Область техники, к которой относится изобретение
Изобретение относится к области органической химии, а именно к промышленному способу получения соединения формулы Ia, которое может быть использовано в качестве прямого ингибитора фактора Ха для контроля свертываемости крови у человека и других млекопитающих, а также для лечения заболеваний, связанных со свертываемостью крови.
Уровень техники
Контроль свертываемости крови может осуществляться с помощью антикоагулянтов, которые являются прямыми и косвенными ингибиторами тромбина. Протромбиназный комплекс, включающий в себя белок (фактор) Ха, преобразует профермент протромбин в активированный тромбин. Фактор Ха относится к семейству сериновых протеаз и образуется из белка (фактора) X. В отличие от тромбина, который действует на множество белков-субстратов и специфических рецепторов, белок Ха, по всей видимости, действует только на единственный субстрат, а именно на протромбин. Поскольку одна молекула белка Ха способна генерировать до 138 молекул тромбина, прямые ингибиторы белка Ха, как ингибиторы образования тромбина, могут быть использованы для эффективного способа контроля свертываемости крови. Поэтому очевидно, что вещества, которые селективно ингибируют белок Ха, могут быть использованы для диагностики свертываемости крови in vitro, а также в качестве лекарств при заболеваниях, связанных со свертываемостью крови.
Известен ряд ингибиторов фактора Ха, относящихся к азотсодержащим гетероциклам, содержащим замещенные амидиновые группы с двумя функциональными группами, которые способны связываться с фактором Ха в двух активных сайтах. Например, в WO 98/28269 описан пиразол с концевыми C(=NH)-NH2 группами; в WO 97/21437 описаны бензимидазольные соединения, замещенные группами, которые соединены с нафталиновой группой напрямую либо через цепь алкилов и -С(=O) или -S(=O)2 группы; в WO 99/10316 описаны соединения, имеющие 4-фенил-н-алкиламидинопиперидин, соединенный с 3-амидинофениловой группой через карбоксиалкиленаминогруппу; а в ЕР 798295 раскрыты соединения, имеющие в своем составе 4-фенокси-н-алкиламидинопиперидиновую группу, соединенную с амидиновой группой через замещенные или незамещенные сульфонамиды.
В патенте ЕА 015918 описано соединение формулы I

которое отличается высокой активностью и продолжительностью действия по отношению к фактору Ха.
Предложенный в патенте ЕА 015918 способ получения соединения формулы I включает следующие этапы:
1) взаимодействие 5-метил-2-нитробензойной кислоты с SOCl2 и 2-амино-5-хлорпиридином с получением N-(5-хлорпиридин-2-ил)-5-метил-2-нитробензамида;
2) восстановление N-(5-хлорпиридин-2-ил)-5-метил-2-нитробензамида хлоридом олова с получением 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида;
3) взаимодействие 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида с трифторацетатом 4-метиламинобензойной кислоты и агентом амидного сочетания EDCI с получением N-(5-хлорпиридин-2-ил)-2-[(4-метил(трифторацетил)аминофенилкарбонил)амино]-5-метилбензамида;
4) снятие трифторацетильной защитной группы путем щелочного гидролиза с получением N-(5-хлорпиридин-2-ил)-2-[(4-метиламинофенилкарбонил)амино]-5-метилбензамида;
5) синтез амидина из N-(5-хлорпиридин-2-ил)-2-[(4-метиламинофенилкарбрнил)амино]-5-метилбензамида путем взаимодействия с ацетонитрилом и пропускания газообразного HCl с получением N-(5-хлорпиридин-2-ил)-2-[(4-этанимидоилметиламинофенилкарбонил)амино]-5-метилбензамида (соединения формулы I).
К недостаткам способа, раскрытого в патенте ЕА 015918, относятся большое количество стадий, необходимость в использовании хлорида олова и соляной кислоты для восстановления нитрогруппы, в введении и снятии защитных групп по атому азота на промежуточных стадиях, а также необходимость выделения и очистки продуктов, получаемых на каждой стадии.
Раскрытие изобретения
Задачей данного изобретения является разработка нового, более простого, экономичного и пригодного для промышленного использования способа получения наиболее фармацевтически приемлемой соли соединения формулы I - гидрохлорида формулы Ia - с чистотой более 99% и высоким выходом. При осуществлении способа по изобретению образуется значительно меньшее количество кислых отходов и отсутствуют отходы, содержащие металлы.
Данное изобретение относится к способу получения гидрохлорида N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(N-метилацетимидамидо)бензамидо)бензамида формулы Ia
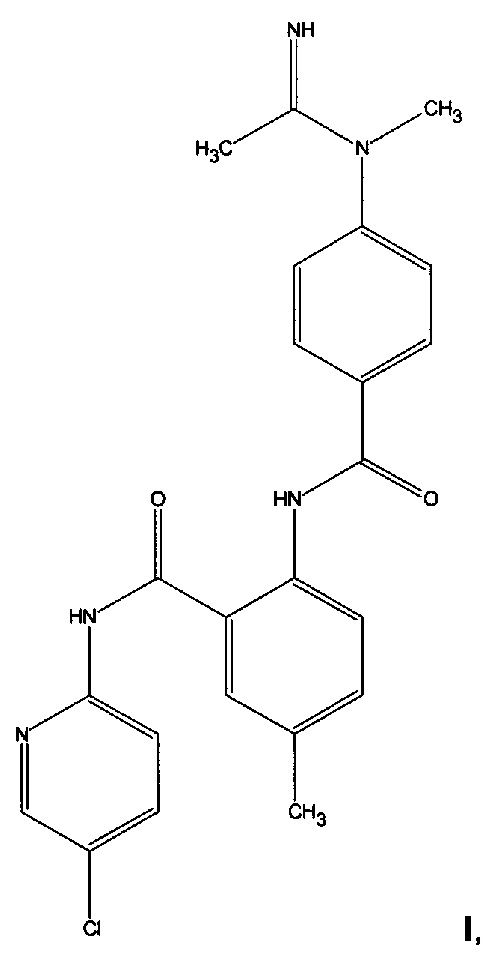
при котором осуществляют:
а) взаимодействие 5-метилантраниловой кислоты и 2-амино-5-хлорпиридина с получением 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида формулы III;
б) взаимодействие 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида формулы III с 4-[(трет-бутоксикарбонил)-N-метил]аминобензойной кислотой формулы II;
в) удаление трет-бутоксикарбонил-защитной группы у продукта реакции стадии (б) с получением N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо) бензамида формулы IV;
г) взаимодействие N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо) бензамида формулы IV с хлорангидридом имидоуксусной кислоты с получением соединения формулы Ia.
В одном из воплощений изобретения удаление трет-бутоксикарбонил-защитной группы на стадии (в) осуществляют путем взаимодействия продукта реакции стадии (б) с трифторуксусной кислотой.
В одном из воплощений изобретения на стадии (в) дополнительно проводят нейтрализацию трифторуксусной кислоты. Нейтрализация может быть осуществлена с использованием любой щелочи, но, предпочтительно, с использованием гидроксида калия или гидроксида натрия, наиболее предпочтительно, гидроксида натрия.
В одном из воплощений изобретения на стадии (г) способа применяют хлорангидрид имидоуксусной кислоты, который образуется in situ из ацетонитрила, хлористого тионила и воды.
Предложенный способ получения соединения формулы 1а отличается от способа, раскрытого в патенте ЕА 015918, более простой синтетической последовательностью, отсутствием необходимости выделять и очищать промежуточные продукты, отсутствием отходов, содержащих металлы, меньшим количеством кислых отходов и более высокой степенью чистоты целевого соединения, а также приводит к наиболее фармацевтически приемлемой соли соединения формулы I, а именно к гидрохлориду, получение которого в патенте ЕА 015918 не раскрыто.
В качестве дополнительного преимущества способа по изобретению может также рассматриваться возможность использования генерируемого in situ хлороводорода, необходимого для получения хлорангидрида имидоуксусной кислоты при осуществлении стадии (г).
Осуществление изобретения
Более подробно с учетом вышеописанных воплощений изобретения и приведенных ниже примеров реакций предложенный способ может быть представлен как включающий следующие этапы:
Реакция 1 - взаимодействие 4-(метиламино)бензойной кислоты и ди-трет-бутилдикарбоната с получением 4-[(трет-бутоксикарбонил)-N-метил]аминобензойной кислоты (соединения формулы II) согласно WO 2004/069792;
Реакция 2 - взаимодействие 5-метилантраниловой кислоты с SOCl2 с получением промежуточного хлорангидрида, который без выделения и очистки вводят в реакцию с 2-амино-5-хлорпиридином с получением 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида (соединения формулы III);
Реакция 3 - взаимодействие соединения формулы II с SOCl2 и соединением формулы III с получением промежуточного продукта, который затем вводят в реакцию с трифторуксусной кислотой, нейтрализуемой щелочью, с получением N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо)бензамида (соединения формулы IV);
Реакция 4 - взаимодействие соединения формулы IV с хлорангидридом имидоуксусной кислоты, образующимся in situ из ацетонитрила, хлористого тионила и воды, с получением соединения формулы Ia.
Общая схема синтеза может быть представлена следующим образом:
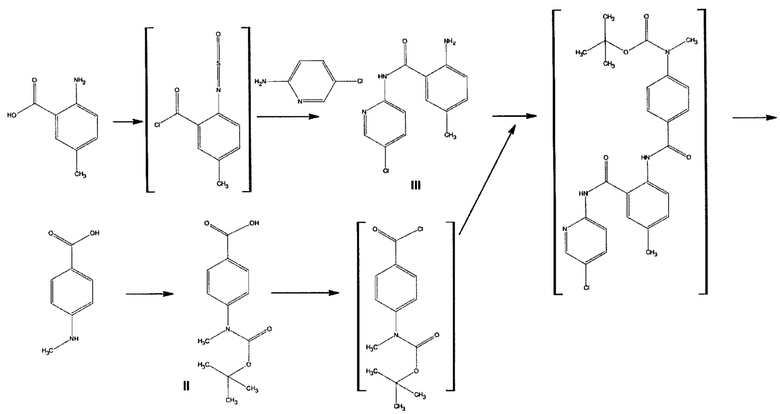

Реакция 1. Получение 4-[(трет-бутоксикарбонил)-N-метил]аминобензойной кислоты (соединения формулы II) раскрыто в WO 2004/069792:

5 кг (33 моль) 4-(метиламино)бензойной кислоты растворяли в 35 л (35 моль) 1Н NaOH (водн.). К полученному раствору добавляли 15 л 1,4-диоксана, 8 кг (36,7 моль) ди-трет-бутилдикарбоната и перемешивали реакционную смесь при комнатной температуре 6 ч. Добавляли дополнительно 2,7 кг (12,4 моль) ди-трет-бутилдикарбоната, после чего продолжали перемешивание еще 18 ч. Добавляли последнюю порцию 1,5 кг (6,8 моль) ди-трет-бутилдикарбоната и перемешивали еще 24 ч. Разбавляли смесь 100 л воды, охлаждали до +5°С и добавляли 1Н HCl (водн.) до рН 5-6. Выпавший осадок фильтровали, промывали водой и сушили на воздухе с получением 6,8 кг продукта белого цвета.
Выход: 82%
Чистота: более 98% (ЯМР)
1Н NMR (CDCl3): 1,51 (s, 9Н), 3,34 (s, 3Н), 7,40 (d, 2Н), 8,09 (d, 2Н) и 11,5 (br s, 1Н).
Реакция 2. Получение 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида (соединения формулы III)

К смеси 30 л толуола и 6 л хлористого тионила порциями добавляли 3,0 кг (20 моль) 5-метилантраниловой кислоты и при перемешивании нагревали суспензию до кипения. После полного растворения осадка продолжали нагрев светло-желтого раствора в течение еще 2 ч. Реакционную смесь охлаждали, упаривали на роторном испарителе, добавляли 10 л толуола и снова упаривали с получением маслянистого остатка промежуточного хлорангидрида. Полупродукт использовали далее без дополнительной очистки.
Полученный хлорангидрид растворяли в 25 л сухого тетрагидрофурана, раствор охлаждали до +10°С и добавляли к нему по каплям смесь 2,31 кг (18 моль) 2-амино-5-хлорпиридина и 1,42 кг (18 моль) пиридина в 20 л тетрагидрофурана. После прекращения охлаждения перемешивали реакционную смесь 2 ч при комнатной температуре. Затем добавляли 70 л холодной воды (+5°С), перемешивали 10 мин, добавляли 5 кг (62,5 моль) 50% NaOH (водн.), интенсивно перемешивали 30 мин, добавляли 1,5 кг NaHC03, перемешивали до растворения и упаривали из реакционной смеси тетрагидрофуран в роторном испарителе. Получившуюся водную суспензию фильтровали, осадок последовательно промывали 20 л воды, 10 л изопропанола и сушили в вакууме с получением 2,5 кг продукта светло-желтого цвета.
Выход: 48%
Чистота: более 98% (ЯМР)
1Н NMR (DMSO-d6): 2,19 (s, 3Н); 6,24 (s, 2Н); 6,70 (d, 1Н); 7,05 (d, 1Н); 7,59 (s, 1Н); 7,91 (d, 1Н); 8,15 (d, 1Н); 8,40 (s, 1Н); 10,52 (s, 1Н).
Реакция 3. Получение N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо)бензамида (соединения формулы IV)

2,76 кг (11 моль) 4-[(трет-бутоксикарбонил)-N-метил]аминобензойной кислоты (соединения формулы II) растворяли в 30 л сухого тетрагидрофурана, добавляли 0,91 кг (11,5 моль) пиридина и к полученному раствору добавляли по каплям за 10 мин 1,31 кг (11 моль) хлористого тионила при комнатной температуре. Перемешивали 30 мин. В другом сосуде смешивали 2,6 кг (10 моль) 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида формулы III и 0,91 кг (11,5 моль) пиридина в 20 л сухого тетрагидрофурана. К этому раствору добавляли по порциям полученную ранее суспензию, поддерживая температуру не выше 25°С, и перемешивали реакционную смесь 3 ч при комнатной температуре. К смеси добавляли 50 л 13% раствора хлорида натрия (водн.), 50 л этилацетата и отделяли водный слой. Органическую фазу промывали 40 л 20% раствора карбоната натрия (водн.), 40 л 26% раствора хлорида натрия (водн.), сушили над безводным сульфатом натрия и упаривали на роторном испарителе. Маслянистый остаток сушили в вакууме с получением 5,0 кг твердого полупродукта. Полупродукт использовали далее без дополнительной очистки.
5,0 кг полученного полупродукта растворяли в 10 л дихлорметана, при перемешивании добавляли 6 л трифторуксусной кислоты и перемешивали смесь 2 ч при комнатной температуре. Реакционную смесь вливали в заранее приготовленный раствор из 6 кг гидроксида натрия и 60 л воды при интенсивном перемешивании и охлаждении (<30°С). К образовавшейся суспензии добавляли 10 л петролейного эфира и фильтровали для отделения осадка от органической и водной фаз. Осадок промывали 50 л воды, 15 л этанола и сушили в вакууме с получением 3,6 кг твердого светло-желтого продукта.
Выход: 81%
Чистота: более 98% (ЯМР)
1Н NMR (DMSO-d6): 2,34 (s, 3Н); 2,74 (s, 3Н); 6,41 (br s, 1Н); 6,62 (d, 2Н); 7,39 (d, 1Н); 7,70 (d, 2Н); 7,80 (s, 1Н); 7,98 (d, 1Н); 8,17 (d, 1Н); 8,35 d (1Н); (8,46 s, 1Н), 11,05 (s, 1Н); 11,20 (s, 1Н).
Реакция 4. Получение гидрохлорида N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(N-метилацетимидамидо)бензамидо)бензамида (соединения формулы Ia)

В 20 л сухого ацетонитрила растворяли 4,160 кг (35 моль) хлористого тионила, раствор охлаждали до +10°С и добавляли к нему по каплям 0,630 кг (35 моль) дистиллированной воды. Смесь перемешивали при +10°С 30 мин и добавляли 1 кг (2,53 моль) N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо)бензамида (соединения формулы IV). После прекращения охлаждения реакционную смесь перемешивали в закрытом сосуде около 30 ч до исчезновения исходного (по данным тонкослойной хроматографии). Реакционную смесь упаривали на роторном испарителе при 35°С. Темный остаток (~3 кг) растворяли в 5 л изопропанола, добавляли 200 г активированного угля, нагревали до кипения, затем уголь отфильтровывали и промывали 1 л горячего (+70°С) изопропанола. После охлаждения полученный осадок отфильтровывали и сушили в вакууме с получением 1,05 кг неочищенного продукта. Полученный продукт суспендировали при перемешивании в изопропаноле и нагревали до кипения, затем охлаждали, фильтровали и сушили в вакууме с получением 0,91 кг целевого соединения.
Выход: 78%
Чистота: 99,7% (ВЭЖХ, градиентный режим 0,1% трифторуксусная кислота - 0,1% трифторуксусная кислота/ ацетонитрил (95/5), колонка С18 (3u, 2.0*150))
1Н NMR (DMSO-d6): 2,12 (br s, 3Н); 2,37 (s, 3Н); 3,43 (s, 3Н); 7,43(d, 1Н); 7,65 (d, 2Н); 7,77 (s, 1Н); 7,96 (d, 1Н); 8,05 (d, 1Н); 8,12 (d, 2Н); (8,44 s, 1Н), 10,0 (br s, 1Н); 11,07 (brs, 1Н); 11,29 (brs, 1Н).
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗИНОНА И ИХ ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЛЕГОЧНЫХ ЗАБОЛЕВАНИЙ | 2008 |
|
RU2479580C2 |
| (3-ЦИКЛОАЛКИЛ-2,3,4,5-ТЕТРАГИДРО-1Н-БЕНЗО[d]АЗЕПИН-7-ИЛОКСИ)ПРОИЗВОДНЫЕ, ИХ ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ Н3 РЕЦЕПТОРОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ПОЛУЧЕНИЯ | 2003 |
|
RU2388752C2 |
| ПРОИЗВОДНЫЕ АМИДА, СПОСОБ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ), ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ | 2000 |
|
RU2260007C2 |
| СОЕДИНЕНИЯ ДЕЙТЕРИРОВАННОГО ДЕФАКТИНИБА И ИХ ПРИМЕНЕНИЕ | 2019 |
|
RU2761825C1 |
| Производные N-пиперидин-3-илбензамида для лечения сердечно-сосудистых заболеваний | 2014 |
|
RU2618628C1 |
| ТРИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ, СПОСОБ ПОЛУЧЕНИЯ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБ ИНГИБИРОВАНИЯ ВАЗОПРЕССИНА | 1994 |
|
RU2149160C1 |
| ПРОИЗВОДНОЕ БЕНЗАМИДА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ ДЛЯ ПРОФИЛАКТИКИ ИЛИ ЛЕЧЕНИЯ РАКА, СОДЕРЖАЩАЯ ЕГО В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2022 |
|
RU2841260C2 |
| ЗАМЕЩЕННЫЕ БЕНЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2658919C2 |
| СОЕДИНЕНИЯ, ОБЛАДАЮЩИЕ АКТИВНОСТЬЮ АНТАГОНИСТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ И АГОНИСТОВ БЕТА-2-АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2013 |
|
RU2661877C2 |
| БИАРИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ БЕЛОК-БЕЛКОВОГО ВЗАИМОДЕЙСТВИЯ YAP/TAZ-TEAD | 2021 |
|
RU2830596C1 |
Изобретение относится к промышленному способу получения соединения формулы Iа
 ,
,
который включает стадии а) взаимодействия 5-метилантраниловой кислоты и 2-амино-5-хлорпиридина с получением 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида; б) взаимодействия 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида с 4-[(трет-бутоксикарбонил)-N-метил]аминобензойной кислотой; в) удаления трет-бутоксикарбонил-защитной группы у продукта реакции стадии (б) с получением N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо)бензамида; г) взаимодействия N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо)бензамида с хлорангидридом имидоуксусной кислоты с получением соединения формулы Iа. Полученные соединения могут быть использованы как эффективный и селективный ингибитор фактора Ха для контроля свертываемости крови у человека и других млекопитающих, а также для лечения заболеваний, связанных со свертываемостью крови. 5 з.п. ф-лы.
1. Способ получения гидрохлорида N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(N-метилацетимидамидо)бензамидо)бензамида формулы Iа
 ,
,
при котором осуществляют:
а) взаимодействие 5-метилантраниловой кислоты и 2-амино-5-хлорпиридина с получением 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида;
б) взаимодействие 2-амино-N-(5-хлорпиридин-2-ил)-5-метилбензамида с 4-[(трет-бутоксикарбонил)-N-метил]аминобензойной кислотой;
в) удаление трет-бутоксикарбонил-защитной группы у продукта реакции стадии (б) с получением N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо)бензамида;
г) взаимодействие N-(5-хлорпиридин-2-ил)-5-метил-2-(4-(метиламино)бензамидо)бензамида с хлорангидридом имидоуксусной кислоты с получением соединения формулы Iа.
2. Способ по п. 1, где удаление трет-бутоксикарбонил-защитной группы на стадии (в) осуществляют путем взаимодействия продукта реакции стадии (б) с трифторуксусной кислотой.
3. Способ по п. 2, где на стадии (в) дополнительно проводят нейтрализацию трифторуксусной кислоты.
4. Способ по п. 3, где нейтрализацию проводят с использованием гидроксида калия или гидроксида натрия.
5. Способ по п. 4, где нейтрализацию проводят с использованием гидроксида натрия.
6. Способ по п. 1, где на стадии (г) применяют хлорангидрид имидоуксусной кислоты, который образуется in situ из ацетонитрила, хлористого тионила и воды.
| Первичные электрические часы | 1929 |
|
SU15918A1 |
| WO 2001064643 A2, 07.09.2001 | |||
| WO 2017217895 A1, 21.12.2017. | |||

