интеграционном векторе. Для того, чтобы облегчить последующее клонирование кассета nat1 должна быть фланкирована с помощью сайтов XbaI (подчеркнут в праймере EV3643) и MluI (подчеркнут в праймер EV3644) посредством ПЦР с помощью:
- праймера EV3643
 и
и
- праймера EV3644 (CACTACGCGTAAAGGTGAGGAAGTATATG) (SEQ ID No 32).
Праймер EV3643 содержит дополнительный сайт ClaI (подчеркнут пунктирной линией) с последующим сайтом XbaI. pEVE2798 служил в качестве матрицы (фигура 25).
Амплификацию осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. ПЦР выполняли с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/10 сек при 54°C/25 сек при 72°C и конечной стадией удлинения в течение 5 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Кассету экспрессии nat1 длиной 1,3 т.п.о. расщепляли посредством рестрикции с помощью ферментов MluI и XbaI (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2787 длиной 2,6 т.п.о. (фигура 23), линеаризованным при помощи ферментов MluI и XbaI (New England Biolabs, Ипсуич, Массачусетс) и очищенным от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 26).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2852 (фигура 27) содержит маркер отбора nat1 под контролем промотора рибулозоредуктазы (poRR) и терминатора оротидин-5'-фосфатдекарбоксилазы (poURA3) и фланкированный двумя сайтами loxP.
Плазмида интеграции на данном этапе не содержит ни одного из гомологичных фрагментов P. ohmeri, необходимых для сайт-специфической интеграции в геном. Данные сайты присоединяли на следующих стадиях.
Гомологичную 5’-область, расположенную выше открытой рамки считывания LEU2, амплифицировали из 50 нг вектора poARS (фигура 6) с помощью:
- праймера EV3548 (CACTCTGCAGGATCCAAGCTATCAACGAGA) (SEQ ID No 33), содержащего сайт PstI (подчеркнут), и
- праймера EV3549 (CACTGCATGCGTTGCGGAAAAAACAGCC) (SEQ ID No 34), содержащего сайт SphI (подчеркнут).
ПЦР осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. Амплификацию выполняли с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/10 сек при 61°C/15 сек при 72°C и конечной стадией удлинения в течение 5 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Фрагмент длиной 567 п.о. расщепляли посредством рестрикции с помощью ферментов PstI и SphI (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2852 длиной 3,9 т.п.о. (фигура 27), линеаризованным при помощи рестрикционных ферментов PstI и SphI (New England Biolabs, Ипсуич, Массачусетс) и очищенным от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 29).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2855 (фигура 28) содержит фрагмент, гомологичный 5’-области, расположенной выше открытой рамки считывания LEU2, и маркер nat1, фланкированным двумя сайтами loxP.
Гомологичную 3’-область, расположенную ниже открытой рамки считывания LEU2, амплифицировали из 50 нг вектора poARS (фигура 6) с помощью:
- праймера EV3550 (CACT CCATGG AGTAGGTATATAAAAATATAAGAG) (SEQ ID No 35), содержащего сайт NcoI (подчеркнут), и
- праймера EV3551 (CACTGCTAGCGTCGACAACAGCAACTAG) (SEQ ID No 36), содержащего сайт NheI (подчеркнут).
ПЦР осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. Амплификацию выполняли с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/10 сек при 51°C/25 сек при 72°C и конечной стадией удлинения в течение 5 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Фрагмент длиной 1,3 п.о. расщепляли посредством рестрикции с помощью ферментов NcoI и NheI (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2855 длиной 4,4 т.п.о. (фигура 28), линеаризованным при помощи рестрикционных ферментов NcoI и NheI (New England Biolabs, Ипсуич, Массачусетс) и очищенным от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 29).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2864 для делеции LEU2 (фигура 30) содержит фрагмент, гомологичный 5’-области, расположенной выше и фрагмент, гомологичный 3’-области, расположенной ниже открытой рамки считывания LEU2, и маркер nat1, фланкированный двумя сайтами loxP.
Пример 16. Получение мутантного штамма P. ohmeri, ауксотрофного в отношении лейцина
Поскольку полученный штамм CNCM I-4605 P. ohmeri до этого не проявлял никакой ауксотрофности, выполняли делецию открытой рамки считывания LEU2, чтобы иметь возможность использовать маркер отбора LEU2 для генной интеграции.
Для этой цели плазмиду pEVE2864 (фигура 30) расщепляли посредством рестрикции с помощью ферментов EcoRV и PstI (New England Biolabs, Ипсуич, Массачусетс) в течение 2,5 ч при 37°C и смесь использовали для трансформации штамма Mut165 согласно процедуре, описанной в примере 12.
К регенерированным клеткам добавляли 7 мл 50°C теплого агара для верхнего слоя (1% дрожжевого экстракта, 2% пептона, 2% глюкозы, 1 M сорбита, pH 5,8 и 2,5% нобль-агара) с 25 мкг/мл натамицина и смесь равномерно выливали на предварительно нагретые планшеты для отбора с сорбитолом (1% дрожжевого экстракта, 2% пептона, 2% глюкозы, 1 M сорбита, pH 5,8 и 2% агара) с 25 мкг/мл натамицина. Планшеты инкубировали в течение 4 дней при 30°C. Делецию открытой рамки считывания LEU2 оценивали по отсутствию роста на планшетах для отбора без лейцина и подтверждали посредством метода молекулярных колоний ПЦР с помощью:
- праймера EV3393 (CAAGCATGCGGGAATGATAAGAGACTTTG) (SEQ ID No 29) и
- праймера EV3795 (CAAGTCGTGGAGATTCTGC) (SEQ ID No 37).
Фрагмент длиной 1,6 т.п.о. амплифицировали с начальной стадией денатурации в течение 30 сек при 98°C, с последующими 30 циклами в течение 10 сек при 98°C/10 сек при 51°C/25 сек при 72°C и конечной стадией удлинения в течение 5 минут при 72°C.
Полученный в результате штамм содержит делецию всей открытой рамки считывания гена LEU2 у CNCM I-4605, фоново, и был депонирован во Франции 5 февраля 2015 года в Collection Nationale de Cultures de Microorganismes [Национальной коллекции культур микроорганизмов] Института Пастера (CNCM), 25 rue du Docteur Roux, 75724 PARIS cedex 15, под номером I- 4955.
Пример 17. Конструирование двойных плазмид для экспрессии, содержащих NADPH-специфическую ксилитдегидрогеназу P. Stipitis и NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli
Для возможности экспрессии NADPH-специфической ксилитдегидрогеназы P. stipitis и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli в мутантном штамме P. ohmeri, ауксотрофном только в отношении лейцина, необходимо конструирование двойной плазмиды для экспрессии.
Кассету экспрессии, содержащую NADPH-специфическую ксилитдегидрогеназу P. stipitis, выделяли из pEVE2562 (фигура 12) посредством рестрикционного вырезания при помощи ферментов SpeI и SacII (New England Biolabs, Ипсуич, Массачусетс). Фрагмент длиной 1,9 т.п.о. очищали от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и у него «затупляли концы» с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10 мин. при 70°C. Вставку затем лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE3157 из 12,1 т.п.о., линеаризованным с помощью SpeI, «с затупленными концами», дефосфорилированным (1 ч. при 37°C с применением фосфатазы Antarctic - New England Biolabs, Ипсуич, Массачусетс) и очищенным от геля (фигура 21), который содержит NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 31).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью, плазмидную ДНК выделяли при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE3318 (фигура 32) содержит двойную конструкцию для экспрессии NADPH-специфической ксилитдегидрогеназы P. stipitis, фланкированной промотором и терминатором рибулозоредуктазы P. ohmeri (poRR) и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы P. ohmeri (poPGK1) и терминатора рибулозоредуктазы (poRR), и маркер отбора poLEU2.
Пример 18. Конструирование интеграционных векторов для экспрессии гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и гена NADPH-специфической ксилитдегидрогеназы P. stipitis в P. ohmeri
Ген NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и ген NADPH-специфической ксилит-дегидрогеназы P. stipitis в конечном счете должны стать неотъемлемой частью генома P. ohmeri. Следовательно, необходимо сконструировать вектор интеграции с маркером отбора LEU2 посредством замены маркера отбора nat1 pEVE2852 и введения двойную конструкцию для экспрессии арабит-оксидоредуктазы и ксилит-дегидрогеназы.
Для этой цели открытую рамку считывания LEU2 P. ohmeri, фланкированную сайтами AscI и SphI, получали посредством ПЦР с помощью:
- праймера EV3645 (CAAGGCGCGCCAAAATGTCTACCAAAACCATTAC) (SEQ ID No 38) и
- праймера EV3646 (GGAGCATGCCTACTTTCCCTCAGCCAAG) (SEQ ID No 39).
Амплификацию осуществляли с 50 нг матрицы poARS (фигура 6) в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. ПЦР выполняли с начальной стадией денатурации в течение 30 с при 98°C с последующими 30 циклами в течение 10 с при 98°C/10 с при 57°C/20 с при 72°C и конечной стадией удлинения в течение 5 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Амплифицированную открытую рамку считывания LEU2 впоследствии расщепляли посредством рестрикции с помощью ферментов AscI и SphI (New England Biolabs, Ипсуич, Массачусетс).
Кроме того, «затупление концов» сайта SphI с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) в течение15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10 мин. при 70°C проводили в промежутке между расщеплением SphI и AscI. Очищенный от геля фрагмент длиной 1,1 т.п.о. затем лигировали с очищенным от геля остовом вектора pEVE2811 длиной 11 т.п.о., вырезанным при помощи рестрикционных ферментов SphI и AscI (New England Biolabs, Ипсуич, Массачусетс). Также проводили «затупление концов» сайта SphI вектора в течение 15 мин. при комнатной температуре с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) с последующей стадией термоинактивации в течение 10 мин. при 70°C до расщепления с помощью AscI. Кроме того, вектор дефосфорилировали в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс). Лигирование открытой рамки считывания LEU2 и остова вектора осуществляли в течение 2 ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2862 (фигура 33) содержит маркер LEU2 P. ohmeri, фланкированный промотором рибулозоредуктазы P. ohmeri (poRR) и терминатором оротидин-5'-фосфатдекарбоксилазы (poURA3).
Впоследствии маркер LEU2 амплифицировали посредством ПЦР с помощью:
- праймера EV3643 (CACTATCGATGGATCCGTAGAAATCTTG) (SEQ ID No 31), содержащего сайт ClaI, и
- праймер EV3644 (CACTACGCGTAAAGGTGAGGAAGTATATG) (SEQ ID No 32), содержащий сайт MluI (подчеркнут), и pEVE2862 (фигура 33) в качестве матрицы.
Амплификацию осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. ПЦР выполняли с начальной стадией денатурации в течение 30 с при 98°C с последующими 30 циклами в течение 10 с при 98°C/10 с при 54°C/30 с при 72°C и конечной стадией удлинения в течение 5 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Амплифицированный фрагмент LEU2 длиной 1,8 т.п.о. расщепляли посредством рестрикции с помощью ферментов ClaI и MluI (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре с остовом вектора pEVE2852 (фигура 27) длиной 2,6 т.п.о., расщепленным посредством ClaI и MluI (New England Biolabs, Ипсуич, Массачусетс) и очищенным от геля, с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 34).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE2865 (фигура 35) содержит маркер LEU2 P. ohmeri, фланкированный двумя сайтами loxP.
Для клонирования вектора интеграции pEVE2865 расщепляли посредством рестрикции с помощью фермента SalI (New England Biolabs, Ипсуич, Массачусетс), «с затупленными концами» с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10 мин. при 70°C и дефосфорилированным в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс).
Очищенный от геля фрагмент остова вектора длиной 4,5 т.п.о. использовали для лигирования. В качестве вставки служила двойная конструкция для экспрессии генов NADPH-специфической ксилитдегидрогеназы P.stipitis и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli, выделенных из pEVE3318 (фигура 32) посредством рестрикционного вырезания при помощи ферментов NdeI и SacII (New England Biolabs, Ипсуич, Массачусетс).
Фрагмент длиной 4,4 т.п.о. очищали от геля с помощью набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и «затупляли концы» с помощью набора ферментативной смеси для «затупления концов» (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре, с последующей термоинактивацией ферментов в течение 10 мин. при 70°C, с последующей дополнительной очисткой от геля. Остов вектора pEVE2865 и вставку pEVE3318 лигировали в течение 2 ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 34).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE3387 (фигура 36) содержит двойную конструкцию для экспрессии генов NADPH-специфической ксилитдегидрогеназы P. stipitis, фланкированной промотором и терминатором рибулозоредуктазаы P. ohmeri (poRR), и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы (poPGK1) и терминатора транскетолазы P. ohmeri (poTKL). В качестве маркера отбора служил ген LEU2 P. ohmeri, фланкированный двумя сайтами loxP.
Пример 19. Конструирование штамма P. ohmeri первого поколения с интегрированием, секретирующего ксилит в среду
Ранее описанный вектор использовали для произвольной интеграции гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и гена NADPH-специфической ксилитдегидрогеназы P. stipitis в геном P. ohmeri.
Для этого штамм CNCM I-4955 (пример 16), ауксотрофный в отношении лейцина, трансформировали при помощи pEVE3387 (фигура 36), расщепленного посредством рестрикции с помощью NotI (New England Biolabs, Ипсуич, Массачусетс), в течение 3 ч. при 37°C согласно процедуре, описанной в примере 12. Трансформанты отбирали на планшетах с сорбитом без какого-либо присутствия лейцина.
Полученный в результате штамм содержит ген NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и ген NADPH-специфической ксилитдегидрогеназы P. stipitis, произвольно интегрированные в геном P. ohmeri, и его депонировали во Франции 20 мая 2015 года в Collection Nationale de Cultures de Microorganismes [Национальной коллекции культур микроорганизмов] Института Пастера (CNCM), 25 rue du Docteur Roux, 75724 Cedex 15, под номером I-4982.
Пример 20. Конструирование двойной/тройной плазмиды экспрессии, содержащей NADPH-специфическую ксилитдегидрогеназу G. oxydans и NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli
Для возможности экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli в мутантном штамме P. ohmeri, ауксотрофном только в отношении лейцина, необходимо конструирование двойной плазмиды для экспрессии.
Кассету экспрессии, содержащую NADPH-специфическую ксилитдегидрогеназу G. oxydans, выделяли из pEVE3284 (фигура 10) посредством рестрикционного вырезания при помощи ферментов SpeI и SacII (New England Biolabs, Ипсуич, Массачусетс). Фрагмент с длиной 1,6 т.п.о. очищали от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и у него "затупляли концы" с помощью набора ферментативной смеси для "затупления концов" (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10 мин. при 70°C. Используемый остов вектора, состоял из 12,1 т.п.о. линеаризованного с помощью SpeI (New England Biolabs, Ипсуич, Массачусетс) и очищенного от геля (набор для извлечения ДНК из геля ZymocleanTM - Zymo Research Corporation, Ирвайн, Калифорния) остова pEVE3157 (фигура 21), содержащего NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli.
Остов дополнительно подвергали "затуплению концов" в течение 15 мин. при комнатной температуре с помощью набора ферментативной смеси для "затупления концов" (New England Biolabs, Ипсуич, Массачусетс) с последующей термоинактивацией ферментов в течение 10 мин. при 70°C и дефосфорилировали в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс). Лигирование осуществляли в течение 2 ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 37).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученные в результате плазмиды pEVE3322 и pEVE3324 (фигура 38) содержат либо двойную конструкцию для экспрессии NADPH-специфической ксилитдегидрогеназы G. Oxydans,, фланкированной промотором и терминатором рибулозоредуктазы P. ohmeri (poRR), и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы (poPGK1) и терминатора транскетолазы (poTKL) P. ohmeri, либо тройную конструкцию для экспрессии двух генов NADPH-специфической ксилитдегидрогеназы G. oxydans, фланкированной промотором и терминатором рибулозоредуктазы P. ohmeri (poRR), и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы (poPGK1) и терминатора транскетолазы (poTKL) P. ohmeri и маркер отбора poLEU2.
Пример 21. Конструирование интеграционных векторов для экспрессии гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и гена NADPH-специфической ксилитдегидрогеназы G. oxydans в P. ohmeri
Помимо интеграционного вектора, содержащего NADPH-специфическую ксилитдегидрогеназу P. stipitis и ген NAD+-специфической D-арабит-4-оксидоредуктазы E. coli, также получали плазмиды, содержащие NADPH-специфическую ксилитдегидрогеназу G. oxydans.
Для этого двойные и тройные кассеты экспрессии, содержащие либо одну, либо две NADPH-специфические ксилитдегидрогеназы G. oxydans и NAD+-специфическую D-арабит-4-оксидоредуктазу E. coli, выделяли соответственно из pEVE3322 и pEVE3324 (фигура 38) посредством рестрикционного вырезания при помощи ферментов NdeI и SacII (New England Biolabs, Ипсуич, Массачусетс).
Фрагменты с длиной 4,1т.п.о. и 5,7т.п.о. очищали от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и у них "затупляли концы" с помощью набора ферментативной смеси для "затупления концов" (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10мин. при 70°C. В качестве вектора служил очищенный от геля (набор для извлечения ДНК из геля ZymocleanTM - Zymo Research Corporation, Ирвайн, Калифорния) линеаризованный с помощью SalI pEVE2865 с длиной 5,7т.п.о. (фигура 35).
Остов вектора дополнительно подвергали "затуплению концов" в течение 15мин. при комнатной температуре с помощью набора ферментативной смеси для "затупления концов" (New England Biolabs, Ипсуич, Массачусетс) с последующей термоинактивацией ферментов в течение 10мин. при 70°C и дефосфорилированием в течение 1ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс). Лигирование вектора и вставку осуществляли в течение 2ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 39).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученные в результате плазмиды pEVE3390 и pEVE3392 (фигура 40) содержат двойные или тройные конструкции для экспрессии либо одного, либо двух генов NADPH-специфической ксилитдегидрогеназы G. oxydans, фланкированных промотором и терминатором рибулозоредуктазаы P. ohmeri (poRR), и NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы (poPGK1) и терминатора транскетолазы P. ohmeri (poTKL). В качестве маркера отбора служил ген LEU2 P. ohmeri, фланкированный двумя сайтами loxP.
Пример 22. Конструирование штаммов второго поколения с интегрированием, способных секретировать более чем 100 г/л ксилита
Штамм CNCM I-4982 первого поколения, содержащий произвольно интегрированную копию гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и ген NADPH-специфической ксилитдегидрогеназы P. stipitis, использовали для дальнейшей интеграции дополнительных копий двух гетерологичных ферментов.
Однако, для возможности интегрирования указанных выше конструкций следовало удалить маркер отбора LEU2. Для этого штамм CNCM I-4982 первого поколения трансформировали вектором pEVE3163 согласно процедуре, описанной в примере 12. Вектор pEVE3163 содержит CRE-рекомбиназу бактериофага P1 (кодон-оптимизированную согласно таблице 7), фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri (poRR). Удаление маркера отбора LEU2 подтверждали отсутствием роста клонов на планшетах без лейцина.
Полученный в результате штамм EYS3842 трансформировали при помощи pEVE3390 или pEVE3392 (фигура 40), расщепленным посредством рестрикции с помощью NotI (New England Biolabs, Ипсуич, Массачусетс), в течение 3 ч. при 37°C согласно процедуре, описанной в примере 12. Трансформанты отбирали на планшетах с сорбитом без какого-либо присутствия лейцина.
Полученный в результате штамм EYS3929 второго поколения содержит два гена NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и два гена NADPH-специфической ксилитдегидрогеназы, один из G. oxydans и второй из P. stipitis, произвольно интегрированные в геном. Штамм EYS3930, напротив, содержит дополнительный ген NADPH-специфической ксилитдегидрогеназы G. oxydans.
Пример 23. Конструирование дополнительного вектора, используемого для интеграции дополнительных копий генов NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и NADPH-специфической ксилитдегидрогеназы G. oxydans
Для конструирования дополнительного вектора интеграции двойную кассету экспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli и NADPH-специфической ксилитдегидрогеназы G. oxydans амплифицировали с помощью ПЦР с использованием:
- праймера EV4904 (ATATCCCGGGCACCGTCATCACCGAAACGC) (SEQ ID NO 40), содержащего сайт SmaI, и
- праймера EV4905 (ATATCCCGGGCACGACCACGCTGATGAGC) (SEQ ID NO 41), содержащего сайт SmaI (подчеркнутый), и
pEVE3321 в качестве матрицы.
Амплификацию осуществляли в реакционной смеси, состоящей из 200 мкM каждого dNTP и 0,5 мкM каждого праймера с 0,02 ЕД/мкл полимеразы iProofTM (BIO-RAD, Геркулес, Калифорния), в подходящем буфере 1X. ПЦР выполняли с начальной стадией денатурации в течение 30 с при 98°C с последующими 30 циклами в течение 10 с при 98°C/10 с при 68°C/75 с при 72°C и конечной стадией удлинения в течение 5 минут при 72°C. Продукт ПЦР отделяли на 1% агарозном геле, экстрагировали и очищали при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния).
Амплифицированный фрагмент 3,9 т.п.о. в длину расщепляли посредством рестрикции с помощью SmaI (New England Biolabs, Ипсуич, Массачусетс) и лигировали в течение 2 ч. при комнатной температуре до линеаризованного с помощью PvuII (New England Biolabs, Ипсуич, Массачусетс), дефосфорилированного фосфатазой Antarctic (New England Biolabs, Ипсуич, Массачусетс) и очищенного от геля остова вектора pEVE2865 с длиной 4,4 т.п.о. (фигура 35) с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 41).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE4390 (фигура 42) содержит двойную конструкцию для экспрессии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli под контролем промотора фосфоглицераткиназы (poPGK1) и терминатора транскетолазы (poTKL) P. ohmeri и гена NADPH-специфической ксилитдегидрогеназы G. oxydans, фланкированного промотором и терминатором рибулозоредуктазы P. ohmeri (poRR). В качестве маркера отбора служил ген LEU2 P. ohmeri, фланкированный двумя сайтами loxP.
Пример 24. Конструирование вектора, используемого для интеграции NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum
Дополнительный интеграционный вектор для экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum конструировали описанным ниже образом. На первой стадии получали двойной вектор экспрессии, содержащий два указанных выше гена. Эту двойную кассету экспрессии клонировали в интеграционный вектор loxP.
Фрагмент ДНК, кодирующий ген NAD+-специфической D-арабит-4-оксидоредуктазы Ralstonia solanacearum, синтезировали химическим путем посредством синтеза генов при помощи GeneArt® (Life Technologies, Регенсбург, Германия) согласно предоставленной последовательности SEQ ID NO 42.
Нуклеотиды 2310548-2309151 последовательности AL646052.1 (полученной из http://www.ncbi.nlm.nih.gov/nuccore/AL646052), кодирующие ген dalD, использовали в качестве матрицы и подвергали оптимизации кодонов для использования в ATCC 20209 P. ohmeri согласно таблице 7 (изложенной выше) с использованием программы для оптимизатора, полученной с http://genomes.urv.es/OPTIMIZER/. На 5’- и 3’-концах полученной в результате последовательности нуклеотиды, кодирующие сайты распознавания рестрикционными ферментами AscI (GGCGCGCC) и SphI (GCATGC) соответственно, добавляли вручную в текстовом файле для облегчения дальнейшего клонирования. Дополнительно, включали аденозиновый триплет спереди от стартового ATG для обеспечения аденозина в положении -3 в Kozak-подобной последовательности у дрожжей.
Конечную последовательность (SEQ ID NO 42) затем предоставляли для синтеза с помощью GeneArt (Регенсбург, Германия). Синтезированный фрагмент ДНК, кодирующий ген dalD, доставляли как 5 мкг лиофилизированной плазмидной ДНК в полученный из pMA-RQ вектор (13AB2EGP, фигура 43).
Фрагмент с длиной 1,4 т.п.о. D-арабит-4-оксидоредуктазы из R. solanacearum выделяли из вектора 13AB2EGP (фигура 43) посредством рестрикционного расщепления с помощью AscI и SphI (New England Biolabs, Ипсуич, Массачусетс) и очищали от геля с помощью набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния). Вставку затем лигировали с остовом pEVE2560 с длиной 11,8 т.п.о. (фигура 8), линеаризованного с помощью AscI и SphI (New England Biolabs, Ипсуич, Массачусетс) и очищенного от геля с помощью ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 44).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE3898 (фигура 45) содержит кодон-оптимизированную NAD+-специфическую D-арабит-4-оксидоредуктазу R. solanacearum, фланкированную промотором и терминатором рибулозоредуктазы P. ohmeri, и маркер отбора poLEU2.
На следующей стадии кассету экспрессии, содержащую NADPH-специфическую ксилитдегидрогеназу G. oxydans, фланкированную промотором фосфоглицераткиназы (poPGK) и терминатором рибулозоредуктазы (poRR), выделяли из pEVE3960 при помощи расщепления посредством рестрикции с помощью SpeI и SacII (New England Biolabs, Ипсуич, Массачусетс). Фрагмент с длиной 1,8 т.п.о. очищали от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и у него "затупляли концы" с помощью набора ферментативной смеси для "затупления концов" (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10 мин. при 70°C. В качестве вектора служил очищенный от геля (набор для извлечения ДНК из геля ZymocleanTM - Zymo Research Corporation, Ирвайн, Калифорния) линеаризованный с помощью SalI pEVE3898 с длиной 13,2 т.п.о. Остов вектора дополнительно подвергали "затуплению концов" в течение 15 мин. при комнатной температуре с помощью набора ферментативной смеси для "затупления концов" (New England Biolabs, Ипсуич, Массачусетс) с последующей термоинактивацией ферментов в течение 10 мин. при 70°C и дефосфорилированием в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс). Лигирование вектора и вставку осуществляли в течение 2 ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 44).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE4077 (фигура 46) содержит двойную конструкцию для экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans, фланкированной промотором фосфоглицераткиназы (poPGK) и терминатором рибулозоредуктазы (poRR) P. ohmeri, и NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum под контролем промотора и терминатора рибулозоредуктазы P. ohmeri (poRR) и маркер отбора poLEU2.
В конечном счете, двойную кассету экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum выделяли из pEVE4077 (фигура 46) посредством рестрикционного вырезания при помощи SapI (New England Biolabs, Ипсуич, Массачусетс). Фрагмент с длиной 5,9 т.п.о. очищали от геля при помощи набора для извлечения ДНК из геля ZymocleanTM (Zymo Research Corporation, Ирвайн, Калифорния) и у него "затупляли концы" с помощью набора ферментативной смеси для "затупления концов" (New England Biolabs, Ипсуич, Массачусетс) в течение 15 мин. при комнатной температуре с последующей термоинактивацией ферментов в течение 10 мин. при 70°C. В качестве вектора служил очищенный от геля (набор для извлечения ДНК из геля ZymocleanTM - Zymo Research Corporation, Ирвайн, Калифорния) линеаризованный с помощью EcoRV pEVE2865 с длиной 4,4 т.п.о. (фигура 35), дефосфорилированный в течение 1 ч. при 37°C с применением фосфатазы Antarctic (New England Biolabs, Ипсуич, Массачусетс). Лигирование вектора и вставку осуществляли в течение 2 ч. при комнатной температуре с применением ДНК-лигазы T4 (New England Biolabs, Ипсуич, Массачусетс) (фигура 44).
После трансформации ультракомпетентных клеток XL10 Gold (Agilent Technologies, Санта-Клара, Калифорния) лигирующей смесью выделяли плазмидную ДНК при помощи набора для выделения плазмидной ДНК ZyppyTM (Zymo Research Corporation, Ирвайн, Калифорния) и дополнительно исследовали посредством рестрикционного расщепления и секвенирования (Microsynth, Бальгах, Швейцария).
Полученная в результате плазмида pEVE4377 (фигура 47) содержит двойную конструкцию для экспрессии NADPH-специфической ксилитдегидрогеназы G. oxydans и NAD+-специфической D-арабит-4-оксидоредуктазы R. solanacearum и маркер отбора poLEU2, фланкированный двумя сайтами loxP.
Пример 25. Конструирование штаммов третьего поколения с интегрированием с повышенной производительностью ксилита
У маркера LEU2 штаммов EYS3929 и EYS3930 второго поколения (пример 22) удаляли сайты lox как описано в примере 18 с использованием вектора pEVE3163. Полученные в результате штаммы EYS4118 и EYS4119 трансформировали при помощи pEVE4377 (фигура 47) и pEVE4390 (фигура 42) соответственно. Векторы расщепляли посредством рестрикции с помощью NotI (New England Biolabs, Ипсуич, Массачусетс) в течение 3 ч. при 37°C согласно процедуре, описанной в примере 12. Трансформанты отбирали на планшетах с сорбитом без какого-либо присутствия лейцина.
Полученный в результате штамм EYS4353 третьего поколения содержит три гена NAD+-специфической D-арабит-4-оксидоредуктазы, два из E. coli и один из R. solanacearum, и три гена NADPH-специфической ксилитдегидрогеназы, два из G. oxydans и один из P. stipitis, произвольно интегрированные в геном.
Второй штамм третьего поколения, напротив, содержит три копии NAD+-специфической D-арабит-4-оксидоредуктазы E. coli, три копии NADPH-специфической ксилитдегидрогеназы G. oxydans и одну копию из P. stipitis, фоново, и был депонирован во Франции 5 марта 2015 года в Collection Nationale de Cultures de Microorganismes [Национальной коллекции культур микроорганизмов] Института Пастера (CNCM), 25 rue du Docteur Roux, 75724 PARIS Cedex 15, под номером I-4960.
Пример 26. Конструирование штаммов с интегрированием четвертого поколения
У маркера LEU2 штаммов CNCM I-4960 третьего поколения (пример 25) удаляли сайты lox как описано в примере 18 с использованием вектора pEVE3163. Полученный в результате штамм EYS4955 трансформировали при помощи pEVE4377 (фигура 47), расщепленным посредством рестрикции с помощью NotI (New England Biolabs, Ипсуич, Массачусетс), в течение 3 ч. при 37°C согласно процедуре, описанной в примере 12. Трансформанты отбирали на планшетах с сорбитом без какого-либо присутствия лейцина.
Полученный в результате штамм четвертого поколения содержит четыре гена NAD+-специфической D-арабит-4-оксидоредуктазы, три из E. coli и один из R. solanacearum, и четыре гена NADPH-специфической ксилитдегидрогеназы, три из G. oxydans и один из P. stipitis, произвольно интегрированные в геном, и был депонирован во Франции 20 мая 2015 года в Collection Nationale de Cultures de Microorganismes [Национальной коллекции культур микроорганизмов] Института Пастера (CNCM), 25 rue du Docteur Roux, 75724 PARIS Cedex 15, под номером I-4981.
Пример 27. Получение полиола при помощи штаммов Pichia ohmeri (синтетическая среда)
Штаммы CNCM I-4605, CNCM I-4982, CNCM I-4960 и CNCM I-4981 дрожжей, сконструированные как описано выше, подвергали ферментации согласно изложенному ниже протоколу.
Процесс ферментации проходит в условиях с ограничениями азота и может быть разделен на фазу роста и фазу продуцирования. В ходе фазы роста аммиак в среде полностью используется при увеличении биомассы, как только образование биомассы останавливается, начинается фаза продуцирования и уровни полиола возрастают. Платформой, используемой для описанного процесса ферментации, была Multifors 2 от INFORS HT с использованием сосудов с рабочим объемом, составляющим 1 л. Ферментеры были оснащены двумя дисковыми турбинами Раштона с шестью лопастями. Воздух использовали для продувания ферментеров.
Температура, pH, перемешивание и скорость аэрации контролировали на всем протяжении культивирования. Температуру поддерживали при 36°C. pH удерживали на уровне 3 путем автоматического добавления 5 M KOH.
Скорость аэрации удерживали на уровне 1,0 vvm и первоначальная скорость смесителя составляла установленную на уровне 300 об./мин. Для предупреждения падения растворенного кислорода (DO) ниже 20% применяли каскад автоматического перемешивания. Рабочие условия, используемые в процессе ферментации, подытожены в таблице 10.
Таблица 10. Рабочие условия для ферментеров для получения полиола
контролировала каскад в смесителе
Для внесения в ферментеры использовали культуру 1 стадии для размножения. Композиция использованной для культуры для размножения среды описана в таблице 11. Культуры для размножения получали путем внесения 100 мл среды в 500-мл встряхиваемые колбы с 4 перегородками (с зазубринами). Встряхиваемые колбы инкубировали на вибрационном столе при 30°C и 150 об./мин. Клетки росли в течение ~24 ч. в средне-экспоненциальной фазе.
Таблица 11. Композиция среды для культуры для размножения.
Перед внесением удаляли количество среды в ферментере, эквивалентное количеству инокулята, и аликвоту культуры для размножения использовали для внесения в ферментер до конечного объема, составляющего 1л, и OD600 при запуске составляла приблизительно 0,2 (CDW приблизительно 0,03 г/л). Композиция среды, используемой в ферментере, описана в таблице 12.
Таблица 12. Композиция среды для ферментирования.
Образцы отбирали через равные промежутки времени и весь ферментативный бульон анализировали в отношении потребления глюкозы и образования внеклеточного полиола (ксилита, арабита и рибита). Кроме того, определяли общие метаболиты ферментации (глицерин, ацетат, этанол, пируват, малат, фумарат и сукцинат). После возрастания биомассы, с одной стороны, следовало определение OD600 и, с другой стороны, сухого веса клеток (CDW). Упомянутые выше измерения использовали для определения продуцирования полиола, выхода арабита или ксилита и производительности; результаты показаны в таблице 13.
Таблица 13. Получение полиола с помощью штаммов Pichia ohmeri (синтетическая среда).
I-4982
I-4981
CNCM I-4605 Pichia ohmeri продуцировал только арабит.
CNCM I-4982 Pichia ohmeri продуцировал арабит, ксилит и рибит. В этот штамм интегрировали одну копию гена NAD+-специфической D-арабит-4-оксидоредуктазы и одну копию гена NADPH-специфической ксилитдегидрогеназы. Модифицированный штамм в настоящее время способен потреблять арабит. Следовательно, после всего потребления глюкозы, арабит и рибит повторно потребляются CNCM I-4982 для продуцирования большего количества ксилита.
CNCM I-4960 (третье поколение) и CNCM I-4981 (четвертое поколение) Pichia ohmeri продуцируют ксилит и рибит, но не большее количество арабита. Внутриклеточное превращение арабита в ксилулозу и ксилит является достаточно эффективным для избегания выделения арабита в бульон.
Чем больше копий генов, кодирующих NAD+-специфическую D-арабитоксидоредуктазу и NADPH-специфическую ксилитдегидрогеназу, вводили в P. ohmeri, тем выше были титр, выход ксилита и способность продуцировать его.
| название | год | авторы | номер документа |
|---|---|---|---|
| МИКРООРГАНИЗМ, ЭКСПРЕССИРУЮЩИЙ КСИЛОЗОИЗОМЕРАЗУ | 2009 |
|
RU2553537C2 |
| СПОСОБ ПОЛУЧЕНИЯ КСИЛИТА | 1993 |
|
RU2142999C1 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕНЕТИЧЕСКИ МОДИФИЦИРОВАННЫХ БАКТЕРИЙ SORANGIUM / POLYANGIUM И РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ МОЛЕКУЛА ДНК | 1992 |
|
RU2124053C1 |
| ПОЛИПЕПТИДЫ ДЛЯ ЭНАНТИОСЕЛЕКТИВНОГО ФЕРМЕНТАТИВНОГО ВОССТАНОВЛЕНИЯ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2008 |
|
RU2486239C2 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ 1-БУТАНОЛА | 2006 |
|
RU2429295C2 |
| ПРОДУКТ И СПОСОБ | 2006 |
|
RU2385931C1 |
| СПОСОБ СОЗДАНИЯ РЕКОМБИНАНТНОГО АДЕНОВИРУСА ПТИЦ ДЛЯ ВАКЦИНАЦИИ И ГЕННОЙ ТЕРАПИИ | 2007 |
|
RU2326942C1 |
| РЕКОМБИНАНТНАЯ ПЛАЗМИДНАЯ ДНК pMALTEV-legumain, КОДИРУЮЩАЯ ПОЛИПЕПТИД, ОБЛАДАЮЩИЙ АНТИГЕННЫМИ СВОЙСТВАМИ БЕЛКА ЛЕГУМАИН Opisthorchis felineus, И ШТАММ E.coli BL 21(DE3)pLysS-pMALTEV-legumain - ПРОДУЦЕНТ РЕКОМБИНАНТНОГО ПОЛИПЕПТИДА, ОБЛАДАЮЩЕГО АНТИГЕННЫМИ СВОЙСТВАМИ БЕЛКА ЛЕГУМАИН Opisthorchis felineus | 2012 |
|
RU2496876C1 |
| ГЕНЫ РЕЦЕПТОРА ТРАНСФЕРРИНА MORAXELA | 1997 |
|
RU2235128C2 |
| ПРОДУКЦИЯ ГЛИКОПРОТЕИНОВ С МОДИФИЦИРОВАННЫМ ФУКОЗИЛИРОВАНИЕМ | 2008 |
|
RU2479629C2 |




































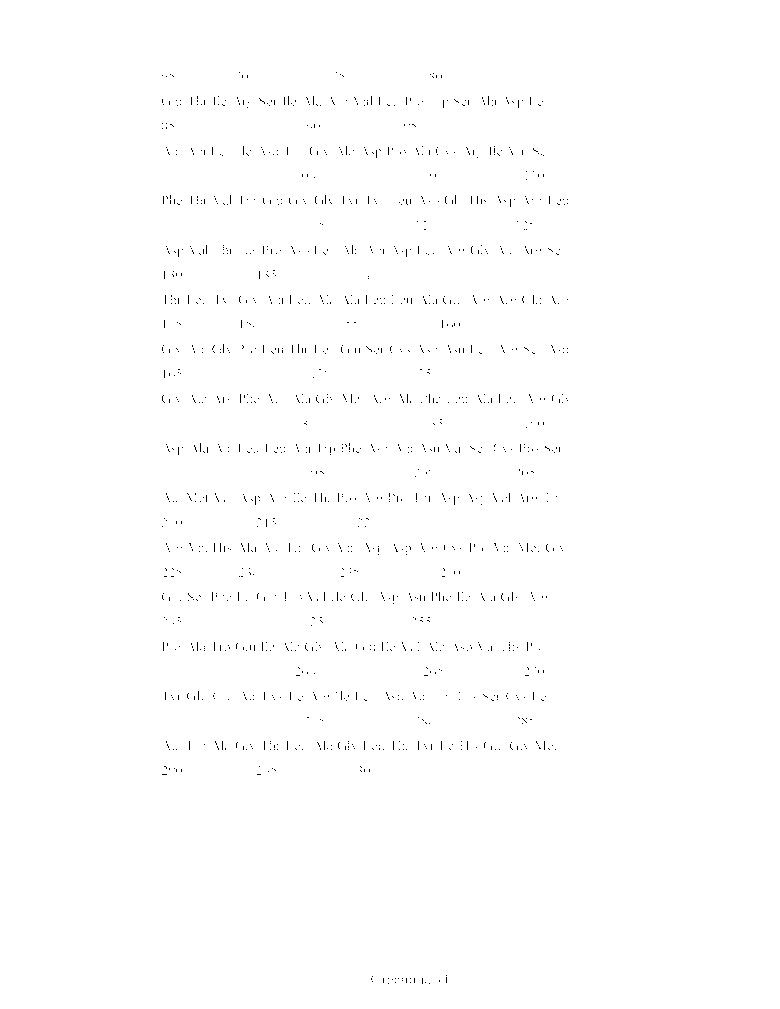

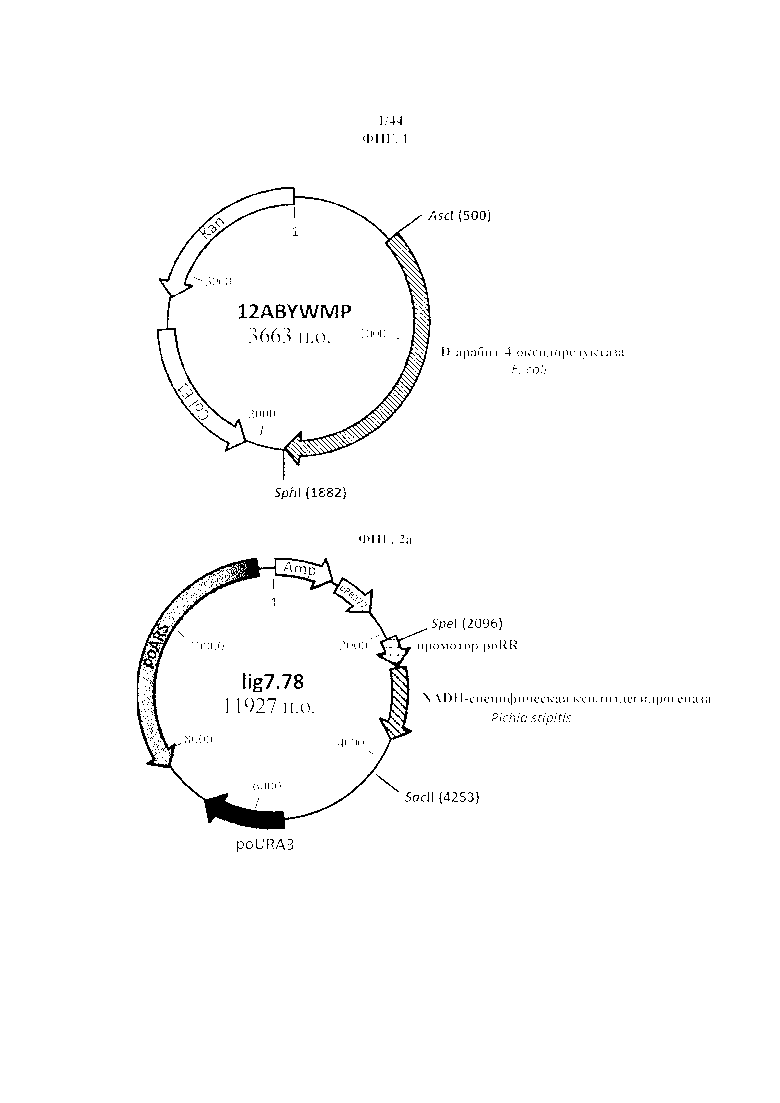
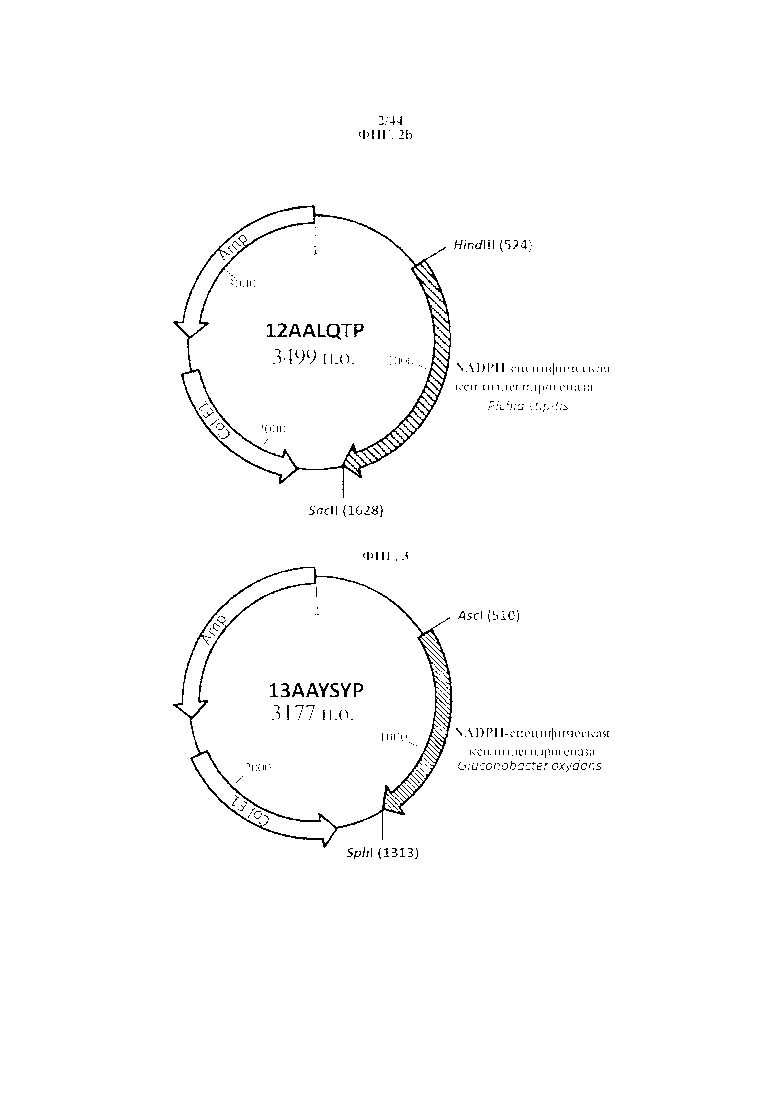
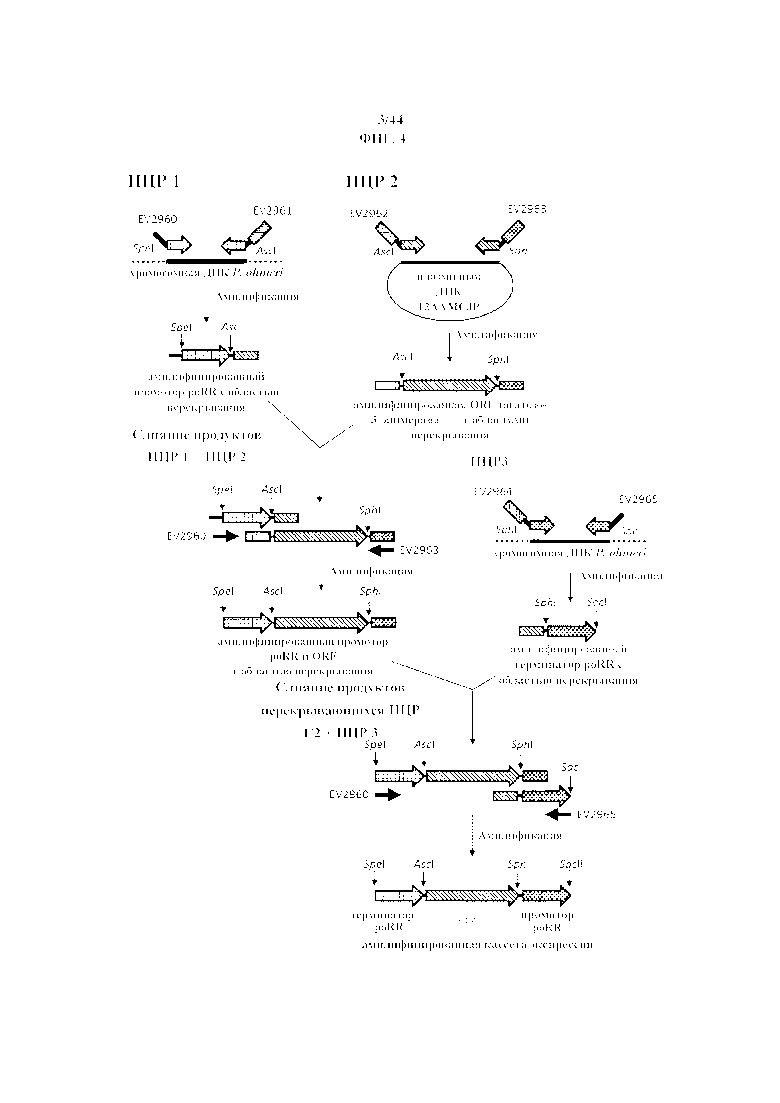
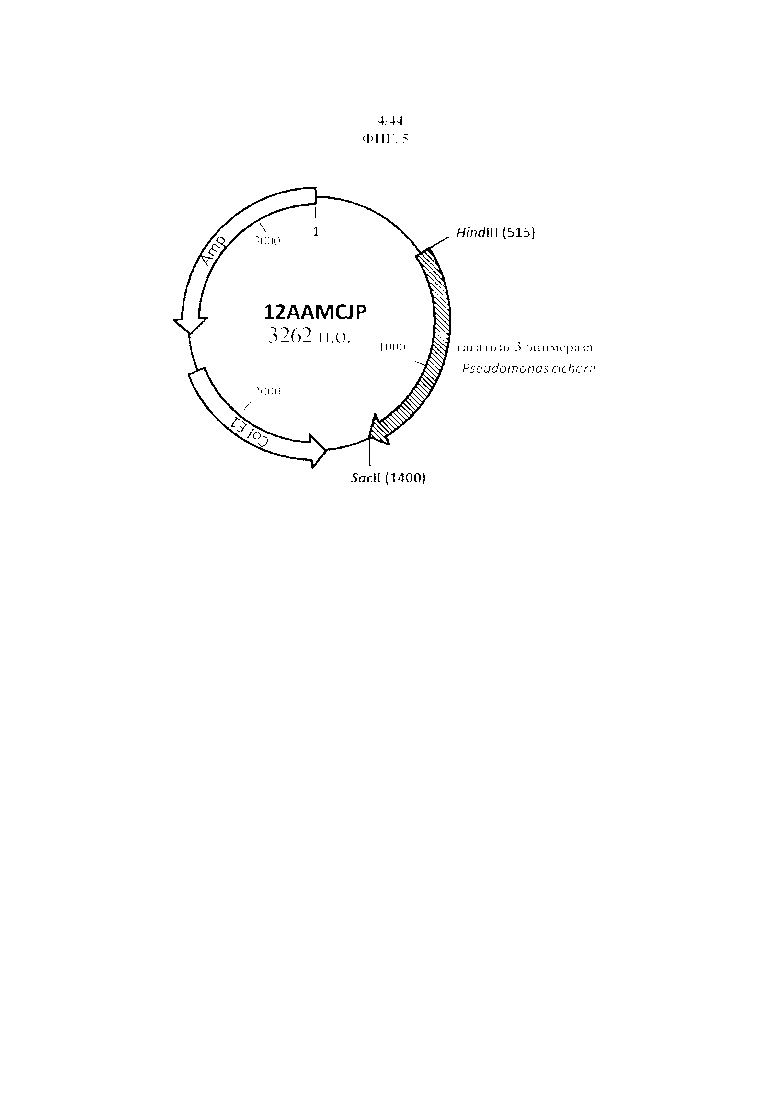
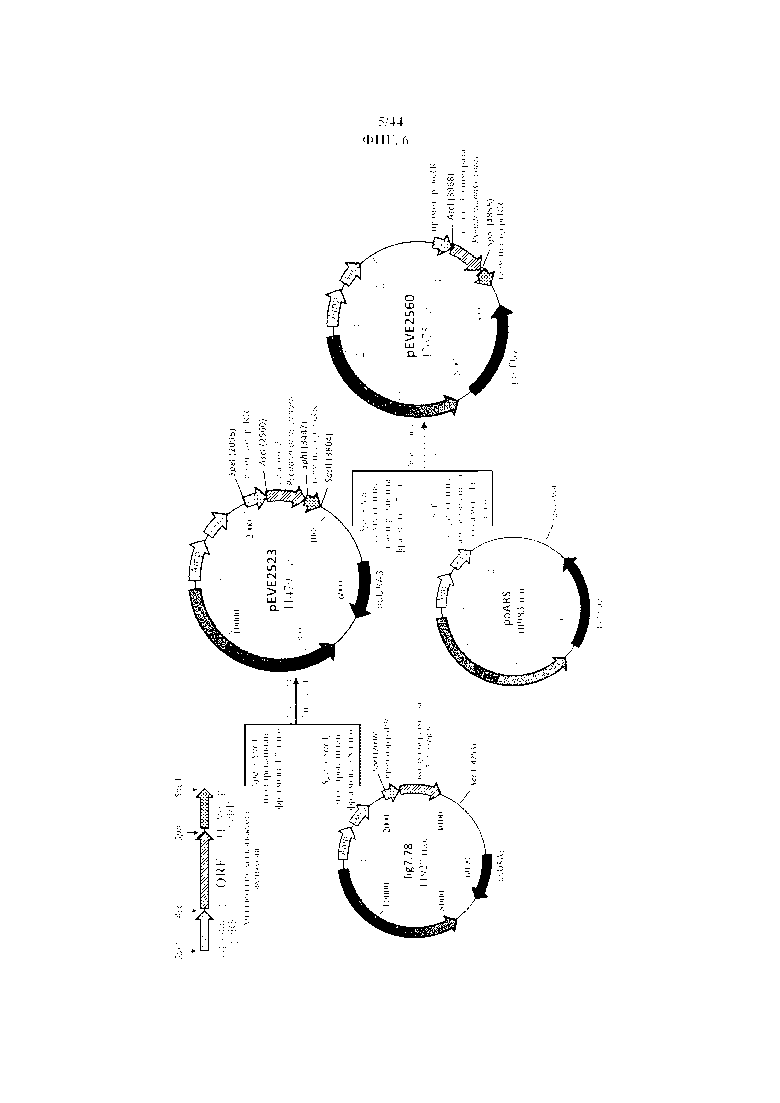
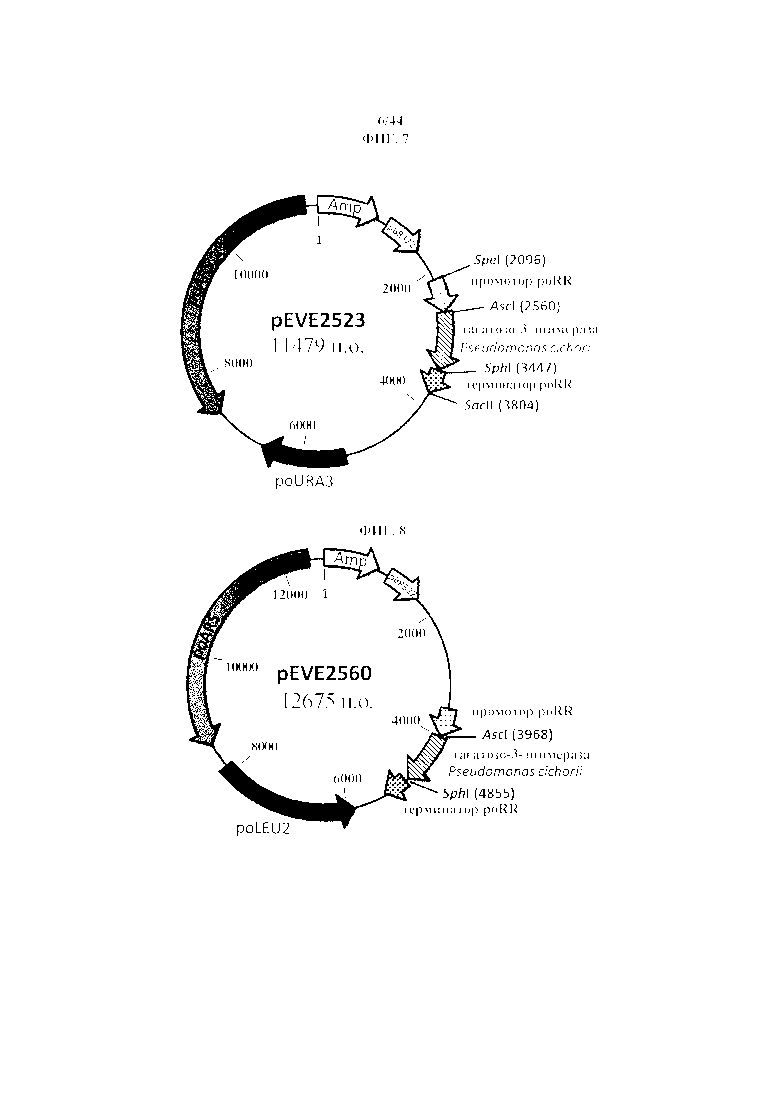


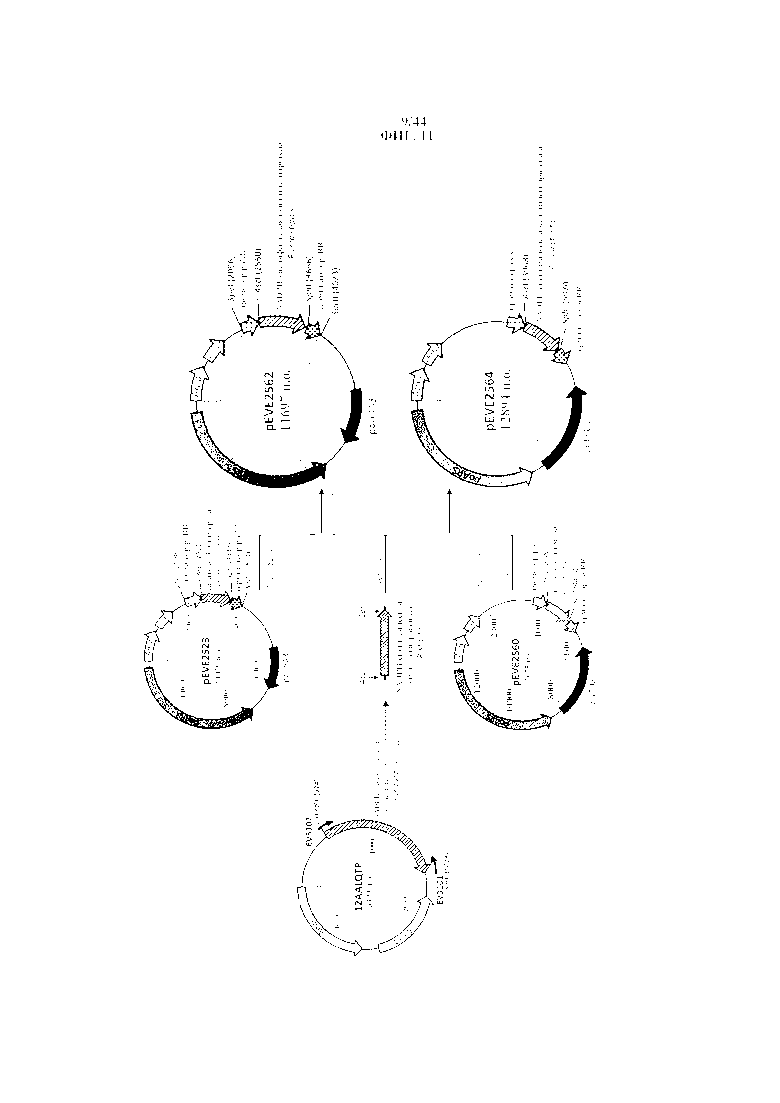

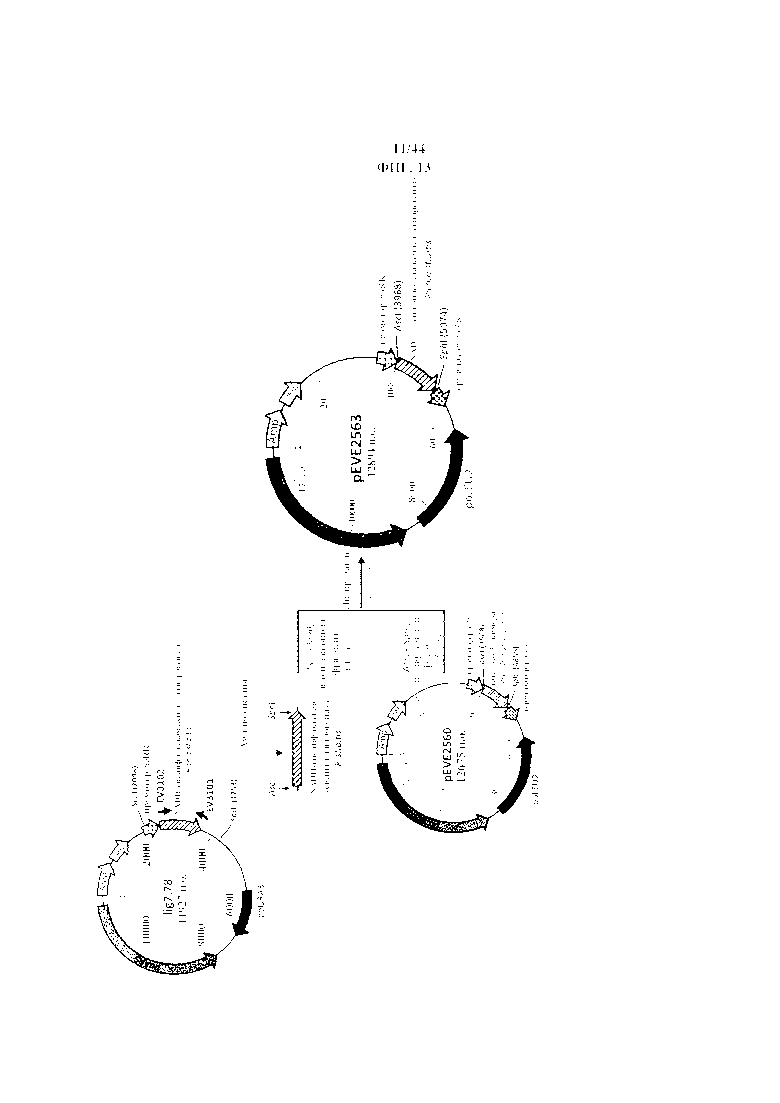
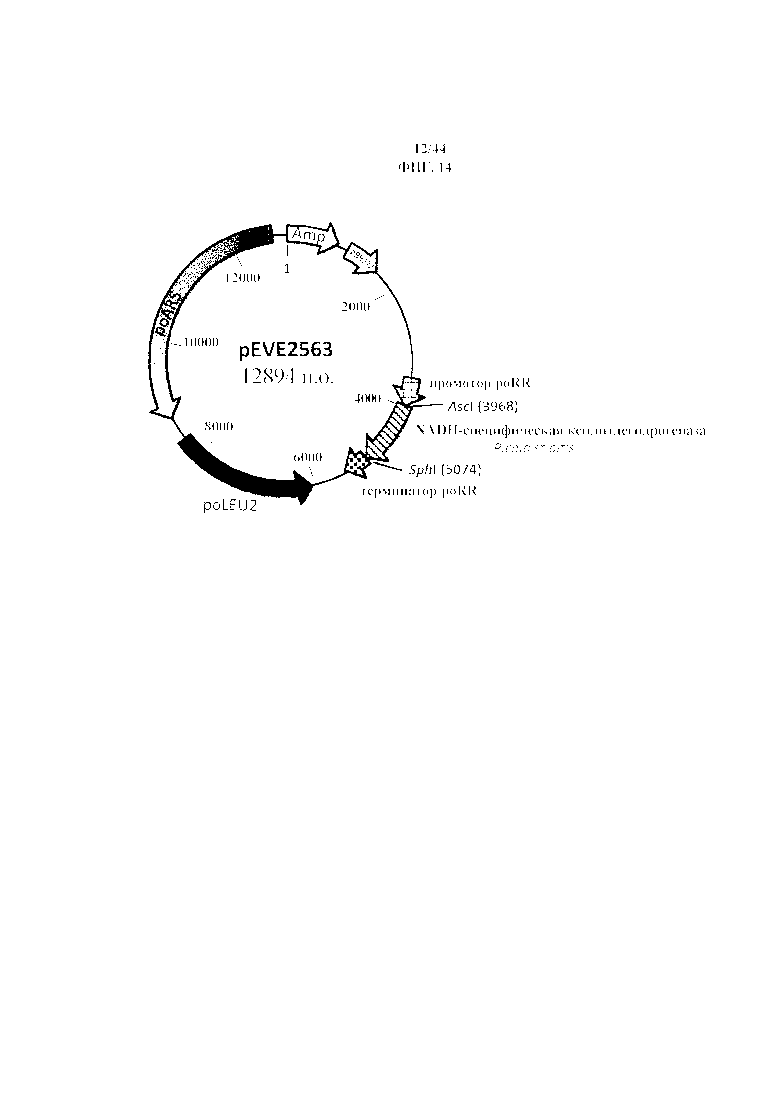
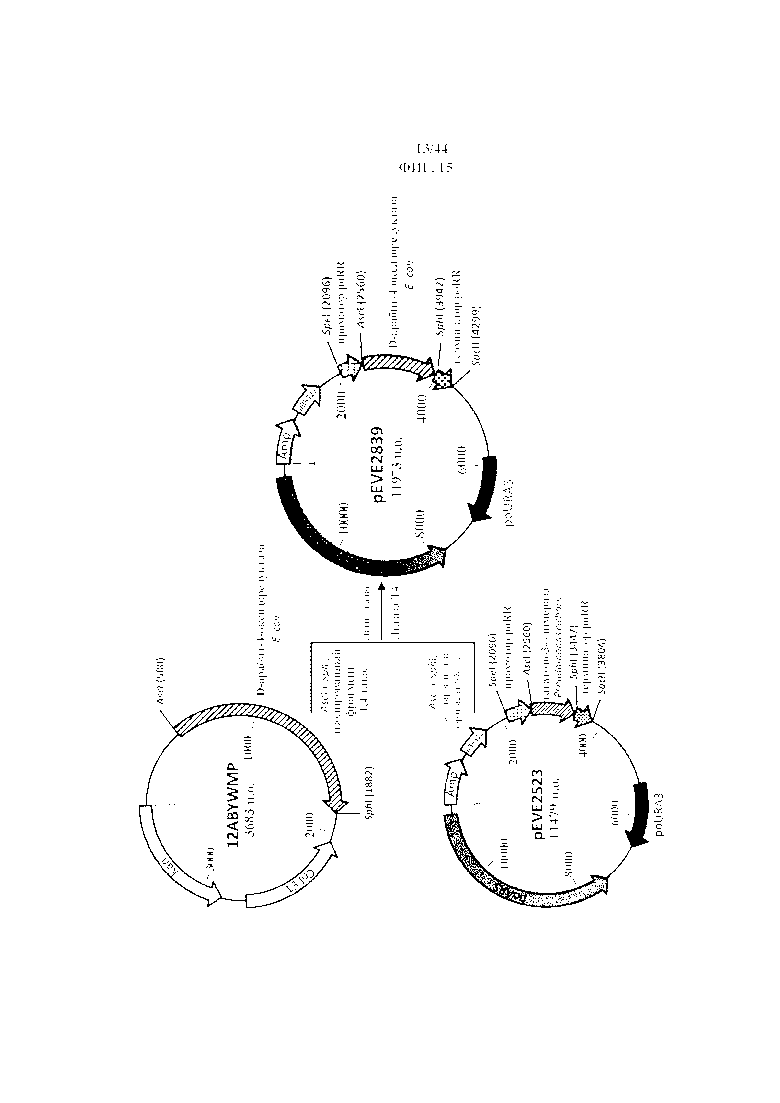
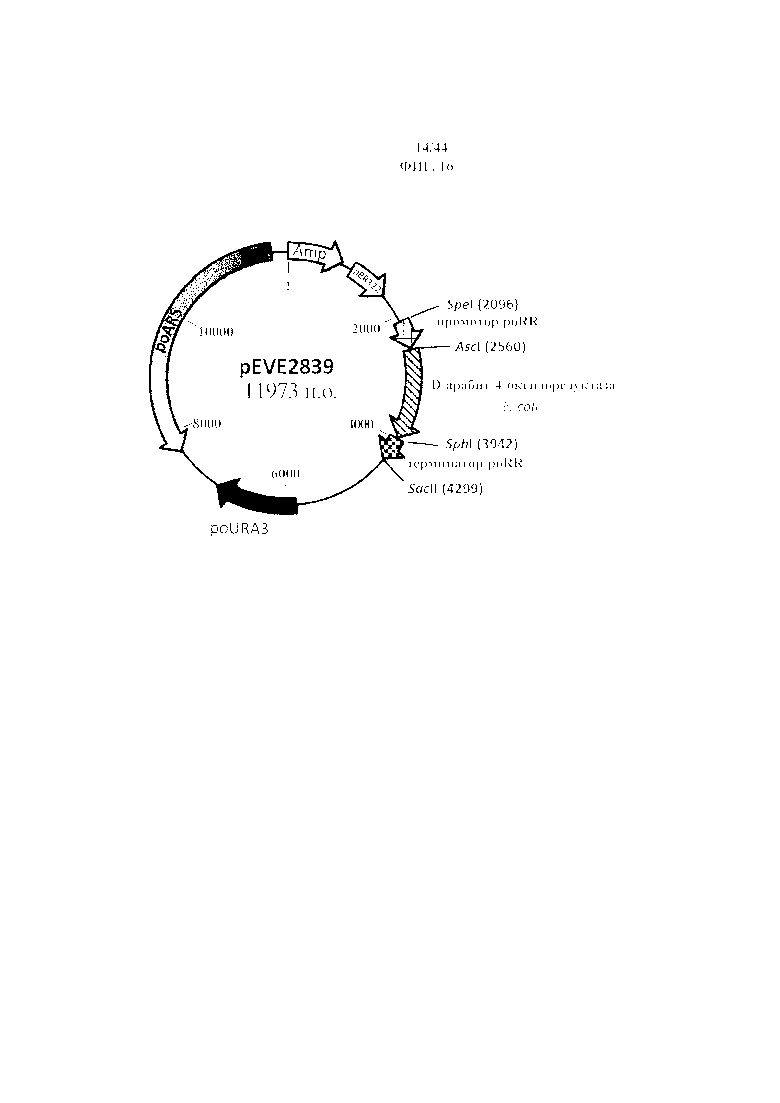
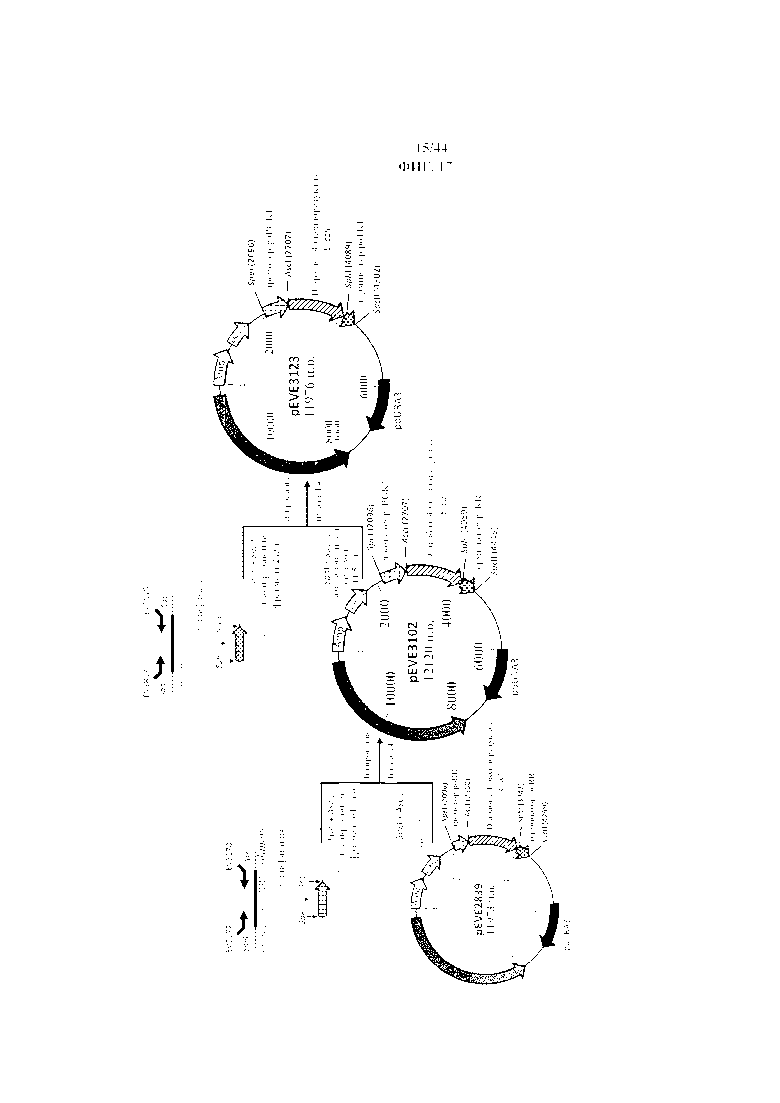
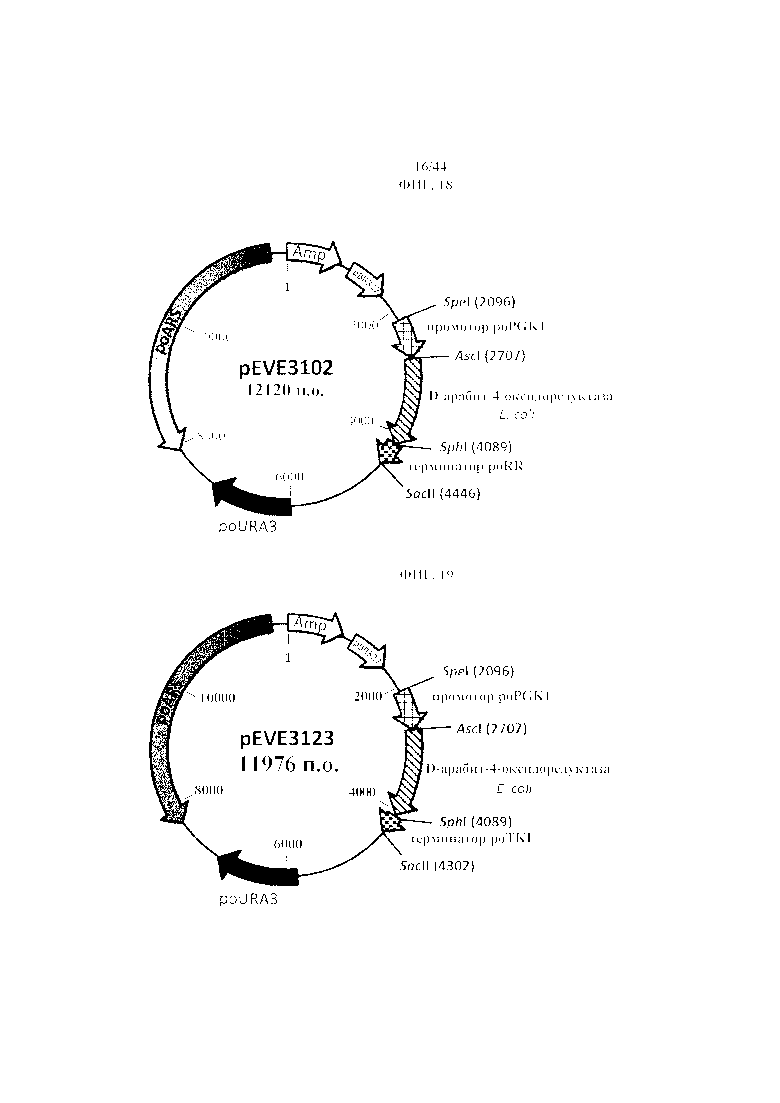
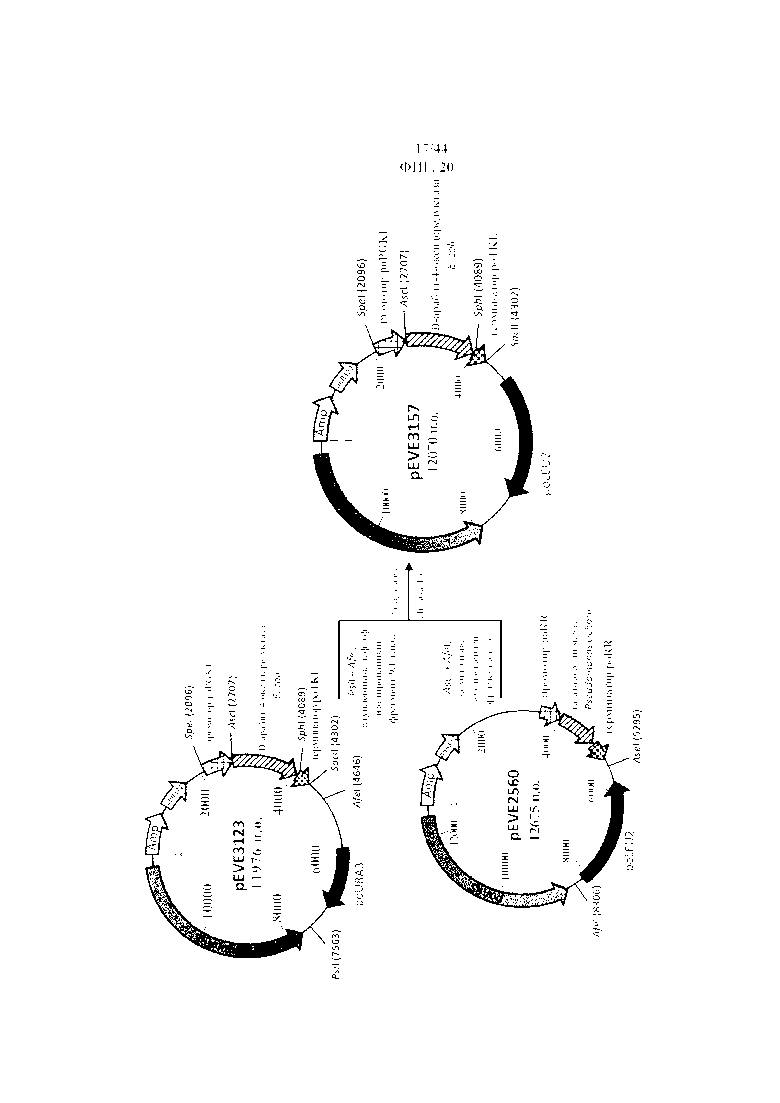

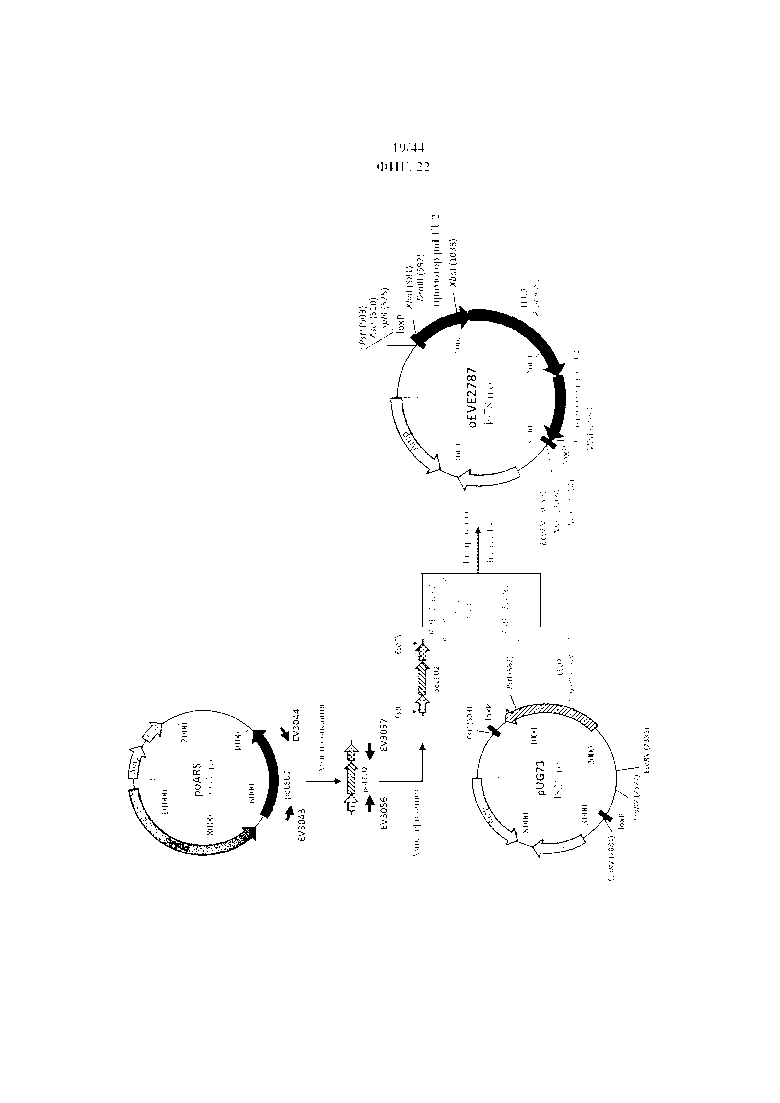


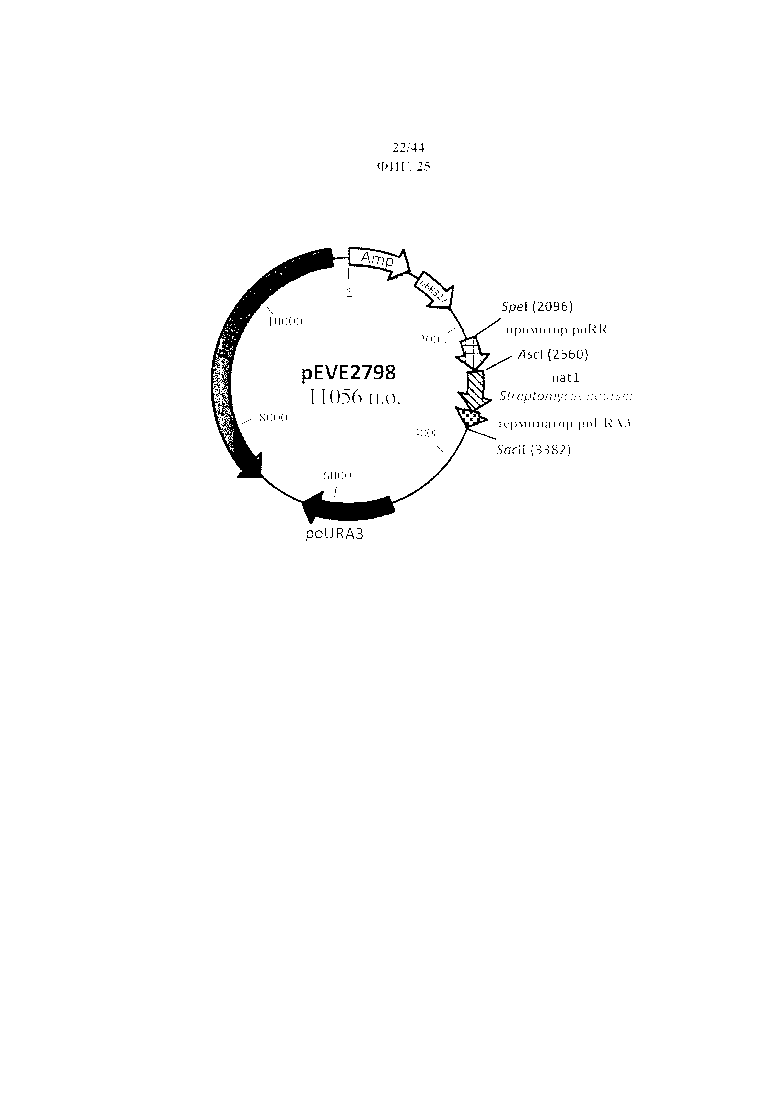



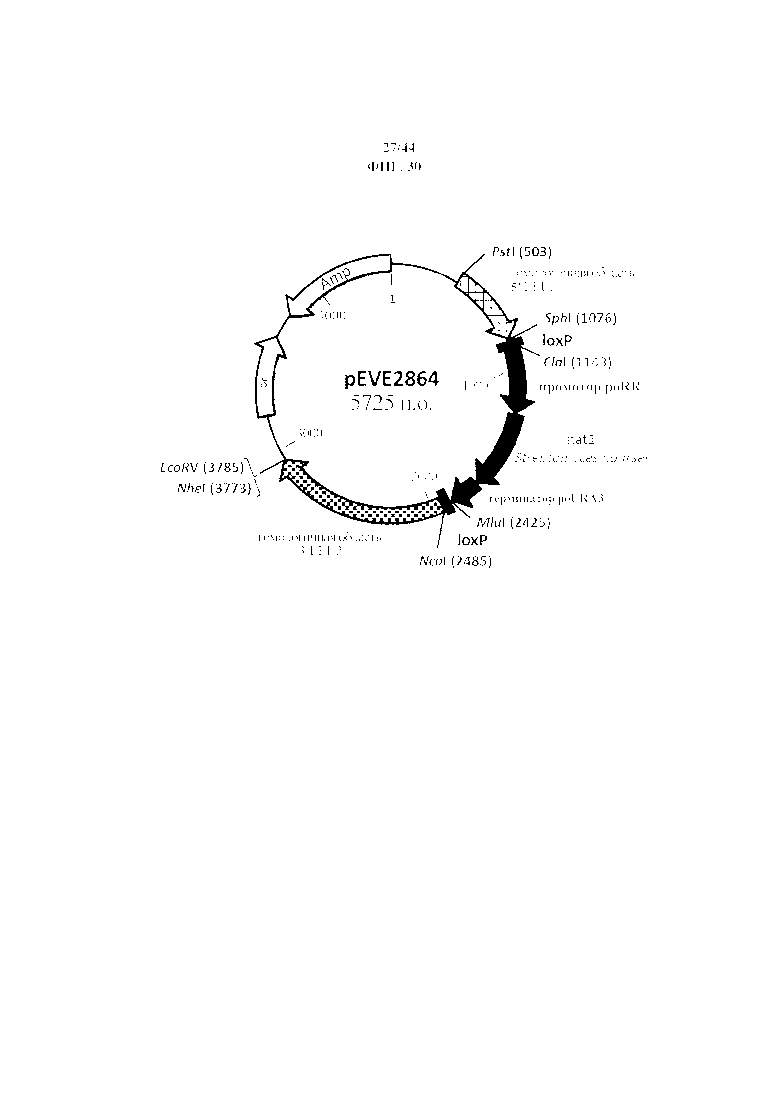

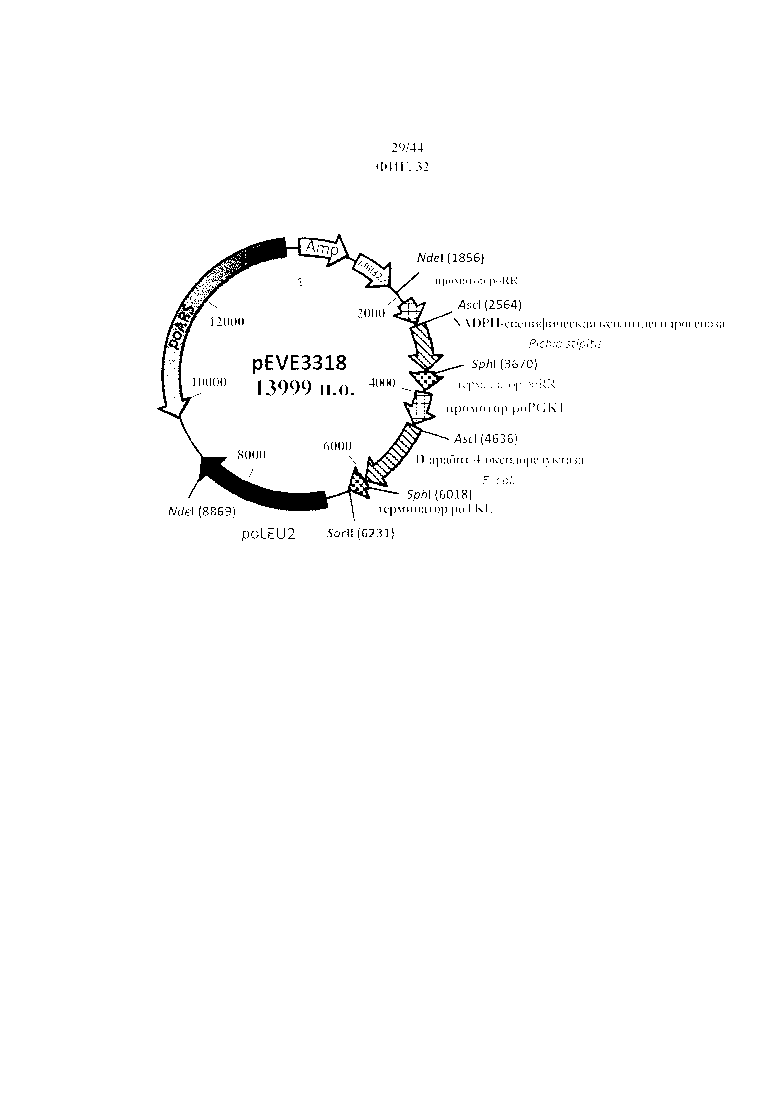
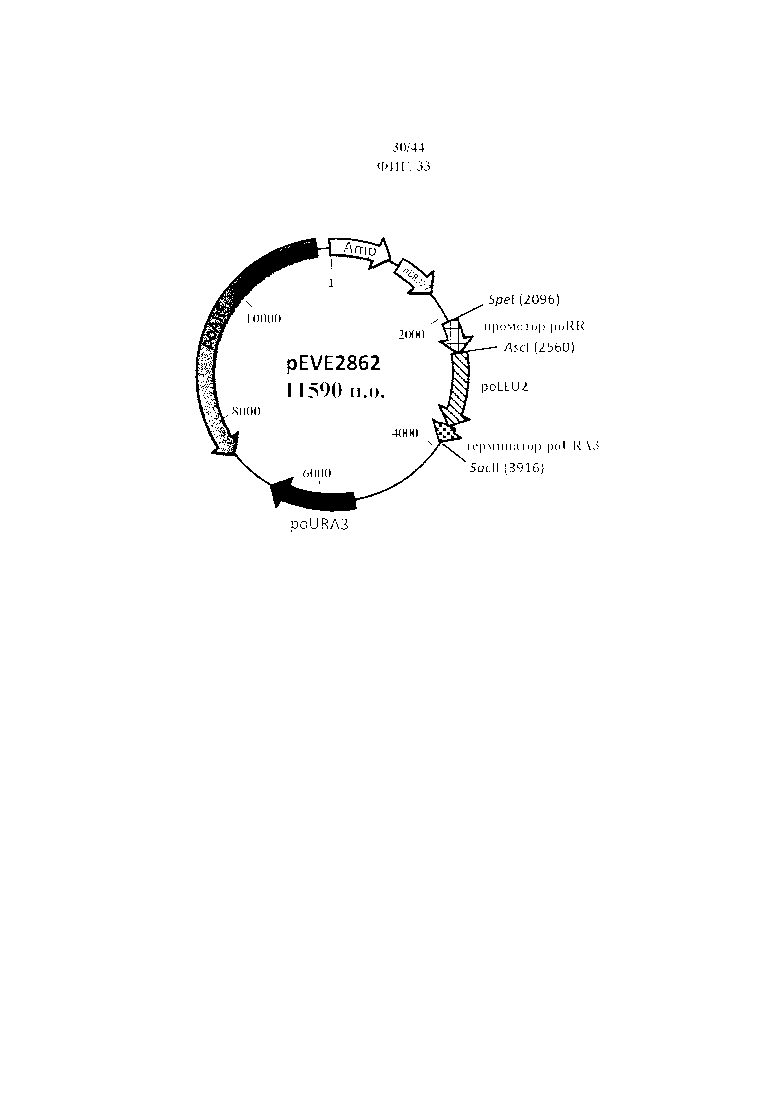
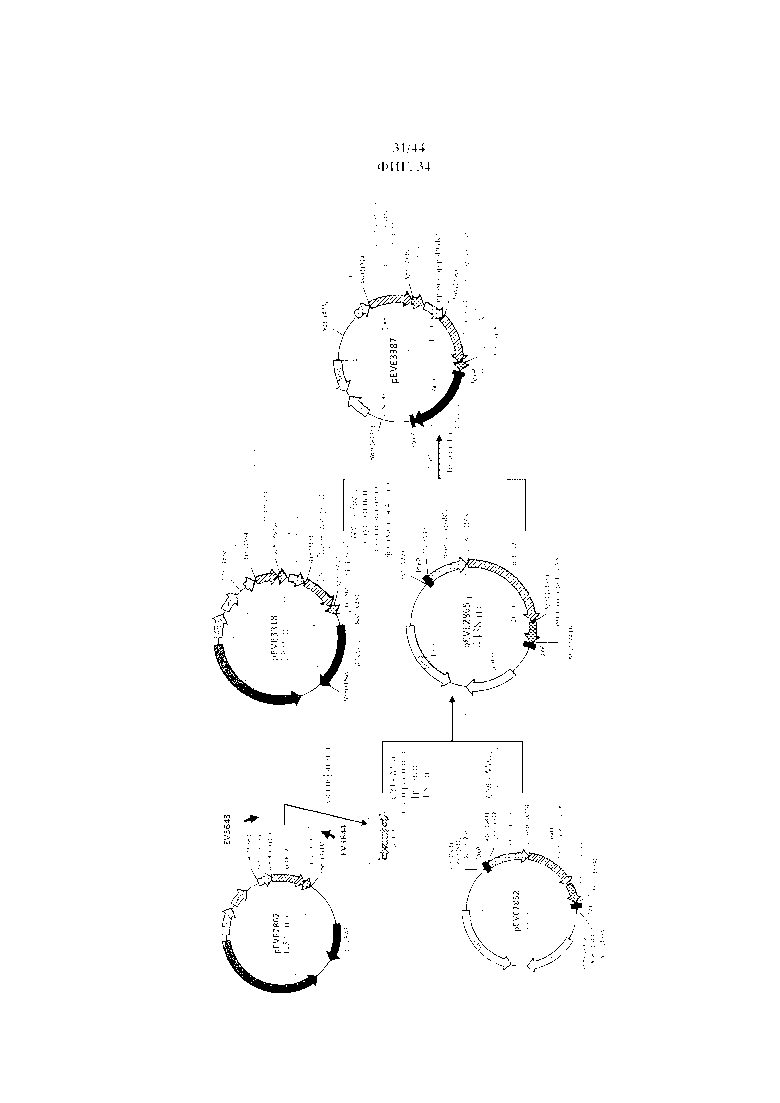
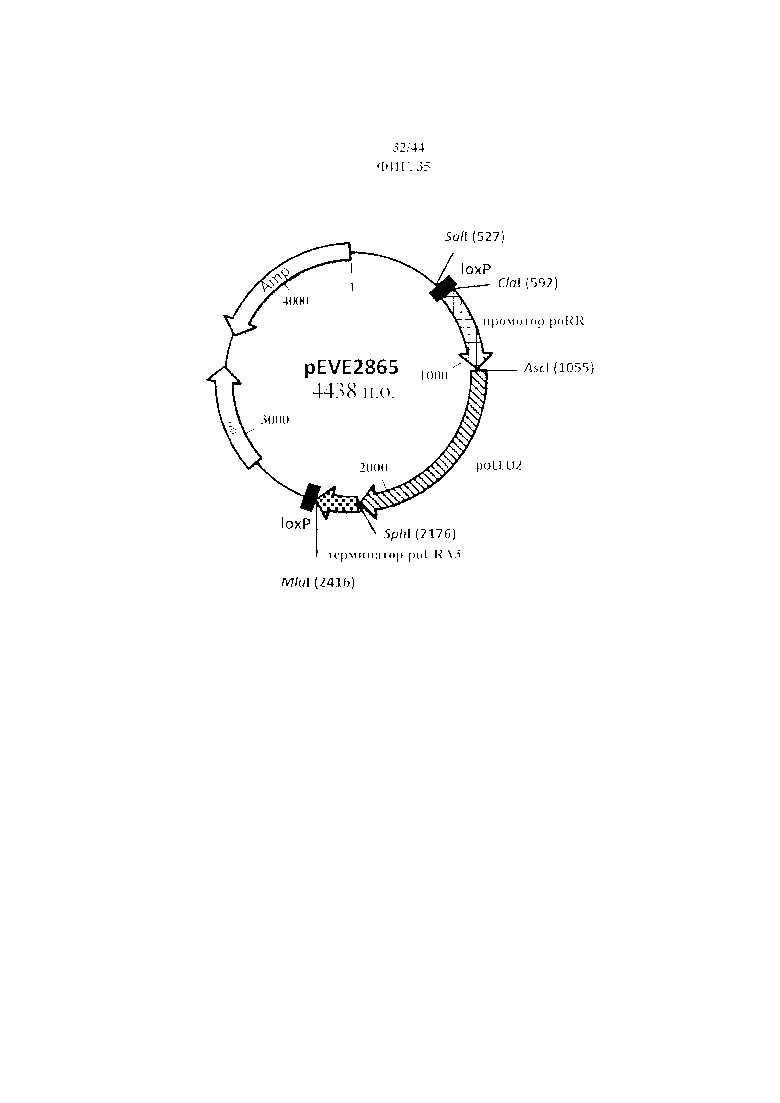
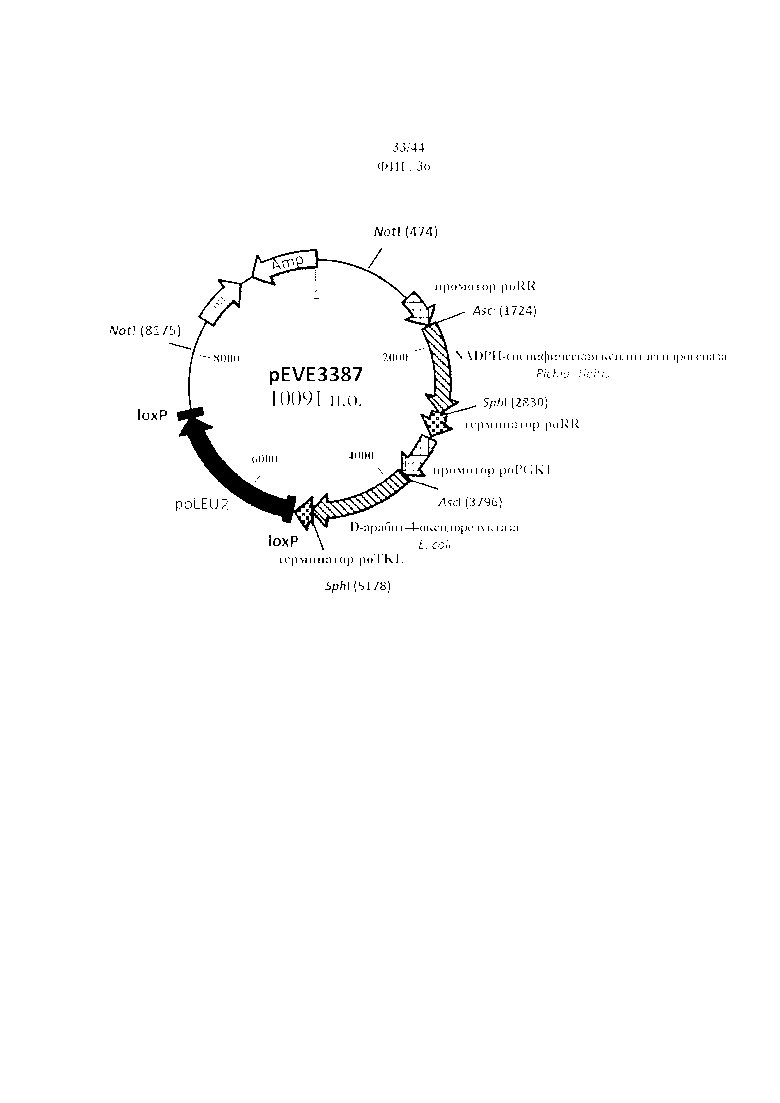

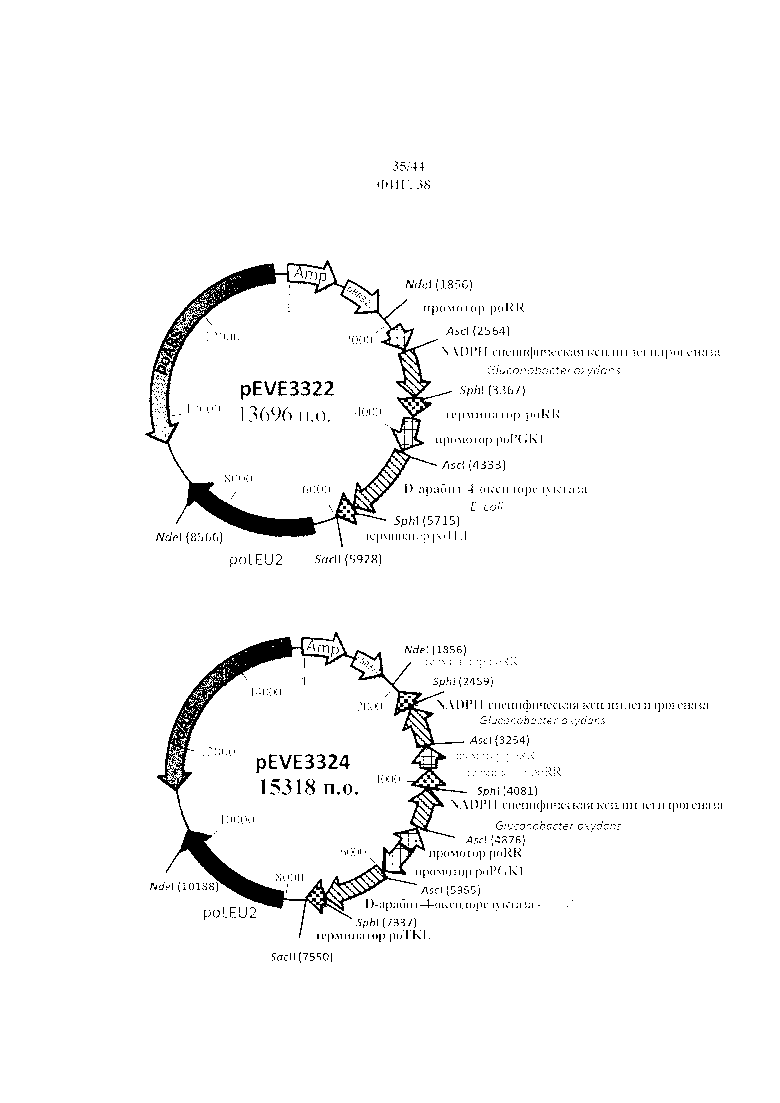



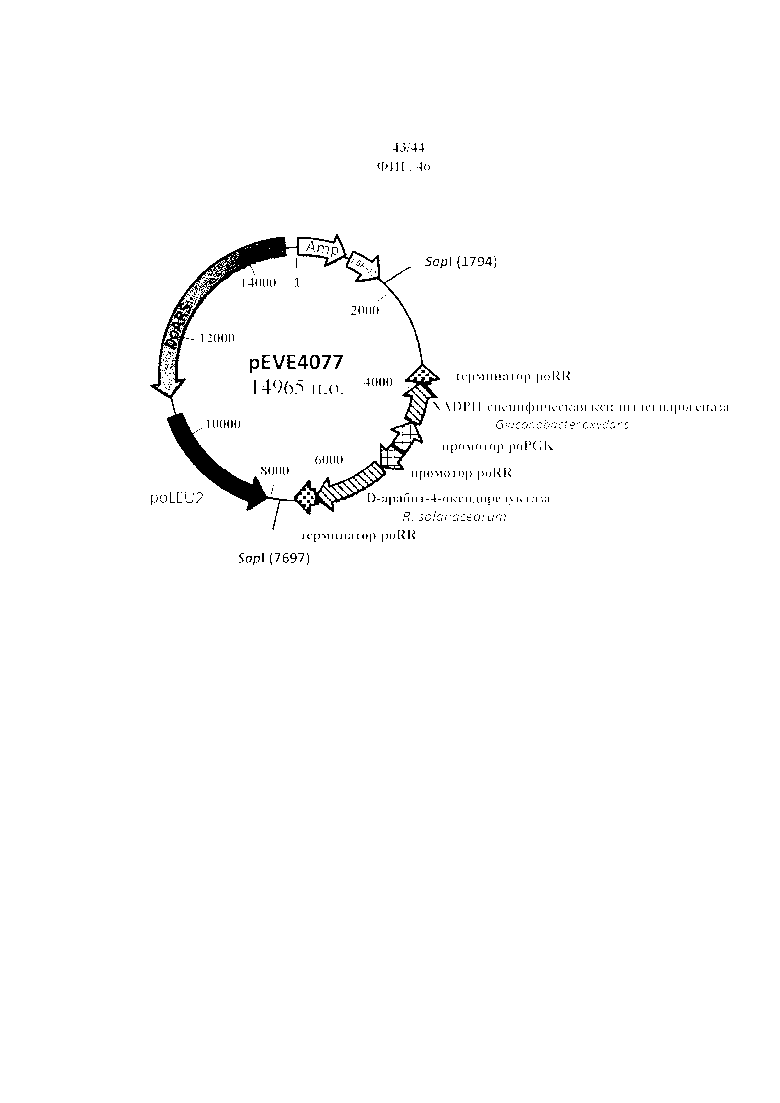
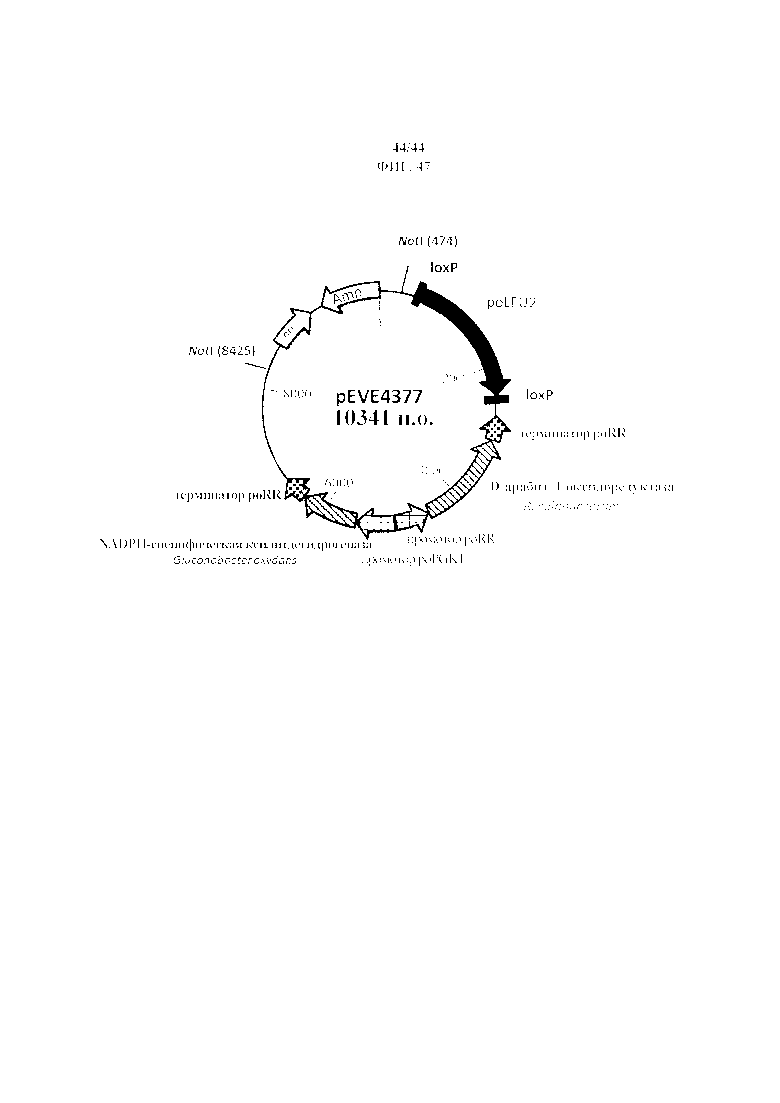
Группа изобретений относится к рекомбинантной дрожжевой клетке-хозяину для продуцирования ксилита и ее применению. Указанная клетка-хозяин продуцирует D-арабит из глюкозы и содержит последовательность гетерологичной нуклеиновой кислоты, кодирующую NAD+-специфическую D-арабит-4-оксидоредуктазу (EC 1.1.1.11), использующую D-арабит в качестве субстрата и дающую D-ксилулозу в качестве продукта, и последовательность гетерологичной нуклеиновой кислоты, кодирующую NADPH-специфическую ксилитдегидрогеназу, использующую D-ксилулозу в качестве субстрата и дающую ксилит в качестве продукта. Предложены также способ получения ксилита с использованием указанной клетки-хозяина и применение указанной клетки-хозяина для получения ксилита. Группа изобретений обеспечивает повышенную продукцию ксилита. 3 н. и 13 з.п. ф-лы, 47 ил., 16 табл., 27 пр.
1. Рекомбинантная дрожжевая клетка-хозяин для продуцирования ксилита, которая продуцирует D-арабит из глюкозы, где указанная клетка-хозяин содержит:
последовательность гетерологичной нуклеиновой кислоты, кодирующую NAD+-специфическую D-арабит-4-оксидоредуктазу (EC 1.1.1.11), использующую D-арабит в качестве субстрата и дающую D-ксилулозу в качестве продукта; и
последовательность гетерологичной нуклеиновой кислоты, кодирующую NADPH-специфическую ксилитдегидрогеназу, использующую D-ксилулозу в качестве субстрата и дающую ксилит в качестве продукта.
2. Рекомбинантная клетка-хозяин по п.1, где клетка-хозяин продуцирует D-арабит из D-глюкозы в условиях среды с высоким осмотическим давлением.
3. Рекомбинантная клетка-хозяин по п.2, где клетка-хозяин не использует D-арабит в качестве единственного источника углерода.
4. Рекомбинантная клетка-хозяин по п.1, где клетка-хозяин является клеткой осмофильных или осмоустойчивых дрожжей.
5. Рекомбинантная клетка-хозяин по п.1, где клетка-хозяин представляет собой клетку Pichia ohmeri.
6. Рекомбинантная клетка-хозяин по любому из пп.1-5, где NAD+-специфическая D-арабит-4-оксидоредуктаза (EC 1.1.1.11) происходит из E. coli и/или Ralstonia solanacearum.
7. Рекомбинантная клетка-хозяин по п.6, где NAD+-специфическая D-арабит-4-оксидоредуктаза (EC 1.1.1.11) представлена последовательностью SEQ ID NO 2 или 43.
8. Рекомбинантная клетка-хозяин по п.7, где последовательность, кодирующая NAD+-специфическую D-арабит-4-оксидоредуктазу (EC 1.1.1.11), представлена последовательностью SEQ ID NO 3 или SEQ ID NO 42.
9. Рекомбинантная клетка-хозяин по любому из пп.1-8, где NADPH-специфическая ксилитдегидрогеназа является ксилитдегидрогеназой из Pichia stipitis или Gluconobacter oxydans, подвергнутой мутации для изменения специфичности в отношении кофактора с NADH на NADPH.
10. Рекомбинантная клетка-хозяин по п.9, где NADPH-специфическая ксилитдегидрогеназа представлена последовательностью SEQ ID NO 5 или 8.
11. Рекомбинантная клетка-хозяин по п.10, где последовательность, кодирующая NADPH-специфическую ксилитдегидрогеназу, представлена последовательностью SEQ ID NO 6 или 9.
12. Рекомбинантная клетка-хозяин по любому из пп.1-11, где клетка-хозяин способна продуцировать титр ксилита, составляющий по меньшей мере 15 г/л в надосадочной жидкости после 48 ч культивирования.
13. Рекомбинантная клетка-хозяин по любому из пп.1-12, где клетка-хозяин представляет собой клетку штамма Pichia ohmeri, выбранного из группы, состоящей из штаммов, депонированных в CNCM под номерами I-4982, I-4960 и I-4981.
14. Рекомбинантная клетка-хозяин по любому из пп.1-12, где клетка-хозяин содержит несколько копий последовательности, кодирующей NAD+-специфическую D-арабит-4-оксидоредуктазу, и/или несколько копий последовательности, кодирующей NADPH-специфическую ксилитдегидрогеназу.
15. Способ получения ксилита, включающий культивирование рекомбинантной клетки-хозяина по любому из пп.1-14 и извлечение ксилита.
16. Применение рекомбинантной клетки-хозяина по любому из пп.1-14 для получения ксилита.
| СПОСОБ ПОЛУЧЕНИЯ КСИЛИТА | 1993 |
|
RU2142999C1 |
| 0 |
|
SU153306A1 | |
| FONSECA C | |||
| ET AL | |||
| Насос | 1917 |
|
SU13A1 |
| Applied and environmental microbiology, 2008, V | |||
| Приспособление в центрифугах для регулирования количества жидкости или газа, оставляемых в обрабатываемом в формах материале, в особенности при пробеливании рафинада | 0 |
|
SU74A1 |
| Приспособление для точного наложения листов бумаги при снятии оттисков | 1922 |
|
SU6A1 |
| Устройство для нейтрализирования статических зарядов, образующихся при выделке и обработке тканей и т.п. изделий из диэлектриков | 1922 |
|
SU1845A1 |
| PAKULA A | |||
| ET AL | |||
| Genetic analysis of protein stability and function | |||
| Annual review of genetics, 1989, V.23, No.1, P.289-310 | |||
| FRANKEL A.E | |||
| Переносная печь для варки пищи и отопления в окопах, походных помещениях и т.п. | 1921 |
|
SU3A1 |
| Protein engineering, 2000, V.13, No.8, P.575-581. | |||

