ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к ингибиторам взаимодействия HDM2 с p53 для применения в лечении рака, где лекарственное средство вводят в соответствии с режимом с прерывистым введением высоких доз.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
p53 индуцируется и активируется рядом потенциально онкогенных процессов, включая аберрантные сигналы роста, повреждение ДНК, ультрафиолетовое излучение и ингибиторы протеинкиназ (Millard M, et al. Curr Pharm Design 2011;17:536-559), и регулирует гены, контролирующие арест клеточного роста, репарацию ДНК, апоптоз и ангиогенез (Bullock AN & Fersht AR. Nat Rev Cancer 2001;1:68-76; Vogelstein B, et al. Nature Education 2010;3(9):6).
Human Double Minute-2 (HDM2) является одним из наиболее важных регуляторов p53. Он связывается непосредственно с p53, ингибируя его трансактивацию, и затем направляет его к цитоплазматической деградации (Zhang Y, et al. Nucleic Acids Res 2010;38:6544-6554).
При раке у человека p53 является одним из наиболее часто инактивируемых белков, при этом инактивация происходит либо путем прямой мутации гена TP53 (обнаруживается примерно в 50% всех случаев рака у человека) (Vogelstein, B et al. Nature 2000;408:307-310), либо посредством супрессивных механизмов, таких как сверхэкспрессия HDM2 (Zhao Y, et al. BioDiscovery 2013;8:4).
В доклинических клеточных моделях и моделях in vivo было показано, что эффективные и селективные ингибиторы взаимодействия HDM2 с p53 (также называемые ингибиторами HDM2 или ингибиторами MDM2), например NVP-HDM201, восстанавливают функцию p53 (Holzer P, et al. Постер, представленный на AACR 2016, тезисы доклада №4855).
Различные режимы введения доз были описаны для ингибиторов HDM2 и протестированы в клинических исследованиях.
Например, в US2013/0245089 раскрыт способ лечения пациента, страдающего от рака, путем введения пациенту 4-{[(2R,3S,4R,5S)-4-(4-хлор-2-фторфенил)-3-(3-хлор-2-фторфенил)-4-циано-5-(2,2-диметилпропил)-пирролидин-2-карбонил]амино}-3-метоксибензойной кислоты в количестве от приблизительно 800 до приблизительно 3000 мг/сутки в течение периода введения, составляющего не более приблизительно 7 дней, в дни 1-7 из 28-дневного цикла лечения, за которым следует период отдыха, составляющий от приблизительно 21 до приблизительно 23 дней.
В статье B. Higgins et al. в Clinical Cancer Research (май 2014) раскрыта схема 28-дневного цикла, где RG7388 вводят один раз в неделю три раза с последующим 13-дневным отдыхом (график 28-дневного цикла) или где лекарственное средство вводят в течение 5 последовательных дней 28-дневного графика. Дополнительные режимы введения доз для ингибиторов HDM2 раскрыты в WO 2015/198266.
Ингибиторы HDM2 и как их получать были раскрыты, например, в WO2013/111105 или WO2011/076786.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Одна из целей в разработке лекарственных средств на основе ингибитора HDM2 состоит в том, чтобы найти режим введения доз, который позволит вводить высокую дозу, которая обеспечивает эффективность, но в то же время снижает риск возникновения нежелательных явлений.
Неожиданно было обнаружено, что один тип режима введения доз является особенно применимым для лечения солидных опухолей ингибиторами HDM2. Было обнаружено, что этот режим введения доз также применим для лечения гематологических опухолей ингибиторами MDM2.
Конкретно, настоящее изобретение предусматривает следующие аспекты, преимущественные признаки и конкретные варианты осуществления, соответственно отдельно или в комбинации, перечисленные в следующих пунктах:
1. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака,
где лекарственное средство вводят в два разных дня введения в пределах цикла лечения,
где первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и
где лечение состоит из по меньшей мере 2 циклов лечения.
2. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 1, где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 18-22 дней.
3. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 1, где короткий период без введения состоит из 5-7 дней, и продолжительный период без введения состоит из 19-21 дня.
4. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 1, где короткий период без введения состоит из 6 дней, и продолжительный период без введения состоит из не менее 20 дней.
5. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из пунктов 1-4, где указанный ингибитор выбран из группы, состоящей из идасанутлина (RG7388, RO5503781), RG7775 (RO6839921), AMG232, DS3032 (DS3032b), ALRN-6924, ATSP-7041, CGM097 и HDM201.
6. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из пунктов 1-4, где указанный ингибитор выбран из группы, состоящей из CGM097 и HDM201.
7. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из пунктов 1-4, где указанный ингибитор представляет собой CGM097.
8. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из пунктов 1-4, где указанный ингибитор представляет собой HDM201.
9. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 7, где указанный ингибитор представляет собой сульфат CGM097.
10. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 8, где указанный ингибитор представляет собой HDM201 с янтарной кислотой.
11. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 8 или пункту 10, где суточная доза в дни введения составляет от 100 мг до 200 мг.
12. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 8 или пункту 10, где суточная доза в дни введения составляет от 100 мг до 150 мг.
13. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 8 или пункту 10, где суточная доза в дни введения составляет 120 мг.
14. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из предыдущих пунктов, где рак представляет собой опухоль с TP53 дикого типа.
15. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из предыдущих пунктов, где рак представляет собой солидную опухоль.
16. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 15, где солидная опухоль выбрана из типов саркомы, например липосаркомы или саркомы мягких тканей, типов меланомы, например меланомы кожи или увеальной меланомы, типов бластомы (например, нейробластомы), опухоли толстой кишки, колоректальной опухоли, опухоли почки и опухоли печени.
17. Ингибитор взаимодействия HDM2 с p53 в комбинации с агонистом рецептора тромбопоэтина для применения в лечении рака согласно любому из предыдущих пунктов.
18. Ингибитор взаимодействия HDM2 с p53 в комбинации с агонистом рецептора тромбопоэтина для применения в лечении рака согласно пункту 17, где агонист рецептора тромбопоэтина представляет собой элтромбопаг.
19. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из предыдущих пунктов, где лечение обеспечивает снижение риска развития гематологических токсических явлений.
20. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 19, где гематологические токсические явления выбраны из группы, состоящей из тромбоцитопении, нейтропении, лейкопении, лимфопении, анемии.
21. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 19, где гематологическое токсическое явление представляет собой тромбоцитопению.
22. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из пунктов 1-14, где рак представляет собой гематологическую опухоль.
23. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 22, где гематологическая опухоль представляет собой лейкемию.
24. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно пункту 22, где гематологическая опухоль выбрана из острого миелоидного лейкоза (AML), миелодиспластического синдрома (MDS) и острого лимфобластного лейкоза (ALL).
25. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из пунктов 22-24, где гематологическая опухоль представляет собой рецидивирующую/рефрактерную гематологическую опухоль.
26. Ингибитор взаимодействия HDM2 с p53 для применения в лечении рака согласно любому из пунктов 1-4, где ингибитор представляет собой CGM097 или HDM201, предпочтительно HDM201, где суточная доза в дни введения составляет от 100 мг до 150 мг, предпочтительно 120 мг, и где рак представляет собой рецидивирующие/рефрактерные гематологические опухоли с TP53 дикого типа, выбранные из острого миелоидного лейкоза (AML), миелодиспластического синдрома (MDS) и острого лимфобластного лейкоза (ALL), предпочтительно AML.
Предпочтительным вариантом осуществления настоящего изобретения является
лекарственное средство на основе ингибитора взаимодействия HDM2 с p53, HDM201, для применения в лечении солидных опухолей, где лекарственное средство вводят в день 1 и день 8 4-недельного (28 дней) цикла лечения, где лечение состоит из по меньшей мере 2 циклов лечения, и доза лекарственного средства в каждый день введения составляет приблизительно 120 мг.
Другим предпочтительным вариантом осуществления настоящего изобретения является
лекарственное средство на основе ингибитора взаимодействия HDM2 с p53, HDM201, для применения в лечении гематологических опухолей, где лекарственное средство вводят в день 1 и день 8 4-недельного (28 дней) цикла лечения, где лечение состоит из по меньшей мере 2 циклов лечения, и доза лекарственного средства на каждый день введения составляет приблизительно 120 мг.
Режимы введения доз по настоящему изобретению, описанные выше, обеспечивают очень благоприятный терапевтический индекс, низкую частоту возникновения тромбоцитопении 3/4 степени при достижении терапевтически значимых значений содержания в плазме крови, активацию пути p53 (повышение экспрессии GDF-15) и клиническую активность.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Далее настоящее изобретение подробно описано со ссылкой на прилагаемые фигуры, на которых изображено следующее.
На фигуре 1 проиллюстрирован наиболее предпочтительный режим введения доз 1B (строка "Предп.") по настоящему изобретению и некоторые альтернативные режимы A1-E2. X1 и X2 представляют собой те дни, в которые вводят лекарственное средство на основе ингибитора HDM2.
На фигуре 2 продемонстрирована средняя концентрация на цикл, рассчитанная для пациентов, получавших 120 мг при режиме 1B. Когорта 1 - 120 мг; когорта 2 - 120 мг, новый вариант. Пунктирная линия - остановка роста опухоли (линия клеток SJSA-1); точечная линия - остановка роста опухоли (линия клеток липосаркомы). Каждый отдельный пациент представлен кругом.
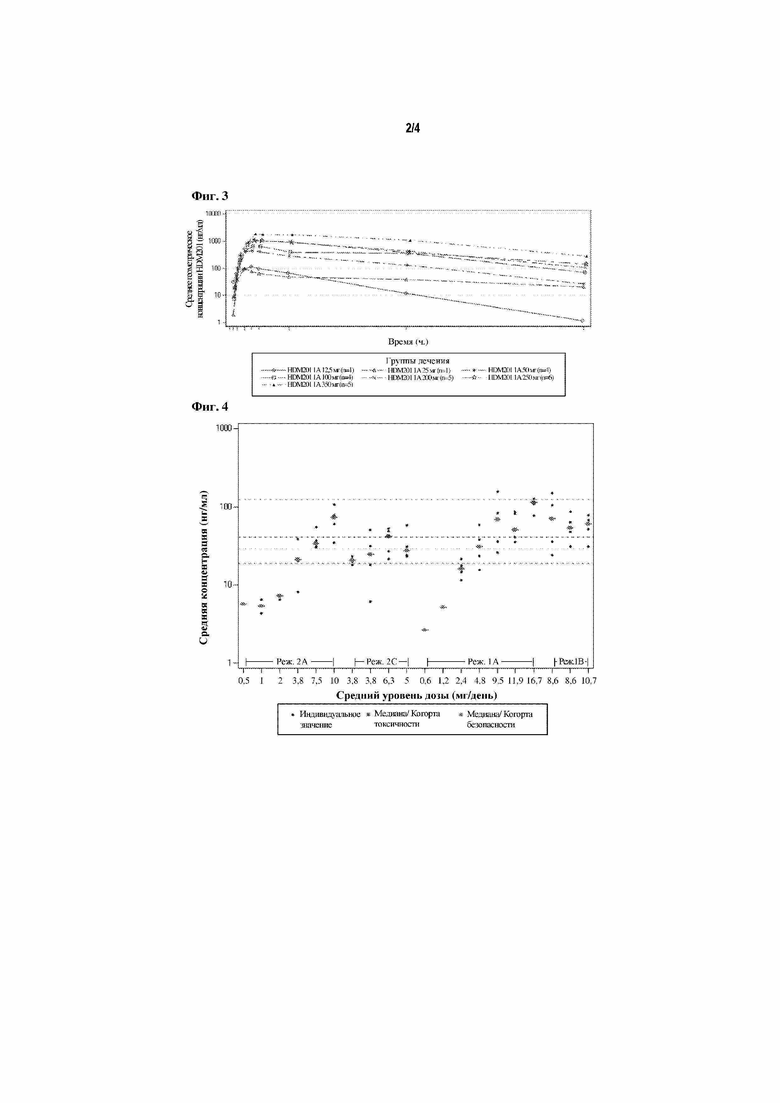
На фигуре 3 продемонстрирована кривая зависимости среднего геометрического концентрации от времени (режим 1A, цикл 1, день 1) (PAS).
На фигуре 4 продемонстрирована средняя концентрация NVP-HDM201 для отдельного человека в течение первого цикла (DDS). Индивидуальная C (среднее)=индивидуальная мода AUC в конце цикла 1, деленная на продолжительность цикла 1 в часах. Средний уровень дозы=суммарная кумулятивная доза в конце цикла 1, деленная на продолжительность цикла 1 в днях.
На фигуре 5 продемонстрированы кинетические кривые зависимости для тромбоцитов, смоделированные на основе следующих доз, проверенных при каждом режиме (по порядку сверху вниз): реж. 2C (D1-7 Q4wk): 25 мг (6,25 мг/сутки); реж. 2A (D1-14 Q4wk): 20 мг (10 мг/сутки); реж. 1B (дни 1, 8 Q4wk): 150 мг (10,7 мг/сутки); реж. 1A (D1 Q3wk): 350 мг (16,7 мг/сутки).
На фигуре 6 проиллюстрировано наилучшее процентное изменение процентной доли бластных клеток в аспирате костного мозга (BM) у пациентов с AML (для пациентов, аспират костного мозга которых имелся в наличии).
*Текущее лечение; #наилучшее процентное изменение составляет ≥100; TF: неэффективное лечение; CR: полный ответ; CRi: морфологический CR с неполным восстановлением формулы крови. Суточные дозы: режим 1A - 250, 350 или 400 мг; режим 1B - 150 мг; режим 2A - 20, 30 мг; режим 2C - 45 мг.
На фигуре 7 продемонстрирована зависимость индивидуальной средней концентрации в течение первого цикла лечения от дозы для разных режимов для пациентов с гематологическими опухолями.
Линия на уровне 120 нг/мл=регрессия опухоли на 95% по сравнению с ксенотрансплантатом SJSA-1 человека у крысы. Линия на уровне 41 нг/мл=средняя концентрация для остановки роста опухоли, полученная при моделировании PK/PD TGI у крысы с ксенотрансплантатом SJSA-1 человека (остеосаркомой). Линия на уровне 19 нг/мл=средняя концентрация для остановки роста опухоли, полученная при моделировании PK/PD TGI у крысы с HSAX2655 (липосаркомой) человека, представляющей собой PDX.
Подсчет уровня средней дозы (мг/сутки):
На фигуре 8 продемонстрировано наилучшее процентное изменение по сравнению с исходным значением для суммы диаметра и наилучшего общего ответа для пациентов с саркомой (липосаркомой и другими типами саркомы), получавших HDM201 в соответствии с режимом 1B (сентябрь 2017). PD: прогрессирующее заболевание, SD: стабильное заболевание, PR: частичный ответ.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В данном документе далее настоящее изобретение описано более подробно и проиллюстрировано в примерах.
В одном аспекте настоящее изобретение предусматривает
ингибитор взаимодействия HDM2 с p53 для применения в лечении рака,
где лекарственное средство вводят в два разных дня введения в пределах цикла лечения,
где первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и
где лечение состоит из по меньшей мере 2 циклов лечения.
"День введения" означает, что это день, когда пациент принимает лекарственное средство на основе ингибитора HDM2. Тогда в этот день пациент может принимать лекарственное средство в виде одной разовой дозы, или суточная доза делится на более мелкие части, например одна половина суточной дозы утром и другая половина вечером. Предпочтительно дозу принимают в виде одной разовой дозы.
"Дни без введения" являются теми днями, на протяжении которых пациент не получает лекарственное средство на основе ингибитора HDM2. Ряд дней без введения, идущих непосредственно один за другим, не прерываемый днем введения, образует "период без введения". Пациент может получать другие лекарственные средства в день без введения. Таким образом, отсутствие введения означает только отсутствие введения лекарственного средства на основе ингибитора HDM2.
"Период, состоит из x-y дней" означает, что продолжительность указанного периода составляет от x до y дней. Например, "период, состоящий из 6 дней" представляет собой период времени длительностью 6 дней.
Под "введением в два разных дня введения в пределах цикла лечения" подразумевается, что в каждом цикле лечения есть только два дня введения, а не, например, три или более дня введения, и не только один день введения. Другими словами, цикл лечения состоит из двух разных дней введения. "Два разных дня введения" означает, что два дня не приходятся на один день введения, а представляют собой два разных, отдельных дня, например, день 1 и день 8 из 28-дневного цикла лечения.
Термин "цикл лечения" обозначает количество и порядок дней, которые образуют одну схему лечения с днями введения и днями без введения до повторения этой схемы лечения снова. Например, цикл лечения из 28 дней с днями введения 1 и 8 означает, что лекарственное средство вводят в день 1, день 8, день 29, день 36, день 57, день 64, день 85, день 92, и т. д.
Настоящее изобретение предусматривает лекарственное средство на основе ингибитора взаимодействия HDM2 с p53 или любой его фармацевтически приемлемой соли для применения в лечении рака,
где лекарственное средство вводят в два разных дня введения в пределах цикла лечения,
где первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и
где лечение состоит из по меньшей мере 2 циклов лечения.
Альтернативно, настоящее изобретение предусматривает способ лечения рака у пациентов, представляющих собой людей, нуждающихся в таком лечении, который включает введение эффективного количества лекарственного средства на основе ингибитора взаимодействия HDM2 с p53 или любой его фармацевтически приемлемой соли,
где указанное лечение характеризуется тем, что лекарственное средство вводят в два разных дня введения в пределах цикла лечения,
и первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
и короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и
лечение состоит из по меньшей мере 2 циклов лечения.
В качестве дополнительной альтернативы настоящее изобретение предусматривает применение лекарственного средства на основе ингибитора взаимодействия HDM2 с p53 или любой его фармацевтически приемлемой соли для получения лекарственного препарата для лечения рака,
характеризующегося тем, что лекарственное средство вводят в два разных дня введения в пределах цикла лечения,
и первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
и короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и
лечение состоит из по меньшей мере 2 циклов лечения.
В качестве дополнительной альтернативы настоящее изобретение предусматривает лекарственный препарат для лечения рака, содержащий лекарственное средство на основе ингибитора взаимодействия HDM2 с p53 или любую его фармацевтически приемлемую соль,
характеризующийся тем, что лекарственное средство вводят в два разных дня введения в пределах цикла лечения,
и первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
и короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и
лечение состоит из по меньшей мере 2 циклов лечения.
Лечение повторяют при условии, что оно имеет клиническую значимость, т. е. рост опухоли по меньшей мере снижается или контролируется, а нежелательные эффекты являются переносимыми. Лечение по настоящему изобретению состоит из по меньшей мере 2 циклов лечения, предпочтительно от 2 до 20 циклов лечения. Однако, если это имеет клиническую значимость, терапию продолжают после 20-го цикла лечения.
На фигуре 1 проиллюстрированы некоторые конкретные режимы введения доз, которые подпадают под настоящее изобретение. В строке "Предп." представлен наиболее предпочтительный режим введения доз. Цикл лечения по указанному режиму введения доз состоит из 4 недель, т. е. 28 дней (день 1 - день 28). Дни введения, указанные на этой фигуре как "X1" (первый день введения) и "X2" (второй день введения), приходятся на день 1 и день 8. При этом остается период без введения из 6 дней между X1 и X2, от дня 2 до дня 6 (короткий период без введения), и период без введения из 20 дней между X2 и X1 следующего цикла лечения, от дня 9 до дня 28 (продолжительный период без введения). День 29 в этом примере в таком случае будет днем 1 следующего цикла лечения.
В вариантах осуществления настоящего изобретения дозы в дни введения X1 и X2 являются предпочтительно одинаковыми, например 120 мг в день 1 (X1) и 120 мг в день 8 (X2).
При наиболее предпочтительном режиме введения доз по настоящему изобретению цикл лечения длится 28 дней, т. е. 4 недели, и дозу лекарственного средства, ингибитора HDM2, вводят в день 1 и день 8. Этот режим также называется "d1, d8 q4w" или подобными версиями аббревиатуры. Этот наиболее предпочтительный режим введения доз также называется в данном документе "режим 1B".
Вместо наиболее предпочтительного дня 8 в качестве второго дня введения, этот второй день введения может альтернативно приходиться на день 6, 7, 9 или 10.
В зависимости от продолжительности цикла лечения, предпочтительно 3, 4, или 5 недель, это приводит к более коротким или более длинным периодам без введения. Например, если диапазон продолжительности цикла лечения составляет от 3 до 5 недель, диапазон короткого периода без введения составляет от 4 до 8 дней, и диапазон длинного периода введения составляет от 13 до 27 дней. Наиболее предпочтительным является цикл лечения, составляющий 4 недели.
Из всех возможных режимов, полученных на основе этих диапазонов для коротких и длинных периодов без введения, определенных выше, на фигуре 1 в строках A1, A2, B1, B2, C1, C2, C3, D1, D2, E1, E2 продемонстрированы предпочтительные варианты. Более предпочтительными режимами являются A2, B1, B2, C1, C2, C3, D1, D2, E1. Еще более предпочтительными режимами являются A2, B2, C2, D1, E1. Еще более предпочтительными режимами являются B2, C2, D1. Наиболее предпочтительным является режим C2, который эквивалентен "режиму 1B", упомянутому выше.
Термин "ингибитор HDM2", также указанный как "HDM2i", "Hdm2i", "ингибитор MDM2", "MDM2i", "Mdm2i", обозначает в данном документе любое соединение, ингибирующее взаимодействие HDM-2 с p53 или HDM-4 с p53 при IC50 менее 10 мкМ, предпочтительно менее 1 мкМ, предпочтительно в диапазоне нМ, измеренной с помощью анализа переноса энергии флуоресценции с временным разрешением (TR-FRET). Ингибирование взаимодействий p53 с Hdm2 и p53 с Hdm4 измеряют с помощью переноса энергии флуоресценции с временным разрешением (TR-FRET). Перенос энергии флуоресценции (или ферстеровский резонансный перенос энергии) характеризует перенос энергии между донорной и акцепторной 5 флуоресцентными молекулами. Для этого анализа белок MDM2 (аминокислоты 2-188) и белок MDM4 (аминокислоты 2-185), меченные C-концевым биотиновым фрагментом, используют в комбинации с стрептавидином, меченым европием (Perkin Elmer, Inc., Уолтем, Массачусетс, США), который служит в качестве донора флуорофора. Меченный Cy5 пептид, полученный из p53, Cy5-TFSDLWKLL (aa18-26 p53), является акцептором энергии. При возбуждении молекулы донора 10 при 340 нм взаимодействие, представляющее собой связывание, между MDM2 или MDM4 и пептидом р53 вызывает передачу энергии и усиление ответа на длине волны излучения акцептора, составляющей 665 нм. Нарушение образования комплекса p53-MDM2 или p53-MDM4 вследствие связывания молекулы, представляющей собой ингибитор, с сайтом связывания p53 в MDM2 или MDM4 приводит к усилению излучения донора при 615 нм. Показания FRET-анализа при логометрическом измерении рассчитывают из 15 необработанных данных двух отдельных сигналов флуоресценции, измеренных в режиме с временным разрешением (интенсивность излучения при 665 нм/интенсивность излучения при 615 нм x 1000). Анализ может быть выполнен в соответствии со следующей процедурой. Испытание проводят в белых микропланшетах 1536w (Greiner Bio-One GmbH, Фриккенхаузен, Германия) в общем объеме 3,1 мкл путем объединения 100 нл соединений, разбавленных в 90% DMSO/10% Н2О (конечная концентрация DMSO 3,2%), с 2 мкл стрептавидина, меченого европием 20 (конечная концентрация 2,5 нМ), в реакционном буфере (PBS, 125 мМ NaCl, 0,001% новексин (состоит из углеводных полимеров (полимеры новексина), предназначенных для повышения растворимости и стабильности белков; Novexin Ltd., Амбриджшир, Великобритания), 0,01% желатин, 0,2% плюроник (блок-сополимер этиленоксида и пропиленоксида, BASF, Людвигсхафен, Германия), 1 мМ DTT) с последующим добавлением 0,5 мкл MDM2-Bio или MDM4-Bio, разбавленных в буфере для анализа (конечная концентрация 10 нМ). Обеспечивают предварительное инкубирование раствора в течение 15 минут при комнатной температуре с последующим добавлением 0,5 мкл пептида Cy5-p53 в буфере для анализа (конечная концентрация 20 нМ). Перед считыванием планшет инкубируют при комнатной температуре в течение 10 минут. Для измерения образцов используют многорежимный микропланшет-ридер Analyst GT (Molecular Devices) со следующими настройками 30: дихроичное зеркало 380 нм, возбуждение при 330 нм, излучение донора при 615 нм и излучение акцептора при 665 нм. Значения IC50 рассчитываются путем аппроксимации кривой с применением XLfit. Если не указано иное, реагенты приобретаются у Sigma Chemical Co, Сент-Луис, Миссури, США.
В аспектах изобретения ингибитор HDM2 может быть выбран из (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(3-оксо-пиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она;
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксо-пиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она;
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(6-{метил-[4-(4-метил-3-оксо-пиперазин-1-ил)-транс-циклогексилметил]-амино}-пиридин-3-ил)-1,4-дигидро-2H-изохинолин-3-она;
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(6-{метил-[4-(3-метил-4-оксо-имидазолидин-1-ил)-транс-циклогексилметил]-амино}-пиридин-3-ил)-1,4-дигидро-2H-изохинолин-3-она;
(S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(5-{метил-[4-(3-метил-4-оксо-имидазолидин-1-ил)-транс-циклогексилметил]-амино}-пиразин-2-ил)-1,4-дигидро-2H-изохинолин-3-она;
1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксо-пиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она;
(S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидро-пиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-пиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-она;
4-[(S)-5-(3-хлор-2-фтор-фенил)-2-(2,4-диметокси-пиримидин-5-ил)-3-изопропил-6-оксо-3,4,5,6-тетрагидро-пирроло[3,4-d]имидазол-4-ил]-бензонитрила;
(S)-5-(5-хлор-2-оксо-1,2-дигидро-пиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-пиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-она;
(S)-5-(3-хлор-4-фторфенил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-((R)-1-метоксипропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она;
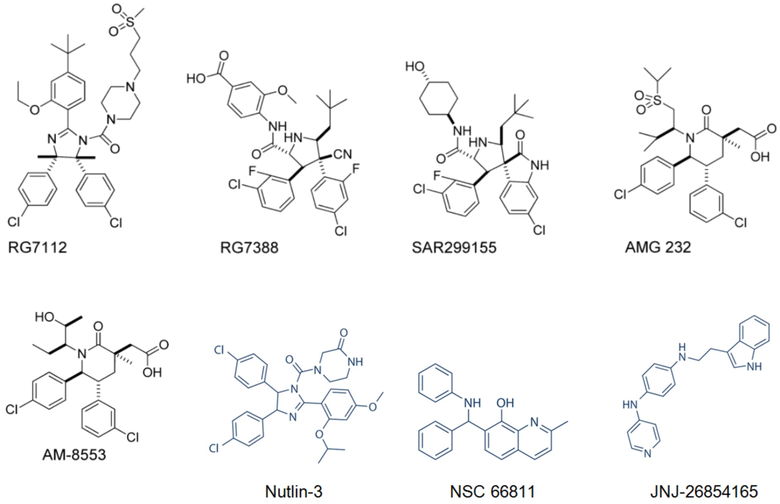
DS3032b
и
(S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-d6-пиримидин-5-ил)-1-((R)-1-метоксипропан-2-ил)-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-она; идасанутлина (RG7388, RO5503781); RG7775 (RO6839921); AMG232; DS3032 (также называемого DS3032b); ALRN-6924; ATSP-7041; CGM097; и HDM201.
Предпочтительно ингибитор HDM2 выбран из идасанутлина (RG7388, RO5503781), RG7775 (RO6839921), AMG232, DS3032 (DS3032b), ALRN-6924, ATSP-7041, CGM097 и HDM201.
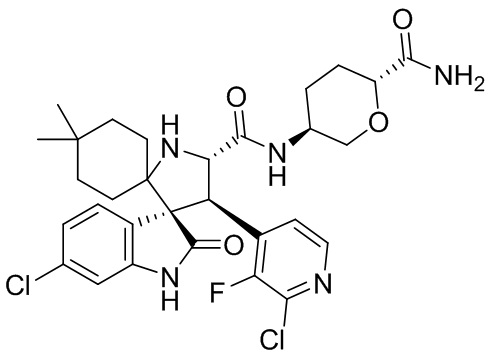
Более предпочтительно ингибитор HDM2 выбран из HDM201, т. е. (S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидро-пиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметокси-пиримидин-5-ил)-1-изопропил-5,6-дигидро-1H-пирроло[3,4-d]имидазол-4-она, также называемого (6S)-5-(5-хлор-1-метил-2-оксо-1,2-дигидропиридин-3-ил)-6-(4-хлорфенил)-2-(2,4-диметоксипиримидин-5-ил)-1-изопропил-5,6-дигидропирроло[3,4-d]имидазол-4(1H)-оном,
 ,
,
и CGM097, т. е. (S)-1-(4-хлорфенил)-7-изопропокси-6-метокси-2-(4-{метил-[4-(4-метил-3-оксо-пиперазин-1-ил)-транс-циклогексилметил]-амино}-фенил)-1,4-дигидро-2H-изохинолин-3-она, также называемого транс-1(S)-(4-хлорфенил)-7-изопропокси-6-метокси-2-[4-[N-метил-N-[4-(4-метил-3-оксопиперазин-1-ил)циклогексилметил]амино]фенил]-1,2,3,4-тетрагидроизохинолин-3-оном,
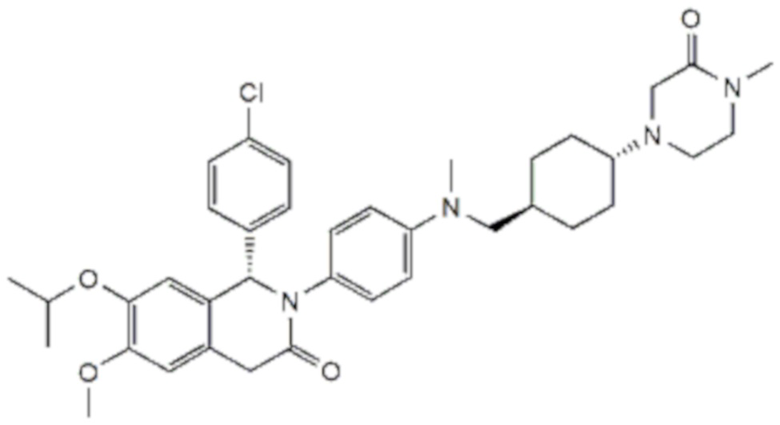 .
.
CGM097 может присутствовать в виде свободного основания или фармацевтически приемлемой соли, предпочтительно присутствует в виде сульфатной соли, более предпочтительно в виде бисульфатной соли. Соли CGM097 описаны в WO2012/066095.
HDM201 может присутствовать в виде свободной молекулы или в виде кислотного варианта. Кислотный вариант может представлять собой соль, образованную из HDM201 и кислоты, или комплекс HDM201 с кислотой, или находиться в виде сокристалла HDM201 с кислотой, предпочтительно HDM201 присутствует в виде сокристалла. Предпочтительной кислотой является янтарная кислота. Наиболее предпочтительно HDM201 присутствует в виде сокристалла с янтарной кислотой. Варианты HDM201 описаны в WO2013/111105.
Наиболее предпочтительно ингибитор HDM2 представляет собой HDM201. Режимы введения доз по настоящему изобретению особенно хорошо подходят для HDM201.
Доза в оба дня введения режима введения доз по настоящему изобретению особенно хорошо подходит для HDM201. Для других ингибиторов HDM2 может потребоваться сначала адаптировать эту дозу на основе рекомендуемой суммарной клинической дозы на цикл лечения для таких других ингибиторов HDM2, деленной на два. Эту первично адаптированную дозу можно затем дополнительно адаптировать на основе наблюдаемой клинической эффективности и токсикологических результатов, полученных с использованием этой первично адаптированной дозы.
Суточная доза ингибитора HDM2 в дни введения по настоящему изобретению может составлять от 50 мг до 400 мг, предпочтительно от 80 мг до 300 мг, более предпочтительно от 100 мг до 200 мг, еще более предпочтительно от 100 мг до 180 мг, еще более предпочтительно от 100 мг до 150 мг, еще более предпочтительно от 100 мг до 130 мг, еще более предпочтительно от 110 мг до 130 мг, наиболее предпочтительно приблизительно 120 мг. Эти дозы относятся к свободной молекуле лекарственного средства без учета какого-либо солеобразователя, комплексообразователя или образователя сокристаллов. Такие суточные дозы особенно подходят для HDM201 в качестве ингибитора HDM2.
В вариантах осуществления настоящего изобретения дозы в дни введения X1 и X2 являются предпочтительно одинаковыми, например 120 мг в день 1 (X1) и 120 мг в день 8 (X2).
Дополнительно в вариантах осуществления настоящего изобретения дозы в дни введения X1 и X2 являются предпочтительно разовыми дозами, которые принимаются пациентом в виде единиц разовой дозы, например, 120 мг в одной капсуле или одной таблетке в день 1 (X1) и 120 мг в одной капсуле или одной таблетке в день 8 (X2).
Режимы введения доз по настоящему изобретению особенно хорошо подходят для солидных опухолей, особенно для солидных опухолей с TP53 дикого типа. Указанные солидные опухоли могут представлять собой, например, типы саркомы, например липосаркому или саркому мягких тканей, типы меланомы или типы остеосаркомы, при этом типы меланомы могут представлять собой, например, меланому кожи (кожную меланому) или увеальную меланому, типы бластомы могут представлять собой, например, нейробластому, опухоль толстой кишки, колоректальную опухоль, опухоль почек (почечно-клеточную карциному), опухоль печени (карциному клеток печени), рак яичка. Режимы введения доз по настоящему изобретению являются в еще большей степени особенно хорошо подходящими для типов саркомы, например типов липосаркомы и других типов саркомы.
Режимы введения доз по настоящему изобретению имеют то преимущество, что они снижают риск гематологических токсических явлений, например, тромбоцитопении, нейтропении, лейкопении, лимфопении, анемии. Режимы введения доз по настоящему изобретению особенно подходят для снижения риска тромбоцитопении.
Лечение, включающее ингибиторы взаимодействия HDM2 с p53, может осуществляться в комбинации с агонистом рецептора тромбопоэтина, предпочтительно указанный агонист рецептора тромбопоэтина представляет собой элтромбопаг (INN), который доступен под торговыми марками PROMACTA или REVOLADE. Эта комбинация может дополнительно снижать риск развития типов цитопении, в частности тромбоцитопении и/или нейтропении.
В соответствии с настоящим изобретением суточная доза в дни введения составляет от 100 мг до 150 мг. Например, суточная доза может составлять 100 мг, 105 мг, 110 мг, 120 мг, 125 мг, 130 мг, 135 мг, 140 мг, 145 мг или 150 мг. Предпочтительно суточная доза выбрана из 120 мг, 125 мг, 130 мг, 135 мг, 140 мг, 145 мг или 150 мг. Более предпочтительно суточная доза выбрана из 120 мг или 150 мг. Еще более предпочтительно суточная доза составляет 120 мг.
Как дополнительный аспект настоящего изобретения предусмотрено следующее:
комбинация ингибитора взаимодействия HDM2 с p53 с одним или более другими терапевтически активными средствами для применения в лечении рака,
где ингибитор взаимодействия HDM2 с p53 вводят в два разных дня введения в пределах цикла лечения,
где первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней,
где лечение состоит из по меньшей мере 2 циклов лечения.
Другое терапевтически активное средство представляет собой предпочтительно противораковое средство.
При лечении гематологических опухолей (например, AML, MDS) указанное противораковое средство может быть выбрано из
ингибиторов FLT3 (например, гилтериниба, квизартиниба),
ингибиторов BCL2 (например, навитоклакса, венетоклакса),
других ингибиторов HDM2 (например, идасанутлина),
гипометилирующих средств (HMA) (например, вайдазы [азацитидин, 5-азацитидин], дакогена [децитабин], гуадецитабин),
антрациклинов (например, идарубицина, даунорубицина, доксорубицина, эпирубицина);
антител к CD33 (например, милотарга [гемтузумаб], вадастуксимаба)
и других средств (например, AraC [цитарабин, арацитин]).
При лечении солидных опухолей (например, типов липосаркомы или других типов саркомы, типов меланомы или типов увеальной меланомы) указанное противораковое средство может быть выбрано из
ингибиторов CDK-4/6 (например, рибоциклиба, палбоциклиба),
ингибиторов протеинкиназы C (например, селинексора, одного или более PKCi, раскрытых в WO2017/029588, т. е. 3-(1.H.-индол-3-ил)-4-[2-(4-метил-пиперазин-1-ил)-хиназолин-4-ил]-пиррол-2,5-диона или его фармацевтически приемлемой соли или 3-амино-N-(3-(4-амино-4-метилпиперидин-1-ил)пиридин-2-ил)-6-(3-(трифторметил)пиридин-2-ил)пиразин-2-карбоксамида или его фармацевтически приемлемой соли, 3-амино-N-(3-(4-аминопиперидин-1-ил)пиридин-2-ил)-6-(3-(трифторметокси)пиридин-2-ил)пиразин-2-карбоксамида или его фармацевтически приемлемой соли, и 3-амино-N-(3-(4-амино-4-метилпиперидин-1-ил)пиридин-2-ил)-6-(3-(трифторметокси)пиридин-2-ил)пиразин-2-карбоксамида),
и других средств (например, халавена [эрибулин], йонделиса [трабектедин]).
ПРИМЕРЫ
Пример 1
Этот пример предусматривает краткое описание данных по клинической безопасности и фармакокинетике (PK), которые служат обоснованием дозы и режима по настоящему изобретению для отдельного средства HDM201 для пациентов с солидными опухолями в фазе 1 испытания CHDM201X2101.
В данном документе раскрыты данные этого многоцентрового, открытого, впервые проводимого с участием людей исследования I фазы, осуществляемого в отношении HDM201 у пациентов с прогрессирующими солидными опухолями с TP53 дикого типа (WT), прогрессирующими при стандартной терапии или для которых не существует стандартной терапии (NCT02143635).
Было обнаружено, что предпочтительным является применение 120 мг HDM201 в день 1 и день 8 4-недельного цикла (режим 1B). Данные взяты из испытания монотерапии с датой завершения сбора данных 19 сентября 2016 г.
Основной целью части исследования, представляющей собой фазу I, было определение максимальной переносимой дозы (MTD) и/или идентификация предпочтительной дозы HDM201. Дизайн исследования позволил провести параллельное исследование безопасности, переносимости и клинической активности двух общих стратегий введения доз HDM201 при солидных злокачественных новообразованиях: режимов с прерывистым введением высоких доз (режим 1A и 1B) и режимов с длительным введением низких доз (режим 2A и 2C). В таблице 1 кратко описаны режимы введения доз в каждой категории, которые оценивали у пациентов с солидными опухолями. В таблице 2 приведены основные характеристики пациентов, вовлеченных в это исследование.
Конечным критерием оценки для основной цели является частота возникновения дозолимитирующих токсических явлений (DLT) в течение первого цикла лечения. Хотя при первичном анализе MTD оценивали на основе значения DLT, при окончательном определении предпочтительной дозы помимо значения DLT цикла 1 использовали дополнительные данные, включая переносимость на более позднем этапе цикла, PK, PD и противоопухолевую активность.
Таблица 1. Режимы введения доз HDM201 и уровни доз, оцененные в группе с солидной опухолью
Популяция пациентов
Пациентов, вовлеченных в это исследование, характеризовали с применением следующих критериев:
пациенты в возрасте ≥18 лет с местно распространенными или метастатическими солидными злокачественными новообразованиями, которые прогрессировали, несмотря на стандартную терапию, или для которых не существует эффективной стандартной терапии;
опухоли с документированным статусом TP53 WT (как минимум, без мутаций в экзонах 5-8), полученные в результате биопсии опухоли, собранные не более чем за 36 месяцев до скрининга;
заболевание c измеряемыми или неизмеряемыми (но определяемыми) проявлениями в соответствии с критериями оценки ответа при солидных опухолях (RECIST) v1.1;
показатель общего состояния по шкале Восточной объединенной онкологической группы (ECOG) ≤2;
отсутствие предыдущего лечения соединениями, которые ингибируют взаимодействие p53 с HDM2, например RG7388 или NVP-CGM097;
отсутствие лечения факторами роста, нацеленными на миелоидную линию, например
G-CSF, ≤2 недель перед исследуемым лечением;
абсолютное количество нейтрофилов >1500/мкл, количество тромбоцитов >100000/мкл, гемоглобин >9,0 г/дл.
В таблице 2 приведены основные характеристики пациентов, вовлеченных в это исследование.
Таблица 2. Основные характеристики (FAS)
(n=26)
(n=20)
(n=20)
(n=19)
(N=85)
Европеоид
Негроид
Азиат
Другая
Отсутствуют данные
1 (4)
8 (31)
2 (8)
1 (4)
0
5 (25)
1 (5)
0
0
4 (20)
2 (10)
0
0
4 (21)
0
0
1 (1)
21 (25)
5 (6)
1 (1)
n (%)
*PS по WHO/ECOG: показатель общего состояния по шкале Восточной объединенной онкологической группы/Всемирной организации здравоохранения
Статистические анализы
При принятии решений о повышении дозы руководствовались байесовской моделью логистической регрессии (BLRM) в комбинации с принципом повышения с контролем передозировки (EWOC).
При принятии решений основывались на обобщении данных, полученных для всех уровней доз и режимов, оцененных в исследовании, включая дозолимитирующие токсические явления, все данные по токсичности ≥2 степени по шкале общих терминологических критериев нежелательных явлений (CTCAE) в течение первого цикла лечения, а также фармакокинетические и фармакодинамические данные от оцениваемых пациентов.
Гематологические токсические явления цикла 2 также принимали во внимание при повышении дозы и выборе режима.
Обоснование дозы/режима
Было обнаружено, что из 4 режимов введения доз, оценку которых осуществляли в солидных опухолях с применением отдельного средства HDM201, режим с прерывистым введением высоких доз 1B (день 1 и день 8 4-недельного цикла) характеризовался наиболее благоприятным терапевтическим индексом. При этом режиме при всех испытанных дозах тромбоцитопения 3/4 степени характеризовалась наименьшей частотой возникновения и не наблюдалась у пациентов, которых лечили при выбранной RDE, составляющей 120 мг (см. таблицу 3-1). Наиболее частые негематологические токсические явления относились к желудочно-кишечной токсичности, но они не являлись дозолимитирующими ни при одном из уровней дозы, оцениваемых при 4 режимах. Фармакокинетические данные демонстрировали, что терапевтически значимые значения содержания в плазме крови достигались при уровне дозы 120 мг для режима 1B, исходя из моделирования PK/PD по данным доклинических исследований и с дополнительным подтверждением на основании наблюдения клинической эффективности у пациентов, лечение которых осуществляли при этой дозе (1 пациент с длительным PR, 1 пациент с неподтвержденным PR и 1 пациент с SD). Доза 120 мг также находилась в диапазоне благоприятных доз, рекомендуемых на основании байесовской модели логистической регрессии (BLRM), свидетельствующей в пользу повышения дозы. Поэтому режим 1В с дозой 120 мг рассматривали как наиболее предпочтительную дозу и режим.
Детальное резюме клинических исследований
На момент даты завершения сбора данных (19 сентября 2016 г.) 85 пациентов с солидными опухолями прошли лечение с помощью HDM201 при 4 оцениваемых режимах введения доз (см. таблицу 1). Дозолимитирующие токсические явления при всех оцениваемых режимах были связаны главным образом с миелосупрессией.
При разных режимах лечения из всех дозолимитирующих типах цитопении чаще всего наблюдали нейтропению и тромбоцитопению 3/4 степени (таблица 3). Следовательно, сравнительная частота возникновения типов цитопении 3/4 степени (что особенно важно, тромбоцитопении) при всех 4 режимах лечения была ключевым фактором, определяющим выбор режима и дозы для применения в расширенной популяции.
Было обнаружено, что во время исследования такая миелосупрессия, индуцированная HDM201, может характеризоваться отсроченным началом (после цикла 1). Следовательно, дозолимитирующие гематологические токсические явления, возникающие в цикле 2, также учитывались при принятии решения о повышении дозы в течение исследования с использованием факультативной модели чувствительности. В таблице 4 представлены сводные данные по количеству дозолимитирующих токсических явлений в течение цикла 1 и дозолимитирующих гематологических токсических явлений в цикле 2 при всех режимах, оцениваемых в случае солидных опухолей.
При повышении дозы сначала оценивали режим с прерывистым введением высоких доз 1A и режим с длительным введением низких доз 2A. Оба режима характеризовались неблагоприятными процентами DLT и отсроченными гематологическими явлениями при уровнях дозы, достигающих предсказанных терапевтически значимых значений содержания в плазме крови. Потому были введены когорты для исследования двух дополнительных режимов: режима 1B с прерывистым введением высоких доз и режима 2C с длительным введением низких доз. При режиме 2C DLT наблюдали при уровнях дозы, при которых значения содержания в плазме крови были ниже значений, эффективность которых была предсказана на основе моделирования PK/PD.
Лечение двадцати пациентов осуществляли в соответствии с режимом 1B при 3 разных уровнях дозы (120 мг, 150 мг и 200 мг). Наиболее частые AE (все степени), в отношении которых было отмечено, что их предположительной причиной является исследуемое лечение в режиме 1B, представляли собой тошноту (12 пациентов, 60,0%), тромбоцитопению/снижение количества тромбоцитов (9 пациентов, 45,0%), нейтропению/снижение количества нейтрофилов (8 пациентов, 40,0%) и рвоту (5 пациентов, 25,0%). Девять пациентов (45,0%) из этой группы имели по меньшей мере по одному AE 3/4 степени по CTCAE, предположительно связанному с лечением. Три наиболее частых AE 3/4 степени по CTCAE, считающиеся предположительно связанными с исследуемым лечением, представляли собой следующие: нейтропению/снижение количества нейтрофилов (6 пациентов, 30,0%), повышение уровня липазы (3 пациента, 15%) и тромбоцитопению/снижение количества тромбоцитов (2 пациента, 10,0%). Один случай продолжительной нейтропении (с началом в день 22 и продолжительностью 18 дней), отвечающий критериям DLT, наблюдали у одного пациента, получавшего дозу 150 мг. Для получения более подробной информации см. таблицу 5. Из 4 оцененных режимов, режим 1B характеризовался наиболее низкой общей частотой возникновения тромбоцитопении 3/4 степени (таблица 3).
При предпочтительной дозе 120 мг (режим 1B) не было ни одного случая AE, представляющего собой тромбоцитопению 3/4 степени (см. таблицу 3-1). При этом уровне дозы не было прерывания или прекращения приема дозы вследствие тромбоцитопении, и ни один пациент не нуждался в переливании тромбоцитов. Частота возникновения нейтропении 3/4 степени была сходной при всех режимах и при уровне дозы 120 мг наблюдалась у 2 из 9 пациентов. При этом уровне дозы не было никаких негематологических дозолимитирующих токсических явлений или AE 3/4 степени.
Важно отметить, что значимая клиническая активность наблюдалась при предпочтительной дозе 120 мг (режим 1B). У 9 пациентов, получавших эту дозу, наблюдали 1 PR (продолжительностью 18 недель и продолжающийся на дату прекращения сбора данных), у пациента с саркомой мягких тканей, 1 неподтвержденный PR и 1 SD (продолжительностью 8 недель), оба у пациентов с липосаркомой, что свидетельствует о достижении терапевтически значимых значений содержания в плазме крови при этой дозе и схеме.
Таблица 3. Все нежелательные явления в виде типов цитопении, предположительно связанные с исследуемым лекарственным средством - солидные опухоли
пониженное количество нейтрофилов*
пониженное количество лейкоцитов*
n (%)
n (%)
n (%)
n (%)
n (%)
n (%)
n (%)
n (%)
Таблица 4. DLT цикла лечения 1 и гематологические дозолимитирующие токсические явления цикла 2 при солидных опухолях
(цикл 1)
(n=9 )
(n=8 )
(n=3 )
Таблица 5. Нежелательные явления всех степеней и степени 3/4, предположительно связанные с исследуемым лекарственным средством, по предпочтительным терминам и лечению - солидные опухоли - режим 1B
- Пациента с несколькими случаями возникновения AE при одном лечении учитывали только один раз в категории AE
для этого лечения.
- Пациента с несколькими нежелательными явлениями учитывали только один раз в строке "Всего".
- Описаны только AE, возникшие в течение лечения или в пределах 30 дней после последнего приема исследуемого лекарственного препарата.
Безопасность
Дозолимитирующие токсические явления, обычно возникающие в течение цикла 2, представляли собой нейтропению и тромбоцитопению.
Нежелательные явления всех степеней, связанные с исследуемым лекарственным средством (AE; возникающие у ≥10% всех пациентов), представлены в таблице 6.
Таблица 6. Нежелательные явления, предположительно связанные с исследуемым лекарственным средством, суммарно по всем режимам лечения (все степени, возникающие в ≥10%)
(N=85)
Наиболее частые негематологические токсические явления относились к желудочно-кишечной токсичности, но они не являлись дозолимитирующими ни при одном из уровней дозы, оцениваемых при 4 режимах; при рассмотрении всех степеней наиболее распространенным желудочно-кишечным AE являлась тошнота (44/85; 52%), которая в основном характеризовалась легкой или средней тяжестью.
Представляющие интерес AE 3/4 степени, связанные с исследуемым лекарственным средством, показаны в таблице 3. Гематологические токсические явления 3/4 степени, предположительно связанные с исследуемым лекарственным средством, наблюдались при всех режимах лечения, при этом они возникали не более чем у ~35% пациентов. Тромбоцитопения 3/4 степени характеризовалась наименьшей частотой возникновения при режиме 1B.
Клиническая PK
Фармакокинетические данные оценивали в течение всего повышения дозы. Во время исследования оценивали два варианта лекарственного средства HDM201 (для получения более подробной информации см. протокол). В результате некомпартментного анализа PK показали, что для всего диапазона доз (от 2 до 350 мг) медиана времени, необходимого для достижения максимальных концентраций в плазме, находилась в диапазоне от 2,0 до 5,8 ч. Предварительная оценка пропорциональности дозы свидетельствовала о приблизительно дозопропорциональной PK (AUClast и Cmax) в исследованном диапазоне доз. Для большинства когорт, получавших разные дозы, межиндивидуальная вариабельность (среднее геометрическое CV%) в отношении AUClast и Cmax являлась низкой или средней (от 6 до 58,5%). Кроме того, был проведен комплексный анализ всех предусмотренных концентраций HDM201 с использованием популяционного подхода. Наилучшее описание PK HDM201 было получено с помощью 1-компартментной модели PK с отсроченным процессом абсорбции нулевого и первого порядка и линейным клиренсом. Вес тела идентифицировали в качестве статистически значимой независимой переменной, влияющей на кажущийся центральный объем распределения (Vc/F), при этом Vc/F увеличивался с увеличением веса тела.
Чтобы получить дополнительные сведения в пользу предпочтительной дозы для HDM201, применяли компартментное моделирование PK для оценки индивидуальной средней концентрации на цикл для 9 пациентов, получавших 120 мг при режиме 1B (фигура 2). Для большинства пациентов (7 из 9) оцененные средние концентрации лекарственного средства на цикл находились рядом с наиболее консервативной средней концентрацией для остановки роста опухоли, составляющей ≈41 нг/мл на цикл, или выше нее, как определено с помощью моделирования PKPD по данным доклинических исследований (крысиная модель с ксенотрансплантатом SJSA-1 человека).
Репрезентативные кривые зависимости среднего геометрического концентрации в плазме от времени для NVP-HDM201 после введения разовой дозы (день 1) для режима лечения 1А (12,5-350 мг) представлены на фигуре 3.
Абсорбция в ротовой полости была быстрой (медиана Tmax 2-5,8 часов) и не варьировала в зависимости от дозы, получаемой группой (2-350 мг).
Средние значения содержания в плазме крови (AUClast и Cmax) увеличивались с увеличением дозы, без значительных отклонений от пропорциональности дозы после однократных и повторных доз.
Стабильное состояние NVP-HDM201 обычно достигалось ко дню 8 благодаря ограниченному накоплению в результате ежедневного введения доз.
Медиана периода полувыведения, оцениваемая после введения разовой дозы в день 1 (50-350 мг), изменялась в диапазоне от 13,7 до 23,1 ч.
Межиндивидуальная вариабельность содержания в плазме крови (среднее геометрическое CV%) обычно находилась в диапазоне от низкой до умеренной. Компартментное популяционное моделирование PKNVP-HDM201 применяли для оценки индивидуальной средней концентрации в плазме для цикла 1 и для сравнения со средней концентрацией для остановки роста опухоли, полученной в доклинических исследованиях с помощью моделирования PK/PD роста опухоли. Результаты представлены на фигуре 4.
По сравнению с режимами 2A/2C, средняя концентрация в плазме, которая достигалась при режимах 1A/1B, находилась ближе к прогнозируемым на основе доклинических исследований целевым эффективным уровням (125 нг/мл), необходимым для регрессии опухоли на 95% (верхняя пунктирная линия на фигуре 5), и рядом с оцененными средними концентрациями для наиболее консервативной средней концентрации для остановки роста опухоли, составляющей ≈41 нг/мл (пунктирная линия), или выше них, как определено с помощью моделирования PKPD в крысиной модели с ксенотрансплантатом SJSA-1 человека (фигура 4).
Пунктирная линия на уровне концентрации ≈19 нг/мл представляет среднюю концентрацию для остановки роста опухоли, определенную с помощью моделирования PK/PD по данным доклинических исследований на крысиной модели с ксенотрансплантатом липосаркомы (HSAX2655), полученным от пациента.
Пунктирная линия на уровне концентрации 29,4 нг/мл представляет значение IC50, определенное по клеточной активности в клеточной линии SJSA-1.
Статистический анализ
В этом исследовании для получения сведений в пользу повышения дозы и для оценки MTD и/или определения предпочтительной дозы HDM201 применяли байесовскую модель логистической регрессии (BLRM). BLRM в комбинации с принципом повышения с контролем передозировки (EWOC) позволяет включать доступную предварительную информацию и обновлять параметры модели на основе новой информации по установленным дозолимитирующим токсическим явлениям (DLT), наблюдаемым в клиническом исследовании. В ходе повышения дозы для режима 1A и 1B для обновления модели и подтверждения решения о следующей дозе использовали частоту возникновения DLT. DLT определяются в соответствии с таблицей 6-3 протокола и представляют собой явления, возникающие во время первого цикла лечения. Когда в ходе исследования стало очевидно, что индуцируемая HDM201 токсичность в отношении костного мозга возникала преимущественно во время цикла 2, для принятия решений в отношении повышения дозы/RDE применили факультативную модель чувствительности, учитывающую DLT цикла 1 и гематологические дозолимитирующие AE в цикле 2 (при этом всем типам цитопении приписывали одинаковый вес). Кроме того, решения всегда основывались на обобщении соответствующих данных, полученных для всех уровней дозы, оцениваемых в исследовании, включая данные о токсичности низкой степени, данные по PK и PD (при их наличии) от оцениваемых пациентов.
Результаты BLRM с использованием данных о явлениях DLT цикла 1 от пациентов, получавших лечение согласно режиму 1B (уровень дозы 120 мг, 150 мг и 200 мг), свидетельствовали в пользу повышения до 400 мг HDM201. Медиана уровня DLT при 120 мг составляла 3,5% и 25,7% согласно анализу по протоколу и анализу чувствительности соответственно. Таким образом, дозу 120 мг установили в качестве предпочтительной, учитывая более низкую частоту возникновения клинически значимой тромбоцитопении 3/4 степени, поддающуюся коррекции нейтропению и значимую клиническую активность, наблюдаемые при этой дозе.
Эффективность
На момент даты завершения сбора данных у 2 из 46 (4%) пациентов, для которых применяли режимы с прерывистым введением высоких доз, достигли PR (1 пациент с STS, представляющей собой интимальную саркому, для которого применяли режим 1А; 1 пациент с STS, представляющей собой гемангиоперицитому, для которого применяли режим 1B) (таблица 7). У 15 из 46 (33%) пациентов, для которых применяли режимы с прерывистым введением высоких доз, и у 14 из 39 (36%) пациентов, для которых применяли режимы с длительным введением низких доз, достигли SD (таблица 7).
Хотя при всех режимах введения доз отмечали существенный уровень контроля заболевания (DCR: 34%), PR были установлены только при режимах 1A и 1B, что позволяет предположить, что режимы с прерывистым введением высоких доз являются более активными.
На сентябрь 2017 года в случае пациентов с саркомой (липосаркомой и другими типами саркомы) наблюдали высокую противоопухолевую эффективность. Из 21 пациента с саркомой, получавших HDM201 в соответствии с режимом 1B, у 5 пациентов наблюдали частичный ответ (PR) и у 11 - стабильное заболевание (SD). Заболевание прогрессировало (PD) только у 5 пациентов (см. фиг. 8).
Таблица 7. Наилучший общий ответ (FAS) (ноябрь 2016 г.)
(n=26)
(n=20)
(n=20)
(n=19)
95% CI
0,1-19,6
0,1-24,9
0,0-16,8
0,0-17,6
95% CI
17,2-55,7
19,1-63,9
15,4-59,2
16,3-61,6
BOR - наилучший общий ответ; CI - доверительный интервал; CR - полный ответ; DCR - уровень контроля заболевания (CR, или PR, или SD); FAS - анализ с полной популяцией; ORR - общий уровень ответа (CR или PR); PD - прогрессирующее заболевание; PR - подтвержденный частичный ответ; SD - стабильное заболевание; BOR основан на оценке состояния болезни исследователем с применением RECIST 1.1; CR и PR подтверждены повторными оценками, проведенными не менее чем через 4 недели после первого соответствия критериям ответа. 95% CI рассчитывали с использованием точного интервала (Клоппера-Пирсона).
Медиана относительной интенсивности дозы (RDI) для пациентов с по меньшей мере стабильным заболеванием или улучшением на конец 32 недель лечения была аналогичной при режимах с длительным введением низких доз 2А и 2С. Из 2 режимов с прерывистым введением высоких доз режим 1В характеризовался более благоприятным RDI, подтверждая его общую лучшую переносимость при терапевтически значимых дозах (таблица 8).
Таблица 8. Сводные данные по относительной интенсивности дозы для пациентов с по меньшей мере стабильным заболеванием в конце 32 недель лечения (SAS)
(n=20)
(n=20)
(n=13)
(n=19)
SAS - популяция для анализа безопасности.
n=общее количество пациентов, получавших лечение, включающее только группы лечения с соответствующими режимами:
Режим 1A: ≥100 мг; Режим 1B: ≥120 мг; Режим 2A: ≥7,5 мг; Режим 2C: ≥15 мг
N=количество пациентов с по меньшей мере одним SD, или PR, или CR, или пациенты, которые прекратили лечение по причинам, отличным от PD.
Пример 2. Подробные фармакокинетические данные
5.1.2.1.1 Монотерапия у взрослых пациентов [HDM201X2101]
PK HDM201 оценивали в ходе продолжающегося клинического испытания фазы I с применением отдельного средства у пациентов с солидными опухолями и гематологическими злокачественными новообразованиями (HDM201X2101). Пациенты получали разовую дозу в день 1 трехнедельного цикла (q3w, режим 1A), разовую дозу в день 1 и день 8 четырехнедельного цикла (d1, d8 4-недельного цикла, режим 1B) или ежедневную дозу в течение первых 7 или 14 дней четырехнедельного цикла (режим 2C [q.d., 1 неделя введения/3 недели без введения] или режим 2A [q.d., 2 недели введения/2 недели без введения], соответственно). Оценки PK проводили после разовой дозы и повторного введения. Предварительные параметры PK, полученные в результате некомпартментного анализа кривых зависимости среднего геометрического концентрации в плазме от времени, обобщены по режиму введения доз и типу опухоли (дата завершения сбора данных 01 апреля 2016 года). Параметры PK для режима 2A (день 1 и 14 цикла 1 для доз от 1 до 20 мг) и режима 1A (день 1 цикла 1 для доз от 12,5 до 350 мг) у пациентов с солидными опухолями обобщены в виде репрезентативных значений PK для режимов с ежедневным и менее частым введением доз (таблицы 9, 10 и 11).
Таблица 9. Сводные данные по основным параметрам PK для режима 2A с ежедневным введением HDM201 после разовой дозы (солидные злокачественные новообразования)
Лечение (день 1) AUClast (нг*ч./мл), Cmax (нг/мл), Tmax (ч.)
HDM201 2A, 1 мг (N=1) N=1 N=1 N=1
134,3 7,9 8,0
HDM201 2A, 2 мг (N=2) N=2 N=2 N=2
169,1 (31,5) 12,0 (23,7) 3,4
HDM201 2A, 4 мг (N=4) N=4 N=4 N=4
192,6 (29,1) 17,6 (22,8 ) 2,5
HDM201 2A, 7,5 мг (N=4) N=4 N=4 N=4
598,1 (49,8) 39,0 (43,9) 5,8
HDM201 2A, 15 мг (N=4) N=4 N=4 N=4
1301,6 (71,0) 91,5 (56,9) 3,1
HDM201 2A, 20 мг (N=5) N=5 N=5 N=5
2300,9 (33,3) 163,6 (15,9) 2,0
Значения представляют собой среднее геометрическое (%CV), кроме Tmax, где представлена медиана.
AUClast подсчитано за период 0-24 ч.
Таблица 10. Сводные данные по основным параметрам PK для режима 2A с ежедневным введением HDM201 в день 14 (солидные злокачественные новообразования)
Лечение (день 14) AUClast (нг*ч./мл) Cmax (нг/мл) Tmax (ч.)
HDM201 2A, 1 мг (N=1) N=1 N=1 N=1
99,5 14,9 4,0
HDM201 2A, 2 мг (N=2) N=2 N=2 N=2
109,3 (6,0) 17,5 (4,9) 3,9
HDM201 2A, 4 мг (N=4) N=3 N=3 N=3
155,0 (21,7) 25,6 (23,9) 4,0
HDM201 2A, 7,5 мг (N=4) N=2 N=2 N=2
221,7 (44,7) 36,1 (43,6) 3,9
HDM201 2A, 15 мг (N=4) N=4 N=4 N=4
688,8 (29,2) 106,0 (27,9) 3,0
HDM201 2A, 20 мг (N=5) N=4 N=4 N=4
1264,0 (26,3) 214,5 (21,4) 4,0
Значения представляют собой среднее геометрическое (%CV), кроме Tmax, где представлена медиана.
AUClast подсчитано за период 0-24 ч.
Таблица 11. Сводные данные по основным параметрам PK для режима 1A с введением HDM201 q3w после
разовой дозы (солидные злокачественные новообразования)
Лечение (день 1) AUClast (нг*ч./мл) Cmax (нг/мл) T1/2 (ч.) Tmax (ч.)
HDM201 1A, 12,5 мг (N=1) N=1 N=1 N=1 N=1
1483,1 118,0 7,2 3,2
HDM201 1A, 25 мг (N=1) N=1 N=1 N=1 N=1
1773,6 100,0 31,1 2,1
HDM201 1A, 50 мг (N=4) N=4 N=4 N=4 N=4
8028,8 (25,6) 467,0 (13,0) 11,8 (27,9) 3,0
HDM201 1A, 100 мг (N=4) N=4 N=4 N=3 N=4
14287,0 (58,5) 663,3 (30,1) 16,3 (21,6) 3,5
HDM201 1A, 200 мг (N=5) N=5 N=5 N=4 N=5
26255,2 (56,2) 1168,8 (43,7) 15,1 (102,7) 3,0
HDM201 1A, 250 мг (N=6) N=6 N=6 N=5 N=6
23850,1 (38,5) 1072,6 (36,3) 15,3 (43,8) 3,5
HDM201 1A, 350 мг (N=5) N=5 N=5 N=4 N=5
50527,1 (24,3) 1936,4 (40,5) 15,1 (47,1) 4,1
Значения представляют собой среднее геометрическое (%CV), кроме Tmax, где представлена медиана.
AUClast подсчитано за период 0-24 ч.
При введении доз пероральным путем (капсула HDM201, натощак) медиана времени, необходимого для достижения максимальных концентраций в плазме, изменялась в диапазоне от 2,0 до 5,8 ч. для всего диапазона доз (от 2 до 350 мг). При режиме с ежедневным введением доз устойчивое состояние HDM201 обычно достигалось к 8 дню, и накопление являлось менее чем 2-кратным. Среднее значение T1/2, оценку которого осуществляли после введения разовой дозы (от 50 до 350 мг), изменялось в диапазоне от 11,8 до 16,3 ч. Предварительная оценка пропорциональности дозы свидетельствовала о приблизительно дозопропорциональной PK (AUClast и Cmax) в исследуемом диапазоне доз после разовой дозы (от 1 до 350 мг) в день 1 и после нескольких доз (от 1 до 30 мг q.d.) ко дню 14. Для большинства когорт, получавших разные дозы, межиндивидуальная вариабельность (среднее геометрическое CV%) в отношении AUClast и Cmax являлась низкой или средней (от 6 до 58,5%).
Кроме того, наилучшее описание индивидуальных данных PK получили с помощью 1-компартментной модели РК с отсроченным процессом абсорбции нулевого и первого порядка (дата завершения сбора данных 01 апреля 2016 года). Оценка среднего по популяции для кажущегося клиренса при пероральном введении (CL/F) и кажущегося объема распределения для центрального компартмента (Vc/F) составила 6,18 л/ч. и 119 л соответственно (относительная стандартная ошибка 7%), при этом межиндивидуальная вариабельность составляла 48% (CL/F) и 30% (Vc/F). Вес тела идентифицировали в качестве статистически значимой независимой переменной, влияющей на Vc/F.
Пример 3. Модель PK/PD тромбоцитопении
Модель PK/PD была создана на основе индивидуальных данных по PK и количеству тромбоцитов с течением времени.
Модель PK: 1-компартментная с двухфазной абсорбцией.
Модель PD: скорректированная модель Фриберга для тромбоцитопении, учитывающая переливания PLT и влияние HDM201 на пролиферативные клетки и пути регуляции.
База данных:
n=73 пациента
1301 наблюдение PK
1023 наблюдения PD тромбоцитов
427 наблюдений PD GDF15
Кинетические кривые зависимости для тромбоцитов, продемонстрированные на фигуре 5, смоделированы на основе следующих доз, проверенных при каждом режиме (по порядку сверху вниз на фигуре 5):
реж. 2C (D1-7 Q4wk): 25 мг ((25 мг x 7 дней введения)/28-дневный цикл=6,25 мг/сутки);
реж. 2A (D1-14 Q4wk): 20 мг ((20 мг x 14 дней введения)/28-дневный цикл=10 мг/сутки);
реж. 1B (дни 1, 8 Q4wk): 150 мг ((150 мг x 2 дня введения)/28-дневный цикл=10,7 мг/сутки);
реж. 1A (D1 Q3wk): 350 мг ((350 мг x 1 день введения)/21-дневный цикл=16,7 мг/сутки).
Основываясь на этом моделировании, 1B характеризуется наилучшей общей кинетической кривой зависимости для тромбоцитов из всех режимов, которые продемонстрировали активность отдельного средства.
Первое появление тромбоцитопении G4 в случае 150 мг при режиме 1B в клиническом исследовании произошло только через 100 дней.
Добавление элтромбопага к 1B может уменьшить уровень относительной задержки и снижения пика восстановления тромбоцитов в последующих циклах.
Пример 4. Лекарственный препарат
Лекарственный препарат состоит из лекарственного средства, представляющего собой HDM201 с янтарной кислотой, расфасованного непосредственно в твердые желатиновые капсулы (HGC), и не содержит каких-либо других вспомогательных веществ. Лекарственный препарат предусмотрен в четырех дозировках: 1 мг, 2,5 мг, 10 мг и 100 мг (в пересчете на вес свободной формы), предназначенных для перорального применения. Капсула с дозировкой 1 мг представляет собой желтую HGC с "размером 3", капсула с дозировкой 2,5 мг представляет собой оранжево-красную HGC с "размером 3", капсула с дозировкой 10 мг представляет собой серую HGC с "размером 1", а с дозировкой 100 мг представляет собой оранжево-красную HGC с "размером 0". Лекарственный препарат упакован в бутылки из полиэтилена высокой плотности (HDPE) с индукционной запайкой с защитой от детей.
Пример 5. Режим 1B при гематологических опухолях
В этом примере представлено краткое описание данных клинических исследований из испытания CHDM201X2101 фазы 1 (дата завершения сбора данных 07 декабря 2016 г.), которые демонстрируют, что режим введения доз "1B" также подходит для лечения пациентов с гематологическими опухолями.
В данном документе раскрыты данные этого многоцентрового, открытого, впервые проводимого с участием людей исследования I фазы, осуществляемого в отношении HDM201 у пациентов с прогрессирующими типами лейкемии с TP53 дикого типа (WT).
Дизайн клинического исследования позволил провести параллельное исследование безопасности, переносимости и клинической активности (эффективности) двух общих стратегий введения доз HDM201 при повышении доз: режимов с прерывистым введением высоких доз (режим 1A и 1B) и режимов с длительным введением низких доз (режим 2A и 2C). В таблице 1 кратко описаны режимы введения доз в каждой категории, оценку которых осуществляли у пациентов с гематологическими опухолями.
Таблица 1.Режимы введения доз HDM201 и уровни дозы, оценку которых осуществляли при гематологических злокачественных новообразованиях
На момент даты завершения сбора данных все из 37 пациентов (35 AML и 2 ALL) прошли лечение с применением HDM201 при 4 оцениваемых режимах введения доз (см. таблицу 1). При режиме 1B 6 пациентов с AML получали HDM201 в дозе 150 мг в день 1 и день 8 28-дневного цикла лечения.
Характеристики этих пациентов представлены в таблице 2.
Таблица 2. Характеристики пациентов
Популяцию пациентов дополнительно характеризовали по следующим критериям включения:
Пациент (мужчина или женщина) в возрасте ≥ 18 лет.
Показатель общего состояния 0-2 по ECOG.
Пациенты с рецидивирующим/рефрактерным AML (как de novo, так и вторичным AML), кроме острого промиелоцитарного лейкоза (APL) с t(15; 17), или пациенты, ранее не получавшие лечение, которые считаются неподходящими кандидатами для стандартной индукционной терапии.
Только при повышении дозы, MDS высокого и очень высокого риска в соответствии с последней редакцией Международной прогностической балльной системы (IPSS-R), в отношении которых предшествующая терапия, такая как терапия с применением азацитидина и децитабина, была неэффективной (пациенты с оценкой IPSS-R > 4,5).
Только при повышении дозы, рецидивирующий/рефрактерный острый лимфобластный лейкоз (B-ALL или T-ALL), включая Ph+ ALL, или пациенты, ранее не получавшие лечение, которые считаются неподходящими кандидатами для стандартной индукционной терапии. Включение пациентов с Ph+ALL, у которых при надзоре за MRD обнаруживают ранние маркеры рецидива, может рассматриваться при условии, что другие методы лечения, такие как TKI, исчерпаны или их применение невозможно.
Опухоль у пациента представляет собой опухоль с TP53wt, характеризующимся как минимум отсутствием мутаций в экзонах 5, 6, 7 и 8, и при этом статус p53 был установлен на образце костного мозга, отобранного не более чем за 3 месяца до подписания основного ICF.
Профиль безопасности и переносимости режима 1В при гематологических опухолях
В таблицах 3 и 4 представлена информация о безопасности и переносимости режима введения доз 1В по сравнению с другими режимами с прерывистым введением высоких доз и режимами с длительным введением низких доз.
Для режима 1B было обнаружено только 1 дозолимитирующее токсическое явление (DLT). Обнаружено, что частота возникновения нежелательных явлений всех степеней и 3/4 степени была сравнительно низкой.
Таблица 3. DLT цикла 1 при гематологических опухолях
-Инфекция G3, реактивация GVHd G3, стоматит G3
-Гипофосфатемия G4
-Субарахноидальное кровоизлияние (фатальное)
-Гипофосфатемия G4
- Острая почечная недостаточность G4
Таблица 4. Нежелательные явления всех степеней и 3/4 степени, предположительно связанные с исследуемым лекарственным средством, по предпочтительным терминам и режимам - гематологические опухоли
- Пациента с несколькими случаями возникновения AE при одном лечении учитывали только один раз в категории AE
для этого лечения.
- Пациента с несколькими нежелательными явлениями учитывали только один раз в строке "Всего".
- Описаны только AE, возникшие в течение лечения или в пределах 30 дней после последнего приема исследуемого лекарственного препарата.
Противоопухолевая активность/эффективность режима 1B при гематологических опухолях
В таблицах 5 и 6 и на фигуре 6 представлена информация о режиме введения доз 1B по сравнению с другими режимами. У одного пациента, который получал лечение согласно режиму 1B, установили CRi и таким образом продемонстрировали, что режим 1B является эффективным также при гематологических опухолях. Этот пациент с CRi продемонстрировал наиболее сильный положительный эффект, что проявилось в лучшем процентном изменении процентной доли бластных клеток в аспирате костного мозга (BM) (фигура 6).
Таблица 5a. Противоопухолевая активность (март 2017 г.)
(N=35)
BOR - наилучший общий ответ; CR - полный ответ; CRi - морфологический CR с неполным восстановлением показателей крови; ORR - общий уровень ответа.
Таблица 5b. Противоопухолевая активность (сентябрь 2017 г.)
350 мг (4): -
400 мг (9): 1 CR
120 мг (6): 1 CR
4 пациента: 30 мг (4): -
1 CR, 3 CRi
1 CRi
1 CR, 1CRi
Количество в скобках - количество пациентов, пациенты; CR - полный ответ; CRi - морфологический CR с неполным восстановлением показателей крови.
Таблица 6a. Характеристики пациентов с AML с CR/CRi (март 2017 г.)
(года)
тельность CR/CRi
(дни)
*Пациент отозвал свое согласие в то время как наблюдался CRi
†Пациент, продолжающий лечение в момент даты завершения сбора данных
Таблица 6b. Обновленная таблица (сентябрь 2017 г.), характеристики пациентов с AML с CR/CRi
HDM
/81
/71
/67
/76
/71
/83
/73
/80
/70
/79
/72
То, что для небольшого числа пациентов, прошедших лечение до настоящего времени, уже установлено 2 результата, представляющие собой CR/CRi, свидетельствует о сильной противоопухолевой активности HDM201 у пациентов с AML при введении в соответствии с режимом 1B.
Клиническая PK
Фармакокинетические данные оценивали в течение всего клинического исследования. В результате некомпартментного анализа PK показали, что для всего диапазона доз (от 2 до 350 мг) медиана времени, необходимого для достижения максимальных концентраций в плазме, находилась в диапазоне от 2,0 до 5,8 ч. Предварительная оценка пропорциональности дозы свидетельствовала о приблизительно дозопропорциональной PK (AUClast и Cmax) в исследованном диапазоне доз. Для большинства когорт, получавших разные дозы, межиндивидуальная вариабельность (среднее геометрическое CV%) в отношении AUClast и Cmax являлась низкой или средней (от 6 до 58,5%). Кроме того, был проведен комплексный анализ всех предусмотренных концентраций HDM201 с применением популяционного подхода. Наилучшее описание PK HDM201 было получено с помощью 1-компартментной модели PK с отсроченным процессом абсорбции нулевого и первого порядка и линейным клиренсом. Вес тела идентифицировали в качестве статистически значимой независимой переменной, влияющей на кажущийся центральный объем распределения (Vc/F), при этом Vc/F увеличивался с увеличением веса тела.
Для оценки индивидуальной средней концентрации в течение цикла 1 у пациентов с гематологическими опухолями, получавших лечение по четырем различным режимам, применяли компартментное моделирование PK (фигура 7). Для всех пациентов с измеренной PK оцененные средние концентрации лекарственного средства в течение цикла 1 превышали наиболее консервативную среднюю концентрацию для остановки роста опухоли, составляющую ≈41 нг/мл на цикл, как определено с помощью моделирования PKPD по данным доклинических исследований (крысиная модель с ксенотрансплантатом SJSA-1 человека).
| название | год | авторы | номер документа |
|---|---|---|---|
| ДОЗА И СХЕМА ВВЕДЕНИЯ ИНГИБИТОРА ВЗАИМОДЕЙСТВИЯ HDM2 С p53 ПРИ ГЕМАТОЛОГИЧЕСКИХ ОПУХОЛЯХ | 2018 |
|
RU2753527C2 |
| КОМБИНАЦИИ ИНГИБИТОРА ВЗАИМОДЕЙСТВИЯ HDM2-P53 И ИНГИБИТОРА BCL2 И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ РАКА | 2019 |
|
RU2793123C2 |
| ПРЕРЫВИСТОЕ ВВЕДЕНИЕ ИНГИБИТОРА MDM2 | 2015 |
|
RU2695228C2 |
| GDF-15 КАК ГЕМАТОЛОГИЧЕСКИЙ БИОМАРКЕР ТОКСИЧНОСТИ | 2016 |
|
RU2741390C2 |
| СХЕМА ПРИЕМА ДЛЯ СЕЛЕКТИВНОГО ПО АЛЬФА-ИЗОФОРМЕ ИНГИБИТОРА ФОСФАТИДИЛИНОЗИТОЛ-3-КИНАЗЫ | 2014 |
|
RU2680246C1 |
| РЕЖИМЫ ДОЗИРОВАНИЯ МЕЛФЛУФЕНА ДЛЯ РАКОВЫХ ЗАБОЛЕВАНИЙ | 2016 |
|
RU2734930C2 |
| ИНГИБИТОРЫ ВЗАИМОДЕЙСТВИЯ МЕЖДУ MDM2 И Р53 | 2007 |
|
RU2436784C2 |
| ДВОЙНЫЕ ИНГИБИТОРЫ ATM И DNA-PK ДЛЯ ПРИМЕНЕНИЯ В ПРОТИВООПУХОЛЕВОЙ ТЕРАПИИ | 2020 |
|
RU2800756C1 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2805145C2 |
| КОМБИНИРОВАННАЯ ТЕРАПИЯ ДЛЯ ЛЕЧЕНИЯ РАКА | 2014 |
|
RU2680714C2 |



Изобретение относится к способу лечения рака с TP53 дикого типа, включающему введение HDM201 в два разных дня введения в пределах цикла лечения, где первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения, где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и где лечение состоит из по меньшей мере 2 циклов лечения, и при этом суточная доза в дни введения составляет от 100 до 200 мг. Заявленный способ позволяет вводить высокую дозу, обеспечивающую эффективность, и снизить риск возникновения нежелательных явлений. 8 з.п. ф-лы, 8 ил., 17 табл., 5 пр.
1. Способ лечения рака с TP53 дикого типа,
включающий введение HDM201 в два разных дня введения в пределах цикла лечения,
где первый день введения и второй день введения разделены коротким периодом без введения, и второй день введения первого или более раннего цикла лечения и первое введение следующего цикла разделены продолжительным периодом без введения,
где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 13-27 дней, и
где лечение состоит из по меньшей мере 2 циклов лечения, и
при этом суточная доза в дни введения составляет от 100 до 200 мг.
2. Способ по п. 1, где короткий период без введения состоит из 4-8 дней, и продолжительный период без введения состоит из 18-22 дней.
3. Способ по п. 1, где короткий период без введения состоит из 5-7 дней, и продолжительный период без введения состоит из 19-21 дня.
4. Способ по п. 1, где короткий период без введения состоит из 6 дней, и продолжительный период без введения состоит из не менее 20 дней.
5. Способ по п. 1, где указанный ингибитор представляет собой HDM201 с янтарной кислотой.
6. Способ по п. 1 или 5, где суточная доза в дни введения составляет от 100 до 150 мг.
7. Способ по п. 1 или 5, где суточная доза в дни введения составляет 120 мг.
8. Способ по любому из предыдущих пунктов, где рак представляет собой солидную опухоль.
9. Способ по п. 8, где солидная опухоль выбрана из типов саркомы например, липосаркомы или саркомы мягких тканей, типов меланомы, например меланомы кожи или увеальной меланомы, типов бластомы, например, нейробластомы, опухоли толстой кишки, колоректальной опухоли, опухоли почки и опухоли печени.
| WO 2015198266 A1, 30.12.2015 | |||
| US 20130245089 A1, 19.09.2013 | |||
| RU 2014141365 A, 10.05.2016 | |||
| WO 2011076786 A1, 30.06.2011 | |||
| WO 2013111105 A1, 01.08.2013. |

