Изобретение касается способов применения генетически модифицированных микроорганизмов для производства ценных химических соединений (метаболической инженерии) и, более конкретно, конструирования микробиологических штаммов посредством генетической манипуляции, способных превращать легко доступные источники углерода, такие, как D-глюкоза, в более ценный продукт, например, ксилит.
Ксилит представляет собой химическое соединение значительной ценности в качестве специального подслащивающего вещества. Он приблизительно так же сладок, как сахароза, и является нетоксичным и некариесогенным.
В настоящее время ксилит получают химической гидрогенизацией D-ксилозы. D-ксилозу получают из гидролизатов различных растительных материалов, в которых она всегда присутствует в смеси с другими пентозами и гексозами. Поэтому очистка ксилозы и также ксилита представляет собой значительную проблему. Известен ряд процессов такого рода. В качестве примеров можно упомянуть U.S. patents 3 784 408, 4 066 711, 4 075 406 и 4 008 285.
Восстановление D-ксилозы до ксилита может быть также достигнуто в микробиологическом процессе с применением либо природных штаммов (Barbosa, M.F.S. et al., J. Industrial Microbiol. 3:241 - 251 (1988)), либо полученных генной инженерией штаммов (Hallborn, J. et. al. , Biotechnology 9:1090 - 1095 (1991)). Однако получение субстрата D-ксилозы в форме, пригодной для дрожжевой ферментации, также представляет собой значительную проблему, вследствие того, что недорогие источники ксилозы, такие как сульфитно-спиртовая барда из таких процессов, как способ превращения в пульпу (сульфитный способ) и способ производства бумаги, содержит примеси, ингибирующие рост дрожжей.
Привлекательным альтернативным способом для производства ксилита было бы получение его ферментацией дешевого и легко доступного субстрата, такого как D-глюкоза. Однако неизвестны микроорганизмы, которые продуцируют ксилит в значительных количествах во время одноступенчатой ферментации какого-либо обычного источника углерода, иного, чем D-ксилоза и D-ксилулоза, которые структурно очень близки к ксилиту.
С другой стороны, многие микроорганизмы, в частности, осмофильные дрожжи, например, Zygosaccharomyces rouxii, Candida polymorpha и Torulopsis candida, продуцируют значительные количества близко родственного пентита, D-арабита, из D-глюкозы (Lewis D.H. and Smith D.C., New Phytol. 66:143 - 184 (1967)). Используя это свойство осмофильных дрожжей, H. Onishi and T. Suzuki разработали способ превращения D-глюкозы в ксилит посредством трех последовательных ферментаций (Appl. Microbiol. 18: 1031 - 1035 (1969)). В этом процессе D-глюкозу сначала превращают в D-арабит путем ферментации с осмофильным штаммом дрожжей. Затем D-арабит окисляют в D-ксилулозу ферментацией с Acetobacter auboxidans. Наконец, D-ксилулозу восстанавливают до ксилита в третьей ферментации с применением одного из многих штаммов дрожжей, способных восстанавливать D-ксилулозу в ксилит.
Очевидным недостатком этого способа является то, что он предусматривает три различные стадии ферментации, каждая из которых занимает от 2 до 5 дней; далее необходимы дополнительные стадии, такие как стерилизация и удаление клеток, что увеличивает стоимость технологии. Выход каждой стадии этого ферментационного процесса является низким, а количество побочных продуктов высоким. Таким образом, все еще существует необходимость в способах экономического получения ксилита в микробных системах из легко доступных субстратов.
Сущность изобретения.
Данное изобретение обеспечивает способы конструирования рекомбинантных хозяев и полученных этими способами рекомбинантных хозяев, способных продуцировать ксилит при росте на источниках углерода, отличающихся от D-ксилулозы или D-ксилозы, и отличающихся от их полимеров или олигомеров, или их смесей. Источники углерода, используемые этими хозяевами, являются недорогими и легко доступными. Микроорганизмы данного изобретения также способны секретировать синтезированный ксилит в культуральную среду.
Эта цель достигается посредством модификации метаболизма желательного микроорганизма, предпочтительно встречающегося в природе дрожжевого микроорганизма, путем введения и экспрессии желательных гетерологических генов. Эта цель также достигается посредством дальнейшей модификации метаболизма такого желательного микроорганизма таким образом, чтобы он избыточно экспрессировал и (или) имел инактивированную активность или экспрессию определенных генов, гомологичных для этого микроорганизма в его нативном состоянии.
Таким образом, целью данного изобретения является обеспечение способа получения ксилита, причем такой способ использует новый и неизвестный микробный штамм, рекомбинантного хозяина, называемого здесь также генетически сконструированным микроорганизмом, в качестве продуцента ксилита, причем такой генетически сконструированный микроорганизм продуцирует ксилит либо de novo, либо в увеличенных количествах по сравнению с нативным микроорганизмом.
Следующей целью данного изобретения является обеспечение способа получения ксилита, использующего новый метаболический путь, который был сконструирован в микроорганизме и который приводит к de novo или увеличенному продуцированию ксилита таким микроорганизмом.
Следующей целью данного изобретения является обеспечение способа получения ксилита, использующего указанный выше новый метаболический путь, модифицирующий путь и (или) метаболизм биосинтеза D-арабита, причем путь модифицирован таким образом, что микроорганизм теперь продуцирует ксилит посредством ферментации источников углерода, которые немодифицированный хозяин использует для биосинтеза D-арабита.
Дальнейшей целью изобретения является обеспечение способа получения ксилита, который использует упомянутый выше измененный путь D-арабита, причем такой путь был изменен путем удлинения предшествующего пути биосинтеза D-арабита) или путем замены одной или нескольких стадий пути D-арабита подобными стадиями, ведущими к образованию ксилита.
Дальнейшей целью изобретения является обеспечение способа получения ксилита, использующего описанный выше измененный путь биосинтеза D-арабита, причем этот путь изменяют удлинением предшествующего пути D-арабита путем введения и избыточной экспрессии генов, кодирующих образующую D-ксилулозу D-арабитолдегидрогеназу (EC 1.1.1.11) и ксилитолдегидрогеназу (EC 1.1.1.9) в продуцирующем D-арабит микроорганизме.
Дальнейшей целью изобретения является обеспечение способа получения ксилита при помощи указанного выше нового микроорганизма, использующего измененный путь биосинтеза D-арабита, упомянутого выше, причем этот путь изменен далее путем инактивации при помощи химически индуцированного мутагенеза или разрушения гена, кодирующего транскетолазу (EC 2.2.1.1), или гена, кодирующего D-ксилулокиназу (EC 2.7.1.17), в таком микроорганизме.
Дальнейшей целью изобретения является обеспечение способа получения ксилита при помощи описанного выше нового микроорганизма, причем этот способ использует генетически конструированную измененную избыточную экспрессию генов, кодирующих ферменты окислительной ветви пентозофосфатного пути и, в частности, D-глюкозо-6-фосфатдегидрогеназу (EC 1.1.1.49) и (или) 6-фосфо-D-глюконатдегидрогеназу (EC 1.1.1.44) в этом микроорганизме.
Следующей целью изобретения является обеспечение способа получения ксилита при помощи нового микроорганизма, описанного выше, причем этот способ использует генетически сконструированную измененную избыточную экспрессию генов, кодирующих ферменты окислительной ветви пентозофосфатного пути, а также гена D-рибулозо-5-фосфатэпимеразы (EC 5.1.3.1).
Фиг. 1 является рестрикционной картой инсерции в плазмиде pARL2. Эта инсерция является инсерцией хромосомного локуса Klebsiella terrigena Php 1 и содержит ген D-арабитолдегидрогеназы K. terrigena. Открытый блок представляет хромосомную ДНК K. terrigena. Стрелка указывает местоположение и направление гена D-арабитолдегидрогеназы (EC 1.1.1.11) в этой ДНК.
Фиг. 2 показывает конструирование pYARD из pADH и pAAH5. На схемах плазмид одиночная линия (-) указывает бактериальные последовательности; волнистая линия указывает 2 мкм ДНК S. cerevisiae; открытая стрелка (⇒) указывает промотор ADCI (ADCI ген кодирует алкогольдегидрогеназу S. cerevisiae (ADCI, прежде называющуюся ADHI); открытый ромб  указывает терминатор транскрипции ADCI; прямоугольный блок указывает ген LEU2 и заштрихованная стрелка указывает ген D-арабитолдегидрогеназы.
указывает терминатор транскрипции ADCI; прямоугольный блок указывает ген LEU2 и заштрихованная стрелка указывает ген D-арабитолдегидрогеназы.
Фиг. 3 изображает конструирование плазмиды pJDB(AX)-16. XYL2 является геном ксилитдегидрогеназы из Pichia stipitis. daLD является геном D-арабитолдегидрогеназы. ADCI представляет собой зону регуляции транскрипции (промотор) гена ADCI, который предшествует кодирующей последовательности daLD и оперативно связан с ней. Эти символы неодинаковы с символами на фиг. 4. На схемах плазмид одиночная линия (-) обозначает бактериальные последовательности 2 мкм ДНК, где отмечено: темная стрелка обозначает промотор ADCI; затененный ромб  обозначает терминатор транскрипции ADCI; прямоугольный блок обозначает маркерный ген LEU2; заштрихованная стрелка обозначает ген XYL2 и блокированный прямоугольник обозначает daLD ген.
обозначает терминатор транскрипции ADCI; прямоугольный блок обозначает маркерный ген LEU2; заштрихованная стрелка обозначает ген XYL2 и блокированный прямоугольник обозначает daLD ген.
Фиг. 4 показывает конструирование челночного вектора pSRT(AX)-9 E. coli-Z.rouxii. Символы такие же, что и в фиг. 3.
Фиг. 5 изображает рестрикционную карту клонированного фрагмента рДНК T. candida.
Фиг. 6 показывает конструирование плазмиды pTC(AX).
Фиг. 7 показывает рестрикционную карту клонированного фрагмента рДНК T. candida.
Фиг. 7a показывает конструирование плазмиды pCPU(AX).
Фиг. 8 изображает клонирование ZWF1 и gnd гена.
Фиг. 8a показывает конструирование плазмиды PAAH (gnd).
Фиг. 9 показывает конструирование плазмиды pSRT(ZG).
Фиг. 10 изображает культивирование штамма Z.rouxii ATCC 13356 (pSRT(AX)-09) в ферментере.
Фиг. 11 изображает культивирование мутанта, полученного из штамма Z. rouxii ATCC 13356 [pSRT(AX)-9] в ферментере.
Детальное описание предпочтительных вариантов.
В следующем далее описании широко используются термины, применяемые в технологии рекомбинантных ДНК (генной, или генетической инженерии). Для обеспечения ясного и согласующегося понимания данной специфики и формулы изобретения, в том числе сферы действия, давшей такие термины, даются следующие определения.
Источник углерода иной, чем ксилоза или ксилулоза.
В применении здесь "источник углерода иной, чем D-ксилоза и D-ксилулоза" обозначает углеродный субстрат для получения ксилита иной, чем D-ксилоза и D-ксилулоза или их полимеры или олигомеры, или их смеси (такие как ксилан и гемицеллюлоза). Такой источник углерода предпочтительно поддерживает рост генетически сконструированных микробных хозяев данного изобретения и ферментацию в дрожжевых хозяевах. Многие дешевые и легко доступные соединения можно использовать в качестве источников углерода для получения кслиита в микробных хозяевах данного изобретения, в том числе D-глюкозу и различные содержащие D-глюкозу сиропы и смеси D-глюкозы с другими сахарами. Другие сахара, ассимилируемые хозяевами данного изобретения, в том числе дрожжами и грибами, такие как различные альдо- и кетогексозы (например, D-фруктоза, D-галактоза и D-манноза) и их олигомеры и полимеры (например, сахароза, лактоза, крахмал, инулин и мальтоза), включаются в этот термин. Пентозы, иные, чем ксилоза и ксилулоза, и неуглеводные источники углерода, такие как глицерин, этанол, различные растительные масла или углеводороды (предпочтительно н-алканы, содержащие 14-16 атомов углерода) также включаются в этот термин. Спектр источников углерода, применимых в качестве субстратов для получения ксилита хозяевами данного изобретения, будут варьировать в зависимости от микробного хозяина. Например, глюкоза и содержащие глюкозу сиропы являются предпочтительным источником углерода для получения ксилита с применением генетически манипулируемых Zygosaccharomyces rouxii этого изобретения, тогда как н-алканы, предпочтительно имеющие 14-16 атомов углерода, являются предпочтительным источником для модифицированных штаммов Candida tropicalis.
Ген. Последовательность ДНК, содержащая матрицу для РНК-полимеразы. РНК, транскрибируемая с гена, может кодировать и не кодировать белок. РНК, которая кодирует белок, называют мессенджер РНК (мРНК) и, в эукариотах, она транскрибируется РНК-полимеразой II. Можно также сконструировать ген, содержащий матрицу РНК-полимеразы II (в результате присутствия промотора РНК-полимеразы II), с которой транскрибируется последовательность РНК, имеющая последовательность, комплементарную последовательности специфической мРНК, но не нетранслируемую нормально. Такая генная конструкция названа здесь "геном антисмысловой РНК", а такой РНК-транскрипт назван "антисмысловой РНК". Антисмысловые РНК не транслируются нормально, вследствие присутствия стоп-кодонов трансляции в антисмысловой последовательности РНК.
"Комплементарная ДНК" или ген "кДНК" обозначает рекомбинантные гены, синтезированные, например, обратной транскрипцией мРНК и не имеющие поэтому расположенных внутренних интронов. Клоны генов из геномной ДНК обычно содержат интроны.
Клонирующий вектор. Плазмидная или фаговая ДНК или другая последовательность ДНК, которая способна переносить генетическую информацию, в частности ДНК, в клетку хозяина. Клонирующий вектор часто характеризуется одним или небольшим числом сайтов узнавания эндонуклеазами, в которых такие последовательности ДНК могут быть разрезаны определенным образом без потери основной биологической функции вектора и в которые может быть сплайсирована желаемая ДНК, чтобы вызвать ее клонирование в клетке хозяина. Клонирующий вектор может, кроме того, содержать маркер, пригодный для использования в идентификации клеток, трансформированных этим клонирующим вектором, и затравки репликации, делающие возможными сохранение и репликацию вектора в одном или нескольких прокариотических или эукариотических хозяевах. Маркерами могут быть, например, устойчивость к тетрациклину или устойчивость к ампициллину. Для "клонирующего вектора" иногда используют слово "вектор". "Плазмида" представляет собой клонирующий вектор, обычно кольцевую ДНК, которая сохраняется и реплицируется автономно по меньшей мере в клетке одного хозяина.
Экспрессирующий вектор. Переносчик или вектор, подобный клонирующему вектору, но такой вектор, который поддерживает экспрессию клонированного в него гена после трансформации в хозяина. Этот клонированный ген обычно помещают под контроль (то есть оперативно соединяют) определенных регуляторных последовательностей, таких как промоторные последовательности, которые могут быть обеспечены вектором или рекомбинантной конструкцией клонированного гена. Регуляторные последовательности экспрессии будут варьировать в зависимости от того, предназначен ли этот вектор для экспрессии оперативно связанного гена в прокариотическом или в эукариотическом хозяине, и могут дополнительно содержать элементы транскрипции, такие как энхансерные элементы (активирующие последовательности, расположенные в направлении 3'-5', то есть в обратном направлении от гена) и терминирующие последовательности, и (или) сайты инициации трансляции и сайты терминации трансляции.
Хозяин. Хозяином называют клетку, прокариотическую или эукариотическую, которую используют в качестве реципиента и носителя рекомбинантного материала.
Хозяин данного изобретения. "Хозяин данного изобретения" представляет собой микробного хозяина, который в природе не продуцирует ксилит в значительных количествах во время ферментации из обычных источников углерода, иных чем D-ксилоза или D-ксилулоза, или их полимеры, или олигомеры, или их смеси, но сконструирован таким образом, что он может продуцировать ксилит из таких источников углерода согласно способам данного изобретения. Под "значительным количеством" подразумевают количество, которое достаточно для выделения ксилита в чистом виде, или количество, которое можно надежно измерить аналитическими способами, обычно применяемыми для анализа углеводов в микробном ферментационном бульоне.
Арабитолдегидрогеназа. Существуют два типа D-арабитолдегидрогеназ: образующая D-ксилолозу (EC 1.1.11) и образующая D-рубилозу. Образующие D-рубилозу дегидрогеназы обнаружены в дрожжах и грибах дикого типа. Образующие D-ксилулозу арабитолдигидрогеназы известны только в бактериях. Если нет других указаний, здесь имеют в виду образующую D-ксилулозу арабитолдегидрогеназу и называют ее арабитолдегидрогеназой.
Окислительная ветвь пентозофосфатного пути. "Окислительная ветвь пентозофосфатного пути" включает ту часть пентозофосфатного шунта, которая катализирует окислительные реакции, такие как реакции, катализируемые D-глюкозо-6-фосфатдегидрогеназой (EC 1.1.1.49) и 6-фосфо-D-глюконатдегидрогеназой (EC 1.1.1.44), и которая использует гексозные субстраты для образования пентозофосфатов. "Неокислительная" часть пентозофосфатного пути (которая также катализирует нетто-образование рибозы из D-глюкозы) характеризуется неокислительными реакциями изомеризации, например, реакциями, катализируемыми рибозо-5-фосфатизомеразой, D-рибулозо-5-фосфат-3- эпимеразой и трансальдолазой. См. Biological Chemistry, H.R. Mahler and E.H. Cordes, Harper and Row, publishers, New York, 1966, pp. 448-454.
Функциональное производное. "Функциональное производное" белка или нуклеиновой кислоты представляет собой молекулу, которая была химически или биохимически произведена из (получена из) такого белка или нуклеионовой кислоты и которая сохраняет биологическую активность (либо функциональную, либо структурную), которая является характеристикой нативных белка или нуклеиновой кислоты. Термин "функциональное производное" охватывает "фрагменты", "варианты", "аналоги" или "химические производные" молекулы, которые сохраняют желаемую активность нативной молекулы.
В применении здесь, молекула, которую называют "химическим производным" другой молекулы, если она содержит дополнительные химические части молекулы, в норме не являющиеся частью этой молекулы. Такие части молекулы могут улучшать растворимость молекулы, период биологического полупревращения и т.д. Такие части молекул могут уменьшать токсичность молекулы или элиминировать или аттенуировать (ослаблять) какое-либо нежелательное действие молекулы и т. д. Молекулярные части, способные медиировать такие действия, описаны в Remington's Pharmaceutical Sciences (1980). Способы соединения таких молекулярных частей с молекулой хорошо известны в данной области.
Фрагмент. Термин "фрагмент" молекулы, такой как белок или нуклеиновая кислота, относится к части нативной аминокислотной или нуклеотидной генетической последовательности и, в частности, к функциональным производным данного изобретения.
Вариант или аналог. Термин "вариант" или "аналог" белка или нуклеиновой кислоты относится к молекуле, в основном сходной по структуре и биологической активности с нативной молекулой, такой, которая кодируется функциональным аллелем.
Конструирование метаболических путей для биосинтеза ксилита.
Согласно данному изобретению нативные метаболические пути микробного хозяина манипулируются таким образом, чтобы уменьшить или элиминировать (исключить) использование углерода в целях других, чем образование ксилита. Все хозяева изобретения продуцируют ксилит в одной ферментационной стадии. В одном варианте хозяева данного изобретения могут обладать ксилитолдегидрогеназной (EC 1.1.9) активностью, достаточной для получения ксилита. Однако, как описано ниже, в тех хозяевах, в которых желательно перепроизводство ксилитолдегидрогеназной активности, в клетку хозяина могут быть трансформированы рекомбинантных гены, кодирующие ксилитолдегидрогеназу.
В практической реализации изобретения все хозяева данного изобретения отличаются способностью синтезировать ксилит из структурно неродственных источников углерода, таких как D-глюкоза, а не из D-ксилозы и (или) D-ксилулозы. Хозяева данного изобретения способны также секретировать синтезированный ксилит в среду.
В частности, в приведенных в качестве примеров и предпочтительных вариантах хозяева изобретения характеризуются наличием у них одного из двух путей. Во-первых, путь, в котором арабит является промежуточным продуктом в образовании ксилита, и, во-вторых, путь, в котором ксилулозо-5-фосфат направляется в образование ксилита через дефосфорилирование и восстановление. В соответствии с этим хозяева изобретения характеризуются по меньшей мере одним из следующих генетических изменений:
(1) ген, кодирующий белок, обладающий D-арабитолдегидрогеназной активностью (EC 1.1.1.11) был клонирован в хозяине, что обеспечивает, таким образом, превращение D-арабита в D-ксилозу (характеристика пути I); и (или)
(2) ген нативного хозяина, кодирующий транскетолазную активность, был инактивирован (характеристика пути II).
Кроме того, может быть выполнено множество других дополнительных модификаций хозяина, так чтобы усилить способности продуцирования ксилита такими хозяевами. Например, хозяева, описанные в (1) и (2), могут быть далее модифицированы таким образом, что:
(3) ген, кодирующий белок, обладающий ксилитолдегирогеназной активностью (EC 1.1.1.9), был клонирован в хозяине;
(3') ген нативного хозяина, кодирующий D-ксилулокиназу (EC 2.7.1.17), был инактивирован;
(4) ген, кодирующий белок, обладающий D-глюкозо-6-фосфат- дегидрогеназной активностью (EC 1.1.1.49), был клонирован в хозяине;
(4') ген, кодирующий белок, обладающий 6-фосфо-D-глюконат- дегидрогеназной активностью (EC 1.1.1.44), был клонирован в хозяине;
(5) ген, кодирующий белок, обладающий D-рибулозо-5-фосфат-3- эпимеразной (EC 5.1.3.1) активностью, был клонирован в хозяине.
В предпочтительном варианте хозяева данного изобретения обладают более чем одним из описанных выше генетических изменений. Например, в предпочтительном варианте углерод идет "прямо" (то есть в одной стадии) от D-арабита к D-ксилулозе и от D-ксилулозы "прямо" к ксилиту. Таким образом, в таком варианте хозяин данного изобретения изменен таким образом, что в хозяина были клонированы ген, кодирующий белок, обладающий образующей D-ксилулозу D-арабитолдегидрогеназной активностью, и ген, кодирующий ксилитолдегидронагеназу (EC 1.1.1.9). Следует отметить, что в то время как во многих вариантах, D-арабит синтезируется внутри клетки из других источников углерода хозяевам изобретения, D-арабит можно было бы добавить извне непосредственно к среде.
В другом предпочтительном варианте путь биосинтеза ксилита не содержит арабит в качестве промежуточного продута. Предпочтительнее углерод идет из D-ксилулозо-5-фосфата к D-ксилулозе и далее ко ксилиту. При применении D-глюкозы в качестве источника углерода углерод идет через окислительную часть пентозофосфатного пути, из D-глюкозы к D-глюкозо-6-фосфату- к 6-фосфо-D-глюконату - к D-рибулозо-5-фосфату. D-рибулозо-5-фосфат эпимеризуется далее до D-ксилулозо-5-фосфата, дефосфорилируется до D-ксилулозы и восстанавливается до ксилита.
Таким образом, хозяин данного изобретения для применения этого варианта является хозяином, в котором:
(a1) ген, кодирующий белок, имеющий D-глюкозо-6-фосфат-дегидрогеназную (EC 1.1.1.49) активность, был клонирован в хозяина или нативный ген хозяина избыточно экспрессируется; и/или)
(a2) ген, кодирующий белок, обладающий D-фосфо-D-глюконат-дегидрогеназной (EC 1.1.1.44) активностью, был клонирован в хозяина или нативный ген этого хозяина избыточно экспрессируется; и/или)
(a3) ген, кодирующий белок, обладающий D-рибулозо-5-фосфат-3- эпимеразной активностью, был клонирован в хозяина или ген этого хозяина избыточно экспрессируется; и/или)
(a4) ген, кодирующий белок, обладающий ксилитолдегидрогеназной (EC 1.1.1.9) активностью, был клонирован в хозяина или нативный ген этого хозяина избыточно экспрессируется;
(b) нативный ген транскетолазы был инактивирован; и (или)
(c) нативный ген хозяина, кодирующий ксилулокиназу (EC 2.7.1.17) было инактивирован.
Стадия дефосфорилирования (превращение D-ксилулозофосфата до D-ксилулозы) является единственной стадией, катализируемой ферментом, который не был охарактеризован в чистом виде. Однако предварительно было показано, что ферментная активность, ответственная за подобную стадию (превращение D-рибулозо-5-фосфата в D-рибулозу) в нативном образующем D-арабит пути осмофильных дрожжей, не является специфичной и способна также дефосфорилировать ксилулозо-5-фосфат (Ingram J. M. и W.A. Wood, J. Bacteriol 89: 1186 - 1194 (1965)). Мутация транскетолазы и избыточная экспрессия этих двух дегидрогеназ окислительного пентозофосфатного пути служит двойной цели. Во-первых, они могут увеличивать эффективность пути I путем увеличения количества рибулозо-5-фосфата в клетке и, следовательно, образования арабита и ксилита. Во-вторых, избыточное накопление ксилузозо-5-фосфата, который необходим для действия пути II, должно также вытекать из той же самой комбинации модификаций.
Таким образом, способы, использующие природно встречающийся путь, ведущий к образованию D-арабита из различных источников углерода и удлинение этого пути посредством еще двух реакций для превращения D-арабита в ксилит, не являются единственным возможным путем в данном изобретении. Могут быть сконструированы другие пути, ведущие ко ксилиту как конечному метаболическому продукту и не включающие в себя D-арабит в качестве промежуточного продукта. Таким образом, путь ко ксилиту от D-рибулозо-5-фосфата может быть реализован через более чем одну цепь реакций. D-рибулозо-5-фосфат может быть эффективно превращен в D-рибулозо-5-фосфат D-рибулозо-5-фосфат-3-эпимеразой и, если дальнейшее превращение D-рибулозо-5-фосфата предотвращается мутацией в гене транскетолазы, но накопленный D-рибулозо-5-фосфат может дефосфорилизоваться той же самой неспецифической фосфатазой, что и D-рибулозо-5-фосфат (Ingram, J.M. et al., J. Bacteriol. 89: 1186 - 1194 (1965)), и восстанавливаться в ксилит ксилитолдегидрогеназой. Реализация этого пути может далее требовать инактивации гена D-ксилулокиназы для того, чтобы уменьшить энергические потери, обусловленные бесполезной петлей: D-ксилулозо-5-фосфат ---> D-ксилулоза ---> D-ксилулозо-5-фосфат. Дополнительное генетическое изменение - введение и (избыточная) экспрессия гена D-рибулоксиназы (EC 2.7.1.47) могло бы уменьшить одновременное образование D-арабита такими штаммами путем улавливания D-рибулозы, продуцируемой неспецифической фосфатазой. D-рибулоза может превращаться опять в D-рибулозо-5-фосфат и далее в D-ксилулозо-5-фосфат.
Конструирование хозяев данного изобретения.
Способ генетического конструирования хозяев данного изобретения, согласно изобретению, облегчается путем выделения и частичного секвенирования чистого белка, кодирующего целевой фермент или клонированием генетических последовательностей, способных кодировать такой белок с применением технологии полимеразной цепной реакции (PCR); и путем экспрессии таких генетических последовательностей. В применении здесь, термин "генетические последовательности" относится к молекуле нуклеиновой кислоты (предпочтительно ДНК). Генетические последовательности, способные кодировать белок, получены из множества источников. Эти источники включают в себя геномную ДНК, кДНК, синтетическую ДНК и их комбинации. Предпочтительным источником геномной ДНК является геномная библиотека дрожжей. Предпочтительным источником кДНК является библиотека кДНК, приготовленная из мРНК дрожжей, росших в условиях, о которых известно, что они индуцируют экспрессию целевой мРНК или белка.
кДНК изобретения не содержит природно встречающихся интронов, если эта кДНК получена с применением зрелой мРНК в качестве матрицы. Геномная ДНК изобретения может содержать и может не содержать природно встречающиеся интроны. Кроме того, такая геномная ДНК может быть получена в ассоциации с 5' промоторным районом последовательностей гена и (или) с 3'-районом терминации транскрипции. Кроме того, такая геномная ДНК может быть получена в ассоциации с генетическими последовательностями, кодирующими 5' нетранслируемый район этой мРНК, и (или) с генетическими последовательностями, кодирующими 3' нетранслируемый район. В том смысле, что клетка хозяина может узнавать регуляторные сигналы транскрипции и(или) трансляции, ассоциированные с экспрессией мРНК и белка, 5' и (или) 3' нетранскрибируемые районы нативного гена и (или) 5' и (или) 3' нетранслируемые районы этой мРНК могут сохраняться и применяться для регуляции транскрипции и трансляции. Геномная ДНК может быть экстрагирована и очищена из какой-либо клетки хозяина, в частности, грибного хозяина, которая природно экспрессирует целевой белок способами, хорошо известными в данной области (например, см. Guide of Molecular Cloning Techniques, S.L. Berger et al., eds. Academic Precc (1987)). Предпочтительно применяемые препараты мРНК обогащены мРНК, кодирующей целевой белок, либо природно, путем выделения из клеток, продуцирующих большие количества этого белка, либо in vitro, путем применения способов, обычно используемых для обогащения препаратов мРНК специфическими последовательностями, такими как центрифугирование в градиенте сахарозы, или обоими путями.
Для клонирования в векторе такие подходящие препараты ДНК (геномной ДНК или кДНК) фрагментируют гидродинамически или расщепляют ферментами соответственно и лигируют в подходящие векторы для образования библиотеки рекомбинантных генов (геномной библиотеки или библиотеки кДНК).
Последовательность ДНК, кодирующая целевой белок или его функциональные производные, может быть встроена в ДНК вектор в соответствии с обычными способами, такими как получение тупых концов или выступающих концов для лигирования, расщепление рестриктирующими ферментами для обеспечения подходящих концов, заполнение липких концов, если нужно, обработка щелочной фосфатазой для того, чтобы избежать нежелательного соединения, и лигирование при помощи соответствующих подходящих лигаз. Способы для таких манипуляций описаны Maniatis, T. , (Maniatis, T. et al., Molecular Cloning (A Laboratory Manual), Cold Spring Harbor Laboratory, second edition, 1988) и хорошо известны в этой области.
Библиотеки, содержащие последовательности, кодирующие целевой ген, можно скринировать и целевую последовательность можно идентифицировать при помощи средств, которые специфически выбирают последовательность, кодирующую такой ген или белок, таких, например, как
a) гибридизация с подходящим зондом нуклеиновой кислоты (зондами), содержащим последовательность, специфическую для этой ДНК или этого белка, или
b) гибридизационно-выбранный трансляционный анализ, в котором нативную мРНК, гибридизующуюся с целевым клоном, транслируют in vitro и продукты трансляции характеризуются далее, или
с) если клонированные генетические последовательности сами способны экспрессировать мРНК, иммунопреципитация транслированного белкового продукта, продуцированного хозяином, содержащим этот клон.
Олигонуклеотидные зонды, специфические для определенного белка, которые могут быть использованы для идентификации клонов для этого белка, могут быть построены на основе знания аминокислотной последовательности этого белка или на основе знания последовательности нуклеиновой кислоты ДНК, кодирующей этот белок или родственный белок. Альтернативно, можно получить антитела против очищенных форм данного белка и использовать их для идентификации присутствия уникальных белковых детерминант в трансформантах, экспрессирующих целевой клонированный белок. Последовательность аминокислотных остатков в пептиде обозначается здесь либо путем применения их обычных обозначений из трех букв, либо путем применения обозначений из одной буквы. Перечень этих трехбуквенных и однобуквенных обозначений можно найти в учебниках, таких как Biochemistry, Lehninger, A. , Worth Publishers, New York, NY (1970). Если аминокислотная последовательность написана горизонтально, если нет других указаний, амино-конец предполагается на левом конце, а карбокси-конец находится на правом конце. Подобным образом, если нет иных указаний или сообщений в контексте, последовательность нуклеиновой кислоты представлена с 5'-концом слева.
Поскольку генетический код является вырожденным, более чем один кодон может использоваться для кодирования определенной аминокислоты (Watson, J.D. , In: Molecular Biology of the gene, 3rd Ed., W.A. Benjamin, Inc., Menlo Park, CA (1977), pp. 356 - 357). Пептидные фрагменты анализируют для идентификации последовательностей аминокислот, которые могут кодироваться олигонуклеотидами, имеющими самую низкую степень вырожденности. Это выполняют предпочтительно путем идентификации последовательностей, содержащих аминокислоты, которые кодируются только одним кодоном.
Хотя иногда аминокислотная последовательность может кодироваться только одной олигонуклеотидной последовательностью, часто аминокислотная последовательность может кодироваться любым из набора сходных олигонуклеотидов. Важно, что хотя все члены этого набора содержат олигонуклеотидные последовательности, способные кодировать один и тот же пептидный фрагмент и, следовательно, потенциально содержат ту же самую олигонуклеотидную последовательность, что и этот ген, который кодирует данный пептидный фрагмент, только один член этого набора содержит нуклеотидную последовательность, которая идентичная кодирующей экзон последовательности этого гена. Поскольку этот член присутствует внутри данного набора олигонуклеотидов и способен гибридизоваться с ДНК, даже в присутствии других членов этого набора, можно применять нефракционированный набор олигонуклеотидов тем же образом, каким применяли бы единственный олигонуклеотид, для клонирования гена кодирующего данный пептид.
С применением генетического кода один или несколько олигонуклеотидов могут быть идентифицированы из аминокислотной последовательности, каждый из которых был бы способен кодировать целевой белок. Вероятность того, что конкретный олигонуклеотид будет действительно составлять (образовывать) истинную кодирующую белок последовательность, можно оценить путем рассмотрения ненормального спаривания оснований и частоты, с которой конкретный кодон действительно используется (для кодирования конкретной аминокислоты) в эукариотических клетках. С применением "правил использования кодонов" можно идентифицировать одну олигонуклеотидную последовательность или набор олигонуклеотидных последовательностей, которые содержат теоретически "наиболее вероятную" нуклеотидную последовательность, способную кодировать эти белковые последовательности.
Пригодные для этого олигонуклеотиды или набор олигонуклеотидов, которые способны кодировать фрагмент определенного гена (или которые комплементарны такому олигонуклеотиду или набору олигонуклеотидов), могут быть синтезированы при помощи средств, хорошо известных в данной области (см. например, Synthesis and Application of DNA and RNA, S.A. Narang, ed., 1987, Academic Press, San Diego, CA) и применимы в качестве зонда для идентификации и выделения клона этого гена способами, известными в этой области. Способы гибридизации нуклеиновых кислот и идентификации клонов описаны Maniatis, T., et al. , in.: Molecular Cloning, A Laboratory Mannul, Cold Spring Harbor Laboratories, Cold Spring Harbor, NY (1982) и Hames, B.D., et al., in: Nucleic Acid Hybridization, A Practical Approach, IRL Press, Washington, DC (1985)). Те члены описанной выше библиотеки генов, которые способны к такой гибридизации, затем анализируют для определения протяженности и природы кодирующих последовательностей, которые они содержат.
Для облегчения детектирования целевой кодирующей последовательности ДНК описанный выше зонд ДНК метят деткектируемой группой. Такой детектируемый группой может быть любой материал, имеющий детектируемое физическое или химическое свойство. Такие материалы были хорошо разработаны в области гибридизации нуклеиновых кислот и в большинстве случаев любая метка, применимая в таких способах, может быть использована для данного изобретения. Особенно применимы радиоактивные метки, такие как 32P, 3H, 14C, 35S, 125I и т.п. Может быть применима любая радиоактивная метка, которая обеспечивает адекватный сигнал и имеет достаточный период полураспада. В случае одноцепочечного олигонуклеотида он может быть радиоактивно помечен при помощи киназных реакций. Альтернативно, в качестве гибридизационных зондов нуклеиновых кислот применимы также полинуклеотиды, которые метят нерадиоактивным маркером, таким как биотин, фермент или флуоресцентная группа.
Таким образом, выяснение частичной последовательности белка делает возможной идентификацию теоретической "наиболее вероятной" последовательности ДНК или набора таких последовательностей, способной кодировать такой пептид. Путем конструирования олигонуклеотида, комплементарного этой теоретической последовательности (или путем конструирования набора олигонуклеотидов, комплементарных набору "наиболее вероятных" олигонуклеотидов), получают молекулу ДНК (или набор молекул ДНК), способную функционировать в качестве зонда (зондов) для идентификации и выделения клонов, содержащих ген.
В альтернативном способе клонирования гена библиотеку получают с применением экспрессирующего вектора путем клонирования ДНК или, более предпочтительно, кДНК, полученной из клетки, способной экспрессировать данный белок, в экспрессирующем векторе. Затем эту библиотеку скринируют на ее члены, которые экспрессируют целевой белок, например, путем скрининга этой библиотеки при помощи антител к этому белку.
Таким образом, обсужденные выше способы способны идентифицировать генетические последовательности, которые способны кодировать белок или биологически активные или антигенные фрагменты этого белка. Для дальнейшей характеристики таких генетических последовательностей и для получения рекомбинантного белка желательно экспрессировать белки, которые эти последовательности кодируют. Такая экспрессия идентифицирует те клоны, которые экспрессируют белки, обладающие свойствами целевого белка. Такими свойствам являются, среди прочих, способность специфически связывать антитела, способность вызывать образование антител, способных связываться с нативным, нерекомбинантным белком, способность обеспечивать в клетке ферментативную активность, являющуюся свойством этого белка, и способность обеспечивать неферментативную (но специфическую) функцию реципиентной клетки.
Последовательность ДНК может быть укорочена известными в данной области средствами для выделения целевого гена из района хромосомы, который содержит больше информации, чем это необходимо для утилизации этого гена в хозяевах данного изобретения. Например, для расщепления последовательности полной длины в желаемом положении можно использовать расщепление рестриктазами. Альтернативно или в добавление можно использовать нуклеазы, которые расщепляют от 3'-конца молекулы ДНК, для расщепления определенной последовательности до укороченной формы, причем желательную длину затем идентифицируют и очищают полученную последовательность при помощи гельэлектрофореза и секвенирования ДНК. Такие нуклеазы включают в себя, например, Экзонуклеазу III и Bal31. Другие нуклеазы также хорошо известны в этой области.
В практической реализации данного изобретения в качестве модели использовали осмофильные дрожжи Z.rouxii. Z.rouxii совместим с производством пищевых продуктов, так как его традиционно используют в Японии для приготовления соевого соуса. Эти дрожжи были описаны, например, в: The Yeasts, A Taxonomic Study, Kreger-van Rij (ed.), Elsevier Science publishers B.V., Amsterdam 3984, где эти дрожжи описаны на страницах 462 - 465. Другие продуцирующие D-арабит дрожжи, такие как Candida polymorpha, Torulopsis candida, Candida tropicalis, Pichia farinosa, Torulaspora hansenii и т.д., так же как и продуцирующие D-арабит грибы, подобные Dendryphiella salina или Sshizophyllum commune, также могут применяться в качестве хозяйских организмов для целей данного исследования.
Известно, что ферменты, окисляющие D-арабит до D-ксилулозы (EC 1.1.1.11), встречаются в бактериях, но не в дрожжах или грибах. Для целей данного изобретения Klebsiella teriigena представляет собой предпочтительный источник гена D-арабитолдегидрогеназы (образующей D-ксилулозу), поскольку этот микроорганизм является непатогенной почвенной бактерией и имеет высокоиндуцируемую D-арбитолдегидрогеназную активность. Штамм Klebsiella terrigena Php1, применяемый в примерах, был получен от K.Haahtela, Helsinki University. Выделение этого штамма описано Haahtela et al., Appl. Env. Microbiol. 45: 563 - 570 (1983)). Клонирование гена D-арабитолдегидрогеназы может быть легко достигнуто конструированием генетической библиотеки хромосомной ДНК. K. terrigena и в подходящем векторе, например, в хорошо известной и коммерчески доступной плазмиде pUC19. Эту библиотеку трансформируют в один из многих штаммов E.coli, которые способны использовать D-ксилулозу, но не D-арабит в качестве единственного источника углерода. Штамм E. coli SCS1, доступный в Stratagene, является примером подходящего штамма. Затем трансформанты высевают на среду, содержащую D-арабит в качестве единственного источника углерода, и выделяют клоны, способные расти на этой среде. Кодирующий район D-арабитолдегидрогеназы K. terrigena может быть легко выделен в форме фрагмента 1,38 т.п.н. BclI-ClaI и слит с подходящими промоторной и терминирующей последовательностями транскрипции. ADCI промотор Saccharomyces cerevisiae и терминатор транскрипции этих дрожжей являются примерами регуляторных элементов транскрипции, пригодными для целей данного изобретения при применении дрожжей Z. rouxii в качестве организма-хозяина. Последовательность ADCI доступна из Gen Bank.
Хотя большинство дрожжей и грибов обладают ксилитолдегидрогеназным геном (EC 1.1.1.9), избыточная экспрессия этого гена обычно необходима для приведения в действие данного изобретения. Клонирование XYL2 гена Pichia stipitis, кодирующего ксилитолдегидрогеназу (EC 1.1.1.9), может быть легко достигнуто технологией полимеразной цепной реакции с применением опубликованной информации о нуклеотидной последовательности XYL2 гена (Kotter et al., Сurr. Genet. 18: 493 - 8500 (1990)). Ген может быть введен в другие виды дрожжей без каких-либо модификаций и экспрессирован под контролем его собственного промотора или промотор может быть заменен на другой сильный дрожжевой промотор.
Генетически стабильные трансформанты можно сконструировать с векторными системами или трансформационными системами, в результате чего желаемая ДНК интегрируется в хромосому хозяина. Такая интеграция может происходить de novo внутри клетки или ей можно содействовать путем трансформации вектором, который функционально встраивается в хромосому хозяина, например, фагом, ретровирусными векторами, транспозонами или другими элементами ДНК, которые усиливают интеграцию последовательностей ДНК в хромосомы.
Гены, кодирующие D-арабитолдегидрогеназу и ксилитолдегидрогеназу (EC 1.1.1.9) под контролем подходящих промоторов, могут быть объединены в одной плазмидной конструкции и введены в клетки хозяйского продуцируемого D-арабит организма путем трансформации. Природа такого плазмидного вектора будет зависеть от хозяйского организма. Так, в предпочтительном варианте данного изобретения для вектора Z. roxii применяют включение ДНК криптической плазмиды pSR1 (Ushio, K. et al., J. Ferment Technol 66: 481 - 488 (1988)). Для других видов дрожжей и грибов, для которых неизвестны автономно реплицирующиеся плазмиды, может быть использована интеграция генов ксилитолдегидрогеназы (EC 1.1.1.9) и D-арабитолдегидрогеназы в хромосому хозяина. Направление интеграции к локусу рибосомной ДНК (ДНК, кодирующей рибосомную РНК) является предпочтительным способом получения интеграции с высоким числом копий и высокого уровня экспрессии этих двух дегидрогеназных генов. Такое нацеливание может быть достигнуто обеспечением рекомбинантных последовательностей ДНК на ремомбинантной конструкции, достаточной для направления интеграции в этот локус. Генетическими маркерами, применяемыми для трансформации продуцирующих D-арабит микроорганизмов, являются преимущественно доминантные маркеры, придающие устойчивость к различным антибиотикам, таким как гентамицин или флеомицин, или к тяжелым металлам, таким как медь, и т.п. Селектируемый маркерный ген может быть либо непосредственно соединен с генными последовательностями ДНК, которые должны быть экспрессированы, либо введен в ту же самую клетку путем котрансформации.
Кроме введения генов D-арабитолдегидрогеназы и ксилитолдегидрогеназы (EC 1.1.1.9), можно использовать другие генетические модификации для конструирования новых продуцирующих ксилит штаммов. Так, гены, кодирующие ферменты оксилительного пентозофосфатного пути, могут быть избыточно экспрессированы для увеличения скорости синтеза предшественника D-арабита D-рибулозо-5-фосфата. Также ген, кодирующий транскетолазу, фермент, катализирующий катаболизм пентулозо-5-фосфатов или пентозо-5-фосфатов, может быть инактивирован при помощи общепринятого мутагенеза или способами разрушения гена, ведущими к повышенному накоплению 5-углеродных сахарофосфатов. Инактивирование гена D-ксилулокиназы может увеличить выход ксилита вследствие элиминирования потери D-ксилулозы или фосфорилировании. Комбинирование инактивирующей транскетолазу мутации с избыточной экспрессией D-рибулозо-5-эпимеразы можно использовать для создания отливающегося типа пути образования ксилита, в котором D-арабит не является промежуточным продуктом.
Для экспрессии целевого белка и (или) его активных производных необходимы транскрипционные и трансляционные сигналы, узнаваемые подходящим хозяином. Клонированные кодирующие последовательности, полученные описанными выше способами и предпочтительно в двухцепочечной форме, могут быть оперативно соединены с последовательностями, контролирующими транскрипционную экспрессию в экспрессирующем векторе, и введены в клетку хозяина, прокариотическую или эукариотическую, для получения рекомбинантного белка или его функционального производного. В зависимости от того, какая цепь кодирующей последовательности оперативно соединена с последовательностью, контролирующей транскрипционную экспрессию, можно также экспрессировать антисмысловую РНК или ее функциональное производное.
Экспрессия белка в разных хозяевах может привести к различным пост-трансляционным модификациям, которые могут изменять свойства этого белка. Предпочтительно, данное изобретение предусматривает экспрессию белка или его функционального производного в эукариотических клетках и, в частности, в дрожжах.
Предположительно, молекула нуклеиновой кислоты, такая как ДНК, "способна экспрессировать" полипептид, если она содержит регуляторные последовательности экспрессии, которые содержат транскрипционную регуляторную информацию, и такие последовательности "оперативно соединены" с нуклеотидной последовательностью, кодирующей полипептид.
Оперативная связь является такой связью, в которой последовательность соединяется с регуляторной последовательностью (или последовательностями) таким образом, чтобы поставить экспрессию этой последовательности под влияние или контроль регуляторной последовательности. Две последовательности ДНК (например, кодирующая последовательность и последовательность промоторного района, соединенная с 5'-концом этой кодирующей последовательности) будут соединены оперативно, если индукция промоторной функции приводит к транскрипции мРНК, кодирующей целевой белок, и если природа связи между двумя последовательностями (I) не изменяет рамки считывания кодирующей
последовательности, (2) не мешает способности регуляторных последовательностей экспрессии направлять экспрессию данного белка, антисмысловой РНК, или (3) не мешает способности матрицы ДНК к транскрибированию. Таким образом, промоторный район является оперативно соединенным с последовательностью ДНК, если он способен воздействовать на транскрипцию этой последовательности ДНК.
Точная природа регуляторных районов, необходимых для экспрессии генов, может варьировать в зависимости от вида или типов клеток, но, как правило, содержит обязательно 5'-нетранскрибируемую и 5'-нестранслируемую (Некодируемые) последовательности, участвующие в инициации транскрипции и инициации трансляции, соответственно, такие как TATA-блок, копирующая последовательность, CAAT последовательность и т.п. В частности, такие 5'-нетранскрибируемые регуляторные последовательности содержат район, который содержит промотор для транскрипционного контроля оперативно связанного гена. Такие последовательности регуляции транскрипции могут также содержать энхансерные последовательности или активаторные (3'-5') последовательности, если желательно.
Экспрессия белка в эукариотических хозяевах, таких как дрожжи, требует применения регуляторных районов, функциональных в таких хозяевах, и предпочтительно дрожжевых регуляторных систем. В зависимости от природы хозяина можно использовать большое разнообразие регуляторных последовательностей транскрипции и трансляции. Предпочтительно, эти регуляторные сигналы связаны в их нативном состоянии с конкретным геном, который способен к высокому уровню экспрессии в клетке-хозяине.
В эукариотах, где транскрипция не связана с трансляцией, такие регуляторные районы могут обеспечивать (и могут не обеспечивать) инициаторный кодон (AUG) метионина, в зависимости от того, содержит ли клонированная последовательность метионин. Такие районы, как правило, содержат промоторный район, достаточный для направления инициации синтеза РНК в клетке хозяина. Можно предпочтительно использовать промоторы из дрожжевых генов, кодирующих продукт мРНК, способный к трансляции, и, в частности, можно применять сильные промоторы, при условии, что они также функционируют как промоторы в клетке хозяина. Предпочтительными сильными дрожжевыми промоторами являются промотор GAL1 гена, промоторы гликолитических генов, таких как ген фосфоглицерокиназы (PGK), или промотор конститутивной алкогольдегидрогеназы (ACD1) (Ammerer, G. Meth. Enzymol. 101C: 192 - 201 (1983); Aho, FFEBS Lett. 291: 45 - 49 (1991)).
Как хорошо известно, трансляция эукариотической мРНК инициируется на кодоне, кодирующем первый метионин. По этой причине предпочтительно гарантировать, что связь между эукариотическим промотором и последовательностью ДНК, кодирующей целевой белок или его функциональное производное, не содержит расположенного внутри нее кодона, способного кодировать метионин. Присутствие таких кодонов приводит к образованию слитого белка (если AUG кодон находится в той же рамке считывания, что и кодирующая белок последовательность) или к мутации со сдвигом рамки (если AUG кодон находится не в одной рамке считывания с кодирующей белок последовательностью).
Могут быть выбраны регуляторные сигналы инициации транскрипции. которые делают возможными репрессию или активацию, так что можно модулировать экспрессию оперативно соединенных генов. Интересны регуляторные сигналы, которые чувствительны к температуре, так что изменением температуры можно репрессировать или инициировать экспрессию, или эти сигналы регулируются химически, например, метаболитом. Если нужно экспрессировать антисмысловую РНК, трансляционные сигналы не являются необходимыми.
Если нужно, нетранскрибируемые и (или) нетранслируемые районы, расположенные в направлении 3' от последовательности, кодирующей целевой белок, могут быть получены описанными выше способами клонирования. 3'-нетранскрибируемый район может быть сохранен ради его элементов регуляторной последовательности терминации транскрипции; 3'-нетранслируемый район может быть сохранен ради его элементов регуляторной последовательности терминации трансляции или ради тех элементов, которые направляют полиаденилирование в эукариотических клетках. Если нативные регуляторные последовательности экспрессии не функционируют удовлетворительно в клетке хозяина, то они могут быть заменены последовательностями, функциональными в клетке хозяина.
Кроме того, векторы этого изобретения могут содержать другие оперативно связанные регуляторные элементы, такие как элементы ДНК, которые придают устойчивость к антибиотикам, или затравки репликации для сохранения вектора в одной или нескольких клетках хозяина.
В другом предпочтительном варианте, в частности для Z. rouxii, вводимую последовательность встраивают в плазмидный вектор, способный автономно реплицироваться в реципиенте-хозяине. Для этой цели могут быть применены многочисленные векторы.
Важными факторами в выборе конкретного плазмидного или вирусного вектора являются легкость, с которой реципиентные клетки, содержащие этот вектор, могут распознаваться и отбираться среди тех реципиентных клеток, которые не содержат этот вектор; число копий вектора, которое желательно в конкретном хозяине; желательно ли, чтобы вектор мог использоваться в клетках хозяев разных видов.
Предпочтительные дрожжевые плазмиды будут зависеть от хозяина. Для Z. rouxii предпочтительны основания на нативных криптических плазмидах векторы pSR1 (Toh, E. et al., J. Bacteriol. 151: 1380 - 1390 (1982), pSB1, pSB2, pSB3, или pSB4 (Toh, E. et al., Gen. Microbiol. 130: 2527 - 2534 (1984)). Плазмида pSRT303D (Jearnpipatkul, A., et al., Mol. Gen. Genet. 206: 88 - 84 (1987) является примером плазмидного вектора, применимого для дрожжей Zygosaccharomyces.
После приготовления векторной или ДНК последовательности, содержащей векторную конструкцию (конструкции) для экспрессии, конструкцию (конструкции) ДНК вводят в подходящую клетку хозяина любым из множества пригодных способов, в том числе путем трансформации. После введения вектора реципиентные клетки выращивают в селективной среде, которая селективна для роста содержащих вектор клеток. Экспрессия клонированной последовательности (последовательностей) гена приводит к образованию целевого белка или к образованию фрагмента этого белка. Эта экспрессия происходит непрерывно в трансформированных клетках или регулируемым способом, например, при помощи индуцирования экспрессии.
Для конструирования хозяев данного изобретения, которые изменяются таким образом, что они больше не экспрессируют определенный генный продукт, может быть проведен сайт-направленный мутагенез при помощи способов, известных в данной области, таких как разрушение гена (Rothstein, R.S., Meth. Enzymol. 101C: 202 - 211 (1983)).
Получение ксилита.
При использовании рекомбинантных продуцирующих арабит дрожжей, предпочтительно осмофильных, в качестве хозяев данного изобретения они могут расти в среде с высоким осмотическим давлением, например, в среде, содержащей 10-60%, D-глюкозы и предпочтительно 25% D-глюкозы. ("Нормальная" среда обычно содержит только 2-3% глюкозы). Среда с высоким осмотическим давлением индуцирует образование D-арабита в штаммах дикого типа осмофильных дрожжей, таких как Z. rouxii. Эту культуральную среду рекомбинантного и контрольного (дикого типа) штаммов анализируют согласно известным способам в различные временные точки культивирования на присутствие ксилита. В условиях культивирования, не оптимизированных для максимального выхода D-арабита, экспериментальный штамм Z. rouxii ATCC 13356 [pSRT(AX)-9] продуцировал и секретировал в культуральные среды как ксилит, так и D-арабит. В культуральной среде контрольного штамма был обнаружен только D-арабит. Выход ксилита в первых экспериментах (см. табл. 4 в примере 4) был приблизительно 7,7 г/л после 48 часов культивирования.
Ксилит мог быть очищен из среды хозяйских клеток данного изобретения в соответствии с любым известным в этой области способом. Например, US 5081026, включенный здесь в виде ссылки, описал хроматографическое отделение ксилита от дрожжевых культур. Таким образом, из стадии ферментации ксилит может быть очищен от культуральной среды при помощи хроматографических стадий, описанных в US 5081026, с последующей кристаллизацией.
После описания в общем виде данного изобретения то же самое станет более понятным при ссылке на определенные конкретные примеры, которые включены здесь для иллюстрации и не являются ограничивающими изобретение, если нет других указаний.
Пример 1. Клонирование бактериального гена D-арабитолдегидрогеназы Klebsiella terrigena Php1 (полученная от K. Haahtela, Helsinki University, см. Haahtela et al., Appl. Env. Microbiol. 45: 563 - 570 (1983) выращивали в 1 л LB среды (1% триптон, 0,5% дрожжевой экстракт 1% NaCl) в течение ночи при 30oC. Бактериальные клетки (приблизительно 5 г) собирали центрифугированием, промывали один раз в TE (10 мМ трис-HCl, 1 мМ ЭДТА, pH 7,5) и ресуспендировали в TE, содержащем 1% додецилсульфат натрия и 200 мкг/мл протеиназы K. Суспензию инкубировали при 37oC в течение 30 мин и затем экстрагировали один раз равным объемом фенола и два раза хлороформом. 3 М ацетат натрия (1/10 по объему) и этанол (3 объема) добавляли и осажденные нуклеиновые кислоты собирали центрифугированием и перерастворяли в 5 мл TE. РНК удаляли центрифугированием раствора через 25 мл 1 М MgCl2 в течение ночи при 30000 об/мин в Ti 50.2 роторе Beckmann. Хромосомная ДНК K. terrigena, полученная этим способом, имела средний размер фрагментов более 50000 п.н. (bp). Эту ДНК затем расщепляли рестриктирующей эндонуклеазой Sau3A в буфере поставщика (Boehringer) при соотношении фермент: ДНК 5 E/мг до тех пор, пока средний размер фрагментов ДНК не уменьшался приблизительно до 5-10 т.п.н. (оценивали при помощи агарозного гель-электрофореза). Продукт расщепления фракционировали при помощи электрофореза через 0,6% агарозный гель (20 х 10 х 0,6 см) в TBE буфере (0,09 М трис-борная кислота. 1 мМ ЭДТА, pH 8,3) при 5 в/см в течение ночи, вырезали лунку в агарозной пластинке в положении, соответствующем размеру фрагмента приблизительно 5 т.п.н., фиксировали вдоль лунки кусок диализной мембраны и продолжали электрофорез до тех пор, пока все фрагменты, большие 5 т.п.н., не адсорбировались на мембране. Плазмидную ДНК pUC19 (купленную в Pharmacia) расщепляли рестриктирующей эндонуклеазой BamH1 и бактериальной щелочной фосфатазой с применением буфера и условий реакции поставщика. Линейную форму pUC19 очищали при помощи препаративного гель-электрофореза с применением мембранного способа электроэлюции, описанного выше, и лигировали с 5-15 т.п.н. фракцией хромосомной ДНК Klebsiella terrigena. Лигированную смесь использовали для трансформации компетентных клеток E. Coli SCS1 (приобретенной в Stratagene) для выработки устойчивости к ампициллину. В этом эксперименте получили приблизительно 10000 рекомбинантных клонов. Объединенные клетки из чашек для трансформации распределяли на чашки с минимальной средой, содержащей D-арабит (1%) в качестве единственного источника углерода. После двух дней инкубирования при 37oC получили несколько колоний. Плазмидную ДНК выделяли из двух (наиболее быстро растущих) этих клонов и использовали для повторной трансформации штамма E.coli HB 101, который не способен катаболизировать D-арабит, но способен использовать D-ксилулозу. Все трансформанты оказались положительными в отношении использования D-арабита на полученных из Difco чашках с содержанием D-арабит агаром McConkey, тогда как все контрольные клоны (тот же самый штамм, трансформированный pUC19) были отрицательными. Рестрикционный анализ показал, что два изолированных клона содержали идентичные плазмиды. Одну из двух изолированных плазмид (названную pARL2) применяли для дальнейшей характеристики. Рестрикционная карта клонированного 9,5 т.п.н. (приблизительно) фрагмента ДНК K. terrigena в pARL2, несущего ген D-арабитолдегидрогеназы, представлена на фиг. 1.
Пример 2. Экспрессия бактериального гена D-арабитолдегидрогеназы в дрожжах.
Из исходного клона ДНК 9,5 т.п.н. K. terrigena, содержащего ген D-арабитолдегидрогеназы, фрагмент Sac1-HinDIII приблизительно 1,8 т.п.н. был субклонирован при помощи общепринятых способов рекомбинантных ДНК, и было обнаружено, что он содержит ген D-арабитолдегидрогеназы. Плазмида, содержащая этот фрагмент ДНК в векторе pUC19 (pADH, где ADH обозначает D-арабитолдегидрогеназу), была выделена из штамма E.coli JM110, расщеплена рестриктирующими эндонуклеазами Bcl1 и ClaI и фрагмент ДНК 1,38 т.п.н. был выделен при помощи препаративного агарозного гель-электрофореза. Этот фрагмент ДНК обрабатывали фрагментом Кленова ДНК-полимеразы I в присутствии всех четырех дезоксинуклеотидтрифосфатов и лигировали с дрожжевым экспрессирующим вектором pAAH 5 (Ammerer, Meth. Enzymol. 101: 192 - 203 (1983)), который разрезали HindIII и обрабатывали фрагментом Кленова (фиг. 2). Полученная экспрессирующая плазмида, pYARD, представляет собой челночный вектор E.coli - Saccharomyces cerevisiea, содержащий бактериальную (E.coli) затравку репликации и ген устойчивости к ампициллину, дрожжевую (S. cerevisiae) затравку репликации из 2 мкм ДНК и дрожжевой (S. cerevisiae) ген LEU2 для отбора в дрожжах. Эта экспрессионная кассета содержит дрожжевой промотор алкогольдегидрогеназы I (ADCI) и терминатор транскрипции (ADCI), фланкирующие и оперативно связанные с геном D-арабитолдегидрогеназы K. terrigena.
Штамм Saccharomyces cerevisiae GRF18 (MATα, leu2-3, 112m his3-11, 15) и штамм S. cerevisiae DBY746 (ATCC 44773; MATα, leu22/, 112, his3-A1 ura 3-52 trp1-189) были использованы в качестве хозяев для трансформации описанным выше экспрессирующим D-арабитолдегидрогеназу вектором pYARD с теми же самыми результатами. Трансформацию проводили по стандартному способу с хлоридом лития (Ito et al. , Bacteriol. 153: 163 - 160 (1983)) с применением LEU2 маркера pYARD для отбора трансформантов. Трансформанты росли в жидкой культуре в минимальной среде: 0,67% дрожжевого азотистого основания ("Difco:), 2% D-глюкозы, 100 мг/л гистидина и триптофана, при 30oC в течение ночи при качании. Клетки собирали центрифугированием, суспендировали в минимальном объеме буфера с 0,1 М фосфатом калия, pH 6,8, содержащего 1 мМ НАД+ и разрушали стеклянными бусинами 0,5 мм в Beat beater apparatus ("Biospec products") в течение 6 мин с охлаждением на льду. D-арабитолдегидрогеназную активность измеряли, как описано выше. Экстракт клеток K. terrigena, росших на D-арабите, использовали в качестве положительного контроля в этих экспериментах, а DBY746, трансформированный pAAH5, использовали в качестве отрицательного контроля. Результаты, представленные в табл. 1, показывают, что ген D-арабитолдегидрогеназы, выделенный из K. terrigena, эффективно экспрессировался в дрожжах.
Пример 3. Конструирование дрожжевых векторов для избыточной экспрессии генов ксилитолдегидрогеназы и D-арабитолдегидрогеназы.
Известную нуклеотидную последовательность дрожжевого (Pichia stipitis) гена, XYL2, кодирующего ксилитолдегидрогеназу (Kotter et al., Curr. Genet. 18: 493 - 500 (1990)), использовали для синтеза олигонуклеотидов для клонирования этого гена при помощи полимеразной цепной реакции. Были сконструированы два олигонуклеотида: CGAATTCTAGACCACCCTAAGTCGTCCC (5'-олигонуклеотид) (SEQ ID N1) и TTCAAGAATTCAAGAAACTCACGTGATGC (3'-олигонуклеотид) (SEQ ID N2) для включения удобных рестрикционных сайтов Xbal и EcoRI при 5' - и 3'-концах продукта PCR. 5'-олигонуклеотид гибридизируется при положении 1-24 XYL2 и 3'-олигонуклеотид гибридизуется при положении 1531 - 1560 в соответствии с нумерацией, используемой Kotter et al., Curr. Genet. 18: 493 - 500 (1990).
Pichia stipitis CBS6054 (Centraalbureau voor Schimmelcultures, Oosterstraat 1, PO Box 273, 3710 AG Baarn, The Netherlands) выращивали в течение ночи в среде YEPD (1% дрожжевой экстракт, 2% пептон, 2% D-глюкоза), клетки собирали центрифугированием, промывали один раз 1 М раствором сорбита, содержащим 1 мМ ЭДТА, pH 7,5, ресуспендировали в том же самом растворе и расщепляли при помощи Lyticase (Sigma). Расщепление контролировали мониторингом оптической плотности при 600 нм 1:100 разведения клеточной суспензии в 1% SDS. Расщепление заканчивали, когда эта величина падала до приблизительно 1/7 исходной величины. Суспензию сферопластов лизировали в 1% додецилсульфате натрия (SDS) и обрабатывали 200 мкг/мл протеиназой K при 37oC в течение 30 мин. После одной фенольной и двух экстракций хлороформом нуклеиновой кислоты осаждали этанолом и перерастворяли в небольшом объеме TE буфера. Целостность хромосомной ДНК проверяли при помощи электрофореза в агарозном геле. Средний размер фрагментов ДНК был более 50 т.п.н., PCR проводили с применением Taq ДНК-полимеразы (Boehringer) в буфере поставщика. Температурный цикл был: 93oC - 30 с, 55oC - 30 с, 72oC - 60 с. Продукт PCR экстрагировали хлороформом, осаждали этанолом и расщепляли EcoRI и Xbal при стандартных условиях. После очистки на агарозном геле фрагмент ДНК клонировали в разрезанную Xbal и EcoRI (плазмида pUC(XYL2)). Последующий рестрикционный анализ подтвердил, что рестрикционная карта клонированного фрагмента соответствует нуклеотидной последовательности гена XYL2 P. stipitis.
Дрожжевые плазмиды для избыточной экспрессии D-аработолдегидрогеназы и ксилитодегидрогеназы были сконструированы, как показано фиг. 3 и 4. Для изменения фланкирующих сайтов рестрикции экспрессирующей D-арбитолдигидрогеназу кассеты плазмиды pYARD всю кассету вырезали расщеплением BamHI и клонировали в расщепленную BamHI плазмиду pUC18. Полученную плазмиду pUC(YARD) расщепляли при помощи SalI и EcoRI и только фрагмент ДНК 2,0 т.п. н. выделяли препаративным гель-электрофорезом. Этот фрагмент лигировали с фрагментом ДНК HindIII-EcoRI 1,6 т.п.н., выделенным из плазмиды pUC(XYL2), и фрагмент 6,6 т.п.н. дрожжевого (E. coli) челночного вектора pJDB207 (Beggs, J. D. Nature 275: 104 - 109 (1978)) переваривали при помощи HindIII и SalI. Плазмиду pJDB(AX)-16 выделяли после трансформации E. coli описанной выше смесью лигирования. Эта плазмида способна реплицироваться как в E. coli, так и в Saccharomyces cerevisiae. В S. cerevisiae она направляет синтез высокого уровня как D-арабитолдегидрогеназы, так и ксилитолдегидрогеназы. Плазмиду pSRT(AX)-9 синтезировали лигированием фрагмента SalI 4,7 т.п.н. из плазмиды pJDB(AX)-9 и линейной формы плазмиды pSRT303D (Jearnpipatkul et al., Mol. Gen. Genet. 206: 88 - 94 (1987)), полученной частичным гидролизом при помощи SalI.
Пример 4. Конструирование дрожжевых штаммов, секретирующих ксилит.
Zygosaccharomayces rouxii ATCC 13356 трансформировали плазмидой pSRT(AX)-9 слегка измененным описанным ранее способом (Ushio, K. et al., J. Ferment. Technol. 66: 481 - 488)). Клетки Z. rouxii росли в течение ночи в среде YEPD (давая культуру с оптической плотностью при 600 нм 3-5), собирали низкоскоростным центрифугированием, промывали дважды в 1 М сорбите, 1 мМ ЭДТА pH 7,5, ресуспендировали в 1/5 исходного объема культуры того же раствора, содержащего 1% 2-меркаптоэтанол, и расщепляли при комнатной температуре литиказой (Sigma). После расщепления разводили подходящую аликвоту клеточной суспензии в 1%-ном растворе додецилсульфата натрия и измеряли оптическую плотность разведенной пробы при 600 нм. Когда эта величина падала до 1/7 от исходной, расщепление заканчивали путем охлаждения суспензии на льду и промыванием (10 мин, 1000 об/мин, центрифугирование при 0oC) раствором сорбита до тех пор, пока не исчезал запах меркаптоэтанола. Сферопласты промывали один раз холодным раствором 0,3 М хлорида кальция в 1 М сорбите и ресуспендировали в том же растворе приблизительно в 1/4 исходного объема культуры. 200 мкл аликвоты этой суспензии и 10 - 20 мкг плазмидной ДНК смешивали и инкубировали при 0oC в течение 40 мин. 0,8 мл охлажденного на льду 50% раствора PEG-6000, содержащего 0,3 М хлорид кальция, добавляли к суспензии сферопластов и инкубирование на холоду продолжали в течение 1 ч. Сферопласты концентрировали центрифугированием при 4000 об/мин в настольной центрифуге, ресуспендировали в 2 мл YEPD, содержащей 1 М сорбит, и оставляли для регенерации на ночь при комнатной температуре. Это регенерированные клетки высевали на чашки YEPD, содержащие 50-100 мкг/мл гентамицина, и инкубировали при 30oC в течение 4-6 дней.
Трансформанты росли в жидкой среде YEPD, клеточные экстракты готовили, как описано для S. cerevisiae (пример 2) и измеряли активности D-арабитолдегидрогеназы и ксилитолдегидрогеназы. Результаты этих измерений сравнивали с подобными измерениями, сделанными в других организмах в табл. 2. Они показывают, что оба гена эффективно экспрессировались в Z. rouxii.
Клетки Z. rouxii ATCC 13356, трансформированные pSRT(AX)-9, росли в течение 2 дней в 50 мл YEPD, содержащей 50 мкг/мл гентамицина, собирали центрифугированием и использовали для инокуляции 100 мл YEPD, содержащей 25% D-глюкозы и 50 мкг/мл гентамицина. Эта культура росла в колбе на 1000 мл на ротационном шейке (200 об/мин) при 30oC. В другом варианте (Эксперимент 2) использовали ту же самую среду без дрожжевого экстракта (низко-фосфатная среда). Содержание ксилита в культуральном бульоне анализировали стандартной HPLC и газовой хроматографией. Результаты представлены в табл. 3. В культуральной среде нетрансформированного штамма Z. rouxii, росшего в той же самой среде (без гентамицина), не обнаружили ксилита.
С применением подобного подхода можно сконструировать штаммы, продуцирующие ксилит из других источников углерода. Например, Candida tropicalis способен превращать н-алканы в D-арабит (Hattori, K. and Suzuki T., Agric. Biol. Chem. 38: 1875 - 1881 (1974)) с хорошим выходом. Фрагмент SalI 4,7 т. п. н. из плазмиды pJDB(AX)-9 может быть введен в плазмиду pCUl (Hass, L. et al. , J. Bacter. 172: 4571 - 4577 (1990)) и использован для трансформации штамма C. tropicalis SU-2 (ura 3). Альтернативно, ту же самую экспрессионную кассету можно трансформировать в фототрофный штамм C. tropicalis на плазмидном векторе, несущем доминантный селективный маркер.
Пример 5. Конструирование объединенного вектора доминантного отбора для экспрессии арабитолдегидрогеназы и ксилитолдегидрогеназы и трансформации Torulopsis candida.
Хромосомную ДНК выделяли из T. candida (ATCC 20214) способом, подобным стандартному способу, примененному для S. cerevisiae. Однако получение сферопластов T. candida требовало более высокой концентрации литиказы (Sigma) - приблизительно 50000 E на 10 г клеток и длительного периода инкубирования - от нескольких часов до ночи, при комнатной температуре, для достижения эффективного лизиса клеточных стенок. Буфер для получения сферопластов содержал 1 мМ ЭДТА, pH 8,1 М сорбит и 1% меркаптоэтанол. Сферопласты промывали три раза тем же буфером (без меркаптоэтанола) и лизировали в 15 мл 1% SDS. Две фенольные экстракции проводили сразу же после лизиса клеток и ДНК осаждали добавлением 2 объемов этанола и центрифугированием (5 мин при 10000 об/мин). ДНК промывали дважды 70% этанолом и сушили под вакуумом. Эту ДНК растворяли в 5 мл 10 мМ трис-HCl буфера, содержащего 1 мМ ЭДТА, добавляли РНКазу до концентрации 10 мкг/мл и раствор инкубировали в течение 1 ч при 37oC. Целостность ДНК подтверждали электрофорезом в агарозном геле при помощи нерасщепленной ДНК вируса лямбда в качестве стандарта молекулярной массы.
200 мкг хромосомной ДНК T. candida расщепляли HindIII и EcoRI, продукт расщепления наносили в лунку 6 см на 0,7% (8 х 15 х 0,8 см) препаративном агарозном геле и разделяли при помощи электрофореза. Полоску 1 см вырезали из геля и блоттировали на положительно заряженную найлоновую мембрану (Boehringer 1209 299). Блот гибридизовали с фрагментом ДНК 10 т.п.н. рДНК Zygosaccharomyces bailii, вырезанного при помощи SalI из плазмиды pAT68 (K. Sugihara et al., Agric. Biol. Chem. 50(6): 1503 - 151 (1986). Зонд метили и блоты проявляли с применением набора GIG DNA Labeling and Detection Kit (Boehringer 1093 657) согласно инструкциям изготовителя. Наблюдали три гибридизационные полосы, соответствующие фрагментам ДНК приблизительно 4,5, 2,7 и 1,1 т.п.н. С применением этого блока в качестве стандарта полосу, соответствующую самому большому гибридизующемуся фрагменту ДНК (4,5 т.п.н.), вырезали из оставшейся части препаративного геля, ДНК электроэлюировали и лигировали с pUC19, расщепленной EcoRI и HindIII.
Смесь для лигирования использовали для трансформации E. coli. Трансформированные бактерии высевали на заряженную найлоновую мембрану (Bio-Rad 162-0164), лежащую на поверхности агара чашки, содержащей среду LB с ампициллином. После 24 ч инкубирования при 37oC мембрану поднимали и проводили in situ лизис бактериальных колоний согласно инструкциям изготовителя. Не было необходимости в чашках-репликах, поскольку E. coli проникает сквозь этот тип мембраны и после поднятия фильтра имеется видимый след каждой бактериальной колонии на поверхности агара. Мембрану гибридизовали с тем же самым фрагментом рДНК Z.bailii с применением того же набора DIG Detection, описанного выше. Идентифицировали ряд положительных клонов (приблизительно 2-5% от всех клонов). Рестрикционный анализ плазмидных минипрепаратов из 8 гибридизационно-положительных клонов и 4 гибридизационно-отрицательных клонов проводили с применением смеси EcoRI, HindIII и EcoRV. Все гибридизационно-положительные клоны продуцировали идентичные распределения рестрикционных фрагментов (с характерными фрагментами 0,55 и 1,5 т.п.н.), тогда как эти распределения гибридизационно-отрицательных клонов все были разными. Плазмидную ДНК из одного из гибридизационно-положительных клонов выделили в препаративном масштабе и назвали PTCrDNA. Был сделан вывод, что этот клонированный кусок был фрагментом рДНК T.candida, поскольку 1) он сильно гибридизовался с рДНК Z.bailii и 2) он был клонирован из частично обогащенного продукта расщепления хромосомной ДНК T.candida с высокой частотой (копийностью) (известно, что рДНК представлена в геноме дрожжей примерно 100 копиями). Частичная рестрикционная карта этого клонированного фрагмента ДНК показана на фиг. 5.
Плазмиду, объединяющую рДНК фрагмента T.candida с доминантным селектируемым маркером, конструировали следующим образом. Плазмиду pUT332 (Gatignol, A., et al., Gene 91: 35 - 41 (1990)) расщепляли HindIII и Kpnl, фрагмент ДНК 1,3 т. п. н. выделяли агарозным электрофорезом и лигировали с фрагментом HindIII-EcoRI 4,5 т.п.н. из PTCrDNA и pUC19, расщепленным EcoRI и Kpnl (фиг. 6). Смесь для лигирования трансформировали в E.coli и клон, несущий плазмиду pTC (PHLE), идентифицировали рестрикционным анализом.
pTC (PHLE) частично расщепляли SalI и линейную форму этой плазмиды очищали электрофорезом на агарозном геле. Ее лигировали с фрагментом SalI 56 т. п. н., выделенным из pJDB(AX)-16 (Пример 2). Плазмиду pTC(AX) идентифицировали среди клонов, полученных после трансформации этой лигирующей смеси и E.coli (фиг. 6). Эта плазмида содержит следующие функциональные элементы:
a) бактериальный ген устойчивости к флеомицину под контролем дрожжевого промотора и терминатора транскрипции, позволяющий прямой отбор дрожжевых клеток, трансформированных этой плазмидой;
b) кусок рДНК T.candida, обеспечивающий мишень для гомологичной рекомбинации с хромосомой T.candida и улучшающий эффективность трансформации;
c) экспрессионную кассету для генов арабитолдегидрогеназы и ксилитолдегидрогеназы, обеспечивающую синтез этих двух ферментов пути превращения арабита в ксилит.
Пример 6. Трансформация T.candida и анализ продуцирования ксилита.
Плазмиду pTC(AX) применяли для трансформации штамма Torulopsis candida ATCC 20214. T.candida выращивали в течение 36 ч в среде YEPD, содержащей 10% глюкозу. Клетки собирали центрифугированием (2000 об/мин, в течение 10 мин при 4oC) и промывали три раза стерильным раствором 1 М сорбита. Осадок клеток суспендировали в равном объеме холодного 1 М сорбита, аликвоты по 200 мкл смешивали с pUT(AX)DNA (20-100 мкг) и затем переносили в охлажденные на льду кюветы для электропорации (2 мм electrode gap, то есть с промежутком 2 мм между электродами) и подвергали электропорации с применением устройства Invitrogen Electroporator apparatus со следующими установленными параметрами: напряжение 1800 В, емкость 50 микрофарад, параллельное сопротивление 150 Ом. Клетки переносили в 2 мл YEPD, содержащего 1 М сорбит, и инкубировали в течение ночи при 30oC на шейкере. Трансформированные клетки собирали центрифугированием при низкой скорости и высевали на чашки, содержащие среду YEPD, титрованную до pH 7,5 и содержащую 30 мкг/мл флеомицина. Чашки инкубировали при 30oC в течение 7-10 дней. Большая часть колоний, которые проявились в течение этого времени, представляла собой "фоновые" мутанты, поскольку подобное число колоний появилось также на контрольных чашках (содержащих клетки, обработанные подобным образом, но без добавления ДНК). Для того, чтобы отличить истинные трансформанты от спонтанных мутантов, выделяли хромосомную ДНК из 72 индивидуальных дрожжевых колоний при помощи снижающейся процедуры (scale down) для выделения хромосомной ДНК T.candida, описанной выше. 10 мкг каждого из этих препаратов ДНК расщепляли смесью EcoRI и BamHI. Продукты расщепления разделяли на 1% агарозных гелях и затем подвергали блоттингу на положительно заряженную найлоновую мембрану, как описано в примере 5. Блоты гибридизовали с ДНК из плазмиды pADH (пример 2), которая содержит арабитолдегидрогеназные последовательности и pUS последовательности, но не фрагменты ДНК дрожжевого происхождения (чтобы избежать гибридизации между возможными гомологичными дрожжевыми последовательностями. Зонд метили и блоты проявляли с применением DIG DNA Labeling and Detection Kit (Boehringer 1093657). Только один клон (T.candida: pTC(AX)) с гибридизационным сигналом, совместимым со структурой трансформирующей плазмиды (три полосы в районе 2-3 т. п. н. ) был обнаружен, что свидетельствует об очень низкой эффективности трансформации. Положительный сигнал был обнаружен еще для одного клона, однако положение единственной гибридизирующейся полосы (примерно 5 т.п.н.) указывало, что только фрагмент pTC(AX) интегрировался в дрожжевую хромосому или в сайте интеграции произошла некоторая реаранжировка. Мы предложили, что эта плазмида интегрировалась в хромосому T.candida. Это предположение находится в согласии с наблюдением, что после роста в неселективной среде и клонирования все клоны T.candida: :pTC(AX) сохраняют свой устойчивый к флеомицину фенотип.
Трансформант T.candida: :pTC(AX) выращивали в среде YEPD, содержащей 10% глюкозу, в течение 36 ч и арабитолдегидрогеназную и ксилитолдегидрогеназную активность измеряли, как описано в примере 4. Результаты представлены в табл. 4.
Активность ксилитолдегидрогеназы не увеличивалась значительно над уровнем активности эндогенной ксилитолдегидрогеназы T.candida. Активность кодируемой плазмидой арабитолдегидрогеназы (EC 1.1.1.11) было трудно отличить от активности эндогенной арабитолдегидрогеназы, образующей рибулозу (EC 1.1.1). Единственным определенным заключением из этого эксперимента было то, что уровень экспрессии обоих ферментов был гораздо ниже, чем в Z.rouxii. Попытки увеличить число плазмидных копий на геном (копийность) и уровень экспрессии этих двух дегидрогеназ интегрированной плазмиды путем культивирования T.candida: : pTC(AX) на средах с увеличенными концентрациями флеомицина были неуспешными.
Образование ксилита T.candida: :pTC(AX) тестировали после выращивания на YEPD, содержащей 10% глюкозу, в течение 5-7 дней. В трех отдельных экспериментах этот трансформант продуцировал 1,1; 1,6 и 0,9 г/л ксилита, в то время как в культуральной среде T.candida дикого типа не был обнаружен ксилит при помощи HPLC. Предел детектирования этого аналитического способа, примененного нами, ниже 0,1 г/л. Таким образом, можно сделать вывод, что образование ксилита T.candida: : pT(AX) действительно определяется этой плазмидой.
Пример 7. Трансформация Candida polymorpha генами арабитолдегидрогеназы и ксилитолдегидрогеназы.
Для того, чтобы ввести гены арабитолдегидрогеназы и ксилитолдегидрогеназы в Candida polymorpha, был выделен мутант по гену оротидинфосфатдекарбоксилазы (далее называемый URA3) при помощи модификации способа Boeke et al. , (Boeke, J.D. et al., Mol. Gen. Genet. 197: 345 - 346 (1984)). Штамм C. polymorpha ATCC 20213 выращивали в течение 24 ч в среде YEPD, клетки собирали центрифугированием (2000 об/мин, 10 мин), промывали водой два раза и суспендировали в трех объемах стерильного содержащего 0,1 М фосфат натрия буфера, pH 7,0. Этилметансульфонат добавляли до 1% концентрации и клетки инкубировали 2 ч при комнатной температуре. Реакцию останавливали перенесением клеток в 0,1 М раствор тиосульфата натрия и промыванием их три раза стерильной водой. Мутагенезированные клетки переносили в 0,5 л YEPD и выращивали при 30oC с качанием в течение двух дней. Дрожжи собирали центрифугированием, промывали два раза водой и переносили в 1 л среды, содержащей 0,7% дрожжевого азотистого основания (Difco), 2% глюкозу (среда SC) и инкубировали на ротационном шейкере в течение 24 ч при 30oC. К культуре добавляли 1 мг нистатина и инкубирование продолжали в течение 4 ч. Обработанные нистатином клетки отделяли от среды центрифугированием, промывали два раза водой и переносили в 1 л среды SC, содержащей 50 мг/л урацила. Клетки инкубировали на ротационном шейкере в течение 5 дней и затем высевали на чашки со средой SC, содержащей 50 мг/л урацила и 1 г/л фтороротовой кислоты. После инкубирования чашек в течение двух недель при 30oC приблизительно 400 устойчивых к фтороротовой кислоте колоний было получено. Все эти колонии тестировали на урациловую ауксотрофию. Были выделены пять урацил-зависимых клона. Однако три клона действительно росли на не содержащей урацила среде, хотя и при пониженной скорости. Два клона (названные C.polymorpha U-2 и C.polymorpha U-5), имеющие явный урацил-зависимый фенотип, использовали для экспериментов по трансформации.
Клонирование гена C.polymorpha URA3 было достигнуто при помощи общепринятой стратегии. Хромосомную ДНК выделяли по способу, описанному в примере 2. ДНК частично расщепляли Sau3A и фракционировали на агарозных гелях. Собирали отдельные фракции и распределение их по молекулярному размеру проверяли при помощи аналитического гель-электрофореза. Фракции в диапазоне 5-10 т.п.н. применяли для экспериментов по клонированию. Вектор, примененный для конструирования библиотеки, pYEp13 (Broach et al. , Gene 8: 121 - 133 (1979)), содержит LEU2 ген S. cerevisiae, 2 микрон затравку репликации и уникальный сайт рестрикции для BamHI. Вектор разрезали BamHI, очищали электрофорезом в агарозном геле и дефосфорилировали бактериальной щелочной фосфатазой. Несколько независимых векторных препаратов и условий лигирования, изменяющих отношение вектора к инсерции и реакционный объем, тестировали в небольших экспериментах для оптимизации по наибольшему числу трансформантов и наивысшему проценту рекомбинантных клонов (путем изучения при помощи рестрикционного анализа плазмидных минипрепаратов из произвольных клонов). Лигирования большого масштаба выполняли с использованием оптимизированных условий и трансформировали продукт лигирования в Штамм E.coli XLI-BLUE. Сконструированная библиотека содержала приблизительно 15000 первичных трансформантов и приблизительно 90% из них содержали инсерции.
Дрожжевой штамм DEY746 (MATalpha, leu2-3, 112, his3-A1, trp1-289, ura3-52) был трансформирован библиотекой генов C.polymorpha. Каждый пул библиотеки трансформировался в S. cerevisiae отдельно с применением приблизительно 20 мкг плазмидной ДНК и посевом на чашку с добавлением урацила, триптофана и гистидина (то есть с применением только лейциновой селекции). На чашку получили 3000 - 10000 дрожжевых трансформантов. Способом реплик эти дрожжевые трансформанты высевали на чашки с минимальной средой, к которой добавляли триптофан и гистидин (чашки - минус урацил). Контрольные реплики были на гистидин-гистидине (чашки - минус урацил). Были также сделаны контрольные реплики на чашках "минус гистидин" и чашках "минус триптофан". Один из четырех урацил-независимых клонов появлялся почти на каждой чашке. Были также получены гистидин-независимые и триптофан-независимые изоляты, однако число этих изолятов было в 2-3 раза ниже, чем для изолятов URA3.
Плазмиды из шести урацил-независимых клонов были помещены в E.coli, выделены в препаративном масштабе и использованы для повторной трансформации DBY746 до лейциновой прототрофии. 10-20 произвольных колоний из каждой трансформации проверяли на зависимость от урацила. Пять из шести "спасенных" плазмид трансформировали DBY746 до фенотипа Leu+ Ura+. Рестрикционный анализ этих пяти плазмид выявил очень сходные рестрикционные распределения фрагментов. Эти распределения были слишком сложными для того, чтобы создать не оставляющую сомнений рестрикционную карту клонированного фрагмента. Однако, было обнаружено, что расщепление при помощи HindIII дает во всех клонах фрагмент, покрывающий большую часть инсерционной ДНК (приблизительно 4,5 т. п. н. ). Этот фрагмент из одного изолята C.polymorpha, pCP291, был очищен электрофорезом в агарозном геле и субклонирован в pJDB(AX), частично расщепленную HindIII. Структура плазмиды pCPU(AX), выделенной в результате этого клонирования, показана на фиг. 7A.
Была сделана попытка трансформации как C.polymorpha U-2, так и C.polymorpha U-5 с применением тех же самых условий электропорации, которые описаны в примере 6. Электропарированные клетки, после качания в течение носи в YEPD, содержащей 1 М сорбит, высевали на чашки со средой SC. После 10 дней инкубирования при 30oC приблизительно 100 колоний обнаружили на чашке, содержащей C.polymorpha U-2, трансформированного pCPU(AX), в то время как только 14 колоний было на контрольной чашке (содержащей подобным образом обработанные клетки этого штамма без добавленной ДНК). C.polymorpha U-5 не обнаружила значительного эффекта по сравнению с контролем без ДНК в опыте по трансформации. Три произвольно выбранные клона из чашки трансформации C.polymorpha U-2 сначала высевали штрихами на чашку со свежей средой SC и эти штрихи использовали для инокуляции 100 мл-культур с содержащей 15% глюкозу средой YEPD. Контрольную культуру засевали C.polymorpha U-2. После инкубирования на ротационном шейкере (200 об/мин) при 30oC в течение 10 дней содержание ксилита в культуральной среде анализировали при помощи HILC. Результаты этого эксперимента представлены в табл. 5.
Наблюдали значительные вариации в уровне образования ксилита между различными клонами. Это может быть следствием интегрирования плазмиды pCPU(AX) при различных локусах хромосомы C. polymorpha. Штамм C.polymorpha U-2: : pCP(AX)-2, возможно, является ревертантом, а не истинным трансформантом. Однако эти эксперименты ясно показали, что путь арабит-ксилит может быть введен также в C.polymorpha.
Пример 8. Клонирование ферментов окислительного пентозофосфатного пути и их избыточная экспрессия в осмофильных дрожжах.
Первый фермент окислительного пентозофосфатного пути, D-глюкозо-6-фосфатдегидрогеназа, кодируется в S. cerevisiae геном ZWF1. Последовательность этого гена известна (Nogae T., and Johnston, M. Gene 96: 161 - 19 (1990)); Thomas D. et al., The EMBO J. 10: 547 - 553 (1991)). Этот ген, содержащий полный кодирующий район и 450 т.п.н. 3'-некодирующего района, был клонирован при помощи PCR с применением двух олигонуклеотидов: CAGGCCGTCGACAAGGATCTCGTCTC 5'-олигонуклеотид) (SEQ 1D N 3) и AATTAGTCGACCGTTAATTGGGGCCACTGAGGC (3'-олигонуклеотид) (GEQ 1 N 4). 5'-олигонуклеотид гибридизуется при положениях 982 - 1007 и 3'-олигонуклеотид гибридизуется при положении 3523 - 3555 в нумерации D-глюкозо-6-фосфатдегидрогеназы, описанной Nogae T, and Johnston, M. Gene 96: 161 - 19 (1990). Хромосомную ДНК выделяли из штамма S. cerevisiae GRF18 способом, описанным в примере 3. Параметры PCR были такими же, что и в примере 3. Амплифицированный фрагмент ДНК, содержащий ZWF1 ген, расщепляли SalI и клонировали в pUC19, расщепленную той же рестриктазой, что приводило к образованию плазмиды pUC(ZWF). Идентичность клонированного гена проверяли рестрикционным анализом.
Второй фермент пентозофосфатного пути, дегидрогеназа 6-фосфоглюконовой кислоты, кодируется в E.coli gnd геном. Нуклеотидная последовательность этого гена известна (Nasoff, M.S. et al., Gene 27: 253 - 264 (1984)). Для клонирования gnd гена из E.coli хромосомную ДНК выделяли из штамма E.coli HB101 способом, идентичным способу, примененному для выделения ДНК Klebsiella terrigena (пример 1). Были сконструированы олигонуклеотиды (GCGAAGCTTAAAAATGTCCAAGCAACAGATCGGCG [SEQ 1D N 5] и GCGAAGCTTAGATTAATCCAGCCATTCGGTATGG [SEQ 1D N 6] для PCR амплификации gnd гена, чтобы амплифицировать только кодирующий район и ввести сайты HindIII сразу же слева от инициирующего кодона и непосредственно справа от стопкодона. 5'-олигонуклеотид гибридизуется при положениях 56 - 78 и 3'-олигонуклеотид гибридизуется в положении 1442 - 1468 в нумерации дегидрогеназы 6-фосфоглюконовой кислоты, описанной Nasoff, M.S. et al., Gene 27: 253 - 264 (1984). Амплифицированный фрагмент ДНК расщепляли HindIII и лигировали с расщепленным HindIII вектором pUC19. Десять независимых явно идентичных клонов полученной плазмиды pUC(gnd) объединяли вместе. Это было сделано, чтобы избежать возможных проблем, связанных с ошибками последовательности, которые могут быть введены во время PCR амплификации. Кодирующий район gnd гена сливали с ADCI промотором S. cerevisiae и терминатором транскрипции этого организма путем перенесения фрагмента HindIII 1,4 т.п.н. из пула pUC(gnd) в экспрессирующий вектор PAAH5 (Ammerer, Meth. Enzymol. 101: 192-203 (1983)). Несколько независимых клонов полученной плазмиды pAAH(gnd) трансформировали в штамм S. cerevisiae GRF18 способом с применением хлорида лития (Ito et al., J. Bacteriol. 153: 163 - 168 (1983)) и активность дегидрогеназы 6-фосфоглюконовой кислоты измеряли в трансформаторах. Во всех трансформантах активность дегидрогеназы 6-фосфоглюконовой кислоты была в несколько раз выше, чем в нетрансформированном хозяине, свидетельствуя о том, что бактериальная дегидрогеназа 6-фосфоглюконовой кислоты может эффективно экспрессироваться в дрожжах. Клон pAAH(gnd), дающий наивысшую активность в дрожжах, был выбран для дальнейших конструирований. Клонирование ZWF1 и gnd генов, а также конструирование плазмиды pAAH(gnd) показаны на фиг. 8 и 8a.
Для избыточной экспрессии одновременно как гена D-глюкозо-6-фосфатдегидрогеназы, так и гена 6-фосфо-6-глюконатдегидрогеназы в осмофильном дрожжевом хозяине была сконструирована плазмида pSRT(ZG). Способ конструирования этой плазмиды показан на фиг. 9. Экспрессионную кассету 6-фосфо-D-глюконатдегидрогеназы из плазмиды pAAG(gnd) переносили в виде фрагмента ДНК BamHI 3,1 т.п.н. в расщепленную при помощи BamHI плазмиду pUC19. Полученную плазмиду pUC(ADHgnd) расщепляли Sacl и Xbal и фрагмент ДНК 3,1 т.п.н. очищали при помощи электрофореза в агарозном геле. Этот фрагмент одновременно лигировали с двумя другими фрагментами ДНК: Фрагментом 2,5 т.п.н. pUC(ZWF), полученным расщеплением при помощи Sacl и частичным расщеплением при помощи Pstl, и векторным фрагментом плазмиды pSRT(AX)-9 (пример 3), расщепленным при помощи Pstl и Xbal. Структуру полученной плазмиды pSRT(ZG) подтверждали рестрикционным анализом. После трансформации Z.rouxii ATCC 13356 плазмидой pSRT(ZG) (по способу, описанному в примере 4) в трансформированном штамме измеряли активность D-глюкозо-6-фосфатдегидрогеназы и 6-фторо-D-глюконатдегидрогеназы. Оба фермента были приблизительно в 20 раз более активны в трансформированном штамме, чем в нетрансформированном контроле (табл. 6). Подобные способы могут быть использованы для достижения избыточной экспрессии двух этих генов, кодирующих ферменты окислительной части пентозофосфатного пути, в других видах дрожжей. Однако, поскольку нет векторов, способных сохраняться в виде экстрахромомных плазмид в большинстве дрожжевых видов, отличающихся от Saccharomyces или Zygosaccharomyces, интегративная (с включением в хромосомы) трансформация является единственным применимым способом для таких хозяев. Предпочтительным типом интегральных векторов являются векторы, нацеленные для интеграции в локусе рибосомной ДНК, поскольку векторы этого типа обеспечивают интеграцию высокого числа копий и, следовательно, высокий уровень экспрессии (Lopes, T.S. et al., Gene 79: 199 - 206 (1989)).
Пример 9. Конструирование транскетолазных мутантов в дрожжах.
Транскетолазные дрожжевые мутанты могут быть наиболее легко получены способом сайт-направленного разрушения генов, хотя применимы также общепринятые способы химического мутагенеза. Гомологичный ген транскетолазы, клонированный из дрожжевых видов, в
которых желательно получить мутацию, обычно необходим для применения технологии разрушения гена, хотя иногда можно применять также гетерологичный клон из близкородственных видов. Последовательность гена транскетолазы из S. cerevisiae известна (Fletcher, T.S., and Kwee, I.L., EMBL DNA sequence library, ID:SCTRANSK, accession number M63302). При помощи этой последовательности ген транскетолазы S. cerevisiae может быть клонирован при помощи PCR. Олигонуклеотиды AGCTCTAGAAATGACTCAATTCACTGACATTGATAAGCTAGCCG (SEQ 1D N 7) и GGAGAATTCAGCTTGTCACCCTTATAGAATGCAATGGTCTTTTG (SEQ 1D N 8) и хромосомную ДНК из S. cerevisiae (выделенную как описано выше) применяли для амплификации фрагмента ДНК, содержащего ген транскетолазы. 5'-олигонуклеотид гибридизуется при положениях 269 - 304 и 3'-олигонуклеотид при положении 2271 - 2305 в нумерации транскетолазы, описанной в (Fletcher, T.S., and Kwee. I.L., EBVL DNA sequence library, ID:SCTRANSK, accession number M63302). Этот фрагмент расщепляли Xbal и EcoRI и клонировали в pUC19, расщепленную теми же ферментами. Рестрикционный анализ полученной плазмиды pUS (ТКТ) подтвердил идентичность этого клонированного фрагмента ДНК. Эту плазмиду разрезали Bgll1 и Clal и большой фрагмент ДНК очищали при помощи агарозного электрофореза. Этот фрагмент лигировали с двумя фрагментами ДНК, выделенными из тплазмиды pUT332 (Gattingol, A. et al., Gene 91: 35 - 41 (1990)): Clal-Patl фрагментом, несущим ген URa3 и 3'-часть гена устойчивости к блеомицину, и BamHI-PstI фрагментом ДНК, несущим дрожжевой TEFI промотор (фактор элонгации транскрипции 1) и 5'-часть кодирующей последовательности гена устойчивости к блеомицину. После трансформации E.coli описанной выше смесью лигирования и рестрикционного анализа плазмидных клонов была выделена плазмида pTRT (BLURA). Эта плазмида содержит кодирующую последовательность гена транскетолазы S. cerevisiae, в которой фрагмент 90 п.н. между сайтами ClaI и BglII заменен фрагментом pUT332, содержащим два маркера, селектируемых в S.cerevisiae: ген URA3 и ген устойчивости к блеомицину под контролем TEFI промотора и CYC1 терминатора транскрипции. Эту плазмиду применяли для трансформации штамма S. cerevisiae DBY746 (ATCC 44773; MATα his3A1 leu2-3, 112 ura3-52 trpL-289) для устойчивости к флеомицину (10 мкг/мл флеомицина в среде YEPD) по способу с применением хлорида лития (Ito et al., J. Bacteriol. 153: 163 - 168 (1983)). Трансформанты тестировали на урациловую прототрофию и на способность расти на D-ксилулозе. Пять клонов URA3, три из которых обнаружили пониженный рост на D-ксилулозе, выращивали в 100 мл-культурах, клеточные экстракты готовили качанием со стеклянными бусинами и транскетолазную активность измеряли в неочищенных экстрактах. Тест выполняли в 0,1 М глицил-глициновом буфере pH 7,6, содержащем 3 мМ MgCl2, 0,1 мМ тиаминпирофосфат, 0,25 мМ НАДН и 0,2 мг/мл бычьего сывороточного альбумина. Непосредственно перед измерением 3 мкл раствора, содержащего 0,5 М D-ксилулозо-5-фосфат и 0,5 М рибозо-5-фосфат, добавляли к 1 мл указанного выше буфера с последующим добавлением 7,5 E триозофосфатизомеразы и 1,5 E D-глицерофосфатдегидрогеназы (оба фермента из Sigma). Реакцию инциировали добавлением подходящей аликвоты неочищенного экстракта и регистрировали снижение оптической плотности при 340 нм. Клеточный экстракт штамма DBY746 применяли в качестве контроля. Транскетолазную активность в неочищенном экстракте DBY746 и в двух трансформантах с незамедленным ростом на D-ксилулозе можно было легко измерить при приблизительно 0,25 E/мг белка. Три трансформанта с замедленным ростом на D-ксилулозе имели транскетолазную активность ниже предела детектирования нашего способа (по меньшей мере в 20 раз ниже, чем в случае дикого типа). Активности D-глюкозо-6-фосфатдегидрогеназы и 6-фосфо-6-глюконатдегидрогеназы также измеряли в качестве контроля на возможную инактивацию ферментов во время приготовления клеточных экстрактов. Активности этих ферментов были очень сходными во всех шести штаммах. Следовательно, был сделан вывод, что три клона, которые плохо росли на D-ксилулозе, содержали мутацию в гене транскетолазы. Рост штаммов с разрушенным геном транскетолазы несколько замедлялся также на синтетической среде, не содержащей ароматических аминокислот фенилаланина и тирозина, хотя этот эффект был меньше выражен, чем влияние этой мутации на использование D-ксилулозы.
Гены транскетолазы из других видов дрожжей можно легко клонировать путем комплементации транскетолазной мутации в штаммах S.cerevisiae, описанной выше. Предпочтительно, клонирование может быть выполнено путем конструирования библиотеки генов отличающегося от Saccharamyces штамма дрожжей в подходящем векторе (например, в хорошо известной плазмиде YEp13), трансформации этой библиотеки в штамм S.cerevisiae, несущий транскетолазную мутацию (например, мутанты, полученные описанным выше разрушением гена) и отбором трансформантов с замедленным ростом на D-ксилулозе. Эта плазмидная ДНК может быть выделена методом "спасения" из таких D-ксилулоза-положительных трансформантов и применена для трансформации того же самого реципиентного штамма. Все клоны из такой трансформации должны быть способны хорошо расти на D-ксилулозе. Транскетолазную активность можно измерить в этих трансформантах и ее появление на значительном уровне может служить доказательством идентичности клонированного гена. Дополнительное и окончательное доказательства можно получить секвенированием коротких участков клонированной дНК и нахождением кусков последовательности, гомологичных последовательности гена транскетолазы S. cerevisiae, или демонстрацией гибридизации клонированного фрагмента ДНК с аутентичным транскетолазным клоном из S. cerevisiae. Альтернативно процедура клонирования может быть основана на гибридизации ДНК как первичном способе для отбора клонов, содержащих транскетолазный ген из библиотеки генов отличающегося от Saccharomyces вида дрожжей. Фрагмент гена транскетолазы S.cerevisiae можно использовать в качестве зонда для эксперимента с гибридизацией колоний или бляшек, и клоны, дающие наиболее сильный гибридизационный сигнал, могут быть анализированы далее частичным секвенированием.
Какой бы способ ни был использован для клонирования гена транскетолазы из выбранного вида дрожжей, последующие стадии получения мутации в гене транскетолазы в этих дрожжах являются теми же самыми. Клонированный фрагмент ДНК должен быть охарактеризован конструированием частичной рестрикционной карты и предпочтительно определением местоположения кодирующего района гена транскетолазы. Затем кусок ДНК, который может функционировать в качестве селектируемого маркера, встраивают во фрагмент ДНК, содержащий ген транскетолазы, не ближе, чем на расстоянии нескольких сотен п.н. от каждого из концов этого фрагмента. Для многих дрожжевых видов можно, например, использовать в качестве селектируемого маркера кассету, содержащую бактериальный ген устойчивости к флеомицину под контролем сильного дрожжевого промотора, такую как описанная выше кассета из плазмиды pUT332. Существенно, что встраивание фрагмента ДНК, несущего селектируемый маркер, выполняют таким образом, что кодирующий район гена транскетолазы либо разрушается встроенной ДНК, либо, предпочтительно, встроенный фрагмент ДНК вытесняет часть кодирующего района. Такую конструкцию ДНК можно затем использовать для разрушения хромосомной копии гена транскетолазы в отобранных дрожжах согласно способу, похожему на способ, описанный выше для получения транскетолазной мутации в S. cerevisiae. Может быть применен любой подходящий способ трансформации, предпочтительными способами являются трансформация протопластов и электропорация. Отбор клонов, несущих разрушенный ген транскетолазы, может быть выполнен по способу, подобному описанному выше для S.cerevisiae. В качестве альтернативного способа или в дополнение к другим способам может быть применен анализ структуры хромосомного района транскетолазы при помощи гибридизации по Саузерну.
Пример 10. Клонирование гена D-рибулокиназы.
Предпочтительный способ клонирования гена D-рибулокиназы сходен со способом, описанным в примере 1 для клонирования D-арабитолдегидрогеназы. Известно, что ген D-рибулокиназы в некоторых бактериях, таких как E.coli или Klebsiella aerogenes, является частью оперона утилизации рибита (Loviny T., et al., Biochem. J. 230: 579 - 585 (1985)). Также известно, что штаммы E.coli B не содержат этого оперона и поэтому не способны использовать рибит в качестве источника углерода. Поэтому штамм E.coli B (например, обычные лабораторные штаммы HB101 и JM103 или штаммы, которые могут быть трансформированы с высокой эффективностью, такие как SCS1 или XLI-Blue из Stratagene) можно трансформировать библиотекой генов, утилизирующих рибит бактерий, сконструированных в любом подходящем векторе, предпочтительно в pUC19. Непатогенные виды бактерий, такие как Klebsiella terrigena, являются предпочтительными исходными организмами для выделения гена D-рибулокиназы. Затем можно отобрать трансформанты E.coli, способные расти на минимальной среде, содержащей рибит в качестве единственного источника углерода. Плазмидная ДНК из таких рибит-положительных клонов может быть выделена и применена для повторной трансформации штамма E. coli B. Все трансформанты из такой повторной трансформации будут расти на рибите в качестве единственного источника углерода. Затем можно сконструировать рестрикционную карту клонированной инсерции. С применением этой карты можно получить исходный клон и проанализировать его на сохранение оперона рибита при помощи вышеупомянутого функционального теста. Могут быть выполнены несколько последовательных делеций для уменьшения размера фрагмента ДНК, несущего оперон рибита, до 3,5 - 4 т. п. н. (размер этого оперона в K. aerogenes). Наконец, (частичную) нуклеотидную последовательность гена D-рибулокиназы можно определить и использовать для вырезания кодирующего района этого гена при помощи подходящих природных сайтов рестрикции или при помощи PCR технологии для введения таких сайтов. Ген D-рибулокиназы может экспрессироваться в других хозяевах, предпочтительно в дрожжах, при помощи способа, предусматривающего стандартные стадии, такие как слияние кодирующего района гена D-рибулокиназы с подходящим промотором и терминатором транскрипции, перенос этой экспрессионной кассеты в вектор, пригодный для транcформации выбранного хозяина, получение трансформатор и, наконец, проверка эффективности экспрессии D-рибулокиназы.
Пример 11. Клонирование и избыточная экспрессия гена D-рибулозо-5-фосфат-3-эпимеразы.
Способ выделения гомогенной D-рибулозо-5-фосфат-3-эпимеразы из хлебопекарных дрожжей Saccharomyces cerevisiae) известен (Williamson, W.T. et al., Meth. Enzymol. 9: 605 - 508 (1966)). Фрагмент можно выделить и можно определить N-концевую, а также частичную внутреннюю аминокислотные последовательности при помощи известных способов. Полученные таким образом частичные аминокислотные последовательности можно затем использовать для создания (способом известным как обратная трансляция) последовательностей олигонуклеотидов, которые затем можно использовать в качестве праймеров полимеразной цепной реакции. Фрагменты ДНК, генерируемые PCR, можно использовать в качестве гибридизационных зондов для скрининга библиотеки генов дрожжей на полноразмерную копию гена D-рибулозо-5-фосфат-3-эпимеразы. Предпочтительным способом для избыточной экспрессии гена D-рибулозо-5-фосфат-3-эпимеразы в других дрожжевых хозяевах является клонирование его в вектор, который имеет высокую копийность в желательном хозяине (например, в вектор pSRT303D для Z. rouxii). Альтернативным и более эффективным путем избыточной экспрессии этого гена является определение по меньшей мере частичной нуклеотидной последовательности гена D-рибулозо-5-фосфат-3-эпимеразы около стартового кодона трансляции и применение этой информации для выделения кодирующей последовательности этого гена и слияние ее с промотором, функционирующим эффективно в выбранном хозяине.
Пример 12. Клонирование гена D-ксилулокиназы и конструирование D-ксидлулокиназных мутантов.
Были описаны способы клонирования гена D-ксилулокиназы (EC 2.7.1.17) из различных дрожжевых видов (Ho, N.W.Y. et al., Enzyme Microbiol. Technol. 11: 417 - 421 (1989); Stevis, P.E. et al., Applied and Environmental Microbiol. 53: 2975 - 2977 (1987)). Также был описан способ конструирования D-ксилулокиназной мутации в S. cerevisiae при помощи разрушения гена (Stevis, P.E. et al., Appl. Biochem. Biotechnol. 20: 327 - 334 (1989)). Подобные способы можно использовать для конструирования D-ксилулокиназных мутантов в других дрожжах. Для видов дрожжей, иных, чем S.cerevisiae, генетическими маркерами, применяемыми для разрушения гена D-ксилулокиназы, являются предпочтительно доминантные маркеры устойчивости к антибиотикам (см. пример 9). Альтернативно, могут быть применены классические способы получения мутантов, основанные на химическом (например, на обработке этилметансульфонатом или акрифлавином) или физическом мутагенезе (УФ, рентгеновское излучение). Обогащение мутантов можно проводить выращиванием мутагенезированных клеток на D-ксилулозе в качестве единственного источника углерода в присутствии антибиотика (такого, как нистатин), убивающего только растущие клетки. Неспособность D-ксилулокиназных мутантов использовать D-ксилулозу в качестве единственного источника углерода для роста может быть использована для отбора мутантов.
Пример 13. Штаммы, продуцирующие ксилит через D-арабит с улучшенным выходом.
Пример 4 описывает способ конструирования дрожжевого штамма, способного продуцировать ксилит из структурно неродственных источников углерода, таких как D-глюкоза, при помощи пути, который используют D-арабит в качестве ключевого (центрального) промежуточного продукта. Для улучшения выхода ксилита в ферментационных процессах со штаммами, использующими этот "путь D-арабита", выход D-арабита должен быть улучшен. Был описан путь, ведущий от D-глюкозы до D-арабита в продуцирующих D-арабит дрожжах (Ingram, J.M. et al., J. Bacteriol. 89: 1186 - 1194 (1965)). D-арабит образуется из D-рибулозо-5-фосфата через дефосфорилирование НАДФН-зависимой D-рибулозоредуктазой. Образование D-рибулозо-5-фосфата из D-глюкозо-5-фосфата при помощи двух последовательных необратимых стадий дегидрогенизации ферментами D-глюкозо-5-фосфатдегидрогеназой и 6-фосфо-D-глюконатдегидрогеназой является универсально встречающимся путем, известным как окислительная ветвь пентозофосфатного пути (или гексозомонофосфатный шунт). В этой неокислительной ветви пентозофосфатного пути D-рибулозо-5-фосфат обратимо изомеризуется в рибозо-5-фосфат и D-ксилулозо-5-фосфат. Рибозо-5-фосфат и D-ксилулозо-5-фосфат метаболизируются далее транскетолазой. Поэтому при мутировании транскетолазы в продуцирующем D-арабит микробном штамме фракция D-рибулозо-5-фосфата, превращаемая в D-арабит, будет увеличиваться. Пример 9 описывает способ получения транскетолазных мутантов. Дальнейшее увеличение выхода D-арабита может достигаться, если скорость биосинтеза D-рибулозо-5-фосфата увеличивается через избыточную экспрессию двух генов, кодирующих ферменты окислительной ветви пентозофосфатного пути, как описано выше (пример 8). Штаммы, оптимизированные этим способом относительно выхода D-арабита, могут быть далее трансформированы рекомбинантными конструкциями ДНК, несущими гены ксилитолдегидрогеназы и D-арабитолдегидрогеназы (примеры 3 и 4), что приводит к получению штаммов с улучшенной эффективностью образования ксилита.
Пример 14. Штаммы, продуцирующие ксилит альтернативными путями.
Способы согласно примерам 4 и 13 являются наиболее прямыми способами конструирования микробных штаммов, способных превращать D-глюкозу и другие источники углерода в ксилит. Эти способы используют природно встречающийся путь, ведущий к образованию D-арабита из различных углеродных источников, и удлиняют этот путь при помощи еще двух реакций для превращения D-арабита в ксилит. Однако этот путь не является единственным возможным путем. Могут быть сконструированы пути, приводящие ко ксилиту как конечному метаболическому продукту и не включающие в себя D-арабит в качестве промежуточного продукта. Так, путь к ксилиту от того же самого предшественника, D-рибулозо-5-фосфата, может быть реализован через другую цепь реакций. D-рибулозо-5-фосфат может эффективно превращаться в D-ксилулозо-5-фосфат ферментом D-рибулозо-5-фосфат-3-эпимеразой (пример 11) и, если дальнейшее превращение D-ксилулозо-5-фосфата предотвращается мутацией в гене транскетолазы, то накопленный D-ксилулозо-5-фосфат может дефосфорилироваться той же самой неспецифической фосфатазой, что и D-рибулозо-5-фосфат (Ingram, J.M. et al., J. Bacteriol. 89: 1186 - 1194 (1965)) и восстанавливаться в ксилит ксилитолдегидрогеназой (пример 3). Реализация этого пути может далее требовать инкативации гена D-ксилулокиназы (пример 12) для уменьшения потери энергии, связанной с бесполезной петлей: D-ксилулозо-5-фосфат ---> D-ксилулоза ---> D-ксилулозо-5-фосфат. Дополнительное генетическое изменение, введение и (избыточная) экспрессия гена D-рибулокиназы (EC 2.7.1.47) могли бы уменьшить одновременное продуцирование D-арабита такими штаммами путем улавливания D-рибулозы, продуцируемой неспецифической фосфатазой. D-рибулоза будет превращаться опять в D-рибулозо-5-фосфат и далее в D-ксилулозо-5-фосфат.
Пример 15. Стабильность рекомбинантного штамма Z. rouxii и образование ксилита в условиях культивирования в ферментере.
Стабильность образования ксилита во время длительного культивирования проверяли как в селективных условиях (при помощи селективной среды: YEPD, содержащей 50 мг/л G418 и 30% глюкозу), так и неселективных условиях (с применением той же самой среды без G418). Один только что полученный трансформант Z. rouxii ATCC 13356 [pSRT(AX)-9] выращивали в объеме 200 мл YEPD, содержащей G418. Клетки переносили в 50% раствор глицерина и замораживали при -70oC в аликвотах по 1 мл. Четыре замороженные аликвоты Z. rouxii [pSRT(AX)-9] применяли для инокуляции культур объемом 50 мл в селективной среде и две аликвоты в неселективной среде. После достижения этими культурами стационарной фазы роста (50-60 ч при 30oC и 200 об/мин) брали пробу для HPLC анализа содержания пентита и 1 мл культуры использовали для инокуляции еще 50 мл той же самой среды (селективной или неселективной). Этот цикл рост-разбавление повторяли еще 4 раза. Условия этого эксперимента приближаются к размножению рекомбинантного штамма из стандартного замороженного инокулята в случае широкомасштабной ферментации. Результаты этого эксперимента представлены в табл. 7. Предсказуемо, стабильность рекомбинантного штамма выше на селективной среде. Однако, даже в случае неселективной среды снижение в выходе ксилита обнаруживали только приблизительно после 20 генераций. При селективных условиях образование ксилита было стабильным приблизительно в течение 30 генераций.
Аликвоту замороженного исходного материала трансформированного штамма Z. rouxii использовали для инокуляции 2-литрового ферментера, содержащего 1 л среды, имеющей следующий состав (на 1 л): 0,1 г NaCl, 6,8 г фосфата калия, 0,5 г сульфата аммония, 20 г дрожжевого экстракта и 400 г глюкозы, 50 мг G481, pH 6,0. Условия культивирования были: скорость аэрации 0,5 /мин; перемешивание 400 об/мин.; температура 30oC.
Фиг. 10 показывает временной ход потребления глюкозы и накопления ксилита в этой ферментации. Показана также концентрация растворенного кислорода, которая отражает дыхательную активность культуры дрожжей. Наблюдали видимый двухфазный рост: в первой фазе плато концентраций глюкозы и ксилита достигалось приблизительно за 40 ч (в этой временной точке потреблялось менее половины доступной глюкозы), вторую фазу наблюдали приблизительно после 200 ч культивирования, когда возобновлялись потребление глюкозы и образование ксилита. Конечная концентрация ксилита была 15 г/л, почти в два раза выше, чем концентрация, полученная при ферментации в колбах. Двухфазный рост с длительным лаг-периодом показывал, что был отобран спонтанный мутант (предположительно имеющий более высокую устойчивость к спирту, чем родительский штамм). Для проверки этой гипотезы один клон был выделен из этой культуры в конечной точке работы ферментера. Этот изолят выращивали в ферментере при условиях, идентичных условиям, описанным выше. Результаты этого эксперимента (фиг. 11) подтвердили, что действительно был выделен мутант, способный завершить ассимиляцию 400 г/л глюкозы приблизительно за 60 ч. Этот эксперимент также показывает, что продуцирующий ксилит штамм Z.rouxii может также ассимилировать ксилит, когда потреблена вся глюкоза в культуральной среде.
Все ссылки даны в тексте в виде неполных ссылок. После полного описания изобретения специалистам данной области будет понятно, что могут быть использованы широкие и эквивалентные диапазоны концентраций, параметры и т.п. без отхода от сущности и сферы действия изобретения или любого его варианта.
Перечень последовательностей приведен в конце текста описания.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПОЛУЧЕНИЕ КСИЛИТА ИЗ ГЛЮКОЗЫ ПРИ ПОМОЩИ РЕКОМБИНАНТНОГО ШТАММА | 2015 |
|
RU2693593C2 |
| МИКРООРГАНИЗМ, ЭКСПРЕССИРУЮЩИЙ КСИЛОЗОИЗОМЕРАЗУ | 2009 |
|
RU2553537C2 |
| ВЕКТОР ДЛЯ НОКАУТА ГЕНА АЦЕТАТКИНАЗЫ В Clostridium thermocellum, КЛЕТКА-ХОЗЯИН, ГЕНЕТИЧЕСКИ МОДИФИЦИРОВАННЫЙ МИКРООРГАНИЗМ Clostridium thermocellum, СПОСОБ ПОЛУЧЕНИЯ ТАКОГО МИКРООРГАНИЗМА И СПОСОБ ПРЕОБРАЗОВАНИЯ ЛИГНОЦЕЛЛЮЛОЗНОЙ БИОМАССЫ В ЭТАНОЛ. | 2008 |
|
RU2541785C2 |
| ГЕНЫ АГГЛЮТИНАЦИИ, ДРОЖЖИ | 1994 |
|
RU2159807C2 |
| ШТАММ ДРОЖЖЕЙ SACCHAROMYCES CEREVISIAL, СОДЕРЖАЩИЙ РЕКОМБИНАНТНУЮ ПЛАЗМИДУ YEP 63/AB, - ПРОДУЦЕНТ ПРОИЗВОДНОГО М-БЕЛКА ВИРУСА ГЕПАТИТА В ЧЕЛОВЕКА | 1994 |
|
RU2082759C1 |
| ФЕРМЕНТАТИВНОЕ ПОЛУЧЕНИЕ 1-БУТАНОЛА | 2006 |
|
RU2429295C2 |
| СПОСОБ СНИЖЕНИЯ СТЕПЕНИ ГЛИКОЗИЛИРОВАНИЯ БЕЛКОВ, СПОСОБЫ И БЕЛКИ | 2010 |
|
RU2575607C2 |
| СПОСОБ ПОЛУЧЕНИЯ ГЕТЕРОЛОГИЧНОГО ПОЛИПЕПТИДА В ЭУКАРИОТИЧЕСКИХ МИКРООРГАНИЗМОВ | 1986 |
|
RU2091490C1 |
| СПОСОБ ПОЛУЧЕНИЯ L-АМИНОКИСЛОТ МЕТОДОМ ФЕРМЕНТАЦИИ С ИСПОЛЬЗОВАНИЕМ БАКТЕРИЙ, ОБЛАДАЮЩИХ ПОВЫШЕННОЙ ЭКСПРЕССИЕЙ ГЕНОВ УТИЛИЗАЦИИ КСИЛОЗЫ | 2005 |
|
RU2283346C1 |
| ПОЛИПЕПТИД, ИМЕЮЩИЙ NADH-ЗАВИСИМУЮ HMF-РЕДУКТАЗНУЮ АКТИВНОСТЬ | 2008 |
|
RU2483107C2 |



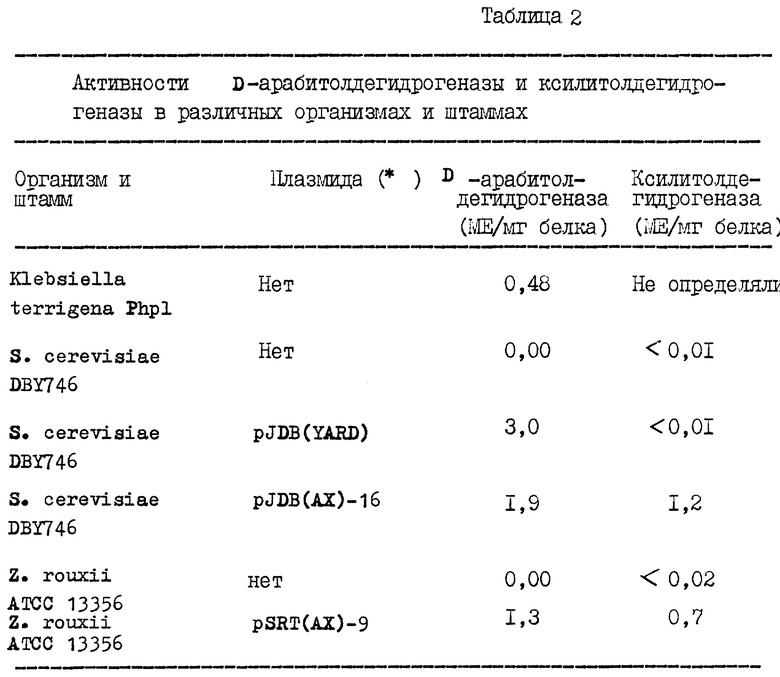






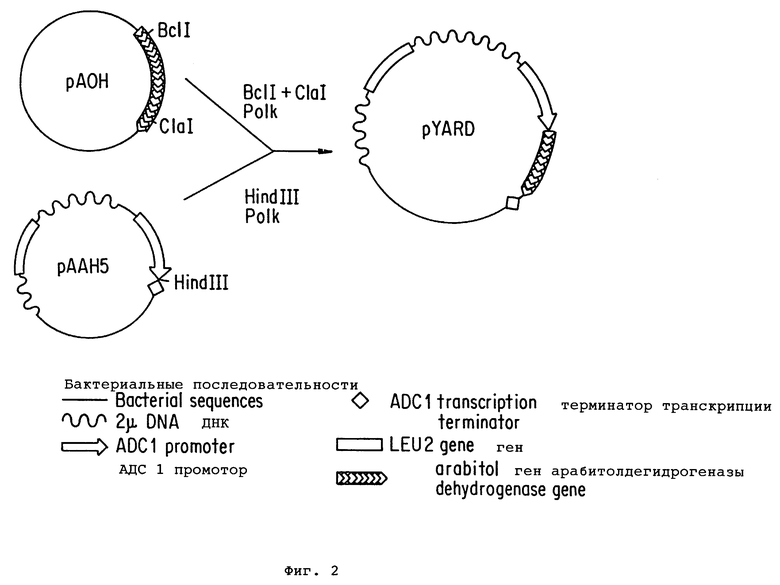


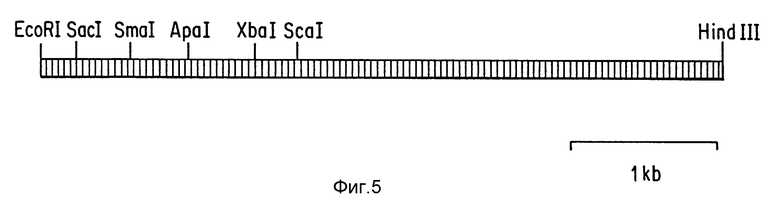

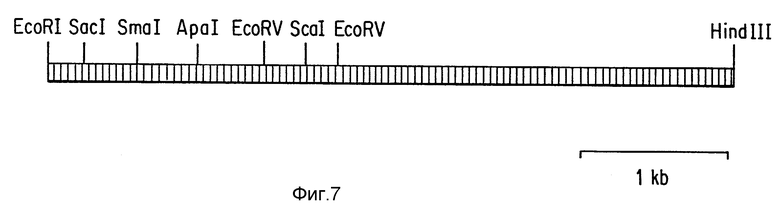
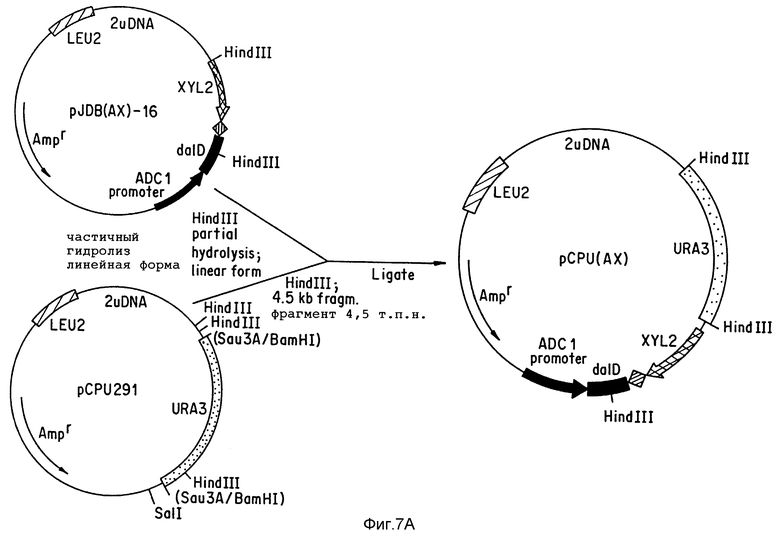
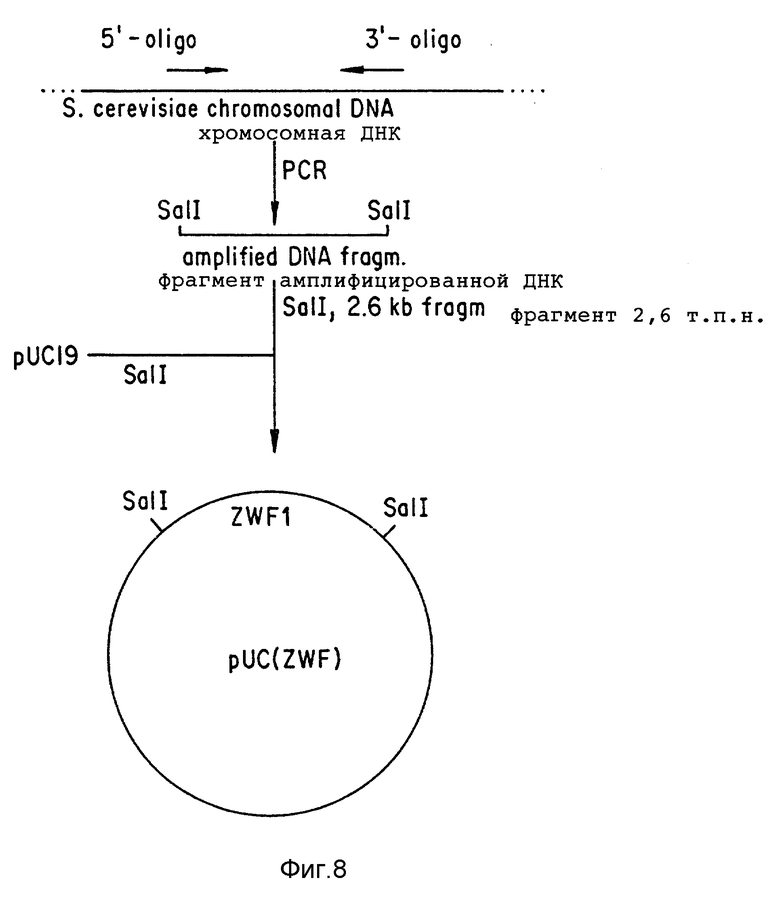
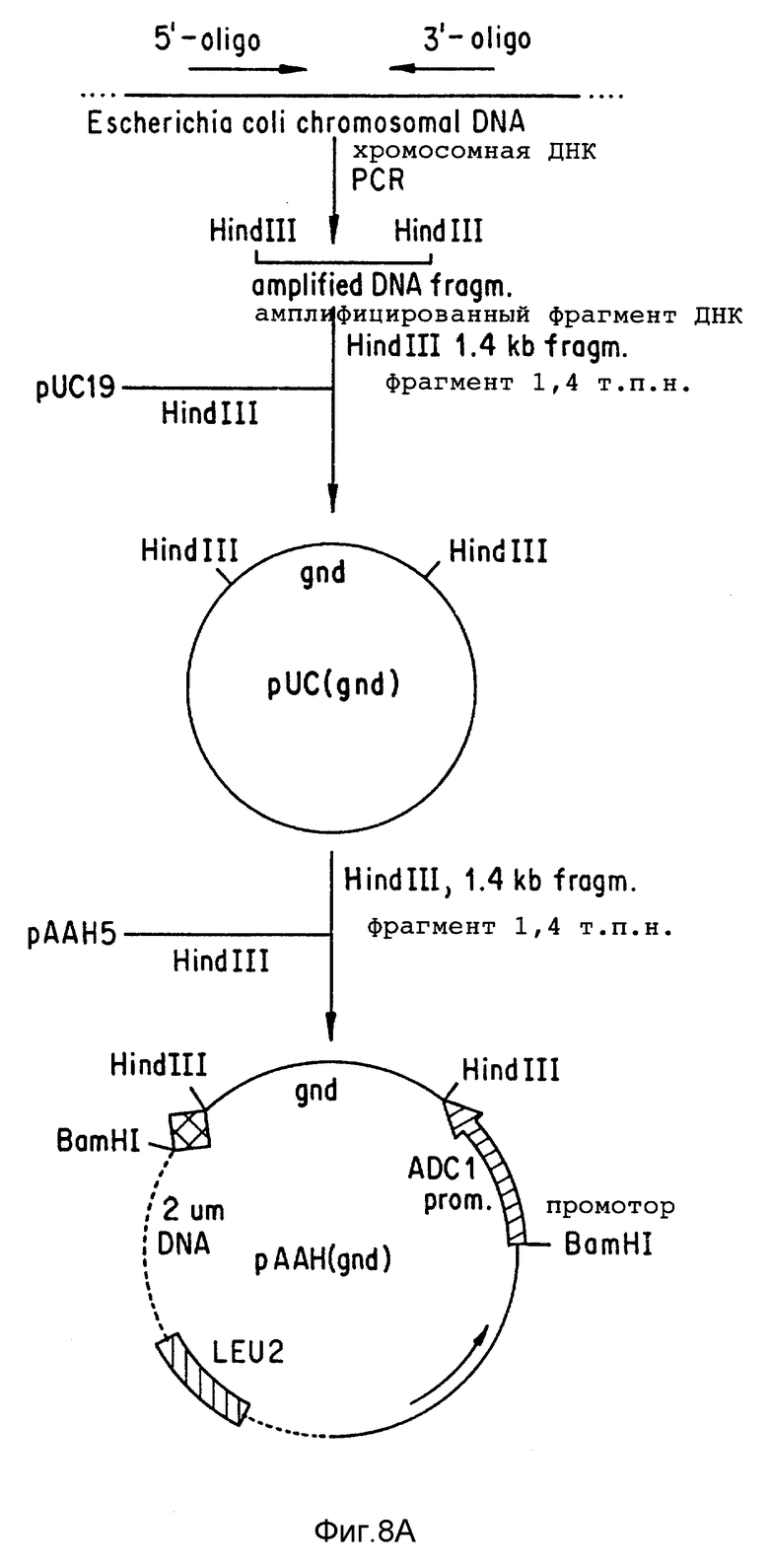
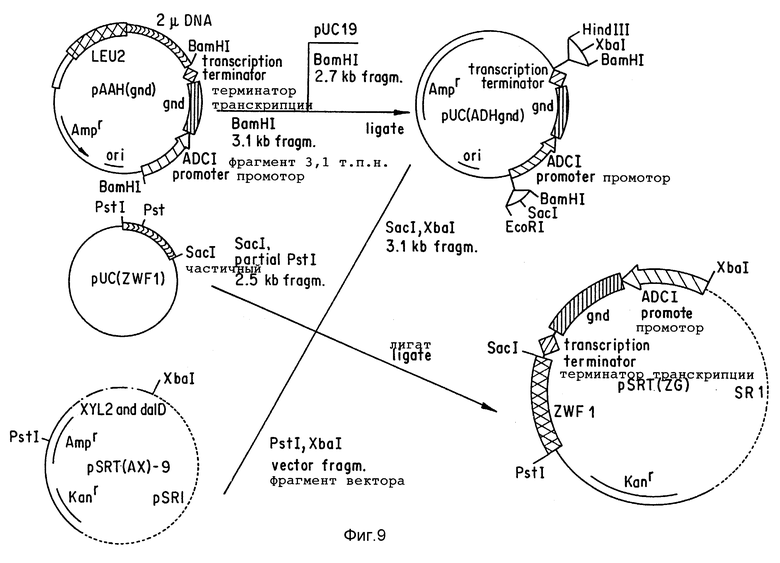
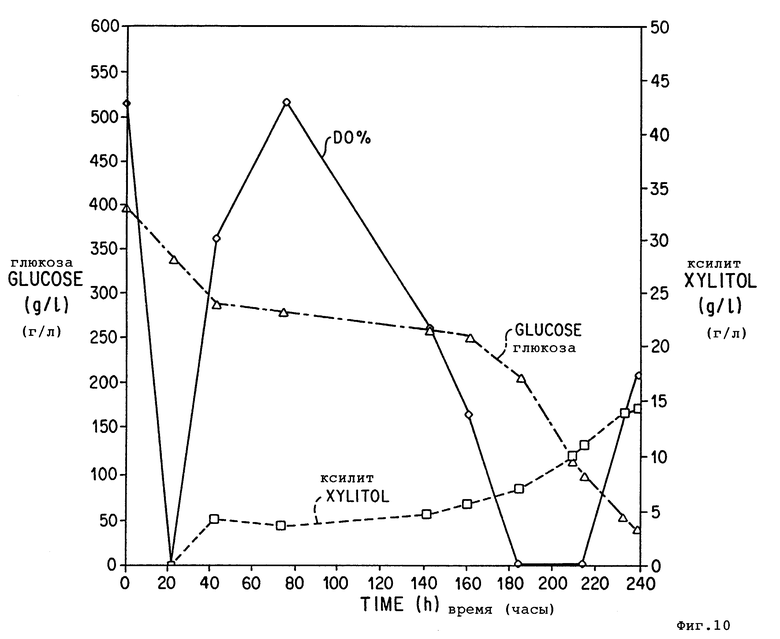
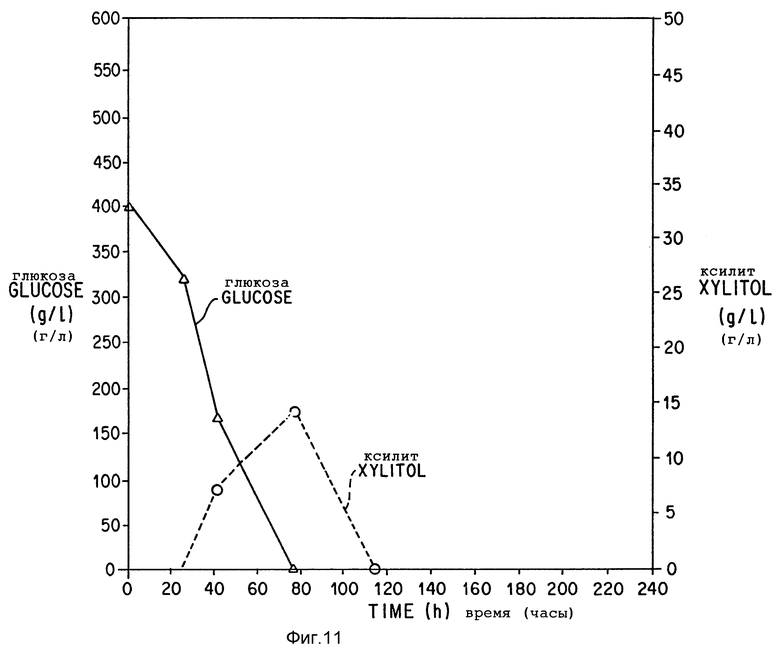
Способ относится к генной инженерии и может быть использован в микробиологической промышленности. Продуцирующие арабит дрожжи или грибы трансформируют ДНК, кодирующей образующую D-ксилозу D-арабитолдегидрогеназу, и ДНК, кодирующей ксилитолдегидрогеназу. Затем выращивают трансформированные дрожжи или грибы в условиях, обеспечивающих синтез ксилита. Ксилит извлекают. Дрожжи выбирают из Lygosaccharomyces rouxii, Candida polymorpha, Torulopsis candida, Pichia farinosa, Torulaspora hansenii. Грибы выбирают из Dendryphiella salina и Schizophyllum commune. Способ позволяет превращать легко доступные источники углерода, такие, как D-глюкоза в ксилит. 20 з.п. ф-лы, 7 табл., 11 ил.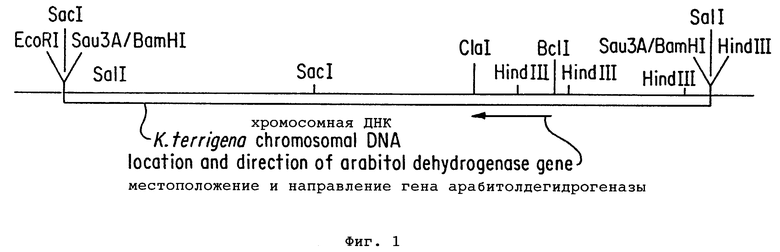
а) выращивание продуцирующих арабит дрожжей или продуцирующих арабит грибов в условиях, обеспечивающих синтез ксилита, причем продуцирующие арабит дрожжи или продуцирующие арабит грибы модифицируют для обеспечения синтеза ксилита в качестве конечного продукта в одной ферментационной стадии при выращивании на D-арабите или источнике углерода, который немодифицированные продуцирующие арабит дрожжи или немодифицированные продуцирующие арабит грибы используют для биосинтеза D-арабита, при этом источник углерода является другим, чем D-ксилоза, D-ксилулоза, смеси D-ксилозы и D-ксилулозы, и полимеры и олигомеры, содержащие D-ксилозу или D-ксилулозу в качестве основных компонентов, в условиях, которые обеспечивают синтез ксилита, причем до упомянутой модификации хозяин продуцировал D-арабит, но не использовал D-арабит для синтеза ксилита, и хозяин трасформирован ДНК, кодирующей образующую D-ксилулозу D-арабитолдегидрогеназу, и ДНК, кодирующей ксилитолдегидрогеназу, и
в) извлечение ксилита, продуцированного на стадии а).
| Огнетушитель | 0 |
|
SU91A1 |
| Устройство электропунктуры | 1973 |
|
SU450430A1 |
| Bio/technology, vol.9, 1991, Johan Hallborn et., Xylitol production by recombinant Saccharomyces cerevisial, page, 1090-1095 | |||
| US 4008285 A, 15.02.77 | |||
| US 4066711 A, 03.01.78 | |||
| US 4075406 A, 21.02.78 | |||
| US 3784407 A, 08.01.74. | |||
