ОБЛАСТЬ ТЕХНИКИ
Изобретение относится к области медицины, а именно к фармакологии и токсикологии, и может быть использовано для определения N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфоната в плазме крови.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Производное аминогуанидина - N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфонат является действующим веществом разрабатываемого противовоспалительного средства для лечения воспалительных и дегенеративных заболеваний опорно-двигательного аппарата.
Одной из актуальных задач при создании нового лекарственного средства является проведение исследований фармакокинетики с целью оценки всасывания, распределения, метаболизма и экскреции. Для проведения исследований фармакокинетики требуется разработать биоаналитическую методику количественного определения действующего вещества в биологических средах, в частности в плазме крови.
В настоящее время не известно способов количественного определения N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфоната в плазме крови и других биологических средах организма. Для поиска возможных прототипов проведен анализ сведений о методах определения веществ с подобной химической структурой, в частности - производных аминогуанидина и производных индола, используемых в качестве действующих веществ лекарственных средств.
Известен способ количественного определения аминогуанидина в плазме крови и других биологических образцах с использованием экстракции вещества из образцов трихлоруксусной кислотой, реакции с 6-метил-2-пиридин-карбоксальдегидом при температуре 95-100°С, и определением вещества методом ВЭЖХ с использованием в качестве подвижной фазы смеси ацетонитрил (15%) : метанол (10%) : 0,01 М гептансульфоната в растворе натри фосфорнокислого однозамещенного и УФ-детектированием при длине волны 302 нм [патент US 5108930, МПК G01N 33/00, опубл. 28.04.1992].
В качестве прототипа использован описанный ранее способ количественного определения производного индола-сертиндола (1-[-2-[4-[5-Хлор-1-(4-фторфенил)-1Н-индол-3-ил]-1-пиперидинил]этил]-2-имидазолидинона). Данный способ основан на методе ВЭЖХ с УФ-детектированием при длине волны 230 нм, с использованием в качестве подвижной фазы А-0,1% раствора кислоты трифторуксусной, фазы Б - ацетонитрила с градиентом концентраций 10-90% [Ремезова И.П. и соавт. Разработка методик обнаружения некоторых атипичных нейролептиков для целей химико-токсикологического анализа. Фармация и фармакология. 2014, №6 (7), С. 54-58].
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Предлагаемое изобретение решает задачу разработки нового способа количественного определения действующего вещества противовоспалительного лекарственного средства - N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидина метансульфоната в плазме крови.
ОСУЩЕСТВЛЕНИЕ ИЗОБРЕТЕНИЯ
Поставленная задача решается способом количественного определения N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфоната в плазме крови, включающим экстракцию определяемого вещества ацетонитрилом с добавлением натрия хлорида с последующим определением его методом высокоэффективной жидкостной хроматографии с градиентным режимом элюирования с использованием в качестве подвижной фазы А - смеси 0,2 М раствора лития перхлората в 0,005 М растворе хлорной кислоты, подвижной фазы Б - ацетонитрила, с последующим УФ-детектированием при длине волны 330 нм.
Наиболее оптимальными, но не необходимыми хроматографическими условиями при этом являются:


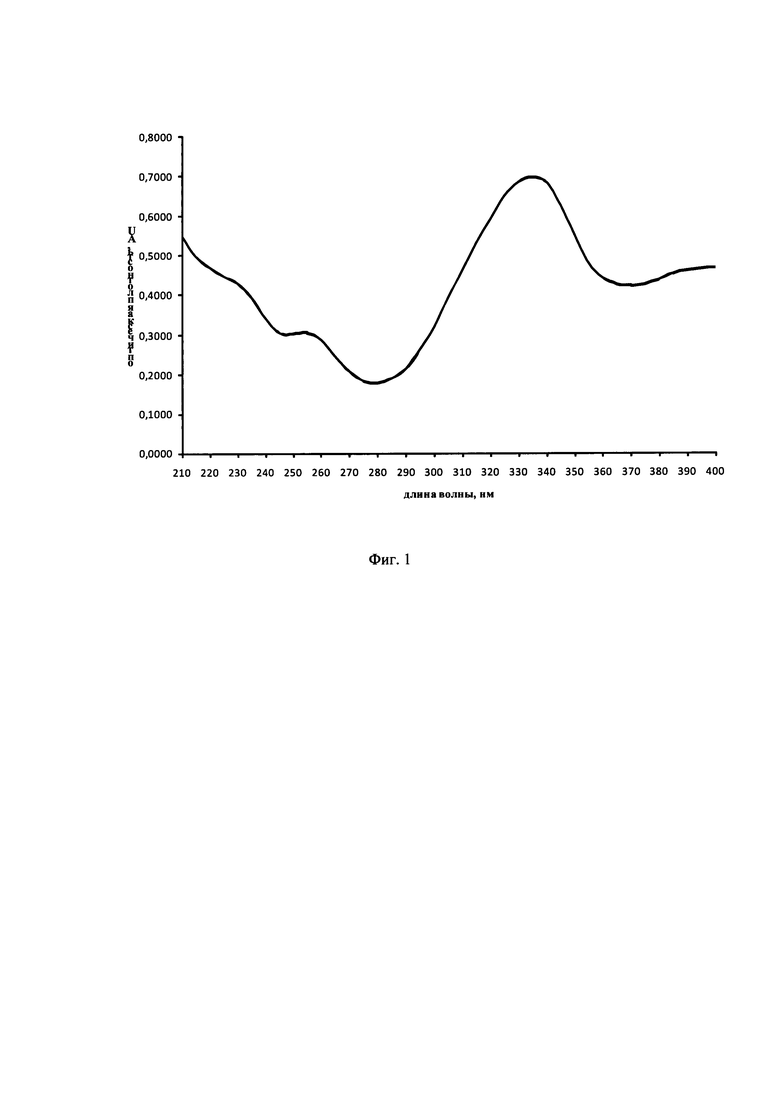
Аналитическую длину волны подбирали по результатам анализа УФ-спектра N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфоната (далее по тексту - субстанция LIS-M) в смеси ацетонитрил-ПФ А в соотношении (1:1) (Фиг. 1) и особенностей матрицы, в которой необходимо проводить количественное определение. На УФ-спектре субстанции LIS-M в области от 210 до 400 нм наблюдается 3 максимума поглощения - 230, 255 и 330 нм. 330 нм выбрана основной аналитической длиной волны, так как она является наиболее селективной и характеризуется практически полным отсутствием поглощения другими компонентами матрицы.
Отсутствие источников информации, содержащих ту же совокупность признаков, что и в разработанном способе, сообщает ему соответствие критерию «новизна».
Та же совокупность признаков позволяет получить новый непредсказуемый эффект - разработка высокоэффективного способа количественного определения в плазме крови нового лекарственного средства - N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфоната и, таким образом, сообщает ему соответствие критерию «изобретательский уровень».
Проведение количественного определения нового лекарственного средства с использованием известного оборудования с помощью известных препаратов сообщает разработанному способу соответствие критерию «промышленная применимость».
Таким образом, разработанный способ соответствует всем 3 критериям охраноспособности изобретения и может быть квалифицирован как изобретение.
На Фиг. 1 представлен УФ-спектр субстанции LIS-M.
Пример 1. Подбор условий извлечения определяемого вещества
Для извлечения определяемого вещества из плазмы крови используется несколько подходов. Наиболее простой и быстрый способ пробоподготовки - осаждение белков органическими растворителями с последующим центрифугированием. Кратность разбавления образца при этом варьируется от 1 до 5, а степень извлечения определяемого вещества обычно находится в интервале 90-100%. После центрифугирования надосадочную жидкость (супернатант) вводят в хроматографическую систему. После осаждения белка в пробе остается большое количество компонентов, которые могут помешать при определении исследуемого вещества. Также при осаждении белков очень проблематично сконцентрировать образец, что не позволяет снизить предел обнаружения.
Другой метод подготовки проб - жидкостно-жидкостная экстракция. Данный метод удобно применять, если требуется концентрирование исследуемого вещества перед определением. Степень извлечения методом жидкостно-жидкостной экстракции составляет обычно 70-90% и при этом концентрацию определяемого ЛВ в инжектируемом образце можно повысить в 5-10 раз по сравнению с исходной пробой, что позволяет существенно снизить предел обнаружения. Таким образом, для проведения фармакокинетических исследований нового лекарственного средства LIS-M более оптимально использовать метод жидкостно-жидкостной экстракции.
Исходя из физико-химических свойств субстанции LIS-M были использованы два экстрагента: ацетонитрил и этилацетат.
Экстракция LIS-M из раствора этилацетатом
В 3 пластиковых центрифужных микропробирки типа «Эппендорф» вместимостью 2 мл помещали по 500 мкл раствора LIS-M, с содержанием 1 мкг/мл, прибавляли 50 мкл 20% раствора натрия гидроксида и перемешивали на встряхивателе типа «Vortex» при 3000 об/мин в течение 1 мин. Экстрагировали на встряхивателе типа «Vortex» при 3000 об/мин в течение 10 мин. Центрифугировали при 16060 g в течение 10 мин. Органический слой отбирали и упаривали под вакуумом при температуре 50°С до сухого остатка. К сухому остатку прибавили 500 мкл смеси ПФ А - ПФ Б в соотношении (1:1), перемешали на встряхивателе типа «Vortex» при 3000 об/мин в течение 10 мин.
Экстракция LIS-M из раствора ацетонитрилом
В 3 пластиковых центрифужных микропробирки типа «Эппендорф» вместимостью 2 мл помещали по 500 мкл раствора LIS-M, с содержанием 1 мкг/мл. Экстрагировали на встряхивателе типа «Vortex» при 3000 об/мин в течение 10 мин. Центрифугировали при 16060 g в течение 10 мин. Органический слой отбирали и упаривали под вакуумом при температуре 50°С до сухого остатка. К сухому остатку прибавили 500 мкл смеси ПФ А - ПФ Б в соотношении (1:1), перемешали на встряхивателе типа «Vortex» при 3000 об/мин в течение 10 мин.
Для определения степени извлечения полученные результаты сравнивали с аналитическим сигналом, полученным для раствора LIS-M, с содержанием 1 мкг/мл.
Результаты определения степени извлечения представлены в таблице 1.
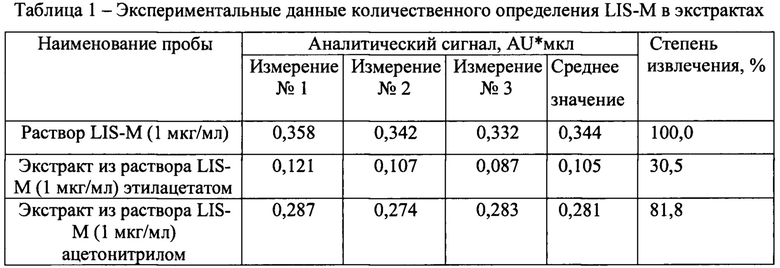
Как видно из таблицы 1, наибольшая степень извлечения достигается ацетонитрилом. Таким образом, данный экстрагент можно использовать для экстракции LIS-M из биологических сред.
Были проведены валидационные тесты, в ходе которых не обнаружено отклонений от установленных критериев приемлемости.
Пример 2. Подтверждение точности.
Точность биоаналитической методики характеризуется близостью полученных с помощью нее значений к номинальным концентрациям аналита.
Точность внутри цикла.
Был проанализирован один аналитический цикл, состоящий из четырех уровней концентраций, по 5 образцов на каждом уровне. Уровни концентрации: 50 нг/мл (нижний предел количественного определения - НПКО), 150 нг/мл (тройная величина НПКО), 1000 нг/мл (50% диапазона калибровочной кривой - КК) и 1500 нг/мл (75% от верхнего диапазона калибровочной кривой).
Критерий приемлемости:
- отклонение среднего значения каждого уровня концентрации от номинального содержания должно быть в пределах 15% для всех уровней концентрации, кроме НПКО для которой допускается отклонение 20%.
Результаты определения точности представлены в таблице 2.
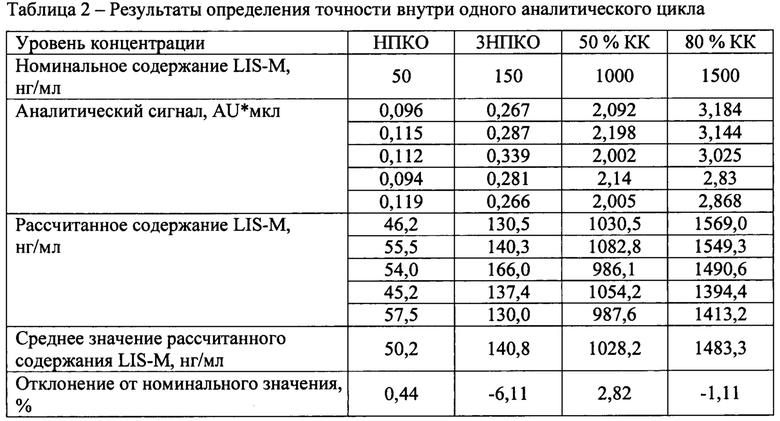
Отклонение от номинального значения средних значений рассчитанных концентраций находится в диапазоне от -6,11 до 2,82%, что удовлетворяет критерию приемлемости.
Точность между циклами
В течение не менее двух дней были проанализированы три аналитических цикла, состоящий из четырех уровней концентраций, по 5 образцов на каждом уровне. Уровни концентрации: НПКО, тройная величина НПКО, около 50% диапазона калибровочной кривой и не менее 75% от верхнего диапазона калибровочной кривой.
Критерий приемлемости:
Отклонение среднего значения каждого уровня концентрации от номинального содержания должно быть в пределах 15% для всех уровней концентрации, кроме НПКО для которой допускается отклонение 20%.
Результаты определения точности между циклами представлены в таблицах 3-6.
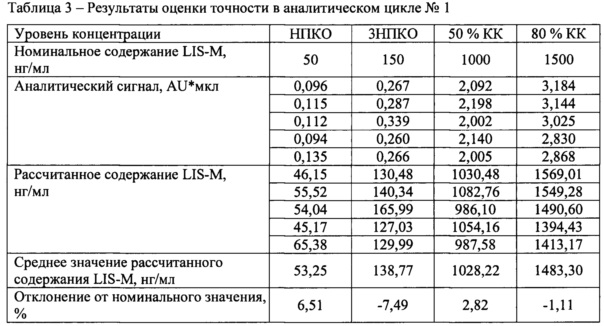
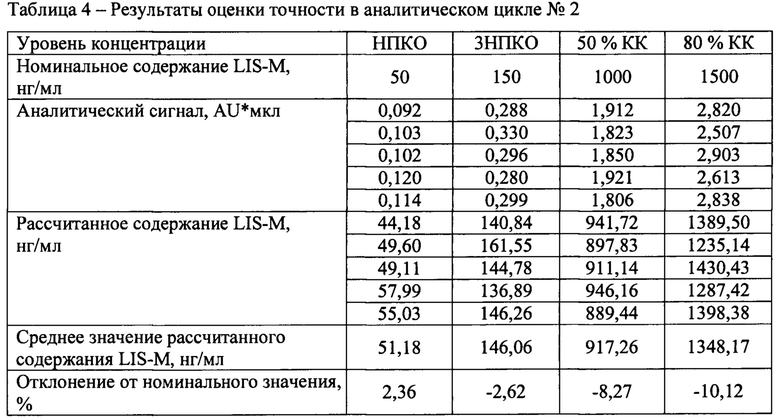
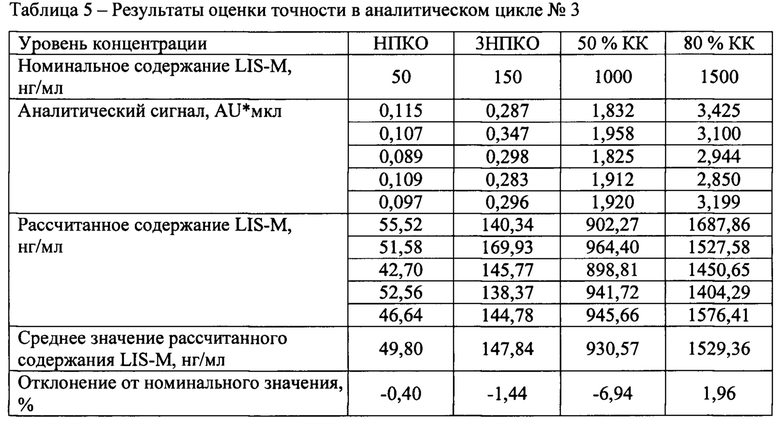
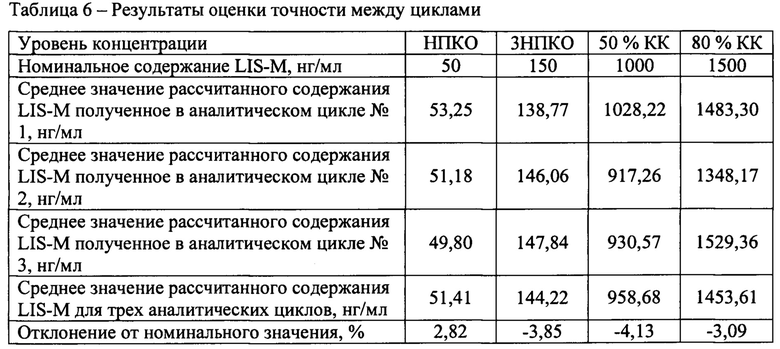
Отклонение от номинального значения средних значений рассчитанных концентраций для всех аналитических циклов находится в диапазоне от -4,13 до 2,82%, что удовлетворяет критерию приемлемости.
Как видно из приведенных примеров, предлагаемое изобретение позволяет получить необходимую и достоверную информацию о способе количественного определения N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метан-сульфоната в биологических средах.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения 2-изопропил-5-метилциклогексил 2-(1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил)ацетата и индометацина в плазме крови | 2021 |
|
RU2781342C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 4-АМИНО-1-(3-НИТРО-2-ОКСО-1-ФЕНИЛ-1,2-ДИГИДРО-1,6-НАФТИРИДИН-5-ИЛ)ПИРИДИНИЙ ХЛОРИДА В БИОЛОГИЧЕСКИХ СРЕДАХ | 2018 |
|
RU2686116C1 |
| НОВОЕ БИОЛОГИЧЕСКИ АКТИВНОЕ СОЕДИНЕНИЕ N-[3-(4-НИТРОФЕНИЛАМИНО)-ИНДОЛ-2-ИЛМЕТИЛЕН] АМИНОГУАНИДИНА ГИДРОХЛОРИД С ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2478618C2 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 8-(ТРИФТОРМЕТИЛ)БЕНЗО[F][1,2,3,4,5]ПЕНТАТИЕПИН-6-АМИНА ГИДРОХЛОРИДА В БИОЛОГИЧЕСКИХ СРЕДАХ | 2018 |
|
RU2676487C1 |
| Способ количественного определения дисульфирама в биологических средах | 2019 |
|
RU2701524C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 2-ФЕНОКСИЭТАНОЛА В БИОЛОГИЧЕСКИХ СРЕДАХ | 2021 |
|
RU2776730C1 |
| Способ количественного определения 2,2,6,6-тетраметил-N-{ 1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил} пиперидин-4-амина дигидрохлорида в биологических средах | 2016 |
|
RU2636231C1 |
| Способ количественного определения дексаметазона в биологических средах с помощью ВЭЖХ с ультрафиолетовым детектированием | 2022 |
|
RU2792274C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЭНАНТИОМЕРОВ ВЕРАПАМИЛА В СУБСТАНЦИЯХ, ТАБЛЕТКАХ И ОБРАЗЦАХ ПЛАЗМЫ КРОВИ МЕТОДОМ ВЭЖХ | 2008 |
|
RU2395807C1 |
| Способ количественного определения доксорубицина в биологических образцах | 2024 |
|
RU2839330C1 |
Изобретение относится к медицине, а именно фармакологии и токсикологии, и может быть использовано для определения N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфоната в плазме крови. Для этого образец экстрагируют ацетонитрилом с добавлением натрия хлорида. Затем препарат количественно определяют методом высокоэффективной жидкостной хроматографии на колонке ProntoSIL, 75х2,0 мм, 5 мкм в градиентном режиме элюирования, в качестве подвижной фазы А смеси используют 0,2 М раствор лития перхлората в 0,005 М растворе хлорной кислоты, в качестве подвижной фазы Б - ацетонитрил с последующим УФ-детектированием при длине волны 330 нм. Изобретение обеспечивает количественное определение N-[3-(4-нитрофениламино)-индол-2-илметилен]аминогуанидина метансульфоната в биологических средах. 1 ил., 6 табл., 2 пр.
Способ количественного определения N-[3-(4-нитрофениламино)-индол-2-илметилен] аминогуанидина метансульфоната в плазме крови, включающий экстракцию определяемого вещества ацетонитрилом с добавлением натрия хлорида с последующим разделением методом высокоэффективной жидкостной хроматографии на колонке ProntoSIL, 75×2,0 мм, 5 мкм с градиентным режимом элюирования с использованием в качестве подвижной фазы А смеси 0,2 М раствора лития перхлората в 0,005 М растворе хлорной кислоты, подвижной фазы Б - ацетонитрила, с последующим УФ-детектированием при длине волны 330 нм.
| НОВОЕ БИОЛОГИЧЕСКИ АКТИВНОЕ СОЕДИНЕНИЕ N-[3-(4-НИТРОФЕНИЛАМИНО)-ИНДОЛ-2-ИЛМЕТИЛЕН] АМИНОГУАНИДИНА ГИДРОХЛОРИД С ПРОТИВОВОСПАЛИТЕЛЬНОЙ АКТИВНОСТЬЮ | 2010 |
|
RU2478618C2 |
| EA 201201286 A1, 30.04.2013 | |||
| US 5108930 A, 28.04.1992 | |||
| РЕМЕЗОВА И.П | |||
| и др | |||
| Разработка методик обнаружения некоторых атипичных нейролептиков для целей химико-токсикологического анализа, Фармация и фармакология, 2014, 6, 7, стр | |||
| Видоизменение прибора для получения стереоскопических впечатлений от двух изображений различного масштаба | 1919 |
|
SU54A1 |
| АРТАСЮК Е.М | |||
| Совершенствование методов анализа лекарственных средств, обладающих противовоспалительной активностью, Улан-Уде, 2009, автореф | |||
| дисс | |||
| кфн, найдено в Интернете 08.04.2019 [on line] на сайте https://www.dissercat.com/content/sovershenstvovanie-metodov-analiza-lekarstvennykh-sredstv-obladayushchikh-protivovospaliteln. | |||

