Список последовательностей по настоящей заявке прислан в электронном посредством EFS-Web в виде отформатированного в ASCII списка последовательностей с названием файла "23744-US-PSP-SEQLIST-01APR2014", с датой создания 1 апреля 2014 года и размером 1,92 кбайт. Этот список последовательностей, предоставленный посредством EFS-Web, представляет собой часть описания, полностью включен в настоящий документ в качестве ссылки.
УРОВЕНЬ ТЕХНИКИ, ПРЕДШЕСТВУЮЩИЙ ИЗОБРЕТЕНИЮ
Ретровирус, обозначаемый как вирус иммунодефицита человека (ВИЧ), в частности штаммы, известные как ВИЧ типа 1 (ВИЧ-1) и типа 2 (ВИЧ-2), этиологически связан с иммуносупрессорным заболеванием, известным как синдром приобретенного иммунодефицита (СПИД). Сначала у серопозитивных по ВИЧ индивидуумов симптомы отсутствуют, но, как правило, развивается ассоциированный со СПИДом комплекс с последующим СПИДом. У пораженных индивидуумов наблюдают тяжелую иммуносупрессию, которая делает их крайне чувствительными к истощению и, в конечном итоге, к смертельным оппортунистическим инфекциям. Репликация ВИЧ клеткой-хозяином требует интеграции вирусного генома в ДНК клетки-хозяина. Так как ВИЧ представляет собой ретровирус, цикл репликации ВИЧ требует транскрипции генома вирусной РНК в ДНК посредством фермента, известного как обратная транскриптаза (RT).
У обратной транскриптазы существуют три известные ферментативные функции: фермент действует в качестве РНК-зависимой ДНК-полимеразы в качестве рибонуклеазы и в качестве ДНК-зависимой ДНК-полимеразы. В ее роли в качестве РНК-зависимой ДНК-полимеразы RT транскрибирует с вирусной РНК одноцепочечную копию ДНК. В качестве рибонуклеазы, RT разрушает исходную вирусную РНК и высвобождает ДНК, только что продуцированную из исходной РНК. И в качестве ДНК-зависимой ДНК-полимеразы RT синтезирует вторую комплементарную цепь ДНК с использованием первой цепи ДНК в качестве матрицы. Две цепи формируют двухцепочечную ДНК, которую интегрирует в геном клетки-хозяина фермент интеграза.
Известно, что соединения, которые ингибируют ферментативные функции RT ВИЧ, ингибируют репликацию ВИЧ в инфицированных клетках. Эти соединения пригодны для профилактики или лечения инфекции ВИЧ у людей. В числе соединений, одобренных для применения при лечении инфекции ВИЧ и СПИДа, находятся ингибиторы RT 3'-азидо-3'-дезокситимидин (AZT), 2',3'-дидезоксиинозин (ddI), 2',3'-дидезоксицитидин (ddC), d4T, 3TC, невирапин, делавирдин, эфавиренз, абакавир, эмтрицитабин и тенофовир. Современный стандарт лечения заключается в проведении высокоактивной антиретровирусной терапии (HAART). Терапия HAART определена как комбинация 3 средств по меньшей мере из 2 классов с различными механизмами действия. Хотя основанные на HAART схемы лечения с применением ингибиторов RT являются эффективными при лечении инфекции ВИЧ и СПИДа, остается необходимость в разработке дополнительных противовирусных лекарственных средств против ВИЧ, включая дополнительные ингибиторы RT. Конкретной проблемой является развитие мутантных штаммов ВИЧ, которые устойчивы к известным ингибиторам. Использование ингибиторов RT для лечения СПИДа часто приводит к развитию вирусов, которые менее чувствительны к ингибиторам. Как правило, эта устойчивость является результатом мутаций, которые происходят в участке обратной транскриптазы пула генов. Продолжающееся применение при лечении инфекции ВИЧ противовирусных соединений неминуемо приводит появлению новых устойчивых штаммов ВИЧ. Таким образом, существует конкретная необходимость в новых ингибиторах RT, которые эффективны против мутантных штаммов ВИЧ.
В WO 2009/067166 и WO 2011/126969 описаны определенные пролекарственные средства ингибиторов RT. В Clemo et al., J. Chem. Soc. 1954, pp. 2693-2702 описаны определенные производные 4-оксо-3-(2-пиридил)пиридоколиновой системы и, в частности, описан 6-метил-6'-фенокси-2,2'-метилендипиридин. В Sweeney et al., Bioorganic & Medicinal Chem. Letters 2008, vol. 18, pp. 4348-4351 описан ряд триазолинонов, которые описаны как ненуклеозидные ингибиторы обратной транскриптазы ВИЧ. В WO 2001/034578 описаны определенные замещенные азолы (включая, например, определенные имидазолы и бензимидазолы), обладающие активностью против Helicobacter pylori. В частности, в WO '578 описан 1-[(3-метил-4-фенокси-2-пиридинил)метил]-1H-бензимидазол (см. соединение 91 на странице 40). В WO 2004/085406 и соответствующем US 7189718 в качестве ингибиторов обратной транскриптазы описаны определенные бензилпиридазиноны. В WO 2005/102989 и соответствующем US 7166738 в качестве ненуклеозидных ингибиторов обратной транскриптазы описаны определенные N-фенил-2-фенилацетамиды. В WO 2006/067587 в качестве модуляторов фермента обратной транскриптазы описаны определенные биарильные эфирные производные. В WO 2007/045572 и WO 2007/045573 в качестве ненуклеозидных ингибиторов обратной транскриптазы описаны определенные 2-(2-феноксифенил)-N-фенилацетамиды. В WO 2008/076225 в качестве ингибиторов обратной транскриптазы ВИЧ описаны определенные индазолы, бензотриазолы и родственные бициклические соединения. В WO 2009/067166 описаны определенные арилокси-, циклоалкилокси- и гетероциклилоксипиридины и родственные соединения. Соединения представляют собой ингибиторы обратной транскриптазы ВИЧ, подходящие, например, для лечения инфекции ВИЧ. В числе описанных соединений находятся определенные 3-(3,5-двузамещенный фенокси)-1-(1H-пиразоло[3,4-b]пиридин-3-илметил)-4-(замещенный)пиридин-2(1H)-оны. В US 2004/0192704 описаны определенные замещенные 3-(фенокси)бензилом 5-членные триазолоны, оксадиазолoны и тиадиазолoны. Соединения описаны в качестве ненуклеозидных ингибиторов обратной транскриптазы, пригодных для лечения или профилактики обусловленных ВИЧ заболеваний. В US 2007/0021442 и WO 2007/015812 описаны определенные замещенные ароматические соединения. Соединения представляют собой ингибиторы обратной транскриптазы ВИЧ, подходящие, например, для лечения инфекции ВИЧ. В WO 2009/067166 и в WO2011/120133 описан ненуклеозидные ингибиторы обратной транскриптазы ВИЧ. В WO 2011/126969 описаны пролекарственные средства ненуклеозидных ингибиторов обратной транскриптазы ВИЧ.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к определенным производным 4-пиримидинонов. Полагают, что соединения формулы I являются пролекарственными средствами, которые in vivo могут подвергаться метаболизму до соединений формулы I' (определенных ниже), которые являются ингибиторами обратной транскриптазы ВИЧ. Соединения формулы I' ингибируют полимеразную функцию обратной транскриптазы ВИЧ-1, а более конкретно, ингибируют действие обратной транскриптазы ВИЧ-1 в качестве РНК-зависимой ДНК-полимеразы. Соединения формулы I' также проявляют активность против устойчивых к лекарственным средствам форм ВИЧ (например, мутантных штаммов ВИЧ-1, в которых обратная транскриптаза содержит мутацию лизин 103 → аспарагин (K103N) и/или тирозин 181 → цистеин (Y181C)). Таким образом, соединения формулы I, которые можно использовать для облегчения введения соединений формулы I', могут проявлять сниженную перекрестную устойчивость к одобренным в настоящее время противовирусным лекарственным средствам. Таким образом, соединения формулы I (включая их гидраты и сольваты) пригодны, например, для ингибирования обратной транскриптазы ВИЧ, профилактики инфекции ВИЧ, лечения инфекции ВИЧ и в профилактике, лечении и задержке начала или прогрессирования СПИДа и/или ARC, или в качестве самостоятельных соединений, или в качестве ингредиентов фармацевтических композиций, в комбинации с другими противовирусными средствами против ВИЧ, противоинфекционными средствами, иммуномодуляторами, антибиотиками или вакцинами, или без.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям структурной формулы I или их фармацевтически приемлемым солям:
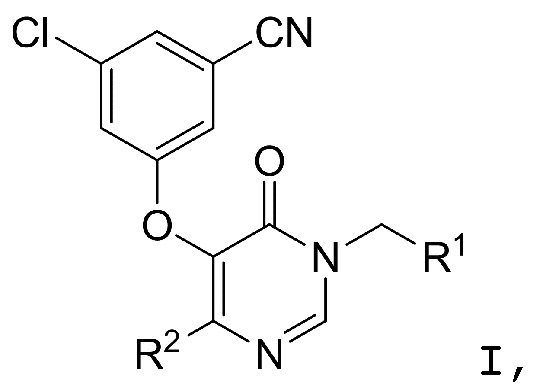 где R1 представляет собой
где R1 представляет собой 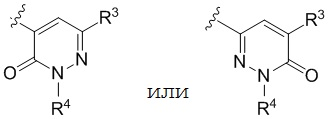 ;
;
R2 представляет собой галоген или -C1-3-алкил, замещенный 1-3 -F;
R3 представляет собой (a) галоген, (b) -C1-3-алкил, замещенный от 1 до 3 -F, или (3) фенил, замещенный галогеном; и
R4 представляет собой 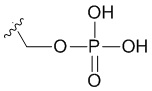 .
.
В варианте осуществления настоящего изобретения A предоставлены соединения формулы I или их фармацевтически приемлемые соли, где R1 представляет собой 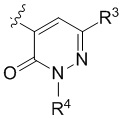 .
.
В варианте осуществления настоящего изобретения B предоставлены соединения формулы I или их фармацевтически приемлемые соли, где R1 представляет собой 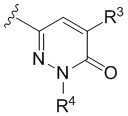 .
.
В другом варианте осуществления настоящего изобретения предоставлены соединения формулы I, или варианта осуществления A, или варианта осуществления B, или их фармацевтически приемлемые соли, где R2 представляет собой метил, замещенный 1, 2 или 3 -F; или этил, замещенный 1, 2 или 3 -F; а более конкретно R2 представляет собой -CHF2, -CF3 или -CF2CH3.
В другом варианте осуществления настоящего изобретения предоставлены соединения формулы I, или варианта осуществления A, или варианта осуществления B, или их фармацевтически приемлемые соли, где R3 представляет собой -F; -Cl; метил, замещенный 1, 2 или 3 -F; этил, замещенный 1, 2 или 3 -F; или фенил, замещенный -F; а более конкретно R3 представляет собой -Cl, -CHF2, -CF3, -CF2CH3 или фенил, замещенный -F.
В другом варианте осуществления настоящего изобретения предоставлены соединения формулы I, варианта осуществления A или варианта осуществления B, или их фармацевтически приемлемой соли, где:
R2 представляет собой метил, замещенный 1, 2 или 3 -F; или этил, замещенный 1, 2 или 3 -F; а более конкретно R2 представляет собой -CHF2, -CF3 или -CF2CH3;
R3 представляет собой -F, -Cl, метил, замещенный 1, 2 или 3 -F; этил, замещенный 1, 2 или 3 -F; или фенил, замещенный -F; а более конкретно R3 представляет собой -Cl, -CHF2, -CF3, -CF2CH3 или фенил, замещенный -F; и
R4 представляет собой .
В контексте настоящего документа, термин "алкил" относится к насыщенному алифатическому углеводородному радикалу с неразветвленной или разветвленной цепью, содержащему ряд атомов углерода в указанном диапазоне. Таким образом, например, "-C1-3 алкил" означает алкильные группы с линейной или разветвленной цепью, включающие все изомеры, содержащие указанное количество атомов углерода, т.е., н- и изо-пропил (Pr=пропил), этил (Et) и метил (Me).
Термин "галоген" относится к фтору, хлору, брому или йоду. Предпочтительными являются фтор или хлор.
Полагают, что соединения формулы I действуют в качестве пролекарственных средств, которые in vivo преобразуются в их фармацевтически активные модификации формулы I', где формула I' идентична формуле I за исключением того, что R4 замещен -H. Для конкретного соединения формулы I, соответствующее соединение формулы I' может быть в настоящем документе указано как "исходное" соединение (вне зависимости от того, находятся ли или нет или одно или оба из соответствующих соединений в форме соли, если не указано иначе), например,
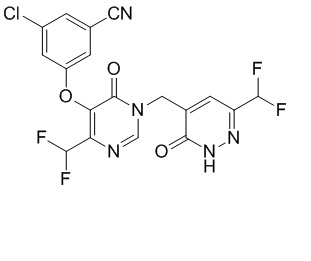
Исходное соединение для примера 1 (в рамках формулы I')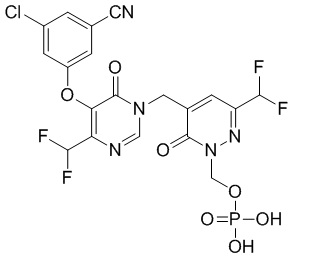
Пример 1 (в рамках формулы I)
Соединения формулы I' являются ингибиторами обратной транскриптазы ВИЧ. В таблице 3 ниже для соединений из примеров 1-8 предоставлены данные AUC для крыс.
Все структурные формулы, варианты осуществления и их классы, описываемые в настоящем документе, включают фармацевтически приемлемые соли соединений, определенных в них. Указание соединений формулы I в настоящем документе включает соединения формул I и все их варианты осуществления и классы. Если не указано иначе, указание соединений по настоящему изобретению, как соединений конкретной формулы или варианта осуществления, например, формулы I или ее вариантов осуществления, или любой другой общей структурной формулы или конкретного соединения, описанного или заявляемого в настоящем документе, предназначено для включения конкретного соединения или соединений, попадающих в объем формулы или варианта осуществления, включая их соли, в частности фармацевтически приемлемые соли, сольваты (включая гидраты) таких соединений и формы их сольватированных солей, где такие формы возможны.
Настоящее изобретение относится к каждому из примеров, описываемых в настоящем документе, и их фармацевтически приемлемым солям. Изобретение также относится к фармацевтическим композициям, содержащим эффективное количество соединения по изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
Если явно не указано противоположного, замещение указанным заместителем допустимо по любому атому в цепи или кольце, при условии, что такое замещение химически допустимо и приводит к стабильному соединению. "Стабильное" соединение представляет собой соединение, которое можно получать и выделять, и структура и свойства которого остаются или их можно заставить оставаться по существу неизменными в течение периода времени, достаточного для того, чтобы обеспечить применение соединения в целях, описываемых в настоящем документе (например, терапевтическое или профилактическое введение индивидууму). Соединения по настоящему изобретению ограничены стабильными соединениями, охватываемыми формулой I и вариантами ее осуществления.
В тех случаях, когда заместители и комбинации заместителей обеспечивают существование таутомеров (например, кето-енольные таутомеры) в соединениях по изобретению, все таутомерные формы этих соединений, существующие отдельно или в смесях, входят в объем настоящего изобретения. Следует понимать, что указание соединения, способного к таутомерии, включает в его рамки указание каждого из конкретных таутомеров и их сочетаний, например, кето- и енольной форм.
В соединениях формулы I атомы могут присутствовать в их природных изотопных составах, или у одного или нескольких из атомов можно искусственным способом обогащать конкретный изотоп с тем же атомным номером, но атомной массой или массовым числом, отличающимися от атомной массы или массового числа, преимущественно существующих в природе. Подразумевается, что в настоящее изобретение включены все соответствующие изотопные варианты соединений формулы I. Например, различные изотопные формы водорода (H) включают протий (1H) и дейтерий (2H). Протий представляет собой преобладающий изотоп водорода, существующий в природе. Обогащение дейтерием может обеспечивать определенные терапевтические преимущества, такие как увеличение времени полувыведения in vivo или снижение требуемых количеств доз, или может обеспечивать соединение, пригодное в качестве стандарта для характеристики биологических образцов. Обогащенные изотопами соединения в рамках формулы I можно получать без излишнего экспериментирования общепринятыми способами, хорошо известными специалистам в данной области, или аналогичными им способами, описанными в схемах и примерах в настоящем документе с использованием соответствующих обогащенных изотопами реагентов и/или промежуточных соединений.
Соединения можно вводить в форме фармацевтически приемлемых солей. Термин "фармацевтически приемлемая соль" относится к соли, которая не является биологически или иным образом нежелательной (например, не является ни токсической, ни иным образом вредной для ее реципиента). Когда соединение формулы I содержит одну или несколько кислотных или основных групп, изобретение также включает соответствующие фармацевтически приемлемые соли. Таким образом, соединения формулы I, которые содержат кислотные группы можно использовать по изобретению, например, но не ограничиваясь ими, в виде солей щелочных металлов, солей щелочноземельных металлов или в виде аммонийных солей. Примеры таких солей в качестве неограничивающих примеров включают натриевые соли, калиевые соли, кальциевые соли, магниевые соли или соли с аммиаком или органическими аминами, такими как, например, этиламин, этаноламин, триэтаноламин или аминокислоты. Соединения формулы I, которые содержат одну или несколько основных групп, т.е. группы, которые могут быть протонированы, можно использовать по изобретению в форме их солей присоединения кислот с неорганическими или органическими кислотами, например, но не ограничиваясь ими, в виде солей с соляной кислотой, бромистым водородом, фосфорной кислотой, серной кислотой, азотной кислотой, бензолсульфоновой кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, нафталиндисульфоновыми кислотами, щавелевой кислотой, уксусной кислотой, трифторуксусной кислотой, винной кислотой, молочной кислотой, салициловой кислотой, бензойной кислотой, муравьиной кислотой, пропионовой кислотой, пиваловой кислотой, диэтилуксусной кислотой, малоновой кислотой, янтарной кислотой, пимелиновой кислотой, фумаровой кислотой, малеиновой кислотой, яблочной кислотой, сульфаминокислотой, фенилпропионовой кислотой, глюконовой кислотой, аскорбиновой кислотой, изоникотиновой кислотой, лимонной кислотой, адипиновой кислотой и т.д. Если соединения формулы I одновременно содержат в молекуле и кислотные, и основные группы, изобретение, в дополнение к указанным формам солей, также включает, фармацевтически приемлемые внутренние соли или бетаины (цвиттерионы). Соли можно получать из соединений формулы I стандартными способами, известными специалисту в данной области, например, посредством комбинации с органической или неорганической кислотой или основанием в растворителе или дисперсанте, или посредством анионного обмена или катионного обмена с другими солями. Настоящее изобретение также относится ко всем солям соединений формулы I, которые вследствие низкой физиологической совместимости не пригодны для прямого применения в фармацевтическом препарате, но которые можно использовать, например, в качестве промежуточных соединений для химических реакций или для получения фармацевтически приемлемых солей.
В качестве примера, соединения формулы I в качестве неограничивающих примеров включают такие соединения, где алкилфосфат R4 может представлять собой соль основания, которая относится к фармацевтически приемлемой соли, которая реализуется при потере по меньшей мере одного протона из группы, сбалансированной одним или несколькими положительными противоионами (например, катионом щелочного металла). Соль основания R4 можно представить в виде:
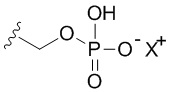 или
или 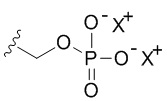 или
или 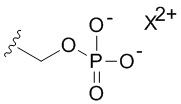 , где X+ и X2+ представляют собой положительные противоионы. Соль основания можно получать, обрабатывая свободную форму соединения формулы I подходящим неорганическим или органическим основанием. Подходящие неорганические основания в качестве неограничивающих примеров включают гидроксид аммония, гидроксиды щелочных металлов (например, NaOH или KOH), гидроксиды щелочноземельных металлов и т.п. Подходящие органические основания включают алкилкарбоксилаты щелочных металлов (например, ацетат калия или ацетат натрия), алкилгидроксиды аммония и т.п.
, где X+ и X2+ представляют собой положительные противоионы. Соль основания можно получать, обрабатывая свободную форму соединения формулы I подходящим неорганическим или органическим основанием. Подходящие неорганические основания в качестве неограничивающих примеров включают гидроксид аммония, гидроксиды щелочных металлов (например, NaOH или KOH), гидроксиды щелочноземельных металлов и т.п. Подходящие органические основания включают алкилкарбоксилаты щелочных металлов (например, ацетат калия или ацетат натрия), алкилгидроксиды аммония и т.п.
Другим вариантом осуществления настоящего изобретения является соединение формулы I, где соединение или его соль находятся по существу в чистой форме. Как используют в настоящем документе "по существу чистый" означает, соответственно, по меньшей мере приблизительно 60% масс., как правило по меньшей мере приблизительно 70% масс., предпочтительно по меньшей мере приблизительно 80% масс., более предпочтительно по меньшей мере приблизительно 90% масс. (например, приблизительно от 90% масс. до приблизительно 99% масс.), даже более предпочтительно по меньшей мере приблизительно 95% масс. (например, приблизительно от 95% масс. до приблизительно 99% масс. или приблизительно от 98% масс. до 100% масс.), а наиболее предпочтительно по меньшей мере приблизительно 99% масс. (например, 100% масс.) продукта, содержащего соединение формулы I или его соль (например, продукта, выделяемого из реакционной смеси, обеспечивающей получение соединения или соли), состоит из соединения или соли. Уровень чистоты соединений и солей можно определять стандартным способом анализа, таким как тонкослойная хроматография, электрофорез в геле, высокоэффективная жидкостная хроматография и/или масс-спектрометрия. Если используют более одного способа анализа и способы приводят к экспериментально значимым отличиям определяемого уровня чистоты, тогда руководствуются способом, позволяющим получать наибольший уровень чистоты. Соединение или соль со 100% чистотой представляют собой соединение или соль, которые не содержат детектируемых примесей, как определяют стандартными способами анализа.
Кроме того, соединения по настоящему изобретению могут существовать в аморфной форме и/или в одной или нескольких кристаллических формах, и по существу все аморфные и кристаллические формы и их смеси соединений формулы I предназначены для включения в объем настоящего изобретения. Кроме того, соединения по настоящему изобретению могут формировать сольваты с водой (т.е., гидраты) или общеизвестными органическими растворителями. Сольваты включают стехиометрические и нестехиометрические сольваты. Такие сольваты и гидраты, в частности фармацевтически приемлемые сольваты и гидраты, соединений по настоящему изобретению, подобным образом, включены в объем настоящего изобретения наряду с несольватированными и безводными формами.
Таким образом, если не указано иначе, соединения в рамках общих структурных формул, вариантов осуществления и конкретных соединений, описанные и заявляемые в настоящем документе, включают соли, все возможные стереоизомеры и таутомеры, физические формы (например, аморфные и кристаллические формы), формы их сольватов и гидратов и любые комбинации этих форм, а также их соли, если такие формы возможны.
Изобретение также относится к способам лечения или профилактики инфекции ВИЧ, для ингибирования обратной транскриптазы ВИЧ или для лечения, профилактики или задержки начала СПИДа у нуждающегося в этом индивидуума, которые включают введение индивидууму эффективного количества соединения по изобретению или его фармацевтически приемлемой соли.
Изобретение также относится к соединению по изобретению или его фармацевтически приемлемой соли для применения в получении лекарственного средства для лечения или профилактики инфекции ВИЧ, для ингибирования обратной транскриптазы ВИЧ или для лечения, профилактики или задержки начала СПИДа у нуждающегося в этом индивидуума.
Изобретение также относится к фармацевтической композиции, содержащей эффективное количество соединения по изобретению или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель и дополнительно содержащей эффективное количество средства против ВИЧ, выбранного из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств. В рамках этого варианта осуществления средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ, ингибиторов вхождения ВИЧ и ингибиторов созревания ВИЧ.
Другие варианты осуществления настоящего изобретения включают следующее:
(a) Фармацевтическая композиция, содержащая эффективное количество соединения формулы I или вариантов осуществления A или B, как определено выше, или пролекарственного средства или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
(b) Фармацевтическая композиция, содержащая продукт, получаемый посредством комбинации (например, смешивания) эффективного количества соединения формулы I или вариантов осуществления A или B, как определено выше, или пролекарственного средства или его фармацевтически приемлемой соли, и фармацевтически приемлемый носитель.
(c) Фармацевтическая композиция по (a) или (b), дополнительно содержащая эффективное количество средства против ВИЧ, выбранного из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств.
(d) Фармацевтическая композиция по (c), где средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ и ингибиторов вхождения ВИЧ.
(e) Комбинация, которая представляет собой (i) соединение формулы I или вариантов осуществления A или B, как определено выше, или их пролекарственные средства или фармацевтически приемлемые соли и (ii) средство против ВИЧ, выбранное из группы, состоящей из противовирусных средств против ВИЧ, иммуномодуляторов и противоинфекционных средств; где каждое из соединения и средства против ВИЧ применяют в количестве, которое обеспечивает эффективность комбинации в отношении ингибирования обратной транскриптазы ВИЧ, для лечения или профилактики инфекции ВИЧ или для лечения, профилактики или задержки начала или прогрессирования СПИДа.
(f) Комбинация (e), где средство против ВИЧ представляет собой противовирусное средство, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов интегразы ВИЧ, ингибиторов слияния ВИЧ и ингибиторов вхождения ВИЧ.
(g) Способ ингибирования обратной транскриптазы ВИЧ у нуждающегося в этом индивидуума, который включает введение индивидууму эффективного количества соединений формулы I или вариантов осуществления A или B или их пролекарственных средств или фармацевтически приемлемой соли.
(h) Способ профилактики или лечения инфекции ВИЧ (например, ВИЧ-1) у нуждающегося в этом индивидуума, который включает введение индивидууму эффективного количества соединений формулы I или вариантов осуществления A или B или их пролекарственных средств или фармацевтически приемлемой соли.
(i) Способ (h), где соединение формулы I или вариантов осуществления A или B вводят в комбинации с эффективным количеством по меньшей мере одного другого противовирусного средства против ВИЧ, выбранного из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов слияния ВИЧ и ингибиторов вхождения ВИЧ.
(j) Способ профилактики, лечения или задержки начала или прогрессирования СПИДа у нуждающегося в этом индивидуума, который включает введение индивидууму эффективного количества соединений формулы I или вариантов осуществления A или B или их пролекарственных средств или фармацевтически приемлемой соли.
(k) Способ (j), где соединение вводят в комбинации с эффективным количеством по меньшей мере одного другого противовирусного средства против ВИЧ, выбранного из группы, состоящей из ингибиторов протеазы ВИЧ, ингибиторов интегразы ВИЧ, ненуклеозидных ингибиторов обратной транскриптазы ВИЧ, нуклеозидных ингибиторов обратной транскриптазы ВИЧ, ингибиторов слияния ВИЧ и ингибиторов вхождения ВИЧ.
(l) Способ ингибирования обратной транскриптазы ВИЧ у нуждающегося в этом индивидуума, который включает введение индивидууму фармацевтической композиции (a), (b), (c) или (d) или комбинации (e) или (f).
(m) Способ профилактики или лечения инфекции ВИЧ (например, ВИЧ-1) у нуждающегося в этом индивидуума, который включает введение индивидууму фармацевтической композиции (a), (b), (c) или (d) или комбинации (e) или (f).
(n) Способ профилактики, лечения или задержки начала или прогрессирования СПИДа у нуждающегося в этом индивидуума, который включает введение индивидууму фармацевтической композиции (a), (b), (c) или (d) или комбинация (e) или (f).
Настоящее изобретение также относится к соединению формулы I или вариантов осуществления A или B или их фармацевтически приемлемой соли, (i) для применения, (ii) для применения в качестве лекарственного средства или (iii) для применения в производстве/получении лекарственного средства для: (a) лечения (например, организма человека), (b) применения лекарственного средства, (c) ингибирования обратной транскриптазы ВИЧ, (d) лечения или профилактики инфекции ВИЧ или (e) лечения, профилактики или задержки начала или прогрессирования СПИДа. В этих применениях соединения по настоящему изобретению необязательно можно применять в комбинации с одним или несколькими другими средствами против ВИЧ, выбранными из противовирусных средств против ВИЧ, противоинфекционных средств и иммуномодуляторов.
Дополнительные варианты осуществления изобретения включают фармацевтические композиции, комбинации и способы, указанные в (a)-(n) выше, и применения от (i)(a)-(e) до (iii)(a)-(e), указанные в предыдущем абзаце, где соединение по настоящему изобретению, применяемое в них, представляет собой соединение по одному из вариантов осуществления, аспектов, классов, подклассов или характеристик, описанных выше. Во всех этих вариантах осуществления и т.д. соединение необязательно можно использовать в форме фармацевтически приемлемой соли.
Дополнительные варианты осуществления настоящего изобретения включают каждое из фармацевтических композиций, комбинаций, способов и применений, указанных в предшествующих абзацах, где соединение по настоящему изобретению или его соль, применяемые в них, являются по существу чистыми. В отношении фармацевтической композиции, содержащей соединение формулы I или фармацевтически приемлемый носитель и необязательно один или несколько эксципиентов, следует понимать, что термин "по существу чистый" относится к самим соединению формулы I или его соли.
Дополнительные варианты осуществления настоящего изобретения включают фармацевтические композиции, комбинации и способы, указанные в (a)-(n) выше и применения от (i)(a)-(e) до (iii)(a)-(e), указанные выше, где соответствующий ВИЧ представляет собой ВИЧ-1. Таким образом, например, в фармацевтической композиции (d) соединение формулы I применяют в количестве, эффективном против ВИЧ-1 и средство против ВИЧ представляет собой противовирусное средство против ВИЧ-1, выбранное из группы, состоящей из ингибиторов протеазы ВИЧ-1, ингибиторов обратной транскриптазы ВИЧ-1, ингибиторов интегразы ВИЧ-1, ингибиторов слияния ВИЧ-1 и ингибиторов вхождения ВИЧ-1.
Если явно не указано противоположного, все диапазоны, приводимые в настоящем документе, являются включительными. Также следует понимать, что любой приводимый в настоящем документе диапазон включает в свои пределы все поддиапазоны этого диапазона. Например, функциональная группа, описанная как необязательно замещенная "заместителями в количестве от 1 до 3", предназначена для включения в качестве ее разновидностей, такой функциональной группы, замещенной заместителями в количестве от 1 до 3, 2 или 3 заместителями, 3 заместителями, 1 или 2 заместителями, 2 заместителями или 1 заместителем. В качестве другого примера, дозировка в диапазоне от 1 до 500 миллиграммов означает, что дозировка может составлять 1 мг, или 500 мг, или любое количество между ними.
Способы по настоящему изобретению включают применение соединений по настоящему изобретению для ингибирования обратной транскриптазы ВИЧ (например, ВИЧ-1 дикого типа и других штаммов), профилактики или лечения инфекции ВИЧ и профилактики, лечения или задержки начала или прогрессирования последующих патологических состояний, таких как СПИД. Профилактика СПИДа, лечение СПИДа, задержка начала или прогрессирования СПИДа или лечение или профилактика инфекции ВИЧ определены, но не ограничиваясь этим, как лечение широкого диапазона состояний инфекции ВИЧ: СПИД, ARC, симптоматических и бессимптомных, и фактическое или потенциальное воздействие ВИЧ. Например, настоящее изобретение можно применять для лечения инфекции ВИЧ при подозрении на риск ВИЧ при применении таких воздействий, как переливание крови, обмен жидкостями тела, укусы, случайные уколы иглами или соприкосновение с кровью пациента при хирургии. В качестве другого примера настоящее изобретение также можно применять для подавления передачи ВИЧ от беременной женщины, инфицированной ВИЧ, ее нерожденному ребенку или от ВИЧ-инфицированной женщины, ухаживающей за ребенком (т.е., грудное вскармливание), ребенку посредством введения эффективного количества соединения формулы I.
Термин "введение" и его варианты (например, "ввод" соединения) по отношению к соединению формулы I означают предоставление соединения индивидууму, нуждающемуся в лечении или профилактике, и включает самостоятельное введение и введение пациенту другим индивидуумом. Когда соединение предоставляют в комбинации с одним или несколькими другими активными средствами (например, противовирусными средствами, пригодными для лечения или профилактики инфекции ВИЧ или СПИДа), каждый из "введения" и его вариантов следует понимать, как включающие предоставление соединения и других средств одновременно или в различное время. Когда средства комбинации вводят одновременно, их можно вводить совместно в одной композиции или их можно вводить раздельно.
В контексте настоящего документа, термин "композиция" предназначен для включения продукта, содержащего указанные ингредиенты, а также любого продукта, который получают, комбинируя указанные ингредиенты.
Под "фармацевтически приемлемым" подразумевают, что ингредиенты фармацевтической композиции должны быть совместимы друг с другом и не вредны для их реципиента.
В контексте настоящего документа, термин "индивидуум" или "пациент" относится к животному, предпочтительно млекопитающему, наиболее предпочтительно человеку, которое является объектом лечения, наблюдения или эксперимента.
В контексте настоящего документа, термин "эффективное количество" означает количество, достаточное для ингибирования обратной транскриптазы ВИЧ, ингибирования репликации ВИЧ, оказания профилактического действия и/или оказания терапевтического действия после введения. Одним из вариантов "эффективного количества" является "терапевтически эффективное количество", которое представляет собой количество соединения, которое эффективно для ингибирования репликации ВИЧ (которое также может быть указано в настоящем документе как "эффективное для ингибирования количество"), лечения инфекции ВИЧ, лечения СПИДа, задержки начала СПИДа и/или замедления прогрессирования СПИДа у пациента. Другой вариант "эффективного количества" представляет собой "профилактически эффективное количество", которое представляет собой количество соединения, которое эффективно для профилактики инфекции ВИЧ или профилактики СПИДа у пациента. Следует понимать, что эффективное количество может быть одновременно терапевтически эффективным количеством, например, для лечения инфекции ВИЧ, и профилактически эффективным количеством, например, для профилактики или снижения риска развития СПИДа. Когда соединение формулы I вводят в виде соли, указание количества соединения приводят для свободной формы (т.е., не являющейся солью формы) соединения.
В способах по настоящему изобретению (например, ингибирования обратной транскриптазы ВИЧ, лечения или профилактики инфекции ВИЧ, ингибирования репликации ВИЧ, лечения или профилактики СПИДа, задержки начала СПИДа или задержки или замедления прогрессирования СПИДа), соединения формулы I, необязательно в форме соли, можно вводить средствами, которые обеспечивают контакт активного средства с участком действия средства. Их можно вводить общепринятыми способами, доступными для применения в сочетании с фармацевтическими средствами, отдельно, в виде индивидуальных терапевтических средств, или в комбинации терапевтических средств. Их можно вводить отдельно, но, как правило, вводят с фармацевтическим носителем, выбираемым на основании выбранного пути введения и стандартной фармацевтической практики. Например, соединения по изобретению можно вводить перорально, парентерально (включая подкожные инъекции, внутривенные, внутримышечные, внутригрудинные инъекции или инфузионными способами), посредством ингаляционного спрея или ректально, в форме единичной дозы фармацевтической композиции, содержащей эффективное количество соединения и общепринятые нетоксические фармацевтически приемлемые носители, адъюванты и носители. Жидкие препараты, подходящие для перорального введения (например, суспензии, сиропы, эликсиры и т.п.) можно получать известными в данной области способами и можно применять любые из обычных носителей, такие как вода, гликоли, масла, спирты и т.п. Твердые препараты, подходящие для перорального введения (например, порошки, пилюли, капсулы и таблетки), можно получать известными в данной области способами и можно применять такие твердые эксципиенты как различные виды крахмала, сахара, каолин, смазочные средства, связывающие средства, дезинтегрирующие средства и т.п. Парентеральные композиции можно получать известными в данной области способами и, как правило, в качестве носителя применяют стерильную воду и необязательно другие ингредиенты, такие как способствующее растворению средство. Инъецируемые растворы можно получать известными в данной области способами, где носитель содержит солевой раствор, раствор глюкозы или раствор, содержащий смесь солевого раствора и глюкозы. Дополнительное описание способов, пригодных для использования в получении фармацевтических композиций для применения по настоящему изобретению, и ингредиентов, пригодных для использования в указанной композиции, находится в Remington's Pharmaceutical Sciences, 18th edition, edited by A. R. Gennaro, Mack Publishing Co., 1990 и в Remington - The Science and Practice of Pharmacy, 22nd Edition, опубликовано Pharmaceutical Press and Philadelphia College of Pharmacy at University of the Sciences, 2012, ISBN 978 0 85711-062-6 и предшествующие издания.
Соединения формулы I можно вводить перорально в диапазоне доз от 0,001 до 1000 мг/кг массы тела млекопитающего (например, человека) в сутки в однократной дозе или в дробных дозах. Один из предпочтительных диапазонов доз представляет собой от 0,01 до 500 мг/кг масса тела в сутки перорально в однократной дозе или в дробных дозах. Другим предпочтительным диапазоном доз является от 0,1 до 100 мг/кг масса тела в сутки перорально в одной или дробных дозах. Для перорального введения композиции можно предоставлять в форме таблеток или капсул, содержащих от 1 до 500 миллиграммов соединения по изобретению, в частности 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400 и 500 миллиграммов для симптоматического подбора дозировки пациенту, подвергаемому лечению. Указанный уровень дозирования и частота дозирования для каждого конкретного пациента могут варьировать и зависят от ряда факторов, включая активность конкретного применяемого соединения, метаболической стабильности и длительности действия этого соединения, возраста, массы тела, общего состояния здоровья, пола, диеты, способа и времени введения, скорости выведения, комбинации лекарственных средств, тяжести конкретного состояния, и хозяина, проходящего терапию. Соединения по изобретению можно вводить в виде однократной дозы, раз в сутки или менее часто.
Если явно не указано противоположного, указания в предшествующих абзацах или в другом месте настоящего документа на введение количества соединения по изобретению представляют собой указания на количество (т.е., дозировку) соответствующего соединения формулы I, не содержащего соли. Как указано выше, настоящее изобретение также относится к применению соединений формулы I с одним или несколькими средствами против ВИЧ. "Средство против ВИЧ" представляет собой любое средство, которое прямо или опосредованно эффективно для ингибирования обратной транскриптазы ВИЧ или другого фермента или белка, необходимого для репликации или инфицирования ВИЧ, для лечения или профилактики инфекции ВИЧ и/или лечения, профилактики или задержки начала или прогрессирования СПИДа. Следует понимать, что средство против ВИЧ эффективно в лечении, профилактике или задержке начала или прогрессирования инфекции ВИЧ или СПИДа и/или заболеваний или состояний, возникающих вследствие них или ассоциированных с ними. Например, соединения по настоящему изобретению можно эффективно вводить, в периоды перед воздействием и/или после воздействия, в комбинации с эффективными количествами одного или нескольких средств против ВИЧ, выбранных из противовирусных средств против ВИЧ, иммуномодуляторов, противоинфекционных средств или вакцин, пригодных для лечения инфекции ВИЧ или СПИДа. Подходящие противовирусные средства против ВИЧ для применения в комбинации с соединениями по настоящему изобретению включают, например, средства, перечисленные в таблице 1, ниже:
Таблица 1
EI=ингибитор вхождения; FI=ингибитор слияния; InI=ингибитор интегразы; PI=ингибитор протеазы; nRTI=нуклеозидный ингибитор обратной транскриптазы; nnRTI=ненуклеозидный ингибитор обратной транскриптазы. Некоторые из лекарственных средств, перечисленных в таблице, используют в форме соли; например, сульфат абакавира, мезилат делавирдина, сульфат индинавира, сульфат атазанавира, мезилат нелфинавира, мезилат саквинавира.
Следует понимать, что область комбинаций соединений по настоящему изобретению со средствами против ВИЧ не ограничена противовирусными средствами против ВИЧ, приведенными в таблице A, но в принципе включает любую комбинацию с любой фармацевтической композицией, пригодной для лечения или профилактики СПИДа. Как правило, противовирусные средства против ВИЧ и другие средства применяют в этих комбинациях в их общепринятых диапазонах доз и со схемами лечения, как опубликовано в данной области, включая, например, дозировки, описанные в Physicians' Desk Reference, Thomson PDR, Thomson PDR, издание 57 (2003), издание 58 (2004) или издание 59 (2005) и current Physicians' Desk Reference (68th ed.). (2014), Montvale, NJ: PDR Network. Диапазоны доз соединения по изобретению в этих комбинациях могут быть такими, как диапазоны, указанные выше.
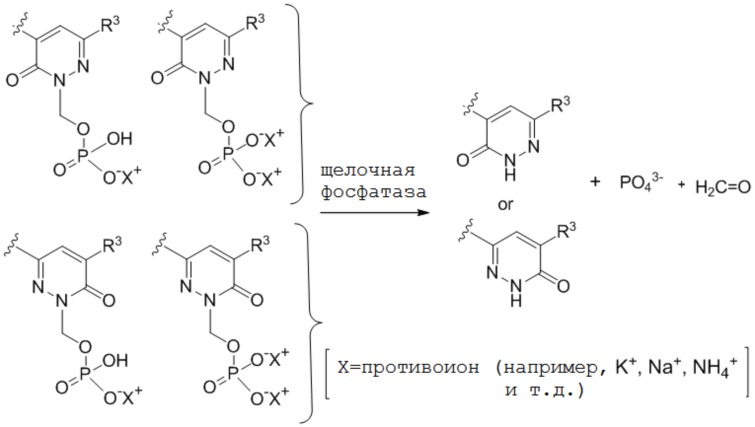
Не желая быть связанными какой либо конкретной теорией, полагают, что соединения по настоящему изобретению действуют в качестве пролекарственных средств, где соединение является относительно стабильным при низком pH (например, pH от 1 до 3), но посредством гидролиза или реакции замыкания цикла преобразуется в свое свободное основание при физиологическом pH (например, pH приблизительно 7), таким образом, высвобождая in vivo активное вещество. Полагают, что в просвете кишечника преимущественно отщепляется фосфатная группа R4 ферментами фосфатазами и вторично на кисточковой каемке фосфатазами, высвобождая in vivo активное вещество. Преобразование можно изобразить следующим образом:
Аббревиатуры и сокращения, используемые в настоящем документе включают следующие:
Приводимые ниже примеры служат только для иллюстрации изобретения и его практического применения. Примеры не следует рассматривать, как ограничение объема или сущности изобретения. В этих примерах термин "комнатная температура" относится к температуре в диапазоне приблизительно от 20°C до приблизительно 25°C.
Промежуточное соединение A
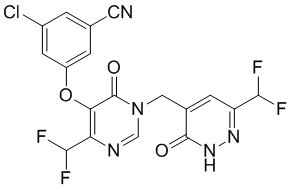
3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1: 6-(дифторметил)пиримидин-4(3H)-он
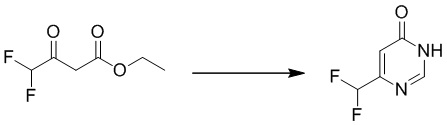
Смесь натрия (2,91 г, 126,5 ммоль) в метаноле (70 мл) перемешивали при комнатной температуре в течение 30 минут, затем добавляли ацетат формамидина (6,3 г, 60 ммоль) и этил 4,4-дифтор 3-оксобутаноат (5,0 г, 30,1 ммоль). Смесь перемешивали при 80°C в течение 4 часов. После охлаждения до температуры окружающей среды смесь подкисляли HCl до pH=6 и экстрагировали этилацетатом (200 мл×5). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 6-(дифторметил)пиримидин-4(3H)-она. MS (ESI): масса/заряд 147 (M+H) +
Этап 2: 6-(дифторметил)пиримидин-4(3H)-он
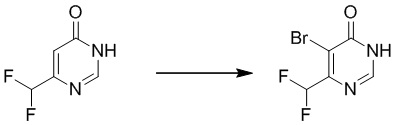
К смеси соединения 6-(дифторметил)пиримидин-4(3H)-она (2,0 г, 13,7 ммоль) и ацетата калия (4,0 г, 41,4 ммоль) в уксусной кислоте (20 мл) добавляли бром (3,3 г, 20,5 ммоль) в атмосфере азота. Полученную смесь перемешивали при 80°C в течение 4 часов. Затем смесь выливали в ледяную воду, и осадок собирали посредством фильтрования с получением 5-бром-6-(дифторметил)пиримидин-4(3H)-она. MS (ESI): масса/заряд 225, 227 (M+H) +
Этап 3: 5-бром-6-(дифторметил)-3-(4-метоксибензил)пиримидин-4(3H)-он
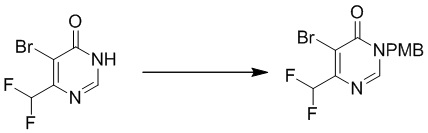
Смесь 5-бром-6-(дифторметил)пиримидин-4(3H)-она (1,01 г, 4,49 ммоль), PMBCl (735 мг, 4,71 ммоль), карбоната калия (1,24 г, 8,98 ммоль) в DMF (10 мл) перемешивали при температуре окружающей среды в течение 4 часов в атмосфере азота. Добавляли 15 мл воды, и осадок собирали посредством фильтрования с получением 5-бром-6-(дифторметил)-3-(4-метоксибензил)пиримидин-4(3H)-она. MS (ESI): масса/заряд 345, 347 (M+H) +
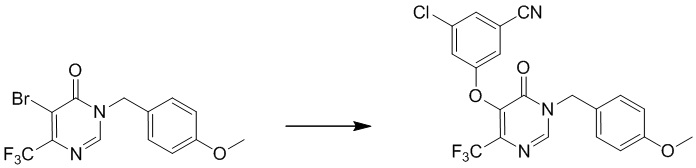
Этап 4: 3-хлор-5-((4-(дифторметил)-1-(4-метоксибензил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
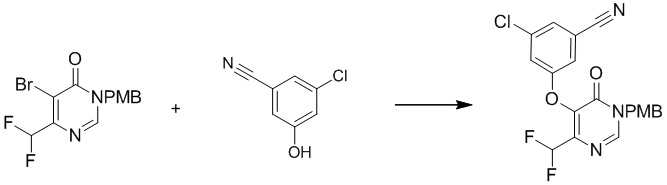
Смесь 3-хлор-5-гидроксибензонитрила (1,57 г, 11,6 ммоль), 5-бром-6-(дифторметил)-3-(4-метоксибензил)пиримидин-4(3H)-она (2,0 г, 5,81 ммоль) и t-BuOK (1,43 г, 12,8 ммоль) в NMP (10 мл) перемешивали при 120°C в течение ночи. После охлаждения до температуры окружающей среды смесь разбавляли 20 мл воды и экстрагировали этилацетатом (100 мл×3). Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Затем добавляли метанол (10 мл), и осадок собирали посредством фильтрования с получением 3-хлор-5-((4-(дифторметил)-1-(4-метоксибензил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI): масса/заряд 418, 420 (M+H) +
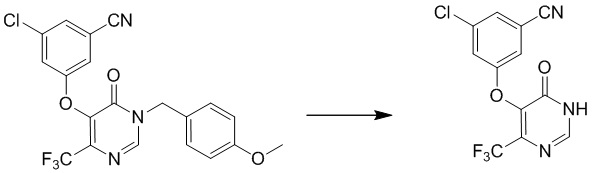
Этап 5: 3-хлор-5-((4-(дифторметил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
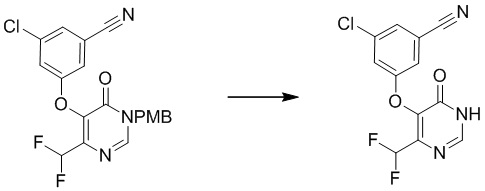
Раствор соединения 3-хлор-5-((4-(дифторметил)-1-(4-метоксибензил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила (400 мг, 0,96 ммоль) в TFA (5 мл) перемешивали при микроволновом излучении при 100°C в течение 10 мин. После охлаждения до температуры окружающей среды смесь концентрировали при пониженном давлении. Затем добавляли метанол (10 мл), и осадок собирали посредством фильтрования с получением 3-хлор-5-((4-(дифторметил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI): масса/заряд 298, 300 (M+H) +
Этап 6: 6-метоксипиридазин-3-карбальдегид
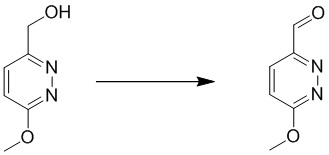
К перемешиваемому раствору (6-метоксипиридазин-3-ил)метанола (13 г, 93 ммоль) в 500 мл безводного дихлорметана добавляли периодинан Десса-Мартина (59 г, 139 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре. Смесь разбавляли дихлорметаном, промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир:этилацетат (от 15:1 до 10:1) в качестве элюента) с получением 6-метоксипиридазин-3-карбальдега. MS (ESI) масса/заряд 139 (M+H)+
Этап 7: 3-(дифторметил)-6-метоксипиридазин
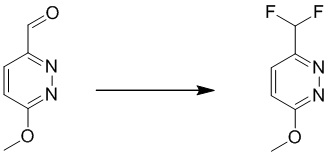
К перемешиваемому раствору 6-метоксипиридазин-3-карбальдегида (6,0 г, 43,4 ммоль) в 100 мл безводного дихлорметана добавляли DAST (22,7 г, 141,3 ммоль). Смесь перемешивали в течение 1 часа при комнатной температуре. Смесь разбавляли дихлорметаном, промывали водным бикарбонатом натрия (0,5 Н, 100 мл), водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат (15:1 до 10: 1) в качестве элюента) с получением 3-(дифторметил)-6-метоксипиридазин. MS (ESI) масса/заряд 161 (M+H)+
Этап 8: 4-(трет-бутоксиметил)-6-(дифторметил)-3-метоксипиридазин
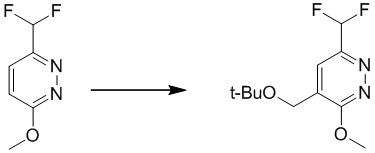
К раствору трет-бутоксиуксусной кислоты (0,92 г, 6,88 ммоль) в THF/воде (20 моль%, 7,76 мл) добавляли 3-(дифторметил)-6-метоксипиридазин (0,7 г, 4,3 ммоль) и AgNO3 (74 мг, 0,43 ммоль). Смесь дегазировали N2 с перемешиванием при комнатной температуре. Затем смесь нагревали до 70°C, а затем по каплям добавляли (NH4)2S2O8(1,7 г, 7,31 ммоль) в воде (10 мл). После добавления смесь перемешивали при 70-80°C в течение 40 минут. После охлаждения до комнатной температуры смесь экстрагировали этилацетатом (10 мл × 3). Объединенные органические слои отмывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат (15:1 до 10:1) в качестве элюента) с получением 4-(трет-бутоксиметил)-6-(дифторметил)-3-метоксипиридазина.
MS (ESI) масса/заряд 247 (M+H)+
Этап 9: (6-(дифторметил)-3-метоксипиридазин-4-ил)метанол
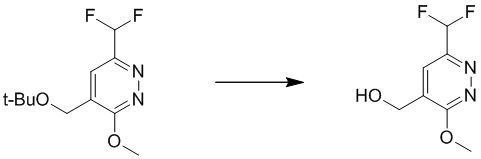
Раствор 4-(трет-бутоксиметил)-6-(дифторметил)-3-метоксипиридазина (480 мг, 1,95 ммоль) в THF/DCE (1,3 мл/4,5 мл) перемешивали при 60°C в течение 1 часа. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении. Остаток очищали посредством препаративной TLC (петролейный эфир/этилацетат (2:1) в качестве элюента) с получением (6-(дифторметил)-3-метоксипиридазин-4-ил)метанола. MS (ESI) масса/заряд 191 (M+H)+
Этап 10: 4-(хлорметил)-6-(дифторметил)-3-метоксипиридазин
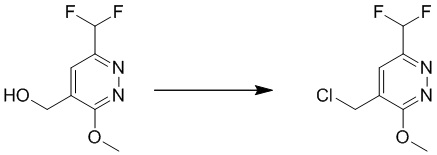
К раствору соединения (6-(дифторметил)-3-метоксипиридазин-4-ил)метанола (600 мг, 3,1 ммоль) в безводном дихлорметане (20 мл) по каплям добавляли метансульфонилхлорид (1,08 г, 9,4 ммоль) и DIPEA (1,22 г, 9,4 ммоль), соответственно, при 0°C. Смесь перемешивали при комнатной температуре в течение 4 часов. Затем смесь гасили водой и экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 4-(хлорметил)-6-(дифторметил)-3-метоксипиридазина. MS (ESI) масса/заряд 209, 211 (M+H) +
Этап 11: 3-хлор-5-((4-(дифторметил)-1-((6-(дифторметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
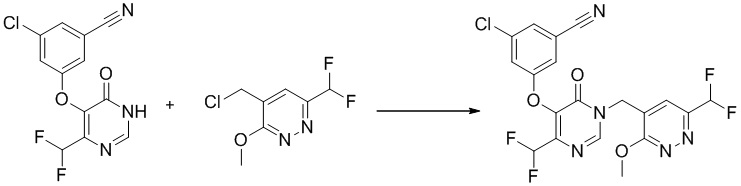
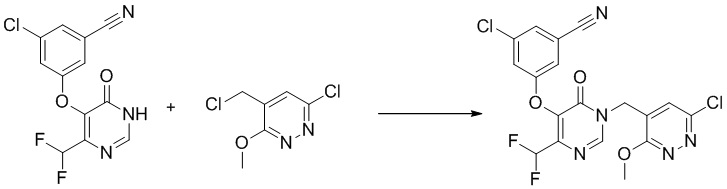
К раствору 3-хлор-5-((4-(дифторметил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила (150 мг, 0,5 ммоль) в DMF (15 мл) добавляли K2CO3 (139 мг, 1,0 ммоль), LiBr (88 мг, 1,0 ммоль) и 4-(хлорметил)-6-(дифторметил)-3-метоксипиридазин (105 мг, 0,5 ммоль). Полученную смесь перемешивали при комнатной температуре в течение ночи, разбавляли водой и экстрагировали EtOAc. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали при пониженном давлении с получением 3-хлор-5-((4-(дифторметил)-1-((6-(дифторметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила без дополнительной очистки. MS (ESI) масса/заряд 470, 472 (M+H)+
Этап 12: 3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
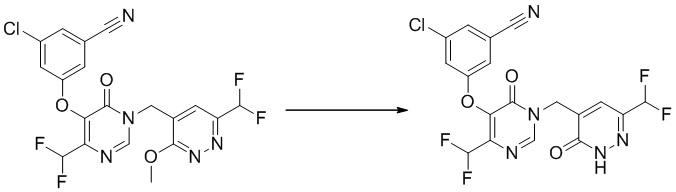
К смеси соединения 3-хлор-5-((4-(дифторметил)-1-((6-(дифторметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила (200 мг, 0,42 ммоль) и KI (142 мг, 0,84 ммоль) в ацетонитриле (3 мл) добавляли TMSCl (93 мг, 0,84 ммоль) при комнатной температуре. Полученную смесь перемешивали при 70°C в течение 1,5 часа. После охлаждения до комнатной температуры смесь разбавляли EtOAc и промывали водным Na2S2O3 и насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ с получением желаемого продукта 3-хлор-5-((4-(дифторметил)-1-((6-(дифторметил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила. 1H ЯМР: (метанол-d4, 400 МГц) δ13,60 (с, 1H), 8,66 (с, 1H), 7,71 (с, 1H), 7,62 (с, 2H), 7,59 (с, 1H), 6,98 (т, J=52,0 Гц, 1H), 6,77 (т, J=54,0 Гц, 1H), 4,97 (с, 2H).
MS (ESI) масса/заряд 456, 458 (M+H)+
Пример 1
(5-((5-(3-хлор-5-цианофенокси)-4-(дифторметил)-6-оксопиримидин-1(6 H)-ил )метил)-3-(дифторметил)-6-оксопиридазин-1(6 H )-ил)метилдигидрофосфат
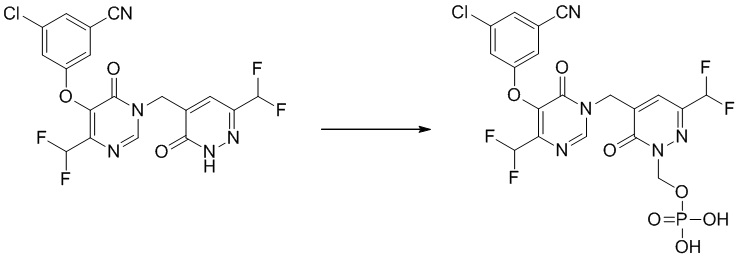
Указанное выше соединение получали, следуя процедурам, сходным с описанными в примере 7 на этапах 1-2. 1H ЯМР: (500 МГц, DMSO-d6) δ 8,70 (с, 1H), 7,65-7,75 (м, 4H), 6,74-7,02 (м, 2H), 5,71 (д, J=7,8 Гц, 2H), 5,06 (с, 2H). MS: 566 (M+H)+
Промежуточное соединение B
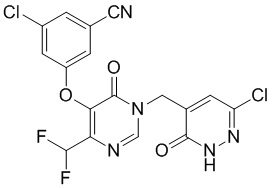
3-хлор-5-((1-((6-хлор-3-оксо-2,3-дигидропиридазин-4-ил)метил)-4-(дифторметил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1: Метил-3,6-дихлорпиридазин-4-карбоксилат

В суспензию 3,6-дихлорпиридазин-4-карбоновой кислоты (5,5 г, 28,5 ммоль) в DCM (50,0 мл) и MeOH (10 мл) медленно добавляли триметилсилилдиазометан (2 М в гексане, 15 мл, 30,0 ммоль) при 0°C. После добавления она становилась прозрачным раствором. Его перемешивали в течение 30 минут и добавляли еще 15 мл триметилсилилдиазометана и перемешивали в течение 30 минут. Его гасили 2 мл уксусной кислоты, концентрировали и очищали посредством ISCO (80 г, 0-40% этилацетат в гексане) с получением указанного в заголовке соединения. MS (ESI): масса/заряд 206 (M+H) +
Этап 2: Метил-6-хлор-3-метоксипиридазин-4-карбоксилат
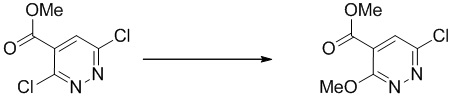
Метил-3,6-дихлорпиридазин-4-карбоксилат (2 г, 9,66 ммоль) навешивали в чистую сухую колбу, снабженную магнитной мешалкой. Ее герметизировали и дважды продували азотом и растворяли в безводном THF (40 мл). Раствор охлаждали в ледяной водяной бане и одной порцией добавляли метоксид натрия (0,69 г, 12,77 ммоль). Смесь перемешивали в течение 30 минут. LC-MS демонстрировала завершение реакции. Ее гасили насыщенным водным хлоридом аммония (20 мл). Смесь экстрагировали этилацетатом (3 × 40 мл). Объединенные органические слои отмывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством ISCO (80 г, 0-30% этилацетат в гексане) с получением указанного в заголовке соединения. MS (ESI): масса/заряд 203 (M+H) +
Этап 3: 6-хлор-3-метоксипиридазин-4-карбоновая кислота
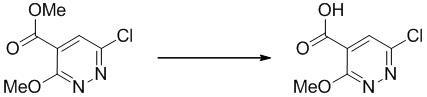
Раствор метил-6-хлор-3-метоксипиридазин-4-карбоксилата (510 мг, 2,52 ммоль) в тетрагидрофуране (5 мл) и метаноле (5,00 мл) обрабатывали 4 M водным LiOH (5 мл, 20,00 ммоль) в течение 15 минут. Его нейтрализовали 1 Н HCl и концентрировали. Остаток сушили в вакууме и использовали без очистки. MS (ESI): масса/заряд 189 (M+H) +
Этап 4: (6-хлор-3-метоксипиридазин-4-ил)метанол
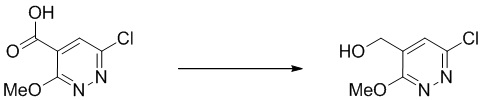
Смесь 6-хлор-3-метоксипиридазин-4-карбоновой кислоты (270 мг, 1,432 ммоль) и карбонилдиимидазола (697 мг, 4,30 ммоль) в THF (12 мл) перемешивали в течение 1 часа при комнатной температуре. Раствор охлаждали до 0°C и добавляли боргидрид натрия (271 мг, 7,16 ммоль) с последующим добавлением воды (4 мл). Смесь перемешивали в течение 15 минут и гасили 5 мл насыщенным водным хлоридом аммония, экстрагировали этилацетатом (4 × 20 мл). Объединенные органические слои отмывали насыщенным солевым раствором (5 мл), сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали посредством ISCO (40 г, 0-50% этилацетат в гексане) с получением указанного в заголовке соединения. MS (ESI): масса/заряд 175 (M+H) +
Этап 5: 6-хлор-4-(хлорметил)-3-метоксипиридазин

К раствору (6-хлор-3-метоксипиридазин-4-ил)метанола (100 мг, 0,573 ммоль) в DCM (5 мл) при 0°C добавляли метансульфонилхлорид (0,134 мл, 1,718 ммоль) и основание Хунига (0,300 мл, 1,718 ммоль). Смесь перемешивали при 0°C в течение 15 минут и позволяли нагреваться до комнатной температуры в течение ночи. Ее концентрировали при пониженном давлении. Остаток очищали посредством ISCO (24 г, 0-30% градиент EtOAc/гексан) с получением указанного в заголовке соединения. MS (ESI): масса/заряд 192 (M+H)
Этап 6: 3-хлор-5-((1-((6-хлор-3-метоксипиридазин-4-ил)метил)-4-(дифторметил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Соединение получали тем же способом, как промежуточное соединение A, этап 11.
Этап 7: 3-хлор-5-((1-((6-хлор-3-оксо-2,3-дигидропиридазин-4-ил)метил)-4-(дифторметил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
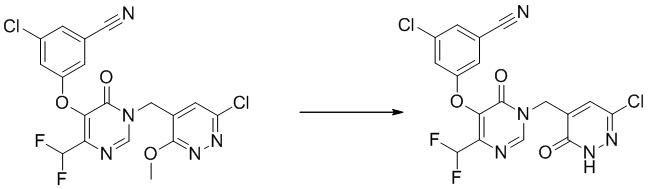
Соединение получали тем же способом как промежуточное соединение A, этап 12.
1 H ЯМР: (DMSO-d6, 500 МГц) δ8,66 (с, 1H), 7,75 (с, 1H), 7,69 (с, 1H), 7,65 (м, 1H), 7,54 (с, 1H), 7,02 (т, J=52,0 Гц, 1H), 4,96 (с, 2H). MS (ESI) масса/заряд 440,1 (M+H)+
Пример 2
(3-хлор-5-((5-(3-хлор-5-цианофенокси)-4-(дифторметил)-6-оксопиримидин-1(6 H )-ил)метил)-6-оксопиридазин-1(6 H )-ил)метилдигидрофосфат
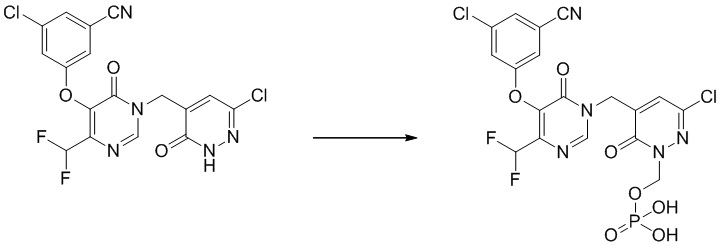
Указанное выше соединение получали, следуя процедурам, сходным с описанными в примере 7 на этапах 1-2. 1H ЯМР: (500 МГц, DMSO-d6) δ 8,66 (с, 1H), 7,64-7,75 (м, 4H), 7,03 (т, J=55 Гц, 1H), 5,63 (д, J=8,05 Гц, 2H), 5,01 (с, 2H). MS: 550 (M+H)+
Промежуточное соединение C
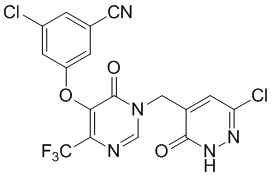
3-хлор-5-((1-((6-хлор-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1:
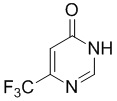
Eur. J. Org. Chem. 2004, 3714-37188.
Этап 2: 5-бром-6-(трифторметил)-4(3H)-пиримидинон
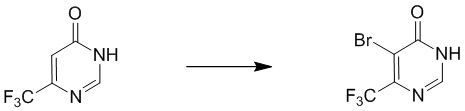
К раствору 6-(трифторметил)пиримидин-4(3H)-она (0,3 г, 1,8 ммоль) в уксусной кислоте (2 мл) добавляли CH3COOK (0,54 г, 5,5 ммоль). Затем к смеси по каплям добавляли раствор Br2 в уксусной кислоте (1 мл). Смесь нагревали до 80°C и перемешивали в течение ночи. После охлаждения до комнатной температуры смесь разбавляли EtOAc, промывали водой и насыщенным солевым раствором, сушили над Na2SO4 и выпаривали с получением 5-бром-6-(трифторметил)-4(3H)-пиримидинона.
Этап 3: 5-бром-3-(4-метоксибензил)-6-(трифторметил)пиримидин-4(3H)-он
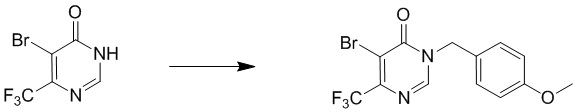
К раствору 5-бром-6-(трифторметил)пиримидин-4(3H)-она (190 мг, 0,91 ммоль) в DMF (2 мл) добавляли K2CO3 (250 мг, 1,82 ммоль) и PMBCl (210 мг, 1,3 ммоль). Смесь перемешивали при комнатной температуре в течение 5 часов. Смесь выливали в воду и экстрагировали EtOAc (40 мл × 3). Органический слой отмывали водой и насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (5:1 до 1:1) в качестве элюента) с получением 1 5-бром-3-(4-метоксибензил)-6-(трифторметил)пиримидин-4(3H)-он. 1H ЯМР: J000159069 H11896-016-3 CDCl3, 400 МГц δ 7,97 (с, 1H, ArH), 7,27 (д, J=8,8, 2H, ArH), 6,87 (д, J=8,8, 2H, ArH), 5,04 (с, 2H, CH), 3,78 (с, 3H, CH).
Этап 4: 3-хлор-5-(1-(4-метоксибензил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-илокси)бензонитрил
К раствору 5-бром-3-(4-метоксибензил)-6-(трифторметил)пиримидин-4(3H)-она (5 г, 13,8 ммоль) в NMP (50 мл) добавляли K2CO3 (5,7 г, 41,3 ммоль) и 3-хлор-5-гидрокси-бензонитрил (3,2 г, 20,7 ммоль). Смесь перемешивали при 120°C в течение 20 часов. Смесь выливали в воду и экстрагировали EtOAc (60 мл×3). Органический слой отмывали водой и насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии (петролейный эфир/этилацетат (5:1 до 1:1) в качестве элюента) с получением 3-хлор-5-(1-(4-метоксибензил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-илокси)бензонитрила. 1H ЯМР: J000169946 H11896-128-3 DMSO, 400 МГц δ 8,86 (с, 1H, ArH), 7,76 (с, 1H, ArH), 7,70 (с, 1H, ArH), 7,68 (с, 1H, ArH), 7,34 (д, J=8,6, 2H, ArH), 6,90 (д, J=8,6, 2H, ArH), 5,10 (с, 2H, CH), 3,72 (с, 3H, CH).
Этап 5: 3-хлор-5-(6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-илокси)бензонитрил
К раствору 3-хлор-5-(1-(4-метоксибензил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-илокси)бензонитрила (2 г, 4,6 ммоль) в CH3CN (20 мл) и H2O (8 мл) частями добавляли Ce(NH4)2(NO3)6 (10 г, 18,4 ммоль). Смесь перемешивали при комнатной температуре в течение ночи, а затем выливали в воду и экстрагировали EtOAc (60 мл × 3). Органический слой отмывали водой и насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (5:1 до 1:1) в качестве элюента) с получением 3-хлор-5-(6-оксо-4-(трифтор метил)-1,6-дигидропиримидин-5-илокси)бензонитрила. 1H ЯМР: J000170654 H11896-138-3 DMSO, 400 МГц δ 13,59 (с, 1H, NH), 8,36 (с, 1H, ArH), 7,76 (с, 1H, ArH), 7,73 (с, 1H, ArH), 7,70 (с, 1H, ArH).
Этап 6: 3-хлор-5-((1-((6-хлор-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
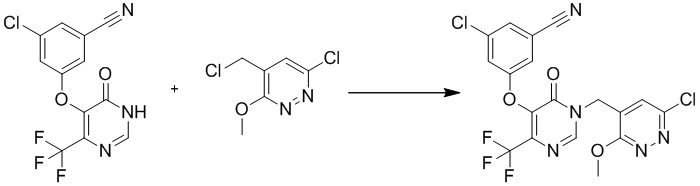
К раствору 3-хлор-5-((6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (100 мг, 0,32 ммоль) в DMF (5 мл) добавляли 6-хлор-4-(хлорметил)-3-метоксипиридазин (55 мг, 0,29 ммоль, пример 2, этап 5) и K2CO3 (80 мг, 0,58 ммоль). Полученную смесь перемешивали при 80°C в течение 2 часов. После охлаждения до комнатной температуры смесь разбавляли водой и экстрагировали EtOAc. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением желаемого продукта 3-хлор-5-((1-((6-хлор-3-метокси-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила, который использовали для следующего этапа без дополнительной очистки. MS (ESI) масса/заряд 472, 474, 476 (M+H)+
Этап 7: 3-хлор-5-((1-((6-хлор-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
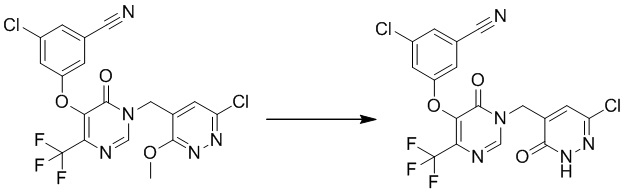
К смеси соединения 3-хлор-5-((1-((6-хлор-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (110 мг, 0,23 ммоль) и KI (77 мг, 0,46 ммоль) в ацетонитриле (10 мл) добавляли TMSCl (50 мг, 0,46 ммоль) при комнатной температуре. Полученную смесь перемешивали в течение 1 часа при 70°C. После охлаждения до комнатной температуры смесь разбавляли EtOAc и промывали водным Na2S2O3 и насыщенным солевым раствором, сушили над безводным Na2SO4 и концентрировали при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ с получением 3-хлор-5-((1-((6-хлор-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. 1H ЯМР: (метанол-d4, 400 МГц):δ 8,60 (с, 1H), 7,68 (с, 1H), 7,47 (с, 1H), 7,28 (с, 1H), 7,24 (с, 1H), 5,07 (с, 2H), 1,92 (т, J=18,4 Гц, 6H).
MS (ESI) масса/заряд 458, 460, 462 (M+H)+
Пример 3
(3-хлор-5-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6 H )-ил)метил)-6-оксопиридазин-1(6 H )-ил)метилдигидрофосфат
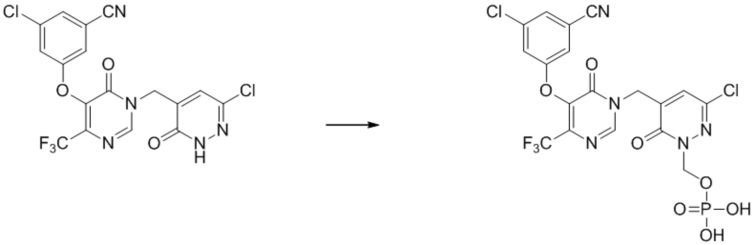
Указанное выше соединение получали, следуя процедурам, сходным с описанными в примере 7 на этапах 1-2. 1H ЯМР: (500 МГц, DMSO-d6) 8,79 (с, 1H), 7,80-7,70 (м, 3H), 7,70 (с, 1H), 5,60 (д, 2H), 5,00 (с, 2H). MS:568 (M+H)+
Промежуточное соединение D
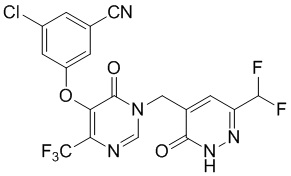
3-хлор-5-((1-((6-(дифторметил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1: метил-5-(гидроксиметил)-6-метоксипиридазин-3-карбоксилат
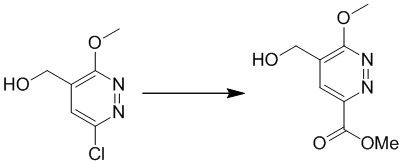
К раствору (6-хлор-3-метоксипиридазин-4-ил)метанола (4,6 г, 26,4 ммоль), триэтиламина (7,4 мл) и Pd(dppf)2Cl2 (0,5 г, 1 ммоль) в 30 мл метанола и этилацетата (10 мл) перемешивали в атмосфере монооксида углерода (344,7 кПа) при 70°C в течение ночи. Затем реакционную смесь выливали в воду, экстрагировали этилацетатом (15 мл × 3). Органические экстракты отмывали водой и насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке (петролейный эфир/этилацетат (5:1 до 2:1) в качестве элюента) с получением метил-5-(гидроксиметил)-6-метоксипиридазин-3-карбоксилата. MS (ESI) масса/заряд 199 (M+H)+
Этап 2: метил-5-(((трет-бутилдиметилсилил)окси)метил)-6-метоксипиридазин-3-карбоксилат
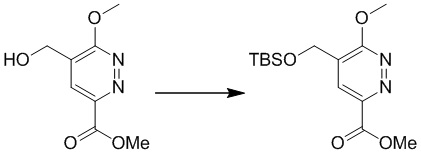
К раствору метил-5-(гидроксиметил)-6-метоксипиридазин-3-карбоксилата (2,1 г, 10,6 ммоль) в THF (150 мл) добавляли TBSCl (4,55 г, 30,2 ммоль) и имидазол (2,05 г, 30,2 ммоль) при комнатной температуре. Затем полученную реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь фильтровали, и фильтрат промывали водой. Органический слой сушили над сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением метил-5-(((трет-бутилдиметилсилил)окси)метил)-6-метоксипиридазин-3-карбоксилата. MS (ESI) масса/заряд 313 (M+H)+
Этап 3: (5-(((трет-бутилдиметилсилил)окси)метил)-6-метоксипиридазин-3-ил)метанол
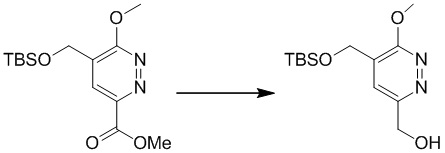
К раствору метил-5-(((трет-бутилдиметилсилил)окси)метил)-6-метоксипиридазин-3-карбоксилата (2,1 г, 6,7 ммоль) в этаноле (15 мл) добавляли NaBH4 (0,38 г, 10,0 ммоль) и CaCl2 (0,37 г, 3,4 ммоль) при 0°C. Смесь перемешивали в течение 1 часа при комнатной температуре, затем гасили посредством добавления воды (20 мл), подкисляли до pH=8 с использованием раствора HCl (2 M) и экстрагировали этилацетатом (15 мл × 3). Комбинированные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (5-(((трет-бутилдиметилсилил)окси)метил)-6-метоксипиридазин-3-ил)метанола. MS (ESI) масса/заряд 285 (M+H)+
Этап 4: 4-(((трет-бутилдиметилсилил)окси)метил)-3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазин
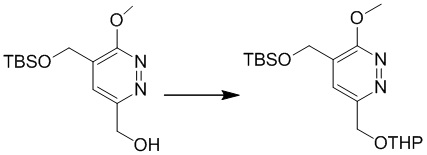
К раствору (5-(((трет-бутилдиметилсилил)окси)метил)-6-метоксипиридазин-3-ил)метанола (1,5 г, 5,3 ммоль) в ацетонитриле (10 мл) добавляли DHP (0,53 г, 6,3 ммоль) и PPTS (126 мг, 0,5 ммоль) при комнатной температуре Смесь перемешивали при 80°C в течение 16 часов. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (10:1) в качестве элюента) с получением 4-(((трет-бутилдиметилсилил)окси)метил)-3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазина. MS (ESI) масса/заряд 369 (M+H)+
Этап 5: (3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазин-4-ил)метанол
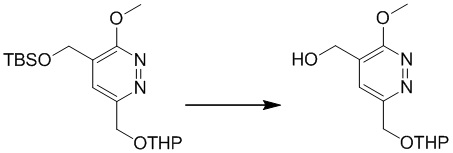
Раствор 4-(((трет-бутилдиметилсилил)окси)метил)-3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазина (0,9 г, 2,4 ммоль) и TBAF (3,2 г, 12,2 ммоль) в THF (20,0 мл) перемешивали в течение 1,0 часа при комнатной температуре. Добавляли воду, и полученную смесь экстрагировали этилацетатом. Комбинированные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной TLC (петролейный эфир/этилацетат (1:2) в качестве элюента) с получением (3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазин-4-ил)метанола. MS (ESI) масса/заряд 255 (M+H)+
Этап 6: 3-хлор-5-((1-((3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
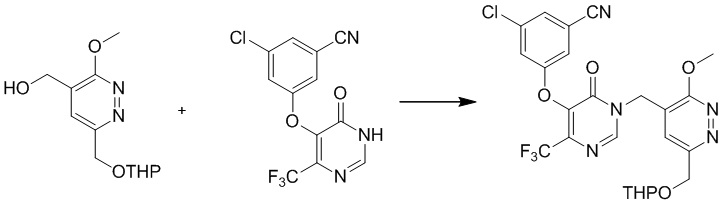
К раствору (3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазин-4-ил)метанола (0,6 г, 2,4 ммоль), 3-хлор-5-((6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (0,76 г, 2,4 ммоль, пример 3, этап 5) и трифенилфосфина (1,3 г, 4,8 ммоль) в дихлорметане (10,0 мл) добавляли DEAD (0,84 г, 4,8 ммоль) при 0°C в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 1 часа, гасили водой (10 мл) и экстрагировали дихлорметаном (20 мл × 3). Комбинированные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством препаративной TLC (петролейный эфир/этилацетат (1:1) в качестве элюента) с получением 3-хлор-5-((1-((3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 552, 554 (M+H)+
Этап 7: 3-хлор-5-((1-((6-(гидроксиметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
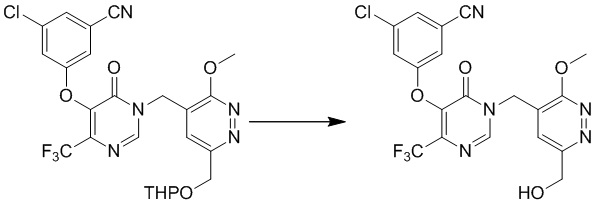
К раствору 3-хлор-5-((1-((3-метокси-6-(((тетрагидро-2H-пиран-2-ил)окси)метил)пиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (1,2 г, 2,2 ммоль) в метаноле (10 мл) добавляли HCl/метанол (1 N, 10 мл) при комнатной температуре Полученную смесь перемешивали при комнатной температуре в течение 1 часа и концентрировали при пониженном давлении с получением 3-хлор-5-((1-((6-(гидроксиметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 468, 470 (M+H)+
Этап 8: 3-хлор-5-((1-((6-формил-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
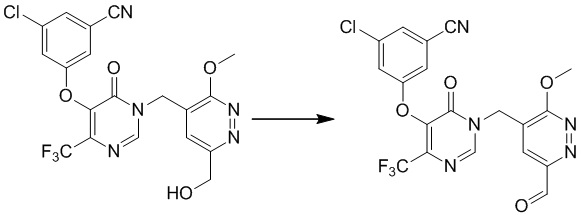
К раствору 3-хлор-5-((1-((6-(гидроксиметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (1,0 г, 2,1 ммоль) в дихлорметане (20 мл) добавляли периодинан Десса-Мартина (1,36 г, 3,2 ммоль) при 0°C в атмосфере азота. Смесь перемешивали при комнатной температуре в течение 1 часа, гасили водой (10 мл) и экстрагировали дихлорметаном (20 мл × 3). Комбинированные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством препаративной TLC (петролейный эфир/этилацетат (1:1) в качестве элюента) с получением 3-хлор-5-((1-((6-формил-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 466, 468 (M+H)+
Этап 9: 3-хлор-5-((1-((6-(дифторметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
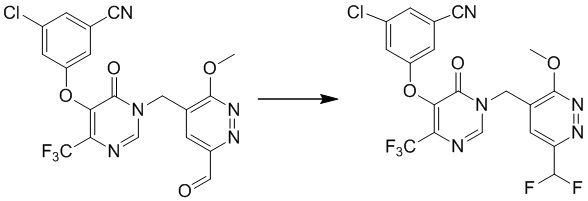
К перемешиваемой смеси 3-хлор-5-((1-((6-формил-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (0,14 г, 0,3 ммоль) в дихлорметане (5 мл) добавляли DAST (0,43 г, 1,6 ммоль) при комнатной температуре и смесь перемешивали в атмосфере азота в течение 16 часов. Смесь гасили H2O и экстрагировали дихлорметаном. Органический слой отмывали водой, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством хроматографии на силикагеле (петролейный эфир/этилацетат (2:1) в качестве элюента) с получением 3-хлор-5-((1-((6-(дифторметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 488, 490 (M+H)+
Этап 10: 3-хлор-5-((1-((6-(дифторметил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
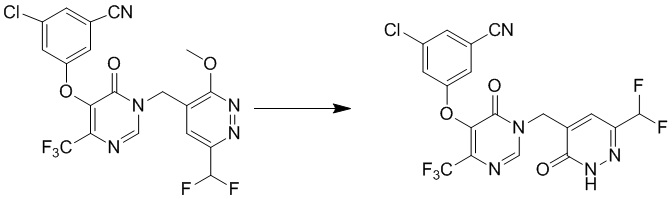
К смеси 3-хлор-5-((1-((6-(дифторметил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (90 мг, 0,2 ммоль) и KI (100 мг, 0,6 ммоль) в ацетонитриле (3 мл) добавляли TMSCl (33 мг, 0,3 ммоль). Смесь перемешивали при комнатной температуре в течение 1 часа, гасили водой и экстрагировали этилацетатом. Комбинированные органические экстракты отмывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ с получением 3-хлор-5-((1-((6-(дифторметил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. 1H ЯМР: (метанол-d4, 400 МГц) δ 13,62 (с, 1H), 8,72 (с, 1H), 7,73 (с, 1H), 7,71 (с, 1H), 7,68 (с, 1H), 7,66 (с, 1H), 6,78 (т, J=56,0 Гц, 1H), 4,99 (с, 2H). MS (ESI) масса/заряд 474, 476 (M+H)+
Пример 4
(5-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6 H )-ил)метил)-3-(дифторметил)-6-оксопиридазин-1(6 H )-ил)метилдигидрофосфат
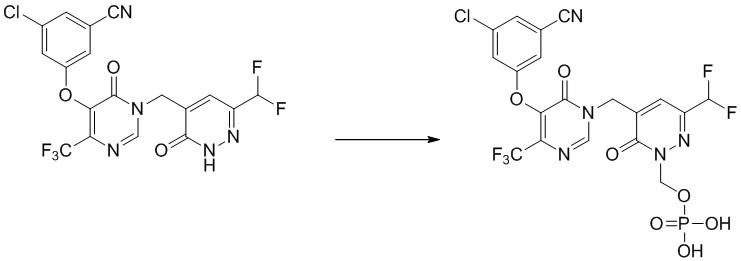
Указанное выше соединение получали, следуя процедурам, сходным с описанными в примере 7 на этапах 1-2. 1H ЯМР: (500 МГц, DMSO-d6) δ 8,76 (с, 1H), 7,72-7,78 (м, 4H), 6,85 (т, J=53 Гц, 1H), 5,71 (д, J=7,81 Гц, 2H), 5,08 (с, 2H). MS: 584 (M+H)+
Промежуточное соединение E
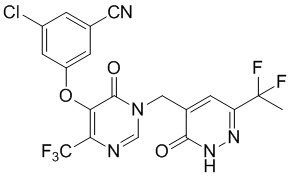
3-хлор-5-((1-((6-(1,1-дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1: 4-(трет-бутоксиметил)-6-(1-этоксивинил)-3-метоксипиридазин
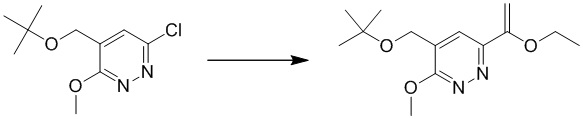
К смеси 4-(трет-бутоксиметил)-6-хлор-3-метоксипиридазина (1 г, 5,7 ммоль), трибутил(1-этоксивинил)станнана (6,2 г, 17,2 ммоль) в толуоле (10 мл) добавляли Pd(PPh3)4 (0,6 г, 0,57 ммоль) в атмосфере N2. Полученную суспензию перемешивали при 120°C в течение ночи в атмосфере азота. После охлаждения до комнатной температуры смесь выливали в ледяную воду, экстрагировали этилацетатом и комбинированные органические экстракты промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (5:1 до 2:1) в качестве элюента) с получением 4-(трет-бутоксиметил)-6-(1-этоксивинил)-3-метоксипиридазина. MS (ESI): масса/заряд 267 (M+H)+
Этап 2: 1-(5-(трет-бутоксиметил)-6-метоксипиридазин-3-ил)этанон
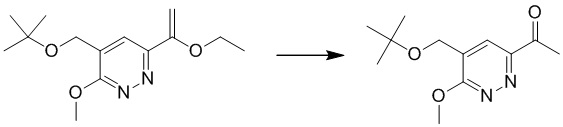
К раствору 4-(трет-бутоксиметил)-6-(1-этоксивинил)-3-метоксипиридазина (400 мг, 1,5 ммоль) в 1,4-диоксане (6 мл) добавляли HCl/1,4-диоксан (3 Н, 6 мл), раствор перемешивали при комнатной температуре в течение ночи. Смесь разбавляли водой и экстрагировали этилацетатом. Комбинированные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной TLC (петролейный эфир/этилацетат (1:1) в качестве элюента) с получением 1-(5-(трет-бутоксиметил)-6-метоксипиридазин-3-ил)этанона. MS (ESI) масса/заряд 239 (M+H)+
Этап 3: 4-(трет-бутоксиметил)-6-(1,1-дифторэтил)-3-метоксипиридазин
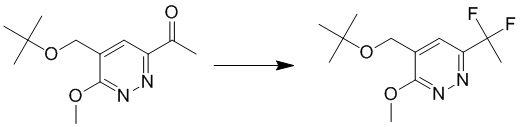
К раствору 1-(5-(трет-бутоксиметил)-6-метоксипиридазин-3-ил)этанона (240 мг, 1,0 ммоль) в дихлорметане (8 мл) добавляли DAST (0,8 мл, 6,1 ммоль). Смесь перемешивали при комнатной температуре в течение 4 часов. LCMS демонстрировала завершение реакции. Смесь гасили водой и экстрагировали этилацетатом. Комбинированные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 4-(трет-бутоксиметил)-6-(1,1-дифторэтил)-3-метоксипиридазина. MS (ESI) масса/заряд 261 (M+H)+
Этап 4: (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метанол
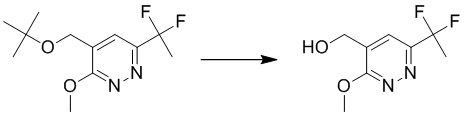
К раствору 4-(трет-бутоксиметил)-6-(1,1-дифторэтил)-3-метоксипиридазина (150 мг, 0,58 ммоль) в дихлорметане (8 мл) добавляли 4N HCl/метанол (3 мл). Смесь перемешивали при комнатной температуре в течение 3 часов, затем гасили водой и экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, концентрировали при пониженном давлении и очищали посредством препаративной TLC (петролейный эфир/этилацетат (1:1,5) в качестве элюента) с получением (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метанола. MS (ESI) масса/заряд 205 (M+H)+
Этап 5: (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метилметансульфонат
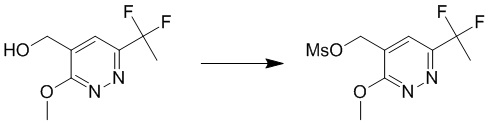
К раствору (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метанола (110 мг, 0,54 ммоль) в дихлорметане (6 мл) по каплям добавляли DIPEA (209 мг, 1,6 ммоль) и метансульфонилхлорид (75 мг, 0,62 ммоль). Смесь перемешивали при комнатной температуре в течение 2 часов. Смесь разбавляли водой и экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метилметансульфоната. MS (ESI) масса/заряд283 (M+H)+
Этап 6: 3-хлор-5-((1-((6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
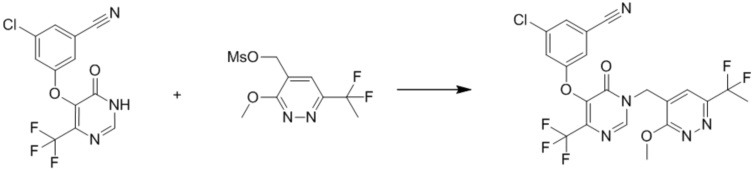
К раствору (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метилметансульфоната (120 мг, 0,54 ммоль) в DMF (5 мл) добавляли 3-хлор-5-((6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил (187 мг, 0,59 ммоль, пример 3, этап 5), TEA (0,23 мл, 1,6 ммоль). Смесь перемешивали при 30°C в течение 2 часов. После охлаждения до комнатной температуры смесь разбавляли водой, экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3-хлор-5-((1-((6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 502, 504 (M+H)+
Этап 7: 3-хлор-5-((1-((6-(1,1-дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
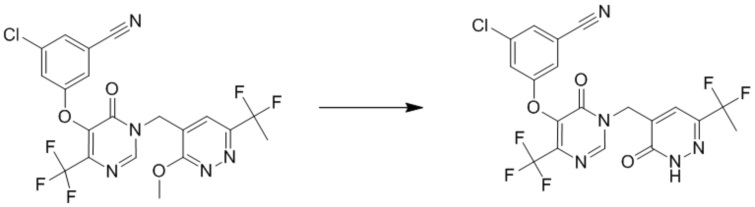
К смеси 3-хлор-5-((1-((6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (120 мг, 0,24 ммоль) и KI (79,5 мг, 0,48 ммоль) в ацетонитриле (4 мл) по каплям добавляли TMSCl (51,7 мг, 0,48 ммоль) при комнатной температуре. После добавления смесь перемешивали при 30°C в течение 3 часов. После охлаждения до комнатной температуры смесь гасили MeOH и концентрировали при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ с получением 3-хлор-5-((1-((6-(1,1-дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. 1H ЯМР: (DMSO-d6, 400MHz): δ 13,53 (с, 1H), 8,72 (с, 1H), 7,67-7,72 (м, 4H), 5,10 (с, 2H), 1,85-1,90 (м, 3H). MS (ESI): масса/заряд 488, 490 (M+H) +
Пример 5
(5-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6 H )-ил)метил)-3-(1,1-дифторэтил)-6-оксопиридазин-1(6 H )-ил)метилдигидрофосфат
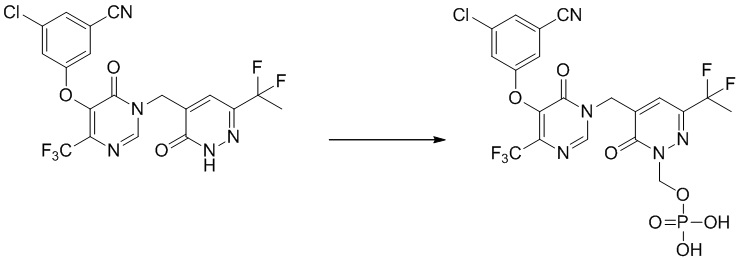
Указанное выше соединение получали, следуя процедурам, сходным с описанными в примере 7 на этапах 1-2. 1H ЯМР: (500 МГц, DMSO-d6)8,90 (с, 1H), 7,80-7,70 (м, 4H), 5,70 (д, 2H), 5,05 (с, 2H), 2,0-1,95 (т, 3H). MS: 598 (M+H)+
Промежуточное соединение F
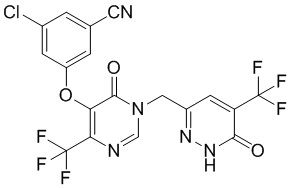
3-хлор-5-((6-оксо-1-((6-оксо-5-(трифторметил)-1,6-дигидропиридазин-3-ил)метил)-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1: 6-(бромметил)-4-(трифторметил)пиридазин-3(2H)-он
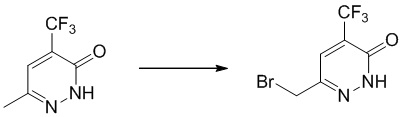
К смеси 6-метил-4-(трифторметил)пиридазин-3(2H)-она (2 г, 11,2 ммоль) в 20 мл CCl4 добавляли NBS (3 г, 17,2 ммоль) и бензоилпероксид (100 мг) при комнатной температуре. Полученную смесь нагревали при кипячении с обратным холодильником в течение 18 часов. LCMS демонстрировала завершение реакции, смесь выливали в ледяную воду и экстрагировали дихлорметаном. Комбинированные экстракты сушили и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (5: 1) в качестве элюента) с получением 6-(бромметил)-4-(трифторметил)пиридазин-3(2H)-она.
Этап 2: 3-хлор-5-((6-оксо-1-((6-оксо-5-(трифторметил)-1,6-дигидропиридазин-3-ил)метил)-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
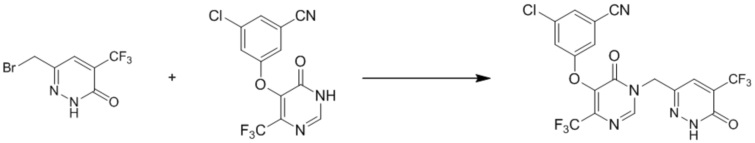
Смесь 3-хлор-5-(6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-илокси)бензонитрила (400 мг, 1,27 ммоль, пример 3, этап 5), 6-(бромметил)-4-(трифторметил)пиридазин-3(2H)-она (260 мг, 1,0 ммоль) и карбоната калия (170 мг, 1,23 ммоль) в DMF (6 мл) перемешивали при комнатной температуре в течение 18 часов. Смесь разбавляли водой, экстрагировали этилацетатом. Объединенные органические слои отмывали водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ с получением 3-хлор-5-((6-оксо-1-((6-оксо-5-(трифторметил)-1,6-дигидропиридазин-3-ил)метил)-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. 1H ЯМР: (DMSO-d6, 400 МГц): δ 13,72 (с, 1H), 8,77 (с, 1H), 8,02(с, 1H), 7,78 (с, 1H), 7,71 (с, 1H), 7,68 (с, 1H), 5,19 (с, 2H). MS (ESI) масса/заряд 492, 494 (M+H)+
Пример 6
(3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6 H )-ил)метил)-6-оксо-5-(трифторметил)пиридазин-1(6 H )-ил)метилфосфат натрия
Этап 1: ди-трет-бутил ((3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-6-оксо-5-(трифторметил)пиридазин-1(6H)-ил)метил)фосфат
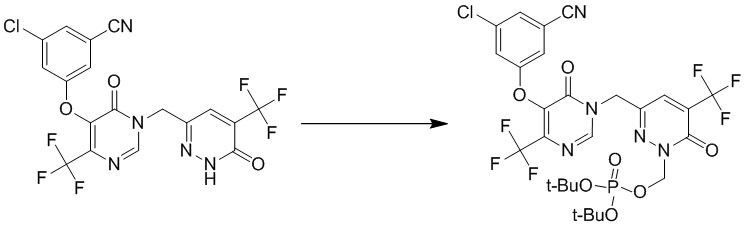
К раствору 3-хлор-5-((6-оксо-1-((6-оксо-5-(трифторметил)-1,6-дигидропиридазин-3-ил)метил)-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (1,5 г, 3,05 ммоль) в диоксане:DMF (5:1, 25 мл) добавляли карбонат калия (0,843 г, 6,1 моль). Реакционную смесь перемешивали при комнатной температуре в течение 0,5 часов. Добавляли сложный хлорметиловый эфир ди-трет-бутилового эфира фосфорной кислоты (1,19 г, 4,58 ммоль) при -30°C. Полученную смесь перемешивали при 40°C в течение 2,5 часов при микроволновом излучении. После охлаждения до комнатной температуры, реакционную смесь выливали в воду и экстрагировали этилацетатом. Объединенный органический слой сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ с получением желаемого продукта ди-трет-бутил ((3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-6-оксо-5-(трифторметил)пиридазин-1(6H)-ил)метил)фосфата. MS (ESI): масса/заряд 736 (M+Na)+. 1H ЯМР: (400 МГц, DMSO-d6) δ 8,75 (с, 1H), 8,13 (с, 1H), 7,63-7,74 (м, 3H), 5,59 (д, J=8,0 Гц, 2H), 5,21 (с, 2H).
Этап 2: (3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-6-оксо-5-(трифторметил)пиридазин-1(6H)-ил)метилфосфат натрия
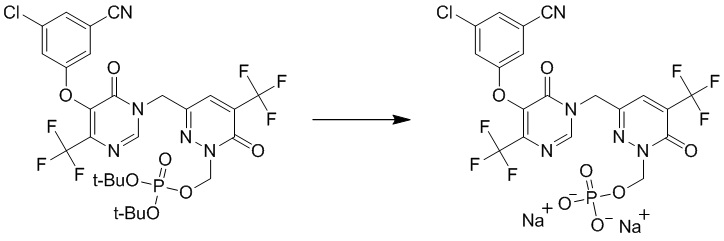
К раствору ди-трет-бутил ((3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-6-оксо-5-(трифторметил)пиридазин-1(6H)-ил)метил)фосфата (260 мг, 0,36 ммоль) в дихлорметане (7 мл) добавляли раствор CF3COOH (0,14 мл) в дихлорметане (0,5 мл). После перемешивания при комнатной температуре в течение 2 часов смесь концентрировали при пониженном давлении. Остаток растворяли в метаноле (5 мл), затем добавляли ацетат натрия (59,8 мг, 0,73 ммоль) в метаноле (0,6 мл). Полученную смесь перемешивали в течение ночи при комнатной температуре. Смесь лиофилизировали, и твердое вещество отмывали метанолом и сушили при пониженном давлении с получением (3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-6-оксо-5-(трифторметил)пиридазин-1(6H)-ил)метилфосфата натрия. 1H ЯМР: (DMSO&D2O, 400 МГц) δ 8,65 (с, 1H), 7,95 (с, 1H), 7,60-7,69 (м, 3H), 5,59 (д, J=8,4 Гц, 2H), 5,14 (с, 2H).
Промежуточное соединение G
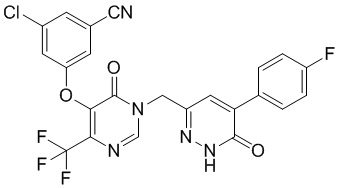
3-хлор-5-((1-((5-(4-фторфенил)-6-оксо-1,6-дигидропиридазин-3-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1: этил 2-(4-фторфенил)-2-оксоацетат
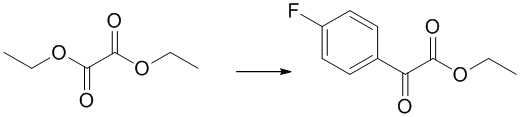
В 10 л круглодонную колбу через которую пропустили азот и которую поддерживали в инертной атмосфере азота, помещали раствор диэтилоксалата (360 г, 2,46 моль, 1,00 эквив.) в тетрагидрофуране (3000 мл). После этого по каплям добавляли раствор бромид 4-фторфенилмагния в тетрагидрофуране (1,9 л, 1 Н 0,78 эквив.) с перемешиванием при -78°C в течение 2,5 часов. Полученный раствор перемешивали в течение 30 мин при -78°C, затем медленно нагревали до -20°C. Затем реакцию гасили добавлением 500 мл 2 M HCl. Полученный раствор экстрагировали 2 × 500 мл этилацетата и органические слои комбинировали. Полученную смесь отмывали 2 × 200 мл насыщенного солевого раствора. Смесь сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Неочищенный продукт очищали посредством возгонки при пониженном давлении (0,67 кПа) и собирали фракцию при 106°C с получением этил 2-(4-фторфенил)-2-оксоацетат.
Этап 2: этил 5-((трет-бутилдифенилсилил)окси)-2-(4-фторфенил)-2-гидрокси-4-оксопентаноат
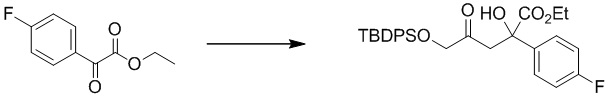
В 250 мл запаянную трубку, через которую пропускали азот и которую поддерживали в инертной атмосфере азота, помещали этил 2-(4-фторфенил)-2-оксоацетат (55 г, 280 ммоль, 1,00 эквив.), 1-[(трет-бутилдифенилсилил)окси]пропан-2-он (110 г, 352 ммоль, 1,26 эквив.), уксусную кислоту (33 г, 550 ммоль, 1,96 эквив.), пирролидин (7,8 г, 93 ммоль, 0,33 эквив.). Полученный раствор перемешивали в течение ночи при 85°C, а затем вносили в колонку с силикагелем с этилацетатом/петролейным эфиром (1:60-1:10) с получением этил 5-[(трет-бутилдифенилсилил)окси]-2-(4-фторфенил)-2-гидрокси-4-оксопентаноата.
Этап 3: 6-[[(трет-бутилдифенилсилил)окси]метил]-4-(4-фторфенил)-2,3-дигидропиридазин-3-он
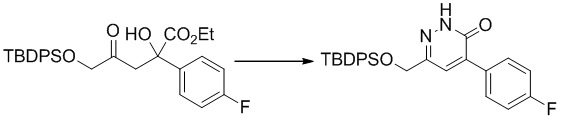
В 1000 мл 4-горлую круглодонную колбу, через которую пропускали азот и которую поддерживали в инертной атмосфере азота, помещали раствор этил 5-[(трет-бутилдифенилсилил)окси]-2-(4-фторфенил)-2-гидрокси-4-оксопентаноат а(292 г, 574 ммоль) в уксусной кислоте (520 мл). После этого по каплям добавляли гидрат гидразина (115 г, 2,30 моль) с перемешиванием при температуре ниже 30°C в течение 30 мин. Полученный раствор перемешивали в течение 3 часов при комнатной температуре, затем нагревали до 80°C в течение 2 часов. Затем реакционную смесь выливали в 2000 мл воды/льда. Полученный раствор экстрагировали 3 × 1000 этилацетата и органические слои комбинировали. Полученную смесь отмывали 2000 мл 5% NaHCO3 и 1000 мл насыщенного солевого раствора. Смесь сушили над безводным сульфатом натрия и концентрировали в вакууме. Неочищенный продукт очищали посредством перекристаллизации из н-гексана с получением 6-[[(трет-бутилдифенилсилил)окси]метил]-4-(4-фторфенил)-2,3-дигидропиридазин-3-она.
Этап 4: 6-[[(трет-бутилдифенилсилил)окси]метил]-4-(4-фторфенил)-2-(оксан-2-ил)-2,3-дигидропиридазин-3-он
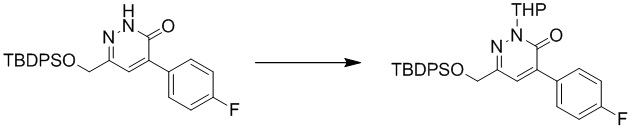
В 2000 мл круглодонную колбу, через которую пропускали азот и которую поддерживали в инертной атмосфере азота, помещали раствор 6-[[(трет-бутилдифенилсилил)окси]метил]-4-(4-фторфенил)-2,3-дигидропиридазин-3-она (150 г, 327 ммоль, 1,00 эквив.) в толуоле (1,2 л), 3,4-дигидро-2H-пиран (80 г, 951 ммоль, 2,91 эквив.), PPTS (15 г, 59,8 ммоль, 0,18 эквив.). Полученный раствор перемешивали в течение 5 часов при 90°C. К этому добавляли дополнительный DHP (55 г, 654 ммоль), и смесь перемешивали в течение ночи при 90°C. Реакционную смесь охлаждали до комнатной температуры, а затем выливали в 1000 мл 5% NaHCO3. Полученный раствор экстрагировали 2×500 мл этилацетата, и органические слои комбинировали. Полученную смесь отмывали 500 мл насыщенного солевого раствора. Смесь сушили над безводным сульфатом натрия и концентрировали в вакууме. Это приводило к получению (неочищенного) 6-[[(трет-бутилдифенилсилил)окси]метил]-4-(4-фторфенил)-2-(оксан-2-ил)-2,3-дигидропиридазин-3-она.
Этап 5: 4-(4-фторфенил)-6-(гидроксиметил)-2-(оксан-2-ил)-2,3-дигидропиридазин-3-он
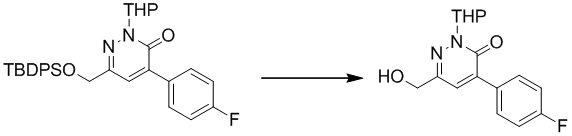
В 2000 мл круглодонную колбу, через которую пропускали азот и которую поддерживали в инертной атмосфере азота, помещали раствор 6-[[(трет-бутилдифенилсилил)окси]метил]-4-(4-фторфенил)-2-(оксан-2-ил)-2,3-дигидропиридазин-3-она (220 г, 324 ммоль, 1,00 эквив., 80%) в тетрагидрофуране (1,1 л). После этого несколькими частями добавляли TBAF (87 г, 333 ммоль, 1,03 эквив.) при 20°C в течение 5 минут. Полученный раствор перемешивали в течение 30 минут при комнатной температуре, а затем выливали в 1000 мл 5% NaHCO3. Полученный раствор экстрагировали 2×500 мл этилацетата, и органические слои комбинировали. Полученную смесь отмывали 1000 мл насыщенного солевого раствора. Смесь сушили над безводным сульфатом натрия и концентрировали в вакууме. Остаток вносили в колонку с силикагелем с этилацетатом/ петролейным эфиром (1:10-1:1) с получением 4-(4-фторфенил)-6-(гидроксиметил)-2-(оксан-2-ил)-2,3-дигидро пиридазин-3-она. MS (ESI) масса/заряд 305 (M+H)+
Этап 6: 6-(бромметил)-4-(4-фторфенил)-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2H)-он
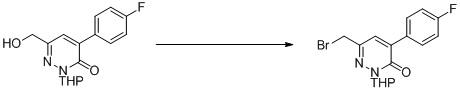
К перемешиваемому раствору 4-(4-фторфенил)-6-(гидроксиметил)-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2H)-она (10 г, 32,9 ммоль) в DCM (40 мл) при 0°C добавляли тетрабромид углерода (13,08 г, 39,4 ммоль) с последующим медленным добавлением раствора трифенилфосфина (10,34 г, 39,4 ммоль) в DCM (10 мл). Полученную смесь оставляли перемешиваться при 0°C в течение 1 часа, а затем концентрировали при пониженном давлении. К неочищенной смеси добавляли диэтиловый эфир (500 мл), и отфильтровывали твердые вещества. Фильтрат концентрировали при пониженном давлении и неочищенный продукт очищали посредством колоночной хроматографии на силикагеле (этилацетат/гексан (0% - 60%) в качестве элюента) с получением 6-(бромметил)-4-(4-фторфенил)-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2H)-она. MS (ESI) масса/заряд 367, 369 (M+H)+
Этап 7: 3-хлор-5-((1-((5-(4-фторфенил)-6-оксо-1-(тетрагидро-2H-пиран-2-ил)-1,6-дигидропиридазин-3-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
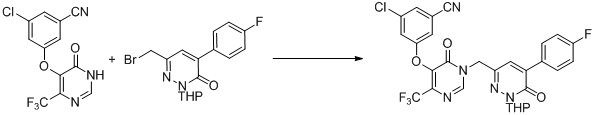
К смеси 6-(бромметил)-4-(4-фторфенил)-2-(тетрагидро-2H-пиран-2-ил)пиридазин-3(2H)-она (10,59 г, 28,8 ммоль) и 3-хлор-5-((6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (9,10 г, 28,8 ммоль, пример 3, этап 5) в DMF (35 мл) добавляли DIPEA (6,55 мл, 37,5 ммоль) при 0°C. Через 30 минут реакционную смесь нагревали до комнатной температуры и перемешивание продолжали в течение 1 дополнительного часа. Смесь концентрировали при пониженном давлении и добавляли воду (200 мл). Полученный осадок собирали посредством фильтрования и промывали водой (2 × 50 мл), с последующим промыванием диэтиловым эфиром (3 × 50 мл) с получением 3-хлор-5-((1-((5-(4-фторфенил)-6-оксо-1-(тетрагидро-2H-пиран-2-ил)-1,6-дигидропиридазин-3-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 601, 602 (M+H)+
Этап 8: 3-хлор-5-((1-((5-(4-фторфенил)-6-оксо-1,6-дигидропиридазин-3-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрил
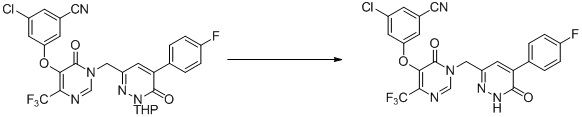
Раствор 3-хлор-5-((1-((5-(4-фторфенил)-6-оксо-1-(тетрагидро-2H-пиран-2-ил)-1,6-дигидропиридазин-3-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (15,78 г, 26,2 ммоль) в TFA (40,4 мл, 524 ммоль) перемешивали при комнатной температуре в течение 1 часа. TFA удаляли при пониженном давлении, и добавляли диэтиловый эфир (250 мл). Полученное твердое вещество собирали посредством фильтрования и промывали диэтиловым эфиром (2 × 125 мл) с получением 3-хлор-5-((1-((5-(4-фторфенил)-6-оксо-1,6-дигидропиридазин-3-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 518, 520 (M+H)+; 1H ЯМР: (DMSO-d6, 400 МГц) δ 13,13 (с, 1H), 8,74 (с, 1H), 7,87 (т, J =6,8 Гц, 2H), 7,62-7,72 (м, 4H), 7,26 (т, J=6,8 Гц, 2H), 5,14 (с, 2H).
Пример 7
(3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6 H )-ил)метил)-5-(4-фторфенил)-6-оксопиридазин-1(6 H )-ил)метилдигидрофосфат
Этап 1: ди-трет-бутил ((3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-5-(4-фторфенил)-6-оксопиридазин-1(6H)-ил)метил)фосфат
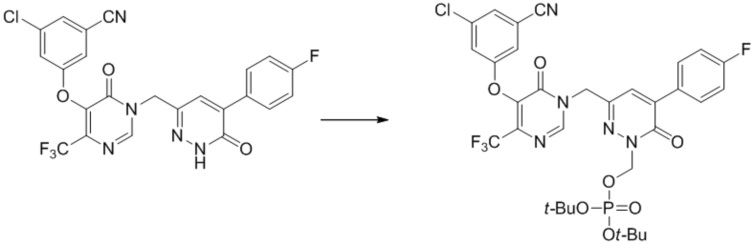
К взвеси 3-хлор-5-((1-((5-(4-фторфенил)-6-оксо-1,6-дигидропиридазин-3-ил)метил)-6-оксо-4-(трифторметил)-1,6-дигидропиримидин-5-ил)окси)бензонитрила (250 мг, 0,48 ммоль) и йодида натрия (94 мг, 0,63 ммоль) в THF (3,8 мл) при 0°C добавляли трет-бутоксид лития (1M в THF) (0,56 мл, 0,56 ммоль) с последующим добавлением ди-трет-бутилхлорметилфосфата (0,13 мл 0,63 ммоль). Реакционную смесь нагревали до комнатной температуры и дополнительно перемешивали в течение 16 часов. После завершения реакции ее охлаждали до 0°C и гасили несколькими каплями воды. Растворитель удаляли посредством испарения с применением роторного испарителя при поддержании температуры водяной бани не более 25°C. Остаток растворяли в DCM (25 мл) и промывали насыщенным NaHCO3 (1 × 5 мл), насыщенным солевым раствором (1 × 5 мл,) сушили над безводным MgSO4, фильтровали и концентрировали посредством испарения с применением роторного испарителя при поддержании температуры водяной бани не более 25°C с получением ди-трет-бутил ((3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-5-(4-фторфенил)-6-оксопиридазин-1(6H)-ил)метил)фосфата, который использовали на следующем этапе без очистки.
Этап 2: (3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-5-(4-фторфенил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат
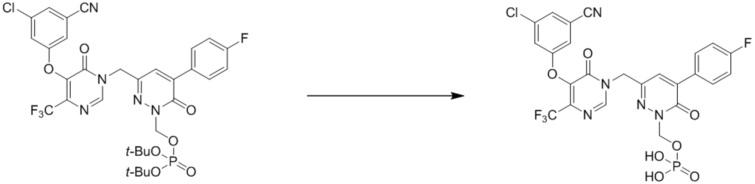
К неочищенному раствору ди-трет-бутил ((3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-5-(4-фторфенил)-6-оксопиридазин-1(6H)-ил)метил)фосфата (357 мг, 0,48 ммоль) в дихлорметане (5,8 мл) при 0°C добавляли трифторуксусную кислоту (149 мкл, 1,932 ммоль). Реакционную смесь оставляли нагреваться до температуры окружающей среды и дополнительно перемешивали в течение 16 часов. После завершения реакции смесь фильтровали и очищали посредством ВЭЖХ с обращенной фазой (ACN/вода с модификатором 0,1% TFA) с получением (3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-5-(4-фторфенил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфата. MS: 628 (M+H)+
1 H ЯМР: (500 МГц, DMSO-d6) δ 8,76 (с, 1H), 7,71-7,86 (м, 6H), 7,33 (т, J=8,7 Гц, 2H), 5,7 1(д, J=7,7 Гц, 2H), 5,19 (с, 2H).
Промежуточное соединение H
3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(1,1-дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
Этап 1: этил 4,4-дифтор-3-оксопентаноат
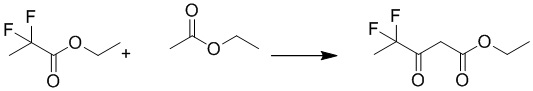
К раствору соединения этил 2,2-дифторпропаноата (10,6 г, 76,8 ммоль) и этилацетата (8,11 г, 92,1 ммоль, сушили над MgSO4) в THF (100 мл) добавляли LiHMDS (92 мл, 92,1 ммоль) при -78°C с защитой N2. Смесь перемешивали при -78°C в течение 1 часа. Затем смесь перемешивали при 20°C в течение дополнительных 1,5 часов. Реакцию медленно гасили раствором HCl (1 Н). Смесь экстрагировали EtOAc (100 мл × 3), промывали насыщенным солевым раствором (200 мл), сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением желаемого продукта. Остаток использовали непосредственно без дополнительной очистки.
Этап 2: 6-(1,1-дифторэтил)пиримидин-4(3H)-он
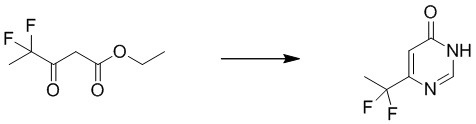
Раствор ацетата формимидамида (16,0 г, 153,6 ммоль) и метоксида натрия (16,6 г, 307 ммоль) в метаноле (140 мл) перемешивали при комнатной температуре в течение 20 минут, затем добавляли этил 4,4-дифтор-3-оксопентаноат (14 г неочищенный, 76,8 ммоль). Полученную смесь перемешивали при 70°C в течение 14 часов. После охлаждения до комнатной температуры смесь разбавляли водой (200 мл), экстрагировали EtOAc (200 мл × 3). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением желаемого продукта. 1H ЯМР: (400 МГц, CDCl3) δ 8,20 (с, 1H), 6,77 (с, 1H), 1,90 (т, J=18,8 Гц, 3H). MS (ESI) масса/заряд 161,21 (M+H)+
Этап 3: 5-бром-6-(1,1-дифторэтил)пиримидин-4(3H)-он
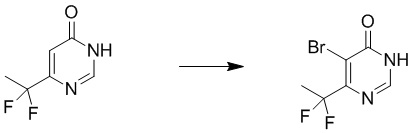
Раствор соединения 6-(1,1-дифторэтил)пиримидин-4(3H)-она (9,5 г, 59 ммоль) и ацетата калия (11,6 г, 119 ммоль) в уксусной кислоте (100 мл) перемешивали при комнатной температуре в течение 30 минут, затем по каплям добавляли Br2 (11,2 г, 71 ммоль) при комнатной температуре. Полученную смесь перемешивали при кипячении с обратным холодильником в течение 2 часов. После охлаждения до комнатной температуры смесь гасили Na2SO3 (насыщенным) до превращения цвета в светло-желтый и экстрагировали EA (200 мл × 3). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении с получением желаемого продукта. MS (ESI) масса/заряд 238,8, 240,8 (M+H)+.
Этап 4: 5-бром-6-(1,1-дифторэтил)-3-(4-метоксибензил)пиримидин-4(3H)-он
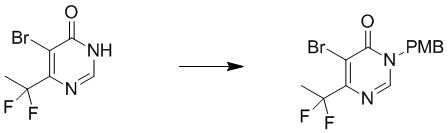
К раствору соединения 5-бром-6-(1,1-дифторэтил)пиримидин-4(3H)-она (15,0 г, 59 ммоль) в DMF (150 мл) добавляли K2CO3 (17,4 г,126 ммоль) и PMBCl (108. г, 69 ммоль). Реакционную смесь перемешивали при 80°C в течение 3 часов. После охлаждения до комнатной температуры смесь разбавляли H2O (100 мл) и экстрагировали EtOAc (200 мл × 3). Объединенные органические слои сушили над MgSO4, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (PE/EA=10/1 до 5/1) с получением желаемого продукта. 1H ЯМР: (400 МГц, CDCl3) δ 8,09 (с, 1H), 7,96 (с, 1H), 7,28 (д, 2H, J=8,4 Гц), 6,85 (с, 2H, J=8,4 Гц), 5,04 (с, 2H), 3,75 (с, 2H), 1,92 (т, J=18,4 Гц, 3H). MS (ESI) масса/заряд 359,1, 361,1 (M+H)+
Этап 5: 3-хлор-5-((4-(1,1-дифторэтил)-1-(4-метоксибензил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
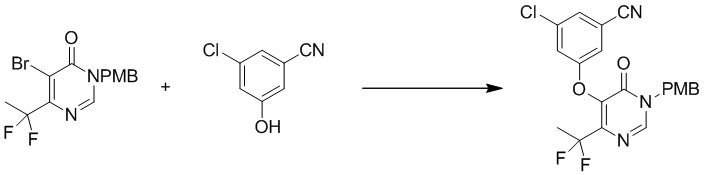
К раствору соединения 5-бром-6-(1,1-дифторэтил)-3-(4-метоксибензил)пиримидин-4(3H)-он (3,0 г, 8,56 ммоль) в NMP (50 мл) добавляли карбонат калия (2,31 г, 16,70 ммоль) и 3-хлор-5-гидроксибензонитрил (3,86 г, 25,07 ммоль). Смесь нагревали до 140°C в течение 6 часов, а затем охлаждали до 130°C, смесь перемешивали при 130°C в течение ночи. После охлаждения, смесь разбавляли водой (200 мл) и экстрагировали этилацетатом (100 мл × 3). Объединенные органические слои отмывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир:этилацетат (20:1 до 8:1) в качестве элюента) с получением 3-хлор-5-((4-(1,1-дифторэтил)-1-(4-метоксибензил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила.
MS (ESI) масса/заряд 432, 434 (M+H)+
Этап 6: 3-хлор-5-((4-(1,1-дифторэтил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
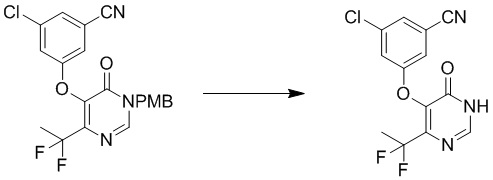
Раствор 3-хлор-5-((4-(1,1-дифторэтил)-1-(4-метоксибензил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила (10 г, 23,2 ммоль) в смешанном растворителе (TFA/TFAA=48 мл/24 мл) перемешивали при 100°C в течение 4 часов. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении с получением неочищенного продукта 3-хлор-5-((4-(1,1-дифторэтил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила, который использовали без дополнительной очистки. MS (ESI) масса/заряд 312, 314 (M+H)+
Этап 7: 3-(1-этоксивинил)-6-метоксипиридазин
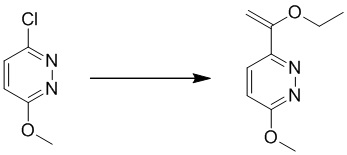
К смеси 3-хлор-6-метоксипиридазина (15 г, 103,8 ммоль), трибутил(1-этоксивинил)станнана (82,44 г, 228,3 ммоль) в толуоле (200 мл) добавляли Pd(PPh3)4 (6 г, 5,19 ммоль) в атмосфере азота. Через полученную суспензию три раза пропускали азот, а затем перемешивали при 110°C в течение 36 часов. После охлаждения до комнатной температуры смесь выливали в ледяную воду, фильтровали через слой целита®. Фильтрат экстрагировали этилацетатом и объединенные органические слои отмывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (15:1 до 10:1) в качестве элюента) с получением желаемого продукта 3-(1-этоксивинил)-6-метоксипиридазина. MS (ESI) масса/заряд 181 (M+H)+
Этап 8: 1-(6-метоксипиридазин-3-ил)этанон
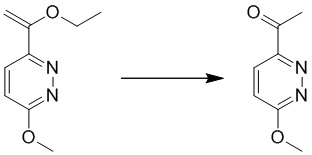
К раствору 3-(1-этоксивинил)-6-метоксипиридазина (12 г, 66,59 ммоль) в 1,4-диоксане (120 мл) по каплям добавляли HCl/1,4-диоксан (24 мл, 4 M) при 0°C. Смесь перемешивали при комнатной температуре в течение 1 часа. Смесь гасили водой и экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (10:1 до 8:1) в качестве элюента) с получением 1-(6-метоксипиридазин-3-ил)этанона. MS (ESI) масса/заряд 153 (M+H)+
Этап 9: 3-(1,1-дифторэтил)-6-метоксипиридазин
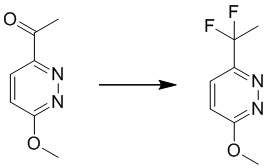
К раствору 1-(6-метоксипиридазин-3-ил)этанона (4,2 г, 27,6 ммоль) в дихлорметане (45 мл) по каплям добавляли DAST (13,35 мг, 82,81 ммоль) при 0°C. Смесь перемешивали при 40°C в течение 24 часов. После охлаждения до комнатной температуры смесь гасили водой и экстрагировали этилацетатом. Объединенные органические слои сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (15:1 до 10:1) в качестве элюента) с получением 3-(1,1-дифторэтил)-6-метоксипиридаза. MS (ESI) масса/заряд 175 (M+H)+
Этап 10: (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метанол
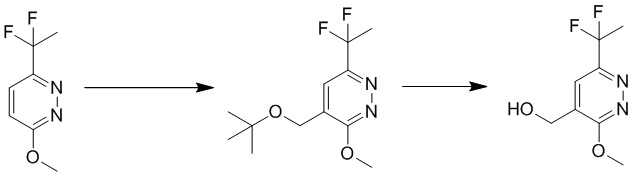
К смеси трет-бутоксиуксусной кислоты (4,23 г, 32,04 ммоль) в TFA/воде (20 моль%, 30 мл) добавляли 3-(1,1-дифторэтил)-6-метоксипиридазин (3,1 г, 17,8 ммоль) и AgNO3 (303 мг, 1,78 ммоль). Через смесь пропускали азот с перемешиванием при комнатной температуре, затем смесь нагревали до 70°C и по каплям добавляли (NH4)2S2O8(8,12 г, 35,6 ммоль) в воде (40 мл). После добавления смесь перемешивали при 75°C в течение 40 минут. После охлаждения до комнатной температуры смесь экстрагировали этилацетатом (20 мл × 3), объединенные органические слои отмывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта. Раствор 4-(трет-бутоксиметил)-6-(1,1-дифторэтил)-3-метоксипиридазина в TFA/DCE (10 мл/40 мл) перемешивали при 60°C в течение 1 часа. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении. Остаток растворяли в этилацетате (20 мл), промывали водным карбонат калия, насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством колоночной хроматографии на силикагеле (петролейный эфир/этилацетат (8:1 до 5:1) в качестве элюента) с получением (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метанола. MS (ESI) масса/заряд 205 (M+H)+
Этап 11: 4-(хлорметил)-6-(1,1-дифторэтил)-3-метоксипиридазин
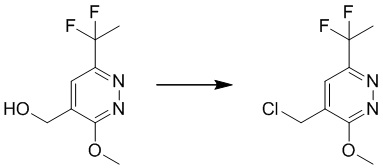
К раствору (6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метанола (180 мг, 0,88 ммоль) в дихлорметане (3 мл) добавляли триэтиламин (268 мг, 2,64 ммоль) и метансульфонилхлорид (303 мг, 2,64 ммоль) при 0°C. Смесь перемешивали при комнатной температуре в течение 24 часов, гасили водой и экстрагировали дихлорметаном. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали посредством препаративной TLC (петролейный эфир/этилацетат (2:1) в качестве элюента) с получением 4-(хлорметил)-6-(1,1-дифторэтил)-3-метоксипиридаза. MS (ESI) масса/заряд 223 (M+H)+
Этап 12: 3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
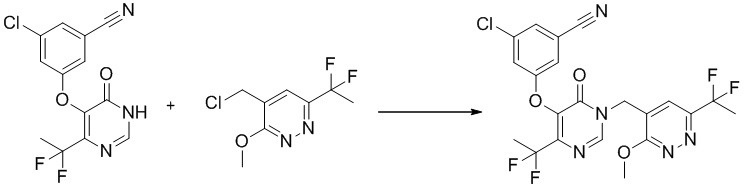
К раствору 3-хлор-5-((4-(1,1-дифторэтил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила (500 мг, 1,60 ммоль) в DMF (2 мл) добавляли карбонат калия (443 мг, 3,21 ммоль) и 4-(хлорметил)-6-(1,1-дифторэтил)-3-метоксипиридазин (как описано на этапах 1-5 примера 178) (82 мг, 0,27 ммоль). Полученную смесь перемешивали при 80°C в течение 3 часов. После охлаждения до комнатной температуры смесь разбавляли водой и экстрагировали EtOAc. Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали при пониженном давлении. Остаток очищали посредством препаративной TLC (петролейный эфир/EtOAc =1:1) с получением желаемого продукта 3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила. MS (ESI) масса/заряд 498, 500 (M+H)+
Этап 13: 3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(1,1-дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрил
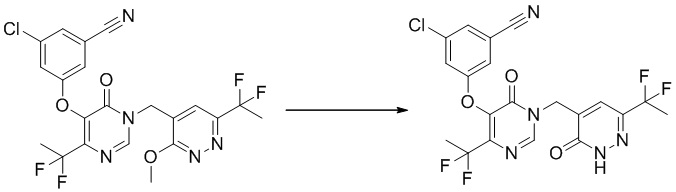
К смеси соединения 3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(1,1-дифторэтил)-3-метоксипиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила (250 мг, 0,50 ммоль) и KI (166 мг, 1,0 ммоль) в ацетонитриле (3 мл) добавляли TMSCl (109 мг, 1 ммоль) при комнатной температуре. Полученную смесь нагревали и перемешивали при 70°C в течение 1 часа. После охлаждения до комнатной температуры смесь разбавляли EtOAc и промывали водным Na2S2O3 и насыщенным солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали посредством препаративной ВЭЖХ с получением 3-хлор-5-((4-(1,1-дифторэтил)-1-((6-(1,1-дифторэтил)-3-оксо-2,3-дигидропиридазин-4-ил)метил)-6-оксо-1,6-дигидропиримидин-5-ил)окси)бензонитрила. 1H ЯМР: (метанол-d4, 400 МГц): δ 8,60 (с, 1H), 7,68 (с, 1H), 7,47 (с, 1H), 7,28 (с, 1H), 7,24 (с, 1H), 5,07 (с, 2H), 1,92 (м, 6H). MS (ESI) масса/заряд 484, 486 (M+H)+
Пример 8
(5-((5-(3-хлор-5-цианофенокси)-4-(1,1-дифторэтил)-6-оксопиримидин-1(6 H )-ил)метил)-3-(1,1-дифторэтил)-6-оксопиридазин-1(6 H )-ил)метилдигидрофосфат
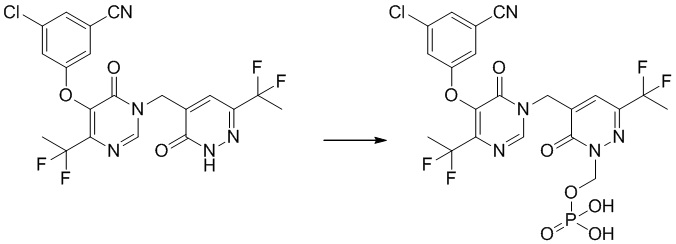
Указанное выше соединение получали, следуя процедурам, сходным с описанными в примере 7 на этапах 1-2. 1H ЯМР: (500 МГц, DMSO-d6) δ 8,68 (с, 1H), 7,61-7,75 (м, 4H), 5,70 (д, J=8,2 Гц, 2H), 5,06 (с, 2H), 1,90-1,99 (м, 6H). MS: 594 (M+H)+
Определение ингибирующей активности в отношении обратной транскриптазы ВИЧ-1
Субстрат гетеродимерной нуклеиновой кислоты, используемый в реакциях с полимеразой ВИЧ-1 RT получали посредством отжига праймера ДНК, биотинилированного pD500 (Sigma Aldrich, USA, 5'-биотин-ttg aaa tga ctg cgg tac ggc-3'), (SEQ ID NO: 1) с матрицей нуклеотидов РНК t500 (получаемой из последовательности вируса гепатита C [HCV], IBA, Germany, 5'-GAG GUU CAG GUG GUU UCC ACC GCA ACA CAA UCC UUC CUG GCG ACC UGC GUC AAC GGC GUG UGU UGG ACC GUU UAC CAU GGU GCU GGC UCA AAG ACC UUA GCC GGC CCA AAG GGG CCA AUC ACC CAG AUG UAC ACU AAU GUG GAC CAG GAC CUC GUC GGC UGG CAG GCG CCC CCC GGG GCG CGU UCC UUG ACA CCA UGC ACC UGU GGC AGC UCA GAC CUU UAC UUG GUC ACG AGA CAU GCU GAC GUC AUU CCG GUG CGC CGG CGG GGC GAC AGU AGG GGG AGC CUG CUC UCC CCC AGG CCU GUC UCC UAC UUG AAG GGC UCU UCG GGU GGU CCA CUG CUC UGC CCU UCG GGG CAC GCU GUG GGC AUC UUC CGG GCU GCC GUA UGC ACC CGG GGG GUU GCG AAG GCG GUG GAC UUU GUG CCC GUA GAG UCC AUG GAA ACU ACU AUG CGG UCU CCG GUC UUC ACG GAC AAC UCA UCC CCC CCG GCC GUA CCG CAG UCA UUU CAA-3'), (SEQ ID NO: 2). Фермент RT ВИЧ-1 дикого типа (конечная концентрация 83 пМ) комбинировали с ингибитором или диметилсульфоксидом (DMSO, 10% в конечной реакционной смеси) в буфере для анализа (62,5 мМ Tris-HCl [pH 7,8], 1,25 мМ дитиотреитол, 7,5 мМ MgCl2, 100 мМ KCl, 0,03% CHAPS и 125 мкМ EGTA). Затем смесь в планшетах для микротитрования (Costar 3365, Corning, USA) предварительно инкубировали на орбитальном шейкере в течение 30 минут при комнатной температуре. Реакцию полимеризации инициировали посредством добавления матрицы РНК/гибрида pD500-праймера ДНК (16,6 нМ конечного гибрида РНК/ДНК) и dNTP (2 мкМ dATP, dGTP, dCTP и 66,6 нМ Ru-dUTP (Meso Scale Discovery, USA)). Планшет герметизировали и инкубировали в течение 5-10 минут при комнатной температуре на орбитальном шейкере. Затем планшет инкубировали в течение 90 мин при 37°C и реакции гасили 60 мкл гасящего буфера (50 мМ ЭДТА, 0,7% BSA, 0,7% Tween-20, 0,017% азид натрия в PBS). Полученный раствор инкубировали при комнатной температуре в течение дополнительных 5 минут, а затем 50 мкл переносили в предварительно блокированные планшеты с авидином (L15AA, Meso Scale Discovery). Каждую лунку планшета с авидином блокировали в течение 1 часа при комнатной температуре 100 мкл 5% BSA в PBS. Блокирующий раствор удаляли посредством энергичного постукивания по фильтровальной бумаге с удалением избытка жидкости. Реакцию на предварительно блокированном планшете с авидином проводили в течение 60 минут при комнатной температуре, а затем содержимое удаляли посредством энергичного постукивания по фильтровальной бумаге с удалением всего избытка жидкости. После отмывки планшета в количестве 3 раз 150 мкл 1× PBS и промоканием насухо между циклами, добавляли 150 мкл 1× буфера Read Buffer T (4× Read Buffer T, Meso Scale Discovery) и инкубировали в течение 5 минут при комнатной температуре с последующим подсчетом на Sector Imager S6000 (Meso Scale Discovery). Кривые титрования и значения IC50 рассчитывали с использованием четырехпараметрической логистической аппроксимации стандартными способами. В кратком изложении, % ингибирования=100 × ((исходное значение для образца) -(среднее значение нижнего контроля или 0% ингибирования))/((среднее значение для лунок, представляющих 100% ингибирования) -(среднее значение 0% ингибирования)). В этом анализе лунки нижнего контроля содержали DMSO (0% ингибирования), а лунки со 100% ингибирования содержали 1 мкМ эфавиренз.
Результаты для тестируемых соединений по изобретению в указанном выше анализе приведены в таблице 2, ниже.
Таблица 2
Фармакокинетические исследования
Все исследования на животных проводили в соответствии с правилами надлежащей лабораторной практики для неклинических лабораторных исследований. Всех животных содержали, и процедуры ухода проводили в соответствии с законом Federal Animal Welfare Act и Institute for Laboratory Animal Resources. Все способы, проводимые на животных, были рассмотрены и одобрены комитетом по уходу и использованию животных института. Виварий полностью аккредитован в международной ассоциации Association for Assessment and Accreditation of Laboratory Animal Care International.
В Charles River (Raleigh, North Carolina, U.S.A.) получили двух или трех самцов крыс Wistar-Hannover с массой от 250 до 350 грамм с вставленным продавцом катетером в сонную артерию. Животных акклиматизировали с циклом света:темноты по 12 часов в течение минимум трех суток в вентилируемых пластиковых клетках со свободным доступом к пище и воде. Образцы крови собирали автоматически с использованием устройства для сбора образцов Instech Automated Blood sampler (ABS, Plymouth Meeting, PA) c определенными интервалами до 24 часов и собирали вручную после 24 часов.
Перед введением крысам исходных соединений из примеров 1-8 (формула I') или соединений из примеров 1-8 (формула I) посредством перорального принудительного кормления в дозах и с носителями, указанными в таблице 3 животные голодали в течение ночи. Животным обеспечивали свободный доступ к воде, тогда как кормление возобновили через 4 часа после дозирования.
Кровь отбирали из катетеров, помещенных в сонную артерию, перед дозированием и через 0,25, 0,5, 1, 2, 4, 8, 12, 18, 24, 30 и 48 часа после дозирования для всех соединений и дополнительно через 72 часа для соединений из примеров 1, 3, 4, 5 и 7. Плазму отделяли посредством центрифугирования (2 минуты при 10000 об./мин.) и хранили при -70°C до последующего анализа.
Количественный анализ соединений проводили с использованием способов жидкостной хроматографии и тандемной масс-спектрометрии (LC-MS/MS). По мере необходимости образцы плазмы перед экстракцией стабилизировали. Экстрагировали по пятьдесят микролитров каждого из неизвестных образцов вместе с образцами стандартов и контроля качества способом автоматического осаждения белков на рабочей станции Hamilton. Экстракт центрифугировали, и супернатант переносили для анализа. Разделение обеспечивали посредством системы LC Thermo Aria LX-2 на колонках для хроматографии с обращенной фазой, например, колонки Waters XSelect HSS T3 XP (50 × 2,1 мм × 2,5u) или Phenomenex Beta Test Column (50 × 2,1 мм). Соединения детектировали выбранными способами мониторинга реакций (SRM) на масс-спектрометре AB Sciex API5000 в режиме положительной и отрицательной ионизации. Кроме того, хроматографические пики для исходных соединений из примеров 1-8 (формула I') и соединений из примеров 1-8 (формула I) разделяли хроматографически с исходного уровня до разделения исходных уровней.
Фармакокинетические параметры получали некомпартментными способами (Watson®). Площадь под кривой концентрации в плазме-времени (AUC0-t) рассчитывали, начиная с первого момента времени (0 минут) до последнего момента времени с измеримой концентрацией лекарственного средства (т.е., концентрацией исходного соединения (формула I' соединение), получаемого после введения каждого из исходного соединения и соединения из примера) с использованием линейного правила трапеций или линейного/лог-линейного правила трапеций. Оставшуюся область под кривой концентрации в плазме-времени (AUCt-∞) определяли, деля наблюдаемую концентрацию в последний момент времени на константу скорости выведения. Это значение добавляли к AUC0-t для расчета AUC0-∞. Максимальную концентрацию в плазме (Cмакс) и время до достижения максимальной концентрации (Tмакс) получали посредством исследования данных концентрации в плазме-времени.
В таблице 3 предоставлены значения AUC, получаемые в этих исследованиях. В таблице 3, исходное соединение для каждого номера примера относится к исходному варианту соединения из указанного номера примера соединения, где R4 замещен -H.
Таблица 3 - Результаты исследований PK
Хотя в приведенном выше описании указаны основы настоящего изобретения с примерами, предоставленными с целью иллюстрации, практическое осуществление настоящего изобретения относится ко всем обычным вариациям, адаптациям и/или модификациям, которые входят в объем приводимой ниже формулы изобретения. Перечисление или изображение конкретного соединения в формуле изобретения (т.е., видовые признаки) без конкретного указания пространственной конфигурации или с таким указанием не для всех хиральных центров, предназначено для включения рацематов, рацемических смесей, каждых конкретных энантиомеров, диастереоизомерических смесей и каждого конкретного диастереомера соединений, где такие формы возможны вследствие присутствия одного или нескольких центров ассиметрии. Все публикации, патенты и патентные заявки, цитируемые в настоящем документе, полностью включены в описание в качестве ссылки.
| название | год | авторы | номер документа |
|---|---|---|---|
| СОЕДИНЕНИЯ ПИРИДИНОНА В КАЧЕСТВЕ СЕЛЕКТИВНЫХ ЦИТОТОКСИЧЕСКИХ АГЕНТОВ ПРОТИВ КЛЕТОК, ИНФИЦИРОВАННЫХ ВИЧ | 2020 |
|
RU2816090C2 |
| НОВЫЕ ПИРИДАЗОНЫ И ТРИАЗИНОНЫ ДЛЯ ЛЕЧЕНИЯ И ПРОФИЛАКТИКИ ЗАРАЖЕНИЯ ВИРУСОМ ГЕПАТИТА B | 2015 |
|
RU2664329C1 |
| ПРОИЗВОДНОЕ 6-ОКСО-1,6-ДИГИДРОПИРИДАЗИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ В МЕДИЦИНЕ | 2020 |
|
RU2791534C2 |
| СОЕДИНЕНИЯ 2,3-ДИГИДРОХИНАЗОЛИНА В КАЧЕСТВЕ ИНГИБИТОРОВ NaV1.8 | 2020 |
|
RU2833870C2 |
| ИНГИБИТОРЫ РЕПЛИКАЦИИ ВИРУСА ИММУНОДЕФИЦИТА ЧЕЛОВЕКА | 2019 |
|
RU2812128C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ MAT2A И СПОСОБЫ ПРИМЕНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2809987C2 |
| ТИАЗОЛОПИРИМИДИНОНЫ В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ РЕЦЕПТОРОВ NMDA | 2014 |
|
RU2703273C2 |
| ПРОИЗВОДНЫЕ ПИРИДИНОНА И ПИРИДАЗИНОНА | 2012 |
|
RU2632915C2 |
| ПРОИЗВОДНЫЕ СЕРИНА В КАЧЕСТВЕ АГОНИСТОВ ГРЕЛИНОВЫХ РЕЦЕПТОРОВ | 2015 |
|
RU2695649C2 |
| ПРОИЗВОДНЫЕ ДИАРИЛПИРИДАЗИНОНА, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЛЮДЕЙ | 2011 |
|
RU2584680C2 |
Изобретение относится к новому соединению формулы I или его фармацевтически приемлемой соли. Соединения обладают свойствами ингибитора обратной транскриптазы ВИЧ-1 и могут найти применение для лечения или профилактики инфекции ВИЧ или лечения, профилактики или задержки начала СПИДа. В формуле I
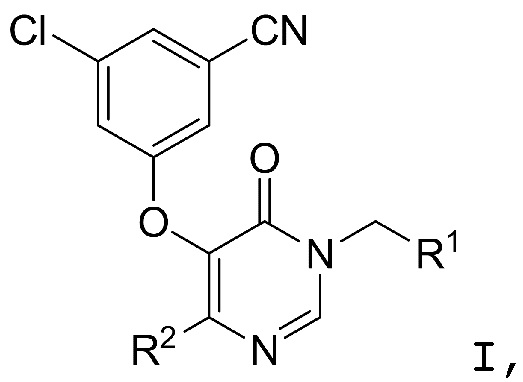
R1 представляет собой 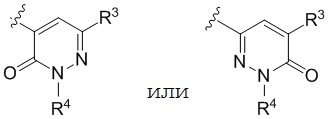 ;
;
R2 представляет собой галоген или -C1-3-алкил, замещенный 1-3 -F; R3 представляет собой (a) галоген, (b) -C1-3-алкил, замещенный 1-3 -F, или (3) фенил, замещенный галогеном; и R4 представляет собой 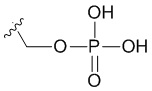 ,
, 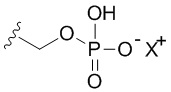 или
или 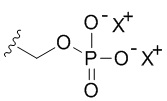 , где X+ представляют собой ион щелочного металла. 5 н. и 7 з.п. ф-лы, 3 табл., 8 пр.
, где X+ представляют собой ион щелочного металла. 5 н. и 7 з.п. ф-лы, 3 табл., 8 пр.
1. Соединение структурной формулы I или его фармацевтически приемлемая соль:
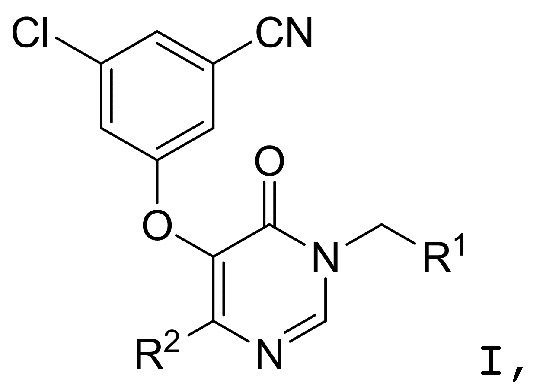
где
R1 представляет собой 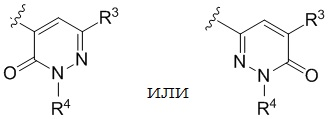 ;
;
R2 представляет собой галоген или -C1-3-алкил, замещенный 1-3 -F;
R3 представляет собой (a) галоген, (b) -C1-3-алкил, замещенный 1-3 -F, или (3) фенил, замещенный галогеном; и
R4 представляет собой 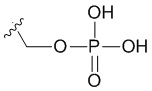 ,
, 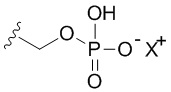 или
или 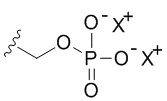 , где X+ представляют собой ион щелочного металла.
, где X+ представляют собой ион щелочного металла.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой 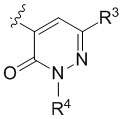 .
.
3. Соединение по п. 1 или его фармацевтически приемлемая соль, где R1 представляет собой 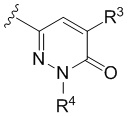 .
.
4. Соединение по любому из пп. 1-3 или его фармацевтически приемлемая соль, где R2 представляет собой метил, замещенный 1, 2 или 3 -F; или этил, замещенный 1, 2 или 3 -F.
5. Соединение по п. 4 или его фармацевтически приемлемая соль, где R2 представляет собой -CHF2, -CF3 или -CF2CH3.
6. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, где R3 представляет собой -F; -Cl; метил, замещенный 1, 2 или 3 -F; этил, замещенный 1, 2 или 3 -F; или фенил, замещенный -F.
7. Соединение по п. 6 или его фармацевтически приемлемая соль, где R3 представляет собой -Cl, -CHF2, -CF3, -CF2CH3 или фенил, замещенный -F.
8. Соединение по п. 1, которое представляет собой:
1) (5-((5-(3-хлор-5-цианофенокси)-4-(дифторметил)-6-оксопиримидин-1(6H)-ил)метил)-3-(дифторметил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат;
2) (3-хлор-5-((5-(3-хлор-5-цианофенокси)-4-(дифторметил)-6-оксопиримидин-1(6H)-ил)метил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат;
3) 3-хлор-5-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат;
4) (5-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-3-(дифторметил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат;
5) (5-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-3-(1,1-дифторэтил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат;
6) (3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-6-оксо-5-(трифторметил)пиридазин-1(6H)-ил)метилдигидрофосфат;
7) (3-((5-(3-хлор-5-цианофенокси)-6-оксо-4-(трифторметил)пиримидин-1(6H)-ил)метил)-5-(4-фторфенил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат; или
8) (5-((5-(3-хлор-5-цианофенокси)-4-(1,1-дифторэтил)-6-оксопиримидин-1(6H)-ил)метил)-3-(1,1-дифторэтил)-6-оксопиридазин-1(6H)-ил)метилдигидрофосфат;
или его фармацевтически приемлемая соль.
9. Фармацевтическая композиция, обладающая свойствами ингибитора обратной транскриптазы ВИЧ-1, содержащая эффективное количество соединения по любому из пп. 1-8 или его фармацевтически приемлемой соли и фармацевтически приемлемый носитель.
10. Способ ингибирования репликации ВИЧ у нуждающегося в этом индивидуума, который включает введение индивидууму эффективного количества соединения по любому из пп. 1-8 или его фармацевтически приемлемой соли.
11. Способ лечения или профилактики инфекции ВИЧ или лечения, профилактики или задержки начала СПИДа у нуждающегося в этом индивидуума, который включает введение индивидууму эффективного количества соединения по любому из пп. 1-8 или его фармацевтически приемлемой соли.
12. Применение соединения по любому из пп. 1-8 или его фармацевтически приемлемой соли в производстве лекарственного средства, пригодного для ингибирования репликации ВИЧ, для лечения или профилактики инфекции ВИЧ или для лечения, профилактики или задержки начала СПИДа.
| US 2013296382 A1, 07.11 | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| US 2005215554 A1, 29.09.2005 | |||
| WO 2014058747 A1, 17.04.2014 | |||
| Lei Ji; et al., Synthesis and Anti-HIV-1 Activity Evaluation of 5-Alkyl-2-alkylthio-6-(arylcarbonyl or α-cyanoarylmethyl)-3,4-dihydropyrimidin-4(3 H )-ones as Novel Non-nucleoside HIV-1 Reverse Transcriptase Inhibitors, Journal of Medicinal Chemistry, 2007, Vol:50, Nr:8, Page(s):1778 - 1786 | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Способ получения сложных фосфорсодержащих удобрений | 1928 |
|
SU17281A1 |

