Перекрестная ссылка на родственную заявку
Данная заявка заявляет приоритет предварительной заявки США с регистрационным номером 60/532031, поданной 12 декабря 2003 г, полное раскрытие которой включено в данную работу посредством ссылки.
Область техники, к которой относится изобретение
Данное изобретение относится в основном к антагонистам CRF-рецепторов и к способам лечения расстройств введением таких антагонистов млекопитающему, нуждающемуся в этом.
Уровень изобретения
Впервые кортикотропинрилизинг-фактор (CRF) был выделен из овечьего гипоталамуса и идентифицирован как 41-аминокислотный пептид (Vale et al., Science 213:1394-1397, 1981). Затем были выделены последовательности CRF человека и крысы и определена их идентичность, но с отличием от овечьего CRF в 7 из 41 аминокислотного остатка (Rivier et al., Proc. Natl. Acad. Sci. USA 80:4851, 1983; Shibahara et al., EMBO J. 2:775, 1983).
Было установлено, что CRF вызывает глубокие изменения в функции эндокринной, нервной и иммунной систем. Полагают, что CRF является главным физиологическим регулятором базального и высвобождаемого при стрессе адренокортикотропного гормона (“АСТН”), β-эндорфина и других пептидов, производных проопиомеланокортина (“РОМC”) из аденогипофиза (Vale et al., Science 213:1394-1397, 1981). Короче, предполагается, что CRF инициирует свои биологические эффекты связыванием с плазменным мембранным рецептором, который, как было установлено, распределен в головном мозге (DeSouza et al., Science 224:1449-1451, 1984), гипофизе (DeSouza et al., Methods Enzymol. 124:560, 1986; Wynn et al., Biochem. Biophys. Res. Comm. 110:602-608, 1983), надпочечниках (Udelsman et al., Nature 319:147-150, 1986) и селезенке (Webster, E.L. and E.B. DeSouza, Endocrinology 122:609-617, 1988). СRF-рецептор сочетается с GTP-связывающим протеином (Perrin et al., Endocrinology 118:1171-1179, 1986), который опосредует CRF-стимулируемое повышение внутриклеточного образования сАМР (Bilezikjian, L.M., and W.W. Vale, Endocrinology 113:657-662, 1983). Рецептор для CRF в настоящее время клонирован от крысы (Perrin et al., Endo 133(6):3058-3061, 1993) и головного мозга человека (Chen et al., PNAS 90(19):8967-8971, 1993; Vita et al., FEBS 335(1):1-5, 1993). Данный рецептор представляет собой протеин из 415 аминокислот, содержащий семь мембранных протяженных доменов. Сравнение идентичности между последовательностями крысы и человека показывает высокую степень гомологии (97%) на аминокислотном уровне.
В дополнение к своей роли в стимулировании образования АСТН и РОМС считается, что CRF координирует многие эндокринные, автономные и поведенческие реакции на стресс и может быть включен в патофизиологию аффективных расстройств. Кроме того, полагают, что CRF является ключевым промежуточным звеном в связи между иммунной, центральной нервной, эндокринной и сердечно-сосудистой системами (Crofford et al., J. Clin. Invest. 90:2555-2564, 1992; Sapolsky et al., Science 238:522-524, 1987; Tilders et al., Regul. Peptides 5:77-84, 1982). В целом, CRF, по-видимому, является одним из основных нейротрансмиттеров центральной нервной системы и играет решающую роль в объединении общего ответа организма на стресс.
Введение CRF непосредственно в головной мозг вызывает поведенческие, физиологические и эндокринные реакции, идентичные реакциям, наблюдаемым у животных под воздействием стрессов в окружающей среде. Например, интрацеребровентрикулярная инъекция CRF приводит к активации поведения (Sutton et al., Nature 297:331, 1982), стойкой активации электроэнцефалограммы (Ehlers et al., Brain Res. 278: 332, 1983), cтимулированию симпатоадреномедуллярного пути (Brown et al., Endocrinology 110:928, 1982), повышению частоты сердечных сокращений и кровяного давления (Fisher et al., Endocrinology 110:2222, 1982), повышению потребления кислорода (Brown et al., Life Sciences 30:207, 1982), изменению активности желудочно-кишечного тракта (Williams et al., Am. J. Physiol. 253:G582, 1987), подавлению потребления пищи (Levine et al., Neuropharmacology 22:337, 1983), изменению сексуального поведения (Sirinathsinghji et al., Nature 305:232, 1983) и иммунной функциональной недостаточности (Irwin et al., Am. J. Physiol. 255:R744, 1988). Кроме того, клинические данные свидетельствуют о том, что CRF может выделяться в повышенных количествах в головном мозге при депрессии, нарушениях, связанных с состояниями тревоги/страха, и анорексии невроза (DeSouza, Ann. Reports in Med. Chem. 25:215-223, 1990). В соответствии с этим клинические данные свидетельствуют о том, что антагонисты CRF-рецепторов могут представлять собой новые антидепрессантные и/или анксиолитические лекарственные средства, которые могут быть применимы в лечении нервно-психических расстройств, проявляющихся из-за гиперсекреции CRF.
Первые антагонисты CRF-рецепторов представляли собой пептиды (см., например, Rivier et al., U.S. Patent No. 4605642; Rivier et al., Science 224:889, 1984). Хотя данные пептиды показали, что антагонисты CRF-рецепторов могут ослаблять фармакологические реакции на CRF, пептидным антагонистам CRF-рецепторов свойственны обычные недостатки пептидных медикаментов, включающие недостаток стабильности и ограниченную пероральную активность. Некоторые из опубликованных патентных документов включают US 6313124, WO 01/23388 и WO 97/29109, все из которых раскрывают пиразолопиримидиновые соединения в качестве CRF-антагонистов. Опубликованная заявка WO 98/54093 описывает некоторые пиразолопиримидиновые соединения в качестве ингибиторов тирозинкиназы.
Благодаря физиологической роли CRF разработка биологически активных малых молекул, обладающих значительной связывающей активностью CRF-рецепторов и способных быть антагонистами CRF-рецепторов, остается актуальной задачей. Такие антагонисты CRF-рецепторов были бы полезны в лечении эндокринных, психических и неврологических состояний или заболеваний, включающих нарушения, связанные со стрессами вообще.
Хотя были сделаны значительные шаги в отношении достижения регулирования CRF путем введения антагонистов CRF-рецепторов, остается потребность в области эффективных малых молекул антагонистов CRF-рецепторов. Существует также потребность в отношении фармацевтических композиций, содержащих такие антагонисты CRF-рецепторов, а также способов, относящихся к их применению, для лечения, например, нарушений, связанных со стрессами. Данное изобретение восполняет указанные потребности и предоставляет другие сопутствующие преимущества.
Сущность изобретения
Вкратце, данное изобретение относится к антагонистам CRF-рецепторов и конкретнее к антагонистам CRF-рецепторов, имеющим следующую общую структуру (I):
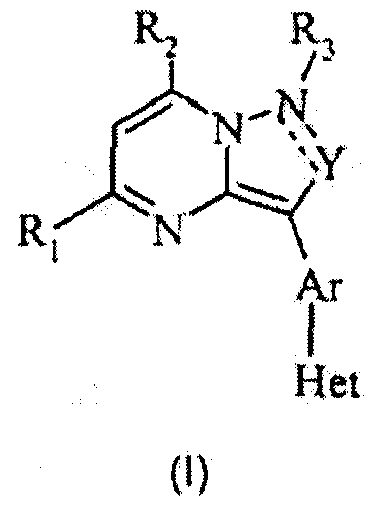
и их фармацевтически приемлемым солям, эфирам, сольватам, стереоизомерам и пролекарствам, где R1, R2, R3, Y, Ar и Het принимают значения, определенные ниже.
Антагонисты CRF-рецепторов могут найти применение в широком диапазоне терапевтических аппликаций и могут быть использованы для лечения многих расстройств или заболеваний, включающих нарушения, связанные со стрессами. Такие способы включают введение фармацевтически активного количества антагониста CRF-рецепторов, предложенного в данном изобретении, предпочтительно в форме фармацевтической композиции, животному, нуждающемуся в этом. Соответственно в другом варианте осуществления раскрываются фармацевтические композиции, содержащие один или несколько антагонистов CRF-рецепторов, предложенных в данном изобретении, и фармацевтически приемлемый носитель и/или разбавитель.
Данные и другие аспекты изобретения будут очевидными после отсылки к следующему подробному описанию. С этой целью в данной работе приводятся различные ссылки, которые описывают более подробно некоторые процедуры, соединения и/или композиции, и, таким образом, включаются ссылкой полностью.
Подробное описание изобретения
Данное изобретение относится в основном к антагонистам рецепторов кортикотропинрилизинг-фактора (CRF).
В первом варианте осуществления антагонисты CRF-рецепторов, предложенные в данном изобретении, имеют следующую структуру (I):
или представляют собой их фармацевтически приемлемую соль, эфир, сольват, стереоизомер или пролекарство,
где:
“---” представляет вторую связь необязательной двойной связи;
R1 представляет собой водород, алкил, замещенный алкил, гетероарил, замещенный гетероарил, -NH2 или галоген;
R2 представляет собой алкил, замещенный алкил, -С(О)NR7R8, арил, замещенный арил, арилоксиалкил, замещенный арилоксиалкил, гетероарилалкоксиалкил, замещенный гетероарилалкоксиалкил, гетероциклоалкил, замещенный гетероциклоалкил, арилалкил, замещенный арилалкил, гетероарил или замещенный гетероарил, где указанный гетероарил или замещенный гетероарил присоединен к пиримидиновому циклу через углерод-углеродную связь;
R3 отсутствует или представляет собой водород или алкил;
Y представляет собой =(СR4)- или -(С=О)-;
R4 представляет собой водород, алкил, замещенный алкил, тиоалкил, алкилсульфинил или алкилсульфонил;
Ar представляет собой фенил, фенил, замещенный 1 или 2 R5, пиридил или пиридил, замещенный 1 или 2 R5;
R5 в каждом случае представляет собой гидрокси, алкил, замещенный алкил, алкокси, замещенный алкокси, циано, галоген, алкилсульфонил или алкилсульфинил;
Het представляет собой гетероарил, необязательно замещенный 1 или 2 R6;
R6 в каждом случае представляет собой гидрокси, алкил, замещенный алкил, алкокси, замещенный алкокси, циано или галоген; и
R7 и R8 независимо представляют собой водород, алкил, замещенный алкил, арил, замещенный арил, гетероцикл, замещенный гетероцикл, арилалкил, замещенный арилалкил, гетероциклоалкил или замещенный гетероциклоалкил; или
R7 и R8 вместе с азотом, к которому они присоединены, образуют гетероциклическое кольцо или замещенное гетероциклическое кольцо.
Вышеупомянутые термины, использованные в данной работе, имеют следующее значение:
“Алкил” означает углеводород с прямой или разветвленной цепью, ациклический или циклический, насыщенный или ненасыщенный, содержащий от 1 до 10 атомов углерода, хотя термин “низший алкил” имеет то же значение, как и алкил, но содержит от 1 до 6 атомов углерода. Типичные насыщенные алкилы с прямой цепью включают метил, этил, н-пропил, н-бутил, н-пентил, н-гексил и тому подобное; в то время как насыщенные разветвленные алкилы включают изопропил, втор-бутил, изобутил, трет-бутил, изо-пентил и тому подобное. Типичные насыщенные циклические алкилы включают циклопропил, циклобутил, циклопентил, циклогексил, -СН2-циклопропил, -СН2-циклобутил, -СН2-циклопентил, -СН2-циклогексил и тому подобное; в то время как ненасыщенные циклические алкилы включают циклопентенил и циклогексенил и тому подобное. Циклические алкилы, также называемые как “гомоциклические кольца”, включают ди- и полигомоциклические кольца, такие как декалин и адамантил. Ненасыщенные алкилы содержат, по меньшей мере, одну двойную или тройную связь между соседними атомами углерода (называемые как “алкенил” или “алкинил” соответственно). Типичные алкенилы с прямой и разветвленной цепью включают этиленил, пропиленил, 1-бутенил, 2-бутенил, изобутиленил, 1-пентенил, 2-пентенил, 3-метил-1-бутенил, 2-метил-2-бутенил, 2,3-диметил-2-бутенил и тому подобное; в то время как типичные алкинилы с прямой и разветвленной цепью включают ацетиленил, пропинил, 1-бутинил, 2-бутинил, 1-пентинил, 2-пентинил, 3-метил-1-бутинил и тому подобное.
“Алкилиденил” представляет собой дивалентный алкил, для образования которого два атома водорода взяты от одного и того же атома углерода, такой как =СН2, =СНСН3, =СНСН2СН3, =С(СН3)СН2СН3 и тому подобное.
“Арил” означает ароматический карбоциклический фрагмент, такой как фенил или нафтил.
“Арилалкил” означает алкил, содержащий, по меньшей мере, один алкильный атом водорода, замещенный арилом, такой как бензил (т.е. -СН2фенил, -СН2-(1- или 2-нафтил), -(СН2)2фенил, -(СН2)3фенил, -СН(фенил)2 и тому подобное.
“Арилоксиалкил” означает арил, присоединенный через кислородный мостик к алкилу (т.е. арил-О-алкил-), такой как -метил-О-фенил и т.п.
“Гетероарил” означает ароматическое гетероциклическое кольцо из 5-10 членов, содержащее, по меньшей мере, один гетероатом, выбранный из азота, кислорода и серы, и содержащее, по меньшей мере, один атом углерода, включающее как моно-, так и бициклические кольцевые системы. Типичные гетероарилы включают (но без ограничения только ими) фурил, бензофуранил, тиофенил, бензотиофенил, пирролил, индолил, изоиндолил, азаиндолил, пиридил, хинолинил, изохинолинил, оксазолил, изооксазолил, бензоксазолил, пиразолил, имидазолил, бензимидазолил, тиазолил, бензотиазолил, изотиазолил, пиридазинил, пиримидинил, пиразинил, триазинил, циннолинил, фталазинил и хиназолинил.
“Гетероарилалкил” означает алкил, содержащий, по меньшей мере, один алкильный атом водорода, замещенный гетероарилом, такой как -СН2-пиридинил, -СН2-пиримидинил и тому подобное.
“Гетероцикл” (также называемый в настоящей работе как “гетероциклическое кольцо”) означает 5-7-членное моноциклическое, или 7-14-членное полициклическое, гетероциклическое кольцо, которое является либо насыщенным, либо ненасыщенным или ароматическим и которое содержит от 1 до 4 гетероатомов, независимо выбранных из азота, кислорода и серы, и в котором гетероатомы азота и серы могут быть необязательно окисленными, и гетероатом азота может быть необязательно кватернизованным, включающий бициклические кольца, в которых любые из вышеуказанных гетероциклов являются конденсированными с бензольным кольцом, а также трициклическими (и более) гетероциклическими кольцами. Гетероцикл может быть присоединен через гетероатом или атом углерода. Гетероциклы включают гетероарилы, определенные выше. Таким образом, в дополнение к ароматическим гетероарилам, перечисленным выше, гетероциклы также включают (но без ограничения только ими) морфолинил, пирролидинонил, пирролидинил, пиперидинил, пиперазинил, гидантоинил, валеролактамил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, тетрагидропиридинил, тетрагидропиримидинил, тетрагидротиофенил, тетрагидротиопиранил и тому подобное.
“Гетероциклоалкил” означает алкил, содержащий, по меньшей мере, один алкильный атом водорода, замещенный гетероциклом, такой как -СН2-морфолинил, и тому подобное.
Термин “замещенный”, используемый в данной работе, относится к любой группе (например, алкилу, арилу, арилалкилу, гетероарилу, гетероарилалкилу, гетероциклу или гетероциклоалкилу), в которой, по меньшей мере, один атом водорода замещен заместителем. В случае кетозаместителя (“-С(=О)-”) замещены два атома водорода. “Заместители” по тексту данного изобретения включают галоген, гидрокси, циано, нитро, амино, алкиламино, диалкиламино, алкил, алкокси, тиоалкил, галогеналкил, гидроксиалкил, арил, замещенный арил, арилалкил, замещенный арилалкил, гетероарил, замещенный гетероарил, гетероарилалкил, замещенный гетероарилалкил, гетероцикл, замещенный гетероцикл, гетероциклоалкил, замещенный гетероциклоалкил, -NRaRb, -NRaC(=O)Rb, -NRaC(=O)NRaRb, -NRaC(=O)ORb, -NRaSO2Rb, -ORa, -C(=O)Ra, -C(=O)ORa, -C(=O)NRaRb, -OC(=O)NRaRb, -SH, -SRa, -SORa, -S(=O)2Ra, -OS(=O)2Ra, -S(=O)2ORa, где Ra и Rb одинаковые или различные и независимо представляют собой водород, алкил, галогеналкил, замещенный алкил, арил, замещенный арил, арилалкил, замещенный арилалкил, гетероарил, замещенный гетероарил, гетероарилалкил, замещенный гетероарилалкил, гетероцикл, замещенный гетероцикл, гетероциклоалкил или замещенный гетероциклоалкил.
“Галоген” означает атом фтора, хлора, брома или иода.
“Галогеналкил” означает алкил, содержащий, по меньшей мере, один атом водорода, замещенный галогеном, такой как трифторметил и тому подобное. Галогеналкил представляет собой конкретный вариант осуществления замещенного алкила, где алкил замещен одним или несколькими атомами галогена.
“Алкокси” означает алкил, присоединенный через кислородный мостик (т.е. -О-алкил), такой как -О-метил, -О-этил, и тому подобное.
“Тиоалкил” означает алкил, присоединенный через серный мостик (т.е. -S-алкил), такой как -S-метил, -S-этил, и тому подобное.
“Алкиламино” и “диалкиламино” означают одну или две алкильные группы, присоединенные через азотный мостик (т.е. -NHалкил или -N(алкил)(алкил)), такие как метиламино, этиламино, диметиламино, диэтиламино, и тому подобное.
“Гидроксиалкил” означает алкил, замещенный, по меньшей мере, одной гидроксигруппой.
“Моно- или ди(циклоалкил)метил” представляет собой метильную группу, замещенную одной или двумя циклоалкильными группами, такой как циклопропилметил, дициклопропилметил и тому подобное.
“Алкилкарбонилалкил” представляет собой алкил, замещенный -С(=О)алкильной группой.
“Алкилкарбонилоксиалкил” представляет собой алкил, замещенный -С(=О)Оалкильной группой или -ОС(=О)алкильной группой.
“Алкоксиалкил” представляет собой алкил, замещенный -О-алкильной группой.
“Алкилтиоалкил” представляет собой алкил, замещенный -S-алкильной группой.
“Моно- или ди(алкил)амино” представляет собой аминогруппу, замещенную одним или двумя алкилами соответственно.
“Моно- или ди(алкил)аминоалкил” представляет собой алкил, замещенный моно- или ди(алкил)аминогруппой.
“Алкилсульфонил или алкилсульфинил” представляет собой алкил, замещенный (-S(=O)2-) или (-S(=O)-) функциональными группами соответственно.
Варианты осуществления данного изобретения даются как пример и не предназначены для ограничения. В первом варианте осуществления изобретения R3 отсутствует и Y означает =(СR4)- в приведенной структуре (II), и в следующем варианте осуществления Y означает -(С=О)- в приведенной структуре (III).
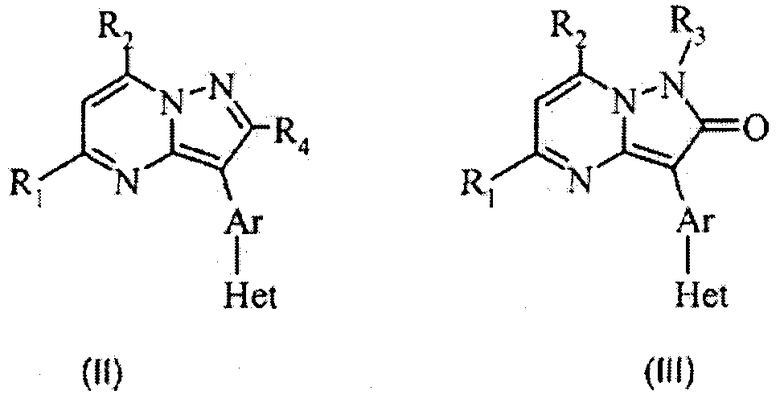
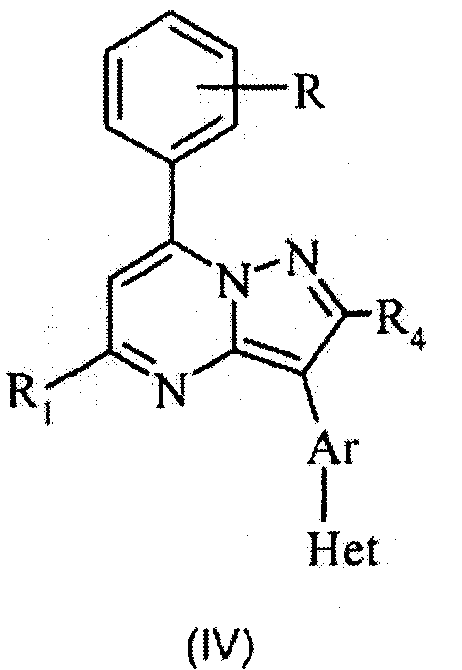
Следующие варианты осуществления относятся к структуре (IV), где R2 означает фенил, R представляет собой необязательный заместитель указанного фенила, и Y означает =(СR4)-.
В следующих вариантах осуществления данного изобретения, где Y означает =(СR4)-, Ar представляет собой фенил, замещенный на 2 R5 в структуре (V), и Het представляет собой пиридил, замещенный на 1 R6 в структуре (VI).

Cоединения данного изобретения, в основном, могут быть применены в виде свободного основания. В альтернативном случае соединения данного изобретения могут быть использованы в виде кислотно-аддитивных солей. Кислотно-аддитивные соли свободно-основных аминосоединений данного изобретения могут быть приготовлены способами, хорошо известными в данной области, и могут быть получены с органическими и неорганическими кислотами. Подходящие органические кислоты включают малеиновую, фумаровую, бензойную, аскорбиновую, янтарную, метансульфоновую, уксусную, щавелевую, пропионовую, винную, салициловую, лимонную, глюконовую, молочную, миндальную, коричную, аспаргиновую, стеариновую, пальмитиновую, гликолевую, глутаминовую и бензолсульфоновую кислоты. Подходящие неорганические кислоты включают хлористоводородную, бромистоводородную, серную, фосфорную и азотную кислоты. Таким образом, термин “фармакологически приемлемая соль” структуры (I) предназначена для охвата любых и всяких фармацевтически приемлемых солевых форм.
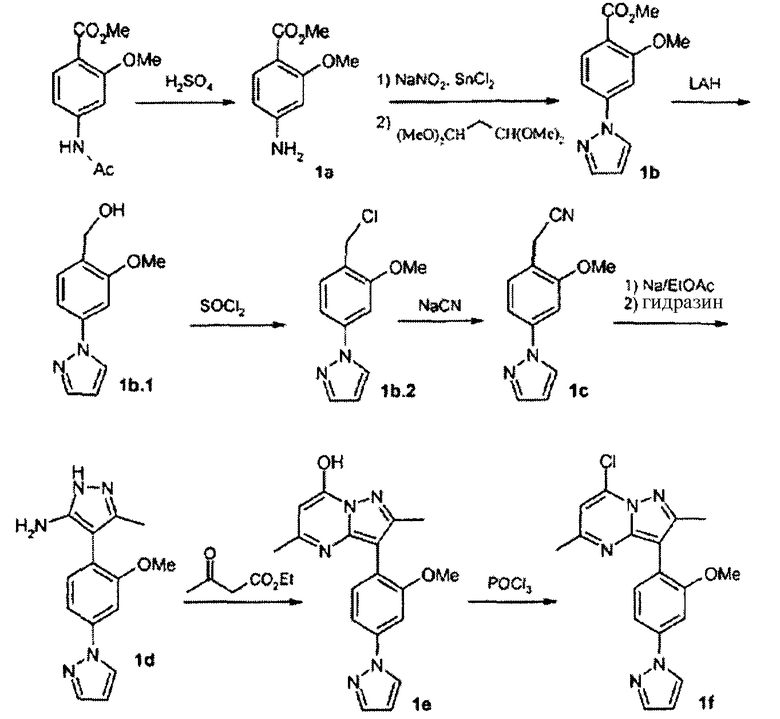
Вообще, соединения структуры (I) могут быть сделаны по методикам органического синтеза, известным специалистам в данной области, а также представленными способами, изложенными в примерах. Примеры синтетических процедур, которые могут быть использованы для приготовления соединений по данному изобретению, проиллюстрированы на схемах реакций 1-3.
Схема реакций 1
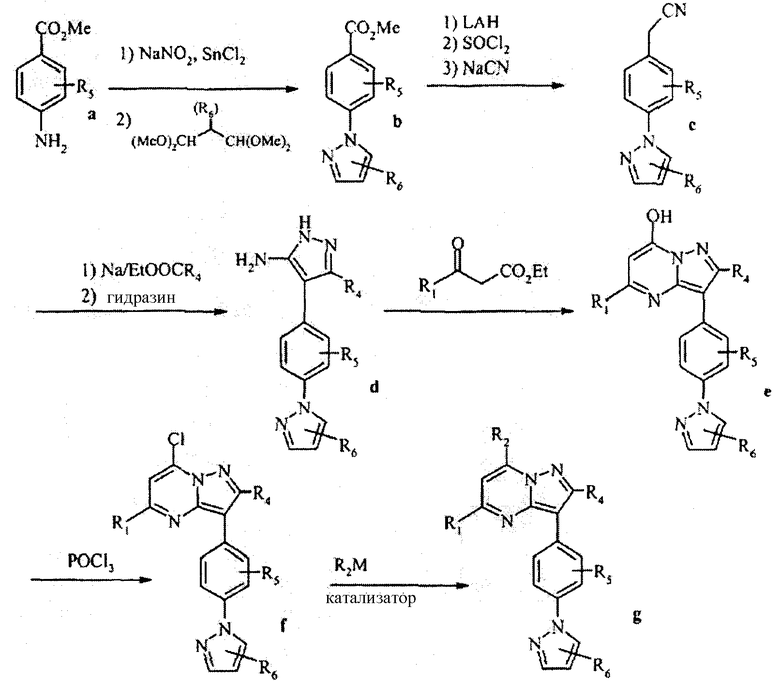
Функциональная аминогруппа 4-аминобензоата а может конденсироваться с необязательно замещенным малоновым альдегидом, давая соответствующий 4-пиразол-1-илбензоат b. После взаимодействия последнего соединения с LAH, SOCl2 и NaCN с превращением его в пиразолофенилацетонитрильное соединение с, реакция с Na/этиловым эфиром карбоновой кислоты и гидразином приводит к бис-пиразолу d. Взаимодействие с соответственно замещенным сложным β-кетоэфиром дает пиразолопиримидин е, который реагирует с POCl3 с образованием хлорида f. Взаимодействие хлорида f с соответствующим металлоорганическим реагентом R2M в присутствии подходящего катализатора или промотора дает соединение g. Примеры подходящих металлоорганических реагентов и подходящих катализаторов/промоторов включают:
1. (замещенные)алкилреагенты Гриньяра R2MgX (Fe(acac)3промотор);
2. арил, гетероарил или алкенилбороновые кислоты или сложные эфиры (Pd(PhP)4 катализатор); и
3. арил или гетероарил цинксодержащие реагенты (Pd(PhP)4 катализатор).
R2 группы, введенные таким образом, затем могут быть подвержены превращению или взаимодействию при использовании стандартных способов, известных специалистам в данной области (например, окисление/восстановление, гидролиз и тому подобное), для обеспечения следующих примеров данного изобретения.
Схема реакций 2
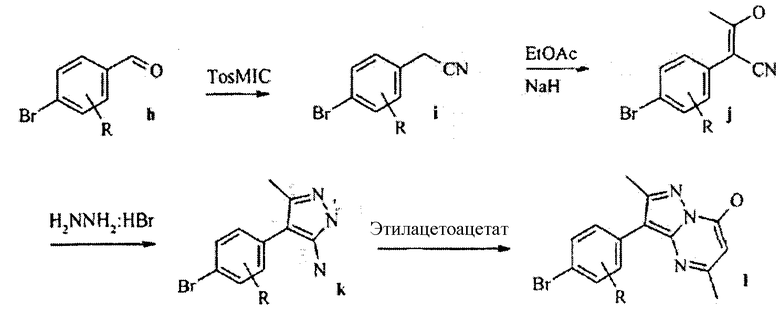
Многостадийные синтетические пути к пиразолопиримидиновому ядру изобретения являются доступными. По схеме реакций 2 необязательно замещенный галогенбензальдегид h реагирует с тозилметилизоцианидом (TosMIC) с образованием фенилацетонитрила i. Взаимодействие i с NaH и EtOAc дает 3-гидроксибут-2-еннитрил j, который претерпевает замыкание цикла в реакции с гидразином HBr, образуя 3-амино-2-фенилпиразол k. Добавление сложного β-кетоэфира дает пиразоло[1,5-a]пиримидин-7-ол l. Замещение кислорода так же, как по схеме реакций 1, и замещение периферического брома на Het приводит к соединениям по данному изобретению.
Схема реакций 3
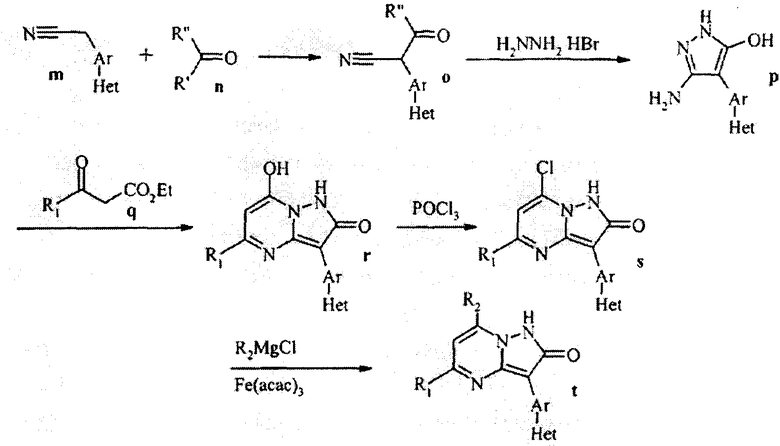
Взаимодействие замещенного ацетонитрила m с кетоном n, где R' представляет собой легко удаляемую группу, такую как алкокси, циано, или галоген, и где R'' представляет собой группу, такую как гидрокси или алкокси, дает цианокетон о, который реагирует с гидразином с образованием замещенного пиразола р. Взаимодействие р со сложным β-кетоэфиром q дает пиразолопиримидин r. Реакция с POCl3 приводит к хлориду s, и замещение хлорида на R2 дает соединение t.
Эффективность соединения как антагониста CRF-рецепторов может быть определена различными способами анализа. Подходящие CRF-антагонисты данного изобретения могут быть способны ингибировать специфическое связывание CRF с его рецептором и противодействовать эффектам, связанным с CRF. Соединение структуры (I) может быть оценено по активности в качестве CRF-антагониста одним или несколькими обычно принятыми для данной цели анализами, включающими (но без ограничения только ими) анализы, раскрытые в публикациях DeSouza et al. (J. Neuroscience 7:88, 1987) и Battaglia et al. (Synapse 1:572, 1987). Как упомянуто выше, подходящие CRF-антагонисты включают соединения, которые проявляют сродство к CRF-рецептору. Сродство к CRF-рецептору может быть определено исследованиями связывания, которые измеряют способность соединения ингибировать связывание меченного радиоактивным изотопом CRF (например, [125I]тирозин-CRF) с его рецептором (например, рецепторы, приготовленные из мембран церебрального кортекса крысы). Анализ радиолигандного связывания, описанный DeSouza et al. (см. выше, 1987), обеспечивает количественные данные для определения сродства соединения к CRF-рецептору. Такую активность обычно рассчитывают из IC50, как концентрации соединения, необходимой для замещения 50% меченного радиоактивным изотопом лиганда рецептора, и публикуют в виде “Ki” величины, вычисленной по следующему уравнению:
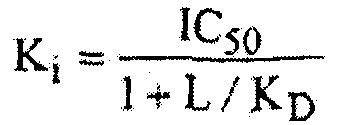
где L = радиолиганд и KD = cродство радиолиганда к рецептору (Cheng and Prussoff, Biochem. Pharmacol. 22:3099, 1973).
В дополнение к ингибированию связывания CRF-рецепторов активность соединения-антагониста CRF-рецепторов может быть установлена по способности соединения препятствовать активности, связанной с CRF. Например, известно, что CRF стимулируют различные биохимические процессы, включающие активность аденилатциклазы. Поэтому соединения могут быть оценены как CRF-антагонисты по их способности препятствовать СRF-стимулируемой активности аденилатциклазы, например, измерением уровней сАМР. Анализ по оценке СRF-стимулируемой активности аденилатциклазы, описанный Battaglia et al. (см. выше, 1987), обеспечивает количественные данные для определения способности соединения препятствовать CRF-активности. Соответственно активность антагониста CRF-рецепторов может быть определена оценочной методикой, которая обычно включает первоначальное определение связывания (такое, которое описано DeSouza (см. выше, 1987)), сопровождаемое сАМР скрининговым протоколом (таким, как описан Battaglia (см. выше, 1987)).
Что касается сродства к связыванию CRF-рецепторов, то антагонисты CRF-рецепторов, предложенные в данном изобретении, характеризуются величиной Кi менее чем 10 мкМ. В предпочтительном варианте осуществления данного изобретения антагонист CRF-рецепторов имеет Ki менее чем 1 мкМ, и более предпочтительно менее чем 0,25 мкМ (т.е., 250 нМ). Как показано подробнее ниже, величины Кi могут быть оценены способами, данными в примере 27.
Антагонисты CRF-рецепторов, предложенные в данном изобретении, могут проявлять активность в CRF-рецепторном сайте и могут быть применены как терапевтические средства для лечения широкого спектра расстройств и заболеваний, включающих эндокринные, психические и неврологические расстройства или заболевания. Конкретнее, антагонисты CRF-рецепторов, предложенные в данном изобретении, могут быть применимы в лечении физиологических состояний или расстройств, возникающих от гиперсекреции CRF. Так как считается, что CRF является основным нейротрансмиттером, который активирует и координирует эндокринные, автономные и поведенческие реакции на стресс, то антагонисты CRF-рецепторов, предложенные в данном изобретении, могут быть применены для лечения нервно-психических расстройств. Нервно-психические расстройства, которые можно лечить антагонистами CRF-рецепторов, предложенными в данном изобретении, включают аффективные расстройства, такие как депрессия; нарушения, связанные с состояниями тревоги/страха, такие как генерализованное тревожное расстройство, паническое расстройство, обсессивно-компульсивное расстройство, атипичная агрессия, сердечно-сосудистые нарушения, такие как нестабильная стенокардия и реактивная гипертензия; расстройства питания, такие как анорексия невроза, булимия и сидром раздраженной толстой кишки. CRF-антагонисты могут быть также применимы в лечении подавления иммунной системы, вызванного стрессом, связанным с различными болезненными состояниями, а также внезапного приступа. Другие применения CRF-антагонистов данного изобретения включают лечение воспалительных состояний (таких как ревматоидный артрит, увеит, астма, воспалительное заболевание кишечника и воспалительная G.I. перистальтика), боли, болезни Кушинга, младенческих судорог, эпилепсии и других припадков как у младенцев, так и у взрослых, и злоупотребления различными веществами и абстиненции (включая алкоголизм).
В другом варианте осуществления изобретения раскрыты фармацевтические композиции, содержащие один или несколько антагонистов CRF-рецепторов. Для введения соединения данного изобретения могут быть приготовлены фармацевтические композиции. Фармацевтические композиции данного изобретения содержат антагонист CRF-рецепторов, предложенный в данном изобретении (т.е. соединение структуры (I)), и фармацевтически приемлемый носитель и/или разбавитель. Антагонист CRF-рецепторов присутствует в композиции в количестве, эффективном для лечения конкретного расстройства, т.е. в количестве, достаточном для достижения антагонистической активности для CRF-рецепторов, и предпочтительно с приемлемой токсичностью для пациента. Предпочтительно, фармацевтические композиции данного изобретения могут включать антагонист CRF-рецепторов в количестве от 0,1 мг до 250 мг на дозировку, зависящую от пути введения, и более предпочтительно от 1 мг до 60 мг. Соответствующие концентрации и дозировки могут быть легко определены специалистом в данной области.
Фармацевтически приемлемый носитель и/или разбавители известны специалистам в данной области. Для композиций, приготовленных как жидкие растворы, приемлемые носители и/или разбавители включают физиологический раствор и стерильную воду и могут необязательно включать антиоксиданты, буферы, бактериостатики и другие обычные добавки. Композиции также могут быть приготовлены в виде пилюль, капсул, гранул или таблеток, которые содержат, в дополнение к антагонисту CRF-рецепторов, разбавители, диспергаторы, поверхностно-активные вещества, связующие вещества и замасливатели. Специалист в данной области, кроме того, может приготовить антагонист CRF-рецепторов целесообразным способом и в соответствии с приемлемыми методиками, такими как методики, описанные в Remington's Pharmaceutical Sciences, Gennaro, Ed., Mack Publishing Co., Easton, PA 1990.
Кроме того, в охват данного изобретения также включены пролекарства. Пролекарства представляют собой любые ковалентно связанные носители, которые высвобождают соединение структуры (I) in vivo, если такое пролекарство введено пациенту. Пролекарства обычно готовят модификацией функциональных групп таким способом, чтобы модификация расщеплялась, либо рутинной манипуляцией, либо in vivo, с образованием исходного соединения.
Что касается стереоизомеров, то соединения структуры (I) могут иметь хиральные центры и могут существовать как рацематы, рацемические смеси и как индивидуальные энантиомеры или диастереомеры. Все такие изомерные формы включены в данное изобретение, в том числе их смеси. Кроме того, некоторые из кристаллических форм соединений структуры (I) могут существовать в альтернативной кристаллической, аморфной или полиморфной формах как полиморфные модификации, которые все включены в данное изобретение. Кроме того, некоторые из соединений структуры (I) также могут образовывать сольваты с водой или другими органическими растворителями. Такие сольваты также включены в охват данного изобретения.
В другом варианте осуществления данное изобретение относится к способу лечения различных расстройств и заболеваний, включающих эндокринные, психические и неврологические расстройства или заболевания. Такие способы включают введение соединения данного изобретения теплокровному животному в количестве, достаточном для лечения расстройства или заболевания. Такие способы включают системное введение антагониста CRF-рецепторов, предложенного в данном изобретении, предпочтительно в форме фармацевтической композиции. Использованное в данной работе системное введение включает пероральный и парентеральный способы введения. Для перорального введения подходящие фармацевтические композиции антагонистов CRF-рецепторов включают порошки, гранулы, пилюли, таблетки и капсулы, а также жидкости, сиропы, суспензии и эмульсии. Указанные композиции могут также включать корригенты, консерваторы, суспендирующие средства, загустители и эмульгаторы и другие фармацевтически приемлемые добавки. Для парентерального введения соединения данного изобретения могут быть приготовлены в виде водных растворов для инъекций, которые могут содержать, в дополнение к антагонисту CRF-рецепторов, буферы, антиоксиданты, бактериостатики и другие добавки, обычно используемые в таких растворах.
В другом варианте осуществления данное изобретение позволяет проводить диагностическую визуализацию определенных сайтов внутри организма применением радиоактивных или нерадиоактивных фармацевтических средств. Применение соединения данного изобретения может обеспечить физиологическую, функциональную или биологическую оценку пациента или обеспечить выявление болезни или патологии и оценку. Радиоактивные фармацевтические средства применяются в сцинтиграфии, позитронной эмиссионной томографии (РЕТ), компьютерной томографии (СТ) и однофотонной эмиссионной компьютерной томографии (ОЭКТ, SPECT). Для данных применений вводятся радиоизотопы таких элементов, как иод (I), включающий 123I (PET), 125I(SPECT) и 131I, технеций (Tc), включающий 99Тс (РЕТ), фосфор (Р), включающий 31Р и 32Р, хром (Cr), включающий 51Cr, углерод (С), включающий 11С, фтор (F), включающий 18F, таллий (Tl), включающий 201Tl, и подобные эмиттеры позитронного и ионизирующего излучения. Нерадиоактивные фармацевтические средства применяются в магнитно-резонансной томографии (МРТ, MRI), флюороскопии и ультразвуковой диагностике. Для данных применений вводятся изотопы таких элементов, как гадолиний (Gd), включающий 153Gd, железо (Fe), барий (Ba), марганец (Mn) и таллий (Tl). Такие частицы также применимы для идентификации присутствия конкретных целевых участков в смеси и для мечения молекул в смеси.
Как отмечено выше, введение соединения данного изобретения может быть использовано для лечения широкого спектра расстройств и заболеваний. В частности, соединения данного изобретения могут быть введены теплокровному животному для лечения депрессии, тревожного расстройства, панического расстройства, обсессивно-компульсивного расстройства, атипичной агрессии, нестабильной стенокардии, реактивной гипертензии, анорексии невроза, булимии, синдрома раздраженной толстой кишки, подавления иммунной системы, вызванного стрессом, удара, воспаления, боли, болезни Кушинга, младенческих судорог, эпилепсии и злоупотребления различными веществами или абстиненции.
Следующие примеры приведены для иллюстративных целей, а не для ограничения.
Примеры
Антагонисты CRF-рецепторов данного изобретения могут быть получены способами, раскрытыми в примерах от 1 до 26. Пример 27 представляет способ для определения сродства связывания с рецептором, и пример 28 раскрывает оценку скрининговых соединений данного изобретения по активности на CRF-стимулируемой аденилатциклазе.
Аналитический ВЭЖХ-МС (HPLC-MS) метод 1
Платформа: Agilent 1100 серии: оснащенные автопробоотборником, УФ-детектором (220 нМ и 254 нМ), МС детектором (APCI);
ВЭЖХ колонка: YMC ODS AQ, S-5, 5 мк, 2,0 x 50 мм картридж;
ВЭЖХ градиент: 1,0 мл/минута, от 10% ацетонитрила в воде до 90% ацетонитрила в воде в 2,5 минуты, поддерживание 90% в течение 1 минуты. И ацетонитрил, и вода содержит 0,025% TFA.
Аналитический ВЭЖХ-МС (HPLC-MS) метод 2
Платформа: Agilent 1100 серии: оснащенные автопробоотборником, УФ-детектором (220 нМ и 254 нМ), МС детектором (APCI);
ВЭЖХ колонка: Phenomenex Synergi-Max RP, 2,0 x 50 мм колонка;
ВЭЖХ градиент: 1,0 мл/минута, от 5% ацетонитрила в воде до 95% ацетонитрила в воде в 13,5 минут, поддерживание 95% в течение 2 минут. И ацетонитрил, и вода содержит 0,025% TFA.
Аналитический ВЭЖХ-МС (HPLC-MS) метод 3
Платформа: Agilent 1100 серии: оснащенные автопробоотборником, УФ-детектором (220 нМ и 254 нМ), МС-детектором (электрораспыление);
ВЭЖХ колонка: XТerra MS, C18, 5 мк, 3,0 x 250 мм колонка;
ВЭЖХ градиент: 1,0 мл/минута, от 10% ацетонитрила в воде до 90% ацетонитрила в воде в 46 минут, скачок до 99% ацетонитрила и поддерживание 99% ацетонитрила в течение 8,04 минуты. И ацетонитрил, и вода содержит 0,025% TFA.
Аналитический ВЭЖХ-МС (HPLC-MS) метод 4
Платформа: Agilent 1100 серии: снабженные автопробоотборником, УФ-детектором (220 нМ и 254 нМ), МС-детектором (APCI) и Berger FCM 1200 CO2 насосным модулем;
ВЭЖХ колонка: Berger Pyridine, PYR 60A, 6 мк, 4,6 x 150 мм колонка;
ВЭЖХ градиент: 4,0 мл/минута, 120 бар; от 10% метанола в надкритическом СО2 до 60% метанола в надкритическом СО2 в 1,67 минуты, поддерживание 60% в течение 1 минуты. Метанол содержит 1,5% воды. Регулируемое противодавление 140 бар.
Препаративная ВЭЖХ-МС
Платформа: Shimadzu HPLC, снабженный Gilson 215 авто-пробоотборником/коллектором фракций, УФ-детектором и РЕ Sciez API150EX масс-детектором;
ВЭЖХ колонка: ВНК ODS-O/B, 5 мк, 30 x 75 мм;
ВЭЖХ градиент: 35 мл/минута, от 10% ацетонитрила в воде до 100% ацетонитрила в 7 минут, поддерживание 100% ацетонитрила в течение 3 минут с 0,025% TFA.
Сокращения:
АА: ацетилацетат
LAН: литийалюминийгидрид
DCM: дихлорметан
DMSO: диметилсульфоксид
ЕАА: этилацетоацетат
LC-MS: жидкостная хроматография-масс-спектроскопия
NaBH(OAc)3: натрийтриацетоксиборогидрид
Pd-C: палладий (10%) на угле
TFA: трифторуксусная кислота
Tosmic: тозилметилизоцианид
асас: ацетилацетонат
EDCI: N-этил-N'-(диметиламинопропил)карбодиимид гидрохлорид
THF: тетрагидрофуран
ТЕА: триэтиламин
tR: время удерживания
Пример 1
7-(2-метоксифенил)-3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин

Стадия 1А:
К охлажденной суспензии метил 4-амино-2-метоксибензоата (6,82 г, 37,7 ммоль) в 6 N HCl (водная) добавляли по каплям раствор нитрита натрия (2,60 г, 37,7 ммоль). После перемешивания при 0°С в течение 20 мин добавляли порциями дигидрат хлорида двухвалентного олова (24,7 г, 109,3 ммоль). Полученную суспензию перемешивали при 0°С в течение 1,5 ч перед фильтрованием. Отделенное твердое вещество суспендировали в EtOH, к которому добавляли бис(диметилацеталь)малональдегид (7,5 мл, 45,7 ммоль), и данную реакционную смесь подвергали кипячению с обратным холодильником в течение ночи. После выпаривания EtOH остаток распределяли между EtOAc и водой и органическую фазу сушили и упаривали досуха. Остаток пропускали через силикагелевый слой (25% EtOAc/гексан), получая соединение 1b (7,43 г) в виде смеси метил и этилбензоата.
Стадия 1В:
К раствору 1b (10,6 г) в сухом диэтиловом эфире (200 мл) при 0°С медленно добавляли порошок LAH (1,74 г). После перемешивания в течение 45 мин при 0°С реакционную смесь декантировали в лед-воду и водную фазу подкисляли до рН 4,0. После выделения полученный спирт (8,8 г) вместе с тионилхлоридом (10 мл) в DCM кипятили с обратным холодильником в течение 2,5 ч, смесь декантировали в лед-воду и экстрагировали с помощью DCM. Неочищенный бензилхлорид (8,26 г) нагревали с NaCN (3,65 г, 74,4 ммоль) в ДМСО (100 мл) при 80°С в течение 45 мин. После удаления ДМСО и колоночной хроматографии с применением 30% EtOAc/гексан получали соединение 1с (5,98 г).
Стадия 1С:
К раствору 1с (5,98 г, 28,1 ммоль) в EtOAc (150 мл) порциями добавляли металлический натрий (1,0 г, 43,5 ммоль) и смесь кипятили с обратным холодильником в течение ночи. Полученную суспензию декантировали в лед-воду и подкисляли до рН 4,0. Органическую фазу сушили и упаривали досуха. Полученное соединение (9,5 г) смешивали с моногидробромидом гидразина (15,3 г, 135,4 ммоль) и кипятили с обратным холодильником в EtOН/H2O (6:1) в течение 5 ч. После выпаривания EtOH и экстракции с EtOAc органическую фазу сушили и упаривали досуха, получая соединение 1d (7,5 г).
Стадия 1D:
Cмесь 1d (7,5 г, 27,9 ммоль) с этилацетоацетатом (5,0 мл) в АсОН (100 мл) кипятили с обратным холодильником в течение 3 ч. После выпаривания АсОН и образования осадка в диэтиловом эфире и последующего фильтрования получали 1е (10,4 г).
Стадия 1Е:
К суспензии 1е (2,1 г, 6,3 ммоль) в ацетонитриле добавляли POCl3 (2,2 мл, 24,1 ммоль) и данную смесь кипятили с обратным холодильником в течение 5 ч, декантировали в лед-воду и экстрагировали EtOAc, получая после хроматографической очистки соединение 1f (1,88 г).
Стадия 1F:
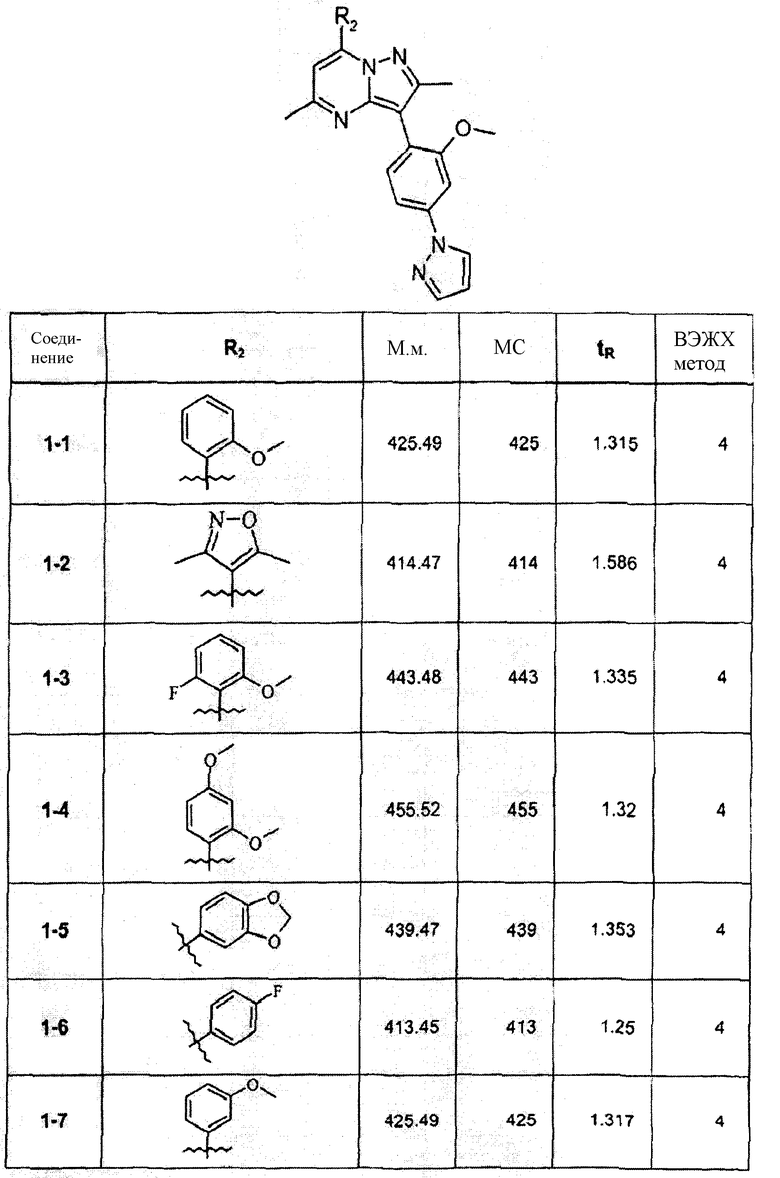
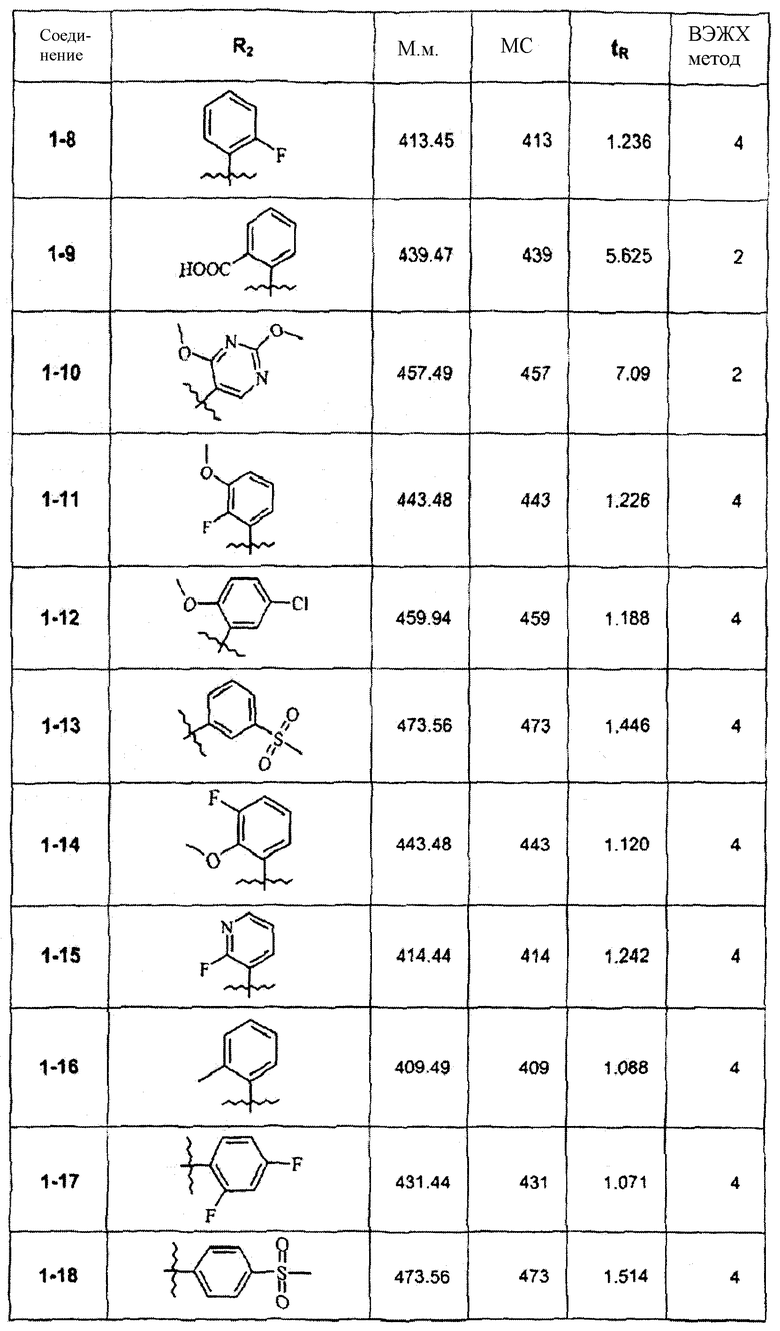
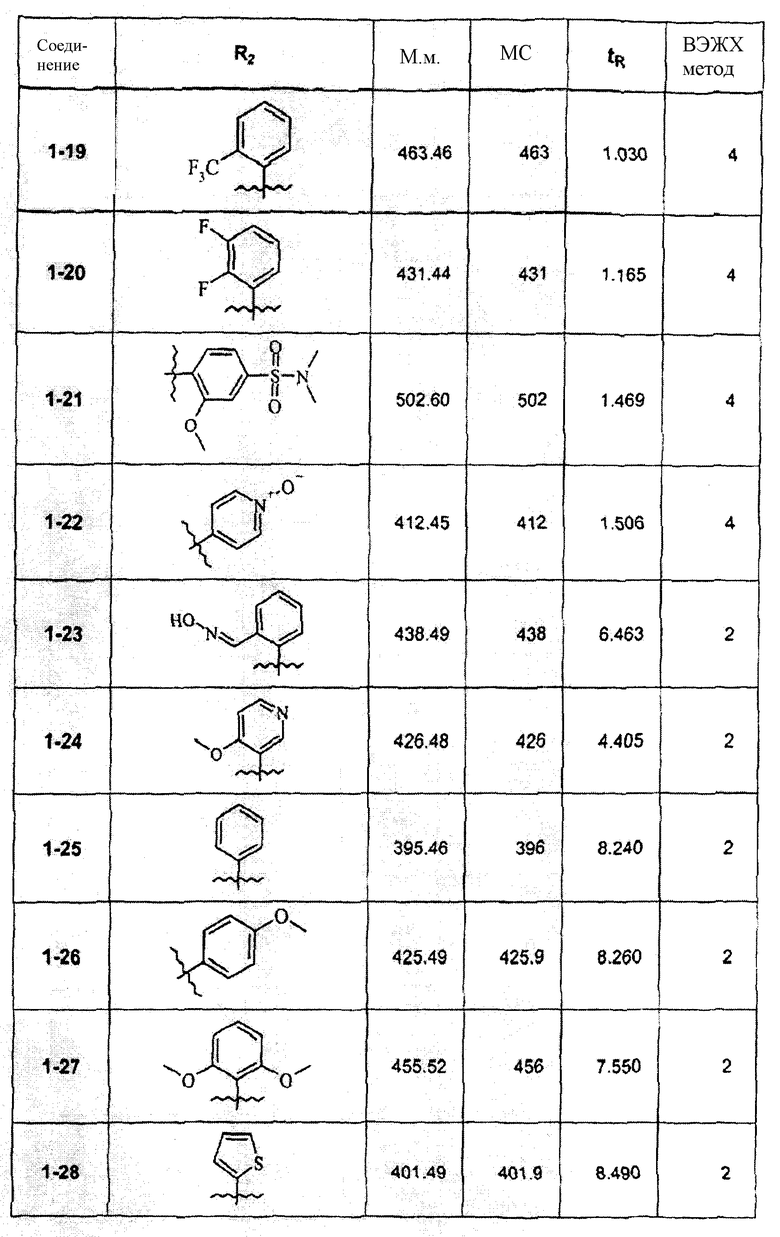
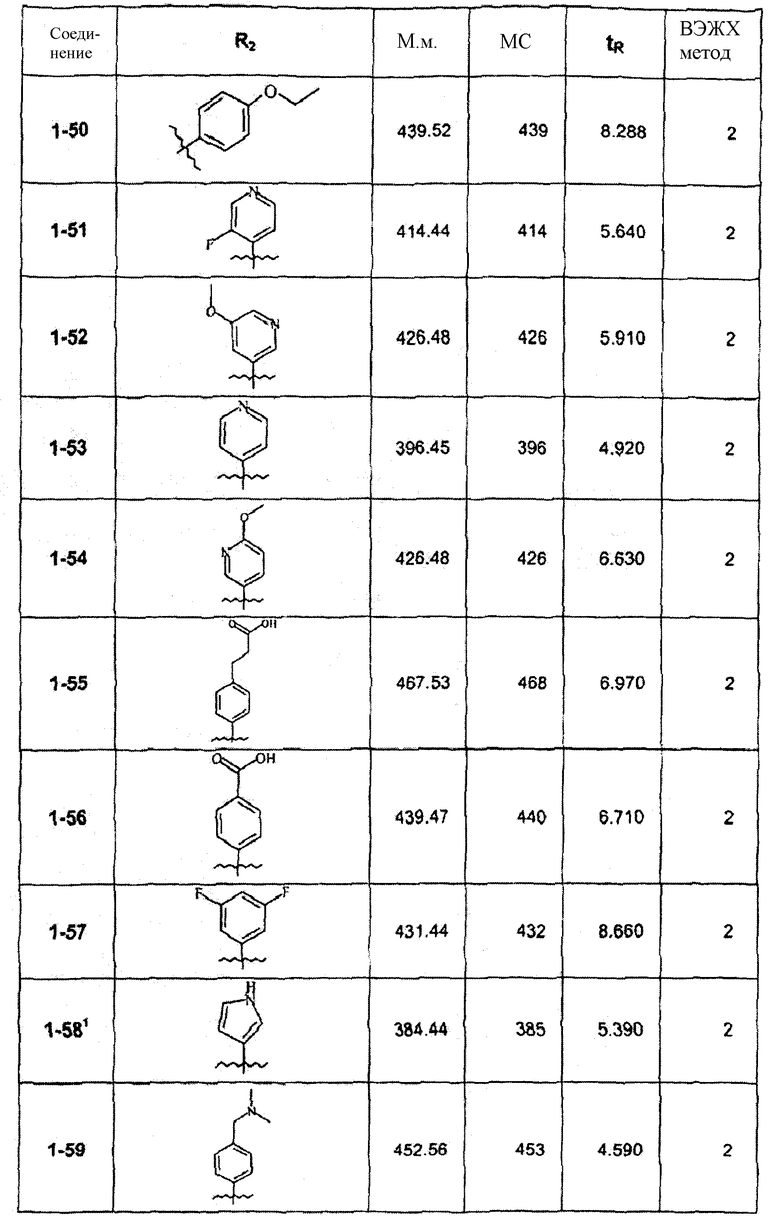
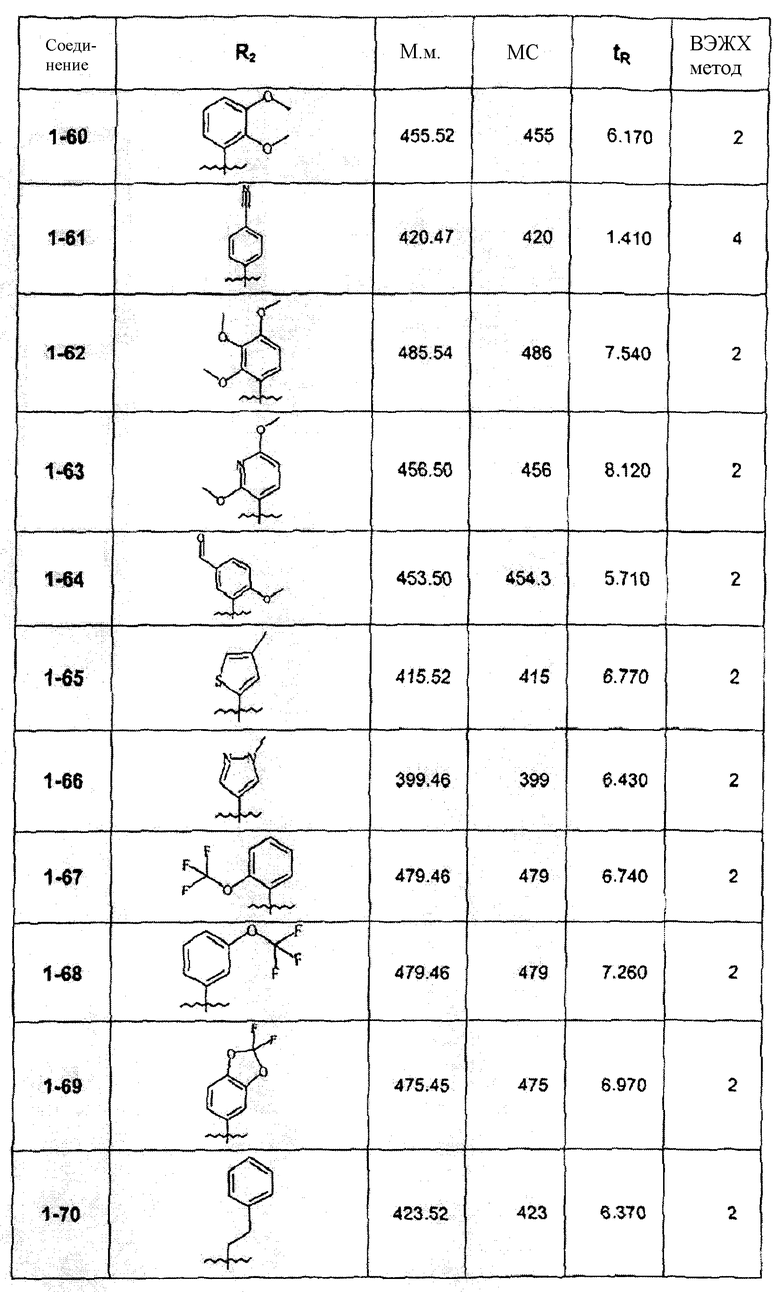
Смесь соединения 1f (1,0 ммоль), 2-метоксифенилбороновой кислоты (1,2 ммоль), К2СО3 (2,0 ммоль) и Pd(PPh3)4 (0,05 ммоль) нагревали в смеси 1,4-диоксан/Н2О (2:1) при 110°С в течение ночи. После выпаривания растворителя смесь экстрагировали CHCl3/H2O, органическую фазу сушили и упаривали досуха. После колоночной хроматографии получали соединение 1-1 (402 мг). В зависимости от функциональности арила в реагенте-арилбороновой кислоте были синтезированы и очищены препаративной ЖХ-МС соединения, перечисленные в следующей таблице:


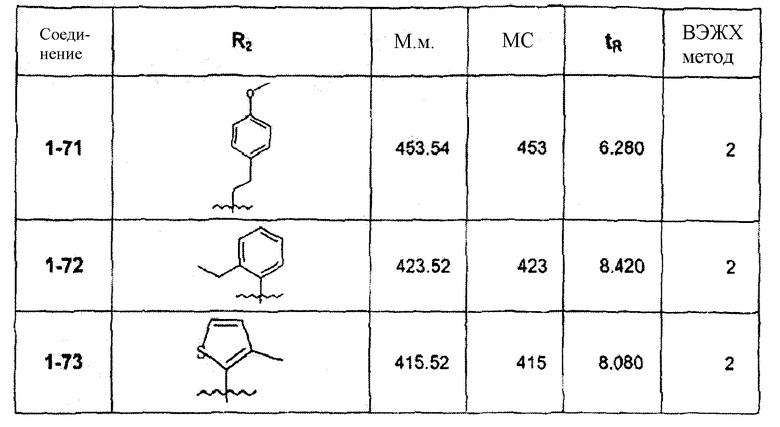
Пример 1А
Альтернативный синтез промежуточного продукта 1f
Стадия 1А-А
В 3-горлую колбу, снабженную механической мешалкой, загружали 250 г (1,12 моль) метилового эфира 2-метокси-4-ацетиламинобензойной кислоты, затем 1 л метанола. Начинали перемешивание и медленно добавляли 94 мл (3,36 ммоль, 3 экв.) концентрированной серной кислоты, допуская слабое кипение с обратным холодильником. Смесь перемешивали в течение 24 ч. Смесь концентрировали в вакууме до образования густой взвеси. Взвесь фильтровали, используя воронку Бюхнера, и промывали 300 мл охлажденного метанола. Собирали осадок с фильтра и сушили в вакууме при 45°С в течение 24 ч, получая 302 г 1а в виде полусульфатной соли с выходом 96%.
Стадия 1А-В
В 2 л трехгорлую колбу Мортона, снабженную механической мешалкой и термопарой, загружали 200 г (716 ммоль) метил 4-амино-2-метоксибензоата 1а. Твердое вещество суспендировали с 700 мл 6 N хлористоводородной кислоты и охлаждали на ледяной бане. К смеси добавляли по каплям 54,3 г (788 ммоль, 1,1 экв.) нитрита натрия в 100 мл воды, поддерживая температуру <15°С в течение добавления. Смесь перемешивали дополнительно 1,5 ч, при этом образовывался светло-желтый гомогенный раствор. К данной смеси осторожно добавляли 272 г (1432 ммоль, 2 экв.) безводного хлорида двухвалентного олова. Температуру в течение добавления поддерживали при <10°С. Смесь перемешивали при 0°С в течение 1 ч и затем хранили при 5°С в течение 16 ч. Осадок собирали фильтрованием на воронке Бюхнера и отфильтрованный осадок сушили на воздухе в течение 2 ч. Отфильтрованный осадок переносили в 2 л круглодонную колбу, снабженную магнитной мешалкой, и разбавляли 600 мл этанола. Во взвесь помещали 142 мл (859 ммоль, 1,2 экв.) бис(диметилацеталь)малональдегида и смесь кипятили с обратным холодильником в течение 6 ч. После выпаривания этанола остаток разбавляли этилацетатом и нейтрализовали гидроксидом натрия. Органическую фазу отделяли, сушили и концентрировали в вакууме. Неочищенный продукт пропускали через слой силикагеля с элюированием 25% этилацетатом в гексане, получая 96 г соединения 1b с выходом 58% в виде смеси метилового и этилового эфиров.
Стадия 1А-С
В 1 л круглодонную колбу, содержащую 500 мл сухого ТГФ, добавляли LAH (14,5 г, 380 ммоль, 0,95 экв.) и смесь охлаждали до 0°С. К данной смеси добавляли по каплям раствор 1b (96 г, 400 ммоль, 1,0 экв.) в 300 мл ТГФ. Температуру в течение добавления поддерживали ниже 15°С. После окончания добавления смесь перемешивали в течение 1 ч, затем реакционную смесь осторожно гасили водой (14,5 мл), 10% водн. гидроксидом натрия (14,5 мл) и водой (43,5 мл). Полученную смесь фильтровали через рыхлый слой целита (Celite®) и концентрировали, получая 1b.1 в виде светло-желтого масла (63,9 г, 75,7%), которое использовали без дальнейшей очистки.
Стадия 1А-D
Тионилхлорид (95 мл, 1,30 моль, 3,1 экв.) добавляли по каплям в течение 1 ч к раствору 1b.1 (85,0 г, 0,42 моль) в 400 мл DCM, устанавливая такую скорость добавления, чтобы поддерживалось слабое кипение с обратным холодильником. Образовывался осадок, который перерастворялся по окончании добавления. Полученный темный раствор кипятили с обратным холодильником в течение 4 ч. Охлажденную реакционную смесь выливали в 500 г льда и полученную смесь экстрагировали 2 х 700 мл DCM. Объединенные органические слои промывали насыщенным водным бикарбонатом натрия, сушили над сульфатом натрия, фильтровали и концентрировали, получая 1b.2 (76,5 г) в виде коричневого твердого вещества, которое использовали без дальнейшей очистки.
Стадия 1А-Е
Раствор 1b.2 (76 г, 340 ммоль, 1,0 экв.) в ДМФА (100 мл) добавляли по каплям в течение 20 мин к смеси цианида натрия (24,5 г, 500 ммоль, 1,5 экв.) и ДМФА (300 мл), нагретой до 100°С. Смесь нагревали при 100°С в течение 4 ч, затем охлажденную смесь фильтровали через целит (Celite®). Фильтрат концентрировали, затем остаток поглощали 300 мл DCM и промывали насыщенным водным раствором бикарбоната натрия (200 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали в вакууме, получая темно-коричневый твердый остаток. Остаток суспендировали в этаноле (100 мл), затем твердое вещество собирали фильтрованием и промывали холодным этанолом и эфиром, получая 1с (48,0 г) в виде не совсем белого твердого вещества. Маточный раствор концентрировали и очищали хроматографией на силикагеле при элюировании смесью гексан/этилацетат 1:1 для получения дополнительных 15,4 г 1с в виде белого твердого вещества. Общий выход 63,4 г.
Стадия 1А-F
К раствору 1с (63,4 г, 0,30 моль, 1 экв.) в этилацетате (800 мл) порциями добавляли металлический натрий (10,3 г, 0,45 ммоль, 1,5 экв.) и смесь кипятили с обратным холодильником в течение 16 ч. Охлажденную суспензию выливали в 500 г льда, подкисляли до рН 5, затем экстрагировали 2 х 300 мл этилацетатом. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали до сырого желтого масла (86,5 г).
Сырое желтое масло (86,5 г) растворяли в этаноле (480 мл) и воде (80 мл), затем добавляли моногидробромид гидразина (100 г, 0,88 моль, 3 экв.) и смесь нагревали при 85°С в течение 16 ч. Растворители выпаривали, добавляли насыщенный солевой раствор (200 мл) и затем смесь экстрагировали 2 х 300 мл этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали, получая 1d (68 г) в виде неочищенной коричневой пены, которую использовали без дальнейшей очистки.
Стадия 1А-G
Смесь 1d (68 г, 250 ммоль, 1,0 экв.), этилацетоацетата (100 мл), уксусной кислоты (150 мл) и этанола (150 мл) кипятили с обратным холодильником в течение 24 ч. Охлажденную смесь концентрировали до образования твердого остатка, который затем помещали на фильтр с пористой стеклянной пластинкой и промывали эфиром, получая 1е (52,0 г, 51,2%) в виде не совсем белого твердого вещества. Маточную жидкость концентрировали, затем хроматографировали на силикагеле, используя 10% метанол в DCM как элюент. Полученный таким образом твердый продукт промывали эфиром для получения дополнительных 17,0 г 1е в виде не совсем белого твердого вещества (общий выход 69 г).
Стадия 1А-Н
К суспензии 1е (41,2 г, 123 ммоль) в ацетонитриле (200 мл) добавляли POCl3 (45,0 мл, 493 ммоль) и данную смесь кипятили с обратным холодильником в течение 16 ч. Охлажденную реакционную смесь выливали в лед-воду и полученную смесь экстрагировали хлороформом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле с элюированием смесью гексаны/этилацетат 3:1, получая 1f (29,0 г) в виде желтовато-коричневого твердого вещества.
Пример 2
7-изопропил-3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин
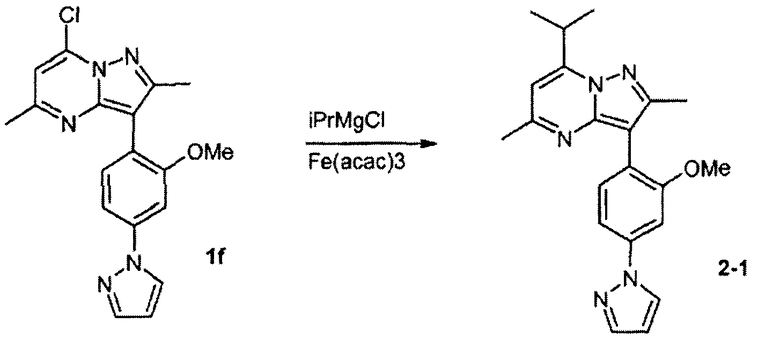
Стадия 2А:
К раствору соединения 1f (1,41 г, 4,0 ммоль) и Fe(acac)3 (424 мг, 1,2 ммоль) в THF/NMP (об./об. = 8:1) медленно добавляли iPrMgCl (2,0 M в THF, 4,0 мл) при комнатной температуре. Реакционную смесь перемешивали в течение 1,5 ч перед гашением с помощью 1 N HCl (водн.). После экстракции с помощью EtOAc cырой продукт очищали колоночной хроматографией (25% EtOAc/гексан), получая соединение 2-1 (628 мг).
В зависимости от функциональности алкила в алкилмагнийгалогениде были синтезированы соединения, перечисленные в следующей таблице:
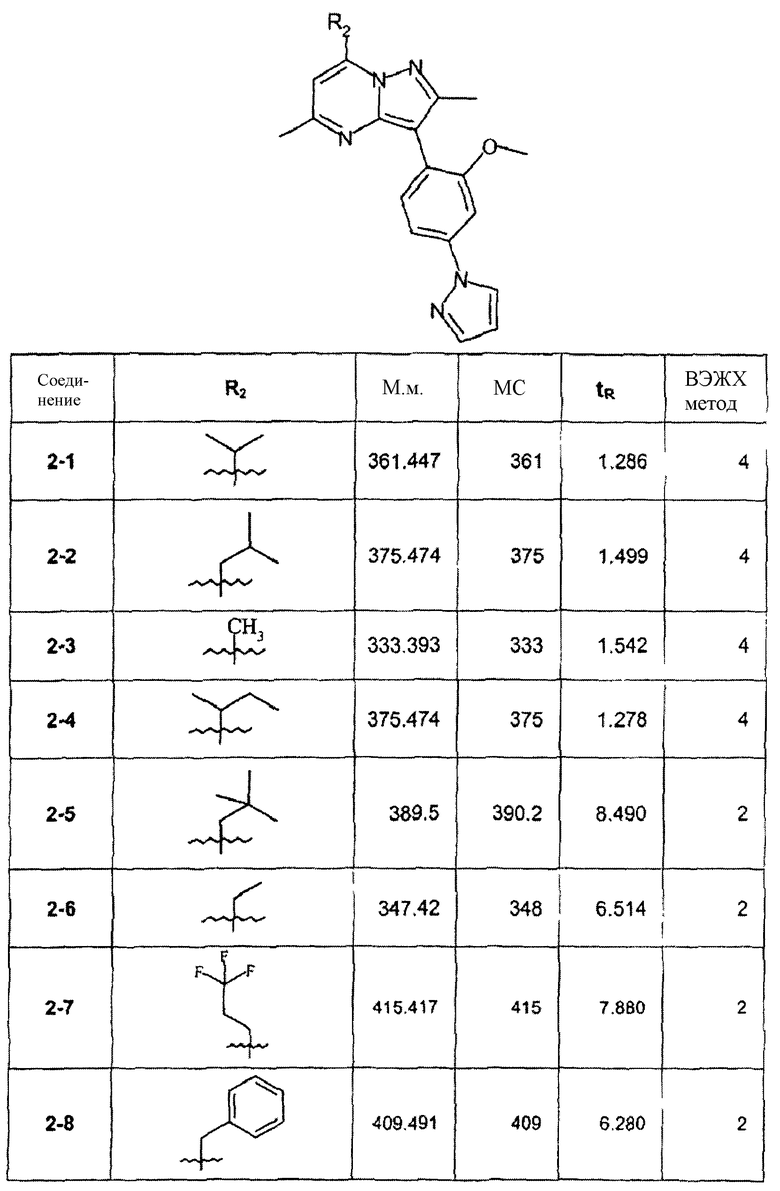
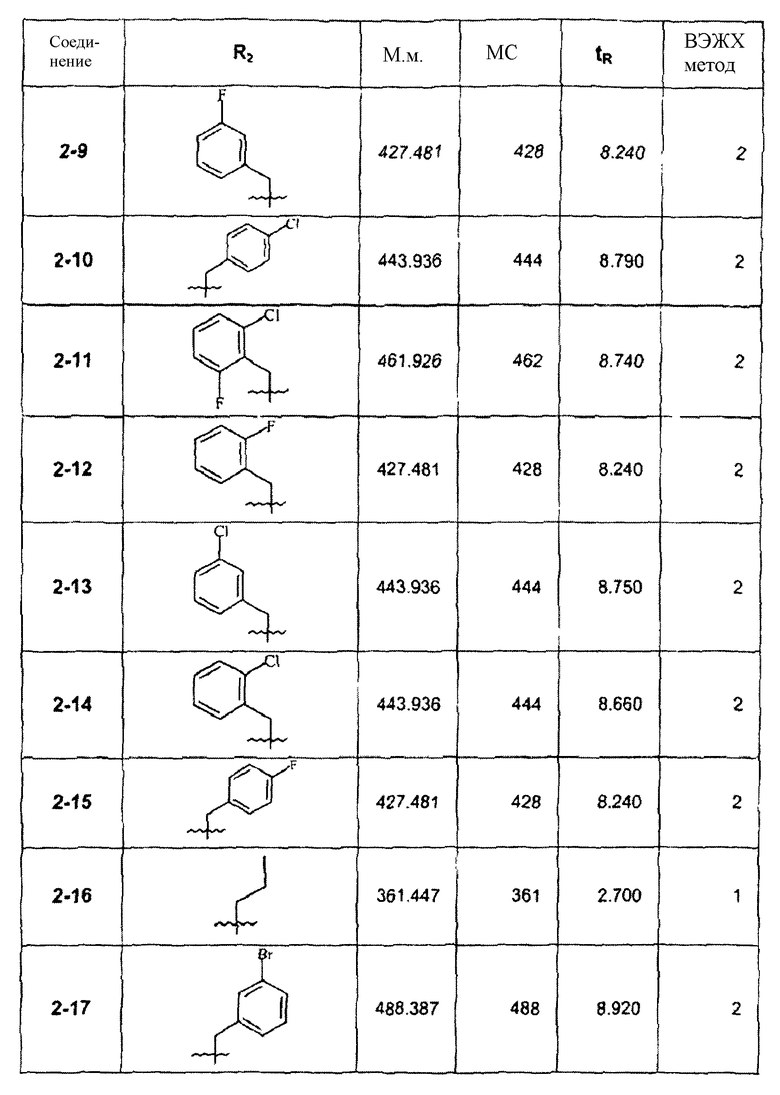
Пример 3
Этиловый эфир 3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин-7-карбоновой кислоты
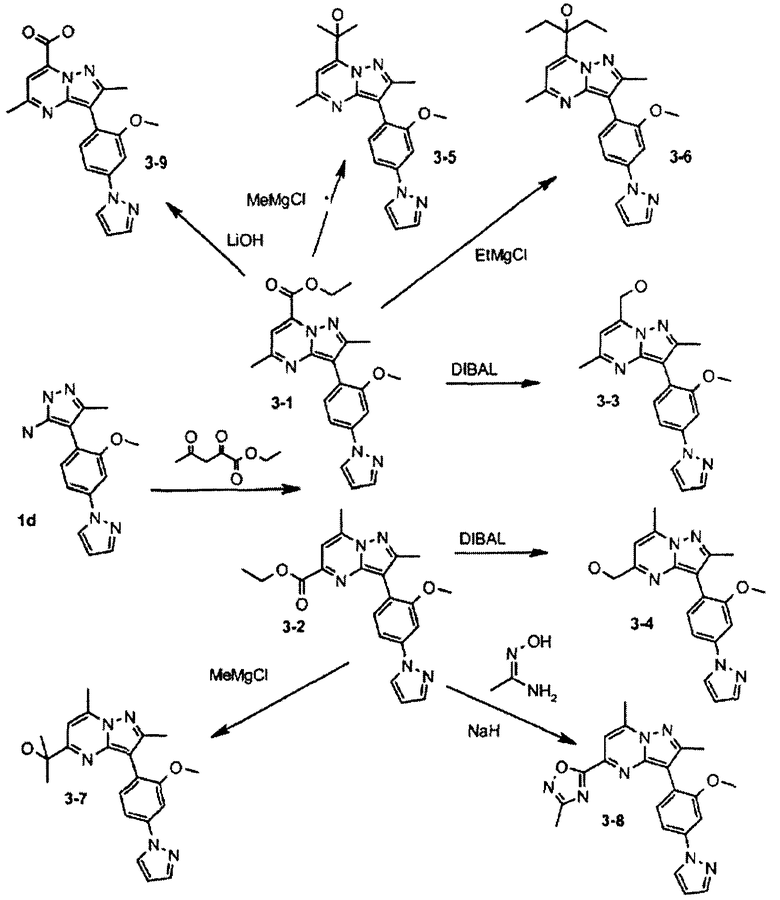
Стадия 3А:
К 20 мл EtOH добавляли соединение 1d (1,0 г, пример 1, стадия 1С) и этил 2,4-диоксовалерат (0,82 г) с последующим добавлением 0,5 мл уксусной кислоты. Реакционную смесь нагревали при 80°С в течение 12 ч. Концентрирование и очистка колоночной хроматографией на силикагеле давала соединение 3-1 (0,66 г, выход 46,1%) и инвертированное дополнительное соединение 3-2 (0,47 г, выход 32,2%).
Стадия 3В:
К раствору соединения 3-1 (30 мг) в ТГФ (1,5 мл) добавляли DIBAL (150 мкл 2 М DIBAL в гексане). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и гасили водой (0,4 мл). После очистки с помощью ЖХ-МС (LC-MS) получали соединение 3-3 (3,3 мг). По той же самой процедуре восстановление соединения 3-2 давало после очистки соединение 3-4 (2,6 мг).
Стадия 3С:
К 1,5 мл ТГФ добавляли соединение 3-1 (30 мг) с последующим добавлением CH3MgBr (150 мкл 2 М CH3MgBr в ТГФ). Реакционную смесь перемешивали при комнатной температуре в течение 2 ч и гасили водой. Образовавшийся продукт очищали ЖХ-МС для получения соединения 3-5 (3,8 мг). Следуя данной процедуре, из соединения 3-1 и CH3СН2MgBr получали после очистки соединение 3-6 (4,1 мг). По той же самой процедуре взаимодействия применение соединения 3-2 в качестве исходного реагента и CH3MgBr в качестве нуклеофила давало после очистки соединение 3-7 (4,0 мг).
Стадия 3D:
К ТГФ (1,5 мл) добавляли ацетамидоксим (20 мг) и NaH (10 мг) с перемешиванием при комнатной температуре в течение 30 мин. Добавляли соединение 3-2 (40 мг) и смесь нагревали при 90°С в течение 2 ч в запаянной трубке. После очистки с помощью ЖХ-МС (LC-MS) получали соединение 3-8 (5,5 мг).
Стадия 3Е:
К соединению 3-1 (200 мг) в смеси диоксан/вода (9:1) добавляли LiOH (30 мг). Взаимодействие осуществляли с перемешиванием в течение 6 ч при комнатной температуре с последующим гашением до рН 4 (HCl, 4 N) и экстрагированием Н2О (20 мл) и EtOAc (20 мл). Органическую фазу сушили над Na2SO4 и концентрировали. Полученный концентрат очищали колоночной хроматографией (50:50 EtOAc/гексан) на силикагеле, выделяя соединение 3-9 (180 мг). Соединения, полученные по примеру 3, представлены в следующей таблице:
Пример 4
3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметил-7-трифторметилпиразоло[1,5-a]пиримидин
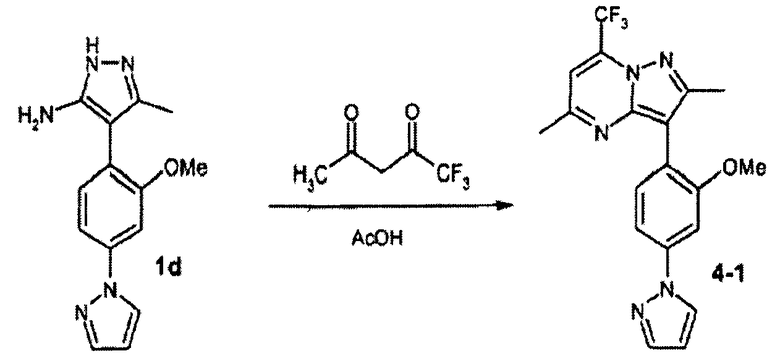
Стадия 4А:
Смесь соединения 1d (40 мг, пример 1, стадия 1С) и 1,1,1-трифторпентан-2,4-диона (избыток) нагревали в АсОН при 150°С в течение 15 мин в микроволновом реакторе, получая после очистки с помощью ЖХ-МС соединение 4-1 (29 мг). В зависимости от трифтордиона были синтезированы соединения в следующей таблице:
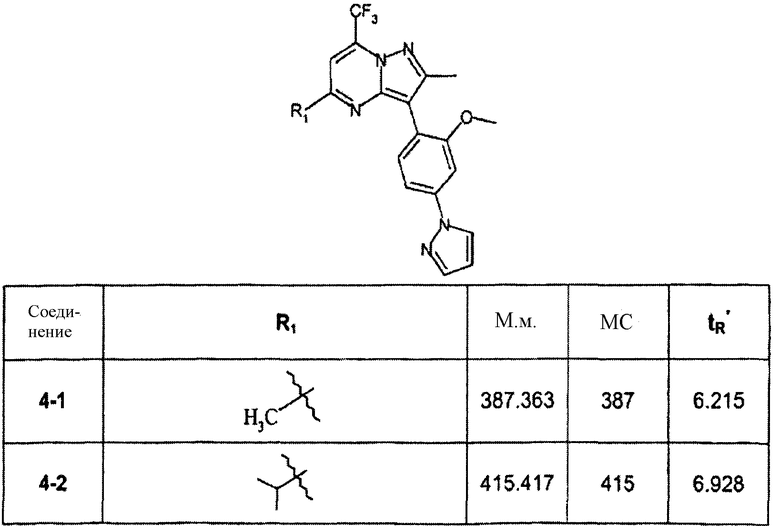
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 5
Диметиламид 3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин-7-карбоновой кислоты
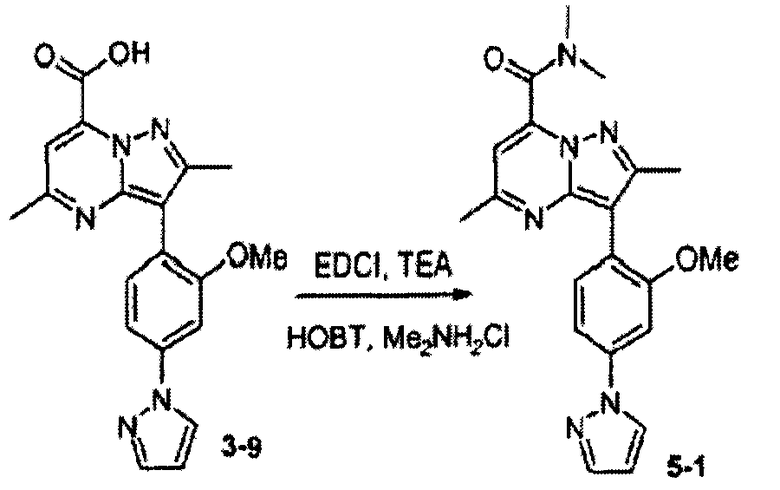
Стадия 5А:
К раствору соединения 3-9 (50 мг, 0,14 ммоль, 1 экв.) в DCM (1 мл) добавляли НОВТ (57 мг, 0,42 ммоль, 3 экв.), ТЕА (0,12 мл, 0,84 ммоль, 6 экв.), гидрохлорид диметиламина (34 мг, 0,42 ммоль, 3 экв.) и EDCI (79 мг, 0,42 ммоль, 3 экв.). Смесь перемешивали при комнатной температуре в течение 16 ч, затем растворитель выпаривали и сырую реакционную смесь очищали препаративной ВЭЖХ/МС, получая соединение 5-1 (10 мг) в виде TFA соли. В зависимости от амина, примененного на стадии амидирования выше, были синтезированы соединения следующей таблицы:
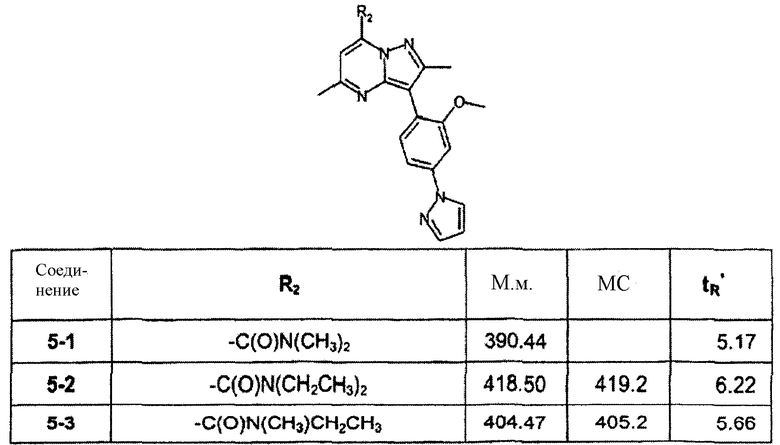
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 6
Циклопентил{2[3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин-7-ил]бензил}амин
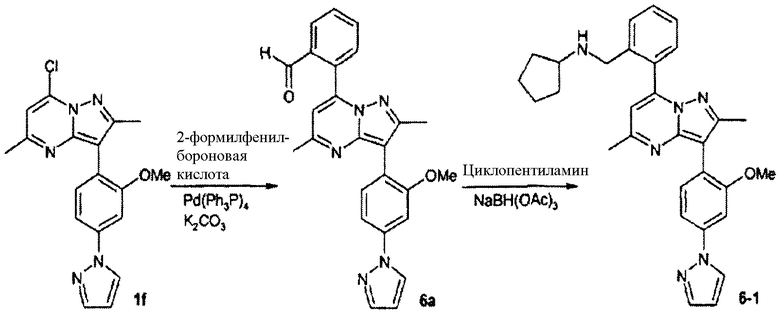
Стадия 6А:
К раствору 1f (500 мг, 1,4 ммоль, 1 экв.) в смеси диоксан/вода 1:1 (6 мл) добавляли 2-формилфенилбороновую кислоту (255 мг, 1,7 ммоль, 1,2 экв.) с последующим добавлением карбоната калия (390 мг, 2,8 ммоль, 2,0 экв.) и тетракис(трифенилфосфин)палладия(0) (82 мг, 0,07 ммоль, 0,05 экв.). Смесь нагревали при 100°С в течение 3 ч в запаянной трубке, затем растворитель удаляли в вакууме. Остаток брали в этилацетат и промывали водой и насыщенным солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали, концентрировали и остаток очищали колоночной хроматографией на силикагеле, используя в качестве элюента 1:1 гексаны/этилацетат, и получали 6а (500 мг, 85%) в виде желтого твердого вещества.
Стадия 6В:
Натрийтриацетоксиборогидрид (80 мг, 0,38 ммоль, 2 экв.) добавляли при комнатной температуре к раствору 6а (80 мг, 0,19 ммоль, 1 экв.) и уксусной кислоты (0,011 мл, 0,19 ммоль, 1 экв.) в дихлорэтане (1 мл). Смесь перемешивали при комнатной температуре в течение 16 ч, затем смесь концентрировали, брали в метанол и cразу очищали препаративной ВЭЖХ/МС, получая 6-1 (36 мг, выход 38%) в виде TFA соли.
В зависимости от амина, примененного на вышеуказанной стадии восстановительного аминирования, были синтезированы соединения следующей таблицы:
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 7
3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметил-7-[2-(2-морфолин-4-илэтил)фенил]пиразоло[1,5-a]пиримидин
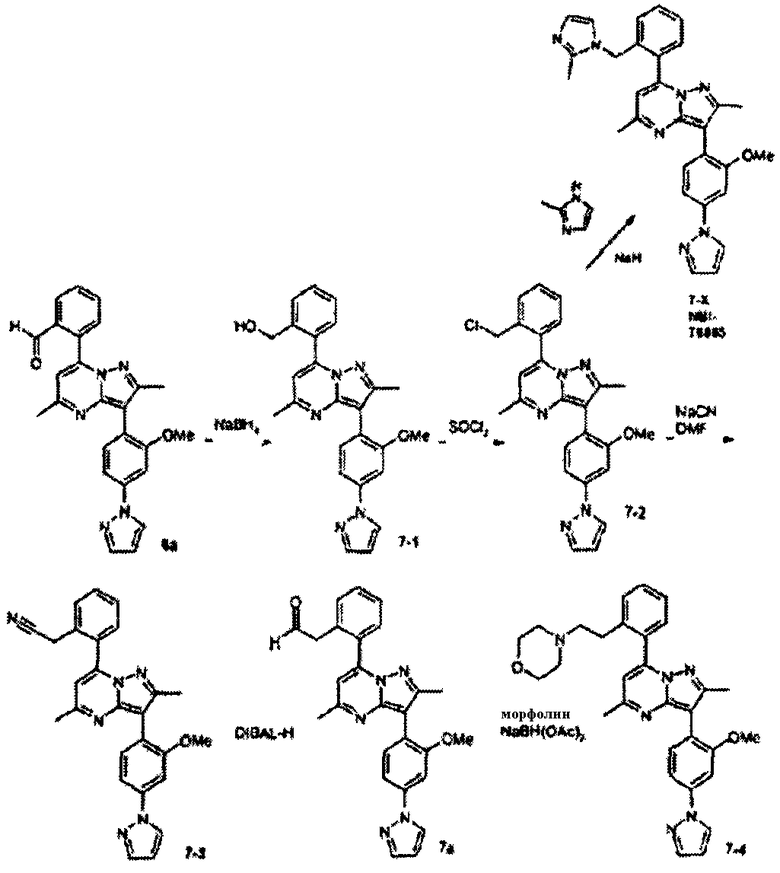
Стадия 7А:
К суспензии 6а (345 мг, 0,82 ммоль) в смеси ТГФ/метанол 1:1 (4 мл) при комнатной температуре осторожно добавляли натрийборогидрид (62 мг, 1,6 ммоль, 2 экв.). Смесь перемешивали в течение 30 мин, затем добавляли воду и смесь экстрагировали с помощью DCM. Объединенные органические слои промывали водой и насыщенным солевым раствором, затем сушили над сульфатом натрия, фильтровали и концентрировали, получая 7-1 (450 мг, 90%) в виде твердого вещества, которое использовали без дальнейшей очистки.
Стадия 7В:
Тионилхлорид (0,17 мл, 2,3 ммоль, 2,2 экв.) добавляли к раствору 7-1 (450 мг, 1,05 ммоль, 1 экв.) в DCM (5 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 30 мин, затем добавляли воду и смесь экстрагировали с помощью DCM. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая 7-2 (420 мг, 90%) в виде желтого твердого вещества.
Стадия 7С:
Гидрид натрия (11 мг 60% дисперсии в минеральном масле, 0,28 ммоль, 4 экв.) добавляли к раствору 2-метилимидазола (17 мг, 0,21 ммоль, 3 экв.) в 2 мл ДМФА при комнатной температуре. Смесь перемешивали в течение 10 мин, затем добавляли раствор 7-2 (30 мг, 0,07 ммоль, 1 экв.) в 0,2 мл ДМФА и смесь перемешивали при комнатной температуре в течение 17 ч. Смесь разбавляли метанолом, затем сразу очищали препаративной ВЭЖХ/МС, получая 7-Х (6 мг) в виде TFA соли.
Стадия 7D:
Цианид натрия (3,3 мг, 0,067 ммоль, 3 экв.) добавляли к раствору 7-2 (10 мг, 0,023 ммоль, 1 экв.) в ДМСО (3 мл) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 2 ч, затем добавляли воду и смесь экстрагировали с помощью DCM. Объединенные органические слои промывали водой и насыщенным солевым раствором, затем сушили над сульфатом натрия, фильтровали и концентрировали, получая неочищенный 7-3 (8 мг, выход 80%) в виде твердого вещества.
Стадия 7Е:
DIBAL-H (0,23 мл 1,5 М раствора в толуоле, 0,35 ммоль, 3 экв.) добавляли к раствору 7-3 (50 мг, 0,11 ммоль) в DCM (1 мл) при -78°С. Смесь перемешивали при -78°С в течение 20 мин, затем ей позволяли нагреваться до комнатной температуры. Добавляли воду и смесь перемешивали в течение 10 мин, затем водный слой экстрагировали двумя дополнительными порциями DCM. Объединенные органические экстракты промывали водой и насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали через целит (Celite®) и концентрировали. Остаток очищали препаративной ВЭЖХ/МС, получая 7а (15 мг) в виде TFA соли.
Стадия 7F:
Натрийтриацетоксиборогидрид (15 мг, 0,069 ммоль, 2 экв.) добавляли при комнатной температуре к раствору 7а (15 мг, 0,034 ммоль, 1 экв.) и уксусной кислоты (0,002 мл, 0,034 ммоль, 1 экв.) в DCM (1 мл). Смесь перемешивали при комнатной температуре в течение 16 ч, затем смесь концентрировали, брали в метанол и cразу очищали препаративной ВЭЖХ/МС, получая 7-4 (11 мг, выход 50%) в виде TFA соли.
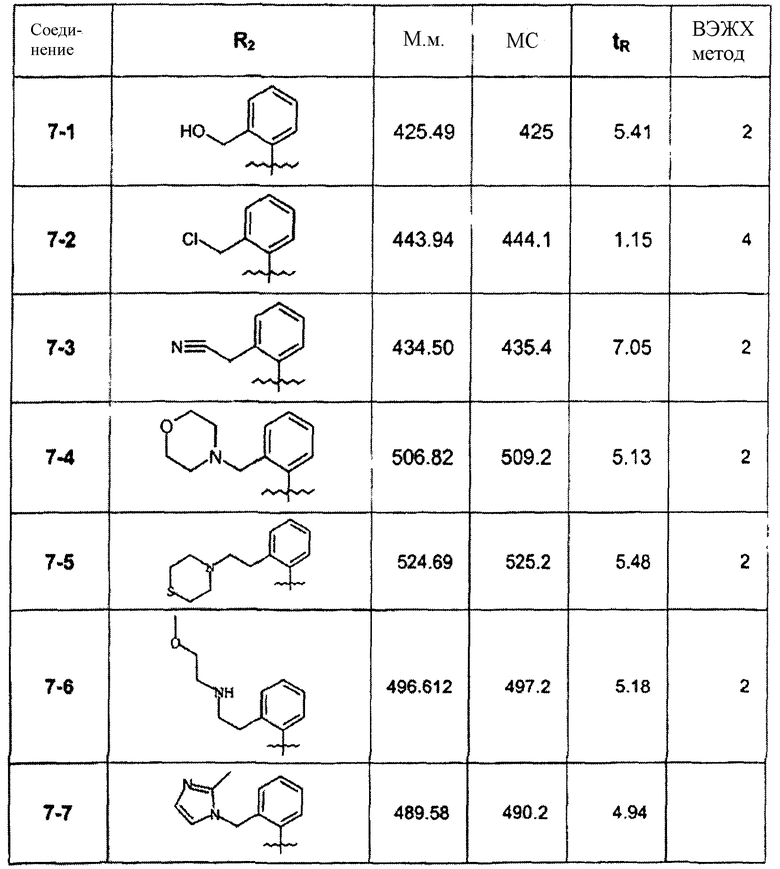
Cледующая таблица суммирует соединения примера 7. Меняя амин, использованный на вышеуказанной стадии восстановительного аминирования, соединения 7-5 и 7-6, включенные в таблицу, были синтезированы способами стадии 7Е:

Пример 8
3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметил-7-(3-метилпиридин-2-ил)пиразоло[1,5-a]пиримидин
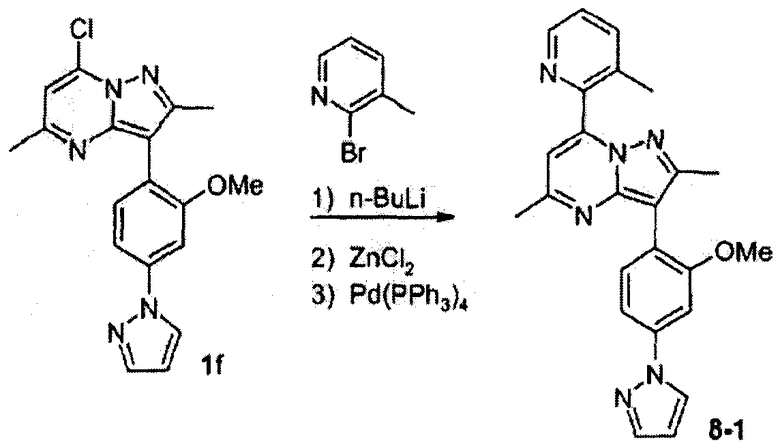
Стадия 8А:
К раствору 2-бром-3-метилпиридина (4,85 г, 28,2 ммоль) в сухом ТГФ (8,0 мл), охлажденному до -70°С, добавляли по каплям n-BuLi (1,6 М раствор в гексане, 17,6 мл, 28,2 ммоль). Реакционную смесь перемешивали при -70°С в течение 30 мин, затем за 5 мин добавляли ZnCl2 (0,5 М раствор в ТГФ, 66,0 мл, 34 ммоль). Смеси позволяли нагреваться до 0°С в течение 1 часа, затем к ней добавляли соединение 1f (1,66 г, 4,70 ммоль) и тетракис(трифенилфосфин)палладий(0) (326 мг, 0,28 ммоль). Смесь затем нагревали при кипении с обратным холодильником в течение 4 ч. Охлажденную реакционную смесь гасили водой, ТГФ выпаривали и полученную водную смесь экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали и остаток хроматографировали на силикагеле, используя 1:3 гексаны/этилацетат, и получали 8-1 свободное основание (1,6 г, 83%) в виде желтого твердого вещества. К раствору 8-1 (1,6 г, 3,9 ммоль) в смеси 7:1 этилацетат/хлороформ (100 мл) добавляли хлористый водород (4,0 мл 2,0 М раствора в простом эфире, 8,0 ммоль) при 0°С. Суспензию разбавляли простым эфиром, затем твердое вещество собирали на фильтре с пористой стеклянной пластинкой и промывали простым эфиром, получая 8-1 HCl соль (1,7 г, 98%) после высушивания в высоком вакууме.
В зависимости от галогенида, примененного на вышепредставленной стадии 8А, были синтезированы соединения следующей таблицы:
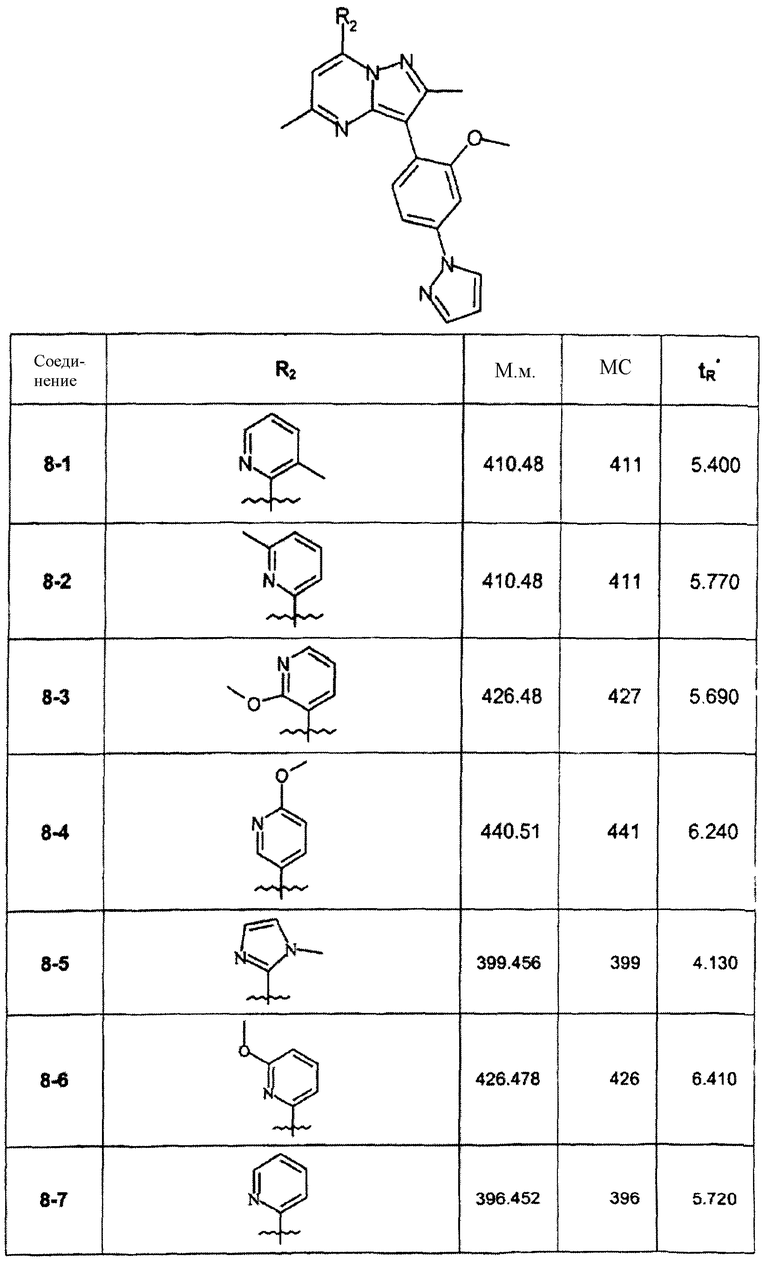
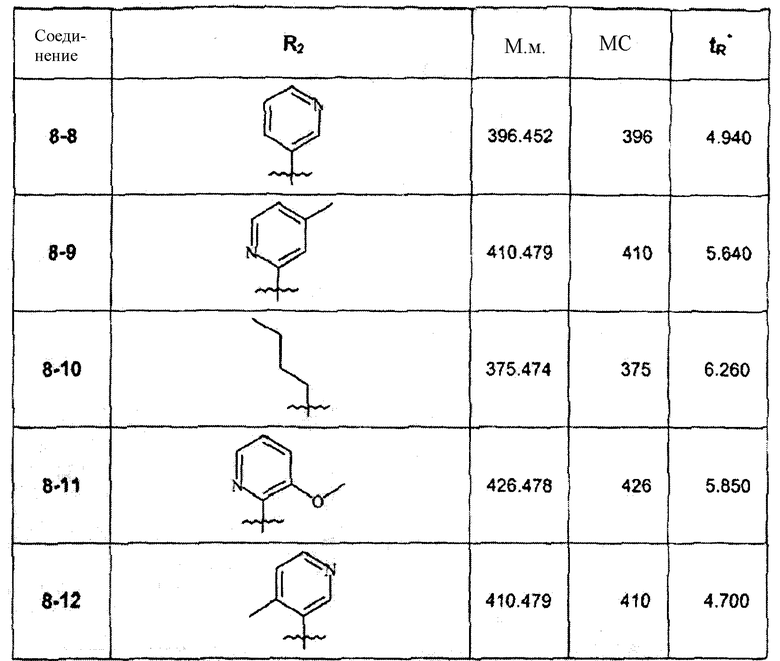
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 9
Синтез реагента - пинаконовый эфир 2-метил-4-(пиразол-1-ил)фенилбороновой кислоты
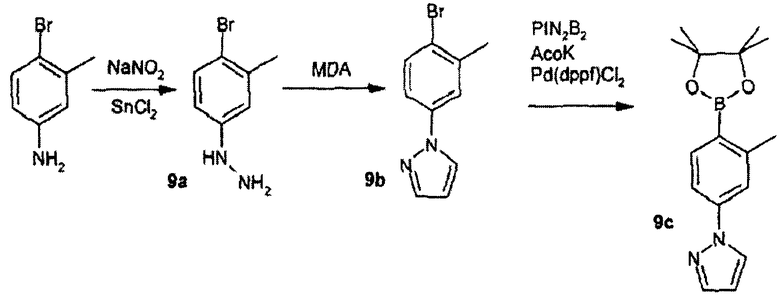
Стадия 9А:
4-Бром-3-метиланилин (10,2 г) суспендировали в 6 N HCl (85 мл) и охлаждали до 0°С. Добавляли раствор нитрита натрия (4 г в 40 мл Н2О) за 10 мин. Реакционную смесь перемешивали в течение 15 мин при 0°С с последующим добавлением дигидрата хлорида двухвалентного олова (36 г в 25 мл 12 N HCl). Реакционную смесь перемешивали в течение 2 ч при 0°С. Реакционную смесь фильтровали и осадок на фильтре промывали холодной Н2О, получая гидрохлорид 4-бром-3-метилфенилгидразина (соединение 9а, 20 г) в виде желтовато-коричневого твердого вещества.
Стадия 9В:
Соединение, полученное на стадии 9А (20 г), суспендировали в 50 мл этанола. Добавляли бис-диметилацеталь малондиальдегида (11,0 мл, 67 ммоль) и реакционную смесь нагревали при 85°С в течение 2 ч. Реакционную смесь нейтрализовали бикарбонатом натрия и экстрагировали промываниями с помощью DCM. Объединенные органические слои сушили над сульфатом натрия и концентрировали. Остаток брали в этилацетат и смесь фильтровали через пад целита (Celite®). Фильтрат упаривали и маслянистый остаток очищали колоночной хроматографией (1:1 этилацетат:гексаны), получая 1-(4-бром-3-метилфенил)пиразол (соединение 9b, 9,6 г, 73%) в виде янтарного масла.
Стадия 9С:
К раствору соединения 9b (2,0 г в 15 мл диоксана) добавляли бис(пинаконато)дибор (2,4 г), ацетат калия (2,4 г) и 1,1'-бис(дифенилфосфино)ферроцендихлорпалладий (II) (500 мг). Реакционную смесь нагревали при 85°С в течение 12 ч. Реакционную смесь фильтровали через пад целита (Celite®) и осадок на фильтре промывали этилацетатом. Фильтрат концентрировали до коричневой жидкости, которую очищали колоночной хроматографией (20% этилацетат/гексаны), получая пинаконовый эфир 2-метил-4-(пиразол-1-ил)фенилбороновой кислоты (соединение 9с, 1,8 г, 75%) в виде желтого масла; ЖХ/МС: [M+H] = 285,0.
Вышеприведенными способами были также приготовлены пинаконовый эфир 2-хлор-4-(пиразол-1-ил)фенилбороновой кислоты (9d) и пинаконовый эфир 2-метил-3-(пиразол-1-ил)фенилбороновой кислоты (9е).
Пример 10
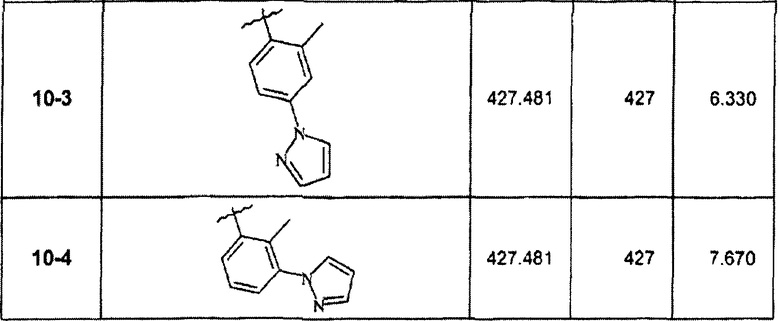
7-(2-Фтор-3-метоксифенил)-2,5-диметил-3-(4-метил-6-пиразол-1-илпиридин-3-ил)пиразоло[1,5-a]пиримидин
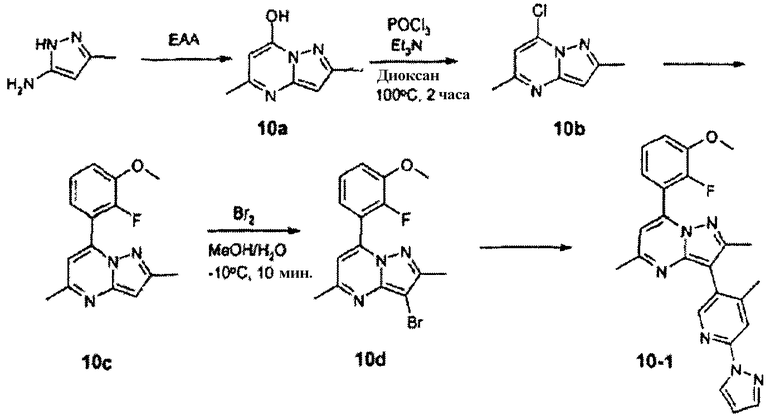
Стадия 10А:
Раствор 3-амино-5-метилпиразола (20,0 г, 206 ммоль), этилацетоацетата (32,0 г, 247 ммоль), уксусной кислоты (6 мл) и диоксана (150 мл) кипятили с обратным холодильником в течение 16 ч. Выделялся белый твердый осадок, который собирали фильтрованием. Осадок на фильтре промывали простым эфиром, получая 10а (29,0 г, 86%) в виде белого твердого вещества.
Стадия 10В:
К суспензии соединения 10а (5,0 г, 31 ммоль) в 1,4-диоксане (30 мл) добавляли триэтиламин (8,50 мл, 62 ммоль) и оксихлорид фосфора (7,4 мл, 77 ммоль). Реакционную смесь нагревали в азоте при 100°С в течение 2 ч. Реакционную смесь охлаждали на ледяной бане, затем последовательно обрабатывали водой и водным раствором бикарбоната натрия (до рН 8). Добавляли дихлорметан и смесь промывали 3×водой. Объединенные органические слои сушили над сульфатом магния, фильтровали и концентрировали до темно-коричневого масла. Сырой продукт очищали хроматографией на силикагеле, используя в качестве элюента 30% этилацетат в гексанах, и получали 10b (3,8 мг, 70%) в виде белого твердого вещества.
Стадия 10С:
К смеси 80 мл диоксана и 8 мл воды добавляли соединение 10b (3,3 г, 18 ммоль, 1 экв.), 2-фтор-3-метоксифенилбороновую кислоту (4,3 г, 26 ммоль, 1,4 экв.), карбонат калия (5,0 г, 36 ммоль, 2 экв.) и тетракис(трифенилфосфин)палладий(0) (1,5 г, 1,3 ммоль, 0,07 экв.). Смесь перемешивали и нагревали при 100°С в течение 16 ч, затем ей позволяли охлаждаться и добавляли воду (75 мл). Смесь экстрагировали этилацетатом, затем объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, используя в качестве элюента 4:1 гексан/этилацетат, и получали cоединение 10с (3,78 г, 76%) в виде белого твердого вещества.
Стадия 10D:
Бром (1,77 г, 11 ммоль) добавляли к раствору 10с (3,0 г, 11 ммоль) в метаноле (30 мл) при -10°С. Через 10 мин смесь фильтровали для отделения образовавшегося осадка. Осадок на фильтре промывали холодным метанолом и затем сушили в вакууме, получая 10d (3,15 г, 83%) в виде желтого твердого вещества.
Стадия 10Е:
Реакция Сузуки с соединением 10d (460 мг, 1,3 ммоль) по вышеприведенной методике стадии 10С при использовании соединения 12-1 вместо 2-фтор-3-метоксифенилбороновой кислоты давала соединение 10-1 (15 мг, твердое вещество) после очистки препаративной ВЭЖХ/МС и хроматографией на силикагеле (элюент 4:1 гексан/этилацетат).
В зависимости от примененных боронатного эфира или кислоты в конечной реакции Сузуки были синтезированы и очищены препаративной ЖХ-МС соединения, перечисленные в следующей таблице:
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 11
7-(2-Фтор-3-метоксифенил)-2,5-диметил-3-(3-метил-5-пиразол-1-илпиридин-2-ил)пиразоло[1,5-a]пиримидин

Стадия 11А:
Гидрид натрия (1,54 г 60% дисперсии в масле, 38,5 ммоль, 2 экв.) добавляли к раствору натриевой соли цианоацетона (2,5 г, 23 ммоль, 1,2 экв.) в ДМФА (40 мл) при комнатной температуре. Смесь перемешивали в течение 15 мин, затем к ней добавляли по каплям раствор 2-фтор-3-метил-5-нитропиридина (3,0 г, 19,2 ммоль, 1,0 экв.) в 10 мл ДМФА. Реакционную смесь перемешивали при комнатной температуре в течение 6 ч. Реакцию гасили 5 г льда, затем 150 мл воды и 10 мл уксусной кислоты. Смесь экстрагировали этилацетатом, затем объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, используя в качестве элюента 30% этилацетат в гексанах, и получали 11а (1,85 г, выход 44%) в виде оранжевого масла.
Стадия 11В:
Смесь 11а (1,8 г, 8,2 ммоль, 1,0 экв.), моногидробромида гидразина (1,0 г, 8,8 ммоль, 1,1 экв.), этанола (30 мл) и воды (3 мл) нагревали при кипении с обратным холодильником в течение 17 ч. Растворитель выпаривали, остаток сразу очищали хроматографией на силикагеле, используя в качестве элюента 1:1 гексаны/этилацетат, и получали 11b (1,8 г, выход 94%) в виде желтой пены.
Стадия 11С:
Смесь 11b (1,8 г, 7,7 ммоль, 1,0 экв.), этанола (15 мл), уксусной кислоты (15 мл) и этилацетоацетата (1,6 г, 12,4 ммоль, 1,6 экв.) нагревали в запаянной трубке при 105°С в течение 19 ч. Растворитель выпаривали, остаток отделяли через фильтр с пористой стеклянной пластинкой, промывали простым эфиром, получая 11с (1,0 г, выход 43%) в виде желтого твердого вещества.
Стадия 11D:
Смесь 11с (800 мг, 2,7 ммоль, 1,0 экв.), оксихлорида фосфора (900 мг, 5,9 ммоль, 2,2 экв.) и ацетонитрила (15 мл) кипятили с обратным холодильником в течение 3 ч. Реакционную смесь выливали в лед, затем смесь экстрагировали этилацетатом. Объединенные этилацетатные экстракты промывали водным бикарбонатом натрия, сушили над сульфатом натрия, фильтровали и концентрировали, получая 11d (640 мг, 76%) в виде желтого твердого вещества.
Стадия 11E:
Суспензию 11d (640 мг, 2,0 ммоль, 1 экв.), 2-фтор-3-метоксифенилбороновой кислоты (480 мг, 3,8 ммоль, 1,4 экв.), карбоната калия (555 мг, 4,0 ммоль, 2 экв.) и тетракис(трифенилфосфин)палладия(0) (230 мг, 0,2 ммоль, 0,1 экв.) в 20 мл диоксана и 2 мл воды перемешивали и нагревали при 100°С в течение 16 ч. Добавляли воду (50 мл) и смесь экстрагировали этилацетатом (50 мл). Органический слой сушили над сульфатом натрия, фильтровали и концентрировали. Остаток растирали в метаноле, получая 11е (300 мг, 37%) в виде желтого твердого вещества.
Стадия 11F:
10% Pd/С (100 мг) добавляли к раствору 11е (300 мг, 0,74 ммоль, 1,0 экв.) с барботируемым азотом в 20 мл этанола и 10 мл ТГФ. Смесь встряхивали в вибраторе Парра при 40 фунтах на квадратный дюйм водорода-газа при комнатной температуре в течение 6 ч. Смесь продували азотом и фильтровали. Фильтрат концентрировали, получая 11f (260 мг, выход 94%) в виде желтого масла.
Стадия 11G:
Раствор нитрита натрия (60 мг, 0,87 ммоль, 1,3 экв.) в воде (10 мл) добавляли по каплям к охлажденному льдом раствору 11f (260 мг, 0,69 ммоль, 1,0 экв.) в 4 N хлористоводородной кислоте (5 мл). Смесь перемешивали при 0°С в течение 1 ч, затем добавляли 10 мл полунасыщенного водного иодида калия. Смесь перемешивали при комнатной температуре в течение 16 ч, затем добавляли 50 мл насыщенного водного раствора бикарбоната натрия и смесь экстрагировали 2х 50 мл этилацетата. Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали и остаток очищали хроматографией на силикагеле, используя в качестве элюента 4:1 гексаны/этилацетат, и получали cоединение 11g (170 мг, выход 51%) в виде желтого твердого вещества.
Стадия 11Н:
К раствору 11g (170 мг, 0,35 ммоль, 1,0 экв.) в диоксане (6 мл) добавляли карбонат калия (200 мг, 1,45 ммоль, 4,1 экв.), пиразол (60 мг, 0,89 ммоль, 2,5 экв.), иодид меди(I) (60 мг, 0,32 ммоль, 0,9 экв.), транс-1,2-диаминоциклогексан (36 мг, 0,32 ммоль, 0,9 экв.) и N,N'-диметилэтилендиамин (28 мг, 0,32 ммоль, 0,9 экв.). Смесь перемешивали и нагревали в запаянной трубке при 100°С в течение 19 ч. Реакционную смесь фильтровали через пад целита (Celite®), концентрировали и очищали препаративной ВЭЖХ/МС, получая соединение 11-1 (70 мг, выход 37%) в виде TFA соли; М.м.: 428,47; ЖХ/МС: 429 [MH]+; tR: 5,390, аналитический метод 2.
Пример 12
4-метил-2-пиразол-1-ил-5-пиридилбороновая кислота
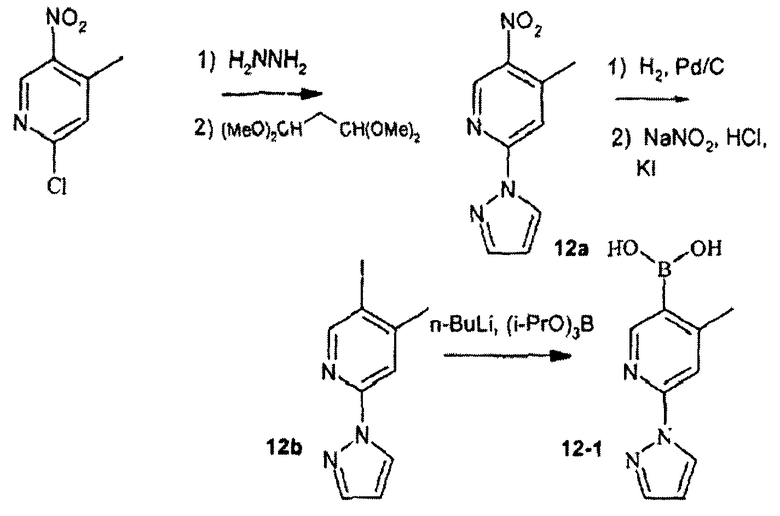
Стадия 12А:
2-Хлор-4-метил-5-нитропиридин (5,0 г, 29 ммоль, 1,0 экв.) растворяли в 50 мл раствора гидразина (1 М раствор в ТГФ) и смесь перемешивали и нагревали в запаянной трубке при 80°С в течение 22 ч. Охлажденную реакционную смесь фильтровали, полученное твердое вещество промывали простым эфиром, получая 5,7 г зеленовато-коричневого твердого вещества.
Смесь данного твердого вещества (5,7 г, 24 ммоль, 1,0 экв.), бис(диметилацеталь)малональдегида (5,9 г, 31 ммоль, 1,3 экв.) и уксусной кислоты (50 мл) перемешивали и нагревали в запаянной трубке при 80°С в течение 5 ч. Растворитель выпаривали, затем добавляли водный раствор бикарбоната натрия (200 мл) и смесь экстрагировали 2×200 мл этилацетата. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток перекристаллизовывали из этанола, получая 12а (2,6 г, выход 53%) в виде желтого твердого вещества.
Стадия 12В:
Смесь 12а (2,6 г, 13 ммоль) и 10% Pd/С (200 мг) в 30 мл 1:1 ТГФ/метанола встряхивали в вибраторе Парра при 40 фунтах на квадратный дюйм водорода при комнатной температуре в течение 2 ч. Реакционную смесь фильтровали через пад целита (Celite®) и фильтрат концентрировали до светло-зеленого масла. Масло суспендировали в 10 мл 3 N бромистоводородной кислоты, охлаждали до 0°С, затем обрабатывали по каплям раствором нитрита натрия (835 мг, 12 ммоль, 1,1 экв.) в 2 мл воды. Смесь перемешивали при 0°С в течение 1 ч, затем добавляли 2 мл полунасыщенного иодида калия и смесь перемешивали при комнатной температуре в течение 22 ч. Добавляли насыщенный водный раствор бикарбоната натрия, затем смесь экстрагировали 2 х 100 мл этилацетата и объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, используя в качестве элюента 4:1 гексаны/этилацетат, и получали 12b (1,23 г, выход 33%) в виде желтого твердого вещества.
Стадия 12С:
н-Бутиллитий (1,8 мл 2,0 М раствора в пентане, 3,6 ммоль) добавляли по каплям к раствору соединения 12b (600 мг, 2,1 ммоль) и триизопропилбората (900 мг, 4,8 ммоль) в 5 мл ТГФ при -78°С. Смеси позволяли нагреваться до комнатной температуры в течение 1 ч, затем смесь охлаждали до -78°С и обрабатывали дополнительно триизопропилборатом (400 мг, 2,1 ммоль), затем дополнительно н-бутиллитием (0,5 мл 2,0 М раствора в пентане, 1,0 ммоль). Смеси снова позволяли нагреваться до комнатной температуры в течение 1 ч, затем добавляли 0,8 мл 1N хлористоводородной кислоты и смесь перемешивали в течение 1 ч. Смесь фильтровали, твердое вещество промывали метанолом и этилацетатом, затем фильтрат концентрировали. Остаток хроматографировали на силикагеле, элюируя смесью 1:1 гексаны/этилацетат, и получали соединение 12-1 (220 мг, выход 52%) в виде красного твердого вещества.
Пример 13
7-(4-хлорфеноксиметил)-3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин
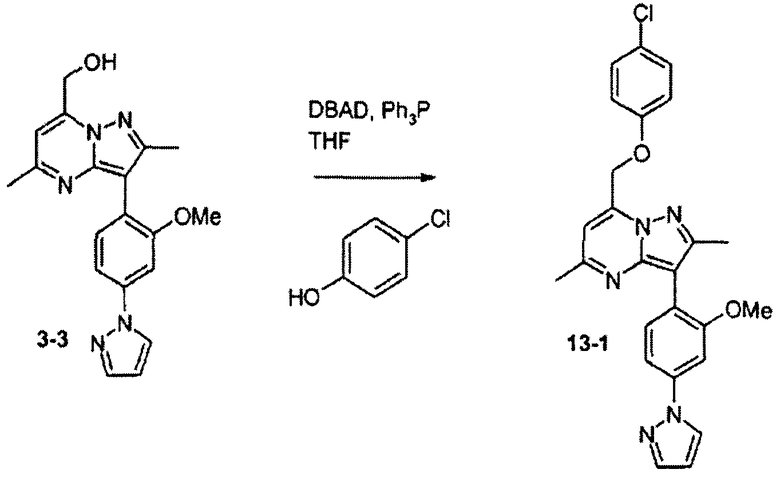
Стадия 13А:
К раствору соединения 3-3 (25 мг, 0,072 ммоль, 1 экв.) в ТГФ (1,5 мл) добавляли ди-трет-бутилазодикарбоксилат (30 мг, 0,11 ммоль, 1,5 экв.), трифенилфосфин (30 мг, 0,11 ммоль, 1,5 экв.) и 4-хлорфенол (30 мг, 0,023 ммоль, 3,3 экв.). Смесь перемешивали при комнатной температуре в течение 17 ч, затем растворитель выпаривали и остаток очищали хроматографией на силикагеле, элюируя смесью гексаны/этилацетат, и получали соединение 13-1 (8 мг) в виде твердого вещества.
В зависимости от примененного фенола были синтезированы и очищены препаративной ЖХ-МС соединения, перечисленные в следующей таблице:
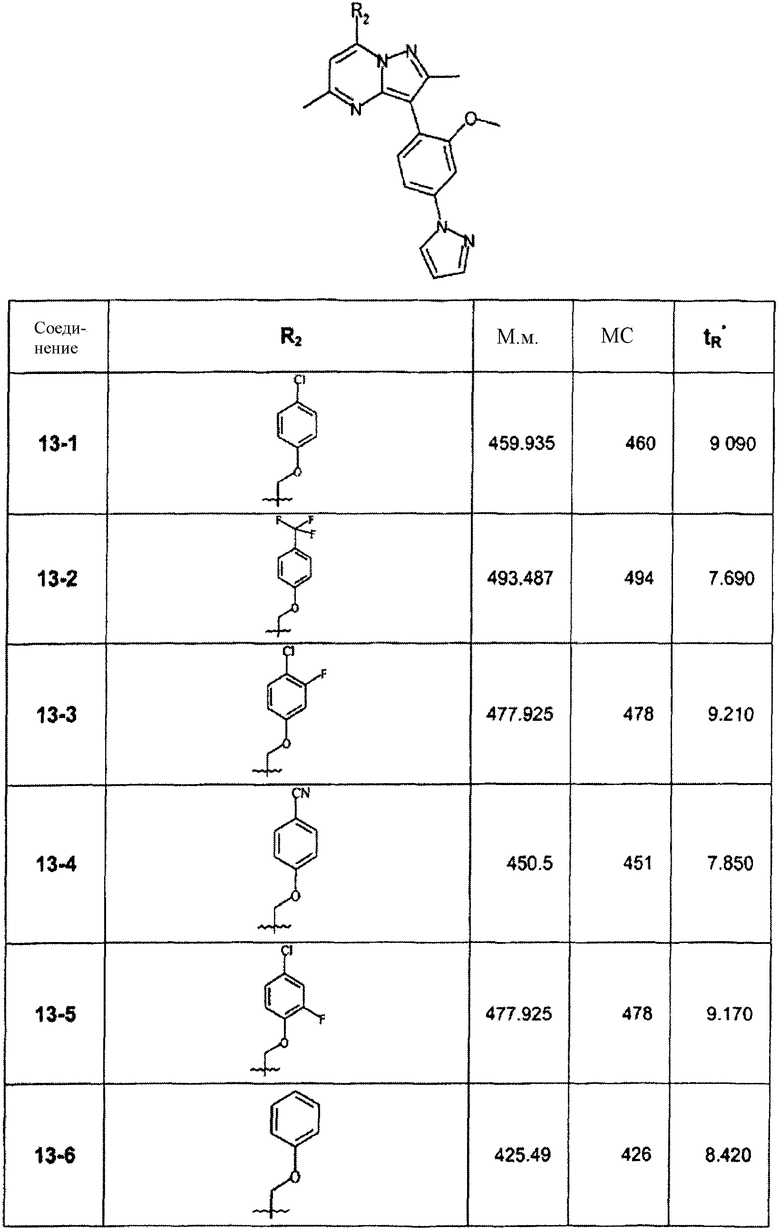
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 14
6-{3-[3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин-7-ил]пропокси}никотинонитрил
Стадия 14А:
К раствору соединения 1f (1,06 г, 3,0 ммоль) и ацетилацетоната железа(III) (353 мг, 1,0 ммоль) в 10 мл безводной смеси THF/NMP (7:1) медленно добавляли 3-бутенилмагнийхлорид (9,0 мл 0,5 М раствора в ТГФ, 4,5 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 1 ч, затем дополнительно добавляли ацетилацетонат железа(III) (1,0 г, 2,8 ммоль) и реагент Гриньяра (6,0 мл, 3,0 ммоль). Реакционную смесь перемешивали в течение 2 ч, затем добавляли воду. Смесь экстрагировали этилацетатом, затем объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток хроматографировали на силикагеле, используя гексаны/этилацетат в качестве элюента, и получали 14а (538 мг, выход 48%).
Стадия 14В:
К раствору 14а (380 мг, 1,02 ммоль) в 10 мл ТГФ/вода (4:1) добавляли тетроксид осмия (26 мг, 0,10 ммоль), затем периодат натрия (642 мг, 3,0 ммоль) при комнатной температуре. Смесь перемешивали при комнатной температуре в течение 1 ч, затем добавляли этилацетат и воду. Органический слой сушили над сульфатом натрия, фильтровали и упаривали, получая неочищенный альдегид, который растворяли в метаноле (20 мл). К данному раствору добавляли порциями натрийборогидрид (152 мг, 4,0 ммоль). После перемешивания при комнатной температуре в течение 20 мин реакционную смесь концентрировали. Остаток очищали хроматографией на силикагеле, элюируя смесью гексаны/этилацетат, и получали соединение 14-1 (230 мг, выход 60%).
Стадия 14С:
Смесь 14-1 (30 мг, 0,08 ммоль, 1 экв.), иодида меди(I) (15 мг, 0,08 ммоль, 1 экв.), карбоната цезия (52 мг, 0,16 ммоль, 2 экв.) и 1,10-фенантролина (14 мг, 0,08 ммоль, 1 экв.) нагревали в 1 мл толуола в запаянном сосуде при 110°С в течение 17 ч. Охлажденную смесь фильтровали через целит (Celite®), затем концентрировали. Остаток очищали хроматографией на силикагеле, используя гексан/этилацетат в качестве элюента, и получали соединение 14-2 (5 мг) в виде твердого вещества.
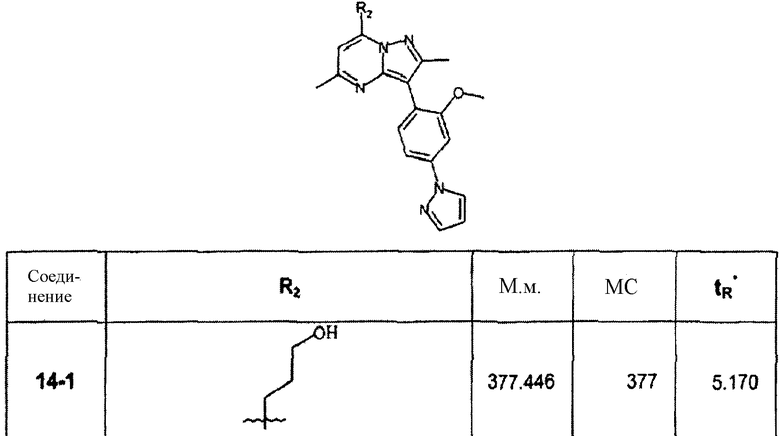
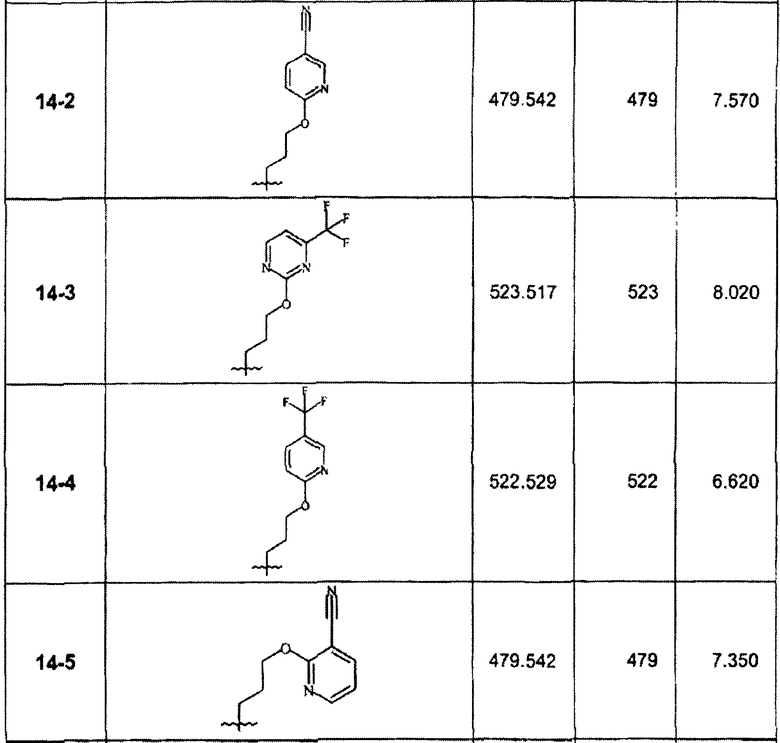
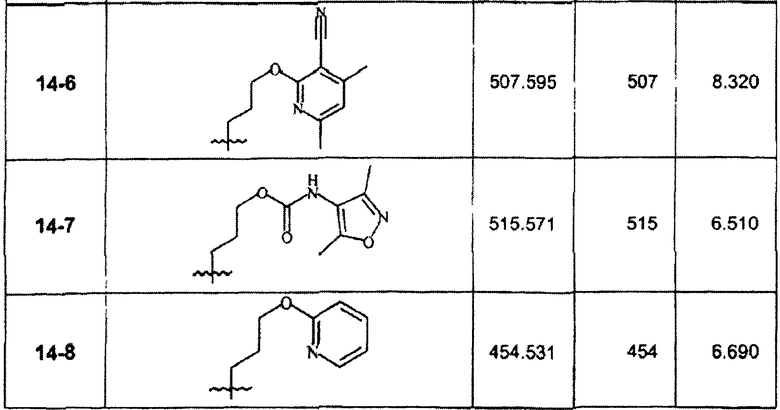
В зависимости от примененного арилгалогенида по способу стадии 14С были синтезированы в дополнение к соединению 14-1 и очищены препаративной ЖХ-МС соединения, перечисленные в следующей таблице:
∗ Для всех ВЭЖХ определений использовали аналитический метод 2.
Пример 15
7-имидазол-1-илметил-3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин

Стадия 15А:
Раствор метансульфонилхлорида (100 мг, 0,86 ммоль, 1,5 экв.) в DCM (0,5 мл) добавляли по каплям при 0°С к раствору соединения 3-3 (200 мг, 0,57 ммоль, 1 экв.) в 5 мл DCM. Смеси позволяли нагреваться до комнатной температуры в течение 1 ч, затем добавляли насыщенный водный раствор бикарбоната натрия и смесь экстрагировали 2×20 мл DCM. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали, получая 15а (180 мг, выход 49%) в виде желтой пены.
Стадия 15В:
Карбонат калия (20 мг, 0,14 ммоль, 2,6 экв.) и имидазол (20 мг, 0,30 ммоль, 5,5 экв.) добавляли к раствору 15а (23 мг, 0,054 ммоль, 1 экв.) в ДМФА (1 мл). Реакционную смесь перемешивали при комнатной температуре в течение 16 ч, затем добавляли метанол (1 мл) и реакционную смесь очищали сразу препаративной ВЭЖХ/МС, получая 15-1 (10 мг) в виде TFA соли.
В зависимости от примененного нуклеофильного гетероцикла или амина были синтезированы и очищены препаративной ЖХ-МС соединения, перечисленные в следующей таблице:

∗ Для всех ВЭЖХ определений использовали аналитический метод 2.
Пример 16
4-метил-2-пиррол-1-ил-5-пиридилбороновая кислота

Стадия 16А:
Раствор 2-амино-5-бром-4-метилпиридина (1 г, 5,4 ммоль) и 2,5-дигидрокситетрагидрофурана (2,8 г, 27 ммоль) в уксусной кислоте (10 мл) нагревали при 90°С в запаянной трубке в течение 2 ч. Реакционную смесь концентрировали и остаток очищали хроматографией на силикагеле, используя 4:1 гексан/этилацетат и получая соединение 16а (900 мг, выход 71%) в виде светло-желтого масла.
Стадия 16В:
н-Бутиллитий (3,6 мл 2,0 М раствора в пентане, 7,2 ммоль) добавляли по каплям к раствору соединения 16а (860 мг, 3,6 ммоль) и триизопропилбората (1,4 г, 7,3 ммоль) в 6 мл ТГФ при -78°С. Смеси позволяли нагреваться до комнатной температуры в течение 1 ч, затем добавляли 0,5 мл 4 N хлористоводородной кислоты и смесь перемешивали в течение 10 мин. Смесь экстрагировали 2×25 мл DCM, затем органический слой сушили над сульфатом натрия, фильтровали и концентрировали, получая 16-1 (250 мг) в виде желтого масла. Водный слой концентрировали, затем твердый остаток промывали этанолом. Объединенные этанольные фильтраты концентрировали, получая дополнительное количество 16-1 (500 мг) в виде желтого масла.
Пример 17
7-этил-2,5-диметил-3-{2-[2-(1-метилпирролидин-2-ил)этокси]-4-пиразол-1-илфенил}пиразоло[1,5-a]пиримидин
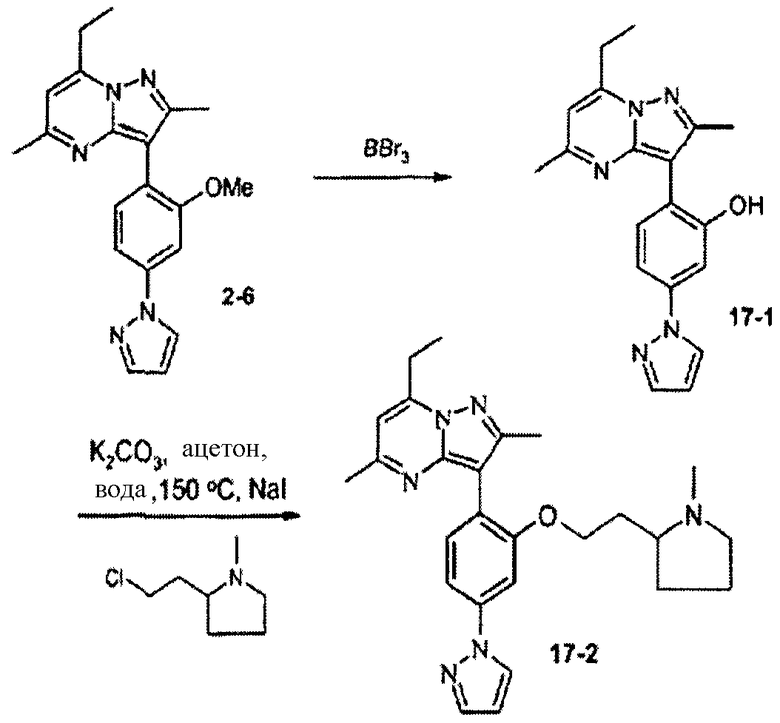
Стадия 17А:
К раствору соединения 2-6 (350 мг) в хлороформе (5 мл) добавляли ВВr3 (1,0 М в DCM, 5 мл). Смесь перемешивали ночь при комнатной температуре и гасили водой. Смесь экстрагировали хлороформом (2×10 мл), затем объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая соединение 17-1 (280 мг) в виде масла. Аликвоту (10 мг) очищали препаративной ВЭЖХ/МС, получая очищенное соединение 17-1 (2,9 мг).
Стадия 17В:
Смесь соединения 17-1 (45 мг, 0,14 ммоль, 1 экв.), карбоната калия (56 мг, 0,41 ммоль, 3 экв.), иодида натрия (20 мг, 0,13 ммоль, 1 экв.), гидрохлорида 2-(2-хлорэтил)-1-метилпирролидина (39 мг, 0,21 ммоль, 1,5 экв.), ацетона (1 мл) и воды (1 мл) нагревали в запаянной трубке в микроволновом реакторе при 150°С в течение 25 мин. Ацетон выпаривали, затем остаток разбавляли метанолом, фильтровали и сразу подвергали препаративной ВЭЖХ/МС очистке, получая соединение 17-2 (14 мг, 20%) в виде TFA соли; М.м.: 444,58; ЖХ/МС: 444 [MH]+; tR: 6,010. Аналитический метод 2.
Пример 18
7-(3-метоксипропил)-3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин
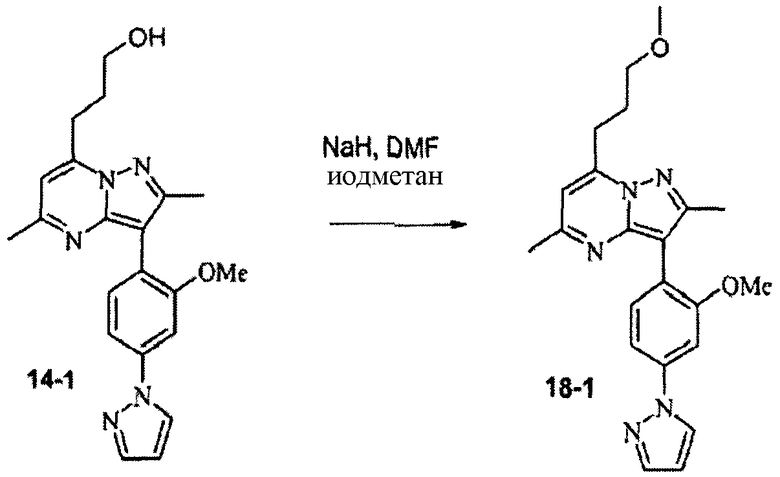
Стадия 18А:
К раствору 14-1 (30 мг) в сухом ДМФА добавляли NaH (10 мг, 60% дисперсия). После перемешивания при комнатной температуре в течение 10 мин добавляли метилиодид (0,015 мл). Смесь перемешивали в течение 1 ч, затем добавляли метанол (1 мл) и смесь сразу подвергали препаративной ВЭЖХ/МС очистке, получая соединение 18-1 (12 мг) в виде TFA соли; М.м.: 391,47; ЖХ/МС: 391 [MH]+; tR: 7,050. Аналитический метод 2.
Пример 19
2-[7-(2-метоксиметилфенил)-2,5-диметилпиразоло[1,5-a]пиримидин-3-ил]-5-пиразол-1-илфенол

Стадия 19А:
Процедуру примера 18 проводили, используя соединение 7-1 в качестве исходного продукта.
В зависимости от примененного алкилгалогенида были синтезированы и очищены препаративной ЖХ-МС соединения, перечисленные в следующей таблице:
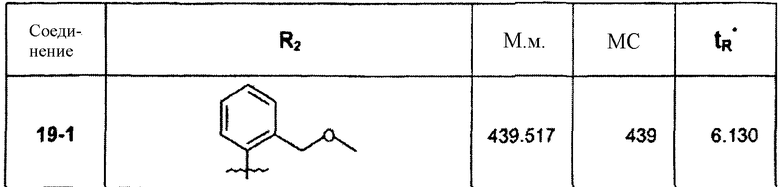
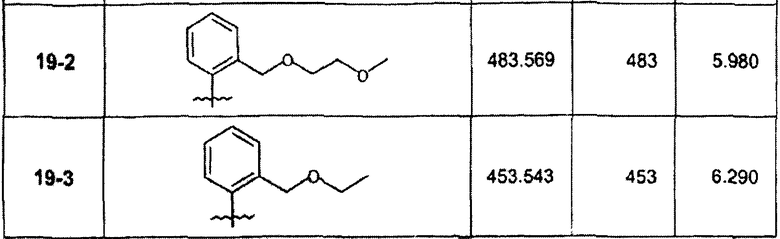
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 20
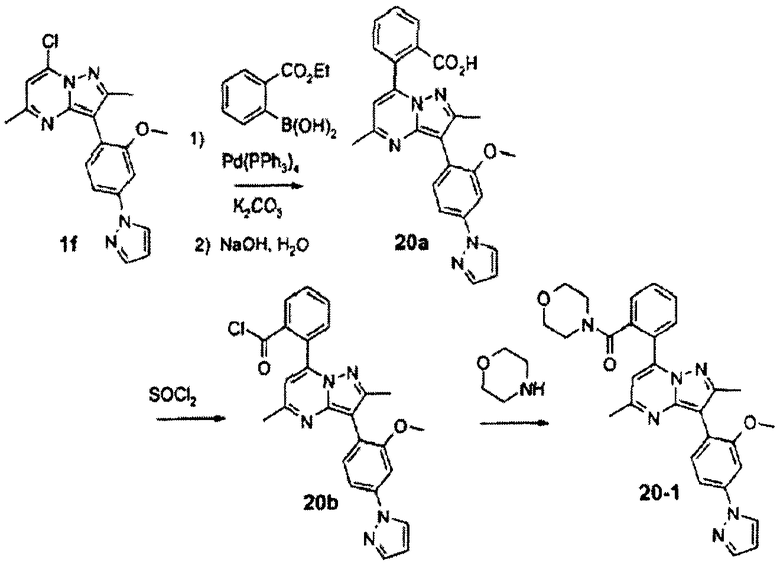
Стадия 20А:
Смесь соединения 1f (710 мг, 2,0 ммоль), (2-этоксикарбонил)фенилбороновой кислоты (470 мг, 2,4 ммоль), тетракис(трифенилфосфин)палладия(0) (116 мг, 0,1 ммоль) и карбоната калия (550 мг, 4,0 ммоль) нагревали в смеси 9:1 диоксан/вода (10 мл) при 100°С в течение 2,5 ч. Добавляли раствор гидроксида натрия (3 N, 10 мл) и смесь перемешивали при 100°С в течение дополнительных 30 мин. Охлажденную смесь концентрировали, затем добавляли воду и рН устанавливали до 2 с помощью хлористоводородной кислоты. Смесь экстрагировали хлороформом, затем объединенные экстракты с хлороформом сушили над сульфатом натрия, фильтровали и концентрировали, получая неочищенное твердое вещество, которое перекристаллизовывали из хлороформа, получая соединение 20а (420 мг, выход 48%) в виде желтого твердого вещества.
Стадия 20В:
Соединение 20а (420 мг, 0,96 ммоль) нагревали в 10 мл хлороформа с тионилхлоридом (1,0 мл, 14 ммоль) при 70°С в течение 2 ч. Летучие компоненты выпаривали, получая соединение 20b (450 мг) в виде темного твердого вещества.
Стадия 20С:
Раствор 20b (32 мг, 0,07 ммоль) в хлороформе (1 мл) обрабатывали морфолином (0,1 мл, 1 ммоль) при комнатной температуре. Смеси позволяли оставаться при комнатной температуре в течение 30 мин, затем растворитель выпаривали. Остаток переводили в метанол, фильтровали и сразу очищали препаративной ВЭЖХ/МС, получая 20-1 (13 мг, 30%) в виде TFA соли. В зависимости от примененного амина были синтезированы и очищены препаративной ВЭЖХ-МС соединения, перечисленные в следующей таблице:
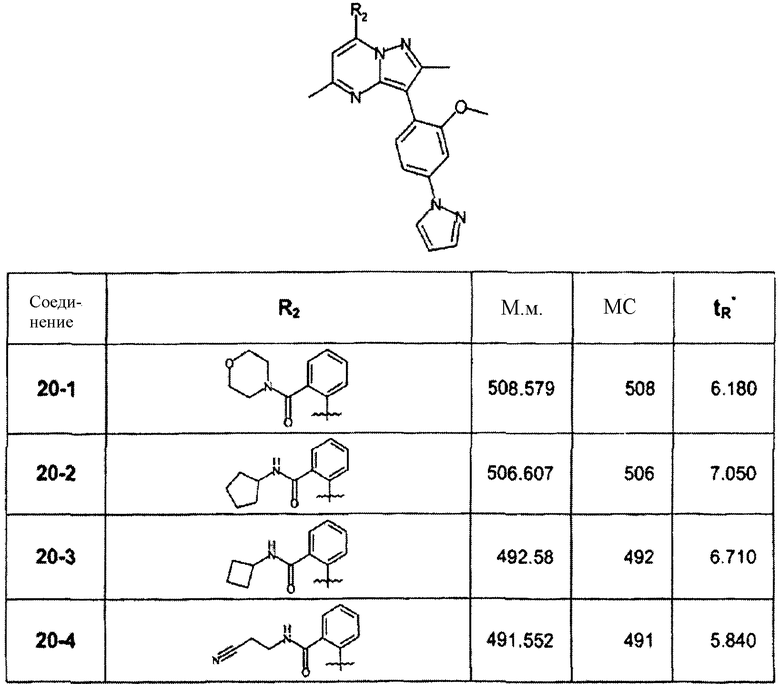
∗ Для всех ВЭЖХ-определений использовали аналитический метод 2.
Пример 21
7-(1-этил-1Н-пиррол-2-ил)-3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин
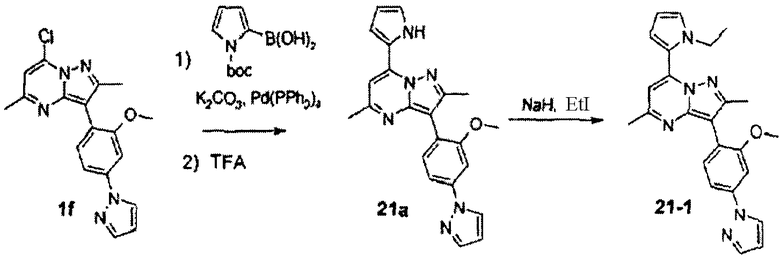
Стадия 21А:
Смесь соединения 1f (210 мг, 0,6 ммоль), N-Boc-пиррол-2-бороновой кислоты (158 мг, 0,75 ммоль), тетракис(трифенилфосфин)палладия(0) (40 мг, 0,035 ммоль) и карбоната калия (166 мг, 1,2 ммоль) нагревали в смеси 9:1 диоксан/вода (5 мл) при 110°С в течение 3 ч в запаянной трубке. Охлажденную смесь концентрировали, затем добавляли воду и смесь экстрагировали хлороформом. Объединенные экстракты с хлороформом сушили над сульфатом натрия, фильтровали и концентрировали, получая неочищенное твердое вещество, которое перемешивали в 1:1 TFA/DCM (3 мл) в течение 16 ч. Смесь разбавляли этилацетатом, затем обрабатывали водным аммиаком. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, затем остаток хроматографировали на силикагеле, используя гексаны/этилацетат в качестве элюента, и получали 21а (110 мг, выход 48%) в виде желтого твердого вещества.
Стадия 21В:
К раствору 21а (110 мг, 0,28 ммоль)) в сухом ДМФА (2 мл) добавляли гидрид натрия (20 мг, 60% дисперсия в минеральном масле, 0,5 ммоль) при комнатной температуре. Смесь перемешивали в течение 5 мин, затем добавляли этилиодид (0,050 мл, 0,60 ммоль) и смесь перемешивали при комнатной температуре в течение 2 ч. Добавляли воду и этилацетат, затем этилацетатный слой промывали водой и насыщенным солевым раствором, затем сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, используя гексаны/этилацетат в качестве элюента, и получали соединение 21-1 (84 мг, выход 73%) в виде желтого твердого вещества; М.м.: 412,50; ЖХ/МС: 412 [MH]+; tR: 7,630. Аналитический метод 2.
Пример 22
7-(3-этил-3Н-имидазол-4-ил)-3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметилпиразоло[1,5-a]пиримидин
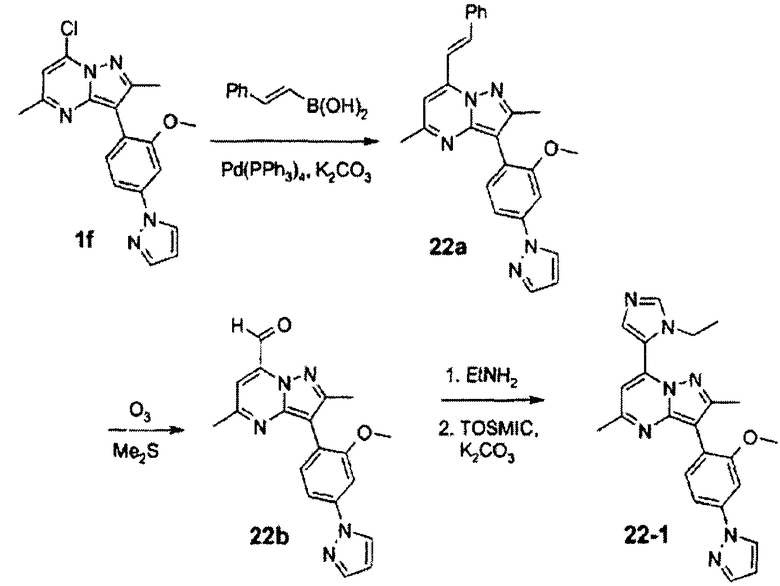
Стадия 22А:
Смесь соединения 1f (1,50 г, 4,25 ммоль), 2-фенилэтенилбороновой кислоты (692 мг, 4,68 ммоль), карбоната калия (1,17 г, 8,50 ммоль) и тетракис(трифенилфосфин)палладия(0) (250 мг, 0,22 ммоль) в диоксане (9 мл) и воде (1 мл) нагревали при 105°С в течение 16 ч. Смесь разбавляли этилацетатом и промывали насыщенным солевым раствором. Органический слой сушили над сульфатом натрия, фильтровали и концентрировали, и остаток хроматографировали на силикагеле, используя гексаны/этилацетат в качестве элюента, и получали 22а (1,60 г, выход 89%) в виде желтого твердого вещества.
Стадия 22В:
Смесь озон/кислород барботировали через раствор 22а (1,60 г, 3,8 ммоль) в сухой смеси 2:1 DCM/метанол (20 мл) при -70°С в течение 8 минут. Добавляли диметилсульфид (1,5 мл) и смесь перемешивали и позволяли ей нагреваться при комнатной температуре в течение 16 ч. Растворитель выпаривали и остаток хроматографировали на силикагеле, используя гексаны/этилацетат в качестве элюента и получая соединение 22b (1,0 г, выход 76%) в виде желтого твердого вещества.
Стадия 22С:
Смесь 22b (35 мг, 0,10 ммоль), этиламина (1,0 мл 2,0 М раствора в ТГФ, 2,0 ммоль) и сульфата магния в 1,2-дихлорэтане перемешивали при комнатной температуре в течение 15 ч. Смесь фильтровали, затем фильтрат упаривали досуха. Остаток переводили в 1:1 этанол/DME (2 мл), затем добавляли TOSMIC (38 мг, 0,19 ммоль) и карбонат калия (55 мг, 0,4 ммоль) и смесь кипятили с обратным холодильником в течение 17 ч. Добавляли воду и смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, используя гексаны/этилацетат в качестве элюента и получая соединение 22-1 (5 мг) в виде масла; М.м.: 413,48; ЖХ/МС: 413 [MH]+; tR: 5,000. Аналитический метод 2.
Пример 23
3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметил-7-(4-метилоксазол-5-ил)пиразоло[1,5-a]пиримидин
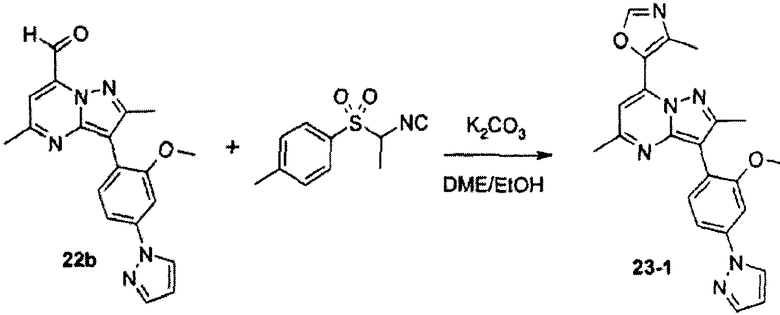
Смесь 22b (208 мг, 0,60 ммоль), альфа-метил-TOSMIC (251 мг, 1,2 ммоль) и карбоната калия (248 мг, 1,8 ммоль) нагревали в 5 мл смеси 1:1 DME/этанол при 80°С в течение 14 ч. Добавляли воду и смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, используя гексаны/этилацетат в качестве элюента и получая 23-1 (60 мг, 23%) в виде масла; М.м.: 400,44; ЖХ/МС: 400 [MH]+; tR: 5,250. Аналитический метод 2.
Пример 24
7-(4-фторбензил)-2,5-диметил-3-(4-метил-6-пиррол-1-илпиридин-3-ил)пиразоло[1,5-a]пиримидин

Стадия 24А:
К раствору 4-фторфенилцинкхлорида (20 мл 0,5 М раствора в ТГФ, 10 ммоль) добавляли соединение 10b (1,0 г, 5,5 ммоль) и тетракис(трифенилфосфин)палладий(0) (300 мг, 0,26 ммоль). Реакционную смесь нагревали в запаянной трубке при 90°С в течение 3 ч. Охлажденную реакционную смесь обрабатывали 4 N хлористоводородной кислотой (4 мл), затем добавляли воду и смесь экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищали хроматографией на силикагеле, элюируя 30% этилацетатом в гексанах, и получали 24а (1,0 г, выход 71%) в виде не совсем белого твердого вещества.
Стадия 24В:
Соединение 24а (1,0 г, 3,9 ммоль) растворяли в 15 мл метанола. К данному раствору добавляли по каплям бром (0,62 г, 3,9 ммоль), что приводило к образованию белого осадка. Твердое вещество собирали на фильтре со стеклянной пористой пластинкой и промывали метанолом. Данное соединение затем очищали колоночной хроматографией на силикагеле, элюируя смесью 4:1 гексаны/этилацетат, и получали первый продукт дибромирования (110 мг, выход 7%) и затем соединение 24b (1,0г, выход 77%) в виде белого твердого вещества.
Стадия 24С:
Смесь соединения 24b (800 мг, 2,4 ммоль), соединения 16-1 (500 мг, 2,5 ммоль), тетракис(трифенилфосфин)палладия(0) (280 мг, 0,24 ммоль) и карбоната калия (600 мг, 4,3 ммоль) нагревали в смеси 9:1 диоксан/вода (3,5 мл) при 95°С в течение 3 ч в запаянной трубке. К охлажденной смеси добавляли водный раствор бикарбоната натрия (5 мл) и затем смесь экстрагировали дважды с помощью DCM. Объединенные DCM-экстракты сушили над сульфатом натрия, фильтровали и концентрировали, получая неочищенное масло, которое частично очищали препаративной ВЭЖХ/MC. Частично очищенный продукт затем хроматографировали на силикагеле, используя 4:1 гексаны/этилацетат в качестве элюента и получая соединение 24-1 (3 мг) в виде желтого твердого вещества; М.м.: 411,48; ЖХ/МС: 412 [MH]+; tR: 9,160. Аналитический метод 2.
Пример 25
3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметил-7-(1-метил-1Н-имидазол-2-ил)пиразоло[1,5-a]пиримидин
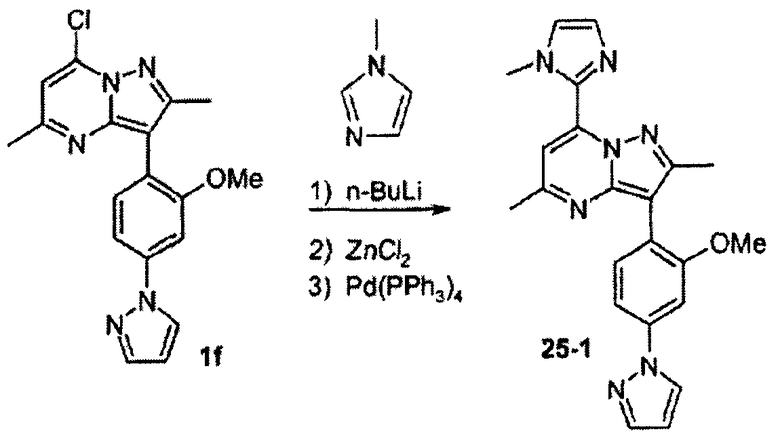
Стадия 25А:
К раствору 1-метилимидазола (246 мг, 3,0 ммоль) в сухом ТГФ (3 мл), охлажденному до -70°С, добавляли по каплям n-BuLi (2,5 M раствор в гексане, 1,7 мл, 4,2 ммоль). Реакционную смесь перемешивали при -70°С в течение 10 мин, затем добавляли ZnCl2 (0,5 M раствор в ТГФ, 20 мл, 10 ммоль) за 5 мин. Смесь перемешивали при -70°С в течение 1 ч, затем нагревали до 0°С. Добавляли соединение 1f (106 мг, 0,30 ммоль) и тетракис(трифенилфосфин)палладий(0) (70 мг, 0,06 ммоль). Смесь нагревали до кипения с обратным холодильником в течение 3 ч. Охлажденную реакционную смесь гасили водой, выпаривали ТГФ и полученную водную смесь экстрагировали этилацетатом. Объединенные органические слои сушили над сульфатом натрия, фильтровали, концентрировали и остаток хроматографировали на силикагеле, используя этилацетат в качестве элюента, и получали 25-1 (15 мг) в виде желтого твердого вещества; ВЭЖХ время удерживания 4,13 мин (метод 2); М.м. 399,5; наблюдаемая МС 399.
Пример 26
3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметил-7-(2-метил-2Н-пиразол-3-ил)пиразоло[1,5-a]пиримидин
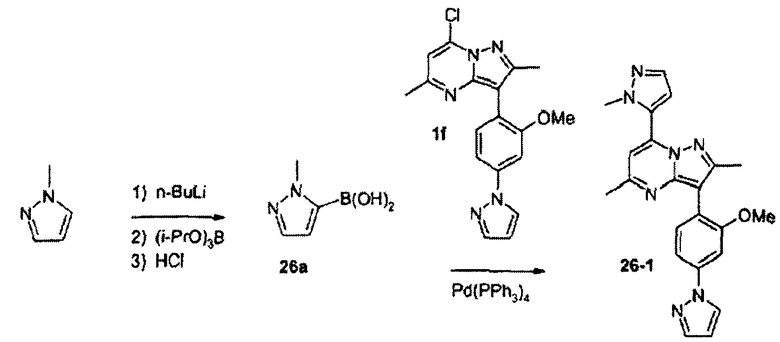
Стадия 26А:
К раствору 1-метилпиразола (820 мг, 10 ммоль) в сухом ТГФ (20 мл), охлажденному до -70°С, добавляли по каплям n-BuLi (1,6 M раствор в гексане, 6,3 мл, 10 ммоль). Реакционную смесь перемешивали при -70°С в течение 5 мин, затем добавляли триизопропилборат (2,5 мл, 11 ммоль) за 5 мин. Смеси позволяли нагреваться до комнатной температуры около 1 ч, затем добавляли 6 N хлористоводородную кислоту (5 мл). Смесь перемешивали в течение 30 мин, затем смесь упаривали досуха, получая сырое 26а в виде твердого вещества, которое применяли без дальнейшей очистки.
Стадия 26В:
Соединение 1f (530 мг, 1,5 ммоль) и неочищенное 26а (все количество, приблизительно 10 ммоль) были введены в реакцию Сузуки по процедуре примера 1. Реакционную смесь концентрировали, затем добавляли воду и смесь экстрагировали хлороформом. Объединенные органические экстракты сушили над сульфатом натрия, фильтровали и концентрировали, затем остаток очищали хроматографией на силикагеле, используя гексаны/этилацетат в качестве элюента. Продукт затем очищали кристаллизацией из ацетонитрила, получая соединение 26-1 (280 мг) в виде желтого твердого вещества; ВЭЖХ время удерживания 6,42 мин (метод 2); М.м. 399,5; наблюдаемая МС 399.
Пример 27
Активность на связывание с CRF-рецептором
Соединения данного изобретения могут быть оценены на связывающую активность с CRF-рецептором стандартным анализом радиолигандного связывания, как в общем описано Grigoriadis et al. (Mol. Pharmacol. Vol. 50, pp 679-686, 1996) и Hoare et al. (Mol. Pharmacol. Vol. 63, pp 751-765, 2003). Использованием меченных радиоактивным изотопом CRF-лигандов данный анализ может быть применен для оценки активности на связывание соединений данного изобретения с любым подтипом CRF-рецепторов.
Вкратце, анализ на связывание включает замещение меченного радиоактивным изотопом CRF-лиганда от CRF-рецептора. Более конкретно, анализ на связывание выполняют в 96-луночных оценочных планшетах использованием 1-10 мкг клеточных мембран от клеток, трансфицированных CRF-рецепторами человека. Каждая лунка получает примерно 0,05 мл оценочного буфера (например, фосфатный буферный физиологический раствор Дулбекко (Dulbecco's), 10 мМ хлорид магния, 2 мМ EGTA), содержащего соединение, представляющее интерес, или эталонный лиганд (например, соувагин/sauvagine, урокортин I или CRF), 0,5 мл [125I]тирозин-соувагин (конечная концентрация ~150 пМ или приблизительно КD, определенная по анализу Скэтчарда/Scatchard) и 0,1 мл суспензии клеточных мембран, содержащей CRF-рецептор. Смесь инкубируют в течение 2 ч при 22°С с последующим отделением связанного и свободного радиолиганда быстрым фильтрованием через стекловолокнистые фильтры. После трех промываний фильтры сушат и считают радиоактивность (электроны Оже от 125I), используя сцинтилляционный счетчик. Все данные по радиолигандному связыванию могут быть проанализированы с помощью нелинейных выравнивающих методом наименьших квадратов аппроксимирующих программ Prism (GraphPad Software Inc) или XLfit (ID Business Solution Ltd).
Пример 28
CRF-стимулируемая активность аденилатциклазы
Соединения данного изобретения могут быть оценены различными функциональными тестами. Например, соединения данного изобретения могут быть отобраны по CRF-стимулируемой активности аденилатциклазы. Количественная оценка по определению CRF-стимулируемой активности аденилатциклазы может быть выполнена, как описано в целом у Battaglia et al. (Synapse 1:572, 1987), c модификациями для пригодности анализа к препаратам с целыми клетками.
Более конкретно, стандартная оценочная смесь может содержать в конечном объеме из 0,1 мл следующие компоненты: 2 мМ L-глутамин, 20 мМ HEPES и 1 мМ IMBX в DMEM буфере. В исследованиях по стимулированию целые клетки с трансфицированными CRF-рецепторами помещают в 96-луночные планшеты и инкубируют в течение 30 мин при 37°С с различными концентрациями CRF-родственных и неродственных пептидов для установления фармакологического оценочно-порядкового профиля конкретного рецепторного субтипа. После инкубации измеряют сАМР в образцах, используя стандартные коммерчески доступные комплекты, такие как сАМР-Screen™ от Applied Biosystems. Для функциональной оценки соединений клетки и единичные концентрации CRF или родственных пептидов, вызывающих 50% стимулирование образования сАМР, инкубируют вместе с различными концентрациями конкурирующих соединений в течение 30 мин при 37°С и определяют сАМР, как описано выше.
Следует отметить, что хотя определенные варианты осуществления изобретения были описаны в данной работе для иллюстративных целей, тем не менее могут быть сделаны различные модификации без отклонения от сущности и объема изобретения. В соответствии с этим изобретение не ограничено только заявленными пунктами изобретения.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ГАЛОГЕНАЛЛИЛАМИНА И ИХ ПРИМЕНЕНИЕ | 2020 |
|
RU2793850C2 |
| ИНГИБИТОРЫ IAP | 2005 |
|
RU2401840C2 |
| ПРОИЗВОДНЫЕ БЕНЗИМИДАЗОЛИДИНОНА В КАЧЕСТВЕ АГЕНТОВ МУСКАРИНОВЫХ РЕЦЕПТОРОВ | 2002 |
|
RU2288919C2 |
| ТРИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ОБЕСПЕЧИВАЮЩИЕ РАЗРУШЕНИЕ БЕЛКА IKAROS И БЕЛКА AIOLOS | 2020 |
|
RU2833608C2 |
| МОДУЛЯТОРЫ КАЛЬПАИНА И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2773288C2 |
| АМИНОЗАМЕЩЕННЫЕ ЭТИЛАМИНО-АГОНИСТЫ β-АДРЕНЕРГИЧЕСКИХ РЕЦЕПТОРОВ | 2004 |
|
RU2385317C2 |
| ПРОТИВОФИБРОЗНЫЕ ПИРИДИНОНЫ | 2015 |
|
RU2692485C2 |
| НОВЫЙ АГОНИСТ β-РЕЦЕПТОРА ТИРЕОИДНЫХ ГОРМОНОВ | 2021 |
|
RU2839610C1 |
| ПРОЛЕКАРСТВА АНТАГОНИСТОВ ОПИОИДНЫХ РЕЦЕПТОРОВ | 2018 |
|
RU2785777C2 |
| ИНГИБИТОРЫ IAP | 2013 |
|
RU2728789C2 |
Изобретение относится к 3-(2-метокси-4-пиразол-1-илфенил)-2,5-диметил-7-(3-метилпиридин-2-ил)пиразоло[1,5-а]пиримидину или его фармацевтически приемлемым солям, сольвату, стереоизомеру, имеющему приведенную ниже структурную формулу, которые являются антагонистами CRF-рецепторов и могут быть применены в лечении разнообразных расстройств, вызывающих гиперсекрецию CRF у теплокровных животных, такую как при внезапном приступе. Также изобретение относится к промежуточным соединениям, фармацевтической композиции на основе указанного соединения и способу лечения расстройства, вызывающего гиперсекрецию CRF у млекопитающего. 9 н. 6 з.п. ф-лы, 14 табл.
1. 3-(2-Метокси-4-пиразол-1-илфенил)-2,5-диметил-7-(3-метилпиридин-2-ил)пиразоло[1,5-а]пиримидин или его фармацевтически приемлемая соль, сольват, стереоизомер, представленный формулой:
2. 3-(2-Метокси-4-пиразол-1-илфенил)-2,5-диметил-7-(3-метилпиридин-2-ил)пиразоло[1,5-а]пиримидин.
3. Соединение, представленное формулой
в качестве промежуточного соединения для получения соединения по п.1 или 2.
4. Соединение, представленное формулой

в качестве промежуточного соединения для получения соединения по п.1 или 2.
5. Соединение, представленное формулой
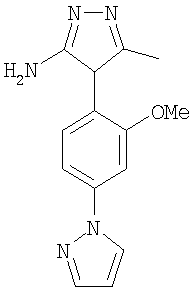
в качестве промежуточного соединения для получения соединения по п.1 или 2.
6. Соединение, представленное формулой
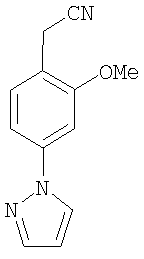
в качестве промежуточного соединения для получения соединения по п.1 или 2.
7. Соединение, представленное формулой
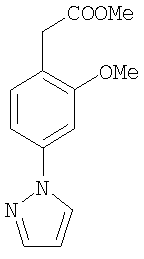
в качестве промежуточного соединения для получения соединения по п.1 или 2.
8. Фармацевтическая композиция, обладающая свойствами антагониста CRF-рецепторов, содержащая соединение по п.1 в сочетании с фармацевтически приемлемым носителем или разбавителем.
9. Способ лечения расстройства, вызывающего гиперсекрецию CRF у млекопитающих, включающий введение животному эффективного количества фармацевтической композиции по п.8.
10. Способ по п.9, в котором расстройство представляет собой удар.
11. Способ по п.9, в котором расстройство представляет собой депрессию.
12. Способ по п.9, в котором расстройство представляет собой расстройство, связанное с состоянием тревоги/страха.
13. Способ по п.9, в котором расстройство представляет собой обсессивно-компульсивное расстройство.
14. Способ по п.9, в котором расстройство представляет собой синдром раздраженной толстой кишки.
15. Способ по п.9, в котором расстройство представляет собой анорексию невроза.
| US 6313124 B1, 06.11.2001 | |||
| Дезинтегратор | 1922 |
|
SU803A1 |
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Система автоматического управления процессом замочки зерна | 1986 |
|
SU1344779A1 |
| Дорожная спиртовая кухня | 1918 |
|
SU98A1 |

