Область техники
Настоящее изобретение принадлежит к области медицины и относится к производному 6-оксо-1,6-дигидропиридазина, способу его получения и его применению в медицине. В частности, настоящее изобретение относится к производному 6-оксо-1,6-дигидропиридазина формулы (I), способу его получения, содержащей его фармацевтической композиции, его применению в качестве ингибитора потенциал-зависимых натриевых каналов (NaV) и его применению для получения лекарственного средства для лечения и/или ослабления боли и связанных с болью заболеваний.
Предшествующий уровень техники
Боль - это комплексная физическая и психологическая деятельность, которая является одним из наиболее часто встречающихся клинических симптомов. Международная ассоциация по изучению боли определяет боль как "неприятное сенсорное и эмоциональное переживание, связанное с реальным или предполагаемым повреждением тканей, которое является субъективным ощущением". Боль может действовать как предупреждающий сигнал, напоминающий телу о необходимости обратить внимание на возможные опасности, и оказывает необходимый защитный эффект в отношении нормальной жизнедеятельности организма. Более того, боль также является часто встречающимся клиническим симптомом. После исчезновения внешнего раздражителя, вызывающего боль, сильная или постоянная боль может привести к нарушению физиологической функции и серьезно повлиять на качество жизни живого организма. Статистика показывает, что примерно пятая часть людей в мире страдает хронической болью в степени от умеренной до тяжелой.
Боль генерируется ноцицепторами в периферической нервной системе. Ноцицепторы представляют собой разновидность свободных нервных окончаний и широко распространены в коже, мышцах, суставах и висцеральных тканях всего организма. Ноцицепторы могут преобразовывать тепловые, механические или химические раздражители в нервные импульсы (потенциалы действия), передавать их телу клетки в дорсальных корешковых ганглиях (DRG, от англ. dorsal root ganglia) через афферентные нервные волокна и в конечном итоге в специальный нервный центр, вызывая, таким образом, боль. Возникновение и распространение потенциалов действия в нейронах зависят от потенциал-зависимых натриевых каналов (NaV), расположенных на клеточной мембране. Когда клеточная мембрана деполяризуется, происходит активация натриевого ионного канала. Канал открывается, вызывая приток ионов натрия и дальнейшую деполяризацию клеточной мембраны, что приводит к возникновению потенциалов действия. Ввиду этого, ингибирование аномальной активности натриевых ионных каналов способствует лечению и облегчению боли.
NaV представляет собой разновидность трансмембранного, образующего ионный канал белка. Данный белок состоит из альфа-субъединицы с молекулярной массой 260 кДа (килодальтон) и бета-субъединицы с молекулярной массой 30-40 кДа. Сообразно с разными α-субъединицами его можно подразделить на 9 подтипов, а именно, от NaV1.1 до NaV1.9. Для разных подтипов продемонстрированы различное распределение в тканях и разные электрофизиологические и фармакологические характеристики (Rush A.M., et al. J. Physiol., 2007, 579, 1-14). В соответствии с тем, возможно ли эффективное ингибирование тетрод ото кси ном (ТТХ, от англ. tetrodotoxin) в наномолярных концентрациях, натриевые ионные каналы подразделяют на каналы ТТХ чувствительного типа (TTX-S, от англ. TTX-sensitive) и ТТХ устойчивого типа (ТТХ-R, TTX-resistant). Среди них NaV1.1, NaV1.2, NaV1.3 и NaV1.7 относятся к TTX-S типу, кодирующие их гены расположены в хромосоме 2q23-24 человека, и они экспрессируются в больших количествах в нейронах. NaV1.5, NaV1.8 и NaV1.9 относятся к TTX-R типу, и кодирующие их гены расположены в хромосоме 3р21-24 человека. Среди них NaV1.5 присутствует главным образом в кардиомиоцитах, а NaV 1.8 и NaV 1.9 присутствуют в периферической нервной системе (Goldin A.L., et al. Аппи. Rev. Physiol., 2001, 63, 871-894). NaV1.4 и NaV1.6 относятся к TTX-S типу и присутствуют в больших количествах в скелетных мышцах и центральной нервной системе, соответственно (Fozzard Н.А., et al. Physiol. Rev., 1996, 76, 887-926). Местный анестетик лидокаин снимает боль посредством ингибирования NaV. Неселективные ингибиторы NaV, такие как ламотриджин, лакосамид и мексилетин, с успехом используются для лечения хронической боли.
NaV1.8 относится к TTX-R типу, кодирующим его геном является SCN10A. Он присутствует главным образом в нейронах тригеминального ганглия и нейронах DRG и имеет электрофизиологические характеристики, соответствующие медленной инактивации и быстрому восстановлению (Dib-Hajj S.D., et al. Аппи. Rev. Neurosci., 2010, 33, 325-347). В нейронах, экспрессирующих NaV1.8, нарастание потенциала действия обусловлено главным образом током через NaV1.8. В некоторых моделях исследования нейропатической боли показано, что при повреждении нервов может повышаться уровень экспрессии NaV1.8 в аксонах и телах нейронных клеток (Sleeper А.А., et al. J. Neurosci,, 2000, 20, 7279-7289). Применение NaV1.8-антисмыслового олигонуклеотида может значительно облегчать боль при одновременном ослаблении экспрессии NaV1.8 (Yoshimura N., et al., J. Neurosci,, 2001, 21, 8690-8696). После введения каррагинана в лапы крыс экспрессия NaV1.8 в нейронах DRG возрастала (Tanaka М., et al. G, NeuroReport, 1998, 9, 967-972). Мыши с нокаутом гена NaV1.8 не могут проявлять признаков боли при обычном воспалении внутренних органов (Kerr B.J., et al. NeuroReport, 2001, 12, 3077-3080). Наличие в гене NaV1.8 человека мутации, приводящей к усилению функциональной активности, будет вызывать периферическую невралгию (Faber C.G., et al., Proa Natl. Acad. Sci. USA, 2012, 109, 19444-19449). Согласно результатам серии экспериментов на животных и генетическим данным для человека снлективное ингибирование NaV1.8 может стать новым типом аналгезирующей терапии, которую можно использовать для лечения различных типов боли, такой как воспалительная боль, нейропатическая боль, послеоперационная боль и боль при раковом заболевании.
Используемые в клиниках ингибиторы NaV могут ингибировать натриевые каналы, экспрессируемые в сердце и центральной нервной системе, вследствие недостаточной избирательности в отношении подтипов. Поэтому, их терапевтическое окно является узким и область применения ограниченной. NaV1.8 присутствует в основном в периферической нервной системе, поэтому избирательное ингибирование NaV1.8 может эффективно снижать побочные эффекты. Таким образом, существует необходимость в разработке ингибиторов NaV1.8 с более высокой активностью, улучшенной селективностью, улучшенными фармакокинетическими свойствами и меньшим количеством побочных эффектов.
Краткое описание сущности изобретения
Целью настоящего изобретения является получение соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли,
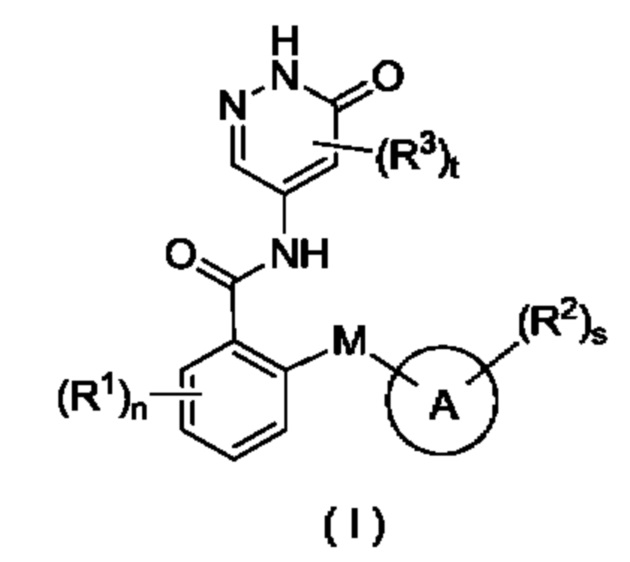
где:
М выбран из группы, состоящей из атома О, CR4R5 и атома S;
кольцо А представляет собой арил или гетероарил, при этом арил или гетероарил возможно конденсированы с циклоалкилом или гетероциклилом;
все R1 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, дейтерированного алкила, дейтерированного алкокси, алкокси, галогеналкила, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетеро цикл ила, арила и гетероарила;
все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, дейтерированного алкила, дейтерированного алкокси, галогеналкила, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, циклоалкилокси, гетеро цикл ил а, арила и гетероарила, при этом алкил, циклоалкил, гетероциклил, арил и гетероарил в каждом случае возможно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, галогеналкила, галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетеро цикл ила, арила и гетероарила;
все R3 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетеро цикл ила, арила и гетероарила;
R4 и R5 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, атома дейтерия, галогена, алкила, алкокси, галогеналкила, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетеро цикл ила, арила и гетероарила;
n равно 0, 1, 2, 3 или 4;
s равно 0, 1, 2, 3 или 4; и
t равно 0, 1 или 2.
В некоторых воплощениях настоящего изобретения предложены соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль:
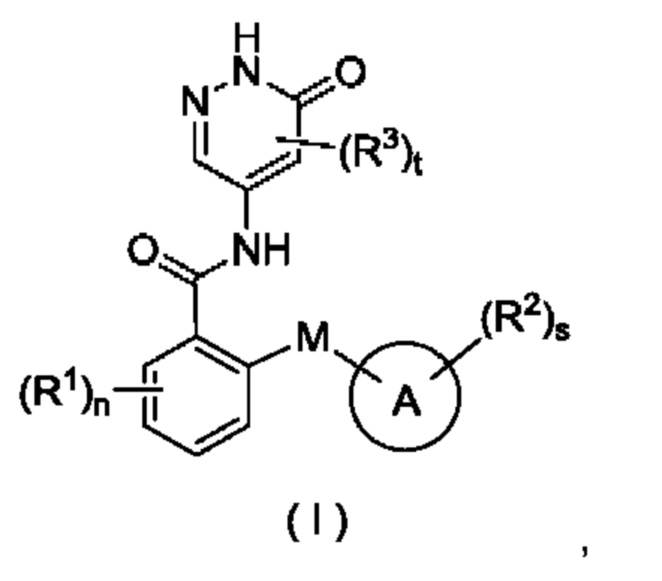
где:
М выбран из группы, состоящей из атома О, CR4R5 и атома S;
кольцо А представляет собой арил или гетероарил;
R1 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R2 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, циклоалкилокси, гетероциклила, арила и гетероарила, при этом алкил, циклоалкил, гетероциклил, арил и гетероарил в каждом случае возможно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, галогеналкила, галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R3 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R4 и R5 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
n равно 0, 1, 2, 3 или 4;
s равно 0, 1, 2, 3 или 4; и
t равно 0, 1 или 2.
В некоторых воплощениях настоящего изобретения предложены соединение
формулы (I):
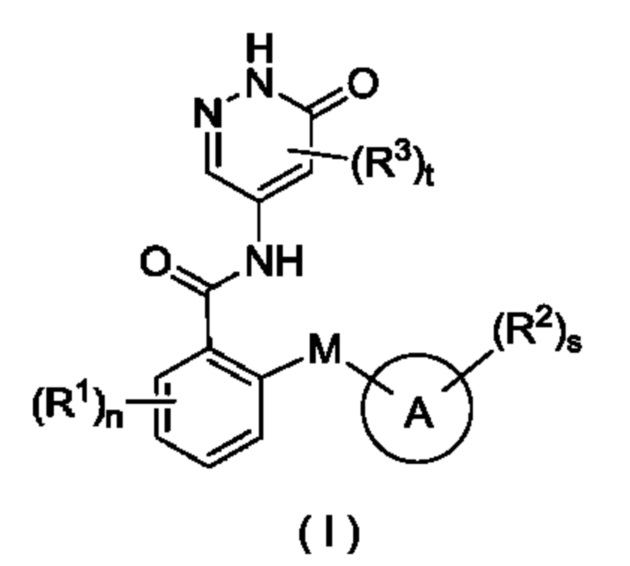
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль,
где:
М выбран из группы, состоящей из О, CR4R5 и S;
кольцо А представляет собой арил или гетероарил;
R1 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R2 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила, при этом алкил, циклоалкил, гетероциклил, арил и гетероарил в каждом случае возможно замещен одним или более заместителями, выбранными из группы, состоящей из алкила, галогеналкила, галогена, амино, нитро, циано, гидрокси, алкокси, галогеналкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R3 выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R4 и R5 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, циано, амино, нитро, гидрокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
n равно 0, 1, 2, 3 или 4;
s равно 0, 1, 2, 3 или 4; и
t равно 0, 1 или 2.
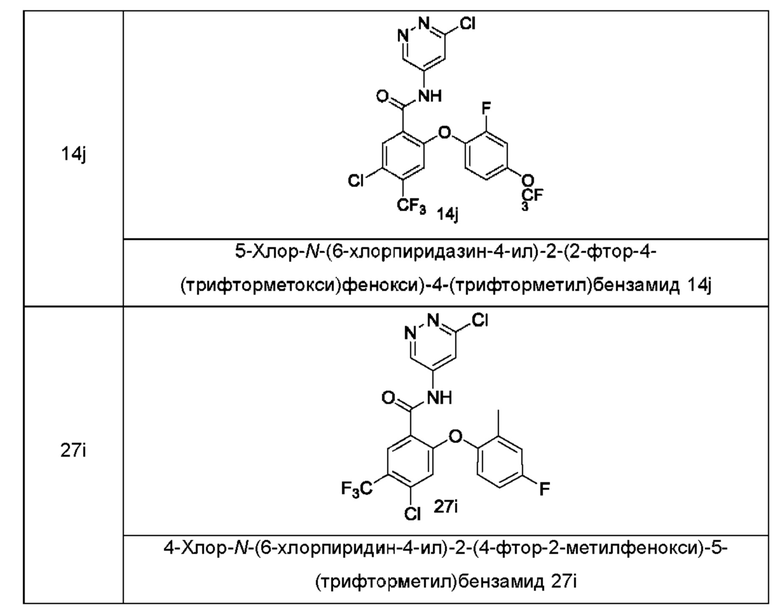
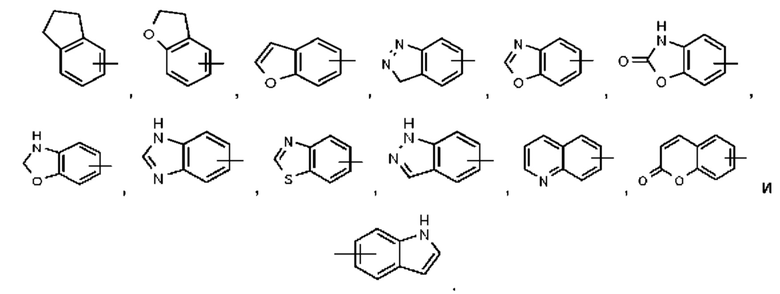
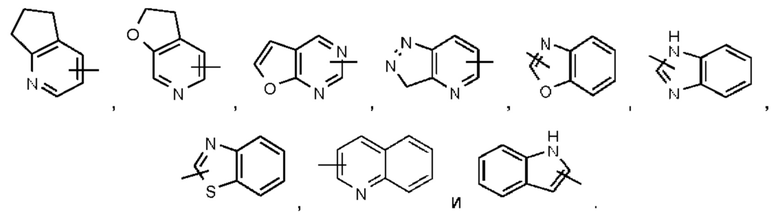
В некоторых воплощениях настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли кольцо А выбрано из группы, состоящей из фенила,  и пиридила.
и пиридила.
В некоторых воплощениях настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли кольцо А представляет собой фенил или пиридил.
В некоторых воплощениях настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли М выбран из группы, состоящей из атома О, СН2 и атома S.
В некоторых воплощениях настоящего изобретения в соединении формулы
(I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли М представляет собой атом О.
В некоторых воплощениях настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль представляет собой соединение формулы
(II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль:
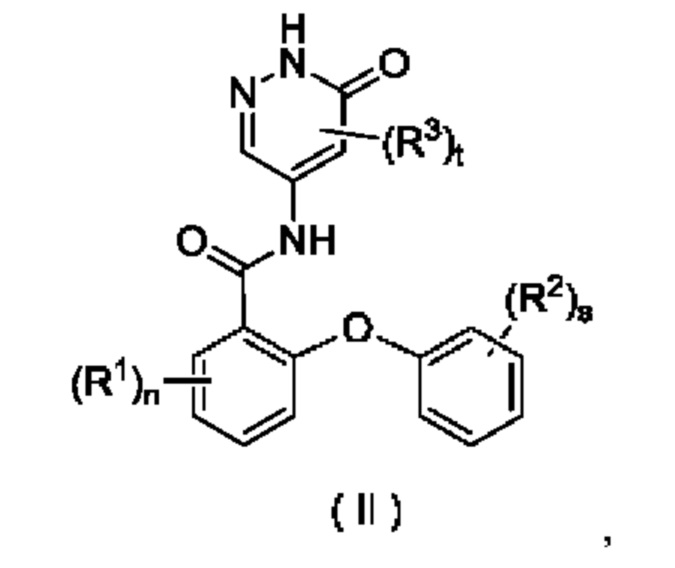
где:
R1, R2, R3, n, s и t являются такими, как определено в формуле (I).
В некоторых воплощениях настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль представляет собой соединение формулы (III) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемую соль:
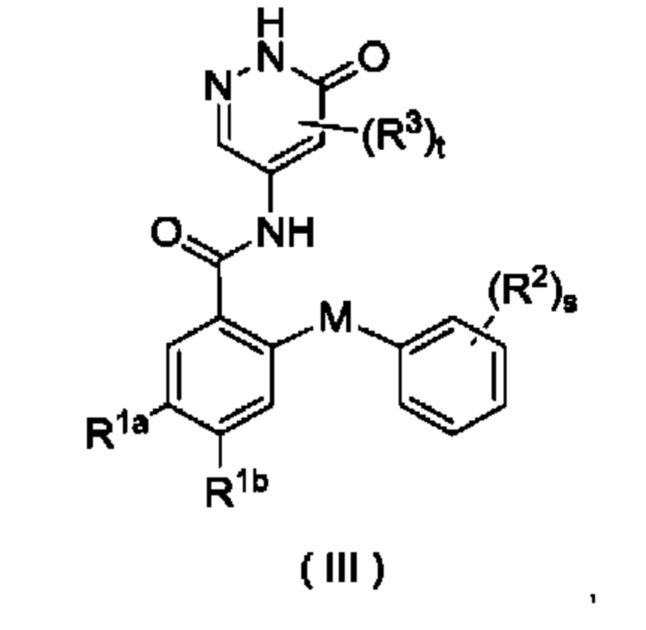
где:
М выбран из группы, состоящей из атома О, СН2 и атома S;
R1a представляет собой галоген и предпочтительно выбран из группы, состоящей из Cl, Br и F;
R1b выбран из группы, состоящей из галогена, алкила, алкокси, галогеналкила и галогеналкокси, и предпочтительно галогеналкила; и
R2, R3, s и t являются такими, как определено в формуле (I).
В некоторых воплощениях настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли, все R1 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила и галогеналкокси.
В некоторых воплощениях настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли, все R1 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила и галогеналкила.
В некоторых воплощениях настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли, все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, дейтерированного алкила, алкокси, дейтерированного алкокси, гидрокси, галогеналкила, галогеналкокси, циклоалкила и циклоалкилокси; предпочтительно, все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6алкила, дейтерированного С1-6алкила, С1-6алкокси, дейтерированного С1-6алкокси, галогенС1-6алкила, галогенС1-6алкокси, гидрокси, С3-6циклоалкила и С3-6циклоалкилокси; и более предпочтительно, все R2 являются одинаковыми или cразными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6алкила, С1-6алкокси и дейтерированного С1-6алкокси.
В некоторых воплощениях настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли, все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, дейтерированного алкокси, гидрокси, галогеналкила, галогеналкокси, циклоалкила и циклоалкилокси; и предпочтительно, все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила, галогеналкокси, циклоалкила и циклоалкилокси.
В некоторых воплощениях настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли, все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси, галогеналкила и галогеналкокси.
В некоторых воплощениях настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли s равно 2.
В некоторых воплощениях настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль представляет собой соединение формулы (IV) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемую соль:
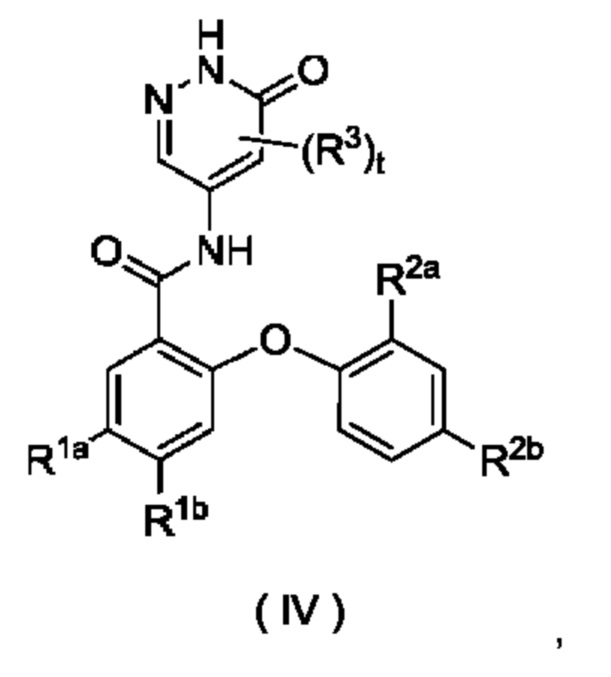
где:
R1aa представляет собой галоген;
R1b выбран из группы, состоящей из галогена, алкила, алкокси, галогеналкила и галогеналкокси;
R2a представляет собой алкокси или дейтерированный алкокси;
R2b выбран из группы, состоящей из атома водорода, галогена, алкила, алкокси и галогеналкокси; и
R3 и t являются такими, как определено в формуле (I).
В некоторых воплощениях настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли, R3 представляет собой атом водорода.
В некоторых воплощениях настоящего изобретения, в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли, R1a представляет собой атом хлора, и R1b представляет собой трифторметил.
Типичные соединения формулы (I) включают приведенные ниже соединения, но не ограничиваются ими.
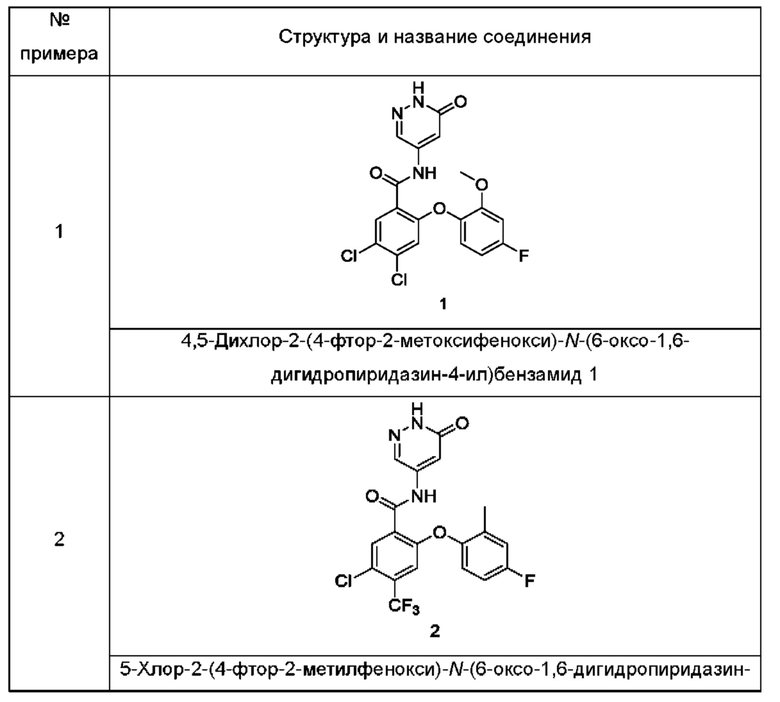
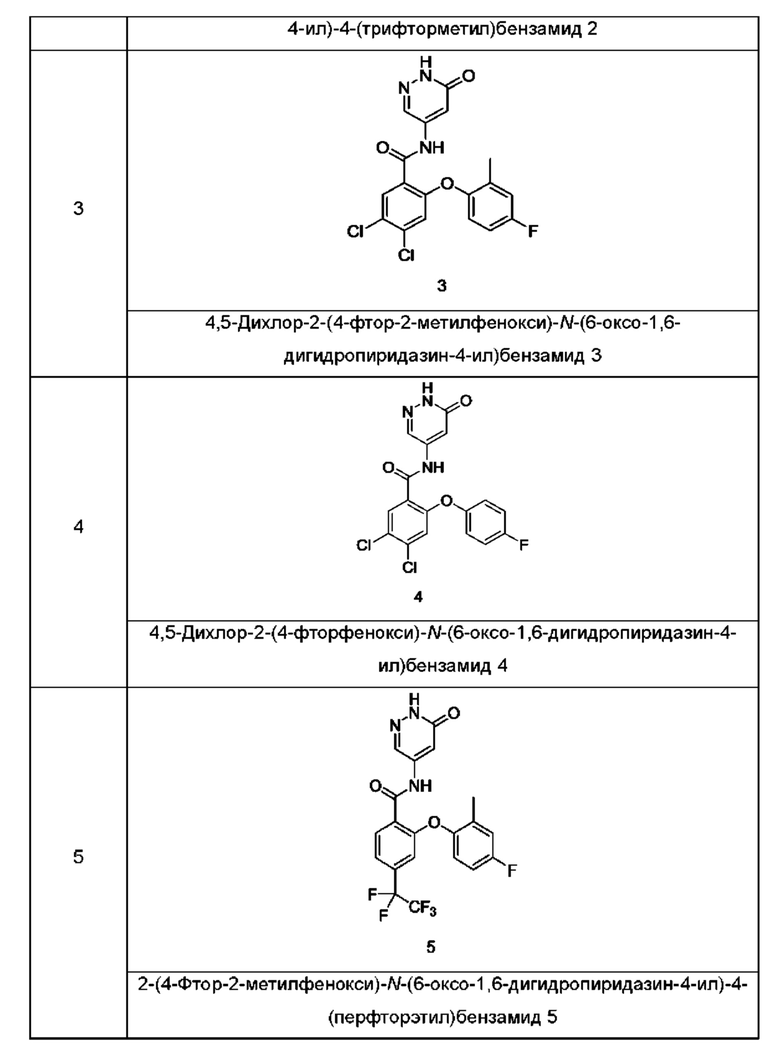
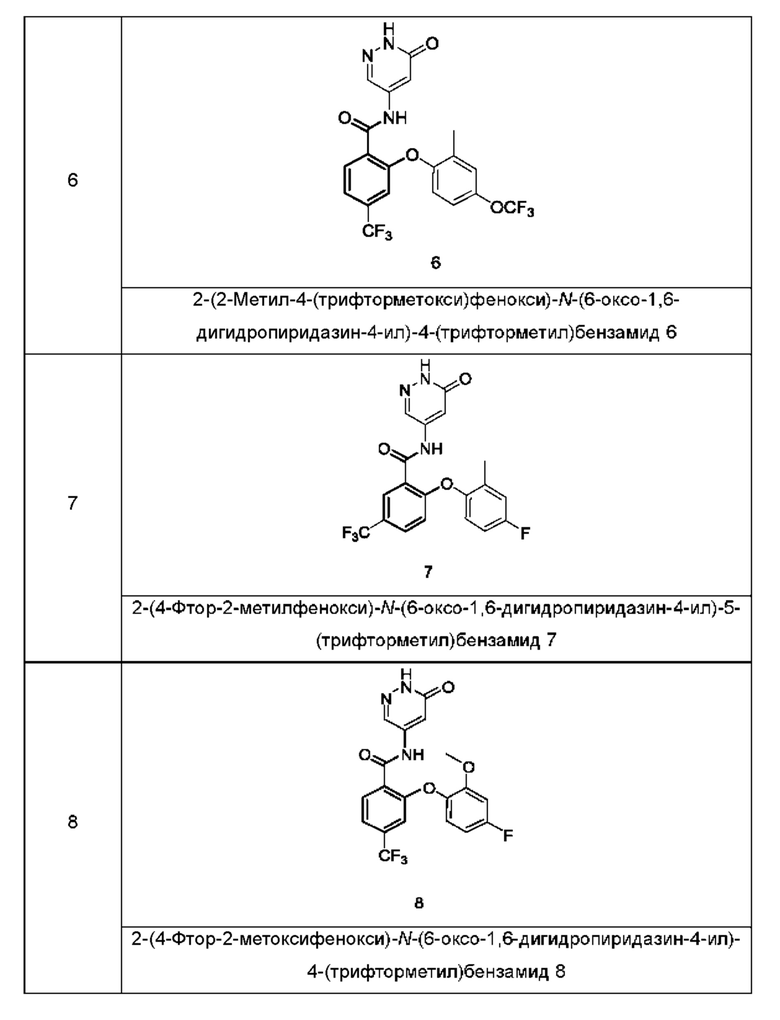
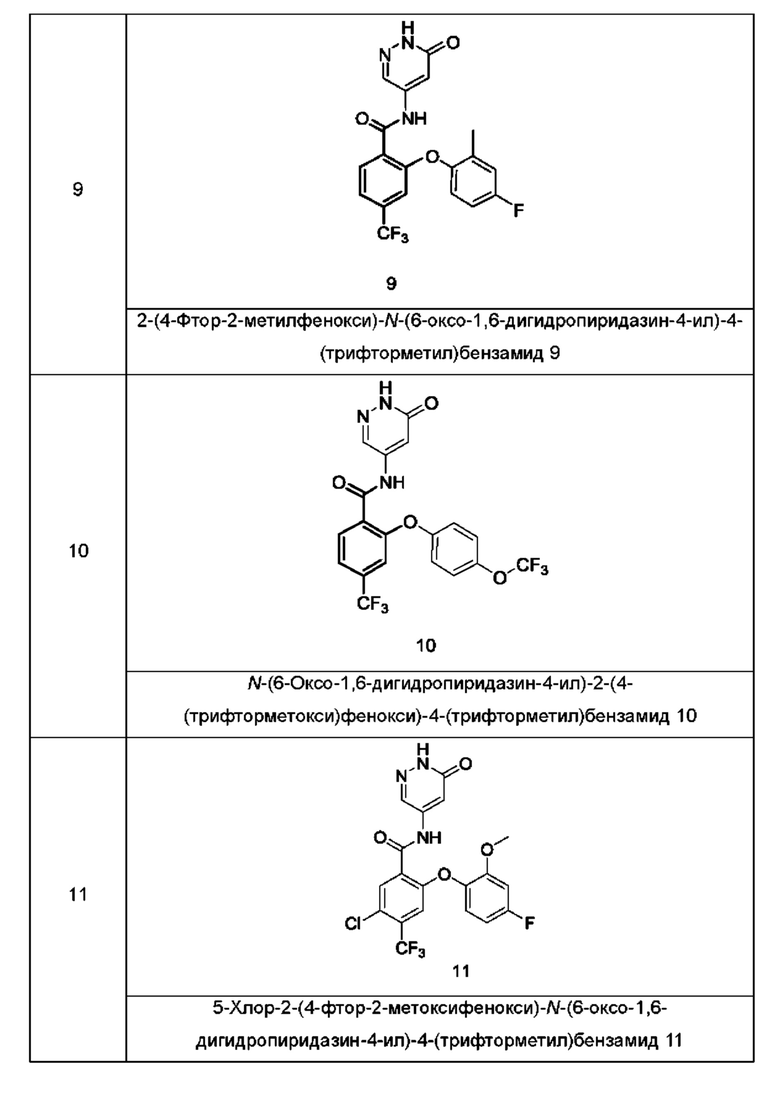
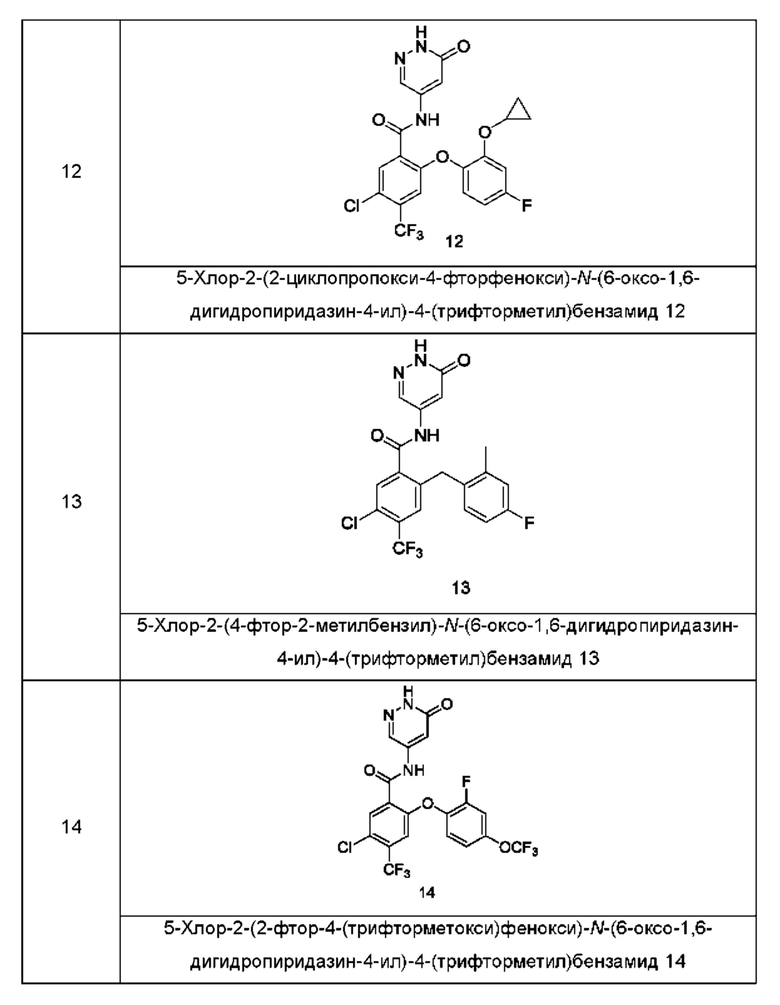
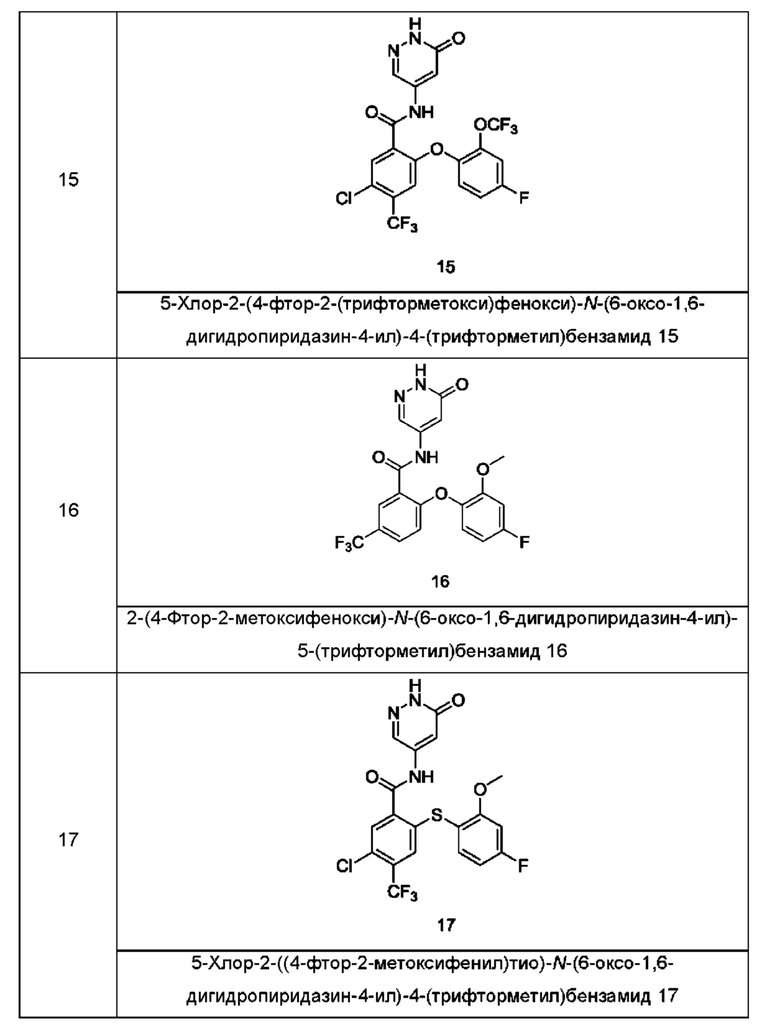
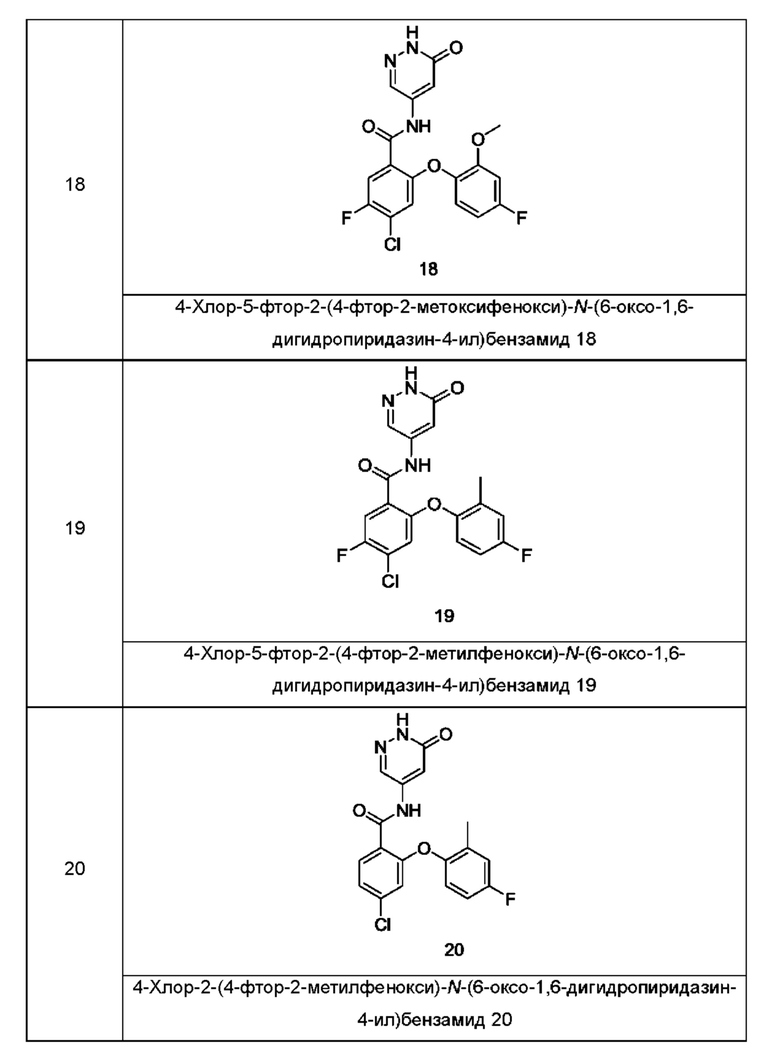
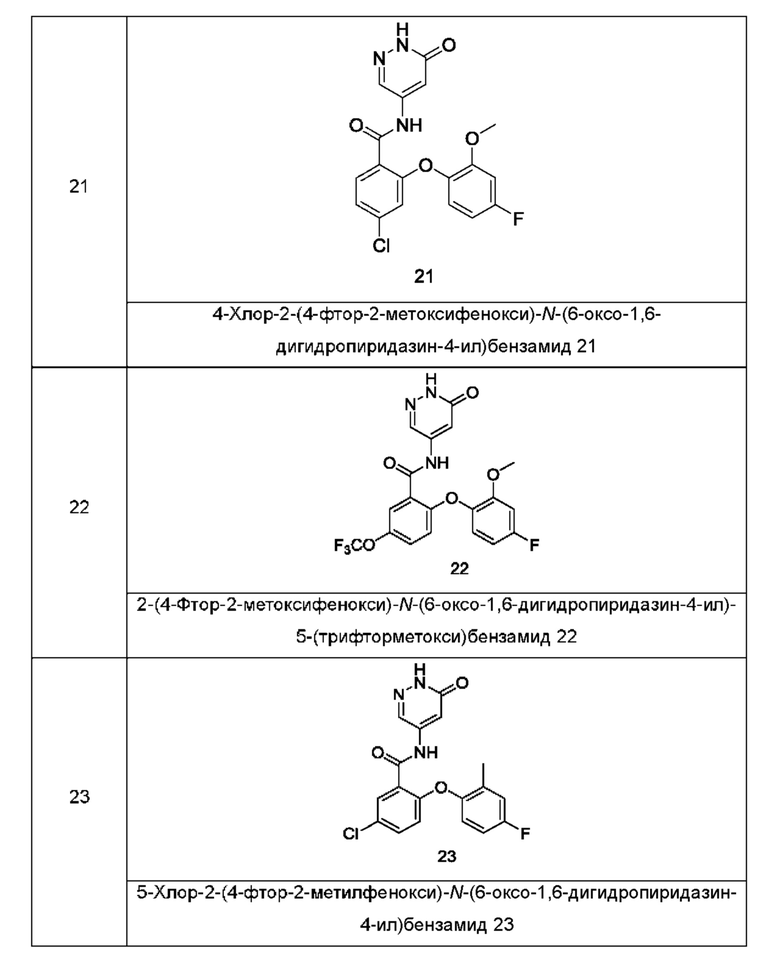
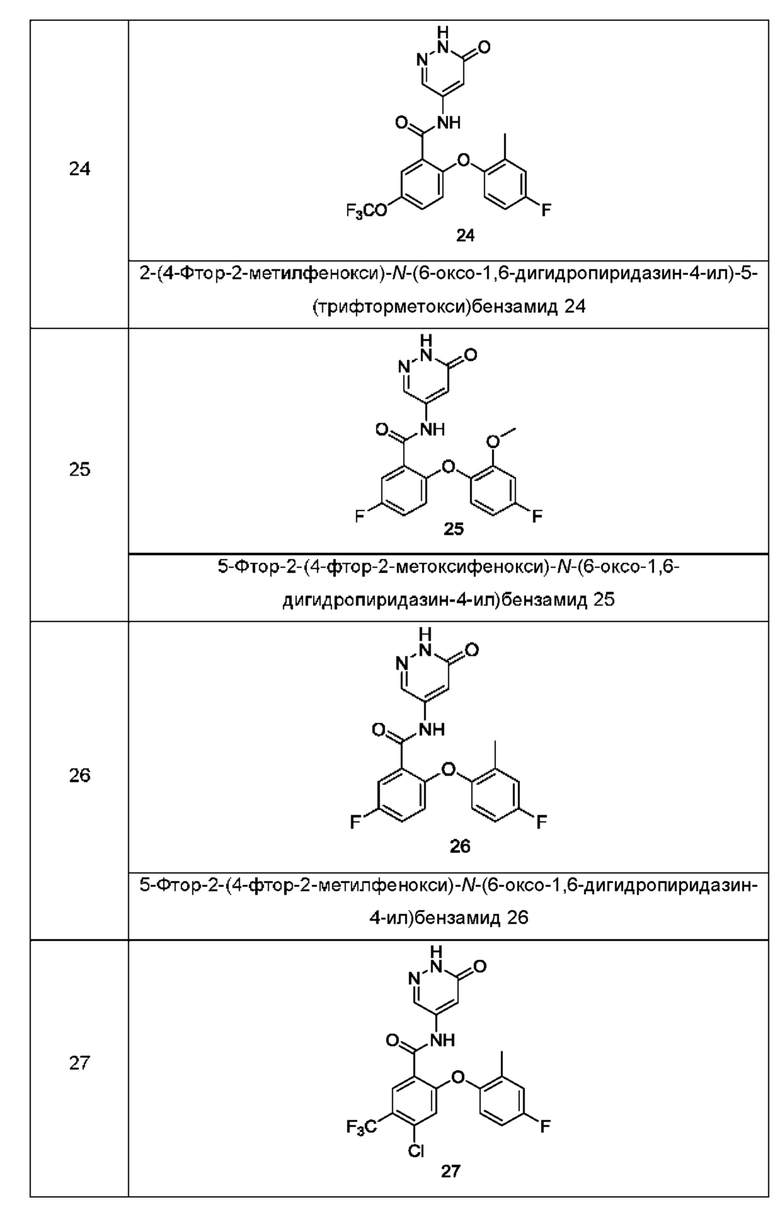
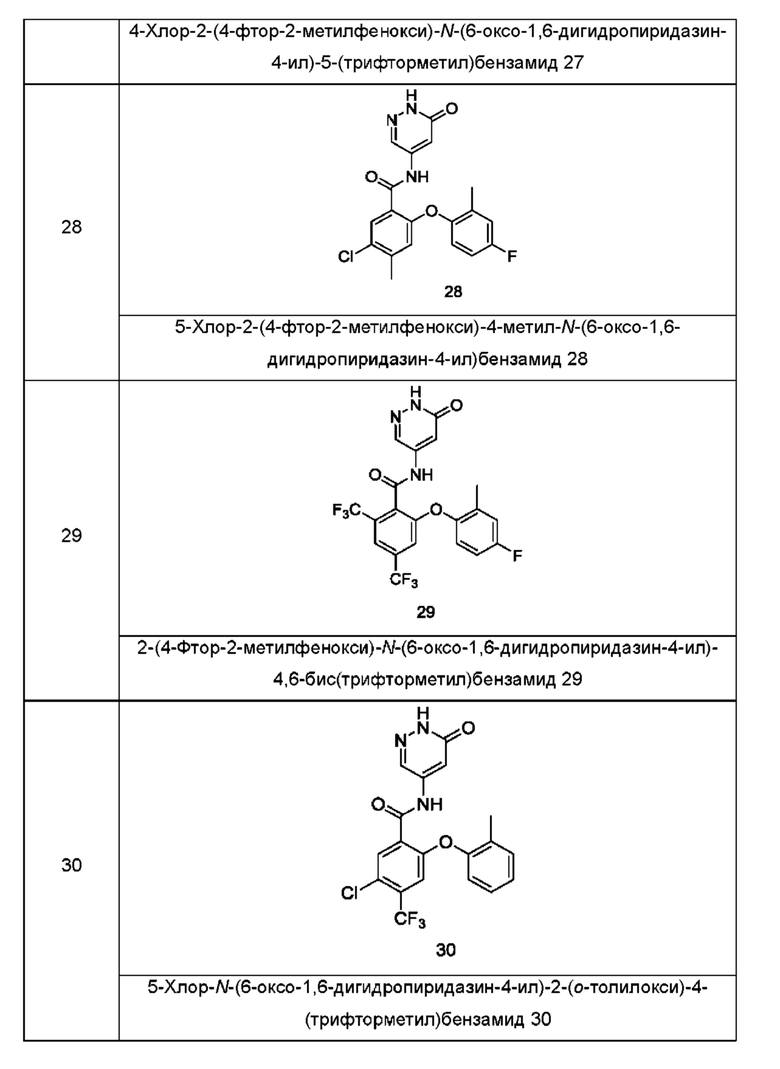
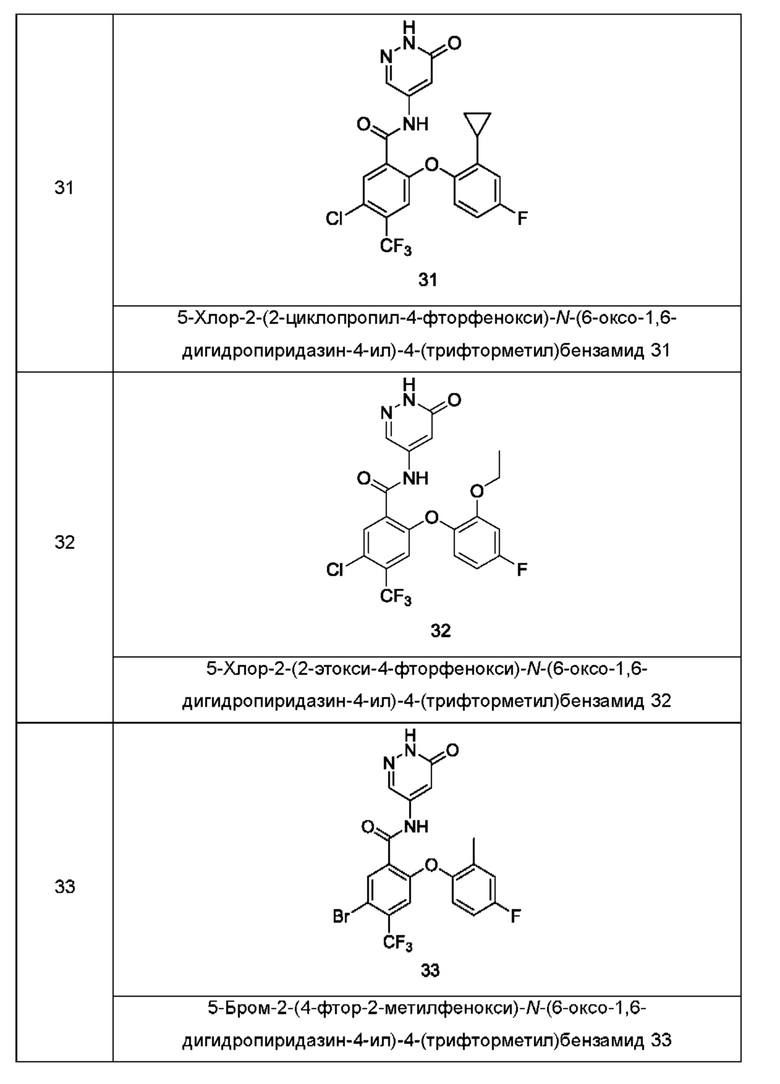
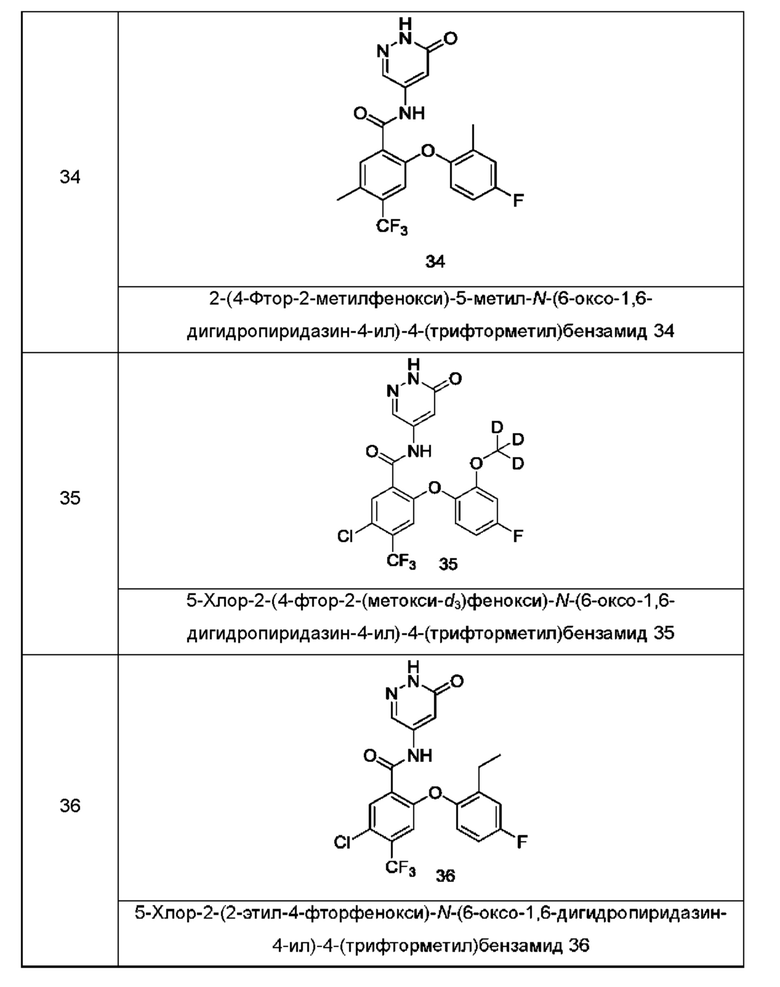
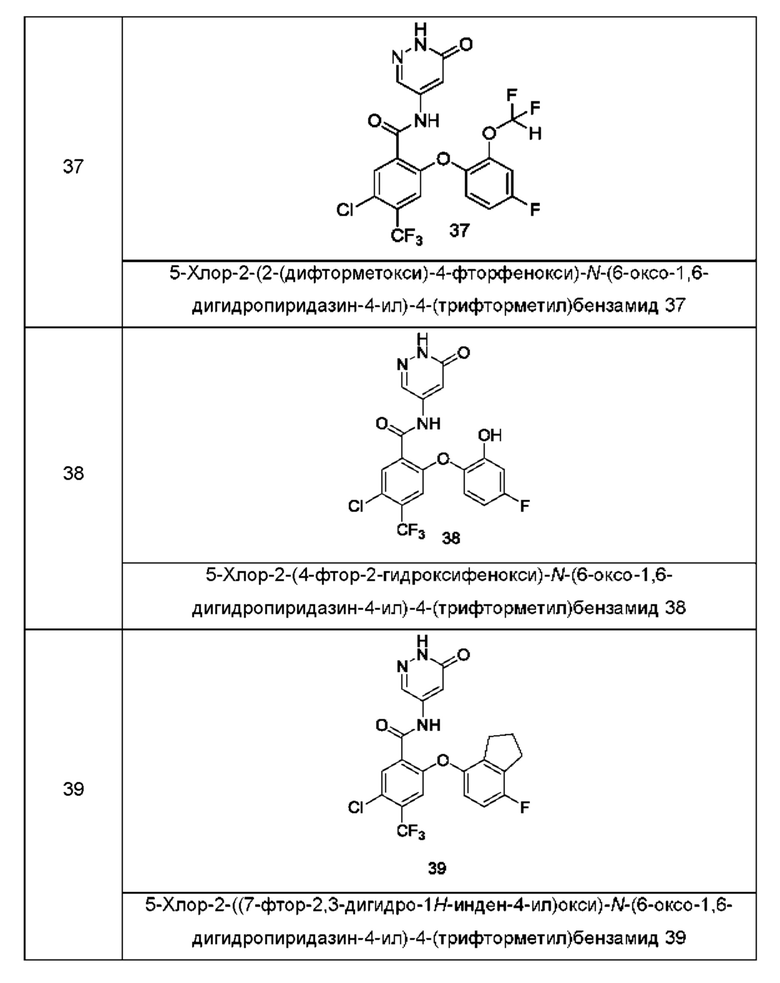
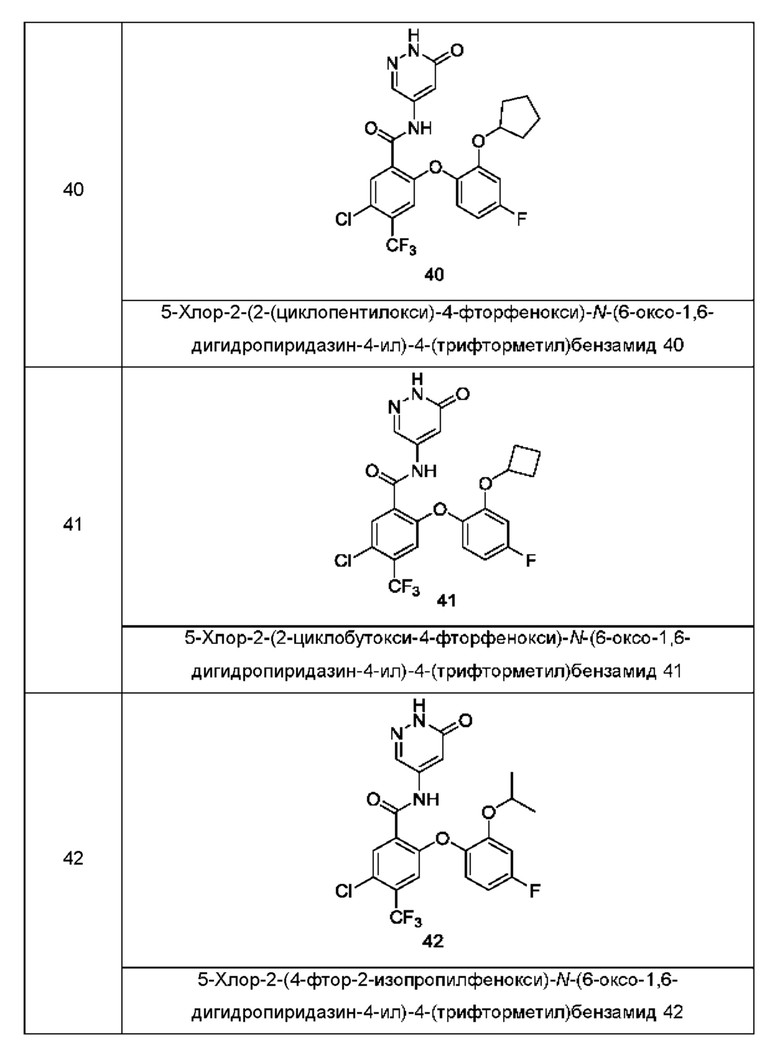
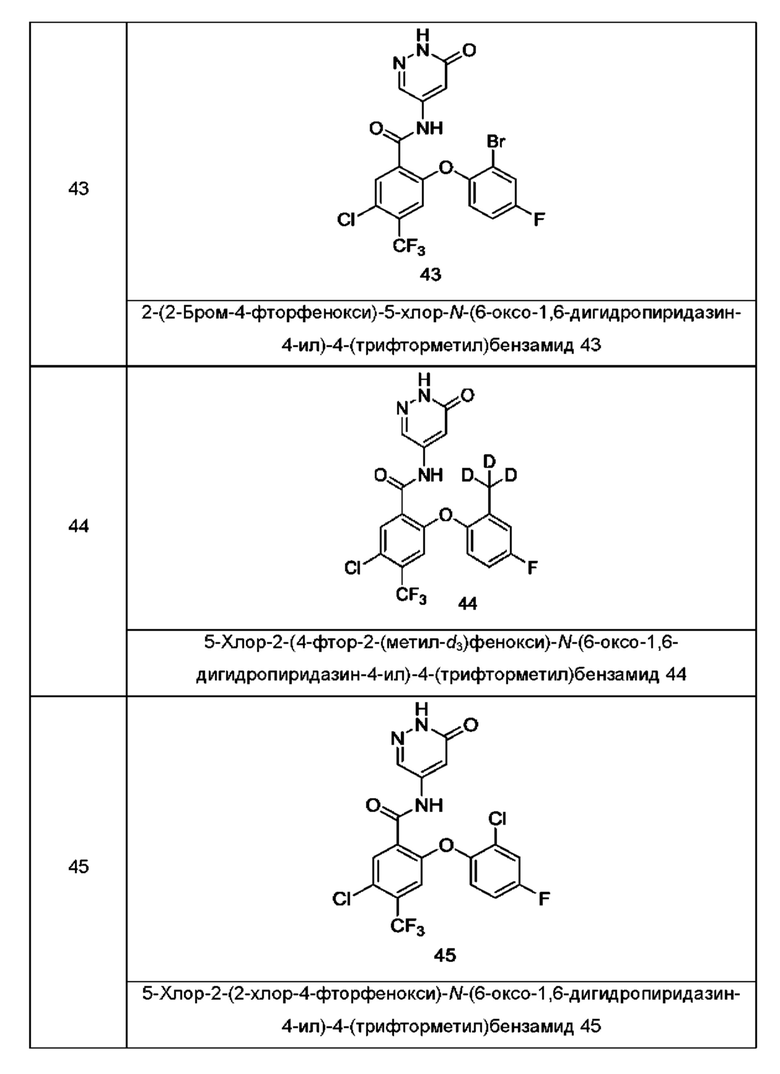
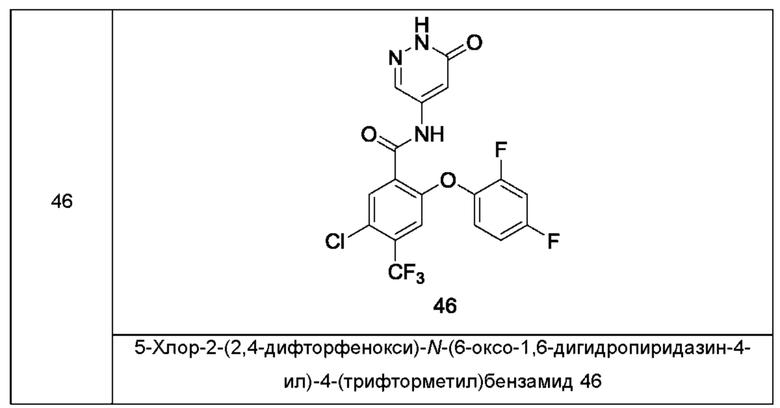
или их таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль.
Согласно другому аспекту настоящего изобретения предложены соединение формулы (IA)
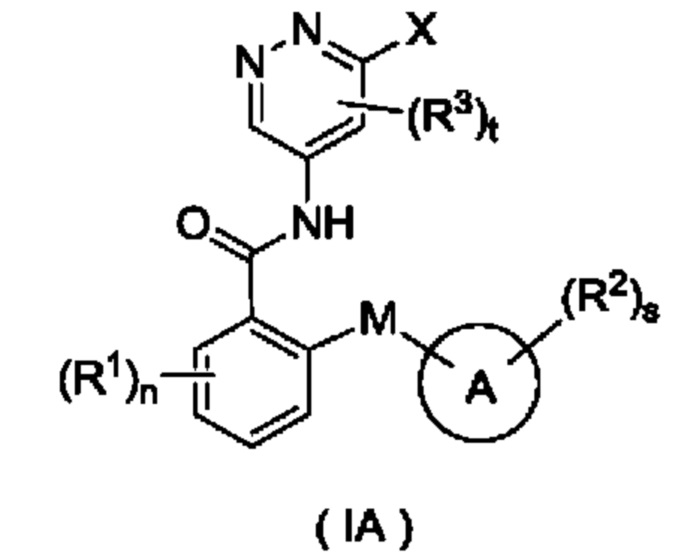
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль,
где:
X представляет собой галоген и предпочтительно Cl; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (I). Соединение формулы (IA) представляет собой промежуточное соединение для получения соединения формулы (I).
Согласно другому аспекту настоящего изобретения предложены соединение формулы (IIA)
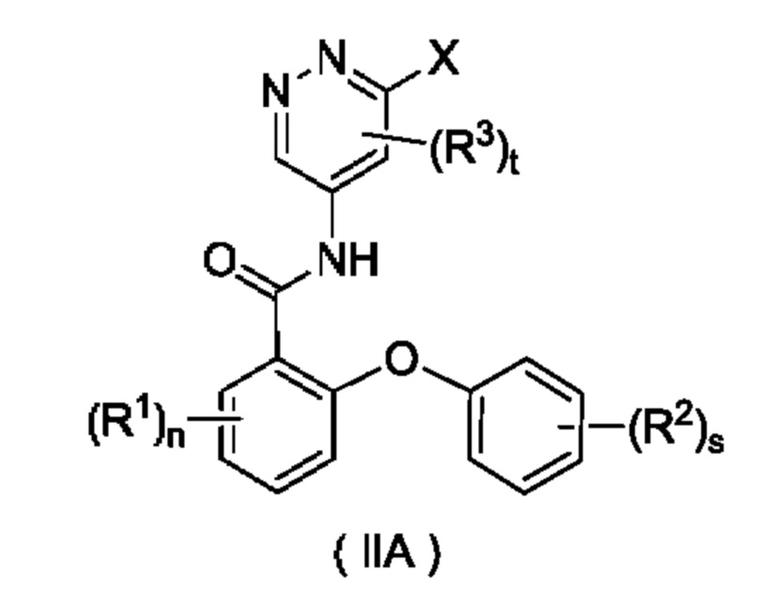
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль,
где:
X представляет собой галоген и предпочтительно Cl; и
кольцо A, R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (II). Соединение формулы (IIA) представляет собой промежуточное соединение для получения соединения формулы (II).
Согласно другому аспекту настоящего изобретения предложены соединение формулы (IB)
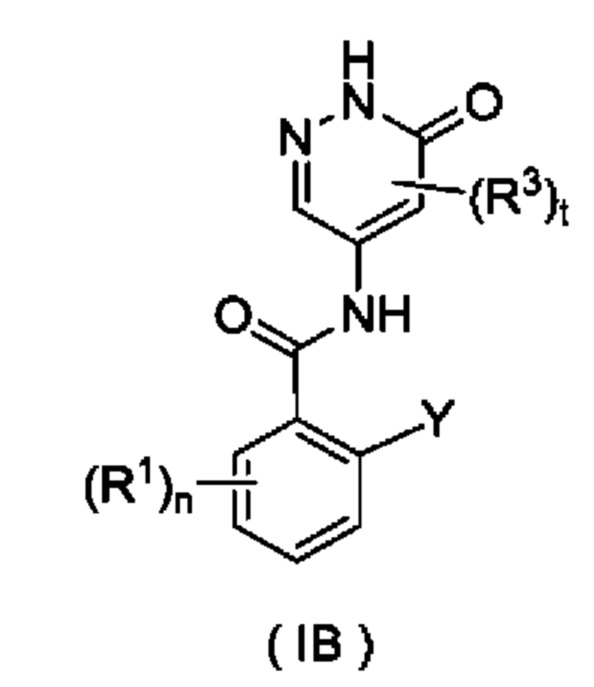
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль,
где:
Y представляет собой галоген и предпочтительно F; и
R1, R3, n и t являются такими, как определено для соединения формулы (I).
Соединение формулы (IB) представляет собой промежуточное соединение для получения соединения формулы (I).
Согласно другому аспекту настоящего изобретения предложены соединение формулы (IIIA)
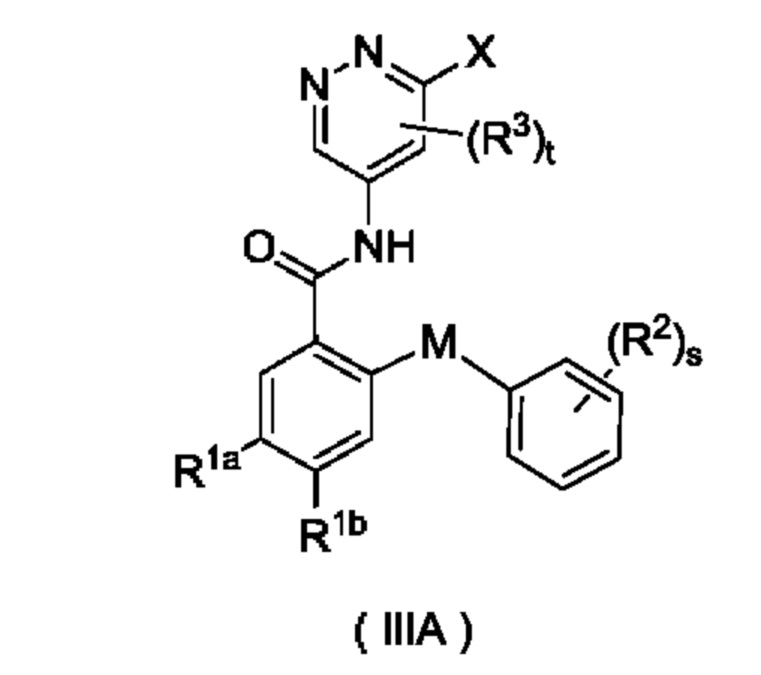
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль, где:
X представляет собой галоген и предпочтительно Cl; и
М, R1a, R1b, R2, R3, s и t являются такими, как определено для соединения формулы (III). Соединение формулы (IIIA) представляет собой промежуточное соединение для получения соединения формулы (III).
Согласно другому аспекту настоящего изобретения предложены соединение формулы (IVA)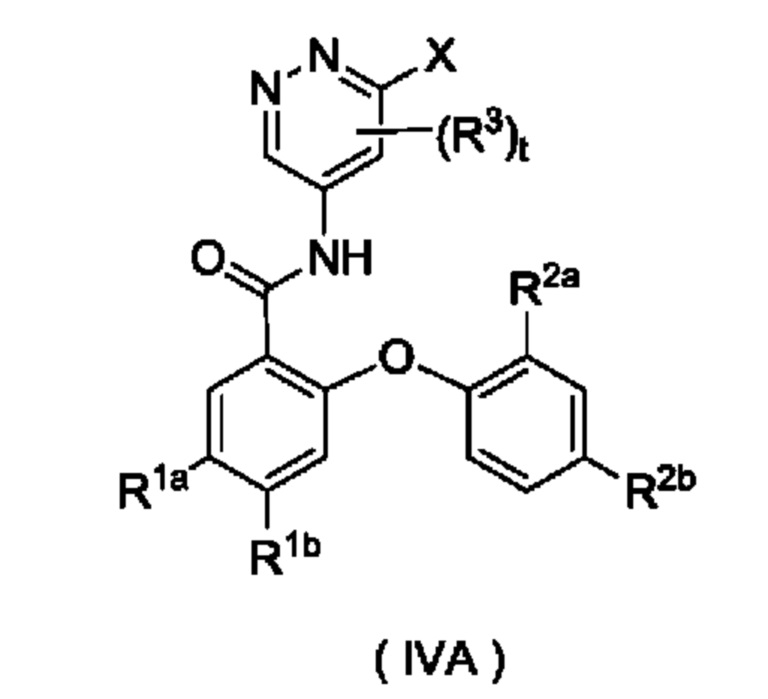
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль,
где:
X представляет собой галоген и предпочтительно Cl; и
R1a, R1b, R2a, R2b, R3, s и t являются такими, как определено для соединения формулы (IV). Соединение формулы (IVA) представляет собой промежуточное соединение для получения соединения формулы (IV).
Согласно другому аспекту настоящего изобретения предложены соединение формулы (IIIB)
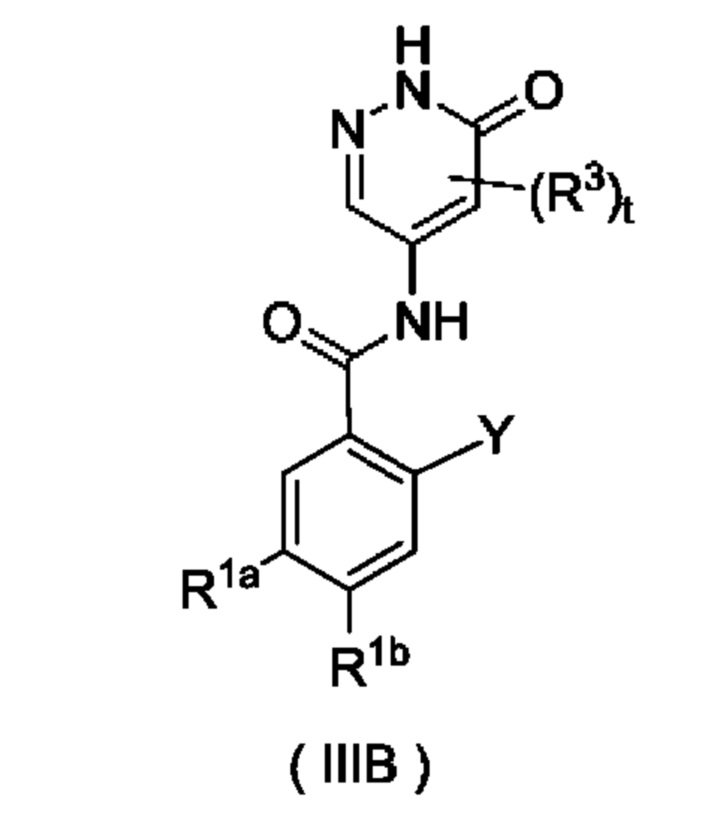
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемая соль,
где:
Y представляет собой галоген и предпочтительно F; и
R1a, R1b, R3 и t являются такими, как определено для соединения формулы (III). Соединение формулы (IIIB) представляет собой промежуточное соединение для получения соединения формулы (III).
Типичные промежуточные соединения включают приведенные ниже соединения, но не ограничиваются ими.
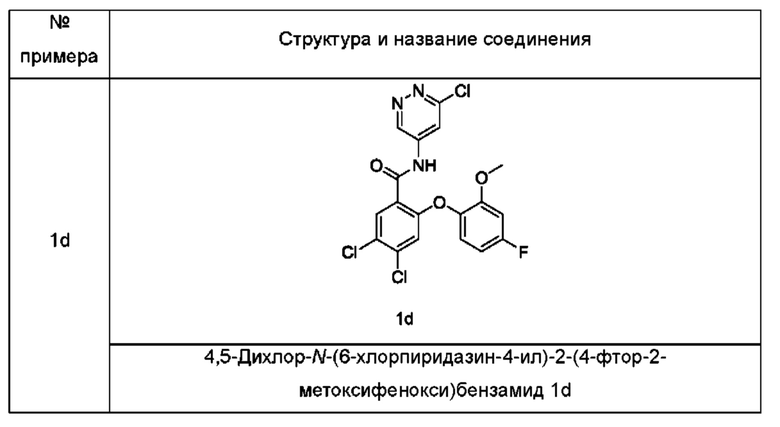
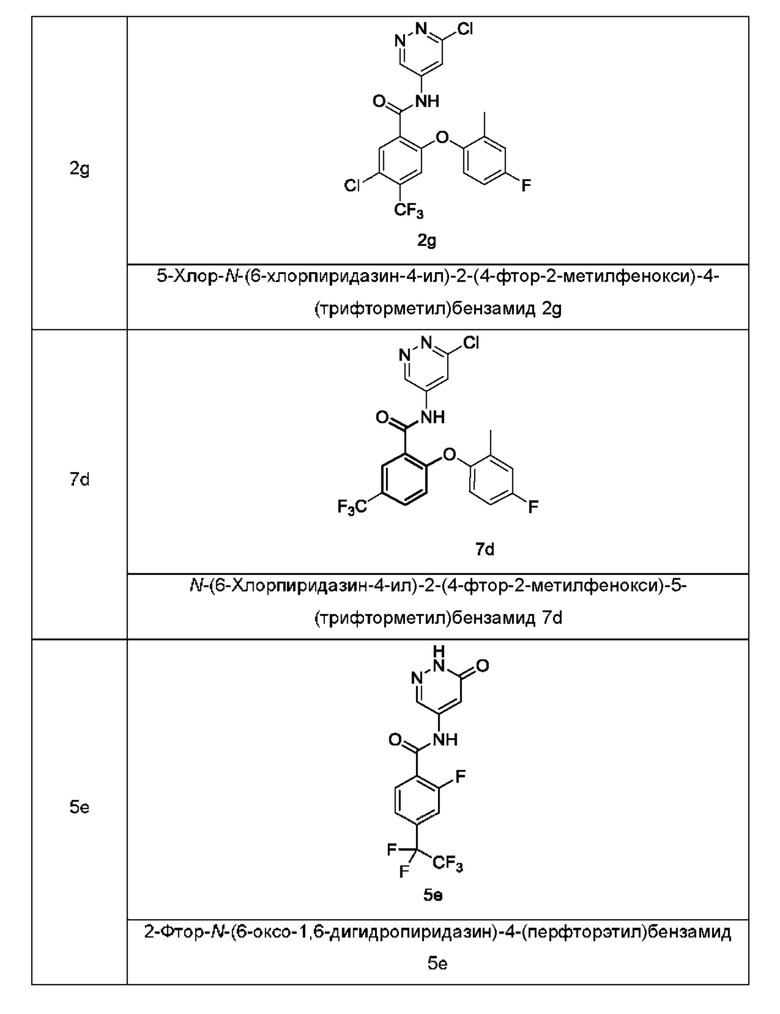
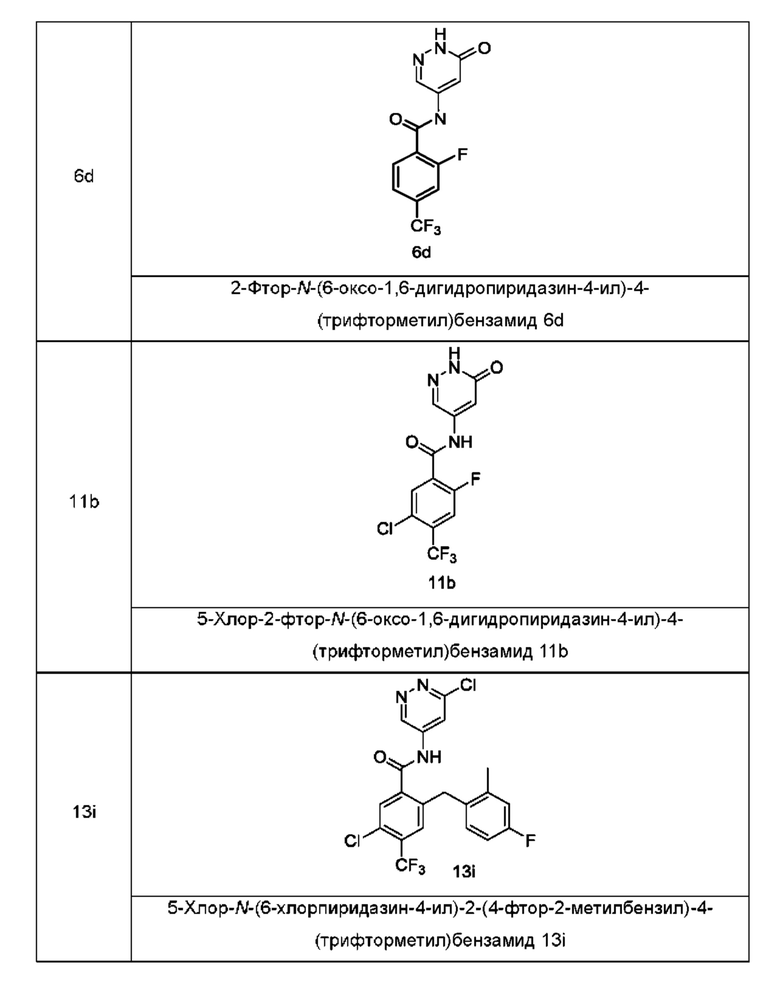
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (I), включающему стадию:
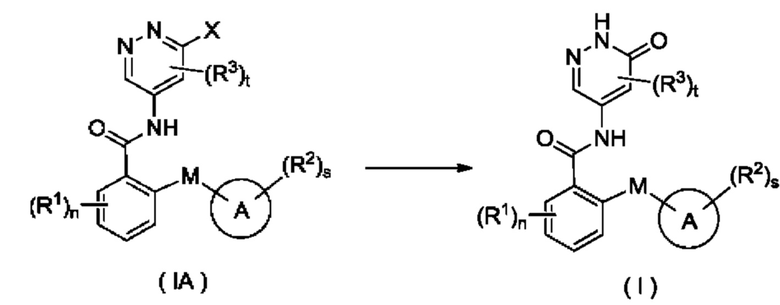
осуществления взаимодействия с участием соединения формулы (IA) с получением соединения формулы (I);
где:
X представляет собой галоген и предпочтительно Cl; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (I).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (I), включающему стадию:
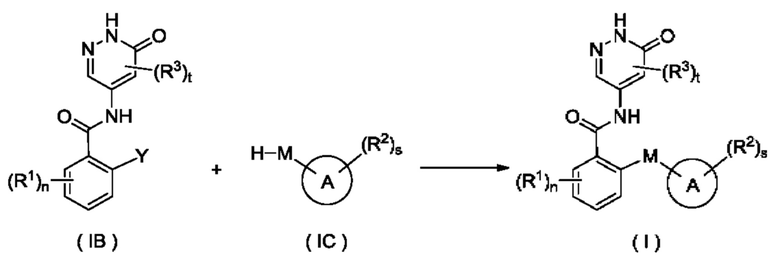
приведения во взаимодействие соединения формулы (IA) и соединения формулы (IC) с получением соединения формулы (I);
где:
Y представляет собой галоген и предпочтительно F; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (I).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (II), включающему стадию:
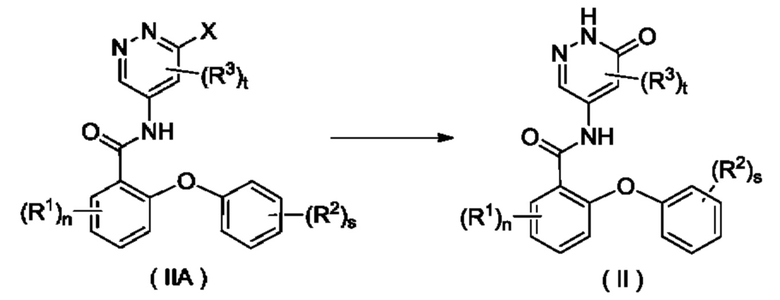
осуществления взаимодействия с участием соединения формулы (МА) с получением соединения формулы (II);
где:
X представляет собой галоген и предпочтительно Cl; и
R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (II).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (II), включающему стадию:
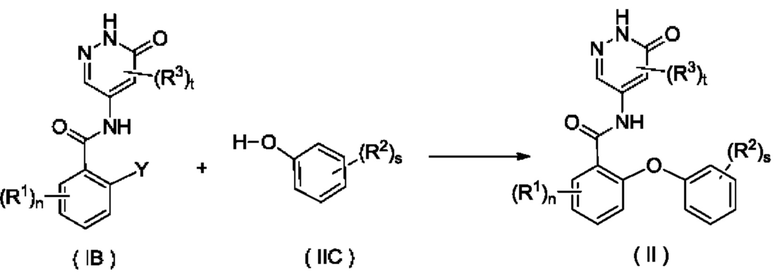
приведения во взаимодействие соединения формулы (IB) и соединения формулы (IIC) с получением соединения формулы (II);
где:
Y представляет собой галоген и предпочтительно F; и
R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (II).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (III), включающему стадию:
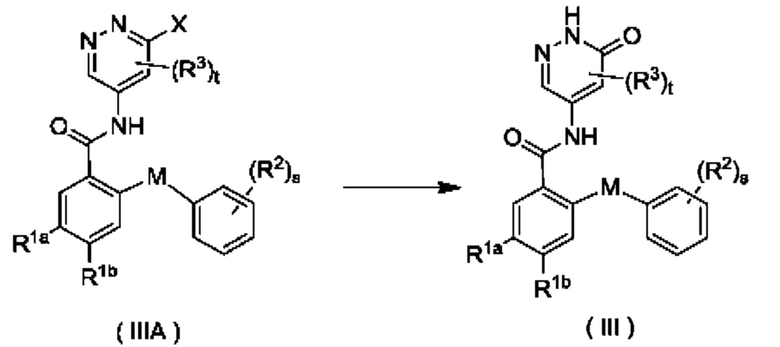
осуществления взаимодействия с участием соединения формулы (IIIA) с получением соединения формулы (III);
где:
X представляет собой галоген и предпочтительно Cl; и
М, R1a, R1b, R2, R3, s и t являются такими, как определено для соединения формулы (III).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (III), включающему стадию:
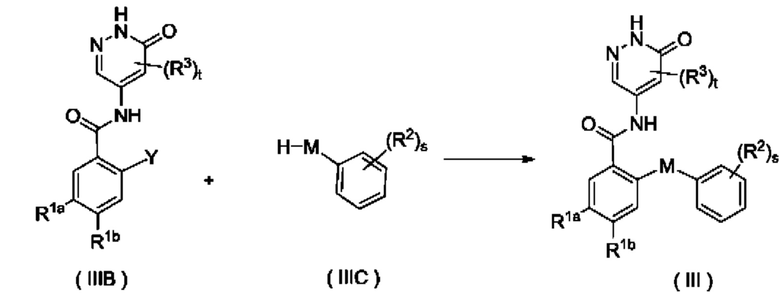
приведения во взаимодействие соединения формулы (IIIB) и соединения формулы (NIC) с получением соединения формулы (III);
где:
Y представляет собой галоген и предпочтительно F; и
М, R1a, R1b, R2, R3, s и t являются такими, как определено для соединения формулы (III).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (IV), включающему стадию:
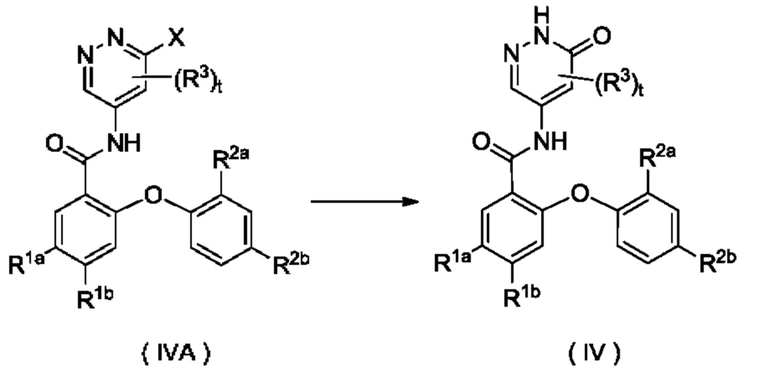
осуществления взаимодействия с участием соединения формулы (IVA) с получением соединения формулы (IV); где:
X представляет собой галоген и предпочтительно Cl; и
R1a, R1b, R2a, R2b, R3 и t являются такими, как определено для соединения формулы (IV).
Согласно другому аспекту настоящее изобретение относится к способу получения соединения формулы (IV), включающему стадию:
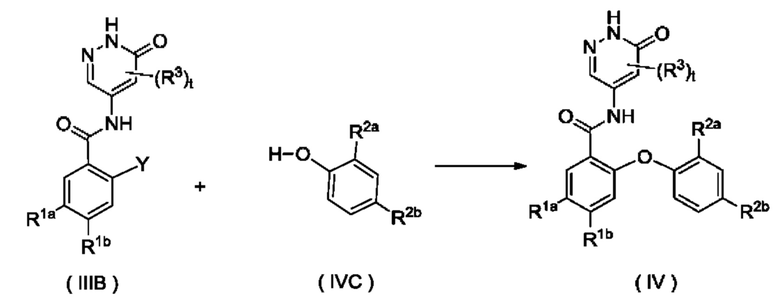
приведения во взаимодействие соединения формулы (IIIB) и соединения формулы (IVC) с получением соединения формулы (IV);
где:
Y представляет собой галоген и предпочтительно F; и
R1a, R1b, R2a, R2b, R3 и t являются такими, как определено для соединения формулы (IV).
Согласно другому аспекту настоящее изобретение относится к фармацевтической композиции, содержащей вышеупомянутые соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь либо их фармацевтически приемлемую соль и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
Кроме того, настоящее изобретение относится к способу получения вышеупомянутой фармацевтической композиции, включающему стадию смешивания вышеупомянутых соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли с фармацевтически приемлемыми носителями, разбавителями или эксципиентами.
Настоящее изобретение также относится к применению вышеупомянутых соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции для получения лекарственного средства для ингибирования потенциал-зависимого натриевого канала у субъекта. Потенциал-зависимый натриевый канал предпочтительно представляет собой NaV1.8.
Настоящее изобретение также относится к применению вышеупомянутых соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции для получения лекарственного средства для лечения и/или ослабления боли и связанных с болью заболеваний, рассеянного склероза, синдрома Шарко-Мари-Тута, недержания или нарушения сердечного ритма. Боль предпочтительно выбрана из группы, состоящей из хронической боли, острой боли, воспалительной боли, боли при раковом заболевании, нейропатической боли, мышечно-скелетной боли, первичной боли, боли в кишечнике и идиопатической боли.
Настоящее изобретение также относится к способу ингибирования потенциал-зависимого натриевого канала у субъекта, включающему стадию введения пациенту, нуждающемуся в этом, вышеупомянутых соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции по настоящему изобретению. Потенциал-зависимый натриевый канал предпочтительно представляет собой NaV1.8.
Настоящее изобретение также относится к способу лечения и/или ослабления боли и связанных с болью заболеваний, рассеянного склероза, синдрома Шарко-Мари-Тута, недержания или нарушения сердечного ритма, включающему стадию введения пациенту, нуждающемуся в этом, вышеупомянутых соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции по настоящему изобретению. Боль предпочтительно выбрана из группы, состоящей из хронической боли, острой боли, воспалительной боли, боли при раковом заболевании, нейропатической боли, мышечно-скелетной боли, первичной боли, боли в кишечнике и идиопатической боли.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо их фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции для применения в качестве лекарственного средства.
Настоящее изобретение также относится к соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо их фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции для применения в качестве лекарственного средства для ингибирования потенциал-зависимого натриевого канала у субъекта. Потенциал-зависимый натриевый канал предпочтительно представляет собой NaV1.8.
Настоящее изобретение также относится к вышеупомянутым соединению формулы (I) или его таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, либо их фармацевтически приемлемой соли или вышеупомянутой фармацевтической композиции для применения в лечении и/или ослаблении боли и связанных с болью заболеваний, рассеянного склероза, синдрома Шарко-Мари-Тута, недержания или нарушения сердечного ритма. Боль предпочтительно выбрана из группы, состоящей из хронической боли, острой боли, воспалительной боли, боли при раковом заболевании, нейропатической боли, мышечно-скелетной боли, первичной боли, боли в кишечнике и идиопатической боли.
Нейропатическая боль в описании настоящего изобретения предпочтительно выбрана из группы, состоящей из невралгии тройничного нерва, постгерпетической невралгии, диабетической невралгии, болезненной ассоциированной с вирусом иммунодефицита (ВИЧ) сенсорной невропатии, синдрома жжения, боли после ампутации, боли после травмы спинного мозга, фантомной боли, болезненной невромы, ампутационной невромы, невромы Мортона, повреждения нервов, стеноза позвоночного канала, запястного синдрома, корешковой боли, боли в пояснично-крестцовом отделе, авульсии нерва, разрыва плечевого сплетения, комплексного регионарного болевого синдрома, невралгии, вызываемой лекарственной терапией, невралгии, вызываемой химиотерапией рака, невралгии, вызываемой антиретровирусной терапией, первичной невропатии тонких волокон, первичной сенсорной невралгии и тригеминальной вегетативной головной боли.
Мышечно-скелетная боль в описании настоящего изобретения предпочтительно выбрана из группы, состоящей из боли при остеоартрите, боли в пояснице, боли от холода, жгучей боли и зубной боли.
Боль в кишечнике в описании настоящего изобретения предпочтительно выбрана из группы, состоящей из боли при воспалительном заболевании кишечника, боли при болезни Крона и боли при интерстициальном цистите.
Воспалительная боль в описании настоящего изобретения предпочтительно выбрана из группы, состоящей из боли при ревматоидном артрите и вульварной боли.
Идиопатическая боль в описании настоящего изобретения включает фибромиалгию.
Дозировка соединения или композиции, используемых в способе лечения по настоящему изобретению, обычно будет варьировать в зависимости от тяжести заболевания, массы пациента и относительной эффективности соединения. Однако, как правило, подходящая стандартная доза может составлять от 0,1 до 1000 мг.
Помимо активного соединения фармацевтическая композиция по настоящему изобретению также может содержать одно или более чем одно вспомогательное вещество, включая наполнитель (разбавитель), связующее вещество, увлажняющий агент, разрыхлитель, эксципиент и тому подобное. В зависимости от способа введения композиция может содержать активное соединение в количестве от 0,1 до 99 мас. %.
Фармацевтическая композиция, содержащая активный ингредиент, может быть в форме, подходящей для перорального введения, например, в форме таблетки, лепешки, пастилки, водной или масляной суспензии, диспергируемого порошка или диспергируемой гранулы, эмульсии, твердой или мягкой капсулы, сиропа или эликсира. Композиция для перорального введения может быть получена в соответствии с любым способом, известным в данной области техники для получения фармацевтической композиции. Такая композиция может содержать один ингредиент или несколько ингредиентов, выбранных из группы, состоящей из подсластителей, корригентов, красителей и консервантов, для придания фармацевтической композиции привлекательного и приятного вкуса. Таблетка содержит активный ингредиент в смеси с нетоксичными фармацевтически приемлемыми эксципиентами, подходящими для изготовления таблеток. Такими эксципиентами могут быть инертные эксципиенты, гранулирующие агенты, разрыхлители и смазывающие вещества. Таблетка может не иметь покрытия или быть покрыта с использованием метода, известного для маскировки вкуса лекарственного средства или задержки распадаемости и всасывания активного ингредиента в желудочно-кишечном тракте, что будет обеспечивать замедленное высвобождение в течение длительного периода времени.
Композиция для перорального применения также может быть представлена в виде мягких желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем либо активный ингредиент смешан с водорастворимым носителем, содержащей масло средой или оливковым маслом.
Водная суспензия содержит активный ингредиент в смеси с эксципиентами, подходящими для изготовления водной суспензии. Такими эксципиентами являются суспендирующие агенты, диспергирующие вещества или увлажняющие агенты. Водная суспензия также может содержать один или несколько консервантов, таких как этилпарабен или н-пропилпарабен, один или несколько красителей, один или несколько корригентов и один или несколько подсластителей.
Масляная суспензия может быть получена путем суспендирования активного ингредиента в растительном масле или минеральном масле. Масляная суспензия может содержать загуститель. Для придания композиции приятного вкуса могут быть добавлены вышеупомянутые подсластители и корригенты. Сохранение этих композиций может быть обеспечено добавлением антиоксиданта.
На основе активного ингредиента в смеси с диспергирующими веществами или увлажняющими агентами, суспендирующими агентами либо с одним или несколькими консервантами можно получить диспергируемые порошки или гранулы, подходящие для получения водной суспензии путем добавления к ним воды. Примеры подходящих диспергирующих веществ или увлажняющих агентов и суспендирующих агентов уже упомянуты выше. Также можно добавлять дополнительные эксципиенты, такие как подсластители, корригенты и красители.
Фармацевтическая композиция по настоящему изобретению также может быть в форме эмульсии типа масло-в-воде. Масляная фаза может представлять собой растительное масло или минеральное масло (такое как вазелиновое масло) либо их смесь. Подходящими эмульгирующими агентами могут быть природные фосфолипиды или неполные сложные эфиры. Эмульсия также может содержать подсластитель, корригент, консервант и антиоксидант.
Фармацевтическая композиция по настоящему изобретению может быть в форме стерильного водного раствора для инъекций. Приемлемыми разбавителями или растворителями, которые могут быть использованы, являются вода, раствор Рингера или изотонический раствор хлорида натрия. Стерильная композиция для инъекций может представлять собой стерильную инъекционную микроэмульсию типа масло-в-воде, в которой активный ингредиент растворен в масляной фазе. Раствор или микроэмульсия для инъекций могут быть введены в кровоток пациента посредством местной болюсной инъекции.
Фармацевтическая композиция по настоящему изобретению может быть в форме стерильной инъекционной водной или масляной суспензии для внутримышечного и подкожного введения. При получении такой суспензии могут быть использованы подходящие диспергирующие вещества или увлажняющие агенты и суспендирующие агенты, которые описаны выше, согласно известным методам. Стерильная композиция для инъекций также может представлять собой стерильный раствор или стерильную суспензию для инъекций, приготовленные в нетоксичном парентерально приемлемом разбавителе или растворителе. Кроме того, в качестве растворителя или суспендирующей среды вполне могут быть использованы стерильные нелетучие масла.
Соединение по настоящему изобретению может быть введено в форме суппозитория для ректального введения. Такие фармацевтические композиции могут быть получены путем смешивания лекарственного средства с подходящим не вызывающим раздражения эксципиентом, который является твердым при обычных температурах, но жидким в прямой кишке, в результате чего претерпевает плавление в прямой кишке с высвобождением лекарственного средства.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от ряда факторов, включая, но не ограничиваясь этим, следующие факторы: активность конкретного соединения, возраст пациента, масса пациента, общее состояние здоровья пациента, поведение пациента, рацион пациента, время введения, путь введения, скорость экскреции, комбинация лекарственных средств и тому подобное. Помимо этого, оптимальное лечение, например, способ лечения, суточную дозу соединения формулы (I) или тип его фармацевтически приемлемой соли, можно контролировать с учетом традиционных схем лечения.
Определения терминов
Если не указано иное, то термины, использованные в данном описании и формуле изобретения, имеют описанные ниже значения.
Термин "алкил" относится к насыщенной алифатической углеводородной группе, которая представляет собой группу с прямой или разветвленной цепью, содержащую 1-20 атомов углерода, предпочтительно алкил, имеющий 1-12 атомов углерода, и более предпочтительно алкил, имеющий 1-6 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их различные разветвленные изомеры. Более предпочтительно, алкильная группа представляет собой низший алкил, имеющий 1-6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, етор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и тому подобное. Алкил может быть замещенным или незамещенным. При наличии замещения замещаемая(ые) группа(ы) может/могут быть замещена(ы) по любому доступному месту присоединения. Замещающей(ими) группой(ами) являет(ют)ся одна или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетеро цикл ил а, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин "алкокси" относится к группе -О-(алкил) или -О-(незамещенный циклоалкил), где алкил является таким, как определено выше. Неограничивающие примеры алкокси включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Группа алкокси возможно может быть замещенной или незамещенной. При наличии замещения замещающей(ими) группой(ами) предпочтительно являет(ют)ся одна или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетеро цикл ила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин "циклоалкил" относится к насыщенной или частично ненасыщенной моноциклической или полициклической углеводородной замещаемой группе, имеющей 3-20 атомов углерода, предпочтительно 3-12 атомов углерода, более предпочтительно 3-6 атомов углерода (например, 3, 4, 5 или 6 атомов углерода) и наиболее предпочтительно 5-6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и тому подобное. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
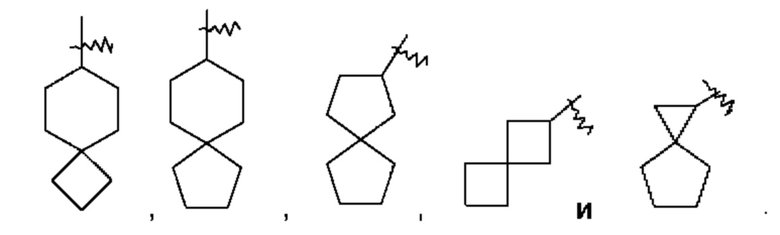
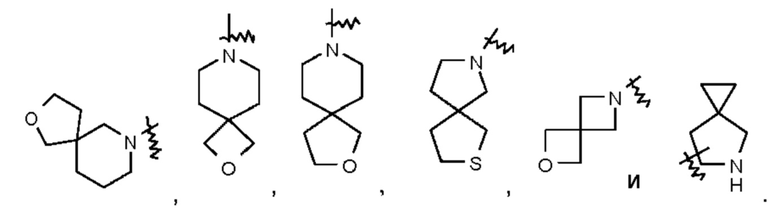
Термин "спиро-цикпоалкил" относится к 5-20-членной полициклической группе, состоящей из отдельных колец, соединенных через один общий атом углерода (называемый спиро-атомом), при этом данные кольца могут содержать одну или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной тг-электронной системы. Спиро-циклоалкилом предпочтительно является 6-14-членный спиро-цикпоалкил и более предпочтительно 7-10-членный спиро-цикпоалкил (например, 7-, 8-, 9- или 10-членный спиро-циклоалкил). В зависимости от количества спиро-атомов, общих для этих колец, спиро-циклоалкилы можно подразделять на моно-спиро-циклоалкил, ди-спиро-циклоалкил или поли-спиро-циклоалкил, и спиро-циклоалкилом предпочтительно является моно-спиро-циклоалкил или ди-спиро-циклоалкил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-циклоалкил. Неограничивающие примеры спиро-циклоалкила включают:
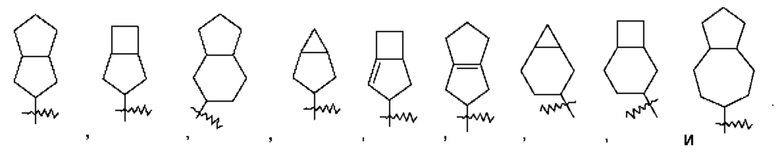
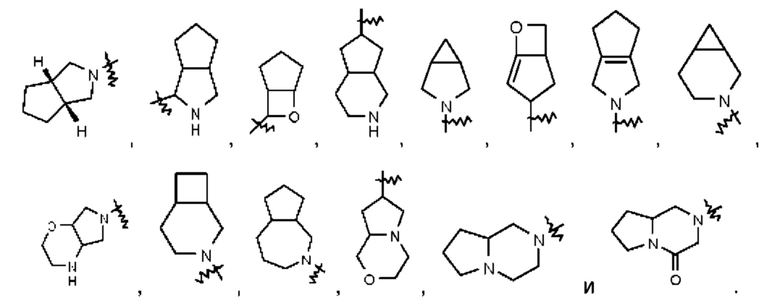
Термин "конденсированный циклоалкил" относится к 5-20-членной состоящей полностью из атомов углерода полициклической группе, при этом каждое кольцо в такой системе имеет общую с другим кольцом соседнюю пару атомов углерода, причем одно кольцо или несколько колец могут содержать одну или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной тг-электронной системы. Конденсированный циклоалкил предпочтительно представляет собой 6-14-членный конденсированный циклоалкил и более предпочтительно 7-10-членный конденсированный циклоалкил. В зависимости от количества указанных колец конденсированные циклоалкилы можно подразделять на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, и конденсированный циклоалкил предпочтительно представляет собой бициклический или трициклический конденсированный циклоалкил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
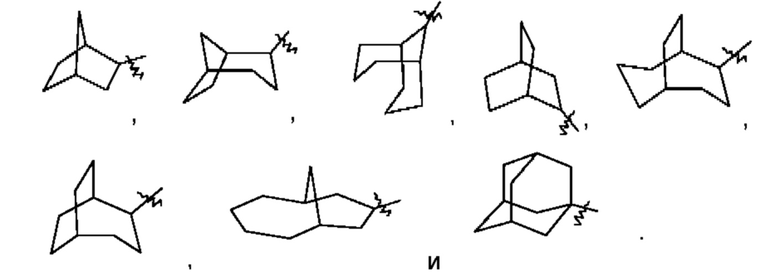
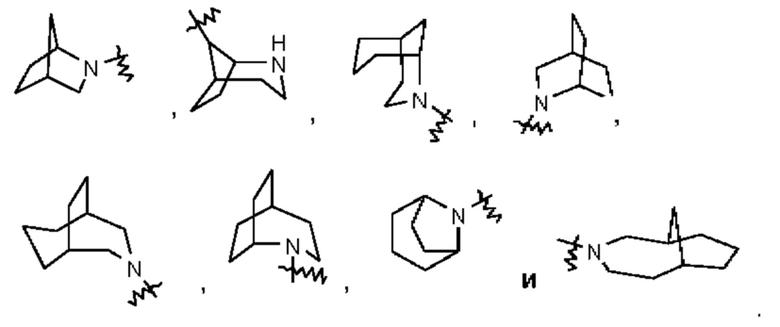
Термин "мостиковый циклоалкил" относится к 5-20-членной состоящей полностью из атомов углерода полициклической группе, при этом каждые два кольца в этой системе имеют два общих несопряженных атома углерода, причем данные кольца могут иметь одну или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной тт-электронной системы. Мостиковый циклоалкил предпочтительно представляет собой 6-14-членный мостиковый циклоалкил и более предпочтительно 7-10-членный мостиковый циклоалкил. В зависимости от количества указанных колец мостиковые циклоалкилы можно подразделять на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, и мостиковый циклоалкил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостикового циклоалкила включают:
Кольцо такого циклоалкила (в том числе циклоалкила, спиро-циклоалкила, конденсированного циклоалкила и мостикового циклоалкила) может быть конденсировано с кольцом арила, гетероарила или гетероциклила, при этом кольцом, связанным с исходной структурой, является циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и тому подобное и предпочтительно бензоциклопентил, тетрагидронафтил. Циклоалкил возможно может быть замещенным или незамещенным. При наличии замещения замещающей(ими) группой(ами) предпочтительно являет(ют)ся одна или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероцикл о алкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин "гетероциклил" относится к 3-20-членной насыщенной или частично ненасыщенной моноциклической либо полициклической углеводородной замещающей группе, при этом один или более атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(О)m (где m представляет собой целое число от 0 до 2), но за исключением -О-О-, -O-S- или -S-S- в кольце, и при этом остальные атомы в кольце представляют собой атомы углерода. Предпочтительно, гетероциклил имеет 3-12 атомов в кольце, при этом 1-4 атома представляют собой гетероатомы; наиболее предпочтительно 3-8 атомов в кольце, при этом 1-3 атома представляют собой гетероатомы; и наиболее предпочтительно 5-6 атомов в кольце, при этом 1-2 или 1-3 атома представляют собой гетероатомы. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидропиранил, тетрагидротиенил, дигидроимидазолил, дигидрофуранил, д и гид ро пиразол ил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и тому подобное и предпочтительно тетрагидропиранил, пиперидинил, пирролидинил. Полициклический гетероциклил включает гетероциклил, имеющий спиро-кольцо, конденсированное кольцо или мостиковое кольцо.
Термин "спиро-гетероциклил" относится к 5-20-членной полициклической гетероцикл ильной группе, содержащей отдельные кольца, соединенные через один общий атом (называемый спиро-атомом), при этом один или более атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), и при этом остальные атомы в кольце представляют собой атомы углерода, причем кольца могут содержать одну или несколько двойных связей, но ни одно из колец не имеет полностью сопряженной тг-электронной системы. Спиро-гетероциклил предпочтительно представляет собой 6-14-членный спиро-гетероциклил и более предпочтительно 7-10-членный спиро-гетероциклил. В зависимости от количества спиро-атомов, общих для этих колец, спиро-гетероциклилы можно подразделять на моно-спиро-гетероциклил, ди-спиро-гетероциклил или поли-спиро-гетероциклил, и спиро-гетероциклил предпочтительно представляет собой моно-спиро-гетероциклил или ди-спиро-гетероциклил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моно-спиро-гетероциклил. Неограничивающие примеры спиро-гетероциклила включают:
Термин "конденсированный гетероциклил" относится к 5-20-членной полициклической гетеро цикл ильной группе, при этом каждое кольцо в такой системе имеет общую с другим кольцом соседнюю пару атомов, причем одно кольцо или несколько колец могут содержать одну или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной тт-электронной системы, и при этом один или более атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(О)m (где m представляет собой целое число от 0 до 2), при этом остальные атомы в кольце представляют собой атомы углерода. Конденсированный гетероциклил предпочтительно представляет собой 6-14-членный конденсированный гетероциклил и более предпочтительно 7-10-членный конденсированный гетероциклил. В зависимости от количества указанных колец конденсированные гетероциклилы можно подразделять на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, и конденсированный гетероциклил предпочтительно представляет собой бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
Термин "мостиковый гетероциклил" относится к 5-14-членной полициклической гетеро цикл ильной группе, при этом каждые два кольца в этой системе имеют два общих несопряженных атома, причем данные кольца могут иметь одну или несколько двойных связей, но ни одно из этих колец не имеет полностью сопряженной тт-электронной системы, и при этом один или более атомов в кольце представляют собой гетероатомы, выбранные из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), причем остальные атомы в кольце представляют собой атомы углерода. Мостиковый гетероциклил предпочтительно представляет собой 6-14-членный мостиковый гетероциклил и более предпочтительно 7-10-членный мостиковый гетероциклил. В зависимости от количества указанных колец мостиковые гетероциклилы можно подразделять на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и мостиковый гетероциклил предпочтительно представляет собой бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостикового гетероциклила включают:
Кольцо такого гетероциклила (в том числе гетероциклила, спиро-гетероциклила, конденсированного гетероциклила и мостикового гетероциклила) может быть конденсировано с кольцом арила, гетероарила или циклоалкила, при этом кольцом, связанным с исходной структурой, является гетероциклил. Его неограничивающие примеры включают:
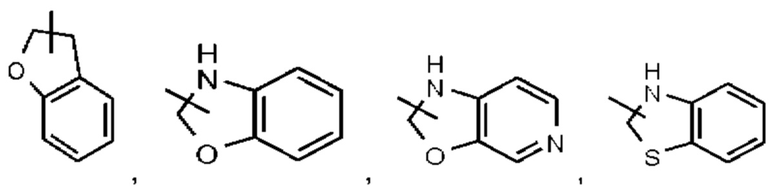 и тому подобное.
и тому подобное.
Гетероциклил возможно может быть замещенным или незамещенным. При наличии замещения замещающей(ими) группой(ами) предпочтительно являет(ют)ся одна или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, оксо, карбокси и алкоксикарбонила.
Термин "арил" относится к 6-14-членному состоящему полностью из атомов углерода моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в такой системе имеет общую с другим кольцом в данной системе соседнюю пару атомов углерода), имеющему сопряженную тт-электронную систему, предпочтительно относится к 6-10-членному арилу, например, фенилу и нафтилу. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, при этом кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Его неограничивающие примеры включают:
Арил может быть замещенным или незамещенным. При наличии замещения замещающей(ими) группой(ами) предпочтительно являет(ют)ся одна или более групп, независимо возможно выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила.
Термин "гетероарил" относится к 5-14-членной гетероароматической системе, имеющей 1-4 гетероатома, выбранных из группы, состоящей из О, S и N. Гетероарил предпочтительно представляет собой 5-10-членный гетероарил, имеющий 1-3 гетероатома, более предпочтительно 5- или 6-членный гетероарил, имеющий 1-2 гетероатома; предпочтительно, например, имидазолил, фурил, тиенил, тиазолил, пиразолил, оксазолил, пирролил, тетразолил, пиридил, пиримидинил, тиадиазолил, пиразинил, пиридазинил и тому подобное, предпочтительно пиридазинил и пиридинил и более предпочтительно пиридазинил. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, при этом кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Его неограничивающие примеры включают:
Гетероарил возможно может быть замещенным или незамещенным. При наличии замещения замещающей(ими) группой(ами) предпочтительно являет(ют)ся одна или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, галогена, тиола, гидрокси, оксо, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклоалкокси, циклоалкилтио, гетероциклилтио, карбокси и алкоксикарбонила, и его неограничивающие примеры включают 
Термин "гидроксиалкил" относится к алкильной группе, замещенной группой(ами) гидрокси, при этом алкил является таким, как определено выше.
Термин "галогеналкил" относится к алкильной группе, замещенной одним атомом или несколькими атомами галогена, при этом алкил является таким, как определено выше.
Термин "галогеналкокси" относится к группе алкокси, замещенной одним атомом или несколькими атомами галогена, при этом группа алкокси является такой, как определено выше.
Термин "дейтерированный алкил" относится к алкильной группе, замещенной одним атомом или несколькими атомами дейтерия, при этом алкил является таким, как определено выше.
Термин "дейтерированный алкокси" относится к группе алкокси, замещенной одним или несколькими атомами дейтерия, при этом группа алкокси является такой, как определено выше.
Термин "циклоалкилалкил" относится к алкильной группе, замещенной одним или несколькими циклоалкилами, при этом циклоалкил и алкил являются такими, как определено выше.
Термин "циклоалкилокси" относится к -О-циклоалкильной группе, где циклоалкил является таким, как определено выше.
Термин "гетероциклилалкил" относится к алкильной группе, замещенной одним или несколькими гетероциклилами, при этом гетероциклил и алкил являются такими, как определено выше.
Термин "арилалкил" относится к алкильной группе, замещенной одним или несколькими арилами, при этом арил и алкил являются такими, как определено выше.
Термин "гидрокси" относится к группе -ОН.
Термин "галоген" относится к фтору, хлору, брому или йоду.
Термин "амино" относится к группе -NH2.
Термин "циано" относится к группе -CN.
Термин "нитро" относится к группе -NO2.
Термин "карбокси" относится к группе -С(O)ОН.
Термин "алкокси карбон ил" относится к группе -С(O)O(алкил) или -С(O)O(циклоалкил), при этом алкил и циклоалкил являются такими, как определено выше.
Термин "ацилгалогенид" относится к соединению, содержащему группу -С(O)-галоген.
Соединение по настоящему изобретению также может представлять собой свои изотопные производные. Термин "изотопные производные" относится к соединениям, которые различаются по структуре только наличием одного или более чем одного обогащенного изотопом атома. Например, соединение, имеющее структуру соединения по настоящему изобретению, за исключением замены атома водорода на "дейтерий" или "тритий", либо замены атома фтора на метку 18F-фтор (18F-изотоп), либо замены атома углерода на 11С-, 13С- или 14С-обогащенный атом углерода (метку 11С-, 13С- или 14С-углерод; 11С-, 13С- или 14С-изотоп), находится в объеме настоящего изобретения. Такие соединения можно использовать, например, в качестве аналитических инструментов либо зондов в биологических анализах, или в качестве индикаторов для диагностической визуализации заболевания in vivo, или в качестве индикаторов для исследований фармакодинамики, фармакокинетики или рецепторов.
Настоящее изобретение также включает соединения формулы (I) в различных дейтерированных формах. Каждый из доступных атомов водорода, присоединенных к атому углерода, может быть независимо заменен на атом дейтерия. Специалисты в данной области техники могут синтезировать соединение формулы (I) в дейтерированной форме, руководствуясь релевантными литературными источниками. Соединение формулы (I) в дейтерированной форме может быть получено путем применения имеющихся в продаже дейтерированных исходных веществ, или они могут быть синтезированы традиционными методами с использованием дейтерированных реагентов, включая, но не ограничиваясь этим, дейтерированный боргидрид, содержащий три атома дейтерия боргидрид в тетрагидрофуране, дейтерированный алюмогидрид лития, дейтерированный иодэтан, дейтерированный подметан и тому подобное.
"Возможный" или "возможно" означает, что событие или обстоятельство, описываемое впоследствии, может произойти, но происходит не обязательно, и такое описание включает ситуацию, в которой данное событие или обстоятельство происходит или не происходит.Например, "гетероциклил, возможно замещенный алкилом", означает, что алкильная группа может присутствовать, но присутствует не обязательно, и такое описание включает ситуацию замещения гетероциклила алкилом и ситуацию, когда гетероциклил не замещен алкилом.
Термин "замещенный" относится к ситуации, когда один или более атомов водорода в группе, предпочтительно до 5 включительно и более предпочтительно от 1 до 3 атомов водорода, независимо замещены соответствующим количеством заместителей. Само собой разумеется, что, что заместители находятся только в своем возможном с точки зрения химии положении. Специалист в данной области техники способен определить, экспериментально или теоретически, возможно или невозможно такое замещение, без приложения чрезмерных усилий. Например, структура, в которой группа амино или гидрокси со свободным атомом водорода, связана с атомами углерода, имеющими ненасыщенные связи (такие как олефиновые), может оказаться нестабильной.
Термин "фармацевтическая композиция" относится к смеси одного или более чем одного из соединений, описанных в данной заявке, или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами и другими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Задачей фармацевтической композиции является облегчение введения соединения в организм, что способствует всасыванию активного ингредиента с целью проявления биологической активности.
Термин "фармацевтически приемлемая соль" относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной для млекопитающих и обладает желаемой биологической активностью.
Способ синтеза соединения по настоящему изобретению
Чтобы достичь цели настоящего изобретения, в настоящем изобретении применены приведенные далее технические решения.
Схема I
Согласно настоящему изобретению предложен способ получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенные далее стадии:
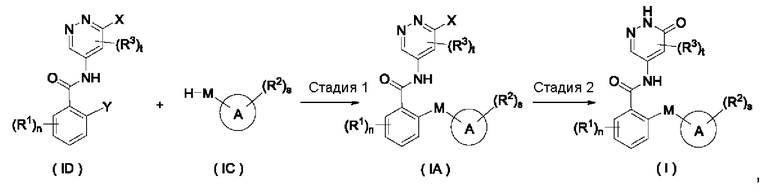
где:
X представляет собой галоген и предпочтительно Cl;
Y представляет собой галоген и предпочтительно F; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (I);
на стадии 1 соединение формулы (ID) и соединение формулы (IC) приводят во взаимодействие в щелочных условиях с получением соединения формулы (IA);
на стадии 2 с участием соединения формулы (IA) осуществляют взаимодействие в щелочных условиях с получением соединения формулы (I).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенные выше реакции предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Схема II
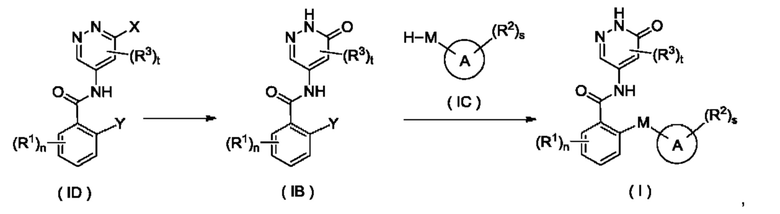
Согласно настоящему изобретению предложен способ получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенные далее стадии:
где:
X представляет собой галоген и предпочтительно Cl;
Y представляет собой галоген и предпочтительно F; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (I);
на стадии 1 с участием соединения формулы (ID) осуществляют взаимодействие в щелочных условиях с получением соединения формулы (IB);
на стадии 2 соединение формулы (IB) и соединение формулы (IC) приводят во взаимодействие в щелочных условиях с получением соединения формулы (I).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенные выше реакции предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Схема III
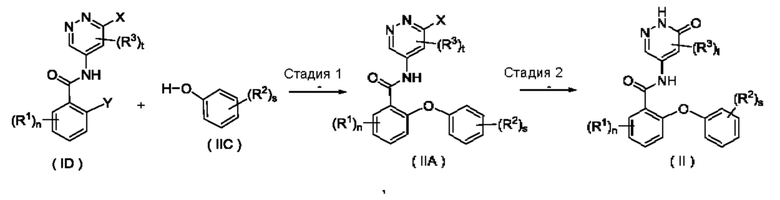
Согласно настоящему изобретению предложен способ получения соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенные далее стадии:
где:
X представляет собой галоген и предпочтительно Cl;
Y представляет собой галоген и предпочтительно F; и
R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (II);
на стадии 1 соединение формулы (ID) и соединение формулы (IIC) приводят во взаимодействие в щелочных условиях с получением соединения формулы (IIA);
на стадии 2 с участием соединения формулы (IIA) осуществляют взаимодействие в щелочных условиях с получением соединения формулы (II).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенные выше реакции предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Схема IV
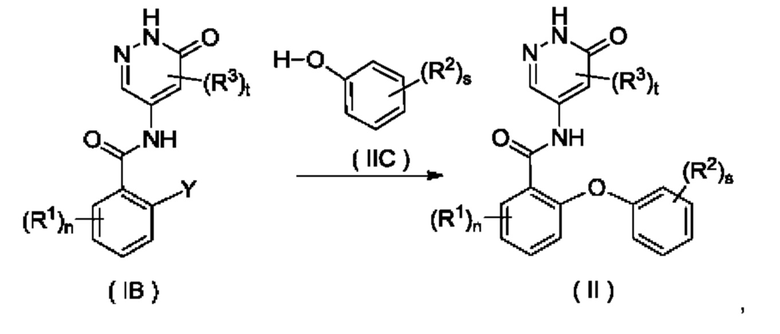
Согласно настоящему изобретению предложен способ получения соединения формулы (II) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенную далее стадию:
где:
Y представляет собой галоген и предпочтительно F; и
R1, R2, R3, n, s и t являются такими, как определено для соединения формулы (II);
соединение формулы (IB) и соединение формулы (IIC) приводят во взаимодействие в щелочных условиях с получением соединения формулы (II).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенную выше реакцию предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Схема V
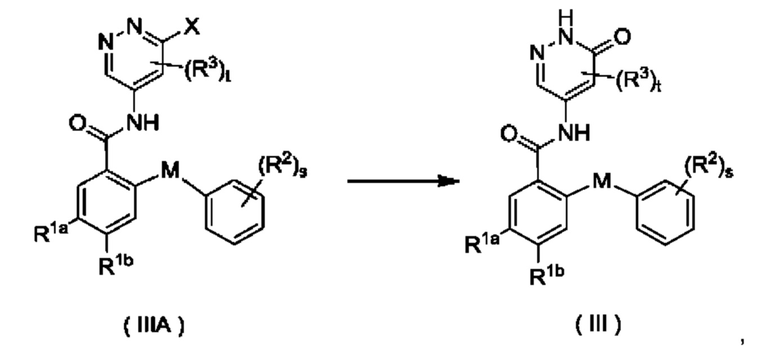
Согласно настоящему изобретению предложен способ получения соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенную далее стадию:
 где:
где:
X представляет собой галоген и предпочтительно Cl; и
М, R1a, R1b, R2, R3, s и t являются такими, как определено для соединения формулы (III);
с участием соединения формулы (IIIA) осуществляют взаимодействие в щелочных условиях с получением соединения формулы (III).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенную выше реакцию предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Схема VI
Согласно настоящему изобретению предложен способ получения соединения формулы (III) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенную далее стадию:
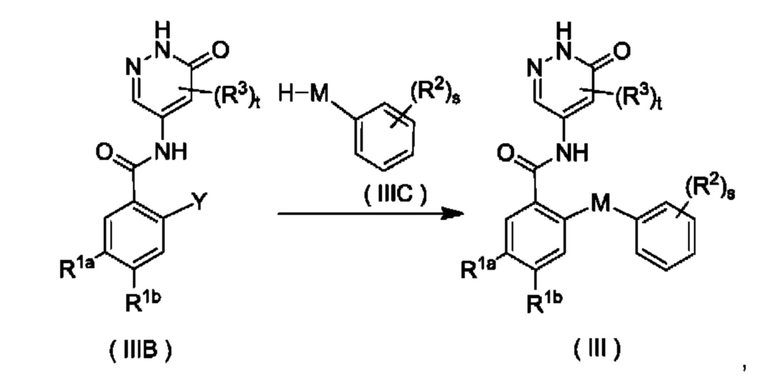
где:
Y представляет собой галоген и предпочтительно F; и
М, R1a, R1b, R2, R3, s и t являются такими, как определено для соединения формулы (III);
соединение формулы (IIIB) и соединение формулы (IIIC) приводят во взаимодействие в щелочных условиях с получением соединения формулы (III).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенную выше реакцию предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Схема VII
Согласно настоящему изобретению предложен способ получения соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенную далее стадию:
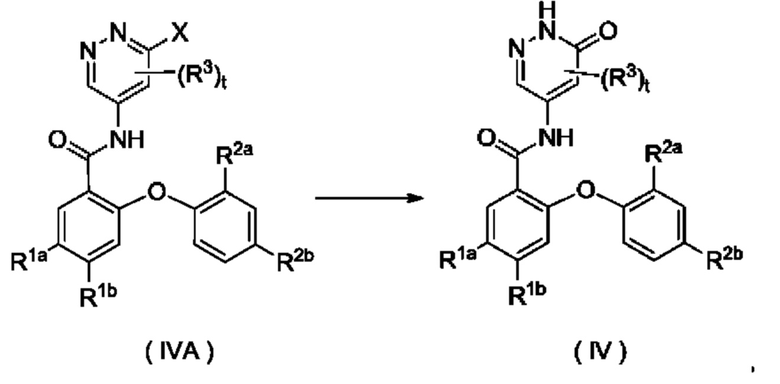
где:
X представляет собой галоген и предпочтительно Cl; и
R1a, R1b, R2a, R2b, R3 и t являются такими, как определено для соединения формулы (IV);
с участием соединения формулы (IVA) осуществляют взаимодействие в щелочных условиях с получением соединения формулы (IV).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенную выше реакцию предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Схема VIII
Согласно настоящему изобретению предложен способ получения соединения формулы (IV) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий приведенную далее стадию:
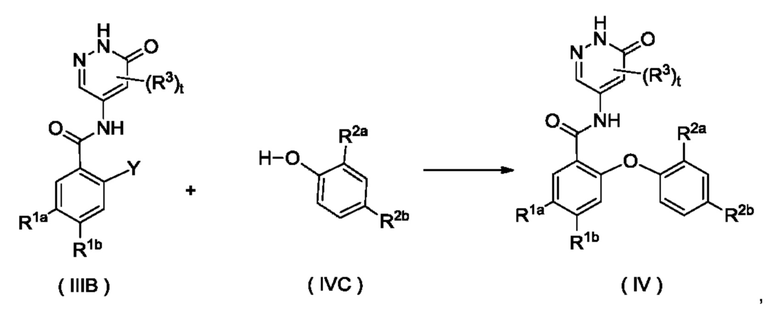
где:
Y представляет собой галоген и предпочтительно F; и
кольцо A, R1a, R1b, R2a, R2b, R3 и t являются такими, как определено для соединения формулы (IV);
соединение формулы (IIIB) и соединение формулы (IVC) приводят во взаимодействие в щелочных условиях с получением соединения формулы (IV).
Реагент, обеспечивающий щелочные условия, включает органические основания и неорганические основания. Органические основания включают, но не ограничиваются этим, пиридин, гексагидропиридин, триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутилат натрия и трет-бутилат калия. Неорганические основания включают, но не ограничиваются этим, гидрид натрия, фосфат калия, карбонат натрия, ацетат натрия, карбонат калия, ацетат калия, карбонат цезия, гидроксид натрия и гидроксид лития.
Приведенную выше реакцию предпочтительно осуществляют в растворителе. Использованные растворители включают, но не ограничиваются этим, уксусную кислоту, трифторуксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, петролейный эфир, этилацетат, н-гексан, диметилсульфоксид, 1,4-диоксан, воду, N-метилпирролидон, N,N-диметилформамид и их смеси.
Подробное описание
Далее настоящее изобретение будет описано со ссылкой на приведенные ниже примеры, но эти примеры не следует рассматривать как ограничивающие объем настоящего изобретения.
Примеры
Структуры соединений устанавливали посредством ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (MS). ЯМР сдвиги (δ) приведены в 10-6 (млн-1). Спектры ЯМР определяют на приборе AVANCE-400 от Bruker. Для определения в качестве растворителей используют дейтерированный диметилсульфоксид (DMSO-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), а в качестве внутреннего стандарта используют тетраметилсилан (TMS).
MS-спектры определяли на масс-спектрометре FINNIGAN LCQAd (с электрораспылительной ионизацией (ESI)) (производитель: Thermo, тип: Finnigan LCQ Advantage MAX).
Анализ методом высокоэффективной жидкостной хроматографии (HPLC) проводили на хроматографах для жидкостной хроматографии высокого давления Agilent HPLC 1200DAD, Agilent HPLC 1200VWD и Waters HPLC e2695-2489.
Анализ методом хиральной HPLC проводили на хроматографе для высокоэффективной жидкостной хроматографии Agilent 1260 DAD.
Препаративную высокоэффективную жидкостную хроматографию осуществляли на препаративных хроматографах Waters 2767, Waters 2767-SQ Detecor2, Shimadzu LC-20AP и Gilson-281.
Хиральное разделение осуществляли на препаративном хроматографе LC-20АР от Shimadzu.
В качестве прибора для быстрой препаративной комби-флеш-хроматографии использовали систему CombiFlash Rf200 (TELEDYNE ISCO).
В качестве пластинки для тонкослойной хроматографии (TLC) на силикагеле использовали пластинку с силикагелем HSGF254 от Yantai Huanghai или GF254 от Qingdao. Размер слоя силикагеля на пластинке, использованной для TLC, составлял от 0,15 мм до 0,2 мм, а размер слоя силикагеля на пластинке, использованной для очистки продуктов, составлял от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии на силикагеле обычно использовали силикагель от Yantai Huanghai с размером частиц 200-300 меш.
Средние значения скоростей ингибирования киназ и значения концентрации, вызывающей 50%-ное ингибирование, (IC50) определяли, используя микропланшетный ридер NovoStar (BMG Co., Germany).
Известные исходные вещества для осуществления настоящего изобретения могут быть получены известными в данной области техники методами или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organics, химической компании Aldrich, Accela ChemBio Inc., химической компании Dari и так далее.
Если не указано иное, реакции проводили в атмосфере аргона или в атмосфере азота.
"Атмосфера аргона" или "атмосфера азота" означает, что реакционная колба оснащена баллоном с аргоном или азотом (примерно 1 л).
"Атмосфера водорода" означает, что реакционная колба оснащена баллоном с водородом (примерно 1 л).
Реакцию гидрирования под давлением проводили на установке Парра для гидрирования 3916EKX и с использованием генератора водорода Qinglan QL-500 или на установке для гидрирования HC2-SS.
Для проведения реакций гидрирования реакционную систему обычно вакуумировали и заполняли водородом, и указанную выше операцию повторяли три раза.
Для проведения реакций в условиях облучения микроволнами использовали микроволновой реактор СЕМ Discover-S 908860 типа.
Если не указано иное, раствор означает водный раствор.
Если не указано иное, температура реакции означает комнатную температуру от 20°С до 30°С.
Мониторинг протекания реакций, приведенных в разделе Примеры, проводили по тонкослойной хроматографии (TLC). Система растворителей, используемая при проведении реакций, система элюентов для колоночной хроматографии и система растворителей для тонкослойной хроматографии, применяемые с целью очистки соединений, включали: А: систему дихлорметан/метанол, В: систему н-гексан/этилацетат, С: систему петролейный эфир/этилацетат, D: ацетон, Е: систему дихлорметан/ацетон, F: систему этилацетат/дихлорметан, G: систему этилацетат/дихлорметан/н-гексан и Н: систему этилацетат/дихлорметан/ацетон. Соотношение объемов растворителей корректировали в соответствии с полярностью соединений, и для корректировки также может быть добавлено небольшое количество щелочного реагента, такого кактриэтиламин, или кислотного реагента, такого как уксусная кислота.
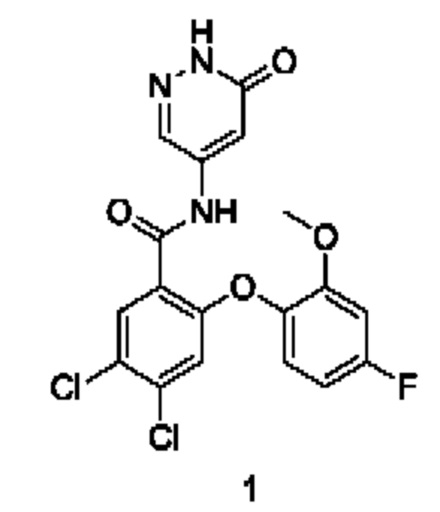
Пример 1
4,5-Дихлор-2-(4-фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 1
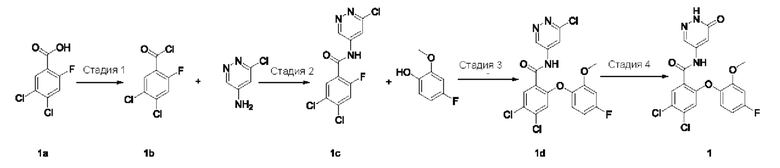
Стадия 1
4,5-Дихлор-2-фторбензоилхлорид 1b
Соединение 4,5-дихлор-2-фторбензойную кислоту 1а (1,5 г; 7,18 ммоль; Accela ChemBio (Shanghai) Inc.) растворяли в тионилхлориде (10 мл) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении, получая указанное в заголовке соединение 1b (1,6 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
4,5-Дихлор-N-(6-хлорпиридазин-4-ил)-2-фторбензамид 1с
Неочищенное соединение 1b (1,6 г; 7,03 ммоль) и 6-хлорпиридазин-4-амин (500 мг; 3,86 ммоль; Pharmablock Sciences (Nanjing), Inc.) растворяли в пиридине (10 мл) и реакционный раствор перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 1 с (650 мг; выход: 53%) в виде белого твердого вещества.
MS m/z (ESI): 321,9 [М+1].
Стадия 3
4,5-Дихлор-N-(6-хлорпиридазин-4-ил)-2-(4-фтор-2-метоксифенокси)бензамид 1d
Соединение 1 с (100 мг; 0,31 ммоль), 4-фтор-2-метоксифенол (50 мг; 0,35 ммоль; Accela ChemBio (Shanghai) Inc.) и карбонат цезия (153 мг; 0,47 ммоль) добавляли к N,N-диметилформамиду (10 мл) и в реакционном растворе осуществляли взаимодействие при 100°С в течение 2 часов. Реакционный раствор охлаждали и фильтровали через диатомовую землю. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с использованием системы элюентов А, получая указанное в заголовке соединение 1d (100 мг; выход: 72%).
MS m/z (ESI): 443,7 [М+1].
Стадия 4
4,5-Дихлор-2-(4-фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 1
Соединение 1d (100 мг; 0,22 ммоль) и ацетат калия (45 мг; 0,46 ммоль) добавляли к уксусной кислоте (5 мл) и в реакционном растворе осуществляли взаимодействие при 120°С в течение 1,5 часа. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с использованием системы элюентов А, получая указанное в заголовке соединение 1 (70 мг; выход: 73%).
MS m/z (ESI): 425,8 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.81 (s, 1Н), 10.85 (s, 1Н),7.93 (s, 1Н), 7.91 (d, 1Н), 7.25-7.20 (m, 1Н), 7.11 (dd, 1Н), 6.88 (s, 1Н), 6.82-6.82 (m, 1Н), 5.73 (s, 1Н), 3.73 (s, 3Н).
Пример 2
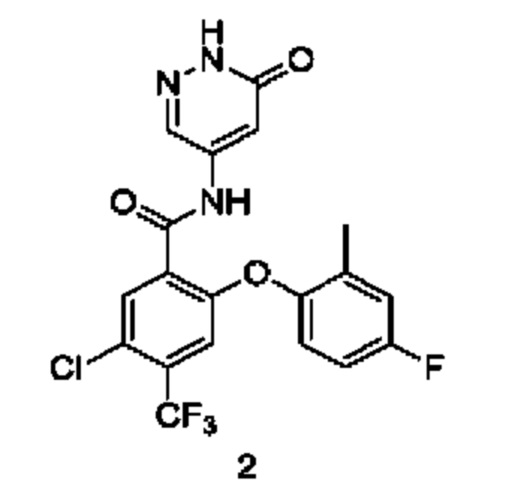
5-Хлор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 2
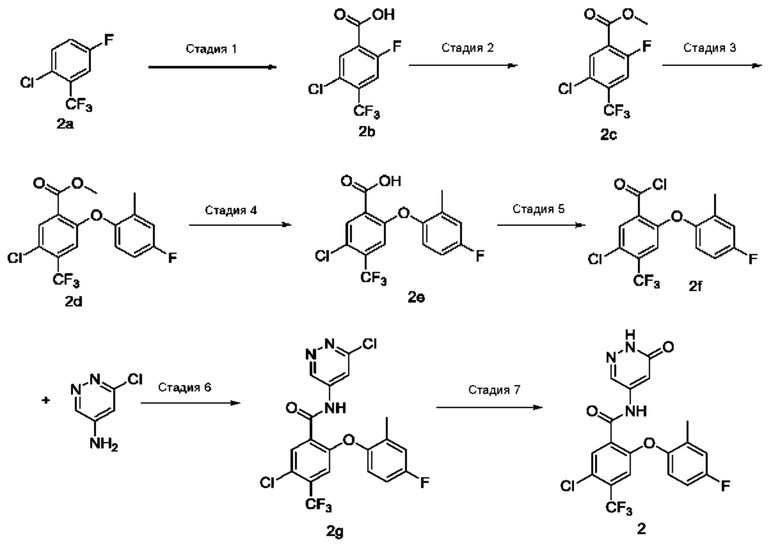
Стадия 1
5-Хлор-2-фтор-4-(трифторметил)бензойная кислота 2b
2,2,6,6-Тетраметилпиперидин (19,2 г; 135,93 ммоль; Accela ChemBio (Shanghai) Inc.) добавляли к тетрагидрофурану (200 мл) в атмосфере аргона. Реакционный раствор охлаждали до 0°С, затем по каплям в течение примерно 45 минут добавляли н-бутиллитий (1,6 М раствор; 85,1 мл) при температуре, поддерживаемой ниже 3°С. В реакционном растворе осуществляли взаимодействие при 0°С в течение 1 часа и затем охлаждали до -78°С. По каплям добавляли соединение 1-хлор-4-фтор-2-(трифторметил)бензол 2а (18 г; 90,66 ммоль; Shanghai Titan Scientific Co., Ltd.) и в реакционном растворе осуществляли взаимодействие в течение 3 часов. Добавляли избыточное количество сухого льда и реакционный раствор оставляли нагреваться естественным образом до 0°С, затем добавляли 150 мл ледяной воды. Проводили разделение реакционного раствора на две фазы. Значение рН водной фазы подводили до 5-6 концентрированной соляной кислотой и экстрагировали этилацетатом (50 мл), а органическую фазу концентрировали при пониженном давлении. Неочищенный продукт промывали н-гексаном (50 мл), затем очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 2b (15 г; выход: 68%).
MS m/z (ESI): 241,1 [М-1].
Стадия 2
Метил-5-хлор-2-фтор-4-(трифторметил)бензоат 2c
Соединение 2b (5 г; 20,61 ммоль) добавляли к тионилхлориду (49,2 г; 413,55 ммоль) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. Полученное масло по каплям добавляли к метанолу (100 мл) и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 2с (2,78 г; выход: 52%).
Стадия 3
Метил-5-хлор-2-(4-фтор-2-метилфенокси)-4-(трифторметил)бензоат 2d Соединение 2с (2,78 г; 10,83 ммоль), 4-фтор-2-метил-фенол (1,5 г; 11,89 ммоль; Shanghai Bide Pharmatech Ltd.) и карбонат цезия (6 г; 18,41 ммоль) добавляли к N,N-диметилформамиду (20 мл) и в реакционном растворе осуществляли взаимодействие при 100°С в течение 1 часа. Реакционный раствор охлаждали и фильтровали. Фильтрат концентрировали, получая целевое соединение 2d (3,92 г), которое использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 363,1 [М+1].
Стадия 4
5-Хлор-2-(4-фтор-2-метилфенокси)-4-(трифторметил)бензойная кислота 2е
Соединение 2d (3,92 г; 10,81 ммоль) растворяли в метаноле (30 мл), затем добавляли воду (10 мл) и гидроксид натрия (1,3 г; 32,5 ммоль) и в реакционном растворе осуществляли взаимодействие в течение 16 часов. Реакционный раствор концентрировали, затем добавляли 10 мл воды и рН подводили до 1 концентрированной соляной кислотой. Полученный раствор экстрагировали этилацетатом (20 мл × 3), органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 2е (3,67 г; выход: 97%).
MS m/z (ESI): 346,8 [М-1].
Стадия 5
5-Хлор-2-(4-фтор-2-метилфенокси)-4-(трифторметил)бензоилхлорид 2f
Соединение 2е (3,67 г; 10,52 ммоль) добавляли к тионилхлориду (20 г; 168,1 ммоль) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 2 часов. Реакционный раствор концентрировали, получая указанное в заголовке соединение 2f (3,86 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 6
5-Хлор-N-(6-хлорпиридазин-4-ил)-2-(4-фтор-2-метилфенокси)-4-(трифторметил)бензамид 2д
4-Диметиламинопиридин (130 мг; 1,05 ммоль) и 6-хлорпиридазин-4-амин (1,51 г; 11,57 ммоль; Pharmablock Sciences (Nanjing), Inc.) растворяли в пиридине (40 мл) и полученный раствор сушили над молекулярными ситами. Добавляли соединение 2f (3,86 г; 10,51 ммоль) и в реакционном растворе осуществляли взаимодействие в течение 16 часов в атмосфере аргона. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 2д (1,3 г; выход: 39%).
MS m/z (ESI): 460,0 [М+1].
Стадия 7
5-Хлор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 2
Соединение 2g (1,3 г; 2,82 ммоль) и ацетат калия (555 мг; 5,65 ммоль) добавляли к уксусной кислоте (20 мл) и в реакционном растворе осуществляли взаимодействие при 130°С в течение 3 часов. Реакционный раствор концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 2 (800 мг; выход: 64%).
MS m/z (ESI): 442,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.83 (s, 1Н), 11.03 (s, 1Н), 8.07 (s, 1Н), 7.86 (s, 1Н), 7.05-7.25 (m, 5Н), 2.14 (s, 3Н).
Пример 3
4,5-Дихлор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 3
В соответствии с путем синтеза из примера 1 исходное соединение 4-фтор-2-метоксифенол на стадии 3 заменяли на соединение 4-фтор-2-метилфенол, получая таким образом указанное в заголовке соединение 3 (20 мг).
MS m/z (ESI): 407,8 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 2.82 (s, 1Н), 10.92 (s, 1Н), 7.99 (s, 1Н), 7.90 (d, 1Н), 7.21-7.18 (m, 2Н), 7.08-7.05 (m, 2Н), 6.99 (s, 1Н), 2.14 (s, 3Н).
Пример 4
4,5-Дихлор-2-(4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 4
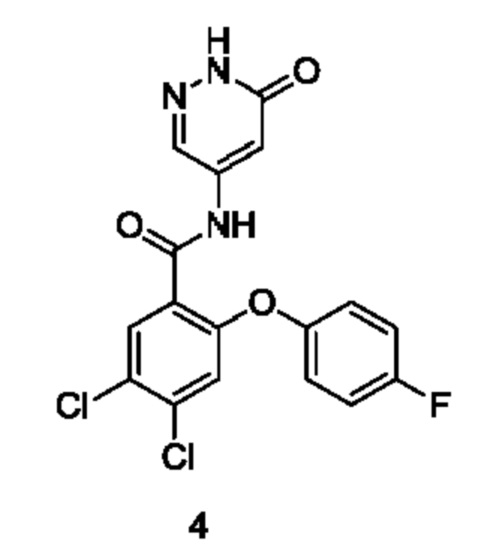
В соответствии с путем синтеза из примера 1 исходное соединение 4-фтор-2-метоксифенол на стадии 3 заменяли на соединение 4-фторфенол, получая таким образом указанное в заголовке соединение 4 (55 мг).
MS m/z (ESI): 395,8 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.81 (s, 1Н), 10.89 (s, 1Н),7.98 (s, 1Н), 7.88 (d, 1Н), 7.26-7.22 (m, 2Н), 7.19 (s, 1Н), 7.16-7.13 (m, 3Н).
Пример 5
2-(4-Фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(перфторэтил)бензамид 5
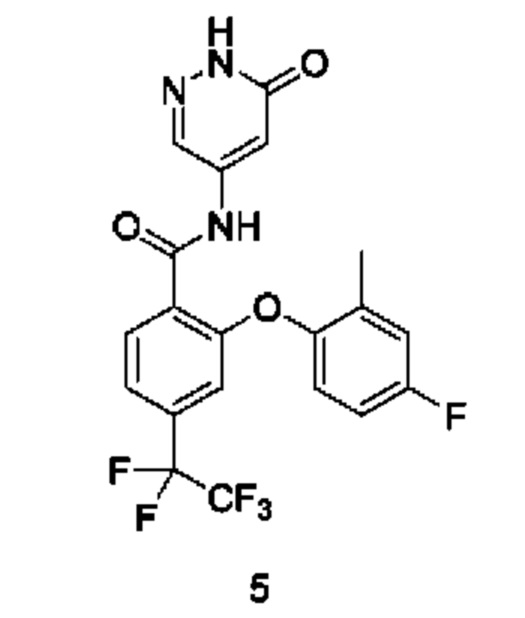
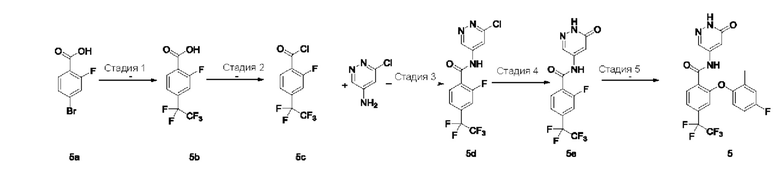
Стадия 1
2-Фтор-4-(перфторэтил)бензойная кислота 5b
4-Бром-2-фторбензойную кислоту 5а (2,19 г; 10 ммоль; J&K Scientific Ltd.) растворяли в диметилсульфоксиде (41 мл) в атмосфере аргона. Добавляли порошок меди (6,36 г; 100 ммоль) и пентафторйодэтан (17,22 г; 70 ммоль; 8,24 мл; Sichuan Shang Fluoro Technology Co., Ltd.), реакционный раствор герметично закрывали в пробирке и осуществляли взаимодействие при 120°С в течение 72 часов. Реакционный раствор охлаждали, затем добавляли 100 мл воды и 100 мл этилацетата, хорошо перемешивали и фильтровали. Фильтрат разделяли на две фазы и водную фазу экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 5b (2,4 г; выход: 93%).
MS m/z (ESI): 257,0 [М-1].
Стадия 2
2-Фтор-4-(перфторэтил)бензоилхлорид 5с
Соединение 5b (500 мг; 1,93 ммоль) добавляли к тионилхлориду (8,19 г; 68,8 ммоль) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 16 часов. Реакционный раствор концентрировали, получая неочищенное указанное в заголовке соединение 5с (535 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
N-(6-Хлорпиридазин-4-ил)-2-фтор-4-(перфторэтил)бензамид 5d
Неочищенное соединение 5 с (535 мг; 1,93 ммоль), 6-хлорпиридазин-4-амин (150 мг; 1,16 ммоль) и пиридин (916 мг; 11,58 ммоль) растворяли в дихлорметане (5 мл) и в реакционном растворе осуществляли взаимодействие в течение 16 часов. Добавляли 50 мл этилацетата и реакционный раствор промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 5d (664 мг; выход: 93%).
MS m/z (ESI): 370,0 [М+1].
Стадия 4
2-Фтор-N-(6-оксо-1,6-дигидропиридазин)-4-(перфторэтил)бензамид 5е
Соединение 5d (400 мг; 1,08 ммоль) и ацетат калия (213 мг; 2,17 ммоль) добавляли к уксусной кислоте (5 мл) и в реакционном растворе осуществляли взаимодействие при 130°С в течение 5 часов. Реакционный раствор концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 5е (190 мг; выход: 50%).
MS m/z (ESI): 352,0 [М+1].
Стадия 5
2-(4-Фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(перфторэтил)бензамид 5
Соединение 5е (190 мг; 0,54 ммоль), 4-фтор-2-метилфенол (103 мг; 0,82 ммоль) и карбонат цезия (265 мг; 0,81 ммоль) добавляли к N,N-диметилформамиду (3 мл) и в реакционном растворе осуществляли взаимодействие при 100°С в течение 2 часов. Реакционный раствор охлаждали, фильтровали и очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2, система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 5 (89 мг; выход: 36%).
MS m/z (ESI): 458,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.05 (d, 1Н), 7.92 (d, 1Н), 7.52 (d, 1Н), 7.49 (d, 1Н), 7.12 (dd, 1Н), 6.99-7.07 (m, 2Н), 6.93 (s, 1Н), 2.21 (s, 3Н).
Пример 6
2-(2-Метил-4-(трифторметокси)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 6
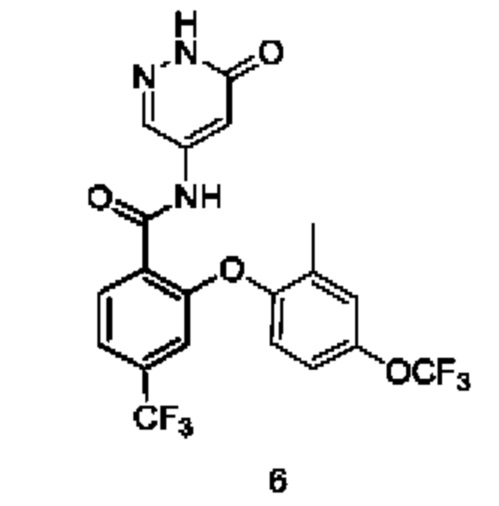
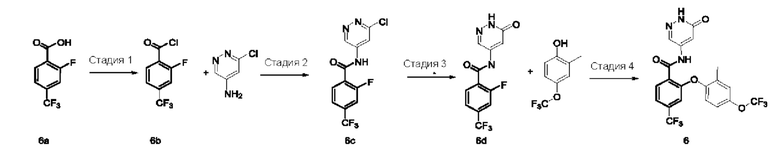
Стадия 1
2-Фтор-4-(трифторметил)бензоилхлорид 6b
2-Фтор-4-(трифторметил)бензойную кислоту 6а (1,2 г; 5,76 ммоль; Accela ChemBio (Shanghai) Inc.) добавляли к тионилхлориду (12 мл) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 16 часов. Реакционный раствор концентрировали, получая неочищенное указанное в заголовке соединение 6b (1,3 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
N-(6-Хлорпиридазин-4-ил)-2-фтор-4-(трифторметил)бензамид 6с
Неочищенное соединение 6b (1,3 г; 5,74 ммоль), 6-хлор-4-аминопиридазин (300 мг; 2,31 ммоль) и пиридин (916 мг; 11,58 ммоль) растворяли в дихлорметане (5 мл) и в реакционном растворе осуществляли взаимодействие в течение 16 часов. Реакционный раствор концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 6 с (475 мг; выход: 64%).
MS m/z (ESI): 320,0 [М+1].
Стадия 3
2-Фтор-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 6d
Соединение 6 с (475 мг; 1,49 ммоль) и ацетат калия (292 мг; 2,98 ммоль) добавляли к уксусной кислоте (5 мл) и в реакционном растворе осуществляли взаимодействие при 120°С в течение 16 часов. Реакционный раствор концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 6d (300 мг; выход: 67%).
MS m/z (ESI): 302,0 [М+1].
Стадия 4
2-(2-Метил-4-(трифторметокси)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 6
Соединение 6d (100 мг; 0,33 ммоль), 2-метил-4-(трифторметокси)фенол (78 мг; 0,4 ммоль) и карбонат цезия (216 мг; 0,66 ммоль) добавляли к N,N-диметилформамиду (3 мл) и в реакционном растворе осуществляли взаимодействие при 100°С в течение 8 часов. Реакционный раствор охлаждали и фильтровали. Фильтрат концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 6 (60 мг; выход: 38%).
MS m/z (ESI): 473,8 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.05 (d, 1Н), 7.92 (d, 1Н), 7.52 (d, 1Н), 7.49 (d, 1Н), 7.12 (dd, 1Н), 6.99-7.07 (m, 2Н), 6.93 (s, 1Н), 2.29 (s, 3Н).
Пример 7
2-(4-Фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-5-(трифторметил)бензамид 7
Стадия 1
2-Фтор-5-(трифторметил)бензоилхлорид 7b
2-Фтор-5-(трифторметил)бензойную кислоту 7а (382 мг; 1,83 ммоль; Shanghai Bide Pharmatech Ltd.) добавляли к тионилхлориду (5 мл) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 16 часов. Реакционный раствор концентрировали, получая неочищенное указанное в заголовке соединение 7b (400 мг), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
N-(6-Хлорпиридазин-4-ил)-2-фтор-5-(трифторметил)бензамид 7с
Неочищенное соединение 7b (393 мг; 1,73 ммоль), 6-хлор-4-аминопиридазин (150 мг; 1,15 ммоль) и пиридин (458 мг; 5,79 ммоль) растворяли в дихлорметане (5 мл) и в реакционном растворе осуществляли взаимодействие в течение 24 часов. Добавляли 50 мл этилацетата и реакционный раствор промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 7с (90 мг; выход: 24%).
MS m/z (ESI): 320,0 [М+1].
Стадия 3
N-(6-Хлорпиридазин-4-ил)-2-(4-фтор-2-метилфенокси)-5-(трифторметил)бензамид 7d
Соединение 7 с (90 мг; 0,28 ммоль), 4-фтор-2-метилфенол (43 мг; 0,34 ммоль) и карбонат цезия (183 мг; 0,56 ммоль) добавляли к N,N-диметилформамиду (3 мл) и в реакционном растворе осуществляли взаимодействие при 100°С в течение 1 часа. Реакционный раствор фильтровали и фильтрат концентрировали. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 7d (46 мг; выход: 37%).
MS m/z (ESI): 426,1 [М+1].
Стадия 4
2-(4-Фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-5-(трифторметил)бензамид 7
Соединение 7d (46 мг; 0,11 ммоль) и ацетат калия (21 мг; 0,21 ммоль) добавляли к уксусной кислоте (1 мл) и в реакционном растворе осуществляли взаимодействие при 130°С в течение 4 часов. Реакционный раствор концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 7 (16 мг; выход: 36%).
MS m/z (ESI): 407,9 [М+1].
1H ЯМР (400 МГц, CD3OD): δ 8.08-8.07 (m, 2H), 7.77 (dd, 1H), 7.52 (d, 1H), 7.14-7.11 (m, 2H), 7.05-7.00 (m, 1H),6.89 (d, 1H), 2.21 (s, 3H).
Пример 8
2-(4-Фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 8
В соответствии с путем синтеза из примера 6 исходное соединение 2-метил-4-(трифторметокси)фенол на стадии 3 заменяли на соединение 4-фтор-2-метоксифенол, получая таким образом указанное в заголовке соединение 8 (20 мг).
MS m/z (ESI): 424,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.07 (d, 1Н), 7.94 (d, 1Н), 7.53-7.49 (m, 2Н), 7.26 (q, 1Н), 7.03 (dd, 1H), 6.97 (s, 1Н), 6.83-6.79 (m, 1Н), 2.89 (s, 3Н).
Пример 9
2-(4-Фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 9
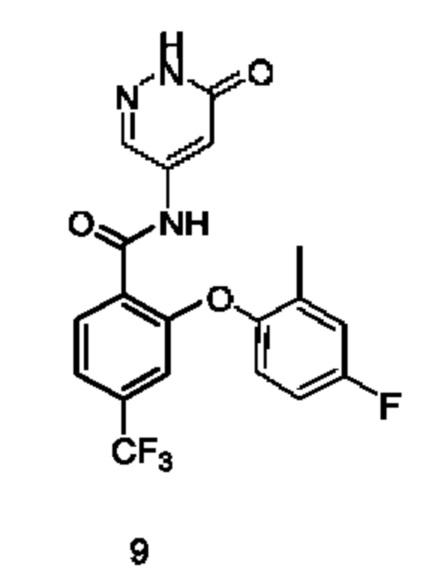
В соответствии с путем синтеза из примера 6 исходное соединение 2-метил-4-(трифторметокси)фенол на стадии 3 заменяли на соединение 4-фтор-2-метилфенол, получая таким образом указанное в заголовке соединение 9 (65 мг).
MS m/z (ESI): 408,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.05 (d, 1Н), 7.90 (d, 1Н), 7.54 (d, 1Н), 7.49 (d, 1Н), 7.11 (dd, 1Н), 6.99-7.07 (m, 2Н), 6.97 (s, 1Н), 2.22 (s, 3Н).
Пример 10
N-(6-Оксо-1,6-дигидропиридазин-4-ил)-2-(4-(трифторметокси)фенокси)-4-(трифторметил)бензамид 10
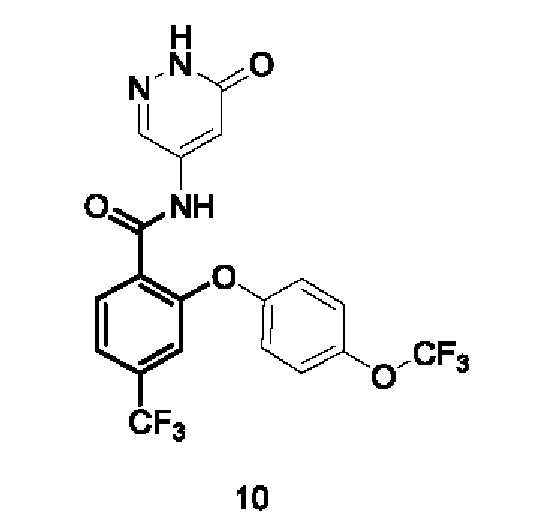
В соответствии с путем синтеза из примера 6 исходное соединение 2-метил-4-(трифторметокси)фенол на стадии 3 заменяли на соединение 4-трифторметоксифенол, получая таким образом указанное в заголовке соединение 10 (18 мг).
MS m/z (ESI): 460,0 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.03 (d, 1Н), 7.93 (d, 1Н), 7.65-7.63 (m, 1Н), 7.43 (d, 1Н), 7.35-7.32 (m, 3Н), 7.19-7.16 (m, 2Н).
Пример 11
5-Хлор-2-(4-фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 11
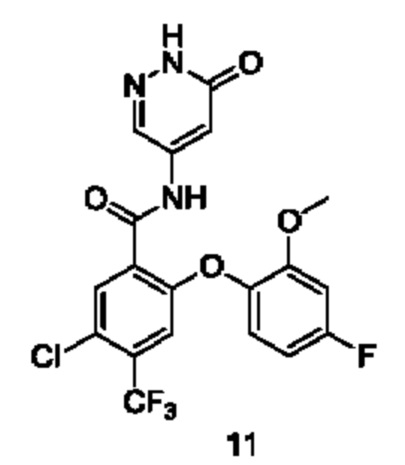
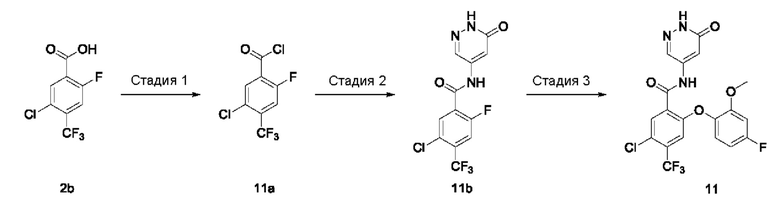
Стадия 1
5-Хл ор-2-фтор-4-(трифторметил)бензоилхлорид 11а
Соединение 2b (5,00 г; 20,6 ммоль) добавляли к 15 мл тионилхлорида и в реакционном растворе осуществляли взаимодействие при 80°С в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, получая неочищенное указанное в заголовке соединение 11а (5,38 г), которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
5-Хлор-2-фтор-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 11b
5-Аминопиридазин-3-он (3,06 г; 24,8 ммоль; Shanghai Medicilon Inc.) растворяли в 40 мл N-метилпирролидона. Полученный раствор охлаждали до 0°С и медленно порциями добавляли гидрид натрия (2,06 г; 51,5 ммоль; чистота: 60%). Реакционный раствор перемешивали при 0°С в течение 30 минут. Соединение 11а (5,38 г; 20,6 ммоль) растворяли в 3 мл N-метилпирролидона и полученный раствор медленно по каплям добавляли к указанному выше реакционному раствору, который затем перемешивали при комнатной температуре в течение ночи. Реакционный раствор, содержащий указанное в заголовке соединение 11b, использовали непосредственно на следующей стадии без очистки.
Стадия 3
5-Хлор-2-(4-фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 11с
4-Фтор-2-метоксифенол (2,34 г; 16,5 ммоль; Tokyo Chemical Industry (Shanghai) Co., Ltd.) и карбонат цезия (6,71 г; 20,6 ммоль; Accela ChemBio (Shanghai) Inc.) добавляли непосредственно в реакционный раствор, содержащий соединение 11b. В реакционном растворе осуществляли взаимодействие при 60°С в течение ночи и затем проводили охлаждение до комнатной температуры. Добавляли этилацетат (250 мл) и реакционный раствор промывали водой (100 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 11 с (3,0 г; выход: 32%).
MS m/z (ESI): 458,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.87 (s, 1Н), 11.03 (s, 1Н), 8.05 (s, 1Н), 7.92 (s, 1Н), 7.27 (dd, 1Н), 7.22 (s, 1Н), 7.15 (dd, 1Н), 7.00 (s, 1Н), 6.87-6.82 (m, 1Н), 3.71 (s, 3Н).
Пример 12
5-Хлор-2-(2-циклопропокси-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 12
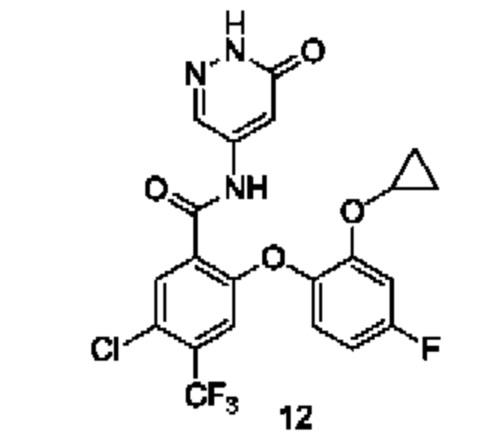
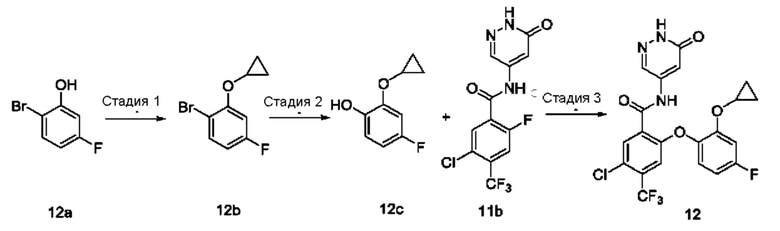
Стадия 1
1-Бром-2-циклопропокси-4-фторбензол 12b
2-Бром-5-фторфенол 12а (2 г; 10,5 ммоль; Accela ChemBio (Shanghai) Inc.), циклопропилбромид (5 г; 41,3 ммоль; Shanghai Titan Scientific Co., Ltd.), карбонат цезия (7 г; 21,5 ммоль; Accela ChemBio (Shanghai) Inc.) и иодид калия (180 мг; 1,1 ммоль) добавляли к N,N-диметилформамиду (10 мл). В реакционном растворе осуществляли взаимодействие, используя микроволновой реактор при 130°С в течение 1,5 часа, и затем охлаждали до комнатной температуры. Добавляли этилацетат (20 мл) и реакционный раствор промывали водой (20 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 12b (3,0 г; выход: 70%).
Стадия 2
2-Циклопропокси-4-фторфенол 12с
Соединение 12b (1,85 г; 8 ммоль) и триизопропилборат (1,96 г; 10,4 ммоль; Shanghai Titan Scientific Co., Ltd.) добавляли к 20 мл тетрагидрофурана. Воздух в реакционной колбе заменяли на аргон. Реакционный раствор охлаждали до -78°С, затем медленно по каплям в течение 20 минут добавляли н-бутиллитий (1,6 М раствор; 7,5 мл; 12 ммоль). Реакционный раствор оставляли нагреваться естественным образом до комнатной температуры и перемешивали в течение ночи. Реакционный раствор охлаждали до 0°С в ледяной бане. Добавляли 50 мл метанола и перекись водорода (30 масс. %-ную; 11 мл) и по каплям добавляли 10%-ный раствор гидроксида натрия (50 мл). По завершении добавления добавляли 400 мл насыщенного раствора хлорида натрия и реакционный раствор экстрагировали этилацетатом (200 мл × 3). Органическую фазу промывали насыщенным раствором бикарбоната натрия (150 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 12b (1,0 г; выход: 74%).
Стадия 3
5-Хлор-2-(2-циклопропокси-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 12
Соединение 11b (700 мг; 2,1 ммоль), соединение 12с (300 мг; 1,78 ммоль) и карбонат цезия (700 мг; 2,1 ммоль; Accela ChemBio (Shanghai) Inc.) добавляли к 7 мл N-метилпирролидона. В реакционном растворе осуществляли взаимодействие при 80°С в течение 1 часа и затем охлаждали до комнатной температуры. Добавляли этилацетат (20 мл) и реакционный раствор промывали водой (10 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 12 (300 мг; выход: 30%).
MS m/z (ESI): 484,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.84 (s, 1Н), 10.99 (s, 1Н), 8.01 (s, 1Н), 7.87 (s, 1Н), 7.30-7.19 (m, 3Н), 6.96 (s, 1Н), 6.85-6.83 (m, 1Н), 3.90-3.88 (m, 3Н), 0.74-0.70 (m, 1Н), 0.40-0.38 (m, 1Н).
Пример 13
5-Хлор-2-(4-фтор-2-метилбензил)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 13
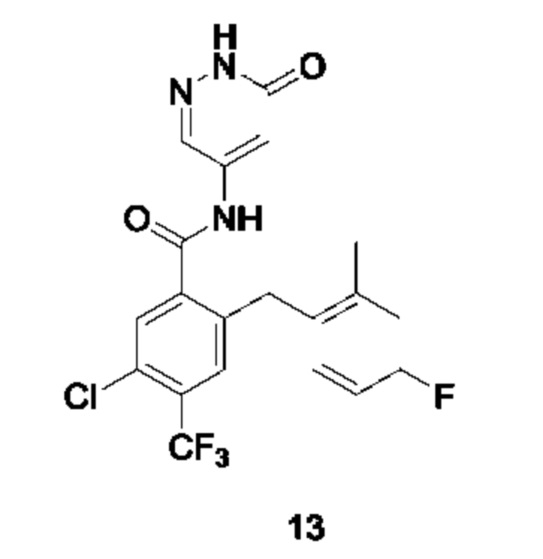
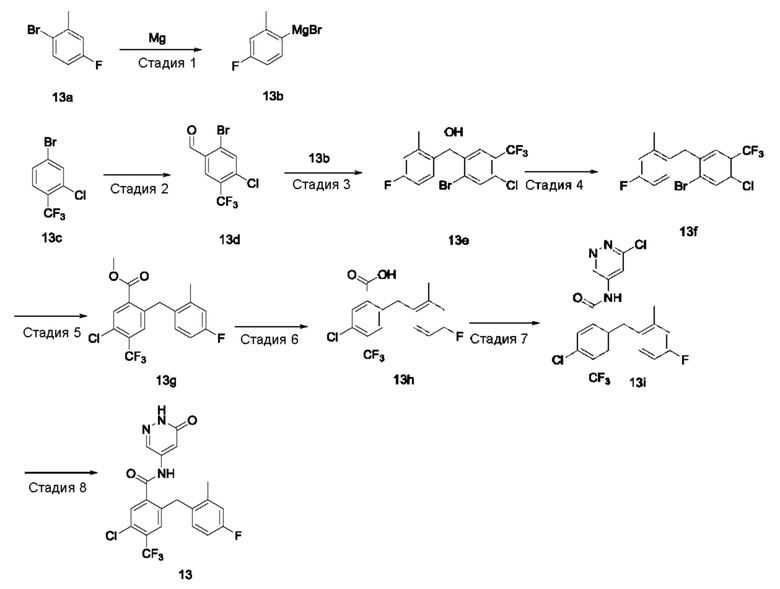
Стадия 1
(4-Фтор-2-метилфенил)магния бромид 13b
Очищенную пластинку магния (760 мг; 31,7 ммоль; Shanghai Sinopharm Chemical Reagent Co., Ltd.) разрезали на небольшие кусочки и добавляли к тетрагидрофурану (80 мл) в атмосфере аргона. По каплям при комнатной температуре добавляли триметилхлорсилан (345 мг; 3,17 ммоль; Accela ChemBio (Shanghai) Inc.), затем добавляли 1-бром-4-фтор-2-метилбензол 13а (1,5 г; 7,9 ммоль; Accela ChemBio (Shanghai) Inc.). После инициации реакции посредством нагревания, добавляли дополнительное количество соединения 13а (4,5 г; 23,7 ммоль; Accela ChemBio (Shanghai) Inc.). Реакционный раствор нагревали до 45°С и взаимодействие осуществляли в течение 1 часа. Пластинка магния полностью исчезала, и получали серую гомогенную жидкость, т.е. раствор указанного в заголовке соединения 13b (0,4 М; 80 мл), который использовали непосредственно на следующей стадии без очистки.
Стадия 2
2-Бром-4-хлор-5-(трифторметил)бензальдегид 13d
Тетрагидрофуран (100 мл) и гексаметилдисилазид лития (1 М; 120 мл; 120 ммоль; Titan Scientific Co., Ltd.) охлаждали до -78°C в атмосфере аргона. По каплям добавляли 4-бром-2-хлор-1-(трифторметил)бензол 13с (25 г; 96,36 ммоль; Accela ChemBio (Shanghai) Inc.), реакционный раствор выдерживали при этой низкой температуре и осуществляли взаимодействие в течение 2 часов. По каплям добавляли N,N-диметилформамид (14,1 г; 192,9 ммоль; J&K Scientific Ltd.), реакционный раствор постепенно нагревали до комнатной температуры и осуществляли взаимодействие в течение 16 часов. Добавляли воду и реакционный раствор экстрагировали этилацетатом (50 мл ×3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 13d (3,14 г; выход: 28%).
1Н ЯМР (400 МГц, CDCl3) δ 10.32 (s, 1H),8.23(s, 1Н),7.87 (s, 1Н).
Стадия 3
(2-Бром-4-хлор-5-(трифторметил)фенил)(4-фтор-2-метилфенил)метанол 13е Соединение 13d (900 мг; 3,13 ммоль) растворяли в тетра гидрофура не (10 мл). По каплям добавляли свежеприготовленный раствор соединения 13b (7,97 ммоль; 19,92 мл) и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 1 часа. Добавляли насыщенный раствор хлорида аммония и реакционный раствор экстрагировали этилацетатом (10 мл*3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 13е (900 мг; выход: 72%).
1Н ЯМР (400 МГц, CD3OD) δ 8.04 (s,1H), 7.93 (s, 1Н), 7.0-7.03 (m, 1Н), 6.87-6.90 (m, 2Н), 6.17 (s, 1H),2.5(s, 3Н).
Стадия 4
1-Бром-5-хлор-2-(4-фтор-2-метилбензил)-4-(трифторметил)бензол 13f
Соединение 13е (4 г; 10,1 ммоль) растворяли в дихлорметане (50 мл). Полученный раствор охлаждали до 0°С, добавляли трифторуксусную кислоту (10 мл; Titan Scientific Co., Ltd.) и затем по каплям добавляли триэтилсилан (6 мл; Accela ChemBio (Shanghai) Inc.). В реакционном растворе осуществляли взаимодействие при 0°С в течение 1 часа. Добавляли воду и реакционный раствор экстрагировали этилацетатом (10 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 13f (3,2 г; выход: 83%).
Стадия 5
Метил-5-хлор-2-(4-фтор-2-метилбензил)-4-(трифторметил)бензоат 13g
Соединение 13f (3,3 г; 8,64 ммоль) растворяли в метаноле (60 мл). Добавляли ацетат палладия (388,31 мг; 1,73 ммоль; J&K Scientific Ltd.), 1,1'-бис(дифенилфосфино)ферроцен (960 мг; 1,73 ммоль; Accela ChemBio (Shanghai) Inc.) и триэтиламин (2,63 г; 25,94 ммоль; Shanghai Sinopharm Chemical Reagent Co., Ltd.). Реакционную систему подсоединяли к баллону с окисью углерода и взаимодействие осуществляли при 60°С в течение 16 часов. Реакционный раствор фильтровали через диатомовую землю. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 13g (2,2 г; выход: 71%).
MS m/z (ESI): 359,1 [М-1].
Стадия 6
5-Хлор-2-(4-фтор-2-метилбензил)-4-(трифторметил)бензойная кислота 13h
Соединение 13g (2,2 г; 6,1 ммоль) растворяли в метаноле (40 мл) и воде (20 мл), затем добавляли раствор гидроксида натрия (5 М; 6 мл; 30 ммоль). Реакционный раствор нагревали до 40°С и осуществляли взаимодействие в течение 3 часов. Реакционный раствор охлаждали, рН подводили до 2, используя 4 М раствор соляной кислоты, и экстрагировали дихлорметаном (10 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая неочищенное указанное в заголовке соединение 13h (2,1 г), которое использовали непосредственно на следующей стадии без очистки. MS m/z (ESI): 345,1 [М-1].
Стадия 7
5-Хлор-N-(6-хлорпиридазин-4-ил)-2-(4-фтор-2-метилбензил)-4-(трифторметил)бензамид 13i
Соединение 13h (300 мг; 0,87 ммоль) растворяли в дихлорметане (15 мл), затем добавляли одну каплю N,N-диметилформамида. По каплям добавляли тионилхлорид (2 мл; Shanghai Sinopharm Chemical Reagent Co., Ltd.) в ледяной бане. В реакционном растворе осуществляли взаимодействие при комнатной температуре в течение ночи и затем выполняли концентрирование при пониженном давлении. Полученный остаток растворяли в пиридине (2 мл), затем добавляли 4 амино-6-хлорпиридазин (168 мг; 1,3 ммоль; Accela ChemBio (Shanghai) Inc.). В реакционном растворе осуществляли взаимодействие при комнатной температуре в течение ночи и затем выполняли концентрирование при пониженном давлении. Добавляли воду (20 мл) и полученный раствор экстрагировали дихлорметаном (10 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая неочищенное соединение 13i (260 мг), которое использовали непосредственно на следующей стадии без очистки.
MS m/z (ESI): 458,1 [М+1].
Стадия 8
5-Хлор-2-(4-фтор-2-метилбензил)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 13
Соединение 13i (260 мг; 0,57 ммоль) растворяли в уксусной кислоте (5 мл), затем добавляли ацетат калия (112 мг; 1,14 ммоль) и в реакционном растворе осуществляли взаимодействие при 130°С в течение ночи. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Voters 2767-SQ Detecor2; система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 13 (10 мг; выход за две стадии: 2,6%).
MS m/z (ESI): 440,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 7.90-7.91 (m, 1Н), 7.81 (m, 1Н), 7.59 (m, 1Н), 7.39 (m, 1H), 6.95-6.97 (m, 1Н), 6.85-6.88 (m, 1Н), 6.78-6.79 (m, 1Н), 4.63 (m, 1Н), 4.19 (s, 2Н), 2.20 (s, 3Н).
Пример 14
5-Хлор-2-(2-фтор-4-(трифторметокси)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 14
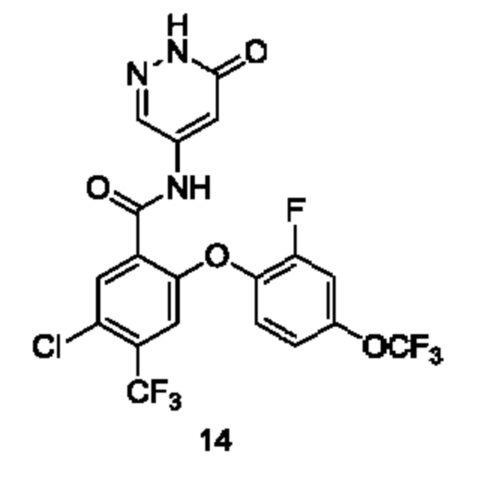
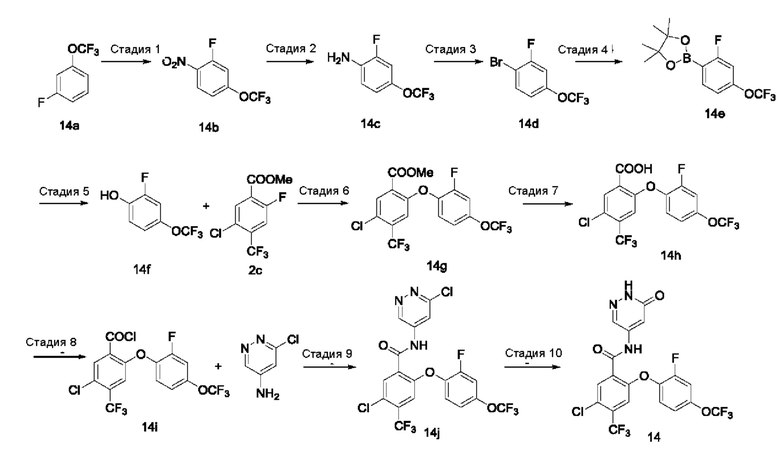
Стадия 1
2-Фтор-1-нитро-4-(трифторметокси)бензол 14b
1-Фтор-3-(трифторметокси)бензол 14а (5 г; 27,76 ммоль; Accela ChemBio (Shanghai) Inc.) растворяли в серной кислоте (20 мл) и полученный раствор охлаждали в ледяной бане. Порциями добавляли нитрат калия (7 г; 69,2 ммоль), реакционный раствор оставляли нагреваться естественным образом до комнатной температуры и взаимодействие осуществляли в течение ночи. Реакционный раствор выливали в ледяную воду, перемешивали в течение 30 минут, трижды экстрагировали этилацетатом, сушили над сульфатом натрия и концентрировали досуха путем упаривания на роторном испарителе, получая неочищенную смесь (5,8 г), содержащую соединение 14b.
Стадия 2
2-Фтор-4-(трифторметокси)анилин 14с
Неочищенную смесь (5,8 г; 25,7 ммоль), содержащую соединение 14b, растворяли в метаноле (80 мл). Полученный раствор продували азотом и добавляли катализатор Pd/C (1,54 г; 14,47 ммоль). Полученный раствор трижды продували водородом и реакцию гидрирования осуществляли при комнатной температуре в течение ночи. Реакционный раствор фильтровали и фильтрат концентрировали. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая неочищенную смесь (3,5 г), содержащую указанное в заголовке соединение 14с.
Стадия 3
1-Бром-2-фтор-4-(трифторметокси)бензол 14d
Смесь (3,4 г; 17,4 ммоль), содержащую соединение 14с, растворяли в ацетонитриле (40 мл), затем добавляли бромид меди (4,67 г; 20,9 ммоль; Accela ChemBio (Shanghai) Inc.) и трет-бутилнитрит (2,16 г; 20,9 ммоль). В реакционном растворе осуществляли взаимодействие при 60°С в течение 0,5 часа и затем фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая смесь (1,3 г), содержащую указанное в заголовке соединение 14d.
Стадия 4
2-(2-Фтор-4-(трифторметокси)фенил)-4,4,5,5-тетраметил-1,3,2-диоксаборолан 14е
Смесь (3 г; 11,6 ммоль), содержащую соединение 14d, растворяли в 1,4-диоксане (50 мл), затем добавляли пинаколдиборат (4,4 г; 17,3 ммоль; Accela ChemBio (Shanghai) Inc.), [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий(М) (424 мг; 0,58 ммоль) и ацетат калия (3,41 г; 34,7 ммоль). Реакционный раствор трижды продували аргоном и взаимодействие осуществляли при 100°С в течение ночи. Реакционный раствор фильтровали через диатомовую землю и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая смесь (950 мг), содержащую указанное в заголовке соединение 14е.
Стадия 5
2-Фтор-4-(трифторметокси)фенол 14f
Смесь (950 мг; 3,1 ммоль), содержащую соединение 14е, растворяли в тетрагидрофуране (20 мл), затем добавляли раствор гидроксида натрия (621 мг; 15,5 ммоль; 5 мл). Полученный раствор охлаждали в ледяной бане. По каплям добавляли перекись водорода (3,1 мл) и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение ночи. Органическую фазу промывали насыщенным раствором бикарбоната натрия (50 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая смесь (220 мг), содержащую указанное в заголовке соединение 14f.
Стадия 6
Метил-5-хлор-2-(2-фтор-4-(трифторметокси)фенокси)-4-(трифторметил)бензоат 14g
Соединение 2 с (300 мг; 1,2 ммоль) и смесь (229 мг; 1,2 ммоль), содержащую соединение 14f, добавляли к N,N-диметилформамиду (5 мл), затем добавляли карбонат цезия (571 мг; 1,8 ммоль). В реакционном растворе осуществляли взаимодействие при 100°С в течение 1 часа, охлаждали и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая смесь (440 мг), содержащую указанное в заголовке соединение 14д.
Стадия 7
5-Хлор-2-(2-фтор-4-(трифторметокси)фенокси)-4-(трифторметил)бензойная кислота 14h
Соединение 14g (400 мг; 0,92 ммоль) растворяли в тетрагидрофуране (5 мл), затем добавляли раствор гидроксида лития (194 мг; 4,6 ммоль; 1 мл). В реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 4 часов, концентрировали при пониженном давлении, затем добавляли воду (5 мл). Значение рН полученного раствора подводили до 4, используя разбавленную соляную кислоту, и трижды экстрагировали этилацетатом (20 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая смесь (340 мг), содержащую указанное в заголовке соединение 14h, которую использовали непосредственно на следующей стадии.
Стадия 8
5-Хлор-2-(2-фтор-4-(трифторметокси)фенокси)-4-(трифторметил)бензоилхлорид 14i
Соединение 14h (80 мг; 0,19 ммоль) растворяли в тионилхлориде (2 мл) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, получая смесь (80 мг), содержащую указанное в заголовке соединение 14i, которую использовали непосредственно на следующей стадии.
Стадия 9
5-Хлор-N-(6-хлорпиридазин-4-ил)-2-(2-фтор-4-(трифторметокси)фенокси)-4-(трифторметил)бензамид 14j
4-Амино-6-хлорпиридазин (31 мг; 0,24 ммоль; Accela ChemBio (Shanghai) Inc.) растворяли в тетрагидрофуране (3 мл) и полученный раствор охлаждали в ледяной бане. Добавляли гидрид натрия (13 мг; 0,34 ммоль; чистота: 60%) и в реакционном растворе осуществляли взаимодействие в течение 30 минут. По каплям добавляли раствор (2 мл) соединения 14i в тетрагидрофуране и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение ночи в атмосфере аргона. Добавляли воду (10 мл) и этилацетат (20 мл), органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая смесь (65 мг), содержащую указанное в заголовке соединение 14j.
Стадия 10
5-Хлор-2-(2-фтор-4-(трифторметокси)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 14
Смесь (65 мг; 0,12 ммоль), содержащую соединение 14j, растворяли в уксусной кислоте (3 мл), затем добавляли ацетат калия (60 мг; 0,61 ммоль) и в реакционном растворе осуществляли взаимодействие при 130°С в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2, система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 14 (8 мг; выход: 11%).
MS m/z (ESI): 512,0 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.02 (d, 1Н), 8.00 (s, 1Н), 7.43 (d, 1Н), 7.40 (s, 1Н), 7.37-7.27 (m, 2Н), 7.19 (d, 1Н).
Пример 15
5-Хлор-2-(4-фтор-2-(трифторметокси)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 15
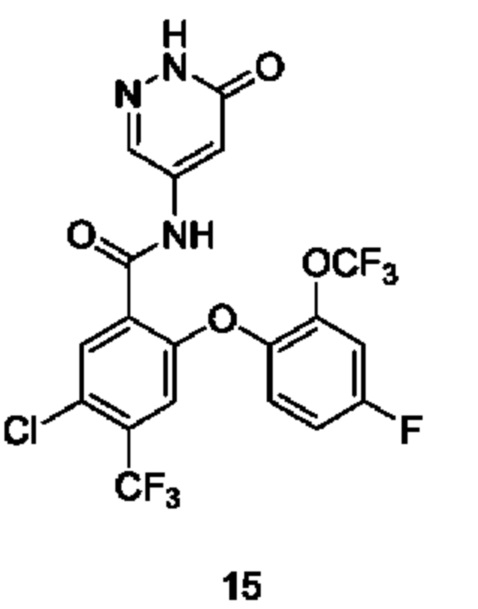
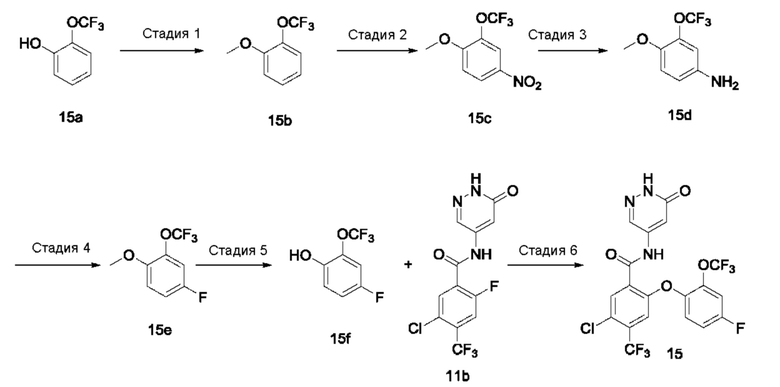
Стадия 1
1-Метокси-2-(трифторметокси)бензол 15b
2-(Трифторметокси)фенол 15а (5 г; 28,1 ммоль; Accela ChemBio (Shanghai) Inc.) растворяли в N,N-диметилформамиде (50 мл), затем добавляли метилиодид (4,78 г; 33,7 ммоль) и карбонат калия (7,75 г; 56,1594 ммоль). В реакционном растворе осуществляли взаимодействие при 80°С в течение 1 часа. К реакционному раствору добавляли этилацетат и воду и водную фазу трижды экстрагировали этилацетатом. Органические фазы объединяли, четыре раза промывали водой, сушили над сульфатом натрия и концентрировали досуха путем упаривания на роторном испарителе, получая указанное в заголовке соединение 15b (5,1 г; выход: 94%).
Стадия 2
1-Метокси-4-нитро-2-(трифторметокси)бензол 15с
Нитрат натрия (2,26 г; 26,6 ммоль) растворяли в трифторуксусной кислоте (50 мл). Полученный раствор охлаждали в ледяной бане и добавляли раствор (10 мл) соединения 15b (5,1 г; 26,5 ммоль) в трифторуксусной кислоте. В реакционном растворе осуществляли взаимодействие в ледяной бане в течение 1 часа и при комнатной температуре в течение 3 часов. К реакционному раствору добавляли воду (50 мл) и этилацетат (50 мл). Органическую фазу промывали насыщенным раствором бикарбоната натрия до нейтрального значения рН, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, получая указанное в заголовке соединение 15 с (5,3 г; выход: 84%).
Стадия 3
4-Метокси-3-(трифторметокси)анилин 15d
Соединение 15с (5,3 г; 22,3 ммоль) растворяли в метаноле (100 мл), затем добавляли концентрированную соляную кислоту (0,5 мл) и катализатор Pd/C (500 мг). Полученный раствор трижды продували водородом и осуществляли реакцию гидрирования при комнатной температуре в течение 4 часов. Реакционный раствор фильтровали через диатомовую землю и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 15d (3,1 г; выход: 67%).
Стадия 4
4-Фтор-1-метокси-2-(трифторметокси)бензол 15е Соединение 15d (800 мг; 3,9 ммоль) добавляли к перемешиваемому раствору воды (15 мл), фтороборной кислоты (1,51 г; 7,0 ммоль; чистота: 40%) и соляной кислоты (687 мг; 7,0 ммоль). Полученный раствор охлаждали в ледяной бане. По каплям добавляли раствор нитрита натрия (280 мг; 4,06 ммоль; 2 мл) и в реакционном растворе осуществляли взаимодействие в ледяной бане в течение 2 часов. Реакционный раствор фильтровали и осадок на фильтре сушили. Твердое вещество нагревали до 130°С в атмосфере азота, оно постепенно плавилось и становилось красным, и осуществляли взаимодействие в течение 1 часа. После охлаждения системы, в которой проводили реакцию, полученный продукт растворяли в дихлорметане, сушили над сульфатом натрия и концентрировали при пониженном давлении, получая указанное в заголовке соединение 15е (600 мг; выход: 74%).
Стадия 5
4-Фтор-2-(трифторметокси)фенол 15f
Соединение 15е (300 мг; 1,4 ммоль) растворяли в дихлорметане (5 мл) и полученный раствор охлаждали в ледяной бане. Добавляли раствор трибромида бора (1 М; 7 мл), реакционный раствор оставляли нагреваться естественным образом до комнатной температуры и осуществляли взаимодействие в течение ночи. Реакционный раствор по каплям добавляли к метанолу и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 15f (100 мг; выход: 36%).
Стадия 6
5-Хлор-2-(4-фтор-2-(трифторметокси)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 15
Соединение 15f (300 мг; 0,89 ммоль) добавляли к N-метилпирролидону (3 мл), затем добавляли соединение 11b (140 мг; 0,71 ммоль) и карбонат цезия (291 мг; 0,9 ммоль). В реакционном растворе осуществляли взаимодействие при 80°С в течение 1 часа. К реакционному раствору добавляли воду (10 мл) и этилацетат (20 мл), органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2; система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 15 (40 мг; выход: 11%).
MS m/z (ESI): 512,0 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.03 (d, 1Н), 8.01 (s, 1Н), 7.46 (d, 1Н), 7.37-7.35 (m, 1H), 7.32-7.22 (m, 3Н).
Пример 16
2-(4-Фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-5-(трифторметил)бензамид 16
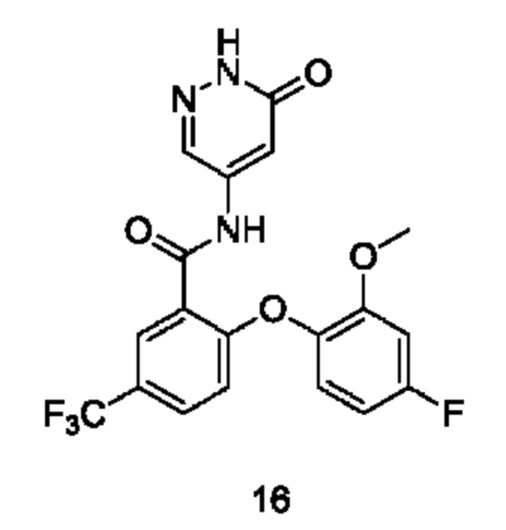
В соответствии с путем синтеза из примера 2 исходное соединение 5-хлор-2-фтор-4-(трифторметил)бензойную кислоту на стадии 2 заменяли на соединение 2-фтор-5-(трифторметил)бензойную кислоту, а исходное соединение 4-фтор-2-метил-фенол на стадии 3 заменяли на 4-фтор-2-метокси-фенол, получая таким образом указанное в заголовке соединение 16 (27 мг).
MS m/z (ESI): 424,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.81 (s, 1Н), 10.90 (s, 1Н), 7.95 (d, 2Н), 7.76 (d, 1Н), 7.21-7.33 (m,2H), 7.15 (d, 1H), 6.72-6.90 (m, 2Н), 3.71 (s, 3Н).
Пример 17
5-Хлор-2-((4-фтор-2-метоксифенил)тио)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 17
С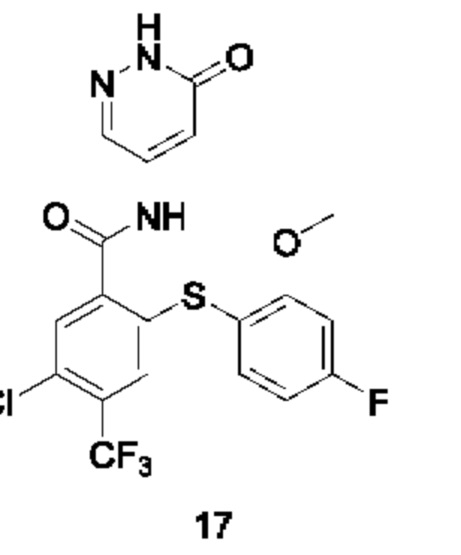
В соответствии с путем синтеза из примера 2 исходное соединение 2-метил-4-фторфенол на стадии 3 заменяли на 2-метоксибензолтиол, получая таким образом указанное в заголовке соединение 17 (35 мг).
MS m/z (ESI): 474,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.90 (s, 1Н), 11.06 (s, 1Н), 8.12 (s, 1Н), 7.97 (s, 1Н), 7.59-7.55 (m, 1H), 7.25 (s, 1H), 7.16-7.13 (m, 2Н), 6.93-6.89 (m, 1Н), 3.74 (s, 3Н).
Пример 18
4-Хлор-5-фтор-2-(4-фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 18
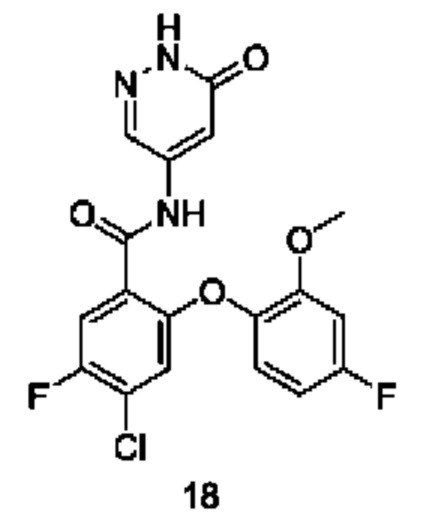
В соответствии с путем синтеза из примера 1 исходное соединение 4,5-дихлор-2-фторбензойную кислоту на стадии 1 заменяли на соединение 4-хлор-2,5-дифторбензойную кислоту, получая таким образом указанное в заголовке соединение 18 (22 мг).
MS m/z (ESI): 408,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.81 (s, 1Н), 10.83 (s, 1Н), 7.90-7.89 (d, 1Н), 7.77-7.75 (d, 1Н), 7.19-7.16 (m, 2Н), 7.10-7.06 (m, 1Н), 6.90-6.81 (m, 1Н), 6.81-6.77 (m, 1Н), 3.71 (s, 3Н).
Пример 19
4-Хлор-5-фтор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 19
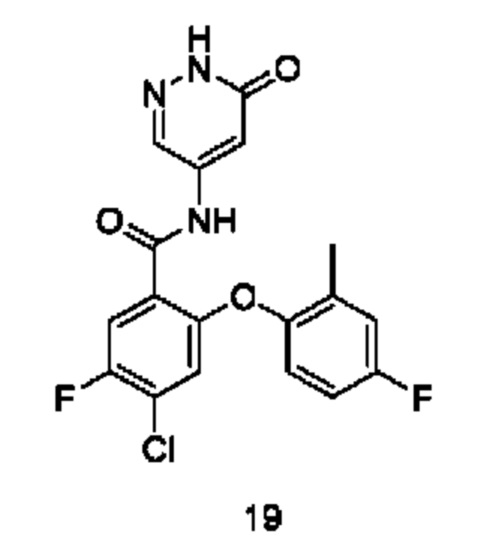
В соответствии с путем синтеза из примера 1 исходное соединение 4,5-дихлор-2-фторбензойную кислоту на стадии 1 заменяли на соединение 4-хлор-2,5-дифторбензойную кислоту, а исходное соединение 2-метокси-4-фторфенол на стадии 3 заменяли на 2-метил-4-фторфенол, получая таким образом указанное в заголовке соединение 19 (9 мг) в виде белого твердого вещества.
MS m/z (ESI): 392,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.81 (s, 1Н), 10.89 (s, 1Н), 7.87-7.86 (d, 1H), 7.82-7.80 (d, 1H), 7.18-7.14 (m, 2H), 7.04-7.03 (m, 2H), 7.00-6.93 (m, 1 H), 2.14 (s, 3H).
Пример 20
4-Хлор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 20
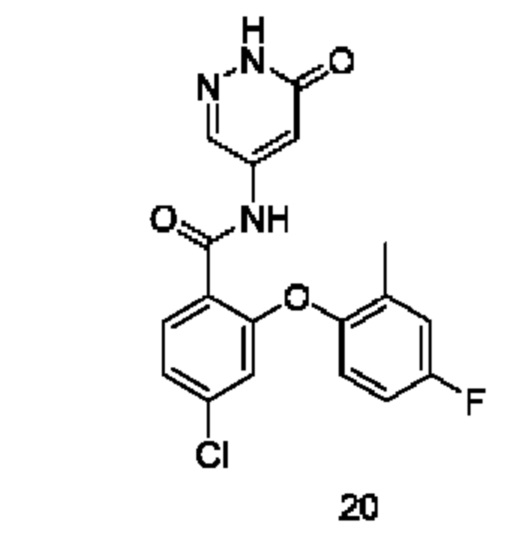
В соответствии с путем синтеза из примера 2 исходное соединение 5-хлор-2-фтор-4-(трифторметил)бензойную кислоту на стадии 2 заменяли на соединение 4-хлор-2-фторбензойную кислоту, получая таким образом указанное в заголовке соединение 20 (40 мг).
MS m/z (ESI): 374,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.79 (s, 1Н), 10.83 (s, 1H),7.89(s, 1Н), 7.67 (d, 1Н), 7.30 (d,1H), 7.14-7.22 (m, 2H), 7.00-7.12 (m, 2H), 6.74 (s, 1H), 2.13 (s, 3Н).
Пример 21
4-Хлор-2-(4-фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 21
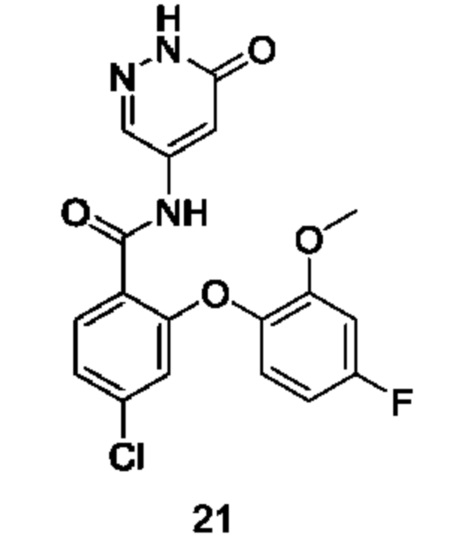
В соответствии с путем синтеза из примера 2 исходное соединение 5-хлор-2-фтор-4-(трифторметил)бензойную кислоту на стадии 2 заменяли на соединение 4-хлор-2-фторбензойную кислоту, а исходное соединение 2-метил-4-фторфенол на стадии 3 заменяли на соединение 2-метокси-4-фторфенол, получая таким образом указанное в заголовке соединение 21 (40 мг).
MS m/z (ESI): 390,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.79 (s, 1Н), 10.75 (s, 1H),7.92(s, 1H), 7.63 (d, 1H), 7.18-7.30 (m, 3Н), 7.10 (dd, 1H), 6.78-6.88 (m, 1H), 6.64 (s, 1H), 3.72 (s, 3Н).
Пример 22
2-(4-Фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-5-(трифторметокси)бензамид 22
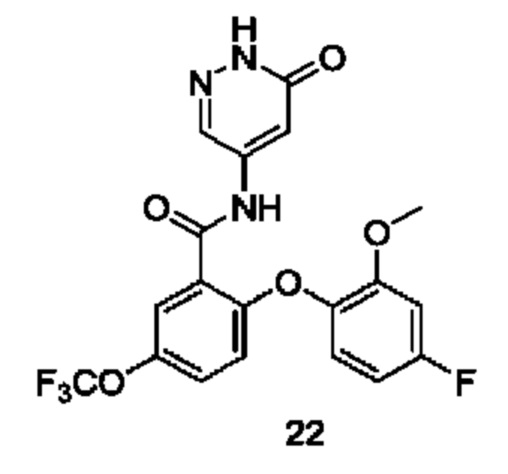
В соответствии с путем синтеза из примера 1 исходное соединение 4,5-дихлор-2-фторбензойную кислоту на стадии 1 заменяли на соединение 2-фтор-5-(трифторметокси)бензойную кислоту, получая таким образом указанное в заголовке соединение 22 (5 мг).
MS m/z (ESI): 440,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.05 (d, 1Н), 7.74 (d, 1Н), 7.54 (d, 1Н), 7.41-7.38 (m, 1Н), 7.26 (q, 1H), 7.03-7.00 (m, 1Н), 6.89 (d, 1Н), 6.81-6.77 (m, 1Н), 3.81 (s, 3Н).
Пример 23
5-Хлор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 23
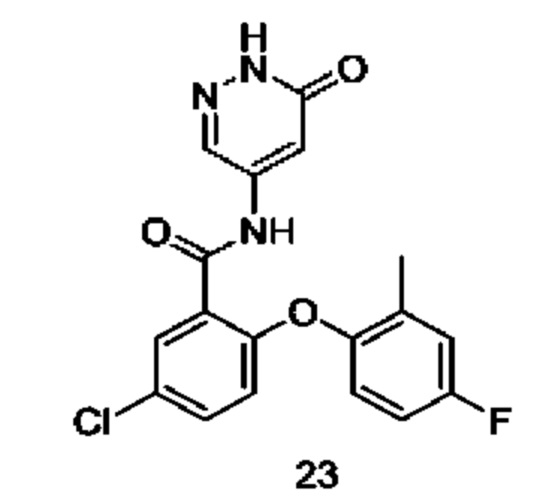
В соответствии с путем синтеза из примера 1 исходное соединение 4,5-дихлор-2-фторбензойную кислоту на стадии 1 заменяли на соединение 5-хлор-2-фторбензойную кислоту, получая таким образом указанное в заголовке соединение 23 (14 мг).
MS m/z (ESI): 374,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.05 (d, 1Н), 7.76 (d, 1Н), 7.49-7.46 (m, 2Н), 7.07 (d, 1Н), 7.01-6.98 (m, 2Н), 6.78 (d, 1Н), 2.22 (s, 3Н).
Пример 24
2-(4-Фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-5-(трифторметокси)бензамид 24
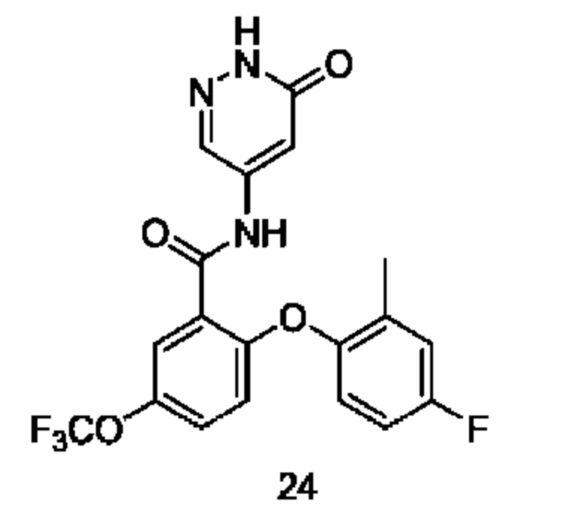
В соответствии с путем синтеза из примера 1 исходное соединение 4,5-дихлор-2-фторбензойную кислоту на стадии 1 заменяли на соединение 2-фтор-5-(трифторметокси)бензойную кислоту, получая таким образом указанное в заголовке соединение 24 (7 мг).
MS m/z (ESI): 424,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.05 (d, 1Н), 7.71 (d, 1Н), 7.50 (d, 1Н), 7.44-7.41 (m, 1Н), 7.11-6.97 (m, 3Н), 6.87 (d, 1Н), 2.23 (s, 3Н).
Пример 25
5-Фтор-2-(4-фтор-2-метоксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 25
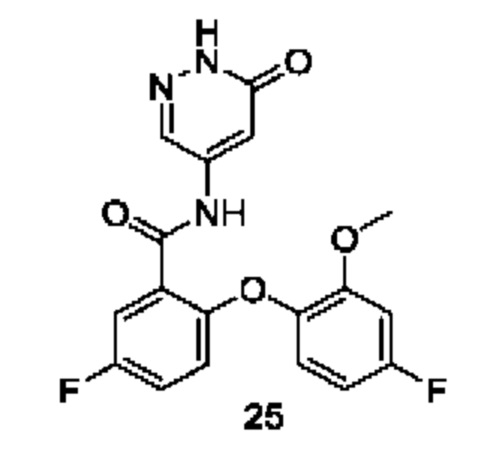
В соответствии с путем синтеза из примера 1 исходное соединение 4,5-дихлор-2-фторбензойную кислоту на стадии 1 заменяли на соединение 2,5-дифторбензойную кислоту, получая таким образом указанное в заголовке соединение 25 (20 мг).
MS m/z (ESI): 374,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.78 (s, 1Н), 10.77 (s, 1Н), 7.91 (s, 1Н), 7.45-7.55 (m, 1Н), 7.00-7.32 (m, 4Н), 6.65-6.85 (m, 2Н), 3.70 (s, 3Н).
Пример 26
5-Фтор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 26
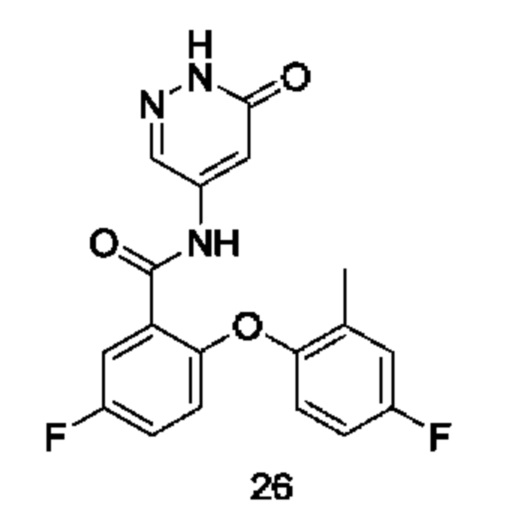
В соответствии с путем синтеза из примера 1 исходное соединение 4,5-дихлор-2-фторбензойную кислоту на стадии 1 заменяли на соединение 2,5-дифторбензойную кислоту, а исходное соединение 2-метокси-4-фторфенол на стадии 3 заменяли на 2-метил-4-фторфенол, получая таким образом указанное в заголовке соединение 26 (20 мг).
MS m/z (ESI): 358,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.79 (s, 1Н), 10.85 (s, 1Н), 7.88 (s, 1Н), 7.50-7.60 (m, 1Н), 7.30-7.40 (m, 1Н), 7.08-7.19 (m, 2Н), 6.95-7.05 (m, 1Н), 6.80-6.95 (m, 2Н), 2.14 (s, 3Н).
Пример 27
4-Хлор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-5-(трифторметил)бензамид 27
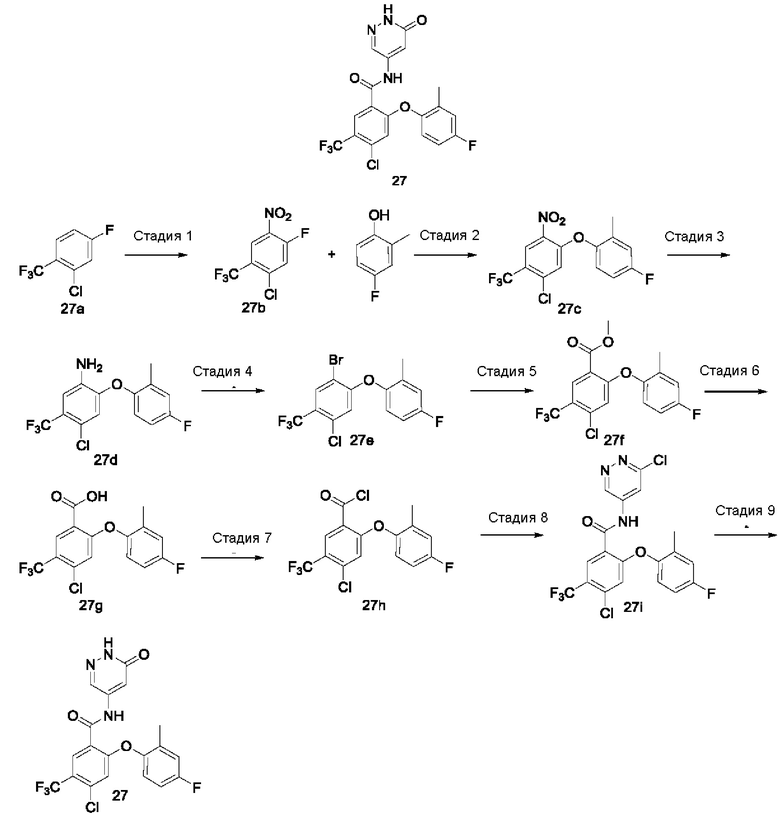
Стадия 1
1-Хлор-5-фтор-4-нитро-2-(трифторметил)бензол 27b
2-Хлор-4-фтор-1-(трифторметил)бензол 27а (3 г; 15,1 ммоль; Adamas Reagent Co., Ltd.) растворяли в серной кислоте (30 мл) и полученный раствор охлаждали до -10°С. Порциями добавляли нитрат калия (1,83 г; 18,1 ммоль), в реакционном растворе осуществляли взаимодействие при -10°С в течение 1 часа и при комнатной температуре в течение ночи. Реакционный раствор выливали на лед и экстрагировали этилацетатом (50 млх2). Органическую фазу сушили над
безводным сульфатом натрия и фильтровали. Фильтр концентрировали при пониженном давлении, получая указанное в заголовке соединение 27b (3,46 г; выход: 94%)
Стадия 2
1-Хлор-5-(4-фтор-2-метилфенокси)-4-нитро-2-(трифторметил)бензол 27с
Соединение 27b (1 г; 4,1 ммоль) растворяли в N,N-диметилформамиде (10 мл), затем добавляли 4-фтор-2-метилфенол (518 мг; 4,1 ммоль) и фосфат калия (2,61 г; 12,3 ммоль). В реакционном растворе осуществляли взаимодействие при 80°С в течение 1 часа. Реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 27с (1,27 г; выход: 88%).
Стадия 3
4-Хлор-2-(4-фтор-2-метилфенокси)-5-(трифторметил)анилин 27d
Соединение 27с (1,2 г; 3,4 ммоль) растворяли в этаноле (20 мл) и воде (10 мл), затем добавляли порошок восстановленного железа (1,15 г; 20,6 ммоль) и хлорид аммония (1,10 г; 20,6 ммоль). В реакционном растворе осуществляли взаимодействие при 80°С в течение 3 часов. Реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 27d (900 мг; выход: 82%).
Стадия 4
1-Бром-4-хлор-2-(4-фтор-2-метилфенокси)-5-(трифторметил)бензол 27е
Соединение 27d (1 г; 3,1 ммоль) растворяли в ацетонитриле (10 мл), затем добавляли бромид меди (839 мг; 3,7 ммоль; Adamas Reagent Co., Ltd.) и трет-бутил-нитрит (387 мг; 3,7 ммоль). Реакционный раствор кипятили с обратным холодильником в течение 2 часов. Реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении, получая указанное в заголовке соединение 27е (1 г; выход: 83%).
Стадия 5
Метил-4-хлор-2-(4-фтор-2-метилфенокси)-5-(трифторметил)бензоат 27f
Соединение 27е (400 мг; 1,04 ммоль) растворяли в метаноле (10 мл), затем добавляли ацетат палладия (47 мг; 0,21 ммоль), 1,1'-бис(дифенилфосфино)ферроцен (116 мг; 0,21 ммоль) и триэтиламин (317 мг; 3,1 ммоль). Полученный раствор трижды продували окисью углерода. В реакционном растворе осуществляли взаимодействие при 70°С в течение ночи в атмосфере окиси углерода. Реакционный раствор фильтровали и фильтрат концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 27f (400 мг; выход: 42%).
Стадия 6
4-Хлор-2-(4-фтор-2-метилфенокси)-5-(трифторметил)бензойная кислота 27g
Соединение 27f (480 мг; 1,3 ммоль) растворяли в тетрагидрофуране (5 мл) и воде (1 мл), затем добавляли гидроксида лития моногидрат (170 мг; 4,0 ммоль). В реакционном растворе осуществляли взаимодействие при комнатной температуре в течение ночи. Реакционный раствор концентрировали при пониженном давлении. Полученный остаток растворяли, добавляя небольшое количество воды и рН подводили до 4 разбавленной соляной кислотой. Полученный раствор экстрагировали этилацетатом (20 мл × 3), органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, получая указанное в заголовке соединение 27g (430 мг; выход: 93%).
Стадия 7
4-Хлор-2-(4-фтор-2-метилфенокси)-5-(трифторметил)бензоилхлорид 27h
Соединение 27g (200 мг; 0,57 ммоль) добавляли к тионилхлориду (2 мл) и в реакционном растворе осуществляли взаимодействие при 80°С в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении, получая неочищенное указанное в заголовке соединение 27h (210 мг), которое использовали непосредственно на следующей стадии.
Стадия 8
4-Хлор-N-(6-хлорпиридин-4-ил)-2-(4-фтор-2-метилфенокси)-5-(трифторметил)бензамид 27i
Соединение 27h (230 мг; 0,73 ммоль) растворяли в пиридине (3 мл), затем добавляли 4-амино-6-хлорпиридазин (95 мг; 0,73 ммоль) и 4-диметиламинопиридин (10 мг; 0,08 ммоль). В реакционном растворе осуществляли взаимодействие при комнатной температуре в течение ночи. Реакционный раствор концентрировали и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 27i (60 мг; выход: 20%).
Стадия 9
4-Хлор-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-5-(трифторметил)бензамид 27
Соединение 27i (60 мг; 0,13 ммоль) растворяли в уксусной кислоте (2 мл), затем добавляли ацетат калия (25 мг; 0,25 ммоль). В реакционном растворе осуществляли взаимодействие при 130°С в течение 4 часов и затем концентрировали при пониженном давлении. Полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2, система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 27 (14 мг; выход: 24%).
MS m/z (ESI): 442,1 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.18 (s, 1Н), 8.06 (d, 1Н), 7.52 (d, 1Н), 7.18-7.15 (m, 2Н), 7.09-7.06 (m, 1Н), 6.87 (s, 1Н), 2.21 (s, 3Н).
Пример 28
5-Хлор-2-(4-фтор-2-метилфенокси)-4-метил-N-(6-оксо-1,6-дигидропиридазин-4-ил)бензамид 28
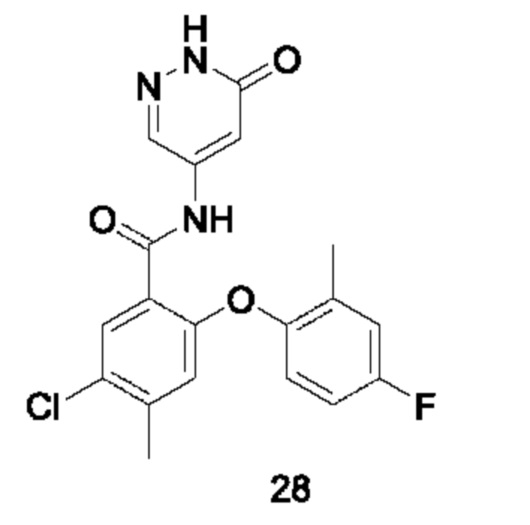
В соответствии с путем синтеза из примера 2 исходное соединение 5-хлор-2-фтор-4-(трифторметил)бензойную кислоту на стадии 2 заменяли на соединение 5-хлор-2-фтор-4-метилбензойную кислоту, получая таким образом указанное в заголовке соединение 28 (3 мг).
MS m/z (ESI): 388,2 [М+1].
1Н ЯМР (400 МГц, CD3OD): δ 8.03 (d, 1Н), 7.79 (s, 1Н), 7.48 (d, 1Н), 7.09-7.06 (m, 1H), 7.00-6.96 (s, 2Н), 6.72 (s, 1Н), 2.33 (s, 3Н), 2.22 (s, 3Н).
Пример 29
2-(4-Фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4,6-бис(трифторметил)бензамид 29
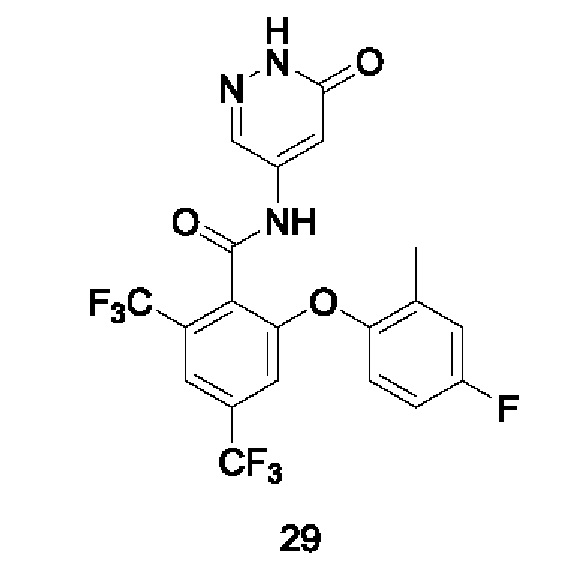
В соответствии с путем синтеза из примера 1 исходное соединение 5-хлор-2-фтор-4-(трифторметил)бензойную кислоту на стадии 1 заменяли на соединение 2-фтор-4,6-бис(трифторметил)бензойную кислоту, получая таким образом указанное в заголовке соединение 29 (2,4 мг).
MS m/z (ESI): 476,0 [М+1].
1Н ЯМР (400 МГц, CD3OD) δ 8.02 (s, 1Н), 7.82 (s, 1Н), 7.50 (s, 1Н), 7.00-7.20 (m, 4Н), 2.19 (s, 3Н).
Пример 30
5-Хлор-N-(6-оксо-1,6-дигидропиридазин-4-ил)-2-(о-толилокси)-4-(трифторметил)бензамид 30
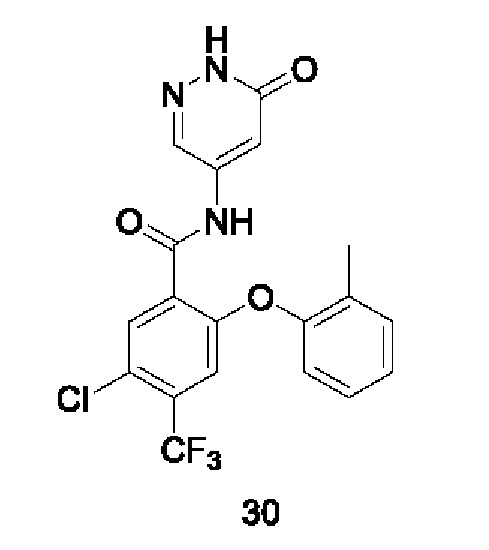
В соответствии с путем синтеза из примера 2 исходное соединение 2-метил-4-фторфенол на стадии 3 заменяли на 2-метилфенол, получая таким образом указанное в заголовке соединение 30 (75 мг).
MS m/z (ESI): 424,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.87 (s, 1Н), 11.08 (s, 1Н), 8.11 (s, 1Н), 7.88 (d, 1Н), 7.34-7.32 (m, 1Н), 7.27-7.23 (m, 1Н), 7.17-7.13 (m, 3Н), 7.02-7.00 (m, 1Н), 2.16 (s, 3Н).
Пример 31
5-Хлор-2-(2-циклопропил-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 31
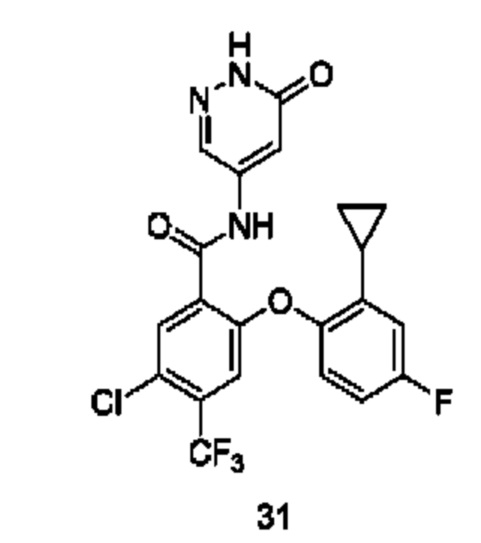
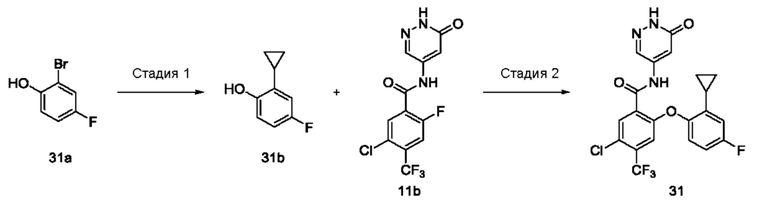
Стадия 1
2-Циклопропил-4-фторфенол 31b
2-Бром-4-фторфенол 31а (1,77 г; 9,26 ммоль; Shanghai Bide Pharmatech Ltd.), трифосфат калия (6,89 г; 32,46 ммоль), трициклогексилфосфин (260 мг; 0,93 ммоль) и циклопропилбороновую кислоту (1,20 г; 13,97 ммоль; Shanghai Bide Pharmatech Ltd.) добавляли к перемешиваемому раствору толуола (40 мл) и воды (2 мл). Полученный раствор трижды продували аргоном. Добавляли ацетат палладия (105 мг; 0,46 ммоль), реакционный раствор трижды продували аргоном и осуществляли взаимодействие при 100°С в течение ночи. Реакционный раствор охлаждали, затем добавляли этилацетат (50 мл) и промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая неочищенное указанное в заголовке соединение 31b (1,41 г).
MS m/z (ESI): 151,1 [М-1].
Стадия 2
5-Хлор-2-(2-циклопропил-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 31
Соединение 11b (1,03 г; 3,06 ммоль), соединение 31b (0,47 г; 3,08 ммоль) и карбонат цезия (1,01 г; 3,09 ммоль) добавляли к N-метилпирролидону (10 мл). В реакционном растворе осуществляли взаимодействие при 60°С в течение ночи. Реакционный раствор охлаждали, затем добавляли этилацетат (150 мл) и промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2, система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 31 (350 мг; выход: 24%).
MS m/z (ESI): 468,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.87 (s, 1H), 11.08 (s, 1Н), 8.10 (s, 1Н), 7.90 (d, 1Н), 7.21 (s, 1Н), 7.14 (dd, 1Н), 7.08-7.03 (m, 2Н), 6.87 (dd, 1Н), 2.01-1.94 (m, 1Н), 0.87-0.83 (m, 2Н), 0.71-0.67 (m, 2Н).
Пример 32
5-Хлор-2-(2-этокси-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 32
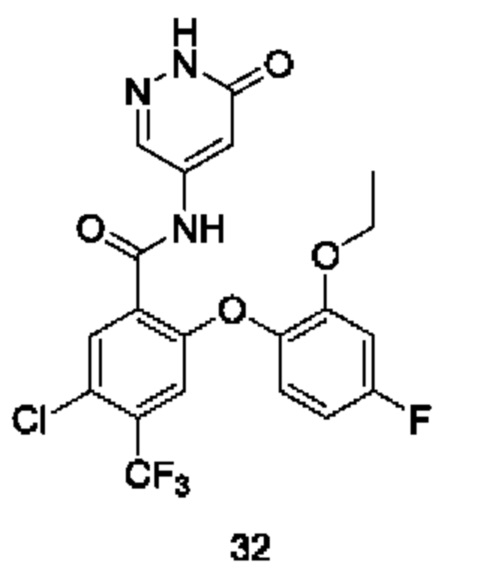
В соответствии с путем синтеза из примера 12 исходное соединение циклопропилбромид на стадии 1 заменяли на соединение иодэтан для осуществления взаимодействия при комнатной температуре, получая таким образом указанное в заголовке соединение 32 (200 мг).
MS m/z (ESI): 472,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.83 (s, 1Н), 10.98 (s, 1Н), 8.02 (s, 1Н),7.89 (s, 1Н), 7.00-7.28 (m,4H), 6.73-6.83 (m,1H), 3.99 (q, 2Н), 1.07 (t, 3Н).
Пример 33
5-Бром-2-(4-фтор-2-метилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 33
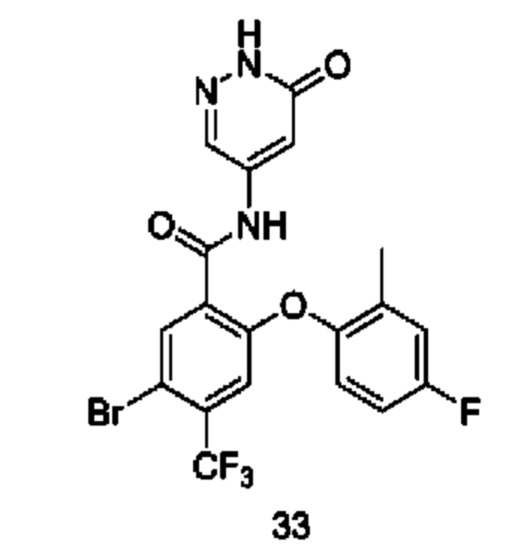
В соответствии с путем синтеза из примера 11 исходное соединение 5-хлор-2-фтор-4-(трифторметил)бензойную кислоту на стадии 1 заменяли на соединение 5-бром-2-фтор-4-(трифторметил)бензойную кислоту, получая таким образом указанное в заголовке соединение 33 (126 мг).
MS m/z (ESI): 486,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.84 (s, 1Н), 11.03 (s, 1Н), 8.19 (s, 1Н), 7.86-7.85 (d, 1Н), 7.21-7.16 (m, 2Н), 7.07-7.06 (d, 1Н), 2.13 (s, 3Н).
Пример 34
2-(4-Фтор-2-метилфенокси)-5-метил-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 34
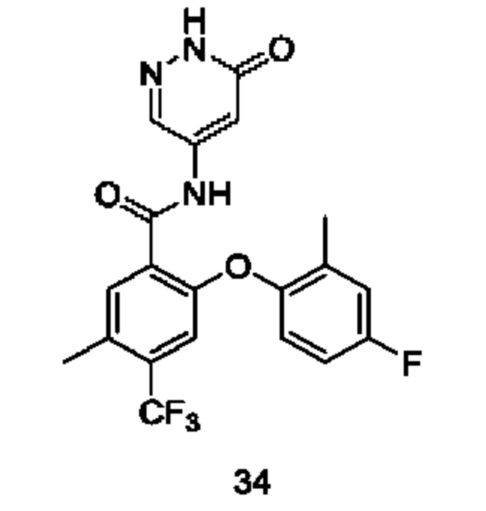
В соответствии с путем синтеза из примера 11 исходное соединение 5-хлор-2-фтор-4-(трифторметил)бензойную кислоту на стадии 1 заменяли на соединение 5-метил-2-фтор-4-(трифторметил)бензойную кислоту, а исходное соединение 2-метокси-4-фторфенол на стадии 3 заменяли на 2-метил-4-фторфенол, получая таким образом указанное в заголовке соединение 34 (15 мг).
MS m/z (ESI): 422,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.83 (s, 1Н), 11.97 (s, 1Н), 7.89-7.88 (d, 1Н), 7.74 (d, 1Н), 7.19-7.18 (m, 2Н), 7.08-7.04 (m, 1Н), 7.00-6.97 (m, 2Н), 2.44 (s, 3Н), 2.15 (s, 3Н).
Пример 35
5-Хлор-2-(4-фтор-2-(метокси-d3)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 35
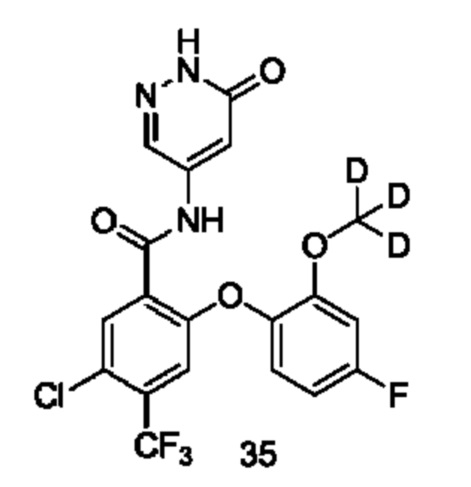
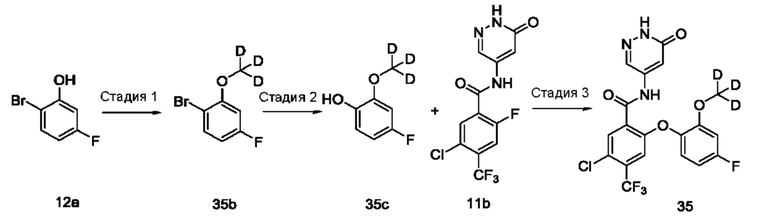
Стадия 1
1-Бром-4-фтор-2-(метокси-d3)бензол 35b
2-Бром-5-фторфенол 12а (1 г; 5,2 ммоль; Accela ChemBio (Shanghai) Inc.), дейтерированный метилиодид (911 мг; 6,3 ммоль; Sun Chemical Technology (Shanghai) Co., Ltd.) и карбонат калия (1,45 г; 10,5 ммоль) добавляли к N,N-диметилформамиду (10 мл). Реакционный раствор перемешивали для осуществления взаимодействия в течение 6 часов. Реакционный раствор охлаждали до комнатной температуры. Добавляли этилацетат (20 мл) и реакционный раствор промывали водой (20 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 35b (840 мг; выход: 71%).
1Н ЯМР (400 МГц, CDCl3): δ 7.49-7.45 (m, 1H), 6.66-6.57 (m, 2Н).
Стадия 2
4-Фтор-2-(метокси-d3)фенол 35с
Соединение 35b (840 мг; 4 ммоль) и триизопропилборат (987 мг; 5,25 ммоль; Shanghai Titan Scientific Co., Ltd.) добавляли к перемешиваемому раствору тетрагидрофурана/толуола (150 мл/30 мл). Воздух в реакционной колбе заменяли на аргон. Реакционный раствор охлаждали до -78°С, затем медленно по каплям в течение 20 минут добавляли н-бутиллитий (1,6 М раствор; 3,8 мл; 6,1 ммоль). Реакционный раствор оставляли нагреваться естественным образом до комнатной температуры и перемешивали в течение ночи. Реакционный раствор охлаждали до 0°С в ледяной бане. Добавляли метанол (50 мл) и перекись водорода (30 масс. %-ную; 10 мл) и по каплям добавляли 10%-ный раствор гидроксида натрия (40 мл). Реакционный раствор перемешивали при комнатной температуре в течение 1 часа. Медленно по каплям добавляли насыщенный раствор тиосульфата натрия (50 мл) и реакционный раствор экстрагировали этилацетатом (200 мл × 3). Органическую фазу промывали насыщенным раствором бикарбоната натрия (150 мл), сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 35с (570 мг; выход: 97%).
MS m/z (ESI): 144,0 [М-1].
1Н ЯМР (400 МГц, DMSO-d6): δ 8.89 (s, 1Н), 6.85-6.82 (m, 1Н), 6.76-6.72 (m, 1Н), 6.59-6.54 (m, 1Н).
Стадия 3
5-Хлор-2-(4-фтор-2-(метокси-d3)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 35
Соединение 11b (1 г; 2,98 ммоль), соединение 35с (433 мг; 2,98 ммоль) и карбонат цезия (1,02 г; 3,13 ммоль; Accela ChemBio (Shanghai) Inc.) добавляли к N-метилпирролидону (10 мл). В реакционном растворе осуществляли взаимодействие при 80°С в течение 3 часов и проводили охлаждение до комнатной температуры. Добавляли этилацетат (20 мл) и реакционный раствор промывали водой (10 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов В, получая указанное в заголовке соединение 35 (280 мг; выход: 20%).
MS m/z (ESI): 461,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.87 (s, 1H), 11.03 (s, 1Н), 8.06 (s, 1Н), 7.93 (d, 1Н), 7.29-7.23 (m, 2H), 7.16-7.13 (m, 1H), 7.01 (s, 1H), 6.88-6.83 (m, 1Н).
Пример 36
5-Хлор-2-(2-этил-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 36
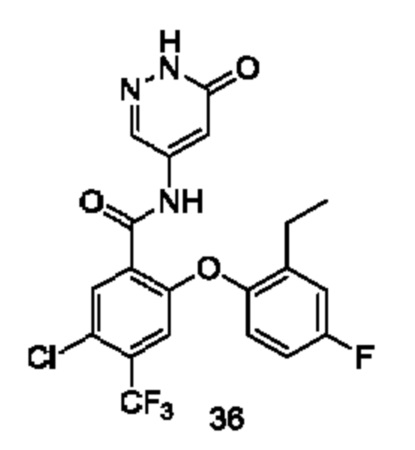
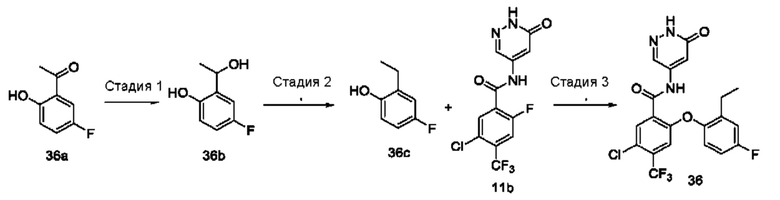
Стадия 1
4-Фтор-2-(1-гидроксиэтил)фенол 36b
Соединение 1-(5-фтор-2-гидроксифенил)этан-1-он 36а (3 г; 19,5 ммоль; Accela ChemBio (Shanghai) Inc.) растворяли в безводном метаноле (20 мл). Медленно порциями добавляли боргидрид натрия (1,1 г; 29,1 ммоль) и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении и добавляли этилацетат и воду. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая неочищенное указанное в заголовке соединение 36b (3,2 г; выход: 100%).
MS m/z (ESI): 155,1 [М-1].
Стадия 2
2-Этил-4-фторфенол 36с
Соединение 36b (1,5 г; 9,6 ммоль) растворяли в дихлорметане (15 мл), затем добавляли трифторуксусную кислоту (11 г; 96,5 ммоль; 7,2 мл). По каплям добавляли триэтилсилан (11,2 г; 96,3 ммоль; 15,4 мл) и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении и добавляли дихлорметан и воду. Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая неочищенное указанное в заголовке соединение 36с (1,38 г; выход: 100%).
MS m/z (ESI): 139,1 [М-1].
Стадия 3
5-Хлор-2-(2-этил-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 36
Соединение 11b (100 мг; 0,3 ммоль), соединение 36с (42 мг; 0,3 ммоль) и карбонат цезия (100 мг; 0,33 ммоль) добавляли к N-метилпирролидону (2 мл). В реакционном растворе осуществляли взаимодействие при 60°С в течение ночи. Реакционный раствор охлаждали, затем добавляли этилацетат (150 мл) и промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2, система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 36 (18 мг; выход: 13%).
MS m/z (ESI): 456,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.88 (s, 1Н), 11.09 (s, 1Н), 8.12 (s, 1H), 7.90-7.90 (d, 1H), 7.25-7.20 (m, 2H), 7.14 (m, 1H), 7.11-7.10 (m, 2H), 2.58-2.54 (m, 2H), 1.10-1.06 (m, 3H).
Пример 37
5-Хлор-2-(2-(дифторметокси)-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 37
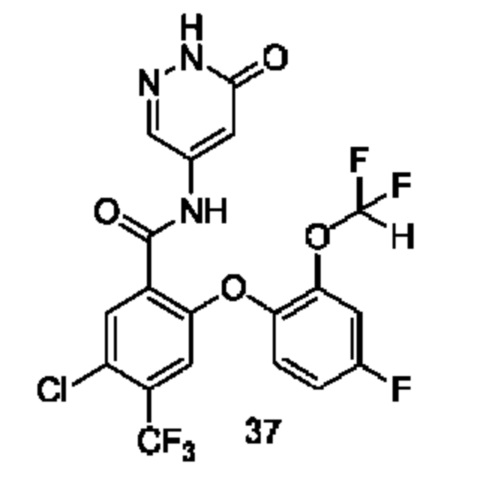
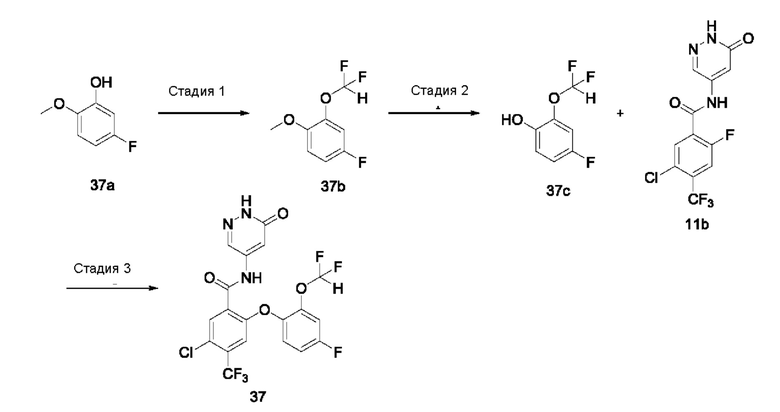
Стадия 1
2-(Дифторметокси)-4-фтор-1-метоксибензол 37b
5-Фтор-2-метоксифенол 37а (1 г; 7,03 ммоль; Accela ChemBio (Shanghai) Inc.), дифторхлорацетат натрия (2,68 г; 17,57 ммоль; Accela ChemBio (Shanghai) Inc.) и карбонат цезия (4,58 г; 14,05 ммоль) добавляли к смеси растворителей N,N-диметилформамид (14 мл)/вода (1,5 мл). В реакционном растворе осуществляли взаимодействие при 100°С в течение ночи. Реакционный раствор охлаждали, затем добавляли дихлорметан (100 мл) и промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении, получая неочищенное указанное в заголовке соединение 37b (1,35 г).
Стадия 2
2-(Дифторметокси)-4-фторфенол 37с
Иодид натрия (5,27 г; 35,15 ммоль; Shanghai Sinopharm Chemical Reagent Co., Ltd.) растворяли в ацетонитриле (20 мл), затем добавляли триметилхлорсилан (3,82 г; 35,16 ммоль; Shanghai Sinopharm Chemical Reagent Co., Ltd.). В реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 20 минут. Добавляли соединение 37b (1,35 г; 7,0263 ммоль) и в реакционном растворе осуществляли взаимодействие при 80°С в течение ночи. Реакционный раствор охлаждали, затем добавляли воду (50 мл) и экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 37 с (540 мг; выход: 43%).
MS m/z (ESI): 177,1 [М-1].
Стадия 3
5-Хлор-2-(2-(дифторметокси)-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 37
Соединение 11b (1,02 г; 3,03 ммоль), соединение 37с (0,54 г; 3,03 ммоль) и карбонат цезия (0,99 г; 3,13 ммоль) добавляли к N-метилпирролидону (10 мл). В реакционном растворе осуществляли взаимодействие при 60°С в течение ночи. Реакционный раствор охлаждали, затем добавляли этилацетат (150 мл) и промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2; система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 37 (450 мг; выход: 30%).
MS m/z (ESI): 493,9 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.86 (s, 1Н), 11.0 6 (s, 1Н), 8.11 (s, 1Н), 7.89 (d, 1Н), 7.38 (dd, 1Н), 7.33 (dd, 1Н),7.27 (s, 1Н), 7.23 (t, 1Н), 7.22-7.17 (m, 2Н).
Пример 38
5-Хлор-2-(4-фтор-2-гидроксифенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 38
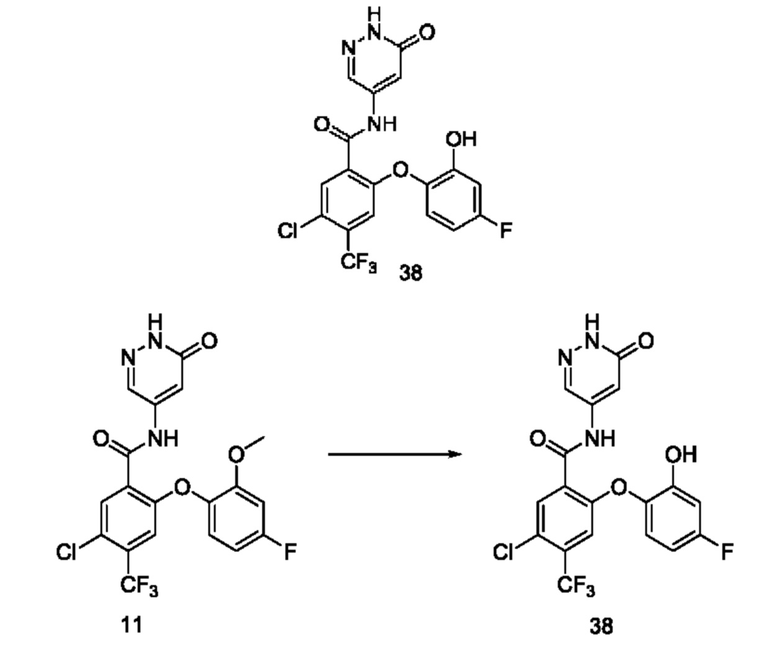
Соединение 11 (100 мг; 0,22 ммоль) добавляли к дихлорметану (2 мл). Полученный раствор охлаждали до -78°С и по каплям добавляли трибромид бора (1 М раствор; 1,09 мл; Adamas Beta (Shanghai) Reagent Co., Ltd.). Реакционный раствор нагревали до 0°С и осуществляли взаимодействие в течение 3 часов. Добавляли дополнительное количество трибромида бора (1 М раствора; 1,09 мл) при 0°С и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение ночи. Реакционный раствор концентрировали и к полученному остатку по каплям добавляли 10 мл метанола. Полученный раствор хорошо перемешивали и концентрировали. Полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2; система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 38 (50 мг; выход: 51%).
MS m/z (ESI): 444,1 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.88 (s, 1Н), 10.99 (s, 1Н), 10.47 (s, 1Н), 8.04 (s, 1H), 7.94 (s, 1H), 7.26-7.22 (m, 2Н), 6.97 (s, 1H), 6.80 (dd, 1H), 6.75-6.70 (m, 1Н).
Пример 39
5-Хлор-2-((7-фтор-2,3-дигидро-1Н-инден-4-ил)окси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 39
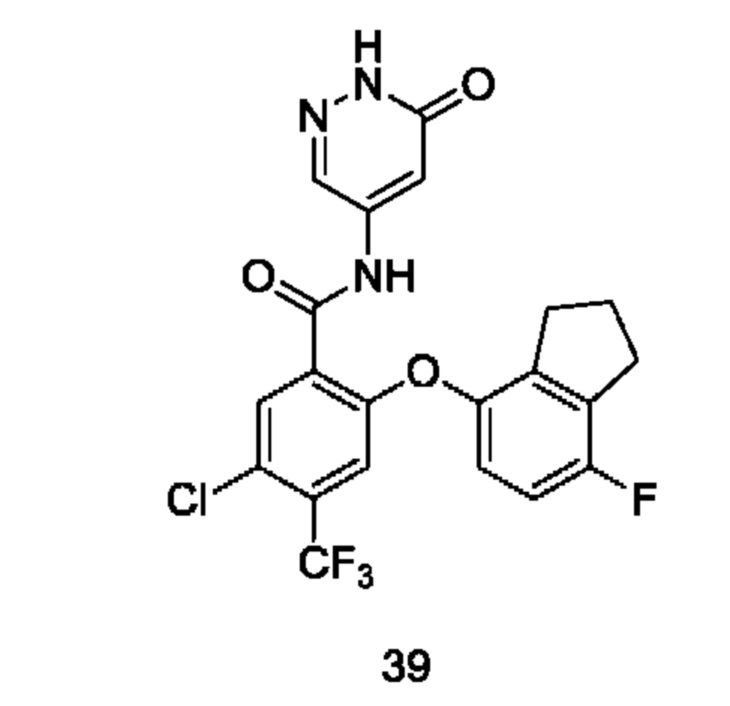
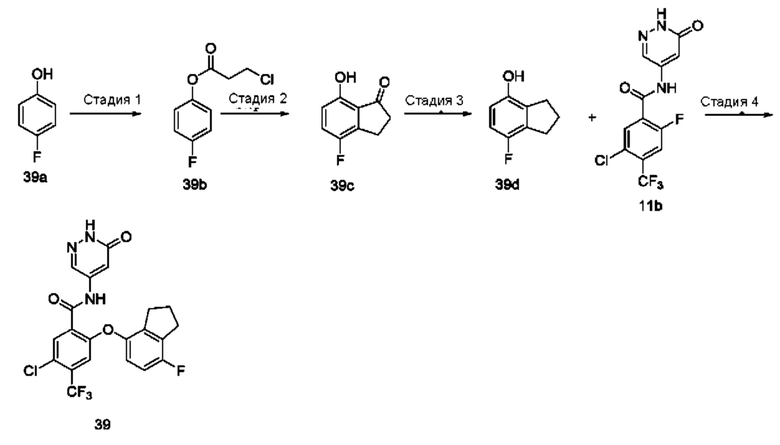
Стадия 1
4-Фторфенил-3-хлорпропаноат 39b
Исходное соединение 4-фторфенол 39а (15 г; 133,8 ммоль; Accela ChemBio (Shanghai) Inc.) растворяли в дихлорметане (80 мл), затем добавляли пиридин (12 г; 151,7 ммоль). Медленно по каплям добавляли 3-хлорпропионилхлорид (18,9 г; 134 ммоль; Accela ChemBio (Shanghai) Inc.) в ледяной бане. По завершении добавления реакционный раствор перемешивали при комнатной температуре в течение 1 часа. Добавляли насыщенный раствор бикарбоната натрия (60 мл) и реакционный раствор экстрагировали этилацетатом (80 мл × 2). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 39b (25 г; выход: 86%).
MS m/z (ESI): 203,1 [М+1].
Стадия 2
4-Фтор-7-гидрокси-2,3-дигидро-1H-инден-1-он 39с
Соединение 39b (10 г; 49,4 ммоль) и трихлорид алюминия (20 г; 150 ммоль; Sinopharm Chemical Reagent Co., Ltd.) хорошо смешивали, реакционную смесь нагревали до 100°С и перемешивали в течение 15 минут. Реакционную смесь нагревали до 180°С и осуществляли взаимодействие в течение 3 часов. Реакционную смесь охлаждали до комнатной температуры и медленно добавляли в ледяную воду. Добавляли этилацетат и полученный раствор перемешивали в течение 2 часов. Органическую фазу сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 39 с (3 г; выход: 37%).
MS m/z (ESI): 167,3 [М+1].
Стадия 3
7-Фтор-2,3-дигидро-1Н-инден-4-ол 39d
Соединение 39с (8 г; 48,2 ммоль) растворяли в трифторуксусной кислоте (80 мл), затем добавляли триэтилсилан (14 г; 120,4 ммоль). Реакционный раствор перемешивали при 80°С в течение ночи. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 39d (6 г; выход: 82%).
MS m/z (ESI): 151,3 [М-1].
Стадия 4
5-Хлор-2-((7-фтор-2,3-дигидро-1Н-инден-4-ил)окси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 39
Соединение 11b (1,02 г; 3,03 ммоль), соединение 39d (0,54 г; 3,03 ммоль) и карбонат цезия (0,99 г; 3,13 ммоль) добавляли к N-метилпирролидону (10 мл). В реакционном растворе осуществляли взаимодействие при 60°С в течение ночи. Реакционный раствор охлаждали, затем добавляли этилацетат (150 мл) и промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2; система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 39 (400 мг; выход: 29%).
MS m/z (ESI): 468,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.86 (s, 1Н), 11.03 (s, 1Н), 8.08 (s, 1Н), 7.874-7.868 (d, 1Н), 7.27 (m,1H), 7.15 (m, 1Н), 7.04-7.00 (m, 1Н), 6.92-6.89 (m, 1H), 2.92-2.88 (m, 2Н), 2.77-2.73 (m, 2Н), 2.05-1.99 (m, 2Н).
Пример 40
5-Хлор-2-(2-(циклопентилокси)-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 40
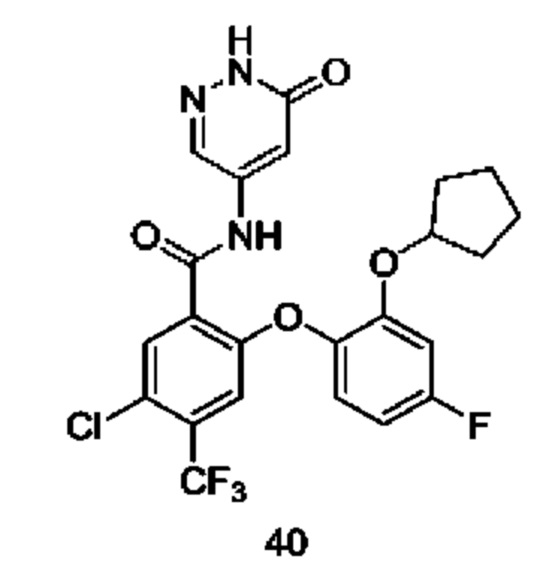
В соответствии с путем синтеза из примера 35 исходное соединение дейтерированный метилиодид на стадии 1 заменяли на соединение бромциклопентан, получая таким образом указанное в заголовке соединение 40 (30 мг).
MS m/z (ESI): 511,9 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.87 (s, 1Н), 10.99 (s, 1Н), 8.05 (s, 1Н),7.95 (s, 1Н), 7.30-7.35 (m, 1Н), 7.29 (s, 1Н), 7.11 (dd, 1Н), 6.98 (s, 1Н), 6.77-6.86 (m, 1Н), 4.80-4.90 (m, 1Н), 1.72-1.85 (m, 2Н), 1.35-1.55 (m, 4Н), 1.15-1.35 (m, 2Н).
Пример 41
5-Хлор-2-(2-циклобутокси-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 41
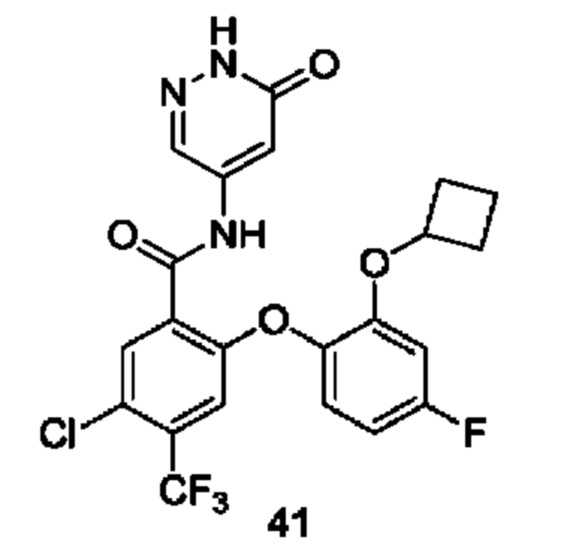
В соответствии с путем синтеза из примера 35 исходное соединение дейтерированный метилиодид на стадии 1 заменяли на соединение бромциклобутан, получая таким образом указанное в заголовке соединение 41 (110 мг).
MS m/z (ESI): 498,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.87 (s, 1H), 11.02 (s, 1Н), 8.06 (s, 1Н), 7.94 (s, 1Н), 7.29-7.23 (m, 2Н), 7.09 (s, 1Н), 6.95-6.92 (m, 1Н), 6.92-6.83 (m, 1Н), 4.73-4.70 (m, 1H), 2.37-2.33 (m, 2H), 1.79-1.72 (m, 2H), 1.68-1.52 (m, 2H).
Пример 42
5-Хлор-2-(4-фтор-2-изопропилфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 42
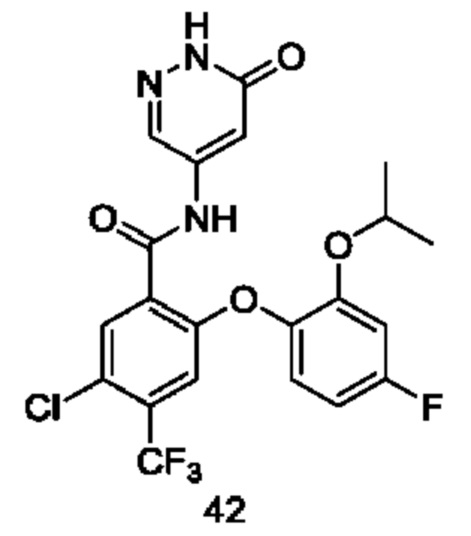
В соответствии с путем синтеза из примера 35 исходное соединение дейтерированный метилиодид на стадии 1 заменяли на соединение иодизопропан, получая таким образом указанное в заголовке соединение 42 (100 мг).
MS m/z (ESI): 485,9 [М+1].
1Н ЯМР (400 МГц, DMSO-d6): δ 12.87 (s, 1Н), 11.00 (s, 1Н), 8.05 (s, 1Н), 7.95 (s, 1Н), 7.32-7.16 (m, 3Н), 7.01 (s, 1Н), 6.86-6.82 (m, 1Н), 4.67-4.62 (m, 1Н), 1.08 (s, 3Н), 1.06 (s, 3Н).
Пример 43
2-(2-Бром-4-фторфенокси)-5-хлор-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 43
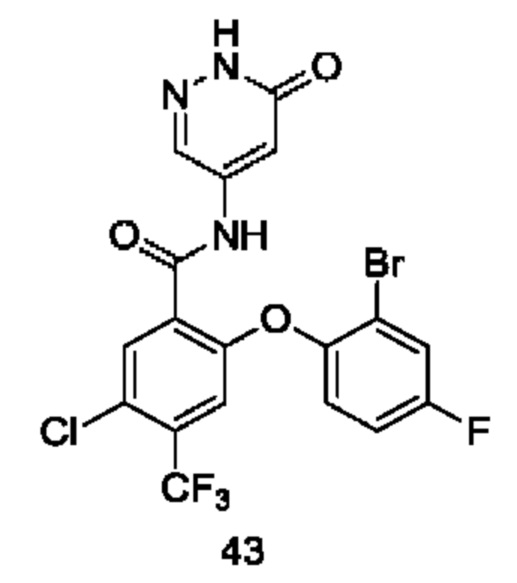
В соответствии с путем синтеза из примера 11 исходное соединение 2-метокси-4-фторфенол на стадии 3 заменяли на 2-бром-4-фторфенол, получая таким образом указанное в заголовке соединение 43 (100 мг).
MS m/z (ESI): 505,7 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.86 (s, 1Н), 11.08 (s, 1Н), 8.14 (s, 1Н),7.90 (d, 1Н), 7.75 (dd, 1Н), 7.36-7.26 (m, 3Н), 7.17 (s, 1Н).
Пример 44
5-Хлор-2-(4-фтор-2-(метил-с(з)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 44
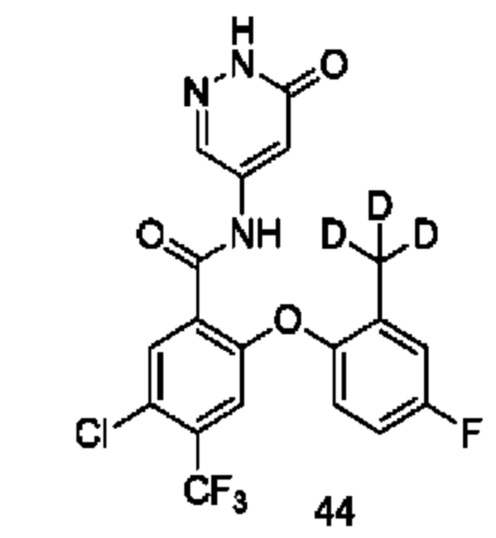
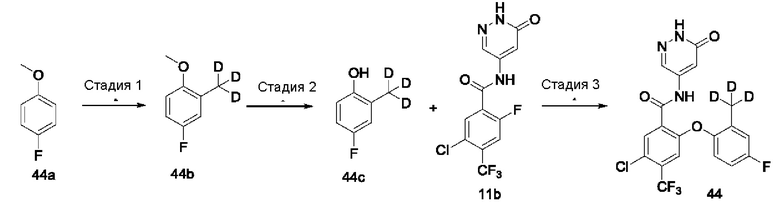
Стадия 1
4-Фтор-1-метокси-2-(метил-d3)бензол 44b
Соединение 1-фтор-4-метоксибензол (5 г; 39,6 ммоль; Accela ChemBio (Shanghai) Inc.) растворяли в тетрагидрофуране (50 мл). Полученный раствор охлаждали до -10°С и по каплям добавляли н-бутиллитий (2,5 М раствор; 43,7 ммоль; 17,5 мл). Реакционный раствор оставляли нагреваться естественным образом до комнатной температуры и осуществляли взаимодействие в течение 1 часа. Медленно по каплям добавляли иодметан-d3 (5,8 г; 40 ммоль; Accela ChemBio (Shanghai) Inc.) в ледяной бане и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 2 часов. Медленно добавляли воду (50 мл) и реакционный раствор экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 44b (1,5 г; выход: 26%).
1Н ЯМР (400 МГц, CDCl3) δ 6.86-6.85 (m, 2Н), 6.73-6.69 (m, 1H), 3.78 (s, 3Н).
Стадия 2
4-Фтор-2-(метил-d3)фенол 44с
Соединение 44а (1,5 г; 10,5 ммоль) добавляли к дихлорметану (30 мл). По каплям добавляли раствор трибромида бора в дихлорметане (1 М раствор; 21,2 ммоль; 21,2 мл) при комнатной температуре и в реакционном растворе осуществляли взаимодействие при комнатной температуре в течение 1 часа. Реакционный раствор концентрировали при пониженном давлении и полученный остаток очищали хроматографией на колонке с силикагелем с использованием системы элюентов А, получая указанное в заголовке соединение 44 с (900 мг; выход: 67%).
Стадия 3
5-Хлор-2-(4-фтор-2-(метил-d3)фенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 44
Соединение 11b (100 мг; 0,3 ммоль), соединение 44с (39 мг; 0,3 ммоль) и карбонат цезия (100 мг; 0,33 ммоль) добавляли к N-метилпирролидону (2 мл). В реакционном растворе осуществляли взаимодействие при 60°С в течение ночи. Реакционный раствор охлаждали, затем добавляли этилацетат (150 мл) и промывали водой (50 мл × 3). Органическую фазу сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении и полученный остаток очищали препаративной высокоэффективной жидкостной хроматографией (Waters 2767-SQ Detecor2; система элюентов: бикарбонат аммония, вода, ацетонитрил), получая указанное в заголовке соединение 44 (21 мг; выход: 16%).
MS m/z (ESI): 445,0 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 8.09 (s, 1Н), 7.89-7.88 (d, 1Н), 7.23-7.21 (d, 1Н), 7.18-7.18 (dd, 1H),7.13(m, 1Н), 7.10-7.08 (m, 2Н).
Пример 45
5-Хлор-2-(2-хлор-4-фторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 45
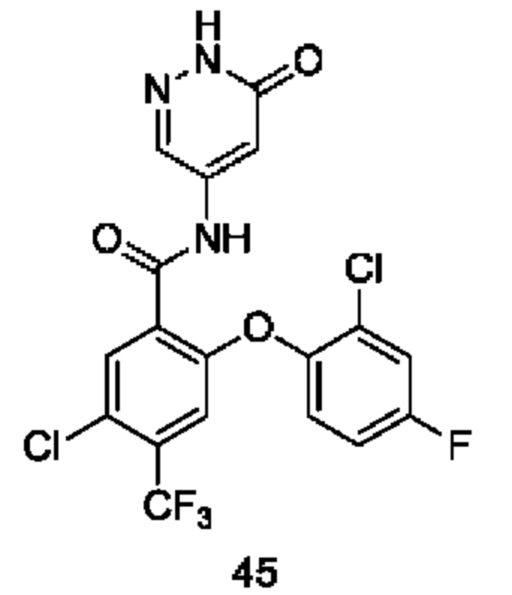
В соответствии с путем синтеза из примера 11 исходное соединение 2-метокси-4-фторфенол на стадии 3 заменяли на 2-хлор-4-фторфенол, получая таким образом указанное в заголовке соединение 45 (40 мг).
MS m/z (ESI): 461,8 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.87 (s, 1Н), 11.08 (s, 1Н), 8.14 (s, 1Н), 7.89 (d, 1Н), 7.64 (dd, 1Н), 7.33-7.26 (m, 3Н), 7.17 (s, 1Н).
Пример 46
5-Хлор-2-(2,4-дифторфенокси)-N-(6-оксо-1,6-дигидропиридазин-4-ил)-4-(трифторметил)бензамид 46
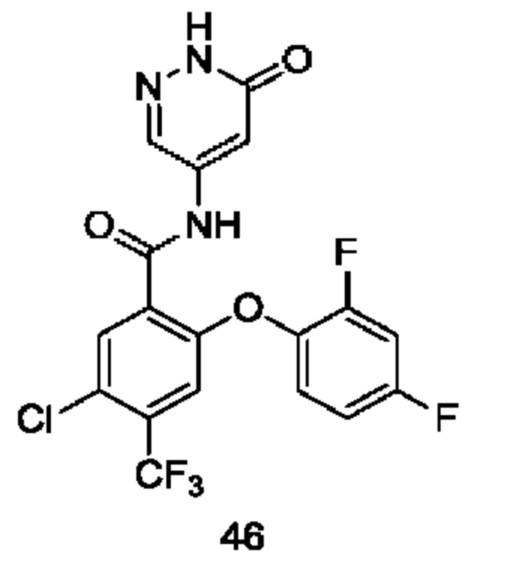
В соответствии с путем синтеза из примера 11 исходное соединение 2-метокси-4-фторфенол на стадии 3 заменяли на 2,4-дифторфенол, получая таким образом указанное в заголовке соединение 46 (70 мг).
MS m/z (ESI): 445,8 [М+1].
1Н ЯМР (400 МГц, DMSO-d6) δ 12.87 (s, 1Н), 11.10 (s, 1Н), 8.12 (s, 1Н), 7.89 (d, 1Н), 7.53-7.47 (m, 1Н), 7.38-7.32 (m, 2Н), 7.18-7.12 (m, 2Н).
Биологическое исследование
Далее настоящее изобретение будет описано со ссылкой на приведенные ниже примеры тестирования, но эти примеры не следует рассматривать как ограничивающие объем настоящего изобретения.
Пример тестирования 1. Определение ингибирующей активности соединений по настоящему изобретению в отношении NaV1.8
Цель данного эксперимента заключается в исследовании влияния соединений на ионный канал NaV1.8 в эксперименте in vitro, при этом ионный канал NaV1.8 стабильно экспрессируется в клетках почки эмбриона человека, линии 293 (HEK293). После установления стабильного тока через NaV1.8 проводят сравнение токов через NaV1.8 до и после введения соединения, чтобы оценить влияние данного соединения на ионный канал NaV1.8.
1. Материалы и приборы для проведения эксперимента
1) Усилитель для пэтч-кламп: РС-505 В (WARNER lnstruments)/MultiClamp 700A(Axon Instruments) для пэтч-кламп.
2) Цифро-аналоговый преобразователь: Digidata 1440А (Axon CNS)/Digidata 1550A(Axon Instruments).
3) Микроманипулятор: MP-225 (SUTTER Instrument).
4) Инвертированный микроскоп: TL4 (Olympus).
5) Приспособление для вытягивания стеклянных микроэлектродов: РС-10 (NARISHIGE).
6) Микроэлектродный стеклянный капилляр: B12024F (Wuhan Weitan Scientific Instrument Co., Ltd.).
7) Диметилсульфоксид (DMSO): D2650 (Sigma-Aldrich).
8) TTX: AF3014 (Affix Scientific).
2. Экспериментальные методики
2.1. Подготовка соединений
За исключением NaOH и КОН, применяемых для проведения кислотного титрования и щелочного титрования, все соединения, использованные для подготовки внеклеточной жидкости и внутриклеточной жидкости, приобретали у Sigma (St. Louis, МО). Внеклеточная жидкость (мМ): NaCl, 137; KCl, 4; CaCl2, 1,8; MgCl2, 1; HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота), 10; глюкоза, 10; рН 7,4 (NaOH для титрования). Внутриклеточная жидкость (мМ): аспарагиновая кислота, 140; MgCl2, 2; EGTA (этиленгликольтетрауксусная кислота), 11; HEPES, 10; рН 7,2 (CsOH для титрования). Все растворы с тестируемым соединением и контрольным соединением содержали 1 мкМ ТТХ.
Тестируемое соединение растворяли в диметилсульфоксиде (DMSO) в исходной концентрации 9 мМ. Этот исходный раствор тестируемого соединения растворяли во внеклеточной жидкости в необходимой концентрации в день проведения тестирования.
2.2. Метод тестирования с применением ручного варианта метода пэтч-кламп
1) Соединение готовили в виде растворов заданных концентраций, растворы добавляли в трубопроводы в порядке, соответствующем от низкой к высокой концентрации, и трубопроводы маркировали.
2) Клетку переносили в камеру для перфузии. К электроду прикладывали положительное давление. Кончик электрода касался клетки. Трехходовой кран воздуховыпускного устройства переводили в трехходовое положение. К электроду прикладывали отрицательное давление, в результате чего между электродом и клеткой образовывался герметичный контакт с высоким сопротивлением. Отрицательное давление прикладывали непрерывно, что приводило к разрыву клеточной мембраны и формированию цепи тока.
3) После того, как ток через оторванный участок клеточной мембраны стабилизировался, поочередно осуществляли перфузию различными концентрациями. Перфузию следующей концентрацией осуществляли после того, как ток оставался стабильным в течение по меньшей мере одной минуты. Продолжительность перфузии в каждой концентрации не превышала пяти минут.
4) Камеру для перфузии освобождали. Камеру для перфузии промывали растворами лекарственного средства в порядке от высокой к низкой концентрации, и продолжительность промывки растворами лекарственного средства в каждой концентрации составляла 20 секунд. В конце, камеру для перфузии промывали внеклеточной жидкостью в течение 1 минуты.
2.3. Выравнивание тестируемой разности потенциалов (в состоянии покоя) и результаты
Клетку фиксировали при -80 мВ. Чтобы получить ток NaV1.8, проводили деполяризацию клетки до 10 мВ, используя прямоугольный импульс длительностью 10 миллисекунд. Эту методику воспроизводили каждые 5 секунд. Измеряли максимальный ток, вызываемый прямоугольным импульсом. После того, как ток стабилизировался, осуществляли перфузию тестируемым соединением. Когда ответ становился стабильным, рассчитывали интенсивность блокирования канала.
3. Анализ данных
Данные записывали в компьютерную систему для анализа. Сбор и анализ данных осуществляли с использованием pCLAMP 10 (Molecular Devices, Union City, CA), и результаты анализа просматривались администратором. Термин "стабильный ток" означает, что ток со временем изменяется в ограниченном диапазоне. Величину стабильного тока использовали для расчета влияния соединения в определенной концентрации.
Ингибирующую активность соединений по настоящему изобретению в отношении NaV1.8 определяли с использованием описанного выше теста, и полученные значения IC50 показаны в Таблице 1.
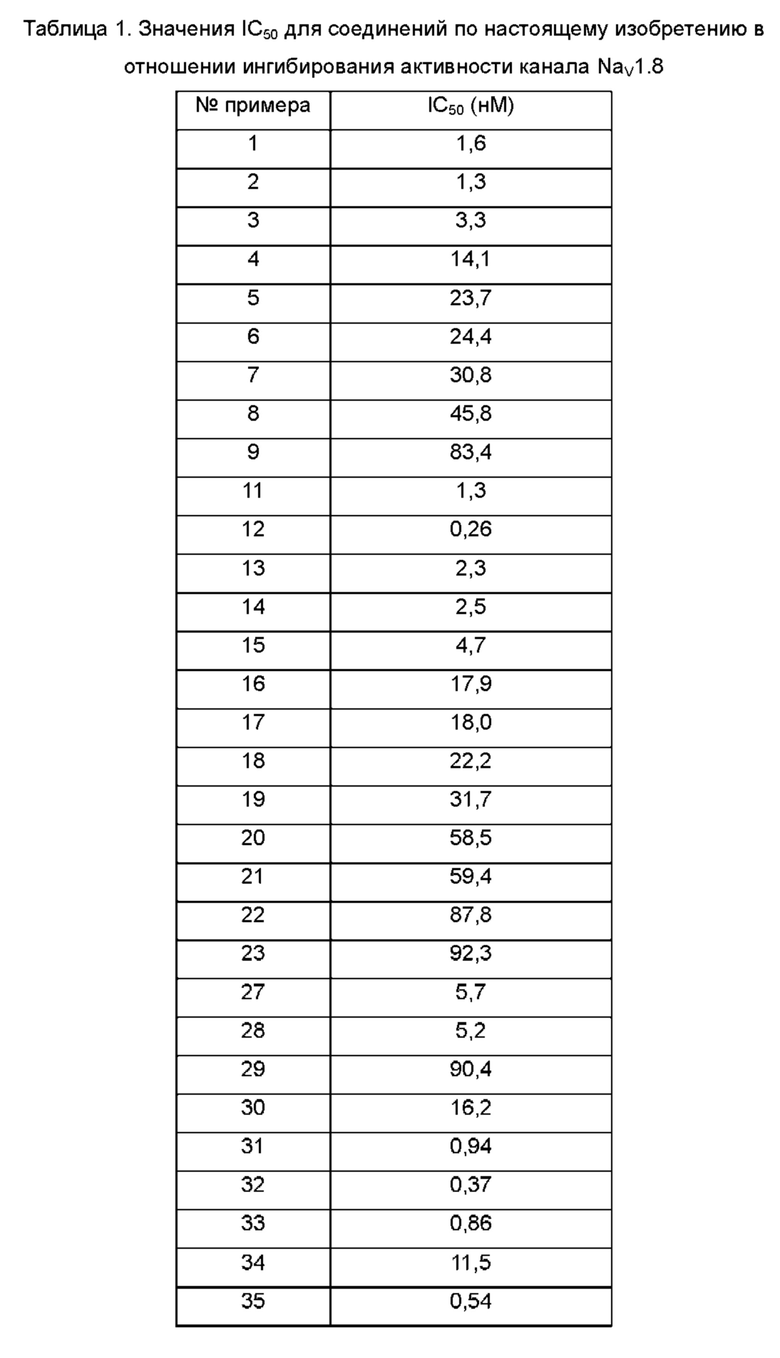
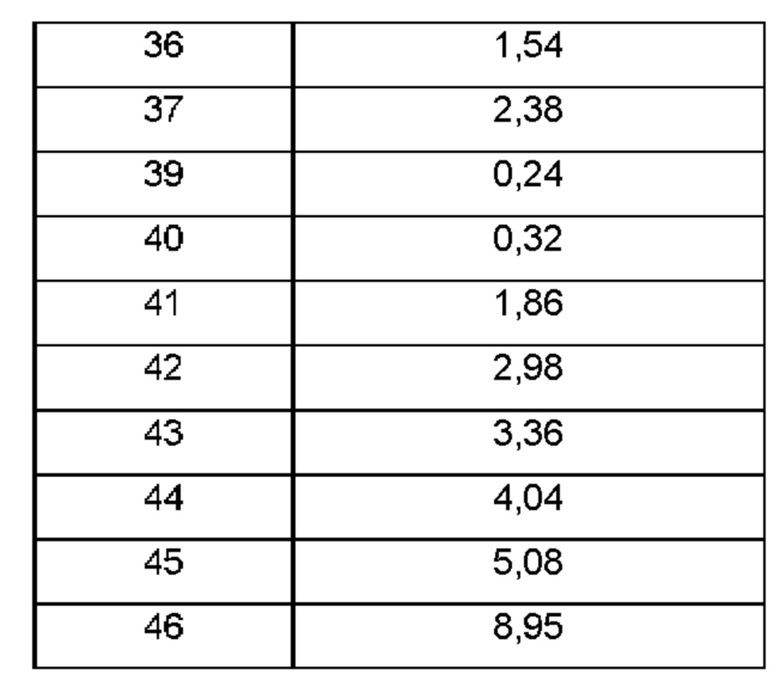
Вывод: соединения по настоящему изобретению обладают значительным ингибирующим влиянием на активность канала NaV1.8.
Оценка фармакокинетических параметров
Пример тестирования 2. Анализ фармакокинетических параметров соединений по настоящему изобретению
1. Краткое описание
В качестве тестируемых животных использовали крыс. Концентрацию лекарственных средств в плазме крови в разные моменты времени определяли методом жидкостной хроматографии в сочетании с тандемной масс-спектрометрией (LC/MS/MS) после внутрижелудочного введения крысам соединений из примера 2, примера 11, примера 12, примера 15, примера 31 и примера 33. Фармакокинетические характеристики соединений по настоящему изобретению изучали и оценивали на крысах.
2. Протокол тестирования
2.1. Тестируемые соединения
Соединения из примера 2, примера 11, примера 12, примера 15, примера 31 и примера 33.
2.2. Тестируемые животные
Двадцать четыре здоровых взрослых крысы Sprague Dawley (SD) (половина самцов и половина самок, поровну разделенных на шесть групп по четыре крысы в каждой группе) приобретали у Shanghai Jiesijie Laboratory Animal Co., LTD.
2.3. Подготовка тестируемого соединения
Взвешивали определенное количество тестируемого соединения, к нему добавляли смесь из 5% DMSO, 5% твина 80 и 90% физиологического раствора, получая бесцветный, чистый и прозрачный раствор 0,2 мг/мл.
2.4. Введение
После голодания в течение ночи крысам SD внутрижелудочно вводили тестируемое соединение в дозе 2,0 мг/кг и в объеме введения из расчета 10,0 мл/кг.
3. Процедура
Крысам внутрижелудочно вводили соединения из примера 2, примера 11, примера 12, примера 15, примера 31 и примера 33. Из глазницы отбирали по 0,2 мл крови в моменты времени до введения и через 0,5; 1,0; 2,0; 4,0; 6,0; 8,0; 11,0 и 24,0 часа после введения. Образцы помещали в гепаринизированные пробирки и центрифугировали в течение 10 минут при 10000 об/мин и 4°С для отделения плазмы крови. Образцы плазмы крови хранили при -20°С. Крыс кормили через 2 часа после введения.
Определяли содержание тестируемого соединения в плазме крови крыс после внутрижелудочного введения тестируемого соединения в разных концентрациях: отбирали по 25 мкл плазмы крови крыс в каждый момент времени после введения, куда добавляли по 30 мкл раствора внутреннего стандарта и по 175 мкл ацетонитрила. Полученный раствор перемешивали вихревым способом в течение 5 минут и центрифугировали в течение 10 минут (3700 об/мин). Из образцов плазмы крови отбирали по 0,5 мкл супернатанта для проведения анализа посредством LC/MS/MS.
4. Результаты измерения фармакокинетических параметров Фармакокинетические параметры для соединений по настоящему
изобретению приведены ниже.
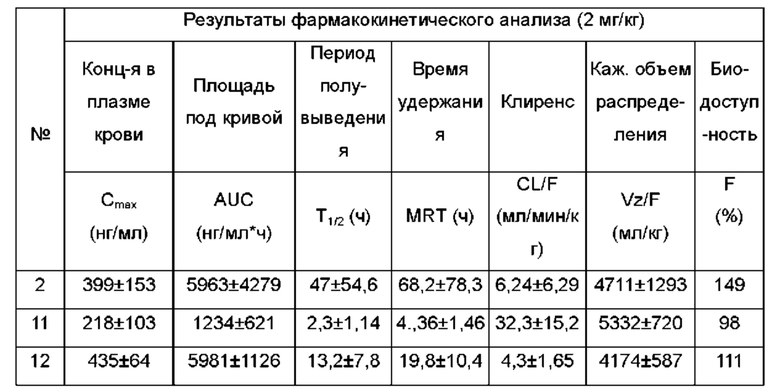
Вывод: соединение по настоящему изобретению хорошо всасывается и обладает значительным фармакокинетическим преимуществом.
Группа изобретений относится к фармацевтической химии и включает производное 6-оксо-1,6-дигидропиридазина формулы (I), промежуточные соединения формул (IA) и (IB) для получения соединения формулы (I), способы получения соединения формулы (I), фармацевтическую композицию, содержащую соединение формулы (I), и применение соединения формулы (I). В формуле (I) М выбран из группы, состоящей из атома О, CН2 и S; кольцо А представляет собой 6-10-членный арил или  ; все R1 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси, галоген-С1-6 алкила и галоген-С1-6 алкокси; все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси, дейтерированного С1-6 алкила, дейтерированного С1-6 алкокси, галоген-С1-6 алкила, галоген-С1-6 алкокси, гидрокси, С3-6 циклоалкила и С3-6 циклоалкилокси; все R3 представляют собой атом водорода; n и s равны 0, 1, 2, 3 или 4; и t равно 0, 1 или 2. Технический результат - производное 6-оксо-1,6-дигидропиридазина формулы (I), обладающее свойствами ингибитора потенциал-зависимых натриевых каналов (NaV). 8 н. и 10 з.п. ф-лы, 4 табл., 48 пр.
; все R1 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси, галоген-С1-6 алкила и галоген-С1-6 алкокси; все R2 являются одинаковыми или разными, и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси, дейтерированного С1-6 алкила, дейтерированного С1-6 алкокси, галоген-С1-6 алкила, галоген-С1-6 алкокси, гидрокси, С3-6 циклоалкила и С3-6 циклоалкилокси; все R3 представляют собой атом водорода; n и s равны 0, 1, 2, 3 или 4; и t равно 0, 1 или 2. Технический результат - производное 6-оксо-1,6-дигидропиридазина формулы (I), обладающее свойствами ингибитора потенциал-зависимых натриевых каналов (NaV). 8 н. и 10 з.п. ф-лы, 4 табл., 48 пр.
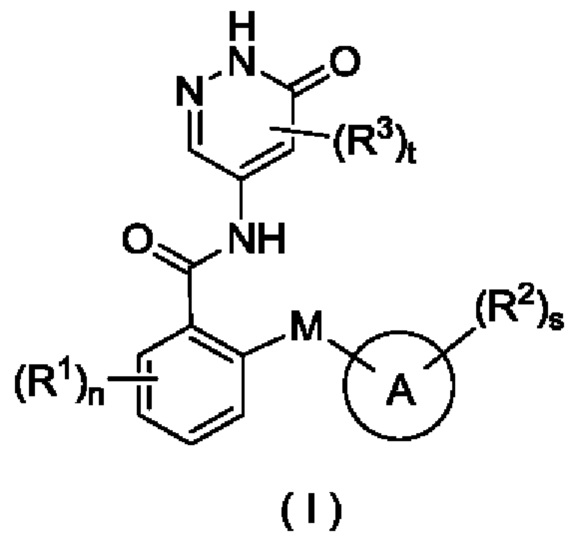 ,
,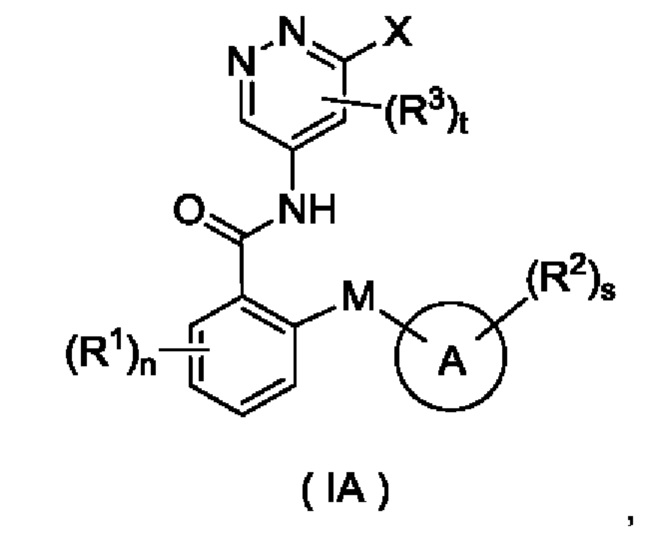
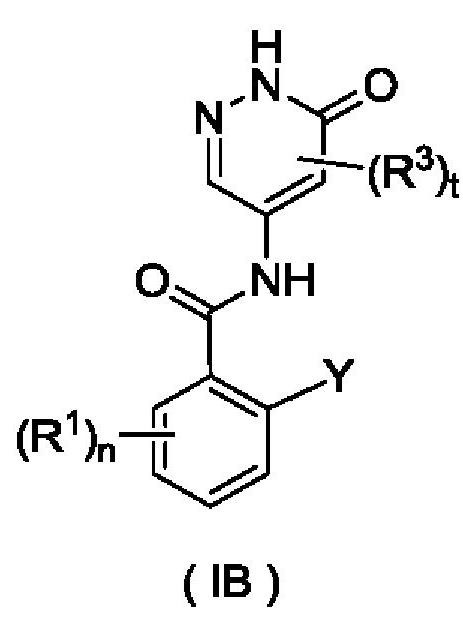
1. Соединение формулы (I) или его рацемат либо их фармацевтически приемлемая соль:
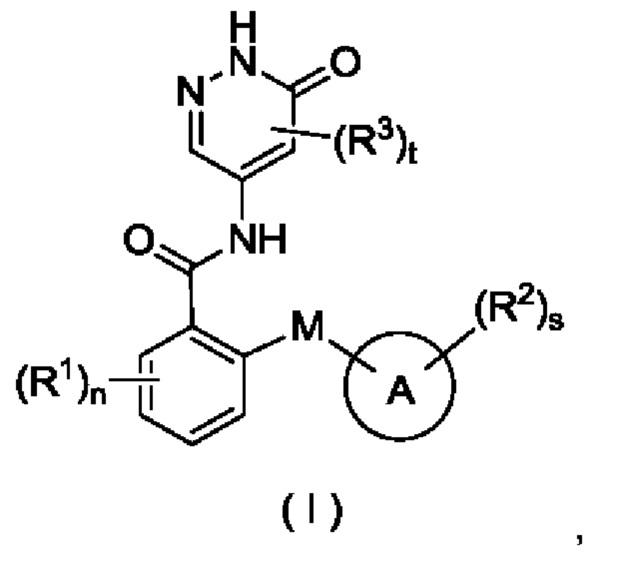
где
М выбран из группы, состоящей из атома О, CН2 и атома S;
кольцо А представляет собой 6-10-членный арил или  ;
;
все R1 являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси, галоген-С1-6 алкила и галоген-С1-6 алкокси;
все R2 являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси, дейтерированного С1-6 алкила, дейтерированного С1-6 алкокси, галоген-С1-6 алкила, галоген-С1-6 алкокси, гидрокси, С3-6 циклоалкила и С3-6 циклоалкилокси;
все R3 представляют собой атом водорода;
n равно 0, 1, 2, 3 или 4;
s равно 0, 1, 2, 3 или 4; и
t равно 0, 1 или 2.
2. Соединение формулы (I) или его рацемат либо их фармацевтически приемлемая соль по п. 1, представляющее собой соединение формулы (II) или его рацемат либо их фармацевтически приемлемую соль:
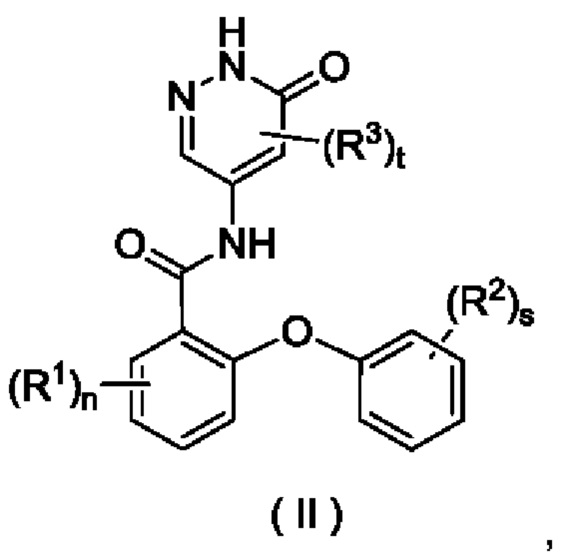
где R1, R2, R3, n, s и t являются такими, как определено в п. 1.
3. Соединение формулы (I) или его рацемат либо их фармацевтически приемлемая соль по п. 1 или 2, представляющее собой соединение формулы (III) или его рацемат либо их фармацевтически приемлемую соль:
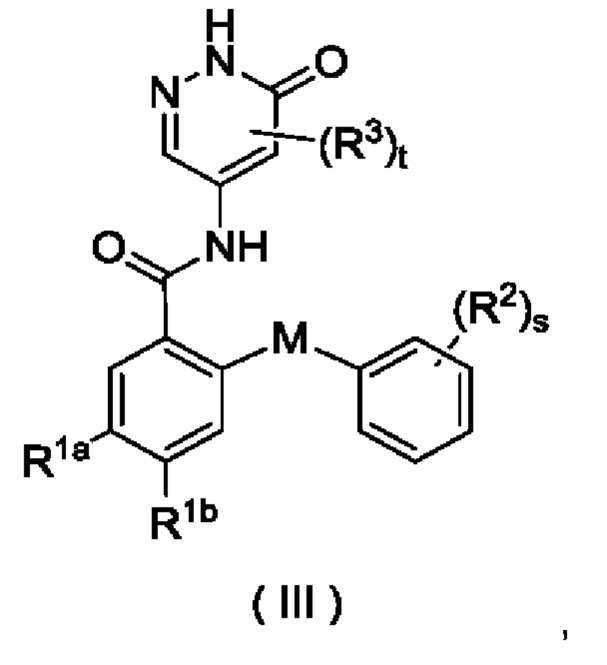
где
М выбран из группы, состоящей из атома О, СН2 и атома S;
R1a представляет собой галоген;
R1b выбран из группы, состоящей из галогена, С1-6 алкила, С1-6 алкокси, галоген-С1-6 алкила и галоген-С1-6 алкокси; и
R2, R3, s и t являются такими, как определено в п. 1.
4. Соединение формулы (I) или его рацемат либо их фармацевтически приемлемая соль по любому из пп. 1-3, где все R2 являются одинаковыми или разными и каждый независимо выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси и дейтерированного С1-6 алкокси.
5. Соединение формулы (I) или его рацемат либо их фармацевтически приемлемая соль по любому из пп. 1-4, где s равно 2.
6. Соединение формулы (I) или его рацемат либо их фармацевтически приемлемая соль по любому из пп. 1-5, представляющее соединение формулы (IV) или его рацемат либо их фармацевтически приемлемую соль:
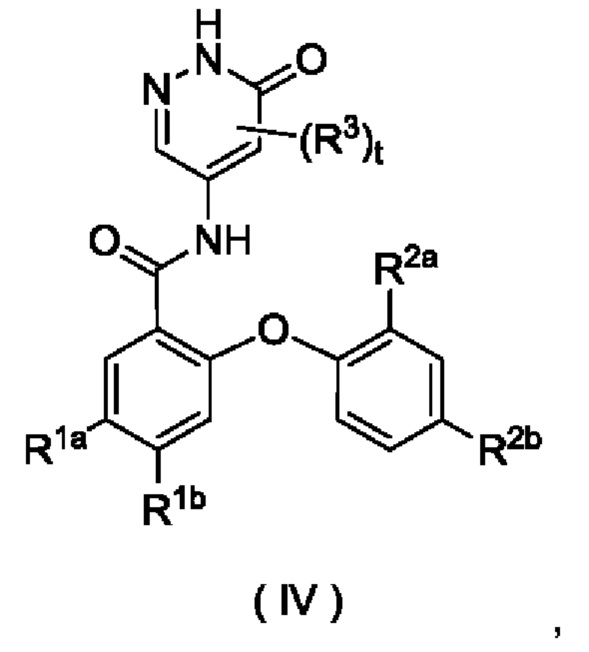
где
R1a представляет собой галоген;
R1b выбран из группы, состоящей из галогена, С1-6 алкила, С1-6 алкокси, галоген-С1-6 алкила и галоген-С1-6 алкокси;
R2a представляет собой С1-6 алкокси или дейтерированный С1-6 алкокси;
R2b выбран из группы, состоящей из атома водорода, галогена, С1-6 алкила, С1-6 алкокси и галоген-С1-6 алкокси; и
R3 и t являются такими, как определено в п. 1.
7. Соединение формулы (I) или его рацемат либо их фармацевтически приемлемая соль по любому из пп. 1-6, выбранное из группы, состоящей из:
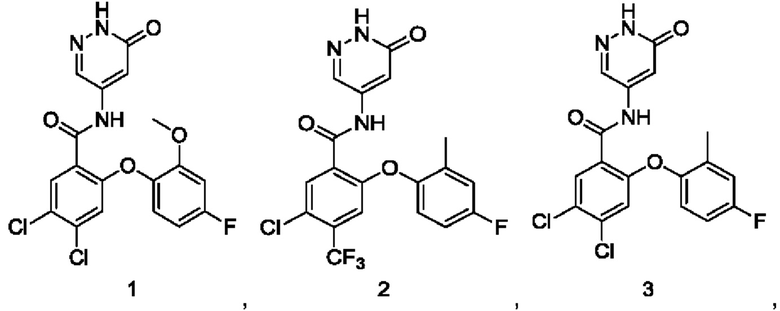
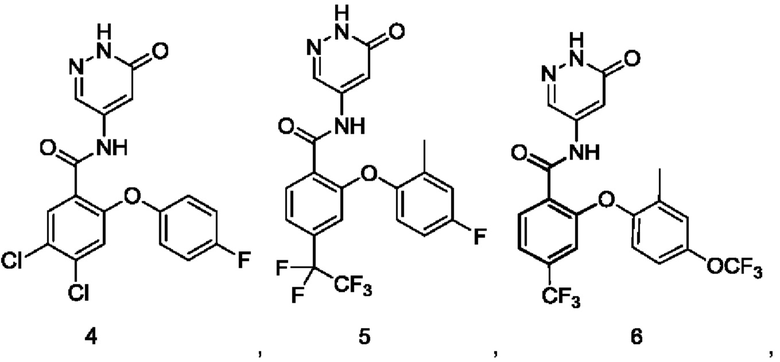
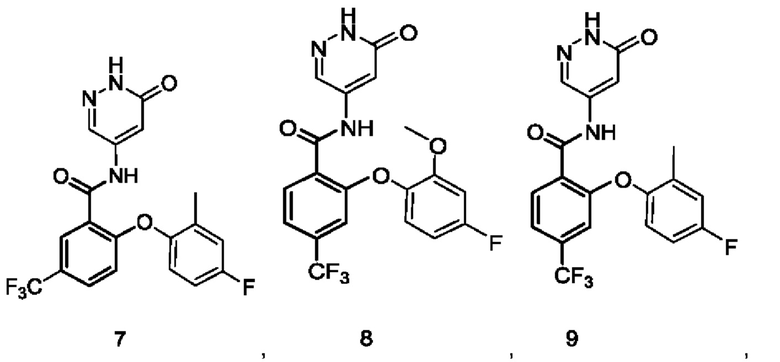
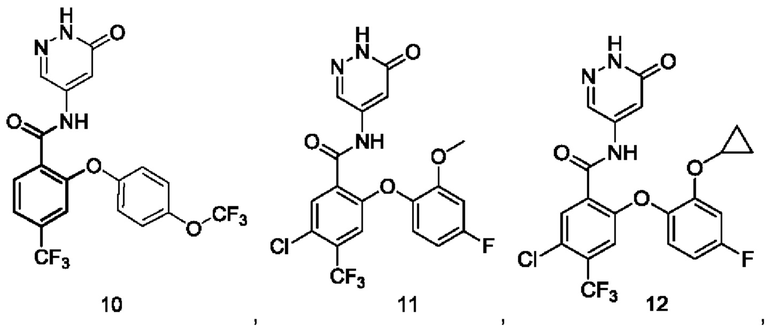
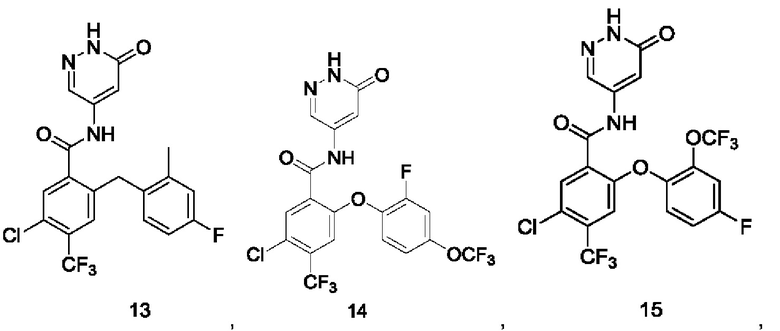
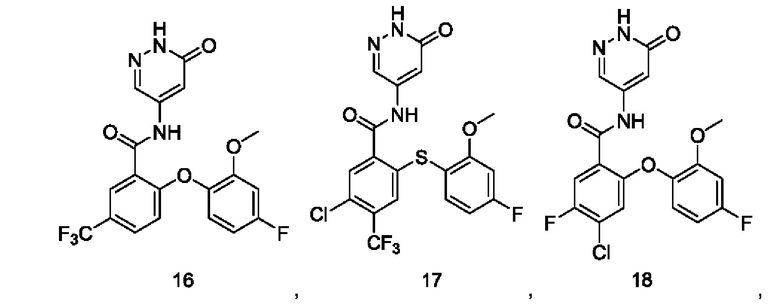
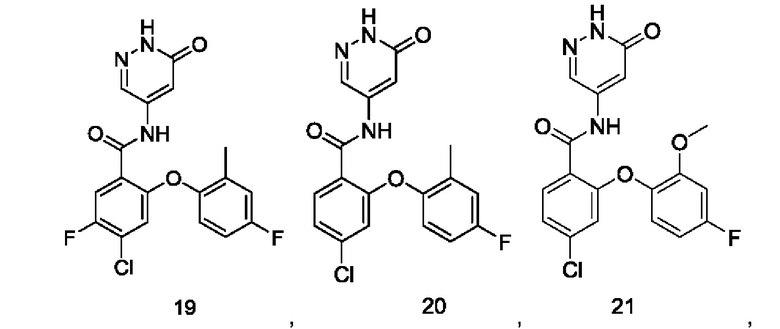
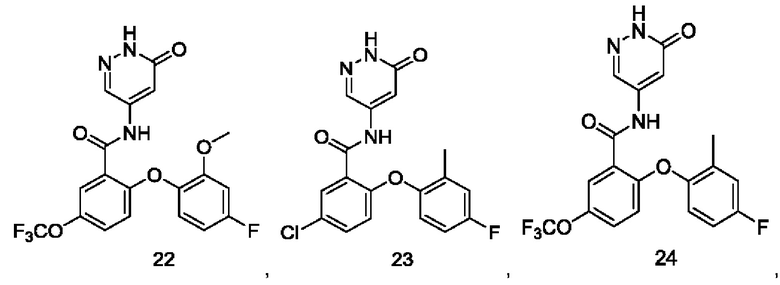
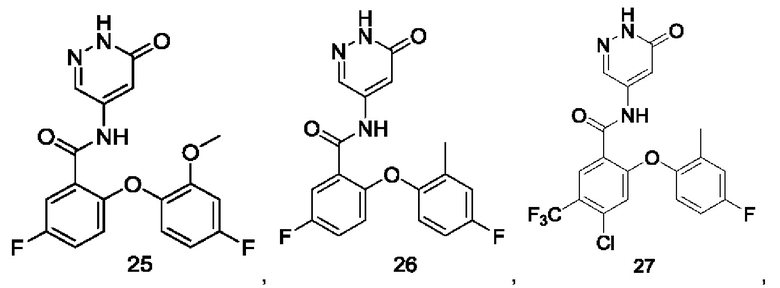
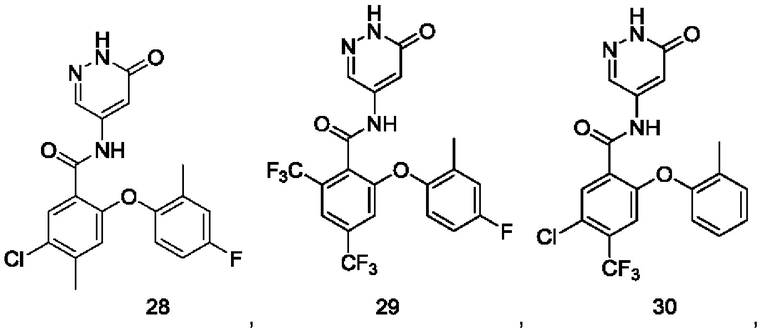
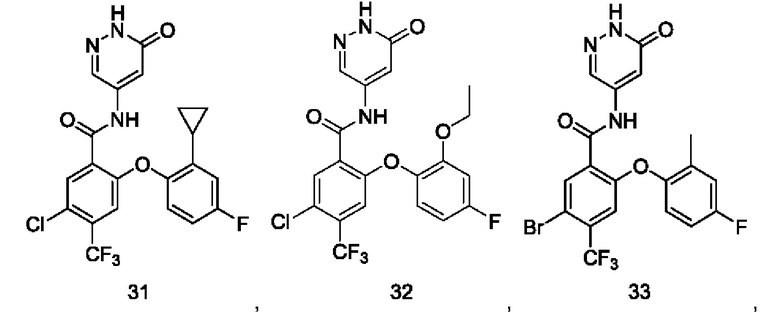
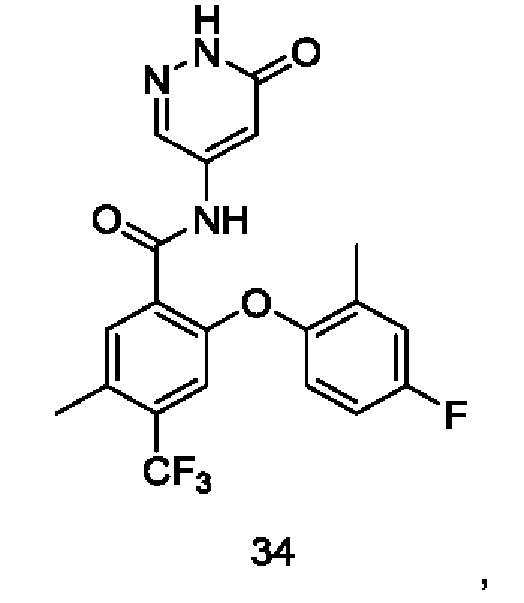
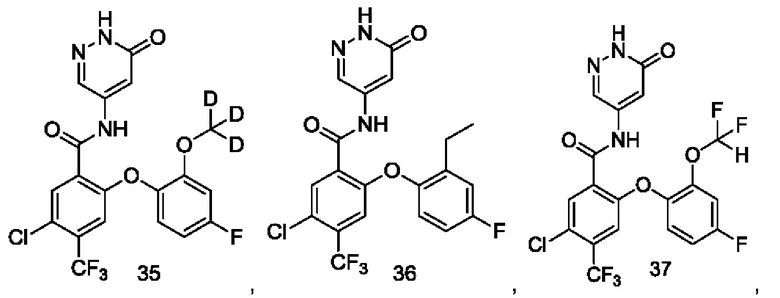
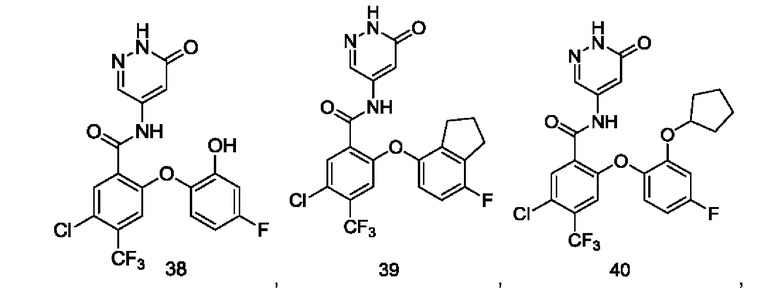
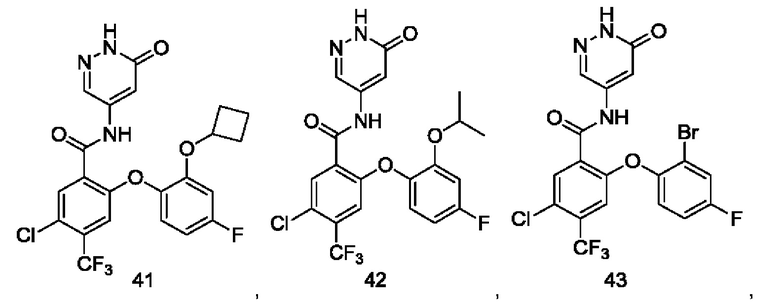
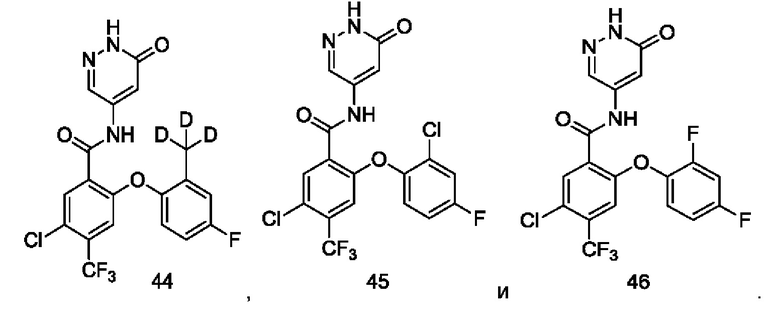
8. Соединение формулы (IA) или его рацемат либо их фармацевтически приемлемая соль:
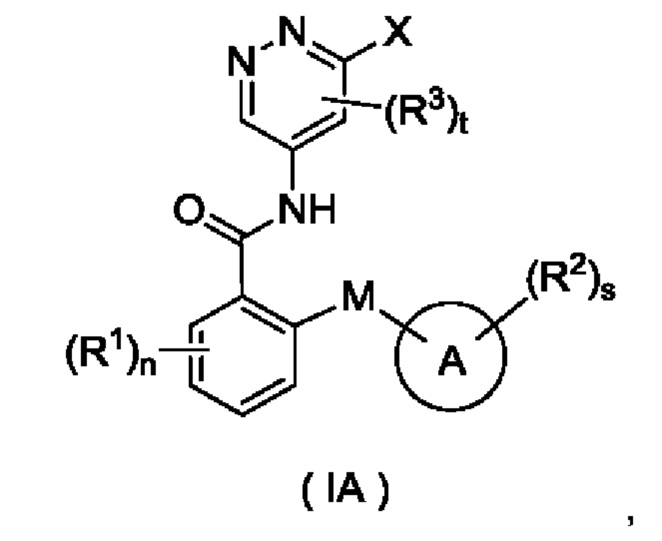
где
X представляет собой галоген; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено в п. 1.
9. Соединение формулы (IA) или его рацемат либо их фармацевтически приемлемая соль по п. 8, выбранное из группы, состоящей из:
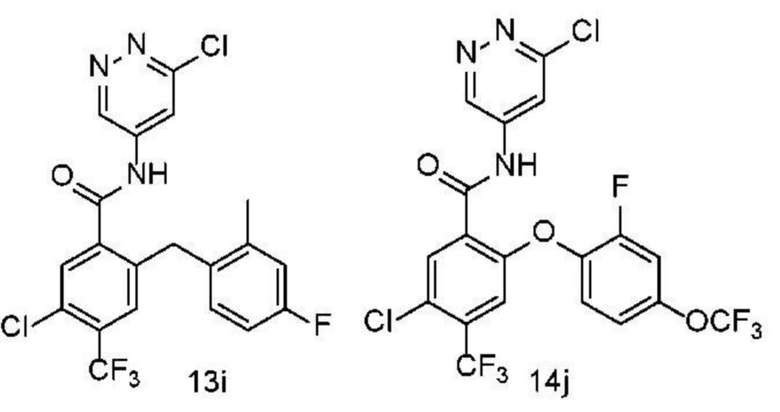 и
и 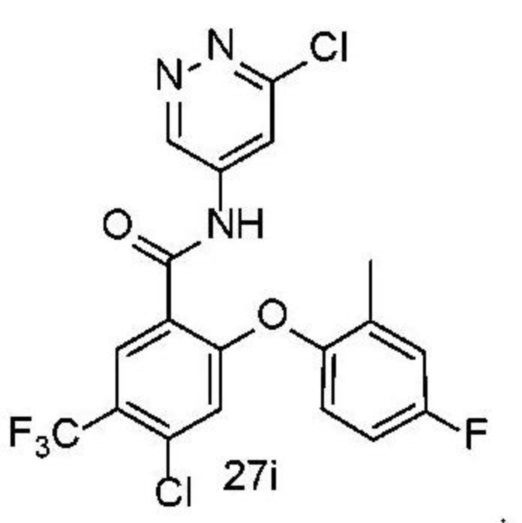
10. Соединение формулы (IB) или его рацемат либо их фармацевтически приемлемая соль:
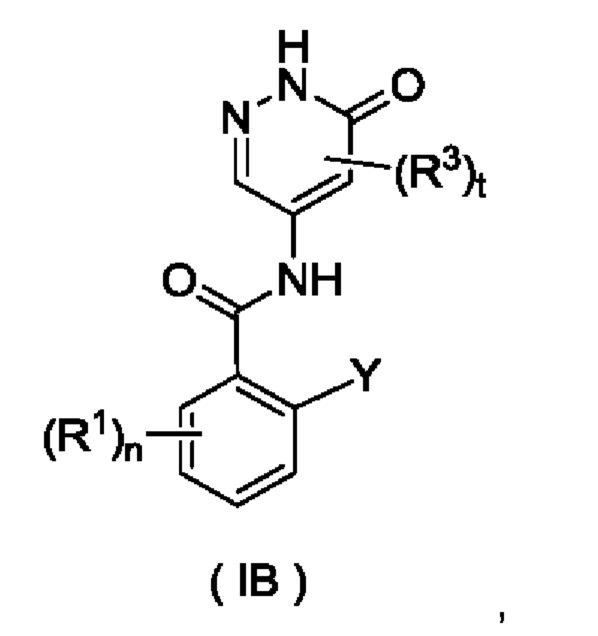
где
Y представляет собой галоген; и
R1, R3, n и t являются такими, как определено в п. 1.
11. Соединение формулы (IB) или его рацемат либо их фармацевтически приемлемая соль по п. 10, выбранное из группы, состоящей из:
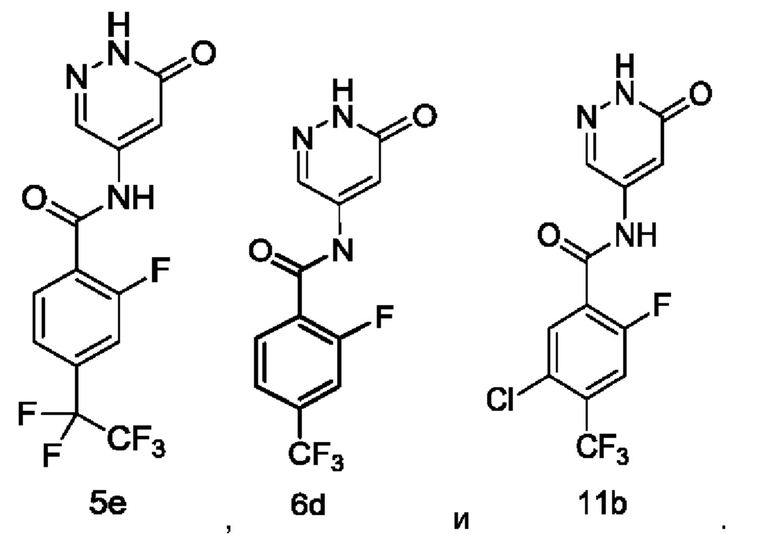
12. Способ получения соединения формулы (I) или его рацемата либо их фармацевтически приемлемой соли по п. 1, включающий стадию:
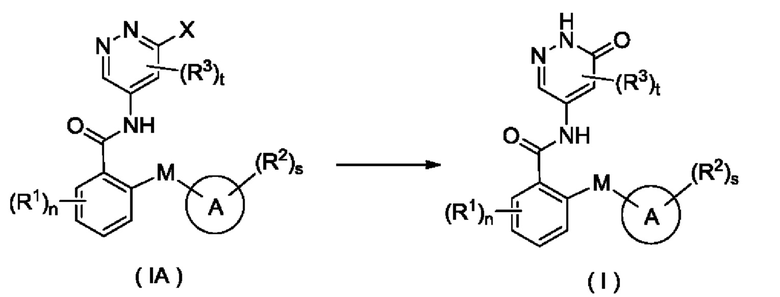
осуществления взаимодействия с участием соединения формулы (IA) с получением соединения формулы (I);
где
X представляет собой галоген; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено в п. 1.
13. Способ получения соединения формулы (I) или его рацемата либо их фармацевтически приемлемой соли по п. 1, включающий стадию:
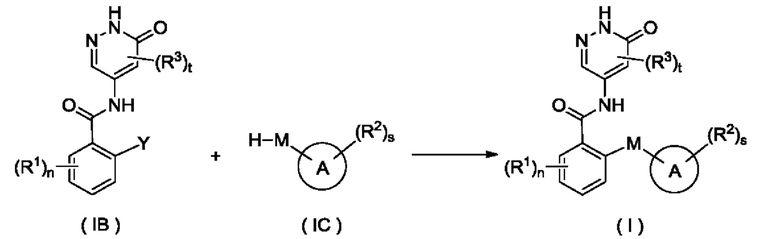
приведения во взаимодействие соединения формулы (IB) и соединения формулы (IС) с получением соединения формулы (I);
где
Y представляет собой галоген; и
кольцо А, М, R1, R2, R3, n, s и t являются такими, как определено в п. 1.
14. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении канала NaV1.8, содержащая соединение формулы (I) или его рацемат либо их фармацевтически приемлемую соль по любому из пп. 1-7 и один или более чем один фармацевтически приемлемый носитель, разбавитель или эксципиент.
15. Применение соединения формулы (I) или его рацемата либо их фармацевтически приемлемой соли по любому из пп. 1-7 или фармацевтической композиции по п. 14 для получения лекарственного средства для ингибирования потенциал-зависимого натриевого канала у субъекта.
16. Применение по п. 15, где потенциал-зависимый натриевый канал представляет собой NaV1.8.
17. Применение соединения формулы (I) или его рацемата либо их фармацевтически приемлемой соли по любому из пп. 1-7 или фармацевтической композиции по п. 14 для получения лекарственного средства для лечения и/или ослабления боли и связанных с болью заболеваний, рассеянного склероза, синдрома Шарко-Мари-Тута, недержания или нарушения сердечного ритма.
18. Применение по п. 17, где боль выбрана из группы, состоящей из хронической боли, острой боли, воспалительной боли, боли при раковом заболевании, нейропатической боли, мышечно-скелетной боли, первичной боли, боли в кишечнике и идиопатической боли.
| CN 105026373 A, 04.11.2015 | |||
| Способ получения цианистых соединений | 1924 |
|
SU2018A1 |
| Зефирова О.Н | |||
| и др | |||
| Об истории возникновения и развития концепции биоизостеризма, Вестник Московского университета, Серия 2 | |||
| Химия, 2002, т.43, 4, с.251-256 | |||
| CN 104968647 A, 07.10.2015 | |||
| CN 101883758 A, 10.11.2010 | |||
| RU 2010118481 A, 20.11.2011. | |||

