ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
[001] Эта заявка заявляет преимущества и приоритет заявки США № 14/792414, поданной 6 июля 2015 года, и предварительной заявки США № 62/171090, поданной 4 июня 2015 года, которые заявляют преимущества и приоритет непредварительной заявки США № 14/686640, поданной 14 апреля 2015 года, которая заявляет преимущество и приоритет, предварительной заявки США № 61/979351, поданной 14 апреля 2014 года, под названием «Модуляторы протеолиза на основе имидов и связанные с ними способы применения», а также заявляет преимущества и приоритет предварительной заявки США № 62/171090, поданной 4 июня 2015 года, под названием «Модуляторы протеолиза на основе имидов и связанные с ними способы применения», полное содержание которых включено в данное описание путем ссылки.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
[002] Описание предоставляет соединения на основе имида, включая содержащие их бифункциональные соединения, и связанные с ними способы применения. Эти бифункциональные соединения являются полезными в качестве модуляторов целевого убиквитинирования, особенно в отношении различных полипептидов и других белков, которые деградируют (разлагаются) и/или иным образом ингибируются бифункциональными соединениями в соответствии с настоящим изобретением.
УРОВЕНЬ ТЕХНИКИ
[003] Большинство лекарственных веществ на основе малых молекул (низкомолекулярные лекарственные вещества) связывают ферменты или рецепторы в узких и строго определенных карманах. С другой стороны, белок-белковые взаимодействия, как известно, являются трудной мишенью при использовании низкомолекулярных соединений из-за больших поверхностей для контакта и неглубоких карманов, или из-за наличия плоских областей контакта. Убиквитинлигазы E3 (из которых у людей известны сотни) обеспечивают субстратную специфичность для убиквитинирования, и, следовательно, они являются более привлекательными терапевтическими мишенями, чем общие протеасомные ингибиторы, из-за их специфичности в отношении некоторых белковых субстратов. Исследования в области лигандов лигаз E3 оказались достаточно сложными, отчасти из-за того, что эти лиганды должны нарушать белок-белковые взаимодействия. Однако недавние исследования привели к обнаружению специфических лигандов, которые связываются с этой лигазой. Так, например, с момента открытия нутлинов, первых низкомолекулярных ингибиторов лигазы Е3, сообщалось о дополнительных соединениях, для которых мишенью являлись лигазы E3, однако эта область остается недостаточно исследованной.
[004] Одной из лигаз E3 с терапевтическими возможностями является супрессор опухолей фон Гиппель-Линдау (VHL). VHL содержит субъединицу субстрата для распознавания комплексом VCB лигазы Е3, который включает элонгины B и C, а также комплекс, включающий Cullin-2 и Rbx1. Основной субстрат VHL представляет собой индуцируемый гипоксией фактор 1α (HIF-1α), фактор транскрипции, который положительно регулирует гены, такие как проангиогенный фактор роста VEGF и индуцирующий красные кровяные тельца цитокин эритропоэтин, в ответ на низкий уровень кислорода. Авторы изобретения впервые получили небольшие молекулы лигандов белка фон Гиппель-Линдау (VHL) для распознавания субстратной субъединицей лигазы Е3, VCB, важной мишенью при раке, хронической анемии и ишемии, а также полученных кристаллических структур, подтверждающих, что соединение имитирует режиме связывания фактор транскрипции HIF-1α, являющегося основным субстратом VHL.
[005] Цереблон представляет собой белок, который у человека кодируется геном CRBN. Ортологи CRBN, от растений до человека, являются высоко консервативными, что подчеркивает его физиологическое значение. Цереблон образует комплекс из убиквитинлигазы E3 со связывающим поврежденную ДНК белком-1 (DDB1), Cullin-4A (CUL4A) и регулятором куллинов 1 (ROC1). Этот комплекс убиквитинирует ряд других белков. Через механизм, который еще полностью не выяснен, цереблон убиквитинирует целевые белки-мишени, что приводит к повышению уровня фактора роста фибробластов 8 (FGF8) и фактора роста фибробластов 10 (FGF10). FGF8, в свою очередь, регулирует ряд процессов развития, таких как формирование конечностей и слухового пузырька. Конечным результатом является то, что этот комплекс убиквитинлигазы важен для роста конечности у эмбрионов. В случае отсутствия цереблон, DDB1 образует комплекс с DDB2, который функционирует как связывающий поврежденную ДНК белок.
[006] Талидомид, который был одобрен для лечения ряда иммунологических состояний, также был одобрен для лечения некоторых опухолевых заболеваний, в том числе и для множественной миеломы. В дополнение к множественной миеломе, талидомид и некоторые его аналоги в настоящее время также проходят исследования по применению их для лечения других различных типов рака. Несмотря на то, что точный механизм противоопухолевой активности талидомида еще только разрабатывается, однако известно, что он ингибирует ангиогенез. В недавних публикациях обсуждается биология имидов, и эти публикации включают Lu et al Science 343, 305 (2014) и Krönke et al Science 343, 301 (2014).
[007] Важно отметить, что талидомид и его аналоги, например помолинамид и леналинамид, как известно, связывают цереблон. Эти агенты связываются с цереблоном, изменяя специфичность комплекса, вызвая убиквитинирование и деградацию факторов транскрипции Ikaros (ikzf1) и Aiolos (IKZF3), необходимых для роста множественной миеломы. Действительно, высокая экспрессия цереблона связана с увеличением эффективности лекарственных препаратов на основе имида при лечении множественной миеломы.
[008] BRD4 привлекает значительное внимание в рамках академических научных исследований и исследований, проводимых в фармацевтической промышленности, из-за того, что он является новой потенциальной мишенью при различных заболеваниях и патологиях, особенно при раке. BRD4 принадлежит к семейству белков с бромо- и экстратерминальным доменами (BET), который характеризуется наличием двух бромодоменов (домены BD) на N-конце и экстратерминальным доменом (доменом ЕТ) на С-конце (J. Shi, et al. Molecular cell, 54 (2014) 728-736; и A.C. Belkina, et al., Nat. Rev. Cancer, 12 (2012) 465-477). Эти два домена BD распознают и взаимодействуют с ацетилированными остатками лизина на N-конце хвоста гистона белка; домен ET еще не полностью охарактеризован, однако считается, что он в значительной степени выполняет функции каркаса при рекрутинге различных регуляторов транскрипции. Таким образом, BRD4 играет ключевую роль в регуляции экспрессии генов путем рекрутинга соответствующих транскрипционных модуляторов в специфические геномные локусы. В ряде исследований установлено, что BRD4 преимущественно локализуется в супер-энхансерных областях, которые часто находятся в начале сигнального пути важных онкогенов, таких как с-Мус, Bcl-хL и BCL-6, а также он играет ключевую роль в регулировании их экспресии (J. Loven, et al., Cell, 153 (2013) 320-334; и B. Chapuy, et al., Cancer Cell, 24 (2013) 777-790). Благодаря своей центральной роли в модуляции экспрессии основных онкогенов, BRD4 рассматривается в качестве многообещающей терапевтической мишени для нескольких типов рака, в том числе для срединной карциномы, AML, MM, BL и рака предстательной железы (J. Loven, et al., Cell, 153 (2013) 320-334; J. Zuber, et al., Nature, 478 (2011) 524-528; J.E. Delmore, et al., Cell, 146 (2011) 904-917; J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674; A. Wyce, et al., Oncotarget, 4 (2013) 2419-2429; I.A. Asangani, et al., Nature, 510 (2014) 278-282; и C.A. French, et al., Oncogene, 27 (2008) 2237-2242). BRD4 характеризуется высоким уровнем в геномных локусах, проксимальным конкретным онкогенам, что обеспечивает потенциальное терапевтическое окно, которое позволит специфически нацеливаться на клетки опухоли, щадя нормальные ткани. В частности, BRD4 может рассматриваться в качестве альтернативной стратегии для нацеливания в качестве мишени с-Мус, который вносит определенный вклад в развитие и сохранение большинства злокачественных опухолей человека, но который не поддается убиквитинированию (J.E. Delmore, et al., Cell, 146 (2011) 904-917; J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674; M.G. Baratta, et al., PNAS, 112 (2015) 232-237; и M. Gabay, et al., Cold Spring Harb Perspect Med. (2014) 4:a014241).
[009] Исследования в области низкомолекулярных ингибиторов BRD4, таких как JQ1, iBET и OTX15, показали их перспективный терапевтический потенциал в доклинических исследованиях на моделях различных видов рака, включая BL (J. Loven, et al., Cell, 153 (2013) 320-334; B. Chapuy, et al., Cancer Cell, 24 (2013) 777-790; J.E. Delmore, et al., Cell, 146 (2011) 904-917; J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674; I.A. Asangani, et al., Nature, 510 (2014) 278-282; M.G. Baratta, et al., PNAS, 112 (2015) 232-237; M. Boi, et al., Clin. Cancer Res., (2015) 21(7):1628-38; и A. Puissant, et al., Cancer discovery, 3 (2013) 308-323). Действительно, ингибиторы BRD4 показали различные противоопухолевые активности с хорошей переносимостью на различных моделях опухолей у мышей, и не удивительно, что высокая чувствительность к ингибиторам BRD4, таким как JQ1, была связана с высокими уровнями с-Мус и N-Myc в различных типах опухолей, включая c-Myc при BL. Почти во всех случаях BL имело место транслокация гена с-Мус под контролем супер-энхансера, расположенного выше по ходу сигнального пути от IgH, что, таким образом, приводило к аномально высокому уровню экспрессии с-Мус, развитию и поддержанию опухоли (K. Klapproth, et al., British journal of haematology, 149 (2010) 484-497).
[0010] В настоящее время четыре ингибитора BET бромодомена находятся в фазе клинических испытаний, преимущественно направленных на срединную карциному и гематологические новообразования (CPI-0610, NCT01949883; GSK525762, NCT01587703; OTX-015, NCT01713582; ТЕN-010, NCT01987362). Доклинические исследования, выполненные с ингибиторами BRD4, показали их значение в супрессии с-Мус и подавлении пролиферации в клеточных линиях BL, хотя и с величиной IC50 преимущественно в диапазоне от 100 нМ до 1 мкМ (J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674; и M. Ceribelli, et al., PNAS, 111 (2014) 11365-11370). Таким образом, несмотря на быстрый прогресс в разработке ингибиторов BRD4, эффект ингибирования BRD4 рассматривался как обнадеживающий, но не идеальный, поскольку эффект действия оказался в основном цитостатическим, и его достижение требовало относительно высокой концентрации ингибиторов.
[0011] В данной области существует необходимость эффективного лечения заболеваний, особенно гиперплазии и рака. Тем не менее, неспецифические действия и неспособность нацеливания на определенные классы белков и модулировать эти белки, таких как факторы транскрипции, остаются препятствием для разработки эффективных противораковых средств. Такие низкомолекулярные терапевтические агенты, которые воздействуют или потенцируют субстратную специфичность цереблона, и при этом являются «перестраиваемыми» на действие в отношении широкого спектра и классов белков, на которые они специфически нацелены и которые ими модулируются, были бы очень полезны в качестве терапевтических средств.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
[0012] Настоящее описание раскрывает бифункциональные соединения, которые действуют по вовлечению эндогенных белков к действию убиквитинлигаз E3 для их деградации, и к способами их применения. В частности, настоящее изобретение обеспечивает бифункциональные или химерные, нацеленные на протеолиз, (PROTAC) соединения, которые находят применение в качестве модуляторов убиквитинирования различных целевого полипептидов-мишеней и других белков, которые затем деградируют и/или иным образом ингибируются бифункциональными соединениями, как описано здесь. Преимущество представленных здесь соединений состоит в том, что они могут иметь широкий спектр фармакологической активности, состоящий в деградации/ингибировании целевых полипептидов практически любого класса или семейства белков. Кроме того, описание раскрывает способы применения эффективного количества описанных здесь соединений для лечения заболеваний или улучшения состояния при заболеваниях, таких как рак, например, при множественной миеломе.
[0013] Таким образом, в одном аспекте изобретение обеспечивает новые соединения на основе имидов, как описано здесь.
[0014] В дополнительном аспекте данное изобретение обеспечивает бифункциональные соединения или соединения Protac (PROteolysis TArgeting Chimera (нацеленная на протеолиз химера)), которые содержат фрагмент связывания убиквитинлигазы E3 (т.е. лиганд для убиквитинлигазы E3 или группу «ULM»), и фрагмент, который связывает белок-мишень (т.е. лиганд, нацеленный на белок/полипептид или группа «PTM»), так чтобы целевой белок/полипептид располагался бы в непосредственной близости от убиквитинлигазы, чтобы этот белок эффективно подвергался деградированию (и ингибированию). В предпочтительном варианте осуществления изобретения ULM представляет собой связывающий фрагмент убиквитинлигазы Е3 цереблона (т.е. «CLM»). Например, структура бифункционального соединения может быть изображена следующим образом:
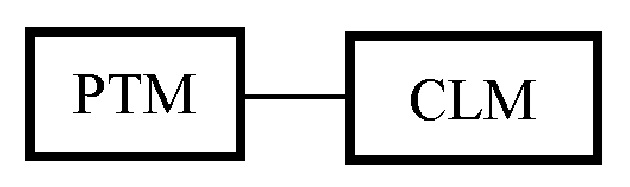
[0015] Соответствующие положения фрагментов PTM и CLM, а также их количество, как показано в настоящем документе, приведены только в качестве примера, и в любом случае они не предназначены для ограничения соединений. Специалисту в данной области понятно, что бифункциональные соединения, описанные здесь, могут быть синтезированы таким образом, что количество и расположение соответствующих функциональных фрагментов может варьировать по желанию.
[0016] В некоторых вариантах осуществления изобретения, бифункциональное соединение дополнительно содержит химический линкер («L»). В этом примере, структура бифункционального соединения может быть изображена следующим образом:
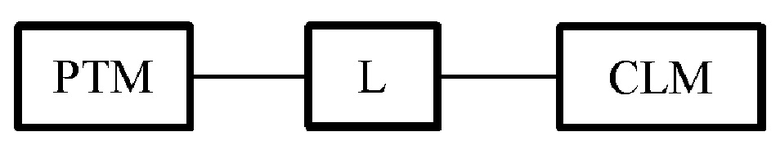
где PTM представляет собой нацеливающий на белок/полипептид фрагмент, L представляет собой линкер и СLM представляет собой связывающий фрагмент убиквитинлигазы Е3 цереблона.
[0017] В некоторых предпочтительных вариантах осуществления убиквитинлигаза Е3 является цереблоном. Таким образом, в некоторых дополнительных вариантах осуществления изобретения, CLM бифункционального соединения включает химические структуры, такие как фрагменты амида, имида, тиоамида, тиоимида. В дополнительных вариантах осуществления CLM содержит фталимидную группу, или ее аналог или производное. В других дополнительных вариантах осуществления изобретения, CLM содержит фталимидо-глутаримидную группу или ее аналог или производное. В других вариантах осуществления изобретения CLM содержит элемент из группы, состоящей из талидомида, леналидомида, помалидомида и их аналогов или производных.
[0018] В некоторых вариантах осуществления изобретения, соединение, описанное здесь, включает несколько CLM, несколько PTM, несколько химических линкеров или их комбинацию.
[0019] В дополнительном аспекте настоящее изобретение предоставляет терапевтические композиции, содержащие эффективное количество соединения, описанного здесь, или его соли, и фармацевтически приемлемый носитель. Терапевтические композиции модулируют деградацию белков у пациента или у субъекта, например, животного, такого как человек, и могут быть использованы для лечения или облегчения состояния или состояний, которые модулируются за счет деградированного белка. В некоторых вариантах осуществления изобретения, терапевтические композиции, описанные здесь, могут быть использованы для деградации белков, представляющих интерес, для лечения или облегчения заболевания, такого, как, например, рак. В еще одном аспекте, настоящее изобретение относится к способу убиквитинилирования/деградации белка-мишени в клетке. В некоторых вариантах осуществления изобретения, способ включает введение бифункционального соединения, описанного в настоящем документ, содержащего CLM и PTM, которые предпочтительно связаны через линкер, как описано здесь, или где CLM соединен с PTM, и где CLM распознает белок пути убиквитина (т.е. убиквитинлигазу, предпочтительно, убиквитинлигазу E3, такую как цереблон), и PTM распознает целевой белок-мишень, что приводит к тому, что деградация белка-мишени будет происходить, когда белок-мишень находится в непосредственной близости от убиквитинлигазы, что в свою очередь приводит к эффекту деградации/ингибирования белка-мишени и к контролю уровня белка. Контроль уровней белка, обеспечиваемый настоящим изобретением, обеспечивает возможность лечения заболевания или состояния, которое модулируется посредством белка-мишени, за счет снижения уровня этого белка в клетках пациента.
[0020] В дополнительном аспекте настоящее изобретение предоставляет способ оценки (т.е. определения и/или измерения) аффинности связывания CLM. В некоторых вариантах осуществления изобретения способ включает получение испытуемого агента или соединения, представляющего интерес, например, агента или соединения, имеющего имидный фрагмент, например, фталимидную группу, фталимидо-глутаримидную группу, дериватизированный талидомид, дериватизированный леналидомид или дериватизированный помалидомид, и сравнения аффинности связывания цереблона и/или ингибирования активности цереблона испытуемым агентом или соединением по отношению с агентом или соединением, у которого известна активность по связыванию цереблона и/или ингибирования цереблона.
[0021] В еще одном аспекте данное описание раскрывает способы лечения или облегчения заболевания, расстройства или его симптомов у субъекта или пациента, например, у животного, такого как человек, включающие введение субъекту, нуждающемуся в этом, композиции, содержащей эффективное количество, например, терапевтически эффективное количество, соединения, раскрытого в данном описании, или его соли, и фармацевтически приемлемый носитель, где композиция является эффективной для лечения или облегчения заболевания или расстройства, или его симптомов у субъекта.
[0022] В другом аспекте данное описание раскрывает способы идентификации эффектов деградации представляющих интерес белков в биологической системе с использованием соединений по изобретению.
[0023] Представленные выше общие указания в отношении области применения представлены только в качестве примера, и они не предназначены для ограничения объема настоящего изобретения и прилагаемой формулы изобретения. Дополнительные цели и преимущества, связанные со структурами, способами и процессами настоящего изобретения понятны любому специалисту в данной области техники в свете настоящей формулы изобретения, описания и примеров. Например, различные аспекты и варианты осуществления настоящего изобретения могут быть использованы в различных комбинациях, все из которых обязательно охватываются настоящим описанием. Эти дополнительные аспекты и варианты осуществления непосредственно включены в объем настоящего изобретения. Публикации и другие материалы, используемые в настоящем описании для освещения уровня техники изобретения, а также в отдельных случаях, для предоставления дополнительных деталей практического выполнения, включены сюда посредством ссылки.
КРАТКОЕ ОПИСАНИЕ ФИГУР
[0024] Прилагаемые фигуры, которые включены в описание и составляют его часть, иллюстрируют несколько вариантов осуществления настоящего изобретения, и, вместе с описанием, служат для объяснения принципов настоящего изобретения. Фигуры предназначены только для целей иллюстрации варианта осуществления изобретения и не должны рассматриваться как ограничивающее настоящее изобретение. Другие цели, отличительные признаки и преимущества настоящего изобретения станут очевидными из последующего подробного описания, рассматриваемого в сочетании с прилагаемыми фигурами, где показаны иллюстративные варианты осуществления настоящего изобретения.
[0025] Фигура 1А и Фигура 1В: иллюстрация общего принципа действия Protac. (A): показаны типовые PROTAC, содержащие фрагмент, нацеливающий на белок (PTM; темный прямоугольник), и фрагмент, связывающий убиквитинлигазу (ULM; светлый затененный треугольник), и, необязательно, линкер (L; черная линия) соединяющий или связывающий PTM с ULM. (В): показано функциональное использование PROTAC, описанных здесь. Вкратце, ULM распознает и связывается с конкретной убиквитинлигазой E3, а PTM связывает и рекрутирует целевой белок, помещая его в непосредственной близости от убиквитинлигазы E3. Как правило, убиквитинлигаза Е3 образует комплекс с убиквитин-конъюгирующим белком Е2, и, либо самостоятельно, либо с помощью белка Е2 катализирует присоединение убиквитина (темные кружки) к лизину белка-мишени посредством изопептидной связи. После этого поли-убиквитинированный белок (крайний справа) становится мишенью для деградации по протеасомному механизму в клетке.
[0026] Фигура 2: Химерное соединение A825, полученное с использованием технологии Protac. A825 содержит связывающий фрагмент BRD4 (производное OTX-15), который соединен с фрагментом рекрутинга убиквитинлигазы E3 цереблона (производное помалидомида) через тетраоксатетрадекановый линкер.
[0027] Фигуры 3A, 3B, 3C, 3D, 3Е, 3F, 3G, 3Н и 3I: изображения вестерн-блотов, показывающие эффекты действия низкомолекулярных ингибиторов BRD4 (JQ1 и OTX-15) на линии клеток BL. JQ1 и OTX-15 приводят к накоплению BRD4 в клетках NAMALWA (А) и в клетках Ramos (B) в зависимости от дозы. OTX-15 приводит к накоплению BRD4 в клетках СА-46 (С) и в клетки DAUDI (D) в зависимости от дозы. JQ1 и OTX-15 приводят к значительному, но не к полному, подавлению с-Мус в клетках NAMALWA (Е) и в клетках Ramos (F). (G) Эффект подавления с-Мус с помощью JQ1 является обратимым. Эффект подавления с-Мус с помощью JQ1 и OTX-15 в клетках NAMALWA (H) и в клетках Ramos (I) является обратимым.
[0028] Фигуры 4A, 4B, 4C, 4D, 4E, 4F и 4G: изображения вестерн-блотов, показывающие эффекты действия A825 на линию клеток BL. Деградация BRD4 под действием A825 в клетках NAMALWA (А) и в клетках СА-46 (B) происходит колоколообразным образом в зависимости от дозы. (С) и (D): деградации BRD4 под действием A825 происходит быстро. (Е) и (F): деградация BRD4, индуцированная обработкой A825, зависит от цереблона. (G): деградация BRD4 под действием A825, опосредованная протеасомой.
[0029] Фигуры 5А, 5В, 5С, 5D, 5E и 5F: сравнение эффектов действия A825, JQ1 и OTX-15 при обработке клеток. (А) и (В): подавление с-Мус под действием A825 является более значительным, чем в случае JQ1 и OTX-15. (С): после обработки A825 уровни белка с-Мус подавлены в большей степени, чем в случае JQ1 и OTX-15. (D), (Е) и (F): функция белка с-Мус (по оценке экспрессии гена SLC19A1) подавляются в большей степени при обработке A825, по сравнению с JQ1 и OTX-15.
[0030] Фигуры 6А, 6В, 6С, 6D, 6Е, 6F, 6G и 6Н: сравнение анти-пролиферативного эффекта на линиях клеток BL при обработке A825, JQ1 и OTX-15. (А)-(D): A825 показал превосходный анти-пролиферативный эффект на линии клеток BL, по сравнению с JQ1 и OTX-15. (Е) A825 приводит к более сильному подавлению пролиферации по сравнению с JQ1 и OTX-15. (G): помалидомид защищает клетки от анти-пролиферативного действия небольших доз A825. (F): помалидомид частично защищает клетки от анти-пролиферативного действия больших доз A825. (H): помалидомид, применяемый отдельно, не оказывает существенного влияния на пролиферацию клеток BL.
[0031] Фигуры 7А и 7В: сравнение эффекта действия A825, JQ1 и OTX-15 на апоптоз клеток BL. (А) A825 приводит к более значительной индукции апоптоза в клетках BL (контроль по каспазной активности), по сравнению с JQ1 и OTX-15. (В) A825 приводит к боле значительной индукции апоптоза в клетках BL (контроль по расщеплению PARP), по сравнению с JQ1 и OTX-15.
[0032] Фигуры 8A и 8B: схема, показывающая механизм действий модели по деградации BRD4 при обработке соединением A825. (А): клетки, обработанные низкими концентрациями A825, эффективно связываются с BRD4 и цереблоном, образуя тримерный комплекс «BRD4-A825-цереблон», который приводит к эффективной деградации BDR4 в клетке. (В): клетки, обработанные высокими концентрациями A825, образуют димеры «BRD4-A825» и «A825-цереблон», которые затрудняют образование оптимального тримера и деградацию BRD4.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
[0033] Ниже приведено подробное описание изобретения с целью помощи специалистам в данной области техники понимания настоящего изобретения при его осуществлении. Специалисты в данной области техники могут вносить модификации и изменения в описанные здесь варианты осуществления изобретения без отхода от сущности или объема настоящего изобретения. Все публикации, патентные заявки, патенты, рисунки и другие документы, упомянутые в настоящем описании, включены во всей полноте путем ссылки.
[0034] В настоящем описании раскрыты структуры и способы, которые связаны с удивительным и неожиданным открытием, что белки убиквитинлигазы Е3, например, цереблон, подвергают убиквитинированию белок-мишень, когда белок-мишень расположен в непосредственной близости от бифункционального соединения или химерной конструкции, которая связывает белок убиквитинлигазу E3 и целевой белок. Соответственно, настоящее изобретение обеспечивает такие соединения и структуры, содержащие связывающий убиквитинлигазу E3 фрагмент («ULM»), соединенный с нацеливающим на белок-мишень фрагментом («PTM»), которые обеспечивают убиквитинирование выбранного белка-мишени, что приводит к деградации белка-мишени с помощью протеасомы (см Фигуру 1). Настоящее изобретение также предоставляет библиотеку композиций и их применения.
[0035] Если не определено иначе, то все используемые в изобретении технические и научные термины имеют такие же значения, которые является общеупотребительными для обычного специалиста в области, к которой принадлежит изобретение. Используемая в изобретении терминология применяется только для описания конкретных вариантов осуществления изобретения, и ее не следует рассматривать как ограничение изобретения.
[0036] В тех случаях, когда указывается диапазон значений, следует иметь в виду, что изобретение охватывает каждое промежуточное значение до десятой доли единицы нижнего предела, если из контекста в явном виде не следует иное (как в случае группы, содержащей некоторое количество углеродных атомов, когда указывается каждый номер углеродного атома, попадающий в диапазон), между верхним и нижним пределом этого диапазона и любым другим заявленным или промежуточным значением в указанном диапазоне. Изобретение также охватывает верхний и нижний пределы этих меньших диапазонов, которые могут быть независимо включены в меньшие диапазоны, с учетом любого специально исключаемого предела в указанном диапазоне. Если указанный диапазон включает в себя один или оба предела, диапазоны, исключающие любой из этих двух включенных пределов, также охватываются изобретением.
[0037] Следующие термины используются для описания настоящего изобретения. В тех случаях, когда термин конкретно не определен в описании, этот термин имеет значение, являющееся общепринятым для обычных специалистов в данной области, которые применяли бы этот термин в случаях его использования при описании настоящего изобретения.
[0038] Используемая в описании изобретения и в формуле изобретения форма единственного числа используются для обозначения одного или более чем одного (то есть, по меньшей мере одного) объекта, если из контекста явным образом не следует иное. В качестве примера, «признак», выраженный единственным числом, означает один признак или более чем один признак.
[0039] Следует понимать, что используемый в описании изобретения и в формуле изобретения термин "и/или" означает "любой из двух или оба вместе" соединенных таким образом элемента, то есть, элементы, которые присутствуют совместно в одних случаях и присутствуют раздельно в других случаях. Множество элементов, перечисленных с использованием "и/или", следует истолковывать аналогичным образом, то есть, "один или более" из соединенных таким образом элемента. Могут необязательно присутствовать и другие элементы, не являющиеся элементами, специально определяемыми условием "и/или", независимо от того, относятся они или не относятся к определяемым элементам. Таким образом, в качестве неограничивающего примера, упоминание "A и/или B", при использовании в сочетании с выражением расширения, например "содержащий", может иметь в виду, в одном варианте осуществления, только A (необязательно включая элементы, не являющимися B), в другом варианте осуществления, только В необязательно включая элементы, не являющимися А), и, в еще одном варианте осуществления, как А, так и В (необязательно включая другие элементы), и т.п.
[0040] Следует понимать, что используемый в описании изобретения и в формуле изобретения термин "или" имеет такое же значение, как термин "и/или", определенный выше. Например, при разделении элементов в списке, термины "или" или "и/или" следует толковать как инклюзивные, то есть, как включающие по меньшей мере одно, но и также включающие более чем одно из ряда или списка элементов, и, необязательно, дополнительных, не включенных в список, элементов. Только термины, которые используют для передачи противоположного смысла, такие как "только один из" или "точно один из", или, при использовании в пунктах формулы изобретения выражения "состоящий из", будут относиться к включению только одного элемента из ряда или списка элементов. Обычно, используемый в изобретении термин "или" следует интерпретировать только как указание на исключающие альтернативы (то есть, "один или другой, но не оба"), когда перед ним используют термины исключительности, такие как "один из двух", "один из", "только один из" или "точно один из".
[0041] Следует понимать, что в формуле изобретения, так же как и в описании изобретения, все фразы переходного типа, такие как "содержащий", "включающий", "несущий", "имеющий", "вмещающий", "включающий в себя", "сохраняющий", "состоящий из" и другие подобные фразы являются неограничивающими, то есть, обозначают включение, но без ограничения. Только переходные фразы "состоящий из" и "состоящий в основном из" должны являться, соответственно, ограничивающими или частично ограничивающими переходными фразами, как указано в "Руководстве по проведению патентной экспертизы в Патентном ведомстве США; Раздел 2111.03." (United States Patent Office Manual of Patent Examining Procedures, Section 2111.03).
[0042] Следует понимать, что используемая в описании изобретения и в формуле изобретения фраза "по меньшей мере, один" в отношении списка одного или более элементов, означает, по меньшей мере, один элемент, выбранный из любого элемента или более элементов в списке элементов, но необязательно включающий, по меньшей мере, один из всех без исключения элементов, специально перечисленных в списке элементов, и не исключающий любые комбинации элементов в списке элементов. Это определение также допускает, что могут необязательно присутствовать элементы, которые не являются элементами, конкретно указанными в списке элементов, к которым относится фраза "по меньшей мере, один", независимо от того, связаны ли они или не связаны с теми элементами, которые специально указаны. Так, в качестве неограничивающего примера, выражение "по меньшей мере, один из A и B" (или, что эквивалентно, "по меньшей мере, один из A или B", или, что эквивалентно, "по меньшей мере, один из A и/или B"), может относится, в одном варианте осуществления, по меньшей мере, к одному А, необязательно включающему более одного A, без присутствия B (и необязательно включающему элементы, не являющиеся B), в другом варианте осуществления, по меньшей мере, к одному В, необязательно включающему более одного, B, без присутствия A (и необязательно включающему элементы, не являющиеся A), и в еще одном варианте осуществления, по меньшей мере, к одному А, необязательно включающему более одного A, и, по меньшей мере, одному В, необязательно включающий более одного B (и необязательно включающему другие элементы), и так далее.
[0043] Следует также понимать, что в описанных в изобретении конкретных способах, которые включают более чем одну стадию или действие, порядок стадий или действий в этом способе не обязательно ограничивается порядком, в котором эти стадии или действия перечислены в описываемом способе, если из контекста не следует иное.
[0044] Термины "совместное введение" или "комбинированная терапия" могут относиться как к одновременному введению (введению двух или более терапевтических средств (агентов) в одно и то же время), так и к введению в разные моменты времени (введение одного или более терапевтических средств в момент времени, который отличается от момента времени введения дополнительного терапевтического средства или средств), при условии, что терапевтические средства будут одновременно присутствовать в организме пациента в течение некоторого времени, предпочтительно, в эффективных количествах. В конкретных предпочтительных аспектах, одно или более из настоящих соединений, описанных в изобретении, вводят совместно в комбинации, по меньшей мере, с одним дополнительным биологически активным агентом, в частности, в том числе с противоопухолевым агентом. В наиболее предпочтительных аспектах, совместное введение соединений позволяет достигать синергетического эффекта и/или терапии, в том числе синергетической противораковой активности.
[0045] Термин «соединение», как он используется здесь, если не указано иное, относится к любому конкретному химическому соединению, раскрытому здесь, и он охватывает таутомеры, региоизомеры, геометрические изомеры и, где это применимо, стереоизомеры, включая оптические изомеры (энантиомеры) и другие стереоизомеры (например, диастереомеры), а также фармацевтически приемлемые соли и производные (в том числе пролекарственные формы), где это применимо в контексте описания. В рамках использования в контексте описания, термин "соединение" обычно относится к одному соединению, но он может также охватывать и стереоизомеры, региоизомеры и/или оптические изомеры (включая рацемические смеси), а также конкретные энантиомеры или энантиомерно обогащенные смеси описанных соединений. Этот термин в контексте описания также относится к пролекарственным формам соединений, которые были модифицированы с целью облегчения введения и доставки соединений к месту их действия. Следует отметить, что при описании соединений настоящего изобретения, приведены описания множества заместителей и переменных, связанных с этими соединениями. Обычным специалистам понятно, что молекулы, которые описанные здесь, относятся к стабильным соединениям, которые в целом описаны здесь ниже. Когда показана химическая связь, то она может быть двойной связью и одинарной связью в зависимости от контекста описания показанного соединения.
[0046] Термин "убиквитинлигаза" относится к семейству белков, которые облегчают перенос убиквитина к определенному субстратному белку, нацеливая и подготавливая белок для деградации. Например, цереблон представляет собой белок убиквитинлигазу Е3, который сам по себе или в сочетании с конъюгирующим ферментом убиквитин E2 вызывают присоединение убиквитина к лизину белка-мишени, а затем нацеливают специфические белки-субстраты для деградации с помощью протеасом. Таким образом, убиквитинлигаза E3 сама по себе или в комплексе с конъюгирующим ферментом убиквитин E2 отвечает за перенос убиквитина к целевым белкам-мишеням. В общем, убиквитин-лигаза принимает участие в полиубиквитинировании, в результате чего второй убиквитин присоединяется к первому; третий убиквитин присоединяется ко второму, и так далее. Полиубиквитинирование маркирует белки для деградации под действием протеасом. Однако, в некоторых случаях, убиквитинирование ограничивается только моно-убиквитинированием, при которых только один убиквитин добавляется убиквитин-лигазой к молекуле субстрата. Моно-убиквитинированные белки не являются мишенями, для воздействия протеасомы с целью деградации, но вместо этого может происходить изменения их клеточной локализации или функции, например, путем связывания других белков, которые имеют домены, способные связываться с убиквитином. Сложности возникают в результате того, что различные лизины на убиквитине могут быть нацелены с помощью E3 для образования цепей. Наиболее распространенным лизином является Lys48 на убиквитиновой цепи. Он представляет собой лизин, используемый для образования полиубиквитина, который распознается протеасомой.
[0047] Термин "пациент" или "субъект" используется по всему описанию для описания животного, предпочтительно человека, или домашнего животного, которое подвергается лечению, включая профилактическое лечение, с использованием композиций настоящего изобретения. Для лечения этих инфекций, состояний или заболеваний, которые являются специфическими для конкретного животного, такого как больной человек, термин "пациент" относится к этому конкретному животному, включая домашнее животное, такое как собаки или кошки, или сельскохозяйственное животное, такое как лошадь, коровы, овцы и т.п. В общем, в настоящем изобретении, термин "пациент" относится к больному человеку, если иное не указано или не подразумевается из контекста при использовании этого термина.
[0048] Термин "эффективный" используются в описании для указания количества соединения, композиции или компонента, который, при использовании его в контексте описания, предполагает достижение эффекта или предполагаемого результата. Термин "эффективный" относится к эффективному количеству или эффективной концентрации, которые указаны в настоящей заявке, если не подразумевается иное.
Соединения и структуры
[0049] В одном аспекте настоящее изобретение предоставляет соединения, содержащие фрагмент, связывающий с убиквитинлигазой E3 («ULM»), который представляет собой фрагмент связывания с убиквитинлигазой Е3 цереблона («CLM») в IAP. В одном варианте осуществления изобретения CLM соединен с химическим линкером (L), согласно следующей структуры:
(I) L-CLM
где L представляет собой связь или группу химического линкера, и CLM представляет собой фрагмент связывания с убиквитинлигазой Е3 цереблона. Количество и/или относительные положения фрагментов и остатков в соединениях, показанных в настоящем описании, приведены только в качестве примера. Специалисту в данной области понятно, что соединения, такие как описанные здесь, могут быть синтезированы с любым желаемым количеством и/или с относительным положением соответствующих функциональных фрагментов.
[0050] Термины "ULM" и "CLM" используются в их инклюзивном смысле, если контекст не указывает иное. Например, термин "ULM" включает все ULM, в том числе и те, которые обеспечивают связывание цереблона (т.е. CLM). Кроме того, термин CLM включает все возможные связывающие убиквитинлигазу E3 фрагменты.
[0051] В другом аспекте настоящее изобретение относится к бифункциональным соединениям или к мультифункциональным соединениям PROTAC, которые полезны для регуляции активности белка путем индукции деградации белка-мишени. В некоторых вариантах осуществления изобретения, соединение содержит связанный CLM, например, ковалентно связанный, прямо или опосредованно, с остатком, который связывает белок-мишень (например, нацеливающий на белок фрагмент или "PTM"). В некоторых вариантах осуществления изобретения CLM и PTM соединены или связаны с помощью химического линкера (L). CLM распознает убиквитинлигазу E3 цереблона, а PTM распознает белок-мишень, и взаимодействие соответствующих фрагментов, согласно их назначению, облегчает деградацию белка-мишени путем расположения целевого белка-мимшени в непосредственной близости от белка убиквитинлигазы. Иллюстративное бифункциональное соединение может быть изображено следующим образом:
(II) PTM-СLM
[0052] В некоторых вариантах изобретения бифункциональное соединение дополнительно содержит химический линкер ("L"). Например, бифункциональное соединение может быть изображено следующим образом:
(III) PTM-L-СLM
где PTM представляет собой фрагмент, нацеливающий на полипептид/белок-мишень, L представляет собой химический линкер, и CLM представляет собой фрагмент, обеспечивающий связывание с убиквитинлигазой Е3 цереблона.
[0053] В некоторых вариантах осуществления изобретения, соединения, описанные здесь, содержат несколько PTM (нацеленные на одинаковые или разные цели белка), несколько CLM, один или более ULM (то есть фрагменты, которые специфически связываются с другой убиквитинлигазой E3, например, VHL) или их сочетания. В любом из вариантов осуществления, описанных здесь, PTM, CLM и ULM могут быть соединены непосредственно или через один или несколько химических линкеров или их комбинации. В дополнительных вариантах, где соединение имеют несколько ULM, эти ULM могут быть для одной и той же убиквитинлигазы E3, или же каждый соответствующий ULM может специфический связываться с различными убиквитинлигазами Е3. В других вариантах, где соединение имеет несколько PTM, эти PTM могут связываться с одним и тем же белком-мишенью или каждый соответствующий PTM может специфически связываться с различными белками-мишенями.
[0054] В другом варианте осуществления изобретения, описание раскрывает соединение, которое содержит множество CLM, соединенных непосредственно или с помощью химического линкера (L). Так, например, соединение, имеющее два CLM, может быть изображено следующим образом:
(IV) CLM-CLM
или
(V) CLM-L-CLM
[0055] В некоторых вариантах, когда соединение содержит множество CLM, и эти CLM являются идентичными. В дополнительных вариантах осуществления изобретения, соединение, содержащее множество CLM дополнительно содержит, по меньшей мере, один PTM, соединенный с CLM непосредственно или через химический линкер (L), или за счет обоих вариантов связывания. В некоторых дополнительных вариантах осуществления изобретения, соединения, содержащее множество CLM, дополнительно содержат множество PTM. В других дополнительных вариантах осуществления изобретения, PTM являются одинаковыми или, необязательно, различными, при этом в вариантах, где PTM являются различными, соответствующие PTM могут связываться с одним и тем же белком-мишенью, или они могут специфически связываться с различными белками-мишенями.
[0056] В дополнительных вариантах осуществления изобретения, описание раскрывает соединение, содержащее, по меньшей мере, два различных CLM, соединенных непосредственно или через химический линкер (L), или за счет обоих вариантов соединения. Например, такое соединение, имеющее два различных CLM, может быть изображено следующим образом:
(VI) CLM-CLM
или
(VII) CLM-L-CLM';
где CLM' обозначает фрагмент связывания с убиквитинлигазой E3, который структурно отличается от CLM. В некоторых вариантах осуществления изобретения, соединение может включать несколько (множество) CLM и/или несколько (множество) CLM'. В дополнительных вариантах, соединение, содержащее по меньшей мере два различных CLM, множество CLM и/или множество CLM', дополнительно содержит по меньшей мере один PTM, связанный с CLM или CLM' непосредственно или через химического линкер, или за счет обоих вариантов соединения. В любом из описанных здесь вариантов, соединение, содержащее по меньшей мере два различных CLM, может дополнительно включать несколько PTM. В еще одном дополнительном варианте осуществления изобретения, PTM являются одинаковыми или, необязательно, различными. В других вариантах изобретения, где PTM являются различными, соответствующие PTM могут связываться с одним и тем же белком-мишенью или специфически связываться с различными белками-мишенями. В других вариантах, PTM, как таковой, может представлять собой ULM или CLM (CLM или CLM').
[0057] В предпочтительном варианте осуществления изобретения, CLM содержит фрагмент, который представляет собой лиганд Е3 убиквитинлигазы цереблона (CRBN). В некоторых вариантах осуществления изобретения CLM содержит хемотип молекул класса «имид». В некоторых дополнительных вариантах осуществления изобретения CLM содержит фталимидную группу или ее аналог или производное. В других дополнительных вариантах осуществления изобретения, CLM содержит фталимидо-глутаримидную группу или ее аналог или производное. В других вариантах осуществления изобретения CLM содержит группу, выбранную из талидомида, леналидомида, помалидомида и их аналогов или производных.
[0058] В дополнительных вариантах осуществления изобретения, описание раскрывает соединения, описанные здесь, включая их энантиомеры, диастереоизомеры, сольваты и полиморфы, в том числе фармацевтически приемлемые соли, например, соли с кислотами и основаниями.
Новые имидные соединения
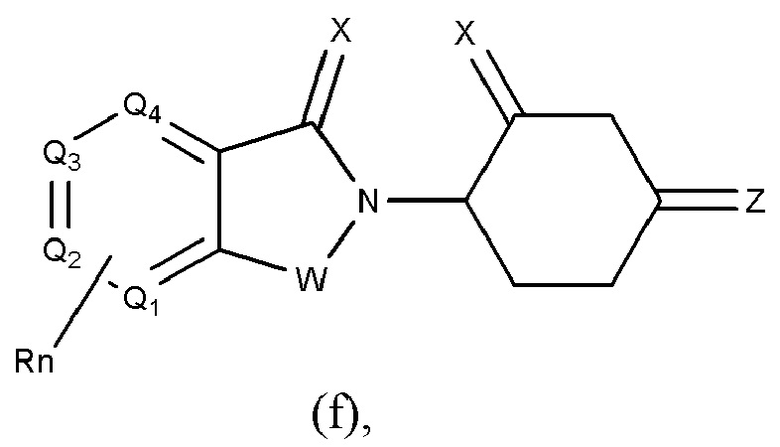
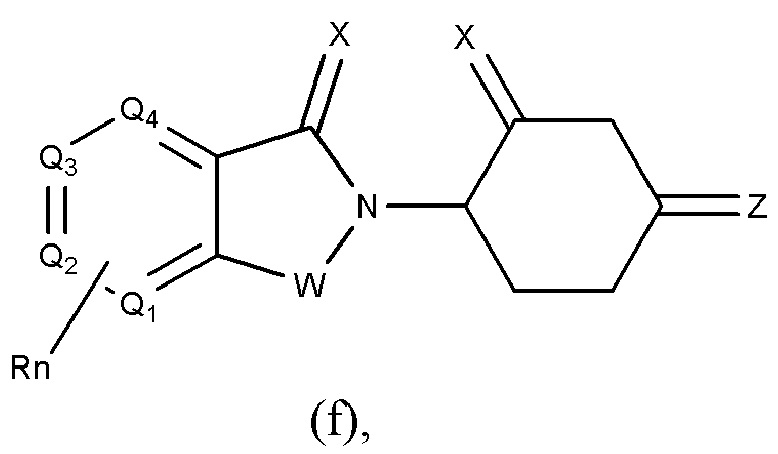
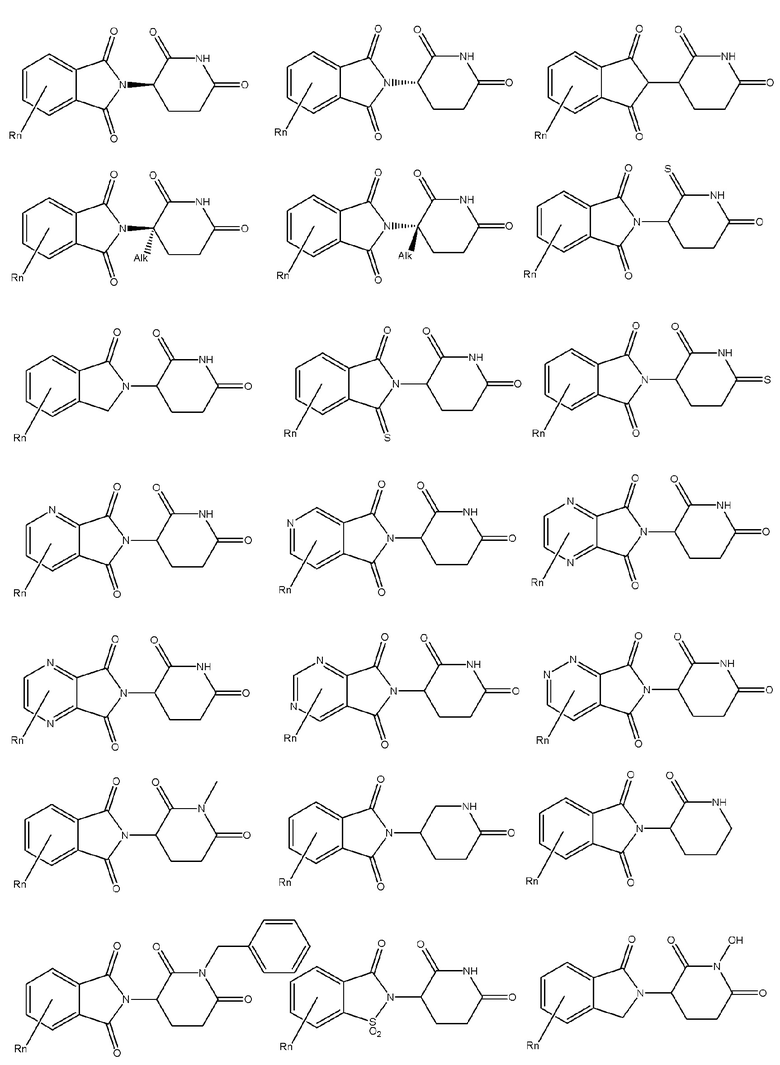
[0059] В одном аспекте изобретение предоставляет соединения, полезные для связывания и/или ингибирования цереблона. В некоторых вариантах осуществления соединение выбрано из группы, состоящей из следующих химических структур:
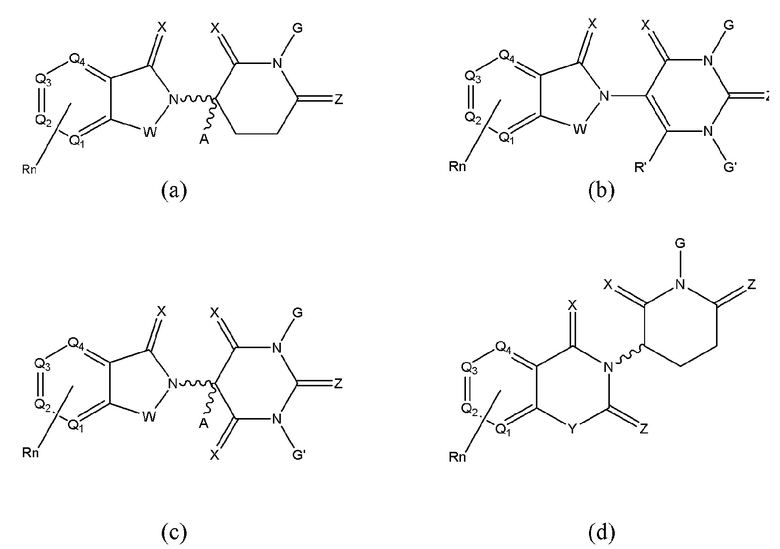
и
где
W независимо выбран из группы CH2, CHR, C=O, SO2, NH и N-алкила;
Х независимо выбран из группы О, S и H2;
Y независимо выбран из группы, NH, N-алкила, N-арила, N-het-арила, N-циклоалкила, N-гетероциклила, О и S;
Z независимо выбран из группы O и S или H2, при этом исключается случай, когда Х и Z одновременно представляют собой Н2;
G и G' независимо друг от друга выбраны из группы Н, алкила, ОН, CH2-гетероциклила, необязательно замещенного R', и бензила, необязательно замещенного R';
Q1-Q4 представляют собой углерод C, замещенный группой, независимо выбранной из R', N или N-оксида;
А независимо выбран из группы алкила, циклоалкила, Cl и F;
R включает, без не ограничения: -CONR'R", -OR', -NR'R", -SR', -SO2R', -SO2NR'R", -CR'R"-, -CR'NR'R"-, арила, -het-арила, алкила, циклоалкила, гетероциклила, -P(O)(OR')R", P(O)R'R", -OP(O)(OR')R", -OP(O)R'R", -Cl, -F, -Br, -I, -CF3, -CN, -NR'SO2NR'R", -NR'CONR'R", -CONR'COR", -NR'C(=N-CN)NR'R", -C(=N-CN)NR'R", -NR'C(=N-CN)R", -NR'C(=C-NO2)NR'R", -SO2NR'COR", -NO2, -CO2R', -C(C=N-OR')R", -CR'=CR'R", -CCR', -S(C=O)(C=N-R')R", -SF5 и -OCF3;
R'и R" независимо выбраны из связи, Н, алкила, циклоалкила, арила, het-арила, гетероциклила;
n является целым числом от 1 до 4;
 представляет собой связь, которая может быть стереоспецифичной ((R) или (S)) или нестереоспецифичной; и
представляет собой связь, которая может быть стереоспецифичной ((R) или (S)) или нестереоспецифичной; и
Rn содержит 1-4 независимые функциональные группы или атомы.
Типовые CLM
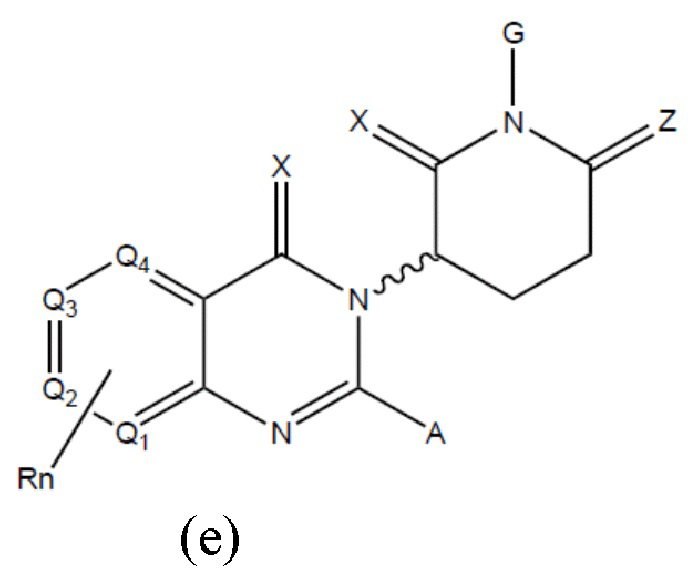
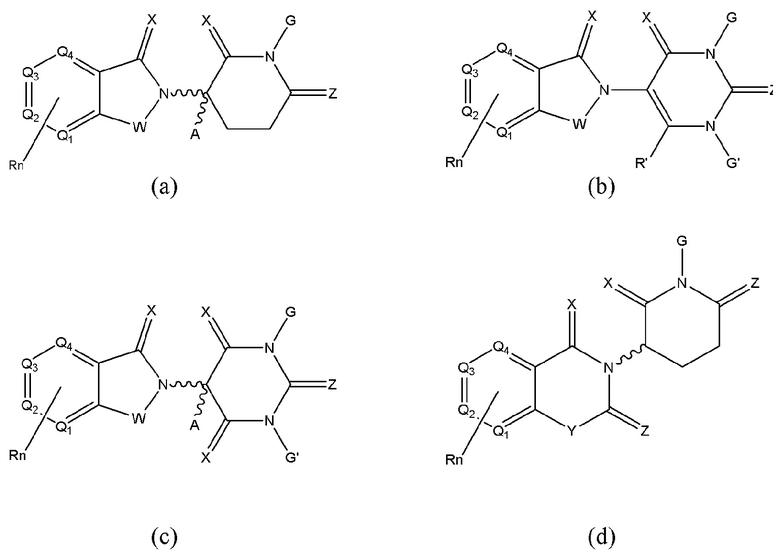
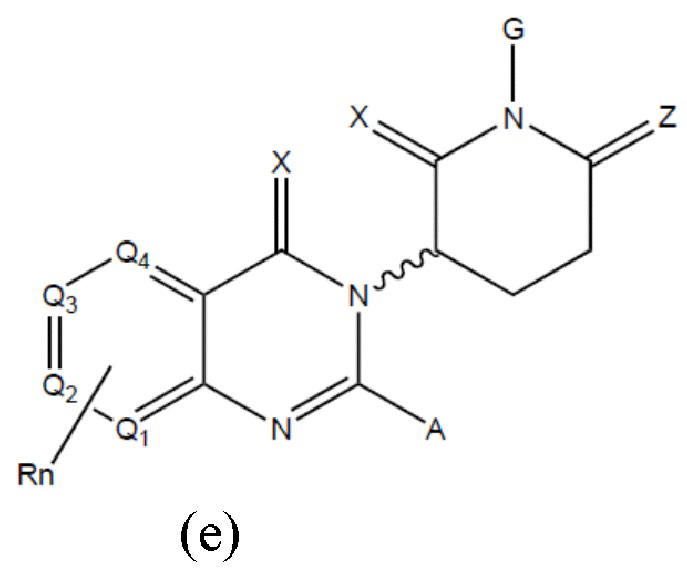
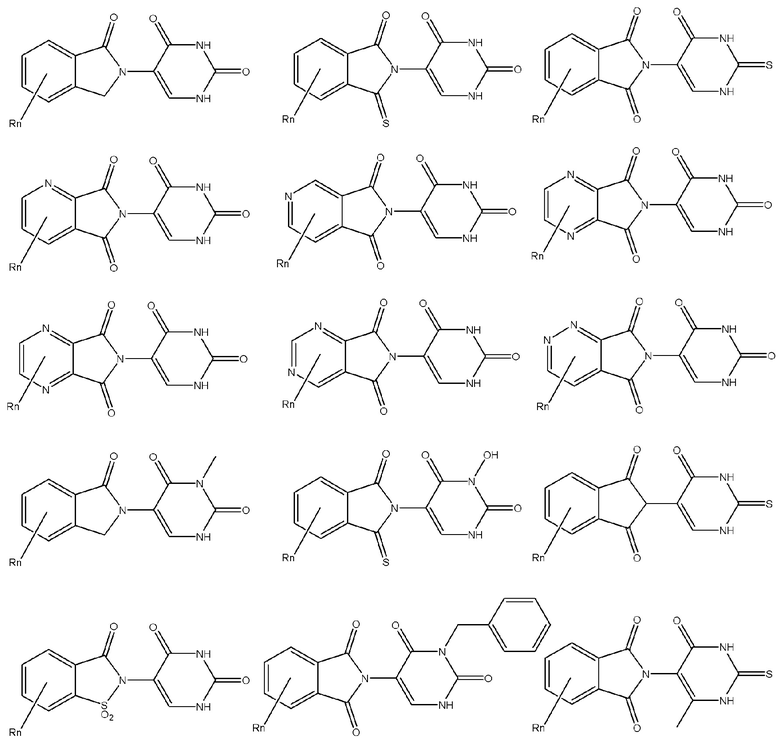
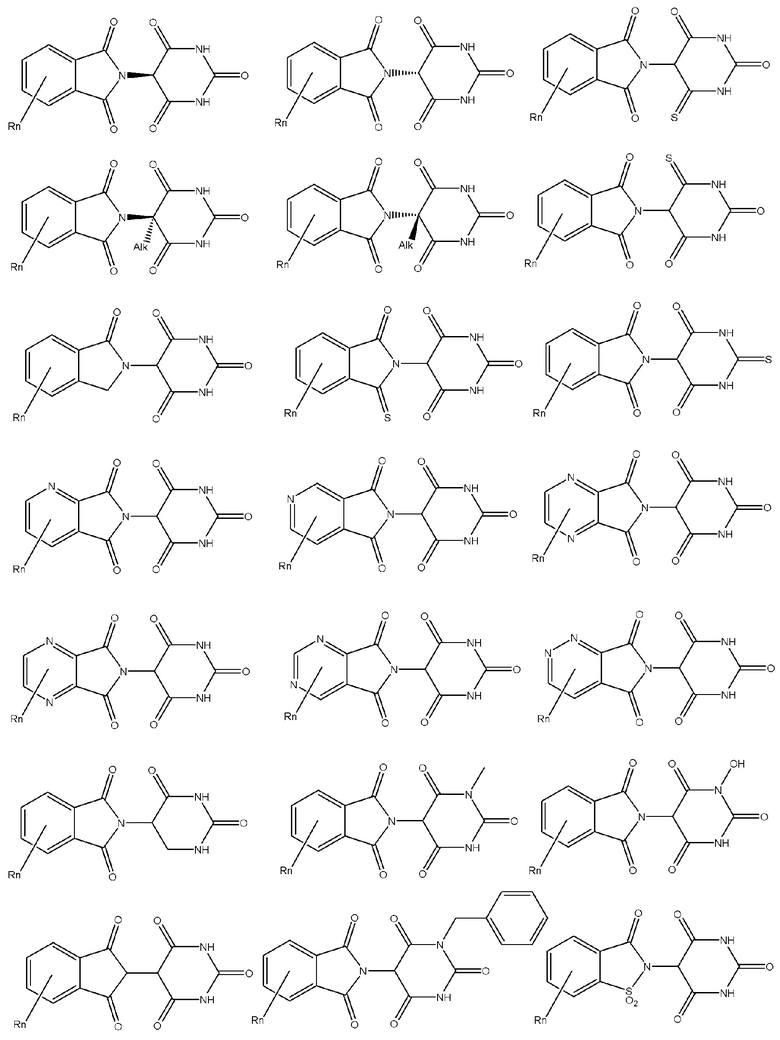
[0060] В любом из описанных здесь соединений, CLM включает химическую структуру, выбранную из группы:
и
где
W независимо выбран из группы CH2, CHR, C=O, SO2, NH и N-алкила;
Х независимо выбран из группы О, S и H2;
Y независимо выбран из группы, NH, N-алкила, N-арила, N-het-арила, N-циклоалкила, N-гетероциклила, О и S;
Z независимо выбран из группы O и S или H2, но при этом исключается случай, когда Х и Z одновременно представляют собой Н2;
G и G' независимо друг от друга выбраны из группы Н, алкила, ОН, CH2-гетероциклила, необязательно замещенного R', и бензила, необязательно замещенного R';
Q1-Q4 представляют собой углерод C, замещенный группой, независимо выбранной из R', N или N-оксида;
А независимо выбран из группы алкила, циклоалкила, Cl и F;
R включает, без не ограничения: -CONR'R", -OR', -NR'R", -SR', -SO2R', -SO2NR'R", -CR'R"-, -CR'NR'R"-, арила, -het-арила, алкила, циклоалкила, гетероциклила, -P(O)(OR')R", P(O)R'R", -OP(O)(OR')R", -OP(O)R'R", -Cl, -F, -Br, -I, -CF3, -CN, -NR'SO2NR'R", -NR'CONR'R", -CONR'COR", -NR'C(=N-CN)NR'R", -C(=N-CN)NR'R", -NR'C(=N-CN)R", -NR'C(=C-NO2)NR'R", -SO2NR'COR", -NO2, -CO2R', -C(C=N-OR')R", -CR'=CR'R", -CCR', -S(C=O)(C=N-R')R", -SF5 и -OCF3;
R'и R" независимо выбраны из связи, Н, алкила, циклоалкила, арила, het-арила, гетероциклила;
n является целым числом от 1 до 4;
 представляет собой связь, которая может быть стереоспецифичной ((R) или (S)) или нестереоспецифичной; и
представляет собой связь, которая может быть стереоспецифичной ((R) или (S)) или нестереоспецифичной; и
Rn содержит 1-4 независимые функциональные группы или атомы, и, необязательно, одна из групп или один из атомов может быть модифицирован, чтобы быть ковалентно соединен с PTM, группой химического линкера (L), ULM, CLM (или CLM') или с их комбинацией.
[0061] Термин «независимо» используется здесь для указания, что переменная, которая используется независимо, применяется независимо от места ее использования.
[0062] Термин «алкил» означает линейный, разветвленный или циклический полностью насыщенный углеводородный радикал или алкильную группу, предпочтительно, C1-C10, более предпочтительно, C1-C6, в качестве варианта, C1-C3 алкильную группу, которые могут быть необязательно замещенными. Примерами алкильных групп наряду с прочими являются метил, этил, н-бутил, вторбутил, н-гексил, н-гептил, н-октил, н-нонил, н-децил, изопропил, 2-метил пропил, циклопропил, циклопропил метил, циклобутил, циклопентил, циклопентилэтил, циклогексилэтил и циклогексил. В некоторых предпочтительных вариантах, алкильная группа содержит на своем конце галоген (At, Br, Cl, F, or I). В некоторых предпочтительных вариантах осуществления, соединения по настоящему изобретению могут быть использованы для ковалентного связывания дегалогеназных ферментов. Эти соединения обычно содержат боковую цепь (часто присоединенную через полиэтиленгликолевую группу), которая заканчивается алкильной группой, имеющей галогеновый заместитель (часто хлор или бром) на ее свободном конце, что делает возможным ковалентное связывание соединения, содержащего такой фрагмент, с белком.
[0063] Термин «алкенил» относится к линейным, разветвленным или циклическим C2-C10 (предпочтительно, C2-C6) углеводородным радикалам, содержащим, по меньшей мере, одну C=C связь.
[0064] Термин «алкинил» относится к линейным, разветвленным или циклическим C2-C10 (предпочтительно, C2-C6) углеводородным радикалам, содержащим, по меньшей мере, одну C≡C связь.
[0065] Используемый в изобретении термин «алкилен» относится к -(CH2)n- группе (n представляет собой целое число, обычно от 0 до 6), которая может быть необязательно замещенной. В случае, когда алкиленовая группа является замещенной, предпочтительно, чтобы она была замещена по одной или более метиленовым группам с помощью C1-C6 алкильной группы (в том числе с помощью циклопропильной группы или третбутильной группы), более предпочтительно, с помощью метильной группы, но она может быть также замещена с помощью одной или более галогеновых групп, предпочтительно, с помощью от 1 до 3 галогеновых групп или одной или двух гидроксильных групп, O-(C1-C6 алкил) групп или боковых аминокислотных цепей, раскрытых в изобретении. В конкретных вариантах осуществления, алкиленовая группа может быть замещена с помощью уретана или алкоксильной группы (или другой группы), которая дополнительно замещена с помощью полиэтиленгликолевой цепи (содержащей от 1 до 10, предпочтительно, от 1 до 6, чаще всего, от 1 до 4 этиленгликолевых звеньев), которая замещена (предпочтительно, но не исключительно, на свободном кольце полиэтиленгликолевой цепи) алкильной цепью, замещенной с помощью одной галогеновой группы, предпочтительно, хлорной группой. В еще одних вариантах осуществления, алкиленовая (чаще всего, метиленовая) группа может быть замещена с помощью аминокислотной амино группы боковой цепи, такой как группа боковой цепи из природной или не встречающейся в природе аминокислоты, например, аланина, β-аланина, аргинина, аспарагина, аспарагиновой кислоты, цистеина, цистина, глутаминовой кислоты, глутамина, глицина, фенилаланина, гистидина, изолейцина, лизина, лейцина, метионина, пролина, серина, треонина, валина, триптофана или тирозина.
[0066] Термин «незамещенный» означает замещенный только атомами водорода. Ряд углеродных атомов, который включает C0, означает, что углерод отсутствует и заменен на H. Так, например, ряд углеродных атомов, который представляет собой C0-C6, включает углеродные атомы 1, 2, 3, 4, 5 и 6, и, в случае C0, H стоит вместо углерода.
[0067] Термин «замещенный» или «необязательно замещенный» обозначает независимо (то есть, когда имеется более чем один заместитель, каждый заместитель является независимым от другого заместителя) один или более заместителей (независимо, до пяти заместителей, предпочтительно, до трех заместителей, чаще всего, 1 или 2 заместителя на фрагменте в соединении по настоящему изобретению и может включать заместители, которые сами могут быть дополнительно замещены) на углероде (или азоте) в любом положении в молекуле в контексте, и включает в качестве заместителей гидроксил, тиол, карбоксил, циано (C≡N), нитро (NO2), галоген (предпочтительно, 1, 2 или 3 галогена, особенно на алкиле, особенно, на метильной группе, такой как трифторметил), алкильную группу (предпочтительно, C1-C10, более предпочтительно, C1-C6), арил (особенно, фенил и замещенный фенил, например бензил или бензоил), алкоксильную группу (предпочтительно, C1-C6 алкил или арил, в том числе фенил и замещенный фенил), тиоэфир (C1-C6 алкил или арил), ацил (предпочтительно, C1-C6 ацил), эфир или тиоэфир (предпочтительно, C1-C6 алкил или арил), в том числе алкиленовый эфир (в котором присоединение происходит на алкиленовой группе, а не по эфирной группе, которая предпочтительно замещена с помощью C1-C6 алкильной или арильной группы), предпочтительно, C1-C6 алкил или арил, галоген (предпочтительно, F или Cl), амин (в том числе пяти- или шестичленный циклический алкиленамин, дополнительно включающий C1-C6 алкиламин или C1-C6 диалкиламин, алкильные группы которого могут быть замещены с помощью одной или двух гидроксильных групп) или необязательно замещенную группу -N(C0-C6 алкил)C(O)(O-C1-C6 алкил) (которая может быть необязательно замещена с помощью полиэтиленгликолевой цепи, к которой дополнительно присоединена алкильная группа, содержащая один галогеновый заместитель, предпочтительно, хлор), гидразин, амидогруппу, которая предпочтительно замещена с помощью одной или двух C1-C6 алкильных групп (в том числе карбоксамид, который необязательно замещен с помощью одной или двух C1-C6 алкильных групп), алканол (предпочтительно, C1-C6 алкил или арил), или алкановую кислоту (предпочтительно, C1-C6 алкил или арил). Заместители по настоящему изобретению могут включать, например -SiR1R2R3 группы, где каждый из R1 и R2 описаны в изобретении, и R3 представляет собой H или C1-C6 алкильную группу, предпочтительно, чтобы R1, R2, R3 в этом контексте представляли C1-C3 алкильную группу (в том числе изопропильную или третбутильную группу). Каждая из описанных выше групп может быть присоединена непосредственно к замещенному фрагменту, или, в качестве варианта, заместитель может быть присоединен к замещенному фрагменту (предпочтительно, в случае арильного или гетероарильного фрагмента) через необязательно замещенную группу -(CH2)m-, или, в качестве варианта, через необязательно замещенную группу -(OCH2)m-, -(OCH2CH2)m- или -(CH2CH2O)m-, которая может быть замещена с помощью любого одного или более из описанных выше заместителей. Алкиленовые группы -(CH2)m- или алкиленовые группы -(CH2)n- или другие цепи, такие как этиленгликолевые цепи, указанные выше, могут быть замещены в любом месте цепи. Предпочтительные заместители на алкиленовых группах включают галоген или C1-C6 (предпочтительно C1-C3) алкильные групп, которые могут быть необязательно замещены с помощью одной или двух гидроксильных групп, одной или двух эфирных групп (O-C1-C6 групп), до трех галогеновых групп (предпочтительно, F), или боковой цепи из аминокислоты, описанной в изобретении, и необязательно замещенного амида (предпочтительно, карбоксамида, замещенного как описано выше) или уретановых групп (часто с одним или двумя C0-C6 алкильными заместителями, группа (группы) в которых может быть дополнительно замещена). В конкретных вариантах осуществления, алкиленовая группа (часто, одна метиленовая группа) замещена с помощью одной или двух необязательно замещенных C1-C6 алкильных групп, предпочтительно, с помощью C1-C4 алкильной группы, наиболее часто, метильной или O-метильной групп или боковой цепи из аминокислоты, описанной в изобретении. В настоящем изобретении, фрагмент в молекуле может быть необязательно замещен заместителями в количестве до пяти, предпочтительно, до трех. Наиболее часто, в настоящем изобретении, фрагменты, которые являются замещенными, замещены одним или двумя заместителями.
[0068] Термин «замещенный» (причем каждый заместитель может быть независимым от любого другого заместителя) также обозначает, в контексте его использования, C1-C6 алкил, C1-C6 алкокси, галоген, амидо, карбоксамидо, сульфон, в том числе сульфонамид, кето, карбокси, C1-C6 эфир (оксиэфир или карбонилэфир), C1-C6 кето, уретан -O-C(O)-NR1R2 или -N(R1)-C(O)-O-R1, нитро, циано и амин (особенно, в том числе C1-C6 алкилен-NR1R2, моно- или ди-C1-C6 алкилзамещенные амины, которые могут быть необязательно замещены с помощью одной или двух гидроксильных групп). Каждая из этих групп содержит, если в тексте не указано иначе, от 1 и 6 углеродных атомов. В конкретных вариантах осуществления, предпочтительные заместители включают, например, -NH-, -NHC(O)-, -O-, =O, -(CH2)m-(где m и n представляют собой, исходя из контекста, 1, 2, 3, 4, 5 или 6), -S-, -S(O)-, SO2- или -NH-C(O)-NH-, -(CH2)nOH, -(CH2)nSH, -(CH2)nCOOH, C1-C6 алкил, -(CH2)nO-(C1-C6 алкил), -(CH2)nC(O)-(C1-C6 алкил), -(CH2)nOC(O)-(C1-C6 алкил), -(CH2)nC(O)O-(C1-C6 алкил), -(CH2)nNHC(O)-R1, -(CH2)nC(O)-NR1R2, -(OCH2)nOH, -(CH2O)nCOOH, C1-C6 алкил, -(OCH2)nO-(C1-C6 алкил), -(CH2O)nC(O)-(C1-C6 алкил), -(OCH2)nNHC(O)-R1, -(CH2O)nC(O)-NR1R2, -S(O)2-RS, -S(O)-RS (RS представляет собой C1-C6 алкил или -(CH2)m-NR1R2 группу), NO2, CN или галоген (F, Cl, Br, I, предпочтительно, F или Cl), в зависимости от контекста применения заместителя. R1 и R2 каждый представляет собой, в зависимости от контекста, H или C1-C6 алкильную группу (которая может быть необязательно замещена одной или двумя гидроксильными группами или до трех галогеновыми группами, предпочтительно, фтором). Термин "замещенный" также обозначает, в зависимости от химического строения определенного соединения и используемого заместителя, необязательно замещенную арильную или гетероарильную группу или необязательно замещенную гетероциклическую группу, описанные выше. Алкиленовые группы могут быть также замещены, как это раскрыто в изобретении, предпочтительно с помощью необязательно замещенных C1-C6 алкильных групп (метил, этил или гидроксиметил или гидроксиэтил являются предпочтительными, в результате чего образуется хиральный центр), с помощью группы боковой цепи из аминокислоты, описанной в изобретении, амидогруппы, описанной выше, или уретановой группы O-C(O)-NR1R2, где R1 и R2 описаны в изобретении, хотя в качестве заместителей может быть использовано большое число и других групп. Различные необязательно замещенные фрагменты могут быть замещены тремя или более заместителями, предпочтительно, не более чем тремя заместителями, и предпочтительно, одним или двумя заместителями. Следует отметить, что в случаях, когда в соединении в конкретном месте молекулы требуется замещение (главным образом, исходя из требований валентности), но замещение не указано, то подразумевается или допускается, что заместитель представляет собой H, если в контексте замещения не предполагается иное.
[0069] Термин «арил» или «ароматический», в контексте описания, относится к замещенному (как описано в изобретении) или незамещенному одновалентному ароматическому радикалу, имеющему одно кольцо (например, бензольное, фенильное, бензильное) или конденсированные кольца (например, нафтильные, антраценильные, фенантренильные и другие), который может быть присоединен к соединению по настоящему изобретению в любом доступном стабильном положении на кольце (кольцах) или же на указанном положении в представленной химической структуре. Другие примеры используемых в изобретении арильных групп могут включать, наряду с прочими, "гетероарильные" группы на основе ароматических кольцевых систем, имеющие один или более атомов азота, кислорода или серы в кольце (моноциклическом), такие как имидазол, фурил, пиррол, фуранил, тиен, тиазол, пиридин, пиримидин, пиразин, триазол, оксазол, или на основе конденсированных кольцевых систем, такие как индол, хинолин, индолизин, азаиндолизин, бензофуразан, и другие, которые могут быть необязательно замещенными, как описано выше. Гетероарильные группы, которые могут быть упомянуты помимо прочих, включают азотсодержащие гетероарильные группы, такие как пиррол, пиридин, пиридон, пиридазин, пиримидин, пиразин, пиразол, имидазол, триазол, триазин, тетразол, индол, изоиндол, индолизин, азаиндолизин, пурин, индазол, хинолин, дигидрохинолин, тетрагидрохинолин, изохинолин, дигидроизохинолин, тетрагидроизохинолин, хинолизин, фталазин, нафтиридин, хиноксалин, хиназолин, циннолин, птеридин, имидазопиридин, имидазотриазин, пиразинопиридазин, акридин, фенантридин, карбазол, карбазолин, перимидин, фенантролин, фенацен, оксадиазол, бензимидазол, пирролопиридин, пирролопиримидин и пиридопиримидин; серосодержащие ароматические гетероциклы, такие как тиофен и бензотиофен; кислородсодержащие ароматические гетероциклы, такие как фуран, пиран, циклопентапиран, бензофуран и изобензофуран; и ароматические гетероциклы, включающие два или более гетероатомов, выбранных, наряду с прочими, из азота, серы и кислорода, такие как тиазол, тиадизол, изотиазол, бензоксазол, бензотиазол, бензотиадиазол, фенотиазин, изоксазол, фуразан, феноксазин, пиразолоксазол, имидазотиазол, тиенофуран, фуропиррол, пиридоксазин, фуропиридин, фуропиримидин, тиенопиримидин и оксазол, каждый из которых может быть необязательно замещенным.
[0070] Термин «замещенный арил» относится к ароматической карбоциклической группе, состоящей, по меньшей мере, из одного ароматического кольца или из множества конденсированных колец, по меньшей мере, одно из которых является ароматическим, где кольцо (кольца) замещены одним или более заместителями. Например, арильная группа может включать заместитель (заместители), выбранные из следующих: -(CH2)nOH, -(CH2)n-O-(C1-C6)алкил, -(CH2)n-O-(CH2)n-(C1-C6)алкил, -(CH2)n-C(O)(C0-C6)алкил, -(CH2)n-C(O)O(C0-C6)алкил, -(CH2)n-OC(O)(C0-C6)алкил, амин, моно- или ди-(C1-C6 алкил)амин, где алкильная группа на амине необязательно замещена одной или двумя гидроксильными группами или до трех галогеновыми (предпочтительно F, Cl) группами, OH, COOH, C1-C6 алкилом, предпочтительно CH3, CF3, OMe, OCF3, NO2 или CN группами (каждая из которых может быть замещена в орто-, мета- и/или пара-положениях фенильного кольца, предпочтительно, в пара-положении), необязательно замещенный фенильная группа (предпочтительно, чтобы сама фенильная группа была замещена линкерной группой, присоединенной к ABM группе, в том числе ULM групп), и/или по меньшей мере, одной из F, Cl, OH, COOH, CH3, CF3, OMe, OCF3, NO2 или CN групп (в орто-, мета- и/или пара-положениях фенильного кольца, предпочтительно, в пара-положении), нафтильная группа, которая может быть необязательно замещена, необязательно замещенный гетероарил, предпочтительно, необязательно замещенный изоксазол, в том числе метилзамещенный изоксазол, необязательно замещенный оксазол, в том числе метилзамещенный оксазол, необязательно замещенный тиазол, в том числе метил замещенный тиазол, необязательно замещенный изотиазол, в том числе метил замещенный изотиазол, необязательно замещенный пиррол, в том числе метилзамещенный пиррол, необязательно замещенный имидазол, в том числе метилимидазол, необязательно замещенный бензимидазол или метоксибензилимидазол, необязательно замещенный оксимидазол или метилоксимидазол, необязательно замещенная диазольная группа, в том числе метилдиазольная группа, необязательно замещенная триазольная группа, в том числе метилзамещенная триазольная группа, необязательно замещенная пиридиновая группа, в том числе галоген-(предпочтительно, F) или метилзамещенная пиридиновая группа или оксапиридиновая группа (где пиридиновая группа соединена с фенильной группой через кислород), необязательно замещенный фуран, необязательно замещенный бензофуран, необязательно замещенный дигидробензофуран, необязательно замещенный индол, индолизин или азаиндолизин (2-, 3- или 4-азаиндолизин), необязательно замещенный хинолин и их комбинации.
[0071] «Карбоксил» обозначает группу --C(O)OR, где R представляет собой водород, алкил, замещенный алкил, арил, замещенный арил, гетероарил или замещенный гетероарил, при этом эти характерные заместители имеют значения, которые идентичны определениям соответствующих групп, представленным в описании.
[0072] Термин «гетероарил» или «het-арил» может означать, но никоим образом не органичивать, необязательно замещенный хинолин (который может быть присоединен к фармакофору или замещен на любом углеродном атоме в хинолиновом кольце), необязательно замещенный индол (в том числе дигидроиндол), необязательно замещенный индолизин, необязательно замещенный азаиндолизин (2-, 3- или 4-азаиндолизин), необязательно замещенный бензимидазол, бензодиазол, бензоксофуран, необязательно замещенный имидазол, необязательно замещенный изоксазол, необязательно замещенный оксазол (предпочтительно, метилзамещенный), необязательно замещенный диазол, необязательно замещенный триазол, тетразол, необязательно замещенный бензофуран, необязательно замещенный тиофен, необязательно замещенный тиазол (предпочтительно, метил- и/или тиолзамещенный), необязательно замещенный изотиазол, необязательно замещенный триазол (предпочтительно, 1,2,3-триазол, замещенный метильной группой, триизопропилсилильную группу, необязательно замещенную -(CH2)m-O-C1-C6 алкильную группу или необязательно замещенную -(CH2)m-C(O)-O-C1-C6 алкильную группу), необязательно замещенный пиридин (2-, 3- или 4-пиридин) или группу с химической структурой:
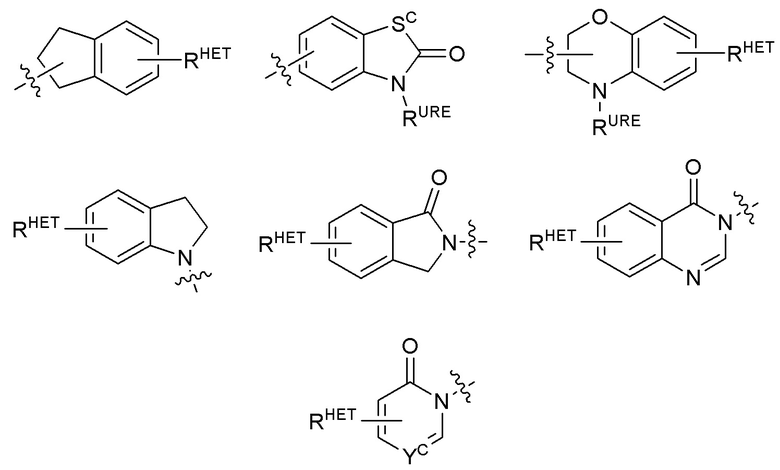
где Sc представляет собой CHRSS, NRURE или O;
RHET представляет собой H, CN, NO2, галоген (предпочтительно Cl или F), необязательно замещенный C1-C6 алкил (предпочтительно замещенный одной или двумя гидроксильными группами или до трех галогеновыми группами (например CF3), необязательно замещенный O(C1-C6 алкил) (предпочтительно замещенный одной или двумя гидроксильными группами или до трех галогеновыми группами) или необязательно замещенную ацетиленовую группу -C≡C-Ra, где Ra представляет собой H или C1-C6 алкильную группу (предпочтительно, C1-C3 алкил);
RSS представляет собой H, CN, NO2, галоген (предпочтительно F или Cl), необязательно замещенный C1-C6 алкил (предпочтительно, замещенный одной или двумя гидроксильными группами или до трех галогеновыми группами), необязательно замещенный O-(C1-C6 алкил) (предпочтительно, замещенный одной или двумя гидроксильными группами или до трех галогеновыми группами) или необязательно замещенный -C(O)(C1-C6 алкил) (предпочтительно, замещенный одной или двумя гидроксильными группами или до трех галогеновыми группами);
RURE представляет собой H, C1-C6 алкил (предпочтительно, H или C1-C3 алкил) или -C(O)(C1-C6 алкил), каждый из которых необязательно замещен одной или двумя гидроксильными группами или до трех галогеновыми группами, предпочтительно, фторными группами, или необязательно замещенный гетероцикл, например пиперидин, морфолин, пирролидин, тетрагидрофуран, тетрагидротиофен, пиперидин, пиперазин, каждый из которых необязательно замещен, и
YC представляет собой N или C-RYC, где RYC представляет собой H, OH, CN, NO2, галоген (предпочтительно Cl или F), необязательно замещенный C1-C6 алкил (предпочтительно, замещенный одной или двумя гидроксильными группами или до трех галогеновыми группами (например CF3), необязательно замещенный O(C1-C6 алкил) (предпочтительно, замещенный одной или двумя гидроксильными группами или до трех галогеновыми группами) или необязательно замещенную ацетиленовую группу -C≡C-Ra, где Ra представляет собой H или C1-C6 алкильную группу (предпочтительно, C1-C3 алкил).
[0073] Термины "аралкил" и "гетероарилалкил" относятся к группам, которые включают как арил или, соответственно, гетероарил, так и алкил и/или гетероалкил и/или карбоциклические и/или гетероциклоалкильные кольцевые системы, в соответствии с приведенными выше определениями.
[0074] Используемый в изобретении термин "арилалкил" относится к определенной выше арильной группе, присоединенной к определенной выше алкильной группе. Арилалкильная группа присоединена к основному фрагменту через алкильную группу, где алкильная группа имеет от одного до шести углеродных атомов. Арильная группа в арилалкильной группе может быть замещена, как это определено выше.
[0075] Термин "гетероцикл" относится к циклической группе, которая содержит, по меньшей мере, один гетероатом, то есть, O, N или S, и которая может быть ароматической (гетероарил) или неароматической. Таким образом, гетероарильные группы охватываются определением "гетероцикл", в зависимости от контекста его использования. Типичные гетероарильные группы описаны выше.
[0076] Примеры гетероциклов включают, наряду с прочими, азетидинил, бензимидазолил, 1,4-бензодиоксанил, 1,3-бензодиоксолили, бензоксазолил, бензотиазолил, бензотиенил, дигидроимидазолил, дигидропиранил, дигидрофуранил, диоксанил, диоксоланил, этиленмочевину, 1,3-диоксолан, 1,3-диоксан, 1,4-диоксан, фурил, гомопиперидинил, имидазолил, имидазолинил, имидазолидинил, индолинил, индолил, изохинолинил, изотиазолидинил, изотиазолил, изоксазолидинил, изоксазолил, морфолинил, нафтиридинил, оксазолидинил, оксазолил, пиридон, 2-пирролидон, пиридин, пиперазинил, N-метилпиперазинил, пиперидинил, фталимид, сукцинимид, пиразинил, пиразолинил, пиридил, пиримидинил, пирролидинил, пирролинил, пирролил, хинолинил, тетрагидрофуранил, тетрагидропиранил, тетрагидрохинолин, тиазолидинил, тиазолил, тиенил, тетрагидротиофен, оксан, оксетанил, оксатиоланил, тиан.
[0077] Гетероциклические группы могут быть необязательно замещены заместителем, выбранным из группы, состоящей из алкокси, замещенного алкокси, циклоалкила, замещенного циклоалкила, циклоалкенила, замещенного циклоалкенила, ацила, ациламино, ацилокси, амино, замещенного амино, аминоацила, аминоацилокси, оксиаминоацила, азидо, циано, галогена, гидроксила, кето, тиокето, карбокси, карбоксиалкила, тиоарилокси, тиогетероарилокси, тиогетероциклоокси, тиола, тиоалкокси, замещенного тиоалкокси, арила, арилокси, гетероарила, гетероарилокси, гетероцикла, гетероциклоокси, гидроксиамино, алкоксиамино, нитро, -SO-алкила, -SO-замещенного алкила, -SO-арила, -SO-гетероарила, -SO2-алкила, -SO2-замещенного алкила, -SO2-арила, оксо (=O) и -SO2-гетероарила. Такие гетероциклические группы могут иметь одно кольцо или множество конденсированных колец. Примеры азотных гетероциклов и гетероарилов включают, но этим не ограничивая, пиррол, имидазол, пиразол, пиридин, пиразин, пиримидин, пиридазин, индолизин, изоиндол, индол, индазол, пурин, хинолизин, изохинолин, хинолин, фталазин, нафтилпиридин, хиноксалин, хиназолин, циннолин, птеридин, карбазол, карболин, фенантридин, акрилин, фенантролин, изотиазол, феназин, изоксазол, феноксазин, фенотиазин, имидазолидин, имидазолин, пиперидин, пиперазин, индолин, морфолино, пиперидинил, тетрагидрофуранил, и другие подобные гетероциклы, а также N-алкокси-азотсодержащие гетероциклы. Термин "гетероциклический" также включает бициклические группы, в которых любое из гетероциклических колец является конденсированным с бензольным кольцом или циклогексановым кольцом или другим гетероциклическим кольцом (например, индолил, хинолил, изохинолил, тетрагидрохинолил и другие подобные кольца).
[0078] Термин "циклоалкил" может обозначать, но никоим образом не ограничивать, одновалентные группы, полученные из моноциклических или полициклических алкильных групп или циклоалканов, определенных в изобретении, например, насыщенные моноциклические углеводородные группы, имеющие от трех до двадцати углеродных атомов в кольце, включающие, но этим не ограничивая, циклопропил, циклобутил, циклопентил, циклогексил, циклогептил и другие подобные группы. Термин "замещенный циклоалкил" может обозначать, но никоим образом не ограничивать, моноциклическую или полициклическую алкильную группу, замещенную одним или более заместителями, например, амино, галогеном, алкилом, замещенным алкилом, карбонилокси, карбонилмеркапто, арилом, нитро, меркапто или сульфо, причем эти типичные замещающие группы имеют значения, которые идентичны определениям соответствующих групп, приведенным в изобретении.
[0079] "Гетероциклоалкил" относится к моноциклической или полициклической алкильной группе, в которой, по меньшей мере, один кольцевой углеродный атом ее циклической структуры заменен на гетероатом, выбранный из группы, состоящей из N, O, S или P. "Замещенный гетероциклоалкил" относится к моноциклической или полициклической алкильной группе, в которой, по меньшей мере, один кольцевой углеродный атом ее циклической структуры заменен на гетероатом, выбранный из группы, состоящей из N, O, S или P, и группа содержит один или более заместителей, выбранных из группы, состоящей из галогена, алкила, замещенного алкила, карбонилокси, карбонилмеркапто, арила, нитро, меркапто или сульфо, причем эти типичные замещающие группы имеют значения, которые идентичны определениям соответствующих групп, приведенным в изобретении.
[0080] Термин "гидрокарбил" означает соединение, которое содержит углерод и водород, и которое может быть полностью насыщено, частично ненасыщенно или ароматическим, и включает арильные группы, алкильные группы, алкенильные группы и алкинильные группы.
[0081] В любом из описанных здесь вариантов осуществления изобретения, W, X, Y, Z, G, G', R, R', R", Q1-Q4, A и Rn независимо друг от друга могут быть ковалентно связаны с линкером и/или с линкером, к которому присоединена одна или несколько групп PTM, ULM, CLM или CLM'.
[0082] Более конкретно, неограничивающие примеры CLM включают CLM, которые показаны ниже, а также "гибридные" молекулы, которые получают из комбинации одной или нескольких структур, представленных в показанных ниже молекулах.
 м
м
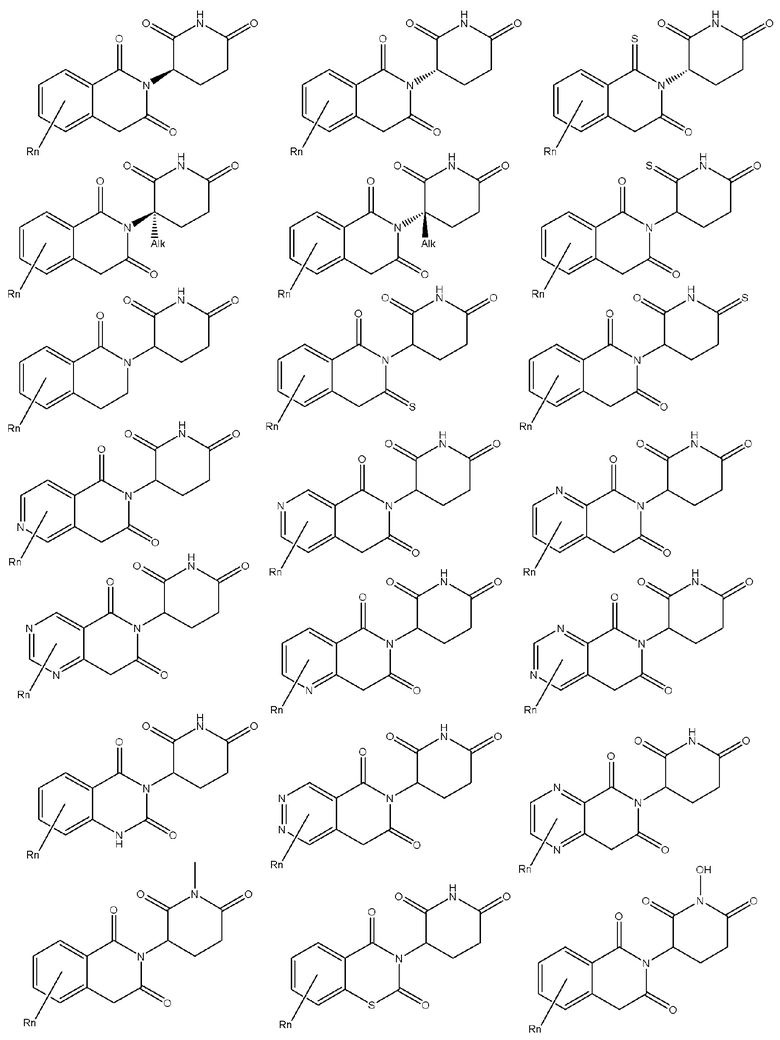
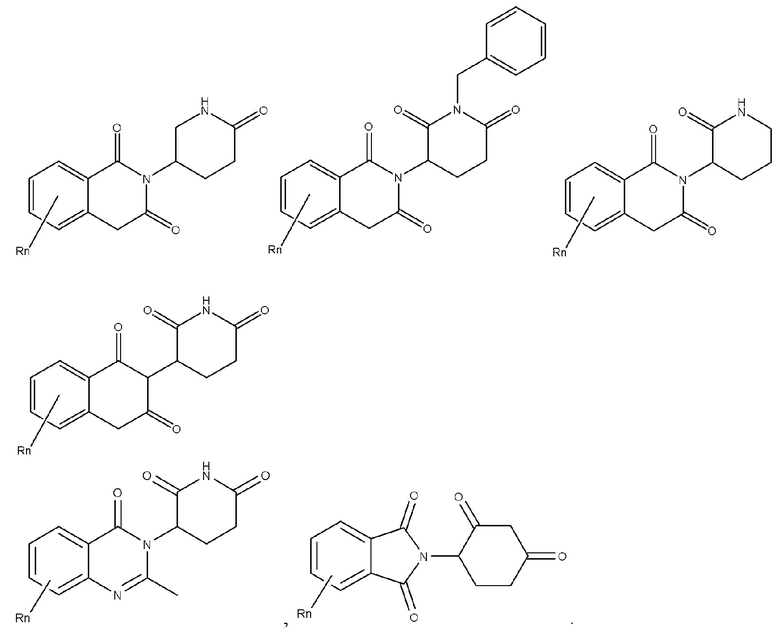
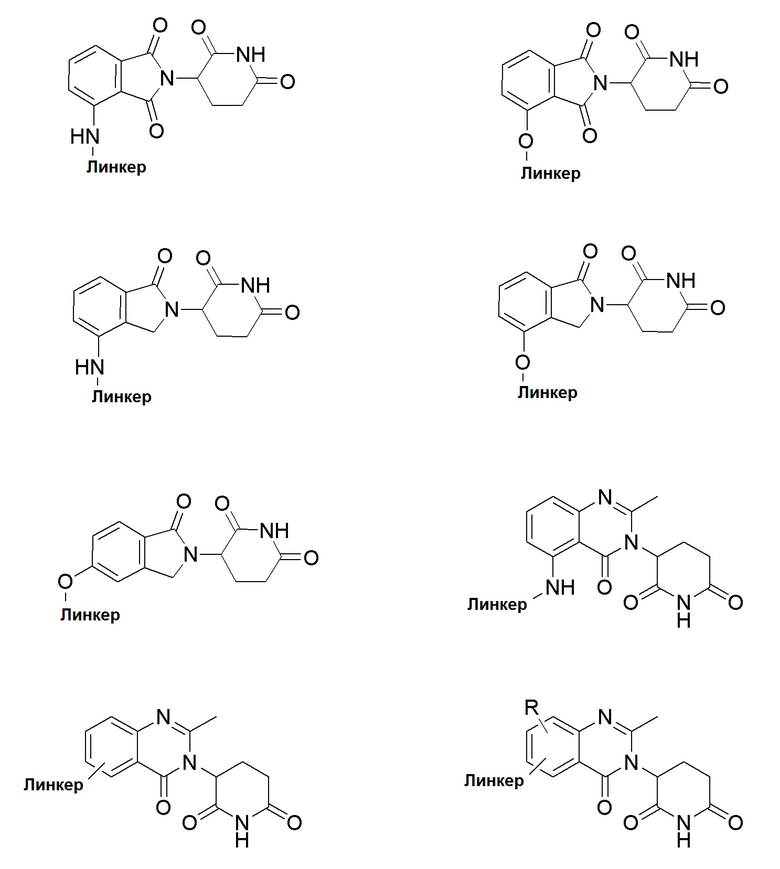
[0083] В некоторых случаях, «CLM» может быть имидами, которые связываются с лигазое E3 цереблона. Эти имиды и места присоединения линкера мгут быть, но без ограничения, представлены следующими структурами:
Типовые линкеры
[0084] В некоторых вариантах осуществления изобретения, соединения, как описано здесь, могут быть химически связаны или соединены с помощью химического линкера (L). В некоторых вариантах осуществления изобретения, линкерная группа L представляет собой группу, содержащую одну или несколько ковалентно соединеных структурных единиц A (например, -A1... Aq-), где A1 является группой, связанной по меньшей мере c одним ULM, PTM или их сочетанием. В некоторых вариантах A1 связывает ULM, PTM или их сочетание непосредственно с другим ULM, PTM, или их сочетанием. В других вариантах осуществления изобретения, A1 связывает ULM, PTM или их сочетание опосредованно с другим ULM, PTM или их сочетанием через Aq.
[0085] В некоторых вариантах осуществления изобретения, A1 связывает ULM, PTM или их сочетание опосредованно с другим ULM, PTM или их сочетанием через Aq. В определенных вариантах, A1 - Aq каждый независимо является связью, CRL1RL2, O, S, SO, SO2, NRL3, SO2NRL3, SONRL3, CONRL3, NRL3CONRL4, NRL3SO2NRL4, CO, CRL1=CRL2, C≡C, SiRL1RL2, P(O)RL1, P(O)ORL1, NRL3C(=NCN)NRL4, NRL3C(=NCN), NRL3C(=CNO2)NRL4, C3-11циклоалкилом, необязательно замещенным 0-6 RL1 и/или RL2 группами, C3-11гетероциклилом, необязательно замещенным 0-6 RL1 и/или RL2 группами, арилом, необязательно замещенным 0-6 RL1 и/или RL2 группами, гетероарилом, необязательно замещенным 0-6 RL1 и/или RL2 группами, где RL1 или RL2, каждый независимо может быть связан с другими группами A, с получением циклоалкильной и/или гетероциклильной части, которая может быть дополнительно замещена 0-4 RL5 группами;
где каждый RL1, RL2, RL3, RL4 и RL5 независимо представляет собой H, галоген, C1-8алкил, OC1-8алкил, SC1-8алкил, NHC1-8алкил, N(C1-8алкил)2, C3-11циклоалкил, арил, гетероарил, C3-11гетероциклил, OC1-8циклоалкил, SC1-8циклоалкил, NHC1-8циклоалкил, N(C1-8циклоалкил)2, N(C1-8циклоалкил)(C1-8алкил), OH, NH2, SH, SO2C1-8алкил, P(O)(OC1-8алкил)(C1-8алкил), P(O)(OC1-8алкил)2, CC-C1-8алкил, CCH, CH=CH(C1-8алкил), C(C1-8алкил)=CH(C1-8алкил), C(C1-8алкил)=C(C1-8алкил)2, Si(OH)3, Si(C1-8алкил)3, Si(OH)(C1-8алкил)2, COC1-8алкил, CO2H, галоген, CN, CF3, CHF2, CH2F, NO2, SF5, SO2NHC1-8алкил, SO2N(C1-8алкил)2, SONHC1-8алкил, SON(C1-8алкил)2, CONHC1-8алкил, CON(C1-8алкил)2, N(C1-8алкил)CONH(C1-8алкил), N(C1-8алкил)CON(C1-8алкил)2, NHCONH(C1-8алкил), NHCON(C1-8алкил)2, NHCONH2, N(C1-8алкил)SO2NH(C1-8алкил), N(C1-8алкил) SO2N(C1-8алкил)2, NHSO2NH(C1-8алкил), NHSO2N(C1-8алкил)2, NHSO2NH2.
[0086] В некоторых вариантах осуществления изобретения, q представляет собой целое число, большее или равное 0. В некоторых вариантах осуществления, q представляет собой целое число, большее или равное 1.
[0087] В некоторых вариантах осуществления изобретения, например, где q больше 2, Аq представляет собой группу, которая соединена с фрагментом ULM или ULM', и A1 и Аq соединены с помощью структурных единиц A (количество таких структурных единиц А равно q-2).
[0088] В некоторых вариантах осуществления изобретения, например, где q равно 2, Аq представляет собой группу, которая соединена с A1 и с ULM или ULM' фрагментом.
[0089] В некоторых вариантах осуществления изобретения, например, где q равно 1, структурой линкерной группы L является -A1- и A1 является группой, которая соединена с ULM или ULM' фрагментом и PTM фрагментом.
[0090] В дополнительных вариантах осуществления изобретения, q равно целому числу от 1 до 100, от 1 до 90, от 1 до 80, от 1 до 70, от 1 до 60, от 1 до 50, от 1 до 40, от 1 до 30, от 1 до 20 или от 1 до 10.
[0091] В некоторых вариантах осуществления изобретения, линкер (L) выбирают из группы, состоящей из:
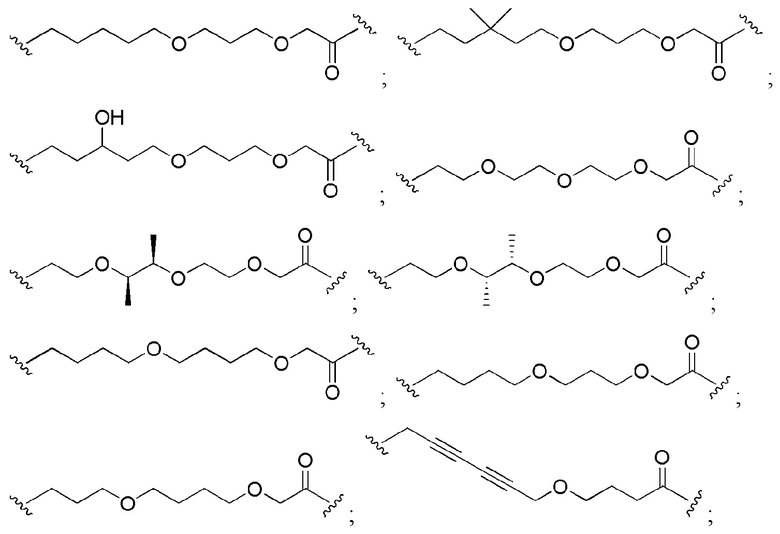
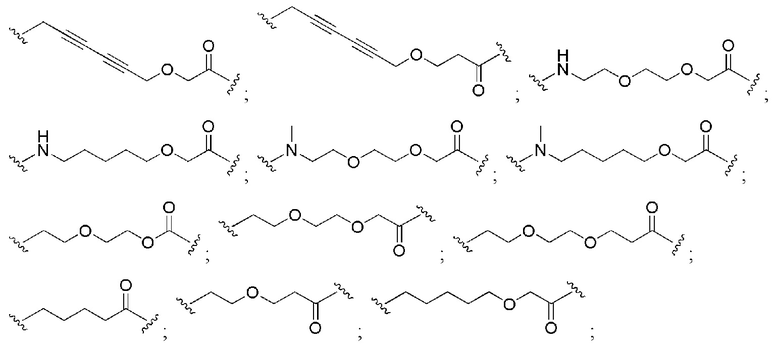
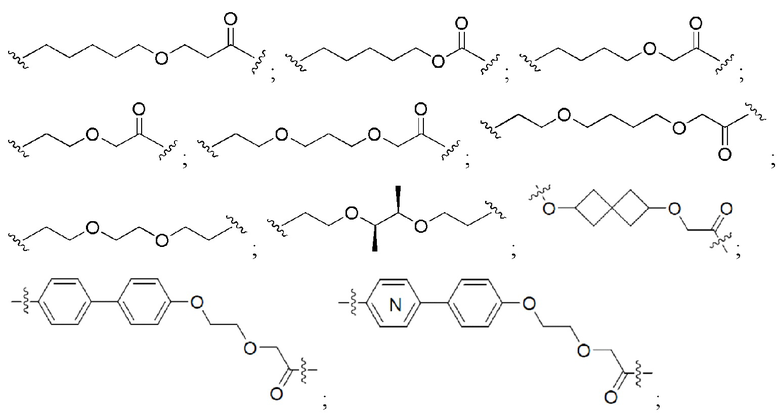
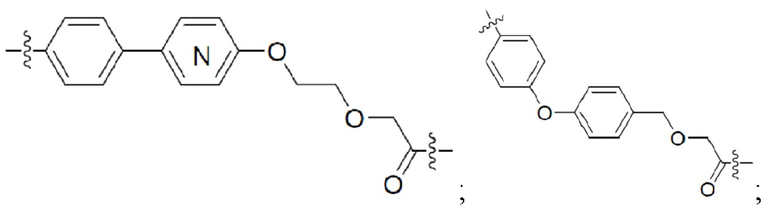
и
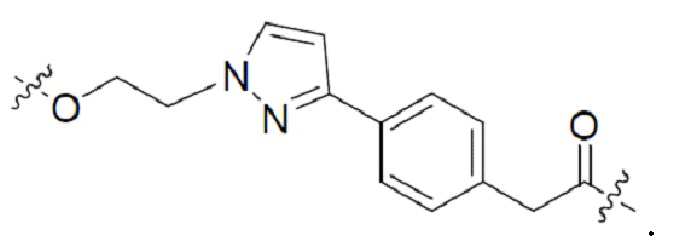
[0092] В дополнительных вариантах осуществления изобретения, линкерная группа необязательно замещенный (поли)этиленгликоль, содержащий от 1 до около 100 единиц этиленгликоля, от около 1 до около 50 единиц этиленгликоля, от 1 до около 25 единиц этиленгликоля, от около 1 до 10 единиц этиленгликоля, от 1 до около 8 единиц этиленгликоля и от 1 до 6 единиц этиленгликоля, от 2 до 4 единиц этиленгликоля, или необязательно замещенными алкильными группами, перемежающими с необязательно замещенными атомами O, N, S, P или Si. В некоторых вариантах линкер замещен арилом, фенилом, бензилом, алкилом, алкиленом или гетероциклом. В некоторых вариантах линкер может быть асимметричным или симметричным.
[0093] В любом из вариантов осуществления изобретения, в описанных здесь соединениях, линкерная группа может представлять собой любой подходящий фрагмент, как описано здесь. В одном варианте осуществления, линкер представляет собой замещенную или незамещенную полиэтиленгликолевую группу, содержащую от приблизительно 1 до приблизительно 12 звеньев этиленгликоля, от 1 до приблизительно 10 звеньев этиленгликоля, от приблизительно 2 до приблизительно 6 звеньев этиленгликоля, от приблизительно от 2 до приблизительно 5 звеньев этиленгликоля, от приблизительно от 2 до приблизительно 4 звеньев этиленгликоля.
[0094] Несмотря на то, что фрагмент CLM (или ULM) и фрагмент PTM могут быть ковалентно связаны с группой линкера через любую группу, которая является подходящей и устойчивой к химии линкера, в предпочтительных аспектах настоящего изобретения линкер независимо ковалентно связан с CLM и PTM предпочтительно через амидную, эфирную, тиоэфирную, кето-группу, карбамат (уретан), углерод или простой эфир, где каждая из указанных групп может быть вставлена в любом месте в фрагменте CLM и фрагменте PTM, чтобы обеспечить максимальное связывание фрагмента CLM на убиквитинлигазе и фрагмент PTM на белке-мишени, подлежащего деградированию. (Следует отметить, что в некоторых вариантах, где фрагмент PTM представляет собой фрагмент ULM, белок-мишень для деградации может быть убиквитинлигазой как таковой). В некоторых предпочтительных вариантах, линкер может быть связан с необязательно замещенной алкильной, алкиленильной, алкенильной или алкинильной группами, арильной группой или гетероциклической группой на фрагментах CLM и/или PTM.
[0095] В некоторых вариантах осуществления изобретения, «L» может представлять собой линейную цепь с количеством атомов в цепи от 4 до 24, при этом атом углерода в линейной цепи может быть замещен кислородом, азотом, амидом, фторированным углеродом и т.п., и представлять собой, например, одно из следующего:
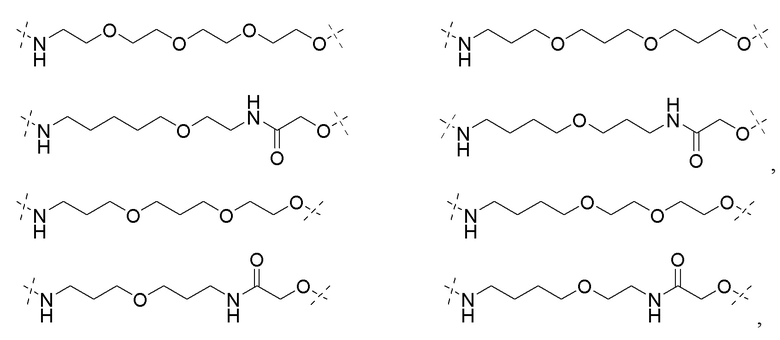
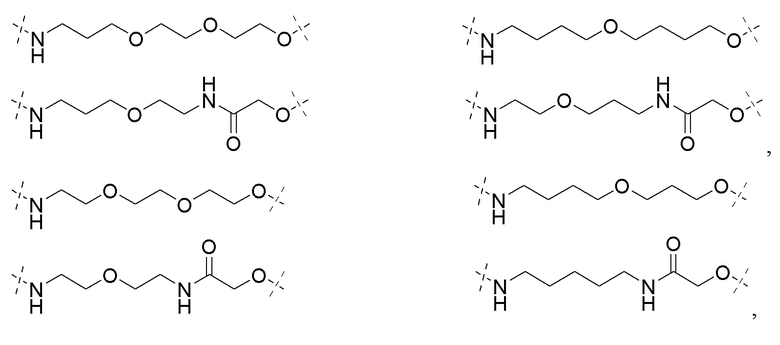
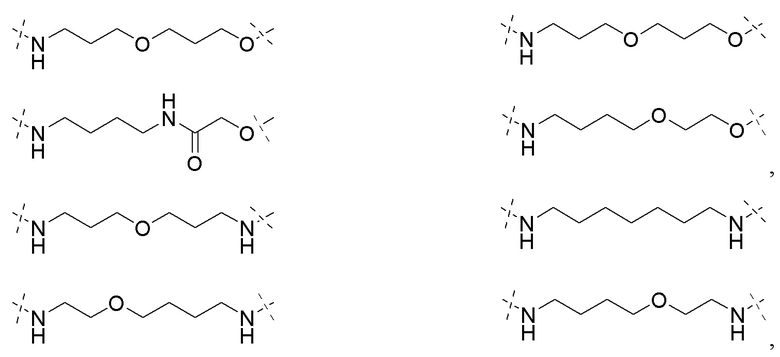
или
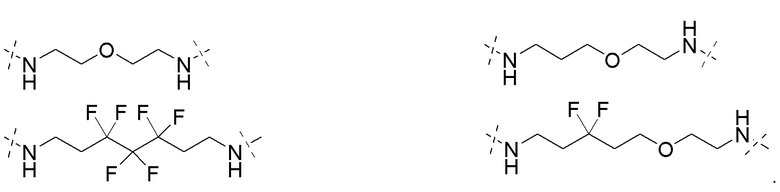
[0096] В некоторых вариантах осуществления изобретения «L» может представлять собой нелинейные цепи, и может быть алифатическими или ароматическими или гетероароматическими циклическими группами. Некоторые примеры «L» включает, но без ограничения, следующее:
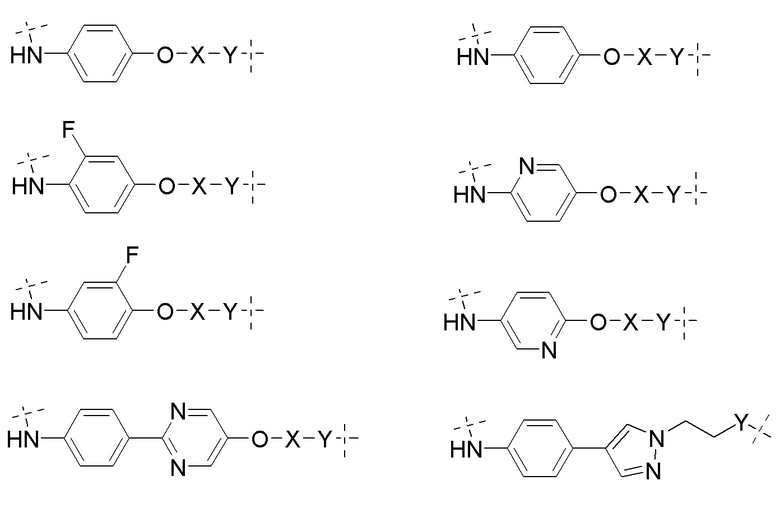
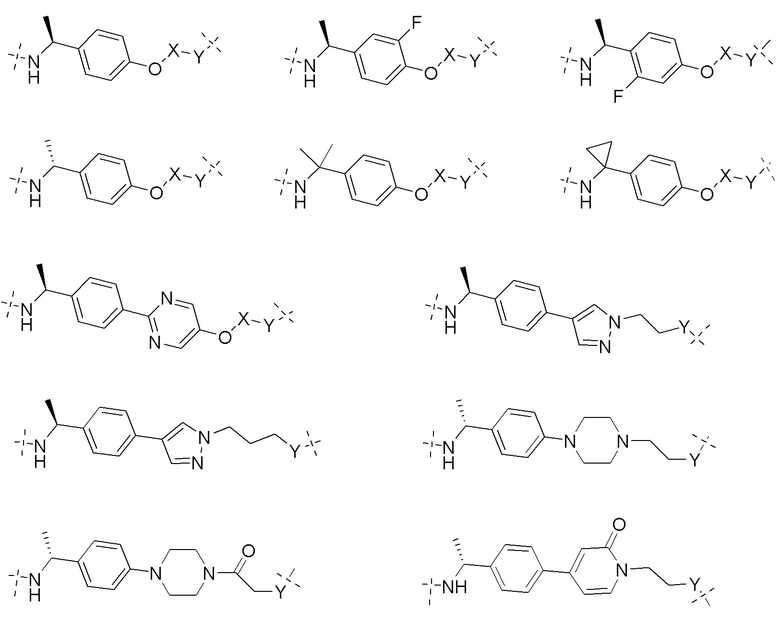
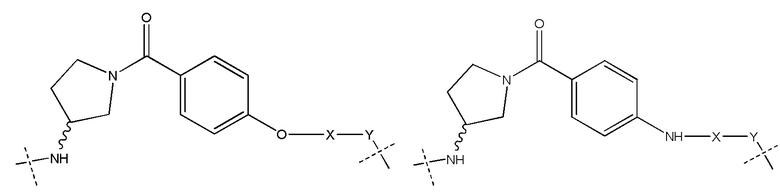
[0097] где:
"Х" в указанных выше структурах может быть линейной цепью с числом атомов в цепи от 2 до 14, и указанная цепь может содержать гетероатомы, такие как кислород; и
[0098] "Y" в указанных выше структурах может представлять собой O, N, S(О)n (n=0, 1, 2).
Типовые PTM
[0099] В предпочтительных вариантах настоящего изобретения, группа PTM представляет собой группу, которая связывается с белками-мишенями. Мишени для группы PTM многочисленны и их выбирают из белков, которые экспрессируются в клетках, таких, которые, по крайней мере, являются частью последовательностей, найденных в клетках, и могут связываться с группой PTM. Термин ʺбелокʺ включает олигопептидные и полипептидные последовательность достаточной длины, позволяющей связываться с группой PTM в соответствии с данным изобретением. Любой белок в эукариотной системе или микробной системе, включая вирус, бактерии или грибы, является целью для убиквитинирования, опосредованного соединениями в соответствии с данным изобретением. Предпочтительно, целевым белком является эукариотный белок. В определенных аспектах, фрагментом, связывающим белок, является галогеналкан (предпочтительно, C1-C10 алкильная группа, которая замещена, по крайней мере, одной галогруппой, предпочтительно, галогруппой на дальнем конце алкильной группы (т.е. далеко от линкера или СLM), который может ковалентно связываться с ферментом дегалогеназой у пациента или в диагностическом исследовании.
[00100] Группы PTM в соответствии с данным изобретением включают, например, любые фрагменты, которые специфически связываются с белком (связывается с целевым белком) и включают следующие неограничивающие примеры низкомолекулярных фрагментов целевых белков: ингибиторы Hsp90, ингибиторы киназы, ингибиторы HDM2 и MDM2, соединения, нацеленные на человеческие BET бромодомен-содержащие белки, ингибиторы HDAC, ингибиторы человеческой лизинметилтрансферазы, ингибиторы ангиогенеза, соединения рецепторов ядерных гормонов, иммунодепрессивные соединения и соединения, нацеленные на арилуглеводородный рецептор (AHR), среди прочих. Описанные ниже структуры представляют некоторые члены этих девяти типов низкомолекулярных фрагментов, связывающих белки-мишени. Такие низкомолекулярные фрагменты, связывающие белки-мишени, также включают фармацевтически приемлемые соли, энантиомеры, сольваты и полиморфы этих структур, а также другие малые молекулы, которые могут быть нацелены на белки-мишени. Такие связывающие фрагменты соединены с фрагментом, связывающим убиквитинлигазу, предпочтительно, через линкер для презентирования белка-мишени (с которым связан фрагмент белка-мишень) рядом с убиквитинлигазой для убиквитинирования и деградации.
[00101] Любой белок, который может быть связан с фрагментом белка-мишени или группой PTM, и который подвергается воздействию или расщеплению убиквитинлигазой, является белком-мишенью в соответствии с данным изобретением. В общем, белки-мишени могут включать, например, структурные белки, рецепторы, ферменты, белки поверхности клетки, белки, имеющие отношение к интегрированной функции клетки, включая белки, вовлеченные в каталитическую активность, активность ароматазы, двигательную активность, активность геликазы, метаболические процессы (анаболизм и катаболизм), антиокислительную активность, протеолиз, биосинтез, белки с активностью киназы, активностью оксидоредуктазы, активностью трансферазы, активностью гидролазы, активностью лиазы, активностью изомеразы, активностью лигазы, активностью ферментного регулятора, активностью переносчика сигнала, активностью структурной молекулы, связывающей активностью (белок, жир, углевод), активностью рецептора, вовлеченные в подвижность клетки, слияние мембраны, клеточную коммуникацию, регулирование биологических процессов, развитие, дифференциацию клетки, ответ на стимулы, поведенческие белки, белки адгезии клеток, белки, вовлеченные в гибель клеток, белки, вовлеченные в транспорт (включая активность белка-транспортера), ядерный транспорт, активность транспортера ионов, активность транспортера канала, активность носителя, активность пермеазы, активность секреции, активность транспортера электрона, патогенез, активность регулятора шаперона, активность связывания нуклеиновой кислоты, активность регулятора транскрипции, активность внеклеточной организации и биогенеза, активность регулятора трансляции. Целевые белки могут включать белки из эукариотов и прокариотов, включая человека в качестве объекта лекарственной терапии, других животных, включая домашних животных, микробов для определения мишеней для антибиотиков и других противомикробных средств, и растений и даже вирусов, среди прочих.
[00102] В других вариантах, группой PTM является галогеналкильная группа, где указанная алкильная группа, в общем, варьирует по размеру от около 1 или 2 углеродов до около 12 углеродов в длину, часто от около 2 до 10 углеродов в длину, часто от около 3 углеродов до около 8 углеродов в длину, более часто, от около 4 углеродов до около 6 углеродов в длину. Галогеналкильными группами обычно являются линейные алкильные группы (хотя также могут применяться разветвленные алкильные группы) и они имеют на конце, по крайней мере, одну галогеновую группу, предпочтительно, одинарную галогеновую группу, часто, одинарную хлоридную группу. Галогеналкильные группы PTM для применения в соответствии с данным изобретением, предпочтительно, представлены химической структурой -(CH2)v-галоген, где v является любым целым числом от 2 до около 12, часто, от около 3 до около 8, и наиболее часто, от около 4 до около 6. Галогеном может быть любой галоген, но, предпочтительно, Cl или Br, и более часто Cl.
[00103] В другом варианте осуществления настоящее изобретение предоставляет библиотеку соединений. Библиотека содержит более одного соединения, где каждая структура соединений имеет формулу А-В, где A является связывающим фрагментом белка убиквитинового пути (предпочтительно, фрагментом E3 убиквитинлигазы, как описано здесь), и B является связывающим белок членом молекулярной библиотеки, где A связан (предпочтительно, через линкерную группу) с B, и где связующий фрагмент белка убиквитинового пути распознает белок убиквитинового пути, в частности, убиквитинлигазу E3, такую как цереблон. В конкретном варианте, библиотека содержит специфический фрагмент связывания убиквитинлигазы E3 цереблона, связывающийся с произвольными связующими элементами белка-мишени (например, с библиотекой химических соединений). Как таковой, белок-мишень не определен заранее, и способ может применяться для определения активности гипотетического элемента, связывающего белок, и его фармакологической ценности в качестве цели при разложении убиквитинлигазы.
[00104] Настоящее изобретение может быть использовано для лечения ряда заболеваний и/или патологических состояний, в том числе любого заболевания и/или патологического состояния, при котором белки дезрегулированы, и где пациент получит пользу от деградации белков.
[00105] В дополнительном аспекте, настоящее изобретение предоставляет терапевтические композиции, содержащие эффективное количество соединения, раскрытого в данном описании, или его соли, и фармацевтически приемлемый носитель, добавку или эксципиент, и необязательно дополнительный биологически активный агент. Терапевтические композиции модулируют деградацию белков у пациента или субъекта, например, у животного, такого как человек, и могут быть использованы для лечения или облегчения заболеваний или состояний, которые модулируются путем деградации белка. В некоторых вариантах, терапевтические композиции, описанные здесь, могут быть использованы для осуществления деградации белков, представляющих интерес, для лечения или облегчения заболевания, такого как, например, рак. В некоторых дополнительных вариантах осуществления изобретения заболевание представляет собой множественную миелому.
[00106] В альтернативных вариантах настоящее изобретение относится к способу лечения заболевания или ослабления симптомов заболевания или состояния у субъекта, нуждающегося в этом, путем деградации белка или полипептида, посредством чего модулируется состояние или заболевание, где способ включает введение указанному пациенту или субъекту эффективного количества, например, терапевтически эффективного количества, по меньшей мере, одного соединения, как описано выше, необязательно в сочетании с фармацевтически приемлемым носителем, добавкой или эксципиентом, и, необязательно, дополнительным биологически активным агентом, где композиция является эффективной для лечения или облегчения заболевания или расстройства, или его симптомов у субъекта. Способ в соответствии с настоящим изобретением может быть использован для лечения большого числа заболеваний или состояний, включая рак, за счет введения эффективных количеств по меньшей мере одного соединения, из описанных в настоящем документе. Состояние или заболевания может быть заболеванием, вызываемое микробиологическим агентом или другим экзогенным агентом, таким как вирус, бактерия, грибы, простейшие или другие микробы, или может быть заболеванием, которое вызвано избыточной экспрессией белка, приводящей к заболеванию и/или состоянию.
[00107] В другом аспекте, в изобретении предлагаются способы идентификации эффектов деградации представляющих интерес белков в биологической системе с использованием соединений настоящего изобретения.
[00108] Термин «целевой белок» или «белок-мишень» применяется для описания белка или полипептида, который является мишенью для связывания с соединением в соответствии с данным изобретением и расщепления его убиквитинлигазой. Такие низкомолекулярные фрагменты, связывающие целевые белки, также включают фармацевтически приемлемые соли, энантиомеры, сольваты и полиморфы этих структур, а также другие малые молекулы, которые могут быть нацелены на белок-мишень. Такие связывающие фрагменты связаны с фрагментами СLM или ULM через линкерные группы L.
[00109] Целевые белки-мишени, которые могут связываться с фрагментом белка-мишени и которые подвергаются деградации с помощью лигазы, с которой связывается фрагмент связывания с убиквитинлигазой, включает любой белок или пептид, в том числе их фрагменты, аналоги и/или их гомологи. Целевые белки могут включать белки и пептиды, обладающие любыми биологическими функциями или активностями, включая структурные, регулирующие функции, гормональную, ферментативную активности, генетические, иммунологические, сократительные функции, функции хранения, транспортировки и передачи сигнала. В некоторых вариантах целевые белки включают, например, структурные белки, рецепторы, ферменты, белки поверхности клетки, белки, имеющие отношение к интегрированной функции клетки, включая белки, вовлеченные в каталитическую активность, активность ароматазы, двигательную активность, активность геликазы, вовлеченные в метаболические процессы (анаболизм и катаболизм), антиокислительную активность, протеолиз, биосинтез, белки с киназной активностью, с активностью оксидоредуктазы, активностью трансферазы, активностью гидролазы, активностью лиазы, активностью изомеразы, активностью лигазы, активностью ферментного регулятора, активностью переносчика сигнала, активностью структурной молекулы, связывающей активностью (белок, жир, углевод), активностью рецептора, вовлеченные в подвижность клетки, слияние мембраны, клеточную коммуникацию, регулирование биологических процессов, развитие, дифференциацию клетки, ответ на стимулы, поведенческие белки, белки адгезии клеток, белки, вовлеченные в гибель клеток, белки, вовлеченные в транспорт (включая активность белка-транспортера, ядерный транспорт, активность транспортера ионов, активность транспортера канала, активность носителя, активность пермеазы, активность секреции, активность транспортера электрона, патогенез, активность регулятора шаперона, активность связывания нуклеиновой кислоты, активность регулятора транскрипции, активность внеклеточной организации и биогенеза, активность регулятора трансляции. Целевые белки могут включать белки из эукариотов и прокариотов, включая человека в качестве объекта лекарственной терапии, других животных, включая домашних животных, микробов для определения мишеней для антибиотиков и других противомикробных средств, и растений и даже вирусов, среди прочих.
[00110] Более конкретно, ряд мишеней для лекарств, используемых для терапевтических целей у человека, представляют собой белковые мишени, с которыми может связываться нацеливающий на белок фрагмент соединений по изобретению. Они включают белки, которые могут быть использованы для восстановления функции при многочисленных полигенных заболеваниях, включая, например, B7.1 и B7, TINFRlm, TNFR2, NADPH-оксидазу, BclIBax и другие партнеры пути апоптоза, рецептор C5a-, HMG-CoA-редуктазу, PDE V фосфодиэстеразу типа 5, PDE IV фосфодиэстеразу типа 4, PDE I, PDE II, PDE III, ингибитор сквален-циклазы, CXCR1, CXCR2, синтазу оксида азота (NO), циклооксигеназу 1, циклооксигеназу 2, рецепторы 5HT, рецепторы допамина, G-белки, т.е. Gq, гистаминовые рецепторы, 5-липоксигеназу, триптазосеринпротеазу, тимидилатсинтазу, пуринонуклеозидфосфорилазу, трипаносомьный GAPDH, гликогенфосфорилазу, карбоангидразу, хемокиновые рецепторы, JAW, STAT, RXR и аналогичные, HIV-1 протеазe, HIV-1 интегразу, нейраминидазу гриппа, обратную транскриптазу гепатита B, натриевый канал, мультилекарственно устойчивый (MDR) белок, P-гликопротеин (и MRP), тирозинкиназу, CD23, CD124, тирозинкиназу p56 lck, CD4, CD5, рецептор IL-2, рецептор IL-1, TNF-альфа R, ICAM1, Cat+ каналы, VCAM, VLA-4 интегрин, селектины, CD40/CD40L, невокинины и рецепторы, инозинмонофосфат дегидрогеназу, p38 MAP киназу, путь метаболизма Ras1Raf1MEWERK, интерлейкин-1 преобразующий фермент, каспазу, HCV, NS3 протеазу, HCV NS3 RNA геликазу, глицинамидрибонуклеотид формилтрансферазу, риновирусную 3C протеазу, протеазу вируса простого герпеса 1 типа (HSV-I), цитомегаловирусную (CMV) протеазу, поли(ADP-рибоза) полимеразу, циклинзависимые киназы, фактор роста эндотелия сосудов, рецептор окситоцина, ингибитор переноса белков через микросомы, ингибитор транспорта желчных кислот, ингибиторы 5 альфа редуктазы, ангиотензин 11, рецептор глицина, рецептор обратного захвата норадреналина, рецепторы эндотелина, нейропептид Y и рецептор, рецепторы эстрогена, рецепторы андрогена, рецепторы аденозина, аденозинкиназу и AMP деаминазу, пуринергические рецепторы (P2Y1, P2Y2, P2Y4, P2Y6, P2X1-7), фарнезилтрансферазы, геранилгеранил трансферазу, TrkA рецептор для NGF, бета-амилоид, тирозинкиназу Flk-IIKDR, рецептор витронектина, рецептор интегрина, Her-21 neu, ингибитор теломеразы, цитозольной фосфолипазы A2 и EGF рецептор тирозинкиназы. Дополнительные целевые белки-мишени включают, например, экдизон 20-монооксигеназу, ионный канал GABA воротного хлоридного канала, ацетилхолинэстеразу, белок потенциалзависимого натриевого канала, кальцийвысвобождающий канал и хлоридные каналы. Помимо того, дополнительные целевые белки-мишени включают ацетил-CoA карбоксилазу, аденилосукцинат синтазу, протопорфириноген оксидазу и енолпирувилшикимат-фосфат синтазу.
[00111] Ферменты галогеналкандегалогеназы представляют собой другую цель конкретных соединений в соответствии с настоящим изобретением. Соединения в соответствии с настоящим изобретением, которые содержат фрагменты, связывающие хлоралкан пептид (C1-C12, часто примерно C1-C10, алкилгалогеновые группы), могут быть использованы для ингибирования и/или деградации ферментов галогеналкандегалогеназы, которые используются в слитых белках или связанных диагностических белках, как описано в PCT/US 2012/063401, поданной 6 декабря 2011г., и опубликованной как WO 2012/078559 14 июня 2012г., содержание которой включено в данный документ путем ссылки.
[00112] Эти различные целевые белки-мишени могут быть использованы в скрининговых исследованиях, в которых идентифицируют фрагменты соединения, связывающегося с белком, и при этом путем введения фрагмента в соединения в соответствии с настоящим изобретением уровень активности белка может быть изменен для конечного терапевтического результата.
[00113] Термин «фрагмент белок-мишень» или PTM используется для описания небольшой молекулы, которая связывается с целевым белком-мишенью или другим белком или полипептидом, представляющим интерес, и которая помещает/презентирует этот белок или полипептид в непосредственной близости к убиквитинлигазе таким образом, что может произойти деградация белка или полипептида за счет действия убиквитинлигазы. Не ограничивающие примеры малых молекул белка-мишени фрагментов связывания включают ингибиторы HSP90, ингибиторы киназы, ингибиторы MDM2, соединения, нацеленные на человеческие BET бромодомен-содержащие белки, ингибиторы HDAC, ингибиторы человеческой лизинметилтрансферазы, ингибиторы ангиогенеза, иммунодепрессивные соединения и соединения, нацеленные на арилуглеводородный рецептор (AHR), среди прочих. Описанные ниже структуры представляют некоторые члены этих девяти типов низкомолекулярных фрагментов, связывающих целевые белки.
[00114] Типовые фрагменты белка-мишени в соответствии с настоящим изобретением включают например, ингибиторы галогеналкангалогеназы, ингибиторы Hsp90, ингибиторы киназы, ингибиторы MDM2, соединения, нацеленные на человеческие BET бромодомен-содержащие белки, ингибиторы HDAC, ингибиторы человеческой лизинметилтрансферазы, ингибиторы ангиогенеза, иммунодепрессивные соединения и соединения, нацеленные на арилуглеводородный рецептор (AHR).
[00115] Описанные ниже структуры представляют некоторые из членов таких типов низкомолекулярных фрагментов, связывающих целевой белок. Такие фрагменты, связывающие целевой белок, также включают фармацевтически приемлемые соли, энантиомеры, сольваты и полиморфы этих структур, а также другие маленькие молекулы, которые могут быть нацелены на целевой белок, представляющий интерес. Приведенные ниже ссылки на цитируемые документы предполагают включение содержания этих документов во всей полноте путем ссылки.
I. Ингибиторы белка теплового шока 90 (HSP90)
[00116] Ингибиторы HSP90 в данном описании включают, но без ограничения, следующие.
[00117] Ингибиторы HSP90, идентифицированные в Vallee, et al., "Tricyclic Series of Heat Shock Protein 90 (HSP90) Inhibitors Part I: Discovery of Tricyclic Imidazo[4,5-C]Pyridines as Potent Inhibitors of the HSP90 Molecular Chaperone (2011) J.Med.Chem. 54: 7206, включая YKB: N-[4-(3Н-имидазо[4,5-c]пиридин-2-ил)-9Н-флуорен-9-ил]сукцинамид
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена через концевую амидную группу;
[00118] 2. Ингибитор HSP90, p54 (модифицированный)
8-[(2,4-диметилфенил)сульфанил]-3-пент-4-ин-1-ил-3Н-пурин-6-амин
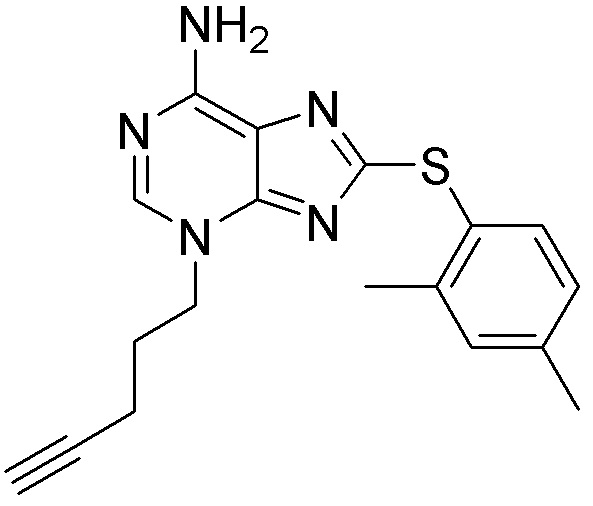
где линкерная группа L или группа -(L-CLM) присоединена, например, через концевую ацетиленовую группу;
[00119] 3. Ингибиторы HSP90 (модифицированные) идентифицированные в Brough, et al., "4,5-Diarylizoxazole HSP90 Chaperone Inhibitors: Potential Therapeutic Agents for the Treatment of Cancer", J.MED.CHEM. vol: 51, pag:196 (2008), включая соединение 2GJ (5-[2,4-дигидрокси-5-(1-метилэтил)фенил]-N-этил-4-[4-(морфолин-4-илметил)фенил]изоксазол-3-карбоксамид), имеющее структуру:
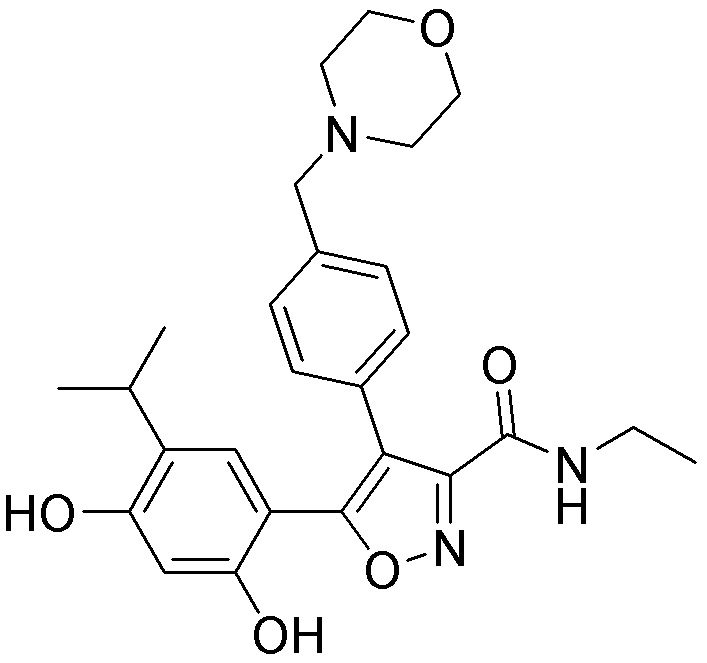
дериватизированное, где линкерная группа L или группа -(L-CLM) присоединена через амидную группу (на амине или на алкильной группе амина);
[00120] 4. Ингибиторы HSP90 (модифицированные), идентифицированные в Wright, et al., Structure-Activity Relationships in Purine-Based Inhibitor Binding to HSP90 Isoforms, Chem Biol. 2004 Jun;11(6):775-85, включая ингибитор HSP90 PU3, имеющий структуру:
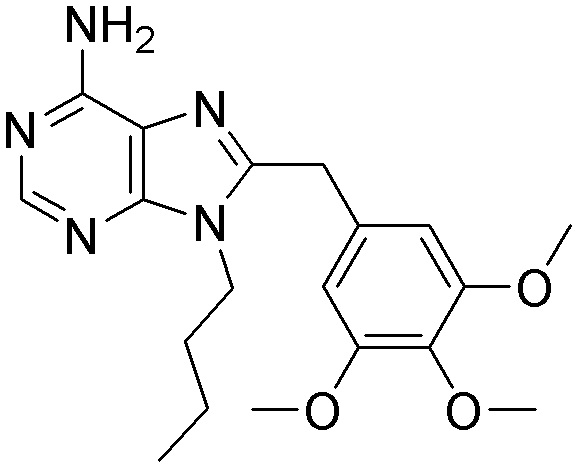
где линкерная группа L или группа -(L-CLM) присоединена, например, через бутильную группу; и
[00121] 5. Ингибитор HSP90 гелданамицин ((4E,6Z,8S,9S,10E,12S,13R,14S,16R)-13-гидрокси-8,14,19-триметокси-4,10,12,16-тетраметил-3,20,22-триоксо-2-азабицикло[16.3.1] (дериватизированный) или любое из его производных (например, 17-алкиламино-17-дезметоксигелданамицин ("17-AAG") или 17-(2-диметиламиноэтил)амино-17-дезметоксигелданамицин ("17-DMAG")) (дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через амидную группу).
II. Ингибиторы киназы и фосфатазы
[00122] Ингибиторы киназ, используемые здесь, включают, но без ограничения, следующие, указанные ниже.
[00123] 1. Ингибитор тирозинкиназы эрлотиниб (производное)
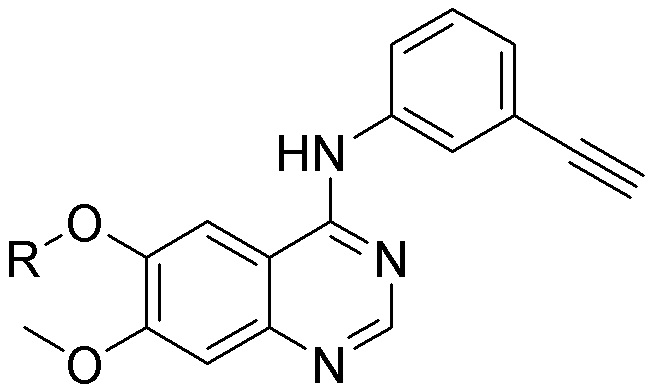
где R представляет собой линкерную группу L или группу -(L-CLM), присоединенную, например, через простую эфирную группу;
[00124] 2. Ингибитор киназы сунитиниб (дериватизированный):
дериватизированный, где R является линкерной группой L или группой -(L-МLM), присоединенной, например, к пиррольной части;
[00125] 3. Ингибитор киназы сорафениб (дериватизированный):
дериватизированный, где R является линкерной группой L или группой -(L-МLM), присоединенной, например, к амидной части;
[00126] 4. Ингибитор киназы дезатиниб (дериватизированный):
дериватизированный, где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к пиримидину;
[00127] 5. ингибитор киназы лапатиниб (дериватизированный):
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена через концевой метил сульфонилметильной группы;
[00128] 6. ингибитор киназы U09-CX-5279 (дериватизированный):
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена через амин (анилин), карбоновую кислоту или амин альфа к циклопропильной группе, или к циклопропильной группе;
[00129] 7. Инигибторы киназы, идентифицированные в Millan, et al., Design and Synthesis of Inhaled P38 Inhibitors for the Treatment of Chronic Obstructive Pulmonary Disease, J.MED.CHEM. vol:54, pag:7797 (2011), включая ингибиторы киназы Y1W и Y1X (дериватизированные), имеющие структуры:
[00130] 1-этил-3-(2-{[3-(1-метилэтил)[1,2,4]триазоло[4,3-a]пиридин-6-ил]сульфанил}бензил)мочевина
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через изо-пропильную группу;
1-(3-трет-бутил-1-фенил-1Н-пиразол-5-ил)-3-(2-{[3-(1-метилэтил)[1,2,4]триазоло[4,3-a]пиридин-6-ил]сульфанил}бензил)мочевина
дериватизированная, где линкерная группа L или группа -(L-CLM) присоединена, например, предпочтительно через изо-пропильную группу или трет-бутильную группу;
[00131] 8. Ингибиторы киназы, идентифицированные в Schenkel, et al., Discovery of Potent and Highly Selective Thienopyridine Janus Kinase 2 Inhibitors J. Med. Chem., 2011, 54 (24), pp 8440-8450, включая соединения 6TP и 0TP (дериватизированные), имеющие структуры:
4-амино-2-[4-(трет-бутилсульфамоил)фенил]-N-метилтиено[3,2-c]пиридин-7-карбоксамид
Тиенопиридин 19
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через концевую метильную группу, связанную с амидной частью;
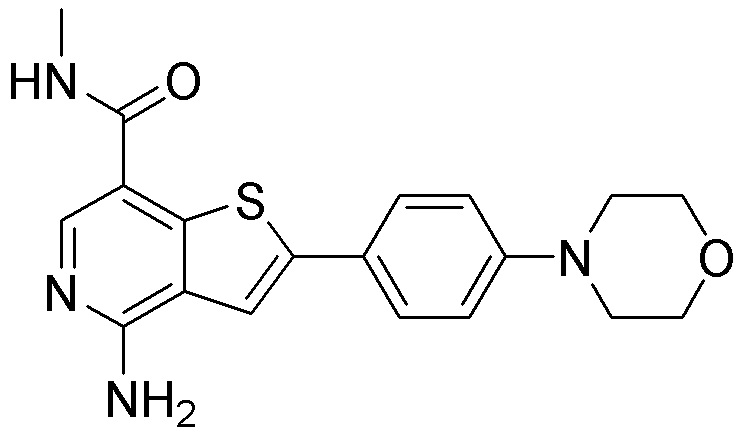
0ТР
4-амино-N-метил-2-[4-(морфолин-4-ил)фенил]тиено[3,2-c]пиридин-7-карбоксамид
Тиенопиридин 8
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через концевую метильную группу, связанную с амидной частью;
[00132] 9. Ингибиторы киназы, идентифицированные в Van Eis, et al., "2,6-Naphthyridines as potent and selective inhibitors of the novel protein kinase C isozymesʺ, Biorg. Med. Chem. Lett.2011 Dec 15; 21(24):7367-72, включая ингибитор киназы 07U, имеющий структуру:
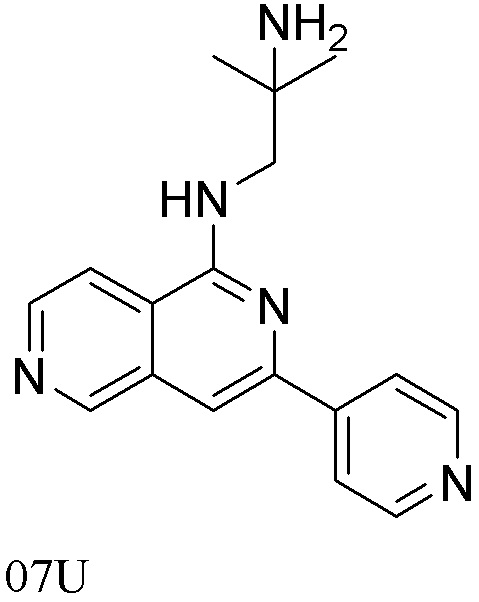
2-метил-N~1~-[3-(пиридин-4-ил)-2,6-нафтиридин-1-ил]пропан-1,2-диамин
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через вторичный амин или концевую аминогруппу;
[00133] 10. Ингибиторы киназы, идентифицированные в Lountos, et al., "Structural Characterization of Inhibitor Complexes with Checkpoint Kinase 2 (Chk2), a Drug Target for Cancer Therapy", J.STRUCT.BIOL. vol:176, pag:292 (2011), включая ингибитор киназы YCF, имеющий структуру:
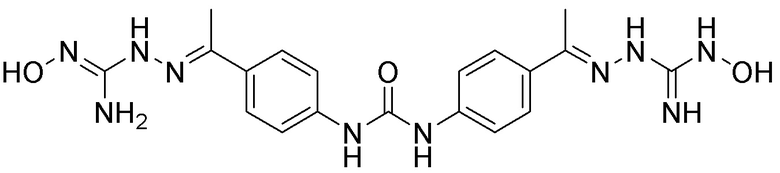
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через простой эфир концевых гидроксильных групп;
[00134] 11. Ингибиторы киназы, идентифицированные в Lountos, et al., "Structural Characterization of Inhibitor Complexes with Checkpoint Kinase 2 (Chk2), a Drug Target for Cancer Therapy", J.STRUCT.BIOL. vol:176, pag:292 (2011), включая ингибиторы киназы XK9 и NXP (дериватизированные), имеющие структуры:
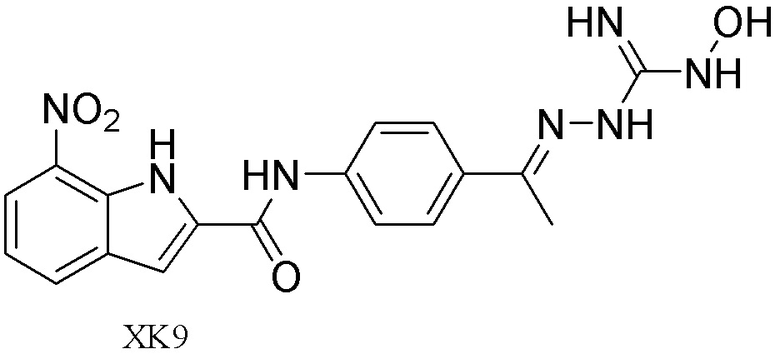
N-{4-[(1Е)-N-(N-гидроксикарбамимидоил)этангидразоноил]фенил}-7-нитро-1Н-индол-2-карбоксамид
NXP
N-{4-[(1Е)-N-карбамимидоилэтангидразоноил]фенил}-1Н-индол-3-карбоксамид
дериватизированные, где линкерная группа L или группа -(L-CLM) присоединена, например, через концевую гидроксильную группу (XK9) или гидразоновую группу (NXP);
[00135] 12. Ингибитор киназы афатиниб (дериватизированный) (N-[4-[(3-Хлор-4-фторфенил)амино]-7-[[(3S)-тетрагидро-3-фуранил]окси]-6-хиназолинил]-4(диметиламино)-2-бутенамид) (дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через алифатическую аминогруппу);
[00136] 13. Ингибитор киназы фостаматиниб (дериватизированный) (гексагидрат динатрийфосфата [6-({5-фтор-2-[(3,4,5-триметоксифенил)амино]пиримидин-4-ил}амино)-2,2-диметил-3-оксо-2,3-дигидро-4H-пиридо[3,2-b]-1,4-оксазин-4-ил]метила) (дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через метоксигруппу);
[00137] 14. Ингибитор киназы гефитиниб (дериватизированный) (N-(3-хлор-4-фторфенил)-7-метокси-6-(3-морфолин-4-илпропокси)хиназолин-4-амин):
дериватизированный, где линкерная группа L или, через метокси или простую эфирную группу;
[00138] 15. Ингибитор киназы ленватиниб (дериватизированный) (4-[3-хлор-4-(циклопропилкарбамоиламино)фенокси]-7-метоксихинолин-6-карбоксамид) (дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через циклопропильную группу);
[00139] 16. Ингибитор киназы вандетаниб (дериватизированный) (N-(4-бром-2-фторфенил)-6-метокси-7-[(1-метилпиперидин-4-ил)метокси]хиназолин-4-амин) (дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через метокси или гидроксильную группу);
[00140] 17. Ингибитор киназы вемурафениб (дериватизированный) ({3-[5-(4-хлорфенил)-1H-пирроло[2,3-b]пиридин-3-карбонил]-2,4-дифторфенил}амид пропан-1-сульфоновой кислоты), дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через сульфонилпропильную группу;
[00141] 18. Ингибитор киназы глеевек (дериватизированный):
дериватизированный, где R в качестве линкерной группы L или группы -(L-CLM) присоединен, например, через амидную группу или через анилинаминовую группу;
[00142] 19. Ингибитор киназы пазопаниб (дериватизированный) (ингибитор VEGFR3):
дериватизированный, где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части или через анилинаминовую группу;
[00143] 20. Ингибитор киназы AT-9283 (дериватизированный) ингибитор аврора-киназы
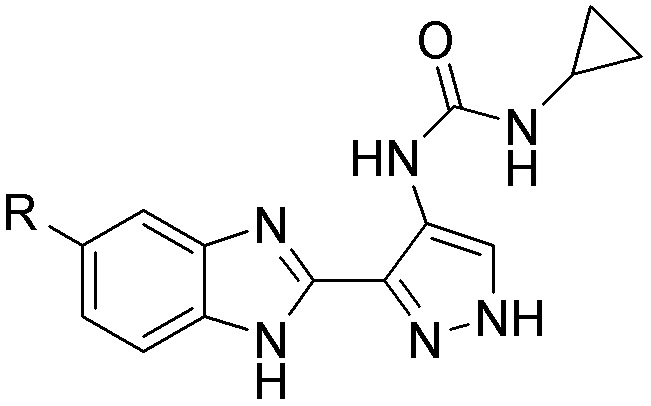
где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части;
[00144] 21. Ингибитор киназы TAE684 (дериватизированный) ингибитор ALK
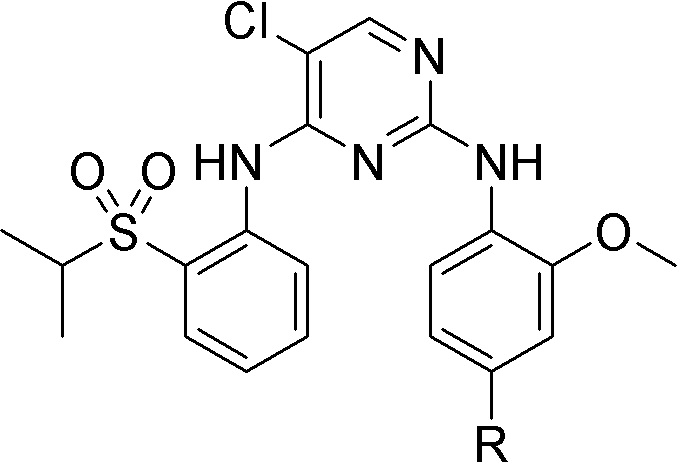
где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части;
[00145] 22. Ингибитор киназы Нилотаниб (дериватизированный), ингибитор Abl:
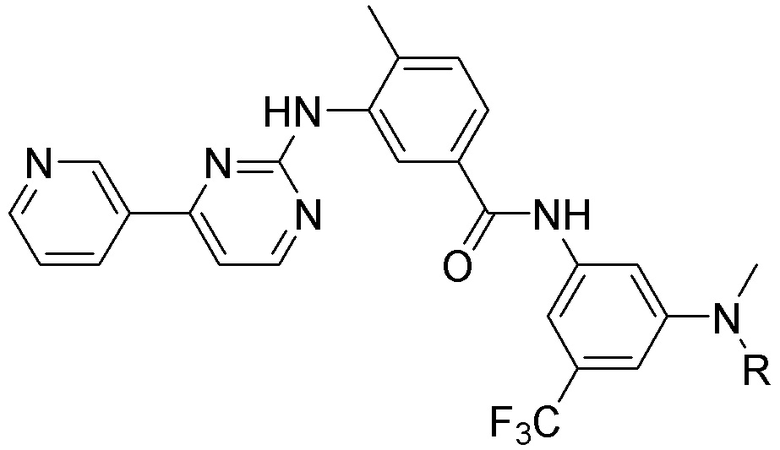
(дериватизированный, где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части или анилинаминовой группе);
[00146] 23. Ингибитор киназы NVP-BSK805 (дериватизированный), ингибитор JAK2
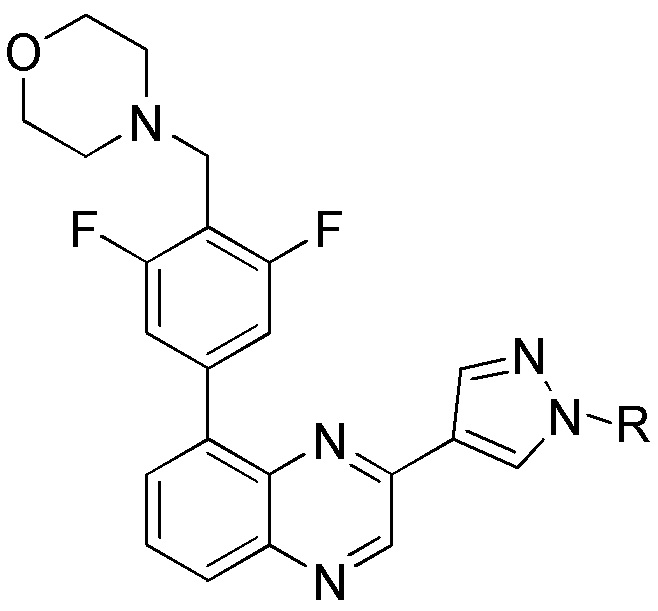
дериватизированный, где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части или к диазольной группе;
[00147] 24. Ингибитор киназы кризотиниб, дериватизированный ингибитор Alk
дериватизированный, где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части или диазольной группе;
[00148] 25. Ингибитор киназы JNJ FMS (дериватизированный) ингибитор
дериватизированный, где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части;
[00149] 26. Ингибитор киназы форетиниб (дериватизированный), ингибитор Met
дериватизированный, где R является линкерной группой L или группой -(L-CLM), присоединенной, например, к фенильной части, или к гидроксильной группе, или к простой эфирной группе на хинолиновой части;
[00150] 27. Аллостерический ингибитор протеинтирозинфосфатазы PTP1B (дериватизированный):
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, на R, как указано.
[00151] 28. Ингибитор домена SHP-2 тирозинфосфатазы (дериватизированный):
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, на R.
[00152] 29. Ингибитор (дериватизированный) BRAF (BRAFV600E)/MEK:
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, на R;
[00153] 30. Ингибитор (дериватизированный) тирозинкиназы ABL
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, на R;
[00154] 31. Ингибитор OSI-027 (дериватизированный), ингибитор mTORC1/2
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, на R;
[00155] 32. Ингибитор киназы OSI-930 (дериватизированный), ингибитор с-Kit/KDR
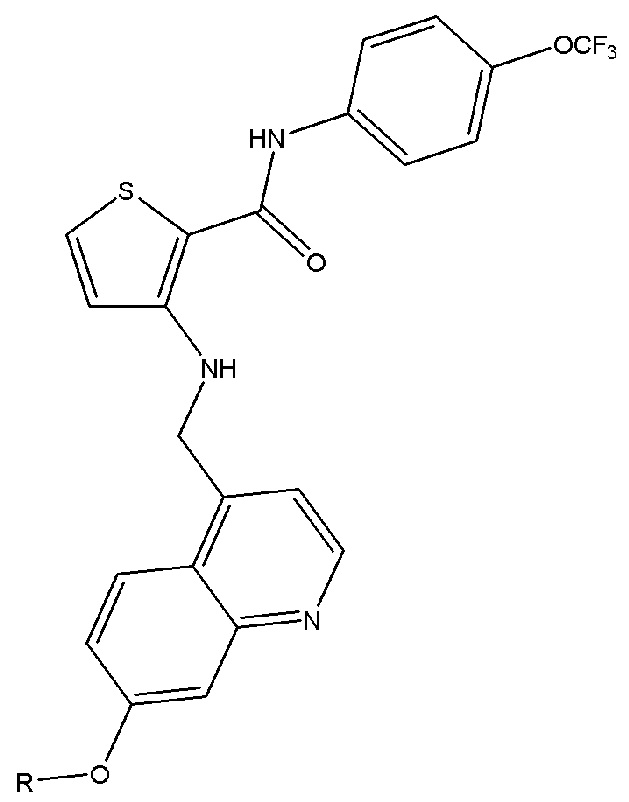
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, на R;
[00156] 33. Ингибитор киназы OSI-906 (дериватизированный) ингибитор IGF1R/IR
дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, на R;
[00157] Где в любом из вариантов, описанных в разделах I-XVII, "R" обозначает участок для присоединения линкерной группы L или группы -(L-CLM) на фрагмент пиперазина.
III. Ингибиторы HDM2/MDM2
[00158] Ингибиторы HDM2/MDM2 в данном описании включают, без ограничения, перчисленные ниже.
[00159] 1. Игибиторы HDM2/MDM2, идентифицированные в Vassilev, et al., In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, SCIENCE vol:303, pag:844-848 (2004) и Schneekloth, et al., Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics, Bioorg. Med. Chem. Lett. 18 (2008) 5904-5908, включая (или дополнительно) соединения нутлин-3, нутлин-2 и нутлин-1 (дериватизированные) как описано ниже, а также все их производные и аналоги:
(дериватизированный, где линкерная группа L или группа -(L-МLM) присоединена, например, на метоксигруппе или гидроксильной группе);
(дериватизированный, где линкерная группа L или группа -(L-МLM) присоединена, например, на метоксигруппе или гидроксильной группе);
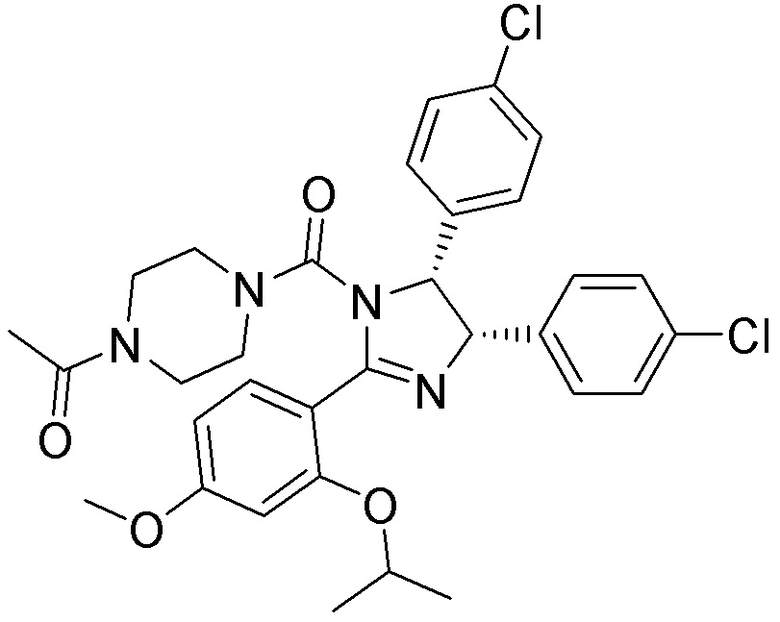
(дериватизированный, где линкерная группа L или группа -(L-МLM) присоединена, например, на метоксигруппе или гидроксильной группе); и
[00160] 2. Транс-4-иод-4'-боранил-халькон
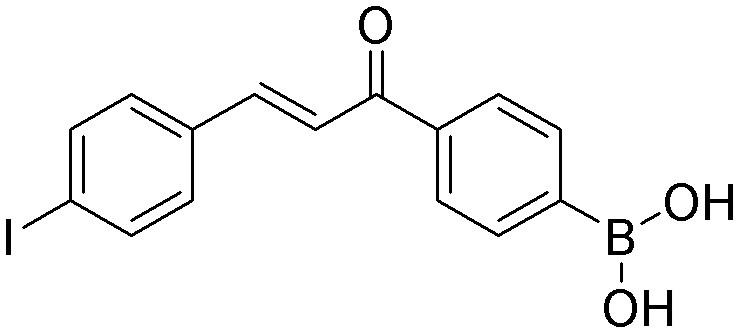
(дериватизированный, где линкерная группа L или группа -(L-МLM) присоединена, например, через гидроксигруппу).
IV. Соединения, нацеленные на человеческие BET бромодомен-содержащие белки
[00162] В некоторых вариантах осуществления «PTM» может представлять собой лиганды, связывающиеся с бромо- и экстра-терминальными (BET) доменами белков BRD2, BRD3 и BRD4. Соединения, нацеленные на человеческие BET бромодомен-содержащие белки, включают, но не ограничены ими, соединения, связанные с мишенями, которые описаны ниже, где ʺRʺ означает место присоединения линкерной группы L или группы -(L-МLM).
[00163] 1. JQ1, Filippakopoulos et al. Selective inhibition of BET bromodomains. Nature (2010):
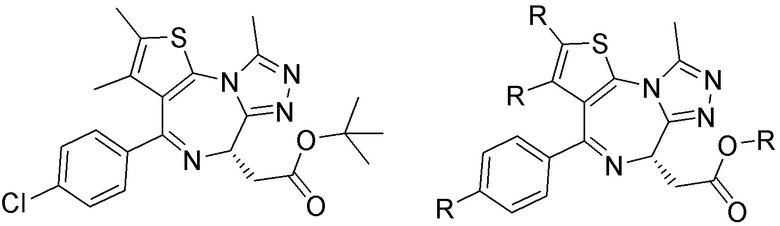
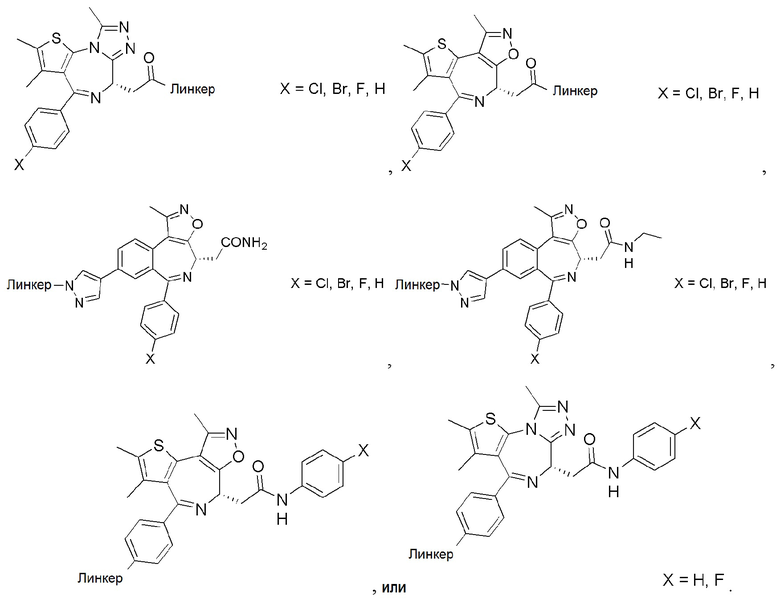
[00164] 2. I-BET, Nicodeme et al. Supression of Inflammation by a Synthetic Histone Mimic. Nature (2010). Chung et al. Discovery and Characterization of Small Molecule Inhibitors of the BET Family Bromodomains. J. Med Chem. (2011):
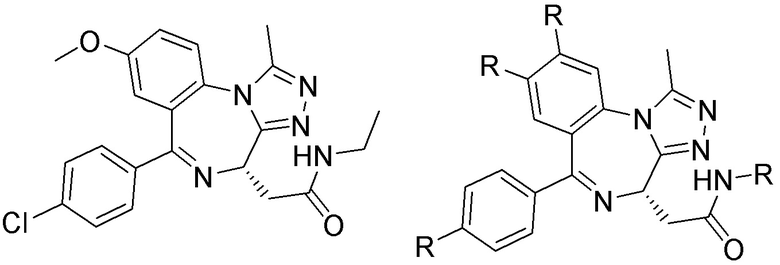
[00165] 3. Соединение, описанное в Hewings et al. 3,5-Dimethylisoxazoles Act as Acetyl-lysine Bromodomain Ligands. J. Med. Chem. (2011) 54 6761-6770:
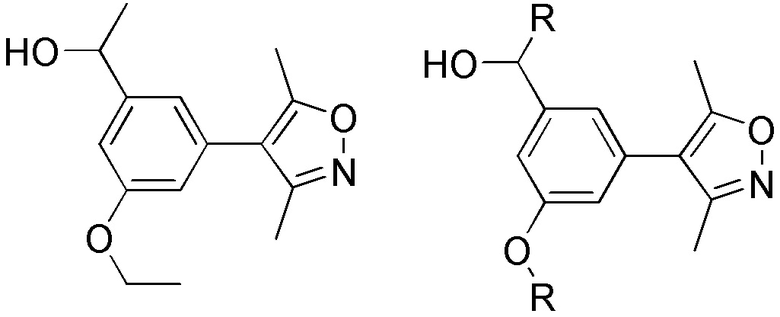
[00166] 4. I-BET151, Dawson et al. Inhibition of BET Recruitment to Chromatin as an Efective Treatment for MLL-fusion Leukemia. Nature (2011):
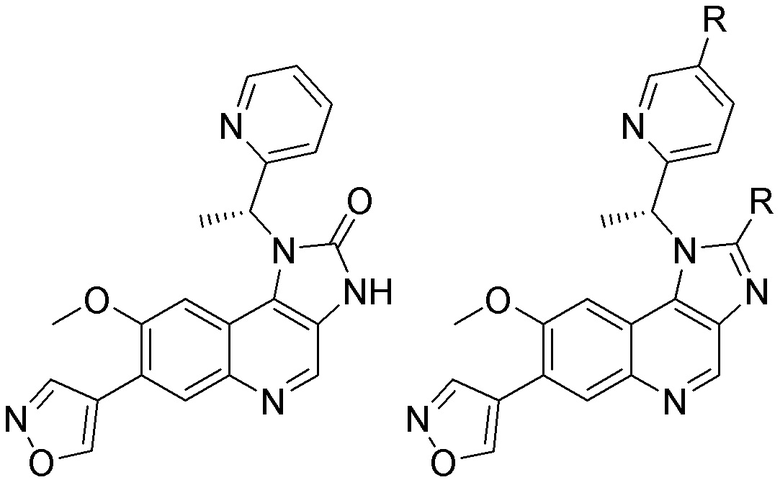
[00167] 5. Тип карбазола (US 2015/0256700)
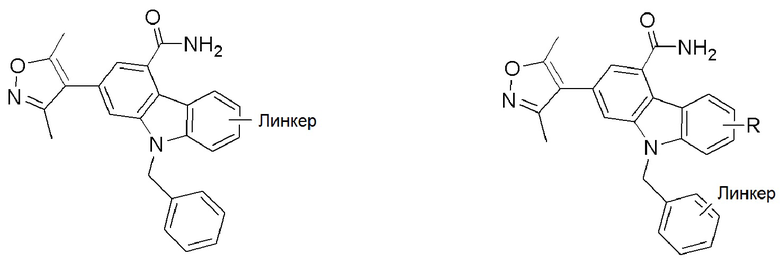
[00168] 6. Тип пирролопиридона (US 2015/0148342)
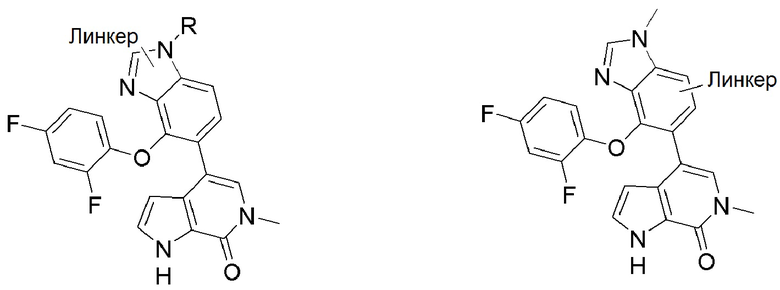
[00169] 7. Тип тетрагидрохинолина (WO 2015/074064)
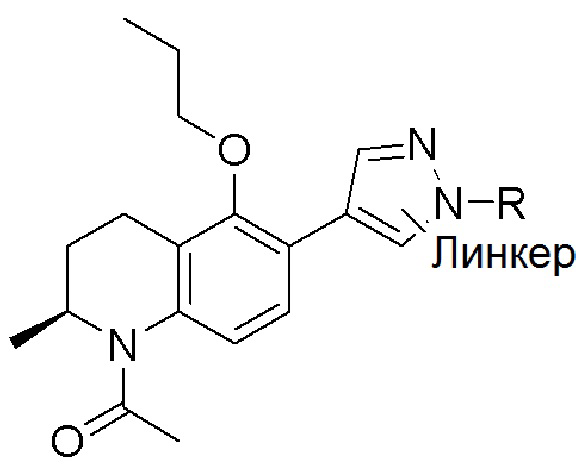
[00170] 8. Тип триазолопиразина (WO 2015/067770)
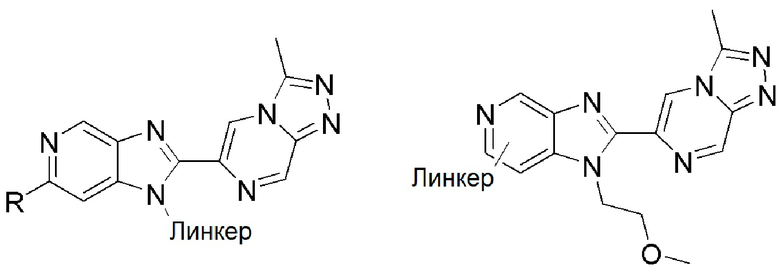
[00171] 9. Тип пиридона (WO 2015/022332)
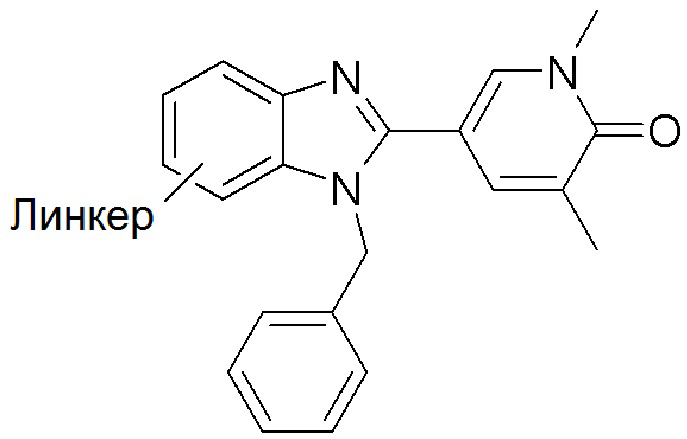
[00172] 10. Тип хиназолинона (WO 2015/015318)
[00173] 11. Тип дигидропиридопиразинона (WO 2015/011084)
[00174] Где в любом из вариантов R, L или линкер обозначает участок для присоединения линкерной группы L или группы -(L-CLM).
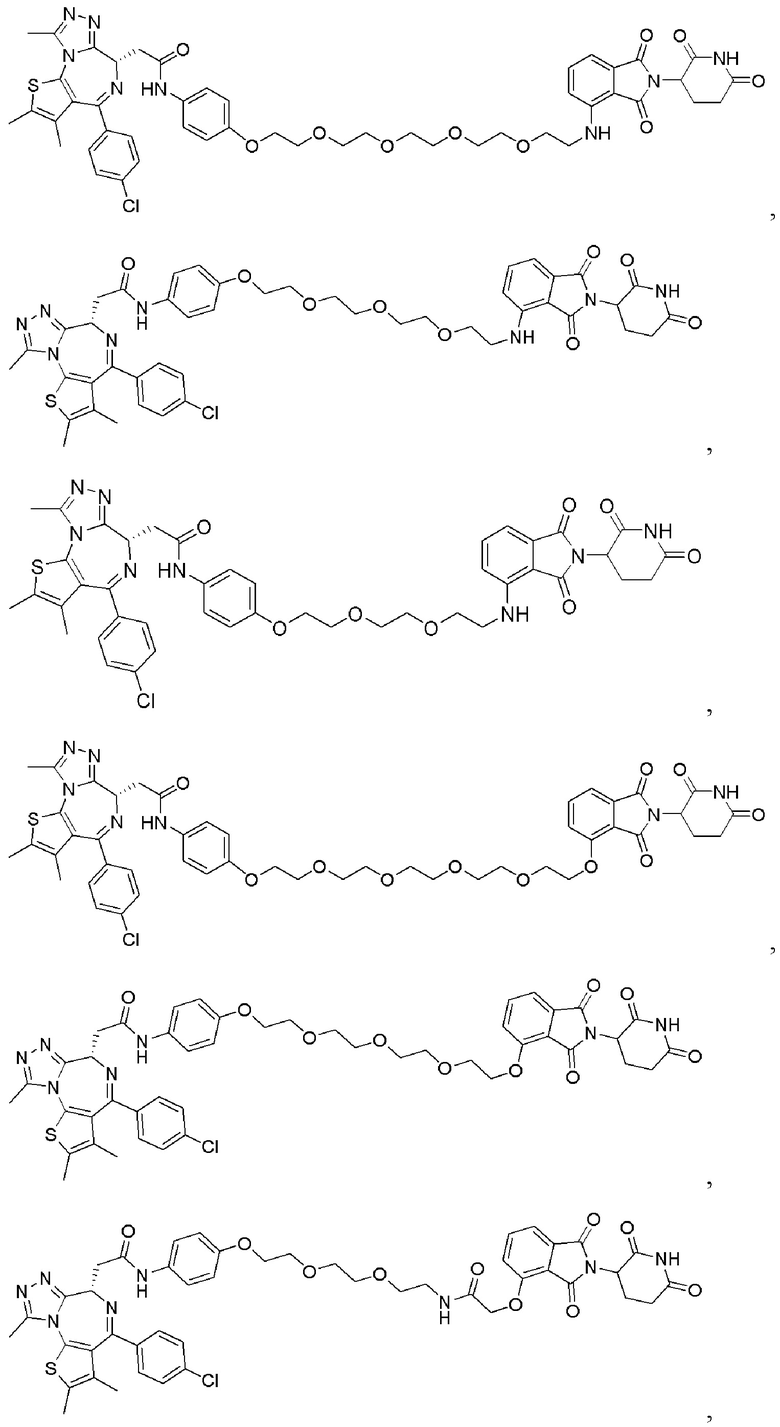
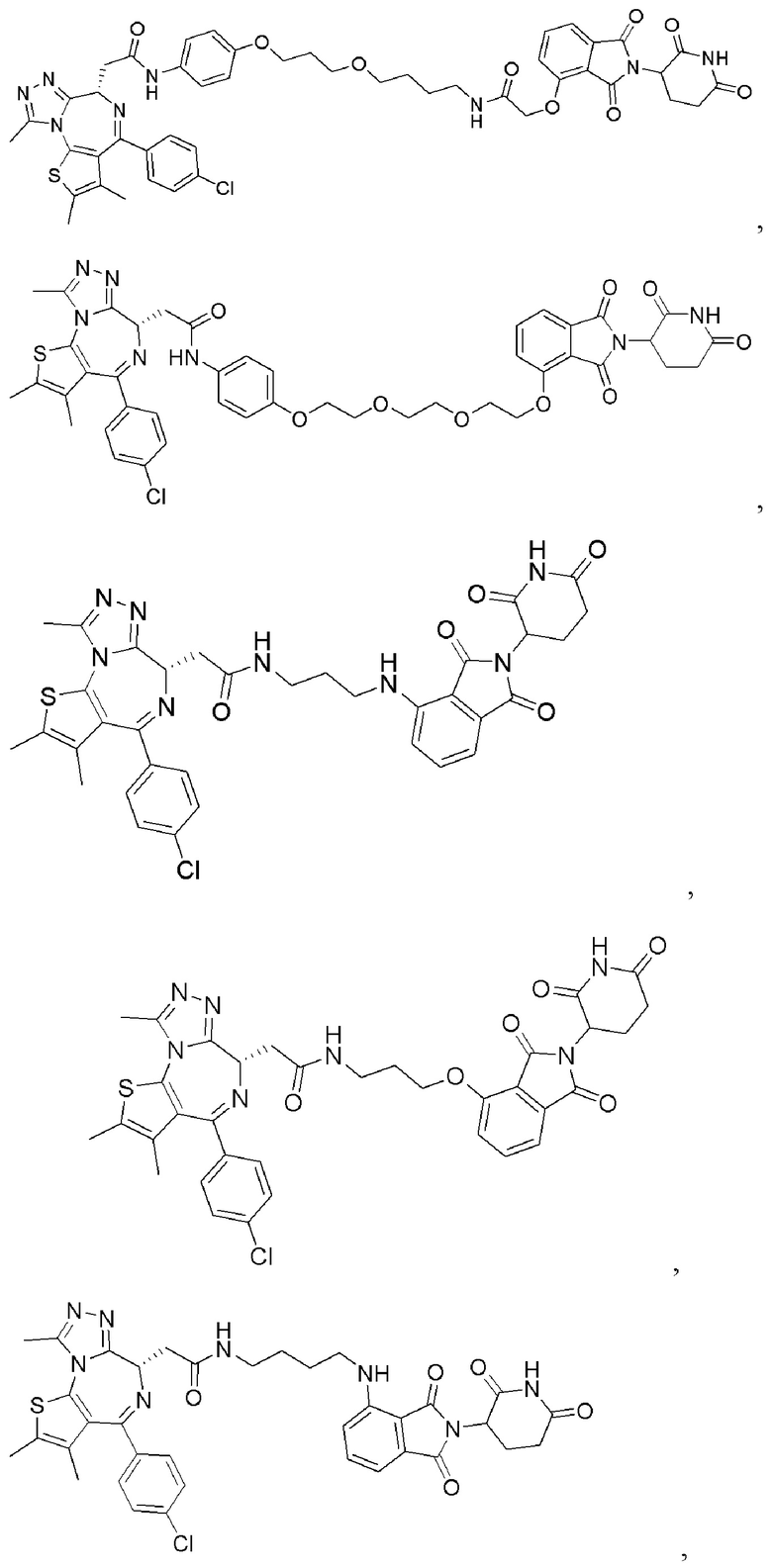
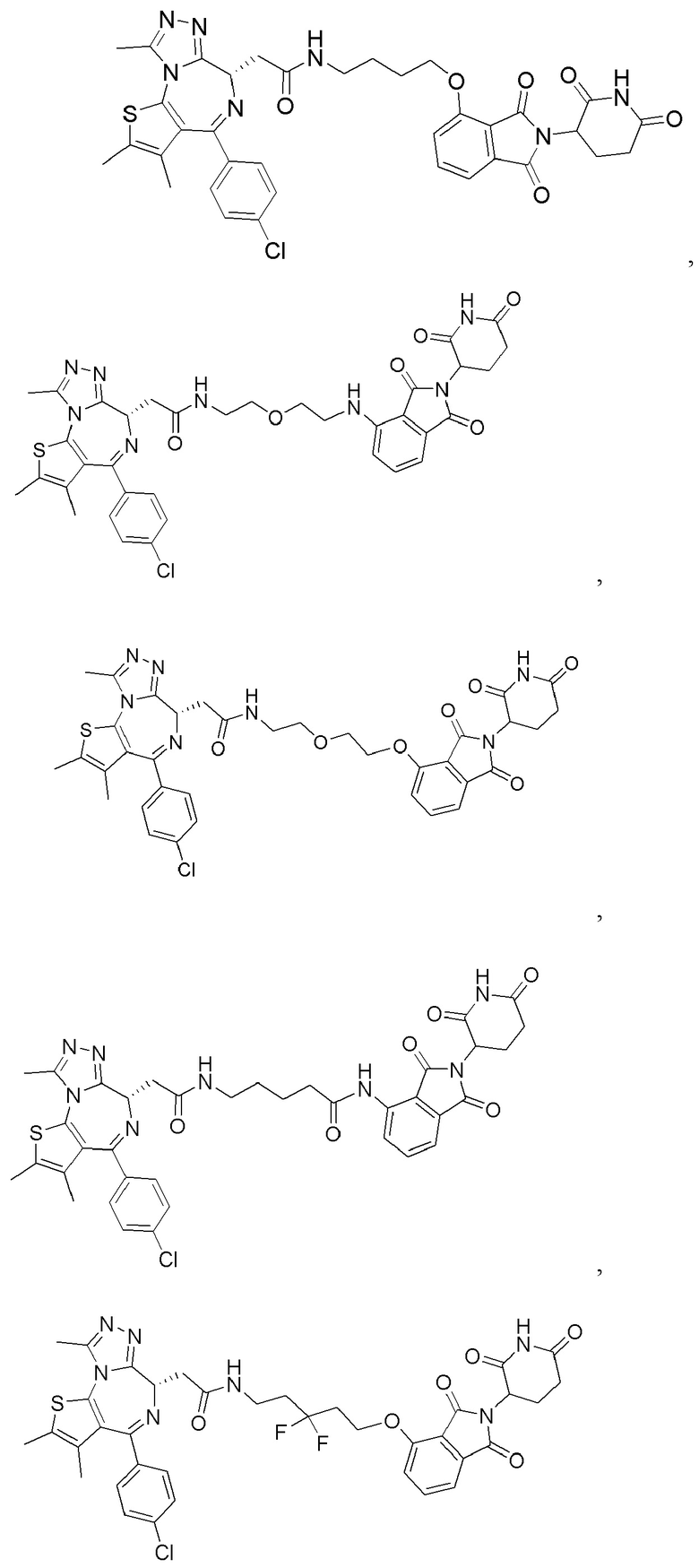
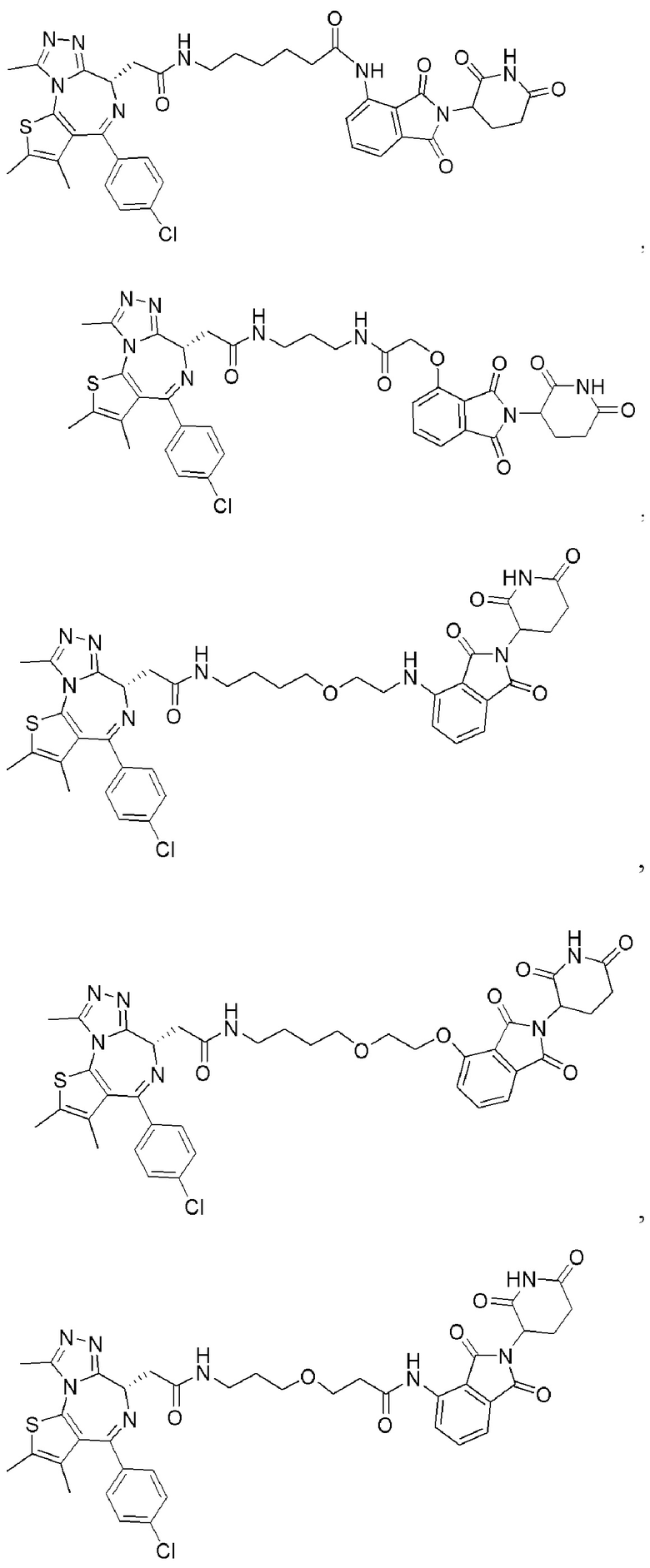
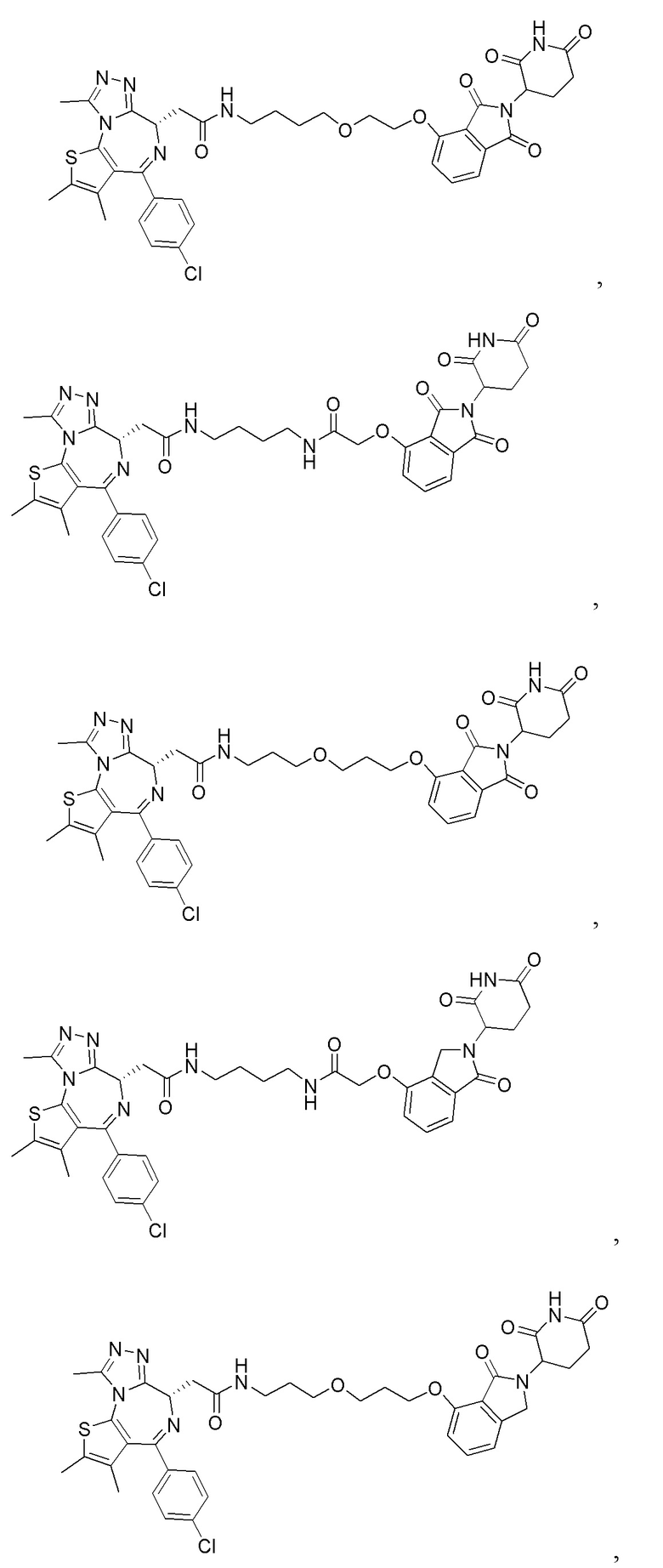
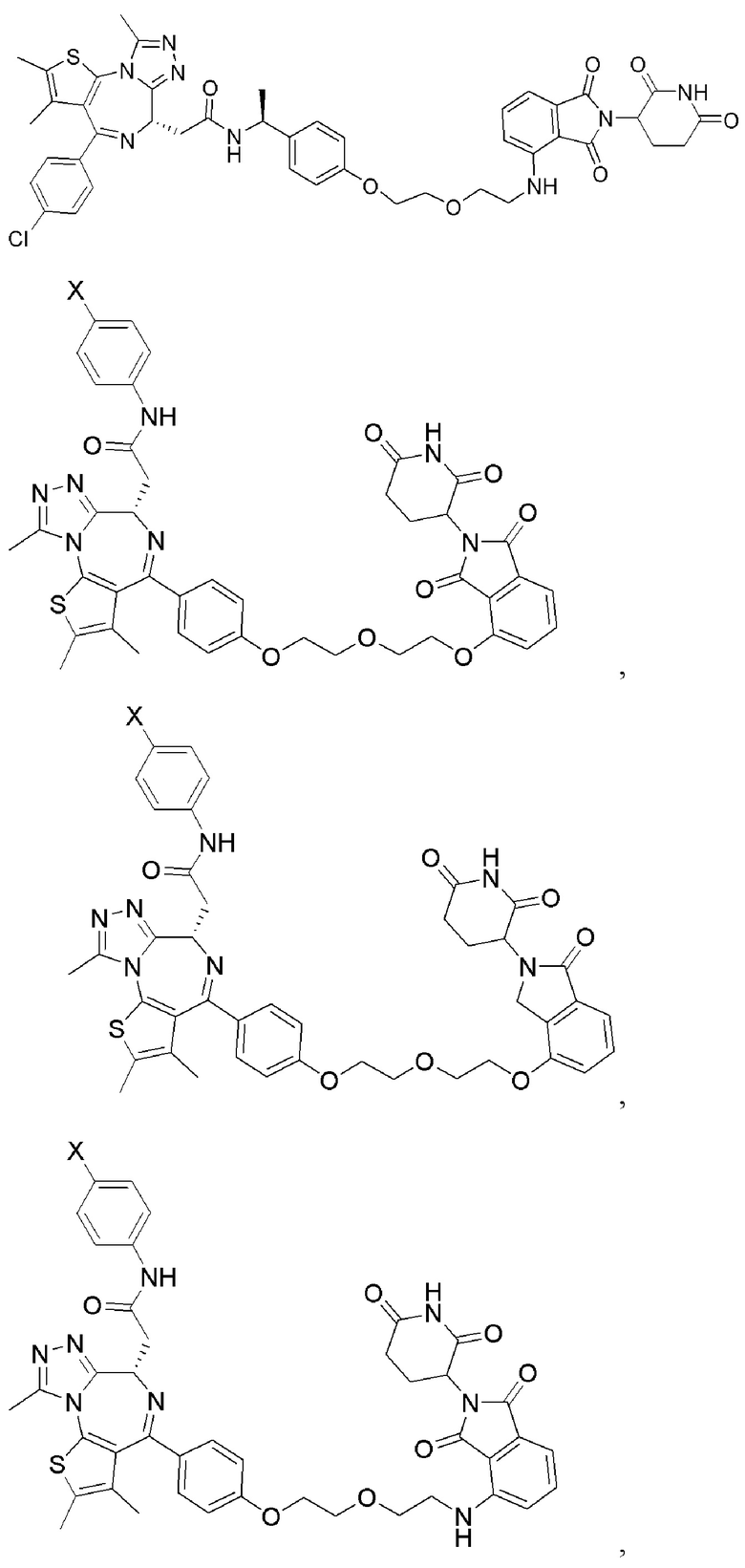
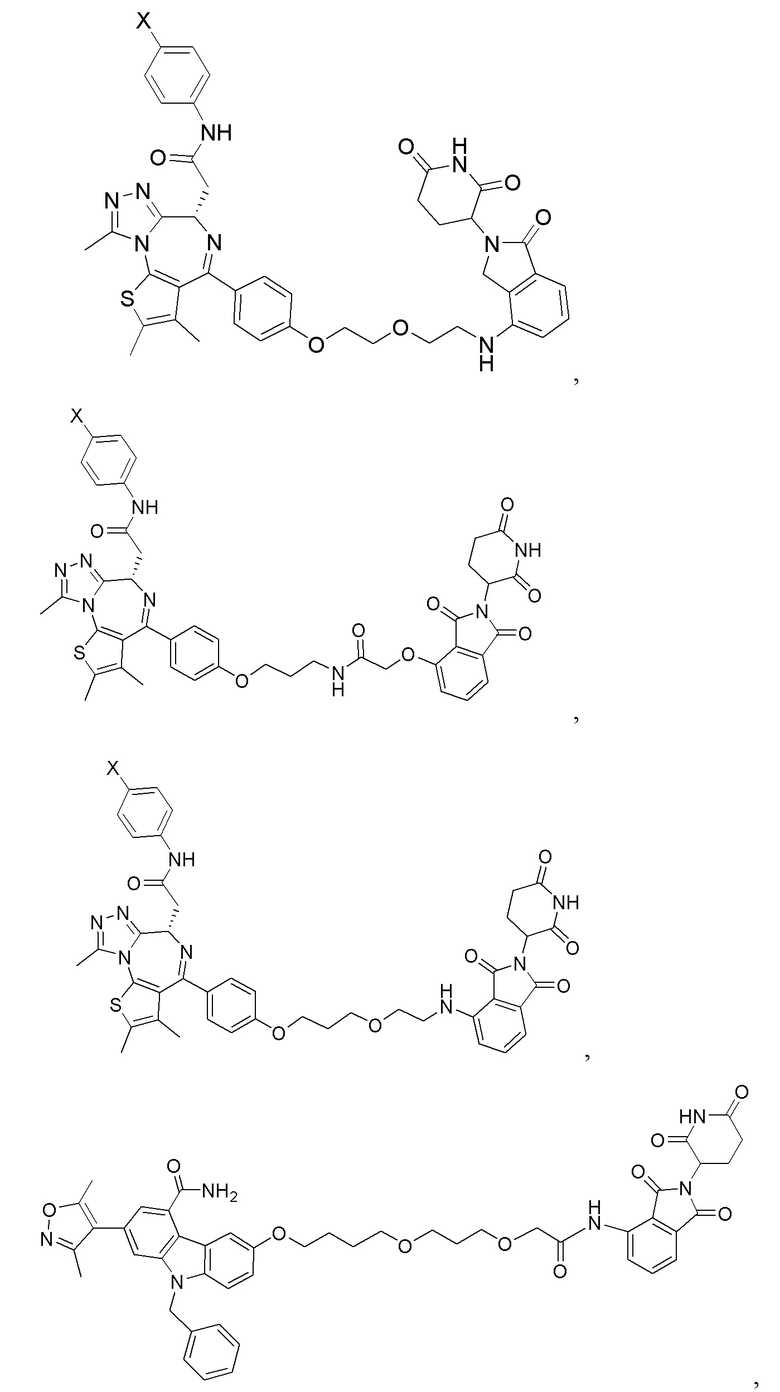
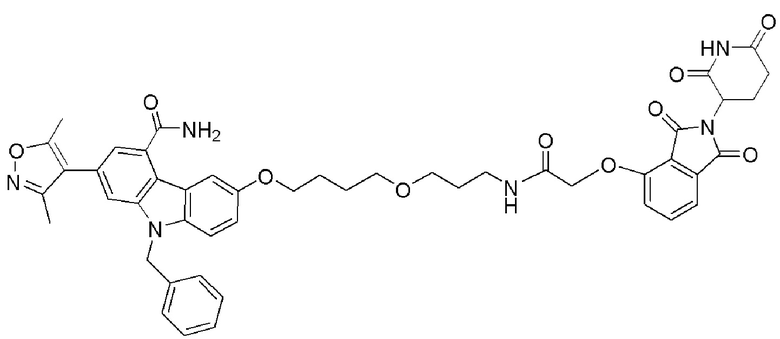
и
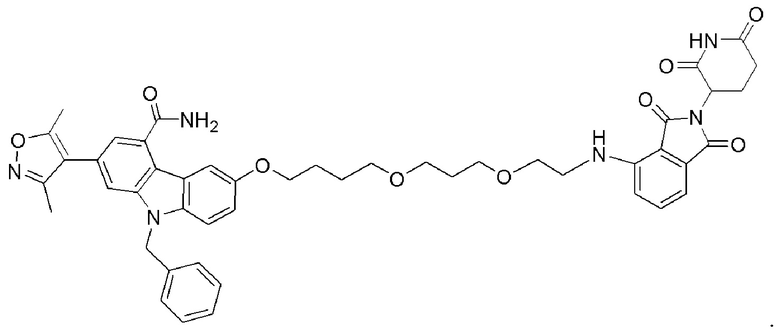
V. Ингибиторы HDAC
[00176] Ингибиторы HDAC (дериватизированные) включают, но без ограничения, следующее.
[00177] 1. Finnin, M.S. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188-193 (1999):
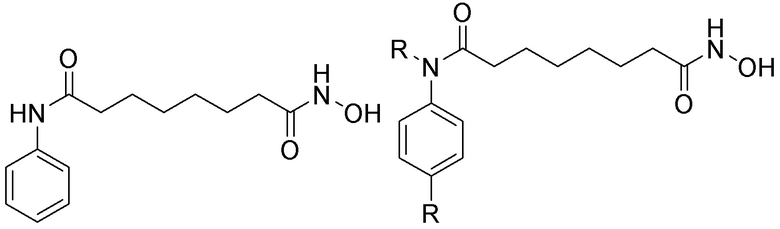
(Дериватизированные, где "R", в каждом случае, означает место присоединения, например, линкерной группы L или группы -(L-CLM)), и
[00178] 2. Соединения, определенные формулой (I) из PCТ WO 2002/22577 (ʺИНГИБИТОРЫ ДЕАЦЕТИЛАЗЫʺ). Дериватизированные, где линкерная группа L или группа -(L-CLM) присоединена, например, через гидроксильную группу.
VI. Ингибиторы лизинметилтрансферазы человека
[00179] Ингибиторы лизинметилтрансферазы человека включают, без ограничения, следующее.
[00180] 1. Chang et al. Structural basis for G9a-like protein lysine methyltransferase inhibition by BIX-01294. Nat Struct Biol (2009) 16(3) 312:
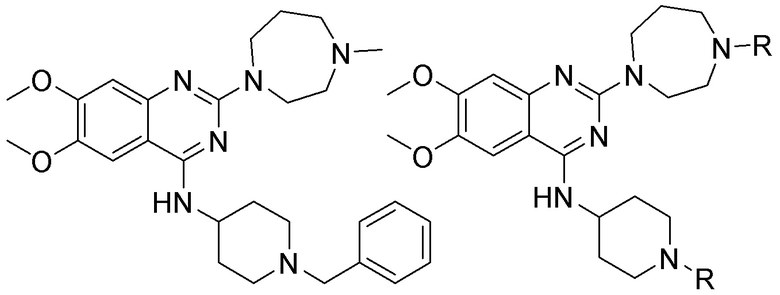
[00181] (Дериватизированные, где "R", в каждом случае, означает место присоединения, например, линкерной группы L или группы -(L-CLM));
[00182] 2. Liu F. et al. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J Med Chem (2009) 52(24) 7950:
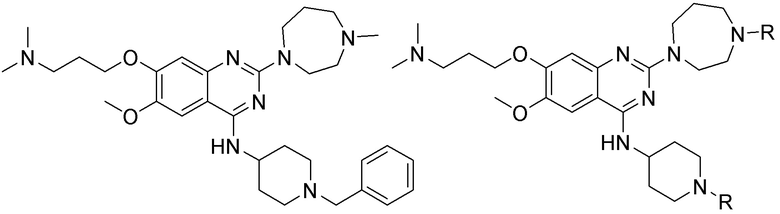
[00183] (Дериватизированные, где "R" означает возможное место присоединения, например, линкерной группы L или группы -(L-CLM));
[00184] 3. Азацитидин (дериватизированный) (4-амино-1-β-D-рибофуранозил-1,3,5-триазин-2(1H)-он) (Дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через гидрокси или аминогруппы); и
[00185] 4. Децитабин (дериватизированный) (4-амино-1-(2-деокси-b-D-эритропентофуранозил)-1,3,5-триазин-2(1H)-он) (Дериватизированный, где линкерная группа L или группа -(L-CLM) присоединена, например, через простую эфирную или гидроксигруппы или на аминогруппе).
VII. Ингибиторы ангиогенеза
[00186] Ингибиторы ангиогенеза включают, без ограничения, следующее.
[00187] 1. GA-1 (дериватизированный) и его производные и аналоги, имеющие структуры и связывающиеся с линкерами как описано в Sakamoto, et al., Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation, Mol Cell Proteomics 2003 Dec;2(12):1350-8;
[00188] 2. Эстрадиол (дериватизированный), который может быть связан с линкерной группой L или с группой -(L-CLM), как в целом описано в Rodriguez-Gonzalez, et al., Targeting steroid hormone receptors for ubiquitination and degradation in breast and prostate cancer, Oncogene (2008) 27, 7201-7211;
[00189] 3. Эстрадиол, тестостерон (дериватизированный) и родственные производные, включая, но не ограничиваясь ими, DHT и его производные и аналоги, имеющие структуры и связывающиеся с линкерной группой L или с группой -(L-CLM), как в целом описано в Sakamoto, et al., Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation, Mol Cell Proteomics 2003 Dec; 2(12):1350-8; и
[00190] 4. Овалицин, фумагиллин (дериватизированный) и их производные и аналоги, имеющие структуры и связывающиеся с линкерной группой L или с группой -(L-CLM), как в целом описано в Sakamoto, et al., Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation Proc Natl Acad Sci USA. 2001 Jul 17;98(15):8554-9 и в патенте США No. 7208157.
VIII. Иммунодепрессивные соединения
[00191] Иммунодепрессивные соединения включают, но без ограничения, следующее.
[00192] 1. AP21998 (дериватизированный), имеющий структуру и связывающийся с линкерной группой L или с группой -(L-CLM), как в целом описано в Schneekloth, et al., Chemical Genetic Control of Protein Levels: Selective in Vivo Targeted Degradation, J. AM. CHEM. SOC. 2004, 126, 3748-3754;
[00193] 2. Глюкокортикоиды (например, гидрокортизон, преднизон, преднизолон и метилпреднизолон) (Дериватизированные, где линкерная группа L или группа -(L-CLM) должна быть связана, например, с любым из гидроксилов) и дипропионат беклометазона (дериватизированный, где линкерная группа или группа -(L-CLM) связана, например, с пропионатом);
[00194] 3. Метотрексат (Дериватизированный, где линкерная группа или группа -(L-CLM) может быть связана, например, с любым из концевых гидроксилов);
[00195] 4. Циклоспорин (Дериватизированный, где линкерная группа или группа -(L-CLM) может быть связана, например, на любой из бутильных групп);
[00196] 5. Такролимус (FK-506) и рапамицин (Дериватизированные, где линкерная группа L или группа -(L-CLM) может быть связана, например, на одной из метоксигрупп); и
[00197] 6. Актиномицины (Дериватизированные, где линкерная группа L или группа -(L-CLM) может быть связана, например, на одной из изопропильных групп).
IX. Соединения, нацеленные на арилуглеводородный рецептор (AHR)
[00198] Соединения, нацеленные на арилуглеводородный рецептор (AHR) включают, но без ограничения, следующее.
[00199] 1. Апигенин (дериватизированный так, чтобы связываться с линкерной группой L или с группой -(L-CLM), как в целом показано в Lee, et al., Targeted Degradation of the Aryl Hydrocarbon Receptor by the PROTAC Approach: A Useful Chemical Genetic Tool, ChemBioChem Volume 8, Issue 17, pages 2058-2062, November 23, 2007); и
[00200] 2. SR1 и LGC006 (дериватизированные так, что линкерная группа L или группа -(L-CLM) является связанной), описанные в Boitano, et al., Aryl Hydrocarbon Receptor Antagonists Promote the Expansion of Human Hematopoietic Stem Cells, Science 10 September 2010: Vol. 329 no. 5997 pp. 1345-1348.
X. Соединения, нацеленные на RAF рецептор (киназу)
[00201]
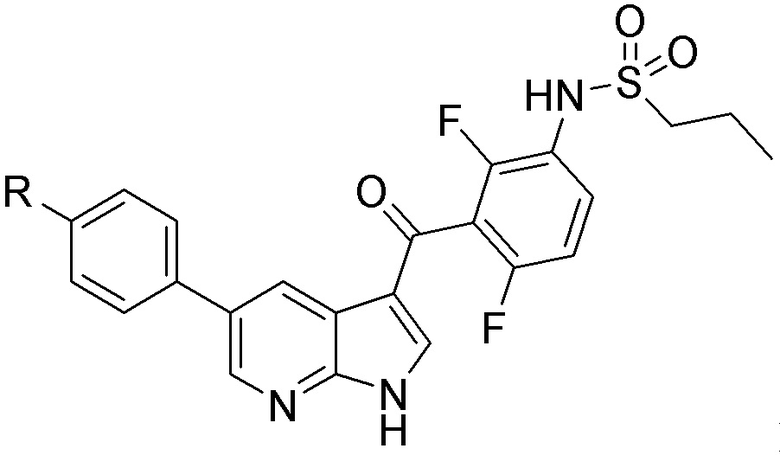
PLX4032
[00202] (Дериватизированный, где ʺRʺ означает место присоединения, например, линкерной группы L или группы-(L-CLM)).
XI. Соединения, нацеленные на FKBP
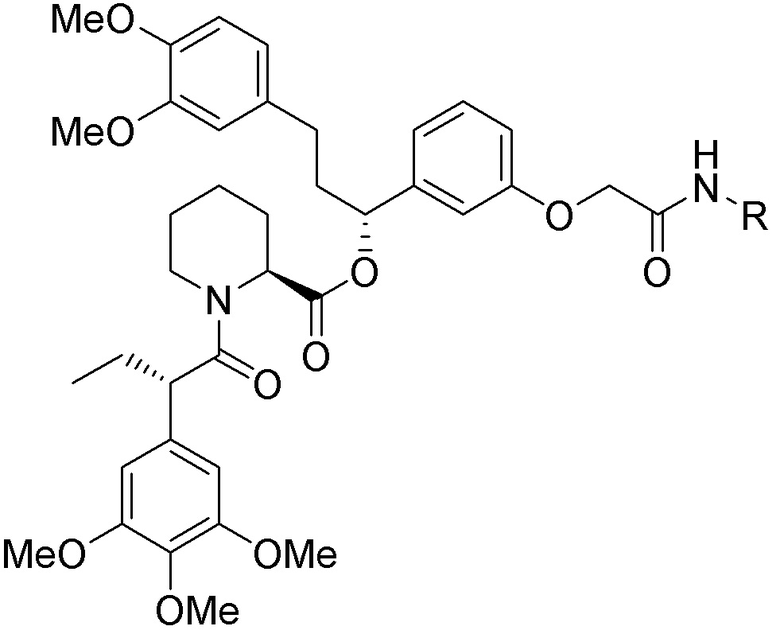
[00203] (Дериватизированный, где ʺRʺ означает место присоединения, например, линкерной группы L или группы-(L-CLM)).
XII. Соединения, нацеленные на андрогеновый рецептор (AR)
[00204] 1. RU59063 лиганд (дериватизированный) андрогенового рецептора
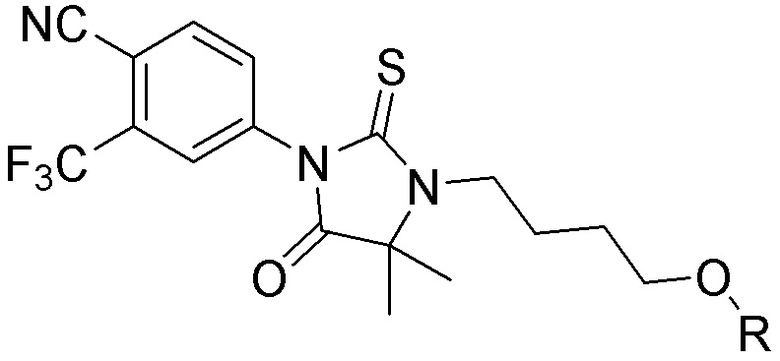
[00205] (Дериватизированный, где ʺRʺ означает место присоединения, например, линкерной группы L или группы-(L-CLM)).
[00206] 2. SARM лиганд (дериватизированный) андрогенового рецептора
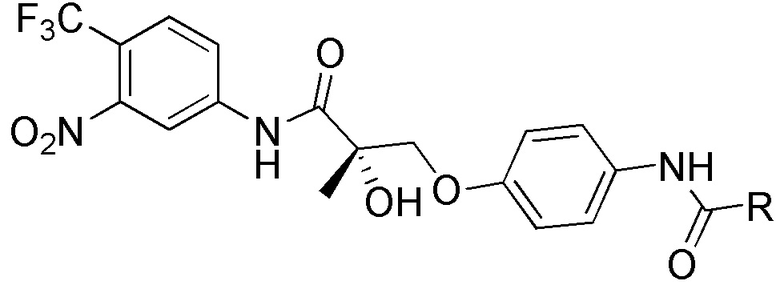
[00207] (Дериватизированный, где ʺRʺ означает место присоединения, например, линкерной группы L или группы-(L-CLM)).
[00208] 3. Лиганд андрогенового рецептора DHT (дериватизированный)
[00209] (Дериватизированный, где ʺRʺ означает место присоединения, например, линкерной группы L или группы-(L-CLM)).
[00210] 4. Лиганд MDV3100 (дериватизированный)
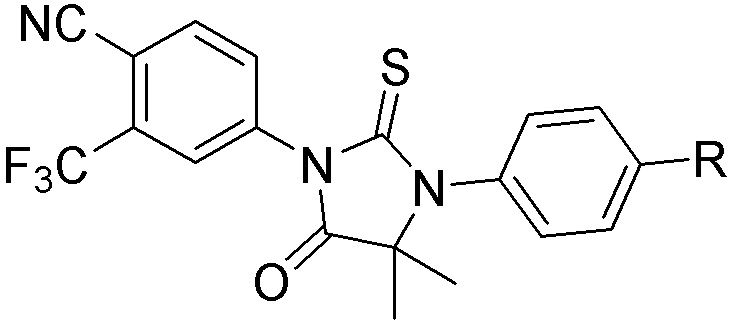
[00211] 5. Лиганд ARN-509 (дериватизированный)
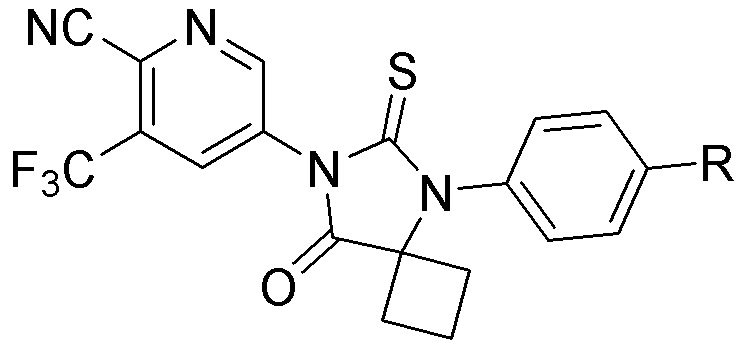
[00212] 6. Гексагидробензизоксазолы
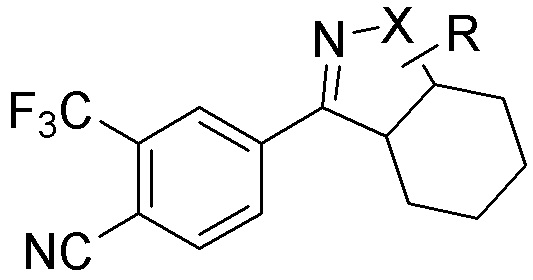
[00213] 7. Тетраметилциклобутаны
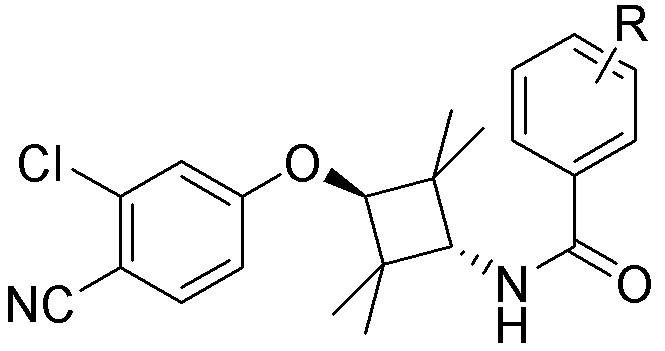
XIII. Соединения, нацеленные на эстрогеновый рецептор (ER) ICI-182780
[00214] 1. Лиганд эстрогенового рецептора
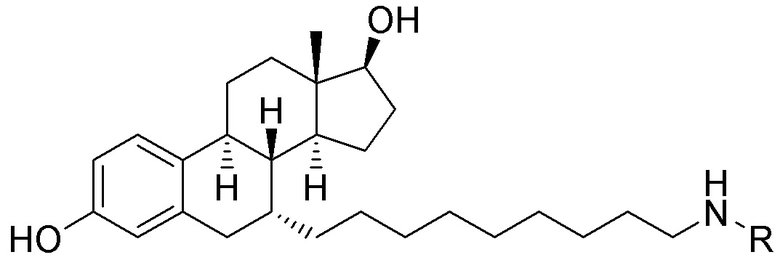
[00215] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM)).
XIV. Соединения, нацеленные на рецептор тироидного гормона (TR)
[00216] Лиганд рецептора тироидного гормона (дериватизированный)
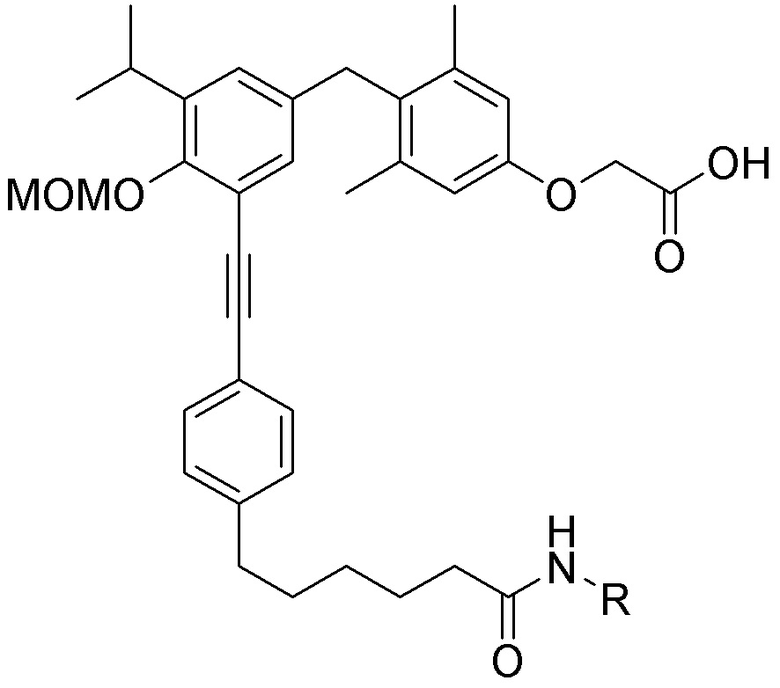
[00217] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM) и MOMO означает метоксиметоксигруппу).
XV. Соединения, нацеленные на HIV-протеазу
[00218] 1. Ингибитор HIV протеазы (дериватизированный)
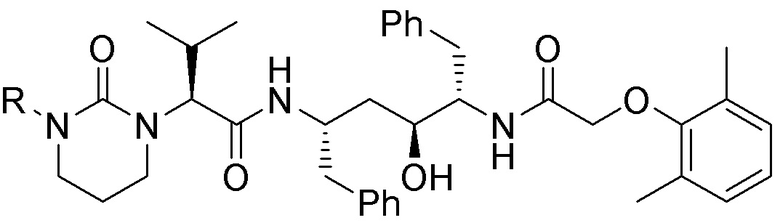
[00219] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM)). См. J. Med. Chem. 2010, 53, 521-538.
[00220] 2. Ингибитор HIV протеазы
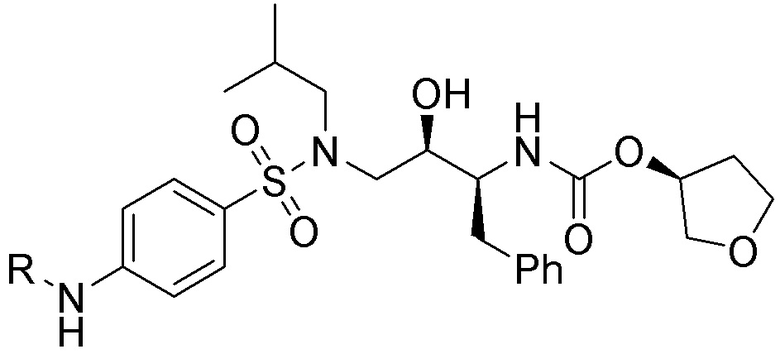
[00221] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM)). См. J. Med. Chem. 2010, 53, 521-538.
XVI. Соединения, нацеленные на HIV интегразу
[00222] 1. Ингибитор HIV интегразы (дериватизированный)
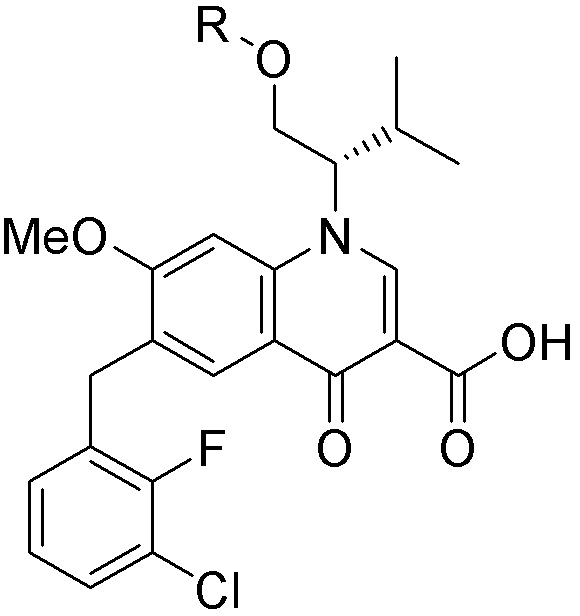
[00223] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM)). См. J. Med. Chem. 2010, 53, 6466.
[00224] 2. Ингибитор HIV интегразы (дериватизированный)
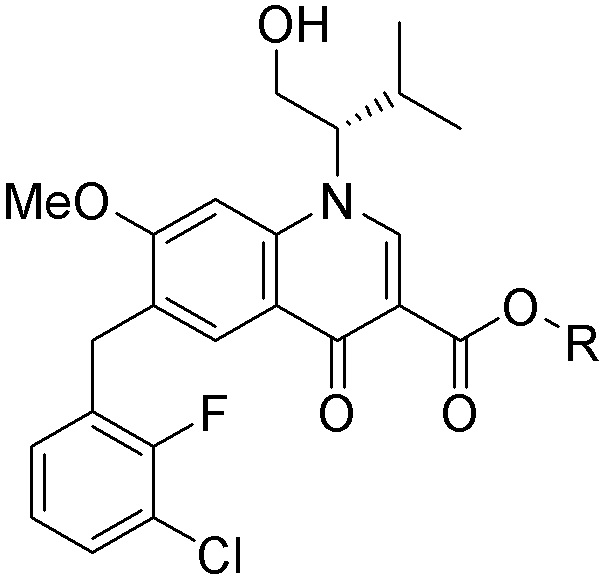
[00225] 3. Ингибитор HIV интегразы изетентресс (дериватизированный)
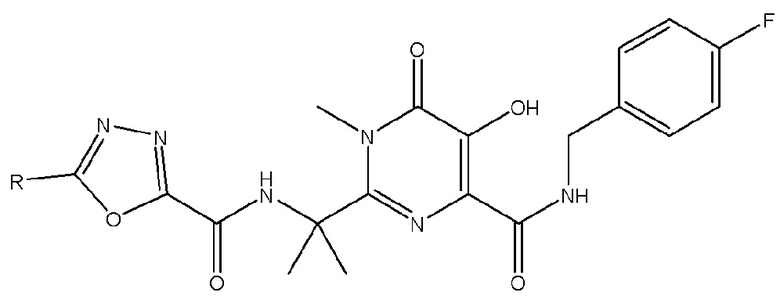
[00226] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM)). См. J. Med. Chem. 2010, 53, 6466.
XVII. Соединения, нацеленные на HCV протеазу
[00227] 1. Ингибиторы HCV протеазы (дериватизированные)
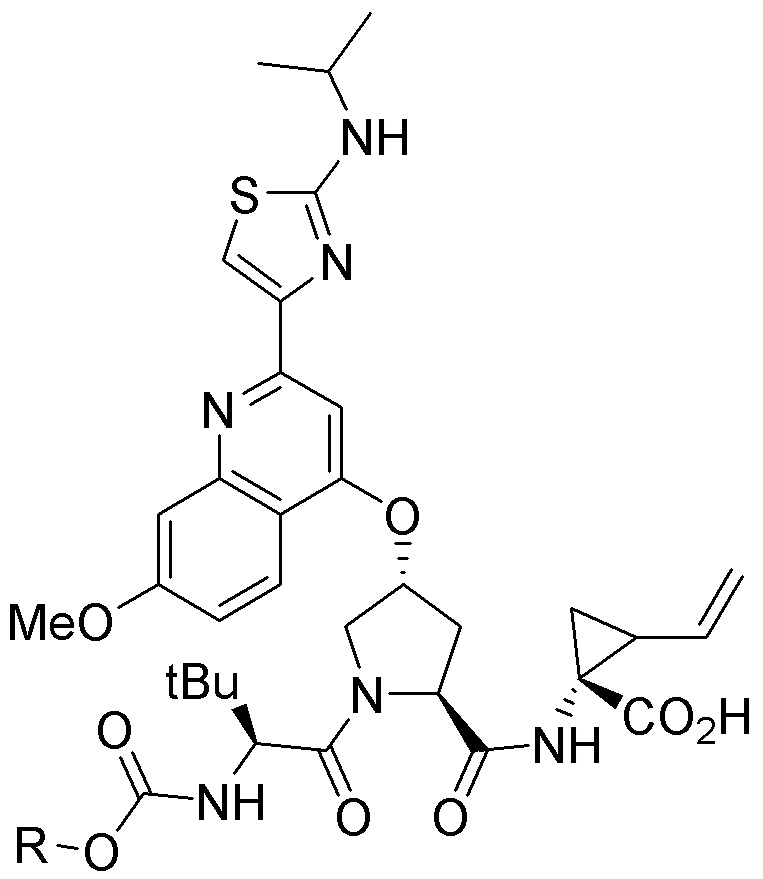
[00228] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM)).
XVIII. Соединения, нацеленные на ацилпротеинтиоэстеразу-1 и ацилпротеинтиоэстеразу-2 (APT1 и APT2)
[00229] 1. Ингибиторы APT1 и APT2 (дериватизированные)
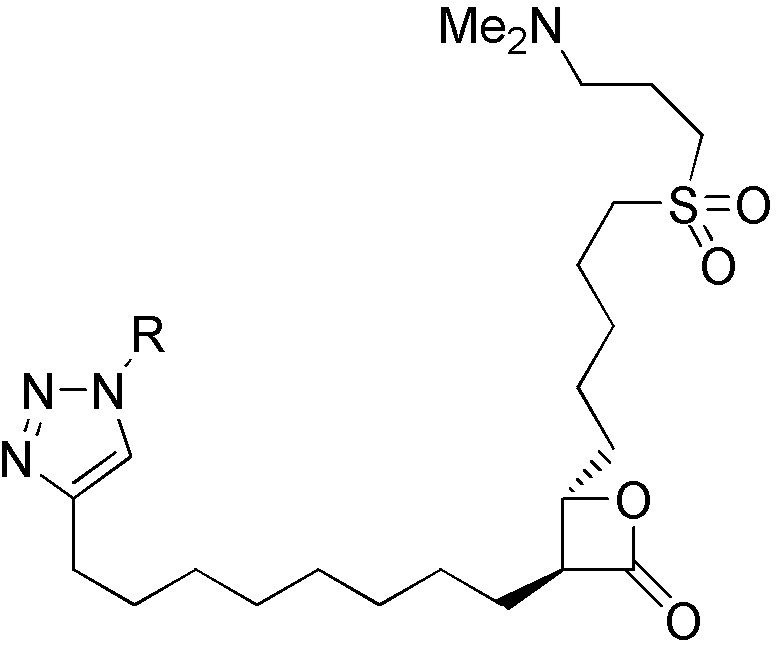
[00230] (Дериватизированный, где ʺRʺ означает место присоединения линкерной группы L или группы-(L-CLM)). См. Angew. Chem. Int. Ed. 2011, 50, 9838-9842, где L является линкерной группой, как описано здесь, и указанная группа CLM является такой, как описано здесь, при этом -(L-CLM) связывает группу CLM с группой PTM, как описано здесь.
Терапевтические композиции
[00231] Фармацевтические композиции, содержащие комбинацию эффективного количества по крайней мере одного бифункционального соединения в соответствии с данным изобретением и одного или более из описанных здесь других соединений, все в эффективных количествах, в сочетании с фармацевтически эффективным количеством носителя, добавки или эксципиента, представляют еще один аспект настоящего изобретения.
[00232] Настоящее изобретение включает, где это применимо, композиции, содержащие фармацевтически приемлемые соли, в частности, кислотные или основные аддитивные соли соединений, как описано здесь. Кислоты, которые применяются для получения фармацевтически приемлемых кислотных аддитивных солей указанных выше основных соединений, включают такие, которые не образуют токсичных кислотных аддитивных солей, т.е. соли, содержащие фармакологически приемлемые анионы, такие как гидрохлорид, гидробромид, гидроиодид, нитрат, сульфат, бисульфат, фосфат, кислый фосфат, ацетат, лактат, цитрат, кислый цитрат, тартрат, битартрат, сукцинат, малеат, фумарат, глюконат, сахарат, бензоат, метансульфонат, этансульфонат, бензолсульфонат, п-толуолсульфонат и памоат [т.е. 1,1'-метиленбис-(2-гидрокси-3-нафтоат)], среди прочих.
[00233] Фармацевтически приемлемые основные аддитивные соли также могут быть использованы для получения фармацевтически приемлемых солей соединений или производных. Химические основания, которые могут применяться для получения фармацевтически приемлемых основных солей соединений, которые являются кислыми по природе, включают такие, которые не образуют токсичных основных солей с такими соединениями. Такие нетоксичные основные соли включают, но без ограничения, соли, полученные из таких фармакологически приемлемых катионов, как катионы щелочного металла (например, калия и натрия) и катионы щелочноземельного металла (например, кальция, цинка и магния), аддитивные соли аммония и водорастворимого амина, такие как N-метилглюкамин-(меглумин) и низший алканоламмоний, и другие основные соли фармацевтически приемлемых органических аминов, среди прочих.
[00234] Описанные здесь соединения в определенных вариантах могут вводиться одной или несколькими единичными дозами пероральным, парентеральным или местным путем. Введение активного соединения может варьироваться от непрерывного (внутривенное вливание) до нескольких пероральных введений в сутки (например, Q.I.D.), и может включать пероральное, местное, парентеральное, внутримышечное, внутривенное, подкожное, чрескожное (с включением агента для улучшения проникновения), буккальное, подъязычное введение и введение в суппозиториев, среди прочих путей введения. Пероральные таблетки с энтеросолюбильной оболочкой также могут применяться для улучшения биодоступности соединений при пероральном пути введения. Наиболее эффективная лекарственная форма будет зависеть от фармакокинетики конкретного выбранного агента и от тяжести заболевания у пациента. Также может применяться введение соединений в соответствии с данным изобретением в виде спреев, распылений или аэрозолей для интраназального, интратрахеального или легочного введения. Поэтому данное изобретение также относится к фармацевтическим композициям, содержащим эффективное количество соединения в соответствии с данным изобретением, необязательно в сочетании с фармацевтически приемлемым носителем, добавкой или эксципиентом. Соединения в соответствии с данным изобретением могут вводиться в формах для немедленного высвобождения, промежуточного высвобождения или замедленного или контролируемого высвобождения. Формы с замедленным или контролируемым высвобождением предпочтительно вводят перорально, но также в виде суппозиториев и чрескожных или других местных форм. Внутримышечные инъекции в форме липосом также могут применяться для контроля или замедления высвобождения соединения в месте введения.
[00235] Композиции в соответствии с данным изобретением могут быть приготовлены обычным способом с использованием одного или более фармацевтически приемлемых носителей, и также могут вводиться в виде композиций с контролируемым высвобождением. Фармацевтически приемлемые носители, которые могут применяться в этих фармацевтических композициях, включают, без ограничения, ионообменные соединения, окись алюминия, стеарат алюминия, лецитин, сывороточные белки, такие как человеческой сывороточный альбумин, буферные вещества, такие как фосфаты, глицин, сорбиновую кислоту, сорбат калия, частичные глицеридные смеси насыщенных растительных жирных кислот, воду, соли или электролиты, такие как сульфат проламина, гидрофосфат динатрия, гидрофосфат калия, хлорид натрия, соли цинка, коллоидную двуокись кремния, трисиликат магния, поливинилпирролидон, вещества на основе целлюлозы, полиэтиленгликоль, карбоксиметилцеллюлозу натрия, полиакрилаты, воски, блоксополимеры полиэтилена-полиоксипропилена, пропиленгликоль и ланолин.
[00236] Описанные здесь композиции могут вводиться перорально, парентерально, ингаляцией, местно, ректально, назально, буккально, вагинально или через имплантированный резервуар. Термин "парентерально" в данном описании включает подкожные, внутривенные, внутримышечные, внутрисуставные, внутрисиновиальные, внутригрудинные, подоболочечные, внутрипеченочные, внутриочаговые и внутричерепные инъекции или вливания. Предпочтительно, композиции водят перорально, внутрибрюшинно или внутривенно.
[00237] Стерильные формы для инъекций описанных композиций могут быть водными или масляными суспензиями. Эти суспензии могут быть приготовлены способами, известными в данной области техники, с применением подходящих диспергирующих или смачивающих агентов и суспендирующих агентов. Стерильным препаратом для инъекций также может быть стерильный раствор или суспензия для инъекций в нетоксичном, парентерально приемлемом разбавителе или растворителе, например, в виде раствора в 1,3-бутандиоле. В качестве приемлемых носителей и растворителей могут применяться вода, раствор Рингера и изотонический раствор хлорида натрия. Кроме того, стерильные нелетучие масла обычно применяют в качестве растворителя или суспендирующей среды. Для этой цели может применяться любое инертное нелетучее масло, включая синтетические моно- или диглицериды. Жирные кислоты, такие как олеиновая кислота, и их глицеридные производные применяют для получения инъекционных форм в качестве натуральных фармацевтически приемлемых масел, таких как оливковое масло или касторовое масло, особенно в их полиоксиэтилированных вариантах. Такие масляные растворы или суспензии также могут содержать длинноцепочечный спиртовой разбавитель или дисперсант, такой как Ph. Helv, или аналогичный спирт.
[00238] Описанные здесь фармацевтические композиции могут вводиться перорально в любой перорально приемлемой дозированной лекарственной форме, включая, но не ограничиваясь ими, капсулы, таблетки, водные суспензии или растворы. В таблетках для перорального введения применяемые носители обычно включают лактозу и кукурузный крахмал. Также обычно добавляли смазывающие агенты, такие как стеарат магния. Для перорального введения в виде капсул, применяемые разбавители включают лактозу и высушенный кукурузный крахмал. Если для перорального применения требуются водные суспензии, активный ингредиент объединяют с эмульгирующими и суспендирующими агентами. При желании также могут быть добавлены определенные подсластители, вкусовые добавки или красители.
[00239] Альтернативно, описанные здесь фармацевтические композиции могут вводиться в форме суппозиториев для ректального введения. Они могут быть получены смешиванием агента с подходящим нераздражающим эксципиентом, который является твердым при комнатной температуре, но жидким при ректальной температуре и, поэтому, он плавится в прямой кишке с выделением лекарственного средства. Такие материалы включают масло какао, пчелиный воск и полиэтиленгликоли.
[00240] Описанные здесь фармацевтические композиции также могут вводиться местно. Подходящие местные композиции легко получают для каждой из этих областей или органов. Местное введение в нижний кишечный тракт может проводиться в виде ректальных суппозиториев (см. выше) или в виде подходящего препарата для введения с помощью клизмы. Также могут применяться приемлемые для местного введения чрескожные пластыри.
[00241] Для местного введения фармацевтические композиции могут быть приготовлены в виде подходящей мази, содержащей активный ингредиент, суспендированный или растворенный в одном или более носителях. Носители для местного введения соединений по изобретению включают, но не ограничиваясь, минеральное масло, жидкий вазелин, белый вазелин, пропиленгликоль, полиоксиэтилен, соединение полиоксипропилена, эмульгируемый воск и воду. В определенных предпочтительных аспектах соединения могут быть включены в стент, который хирургически имплантируют пациенту для ингибирования или снижения у пациента вероятности возникающей в стенте окклюзии.
[00242] В качестве альтернативы, фармацевтические композиции могут быть приготовлены в виде подходящего лосьона или крема, содержащего активные компоненты, суспендированные или растворенные в одном или нескольких фармацевтически приемлемых носителях. Подходящие носители включают, но не ограничиваются ими, минеральное масло, моностеарат сорбитана, полисорбат 60, воск цетиловых эфиров, цетеариловый спирт, 2-октилдодеканол, бензиловый спирт и воду.
[00243] Для офтальмологического применения фармацевтические композиции могут быть приготовлены в виде микронизированных суспензий в изотоническом, pH скорректированном стерильном солевом растворе или, предпочтительно, в виде растворов в изотоническом, pH-скорректированном стерильном солевом растворе, с или без консерванта, такого как хлорид бензалкония. Альтернативно, для офтальмологического применения, фармацевтические композиции могут быть составлены в мази, такой как вазелин.
[00244] Фармацевтические композиции, описанные здесь, также могут быть введены в виде назального аэрозоля или путем ингаляции. Такие композиции получают способами, хорошо известными в области фармацевтических составов, и они могут быть получены в виде растворов в солевом растворе, с использованием бензилового спирта или других подходящих консервантов, промоторов абсорбции для улучшения биодоступности, фтороуглеродов и/или других обычных солюбилизирующих или диспергирующих агентов.
[00245] Количество соединения в фармацевтической композиции по изобретению, которое можно объединять с носителем для получения единичной дозированной формы, изменяется в зависимости от хозяина и заболевания, которое лечится, и от конкретного способа введения. Предпочтительно, композиции должны быть притовлены таким образом, чтобы они содержали от приблизительно 0,05 миллиграмма до приблизительно 750 миллиграммов или более, более предпочтительно, от приблизительно 1 миллиграмма до приблизительно 600 миллиграммов и даже более предпочтительно - от приблизительно 10 миллиграммов до приблизительно 500 миллиграммов активного ингредиента, отдельно или в сочетании с по крайней мере одним другим соединением в соответствии с настоящим изобретением.
[00246] Следует также понимать, что конкретная доза и режим лечения для любого конкретного пациента будет зависеть от множества факторов, включая активность конкретного применяемого соединения, возраст пациента, массу тела, общее состояние здоровья, пол, питание, время введения, скорость выведения, сочетание лекарственных средств и мнение лечащего врача, а также тяжесть конкретного заболевания или состояния, подлежащего лечению.
[00247] Пациент или субъект, нуждающийся в лечении с применением соединений в соответствии с данным изобретением, может лечиться введением этому пациенту (субъекту) эффективного количества соединения в соответствии с настоящим изобретением, включая его фармацевтически приемлемые соли, сольваты или полиморфы, необязательно в фармацевтически приемлемом носителе или разбавителе, отдельно или в сочетании с другими известными агентами, стимулирующими эритропоэз, как указано здесь.
[00248] Эти соединения могут вводиться любым подходящим путем, например, перорально, парентерально, внутривенно, внутрикожно, подкожно или местно, включая чрескожно, в жидкой, кремовой, гелевой или твердой форме или в виде аэрозоля.
[00249] Активное соединение включают в фармацевтически приемлемый носитель или разбавитель в количестве, достаточном для доставки пациенту терапевтически эффективного количества для требуемого назначения, не вызывающего серьезных токсических эффектов у пациента, подвергаемому лечению. Предпочтительная доза активного соединения для всех приведенных здесь выше состояниях находится в диапазоне от приблизительно 10 нг/кг до 300 мг/кг, предпочтительно от 0,1 до 100 мг/кг в сутки, более обычно от 0,5 до приблизительно 25 мг на килограмм массы тела реципиента/пациента в сутки. Типичная дозировка для местного применения находится в диапазоне 0,01-5% масс./масс. в подходящем носителе.
[00250] Соединение обычно вводят в любой подходящей единичной дозированной лекарственной форме, включая, но без ограничения, форму, содержащую менее 1 мг, от 1 мг до 3000 мг, предпочтительно - от 5 до 500 мг активного ингредиента на единичную дозированную лекарственную форму. Пероральная доза, составляющая приблизительно 25-250 мг часто является удобной.
[00251] Активный ингредиент, предпочтительно, вводят для достижения пиковых концентраций активного соединения в плазме приблизительно 0,00001-30 мМ, предпочтительно - приблизительно 0,1-30 мкМ. Это может быть достигнуто, например, внутривенной инъекцией раствора или композиции активного ингредиента, необязательно в солевом растворе или водной среде, или его вводят в виде болюса активного ингредиента. Пероральное введение также подходит для создания эффективных концентраций активного агента в плазме.
[00252] Концентрация активного соединения в композиции лекарственного средства зависит от абсорбции, распределения, инактивации и скорости выведения лекарственного средства, а также от других факторов, известных специалисту в данной области техники. Также необходимо отметить, что величина доз также варьируются в зависимости от тяжести состояния пациента. Также понятно, что для любого конкретного субъекта конкретные режимы дозирования необходимо корректировать во времени согласно индивидуальной потребности и в соответствии с профессиональным мнением лица, осуществляющего введение или наблюдающего за результатом введения композиций, описанных здесь, и что представленные здесь интервалы концентраций являются только типовыми, и они не ограничивают объем притязаний или практику применения заявленной композиции. Активный ингредиент может вводиться один раз или может быть поделен на несколько меньших доз для введения в разные интервалы времени.
[00253] Пероральные композиции обычно включают в себя инертный разбавитель или съедобный носитель. Они могут быть заключены в желатиновые капсулы или спрессованы в таблетки. Для целей перорального терапевтического введения, активное соединение или его пролекарство может быть смешано с эксципиентами и применяться в форме таблеток, пастилок или капсул. Фармацевтически совместимые связующие агенты и/или адъюванты могут быть включены в качестве составной части композиции.
[00254] Таблетки, пилюли, капсулы, пастилки или подобные могут содержать любой из следующих ингредиентов или соединений подобной природы: связующий агент, такой как микрокристаллическая целлюлоза, трагакант или желатин; эксципиент, такой как крахмал или лактоза, диспергирующий агент, такой как альгиновая кислота, примогель (Primogel) или кукурузный крахмал; смазывающий агент, такой как стеарат магния или стеротес (Sterotes); глидант, такой как коллоидная двуокись кремния; подсластитель, такой как сахароза или сахарин; или вкусовая добавка, такая как перечная мята, метилсалицилат или апельсин. Если единичной дозированной лекарственной формой является капсула, то она может содержать, в дополнение к указанному выше материалу, жидкий носитель, такой как жирное масло. Кроме того, единичные дозированные лекарственные формы могут содержать различные другие материалы, которые модифицируют физическую форму дозированной формы, например, покрытия из сахара, шеллака или энтеросолюбильных агентов.
[00255] Активное соединение или его фармацевтически приемлемая соль может вводиться в виде компонента эликсира, суспензии, сиропа, вафли, жевательной резинки или подобного. Сироп может содержать, в дополнение к активным соединениям, сахарозу в качестве подсластителя, и некоторые консерванты, красители и вкусовые добавки.
[00256] Активное соединение или его фармацевтически приемлемые соли также могут быть смешаны с другими активными материалами, которые не ухудшают желаемое действие, или с материалами, которые дополняют желаемое действе, такими как агенты, стимулирующие эритропоэтин, включая, среди прочих, EPO и альфа дарбапоэтин. В некоторых предпочтительных вариантах изобретения, одно или несколько соединений в соответствии с изобретением вводят совместно с другим биологически активным агентом, таким как агент, стимулирующий эритропоэтин, или ранозаживляющий агент, включая антибиотик, как описано здесь.
[00257] Растворы или суспензии, используемые для парентерального, внутрикожного, подкожного или местного введения, могут включать следующие компоненты: стерильный разбавитель, такой как вода для инъекций, солевой раствор, нелетучие масла, полиэтиленгликоли, глицерин, пропиленгликоль или другие синтетические растворители; антибактериальные агенты, такие как бензиловый спирт или метилпарабены; антиоксиданты, такие как аскорбиновая кислота или бисульфит натрия; хелатирующие агенты, такие как этилендиаминтетрауксусная кислота; буферы, такие как ацетаты, цитраты или фосфаты, и агенты для регулировки тоничности, такие как хлорид натрия или декстроза. Парентеральные препараты могут быть включены в ампулы, одноразовые шприцы или во флаконы из стекла или пластика, содержащие множество доз.
[00258] При внутривенном введении предпочтительными носителями являются физиологический раствор или фосфатно-солевой буфер (PBS).
[00259] В одном варианте осуществления изобретения, активные соединения используют вместе с носителями, которые будут защищать соединение от быстрого удаления из организма, например, в виде композиции с контролируемым высвобождением, включая имплантаты и микроинкапсулированные системы доставки. Могут использоваться биоразлагаемые, биосовместимые полимеры, такие как этиленвинилацетат, полиангидриды, полигликолевая кислота, коллаген, полиортоэфиры и полимолочная кислота. Способы получения таких композиций хорошо известны специалистам в данной области техники.
[00260] Липосомные суспензии также могут быть фармацевтически приемлемыми носителями. Они могут быть получены способами, известными специалистам в данной области техники, например, как описано в патенте США No. 4522811 (который полностью включен сюда путем ссылки). Например, липосомные композиции могут быть получены растворением подходящих жиров (таких как стеароилфосфатидилэтаноламин, стеароилфосфатидилхолин, арахадоилфосфатидилхолин и холестерин) в органическом растворителе, который затем выпаривают с получением тонкой пленки высушенного жира на поверхности контейнера. Водный раствор активного соединения помещают в контейнер. Затем контейнер взбалтывают вручную для высвобождения липидного материала со стенок контейнера и для диспергирования жировых агрегатов с получением липосомной суспензии.
Терапевтические методы
[00261] В дополнительном аспекте, изобретение предоставляет терапевтические композиции, содержащие эффективное количество соединения, описанного здесь, или его соль, и фармацевтически приемлемый носитель. Терапевтические композиции модулируют деградацию белков у пациента или субъекта, например, животного, такого как человек, и они могут быть использованы для лечения или облегчения заболевания или состояния, которые модулируются за счет деградации белка, ассоциированного с этим заболеванием.
[00262] Термины ʺлечитьʺ, ʺобработкаʺ, ʺлечениеʺ и т.п. в данном описании относятся к любому действию, приносящему пользу пациенту, которому могут быть введены данные соединения, включая лечение любого заболевания или состояния, которое модулируется через белок, с которым связываются данные соединения. Заболевания или состояния, включая рак, которые можно лечить с использованием соединений согласно настоящему изобретению, изложены выше.
[00263] В описании представлены терапевтические композиции, как описано здесь, для осуществления эффективной деградации белков, представляющих интерес, для лечения или облегчения заболевания, например рака. В некоторых дополнительных вариантах осуществления изобретения, заболевание представляет собой множественную миелому. В другом аспекте описание раскрывает способ убиквитирования/деградации целевого белка-мишени в клетке. В некоторых вариантах осуществления изобретения, способ включает введение бифункционального соединения, описанного здесь, где соединение включает, например, CLM и PTM, предпочтительно связанные через линкерный фрагмент, как описано здесь, где CLM связан с PTM, и где CLM распознает белок пути убиквитинлигазы (например, убиквитинлигазу, предпочтительно убиквитинлигазу E3, например, такой как, цереблон), и РТМ распознает целевой белок-мишень, и при этом происходит деградация целевого белка-мишени, когда целевой белок будет помещен вблизи убиквитинлигазы, что приводит к деградации/ингибированию действия целевого белка-мишени и к контролю уровней белка. Контроль уровней белка, обеспечиваемый настоящим изобретением, позволяет лечить заболевание или состояния, которые модулируются через целевой белок, снижая уровень этого белка в клетке, например, в клетке пациента. В некоторых вариантах осуществления изобретения, способ включает введение эффективного количества соединения, описанного здесь, с необязательными добавками фармацевтически приемлемых эксципиента, носителя, адъюванта, другого биологически активного агента или их комбинации.
[00264] В дополнительных вариантах осуществления изобретения, в описании раскрыты способы лечения или облегчения заболевания, расстройства или их симптомов у субъекта или пациента, например животного, такого как человек, включающие введение субъекту, нуждающемуся в этом, композиции, содержащей эффективное количество, например, терапевтически эффективное количество, соединения, описанного здесь, или его соли и фармацевтически приемлемый эксципиент, носитель, адъювант, другой биологически активный агент или их комбинацию, где композиция эффективна для лечения или облегчения заболевания, расстройства или их симптомов у субъекта.
[00265] В другом аспекте, в описании представлены способы идентификации эффектов деградации представляющих интерес белков в биологической системе с использованием соединений в соответствии с настоящим изобретением.
[00266] В другом варианте осуществления, настоящее изобретение относится к способу лечения человека, нуждающегося в лечении заболевания или состояния, модулируемого белком, где деградация этого белка будет обеспечивать терапевтический эффект у этого пациента, при этом способ включает введение пациенту эффективного количества соединения в соответствии с настоящим изобретением, необязательно в комбинации с другим биологически активным агентом. Заболевание или состояние может быть заболеванием, вызываемое микробным агентом или другим экзогенным агентом, таким как вирус, бактерия, грибы, простейшие или другой микроб, или может быть заболеванием, которое вызвано избыточной экспрессией белка, что приводит к заболеванию и/или болезненному состоянию.
[00267] Термин ʺзаболевание или состояниеʺ применяется для описания любого заболевания или состояния, где имеет место дисрегуляция белка (т.е. повышение количества белка, экпрессируемого у пациента) и где деградация одного или нескольких белков у пациента может быть полезной терапией или облегчением симптомов у пациента, нуждающегося в этом. В некоторых случаях заболевание или состояние может быть вылечено.
[00268] Заболевания или состояния, которые можно лечить с использованием соединений настоящего изобретения, включают, например, астму, аутоиммунные заболевания, такие как рассеянный склероз, различные виды рака, цилиопатии, расщелины неба (волчья пасть), диабет, сердечные заболевания, гипертензию, воспалительное заболевания кишечника, умственную отсталость, расстройство настроения, ожирение, рефракционную аномалию, бесплодие, синдром Ангельмана, болезнь Канавана, глютеновую болезнь, амиотрофию Шарко-Мари-Тута, муковисцидоз, мышечную дистрофию Дюшенна, гемохроматоз, гемофилию, синдром Клайнфелтера, нейрофиброматоз, фенилкетонурию, поликистозное заболевание почек, (PKD1) или 4 (PKD2), синдром Прадера-Вилли, дрепаноцитарную анемию, болезнь Тея-Сакса, синдром Тернера.
[00269] Другие дополнительные заболевания или состояния, которые можно лечить с помощью соединений настоящего изобретения, включают болезнь Альцгеймера, боковой амиотрофический склероз (болезнь Лу Герига), нервную анорексию, тревожное расстройство, атеросклероз, расстройство гиперактивности при дефиците внимания, аутизм, биполярное расстройство, синдром хронической усталости, хроническую обструктивную болезнь легких, болезнь Крона, ишемическую болезнь сердца, деменцию, депрессию, сахарный диабет 1 типа, сахарный диабет 2 типа, эпилепсию, синдром Гийена-Барре, синдром раздраженной толстой кишки, волчанку, метаболический синдром, рассеянный склероз, инфаркт миокарда, ожирение, обсессивно-компульсивное расстройство, паническое расстройство, болезнь Паркинсона, псориаз, ревматоидный артрит, саркоидоз, шизофрению, инсульт, облитерирующий тромбоангиит, синдром Туретта, васкулит.
[00270] Другие дополнительные заболевания или состояния, которые можно лечить с помощью соединений настоящего изобретения, включают ацероплазминемию, ахондрогенез II типа, ахондроплазию, акроцефалию, болезнь Гоше 2 типа, острую интермиттирующую порфирию, болезнь Канавана, аденоматозный полипоз Коли, дефицит дегидратазы ALA, дефицит аденилозукцинатлиазы, адреногенитальный синдром, адренолеодистрофию, порфирию ALA-D, недостаточность дегидратазы ALL, алкаптонурию, болезнь Александера, алкаптонурический охроноз, дефицит альфа-1-антитрипсина, ингибитора альфа-1-протеиназы, эмфизему, боковой амиотрофический склероз, синдром Алстрема, болезнь Александера, несовершенный амелогенез, недостаточность дегидратазы ALA, болезнь Андерсона-Фабри, синдром нечувствительности к андрогенам, анемию, диффузную ангиокератому туловища, ангиоматоз сетчатки (болезнь фон Гиппель-Линдау), синдром Аперта, арахнодактилию (синдром Марфана), синдром Стиклера, дерматорексис (синдром Эверса-Данлоса типа артрохалазии), атаксию-телеангиэктазию, синдром Ретта, первичную легочную гипертензию, болезнь Сандхоффа, нейрофиброматоз II типа, синдром Бира-Стивенсона, средиземноморскую лихорадку, семейную, синдром Бенджамина, бета-талассемию, двухсторонний акустический нейрофиброматоз (нейрофиброматоз II типа), тромбофилию фактора V Ляйдена, синдром Блоха-Сульцбергера (недержание пигмента), синдром Блума, X-сцепленную сидеробластическую анемию, синдром Бонневи-Ульриха (синдром Тернера), болезнь Борневилля (туберозный склероз), прионную болезнь, синдром Бирта-Хогг-Дубе, болезнь хрупких костей (несовершенный остеогенез), синдром большого пальца стопы (синдром Рубинштейна-Тайби), бронзовый диабет/бронзовый цирроз (гемохроматоз), бульбоспинальную мышечную атрофию (болезнь Кеннеди), синдром Бургера-Грутца (дефицит липопротеинлипазы), хронический гранулематоз CGD, кампомелическую дисплазию, дефицит биотинидазы, кардиомиопатию (синдром Нунана), синдром кошачьего крика, CAVD (врожденное отсутствие семявыносящего протока), кардиотофациальный синдром Кэйлора (CBAVD), CEP (эритропоэтическую уропорфирию), кистозный фиброз, врожденный гипотиреоз, хондродистрофический синдром (ахондроплазию), отоспигломегаэпифизарную дисплазию, синдром Леша-Нихана, галактоземию, синдром Элерса-Данлоса, танатофорическую дисплазию, синдром Коффин-Лоури, синдром Коккейна, (семейный аденоматозный полипоз), эритропоэтическую уропорфирию, врожденный порок сердца, метгемоглобинемию/врожденную метгемоглобинемию, ахондроплазию, Х-сцепленную сидеробластическую анемию, коллагеноз, велокардиофасциальный синдром, анемию Кули (бета-талассемию), болезнь накопления меди (болезнь Вильсона), синдром Менкеса (болезнь Менкеса), наследственную копропорфирию, синдром Коудена, краниофациальный дизартроз (синдром Кроусона), болезнь Крейтцфельдта-Якоба (прионную болезнь), синдром Коккейна, синдром Каудена, болезнь Куршманна-Баттена-Штейнерта (миотоническую дистрофию), синдром Бира-Стивенсона, первичную гипероксалурию, спондилоэпиметафизеальную дисплазию (по Струдвику), мышечную дистрофию, болезни Дюшенна и Беккера (DBMD), синдром Ушера, дегенеративные нервные заболевания, включая синдром де Груши и синдром Дежерина-Соттаса, отклонения в развитии, дистрофическую мышечную атрофию позвоночника V типа, синдром нечувствительности к андрогенам, глобоидно-клеточную лейкодистрофию (болезнь Краббе), синдром Ди Джорджа, дефицит рецептора дигидротестостерона, синдром нечувствительности к андрогенам, синдром Дауна, карликовость, эритропоэтическую протопорфирию, дефицит эритроид 5-аминолевулинатсинтетазы, эритропоэтическую порфирию, эритропоэтическую протопорфирию, эритропоэтическую уропорфирию, атаксию Фридриха, семейный рецидивирующий полисерозит, гемохроматоз, семейную сенсорную невропатию, первичную легочную гипертензию (PPH), фиброзно-кистозную болезнь поджелудочной железы, синдром ломкой X-хромосомы, галактоземию, генетические нарушения головного мозга, врожденный гигантоклеточный гепатит (гемохроматоз новорожденных), синдром Гронблад-Страндберга (эластическую псевдоксантому), болезнь Гюнтера (врожденную эритропоэтическую порфирию), гемохроматоз, синдром Холлгрена, серповидноклеточную анемию, гемофилию, гепатоэритропоэтическую порфирию (HEP), болезнь Гиппель-Линдау (болезнь фон Гиппеля-Линдау), болезнь Хантингтона, синдром прогерии Хатчинсона-Гилфорда (прогерию), гиперандрогенизм, гипохондроплазию, гипохромную анемию, расстройства иммунной системы, включая синдром Х-связанного тяжелого комбинированного иммунодефицита, синдром Инсли-Астли, синдром Джексона-Вейсса, синдром Жубера, синдром Леша-Найхана, синдром Джексона-Вейсса, заболевания почек, включая гипероксалурию, синдром Клайнфелтера, дисплазию Книста, лакунарную деменцию, ахондрогенез Лангера-Салдино, атаксию-телангиэктазию, синдром Линча, синдром Линча, дефицит лизилгидроксилазы, болезнь Мачадо-Джозефа, метаболические расстройства, включая дисплазию Книста, синдром Марфана, расстройства движения, синдром Мовата-Вильсона, кистозный фиброз, синдром Менке, множественный нейрофиброматоз, синдром Нэнси-Инсли, хондродисплазию Нэнси-Суини, болезнь Ниманна-Пика, синдром Ноака (синдром Пфайфера), болезнь Ослера-Вебера-Ренду, синдром Пейтца-Егерса, поликистоз почек, полиотссальную фиброзную остеодисплазию (синдром МакКуна-Олбрайт), синдром Пейтца-Егерса, синдром Прадера-Лабхарта-Вилли, гемохроматоз, синдром первичной гиперурикемии (синдром Леша-Нихана), первичную легочную гипертензию, первичную старческую дегенеративную деменцию, прионную болезнь, прогерию (синдром Хатчинсона-Гилфорда), прогрессирующую хорею, хроническую наследственную болезнь Хантингтона (болезнь Хантингтона), прогрессирующую мышечную атрофию, спинальную мышечную атрофию, пропионовую ацидемию, протопорфирию, проксимальную миотоническую дистрофию, легочную артериальную гипертензию, PXE (эластическую псевдоксантому), Rb (ретинобластому), болезнь Реклингхаузена (нейрофиброматоз I типа), рецидивирующий полисерозит, нарушения сетчатки, ретинобластому, синдром Ретта, RFALS типа 3, синдром Рикера, синдром Райли-Дея, синдром Русси-Леви, тяжелую ахондроплазию с задержкой в развитии и акантокератодермией (SADDAN), синдром Ли-Фраумени, синдром саркомы-молочной железы-лейкоза-надпочечников (SBLA), туберозный склероз (туберозный склероз), SDAT, врожденная SED (врожденная спондилоэпифизарная дисплазия), SED Страдвика (спондилоэпилептическая дисплазия, тип Страдвика), SEDс (врожденная спондилоэпилептическая дисплазия) SEMD типа Страдвика (спондилоэпиметфизеальная дисплазия типа Страдвика), синдром Шпринтцена, расстройства пигментации кожи, синдром Смита-Лемли-Опитца, южноафриканская генетическая порфирия (смешанная порфирия), детский афферентный наследственный спастический паралич, нарушения речи и коммуникации, сфинголипидоз, болезнь Тая-Сакса, спиноцеребеллярная атаксия, синдром Стиклера, удар, синдром нечувствительности к андрогенам, дефицит тетрагидробиоптерина, бета-талассемия, заболевание щитовидной железы, томакулезная невропатия (наследственная компрессионная невропатия), синдром Тревера-Коллинза, синдром тройной X (синдром тройной Х-хромосомы), трисомия 21 (синдром Дауна), трисомию X, синдром VHL (болезнь фон Гиппель-Линдау), ухудшение зрения и слепота (синдром Альстрема), болезнь Вролика, синдром Ваарденбурга, синдром Варбурга-Сьо-Фледелиуса, синдром Вайссенбахера-Цвеймюллера, синдром Вольфа-Хиршхорна, периодическая болезнь Вольфа, синдром Вайссенбахера-Цвеймюллера и пигментная ксеродерма, среди прочих.
[00271] Термин «неоплазия» или «рак» используется в описании для обозначения патологического процесса, который приводит к образованию и росту ракового или злокачественного новообразования, т.е. аномальной ткани, которая растет за счет клеточной пролиферации, часто более быстро, чем обычная ткань, и продолжает расти после того, как стимулы, которые инициировали новый рост, прекращали действие. Злокачественные образования характеризуются частичным или полным отсутствием структурной организации и функциональной координации по сравнению с нормальной тканью, и они способны вторгаться в окружающие ткани, метастазировать в нескольких местах и могут возобновятся после попытки их удаления, и привести к смерти пациента, при отсутствии адекватного лечения. В данном описании термин неоплазия применяется для описания всех раковых состояний, и он охватывает или включает патологический процесс, связанный со злокачественными гематогенезом, асцитными и солидными опухолями. Примеры раковых заболеваний, которые можно лечить с помощью соединения настоящего изобретения используемого отдельно или в сочетании с по меньшей мере одним дополнительным противораковым средством, включают плоскоклеточный рак, базалиому, аденокарциному, печеночно-клеточные карциномы и карциномы клеток почек, рак мочевого пузыря, кишечника, груди, шейки матки, толстой кишки, пищевода, головы, почек, печени, легких, шеи, яичников, поджелудочной железы, предстательной железы и желудка; лейкозы; доброкачественные и злокачественные лимфомы, особенно лимфому Беркитта и неходжкинскую лимфому; доброкачественные и злокачественные меланомы; миелопролиферативные заболевания; саркомы, включая саркому Юинга, гемангиосаркому, саркому Капоши, липосаркому, миосаркомы, периферическую нейроэпителиому, синовиальную саркому, глиомы, астроцитомы, олигодендроглиомы, эпендимомы, глиобластомы, нейробластомы, ганглионевромы, ганглиоглиомы, медуллобластомы, опухоли шишковидных клеток, менингиомы, менингеальные саркомы, нейрофибромы и шванномы; рак толстой кишки, рак молочной железы, рак предстательной железы, рак шейки матки, рак матки, рак легких, рак яичников, рак яичек, рак щитовидной железы, астроцитому, рак пищевода, рак поджелудочной железы, рак желудка, рак печени, рак толстой кишки, меланому; карциносаркому, болезнь Ходжкина, опухоль Вильмса и тератокарциномы. Дополнительные раки, которые могут быть лечены с применением соединений в соответствии с данным изобретением, включают, например, T-клеточный острый лимфобластный лейкоз (T-ALL), T-клеточную лимфобластную лимфому (T-LL), периферическую T-клеточную лимфому, Т-клеточный лейкоз взрослых, Пре-B ALL, Пре-B лимфомы, крупноклеточную B-клеточную лимфому, лимфому Беркитта, B-клеточный ALL, положительный по филадельфийской хромосоме ALL и положительный по филадельфийской хромосоме CML.
[00272] Термин "биологически активный агент" используется здесь для описания агента, другого, чем соединение по изобретению, которое используется в комбинации с соединениями по изобретению в качестве средства с дополнительной биологической активностью, помогающего в проведении назначенной терапии, ингибирования и/или предотвращения/профилактики, где используются соединения настоящего изобретения. Предпочтительные биологически активные агенты для использования в настоящем изобретении включают агенты, которые обладают фармакологической активностью, аналогичной активности соединений по изобретению, которые используют или вводят, и они включают, например, противораковые агенты, противовирусные агенты, особенно, в том числе, агенты против HIV и агенты против HCV, антимикробные агенты, противогрибковые агенты и т.п.
[00273] Термин "дополнительный противораковый агент" используется для описания противоракового агента, который может быть объединен с соединениями по изобретению для лечения рака. Эти агенты включают, например, эверолимус, трабектедин, абраксан, TLK 286, AV-299, DN-101, пазопаниб, GSK690693, RTA 744, ON 0910.Na, AZD 6244 (ARRY-142886), AMN-107, TKI-258, GSK461364, AZD 1152, энзастаурин, вандетаниб, ARQ-197, MK-0457, MLN8054, PHA-739358, R-763, AT-9263, ингибитор FLT-3, ингибитор VEGFR, ингибитор EGFR TK, ингибитор авроракиназы, модулятор PIK-1, ингибитор Bcl-2, ингибитор NDAC, ингибитор c-MET, ингибитор PARP, ингибитор Cdk, ингибитор EGFR TK, ингибитор IGFR-TK, анти-HGF антитело, ингибиторы PI3 киназ, ингибитор AKT, ингибитор mTORC1/2, ингибитор JAK/STAT, ингибитор контрольной точки-1 или 2, ингибитор киназы фокальной адгезии, ингибитор Map киназы (mek), антитело ловушки VEGF, пеметрексед, эрлотиниб, дазатаниб, нилотиниб, декатаниб, панитумумаб, амрубицин, ореговомаб, Lep-etu, нолатрексед, azd2171, батабулин, офатумумаб, занолимумаб, эдотерацин, тетрандрин, рубитекан, тесмилифен, облимерсен, тицилимумаб, ипилимумаб, госсипол, Bio 111, 131-I-TM-601, ALT-110, BIO 140, CC 8490, циленгитид, гиматекан, IL13-PE38QQR, INO 1001, IPdR1 KRX-0402, лукантон, LY317615, нейрадиаб, витеспан, RTA 744, Sdx 102, талампанел, атрасентан, Xr 311, ромидепсин, ADS-100380, сунитиниб, 5-фторурацил, вориностат, этопозид, гемцитабин, доксорубицин, липосомный доксорубицин, 5'-деокси-5-фторуридин, винкристин, темозоломид, ZK-304709, селициклиб; PD0325901, AZD-6244, капецитабин, L-глутаминовую кислоту, гептагидрат динатриевой соли N-[4-[2-(2-амино-4,7-дигидро-4-оксо-1H-пирроло[2,3-d]пиримидин-5-ил)этил]бензоил], камптотецин, PEG-меченный иринотекан, тамоксифен, цитрат тофемифена, анастразол, эксеместан, летрозол, дез(диэтилстилбестрол), эстрадиол, эстроген, конъюгированный эстроген, бевацизумаб, IMC-1C11, CHIR-258); 3-[5-(метилсульфонилпиперадинметил)индолил]хинолон, ваталаниб, AG-013736, AVE-0005, ацетат госерелина, ацетат лейпролида, памоат трипторелина, ацетат гидроксипрогестерона, капроат гидроксипрогестерона, ацетат мегестрола, ралоксифен, бикалутамид, флутамид, нилутамид, ацетат мегестрола, CP-724714; TAK-165, HKI-272, эрлотиниб, лапатаниб, канертиниб, ABX-EGF антитело, эрбитукс, EKB-569, PKI-166, GW-572016, ионафарниб, BMS-214662, типифарниб; амифостин, NVP-LAQ824, субероиланалид гидроксамовой кислоты, вальпроевая кислота, трихостатин A, FK-228, SU11248, сорафениб, KRN951, аминоглутетимид, арнсакрин, анагрелид, L-аспарагиназа, вакцина Bacillus Calmette-Guerin (BCG), адриамицин, блеомицин, бусерелин, бусульфан, карбоплатин, кармустин, хлорамбуцил, цисплатин, кладрибин, клодронат, ципротерон, цитарабин, дакарбазин, дактиномицин, даунорубицин, диэтилстилбестрол, эпирубицин, флударабин, флудрокортизон, флуоксиместерон, флутамид, глеевек, гемцитабин, гидроксимочевина, идарубицин, ифосфамид, иматиниб, лейпролид, левамизол, ломустин, мехлорэтамин, мелфалан, 6-меркаптопурин, месна, метотрексат, митомицин, митотан, митоксантрон, нилутамид, октреотид, оксалиплатин, памидронат, пентостатин, пликамицин, порфимер, прокарбазин, ралтитрексед, ритуксимаб, стрептозоцин, тенипозид, тестостерон, талидомид, тиогуанин, тиотепа, третиноин, виндезин, 13-цис-ретиноевая кислота, мелфалан, урамустин, эстрамустин, альтретамин, флоксуридин, 5-деоксиуридин, цитозин арабинозид, 6-мекаптопурин, деоксикоформицин, кальцитриол, валрубицин, митрамицин, винбластин, винорелбин, топотекан, разоксин, маримастат, COL-3, неофастат, BMS-275291, скваламин, эндостатин, SU5416, SU6668, EMD121974, интерлейкин-12, IM862, ангиостатин, витаксин, дролоксифен, идоксифен, спиронолактон, финастерид, цимитидин, трастузумаб, денилейкин дифтитокс, гефитиниб, бортезимиб, паклитаксел, безкремофорный паклитаксел, доцетаксел, эпитилон B, BMS-247550, BMS-310705, дролоксифен, 4-гидрокситамоксифен, пипендоксифен, ERA-923, арзоксифен, фулвестрант, аколбифен, лазофоксифен, идоксифен, TSE-424, HMR-3339, ZK186619, топотекан, PTK787/ZK 222584, VX-745, PD 184352, рапамицин, 40-O-(2-гидроксиэтил)рапамицин, темсиролимус, AP-23573, RAD001, ABT-578, BC-210, LY294002, LY292223, LY292696, LY293684, LY293646, вортманнин, ZM336372, L-779,450, PEG-филграстим, дарбепоэтин, эритропоэтин, гранулоцитарный колониестимулирующий фактор, преднизон, цетуксимаб, гранулоцитарный макрофаговый колониестимулирующий фактор, гистрелин, пэгилированный интерферон альфа-2a, интерферон альфа-2a, пэгилированный интерферон альфа-2b, интерферон альфа-2b, азацитидин, PEG-1-аспарагиназу, леналидомид, гемтузумаб, гидрокортизон, интерлейкин-11, дексразоксан, алемтузумаб, всетрансетиновая кислота, кетоконазол, интерлейкин-2, мегестрол, иммунноглобулин, азотистый иприт, метилпреднизолон, ибритгумомаб тиуксетан, андрогены, децитабин, гексаметилмеламин, бексаротин, тоцитумомаб, триоксид мышьяка, кортизон, эдитронат, митотан, циклоспорин, липосомальный даунорубицин, аспарагиназа Эдвина, стронций 89, казопитант, нетупитант, антагонист рецептора NK-1, палоносетрон, апрепитант, дифенгидрамин, гидроксизин, метоклопрамид, лоразепам, альпразолам, галоперидол, дроперидол, дронабинол, дексаметазон, метилпреднизолон, прохлорперазин, гранисетрон, ондансетрон, доласетрон, трописетрон, пегфилграстим, эритропоэтин, эпоэтин альфа, дарбепоэтин альфа и их смеси.
[00274] Термин "анти-HIV агент" (средство против ВИЧ) или "дополнительный анти-HIV агент" включает, например, нуклеозидные ингибиторы обратной транскриптазы (NRTI), другие не-нуклеозидные ингибиторы обратной транскриптазы (т.е. такие, которые не являются представителями данного изобретения), ингибиторы протеазы, ингибиторы слияния, среди прочих, типовые соединения которых включают, например, 3TC (ламивудин), AZT (зидовудин), (-)-FTC, ddI (диданозин), ddC (залцитабин), абакавир (ABC), тенофовир (PMPA), D-D4FC (реверсет), D4T (ставудин), рацивир, L-FddC, L-FD4C, NVP (невирапин), DLV (делавирдин), EFV (эфавиренц), SQVM (саквинавир мезилат), RTV (ритонавир), IDV (индинавир), SQV (саквинавир), NFV (нелфинавир), APV (ампренавир), LPV (лопинавир), ингибиторы слияния, такие как T20, среди прочих, фузеон и его смеси, включая соединения против ВИЧ, находящиеся в стадии клинических исследований или разработки.
[00275] Другие агенты против ВИЧ, которые могут быть использованы для совместного введения с соединениями в соответствии с данным изобретением, включают, например, другие NRTI (т.е. отличные от NRTI в соответствии с данным изобретением), выбранные из группы, включающей невирапин (BI-R6-587), делавирдин (U-90152S/T), эфавиренц (DMP-266), UC-781 (N-[4-хлор-3-(3-метил-2-бутенилокси)фенил]-2-метил-3-фуранкарботиамид), этравирин (TMC125), Тровирдин (Ly300046.HCl), MKC-442 (эмивирин, коактинон), HI-236, HI-240, HI-280, HI-281, рилпивирин (TMC-278), MSC-127, HBY 097, DMP266, байкалин (TJN-151), ADAM-II (метил 3',3'-дихлор-4',4ʺ-диметокси-5',5ʺ-бис(метоксикарбонил)-6,6-дифенилгексеноат), метил 3-бром-5-(1-5-бром-4-метокси-3-(метоксикарбонил)фенил)гепт-1-енил)-2-метоксибензоат (аналог алкенилдиарилметана, аналог ADAM), (5-хлор-3-(фенилсульфинил)-2'-индолкарбоксамид), AAP-BHAP (U-104489 или PNU-104489), каправирин (AG-1549, S-1153), атевирдин (U-87201E), ауринтрикарбоновая кислота (SD-095345), 1-[(6-Циано-2-индолил)карбонил]-4-[3-(изопропиламино)-2-пиридинил]пиперазин (производное пиперазин-1-пиридин-4-индолила), 1-[5-[[N-(метил)метилсульфониламино]-2-индолилкарбонил-4-[3-(изопропиламино)-2-пиридинил]пиперазин (производное пиперазин-1-пиридин-5-индолила), 1-[3-(этиламино)-2-[пиридинил]-4-[(5-гидрокси-2-индолил)карбонил]пиперазин, 1-[(6-формил-2-индолил)карбонил]-4-[3-(изопропиламино)-2-пиридинил]пиперазин, 1-[[5-(метилсульфонилокси)-2-индолил)карбонил]-4-[3-(изопропиламино)-2-пиридинил]пиперазин, U88204E, бис(2-нитрофенил)сульфон (NSC 633001), каланолид A (NSC675451), каланолид B, 6-бензил-5-метил-2-(циклогексилокси)пиримидин-4-он (DABO-546), DPC 961, E-EBU, E-EBU-dm, E-EPSeU, E-EPU, фоскарнет (фоскавир), HEPT (1-[(2-гидроксиэтокси)метил]-6-(фенилтио)тимин), HEPT-M (1-[(2-гидроксиэтокси)метил]-6-(3-метилфенил)тио)тимин), HEPT-S (1-[(2-гидроксиэтокси)метил]-6-(фенилтио)-2-тиотимин), инофиллум P, L-737,126, мишелламин A (NSC650898), мишелламин B (NSC649324), мишелламин F, 6-(3,5-диметилбензил)-1-[(2-гидроксиэтокси)метил]-5-изопропилурацил, 6-(3,5-диметилбензил)-1-(этилоксиметил)-5-изопропилурацил, NPPS, E-BPTU (NSC 648400), олтипраз (4-метил-5-(пиразинил)-3H-1,2-дитиол-3-тион), N-{2-(2-хлор-6-фторфенэтил]-N'-(2-тиазолил)тиомочевина (Cl-, F-производное PETT), N-{2-(2,6-дифторфенэтил]-N'-[2-(5-бромпиридил)]тиомочевина (производное PETT), N-{2-(2,6-дифторфенетил]-N'-[2-(5-метилпиридил)]тиомочевина (пиридил-производное PETT), N-[2-(3-фторфуранил)этил]-N'-[2-(5-хлорпиридил)]тиомочевина, N-[2-(2-фтор-6-этоксифенетил)]-N'-[2-(5-бромпиридил)]тиомочевина, N-(2-фенетил)-N'-(2-тиазолил)тиомочевина (LY-73497), L-697,639, L-697,593, L-697,661, 3-[2-(4,7-дифторбензоксазол-2-ил)этил}-5-этил-6-метил(пиридин-2(1H)-тион (2-пиридинон производное), 3-[[(2-метокси-5,6-диметил-3-пиридил)метил]амин]-5-этил-6-метил(пиридин-2(1H)-тион (производное 2-пиридинона), R82150, R82913, R87232, R88703, R89439 (ловирид), R90385, S-2720, супамин натрий, TBZ (тиазолобензимидазол, NSC 625487), тиазолоизоиндол-5-он, (+)(R)-9b-(3,5-диметилфенил-2,3-дигидротиазоло[2,3-a]изоиндол-5(9bH)-он, тивирапин (R86183), UC-38 и UC-84, среди прочих.
[00276] Термин «фармацевтически приемлемая соль» используется в описании для указания, где это применимо, соли одного или нескольких описанных здесь соединений, которая используется для повышения растворимости соединения в желудочном соке желудочно-кишечного тракта пациента, чтобы способствовать растворению и биодоступности соединений. Фармацевтически приемлемые соли включают соли, полученные из фармацевтически приемлемых неорганических или органических оснований и кислот, где применимо. Подходящие соли включают соли, полученные из щелочных металлов, таких как калий и натрий, щелочноземельных металлов, таких как кальций, магний и аммоний, среди прочих оснований и кислот, хорошо известных в области фармацевтики. Соли натрия и калия особенно предпочтительны в качестве нейтрализующих солей фосфатов в соответствии с данным изобретением.
[00277] Термин "фармацевтически приемлемое производное" используется по всему описанию для указания любой фармацевтически приемлемого пролекарства (такого как сложный эфир, амид, другие группы пролекарств), которое, при введении пациенту, предоставляют пациенту, прямо или косвенно, данное соединение или активный метаболит данного соединения.
Общие подходы к синтезу
[00278] Синтез и оптимизация бифункциональных молекул, описанных здесь, могут быть подобраны постадийно или модульно. Например, идентификация соединений, которые связываются с молекулами-мишенями, может включать процедуру скрининга с высокой или средней пропускной способностью, если подходящие лиганды сразу не доступны. Нет ничего необычного в том, чтобы для начальных лигандов осуществлять итеративный дизайн и оптимизировать циклы для улучшения субоптимальных аспектов, с подтверждением данными подходящих анализов в условиях in vitro и фармакологических анализов и/или анализов ADMET. Часть работы по оптимизации/SAR будет состоять в том, чтобы исследовать положения лиганда, которые толерантны к замещению, и которые могут быть подходящими местами для присоединения линкера, указанного здесь выше. Там, где доступны кристаллические или структурные данные ЯМР, их можно использовать для направления условий синтеза.
[00279] Аналогичным образом можно идентифицировать и оптимизировать лиганды для лигазы E3, то есть ULM/CLM.
[00280] При использовании РТМ и ULM (например, CLM), специалист в данной области может использовать известные синтетические способы для их комбинации с применением или без применения линкера. Линкерные фрагменты могут быть синтезированы с использованием ряда композиций, варьируя длину и гибкость, а также функционализировать их таким образом, что группы РТМ и ULM могут быть последовательно присоединены к дистальным концам линкера. Таким образом, библиотека бифункциональных молекул может быть реализована и профилирована в фармакологических исследованиях in vitro и in vivo, а такуже в ADMET/PK исследованиях. Конечные бифункциональные молекулы для групп PTM и ULM могут подвергаться итерационным циклам дизайна и оптимизации, чтобы идентифицировать молекулы с желательными свойствами.
[00281] Некоторые неограничивающие примеры методов получения CLM, описанных здесь, показаны ниже в обобщенном виде.
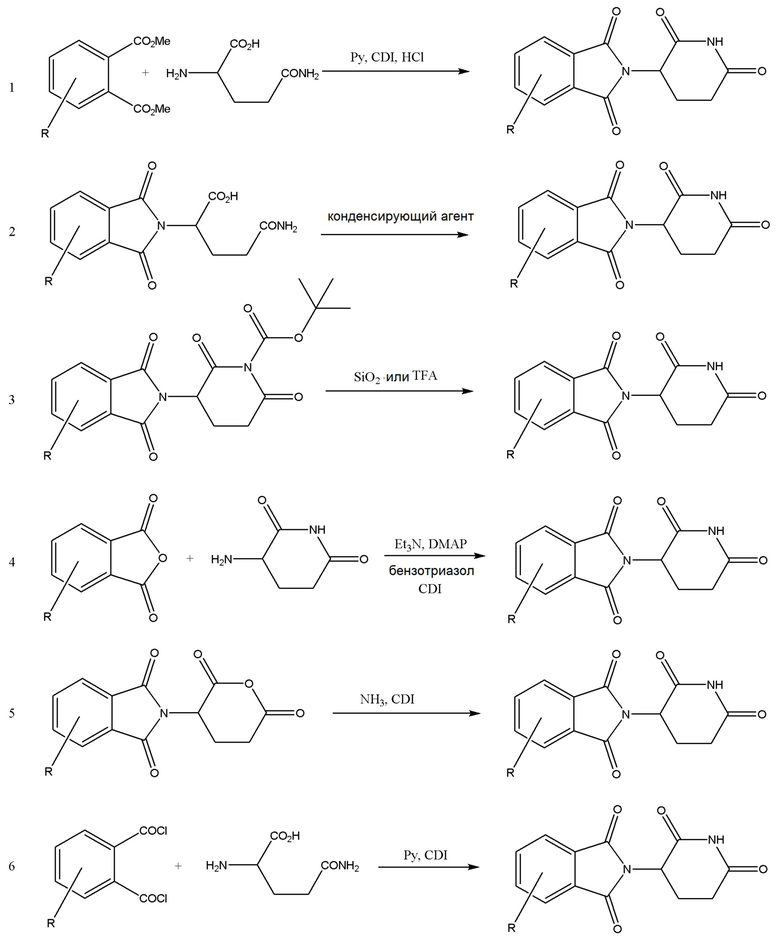
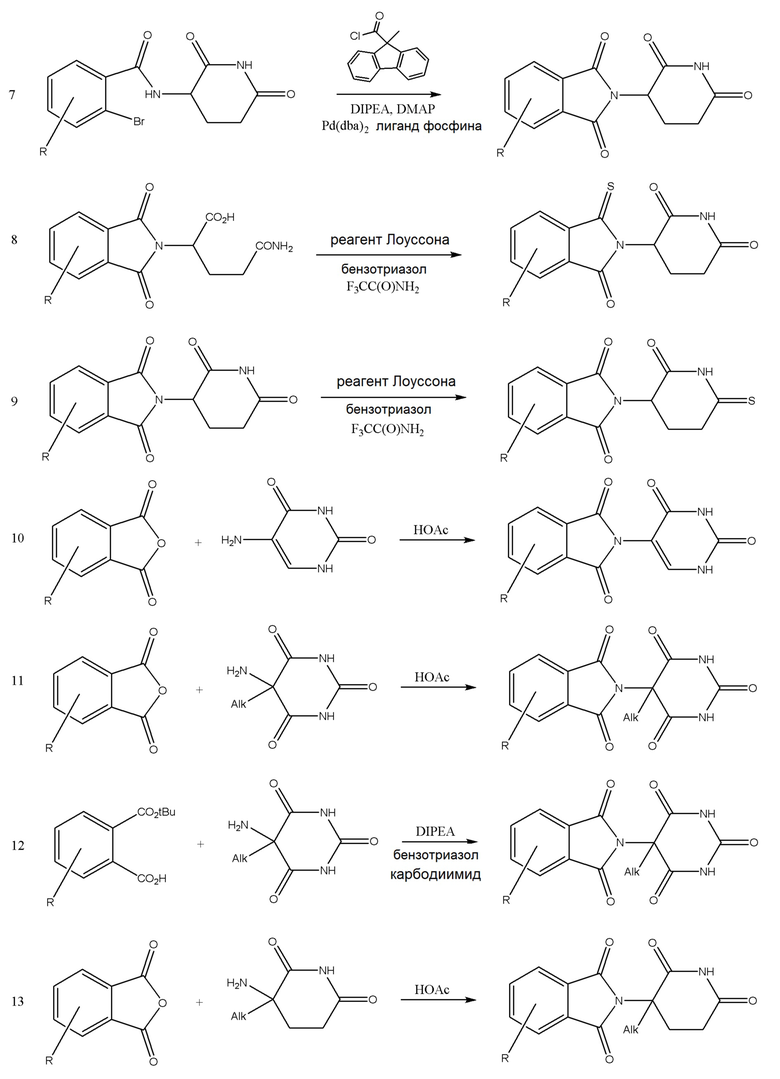
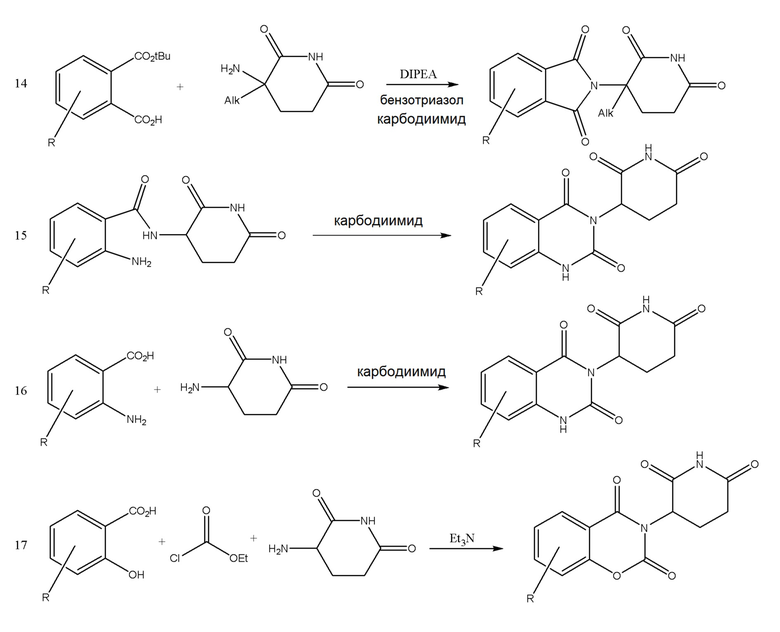
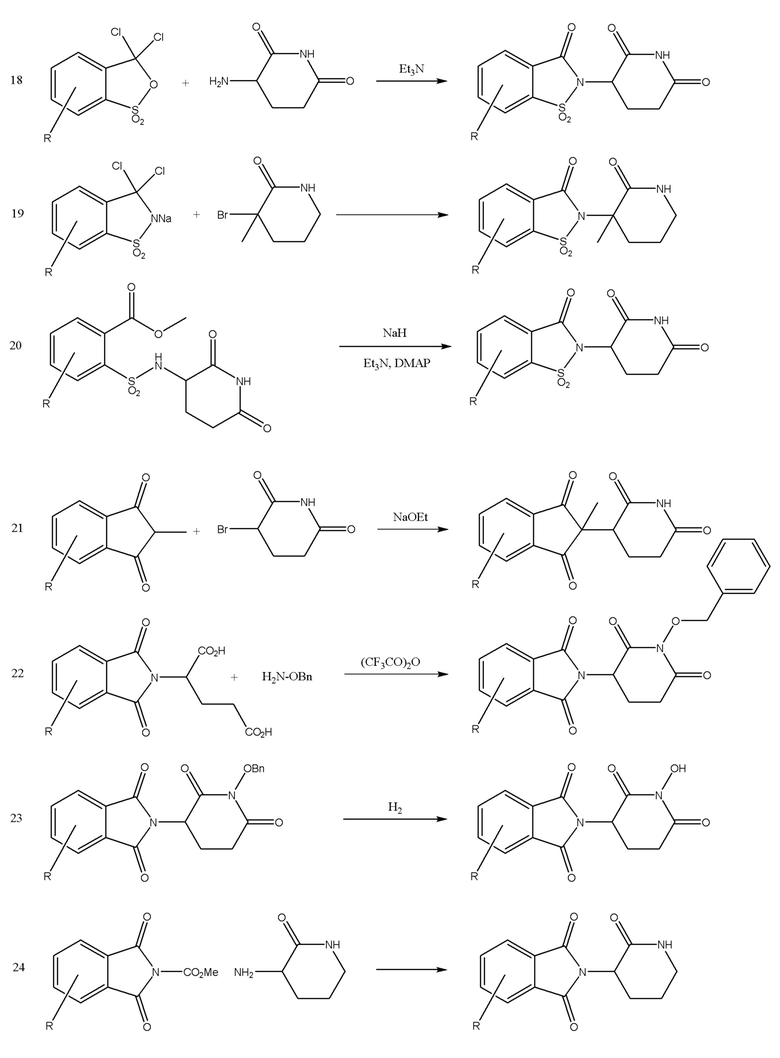
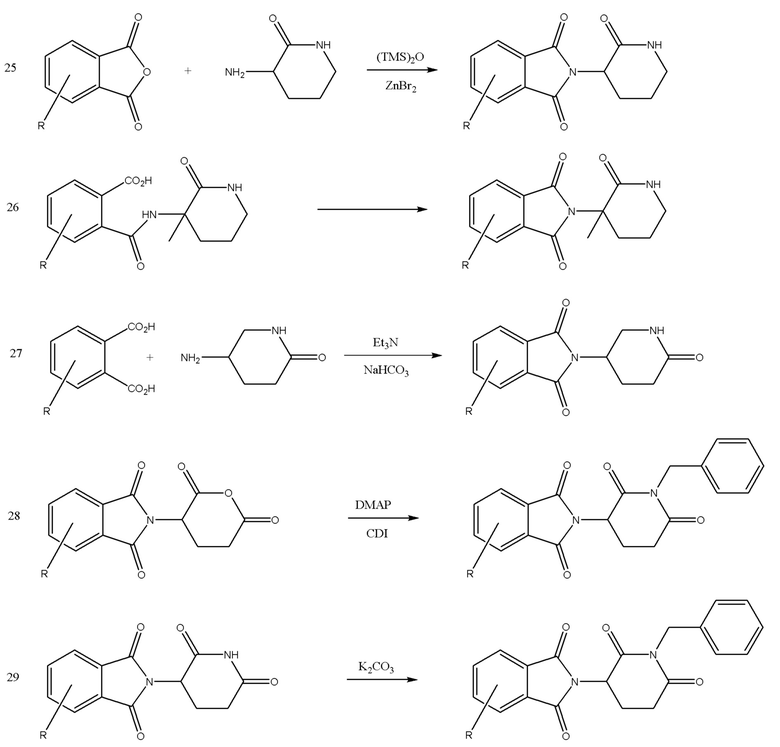
[00282] Как показано на схеме репрезентативной реакции 1, производные диметилфталата можно конденсировать с глутамином (рацематом или энантиомером) или аналогами глутамина, а затем далее провести взаимодействие с реагентами, такими как карбонилдиимидазол, с образованием 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона.
[00283] Альтернативно, как показано на схеме репрезентативной реакции 2, промежуточный фталимид, полученный при начальной конденсации, описанный выше, может быть получен и/или выделен отдельно, а затем он может быть подвергнут взаимодействию с дегидратирующими агентами, такими как трифторацетамид, POCl3 или уксусный ангидрид, с получением целевого 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона. Такой же тип промежуточного фталимида можно также подвергнуть взаимодействию с реагентом Лоуссона до стадии дегидратации, чтобы получить тио-аналоги, такие, которые показаны на схемах репрезентативных реакций 8 и 9.
[00284] У защищенных производных 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона, типа N1-ВОС, которые показаны в илюстративном примере 3, может быть снята защита с получением целевых производных 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона, используя в этом случае реагенты, такие как TFA или диоксид кремния.
[00285] Фталевые ангидриды, которые показаны на схеме репрезентативной реакции 4, могут быть получены путем открытия кольца реакцией с аминами, такими как 3-аминопиперидин-2,6-дион, с образованием промежуточных карбоксилатных видов соединений, которые при обработке карбонилдиимидазолом и бензотриазолом образуют целевые производные 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона. Альтернативно, для получения целевого продукта, два компонента могут быть объединены в присутствии уксусной кислоты, как показано на схеме репрезентативной реакции 13.
[00286] В аналогичной реакции, производные ангидрида, подобные тем, которые показаны на схеме репрезентативной реакции 5, могут быть подвергнуты взаимодействию с аминами (в показанном примере - аммиак), затем карбонилдиимидазол образует целевое производное 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона.
[00287] Когда имеются фталоилхлориды, возможна прямая конденсация с глутамином (рацематом или энантиомером) или аналогами глутамина с последующим взаимодействием с реагентами, такими как карбонилдиимидазол, с образованием производных 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона, как показано на схеме репрезентативной реакции 6.
[00288] O-бромбензамиды могут быть подвергнуты взаимодействию с источником СО, таким как хлорангидрид кислоты, как показано на схеме репрезентативной реакции 7, в присутствии палладиевого катализатора и связанного фосфинового лиганда, с получением целевых производных 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона. Альтернативно, газообразный CO может использоваться в сочетании с катализаторами на основе родия (II) и карбонатом серебра с получением целевых продуктов.
[00289] Производные 2-(2,4-диоксо-1,2,3,4-тетрагидропиримидин-5-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона и 5-(1,3-диоксо-2,3-дигидро-1Н-изоиндол-2-ил)-1,3-диазинин-2,4,6-триона могут быть получены с помощью аналогичных методов, как описано выше для производных 2-(2,6-диоксопиперидин-3-ил)2,3-дигидро-1Н-изоиндол-1,3-диона. На схемах репрезентативных реакций 20 и 21 показано, что, фталевый ангидрид может быть подвергнут взаимодействию с производными 5-амино-1,2,3,4-тетрагидропиримидин-2,4-диона или 5-амино-1,3-диазинин-2,4,6-триона, соответственно, в присутствии уксусной кислоты, с образованием целевых продуктов.
[00290] Альтернативно, производные 5-(1,3-диоксо-2,3-дигидро-1Н-изоиндол-2-ил)-1,3-диазинин-2,4,6-триона могут быть получены в результате реакции производных 5-амино-1,3-диазинин-2,4,6-триона с трет-бутиловыми эфирами монофталевой кислоты в присутствии основания Хюнига, карбодиимида и бензотриазола, как показано на схеме репрезентативной реакции 12. Аналогичные условия могут быть применены для получения производных 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона из фталевой кислоты, моно-трет-бутиловых эфиров, как показано на схеме репрезентативной реакции 14.
[00291] Соединения, такие как 3-(2,6-диоксопиперидин-3-ил)-1,2,3,4-тетрагидрохиназолин-2,4-дион, могут быть получены из производных антраниловой кислоты путем взаимодействия 3-аминопиперидин-2,6-дионов с карбодиимидом, как показано на схеме репрезентативной реакции 16. Промежуточный бензамидный продукт может быть выделен (или получен отдельно), а затем его подвергают взаимодействию с карбодиимидом, с получением производных 3-(2,6-диоксопиперидин-3-ил)-1,2,3,4-тетрагидрохиназолин-2,4-диона, как показано на схеме репрезентативной реакции 15.
[00292] Аналоги 3-(2,6-диоксопиперидин-3-ил)-3,4-дигидро-2Н-1,3-бензоксазин-2,4-диона могут быть получены путем активации салициловых кислот с хлорформиатами с последующей конденсацией с 3-аминопиперидин-2,6-дионами, как показано на на схеме репрезентативной реакции 17.
[00293] 3,3-дихлор-2,1λ6-бензоксатиол-1,1-дионы, как показано на схеме репрезентативной реакции 18, могут быть получены путем взаимодействия 2-сульфобензойных кислот с POCl3 и PCl5. Эти соединения могут быть введены в реакцию с производными аминокислот для получения, например, целевых производных 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1λ6,2-бензотиазол-1,1,3-триона.
[00294] Как показано на схеме репрезентативной реакции 19, анионы производных сахарина можно алкилировать электрофилами, такими как 3-бром-3-метилпиперидин-2-он, для получения целевых производных 2-(3-метил-2-оксопиперидин-3-ил)-2,3-дигидро-1λ6,2-бензотиазол-1,1,3-триона.
[00295] Аналоги 2-(2,6-диоксопиперидин-3-ил)-2,3-дигидро-1λ6,2-бензотиазол-1,1,3-триона также могут быть получены путем взаимодействия метил-2-[(2,6-диоксопиперидин-3-ил)сульфамоил]бензоата с сильными основаниями, такими как гидрид натрия (см схему репрезентативной реакции 20).
[00296] Депротонирование произодных 2-метил-2,3-дигидро-1Н-инден-1,3-диона с этоксидом натрия, с последующим взаимодействием с электрофилами, такими как 3-бромпиперидин-2,6-дион приводит к получению 3-(2-метил-1,3-диоксо-1H-инден-2-ил)пиперидин-2,6-диона, как показано на схеме репрезентативной реакции 21.
[00297] Получение N1-замещенных соединений, таких как 2-[1-(бензилокси)-2,6-диоксопиперидин-3-ил]-2,3-дигидро-1Н-изоиндол-1,4-дион (см. схему репрезентативной реакции 22) может быть достигнуто путем взаимодействия 2-(1,3-диоксо-2,3-дигидро-1Н-изоиндол-2-ил)пентадиоевой кислоты с N-бензилгидроксиламином и ангидридом трифторуксусной кислоты.
[00298] В свою очередь соединения, такие как 2-[1-(бензилокси)-2,6-диоксопиперидин-3-ил]-2,3-дигидро-1Н-изоиндол-1,4-дион (см. схему репрезентативной реакции 23) могут быть подвергнуты удалению бензила в условиях гидрирования, с получением N1-гидрокси-аналогов, таких как 2-(1-гидрокси-2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-дион.
[00299] На схеме репрезентативной реакции 24 показано, что метил-1,3-диоксо-2,3-дигидро-1Н-изоиндол-2-карбоксилат (и его аналоги) подвергают взаимодействие с 3-аминопиперидином-2-оном, с получением 2-(2-оксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-дионов.
[00300] Этот же амин может также быть подвергнут взаимодействию с производным фталевого ангидрида в присутствии кислоты Льюиса, такой как бромид цинка и триметилсилилового эфира, с получением продукта, тип которого показан на схеме репрезентативной реакции 25. Промежуточные продукты этой реакции, если их выделить или получить другим способом (см. схему репрезентативной реакции 26) могут быть включены в полную циклизацию при использовании дегидратирующего реагента.
[00301] Изомерные производные, такие как производные 2-(6-оксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-диона, показанные на схеме репрезентативной реакции 27, могут быть получены путем взаимодействия фталевой кислоты с 5-аминопиперидин-2-оном.
[00302] Получение N1-замещенных соединений, таких как 2-(1-бензил-2,6-диоксопиперидин-3-ил)-2,3-дигидро-1Н-изоиндол-1,4-дион (см. репрезентативные реакции 28 и 29) может быть реализовано несколькими путями. Так, например, ангидрид (2-(2,6-диоксооксан-3-ил)-2,3-дигидро-1Н-изоиндол-1,3-дион) может быть подвергнут конденсации с 3-аминопиперидина-2,6-дионом в присутствии DMAP и карбонилдиимидазола (см. репрезентативную реакцию 28), или производные 2-(2,6-диоксопиперидин-3-ил)2,3-дигидро-1Н-изоиндол-1,3-диона можно алкилировать электрофилами, такими как бензилбромид, в присутствии основания, как показано на схеме репрезентативной реакции 29.
[00303] В некоторых случаях может потребоваться стратегии взаимопревращения с использованием защитной группы и/или функциональной группы (FGI) с целью облегчения получения целевых продуктов. Такие химические процессы хорошо известны в области синтетической органической химии, и многие из них могут быть найдены в справочниках, таких как ʺGreene's Protective Groups in Organic Synthesisʺ Peter G. M. Wuts, Theodora W. Greene (Wiley); и ʺOrganic Synthesis: The Disconnection Approachʺ Stuart Warren, Paul Wyatt (Wiley).
Контроль уровня белка
[00304] Настоящее изобретение также относится к способам контроля уровней белка в клетке. Способ основан на использовании соединений, описанных здесь, которые, как известно, специфически взаимодействуют с конкретным целевым белком-мишенью, и деградация целевого белка in vivo позволяет контролировать количество белка в биологической системе, предпочтительно, до достижения конкретного терапевтического эффекта.
[00305] Следующие примеры представлены для помощи в понимании настоящего изобретения, и они не должны рассматриваться как ограничивающие данное изобретение каким-либо образом.
Конкретные варианты осуществления изобретения
[00306] Настоящее изобретение включает в себя следующие конкретные варианты осуществления. Эти следующие варианты осуществления могут включать все признаки, указанные в выполняемом варианте изобретения, как описано. Там, где это применимо, следующие варианты осуществления могут также включать признаки, указанные в любом выполняемом варианте изобретения включительно, или в альтернативном варианте выполнения изобретения
[00307] Один аспект изобретения раскрывает соединение, имеющее химическую структуру: PTM-L-CLM, где CLM представляет собой фрагмент связывания с убиквитинлигазой Е3 цереблона; PTM представляет собой фрагмент нацеливания на белок-мишень; L представляет собой линкер, выбранный из группы, состоящей из связи (линкер отсутствует) или химической линкерной группы; при этом PTM ковалентно присоединен к CLM через линкер.
[00308] В любом из аспектов или вариантов осуществления изобретения, описанных здесь, PTM представляет собой фрагмент, который связывает бромодомен-содержащий белок, например, BRD4.
[00309] В любом из аспектов или вариантов осуществления изобретения, описанных здесь, описанное соединение может дополнительно содержать второй фрагмент связывания с убиквитинлигазой Е3, присоединенный через линкерную группу.
[00310] В любом из аспектов или вариантов осуществления изобретения, описанных здесь, CLM содержит химическую группу, которая является производным имида, фталимидной группы, талидомида, леналидомида, помалидомида или их аналогов, их изостер, или их производных; при этом CLM представлен следующими химическими структурами.
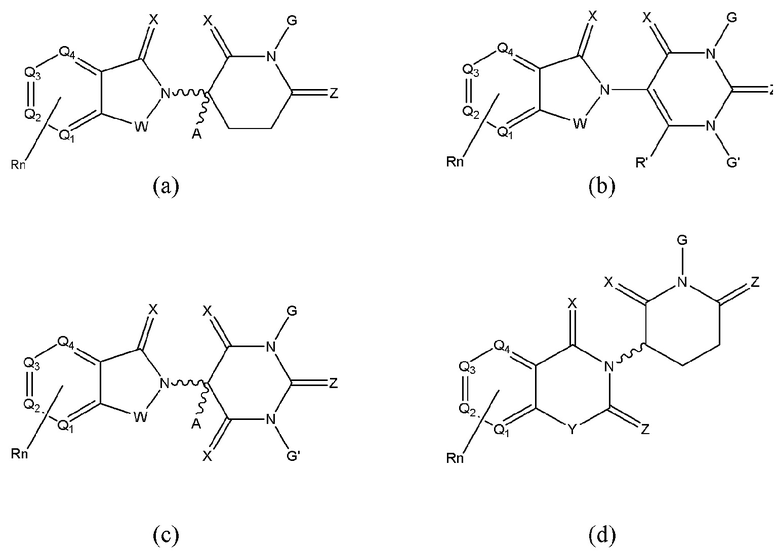
где
W выбран из группы, состоящей из CH2, CHR, C=O, SO2, NH и N-алкила;
каждый Х независимо выбран из группы, состоящей из О, S и H2;
Y независимо выбран из группы, состоящей из NH, N-алкила, N-арила, N-het-арила, N-циклоалкила, N-гетероциклила, О и S;
Z независимо выбран из группы, состоящей из O и S или H2;
G и G' независимо друг от друга выбраны из группы Н, алкила, ОН, CH2-гетероциклила, необязательно замещенного R', и бензила, необязательно замещенного R';
Q1-Q4 представляют собой углерод C, замещенный группой, независимо выбранной из R', N или N-оксида;
А независимо выбран из группы алкила, циклоалкила, Cl и F;
R включает: -CONR'R", -OR', -NR'R", -SR', -SO2R', -SO2NR'R", -CR'R"-, -CR'NR'R"-, арил, -het-арил, алкил, циклоалкил, гетероциклил, -P(O)(OR')R", P(O)R'R", -OP(O)(OR')R", -OP(O)R'R", -Cl, -F, -Br, -I, -CF3, -CN, -NR'SO2NR'R", -NR'CONR'R", -CONR'COR", -NR'C(=N-CN)NR'R", -C(=N-CN)NR'R", -NR'C(=N-CN)R", -NR'C(=C-NO2)NR'R", -SO2NR'COR", -NO2, -CO2R', -C(C=N-OR')R", -CR'=CR'R", -CCR', -S(C=O)(C=N-R')R", -SF5 и -OCF3;
R'и R" независимо выбраны из группы, состоящей из связи, Н, алкила, циклоалкила, арила, het-арила, гетероциклила;
 представляет собой связь, которая может быть стереоспецифичной ((R) или (S)) или нестереоспецифичной; и
представляет собой связь, которая может быть стереоспецифичной ((R) или (S)) или нестереоспецифичной; и
Rn включает функциональную группу или атом, где n является целым числом от 1 до 4; и где
когда n равно 1, то Rn модифицирован за счет ковалентного соединения с линкерной группой (L), и
когда n равно 2, 3 или 4, то один Rn модифицирован за счет ковалентного соединения с линкерной группой (L), и любой другой Rn необязательно может быть модифицирован за счет ковалентного соединения с PTM, ULM, со вторым CLM, имеющим такую же структуру, как CLM, CLM', вторым линкером или с любым числом их или их комбинацией.
[00311] В любом из аспектов или вариантов осуществления изобретения, описанных здесь, CLM имеет химическую структуру, представленную следующими формулами:
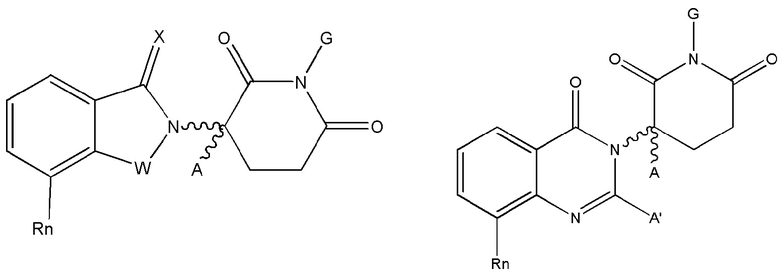
где
W выбран из группы, состоящей из CH2, C=O, SO2 и NH;
каждый Х независимо выбран из группы, состоящей из О и H2;
G независимо выбран из группы Н, алкила, ОН;
А и A' независимо выбраны из группы алкила, циклоалкила, Cl и F;
Rn включает функциональную группу или атом, где n является целым числом от 1 до 4; и где
когда n равно 1, то Rn модифицирован за счет ковалентного соединения с линкерной группой (L), и
когда n равно 2, 3 или 4, то один Rn модифицирован за счет ковалентного соединения с линкерной группой (L), и любой другой Rn необязательно может быть модифицирован за счет ковалентного соединения с PTM, ULM, со вторым CLM, имеющим такую же структуру, как CLM, CLM', вторым линкером или с любым числом их или их комбинацией.
[00312] В другом аспекте данное изобретение обеспечивает соединение, выбранное из группы, состоящей из следующих соединений:
2-[(9R)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[4-({1-[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}окси)фенил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-(4-{5-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]пиримидин-2-ил}фенил)этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-{2-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]этил}ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1R)-1-{4-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[5-(3-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропокси)пиримидин-2-ил]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[3-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)пропокси]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(4-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}бутил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(5-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пентил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(3-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(4-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}бутил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]-3-фторфенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[4-({1-[2-(3-метил-2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}окси)фенил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[4-({1-[2-(1-метил-2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}окси)фенил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1R)-1-[3-(3-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропокси)фенил]этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(3S)-1-{4-[(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этил)амино]бензоил}пирролидин-3-ил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[2-(2-{[3-(2,6-диоксопиперидин-3-ил)-2-метил-4-оксо-3,4-дигидрохиназолин-5-ил]амино}этокси)этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[3-(3-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропокси)пропил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(3S)-1-[4-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)бензоил]пирролидин-3-ил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(4-{2-[2-(2-{[3-(2,6-диоксопиперидин-3-ил)-2-метил-4-оксо-3,4-дигидрохиназолин-5-ил]амино}этокси)этокси]этокси}фенил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(5-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пентил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[3-(3-{[3-(2,6-диоксопиперидин-3-ил)-2-метил-4-оксо-3,4-дигидрохиназолин-5-ил]амино}пропокси)пропил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(3S)-1-[4-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)бензоил]пирролидин-3-ил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1R)-1-[3-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)фенил]этил]ацетамид;
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10,13-тетраокса-1-азапентадекан-15-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил;
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил;
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10,13,16-пентаокса-1-азаоктадекан-18-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил; и
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10,13,16,19-гексаокса-1-азагеникозан-21-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил,
включая их фармацевтические приемлемые соли.
[00313] В любом из описанных здесь аспектов или вариантов осуществления изобретения, линкерная группа (L) включает химическое структурное звено, представленное формулой:
-Aq-,
где:
q представляет собой целое число, большее чем 1; и
A независимо выбран из группы, состоящей из связи, CRL1RL2, O, S, SO, SO2, NRL3, SO2NRL3, SONRL3, CONRL3, NRL3CONRL4, NRL3SO2NRL4, CO, CRL1=CRL2, C≡C, SiRL1RL2, P(O)RL1, P(O)ORL1, NRL3C(=NCN)NRL4, NRL3C(=NCN), NRL3C(=CNO2)NRL4, C3-11циклоалкила, необязательно замещенного 0-6 группами RL1 и/или RL2, C3-11гетероциклила, необязательно замещенного 0-6 группами RL1 и/или RL2, арила, необязательно замещенного 0-6 группами RL1 и/или RL2, гетероарила, необязательно замещенного 0-6 группами RL1 и/или RL2;
где:
RL1, RL2, RL3, RL4 и RL5, каждый, независимо, выбран из группы, состоящей из H, галогена, C1-8алкила, OC1-8алкила, SC1-8алкила, NHC1-8алкила, N(C1-8алкил)2, C3-11циклоалкила, арила, гетероарила, C3-11гетероциклила, OC1-8циклоалкила, SC1-8циклоалкила, NHC1-8циклоалкила, N(C1-8циклоалкил)2, N(C1-8циклоалкил)(C1-8алкил), OH, NH2, SH, SO2C1-8алкила, P(O)(OC1-8алкил)(C1-8алкил), P(O)(OC1-8алкил)2, CC-C1-8алкила, CCH, CH=CH(C1-8алкил), C(C1-8алкил)=CH(C1-8алкил), C(C1-8алкил)=C(C1-8алкил)2, Si(OH)3, Si(C1-8алкил)3, Si(OH)(C1-8алкил)2, COC1-8алкил, CO2H, галогена, CN, CF3, CHF2, CH2F, NO2, SF5, SO2NHC1-8алкила, SO2N(C1-8алкил)2, SONHC1-8алкил, SON(C1-8алкил)2, CONHC1-8алкил, CON(C1-8алкил)2, N(C1-8алкил)CONH(C1-8алкил), N(C1-8алкил)CON(C1-8алкил)2, NHCONH(C1-8алкил), NHCON(C1-8алкил)2, NHCONH2, N(C1-8алкил)SO2NH(C1-8алкил), N(C1-8алкил) SO2N(C1-8алкил)2, NH SO2NH(C1-8алкил), NH SO2N(C1-8алкил)2, и NHSO2NH2; и
когда q больше 1, то RL1 или RL2, каждый, независимо, может быть связан с другой группой A, с образованием циклоалкильной и/или гетероциклильной группы, которая может быть дополнительно замещена 0-4 группами RL5.
[00314] В любом из описанных здесь аспектов или вариантов осуществления изобретения, L включает нелинейные цепи, алифатические, ароматические или гетероароматические циклические группы.
[00315] В любом из описанных здесь аспектов или вариантов выполнения изобретения, линкерная группа (L) выбрана из группы, состоящей из следующих структур:
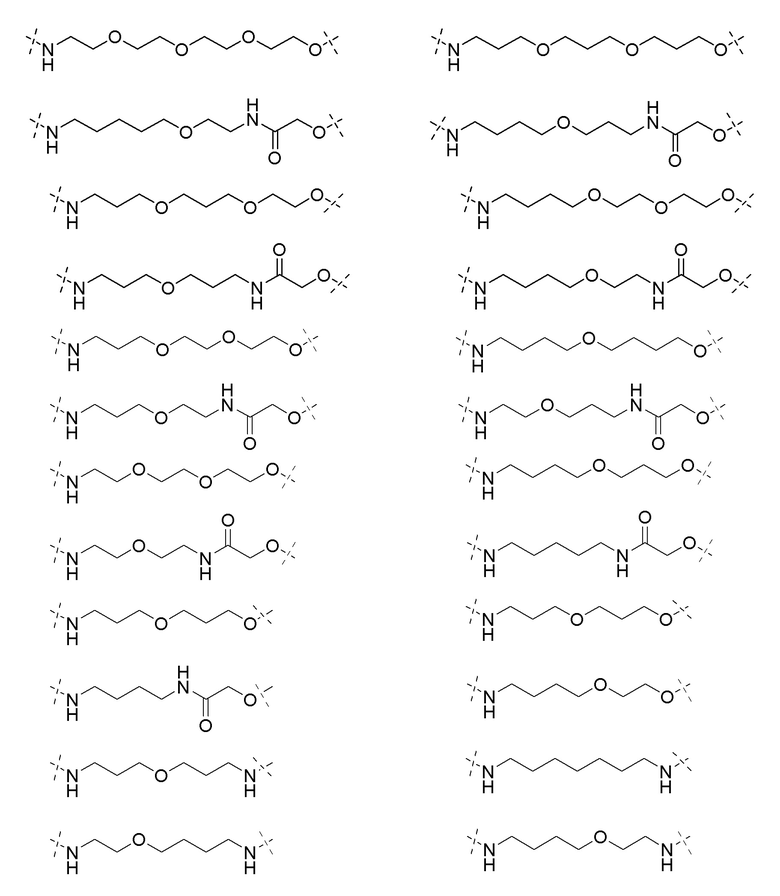
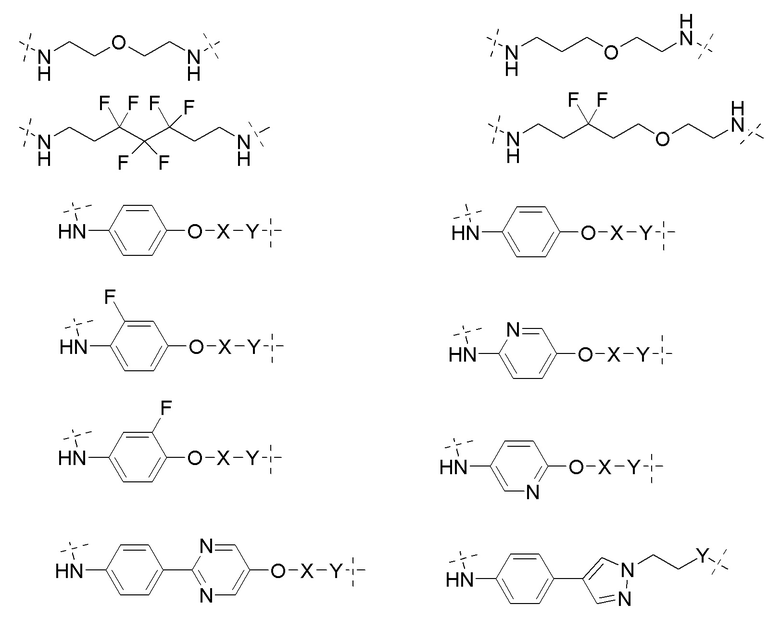
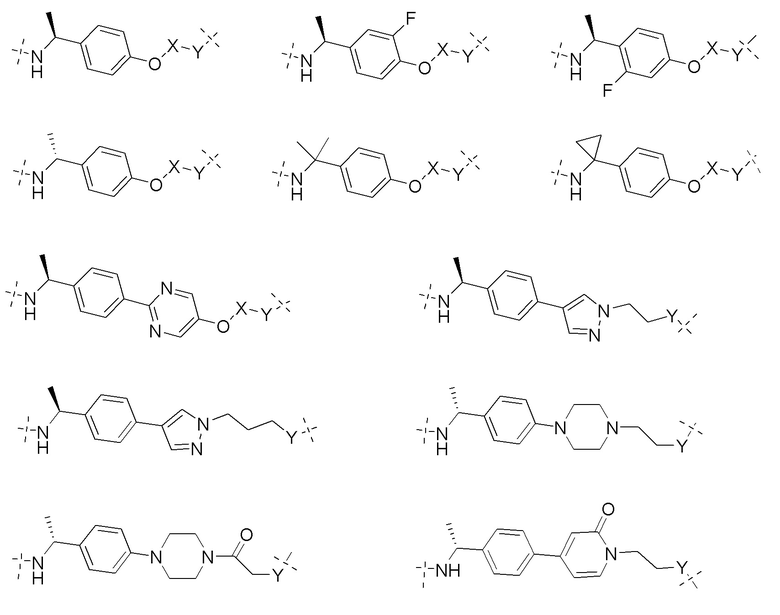
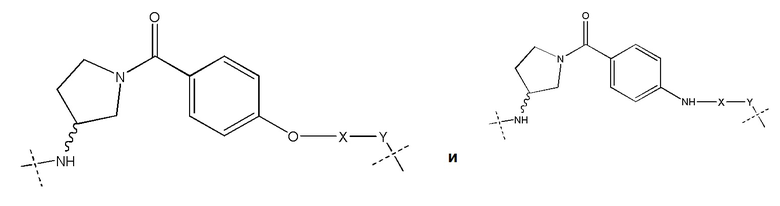
где X представляет собой линейную цепь с 2-14 атомами, необязательно замещенную гетероатомами; и
Y независимо выбран из группы, состоящей из O, N, S(O)n (n=0, 1, 2).
[00316] В дополнительном аспекте изобретение обеспечивает композиции, включающие эффективное количество соединения, описанного здесь
[00317] В других аспектах данное изобретение обеспечивает фармацевтические композиции, включающие эффективное количество соединения, описанного здесь, и фармацевтически приемлемый носитель, добавки и/или эксципиент.
[00318] В любом из описанных здесь аспектов или вариантов осуществления изобретения, фармацевтическая композиция может дополнительно содержать дополнительный биологически активный агент.
[00319] В любом из описанных здесь аспектов или вариантов осуществления изобретения, дополнительный биологически активный агент представляет собой противораковое средство.
[00320] В любом из описанных здесь аспектов или вариантов осуществления изобретения, противораковое средство выбрано из группы, состоящей из следующих средств: эверолимус, трабектин, абраксан, TLK 286, AV-299, DN-101, пазопаниб, GSK690693, RTA 744, ON 0910.Na, AZD 6244 (ARRY-142886), AMN-107, TKI-258, GSK461364, AZD 1152, энзастаурин, вандетаниб, ARQ-197, MK-0457, MLN8054, PHA-739358, R-763, AT-9263, ингибитор FLT-3, ингибитор VEGFR, ингибитор TK EGFR, ингибитор авроракиназы, модулятор PIK-1, ингибитор Bcl-2, ингибитор HDAC, ингибитор c-MET, ингибитор PARP, ингибитор Cdk, ингибитор TK EGFR, ингибитор IGFR-TK, анти-HGF-антитело, ингибиторы PI3-киназы, ингибитор AKT, ингибитор mTORC1/2, ингибитор JAK/STAT, ингибитор контрольной точки 1 или 2, ингибитор киназы фокальной адгезии, ингибитор Map киназы (mek), антитело к ловушке VEGF, пеметрексед, эрлотиниб, дасатаниб, нилотиниб, декатаниб, панитумумаб, амрубицин, ореговомаб, Lep-etu, нолатрексед, azd2171, батабулин, офатумумаб, занолимумаб, эдотерацин, тетрандрин, рубитекан, тесмилифен, облимерсен, тицилимумаб, ипилимумаб, госсипол, Bio 111, 131-I-TM-601, ALT-110, BIO 140, CC 8490, циленгитид, гиматекан, IL13-PE38QQR, INO 1001, IPdR1 KRX-0402, лукантон, LY317615, нейрадиаб, витеспан, Rta 744, Sdx 102, талампанел, атрасентан, Xr 311, ромидепсин, ADS-100380, сунитиниб, 5-фторурацил, вориностат, этопозид, гемцитабин, доксорубицин, липосомный доксорубицин, 5'-деокси-5-фторуридин, винкристин, темозоломид, ZK-304709, селициклиб; PD0325901, AZD-6244, капецитабин, L-глутаминовую кислоту, гептагидрат динатриевой соли N-[4-[2-(2-амино-4,7-дигидро-4-оксо-1H-пирроло[2,3-d]пиримидин-5-ил)этил]бензоил], камптотецин, PEG-меченный иринотекан, тамоксифен, цитрат тофемифена, анастразол, эксеместан, летрозол, дез(диэтилстилбестрол), эстрадиол, эстроген, конъюгированный эстроген, бевацизумаб, IMC-1C11, CHIR-258); 3-[5-(метилсульфонилпиперадинметил)индолил]хинолон, ваталаниб, AG-013736, AVE-0005, уксуснокислая соль [D- Ser(But)6,Azgly10](pyro-Glu-His-Trp-Ser-Tyr-D-Ser(But)-Leu-Arg-Pro-Azgly-NH2 ацетата [C59H84N18O14-(C2H4O2)X, где x=1-2,4], ацетат госерелина, ацетат лейпролида, памоат трипторелина, ацетат гидроксипрогестерона, капроат гидроксипрогестерона, ацетат мегестрола, ралоксифен, бикалутамид, флутамид, нилутамид, ацетат мегестрола, CP-724714; TAK-165, HKI-272, эрлотиниб, лапатаниб, канертиниб, антитело ABX-EGF, эрбитукс, EKB-569, PKI-166, GW-572016, ионафарниб, BMS-214662, типифарниб; амифостин, NVP-LAQ824, субероиланалид гидроксамовой кислоты, вальпроевая кислота, трихостатин A, FK-228, SU11248, сорафениб, KRN951, аминоглутетимид, арнсакрин, анагрелид, L-аспарагиназа, вакцина Bacillus Calmette-Guerin (BCG), адриамицин, блеомицин, бусерелин, бусульфан, карбоплатин, кармустин, хлорамбуцил, цисплатин, кладрибин, клодронат, ципротерон, цитарабин, дакарбазин, дактиномицин, даунорубицин, диэтилстилбестрол, эпирубицин, флударабин, флудрокортизон, флуоксиместерон, флутамид, глеевек, гемцитабин, гидроксимочевина, идарубицин, ифосфамид, иматиниб, лейпролид, левамизол, ломустин, мехлорэтамин, мелфалан, 6-меркаптопурин, месна, метотрексат, митомицин, митотан, митоксантрон, нилутамид, октреотид, оксалиплатин, памидронат, пентостатин, пликамицин, порфимер, прокарбазин, ралтитрексед, ритуксимаб, стрептозоцин, тенипозид, тестостерон, талидомид, тиогуанин, тиотепа, третиноин, виндезин, 13-цис-ретиноевая кислота, мелфалан, урамустин, эстрамустин, альтретамин, флоксуридин, 5-деоксиуридин, цитозин арабинозид, 6-мекаптопурин, деоксикоформицин, кальцитриол, валрубицин, митрамицин, винбластин, винорелбин, топотекан, разоксин, маримастат, COL-3, неофастат, BMS-275291, скваламин, эндостатин, SU5416, SU6668, EMD121974, интерлейкин-12, IM862, ангиостатин, витаксин, дролоксифен, идоксифен, спиронолактон, финастерид, цимитидин, трастузумаб, денилейкин дифтитокс, гефитиниб, бортезимиб, паклитаксел, безкремофорный паклитаксел, доцетаксел, эпитилон B, BMS-247550, BMS-310705, дролоксифен, 4-гидрокситамоксифен, пипендоксифен, ERA-923, арзоксифен, фулвестрант, аколбифен, лазофоксифен, идоксифен, TSE-424, HMR-3339, ZK186619, топотекан, PTK787/ZK 222584, VX-745, PD 184352, рапамицин, 40-O-(2-гидроксиэтил)рапамицин, темсиролимус, AP-23573, RAD001, ABT-578, BC-210, LY294002, LY292223, LY292696, LY293684, LY293646, вортманнин, ZM336372, L-779,450, PEG-филграстим, дарбепоэтин, эритропоэтин, гранулоцитарный колониестимулирующий фактор, преднизон, цетуксимаб, гранулоцитарный макрофаговый колониестимулирующий фактор, гистрелин, пэгилированный интерферон альфа-2a, интерферон альфа-2a, пэгилированный интерферон альфа-2b, интерферон альфа-2b, азацитидин, PEG-L-аспарагиназу, леналидомид, гемтузумаб, гидрокортизон, интерлейкин-11, дексразоксан, алемтузумаб, всетрансетиновая кислота, кетоконазол, интерлейкин-2, мегестрол, иммунноглобулин, азотистый иприт, метилпреднизолон, ибритгумомаб тиуксетан, андрогены, децитабин, гексаметилмеламин, бексаротин, тоцитумомаб, триоксид мышьяка, кортизон, эдитронат, митотан, циклоспорин, липосомальный даунорубицин, аспарагиназа Эдвина, стронций 89, казопитант, нетупитант, антагонист рецептора NK-1, палоносетрон, апрепитант, дифенгидрамин, гидроксизин, метоклопрамид, лоразепам, альпразолам, галоперидол, дроперидол, дронабинол, дексаметазон, метилпреднизолон, прохлорперазин, гранисетрон, ондансетрон, доласетрон, трописетрон, пегфилграстим, эритропоэтин, эпоэтин альфа, дарбепоэтин альфа и их смеси.
[00321] В дополнительном аспекте изобретение предоставляет способы индуцирования деградации целевого белка-мишени в клетке, включающие введение эффективного количества соединения, описанного здесь, в клетку, где соединение обеспечивает эффективную деградацию белка-мишени.
[00322] В других дополнительных аспектах изобретение предоставляет композиции, содержащие эффективное количество соединения, описанного здесь, для применения в способе лечения рака, где указанный способ включает введение композиции пациенту, нуждающемуся в этом, и при этом композиция обеспечивает эффективное лечение рака или облегчение по меньшей мере одного симптома рака у пациента.
[00323] В любом из аспектов или вариантов осуществления изобретения, описанных здесь, рак представляет собой плоскоклеточную карциному, базальноклеточную карциному, аденокарциному, гепатоцеллюлярные карциномы и карциномы клеток почек, рак мочевого пузыря, кишечника, груди, шейки матки, толстой кишки, пищевода, головы, почек, печени, легких, шеи, яичников, поджелудочной железы, предстательной железы и желудка; лейкозы; доброкачественные и злокачественные лимфомы, особенно лимфому Беркитта и неходжкинскую лимфому; доброкачественные и злокачественные меланомы; миелопролиферативные заболевания; саркомы, включая саркому Юинга, гемангиосаркому, саркому Капоши, липосаркому, миосаркомы, периферическую нейроэпителиому, синовиальную саркому, глиомы, астроцитомы, олигодендроглиомы, эпендимомы, глиобластомы, нейробластомы, ганглионевромы, ганглиоглиомы, медуллобластомы, опухоли шишковидных клеток, менингиомы, менингеальные саркомы, нейрофибромы и шванномы; рак толстой кишки, рак молочной железы, рак предстательной железы, рак шейки матки, рак матки, рак легких, рак яичников, рак яичек, рак щитовидной железы, астроцитому, рак пищевода, рак поджелудочной железы, рак желудка, рак печени, рак толстой кишки, меланому; карциносаркому, болезнь Ходжкина, опухоль Вильмса или тератокарциномы, T-клеточный острый лимфобластный лейкоз (T-ALL), T-клеточную лимфобластную лимфому (T-LL), периферическую T-клеточную лимфому, Т-клеточный лейкоз взрослых, пре-B ALL, пре-B лимфомы, крупноклеточную B-клеточную лимфому, лимфому Беркитта, B-клеточный ALL, положительный к филадельфийской хромосоме ALL и положительный к филадельфийской хромосоме CML.
[00324] В любом из аспектов или вариантов осуществления изобретения, описанных здесь, CLM соединен с PTM, имеющим структуру, выбранную из группы, состоящей из следующего:
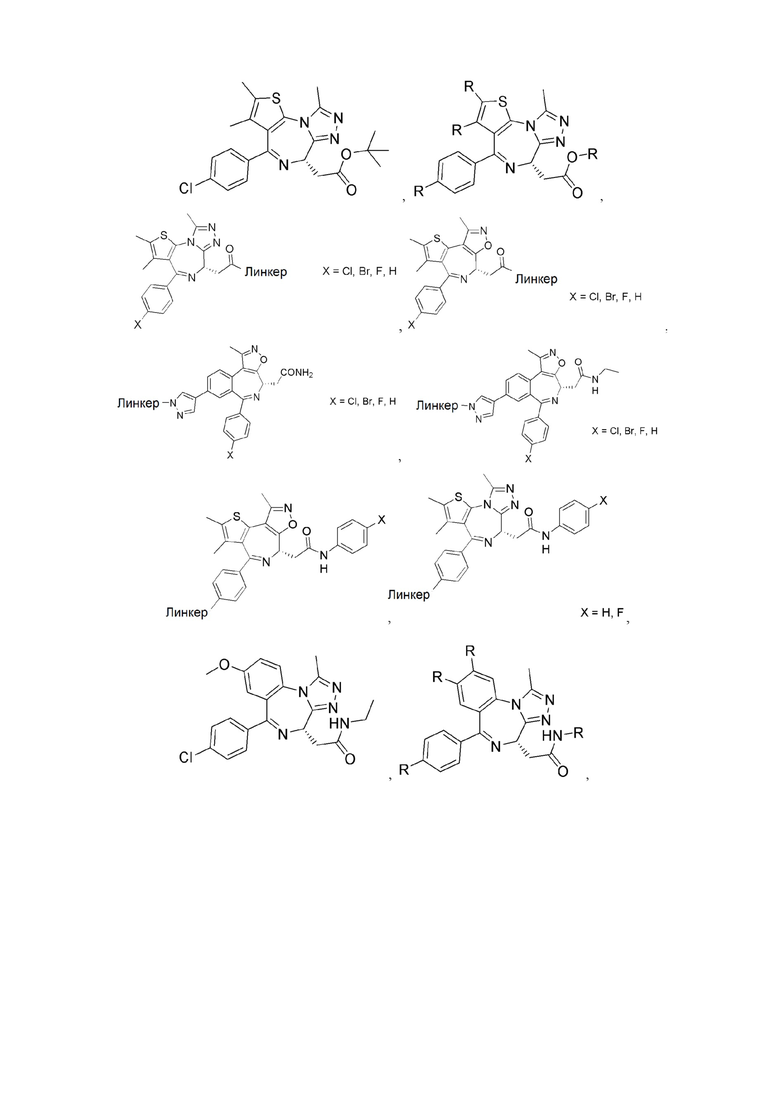
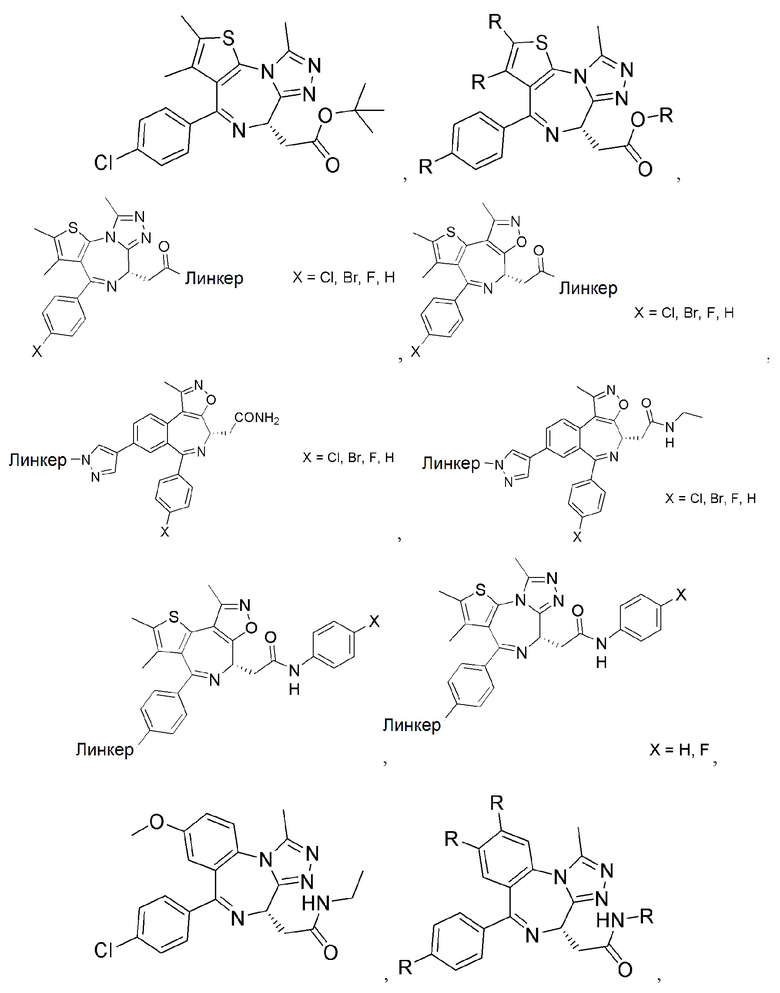
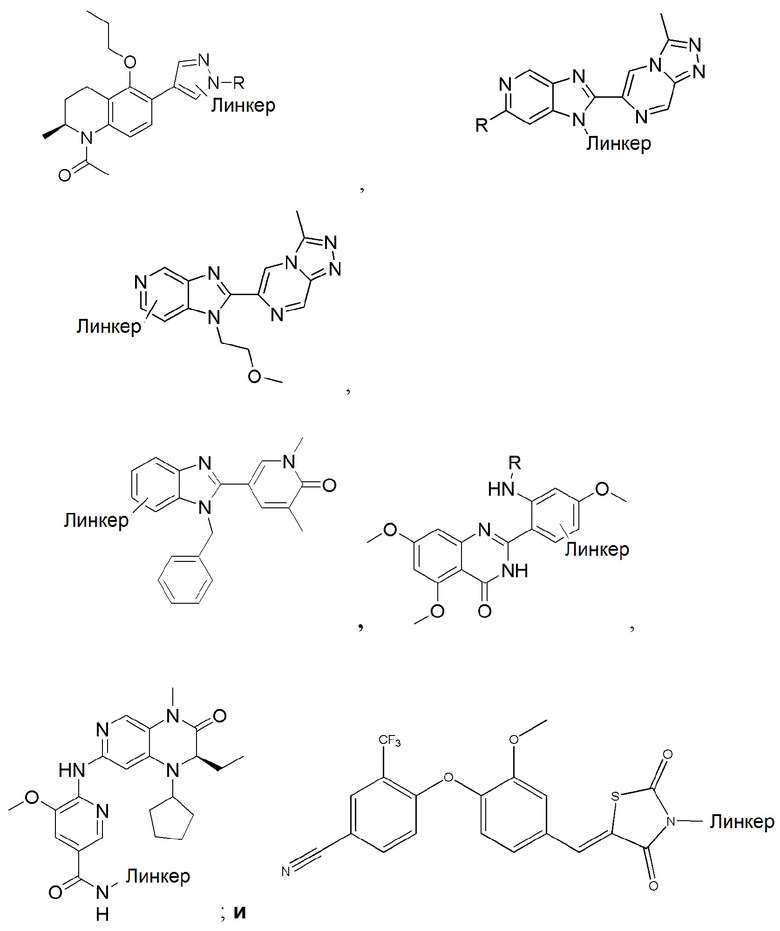
где
R или линкер представляет собой связь или химический линкерный фрагмент, который соединяет CLM с PTM, включая фармацевтически приемлемые соли.
Примеры
[00325] А. Клонирование, экспрессия и очистка CRBN и DDB1 человека
Процедура является стандартным приемом, известным специалистам в данной области техники, поскольку она описана как типовая методика в Lopez-Girona et al. (Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide, A Lopez-Girona, D Mendy, T Ito, K Miller, A K Gandhi, J Kang, S Karasawa, G Carmel, P Jackson, M Abbasian, A Mahmoudi, B Cathers, E Rychak, S Gaidarova, R Chen, P H Schafer, H Handa, T O Daniel, J F Evans and R Chopra, Leukemia 26: 2326-2335, 2012).
[00326] кДНК для генов CRBN и DDB1 могут быть амплифицированы с помощью ПЦР, с использованием в качестве полимеразы Pfusion (NEB) и следующих праймерных последовательностей:
(SEQ ID NO: 1)
(SEQ ID NO: 2)
(SEQ ID NO: 3)
(SEQ ID NO: 4)
(SEQ ID NO: 5)
[00327] CRBN можно клонировать в pBV-ZZ-HT-LIC, pBV-GST-LIC, pMA-HT-LIC, а DDB1 - в pBV-notag-LIC, используя безлигазное клонирование 26. Для клонирования в векторе млекопитающего pMA-HT-LIC, олигонуклеотид CRBN-Flag-обратный добавляет C-концевую метку FLAG для иммунодетекции. DDB1-Rev добавляет метку StrepTag 27. ZZ-tag 28 необходим для достижения высокой экспрессии растворимого CRBN; без него His-CRBN экспрессируется на низком уровне, тогда как GST-CRBN приводит к агрегированному белку. Получали и амплифицировали рекомбинантный бакуловирус из ZZ-His-CRBN и DDB1-StrepTag (ST), используя систему экспрессии бакуловируса Bac-to-Bac от Invitrogen в клетках насекомых Sf9. ZZ-His-CRBN и DDB1-ST совместно экспрессировали в клетках насекомых High Five (Tni) в 10-литровых мешках при 27°C, с использованием недополненной среды ESF921 от Expression Systems. Клетки собирали через 48 часов после заражения путем центрифугирования, и полученную пасту повторно суспендировали в PBS с 5X коктейле протеазных ингибиторов (Roche, Indianapolis, IN).
[00328] Все последующие стадии очистки белка проводили при температуре 4°С. Замороженные клетки оттаивали, суспендировали в 5 объемах буфера для лизиса (50 мМ Трис-HCl, рН 8,0, 0,5М NaCl, 10% глицерина, 2 мМ DTT) плюс 20 мМ имидазола и ингибиторов протеазы, лизировали и центрифугировали с получением чистого супернатанта. CRBN-DDB1 очищали на системе Akta-Xpress (GE Healthcare) с использованием хроматографии на никель-сефарозе и S200 Sephacryl. Затем комплекс дополнительно очищали с помощью анионообменной хроматографии на колонке 8 мл MonoQ и повторно выполняли гель-фильтрацию на геле S-200. CRBN-DDB1 идентифицировали с помощью SDS-PAGE, и объединенные фракции CRBN-DDB1 хранили при -70°С.
2. Флуоресцентный анализ расплава для измерения связывания соединений с рекомбинантным CRBN
[00329] Процедура является стандартным приемом, известным специалистам в данной области техники, поскольку она описана как типовая методика в Lopez-Girona et al. (Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide, A Lopez-Girona, D Mendy, T Ito, K Miller, A K Gandhi, J Kang, S Karasawa, G Carmel, P Jackson, M Abbasian, A Mahmoudi, B Cathers, E Rychak, S Gaidarova, R Chen, P H Schafer, H Handa, T O Daniel, J F Evans and R Chopra, Leukemia 26: 2326-2335, 2012).
[00330] Термическая стабильность CRBN-DDB1 в присутствии или в отсутствии испытуемых соединений испытывалась с использованием Sypro Orange в микропланшетном формате, как описано в Pantoliano et al. (Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E et al. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen 2001; 6: 429-440). Два мг белка в 20 мл буфера для анализа (25 мМ Трис-HCl, рН 8,0, 150 мМ NaCl, 2 мкМ Sypro Orange) подвергали ступенчатому повышению температуры от 20 до 70°С, и при каждом повышении температуры на 1°С регистрировали флуоресценцию на ABIPrism 7900HT (Applied Biosystems, Carlsbad, CA, USA). Соединения растворяли в DMSO (конечная концентрация при анализе 1%) и тестировали в четырех повторностях в диапазоне концентраций от 30 нМ до 1000 мкМ; контроль содержал только 1% DMSO.
3. Метод LCMS
[00331] Анализ проводили на колонке Poroshell 120 EC C18 (50 мм х 3,0 мм (внутренний диаметр), 2,7 мкм (диаметр наполнителя)) при температуре 45оС.
[00332] В качестве растворителей использовали следующее:
A=0,1% об./об. раствор муравьиной кислоты в воде.
В=0,1% об./об. раствор муравьиной кислоты в ацетонитриле.
[00333] Использовали следующие градиенты:
(мин)
(мл/мин)
[00334] УФ-детектирование выполняли по усредненному сигналу с длиной волны от 210 нм до 350 нм, и масс-спектры регистрировали на масс-спектрометре с использованием режима положительной ионизации при электрораспылении.
[00335] Ниже показаны подвижные фазы и градиенты, которые использовали при очистке соединения с помощью препаративной HPLC (ВЭЖХ).
4. Препаративная HPLC (модификатор - муравьиная кислота)
[00336] Анализ HPLC проводили на колонке X Bridge RP18 OBD (150 мм х 19 мм (внутренний диаметр), 5 мкм (диаметр наполнителя)) при температуре окружающей среды.
[00337] В качестве растворителей использовали следующее:
A=0,1% об./об. раствор муравьиной кислоты в воде.
В=ацетонитрил.
5. Препаративная HPLC (модификатор - бикарбонат аммония)
[00338] Анализ HPLC проводили на колонке X Bridge RP18 OBD (150 мм х 19 мм (внутренний диаметр), 5 мкм (диаметр наполнителя)) при температуре окружающей среды.
[00339] В качестве растворителей использовали следующее:
A=10 мМ раствор бикарбоната аммония в воде.
В=ацетонитрил.
[00340] Для каждой из препаративных очисток, независимо от используемого модификатора, применяемый градиент зависел от времени удерживания конкретного соединения, подвергающегося очистке, как указано в описании анализа LCMS. Скорость потока составляла 20 мл/мин.
[00341] УФ-детектирование выполняли по сигналу с длиной волны 254 нм или 220 нм.
[00342] Предпочтительные варианты осуществления настоящего изобретения были показаны и описаны здесь выше, однако следует понимать, что такие варианты осуществления приведены только в качестве примера. Многочисленные вариации, изменения и замены без отхода от сущности настоящего изобретения, являются очевидными для специалистов в данной области техники. Соответственно, предполагается, что прилагаемая формула изобретения охватывает все такие изменения, которые охватываются сущностью и объемом настоящего изобретения.
В. Синтез
[00343] Примеры синтезов, представленные ниже, отражают общие процедуры, которые являются типовыми для более широкого набора примеров синтеза.
[00344] 1. 2-(2,6-диоксопиперидин-3-ил)-4-фтор-2,3-дигидро-1Н-изоиндол-1,3-дион
[00345] Стадия 1: 4-фторизобензофуран-1,3-дион
[00346] Смесь 3-фторфталевой кислоты (50 г, 271,7 ммоль) в уксусном ангидриде (400 мл) кипятили с обратным холодильником в течение 2 часов. Летучие вещества удаляли в вакууме, и остаток выкристаллизовывали из уксусного ангидрида, с получением 4-фторизобензофуран-1,3-диона (40 г, неочищенного) в виде коричневого твердого вещества. LC-MS: 167,1 [МН]+. 1H-ЯМР (400 МГц, CDCl3): δ 7,58 (т, J=8,0 Гц, 1H), 7,86 (д, J=7,2 Гц, 1H), 7,92-7,97 (м, 1H).
[00347] Стадия 2: 5-амино-2-(4-фтор-1,3-диоксоизоиндолин-2-ил)-5-оксопентановая кислота
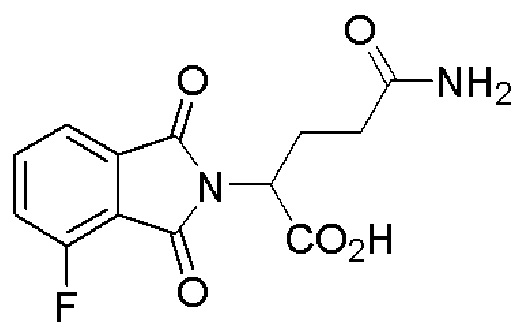
[00348] Смесь указанного выше 4-фторизобензофуран-1,3-диона (40 г, неочищенного) и L-глутамина (35 г, 239 ммоль) в сухом DMF (200 мл) перемешивали при 90°C в течение 8 часов. Растворитель удаляли при пониженном давлении. Остаток повторно растворяли в 4 Н HCl (200 мл) и перемешивали еще в течение 8 часов. Полученный осадок собирали фильтрованием, промывали водой и сушили с получением 5-амино-2-(4-фтор-1,3-диоксоизоиндолин-2-ил)-5-оксопентановой кислоты (37 г, неочищенной) в виде беловатого твердого вещества. LC-MS: 295,2 [МН]+. 1H-ЯМР (400 МГц, CDCl3): δ 2,16-2,20 (м, 2Н), 2,31-2,43 (м, 2H), 4,79-4,83 (м, 1H), 6,79 (шир, 1H), 7,26 (шир, 1H), 7,77-7,85 (м, 2H), 7,98-8,03 (м, 1H), 13,32 (шир, 1H).
[00349] Стадия 3: 2-(2,6-диоксопиперидин-3-ил)-4-фтор-2,3-дигидро-1Н-изоиндол-1,3-дион
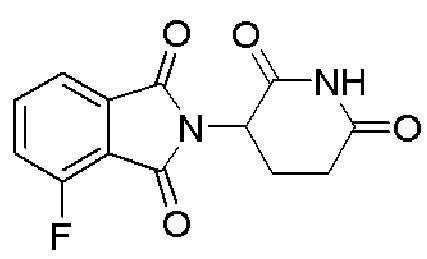
[00350] Смесь полученной выше 5-амино-2-(4-фтор-1,3-диоксоизоиндолин-2-ил)-5-оксопентановой кислоты (37 г, неочищенной), 1,1'-карбонилдиимидазола (CDI), (24,2 г, 149,4 ммоль) и 4-диметиламинопиридин (DMAP) (1,3 г, 11,5 ммоль) в ацетонитриле (80 мл) кипятили с обратным холодильником в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры. Полученное твердое вещество собирали фильтрованием и промывали ацетонитрилом (100 мл) с получением неочищенного продукта, который очищали с помощью хроматографии на силикагеле с использованием 1-10% MeOH в DCM в качестве элюента, с получением 2-(2,6-диоксопиперидин-3-ил)-4-фторизоиндолин-1,3-диона (9,0 г, выход 12% за три стадии) в виде светло-желтого твердого вещества. LC-MS: 277,2 [МН]+. 1H-ЯМР (400 МГц, CDCl3): δ 2,14-2,19 (м, 1H), 2,75-2,95 (м, 3H), 4,97-5,01 (м, 1H), 7,43 (т, J=8,4 Гц, 1H), 7,10-7,81 (м, 2H), 8,08 (шир, 1H).
[00351] 2. N-(3-(5-бром-2-хлорпиримидин-4-ил)пропил)-N-метилциклобутан карбоксамид
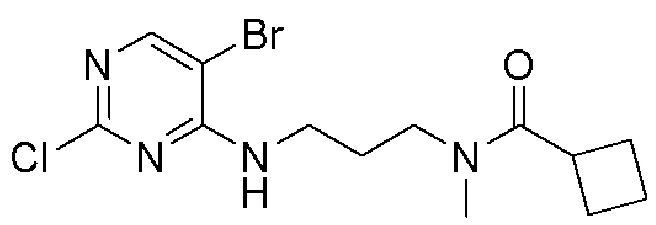
[00352] Стадия 1: трет-бутил-N-{3-[(5-бром-2-хлорпиримидин-4-ил)амино]пропил}-N-метилкарбамат
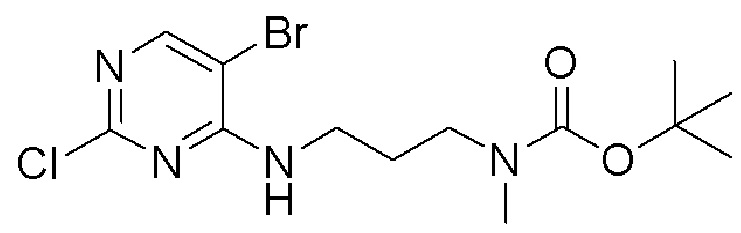
[00353] Смесь трет-бутил-N-(3-аминопропил)-N-метилкарбамата (826 мг, 4,40 ммоль) и 5-бром-2,4-дихлорпиримидина (400 мг, 1,76 ммоль) в МеОН (10 мл) перемешивали при комнатной температуре в течение 1 часа. Затем реакционную смесь концентрировали в вакууме, и остаток ощищали с помощью хроматографии Teledyne ISCO [0 → 35% EtOAc/гептан] с получением трет-бутил-N-{3-[(5-бром-2-хлорпиримидин-4-ил)амино]пропил}-N-метилкарбамата (615 мг, выход 92%). LC-MS (ES+): m/z=381,05/383,05 [МН+], tR=2,55 мин.
[00354] Стадия 2: {3-[(5-бром-2-хлорпиримидин-4-ил)амино]пропил}(метил)амин
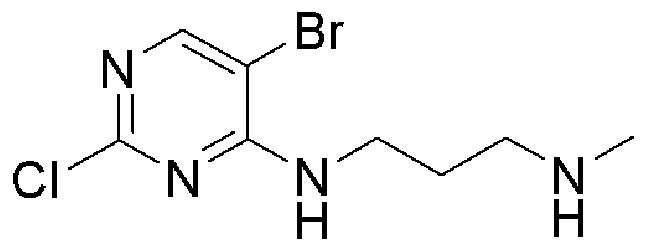
[00355] К раствору трет-бутил-N-{3-[(5-бром-2-хлорпиримидин-4-ил)амино]пропил}-N-метилкарбамата (615 мг, 1,62 ммоль) в DCM (5 мл) добавляли трифторуксусную кислоту (0,54 мл, 6,5 ммоль) при комнатной температуре. После перемешивания смеси в течение 1 часа ее концентрировали в вакууме. Остаток очищали с помощью хроматографии Teledyne ISCO [0 → 15% метанол в DCM], получая {3-[(5-бром-2-хлорпиримидин-4-ил)амино]пропил}(метил)амин (371 мг, выход 82%). LC-MS (ES+): m/z=280,99/282,99 [МН+], tR=1,13 мин.
[00356] Стадия 3: N-{3-[(5-бром-2-хлорпиримидин-4-ил)амино]пропил}-N-метилциклобутан карбоксамид
[00357] К раствору {3-[(5-бром-2-хлорпиримидин-4-ил)амино] пропил}(метил)амина (371 мг, 1,33 ммоль) и циклобутанкарбонилхлорида (188 мг, 1,60 ммоль) в DCM (10 мл) при комнатной температуре добавляли триэтиламин (0,41 мл, 2,92 ммоль). Реакционную смесь оставляли перемешиваться при комнатной температуре в течение 16 часов, затем концентрировали в вакууме. Остаток очищали с помощью хроматографии ISCO Teledyne [0 → 100% EtOAc/гептан] с получением N-{3-[(5-бром-2-хлорпиримидин-4-ил)амино]пропил}-N-метилциклобутан карбоксамида (268 мг, 56%). LC-MS (ES+): m/z=363,04/365,04 [МН+], tR=2,18 мин.
[00358] 3. (S)-2-(4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-а][1,4]диазепин-6-ил)уксусная кислота
[00359] Указанное в заголовке соединение получали в соответствии с процедурами, описанными в WO 2011/143660.
[00360] 4. (Z)-4-(4-((2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрил
[00361] Указанное в заголовке соединение получали в соответствии с процедурами, описанными в Patch, R. J. et al J. Med. Chem. 2011, 54, 788-808.
[00362] 5. 4-[3-(4-гидроксифенил)-4,4-диметил-5-оксо-2-сульфанилиденимдазолидин-1-ил]-2-(трифторметил)бензонитрил
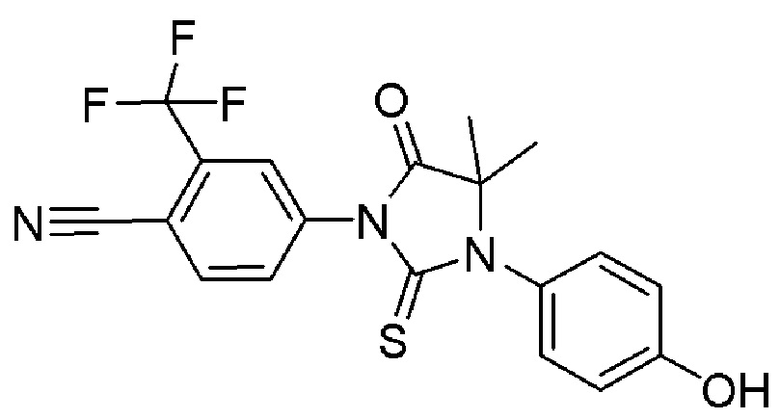
[00363] Указанное в заголовке соединение получали в соответствии с процедурами, описанными в in Jung, M. E. et al J. Med. Chem. 2010, 53, 2779-2796.
[00364] 6. Соль 2-хлор-4-(транс-3-амино-2,2,4,4-тетраметилциклобутокси)бензонитрила с хлористоводородной кислотой
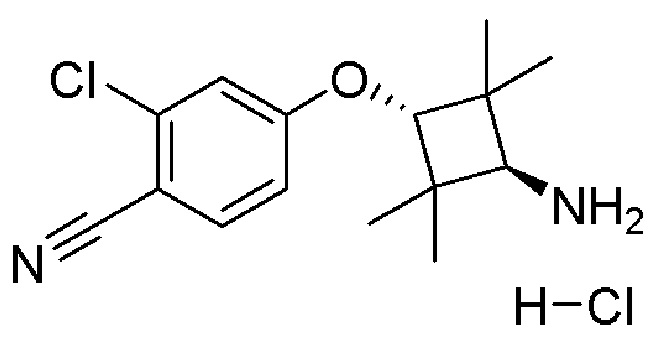
[00365] Указанное в заголовке соединение получали в соответствии с процедурами, описанными в Guo, C. et al J. Med. Chem. 2011, 54, 7693-7704.
[00366] 7. [N-(3-(5-бром-2-(4-(2-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)этокси)этокси)этокси)этокси)фениламино)пиримидин-4-иламино)пропил)-N-метилциклобутан карбоксамид
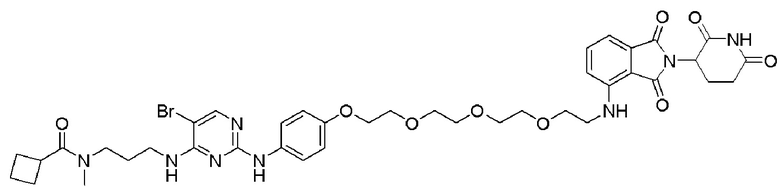
[00367] (Соединение со структурой # 17 показано в таблице 1)
[00368] Стадия 1: 2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этил-4-метилбензолсульфонат
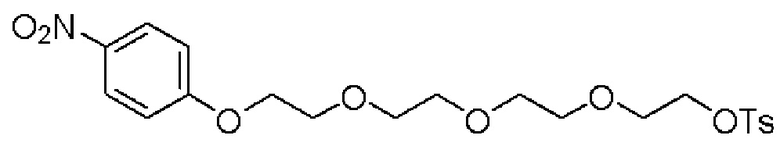
[00369] Смесь 2,2'-(2,2'-окси-бис-(этан-2,1-диил)бис(окси))бис(этан-2,1-диил)бис(4-метилбензолсульфонат) (3 г, 5,96 ммоль), 4-нитрофенола (813 мг, 5,84 ммоль) и карбоната калия (1,65 г, 11,94 ммоль) в сухом N,N-диметилформамиде (20 мл) перемешивали при 50ос в течение ночи. Смесь охлаждали до комнатной температуры и выливали в воду (60 мл), затем экстрагировали этилацетатом (80 мл х 3). Объединенные органические фазы промывали водой (50 мл) и насыщенным раствором соли (50 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 10-20% этилацетат в гексане) с получением 2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этил-4-метилбензолсульфоната (2,65 г, выход 95%) в виде желтого масла. LC-MS (ES+): m/z 470,2 [МН+] (tR=2,83 мин)
[00370] Стадия 2: [1-(2-(2-(2-(2-азидоэтокси)этокси)этокси)этокси)-4-нитробензол]
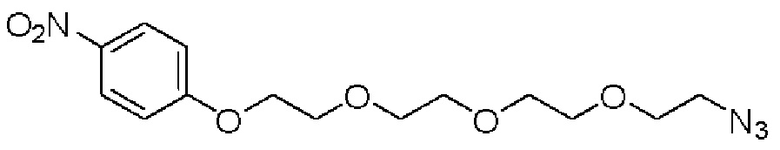
[00371] Смесь 2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этил-4-метилбензолсульфоната (2,65 г, 5,64 ммоль) и азида натрия (734 мг, 11,29 ммоль) в этаноле (30 мл) кипятили с обратным холодильником в течение 16 часов. Смесь охлаждали до комнатной температуры, гасили водой (50 мл) и экстрагировали дихлорметаном (50 мл х 3). Объединенные органические фазы промывали водой (50 мл) и насыщенным раствором соли (40 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного 1-(2-(2-(2-(2-азидоэтокси)этокси)этокси)этокси)-4-нитробензола (865 мг) в виде желтого масла.
[00372] Стадия 3: [2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этанамин]
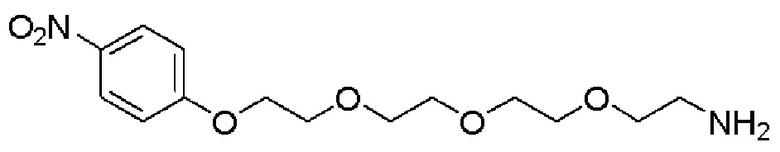
[00373] Смесь полученного выше 1-(2-(2-(2-(2-азидоэтокси)этокси)этокси)этокси)-4-нитробензола (865 мг, 2,54 ммоль), трифенилфосфина (999 мг, 3,81 ммоль) и воды (69 мг, 3,83 ммоль) в тетрагидрофуране (10 мл) перемешивали при комнатной температуре в течение 14 часов в атмосфере азота. Летучие вещества удаляли при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюирование смесью 3-5% метанола в дихлорметане) с получением 2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этанамина (661 мг, выход 83%) в виде желтого масла. 1H-ЯМР (400 МГц, CDCl3): δ 2,86 (т, J=5,2 Гц, 2H), 3,51 (т, J=5,6 Гц, 2H), 3,63-3,75 (м, 8Н), 3,90 (т, J=4,4 Гц, 2H), 4,23 (т, J=4,8 Гц, 2H), 6,97-6,99 (м, 2H), 8,18-8,22 (м, 2H).
[00374] Стадия 4: трет-бутил-2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этилкарбамат
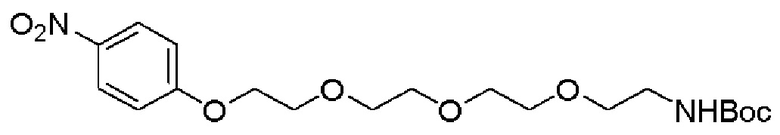
[00375] Смесь 2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этанамина (661 мг, 2,1 ммоль), триэтиламина (449 мг, 4,43 ммоль) и ди-трет-бутилдикарбоната (505 мг, 2,31 ммоль) в дихлорметане (25 мл) перемешивали при комнатной температуре в течение 2 часов. Смесь разбавляли дихлорметаном (100 мл), промывали водой (30 мл х 2) и насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 20-40% этилацетат в гексане) с получением трет-бутил-2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этилкарбамата (818 мг, выход 94%) в виде желтого масла. 1H-ЯМР (400 МГц, CDCl3): δ 1,44 (с, 9H), 3,37 (д, J=5,2 Гц, 2H), 3,54 (т, J=5,2 Гц, 2H), 3,62-3,70 (м, 6H ), 3,73-3,76 (м, 2H), 3,90 (т, J=4,4 Гц, 2H), 4,23 (т, J=4,8 Гц, 2H), 5,01 (шир, 1H), 6,96-7,00 (м, 2H), 8,18-8,22 (м, 2H).
[00376] Стадия 5: трет-бутил-2-(2-(2-(2-(4-аминофенокси)этокси)этокси)этокси)этилкарбамат
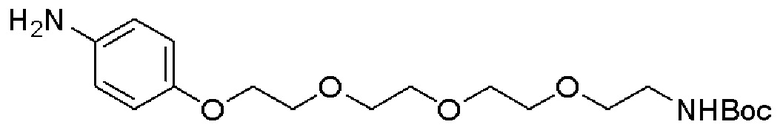
[00377] Смесь 2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этилкарбамата (818 мг, 1,97 ммоль), порошка железа (1,1 г, 0,65 ммоль) и хлорида аммония (528 мг, 9,87 ммоль) в смеси этанола (20 мл) и воды (5 мл) перемешивали при 80°С в течение 1 часа. Смесь охлаждали до комнатной температуры, твердый осадок удаляли фильтрованием и промывали этилацетатом (20 мл х 2). Фильтрат разделяли между этилацетатом (120 мл) и водой (30 мл). Органическую фазу промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью хроматографии на силикагеле (элюент: 30-40% этилацетата в гексане) с получением трет-бутил-2-(2-(2-(2-(4-аминофенокси)этокси)этокси)этокси)этилкарбамата (512 мг, выход 67%) в виде желтого масла.
[00378] Стадия 6: трет-бутил-2-(2-(2-(2-(4-(5-бром-4-(3-(N-метилциклобутанкарбоксамидо)пропиламино)пиримидин-2-иламино)фенокси)этокси)этокси)этокси)этилкарбамат
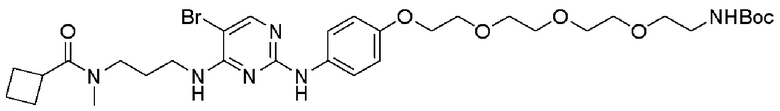
[00379] Смесь трет-бутил-2-(2-(2-(2-(4-аминофенокси)этокси)этокси)этокси)этилкарбамата (130 мг, 0,34 ммоль), N-(3-(5-бром-2-хлорпиримидин-4-ил)пропил)-N-метилциклобутанкарбоксамида (24 мг, 0,06 ммоль) и п-толуолсульфоновой кислоты (11,6 мг, 0,07 ммоль) в диоксане (1,5 мл) нагревали с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, гасили водным раствором бикарбоната натрия (1,0 Н, 30 мл) и экстрагировали этилацетатом (30 мл х 3). Объединенные органические фазы промывали водой (30 мл) и насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 50% этилацетат в гексане) с получением трет-бутил-2-(2-(2-(2-(4-(5-бром-4-(3-(N-метилциклобутанкарбоксамидо)пропиламино)пиримидин-2-иламино)фенокси)этокси)этокси)этокси)этилкарбамата (40 мг, выход 17%) в виде желтого масла.
[00380] Стадия 7: N-(3-(2-(4-(2-(2-(2-(2-аминоэтокси)этокси)этокси)этокси)фениламино)-5-бромпиримидин-4-ил)пропил)-N-метилциклобутанкарбоксамид
[00381] Смесь трет-бутил-2-(2-(2-(2-(4-(5-бром-4-(3-(N-метилциклобутанкарбоксамидо)пропиламино)пиримидин-2-иламино)фенокси)этокси)этокси)этокси)этилкарбамата (40 мг, 0,06 ммоль) в 2,2,2-трифторуксусной кислоте (1 мл) и дихлорметане (1 мл) перемешивали при комнатной температуре в течение 2 часов. Летучие вещества удаляли при пониженном давлении. Остаток распределяли между дихлорметаном (60 мл) и водным раствором бикарбоната натрия (2,0 Н, 30 мл). Органический слой промывали насыщенным раствором соли (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением N-(3-(2-(4-(2-(2-(2-(2-аминоэтокси)этокси)этокси)этокси)фениламино)-5-бромпиримидин-4-ил)пропил)-N-метилциклобутанкарбоксамида (18 мг, выход 52%) в виде желтого масла.
[00382] Стадия 8: N-(3-(5-бром-2-(4-(2-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)этокси)этокси)этокси)этокси)фениламино)пиримидин-4-иламино)пропил)-N- метилциклобутанкарбоксамид
[00383] Смесь N-(3-(2-(4-(2-(2-(2-(2-аминоэтокси)этокси)этокси)этокси)фениламино)-5-бромпиримидин-4-ил)пропил)-N-метилциклобутанкарбоксамида (130 мг, 0,03 ммоль), 2-(2,6-диоксопиперидин-3-ил)-4-фтор-2,3-дигидро-1Н-изоиндол-1,3-диона (8,2 мг, 0,03 ммоль) и N-этил-N-изопропилпропан-2-амина (7,6 мг, 0,06 ммоль) в сухом N,N-диметилформамиде (1 мл) перемешивали при 90°С в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, распределяли между этилацетатом (100 мл) и водой (30 мл). Органическую фазу промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Остаток очищали с помощью препаративной тонкослойной хроматографии (TLC) с получением N-(3-(5-бром-2-(4-(2-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)этокси)этокси)этокси)этокси)фениламино)пиримидин-4-иламино)пропил)-N-метилциклобутанкарбоксамида (10,2 мг, выход 40%) в виде твердого вещества желтого цвета. LC-MS (ES+): m/z=865,27/867,27 (1:1) [МН]+. tR=2,06 мин. 1H-ЯМР (400 МГц, CD3OD): δ 1,68-1,77 (м, 3H), 1,89-1,92 (м, 3H), 2,08-2,15 (м, 3H), 2,60-2,79 (м, 7H), 3,28-3,35 (м, 6H), 3,55-3,61 (м, 10H), 3,69-3,72 (м, 2H), 3,96-3,99 (м, 2H), 4,91-4,95 (м, 1H), 6,75-6,78 (м, 2H), 6,91-6,94 (м, 2H), 7,34-7,42 (м, 3H), 7,76 (д, J=12,8 Гц, 1H).
[00384] 8. 2-((S)-4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазол[4,3-a][1,4]диазепин-6-ил)-N-(4-(2-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)этокси)этокси)этокси)этокси)фенил)ацетамид
[00385] (Соединение со структурой # 14 показано в таблице 1)
[00386] Стадия 1: (2-(2,6-диоксопиперидин-3-ил)-4-(2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этиламино)изоиндолин-1,3-дион
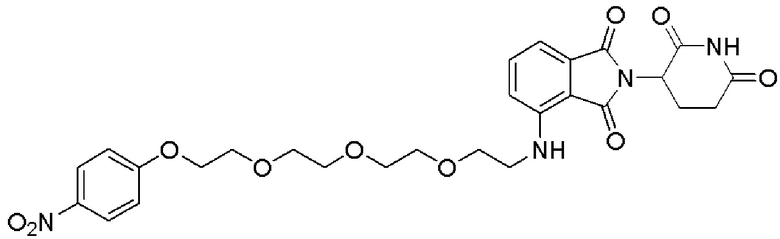
[00387] Смесь 2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этанамина (128 мг, 0,41 ммоль), 2-(2,6-диоксопиперидин-3-ил)-4-фтор-2,3-дигидро-1Н-изоиндол-1,3-диона (112,5 мг, 0,41 ммоль) и N-этил-N-изопропилпропан-2-амина (105 мг, 0,81 ммоль) в сухом N,N-диметилформамиде (2 мл) перемешивали при 90°С в течение 12 часов. Смесь охлаждали до комнатной температуры, выливали в воду (20 мл) и экстрагировали этилацетатом (35 мл х 2). Объединенные органические фазы промывали водой (30 мл) и насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью препаративной TLC с получением 2-(2,6-диоксопиперидин-3-ил)-4-(2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этиламин)изоиндолин-1,3-диона (73 мг, выход 31%) в виде твердого вещества желтого цвета. LC-MS (ES+): m/z 571,3 [МН+], tR=2,46 мин.
[00388] Стадия 2: (4-(2-(2-(2-(2-(4-аминофенокси)этокси)этокси)этокси)этиламино)-2-(2,6-диоксопиперидин-3-ил)изоиндолин-1,3-дион)
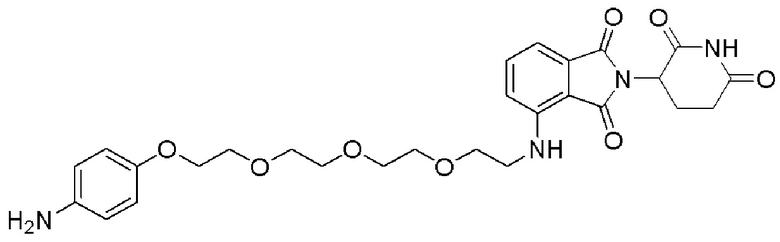
[00389] К суспензии 2-(2,6-диоксопиперидин-3-ил)-4-(2-(2-(2-(2-(4-нитрофенокси)этокси)этокси)этокси)этиламино)изоиндолин-1,3-диона (73 мг, 0,128 ммоль) и порошка железа (71,6 мг, 1,28 ммоль) в этаноле (2 мл) добавляли раствор хлорида аммония (68 мг, 1,26 ммоль) в воде (0,5 мл) при комнатной температуры, полученную смесь перемешивали при 80°С в течение 1 часа. После охлаждения смеси до комнатной температуры твердый осадок отфильтровывали и промывали этилацетатом (10 мл х 2). Фильтрат разделяли между этилацетатом (60 мл) и водой (30 мл). Органический слой промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 4-(2-(2-(2-(2-(4-аминофенокси)этокси)этокси)этокси)этиламино)-2-(2,6-диоксопиперидин-3-ил)изоиндолин-1,3-диона (66,5 мг, неочищенного) в виде желтого масла. LC-MS (ES+): m/z 541,5 [МН+], tR=1,593 мин.
[00390] Стадия 3: 2-((S)-4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазол[4,3-а][1,4]диазепин-6-ил)-N-(4-(2-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)этокси)этокси)этокси)этокси)фенил)ацетамид
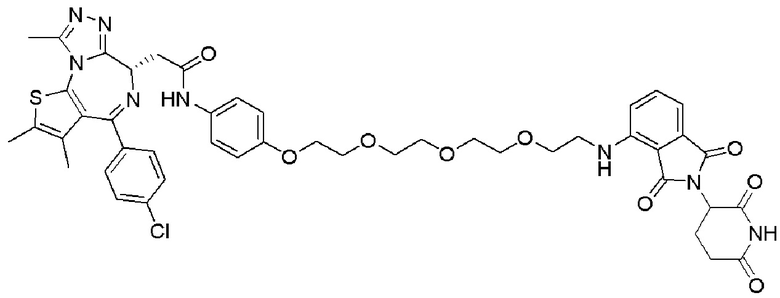
[00391] К перемешиваемому раствору 4-(2-(2-(2-(2-(4-аминофенокси)этокси)этокси)этокси)этиламино)-2-(2,6-диоксопиперидин-3-ил)изоиндолин-1,3-диона (58,4 мг, 0,11 ммоль), (S)-2-(4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-а][1,4]диазепин-6-ил)уксусной кислоты (43,3 мг, 0,11 ммоль) и N-этил-N-изопропилпропан-2-амина (41,8 мг, 0,32 ммоль ) в сухом N,N-диметилформамиде (1 мл) добавляли (2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуронийгексафторфосфат) (82 мг, 0,21 ммоль) при 0°С. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 20 минут. Смесь выливали в воду (25 мл), экстрагировали этилацетатом (35 мл х 2). Объединенные органические фазы промывали водой (20 мл) и насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью препаративной TLC с получением 2-((S)-4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-а][1,4]диазепин-6-ил)-N-(4-(2-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)этокси)этокси)этокси)этокси)фенил)ацетамида (52 мг, выход 52%) в виде твердого вещества желтого цвета. LC-MS (ES+): m/z 923,29/925,29 (3:1) [МН+], tR=2,689 мин. 1H-ЯМР (400 МГц, CDCl3): δ 1,67 (с, 3H), 2,05-2,12 (м, 1H), 2,40 (с, 3H), 2,65-2,85 (м, 6H), 3,41-3,54 (м, 4H), 3,65-3,74 (м, 10H), 3,81-3,85 (м, 2H), 4,06-4,11 (м, 2H), 4,63-4,69 (м, 1H), 4,85-4,93 (м, 1H), 6,38-6,55 (м, 1H), 6,83 (д, J=8,8 Гц, 2H), 6,92 (д, J=8,8 Гц, 1H), 7,09 (д, J=7,2 Гц, 1H), 7,33 (д, J=8,4 Гц, 2H), 7,39-7,51 (м, 5H), 8,59 (д, J=5,2 Гц, 1H), 8,77 (д, J=3,2 Гц, 1H).
[00392] 9. (Z)-4-(4-((3-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)этокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрил
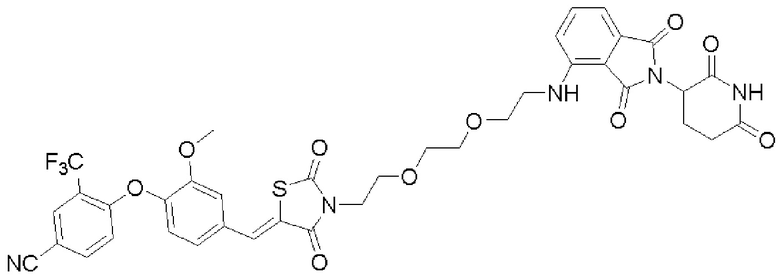
[00393] (Соединение со структурой # 22 показано в таблице 1)
[00394] Стадия 1: (Z)-2-(2-(2-(5-(4-(4-циано-2-(трифторметил)фенокси)-3-метоксибензилиден)-2,4-диоксотиазолидин-3-ил)этокси)этокси)этил-4-метилбензолсульфонат)
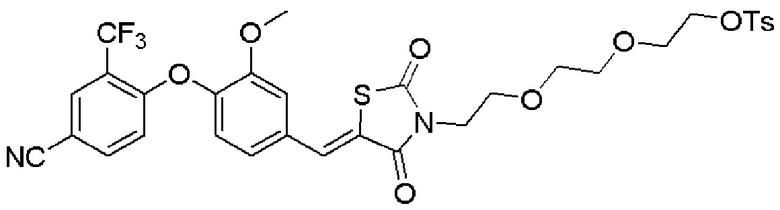
[00395] Смесь (Z)-4-(4-((2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрила (1,0 г, 2,3 ммоль), карбоната калия (1,0 г, 6,9 ммоль) и 2,2'-(этан-1,2-диилбис(окси))бис(этан-2,1-диил)бис(4-метилбензолсульфонат) (1,3 г, 2,7 ммоль) в N,N-диметилформамиде (10 мл) перемешивали при 80°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, гасили водой (10 мл) и экстрагировали этилацетатом (40 мл х 3). Объединенные органические фазы промывали водой (50 мл) и насыщенным раствором соли (50 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Неочищенный остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 10-30% этилацетата в гексане) с получением (Z)-2-(2-(2-(5-(4-(4-циано-2-(трифторметил)фенокси)-3-метоксибензилиден)-2,4-диоксотиазолидин-3-ил)этокси)этокси)этил-4-метилбензолсульфоната (1,0 г, выход 61%) в виде светло-желтого твердого вещества.
[00396] Стадия 2: (Z)-4-(4-((3-(2-(2-(2-азидоэтокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрил
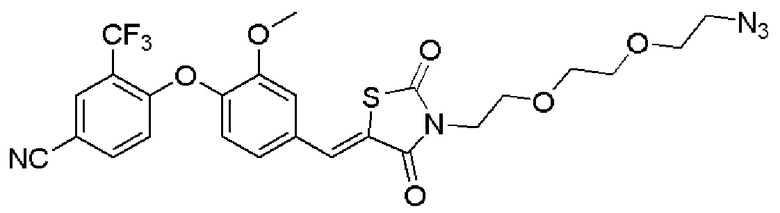
[00397] Смесь (Z)-2-(2-(2-(5-(4-(4-циано-2-(трифторметил)фенокси)-3-метоксибензилиден)-2,4-диоксотиазолидин-3-ил)этокси)этокси)этил-4-метилбензолсульфоната (1,0 г, 1,4 ммоль) и азида натрия (185 мг, 2,8 ммоль) в этаноле (20 мл) кипятили с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры и распределяли между этилацетатом (100 мл) и водой (20 мл). Органический слой промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением (Z)-4-(4-((3-(2-(2-(2-азидоэтокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрила (130 мг, неочищенного) в виде светло-желтого масла, который использовали на следующей стадии без дополнительной очистки.
[00398] Стадия 3: (Z)-4-(4-((3-(2-(2-(2-аминоэтокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрил
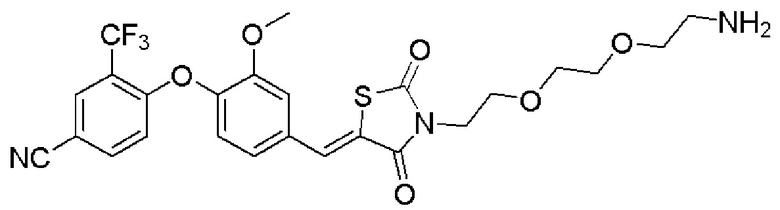
[00399] Смесь полученного выше (Z)-4-(4-((3-(2-(2-(2-азидоэтокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрила (130 мг, неочищенного), трифенилфосфина (100 мг, 0,34 ммоль) в воде (0,2 мл) и тетрагидрофуране (20 мл) перемешивали при комнатной температуре в течение 14 часов. Смесь концентрировали при пониженном давлении. Неочищенный остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюирование смесью 3-5% метанола в дихлорметане) с получением (Z)-4-(4-((3-(2-(2-(2-аминоэтокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрила (60 мг, выход 8% после двух стадий) в виде желтого масла. LC-MS (ES+): m/z 552,1 [МН+], tR=2,15 мин.
[00400] Стадия 4: (Z)-4-(4-((3-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)этокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрил
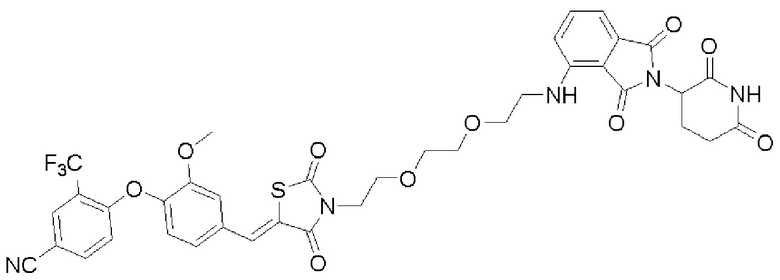
[00401] Смесь (Z)-4-(4-((3-(2-(2-(2-аминоэтокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрила (60 мг, 0,10 ммоль), 2-(2,6-диоксопиперидин-3-ил)-4-фтор-2,3-дигидро-1Н-изоиндол-1,3-диона (30 мг, 0,13 ммоль) и N-этил-N-изопропилпропан-2-амина (50 мг, 0,39 ммоль) в 1-метилпирролидин-2-оне (1 мл) перемешивали при 90°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, гасили водой (5 мл) и экстрагировали этилацетатом (20 мл х 3). Объединенные органические слои промывали водой (10 мл х 2) и насыщенным раствором соли (10 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью препаративной TLC с получением (Z)-4-(4-((3-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)этокси)этокси)этил)-2,4-диоксотиазолидин-5-илиден)метил)-2-метоксифенокси)-3-(трифторметил)бензонитрила (9,5 мг, выход 11,8%) в виде желтого твердого вещества. LC-MS (ES+): m/z 808,19 [МН+], tR=3,022 мин. 1H-ЯМР (400 МГц, CDCl3): δ 2,12-2,16 (м, 1H), 2,73-2,91 (м, 3H), 3,42 (с, 2H), 3,67-3,80 (м, 11H), 3,99 (с, 2H), 4,91-4,95 (м, 1H), 6,51 (с, 1H), 6,76-6,86 (м, 2H), 7,02-7,19 (м, 4H), 7,43 (т, J=7,6 Гц, 1H), 7,68 (д, J=8,0 Гц, 1H), 7,85-8,12 (м, 3H).
[00402] 10. 4-(3-(4-(3-(2-(2-(2-((2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)амино)этокси)этокси)этокси)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил) бензонитрил
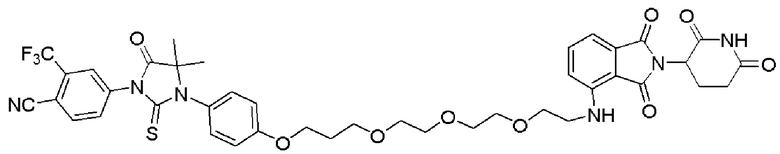
(Соединение cо структурой # 1 показано в Таблице 1)
[00403] Стадия 1: 1,1,1,16-тетрафенилборат-2,5,8,11,15-пентаоксагексадекан
[00404] К раствору 2-(2-(2-(тритилокси)этокси)этокси)этанола (7 г, 17,7 ммоль) в N,N-диметилформамиде (50 мл) медленно добавляли гидрид натрия (60% в минеральном масло, 707 мг, 17,7 ммоль) при 0°С. После перемешивания смеси при комнатной температуре в течение 30 минут к ней добавляли 3-(бензилокси)пропил-4-метилбензолсульфонат (5,8 г 18,0 ммоль) в виде одной порции при 0°С. Полученную смесь оставляли перемешиваться при температуре 70°С в течение ночи. После охлаждения смеси до комнатной температуры, ее осторожно гасили водой (40 мл) и экстрагировали этилацетатом (60 мл х 3). Объединенные органические фазы промывали солевым раствором (80 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью хроматографии на силикагеле (элюирование смесью 5-10% этилацетата в гексане) с получением 1,1,1,16-тетрафенилборат-2,5,8,11,15-пентаоксагексадекана (4,8 г, выход 50%) в виде бесцветного масла. 1H-ЯМР (400 МГц, CDCl3): δ 1,85-1,92 (м, 2H), 3,23 (т, J=5,2 Гц, 2H), 3,53-3,59 (м, 6H), 3,64-3,68 (м, 8H), 4,47 (с, 2H), 7,19-7,33 (м, 15H), 7,45-7,47 (м, 5H).
[00405] Стадия 2: 1-фенил-2,6,9,12-тетраоксатетрадекан-14-ол
[00406] К раствору 1,1,1,16-тетрафенилбората-2,5,8,11,15-пентаоксагексадекана (4,8 г 8,8 ммоль) в метилендихлориде (10 мл) и метаноле (10 мл) добавляли водный раствор хлористоводородной кислоты (37%, 2,5 мл) при 0оС. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь выливали в воду (30 мл) и экстрагировали дихлорметаном (20 мл х 3). Объединенные органические фазы промывали водным раствором бикарбоната натрия (1 Н, 50 мл), водой (30 мл), насыщенным раствором соли, сушили над безводным Na2SO4, и концентрировали при пониженном давлении. Неочищенный остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 20-40% этилацетата в гексане), с получением 1-фенил-2,6,9,12-тетраоксатетрадекан-14-ола (1,9 г, выход 73%) в виде бесцветного масла.
[00407] Стадия 3: 1-фенил-2,6,9,12-тетраоксатетрадекан-14-ил-4-метилбензолсульфонат

[00408] Смесь 1-фенил-2,7,10,13-тетраоксапентадекан-15-ола (1,9 г, 6,3 ммоль), триэтиламина (1,3 мл, 9,5 ммоль), N,N-диметилпиридин-4-амина (75 мг, 0,63 ммоль) и 4-метилбензол-1-сульфонилхлорида (1,45 г, 7,65 ммоль) в дихлорметане (20 мл) перемешивали при комнатной температуре в течение 3 часов. Для гашения реакции добавляли воду (20 мл), и продукт экстрагировали дихлорметаном (40 мл х 3). Объединенные органические фазы промывали насыщенным раствором соли (50 мл), сушили над сульфатом натрия и упаривали при пониженном давлении. Неочищенный остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 10-30% этилацетата в гексане) с получением 1-фенил-2,6,9,12-тетраоксатетрадекан-14-ил-4-метилбензолсульфоната (2,2 г, выход 78%) в виде бесцветного масла. 1H-ЯМР (400 МГц, CDCl3): δ 1,87-1,92 (м, 2H), 2,43 (с, 3H), 3,54-3,60 (м, 12H), 3,67 (т, J=5,2 Гц, 2H), 4,15 (т, J=5,0 Гц, 2H), 4,48 (с, 2H), 7,27-7,33 (м, 7H), 7,79 (д, J=8,4 Гц, 2H).
[00409] Стадия 4: 14-азидо-1-фенил-2,6,9,12-тетраоксатетрадекан
[00410] Смесь 1-фенил-2,6,9,12-тетраоксатетрадекан-14-ил-4-метилбензолсульфоната (2,2 г, 4,9 ммоль) и азида натрия (420 мг, 6,3 ммоль) в этаноле (10 мл) кипятили с обратным холодильником в течение 5 часов. Реакционную смесь охлаждали до комнатной температуры, выливали в воду (10 мл) и экстрагировали дихлорметаном (50 мл х 3). Объединенные органические слои промывали насыщенным солевым раствором (50 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 14-азидо-1-фенил-2,6,9,12-тетраоксатетрадекана (1,4 г, неочищенного) в виде бесцветного масла, который использовали на следующей стадии без дополнительной очистки.
[00411] Стадия 5: трет-бутил-(1-фенил-2,6,9,12-тетраоксатетрадекан-14-ил)карбамат
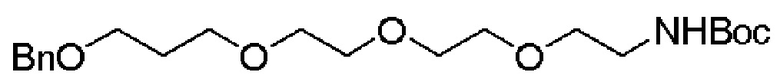
[00412] Смесь указанного выше 14-азидо-1-фенил-2,6,9,12-тетраоксатетрадекана (1,4 г, неочищенного) и трифенилфосфина (1,7 г, 6,5 ммоль) в смеси тетрагидрофурана (15 мл) и воды (0,5 мл) перемешивали при комнатной температуре в течение ночи в атмосфере азота. К реакционной смеси добавляли триэтиламин (0,9 мл, 6,5 ммоль) и ди-трет-бутилдикарбонат (1,1 г, 5,2 ммоль) при 0оС. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 2 часов. Летучие вещества выпаривали при пониженном давлении, и остаток распределяли между дихлорметаном (100 мл) и водой (50 мл). Органическую фазу промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью хроматографии на силикагеле (элюент: 30-50% этилацетата в гексане) с получением трет-бутил-(1-фенил-2,6,9,12-тетраоксатетрадекан-14-ил)карбамата (1,2 г, 50%-ный выход за две стадии) в виде бесцветного масла.
[00413] Стадия 6: трет-бутил-2-(2-(2-(3-гидроксипропокси)этокси)этокси)этилкарбамат
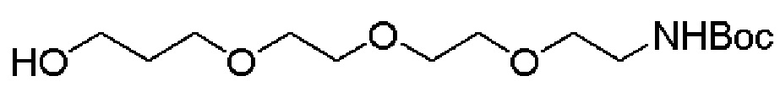
[00414] Смесь трет-бутил-(1-фенил-2,6,9,12-тетраоксатетрадекан-14-ил)карбамата (1,2 г, 3 ммоль) и палладия на угле (10%, 200 мг) в этаноле ( 50 мл) перемешивали при комнатной температуре в атмосфере водорода (подача из баллона с водородом). Палладий на угле удаляли фильтрацией и промывали этанолом (20 мл). Фильтрат концентрировали при пониженном давлении с получением трет-бутил-2-(2-(2-(3-гидроксипропокси)этокси)этокси)этилкарбамата (900 мг, неочищенного) в виде бесцветного масла, который использовали на следующей стадии без дополнительной очистки,
[00415] Стадия 7: 2,2-диметил-4-оксо-3,8,11,14-тетраокса-5-азагептадекан-17-ил-4-метилбензолсульфонат
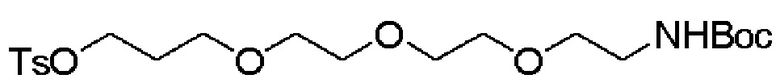
[00416] Смесь полученного выше трет-бутил-2-(2-(2-(3-гидроксипропокси)этокси)этокси)этилкарбамата (900 мг, неочищенный), триэтиламина (0,6 мл, 4,35 ммоль), N,N-диметилпиридин-4-амина (16 мг, 0,14 ммоль) и 4-метилбензол-1-сульфонилхлорида (660 мг, 3,5 ммоль) в безводном дихлорметане (15 мл) перемешивали при комнатной температуре в течение 3 часов. Добавляли воду (20 мл) для гашения реакции, и продукт экстрагировали дихлорметаном (50 мл х 3). Объединенные органические фазы промывали насыщенным раствором соли (50 мл), сушили над безводным сульфатом натрия и упаривали при пониженном давлении. Неочищенный остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 20-30% этилацетата в гексане) с получением 2,2-диметил-4-оксо-3,8,11,14-тетраокса-5-азагептадекан-17-ил-4-метилбензолсульфоната (650 мг, выход 77%) в виде светло-желтого масла. 1H-ЯМР (400 МГц, CDCl3): δ 1,44 (с, 9H), 1,88-1,95 (м, 2H), 2,45 (с, 3H), 3,29-3,33 (м, 2H), 3,48-3,61 (м, 12H), 4,09-4,15 (м, 2H), 5,04 (уш. с, 1H), 7,34 (д, J=8,0 Гц, 2H), 7,79 (д, J=8,0 Гц, 2H).
[00417] Стадия 8: трет-бутил-(2-(2-(2-(3-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)фенокси)пропокси)этокси)этокси)этил)карбамат
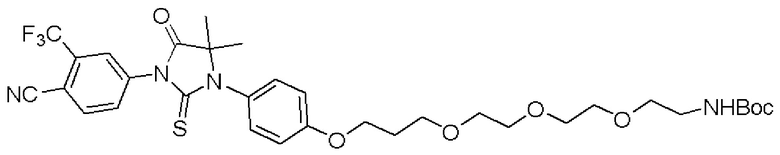
[00418] Смесь 2,2-диметил-4-оксо-3,8,11,14-тетраокса-5-азагептадекан-17-ил-4-метилбензолсульфоната (115 мг, 0,25 ммоль), карбоната калия (69 мг, 0,50 ммоль) и 4-(3-(4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила (100 мг, 0,25 ммоль) в ацетонитриле (5 мл) перемешивали при 80°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, гасили водой (30 мл) и экстрагировали этилацетатом (30 мл х 3). Объединенные органические фазы промывали водой (30 мл) и насыщенным раствором соли (30 мл), сушили над сульфатом магния и упаривали при пониженном давлении. Неочищенный остаток очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 10-30% этилацетата в гексане) с получением трет-бутил-2-(2-(2-(3-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)фенокси)пропокси)этокси)этокси)этилкарбамата (150 мг, выход 82%) в виде желтого масла. LC-MS (ES+): m/z 695,40 [МН+], tR=2,79 мин.
[00419] Стадия 9: 4-(3-(4-(3-(2-(2-(2-аминоэтокси)этокси)этокси)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
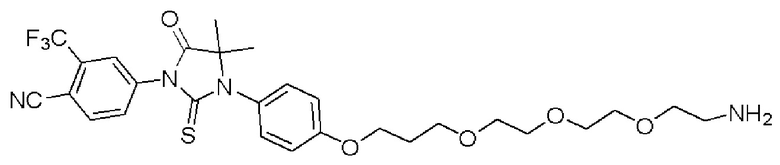
[00420] Смесь трет-бутил-2-(2-(2-(3-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)фенокси)пропокси)этокси)этокси)этилкарбамата (150 мг, 0,21 ммоль) в безводном дихлорметане (2 мл) и 2,2,2-трифторуксусной кислоты (1 мл) перемешивали при комнатной температуре в течение 1 часа. Летучие вещества выпаривали при пониженном давлении, остаток выливали в водный раствор бикарбоната натрия (1 Н, 20 мл) и экстрагировали дихлорметаном (50 мл х 3). Объединенные органические фазы промывали насыщенным раствором соли (50 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением 4-(3-(4-(3-(2-(2-(2-аминоэтокси)этокси)этокси)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила (115 мг, неочищенного) в виде коричневого масла, который использовали на следующей стадии без дальнейшей очистки.
[00421] Стадия 10: 4-(3-(4-(3-(2-(2-(2-((2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)амино)этокси)этокси)этокси)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
[00422] Раствор полученного выше 4-(3-(4-(3-(2-(2-(2-аминоэтокси)этокси)этокси)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила (115 мг, неочищенного), 2-(2,6-диоксопиперидин-3-ил)-4-фтор-2,3-дигидро-1Н-изоиндол-1,3-диона (41 мг, 0,15 ммоль) и N-этил-N-изопропилпропан-2-амина (58 мг, 0,44 ммоль) в N,N-диметилформамиде (2 мл) перемешивали при 90°С в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, гасили водой (3 мл) и экстрагировали этилацетатом (30 мл х 3). Объединенные органические слои промывали водой (30 мл х 2) и насыщенным раствором соли (20 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Неочищенный остаток очищали с помощью препаративной TLC с получением 4-(3-(4-(3-(2-(2-(2-((2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)амино)этокси)этокси)этокси)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила (34,5 мг, выход 27%) в виде желтого твердого вещества. LC-MS (ES+): m/z 851,25 [МН+], tR=2,652 мин. 1H-ЯМР (400 МГц, CD3OD): δ 1,57 (с, 6H), 2,07-2,11 (м, 3H), 2,70-2,90 (м, 3H), 3,46-3,72 (м, 14H), 4,10 (т, J=6,2 Гц, 2H), 4,88-4,92 (м, 1H), 6,48-6,49 (м, 1H), 6,91-7,26 (м, 6H), 7,49 (т, J=7,8 Гц, 1H), 7,83-7,85 (м, 1H), 7,97-8,02 (м, 3H).
[00423] 11. 4-{[5-(3-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1Н-изоиндол-4-ил]амино}пропокси)пентил]окси}-N-[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]бензамид
[00424] Стадия 1: 3-[(5-гидроксипентил)окси]пропаннитрил
[00425] Пентан-1,5-диол (2,98 г, 28,6 ммоль) добавляли к суспензии гидрида натрия (60% дисперсия в минеральном масле, 820 мг, 34,2 ммоль) в THF (50 мл). После этого смесь перемешивали при комнатной температуре в течение 20 минут и ее охлаждали до 0°C, и добавляли по каплям акрилонитрил (1,20 г, 22,8 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 10 часов. Часть растворителя удаляли в вакууме и остаток выливали в воду. Смесь экстрагировали DCM (3 х). Органический слой фильтровали через универсальный сепаратор для разделения фаз Biotage и концентрировали в вакууме. Неочищенный продукт очищали на силикагеле с помощью хроматографии Teledyne Combiflash ISCO, элюируя смесью MeOH/DCM (от 0:100 до 3:97), с получением 3-[(5-гидроксипентил)окси]пропаннитрила (635 мг, выход 18%). 1H-ЯМР (400 МГц, CDCl3) δ 3,60-3,73 (м, 4H), 3,45-3,55 (м, 2H), 2,60 (дт, J=4,1, 6,4 Гц, 2H), 2,06 (д, J=3,9 Гц, 1H), 1,57-1,69 (м, 4H), 1,43-1,50 (м, 2H).
[00426] Стадия 2: трет-бутил-N-{3-[(5-гидроксипентил)окси]пропил}карбамат
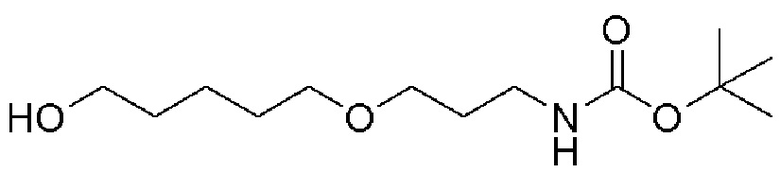
[00427] К раствору 3-[(5-гидроксипентил)окси]пропаннитрила (400 мг, 2,54 ммоль) в MeOH (12 мл) и H2O (2,0 мл) добавляли хлорид никеля (II) (393 мг, 3,04 ммоль), а затем по порциям боргидрид натрия (360 мг, 9,52 ммоль). Смесь перемешивали при комнатной температуре в течение 3 часов, затем гасили МеОН (12 мл). Смесь фильтровали через целит и промывали метанолом. Фильтрат концентрировали в вакууме. К раствору вышеуказанного сырого продукта в THF (5 мл) добавляли 6 М водный раствор NaOH (0,5 мл) и ди-трет-бутилдикарбонат (831 мг, 3,81 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 3 часов, затем концентрировали в вакууме. Неочищенный продукт очищали на силикагеле с помощью хроматографии Teledyne Combiflash ISCO, элюируя смесью MeOH/DCM (от 0:100 до 4:96) с получением трет-бутил-N-{3-[(5-гидроксипентил)окси]пропил}карбамата (366 мг, выход 55%).
[00428] 1H-ЯМР (400 МГц, CDCl3) δ 4,91 (шир. с., 1H), 3,66 (шир. с., 2H), 3,49 (т, J=5,9 Гц, 2H), 3,43 (т, J=6,3 Гц, 2H), 3,24 (д, J=5,9 Гц, 2H), 1,75 (кв., J=6,2 Гц, 2H), 1,57-1,65 (м, 5H), 1,41-1,52 (м, 11H).
[00429] Стадия 3: трет-бутил-N-[3-({5-[(4-метилбензолсульфонил)окси]пентил}окси)пропил]карбамат
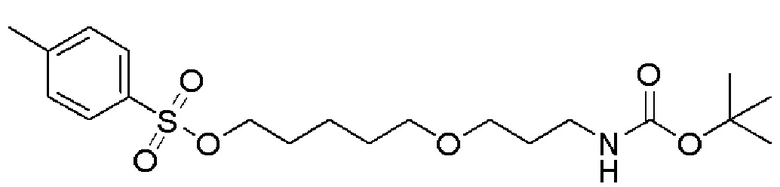
[00430] К раствору трет-бутил-(3-((5-гидроксипентил)окси)пропил)карбамата (300 мг, 3,88 ммоль) в DCM (10 мл) добавляли DIPEA (599,3 мкл, 3,44 ммоль), тозилхлорид (262,3 мг, 1,38 ммоль) и 4-диметиламинопиридин (14,0 мг, 0,115 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 20 часов. Реакционную смесь гасили полунасыщенным раствором бикарбоната натрия, экстрагировали с помощью DCM (2 раза), фильтровали через универсальный сепаратор для разделения фаз Biotage и концентрировали в вакууме. Неочищенный продукт очищали на силикагеле с помощью хроматографии Teledyne Combiflash ISCO, элюируя смесью EtOAc/гептан (от 0:100 до 30:70), с получением трет-бутил-N-[3-({5-[(4-метилбензолсульфонил)окси]пентил}окси)пропил]карбамата (914 мг, выход 26%). 1H-ЯМР (400 МГц, CDCl3) δ 7,78 (д, J=8,2 Гц, 2H), 7,34 (д, J=8,2 Гц, 2H), 4,02 (т, J=6,5 Гц, 2H), 3,44 (т, J=6,1 Гц, 2H), 3,35 (т, J=6,3 Гц, 2H), 3,19 (д, J=5,9 Гц, 2H), 2,44 (с, 3H), 1,64-1,74 (м, 5H), 1,49-1,54 (м, 2H), 1,42 (с, 9H), 1,33-1,40 (м, 2H). LC-MS (ES+): m/z 438,19 [M+Na+], tR=2,65 мин.
[00431] Стадия 4: метил-4-{[5-(3-{[(трет-бутокси)карбонил]амино}пропокси)пентил]окси}бензоат
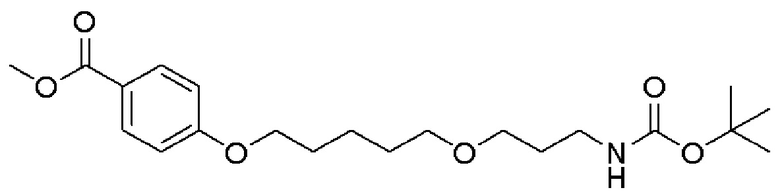
[00432] Смесь трет-бутил-N-[3-({5-[(4-метилбензолсульфонил)окси]пентил}окси)пропил]карбамата (340 мг, 0,82 ммоль), метил-4-гидроксибензоата (117 мг, 0,77 ммоль), карбоната калия (203 мг, 1,47 ммоль) в MeCN (10 мл) перемешивали при 80°С в течение 24 часов. Реакционную смесь разбавляли EtOAc, промывали полунасыщенным раствором бикарбоната натри (1x), водой (2x), насыщенным раствором соли (1 х), а затем фильтровали через универсальный сепаратор для разделения фаз Biotage. Фильтрат концентрировали в вакууме, и остаток очищали на силикагеле с помощью хроматографии Teledyne Combiflash ISCO, элюируя смесью EtOAc/гептан (от 0:100 до 50:50), с получением метил-4-{[5-(3-{[(трет-бутокси)карбонил]амино}пропокси)пентил]окси}бензоата (300 мг, выход 93%). LC-MS (ES+): m/z 418,21 [M+Na+], tR=2,74 мин.
[00433] Стадия 5: 4-{[5-(3-{[(трет-бутокси)карбонил]амино}пропокси)пентил]окси}бензойная кислота
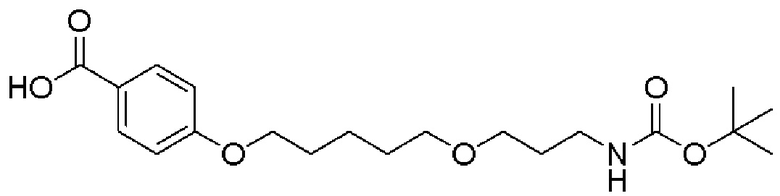
[00434] К раствору 4-{[5-(3-{[(трет-бутокси)карбонил]амино}пропокси)пентил]окси}бензоата (150 мг, 0,38 ммоль) в смеси 1:1:1 THF/вода/MeOH (6,0 мл, об./об./об.) добавляли гидроксид лития (81,6 мг, 3,41 ммоль). Полученную смесь перемешивали в течение ночи при комнатной температуре, затем подкисляли до рН 2-3 с помощью 6 Н водного раствора НСl. Смесь концентрировали в вакууме для удаления большинства растворителей, затем разбавляли EtOAc, промывали водой (2 х), насыщенным раствором соли (2 х), фильтровали через универсальный сепаратор для разделения фаз Biotage и концентрировали в вакууме. Неочищенный продукт использовали на следующей стадии без дополнительной очистки (123 мг). LC-MS (ES+): m/z 404,20 [MNa+], tR=2,40 мин.
[00435] Стадия 6: трет-бутил-N-(3-{[5-(4-{[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]карбамоил}фенокси)пентил]окси}пропил)карбамат
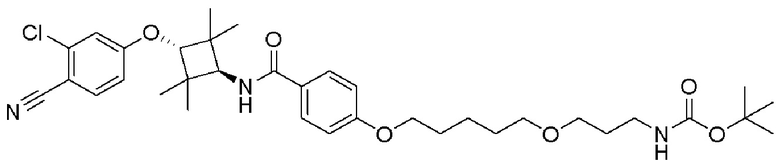
[00436] К раствору 4-{[5-(3-{[(трет-бутокси)карбонил]амино}пропокси)пентил]окси}бензойной кислоты (124 мг, 0,322 ммоль) и 2-хлор-4-(транс-3-амино-2,2,4,4-тетраметилциклобутокси)бензонитрила (89,8 мг, 0,322 ммоль) в DMF (5 мл) добавляли DIPEA (112 мкл, 0,65 ммоль) и TBTU (155 мг, 0,48 ммоль). Полученную смесь перемешивали при комнатной температуре в течение 1 часа, затем разбавляли EtOAc, промывали водой (3 х), насыщенным раствором соли (1 х), фильтровали через универсальный сепаратор для разделения фаз Biotage и концентрировали в вакууме. Остаток очищали на силикагеле с помощью хроматографии Teledyne Combiflash ISCO, элюируя смесью MeOH/DCM (от 0:100 до 5:95), с получением трет-бутил-N-(3-{[5-(4-{[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]карбамоил}фенокси)пентил]окси}пропил)карбамата (169 мг, выход 82%). LC-MS (ES+): m/z 643,32/645,31 (3:1) [МН+], tR=3,04 мин.
[00437] 12. 4-{[5-(3-аминопропокси)пентил]окси}-N-[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]бензамид
[00438] К раствору трет-бутил-N-(3-{[5-(4-{[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]карбамоил}фенокси)пентил]окси}пропил)карбамата (124 мг, 0,192 ммоль) в DCM (5 мл) добавляли трифторуксусную кислоту (372 мкл, 4,86 ммоль) и нагревали при 45°С в течение 1 часа до завершения реакции. Реакционную смесь затем концентрировали в вакууме до твердого состояния, и полученный продукт использовали на следующей стадии без дополнительной очистки (104 мг, выход 99%). LC-MS (ES+): m/z 543,27/545,26 (3:1) [МН+], tR=2,26 мин.
[00439] 13. 4-{[5-(3-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1Н-изоиндол-4-ил]амино}пропокси)пентил]окси}-N-[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]бензамид
[00440] (Соединение со структурой # 11 показано в таблице 1)
[00441] К раствору 4-{[5-(3-аминопропокси)пентил]окси}-N-[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]бензамида (30,0 мг, 0,0553 ммоль) в 1,4-диоксане (2 мл) добавляли диизопропилэтиламин (384 мкл, 2,21 ммоль) и 2-(2,6-диоксопиперидин-3-ил)-4-фтор-2,3-дигидро-1Н-изоиндол-1,3-дион (18,3 мг, 0,0664 ммоль). Полученную смесь кипятили с обратным холодильником в течение 16 часов, затем разбавляли EtOAc, промывали полунасыщенным солевым раствором (2 х), фильтровали через универсальный сепаратор для разделения фаз Biotage и концентрировали в вакууме. Остаток очищали на силикагеле с помощью хроматографии Teledyne Combiflash ISCO, элюируя смесью MeOH/DCM (от 0:100 до 7:93), с получением 4-{[5-(3-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1Н-изоиндол-4-ил]амино}пропокси)пентил]окси}-N-[транс-3-(3-хлор-4-цианофенокси)-2,2,4,4-тетраметилциклобутил]бензамида (12 мг, выход 28%). LC-MS (ES+): m/z 799,31/801,31 (3:1) [МН+], tR=2,97 мин. 1H-ЯМР (400 МГц, CDCl3) δ 8,03 (с, 1H), 7,72 (д, J=9,0 Гц, 2H), 7,58 (д, J=8,6 Гц, 1H), 7,48 (дд, J=7,2, 8,4 Гц, 1H), 7,07 (д, J=7,0 Гц, 1H), 6,98 (д, J=2,3 Гц, 1H), 6,89-6,96 (м, 3H), 6,82 (дд, J=2,5, 8,8 Гц, 1H), 6,18 (д, J=8,2 Гц, 1H), 4,89 (дд, J=5,1, 12,1 Гц, 1H), 4,16 (д, J=7,8 Гц, 1H), 4,06 (с, 1H), 4,02 (т, J=6,7 Гц, 2H), 3,56 (т, J=5,9 Гц, 2H), 3,50 (с, 2H), 3,46-3,48 (м, 1H), 3,41 (т, J=6,5 Гц, 2H), 2,82-2,90 (м, 1H), 2,76-2,81 (м, 1H), 2,67-2,75 (м, 1H), 2,07-2,14 (м, 1H), 1,94 (кв., J=6,1 Гц, 2H), 1,82-1,87 (м, 2H), 1,67-1,73 (м, 2H), 1,53-1,59 (м, 2H), 1,28 (с, 6H), 1,20-1,25 (м, 6H).
[00442] 14. 2-((S)-4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазол[4,3-a][1,4]диазепин-6-ил)-N-((1S)-1-(4-(5-(3-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)пропокси)пиримидин-2-ил)фенил)этил)ацетамид, также известный как 2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[5-(3-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1Н-изоиндол-4-ил]амино}пропокси)пиримидин-2-ил]фенил}этил]ацетамид
(Соединение # 40, Таблица 1)
[00443] Соединение 40 может быть получено по следующей типовой схеме:
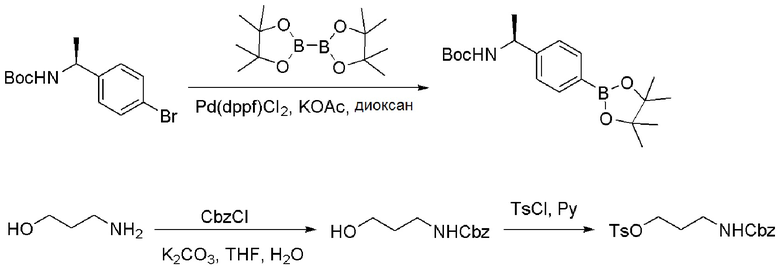
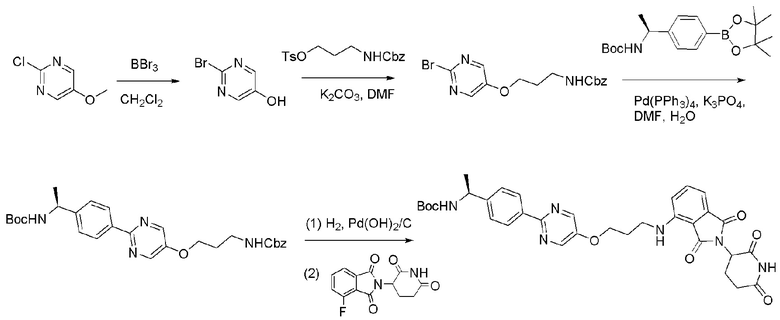
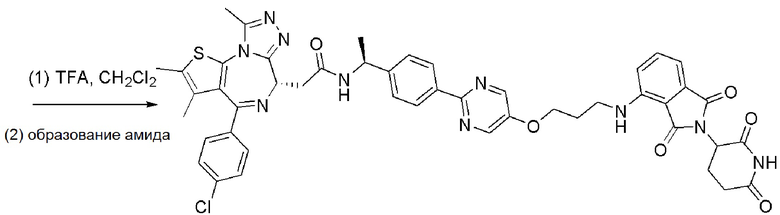
[00444] Стадия 1: Получение (S)-трет-бутил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этилкарбамата
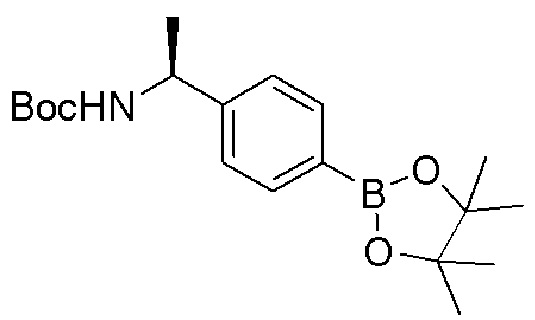
[00445] К перемешиваемому раствору (S)-трет-бутил-1-(4-бромфенил)этилкарбамата (6 г, 20,0 ммоль), 4,4,4',4',5,5,5',5' -октаметил-2,2'-бис(1,3,2-диоксаборолан) (7,6 г, 29,9 ммоль) и ацетата калия (5,9 мг, 60,1 ммоль) в диоксане (50 мл) добавляли 1,1'-бис(дифенилфосфин)ферроцен дихлорпалладий (II), (440 мг, 0,60 ммоль) при комнатной температуре в атмосфере азота. Смесь дегазировали и продували азотом три раза. Полученную смесь перемешивали при 90°С в течение ночи. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом (100 мл) и водой (50 мл). Водный слой отделяли и экстрагировали этилацетатом (50 мл х 2). Объединенные органические слои промывали насыщенным раствором соли (100 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюирование смесью 5-10% этилацетата в гексане) с получением (S)-трет-бутил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этилкарбамата (7,4 г, выход 98%) в виде желтого масла. LC/MS (ES+): m/z 370,0 [M+Na+]; tR=3,165 мин; 1H-ЯМР (400 МГц, CDCl3): δ 1,26 (с, 12H), 1,34 (с, 12H), 4,78 (шир, 1H), 7,30 (д, J=7,6 Гц, 2H), 7,78 (д, J=8,0 Гц, 2H); Химическая формула: С19Н30BNO4; молекулярная масса: 347,26.
[00446] Стадия 2: Получение бензил-3-гидроксипропилкарбамата
[00447] К перемешиваемому раствору 3-аминопропан-1-ола (20 г, 266 ммоль) и карбоната калия (73 г, 529 ммоль) в смеси воды (50 мл) и тетрагидрофурана (100 мл) добавляли бензилхлорформиат (68 г, 398 ммоль) при 0°С. Смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Реакционную смесь распределяли между этилацетатом (200 мл) и водой (100 мл). Органический слой собирали, промывали солевым раствором (100 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 20-50% этилацетата в гексане), получая бензил-3-гидроксипропилкарбамат (26,9 г, выход 52%) в виде бесцветного масла. LC/MS (ES+): m/z 232,1 [M+Na+]; tR=1,697 мин; 1Н-ЯМР (400 МГц, CDCl3): δ 1,67-1,73 (м, 2H), 2,56 (т, J=5,8 Гц, 1H), 3,33-3,38 (м, 2H), 3,65-3,70 (м, 2H), 5,06 (ш, 1H), 5,11 (с, 2H), 7,29-7,36 (м, 5H); Химическая формула: С11Н15 NO3; молекулярная масса: 209,24.
[00448] Стадия 3: Получение 3-(бензилоксикарбониламин)пропил-4-метилбензолсульфоната
[00449] К раствору бензил-3-гидроксипропилкарбамата (26,9 г, 128,6 ммоль) в пиридине (40 мл) добавляли 4-толуолсульфонилхлорид (73 г, 384 ммоль) при 0°С. Смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь распределяли между этилацетатом (120 мл) и водой (80 мл). Органический слой собирали, промывали хлористоводородной кислотой (1 Н, 480 мл) и насыщенным раствором соли (100 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 10-20% этилацетат в гексане), с получением 3-(бензилоксикарбониламино)пропил-4-метилбензолсульфоната (38,5 г, выход 82%) в виде желтого масла. LC/MS (ES+): m/z 386,2 [M+Na+]; tR=2,582 мин; 1H-ЯМР (400 МГц, CDCl3): δ 1,85-1,91 (м, 2H), 2,43 (с, 3H), 3,25 (м, 2H), 4,09 (т, J=6,0 Гц, 2H), 4,83 (шир., 1H), 5,07 (с, 2H), 7,26-7,39 (м, 7H), 7,78 (д, J=8,4 Гц, 2H); Химическая формула: С18Н21NO5S; молекулярная масса: 363,43.
[00450] Стадия 4: Получение 2-бромпиримидин-5-ола
[00451] К перемешиваемому раствору 2-хлор-5-метоксипиримидина (10 г, 69,1 ммоль) в безводном дихлорметане (60 мл) добавляли раствор трибромида бора (34,7 г, 138,5 ммоль) в дихлорметане (100 мл) при -78°С. Смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение ночи. Реакционную смесь гасили добавлением по каплям метанола (80 мл) при -78°С. Растворитель удаляли при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюирование смесью 2-5% метанола в безводном дихлорметане), с получением 2-бромпиримидин-5-ола (6,5 г, выход 54%) в виде желтого твердого вещества. 1H-ЯМР (400 МГц, DMSO-d6): δ 8,26 (с, 2H), 8,49 (с, 1H); химическая формула: C4H3BrN2O; молекулярная масса 174,98.
[00452] Стадия 5: Получение бензил-3-(2-бромпиримидин-5-илокси)пропилкарбамата
[00453] Смесь 2-бромпиримидин-5-ола (5 г, 38,3 ммоль), 3-(бензилоксикарбониламино)пропил-4-метилбензолсульфоната (13,9 г, 38,2 ммоль) и карбоната калия (10,6 г, 76,8 ммоль) в N,N-диметилформамиде (30 мл) перемешивали при 80°С в течение ночи. После охлаждения до комнатной температуры реакционную смесь распределяли между этилацетатом (50 мл) и водой (30 мл). Водный слой отделяли и экстрагировали этилацетатом (50 мл х 2). Объединенные органические слои промывали насыщенным раствором соли (80 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюент: 20-40% этилацетата в гексане), с получением бензил-3-(2-бромпиримидин-5-илокси)пропилкарбамата (2,4 г, выход 23%) в виде бесцветного масла. LC/MS (ES+): m/z 367,9 [М+1] для Br81; tR=2,375 мин; 1H-ЯМР (400 МГц, CDCl3): δ 2,04-2,08 (м, 2H), 3,39-3,43 (м, 2H), 4,08-4,13 (м, 2H), 5,09 (с, 2H), 7,34-7,36 (м, 5H), 8,22 (с, 2H); Химическая формула: С15Н16BrN3О3; молекулярная масса: 366,21.
[00454] Стадия 6: Получение трет-бутил-(S)-(1-(4-(5-(3-(((бензилокси)карбонил)амино)пропокси)пиримидин-2-ил)фенил)этил)карбамата
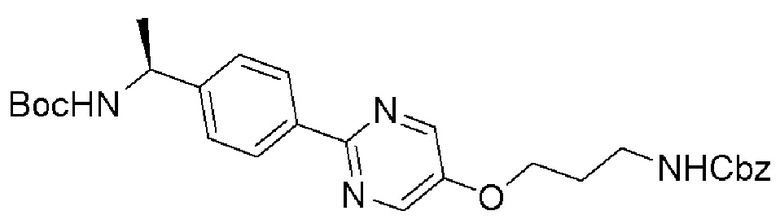
[00455] К перемешиваемому раствору бензил-3-(2-бромпиримидин-5-илокси)пропилкарбамата (2,4 г, 6,6 ммоль), (S)-трет-бутил-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)этилкарбамата (2,3 г, 6,6 ммоль) и тригидрата трехосновного фосфата калия (3,5 г, 13,3 ммоль) в смеси N,N-диметилформамида (30 мл) и воды (5 мл) добавляли хлорид бис(трифенилфосфин)палладия (II) (766 мг, 0,66 ммоль) при комнатной температуре в атмосфере азота. Смесь дегазировали и продували азотом три раза. Полученную смесь перемешивали при 80°С в течение 4 часов. Реакционную смесь распределяли между этилацетатом (70 мл) и водой (30 мл). Органический слой собирали, промывали солевым раствором (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюирование 10-50% этилацетата в гексане), с получением трет-бутил-(S)-(1-(4-(5-(3-(((бензилокси)карбонил)амино)пропокси)пиримидин-2-ил)фенил)этил)карбамата (2,2 г, выход 67%) в виде белого твердого вещества. LC/MS (ES+): m/z 507,5 [М+Н+]; tR=2,841 мин; химическая формула: С28Н34N4O5; молекулярная масса: 506,59.
[00456] Стадия 7: Получение трет-бутил-(1S)-1-(4-(5-(3-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)пропокси)пиримидин-2-ил)фенил)этилкарбамата
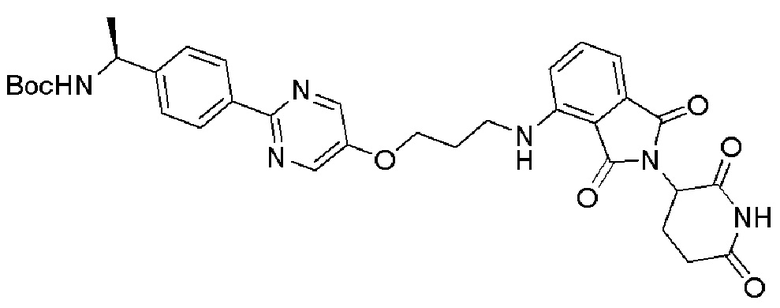
[00457] Смесь трет-бутил-(S)-(1-(4-(5-(3-(((бензилокси)карбонил)амино)пропокси)пиримидин-2-ил)фенил)этил)карбамата (2,2 г, 4,4 ммоль) и гидроксида палладия на угле (10%, 200 мг) в метаноле (5 мл) перемешивали при комнатной температуре в течение ночи в атмосфере водорода (подача из баллона с водородом). Катализатор удаляли фильтрацией и промывали метанолом (50 мл). Объединенный фильтрат концентрировали при пониженном давлении. Остаток повторно растворяли в 1-метил-2-пирролидиноне (20 мл) с последующим последовательным добавлением 2-(2,6-диоксопиперидин-3-ил)-4-фторизоиндолин-1,3-диона (1,2 г, 4,3 ммоль) и N-этил-N-изопропилпропан-2-амина (2,3 г, 17,4 ммоль). Полученную смесь перемешивали при 80°С в течение 2 часов. Реакционную смесь распределяли между этилацетатом (30 мл) и водой (15 мл). Водный слой отделяли и экстрагировали этилацетатом (25 мл х 2). Объединенный органический слой промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали на силикагеле с помощью колоночной флэш-хроматографии (элюирование 1-2% метанолом в дихлорметане), с получением трет-бутил-(1S)-1-(4-(5-(3-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)пропокси)пиримидин-2-ил)фенил)этилкарбамата (710 мг, выход 26%) в виде желтого масла. LC/MS (ES+): m/z 629,3 [М+Н+]; tR=2,660 мин; 1H-ЯМР (400 МГц, CDCl3): δ 1,42-1,48(м, 12H), 2,04-2,07 (м, 2H), 2,11-2,26 (м, 4H), 3,54-3,59 (м, 2H), 4,24-4,26 (м, 2H), 4,90-4,94 (м, 1H), 6,50-6,53 (м, 1H), 6,93-6,95 (м, 1H), 7,11-7,12 (м, 1H), 7,39-7,41 (м, 2H), 7,43-7,48 (м, 3H), 8,08 (шир., 1H), 8,28-8,32 (м, 2H), 8,51 (с, 2H); химическая формула: С33Н36N6O7; молекулярная масса: 628,67.
[00458] Стадия 8: Получение 2-((S)-4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-а][1,4]диазепин-6-ил)-N-((1S)-1-(4-(5-(3-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)пропокси)пиримидин-2-ил)фенил)этил)ацетамид, также известного как 2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.0²,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[5-(3-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1Н-изоиндол-4-ил]амино}пропокси)пиримидин-2-ил]фенил}этил]ацетамид
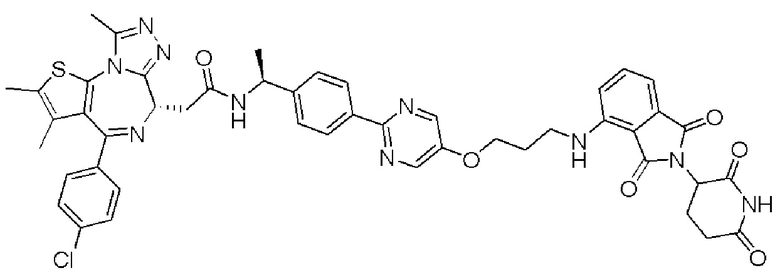
[00459] Смесь трет-бутил-(1S)-1-(4-(5-(3-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)пропокси)пиримидин-2-ил)фенил)этилкарбамата (710 мг, 1,1 ммоль) и 2,2,2-трифторуксусной кислоты (7 мл) в дихлорметане (7 мл) перемешивали при комнатной температуре в течение 1 часа. Летучие вещества выпаривали при пониженном давлении. Остаток повторно растворяли в сухом N,N-диметилформамиде (10 мл), с последующим последовательным добавлением (S)-2-(4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-а][1,4]диазепин-6-ил)уксусной кислоты (407 мг, 1,0 ммоль), N-этил-N-изопропилпропан-2-амина (730 мг, 5,6 ммоль) и HATU (2-(7-аза-1H-бензотриазол-1-ил)-1,1,3,3-тетраметилурония) (1,3 г, 3,3 ммоль) при 0°С. Полученную смесь оставляли нагреваться до комнатной температуры и перемешивали при комнатной температуре в течение 30 минут. Реакционная смесь распределяли между этилацетатом (40 мл) и водой (20 мл). Водный слой отделяли и экстрагировали этилацетатом (25 мл х 2). Объединенные органические слои промывали насыщенным раствором соли (30 мл), сушили над безводным сульфатом натрия и концентрировали при пониженном давлении с получением неочищенного остатка, который очищали с помощью препаративной TLC (элюент: 7% метанола в дихлорметане), получая 2-((S)-4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-а][1,4]диазепин-6-ил)-N-((1S)-1-(4-(5-(3-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-иламино)пропокси)пиримидин-2-ил)фенил)этил)ацетамида (160 мг, выход 15,5% после двух стадий) в виде желтого твердого вещества. LC/MS (ES+): m/z 911,3 [М+Н+]; tR=2,666 мин; 1H-ЯМР (400 МГц, CDCl3): δ 1,58 (д, J=6,8 Гц, 3H), 1,66 (с, 3H), 1,94-2,01 (м, 1H), 2,11-2,14 (м, 1H), 2,22-2,23 (м, 2H), 2,38 (с, 3H), 2,66 (с, 3H), 2,75-2,90 (м, 2H), 3,38-3,43 (м, 1H), 3,55-3,62 (м, 3H), 4,24-4,26 (м, 2H), 4,58-4,61 (м, 1H), 4,89-4,93 (м, 1H), 5,18-5,22 (м, 1H), 6,48-6,55 (м, 1H), 6,89-6,94 (м, 2H), 7,10-7,12 (м, 1H), 7,32-7,41 (м, 6H), 7,50 (т, J=7,6 Гц, 1H), 8,26-8,28 (м, 3H), 8,51 (с, 2H); химическая формула: С47Н43ClN10O6S; молекулярная масса: 911.43.
C. Биоанализ деградации белка
[00460] Для оценки уровня деградации белка в различных типах клеток были выполнены следующие биоанализы с использованием репрезентативных соединений, раскрытых в настоящем описании.
[00461] В каждом биоанализе клетки обрабатывали различным количеством соединений настоящего изобретения, как показано в таблице 1. В этом исследовании были оценена деградация следующих белков: TANK-связывающая киназа 1 (TBK1), рецептор эстрогена α (ERα), бромодомен-содержащий белок 4 (BRD4), андрогенный рецептор (AR) и с-Myc.
1. Протокол вестернблоттинга TBK1
[00462] Клетки Panc02.13, приобретенные в АТСС, культивировали в среде RPMI-1640 (Gibco), дополненной 15% FBS (АТСС) и 10 Ед/мл человеческого рекомбинантного инсулина (Gibco). Обработку контролем (DMSO) и испытуемыми соединениями (0,1 мкМ, 0,3 мкМ и 1 мкМ) проводили в 12-луночных планшетах в течение 16 часов. Агонист TLR3 Poly I:С (Invivogen; tlrl-pic) добавляли в последние 3 часа инкубации. Клетки собирали и лизировали в буфере RIPA (50 мМ Трис, рН 8, 150 мМ NaCl, 1% ТХ-100, 0,1% SDS, 0,5% дезоксихолата натрия) с добавлением ингибиторов протеазы и фосфатазы. Лизаты осветляли при 16000 g в течение 10 минут, и супернатанты разделяли с помощью SDS-PAGE. Иммуноблоттинг проводили с использованием стандартных протоколов. Использовали антитела к TBK1 (Cell Signaling # 3504), pIRF3 (Abcam # ab76493) и GAPDH (Cell Signaling # 5174). Количественную оценку полос проводили с использованием системы визуализации Bio-Rad ChemiDoc MP.
2. Протокол вестернблоттинга ERRα
[00463] Клетки NAMALWA (из ATCC) культивировали в среде RPMI-1640 (Life Technologies) с добавлением 15% FBS (Life Technologies). Обработку контролем (DMSO) и испытуемыми соединениями (0,1 мкМ, 0,3 мкМ и 1 мкМ) проводили в 12-луночных планшетах в течение 16 часов. Клетки собирали и лизировали в буфере для лизиса клеток (Cell Signaling Technologies), содержащем ингибиторы протеазы (Thermo Scientific). Лизаты осветляли при 16000 g в течение 10 минут, и супернатанты разделяли с помощью SDS-PAGE. Иммуноблоттинг проводили с использованием стандартных протоколов. Использовали антитела к ERRα (Cell Signaling # 8644) и GAPDH (Cell Signaling # 5174). Количественную оценку полос проводили с использованием системы визуализации Bio-Rad ChemiDoc MP.
3. Протокол вестернблоттинга BRD4
[00464] Клетки VCaP, приобретенные в АТСС, культивировали в модифицированной по Дульбекко среде Игла (АТСС), с добавлением 10% FBS (АТСС) и смеси пенициллин/стрептомицин (Life Technologies). Обработку контролем (DMSO) и испытуемыми соединениями (0,003 мкМ, 0,01 мкМ, 0,03 мкМ и 0,1 мкМ) проводили в 12-луночных планшетах в течение 16 часов. Клетки собирали и лизировали в буфере RIPA (50 мМ Трис, рН 8, 150 мМ NaCl, 1% ТХ-100, 0,1% SDS, 0,5% дезоксихолата натрия) с добавлением ингибиторов протеазы и фосфатазы. Лизаты осветляли при 16000 g в течение 10 минут, и определяли концентрации белка. Равные количества белка (20 мкг) подвергали анализу SDS-PAGE и с последующим иммуноблоттингом в соответствии со стандартными протоколами. Использовали антитела к BRD4 (Cell Signaling # 13440) и актину (Sigma # 5441). Реагент для обнаружения представлял собой субстрат Clarity Western ECL substrate (Bio-rad #170-5060).
4. Протокол ELISA для AR
[00465] Клетки VCaP, приобретенные в АТСС, культивировали в модифицированной по Дульбекко среде Игла (АТСС), с добавлением 10% FBS (АТСС) и смеси пенициллин/стрептомицин (Life Technologies). Обработку контролем (DMSO) и испытуемыми соединениями (0,0001 мкМ - 1 мкМ) проводили в 96-луночных планшетах в течение 16 часов. Клетки собирали и лизировали в буфере для лизиса клеток (катал. # 9803) (20 мМ Трис-HCl (рН 7,5), 150 мМ NaCl, 1 мМ Na2EDTA, 1 мМ EGTA, 1% Тритон, 2,5 мМ пирофосфата натрия, 1 мМ B-глицерофосфата, 1 мМ Na3VO4, 1 мкг/мл лейпептина). Лизаты осветляли при 16000 g в течение 10 минут, и загружали в PathScan AR ELISA (Cell Signaling, катал. # 12850). Набор PathScan® Total Androgen Receptor Sandwich ELISA Kit обеспечивает твердофазный иммуноферментный сэндвич-анализ (ELISA), который позволяет определить эндогенные уровни общего белка рецептора андрогена. Моноклональные (mAb) кроличьи антитела к рецептору андрогена наносили на поверхность лунок в виде покрытия. После инкубации с клеточными лизатами, белок рецептора андрогена захватывался антителом покрытия. После тщательной промывки добавляли моноклональные мышиные антитела для обнаружения mAb к рецептору андрогена, для обнаружения захваченного белка рецептора андрогена. Затем использовали меченные HRP антитела против мышиного IgG для выявления связанного антитела для обнаружения. Добавляли субстрат для HRP, TBM, и регистрировали развитие окраски. Величина поглощения света при развитии окраски пропорциональна количеству общего белка рецептора андрогена.
[00466] Антитела в наборе представляли собой заказные реагенты, специфические для такого набора.
5. Протокол анализ ELISA для с-Мус
[00467] Клетки 22RV-1, приобретенные в АТСС, культивировали в среде RPMI с добавкой 10% FBS. Клетки собирали с использованием трипсина (Gibco # 25200-114), подсчитывали и высевали из расчета 30000 клеток на лунку в объеме 75 мкл на лунку в среде RPMI+10% FBS в 96-луночные планшеты. К клеткам добавляли соединения, разбавленные в 0,1% DMSO, инкубировали в течение 18 часов, затем промывали и лизировали в 50 мкл буфера RIPA (50 мМ Трис, рН 8, 150 мМ NaCl, 1% ТХ-100, 0,1% SDS, 0,5% дезоксихолата натрия) с добавлением ингибиторов протеазы и фосфатазы. Лизаты осветляли при 4000 об/мин при температуре 4°С в течение 10 минут, а затем аликвоты вносили в 96-луночные планшеты для ELISA набора Novex Human c-myc ELISA Life Technologies (катал. # KHO2041). В каждую лунку добавляли 50 мкл антител для обнаружения с-Мус, планшеты инкубировали при комнатной температуре в течение 3 часов, а затем промывали буфером ELISA для промывки. В каждую лунку добавляли 100 мкл вторичного антитела против кроличьего IgG, меченного HRP, и инкубировали при комнатной температуре в течение 30 минут. Планшеты промывали буфером ELISA для промывки, в каждую лунку добавляли 100 мкл ТМВ, и затем контролировали изменение цвета каждые 5 минут. Добавляли 100 мкл стоп-раствора и планшеты считывали при длине волны 450 нм.
D. Результаты
[00468] В Таблице 1 представлены экспериментальных данные и результаты, полученные для репрезентативных соединений настоящего изобретения. В частности, различные типы клеток были обработаны соединениями, перечисленными в Таблице 1, которые идентифицированы по своей химической структуре, данным масс-спектрометрии и по названиям соединений.
[00469] В Таблице 1 показано следующее. (А): 10-30% деградации была достигнута в клетках, обработанных 1 мкМ соединений 1, 6-9, 12 и 17; (В): деградация 31-50% была достигнута в клетках, обработанных 1 мкМ соединений 2-5, 10 и 20; и (С): деградация >50% была достигнута в клетках, обработанных 1 мкМ соединений 11, 13-16, 18-19, 21 и 22. В Таблице 1 также показано, что (D): соединения 24 и 26-35 имеют IC50<50 нМ, в то время как (Е): соединения 23 и 25 имеют IC50 > 50 нМ.
Пример 2
[00470] Низкомолекулярные ингибиторы являются краеугольным камнем для разработки лекарственных средств для онкологии, и они обычно действуют путем ингибирования активности ферментов (например, как ингибиторы киназы) или путем вмешательства в белок-белковые взаимодействия (например, как ингибиторы BRD4). Учитывая обратимое связывание большинства низкомолекулярных ингибиторов, часто требуется большие системные концентрации лекарственного вещества, чтобы обеспечить достаточное функциональное ингибирование. Кроме того, достижение и поддержание высокого системного уровня лекарственного вещества, которое необходимо для обеспечения требуемой эффективности в условиях in vivo, связано с рядом сложностей для многих целевых мишеней.
[00471] Белок BRD4, член семейства белков с бромо- и экстратерминальным доменом (ВЕТ), представляет собой белок, характеризующийся двумя бромодоменами (домен BD) на N-конце и в экстратерминальным доменом (домен ET) на С-конце. Эти два домена BD распознают и взаимодействуют с остатками ацетилированного лизина на N-конце хвоста гистона белка. Считается, что домен ET выполняет функции каркаса при рекрутинге различных регуляторов транскрипции, но его функция еще не полностью установлена. Было показано, что BRD4 локализован в супер-энхансерных областях, которые часто располагаются вверх по сигнальному пути важных онкогенов, таких как с-Мус, Bcl-XL и BCL-6, и он также играет ключевую роль в регулировании их экспрессии. Принимая во внимание его роль в регулировании экспрессии генов путем рекрутинга соответствующих транскрипционных модуляторов в специфические геномные локусу, BRD4 рассматривается в качестве многообещающей терапевтической мишени для лечения и/или профилактики ряда раковых заболеваний человека, таких как срединная карцинома, острый миелоидный лейкоз (AML), множественная миелома (ММ), лимфома Беркитта (BL) и рак предстательной железы.
[00472] Были разработаны несколько низкомолекулярных ингибиторов BET бромодомена, например, JQ1, IBET и OTX15, которые показали терапевтические перспективы в некоторых доклинических испытаниях на моделях различных видов рака, в том числе и BL. Почти во всех случаях BL имеет место транслокация гена с-Мус, в результате которой он располагается под контролем супер-энхансера, расположенного выше от IgH, что приводит к аномально высокому уровню экспрессии с-Мус, развитию опухоли и новообразований. Доклинические исследования, выполненные с ингибиторами BRTD4, показали их способность к супрессии с-Мус и к подавлению пролиферации в клеточных линиях BL; однако, значения IC50 для этих ингибиторов часто находятся в диапазоне от 100 нМ до 1 мкМ.
Материалы и методы
[00473] Детали выполнения этого исследования представлены ниже.
[00474] 1. Соединения
[00475] Соединение No. 14 (см. Таблицу 1) синтезировали в соответствии с процедурой, описанной выше в Примере 1, синтез # 8. Это соединение упоминается в данном примере как «A825», и оно имеет следующее структуру и название.
[00476] 2-((S)-4-(4-хлорфенил)-2,3,9-триметил-6Н-тиено[3,2-f][1,2,4]триазоло[4,3-а][1,4]диазепин-6-ил)-N-(4-(2-(2-(2-(2-(2-(2,6-диоксопиперидин-3-ил)-1,3-диоксоизоиндолин-4-ил)этокси)этокси)этокси)этокси)фенил)ацетамид
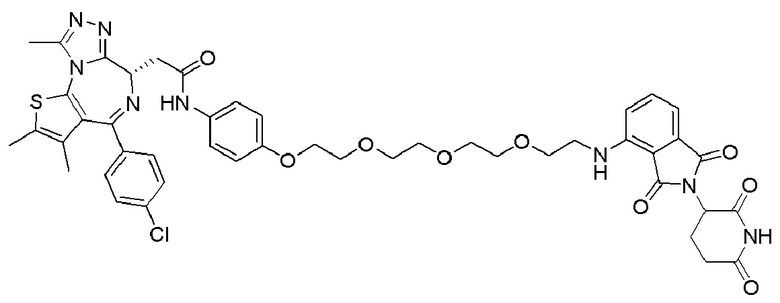
[00477] Как показано на Фигуре 2, соединение A825 содержит фрагмент связывания с BRD4 (производное OTX-15), который соединен с фрагментом рекрутинга убиквитинлигазы E3 цереблона (производное помалидомида) через тетраоксатетрадекановый линкер.
[00478] Эффекты действия A825 на клетки были оценены с использованием различных клеточных линий, и эти эффекты сравнивали с эффектами действия двух известных ингибиторов домена BET, таких как JQ1 и OTX-15. JQ1 является наиболее часто используемым ингибитором домена BET в опубликованных исследованиях, и OTX-15 является ингибитором домена BET, который испытывался на более поздних стадиях клинических разработок.
[00479] Фрагмент рекрутинга цереблона соединением A825 также оценивали на различных клеточных линиях, и его сравнивали с помалидомидом.
[00480] Ингибиторы JQ1, OTX-15 и помалидомид были синтезированы в соответствии с опубликованными методами.
2. Клетки и реагенты
[00481] Клетки NAMALWA, Ramos, CA-46 и DAUDI были приобретены в АТСС и их поддерживали в соответствии с инструкциями. Антитела против BRD4 (# E2A7X), с-Мус (# D84C12), PARP (# 46D11) были приобретены в Cell Signaling Technology. Антитела против актина (# A5441) закупали в Sigma-Aldrich. Вторичные антитела (# 7074, # 7076) были приобретены в Cell Signaling Technology. MG132 (# M7449) приобретено в Sigma-Aldrich. Карфизомиб (carfizomib, # S2853) был приобретен в Selleck.
3. Вестерн-блот анализ
[00482] Культивируемые клетки собирали в буфере для лизиса, содержащем 40 мМ HEPES (рН 7,4), 140 мМ NaCl, 2,5 мМ EDTA, 1% NP-40, 0,1% SDS и коктейль ингибиторов протеазы. Через 10 минут центрифугирования (14000 оборотов в минуту) супернатант собирали для определения концентрации белка по методу BCA и подвергали иммуноблоттингу по стандартному протоколу. Результаты вестерн-блоттинга визуализировали с использованием субстрата из набора Bio-Rad Clarity ECL Western Blotting и системы визуализации Bio-Rad ChemiDocTM MP.
4. RT-PCR (ОТ-ПЦР)
[00483] Выделение РНК проводили с использованием набора Aurum™ Total RNA Mini Kit (#732-6820) от Bio-Rad. Первую цепь кДНК из общей РНК синтезировали с использованием набора High-Capacity cDNA Reverse Transcription Kit (#4368813) от Life Technologies в соответствии с инструкцией изготовителя. Количественную PCR проводили с использованием набора Bio-rad SsoAdvanced™ Universal SYBR® Green Supermix (#172-5271). Использовали следующие праймеры:
(SEQ ID NO: 6)
(SEQ ID NO: 7)
(SEQ ID NO: 8)
(SEQ ID NO: 9)
5. Анализ пролиферации
[00484] Для оценки влияния ингибиторов на пролиферацию, клетки (50000/100 мкл) высевали в 96-луночные планшеты для культивирования тканей с последующим добавлением соединения в указанной концентрации. Через 72 часа в лунки добавляли 100 мкл восстановленного реагента CellTiter-Glo (CTG) (Promega, # G7572) и планшеты считывали на ридере Cytation 3 от BIOTEK. Относительный рост клеток определяли путем сравнения показаний, полученных в результате анализа обработанных клеток и клеток, обработанных DMSO (контроль).
6. Определение Kd
[00485] Аффинность соединений к бромодоменам 1 и 2 в BRD4 определяли с помощью BROMOscan ™ от DiscoverX.
B. Результаты
[00486] Эффекты действия JQ1, OTX-15 и A825 на клетки оценивали и сравнивали в рамках следующих экспериментов.
[00487] 1. Низкомолекулярные ингибиторы доменов BET привели к значительному накоплению белка BRD4 и неэффективному подавлению с-Мус
[00488] а. Дозозависимое накопление BRD4 при обработке JQ1 и OTX-15
[00489] Исследования показали, что клеточные линии лимфомы Беркитта (BL) реагируют на ингибиторы BRD4 из-за зависимости клеточных линий от онкогена с-Mус, который транслоцируется и обеспечивает контроль BRD4 со стороны супер-энзансеров IgH, расположенных ниже BRD4.
[00490] В начальном эксперименте, различные клеточные линии BL (клетки NAMALWA, Ramos, СА-46 и DAUDI) обрабатывали двумя известными ингибиторами домена BET (JQ1 и OTX-15) в различных концентрациях, чтобы подтвердить, что эти ингибиторы являются эффективными для снижения и/или предотвращения деградации белка BRD4. В частности, клетки NAMALWA и Ramos обрабатывали различными концентрациями JQ1 и OTX-15 (3 нМ, 10 нМ, 100 нМ, 300 нМ, 1000 нМ и 3000 nM); и клетки CA-46 и DAUDI обрабатывали JQ1 и OTX-15 в концентрациях 100 нМ и 300 нМ. Отдельный группу клеток обрабатывали аналогичным образом, за исключением того, что вместо ингибитора использовали DMSO. Все клетки обрабатывали в течение ночи увеличивающимися дозами JQ1 и OTX15. После обработки собирали клеточные лизаты и анализировали с помощью иммуноблота для актина и BRD4.
[00491] Эффекты от этих обработок определяли путем оценки количества BRD4, присутствующего в клетках, с помощью вестерн-блот-анализа после обработки (см. Фигуры 3A, 3B, 3C и 3D).
[00492] На Фигурах 3A-3D показано, что JQ1 и OTX-15 приводят к значительному накоплению белка BRD4 дозозависимым образом во всех клеточных линиях, использованных в испытании. Эти результаты согласуются с результатами предыдущих исследований, где было показано, что обработка JQ1 приводит к положительному регулированию уровней BRD4 в некоторых клеточных линиях рака легкого: см. Shimamura, T., Chen, Z., Soucheray, M., Carretero, J., Kikuchi, E., Tchaicha, J.H., Gao, Y., Cheng, K.A., Cohoon, T.J., Qi, J., et al. (2013); J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674; и K. Klapproth, et al., British journal of haematology, 149 (2010) 484-497.
[00493] b. Скорость накопления BRD4 при обработке JQ1 и OTX-15
[00494] Также была определена скорость, с которой происходит накопление BRD4 в клеточных линиях BL после обработки их JQ1 и OTX-15. В частности, клетки NAMALWA и Ramos обрабатывали 300 нМ каждого ингибитора в течение 0, 0,5, 1,0, 2,0, 4,0, 7,0, 24 и 48,0 часов. Клеточные лизаты собирали после обработки и анализировали с помощью иммуноблота для актина и BRD4.
[00495] На Фигуре 3E показано, что клетки NAMALWA содержат регистрируемый уровень белка BRD4 до обработки каким-либо ингибитором (0 часов). Количество белка BRD4, присутствующего в клетках NAMALWA, заметно возросло после 30-минутной обработки JQ1 или OTX-15, и количество белка BRD4 продолжало возрастать при более длительном времени обработки (с 0,5 до 48,0 часов).
[00496] На Фигуре 3F показаны результаты на клетках Ramos, схожие с результатами, полученными на клетках NAMALWA. Клетки Ramos также содержат регистрируемый уровень белка BRD4 до обработки каким-либо ингибитором (0 часов). Однако накопление белка BRD4 в клетках Ramos происходит с более медленной скоростью, чем в клетах NAMALWA. В частности, отмечено заметное увеличение количества BRD4 при обработке соединениями JQ1 или OTX-15 длительностью от примерно 4,0 до примерно 7,0 часов. Заметное увеличение количества BRD4 в клетках Ramos наблюдали после 24,0 часов обработки обеими ингибиторами.
[00497] В совокупности, результаты, представленные на Фигурах 3Е и 3F, показывают, что низкомолекулярные ингибиторы BRD4 приводят к быстрому накоплению белка BRD4 в различных клеточных линиях при концентрации 0,3 мкМ JQ1 или OTX15.
[00498] с. JQ1 и OTX-15 приводит к подавлению с-Мус по ходу сигнального пути
[00499] Как описано выше, белок BRD4 расположен в супер-энхансерных регионах, которые часто расположены по ходу сигнального пути важных онкогенов, таких как с-Мус, Bcl-xL и Bcl-6. Для определения того, могут ли ингибиторы домена BET влиять на экспрессию онкогенов по ходу сигнального пути, клетки NAMALWA обрабатывали более высокими концентрациями (3 нМ, 10 нМ, 100 нМ, 300 нМ, 1000 нм и 3000 нМ) соединениями JQ1 или OTX-15 в течение ночи. Отдельный набор клеток обрабатывали таким же образом, за исключением того, что вместо ингибитора использовали DMSO., После обработки собирали клеточные лизаты и анализировали их при помощи иммуноблота для с-Мус и актина.
[00500] На Фигуре 3G показано, что обработка клеток ингибиторами домена BET может привести к некоторому подавлению с-Мус по ходу сигнального пути, но даже при высоких концентрациях ингибиторы не были способны полностью ингибировать экспрессию с-Myc. В частности, на Фигуре показано, что низкие концентрации (3 нМ до 30 нМ) ингибиторов не оказали заметного влияния на уровень с-Мус, присутствующего в клетках. Тем не менее, количество с-Мус заметно снижается в клетках, обработанных JQ1 или OTX-15 в концентрации 100 нМ, и еще большее снижение наблюдали в клетках, обработанных JQ1 или OTX-15 в концентрациях 300 нМ и 1000 нМ. Хотя JQ1 и OTX-15 значительно подавляли уровни с-Мус уровня в концентрациях от 100 нМ до 1000 нМ, однако результаты показывают, что более высокие дозы этих ингибиторов не приводят к дальнейшему снижению уровня с-Мус (см. Фиг. 3G: сравни результаты для 1000 нМ и для 3000 нМ).
[00501] На основании этих результатов ясно, что обработка клеток ингибиторами домена BET, такими как JQ1 и OTX-15, приводит значительному подавление белка с-Мус по ходу сигнального пути BRD4 при концентрациях от 100 нМ до 1000 нМ. Однако, более высокие концентрации JQ1 и OTX-15 (свыше 1000 нМ) не приводит к дальнейшему подавлению белка с-Myc, и эффект подавления не выходит за пределы, достигнутые при концентрации ингибитора, составляющего 1000 нМ. Кроме того, ни JQ1, ни OTX-15 не смогли полностью подавить экспрессию с-Myc, даже при концентрации 3000 нМ.
[00502] d. Подавление с-Мус при действии JQ1 и OTX-15 является обратимым
[00503] Следующее исследование было проведено с целью определения, является ли обратимым эффект подавления экспрессии с-Мус при действии JQ1 и OTX-15.
[00504] В этом исследовании клетки NAMALWA обрабатывали соединением JQ1 (1000 нМ) в течение 24 часов, с последующими тремя промывками для удаления ингибитора. Клетки повторно высевали и инкубировали без ингибитора в течение 0, 0,5, 1,0, 2,0, 3,0, 4,0 и 6,0 часов. Клеточные лизаты затем собирали в различные моменты времени и анализировали с помощью иммуноблоттинга для с-Мус и актина. В параллельном эксперименте клетки NAMALWA обрабатывали таким же образом, за исключением того, что вместо JQ1 использовали DMSO.
[00505] На Фигуре 3Н показано, что JQ1 в концентрации 1000 нМ значительно подавлял уровни белка с-Myc в клетках NAMALWA (сравни полосу при 0 час для клеток, обработанных JQ1, с полосой при 0 час для клеток контроля, обработанных DMSO), что согласуется с результатами, представленными на Фигурах 3A-3D. Фигура 3H также показывает, что подавление с-Мус при действии JQ1 является быстро обратимым, так как уровни белка с-Myc значительно увеличилось за время от 1,0 до 2,0 часов после удаления ингибитора, и в пределах 3,0 часов после удаления ингибитора уровень белка с-Мус возвращался к уровню контрольного образца.
[00506] В другом эксперименте клетки Ramos обрабатывали соединениями JQ1 (1000 нМ), OTX-15 (1000 нМ) или DMSO (контроль) в течение 24 часов. После обработки клетки либо лизировали (для оценки подавления с-Мус под действием ингибитора) или промывали для удаления ингибитора, и вновь высевали для инкубирования без ингибитора в течение 4,0 часов (для оценки обратимости подавления с-Мус). Клеточные лизаты собирали, и их анализировали при помощи иммуноблота для с-Мус и актина.
[00507] Результаты, полученные для клеток Ramos, согласуются с результатами, полученными для клеток NAMALWA. В частности, на Фигуре 3I (нижняя панель) показано, что JQ1 и OTX15 подавляют с-Myc в клетках Ramos (полосы «JQ1» и «OTX15»), но этот эффект подавления является обратимым, так как уровень с-Myc значительно увеличивался в течение 4,0 часов после удаления ингибиторов (см. полосы «4 часа после вымывания JQ1» и «4 часа после вымывания OTX15»).
[00508] Результаты данного исследования показывают, что низкомолекулярные ингибиторов BRD4 (JQ1 и OTX-15) приводит к подавлению с-Myc, расположенному по ходу сигнального пути, в клеточных линиях BL. Тем не менее, ингибиторы не смогли полностью подавить экспрессию с-Мус в клетках, даже при высоких концентрациях. Кроме того, был обнаружено, что эффект подавления экспрессии с-Мус этими ингибиторами является быстро обратимым, и после удаления ингибиторов уровни белка с-Myc возрастают в течение приблизительно от 2,0 до 4,0 часов. Результаты, полученные в этом исследовании, согласуются с предыдущими результатами, полученными для AML, где было показано, что при обработки JQ1 происходит подавление с-Мус, но этот эффект является быстро обратимым после удаления JQ1 (J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674).
[00509] 2. Захват убиквитинлигазы Е3 цереблона обеспечивает эффективное деградирование BRD4 при действии Protac
[00510] Быстрое и надежное накопление BRD4 при обработке соединениями JQ1 и OTX-15, вместе с обратимой природой связывания ингибитора с BRD4, что показано в предыдущем исследовании, может быть причиной незначительных эффектов в отношении подавления с-Мус по ходу сигнального пути и ингибирования пролиферации для линий клеток BL и других видов рака. Чтобы обойти недостатки низкомолекулярных ингибиторов BRD4, с использованием технологии PROTAC было разработано химерное соединеие A825 (описано выше и показано на Фигуре 2).
[00511] а. Аффинности связывания ингибитора с бромодоменом белка BRD4
[00512] Аффинность связывания соединения A825 с бромодоменом 1 (BD1) и бромодоменом 2 (BD2) белка BRD4 оценивали и сравнивали с аффинностью связывания этих же областей соединениями JQ1 и OTX-15. Аффинности связывания каждого из этих соединений показаны в Таблице, приведенной ниже.
(Kd)
[00513] Исследования по определению аффинности связываниия показали, что аффинность связываниия соединения A825 с BD1 и BD2 белка BRD4 немного ниже, чем аффинности связывания этих же доменов соединениями JQ1 и OTX-15.
[00514] b. A825 обеспечивает эффективную деградацию белка BRD4
[00515] Оценивали эффект действия A825 на уровни белка BRD4 в линии клеток BL.
[00516] В частности, клетки NAMALWA и СА-46 обрабатывали в течение ночи возрастающими концентрациями (0,3 нМ, 1,0 нМ, 3,0 нМ, 10 нМ, 30 нМ, 100 нМ, 300 нМ и 1000 нм) соединения A825. После обработки собирали клеточные лизаты и их анализировали с помощью иммуноблота для актина и BRD4.
[00517] На Фигурах 4A и 4В показано, что обработка клеточных линий BL соединением A825 вызывает полную деградацию белка BRD4 при низких концентрациях этого соединения. В частности, на основании данных, показанных на этой Фигуре, ясно, что DC50 (50% от максимальной деградации) BRD4 в клетках NAMALWA достигается при обработке клеток соединением A825 при концентрациях 1,0 нМ или менее (Фиг. 4A). Аналогично, DC50 для BRD4 в клетках СА-46 достигается при обработке соединением A825 при концентрациях от 0,3 нМ до 1,0 нМ или менее (Фиг. 4B).
[00518] Кроме того, по-видимому, белок BRD4 полностью деградирует в обеих линиях клеток BL, обработанных соединением A825 в концентрациях в диапазоне от приблизительно 3,0 нМ до приблизительно 300 нМ, о чем свидетельствует отсутствие каких-либо заметных полос белка BRD4 для этих концентраций обработки. Интересно отметить, что наблюдали небольшое количество белка BRD4 на полосах лизатов обеих линий клеток BL, обработанных 1000 нМ соединения A825, что свидетельствует о том, что деградация белка BRD4 за счет действия A825 происходит в дозозависимым образом с формой кривой в виде колокола. То есть, полная деградация BRD4 происходит в пределах критического диапазона концентраций A825, поскольку наблюдается неполное разложение BRD4 при действии A825 с концентрацией выше или ниже этого критического диапазона.
[00519] Учитывая, что фрагменты связывания BRD4 и цереблона в соединении A825 имеют Kd 28-90 нМ и 3 мкМ, для соответствующих мишеней, это свидетельствует о том, что A825 действует каталитически в опосредовании деградации BRD4.
[00520] с. A825 приводит к быстрой деградации белка BRD4
[00521] Также определяли скорость разложения BRD4 в линиях клеток BL после их обработки соединением A825. В этом исследовании клетки NAMALWA и Ramos обрабатывали соединением A825 (100 нМ) в течение 0, 0,5, 1,0, 2,0, 4,0, 7,0 и 24 часов. После обработки клеточные лизаты собирали и анализировали с помощью иммуноблота для актина и BRD4.
[00522] На Фигурах 4C и 4D показано, что белок BRD4 присутствует в обеих клеточных линиях BL до начала обработки соединением A825 (0 ч). Уровни белка BRD4 заметно снизились за 1 час обработки соединением A825 и уровни белка продолжали постепенно снижаться в течение периода обработки 24,0 часа. На этой Фигуре также показано, что деградация BRD4 при действии A825 происходит достаточно быстро, что приводит к тому, что более чем 50% белка теряется после 2 часов обработки соединением A825.
[00523] d. Деградация BRD4 при действии A825 зависит от цереблона
[00524] Для подтверждения того, что деградация BRD4 под действием A825 зависит от цереблона, выполнили эксперимент по конкурентному ингибированию, в котором клеточные линии BL обрабатывали A825, помалидомидом или их сочетанием. Как обсуждено выше и показано на Фигуре 2, соединение A825 содержит фрагмент рекрутинга убиквитинлигазы E3 цереблона, который является производным помалидомида, и помалидомид, как таковой, и A825 конкурируют за связывание цереблона. Таким образом, если деградация BRD4 при обработке соединением A825 зависит от цереблона, то клетки, обработанные комбинацией A825 и помалидомида должны показать снижение деградации BRD4 по сравнению с клетками, обработанными только соединением A825.
[00525] В этом исследовании клетки NAMALWA и Ramos обрабатывали в течение ночи различными концентрациями A825 (10 нМ, 100 нМ и 1000 нМ), помалидомида (10 мкМ) по-отдельности, или комбинацией A825 и помалидомида. После обработки клеточные лизаты собирали и анализировали с помощью иммуноблота для актина и BRD4.
[00526] На Фигурах 4E и 4F показана полная деградация BRD4 в клетках, обработанных 10 нМ и 100 нМ соединения A825, в то время как небольшое количество BRD4 присутствовало в клетках, обработанных 1000 нМ соединения A825, что согласуется с результатами, представленными на Фигурах 4А и 4В. Фигуры 4E и 4F также показывают, что уровни BRD4 не изменились в клетках, обработанных только помалидомидом, что явилось ожидаемым, поскольку помалидомид не влияет на деградацию BRD4. И, наконец, на Фигурах 4E и 4F показано, что уровни белка BRD4 были частично спасены от деградации в клетках, обработанных комбинацией A825 и помалидомида.
[00527] Результаты этого исследования подтверждают, что деградация BRD4 под действием соединения A825 опосредована цереблоном.
[00528] е. Ингибиторы протеасом предотвращают деградацию BRD4 при действии A825
[00529] Цереблон является белком убиквитинлигазы Е3, котрый по отдельности или в сочетании с ферментом коннъюгации убиквитина E2 вызывает присоединение убиквитина к лизину на белке-мишени, а затем нацеливает специфические белковые субстраты для деградации протеасомой. Для подтверждения того, что деградация BRD4 под лействием A825 проходит через протеасомный путь, линии клеток BL обрабатывали соединием A825 в присутствии и в отсутствие ингибиторов протеасом.
[00530] В частности, клетки NAMALWA обрабатывали в течение ночи только соединением A825 (10 нМ и 100 нМ); соединением MG132 (5 мкМ); или только карфизомибом (5 мкМ); или сочетанием A825 с MG132 или с карфизомибом. После обработки клеточные лизаты собирали и анализировали с помощью иммуноблота для актина и BRD4.
[00531] На Фигуре 4G показано, что белок BRD4 полностью деградируют в клетках, обработанных 10 нМ или 100 нМ соединения A825, что согласуется с результатами, представленными на Фигурах 4А и 4В. На Фигурах 4E и 4F также показано, что MG132 и карфизомиб полностью предотвращают деградацию BRD4, вызванную воздействием 10 нМ или 100 нМ соединения A825. Эти результаты подтверждают, что деградация BRD4 при действии A825 протекает по обычному пути цереблона через протеасому.
[00532] е. Обобщение результатов и их обсуждение
[00533] Совокупность данных, полученных в экспериментах (а)-(f), описанных выше, показывает, что A825 приводит к быстрой и эффективной деградации белка BRD4 по механизму, опосредованному цереблоном, и зависимым от протеасом образом.
[00534] Наблюдаемый в данном исследовании профиль деградации белка BRD4 поддерживает механизм действия, представленный на модели, показанной на Фигуре 8. В частности, в необработанных клетках не ингибируется экспрессия BRD4 и он функционирует под регулярным клеточным контролем. Однако, когда клетки обрабатывают низкими концентрациями A825, достаточными для присутствия A825 в клетке, он эффективно связывает белок BRD4 на одном конце молекулы и молекулу цереблона на другом конце, образуя тримерный комплекс «BRD4-A825-Цереблон» (см. Фигуру 8A). Этот тримерный комплекс «BRD4-A825-Цереблон» обеспечивает эффективную деградацию белка BRD4 в клетке. Тримерный комплекс образуется в клетках, обработанных A825, в пределах определенного диапазона концентраций, и он может привести к полному истощению белка BRD4 в клетках (см. Фигуру 8А). Однако, когда клетки обрабатывают высокой концентрацией A825, образуются димеры «BRD4-A825» и «A825-Цереблон», что препятствует образованию оптимального тримера, и приводит к менее эффективной деградация белка BRD4 (см. Фигуру 8В).
[00535] 3. A825 приводит к более высокому и более длительному подавлению с-Мус, чем низкомолекулярные ингибиторы
[00536] Как описано выше, обработка клеток низкомолекулярными ингибиторами доменов BET, такими как JQ1 и OTX-15, в концентрациях 100 нМ или выше, привела к значительному, но неполному, подавление белка с-Мус, расположенному ниже по сигнальному пути, и концентрации выше 1000 нМ не приводят к дальнейшему подавлению с-Мус. В следующих исследованиях эффекты действия A825 на экспрессию с-Мус по ходу сигнального пути сравнивались с эффектами действия низкомолекулярных ингибиторов JQ1 и OTX15.
[00537] а. A825 подавляет с-Myc в большей степени, чем JQ1 и OTX-15
[00538] В этом исследовании подавление с-Мус соединением A825 сравнивали с эффектами действия JQ1 и OTX-15.
[00539] В частности, клетки NAMALWA и Ramos обрабатывали в течение ночи различными концентрациями соединения A825 (100 нМ, 300 нМ и 1000 нм), или соединения JQ1 (100нм, 300 нм, 1000 нм, 3000 нм и 10000nM), или соединения OTX-15 (100 нМ, 300 нМ, 1000 нМ, 3000 нМ и 10000 нМ). После обработки клеточные лизаты собирали и анализировали с помощью иммуноблота для BRD4, с-Мус и актина.
[00540] На Фигурах 5А и 5В показано, что JQ1 и OTX-15 приводят к устойчивому накоплению BRD4, и значительному, но неполному, подавлению с-Myc в обеих клеточных линиях BL (см. Фигуру 3G). Фигуры 5А и 5В также показывают, что соединение A825 привело к значительной деградации BRD4 (см. Фигуры 4А-4G) и более сильному подавлению с-Мус, по сравнению с JQ1 и OTX-15 в обеих клеточных линиях BL. Следует отметить, что соединение A825 обеспечивало понижающую регуляцию экспрессии с-Myc в гораздо большей степени, чем JQ1 и OTX-15, и при этом этот эффект наблюдался при существенно меньших концентрациях соединения.
[00541] b. A825 подавляет экспрессию с-Myc в течение более длительного времени, чем JQ1 и OTX-15
[00542] Следующее исследование было проведено с целью сравнения продолжительности подавления с-Мус при действии A825, JQ1 и OTX-15.
[00543] В частности, клетки NAMALWA обрабатывали в течение 24 часов соединением A825 (0,1 мкм), JQ1 (1,0 мкМ) и OTX-15 (1,0 мкМ), с последующими тремя промывками для удаления этих соединений. Клетки повторно высевали в свежей среде и инкубировали в отсутствии какого-либо из соединений в течение 0, 2,0, 4,0, 6,0 и 24,0 часов. В параллельном контрольном эксперименте клетки обрабатывали таким же образом, за исключением того, что вместо ингибитора использовали DMSO. Лизаты собирали и анализировали при помощи иммуноблота для белка BRD4, с-Мус и актина.
[00544] На Фигуре 5С показано, что эффект действия в отношении белка BRD4 после обработки соединением A825 (деградация BRD4) сохраняется значительно дольше, чем наблюдаемый эффект после обработки соединениями JQ1 и OTX-15 (накопление BRD4). Кроме того, после обработки соединением A825 эффект подавления с-Мус также сохранялся намного дольше, чем в случае обработки соединениями JQ1 и OTX-15. В частности, на этой Фигуре показано, что детектируемые уровни белка BRD4 не наблюдались в клетках через 6 часов после их обработки соединением A825.
[00545] Кроме того, даже через 24 часа после обработки соединением A825, с помощью вестерн-блота наблюдалось лишь небольшое количество BRD4, которое было значительно ниже уровня BRD4, наблюдаемого в контрольном образце. В противоположность этому, накопление BRD4 при обработке JQ1 BRD4 и OTX-15 было кратковременным, и уровень белка BRD4 в этих образцах возвращающихся к уровню контрольного образца в течение примерно 4 часов после обработки. На Фигуре также показано, что только небольшой уровень белка с-Myc был обнаружен между 2 и 6 часами после обработки соединением A825, и даже через 24 часа после обработки уровень с-Мус был значительно ниже, чем у контрольного образца. В противоположность этому, на Фигуре показано, что уровни белка с-Myc восстанавливаться до уровня контроля в течение примерно 4 часов после удаления JQ1 и OTX15. Таким образом, эти результаты показывают, что эффекты в отношении BRD4 и с-Мус после обработки соединением A825 сохраняются в течение более длительного периода времени по сравнению с обработкой соединениями JQ1 и OTX-15.
[00546] с. Соединение A825 подавляет функционирование с-Мус дольше, чем JQ1 и OTX-15
Белок с-Мус представляет собой фактор транскрипции, который активирует экспрессию многих генов, включая ген SLC19A1, кодирующий мембранный белок, который является переносчиком фолатов и который участвует в регуляции концентрации внутриклеточной фолиевой кислоты. В предыдущих экспериментах было показано, что A825, JQ1 и OTX-15 подавляют экспрессию с-Мус, и эффект от действия A825, по сравнению с JQ1 и OTX-15, является более сильным и более длительным. Для дальнейшего исследования, как A825, JQ1 и OTX-15 могут воздействовать на путь и события, происходящие по ходу сигнального пути от белка BRD4, клетки обрабатывали каждым соединением и оценивали экспрессию гена SLC19A1 в разное время после этой обработки.
[00547] В частности, клетки NAMALWA обрабатывали в течение 24 часов соединениями A825 (0,1 мкм), JQ1 (1,0 мкМ) и OTX-15 (1,0 мкМ), с последующими тремя промывками для удаления соединений. Клетки повторно высевали в свежей среде и инкубировали без какого-либо ингибитора 0, 6,0 и 24,0 часа. В параллельном эксперименте клетки обрабатывали таким же способом, за исключением того, что вместо ингибитора использовали DMSO. В каждый из указанных моментов времени из лизатов экстрагировали РНК, котрую обратно транскрибировали в кДНК, которую подвергали количественному анализу методом QPCR, используя специфические для SLC19A1 праймеры. Также определяли количество GAPDH с помощью QPCR в качестве внутреннего контроля.
[00548] В соответствии с результатами по подавлению белка с-Myc (как показано на Фигуре 5С), на Фигурах 5D-5F показано, что обработка соединением A825, по сравнению с JQ1 и OTX-15, в более существенной степени и более длительно подавляют функции с-Myc, что определено по экспрессии гена SLC19A1. В частности, на Фигуре показано, что экспрессия гена SLC19A1 значительно снижается при обработке соединением A825, и даже через 24 часа после обработки экспрессия гена SLC19A1 значительно снижена по сравнению с контрольным образцом. В противоположность этому, Фигура показывает, что при обработке соединениями JQ1 и OTX-15 экспрессия гена SLC19A1 уменьшается, но затем возвращается к уровню до обработки, в течение времени от 6,0 до 24,0 часов.
[00549] 4. A825 в превосходной степени подавляет клеточную пролиферацию, по сравнению с другими низкомолекулярными ингибиторами
[00550] Как известно, клетки BL чувствительны к действию ингибиторов BRD4, которые подавляют передачу сигнала с-Myc и индуцируют ингибирование пролиферации клеток (J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674). Эффекты действия соединений A825, JQ1 и OTX-15 на пролиферацию клеток оценивали в следующих экспериментах.
[00551] а. Соединение A825 подавляет клеточную пролиферацию в большей степени, чем JQ1 и OTX-15
[00552] В этом исследовании оценивали пролиферацию различных клеточных линий BL после их обработки соединениями A825, JQ1 и OTX-15.
[00553] В частности, клеточные линии NAMALWA, Ramos, CA-46 и DAUDI высевали при плотности 50000 клеток/100 мкл в 96-луночные планшеты. Клетки обрабатывали возрастающими концентрациями соединений A825 (100 мкМ, 300 мкМ, 1 нМ, 3 нМ, 10 нМ, 30 нМ, 100 нМ, 300 нМ, 1 мкМ), JQ1 и OTX15 (1 нМ, 3 нМ, 10 нМ, 30 нМ, 100 нМ, 300 нМ, 1 мкМ, 3 мкм; соединения JQ1 и OTX-15 для клеток NAMALWA были использованы в концентрации 10 мкМ), как показано на Фигурах 6А-6D. Относительную пролиферацию образцов клеток определяли с помощью анализа CGT через 72 часа после обработки.
[00554] На Фигурах 6А-6D показано, что обработка соединением A825 привела к более сильному подавлению пролиферации по сравнению с обработкой соединениями JQ1 или OTX15 для всех линий клеток BL во всех испытаниях, и этот эффект был достигнут при использовании значительно более низких концентраций соединения. Интересно отметить, что относительный рост линий клеток Ramos и DAUDI, которые были обработаны более высокими концентрациями соединения A825, был близок к 0.
[00555] b. A825 подавляет клеточную пролиферацию более в течение более длительного времени, чем JQ1 и OTX-15
[00556] В этом исследовании, оценивали продолжительность эффекта антипролиферативного действия на клетках NAMALWA после обработки и удаления соединений A825, JQ1 и OTX-15.
[00557] В частности, клетки NAMALWA обрабатывали в течение 24 часов соединениями A825 (0,1 мкМ), JQ1 (1,0 мкМ) и OTX15 (1,0 мкМ) с последующими тремя промывками для удаления соединений. Клетки повторно высевали в свежей среде и инкубировали без каких-либо соединений в течение 0, 24,0 и 48,0 часов. В параллельном эксперименте клетки обрабатывали таким же способом, за исключением того, что вместо ингибитора использовали DMSO. Относительную пролиферацию образцов после обработки определяли с помощью анализа CTG.
[00558] На Фигуре 6E показано, что эффект подавления пролиферации при использовании соединения A825 был устойчивым в течение более 48 часов после обработки, по сравнению с обработкой соединениями JQ1 или OTX15. Этот результат согласуется с результатами экспериментов, описанными выше, где показано, что A825 обеспечивает длительный эффект в отношении процесса деградации белка BRD4 и подавления сигнального пути (например, см. Фигуры 5а-5f).
[00559] с. Помалидомид уменьшает антипролиферативное действие соединения A825
[00560] Как описано выше и как показано на Фигуре 4C, уровни белка BRD4 были частично спасены от деградации, когда помалидомид присутствовал во время обработки соединением A825 за счет конкурентного ингибирования связывания цереблона. Чтобы определить, может ли помалидомид также предотвратить или по крайней мере уменьшить анти-пролиферативный эффект действия соединения A825 на различных клеточных линиях BL, был проведен следующий эксперимент.
[00561] В частности, клетки NAMALWA, CA-46 и DAUDI обрабатывали только соединением A825 (10 нМ или 100 нМ) или в сочетании с помалидомидом (1,0 мкМ или 10,0 мкМ) в течение 72 часов. В параллельном эксперименте клетки обрабатывали таким же способом, за исключением того, что вместо ингибитора использовали DMSO. Относительную пролиферацию образцов после обработки определяли с помощью анализа CTG.
[00562] Обработка клеток только соединением A825 при концентрации 10 нМ привела к значительному подавлению пролиферации по сравнению с контрольными клетками (см. Фигуру 6F), что согласуется с результатами, представленными на Фигурах 6А-6Е. На Фигуре 6F показано, что обработка клеток только соединением A825 при концентрации 10 нМ привела к уменьшенному росту клеток до уровня примерно 40% в клетках NAMALWA и CA-46, и примерно до уровня 65% в клетках DAUDI, по отношению к уровню роста контрольных клеток. Помалидомид уменьшил антипролиферативный эффект действия соединения A825 в концентрации 10 нМ дозозависимым образом. В частности, обработка 1,0 мкМ помалидомида в комбинации с 10 нМ A825 привела к менее резкому снижению роста клеток по сравнению с контрольным образцом (около 80% в клетках NAMALWA, около 90% в клетках СА-46 и около 95% в клетках DAUDI). Повышение концентрации помалидомид до 10,0 мкМ при обработке 10 нМ A825 предотвращало антипролиферативный эффект во всех испытуемых клеточных линиях по сравнению с контрольным образцом.
[00563] Обработка клеток только соединением A825 в концентрации 100 нМ привела к подавлению пролиферации всех типов испытанных клеток в большей степени, по сравнению с обработкой только одним соединением A825 в концентрации 10 нМ (сравни Фигуру 6G и Фигуру 6F), что согласуется с результатами, представленными на Фигурах 6А-6Е. На Фигуре 6G показано, что обработка только одним соединением A825 в концентрации100 нМ привела к уменьшению роста клеток примерно до уровня 25%-27% в линиях клеток NAMALWA и DAUDI, и примерно до 33% в клетках DAUDI, по отношению к росту контрольных клеток. Помалидомид уменьшает антипролиферативный эффект действия соединения A825 в концентрации 100 нМ дозозависимым образом. В частности, обработка 1,0 мкМ помалидомида в сочетании с 100 нМ A825 привела к менее резкому снижению роста клеток (рост сотавил примерно 55% во всех клеточных линиях) по сравнению с ростом контрольного образца. Повышение концентрации помалидомида до 10,0 мкМ при обработке соединением A825 в концентрации 100 нМ дополнительно снизило антипролиферативный эффект во всех клеточных линиях (от примерно 70% до примерно 80% для всех клеточных линиях по сравнению с контрольным образцом).
[00564] В качестве дополнительного контроля, чтобы определить, оказывает ли помалидомид, без A825, влияние на пролиферацию клеток, клетки BL обрабатывали помалидомидом. В частности, клетки BL обрабатывали различными концентрациями одного помалидомида (0,001 мкМ, 0,003 мкМ, 0,01 мкМ, 0,03 мкМ, 0,1 мкМ, 0,3 мкМ, 1 мкМ, 3 мкМ, 10 мкМ, как показано на Фигуре 6H) в течение 72 часов. В параллельном эксперименте клетки обрабатывали таким же образом, за исключением того, что вместо помалидомида использовали DMSO. После обработки относительную пролиферацию образцов, обработанных помалидомидом, определяли с помощью анализа CTG, и сравнивали с контролем (DMSO).
[00565] На Фигуре 6H показано, что обработка клеток одним помалидомидом не приводит к существенному влиянию на пролиферацию этих клеточных линий.
[00566] 5. A825 в превосходной степени индукцирует апоптоз, по сравнению с другими низкомолекулярными ингибиторами
[00567] с-Мус представляет собой плейотропный онкобелок, участвующий во многих проявлениях рака, включая клеточный цикл, старение, пролиферацию и апоптоз, в зависимости от типа различных опухолевых проявлений (M. Gabay, et al., Cold Spring Harb Perspect Med. (2014) 4:a014241). Предшествующие эксперименты показали универсальный эффект действия в отношении подавления пролиферации во всех испытанных линиях клеток BL после обработки их низкомолекулярными ингибиторами белка BRD4 (JQ1 и OTX-15), а также соединением A825. В следующих экспериментах была оценена степень индуцирования апоптоза соединениями JQ1, OTX-15, и A825 в клеточных линиях BL.
[00568] а. A825 приводит к более значительному увеличению активности каспазы по сравнению с JQ1 и OTX-15
[00569] Различные клеточные линии BL обрабатывали соединениями ARV-825 (0,1 мкМ), JQ1 (1,0 мкМ), OTX-015 (1,0 мкМ) или пуромицином (10 мг/мл, в качестве положительного контроля индукции апоптоза), в течение 24 часов, и измеряли активность каспазы 3/7 с помощью набора для анализа Caspase 3/7-Glow.
[00570] На Фигуре 7А показано, что активность каспазы значительно варьирует в зависимости от клеточной линии BL и ингибитора, используемого для обработки. В частности, обработка клеток BL соединением A825 в концентрации 100 нМ привела к статистически значимому увеличению активности каспазы по сравнению с клетками BL, обработанными JQ1 и OTX-15. Увеличение активности каспазы было еще более значительным в клетках DAUDI и NAMALWA, по сравнению с клетками Ramos и СА-56.
[00571] Наблюдали увеличение активности каспазы 3/7 после 24 часов обработки всех клеточных линий BL соединением A825, но не в случае более высоких доз соединений JQ1 и OTX-15 (фиг.5А).
[00572] b. A825 приводит к более значительному увеличению PARP расщепления по сравнению с JQ1 и OTX-15
[00573] Клетки Ramos и СА-46 обрабатывали увеличивающимися дозами соединений АРВ-825 (до 1,0 мкМ) или JQ1 и OTX-015 (до 10,0 мкМ) в течение 48 часов. Лизаты собирали и анализировали при помощи иммуноблота для PARP расщепления, с актином в качестве контроля нагрузки.
[00574] На Фигуре 7В показано, что через 48 часов после обработки 0,1 мкМ A825 клетки Ramos показали значительный апоптоз, о чем свидетельствует заметное PARP расщепление. В противоположность этому, оказалось, что необходимы значительно более высокие дозы ингибиторов JQ1 и OTX-15 чтобы вызвать подобный уровень апоптоза в соответствующих клеточных линиях. Необходимость использования более высоких концентраций JQ1 и OTX-15 вероятно вызвано тем, что эти ингибиторы в недостаточной степени ингибируют BRD4 и подавляют с-Myc по ходу сигнального потока.
[00575] Эти результаты, рассмотренные вместе, являются убедительным доказательством, что деградации белка BRD4, опосредованная Protac, является более эффективной стратегией, нацеленной на BRD4 при BL, по сравнению с традиционными низкомолекулярными ингибиторами.
[00576] 6. Обобщение результатов и их обсуждение
[00577] Как известно, клетки BL чувствительны к действию ингибиторов BRD4, которые подавляют путь передачи сигнала через с-Myc, и вызывают ингибирование пролиферации клеток (J.A. Mertz, et al., PNAS, 108 (2011) 16669-16674). В последнее время наблюдается значительный прогресс в разработке соединений, которые эффективно ингибируют в клетках BRD4. Тем не менее, несмотря на это недавние успехи, ингибиторы BRD4, имеющие значительные функциональные и клинические преимущества, до сих пор не обнаружены, что частично может быть объяснено выраженным накоплением BRD4, наблюдаемым во время лечения ингибитором, и обратимого/временного характера ингибирования, наблюдаемого после лечения, когда ингибитор элиминирован.
[00578] Проведенные эксперименты, описанные в этом примере, показывают, что небольшие низкомолекулярные ингибиторы BRD4, такие как JQ1 и OTX-15, приводят к значительному накоплению белка BRD4 во всех тестированных линиях клеток BL. Хотя оба ингибиторы подавляют уровни с-Myc по ходу сигнального пути, такое подавление требует высокой концентрации этих соединений. Более того, даже при высоких концентрациях этих ингибиторов, подавление с-Мус не было полным. Результаты, полученные в этом примере для JQ1 и OTX-15, согласуются с результатами, полученными с другими клетками клеточных линий рака легких и предстательной железы (Shimamura, T., Chen, Z., Soucheray, M., Carretero, J., Kikuchi, E., Tchaicha, J.H., Gao, Y., Cheng, K.A., Cohoon, T.J., Qi, J., et al. (2013); и не показанные здесь данные). Полученные выше результаты свидетельствуют о том, что устойчивое накопление белка BRD4, вместе с обратимой природой ингибирования связывания BRD4, может быть причиной незначительного эффекта подавления с-Мус по сигнальному пути и связанной с ограниченным ингибированием пролиферации низкомолекулярными ингибиторами. Одним из возможных объяснений данных, полученных для JQ1 и OTX-15, является то, что связывание ингибитора с BRD4 приводит к конформационным изменениям, которые, в свою очередь, приводят к увеличению стабильности или препятствуют доступности BRD4 для деградации. Альтернативно, ингибиторы могут подавляться за счет отрицательной обратной связи цикла BRD4, который регулирует уровни белка BRD4. Тем не менее, заметное увеличение уровня BRD4, вместе с обратимой природой связывания ингибитором, частично может объяснить низкую эффективность ингибирования BRD4 и подавления MYC по ходу сигнального пути.
[00579] Как показали доклинические и клинические исследования, эффекты действия ингибиторов BRD4 были в значительной степени цитостатическими, с ограниченным апоптозом для нескольких клеточных линий и ограниченным числом пациентов, привлеченных к исследованиям I фазы. Это существенно ограничивает потенциальную пользу для будущих пациентов при использовании ингибиторов BRD4 в клинически приемлемых концентрациях.
[00580] Еще одно явление, которое встречается при разработке лекарственных средств на основе низкомолекулярных ингибиторов, является возникновением мутаций в целевых белках-мишенях, которые могут опосредовать их устойчивость, или даже привести к изменению действия активного вещества из антагонистического в агонистическое. Например, считается, что ензалутамид эффективен при лечении рака предстательной железы за счет ингибирования рецептора андрогена, он становится агонистом в опухолевых клетках с рецептором андрогена, содержащего мутацию F876L. Таким образом, у больных раком предстательной железы, у которых опухоли содержат уже существующую мутацию ARF876L или мутацию, вызванную лечением, не получит эффективного лечения при использовании ензалутамида. В противоположность этому, опосредованная за счет Protac деградация конкретной мишени позволит избежать этих ошибок и обеспечить эффективную стратегию целенаправленного воздействия.
[00581] Для того, чтобы обойти ограничения, характерные для низкомолекулярных ингибиторов BRD4, было разработано химерное соединение A825 путем соединения небольшой молекулы фрагмента, связывающего BRD4, с фрагментом связывания лигазы Е3 цереблона, с помощью технологии Protac.
[00582] Эксперименты, описанные выше, показывают, что соединение A825 индуцированных быструю и эффективную деградацию BRD4 путем активного рекрутинга белка BRD4 к лигазе E3 цереблона, за счет чего белок BRD4 направляется к протеасоме для деградации. Эти результаты также показывают, что соединение A825 обеспечивает более эффективное подавление экспрессии и функции с-Мус по ходу сигнального пути, а также пролиферации клеток и индукции апоптоза по сравнению с известными низкомолекулярными ингибиторами BRD4.
[00583] Улучшенные функциональные эффекты действия ингибиторов в отношении деградации белка BRD4 могут быть частично связаны с более полным и устойчивым подавлением с-Мус, который является ведущим онкобелком при BL. Также возможно, что BRD4 обладает функциями «шаперона», поскольку он является большим белком, который может связываться со многими партнерами связывания, которые в дальнейшем будут более точно определены и идентифицированы. Вполне понятно, что элиминирование BRD4 вызывает более широкое действие, чем простое ингибирование связывания с партнерами, содержащими ацетил-лизин. Сравнение фенотипов с нокаутом BRD4 или содержащими BRD4 (например, методом CRISPR и с использованием шпилечных РНК) при ингибировании BRD4 возможно даст ответ на этот вопрос, однако такие исследования выходят за рамки настоящего документа.
[00584] Аффинность связывания OTX-15 и помалидомида с их соответствующими мишенями, BRD4 и цереблоном, составляет, соответственно, ~ 10 нМ и ~ 3 мкМ. Величина DC50 для соединения A825, которое основано на этих двух лигандах, в отношении BRD4 имеет значение ниже 1 нМ. Это наводит на мысль, что Protac обладает каталитической функцией в отношении BRD4, и это открывает огромные возможности в разработке функциональных деградантов, состоящих из целевых лигандов с суб-оптимальной аффинностью к соединениям с неизвестными функциями. Поэтому многие «трудные» мишени, которые обычно не имеют природных сайтов связывания лиганда, могут стать доступными для воздействия лекарственных средств при использовании технологии Protac для опосредования деградации этих мишеней.
[00585] Таким образом, настоящее изобретение обеспечивает новую стратегию для эффективного нацеливания на BRD4 за счет создания мощного деграданта для BRD4 по технологии Protac. Кроме того, изобретение создает почву для разработки нового класса молекул лекарственных веществ, которые активно рекрутируют лигазы E3 к целевым белкам, специфичным для конкретных патологий, для деградации этих белков, и таким образом, многие «трудные» мишени, которые были недоступны для действия традиционной низкомолекулярных соединений, становятся доступными для воздейстия лекарственных средств.
[00586] 7. Промышленная применимость
[00587] Здесь описана новая бифункциональная молекула, которая содержит фрагмент рекрутинга белка BRD4 и фрагмент рекрутинга лигазы Е3 цереблона, полученная с помощью технологии Protac. Соединение A825 активно деградирует белок BRD4, что приводит к значительному и стойкому подавлению по ходу сигнального пути через MYC, устойчивому подавлению клеточной пролиферации и индукции апоптоза при BL. Соединение A825 представляет новую стратегию для эффективного целевого воздействия на белок BRD4, который рассматривается как многообещающая мишень в нескольких видах рака. Соединение A825 является одним из примеров, где соединения Protac опосредуют деградацию белка, обеспечивая многообещающую стратегию в целенаправленном воздействии на белки-мишени, которые ранее рассматривались как ассоциированные с патологическим процессом белки, недоступные для действия традиционных лекарственных веществ.
[00588] Содержание всех ссылок, патентов, заявок на патенты и опубликованных патентов, процитированных в настоящей заявке, включено посредством ссылки.
[00589] Специалист в данной области техники поймет или сможет установить с помощью обычных экспериментов множество эквивалентов конкретных вариантов выполнения описанного здесь изобретения. Такие эквиваленты рассматриваются как охватываемые представленной ниже формулой изобретения. Понятно, что подробные примеры и варианты, описанные здесь, представлены только в качестве примера для иллюстративных целей, и они не должны рассматриваться как ограничивающие настоящее изобретение. Различные модификации или изменения в их свете могут предлагаться специалистами в данной области техники, и они включены в суть и содержание этой заявки и расцениваются как входящие в объем притязаний формулы изобретения. Например, относительные количества ингредиентов могут меняться для оптимизации желаемых эффектов, могут быть добавлены дополнительные ингредиенты и/или один или несколько описанных ингредиентов могут быть заменены на схожие ингредиенты. Дополнительные преимущественные характеристики и функциональности, связанные с системами, способами и процессами данного изобретения, будут очевидны из формулы изобретения. Более того, специалисты в данной области техники увидят или смогут установить с помощью обычных экспериментов множество эквивалентов конкретных вариантов выполнения описанного изобретения. Такие эквиваленты также охватываются следующей формулой изобретения.
Таблица 1
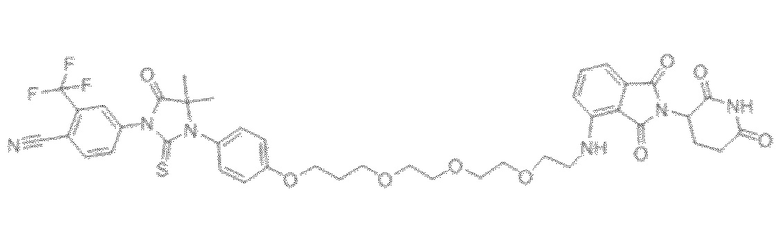
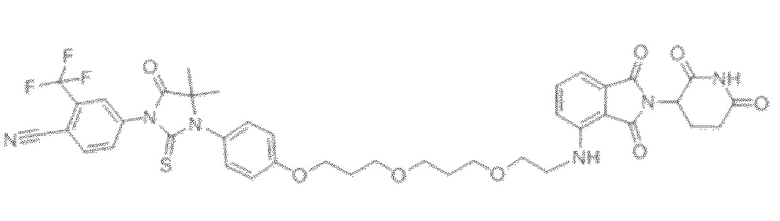
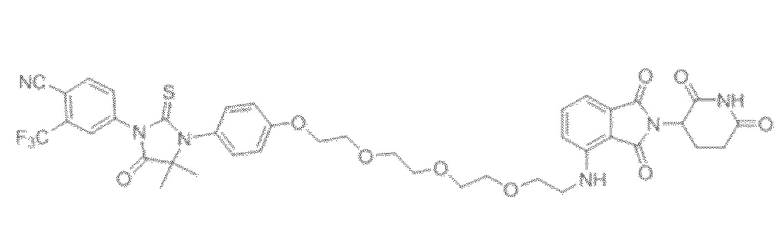
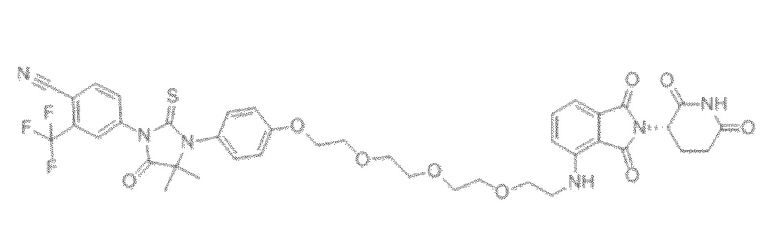
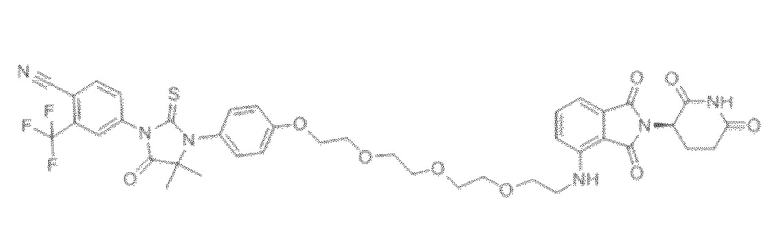
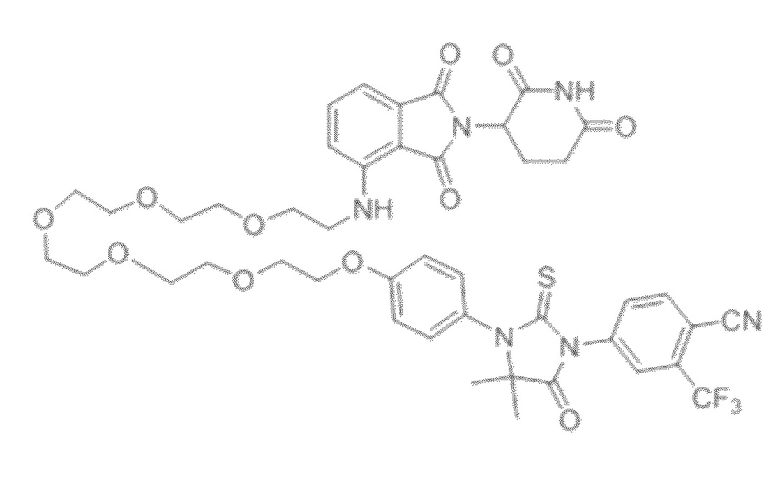
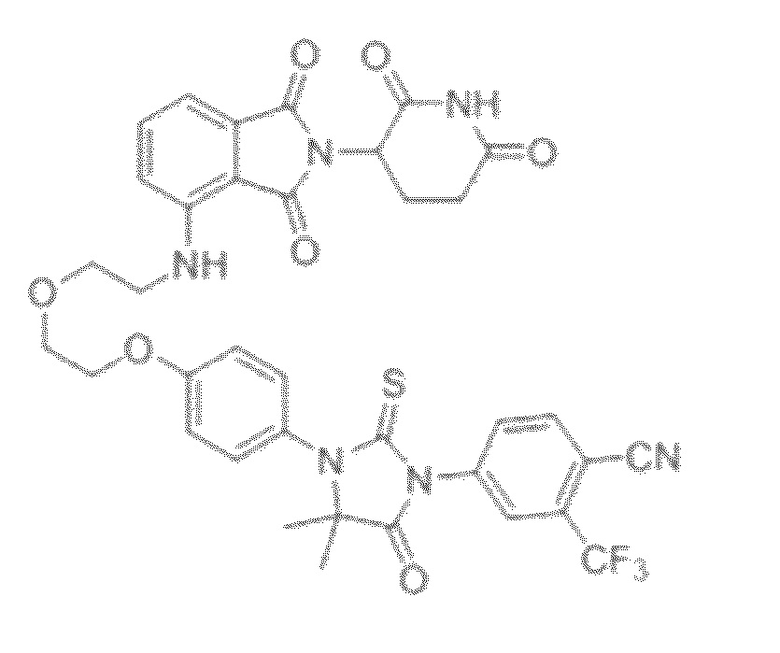
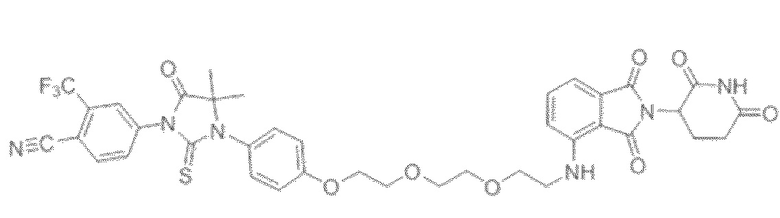
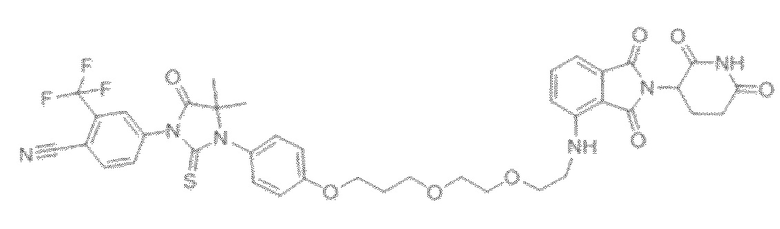
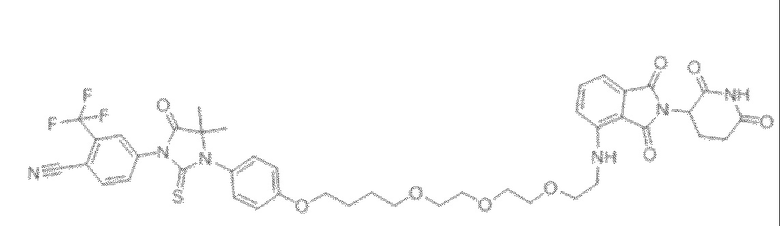
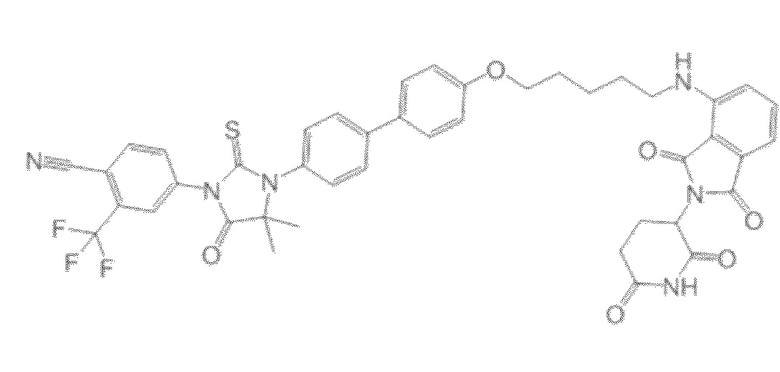
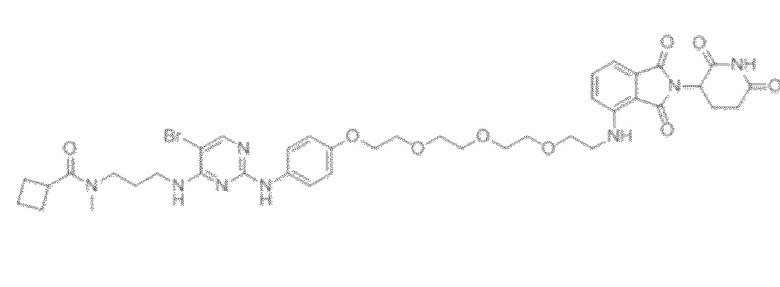
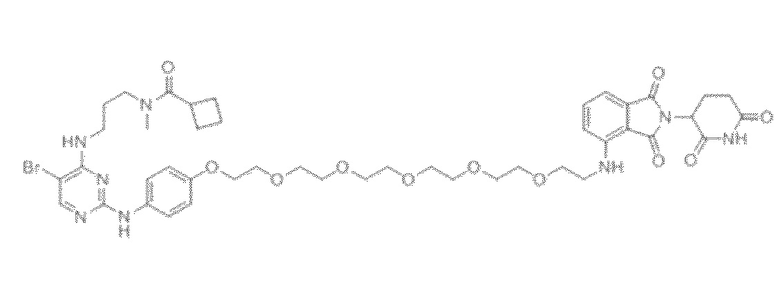
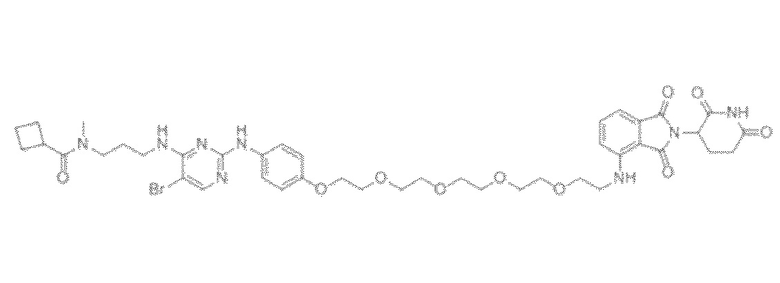
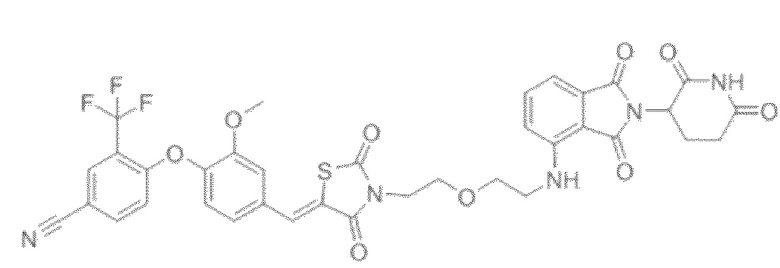
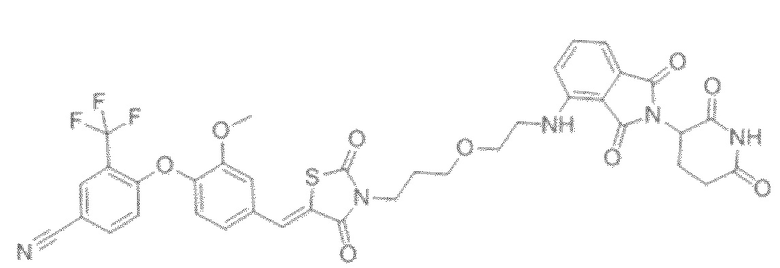
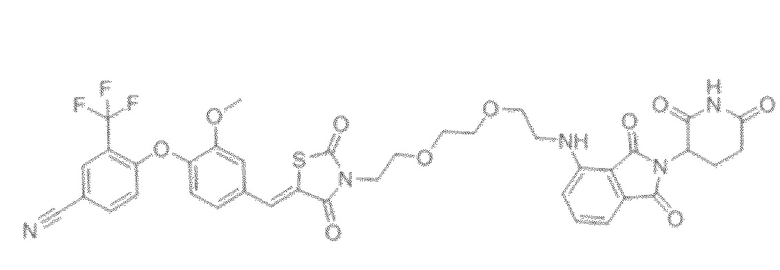
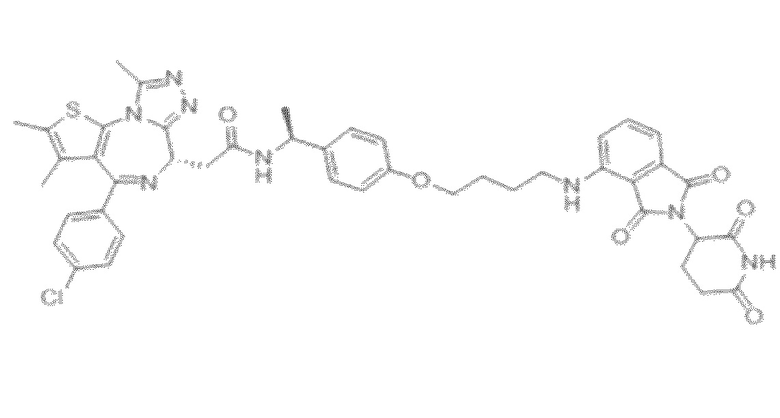
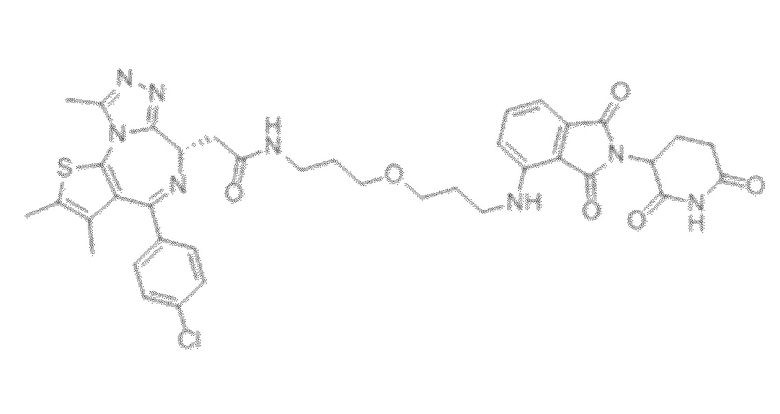
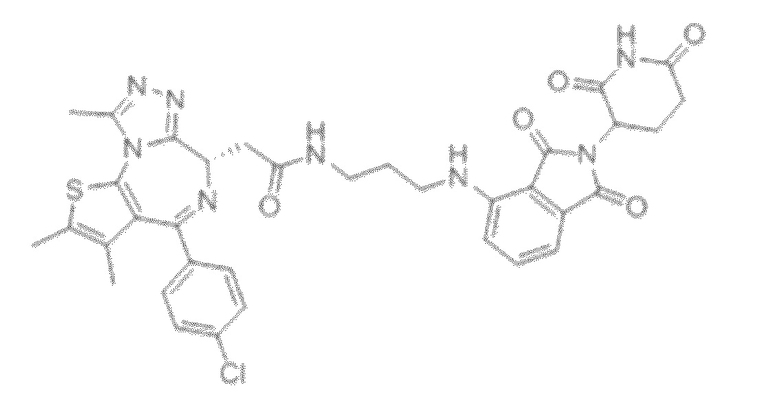
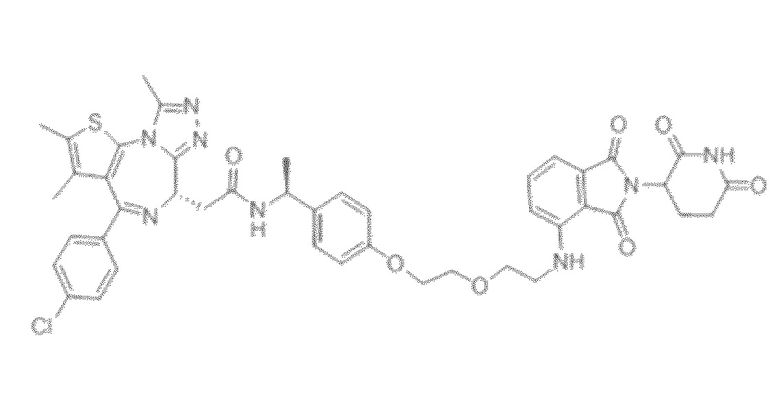
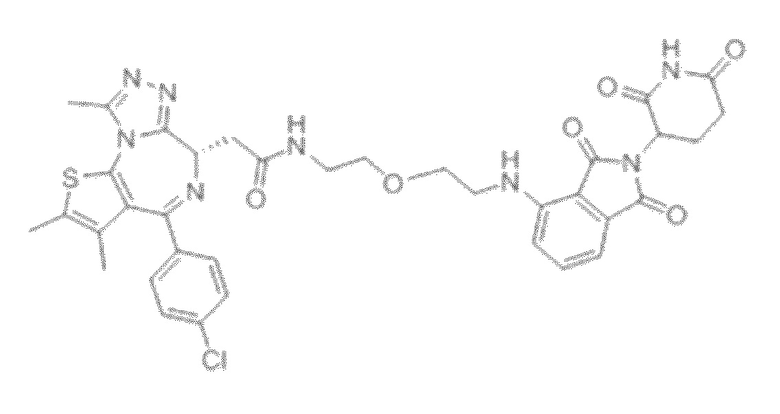
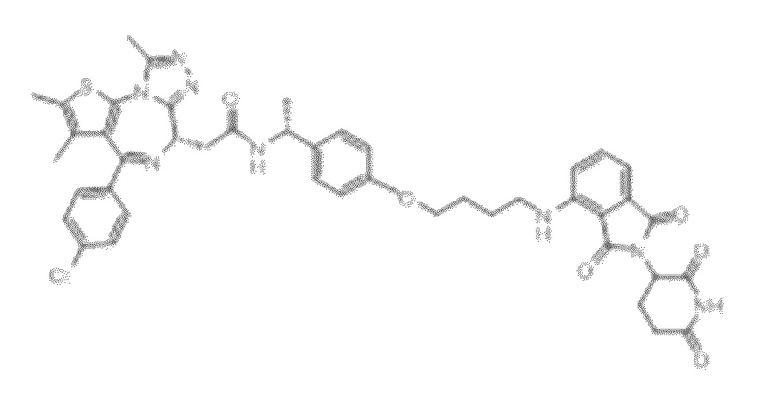
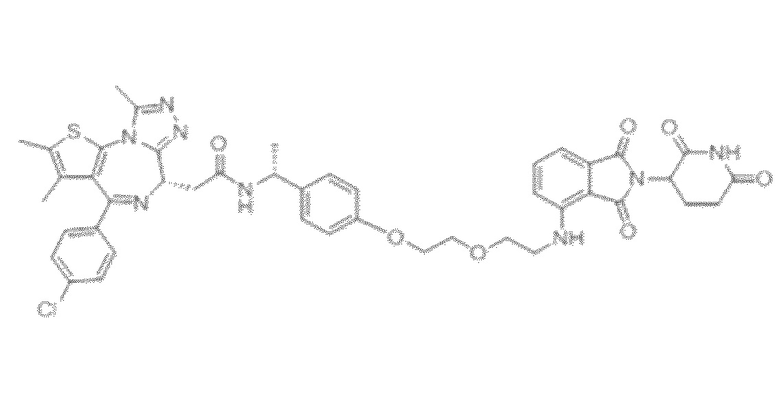
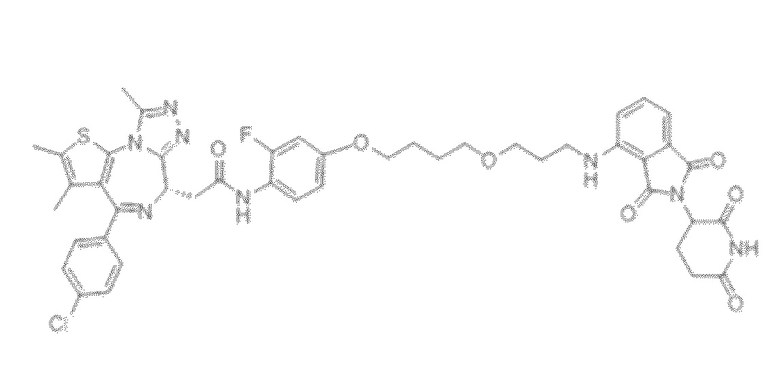
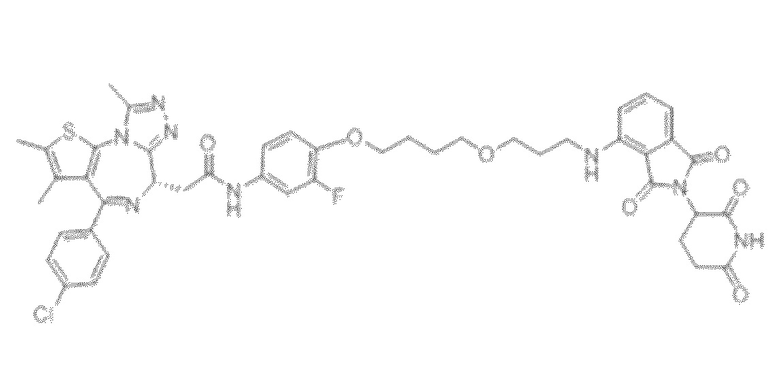
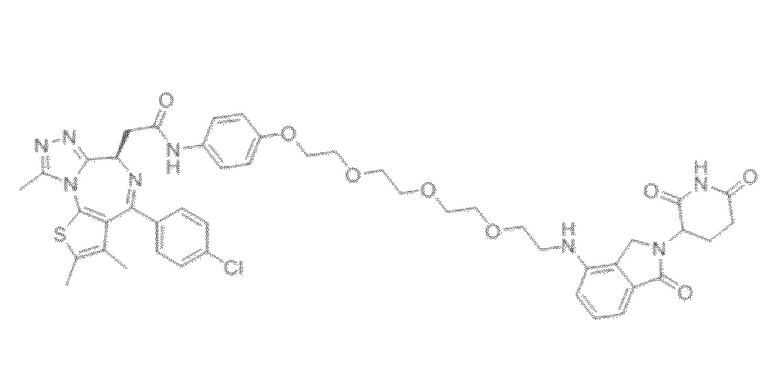
Категории деградирующей активности:
А=деградация 10-30% при 1 мкМ
В=деградация 31-50% при 1 мкМ
С=деградация >50% при 1 мкМ
D=IC50 <50 нМ
E=IC50 >50 нМ
Клетки, использованные для биоанализа
(1) клетки VCaP
(2) клетки Panc02.13
(3) клетки Namalwa
(4) клетки 22RV-1Таблица 1 (продолжение)
Категории активности:
А=деградация 10-30% при 1 мкМ
В=деградация 31-50% при 1 мкМ
С=деградация >50% при 1 мкМ
D=IC50 <50 нМ
E=IC50 >50 нМ
F=не испытывали
IC50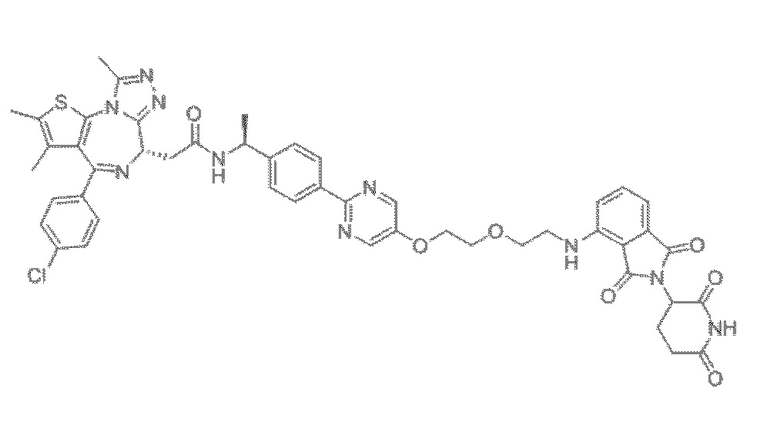
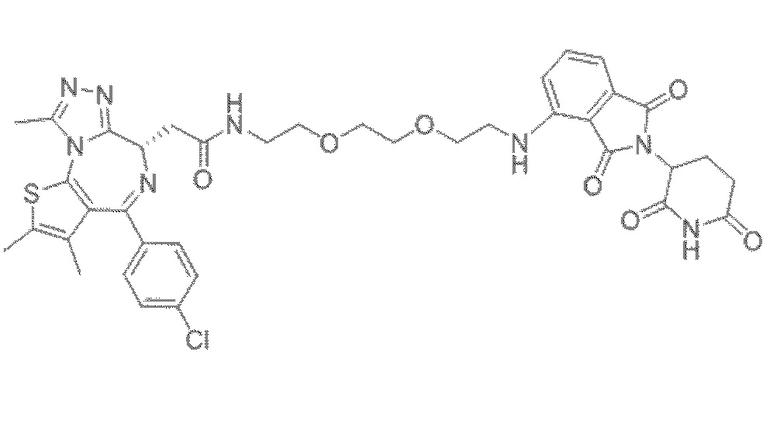
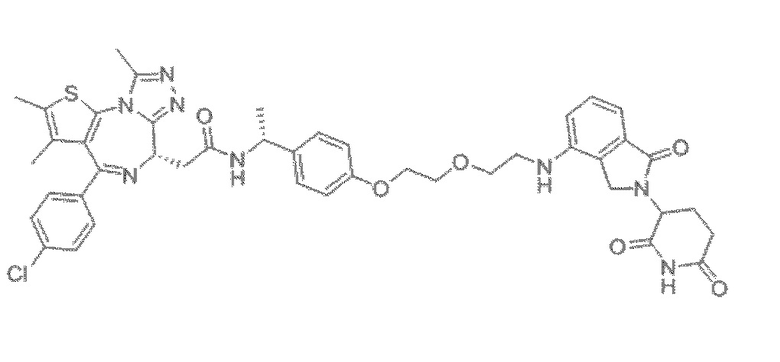
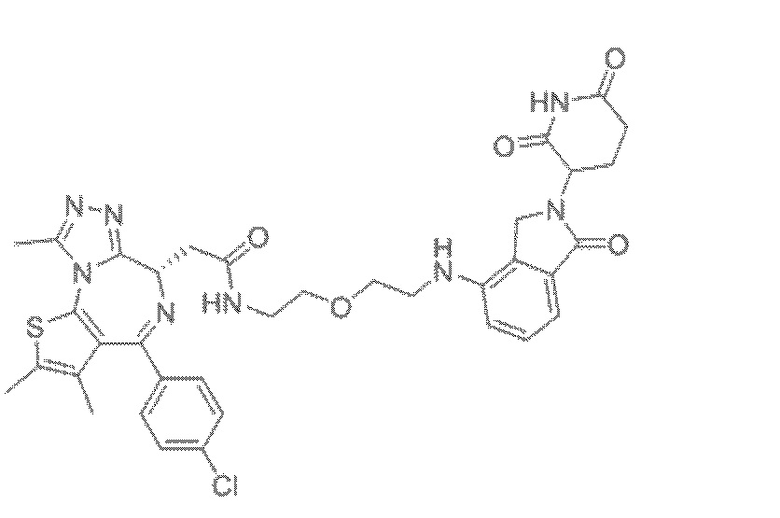
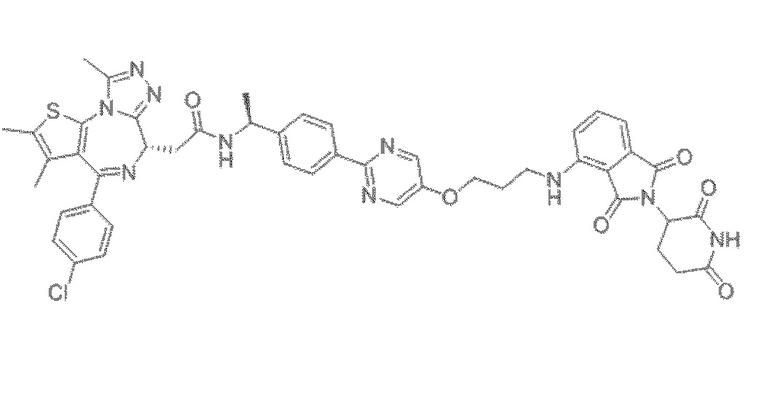
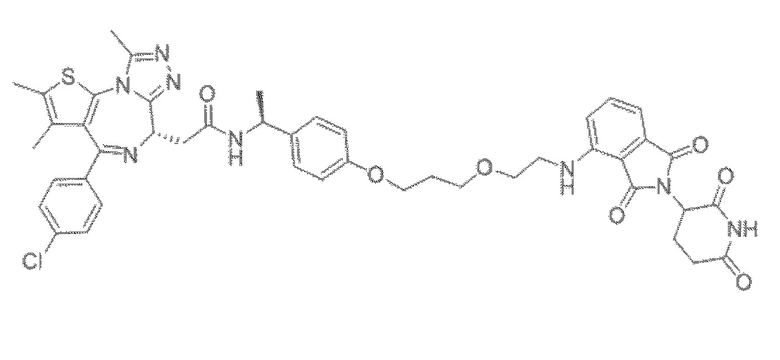
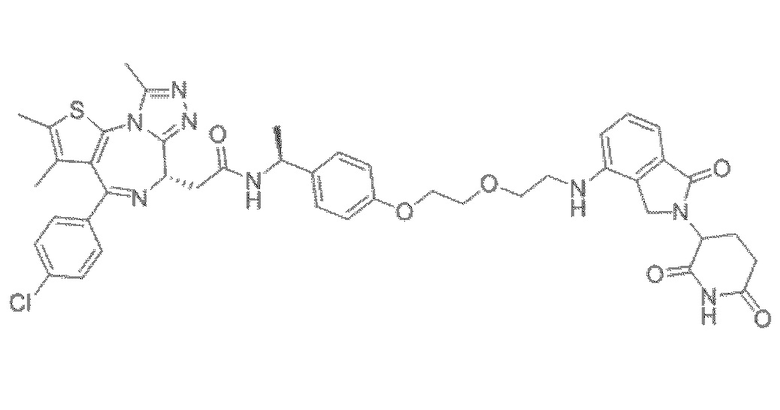
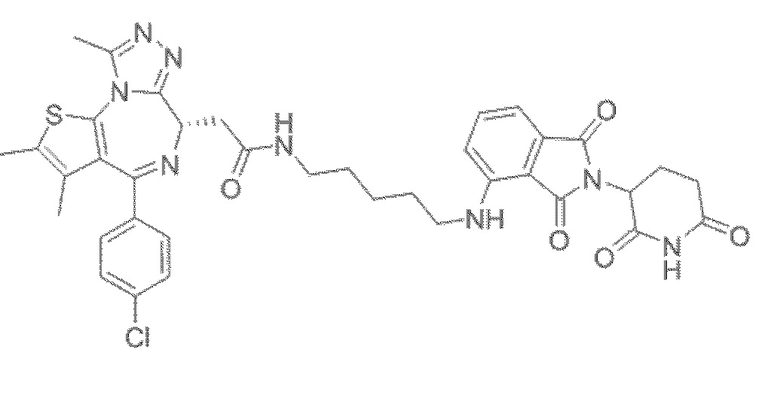
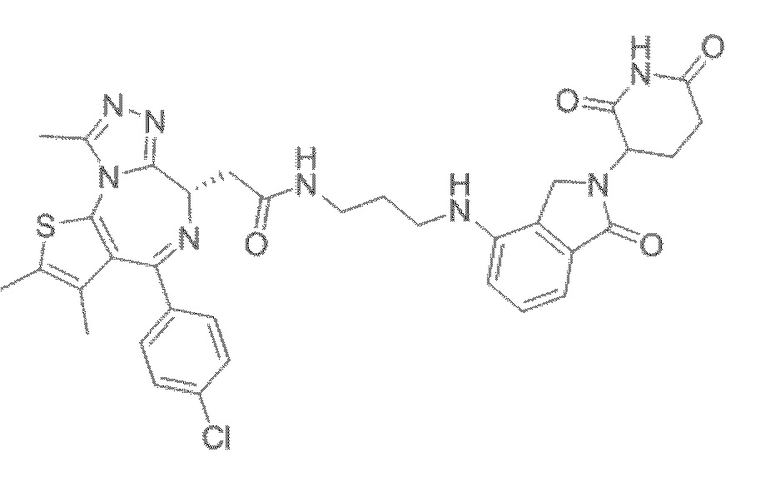
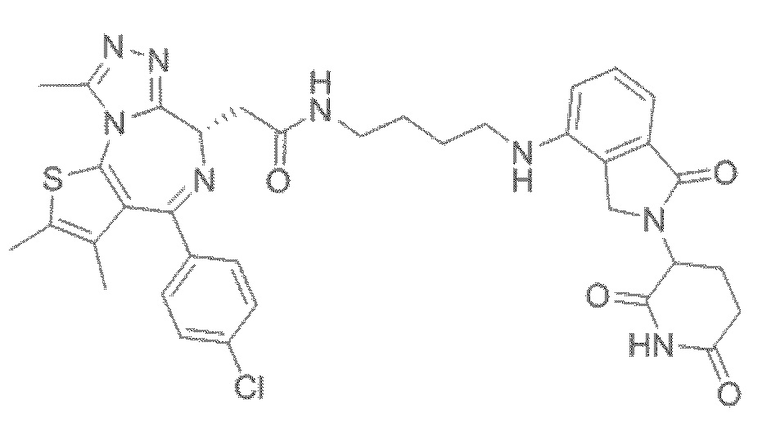
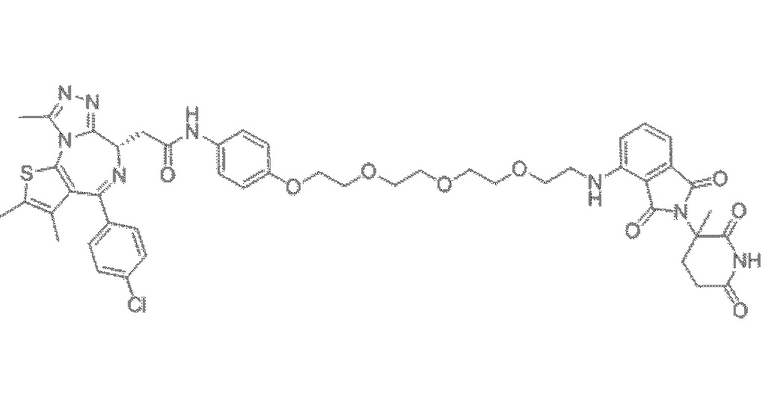
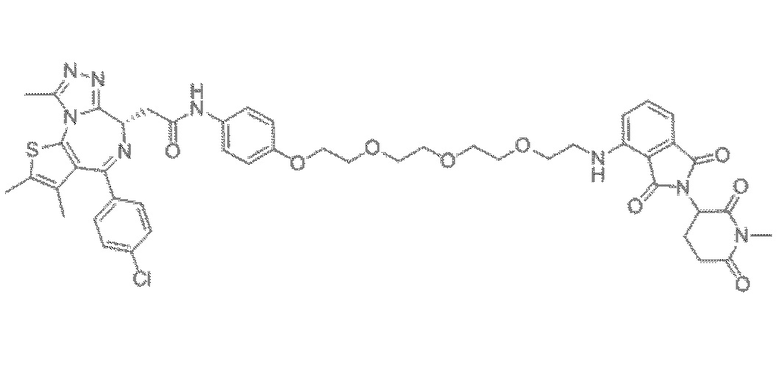
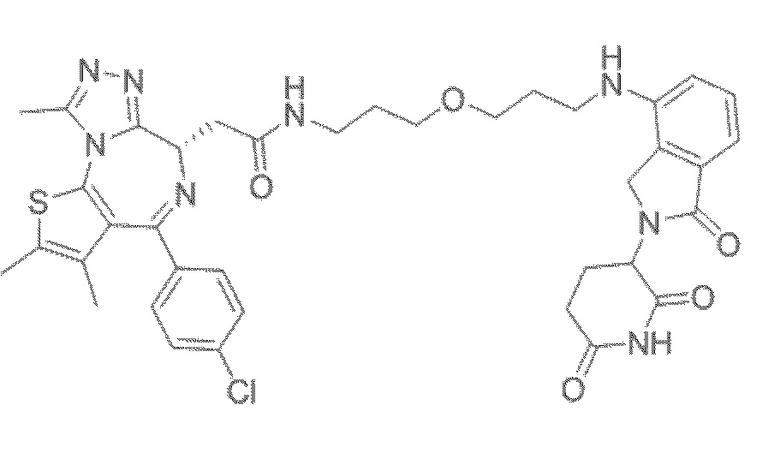
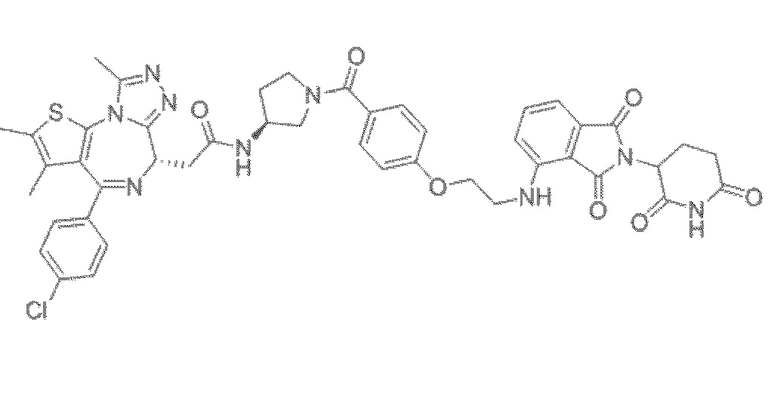
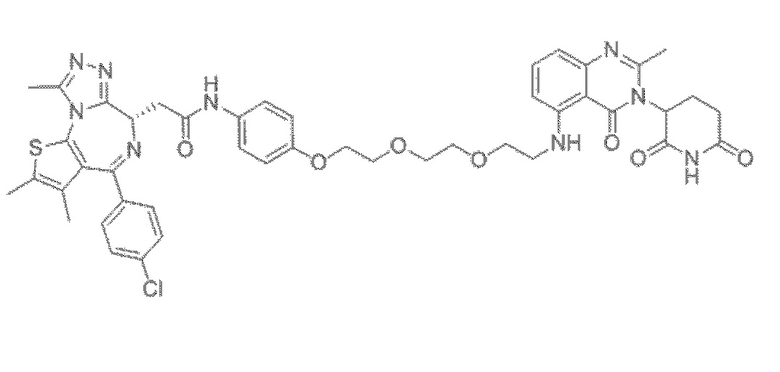
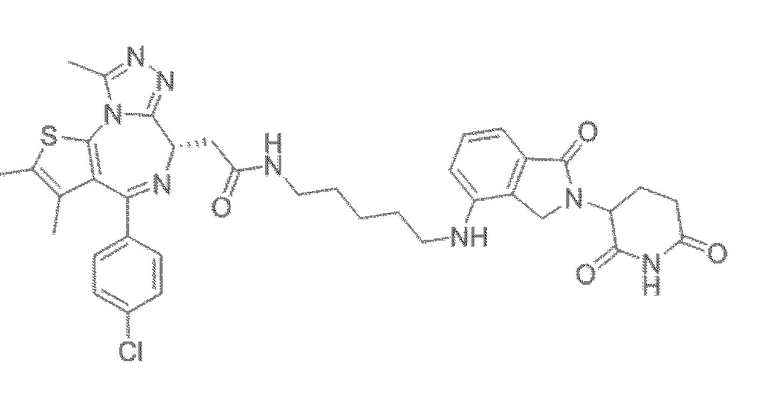
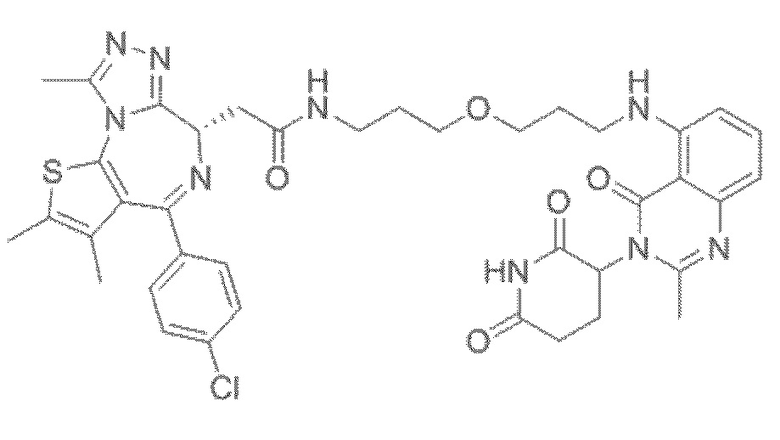
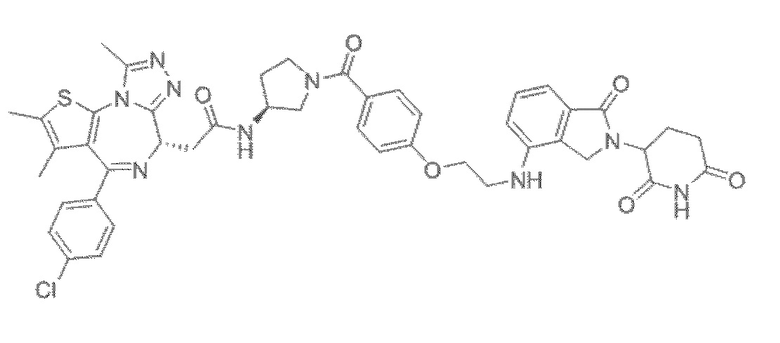
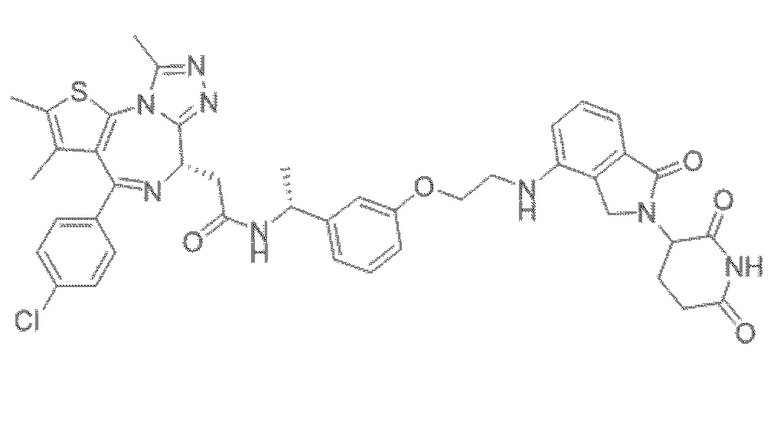
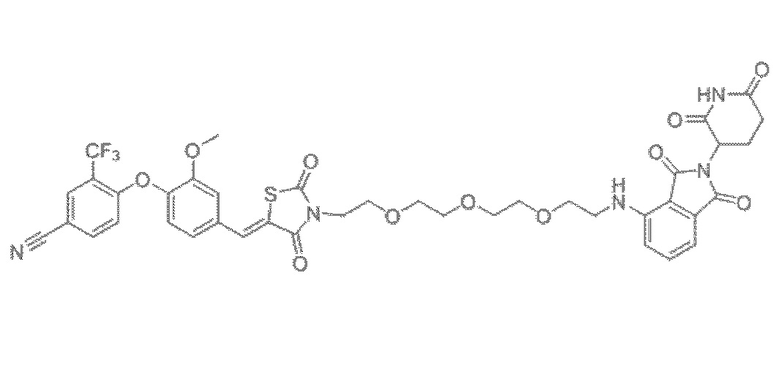
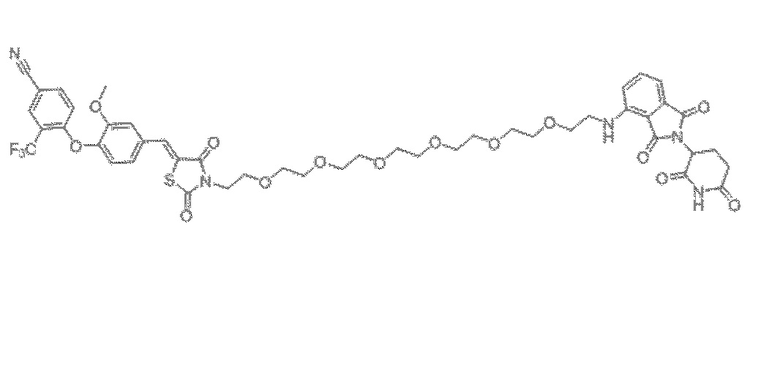
| название | год | авторы | номер документа |
|---|---|---|---|
| ИМИДНЫЕ МОДУЛЯТОРЫ ПРОТЕОЛИЗА И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2015 |
|
RU2738833C2 |
| ЛИГАНДЫ ЦЕРЕБЛОНА И БИФУНКЦИОНАЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ | 2018 |
|
RU2795146C2 |
| ИЗОИНДОЛИНОВЫЕ СОЕДИНЕНИЯ ДЛЯ ПРИМЕНЕНИЯ ПРИ ЛЕЧЕНИИ РАКА | 2009 |
|
RU2527952C2 |
| ПРОИЗВОДНЫЕ ТИЕНОПИРРОЛА ДЛЯ ПРИМЕНЕНИЯ ДЛЯ НАЦЕЛИВАНИЯ НА БЕЛКИ, КОМПОЗИЦИИ С УКАЗАННЫМИ ПРОИЗВОДНЫМИ, СПОСОБЫ И ПРИМЕНЕНИЯ | 2017 |
|
RU2771166C2 |
| СОЕДИНЕНИЕ НА ОСНОВЕ ПИРРОЛАМИДОПИРИДОНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ПРИМЕНЕНИЕ | 2020 |
|
RU2809596C2 |
| ПОЛИЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ И СПОСОБЫ ЦЕЛЕНАПРАВЛЕННОЙ ДЕГРАДАЦИИ ПОЛИПЕПТИДОВ БЫСТРО УСКОРЕННОЙ ФИБРОСАРКОМЫ | 2019 |
|
RU2830173C2 |
| PROTAC, ЦЕЛЕНАПРАВЛЕННО ВОЗДЕЙСТВУЮЩИЕ НА ТАУ-БЕЛОК, И СВЯЗАННЫЕ С НИМИ СПОСОБЫ ПРИМЕНЕНИЯ | 2017 |
|
RU2805523C2 |
| ДЕСТРУКТОРЫ БЕЛКА MDM2 | 2017 |
|
RU2743432C2 |
| СОЕДИНЕНИЯ И СПОСОБЫ УСИЛЕНИЯ ДЕГРАДАЦИИ БЕЛКОВ-МИШЕНЕЙ И ДРУГИХ ПОЛИПЕПТИДОВ С ПОМОЩЬЮ Е3 УБИКВИТИН ЛИГАЗЫ | 2013 |
|
RU2666530C2 |
| БИВАЛЕНТНЫЕ ИНГИБИТОРЫ БРОМОДОМЕНОВ И ПУТИ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2742035C2 |



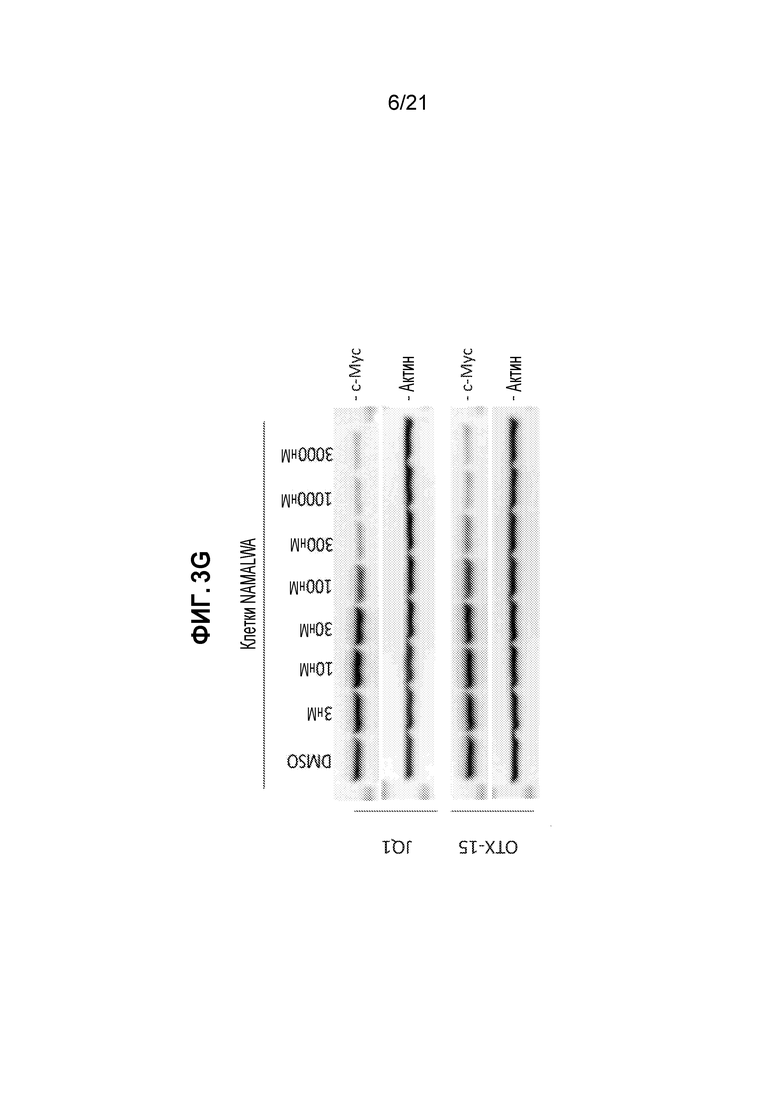
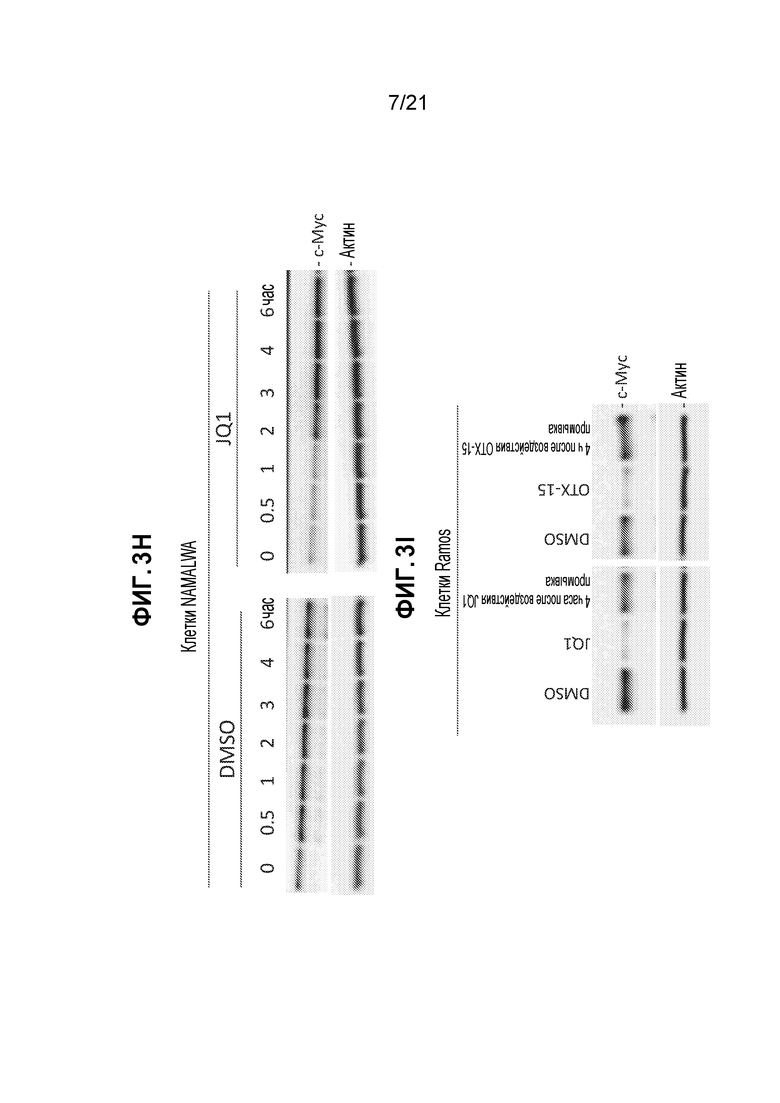

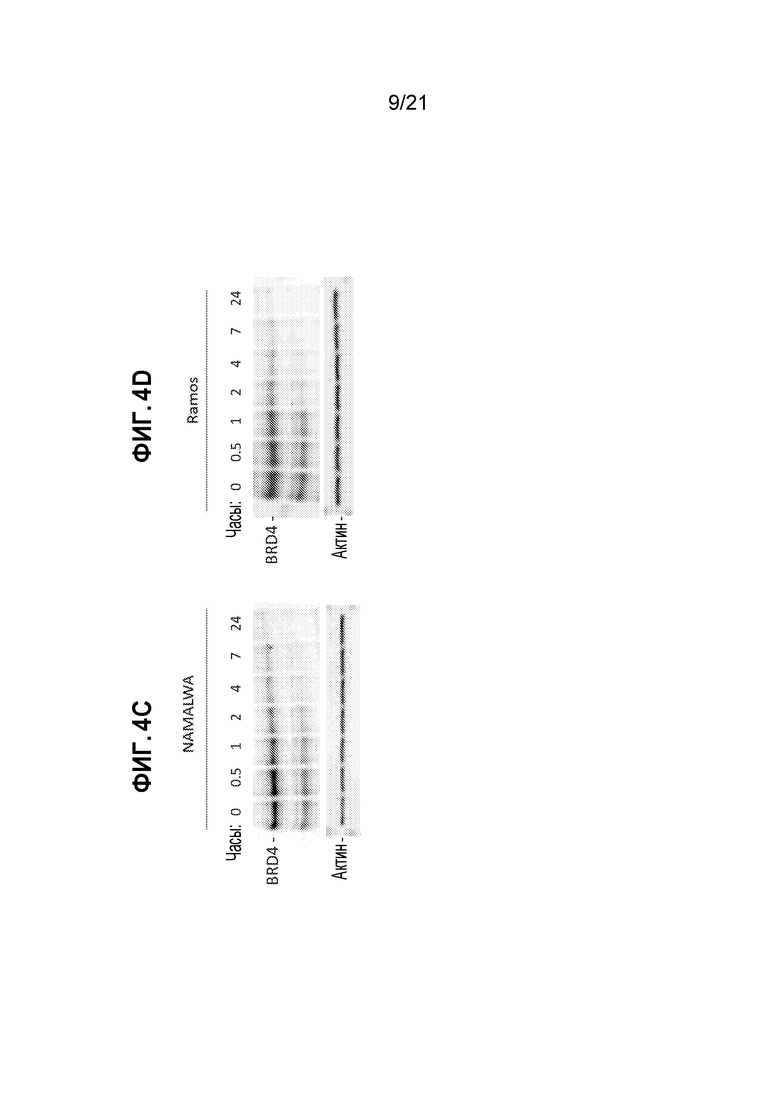
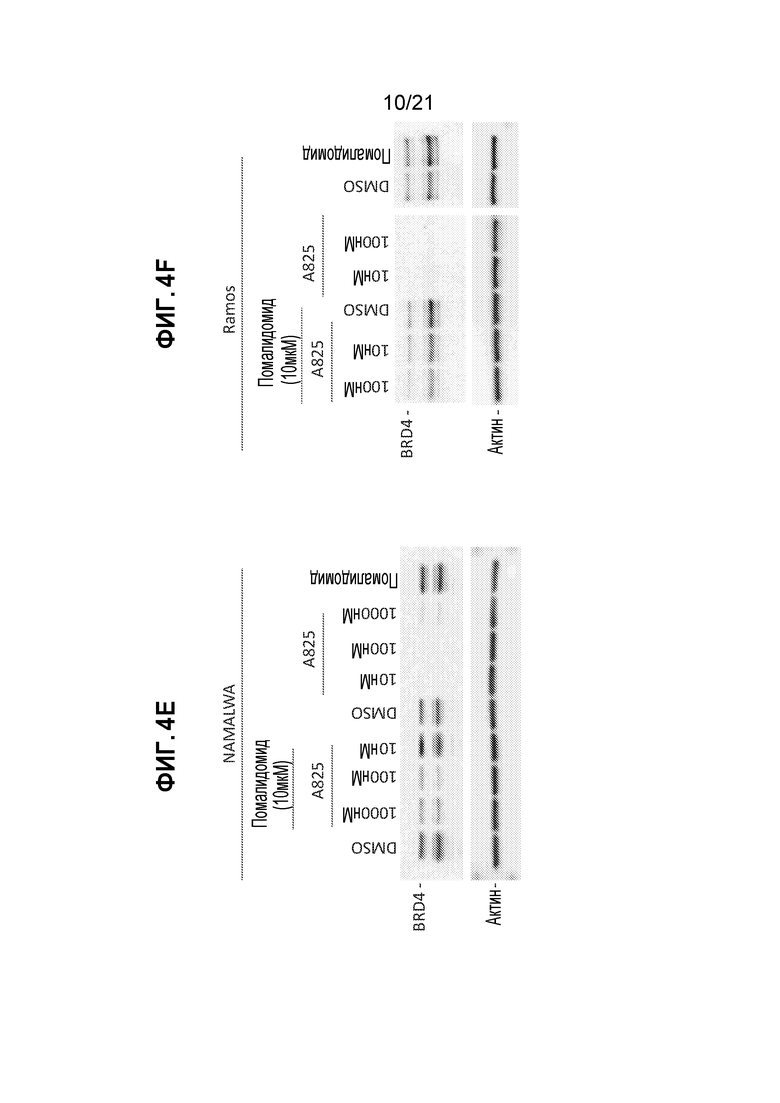


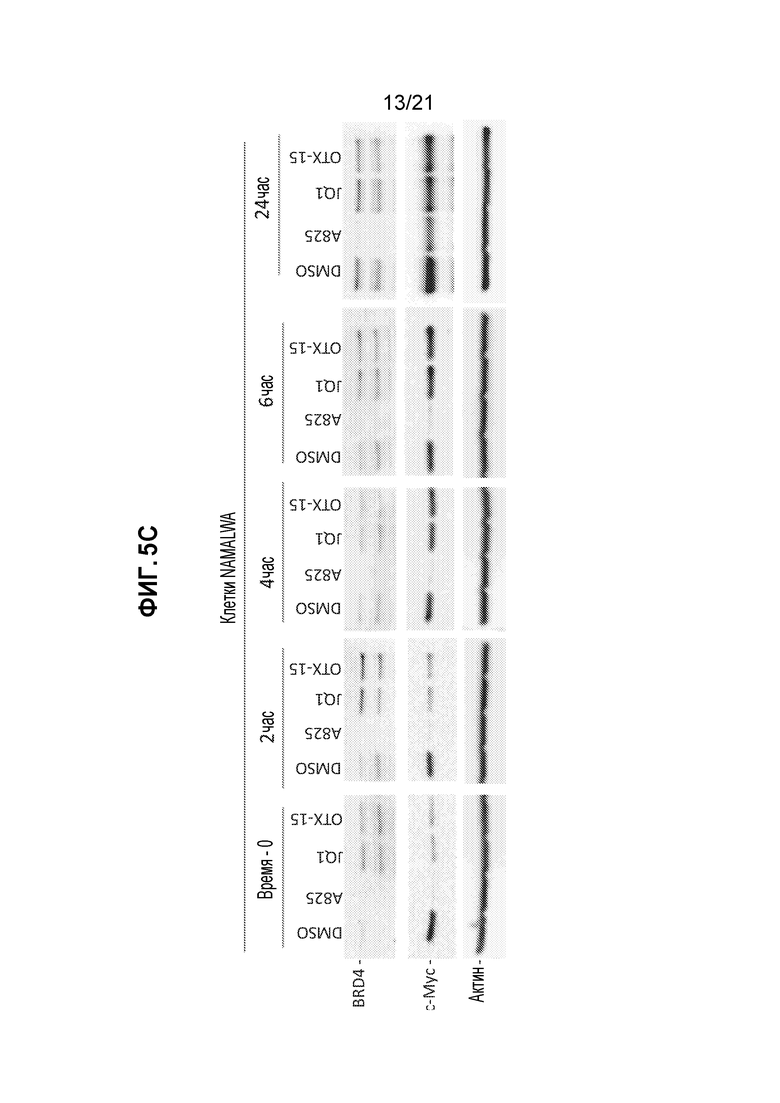
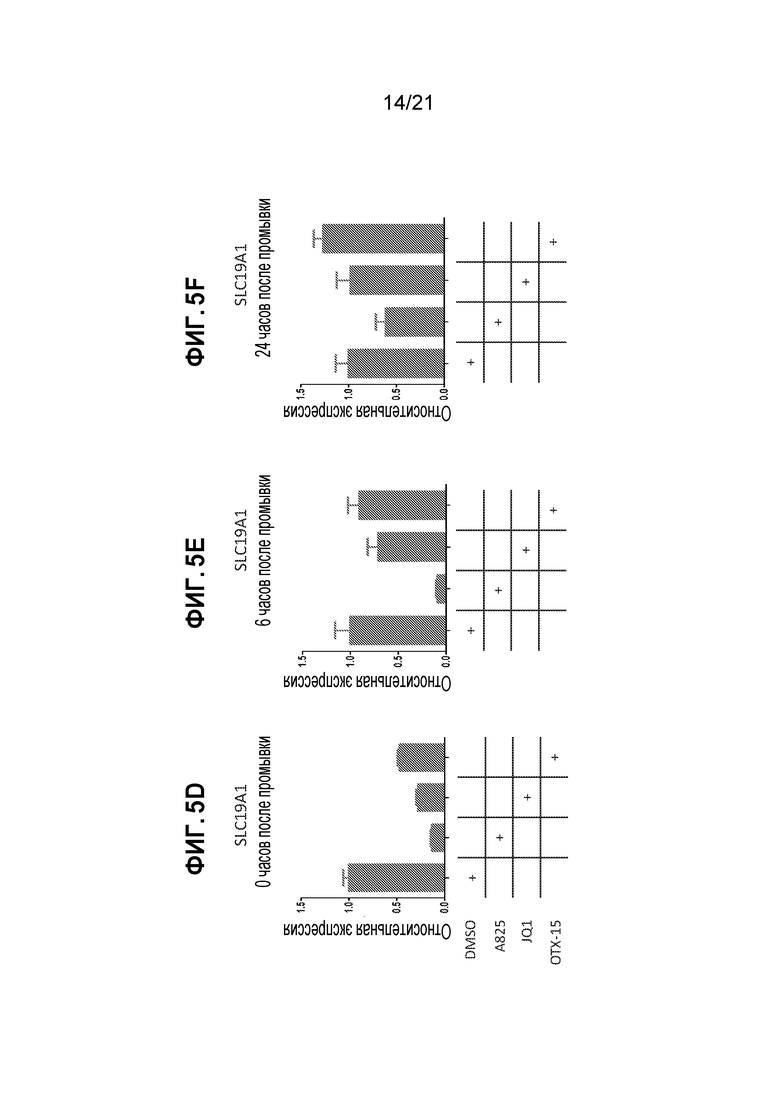
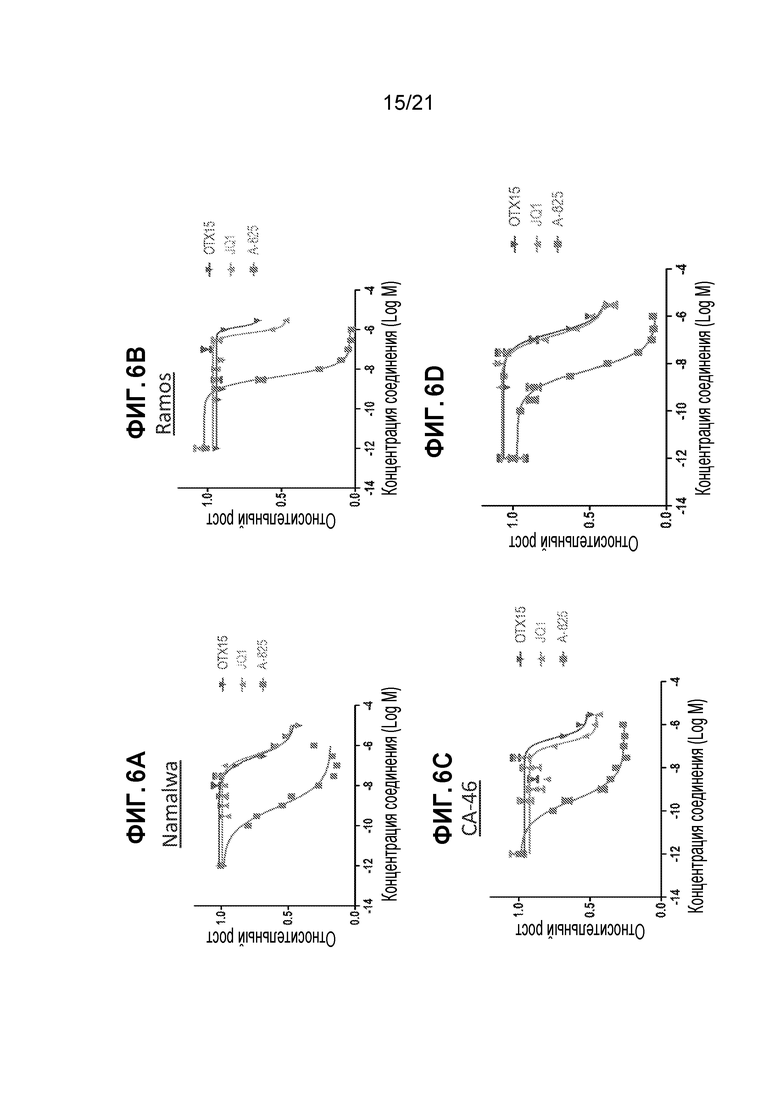
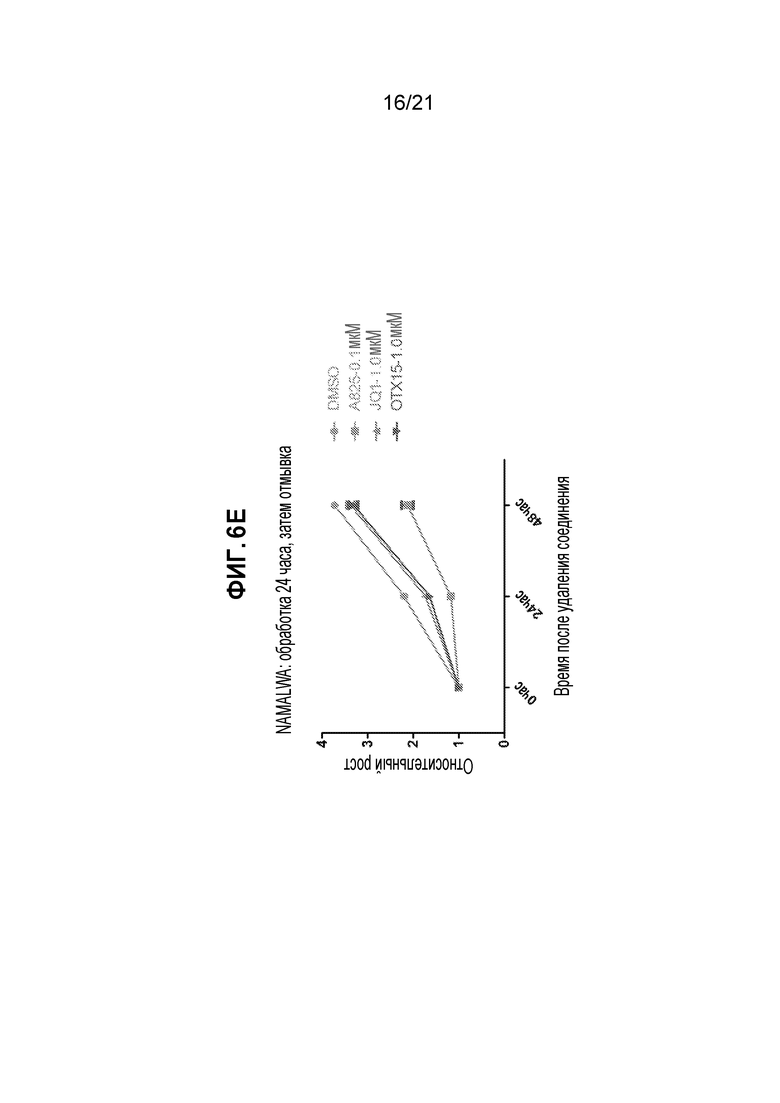
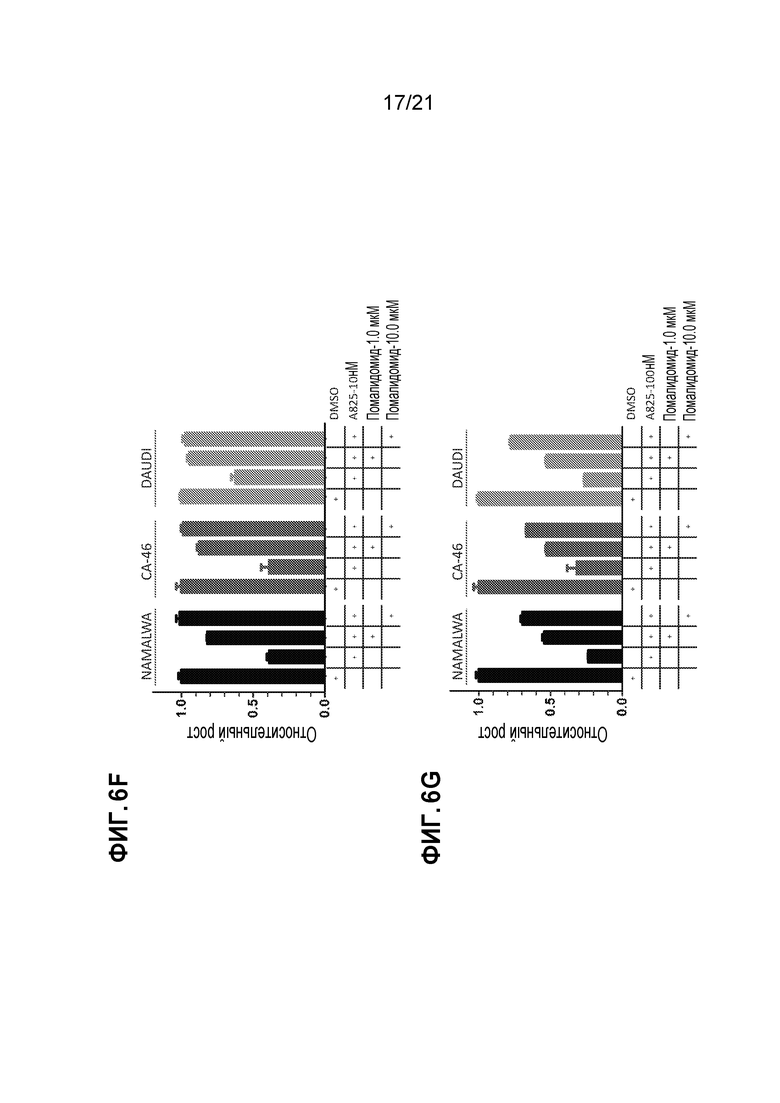

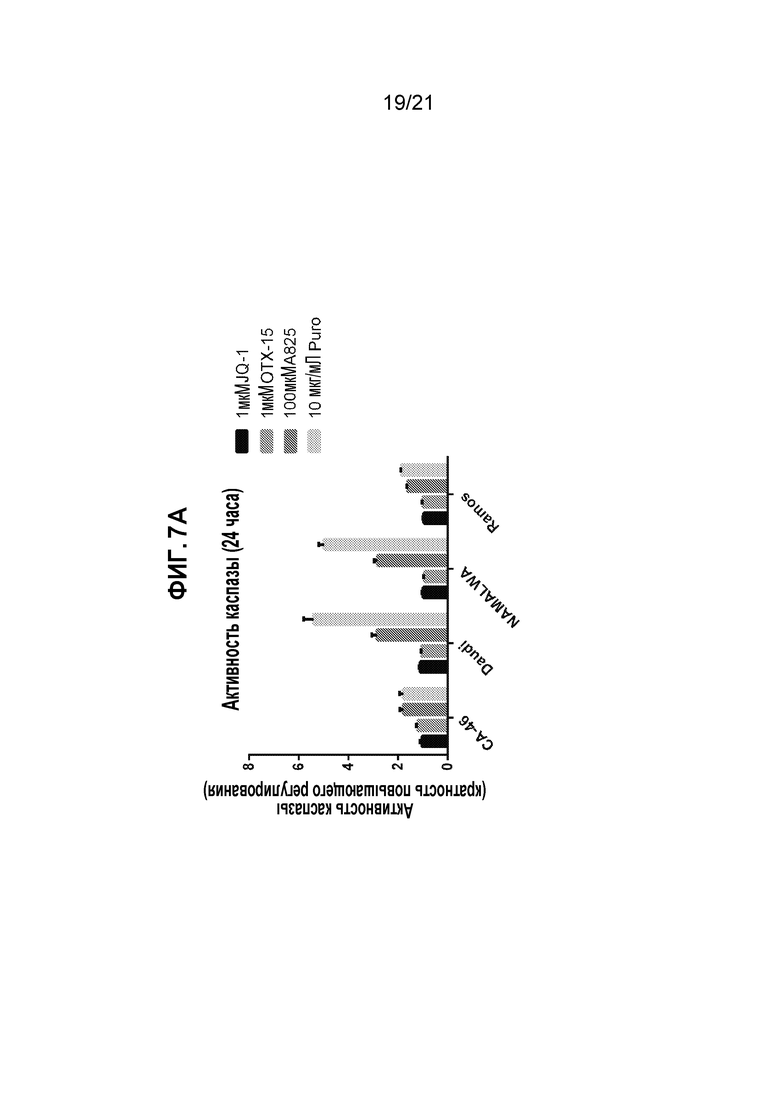
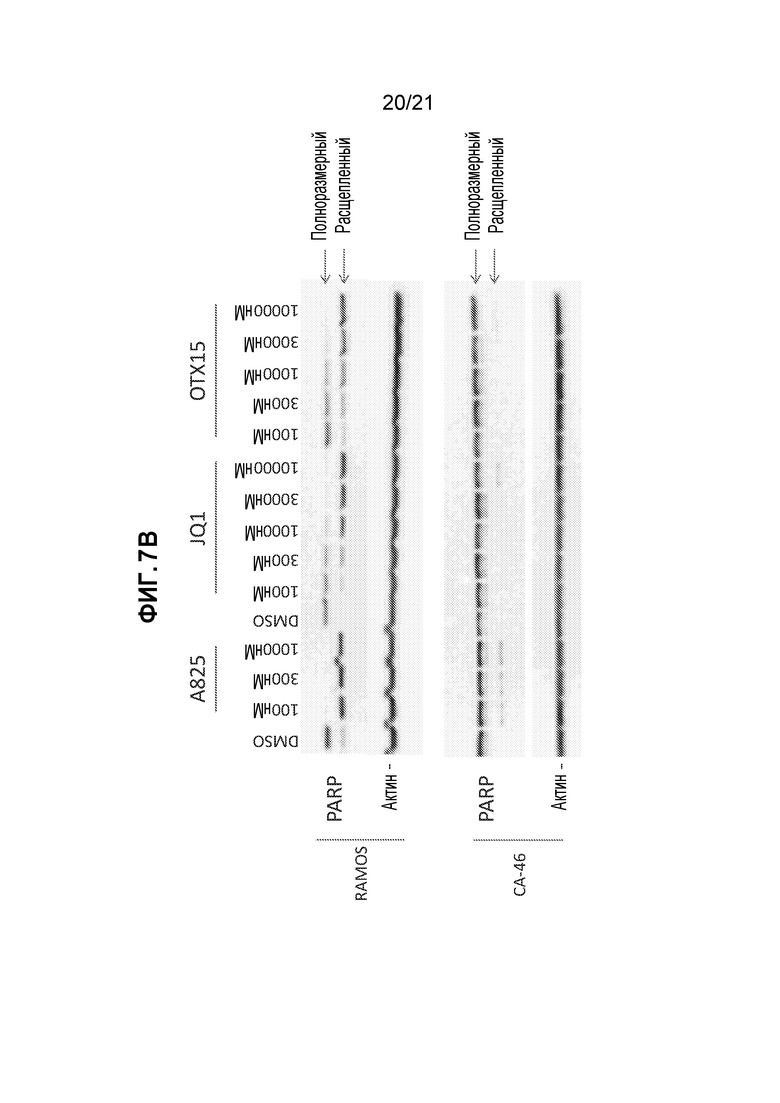
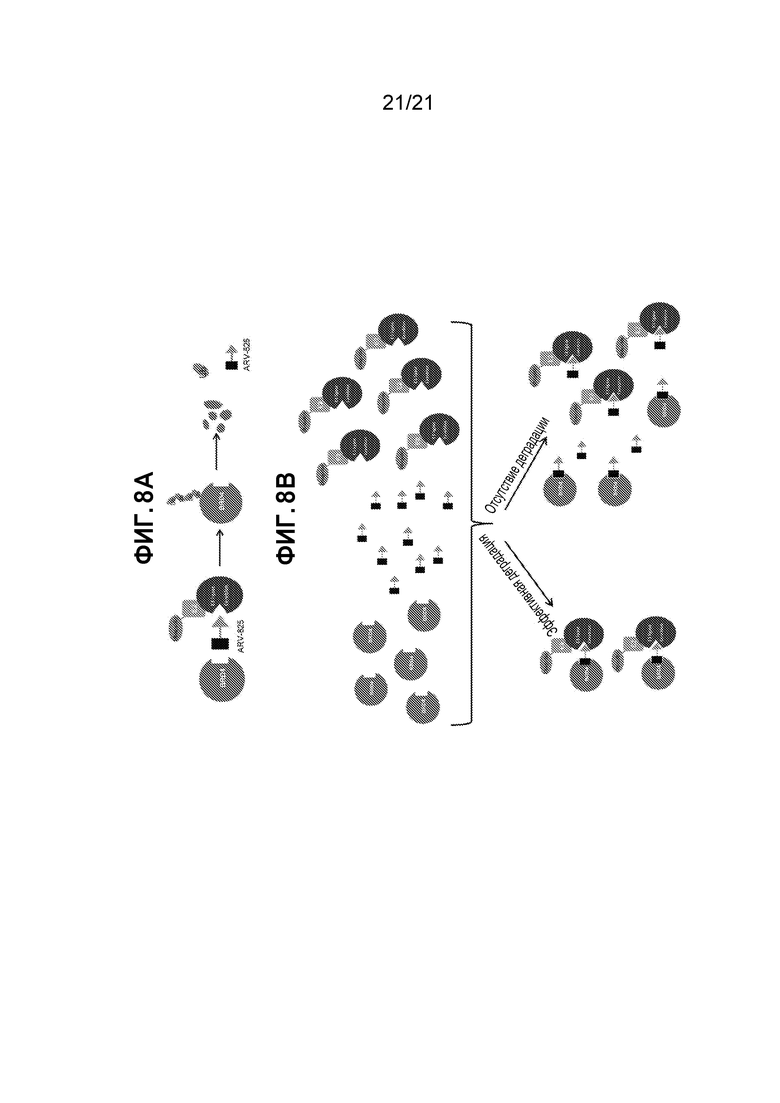
Изобретение относится к бифункциональным соединениям, которые содержат на одном конце лиганд, связывающийся с убиквитинлигазой E3, и на другом конце фрагмент, который связывается с целевым белком-мишенью, так что целевой белок-мишень оказывается вблизи убиквитинлигазы, которая осуществляет деградацию (и ингибирование) этого белка. Соединения предназначены для приготовления композиции для деградации BET бромодомен-содержащего белка или ERRα в клетке. 5 н. и 2 з.п. ф-лы, 21 ил., 1 табл., 2 пр.
1. Соединение, выбранное из группы, состоящей из:
2-[(9R)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[4-({1-[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}окси)фенил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-(4-{5-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]пиримидин-2-ил}фенил)этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-{2-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]этил}ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1R)-1-{4-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[5-(3-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропокси)пиримидин-2-ил]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[3-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)пропокси]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]фенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(4-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}бутил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(5-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пентил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(3-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(4-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}бутил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1S)-1-{4-[2-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)этокси]-3-фторфенил}этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[4-({1-[2-(3-метил-2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}окси)фенил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[4-({1-[2-(1-метил-2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}окси)фенил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1R)-1-[3-(3-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропокси)фенил]этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(3S)-1-{4-[(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этил)амино]бензоил}пирролидин-3-ил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[2-(2-{[3-(2,6-диоксопиперидин-3-ил)-2-метил-4-оксо-3,4-дигидрохиназолин-5-ил]амино}этокси)этил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[3-(3-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пропокси)пропил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(3S)-1-[4-(2-{[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)бензоил]пирролидин-3-ил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(4-{2-[2-(2-{[3-(2,6-диоксопиперидин-3-ил)-2-метил-4-оксо-3,4-дигидрохиназолин-5-ил]амино}этокси)этокси]этокси}фенил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-(5-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}пентил)ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[3-(3-{[3-(2,6-диоксопиперидин-3-ил)-2-метил-4-оксо-3,4-дигидрохиназолин-5-ил]амино}пропокси)пропил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(3S)-1-[4-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)бензоил]пирролидин-3-ил]ацетамид;
2-[(9S)-7-(4-хлорфенил)-4,5,13-триметил-3-тиа-1,8,11,12-тетраазатрицикло[8.3.0.02,6]тридека-2(6),4,7,10,12-пентаен-9-ил]-N-[(1R)-1-[3-(2-{[2-(2,6-диоксопиперидин-3-ил)-1-оксо-2,3-дигидро-1H-изоиндол-4-ил]амино}этокси)фенил]этил]ацетамид;
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10,13-тетраокса-1-азапентадекан-15-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил;
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10-триокса-1-азадодекан-12-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил;
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10,13,16-пентаокса-1-азаоктадекан-18-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил; и
4-(4-{[(5Z)-3-{1-[2-(2,6-диоксопиперидин-3-ил)-1,3-диоксо-2,3-дигидро-1H-изоиндол-4-ил]-4,7,10,13,16,19-гексаокса-1-азагеникозан-21-ил}-2,4-диоксо-1,3-тиазолидин-5-илиден]метил}-2-метоксифенокси)-3-(трифторметил)бензонитрил,
включая их фармацевтические приемлемые соли.
2. Композиция для деградации BET бромодомен-содержащего белка или ERRα, содержащая эффективное количество соединения по п. 1 и носитель и/или эксципиент.
3. Фармацевтическая композиция для деградации BET бромодомен-содержащего белка или ERRα, содержащая эффективное количество соединения по п. 1 и фармацевтически приемлемый носитель, добавку и/или эксципиент.
4. Способ индуцирования деградации BET бромодомен-содержащего белка или ERRα в клетке, включающий введение эффективного количества соединения по п. 1 в клетку, где соединение обеспечивает деградацию BET бромодомен-содержащего белка или ERRα.
5. Композиция, содержащая эффективное количество соединения по п. 1 для применения в способе деградации BET бромодомен-содержащего белка или ERRα в клетке, где композиция обеспечивает деградацию BET бромодомен-содержащего белка или ERRα.
6. Композиция по п. 5, где клетка представляет собой лейкемическую клетку, клетку доброкачественной лимфомы, клетку злокачественной лимфомы, клетку лимфомы Беркитта, клетку неходжкинской лимфомы, клетку рака кишечника, клетку рака молочной железы, клетку рака предстательной железы, клетку рака легкого, клетку рака яичника, клетку рака толстой кишки, клетку болезни Ходжкина, клетку T-клеточного острого лимфобластного лейкоза (T-ALL), клетку T-клеточной лимфобластной лимфомы (T-LL), клетку периферической T-клеточной лимфомы, клетку Т-клеточного лейкоза взрослых, клетку пре-B ALL, клетку пре-B лимфомы, клетку крупноклеточной B-клеточной лимфомы, клетку лимфомы Беркитта, клетку B-клеточного ALL, клетку положительного к филадельфийской хромосоме ALL или клетку положительного к филадельфийской хромосоме CML.
7. Способ по п. 4, где клетка представляет собой лейкемическую клетку, клетку доброкачественной лимфомы, клетку злокачественной лимфомы, клетку лимфомы Беркитта, клетку неходжкинской лимфомы, клетку рака кишечника, клетку рака молочной железы, клетку рака предстательной железы, клетку рака легкого, клетку рака яичника, клетку рака толстой кишки, клетку болезни Ходжкина, клетку T-клеточного острого лимфобластного лейкоза (T-ALL), клетку T-клеточной лимфобластной лимфомы (T-LL), клетку периферической T-клеточной лимфомы, клетку Т-клеточного лейкоза взрослых, клетку пре-B ALL, клетку пре-B лимфомы, клетку крупноклеточной B-клеточной лимфомы, клетку лимфомы Беркитта, клетку B-клеточного ALL, клетку положительного к филадельфийской хромосоме ALL или клетку положительного к филадельфийской хромосоме CML.
| RU 2014133019 A, 27.02.2016 | |||
| WINTER et al., "Phthalimide conjugation as a strategy for in vivo target protein degradation", Science, 2015.05.21, vol | |||
| Телефонная трансляция с местной цепью для уничтожения обратного действия микрофона | 1924 |
|
SU348A1 |
| Способ приготовления состава для замены олифы и связывающих веществ для красок | 1921 |
|
SU6241A1 |

