Изобретение относится к области медицины, а именно к патофизиологии, фармакологии и токсикологии, и может быть использовано для определения 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида как в различных биологических средах, так и в плазме крови, в частности, у больных в условиях различных неблагоприятных воздействий, включая побочное действие лекарственных средств.
2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорид относится к лекарствам нового поколения, являясь мультитаргетным лекарственным средством для коррекции когнитивных нарушений, например болезни Альцгеймера.
Одной из актуальных задач при проведении фармакокинетических исследований нового лекарственного средства является разработка аналитической методики количественного определения действующего вещества в биологических средах, в частности в плазме крови, для чего в первую очередь необходимо выделить его из плазмы крови. Для определения концентрации лекарственного средства в плазме крови могут быть использованы различные методы - физико-химические, иммунологические, микробиологические и т.д., обладающие специфичностью, высокой чувствительностью, точностью и воспроизводимостью.
Одним из самых распространенных и перспективных методов для изучения фармакокинетики лекарственного средства является метод высокоэффективной жидкостной хроматографии (ВЭЖХ). Метод ВЭЖХ обладает рядом преимуществ по сравнению с другими методами, а именно:
- возможностью разделения компонентов пробы при температурах, близких к комнатной, что делает метод ВЭЖХ особенно пригодным, а зачастую единственным методом исследования лабильных соединений;
- высокой скоростью анализа. Обычно разделение компонентов сложной смеси занимает несколько минут;
- эффективностью разделения до 150000 ТТ (теоретическая тарелка) на 1 м хроматографической колонки;
- высокой чувствительностью, которая позволяет определять микроколичества вещества в сложных смесях и т.д
В настоящее время не существует способов определения 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида в биологических средах.
Известен для производных тиадозолов только способ обнаружения остаточного метил-тиадиазола, содержащегося в кислотах табака, фруктов и овощей методом экстракции из твердой фазы и последующей высокоэффективной жидкостной хроматографии (патент CN 103163246 (B), МПК G01 N30/02. Опуб. 2014-07-23).
Однако для определения 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида он не применим.
Известен принятый за прототип способ количественного определения лекарственного средства - гистамина, с использованием метода ВЭЖХ, в котором используют в качестве элюэнта - смесь 0,1 М раствора дигидрофосфата натрия и ацетонитрила при градиенте концентрации последнего от 0 до 50% и скорости подачи потока элюента 90 мкл/мин, объеме элюирования 1500 мкл, температура хроматографической колонки 35°C, детекцию проводят при длине волны 210 нм (патент RU №2302632, МПК G01N 33/50 (2006.01), опуб. 10.07.2007).
Однако для определения 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида он так же не применим.
Предлагаемое изобретение решает задачу разработки нового высокоэффективного способа количественного определения лекарственного средства для коррекции когнитивных нарушений в биологических средах - 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида.
Поставленная задача решается способом количественного определения 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида в биологических средах, включающим экстракцию определяемого вещества ацетонитрилом из щелочной среды, с последующим определением его методом высокоэффективной жидкостной хроматографии с использованием в качестве элюента смеси 0,5% раствора, содержащего калия гидрофосфат однозамещенный:метанол:триэтиламин в пропорции 74:25:1, с метанолом при градиенте концентрации последнего от 10 до 70%, при этом pH смеси составляет 3,3.
Наиболее оптимальными, но не необходимыми режимами высокоэффективной жидкостной хроматографии при этом являются:
скорость подачи потока элюента 0,1 мл/мин;
объем пробы 20 мкл;
температура хроматографической колонки 35°C.
Отсутствие источников информации, содержащих ту же совокупность признаков, что и в разработанном способе, сообщает ему соответствие критерию «новизна».
Та же совокупность признаков позволяет получить новый непредсказуемый эффект - разработка высокоэффективного способа количественного определения в биологических средах нового лекарственного средства - 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида (далее субстанция 1) и, таким образом, сообщает ей соответствие критерию «изобретательский уровень».
Проведение количественного определения нового лекарственного средства с использованием известного оборудования с помощью известных препаратов сообщает разработанному способу соответствие критерию «промышленная применимость».
Итак, разработанный способ соответствует всем 3 критериям охраноспособности изобретения и может быть квалифицирован как изобретение.
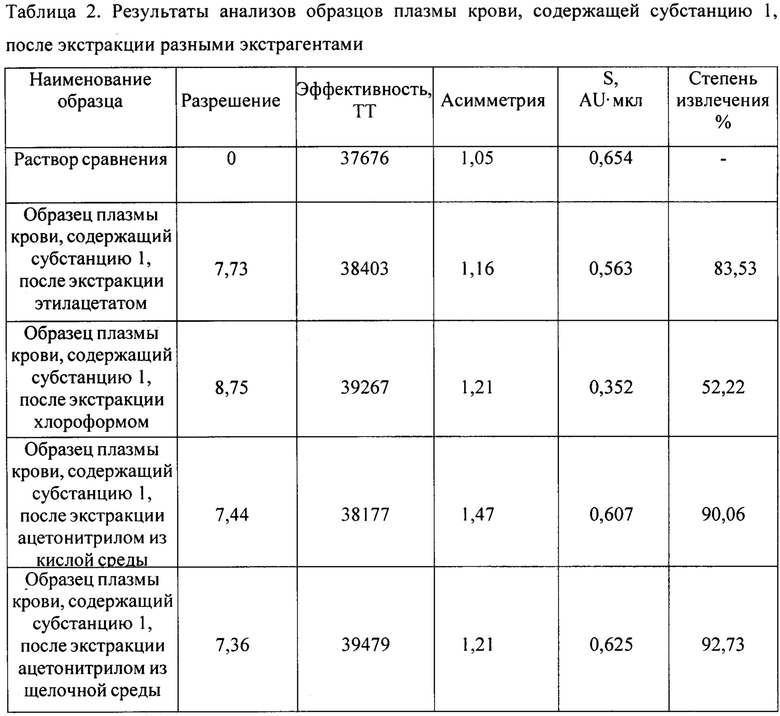
На Фиг. 1 представлена хроматограмма испытуемого раствора субстанции 1 при условиях хроматографирования 1. Время удерживания пика субстанции 1 8,5 мин.
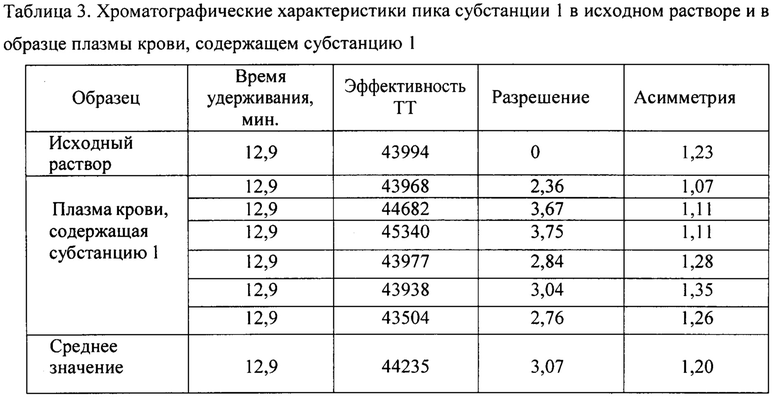
На Фиг. 2 представлена хроматограмма испытуемого раствора субстанции 1 при условиях хроматографирования 2. Время удерживания пика субстанции 1 14,7 мин.
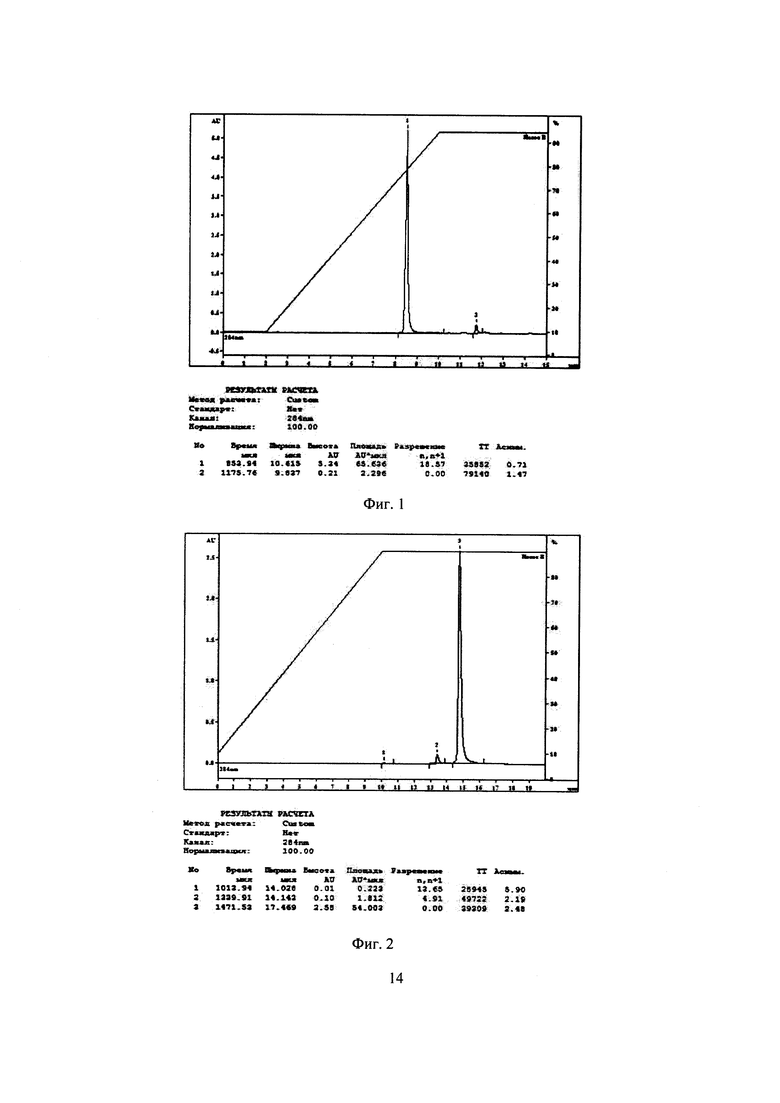
На Фиг. 3 представлена хроматограмма испытуемого раствора субстанции 1 при условиях хроматографирования 3. Время удерживания пика субстанции 1 7,5 мин.
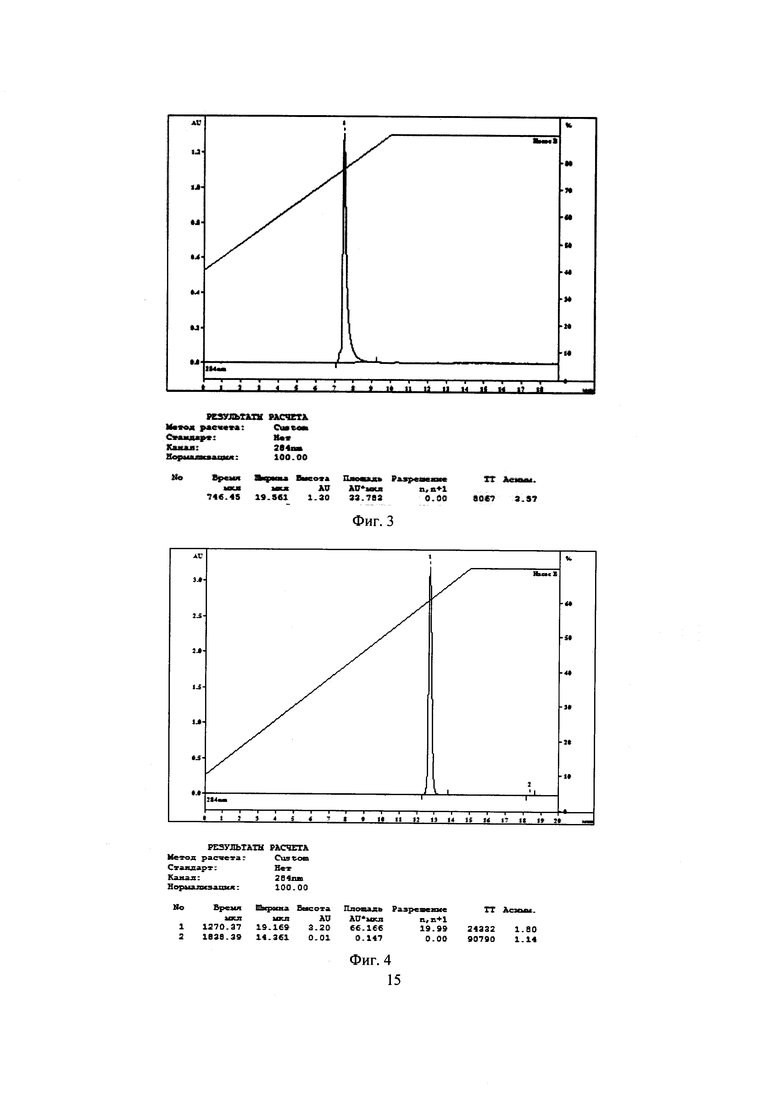
На Фиг. 4 представлена хроматограмма испытуемого раствора субстанции 1 при условиях хроматографирования 4. Время удерживания пика субстанции 1 12,7 мин.
В Таблице 1 представлены результаты анализов по подбору условий хроматографирования.
В Таблице 2 представлены результаты анализов образцов плазмы крови, содержащей субстанцию 1, после экстракции разными экстрагентами, полученные после обработки хроматограмм.
В Таблице 3 приведены хроматографические характеристики пика субстанции 1 в исходном растворе и в образце плазмы крови, содержащем субстанцию 1.
Пример 1. Подбор условий хроматографирования.
Условия хроматографирования считаются приемлемыми, если выполняются стандартные требования пригодности хроматографической системы. К параметрам пригодности хроматографической системы относятся: время удерживания определяемого компонента, которое должно находиться в приемлемом для анализа диапазоне (от 5 до 15 мин), эффективность хроматографической колонки, которая должна быть не менее 2000 ТТ, разрешение между пиками определяемого компонента и матрицы испытуемого образца, которое должно быть не менее 1,5, фактор ассиметрии хроматографического пика, который должен быть от 0,8 до 2,0.
На параметры пригодности хроматографической системы при проведении анализа методом ВЭЖХ влияют состав подвижной фазы (ПФ) и режим элюирования компонентов пробы.
С целью подбора оптимальных условий хроматографирования, отвечающих критериям проверки пригодности хроматографической системы, был приготовлен испытуемый раствор субстанции 1 концентрацией 0,1 мг/мл, и было апробировано несколько составов ПФ, состоящих из смеси растворов А и Б, и режимов элюирования.
Все испытания проводились в одинаковых условиях:
Скорость потока 0,1 мл/мин;
Температура колонки 35°C;
Детектор - спектрофотометрический, 284 нм;
Объем пробы 20 мкл.
1. ПФ - А - 0,025% раствор трифторуксусной кислоты;
- Б - ацетонитрил.
Режим элюирования - градиентный:
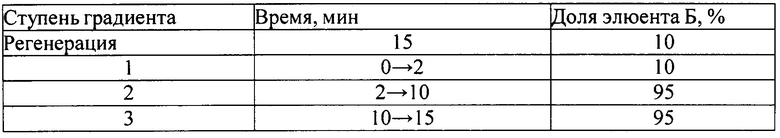
2. ПФ - А - 1% раствор натрия гидрофосфата однозамещенного (pH=8,3)
- Б - метанол.
Режим элюирования - градиентный:
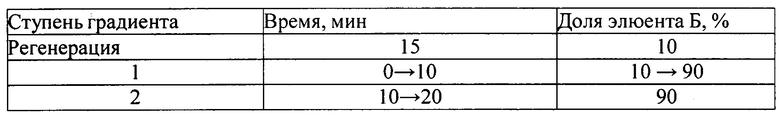
3. ПФ - А - 0,2 М раствор лития перхлората в 0,005 М растворе хлорной кислоты;
- Б - ацетонитрил.
Режим элюирования - градиентный:
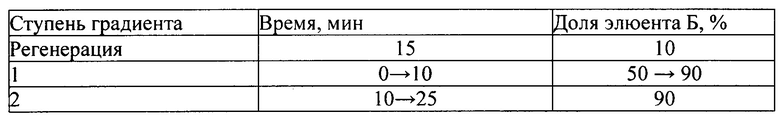
4. ПФ - А - 0,5% раствора, содержащий калия гидрофосфат однозамещенный:метанол: триэтиламин как 74:25:1;
- Б - метанол (pH=3,3).
Режим элюирования - градиентный:
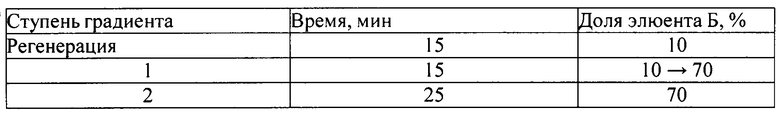
На Фиг. 1-4 приведены полученные хроматограммы, результаты анализов представлены в таблице 1.

Как видно из таблицы 1, параметры пика субстанции 1 при условиях хроматографирования в опытах 1-3 отвечают критериям приемлемости пригодности хроматографической системы, таким как время удерживания пика определяемого компонента и эффективность хроматографической колонки, но не отвечает критерию приемлемости пригодности хроматографической системы по фактору ассиметрии хроматографического пика, который должен быть от 0,8 до 2,0. При условиях хроматографирования опыта 4 параметры пригодности хроматографической системы отвечают критериям приемлемости: время удерживания пика определяемого компонента - 12,7 мин при норме от 5 до 15 мин, эффективность хроматографической колонки - 24332 ТТ при норме не менее 2000 ТТ, ассиметрия хроматографического пика - 1,8 при норме от 0,8 до 2,0.
Следовательно, условия хроматографирования в опыте 4 являются оптимальными для определения действующего вещества - субстанции 1 - в испытуемом растворе. Данные условия были нами использованы в дальнейших исследованиях по подбору условий пробоподготовки.
Пример 2. Подбор условий пробоподготовки.
Выбор оптимальных условий пробоподготовки основывался на показателе степени извлечения субстанции 1 из плазмы крови.
Для оценки степени извлечения субстанции 1 из плазмы крови в качестве раствора сравнения использовали раствор субстанции 1 с концентрацией 1000 нг/мл. В качестве экстрагентов использовали: этилацетат, хлороформ и ацетонитрил в кислой и щелочной средах.
Результаты анализов образцов плазмы крови, содержащей субстанцию 1, после экстракции разными экстрагентами, полученные после обработки хроматограмм, приведены в Таблице 2.
Как видно из таблицы 2, наилучшее извлечение субстанции 1 из плазмы крови достигается ацетонитрилом из щелочной среды, показатель степени извлечения составил 92,73%.
Пример 3. Количественное определение субстанции 1 в плазме крови.
Приготовление подвижной фазы А (ПФ А). 0,5 г (точная навеска) калия фосфорнокислого однозамещенного помещают в мерную колбу вместимостью 100 мл, прибавляют 50 мл воды очищенной и перемешивают до полного растворения, доводят объем раствора тем же растворителем до метки, перемешивают.
Берут 74 мл 0,5% раствора калия фосфорнокислого однозамещенного, помещают в стеклянную емкость вместимостью 150 мл, прибавляют 25 мл метанола и перемешивают. Затем прибавляют 1 мл триэтиламина и тщательно перемешивают. Таким образом соотношение компонентов в ПФ А составляет: 0,5% раствор калия гидрофосфата однозамещенного:метанол: триэтиламин в пропорции 74:25:1. pH полученного раствора доводят потенциометрически до значения 3,3 ортофосфорной кислотой.
Приготовление подвижной фазы Б (ПФ Б). 100 мл метанола помещают в стеклянную емкость объемом 250 мл, pH метанола доводят потенциометрически до значения 3,3 ортофосфорной кислотой.
Для приготовления ацетонитрила в щелочной среде готовят 20% раствор натрия гидроксида. Берут 20 г (точная навеска) натрия гидроксида, помещают в мерную колбу вместимостью 100 мл, прибавляют 50 мл воды очищенной, перемешивают до полного растворения, доводят объем раствора тем же растворителем до метки, перемешивают.
Приготовление растворов субстанции 1. 0,01 г (точная навеска) субстанции 1 помещают в мерную колбу вместимостью 10 мл, прибавляют 5 мл подвижной фазы А, перемешивают до полного растворения, доводят объем раствора до метки тем же растворителем, перемешивают. 1 мл полученного раствора помещают в мерную колбу вместимость 10 мл, доводят до метки ПФ А, перемешивают.
Из раствора субстанции 1 концентрацией 0,1 мг/мл методом разведения готовят растворы субстанции 1 концентрацией: 10 мкг/мл, 7,5 мкг/мл, 5,0 мкг/мл, 2,5 мкг/мл, 0,5 мкг/мл. Растворы используют свежеприготовленными.
Приготовление калибровочных растворов. В пластиковую микропробирку типа «Эппендорф» вместимостью 2 мл помещают 450 мкл плазмы крови и прибавляют 50 мкл раствора субстанции 1, соответствующей концентрации, каждый раствор перемешивают в течение 5 мин. Затем вносят 50 мкл 20% раствора натрия гидроксида, перемешивают в течение 3 мин. Прибавляют 1000 мкл ацетонитрила, перемешивают в течение 10 мин. К раствору добавляют 0,5 г натрия хлорида и перемешивают в течение 5 мин. Полученный раствор центрифугируют при 13000 об/мин в течение 10 мин. Водную фазу отбрасывают, а органическую упаривают на вакуумном концентраторе при температуре 45°C в течение 90 мин. К сухому остатку прибавляют 200 мкл ПФ А, перемешивают в течение 5 мин и центрифугируют при 13000 об/мин в течение 15 мин.
Для хроматографического анализа используют надосадочную жидкость.
Полученные калибровочные растворы, содержащие субстанцию 1 в концентрации: 0,05 мкг/мл, 0,25 мкг/мл, 0,5 мкг/мл, 0,75 мкг/мл, 1,0 мкг/мл, анализируют методом ВЭЖХ.
Для проверки пригодности хроматографической системы используют раствор субстанции 1 концентрацией 0,5 мкг/мл. Подготовку пробы проводят также, как и приготовление калибровочных растворов.
Условия хроматографирования.
Хроматограф - жидкостной с УФ-детектором с переменной длиной волны;
Колонка - 75×2 мм с октадецил-модифицированным силикагелем (С18), 5 мкм;
ПФ - А - 0,5% раствор калия гидрофосфата однозамещенного:метанол:триэтиламин в соотношении (74:25:1) (pH=3,3);
- Б - метанол (pH=3,3).
Режим элюирования - градиентный:

Скорость потока 0,1 мл/мин;
Температура колонки 35°C;
Детектор - спектрофотометрический, 284 нм;
Объем пробы 20 мкл.
Хроматографируют раствор для проверки пригодности хроматографической системы. Калибровочные растворы хроматографируют не менее 3 раз.
По результатам анализа калибровочных растворов строят калибровочные кривые зависимости площади пика анализируемого вещества от его концентрации в растворе. Построение калибровочной кривой осуществляют на 3-х сериях калибровочных растворов.
Проверку пригодности хроматографической системы определяют по совпадению времени удерживания пика субстанции 1 на хроматограмме образца плазмы крови, содержащего субстанцию 1, и исходного раствора субстанции 1.
Хроматографические характеристики пика субстанции 1 в исходном растворе и в образце плазмы крови, содержащем субстанцию 1, приведены в таблице 3.
Концентрацию действующего вещества субстанции 1 в исследуемых образцах определяют по калибровочной кривой.
Как видно из таблицы 3, эффективность хроматографической колонки при анализе образцов плазмы крови, содержащих субстанцию 1, изменяется от 43504 до 45340 теоретических тарелок (ТТ), при норме не менее 2000 ТТ. Разрешение для пика субстанции 1 относительно ближайшего пика варьируется от 2,36 до 3,75 единиц, при норме не менее 1,5 единиц, то есть пик субстанции 1 достаточно отделен от других компонентов плазмы крови. Показатель асимметрии пика субстанции 1 находится в допустимых пределах от 1,07 до 1,35 единиц, при норме от 0,8 до 2,0 единиц.
Валидация аналитической методики количественного определения субстанции 1 в плазме крови.
Валидация - это процесс экспериментального подтверждения того, что аналитическая методика обеспечивает получение необходимой и достоверной информации об объекте анализа и пригодна для практического использования.
В результате проведенных исследований по стандартным для ВЭЖХ методикам доказаны:
- селективность предлагаемого способа по отсутствию пиков веществ, совпадающих по времени удерживания с пиком субстанции 1;
- установлен предел обнаружения субстанции 1 в плазме крови по разработанной аналитической методике, который составляет 0,03 мкг/мл,
- и нижний предел количественного определения, который составляет 0,05 мкг/мл;
- линейность аналитической методики подтверждена в диапазоне концентраций от 0,05 мкг/мл до 1,0 мкг/мл. Результаты обратного расчета концентраций калибровочных растворов показали, что разработанная аналитическая методика достоверна и пригодна для количественного определения субстанции 1 в рассматриваемом диапазоне;
- прецизионность разработанной методики подтверждена коэффициентом вариации от 1,64% до 5,65%, что удовлетворяет критерию приемлемости методики. Коэффициент вариации не должен превышать 15% для всех калибровочных точек, кроме точки на нижнем пределе количественного определения, для которой коэффициент вариации не должен превышать 20%;
- точность определений находится в диапазоне 93,5-112,9%, как и требуется по критериям оценки 100±15% для всех калибровочных точек, кроме точки на нижнем пределе количественного определения, для которой допускается отклонение 100±20%;
- показано отсутствие переноса субстанции 1 на образец холостой плазмы;
- стабильность субстанции 1 в плазме крови подтверждена тем, что хранение образцов плазмы крови, содержащих субстанцию 1, в течение 24 ч в холодильнике, при комнатной температуре, при многократном замораживании и размораживании не приводит к деградации субстанции 1 в образцах плазмы крови.
Проведенные валидационные тесты доказывают, что заявляемый способ обеспечивает возможность его эффективного использования при проведении фармакокинетических исследований.
Как видно из приведенных примеров, предлагаемое изобретение позволяет получить с высокой точностью и селективностью необходимую и достоверную информацию количественного определения субстанции 1 - 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида в биологических средах.
| название | год | авторы | номер документа |
|---|---|---|---|
| Способ количественного определения хлорида 2-[(Z)-1-(3,5-дифенил-1,3,4-тиадиазол-2(3Н)-илиден)метил]-3,5-дифенил-1,3,4-тиадиазол-3-ия в биологических объектах | 2019 |
|
RU2702330C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ N-[3-(4-НИТРОФЕНИЛАМИНО)-ИНДОЛ-2-ИЛМЕТИЛЕН]АМИНОГУАНИДИНА МЕТАНСУЛЬФОНАТА В ПЛАЗМЕ КРОВИ | 2018 |
|
RU2699550C1 |
| СПОСОБ КОЛИЧЕСТВЕННОГО ОПРЕДЕЛЕНИЯ 4-АМИНО-1-(3-НИТРО-2-ОКСО-1-ФЕНИЛ-1,2-ДИГИДРО-1,6-НАФТИРИДИН-5-ИЛ)ПИРИДИНИЙ ХЛОРИДА В БИОЛОГИЧЕСКИХ СРЕДАХ | 2018 |
|
RU2686116C1 |
| Способ количественного определения 2-изопропил-5-метилциклогексил 2-(1-(4-хлорбензоил)-5-метокси-2-метил-1Н-индол-3-ил)ацетата и индометацина в плазме крови | 2021 |
|
RU2781342C1 |
| СОКРИСТАЛЛИЧЕСКАЯ ФОРМА 1-[5-(4-ХЛОРФЕНИЛАМИНО)-1,2,4-ТИАДИАЗОЛ-3-ИЛ]-ПРОПАН-2-ОЛА | 2018 |
|
RU2712443C1 |
| Способ количественного анализа 3-нитро-1,2,4-триазола | 1986 |
|
SU1383200A1 |
| 5-АМИНО-3-(2-АМИНОПРОПИЛ)-[1,2,4]ТИАДИАЗОЛЫ | 2011 |
|
RU2449997C1 |
| СОКРИСТАЛЛИЧЕСКАЯ ФОРМА 1-[(5-ПАРА-МЕТИЛ-МЕТА-ХЛОР-ФЕНИЛАМИНО)-1,2,4-ТИАДИАЗОЛ-3-ИЛ]-ПРОПАН-2-ОЛА | 2019 |
|
RU2721335C1 |
| Способ количественного определения леводопы в плазме крови | 2017 |
|
RU2665164C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ ЭНАНТИОМЕРОВ ВЕРАПАМИЛА В СУБСТАНЦИЯХ, ТАБЛЕТКАХ И ОБРАЗЦАХ ПЛАЗМЫ КРОВИ МЕТОДОМ ВЭЖХ | 2008 |
|
RU2395807C1 |
Изобретение относится к области медицины, а именно к патофизиологии, фармакологии и токсикологии, и касается определения 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-л}пиперидин-4-амина дигидрохлорида в различных биологических средах, в частности в плазме крови у больных в условиях различных неблагоприятных воздействий, включая побочное действие лекарственных средств. Способ включает экстракцию определяемого вещества ацетонитрилом из щелочной среды с последующим определением его методом высокоэффективной жидкостной хроматографии с использованием в качестве элюента смеси 0,5% раствора, содержащего калия гидрофосфат однозамещенный:метанол:триэтиламин в пропорции 74:25:1, с метанолом при градиенте концентрации последнего от 10 до 70%, при этом pH смеси составляет 3,3. 3 з.п. ф-лы, 4 ил., 3 табл.
1. Способ количественного определения 2,2,6,6-тетраметил-N-{1-[5-(4-метил-3-хлоранилино)-1,2,4-тиадиазол-3-ил]пропан-2-ил}пиперидин-4-амина дигидрохлорида в биологических средах, включающий экстракцию определяемого вещества ацетонитрилом из щелочной среды, с последующим определением его методом высокоэффективной жидкостной хроматографии с использованием в качестве элюента смеси 0,5% раствора, содержащего калия гидрофосфат однозамещенный:метанол:триэтиламин в пропорции 74:25:1, с метанолом при градиенте концентрации последнего от 10 до 70%, при этом рН смеси составляет 3,3.
2. Способ по п. 1, отличающийся тем, что скорость подачи потока элюента составляет 0,1 мл/мин.
3. Способ по п. 1, отличающийся тем, что объем пробы составляет 20 мкл.
4. Способ по п. 1, отличающийся тем, что температура хроматографической колонки составляет 35°C.
| СПОСОБ ОПРЕДЕЛЕНИЯ ХЛОРАНИЛИНОВ В ВОДНЫХ СРЕДАХ | 2010 |
|
RU2458343C2 |
| CN 103163246 A, 19.06.2013 | |||
| СПОСОБ ОПРЕДЕЛЕНИЯ ГИСТАМИНА В ПЛАЗМЕ КРОВИ | 2005 |
|
RU2302632C2 |

