Область техники
Настоящее изобретение относится к соединениям дейтерированного дефактиниба и их применению.
Уровень техники
Дефактиниб (VS-6063), разработанный компанией Verastem Oncology, представляет собой селективный и перорально эффективный ингибитор FAK, имеет структуру 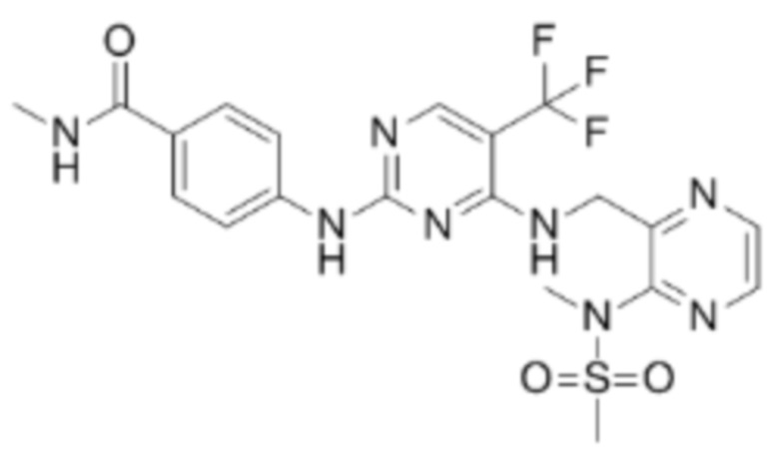 и в настоящее время проходит клинические испытания.
и в настоящее время проходит клинические испытания.
Дейтерированные лекарственные средства характеризуются заменой части атомов водорода в молекулах лекарственного средства на дейтерий. Поскольку форма и объем дейтерия в молекуле лекарственного средства очень близки к водороду, дейтерированное лекарственное средство сохраняет биологическую активность и селективность in vitro исходного лекарственного средства. Поскольку связь C-D является более стабильной по сравнению со связью C-H, связь C-D с меньшей вероятностью разрушается во время метаболической реакции дейтерированных лекарственных средств, и период полувыведения может быть продлен.
Тем не менее, из-за сложных метаболических процессов в биологической системе фармакокинетические свойства лекарственных средств в организмах подвергаются воздействию многих факторов и также демонстрируют соответствующую сложность. По сравнению с соответствующими недейтерированными лекарственными средствами изменения фармакокинетических свойств дейтерированных лекарственных средств являются непредсказуемыми и в значительной степени являются случайными. Дейтерирование определенных участков может не только не продлевать период полувыведения, но может сокращать его (Scott L. Harbeson, Roger D. Tung. Deuterium in Drug Discovery and Development, P405-406) и ухудшать фармакокинетические свойства; с другой стороны, также крайне сложно заменить водород в определенных положениях молекулы лекарственного средства на дейтерий. Следовательно, положение в молекуле лекарственного средства, которое подходит для дейтерирования, является неочевидным, а эффект дейтерирования является непредсказуемым.
Таким образом, молекулу дефактиниба дейтерировали для получения класса дейтерированных лекарственных средств с улучшенными фармакокинетическими свойствами, уменьшенной дозировкой и пониженной токсичностью и побочными эффектами метаболитов, что является важным для получения новых, более эффективных и безопасных лекарственных средств.
Описание изобретения
Задача настоящего изобретения заключается в обеспечении нового эффективного и безопасного лекарственного средства для лечения рака, обладающего улучшенными метаболической стабильностью и фармакокинетическими свойствами.
В первом аспекте, в настоящем изобретении предложено соединение формулы (I) или его оптический изомер, фармацевтически приемлемая соль, гидрат или сольват:
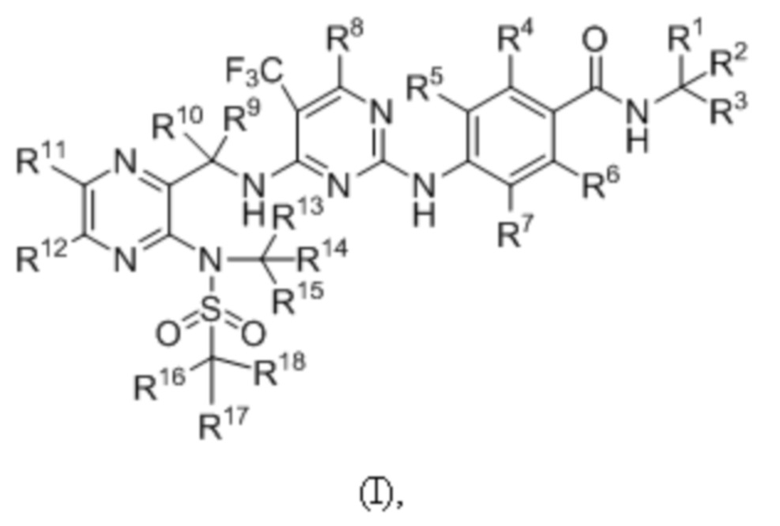
где каждый из R1~R18 независимо выбран из водорода и дейтерия, и не все из них представляют собой водород.
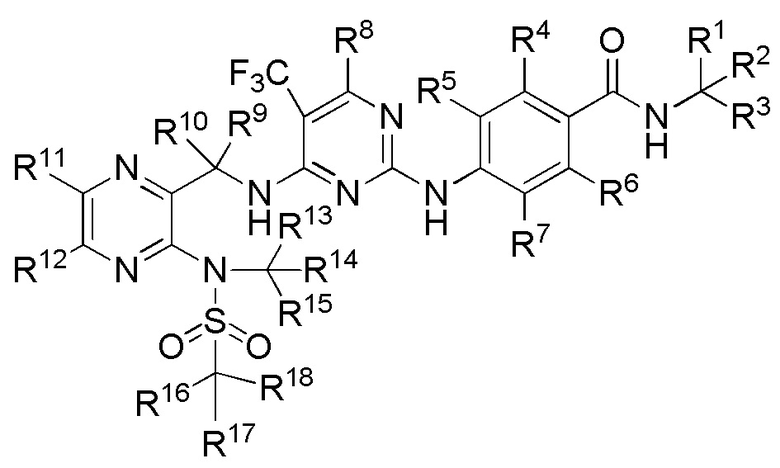
В одном из вариантов реализации настоящего изобретения указанное соединение имеет структуру формулы (II):
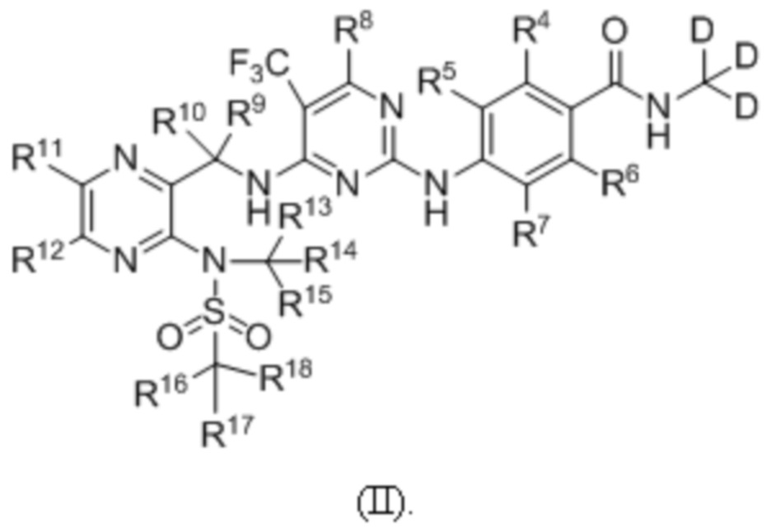
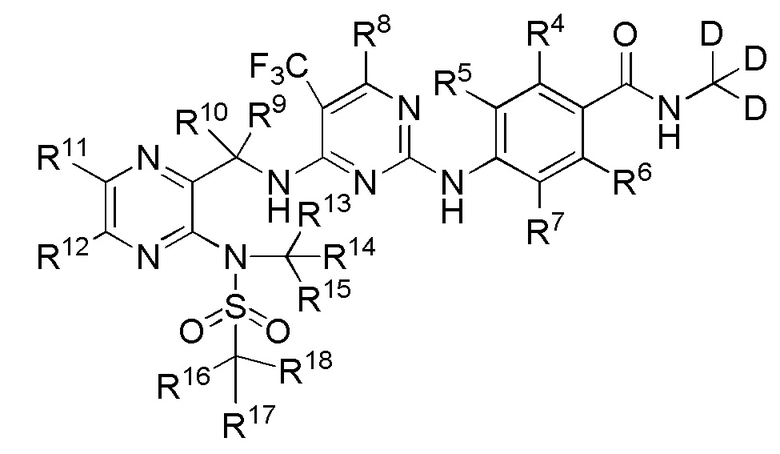
В одном из вариантов реализации настоящего изобретения указанное соединение имеет структуру формулы (III):
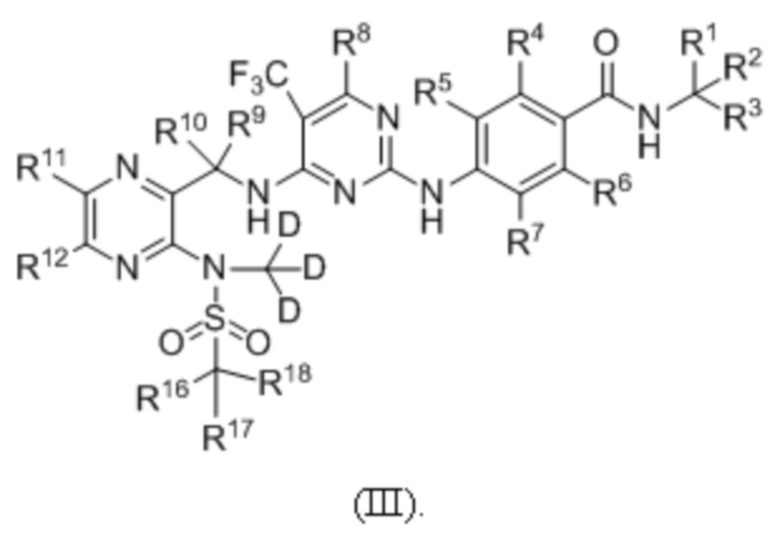
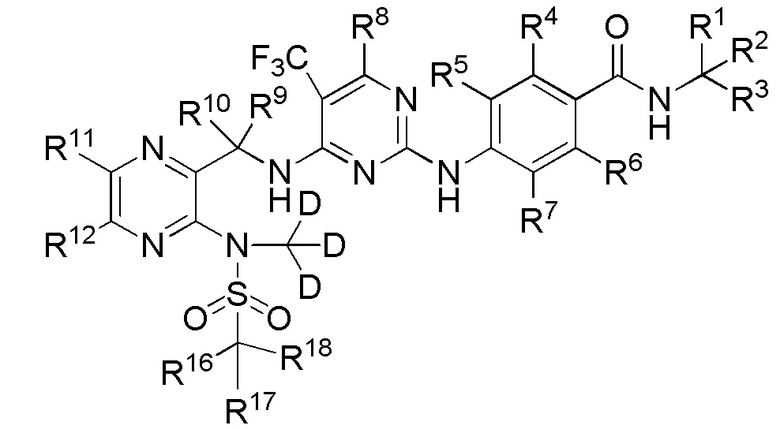
В одном из вариантов реализации настоящего изобретения указанное соединение имеет структуру формулы (IV):
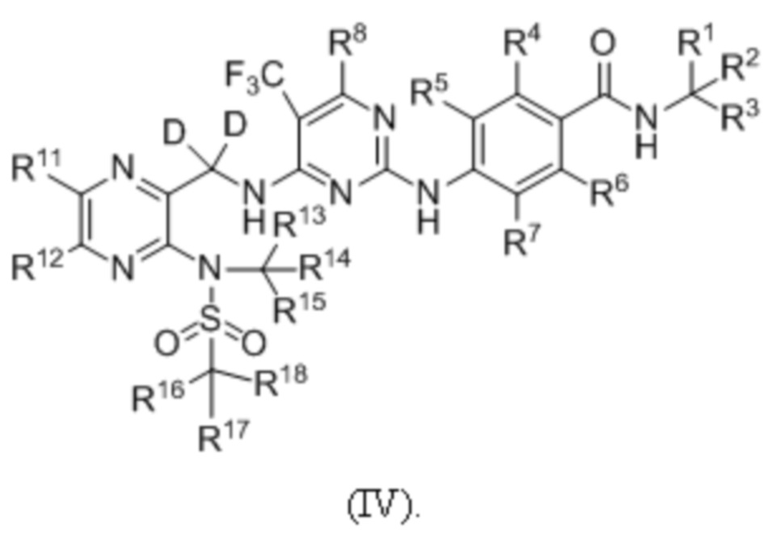
В одном из вариантов реализации настоящего изобретения указанное соединение имеет структуру формулы (V):
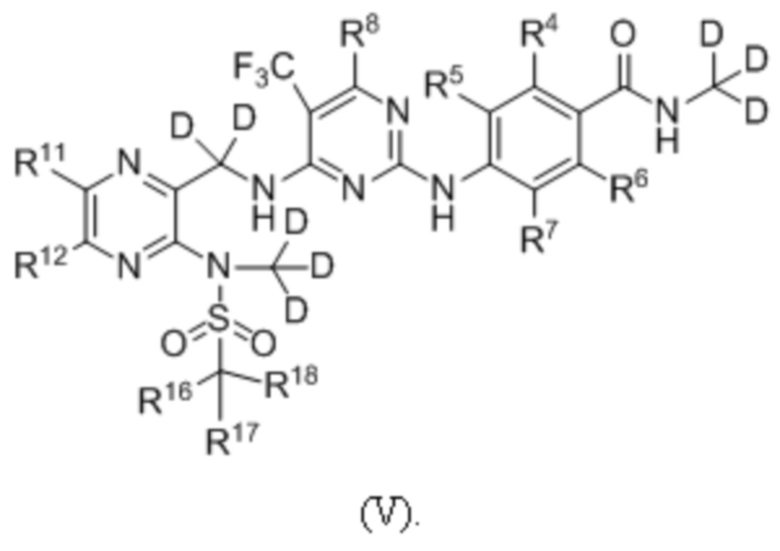
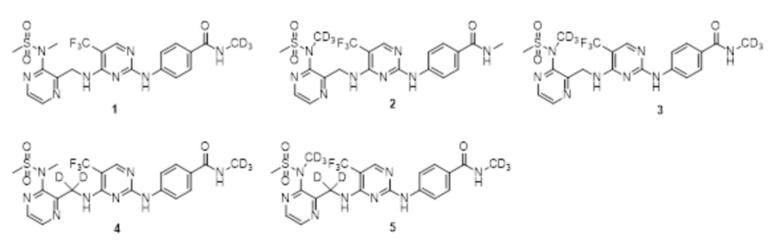
В одном из вариантов реализации настоящего изобретения указанное соединение представляет собой одно из следующих соединений:
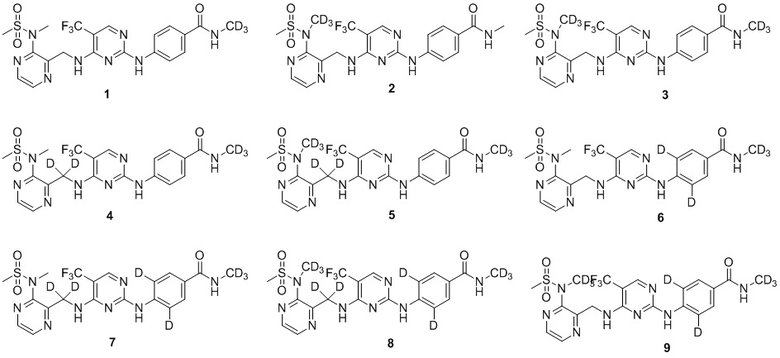
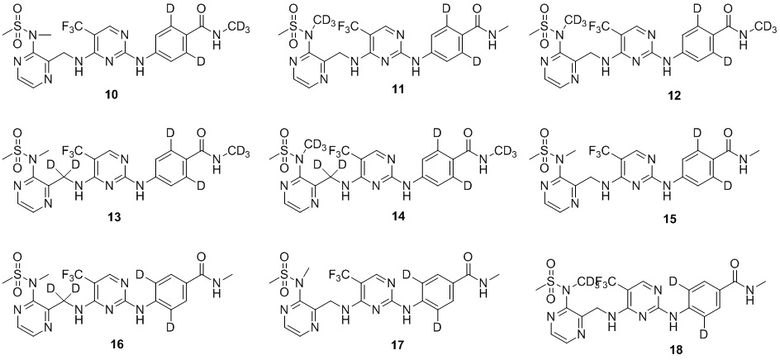 .
.
В одном из вариантов реализации настоящего изобретения указанная фармацевтически приемлемая соль представляет собой фосфат, d-камфорсульфонат, гидрохлорид, гидробромид, гидрофторид, сульфат, нитрат, формиат, ацетат, пропионат, оксалат, малонат, сукцинат, фумарат, малеат, лактат, малат, тартрат, цитрат, пикрат, метансульфонат, безилат, бензолсульфонат, аспартат или глутамат соединения и предпочтительно гидрохлорид соединения.
В настоящем изобретении также предложено применение указанного выше соединения или его оптического изомера, фармацевтически приемлемой соли, гидрата или сольвата для получения лекарственных средств для лечения рака.
В одном из вариантов реализации настоящего изобретения рак выбран из рака поджелудочной железы, солидной опухоли, немелкоклеточного рака легких, мезотелиомы и рака яичников.
В настоящем изобретении также предложено применение указанного выше соединения или его оптического изомера, фармацевтически приемлемой соли, гидрата или сольвата для получения ингибиторов FAK.
В настоящем изобретении также предложено лекарственное средство для лечения рака, которое представляет собой препарат, полученный с применением соединения, указанного выше, или его оптического изомера, фармацевтически приемлемой соли, гидрата или сольвата в качестве активных ингредиентов с добавлением фармацевтически приемлемых вспомогательных веществ.
Эксперименты доказали, что соединения, предложенные в настоящем изобретении, и их соли, гидраты или сольваты можно применять в качестве ингибиторов FAK для получения противораковых лекарственных средств, и по сравнению с контрольным недейтерированным соединением дефактиниба метаболическая стабильность и фармакокинетические свойства соединения согласно настоящему изобретению значительно улучшены, и перспективы применения превосходны.
Применяемый в настоящем документе термин “дейтерированный” обозначает замену одного или более водородов в соединении или группе на дейтерий. Дейтерирование может представлять собой моно-, ди-, поли- или полное замещение. В другом предпочтительном примере содержание изотопа дейтерия в положении замещения дейтерием превышает естественное содержание изотопа дейтерия (0,015%), предпочтительно составляет более 50%, предпочтительно более 75%, предпочтительно более 95%, предпочтительно более 97%, предпочтительно более 99% и предпочтительно более 99,5%.
Применяемый в настоящем документе термин “соединение согласно настоящему изобретению” обозначает соединение формулы (I). Термин также включает различные оптические изомеры, фармацевтически приемлемые соли, гидраты или сольваты соединения формулы (I).
Применяемый в настоящем документе термин “фармацевтически приемлемая соль” обозначает фармацевтически приемлемые соли, образованные соединением согласно настоящему изобретению и кислотой или основанием. Фармацевтически приемлемые соли включают неорганические соли и органические соли. Предпочтительным классом солей являются соли, образованные соединениями согласно настоящему изобретению и кислотами. Кислоты, подходящие для образования солей, включают, но не ограничиваются следующими:
фосфорную кислоту, D-камфорсульфоновую кислоту, хлористоводородную кислоту, бромистоводородную кислоту, фтористоводородную кислоту, серную кислоту, азотную кислоту, муравьиную кислоту, уксусную кислоту, пропионовую кислоту, щавелевую кислоту, малоновую кислоту, янтарную кислоту, фумаровую кислоту, малеиновую кислоту, молочную кислоту, яблочную кислоту, винную кислоту, лимонную кислоту, пикриновую кислоту, метансульфоновую кислоту, толуолсульфоновую кислоту, бензолсульфоновую кислоту, аспарагиновую кислоту или глутаминовую кислоту.
В одном из вариантов реализации настоящего изобретения кислота, образующая фармацевтически приемлемую соль, представляет собой хлористоводородная кислота.
Фармацевтически приемлемые вспомогательные вещества, описанные в настоящем документе, обладают конкретными физиологическими активностями, но добавление ингредиентов не изменяет доминирующего положения указанной выше фармацевтической композиции в ходе лечения заболевания, а только выполняет вспомогательные функции. Указанные вспомогательные функции представляют собой только применение известной активности ингредиентов, которые обычно используют в качестве вспомогательного лечения в области медицины. Если указанные выше вспомогательные компоненты применяют в сочетании с фармацевтической композицией согласно настоящему изобретению, они все равно должны попадать под объем правовой охраны настоящего изобретения.
Очевидно, что на основе приведенного выше описания настоящего изобретения можно дополнительно проводить различные другие модификации, доработки или изменения в соответствии с общими техническими знаниями и обычными средствами в данной области техники без отступления от указанных выше основных технических аспектов.
Приведенное выше описание настоящего изобретения дополнительно проиллюстрировано следующими конкретными примерами указанных вариантов реализации. Но не следует считать, что объем указанного выше объекта настоящего изобретения ограничивается следующими примерами. Все способы, реализованные на основе приведенного выше содержания настоящего изобретения, входят в объем настоящего изобретения.
Примеры
Все исходные материалы и оборудование, применяемые в настоящем изобретении, являются хорошо известными продуктами и могут быть куплены в виде коммерчески доступных продуктов.
Ниже соединение согласно настоящему изобретению получали при помощи способа, указанного в способе синтеза 1 или 2:
Способ синтеза 1:
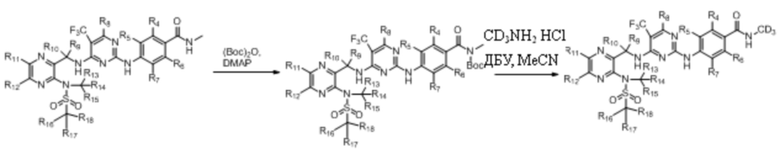
Способ синтеза 2:
Пример 1. Синтез N-(тридейтерометил)-4-((4-(((3-(N-метилметилсульфонил)пиразин-2-ил)метил) амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 1)
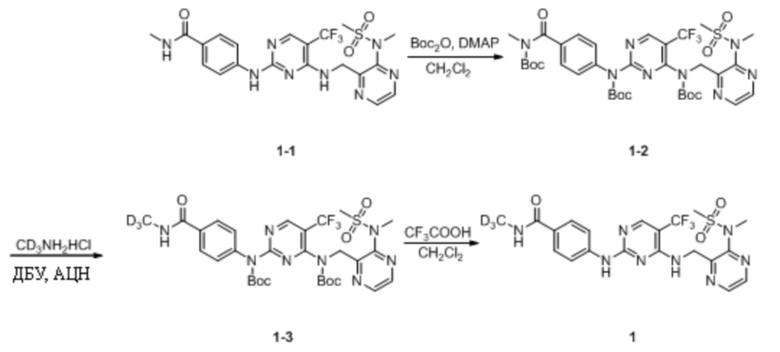
(1) Синтез трет-бутил-(4-((трет-бутоксикарбонил)(4-((трет-бутоксикарбонил)((3-(N- метилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензоил)(метил)карбамата
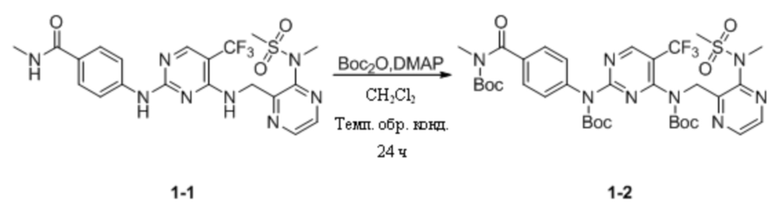
N-Метил-4-((4-(((3-(N-метилметилсульфонил)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамид (200,0 мг, 0,39 ммоль; а именно соединение 1-1, приобретенное в Chengdu Henghui Chemical Pharmaceutical Technology Co., Ltd.) и DMAP (1,3 г, 10,57 ммоль) растворяли в дихлорметане (10 мл), а затем по каплям добавляли (Boc)2O (1,7 г, 7,83 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 24 ч на масляной бане при 45°C. На следующий день реакционную смесь охлаждали до комнатной температуры, добавляли дихлорметан и раствор HCl (0,1 М) и экстрагировали, а затем выдерживали для разделения слоев. Органическую фазу промывали насыщенным солевым раствором, сушили с безводным сульфатом натрия, фильтровали в вакууме и выпаривали на роторном испарителе для удаления растворителя. Неочищенный продукт отделяли путем колоночной хроматографии с получением трет-бутил-(4-((трет-бутоксикарбонил)(4-((трет-бутоксикарбонил)((3-(N-метилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензоил)(метил)карбамата (136,0 мг, выход 42,8%) в виде беловатого твердого вещества. МС (ИЭР) m/z 811,2 [M+H]+.
(2) Синтез трет-бутил-(4-((трет-бутоксикарбонил)((3-(N-метилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)(4-(метилкарбамоил)фенил)карбамата
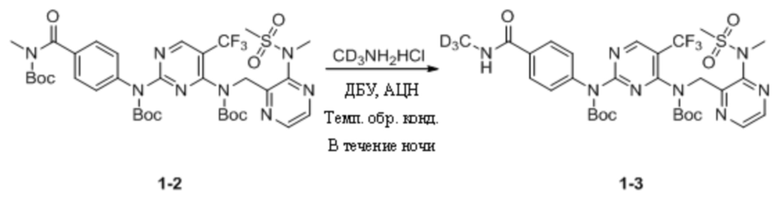
Трет-бутил-(4-((трет-бутоксикарбонил)(4-((трет-бутоксикарбонил)((3-(N-метилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензоил)(метил)карбамат (136,0 мг, 0,17 ммоль) и дейтерированный гидрохлорид метиламина (189,0 мг, 2,68 ммоль) добавляли к ацетонитрилу (5 мл) и перемешивали при комнатной температуре. Затем также добавляли ДБУ (613,0 мг, 4,03 ммоль), постепенно растворяли, и раствор становился прозрачным. Затем реакционную смесь кипятили с обратным холодильником в течение ночи на масляной бане. На следующий день реакционную смесь охлаждали до комнатной температуры и выпаривали на роторном испарителе для удаления растворителя, добавляли дихлорметан и раствор HCl (0,1 М), энергично перемешивали, а затем выдерживали для разделения слоев. Органическую фазу промывали чистой водой и насыщенным солевым раствором, соответственно, сушили с безводным сульфатом натрия и выпаривали на роторном испарителе для удаления растворителя. Неочищенный продукт отделяли и очищали путем преп. ТСХ (ПЭ/ЭА = 2:1) с получением трет-бутил-(4-((трет-бутоксикарбонил)((3-(N-метилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)(4-(метилкарбамоил)фенил)карбамата (42,0 мг, выход 35,3%) в виде белого твердого вещества. МС (ИЭР) m/z 614,2 [M+H]+.
(3) Синтез N-(тридейтерометил)-4-((4-(((3-(N-метилметилсульфонил)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 1)
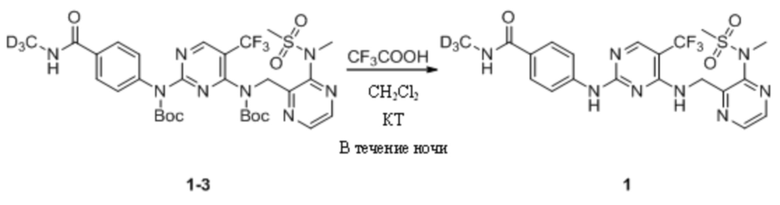
Трет-бутил-(4-((трет-бутоксикарбонил)((3-(N-метилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)(4-(метилкарбамоил)фенил)карбамат (42,0 мг, 0,06 ммоль) растворяли в дихлорметане (2 мл) и перемешивали при комнатной температуре (не смог раствориться и стать прозрачным), а затем добавляли трифторуксусную кислоту (0,1 мл). Реакционная смесь постепенно становилась прозрачной, и ее оставляли для протекания реакции в течение ночи при комнатной температуре при перемешивании. На следующий день реакционную смесь выпаривали на роторном испарителе для удаления растворителя, добавляли этилацетат и насыщенный раствор NaHCO3 и смесь энергично перемешивали, а затем выдерживали для разделения слоев. Было определено, что значение pH водной фазы составляло примерно 7~8. Органическую фазу промывали водой и насыщенным солевым раствором два раза, соответственно, сушили с безводным сульфатом натрия и выпаривали на роторном испарителе для удаления растворителя с получением N-дейтерометил-4-((4-(((3-(N-метилметилсульфонил)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-)амино)бензамида в виде беловатого твердого вещества (24,0 мг, выход 80,0%). МС (ИЭР) m/z 514,2 [M+H]+.
1H-ЯМР (400 МГц, ДМСО-d6): δ 9,86 (s, 1H), 8,69 (d, J = 2,4 Гц, 1H), 8,58 (d, J = 2,8 Гц, 1H), 8,32 (s, 1H), 8,18 (s, 1H), 7,67-7,59 (dd, J = 19,6, 8,8 Гц, 4H), 7,48-7,45 (t, J = 5,2 Гц, 1H), 5,00 (d, J = 4,8 Гц, 2H), 3,22 (s, 3H), 3,20 (s, 3H).
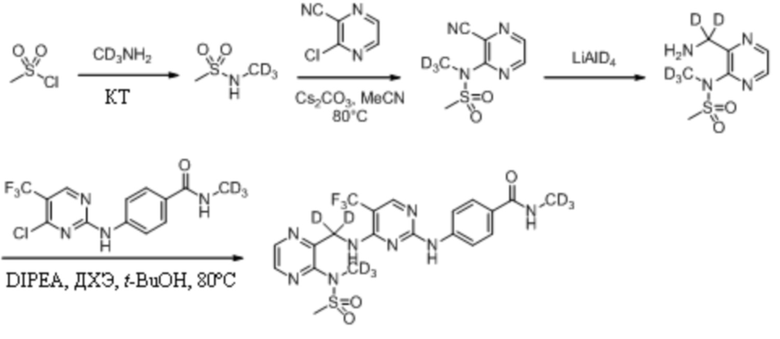
Пример 2. Синтез N-метил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 2)
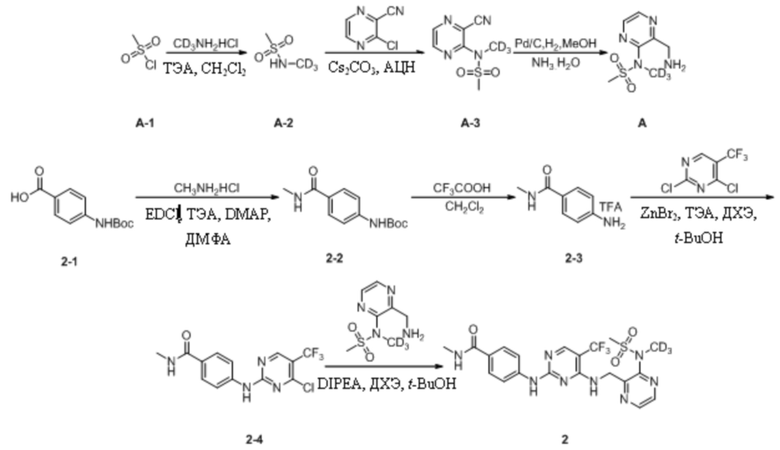
(1) Синтез N-дейтерометилметансульфонамида
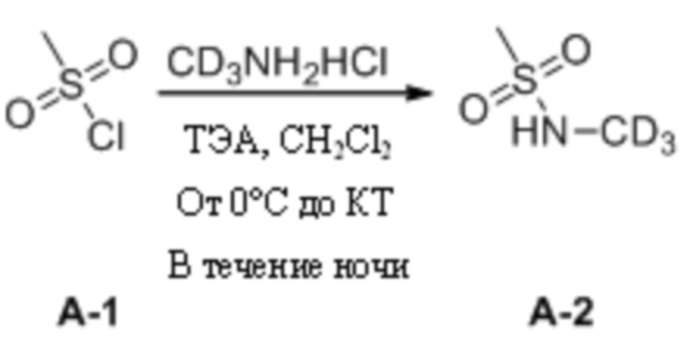
Метилсульфонилхлорид (3,0 г, 26,19 ммоль) взвешивали и помещали в 100 мл одногорлую круглодонную колбу, в которую добавляли дихлорметан (30 мл), а затем перемешивали для получения прозрачного раствора при комнатной температуре. Затем систему перемещали на баню с ледяной водой для продолжения охлаждения и перемешивания. Через 15 мин к системе медленно добавляли триэтиламин (6,1 г, 60,24 ммоль). После добавления систему дополнительно перемешивали в течение 10 мин под изоляцией. Затем к системе порциями медленно добавляли дейтерированный гидрохлорид метиламина (2,0 г, 28,81 ммоль). Затем ледяную баню убирали и систему нагревали до комнатной температуры и оставляли для протекания реакции в течение ночи при перемешивании. На следующий день, после завершения реакции, определяемого путем мониторинга, удаляли растворитель путем ротационного выпаривания, и к системе дополнительно добавляли этилацетат (30 мл). Реакционную смесь перемешивали в течение 10 мин и подвергали фильтрованию с отсасыванием, а затем осадок на фильтре промывали небольшим количеством этилацетата. Фильтрат объединяли и концентрировали при пониженном давлении с получением неочищенного продукта, который затем отделяли путем колоночной хроматографии с получением N-дейтерометилметансульфонамида в виде бесцветной прозрачной маслянистой жидкости (2,1 г, выход 71,4%).
(2) Синтез N-(3-цианопиразин-2-ил)-N-дейтерометилметансульфонамида
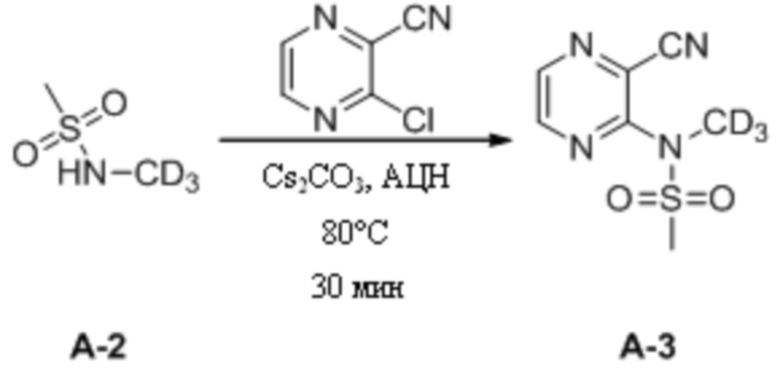
3-Хлорпиразин-2-нитрил (1,7 г, 12,48 ммоль) взвешивали и помещали в 100 мл одногорлую круглодонную колбу, в которую добавляли 40 мл ацетонитрила, и смесь перемешивали для получения прозрачного раствора при комнатной температуре. Затем к системе порциями медленно добавляли карбонат цезия (8,1 г, 24,96 ммоль). После добавления к системе по каплям добавляли раствор N-дейтерометилметансульфонамида (2,1 г, 18,72 ммоль) в ацетонитриле (10 мл). Затем систему переносили на масляную баню при 80°C для кипячения с обратным холодильником и протекания реакции при перемешивании. Через 30 мин при помощи ТСХ определяли полное израсходование исходных материалов. Масляную баню убирали и систему оставляли охлаждаться до комнатной температуры, а затем проводили фильтрование с отсасыванием. Осадок на фильтре несколько раз промывали ацетонитрилом (100 мл). Фильтрат объединяли и растворитель удаляли путем ротационного выпаривания с получением неочищенного продукта, который затем отделяли путем колоночной хроматографии с получением N-(3-цианопиразин-2-ил)-N-дейтерометилметансульфонамида в виде светло-коричневой маслянистой жидкости (1,1 г, выход 42,3%). МС (ИЭР) m/z 233,1 [M+H2O]+.
1H-ЯМР (400 МГц, CDCl3) δ 8,65-8,63 (dd, J = 6,0, 2,4 Гц, 2H), 3,26 (s, 3H).
(3) Синтез N-(3-(аминометил)пиразин-2-ил)-N-дейтерометилметансульфонамида
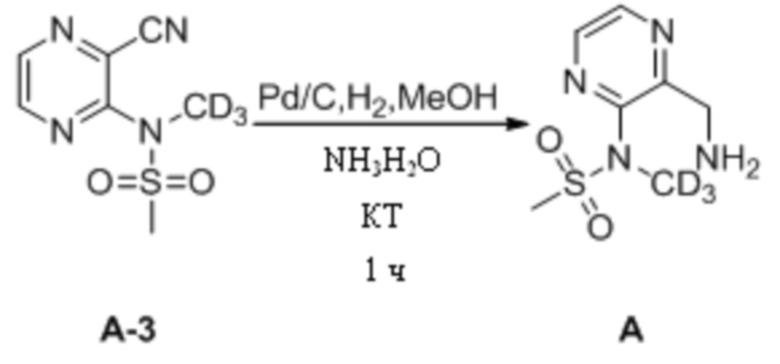
N-(3-Цианопиразин-2-ил)-N-дейтерометилметансульфонамид (42 мг, 0,20 ммоль) взвешивали и помещали в 25 мл одногорлую круглодонную колбу, в которую добавляли 4 мл метанола и 1 мл раствора аммиака и смесь перемешивали при комнатной температуре. Затем к системе добавляли влажный палладированный уголь (10,0 мг) и систему подвергали операции замены водорода, которую повторяли десять раз. Затем систему перемешивали и подвергали протеканию реакции при комнатной температуре. Через 1 ч детектировали завершение реакции. Систему подвергали фильтрованию с отсасыванием и осадок на фильтре несколько раз промывали небольшим количеством метанолом (25 мл). Фильтрат объединяли, концентрировали при пониженном давлении для удаления растворителя и остаточную воду в системе удаляли путем повторного выпаривания с метанолом с получением N-(3-(аминометил)пиразин-2-ил)-N- дейтерометилметансульфонамида в виде светлой желто-коричневой маслянистой жидкости, которую непосредственно применяли на следующей стадии без дополнительной очистки. МС (ИЭР) m/z 220,1 [M+H]+.
(4) Синтез трет-бутил-(4-(метилкарбамоил)фенил)карбамата
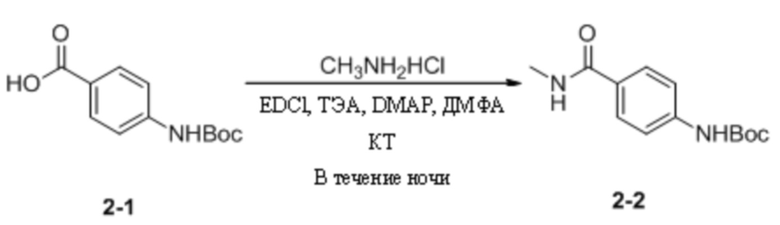
4-((Трет-бутоксикарбонил)амино)бензойную кислоту (3,0 г, 12,65 ммоль) взвешивали и помещали в 250 мл одногорлую круглодонную колбу, в которую добавляли 50 мл ДМФА, и смесь перемешивали при комнатной температуре. Затем к системе последовательно добавляли EDCI (4,8 г, 25,29 ммоль), ТЭА (4,5 г, 44,28 ммоль), гидрохлорид метиламина (1,3 г, 18,98 ммоль) и DMAP (16,0 мг, 0,13 ммоль). Затем систему перемешивали и оставляли для протекания реакции в течение ночи при комнатной температуре. На следующий день, когда детектировали израсходование исходных материалов, к системе добавляли этилацетат (70 мл) и воду (50 мл). Смесь энергично перемешивали и выдерживали для разделения слоев. Водную фазу подвергали обратной экстракции этилацетатом (20 мл * 3) и органические слои объединяли, последовательно промывали водой (20 мл * 3) и насыщенным солевым раствором (30 мл), сушили с безводным сульфатом натрия. Растворитель удаляли путем ротационного выпаривания с получением неочищенного продукта, который затем отделяли путем колоночной хроматографии с получением трет-бутил-(4-(метилкарбамоил)фенил)карбамата в виде беловатого твердого вещества (2,1 г, выход 66,5%). МС (ИЭР) m/z 251,2 [M+H]+.
(5) Синтез трифторацетата 4-амино-N-метилбензамида
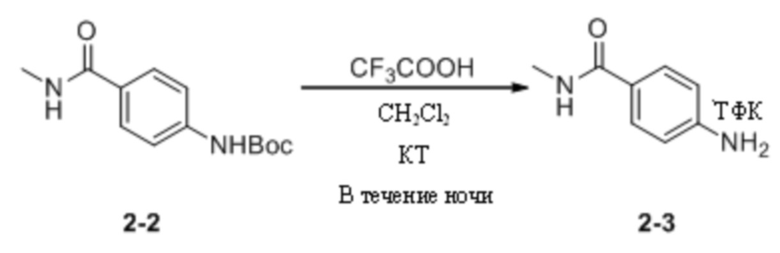
Трет-бутил-(4-(метиламиноформил)фенил)карбамат (500 мг, 2,00 ммоль) взвешивали и помещали в 50 мл одногорлую круглодонную колбу, в которую добавляли дихлорметан (10 мл), и смесь перемешивали при комнатной температуре. Затем к системе добавляли трифторуксусную кислоту (1 мл). После добавления систему перемешивали и оставляли для протекания реакции в течение ночи при комнатной температуре. На следующий день завершение реакции детектировали путем ТСХ. Реакционный раствор концентрировали для удаления растворителя и избытка трифторуксусной кислоты и остаточную трифторуксусную кислоту в системе удаляли путем многократного совместного пропаривания с дихлорметаном до полного отверждения системы с получением трифторацетата 4-амино-N-метилбензамида (510 мг) в виде беловатого твердого вещества, которое непосредственно применяли на следующей стадии без дополнительной очистки.
(6) Синтез 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-метилбензамида
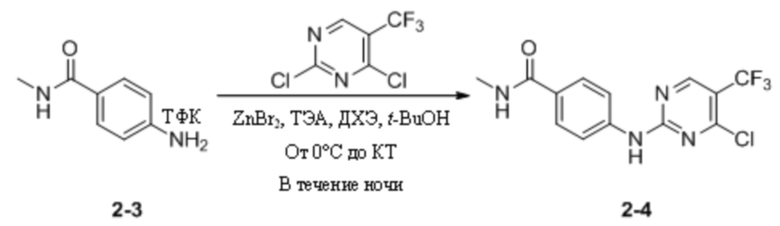
ТФК
2,4-Дихлор-5-(трифторметил)пиримидин (499 мг, 2,30 ммоль) взвешивали и помещали в 50 мл одногорлую круглодонную колбу, в которую добавляли 1,2-дихлорэтан (5 мл) и трет-бутанол (5 мл), и смесь перемешивали при комнатной температуре для получения прозрачного раствора. Затем систему переносили в баню с ледяной водой для продолжения охлаждения и перемешивания. Через 15 мин к системе добавляли бромид цинка (1,4 г, 6,00 ммоль). После добавления систему дополнительно выдерживали в бане с ледяной водой и перемешивали в течение 30 мин. Затем к системе добавляли трифторацетат 4-амино-N-метилбензамида и триэтиламин (648 мг, 6,40 ммоль), полученные на предыдущей стадии. После добавления ледяную баню убирали и систему перемешивали и оставляли для протекания реакции в течение ночи при комнатной температуре. На следующий день детектировали завершение реакции и после удаления растворителя путем ротационного выпаривания к системе добавляли этилацетат (30 мл) и воду (20 мл). Реакционную смесь энергично перемешивали и выдерживали для разделения слоев. Водный слой подвергали обратной экстракции этилацетатом (10 мл * 3) и органические фазы объединяли, последовательно промывали водой (15 мл * 3) и насыщенным солевым раствором (15 мл), сушили с безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта, который затем отделяли путем колоночной хроматографии с получением 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-метилбензамида в виде беловатого твердого вещества (280 мг, выход 42,3%). МС (ИЭР) m/z 331,0 [M+H]+.
(7) Синтез N-метил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида
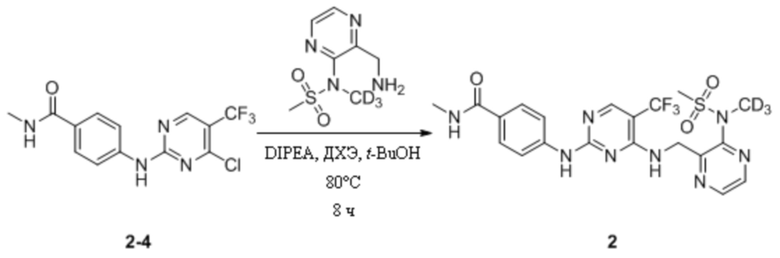
В 25 мл одногорлую круглодонную колбу, содержащую N-(3-(аминометил)пиразин-2-ил)-N-дейтерометилметансульфонамид (65,8 мг, 0,30 ммоль), добавляли 1,2-дихлорэтан (5 мл) и трет-бутанол (5 мл) и смесь перемешивали при комнатной температуре для получения прозрачного раствора. Затем к системе последовательно добавляли 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-метилбензамид (100,0 мг, 0,30 ммоль) и диизопропилэтиламин (116,3 мг, 0,90 ммоль) и, после добавления, систему переносили на масляную баню при 80°C и кипятили с обратным холодильником для протекания реакции. Через 8 ч при помощи ТСХ определяли полное израсходование исходных материалов. После прекращения нагревания систему охлаждали до комнатной температуры и растворитель удаляли путем ротационного выпаривания с получением неочищенного продукта, который затем отделяли и очищали путем преп. ТСХ с получением N-метил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида в виде беловатого твердого вещества (12 мг, выход 7,8%). МС (ИЭР) m/z 514,2 [M+H]+.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,83 (s, 1H), 8,69 (s, 1H), 8,59 (s, 1H), 8,32 (s, 1H), 8,20 (d, J = 4,0 Гц, 1H), 7,68-7,61 (dd, J = 14,4, 8,4 Гц, 4H), 7,41-7,39 (t, J = 4,4 Гц, 1H), 5,01 (d, J = 3,6 Гц, 2H), 3,20 (s, 3H), 2,76 (d, J = 4,0 Гц, 3H).
Пример 3. Синтез N-(тридейтерометил)-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил) метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 3)
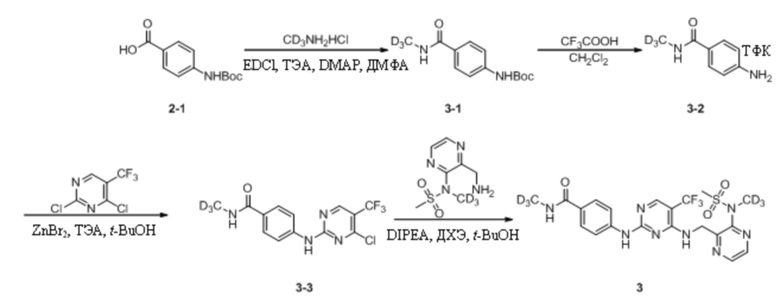
(1) Синтез трет-бутил-(4-(дейтерометиламиноформил)фенил)карбамата
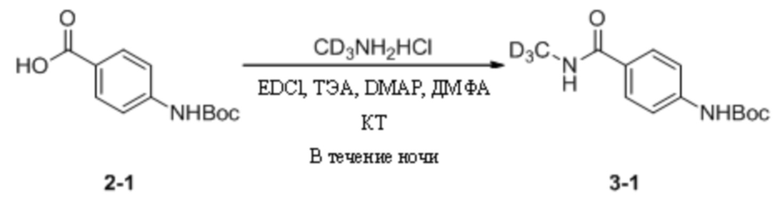
4-((Трет-бутоксикарбонил)амино)бензойную кислоту (1,1 г, 4,64 ммоль) взвешивали и помещали в 100 мл одногорлую круглодонную колбу, в которую добавляли 20 мл ДМФА, и смесь перемешивали при комнатной температуре. Затем к системе последовательно добавляли EDCI (1,3 г, 6,96 ммоль), ТЭА (1,2 г, 11,6 ммоль), дейтерированный гидрохлорид метиламина (327,2 мг, 4,64 ммоль) и DMAP (28 мг, 0,23 ммоль). Затем систему перемешивали и оставляли для протекания реакции в течение ночи при комнатной температуре. На следующий день, когда детектировали израсходование исходных материалов, к системе добавляли этилацетат (30 мл) и воду (20 мл), энергично перемешивали и выдерживали для разделения слоев. Водную фазу подвергали обратной экстракции этилацетатом (15 мл * 3) и органические слои объединяли, промывали водой (15 мл * 3) и насыщенным солевым раствором (20 мл), а затем сушили с безводным сульфатом натрия. Растворитель удаляли путем ротационного выпаривания с получением неочищенного продукта, который затем отделяли путем колоночной хроматографии с получением трет-бутил-(4-(дейтерометиламиноформил)фенил)карбамата в виде беловатого твердого вещества (953 мг, выход 81,2%). МС (ИЭР) m/z 254,2 [M+H]+.
(2) Синтез трифторацетата 4-амино-N-дейтерометилбензамида
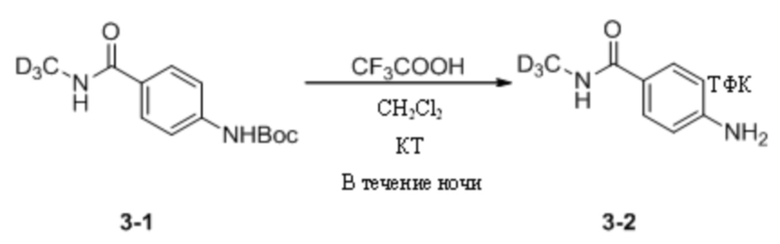
Трет-бутил-(4-(дейтерометиламиноформил)фенил)карбамат (953,0 мг, 3,76 ммоль) взвешивали и помещали в 50 мл одногорлую круглодонную колбу, в которую добавляли дихлорметан (20 мл), и смесь перемешивали при комнатной температуре. Затем к системе добавляли трифторуксусную кислоту (5 мл). После добавления систему перемешивали и оставляли для протекания реакции в течение ночи при комнатной температуре. На следующий день завершение реакции детектировали путем ТСХ. Реакционный раствор концентрировали для удаления растворителя и избытка трифторуксусной кислоты и остаточную трифторуксусную кислоту в системе удаляли путем многократного совместного пропаривания с дихлорметаном до полного отверждения системы с получением трифторацетата 4-амино-N-дейтерометилбензамида (941 мг) в виде беловатого твердого вещества, которое непосредственно применяли на следующей стадии без дополнительной очистки. МС (ИЭР) m/z 154,1 [M+H]+.
(3) Синтез 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-дейтерометилбензамида
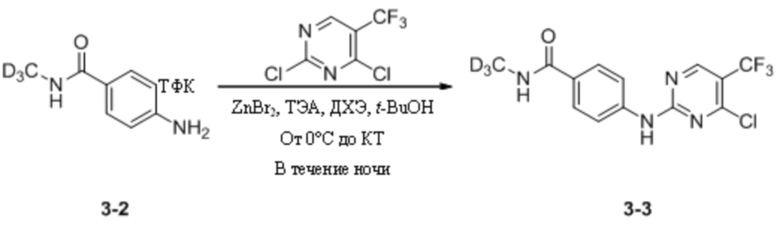
2,4-Дихлор-5-(трифторметил)пиримидин (891,8 мг, 4,11 ммоль) взвешивали и помещали в 100 мл одногорлую круглодонную колбу, в которую добавляли 1,2-дихлорэтан (10 мл) и трет-бутанол (10 мл), и смесь перемешивали при комнатной температуре для получения прозрачного раствора. Затем систему переносили в баню с ледяной водой для продолжения охлаждения и перемешивания. Через 15 мин к системе добавляли бромид цинка (2,3 г, 10,71 ммоль). После добавления систему дополнительно выдерживали в бане с ледяной водой и перемешивали в течение 30 мин. Затем к системе добавляли трифторацетат 4-амино-N-дейтерометилбензамида и триэтиламин (1,2 г, 11,41 ммоль), полученные на предыдущей стадии. После добавления ледяную баню убирали и систему перемешивали и оставляли для протекания реакции в течение ночи при комнатной температуре. На следующий день детектировали завершение реакции и, после удаления растворителя путем ротационного выпаривания, к системе добавляли этилацетат (50 мл) и воду (20 мл). Реакционную смесь энергично перемешивали и выдерживали для разделения слоев. Водный слой подвергали обратной экстракции этилацетатом (20 мл * 3) и органические фазы объединяли, последовательно промывали водой (20 мл * 3) и насыщенным солевым раствором (20 мл), сушили с безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного продукта, который затем отделяли путем колоночной хроматографии с получением 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-дейтерометилбензамида в виде беловатого твердого вещества (467 мг, выход 39,2%). МС (ИЭР) m/z 334,0 [M+H]+.
(4) Синтез N-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил)метил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида
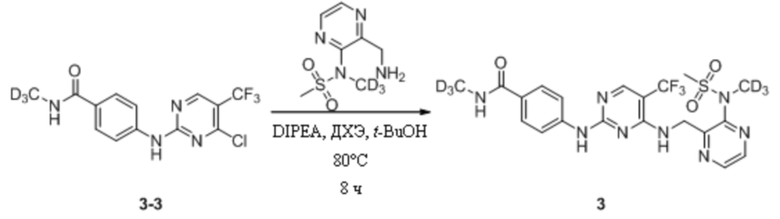
В 25 мл одногорлую круглодонную колбу, содержащую N-(3-(аминометил)пиразин-2-ил)-N-дейтерометилметансульфонамид (43,8 мг, 0,20 ммоль), добавляли 1,2-дихлорэтан (3 мл) и трет-бутанол (3 мл) и смесь перемешивали при комнатной температуре для получения прозрачного раствора. Затем к системе последовательно добавляли 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-дейтерометилбензамид (66,7 мг, 0,20 ммоль) и диизопропилэтиламин (77,6 мг, 0,60 ммоль) и после добавления систему переносили на масляную баню при 80°C и кипятили с обратным холодильником для протекания реакции. Через 8 ч при помощи ТСХ определяли полное израсходование исходных материалов. После прекращения нагревания систему охлаждали до комнатной температуры и растворитель удаляли путем ротационного выпаривания с получением неочищенного продукта, который затем отделяли и очищали путем преп. ТСХ с получением N-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил)метил) амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида в виде беловатого твердого вещества (35,5 мг, выход 34,4%). МС (ИЭР) m/z 517,2 [M+H]+.
1H-ЯМР (400 МГц, ДМСО-d6) δ 9,83 (s, 1H), 8,69 (d, J = 2,8 Гц, 1H), 8,58 (d, J = 2,4 Гц, 1H), 8,31 (s, 1H), 8,17 (s, 1H), 7,67-7,60 (dd, J = 15,4, 8,6 Гц, 4H), 7,42-7,39 (t, J = 5,2 Гц, 1H), 5,00 (d, J = 4,8 Гц, 2H), 3,20 (s, 3H).
Пример 4. Синтез N-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил) дейтерометил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида (соединение 5)
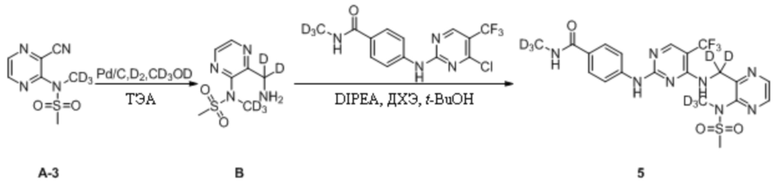
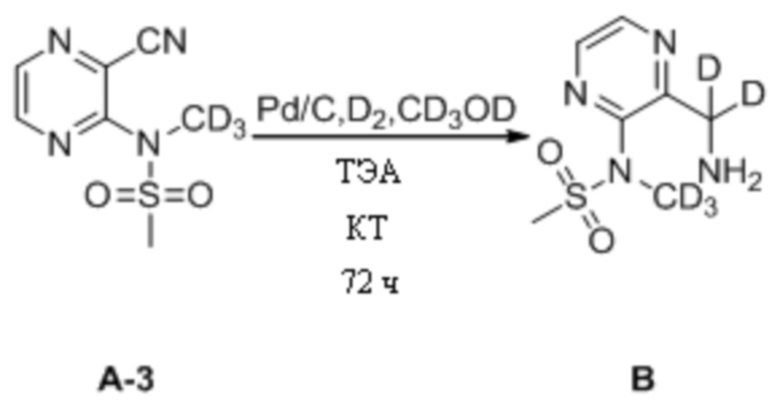
(1) Синтез N-(3-(аминодейтерометил)пиразин-2-ил)-N-дейтерометилметансульфонамида
ТЭА
КТ
72 ч
N-(3-Цианопиразин-2-ил)-N-дейтерометилметансульфонамид (100,0 мг, 0,46 ммоль) взвешивали и помещали в 25 мл одногорлую круглодонную колбу, в которую добавляли 5 мл дейтерированного метанола, и смесь перемешивали при комнатной температуре для получения прозрачного раствора. Затем к системе последовательно добавляли влажный палладированный уголь (20,0 мг) и триэтиламин (188,2 мг, 1,86 ммоль) и систему подвергали операции замены водорода, которую повторяли десять раз. Затем систему перемешивали и оставляли для протекания реакции при комнатной температуре. Через 72 ч детектировали завершение реакции. Систему подвергали фильтрованию с отсасыванием и осадок на фильтре несколько раз промывали небольшими количествами дейтерированного метанола (10 мл). Фильтрат объединяли и концентрировали при пониженном давлении для удаления растворителя с получением N-(3-(аминодейтерометил)пиразин-2-ил)-N-дейтерометилметансульфонамида в виде светлой желто-коричневой маслянистой жидкости, которую непосредственно применяли на следующей стадии без дополнительной очистки. МС (ИЭР) m/z 222,2 [M+H]+.
(2) Синтез N-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил) дейтерометил)амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида
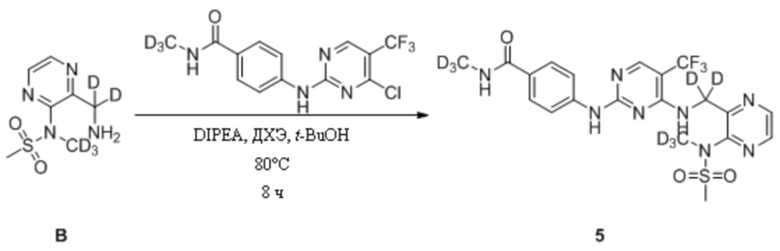
В 25 мл одногорлую круглодонную колбу, содержащую N-(3-(аминодейтерометил)пиразин-2-ил)-N-дейтерометилметансульфонамид (22,1 мг, 0,10 ммоль), добавляли 1,2-дихлорэтан (2 мл) и трет-бутанол (2 мл) и смесь перемешивали при комнатной температуре для получения прозрачного раствора. Затем к системе последовательно добавляли 4-((4-хлор-5-(трифторметил)пиримидин-2-ил)амино)-N-дейтерометилбензамид (33,4 мг, 0,10 ммоль) и диизопропилэтиламин (30,6 мг, 0,30 ммоль) и после добавления систему переносили на масляную баню при 80°C и кипятили с обратным холодильником для протекания реакции. Через 8 ч при помощи ТСХ определяли полное израсходование исходных материалов. После прекращения нагревания систему охлаждали до комнатной температуры и растворитель удаляли путем ротационного выпаривания с получением неочищенного продукта, который затем отделяли и очищали путем преп. ТСХ с получением N-дейтерометил-4-((4-(((3-(N-дейтерометилметансульфонамидо)пиразин-2-ил)метил) амино)-5-(трифторметил)пиримидин-2-ил)амино)бензамида в виде беловатого твердого вещества (8,1 мг, выход 15,6%). МС (ИЭР) m/z 519,2 [M+H]+. 1H-ЯМР (400 МГц, ДМСО-d6) δ 9,83 (s, 1H), 8,69 (d, J = 2,0 Гц, 1H), 8,58 (d, J = 2,4 Гц, 1H), 8,31 (s, 1H), 8,17 (s, 1H), 7,67-7,61 (dd, J = 15,2, 8,8 Гц, 4H), 7,39 (s, 1H), 3,20 (s, 3H).
Применяя следующие соединения в качестве исходных материалов в соответствии со способами синтеза, аналогичными способам для соединений 1-3, 5 и соединения 3-3, получали соединения 4 и 6-18 согласно настоящему изобретению: соединение A, 2-3, B, 2-4, 3-3, N-(3-(аминометил)пиразин-2-ил)-N-метилметансульфонамид (ссылка: международная заявка согласно PCT, 2008129380), N-метилметансульфонамид, N-тридейтерометилметансульфонамид, 2,4-дихлор-5-(трифторметил)пиримидин, 4-амино-3,5-дидейтеробензойная кислота (ссылка: Journal of Labelled Compounds and Radiopharmaceuticals, 53(11-12), 668-673; 2010), 4-амино-2,6-дидейтеробензойная кислота (ссылка: Journal of Labelled Compounds and Radiopharmaceuticals, 53(11-12), 668-673; 2010).
Полезный эффект настоящего изобретения проиллюстрирован экспериментальными примерами.
Экспериментальный пример 1. Эксперимент по изучению метаболической стабильности в микросомах печени
Стадия 1: маточный раствор готовили в соответствии с отношением компонентов, приведенным в таблице 1:
Стадия 2: проводили два эксперимента следующим образом:
A) добавление восстановленного кофермента II (NADPH): в инкубационный эксперимент добавляли 20 мг/мл микросомы печени (10 мкл) и 10 мМ раствор NADPH (40 мкл). Конечные концентрации микросом печени и NADPH составляли 0,5 мг/мл и 1 мМ, соответственно.
B) без NADPH: в инкубационный эксперимент добавляли 20 мг/мл микросомы печени (10 мкл) и воду высокой степени очистки (40 мкл). Конечная концентрация микросом печени составляла 0,5 мг/мл.
Стадия 3: реакция начиналась после добавления 200 мкМ положительного контроля (4 мкл) или испытываемого соединения. В указанном эксперименте положительный контроль представлял собой верапамил. Конечная концентрация испытываемого соединения составляла 2 мкМ.
Стадия 4: в моменты времени 0 мин, 15 мин, 30 мин, 45 мин и 60 мин из каждого реакционного раствора отбирали 50 мкл раствора. В реакционный раствор добавляли ацетонитрил (4 объема реакционного раствора) и IS (100 нМ алпразолама, 200 нМ лабеталола, 200 нМ кофеина и 2 мкМ кетопрофена). Образец центрифугировали при гравитации 3220 g в течение 40 мин. К 100 мкл надосадочной жидкости добавляли 100 мкл воды высокой степени очистки и анализировали путем ЖХ-МС/МС.
Стадия 5: анализ данных: площадь пика определяли по экстракционной ионной хроматограмме. Величину наклона k определяли путем линейной регрессии натурального логарифма, полученного из кривой зависимости между остаточным процентом исходного лекарственного средства и временем инкубирования.
Период полувыведения in vitro (In vitro t1/2) определяли по величине наклона: 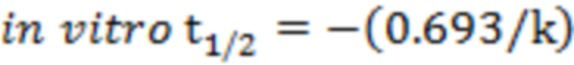 .
.
Собственный клиренс in vitro (in vitro CLint, в мкл/мин/мг) рассчитывали из периода полувыведения in vitro t1/2 (мин) с применением следующего уравнения (среднее значение повторных определений):
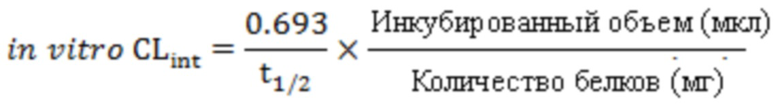
Масштабированный собственный клиренс (масштабированный CLint, в мл/мин/кг) рассчитывали из периода полувыведения in vitro t1/2 (мин) с применением следующего уравнения (среднее значение повторных определений):

Результаты приведены в таблице 2.
Таблица 2. Результаты экспериментов по изучению метаболической стабильности в микросомах печени мышей, крыс и человека
Как показано в приведенной выше таблице, метаболическая стабильность соединений 1 и 3 согласно настоящему изобретению в микросомах печени являлась значительно улучшенной по сравнению с недейтерированным контрольным соединением дефактинибом, в то время как метаболическая стабильность соединения 2 в микросомах печени крыс и человека была немного хуже по сравнению с недейтерированным контрольным соединением дефактинибом, что указывает на то, что соединения согласно настоящему изобретению, особенно соединения 1 и 3, обладают улучшенной метаболической стабильностью. Кроме того, они могут иметь улучшенную фармакокинетику, улучшенную безопасность и эффективность, что дополнительно подтверждается в следующем экспериментальном примере.
Экспериментальный пример 2. Фармакокинетика соединения согласно настоящему изобретению у крыс
1) материалы и инструменты для исследования:
Система LC-20AD HPLC, приобретенная в компании SHIMADZU в Японии
Тройной квадрупольный масс-спектрометр API4000, приобретенный в компании Applied Biosystem в США
Фармакокинетическое программное обеспечение PhenixWinnolin (версия 6.3), приобретенное в Certara в США,
Высокоскоростная центрифуга с замораживанием, приобретенная в Thermo Fisher Scientific
Аналитические весы, приобретенные в Sartorius, SECURA225D-1CN
Крысы линии Спрег-Доули, приобретенные в Chengdu Dashuo Experimental Animal Co., Ltd
N, N-Диметилацетамид (ДМАА) (Sigma)
Карбоксиметилцеллюлоза натрия (Na-КМЦ) и гепарин, приобретенные в Chengdu Kelong Chemical Co., Ltd
2) Экспериментальные способы и результаты
Соответствующее количество лекарственного средства (эквивалентное 10 мг исходного лекарственного средства) точно взвешивали, добавляли 0,25 мл ДМАА для растворения лекарственного средства и медленно добавляли 0,5% Na-КМЦ до 5 мл, а затем тщательно перемешивали с применением ультразвука и вортекса. Отбирали 0,2 мл полученного конечного раствора и хранили при -20°C для определения концентрации. Трем здоровым взрослым самцам крыс линии Спрег-Доули (180-250 г) давали лекарственные средства в дозе 5 мл/кг при помощи желудочного зонда после ночного голодания (со свободным доступом к питьевой воде); отбирали по 0,1 мл крови из ретроорбитального венозного сплетения перед введением и через 0,5, 1, 2, 4, 6, 8, 12 и 24 ч после введения и плазму отделяли путем центрифугирования при 4°C в течение 5 мин и хранили при -20°C до применения. Затем для определения концентрации соединений в плазме применяли способ ЖХ/МС/МС.
Таблица 3. Фармакокинетические параметры согласно настоящему изобретению
tmax (ч)
Cmax (нг/мл)
t1/2 (ч)
MRTinf (ч)
Как показано в таблице 3, по сравнению с недейтерированным контрольным соединением дефактинибом соединение согласно настоящему изобретению обладает более высокой пиковой концентрацией лекарственного средства в крови, более высокой дозой в плазме и более долгим периодом полувыведения, что указывает на то, что соединение, предложенное в настоящем изобретении, обладает улучшенной фармакокинетической эффективностью. Соединения согласно настоящему изобретению являются перспективными в лечении рака.
Таким образом, различные дейтерированные соединения и их соли, гидраты или сольваты, предложенные в настоящем изобретении, можно применять в качестве ингибиторов FAK для получения противораковых лекарственных средств. Кроме того, по сравнению с недейтерированным контрольным соединением дефактинибом метаболическая стабильность и фармакокинетические свойства соединений согласно настоящему изобретению являются значительно улучшенными, и они обладают хорошими перспективами применения.
Изобретение относится к соединению формулы (I) или к его фармацевтически приемлемой соли, где каждый из R1-R3, R9, R10 и R13-R15 независимо выбран из водорода или дейтерия, и R4–R8, R11, R12 и R16-R18 представляют собой водород. Изобретение также относится к фармацевтической композиции для ингибирования FAK на основе указанного соединения формулы (I). Технический результат – получены новые соединения и фармацевтическая композиция на их основе, которые могут найти применение в медицине для получения противораковых лекарственных средств. 4 н. и 6 з.п. ф-лы, 3 табл., 4 пр.
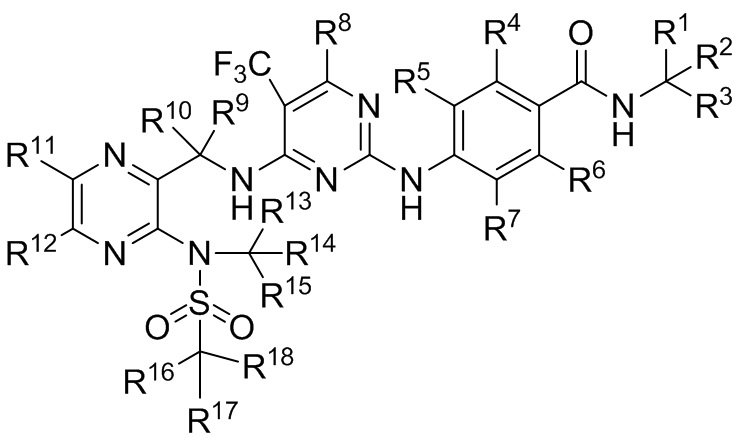 (I)
(I)
1. Соединение формулы (I) или его фармацевтически приемлемая соль:
(I),
где каждый из R1-R3, R9, R10 и R13-R15 независимо выбран из водорода или дейтерия, и R4–R8, R11, R12 и R16-R18 представляют собой водород.
2. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (II):
(II).
3. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (III):
(III).
4. Соединение по п. 1 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (IV):
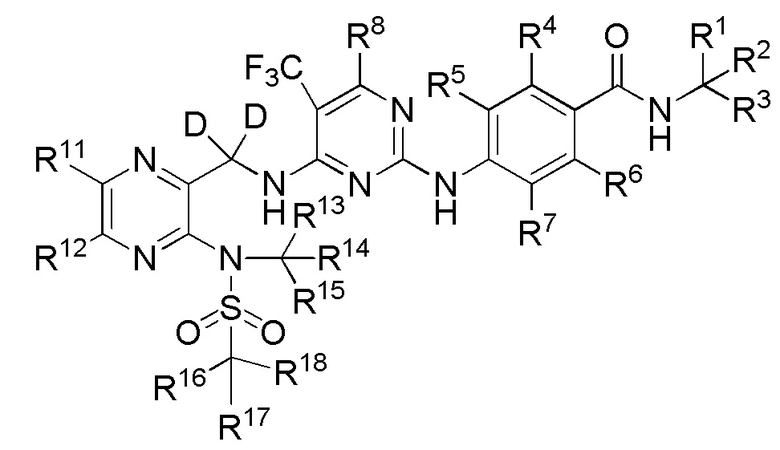
(IV).
5. Соединение по любому из пп. 1-4 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение имеет структуру формулы (V):
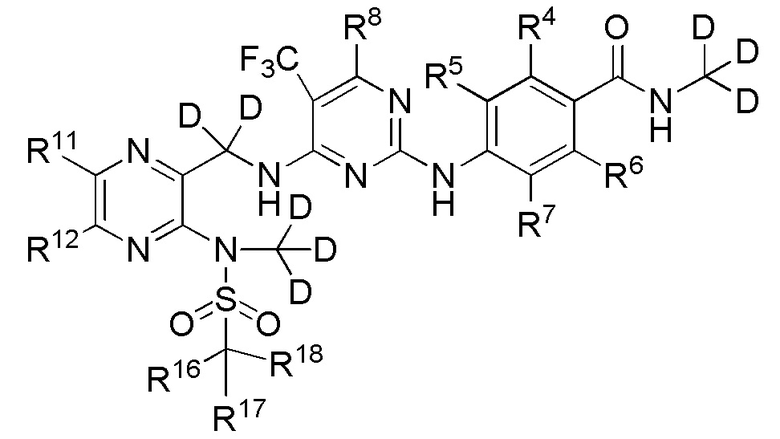
(V).
6. Соединение по любому из пп. 1-5 или его фармацевтически приемлемая соль, отличающееся тем, что указанное соединение представляет собой одно из следующих соединений:
7. Соединение по любому из пп. 1-6 или его фармацевтически приемлемая соль, отличающееся тем, что указанная фармацевтически приемлемая соль представляет собой фосфат, d-камфорсульфонат, гидрохлорид, гидробромид, гидрофторид, сульфат, нитрат, формиат, ацетат, пропионат, оксалат, малонат, сукцинат, фумарат, малеат, лактат, малат, тартрат, цитрат, пикрат, метансульфонат, безилат, бензолсульфонат, аспартат или глутамат соединения и предпочтительно гидрохлорид соединения.
8. Применение соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли для получения лекарственных средств, ингибирующих FAK, для лечения рака, выбранного из рака поджелудочной железы, солидной опухоли, немелкоклеточного рака легких, мезотелиомы и рака яичников.
9. Применение соединения по любому из пп. 1-7 или его фармацевтически приемлемой соли для получения ингибиторов FAK.
10. Фармацевтическая композиция для ингибирования FAK, содержащая соединение по любому из пп. 1-7 или его фармацевтически приемлемую соль в качестве активного ингредиента и фармацевтически приемлемое вспомогательное вещество.
| Устройство для сигнализации о неправильной работе нумераторов на типографских машинах | 1928 |
|
SU16679A1 |
| CN 106146406 A, 23.11.2016 | |||
| US 20110053968 A1, 03.03.2011 | |||
| WO 2010144499 A2, 16.12.2010 | |||
| Аппарат для разливки варенья в мелкую упаковку | 1929 |
|
SU19941A1 |

