ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым мультивиниламиносиланам, которые являются полезными в качестве разветвляющих агентов при полимеризации сопряженных диеновых мономеров, необязательно вместе с ароматическими виниловыми мономерами, в результате чего получают полимеры, в частности, эластомерные полимеры, которые можно выгодно использовать в резиновых изделиях, например, в шинах.
УРОВЕНЬ ТЕХНИКИ
Повышение цен на нефть и внутригосударственное законодательство, требующее сокращения автомобильных выбросов углекислого газа, заставляют производителей шин и каучука производить «топливоэкономичные» и, таким образом, топливосберегающие шины. Один подход для получения топливоэкономичных шин заключается в производстве составов шин, имеющих пониженные гистерезисные потери. Гистерезисные потери сшитой эластомерной полимерной композиции связаны с ее значением tan δ при 60°C (см. ISO 4664-1:2005; Rubber, Vulcanized or thermoplastic; Determination of dynamic properties - part 1: General guidance). В целом, вулканизированные эластомерные полимерные композиции, имеющие относительно низкие значения tan δ при 60°C являются предпочтительными, как имеющие более низкие гистерезисные потери. Для готовой произведенной шины это обуславливает более низкое сопротивление качению и лучшую экономию топлива. В противоположность этому, более низкое значение tan δ при 0°C соответствует ухудшенному сцеплению готовой шины с мокрым дорожным покрытием. Таким образом, общепризнанно, что более низкое сопротивление шины качению может быть достигнуто за счет ухудшенных характеристик сцепления шины с мокрым дорожным покрытием. Например, если в произвольном растворном бутадиен-стирольном каучуке (произвольном SSBR) концентрация звеньев полистирола снижается по отношению к общей концентрации звеньев полибутадиена, то температура стеклования SSBR уменьшается и в результате этого оба значения tan δ при 60°C и tan δ при 0°C уменьшаются, в целом соответствуя техническим характеристикам улучшенного сопротивления шины качению и ухудшенного сцепления шины с мокрым дорожным покрытием. Соответственно, при правильной оценке технических характеристик каучукового вулканизата необходимо контролировать оба значения tan δ при 60°C и tan δ при 0°C наряду с теплообразованием в шине.
WO 2012/091753 относится к силан-функционализированным полимерам и полученным из них каучуковым вулканизатам. Авторы описывают использование некоторых алкениламиносиланов для использования при инициировании анионной полимеризации.
US 8299167 B2 относится к сопряженному диеновому полимеру, полученному посредством полимеризации сопряженного диенового мономера и виниламиносилана в присутствии катализатора на основе щелочного металла.
WO 2011/028523 относится к способу синтеза полидиена, включающему полимеризацию сопряженного диенового мономера с каталитической системой на основе лантанида в присутствии винилсилана, аллилсилана или аллилвинилсилана.
US 3485857 относится к классу соединений, имеющих как кремний-азотную связь, так и металл-углеродную связь, применимых в качестве промежуточных соединений при синтезе кремнийорганических соединений, имеющих кремниевую функциональную группу, углеродную функциональную группу или обе. В данном документе описана реакция метиламина с винилдиметилхлорсиланом для синтезирования сим-дивинилтетраметил-N-метилдисилазана и сополимеризация продукта реакции и стирола в присутствии н-бутилллития.
Настоящее изобретение направлено на получение отвержденных эластомерных полимерных (каучуковых) композиций, демонстрирующих улучшенные технические характеристики по показателю теплообразования (HBU), по упругому восстановлению после деформации 60, по сопротивлению качению при сохранении сцепления шины с мокрым и обледеневшим дорожным покрытием (при такой же микроструктуре, Tg отраженный посредством tan δ) в равновесии с хорошими технологическими харатеристиками (CML-ML) и относительно низкой вязкостью по Муни. Вязкость полимера по Муни не показывает увеличения при длительном времени хранения. В целом, изобретение направлено на улучшение баланса экономии топлива и технологических характеристик.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение, в частности, основывается на обнаружении того, что решение по вышеуказанным объектам может быть найдено посредством применения нового полимеризуемого мультвиниламиносилана в качестве разветвляющего агента, в частности, посредством применения такого соединения в полимеризации одного или нескольких сопряженных диенов, например, 1,3-бутадиена («бутадиена») и изопрена и необязательно одного или нескольких ароматических виниловых соединений, например, стирола.
Таким образом, в первом аспекте настоящее изобретение предлагает мультивиниламиносилан следующей Формулы 1:

где
A представляет собой органическую группу, имеющую по меньшей мере две аминогруппы;
каждый В независимо выбран из группы -Si(R1)(R2)(R3), при этом каждый из R1, R2 и R3 независимо выбраны из винила, бутадиенила, метила, этила, пропила, бутила, гексила, октила, фенила и бензила при условии, что по меньшей мере один из R1, R2 и R3 выбран из винила и бутадиенила, причем каждая группа В является заместителем аминогруппы группы A;
по меньшей мере каждая из двух аминогрупп в группе A является замещенной по меньшей мере одной группой B;
n равен целому числу, по меньшей мере 2, предпочтительно целому числу, выбранному из чисел от 2 до 6; и
все аминогруппы в группе A являются третичными аминогруппами.
В альтернативном первому аспекту варианте, настоящее изобретение предлагает инициирующее соединение, подходящее для использования в качестве инициатора при полимеризации сопряженных диенов, при этом указанное инициирующее соединение может быть получено посредством реакции мультивиниламиносилана Формулы 1 с органическим соединением щелочного металла.
Во втором аспекте настоящее изобретение предлагает способ получения мультивиниламиносилана Формулы 1, причем указанный способ включает приведение в контакт амина с силаном следующей Формулы 2 в присутствии основания:

где
X выбран из Cl, Br, I, трифторметансульфоната (OTf) и тозилата (OTos);
R1, R2 и R3 каждый независимо выбран из винила, бутадиенила, метила, этила, пропила, бутила, гексила, октила, бензила и фенила, при условии, что по меньшей мере один из R1, R2 и R3 выбран из винила и бутадиенила; и
амин представляет собой соединение, имеющее по меньшей мере две группы, независимо выбранные из первичной аминогруппы и вторичной аминогруппы.
В третьем аспекте настоящее изобретение предлагает использование мультивиниламиносилана Формулы 1 в качестве разветвляющего агента для начала разветвления в эластомерном полимере.
В четвертом аспекте настоящее изобретение предлагает способ синтеза разветвленного эластомерного полимера, указанный процесс включает (i) полимеризацию по меньшей мере одного сопряженного диена с мультивиниламиносиланом Формулы 1 в присутствии инициирующего соединения, или (ii) полимеризацию по меньшей мере одного сопряженного диена в присутствии инициирующего соединения, получаемого посредством приведения в контакт мультивиниламиносилана Формулы 1 с органическим соединением щелочного металла.
В пятом аспекте настоящее изобретение предлагает другой способ получения разветвленного эластомерного полимера, указанный способ включает приведение в контакт живого полимера, получаемого посредством анионной полимеризации по меньшей мере одного сопряженного диена, с мультивиниламиносиланом Формулы 1.
В шестом аспекте настоящее изобретение предлагает разветвленный эластомерный полимер, получаемый посредством способа согласно четвертому или пятому аспекту изобретения.
В седьмом аспекте настоящее изобретение предлагает невулканизированную (неотвержденную) полимерную композицию, содержащую разветвленный эластомерный полимер согласно шестому аспекту изобретения и один или несколько дополнительных компонентов, выбранных из (i) компонентов, которые добавлены в или образованы в результате процесса полимеризации, используемого для получения указанного полимера, (ii) компонентов которые остаются после удаления растворителя из процесса полимеризации и (iii) компонентов, которые добавляют в полимер после завершения процесса изготовления полимера, таким образом включая компоненты, которые добавляют в полимер «без растворителя» посредством применения (но не ограничиваясь только им) механического смесителя.
В восьмом аспекте настоящее изобретение предлагает вулканизированную (отвержденную) полимерную композицию, которую получают посредством вулканизации (отверждения) неотвержденной полимерной композиции согласно седьмому аспекту изобретения, которая содержит одну или несколько вулканизирующих (отверждающих) добавок.
В девятом аспекте настоящее изобретение предлагает способ получения вулканизированной полимерной композиции, указанный способ включает вулканизацию невулканизированной полимерной композиции согласно седьмому аспекту изобретения, которая содержит одну или несколько вулканизирующих добавок.
В десятом аспекте настоящее изобретение предлагает изделие, содержащее по меньшей мере один компонент, образованный из вулканизированной полимерной композиции согласно восьмому аспекту изобретения.
Мультивиниламиносиланы Формулы 1 имеют общую способность одновременно и обратимо разветвлять и функционализировать две или несколько полимерных цепей, при этом «функционализировать» означает, что таким образом функционализированный полир может вступать в реакцию с другими компонентами в полимерной композиции, в частности, с наполнителем. В зависимости от способности к сшиванию, т.е. степени присоединения функциональной группы, одна молекула Формулы 1 может ввести в цепь полимера несколько и, предпочтительно, вплоть до шести функциональных групп, таким образом уменьшая общее количество разветвляющего агента, необходимого для желаемой степени функционализации, с соответствующими затратами и результатами.
Эластомерный полимер согласно шестому аспекту изобретения, как было обнаружено, не демонстрирует значительное увеличение вязкости во время длительного хранения, как проиллюстрировано на Фиг 1. Невулканизированная полимерная композиция согласно седьмому аспекту изобретения, особенно когда она содержит кремнезем в качестве наполнителя, как было обнаружено, демонстрирует сравнительно низкую вязкость соединения по Муни (CML1+4). Более того, вулканизированная полимерная композиция согласно восьмому аспекту изобретения, как было обнаружено, демонстрирует уменьшенное теплообразование и улучшенные значения tan δ, соответствующие уменьшенному сопротивлению качению и улучшенному сцеплению шины с обледеневшим дорожным покрытием.
В целом, настоящее изобретение достигает цели в получении улучшенного баланса технологических характеристик полимерной композиции и топливосберегающих характеристик полимерной композиции после вулканизации и формовки шины.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
На Фиг. 1 проиллюстрирована диаграмма, показывающая изменение вязкости по Муни MU эластомерного полимера Y по изобретению, модифицированного (разветвленного) с использованием мультивиниламиносилана Примера M12.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
МУЛЬТИВИНИЛАМИНОСИЛАН ФОРМУЛЫ 1
Мультивиниламиносилан Формулы 1 имеет по меньшей мере две аминогруппы, замещенные по меньшей мере одной этиленненасыщенной силильной группой B. В контексте настоящего изобретения используют выражение «группа В является заместителем аминогруппы» или «аминогруппа, замещенная группой B» для описания образования химических связей группы В с атомом азота аминогруппы, т.е. > N-Si(R1)(R2)(R3). Аминогруппа группы A может быть замещенной 0, 1 или 2 группами B. Все аминогруппы группы A представляют собой третичные аминогруппы, т.е. аминогруппы, не содержащие атома водорода.
Органическая группа A предпочтительно представляет собой группу, не имеющую активного водорода. Выражение «активный водород» используют в контексте настоящего изобретения для обозначения атома водорода, который не является инертным, т.е. будет вступать в реакцию, при анионной полимеризации сопряженных диенов, например, бутадиена или изопрена.
Органическая группа A также предпочтительно представляет собой группу, не имеющую электрофильных групп. Выражение «электрофильная группа» используют в контексте настоящего изобретения для обозначения группы, которая будет вступать в реакцию с н-бутиллитием в качестве инициатора модели и/или с живой цепью при анионной полимеризации сопряженных диенов, например, бутадиена или изопрена. Электрофильные группы содержат: алкины, (карбо)катионы, атомы галогена, Si-O, Si-S, Si-галогеновые группы, металл-C- группы, нитрилы, (тио)карбоксилаты, (тио)карбоксильные эфиры, (тио)ангидриды, (тио)кетоны, (тио)альдегиды, (тио)цианаты, (тио)изоцианаты, спирты, тиолы, (тио)сульфаты, сульфонаты, сульфаматы, сульфоны, сульфоксиды, имины, тиокетали, тиоацетали, оксимы, карбазоны, карбодиимиды, мочевины, уретаны, соли диазония, карбаматы, амиды, нитроны, нитрогруппы, нитрозамины, ксантогенаты, фосфаны, фосфаты, фосфины, фосфонаты, бороновые кислоты, бороновые эфиры и т.д.
Более предпочтительно, органическая группа A представляет собой группу, не имеющую ни активного водорода, ни электрофильных групп.
В предпочтительных вариантах реализации по первому аспекту изобретения, мультивиниламиносилан Формулы 1 выбран из следующих соединений Формул от 1-1 до 1-5, в которых в отношении группы В и индекса n применяются те же ограничения и условия, как для Формулы 1, т.е. мультивиниламиносилан имеет по меньшей мере две группы B, в то время как ограничения и условия Формулы 1 для группы A по существу удовлетворены.
Вариант реализации изобретения 1;
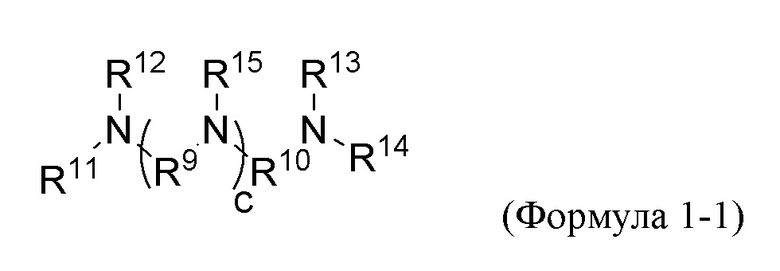
где
каждый из R11, R12, R13, R14 и R15 независимо выбран из группы B, C1-C18 алкила, C6-C18 арила, необязательно C1-C4 алкилзамещенного C3-C12 гетероарила, C7-C18 аралкила, (R4)a-O-(R5)b, при этом каждый из R4 и R5 независимо выбран из C1-C6 алкила и С6-С18 арила и каждый из a и b равен целому числу, независимо выбранному из чисел от 0 до 4, и -Si(R6)(R7)(R8), при этом каждый из R6, R7 и R8 независимо выбран из метила, этила, пропила, бутила, гексила, октила, фенила и бензила;
каждый из R9 и R10 независимо выбран из бивалентного этила, пропила, бутила, фенила и -(СН2)a'-С6Н5-(СН2)b'-, при этом каждый из a' и b' равен целому числу, независимо выбранному из 0 и 1; и
c равен целому числу, выбранному из 0, 1, 2 и 3.
Предпочтительно, в Формуле 1-1 каждый из R11, R12, R13, R14 и R15 независимо выбран из группы В и метила; каждый из R9 и R10 представляет собой бивалентный этил; и c равен целому числу, выбранному из 0, 1, 2 и 3.
Конкретные варианты реализации изобретения Формулы 1-1 включают:

где каждый R независимо выбран из В, C1-C6 алкила и бензила.
Вариант реализации изобретения 2:

где группа -N<>N- представляет собой от 5- до 18-членную гетероциклическую группу, которая может быть насыщенной или ненасыщенной и которая может быть замещенной на любом углеродном атоме кольца C1-С6 алкильной группой, при этом гетероатомные группы, кроме двух атомов N, специально проиллюстрированных в Формуле 1-2, выбраны из -N=, >NR16, причем R16 выбран из группы B, C1-С6 алкила, фенила и бензила, -O-, -S- и >SiR17R18, причем каждый из R17 и R18 независимо выбран из C1-С6 алкила, фенила и бензила. Предпочтительно, группа -N<>N- представляет собой пиперазинил.
Конкретные варианты реализации изобретения Формулы 1-2 включают:

где R представляет собой C1-С6 алкильную группу.
Вариант реализации изобретения 3:

где каждый из R20 R21 и R22 независимо выбран из одинарной связи и бивалентной алкильной группы C1-С10, d равен целому числу, выбранному из 0, 1 и 2, d' равен целому числу, выбранному из 0 и 1, причем d равен 0 если d' равен 0, каждая группа -N<>X- независимо выбрана из от 5- до 10- членной гетероциклической группы, которая может быть насыщенной или ненасыщенной и которая может быть замещенной на любом углеродном атоме кольца C1-С6 алкильной группой, при этом каждый X независимо выбран из -N-, -C= и -CH- и гетероатомные группы, кроме двух групп N и X, специально проиллюстрированных в Формуле 1-3, выбраны из -N=, >NR16, при этом R16 выбран из C1-С6 алкила, группы B, фенила и бензила, -O-, -S- и >SiR17R18, причем каждый из R17 и R18 независимо выбран из C1-С6 алкила, фенила и бензила. Предпочтительно, группы -N<>X- содержат пиперидинил и пиперазинил.
Конкретные варианты реализации изобретения Формулы 1-3 включают:

Вариант реализации изобретения 4:

где D представляет собой от 5- до 10-членную карбоциклическую или гетероциклическую группу, которая может быть насыщенной или ненасыщенной и которая может быть замещенной на любом углеродном атоме кольца C1-С6 алкильной группой, при этом гетероатомные группы выбраны из -N=, >NR16, причем R16 выбран из C1-С6 алкила, группы B, фенила и бензила, -О-, -S- и >SiR17R18, при этом каждый из R17 и R18 независимо выбран из C1-С6 алкила и фенила, каждая группа -N<>X- независимо выбрана из от 5- до 10-членной гетероциклической группы, которая может быть насыщенной или ненасыщенной и которая может быть замещенной на любом углеродном атоме кольца C1-С6 алкильной группой, причем каждый X независимо выбран из -N-, -C= и -CH-, R23 выбран из одинарной связи и дивалентной C1-C10 алкильной группы, и e равен целому числу, выбранному из 2, 3 и 4. Предпочтительно, группы D содержат циклопентил, циклогексил, фенил и тетрагидрофуранил. Предпочтительно, группы N-<>X- содержат пиперидинил и пиперазинил.
Предпочтительно, в Формуле 1-4 D выбран из циклопентила, циклогексила, фенила и тетрагидрофуранила; каждая группа -N<>X- выбрана из пиперидинила и пиперазинила; R23 представляет собой одинарную связь; и e равен целому числу, выбранному из 2 и 3.
Конкретные варианты реализации изобретения Формулы 1-4 включают:
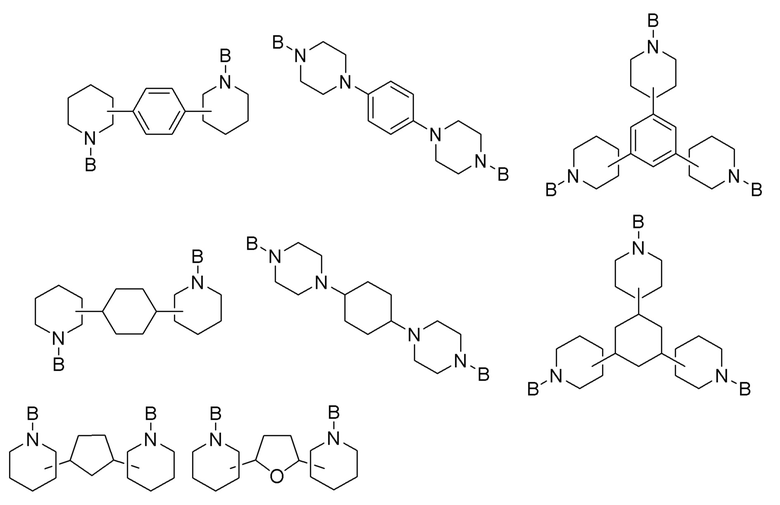
Вариант реализации изобретения 5:

где E представляет собой от 6- до 10-членную циклоалифатическую или ароматическую группу, каждый R' независимо выбран из одинарной связи и C1-C2 алкила, каждый R независимо выбран из В, C1-C4 алкила и бензила, и f равен целому числу, выбранному из 2 и 3.
Предпочтительно, в Формуле 1-5 E выбран из циклогексила и фенила; R' представляет собой одинарную связь, R выбран из B, C1-C4 алкила и бензила; и f равен целому числу, выбранному из 2 и 3.
Конкретные варианты реализации изобретения формулы 1-5 включают:

где каждый R независимо выбран из В, C1-C4 алкила и бензила.
ИНИЦИИРУЮЩЕЕ СОЕДИНЕНИЕ, ПОЛУЧАЕМОЕ ИЗ СОЕДИНЕНИЯ ФОРМУЛЫ 1
Инициирующее соединение, которое составляет часть первого аспекта изобретения, получают посредством приведения в контакт мультивиниламиносилана Формулы 1, как определено выше, включая все его варианты реализации изобретения, с органическим соединением щелочного металла.
Подходящие, типовые органические соединения щелочного металла включают метиллитий, этиллитий, н-бутиллитий, втор-бутиллитий, трет-октиллитий, изопропиллитий, фениллитий, циклогексиллитий, 2-бутиллитий, 4-фенилбутиллитий, трет-бутилдиметилсилилоксипропиллитий, диалкиламинопропиллитий, N-морфолинопропиллитий, натрий бифенилид, натрий нафталенид и калий нафталенид. Более предпочтительно, инициирующее соединение представляет собой монолитийалкил, алкиларил или соединение арила.
Инициирующее соединение, получаемое из мультивиниламиносилана Формулы 1, может быть синтезировано в инертном растворителе и в присутствии рандомизирующих соединений, при этом подходящие растворители такие же, какие используют в полимеризационном растворе, как определено ниже.
ПОЛУЧЕНИЕ МУЛЬТИВИНИЛАМИНОСИЛАНА ФОРМУЛЫ 1
В целом, согласно второму аспекту настоящего изобретения, мультивиниламиносиланы Формулы 1 первого аспекта изобретения получают посредством приведения в контакт, в присутствии основания, амина, имеющего по меньшей мере две группы, независимо выбранные из первичной аминогруппы и вторичной аминогруппы, с силаном Формулы 2, как определено в настоящем описании.
Применимые силаны Формулы 2 для получения мультивиниламиносилана Формулы 1 содержат хлорди(C1-C4 алкил)винилсиланы, в частности, хлордиметилвинилсилан, хлордиэтилвинилсилан и хлордифенилвинилсилан, предпочтительно хлордиметилвинилсилан.
Амин, имеющий по меньшей мере две первичные или вторичные аминогруппы, структурно соответствует группе A в Формуле 1, имеющей еще по меньшей мере две первичные или вторичные аминогруппы вместо соответствующей третичной аминогруппы.
Таким образом, в варианте реализации изобретения 1 амин, используемый для получения соединения Формулы 1-1, может быть выбран из фенилендиамина, C2-C4 моноалкилендиаминов, C2-C4 диалкилентриаминов и C2-C4 триалкилентетрааминов, предпочтительно из моноэтилендиамина, диэтилентриамина, дипропилентриамина, триэтилентетраамина и тетраэтиленпентамина.
В варианте реализации изобретения 2 амин, используемый для получения соединения Формулы 1-2, может быть выбран из пиперазина и 1,7-диокса-4,10-диазациклододекана.
В варианте реализации изобретения 3 амин, используемый для получения соединения Формулы 1-3, может быть выбран из триэтилентетрамина, 4,4'-триметилендипиперидина, 4,4'-этилендипиперидина и 1-(3-пиперазин-1-илпропил)пиперазина.
В варианте реализации изобретения 4 один амин, используемый для получения соединения Формулы 1-4, может быть 1,4-ди(пиперазин-l-ил)бензолом (синтезированным из, например, 1,4-дибромбензола и пиперазина). Насыщенные амины, используемые для получениясоединения Формулы 1-4, могут быть синтезированы посредством катализируемого переходным металлом гидрирования соответствующих ароматических аминов.
В варианте реализации изобретения 5 амин, используемый для получения соединения Формулы 1-5, может быть выбран из 1,2-циклогександиамина, о- или п-фенилендиамина, 1,3,5-триаминобензола, 1,5-диаминонафталена и 1,8-диаминонафталена.
В целом, амин и силан Формулы 2 будут вступать в реакцию в таких пропорциях, чтобы прикрепить по меньшей мере две группы В к амину, например с использованием молярного отношения амина к силану Формулы 2 в диапазоне от 0,5 до 0,1 (в зависимости от количества аминов). Для каждого моля первичной и вторичной аминогруппы используют от 1 до 3 молей силана Формулы 2.
Получения мультивиниламиносилана Формулы 1 может также включать (частичную) реакцию между амином и силаном, отличным от силана Формулы 2. В таком случае, реакцию между амином, силаном Формулы 2 и другим силаном могут проводить последовательно, например, амин приводят в контакт сначала с силаном Формулы 2, а потом с другим силаном или наоборот.
Основание предпочтительно выбирают из третичных алифатических или ароматических аминов, например, триэтиламина, пиридина и 1,4-диазабицикло[2,2,2]октана (DABCO) или замещенных амидинов или гуанидинов, например, 1,8-диазабициклоундекана (DBU), l,5-диазабицикло[4,3,0]нон-5-ена (DBN), 1,1,3,3-тетраметилгуанидина, предпочтительно, триэтиламина и пиридина. Основание, в целом, используют в общем количестве от 0,5 до 5 моль на моль первичных и вторичных аминогрупп, предпочтительно от 1 до 3 моль на моль первичных и вторичных аминогрупп. Также возможно использовать амин, имеющий по меньшей мере две первичные или вторичные аминогруппы в качестве основания, но тогда будет необходимо соответственно увеличить его количество, как правило, посредством 1,5-5 эквивалентов. Важно обеспечить избыток основания по сравнению с силановым реагентом исходя из молярных количеств таким образом, чтобы все HX, образующиеся в реакции, могли быть поглощены (погашены) основанием.
Реакцию можно проводить в растворителе, в частности, в инертном растворителе, например, в углеводородном растворителе, включая пентан, н-гексан, циклогексан, гептан, бензол и толуол, в эфирном растворителе, включая диэтилэфир, тетрагидрофуран и трет-бутилметилэфир, в хлорированном растворителе, включая хлороформ, тетрахлорметан и дихлорметан, в сложноэфирном растворителе, например, в этилацетате и метилацетате, или в других диполярных растворителях, например, в ацетоне, диметилформамиде и ацетонитриле. Предпочтительными растворителями являются дихлорметан, хлороформ, диэтилэфир, этилацетат, толуол и циклогексан. Общая концентрация реагентов в растворителе, как правило, находится в диапазоне от 0,1 до 2 M.
Реакцию между амином и силаном Формулы 2 можно проводить в условиях, которые будут очевидны специалисту в данной области техники, например, из реакций, используемых для приведения в контакт амина с галосиланом.
Реакцию можно проводить соответствующим образом при температуре от -30°C до температуры кипения реакционной смеси, предпочтительно от 0 °C до 25 °C.
Как правило, реакцию проводят посредством добавления по каплям силана Формулы 2, в растворе или без примесей, к раствору амина и основания. Реакционную смесь перемешивают и приводят в контакт в течение достаточного времени, в целом, в течение нескольких часов и предпочтительно в течение по меньшей мере одного часа при температуре, как правило, от 0°C до температуры кипения. После окончания или завершения реакции любые нерастворимые соли, образованные в ходе реакции, могут быть отфильтрованы, растворитель может быть удален посредством перегонки при пониженном давлении, и очисткой, например, посредством вакуумной перегонки или перекристаллизации, получают мультивиниламиносилан Формулы 1.
ИСПОЛЬЗОВАНИЕ В КАЧЕСТВЕ РАЗВЕТВЛЯЮЩЕГО АГЕНТА - ПОЛИМЕРИЗАЦИЯ
Согласно третьему аспекту настоящего изобретения, мультивиниламиносилан Формулы 1 настоящего изобретения соответствующим образом используют в качестве разветвляющего агента для начала обратимого разветвления в эластомерном полимере. Для этой цели мультивиниламиносилан - сам по себе или после реакции с органическим соединением щелочного металла для образования инициирующего соединения - уже может быть использован в реакции полимеризации, которую проводят для получения эластомерного полимера (четвертый аспект изобретения), или он может быть добавлен к прореагировавшим с живым эластомерным полимером (пятый аспект изобретения). Способ по четвертому аспекту изобретения, в частности, включает: (i) полимеризации по меньшей мере одного сопряженного диена с мультивиниламиносиланом Формулы 1 и необязательно с одним или несколькими ароматическими виниловыми мономерами в присутствии инициирующего соединения, или (ii) полимеризацию по меньшей мере одного сопряженного диена, необязательно, с одним или несколькими ароматическими виниловыми мономерами в присутствии инициирующего соединения, получаемого посредством приведения в контакт мультивиниламиносилана формулы 1 с органическим соединением щелочного металла.
Эластомерный полимер может быть получен, в целом, посредством анионной, радикальной или катализируемой переходным металлом полимеризации, но предпочтительно, получен посредством анионной полимеризации. Два или несколько виниламиносилановых соединения Формулы 1 могут быть использованы в комбинации. Полимеризацию можно проводить в растворителе и можно осуществлять с одним или несколькими модифицирующими агентами концевой группы цепи, связующими агентами (включая модифицированные связующие агенты), рандомизирующими соединениями и соединениями, ускоряющими полимеризацию.
В дополнение к следующему конкретному описанию, в отношении всех применимых направлений технологии полимеризации, включая инициирующие полимеризацию соединения, координирующие полярность соединения и ускорители (для повышения/изменения реакционной способности инициатора, для статистического расположения ароматических виниловых мономеров и/или для статистического расположения и/или изменения концентрации звеньев 1,2-полибутадиена, или 1,2-полиизопрена, или 3,4-полиизопрена, внедренных в полимер); количества каждого соединения; мономер(ы); и подходящие для процесса условия описаны в WO 2009/148932, полностью включенном в данный документ посредством ссылки.
СОПРЯЖЕННЫЕ ДИЕНЫ (СОПРЯЖЕННЫЕ ДИЕНОВЫЕ МОНОМЕРЫ)
Типовые сопряженные диеновые мономеры, применимые в настоящем изобретении, содержат 1,3-бутадиен, 2-(C1-C5 алкил)-1,3-бутадиен например, изопрен(2-метил-1,3-бутадиен), 2,3-диметил-1,3-бутадиен, 1,3-пентадиен, 2,4-гексадиен, 1,3-гексадиен, 1,3-гептадиен, 1,3-октадиен, 2-метил-2,4-пентадиен, циклопентадиен, 2,4-гексадиен и 1,3-циклооктадиен. Может быть использована смесь двух или нескольких сопряженных диенов. Предпочтительные сопряженные диены содержат 1,3-бутадиен и изопрен. В одном из вариантов реализации изобретения сопряженным диеном является 1,3-бутадиен.
АРОМАТИЧЕСКИЕ ВИНИЛОВЫЕ МОНОМЕРЫ
Произвольные ароматические виниловые мономеры содержат моновинилароматические соединения, т.е. соединения, имеющие только одну винильную группу, прикрепленную к ароматической группе, и ди- или выше винилароматические соединения, которые имеют две или несколько винильных групп, прикрепленных к ароматической группе. Типовые ароматические виниловые мономеры, необязательно, используют вместе с по меньшей мере одним сопряженным диеном, включая стирол, С1-4 алкилзамещенный стирол, например, 2-метилстирол, 3-метилстирол, 4- метилстирол, 2,4-диметилстирол, 2,4,6-триметилстирол, a-метилстирол, 2,4-диизопропилстирол и 4-трет-бутилстирол, стильбен, винилбензилдиметиламин, (4-винилбензил)диметиламиноэтиловый эфир, N,N-диметиламиноэтилстирол, трет-бутоксистирол, винилпиридин и дивинилароматические соединения, например, 1,2-дивинилбензол, 1,3-дивинилбензол и 1,4- дивинилбензол. Два или несколько ароматических виниловых мономера могут быть использованы в комбинации. Предпочтительный ароматический виниловый мономер представляет собой моновинилароматическое соединение, более предпочтительно - стирол.
Моновинилароматическое(ие) соединение(я), особенно включая стирол, могут быть использованы, в зависимости от области применения, в общих количествах вплоть до 70%мас., более конкретно, 40-70%мас., или 15-40%мас., или 1-15%мас., исходя из общей массы мономеров, используемых в реакции полимеризации. Ди- или выше винилароматические соединения, например, дивинилбензол, включая 1,2-дивинилбензол, 1,3- дивинилбензол и 1,4-дивинилбензол, могут быть использованы в общем количестве 1%мас. или менее (исходя из общей молярной массы мономеров, используемых для получения полимера). В одном из предпочтительных вариантов реализации изобретения 1,2-дивинилбензол используют в комбинации со стиролом и бутадиеном или изопреном.
ДРУГИЕ МОНОМЕРЫ
Сомономеры, кроме мультивиниламиносилана Формулы 1, сопряженный диеновый мономер и произвольный ароматический виниловый мономер, который может быть использован в получения эластомерного полимера по изобретению, содержат акриловые мономеры, например, акрилонитрил, акрилаты, например, акриловую кислоту, метилакрилат, этилакрилат, пропилакрилат и бутилакрилат, и метакрилаты, например, метилметакрилат, этилметакрилат, пропилметакрилат и бутилметакрилат. Такие другие coмономеры, как правило, используют в пропорции не более чем 5%мас., исходя из общей массы мономеров, используемых в реакции полимеризации. Дополнительно могут быть использованы виниламинодисилоксановые или бутадиениламинодисилоксановые мономеры, например, 4-[3-(трет-бутил)-1,3,3- триметил-1-винилдисилоксанил]морфоолин, 3-(трет-бутил)-N,N-диэтил-1,3,3-триметил-1-винил-дисилоксан-1-амин и 3-(трет-бутил)-N,N-дибутил-1,3,3-триметил-1-винилдисилоксан-1-амин
ИНИЦИИРУЮЩИЕ СОЕДИНЕНИЯ
Инициирующее соединение используют в процессе полимеризации по настоящему изобретению, при этом два или несколько инициирующих соединения могут быть использованы в комбинации. Инициирующее соединение может быть моновалентным или мультивалентным (дивалентным, тривалентным и т.д.) инициирующим соединением. Подходящие инициирующие соединения содержат щелочные металлы, органические соединения щелочного металла, комплекс между щелочным металлом и полярным соединением, олигомер, содержащий щелочной металл, и комплексы кислот и оснований Льюиса. Типовые щелочные металлы включают литий, натрий, калий, рубидий и цезий. Типовые органические соединения щелочного металла включают этиллитий, н-бутиллитий, втор-бутиллитий, трет-октиллитий, изопропиллитий, фениллитий, циклогексиллитий, 2-бутиллитий, 4-фенилбутиллитий, трет-бутилдиметилсилилоксипропиллитий, диалкиламинопропиллитий, N-морфолинопропиллитий, литий-диизопропиламид, литий-пиперидид, литий-пирролидид, соединения дилитиированного дифенилэтилена, дилитиированные бис(1-арилэтенил)бензолы, соединения мульти-литиированного тривинилбензола, натрий бифенилид, натрий нафталенид и калий нафталенид. Типовые комплексы между щелочным металлом и полярным соединением включают литий-тетраметилэтилендиаминовый комплекс, литий-тетрагидрофурановый комплекс, литий-дитетрагидрофуранпропановый комплекс и их натриевые и калиевые аналоги. Более предпочтительно, инициирующее соединение представляет собой моно- или дилитийалкил, алкилариловое или ариловое соединение. Дополнительные применимые инициаторы содержат инициаторы аминосилановой полимеризации, описанные в WO2014/040640 и инициаторы полимеризации, описанные в PCT/EP2013/065399.
В конкретном варианте реализации изобретения инициирующее соединение является получаемым посредством приведения в контакт мультвиниламиносилана Формулы 1 с органическим соединением щелочного металла, как определено выше. В этом варианте реализации изобретения инициирующее соединение способно выполнять функции как инициирующего соединения, так и разветвляющего агента.
Общее количество инициатора(ов), в частности, органолитиевого(ых) инициатора(ов), будет скорректировано в зависимости от мономера и целевой молекулярной массы или полимера. Общее количество составляет, как правило, от 0,05 до 5 ммоль, предпочтительно от 0,2 до 3 ммоль на 100 грамм мономера.
Полимеры с низкой молекулярной массой могут быть получены с использованием от 5 до 20 ммоль инициатора на 100 г мономера.
РАСТВОРИТЕЛЬ
Полимеризацию, как правило, проводят как полимеризацию в растворе, при этом образуется полимер, по-существу растворимый в реакционной смеси, или как суспензионную полимеризацию, при этом образуется полимер, по-существу растворимый в реакционной среде. Более предпочтительно, полимер получают при полимеризации в растворе. В качестве полимеризационного растворителя, как правило, используют углеводородный растворитель, который не дезактивирует инициатор, катализатор или активную цепь полимера. Полимеризационный растворитель может быть комбинацией двух или нескольких растворителей. Типовые углеводородные растворители содержат алифатические и ароматические растворители. Конкретные примеры включают (включая все возможные структурные изомеры): пропан, бутан, пентан, гексан, гептан, бутилен, пропилен, пентен, гексан, октан, бензол, толуол, этилбензол и ксилен.
МОДИФИЦИРУЮЩИЕ АГЕНТЫ КОНЦЕВОЙ ГРУППЫ ЦЕПИ
В реакции полимеризации по настоящему изобретению могут быть использованы один или несколько модифицирующих агентов концевой группы цепи для дальнейшего управления свойствами полимера посредством приведения в контакт с концевыми группами полимерных цепей в полимере по изобретению. В целом, для этой цели могут быть использованы силансульфидные модифицирующие агенты концевой омега-группы цепи, например, описанные в документах WO 2007/047943, WO 2009/148932, US 6229036 и US 2013/0131263, каждый из которых в полном объеме включен в данный документ посредством ссылки. Другие модифицирующие агенты концевой группы цепи, подходящие для использования в настоящем изобретении, описаны в WO2014/040640 и PCT/EP2013/065399, и силансульфидные модификаторы описаны в WO2014/040639.
В процессе полимеризации модифицирующие агенты концевой группы цепи можно добавлять с периодичностью (с регулярными или нерегулярными интервалами) или непрерывно, но предпочтительно их добавляют при скорости конверсии полимеризации более чем 80 процентов, и более предпочтительно при скорости конверсии более чем 90 процентов. Предпочтительно, значительное количество концевых групп цепи полимера до реакции с модифицирующим агентом концевой группы цепи являются не оконченными; то есть, присутствуют концевые группы цепи живого полимера, способные вступать в реакцию с модифицирующим агентом.
СВЯЗУЮЩИЕ АГЕНТЫ
Для дальнейшего управления молекулярной массой полимера и свойствами полимера, в процессе по изобретению в качестве необязательного компонента может быть использован связующий агент («сшивающий агент»). Связующий агент будет уменьшать гистерезисные потери посредством уменьшения количества свободных концевых групп цепи эластомерного полимера и/или посредством уменьшения вязкости полимерного раствора, по сравнению с несвязанными по существу линейными макромолекулами полимера с идентичной молекулярной массой. Связующие агенты, например, тетрахлорид олова, могут функционализировать концевую группу полимерной цепи и вступать в реакцию с компонентами эластомерной композиции, например, с наполнителем или с ненасыщенными участками полимера. Типовые связующие агенты описаны в U.S. 3281383, U.S. 3244664 и U.S. 3692874 (например, тетрахлорсилан); U.S. 3978103. U.S. 4048.206, 4.474908 и U.S. 6777569 (блокированые меркаптосиланы); U.S. 3078254 (мульти-галоген-замещенный углеводород, например, 1,3,5-три(бромметил)бензол); U.S. 4616069 (соединение олова и органическое амино- или амин- соединение); и U.S. 2005/0124740.
В целом, модифицирующий агент концевой группы цепи добавляют перед, в процессе или после добавления связующего агента, при этом реакцию модификации проводят предпочтительно после добавления связующего агента.
Общее количество используемых связующих агентов будет влиять на вязкость по Муни связанного полимера и, как правило, находится в диапазоне от 0,001 до 4,5 миллиэквивалентов на 100 грамм эластомерного полимера, например от 0,01 до около 1,5 миллиэквивалентов на 100 грамм полимера.
РАНДОМИЗИРУЮЩИЕ СОЕДИНЕНИЯ
Рандомизирующие соединения, обычно известные в данной области техники (также известные как координирующие полярность соединения), необязательно могут быть добавлены к смеси мономера или в реакцию полимеризации для того, чтобы регулировать микроструктуру (т.е. содержание винильных связей) части сопряженного диена в полимере, или чтобы регулировать композиционное распределение какого-либо ароматического винилового мономера и винильных связей в полимерной цепи. Может быть использована комбинация из двух или нескольких рандомизирующих соединений. Примером рандомизирующих соединений, применяемых в настоящем изобретении, в целом могут служить соединения оснований Льюиса. Подходящими основаниями Льюиса для использования в настоящем изобретении являются, например, эфирные соединения, например, диэтиловый эфир, ди-н-бутиловый эфир, этиленгликоль диэтиловый эфир, этиленгликоль дибутиловый эфир, диэтиленгликоль диметиловый эфир, пропиленгликоль диметилвый эфир, пропиленгликоль диэтиловый эфир, пропиленгликоль дибутиловый эфир, (C1-C8 алкил)тетрагидрофуриловые эфиры (включая метилтетрагидрофуриловый эфир, этилтетрагидрофуриловый эфир, пропилтетрагидрофуриловый эфир, бутилтетрагидрофуриловый эфир, гексилтетрагидрофуриловый эфир и октилтетрагидрофуриловый эфир), тетрагидрофуран, 2,2-(бистетрагидрофурфурил)пропан, бистетра-гидрофурфурилформал, метиловый эфир тетрагидрофурфурилового спирта, этиловый эфир тетрагидрофурфурилового спирта, бутиловый эфир тетрагидрофурфурилового спирта, α-метокситетрагидрофуран, диметоксибензол и диметоксиэтан, и третичные амины, например, триэтиламин, пиридин, N,N,N',N'-тетраметил этилендиамин, дипиперидинoэтан, метиловый эфир N,N-диэтилэтаноламина, этиловый эфир N,N-диэтилэтаноламина, N,N-диэтилэтаноламин и диметил N,N-тетрагидрофурфуриламин Примеры предпочтительных рандомизирующих соединений приведены в WO 2009/148932, включенном в данный документ в полном объеме посредством ссылки.
Рандомизирующее соединение, как правило, добавляют в молярном соотношении рандомизирующего соединения к инициирующему соединению от 0,012:1 до 10:1, предпочтительно от 0,1:1 до 8:1 и более предпочтительно от 0,25:1 до около 6:1.
УСКОРЯЮЩИЕ СОЕДИНЕНИЯ
Полимеризация, необязательно, может включать ускорители для увеличения реакционной способности инициатора (и, таким образом, для увеличения скорости полимеризации), для статистического распределения ароматических виниловых мономеров, внедренных в полимер, или для получения одной цепи ароматических виниловых мономеров, таким образом влияя на распределение ароматических виниловых мономеров в живом анионном эластомерном сополимере. Примеры ускорителей включают натрий алкоксиды или натрий феноксиды и калий алкоксиды или калий феноксиды, предпочтительно калий алкоксиды или калий феноксиды, например, калий изопропоксид, калий трет-бутоксид, калий трет-амилоксид, калий н-гептилоксид, калий бензилоксид, калий феноксид; калиевые соли карбоксильных кислот, например, изовалериановая кислота, каприловая кислота, лауриновая кислота, пальмитиновая кислота, стеариновая кислота, олеиновая кислота, линоленовая кислота, бензойная кислота, фталевая кислота и 2-этилгексансановая кислота; калиевые соли органических сульфоновых кислот, например, додецилбензолсульфоновая кислота, тетрадецилбензолсульфоновая кислота, гексадецилбензолсульфоновая кислота и октадецилбензолсульфоновая кислота; и калиевые соли органических фосфорных кислот, например, диэтилфосфит, диизопропилфосфит, дифенилфосфит, дибутилфосфит, и дилаурилфосфит.
Такие ускоряющие соединения могут быть добавлены в общем количестве от 0,005 до 0,5 моль на 1,0 грамм-атом-эквивалент литиевого инициатора. В случае, если добавлено менее, чем 0,005 моль, то, как правило, достаточного эффекта не достигается. С другой стороны, если количество ускоряющего соединения составляет более чем около 0,5 моль, то производительность и эффективность реакции модификации концевой групповой цепи могут быть значительно снижены.
ДОЗИРОВАНИЕ МУЛЬТИВИНИЛАМИНОСИЛАНА
Мультвиниламиносилан Формулы 1 может быть использован в общем количестве от 0,01 до 10 моль на моль инициирующего(их) соединения(ий). Предпочтительно, его используют в общем количестве от 0,1 до 5 моль, или от 0,1 до 3 моль, или от 0,1 до 1,5 моль. Различные мультвиниламиносиланы Формулы 1 могут быть использованы в комбинации в соответствии с настоящим изобретением. В случае, если полимер по изобретению используют в области применения для шин, например, в каучуковом соединении для протектора шины или боковины покрышек, предпочтительно использовать мультивиниламиносилан Формулы 1 в общем количестве от 0,1 до 5 моль на моль инициирующего(их) соединения(ий), более предпочтительно от 0,1 до 2 моль.
Режим добавления («дозирование») мультивиниламиносилана Формулы 1 в процессе полимеризации по отношению к сопряженному диеновому мономеру и произвольному ароматическому виниловому мономеру, инициирующему соединению и другим компонентам, будет влиять на структуру конечного полимера. Таким образом, могут быть получены статистические сополимеры и блоксополимеры, имеющие блоки мультивиниламиносиланового полимера и блоки других мономеров в желаемых пропорциях и последовательностях. Схемы типового дозирования следующие:
(1) Непрерывное (постепенно нарастающее) добавление мультивиниламиносилана Формулы 1 к смеси, содержащей сопряженный диеновый мономер, необязательно, ароматический виниловый мономер и инициирующее соединение в процессе проведения полимеризации приводит к получению статистического сополимера.
(2) Дозирование мультвиниламиносилана формулы 1 до добавления основного количества инициатора вместе с основным количеством coмономеров. После количественной или близкой к количественной конверсии мономеров может быть выполнено второе добавление мультивиниламиносилана для создания блочной структуры на конце полимера.
(3) Дозирование мультивиниламиносилана Формулы 1 перед добавлением основного количества инициатора вместе с основным количеством coмономеров, которые могут быть добавлены после количественной или близкой к количественной конверсии мультвиниламиносилана. Дополнительно, может быть проведено несколько этапов дозирования мультивиниламиносилана Формулы 1 в переменных пропорциях при определенных степенях конверсии общего мономера для создания n конических или блочных структурных элементов внутри полимерной цепи. После количественной или близкой к количественной конверсии мономеров может быть использовано окончательное добавление мультивиниламиносилана или модифицирующего агента концевой группы цепи или связующего агента для создания блочной структуры или другой функционализации или присоединения к концевой группе полимера.
(4) Может быть проведено несколько этапов дозирования мультивиниламиносилана Формулы 1 в переменных пропорциях при определенных степенях конверсии общего мономера для создания конических или блочных структурных элементов внутри полимерной цепи. После количественной или близкой к количественной конверсии мономеров может быть использовано окончательное добавление мультивиниламиносилана или модифицирующего агента концевой группы цепи или связующего агента для создания блочной структуры или другой функционализации или присоединения к концевой группе полимера.
(5) Дозирование мультивиниламиносилана Формулы 1 перед добавлением основного количества инициатора вместе с основным количеством coмономеров (конической структуры), которые могут быть добавлены после количественной или близкой к количественной конверсии мультивиниламиносилана для создания блочной структуры. После количественной или близкой к количественной конверсии мономеров может быть добавлен модифицирующий агент концевой группы цепи или связывающий агент для функционализации или присоединения полимерных цепей, что является предпочтительным параметром дозирования.
ПОЛИМЕР
Эластомерный полимер согласно шестому аспекту изобретения получают посредством процесса по настоящему изобретению, а именно, посредством полимеризации по меньшей мере одного сопряженного диена с мультивиниламиносиланом Формулы 1 в присутствии инициирующего соединения, полимеризации по меньшей мере одного сопряженного диена в присутствии инициирующего соединения, получаемого посредством приведения в контакт мультивиниламиносилана Формулы 1 с органическим соединением щелочного металла, или посредством приведения в контакт живого полимера, получаемого посредством анионной полимеризации по меньшей мере одного сопряженного диена, с мультивиниламиносиланом Формулы 1. Полимер по изобретению может быть статистическим, блок- или коническим сополимером, или альфа-, или альфа,омега-модифицированным полимером, при этом мультивиниламиносилан Формулы 1 включают в полимерную цепь посредством его винильных функциональных групп. Полученный полимер в целом является разветвленным эластомерным полимером.
В предпочтительных вариантах реализации изобретения полимер по изобретению представляет собой SSBR (растворный бутадиен-стирольный каучук) с содержанием винила предпочтительно 15-80%, более предпочтительно 30-75%, наиболее предпочтительно 40- 70% (в зависимости от конкретной области применения), содержанием стирола (в зависимости от конкретной области применения) в общих количествах 40-70%,мас. или 15-40%,мас. или 1-15%;мас. PBR (полибутадиеновый каучук) с содержанием винила <15%; или 15-40%, или 40-80%; PIR (полиизопреновый каучук); SSIR (растворный стирол-изопреновый каучук); или SSIBR (растворный стирол-изопрен-бутадиеновый каучук); более предпочтительно SSBR или PBR; даже более предпочтительно SSBR, каждый из которых модифицирован посредством включения мультивиниламиносилана Формулы 1. В случае с SSBR, эластомерный полимер характеризуют посредством температуры стеклования (Tg, определяют посредством ДСК (дифференциальной сканирующей калориметрии)) при от -90 до 0 °C, предпочтительно, от -80 до -5 °C, более предпочтительно, от -70 до -10 °C. Наиболее предпочтительно, Tg в области применения для шин грузовых автомобилей составляет от -70 до -40 °C, и наиболее предпочтительно, Tg в области применения для шин легковых автомобилей составляет от -40 до -10 °C.
НЕОТВЕРЖДЕННАЯ ПОЛИМЕРНАЯ КОМПОЗИЦИЯ
Неотвержденная полимерная композиция по седьмому аспекту настоящего изобретения содержит эластомерный полимер по шестому аспекту изобретения и один или несколько дополнительных компонентов, выбранных из: (i) компонентов, которые добавляют к или образуются в результате процесса полимеризации, используемого для получения указанного полимера, (ii) компонентов, которые остаются после удаления растворителя из процесса полимеризации, и (iii) компонентов, которые добавляют к полимеру после завершения процесса изготовления полимера. Более конкретно, такие компоненты от (i) до (iii) могут быть одним или несколькими компонентами, выбранными из масел (масел-наполнителей), наполнителей, стабилизаторов и дополнительных полимеров (которые не являются полимерами по изобретению). В одном из вариантов реализации изобретения полимерная композиция дополнительно содержит одну или несколько вулканизирующих добавок.
В одном из вариантов реализации изобретения неотвержденную (несшитую или невулканизированную) полимерную композицию получают посредством обычной обработки реакционной смеси, полученной в процессе полимеризации. Обработка означает удаление растворителя с использованием технологий перегонки с водяным паром или вакуумного испарения.
В другом варианте реализации изобретения неотвержденную полимерную композицию по изобретению получают в результате дополнительного процесса механического перемешивания, с использованием обработанной реакционной смеси (включая полимер по изобретению), предпочтительно в виде каучукового блока (т.е. продукта обычного процесса компаундирования во внутреннем смесителе и/или посредством двухвальковой мельницы), и по меньшей мере одного наполнителя. Более подробно процесс описан в F. Rothemeyer, F. Sommer, Kautschuk Technologic. Werkstoffe - Verarbeitung - Produkte, 3rd ed., (Hanser Verlag, 2013) и в цитируемых в этом документе ссылках.
В неотвержденные композиции, используемые в шинах, как правило, добавляют следующие компоненты: масла-наполнители, стабилизаторы, наполнители, дополнительные полимеры.
МАСЛА (НАПОЛНИТЕЛИ)
В одном из вариантов реализации изобретения полимерная композиция по настоящему изобретению содержит эластомерный полимер по изобретению в комбинации с одним или несколькими маслами, в частности, минеральными маслами. Репрезентативные примеры и классификацию масел см. в документах WO 2009/148932 и US 2005/0159513, каждый из которых в полном объеме включен в данный документ посредством ссылки. Такие масла содержат, например, общеизвестные масла-наполнители, например, ароматические, нафталеновые и парафиновые масла-наполнители, например MES (сольват слабой экстракции), TDAE (очищенный дистиллированный ароматический экстракт), масла каучук-в-жидкость (RTL), масла биoмасса-в-жидкости (BTL), фактисы, каучук наполнители или жидкие полимеры (например, жидкий BR ), имеющий среднюю молекулярную массу (определенную посредством ГПХ согласно BS ISO 11344:2004) от 500 до 20000 г/моль. В случае, если минеральное масло используют в качестве масла-наполнителя, то предпочтительно это один или несколько выбранных из: DAE (дистилированных ароматических экстрактов), RAE (остаточных ароматических экстрактов), TDAE, MES и нафталиновых масел. Вышеупомянутые масла содержат в различных концентрациях полициклические ароматические соединения, парафиновые, нафтеновые и ароматические соединения и имеют различные температуры стеклования. Вышеупомянутые виды масел были охарактеризованы в "Kautschuk. Gummi Kunststoffe", vol. 52, стр. 799-805. В некоторых вариантах реализации изобретения MES, RAE и TDAE являются предпочтительными маслами-наполнителями для каучука.
До или после окончания процесса полимеризации к полимеру может быть добавлено одно или несколько масел. В случае, если масло-наполнитель добавлено к полимерному раствору, то время осуществления добавления должно быть предпочтительно после модификации полимера или окончания полимеризациии, например, после добавления модифицирующего агента или обрывающего цепь полимеризации агента. После добавления масла-наполнителя наполненная маслом полимерная композиция может быть получена посредством отделения какого-либо полимеризационного растворителя от полимера посредством способа прямой сушки или перегонки с водяным паром, высушивания каучука с использованием вакуумной сушилки, сушилки с горячим воздухом, ролика и т.п.
Полимерная композиция может содержать одно или несколько масел в общем количестве от 0 до 70 м.ч./100 м.ч. каучука, предпочтительно 0,1 до 60 м.ч./100 м.ч. каучука, более предпочтительно 0,1 до 50 м.ч./100 м.ч. каучука. В случае, если жидкие полимеры используют в качестве масел-наполнителей в полимерной композиции по настоящему изобретению, то при расчете не учитывают композицию полимерного матрикса.
В другом варианте реализации изобретения масло добавляют к полимеру «без растворителя» в механическом смесителе по меньшей мере вместе с одним наполнителем, предпочтительно по меньшей мере с одним наполнителем и по меньшей мере с одним дополнительным полимером.
НАПОЛНИТЕЛИ
Полимерная композиция по изобретению, которая, необязательно, содержит одно или несколько масел-наполнителей, как определено выше, может дополнительно содержать один или несколько наполнителей. Наполнитель может быть добавлен к полимеру до или после окончания процесса полимеризации. Примеры подходящих наполнителей включают углеродную сажу (включая электропроводящую углеродную сажу), углеродные нанотрубки (CNT) (включая дискретные CNT, полые углеродные волокна (HCF) и модифицированные CNT, несущие одну или несколько функциональных групп, например, гидроксильную, карбоксильную и карбонильную группы), графит, графен (включая дискретные графеновые пластины), кремнезем, двухслойный углерод-кремнеземный наполнитель, глины (слоистые силикаты, включая отслоившиеся наноглины и органоглины), карбонат кальция, карбонат магния, оксид магния, диоксид титана, каучуковые гели, лигнин, аморфные наполнители, например, основанные на стеклянных частицах наполнители, основанные на крахмале наполнители и их комбинации. Дополнительные примеры подходящих наполнителей описаны в WO 2009/148932, который полностью включен в данный документ посредством ссылки.
Может быть использована углеродная сажа любого типа, обычно известного специалисту в данной области техники. В одном из вариантов реализации изобретения, углеродная сажа имеет йодный индекс согласно ASTM D 1510 от 20 до 250 мг/г, предпочтительно от 30 до 180 мг/г, более предпочтительно от 40 до 180 мг/г, и даже более предпочтительно от 40 до 130 мг/г, и масляное число по ДБФ согласно ASTM D 2414 от 80 до 200 мл/100 г, предпочтительно от 100 до 200 мл/100 г, более предпочтительно от 115 до 200 мл/100 г (масляное число по ДБФ (дибутилфталату) определяет объем избирательного поглощения углеродной сажи или какого-либо яркого наполнителя посредством дибутилфталата).
Может быть использован кремнезем любого типа, обычно известного специалисту в данной области техники и подходящий в качестве наполнителя для смесей шинного каучука. В частности, предпочтительно использовать высокодисперсный, осажденный кремнезем, имеющий удельную поверхность по азоту (удельную поверхность БЭТ; согласно DIN ISO 9277 и DIN 66132) от 35 до 350 м2/г, предпочтительно от 35 до 260 м2/г, более предпочтительно от 100 до 260 м2/г и даже более предпочтительно от 130 до 235 м2/г, и имеющий удельную поверхность по ЦТАБ (цетилтриметиламмоний бромиду)(согласно ASTM D 3765) от 30 до 400 м2/г, предпочтительно от 30 до 250 м2/г, более предпочтительно от 100 до 250 м2/г и даже более предпочтительно от 125 до 230 м2/г. Такой кремнезем, например, в каучуковых смесях для протекторов шин, приводит к особенно полезным физическим свойствам вулканизатов. К тому же, это может привнести преимущества при обработке смеси, а именно, уменьшению времени, требуемого для смешивания, одновременно сохраняя свойства продукции, таким образом, улучшая производительность. Применимые кремнеземы содержат кремнеземы типа Ultraсил® VN3 (торговая марка принадлежит Evonik Industries), а также кремнеземы высоко диспергированного типа, так называемые HD кремнеземы (например, Zeoсил® 1165 MP от Rhodia)
СТАБИЛИЗАТОРЫ
Один или несколько стабилизаторов («противоокислителей») необязательно могут быть добавлены к полимеру до или после окончания процесса полимеризации для предотвращения разрушения эластомерного полимера молекулярным кислородом. Как правило, используют противоокислители, основанные на стерически затрудненных фенолах, например, 2,6-ди-трет-бутил-4-метилфенол, 6,6'-метиленбис-(2-трет-бутил-4-метилфенол), изо-октил-3-(3,5-ди-трет-бутил-4-гидроксифенил)-пропионат, гексаметиленбис-[3-(3,5-ди-трет-бутил-4-гидрокси-фенил)-пропионат], октадецил-3-(3,5-ди-трет-бутил-4-гидроксифенил)-пропионат, изотридецил-3-(3,5-ди-трет-бутил-4-гидроксифенил)-пропионат, 1,3,5-триметил-2,4,6-трис(3,5-ди-трет-бутил-4-гидроксибензил)-бензол, 2,2'-этилиденбис-(4,6-ди-трет-бутилфенол), тетракис-[метилен-3-(3,5-ди-трет-бутил-4-гидроксифенил)-пропионат]-метан, 2-[1-(2-гидрокси-3,5-ди-трет-пентил-фенил)этил]-4, 6-ди-трет-пентилфенилакрилат и 2-трет-бутил-6-(3-трет-бутил-2-гидрокси-5-метилбензил)-4-метилфенилакрилат, а также противоокислители, основанные на тио-эфирах, например, 4,6-бис-(октилтиометил)-o-крезол и пентаэритритил-тетракис-(3-лаурилтиопропионат). Дополнительные примеры подходящих стабилизаторов могут быть обнаружены в F. Röthemeyer, F. Sommer, Kautschuk Technologie, 2nd ed., (Hanser Verlag, 2006) стр. 340-344 и цитируемых в этом документе ссылках.
ДОПОЛНИТЕЛЬНЫЕ ПОЛИМЕРЫ
Помимо полимера по изобретению, масла(ел)-наполнителя(ей), наполнителя(ей) и т.д., полимерная композиция по изобретению может также содержать дополнительный полимер, в частности, дополнительный эластомерный полимер. Дополнительные полимеры могут быть добавлены в качестве раствора к раствору полимера по изобретению до обработки полимерной смеси или могут быть добавлены в процессе механического перемешивания, например, в смеситель типа Брабендер.
Дополнительные (эластомерные) полимеры, как указано в данном документе, представляют собой эластомерные полимеры, которые не соответствуют полимеру по изобретению, т.е. которые не содержат повторяющихся звеньев, извлеченных из мультивиниламиносилана Формулы 1.
ВУЛКАНИЗИРУЮЩИЕ ДОБАВКИ И УСКОРИТЕЛИ ВУЛКАНИЗАЦИИ
Полимерная композиция по изобретению, необязательно, может дополнительно содержать по меньшей мере одну вулканизирующую добавку. В изобретении может быть использована любая вулканизирующая добавка, обычно используемая в изготовлении каучуковой продукции, а также может быть использована комбинация из двух или нескольких вулканизирующих добавок.
Сера, серосодержащие соединения, действующие как доноры серы, например, дитиолы, системы серных ускорителей и пероксиды являются наиболее распространенными вулканизирующими добавками. Примеры серосодержащих соединений, действующих как доноры серы, включают дитиодиморфолин (DTDM), тетраметилтиурамдисульфид (TMTD), тетраэтилтиурамдисульфид (TETD) и дипентаметилентиурамтетрасульфид (DPTT). Примеры серных ускорителей включают производные амина, производные гуанидина, продукты альдегидаминовой конденсации, тиазолы, ксантогенаты, тиурамсульфиды, дитиокарбаматы и тиофосфаты. Предпочтительно использовать один или несколько сульфонамидных ускорителей, выбранных из: N-циклогексил-2-бензотиазола сульфенамида (CBS), N,N-дициклогексилбензотиазол-2-сульфенамида (DCBS), бензотиазил-2-сульфенморфолида (MBS) и N-трет-бутил-2-бензотиазилсульфенамида (TBBS). К полимерной композиции могут быть добавлены дополнительные сшивающие системы, например, доступные под торговыми марками Vulkuren® (1,6-бис(N,N-дибензилтиокарбамоилдитио)-гексан; Lanxess), Duralink® или Perkalink® (1,3-бис-(цитраконимидометил)-бензол; Lanxess) или описанные в WO 2010/049261. Примеры пероксидов включают ди-трет-бутил-пероксиды, ди-(трет-бутил-перокси-триметил-циклогексан), ди-(трет.-бутил-перокси-изопропил-)-бензол, дихлор-бензоилпероксид, дикумилпероксиды, трет.-бутил-кумил-пероксид, диметил-ди-(трет.-бутил-перокси)-гексан, диметил-ди-(трет.-бутил-перокси)-гексан и бутил-ди-(трет.-бутил-перокси)валериат (Rubber Handbook, SGF, The Swedish Institution of Rubber Technolgy 2000).
Ускоритель вулканизации сульфенамидного типа, гуанидинового типа или тиурамового типа может быть использован вместе с вулканизирующей добавкой по требованию.
К тому же, полимерная композиция по изобретению может содержать обычные добавки и вспомогательные материалы для вулканизации в обычно используемых пропорциях. Такие добавки содержат:
a) ингибиторы состаривания, например, N-фенил N'-(1,3-диметилбутил)-п-фенилендиамин (6PPD), N,N'-дифенил-п-фенилендиамин (DPPD), N,N'-дитолил-п-фенилендиамин (DTPD), N-изопропил N'-фенил-п-фенилендиамин (IPPD), 2,2,4-триметил 1,2-дигидрохинолин (TMQ),
b) активаторы, например, оксид цинка и жирные кислоты (например, стеариновую кислоту),
c) воски,
d) смолы, в частности, адгезионные смолы,
e) добавки пластикации, например, 2,2,-дибензамидодифенилдисульфид (DBD) и
f) технологические добавки, например, цинковые соли жирных кислот и эфиры жирных кислот и их производные. Оксид цинка (цинковые белила) предпочтительно используют в качестве компонента серной ускоряющей системы.
Вулканизирующую добавку, как правило, добавляют к полимерной композиции в количестве от 0,5 до 10 частей по массе или, в некоторых вариантах реализации изобретения, от 1 до 6 частей по массе на 100 частей по массе общего полимера. Примеры ускорителей вулканизации и их количества, добавленные по отношению к общему объему полимера, даны в WO 2009/148932, который включен в данный документ в полном объеме посредством ссылки.
ВУЛКАНИЗИРОВАННАЯ ПОЛИМЕРНАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ СИНТЕЗА
Вулканизированную полимерную композицию по восьмому аспекту изобретения получают посредством вулканизации полимерной композиции по седьмому аспекту изобретения, содержащей одну или несколько вулканизирующих добавок, в условиях и в устройствах, обычно известных в данной области техники. Процесс вулканизации составляет девятый аспект настоящего изобретения.
ИЗДЕЛИЕ, СОДЕРЖАЩЕЕ ВУЛКАНИЗИРОВАННУЮ ПОЛИМЕРНУЮ КОМПОЗИЦИЮ
Поскольку вулканизированные полимерные композиции по изобретению обладают низким сопротивлением качению, низкой динамикой теплообразования и повышенным сцеплением шины с мокрым дорожным покрытием, они хорошо подходят для использования в изготовлении, например, шин или частей шин, включая, например, протекторы шин, боковины покрышек и каркасы шин, а также другую промышленную продукцию, например, ремни, шланги, виброгасители и компоненты обуви. Таким образом, изделие по десятому аспекту настоящего изобретения содержит по меньшей мере один компонент, образованный из вулканизированной полимерной композиции по изобретению. Изделие может быть, например, шиной, протектором шины, боковиной покрышки, каркасом шины, ремнем, прокладкой, уплотнением, шлангом, виброгасителем, мячом для игры в гольф или компонентом обуви, например, обувной подошвой.
ОПРЕДЕЛЕНИЯ
Алкильные группы, как определено в настоящем описании, сами по себе или связанные с другими группами, например, с алкиларильной или алкокси группой, содержат одновременно прямоцепные алкильные группы, например, метил (Me), этил (Et), н-пропил (Pr), н-бутил (Bu), н-пентил, н-гексил, и т.д., разветвленные алкильные группы, например, изопропил, трет-бутил и т.д. и циклические алкильные группы, например, циклогексил.
Арильные группы, как определено в настоящем описании, содержат фенил, бифенил и другие бензоидные соединения. Арильные группы предпочтительно содержат только одно ароматическое кольцо и наиболее предпочтительно, содержат C6 ароматическое кольцо.
Алкиларильные группы, как определено в настоящем описании, относятся к комбинации одной или нескольких арильных групп, связанных с одной или несколькими алкильными группами, например, в виде алкил-арил, арил-алкил, алкил-арил-алкил и арил-алкил-арил. Алкиларильные группы предпочтительно содержат только одно ароматическое кольцо, наиболее предпочтительно, содержат C6 ароматическое кольцо.
Настоящее изобретение раскрыто более подробно посредством не ограничивающих настоящее изобретение примеров.
ПРИМЕРЫ
Получения и характеристика разветвляющих агентов (BA) и выбранных модификаторов)
Соединения настоящего изобретения отмечены звездочкой (*).
N-[диметил(винил)силил]-N-(4-метоксифенил)-1,1-диметил-1-винилсиланамин
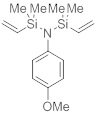 п-Метоксианилин (4,22 г, 34,3 ммоль, 1,0 экв.) добавили к раствору ТЭА (8,68 г, 85,8 ммоль, 2,5 экв.) и хлордиметилвинилсилана (10,3 г,
п-Метоксианилин (4,22 г, 34,3 ммоль, 1,0 экв.) добавили к раствору ТЭА (8,68 г, 85,8 ммоль, 2,5 экв.) и хлордиметилвинилсилана (10,3 г,
85,8 ммоль, 2,5 экв.) в ДХМ (60 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 16 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (7,80 г, 26,8 ммоль, 78%) в виде бесцветного масла.
C15H25NOSi2, Мw=291,54 г/моль
Ткип=130-132°C (6 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,87-6,85 (m, 2 H), 6,70-6,67 (m, 2 H), 6,29 (dd, J=20,3 Гц, J=14,7 Гц, 2 H), 5,91 (dd, J= 14,7 Гц, J= 3,7 Гц, 2 H), 5,72 (dd, J= 20,4 Гц, J=3,8 Гц, 2 H), 3,29 (s, 3 H), 0,19 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ= 156,88 (C), 140,83 (2 CH), 139,74 (C), 131,60 (2 CH2), 131,19 (2 CH), 114,15 (2 CH), 54,77 (CH3), 0,40 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=291 (M+. 76), 276 (M+-CH3, 100), 260 (22), 248 (67), 222 (41), 176 (21), 134 (8), 85 (17).
N-[диметил(винил)силил]-N-(2-метоксиэтил)-1,1-диметил-1-винилсиланамин
 Хлордиметилвинилсилан (20,1 г, 166 ммоль, 2,5 экв.) добавляли по каплям к раствору метоксиэтиламина (5,00 г, 66,6 ммоль, 1,0 экв.) и ТЭА (16,8 г, 166 ммоль, 2,5 экв.) в ДХМ (60 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (11,0 г, 45,2 ммоль, 68%) в виде бесцветного масла.
Хлордиметилвинилсилан (20,1 г, 166 ммоль, 2,5 экв.) добавляли по каплям к раствору метоксиэтиламина (5,00 г, 66,6 ммоль, 1,0 экв.) и ТЭА (16,8 г, 166 ммоль, 2,5 экв.) в ДХМ (60 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 16 часов. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (11,0 г, 45,2 ммоль, 68%) в виде бесцветного масла.
C11H25NOSi2, Mw=243,50 г/моль
Ткип=83-84°C (7 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,25 (dd, J=20,3 Гц, J= 14,6 Гц, 2 H), 5,90 (dd, J=14,7 Гц, J=3,8 Гц, 2 H), 5,69 (dd, J= 20,3 Гц, J= 3,8 Гц, 2 H), 3,21 (t, J=6,6 Гц, 2 H), 3,09 (s, 3 H), 3,08 (t, J=6,5 Гц, 2 H), 0,23 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=141,10 (2 CH), 131,50 (2 CH2), 75,78 (CH2), 58,59 (CH3), 44,90 (CH2), 0,52 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=243 (M+-CH3, 2), 198 (100), 170 (6), 142 (1), 112 (6), 85 (24), 59 (25).
N,N-бис[диметил(винил)силил]-О-метилгидроксиламин
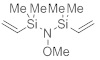 Метоксиамингидрохлорид (4,83 г, 57,8 ммоль, 1,0 экв.) суспендировали в ДХМ (150 мл) при комнатной температуре. Затем добавили ТЭА (17,6 г, 174 ммоль, 3,0 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (16,0 г, 133 ммоль, 2,3 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C втечение 4 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (4,64 г, 21,6 ммоль, 37%) в виде бесцветного масла.
Метоксиамингидрохлорид (4,83 г, 57,8 ммоль, 1,0 экв.) суспендировали в ДХМ (150 мл) при комнатной температуре. Затем добавили ТЭА (17,6 г, 174 ммоль, 3,0 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (16,0 г, 133 ммоль, 2,3 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C втечение 4 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (4,64 г, 21,6 ммоль, 37%) в виде бесцветного масла.
C9H21NOSi2, Mw=215,44 г/моль
Ткип=89-90°C (40 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,28 (dd, J=20,4 Гц, J=14,7 Гц, 2 H), 5,92 (dd, J=14,8 Гц, J=3,8 Гц, 2 H), 5,75 (dd, J= 20,4 Гц, J= 3,8 Гц, 2 H), 3,37 (s, 3 H), 0,23 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=139,23 (2 CH), 132,27 (2 CH2), 65,30 (CH3), -1,33 (4 СНз) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=215 (M+, 100), 200 (M+-CH3, 1), 186 (8), 170 (53), 142 (56), 116(41), 85 (60), 59 (86).
N-[диметил(винил)силил]-1,1-диметил-N-(пиридин-3-илметил)-1-винилсиланамин
 2-Аминометилпиридин (3,92 г, 36,2 ммоль, 1,0 экв.) добавили к раствору ТЭА (9,16 г, 90,5 ммоль, 2,5 экв.) и хлордиметилвинилсилана (10,9 г, 90,5 ммоль, 2,5 экв.) в ДХМ (60 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 5 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (8,78 г, 31,8 ммоль, 88%) в виде бесцветного масла.
2-Аминометилпиридин (3,92 г, 36,2 ммоль, 1,0 экв.) добавили к раствору ТЭА (9,16 г, 90,5 ммоль, 2,5 экв.) и хлордиметилвинилсилана (10,9 г, 90,5 ммоль, 2,5 экв.) в ДХМ (60 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 5 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (8,78 г, 31,8 ммоль, 88%) в виде бесцветного масла.
C14H24N2Si2, Mw=276,53 г/моль
Ткип=110-112°C (7 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=8,67 (dd, J=2,2 Гц, J= 0,8 Гц, 1 H), 8,49-8,47 (m, 1 H), 7,32-7,29 (m, 1 H), 6,78 (ddd, J= 7,8 Гц, J= 4,8 Гц, J= 0,4 Гц, 1 H), 6,13 (dd, J= 20,3 Гц, J= 14,7 Гц, 2 H), 5,83 (dd, J= 14,7 Гц, J= 3,8 Гц, 2 H), 5,61 (dd, J= 20,3 Гц, J= 3,8 Гц, 2 H), 3,89 (s, 2 H), 0,11 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6); δ=149,33 (CH), 148,38 (CH), 140,12 (2 CH), 138,82 (C), 133,70 (CH), 132,02 (2 CH2), 122,88 (CH), 46,79 (CH2), 0,18 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=276 (M+, 30), 261 (M+-CH3, 100), 233 (15), 198 (32), 163 (28), 136 (12), 116 (10), 85 (35), 59 (53).
2,4-диметил-1,3-дипропил-2,4-дивинил-1,3,2,4-диазадисилетидин
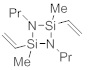 1-Метил-NN'-дипропил-1-винилсиландиамин (2,00 г, 10,7 ммоль, 1,0 экв.) растворили в МТБЭ (25 мл) при 25 °C. После этого добавляли раствор н-BuLi (7,05 г, 22,0 ммоль, 2,05 экв.) в циклогексане (20 мас.%) в течение 30 мин. Через час добавили раствор хлордиметилвинилсилана (1,51 г, 10,7 ммоль, 1,0 экв.) в МТБЭ (10 мл) и перемешивали полученную смесь дополнительно в течение 1 ч при 25 °C. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (1,0 г, 3,9 ммоль, 37%) в виде бесцветного масла.
1-Метил-NN'-дипропил-1-винилсиландиамин (2,00 г, 10,7 ммоль, 1,0 экв.) растворили в МТБЭ (25 мл) при 25 °C. После этого добавляли раствор н-BuLi (7,05 г, 22,0 ммоль, 2,05 экв.) в циклогексане (20 мас.%) в течение 30 мин. Через час добавили раствор хлордиметилвинилсилана (1,51 г, 10,7 ммоль, 1,0 экв.) в МТБЭ (10 мл) и перемешивали полученную смесь дополнительно в течение 1 ч при 25 °C. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (1,0 г, 3,9 ммоль, 37%) в виде бесцветного масла.
C12H26N2Si2, Mw=254,52 г/моль
Ткип=78-80°C (6 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,21 (ddd, J= 20,2 Гц, J=14,6 Гц, J=10,4 Гц, 2 H) часть ABX-системы, 6,02 (ddd, J=14,7 Гц, J=5,2 Гц, J= 4,1 Гц, 2 H) часть ABX-системы, 5,87 (ddd, J=20,2 Гц, J=7,7 Гц, J=4,1 Гц, 2 H) часть ABX-системы, 2,78-2,73 (m, 4 H), 1,41 (sext, J= 7,2 Гц, 4 H), 0,84 (t, J= 7,2 Гц, 6 H), 0,40 (s, 3 H), 0,38 (s, 3 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=140,12 (CH), 139,97 (CH), 134,22 (CH2), 134,18 (CH2), 43,98 (2 CH2) 28,21 (2 CH2), 11,99 (2 CH3), -0,72 (CH3) -0,92 (CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=254 (M+, 1), 225 (M+ 100), 198 (29), 170 (6), 128 (7), 98 (24), 85 (8), 71 (27).
1,3-бис[диметил(винил)силил]-2,2-диметил-1,3,2-диазасилан (*)
 Раствор 1,3-диаминопропана (3,72 г, 50,2 ммоль, 1,0 экв.) в 30 мл ДХМ добавили по каплям к раствору ТЭА (21,8 г, 216ммоль, 4,3 экв.) и дихлордиметилсилана (6,51 г, 50,2 ммоль, 1,0 экв.) в ДХМ (70 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 16 ч. Затем добавили хлордиметилвинилсилан (12,1 г, 100 ммоль, 2,0 экв.) и полученную суспензию перемешивали при 25°C дополнительно в течение16 ч. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (11,1 г, 37,2 ммоль, 74%) в виде бесцветного масла.
Раствор 1,3-диаминопропана (3,72 г, 50,2 ммоль, 1,0 экв.) в 30 мл ДХМ добавили по каплям к раствору ТЭА (21,8 г, 216ммоль, 4,3 экв.) и дихлордиметилсилана (6,51 г, 50,2 ммоль, 1,0 экв.) в ДХМ (70 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 16 ч. Затем добавили хлордиметилвинилсилан (12,1 г, 100 ммоль, 2,0 экв.) и полученную суспензию перемешивали при 25°C дополнительно в течение16 ч. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (11,1 г, 37,2 ммоль, 74%) в виде бесцветного масла.
C13H30N2Si3, Mw=298,65 г/моль
Ткип=115-117°C (7 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,21 (dd, J=20,3 Гц, J =14,7 Гц, 2 H), 5,93 (dd, J=14,6 Гц, J= 4,0 Гц, 2 H), 5,74 (dd, J=20,3 Гц, J=4,0 Гц, 2 H), 2,98-2,95 (m, 4 H), 1,49-1,43 (m, 2 H), 0,30 (s, 6 H), 0,19 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=140,67 (2 CH), 131,55 (2 CH2) 42,50 (2 CH2), 31,93 (CH2), 3,96 (2 CH3), -0,24 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=298 (M+, 14), 283 (M+-CH3, 100), 255 (8), 228 (7), 184 (13), 158(8), 130(5), 100 (1).
l,2-бис{3-[диметил(винил)силил]-2,2-диметил-1,3,2-диазасилoлидин-1-ил}этан (*)
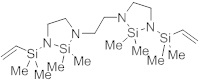 Хлордиметилвинилсилан (4,3 г, 35,9 ммоль, 2,1 экв.) добавили к раствору триэтилентетрамина (2,50 г, 17,1 ммоль, 1,0 экв.) и ТЭА (11,2 г, 111 ммоль, 6,5 экв.) в ДХМ (50 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 1 ч. Затем добавили дихлордиметилсилан (4,41 г, 34,2 ммоль, 2,0 экв.) и полученную суспензию перемешивали при 25°C дополнительно в течение 1 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (772 мг, 1,81 ммоль, 11%) в виде слегка желтого, очень вязкого масла.
Хлордиметилвинилсилан (4,3 г, 35,9 ммоль, 2,1 экв.) добавили к раствору триэтилентетрамина (2,50 г, 17,1 ммоль, 1,0 экв.) и ТЭА (11,2 г, 111 ммоль, 6,5 экв.) в ДХМ (50 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 1 ч. Затем добавили дихлордиметилсилан (4,41 г, 34,2 ммоль, 2,0 экв.) и полученную суспензию перемешивали при 25°C дополнительно в течение 1 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение (772 мг, 1,81 ммоль, 11%) в виде слегка желтого, очень вязкого масла.
C18H42N4Si4, Mw=426,90 г/моль
Ткип=152-154°C (1,7×10-2 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,20 (dd, J=20,2 Гц, J= 14,7 Гц, 2 H), 5,96 (dd, J=14,6 Гц, J=4,0 Гц, 2 H), 5,76 (dd, J=20,2 Гц, J=4,0 Гц, 2 H), 3,06 (t, J=6,2 Гц, 4 H), 2,95-2,85 (m, 8 H), 0,21 (s, 12 H), 0,20 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=139,54 (2 CH), 132,04 (2 CH2), 50,75 (2 CH2), 48,10 (2 CH2), 45,30 (2 CH2), 1,77 (4 CH3), -1,48 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=426 (M+, 4), 411 (M+-CH3, 3), 320 (9), 278 (2), 213 (100), 199 (1), 185 (2), 171 (2), 157 (1), 142 (3), 100 (4), 85 (6).
1,4-бис[диметил(винил)силил]пиперазин (M1) (*)
 Пиперазин (8,00 г, 92,9 ммоль) растворили в ДХМ (150 мл) при комнатной температуре. Добавили ТЭА (21,6 г, 214 ммоль, 2,3 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (25,8 г, 214 ммоль, 2,3 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 7 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение М1 (17,4 г, 68,4 ммоль, 74%) в виде бесцветного масла.
Пиперазин (8,00 г, 92,9 ммоль) растворили в ДХМ (150 мл) при комнатной температуре. Добавили ТЭА (21,6 г, 214 ммоль, 2,3 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (25,8 г, 214 ммоль, 2,3 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 7 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение М1 (17,4 г, 68,4 ммоль, 74%) в виде бесцветного масла.
C12H26N2Si2, Mw=254,52 г/моль
Ткип=92-93°C (5 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,14 (dd, J=20,2 Гц, J=14,7 Гц, 2 H), 5,94 (dd, J=14,7 Гц, J=4,1 Гц, 2 H), 5,73 (dd, J= 20,2 Гц, J= 4,1 Гц, 2 H), 2,97 (s, 8 H), 0,12 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=139,34 (2 CH), 132,15 (2 CH2), 47,36 (4 CH2), -2,43 (4 СНз) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=254 (M+, 100), 239 (M+-CH3, 9), 210 (2), 170 (9), 140 (20), 85 (44).
N,N-бис[диметил(винил)силил]бензиламин (М2)
 Бензиламин (6,00 г, 56,0 ммоль, 1,0 экв.) растворили в ДХМ (110 мл) при комнатной температуре. Добавили ТЭА (14,2 г, 140 ммоль, 2,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (16,9 г, 140 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение М2 (8,96 г, 32,5 ммоль, 58%) в виде бесцветного масла.
Бензиламин (6,00 г, 56,0 ммоль, 1,0 экв.) растворили в ДХМ (110 мл) при комнатной температуре. Добавили ТЭА (14,2 г, 140 ммоль, 2,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (16,9 г, 140 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение М2 (8,96 г, 32,5 ммоль, 58%) в виде бесцветного масла.
C15H25NSi2, Mw=275,54 г/моль
Ткип=109-113°C (5 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=7,28-7,24 (m, 2 H), 7,21-7,17 (m, 2 H), 7,07 (t, J= 7,5 Гц, 1 H), 6,23 (dd, 20,4 Гц, J= 14,7 Гц, 2 H), 5,87 (dd, J=14,7 Гц, J=3,8 Гц, 2 H), 5,66 (dd, J=20,3 Гц, J= 3,8 Гц, 2 H), 4,08 (s, 2 H), 0,19 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=144,14 (C), 140,61 (2 CH), 131,71 (2 CH2), 128,34 (2 CH), 126,75 (2 CH), 126,52 (CH), 49,14 (CH2), 0,29 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=275 (M+, 14), 260 (M+-CH3, 100), 232 (14), 198 (66), 162 (25), 135 (23), 116 (10), 85 (41), 59 (64).
N,N-бис[диметил(винил)силил]толуидин (М3)
 Толуидин (3,89 г, 36,3 ммоль, 1,0 экв.) и ТЭА (9,18 г, 90,7 ммоль, 2,5 экв.) растворили в ДХМ (60 мл) при комнатной температуре. Затем добавили по каплям хлордиметилвинилсилан (10,9 г, 90,7 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Затем добавили вторую порцию хлордиметилвинилсилана (2,20 г, 18,2 ммоль, 0,5 экв.) и полученную смесь перемешивали дополнительно в течение 6 дн. при 25 °C. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение М3 (7,70 г, 27,9 ммоль, 77%) в виде бесцветного масла.
Толуидин (3,89 г, 36,3 ммоль, 1,0 экв.) и ТЭА (9,18 г, 90,7 ммоль, 2,5 экв.) растворили в ДХМ (60 мл) при комнатной температуре. Затем добавили по каплям хлордиметилвинилсилан (10,9 г, 90,7 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Затем добавили вторую порцию хлордиметилвинилсилана (2,20 г, 18,2 ммоль, 0,5 экв.) и полученную смесь перемешивали дополнительно в течение 6 дн. при 25 °C. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение М3 (7,70 г, 27,9 ммоль, 77%) в виде бесцветного масла.
C15H25NSi2, Mw=275,54 г/моль
Ткип=100-102°C (4 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,92-6,88 (m, 4 H), 6,29 (dd, J=20,4 Гц, J=14,7 Гц, 2 H), 5,90 (dd, J= 14,7 Гц, J= 3,7 Гц, 2 H), 5,71 (dd, J=20,3 Гц, J= 3,8 Гц, 2 H), 2,09 (s, 3 H), 0,18 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=144,65 (C), 140,83 (2 CH), 133,39 (C), 131,62 (2 CH2), 130,27 (2 CH), 129,60 (2 CH), 20,84 (CH3), 0,44 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=275 (M+, 74), 260 (М+-СН3, 99), 246 (23), 232 (100), 222 (60), 206 (55), 174 (21), 148 (13), 121 (5), 85 (32), 59 (91).
N,N,N',N'-тетракис[диметил(винил)силил-1,3-диаминопропан (M4) (*)
 1,3-Диаминопропан (1,50 г, 20,2 ммоль, 1,0 экв.) добавили по каплям к раствору ТЭА (12,3 г, 121,4 ммоль, 6,0 экв.) и хлордиметилвинилсилана (13,1 г, 101,2 ммоль, 5,0 экв.) в ДХМ (70 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 4 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M4 (7,15 г, 17,4 ммоль, 86%) в виде бесцветного масла.
1,3-Диаминопропан (1,50 г, 20,2 ммоль, 1,0 экв.) добавили по каплям к раствору ТЭА (12,3 г, 121,4 ммоль, 6,0 экв.) и хлордиметилвинилсилана (13,1 г, 101,2 ммоль, 5,0 экв.) в ДХМ (70 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 4 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M4 (7,15 г, 17,4 ммоль, 86%) в виде бесцветного масла.
C19H42N2Si4, Mw=410,90 г/моль
Ткип=144-147°C (3×10-2 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,27 (dd, J=20,6 Гц, J= 14,6 Гц, 4 H), 5,94 (dd, J=14,6 Гц, J= 3,8 Гц, 4 H), 5,73 (dd, J= 20,3 Гц, J=3,9 Гц, 4 H), 2,67-2,63 (m, 4 H), 1,70- 1,62 (m, 2 H), 0,26 (s, 24 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=141,04 (4 CH), 131,55 (4 CH2), 43,24 (2 CH2), 41,34 (CH2), 0,53 (8 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=410 (M+, 64), 395 (M+-CH3, 12), 309 (3), 198 (100), 142 (6), 85 (26).
N,N-бис[диметил(винил)силил]метиламин (M5)
 Хлордиметилвинилсилан (30,2 г, 251 ммоль, 2,3 экв.) растворили в ДХМ (120 мл) при комнатной температуре. Затем добавили метиламмонийхлорид (6,77 г, 100 ммоль, 1,0 экв.) с последующим добавлением по каплям раствора ТЭА (36,5 г, 361 ммоль, 3,6 экв.) в ДХМ (30 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M5 (13,9 г, 697 ммоль, 70%) в виде бесцветного масла.
Хлордиметилвинилсилан (30,2 г, 251 ммоль, 2,3 экв.) растворили в ДХМ (120 мл) при комнатной температуре. Затем добавили метиламмонийхлорид (6,77 г, 100 ммоль, 1,0 экв.) с последующим добавлением по каплям раствора ТЭА (36,5 г, 361 ммоль, 3,6 экв.) в ДХМ (30 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M5 (13,9 г, 697 ммоль, 70%) в виде бесцветного масла.
C9H21NSi2, Mw=199,44 г/моль
Ткип=75-76°C (30 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,20 (dd, J= 20,2 Гц, J=14,6 Гц, 2 H), 5,92 (dd, J=14,6 Гц, J=3,9 Гц, 2 H), 5,70 (dd, J= 20,3 Гц, 4,0 Гц, 2 H), 2,42 (s, 3 H), 0,19 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=140,52 (2 CH), 131,49 (2 CH2), 31,46 (CH3), -0,26 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=199 (M+, 16), 185 (M+-CH3, 100), 172 (14), 156 (48), 144 (15), 130 (15), 116 (10), 103 (4), 73 (21), 59 (51).
N,N-бис[диметил(винил)силил]пропиламин (M6)
 Пропиламин (4,00 г, 67,7 ммоль, 1,0 экв.) растворили в ДХМ (120 мл) при комнатной температуре. Добавили ТЭА (17,1 г, 169 ммоль, 2,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (20,4 г, 169 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 18 ч. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M6 (10,3 г, 45,3 ммоль, 67%) в виде бесцветного масла.
Пропиламин (4,00 г, 67,7 ммоль, 1,0 экв.) растворили в ДХМ (120 мл) при комнатной температуре. Добавили ТЭА (17,1 г, 169 ммоль, 2,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (20,4 г, 169 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 18 ч. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M6 (10,3 г, 45,3 ммоль, 67%) в виде бесцветного масла.
C11H25NSi2, Mw=227,50 г/моль
Ткип=75-76°C (10 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,24 (dd, J= 20,3 Гц, J= 14,6 Гц, 2 H), 5,91 (dd, J= 14,6 Гц, J=3,9 Гц, 2 H), 5,70 (dd, J= 20,3 Гц, 3,9 Гц, 2 H), 2,75-2,71 (m, 2 H), 1,45- 1,35 (m, 2 H), 0,71 (t, J=7,4 Гц, 3 H), 0,22 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=141,21 (2 CH), 131,36 (2 CH2), 47,98 (CH2), 28,65 (CH2), 11,35 (CH3), 0,53 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=227 (M+, 0,8), 212 (M+-CH3, 5), 198 (100), 184 (3), 170 (7), 156 (2), 142 (3), 114(15), 100 (7), 85 (50), 59 (57).
N,N-бис[диметил(винил)силил)бутиламин (M7)
 Бутиламин (5,00 г, 68,4 ммоль, 1,0 экв.) растворили в ДХМ (120 мл) при комнатной температуре. Добавили ТЭА (17,3 г, 170 ммоль, 2,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (20,6 г, 170 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 2 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Указанное в заголовке соединение M7 (8,87 г, 36,7 ммоль, 54%) получили в виде бесцветного масла.
Бутиламин (5,00 г, 68,4 ммоль, 1,0 экв.) растворили в ДХМ (120 мл) при комнатной температуре. Добавили ТЭА (17,3 г, 170 ммоль, 2,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (20,6 г, 170 ммоль, 2,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 2 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Указанное в заголовке соединение M7 (8,87 г, 36,7 ммоль, 54%) получили в виде бесцветного масла.
C12H27NSi2, Mw=241,52 г/моль
Ткип=75-76°C (6 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,26 (dd, J= 20,3 Гц, J= 14,7 Гц, 2 H), 5,92 (dd, J=14,7 Гц, J=3,9 Гц, 2 H), 5,72 (dd, J=20,2 Гц, J=3,8 Гц, 2 H), 2,82-2,78 (m, 2 H), 1,46-1,39 (m, 2 H), 1,14 (sext, J= 7,5 Гц, 2 H), 0,84 (t, J= 7,4 Гц, 3 H), 0,24 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=141,24 (2 CH), 131,38 (2 CH2), 45,83 (CH2), 37,85 (CH2), 20,58 (CH2), 14,20 (CH3), 0,56 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=241 (M+, 2), 226 (M+-CH3, 17), 198 (100), 170 (21), 154 (6), 128 (32), 112 (12), 85 (100), 59 (100).
N,N-бис[диметил(винил)силил]-2,2-диметил-2,3-дигидро-1Н-benzo[d] [1,3,2]диазасилoл (M8) (*)
 Дихлордиметилсилан (4,02 г, 31,0 ммоль, 1,0 экв.) добавили по каплям к раствору o-фенилендиамина (3,35 г, 31,0 ммоль, 1,0 экв.) и ТЭА (15,7 г, 155 ммоль, 5,0 экв.) в ДХМ (100 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 16 ч. После этого добавили хлордиметилвинилсилан (9,35 г, 77,5 ммоль, 2,5 экв.) и полученную суспензию перемешивали при 25°C дополнительно в течение 3 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M8 (7,20 г, 21,6 ммоль, 70%) в виде бесцветного масла.
Дихлордиметилсилан (4,02 г, 31,0 ммоль, 1,0 экв.) добавили по каплям к раствору o-фенилендиамина (3,35 г, 31,0 ммоль, 1,0 экв.) и ТЭА (15,7 г, 155 ммоль, 5,0 экв.) в ДХМ (100 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 16 ч. После этого добавили хлордиметилвинилсилан (9,35 г, 77,5 ммоль, 2,5 экв.) и полученную суспензию перемешивали при 25°C дополнительно в течение 3 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M8 (7,20 г, 21,6 ммоль, 70%) в виде бесцветного масла.
C16H28N2Si3, Mw=332,67 г/моль
Ткип=135°C (1,3 X 10-2 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,98 (dd, J= 5,8 Гц, J= 3,4 Гц, 2 H), 6,78 (dd, J= 5,8 Гц, J=3,4 Гц, 2 H), 6,24 (dd, J= 20,3 Гц, J= 14,7 Гц, 2 H), 5,90 (dd, J=14,7 Гц, J= 3,6 Гц, 2 H), 5,73 (dd, J= 20,3 Гц, J= 3,6 Гц, 2 H), 0,34 (s, 6 H), 0,30 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=142,45 (2 C), 138,09 (2 CH), 133,16 (2 CH2), 118,51 (2 CH), 114,16 (2 CH), 5,54 (2 CH3), -1,36 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=275 (M+, 14), 260 (M+-CH3, 100), 232 (14), 198 (66), 162 (25), 135 (23), 116 (10), 85 (41), 59 (64).
N,N,N',N'-тетракис[диметил(винил)силил]-1,2-диаминоэтан (M9) (*)
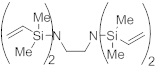 Этилендиамин (1,00 г, 16,6 ммоль) растворили в ДХМ (50 мл) при комнатной температуре. Добавили ТЭА (10,1 г, 99,8 ммоль, 6,0 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (12,0 г, 99,8 ммоль, 6,0 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 2 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M9 (3,71 г, 9,35 ммоль, 56%) в виде бесцветного масла.
Этилендиамин (1,00 г, 16,6 ммоль) растворили в ДХМ (50 мл) при комнатной температуре. Добавили ТЭА (10,1 г, 99,8 ммоль, 6,0 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (12,0 г, 99,8 ммоль, 6,0 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 2 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M9 (3,71 г, 9,35 ммоль, 56%) в виде бесцветного масла.
C18H40N2Si4, Mw=396,87 г/моль
Ткип=155-157°C (1,7 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,28 (dd, J= 20,4 Гц, J=14,7 Гц, 4 H), 5,92 (dd, J=14,7 Гц, J=3,8 Гц, 4 H), 5,71 (dd, J= 20,3 Гц, J=3,8 Гц, 4 H), 2,95 (s, 4 H), 0,28 (s, 24 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=141,59 (4 CH), 131,66 (4 CH2), 49,41 (2 CH2), 1,50 (8 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=396 (M+, 0,8), 381 (M+-CH3, 3), 369 (1), 295 (6), 283 (8), 269 (14), 200 (100), 170 (31), 142 (14), 112 (19), 85 (100), 59 (100).
Трис[диметил(винил)силил]амин (M10)
 Бис[диметил(винил)силил]амин (6,50 г, 35,1 ммоль, 1,0 экв.) растворили в ТГФ (60 мл) при 25 °C. Затем добавили раствор н-BuLi (11,8 г, 36,8 ммоль, 1,05 экв.) в циклогексане (20 мас.%). Через 1 ч при этой температуре добавили хлордиметилвинилсилан (5,29 г, 43,8 ммоль, 1,25 экв.) и полученную смесь перемешивали дополнительно в течение 16 ч при 25 °C. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M10 (7,00 г, 26,0 ммоль, 74%) в виде липкого масла/твердого вещества.
Бис[диметил(винил)силил]амин (6,50 г, 35,1 ммоль, 1,0 экв.) растворили в ТГФ (60 мл) при 25 °C. Затем добавили раствор н-BuLi (11,8 г, 36,8 ммоль, 1,05 экв.) в циклогексане (20 мас.%). Через 1 ч при этой температуре добавили хлордиметилвинилсилан (5,29 г, 43,8 ммоль, 1,25 экв.) и полученную смесь перемешивали дополнительно в течение 16 ч при 25 °C. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M10 (7,00 г, 26,0 ммоль, 74%) в виде липкого масла/твердого вещества.
C12H27NSi3, Mw=269,61 г/моль
Ткип=85°C (4 мбар).
ЯМР 1H (400 МГц, 20 °C, CDCl3): δ=6,26 (dd, J= 20,3 Гц, J= 14,7 Гц, 3 H), 5,88 (dd, J= 14,7 Гц, J= 3,6 Гц, 3 H), 5,66 (dd, J= 20,4 Гц, J= 3,6 Гц, 3 H), 0,24 (s, 18 H) м.д.
ЯМР 13C (100 МГц, 20 °C, CDCl3): δ=143,27 (3 CH), 130,40 (3 CH2), 3,84 (6 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=369 (M+, 0,5), 254 (M+-CH3,100), 226 (M+, 65), 200 (62), 172 (18), 154 (9), 130 (32), 100 (30), 59 (30).
Литература: J. Hu, D. Son, Macromolecules 1998, 31, 8644.
N1,N1,N4,N4-тетракис[диметил(винил)силил]бензол-1,4-диамин (М11) (*)
 Хлордиметилвинилсилан (12,2 г, 101 ммоль, 4,5 экв.) добавляли по каплям к раствору п-фенилендиамина (2,43 г, 22,5 ммоль, 1,0 экв.) и ТЭА (10,5 г, 104 ммоль, 4,6 экв.) в ДХМ (60 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Затем растворитель снизили до 50% и добавили хлордиметилвинилсилан (1,35 г, 11,2 ммоль, 0,5 экв.), чтобы увеличить скорость реакции. Полученную суспензию перемешивали при 25°C дополнительно в течение 21 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M11 (6,40 г, 14,4 ммоль, 64%) в виде бесцветного масла.
Хлордиметилвинилсилан (12,2 г, 101 ммоль, 4,5 экв.) добавляли по каплям к раствору п-фенилендиамина (2,43 г, 22,5 ммоль, 1,0 экв.) и ТЭА (10,5 г, 104 ммоль, 4,6 экв.) в ДХМ (60 мл) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 3 дн. Затем растворитель снизили до 50% и добавили хлордиметилвинилсилан (1,35 г, 11,2 ммоль, 0,5 экв.), чтобы увеличить скорость реакции. Полученную суспензию перемешивали при 25°C дополнительно в течение 21 дн. Фильтрование и удаление растворителя дали остаток, который очистили посредством вакуумной перегонки. Получили указанное в заголовке соединение M11 (6,40 г, 14,4 ммоль, 64%) в виде бесцветного масла.
C22H40N2Si4, Mw=442,92 г/моль
Ткип=162-164°C (3,5×10-2 мбар).
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,81 (s, 4 H), 6,28 (dd, J=20,4 Гц, J= 14,7 Гц, 4 H), 5,91 (dd, J=14,9 Гц, J=3,8 Гц, 4 H), 5,71 (dd, J= 20,3 Гц, J= 3,8 Гц, 4 H), 0,18 (s, 24 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=143,35 (2 C), 140,74 (4 CH), 131,67 (4 CH2), 130,42 (4 CH), 0,23 (8 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=444 (M+, 100), 429 (M+-CH3, 9), 415 (5), 401 (32), 387 (6), 373 (9), 359 (7), 345 (2), 331 (2), 289 (2), 259 (5), 218 (2), 173 (10), 130 (9), 100 (5), 85 (29).
N1-<2-{бис[диметил(винил)силил]амино}этил>N1,N2,N2,-трис(диметил(винил)силил]этан-1,2-диамин (M12) (*)
 Диэтилентриeамин (3,00 г, 29,1 ммоль, 1,0 экв.) растворили в ДХМ (125 мл) при комнатной температуре. Добавили ТЭА (19,1 г, 189 ммоль, 6,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (22,8 г, 189 ммоль, 6,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 7 дн. Фильтрование и удаление всех летучих материалов дали указанное в заголовке соединение M12 (12,0 г, 23,0 ммоль, 79%, чистота: 91%) в виде желтого масла.
Диэтилентриeамин (3,00 г, 29,1 ммоль, 1,0 экв.) растворили в ДХМ (125 мл) при комнатной температуре. Добавили ТЭА (19,1 г, 189 ммоль, 6,5 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (22,8 г, 189 ммоль, 6,5 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 7 дн. Фильтрование и удаление всех летучих материалов дали указанное в заголовке соединение M12 (12,0 г, 23,0 ммоль, 79%, чистота: 91%) в виде желтого масла.
C24H53N3Si5, Mw=524,13 г/моль
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,29 (dd, J= 20,3 Гц, J=14,6 Гц, 5 H), 5,94 (dd, J=14,6 Гц, J= 3,8 Гц, 5 H), 5,74 (dd, J= 20,3 Гц, J= 3,8 Гц, 2 H), 2,99-2,95 (m, 4 H), 2,86- 2,84 (m, 4 H), 0,29 (s, 24 H), 0,24 (s, 6 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=141,26 (4 CH), 140,28 (CH), 132,23 (4 CH2), 131,73 (CH2), 53,43 (2 CH2), 47,25 (2 CH2), 1,09 (8 CH3), -0,64 (2 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): m/z (%)=524 (M+, 0,1), 325 (100), 283 (12), 212 (100), 198 (89), 172 (5), 128 (24), 85 (82).
N1,N6-(этан-1,2-диил)бис{N1,N2,N2-трис[диметил(винил)силил]этан-1,2-диамин} (M13) (*)
 Триэтилентетрамин (2,00 г, 13,7 ммоль, 1,0 экв.) растворили в ДХМ (60 мл) при комнатной температуре. Добавили ТЭА (11,1 г, 109 ммоль, 8,0 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (13,2 г, 109 ммоль, 8,0 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 7 дн. Фильтрование и удаление всех летучих материалов дали указанное в заголовке соединение M13 (6,15 г, 9,44 ммоль, 69%, чистота: 80% основного изомера) в виде желтого масла.
Триэтилентетрамин (2,00 г, 13,7 ммоль, 1,0 экв.) растворили в ДХМ (60 мл) при комнатной температуре. Добавили ТЭА (11,1 г, 109 ммоль, 8,0 экв.) с последующим добавлением по каплям хлордиметилвинилсилана (13,2 г, 109 ммоль, 8,0 экв.) при 25 °C. Реакционную смесь перемешивали при 25°C в течение 7 дн. Фильтрование и удаление всех летучих материалов дали указанное в заголовке соединение M13 (6,15 г, 9,44 ммоль, 69%, чистота: 80% основного изомера) в виде желтого масла.
C30H66N4Si6, Mw=651,40 г/моль
ЯМР 1H (400 МГц, 20 °C, C6D6): δ=6,30 (dd, J=20,3 Гц, J=14,6 Гц, 4 H), 6,25 (dd, J=20,0 Гц, J= 14,8 Гц, 2 H), 5,97 (dd, J= 14,7 Гц, J= 3,9 Гц, 2 H), 5,96 (dd, J= 14,6 Гц, J=3,8 Гц, 4 H), 5,76 (dd, J= 20,2 Гц, J=3,9 Гц, 2 H), 5,75 (dd, J= 20,3 Гц, J= 3,8 Гц, 4 H), 3,00-2,82 (m, 12 H), 0,30 (s, 24 H), 0,26 (s, 12 H) м.д.
ЯМР 13C (100 МГц, 20 °C, C6D6): δ=141,14 (4 CH), 140,15 (2 CH), 132,18 (2 CH2), 131,81 (4 CH2), 52,74 (2 CH2), 50,06 (2 CH2), 46,52 (2 CH2), 0,96 (8 CH3), -0,91 (4 CH3) м.д.
ГХ/МС (ЭИ, 70 эВ): основной изомер m/z (%)=452 (M+-199, 32), 369 (3), 325 (100), 256 (19), 212 (100), 198 (73), 142 (32), 85 (27); минорный изомер m/z (%)=452 (M+-199, 100), 212 (2). Соотношение 4,8:1 (ГХ)
Процедура полимеризации
Основная процедура полимеризации; Примеры A-S, Y, Z и лит. ссылки 1+2
Циклогексан (количество приведено в таблицах 1-5), бутадиен (98,3% от количества, приведенного в таблицах 1-6) и стирол (количество приведено в таблице) загрузили в работающий без доступа воздуха 10 л (или 5 л) реактор, и перемешанную смесь нагрели вплоть до 40°C. Затем добавили TMEDA (тетраметилэтилендиамин) (количество приведено в таблице) и разветвляющий агент (BA) (количество и модификатор даны в таблицах 1-5) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество инициатора в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим в течение 80 мин повышали температуру посредством нагревания или посредством охлаждения стенок реактора, начиная с температуры загрузки инициатора до конечной температуры полимеризации 60 °C. Затем загрузили бутадиен (1,7% от количества, приведенного в таблице). Через 5 мин добавили модификатор концевой группы цепи 3-метокси-3,8,8,9,9-пентаметил-2-окса-7-тиа-3,8-дисиладекан (2f) (количество приведено в таблицах 1-5). Реакция была окончена через 20 мин посредством загрузки метанола. Раствор полимера стабилизировали с использованием Irganox 1520D, полимер извлекли посредством перегонки с водяным паром и сушили до тех пор, пока не получили < 0,6% содержания остаточных летучих веществ. Полный набор данных по образцу представлен в таблицах 1-5.
Процедура полимеризации: Пример T
Циклогексан (количество приведено в таблице 4), бутадиен (100% от количества, приведенного в таблице 4) загрузили в работающий без доступа воздуха 5 л реактор и перемешанную смесь нагрели вплоть до 40 °C. Затем добавили TMEDA (62,4ммоль), фенантролин (индикатор) и разветвляющие агенты М1 и M12 (каждого по 3,25 ммоль) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим в течение 60 мин повышали температуру посредством нагревания или охлаждения стенок реактора, начиная с температуры загрузки инициатора до конечной температуры полимеризации 60 °C. Затем реакция была окончена посредством загрузки метанола. Раствор полимера стабилизировали с использованием Irganox 1520D. Полный набор данных по образцу представлен в таблице 4.
Процедура полимеризации: Пример U
Циклогексан (количество приведено в таблице 4), бутадиен (75% от количества, приведенного в таблице 4) загрузили в работающий без доступа воздуха 5 л реактор и перемешанную смесь нагрели вплоть до 60 °C. Затем добавили TMEDA (количество приведено в таблице 4), фенантролин (индикатор) и разветвляющий агент (BA) (количество и модификатор даны в таблице 4) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим в течение 80 мин повышали температуру (0,8 K/мин) посредством нагревания или охлаждения стенок реактора, начиная с температуры загрузки инициатора до конечной температуры полимеризации 80 °C. Одновременно с этим, в течение более 15 мин медленно загружали бутадиен (25% от количества, приведенного в таблице). Затем добавили модификатор концевой группы цепи N (количество приведено в таблице 4). Реакция была окончена через 20 мин посредством загрузки метанола. Раствор полимера стабилизировали с использованием Irganox 1520D. Полный набор данных по образцу представлен в таблице 4.
Процедура полимеризации: Пример V
Циклогексан (количество приведено в таблице 4), бутадиен (30% от количества, приведенного в таблице 4) и стирол (количество приведено в таблице) загрузили в работающий без доступа воздуха 5 л реактор и перемешанную смесь нагрели вплоть до 60 °C. Затем добавили TMEDA (количество приведено в таблице) и разветвляющий агент М1 (количество приведено в таблице 4) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим повышали температуру (0,5 K/мин) посредством нагревания или охлаждения стенок реактора начиная с температуры загрузки инициатора до конечной температуры полимеризации 85 °C. Одновременно с этим загрузили бутадиен (34% от количества, приведенного в таблице 4, расход 1,6 г/мин), с последующим вторым постепенно нарастающим дозированием бутадиена (21% от количества, приведенного в таблице 4, расход 1,0 г/мин) и с последующим последним постепенно нарастающим дозированием бутадиена (14% от количества, приведенного в таблице 4, расход 0,7 г/мин).
После 30 мин перемешивания при 85°C загрузили бутадиен (1% от количества, приведенного в таблице 4). Через 5 мин добавили модификатор концевой группы цепи P (количество приведено в таблице 4). Реакция была окончена через 10 мин посредством загрузки метанола. Раствор полимера стабилизировали с использованием Irganox 1520D. Полный набор данных по образцу представлен в таблице 4.
Процедура полимеризации: Пример W
Циклогексан (количество приведено в таблице 4), бутадиен (50% от количества, приведенного в таблице 4) и стирол (65% от количества, приведенного в таблице) загрузили в работающий без доступа воздуха 5 л реактор и перемешанную смесь нагрели вплоть до 40 °C. Затем добавили TMEDA (количество приведено в таблице) и разветвляющий агент M12 (количество приведено в таблице 4) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим повышали температуру (0,25 K/мин) посредством нагревания или охлаждения стенок реактора начиная с температуры загрузки инициатора до конечной температуры полимеризации 60 °C. Одновременно с этим загрузили бутадиен (50% от количества, приведенного в таблице 4, расход 2,1 г/мин) и стирол (34% от количества, приведенного в таблице 4, расход 0,6 г/мин). Затем загрузили стирол (1% от количества, приведенного в таблице 4). Реакция была окончена через 5 мин посредством загрузки метанола. Раствор полимера стабилизировали с использованием Irganox 1520D. 112,4 г (37,5 м.ч./100 м.ч. каучука), добавили масло типа TDAE (Vivatec 500) и перемешали раствор механической мешалкой. В результате этого вязкость по Муни была уменьшена от 119,5 MU до 36,9 MU. Полный набор данных по образцу представлен в таблице 4.
Процедура полимеризации: Пример X
Циклогексан (количество приведено в таблице 4), бутадиен 45% от количества, приведенного в таблице 4) и стирол (66% от количества, приведенного в таблице 4) загрузили в работающий без доступа воздуха 5 л реактор и перемешанную смесь нагрели вплоть до 55 °C. Затем добавили TMEDA (количество приведено в таблице) и разветвляющий агент M12 (0,82 ммоль) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим повышали температуру (1 K/мин) посредством нагревания или охлаждения стенок реактора начиная с температуры загрузки инициатора до конечной температуры полимеризации 70 °C. Одновременно с этим загрузили бутадиен (54% от количества, приведенного в таблице 4, расход 3,1 г/мин) и стирол (34% от количества, приведенного в таблице 4, расход 0,3 г/мин). Затем загрузили бутадиен (1% от количества, приведенного в таблице 4). Через 5 мин добавили разветвляющий агент М1 (1,94 ммоль). Спустя 5 мин добавили модификатор концевой группы цепи P (2,30 ммоль). Реакция была окончена через 10 мин посредством загрузки метанола. Раствор полимера стабилизировали с использованием Irganox 1520D. Полный набор данных по образцу представлен в таблице 4.
Сравн. прим. 1
Циклогексан (4652 г), бутадиен (682,3 г) и стирол (186,1 г) загрузили в работающий без доступа воздуха 10 л реактор и перемешанную смесь нагрели вплоть до 40 °C. Затем добавили TMEDA (1,02 г) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB (4,39 ммоль, c=250 моль/кг) в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим в течение 80 мин повышали температуру посредством нагревания или охлаждения стенок реактора начиная с температуры загрузки инициатора до конечной температуры полимеризации 60 °C. Затем через цилиндр загрузили бутадиен (3,6 г) с последующим SnCl4(0,31 ммоль) и 50 г циклогексана. Реакции позволили завершится в пределах 15 минут с последующим последним добавлением бутадиена (12,5 г). Через 5 мин добавили модификатор концевой группы цепи 3-метокси-3,8,8,9,9-пентаметил-2-окса-7-тиа-3,8-дисиладекан (2f) (1,09 г), растворенный в 20 г циклогексана. Реакция была окончена через 20 мин посредством загрузки метанола (10,8 г). Раствор полимера стабилизировали с использованием Irganox 1520D (2,2 г), полимер извлекли посредством перегонки с водяным паром и сушили до тех пор, пока не Получили < 0,6% содержания остаточных летучих веществ. Полный набор данных по образцу представлен в таблице 4.
Сравн. прим. 2
Циклогексан (4739 г), бутадиен (649,2 г) и стирол (176,0 г) загрузили в работающий без доступа воздуха 10 л реактор и перемешанную смесь нагрели вплоть до 40 °C. Затем добавили TMEDA (2,32 г) и DV (0,37 г) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB (4,14 ммоль, c=0,159 моль/кг) в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим в течение 80 мин повышали температуру посредством нагревания или охлаждения стенок реактора начиная с температуры загрузки инициатора до конечной температуры полимеризации 60 °C. Затем загрузили бутадиен (11,2 г). Реакции позволили завершится в пределах 5 минут. Через 5 мин добавили модификатор концевой группы цепи 3-метокси-3,8,8,9,9-пентаметил-2-окса-7-тиа-3,8-дисиладекан (2f) (1,24 г), растворенный в 20 г циклогексана. Реакция была окончена через 20 мин посредством загрузки метанола (10,1 г). Раствор полимера стабилизировали с использованием Irganox 1520D (2,09 г), полимер извлекли посредством перегонки с водяным паром и сушили до тех пор, пока не Получили < 0,6% содержания остаточных летучих веществ. Полный набор данных по образцу представлен в таблице 4.
Сравн. прим. 3
Циклогексан (4111 г), бутадиен (559,7 г) и стирол (152,7 г) загрузили в работающий без доступа воздуха 10 л реактор и перемешанную смесь нагрели вплоть до 40 °C. Затем добавили TMEDA (0,84 г) и загрузили по каплям н-бутиллитий, чтобы прореагировали примеси, пока цвет реакционной смеси не изменится на желтоватый (титрование). После этого для начала полимеризации незамедлительно загрузили дозированное количество NB (3,54 ммоль, c=0,249 моль/кг) в циклогексане, соответствующее целевой молекулярной массе полимера. Время начала загрузки инициатора считалось временем начала полимеризации. Параллельно с этим в течение 80 мин повышали температуру посредством нагревания или охлаждения стенок реактора, начиная с температуры загрузки инициатора до конечной температуры полимеризации 60°C с последующим вторым добавлением бутадиена (2,7 г). Через 1 мин добавили связующий агент SiCl4 (1,98 г), растворенный в 20 г циклогексана. Через 15 мин бутадиен (10,3 г) добавили в третий раз. Через 5 мин добавили модификатор концевой группы цепи 3-метокси-3,8,8,9,9-пентаметил-2-окса-7-тиа-3,8-дисиладекан (2f) (1,08 г), растворенный в 20 г циклогексана. Реакция была окончена через 20 мин посредством загрузки метанола (0,11 г). Раствор полимера стабилизировали с использованием Irganox 1520D (1,81 г), полимер извлекли посредством перегонки с водяным паром и сушили до тех пор, пока не получили < 0,6% содержания остаточных летучих веществ. Полный набор данных по образцу представлен в таблице 4.
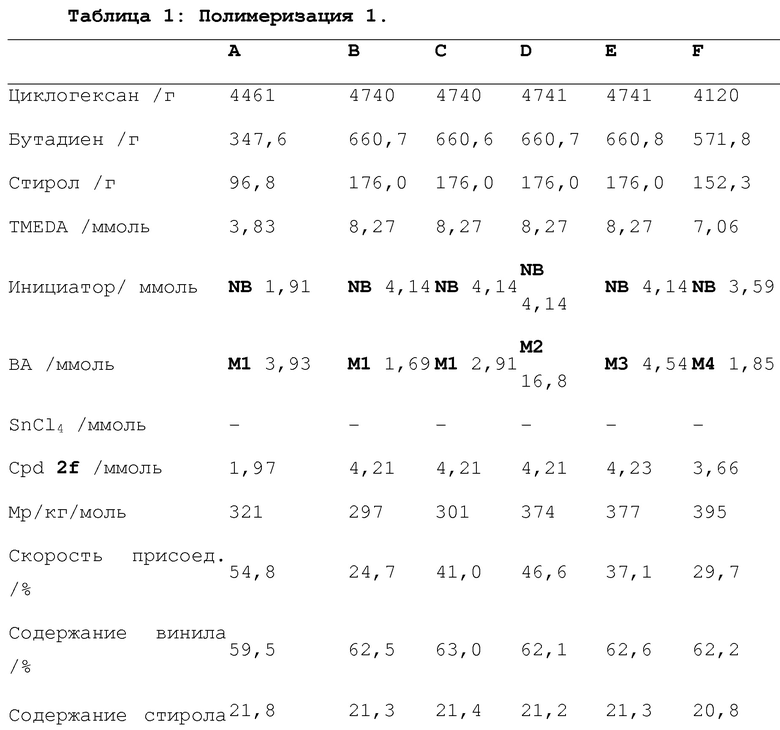



Устойчивость при хранении ненаполненных полимеров по избретению
Как проиллюстрировано на Фиг. 1, ненаполненные полимеры по изобретению демонстрируют необычное, выгодное поведение при хранении. В противоположность большинству функционализированных полимеров, ненаполненные полимеры по изобретению со временем не показывают значительного увеличения вязкости по Муни при 25°C (на воздухе). Не привязываясь к теории, предполагается, что относительно лабильные Si-N связи, благодаря их химической структуре, при увеличении времени хранения обеспечивают незначительное уменьшение скорости присоединения (отраженное вязкостью Муни) в зависимости от молекулярной структуры модификатора. В результате этого, потенциал Муни, увеличенный в результате активированного кислородом присоединения или медленного сшивания за счет реакций конденсации, может находиться в равновесном состоянии.
Состав с кремнеземом в качестве наполнителя:
Таблица 6: Информация о соединении 1.
ссылка 1
Температуры стеклования полимеров; A: -26,9 °C, В: -22,3 °C, C: -22,4 °C, лит.ссылка 1: - 22,5 °C, cравн. прим. 2; ~22,5 °C.
Составные данные по вязкости по Муни (CML) полимеров A, В и C отражают разные количества BA и, таким образом, разные уровни технологических характеристик, т.е. чем больше BA было использовано, тем выше данные CML (но все же более низкие, чем в сравн. прим. 2). Отражено эффективное взаимодействие полимер-наполнитель посредством улучшенных данных Mod300 для A и В по сравнению со всеми ссылками, Mod300 для C является выровненным со ссылками. В противоположность этому, C показывает выровненную прочность на разрыв с образцами по ссылке, при этом A и В обладают немного более низкими значениями прочности на разрыв. По отношению к гистерезисным свойствам (HBU, эластичность по упругому отскоку @ 60 °C, tan δ @ 60 °C), образцы A и В показывают явно уменьшенные гистерезисные потери и, таким образом, менее нежелательное тепловыделение, по сравнению со всеми ссылками, включая состояние примера из литературной статьи по лит. ссылке 1 в том же дозированном количестве модификатора. Из этих данных, улучшенные свойства сопротивления качению по сравнению с лит. ссылкой 1, сравн. примером 1 и сравн. примером 2 могут быть ожидаемыми. Образец C также показывает улучшенные гистерезисные свойства по сравнению со сравн. примером 1 и сравн. примером 2, при этом C является выровненным с лит. ссылкой 1. В зависимости от дозированного количества BA, сцепление шины с обледеневшим дорожным покрытием (отраженное посредством tan δ @ -10 °C) является улучшенным по сравнению со всеми ссылочными примерами. Сопротивление потенциального износа отражается посредством истирания по DIN наравне со всеми шестью образцами.
Таблица 7: Информация о соединении 2.
ссылка 2
*) Сравнительный пример с модификатором согласно US 3.485.857
Температуры стеклования полимеров: D: -22,6 °C, E: -23,6 °C, Lit. Ref,2: -23,5 °C, сравн.пр. 3: -23,8 °C.
В таблице 7 сравниваются модификаторы первичного амина, как описано в US 3485857, с литературным и сравнительным примерами. Гистерезисные свойства (HBU, эластичность по упругому отскоку @ 60 °C, tan δ @ 60 °C) не улучшаются с использованием основанных на первичном амине модификаторов. С более высоким дозированием модификатора М2 (4 моль М2/ инициатор для образца D), гистерезисные свойства являются даже более низкими, чем результаты, полученные в примере E (1,08 моль M3/инициатор). Механические характеристики (Mod 300-Mod 100, удлинение при разрыве, прочность на разрыв) для образцов D и E были наравне со сравн. прим. 3.
Таблица 8: Информация о соединении 3.
Температуры стеклования полимеров: P: -22,9 °C, R: -20,4 °C, cравн. прим. 4: -22,3 °C.
Сравн. прим. 4=коммерческий доступный функционализованный класс SSBR SPRINTAN™ SLR 4602- Schkopau
По отношению к механическим характеристикам, образец Z и особенно образец P превосходят сравн. прим. 4 в прочности на разрыв, при этом образец R показывает слегка более низкую прочность на разрыв. Гистерезисные свойства (HBU, эластичность по упругому отскоку @ 60 °C, tan δ @ 60 °C) являются улучшенными для образцов P, R и Z по сравнению со сравн. прим. 4, следуя порядку Z > R > P >> Сравн. прим. 4, по которому улучшенные свойства сопротивления качению для топливоэкономичных шин могут быть ожидаемыми. В пределах расчетного стандартного отклонения истирания по DIN все четыре образца могут быть оценены наравне.
Состав с углеродной сажей в качестве наполнителя:
Таблица 9: Информация о соединении 1.
1)ML 1+4 для образца Z был измерен вскоре после обработки полимера, при этом ML 1+4 образца Z в таблице 8 был определен шесть недель спустя (снижен на приблиз. 15 MU), сравн. прим. 4=коммерческий доступный функционализованный класс SSBR SPRINTAN™ SLR 4602-Schkopau
Поведение процесса изготовления, отраженное посредством CML немного более сложное для образца P чем для сравн. прим. 4, Z и особенно R, имеет даже более высокие значения CML, соответственно, которые указывают на эффективное взаимодействие каучук-наполнитель. Механические характеристики прочность на разрыв и Mod300-Mod100 выровнены для всех образцов. Гистерезисные свойства, указанные посредством HBU и эластичности по упругому отскоку @ 60°C наравне для всех образцов, при этом tan δ @ 60°C является значительно уменьшенным, т.е. улучшенным для образцов P, R и Z по сравнению со сравн. прим. 4, особенно для образца R. Благодаря меньшему значению tan δ @ 60°C для P, R и Z, в такой области применения как шины может быть ожидаемым уменьшенное сопротивление качению. Истирание по DIN выровнено для всех образцов, т.е. в образцах P, R и Z сопротивление истиранию не ухудшается.
Рецепт смешивания для соединений кремнезема
1-я стадия смешивания:
SSBR 80
Высший цис 1,4-полибутадиен (BUNA™ cis 132-Schkopau, Trinseo Deutschland GmbH) 20
Осажденный кремнезем (Ultraсил 7000 GR, Evonik Industries) 80
Силан (SI 75, бис(триэтоксисилилпропил)дисульфан, Evonik Industries) 6,9
Стеариновая кислота (Cognis GmbH) 1,0
Антиозонат (Dusantox 6 PPD [Н-(1,3-диметилбутил)-N'-фенил-1,4-фенилендиамин], Duslo a.s.) 2,0
Оксид цинка (Grillo-Zinkoxid GmbH) 2,5
Воск для защиты от озона (Antilux 654, Rhein Chemie Rheinau GmbH) 1,5
Смягчитель (TDAE oil, VivaTec500, Hansen & Rosenthal КГ) 20
Сера (Solvay AГ) 1,4
Ускоритель (TBBS, N-трет-бутил-2-бензотиазолсульфенамид Rhein Chemie Rheinau GmbH) 1,5
DPG (дифенилгуанидин, Vulkacit D, Lanxess AГ) 1,5
Рецепт смешивания для соединений углеродной сажи
1я стадия смешивания:
SSBR 100-
IRB 8 50,0
Стеариновая кислота (Cognis GmbH) 1,5
Смягчитель (масло TDAE, VivaTecSOO, Hansen & Rosenthal КГ) 15,0
Сера (Soivay AG) 1,75
Ускоритель (TBBS, N-трет-бутил-2-бензотиазолсульфенамид, Rhein Chemie Rheinau GmbH) 1,0
Методы испытаний
Анализ молекулярной массы проводили посредством SEC/RI с использованием HEWLETT PACKARD HP 1100. Элюент ТГФ дегазировали в оперативном режиме. Расход растворителя составлял 1,0 мл/мин. Ввели 100 мл раствора полимера для анализа. Анализ проводили при 40 °C. Молекулярную массу первоначально рассчитали, основываясь на калибровке полистирола, и привели в таблицах как полистирол. Реальная молекулярная масса (молекулярные массы SSBR) может быть определена посредством деления на коэффициент, извлеченный из более раннего сравнения молекулярных масс SEC/RI и SEC/MALLS. Значение коэффициента зависит от полимерной композиции (содержания стирола и бутадиена). Коэффициент 1,52 может быть использован для SSBR с 21% и 25% содержанием стирола. Коэффициент 1,84 может быть использован для SBR с 0% содержанием стирола. Коэффициент 1,56 может быть использован для SSBR с 16% содержанием стирола. Коэффициент 1,41 может быть использован для SSBR с 45% содержанием стирола.
Спектроскопию ЯМР выполняли на BRUKER Avance 400 в 5 мм BBO ампуле.
Растворители, частоты и температура представлены в характеристических данных.
Для определения содержания винила и содержания стирола была использована ИК фурье-спектроскопия, измеряемая в аттенюированном полном отражении.
Температуру стеклования определили с использованием DSK Q2000 в следующих условиях:
Масса: ca. 10-12 мг
Емкость для образца: Alu/S
Диапазон температур: (-140…80) °С
Скорость нагревания: 20К/мин соответственно 5К/мин
Скорость охлаждения: естественное охлаждение
Продувочный газ: 20 мл Ar/мин
Охлаждающий агент: жидкий азот
Каждый образец был измерен по меньшей мере один раз. Измерения содержали два тепловых испытания. Второе тепловое испытание (2) было использовано для определения температуры стеклования.
Измерения реологических свойств невулканизированного каучука согласно ASTM D 5289-95 проводили с использованием реометра без ротора сдвигового типа (MDR 2000 E), чтобы охарактеризовать параметры отверждения. Испытуемые образцы были вулканизированы посредством t95 при 160 °C, особенно для испытаний твердости и эластичности по упругому отскоку образцы были вулканизированы посредством T95+5 мин при 160 °C. Прочность на разрыв и коэффициенты были измерены согласно ASTM D 412 на Zwick Z010. Истирание по DIN было измерено согласно DIN 53516 (1987-06-01). Твердость по А. Шору (ASTM D 2240) и эластичность по упругому отскоку (ISO 4662) были измерены при 0 °C, RT и 60°. Динамические характеристики tan δ при 0°C и 60°C были измерены с использованием динамического спектрометра Eplexor 150N/500N, изготовленного компанией Gabo Qualimeter Testanlagen GmbH (Германия), применяющего динамическую деформацию сжатия 0,2% при частоте 2 Гц. Теплообразование было измерено согласно ASTM D 623, способом A, на флексометре Doli 'Goodrich'.
| название | год | авторы | номер документа |
|---|---|---|---|
| ВИНИЛСИЛАНЫ ДЛЯ ПРИМЕНЕНИЯ В ФУНКЦИОНАЛИЗИРОВАННЫХ ЭЛАСТОМЕРНЫХ ПОЛИМЕРАХ | 2013 |
|
RU2669799C2 |
| ПРИМЕНЕНИЕ ОПРЕДЕЛЕННЫХ АМИНОСИЛИЛЬНЫХ МОНОМЕРОВ В ПРОИЗВОДСТВЕ КАУЧУКА | 2018 |
|
RU2782265C2 |
| ПОЛИМЕРЫ, МОДИФИЦИРОВАННЫЕ АМИНОСИЛАНОМ | 2012 |
|
RU2609166C2 |
| КОМПОЗИЦИЯ ГЕТЕРОФАЗНОГО ПОЛИПРОПИЛЕНА С УЛУЧШЕННЫМ БАЛАНСОМ СВОЙСТВ | 2019 |
|
RU2754224C1 |
| СПОСОБ ПОЛУЧЕНИЯ АЦИЛФОСФАТОВ | 2013 |
|
RU2719592C2 |
| АМФОТЕРНЫЕ СОЕДИНЕНИЯ ПЕРЕХОДНЫХ МЕТАЛЛОВ, СПОСОБ ИХ ПОЛУЧЕНИЯ, СПОСОБ ПОЛИМЕРИЗАЦИИ ОЛЕФИНОВ И КОМПОНЕНТ КАТАЛИЗАТОРА ДЛЯ ЭТОГО СПОСОБА | 1995 |
|
RU2140922C1 |
| МОДИФИЦИРОВАННЫЕ ПОЛИМЕРНЫЕ КОМПОЗИЦИИ | 2010 |
|
RU2558597C2 |
| ИНГИБИТОРЫ ПУТИ ПЕРЕДАЧИ СИГНАЛА NOTCH И ИХ ПРИМЕНЕНИЕ ПРИ ЛЕЧЕНИИ РАКА | 2012 |
|
RU2631611C2 |
| КОМПОЗИЦИЯ ГЕТЕРОФАЗНОГО ПОЛИПРОПИЛЕНА С ВЫСОКОЙ ГИБКОСТЬЮ И МЯГКОСТЬЮ | 2019 |
|
RU2754212C1 |
| КУМАРИНОПОДОБНОЕ ЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА МЕК И ЕГО ПРИМЕНЕНИЕ | 2018 |
|
RU2742234C1 |

Изобретение относится к новым полимеризуемым мультивиниламиносиланам, которые применимы в качестве разветвляющих агентов для синтетического и натурального каучука. Предложены новые мультивиниламиносиланы, в которых по меньшей мере два атома азота замещены группой –Si(R1)(R2)(R3), где каждый из R1, R2 и R3 независимо выбран из винила, метила, этила, пропила, бутила, гексила и октила при условии, что по меньшей мере один из R1, R2 и R3 является винилом, при этом данный мультивиниламиносилан не является N,N’-бис(этенилдиметилсилил)-N,N’-диметил-1,2-этандиамином. Предложены также способ получения заявленного мультивиниламиносилана, инициирующее соединение для получения разветвленного эластомерного полимера на его основе, применение мультивиниламиносилана в качестве разветвляющего агента, способ получения разветвленного эластомерного полимера с его участием и соответствующий разветвленный эластомерный полимер, невулканизированная полимерная композиция для использования при изготовлении резиновых изделий, содержащая разветвленный эластомерный полимер, способ получения вулканизированной полимерной композиции и соответствующая композиция, а также изделие из нее. Технический результат – использование предложенного мультивиниламиносилана при получении разветвленного эластомерного полимера позволяет получить отвержденные эластомерные каучуковые композиции с улучшенными техническими характеристиками, пригодные для получения изделий, обладающих отличным соотношением экономии топлива и технологических характеристик. 10 н. и 4 з.п. ф-лы, 1 ил., 9 табл.
1. Мультивиниламиносилан, который представляет собой соединение, выбранное из следующих формул 1-1, 1-2 и 1-3:
 ,
,
где каждый из R11, R12, R13, R14 и R15 независимо выбран из группы B и C1-C18 алкила, при этом каждый из В независимо выбран из группы -Si(R1)(R2)(R3), при этом каждый из R1, R2 и R3 независимо выбран из метила, этила, пропила, бутила, гексила и октила при условии, что по меньшей мере один из R1, R2 и R3 является винилом, и где по меньшей мере два из R11, R12, R13, R14 и R15 представляют собой группу В;
R9 независимо выбран из этилена (-С2Н4-), пропилена (-С3Н6-) и бутилена (-С4Н8-);
R10 выбран из этилена (-С2Н4-), пропилена (-С3Н6-), бутилена (-С4Н8-) и фенилена (-С6Н4-); и
c равен целому числу, выбранному из 0, 1, 2 и 3;
 ,
,
где группа -N<>N- представляет собой от 5- до 18-членную гетероциклическую группу, которая может быть насыщенной или ненасыщенной, при этом гетероатомные группы, кроме двух атомов N, специально проиллюстрированных в Формуле 1-2, выбраны из >SiR17R18, причем каждый из R17 и R18 независимо выбран из C1-С6 алкила, фенила и бензила, и где каждый из В независимо выбран из группы -Si(R1)(R2)(R3), где каждый из R1, R2 и R3 независимо выбран из метила, этила, пропила, бутила, гексила и октила при условии, что по меньшей мере один из R1, R2 и R3 является винилом;
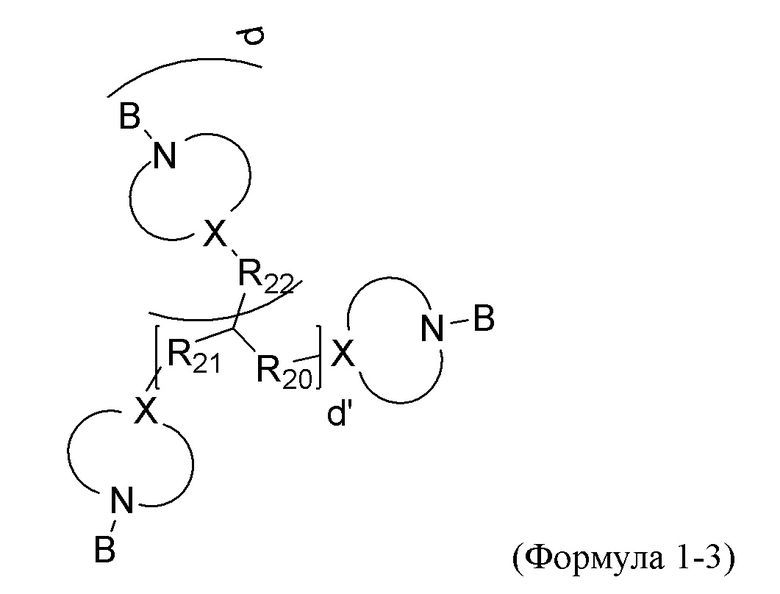
где каждый из R20 и R21 независимо выбран из одинарной связи и алкиленовой группы C1-С10, d равен 0, d' равен целому числу, выбранному из 0 и 1, каждая группа -N<>X- независимо выбрана из от 5- до 10-членной насыщенной гетероциклической группы, при этом каждый X представляет собой -CH-, и гетероатомные группы кроме двух групп N и X, специально проиллюстрированных в Формуле 1-3, выбраны из >SiR17R18, причем каждый из R17 и R18 независимо выбран из C1-С6 алкила, фенила и бензила, и где каждый из В независимо выбран из группы -Si(R1)(R2)(R3), где каждый из R1, R2 и R3 независимо выбран из метила, этила, пропила, бутила, гексила и октила при условии, что по меньшей мере один из R1, R2 и R3 является винилом;
где при этом данный мультивиниламиносилан не является N,N’-бис(этенилдиметилсилил)-N,N’-диметил-1,2-этандиамином.
2. Мультивиниламиносилан по п. 1, отличающийся тем, что в Формуле 1-1 каждый из радикалов R11, R12, R13, R14 и R15 независимо выбран из группы В и метила; каждый из R9 и R10 представляет собой этилен; и c равен целому числу, выбранному из 0, 1, 2 и 3.
3. Мультивиниламиносилан по п. 1, который выбран из:

при этом каждый R независимо выбран из B и C1-C6 алкила;

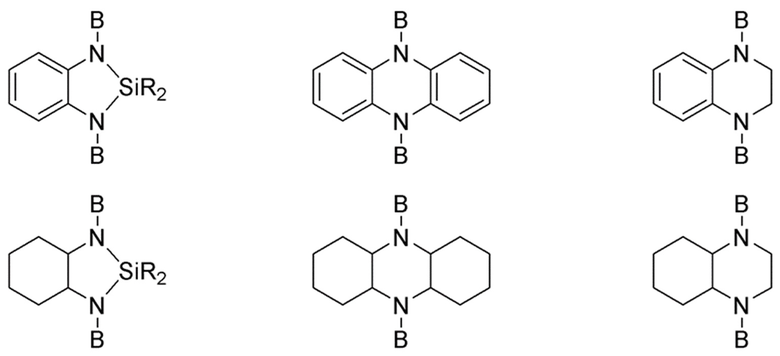

4. Инициирующее соединение для получения разветвленного эластомерного полимера, получаемое посредством взаимодействия мультивиниламиносилана по любому из пп. 1-3 с органическим соединением щелочного металла, выбранным из метиллития, этиллития, н-бутиллития, втор-бутиллития, трет-октиллития, изопропиллития и 2-бутиллития.
5. Способ получения мультивиниламиносилана по п. 1, включающий осуществление взаимодействия амина с силаном следующей Формулы 2 в присутствии основания:

где
X выбран из Cl, Br, I, трифторметансульфoната (OTf) и тозилата (OTos);
R1, R2 и R3 каждый независимо выбран из винила, метила, этила, пропила, бутила, гексила и октила при условии, что по меньшей мере один из R1, R2 и R3 выбран из винила; и
амин представляет собой соединение, имеющее по меньшей мере две группы, независимо выбранные из первичной аминогруппы и вторичной аминогруппы.
6. Применение мультивиниламиносилана по любому из пп. 1-3 в качестве разветвляющего агента для введения разветвления в эластомерный полимер.
7. Способ получения разветвленного эластомерного полимера, включающий:
полимеризацию по меньшей мере одного сопряженного диена с мультивиниламиносиланом по любому из пп. 1-3 в присутствии инициирующего соединения,
полимеризацию по меньшей мере одного сопряженного диена в присутствии инициирующего соединения по п. 4, или
приведение в контакт ʺживогоʺ полимера, получаемого посредством анионной полимеризации по меньшей мере одного сопряженного диена, с мультивиниламиносиланом по любому из пп. 1-3.
8. Разветвленный эластомерный полимер, получаемый способом по п. 7.
9. Невулканизированная полимерная композиция для использования при изготовлении резиновых изделий, содержащая разветвленный эластомерный полимер по п. 8 и один или несколько дополнительных компонентов, выбранных из (i) компонентов, которые добавляют к или образуются в результате процесса полимеризации, используемого для получения указанного полимера, (ii) компонентов, которые остаются после удаления растворителя из процесса полимеризации, и (iii) компонентов, которые добавляют к полимеру после завершения процесса изготовления полимера.
10. Невулканизированная полимерная композиция по п. 9, содержащая один или более наполнителей.
11. Невулканизированная полимерная композиция по п. 9 или 10, содержащая одну или более вулканизирующих добавок.
12. Вулканизированная полимерная композиция для использования при изготовлении резиновых изделий, которую получают посредством вулканизации невулканизированной полимерной композиции по п. 11.
13. Способ получения вулканизированной полимерной композиции, включающий вулканизацию невулканизированной полимерной композиции по п. 11.
14. Изделие, содержащее по меньшей мере один компонент, образованный из вулканизированной полимерной композиции по п. 12, которое предпочтительно выбрано из шины, протектора шины, боковины покрышки, каркаса шины, ремня, прокладки, уплотнения, гибкого рукава, виброгасителя, компонента обуви, мяча для игры в гольф и шланга.
| Piotr Pawluć et al | |||
| Печь для непрерывного получения сернистого натрия | 1921 |
|
SU1A1 |
| Synthesis,2006, No.21, pp.3739-3745 | |||
| СПОСОБ ПОЛУЧЕНИЯ р-АМИНОЭТИЛСИЛАНОВ С ФУНКЦИОНАЛЬНЫМИ АМИНОГРУППАМИ У АТОМАКРЕМНИЯ | 0 |
|
SU323006A1 |
| WO 2015010710 A1, 29.01.2015. | |||

