ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к ингибиторам mdm2 для применения в определенных схемах введения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Белок p53 является фактором транскрипции, который управляет экспрессией большого числа генов-мишеней, вовлеченных в восстановление повреждений ДНК, апоптоз и остановку клеточного цикла, причем все перечисленные явления важны для противодействия злокачественному росту опухолей. Таким образом, p53 имеет ключевое значение для поддержания генетической стабильности и предотвращения развития опухолей. Ген TP53 является одним из генов, наиболее часто подвергающихся мутациям при раковых заболеваниях человека. Сообщается, что примерно в половине случаев раковых заболеваний имеет место инактивация p53, вызванная прямой мутацией. В случае раковых заболеваний, при которых ген p53 не подвергся мутации, была продемонстрирована функциональная инактивация на уровне белка. Один из описанных механизмов инактивации p53 заключается в его взаимодействии с человеческим гомологом MDM2 (белком, кодируемым геном ʺMouse double minute 2ʺ). Таким образом, белок Mdm2 является важным негативным регулятором супрессора опухолей p53. Белок Mdm2 действует как в качестве E3 убиквитин лигазы, приводя к протеасомальному разрушению p53, так и в качестве ингибитора активации транскрипции p53. Часто оказывается, что ген Mdm2 амплифицирован в p53 опухолях дикого типа.
Были разработаны ингибиторы Mdm2, которые подавляют взаимодействие p53-mdm2 и способны оказывать антинеопластическое действие.
В заявке US 2013/0245089 раскрыт способ лечения пациента, страдающего раковым заболеванием, путем введения пациенту 4-{[(2R,3S,4R,5S)-4-(4-хлор-2-фторфенил)-3-(3-хлор-2-фторфенил)-4-циано-5-(2,2-диметилпропил)пирролидин-2-карбонил]амино}-3-метоксибензойной кислоты в количестве от примерно 800 до примерно 3000 мг/сутки в течение периода вплоть до примерно 7 дней в дни 1-7 28-дневного цикла введения, с последующим периодом отдыха от примерно 21 до 23 дней.
В документе за авторством B.Higgins et al., размещенном в Clinical Cancer Research (May 2014), описана 28-дневная циклическая схема введения, в которой RG7388 вводят три раза по одному разу в неделю, с последующим 13-дневным периодом отдыха (схема с 28-дневным циклом), или препарат вводят в течение 5 дней подряд в 28-дневной схеме.
Ингибиторы Mdm2 и способы их получения были раскрыты, например, в WO 2013111105 или WO 2011076786.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Неожиданно было обнаружено, что предпочтительные схемы введения ингибитора Mdm2 (далее по тексту именуемого ʺMdm2iʺ) можно разработать на основе понимания биологии мишени данных препаратов и того, как концентрация Mdm2 может менять передачу сигнала вниз по цепи, оказывая влияние на противоопухолевую эффективность и переносимость. Неожиданно было обнаружено, что в случае применения достаточно мощного Mdm2i или, в качестве альтернативы, достаточно высокой дозировки Mdm2i, препарат может вызвать антинеопластический эффект за счет запуска в клетках долговременного антипролиферативного механизма. Если раковую клетку подвергнуть действию соответствующего Mdm2i в достаточно высокой концентрации в течении такого короткого периода времени, как 8 часов (и пропорционально более продолжительного периода при применении более низкой концентрации), Mdm2i вызывает резкий рост экспрессии мРНК p21 и Puma в течение последующих 48-72 часов, что приводит к существенному стимулированию активности каспазы 3/7 и, за счет этого, к значительному апоптозу. У животных, которым были подкожно имплантированы раковые клетки, наблюдался такой же эффект после введения животным достаточно высокой разовой дозы. Это приводило к значительному уменьшению размеров опухоли. Ни один из этих эффектов не обнаруживался, если содержание Mdm2i было ниже определенного порогового уровня, до которого этот второй путь действия Mdm2i не активируется. Информация о втором пути действия Mdm2i может помочь планированию клинических исследований за счет уменьшения побочных эффектов из-за нацеленного воздействия лекарственного препарата.
Интересно отметить наблюдение, что долговременный эффект может продолжаться в течение нескольких недель после введения одной дозы, что устраняет необходимость в ежедневном введении и дает возможность вводить препарат в прерывистом режиме. Во время перерывов, когда введение препарата не осуществляется, организм может восстанавливаться от потенциальных целевых воздействий или побочных эффектов; в том числе может восстанавливаться количество белых кровяных телец (лейкоцитов, WBC), нейтрофилов и тромбоцитов. Введение Mdm2i в дозировках, которые запускают механизмы долговременного действия, позволяют Mdm2i действовать по крайней мере столь же эффективно, как и при ежедневном введении в более низких дозировках, и такая схема введения может лучше переноситься пациентом. Менее частое введение может также приводить к более доброжелательному отношению пациента, приверженности пациента лечению, в особенности в том случае, когда лекарство вводят внутривенно, что может принести значительную пользу пациенту. Например, раздражение в месте введения может зажить в достаточной степени до срока введения следующей дозы.
Прерывистое введение Mdm2i с устойчивым эффектом можно сочетать со схемой, включающей ежедневное введение более низкой дозы, по сравнению с дозой, применяемой для достижения устойчивого эффекта. Комбинация прерывистого введения первой дозы и ежедневного введения второй дозы приводит к синергетическому эффекту с точки зрения эффективности применения соединения, что наблюдается, например, в форме уменьшения размеров опухоли или регрессии опухоли. Кроме того, из-за лучшей переносимости Mdm2i при прерывистом введении, этот препарат можно применять в комбинации с другими антинеопластическими агентами. Комбинация Mdm2i и другого антинеопластического агента может позволить добиться улучшения переносимости Mdm2i при его прерывистом введении, с одновременным повышением суммарной эффективности комбинированной терапии, включающей второй антинеопластический агент.
Конкретно, настоящее изобретение относится к следующим аспектам, полезным элементам и конкретным вариантам осуществления, соответственно, индивидуально или в комбинации, которые перечислены в следующих пунктах:
1. MDM2i для применения в лечении рака, где MDM2i вводят субъекту в прерывистом режиме в виде как минимум трех последовательных доз, и период между введением каждых двух последовательных доз составляет как минимум 2 недели.
2. MDM2i для применения в лечении рака по п.1, где MDM2i вводят субъекту в прерывистом режиме и период между каждыми двумя последовательными введениями составляет как минимум 3 недели и не превышает 60 дней.
3. MDM2i для применения в лечении рака по пп.1 или 2, где MDM2i вводят субъекту в прерывистом режиме, и промежуток времени между последовательными введениями составляет 3 недели.
4. MDM2i для применения в лечении рака по любому из пп.1-3, где MDM2i вводят внутривенно.
5. MDM2i для применения в лечении рака, где MDM2i вводят субъекту в первой и второй дозе, причем первую дозу вводят в тот же день, что и вторую дозу, в последовательные дни или в другой день относительно второй дозы, где два последовательных введения первой дозы осуществляют в прерывистом режиме по крайней мере один раз в 1 неделю, 2 недели, 3 недели, 4 недели, 6 недель или один раз в 60 дней, причем введение осуществляют не реже одного раза в 60 дней, и первая и вторая дозы не являются одинаковыми.
6. MDM2i для применения в лечении рака по п.5, где вторую дозу вводят ежедневно, необязательно с перерывами.
7. MDM2i для применения в лечении рака по п.6, где перерыв продолжается как минимум 1 день, 2 дня, 3 дня, 4 дня, 1 неделю, 2 недели или 3 недели и его наибольшая продолжительность составляет 26 дней.
8. MDM2i для применения в лечении рака по любому из пп.5-7, где вторую дозу вводят через 1-14 дней после того, как была введена первая доза.
9. MDM2i для применения в лечении рака по любому из пп.5-8, где вторую дозу вводят в течение двух недель после чего следует двухнедельный период без введения препарата и затем описанный цикл повторяется.
10. MDM2i для применения в лечении рака по любому из пп.5-9, где первая доза превышает вторую дозу.
11. MDM2i для применения в лечении рака по любому из пп.5-10, где как минимум одну из первой или второй дозы вводят внутривенно.
12. MDM2i для применения в лечении рака по любому из пп.5-11, где два последовательных введения первой дозы осуществляют в прерывистом режиме как минимум один раз в 2 недели.
13. MDM2i для применения в лечении рака по любому из пп.5-11, где два последовательных введения первой дозы осуществляют в прерывистом режиме как минимум один раз в 3 недели.
14. MDM2i для применения в лечении рака по любому из пп.1-13, где рак является раком мочевого пузыря, груди, мозга, органов головы и шеи, печени, ротовой полости, желчевыводящих путей, острым и хроническим лимфоидным лейкозом, острым и хроническим миелоидным лейкозом, хроническим миеломоноцитарным лейкозом, колоректальным раком, раком желудка, желудочно-кишечной стромальной опухолью, гепатоклеточным раком, глиомой, лимфомой, меланомой, множественной миеломой, миелопролиферативной болезнью, нейроэндокринным раком, раком легких, немелкоклеточным раком легких, раком поджелудочной железы, яичников, простаты, почечноклеточным раком, саркомой, липосаркомой и раком щитовидной железы.
15. MDM2i для применения в лечении рака по любому из пп.1-13, где рак является меланомой, раком легких или нейробластомой.
16. MDM2i для применения в лечении рака по любому из пп.1-13, где рак является меланомой.
17. Применение MDM2i для получения лекарственного средства, предназначенного для лечения рака, где MDM2i вводят в прерывистом режиме в виде как минимум трех последовательных доз, и период между каждыми двумя последовательными введениями составляет как минимум 2 недели, как минимум 3 недели, как минимум 4 недели, как минимум 6 недель или 60 дней, и не превышает 60 дней.
18. Способ лечения рака, в котором MDM2i вводят субъекту, которому это необходимо, в прерывистом режиме, в виде как минимум трех последовательных доз, и период между каждыми двумя последовательными введениями составляет как минимум 2 недели, как минимум 3 недели, как минимум 4 недели, как минимум 6 недель или 60 дней, и не превышает 60 дней.
19. MDM2i для применения в лечении рака по любому из пп.1-16, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по п.17, или способ лечения рака по п.18, где MDM2i вводят человеку.
20. MDM2i для применения в лечении рака по любому из пп.1-16 или 19, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака, по пп.17 или 19, или способ лечения рака по пп.18 или 19, где MDM2i выбран из группы, состоящей из:
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-она,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-она,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(6-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}пиридин-3-ил)-1,4-дигидро-2H-изохинолин-3-она,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(6-{метил-[4-(3-метил-4-оксоимидазолин-1-ил)-транс-циклогексилметил]амино}пиридин-3-ил)-1,4-дигидро-2H-изохинолин-3-она,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(5-{метил-[4-(3-метил-4-оксоимидазолин-1-ил)-транс-циклогексилметил]амино}пиразин-2-ил)-1,4-дигидро-2H-изохинолин-3-она,
1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-она,
(S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-она,
4-[(S)-5-(3-хлор-2-фторфенил)-2-(2,4-диметоксипиримидин-5-ил)-3-изопропил-6-оксо-3,4,5,6-тетрагидропирроло[3,4-d]имидазол-4-ил]бензонитрила,
(S)-5-(5-хлор-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-она,
(S)-5-(3-хлор-4-фторфенил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-((R)-1-метоксипиран-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она,
и
(S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-d6-пиримидин-5-ил)-1-((R)-1-метоксипропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она.
21. MDM2i для применения в лечении рака по любому из пп.1-16 или 19, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по пп.17 или 19, или способ лечения рака по пп.18 или 19, где MDM2i представляет собой:
(S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он или
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он.
22. MDM2i для применения в лечении рака по любому из пп.1-16 или 19, применение MDM2i для получения лекарственного средства для лечения рака по пп.17 или 19, или способ лечения рака по пп.18 или 19, где MDM2i представляет собой (S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он.
23. MDM2i для применения в лечении рака по любому из пп.1-16 или 19-22, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака, по любому из пп.17 или 19-22, или способ лечения рака по пп.18 или 19-22, где MDM2i для применения в лечении рака, применение MDM2i для получения лекарственного средства, или способ лечения рака дополнительно включают другой фармацевтический ингредиент, который вводят пациенту.
24. MDM2i для применения в лечении рака по п.23, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по п.23, или способ лечения рака по п.23, где другой фармацевтический ингредиент представляет собой другой антинеопластический агент.
25. MDM2i для применения в лечении рака по п.24, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по п.24, или способ лечения рака по п.24, где вводят более одного дополнительного антинеопластического агента.
26. MDM2i для применения в лечении рака по любому из пп.23-25, применение MDM2i для получения лекарственного средства для лечения рака по любому из пп.23-25, или способ лечения рака по любому из пп.23-25, где MDM2i вводят в периодическом режиме в виде как минимум трех последовательных доз, и период между каждыми двумя последовательными введениями составляет как минимум 1 неделю.
27. MDM2i для применения в лечении рака по любому из пп.23-25, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по любому из пп.23-25, или способ лечения рака по любому из пп.23-25, где MDM2i вводят в периодическом режиме в виде как минимум трех последовательных доз, и период между каждыми двумя последовательными введениями составляет как минимум 2 недели.
28. MDM2i для применения в лечении рака по любому из пп.23-25, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по любому из пп.23-25, или способ лечения рака по любому из пп.23-25, где MDM2i вводят в периодическом режиме в виде как минимум трех последовательных доз, и период между каждыми двумя последовательными введениями составляет как минимум 3 недели.
29. MDM2i для применения в лечении рака по любому из пп.1-16 или 19-28, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по любому из пп.17 или 19-28, или способ лечения рака по пп.18 или 19-28, где MDM2i представляет собой (S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он.
30. MDM2i для применения в лечении рака по любому из пп.1-16 или 19-28, применение MDM2i для получения лекарственного средства, предназначенного для лечения рака по любому из пп.17 или 19-28, или способ лечения рака по пп.18 или 19-28, где MDM2i представляет собой (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он.
Термин «ингибитор Mdm2» или «Mdm2i» в настоящей заявке означает любое соединение, ингибирующее взаимодействия HDM-2/p53 или HDM-4/p53 с константой IC50 менее 10 мкМ, предпочтительно, менее 1 мкМ, предпочтительно, в диапазоне нМ, при измерении способом резонансного переноса энергии флуоресценции с разделением по времени (TR-FRET). Ингибирование взаимодействий p53-Hdm2 и p53-Hdm4 измеряют способом резонансного переноса энергии флуоресценции с разделением по времени (TR-FRET). Перенос энергии флуоресценции (или Ферстеровский резонансный перенос энергии) описывает перенос энергии между донорной и акцепторной флуоресцентными молекулами. Для проведения анализа, белок MDM2 (аминокислоты 2-188) и белок MDM4 (аминокислоты 2-185) меченые C-концевым биотиновым фрагментом, используются в комбинации со стрептавидином, меченым европием (Perkin Elmer, Inc., Waltham, MA, USA), служащим донорным флуорофором. Полученный из p53, меченый Cy5 пептид Cy5-TFSDLWKLL (аминокислотные остатки 18-26 белка p53) служит акцептором энергии. После возбуждения донорной молекулы на длине волны 340 нм, связывающее взаимодействие между MDM2 или MDM4 и пептидом p53 вызывает перенос энергии и усиливает сигнал на длине волны излучения акцептора при 665 нм. Прекращение образования комплексов p53-MDM2 или p53-MDM4 из-за связывания молекулы ингибитора с сайтом связывания MDM2 или MDM4 приводит к усилению эмиссии донора при 615 нм. Результат анализа FRET, основанного на соотношении сигналов, вычисляют по 15 отдельным результатам по двум разным сигналам флуоресценции, измеренным в режиме временного разделения (интенсивность при 665 нм/интенсивность при 615 нм × 1000). Анализ можно выполнять по следующей методике: тест проводят в белых микропланшетах 1536w (Greiner Bio-One GmbH, Frickenhausen, Germany) в суммарном объеме реакционной смеси 3,1 мкл, которую получают смешиванием 100 нл соединений, разбавленных смесью 90% ДМСО/10%H2O (конечная концентрация ДМСО 3,2%), с 2 мкл стрептавидина, меченого европием 20 (конечная концентрация 2,5 нМ) в реакционном буфере (PBS, 125 мМ NaCl, 0,001% Novexin (состоит из углеводных полимеров (полимеров Novexin), предназначенных для повышения растворимости и стабильности белков; Novexin Ltd., Ambridgeshire, United Kingdom), желатин 0,01%, 0,2% Pluronic (блок-сополимер этиленоксида и пропиленоксида, BASF, Ludwigshafen, Germany), 1 мМ DTT) с последующим добавлением 0,5 мкл MDM2-Bio или MDM4-Bio, разбавленных аналитическим буфером (конечная концентрация 10 нМ). Оставляют раствор для предварительного инкубирования в течение 15 минут при комнатной температуре, с последующим добавлением 0,5 мкл пептида Cy5-p53 в аналитическом буфере (конечная концентрация 20 нМ). Инкубируют при комнатной температуре в течение 10 минут, после чего производят считывание планшета. Для регистрации сигналов образцов используют мультифункциональный прибор для считывания планшетов Analyst GT multimode microplate reader (Molecular Devices) со следующими настройками: дихроичное зеркало 380 нм, возбуждение 330 нм, эмиссия донора 615 нм и эмиссия акцептора 665 нм. Значения IC50 вычисляют способом подгонки кривых, используя программу XLfit. Если не указано иное, реагенты приобретают у Sigma Chemical Co, St.Louis, MO, USA.
Согласно одному из вариантов осуществления, ингибитор Mdm2 может представлять собой, например, любое из перечисленных ниже соединений:
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(6-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}пиридин-3-ил)-1,4-дигидро-2H-изохинолин-3-он,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(6-{метил-[4-(3-метил-4-оксоимидазолин-1-ил)-транс-циклогексилметил]амино}пиридин-3-ил)-1,4-дигидро-2H-изохинолин-3-он,
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(5-{метил-[4-(3-метил-4-оксоимидазолин-1-ил)-транс-циклогексилметил]амино}пиразин-2-ил)-1,4-дигидро-2H-изохинолин-3-он,
1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он,
(S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он,
4-[(S)-5-(3-хлор-2-фторфенил)-2-(2,4-диметоксипиримидин-5-ил)-3-изопропил-6-оксо-3,4,5,6-тетрагидропирроло[3,4-d]имидазол-4-ил]бензонитрил,
(S)-5-(5-хлор-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он,
(S)-5-(3-хлор-4-фторфенил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-((R)-1-метоксипиран-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-он,
или
(S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-d6-пиримидин-5-ил)-1-((R)-1-метоксипропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-он.
В конкретном варианте осуществления, MDM2i представляет собой (S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он (далее по тексту именуемый соединением A) или его фармацевтически приемлемую соль.
В другом варианте осуществления, MDM2i представляет собой (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он (далее по тексту именуемый соединением B).
Термины «субъект» или «пациент» в настоящей заявке включают животных, которые могут болеть или поражаться раком или любым расстройством, включающим, прямо или косвенно, рак. Примеры субъектов включают млекопитающих, например, людей, собак, коров, лошадей, свиней, овец, коз, кошек, мышей, кроликов, крыс и трансгенных животных, помимо человека. В предпочтительном варианте осуществления, субъект представляет собой человека, например, человека, страдающего или у которого имеется риск или потенциальная возможность страдать раком. В конкретном варианте осуществления, субъект или пациент является человеком.
Термин «лечение» в настоящей заявке означает остановку, отсрочку начала (т.е. периода до появления клинических проявлений заболевания) и/или уменьшение риска развития или ухудшения течения заболевания, или этот термин включает ослабление, уменьшение или облегчение по крайней мере одного симптома или отсрочку прогрессирования заболевания у субъекта. Например, лечение может означать ослабление одного или нескольких симптомов расстройства или полную ликвидацию расстройства, например, рака.
Термин «антинеопластический агент» относится к фармацевтическому действующему ингредиенту, который демонстрирует антипролиферативную или противораковую активность. Возможные антинеопластические агенты, подходящие для комбинированного лечения, включают, не ограничиваясь указанными, ингибиторы BRAF (например, (S)-метил-1-(4-(3-(5-хлор-2-фтор-3-(метилсульфонамидо)фенил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамид или вемурафениб); ингибиторы киназы анапластической лимфомы (ALK) (например, серитиниб, AE684, алектиниб, кризотиниб, AP26113, ASP3026, ADZ3463); ингибиторы ароматазы (например, атаместан, экземестан и форместан, аминоглютетимид, роглетимид, пиридоглтетимид, трилостан, тестолактон, кетоконазол, ворозол, фадрозол, анастрозол или летрозол); антиэстрогены (тамоксифен, фульвестрант, ралоксифен или ралоксифена гидрохлорид); антиандрогены (например, бикалутамид); ингибиторы топоизомеразы I (например, топотекан, гиматекан, иринотекан, каптотецин и его аналоги, 9-нитрокамптотецин и макромолекулярный конъюгат камптотецина PNU-166148 (соединение A1 в заявке WO99/17804); ингибиторы топоизомеразы II (например, доксорубицин, даунорубицин, эпирубицин, идарубицин, неморубицин, митоксантрон, лозоксантрон, этопозид или тенипозид); соединения, активирующие микротрубочки (например, паклитаксел, доцетаксел, винбластин, винбластина сульфат, винкристин, винкристина сульфат, винорелбин, дискодермолиды, кохицин); алкилирующие соединения (например, циклофосфамид, ифосфамид, мелфалан или нитрозомочевину); ингибиторы гистон деацетилазы; соединения, которые инициируют процесс дифференцировки клеток; ингибиторы циклооксигеназы; ингибиторы MMP; ингибиторы mTOR; антинеопластические антиметаболиты; соединения платины; соединения нацеливающие/уменьшающие активность протеин- или липидкиназы; антиангиогенные соединения; соединения, которые нацеливают, уменьшают или ингибируют активность протеин- или липид фосфатазы; агонисты гонадорелина (например, абареликс, госерелин и госерелина ацетат); ингибиторы метионин аминопептидазы; бисфосфонаты; модификаторы биологической реакции; антипролиферативные антитела; ингибиторы гепараназы; ингибиторы онкогенных изоформ Ras; ингибиторы теломеразы; ингибиторы протеасомы; соединения, применяемые при лечении гематологических злокачественных заболеваний; соединения, которые нацеливают, уменьшают или ингибируют активность Flt-3; ингибиторы Hsp90; ингибиторы моторного белка кинезина; ингибиторы MEK; лейковорин; вещества, связывающие EDG; антилейкемические соединения; ингибиторы рибонуклеотид редуктазы; ингибиторы S-аденозилметионин декарбоксилазы; ангиостатические стероиды; кортикостероиды; другие химиотерапевтические соединения (которые определены ниже); фотосенсибилизирующие соединения (например, VISUDYNE и порфимер натрия).
КРАТКОЕ ОПИСАНИЕ ИЛЛЮСТРАТИВНОГО МАТЕРИАЛА
Фиг.1 PK (фармакокинетика) соединения A у крыс, несущих опухоль SJSA-1, после однократного внутривенного (i.v.) введения.
Фиг.2 PK и PD (фармакодинамика) соединения у A крыс, несущих опухоль SJSA-1 после однократного i.v. введения.
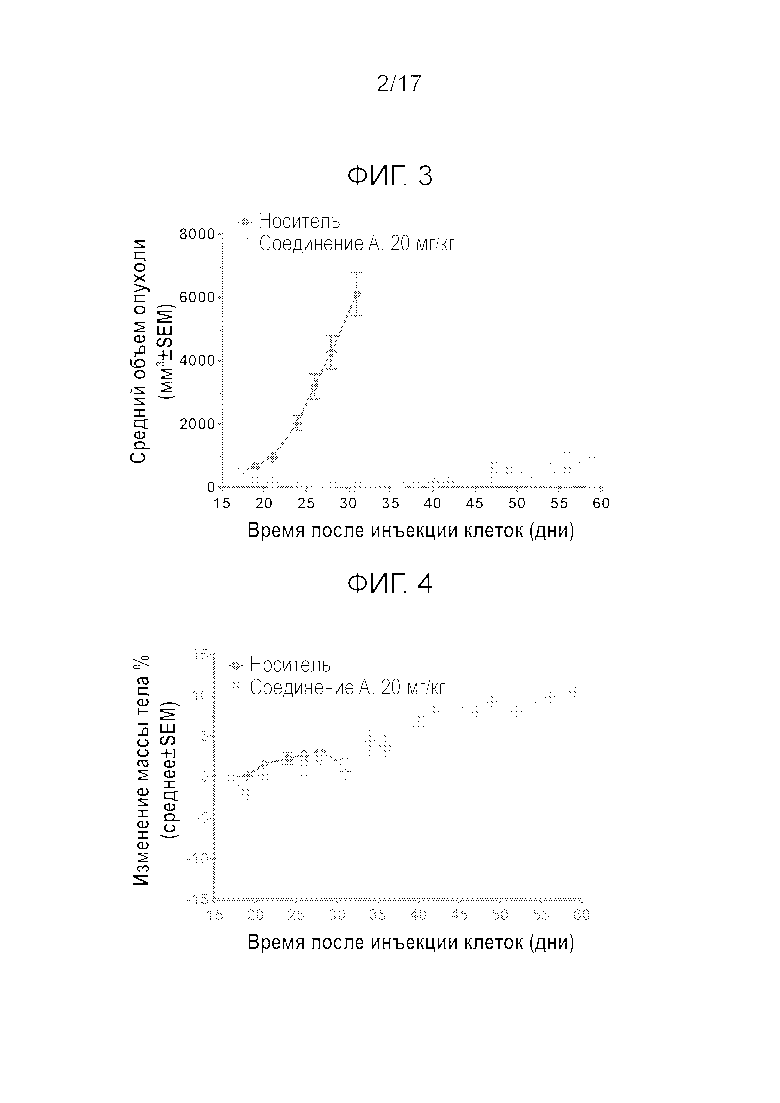
Фиг.3 Рост опухоли после однократного i.v. введения соединения 1 бестимусной крысе, несущей опухоль SJSA-1.
Фиг.4 Изменение массы тела (BW) после однократного i.v. введения соединения A бестимусной крысе, несущей опухоль SJSA-1.
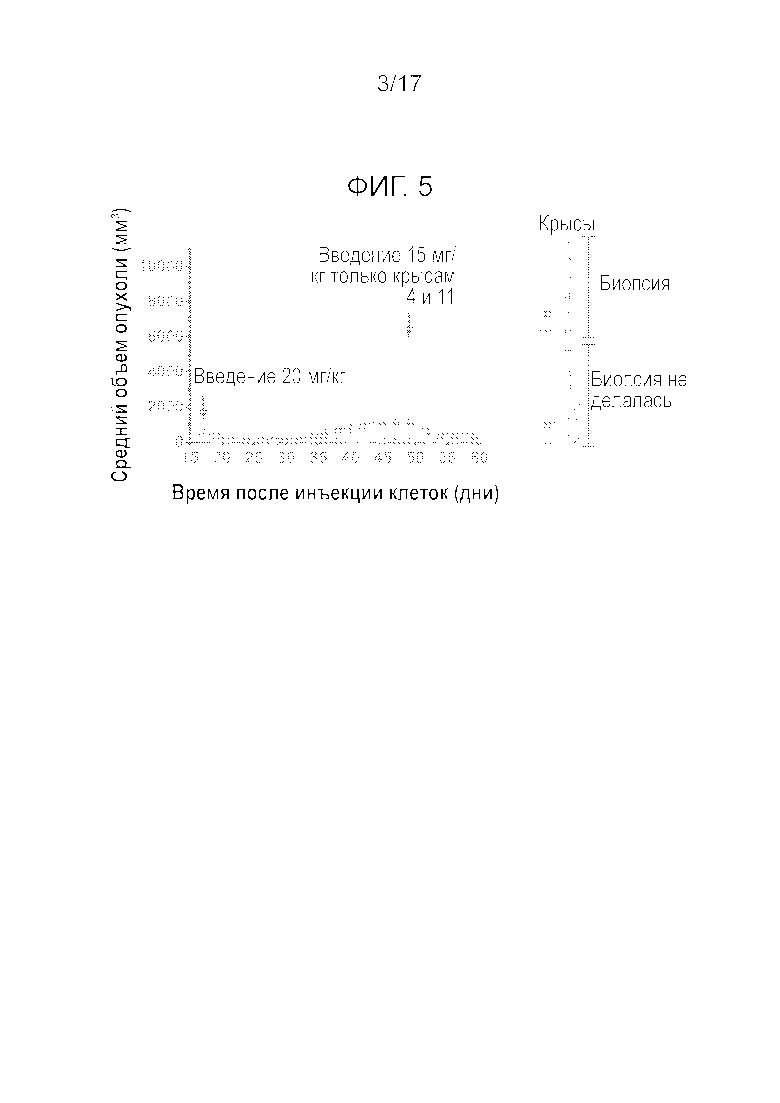
Фиг.5 Эффективность соединения A после однократного i.v. введения бестимусной крысе, несущей опухоль SJSA-1 - индивидуальные данные.
Фиг.6 Восстановление костного мозга после однократного i.v. введения.
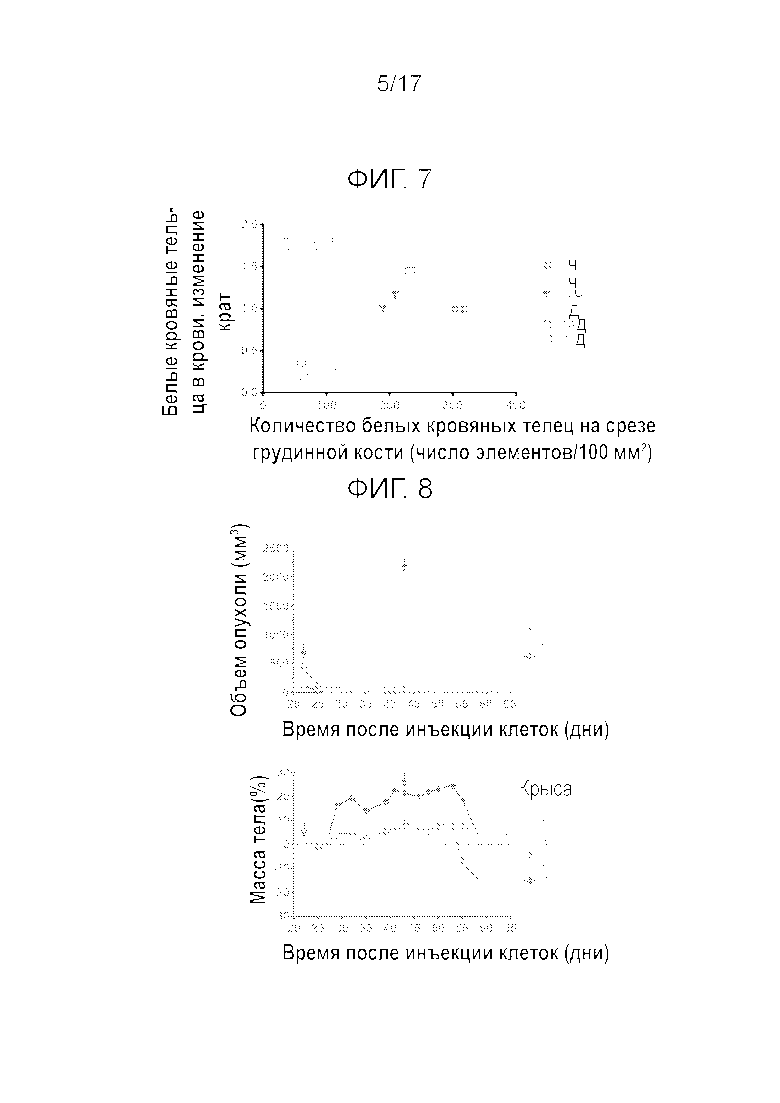
Фиг.7 Корреляция между количеством белых кровяных телец в крови и на срезе костного мозга из грудинной кости.
Фиг.8 Демонстрирует рост опухоли и изменение массы тела у бестимусных крыс в течение 42 дней после i.v. (q3w, т.е. один раз каждые три недели) введения бестимусным крысам, несущим опухоль SJSA-1.
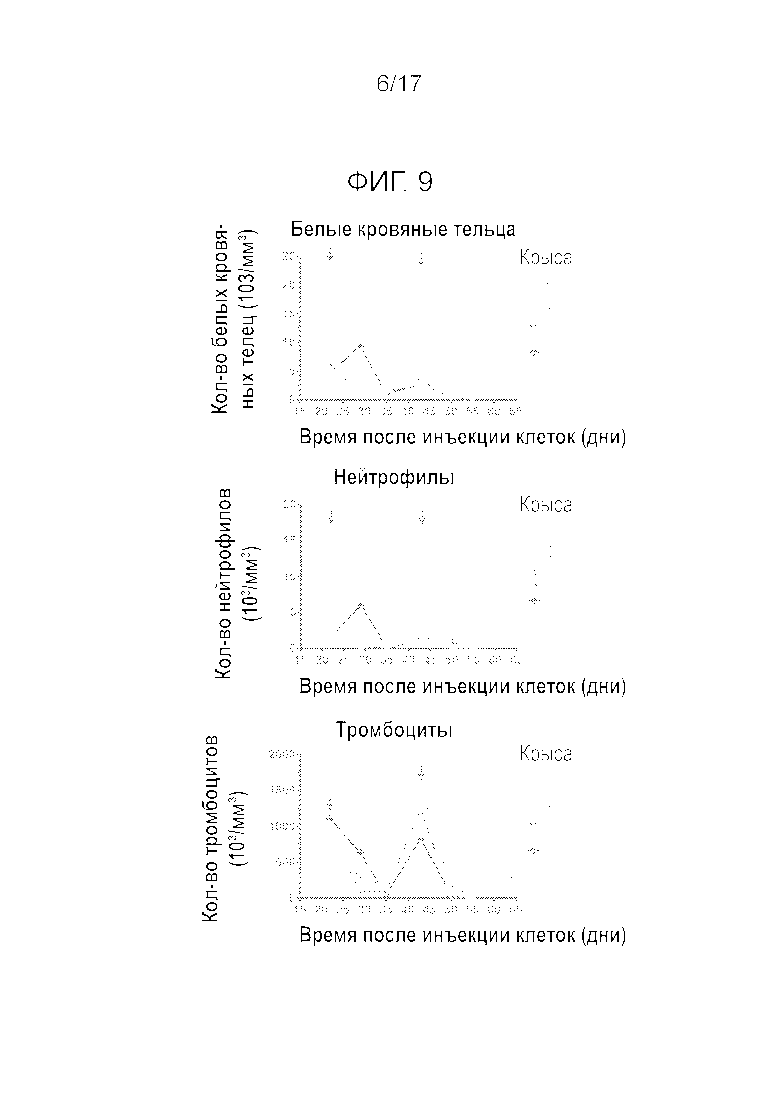
Фиг.9 Влияние i.v. введения (q3w) на количество белых кровяных телец (WBC), нейтрофилов и тромбоцитов.
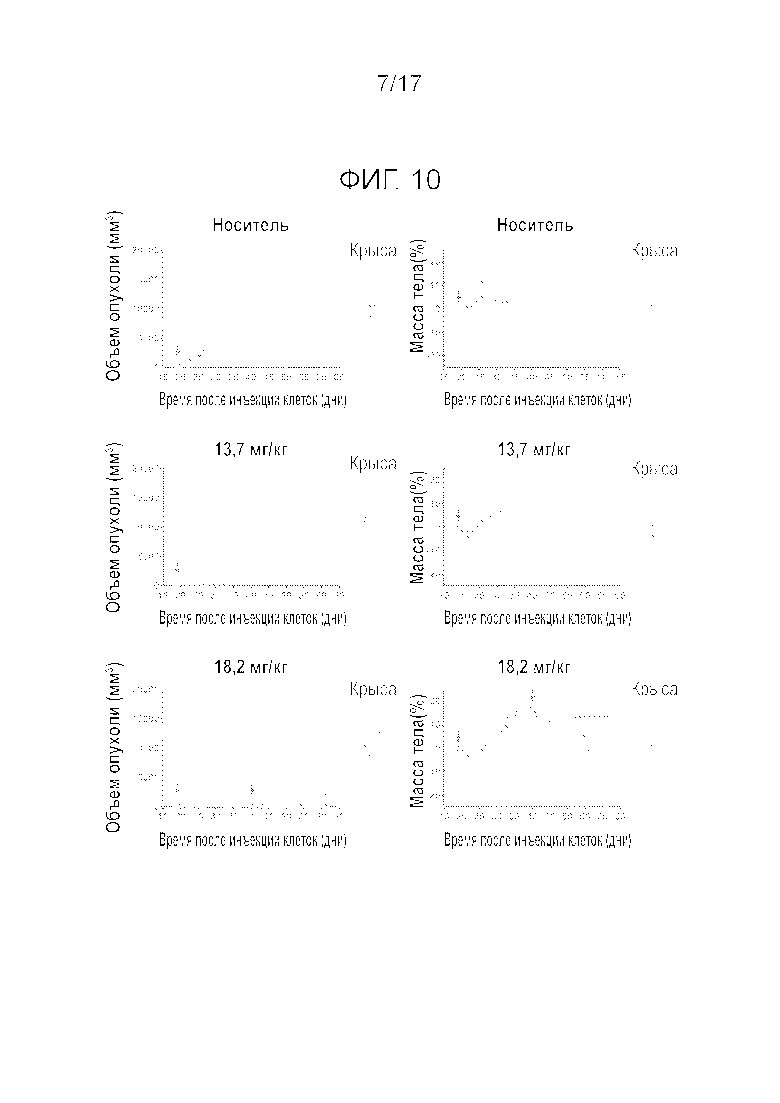
Фиг.10 Демонстрирует рост опухоли и изменение массы тела бестимусных крыс в течение 42 дней q3w i.v. введения соединения A в дозировках 13,7 и 18,2 мг/кг.
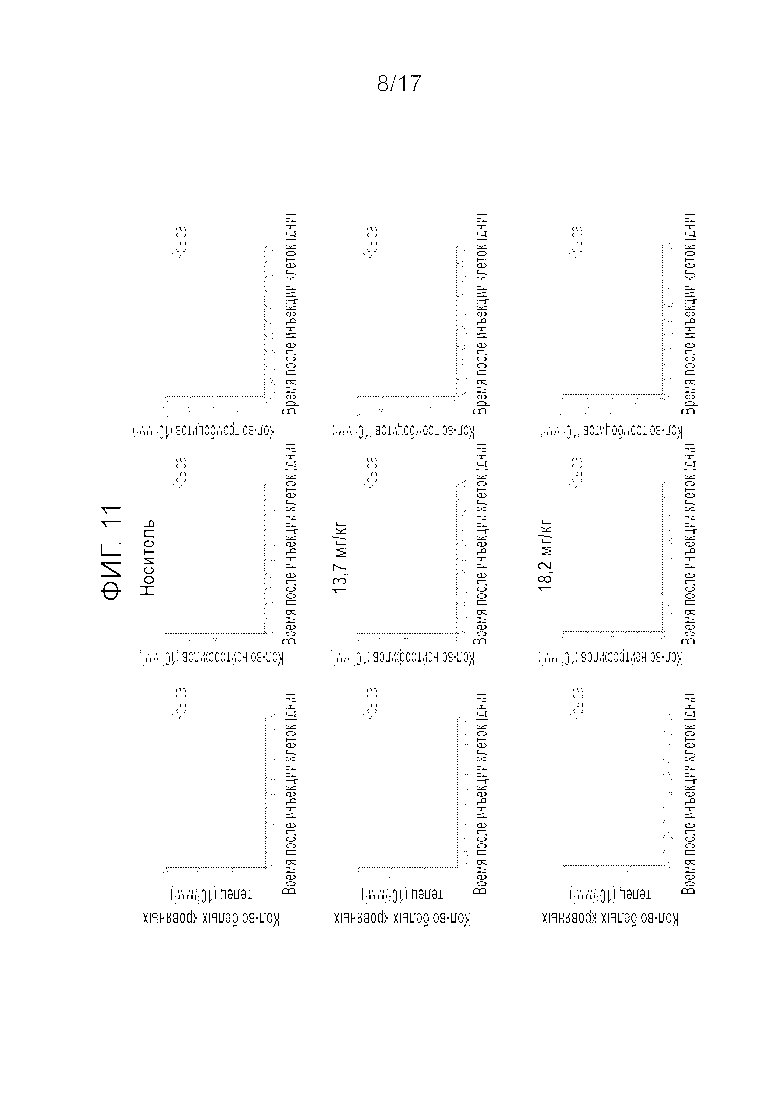
Фиг.11 Влияние i.v. введения (q3w) соединения A в дозировке 13,7 и 18,2 мг/кг на количество белых кровяных телец (WBC), нейтрофилов и тромбоцитов.
Фиг.12 Исследование PK у крыс, несущих опухоль SJSA-1, после однократного введения соединения A per os (перорально).
Фиг.13 Демонстрирует концентрацию лекарственного средства и PD реакцию в опухоли после однократного введения соединения A перорально.
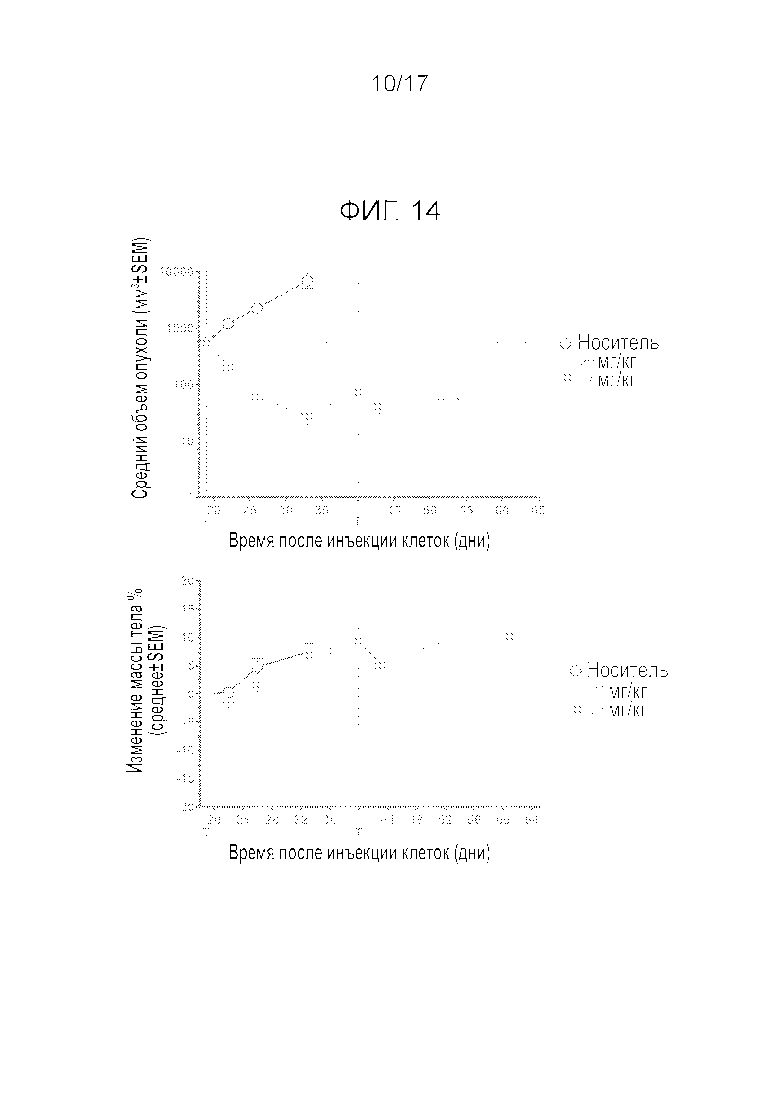
Фиг.14 Демонстрирует рост опухоли и изменение массы тела бестимусных крыс в течение 42 дней при q3w p.o. введении соединения A.
Фиг.15 Демонстрирует количество белых кровяных телец и тромбоцитов в течение 42 дней при q3w p.o. введении соединения A.
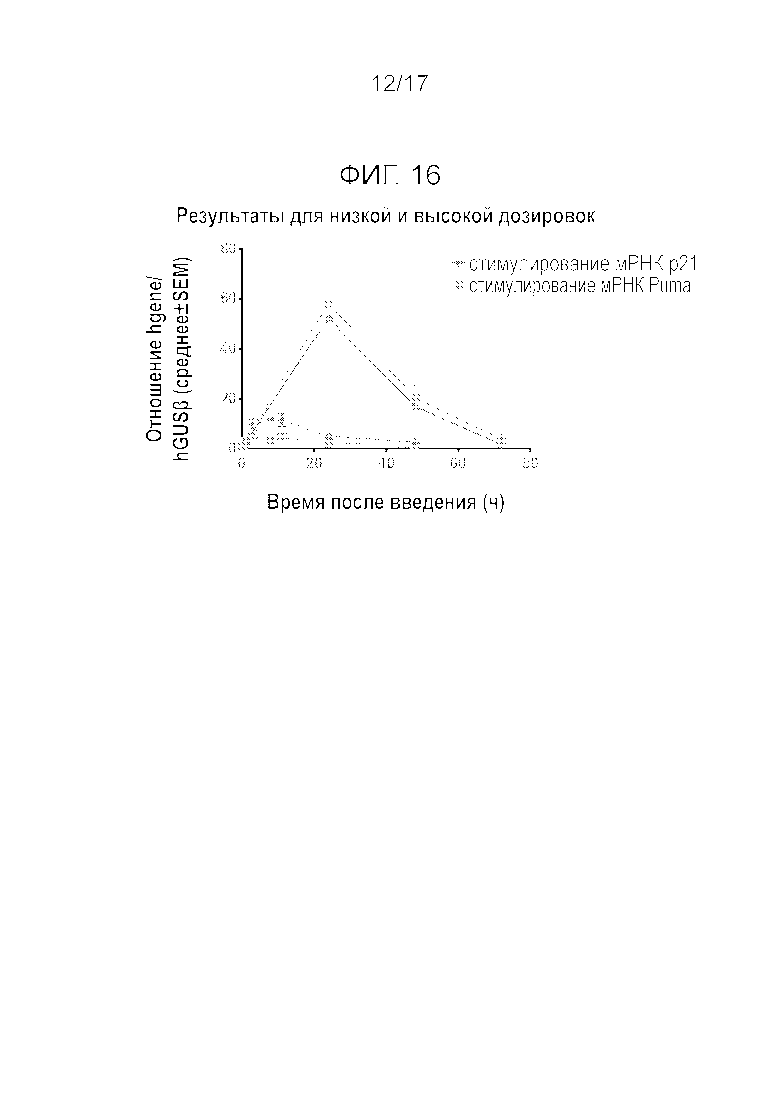
Фиг.16 Низкие дозировки Mdm2i не запускают такой же биохимический эффект, как и высокие дозировки.
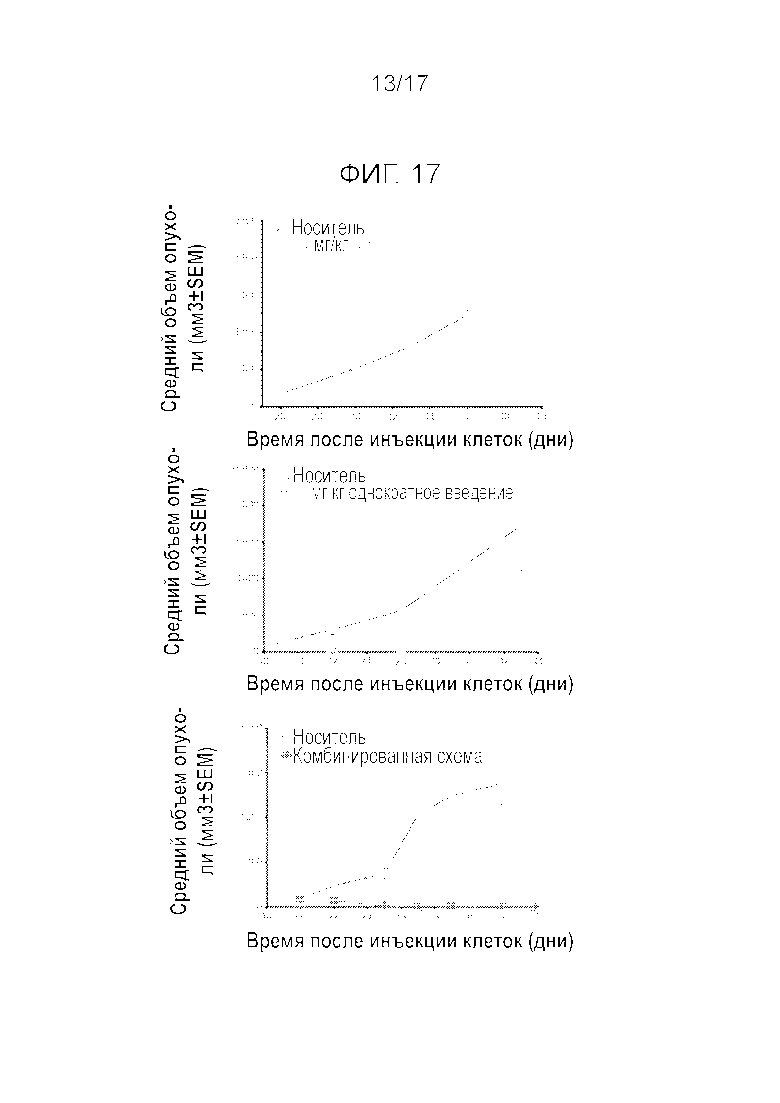
Фиг.17 Комбинация прерывистого и более частого режима введения Mdm2i оказывает синергетическое действие на эффективность.
Фиг.18 Результаты введения соединения A крысе, несущей опухоль SJSA-1, при прерывистом введении высокой дозы, ежедневном введении низкой дозы и комбинированном применении обеих схем введения.
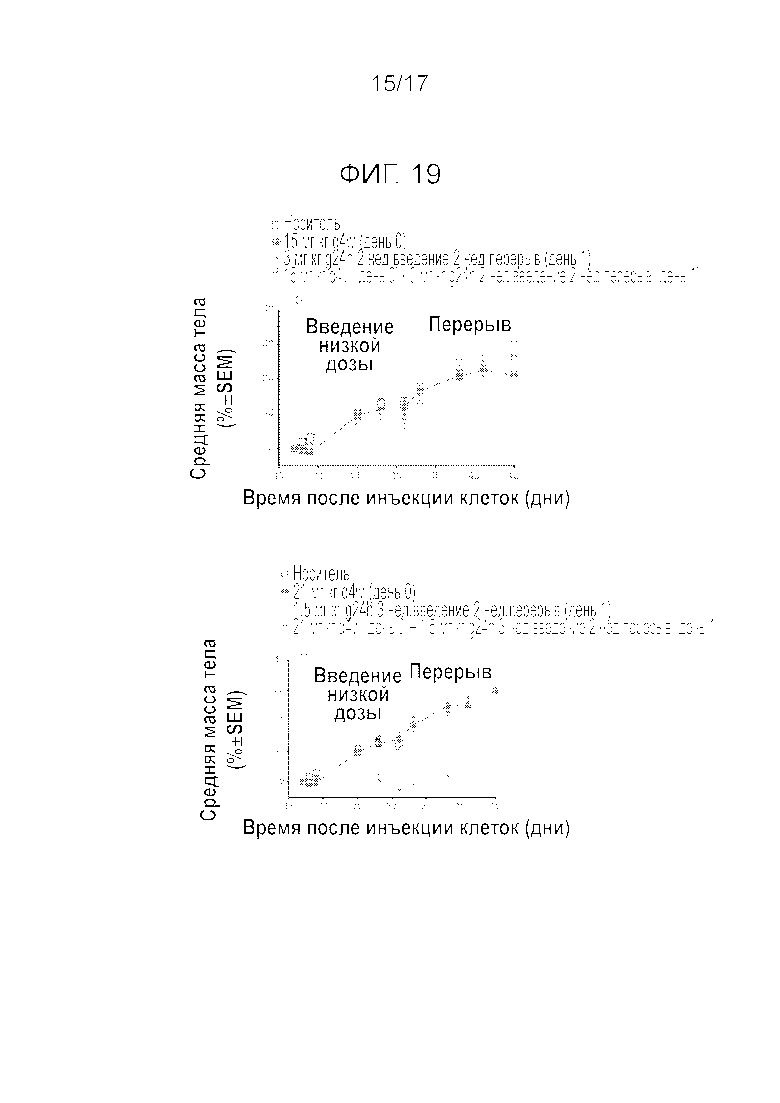
Фиг.19 Переносимость у крыс, несущих опухоль SJSA-1, после введения соединения A в прерывистом режиме в высокой дозировке, ежедневно в низкой дозировке и комбинированном применении обеих схем введения.
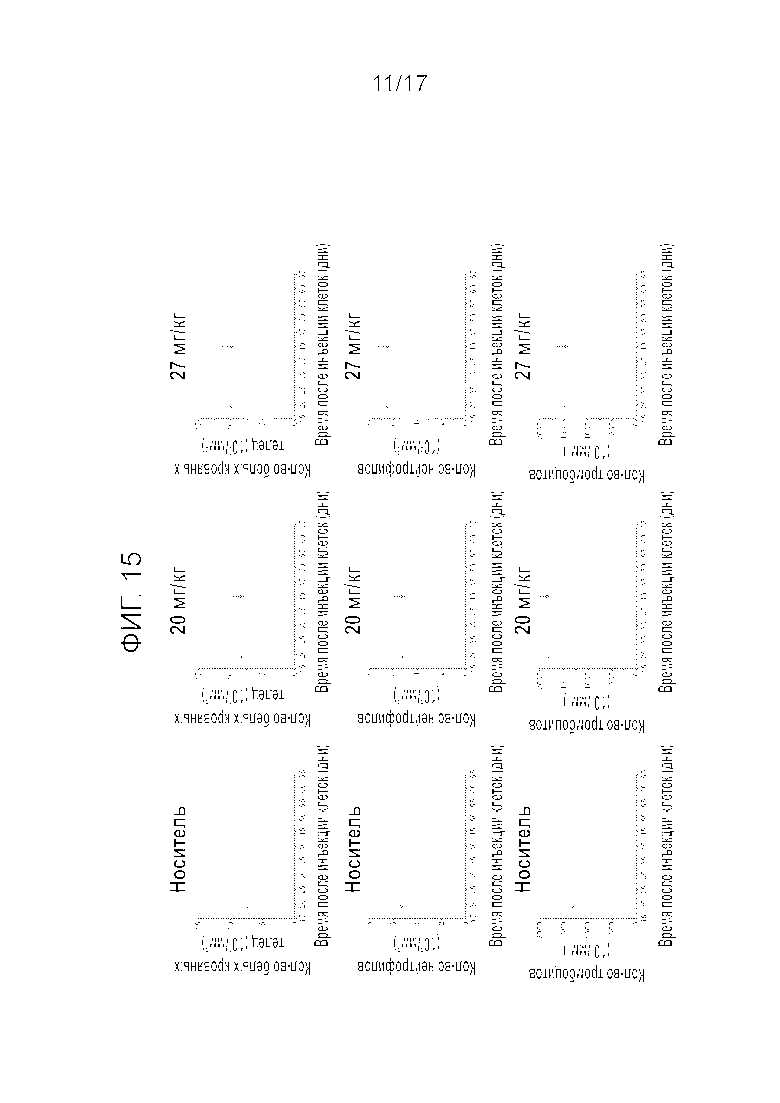
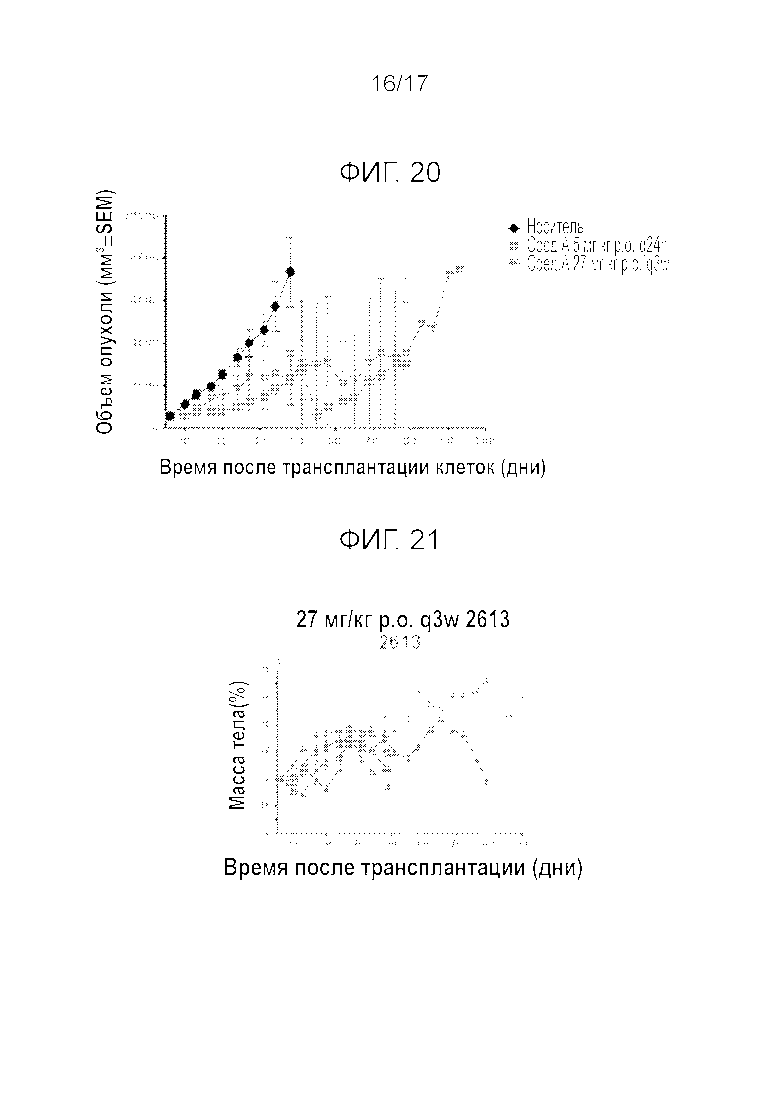
Фиг.20 Эффективность Mdm2i при введении в дозировке 27 мг/кг q3w per os у крыс, несущих меланому PTX. «Cmp A» является сокращением для «Compound A» («Соединение A»).
Фиг.21 Переносимость Mdm2i в дозировке 27 мг/кг q3w per os у крыс, несущих меланому PTX.
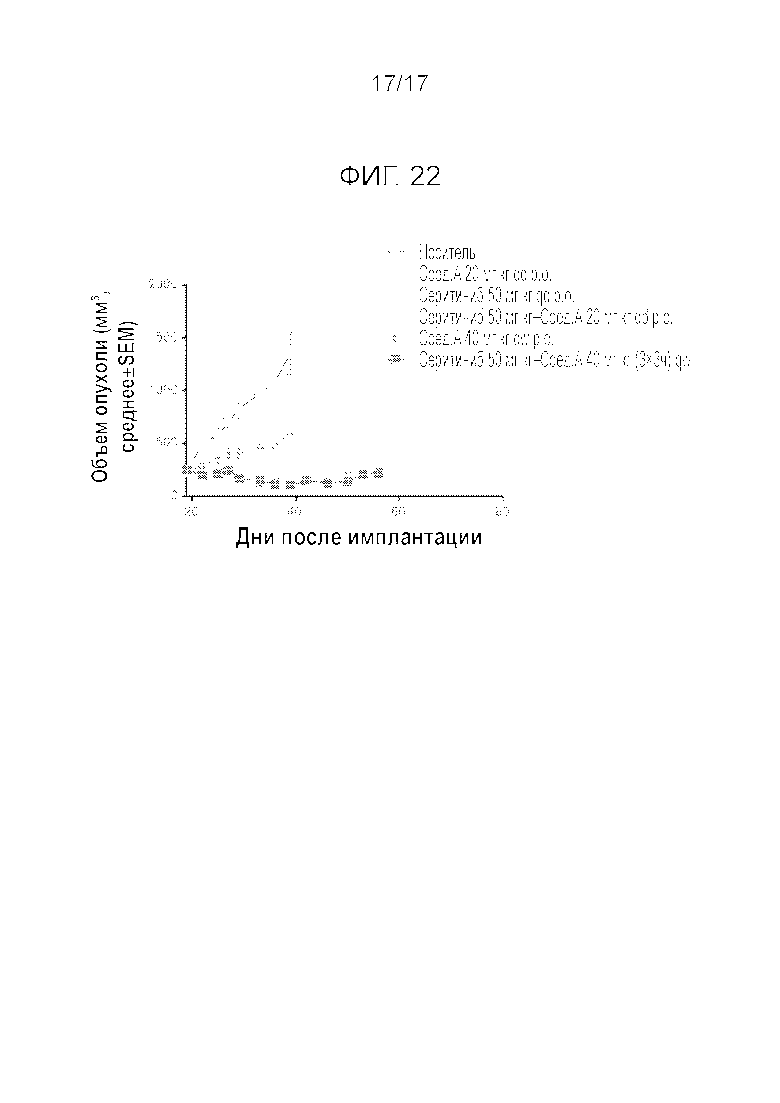
Фиг.22 Эффективность прерывистого введения соединения A в комбинации с серитинибом у мышей, несущих опухоль SHSY5Y. «Cmp A» является сокращением для «Compound A» («Соединение A»).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящее время ингибиторы Mdm2 вводят ежедневно, необязательно с перерывами. Перерыв после серии ежедневных введений в некоторых случаях может оказаться продолжительным из-за проблем с переносимостью. В отдельных случаях ингибиторы Mdm2 вводят с недельными интервалами. В настоящем изобретении было обнаружено, что (S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он (соединение A) при внутривенном или пероральном введении высокой разовой дозы, с первого введения приводит к сильному стимулированию мРНК Puma (стимулирование Emax≥70 крат), чего никогда не наблюдалось ранее при пероральном (per os, p.o.) введении соединения A или (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-она (соединения B). Интересно отметить, что Mdm2, который является ингибитором p53, в наименьшей степени стимулирует индукцию мРНК. После такого сильного стимулирования мРНК Puma в опухоли происходила сильная активация каспазы-3 в течение 24 часов после введения, что приводило к резкому уменьшению плотности опухолевых клеток через 48 и 72 ч после введения. Сильное стимулирование апоптотических путей явно служило основной причиной резкой и неожиданной регрессии опухоли после однократного введения соединения A в высокой дозе. Действительно, однократное i.v. введение соединения A в дозировке 20 мг/кг вызывало полную ремиссию опухоли SJSA-1 (100% регрессию) у 82% (9/11) крыс, получивших препарат, в течение 42 дней. Кроме того, p.o. введение соединения A один раз в три недели (q3w) в дозировке 27 мг/кг приводило к 88% и 27% регрессии опухоли SJSA-1 после одного и двух циклов, соответственно.
Авторы изобретения обнаружили, что для запуска продолжительного апоптоза или устойчивого антипролиферативного эффекта с сильным стимулированием Puma (т.е. стимулированием по крайней мере 20-кратного увеличения экспрессии мРНК по сравнению с экспрессией мРНК в раковых клетках при отсутствии препарата) соединение A необходимо вводить в достаточно высокой дозе. Такая доза обеспечивает возможность введения препарата в прерывистом режиме без значимой потери эффективности и потенциальном улучшении переносимости. Отдельные дозы соединения A можно вводить один раз в две недели. Кроме того, промежутки между введениями, равные 3 неделям, 4 неделям, 6 неделям или даже 60 дням все еще могут демонстрировать значительное воздействие на опухоль. В дозах, меньших, чем указанная высокая доза, соединение A вызывает экспрессию мРНК Puma вплоть до 5-6-кратной, в результате чего необходимо вводить препарат непрерывно, например, ежедневно, для достижения непрерывного антипролиферативного действия. Если лекарственное средство вводят достаточно долго, даже в пониженных дозах, можно сделать перерыв в лечении, но цикл введения необходимо повторять как минимум примерно через 2 недели, в противном случае антипролиферативный эффект больше не наблюдается.
В одном из вариантов осуществления, настоящее изобретение относится к применению Mdm2i для лечения рака, где одиночную дозу Mdm2i необходимо вводить как минимум один раз в две недели, и не реже, чем один раз в 60 дней. В другом варианте осуществления, одиночную дозу Mdm2i необходимо вводить как минимум один раз в три недели, и не реже, чем один раз в 60 дней.
Другие Mdm2i, помимо соединения A, также могут вызывать сильное стимулирование Puma, но дозировка, которую следует применять, зависит от активности соединения. Не ограничиваясь каким-либо конкретной теорией, считается, что дозировка Mdm2i, необходимая для обеспечения продолжительного эффекта, за счет выраженного стимулирования Puma, должна быть тем меньше, чем более активен ингибитор Mdm2i. Но по существу также и Mdm2i с низкой активностью способен активировать этот механизм действия второго уровня, который приводит к долговременному эффекту, только если вводить его в дозировке, при которой достигается значительное содержание действующего вещества в плазме. Примерно 26% регрессии опухоли можно добиться, если концентрация Mdm2i превышает GI80 в течение по крайней мере 8 часов, и более 90%, если содержание Mdm2i устойчиво превышает GI80 в течение по крайней мере 17 часов. GI-80 означает дозировку, которая требуется, чтобы вызвать 80% ингибирование роста опухолевых клеток. Таким образом, как правило, высокая или повышенная доза Mdm2i представляет собой такую дозу, которая позволяет Mdm2i устойчиво сохраняться в плазме in vivo в течение как минимум 8 часов, предпочтительно, как минимум 10 часов, по крайней мере в такой концентрации, которая вызывает GI-80 при воздействии Mdm2i на опухолевые клетки in vitro в течение 8 часов. Концентрацию GI-80 можно измерить с применением любого теста для определения пролиферации. Например, используют люминесцентный анализ для определения выживаемости клеток CellTiter-Glo® Luminescent Cell Viability Assay. Например, клетки in vitro подвергают обработке Mdm2i в течение 8 часов, после чего клетки промывают, удаляя соединение в среде, и через 72 часа проводят определение числа выживших клеток. Описанную методику повторяют для различных концентраций, для определения концентрации GI-80. Указанная высокая доза должна достигать или превышать in vivo определенную описанным способом концентрацию GI-80 Mdm2i в течение как минимум 8 часов. Низкой дозой считается доза, которая меньше самой низкой высокой дозы. К сожалению, введение высоких доз Mdm2i не всегда приводит к достаточно высокому содержанию действующего вещества в плазме просто из-за специфической фармакокинетики соединений, особенно в случае перорального приема, поскольку, например, низкая биодоступность может помешать достижению достаточно высоких уровней препарата в плазме. Этот недостаток перорального пути введения преодолевается в случае внутривенного введения Mdm2i.
Термины «доза», «дозировка» в настоящей заявке в контексте введенной дозы могут также означать силу действия.
Таким образом, одна из целей настоящего изобретения заключается в разработке MDM2i для применения в лечении рака, где MDM2i предполагается вводить субъекту в периодическом режиме, и промежуток времени между как минимум тремя последовательными введениями составляет как минимум 2 недели, как минимум 3 недели, как минимум 4 недели, как минимум 6 недель или 60 дней, и не превышает 60 дней. MDM2i необходимо вводить субъекту в прерывистом режиме в виде как минимум трех последовательных доз, и промежуток времени между введением каждых двух последовательных доз из указанных трех доз составляет как минимум 2 недели, как минимум 3 недели, как минимум 4 недели, как минимум 6 недель или 60 дней. Верхний предел установлен на основании имеющихся данных, но авторы допускают возможность, что еще более редкое введение может привести к клинически приемлемому результату и могло бы оказаться применимыми. Для повышения приверженности пациента лечению, режим введения MDM2i может включать введение один раз в 3 или 4 недели, конкретно один раз в 3 недели.
Авторы обнаружили, что проблему недостаточной концентрации Mdm2i в организме, в особенности, если IC50 для клеточной пролиферации составляет более 1 мкМ, можно решить путем внутривенного введения лекарственного средства. В качестве примера, авторы обнаружили, что внутривенно можно вводить более низкие дозировки соединения A (20 мг/кг), причем для достижения аналогичной реакции необходимо пероральное введение в дозировке 27 мг/кг. Таким образом, внутривенное введение Mdm2i обеспечивает возможность применения Mdm2i с более низкой активностью для достижения упомянутой выше второй стадии реакции с долговременным антипролиферативным эффектом. Таким путем достигается возможность вводить лекарственное средство менее часто, поскольку только внутривенное введение приведет к достижению необходимой концентрации в организме. Кроме того, внутривенное введение Mdm2i в более низкой дозировке по сравнению с той, которая потребовалась бы при пероральном введении, способно обеспечить по крайней мере определенные преимущества с точки зрения переносимости.
Таким образом, в одном из вариантов осуществления, авторы разработали Mdm2i для применения в лечении рака, где Mdm2i следует вводить внутривенно.
В другом варианте осуществления, прерывистое введение MDM2i можно дополнить другой схемой введения второй дозы MDM2i, которая отличается от дозы, применяемой для прерывистого введения. Комбинирование прерывистой схемы введения с другой схемой, предполагающей более частое введение, дает возможность уменьшить дозировку Mdm2i, применяемую в каждой из схем, и за счет этого дополнительно улучшить переносимость. Прерывистое введение высокой дозы Mdm2i при одновременном более частом введении Mdm2i, например, ежедневном, в более низкой дозировке, позволяет уменьшить дозировку в обеих схемах до такого уровня, который при иной схеме введения был бы неэффективным, как минимум в одной из двух схем введения, по сравнению с индивидуальной реализацией указанных схем введения. Комбинирование лечения высокими и низкими дозами по разным схемам также оказывается синергетически эффективным. В одном из вариантов осуществления, прерывистое введение, при котором Mdm2i вводят как минимум один раз в 2 недели, можно дополнить ежедневным введением Mdm2i. Авторы обнаружили, что комбинирование двух схем введения соединения A, а именно введения высокой дозы один раз в 3 недели и 2-недельного ежедневного введения низкой дозы с 2-недельным перерывом каждый 28-дневный цикл, приводит к синергетическому противоопухолевому эффекту обеих схем введения. Вторую схему введения, которую добавляют к прерывистому введению, можно начинать в тот же, следующий или какой-либо другой день. Вторая схема введения может являться, например, ежедневной, необязательно, с перерывом. Перерыв после ряда ежедневных введений может иметь продолжительность как минимум 1 день, 2 дня, 3 дня, 4 дня, 1 неделю, 2 недели или 3 недели, и максимум 26 дней. В одном из вариантов осуществления, введение препарата по второй схеме следует осуществлять с 1 по 14 дни после введения первой дозы. В конкретном варианте осуществления, вторую схему введения с более низкой дозировкой Mdm2i начинают реализовывать на следующий день после введения одиночной высокой дозы. Дозу, которую вводят ежедневно, можно вводить в течение двух недель с последующим двухнедельным перерывом и затем описанный цикл введения можно повторить. Как правило, дозировка, используемая при прерывистом введении, должна быть выше, чем вторая дозировка, используемая при более частом введении, которое проводят в дополнение к прерывистому введению. Mdm2i можно вводить либо перорально, либо внутривенно, или комбинируя эти пути. Например, дозу прерывистого введения можно вводить внутривенно один раз в 2, 3, 4, 6 недель или 60 дней, тогда как вторую ежедневную дозу можно вводить перорально. Однако обе дозы можно вводить внутривенно, или обе перорально. В одном из вариантов осуществления, первую дозу, которую вводят в прерывистом режиме, можно вводить в периоды между двумя последовательными введениями, которые составляют как минимум 2 недели.
В одном из аспектов, вторая схема введения, которую добавляют к прерывистой схеме введения, может включать введение Mdm2i в течение периода как минимум 5 дней с последующим перерывом 1 день или более, и с повторением указанного цикла во время лечения пациента по прерывистой схеме с применением другой дозировки. Однако дополнительная вторая схема введения включает, например, циклы 2 недели введение, 1 или 2 недели без введения; 3 недели введения, 1,2 или 3 недели без введения; 4 недели введения, 1, 2, 3 или 4 недели без введения; 1 неделя введения, 3 недели без введения; 3 недели введения, 1 неделя без введения; 4 недели введения, 1 неделя без введения.
Помимо добавления к прерывистой схеме введения второй схемы, клинический результат лечения MDM2i прерывистым введением можно улучшить путем введения субъекту дополнительного фармацевтического ингредиента. Этот дополнительный фармацевтический ингредиент может являться другим Mdm2i, но чаще всего он будет являться лекарственным средством с другим механизмом действия. В изобретении подразумевается, что введение другого антинеопластического агента в дополнение к прерывистому введению Mdm2i может позволить добиться улучшения противоопухолевого эффекта. Кроме того, прерывистое введение обеспечивает более гибкое комбинирование Mdm2i с другим антинеопластическим агентом поскольку за счет уменьшения частоты введения Mdm2i можно улучшить переносимость, что дает более широкие возможности применения дополнительного противоракового средства. В одном из вариантов осуществления, Mdm2i вводят в прерывистом режиме, как описано в тексте заявки, в комбинации с ингибитором BRAF или ингибитором ALK. Конкретно, другой фармацевтический ингредиент представляет собой (S)-метил-1-(4-(3-(5-хлор-2-фтор-3-(метилсульфонамидо)фенил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат. В другом варианте осуществления, применяют комбинацию с серитинибом. Если Mdm2i применяют в комбинации с другим фармацевтическим ингредиентом, Mdm2i можно вводить в прерывистом режиме с промежутками между отдельными введениями как минимум 1 неделя, как минимум 2 недели, как минимум 3 недели, как минимум 4 недели, как минимум 6 недель или 60 дней, и не более 60 дней.
Кроме того, настоящее изобретение относится к соединению A для применения в лечении, где соединение A вводят в прерывистом режиме например, промежуток между каждыми двумя введениями из как минимум трех введений составляет как минимум 1 неделю, как минимум 2 недели, как минимум 3 недели, как минимум 4 недели, как минимум 6 недель или 60 дней, и не более 60 дней, и, кроме того, применяется (S)-метил-1-(4-(3-(5-хлор-2-фтор-3-(метилсульфонамидо)фенил)-1-изопропил-1H-пиразол-4-ил)пиримидин-2-иламино)пропан-2-илкарбамат или серитиниб. В частности, соединение A вводят в количестве одной дозы как минимум один раз в неделю, или как минимум один раз в три недели.
Mdm2i и дополнительный фармацевтический ингредиент могут применяться или входить в состав отдельных лекарственных форм, и включать или не включать, предпочтительно, включать инструкции по комбинированному применению или комбинированным продуктам. Таким образом, соединения, входящие в комбинацию, могут вводиться полностью раздельно или включаться в отдельные фармацевтические дозированные формы. Партнеры по комбинации могут представлять собой фармацевтические композиции, которые также продаются независимо друг от друга и в этом случае к упаковке прилагаются только инструкции по их комбинированному применению, например, в виде листовки-вкладыша и т.п., или другая информация, например, предназначенная для врача или медицинского персонала (например, устная информация, информация в письменной форме и т.п.) относительно их одновременного или последовательного применения, чтобы они проявляли совместную активность. Mdm2i и другой действующий фармацевтический ингредиент могут применяться в виде фиксированной или не фиксированной комбинации действующих ингредиентов. Термин «фиксированная комбинация» означает, что действующие ингредиенты, например, ингибитор Mdm2 и антинеопластический агент, вводятся пациенту одновременно в виде единой лекарственной или дозированной формы. Другими словами, действующие ингредиенты находятся в одной дозированной форме, например, в одной таблетке или в одной капсуле. Термин «не фиксированная комбинация» означает, что оба действующих ингредиента вводятся пациенту в отдельных лекарственных формах, либо одновременно, параллельно или последовательно без определенных временных ограничений, причем такое введение обеспечивает терапевтически эффективные уровни двух указанных соединений в организме пациента.
Раковые заболевания, которые можно лечить с применением Mdm2i, как описано в настоящей заявке, включают такие разновидности рака, как, но не ограничиваясь перечисленными: рак мочевого пузыря, груди, мозга, органов головы и шеи, печени, ротовой полости, желчевыводящих путей, острый и хронический лимфоидный лейкоз, острый и хронический миелоидный лейкоз, хронический миеломоноцитарный лейкоз, колоректальный рак, рак желудка, желудочно-кишечную стромальную опухоль, гепатоклеточный рак, глиому, лимфому, меланому, множественную миелому, миелопролиферативную болезнь, нейроэндокринный рак, рак легких, немелкоклеточный рак легких, рак поджелудочной железы, яичников, простаты, почечноклеточный рак, саркому, липосаркому и рак щитовидной железы. В конкретных вариантах осуществления, рак является меланомой. В другом варианте осуществления, рак является нейробластомой. В еще одном варианте осуществления, раковое заболевание является лейкемией.
Основываясь на данных, полученных для соединения A, а также с учетом известной биохимической реакции на (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он (соединение B), можно ожидать что предложенные схемы введения могли бы применяться для достижения высокой эффективности или переносимости по крайней мере для перечисленных выше Mdm2i.
Доставку Mdm2i субъекту можно осуществлять в составе фармацевтической композиции. Подходящими для применения пероральными лекарственными формами являются, например, таблетки, капсулы, саше, микрогранулы, гранулы и т.п. Пероральные дозированные формы помимо Mdm2i могут включать другие стандартные носители или эксципиенты, применяемые для изготовления фармацевтических препаратов. Примеры таких носителей или эксципиентов включают, не ограничиваясь перечисленными, дезинтегрирующие средства, связующие вещества, смазывающие компоненты, средства для скольжения, стабилизаторы, а также наполнители, разбавители, красители, вкусоароматические компоненты и консерванты. Рядовой специалист в данной области техники может выбрать одну или несколько из перечисленных выше добавок в соответствии с конкретно желаемыми свойствами дозированной формы в результате проведения стандартных экспериментов и без неприемлемых затрат. Количество каждого из применяемых носителей можно менять в пределах принятых в технике диапазонов. В приведенных ниже источниках раскрыты методики и эксципиенты, применяемые для получения пероральных дозированных форм. Смотрите The Handbook of Pharmaceutical Excipients, 4th edition, Rowe et al., Eds., American Pharmaceuticals Association (2003); и Remington: the Science and Practice of Pharmacy, 20th edition, Gennaro, Ed., Lippincott Williams & Wilkins (2003). Дозированные формы получают, например, смешиванием, гранулированием, прессованием, сжатием, наполнением, просеиванием, смешиванием и/или таблетированием.
Mdm2i могут применяться in vivo внутривенно, например, в форме раствора. Как правило, дозированные формы перед введением можно подвергнуть обработке в автоклаве или стерилизовать другими способами. Внутривенное введение препаратов можно осуществлять путем инъекции или инфузии. Предпочтительно, Mdm2i вводят внутривенной инфузией в течение периода времени менее 3 часов, более предпочтительно в течение периода до 2 часов, в частности, в течение периода примерно 1 час.
Mdm2i можно применять для получения лекарственного препарата, причем препарат изготавливают в виде дозированной лекарственной формы. Последние затем можно поместить в упаковку и снабдить информационной листовкой-вкладышем для пациента.
Mdm2i вводят в терапевтически эффективном количестве. Термин «терапевтически эффективное количество» Mdm2i относится к количеству соединения, которое вызовет биологическую или медицинскую реакцию у субъекта, например, облегчение симптомов, улучшение состояния, замедление или отсрочивание прогрессирования заболевания, замедление роста опухоли, регрессию опухоли или подобную реакцию. В одном из вариантов осуществления, терапевтически эффективное количество in vivo в зависимости от пути введения может находиться в диапазоне примерно 0,1-500 мг/кг, или примерно 1-100 мг/кг. Например, для соединения A эффективное количество in vivo находится в пределах от 100 до 1500 мг один раз в три недели, в частности от 100 до 800 мг один раз в три недели, или от 50 до 600 мг ежедневно при пероральном введении. Для соединения B, эффективное количество находится в пределах от 500 до 4000 мг, в т.ч. от 1500 до 4000 мг, при пероральном введении. Внутривенные дозировки можно было бы снизить соответствующим образом.
Приведенные ниже по тексту примеры иллюстрируют настоящее изобретение.
ПРИМЕРЫ
Методики и материалы, использованные в примерах
Соединение A: (S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-он;
Соединение B: (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он;
Клеточная культура
Клетки остеосаркомы SJSA-1 (CRL-2098, ATCC) являлись клетками дикого типа по белку p53 и амплифицированы по Mdm2 (16,9 копий SNP 6,0), но не по Mdm4. Клетки культивировали в RPMI 1640 (#1-41F01-I, AMIMED) с добавкой 10% FCS (#2-01F16-I, AMIMED), 2 мМ L-глутамина(#5-10K00-H, AMIMED). Пассирование клеток осуществляли, во-первых, промыванием PBS Дульбекко без Ca2+/Mg2+ (#3-05F29-I, AMIMED), трипсинизацией клеток 0,05% трипсином в PBS с EDTA (#5-51F00-H, AMIMED), центрифугированием в соответствующей культуральной среде и разделением клеток в свежей среде в соотношении 1:8, 2 раза в неделю.
Животные
Всем бестимусным крысам (Hsd:RH-Fox1mu, Harlan Sprague Dawley; SF480) давали адаптироваться в течение 4 дней и поселяли в условиях с контролируемым содержанием патогенов (5 особей/клетка типа III) с неограниченным доступом к пище и воде. Животных идентифицировали с помощью транспондеров. Исследования, описанные в данной заявке, проводили в соответствии с методиками, которые подпадают под разрешение №1975, выданное Kantonales Veterinäramt Basel-Stadt, и строго следовали Eidgenössisches Tierschutzgesetz и Eidgenössische Tierschutzverordnung. Все эксперименты проводили с участием 4-7 крыс. Мышей использовали для экспериментов с комбинациями соединений.
Модели опухолей
Образование подкожных опухолей инициировали, помещая 1×107 клеток SJSA-1 в 50% Matrigel® и осуществляя инъекцию полученного препарата в правый бок бестимусных крыс Harlan. Эксперименты по определению эффективности начинали через 14 дней после инъекции клеток. Для каждого введения готовили свежий препарат соединения A. Для i.v. инъекции (4 мл/кг) соединение A растворяли в смеси 30% PEG300, 10% Solutol HS 15, 6% Pluronic F68 и 54% воды. Для перорального (p.o.) введения (5 мл/кг), соединение A растворяли в растворе метилцеллюлозы 0,5% масса/объем в фосфатном буфере pH 6,8 50 мМ. Животным вводили либо высокую дозу (20 мг/кг i.v. или 27 мг/кг p.o.) один раз в три недели (q3w) или высокую дозу (15 мг/кг p.o.) и затем через 24 часа осуществляли ежедневное введение низкой дозы (3 мг/кг p.o., 2 недели введение/2 недели перерыв).
Объем опухоли (TVol) и массу тела (BW) животных определяли три раза в неделю, что позволяло вычислять процентное изменение параметра TVol (Δ%TVol) в любой конкретный момент времени относительно дня начала введения препаратов (дня 0). Реакцию опухоли количественно определяли по изменению объема опухоли (конечное значение минус исходное значение в мм3) как T/C, т.е. 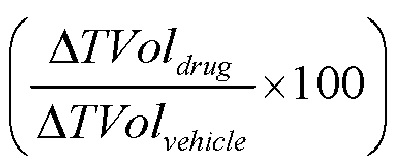 . В случае регрессии опухоли или для получения процентного изменения TVol, реакцию опухоли количественно выражали в процентах регрессии исходного TVol, т.е.
. В случае регрессии опухоли или для получения процентного изменения TVol, реакцию опухоли количественно выражали в процентах регрессии исходного TVol, т.е. 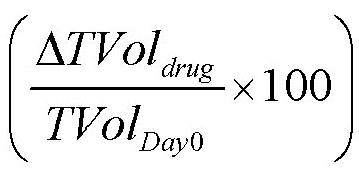 .
.
Аналогично, массу тела (BW) животного измеряли три раза в неделю что позволяло вычислять процентное изменение параметра BW (Δ%BW) в любой конкретный момент времени относительно дня начала введения препаратов (дня 0).
Белые кровяные тельца (WBC), нейтрофилы и тромбоциты подсчитывали с помощью Sysmex (XT-2000i). Кровь собирали в микропробирки, покрытые EDTA, промышленного производства (BD Microtainer, cat#365975).
Фармакокинетика (PK) и фармакодинамика (PD)
В указанные моменты времени, животным проводили анестезию действием 2-3% (объем/объем) изофлурана в медицинском кислороде.
- либо осуществляли умерщвление животных без выхода из анестезии после отбора крови. Кровь собирали в пробирки, покрытые EDTA, промышленного производства (Milian, cat# TOM-14C) для отделения плазмы. Ткани иссекали, взвешивали и быстро замораживали в жидком азоте.
- либо отбирали биопсию опухоли, используя биопсийный пистолет и смывая содержимое иглы буфером RLT в пробирки Barney (Covaris, cat #520048). Кроме того, из хвостовой вены мог осуществляться отбор 20 мкл крови, и образец крови разбавляли 20 мкл воды. После выхода из анестезии, животных переносили в соответствующие клетки.
Образцы тканей, крови и плазмы хранили в замороженном состоянии при -80°C до исследования.
Получение препаратов тканей
Замороженные ткани подвергали криогенному высушиванию, измельчали и образцы биопсий обрабатывали ультразвуком, используя систему CryoPrepTM (модель CP-02) фирмы Covaris. Более конкретно, замороженные ткани переносили в одноразовые пробирки, именуемые TissueTubesTM, помещали в систему CryoPrepTM и затем измельчали, используя подходящие установки силы импульса. Полученный порошок собирали шпателем и взвешивали, после чего подвергали дальнейшей переработке (очистке мРНК или количественному определению соединения в тканях). Биопсии смывали в стеклянные пробирки Barney 350 мкл буфера RLT и помещали в прибор Covaris для обработки ультразвуком (1 мин на биопсию). Полученный лизат переносили в колонку QIAshredder (79654, Qiagen) для выделения РНК.
Фармакодинамика (qRT-PCR)
РНК полностью выделяли из лизата клеток с использованием колонки QIAshredder (79654, Qiagen) и набора RNeasy Mini Kit (74106, Qiagen) согласно инструкциям производителей, за исключением того, что не осуществляли расщепление ДНК. Всю РНК элюировали 50 мкл воды, не содержащей РНКаз. Суммарное количество РНК определяли с помощью спектрофотометра ND-1000 Nanodrop®. qRT-PCR (количественную полимеразную цепную реакцию с обратной транскриптазой) осуществляли в трех повторах для каждого образца, используя One-Step RT qPCR Master Mix Plus (RT-QPRT-032X, Eurogentec) либо с контрольными праймерами, либо с праймерами для мишеней, а именно набором для анализа экспрессии генов TaqMan Gene Expression (20× детектирующий краситель FAMTM (или VIC)-TAMRA (или MGB); Applied Biosystems), перечисленными в таблице 1.
Таблица 1: источник праймеров для qRT-PCR
Фармакокинетика
Получение образцов и методика биоанализа
Концентрации соединения A плазме и тканях определяли одновременно способом UPLC/MS-MS (Сверхпроизводительная ЖХ/МС-МС). Ткани гомогенизировали в равном объеме воды для ВЭЖХ (вода для хроматографии, Merck), используя систему FastPrep®-24 (M.P.Biomedicals, Irvine, CA, USA). После добавления к анализируемым аликвотам (25 мкл) плазмы или гомогенатов тканей 25 мкл смеси, используемой в качестве внутреннего стандарта (1 мкг/мл), белки осаждали добавлением 200 мкл ацетонитрила. Супернатант переносили в новые флаконы. После упаривания досуха, образцы повторно растворяли в 60 мкл смеси ацетонитрил/вода (1/1 объем/объем). Аликвоты (5 мкл) этого раствора разделяли на колонке ACQUITY UPLC BEH C18 (WatersTM, размер частиц 1,7 мкм, 2,1×50 мкм) с использованием подвижной фазы, состоящей из смеси 0,1% раствора муравьиной кислоты в воде (растворитель A) и 0,1% раствора муравьиной кислоты в ацетонитриле (растворитель B). Использовалось программирование градиента при скорости потока 600 мкл/мин. После достижения равновесия при 95% содержании растворителя A, осуществляли введение 5 мкл образца. После периода задержки 0,25 мин, образец элюировали линейным градиентом 5-100% растворителя B в течение 0,65 минут с последующим периодом удерживания 0,35 мин. Подготовку колонки к следующему образцу осуществляли повторным приведением к равновесию в течение 0,25 мин в исходных условиях. Элюат из колонки вводили непосредственно в источник ионов тройного квадрупольного масс-спектрометра TQDTM (Waters Corporation, Milford, MA, USA), который находился под управлением программы MasslynxTM 4.1. Для МС/МС исследования аналита использовали положительную ионизацию электрораспылением (ESI+) и мониторинг множественных реакций. Для определения соединения A использовали превращение иона предшественника в ион продукта m/z 555,3 → m/z 329,2. Предел количественного определения для соединения (LQD) устанавливали равным 0,7 нг/мл (CV и суммарное смещение менее 30%). Проводили регрессионный анализ и дополнительные вычисления, используя QuanLynxTM 4.1 (Micromass) и ExcelTM 2007 (Microsoft). Концентрации неизвестных образцов получали обратным пересчетом на основе соотношения площадей пиков аналит/IS (внутр.стандарт) из калибровочной кривой, полученной с использованием калибровочных образцов, добавленных в холостую плазму или ткань, отобранные у животных, получавших носитель.
Вычисление фармакокинетических параметров
Площади под кривыми зависимости концентрации в плазме от времени (AUC) вычисляли из средних значений с помощью линейного правила трапеций, и другие необходимые параметры рассчитывали с использованием некомпартментной модели для внесосудистого введения (WinNonlin® Professional Version 5.2, Pharsight corp., CA, US).
Иммуно-гистохимия
Все ткани обрабатывали FFPE согласно стандартным методикам, и после фиксации грудинные кости крыс декальцинировали в буфере цитрат/EDTA в течение 5 дней, причем замену буфера осуществляли один раз в 24 часа. Делали срезы толщиной 3 мкм, используя микротом. Иммуногистохимию p21 и расщепленной каспазы-3 осуществляли на автоматизированной установке для иммуноокрашивания Ventana Discovery XT, используя вторичный реагент OmniMap антитело против мышиной или кроличьей HRP и хромогенную систему ChromoMap DAB (Ventana/Roche Diagnostics GmbH, Mannheim, Germany). Демаскирование антигена осуществляли с использованием Cell Conditioning Discovery CC1 (Ventana/Roche Diagnostics) в мягких (95°C 8 мин+100°C 20 мин для расщепленной каспазы-3) или стандартных (95°C 8 мин+100°C 36 мин для p21) условиях. Первичные антитела наносили вручную в желаемом разбавлении в разбавителе для антител Dako, после чего инкубировали в течение 1 часа при комнатной температуре. Соответствующие образцы для отрицательного контроля инкубировали только с AbD. Контрастирующее окрашивание срезов осуществляли с помощью гематоксилина (Ventana/Roche Diagnostics). После осуществления автоматического окрашивания, срезы дегидратировали в серии растворов этанола с постепенно повышающейся концентрацией, осветляли и заключали в гистологическую среду Pertex. Первичные антитела, используемые для иммуногистохимии, описаны в таблице 2
Таблица 2: Антитела, использованные для иммуногистохимии
В приведенной таблице показаны источники антител, применяемых для иммуногистохимии, а также их разбавления.
Гибридизация мРНК in situ
Гибридизацию in situ проводили с использованием аналитического набора QuantiGene ViewRNA FFPE (Affymetrix/Panomics), следуя протоколу производителя. Наборы ген-специфичных зондов для мРНК крысиного Ubc (убиквитина C) и Bbc3 (PUMA) были разработаны специально для данного анализа и синтезированы Affymetrix. Зонды Bbc3 использовались в комбинации с красителем тип 1/прочный красный, и зонды Ubc использовались в комбинации с красителем тип 6/прочный синий. Препараты обрабатывали в строгом соответствии с протоколом QuantiGene. Было обнаружено, что условия предгибридизации являются оптимальными при 10 мин кипячении в растворе для предгибридизации (Affymetrix) и 10 мин расщеплении Protease QF (Affymetrix) при 40°C. Вкратце, нарезали срезы толщиной пять микрометров, фиксировали в 10% формальдегиде, депарафинизировали и регидратировали. Затем для повышения доступности мРНК, препараты кипятили в растворе для предварительной обработки (Affymetrix) и расщепляли protease QF (Affymetrix) в оптимальных условиях. Затем срезы подвергали гибридизации в течение 3 ч при 40°C со специально разработанными зондами QuantiGene ViewRNA для Bbc3 и управляющего гена Ubc. Образец, не содержащий зондов, использовали в качестве отрицательного контроля, следуя рекомендациям инструкции Affymetrix. После гибридизации несвязанные зонды вымывали буфером (Affymetrix), тогда как связанные зонды затем подвергали амплификации, следуя протоколу Affymetrix (разветвленная амплификация ДНК), с использованием PreAmp (25 мин при 40°C), затем молекул Amp (15 мин при 40°C) и, наконец, многокомпонентных олигонуклеотидов Label Probe, конъюгированных со щелочной фосфатазой (LP-AP) в течение 15 мин при 40°C. Детектирование сигналов зонда LP-AP типа 6 осуществляли с помощью субстрата Fast Blue (прочный синий) (синие точки, флуоресценция Cy5) в течение 30 мин при КТ в темноте, и затем детектирование сигналов зонда LP_AP типа 1 с помощью субстрата Fast Red (прочный красный) (красные точки, флуоресценция Cy 3) в течение 30 мин при 40°C. После обнаружения сигнала, стекла с препаратами подвергали контрастному окрашиванию гематоксилином Майера и осуществляли заливку/закрывали покровными стеклами с использованием гистологической среды на водной основе Ultramount aqueous mounting medium (DAKO). Изображения регистрировали на микроскопе Olympus BX51, снабженным цветной камерой ColorView III (Soft Imaging System).
Зонды, использованные для мРНК ISH, описаны в таблице 3.
Таблица 3: Зонды, использованные для мРНК ISH
Пример 1: Фармакокинетика (PK) соединения A после однократной i.v. инъекции в дозировке 20 мг/кг
На Фиг.1 показаны концентрации соединения A в плазме, опухоли и печени в течение 144 часов после однократной i.v. инъекции. Tmax для соединения в плазме и печени составляло 5 мин, а в опухоли равнялось 1 часу. Содержание соединения A в опухоли было в два раза выше (AUC0-144hdn=16,5 ч⋅нмоль/г) по сравнению с содержанием в плазме (AUC0-144hdn=8,1 ч⋅мкмоль). На Фиг.2 показана концентрация соединения A в опухоли и фармакодинамическая (PD) реакция в опухоли. Стимулирование мРНК Puma и p21 было очень близким, достигая максимума экспрессии, равного 180- и 200-кратному увеличению, через 24 ч после введения, соответственно.
Пример 2: PK, PD, эффективность и переносимость соединения A (i.v. однократное введение) у крыс, несущих опухоль SJSA-1
На Фиг.3 и 4, соответственно, показан рост опухоли и изменение массы тела бестимусных крыс в течение 42 дней. Однократное i.v. введение соединения A в дозировке 20 мг/кг (высокая дозировка) вызывало 92% регрессию опухоли через 14 дней после введения. Одну крысу пришлось умертвить в день 9 после введения из-за слишком сильного уменьшения массы тела (BW). Несмотря на небольшое понижение BW через три дня после введения у всех остальных крыс, они быстро восстановились и продемонстрировали увеличение массы тела за время эксперимента. Только у 2 из 11 опухолей наблюдалась неполная реакция и повторный рост (Фиг.5). Этим двум животным осуществили повторное i.v. введение в дозировке 15 мг/кг, к которому опухоли оказались по-прежнему чувствительными. Тем не менее, регрессия опухоли оказалась более слабой, причиной чего мог являться более крупный размер опухоли. После первого введения экспрессия мРНК p21 и Puma в опухолях увеличилась более, чем в 50 раз относительно исходной экспрессии мРНК. Стимулирование экспрессии мРНК Mdm2 было существенно более низким (Emax=10-крат). В день 59 после первого введения препарата, всем крысам осуществляли i.v. введение 20 мг/кг, для оценки влияния препарата на организм хозяина. Содержание соединения A в сердце, тонкой кишке, селезенке, печени и костном мозге было примерно одинаковым, но в два раза выше, чем в плазме (Cmax неизвестна). Максимальное повышение p21, Mdm2 и расщепленной каспазы-3 в тонком кишечнике и костном мозге (грудинной кости) всегда наблюдалось через 3 часа после введения. Все параметры возвращались к исходным значениям через 7 дней после введения. Увеличение содержания расщепленной каспазы-3 на срезах тонкого кишечника показало сильную корреляцию со стимулированием мРНК Puma (Bbc3), которое определяли с помощью ISH (гибридизации in situ) мРНК. Действительно, максимальное увеличение Puma наблюдалось через 3 часа после введения, причем возвращение на исходный уровень имело место через 7 дней после введения. ISH РНК явно показала, что на срезе тонкого кишечника окрашиванию подвергались только клетки кишечных крипт. То же оказалось справедливым для селезенки и сердца. Наконец, через 14 дней после введения могло наблюдаться сильное истощение костного мозга, с частичным восстановлением в день 22 (Фиг.6). Количество белых кровяных телец продемонстрировало значительную корреляцию с истощением костного мозга (R2=0,59, P=0,006, Фиг.7). Приведенные данные показывают, что прерывистое введение может улучшить переносимость благодаря возможности восстановления костного мозга.
Пример 3: PK, PD, эффективность и переносимость соединения A (i.v.,q3w) у крыс, несущих опухоль SJSA-1
На Фиг.8 показан рост опухоли и изменение массы тела бестимусных крыс на протяжении 42 дней. Через три недели после первого введения в дозировке 20 мг/кг, соединение A вызвало в среднем 6% регрессию опухоли. Однако данные для отдельных животных показывают, что у трех крыс наблюдалась полная реакция (100% регрессия), у 2 особей имела место частичная реакция (регрессия более 50%), у 1 особи наблюдалось стабильное состояние заболевания и у 1 особи наблюдалось прогрессирование заболевания, несмотря на первоначальную 80% регрессию через 1 неделю после введения. После второго введения соединение A вызвало 100% регрессию опухоли но только 2 животных из 7 выжили после 2 полных циклов. Действительно, 5 крыс пришлось подвергнуть умерщвлению после второго введения из-за чрезмерной потери BW: первую особь подвергли умерщвлению через 10 дней после второго введения и четыре других особи 8 днями позднее. На фиг.9 показаны количества белых кровяных телец (WBC), нейтрофилов и тромбоцитов на протяжении 42 дней эксперимента. Соединение A вызывало резкое уменьшение количества WBCs, нейтрофилов и тромбоцитов после первого введения, и у большинства крыс эти параметры лишь частично восстанавливались на 21 день после обработки. Вследствие этого, второе введение приводило к снижению количества WBCs, нейтрофилов и тромбоцитов до чрезвычайно низких уровней, близких к 0.
Пример 4: PK, PD, эффективность и переносимость соединения A (i.v.,q3w) у крыс, несущих опухоль SJSA-1
На Фиг.10 показан рост опухоли и изменение массы тела бестимусных крыс на протяжении 42 дней. Введение соединения A в дозировках 13,7 и 18,2 мг/кг вызывало в среднем 66 и 88% регрессию опухолей через одну неделю после обработки. Через две недели все опухоли у животных, получивших 13,7 мг/кг, возобновили рост и авторы решили остановить исследования в этой группе. Через три недели после введения 18,2 мг/кг опухоли претерпели регрессию на 36% при двух случаях полной реакции. Влияние на рост опухоли имело тенденцию к уменьшению после второго введения так что в среднем наблюдалось прогрессирование опухоли на 118% (Таблица 4), и в двух упомянутых выше случаях опухоли продемонстрировали полную реакцию. В результате, второе введение не повышало число полных реакций. Что касается переносимости, у крыс наблюдалась незначительная потеря массы тела BW через три дня после введения, но животные быстро восстанавливались и набирали BW до следующего введения. Действительно, только одна крыса продемонстрировала значительную потерю BW в последний день эксперимента, и невозможно было определить, связана ли эта потеря с введением препарата. На фиг.11 показаны количества белых кровяных телец, нейтрофилов и тромбоцитов на протяжении 42 дней эксперимента. Соединение A вызывало существенное уменьшение количества WBCs, нейтрофилов и тромбоцитов после первого введения, но все обследованные животные полностью восстановились на 21 день после введения. Второе введение оказало аналогичное действие на количество указанных клеток, но крысы лишь частично восстановились на 21 день после второго введения.
Таблица 4: Эффективность и переносимость соединения A после i.v. введения (q3w) бестимусным крысам (SF480), несущим опухоль SJSA-1
недели после введения
Пример 5: PK и PD соединения A у крыс, несущих опухоль SJSA-1, после однократного перорального введения (27 мг/кг)
На Фиг.12 показана концентрация препарата в плазме, опухоли и печени в течение 144 часов после однократного перорального введения соединения A. Tmax для соединения A составляло 3 часа (ч) во всех тканях. Соединение A продемонстрировало более высокое содержание в опухоли (AUC0-144h=277,7 ч⋅нмоль/г) по сравнению с плазмой (AUC0-144h=111,5 ч⋅мкМ). На Фиг.13 показана концентрация лекарственного средства и PD реакция опухоли. Стимулирование мРНК Puma и p21 достигало Emax 162- и 180-крат через 48 ч после введения. При указанной пероральной дозировке, Mdm2 имел существенно более низкое значение Emax (34 крата).
Пример 6: Эффективность для крыс, несущих опухоль SJSA-1, при режиме введения 3qw (p.o.)
На Фиг.4 показан рост опухоли и изменение массы тела бестимусных крыс на протяжении 42 дней q3w p.o. введения соединения A. Через три недели после первого введения обе дозировки смогли вызвать регрессию опухоли (27 и 88% для дозировок 20 и 27 мг/кг соответственно). Однако влияние на рост опухоли имело тенденцию к ослаблению после второго введения, поскольку только большая дозировка по-прежнему была способна вызывать регрессию опухоли (27% при дозировке 27 мг/кг). После первого введения, Cmax и AUC0-24h соединения A в крови, а также экспрессия мРНК p21 и Puma в опухоли значительно и дозозависимым образом возрастали. Обе дозировки вызывали незначительное уменьшение массы тела (BW) через 3 дня после каждого введения, но все животные быстро восстанавливались и демонстрировали увеличение BW через 3 недели после введения. На Фиг.15 показаны количества белых кровяных телец и тромбоцитов на протяжении 42 дней эксперимента. Соединение A вызывало дозозависимое уменьшение количества WBCs, нейтрофилов и тромбоцитов. Количество WBCs и тромбоцитов полностью восстанавливалось перед вторым введением при дозировке 20 мг/кг. Для дозировки 27 мг/кг, тромбоциты также полностью восстанавливались, но WBCs восстанавливались лишь частично.
Пример 7: Результат введения низких доз по сравнению с высокими дозами
Проводили эксперименты для оценки реакции на введение высокой дозы и низкой дозы. Животным вводили низкую дозу 5 мг/кг p.o. или высокую дозу 27 мг/кг p.o. или 20 мг/кг i.v. Низкая доза Mdm2i не вызывала такого же биохимического эффекта, который вызывали высокие дозы (Фиг.16).
Пример 8: Комбинирование введения высокой и низкой дозы является высоко синергетическим
Повторяли эксперименты на крысах, несущих опухоли SJSA-1, комбинируя две схемы введения соединения A, прерывистое введение (15 мг/кг однократно) и ежедневное введение (1,5 мг/кг). На Фиг.17 показано, что комбинация двух схем введения оказывает значительное синергетическое действие. Данный график является схематическим отражением нескольких экспериментов. Кроме того, явное синергетическое действие можно видеть на Фиг.18 и 19. Эффективность (Фиг.18) и переносимость (Фиг.19) для крыс, несущих опухоль SJSA-1, дополнительно исследовали для других дозировок и схем введения (15 мг/кг q4w (день 0)+3 мг/кг q24h 2w введение/2w перерыв (день 1); 21 мг/кг q4w (день 0)+1,5 мг/кг q24h 3w введение/1w перерыв (день 1)). Было показано, что комбинирование схем введения улучшает эффективность и может повысить переносимость, в т.ч. могут применяться более низкие дозы, которые, тем не менее, позволят добиться более сильного уменьшения размеров опухоли.
Пример 9: Эффективность и переносимость у крыс, несущих ксенотрансплантат меланомы, извлеченный из организма пациента (PDX) (пероральное введение)
Аналогичные эксперименты повторяли на крысах, несущих PTX меланомы. Эффективность (Фиг.20) и переносимость (Фиг.21) соединения A тестировали при схеме введения 27 мг/кг q3w. Прерывистое введение также продемонстрировало эффективность в моделях меланомы.
Пример 10: Эффективность прерывистого введения соединения A в комбинации с серитинибом у мышей, несущих опухоль SHSY5Y
Проводили аналогичные эксперименты, вводя мышам комбинацию серитиниба и соединения A. Эти эксперименты показали (Фиг.22), что соединение A можно применять каждую неделю при комбинированном введении с другим соединением. Еженедельное введение серитиниба в комбинации с соединением A в дозировке 120 мг/кг (40 мг/кг × 3 каждые 3 часа) приводило к лучшему противоопухолевому эффекту по сравнению с индивидуальным введением серитиниба или по сравнению с комбинацией серитиниб+соединение A ежедневно в дозировке 20 мг/кг (n=5). У мышей наблюдалась фармакокинетика, отличная от крыс. Поэтому для достижения желаемого воздействия указанную дозу следовало вводить 3 раза каждые три часа. Принимая во внимание эту особенность мышиной модели, в т.ч. значительно более высокий клиренс, эксперименты на мышах можно экстраполировать на крыс и других субъектов, и можно предположить, что мышиная модель доказывает, что как минимум такой же результат мог бы быть достигнут у крыс или других субъектов, в т.ч. людей, даже если соединение A вводить как минимум один раз в три недели.
| название | год | авторы | номер документа |
|---|---|---|---|
| ДОЗА И СХЕМА ВВЕДЕНИЯ ИНГИБИТОРА ВЗАИМОДЕЙСТВИЯ HDM2 С p53 ПРИ ГЕМАТОЛОГИЧЕСКИХ ОПУХОЛЯХ | 2018 |
|
RU2753527C2 |
| ДОЗА И РЕЖИМ ВВЕДЕНИЯ ДЛЯ ИНГИБИТОРОВ ВЗАИМОДЕЙСТВИЯ HDM2 С P53 | 2017 |
|
RU2762573C2 |
| КОМБИНАЦИИ ИНГИБИТОРА ВЗАИМОДЕЙСТВИЯ HDM2-P53 И ИНГИБИТОРА BCL2 И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2793123C2 |
| GDF-15 КАК ГЕМАТОЛОГИЧЕСКИЙ БИОМАРКЕР ТОКСИЧНОСТИ | 2016 |
|
RU2741390C2 |
| ИНГИБИТОРЫ MDM2 И ИХ КОМБИНАЦИИ | 2016 |
|
RU2740091C2 |
| СПИРООКСИНДОЛЬНЫЕ АНТАГОНИСТЫ MDM2 | 2010 |
|
RU2553269C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМБИНАЦИИ | 2014 |
|
RU2684106C2 |
| ИЗОИНДОЛИНОНОВЫЕ ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ MDM2-P53, ОБЛАДАЮЩИЕ ПРОТИВОРАКОВОЙ АКТИВНОСТЬЮ | 2016 |
|
RU2794333C1 |
| СПОСОБЫ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ И НАРУШЕНИЙ, СВЯЗАННЫХ С LSD1, ИНГИБИТОРАМИ LSD1 | 2020 |
|
RU2826217C1 |
| СПОСОБ ВВЕДЕНИЯ ПРОТИВООПУХОЛЕВОГО АГЕНТА | 2013 |
|
RU2638795C2 |



Группа изобретений относится к онкологии и фармакологии. Предложено применение ингибитора MDM2 (MDM2i) для лечения рака, где MDM2i следует вводить один раз каждые 3 недели (q3w), где MDM2i представляет собой (S)-5-(5-хлор- 1-метил-2-оксо-1,2-дигидро-пиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-пиримидин-5-ил)-1-изопропил-5,6-дигидро-1Н-пирроло [3,4-d] имидазол-4-он или (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксо-пиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он; применение первого MDM2i, который вводят в дозе от 100 до 800 мг один раз каждые 3 недели per os; применение второго MDM2i, который вводят в дозе от 500 до 4000 мг один раз каждые 3 недели per os (варианты). Технический результат: изобретения обеспечивают антинеопластический эффект путем запуска существенно более длительно действующих антипролиферативных механизмов в клетках, длительность которых сохраняется в течение нескольких недель после введения одной дозы, что позволяет вводить заявленные MDM2i в прерывистом режиме. Лечение с прерывистой схемой введения ингибитора Mdm2 можно сочетать с ежедневным введением Mdm2i или другого фармацевтически приемлемого ингредиента. 3 н. и 8 з.п. ф-лы, 22 ил., 4 табл., 10 пр.
1. Применение ингибитора MDM2 (MDM2i) для лечения рака, где MDM2i следует вводить один раз каждые 3 недели (q3w), где MDM2i представляет собой (S)-5-(5-хлор- 1-метил-2-оксо-1,2-дигидро-пиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-пиримидин-5-ил)-1-изопропил-5,6-дигидро-1Н-пирроло [3,4-d] имидазол-4-он или (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксо-пиперазин-1-ил)-транс-циклогексилметил]амино}фенил)-1,4-дигидро-2H-изохинолин-3-он.
2. Применение ингибитора MDM2 (MDM2i) для лечения рака, где MDM2i представляет собой (S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2- (2,4-диметокси-пиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло [3,4-d] имидазол-4-он и где указанный MDM2i вводят в дозе от 100 до 800 мг один раз каждые 3 недели per os.
3. Применение ингибитора MDM2 (MDM2i) для лечения рака, где MDM2i представляет собой (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксопиперазин-1-ил)-трансциклогексилметил]амино}фенил)-1,4-дигидро-2Н-изохинолин-3-он и где указанный MDM2i вводят в дозе от 500 до 4000 мг один раз каждые 3 недели per os.
4. Применение ингибитора MDM2 (MDM2i) для лечения рака по пп.1, 2 или 3, где рак является раком мочевого пузыря, груди, мозга, органов головы и шеи, печени, ротовой полости, желчевыводящих путей, острым и хроническим лимфоидным лейкозом, острым и хроническим миелоидным лейкозом, хроническим миеломоноцитарным лейкозом, колоректальным раком, раком желудка, желудочно-кишечной стромальной опухолью, гепатоклеточным раком, глиомой, лимфомой, меланомой, множественной миеломой, миелопролиферативной болезнью, нейроэндокринным раком, раком легких, немелкоклеточным раком легких, раком поджелудочной железы, яичников, простаты, почечноклеточным раком, саркомой, липосаркомой и раком щитовидной железы.
5. Применение ингибитора MDM2 (MDM2i) для лечения рака по пп.1, 2 или 3, где рак представляет собой меланому, рак легкого или нейробластому.
6. Применение ингибитора MDM2 (MDM2i) для лечения рака по п.1, где рак представляет собой меланому.7.
7. Применение ингибитора MDM2 (MDM2i) для лечения рака по п.1, где рак представляет собой нейробластому.
8. Применение ингибитора MDM2 (MDM2i) для лечения рака по любому из пп.1, 2 или 3, где рак представляет собой лейкоз.
9. Применение ингибитора MDM2 (MDM2i) для лечения рака по любому из пп.1, 2 или 3, где лечение дополнительно включает другой фармацевтический ингредиент, который следует вводить пациенту.
10. Применение ингибитора MDM2 (MDM2i) , предназначенного для лечения рака по п.9, где другой фармацевтический ингредиент представляет собой другой антинеопластический агент.
11.11. Применение ингибитора MDM2 (MDM2i) для лечения рака по п.10, где следует вводить более одного дополнительного антинеопластического агента.
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| HIGGINS B | |||
| et al | |||
| Аппарат для очищения воды при помощи химических реактивов | 1917 |
|
SU2A1 |
| Clin | |||
| Cancer Res | |||
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| Способ защиты переносных электрических установок от опасностей, связанных с заземлением одной из фаз | 1924 |
|
SU2014A1 |
| СПРАВОЧНИК | |||
| ПРОТИВООПУХОЛЕВАЯ ХИМИОТЕРАПИЯ | |||
| Под ред | |||
| Переводчиковой Н.И | |||
| М.: Медицина, 1993, с.124, раздел Хемодектомы, с.147 2-й абзац - 149 | |||
| Изложница с суживающимся книзу сечением и с вертикально перемещающимся днищем | 1924 |
|
SU2012A1 |

