Область техники настоящего изобретения
Настоящее изобретение относится к новому имидазольному производному, обладающему ингибирующей c-Jun-N-терминальную киназу (JNK) активностью, и его применению.
Уровень техники настоящего изобретения
Параллельно с современным ростом стареющего населения, наблюдается быстрый рост пациентов, страдающих от дегенеративных заболеваний нервной системы мозга. Дегенеративные заболевания нервной системы мозга могут возникать в результате вызванных старением структурных нарушений нервных клеток мозга; вторичных симптомов, вызванных заболеваниями у взрослых, такими как расстройство кровообращения и т.д.; или физических, механических факторов, таких как дорожные происшествия, производственные травмы, отравление монооксидом углерода и т.д., где болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, множественный склероз, инсульт и подобные известны в качестве соответствующих заболеваний.
Между тем, C-Jun N-терминальную киназу (JNK), которую классифицируют как серин-треониновая киназа, также называют стресс-активируемой протеинкиназой (SAPK), которая представляет собой один из трех подтипов митоген-активируемой протеинкиназы. JNK активируется в реакции с различными стимулами, такими как цитокин, митоген, осмотический стресс, ультрафиолетовое излучение и т.д., где известно, что данная активированная JNK стимулирует фосфорилирование множества транскрипционных факторов, включая C-Jun AP-1, а также фосфорилирование внутриклеточных белков, таких как Bcl2, p53 и т.д., которые связаны с апоптозом. Кроме того, гены JNK образуют различные изоформы белка за счет сплайсинга. Помимо этого, распределение JNK3 сконцентрировано в тканях мозга, в отличие от приблизительно 10 других изоформ белка того же типа, так что имеются многочисленные исследования, проводимые в настоящее время, связи JNK3 и дегенеративных заболеваний нервной системы мозга.
В частности, JNK3 осуществляет фосфорилрование-активацию белка-предшественника амилоида (APP), который представляет собой основную причину болезни Альцгеймера, вследствие того, что APP расположен на клеточной мембране, и стимулируется его превращение в бета-амилоид. При этом образуется бета-амилоид, после чего его возникающая при этом токсичность вызывает некроз нервных клеток. В данном случае, сообщают, что активация JNK3 является основным фактором. Кроме того, наблюдали, что мышь с наследственной болезнью Альцгеймера (FAD) показывала заметное снижение олигомерного бета-амилоида и улучшение когнитивных способностей за счет удаления JNK3, и также обнаружено, что мышь с удаленным JNK3 геном показывала приобретение устойчивости к MPTP, веществу, вызывающему болезнь Паркинсона; приобретала ингибирующий эффект на побочные реакции к глютаматным аналогом, нейротоксическому веществу; и подобные.
На данном уровне техники, существуют активно проводимые исследования по обнаружению JNK3 ингибитора в качестве нового вещества для лечения дегенеративных заболеваний нервной системы мозга (корейская патентная публикация No. 2001-0029352), но они еще недостаточны для обеспечения удовлетворительных результатов.
Подробное описание настоящего изобретения
Техническая проблема
Настоящее изобретение направлено на решение приведенных выше проблем, где изобретатели настоящего изобретения провели типовое исследование для обнаружения нового вещества, обладающего потенциалом для разработки терапевтического агента для дегенеративных заболеваний нервной системы мозга, таким образом, обнаружив новое имидазольное производное, обладающее JNK ингибирующей активностью и, соответственно, завершили настоящее изобретение.
Соответственно, цель настоящего изобретения заключается в обеспечении нового имидазольного производного или его фармацевтически приемлемой соли, обладающих JNK ингибирующей активностью.
Другая цель настоящего изобретения заключается в обеспечении способа получения нового имидазольного производного, обладающего JNK ингибирующей активностью.
Другая цель настоящего изобретения заключается в обеспечении фармацевтической композиции для предотвращения или лечения дегенеративных заболеваний нервной системы мозга, содержащей имидазольное производное выше или его фармацевтически приемлемую соль в качестве эффективного компонента.
Однако техническая цель, которая достигается настоящим изобретением, не ограничивается задачами, приведенными выше, и другие задачи, не приведенные в настоящем изобретении, могут быть ясны специалисту в данной области техники из последующего описания.
Техническое решение
Для достижения целей настоящего изобретения выше, настоящее изобретение обеспечивает имидазольное производное, представленное следующей формулой 1, или его фармацевтически приемлемую соль.
[формула 1]
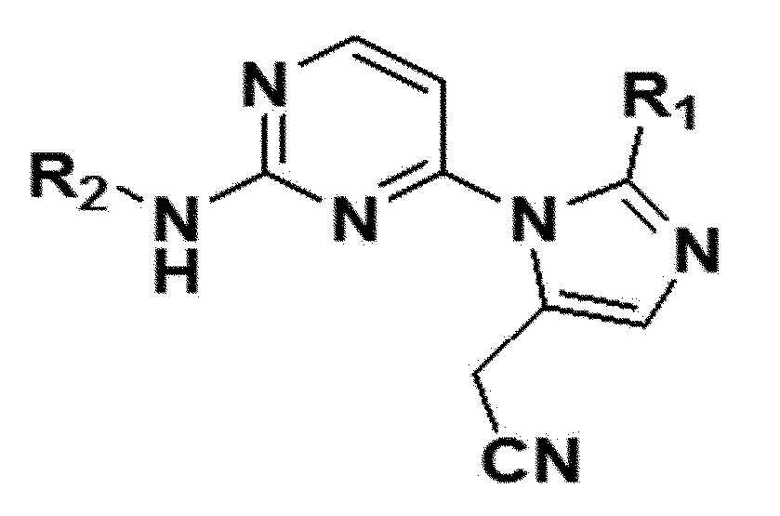
в формуле 1 выше
R1 представляет собой C4-C10 арил, C4-C10 гетероарил или C4-C10 гетероциклоалкил,
где C4-C10 арил и C4-C10 гетероарил представляет собой арил или гетероарил, выбранный из группы, состоящей из фенила, нафтила, пиренила, карбазолила, бензоксазолила, бензодиоксазолила, 1,3-бензодиоксолила, 1,4-бензодиоксинила, бензотиазолила, бензоимидазолила, бензотиофенила, хинолинила, изохинолинила, индолила, бензофуранила, пуринила и индолизинила,
где C4-C10 гетероциклоалкил представляет собой гетероциклоалкил, выбранный из группы, состоящей из тетрагидрофуранила, тетрагидротиофенила, пирролидинила, тетрагидропиранила, тетрагидротиопиранила, пиперидинила и дигидробензодиоксинила,
и где R1 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, C1-C6 алкокси, C1-C6 галогеналкила, гидрокси, амино и галогена; и
R2 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкокси, C1-C6 спирт, C3-C10 циклоалкил или C4-C10 гетероциклоалкил,
где C4-C10 гетероциклоалкил представляет собой гетероциклоалкил, выбранный из группы, состоящей из тетрагидрофуранила, тетрагидротиофенила, пирролидинила, тетрагидропиранила, тетрагидротиопиранила, пиперидинила и дигидробензодиоксинила,
и где R2 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, C1-C6 алкокси, C1-C6 галогеналкила, гидрокси, амино, галогена, C2-C10 алкилкарбонила и C4-C10 циклоалкилкарбонила.
Кроме того, настоящее изобретение, как показано на следующей реакционной формуле 1, обеспечивает способ получения имидазольного производного выше, включающий стадии:
проведения реакции конденсации с аминированием по Бухвальду между соединением формулы I и 4-хлор-2-(метилтио)пиримидином, получая соединение формулы II (стадия 1);
окисления соединения формулы II, полученного на стадии 1 выше, получая соединение формулы III (стадия 2); и
замещение метилсульфонильной группы соединения формулы III, полученного на стадии 2 выше, аминогруппой, получая соединение формулы IV (стадия 3).
[реакционная формула 1]

(в формулах I-IV выше, R1 и R2 представляют собой, как определено в формуле 1 по п. 1.)
Кроме того, настоящее изобретение обеспечивает фармацевтическую композицию для предотвращения или лечения дегенеративных заболеваний нервной системы мозга, содержащую производное формулы 1 выше или его фармацевтически приемлемую соль в качестве эффективного компонента.
В одном примерном варианте осуществления настоящего изобретения, дегенеративные заболевания нервной системы мозга выше можно выбрать из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, болезни Хантингтона, множественного склероза и инсульта.
В другом примерном варианте осуществления настоящего изобретения, композиция выше может ингибировать активность киназы, выбранной из группы, состоящей из C-Jun N-терминальной киназы 1 (JNK 1), C-Jun N-терминальной киназы 2 (JNK 2) и C-Jun N-терминальной киназы 3 (JNK 3).
Кроме того, настоящее изобретение обеспечивает способ лечения дегенеративных заболеваний нервной системы мозга, включающий стадию введения производного формулы 1 выше или его фармацевтически приемлемой соли индивиду.
Более того, настоящее изобретение обеспечивает применение производного формулы 1 выше или его фармацевтически приемлемой соли для лечения дегенеративных заболеваний нервной системы мозга.
Полезные эффекты
Новое имидазольное производное или его фармацевтически приемлемая соль согласно настоящему изобретению обладает превосходной ингибирующей C-Jun N-терминальную киназу активностью (JNK) и, таким образом, предполагается, что фармацевтическая композиция, содержащая производное выше, можно с пользой применять в предотвращении и лечении дегенеративных заболеваний нервной системы мозга.
Лучший способ настоящего изобретения
Далее, настоящее изобретение будет описано более подробно.
Настоящее изобретение обеспечивает имидазольное производное, представленное следующей формулой 1, или его фармацевтически приемлемую соль:
[Формула 1]
В формуле 1 выше,
R1 представляет собой C4-C10 арил, C4-C10 гетероарил или C4-C10 гетероциклоалкил, где C4-C10 арил и C4-C10 гетероарил представляют собой арил или гетероарил, выбранный из группы, состоящей из фенила, нафтила, пиренила, карбазолила, бензоксазолила, бензодиоксазолила, 1,3-бензодиоксолила, 1,4-бензодиоксинила, бензотиазолила, бензоимидазолила, бензотиофенила, хинолинила, изохинолинила, индолила, бензофуранила, пуринила и индолизинила, где C4-C10 гетероциклоалкил представляет собой гетероциклоалкил, выбранный из группы, состоящей из тетрагидрофуранила, тетрагидротиофенила, пирролидинила, тетрагидропиранила, тетрагидротиопиранила, пиперидинила и дигидробензодиоксинила, и где R1 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, C1-C6 алкокси, C1-C6 галогеналкила, гидрокси, амино и галогена; и
R2 представляет собой C1-C6 алкил, C2-C6 алкенил, C2-C6 алкинил, C1-C6 алкокси, C1-C6 спирт, C3-C10 циклоалкил или C4-C10 гетероциклоалкил, где C4-C10 гетероциклоалкил представляет собой гетероциклоалкил, выбранный из группы, состоящей из тетрагидрофуранила, тетрагидротиофенила, пирролидинила, тетрагидропиранила, тетрагидротиопиранила, пиперидинила и дигидробензодиоксинила, и где R2 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, C1-C6 алкокси, C1-C6 галогеналкила, гидрокси, амино, галогена, C2-C10 алкилкарбонила и C4-C10 циклоалкилкарбонила.
В настоящем изобретении, ʺзамещеннаяʺ группа представляет собой группу, в которой, по меньшей мере, один атом водорода замещен, по меньшей мере, одной группой, отличной от атома водорода, но требуется, чтобы удовлетворялись требования валентности и в результате замещения получалось химически стабильное соединение. В настоящем описании, следует понимать, что все заместители можно замещать или не замещать, если явно не описано как ʺнезамещенныйʺ в настоящем изобретении. Каждый заместитель R1 и R2 имидазольного производного согласно настоящему изобретению может быть снова замещен, по меньшей мере, одним из заместителей, определенных выше.
ʺАлкилʺ обычно обозначает линейные и разветвленные насыщенные углеводородные группы, содержащие указанное количество атомов углерода (например, 1-12 атомов углерода). Примеры алкильной группы включают, без ограничения, метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил, трет-бутил, н-пентил, н-гексил, н-гептил и подобные. Алкил может быть соединен с исходной группой или субстратом по любому кольцевому атому, если его присоединение не нарушает требований валентности. Аналогично, алкильная или алкенильная группа может содержать, по меньшей мере, один неводородный заместитель, если его присоединение не нарушает требований валентности.
ʺЦиклоалкилʺ относится к насыщенным моноциклическим и полициклическим углеводородным кольцам, обычно содержащим указанное количество атомов углерода в кольце (т.е., C3-10 циклоалкил относится к циклу, содержащему 3, 4, 5, 6, 7, 8, 9 или 10 атомов углерода в качестве кольцевых членов). ʺГетероциклоалкилʺ относится к моноциклическим и полициклическим гетерокольцам, содержащим 1-4 гетероатома, независимо выбранные из азота, кислорода и серы, где примеры гетероциклоалкила включают, без ограничения, тетрагидрофуранил, тетрагидротиофенил, пирролидинил, тетрагидропиранил, тетрагидротиопиранил, пиперидинил, дигидробензодиоксинил и подобные. Циклоалкил и гетероциклоалкил можно присоединять к исходной группе или субстрату по любому кольцевому атому, если их присоединение не нарушает требований валентности. Аналогично, циклоалкил и гетероциклоалкил может содержать, по меньшей мере, один неводородный заместитель, если их присоединение не нарушает требований валентности.
ʺАрилʺ относится к каждой из моновалентных и дивалентных ароматических групп, включая 5- и 6-членные моноциклические ароматические или полициклические ароматические группы, и ʺгетероарилʺ относится к каждой из моновалентных и двухвалентных ароматических групп, включая 5- и 6-членные моноциклические ароматические группы, содержащие 1-4 гетероатома, независимо выбранные из азота, кислорода и серы. Примеры моноциклической арильной группы и гетероарильной группы включают, без ограничения, фенил, пиридинил, фуранил, пирролил, тиофенил, тиазолил, изотиазолил, имидазолил, триазолил, тетразолил, пиразолил, оксазолил, изоксазолил, пиразинил, пиридазинил, пиримидинл, нафтил и т.д. Арильная группа и гетероарильная группа также включает бициклическую группу, трициклическую группу и т.д., включая конденсированные 5- и 6-членные кольца, определенные выше. Примеры полициклической арильной группы и гетероарильной группы включают, без ограничения, изохинолинил, нафтил, бифенил, антраценил, пиренил, карбазолил, бензоксазолил, бензодиоксазолил, бензодиоксинил, бензотиазолил, бензоимидазолил, бензотиофенил, хинолинил, индолил, бензофуранил, фуринил, индолизинил и т.д. Арильную группу и гетероарильную группу выше можно присоединять к исходной группе или субстрату по любому кольцевому атому, если их присоединение не нарушает требований валентности. Аналогично, арильная группа и гетероарильная группа может содержать, по меньшей мере, один неводородный заместитель, если данные замещения не нарушают требований валентности. Неводородный заместитель арильной группы и гетероарильной группы можно также замещать дополнительным неводородным заместителем.
ʺКарбонилʺ представляет собой -C(O)R'. В настоящем описании, (O) обозначает то, что атом кислорода соединен с атомом, таким как углерод или сера двойной связью. В настоящем изобретении, R' представляет собой неводородный заместитель, такой как низший алкил, низший алкокси и т.д. Примеры карбонильной группы включают, без ограничения, 2-метоксиоксоэтил, 3-метоксиоксопропил и т.д. Карбонил можно присоединять к исходной группе или субстрату по любому кольцевому атому, если его присоединение не нарушает требований валентности. Аналогично, карбонильная группа может содержать, по меньшей мере, один неводородный заместитель, если его присоединение не нарушает требований валентности.
ʺАлкоксиʺ относится к алкил-O-, где алкил определяют выше. Примеры алкокси группы включают, без ограничения, метокси, этокси и т.д. Алкокси можно присоединять к исходной группе или субстрату по любому кольцевому атому, если его присоединение не нарушает требований валентности. Аналогично, алкокси группа может содержать, по меньшей мере, один неводородный заместитель, если его присоединение не нарушает требований валентности.
Кроме того, имидазольное производное формулы 1 выше может содержать его рацемат или соединение изомерной формы.
В имидазольном производном формулы 1 настоящего изобретения, R1 представляет собой фенил, нафтил, 1,3-бензодиоксолил, хинолинил, 2,3-дигидро-1,4 бензодиоксинил или бензофуранил, где R1 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1-C6 алкила, C2-C6 алкенила, C2-C6 алкинила, C1-C6 алкокси, C1-C6 галогеналкила, гидрокси, амино и галогена; и
R2 представляет собой C1-C6 спирт, C3-C10 циклоалкил или C4-C10 гетероциклоалкил,
где C4-C10 гетероциклоалкил представляет собой гетероциклоалкил, выбранный из группы, состоящей из тетрагидрофуранила, тетрагидротиофенила, пирролидинила, тетрагидропиранила, тетрагидротиопиранила, пиперидинила и дигидробензодиоксинила, и где R2 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1-C6 алкила, C1-C6 алкенила, C1-C6 алкинила, C1-C6 алкокси, C1-C6 галогеналкила, гидрокси, амино, галогена, C2-C10 алкилкарбонила и C4-C10 циклоалкилкарбонила.
В другом примерном варианте осуществления настоящего изобретения, R1 представляет собой фенил, нафтил, 1,3-бензодиоксолил, хинолинил, 2,3-дигидро-1,4-бензодиоксинил или бензофуранил, где R1 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1-C6 алкила, C1-C6 галогеналкила и галогена; и
R2 представляет собой C1-C6 спирт, C3-C10 циклоалкил или C4-C10 гетероциклоалкил,
где C4-C10 гетероциклоалкил представляет собой гетероциклоалкил, выбранный из группы, состоящей из тетрагидрофуранила, тетрагидротиофенила, пирролидинила, тетрагидропиранила, тетрагидротиопиранила и пиперидинила, и где R2 может быть незамещенным или замещенным, по меньшей мере, одним заместителем C2-C10 алкилкарбонила или C4-C10 циклоалкилкарбонила.
В другом примерном варианте осуществления настоящего изобретения, R1 представляет собой фенил, нафтил, 1,3-бензодиоксолил, хинолинил, 2,3-дигидро-1,4-бензодиоксинил или бензофуранил, где фенил может быть незамещенным или замещенным, по меньшей мере, одним заместителем C1-C6 галогеналкила или галогена; и
R2 представляет собой 2-гидроксипропил, циклогексил, тетрагидропиранил или пиперидинил, где пиперидинил может быть незамещенным или замещенным, по меньшей мере, одним заместителем C4-C10 циклоалкилкарбонила.
В другом примерном варианте осуществления настоящего изобретения, R1 представляет собой фенил, нафтил, 1,3-бензодиоксолил, хинолинил, 2,3-дигидро-1,4-бензодиоксинил или бензофуранил, где фенил может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из фтора, хлора и трифторметила; и
R2 представляет собой 2-гидроксипропил, циклогексил, тетрагидропиранил или пиперидинил, где пиперидинил может быть незамещенным или замещенным циклопропанкарбонилом.
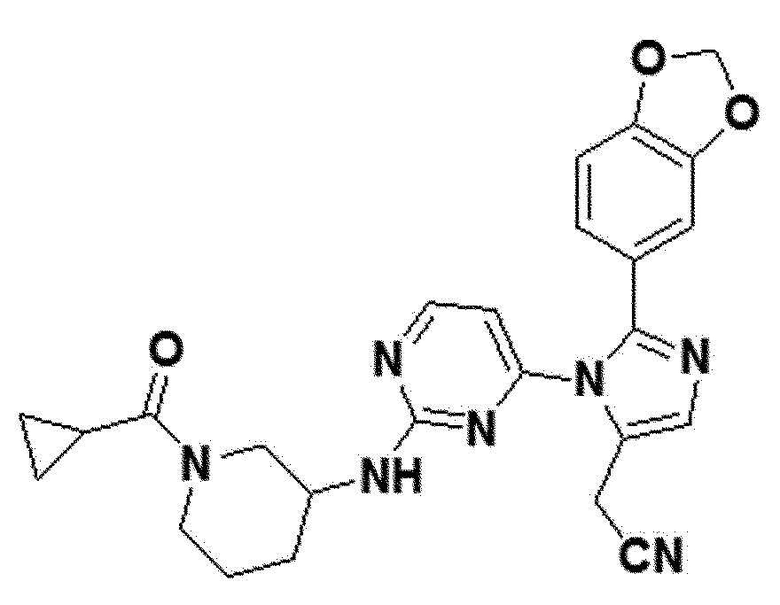
В другом примерном варианте осуществления настоящего изобретения, может быть предусмотрено, что имидазольное производное формулы 1 представляет собой 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(хинолин-2-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(нафталин-2-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(3,4-дихлорфенил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(циклогексиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(3,4-дихлорфенфенил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(3,4-дихлорфенил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(нафталин-2-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(3,4-дихлорфенил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(3,4-дихлорфенил)-1H-имидазол-5-ил)ацетонитрил; 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)ацетонитрил; или 2-(2-(бензофуран-5-ил)-1-(2-(1-циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил.
При этом, соединение выше настоящего изобретения можно применять в виде фармацевтически приемлемой соли, где соль присоединения кислоты, образованная с помощью фармацевтически приемлемой свободной кислоты, является пригодный в качестве соли.
Соль присоединения кислоты, образованная с помощью фармацевтически приемлемой свободной кислоты, является пригодный в качестве соли, термин, применяемый в настоящем изобретении. Соль присоединения кислоты получают из неорганических кислот, таких как хлористоводородная кислота, азотная кислота, фосфорная кислота, серная кислота, бромистоводородная кислота, йодистоводородная кислота, азотистая кислота или фосфористая кислота; и нетоксичных органических кислот, таких как алифатический моно- и дикарбоксилат, фенил-замещенный алканоат, гидроксиалканоат и алкандиоат, ароматические кислоты, алифатические и ароматические сульфокислоты. Данные фармацевтически нетоксичные соли включают сульфат, пиросульфат, бисульфат, сульфит, бисульфит, нитрат, фосфат, моногидрофосфат, дигидрофосфат, метафосфат, пирафосфат, хлорид, бромид, йодид, фторид, ацетат, пропионат, деканоат, каприлат, акрилат, формиат, изобутират, капрат, гептаноат, пропиолат, оксалат, малонат, сукцинат, суберат, себакат, фумарат, малеат, бутин-1,4-диоат, гексан-1,6-диоат, бензоат, хлорбензоат, метилбензоат, динитробензоат, гидроксибензоат, метоксибензоат, фталат, терефталат, бензолсульфонат, толуолсульфонат, хлорбензолсульфонат, ксилолсульфонат, фенилацетат, фенилпропионат, фенилбутират, цитрат, лактат, β-гидроксибутират, гликолят, малат, тартрат, метансульфонат, пропансульфонат, нафталин-1-сульфонат, нафталин-2-сульфонат или манделат.
Соль присоединения кислоты согласно настоящему изобретению можно получить общепринятым способом, например, таким способом, что соединения, представленные формулами 1-4, растворяют в избыточном количестве кислого водного раствора, и затем полученную в результате соль осаждают с помощью смешиваемого с водой органического растворителя, например, метанола, этанола, ацетона или ацетонитрила. Кроме того, данную соль присоединения кислоты можно также получить упариванием растворителя или избыточного количества кислоты из полученной в результате смеси, и затем обезвоживанием полученного в результате остатка или осуществлением фильтрования с отсасыванием полученной в результате осажденной соли.
Кроме того, фармацевтически приемлемую соль металла можно получить с помощью основания. Соль щелочного или щелочноземельного металла получают, например, растворением соединения в избыточном количестве раствора гидроксида щелочного металла или гидроксид щелочноземельного металла, фильтрованием нерастворенной соли соединения, и упариванием и обезвоживанием оставшегося раствора. При этом, в качестве соли металла, фармацевтически приемлемо получать соли натрия, калия или кальция. Кроме того, соответствующую соль серебра получают таким способом, что соль щелочного металла или щелочноземельного металла реагирует с подходящей солью серебра (например, нитратом серебра).
Кроме того, соединение настоящего изобретения включает фармацевтически приемлемую соль, а также все соли, изомеры, гидраты и сольваты, которые можно получить общепринятым способом.
Имидазольное производное формулы 1 выше согласно настоящему изобретению можно получить несколькими способами.
В конкретном варианте осуществления,
как показано на следующей реакционной формуле 1,
имидазольное производное формулы 1 можно получить способом, включающим стадии: проведения реакции конденсации с аминированием по Бухвальду между соединением формулы I и 4-хлор-2-(метилтио)пиримидином, получая соединение формулы II (стадия 1); окисления соединения формулы II, полученного на стадии 1 выше, получая соединение формулы III (стадия 2); и замещения метилсульфонильной группы соединения формулы III, полученного на стадии 2 выше, аминогруппой, получая соединение формулы IV (стадия 3). В настоящем изобретении, если R2 соединения формулы IV представляет собой тетрагидропиран, циклогексан, 2-гидроксипропан или трет-бутилпиперидин-1-карбоксилат, то каждый из них представляет собой соединение формулы IV-1, IV-2, IV-3 или IV-4.
[реакционная формула 1]
В одном примерном варианте осуществления, имидазольное производное формулы 1 можно получить стадиями: проведения реакции конденсации с аминированием по Бухвальду между соединением 7 настоящего изобретения и 4-хлор-2-(метилтио)пиримидином, получая соединение 8 (стадия 1); окисления соединения 8, полученного на стадии 1 выше, получая соединение 9 (стадия 2); и замещения метилсульфонильной группы соединения 9, полученного на стадии 2 выше, аминогруппой, получая соединения 10-13 (стадия 3).
Кроме того, как показано на следующей реакционной формуле 2,
имидазольное производное формулы 1 можно получить способом, дополнительно включающим стадию проведения деблокирования соединения формулы IV-4, полученного на стадии 3 выше, получая соединение формулы V (стадия 4).
[реакционная формула 2]

В одном примерном варианте осуществления, имидазольное производное формулы 1 можно получить стадией растворения соединения 13 настоящего изобретения в 1,4-диоксане, и затем обработкой полученного в результате раствора хлористоводородной кислотой, получая соединение 14 (стадия 4).
Кроме того, как показано на следующей реакционной формуле 3,
имидазольное производное формулы 1 можно получить способом, дополнительно включающим стадию ацилирования соединения формулы V, полученного на стадии 4 выше, получая соединение формулы VI (стадия 5).
[реакционная формула 3]
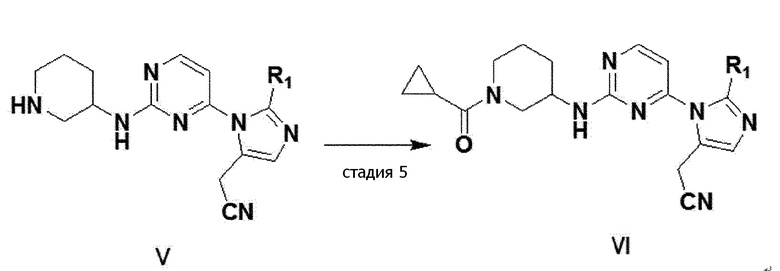
В одном примерном варианте осуществления, имидазольное производное формулы 1 можно получить стадией ацилирования соединение 14 настоящего изобретения, получая соединение 15 (стадия 5).
В одном варианте осуществления настоящего изобретения, обнаружено, что соединения 10a-12f и соединения 14a-15g, полученные согласно стратегии синтеза следующей реакционной формулы 4, показали превосходную ингибирующую JNK3 активность, в частности, что соединение 15d не показало активности по ингибированию других протеинкиназ, но было способно селективно ингибировать активность JNK1/2/3, таким образом, было обнаружено, что они могут с пользой применяться в качестве эффективного вещества для лечения дегенеративных заболеваний нервной системы мозга (смотри экспериментальные примеры 1-2).
[реакционная формула 4]

Соответственно, настоящее изобретение обеспечивает фармацевтическую композицию для предотвращения или лечения дегенеративных заболеваний нервной системы мозга, содержащую имидазольное производное формулы 1 выше или его фармацевтически приемлемую соль в качестве эффективного компонента; применение имидазольного производного формулы 1 выше или его фармацевтически приемлемой соли для лечения заболеваний выше; и способ лечения заболеваний выше, включающий введение терапевтически эффективного количества соединения формулы 1 выше или его фармацевтически приемлемой соли субъекту.
Как применяют в настоящем изобретении, термин ʺпредотвращениеʺ обозначает все действия по ингибированию дегенеративных заболеваний нервной системы мозга или замедлению его возникновения посредством введения фармацевтической композиции согласно настоящему изобретению.
Как применяют в настоящем изобретении, термин ʺлечениеʺ обозначает все действия, с помощью которых симптом дегенеративных заболеваний нервной системы мозга изменяется в лучшую сторону или принимает благоприятный оборот посредством введения фармацевтической композиции согласно настоящему изобретению.
ʺДегенеративные заболевания нервной системы мозгаʺ, которые представляют собой заболевания, которые будут предотвращать или лечить посредством композиции настоящего изобретения, могут включать, без ограничения, любые заболевания, вызванные повреждением мозга, но предпочтительно могут представлять собой болезнь Альцгеймера, болезнь Паркинсона, болезнь Хантингтона, множественный склероз или инсульт.
Фармацевтическая композиция настоящего изобретения может содержать фармацевтически приемлемый носитель в добавление к эффективному компоненту. При этом, фармацевтически приемлемый носитель может представлять собой носитель, обычно применяемый в получении состава, где данный носитель включает лактозу, декстрозу, сахарозу, сорбитол, маннитол, крахмал, гуммиарабик, фосфат кальция, альгинат, желатин, силикат кальция, микрокристаллическую целлюлозу, поливинилпирролидон, целлюлозу, воду, паточный сироп, метилцеллюлозу, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния, минеральное масло и подобными, но не ограничивается ими. В дополнение к компонентам выше, фармацевтическая композиция настоящего изобретения может дополнительно содержать смазывающее вещество, увлажняющее вещество, подслащивающий агент, ароматизатор, эмульгатор, суспендирующий агент, консервант и т. д.
Фармацевтическую композицию настоящего изобретения можно вводить перорально или парентерально (например, внутривенно, подкожно, интраперитонеально или местно) согласно целевому способу, где доза может изменяться в зависимости от состояния и веса пациента, тяжести заболевания, типа лекарственного средства, пути и времени введения, но ее можно подходящим образом выбрать специалист в данной области техники.
Фармацевтическую композицию настоящего изобретения вводят в ее фармацевтически эффективном количестве. Согласно настоящему изобретению, фармацевтически эффективное количество обозначает количество, достаточное для лечения заболевания при приемлемом соотношении риск/польза, пригодном для медицинского лечения, где уровень данного эффективного количества может зависеть от факторов, включающих тип заболевания у пациента, тяжесть заболевания, активность лекарственного средства, чувствительность к лекарственному средству, время введения, путь введения и скорость выведения, период лечения и одновременно применяемые лекарственные средства, а также другие факторы, известные в медицинской области. Фармацевтическую композицию согласно настоящему изобретению можно вводить в виде отдельного терапевтического агента или в комбинации с другими терапевтическими агентами, вводимыми последовательно или одновременно с общепринятыми терапевтическими агентами, и вводимыми в виде одной дозы или множества доз. Важно вводить количество данной композиции, которое способно обеспечивать максимальный эффект с минимальным количеством без побочного эффекта, принимая все факторы выше, где его может легко определить специалист в данной области техники.
В частности, эффективное количество фармацевтической композиции настоящего изобретения может изменяться в зависимости от возраста, пола, состояния и веса пациента, поглощения активного компонента in vivo, неактивного соотношения и скорости выведения, типа заболевания и сопутствующего лекарственного средства, где его обычно вводят в количестве 0,001-150 мг, предпочтительно 0,01-100 мг на 1 кг веса тела, каждый день или раз в два дня, или вводят таким способом, что количество разделяют на введение 1-3 раза в день. Однако его можно повышать или снижать, в зависимости от пути введения, тяжести ожирения, пола, веса, возраста и т.д., таким образом, доза выше не предполагается ограничивающей объем настоящего изобретения любым способом.
В настоящем изобретении, ʺиндивидʺ обозначает объект, который требует лечения заболеваний, более конкретно, людей или млекопитающих, таких как приматы, отличные от человека, мыши, собаки, кошки, лошади, коровы и подобные.
Далее, предпочтительные примеры будут предложены для лучшего понимания настоящего изобретения. Однако следующие примеры приводятся только с целью проиллюстрировать настоящее изобретение и, таким образом, настоящее изобретение не ограничивается ими.
<Пример получения 1> Получение этил 1,3-бензодиоксолил-5-карбоксиимидата (соединение 4a)
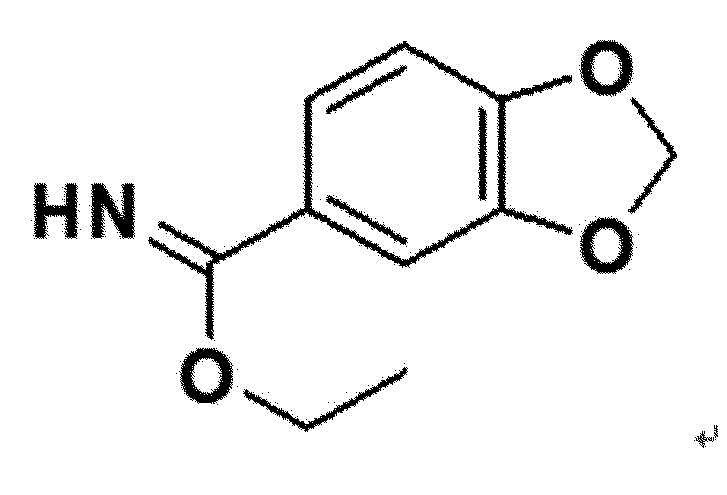
Стадия 1. Получение 1,3-бензодиоксолил-5-карбоксиамида
1,3-бензодиоксолил-5-карбоновую кислоту (соединение 1a, 1 г, 6 ммоль) смешивали с тионилхлоридом (SOCl2, 4,4 мл), и затем полученный в результате раствор нагревали при 80°С до того, как соединение выше не исчезало по ТСХ. После завершения реакции, полученный в результате раствор охлаждали до комнатной температуры, затем растворитель удаляли из него в вакууме, затем полученное из него соединение и метанол (6 мл), растворенный в 7N аммиаке, смешивали в этаноле (9 мл), и затем полученную в результате смесь выше перемешивали при комнатной температуре в течение 12 часов. После обнаружения завершения реакции, полученную в результате смесь концентрировали в вакууме, разбавляли эфиром и перемешивали до того, как полученный в результате продукт не отделялся в виде твердого остатка. Затем, твердый продукт фильтровали, и затем промывали эфиром и гексаном, таким образом получая 1,3-бензодиоксолил-5-карбоксамид (соединение 2a) (95% выход).
белый твердый остаток (95%); 1H ЯМР (400 МГц, DMSO) δ7,83 (с, 1H), 7,46 (дд, J=8,1, 1,7 Гц, 1H), 7,40 (д, J=1,7 Гц, 1H), 6,96 (д, J=8,1 Гц, 1H), 6,08 (с, 2H); LRMS (ESI) m/z рассчитано для C8H7NO3 [M+H]+:166, найдено 166.
Стадия 2. Получение 1,3-бензодиоксолил-5-карбонитрила
Соединение 2a (6,66 ммоль), полученное на стадии 1 выше, смешивали с хлорокисью фосфора (POCl3, 19 мл), и затем смесь выше перемешивали при 95°С в течение 2 часов. После завершения реакции, полученную в результате смесь охлаждали до комнатной температуры, затем растворитель удаляли из нее в вакууме, затем проводили экстракцию из концентрированной смеси выше этилацетатом (EtOAc) и 10% водным раствором K2CO3, и затем полученный в результате органической слой промывали водой и соляным раствором. Затем, полученный в результате осадок обезвоживали безводным сульфат магния (MgSO4), и затем растворитель упаривали из него, таким образом получая 1,3-бензодиоксолил-5-карбонитрил (соединение 3a) (83% выход).
белый твердый остаток (83%); 1H ЯМР (400 МГц, DMSO) δ 7,43 (д, J=1,4 Гц, 1H), 7,40 (дд, J=8,0, 1,7 Гц, 1H), 7,10 (д, 1H), 6,17 (с, 2H); LRMS (ESI) m/z рассчитано для C8H5NO2 [M+H]+:148, найдено 148.
Стадия 3. Получение этил 1,3-бензодиоксолил-5-карбоксиимидата
Соединение 3a, полученное на стадии 2 выше, растворяли в этаноле (3,4 мл), затем ацетилхлорид (AcCl, 3,1 мл) медленно добавляли к полученному в результате раствору при 0°С, и затем полученную в результате смесь перемешивали при комнатной температуре в течение 24-48 часов. После полного исчезновения соединений 3a-3f выше по ТСХ, полученный в результате остаток концентрировали в вакууме, и затем концентрированную смесь выше разбавляли эфиром и перемешивали до отделения твердого продукта. Затем, твердый продукт выше фильтровали и последовательно промывали эфиром и гексаном, таким образом получая этил 1,3-бензодиоксолил-5-карбоксиимидат (соединение 4a) (99% выход).
белый твердый остаток (99%); 1H ЯМР (400 МГц, DMSO) δ 7,75 (дд, J=8,3, 2,0 Гц, 1H), 7,70 (д, J=1,9 Гц, 1H), 7,18 (д, J=8,3 Гц, 1H), 6,23 (с, 2H), 4,58 (кв, J=7,0 Гц, 2H), 1,46 (т, J=7,0 Гц, 3H); LRMS (ESI) m/z рассчитано для C10H11NO3 [M+H]+: 194, найдено 194.
<Пример получения 2> Получение этил 1,4-бензодиоксан-6-карбоксиимидата (соединение 4b)
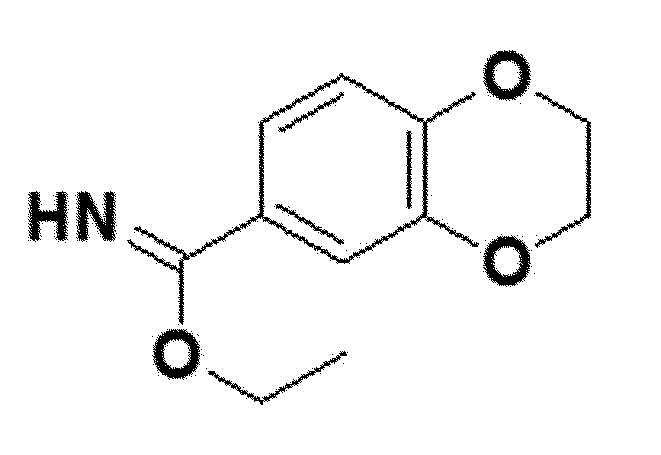
Стадия 1. Получение 1,4-бензодиоксан-6-карбонитрила
1,4-бензодиоксан-6-карбальдегид (соединение 2b, 500 мг, 3,05 ммоль), гидрохлорид гидроксидамина (255 мг, 3,7 ммоль) и сульфат натрия (434 мг, 3,05 ммоль) растворяли в DMF (15,3 мл), и затем полученный в результате раствор перемешивали при 170°С в течение 4 часов. Затем, сульфат натрия фильтровали, затем проводили экстракцию этилацетатом (EtOAc), и затем органический слой промывали водой ил соляным раствором. Полученный в результате продукт обезвоживали безводным сульфатом магния (MgSO4), и затем растворитель упаривали из него, таким образом получая 1,4-бензодиоксан-6-карбонитрил (соединение 3b) (выход 81%).
белый твердый остаток (81%); 1H ЯМР (400 МГц, DMSO) δ 7,39 (д, J=2,0 Гц, 1H), 7,30 (дд, J=8,4, 2,0 Гц, 1H), 7,03 (д, J=8,4 Гц, 1H), 4,35-4,32 (м, 2H), 4,31-4,27 (м, 2H); LRMS (ESI) m/z рассчитано для C9H7NO2 [M+H]+: 162, найдено 162.
Стадия 2. Получение этил 1,4-бензодиоксан-6-карбоксиимидата
Соединение 3b, полученное на стадии 1 выше, применяли для получения этил 1,4-бензодиоксан-6-карбоксиимидата (соединение 4b) тем же способом, как показано в 1-3 выше.
белый твердый остаток (99%); 1H ЯМР (400 МГц, DMSO) δ 11,70 (с, 1H), 7,70 (д, J=2,3 Гц, 1H), 7,64 (дд, J=8,6, 2,3 Гц, 1H), 7,11 (д, J=8,6 Гц, 1H), 4,57 (кв, J=7,0 Гц, 2H), 4,40-4,37 (м, 2H), 4,34-4,31 (м, 2H), 1,46 (т, J=7,0 Гц, 3H); LRMS (ESI) m/z рассчитано для C11H13NO3 [M+H]+:208, найдено 208.
Соединения следующих примеров получения 3-6 получали тем же способом, как показано в примере получения 1 выше (где 1,3-бензодиоксолил заменяли на хинолинил, нафтилил, 3,4-дихлорфенил, 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример получения 3> Получение этил хинолин-2-карбимидата (соединение 4c)

желтый твердый остаток (99%); 1H ЯМР (400 МГц, DMSO) δ 12,03 (с, 1H), 8,76 (д, J=8,4 Гц, 1H), 8,22 (т, J=9,2 Гц, 3H), 7,99 (ддд, J=8,5, 6,9, 1,4 Гц, 1H), 7,86 (ддд, J=8,1, 6,9, 1,2 Гц, 1H), 4,78 (кв, J=7,0 Гц, 2H), 1,55 (т, J=7,0 Гц, 3H); LRMS (ESI) m/z рассчитано для C12H12N2O [M+H]+:201, найдено 201.
<Пример получения 4> Получение этил 2-нафтилимидата (соединение 4d)

белый твердый остаток (99%); 1H ЯМР (400 МГц, DMSO) δ 8,90 (с, 1H), 8,14 (т, J=3,9 Гц, 3H), 8,07 (д, J=8,2 Гц, 1H), 7,79-7,73 (м, 1H), 7,67-7,72 (м, J=8,1, 7,0, 1,2 Гц, 1H), 4,71 (кв, J=7,0 Гц, 2H), 3,70 (с, 1H), 1,53 (т, J=7,0 Гц, 3H); LRMS (ESI) m/z рассчитано для C13H13NO [M+H]+:200, найдено 200.
<Пример получения 5> Получение этил 3,4-дихлорбензимидата (соединение 4e)
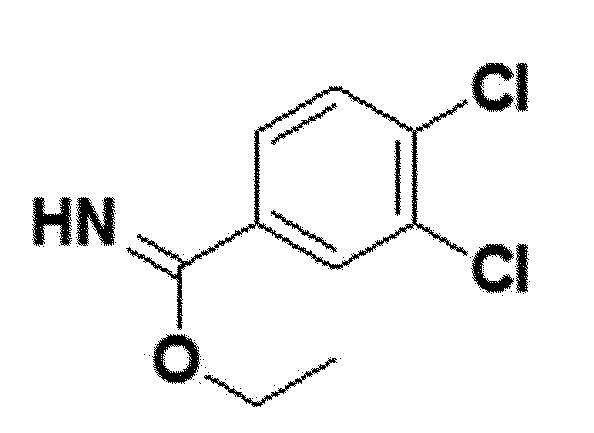
белый твердый остаток (99%); 1H ЯМР (400 МГц, DMSO) δ 8,41 (с, 1H), 8,06 (д, J=8,0 Гц, 1H), 7,93 (д, J=8,2 Гц, 1H), 4,60 (кв, J=6,4 Гц, 2H), 1,46 (т, J=5,9 Гц, 3H). ); LRMS (ESI) m/z рассчитано для C9H9Cl2NO [M+H]+: 219, найдено 219.
<Пример получения 6> Получение этил 4-фтор-3-(трифторметил)бензимидата (соединение 4f)
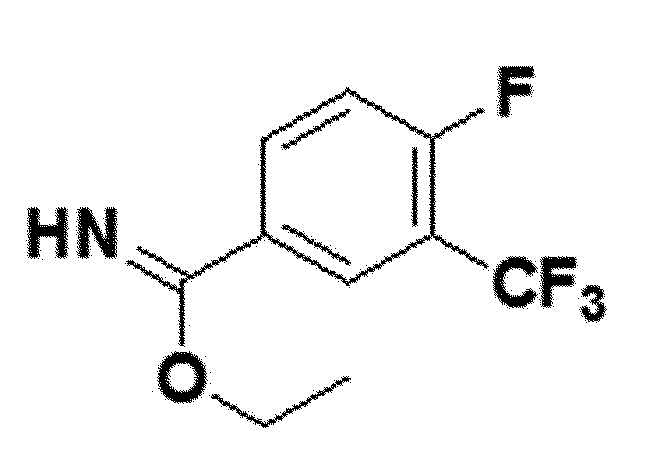
белый твердый остаток (84%); 1H ЯМР (400 МГц, DMSO) δ 8,57 (дд, J=6,6, 2,0 Гц, 1H), 8,54-8,47 (м, 1H), 7,84 (т, 1H), 4,63 (кв, J=7,0 Гц, 2H), 1,48 (т, J=7,0 Гц, 3H); LRMS (ESI) m/z рассчитано для C10H9F4NO [M+H]+: 236, найдено 236.
<Пример получения 7> Получение бензо[d][1,3]диоксол-5-карбоксиимидамида (соединение 5a)
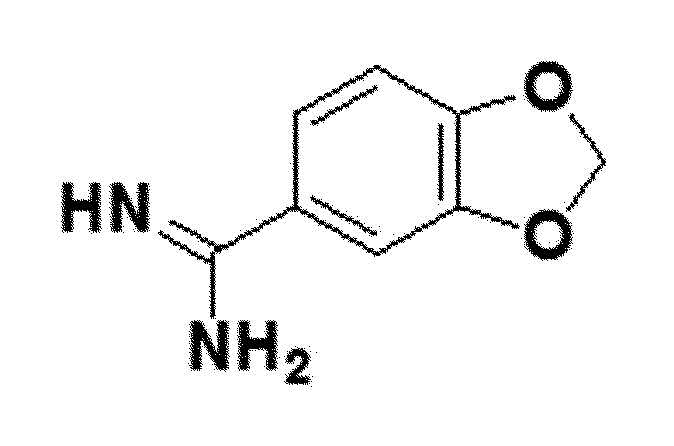
Соединение 4a (4,25 ммоль), полученное в примере получения 1 выше, и метанол (4,25 мл), растворенный в 7N аммиак, смешивали в этаноле (4,25 мл), и затем смесь выше перемешивали при комнатной температуре в течение 12 часов. После полного исчезновения соединения 4a по ТСХ, полученный в результате остаток концентрировали в вакууме, и затем концентрированную смесь выше разбавляли эфиром и перемешивали до того, как продукт не отделялся от нее. Затем, твердый продукт выше фильтровали и последовательно промывали эфиром и гексаном, таким образом получая закристаллизовавшееся соединение 5a.
белый твердый остаток (81%); 1H ЯМР (400 МГц, DMSO) δ 9,07 (с, 4H), 7,46 (дд, J=8,2 Гц, 1H), 7,43 (д, J=1,8 Гц, 1H), 7,16 (д, J=8,2 Гц, 1H), 6,19 (с, 2H); LRMS (ESI) m/z рассчитано для C8H9ClN2O2 [M+H]+: 201, найдено 201.
Соединения следующих примеров получения 8-12 получали тем же способом, как показано в примере получения 7 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4 бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример получения 8> Получение 2,3-дигидробензо[b][1,4]диоксин-6-карбоксиимидамида (соединение 5b)
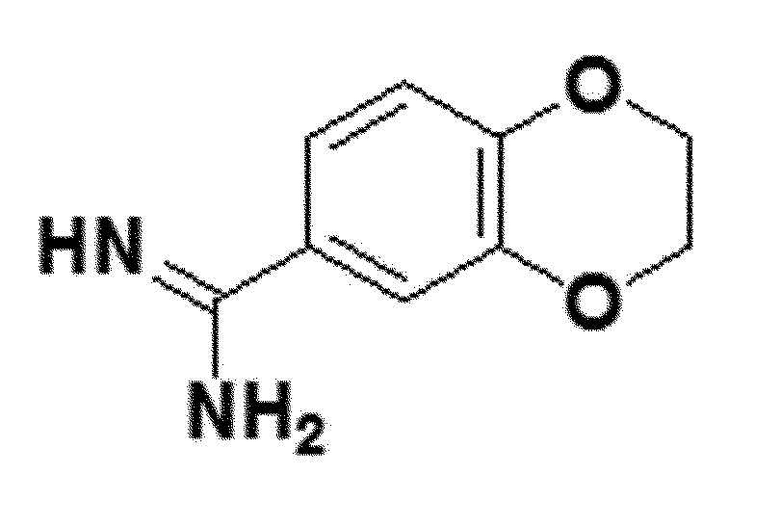
белый твердый остаток (92%); 1H ЯМР (400 МГц, DMSO) δ 9,22 (с, 4H), 9,09 (с, 3H), 7,45 (д, J=2,3 Гц, 2H), 7,40 (дд, J=8,5, 2,4 Гц, 2H), 7,08 (д, J=8,5 Гц, 2H), 4,37-4,33 (м, 4H), 4,33-4,29 (м, 4H); LRMS (ESI) m/z рассчитано для C9H11ClN2O2 [M+H]+: 216, найдено 216.
<Пример получения 9> Получение хинолин-2-карбоксиимидамида (соединение 5c)

белый твердый остаток (93%); 1H ЯМР (400 МГц, DMSO) δ 9,77 (с, 3H), 8,76 (д, J=8,6 Гц, 1H), 8,35 (д, J=8,6 Гц, 1H), 8,23-8,15 (дд, J=12,0, 8,4 Гц, 2H), 8,00-7,93 (м, 1H), 7,83 (м, J=7,5 Гц, 1H), 7,33 (с, 2H); LRMS (ESI) m/z рассчитано для C10H10ClN3 [M+H]+: 208, найдено 208.
<Пример получения 10> Получение 2-нафтилимидамида (соединение 5d)
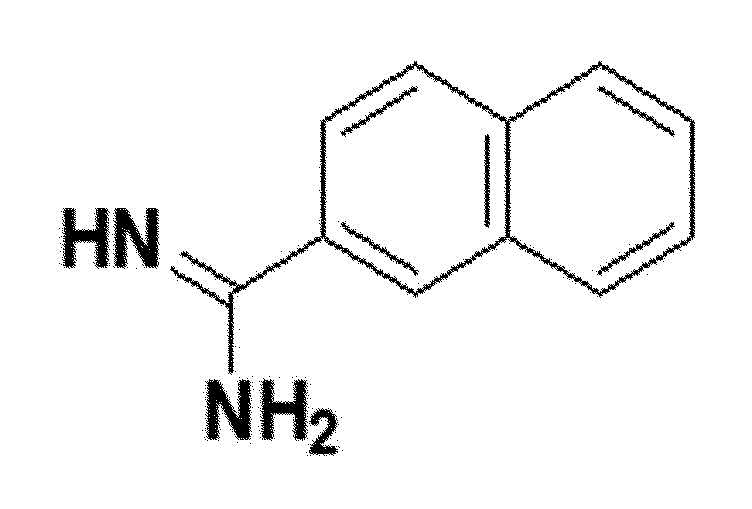
белый твердый остаток (83%); 1H ЯМР (400 МГц, DMSO) δ 9,63 (с, 5H), 9,44 (с, 5H), 8,57 (д, J=1,5 Гц, 3H), 8,15 (д, J=87 Гц, 3H), 8,06-8,10 (т, J=8,4 Гц, 6H), 7,85-7,90 (дд, J=8,6, 1,9 Гц, 3H), 7,66-7,75 (тдд, J=14,6, 6,9, 1,4 Гц, 6H); LRMS (ESI) m/z рассчитано для C11H11N2 [M+H]+: 207, найдено 207.
<Пример получения 11> Получение 3,4-дихлорбензимидамида (соединение 5e)
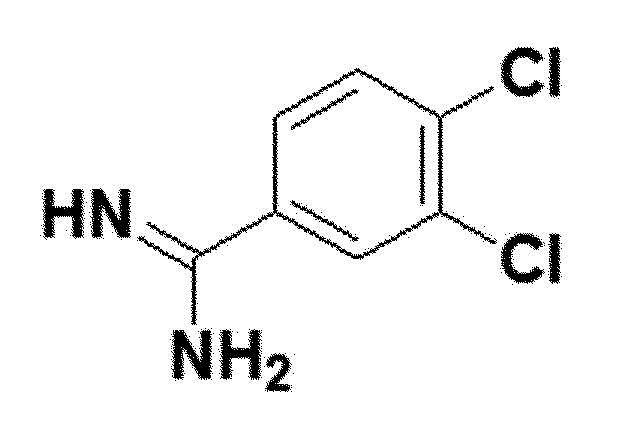
белый твердый остаток (70%); 1H ЯМР (400 МГц, DMSO) δ 9,19 (с, 4H), 8,18 (с, 1H), 7,94 (д, 1H), 7,85 (д, 1H); LRMS (ESI) m/z рассчитано для C7H7Cl3N2 [M+H]+: 226, найдено 226.
<Пример получения 12> Получение 4-фтор-3-(трифторметил)бензимидамида (соединение 5f)

белый твердый остаток (95%); 1H ЯМР (400 МГц, DMSO) δ 9,32 (с, 4H), 8,31 (дд, J=6,6, 2,0 Гц, 1H), 8,29-8,24 (м, 1H), 7,82 (т, 1H); LRMS (ESI) m/z рассчитано для C8H6F4N2 [M+H]+: 207, найдено 207.
<Пример получения 13> Получение (2-(бензо[d][1,3]диоксол-5-ил)-1H-имидазол-5-ил)метанола (соединение 6a)
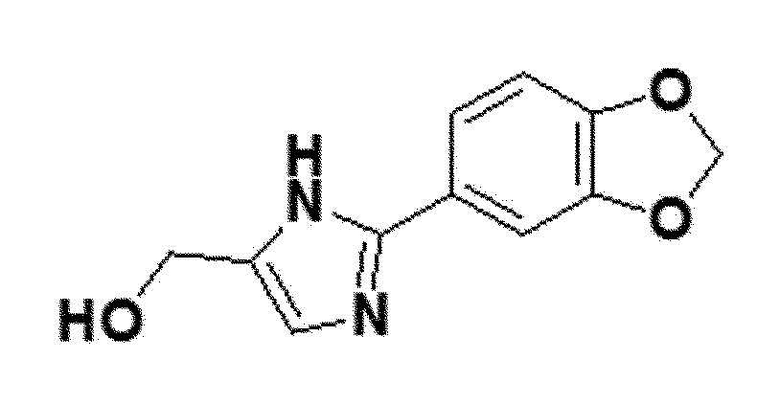
Соединение 5a (3,1 ммоль), полученное в примере получения 7 выше, 1,3-дигидроксиацетоновый димер (3,34 ммоль), NH4OH (12,2 мл) и NH4Cl (16,2 моль) перемешивали при 80°С в течение 2 часов. После полного исчезновения соединения 5a по ТСХ, реакционную смесь охлаждали до комнатной температуры, и затем добавляли к ней хлористый метилен (CH2Cl2), таким образом, отделяя от нее слой. После фильтрования твердого продукта, полученного в органическом слое, полученный в результате твердый продукт промывали хлористым метиленом (CH2Cl2) и кристаллизовали, таким образом получая соединение 6a.
коричневый твердый остаток (38%); 1H ЯМР (400 МГц, DMSO) δ 12,20 (с, 1H), 7,43 (дд, J=5,9, 1,7 Гц, 2H), 6,97 (д, 1H), 6,93 (с, 1H), 6,05 (с, 2H), 4,94 (с, 1H), 4,40 (д, J=3,3 Гц, 2H); LRMS (ESI) m/z рассчитано для C11H10N2O3 [M+H]+: 219, найдено 219.
Соединения следующих примеров получения 14-18 получали тем же способом, как показано в примере получения 13 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4 бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример получения 14> Получение (2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)метанола (соединение 6b)
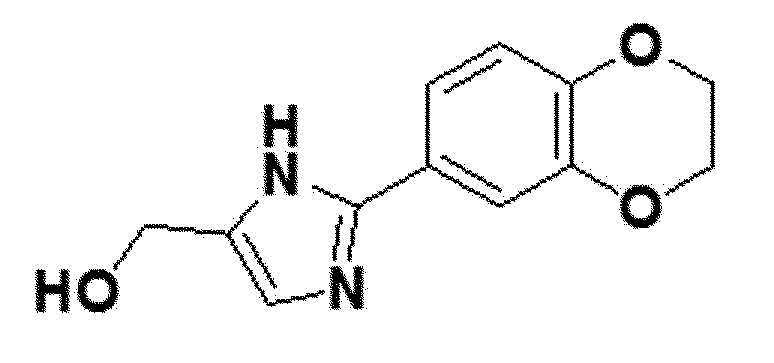
коричневый твердый остаток (54%); +H ЯМР (400 МГц, DMSO) δ 12,17 (с, 1H), 7,41 (д, J=2,1 Гц, 1H), 7,38 (д, J=2,1 Гц, 1H), 6,91 (с, 1H), 6,89 (дд, J=8,2, 0,4 Гц, 1H), 4,93 (с, 1H), 4,39 (с, 2H), 4,26 (с, 4H); LRMS (ESI) m/z рассчитано для C12H12N2O3 [M+H]+: 233, найдено 233.
<Пример получения 15> Получение (2-(хинолин-2-ил)-1H-имидазол-5-ил)метанола (соединение 6c)
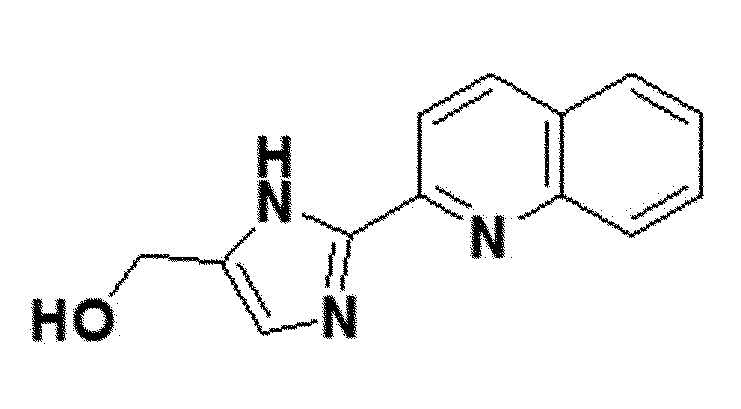
желтый твердый остаток (60%); 1H ЯМР (400 МГц, DMSO) δ 12,76 (с, 1H), 8,41 (д, J=8,6 Гц, 1H), 8,19 (д, J=8,6 Гц, 1H), 8,04 (д, J=8,7 Гц, 1H), 7,98 (д, J=8,2, 1,0 Гц, 1H), 7,81-7,76 (м, J=8,4, 6,9, 1,4 Гц, 1H), 7,62-7,56 (м, J=8,1, 6,9, 1,2 Гц, 1H), 7,15 (с, 1H), 4,98 (с, 1H), 4,50 (дд, J=30,0, 5,1 Гц, 2H). LRMS (ESI) m/z рассчитано для C13H11N3O [M+H]+: 226, найдено 226.
<Пример получения 16> Получение (2-(нафталин-2-ил)-1H-имидазол-5-ил)метанола (соединение 6d)

белый твердый остаток (50%); 1H ЯМР (400 МГц, DMSO) δ 12,56 (с, 1H), 8,44 (с, 1H), 8,10 (дд, J=8,6, 1,3 Гц, 1H), 7,96 (д, J=8,7 Гц, 1H), 7,94-7,89 (м, J=6,1 Гц, 2H), 7,58-7,46 (м, J=6,8, 1,4 Гц, 2H), 7,06 (с, 1H), 4,98 (с, 1H), 4,47 (д, J=5,3 Гц, 2H); LRMS (ESI) m/z рассчитано для C14H12N2O [M+H]+: 225, найдено 225.
<Пример получения 17> Получение (2-(3,4-дихлорфенил)-1H-имидазол-5-ил)метанола (соединение 6e)
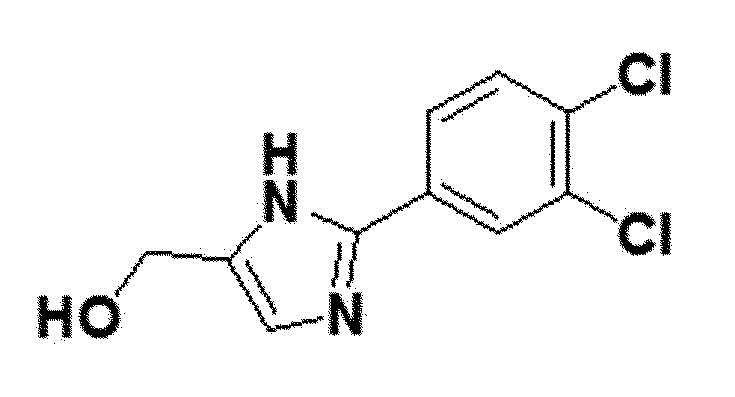
коричневый твердый остаток (58%); 1H ЯМР (400 МГц, DMSO) δ 12,57 (с, 1H), 8,15 (дд, 1H), 7,90 (д, J=1,9 Гц, 1H), 7,69 (д, J=8,5 Гц, 1H), 7,03 (д, J=90,6 Гц, 1H), 5,07 (д, J=103,1 Гц, 1H), 4,43 (д, J=19,6 Гц, 2H). LRMS (ESI) m/z рассчитано для C10H8Cl2N2O [M+H]+: 244, найдено 244.
<Пример получения 18> Получение (2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)метанола (соединение 6f)
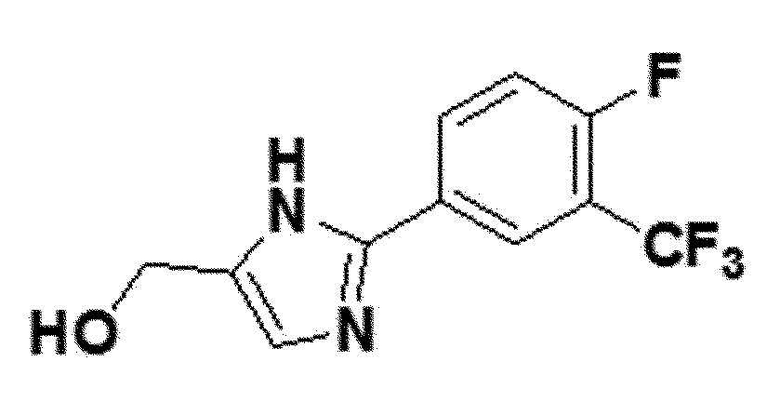
коричневый твердый остаток (80%); 1H ЯМР (400 МГц, DMSO) δ 12,74 (с, 1H), 8,32 (дд, J=6,9, 2,0 Гц, 1H), 8,29-8,23 (м, 1H), 7,60 (дд, J=10,3, 9,1 Гц, 1H), 7,05 (с, 1H), 5,06 (с, 1H), 4,44 (с, 2H); LRMS (ESI) m/z рассчитано для C11H8F4N2O [M+H]+: 261, найдено 261.
<Пример получения 19> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 7a)

Тионилхлорид (3,5 мл) смешивали с соединением 6a (4,8 ммоль), полученным в примере получения 13 выше, и затем полученный в результате раствор нагревали при 80°С до исчезновения соединения выше по ТСХ. После завершения реакции, полученный в результате остаток охлаждали до комнатной температуры, затем растворитель удаляли из него в вакууме, и затем диметилсульфоксид (24 мл) добавляли к полученному в результате соединению и цианиду натрия (24 ммоль) и перемешивали при комнатной температуре в течение 24 часов. После установления завершения реакции, экстракцию из реакционной смеси проводили этилацетатом (EtOAc), и затем органический слой промывали водой и соляным раствором. Затем, полученный в результате остаток обезвоживали безводным сульфатом магния (MgSO4), затем растворитель упаривали, и затем полученный в результате продукт отделяли и очищали колоночной хроматографией (EA:HEX=1:1), таким образом получая соединение 7a.
желтый твердый остаток (43%); 1H ЯМР (400 МГц, DMSO) δ 12,38 (с, 1H), 7,44 (дд, 1H), 7,42 (с, 1H), 7,14 (с, 1H), 6,99 (дд, J=7,5, 1,1 Гц, 1H), 6,06 (с, 2H), 3,87 (с, 2H); LRMS (ESI) m/z рассчитано для C12H9N3O2 [M+H]+: 228, найдено 228.
Соединения следующих примеров получения 20-24 получали тем же способом, как показано в примере получения 19 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4 бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример получения 20> Получение 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 7b)

желтый твердый остаток (32%); 1H ЯМР (400 МГц, DMSO) δ 12,36 (с, 1H), 7,40 (д, 1H), 7,39 (д, 1H), 7,11 (с, 1H), 6,91 (дд, 1H), 4,27 (с, 4H), 3,87 (с, 2H); LRMS (ESI) m/z рассчитано для C13H11N3O2 [M+H]+: 242, найдено 242.
<Пример получения 21> Получение 2-(2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 7c)
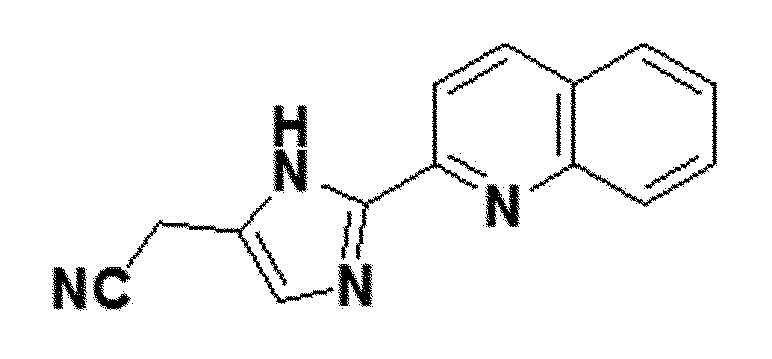
желтый твердый остаток (36%); 1H ЯМР (400 МГц, DMSO) δ 13,00 (с, 1H), 8,45 (д, J=8,4 Гц, 1H), 8,19 (д, J=8,6 Гц, 1H), 8,05 (д, J=8,3 Гц, 1H), 8,00 (д, J=8,2 Гц, 1H), 7,83-7,77 (м, J=8,4, 6,9, 1,5 Гц, 1H), 7,64-7,58 (м, J=8,1, 6,9, 1,1 Гц, 1H), 7,27 (с, 1H), 3,97 (д, J=0,6 Гц, 2H); LRMS (ESI) m/z рассчитано для C14H10N4 [M+H]+: 235, найдено 235.
<Пример получения 22> Получение 2-(2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 7d)

желтый твердый остаток (74%); 1H ЯМР (400 МГц, DMSO) δ 12,76 (с, 1H), 8,46 (с, 1H), 8,09 (дд, J=8,6, 1,6 Гц, 1H), 7,99 (д, J=9,0 Гц, 1H), 7,97-7,90 (м, 2H), 7,59-7,49 (м, 2H), 7,27 (с, 1H), 3,95 (с, 2H); LRMS (ESI) m/z рассчитано для C15H11N3 [M+H]+: 234, найдено 234.
<Пример получения 23> Получение 2-(2-(3,4-дихлорфенил)-1H-имидазол-5-ил)ацетонитрила (соединение 7e)

желтый твердый остаток (75%); 1H ЯМР (400 МГц, DMSO) δ 12,78 (с, 1H), 8,13 (д, J=2,0 Гц, 1H), 7,89 (дд, J=8,5, 2,1 Гц, 1H), 7,72 (д, J=8,5 Гц, 1H), 7,28 (с, 1H), 3,92 (с, 2H); LRMS (ESI) m/z рассчитано для C11H7Cl2N3 [M+H]+: 253, найдено 253.
<Пример получения 24> Получение 2-(2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)ацетонитрила (соединение 7f)
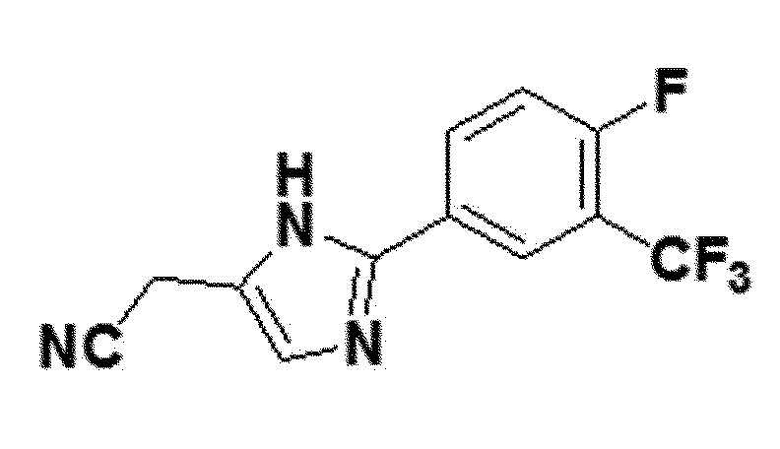
желтый твердый остаток (43%); 1H ЯМР (400 МГц, DMSO) δ 12,83 (с, 1H), 8,29 (д, J=7,0 Гц, 1H), 8,25 (дд, J=5,6, 3,1 Гц, 1H), 7,63 (дд, 1H), 7,29 (с, 1H), 3,92 (с, 2H); LRMS (ESI) m/z рассчитано для C12H7F4N3 [M+H]+: 270, найдено 270.)
<Пример получения 25> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(метилтио)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 8a)
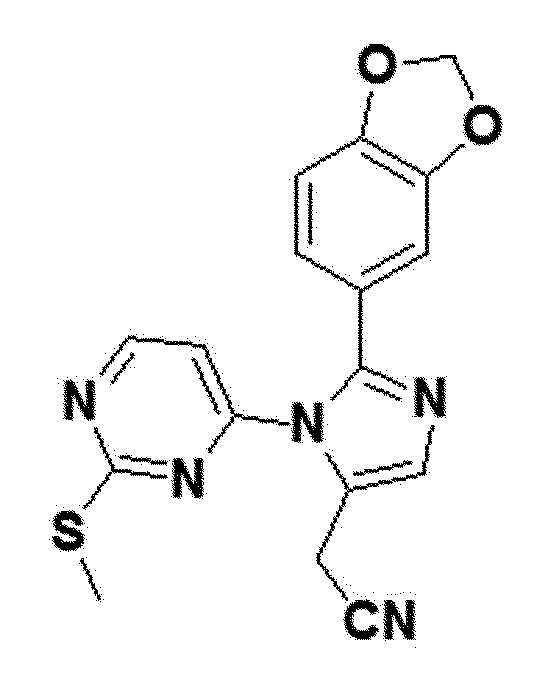
Соединение 7a (1,3 ммоль), полученное в примере получения 19 выше, 4-йод-2-(метилтио)пиримидин (320 мг, 1,3 ммоль), (II) ацетат палладия (Pd(oAc)2, 88 мг, 0,13 ммоль), X-Phos (62 мг, 0,13 ммоль), и Cs2CO3 продували азотом, и затем толуол (13 мл) добавляли к полученному в результате раствору и смешивали. После обработки ультразвуком смеси выше в течение 5 минут, полученную в результате смесь нагревали вплоть до 130°С в таких условиях, что азот присутствовал в смеси, и затем полученную в результате смесь перемешивали без азота при 130°С в течение 3 часов. После охлаждения полученной в результате смеси до комнатной температуры, реакционную смесь фильтровали через слой целита, затем растворитель удаляли в вакууме, и затем полученный в результате остаток отделяли и очищали колоночной хроматографией (DCM:MEOH =40:1), таким образом получая соединение 8a.
желтый твердый остаток (27%); 1H ЯМР (400 МГц, DMSO) δ 8,65 (д, J=5,4 Гц, 1H), 7,79 (с, 1H), 6,99 (дд, J=3,5, 1,9 Гц, 2H), 6,96 (д, J=8,0 Гц, 1H), 6,87 (дд, J=8,1, 1,7 Гц, 1H), 6,08 (с, 2H), 3,99 (д, J=0,8 Гц, 2H), 2,30 (с, 3H); LRMS (ESI) m/z рассчитано для C17H13N5O2S [M+H]+: 352, найдено 352.
Соединения следующих примеров получения 26-30 получали тем же способом, как показано в примере получения 25 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4 бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример получения 26> Получение 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(метилтио)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 8b)

желтый твердый остаток (33%); 1H ЯМР (400 МГц, CDCl3) δ 8,43 (д, J=5,4 Гц, 1H), 7,73 (с, 1H), 7,01 (с, 1H), 6,90 (д, J=1,1 Гц, 2H), 6,58 (д, J=5,4 Гц, 1H), 4,34-4,31 (м, 2H), 4,30-4,26 (м, 2H), 3,86 (с, 2H), 2,54 (с, 3H); LRMS (ESI) m/z рассчитано для C18H15N5O2S [M+H]+: 366, найдено 366.
<Пример получения 27> Получение 2-(1-(2-(метилтио)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 8c)
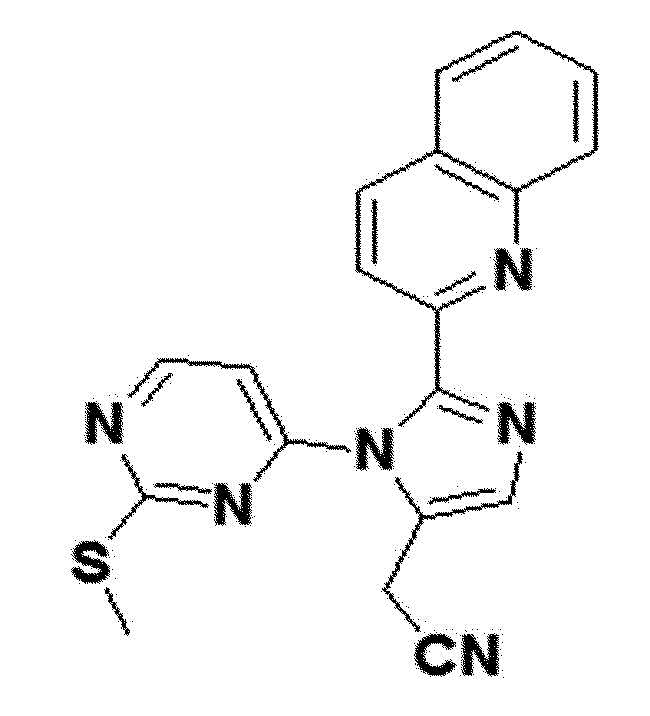
желтый твердый остаток (24%); 1H ЯМР (400 МГц, CDCl3) δ 8,47 (д, J=5,4 Гц, 1H), 8,28 (д, J=8,5 Гц, 1H), 8,10 (д, J=8,5 Гц, 1H), 7,85 (д, J=7,7 Гц, 1H), 7,73 (д, J=8,4 Гц, 1H), 7,70-7,65 (м, 2H), 7,60-7,54 (м, J=8,1, 6,7, 1,4 Гц, 1H), 6,82 (д, J=5,4 Гц, 1H), 3,86 (д, J=0,9 Гц, 2H), 2,41 (с, 3H); LRMS (ESI) m/z рассчитано для C19H14N6S [M+H]+: 359, найдено 359.
<Пример получения 28> Получение 2-(1-(2-(метилтио)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 8d)
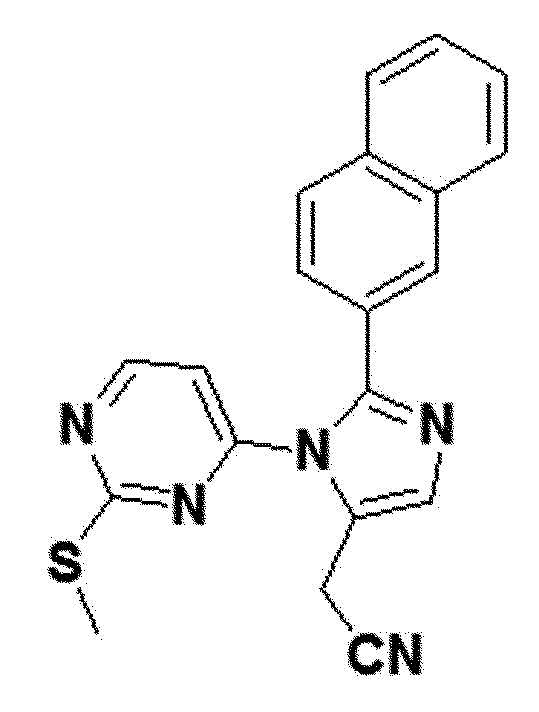
желтый твердый остаток (27%); 1H ЯМР (400 МГц, CDCl3) δ 8,33 (д, J=5,4 Гц, 1H), 8,05 (с, 1H), 7,88 (дд, J=7,0, 1,6 Гц, 2H), 7,85 (с, 1H), 7,80 (с, 1H), 7,60-7,52 (м, 2H), 7,42 (дд, J=8,5, 1,7 Гц, 1H), 6,51 (д, J=5,4 Гц, 1H), 3,88 (д, J=0,9 Гц, 2H), 2,43 (с, 3H); LRMS (ESI) m/z рассчитано для C20H15N5S [M+H]+: 358, найдено 358.
<Пример получения 29> Получение 2-(2-(3,4-дихлорфенил)-1-(2-(метилтио)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 8e)
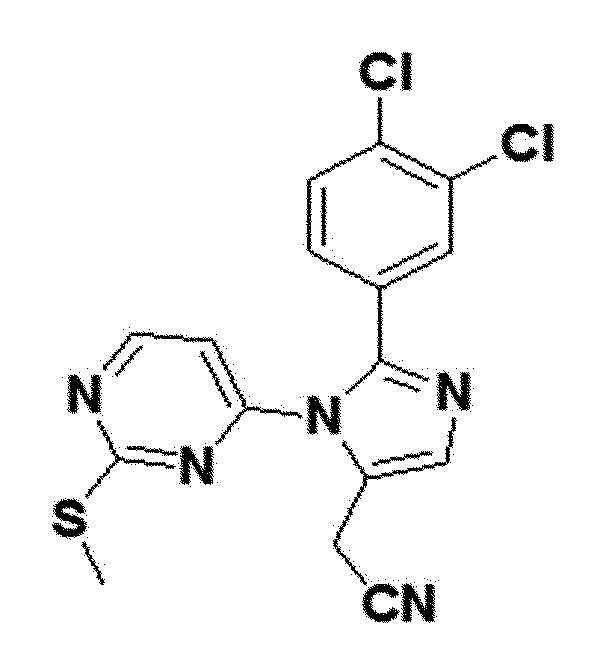
желтый твердый остаток (35%); 1H ЯМР (400 МГц, CDCl3) δ 8,42 (д, J=5,4 Гц, 1H), 7,61 (с, 1H), 7,58 (д, J=2,0 Гц, 1H), 7,40 (д, J=8,3 Гц, 1H), 7,15 (дд, J=8,3, 2,0 Гц, 1H), 6,53 (д, J=5,4 Гц, 1H), 3,74 (д, J=0,8 Гц, 2H), 2,37 (с, 3H); LRMS (ESI) m/z рассчитано для C16H11Cl2N5S [M+H]+: 377, найдено 377.
<Пример получения 30> Получение 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(метилтио)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 8f)

желтый твердый остаток (23%); 1H ЯМР (400 МГц, MeOD) δ 8,61 (д, J=5,4 Гц, 1H), 7,84 (дд, J=6,9, 2,2 Гц, 1H), 7,82 (дд, J=2,2, 1,3 Гц, 1H), 7,73-7,68 (м, 1H), 7,42 (дд, 1H), 7,11 (д, J=5,4 Гц, 1H), 3,93 (д, J=0,9 Гц, 2H), 2,17 (с, 3H); LRMS (ESI) m/z рассчитано для C17H11F4N5S [M+H]+: 394, найдено 394.
<Пример получения 31> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(метилсульфонил)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 9a)
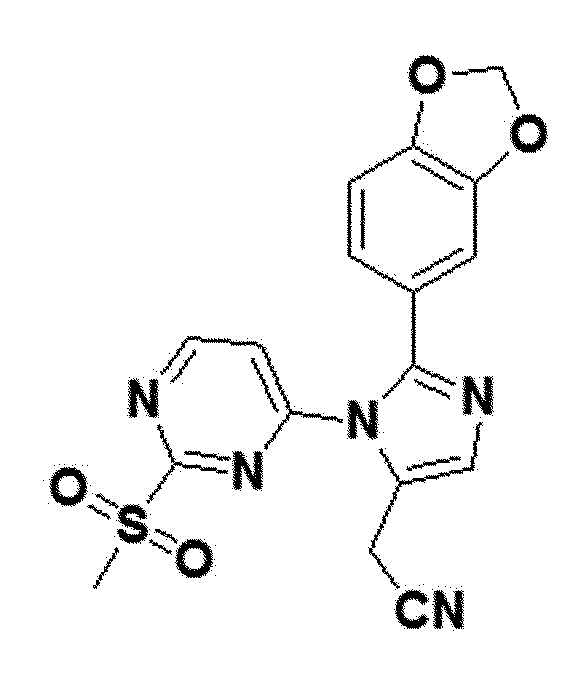
Соединение 8a (1 ммоль), полученное в примере получения 25 выше, и пероксомоносульфат калия (5 ммоль) смешивали в растворителе (5 мл), который смешивали в соотношении метанол:вода=1:1, и затем смесь выше перемешивали при комнатной температуре в течение 1 часа. После полного исчезновения соединения выше по ТСХ, метанол концентрировали в вакууме. Концентрированную смесь выше разбавляли добавлением к ней воды и перемешивали до отделения от нее твердого продукта. Твердый продукт выше фильтровали, последовательно промывали эфиром и гексаном и кристаллизовали соединение.
белый твердый остаток (46%); 1H ЯМР (400 МГц, DMSO) δ 9,06 (д, J=5,6 Гц, 1H), 7,92 (с, 1H), 7,70 (дд, J=8,2 Гц, 1H), 7,61 (д, J=1,7 Гц, 1H), 7,54 (д, J=5,6 Гц, 1H), 6,97 (с, 1H), 6,16 (с, 2H), 4,03 (с, 2H), 3,24 (с, 3H); LRMS (ESI) m/z рассчитано для C17H13N5O4S [M+H]+: 384, найдено 384.
Соединения следующих примеров получения 32-36 получали тем же способом, как показано в примере получения 31 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4 бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример получения 32> Получение 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(метилсульфонил)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 9b)

белый твердый остаток; 1H ЯМР (400 МГц, DMSO) δ 11,57 (с, 1H), 8,90 (д, J=5,8 Гц, 1H), 8,38 (д, J=5,8 Гц, 1H), 7,64 (д, 1H), 7,62 (дд, J=8,5 Гц, 1H), 7,00 (д, J=8,4 Гц, 1H), 4,36-4,32 (м, J=4,6 Гц, 2H), 4,32-4,28 (м, J=4,3 Гц, 2H), 3,60 (с, 2H), 3,41 (с, 3H); LRMS (ESI) m/z рассчитано для C18H15N5O4S [M+H]+: 398, найдено 398.
<Пример получения 33> Получение 2-(1-(2-(метилсульфонил)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 9c)

желтый твердый остаток (73%); 1H ЯМР (400 МГц, DMSO) δ 9,17 (д, J=5,4 Гц, 1H), 8,53 (д, J=8,7 Гц, 1H), 8,20 (д, J=8,6 Гц, 1H), 8,03 (д, J=8,2 Гц, 1H), 7,98 (д, J=5,4 Гц, 1H), 7,94 (с, 1H), 7,73-7,68 (м, 1H), 7,64-7,59 (м, J=10,9, 4,1 Гц, 1H), 7,39 (д, J=7,7 Гц, 1H), 4,14 (д, J=0,8 Гц, 2H), 3,25 (с, 3H); LRMS (ESI) m/z рассчитано для C19H14N6O2S [M+H]+: 391, найдено 391.
<Пример получения 34> Получение 2-(1-(2-(метилсульфонил)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 9d)
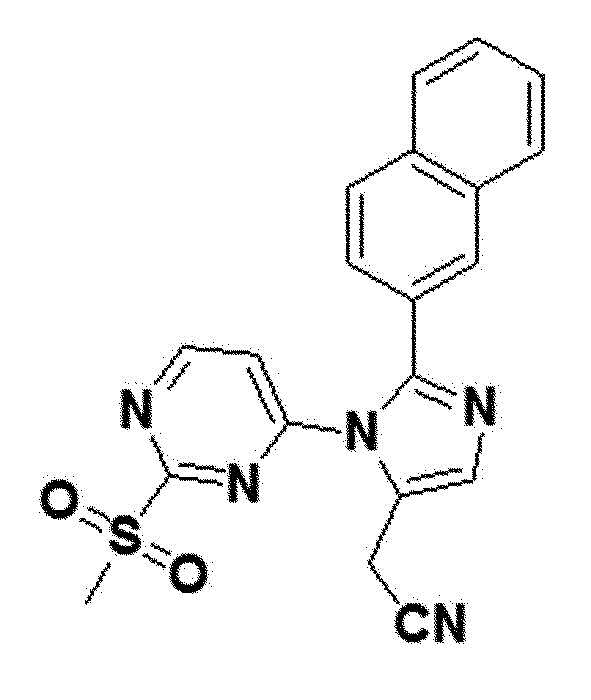
желтый твердый остаток (86%); 1H ЯМР (400 МГц, DMSO) δ 9,05 (д, J=5,6 Гц, 1H), 8,17 (д, J=1,4 Гц, 1H), 8,03 (с, 1H), 8,01-7,97 (м, J=5,1, 4,0 Гц, 2H), 7,96 (д, J=8,6 Гц, 1H), 7,63 (д, J=5,6 Гц, 1H), 7,61-7,54 (м, 3H), 4,10 (д, J=0,8 Гц, 2H), 3,02 (с, 3H); LRMS (ESI) m/z рассчитано для C20H15N5O2S [M+H]+: 390, найдено 390.
<Пример получения 35> Получение 2-(2-(3,4-дихлорфенил)-1-(2-(метилсульфонил)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 9e)

белый твердый остаток (83%); 1H ЯМР (400 МГц, DMSO) δ 9,13 (д, J=5,6 Гц, 1H), 8,03 (с, 1H), 7,84 (д, J=2,0 Гц, 1H), 7,80 (д, J=5,6 Гц, 1H), 7,68 (д, J=8,4 Гц, 1H), 7,47 (дд, J=8,4, 2,1 Гц, 1H), 4,07 (д, J=0,8 Гц, 2H), 3,14 (с, 3H); LRMS (ESI) m/z рассчитано для C16H11Cl2N5O2S [M+H]+: 409, найдено 409.
<Пример получения 36> Получение 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(метилсульфонил)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 9f)
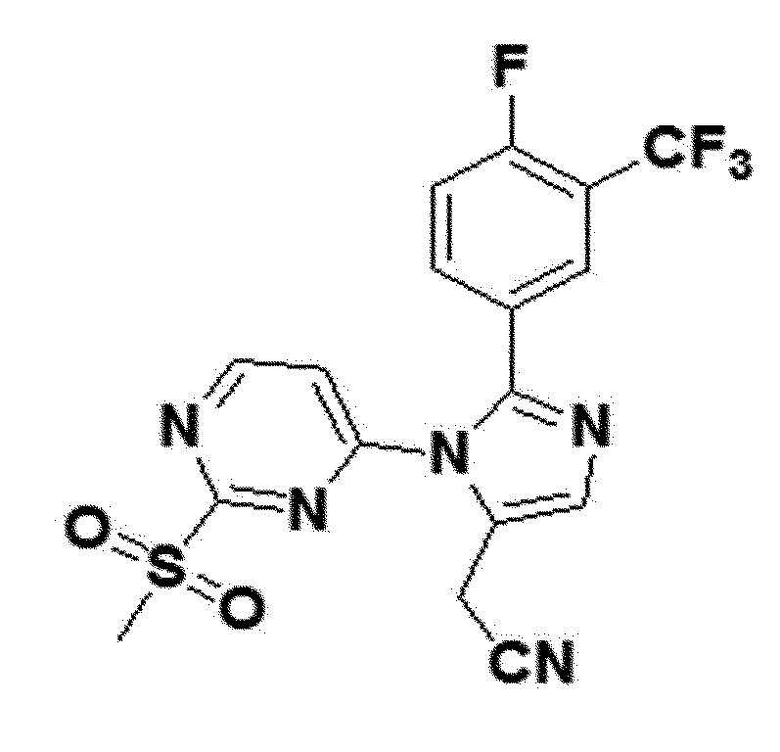
желтый твердый остаток (57%); 1H ЯМР (400 МГц, DMSO) δ 9,14 (д, J=5,6 Гц, 1H), 8,07 (с, 1H), 7,96 (дд, J=6,8, 1,7 Гц, 1H), 7,91-7,82 (м, J=5,2 Гц, 2H), 7,56 (дд, 1H), 4,08 (с, 2H), 3,11 (с, 3H); LRMS (ESI) m/z рассчитано для C17H11F4N5O2S [M+H]+: 426, найдено 426.
<Пример 1> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 10a)

Соединение 9a (0,03 ммоль), полученное в примере получения 31 выше, и тетрагидро-2H-пиран-4-амин (0,06 ммоль) перемешивали в THF (0,3 мл) при 60°С в течение 5 часов. После полного исчезновения соединения 9a, реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Концентрированную смесь выше отделяли и очищали колоночной хроматографией (DCM:MEOH =40:1), таким образом получая соединение 10a.
желтый твердый остаток (84%); 1H ЯМР (400 МГц, DMSO) δ 8,34 (д, 1H), 7,68 (д, 1H), 7,61 (с, 1H), 7,54 (д, 1H), 6,96 (д, J=8,3 Гц, 1H), 6,86 (дд, J=8,0, 1,7 Гц, 1H), 6,06 (с, 2H), 4,20 (с, 1H), 3,96 (с, 2H), 3,84-3,73 (м, 2H), 3,18-3,10 (м, 1H), 1,50-1,41 (м, 2H), 1,40-1,29 (м, 2H), 0,88-0,81 (м, 2H); LRMS (ESI) m/z рассчитано для C21H20N6O3 [M+H]+: 405, найдено 405.
Соединения следующих примеров 2-6 получали тем же способом, как показано в примере 1 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4-бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример 2> Получение 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 10b)

белый твердый остаток (33%); 1H ЯМР (400 МГц, DMSO) δ 8,35 (д, 1H), 7,59 (с, 1H), 7,54 (с, 1H), 6,88 (д, J=8,6 Гц, 1H), 6,83 (дд, J=8,4, 1,8 Гц, 1H), 6,37 (д, J=159,2 Гц, 1H), 4,29-4,19 (м, J=2,9 Гц, 4H), 3,96 (с, 2H), 3,76 (с, 1H), 3,19-3,08 (м, 1H), 1,55-1,41 (м, 2H), 1,39-1,26 (м, 2H), 1,27-1,17 (м, 2H), 0,88-0,77 (м, J=8,7, 5,5, 2,1 Гц, 2H); LRMS (ESI) m/z рассчитано для C22H22N6O3 [M+H]+: 419, найдено 419.
<Пример 3> Получение 2-(2-(хинолин-2-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 10c)

желтый твердый остаток (35%); 1H ЯМР (400 МГц, MeOD) δ 8,46 (д, J=8,5 Гц, 1H), 8,33 (д, J=5,2 Гц, 1H), 7,99 (д, J=8,1 Гц, 1H), 7,89 (с, 1H), 7,84-7,72 (м, 3H), 7,67-7,61 (м, J=7,4 Гц, 1H), 6,73 (д, 1H), 3,97 (д, J=0,8 Гц, 2H), 3,53 (с, 1H), 2,05-2,00 (м, J=9,6 Гц, 2H), 1,92-1,71 (м, J=30,9 Гц, 2H), 1,61-1,47 (м, 2H), 1,20-1,06 (м, 3H), 0,92-0,85 (м, 1H); LRMS (ESI) m/z рассчитано для C23H21N7O [M+H]+: 412, найдено 412.
<Пример 4> Получение 2-(2-(нафталин-2-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 10d)
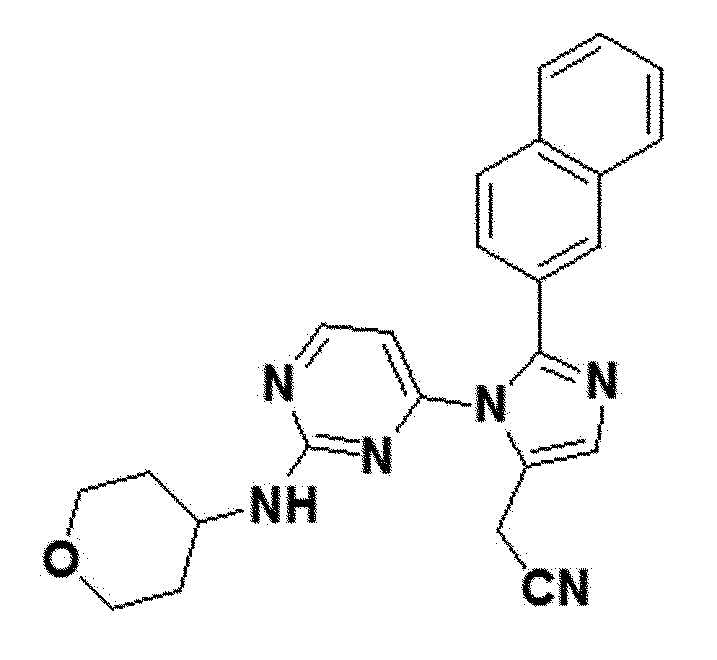
белый твердый остаток (35%); 1H ЯМР (400 МГц, CDCl3) δ 8,10 (д, J=4,7 Гц, 1H), 8,00 (с, 1H), 7,81 (д, J=2,3 Гц, 1H), 7,78 (д, J=8,4 Гц, 2H), 7,57 (с, 1H), 7,53-7,46 (м, J=6,9, 1,7 Гц, 2H), 7,34 (д, J=8,1 Гц, 1H), 6,38 (д, 1H), 4,68 (с, 1H), 3,80 (д, J=0,9 Гц, 2H), 3,65-3,54 (м, 1H), 3,33-3,19 (м,14H), 2,92-2,77 (м, 1H), 1,53-1,39 (м, 2H), 1,34 (д, J=19,3 Гц, 2H), 1,21-1,16 (м, 1H), 0,84-0,75 (м, 1H); LRMS (ESI) m/z рассчитано для C24H22N6O [M+H]+: 411, найдено 411.
<Пример 5> Получение 2-(2-(3,4-дихлорфенил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 10e)

белый твердый остаток (89%); 1H ЯМР (400 МГц, DMSO) δ 8,41 (д, J=4,8 Гц, 1H), 7,73 (д, 1H), 7,68 (с, 1H), 7,54 (д, J=7,8 Гц, 1H), 7,35 (дд, J=8,4, 2,0 Гц, 1H), 6,77 (д, 1H), 4,00 (с, 2H), 3,83 (с, 1H), 3,77-3,67 (м, J=9,7 Гц, 2H), 3,12-2,93 (м, J=9,5 Гц, 3H), 1,38-1,24 (м, J=11,6 Гц, 4H); LRMS (ESI) m/z рассчитано для C20H18Cl2N6O [M+H]+: 430, найдено 430.
<Пример 6> Получение 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 10f)
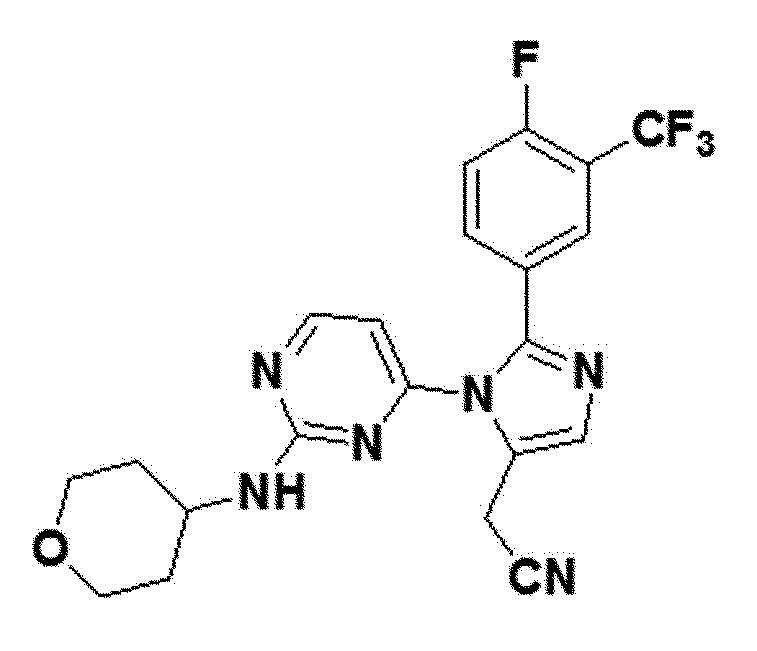
желтый твердый остаток (82%); 1H ЯМР (400 МГц, DMSO) δ 8,42 (д, 1H), 7,77 (д, J=5,1 Гц, 2H), 7,64-7,51 (м, J=22,3, 8,4 Гц, 2H), 6,77 (д, 1H), 4,01 (с, 2H), 3,71 (с, 1H), 3,03-2,93 (м, 2H), 2,10-2,06 (м, J=1,0 Гц, 2H), 1,58-1,42 (м, 1H), 1,37-1,22 (м, J=19,1, 13,5 Гц, 4H); LRMS (ESI) m/z рассчитано для C21H18F4N6O [M+H]+: 447, найдено 447.
<Пример 7> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(циклогексиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 11a)

Соединение 9a (0,03 ммоль), полученное в примере получения 31 выше, и циклогексиламин (0,06 ммоль) перемешивали в THF (0,3 мл) при 60°С в течение 5 часов. После полного исчезновения соединения 9a, реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Концентрированную смесь выше отделяли и очищали колоночной хроматографией (DCM:MEOH =40:1), таким образом получая соединение 11a.
желтый твердый остаток (64%); 1H ЯМР (400 МГц, MeOD) δ 8,22 (с, 1H), 7,62 (с, 1H), 6,93-6,85 (м, J=14,0, 4,7 Гц, 3H), 6,47 (с, 1H), 6,00 (с, 2H), 3,87 (с, 2H), 3,55 (с, 1H), 1,79-1,65 (м, 3H), 1,65-1,58 (м, 1H), 1,36-1,26 (м, 2H), 1,26-1,05 (м, 5H); LRMS (ESI) m/z рассчитано для C22H22N6O2 [M+H]+: 403, найдено 403.
Соединения следующих примеров 8-12 получали тем же способом, как показано в примере 7 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4-бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример 8> Получение 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 11b)
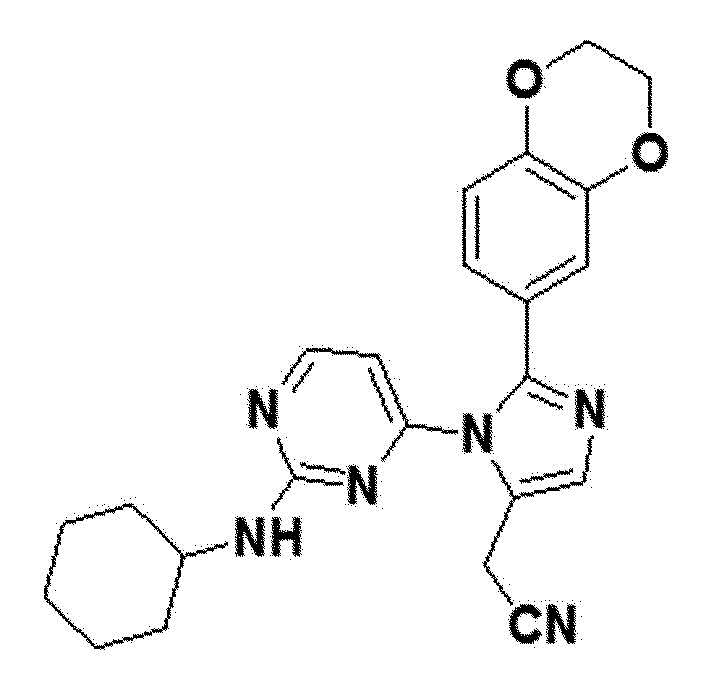
белый твердый остаток (25%); 1H ЯМР (400 МГц, DMSO) δ 8,33 (с, 1H), 7,57 (с, 1H), 7,41 (д, 1H), 6,87 (д, J=8,4 Гц, 1H), 6,83 (дд, J=8,4, 1,8 Гц, 1H), 6,34 (д, J=153,0 Гц, 1H), 4,29-4,19 (м, 4H), 3,95 (с, 2H), 3,18-3,05 (м, 1H), 1,90-1,68 (м, 1H), 1,65-1,49 (м, J=13,7 Гц, 4H), 1,27-1,21 (м, 2H), 1,13-0,99 (м, 4H); LRMS (ESI) m/z рассчитано для C23H24N6O2 [M+H]+: 417, найдено 417.
<Пример 9> Получение 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 11c)
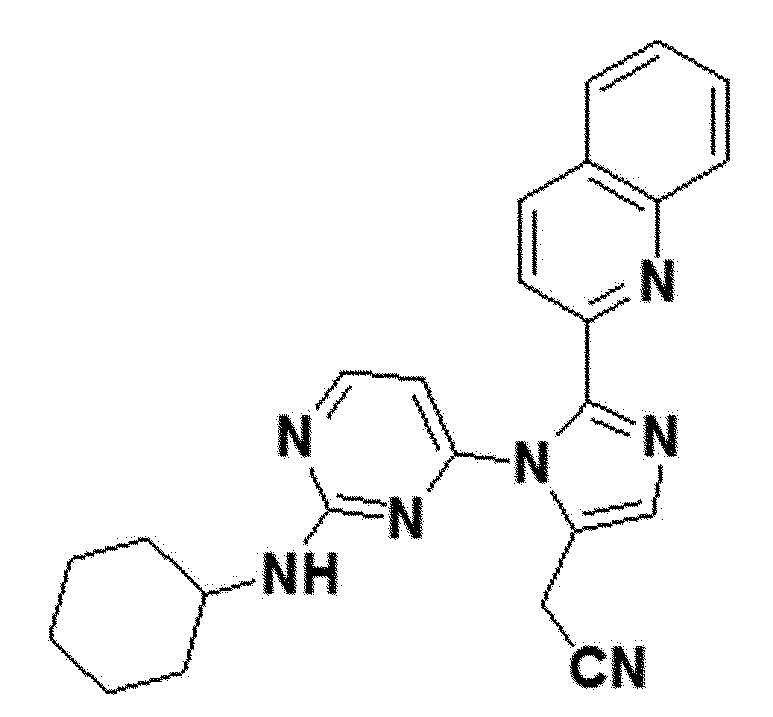
желтый твердый остаток (70%); 1H ЯМР (400 МГц, MeOD) δ 8,47-8,43 (м, J=8,5 Гц, 1H), 8,29 (д, J=4,5 Гц, 1H), 8,05 (д, J=6,0 Гц, 1H), 7,98 (д, J=8,1 Гц, 1H), 7,83-7,77 (м, 1H), 7,76 (с, 1H), 7,73 (д, J=7,2 Гц, 1H), 7,66-7,60 (м, J=11,5, 4,7 Гц, 1H), 6,54 (д, J=6,1 Гц, 1H), 3,97 (д, J=0,8 Гц, 2H), 2,04-1,98 (м, J=10,1 Гц, 2H), 1,82-1,75 (м, J=13,0 Гц, 2H), 1,70-1,64 (м, J=12,8 Гц, 1H), 1,46-1,43 (м, 1H), 1,43-1,40 (м, 1H), 1,40-1,37 (м, 1H), 1,27-1,25 (м, J=3,3 Гц, 1H), 1,24-1,22 (м, J=3,7 Гц, 1H); LRMS (ESI) m/z рассчитано для C24H23N7 [M+H]+: 410, найдено 410.
<Пример 10> Получение 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 11d)

белый твердый остаток (50%); 1H ЯМР (400 МГц, CDCl3) δ 8,06 (д, J=5,6 Гц, 1H), 8,03 (с, 1H), 7,84 (д, 1H), 7,81 (д, J=8,3 Гц, 2H), 7,63 (с, 1H), 7,55-7,47 (м, J=7,0, 3,5 Гц, 2H), 7,40 (д, J=8,3 Гц, 1H), 6,33 (д, J=83,7 Гц, 1H), 4,49 (с, 1H), 3,82 (с, 2H), 3,41-3,19 (м, 1H), 1,77-1,59 (м, 2H), 1,59-1,38 (м, J=42,8 Гц, 3H), 1,19-0,89 (м, 5H); LRMS (ESI) m/z рассчитано для C25H24N6 [M+H]+: 409, найдено 409.
<Пример 11> Получение 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(3,4-дихлорфенфенил)-1H-имидазол-5-ил)ацетонитрила (соединение 11e)

белый твердый остаток (58%); 1H ЯМР (400 МГц, MeOD) δ 8,31 (д, J=5,2 Гц, 1H), 7,71 (с, 1H), 7,66 (д, J=1,4 Гц, 1H), 7,58 (д, J=8,3 Гц, 1H), 7,32 (дд, J=8,3, 2,0 Гц, 1H), 6,47 (д, J=89,7 Гц, 1H), 3,90 (с, 2H), 3,61 (с, 1H), 3,03-2,81 (м, J=39,3 Гц, 2H), 1,36-1,24 (м, 3H), 1,24-1,10 (м, 2H), 1,09-0,95 (м, 3H), 0,93-0,83 (м, 1H); LRMS (ESI) m/z рассчитано для C21H20Cl2N6 [M+H]+: 428, найдено 428.
<Пример 12> Получение 2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)ацетонитрила (соединение 11f)

желтый твердый остаток (90%); 1H ЯМР (400 МГц, DMSO) δ 8,39 (д, J=4,8 Гц, 1H), 7,77-7,73 (м, 2H), 7,58 (дд, J=9,8 Гц, 1H), 7,43 (д, J=7,8 Гц, 1H), 6,75 (д, J=4,4 Гц, 1H), 4,01 (с, 2H), 2,73 (с, 1H), 1,59-1,49 (м, 2H), 1,46-1,36 (м, J=12,3 Гц, 2H), 1,30-1,19 (м, J=20,4 Гц, 2H), 1,04-0,91 (м, J=22,7, 12,9 Гц, 3H), 0,91-0,82 (м, J=14,7, 7,3 Гц, 2H); LRMS (ESI) m/z рассчитано для C22H20F4N6 [M+H]+: 445, найдено 445.
<Пример 13> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 12a)
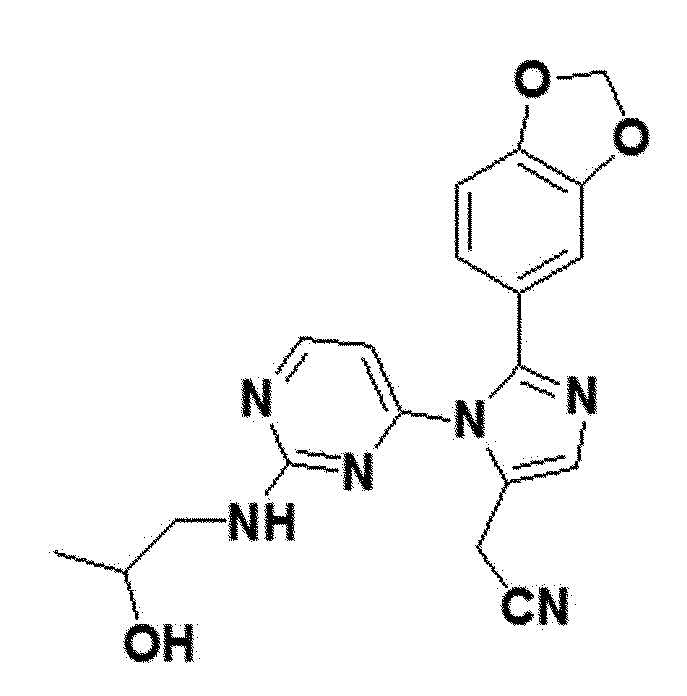
Соединение 9a (0,03 ммоль), полученное в примере получения 31 выше, и 1-аминопропан-2-ол (0,06 ммоль) перемешивали в THF (0,3 мл) при 60°С в течение 5 часов. После полного исчезновения соединения 9a, реакционную смесь охлаждали до комнатной температуры, и концентрировали в вакууме. Концентрированную смесь выше отделяли и очищали колоночной хроматографией (DCM:MEOH =40:1), таким образом получая соединение 12a.
желтый твердый остаток (61%); 1H ЯМР (400 МГц, MeOD) δ 8,22 (д, J=5,2 Гц, 1H), 7,66 (д, J=0,7 Гц, 1H), 6,94-6,84 (м, J=15,4, 10,5, 4,8 Гц, 3H), 6,38 (с, 1H), 6,01 (с, 2H), 3,87 (с, 2H), 3,60 (с, 1H), 3,34 (с, 1H), 3,27-2,99 (м, 2H), 1,37-1,23 (м, 1H), 1,18-1,05 (м, 3H); LRMS (ESI) m/z рассчитано для C19H18N6O3 [M+H]+: 379, найдено 379.
Соединения следующих примеров 14-18 получали тем же способом, как показано в примере 13 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4-бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример 14> Получение 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 12b)
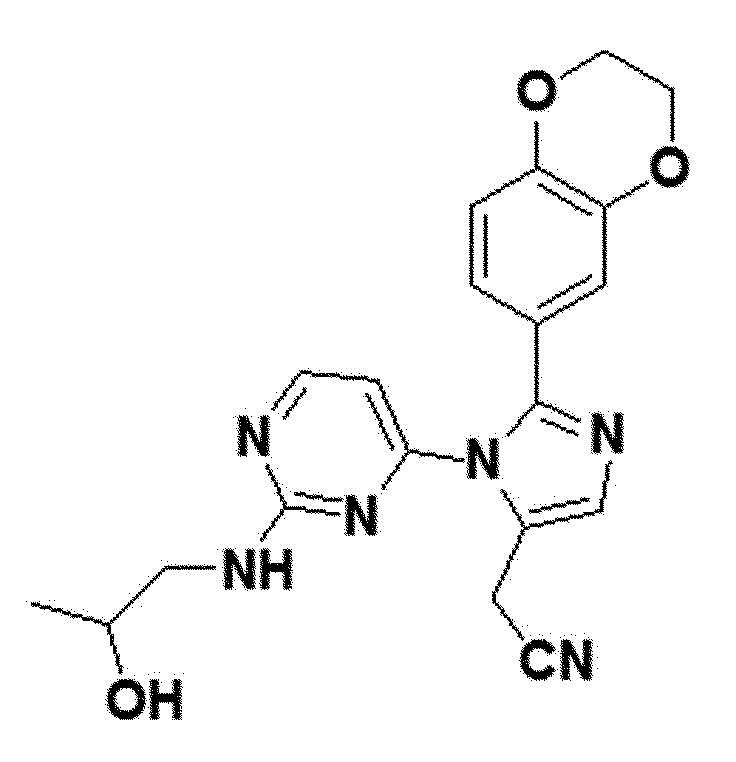
желтый твердый остаток (76%); 1H ЯМР (400 МГц, MeOD) δ 8,22 (д, J=5,3 Гц, 1H), 7,74-7,59 (м, 2H), 6,92 (с, 1H), 6,86 (д, J=1,2 Гц, 1H), 6,37 (д, 1H), 4,31-4,22 (м, J=5,4, 3,7, 1,7 Гц, 4H), 3,86 (д, J=0,9 Гц, 2H), 3,66 (с, 1H), 3,12 (с, 1H), 1,29-1,27 (м, 1H), 1,16-1,08 (м, 2H), 0,92-0,84 (м, 3H); LRMS (ESI) m/z рассчитано для C20H20N6O3 [M+H]+: 393, найдено 393.
<Пример 15> Получение 2-(1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 12c)

белый твердый остаток (38%); 1H ЯМР (400 МГц, MeOD) δ 8,44 (д, J=8,5 Гц, 1H), 8,28 (д, J=5,3 Гц, 1H), 7,97 (д, J=8,5 Гц, 2H), 7,80-7,69 (м, 3H), 7,66-7,59 (м, 1H), 6,61 (с, 1H), 4,26 (с, 27H), 3,97 (д, J=0,9 Гц, 52H), 2,65 (с, 1H), 2,06-1,96 (м, 1H), 1,35-1,24 (м, 2H), 0,93-0,65 (м, 3H); LRMS (ESI) рассчитано для C21H19N7O [M+H]+: 386, найдено 386.
<Пример 16> Получение 2-(1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 12d)

белый твердый остаток (85%); 1H ЯМР (400 МГц, CDCl3) δ 8,04 (д, J=4,7 Гц, 1H), 8,01 (с, 1H), 7,82 (д, J=4,1 Гц, 1H), 7,80 (д, J=8,3 Гц, 2H), 7,64 (с, 1H), 7,54-7,46 (м, 2H), 7,40 (д, J=8,3 Гц, 1H), 6,14 (д, 1H), 3,93 (с, 1H), 3,80 (с, 2H), 3,21 (с, 1H), 1,30-1,18 (м, 3H), 0,99-0,91 (м, J=6,6, 3,1 Гц, 1H), 0,88-0,80 (м, 2H); LRMS (ESI) m/z рассчитано для C22H20N6O [M+H]+: 385, найдено 385.
<Пример 17> Получение 2-(2-(3,4-дихлорфенил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 12e)
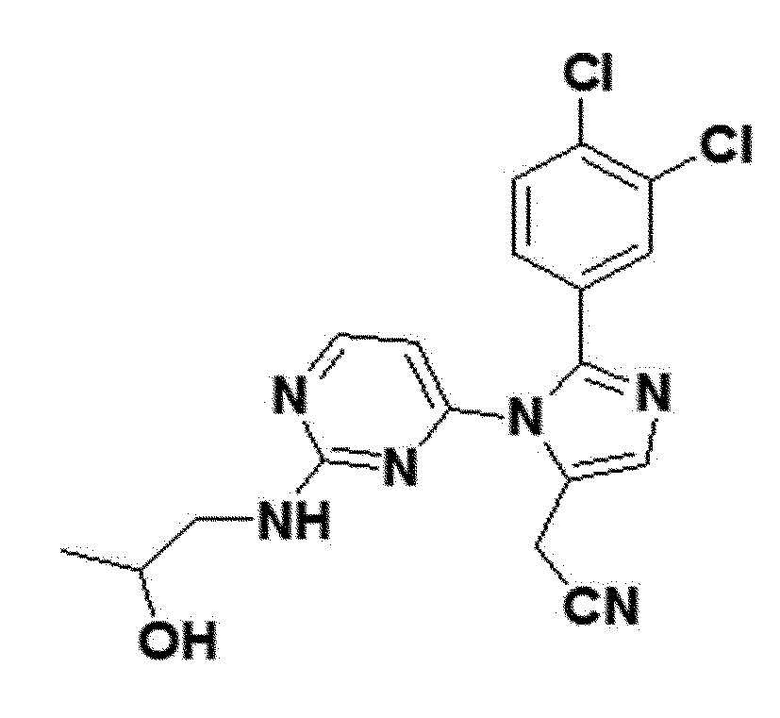
желтый твердый остаток (74%); 1H ЯМР (400 МГц, DMSO) δ 8,37 (д, J=5,1 Гц, 1H), 7,70-7,64 (м, 2H), 7,37-7,32 (м, 2H), 6,52 (д, J=83,1 Гц, 1H), 4,22 (д, J=13,3 Гц, 1H), 4,00 (с, 2H), 2,75 (с, 1H), 1,23 (с, 2H), 1,08-1,01 (м, 1H), 0,84 (д, J=4,0 Гц, 3H); LRMS (ESI) m/z рассчитано для C18H16Cl2N6O [M+H]+: 404, найдено 404.
<Пример 18> Получение 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 12f)
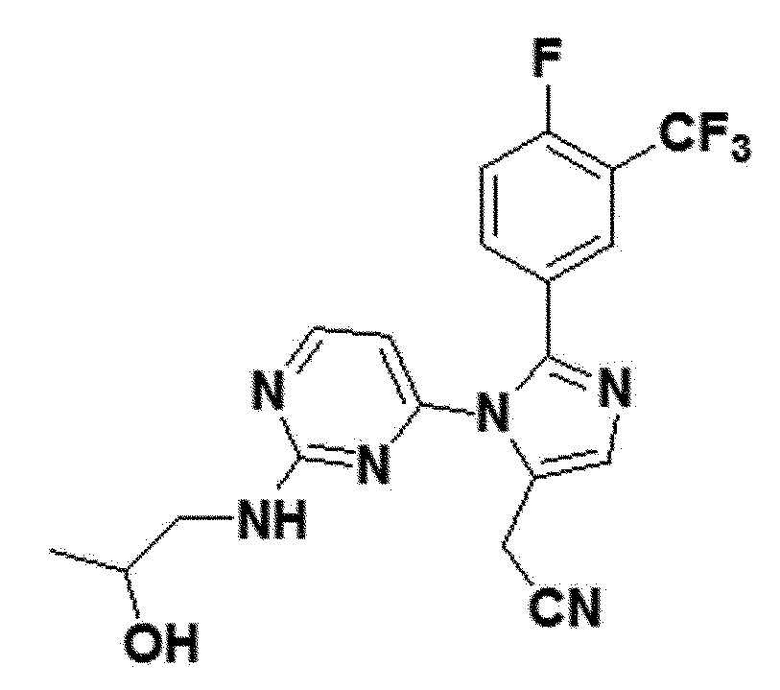
белый твердый остаток (78%); 1H ЯМР (400 МГц, DMSO) δ 8,37 (д, J=5,2 Гц, 1H), 7,79-7,73 (м, 2H), 7,61-7,51 (м, 1H), 7,36 (с, 1H), 6,60 (д, J=47,3, 36,1 Гц, 1H), 4,53 (с, 1H), 4,01 (с, 2H), 2,68 (с, 1H), 1,30-1,14 (м, 2H), 1,10-0,97 (м, 1H), 0,85 (дд, J=9,4, 4,7 Гц, 3H); LRMS (ESI) m/z рассчитано для C19H16F4N6O [M+H]+: 421, найдено 421.
<Пример 19> Получение трет-бутил 3-(4-(2-(бензо[d][1,3]диоксол-5-ил)-5-(цианометил)-1H-имидазол-1-ил)пиримидин-2-иламино)пиперидин-1-карбоксилата (соединение 13a)
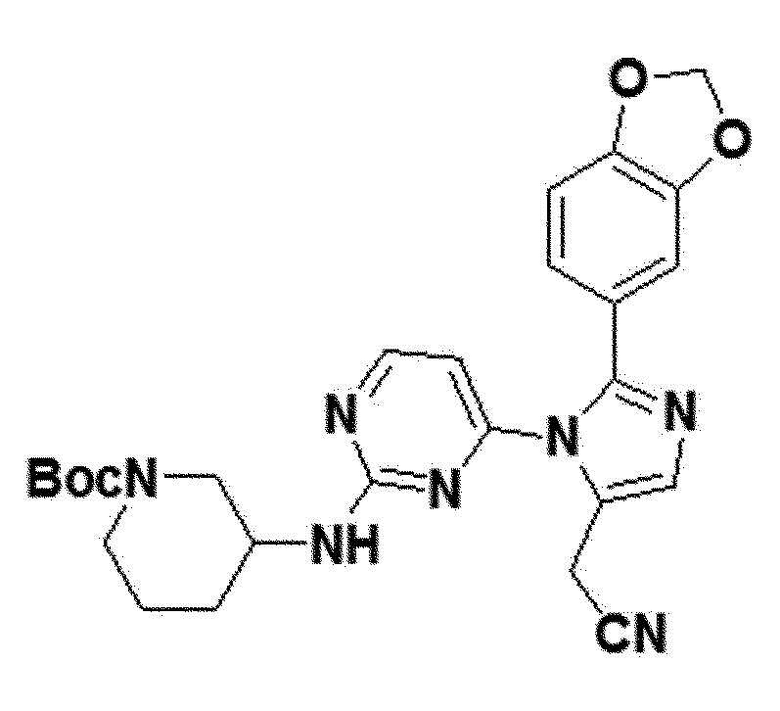
Соединение 9a (0,03 ммоль), полученное в примере получения 31 выше, и трет-бутил 3-аминопиперидин-1-карбоксилат (0,06 ммоль) перемешивали в THF (0,3 мл) при 60°С в течение 5 часов. После полного исчезновения соединения 9a, реакционную смесь охлаждали до комнатной температуры и концентрировали в вакууме. Концентрированную смесь выше отделяли и очищали колоночной хроматографией (DCM:MEOH =40:1), таким образом получая соединение 13a.
желтый твердый остаток (89%); 1H ЯМР (400 МГц, MeOD) δ 8,23 (д, J=5,2 Гц, 1H), 7,74-7,59 (м, J=26,9, 4,4, 2,1 Гц, 1H), 6,93-6,84 (м, 3H), 6,31 (д, J=69,9 Гц, 1H), 6,02 (дд, J=3,5, 1,6 Гц, 2H), 4,03 (с, 1H), 3,86 (д, J=0,5 Гц, 2H), 3,70-3,52 (м, J=15,1 Гц, 1H), 3,52-3,36 (м, J=10,1, 9,3 Гц, 1H), 3,06-2,87 (м, J=2,3 Гц, 1H), 1,93-1,81 (м, 1H), 1,80-1,68 (м, 1H), 1,43 (с, 9H), 1,35-1,28 (м, 4H); LRMS (ESI) m/z рассчитано для C26H29N7O4 [M+H]+: 504, найдено 504.
Соединения следующих примеров 20-24 получали тем же способом, как показано в примере 19 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4-бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример 20> Получение трет-бутил 3-(4-(5-(цианометил)-2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-1-ил)пиримидин-2-иламино)пиперидин-1-карбоксилата (соединение 13b)
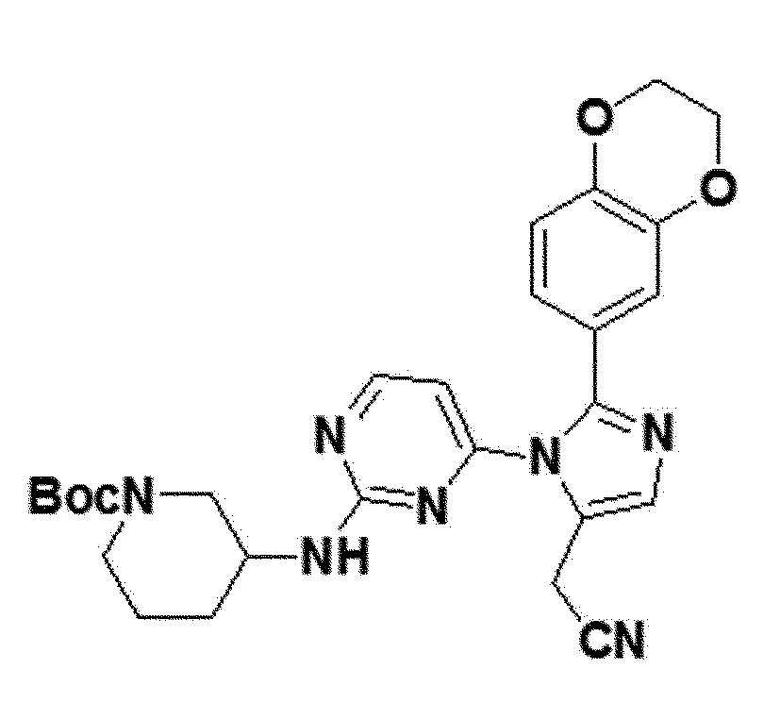
желтый твердый остаток (53%); 1H ЯМР (400 МГц, MeOD) δ 8,24 (д, J=4,7 Гц, 1H), 7,74 (д, J=40,9 Гц, 1H), 6,94 (с, 1H), 6,91-6,84 (м, J=14,3 Гц, 2H), 6,29 (д, J=96,1 Гц, 1H), 4,30-4,26 (м, 2H), 4,26-4,23 (м, 2H), 3,88 (с, 2H), 3,80 (с, 1H), 3,10-2,91 (м, 1H), 1,91-1,80 (м, 1H), 1,80-1,69 (м, 1H), 1,43 (с, 9H), 1,38-1,24 (м, J=15,2 Гц, 6H); LRMS (ESI) m/z рассчитано для C27H31N7O4 [M+H]+: 518, найдено 518.
<Пример 21> Получение трет-бутил 3-(4-(5-(цианометил)-2-(хинолин-2-ил)-1H-имидазол-1-ил)пиримидин-2-иламино)пиперидин-1-карбоксилата (соединение 13c)
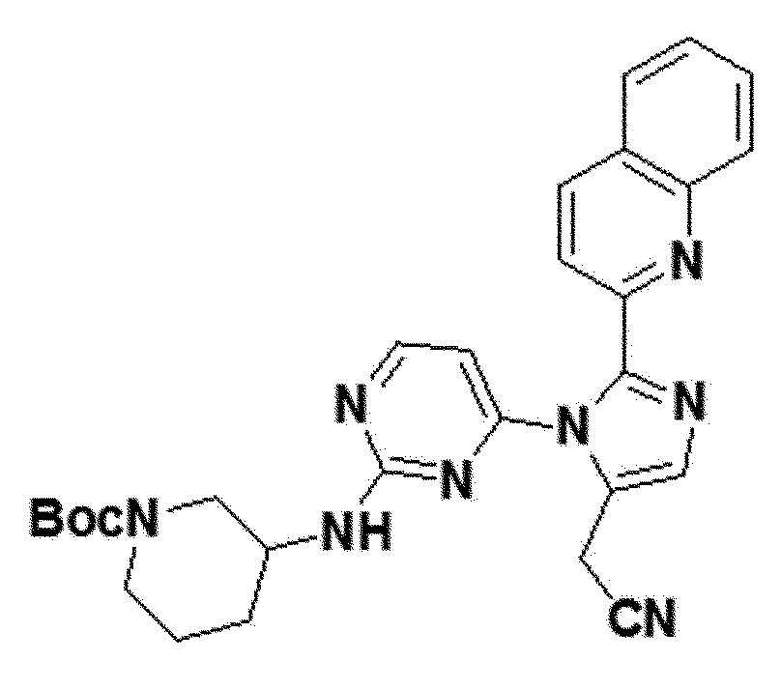
желтый твердый остаток (43%); 1H ЯМР (400 МГц, MeOD) δ 8,43 (д, J=8,5 Гц, 1H), 8,29 (д, J=4,5 Гц, 1H), 7,97 (д, J=8,0 Гц, 1H), 7,93-7,86 (м, 1H), 7,82-7,70 (м, J=13,3, 7,2 Гц, 2H), 7,62 (дд, J=11,0, 5,0 Гц, 1H), 6,65 (д, 1H), 3,97 (д, J=0,6 Гц, 2H), 3,59 (с, 1H), 2,95-2,64 (м, J=80,5 Гц, 2H), 1,48 (с, 8H), 1,34-1,18 (м, J=17,0 Гц, 4H), 1,12 (д, J=6,4 Гц, 1H), 1,01-0,81 (м, J=18,4, 12,0 Гц, 1H); LRMS (ESI) m/z рассчитано для C28H30N8O2 [M+H]+: 511, найдено 511.
<Пример 22> Получение трет-бутил 3-(4-(5-(цианометил)-2-(нафталин-2-ил)-1H-имидазол-1-ил)пиримидин-2-иламино)пиперидин-1-карбоксилата (соединение 13d)
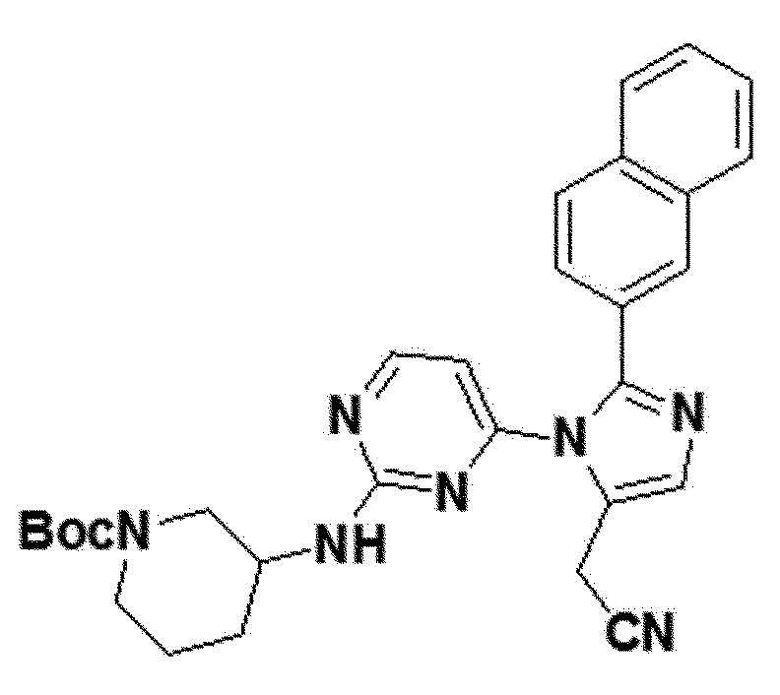
желтый твердый остаток (88%); 1H ЯМР (400 МГц, MeOD) δ 8,17 (с, 1H), 7,99 (с, 1H), 7,88-7,85 (м, 2H), 7,81 (дд, J=20,8 Гц, 2H), 7,56-7,49 (м, 2H), 7,39 (с, 1H), 6,47 (д, 1H), 3,91 (с, 2H), 3,79-3,56 (м, 1H), 3,55-3,33 (м, J=39,4 Гц, 1H), 3,03-2,67 (м, 2H), 2,02-1,97 (м, J=7,0, 3,3 Гц, 1H), 1,38 (с, 9H), 1,34-1,26 (м, 4H), 0,97-0,70 (м, 1H); LRMS (ESI) m/z рассчитано для C29H31N7O2 [M+H]+: 510, найдено 510.
<Пример 23> Получение трет-бутил 3-(4-(5-(цианометил)-2-(3,4-дихлорфенил)-1H-имидазол-1-ил)пиримидин-2-иламино)пиперидин-1-карбоксилата (соединение 13e)
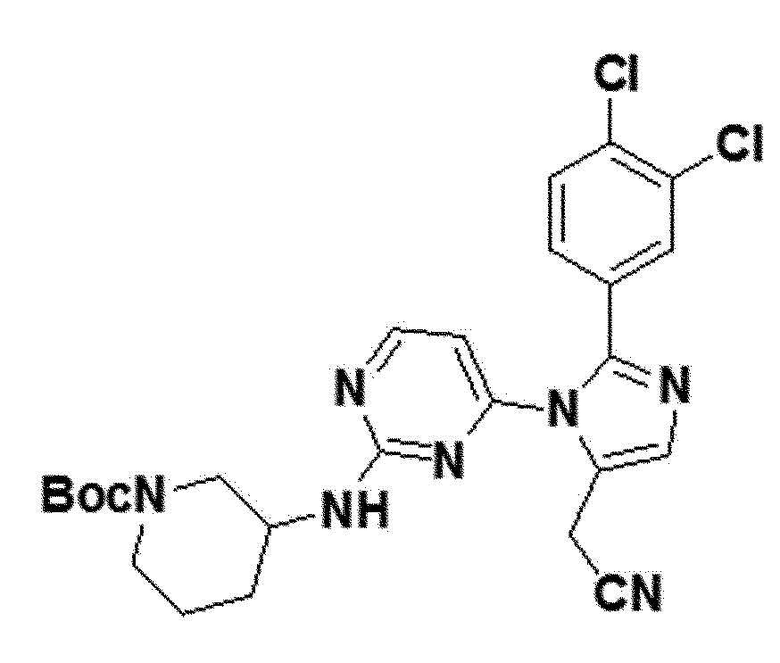
желтый твердый остаток (83%); 1H ЯМР (400 МГц, MeOD) δ 8,33 (д, J=4,6 Гц, 1H), 7,71 (дд, J=5,4, 3,2 Гц, 1H), 7,66 (с, 1H), 7,56 (д, J=8,3 Гц, 1H), 7,32 (д, J=8,5 Гц, 1H), 6,62 (д, 1H), 3,90 (с, 2H), 3,73 (с, 1H), 3,10-2,82 (м, 2H), 2,65 (с, 1H), 1,76-1,66 (м, 2H), 1,28 (с, 9H), 0,92-0,82 (м, J=10,6, 5,9 Гц, 4H); LRMS (ESI) m/z рассчитано для C25H27Cl2N4O2 [M+H]+: 528, найдено 528
<Пример 24> Получение трет-бутил 3-(4-(5-(цианометил)-2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-1-ил)пиримидин-2-иламино)пиперидин-1-карбоксилата (соединение 13f)
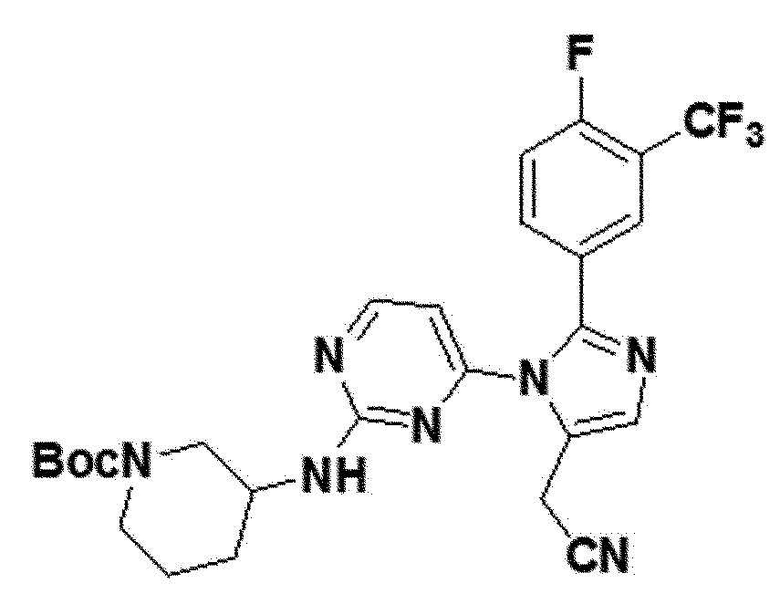
желтый твердый остаток (58%); 1H ЯМР (400 МГц, DMSO) δ 8,43-8,37 (м, J=5,2 Гц, 1H), 7,77 (д, J=5,8 Гц, 1H), 7,71 (д, J=5,9 Гц, 1H), 7,55 (с, 2H), 6,68 (д, J=4,3 Гц, 1H), 4,01 (с, 2H), 3,82-3,69 (м, 1H), 3,68-3,56 (м, J=21,6, 14,3 Гц, 1H), 3,37 (с, 1H), 2,94-2,80 (м, J=4,2 Гц, 1H), 2,79-2,65 (м, 1H), 2,23-2,11 (м, 1H), 1,55-1,45 (м, 2H), 1,37 (с, 8H), 1,24-1,20 (м, 2H); LRMS (ESI) m/z рассчитано для C26H27F4N7O2 [M+H]+: 546, найдено 546.
<Пример 25> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 14a)

Соединение 13a (18 мг, 0,033 ммоль), полученное в примере 19 выше, растворяли в 1,4-диоксане (0,33 мл) и обрабатывали в 1,4-диоксане 4 M-HCl (0,17 мл), и затем реакционную смесь выше перемешивали при комнатной температуре в течение 20 минут. Смесь выше разбавляли добавлением эфира и перемешивали до отделения твердого продукта. Твердый продукт выше фильтровали, последовательно промывали эфиром и гексаном и кристаллизовали, таким образом получая соединение 14a.
белый твердый остаток (61%); 1H ЯМР (400 МГц, MeOD) δ 8,28 (д, J=5,3 Гц, 1H), 7,70 (с, 1H), 6,94-6,85 (м, 3H), 6,38 (д, J=12,5 Гц, 1H), 6,02 (с, 2H), 4,04 (с, 1H), 3,88 (д, J=0,8 Гц, 2H), 3,53-3,35 (м, 1H), 3,35-3,31 (м, 1H), 3,29-3,24 (м, 1H), 3,01-2,80 (м, 2H), 2,12-1,92 (м, J=15,5, 12,9 Гц, 2H), 1,82-1,71 (м, J=7,8 Гц, 1H), 1,69-1,57 (м, 1H), 1,40-0,93 (м, 1H); LRMS (ESI) m/z рассчитано для C21H21N7O2 [M+H]+: 404, найдено 404.
Соединения следующих примеров 26-30 получали тем же способом, как показано в примере 25 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4-бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил и 4-фтор-3-(трифторметил)фенил, соответственно).
<Пример 26> Получение 2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 14b)

белый твердый остаток (91%); 1H ЯМР (400 МГц, MeOD) δ 8,41-8,37 (м, J=4,3 Гц, 1H), 8,01 (д, J=39,3 Гц, 1H), 7,05 (д, J=14,0 Гц, 1H), 7,01-6,97 (м, J=4,9 Гц, 2H), 6,56 (д, J=65,3 Гц, 1H), 4,35-4,31 (м, J=3,6, 1,7 Гц, 2H), 4,31-4,27 (м, J=3,5, 1,6 Гц, 2H), 4,12 (с, 1H), 4,09 (д, J=0,8 Гц, 2H), 3,60-3,57 (м, 1H), 3,47-3,37 (м, J=25,5 Гц, 1H), 3,35-3,32 (м, 1H), 2,94 (м, J=26,5, 11,4 Гц, 2H), 2,08-1,99 (м, 1H), 1,92-1,75 (м, 2H), 1,65 (с, 2H); LRMS (ESI) m/z рассчитано для C22H23N7O2 [M+H]+: 418, найдено 418.
<Пример 27> Получение 2-(1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 14c)
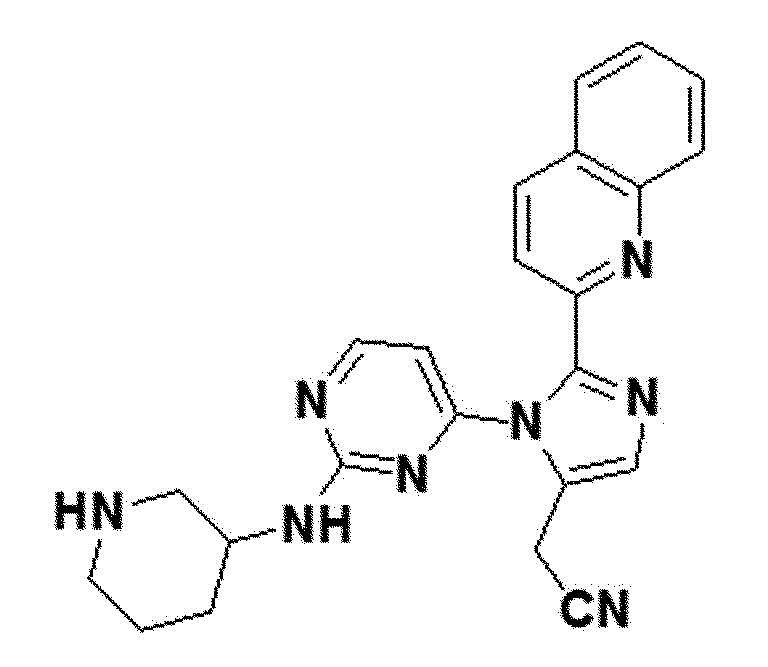
желтый твердый остаток (47%); 1H ЯМР (400 МГц, MeOD) δ 8,45 (д, J=8,5 Гц, 1H), 8,34 (д, J=5,3 Гц, 1H), 8,01 (д, J=8,5 Гц, 1H), 7,97 (д, J=8,1 Гц, 1H), 7,76 (с, 1H), 7,75-7,70 (м, 1H), 7,67 (д, J=7,7 Гц, 1H), 7,64-7,59 (м, J=8,1, 6,6, 1,5 Гц, 1H), 6,59 (с, 1H), 3,97 (д, J=0,8 Гц, 2H), 3,18-3,07 (м, 1H), 2,83 (с, 1H), 2,67 (с, 1H), 2,08-1,96 (м, J=9,6 Гц, 1H), 1,87-1,65 (м, J=10,8 Гц, 2H), 1,60-1,41 (м, J=28,7 Гц, 2H), 1,35-1,22 (м, 2H), 0,99-0,80 (м, J=18,1, 12,1 Гц, 1H); LRMS (ESI) m/z рассчитано для C23H22N8 [M+H]+: 411, найдено 411.
<Пример 28> Получение 2-(2-(нафталин-2-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 14d)
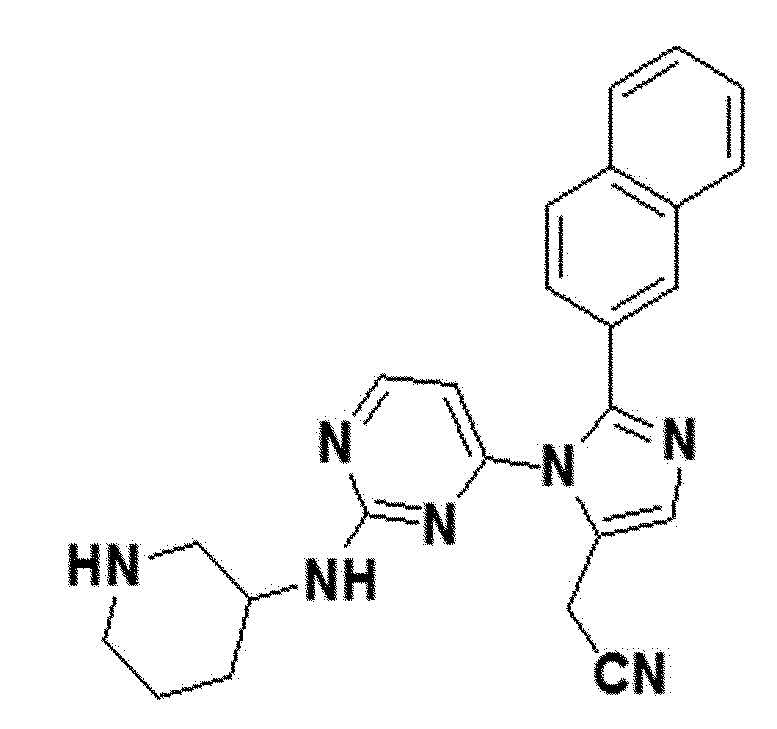
белый твердый остаток (89%); 1H ЯМР (400 МГц, MeOD) δ 8,15 (с, 1H), 7,92 (с, 1H), 7,88-7,77 (м, 4H), 7,52-7,44 (м, 2H), 7,36 (д, J=8,3 Гц, 1H), 6,20 (д, J=93,0 Гц, 1H), 3,91 (с, 2H), 3,62 (с, 1H), 3,21-3,05 (м, 2H), 2,90-2,73 (м, 2H), 2,05-1,90 (м, J=25,3 Гц, 1H), 1,83-1,56 (м, 2H), 1,52-1,25 (м, 3H); LRMS (ESI) m/z рассчитано для C24H23N7 [M+H]+: 418, найдено 418.
<Пример 29> Получение 2-(2-(3,4-дихлорфенил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 14e)

белый твердый остаток (47%); 1H ЯМР (400 МГц, MeOD) δ 8,45 (д, J=5,5 Гц, 1H), 8,10 (с, 1H), 7,83 (д, J=1,5 Гц, 1H), 7,73 (д, J=8,3 Гц, 1H), 7,50 (дд, J=8,6 Гц, 1H), 6,66 (д, 1H), 4,13 (с, 2H), 3,95 (с, 1H), 3,81-3,62 (м, 1H), 3,61-3,56 (м, 1H), 3,02-2,86 (м, 3H), 2,03 (с, 1H), 1,89-1,73 (м, 2H), 1,71-1,55 (м, J=37,0 Гц, 2H). LRMS (ESI) m/z рассчитано для C20H19Cl2N7 [M+H] +: 429, найдено 429.
<Пример 30> Получение 2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 14f)

белый твердый остаток (67%); 1H ЯМР (400 МГц, DMSO) δ 8,38 (д, 1H), 7,74 (с, 2H), 7,56 (д, J=8,9 Гц, 1H), 7,42 (д, 1H), 6,67 (д, J=4,4 Гц, 1H), 4,01 (с, 2H), 3,76 (с, 1H), 3,45-3,35 (м, J=5,6 Гц, 1H), 3,05-2,91 (м, 1H), 2,72-2,59 (м, 1H), 2,42-2,13 (м, 2H), 1,51-1,33 (м, 2H), 1,35-1,20 (м, J=32,9, 17,3 Гц, 2H), 0,88-0,80 (м, 1H); LRMS (ESI) m/z рассчитано для C21H19F4N7 [M+H]+: 446, найдено 446.
<Пример 31> Получение 2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 15a)
Соединение 14a (10 мг, 0,024 ммоль), полученное в примере 25 выше, охлаждали до 0°С в THF (0,24 мл) и обрабатывали TEA (5 мкл, 0,038 ммоль). Циклопропанкарбонилхлорид (6,5 мг, 0,024 ммоль) добавляли к смеси выше при 0°С и перемешивали при комнатной температуре в течение 1 часа. После концентрирования реакционной смеси выше в вакууме, полученный в результате концентрат разбавляли хлористым метиленом и промывали водой и насыщенным водным раствором хлорида натрия. Органический слой обезвоживали сульфатом натрия, и затем полученный в результате остаток концентрировали в вакууме, и отделяли и очищали колоночной хроматографией (DCM:MEOH=40:1), таким образом получая соединение 15a.
белый твердый остаток (49%); 1H ЯМР (400 МГц, MeOD) δ 8,33-8,16 (м, 1H), 7,69 (д, J=40,8 Гц, 1H), 6,90 (дд, J=7,9, 1,7 Гц, 1H), 6,89-6,84 (м, J=7,0 Гц, 2H), 6,44 (д, J=44,0 Гц, 1H), 6,01 (д, J=5,7 Гц, 2H), 4,11 (д, J=12,8 Гц, 1H), 3,87 (д, J=0,8 Гц, 2H), 3,49-3,33 (м, 1H), 3,09-2,70 (м, J=116,5 Гц, 1H), 2,07-1,63 (м, J=75,2, 39,4, 22,7 Гц, 4H), 1,62-1,45 (м, 2H), 1,34-1,20 (м, 1H), 0,97-0,73 (м, 4H), 0,72-0,55 (м, J=32,5, 25,1 Гц, 1H); LRMS (ESI) m/z рассчитано для C25H25N7O3 [M+H]+: 472, найдено 472.
Соединения следующих примеров 32-37 получали тем же способом, как показано в примере 31 выше (1,3-бензодиоксолил заменяли на 2,3-дигидро-1,4-бензодиоксинил, хинолинил, нафтилил, 3,4-дихлорфенил, 4-фтор-3-(трифторметил)фенил и бензофуранил, соответственно).
<Пример 32> Получение 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 15b)
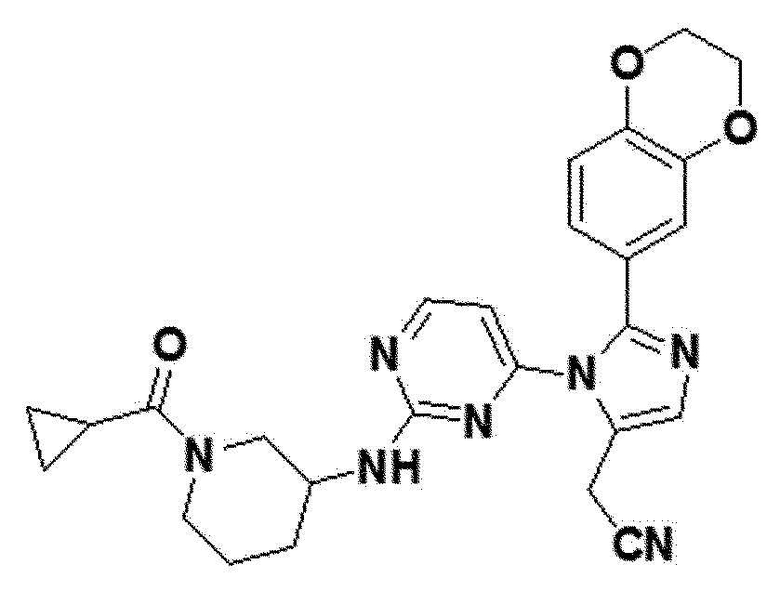
белый твердый остаток (82%); 1H ЯМР (400 МГц, MeOD) δ 8,24 (д, 1H), 7,72 (д, J=5,7, 3,1 Гц, 1H), 7,61 (д, 1H), 6,93-6,83 (м, 2H), 6,45 (д, J=53,2 Гц, 1H), 4,31-4,21 (м, 4H), 3,96 (с, 1H), 3,87 (с, 2H), 3,08-2,94 (м, 1H), 2,06-1,86 (м, 2H), 1,79-1,67 (м, 2H), 1,64-1,52 (м, 2H), 1,38-1,33 (м, J=18,1 Гц, 2H), 0,92-0,85 (м, J=7,4, 6,0, 4,0 Гц, 5H); LRMS (ESI) m/z рассчитано для C26H27N7O3 [M+H]+: 486, найдено 486.
<Пример 33> Получение 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 15c)
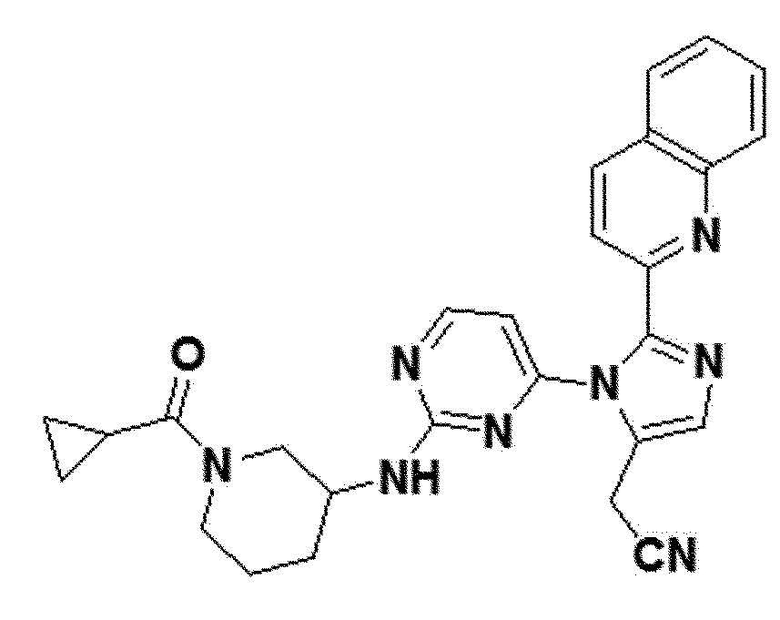
желтый твердый остаток (32%); 1H ЯМР (400 МГц, MeOD) δ 8,44-8,40 (м, J=8,5 Гц, 1H), 8,30 (дд, 1H), 8,02-7,94 (м, J=8,6 Гц, 2H), 7,87 (д, J=23,8 Гц, 1H), 7,75 (с, 1H), 7,73-7,67 (м, 1H), 7,66-7,58 (м, 1H), 6,69 (д, 1H), 3,97 (д, J=0,7 Гц, 2H), 3,89 (с, 1H), 2,95-2,70 (м, 2H), 2,06-1,99 (м, J=9,8 Гц, 1H), 1,97-1,87 (м, 1H), 1,57-1,46 (м, 2H), 1,32-1,26 (м, 2H), 1,00-0,92 (м, 1H), 0,91-0,77 (м, J=20,3, 6,7 Гц, 3H), 0,67-0,42 (м, 2H); LRMS (ESI) m/z рассчитано для C27H26N8O [M+H]+: 479, найдено 479.
<Пример 34> Получение 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 15d)
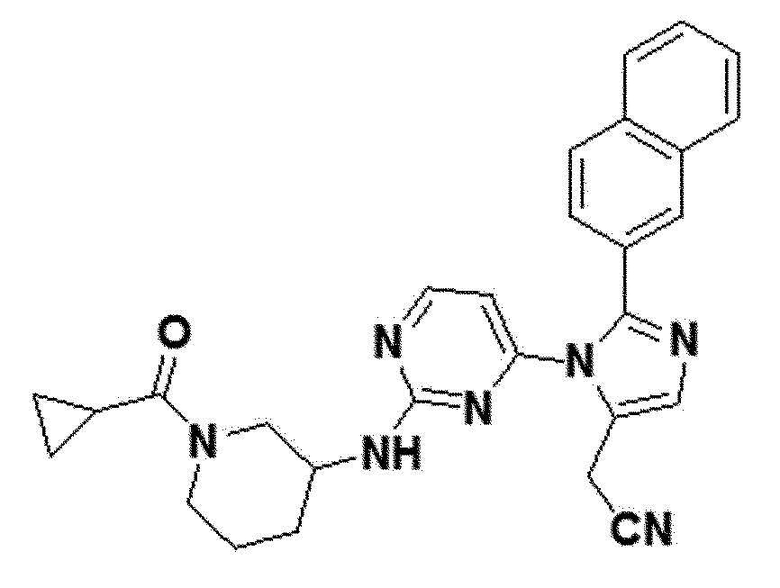
желтый твердый остаток (43%); 1H ЯМР (400 МГц, MeOD) δ 8,23 (д, J=42,0 Гц, 1H), 7,99 (д, J=5,5 Гц, 1H), 7,92-7,81 (м, J=11,6, 8,5 Гц, 3H), 7,71 (с, 1H), 7,56-7,48 (м, 2H), 7,37 (д, J=7,2 Гц, 1H), 6,53 (д, J=56,5 Гц, 1H), 3,91 (с, 2H), 3,86 (с, 1H), 2,92-2,68 (м, J=29,7, 20,7 Гц, 2H), 1,88-1,81 (м, J=7,3, 2,8 Гц, 1H), 1,64-1,50 (м, 2H), 0,98-0,83 (м, 4H), 0,82-0,64 (м, 4H), 0,61-0,51 (м, J=8,2 Гц, 1H); LRMS (ESI) m/z рассчитано для C28H27N7O [M+H]+: 478, найдено 478.
<Пример 35> Получение 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(3,4-дихлорфенил)-1H-имидазол-5-ил)ацетонитрила (соединение 15e)
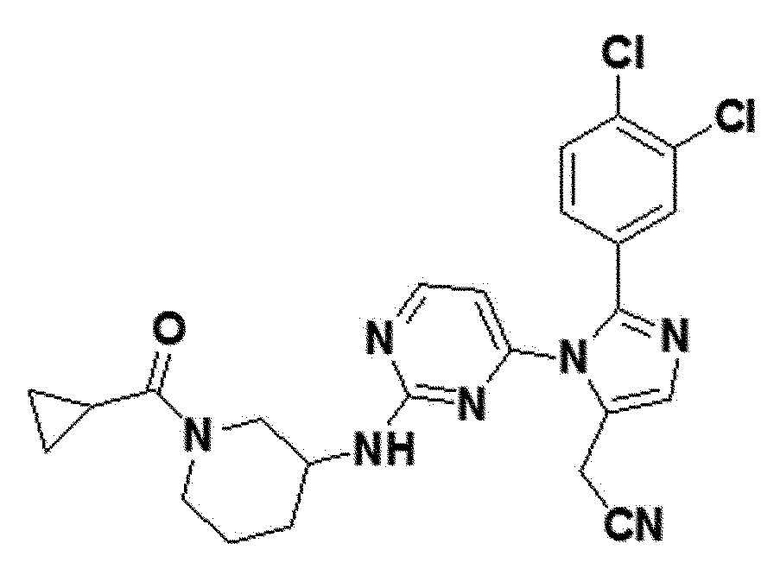
белый твердый остаток (32%); 1H ЯМР (400 МГц, MeOD) δ 8,33 (д, 1H), 7,73 (д, J=24,2 Гц, 1H), 7,65 (с, 1H), 7,56 (дд, J=8,2, 5,3 Гц, 1H), 7,32 (д, J=8,0 Гц, 1H), 6,68 (д, 1H), 4,28 (с, 1H), 3,90 (д, J=0,8 Гц, 2H), 2,68-2,51 (м, 1H), 2,06-1,97 (м, 2H), 1,87-1,73 (м, 2H), 1,59-1,47 (м, 2H), 1,31-1,25 (м, 2H), 0,94-0,76 (м, 4H), 0,68-0,58 (м, 1H); LRMS (ESI) m/z рассчитано для C24H23Cl2N7O [M+H]+: 497, найдено 497.
<Пример 36> Получение 2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)ацетонитрила (соединение 15f)

белый твердый остаток (35%); 1H ЯМР (400 МГц, MeOD) δ 8,32 (д, J=4,8 Гц, 1H), 7,79 (д, 1H), 7,71 (с, 1H), 7,39 (д, J=8,2 Гц, 1H), 6,62 (д, 1H), 4,21 (с, 1H), 3,91 (д, J=0,8 Гц, 2H), 3,08-2,99 (м, 1H), 1,85-1,75 (м, 4H), 1,57-1,50 (м, 4H), 0,91-0,85 (м, J=7,0, 4,6 Гц, 5H); LRMS (ESI) m/z рассчитано для C25H23F4N7O [M+H]+: 514, найдено 514.
<Пример 37> Получение 2-(2-(бензофуран-5-ил)-1-(2-(1-циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрила (соединение 15g)

1H ЯМР (400 МГц, DMSO) δ 8,30 (д, J=22,8 Гц, 1H), 8,03 (с, 1H), 7,93 (м, 1H), 7,72 (д, J=10,2 Гц, 1H), 7,65 (с, 1H), 7,49-7,37 (м, 1H), 6,82 (д, J=7,4 Гц, 1H), 6,58-6,11 (м, 1H), 4,10 (с, NH, 1H), 3,81 (с, 2H), 3,38 (с, 2H), 2,87 (с, 1H), 1,81 (м, 2H), 1,65-1,45 (м, 2H), 1,34 (м, 2H), 0,76-0,58 (м, 5H). LRMS (ESI) m/z рассчитано для C26H25N7O2 [M+H]+: 467, найдено 467.
Экспериментальный пример 1. Измерение активности фермента JNK3
Изменение активности фермента JNK3 в результате обработки имидазольным производным настоящего изобретения (формула 1), показанные в следующей таблицы 1, приведены с помощью IC50.
[Таблица 1]

Прежде всего, после помещения субстрата в приготовленный буферный раствор для основной реакции (20 мМ Hepes (pH 75), 10 мМ MgCl2, 1 мМ EGTA, 0,02% Brij35, 0,02 мг/мл BSA, 0,1 мМ Na3VO4, 2 мМ DTT, 1% DMSO), человеческий JNK3 фермент добавляли в приготовленный раствор субстрата, и затем перемешивали. В качестве субстрата, применяли АТФ (10 мкМ) и ATF2 (3 мкМ), из них АТФ также применяли в качестве стандартного субстрата. Затем, соединения примеров 1-18 и 25-36, которые растворяли в 100% DMSO, вводили в раствор для реакции фермента, и затем выдерживали при комнатной температуре в течение 20 минут. Затем, 33P-АТФ вводили в раствор реакционной смеси выше, вызывая реакцию, затем выдерживали при комнатной температуре в течение 2 часов, и затем активность фермента определяли посредством способа связывания на фильтре.
В частности, после того как 25 мкл полученного в результате раствора медленно прикапывали на P81 бумагу, ее вставляли в сцинтилляционный флакон и промывали 0,75% фосфорной кислотой четыре раза в течение 10 минут каждый и ацетоном один раз в течение 5 минут. 5 мл сцинтилляционного коктейля вводили в сцинтилляционный флакон выше, и полученный в результате сигнал считывали посредством сцинтилляционного счетчика.
IC50 величины соединений примеров 1-18 и 25-36 относительно активности фермента JNK3 показаны в следующей таблице 2. Результаты показали, что имидазольное производное согласно настоящему изобретению обладает превосходной ингибирующей активностью относительно JNK3.
[Таблица 2]
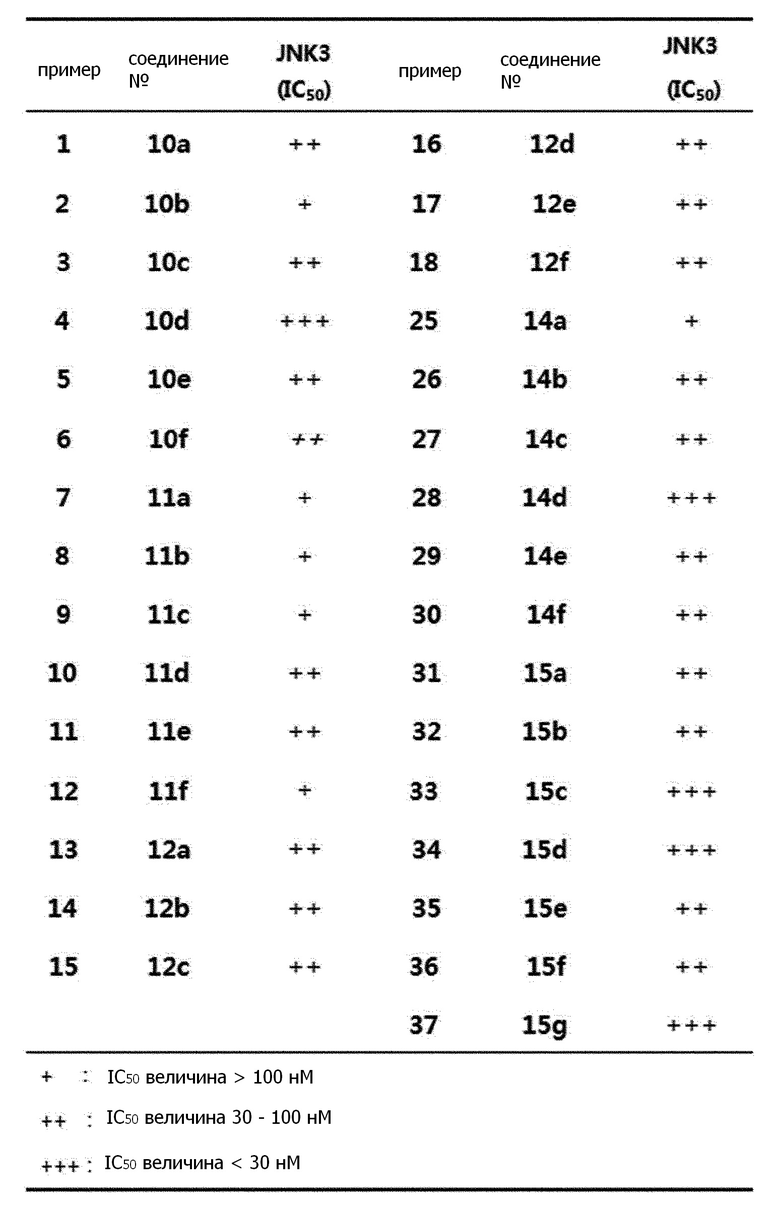
Экспериментальный пример 2. измерение ингибирующей активности относительно различных протеинкиназ
Ингибирующую активность соединения (10 мкМ) примера 34 относительно различных протеинкиназ измеряли посредством службы профилирования киназ (IC50 profiler express) Reaction Biology Corp., где IC50, концентрация, при которой JNK ферментная активность ингибируется на 50%, также измеряли последовательным снижением концентрации соединения.
IC50 величины соединения примера 34 относительно протеинкиназ показаны в следующей таблице 3. Результаты показали, что имидазольное производное согласно настоящему изобретению обладает превосходной ингибирующей активностью относительно JNK1/2/3, но не обладает ингибирующей активностью относительно других протеинкиназ, таким образом, можно видеть, что данное имидазольное производное способно селективно ингибировать активность JNK1/2/3.
[Таблица 3]

Приведенное выше описание настоящего изобретения предполагается для иллюстрации, и специалисту в данной области техники, к которому имеет отношение настоящее изобретение, ясно, что его можно легко модифицировать до других конкретных форм, не изменяя технический объем или необходимые признаки настоящего изобретения. Таким образом, варианты осуществления, описанные выше, следует рассматривать как иллюстративные и неограничивающие во всех аспектах.
| название | год | авторы | номер документа |
|---|---|---|---|
| ГЕМИНАЛЬНО-ЗАМЕЩЕННЫЕ ЦИАНОЭТИЛПИРАЗОЛОПИРИДОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ JANUS КИНАЗ | 2014 |
|
RU2664533C2 |
| ИНГИБИТОРНЫЕ СОЕДИНЕНИЯ | 2013 |
|
RU2673079C2 |
| БИЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ИНГИБИРОВАНИЯ SUV39H2 | 2016 |
|
RU2729187C1 |
| АМИНОТРИАЗОЛОПИРИДИНЫ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗ | 2009 |
|
RU2552642C2 |
| АЗАБИЦИКЛОСОЕДИНЕНИЕ И ЕГО СОЛЬ | 2010 |
|
RU2565078C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ РЕЦЕПТОРА АРОМАТИЧЕСКИХ УГЛЕВОДОРОДОВ | 2021 |
|
RU2826628C1 |
| ПРОИЗВОДНЫЕ N-(ЗАМЕЩЕННЫЙ ФЕНИЛ)-СУЛЬФОНАМИДА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ | 2017 |
|
RU2753520C2 |
| ПРОИЗВОДНЫЕ ХИНОЛИНА И СОДЕРЖАЩИЕ ИХ ИНГИБИТОРЫ MELK | 2011 |
|
RU2582610C2 |
| ПРОИЗВОДНЫЕ 6-[4-(1Н-ИМИДАЗОЛ-2-ИЛ)ПИПЕРИДИН-1-ИЛ]ПИРИМИДИН-4-АМИНА В КАЧЕСТВЕ МОДУЛЯТОРОВ АКТИВНОСТИ КИНАЗ | 2014 |
|
RU2674261C2 |
| ИНГИБИТОРЫ С-FMS КИНАЗЫ | 2007 |
|
RU2475483C2 |
Настоящее изобретение относится к новому имидазольному производному, обладающему ингибирующей C-Jun N-терминальную киназу (JNK) активностью и его применению. Новое имидазольное производное или его фармацевтически приемлемая соль согласно настоящему изобретению обладает превосходной ингибирующей C-Jun N-терминальную киназу (JNK) активностью и, таким образом, предполагается, что более фундаментальный подход и направленное лечение будет возможно при предотвращении или лечении дегенеративных заболеваний нервной системы мозга. 5 н. и 7 з.п. ф-лы, 3 табл., 37 пр.
1. Имидазольное производное следующей формулы 1 или его фармацевтически приемлемая соль, где:
[формула 1]

в формуле 1 выше
R1 представляет собой C6-C10 арил или C7-C9 гетероарил,
где C6-C10 арил представляет собой арил, выбранный из группы, состоящей из фенила, нафтила,
C7-C9 гетероарил представляет собой гетероарил, выбранный из группы, состоящей из 1,3-бензодиоксолила, 1,4-бензодиоксинила, хинолинила или бензофуранила,
и где R1 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из C1 галогеналкила и галогена; и
R2 представляет собой C3 алкил, C6 циклоалкил или C4-C5 гетероциклоалкил,
где C4-C5 гетероциклоалкил представляет собой гетероциклоалкил, выбранный из группы, состоящей из тетрагидропиранила и пиперидинила,
и где R2 может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из гидрокси и C3 циклоалкилкарбонила.
2. Имидазольное производное формулы 1 или его фармацевтически приемлемая соль по п. 1, где:
R1 представляет собой фенил, нафтил, 1,3-бензодиоксолил, хинолинил, 2,3-дигидро-1,4-бензодиоксинил или бензофуранил,
где фенил может быть незамещенным или замещенным, по меньшей мере, одним из C1 галогеналкила или галогена; и
R2 представляет собой 2-гидроксипропил, циклогексил, тетрагидропиранил или пиперидинил,
где пиперидинил может быть незамещенным или замещенным, по меньшей мере, одним из C3 циклоалкилкарбонила.
3. Имидазольное производное формулы 1 или его фармацевтически приемлемая соль по п. 1, где:
R1 представляет собой фенил, нафтил, 1,3-бензодиоксолил, хинолинил, 2,3-дигидро-1,4-бензодиоксинил или бензофуранил,
где фенил может быть незамещенным или замещенным, по меньшей мере, одним заместителем, выбранным из группы, состоящей из фтора, хлора и трифторметила; и
R2 представляет собой 2-гидроксипропил, циклогексил, тетрагидропиранил или пиперидинил,
где пиперидинил может быть незамещенным или замещенным циклопропанкарбонилом.
4. Соединение, представленное формулой 1, или его фармацевтически приемлемая соль по п. 1, где
имидазольное производное формулы 1 выше представляет собой:
2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(хинолин-2-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(нафталин-2-ил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(3,4-дихлорфенил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(тетрагидро-2H-пиран-4-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(циклогексиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(3,4-дихлорфенил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(циклогексиламино)пиримидин-4-ил)-2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(3,4-дихлорфенил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(2-гидроксипропиламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(нафталин-2-ил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(3,4-дихлорфенил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(4-фтор-3-(трифторметил)фенил)-1-(2-(пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(2-(бензо[d][1,3]диоксол-5-ил)-1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(2,3-дигидробензо[b][1,4]диоксин-6-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(хинолин-2-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(нафталин-2-ил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(3,4-дихлорфенил)-1H-имидазол-5-ил)ацетонитрил;
2-(1-(2-(1-(циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-2-(4-фтор-3-(трифторметил)фенил)-1H-имидазол-5-ил)ацетонитрил; или
2-(2-(бензофуран-5-ил)-1-(2-(1-циклопропанкарбонил)пиперидин-3-иламино)пиримидин-4-ил)-1H-имидазол-5-ил)ацетонитрил.
5. Способ получения имидазольного производного по п. 1, как показано на следующей реакционной формуле 1, включающий стадии:
проведения реакции конденсации с аминированием по Бухвальду между соединением формулы I и 4-хлор-2-(метилтио)пиримидином, получая соединение формулы II (стадия 1);
окисления соединения формулы II, полученного на стадии 1 выше, получая соединение формулы III (стадия 2); и
замещения метилсульфонильной группы соединения формулы III, полученного на стадии 2 выше, аминогруппой, получая соединение формулы IV (стадия 3)
[реакционная формула 1]
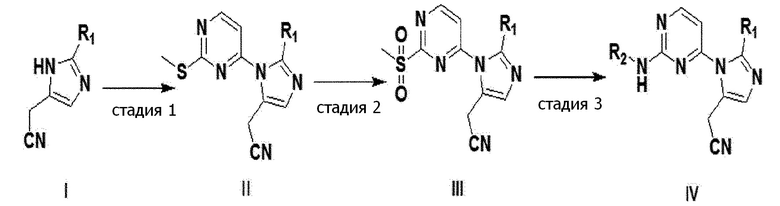
в формуле I-IV выше R1 и R2 представляют собой, как определено в формуле 1 по п. 1.
6. Способ получения имидазольного производного по п. 1 по п. 5, как показано на следующей реакционной формуле 2, дополнительно включающий стадию:
проведения деблокирования соединения формулы IV-4, полученного на стадии 3 выше, получая соединение формулы V (стадия 4)
[реакционная формула 2]

в формуле IV-4 и V выше R1 представляет собой, как определено в формуле 1 по п. 1.
7. Способ получения имидазольного производного по п. 1 по п. 6, как показано на следующей реакционной формуле 3, дополнительно включающий стадию:
ацилирования соединения формулы V, полученного на стадии 4 выше, получая соединение формулы VI (стадия 5)
[реакционная формула 3]
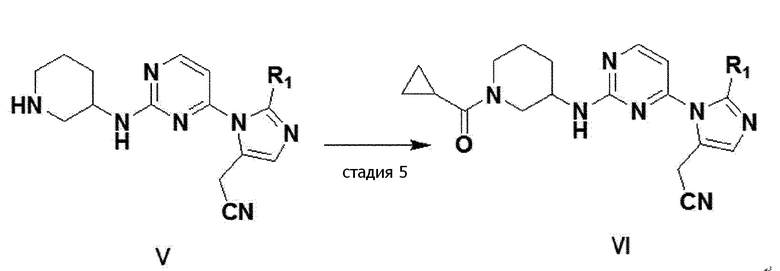
в формуле V и VI выше R1 представляет собой, как определено в формуле 1 по п. 1.
8. Фармацевтическая композиция, содержащая имидазольное производное по одному из пп. 1-4 или его фармацевтически приемлемую соль в качестве эффективного компонента и по меньшей мере один фармацевтически приемлемый носитель, для применения в предотвращении или лечении дегенеративных заболеваний нервной системы мозга.
9. Фармацевтическая композиция по п. 8, характеризующаяся тем, что дегенеративные заболевания нервной системы мозга выше выбраны из группы, состоящей из болезни Альцгеймера, болезни Паркинсона, болезни Хантингтона, множественного склероза и инсульта.
10. Фармацевтическая композиция по п. 9, характеризующаяся тем, что композиция выше ингибирует активность киназы, выбранной из группы, состоящей из C-Jun N-терминальной киназы 1 (JNK 1), C-Jun N-терминальной киназы 2 (JNK 2) и C-Jun N-терминальной киназы 3 (JNK 3).
11. Способ лечения дегенеративных заболевания нервной системы мозга, включающий стадию введения терапевтически эффективного количества имидазольного производного по одному из пп. 1-4 или его фармацевтически приемлемой соли индивиду.
12. Применение имидазольного производного по любому из пп. 1-4 или его фармацевтически приемлемой соли для предотвращения или лечения дегенеративных заболеваний нервной системы мозга.
| US 2004220201 A1, 04.11.2004 | |||
| US 6410726 B1, 25.06.2002 | |||
| US 6200977 B1, 13.03.2001 | |||
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2005 |
|
RU2401265C2 |

