Настоящая заявка испрашивает приоритет на основании:
[1] CN201710488401.X, дата подачи: 23 июня 2017 г.;
CN201810596587.5, дата подачи: 11 июня 2018 г.
Область техники
[2] Настоящее описание относится к классу кумариноподобных циклических соединений в качестве ингибиторов MEK и фармацевтической композиции, содержащей указанные соединения, а также к их применению для получения лекарственного средства для лечения заболевания, связанного с MEK, в частности, относится к соединению, представленному формулой (I), и его фармацевтически приемлемой соли.
Уровень техники
[3] Путь MAPK задействован в ряде клеточных процессов, таких как пролиферация клетки, дифференцировка, апоптоз и реакция на стресс. Существует четыре известных пути MAPK: ERK1/2, JNK, p38 и ERK5. Одним из наиболее важных и широко известных путей MAPK является путь Ras/Raf-киназы. В этом пути, в первую очередь внеклеточные факторы роста (т.е. PDGF или EGF и т. д.) объединяются с трансмембранными рецепторами (т.е., PDGFR или EGFR или ErbB2 и т. д.), активируя указанные рецепторы, и активированные рецепторы индуцируют объединение внутримембранного Ras с GTP и его активацию посредством фактора обмена гуаниновых нуклеотидов (например, SOS); активированный Ras далее опосредованно фосфорилирует и активирует Raf (MAPKKK в данном пути); затем активированный Raf индуцирует фосфорилирование двух сериновых остатков MEK1/2 (MAPKK в данном пути) (MEK1 соответствует S218 и S222; MEK2 соответствует S222 и S226) (Ahn et al., Methods in Enzymology, 2001, 332, 417-431). Фосфорилированный ERK димеризуется и накапливается в ядре (Khokhlatchev et al., Cell, 1998, 93, 605-615). ERK в ядре задействован во многих клеточных функциях, включая, но не ограничиваясь ими, ядерный транспорт, передачу сигнала, репарацию ДНК, сборку и миграцию нуклеосом, а также процессинг и трансляцию мРНК (Ahn et al., Molecular Cell, 2000, 6, 1343-1354).
[4] Путь RAF-MEK-ERK может передавать пролиферативные и антиапоптотические сигналы от факторов роста и онкогенных факторов, тем самым способствуя росту, развитию и метастазированию клеток; мутации генов, участвующих в данном пути, или повышенная экспрессия факторов роста, нисходящих сигнальных белков или протеинкиназ будут приводить к неконтролируемой пролиферации клеток и, в конечном итоге, к онкогенезу. Например, мутации в раковых клетках вызывают повышенную экспрессию факторов роста, что приводит к непрерывной активации внутреннего пути MAPK; или неспособность деактивировать активированные комплексы Ras из-за мутаций также будет вызывать непрерывную активацию пути MAPK; мутации bRaf были недавно выявлены в более чем 60% случаях меланом (Davies, H. et al., Nature, 2002, 417, 949-954). Указанные мутации bRaf приводят к спонтанной активации каскада MAPK. Спонтанную или чрезмерную активацию пути MAPK также наблюдали в исследованиях образцов первичной опухоли и клеточных линий, например, рака поджелудочной железы, рака толстой кишки, рака яичников и рака почки (Hoshino, R. et al., Oncogene, 1999, 18, 813-822). Следовательно, существует сильная корреляция между гиперактивным путем MAPK вследствие мутаций генов и раком.
[5] Поскольку путь MAPK занимает центральное положение в пролиферации и дифференцировке клеток, ингибирование этого пути будет полезным для лечения множества гиперпролиферативных заболеваний, и MEK, расположенный в пути ниже Ras и Raf, стал играть ключевую роль в этом пути. Кроме того, известные в настоящее время субстраты, которые могут фосфорилироваться и активироваться MEK, представляют собой только MAPK, а именно ERK1 и ERK2. Данная строгая селективность и уникальная способность бифункциональной киназы делают ее привлекательной лекарственной мишенью для потенциальных и широко распространенных терапевтических применений, таких как злокачественные и доброкачественные гиперпролиферативные заболевания, иммунная регуляция и воспаление.
[6] В настоящее время на клинической и начальной стадии разрабатываются множество ингибиторов Raf и MEK, нацеленных на сигнальный путь MAPK. Например, сорафениб (Bay 43-9006), одобренный FDA в декабре 2005 года, является неспецифическим ингибитором серин/треонин- и тирозинкиназ, его мишенями являются Raf, MEK, VEGFR2/3, Flt-3, PDGFR, c-Kit и т. д. Специфические ингибиторы B-Raf, такие как дабрафениб (GSK21 18436) и вемурафениб (PLX4032), обладают хорошей клинической эффективностью, но их продолжительность действия невелика, и клинические исследования показали, что длительное лечение ингибиторами B-Raf может привести у пациентов к приобретенной лекарственной устойчивости. Следовательно, в клинической практике ингибиторы MEK обычно используют в комбинации с ингибиторами B-Raf. Траметиниб (GSK-1 120212), специфический ингибитор MEK1/2, был одобрен FDA в мае 2013 года и одобрен для лечения прогрессирующей меланомы в комбинации с дабрафенибом в январе 2014 года; кобиметиниб представляет собой ингибитор, который специфически ингибирует MEK1/2, и был одобрен FDA в 2015 году для лечения меланомы в комбинации с вемурафенибом. Заявка по биниметинибу была подана в FDA в 2016 году для применения для лечения меланомы с мутантным N-RAS. Кроме того, на клинической стадии имеются ингибиторы MEK1/2, такие как селуметиниб и рефаметиниб.
[7] В настоящее время раскрыта серия патентных заявок, касающихся ингибиторов МЕК, включая WO2007/096259, WO2010/003022, WO2012/162293, WO2014/169843, WO2015/058589, и т.д.
Краткое описание изобретения
[8] В настоящем описании предложено соединение, представленное формулой (I), его фармацевтически приемлемая соль или его таутомер:
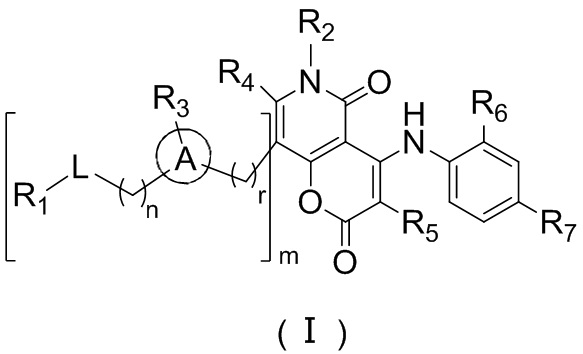
[9] где,
[10] n выбран из 0, 1 или 2;
[11] r выбран из 0, 1, 2 или 3;
[12] m выбран из 0 или 1; когда m представляет собой 0, тогда 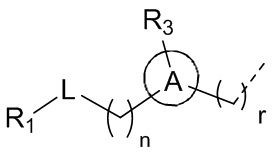 представляет собой H;
представляет собой H;
[13] кольцо A выбрано из фенила или 5-6-членного гетероарила;
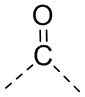
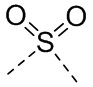
[14] L выбран из одинарной связи, -S(=O)-, -S(=O)2-, -C(=O)-, -NH-, -NH-C(=O)-, -NH-C(=O)-O-, -NH-S(=O)2-, -NH-S(=O)- и -NH-C(=O)-NH-, где -NH-, -NH-C(=O)-, -NH-C(=O)-O-, -NH-S(=O)2-, -NH-S(=O)- и -NH-C(=O)-NH- каждый необязательно замещен 1, 2 или 3 R;
[15] R1 выбран из H, NH2, C1-6 алкила, 3-6-членного гетероциклоалкила, C3-6 циклоалкила и C1-3 гетероалкила, где NH2, C1-6 алкил, 3-6-членный гетероциклоалкил, C3-6 циклоалкил и C1-3 гетероалкил каждый необязательно замещен 1, 2 или 3 R;
[16] R2 выбран из H, C1-6 алкила, C1-6 гетероалкила, C3-6 циклоалкила и 5-6-членного гетероциклоалкила, где C1-6 алкил, C1-6 гетероалкил, C3-6 циклоалкил и 5-6-членный гетероциклоалкил каждый необязательно замещен 1, 2 или 3 R;
[17] R3 выбран из H, F, Cl, Br, I, C1-3 алкила, C1-3 алкокси, C1-4 алкинила, C1-4 алкенила и фенила, где C1-3 алкил, C1-3 алкокси, C1-4 алкинил, C1-4 алкенил и фенил каждый необязательно замещен 1, 2 или 3 R;
[18] R4 и R5 независимо выбраны из H, F, Cl, Br, I, NH2, OH, C1-6 алкила и C1-3 алкокси, где C1-6 алкил и C1-3 алкокси каждый необязательно замещен 1, 2 или 3 R;
[19] или R3 и R4 связаны вместе с образованием 5-7-членного циклоалкила, 5-7-членного гетероциклоалкила, 5-7-членного арила или 5-7-членного гетероарила;
[20] R6 и R7 независимо выбраны из H, F, Cl, Br, I, CH3, Et, CH3-O- и CH3-CH2-O-;
[21] R выбран из F, Cl, Br, I, OH, NH2, C1-3 алкила и C1-3 гетероалкила, где NH2, C1-3 алкил и C1-3 гетероалкил каждый необязательно замещен 1, 2 или 3 R';
[22] R' выбран из F, Cl, Br, I, NH2 или C1-3 алкила;
[23] каждый из «гетеро» в 5-6-членном гетероариле, 5-6-членном гетероциклоалкиле, 3-6-членном гетероциклоалкиле, C1-3 гетероалкиле, 5-7-членном гетероциклоалкиле, 5-7-членном гетероциклоалкиле, 5-7-членном ариле и 5-7-членном гетероариле независимо выбран из -NH-, N, -O-, -S(=O)2-, -S(=O)2-NH-, -NH-S(=O)2-NH-, - C(=O)-NH-, -S(=O)-, -C(=O)-, -S(=O)-NH- и -O-C(=O)-NH-;
[24] В любом из вышеперечисленных случаев количество гетероатомов и гетероатомных групп независимо выбрано из 1, 2, 3 или 4.
[25] Согласно некоторым вариантам реализации настоящего описания R' выбран из F, Cl, Br, I, NH2 или CH3.
[26] Согласно некоторым вариантам реализации настоящего описания R выбран из F, Cl, Br, I, OH, NH2, метила, этила, C1-3 алкил-S(=O)2-NH-, C1-3 алкил-S(=O)2-, C1-3 алкил-C(=O)-NH- и C1-3 алкил-O-, где NH2, метил, этил, C1-3 алкил-S(=O)2-NH-, C1-3 алкил-S(=O)2-, C1-3 алкил-C(=O)-NH- и C1-3 алкил-O- каждый необязательно замещен 1, 2 или 3 R'.
[27] Согласно некоторым вариантам реализации настоящего описания R выбран из F, Cl, Br, I, OH, NH2, CH3,  ,
,  ,
, 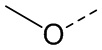 и
и 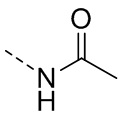 , где NH2, CH3, , , и каждый необязательно замещен 1, 2 или 3 R'.
, где NH2, CH3, , , и каждый необязательно замещен 1, 2 или 3 R'.
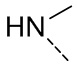
[28] Согласно некоторым вариантам реализации настоящего описания R выбран из F, Cl, Br, I, OH, NH2, CH3, 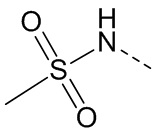 , , ,
, , ,  ,
,  и
и  .
.
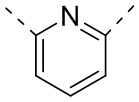
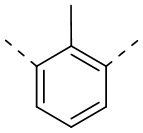
[29] Согласно некоторым вариантам реализации настоящего описания кольцо A выбрано из фенила, пиридила или пиразинила.
[30] Согласно некоторым вариантам реализации настоящего описания кольцо A выбрано из 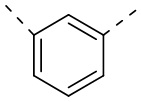 ,
, 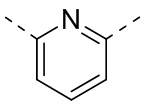 ,
, 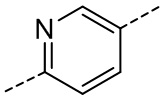 ,
, 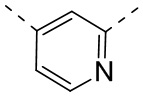 или
или 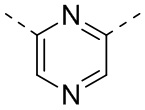 .
.
[31] Согласно некоторым вариантам реализации настоящего описания L выбран из одинарной связи, -NH-, -N(CH3)-, 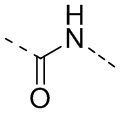 ,
, 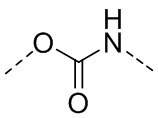 ,
, 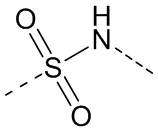 ,
, 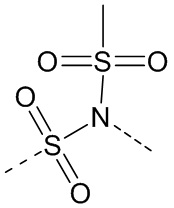 ,
, 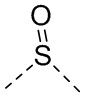 ,
, 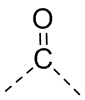 ,
, 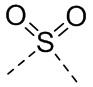 ,
, 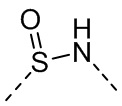 или
или 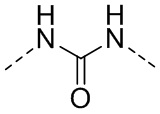 .
.
[32] Согласно некоторым вариантам реализации настоящего описания R1 выбран из H, NH2, метила, этила, изобутила, оксетанила, морфолинила, циклопропила и CH3-O-, где NH2, метил, этил, изобутил, оксетанил, морфолинил, циклопропил и CH3-O- каждый необязательно замещен 1, 2 или 3 R.
[33] Согласно некоторым вариантам реализации настоящего описания R1 выбран из H, NH2, Me, Et, 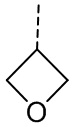 ,
, 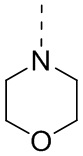 ,
,  ,
, 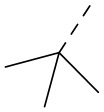 и
и 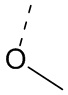 , где NH2, Me, Et, , , , и каждый необязательно замещен 1, 2 или 3 R.
, где NH2, Me, Et, , , , и каждый необязательно замещен 1, 2 или 3 R.
[34] Согласно некоторым вариантам реализации настоящего описания R1 выбран из H, NH2, CH3, CF3, Et, , 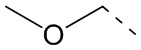 ,
, 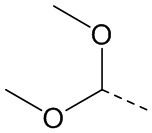 , ,
, , 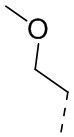 , ,
, , 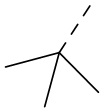 ,
, 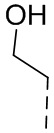 или .
или .
Согласно некоторым вариантам реализации настоящего описания R2 выбран из H, метила, этила, пропила, циклопропила и тетрагидропиранила, где метил, этил, пропил, циклопропил и тетрагидропиранил каждый необязательно замещен 1, 2 или 3 R.
[36] Согласно некоторым вариантам реализации настоящего описания R2 выбран из H, CH3,  ,
, 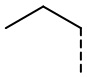 ,
, 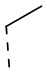 и
и 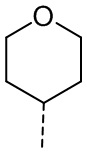 , где CH3, , , и каждый необязательно замещен 1, 2 или 3 R.
, где CH3, , , и каждый необязательно замещен 1, 2 или 3 R.
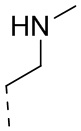
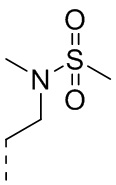
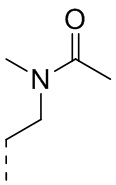
[37] Согласно некоторым вариантам реализации настоящего описания R2 выбран из H, CH3, , 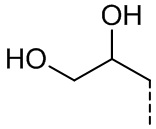 ,
,  ,
,  ,
,  ,
, 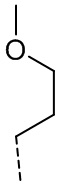 ,
, 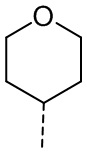 ,
, 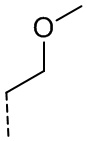 или
или 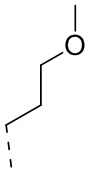 .
.
[38] Согласно некоторым вариантам реализации настоящего описания R3 выбран из H, F, Cl, Br, I, CH3, CF3 или CH3-O-.
[39] Согласно некоторым вариантам реализации настоящего описания R4 и R5 независимо выбраны из H, F, Cl, Br, I, CH3, CH3CH2- и CH3-O-.
[40] Согласно некоторым вариантам реализации настоящего описания структурное звено 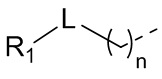 выбрано из H, CH3, NH2,
выбрано из H, CH3, NH2,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
, 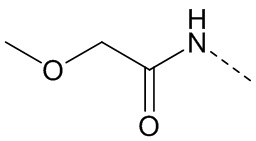 ,
, 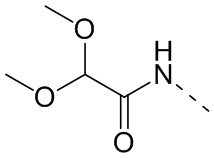 ,
, 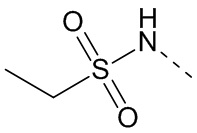 ,
, 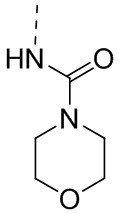 ,
, 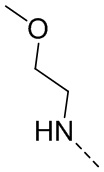 ,
, 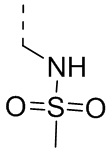 ,
, 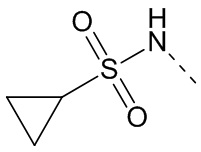 ,
, 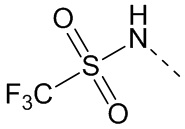 ,
, 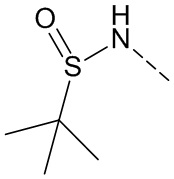 ,
, 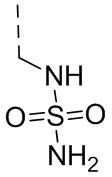 ,
, 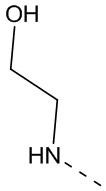 ,
, 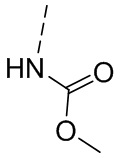 ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  ,
,  или
или  .
.
[41] Согласно некоторым вариантам реализации настоящего описания структурное звено 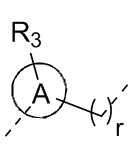 выбрано из
выбрано из  ,
,  ,
,  ,
, 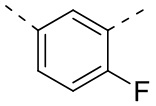 ,
, 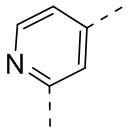 ,
,  ,
, 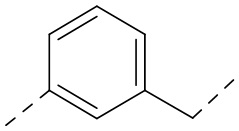 ,
, 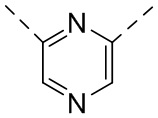 или
или 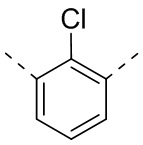 .
.
[42] Согласно некоторым вариантам реализации настоящего описания R' выбран из F, Cl, Br, I, NH2 или CH3, другие варианты являются такими, как определено в настоящем описании.
[43] Согласно некоторым вариантам реализации настоящего описания R выбран из F, Cl, Br, I, OH, NH2, метила, этила, C1-3 алкил-S(=O)2-NH-, C1-3 алкил-S(=O)2-, C1-3 алкил-C(=O)-NH- и C1-3 алкил-O-, где NH2, метил, этил, C1-3 алкил-S(=O)2-NH-, C1-3 алкил-S(=O)2-, C1-3 алкил-C(=O)-NH- и C1-3 алкил-O- каждый необязательно замещен 1, 2 или 3 R', другие варианты являются такими, как определено в настоящем описании.
[44] Согласно некоторым вариантам реализации настоящего описания R выбран из F, Cl, Br, I, OH, NH2, CH3, , , и , где NH2, CH3, , , и каждый необязательно замещен 1, 2 или 3 R', другие варианты являются такими, как определено в настоящем описании.
[45] Согласно некоторым вариантам реализации настоящего описания R выбран из F, Cl, Br, I, OH, NH2, CH3, , , , , или , другие варианты являются такими, как определено в настоящем описании.
[46] Согласно некоторым вариантам реализации настоящего описания кольцо A выбрано из фенила, пиридила или пиразинила, другие варианты являются такими, как определено выше.
[47] Согласно некоторым вариантам реализации настоящего описания кольцо A выбрано из , , , 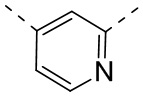 или
или 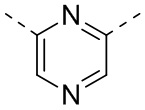 , другие варианты являются такими, как определено в настоящем описании.
, другие варианты являются такими, как определено в настоящем описании.
[48] Согласно некоторым вариантам реализации настоящего описания L выбран из одинарной связи, -NH-, -N(CH3)-, , , , , 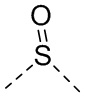 ,
,  ,
,  ,
,  или , другие варианты являются такими, как определено в настоящем описании.
или , другие варианты являются такими, как определено в настоящем описании.
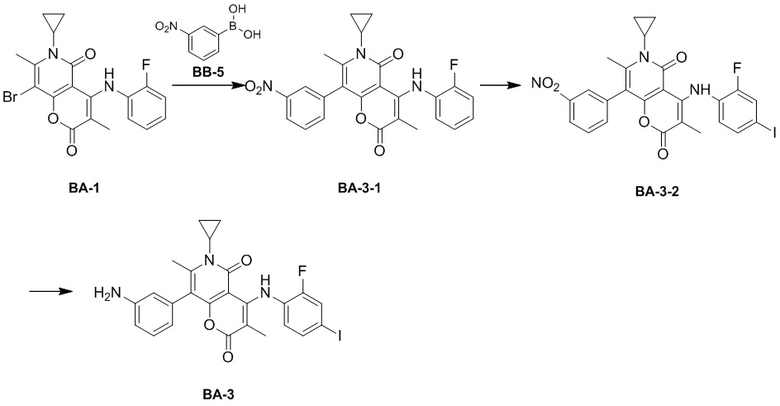
[49] Согласно некоторым вариантам реализации настоящего описания R1 выбран из H, NH2, метила, этила, изобутила, оксетанила, морфолинила, циклопропила и CH3-O-, где NH2, метил, этил, изобутил, оксетанил, морфолинил, циклопропил и CH3-O- каждый необязательно замещен 1, 2 или 3 R, другие варианты являются такими, как определено в настоящем описании.
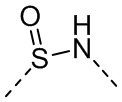
[50] Согласно некоторым вариантам реализации настоящего описания R1 выбран из H, NH2, Me, Et, , 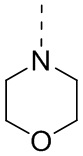 , , и , где NH2, Me, Et, , , , и каждый необязательно замещен 1, 2 или 3 R, другие варианты являются такими, как определено в настоящем описании.
, , и , где NH2, Me, Et, , , , и каждый необязательно замещен 1, 2 или 3 R, другие варианты являются такими, как определено в настоящем описании.
[51] Согласно некоторым вариантам реализации настоящего описания R1 выбран из H, NH2, CH3, CF3, Et, , , , 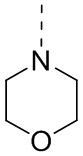 , , , , и , другие варианты являются такими, как определено в настоящем описании.
, , , , и , другие варианты являются такими, как определено в настоящем описании.
[52] Согласно некоторым вариантам реализации настоящего описания R2 выбран из H, метила, этила, пропила, циклопропила и тетрагидропиранила, где метил, этил, пропил, циклопропил и тетрагидропиранил каждый необязательно замещен 1, 2 или 3 R, другие варианты являются такими, как определено в настоящем описании.
[53] Согласно некоторым вариантам реализации настоящего описания R2 выбран из H, CH3, , , и 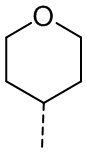 , где CH3, , , и каждый необязательно замещен 1, 2 или 3 R, другие варианты являются такими, как определено в настоящем описании.
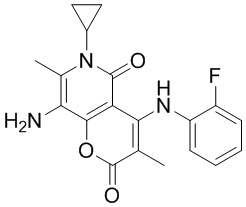
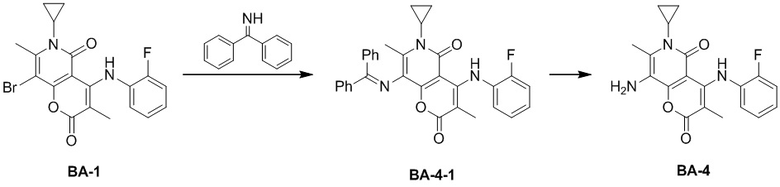
, где CH3, , , и каждый необязательно замещен 1, 2 или 3 R, другие варианты являются такими, как определено в настоящем описании.
[54] Согласно некоторым вариантам реализации настоящего описания R2 выбран из H, CH3, , , , , , ,  , или , другие варианты являются такими, как определено в настоящем описании.
, или , другие варианты являются такими, как определено в настоящем описании.
[55] Согласно некоторым вариантам реализации настоящего описания R3 выбран из H, F, Cl, Br, I, CH3, CF3 или CH3-O-, другие варианты являются такими, как определено в настоящем описании.
[56] Согласно некоторым вариантам реализации настоящего описания R4 и R5 независимо выбраны из H, F, Cl, Br, I, CH3, CH3CH2-, CH3-O-, другие варианты являются такими, как определено в настоящем описании.
[57] Согласно некоторым вариантам реализации настоящего описания структурное звено выбрано из H, CH3, NH2, , , , , , , , , , , , , , , , , , , , , , , , , , , или , другие варианты являются такими, как определено в настоящем описании.
[58] Согласно некоторым вариантам реализации настоящего описания структурное звено выбрано из  , , ,
, , ,  ,
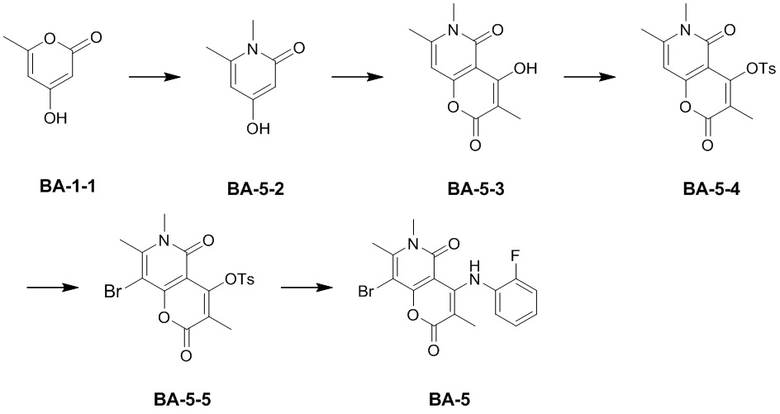
,  , , ,
, , ,  или , другие варианты являются такими, как определено в настоящем описании.
или , другие варианты являются такими, как определено в настоящем описании.
[59] Другие варианты реализации настоящего раскрытия получены путем произвольных комбинаций указанных выше вариантов.
[60] Согласно некоторым вариантам реализации настоящего описания соединение, его фармацевтически приемлемая соль и его изомер выбраны из
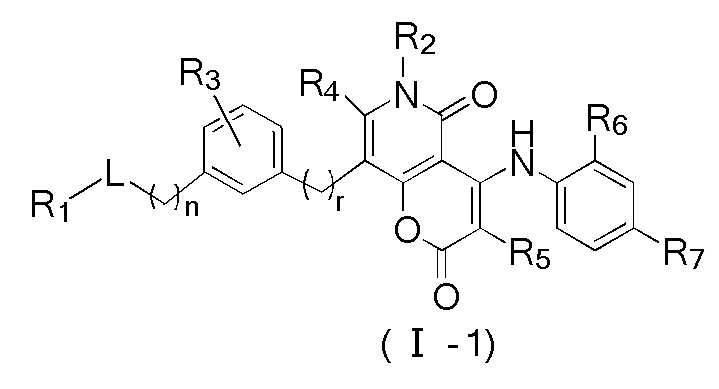
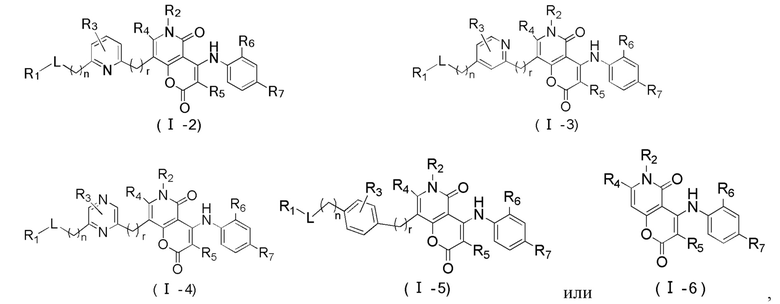
[61] где, R1, R2, R3, R4, R5, R6, R7, L, r и n являются такими, как определено в настоящем описании.
[62] В настоящем описании также предложено соединение, представленное формулой ниже, его фармацевтически приемлемая соль или его таутомер, где соединение выбрано из
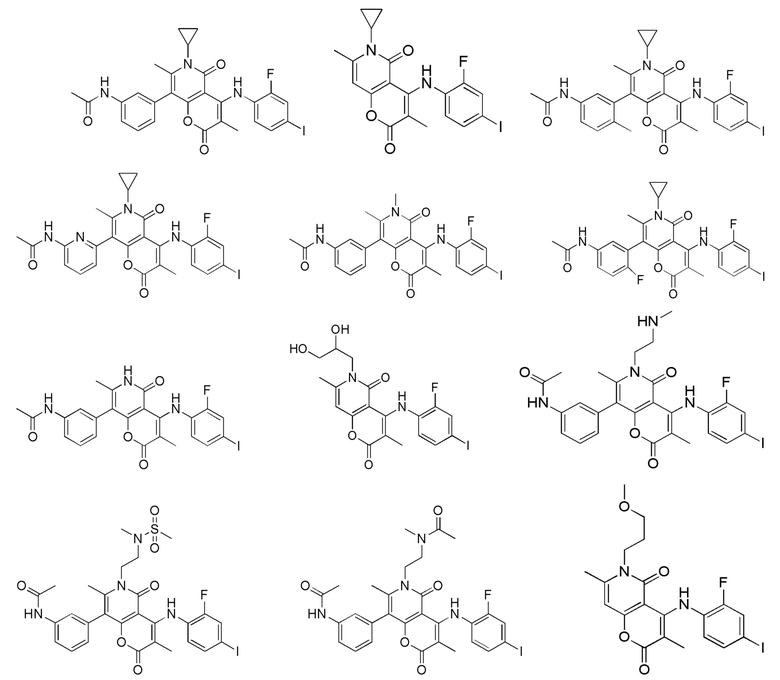
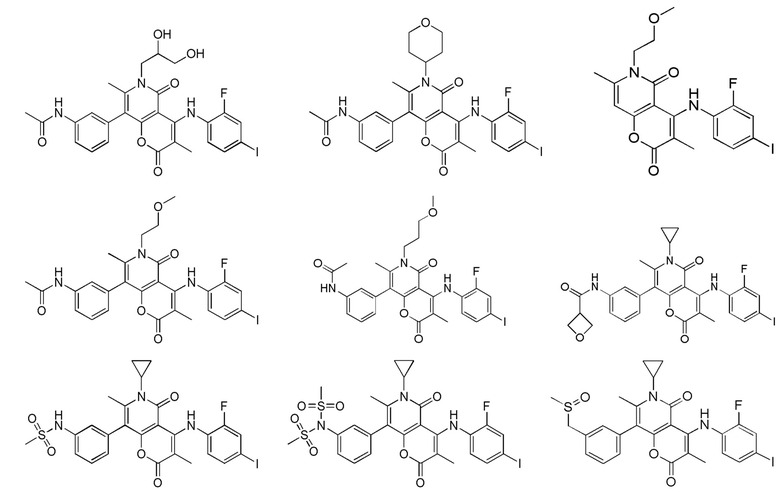
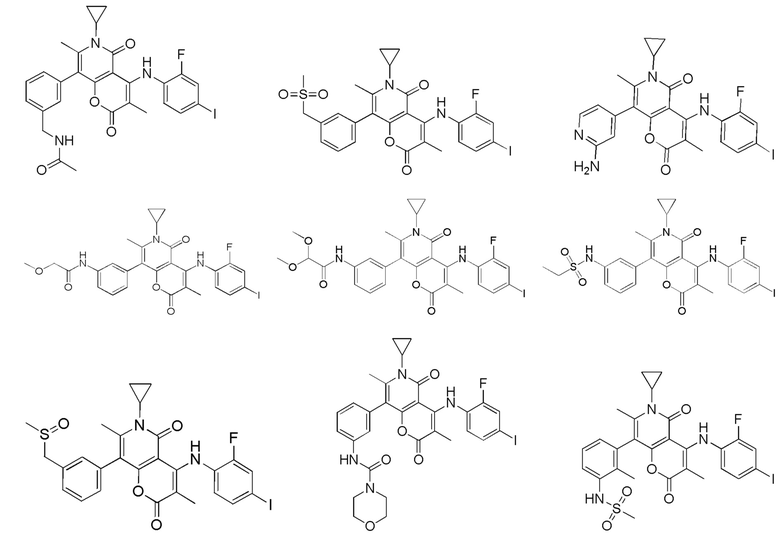
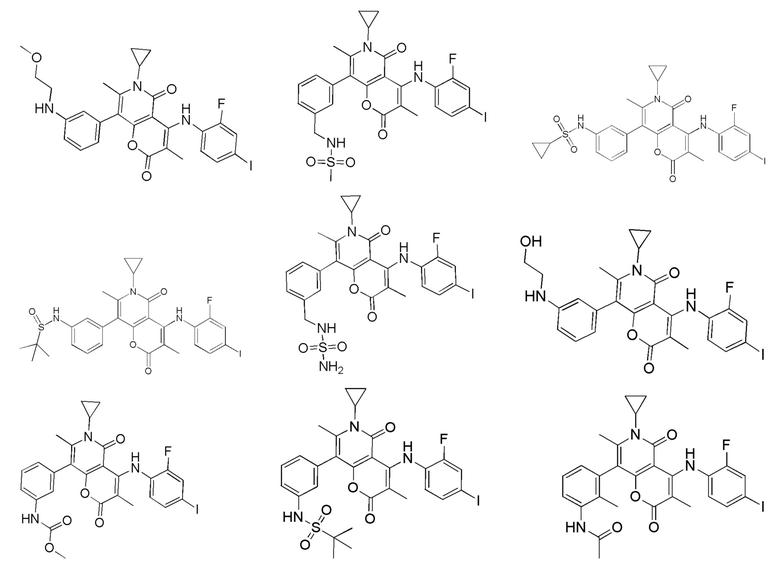
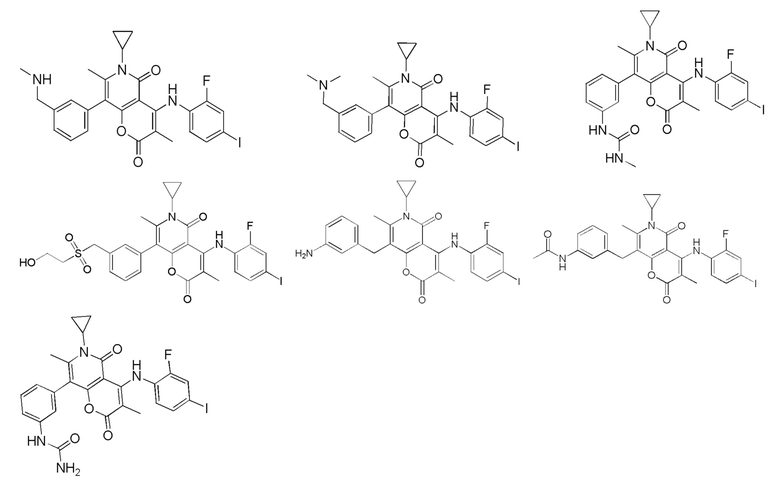
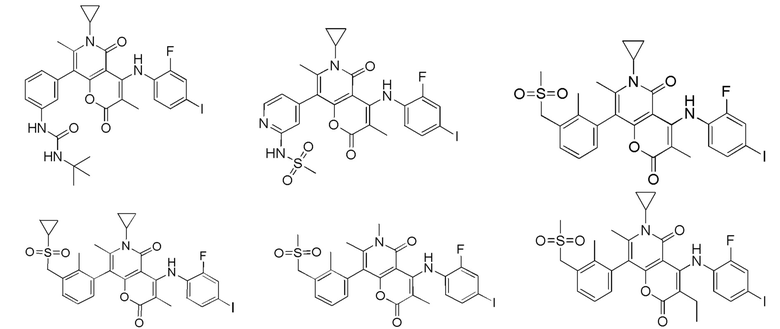
[63] Согласно некоторым вариантам реализации настоящего описания соединение выбрано из
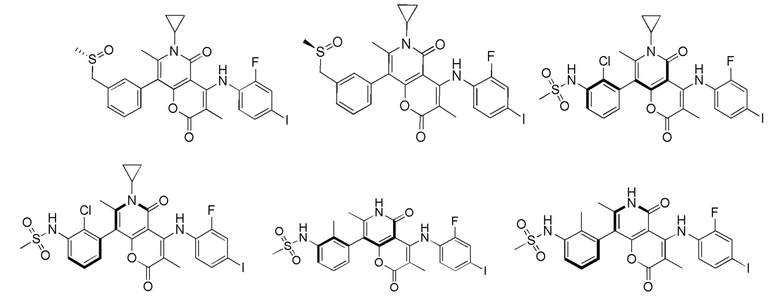
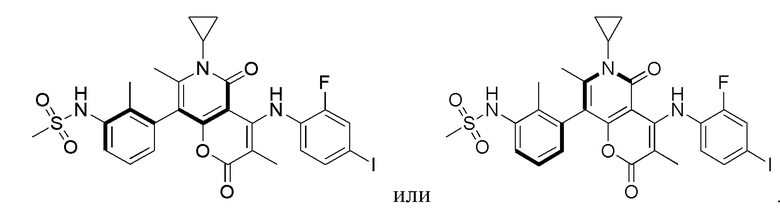
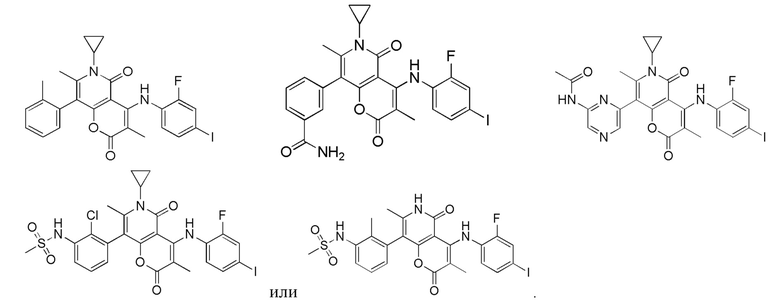
[64] Согласно некоторым вариантам реализации настоящего описания соль выбрана из гидрохлорида или формиата.
[65] Согласно некоторым вариантам реализации настоящего описания гидрохлорид выбран из
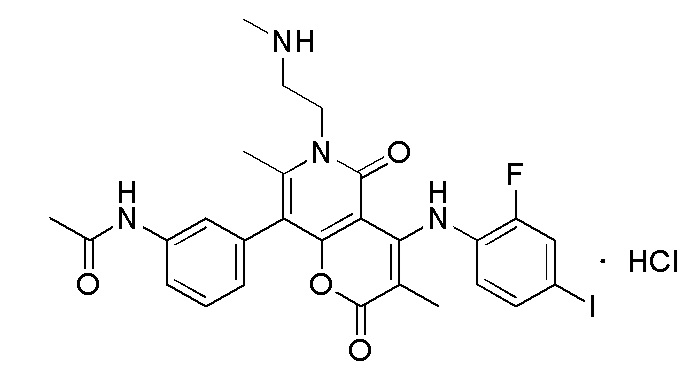 ,
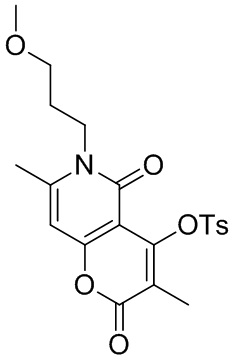
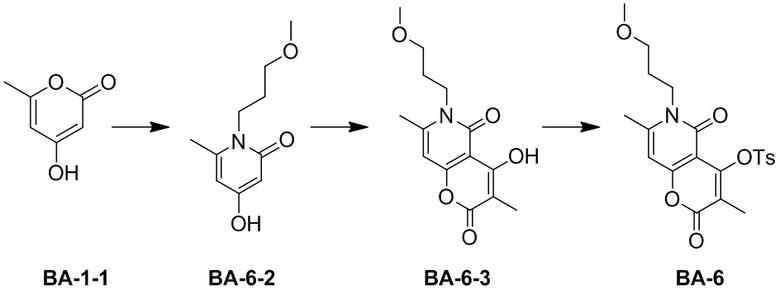
, 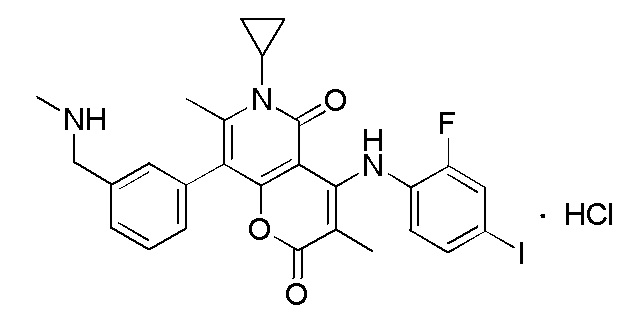 или
или 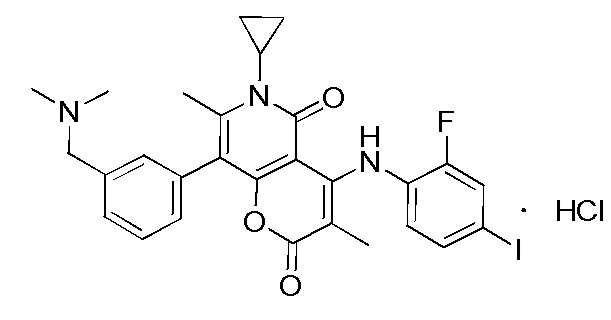 .
.
[66] В настоящем описании также предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли в качестве активного ингредиента и фармацевтически приемлемый носитель.
[67] В настоящем описании также предложено применение соединения или его фармацевтически приемлемой соли или композиции для получения лекарственного средства для лечения заболеваний, связанных с MEK.
Определения и описание
[68] Если не указано иное, следующие термины при использовании в описании и формуле настоящего изобретения имеют следующие значения. Конкретный термин или фраза не должны считаться неопределенными или неясными в отсутствие конкретного определения, но должны пониматься в обычном смысле. Когда в документе появляется торговое наименование, оно предназначено для обозначения соответствующего товара или его активного ингредиента. Термин «фармацевтически приемлемый» используется в настоящем документе в отношении тех соединений, материалов, композиций и/или лекарственных форм, которые подходят для использования в контакте с тканями человека и животных в рамках здравого медицинского суждения без чрезмерной токсичности, раздражения, аллергической реакции или других проблем или осложнений, соразмерно с разумным соотношением пользы/риска.
[69] Термин «фармацевтически приемлемая соль» относится к соли соединения согласно настоящему описанию, которая получена путем взаимодействия соединения, имеющего определенный заместитель согласно настоящему описанию, с относительно нетоксичной кислотой или основанием. Когда соединение согласно настоящему описанию содержит относительно кислотную функциональную группу, соль присоединения основания может быть получена путем приведения нейтральной формы соединения в контакт с достаточным количеством основания в чистом растворе или подходящем инертном растворителе. Фармацевтически приемлемая соль присоединения основания включает соль натрия, калия, кальция, аммония, органического амина или магния или подобные соли. Когда соединение согласно настоящему описанию содержит относительно основную функциональную группу, соль присоединения кислоты может быть получена путем приведения нейтральной формы соединения в контакт с достаточным количеством кислоты в чистом растворе или подходящем инертном растворителе. Примеры фармацевтически приемлемой соли присоединения кислоты включают соль неорганической кислоты, где неорганическая кислота включает, например, соляную кислоту, бромистоводородную кислоту, азотную кислоту, угольную кислоту, бикарбонат, фосфорную кислоту, гидрофосфат, дигидрофосфат, серную кислоту, гидросульфат, йодистоводородную кислоту, фосфористую кислоту и т. п.; и соль органической кислоты, где органическая кислота включает, например, уксусную кислоту, пропионовую кислоту, изомасляную кислоту, малеиновую кислоту, малоновую кислоту, бензойную кислоту, янтарную кислоту, субериновую кислоту, фумаровую кислоту, молочную кислоту, миндальную кислоту, фталевую кислоту, бензолсульфоновую кислоту, п-толуолсульфокислоту, лимонную кислоту, винную кислоту и метансульфоновую кислоту и т. п.; и соль аминокислоты (например, аргинина и тому подобное) и соль органической кислоты, такой как глюкуроновая кислота и тому подобное. Некоторые конкретные соединения согласно настоящему описанию, которые содержат как основные, так и кислотные функциональные группы, могут быть превращены в любую соль присоединения кислоты или основания.
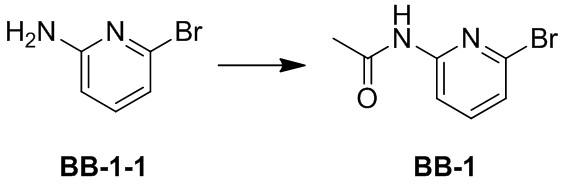
[70] Предпочтительно, соль приводят в контакт с основанием или кислотой традиционным способом, и затем исходное соединение выделяют для получения нейтральной формы соединения. Исходная форма соединения отличается от его различных солевых форм определенными физическими свойствами, такими как различная растворимость в полярных растворителях.
[71] Используемый в настоящем документе термин «фармацевтически приемлемая соль» представляет собой производное соединения согласно настоящему описанию, где исходное соединение модифицируют путем образования соли с кислотой или основанием. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, основные, такие как соли неорганических или органических кислот и аминов, кислотные, такие как щелочные или органические соли карбоновых кислот, и тому подобное. Фармацевтически приемлемые соли включают традиционные нетоксичные соли или четвертичные аммониевые соли исходного соединения, такие как соли, образованные из нетоксичных неорганических или органических кислот. Традиционные нетоксичные соли включают, но не ограничиваются ими, соли, полученные из неорганических и органических кислот, выбранных из группы, состоящей из 2-ацетоксибензойной кислоты, 2-гидроксиэтансульфоновой кислоты, уксусной кислоты, аскорбиновой кислоты, бензолсульфоновой кислоты, бензойной кислоты, бикарбоната, угольной кислоты, лимонной кислоты, этилендиаминтетрауксусной кислоты, этандисульфоновой кислоты, этансульфоновой кислоты, фумаровой кислоты, глюкогептозы, глюконовой кислоты, глутаминовой кислоты, гликолевой кислоты, бромистоводородной кислоты, соляной кислоты, гидроиодата, гидроксила, гидроксинафталина, изетионата, молочной кислоты, лактозы, додецилсульфокислоты, малеиновой кислоты, яблочной кислоты, миндальной кислоты, метансульфоновой кислоты, азотной кислоты, щавелевой кислоты, памовой кислоты, пантотеновой кислоты, фенилуксусной кислоты, фосфорной кислоты, полигалактальдегида, пропионовой кислоты, салициловой кислоты, стеариновой кислоты, уксусной кислоты, янтарной кислоты, аминосульфоновой кислоты, п-аминобензолсульфоновой кислоты, серной кислоты, танина, винной кислоты и п-толуолсульфоновой кислоты.
[72] Фармацевтически приемлемые соли согласно настоящему описанию могут быть синтезированы из исходного соединения, содержащего кислоту или основание, традиционным химическим способом. В целом, такие соли получают взаимодействием указанных соединений в форме свободной кислоты или основания со стехиометрически подходящим основанием или кислотой в воде или органическом растворителе или их смеси. В целом, неводная среда, такая как эфир, этилацетат, этанол, изопропанол или ацетонитрил, является предпочтительной.
[73] Некоторые соединения согласно настоящему описанию могут иметь асимметричные атомы углерода (оптические центры) или двойные связи. Рацематы, диастереомеры, геометрические изомеры и отдельные изомеры включены в объем настоящего описания.
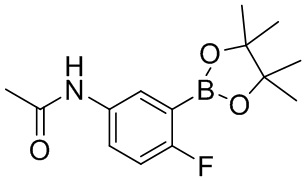
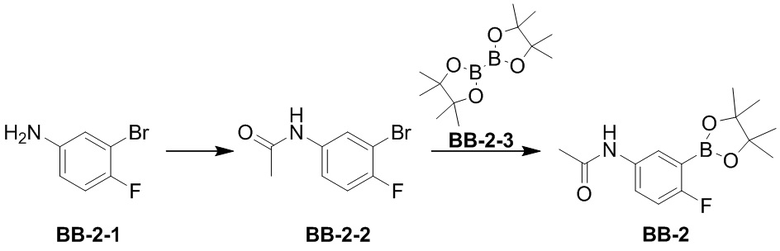
[74] Если не указано иное, абсолютная конфигурация стереогенного центра представлена сплошной клиновидной связью ( ) и пунктирной клиновидной связью (
) и пунктирной клиновидной связью ( ), волнистая линия (
), волнистая линия ( ) представляет собой сплошную клиновидную связь () или пунктирную клиновидную связь () и относительная конфигурация стереогенного центра представлена сплошной прямой связью (
) представляет собой сплошную клиновидную связь () или пунктирную клиновидную связь () и относительная конфигурация стереогенного центра представлена сплошной прямой связью ( ) и пунктирной прямой связью (
) и пунктирной прямой связью ( ). Когда соединения, описанные в настоящем документе, содержат олефиновые двойные связи или другие центры геометрической асимметрии, если не указано иное, они включают геометрические изомеры E, Z. Аналогично, все таутомерные формы включены в объем описания.
). Когда соединения, описанные в настоящем документе, содержат олефиновые двойные связи или другие центры геометрической асимметрии, если не указано иное, они включают геометрические изомеры E, Z. Аналогично, все таутомерные формы включены в объем описания.
[75] Термин «обогащенный одним изомером» относится к содержанию одного из изомеров, составляющего <100%, и ≥60%, предпочтительно ≥70%, предпочтительно ≥80%, предпочтительно ≥90%, предпочтительно ≥95%, предпочтительно ≥96%, предпочтительно ≥97%, предпочтительно ≥98%, предпочтительно ≥99%, предпочтительно ≥99.5%, предпочтительно ≥99.6%, предпочтительно ≥99.7%, предпочтительно ≥99.8%, предпочтительно ≥99.9%.
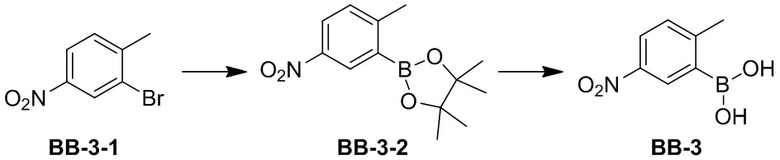
[76] Избыток изомера относится к разнице между относительными процентными содержаниями двух изомеров. Например, когда содержание одного из изомеров составляет 90%, а другого - 10%, тогда избыток изомера составляет 80%.
[77] «(+)» обозначает правое вращение, «(-)» обозначает левое вращение или «(±)» обозначает рацемизацию.
[78] Соединения согласно настоящему описанию могут существовать в определенных геометрических или стереоизомерных формах. В данном описании рассматриваются все такие соединения, включая цис- и транс-изомеры, (-)- и (+)-энантиомеры, (R)- и (S)-энантиомеры, диастереомерные изомеры, (D)-изомеры, (L)-изомеры, и рацемические и другие их смеси, такие как энантиомеры или обогащенные диастереомерами смеси, которые входят в объем настоящего описания. Дополнительные асимметричные атомы углерода могут присутствовать в заместителях, таких как алкил. Все указанные изомеры и их смеси включены в объем настоящего описания.
[79] Оптически активный (R) - и (S)-изомер или D и L изомер может быть получен с использованием хирального синтеза или хиральных реагентов или других традиционных методик. Если необходимо получить один тип энантиомера определенного соединения согласно настоящему описанию, чистый требуемый энантиомер может быть получен асимметричным синтезом или дериватизирующим действием хирального вспомогательного вещества с последующим разделением полученной диастереомерной смеси и отщеплением вспомогательной группы. Альтернативно, когда молекула содержит основную функциональную группу (например, аминогруппу) или кислотную функциональную группу (например, карбоксигруппу), соединение реагирует с подходящей оптически активной кислотой или основанием с образованием соли диастереомерного изомера, которую затем подвергают разделению диастереомеров с помощью традиционного в данной области способа получения чистого энантиомера. Кроме того, энантиомер и диастереоизомер, как правило, выделяют с помощью хроматографии, в которой используется хиральная стационарная фаза и, возможно, в сочетании с методом химической дериватизации (например, карбамат, полученный из амина).
[80] Соединение согласно настоящему описанию может содержать неестественную долю изотопа атома в одном или нескольких атомах, которые составляют соединение. Например, соединение может быть помечено радиоактивным изотопом, таким как тритий (3H), йод-125 (125I) или C-14 (14C). Все превращения, претерпеваемые изотопными композициями настоящего описания, радиоактивными или нет, включены в объем настоящего описания.
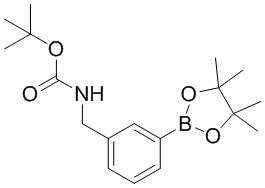
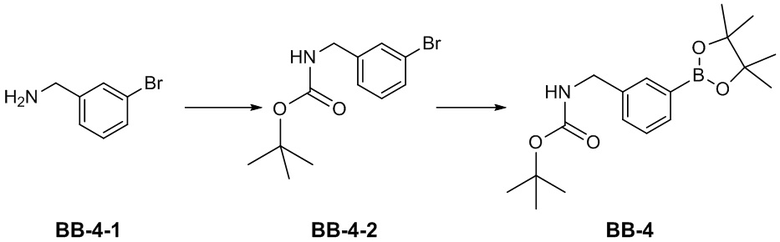
[81] Термин «замещенный» означает, что один или более атомов водорода на конкретном атоме замещены заместителем, включая варианты дейтерия и водорода, при условии, что валентность конкретного атома является нормальной и замещенное соединение является стабильным. Когда заместитель представляет собой кислород (то есть =O), это означает, что замещены два атома водорода. Положения на ароматическом кольце не могут быть замещены кетоном. Термин «необязательно замещенный» означает, что атом может быть замещен или не замещен заместителем, если не указано иное, тип и количество заместителей могут быть произвольными, если они достижимы химически.
[82] Когда какая-либо переменная (например, R) встречается в составе или структуре соединения более одного раза, определение переменной в каждом случае является независимым. Таким образом, например, если группа замещена 0-2 R, группа может быть необязательно замещена до двух R, причем определение R в каждом случае является независимым. Более того, комбинация заместителя и/или его варианта допускается только тогда, когда данная комбинация приводит к стабильному соединению.
[83] Когда количество связывающей группы равно 0, например -(CRR)0-, это означает, что связывающая группа представляет собой одинарную связь.
[84] Когда одна из переменных выбрана из одинарной связи, это означает, что две группы, связанные одинарной связью, связаны непосредственно. Например, когда L в A-L-Z представляет собой одинарную связь, структура A-L-Z фактически представляет собой A-Z.
[85] Когда заместитель является вакантным, это означает, что заместитель не существует, например, когда X является вакантным в A-X, структура A-X фактически представляет собой A. Когда заместитель может быть присоединен к более чем одному атому в кольце заместитель может быть связан с любым атомом в кольце, например, в структурном звене 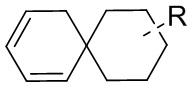 или
или 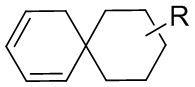 замещение заместителем R может иметь место в любом положении на циклогексиле или циклогексадиене. Когда перечисленные заместители не указывают, посредством какого атома они присоединены к замещенной группе, такие заместители могут быть связаны через любой из атомов, например, пиридил в качестве заместителя может быть присоединен к замещенной группе через любой из атомов углерода на пиридиновом кольце. Когда перечисляющая связывающая группа не указывает направление для связи, направление связи является произвольным, например, если связывающая группа L, содержащаяся в
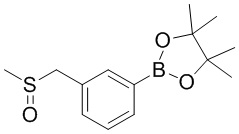
замещение заместителем R может иметь место в любом положении на циклогексиле или циклогексадиене. Когда перечисленные заместители не указывают, посредством какого атома они присоединены к замещенной группе, такие заместители могут быть связаны через любой из атомов, например, пиридил в качестве заместителя может быть присоединен к замещенной группе через любой из атомов углерода на пиридиновом кольце. Когда перечисляющая связывающая группа не указывает направление для связи, направление связи является произвольным, например, если связывающая группа L, содержащаяся в 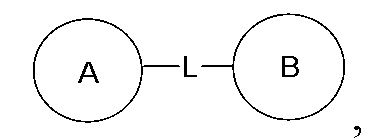 представляет собой -M-W-, тогда -M-W- может связывать кольцо A и кольцо B с образованием
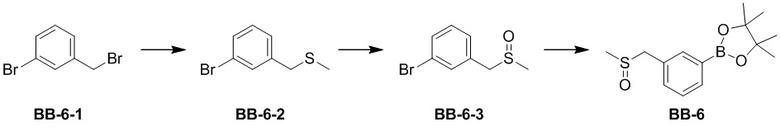
представляет собой -M-W-, тогда -M-W- может связывать кольцо A и кольцо B с образованием 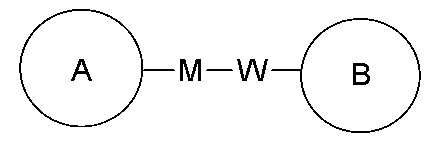 в направлении, аналогичном порядку чтения слева направо, и образованием
в направлении, аналогичном порядку чтения слева направо, и образованием 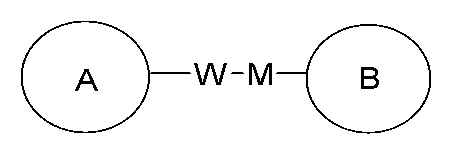 в направлении, противоположном порядку чтения слева направо. Комбинации связывающих групп, заместителей и/или их вариантов допустимы, только если такие комбинации приводят к стабильным соединениям.
в направлении, противоположном порядку чтения слева направо. Комбинации связывающих групп, заместителей и/или их вариантов допустимы, только если такие комбинации приводят к стабильным соединениям.
[86] Если не указано иное, термин «гетеро» представляет собой гетероатом или гетероатомную группу (например, атомную группу, содержащую гетероатом), включая атом, кроме углерода (С) и водорода (Н), и атомную группу, содержащую указанный выше гетероатом, например, кислород (O), азот (N), серу (S), кремний (Si), германий (Ge), алюминий (Al), бор (B), -O-, -S-, =O, =S, -C(=O)O-, -C(=O)-, -C(=S)-, -S(=O), -S(=O)2-, и группу, состоящую из -C(=O)N(H)-, -N(H)-, -C(=NH)-, -S(=O)2N(H)- и -S(=O)N(H)-, каждая из которых необязательно замещена.
[87] Если не указано иное, термин «кольцо» относится к замещенному или незамещенному циклоалкилу, гетероциклоалкилу, циклоалкенилу, гетероциклоалкенилу, циклоалкинилу, гетероциклоалкинилу, арилу или гетероарилу. Так называемое кольцо включает одно кольцо, двойное кольцо, спирокольцо, конденсированное кольцо или кольцо с мостиковой связью. Число атомов в кольце обычно определяется как число членов в кольце, например, «5-7-членное кольцо» означает, что в кольце находится от 5 до 7 атомов. Если не указано иное, кольцо необязательно содержит от 1 до 3 гетероатомов. Следовательно, «5-7-членное кольцо» включает, например, фенил, пиридинил и пиперидинил; с другой стороны, термин «5-7-членное гетероциклоалкильное кольцо» включает пиридил и пиперидинил, но не включает фенил. Термин «кольцо» также включает кольцевую систему, содержащую по меньшей мере одно кольцо, где каждое кольцо независимо соответствует приведенному выше определению.
[88] Если не указано иное, термин «гетероцикл» или «гетероциклил» относится к стабильному моноциклическому, бициклическому или трициклическому кольцу, содержащему гетероатом или гетероатомную группу, которое может быть насыщенным, частично ненасыщенным или ненасыщенным (ароматическим) и может содержать атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, независимо выбранных из N, O и S, где любой из указанных выше гетероциклов может быть конденсирован с бензольным кольцом с образованием бициклического кольца. Гетероатомы азота и серы могут быть необязательно окислены (то есть, NO и S(O)p, p равно 1 или 2). Атом азота может быть замещенным или незамещенным (то есть, N или NR, где R представляет собой Н или другие заместители, уже определенные в настоящем документе). Гетероцикл может быть присоединен к боковой группе любого гетероатома или атома углерода с образованием стабильной структуры. Если полученное соединение является стабильным, гетероцикл, описанный в настоящем документе, может иметь замещение по положению углерода или азота. Атом азота на гетероцикле необязательно кватернизован. В предпочтительном варианте реализации, когда общее число атомов S и O в гетероцикле составляет более 1, гетероатомы не являются соседними друг к другу. В другом предпочтительном варианте общее число атомов S и O в гетероцикле составляет не более 1. Используемый в настоящем документе термин «ароматическая гетероциклическая группа» или «гетероарил» относится к стабильному 5-, 6- или 7-членному моноциклическому или бициклическому или 7-, 8-, 9- или 10-членному бициклическому гетероциклическому ароматическому кольцу, которое содержит атомы углерода и 1, 2, 3 или 4 кольцевых гетероатома, независимо выбранных из N, O и S. Атом азота может быть замещенным или незамещенным (то есть, N или NR, где R представляет собой Н или другие заместители, уже определенные в настоящем документе). Гетероатомы азота и серы могут быть необязательно окислены (то есть, NO и S(O)p, p равно 1 или 2). Стоит отметить, что общее число атомов S и O ароматического гетероцикла не превышает одного. Мостиковое кольцо также включено в определение гетероцикла. Мостиковое кольцо образуется, когда один или несколько атомов (то есть, C, O, N или S) связывают два несмежных атома углерода или азота. Предпочтительное мостиковое кольцо включает, но не ограничивается ими, один атом углерода, два атома углерода, один атом азота, два атома азота и одну группу углерод-азот. Стоит отметить, что мостик всегда превращает моноциклическое кольцо в трициклическое кольцо. В мостиковом кольце заместитель на кольце также может присутствовать на указанном мостике.
[89] Примеры гетероциклического соединения включают, но не ограничиваются ими: акридинил, азоцинил, бензимидазолил, бензофуранил, бензомеркаптофуранил, бензомеркаптофенил, бензоксазолил, бензоксазолинил, бензотиазолил, бензотриазолил, бензотетразолил, бензоизоксазолил, бензоизотиазолил, бензоимидазолинил, карбазолил, 4aH-карбазолил, карболинил, бензодигидропиранил, хромен, циннолинил декагидрохинолинил, 2Н,6Н-1,5,2-дитиазинил, дигидрофуро [2,3-b]тетрагидрофуранил, фуранил, фуразанил, имидазолидинил, имидазолинил, имидазолил, 1H-индазолил, индоленил, индолинил, индолизинил, индолил, 3Н-индолил, изобензофуранил, изоиндолил, изоиндолинил, изохинолинил, изотиазолил, изоксазолил, метилендиоксифенил, морфолинил, нафтиридинил, октагидро-изохинолинил, оксадиазолил, 1,2,3-оксадиазолил, 1,2,4-оксадиазолил, 1,2,5-оксадиазолил, 1,3,4-оксадиазолил, оксазолидинил, оксазолил, гидроксиндолил, пиримидинил, фенантридинил, фенантролинил, феназин, фенотиазин, бензоксантинил, фенолоксазинил, фталазинил, пиперазинил, пиперидинил, пиперидонил, 4-пиперидонил, пиперонил, птеридинил, пуринил, пиранил, пиразинил, пиразолидинил, пиразолинил, пиразолил, пиридазинил, пиридо-оксазолил, пиридо-имидазолил, пиридо-тиазолил, пиридинил, пирролидинил, пирролинил, 2Н-пирролил, пирролил, хиназолинил, хинолинил, 4Н-хинолизинил, хиноксалинил, хинуклидинил, тетрагидрофуранил, тетрагидроизохинолинил, тетрагидрохинолинил, тетразолил, 6H-1,2,5-тиадиазинил, 1,2,3-тиадиазолил, 1,2,4-тиадиазолил, 1,2,5-тиадиазолил, 1,3,4-тиадиазолил, тиантренил, тиазолил, изотиазолилтиенил, тиенооксазолил, тиенотиазолил, тиеноимидазолил, тиенил, триазинил, 1Н-1,2,3-триазолил, 2Н-1,2,3-триазолил, 1Н-1,2,4-триазолил, 4Н-1,2,4-триазолил и ксантенил. Также включены соединения с конденсированными кольцами и спиросоединения.
[90] Если не указано иное, термин «гидрокарбил» или его гипонимы (например, алкил, алкенил, алкинил и арил и т. д.) сами по себе или как часть другого заместителя относятся к линейному, разветвленному или циклическому углеводородному радикалу или любой их комбинации, они могут быть полностью насыщенными (например, алкил), моно- или полиненасыщенными (например, алкенил, алкинил и арил), могут быть моно-, ди- или полизамещенными, могут быть одновалентными (например, метил), двухвалентными (например, метилен) или многовалентными (например, метенил), также могут включать двухвалентную или многовалентную группу, имеют указанное число атомов углерода (например, C1-C12 обозначает от 1 до 12 атомов углерода, C1-C12 выбирают из C1, C2, C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12; C3-12 выбирают из C3, C4, C5, C6, C7, C8, C9, C10, C11 и C12). Термин «гидрокарбил» включает, но не ограничивается ими, алифатический гидрокарбил и ароматический гидрокарбил, алифатический гидрокарбил включает линейный и циклический гидрокарбил, в частности включает, но не ограничивается ими, алкил, алкенил и алкинил. Ароматический гидрокарбил включает, но не ограничивается ими, 6-12-членный ароматический гидрокарбил, такой как фенил, нафтил и тому подобное. В некоторых вариантах реализации термин «гидрокарбил» относится к линейной или разветвленной группе или их комбинации, которая может быть полностью насыщенной, моно- или полиненасыщенной и может включать двухвалентную или многовалентную группу. Примеры насыщенной гидрокарбильной группы включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, трет-бутил, изобутил, втор-бутил, изобутил, циклогексил, (циклогексил)метил, циклопропилметил и гомолог или изомер н-амильной, н-гексильной, н-гептильной, н-октильной и других групп атомов. Ненасыщенный гидрокарбил содержит одну или несколько двойных или тройных связей. Примеры ненасыщенного алкила включают, но не ограничиваются ими, винил, 2-пропенил, бутенил, кротил, 2-изопентенил, 2-(бутадиенил), 2,4-пентадиенил, 3-(1,4-пентадиенил), этинил, 1- и 3-пропинил, 3-бутинил и более высшие гомологи и изомеры.
[91] Если не указано иное, термин «гетерогидрокарбил» или его гипонимы (такие как гетероалкил, гетероалкенил, гетероалкинил и гетероарил и т. д.) сами по себе или как часть другого заместителя относятся к стабильному линейной, разветвленной или циклической углеводородной группе или любая их комбинации, которая содержит указанное число атомов углерода и по меньшей мере один гетероатом. В некоторых вариантах реализации термин «гетероалкил» сам по себе или в сочетании с другим термином относится к стабильной линейной цепи, разветвленному углеводородному радикалу или их комбинации, которая имеет определенное число атомов углерода и по меньшей мере один гетероатом. В конкретном варианте реализации гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены и атом азота необязательно кватернизован. Гетероатом или гетероатомная группа может быть расположена по любому внутреннему положению гетерогидрокарбила, включая положение, по которому гидрокарбил присоединяется к остальной части молекулы. Но термины «алкокси», «алкиламино» и «алкилтио» (или тиоалкил) используются в общепринятом значении и относятся к алкильной группе, соединенной с остальной частью молекулы через атом кислорода, амино или атом серы, соответственно. Примеры включают, но не ограничиваются ими, -CH2-CH2-O-CH3, -CH2-CH2-NH-CH3, -CH2-CH2-N(CH3)-CH3, -CH2-S-CH2-CH3, -CH2-CH2, -S(O)-CH3, -CH2-CH2-S(O)2-CH3, -CH=CH-O-CH3, -CH2-CH=N-OCH3 и -CH=CH-N(CH3)-CH3. Может присутствовать до двух последовательных гетероатомов, например как в -CH2-NH-OCH3.
[92] Если не указано иное, термин «циклогидрокарбил», «гетероциклогидрокарбил» или его гипонимы (такие как арил, гетероарил, циклоалкил, гетероциклоалкил, циклоалкенил, гетероциклоалкенил, циклоалкинил, гетероциклоалкинил и т.д.) сами по себе или в сочетании с другим термином относятся к циклизованному «гидрокарбилу» или «гетерогидрокарбилу». Кроме того, для гетерогидрокарбила или гетероциклогидрокарбила (например, гетероалкила и гетероциклоалкила) один гетероатом может занимать положение, по которому гетероцикл присоединяется к остатку молекулы. Примеры циклогидрокарбила включают, но не ограничиваются ими, циклопентил, циклогексил, 1-циклогексенил, 3-циклогексенил, циклогептил и тому подобное. Неограничивающие примеры гетероциклила включают 1-(1,2,5,6-тетрагидропиридил), 1-пиперидинил, 2-пиперидинил, 3-пиперидинил, 4-морфолинил, 3-морфолинил, тетрагидрофуран-2-ил, тетрагидрофуран-3-ил, тетрагидротиофен-2-ил, тетрагидротиофен-3-ил, 1-пиперазинил и 2-пиперазинил.
[93] Если не указано иное, термин «алкил» относится к линейной или разветвленной насыщенной углеводородной группе, может быть монозамещенным (например, -CH2F) или полизамещенным (например, -CF3), может быть одновалентным (например, метил), двухвалентным (например, метилен) или многовалентным (например, метенил). Примеры алкила включают метил (Me), этил (Et), пропил (например, н-пропил и изопропил), бутил (например, н-бутил, изобутил, втор-бутил, трет-бутил), пентил (например, н-пентил, изопентил, неопентил) и тому подобное.
[94] Если не указано иное, «алкенил» относится к алкильной группе, имеющей одну или несколько углерод-углеродных двойных связей в любом положении в цепи, которая может быть моно- или полизамещенной и может быть одновалентной, двухвалентной или поливалентной. Примеры алкенила включают винил, пропенил, бутенил, пентенил, гексенил, бутадиенил, пентадиенил, гексадиенил и т.д.
[95] Если не указано иное, «алкинил» относится к алкильной группе, имеющей одну или несколько углерод-углеродных тройных связей в любом положении в цепи, которая может быть моно- или полизамещенной и может быть одновалентной, двухвалентной или поливалентной. Примеры алкинила включают этинил, пропинил, бутинил, пентинил и т.д.
[96] Если не указано иное, циклоалкил включает любой стабильный циклический или полициклический гидрокарбил, и любой атом углерода является насыщенным, может быть монозамещенным или полизамещенным и может быть одновалентным, двухвалентным или многовалентным. Примеры циклоалкила включают, но не ограничиваются ими, циклопропил, норборнанил, [2.2.2]бициклооктан, [4.4.0]бициклодеканил и тому подобное.
[97] Если не указано иное, термин «гало» или «галоген» относится к атому фтора, хлора, брома или йода сам по себе или как часть другого заместителя. Кроме того, подразумевается, что термин «галогеналкил» включает моногалогеналкил и полигалогеналкил. Например, подразумевается, что термин «галоген (C1-C4) алкил» включает, но не ограничивается ими, трифторметил, 2,2,2-трифторэтил, 4-хлорбутил, 3-бромпропил и тому подобное. Если не указано иное, примеры галогеналкила включают, но не ограничиваются ими, трифторметил, трихлорметил, пентафторэтил и пентахлорэтил.
[98] Термин «алкокси» обозначает любой алкил, определенный выше, имеющий определенное количество атомов углерода, присоединенных кислородным мостиком. Если не указано иное, C1-6 алкокси включает C1, C2, C3, C4, C5 и C6 алкокси. Примеры алкокси включают, но не ограничиваются ими, метокси, этокси, н-пропокси, изопропокси, н-бутокси, втор-бутокси, трет-бутокси, н-пентилокси и втор-пентокси.
[99] Если не указано иное, термин «арил» относится к полиненасыщенному ароматическому заместителю, может быть моно-, ди- или полизамещенным, может быть одновалентным, двухвалентным или многовалентным, может представлять собой одно кольцо или множество колец ( например, от одного до трех колец; где по меньшей мере одно кольцо является ароматическим), которые конденсированы вместе или ковалентно связаны. Термин «гетероарил» относится к арилу (или кольцу), содержащему от одного до четырех гетероатомов. В иллюстративном примере гетероатом выбран из B, O, N и S, где атомы азота и серы необязательно окислены и атом азота необязательно кватернизован. Гетероарил может присоединяться к остальной части молекулы через гетероатом. Неограничивающие примеры арила или гетероарила включают фенил, нафтил, бифенил, пирролил, пиразолил, имидазолил, пиразинил, оксазолил, фенилоксазолил, изоксазолил, тиазолил, фуранил, тиенил, пиридил, пиримидинил, бензотиазолил, пуринил, бензимидазолил, индолил, изохинолил, хиноксалинил, хинолил, 1-нафтил, 2-нафтил, 4-бифенил, 1-пирролил, 2-пирролил, 3-пирролил, 3-пиразолил, 2-имидазолил, 4-имидазолил, пиразинил, 2-оксазолил, 4-оксазолил, 2-фенил-4-оксазолил, 5-оксазолил, 3-изоксазолил, 4-изоксазолил, 5-изоксазолил, 2-тиазолил, 4-тиазолил, 5-тиазолил, 2-фурил, 3-фурил, 2-тиенил, 3-тиенил, 2-пиридил, 3-пиридил, 4-пиридил, 2-пиримидил, 4-пиримидил, 5-бензотиазолил, пуринил, 2-бензимидазолил, 5-индолил, 1-изохинолил, 5-изохинолил, 2-хиноксалинил, 5-хиноксалинил, 3-хинолил и 6-хинолил. Заместитель любой из указанных выше арильных и гетероарильных кольцевых систем выбран из приемлемого заместителя, описанного ниже.
[100] Если не указано иное, когда арил объединяют с другими терминами (такими как арилокси, арилтио, арилалкил), арил включает арильное и гетероарильное кольцо, как определено выше. Таким образом, подразумевается, что термин «аралкил» включает группу (например, бензил, фенэтил, пиридилметил и т. д.), где арил присоединен к алкилу, включая алкил, в котором атом углерода (например, метилен) был заменен на атом, такой как кислород, например, феноксиметил, 2-пиридилокси, 3-(1-нафтилокси)пропил и тому подобное.
[101] Соединение согласно настоящему описанию может быть получено различными способами синтеза, хорошо известными специалистам в данной области техники, включая следующие перечисленные варианты реализации, варианты реализации, образованные следующими перечисленными вариантами реализации в сочетании с другими способами химического синтеза и эквивалентными заменами, хорошо известными специалисту в данной области техники. Предпочтительные варианты реализации включают, но не ограничивается ими, варианты реализации настоящего описания.
[102] Растворитель, используемый в настоящем описании, является коммерчески доступным. В настоящем описании используют следующие сокращения: водн. обозначает воду; HATU обозначает O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат; EDC обозначает N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид; m-CPBA обозначает 3-хлорпероксибензойную кислоту; экв. обозначает эквивалент, эквивалент; CDI обозначает карбонилдиимидазол; ДХМ обозначает дихлорметан; PE обозначает петролейный эфир; DIAD обозначает диизопропилазодикарбоксилат; ДМФА обозначает N,N-диметилформамид; ДМСО обозначает диметилсульфоксид; EtOAc обозначает этилацетат; EtOH обозначает этанол; МеОН обозначает метанол; CBz обозначает бензилоксикарбонил, защитную группу амина; ВОС обозначает трет-бутилкарбонил, защитную группу амина; HOAc обозначает уксусную кислоту; NaCNBH3 обозначает цианоборогидрид натрия; к.т. обозначает комнатную температуру; O/N обозначает в течение ночи; ТГФ обозначает тетрагидрофуран; Boc2O обозначает ди-трет-бутилдикарбонат; TFA обозначает трифторуксусную кислоту; DIPEA обозначает диизопропилэтиламин; SOCl2 обозначает тионилхлорид; CS2 обозначает сероуглерод; TsOH обозначает п-толуолсульфоновую кислоту; NFSI обозначает N-фтор-N-(фенилсульфонил)бензолсульфонамид; NCS обозначает 1-хлорпирролидин-2,5-дион; n-Bu4NF обозначает фторид тетрабутиламмония; iPrOH обозначает 2-пропанол; т.пл. обозначает температуру плавления; LDA обозначает диизопропиламинолитий; Pd(OAc)2 обозначает ацетат палладия; Pd2(dba)3 обозначает трис(дибензилиденацетон)дипалладий; DPPP обозначает бис(дифенилфосфино)пропан; NIS обозначает N-йодосукцинимид; SPhos обозначает 2-дициклогексилфосфино-2',6'-диметоксибифенил; TBAF обозначает фторид тетрабутиламмония; Pd(PPh3)2Cl2 обозначает дихлорбис(трифенилфосфин)палладий; DMAP обозначает диметиламинопиридин; NBS обозначает N-бромсукцинимид; RuPhos обозначает 2-бисциклогексилфосфино-2',6'-диизопропоксибифенил; EA обозначает этилацетат; Pd(dppf)Cl2.CH2Cl2 обозначает комплекс [1,1-бис(дибензилфосфино)ферроцен]палладия дихлорида и дихлорметана; PBr3 обозначает трибромид фосфора; DEA обозначает диэтаноламин; ЭДТА обозначает этилендиаминтетрауксусную кислоту.
[103] Соединения называли вручную или с помощью программного обеспечения ChemDraw®, для коммерчески доступных соединений использовали названия из каталогов их поставщиков.
Технический результат
[104] В качестве нового ингибитора MEK соединения согласно настоящему описанию обладают хорошей биологической активностью в отношении MEK и ингибирующей активностью в отношении роста клеток в опухолевых клетках, которые связаны с указанным сигнальным путем.
Подробное описание вариантов реализации
[105] Следующие примеры дополнительно иллюстрируют настоящее изобретение, однако настоящее изобретение не ограничивается ими. Настоящее изобретение было подробно описано в тексте, и его конкретные варианты реализации также были раскрыты, для специалистов в данной области техники очевидно, что можно модифицировать и улучшать варианты реализации настоящего описания в пределах сущности и объема настоящего описания.
[106] В настоящем описании обычно использовали следующие методики проведения реакций:
[107] I. Реакция Сузуки
[108] Способ A:
[109] Субстрат + эфир борной кислоты/борная кислота + Pd(dppf)Cl2.CH2Cl2 + SPhos + основание + растворитель → продукт
[110] Субстрат (1,00 экв.) и эфир борной кислоты/борную кислоту (1,00-2,00 экв.) растворяли в растворителе, затем Pd(dppf)Cl2.CH2Cl2 (0,10 экв.) и основание (2,00-3,00 экв.) добавляли в защитной атмосфере азота при 20°C, реакционную смесь перемешивали при 85-100°C. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали смесью дихлорметан/EtOAc. Органическую фазу собирали, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения.
[111] Способ B:
[112] Субстрат + эфир борной кислоты/борная кислота + Pd(dppf)Cl2.CH2Cl2 + основание + растворитель → продукт
[113] Субстрат (1,00 экв.) и эфир борной кислоты/борную кислоту (1,00-2,00 экв.) добавляли в растворитель, затем добавляли Pd(dppf)Cl2.CH2Cl2 (0,10 экв.) и основание (2,00-5,00 экв.), реакционную смесь перемешивали при 60-110°C в защитной атмосфере азота. После завершения реакции реакционную смесь разбавляли водой и экстрагировали смесью EtOAc/дихлорметан. Органические фазы объединяли и сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения.
[114] Способ C:
[115] Субстрат + эфир борной кислоты/борная кислота +Pd2(dba)3+RuPhos+основание + растворитель → продукт
[116] Субстрат (1,00 экв.) добавляли в растворитель при 15°C, затем добавляли эфир борной кислоты/борную кислоту (1,20 экв.), Pd2(dba)3 (0,10 экв.), RuPhos (0,10 экв.) и основание (2,00 экв.). Реакционную смесь перемешивали при 130°C. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой и экстрагировали смесью EtOAc/дихлорметан. Органическую фазу собирали, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии с получением целевого соединения.
[117] Способ D:
[118] Субстрат + эфир борной кислоты/борная кислота +Pd2(dba)3+SPhos+основание + растворитель → продукт
[119] Субстрат (1,00 экв.) и эфир борной кислоты/борную кислоту (2,00 экв.) растворяли в растворителе, затем в защитной атмосфере азота добавляли SPhos (0,10 экв.), Pd2(dba)3 (0,05 экв.) и основание (2,00-2,50 экв.). Реакционную смесь перемешивали при 110-120°C. После завершения реакции реакционную смесь разбавляли водой и экстрагировали смесью дихлорметан/EtOAc. Органическую фазу собирали, выпаривали досуха с помощью роторного испарителя и очищали с помощью колоночной хроматографии с получением целевого соединения.
[120] II. Реакция йодирования
[121] Субстрат + трифторуксусная кислота + N-йодсукцинимид + растворитель → продукт
[122] Субстрат (1,00 экв.) растворяли в N,N-диметилформамиде, трифторуксусную кислоту и N-йодсукцинимид (1,50-3,00eq) добавляли при 0°C в защитной атмосфере азота. Реакционную смесь перемешивали при 30°C. После завершения реакции реакционный раствор последовательно гасили насыщенным раствором тиосульфата натрия и экстрагировали смесью EtOAc/дихлорметан. Объединенную органическую фазу сушили над безводным сульфатом натрия, фильтровали и фильтрационный осадок выпаривали досуха с помощью роторного испарителя. Фильтрационный осадок растирали с растворителем метил-трет-бутиловый эфир/PE = 1/1. После фильтрования фильтрационный осадок промывали петролейным эфиром (PE) с получением целевого соединения.
[123] Ссылочный пример 1: фрагмент BA-1
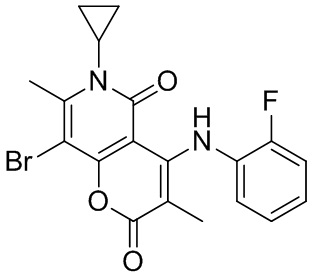
[124] Схема синтеза:
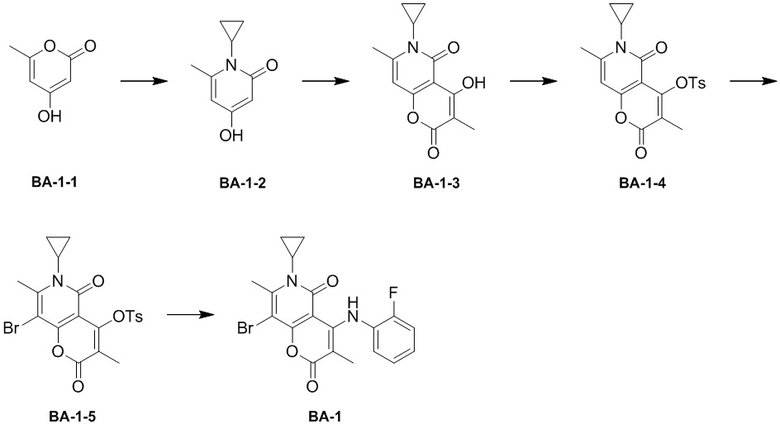
[125] Стадия 1: синтез соединения BA-1-2.
[126] Соединение BA-1-1 (249,70 г, 1,98 моль, 1,00 экв.) растворяли в воде (500,00 мл) и циклопропиламин (113,04 г, 1,98 моль, 1,00 экв.) добавляли при 60-80°C. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 6 часов, и образовался осадок. После завершения реакции реакционную смесь охлаждали до комнатной температуры. Затем добавляли метанол (100 мл) и смесь перемешивали в течение 30 минут. После фильтрования фильтрационный осадок собирали, промывали EtOAc (50 мл*3) и выпаривали досуха с помощью роторного испарителя с получением соединения BA-1-2. MS m/z:205,0 [M+H]+
[127] Стадия 2-способ A: синтез соединения BA-1-3.
[128] Соединение BA-1-2 (31,85 г, 192,81 ммоль, 1,00 экв.) и метилмалоновую кислоту (33,59 г, 192,81 ммоль, 1,00 экв.) смешивали в дифениловом эфире (180,00 мл), температуру реакционной смеси повышали до 220-230°C в защитной атмосфере азота и реакционную смесь перемешивали в течение 6 часов. После завершения реакции реакционный раствор охлаждали и разбавляли петролейным эфиром (1 л). Фильтрационный осадок собирали, промывали петролейным эфиром (50 мл*3), выпаривали досуха с помощью роторного испарителя и затем перемешивали в дихлорметане (500 мл) в течение 30 минут. Смесь фильтровали и фильтрационный осадок промывали дихлорметаном (50 мл*3). Органические фазы объединяли и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (ДХМ/EA=1/1) с получением целевого соединения BA-1-3. 1H ЯМР (400 МГц, CDCl3-d) δ 12,85(s, 1 H), 6,25 (s, 1 H), 2,91-2,89 (m, 1 H), 2,60 (s, 3 H), 1,99 (s, 3 H), 1,37-1,35 (m, 2 H), 1,00-0,99(m, 2 H). MS m/z : 247,9 [M+H]+
[129] Стадия 2-способ B: синтез соединения BA-1-3.
[130] Соединение BA-1-2 (13,60 г, 82,33 ммоль, 1,00 экв.) растворяли в уксусном ангидриде (60,00 мл), с последующим добавлением диэтилметилмалоната (14,58 г, 123,49 ммоль, 1,50 экв.) при 10°C. Температуру реакционной смеси повышали до 80°C и реакционную смесь перемешивали в течение 0,5 часа. После завершения реакции реакционную смесь фильтровали и фильтрационный осадок собирали. Фильтрационный осадок промывали метил-трет-бутиловым эфиром (100 мл) с получением целевого соединения BA-1-3.
[131] 1H ЯМР (400 МГц, CDCl3-d) δ 13,42 (s, 1 H), 6,607 (s, 1 H), 3,31-2,98 (m, 1 H), 2,57 (s, 3 H), 1,81 (s, 3 H), 1,19-1,16 (m, 2 H), 0,96-0,92 (m, 2 H). MS m/z: 247,9 [M+H]+
[132] Стадия 3: синтез соединения BA-1-4.
[133] Соединение BA-1-3 (4,83 г, 19,53 ммоль, 1,00 экв.), триэтиламин (3,95 г, 39,06 ммоль, 2,00 экв.) и DMAP (47,72 мг, 390,60 мкмоль, 0,02 экв.) растворяли в дихлорметане (120,00 мл) с последующим добавлением 4-метилбензолсульфонилхлорида (3,72 г, 19,53 ммоль, 1,00 экв.) при 15°C. Реакционную смесь перемешивали при 15°C в течение 16 часов. После завершения реакции реакционный раствор промывали последовательно водой (50 мл) и насыщенным раствором хлорида натрия (50мл), затем сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (ДХМ, ДХМ/EA=5/1) с получением целевого соединения BA-1-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,96-7,94 (m, 2 H), 7,41-7,39 (m, 2 H), 6,07(s, 1 H), 2,85(s, 1 H), 2,54(m, 3 H), 2,49(s, 3 H), 1,67(s, 3 H), 1,32-1,30(m, 2 H), 0,87-0,86(m, 2 H).
[134] Стадия 4: синтез соединения BA-1-5.
[135] Соединение BA-1-4 (6,97 г, 17,36 ммоль, 1,00 экв.) растворяли в ацетонитриле (25,00 мл) и дихлорметане (25,00 мл), N-бромсукцинимид (4,63 г, 26,04 ммоль, 1,50 экв.) добавляли партиями. Реакционную смесь перемешивали при 15°C в течение 1 часа. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с ацетонитрилом (50 мл) в течение 30 минут, фильтровали и промывали ацетонитрилом (10 мл*3). Фильтрационный осадок собирали и выпаривали досуха с помощью роторного испарителя с получением целевого соединения BA-1-5. 1H ЯМР (400 МГц, CDCl3-d) δ 7,92-7,90(m, 2 H), 7,40-7,38(m, 2 H), 2,95(m, 1 H), 2,75(s, 3 H), 2,47(s, 3 H), 1,64(s, 3 H), 1,40-1,30(m, 2 H), 0,87-0,85(m, 2 H). MS m/z:481,9 [M+H]+
[136] Стадия 5: синтез соединения BA-1.
[137] Соединение BA-1-5 (5,30 г, 11,03 ммоль, 1,00 экв.) и 2-фторанилин (5,30 г, 47,65 ммоль, 4,32 экв.) растворяли в этаноле (120,00 мл). Температуру реакционной смеси повышали до 85°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционный раствор охлаждали и фильтровали, фильтрационный осадок промывали этанолом (30 мл*3). Фильтрационный осадок собирали и выпаривали досуха с помощью роторного испарителя с получением целевого соединения BA-1. 1H ЯМР (400 МГц, CDCl3-d) δ 11,01(s, 1 H), 7,12 -7,10(m, 3 H), 7,03-7,01(m, 1 H), 2,97(s, 3 H), 1,61(m, 3 H), 1,38-1,36(t, 3 H), 0,91-0,90(t, 3 H). MS m/z:420,8 [M+H]+
[138] Ссылочный пример 2: фрагмент BA-2
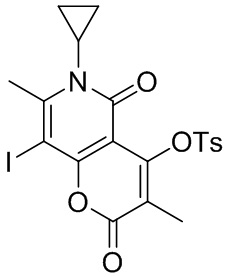
[139] Схема синтеза:
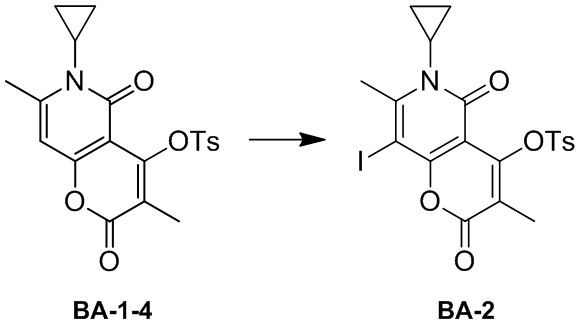
[140] Стадия 1: синтез соединения BA-2.
[141] Соединение BA-1-4 (3,00 г, 7,47 ммоль, 1,00 экв.) растворяли в ацетонитриле (40,00 мл) и дихлорметане (20,00 мл), N-йодсукцинимид (2,52 г, 11,21 ммоль, 1,50 экв.) добавляли в защитной атмосфере азота. Реакционную смесь перемешивали при 15°C в течение 16 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с этанолом (150 мл) при 20°C в течение 16 часов, затем фильтровали и промывали этанолом (20 мл). Фильтрационный осадок собирали и выпаривали досуха с помощью роторного испарителя с получением целевого соединения BA-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,86-7,84(d, J= 8,4 Гц, 2 H), 7,33-7,31(d, J= 8,0 Гц, 2 H), 2,93 -2,87(m, 1 H), 2,81(s, 3 H), 2,41(s, 3 H), 1,58(s, 3 H), 1,57-1,25(m, 2 H), 0,81-0,76(m, 2 H). MS m/z: 527,9 [M+H]+
[142] Ссылочный пример 3: фрагмент BA-3
[143] Схема синтеза:
[144] Стадия 1: синтез соединения BA-3-1.
[145] Соединение BA-1 (4,50 г, 10,73 ммоль, 1,00 экв.), соединение BB-5 (2,15 г, 12,88 ммоль, 1,20 экв.) и раствор бикарбоната натрия (1 M, 21,47 мл, 2,00 экв.) растворяли в диоксане (300,00 мл), затем соединение Pd(dppf)Cl2.CH2Cl2 (876,56 мг, 1,07 ммоль, 0,10 экв.) добавляли в защитной атмосфере азота. В защитной атмосфере азота температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции смесь выпаривали досуха с помощью роторного испарителя, растворяли в дихлорметане (300 мл) и перемешивали в течение 30 минут. Смесь затем фильтровали с помощью колоночной хроматографии (1CM) и промывали смесью дихлорметан/EtOAc =1/1 (200 мл). Органическую фазу удаляли с помощью роторного испарителя, добавляли дихлорметан (10 мл), затем этанол (200 мл) медленно добавляли при перемешивании. Полученный осадок собирали, промывали этанолом (20 мл) и выпаривали досуха с помощью роторного испарителя с получением соединения BA-3-1. 1H ЯМР (400 МГц, CDCl3-d) δ 11,06(s, 1 H), 8,31(m, 1 H), 8,29(m, 1 H), 7,99(m, 1 H), 7,97(m, 1 H), 7,69-7,60(s, 3 H), 7,12-7,02(m, 1 H), 2,96(m, 1 H), 2,39(s, 3 H), 1,59-1,58(s, 3 H), 1,38-1,42(m, 2 H), 0,99-0,98(m, 2 H). MS m/z:484,1 [M+Na]+
[146] Стадия 2: синтез соединения BA-3-2.
[147] Соединение BA-3-1 (3,76 мг, 8,15 ммоль, 1,00 экв.) растворяли в N,N-диметилформамиде (25,00 мл), трифторуксусную кислоту (7,00 мл) и N-йодсукцинимид (3,67 г, 16,30 ммоль, 2,00 экв.) добавляли при 20°C. Реакционную смесь перемешивали при 20°C в течение 16 часов. После завершения реакции воду (400 мл) добавляли для гашения реакционной смеси. Твердое вещество собирали и промывали водой (200 мл*2), и затем сушили. Полученное твердое вещество дважды растирали с этанолом (50 мл), каждый раз по 30 минут. После фильтрования фильтрационный осадок собирали. Фильтрационный осадок растирали с метил-трет-бутиловым эфиром (100 мл) в течение 2 часов и фильтровали. Неочищенный продукт собирали, промывали метил-трет-бутиловым эфиром (20 мл) и выпаривали досуха с помощью роторного испарителя с получением соединения BA-3-2. 1H ЯМР (400 МГц, CDCl3-d) δ 11,03(s, 1 H), 8,31(m, 1 H), 8,29(m, 1 H), 8,1(m, 1 H), 7,69-7,59(m, 1 H), 7,47-7,41(m, 2 H), 6,72-6,68(m, 1 H), 2,95(s, 3 H), 2,39(s, 3 H), 1,60(s, 3 H), 1,40-1,38(m, 2 H), 0,99-0,98(m, 2 H). MS m/z:587,9 [M+H]+
[148] Стадия 3: синтез соединения BA-3.
[149] Соединение BA-3-2 (1,80 мг, 3,06 ммоль, 1,00 экв.) растворяли в уксусной кислоте (25,00 мл), с последующим добавлением цинкового порошка (3,00 г, 45,87 ммоль, 14,99 экв.). Реакционную смесь перемешивали при 10°C в течение 3 часов. После завершения реакции реакционную смесь фильтровали и фильтрационный осадок промывали дихлорметаном (30 мл*3). Фильтрат собирали и затем промывали последовательно водой (30 мл*3), насыщенным раствором бикарбоната натрия (30 мл*3) и насыщенным раствором хлорида натрия (30 мл). Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (ДХМ, ДХМ/EA=10/1) с получением целевого соединения BA-3. 1H ЯМР (400 МГц, CDCl3-d) δ 11,17(s, 1 H), 7,47-7,46(m, 1 H), 7,46-7,44(m, 1 H), 7,21-7,19(m, 1 H), 6,73-6,69(m, 2 H), 6,58(m, 1 H), 6,56-6,52(m, 1 H), 2,94-2,92(m, 1 H), 2,38(s, 3 H), 2,38(s, 3 H), 1,36-1,35(m, 2 H), 0,95-0,94(m, 2 H). MS m/z: 587,9 [M+H]+
[150] Ссылочный пример 4: фрагмент BA-4
[151] Схема синтеза:
[152] Стадия 1: синтез соединения BA-4-1.
[153] Соединение BA-1 (5,00 г, 11,93 ммоль, 1,00 экв.) и бензофенонимин (3,24 г, 17,90 ммоль, 1,50 экв.) добавляли в толуол (10,00 мл), затем карбонат цезия (8,55 г, 26,25 ммоль, 2,20 экв.), Pd2(dba)3 (1,09 г, 1,19 мкмоль, 0,10 экв.) и SPhos (1,04 г, 1,79 ммоль, 0,15 экв.) добавляли в защитной атмосфере азота. Температуру реакционной смеси повышали до 110°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционную смесь разбавляли водой (50мл) и экстрагировали EtOAc (50 мл*3). Органическую фазу собирали, промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=5/1) с получением соединения BA-4-1. 1H ЯМР (400 МГц, CDCl3-d) δ 11,16(s, 1 H), 7,82(m, 2 H), 7,80(m, 1 H), 7,45-7,43(m, 3 H), 7,33-7,31(m, 3 H), 7,20-7,19(m, 2 H), 7,07-7,06(m, 3 H), 6,93(m, 1 H), 2,89-2,84(m, 1 H), 2,43(s, 3 H), 1,57(s, 3 H), 1,54(m, 2 H), 1,20(m, 2 H). MS m/z: 520,3 [M+H]+
[154] Стадия 2: синтез соединения BA-4.
[155] Соединение BA-4-1 (2,10 г, 4,04 ммоль, 1,00 экв.) растворяли в 1 M водном растворе соляной кислоты (20,00 мл) и ацетонитриле (10,00 мл), реакционную смесь перемешивали при 20°C в атмосфере азота в течение 0,5 часа. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя, затем добавляли насыщенный раствор бикарбоната натрия (30 мл) и образовывалось твердое вещество. После фильтрования фильтрационный осадок собирали, растирали с метил-трет-бутиловым эфиром (50 мл) и выпаривали досуха с помощью роторного испарителя с получением целевого соединения BA-4.
[156] 1H ЯМР (400 МГц, CDCl3-d) δ 11,76(s, 1 H), 7,31-7,28(m, 1 H), 7,80(m, 1 H), 7,20-7,17(m, 2 H), 7,13-7,11(m, 1 H), 4,36(s, 2 H), 3,00-2,94(m, 1 H), 2,47(s, 3 H), 1,47(s, 3 H), 1,21-1,16(m, 2 H), 0,81-0,77(m, 2 H). MS m/z: 356,1 [M+H]+
[157] Ссылочный пример 5: фрагмент BA-5
[158] Схема синтеза:
[159] Стадия 1: синтез соединения BA-5-2.
[160] Соединение BA-1-1 (20,00 г, 158,59 ммоль, 1,00 экв.) растворяли в метиламине (30%) (150,00 г, 1,45 моль, 9,14 экв.), реакционную смесь перемешивали при 100°C в течение 2 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, указанный растворитель удаляли с помощью роторного испарителя. Неочищенный продукт добавляли в ацетонитрил (200мл) и смесь фильтровали. Фильтрационный осадок собирали с получением целевого соединения BA-5-2. 1H ЯМР (400 МГц, CDCl3-d) δ 10,22(s, 1 H), 5,76(s, 1 H), 5,49(s, 1 H), 3,30(s, 3 H), 2,27(s, 3 H). MS m/z: 139,8 [M+H]+
[161] Стадия 2: синтез соединения BA-5-3.
[162] Соединение BA-5-2 (5,00 г, 35,93 ммоль, 1,00 экв.) и метилмалоновую кислоту (6,26 г, 35,93 ммоль, 6,14 мл, 1,00 экв.) растворяли в дифениловом эфире (100,00 мл), реакционную смесь перемешивали при нагревании с обратным холодильником в течение 2 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, и образовался осадок. Смесь фильтровали и фильтрационный осадок собирали с получением целевого соединения BA-5-3. 1H ЯМР (400 МГц, CDCl3-d) δ 13,35(s, 1 H), 6,64(s, 1 H), 3,54(s, 1 H), 2,52(s, 3 H), 1,84(s, 3 H). MS m/z: 221,9 [M+H]+
[163] Стадия 3: синтез соединения BA-5-4.
[164] Соединение BA-5-3 (4,80 г, 21,70 ммоль, 1,00 экв.) растворяли в дихлорметане (10,00 мл), добавляли триэтиламин (4,39 г, 43,40 ммоль, 6,02 мл, 2,00 экв.), затем п-толуолсульфонилхлорид (4,96 г, 26,04 ммоль, 1,20 экв.) добавляли при 25°C. Реакционную смесь перемешивали при 25°C в течение 15 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя, неочищенный продукт растирали с этанолом и фильтровали. Фильтрационный осадок собирали с получением целевого соединения BA-5-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,90-7,85(s, 2 H), 7,49-7,47(m, 2 H), 6,39(m, 1 H), 3,36(m, 6 H), 2,45-2,44(m, 6 H). MS m/z: 376,1 [M+H]+
[165] Стадия 4: синтез соединения BA-5-5.
[166] Соединение BA-5-4 (2,80 г, 7,46 ммоль, 1,00 экв.) растворяли в дихлорметане (30 мл) и ацетонитриле (60 мл), медленно добавляли N-бромсукцинимид (1,99 г, 11,19 ммоль, 1,50 экв.). Реакционную смесь перемешивали при 25°C в течение 15 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя, неочищенный продукт растирали с этанолом (100мл) и фильтровали. Фильтрационный осадок собирали с получением целевого соединения BA-5-5. 1H ЯМР (400 МГц, CDCl3-d) δ 7,93-7,91(d, J=8,4, 2 H), 7,40-7,27(d, J=8,0, 2 H), 3,63(s, 3 H), 2,68(s, 3 H), 2,48(s, 3 H), 1,63(s, 3 H). MS m/z: 456,0 [M+H]+
[167] Стадия 5: синтез соединения BA-5.
[168] Соединение BA-5-5 (1,00 г, 2,20 ммоль, 1,00 экв.) и 2-фторанилин (4,89 г, 44,00 ммоль, 4,25 мл, 20,00 экв.) растворяли в этаноле (10,00 мл) и реакционную смесь перемешивали при интенсивном нагреве с обратным холодильником в течение 15 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и появилось твердое вещество. Смесь фильтровали и фильтрационный осадок собирали с получением целевого соединения BA-5. 1H ЯМР (400 МГц, CDCl3-d) δ 11,10(s, 1 H), 7,13-7,12(d, J=5,6, 3 H), 7,11(s, 1 H), 3,69(s, 3 H), 2,74(s, 3 H). MS m/z: 394,9 [M+H]+
[169] Ссылочный пример 6: фрагмент BA-6

[170] Схема синтеза:
[171] Стадия 1: синтез соединения BA-6-2.
[172] Соединение BA-1-1 (10,00 г, 79,30 ммоль, 1,00 экв.) и метоксипропиламин (7,07 г, 79,30 ммоль, 8,13 мл, 1,00 экв.) растворяли в воде (100,00 мл), реакционную смесь нагревали с обратным холодильником и перемешивали в течение 1 часа. После завершения реакции смесь охлаждали до комнатной температуры, указанный растворитель удаляли с помощью роторного испарителя, неочищенный продукт растирали с метил-трет-бутиловым эфиром (150 мл) и фильтровали. Фильтрационный осадок собирали с получением целевого соединения BA-6-2. MS m/z: 527,9 [M+H]+
[173] Стадия 2: синтез соединения BA-6-3.
[174] Соединение BA-6-2 (13,70 г, 69,46 ммоль, 1,00 экв.) и метилмалоновую кислоту (12,30 г, 104,19 ммоль, 8,42 мл, 1,50 экв.) растворяли в уксусном ангидриде (200,00 мл). Реакционную смесь перемешивали при 100°C в течение 1 часа. После завершения реакции смесь охлаждали до комнатной температуры и добавляли метил-трет-бутиловый эфир (200 мл). Через 15 часов образовалось белое твердое вещество и указанную смесь фильтровали. Фильтрационный осадок собирали с получением целевого соединения BA-6-3. MS m/z:279,9 [M+H]+
[175] Стадия 3: синтез соединения BA-6.
[176] Соединение BA-6-3 (10,00 г, 35,81 ммоль, 1,00 экв.) растворяли в дихлорметане (150,00 мл), с последующим добавлением триэтиламина (7,25 г, 71,62 ммоль, 9,93 мл, 2,00 экв.) и п-толуолсульфонилхлорида (13,65 г, 71,62 ммоль, 2,00 экв.). Реакционную смесь перемешивали при 15°C в течение 15 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя, неочищенный продукт растирали с метил-трет-бутиловым эфиром (200 мл) и фильтровали. Фильтрационный осадок собирали с получением целевого соединения BA-6. MS m/z: 434,1 [M+H]+
[177] Ссылочный пример 7: фрагмент BB-1
[178] Схема синтеза:
[179] Стадия 1: синтез соединения BB-1.
[180] Соединение BB-1-1 (3,50 г, 20,23 ммоль, 1,00 экв.) растворяли в дихлорметане (40,00 мл) с последующим добавлением уксусного ангидрида (6,55 г, 64,13ммоль, 3,17 экв.). Реакционную смесь перемешивали при 20°C в течение 16 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растворяли в EtOAc (100 мл) и последовательно промывали водой (200 мл), насыщенным раствором бикарбоната натрия (200 мл) и насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением целевого соединения BB-1. 1H ЯМР (400 МГц, CDCl3-d) δ 8,15 (d, J=8,0 Гц, 1 H), 8,08(br. s., 1 H), 7,55(t, J=8,0 Гц, 2 H), 8,15(d, J=7,6 Гц, 1 H), 2,20(s, 3 H).
[181] Ссылочный пример 8: фрагмент BB-2
[182] Схема синтеза:
[183] Стадия 1: синтез соединения BB-2-2.
[184] Соединение BB-2-1 (5,00 г, 26,31 ммоль, 1,00 экв.) растворяли в дихлорметане (120,00 мл), добавляли триэтиламин (3,19 г, 31,57 ммоль, 1,20 экв.), затем по каплям добавляли ацетилхлорид (2,07 г, 26,31 ммоль, 1,00 экв.) при 0°C. Температуру реакционной смеси повышали до 25°C и реакционную смесь перемешивали в течение 3 часов. После завершения реакции реакционный раствор промывали последовательно насыщенным раствором бикарбоната натрия (80 мл*3), водой (100 мл*2) и насыщенным раствором хлорида натрия (80 мл). Органическую фазу собирали, сушили над безводным сульфатом натрия, выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с петролейным эфиром (30 мл), фильтровали и выпаривали досуха с помощью роторного испарителя с получением соединения BB-2-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,81-7,79(m, 1 H), 7,40-7,36(m, 2 H), 7,06(t, J=8,6 Гц, 1 H), 2,18(s, 3 H). MS m/z: 231,8 [M+H]+
[185] Стадия 2: синтез соединения BB-2.
[186] Соединение BB-2-2 (1,00 г, 4,31 ммоль, 1,00 экв.) растворяли в безводном диоксане (20,00 мл), соединение BB-2-3 (1,42 г, 5,60 ммоль, 1,30 экв.), ацетат калия (1,27 г, 12,93 ммоль, 3,00 экв.) и Pd(dppf)Cl2 (157,68 мг, 215,50 мкмоль, 0,05 экв.) последовательно добавляли при 25°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 15 часов. После завершения реакции твердое вещество удаляли фильтрованием и промывали дихлорметаном (25 мл*3). Фильтрат собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=8/1- 2/1) с получением целевого соединения BB-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,84-7,81(m, 1 H), 7,56-7,54(m, 1 H), 7,22(br. s., 1 H), 7,00(t, J=8,8 Гц, 1 H), 2,15(s, 3 H), 1,35(s, 12 H). MS m/z: 279,9 [M+H]+
[187] Ссылочный пример 9: фрагмент BB-3
[188] Схема синтеза:
[189] Стадия 1: синтез соединения BB-3-2.
[190] Соединение BB-3-1 (20,00 г, 92,58 ммоль, 1,00 экв.), соединение BB-2-3 (28,21 г, 111,10 ммоль, 1,20 экв.), ацетат калия (27,26 г, 277,74 ммоль, 3,00 экв.) и Pd(dppf)Cl2.CH2Cl2 (3,78 г, 4,63 ммоль, 0,05 экв.) растворяли в диоксане (150,00 мл). Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в атмосфере азота в течение 16 часов. После завершения реакции реакционную смесь растворяли в EtOAc (350 мл), последовательно промывали водой (100 мл*3) и насыщенным раствором хлорида натрия (200 мл*2), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя. Остаток растирали с петролейным эфиром (50 мл) в течение 30 минут, затем фильтровали и собирали. Неочищенный продукт промывали петролейным эфиром (20 мл) и выпаривали досуха с помощью роторного испарителя с получением соединения BB-3-2. 1H ЯМР (400 МГц, CDCl3-d) δ 8,61(d, J=2,0 Гц, 1 H), 8,15(dd, J=8,2 Гц, J=2,6 Гц, 1 H), 7,32(d, J=8,4 Гц, 1 H), 2,65(s, 3 H), 1,38(s, 12 H).
[191] Стадия 2: синтез соединения BB-3.
[192] Соединение BB-3-2 (16,00 г, 60,81 ммоль, 1,00 экв.) растворяли в ТГФ (400,00 мл) и воде (200,00 мл) с последующим добавлением периодата натрия (39,02 г, 182,43 ммоль, 3,00 экв.). Реакционную смесь перемешивали при 20°C в течение 1 часа, затем добавляли соляную кислоту (1 M, 200,06 мл, 3,29 экв.) и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционный раствор экстрагировали EtOAc (250 мл*3). Органическую фазу последовательно промывали водой (200 мл*3) и насыщенным раствором хлорида натрия (200 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (100 мл) в течение 1 часа, затем фильтровали с получением целевого соединения BB-3. 1H ЯМР (400 МГц, MeOD) δ 8,16(s, 1 H), 8,11(d, J=8,0 Гц, 1 H), 7,42(d, J=8,0 Гц, 1 H), 2,46(s, 3 H).
[193] Ссылочный пример 10: фрагмент BB-4
[194] Схема синтеза:
[195] Стадия 1: синтез соединения BB-4-2.
[196] Соединение BB-4-1 (4,50 г, 24,19 ммоль, 1,00 экв.) растворяли в диоксане (45,00 мл), добавляли насыщенный раствор бикарбоната натрия (45 мл), затем добавляли Boc2O (7,92 г, 36,28 ммоль, 1,50 экв.) при 0°C. Температуру реакционной смеси повышали до 20°C и реакционную смесь перемешивали в течение 1 часа. После завершения реакции добавляли EtOAc (50 мл*2). Органическую фазу промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=50/1-10/1) и выпаривали досуха с помощью роторного испарителя с получением соединения BB-4-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,44(s, 1 H), 7,42-7,39(m, 1 H), 7,21(d, J=6,4 Гц, 2 H), 7,00(t, J=8,8 Гц, 1 H), 4,86(br. s., 1 H), 4,30(d, J=5,6 Гц, 2 H), 1,47(s, 9 H). MS m/z: 231,8 [M+H]+
[197] Стадия 2: синтез соединения BB-4.
[198] Соединение BB-2-3 (7,45 г, 29,36 ммоль, 1,50 экв.) и соединение BB-4-2 (5,60 г, 19,57 ммоль, 1,00 экв.), ацетат калия (3,84 г, 39,14 ммоль, 2,00 экв.) и Pd(dppf)Cl2.CH2Cl2 (1,60 г, 1,96 ммоль, 0,10 экв.) растворяли в диоксане (110,00 мл). В защитной атмосфере азота температуру реакционной смеси повышали до 80°C и реакционную смесь перемешивали в течение 3 часов. После завершения реакции реакционный раствор фильтровали через диатомовую землю, фильтрат выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=5/1) с получением целевого соединения BB-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,72(d, J=6,0 Гц, 2 H), 7,44-7,40(m, 1 H), 7,35(t, J=8,0 Гц, 1 H), 4,81(br. s., 1 H), 4,31(d, J=5,6 Гц, 2 H), 1,47(s, 9 H), 1,35(s, 12 H). MS m/z: 233,9 [M-Boc+H]+
[199] Ссылочный пример 11: фрагмент BB-6
[200] Схема синтеза:
[201] Стадия 1: синтез соединения BB-6-2.
[202] При 0°C в защитной атмосфере азота растворяли тиометоксид натрия (1,34 г, 18,16 ммоль, 1,22 мл, 2,27 экв.) в N,N-диметилформамиде (5,00 мл) с последующим добавлением соединения BB-6-1 (2,00 г, 8,00 ммоль, 1,00 экв.). Температуру реакционной смеси повышали до 15°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали EtOAc (30 мл*2). Органическую фазу промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением соединения BB-6-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,40(s, 1 H), 7,31(d, J=7,6 Гц, 1 H), 7,16(d, J=7,6 Гц, 1 H), 7,11(t, J=7,8 Гц, 1 H), 3,56(s, 2 H), 1,93(s, 3 H).
[203] Стадия 2: синтез соединения BB-6-3.
[204] Соединение BB-6-2 (1,70 г, 7,83 ммоль, 1,00 экв.) растворяли в гексафторизопропаноле (2. 00 мл), с последующим добавлением пероксида водорода (1,78 г, 15,66 ммоль, 1,50 мл, 2,00 экв.). Реакционную смесь перемешивали при 25°C в течение 2 часов. После завершения реакции реакционную смесь разбавляли водой (10 мл) и экстрагировали EtOAc (20 мл*2). Органические фазы объединяли и промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт перемешивали в петролейном эфире (10 мл) в течение 0,5 часа, затем фильтровали с получением соединения BB-6-3. 1H ЯМР (400 МГц, CDCl3-d) δ 7,52(d, J=7,2 Гц, 1 H), 7,48(s, 1 H), 7,29-7,26(m, 2 H), 3,98(d, J=12,8 Гц, 1 H), 3,92(d, J=12,8 Гц, 1 H), 2,51(s, 3 H).
[205] Стадия 3: синтез соединения BB-6.
[206] Ацетат калия (1,52 г, 15,44 ммоль, 2,00 экв.) и Pd(dppf)Cl2 (564,95 мг, 772,00 мкмоль, 0,10 экв.) добавляли к раствору соединения BB-6-3 (1,80 г, 7,72 ммоль, 1,00 экв.) и соединения BB-2-3 (2,94 г, 11,58 ммоль, 1,50 экв.) в диоксане (10,00 мл). Реакционную смесь перемешивали при 80°C в атмосфере азота в течение 16 часов. После завершения реакции смесь фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (ДХМ/MeOH=1/0-30/1) с получением целевого соединения BB-6. 1H ЯМР (400 МГц, CDCl3-d) δ 7,82-7,78(m, 1 H), 7,71(s, 1 H), 7,44-7,39(m, 2 H), 4,13(d, J=12,8 Гц, 1 H), 3,94(d, J=12,8 Гц, 1 H), 2,48(s, 3 H), 1,37(s, 12 H). MS m/z: 281,1 [M+H]+
[207] Ссылочный пример 12: фрагмент BB-7
[208] Схема синтеза:
[209] Стадия 1: синтез соединения BB-7-2.
[210] Соединение BB-6-1 (1,00 г, 4,00 ммоль, 1,00 экв.) и фталимид калия (643,20 мг, 3,47 ммоль, 0,87 экв.) растворяли в N,N-диметилформамиде (5,00 мл), реакционную смесь обезвоживали хлоридом кальция и перемешивали при 100°C в течение 12 часов. После завершения реакции реакционную смесь разбавляли водой (15 мл) и экстрагировали EtOAc (40 мл*2). Органические фазы объединяли и промывали насыщенным раствором хлорида натрия (20 мл*3), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт перемешивали в петролейном эфире (10 мл) в течение 0,5 часа, затем фильтровали и выпаривали досуха с помощью роторного испарителя с получением соединения BB-7-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,90-7,87(m, 2 H), 7,76-7,74(m, 2 H), 7,60(s, 1 H), 7,46-7,35(m, 2 H), 7,21(t, J=5,2 Гц, 1 H), 4,83(s, 2 H). MS m/z: 315,9 [M+H]+
[211] Стадия 2: синтез соединения BB-7-3.
[212] Соединение BB-7-2 (700,00 мг, 2,21 ммоль, 1,00 экв.) и соединение BB-2-3 (673,45 мг, 2,65 ммоль, 1,20 экв.) растворяли в диоксане (10,00 мл), затем добавляли ацетат калия (433,78 мг, 4,42 ммоль, 2,00 экв.) и Pd(dppf)Cl2 (161,71 мг, 221,00 мкмоль, 0,10 экв.). Реакционную смесь перемешивали при 80°C в атмосфере азота в течение 16 часов. После завершения реакции реакционную смесь фильтровали, выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA = 10/1-2/1) с получением соединения BB-7-3. 1H ЯМР (400 МГц, CDCl3-d) δ 7,89-7,86(m, 3 H), 7,74-7,71(m, 3 H), 7,53(d, J=6,4 Гц, 1 H), 7,34(t, J=8,0 Гц, 1 H), 4,88(s, 2 H), 1,36(s, 12 H). MS m/z: 364,0 [M+H]+
[213] Стадия 3: синтез соединения BB-7-4.
[214] Соединение BB-7-3 (600,00 мг, 1,65 ммоль, 1,00 экв.) растворяли в ТГФ (5,00 мл), затем добавляли гидразин-гидрат (253,15 мг, 4,96 ммоль, 3,00 экв.). Реакционную смесь перемешивали при 80°C в атмосфере азота в течение 12 часов. После завершения реакции реакционную смесь фильтровали и выпаривали досуха с помощью роторного испарителя с получением соединения BB-7-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7. 77(m, 1 H), 7,73-7,71(m, 1 H), 7,45-7,43(m, 1 H), 7,39-7,35(s, 1 H), 3,90(s, 2 H), 1,37(m, 12 H).
[215] Стадия 4: синтез соединения BB-7.
[216] Соединение BB-7-4 (500,00 мг, 1,34 ммоль, 1,00 экв.) и триэтиламин (271,19 мг, 2,68 ммоль, 2,00 экв.) растворяли в дихлорметане (10 мл) с последующим добавлением ацетилхлорида (157,79 мг, 2,01 ммоль, 1,50 экв.) при 0°C. Реакционную смесь перемешивали при комнатной температуре в атмосфере азота в течение 20 минут. После завершения реакции реакционную смесь разбавляли водой (10мл) и экстрагировали EtOAc (20 мл*3). Органические фазы объединяли, промывали водой (30 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=5/1-1/1) с получением целевого соединения BB-7. 1H ЯМР (400 МГц, CDCl3-d) δ 7,67-7,64(m, 2 H), 7,34-7,26(m, 2 H), 4,37(d, J=6,0 Гц, 2 H), 1,95(s, 3 H), 1,28(s, 12 H). MS m/z:276,2 [M+H]+
[217] Ссылочный пример 13: фрагмент BB-8
[218] Схема синтеза:
[219] Стадия 1: синтез соединения BB-8.
[220] Соединение BB-8-1 (60,00 мг, 688,71 мкмоль, 1,00 экв.) и трифосген (214,59 мг, 723,15 мкмоль, 1,05 экв.), пиридин (163,43 мг, 2,07 ммоль, 3,00 экв.) добавляли в дихлорметан (2,00 мл), реакционную смесь перемешивали при 0°C в атмосфере азота в течение 1 часа. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением целевого соединения BB-8.
[221] Ссылочный пример 14: фрагмент BB-9
[222] Схема синтеза:
[223] Стадия 1: синтез соединения BB-9-2.
[224] Соединение BB-9-1 (5,00 г, 26,87 ммоль, 1,00 экв.) растворяли в пиридине (100,00 мл), затем добавляли метансульфонилхлорид (11,87 г, 103,62 ммоль, 3,86 экв.) при 0°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 60°C и реакционную смесь перемешивали в течение 1 часа. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя. Остаток растворяли в дихлорметане (100 мл), промывали соляной кислотой (1 M, 100 мл) и насыщенным раствором бикарбоната натрия (100 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=10/1-2/1) с получением соединения BB-9-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,47(d, J=8,0 Гц, 1 H), 7,43(d, J=8,0 Гц, 1 H), 7,10(t, J=8,0 H, 1 H), 6,39(s, 1 H), 3,03(s, 3 H), 2,45(s, 3 H). MS m/z: 264,0 [M+H]+
[225] Стадия 2: синтез соединения BB-9.
[226] Соединение BB-9-2 (3,00 г, 11,36 ммоль, 1,00 экв.) и соединение BB-2-3 (4,33 г, 17,04 ммоль, 1,50 экв.) растворяли в диоксане (60,00 мл), затем Pd(dppf)Cl2.CH2Cl2 (927,51 мг, 1,14 ммоль, 0,10 экв.) и ацетат калия (3,34 г, 34,08 ммоль, 3,00 экв.) добавляли при 20°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 85°C и реакционную смесь перемешивали в течение 3 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли водой (100 мл) и экстрагировали дихлорметаном (100 мл*2). Органическую фазу собирали, промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ТСХ (PE/EA=1/1) с получением соединения BB-9. 1H ЯМР (400 МГц, CDCl3-d) δ 7,70-7,65(m, 1 H), 7,61-7,56(m, 1 H), 7,25(t, J=7,6 Гц, 1 H), 6,29(s, 1 H), 3,00(s, 3 H), 2,55(s, 3 H), 1,37(s, 12 H). MS m/z: 311,9 [M+H]+
[227] Ссылочный пример 15: фрагмент BB-10
[228] Схема синтеза:
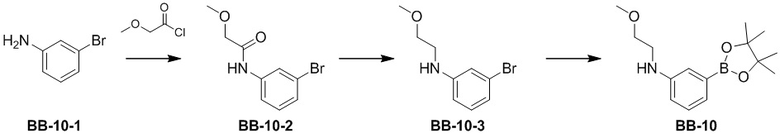
[229] Стадия 1: синтез соединения BB-10-2.
[230] Соединение BB-10-1 (6,00 г, 34,88 ммоль, 1,00 экв.) растворяли в дихлорметане (60,00 мл), затем добавляли триэтиламин (7,06 г, 69,76 ммоль, 9,67 мл, 2,00 экв.) и раствор метоксиацетилхлорида (3,97 г, 36,62 ммоль, 1,05 экв.) в дихлорметане (60 мл) добавляли по каплям при 0°C. Температуру реакционной смеси повышали до 15°C и реакционную смесь перемешивали в течение 2 часов. После завершения реакции добавляли воду (120 мл) для гашения реакционной смеси. Органическую фазу последовательно промывали насыщенным раствором бикарбоната натрия (100 мл *2) и водой (100 мл). Водную фазу экстрагировали дихлорметаном (100 мл*2) и органические фазы объединяли. Объединенную органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (10 мл) и петролейным эфиром (10 мл) с получением соединения BB-10-2. 1H ЯМР (400 МГц, CDCl3-d) δ 8,25(br. s., 1 H), 7,84-7,82(m, 1 H), 7,50(d, J=7,2 Гц, 1 H), 7,26(d, J=4,0 Гц, 1 H), 7,20(t, J=8,0 Гц, 1 H), 4,01(s, 2 H), 3,51(s, 3 H). MS m/z: 245,9 [M+H]+
[231] Стадия 2: синтез соединения BB-10-3.
[232] Соединение BB-10-2 (5,07 г, 20,77 ммоль, 1,00 экв.) растворяли в ТГФ (80,00 мл), раствор борана (8,92 г, 103,85 мл, 1 M, 5,00 экв.) в ТГФ добавляли по каплям при 0°C, затем температуру восстанавливали до 15°C. Реакционную смесь перемешивали при 80°C в течение 17 часов. После завершения реакции температуру понижали до 0°C, и реакционную смесь медленно гасили метанолом (150 мл), затем указанный растворитель удаляли с помощью роторного испарителя. Неочищенный продукт разбавляли EtOAc (200 мл) и промывали соляной кислотой (3 M, 100 мл *3). Водную фазу экстрагировали EtOAc (100 мл). Водную фазу доводили до pH=7 насыщенным раствором бикарбоната натрия и затем экстрагировали EtOAc (150 мл *3). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-10-3. 1H ЯМР (400 МГц, CDCl3-d) δ 7,01(t, J=8,2 Гц, 1 H), 6,82(d, J=7,6 Гц, 1 H), 6,76(s, 1 H), 6,54(dd, J=8,0 Гц, J=2,0 Гц, 1 H), 4,44(br. s., 1 H), 3,60(t, J=5,2 Гц, 2 H), 3,39(s, 3 H), 3,26(t, J=5,2 Гц, 2 H). MS m/z: 231,7 [M+H]+
[233] Стадия 3: синтез соединения BB-10.
[234] Соединение BB-10-3 (3,94 г, 17,12 ммоль, 1,00 экв.) растворяли в диоксане (80,00 мл), добавляли соединение BB-2-3 (5,65 г, 22,26 ммоль, 1,30 экв.) и ацетат калия (5,04 г, 51,36 ммоль, 3,00 экв.), затем Pd(dppf)Cl2.CH2Cl2 (1,40 г, 1,71 ммоль, 0,10 экв.) добавляли при 15°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 120°C и реакционную смесь перемешивали в течение 18 часов. После завершения реакции смесь фильтровали и фильтрационный осадок промывали дихлорметаном (50 мл *4). Фильтрат собирали, выпаривали досуха с помощью роторного испарителя, растворяли в EtOAc (150 мл) и затем промывали водой (120 мл*3). Водные фазы объединяли и экстрагировали EtOAc (80 мл*3). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=20/1-5/1) с получением целевого соединения BB-10. 1H ЯМР (400 МГц, CDCl3-d) δ 7,27(s, 1 H), 7,19(t, J=8,0 Гц, 1 H), 7,09(d, J=2,0 Гц, 1 H), 6,76-6,74(m, 1 H), 4,03(br. s., 1 H), 3,61(t, J=4,8 Гц, 2 H), 3,39(s, 3 H), 3,33(t, J=4,8 Гц, 2 H), 1,34(s, 12 H). MS m/z: 278,0 [M+H]+
[235] Ссылочный пример 16: фрагмент BB-12
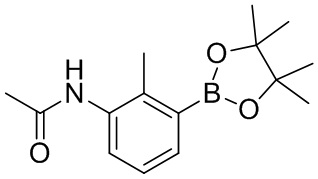
[236] Схема синтеза:
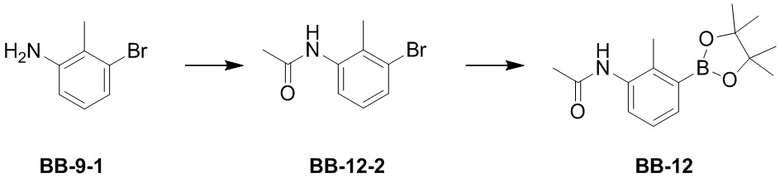
[237] Стадия 1: синтез соединения BB-12-2.
[238] Соединение BB-9-1 (5,00 г, 26,87 ммоль, 1,00 экв.) добавляли в пиридин (50,00 мл) и хлороформ (100,00 мл), затем добавляли ацетилхлорид (2,74 г, 34,93 ммоль, 1,30 экв.) при 0°C в защитной атмосфере азота. Реакционную смесь перемешивали при 25oC в течение 2 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=1/1, Rf=0,5) с получением соединения BB-12-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,64(br. s., 1 H), 7,43(d, J=7,6 Гц, 1 H), 7,09(t, J=8,0 Гц, 2 H), 2,37(s, 3 H), 2,24(s, 3 H). MS m/z:229,7 [M+H]+
[239] Стадия 2: синтез соединения BB-12.
[240] Соединение BB-12-2 (3,00 г, 13,15 ммоль, 1,00 экв.) и соединение BB-2-3 (5,01 г, 19,72 ммоль, 1,50 экв.) добавляли в диоксан (60,00 мл), затем добавляли Pd(dppf)Cl2.CH2Cl2 (1,07 г, 1,32 ммоль, 0,10 экв.) и ацетат калия (3,87 г, 39,45 ммоль, 3,00 экв.) при 20°C в защитной атмосфере азота. Реакционную смесь перемешивали при 85°C в течение 3 часов. После завершения реакции реакционную смесь разбавляли водой (100 мл) и экстрагировали дихлорметаном (100 мл*2). Органическую фазу собирали и промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=2/1) с получением целевого соединения BB-12. 1H ЯМР (400 МГц, CDCl3-d) δ 7,85(d, J=8,0 Гц, 1 H), 7,30(d, J=7,2 Гц, 1 H), 7,23(t, J=7,4 Гц, 1 H), 6,98(br. s., 1 H), 2,48(s, 3 H), 2,24(s, 3 H), 1,37(s, 12 H). MS m/z: 276,0 [M+H]+
[241] Ссылочный пример 17: фрагмент BB-13
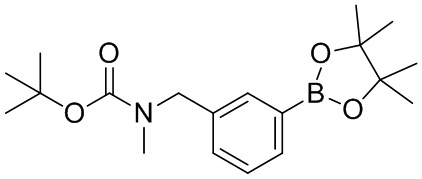
[242] Схема синтеза:
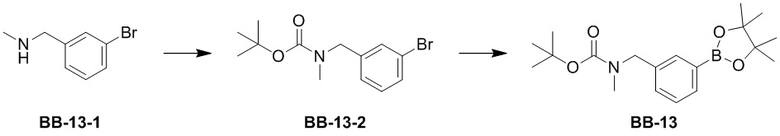
[243] Стадия 1: синтез соединения BB-13-2.
[244] Соединение BB-13-1 (5,00 г, 24,99 ммоль, 1,00 экв.) растворяли в диоксане (50,00 мл) и насыщенном растворе бикарбоната натрия (50,00 мл) с последующим добавлением Boc2O (8,18 г, 37,49 ммоль, 1,50 экв.) при 0°C. Реакционную смесь перемешивали при 20°C в течение 10 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=10/1) с получением соединения BB-13-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,43-7,39(m, 2 H), 7,25-7,17(m, 2 H), 4,40(br. s., 2 H), 2,88-2,81(m, 3 H), 1,49(s, 9 H). MS m/z: 245,8 [M+H]+
[245] Стадия 2: синтез соединения BB-13.
[246] Соединение BB-2-3 (6,34 г, 24,99 ммоль, 1,50 экв.), соединение BB-13-2 (5,00 г, 16,66 ммоль, 1,00 экв.), ацетат калия (3,27 г, 33,32 ммоль, 2,00 экв.) и Pd(dppf)Cl2.CH2Cl2 (1,36 г, 1,67 ммоль, 0,10 экв.) добавляли в высушенную реакционную колбу и добавляли диоксан (100,00 мл). Температуру реакционной смеси повышали до 80°C и реакционную смесь перемешивали в течение 3 часов. После завершения реакции реакционный раствор фильтровали через диатомовую землю. Фильтрат собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=5/1) с получением целевого соединения BB-13.
[247] 1H ЯМР (400 МГц, CDCl3-d) δ 7,73-7,68(m, 2 H), 7,37-7,33(m, 2 H), 4,42(br. s., 2 H), 2,85-2,76(m, 3 H), 1,49(s, 9 H), 1,35(s, 12 H). MS m/z: 291,9 [M+H]+
[248] Ссылочный пример 18: фрагмент BB-14
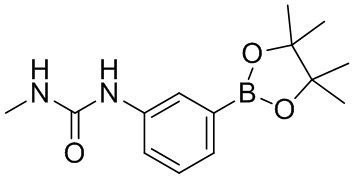
[249] Схема синтеза:
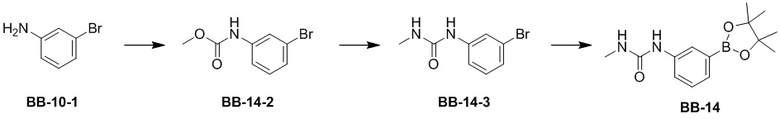
[250] Стадия 1: синтез соединения BB-14-2
[251] Метилхлорформиат (22,90 г, 242,33 ммоль, 5,21 экв.) растворяли в дихлорметане (100,00 мл), затем раствор соединения BB-10-1 (8,00 г, 46,51 ммоль, 1,00 экв.) и триэтиламина (14,12 г, 139,53 ммоль, 3,00 экв.) в дихлорметане (60,00 мл) добавляли при -10°C. Реакционную смесь перемешивали при -10°C в течение 2 часов. После завершения реакции реакционную смесь разбавляли метил-трет-бутиловым эфиром (500 мл) и образовывалось твердое вещество. Твердое вещество фильтровали и промывали метил-трет-бутиловым эфиром (50 мл*3), фильтрат собирали и выпаривали досуха с помощью роторного испарителя. Неочищенный продукт разбавляли метил-трет-бутиловым эфиром (200 мл), затем последовательно промывали 0,5 M раствором соляной кислоты (100 мл*2), насыщенным раствором бикарбоната натрия (100 мл*2) и насыщенным раствором хлорида натрия (80 мл). Органическую фазу собирали, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-14-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,64(s, 1 H), 7,30-7,25(m, 1 H), 7,20-7,15(m, 2 H), 6,67(br. s., 1 H), 3,78(s, 3 H). MS m/z: 231,8 [M+H]+
[252] Стадия 2: синтез соединения BB-14-3
[253] Соединение BB-14-2 (1,04 г, 4,52 ммоль, 1,00 экв.) и метиламин (6,60 г, 53,12 ммоль, 11,75 экв.) добавляли в герметичную пробирку, температуру повышали до 100°C и реакционную смесь перемешивали в течение 40 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с петролейным эфиром (20 мл) и метил-трет-бутиловым эфиром (4 мл), фильтровали и выпаривали досуха с помощью роторного испарителя с получением соединения BB-14-3. 1H ЯМР (400 МГц, CDCl3-d) δ 7,51(s, 1 H), 7,43(s, 1 H), 7,19(d, J=8,0 Гц, 1 H), 7,14-7,09(m, 2 H), 5,54(d, J=3,6 Гц, 1 H), 2,78(d, J=4,8 Гц, 3 H). MS m/z: 230,8 [M+H]+
[254] Стадия 3: синтез соединения BB-14
[255] Соединение BB-14-3 (1,31 г, 5,72 ммоль, 1,00 экв.) растворяли в диоксане (25,00 мл), затем соединение BB-2-3 (1,74 г, 6,86 ммоль, 1,20 экв.), ацетат калия (1,68 г, 17,16 ммоль, 3,00 экв.) и Pd(dppf)Cl2.CH2Cl2 (467,02 мг, 572,00 мкмоль, 0,10 экв.) последовательно добавляли при 25°C в защитной атмосфере азота. Температуру повышали до 100°C и реакционную смесь перемешивали в течение 3 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя и разбавляли EtOAc (50 мл), последовательно промывали водой (30 мл*2) и насыщенным раствором хлорида натрия (20 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (20 мл), фильтровали и выпаривали досуха с помощью роторного испарителя с получением целевого соединения BB-14. 1H ЯМР (400 МГц, CDCl3-d) δ 7,64(s, 1 H), 7,55-7,51(m, 2 H), 7,33(t, J=7,6 Гц, 1 H), 6,88(s, 1 H), 5,12(d, J=4,0 Гц, 1 H), 2,80(d, J=4,4 Гц, 3 H), 1,34(s, 12 H). MS m/z: 276,9 [M+H]+
[256] Ссылочный пример 19: фрагмент BB-15

[257] Схема синтеза:
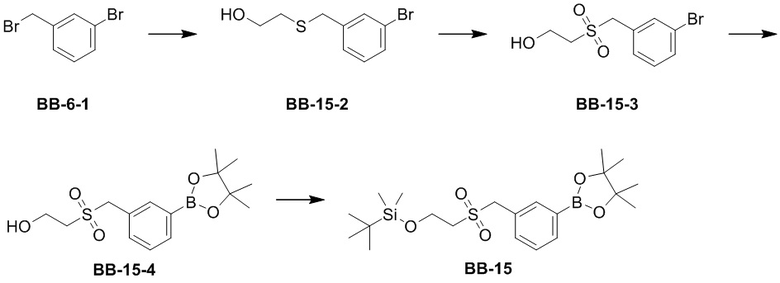
[258] Стадия 1: синтез соединения BB-15-2.
[259] 2-Меркаптоэтанол (2,19 г, 28,08 ммоль, 1,96 мл, 2,34 экв.) растворяли в метаноле (30,00 мл), затем метоксид натрия (1,32 г, 24,00 ммоль, 98% чистота, 2,00 экв.) добавляли при 20°C в защитной атмосфере азота. Реакционную смесь перемешивали при 20°C в течение 1 часа, затем добавляли соединение BB-6-1 (3,00 г, 12,00 ммоль, 1,00 экв.) и реакционную смесь перемешивали при 20°C в течение дополнительных 3 часов. После завершения реакции метанол удаляли с помощью роторного испарителя и для гашения реакционной смеси добавляли воду (10мл). Водную фазу экстрагировали EtOAc (30 мл*2). Органические фазы объединяли и промывали насыщенным раствором хлорида натрия (10 мл). Органическую фазу собирали, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=20/1-5/1) с получением соединения BB-15-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,50(s, 1 H), 7,41(d, J=7,6 Гц, 1 H), 7,27(d, J=8,4 Гц, 1 H), 7,21(t, J=7,6 Гц, 1 H), 3,76-3,62(m, 4 H), 2,66(t, J=5,8 Гц, 2 H).
[260] Стадия 2: синтез соединения BB-15-3
[261] Соединение BB-15-2 (2,90 г, 11,73 ммоль, 1,00 экв.) растворяли в ТГФ (30,00 мл) и воде (30,00 мл), затем комплексную соль бисульфата калия (21,64 г, 17,60 ммоль, 50% чистота, 1,50 экв.) добавляли при 20°C в защитной атмосфере азота. Реакционную смесь перемешивали при 20°C в течение 6 часов. После завершения реакции добавляли насыщенный раствор сульфита натрия (20 мл) и смесь экстрагировали EtOAc (30 мл*3). Органические фазы объединяли и промывали насыщенным раствором хлорида натрия (15 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-15-3.
[262] 1H ЯМР (400 МГц, CDCl3-d) δ 7,62(s, 1 H), 7,55(d, J=8,0 Гц, 1 H), 7,41(d, J=7,6 Гц, 1 H), 7,29(t, J=8,0 Гц, 1 H), 4,33(s, 2 H), 4,14(q, J=4,8 Гц, 2 H), 3,11(t, J=5,0 Гц, 2 H).
[263] Стадия 3: синтез соединения BB-15-4
[264] Соединение BB-15-3 (3,00 г, 10,75 ммоль, 1,00 экв.) и соединение BB-2-3 (4,09 г, 16,12 ммоль, 1,50 экв.) растворяли в диоксане (10,00 мл), затем Pd(dppf)Cl2.CH2Cl2 (877,64 мг, 1,07 ммоль, 0,10 экв.) и ацетат калия (2,11 г, 21,49 ммоль, 2,00 экв.) добавляли при 20°C в защитной атмосфере азота. Температуру повышали до 80°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (ДХМ/MeOH=1/0-20/1) с получением соединения BB-15-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,87-7,80(m, 2 H), 7,58(d, J=7,6 Гц, 1 H), 7,42(t, J=7,6 Гц, 1 H), 4,35(s, 2 H), 4,12-4,05(m, 2 H), 3,14-3,07(m, 2 H), 1,35(s, 12 H).
[265] Стадия 4: синтез соединения BB-15
[266] Соединение BB-15-4 (2,93 г, 8,98 ммоль, 1,00 экв.) растворяли в N,N-диметилформамиде (20,00 мл), затем трет-бутилдиметилсилилхлорид (2,03 г, 13,47 ммоль, 1,65 мл, 1,50 экв.) и имидазол (1,53 г, 22,45 ммоль, 2,50 экв.) добавляли при 25°C в защитной атмосфере азота. Реакционную смесь перемешивали при 25°C в течение 2 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (ДХМ/MeOH=20/1) с получением целевого соединения BB-15. 1H ЯМР (400 МГц, CDCl3-d) δ 7,88(s, 1 H), 7,84(d, J=7,2 Гц, 1 H), 7,60(d, J=8,0 Гц, 1 H), 7,43(t, J=7,6 Гц, 1 H), 4,37(s, 2 H), 4,12(t, J=5,4 Гц, 2 H), 3,08(t, J=5,4 Гц, 2 H), 1,36(s, 12 H), 0,98(s, 9 H), 0,19(s, 6 H).
[267] Ссылочный пример 20: фрагмент BB-16
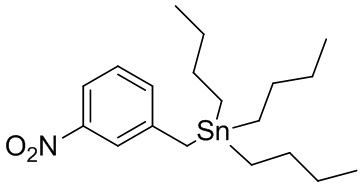
[268] Схема синтеза:
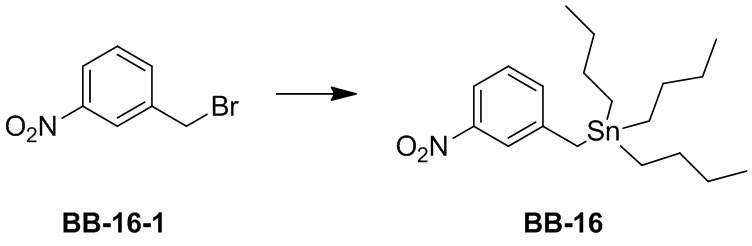
[269] Стадия 1: синтез соединения BB-16.
[270] Соединение BB-16-1 (5,00 г, 23,14 ммоль, 1,00 экв.) растворяли в толуоле (125,00 мл), затем гексабутилдиолово (18,79 г, 31,74 ммоль, 16,20 мл, 98% чистота, 1,37 экв.) и Pd(PPh3)4 (267,45 мг, 231,40 мкмоль, 0,01 экв.) добавляли при 20°C в защитной атмосфере азота. Температуру повышали до 100°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционную смесь промывали 20% водным раствором фторида калия (15 мл). Водную фазу экстрагировали EtOAc (40 мл*2). Органические фазы объединяли и промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=1/0) с получением целевого соединения BB-16. 1H ЯМР (400 МГц, CDCl3-d) δ 7,78-7,73(m, 2 H), 7,27-7,19(m, 2 H), 2,33(s, 2 H), 1,46-1,30(m, 6 H), 1,25-1,13(m, 6 H), 0,85-0,73(m, 15 H).
[271] Ссылочный пример 21: фрагмент BB-17
[272] Схема синтеза:
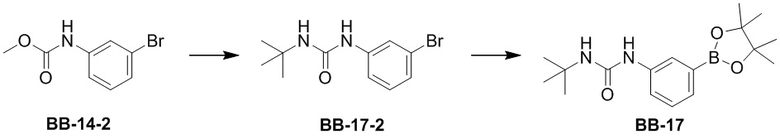
[273] Стадия 1: синтез соединения BB-17-2.
[274] Соединение BB-14-2 (3,15 г, 13,69 ммоль, 1,00 экв.) и трет-бутиламин (17,40 г, 237,90 ммоль, 25,00 мл, 17,38 экв.) добавляли в чашу, реакционную смесь нагревали до 100°C и перемешивали в течение 65 часов. После того, как ЖХМС показала неполное завершение реакции, реакционную смесь перемешивали в течение дополнительных 22 часов при 100°C. После того, как ЖХМС показала неполное завершение реакции, реакционную смесь перемешивали в течение дополнительных 22 часов при 100°C. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с петролейным эфиром (20 мл) и метил-трет-бутиловым эфиром (4 мл) и очищали с получением соединения BB-17-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,56(s, 1 H), 7,22(s, 1 H), 7,20-7,14(m, 1 H), 7,10-7,05(m, 2 H), 5,30(s, 1 H), 1,35(s, 9 H). MS m/z:272,8 [M+H]+
[275] Стадия 2: синтез соединения BB-17.
[276] Соединение BB-17-2 (3,25 г, 11,99 ммоль, 1,00 экв.) и соединение BB-2-3 (3,65 г, 14,39 ммоль, 1,20 экв.) растворяли в диоксане (65,00 мл), Pd(dppf)Cl2.CH2Cl2 (978,82 мг, 1,20 ммоль, 0,10 экв.) и ацетат калия (3,53 г, 35,97 ммоль, 3,00 экв.) добавляли при 25°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 3 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя и неочищенный продукт разбавляли EtOAc (60 мл). После фильтрования фильтрационный осадок промывали EtOAc (20 мл*3). Органические фазы объединяли, промывали последовательно водой (80 мл*2) и насыщенным раствором хлорида натрия (50 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растворяли в дихлорметане (100 мл), затем добавляли петролейный эфир (150 мл) и образовывалось твердое вещество. После фильтрования фильтрационный осадок промывали дихлорметаном (15 мл*4) и собирали, затем выпаривали досуха с помощью роторного испарителя с получением целевого соединения BB-17. 1H ЯМР (400 МГц, CDCl3-d) δ 7,57(s, 1 H), 7,54-7,49(m, 2 H), 7,34(t, J=7,6 Гц, 1 H), 6,19(s, 1 H), 4,64(s, 1 H), 1,38(s, 9 H), 1,35(s, 12 H). MS m/z: 319,1 [M+H]+
[277] Ссылочный пример 22: фрагмент BB-18
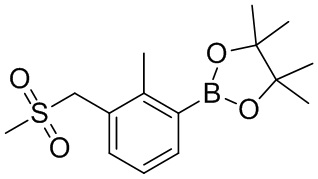
[278] Схема синтеза:
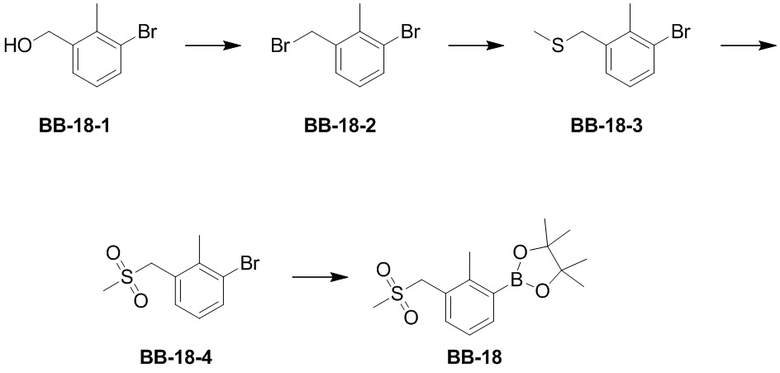
[279] Стадия 1: синтез соединения BB-18-2.
[280] BB-18-1 (15,00 г, 74,60 ммоль, 1,00 экв.) растворяли в дихлорметане (300,00 мл), PBr3 (13,13 г, 48,49 ммоль, 4,61 мл, 0,65 экв.) добавляли по каплям при 0°C, затем реакционную смесь перемешивали при 25°C в течение 15 часов. После завершения реакции смесь охлаждали до 0°C и гасили метанолом (20 мл). Смесь последовательно промывали насыщенным раствором бикарбоната натрия (100 мл*3) и водой (100 мл*2). Органическую фазу собирали, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-18-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,53(d, J=8,0 Гц, 1 H), 7,26(d, J=6,0 Гц, 1 H), 7,03(t, J=7,8 Гц, 1 H), 4,52(s, 2 H), 2,49(s, 3 H).
[281] Стадия 2: синтез соединения BB-18-3.
[282] Соединение BB-18-2 (20,26 г, 76,75 ммоль, 1,00 экв.) растворяли в N,N-диметилформамиде (250,00 мл), добавляли тиометоксид натрия (5,40 г, 77,04 ммоль, 1,00 экв.), затем реакционную смесь перемешивали при 0°C в течение 3 часов. После завершения реакции реакционный раствор выливали в воду (500 мл) и экстрагировали EtOAc (150 мл*3). Органические фазы объединяли, последовательно промывали водой (300 мл*3) и насыщенным раствором хлорида натрия (250 мл). Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-18-3. 1H ЯМР (400 МГц, CDCl3-d) δ 7,49(d, J=8,0 Гц, 1 H), 7,13(d, J=7,2 Гц, 1 H), 7,00(t, J=7,6 Гц, 1 H), 3,73(s, 2 H), 2,49(s, 3 H), 2,05(s, 3 H).
[283] Стадия 3: синтез соединения BB-18-4.
[284] Соединение BB-18-3 (17,30 г, 74,84 ммоль, 1,00 экв.) растворяли в дихлорметане (350,00 мл), партиями добавляли м-хлорпербензойную кислоту (35,52 г, 164,65 ммоль, 2,20 экв.) при 0°C, затем температуру медленно повышали до 25°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции температуру понижали до 0°C и для гашения реакционной смеси добавляли насыщенный раствор сульфита натрия (150 мл). Дихлорметан удаляли с помощью роторного испарителя и смесь фильтровали. Фильтрационный осадок промывали метанолом (30 мл*4) с получением неочищенного продукта 1. Фильтрат концентрировали и экстрагировали EtOAc (100 мл*3), органические фазы объединяли и последовательно промывали насыщенным раствором сульфита натрия (100 мл*2), насыщенным раствором бикарбоната натрия (100 мл*3), водой (100 мл) и насыщенным раствором хлорида натрия (100 мл), затем сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта 2. Неочищенный продукт 1 и неочищенный продукт 2 объединяли и растирали с петролейным эфиром (100 мл) с получением соединения BB-18-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,61(d, J=8,0 Гц, 1 H), 7,29(d, J=7,6 Гц, 1 H), 7,10(t, J=7,8 Гц, 1 H), 4,39(s, 2 H), 2,84(s, 3 H), 2,54(s, 3 H).
[285] Стадия 4: синтез соединения BB-18.
[286] Соединение BB-18-4 (10,00 г, 38,00 ммоль, 1,00 экв.) растворяли в диоксане (100,00 мл), затем последовательно добавляли соединение BB-2-3 (11,58 г, 45,60 ммоль, 1,20 экв.), ацетат калия (11,19 г, 114,00 ммоль, 3,00 экв.) и Pd(dppf)Cl2.CH2Cl2 (3,10 г, 3,80 ммоль, 0,10 экв.). Реакционную смесь перемешивали при 100°C в атмосфере азота в течение 14 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и указанный растворитель удаляли с помощью роторного испарителя. Остаток растворяли EtOAc (500 мл). Органическую фазу последовательно промывали водой (250 мл*2) и насыщенным раствором хлорида натрия (200 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=25/1-5/1) с получением целевого соединения BB-18. 1H ЯМР (400 МГц, CDCl3-d) δ 7,72(d, J=7,2 Гц, 1 H), 7,35(dd, J=7,6 Гц, J=1,2 Гц, 1 H), 7,15(t, J=7,6 Гц, 1 H), 4,30(s, 2 H), 2,71(s, 3 H), 2,57(s, 3 H), 1,28(s, 12 H).
[287] Ссылочный пример 23: фрагмент BB-19
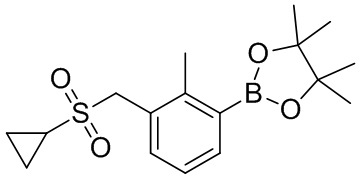
[288] Схема синтеза:
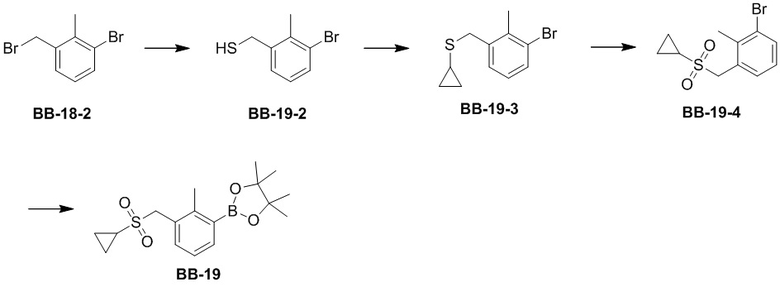
[289] Стадия 1: синтез соединения BB-19-2
[290] Соединение BB-18-2 (10,00 г, 37,88 ммоль, 1,00 экв.) растворяли в метаноле (50,00 мл), затем карбонат калия (6,28 г, 45,46 ммоль, 1,20 экв.) и тиоуксусную кислоту (3,46 г, 45,46 ммоль, 1,20 экв.) последовательно добавляли в защитной атмосфере азота. Затем смесь перемешивали при 25°C в течение 30 минут, добавляли карбонат калия (6,28 г, 45,46 ммоль, 1,20 экв.) и реакционную смесь перемешивали в течение 30 минут. После завершения реакции реакционную смесь выливали в воду (80 мл), водную фазу экстрагировали EtOAc (100 мл*2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (60 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-19-2. 1H ЯМР (400 МГц, CDCl3-d) δ 7,51-7,49(m, 1 H), 7,01-6,97(m, 2 H), 4,47(s, 2 H), 3,41(s, 3 H).
[291] Стадия 2: синтез соединения BB-19-3
[292] Соединение BB-19-2 (5,00 г, 23,03 ммоль, 1,00 экв.) и трет-бутоксид калия (2,58 г, 23,03 ммоль, 1,00 экв.) растворяли в диметилсульфоксиде (30,00 мл), циклопропилбромид (3,06 г, 25,33 ммоль, 1,10 экв.) добавляли в защитной атмосфере азота. Температуру повышали до 90°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции добавляли воду (80 мл) и водную фазу экстрагировали EtOAc (80 мл*2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта соединения BB-19-3.
[293] Стадия 3: синтез соединения BB-19-4
[294] Соединение BB-19-3 (5,20 г, 20,22 ммоль, 1,00 экв.) растворяли в ТГФ (50,00 мл) и воде (50,00 мл), гидросульфат калия (37,29 г, 30,33 ммоль, 50% чистота, 1,50 экв.) добавляли в защитной атмосфере азота. Реакционную смесь перемешивали при 25°C в течение 4 часов. После завершения реакции добавляли насыщенный раствор сульфита натрия (10 мл). Водную фазу экстрагировали EtOAc (20 мл*3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=20/1-3/1) с получением соединения BB-19-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,60(d, J=8,0 Гц, 1 H), 7,40(d, J=7,6 Гц, 1 H), 7,08(t, J=8,0 Гц, 1 H), 4,41(s, 2 H), 2,55(s, 3 H), 2,34-2,28(m, 1 H), 1,20-1,16(m, 2 H), 1,02-1,00(m, 2 H).
[295] Стадия 4: синтез соединения BB-19
[296] Соединение BB-19-4 (2,20 г, 7,61 ммоль, 1,00 экв.) и соединение BB-2-3 (2,90 г, 11,41 ммоль, 1,50 экв.) добавляли в диоксан (20,00 мл), затем Pd(dppf)Cl2.CH2Cl2 (621,26 мг, 760,75 мкмоль, 0,10 экв.) и ацетат калия (1,49 г, 15,21 ммоль, 2,00 экв.) добавляли в защитной атмосфере азота. Температуру повышали до 90°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (PE/EA=20/1-5/1) с получением целевого соединения BB-19. 1H ЯМР (400 МГц, CDCl3-d) δ 7,78(d, J=7,6 Гц, 1 H), 7,46(d, J=7,6 Гц, 1 H), 7,21(t, J=7,6 Гц, 1 H), 4,40(s, 2 H), 2,66(s, 3 H), 2,30-2,25(m, 1 H), 1,36(s, 12 H), 1,17-1,15(m, 2 H), 0,98-0,95(m, 2 H).
[297] Ссылочный пример 24: фрагмент BB-20
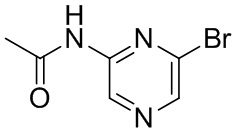
[298] Схема синтеза:
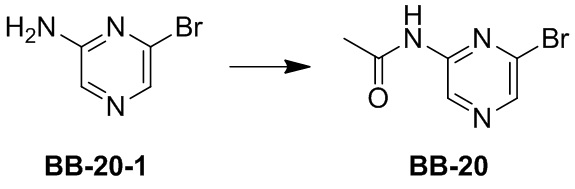
[299] Стадия 1: синтез соединения BB-20
[300] Соединение BB-20-1 (500,00 мг, 2,87 ммоль, 1,00 экв.) добавляли в уксусный ангидрид (5,45 г, 53,38 ммоль, 5,00 мл, 18,60 экв.), затем реакционную смесь перемешивали при 25°C в течение 5 часов. Реакционный раствор концентрировали и разбавляли дихлорметаном (50 мл). Органическую фазу последовательно промывали насыщенным раствором карбоната натрия (50 мл*3) и насыщенным раствором хлорида натрия (50 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением целевого соединения BB-20. 1H ЯМР (400 МГц, CDCl3-d) δ 9,45(s, 1 H), 8,44(s, 1 H), 8,00(br. s., 1 H), 2,26(s, 3 H). MS m/z: 217,7 [M+H]+
[301] Ссылочный пример 25: фрагмент BB-21
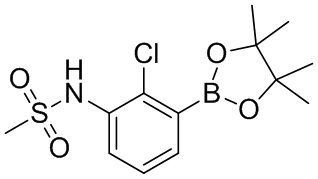
[302] Схема синтеза:
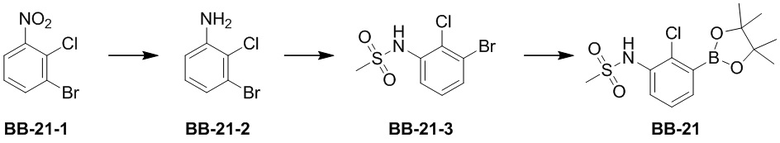
[303] Стадия 1: синтез соединения BB-21-2
[304] Соединение BB-21-1 (4,80 г, 20,30 ммоль, 1,00 экв.) растворяли в этаноле (100,00 мл), затем добавляли дигидрат хлорида олова (22,90 г, 101,50 ммоль, 8,45 мл, 5,00 экв.). Температуру повышали до 80°C и реакционную смесь перемешивали в течение 2 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры и разбавляли водой (50мл). Смесь доводили до pH=8-9 насыщенным раствором бикарбоната натрия, затем водную фазу экстрагировали EtOAc (50мл*3). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-21-2. 1H ЯМР (400 МГц, CDCl3-d) δ 6,92(d, J=7,6 Гц, 1 H), 6,83(t, J=7,8 Гц, 1 H), 6,62(dd, J=8,0 Гц, J=1,2 Гц, 1 H), 4,14(br. s., 2 H). MS m/z: 207,9 [M+H]+
[305] Стадия 2: синтез соединения BB-21-3
[306] При 0°C соединение BB-21-2 (2,75 г, 13,32 ммоль, 1,00 экв.) растворяли в дихлорметане (20,00 мл), затем добавляли пиридин (1,05 г, 13,32 ммоль, 1,08 мл, 1,00 экв.) и метансульфонилхлорид (2,29 г, 19,98 ммоль, 1,55 мл, 1,50 экв.). В условиях защитной атмосферы азота температуру повышали до 12°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционную смесь гасили насыщенным раствором бикарбоната натрия (20 мл) при комнатной температуре, затем смесь разбавляли водой (30 мл) и экстрагировали дихлорметаном (30 мл*3). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-21-3. 1H ЯМР (400 МГц, CDCl3-d) δ 7,57(dd, J=8,4 Гц, J=1,6 Гц, 1 H), 7,40(dd, J=8,0 Гц, J=1,2 Гц, 1 H), 7,11(t, J=8,0 Гц, 1 H), 6,94(br. s., 1 H), 2,96(s, 3 H).
[307] Стадия 3: синтез соединения BB-21
[308] Соединение BB-21-3 (500,00 мг, 1,76 ммоль, 1,00 экв.) растворяли в диоксане (4,00 мл), последовательно добавляли соединение BB-2-3 (581,01 мг, 2,29 ммоль, 1,30 экв.), ацетат калия (207,27 мг, 2,11 ммоль, 1,20 экв.) и Pd(dppf)Cl2 (257,56 мг, 352,00 мкмоль, 0,20 экв.). В условиях защитной атмосферы азота температуру повышали до 100°C и реакционную смесь перемешивали в течение 4 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, разбавляли EtOAc (20 мл), последовательно промывали водой (20 мл*3) и насыщенным раствором хлорида натрия (20 мл). Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=1/1) с получением целевого соединения BB-21.
[309] 1H ЯМР (400 МГц, CDCl3-d) δ 7,66 (dd, J=8,0 Гц, J=1,6 Гц, 1 H), 7,48(dd, J=7,6 Гц, J=1,6 Гц, 1 H), 7,23(t, J=7,8 Гц, 1 H), 6,84(br. s., 1 H), 2,89(s, 3 H), 1,31(s, 12 H).
[310] Ссылочный пример 26: фрагмент BB-22
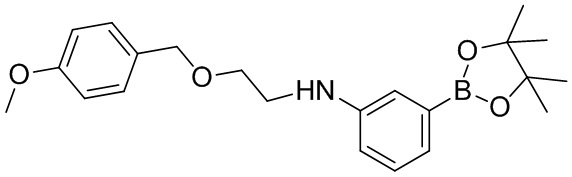
[311] Схема синтеза:
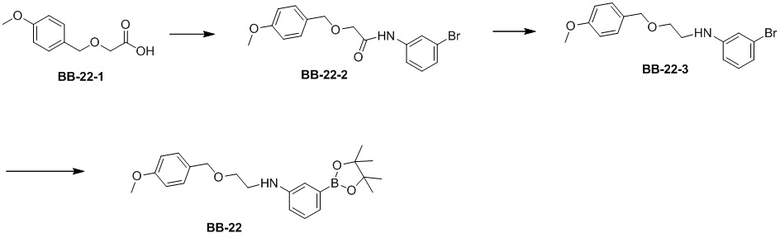
[312] Стадия 1: синтез соединения BB-22-2
[313] Соединение BB-22-1 (4,5 г, 22,94 ммоль, 1,00 экв.) растворяли в ДМФА (90 мл), затем добавляли DIPEA (8,89 г, 68,82 ммоль, 12,01 мл, 3,00 экв.) и HATU (15,70 г, 41,29 ммоль, 1,80 экв.) в защитной атмосфере азота при 0°C. Реакционный раствор подвергали взаимодействию при 20°C в течение 0,5 часа и затем добавляли 3-броманилин (5,92 г, 34,41 ммоль, 3,75 мл, 1,50 экв.). Реакционную смесь перемешивали при 20°C в течение 12 часов. После завершения реакции для гашения реакционной смеси добавляли воду (200 мл) и смесь экстрагировали EtOAc (200 мл) дважды. Органическую фазу промывали насыщенным солевым раствором (200 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (EA/PE=5/1-1/1) с получением соединения BB-22-2. 1H ЯМР (400 МГц, CDCl3-d) δ 8,28(s,1 H), 7,78(s, 1 H), 7,46(d, J=8,0 Гц,1 H), 7,30( m, 3H), 7,18(m, 1H), 6,94(m, 2H),4,59(s,1H), 4,07(d, J=4,0 Гц, 1H), 3,83(s, 3H). MS m/z: 350 [M+H]+
[314] Стадия 2: синтез соединения BB-22-3
[315] При 0°C в защитной атмосфере азота раствор диметилсульфид-борана (10 M, 5,57 мл, 3,00 экв.) добавляли к раствору соединения BB-22-2 (6,5 г, 18,56 ммоль, 1,00 экв.) в безводном ТГФ (130 мл). Температуру медленно повышали до 65°C и реакционную смесь перемешивали при 65°C в течение 12 часов. После завершения реакции реакционную смесь охлаждали до 0°C и гасили метанолом (100 мл). После гашения смесь перемешивали при 20°C в течение 1 часа, затем концентрировали досуха с помощью роторного испарителя. Остаток растворяли в дихлорметане (100,00 мл), промывали водой (100 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения BB-22-3. MS m/z: 337 [M+H]+
[316] Стадия 3: синтез соединения BB-22
[317] Соединение BB-22-3 (5,1 г, 15,17 ммоль, 1,00 экв.) и соединение BB-2-3 (5..78g, 22,76 ммоль, 1,50 экв.) смешивали в диоксане (100 мл), затем последовательно добавляли ацетат калия (3,72 г, 37,92 ммоль, 2,5 экв.) и Pd(dppf)Cl2.CH2Cl2 (1,24 г, 1,52 мкмоль, 0,10 экв.). В условиях защитной атмосферы азота температуру повышали до 80°C и реакционную смесь перемешивали в течение 2 часов. После завершения реакции смесь охлаждали до комнатной температуры, реакционный раствор фильтровали через короткую колонку с силикагелем. Фильтрат концентрировали и очищали с помощью колоночной хроматографии (PE/EA=10/1-5/1) с получением соединения BB-22. 1H ЯМР (400 МГц, CDCl3-d) δ 7,66 (dd, J=8,0 Гц, J=1,6 Гц, 1 H), 7,48(dd, J=7,6 Гц, J=1,6 Гц, 1 H), 7,23(t, J=7,8 Гц, 1 H), 6,84(br. s., 1 H), 2,89(s, 3 H), 1,31(s, 12 H). MS m/z: 384 [M+H]+
[318] Вариант реализации 1: WX040
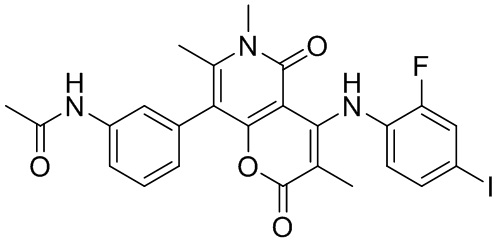
[319] Схема синтеза:
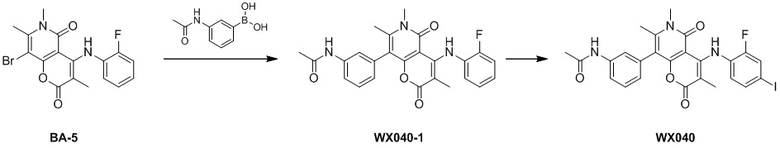
[320] Стадия 1: синтез соединения WX040
[321] Соединение BA-5 (500,00 мг, 1,27 ммоль, 1,00 экв.) и 3-ацетаминофенилбороновую кислоту (454,61 мг, 2,54 ммоль, 2,00 экв.) растворяли в диоксане (20,00 мл), затем в реакционный раствор добавляли Pd(dppf)Cl2.CH2Cl2 (103,71 мг, 127,00 мкмоль, 0,10 экв.) и в реакционный раствор добавляли раствор карбоната натрия (269,21 мг, 2,54 ммоль, 2,00 экв.) в воде (5,00 мл). Температуру повышали до 100°C и реакционную смесь перемешивали в течение 15 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры и фильтровали. Фильтрат собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (EA/PE=10%-50%) с получением целевого соединения WX040-1. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,41(s, 1 H), 10,08(s, 1 H), 7,64-7,58(m, 2 H), 7,43(t, J=7,6 Гц, 1 H), 7,35-7,28(m, 1 H), 7,22-7,12(m, 3 H), 6,96(d, J=7,2 Гц, 1 H), 3,61(s, 3 H), 2,26(s, 3 H), 2,01(s, 3 H), 1,44(s, 3 H). MS m/z: 448,0 [M+H]+
[322] Стадия 2: синтез соединения WX040
[323] Соединение WX040-1 (100,00 мг, 223,48 мкмоль, 1,00 экв.) растворяли в N,N-диметилформамиде (2,00 мл), затем добавляли трифторуксусную кислоту (76,44 мг, 670,45 мкмоль, 49,64 мкл, 3,00 экв.), и в реакционный раствор добавляли N-йодсукцинимид (125,70 мг, 558,70 мкмоль, 2,50 экв.). Реакционную смесь перемешивали при 25°C в течение 15 часов. После завершения реакции реакционную смесь очищали с помощью ВЭЖХ с получением целевого соединения WX040. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,34(s, 1 H), 10,06(s, 1 H), 7,70(dd, J=10,2 Гц, J=1,8 Гц, 1 H), 7,60-7,54(m, 2 H), 7,51(d, J=8,0 Гц, 1 H), 7,40(t, J=8,0 Гц, 1 H), 6,93(d, J=7,6 Гц, 1 H), 6,87(t, J=8,0 Гц, 1 H), 3,58(s, 3 H), 2,24(s, 3 H), 2,04(s, 3 H), 1,44(s, 3 H). MS m/z: 574,1 [M+H]+
[324] Вариант реализации 2: WX049
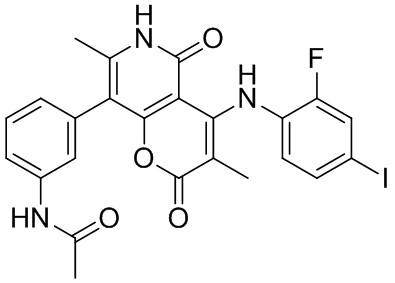
[325] Схема синтеза:
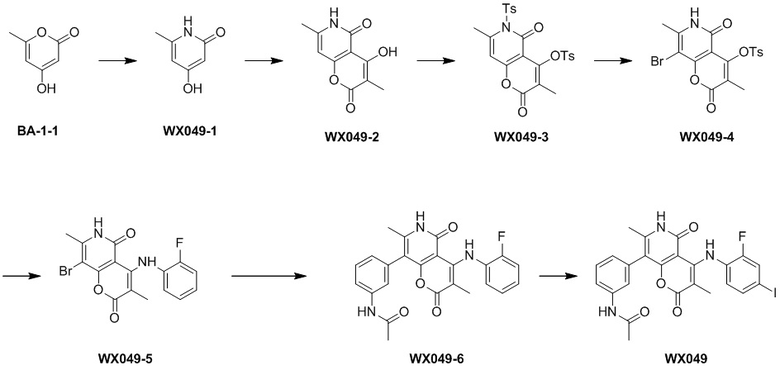
[326] Стадия 1: синтез соединения WX049-1
[327] Соединение BA-1-1 (10,00 г, 79,30 ммоль, 1,00 экв.) растворяли в гидроксиде аммония (91,00 г, 700,98 ммоль, 100,00 мл, 8,84 экв.). В защитной атмосфере азота смесь нагревали до 100°C и реакционную смесь перемешивали при нагревании с обратным холодильником в течение 6 часов. После завершения реакции смесь охлаждали до комнатной температуры и выпаривали досуха с помощью роторного испарителя с получением целевого соединения WX049-1. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,89(br. s., 1 H), 5,56(d, J=1,2 Гц, 1 H), 5,30(d, J=2,4 Гц, 1 H), 2,05(s, 3 H). MS m/z: 125,8 [M+H]+
[328] Стадия 2: синтез соединения WX049-2
[329] Соединение WX049-1 (9,50 г, 75,92 ммоль, 1,00 экв.) и метилмалоновую кислоту (13,22 г, 75,92 ммоль, 12,97 мл, 1,00 экв.) растворяли в дифениловом эфире (100,00 мл). Смесь нагревали до 250°C и реакционную смесь перемешивали при нагревании с обратным холодильником в течение 2 часов до окончания образования газообразного метанола. После завершения реакции добавляли воду (30 мл) и затем смесь экстрагировали EtOAc (30 мл*3). Органическую фазу собирали и выпаривали досуха с помощью роторного испарителя с получением целевого соединения WX049-2. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,26(s, 1 H), 12,75(br. s., 1 H), 6,45(s, 1 H), 2,31(s, 3 H), 1,82(s, 3 H). MS m/z: 207,8 [M+H]+
[330] Стадия 3: синтез соединения WX049-3
[331] Соединение WX049-2 (7,00 г, 33,79 ммоль, 1,00 экв.) растворяли в дихлорметане (200,00 мл), затем добавляли триэтиламин (10,26 г, 101,37 ммоль, 14,05 мл, 3,00 экв.) и диметиламинопиридин (206,39 мг, 1,69 ммоль, 0,05 экв.) с последующим добавлением п-толуолсульфонилхлорида (14,17 г, 74,34 ммоль, 2,20 экв.). Реакционную смесь перемешивали при 25°C в течение 12 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя, затем растирали с этанолом (100 мл) и фильтровали. Фильтрационный осадок растирали с хлороформом (100 мл) и фильтровали. Фильтрат собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта целевого соединения WX049-3. 1H ЯМР (400 МГц, CDCl3-d) δ 7,96(d, J=8,4 Гц, 2 H), 7,89(d, J=8,4 Гц, 2 H), 7,39(d, J=8,4 Гц, 2 H), 7,34(d, J=8,0 Гц, 2 H), 6,94(s, 1 H), 2,49(s, 3 H), 2,46(s, 3 H), 2,42(s, 3 H), 1,81(s, 3 H). MS m/z: 538,1 [M+Na]+
[332] Стадия 4: синтез соединения WX049-4
[333] Соединение WX049-3 (11,00 г, 21,34 ммоль, 1,00 экв.) растворяли в ТГФ (100,00 мл) и ацетонитриле (100. 00 мл), затем добавляли N-бромсукцинимид (7,59 г, 42,68 ммоль, 2,00 экв.). Смесь нагревали до 80°C и реакционную смесь перемешивали в течение 15 часов. Часть исходного вещества осталась. Добавляли дополнительное количество N-бромсукцинимиад (7,59 г, 42,68 ммоль, 2,00 экв.) и реакционную смесь перемешивали при 80°C в течение 5 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры, фильтровали и фильтрационный осадок собирали с получением целевого соединения WX049-4. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,33(s, 1 H), 7,88(d, J=8,8 Гц, 2 H), 7,46(d, J=8,0 Гц, 2 H), 2,42(s, 3 H), 2,37(s, 3 H), 1,50(s, 3 H). MS m/z: 442,0 [M+H]+
[334] Стадия 5: синтез соединения WX049-5
[335] Соединение WX049-4 (7,50 г, 17,04 ммоль, 1,00 экв.) и 2-фторанилин (18,93 г, 170,40 ммоль, 16,46 мл, 10,00 экв.) растворяли в этаноле (100,00 мл). Реакционную смесь перемешивали при нагревании с обратным холодильником в течение 15 часов. После завершения реакции реакционный раствор охлаждали до комнатной температуры, фильтровали и фильтрационный осадок собирали с получением целевого соединения WX049-5. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,84(s, 1 H), 11,17(s, 1 H), 7,32-7,17(m, 4 H), 2,45(s, 3 H), 1,46(s, 3 H). MS m/z: 442,0 [M+H]+
[336] Стадия 6: синтез соединения WX049-6
[337] Соединение WX049-5 (500,00 мг, 1,32 ммоль, 1,00 экв.) и 3-ацетаминофенилбороновую кислоту (472,02 мг, 2,64 ммоль, 2,00 экв.) растворяли в диоксане (10,00 мл) и воде (5,00 мл). В защитной атмосфере азота добавляли SPhos (54,13 мг, 131,86 мкмоль, 0,10 экв.), Pd2(dba)3 (37,91 мг, 65,93 мкмоль, 0,05 экв.) и фосфат калия (699,77 мг, 3,30 ммоль, 2,50 экв.). Температуру повышали до 110°C и реакционную смесь перемешивали в течение 15 часов. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали дихлорметаном (20мл*3). Органическую фазу собирали, выпаривали досуха с помощью роторного испарителя, затем очищали с помощью колоночной хроматографии (PE/EA=5/1-0/1) с получением целевого соединения WX049-6. MS m/z: 434,1 [M+H]+
[338] Стадия 7: синтез соединения WX049
[339] Соединение WX049-6 (250,00 мг, 576,79 мкмоль, 1,00 экв.) растворяли в диметилсульфоксиде (2,00 мл), затем добавляли трифторуксусную кислоту (197,30 мг, 1,73 ммоль, 128,12 мкл, 3,00 экв.) и N-йодсукцинимид (389,30 мг, 1,73 ммоль, 3,00 экв.). Смесь нагревали до 25°C и реакционную смесь перемешивали в течение 15 часов. После завершения реакции реакционный раствор разбавляли водой (20 мл) и экстрагировали EtOAc (20 мл*2). Органическую фазу собирали, сушили над безводным сульфатом натрия, выпаривали досуха с помощью роторного испарителя и очищали с помощью препаративной ВЭЖХ с получением соединения WX049. 1H ЯМР (400 МГц, ДМСО-d6) δ 12,56(s, 1 H), 11,28(s, 1 H), 10,08(s, 1 H), 7,73(dd, J=10,0 Гц, J=1,6 Гц, 1 H), 7,72-7,53(m, 3 H), 7,40(t, J=8,8 Гц, 1 H), 6,99(d, J=7,2 Гц, 1 H), 6,92(t, J=8,8 Гц, 1 H), 2,11(s, 3 H), 2,06(s, 3 H), 1,45(s, 3 H). MS m/z: 560,0 [M+H]+
[340] Вариант реализации 3: WX053
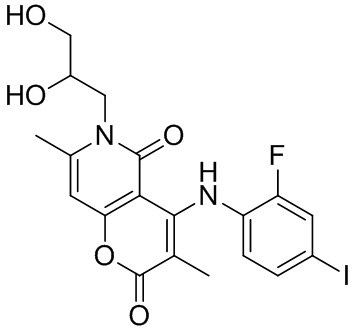
[341] Схема синтеза:
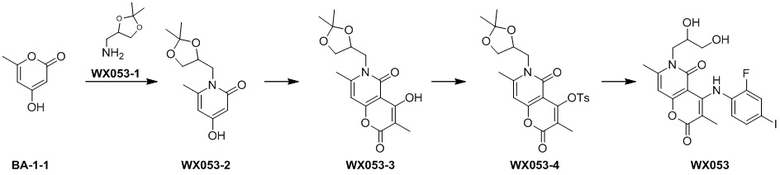
[342] Стадия 1: синтез соединения WX053-2
[343] Соединение BA-1-1 (2,50 г, 19,82 ммоль, 1,00 экв.) растворяли в воде (30,00 мл), затем добавляли соединение WX053-1 (2,86 г, 2,83 мл, 1,10 экв.) при 25°C. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 2 часов, и образовался осадок. После завершения реакции реакционную смесь фильтровали. Фильтрационный осадок собирали, промывали водой (20 мл), сушили при 45°C пока масса не стала постоянной, с получением целевого соединения WX053-2.
[344] 1H ЯМР (400 МГц, CDCl3-d) δ 5,96(d, J=2,4 Гц, 1 H), 5,90(d, J=2,0 Гц, 1 H), 4,44(m, 1 H), 4,34(dd, J=14,0, 2,8 Гц, 1 H), 4,16(dd, J=8,8, 6,4 Гц, 1 H), 3,91(dd, J=14,0, 7,2 Гц, 1 H), 3,69(dd, J=8,4, 7,2 Гц, 1 H), 2,45(s, 3 H), 1,40(s, 3 H), 1,31(s, 3 H). MS m/z: 239,9 [M+H]+
[345] Стадия 2: синтез соединения WX053-3
[346] Соединение WX053-2 (200 мг, 835,88 мкмоль, 1,00 экв.) и диэтилметилмалонат (218 мг, 1,25 ммоль, 214,12 мкл, 1,50 экв.) смешивали в дифениловом эфире (5,00 мл). Температуру повышали до 250°C и реакционную смесь перемешивали при нагревании с обратным холодильником в течение 2 часов до окончания испарения этанола. После завершения реакции реакционный раствор охлаждали и добавляли метил-трет-бутиловый эфир (10 мл) и петролейный эфир (10 мл), затем образовывалось твердое вещество. После завершения реакции смесь фильтровали. Фильтрационный осадок промывали метил-трет-бутиловым эфиром (2 мл) и петролейным эфиром (2 мл) и выпаривали досуха с помощью роторного испарителя с получением целевого соединения WX053-3. 1H ЯМР (400 МГц, CDCl3-d) δ 6,28(s, 1 H), 4,46-4,54(m, 1 H), 4,42(dd, J=14,0, 2,8 Гц, 1 H), 4,21(dd, J=8,8, 6,8 Гц, 1 H), 4,00(dd, J=14,0, 7,6 Гц, 1 H), 3,74(dd, J=8,8, 6,8 Гц, 1 H), 2,60(s, 3 H), 2,00(s, 3 H), 1,42(s, 3 H), 1,32(s, 3 H). MS m/z: 322,1 [M+H]+
[347] Стадия 3: синтез соединения WX053-4
[348] Соединение WX053-3 (1,80 г, 5,60 ммоль, 1,00 экв.) растворяли в дихлорметане (20 мл), затем добавляли триэтиламин (1,70 г, 16,80 ммоль, 2,33 мл, 3,00 экв.), DMAP (34,22 мг, 280 мкмоль, 0,05 экв.) и п-толуолсульфонилхлорид (1,60 г, 8,40 ммоль, 1,50 экв.). Реакционную смесь перемешивали при 25°C в течение 12 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (PE/EA=5/1-1/1) с получением целевого соединения WX053-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,91(d, J=8,4 Гц, 2 H), 7,39(d, J=8,0 Гц, 2 H), 6,13(s, 1 H), 4,42-4,53(m, 2 H), 4,19(dd, J=8,8, 6,4 Гц, 1 H), 3,90-3,97(m, 1 H), 3,71(dd, J=8,8, 7,2 Гц, 1 H), 2,56(s, 3 H), 2,48(s, 3 H), 1,54(s, 3 H), 1,41(s, 3 H), 1,31(s, 3 H). MS m/z: 476,2 [M+H]+
[349] Стадия 4: синтез соединения WX053
[350] Соединение WX053-4 (200 мг, 420,60 мкмоль, 1,00 экв.) растворяли в этаноле (4,00 мл), затем добавляли 2-фтор-4-йоданилин (299 мг, 1,26 ммоль, 3,00 экв.) при 25°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 80°C и реакционную смесь перемешивали в течение 36 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением соединения WX053. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,21(s, 1 H), 7,73(d, J=10,0 Гц, 1 H), 7,54(d, J=8,4 Гц, 1 H), 6,90(t, J=8,4 Гц, 1 H), 6,51(s, 1 H), 5,05(d, J=4,8 Гц, 1 H), 4,78(t, J=5,6 Гц, 1 H), 4,27(d, J=11,2 Гц, 1 H), 3,79-3,92(m, 2 H), 3,39-3,49(m, 2 H), 2,56(s, 3 H), 1,49(s, 3 H). MS m/z: 501,1 [M+H]+
[351] Вариант реализации 4: WX054
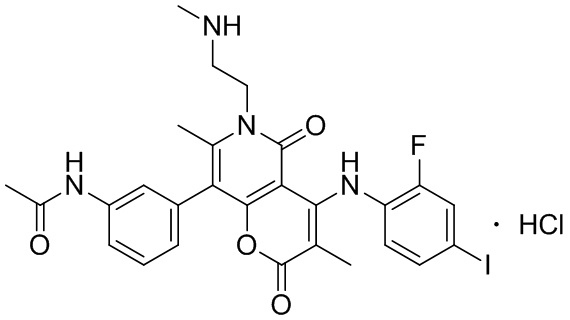
[352] Схема синтеза:
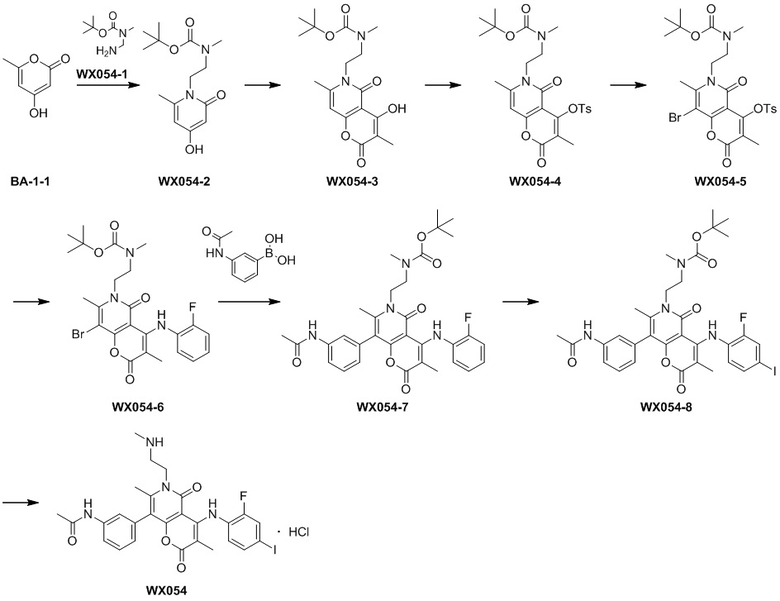
[353] Стадия 1: синтез соединения WX054-2
[354] Соединение BA-1-1 (6,70 г, 53,13 ммоль, 1,00 экв.) растворяли в воде (250,00 мл), затем добавляли соединение WX054-1 (9,26 г, 53,13 ммоль, 9,45 мл, 1,00 экв.) при комнатной температуре. Реакционный раствор нагревали с обратным холодильником и реакционную смесь перемешивали в течение 1 часа, и образовывалось твердое вещество. После завершения реакции смесь фильтровали и фильтрационный осадок собирали с получением целевого соединения WX054-2. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,34(s, 1 H), 5,75(d, J=2,0 Гц, 1 H), 5,48(d, J=2,4 Гц, 1 H), 3,96(s, 2 H), 3,38(s, 2 H), 2,76(s, 3 H), 2,27(s, 3 H), 1,31(s, 9 H). MS m/z: 282,9 [M+H]+
[355] Стадия 2: синтез соединения WX054-3
[356] Соединение WX054-2 (4,60 г, 16,29 ммоль, 1,00 экв.) и метилмалоновую кислоту (2,89 г, 24,44 ммоль, 1,50 экв.) добавляли в уксусный ангидрид (50,00 мл). Температуру повышали до 100°C и реакционную смесь перемешивали в течение 1 часа. После завершения реакции реакционный раствор охлаждали до комнатной температуры, добавляли метил-трет-бутиловыц эфир (200 мл). Затем смесь оставляли отстаиваться в течение 15 часов, образовался осадок и его фильтровали. Фильтрационный осадок собирали с получением целевого соединения WX054-3. 1H ЯМР (400 МГц, ДМСО-d6) δ 13,24(s, 1 H), 6,66(s, 1 H), 4,20(s, 2 H), 3,56-3,50(m, 2 H), 2,80(s, 3 H), 1,82(s, 3 H), 1,26(s, 3 H), 1,16(s, 9 H). MS m/z: 365,0 [M+H]+
[357] Стадия 3: синтез соединения WX054-4
[358] Соединение WX054-3 (4,40g, 12,07 ммоль, 1,00 экв.) растворяли в дихлорметане (100,00 мл), затем добавляли триэтиламин (2,44 г, 24,14 ммоль, 3,35 мл, 2,00 экв.) и DMAP (294,92 мг, 2,41 ммоль, 0,20 экв.) с последующим добавлением п-толуолсульфонилхлорида (3,45 г, 18,11 ммоль, 1,50 экв.) при 0°C. Реакционный раствор нагревали до 20°C и перемешивали в течение 15 часов. После завершения реакции реакционный раствор промывали водой (200 мл) и жидкость отделяли. Органическую фазу сушили над безводным сульфатом натрия и затем выпаривали досуха с помощью роторного испарителя. Твердое вещество растирали с метил-трет-бутиловым эфиром (200 мл) и фильтровали, фильтрационный осадок собирали с получением целевого соединения WX054-4. MS m/z: 541,1 [M+Na]+
[359] Стадия 4: синтез соединения WX054-5
[360] Соединение WX054-4 (2,70 г, 5,21 ммоль, 1,00 экв.) добавляли в смешанный растворитель дихлорметана (20,00мл) и ацетонитрила (40,00мл), затем добавляли N-бромсукцинимид (1,39 г, 7,82 ммоль, 1,50 экв.). Реакционный раствор перемешивали при 20°C в течение 15 часов. После завершения реакции реакционный раствор непосредственно выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который подвергали колоночной хроматографии (PE/EA=3/1, Rf=0,3) с получением WX054-5.
[361] MS m/z: 621,1 [M+Na]+
[362] Стадия 5: синтез соединения WX054-6
[363] Соединение WX054-5 (2,70 г, 4,52 ммоль, 1,00 экв.) растворяли в этаноле (50,00мл), затем добавляли 2-фторанилин (3,01 г, 27,12 ммоль, 2,62 мл, 6,00 экв.). Реакционный раствор нагревали с обратным холодильником и перемешивали в течение 15 часов, затем образовывалось твердое вещество. После завершения реакции смесь фильтровали и фильтрационный осадок собирали с получением целевого соединения WX054-6. MS m/z: 536,1 [M+H]+
[364] Стадия 6: синтез соединения WX054-7
[365] Соединение WX054-6 (700,00 мг, 1,31 ммоль, 1,00 экв.) и 3-ацетаминофенилбороновую кислоту (422,03 мг, 2,36 ммоль, 1,80 экв.) добавляли в диоксан (20,00 мл) и воду (5,00 мл), затем добавляли Pd(dppf)Cl2.CH2Cl2 (106,98 мг, 131,00 мкмоль, 0,10 экв.) и бикарбонат натрия (550,27 мг, 6,55 ммоль, 5,00 экв.). В защитной атмосфере азота реакционную смесь нагревали до 110oC и перемешивали в течение 1,5 часов. После завершения реакции реакционную смесь разбавляли водой (20 мл) и экстрагировали EtOAc (10 мл*3). Органические фазы объединяли, сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (PE/EA=2/1-0/1, Rf=0,31) с получением целевого соединения WX054-7. MS m/z: 591,2 [M+H]+
[366] Стадия 7: синтез соединения WX054-8
[367] Соединение WX054-7 (500,00 мг, 846,54 мкмоль, 1,00 экв.) растворяли в диметилсульфоксиде (10,00 мл), добавляли трифторуксусную кислоту (289,57 мг, 2,54ммоль, 188,03 мкл, 3,00 экв.) с последующим добавлением в темноте N-йодсукцинимида (571,36 мг, 2,54 ммоль, 3,00 экв.) при 15°C. Реакционную смесь перемешивали в течение 15 часов в темном месте. После завершения реакции реакционный раствор разбавляли водой (30 мл) и экстрагировали EtOAc (10 мл*3). Органические фазы объединяли, сушили над безводным сульфатом натрия и фильтровали. Фильтрат выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ (препаративное разделение с получением целевого соединения WX054-8. 1H ЯМР (400 МГц, CDCl3-d) δ 11,23(d, J=8,4 Гц, 1 H), 7,89(s, 1 H), 7,68(s, 1 H), 7,49-7,43(m, 2 H), 7,35-7,25(m, 1 H), 6,89(d, J=7,2 Гц, 1 H), 6,74(t, J=7,2 Гц, 1 H), 4,25(s, 1 H), 2,93(s, 2 H), 2,36(s, 2 H), 2,09(s, 3 H), 1,62(s, 3 H), 1,57(s, 9 H), 1,42(s, 3 H), 1,40(s, 3 H). MS m/z: 717,1 [M+H]+
[368] Стадия 8: синтез соединения WX054
[369] Соединение WX054-8 (160,00 мг, 223,30 мкмоль, 1,00 экв.) растворяли в дихлорметане (5,00 мл), добавляли раствор хлороводорода в EtOAc (4M, 10,00 мл, 179,13 экв.). Реакционную смесь перемешивали 15°C в течение 1 часа. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением целевого соединения WX054. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,18(s, 1 H), 10,19(s, 1 H), 8,64(br. s., 1 H), 7,73(d, J=10,8 Гц, 1 H), 7,68(s, 1 H), 7,56(t, J=9,2 Гц, 2 H), 7,44(t, J=8,0 Гц, 1 H), 6,96(d, J=7,6 Гц, 1 H), 6,90(t, J=8,0 Гц, 1 H), 4,42(br. s., 2 H), 3,24(br. s., 2 H), 2,55(s, 3 H), 2,32(s, 3 H), 2,07(s, 3 H), 1,48(s, 3 H). MS m/z: 617,1 [M+H-HCl]+
[370] Вариант реализации 5: WX055
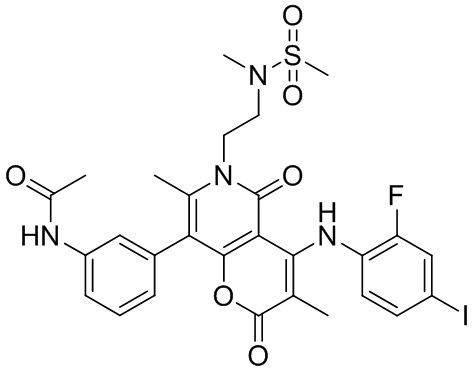
[371] Схема синтеза:
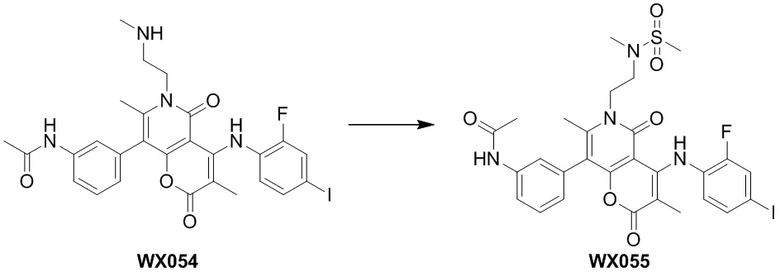
[372] Стадия 1: синтез соединения WX055
[373] Соединение WX054 (30,00 мг, 45,95 мкмоль, 1,00 экв.) растворяли в дихлорметане (5,00 мл), добавляли триэтиламин (18,60 мг, 183,80 мкмоль, 25,48 мкл, 4,00 экв.) с последующим добавлением метансульфонилхлорида (800,00 мг, 6,98 ммоль, 540,54 мкл, 151,99 экв.) при 15°C. Реакционную смесь перемешивали при 15°C в течение 1 часа. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя и очищали с помощью препаративной ТСХ (EA, Rf = 0,3) с получением целевого соединения WX055. 1H ЯМР (400 МГц, CDCl3-d) δ 11,16(s, 1 H), 8,22(s, 1 H), 7,78(br. s., 1 H), 7,50-7,43(m, 2 H), 7,27(d, J=4,8 Гц, 1 H), 7,15(d, J=8,0 Гц, 1 H), 6,87(d, J=8,0 Гц, 1 H), 6,75(t, J=8,8 Гц, 1 H), 4,36(t, J=6,4 Гц, 2 H), 3,47(t, J=6,8 Гц, 2 H), 3,00(s, 3 H), 2,84(s, 3 H), 2,34(s, 3 H), 2,05(s, 3 H), 1,62(s, 3 H). MS m/z: 694,9 [M+H]+
[374] Вариант реализации 6: WX056
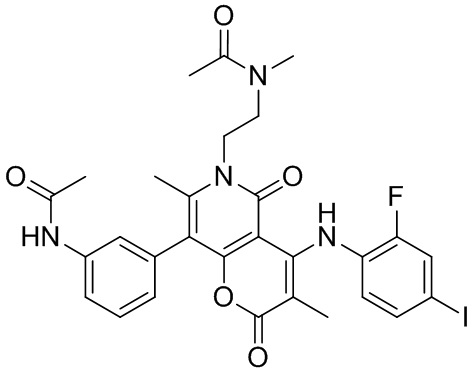
[375] Схема синтеза:
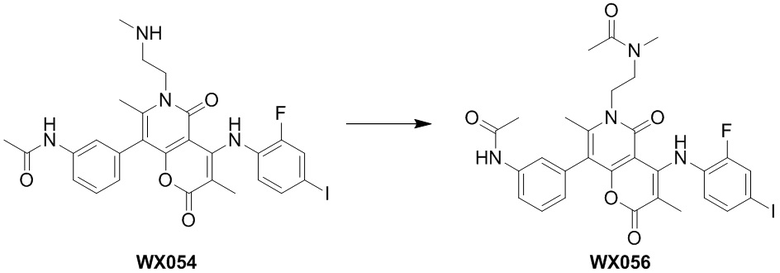
[376] Стадия 1: синтез соединения WX056
[377] Соединение WX054 (30,00 мг, 45,95 мкмоль, 1,00 экв.) растворяли в дихлорметане (5,00 мл), добавляли триэтиламин (18,60 мг, 183,80 мкмоль, 25,48 мкл, 4,00 экв.) с последующим добавлением ацетилхлорида (10,82 мг, 137,85 мкмоль, 9,84 мкл, 3,00 экв.). Реакционную смесь перемешивали при 15°C в течение 1 часа. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя и очищали с помощью препаративной ТСХ (EA, Rf=0,2) с получением целевого соединения WX056. 1H ЯМР (400 МГц, CDCl3-d) δ 11,24(s, 1 H), 8,29(s, 1 H), 7,79(br. s., 1 H), 7,50-7,43(m, 2 H), 7,26(d, J=4,8 Гц, 1 H), 7,14(d, J=8,0 Гц, 1 H), 6,86(d, J=6,8 Гц, 1 H), 6,76(t, J=8,0 Гц, 1 H), 4,28(d, J=7,2 Гц, 2 H), 3,64(t, J=7,2 Гц, 2 H), 3,14(s, 3 H), 2,39(s, 3 H), 2,09(s, 3 H), 2,04(s, 3 H), 1,63(s, 3 H). MS m/z: 659,0 [M+H]+
[378] Вариант реализации 7: WX057
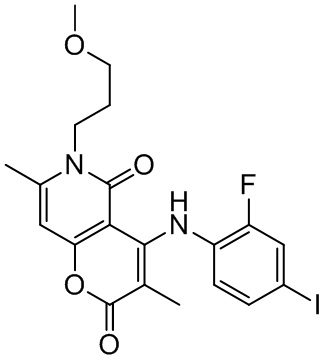
[379] Схема синтеза:
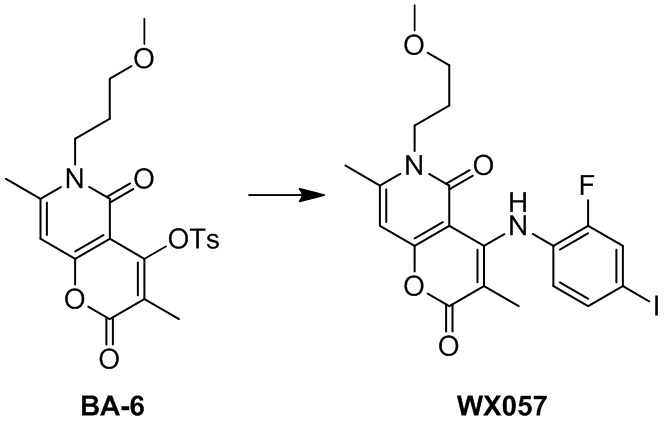
[380] Стадия 1: синтез соединения WX057
[381] Соединение BA-6 (2,00 г, 2,86 ммоль, 1,00 экв.) и 2-фтор-4-йоданилин (3,39 г, 14,30 ммоль, 5,00 экв.) растворяли в этаноле (20,00 мл), смесь нагревали с обратным холодильником и перемешивали в течение 15 часов. Реакционную смесь выпаривали досуха с помощью роторного испарителя и остаток растирали с метил-трет-бутиловым эфиром (200 мл*2). Смесь фильтровали, затем фильтрационный осадок собирали и дважды очищали с помощью колоночной хроматографии (PE/EA=1/1, Rf=0,25). Полученный неочищенный продукт дополнительно очищали с помощью препаративной ВЭЖХ с получением соединения WX057. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,16(s, 1 H), 7,72(dd, J=10,0 Гц, J=2,0 Гц, 1 H), 7,53(d, J=8,0 Гц, 1 H), 6,89(t, J=8,6 Гц, 1 H), 6,53(s, 1 H), 4,07(t, J=7,6 Гц, 2 H), 3,39(t, J=5,8 Гц, 2 H), 3,24(s, 3 H), 2,53(s, 3 H), 1,91-1,83(m, 2 H), 1,47(s, 3 H). MS m/z: 498,9 [M+H]+
[382] Вариант реализации 8: WX058
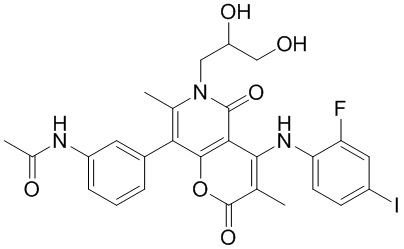
[383] Схема синтеза:
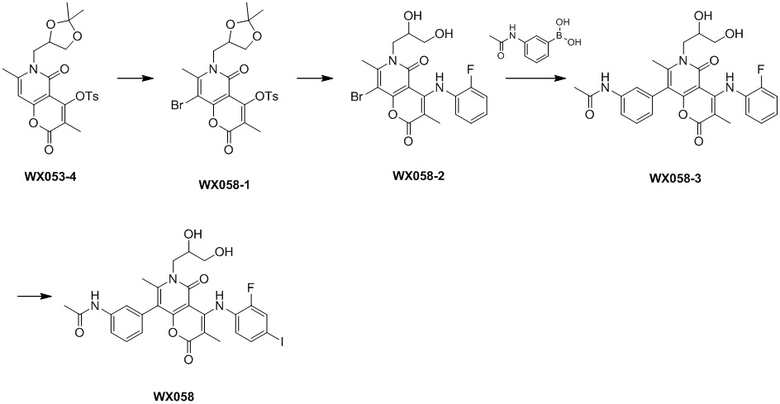
[384] Стадия 1: синтез соединения WX058-1
[385] Соединение WX053-4 (1 г, 2,1 ммоль, 1,00 экв.) добавляли в дихлорметан (10 мл) и ацетонитрил (20 мл), затем добавляли бикарбонат натрия (176,42 мг, 2,1 ммоль, 1,00 экв.) и N-бромсукцинимид (747,52 мг, 4,2 ммоль, 2,00 экв.) при 25°C в защитной атмосфере азота. Реакционную смесь перемешивали при 25°C в течение 12 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=1/1, Rf=0,6) с получением целевого соединения WX058-1. 1H ЯМР (400 МГц, CDCl3-d) δ 7,88(d, J=8,4 Гц, 2 H), 7,38(d, J=8,0 Гц, 2 H), 4,58(dd, J=14,2 Гц, J=2,6 Гц, 1 H), 4,50-4,48(m, 1 H), 4,19(dd, J=8,8 Гц, J=6,4 Гц, 1 H), 4,05(dd, J=14,4 Гц, J=7,6 Гц, 1 H), 3,71(dd, J=8,8 Гц, J=6,8 Гц, 1 H), 2,96(s, 3 H), 2,48(s, 3 H), 1,53(s, 3 H), 1,41(s, 3 H), 1,30(s, 3 H). MS m/z: 555,8 [M+H]+
[386] Стадия 2: синтез соединения WX058-2
[387] Соединение WX058-1 (600 мг, 1,08 ммоль, 1,00 экв.) растворяли в этаноле (6,00 мл), затем добавляли 2-фторанилин (360,03 мг, 3,24 ммоль, 313,07 мкл, 3,00 экв.) при 20°C. Температуру реакционной смеси повышали до 80°C и реакционную смесь перемешивали в течение 24 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (10 мл) и затем фильтровали с получением целевого соединения WX058-2. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,26(s, 1 H), 7,37-7,28(m, 1 H), 7,23-7,12(m, 3 H), 5,11(d, J=4,8 Гц, 1 H), 4,81(t, J=5,8 Гц, 1 H), 4,40(dd, J=14,0 Гц, J=2,8 Гц, 1 H), 4,05-3,96(m, 1 H), 3,93-3,84(m, 1 H), 3,50-3,38(m, 2 H), 2,81(s, 3 H), 1,49(s, 3 H). MS m/z: 477,2 [M+Na]+
[388] Стадия 3: синтез соединения WX058-3
[389] Соединение WX058-2 (250 мг, 551,56 мкмоль, 1,00 экв.) и 3-ацетаминофенилбороновую кислоту (148,08 мг, 827,34 мкмоль, 1,50 экв.) добавляли в диоксан (10,00 мл), затем добавляли насыщенный раствор бикарбоната натрия (2,00 мл, 1,00 экв.) и Pd(dppf)Cl2.CH2Cl2 (45,04 мг, 55,16 мкмоль, 0,10 экв.) при 20°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 80-90°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью препаративной ВЭЖХ (система трифторуксусной кислоты) с получением целевого соединения WX058-3. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,43(s, 1 H), 10,09(d, J=3,6 Гц, 1 H), 7,62-7,56(m, 2 H), 7,45-7,39(m, 1 H), 7,36-7,27(m, 1 H), 7,22-7,12(m, 3 H), 6,94(t, J=8,4 Гц, 1 H), 5,12(d, J=9,2 Гц, 1 H), 4,80(t, J=5,4 Гц, 1 H), 4,44-4,34(m, 1 H), 3,95(br. s., 2 H), 3,45(br. s., 2 H), 2,34(s, 3 H), 2,06(s, 3 H)1,45(s, 3 H). MS m/z: 508,2 [M+H]+
[390] Стадия 4: синтез соединения WX058
[391] При 0°C в защитной атмосфере азота и в темном месте соединение WX058-3 (80 мг, 157,63 мкмоль, 1,00 экв.) и N-йодбромсукцинимид (70,93 мг, 315,26 мкмоль, 2,00 экв.) растворяли в N,N-диметилформамиде (2,00 мл) с последующим добавлением трифторуксусной кислоты (35,95 мг, 315,26 мкмоль, 23,34 мкл, 2,00 экв.). Реакционную смесь перемешивали при 20°C в течение 12 часов. После завершения реакции реакционную смесь очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX058. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,39(s, 1 H), 10,09(d, J=4,0 Гц, 1 H), 7,73(dd, J=10,0 Гц, J=1,6 Гц, 1 H), 7,62-7,52(m, 3 H), 7,42(t, J=8,0 Гц, 1 H), 6,97-6,88(m, 2 H), 5,11(d, J=10,0 Гц, 1 H), 4,80(br. s., 1 H), 4,42-4,34(m, 1 H), 3,95(br. s., 2 H), 3,45(br. s., 2 H), 2,33(s, 3 H), 2,06(s, 3 H), 1,47(s, 3 H). MS m/z: 633,9 [M+H]+
[392] Вариант реализации 9: WX059
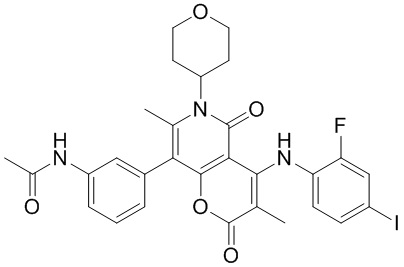
[393] Схема синтеза:
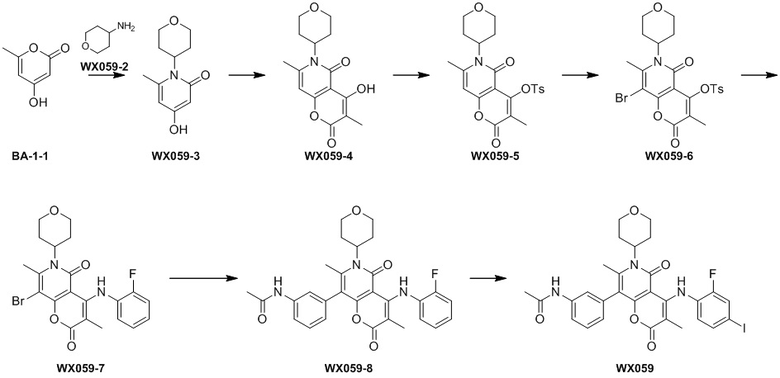
[394] Стадия 1: синтез соединения WX059-3
[395] Соединение BA-1-1 (4 г, 31,72 ммоль, 1,00 экв.) растворяли в H2O (1,60 л), затем соединение WX059-2 (3,37 г, 33,31 ммоль, 1,05 экв.) добавляли по каплям при 25°C. В условиях защитной атмосферы азота температуру реакционной смеси повышали до 120°C и реакционную смесь перемешивали в течение 36 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя, неочищенный продукт очищали с помощью препаративной ВЭЖХ (система трифторуксусной кислоты) с получением целевого соединения WX059-3. 1H ЯМР (400 МГц, ДМСО-d6) δ 10,41(br. s., 1 H), 5,76(br. s., 1 H), 5,46(br. s., 1 H), 4,16(br. s., 1 H), 3,90(dd, J=11,4 Гц, J=4,6 Гц, 2 H), 3,36(t, J=11,0 Гц, 2 H), 2,89(br. s., 2 H), 2,35(s, 3 H), 1,47(d, J=12,0 Гц, 2 H). MS m/z: 210,0 [M+H]+
[396] Стадия 2: синтез соединения WX059-4
[397] Соединение WX059-3 (700 мг, 3,35 ммоль, 1,00 экв.) растворяли в дифениловом эфире, добавляли соединение диэтилметилмалонат (700,24 мг, 4,02 ммоль, 686,51 мкл, 1,20 экв.). Температуру реакционной смеси повышали до 250°C и реакционную смесь перемешивали в течение 2 часов. После завершения реакции добавляли петролейный эфир (20 мл), затем твердое вещество выпадало в осадок. Твердое вещество фильтровали и фильтрационный осадок промывали петролейным эфиром (5 мл) с получением целевого соединения WX059-4. 1H ЯМР (400 МГц, CDCl3-d) δ 12,98(s, 1 H), 6,23(s, 1 H), 4,28(br. s., 1 H), 4,15(dd, J=11,4 Гц, J=4,6 Гц, 2 H), 3,46(t, J=11,8 Гц, 2 H), 3,17(br. s., 2 H), 2,52(s, 3 H), 1,99(s, 3 H), 1,64-1,62(m, 2 H). MS m/z: 292,1 [M+H]+
[398] Стадия 3: синтез соединения WX059-5
[399] Соединение WX059-4 (1,1 г, 3,78 ммоль, 1,00 экв.) растворяли в дихлорметане (20 мл), затем добавляли триэтиламин (1,15 г, 11,34 ммоль, 1,57 мл, 3,00 экв.), DMAP (46,18 мг, 378 мкмоль, 0,10 экв.) и п-толуолсульфонилхлорид (1,08 г, 5,67 ммоль, 1,50 экв.) при 0°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 20°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=1/1) с получением целевого соединения WX059-5. 1H ЯМР (400 МГц, CDCl3-d) δ 7,94(d, J=8,4 Гц, 2 H), 7,40(d, J=8,0 Гц, 2 H), 6,05(s, 1 H), 4,46-4,19(m, 1 H), 4,12(dd, J=11,4 Гц, J=4,2 Гц, 2 H), 3,44(t, J=11,2 Гц, 2 H), 3,07(s, 2 H), 2,47(s, 3 H), 1,73(s, 3 H), 1,61(s, 3 H), 1,55(d, J=12,4 Гц, 2 H). MS m/z: 446,1 [M+H]+
[400] Стадия 4: синтез соединения WX059-6
[401] Соединение WX059-5 (950 мг, 2,13 ммоль, 1,00 экв.) растворяли в ацетонитриле (10 мл) и ТГФ (10 мл), с последующим добавлением N-бромсукцинимида (682,38 мг, 3,83 ммоль, 1,80 экв.) при 0°C. В условиях защитной атмосферы азота температуру реакционной смеси повышали до 25°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (20 мл) и этанолом (5 мл) и фильтровали. Фильтрационный осадок собирали с получением целевого соединения WX059-6. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,87(d, J=8,4 Гц, 2 H), 7,49(d, J=8,0 Гц, 2 H), 4,41(br. s., 1 H), 3,93(dd, J=10,8 Гц, J=4,4 Гц, 2 H), 3,48-3,40(m, 2 H), 2,77(s, 3 H), 2,73-2,66(m, 2 H), 2,44(s, 3 H), 1,71(d, J=11,6 Гц, 1 H), 1,52(br. s., 1 H), 1,49(s, 3 H). MS m/z: 524,0 [M+H]+
[402] Стадия 5: синтез соединения WX059-7
[403] Соединение WX059-6 (650 мг, 1,24 ммоль, 1,00 экв.) растворяли в этаноле (10 мл), затем добавляли 2-фторанилин (688,94 мг, 6,20 ммоль, 599,08 мкл, 5,00 экв.) при 20°C в защитной атмосфере азота. Реакционную смесь нагревали до 80°C и реакционную смесь перемешивали в течение 15 часов. После завершения реакции реакционную смесь фильтровали. Фильтрационный осадок собирали и растирали с метил-трет-бутиловым эфиром (10 мл). Твердое вещество собирали фильтрованием с получением целевого соединения WX059-7. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,21(s, 1 H), 7,36-7,29(m, 1 H), 7,22-7,12(m, 3 H), 4,61(br. s., 1 H), 3,98-3,90(m, 3 H), 3,48-3,39(m, 3 H), 2,83(s, 3 H), 1,68(d, J=10,0 Гц, 2 H), 1,47(s, 3 H). MS m/z: 465,0 [M+H]+
[404] Стадия 6: синтез соединения WX059-8
[405] Соединение WX059-7 (260 мг, 561,19 мкмоль, 1,00 экв.) и соединение 3-ацетаминофенилбороновую кислоту (150,66 мг, 841,79 мкмоль, 1,50 экв.) растворяли в диоксане (10 мл), затем добавляли Pd(dppf)Cl2.CH2Cl2 (45,83 мг, 56,12 мкмоль, 0,10 экв.) и насыщенный раствор бикарбоната натрия (2 мл) при 20°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 80-90°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя. Дихлорметан (20 мл) и воду (10 мл) добавляли для растворения остатка, и жидкость разделяли. Органическую фазу собирали, промывали насыщенным раствором хлорида натрия, сушили над безводным сульфатом натрия, затем выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (дихлорметан/метанол =20/1) с получением целевого соединения WX059-8. 1H ЯМР (400 МГц, CDCl3-d) δ 11,48(s, 1 H), 8,86(s, 1 H), 7,92(s, 1 H), 7,24-7,10(m, 5 H), 6,97(d, J=8,0 Гц, 1 H), 6,82(d, J=7,6 Гц, 1 H), 4,51-4,30(m, 1 H), 4,15(d, J=10,0 Гц, 2 H), 3,46(t, J=11,6 Гц, 2 H), 3,31-3,09(m, 2 H), 2,29(s, 3 H), 2,03-1,96(m, 4 H), 1,64(br. s., 1 H), 1,60(s, 3 H). MS m/z: 518,3 [M+H]+
[406] Стадия 7: синтез соединения WX059
[407] Соединение WX059-8 (200 мг, 386,44 мкмоль, 1,00 экв.) растворяли в N,N-диметилформамиде (3 мл), затем добавляли трифторуксусную кислоту (88,12 мг, 772,88 мкмоль, 57,22 мкл, 2,00 экв.) с последующим добавлением N-йодсукцинимида (173,88 мг, 772,88 мкмоль, 2,00 экв.) при 0°C в защитной атмосфере азота и в темном месте. Температуру реакционной смеси повышали до 20°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционную смесь разбавляли водой (10мл) и экстрагировали дихлорметаном (10 мл*2). Органическую фазу промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX059. 1H ЯМР (400 МГц, CDCl3-d) δ 11,44(br. s., 1 H), 7,77(br. s., 1 H), 7,51-7,42(m, 2 H), 7,36-7,29(m, 1 H), 7,19(br. s., 1 H), 6,92(d, J=7,6 Гц, 1 H), 6,80(br. s., 1 H), 4,15(d, J=11,6 Гц, 2 H), 3,46(t, J=11,6 Гц, 2 H), 3,20(br. s., 1 H), 2,33(s, 3 H), 2,06(s, 3 H), 1,71-1,67(m, 4 H), 1,60(s, 3 H). MS m/z: 644,2 [M+H]+
[408] Вариант реализации 10: WX060
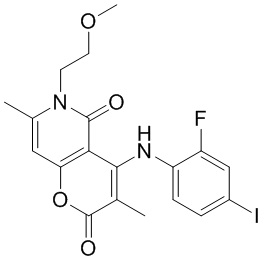
[409] Схема синтеза:
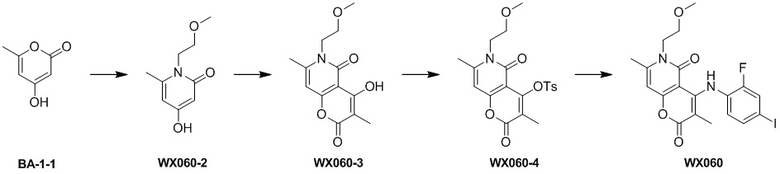
[410] Стадия 1: синтез соединения WX060-2
[411] Соединение BA-1-1 (15 г, 118,94 ммоль, 1,00 экв.) добавляли в воду (200 мл), с последующим добавлением метоксиэтиламина (9,83 г, 130,83 ммоль, 11,43 мл, 1,10 экв.) при 20°C. Реакционную смесь перемешивали при 100°C в течение 1,5 часов, и образовалось белое твердое вещество. После завершения реакции реакционную смесь фильтровали и фильтрационный осадок промывали водой (200 мл) с получением целевого соединения WX060-2. 1H ЯМР (400 МГц, CDCl3-d) δ 5,97(d, J=2,4 Гц, 1 H), 5,87(d, J=2,0 Гц, 1 H), 4,15(t, J=5,0 Гц, 1 H), 3,65(t, J=5,2 Гц, 1 H), 3,30(s, 1 H), 2,40(s, 1 H). MS m/z: 205,9 [M+Na]+
[412] Стадия 2: синтез соединения WX060-3
[413] Соединение WX060-2 (8 г, 43,67 ммоль, 1,00 экв.) добавляли в уксусный ангидрид (20 мл) с последующим добавлением метилмалоновой кислоты (7,74 г, 65,50 ммоль, 1,50 экв.) при 20°C. Реакционную смесь перемешивали при 20°C в течение 0,5 часа, и образовалось белое твердое вещество. После завершения реакции реакционную смесь фильтровали и фильтрационный осадок промывали метил-трет-бутиловым эфиром (100 мл) с получением целевого соединения WX060-3. 1H ЯМР (400 МГц, CDCl3-d) δ 12,78(s, 1 H), 6,25(s, 1 H), 4,26(t, J=5,0 Гц, 1 H), 3,70(t, J=5,0 Гц, 1 H), 3,31(s, 3 H), 2,54(s, 3 H), 2,00(s, 3 H). MS m/z: 265,9 [M+H]+
[414] Стадия 3: синтез соединения WX060-4
[415] Соединение WX060-3 (10,5 г, 39,58 ммоль, 1,00 экв.) добавляли в дихлорметан (200 мл), затем добавляли триэтиламин (10,01 г, 98,95 ммоль, 13,71 мл, 2,50 экв.), DMAP (483,60 мг, 3,96 ммоль, 0,10 экв.) и п-толуолсульфонилхлорид (11,32 г, 59,37 ммоль, 1,50 экв.) при 0°C в защитной атмосфере азота. Реакционную смесь перемешивали при 20°C в течение 12 часов. После завершения реакции реакционную смесь промывали водой (200 мл*2). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (200 мл) с получением целевого соединения WX060-4. 1H ЯМР (400 МГц, CDCl3-d) δ 7,91(d, J=8,4 Гц, 1 H), 7,38(d, J=8,0 Гц, 1 H), 6,09(s, 1 H), 4,22(t, J=4,8 Гц, 2 H), 3,68(t, J=4,8 Гц, 2 H), 3,28(s, 3 H), 2,49(d, J=9,2 Гц, 6 H), 1,55(s, 3 H). MS m/z: 442,2 [M+Na]+
[416] Стадия 4: синтез соединения WX060
[417] Соединение WX060-4 (1 г, 2,38 ммоль, 1,00 экв.) добавляли в этанол (10,00 мл) с последующим добавлением соединения 2-фтор-4-йоданилина (564,09 мг, 2,38 ммоль, 1,00 экв.) при 20°C. Реакционную смесь перемешивали при 80°C в течение 12 часов, и образовалось белое твердое вещество. После завершения реакции реакционную смесь фильтровали и промывали метил-трет-бутиловым эфиром (10 мл) с получением фильтрационного осадка. Фильтрационный осадок растирали с растворителем диметилсульфоксидом/ацетонитрилом =1/1 (10 мл), и затем фильтровали с получением целевого соединения WX060. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,10(s, 1 H), 7,73(dd, J=10,2 Гц, 2,0 Гц, 2 H), 7,54(d, J=9,2 Гц, 1 H), 6,90(t, J=8,8 Гц, 1 H), 6,52(s, 1 H), 4,22(t, J=5,2 Гц, 2 H), 3,61(t, J=5,2 Гц, 2 H), 3,24(s, 3 H), 2,54(s, 2 H), 1,48(s, 3 H). MS m/z: 485,0 [M+H]+
[418] Вариант реализации 11: WX061
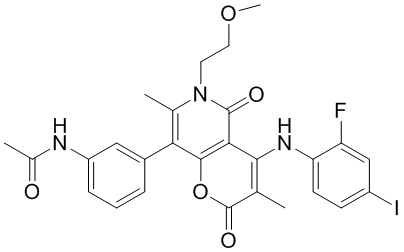
[419] Схема синтеза:
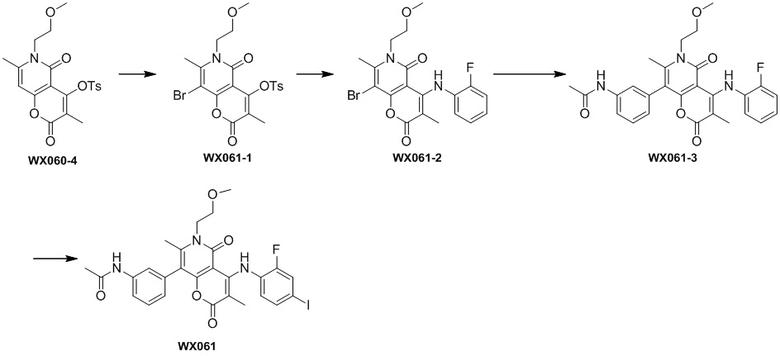
[420] Стадия 1: синтез соединения WX061-1
[421] Соединение WX060-4 (12,50 г, 29,8 ммоль, 1,00 экв.) добавляли в ацетонитрил (130 мл) и дихлорметан (130 мл) с последующим добавлением N-бромсукцинимида (7,96 г, 44,7 ммоль, 1,50 экв.) при 0°C в защитной атмосфере азота. Реакционную смесь перемешивали при 20°C в течение 12 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением твердого вещества. Твердое вещество растирали с метил-трет-бутиловым эфиром (100 мл) с получением целевого соединения WX061-1. 1H ЯМР (400 МГц, ДМСО-d6) δ 7,86(d, J=8,4 Гц, 2 H), 7,46(d, J=8,4 Гц, 2 H), 4,18(t, J=5,4 Гц, 2 H), 3,52-3,45(m, 2 H), 3,24-3,18(m, 3 H), 2,70(s, 3 H), 2,42(s, 3 H), 1,46(s, 3 H). MS m/z: 521,9 [M+H]+
[422] Стадия 2: синтез соединения WX061-2
[423] Соединение WX061-1 (12,00 г, 24,08 ммоль, 1,00 экв.) добавляли в этанол (200 мл) с последующим добавлением 2-фторанилина (13,38 г, 120,40 ммоль, 11,63 мл, 5,00 экв.) при 20°C в защитной атмосфере азота. Реакционную смесь перемешивали при 80°C в течение 12 часов, и образовалось белое твердое вещество. После завершения реакции реакционную смесь фильтровали и фильтрационный осадок промывали метил-трет-бутиловым эфиром (100 мл) с получением целевого соединения WX061-2.
[424] 1H ЯМР (400 МГц, ДМСО-d6) δ 11,16(s, 1 H), 7,38-7,28(m, 1 H), 7,25-7,11(m, 3 H), 4,36(t, J=5,2 Гц, 2 H), 3,63(t, J=5,2 Гц, 2 H), 3,26-3,22(m, 3 H), 2,78(s, 3 H), 1,48(s, 3 H). MS m/z: 437,0 [M+H]+
[425] Стадия 3: синтез соединения WX061-3
[426] Соединение WX061-2 (5,00 г, 11,43 ммоль, 1,00 экв.) и 3-ацетаминофенилбороновую кислоту (3,07 г, 17,15 ммоль, 1,50 экв.) добавляли в диоксан (100 мл) с последующим добавлением насыщенного раствора бикарбоната натрия (20 мл) и Pd(dppf)Cl2.CH2Cl2 (933,82 мг, 1,14 ммоль, 0,10 экв.) при 20°C в защитной атмосфере азота. Реакционную смесь перемешивали при 80-90°C в течение 12 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=0/1) с получением целевого соединения WX061-3. 1H ЯМР (400 МГц, CDCl3-d) δ 11,36(s, 1 H), 8,44(s, 1 H), 7,80(s, 1 H), 7,17-7,05(m, 5 H), 6,88(d, J=7,6 Гц, 1 H), 4,35(s, 2 H), 3,75(t, J=5,2 Гц, 2 H), 3,33(s, 3 H), 2,35(s, 3 H), 2,04(s, 3 H), 1,62(s, 3 H). MS m/z: 492,2 [M+H]+
[427] Стадия 4: синтез соединения WX061
[428] Соединение WX061-3 (2,30 г, 4,68 ммоль, 1,00 экв.) добавляли в N,N-диметилформамид (25 мл), затем добавляли трифторуксусную кислоту (1,07 г, 9,36 ммоль, 693 мкл, 2,00 экв.) и N-йодсукцинимид (2,11 г, 9,36 ммоль, 2,00 экв.) при 0°C в защитной атмосфере азота и в темном месте. Реакционную смесь перемешивали при 20°C в течение 15 часов. После завершения реакции добавляли воду (100мл) и смесь экстрагировали дихлорметаном (100 мл*2). Органическую фазу промывали насыщенным раствором хлорида натрия (100 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и затем растирали с дихлорметаном (10 мл) и метил-трет-бутиловым эфиром (10 мл) с получением целевого соединения WX061. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,25(s, 1 H), 10,07(s, 1 H), 7,71(d, J=10,0 Гц, 1 H), 7,59-7,49(m, 3 H), 7,40(t, J=7,6 Гц, 1 H), 6,93(d, J=7,6 Гц, 1 H), 6,88(t, J=8,4 Гц, 1 H), 4,30(t, J=5,2 Гц, 2 H), 3,64(t, J=5,2 Гц, 2 H), 3,23(s, 3 H), 2,29(s, 3 H), 2,04(s, 3 H), 1,44(s, 3 H). MS m/z: 617,9 [M+H]+
[429] Вариант реализации 12: WX062
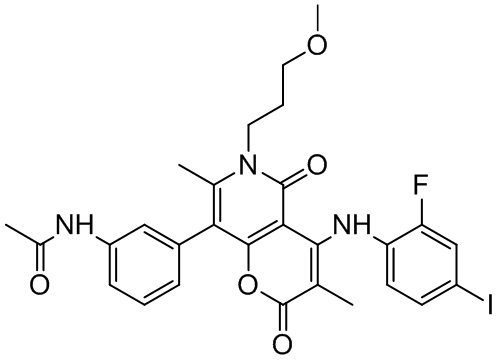
[430] Схема синтеза:
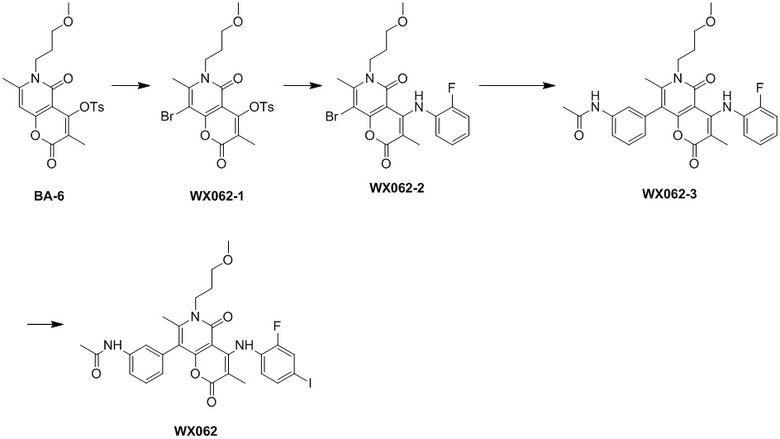
[431] Стадия 1: синтез соединения WX062-1
[432] Соединение BA-6 (20,00 г, 46,14 ммоль, 1,00 экв.) растворяли в ацетонитриле (100,00 мл) и ТГФ (150,00 мл) с последующим добавлением N-бромсукцинимида (12,32 г, 69,21 ммоль, 1,50 экв.). Реакционную смесь перемешивали при 15°C в течение 15 часов. После завершения реакции смесь выпаривали досуха с помощью роторного испарителя. Неочищенный продукт растирали с растворителем метил-трет-бутиловым эфиром/этанолом =2/1 (150мл) и фильтровали. Фильтрационный осадок собирали с получением целевого соединения WX062-1. MS m/z: 536,0 [M+Na]+
[433] Стадия 2: синтез соединения WX062-2
[434] Соединение WX062-1 (7,00 г, 11,59 ммоль, 1,29 экв.) и 2-фторанилин (7,00 г, 62,99 ммоль, 6,09 мл, 7,00 экв.) растворяли в этаноле (100,00 мл), смесь нагревали с обратным холодильником и перемешивали в течение 15 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (200 мл), фильтровали и фильтрационный осадок собирали с получением целевого соединения WX062-2. MS m/z: 453,1 [M+H]+
[435] Стадия 3: синтез соединения WX062-3
[436] Соединение WX062-2 (3,00 г, 6,65 ммоль, 1,00 экв.) и 3-ацетаминофенилбороновую кислоту (1,79 г, 9,98 ммоль, 1,50 экв.) добавляли в диоксан (100,00 мл), затем последовательно добавляли Pd(dppf)Cl2 (486,59 мг, 665,00 мкмоль, 0,10 экв.) и насыщенный раствор бикарбоната натрия (6,65 ммоль, 10,00 мл). В условиях защитной атмосферы азота температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 15 часов. После завершения реакции добавляли воду (100 мл) и смесь экстрагировали дихлорметаном (50 мл *3). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью колоночной хроматографии (PE/EA=1/0-1/5, Rf=0,42) с получением целевого соединения WX062-3. MS m/z: 506,2 [M+H]+
[437] Стадия 4: синтез соединения WX062
[438] Соединение WX062-3 (400,00 мг, 791,23 мкмоль, 1,00 экв.) растворяли в N,N-диметилформамиде (10,00 мл) и дихлорметане (10,00 мл) с последующим добавлением N-йодсукцинимида (534,03 мг, 2,37 ммоль, 3,00 экв.) при 15°C в защитной атмосфере азота. Реакционную смесь перемешивали при 15°C в течение 15 часов. После того, как ЖХМС показала неполное завершение реакции, добавляли дополнительное количество N-йодсукцинимида (534,03 мг, 2,37 ммоль, 3,00 экв.), и реакционную смесь перемешивали при 15°C в течение 15 часов. После того, как ЖХМС показала неполное завершение реакции, реакционную смесь перемешивали при 15°C в течение дополнительных 48 часов. После завершения реакции смесь промывали водой (50мл) и экстрагировали дихлорметаном (50 мл*3). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX062. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,33(s, 1 H), 10,06(s, 1 H), 7,71(dd, J=10,4 Гц, J=2,0 Гц, 1 H), 7,58-7,55(m, 2 H), 7,51(d, J=8,4 Гц, 1 H), 7,40(t, J=8,0 Гц, 1 H), 6,95(d, J=8,0 Гц, 1 H), 6,88(t, J=8,0 Гц, 1 H), 4,16(t, J=7,6 Гц, 2 H), 3,40(t, J=5,8 Гц, 2 H), 3,23(s, 3 H), 2,26(s, 3 H), 2,04(s, 3 H), 1,44(s, 3 H). MS m/z: 632,0 [M+H]+
[439] Вариант реализации 13: WX109
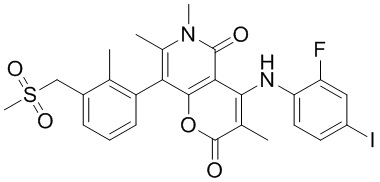
[440] Схема синтеза:
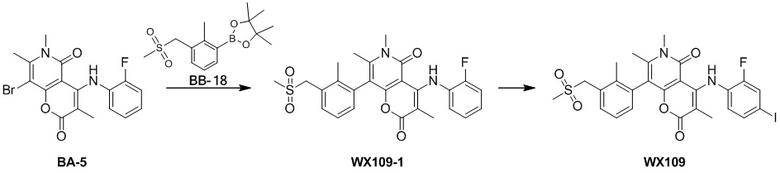
[441] Стадия 1: синтез соединения WX109-1
[442] Соединение BA-5 (400,00 мг, 1,27 ммоль, 0,80 экв.) растворяли в диоксане (8,00 мл) и воде (4,00 мл), затем соединение BB-18 (395,53 мг, 1,28 ммоль, 1,00 экв.), фосфат калия (541,29 мг, 2,55 ммоль, 2,00 экв.), Pd(dppf)Cl2.CH2Cl2 (104,12 мг, 127,50 мкмоль, 0,10 экв.) и SPhos (104,69 мг, 255,00 мкмоль, 0,20 экв.) добавляли в реакционный раствор при 25°C. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 7 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя. Остаток растворяли в EtOAc (20 мл), промывали водой (10 мл* 3) и насыщенным раствором хлорида натрия (5 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX109-1. 1H ЯМР (400 МГц, CDCl3-d) δ 11,21(s, 1 H), 7,46(d, J=6,8 Гц, 1 H), 7,34(t, J=8,8 Гц, 1 H), 7,17-7,08(m, 4 H), 7,06-6,98(m, 1 H), 4,50-4,36(m, 2 H), 3,67(s, 3 H), 2,90(s, 3 H), 2,22(d, J=2,4 Гц, 6 H), 1,58(s, 3 H). MS m/z: 497,3 [M+H]+
[443] Стадия 2: синтез соединения WX109
[444] Соединение WX109-1 (50,00 мг, 100,69 мкмоль, 1,00 экв.) растворяли в N,N-диметилформамиде (1,00 мл), с последующим добавлением N-йодсукцинимида (45,31 мг, 201,38 мкмоль, 2,00 экв.) и трифторуксусной кислоты (500,00 мкл) при 0°C. Реакционную смесь перемешивали при 25°C в течение 16 часов. После завершения реакции реакционный раствор растворяли в EtOAc (20 мл), последовательно промывали насыщенным раствором бикарбоната натрия (20 мл* 3), воду (20 мл* 2) и насыщенным раствором хлорида натрия (20 мл). Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX109. 1H ЯМР (400 МГц, ДМСО-d6) δ 11,38(s, 1 H), 7,72(d, J=10,0 Гц, 1 H), 7,52(d, J=8,4 Гц, 1 H), 7,46(d, J=8,0 Гц, 1 H), 7,34(t, J=7,6 Гц, 1 H), 7,18(d, J=7,6 Гц, 1 H), 6,90(t, J=8,4 Гц, 1 H), 4,70-4,61(m, 2 H), 3,60(s, 3 H), 3,33(s, 3 H), 3,01(s, 3 H), 2,15(s, 3 H), 1,45(s, 3 H). MS m/z: 623,0 [M+H]+
[445] Вариант реализации 14: WX110
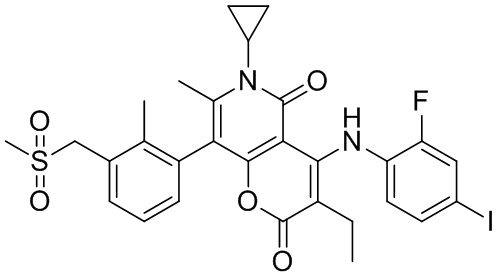
[446] Схема синтеза:
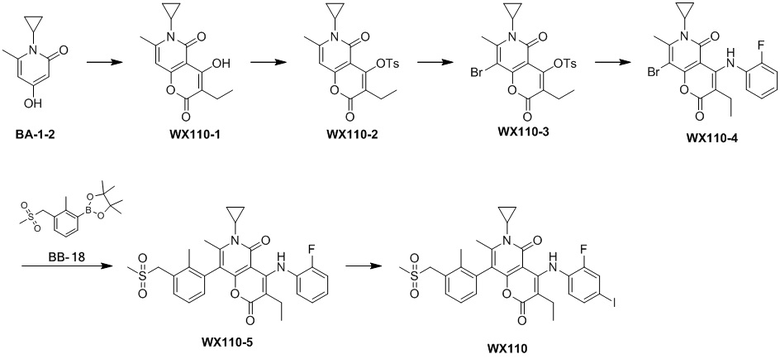
[447] Стадия 1: синтез соединения WX110-1
[448] Соединение BA-1-2 (7,20 г, 42,07 ммоль, 1,00 экв.) растворяли в уксусном ангидриде (70,00 мл) с последующим добавлением 2-этилмалоновой кислоты (8,34 г, 63,11 ммоль, 1,50 экв.) при 25°C . Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 0,5 часа. После завершения реакции реакционную смесь охлаждали до 25°C. Затем смесь оставляли отстаиваться в течение 15 часов, образовывалось твердое вещество. Смесь фильтровали и фильтрационный осадок собирали. Фильтрационный осадок растирали с метил-трет-бутиловым эфиром (50 мл) и сушили с получением целевого соединения WX110-1. 1H ЯМР (ДМСО-d6) δ 13,41(s, 1 H), 6,54(s, 1 H), 3,00-2,92(m, 1 H), 2,53(s, 3 H), 2,30(q, J=7,6 Гц, 2 H), 1,15-0,98(d, J=5,8 Гц, 2 H), 0,96(t, J=7,2 Гц, 3 H), 0,93-0,88(m, 2 H). MS m/z: 261,9 [M+H]+
[449] Стадия 2: синтез соединения WX110-2
[450] Соединение WX110-1 (3,70 г, 14,16 ммоль, 1,00 экв.) растворяли в дихлорметане (100,00 мл), добавляли триэтиламин (3,58 г, 35,40 ммоль, 4,91 мл, 2,50 экв.) и DMAP (432,48 мг, 3,54 ммоль, 0,25 экв.) при 0°C с последующим добавлением п-толуолсульфонилхлорида (6,75 г, 35,40 ммоль, 2,50 экв.). Температуру реакции оставляли повышаться до 25°C и реакционную смесь перемешивали в течение 16 часов. Затем, когда было выявлено, что на субстрате ничего не осталось, добавляли дополнительное количество триэтиламина (1,43 г, 14,16 ммоль, 1,96 мл, 1,00 экв.) и DMAP (86,50 мг, 708,00 мкмоль, 0,50 экв.) с последующим добавлением 4-метилбензолсульфонилхлорида (2,70 г, 14,16 ммоль, 1,00 экв.) при 0°C. Температуру реакции оставляли повышаться до 25°C и реакционную смесь перемешивали в течение 4 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с этанолом (100 мл), затем фильтровали и фильтрационный осадок собирали и сушили с получением соединения WX110-2. 1H ЯМР (ДМСО-d6) δ 7,90(d, J=8,4 Гц, 2 H), 7,50(d, J=8,4 Гц, 2 H), 6,33(s, 1 H), 2,87-2,83(m, 1 H), 2,50(s, 3 H), 2,44(s, 3 H), 2,06(q, J=7,2 Гц, 2 H), 1,13(q, J=6,8 Гц, 2 H), 0,85(t, J=7,2 Гц, 3 H), 0,69(d, J=4,0 Гц, 2 H). MS m/z: 416,1 [M+H]+
[451] Стадия 3: синтез соединения WX110-3
[452] Соединение WX110-2 (2,90 г, 6,98 ммоль, 1,00 экв.) растворяли в дихлорметане (30,00 мл) и ацетонитриле (50,00 мл), затем партиями добавляли N-бромсукцинимид (1,86 г, 10,47 ммоль, 1,50 экв.) при 25°C в защитной атмосфере азота. Реакционную смесь перемешивали при 25°C в течение 15 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с этанолом (50 мл), фильтровали и фильтрационный осадок собирали с получением целевого соединения WX110-3. 1H ЯМР (CDCl3-d) δ 7,93(d, J=8,3 Гц, 2 H), 7,37(d, J=8,0 Гц, 2 H), 3,00-2,85(m, 1 H), 2,73(s, 3 H), 2,46(s, 3 H), 2,17(q, J=7,4 Гц, 2 H), 1,35-1,25(m, 2 H), 0,94(t, J=7,6 Гц, 3 H), 0,89-0,83(m, 2 H). MS m/z: 494,0 [M+H]+
[453] Стадия 4: синтез соединения WX110-4
[454] Соединение WX110-3 (2,5 г, 5,06 ммоль, 1,00 экв.) растворяли в этаноле (100,00 мл), затем добавляли 2-фторанилин (5,62 г, 50,60 ммоль, 10,00 экв.) при 25°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 12 часов. Затем, когда было выявлено, что на субстрате ничего не осталось, добавляли дополнительное количество 2-фторанилина (2,81 г, 25,30 ммоль, 5,00 экв.), реакционную смесь перемешивали в течение дополнительных 12 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя, остаток растирали с этанолом (50 мл) и фильтровали. Фильтрат выпаривали досуха с помощью роторного испарителя и затем растирали с метил-трет-бутиловым эфиром (50 мл). Смесь фильтровали и фильтрат собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=30/1-5/1), затем растирали с метил-трет-бутиловым эфиром (50 мл) и фильтровали. Фильтрационный осадок сушили с получением целевого соединения WX110-4. 1H ЯМР (400 МГц, CDCl3-d) δ 10,99(s, 1 H), 7,18-7,01(m, 4 H), 3,01-2,90(m, 1 H), 2,79(s, 3 H), 2,16(q, J=7,4 Гц, 2 H), 1,41-1,32(m, 2 H), 0,94-0. 85(m, 2 H), 0,81(t, J=7,4 Гц, 3 H). MS m/z: 435,0 [M+H]+
[455] Стадия 5: синтез соединения WX110-5
[456] Соединение WX110-4 (420,00 мг, 969,37 мкмоль, 1,00 экв.) растворяли в диоксане (8,00 мл) и воду (4,00 мл), затем последовательно добавляли соединение BB-18 (375,90 мг, 1,21 ммоль, 1,25 экв.), фосфат калия (514,42 мг, 2,42 ммоль, 2,50 экв.), Pd(dppf)Cl2.CH2Cl2 (98,95 мг, 21,17 мкмоль, 0,02 экв.) и SPhos (99,49 мг, 242,34 мкмоль, 0,25 экв.) при 25°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 15 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя, разбавляли EtOAc (20 мл), затем промывали водой (10 мл* 3) и насыщенным раствором хлорида натрия (5 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX110-5. 1H ЯМР (400 МГц, CDCl3-d) δ 11,13(s, 1 H), 7,44(d, J=7,6 Гц, 1 H), 7,33(t, J=7,6 Гц, 1 H), 7,16-7,10(m, 5 H), 4,49-4. 44(m, 1 H), 4. 41-4. 33(m, 1 H), 2,97-2,91(m, 1 H), 2,89(s, 3 H), 2,30(s, 3 H), 2,21(s, 3 H), 2,10(q, J=7,2 Гц, 2 H), 1,40-1,32(m, 2 H), 0,95(q, J=4,4 Гц, 2 H), 0,76(t, J=7,2 Гц, 3 H). MS m/z: 537,3 [M+H]+
[457] Стадия 6: синтез соединения WX110
[458] Соединение WX110-5 (40,00 мг, 74,54 мкмоль, 1,00 экв.) растворяли в N,N-диметилформамиде (1,00 мл), затем последовательно добавляли N-йодсукцинимид (33,54 мг, 149,08 мкмоль, 2,00 экв.) и трифторуксусную кислоту (1,00 мл) при 0°C. Температуру реакционной смеси повышали до 25°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционную смесь разбавляли EtOAc (10 мл), затем последовательно промывали насыщенным раствором бикарбоната натрия (10 мл*3), водой (10 мл*2) и насыщенным раствором хлорида натрия (20 мл). Органическую фазу сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX110. 1H ЯМР (ДМСО-d6) δ 11,31(s, 1 H), 7,74(d, J=9,8 Гц, 1 H), 7,53(d, J=7,6Гц, 1 H), 7,45(d, J=7,6Гц, 1 H), 7,34(t, J=7,4Гц, 1 H), 7,21(d, J=8,0 Гц, 1 H), 7,02(t, J=8,4 Гц, 1 H), 4,70-4,61(m, 2 H), 3,10(m, 1 H), 3,01(s, 3 H), 2,21(s, 3 H), 2,14(s, 3 H), 2,00(d, J=7,2 Гц, 2 H), 1,23(s, 2 H), 0,91(d, J=10,8 Гц, 2 H), 0,70(t, J=7,2 Гц, 3 H). MS m/z: 663,0 [M+H]+
[459] Вариант реализации 15: WX118&119
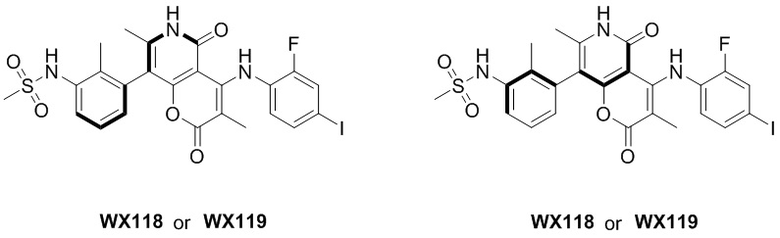
[460] Схема синтеза:
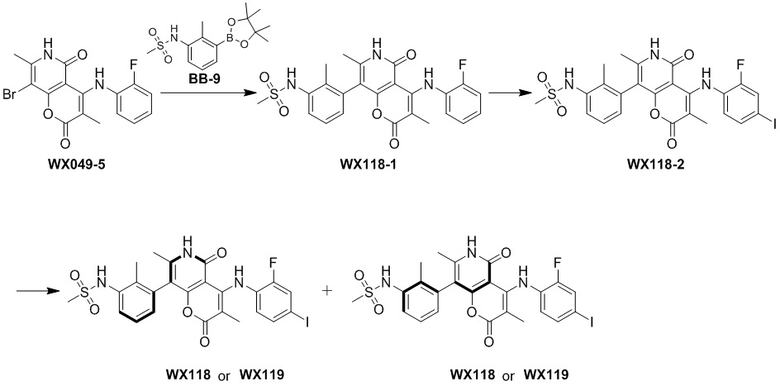
[461] Стадия 1: синтез соединения WX118-1
[462] Соединение WX049-5 (3,15 г, 8,31 ммоль, 1,00 экв.) растворяли в диоксане (50 мл) и воде (10 мл), затем соединение BB-9 (5,17 г, 16,61 ммоль, 2,00 экв.), SPhos (341,04 мг, 830,74 мкмоль, 0,10 экв.), фосфат калия (3,53g, 16,61 ммоль, 2,00 экв.) и ацетат палладия (93,25 мг, 415,37 мкмоль, 0,50 экв.) добавляли последовательно при 15°C. Температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 32 часов. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который затем разбавляли дихлорметаном (50 мл) и фильтровали. Фильтрационный осадок промывали дихлорметаном (15 мл *3). Органические фазы объединяли, промывали водой (40 мл*3), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=20/1-1/1) с получением целевого соединения WX118-1. 1H ЯМР (ДМСО-d6) δ 12,57(s, 1 H), 11,32(m, 1 H), 9,18(m, 1 H), 7. 38-7. 36(m, 1 H), 7,31-7,28(m, 1 H), 7,18-7,11(m, 3 H), 7,09(m, 1 H), 3,00(m, 1 H), 2,06(s, 3 H), 1,98(s, 3 H), 1. 43-1,39(s, 3 H). MS m/z: 484,1 [M+H]+
[463] Стадия 2: синтез соединения WX118-2
[464] Соединение WX118-1 (0,32 г, 661,83 мкмоль, 1,00 экв.) растворяли в дихлорметане (4,80 мл), затем последовательно добавляли трифторуксусную кислоту (1,48 г, 12,97 ммоль, 19,59 экв.) и N-йодсукцинимид (178,68 мг, 794,19 мкмоль, 1,20 экв.) при 0°C в темном месте. Реакционную смесь перемешивали при 15°C в течение 24 часов. После завершения реакции реакционный раствор последовательно промывали насыщенным раствором бикарбоната натрия (50 мл* 3) и водой (50 мл*2), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением целевого соединения WX118-2. 1H ЯМР (ДМСО-d6) δ 12,23-11,34(m, 1 H), 7,72(d, J=10,4 Гц, 1 H), 7,53(d, J=7,8 Гц, 1 H), 7,42-7. 35(m, 1 H), 7,34-7,26(m, 1 H), 7. 10(d, J=7,6Гц, 1 H), 6,93(t, J=8,4 Гц, 1 H), 3,01(s, 3 H), 2,67(s, 1 H), 2,33(s, 1 H), 2,06(s, 3 H), 1,99(s, 3 H), 1,43(s, 3 H). MS m/z: 609,9 [M+H]+
[465] Стадия 3: синтез соединения WX118 и WX119
[466] WX118-2 подвергали сверхкритической жидкостной хроматографии (колонка: Chiralpak AD-3 100 × 4,6 мм ID, 3 мкм; подвижная фаза: A: CO2, B: этанол (0,05% DEA); градиент: от 5% B до 40% B при однородной скорости за 4,5 минуты, затем 40% B в течение 2,5 минут, 5% B в течение 1 минуты; скорость потока: 2,8 мл/мин; температура колонки: 40°C), с получением атропоизомеров WX118 и WX119, с временем удерживания 5,934 мин и 4,958 мин, соответственно, и соотношением 6:7.
[467] WX118
[468] 1H ЯМР (ДМСО-d6) δ 11,33(m, 1 H), 7,72-7. 35(m, 1 H), 7,37-7,10(m, 1 H), 6,92(t, J=8,4 Гц, 1 H), (s, 3 H), 3,01(m, 2 H), 2,50(s, 3 H), 2,06(s, 3 H), 2,00(s, 3 H), 1,43(s, 1 H). MS m/z: 609,9 [M+H]+
[469] WX119
[470] 1H ЯМР (ДМСО-d6) δ 11,35-11,26(m, 1 H), 7,75-7,68(m, 1 H), 7,59-7,25(m, 3 H), 7,10(m, 1 H), 6,89-6,95(m, 1 H), 3,00(s, 3 H), 2,06(s, 3 H), 1,99(s, 3 H), 1,43(s, 3 H). MS m/z: 610,0 [M+H]+
[471] Вариант реализации 16: WX034
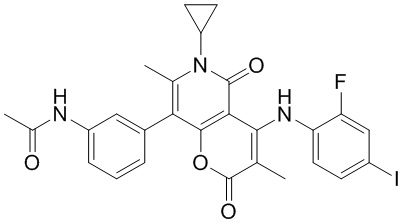
[472] Схема синтеза:
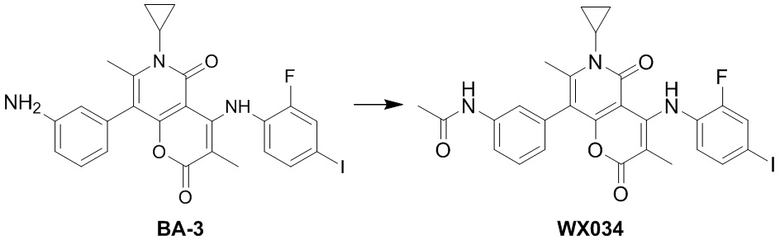
[473] Стадия 1: синтез соединения WX034
[474] Соединение BA-3 (500,00 мг, 897,10 мкмоль, 1,00 экв.) растворяли в хлороформе (5,00 мл) и N,N-диметилформамиде (5,00 мл), добавляли пиридин (4,90 г, 61,94 ммоль, 69,05 экв.) при 0°C с последующим добавлением по каплям ацетилхлорида (211,27 мг, 2,69 ммоль, 3,00 экв.). Реакционную смесь перемешивали при 20°C в течение 12 часов. После завершения реакции указанный растворитель удаляли с помощью роторного испарителя. Остаток разбавляли водой (10 мл) и экстрагировали EtOAc (15 мл*3). Органические фазы объединяли, последовательно промывали разбавленной соляной кислотой (0,5 M, 10 мл*2), насыщенным раствором бикарбоната натрия (10 мл*2), водой (10 мл) и насыщенным раствором хлорида натрия (10 мл). Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растирали с метил-трет-бутиловым эфиром (18 мл) и фильтровали с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX034. 1H ЯМР (ДМСО-d6) δ 11,28(s, 1 H), 10,05(m, 2 H), 7,72-7,69(m, 1 H), 7,56(m, 1 H), 7,52(m, 1 H), 7,40(m, 1 H), 6,97(m, 1 H), 6,86(m, 1 H), 3,03(m, 1 H), 2,31(m, 3 H), 2,04(s, 3 H), 1,44(s, 3 H), 1,19(m, 2 H), 0,90(m, 2 H). MS m/z: 600,1 [M+H]+
[475] Вариант реализации 17: WX035
[476] Схема синтеза:
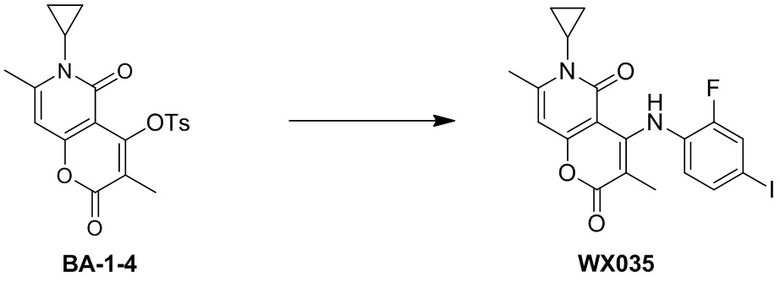
[477] Стадия 1: синтез соединения WX035
[478] Соединение BA-1-4 (300,00 мг, 747,33 мкмоль, 1,00 экв.) и 2-фтор-4-анилин (531,37 мг, 2,24 ммоль, 3,00 экв.) растворяли в этаноле (4,00 мл), реакционную смесь перемешивали при 90°C в течение 16 часов. После завершения реакции смесь охлаждали до комнатной температуры, фильтровали и фильтрационный осадок собирали. Фильтрационный осадок очищали с помощью колоночной хроматографии (метил-трет-бутиловый эфир) с получением неочищенного продукта. Неочищенный продукт дополнительно очищали с помощью препаративной ТСХ (ДХМ) с получением целевого соединения WX035. 1H ЯМР (400 МГц, CDCl3-d) δ 10,99(s, 1 H), 7,48-7,42(m, 2 H), 6,74-6,20(m, 1 H), 6,22(s, 1 H), 2. 91-2,88(m, 1 H), 2,59(m, 3 H), 1,64(s, 3 H), 1,36-1,34(s, 2 H), 0,94-0,93(s, 2 H).
[479] Вариант реализации 18: WX039
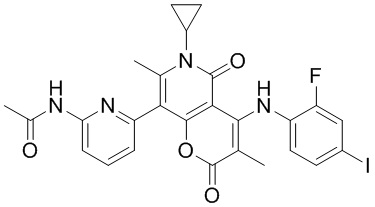
[480] Схема синтеза:
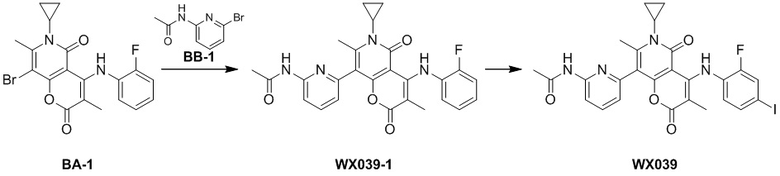
[481] Стадия 1: синтез соединения WX039-1
[482] Соединение BB-1 (500,00 мг, 2,33 мкмоль, 1,00 экв.), Pd(PPh3)2Cl2 (163,54 мг, 233,00 мкмоль, 0,10 экв.) и гексаметилдиолово (839,72 мг, 2,56 ммоль, 1,10 экв.) добавляли в безводный толуол (15,00 мл). Температуру реакционной смеси повышали до 110°C в защитной атмосфере азота и реакционную смесь перемешивали в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры, затем добавляли соединение BA-1 (976,83 мг, 2,33 ммоль, 1,00 экв.) и диоксан (15,00 мл). В условиях защитной атмосферы азота температуру повышали до 100°C и реакционную смесь перемешивали в течение 24 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры с последующим добавлением фторида калия (0,5 г). Смесь перемешивали в течение 30 минут и фильтровали, фильтрационный осадок промывали дихлорметаном (30 мл*3). Органическую фазу собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт перемешивали в метаноле (30 мл) в течение 30 минут и фильтровали. Фильтрационный осадок промывали метанолом (80 мл) и выпаривали досуха с помощью роторного испарителя с возвратом исходного вещества BA-1. Фильтраты объединяли и выпаривали досуха с помощью роторного испарителя. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=3/1-0/1) с получением соединения WX039-1. MS m/z: 475,2 [M+H]+
[483] Стадия 2: синтез соединения WX039
[484] Соединение WX039-1 (44,00 мг, 60,74 мкмоль, 1,00 экв.) растворяли в N,N-диметилформамиде (1,00 мл), затем последовательно партиями добавляли трифторуксусную кислоту (1,00 мл) и N-йодсукцинимид (45,00 мг, 200,02 мкмоль, 3,29 экв.) при 25°C. Реакционную смесь перемешивали при 25°C в течение 32 часов. После того, как ЖХМС показала неполное завершение реакции добавляли дополнительное количество N-йодсукцинимида (40,00 мг) и реакционную смесь перемешивали в течение дополнительных 6 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном (20 мл), карбонат натрия (2 г) добавляли для гашения реакционной смеси до окончания образования газа. Смесь фильтровали и фильтрат собирали, выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и сушили с получением соединения WX039. 1H ЯМР (400 МГц, CDCl3-d) δ 11,04(s, 1 H), 8,27-8,25(s, 1 H), 7,95(m, 1 H), 7,87-7,83(m, 1 H), 7,49-7,47(m, 1 H), 7,46-7,42(m, 1 H), 7,11-7,09(m, 1 H), 6,73-6,70(m, 1 H), 2,98-2,92(s, 1 H), 2,40(s, 3 H), 2,24(s, 3 H), 1,62(m, 3 H), 1,43-1,38(m, 2 H), 0,98-0,97(m, 2 H). MS m/z: 601,1 [M+H]+
[485] Вариант реализации 19: WX048
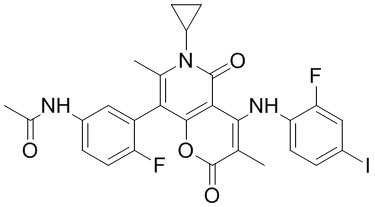
[486] Схема синтеза:
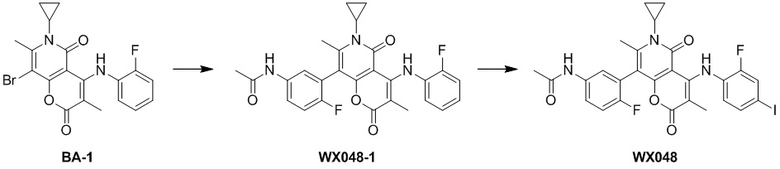
[487] Стадия 1: синтез соединения WX048-1
[488] Соединение BA-1 (75,10 мг, 179,14 мкмоль, 1,00 экв.) растворяли в диоксане (4,00 мл) и воде (1,00 мл), затем добавляли Pd2(dba)3 (8,20 мг, 8,96 мкмоль, 0. 05 экв.), фосфат калия (76,05 мг, 358,28 мкмоль, 2,00 экв.), SPhos (7,35 мг, 17,91 мкмоль, 0,10 экв.) и соединение BB-2 (100,00 мг, 358,28 мкмоль, 2,00 экв.) при 25°C в защитной атмосфере азота. Температуру реакционной смеси повышали до 120°C и реакционную смесь перемешивали в течение 21 часов. После завершения реакции реакционную смесь фильтровали и фильтрационный осадок промывали EtOAc (10 мл*3). Фильтраты объединяли, последовательно промывали водой (15 мл*3) и насыщенным раствором хлорида натрия (15 мл). Объединенную водную фазу экстрагировали EtOAc (15 мл). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ТСХ (PE/EA=1/1) с получением соединения WX048-1. MS m/z :492,1 [M+H]+
[489] Стадия 2: синтез соединения WX048
[490] Соединение WX048-1 (50,00 мг, 101,73 мкмоль, 1,00 экв.) добавляли в смесь трифторуксусной кислоты (250,00 мкл) и N,N-диметилформамида (500,00 мкл) с последующим добавлением N-йодсукцинимида (45,78 мг, 203,46 мкмоль, 2,00 экв.) при 0°C. Реакционную смесь перемешивали при 25oC в течение 16 часов. После завершения реакции реакционную смесь разбавляли EtOAc (20 мл), последовательно промывали насыщенным раствором сульфита натрия (20 мл*2) и насыщенным раствором бикарбоната натрия (20 мл*2). Объединенную водную фазу экстрагировали EtOAc (30 мл*3). Органические фазы объединяли, сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и сушили с получением целевого соединения WX048. 1H ЯМР (400 МГц, CDCl3-d) δ 11,22(s, 1 H), 10,13(s, 1 H), 7,75-7,72(m, 1 H), 7,65-7,64(m, 2 H), 7,54-7,52(m, 1 H), 7,32(m, 1 H), 6,92-6,90(m, 1 H), 3,08-3,07(m, 1 H), 2,37(s, 3 H), 2,06(s, 3 H), 1,46(s, 3 H), 1,23-1,22(m, 2 H), 0,96-0,88(m, 2 H). MS m/z: 618,1 [M+H]+
[491] Вариант реализации 20: WX071
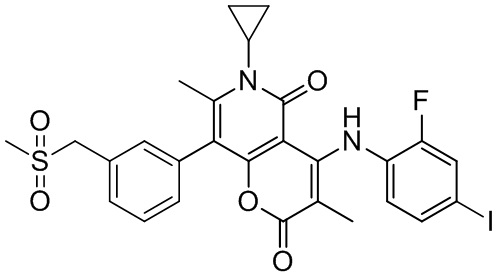
[492] Схема синтеза:
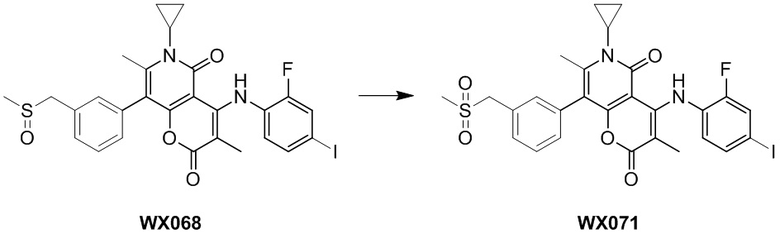
[493] Стадия 1: синтез соединения WX071
[494] Соединение WX068 (55,00 мг, 88,93 мкмоль, 1,00 экв.) растворяли в смешанном растворителе ТГФ (2,00 мл) и воде (2,00 мл), с последующим добавлением гидросульфата калия (54,13 мг, 177,86 мкмоль, 2,00 экв.) в защитной атмосфере азота. Реакционную смесь перемешивали при 15°C в течение 16 часов. После завершения реакции реакционную смесь разбавляли водой (20 мл), водную фазу экстрагировали дихлорметаном (20 мл*3). Органическую фазу собирали и промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и лиофилизировали с получением целевого соединения WX071. 1H ЯМР (400 МГц, CDCl3-d) δ 11,12(s, 1 H), 7,55-7,42(m, 2 H), 7,35-7,30(m, 2 H), 6,74-6,70(m, 1 H), 4,35-4,28(m, 1 H), 2,98-2,93(m, 1 H), 2,87(s, 3 H), 2,44(s, 3 H), 1,61(s, 3 H), 1,40-1,38(m, 2 H), 0. 99-0,90(m, 2 H). MS m/z: 635,0 [M+H]+
[495] Вариант реализации 21: WX083 & WX084
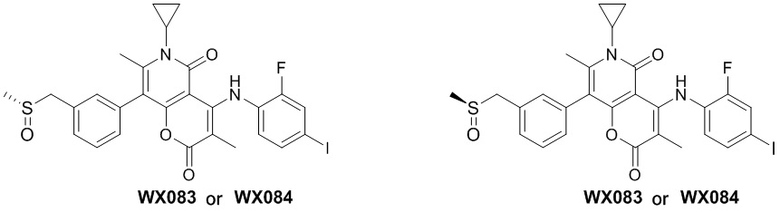
[496] Схема синтеза:
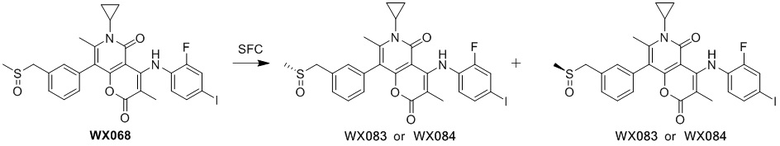
[497] Стадия 1: синтез соединения WX083 и соединение WX084
[498] Соединение WX068 подвергали сверхкритической жидкостной хроматографии (колонка Chiral: Chiralpak OD-3 150 × 4,6 мм ID, 3 мкм; подвижная фаза: A: CO2, B: 40% этанол (0,05% DEA); скорость потока: 2,4 мл/мин; длина волны: 220 нм), с получением атропоизомеров WX083 и WX084, с временем удерживания 5,88 мин и 6,79 мин. Соответственно, и соотношением 1:1.
[499] WX083
[500] 1H ЯМР (400 МГц, CDCl3-d) δ 11,03(s, 1 H), 7,42-7,35(m, 2 H), 7,33-7,28(m, 2 H), 7,19-7,12(m, 2 H), 6,64-6,60(m, 1 H), 4,09-3,86(m, 2 H), 2,89-2,85(m, 1 H), 2,40(s, 3 H), 2,33(s, 3 H), 1,52(s, 2 H), 1,30-1,20(m, 2 H), 1,18-1,13(m, 1 H), 0,89-0,88(m, 2 H). MS m/z: 619,1 [M+H]+
[501] WX084
[502] 1H ЯМР (400 МГц, CDCl3-d) δ 11,08(s, 1 H), 7,49-7,72(m, 2 H), 7,40-7,36(m, 2 H), 7,26-7,19(m, 2 H), 6,72-6,68(s, 1 H), 4,16-3,93(m, 2 H), 2,93(m, 1 H), 2,48(s, 3 H), 2,40(s, 3 H), 1,63-1,59(s, 2 H), 1,38-1,30(m, 2 H), 1,29-1,20(m, 1 H), 0,96(m, 2 H). MS m/z: 619,1 [M+H]+
[503] Вариант реализации 22: WX092
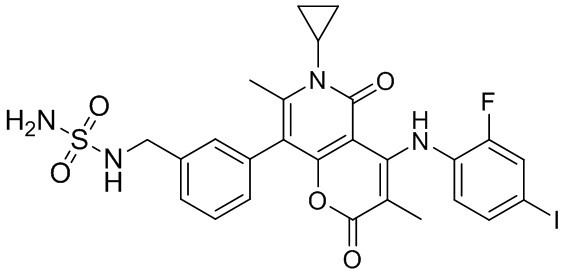
[504] Схема синтеза:
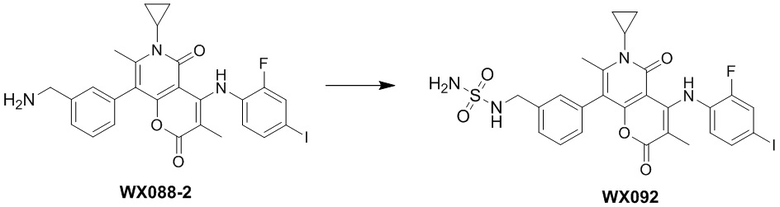
[505] Стадия 1: синтез соединения WX
[506] Соединение WX088-2 (50,00 мг, 87,51 мкмоль, 1,00 экв.) растворяли в дихлорметане (2,00 мл), затем последовательно добавляли триэтиламин (17,71 мг, 175,02 мкмоль, 2,00 экв.), DMAP (2,14 мг, 17,50 мкмоль, 0,20 экв.) и хлорсульфонамид (12,13 мг, 105,01 мкмоль, 1,20 экв.) при 0°C. Температуру реакционной смеси повышали до 25°C и реакционную смесь перемешивали в течение 1 часа. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX092. 1H ЯМР (400 МГц, CDCl3-d) δ 11,19(s, 1 H), 7,50-7,45(m, 5 H), 7,43-7,16(m, 1 H), 6,76-6,71(m, 1 H), 4,91(m, 1 H), 4,83(s, 2 H), 4,39-4,38(s, 2 H), 2,98-2,96(m, 1 H), 2,49(s, 3 H), 1,59(s, 3 H), 1,41(m, 2 H), 1,00(m, 2 H). MS m/z: 651,1 [M+H]+
[507] Вариант реализации 23: WX100
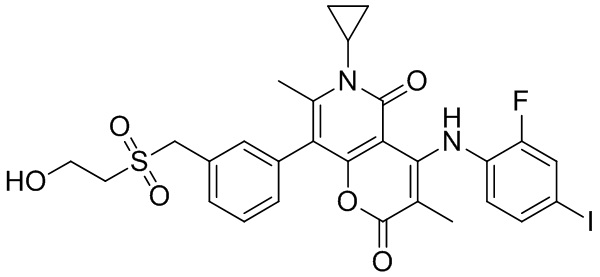
[508] Схема синтеза:
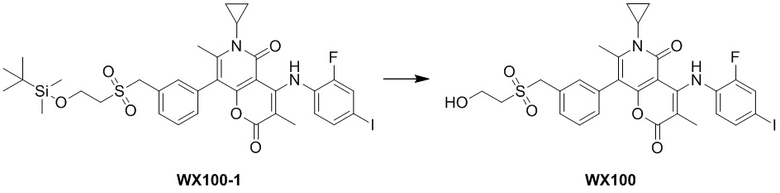
[509] Стадия 1: синтез соединения WX100
[510] Соединение WX100-1 (220,00 мг, 157,71 мкмоль, 1,00 экв.) растворяли в ТГФ (2,00 мл), затем TBAF (1 M, 565,02 мкл, 3,58 экв.) добавляли в защитной атмосфере азота. Реакционную смесь перемешивали при 0°C в течение 2 часов. После завершения реакции реакционную смесь разбавляли водой (30 мл) и экстрагировали EtOAc (40 мл*3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=10/1-1/1), и дополнительно очищали с помощью препаративной ВЭЖХ (система муравьиной кислоты) с получением целевого соединения WX100.
[511] 1H ЯМР (400 МГц, CDCl3-d) δ 11,24(s, 1 H), 7,68-7,66(m, 1 H), 7,52(m, 1 H), 7,49-7,44(m, 2 H), 7,43-7,39(m, 1 H), 7,25(m, 1 H), 6,76-6,71(s, 1 H), 4,47-4,31(m, 2 H), 4,01-3,95(s, 3 H), 3,67(s, 1 H), 3,04-2,95(s, 1 H), 2,48(m, 2 H), 1,58(s, 3 H), 1,43-1,38(m, 2 H), 0,98(m, 2 H). MS m/z: 665,3 [M+H]+
[512] Вариант реализации 24: WX102
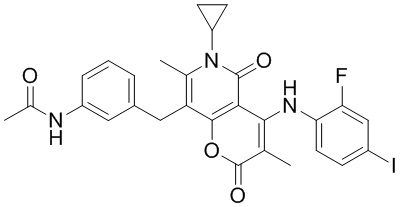
[513] Схема синтеза:
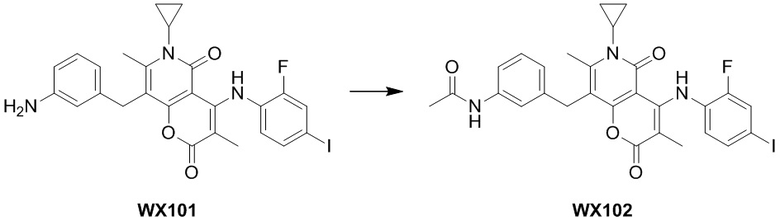
[514] Стадия 1: синтез соединения WX102
[515] Соединение WX101 (100,00 мг, 175,01 мкмоль, 1,00 экв.) растворяли в дихлорметане (2,00 мл), затем добавляли триэтиламин (35,42 мг, 350,03 мкмоль, 2,00 экв.) и ацетилхлорид (20,61 мг, 262,52 мкмоль, 1,50 экв.) при 0°C в защитной атмосфере азота. Реакционную смесь перемешивали при 0°C в течение 20 минут. После завершения реакции реакционную смесь добавляли в воду (20 мл) и водную фазу экстрагировали дихлорметаном (40 мл*3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл*3), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX102. 1H ЯМР (400 МГц, CDCl3-d) δ 11,21(s, 1 H), 7,47-7,44(m, 1 H), 7,40-7,34(m, 3 H), 7,23(s, 1 H), 6,96(m, 1 H), 6,73-6,70(m, 1 H), 4,03(s, 2 H), 2,94-2,88(m, 1 H), 2,60(s, 3 H), 2,16(s, 3 H), 1,62(s, 3 H), 1,37-1,32(m, 2 H), 0,92-0,88(m, 2 H). MS m/z: 614,2 [M+H]+
[516] Вариант реализации 25: WX103
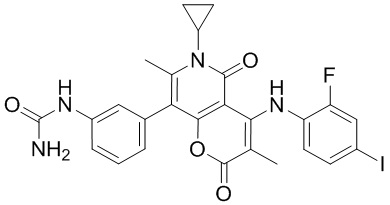
[517] Схема синтеза:
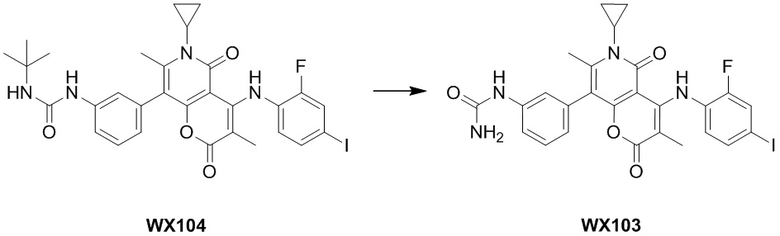
[518] Стадия 1: синтез соединения WX103
[519] Соединение WX104 (120,00 мг, 129,78 мкмоль, 1,00 экв.) растворяли в дихлорметане (2,00 мл), затем трифторуксусную кислоту (2,00 мл) по каплям добавляли при 0°C. Температуру реакционной смеси повышали до 45°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционную смесь разбавляли EtOAc (25 мл), последовательно промывали насыщенным раствором бикарбоната натрия (15 мл*3), водой (15 мл) и насыщенным раствором хлорида натрия (10 мл). Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX103. 1H ЯМР (400 МГц, CDCl3-d) δ 11,28(s, 1 H), 8,66(m, 1 H), 7,70(m, 1 H), 7,51(s, 1 H), 7,73(m, 3 H), 6,83(m, 2 H), 5,88(m, 2 H), 3,02(m, 1 H), 2,31(s, 3 H), 1,43(s, 3 H), 1,19(m, 2 H), 0,89(m, 2 H). MS m/z: 650,5 [M+H]+
[520] Вариант реализации 26: WX105
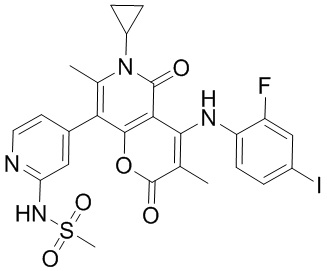
[521] Схема синтеза:
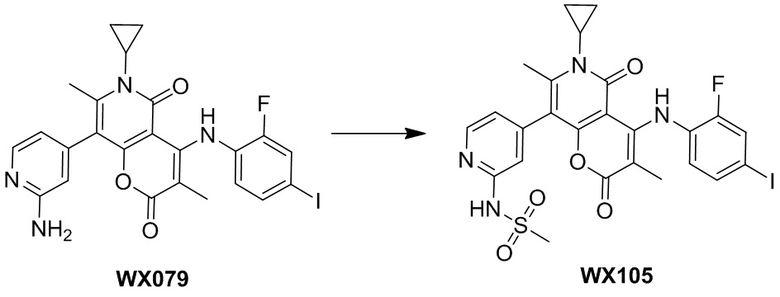
[522] Стадия 1: синтез соединения WX105
[523] Соединение WX079 (65,00 мг, 116,42 мкмоль, 1,00 экв.) растворяли в пиридине (2,00 мл) с последующим добавлением метансульфонилхлорида (200,00 мг, 1,75 ммоль, 15,00 экв.) при 0°C. Температуру реакционной смеси повышали до 25°C и реакционную смесь перемешивали в течение 2 часов. После завершения реакции реакционную смесь разбавляли EtOAc (25 мл), последовательно промывали 0,5 M водным раствором соляной кислоты (15 мл*3), насыщенным раствором бикарбоната аммония (15 мл*2), водой (15 мл) и насыщенным раствором хлорида натрия (10 мл). Органическую фазу сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX105. 1H ЯМР (ДМСО-d6) δ 11,18(s, 1 H), 8,34(m, 1 H), 7,72-7,69(m, 1 H), 7,52-7,50(m, 1 H), 6,99-6,98(m, 1 H), 6,90-6,87(m, 1 H), 6,85-6,83(m, 1 H), 3,31(s, 3 H), 3,02(m, 1 H), 2,35(s, 3 H), 1,44(m, 3 H), 1,21- 1,18(m, 2 H), 0,90(m, 2 H). MS m/z: 637,0 [M+H]+
[524] Вариант реализации 27: WX036
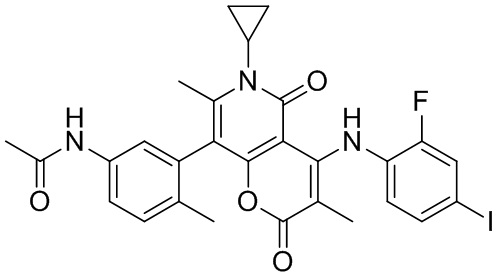
[525] Схема синтеза:
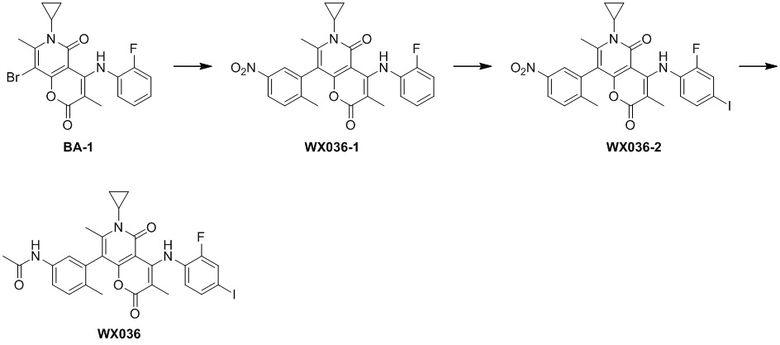
[526] Стадия 1: синтез соединения WX036-1
[527] Соединение BA-1 (2,00 г, 4,77 ммоль, 1,00 экв.), соединение BB-3 (1,73 г, 9,54 ммоль, 2,00 экв.), карбонат цезия (3,11 г, 9,54 ммоль, 2,00 экв.), ацетат палладия (107,10 г, 477,00 мкмоль, 0,10 экв.) и SPhos (445,22 мг, 954,00 мкмоль, 0,20 экв.) растворяли в N,N-диметилформамиде (80,00 мл). В условиях защитной атмосферы азота температуру реакционной смеси повышали до 100°C и реакционную смесь перемешивали в течение 16 часов. Реакционную смесь фильтровали и фильтрационный осадок промывали дихлорметаном (30 мл*3). Фильтрат собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт растворяли в дихлорметане (200 мл), промывали водой (200 мл*3) и насыщенным раствором хлорида натрия (200 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали последовательно с помощью колоночной хроматографии (ДХМ, ДХМ/EA=10/1) и препаративной ТСХ (ДХМ) с получением соединения WX036-1. MS m/z: 476,2 [M+H]+
[528] Стадия 2: синтез соединения WX036-2
[529] Соединение WX036-1 (99,00 мг, 208,22 ммоль, 1,00 экв.) растворяли в N,N-диметилформамиде (2,00 мл), с последующим добавлением трифторуксусной кислоты (2,00 мл) и N-йодсукцинимида (49,49 мг, 218,63 мкмоль, 1,05 экв.) при 0°C. Реакционную смесь перемешивали при 20°C в течение 16 часов. Когда ЖХМС показала неполное завершение реакции, добавляли дополнительное количество N-йодсукцинимида (49,49 мг, 218,63 мкмоль, 1,05 экв.). Реакционную смесь перемешивали в течение дополнительных 16 часов. После завершения реакции реакционную смесь разбавляли дихлорметаном (20 мл), промывали водой (10 мл*3), насыщенным раствором карбоната натрия (20 мл*2) и насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением соединения WX036-2. MS m/z: 602,1 [M+H]+
[530] Стадия 3: синтез соединения WX036
[531] Соединение WX036-2 (100,00 мг, 166,29 ммоль, 1,00 экв.) растворяли в уксусной кислоте (4,00 мл) с последующим добавлением цинкового порошка (108,74 мг, 1,66 ммоль, 10,00 экв.) при 10°C. Реакционную смесь перемешивали при 20°C в течение 1,5 часов, обнаружение показало образование промежуточного вещества. Добавляли уксусный ангидрид (2,00 мл) и реакционную смесь перемешивали при 20°C в течение 1 часа. После завершения реакции реакционную смесь фильтровали и фильтрационный осадок промывали дихлорметаном (30 мл). Фильтрат собирали, последовательно промывали водой (40 мл), насыщенным раствором карбоната натрия (30 мл) и насыщенным раствором хлорида натрия (30 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и дополнительно очищали с помощью препаративной ТСХ (ДХМ/EA=4/1) с получением целевого соединения WX036. 1H ЯМР (400 МГц, CDCl3-d) δ 11,26(s, 1 H), 8,08(m, 1 H), 7,65(m, 1 H), 7,49-7,43(m, 2 H), 7,15-7,13(m, 1 H), 7,03-7,01(m, 1 H), 6,78-3,74(m, 1 H), 2,94(s, 1 H), 2,30(s, 3 H), 2,05-2,03(m, 7 H), 1,62(s, 3 H), 1,36(m, 2 H), 0,97- 0,95(m, 2 H). MS m/z: 602,0 [M+H]+
[532] Соединения или промежуточные соединения примеров из следующей таблицы были синтезированы со ссылкой на способ синтеза стадий 1-2 варианта реализации 19. Структуры в следующей таблице также представляют их возможные изомеры.
[533] Таблица1. Структура соединений
Сузуки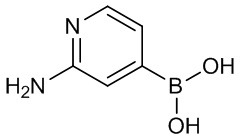
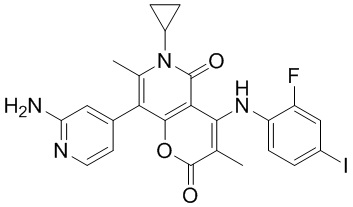
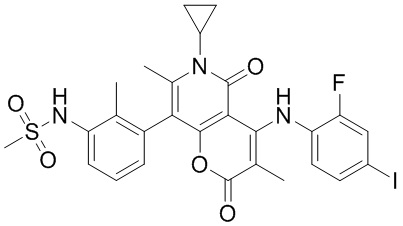
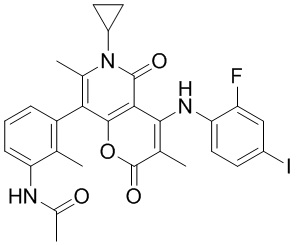
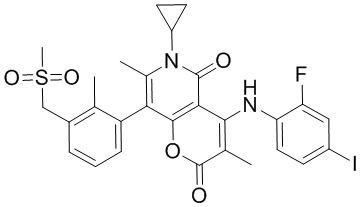
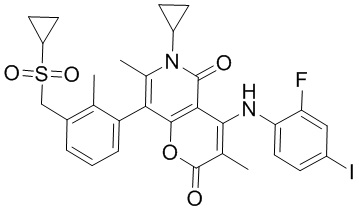
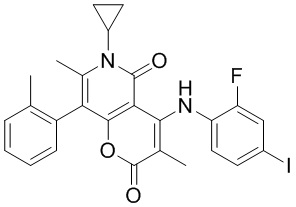
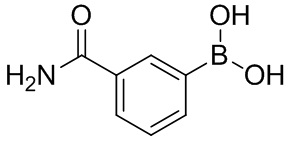
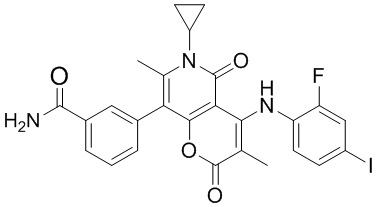
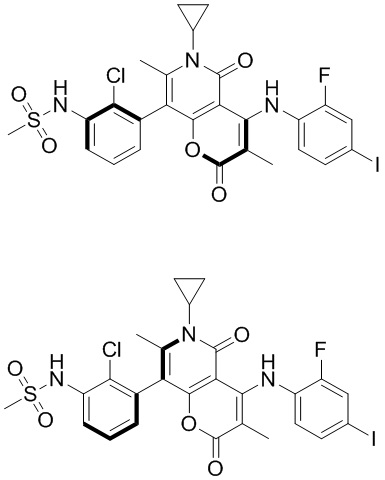
или
WX117
[534] Вариант реализации 36: WX115
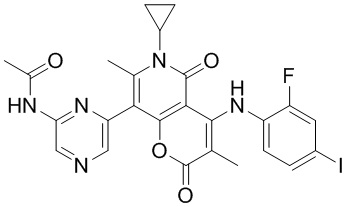
[535] Схема синтеза:
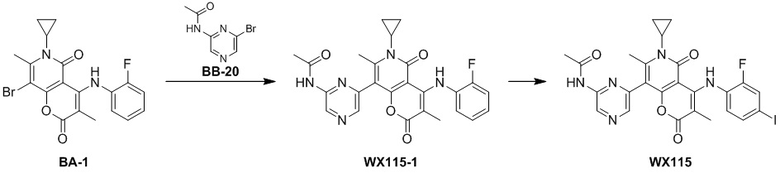
[536] Стадия 1: синтез соединения WX115-1
[537] Соединение BB-20 (420,00 мг, 1,94 ммоль, 1,00 экв.), Pd(PPh3)2Cl2 (272,91 мг, 388,82 мкмоль, 0,20 экв.), гексаметилдиолово (955,41 мг, 2,92 ммоль, 604,69 мкл, 1,50 экв.) добавляли в безводный толуол (12,00 мл). Температуру реакционной смеси повышали до 110°C в защитной атмосфере азота и реакционную смесь перемешивали в течение 20 часов. Затем добавляли соединение BA-1 (650,66 мг, 1,55 ммоль, 0,80 экв.) и диоксан (15,00 мл). В условиях защитной атмосферы азота температуру реакционной смеси повышали до 110°C и реакционную смесь перемешивали в течение 28 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры с последующим добавлением фторида калия (0,5 г). Смесь перемешивали в течение 30 минут, разбавляли дихлорметаном (50 мл) и фильтровали. Органическую фазу собирали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением соединения WX115-1. 1H ЯМР (400 МГц, CDCl3-d) δ 11,03(s, 1 H), 9,52(s, 1 H), 8,37(s, 1 H), 8,14(s, 1 H), 7,18-7,06(m, 3 H),7,18-6,98(m, 1 H), 3,02-2,92(m, 1 H), 2,37(s, 3 H), 2,27(s, 3 H), 1,58(s, 3 H), 1,44-1,34(m, 2 H), 1,00-0,92(m, 2 H). MS m/z: 587,1 [M+H]+
[538] Стадия 2: синтез соединения WX115
[539] Соединение WX115-1 (70,00 мг, 147,22 мкмоль, 1,00 экв.) растворяли в дихлорметане (1,00 мл), затем последовательно и партиями добавляли трифторуксусную кислоту (308,00 мг, 2,70 ммоль, 200,00 мкл, 18,35 экв.) и N-йодсукцинимид (33,12 мг, 147,22 мкмоль, 1,00 экв.) при 0°C. Температуру реакционной смеси повышали до 21°C и реакционную смесь перемешивали в течение 2 часов в темном месте. После завершения реакции реакционную смесь выпаривали досуха с помощью роторного испарителя, остаток разбавляли EtOAc (15 мл), последовательно промывали насыщенным раствором бикарбоната (10 мл*3), насыщенным раствором тиосульфата натрия (10 мл*2), водой (10 мл*2) и насыщенным раствором хлорида натрия (10 мл). Органическую фазу промывали безводным сульфатом натрия и фильтровали, фильтрат выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и сушили с получением целевого соединения WX115. 1H ЯМР (400 МГц, CDCl3-d) δ 10,98(s, 1 H), 9,58(s, 1 H), 8,40(s, 1 H), 7,88(s, 1 H), 7,50-7,43(m, 2 H),6,72(t, J=8,6 Гц, 1 H), 2,99-2,95(m, 1 H), 2,42(s, 3 H), 2,30(s, 3 H), 1,62(s, 3 H), 1,45-1,39(m, 2 H), 1,02-0,95(m, 2 H). MS m/z: 601,8 [M+H]+
[540] Вариант реализации 37: WX113 & WX114
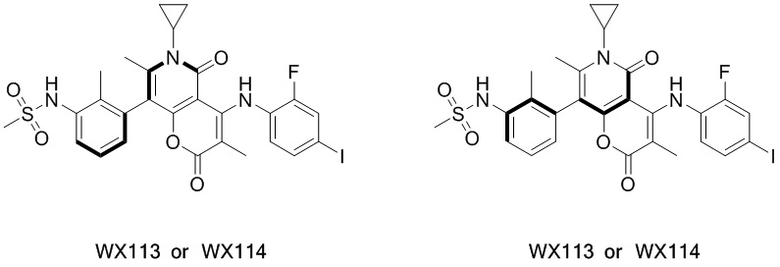
[541] Схема синтеза:
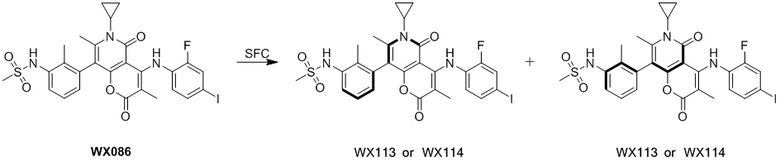
[542] Стадия 1: синтез соединения WX113 и соединение WX114
[543] Соединение WX086 подвергали сверхкритической жидкостной хроматографии (колонка: Chiralpak OJ-3 100 × 4,6 мм ID, 3 мкм; подвижная фаза: A: CO2 B: метанол (0,05% DEA); градиент: от 5% B до 40% B при постоянной скорости за 4,5 минуты, 5% B в течение 2,5 минут, 5% B в течение 1 минуты. Скорость потока: 2,8 мл/мин; температура колонки: 40°C), с получением атропоизомеров WX113 и WX114, с временем удерживания 4,503 мин и 4,166 мин, соответственно, и соотношением 1:1.
[544] Соединение WX113 1H ЯМР (400 МГц, ДМСО-d6) δ 11,31(s, 1 H), 9,22(s, 1 H), 7,73(dd, J=10,0 Гц, J=1,6 Гц, 1 H), 7,53(d, J=8,4 Гц, 1 H), 7,43-7,38(m, 1 H), 7,36-7,30(m, 1 H), 7,12(d, J=7,6 Гц, 1 H), 6,89(t, J=8,0 Гц, 1 H), 3,08-3,04(m, 1 H), 3,02(s, 3 H), 2,24(s, 3 H), 2,07(s, 3 H), 1,45(s, 3 H), 1,27-1,16(m, 2 H), 1,00-0,86(m, 2 H). 650,0 [M+H]+
[545] Соединение WX114 1H ЯМР (400 МГц, ДМСО-d6) δ 11,31(s, 1 H), 9,22(s, 1 H), 7,73(dd, J=10,4 Гц, J=2,0 Гц, 1 H), 7,53(d, J=8,8 Гц, 1 H), 7,43-7,38(m, 1 H), 7,36-7,30(m, 1 H), 7,12(d, J=7,6 Гц, 1 H), 6,89(t, J=8,4 Гц, 1 H), 3,08-3,04(m, 1 H), 3,02(s, 3 H), 2,24(s, 3 H), 2,07(s, 3 H), 1,45(s, 3 H), 1,27-1,15(m, 2 H), 1,01-0,85(m, 2 H). 650,0 [M+H]+
[546] Вариант реализации 38: WX088
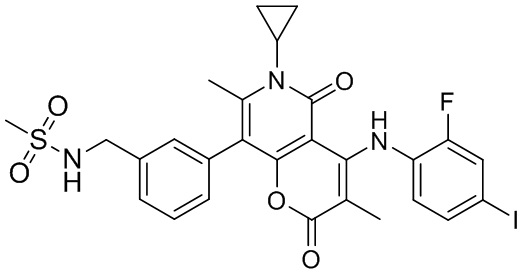
[547] Схема синтеза:
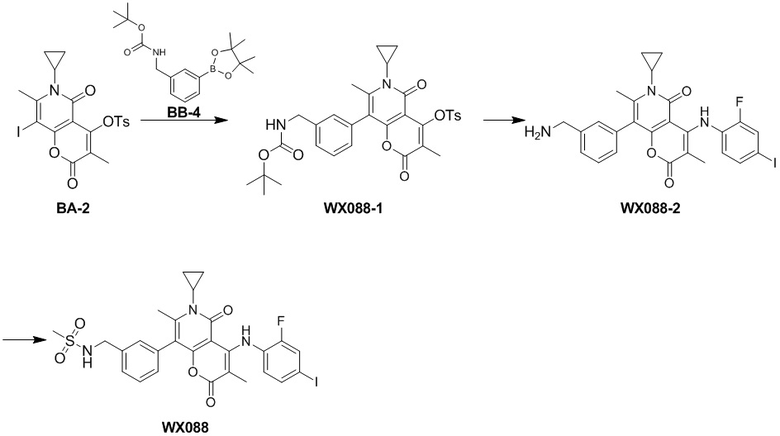
[548] Стадия 1: синтез соединения WX088-1
[549] Соединение BA-2 (5,06 г, 9,60ммоль, 0,80 экв.) и соединение BB-4 (4,00 г, 12,00 ммоль, 1,00 экв.) растворяли в диоксане (80,00 мл), с последующим добавлением Pd(dppf)Cl2.CH2Cl2 (979,97 мг, 1,20 ммоль, 0,10 экв.) и насыщенного раствора бикарбоната натрия (40,00 мл). В условиях защитной атмосферы азота температуру реакционной смеси повышали до 60°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционный раствор фильтровали через диатомовую землю и фильтрат выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=5/1-1/1) с получением соединения WX088-1. 1H ЯМР (400 МГц, CDCl3-d) δ 7,99 -7,94(s, 4 H), 7,41-7,40(m, 3 H), 7,13(m, 1 H), 6,07-6,05(m, 1 H), 4,38-4,37(d, J=6,00, 2 H), 2,86-2,82(m, 1 H), 2,53(m, 3 H), 2,33(m, 3 H), 1,67(s, 3 H), 1,50-1,47(m, 9 H), 0,95-0,90(m, 2 H), 0,87- 0,84(m, 2 H). MS m/z: 551,1 [M+H]+
[550] Стадия 2: синтез соединения WX088-2
[551] Соединение WX088-1 (600,00 мг, 988,99 мкмоль, 1,00 экв.) растворяли в этаноле (10,00 мл), затем последовательно добавляли 2-фтор-4-йоданилин (1,17 г, 4,94 ммоль, 5,00 экв.) в защитной атмосфере азота. Температуру реакционной смеси повышали до 80°C и реакционную смесь перемешивали в течение 12 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=5/1-1/1) с получением соединения WX088-2. 1H ЯМР (400 МГц, CDCl3-d) δ 10,41(s, 1 H), 8,08(m, 2 H), 7,47(m, 1 H), 7,45-7,35(m, 3 H), 7,25(m, 1 H), 7,13(m, 1 H), 6,72-6,67(m, 1 H), 4,00-3,95(m, 2 H), 3,47(s, 3 H), 2,82-2,81(m, 1 H), 2,22(s, 3 H), 1,25-1,22(m, 2 H), 0,90-0,83(m, 2 H). MS m/z: 572,0 [M+H]+
[552] Стадия 3: синтез соединения WX088
[553] Соединение WX088-2 (600,00 мг, 988,99 мкмоль, 1,00 экв.) растворяли в N,N-диметилформамиде (4,00 мл), затем последовательно добавляли триэтиламин (70,84 мг, 700,06 мкмоль, 2,00 экв.) и метансульфонилхлорид (48,12 мг, 420,04 мкмоль, 1,20 экв.) при 0°C. Температуру реакционной смеси повышали до 25°C и реакционную смесь перемешивали в течение 1 часа. После завершения реакции реакционную смесь разбавляли водой (20,00 мл) и экстрагировали дихлорметаном (20 мл*2). Органическую фазу промывали насыщенным раствором хлорида натрия (20 мл), сушили над безводным сульфатом натрия и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX088. 1H ЯМР (400 МГц, CDCl3-d) δ 11,13(s, 1 H), 7,49 -7,38(m, 5 H), 7,18-7,15(m, 1 H), 6,73-6,68(m, 1 H), 4,96-4,93(m, 1 H), 4,39-4,36(m, 2 H), 2,96-2,92(m, 4 H), 1,58(s, 3 H), 1,38-1,36(m, 2 H), 0,97(m, 2 H). MS m/z: 650,5 [M+H]+
[554] Со ссылкой на способ синтеза стадий 1-2 варианта реализации 38 были синтезированы варианты реализации из следующей таблицы. Структуры в указанной таблице также представляют их возможные изомеры.
[555] Замечание: Все условия реакции в данной таблице были следующими (основание: бикарбонат натрия, растворитель: ДМФА).
[556] Таблица 2: Структуры соединений
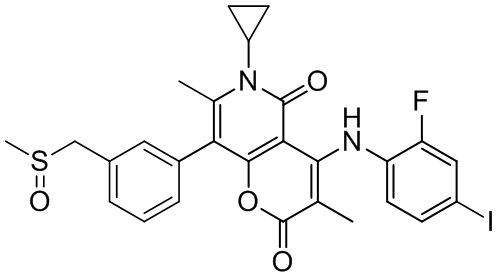
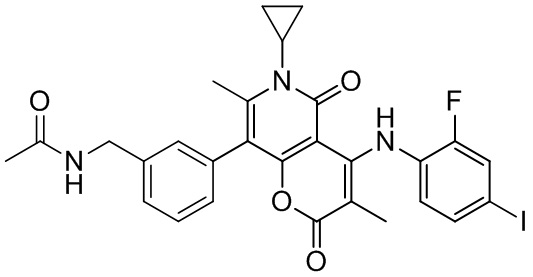
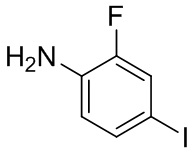
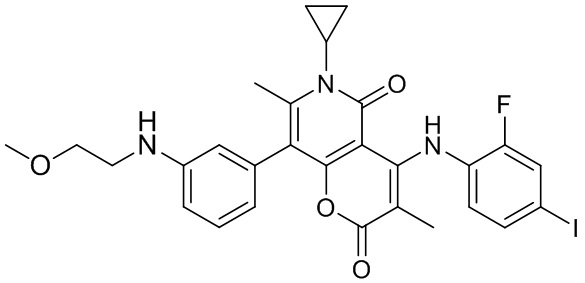
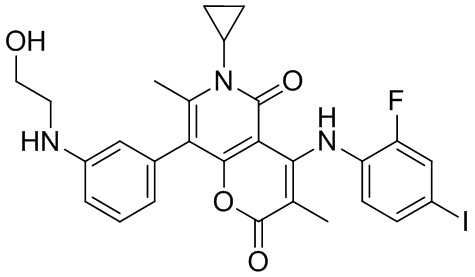
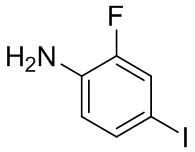
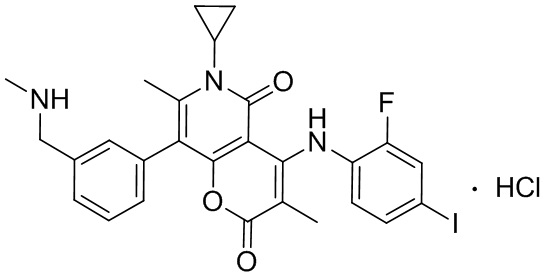
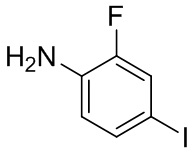
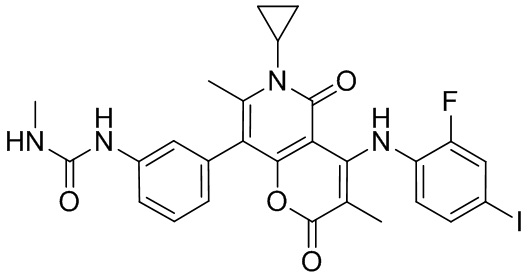
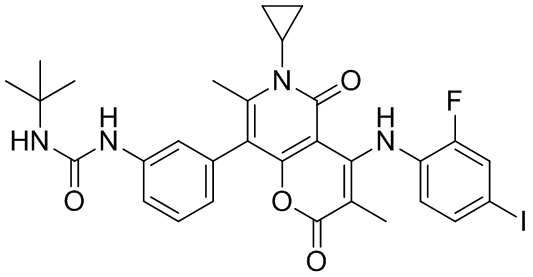
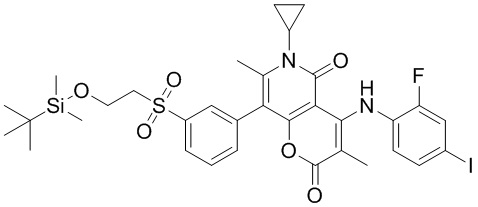
[557] Вариант реализации 46: WX098
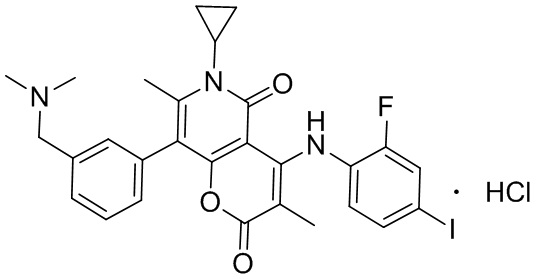
[558] Схема синтеза:
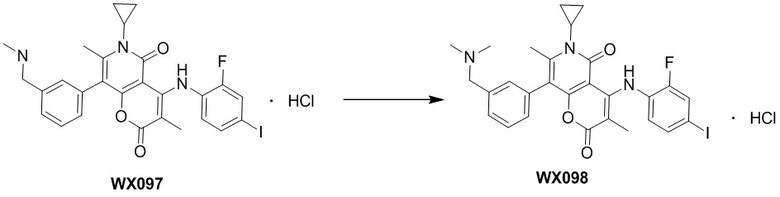
[559] Стадия 1: синтез соединения WX098
[560] В защитной атмосфере азота, формальдегид (130,83 мг, 871,36 мкмоль, 120,03 мкл, 20% чистота, 5,00 экв.), уксусную кислоту (10,46 мг, 174,27 мкмоль, 9,97 мкл, 1,00 экв.) и триацетоксиборгидрид натрия (73,87 мг, 348,54 мкмоль, 2,00 экв.) последовательно добавляли к раствору соединения WX097 (120 мг, 174 мкмоль, 1,00 экв.) в 1,2-дихлорэтане (2мл) при 0°C. Реакционный раствор перемешивали при 20°C в течение 12 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта, который очищали с помощью препаративной ВЭЖХ с получением соединения WX098.
[561] 1H ЯМР (400 МГц, ДМСО-d6) δ 11,25(s, 1 H), 10,16(br. s., 1 H), 7,72(dd, J=9,8 Гц, J=2,0 Гц, 1 H), 7,62-7,56(m, 2 H), 7,55-7,49(m, 2 H), 7,48-7,43(m, 1 H), 6,85(t, J=8,6 Гц, 1 H), 4,37-4,24(m, 2 H), 3,10-3,02(m, 1 H), 2,76(d, J=4,4 Гц, 3 H), 2,71(d, J=4,8 Гц, 3 H), 2,37(s, 3 H), 1,44(s, 3 H), 1,26-1,16(m, 2 H), 0,98-0,84(m, 2 H). MS m/z: 600 [M+H]+
[562] Вариант реализации 47: WX101
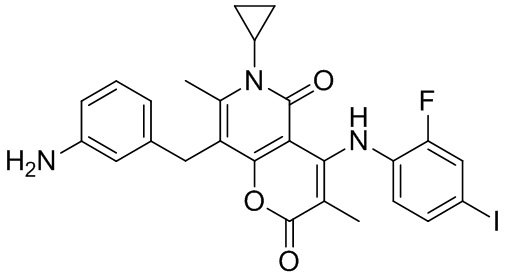
[563] Схема синтеза:
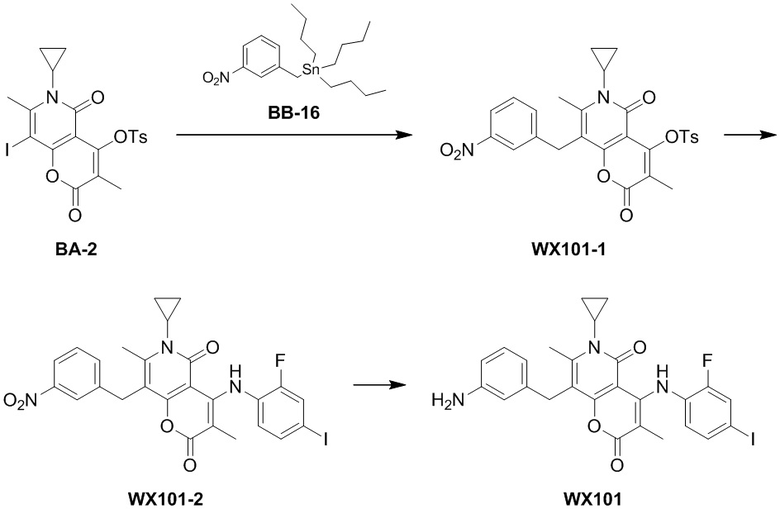
[564] Стадия 1: синтез соединения WX101-1
[565] Соединение BA-2 (2,00 г, 3,79 ммоль, 1,00 экв.) и соединение BB-16 (2,42 г, 5,69 ммоль, 1,50 экв.) растворяли в N,N-диметилформамиде (10,00 мл) с последующим добавлением Pd(PPh3)4 (438,27 мг, 379,27 мкмоль, 0,10 экв.) и фторидк цезия (576,11 мг, 3,79 ммоль, 1,00 экв.) и йодида меди (361,16 мг, 1,90 ммоль, 0,50 экв.). В условиях защитной атмосферы азота температуру реакционной смеси повышали до 60°C и реакционную смесь перемешивали в течение 2 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=10/1-1/1) с получением соединения WX101-1. MS m/z: 537,1 [M+H]+
[566] Стадия 2: синтез соединения WX101-2
[567] Соединение WX101-1 (1,50 г, 749,73 мкмоль, 1,00 экв.) суспендировали в этаноле (10,00 мл), последовательно добавляли 2-фтор-4-йоданилин (4,64 г, 19,58 ммоль, 26,11 экв.) в защитной атмосфере азота. Температуру реакционной смеси повышали до 80°C и реакционную смесь перемешивали в течение 16 часов. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (PE/EA=10/1-1/1) с получением соединения WX101-2. MS m/z: 602,2 [M+H]+
[568] Стадия 3: синтез соединения WX101
[569] Соединение WX101-2 (210,00 мг, 182,98 мкмоль, 1,00 экв.) суспендировали в этаноле (4,00 мл) и воду (2,00 мл), затем последовательно добавляли порошок железа (195,03 мг, 3,49 ммоль, 19,08 экв.) и хлорид аммония (186,79 мг, 3,49 ммоль, 19,08 экв.). В условиях защитной атмосферы азота температуру реакционной смеси повышали до 70°C и реакционную смесь перемешивали в течение 1 часа. После завершения реакции реакционный раствор выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ТСХ (PE/EA=1/1) и дополнительно очищали с помощью препаративной ВЭЖХ с получением целевого соединения WX101. 1H ЯМР (400 МГц, CDCl3-d) δ 11,20(s, 1 H), 7,46(dd, J=9,6 Гц, J=2,0 Гц, 1 H), 7,41(d, J=8,4 Гц, 1 H), 7,05(t, J=7,6 Гц, 1 H), 6,71(t, J=7,6 Гц, 1 H), 6,58-6,49(m, 3 H), 3,98(s, 2 H), 2,91-2,86(m, 1 H), 2,55(s, 3 H), 1,63(s, 3 H), 1,36-1,30(m, 2 H), 0,90-0,84(m, 2 H). MS m/z: 572,2 [M+H]+
[570] Вариант реализации 48: WX091&WX095
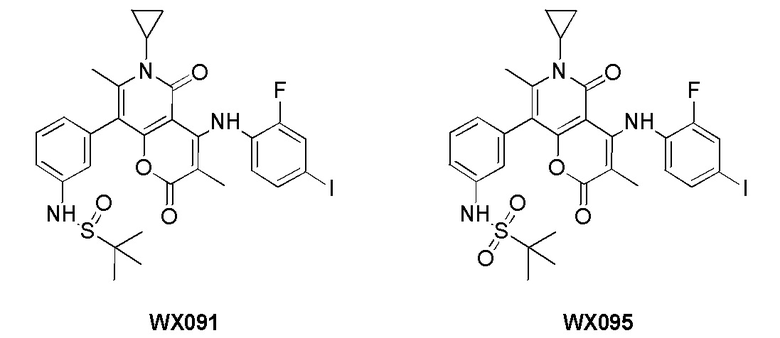
[571] Схема синтеза:
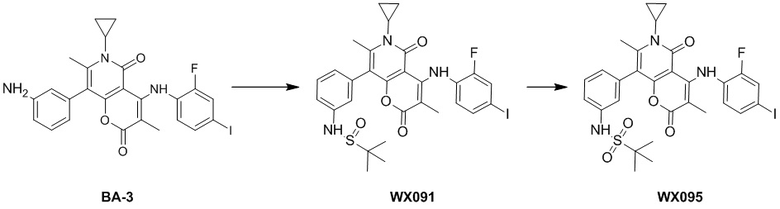
[572] Стадия 1: синтез соединения WX091
[573] Соединение BA-3 (300,00 мг, 538,26 мкмоль, 1,00 экв.) растворяли в дихлорметане (4,00 мл) с последующим добавлением триэтиламина (108,93 мг, 1,08 мкмоль, 2,00 экв.), DMAP (131,52 мг, 1,08 мкмоль, 2,00 экв.) и соединение трет-бутилсульфонилхлорида (90,83 мг, 645,91 мкмоль, 1,20 экв.) последовательно добавляли при 0°C. Реакционную смесь перемешивали при 0°C в течение 15 минут. После завершения реакции реакционную смесь добавляли в воду (30 мл) и экстрагировали дихлорметаном (60 мл*3). Органические фазы объединяли и промывали насыщенным раствором хлорида натрия (40 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ТСХ (PE/EA=1/2) с получением соединения WX091. MS m/z: 662,0 [M+H]+
[574] Стадия 2: синтез соединения WX095
[575] Соединение WX091 (47,00 мг, 71,05 мкмоль, 1,00 экв.) растворяли в ТГФ (2,00 мл) и воде (2,00 мл), затем добавляли гидросульфат калия (174,71 мг, 142,10 мкмоль, 2,00 экв.) при 0°C в защитной атмосфере азота. Реакционную смесь перемешивали при 20°C в течение 6 часов. Реакционную смесь разбавляли водой (20 мл) и водную фазу экстрагировали дихлорметаном (20 мл*3). Органическую фазу собирали и промывали насыщенным раствором хлорида натрия (10 мл), сушили над безводным сульфатом натрия, фильтровали и выпаривали досуха с помощью роторного испарителя с получением неочищенного продукта. Неочищенный продукт очищали с помощью препаративной ВЭЖХ и лиофилизировали с получением целевого соединения WX095. 1H ЯМР (400 МГц, CDCl3-d) δ 11,14(s, 1 H), 7,49 -7,42(m, 1 H), 7. 38-7,36(m, 1 H), 6,99-6,97(m, 1 H), 6,75-6,72(m, 1 H), 2,96-2,94(m, 1 H), 2,40(s, 3 H), 1,61(s, 3 H), 1,45(m, 9 H), 1,39-1,37(m, 2 H), 0,99(m, 2 H). MS m/z: 677,9 [M+H]+
[576] Со ссылкой на способ синтеза стадий 1-2 варианта реализации 48 были синтезированы варианты реализации из следующей таблицы. Структуры в указанной таблице также представляют их возможные изомеры.
[577] Таблица 3: Структуры соединений
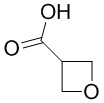
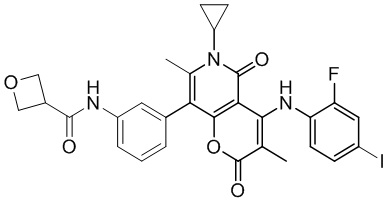
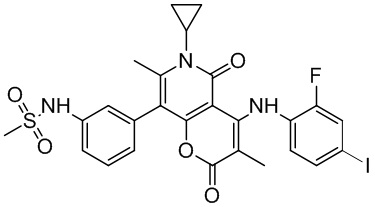
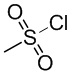
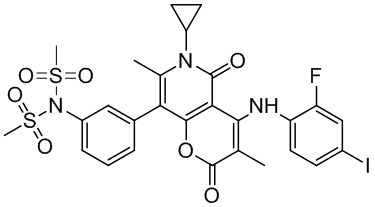
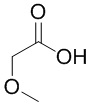
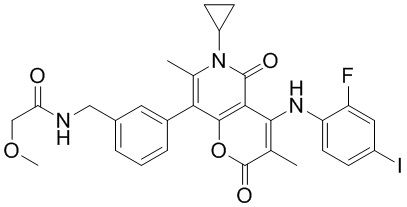
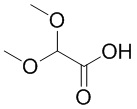
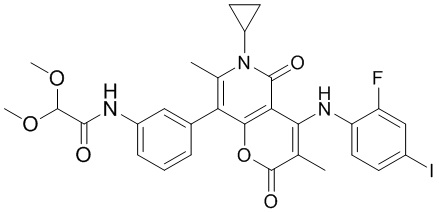
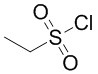
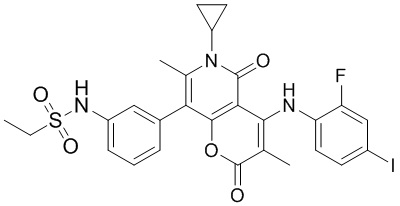
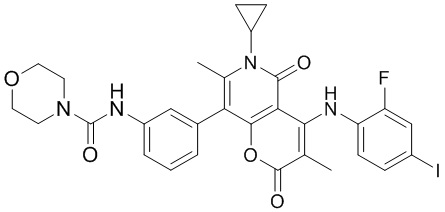
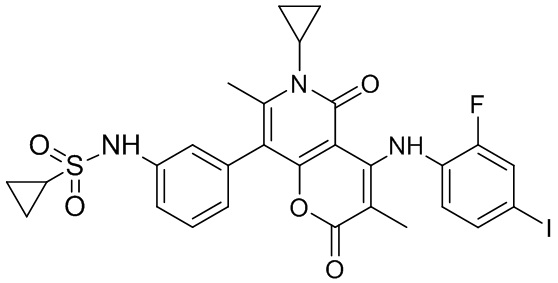
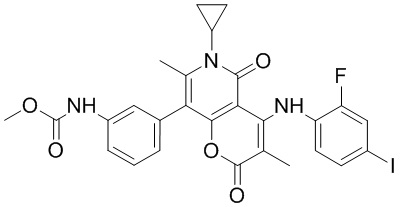
[578] Таблица 4: Данные по ЯМР и MS указанных вариантов реализации
WX119
WX119: 1H ЯМР (400 МГц, ДМСО-d6) δ 11,35-11,26(m, 1 H), 7,75-7,68(m, 1 H), 7,59-7,25(m, 3 H), 7,10(m, 1 H), 6,89-6,95(m, 1 H), 3,00(s, 3 H), 2,06(s, 3 H), 1,99(s, 3 H), 1,43(s, 3 H) способ определения SFC: колонка: Chiralpak AD-3 100 × 4,6 мм I.D., 3 мкм; подвижная фаза: A: CO2 B: метанол (0,05% DEA); градиент: от 5%B до 40% B при постоянной скорости за 4,5 минуты, затем 40% B в течение 2,5 минут, 5%B в течение 1 минуты; скорость потока:2,8 мл/мин, температура колонки: oC. Время удерживания соединения WX118 и WX119 составило 5,934 мин и 4,958 мин, соответственно, соотношение было 6:7.
610,0 [M+H]+
WX084
WX084:1H ЯМР (400 МГц, CDCl3-d) δ 11,08(s, 1 H), 7,49-7,72(m, 2 H), 7,40-7,36(m, 2 H), 7,26-7,19(m, 2 H), 6,72-6,68(s, 1 H), 4,16-3,93(m, 2 H), 2,93(m, 1 H), 2,48(s, 3 H), 2,40(s, 3 H), 1,63-1,59(s, 2 H), 1,38-1,30(m, 2 H), 1,29-1,20(m, 1 H), 0,96(m, 2 H). способ определения SFC: колонка: Chiralpak OD-3 150 × 4,6 мм I.D., 3 мкм; подвижная фаза: A:CO2 B:метанол (0,05% DEA); градиент: от 5% B до 40% B при постоянной скорости за 4,5 минуты, 40% B в течение 2,5 минут, 5% B в течение 1 минуты; скорость потока:2,4 мл/мин; температура колонки:40oC. Время удерживания соединения WX083 и WX084 составило 5,88 мин и 6,79 мин, соответственно, соотношение было 1:1.
619,1 [M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
WX117
1H ЯМР (400 МГц, CDCl3-d) δ 7,99 -7,94(s, 4 H), 7,41-7,40(m, 3 H), 7,13(m, 1 H), 6,07-6,05(m, 1 H), 4,38-4,37(d, J=6,00, 2 H), 2,86-2,82(m, 1 H), 2,53(m, 3 H), 2,33(m, 3 H), 1,67(s, 3 H), 1,50-1,47(m, 9 H), 0,95-0,90(m, 2 H), 0,87- 0,84(m, 2 H).
WX117:1H ЯМР (400 МГц, CDCl3-d) δ 10,97(s, 1 H), 7,69-7,67(d, 1 H), 7,38-7,33(m, 3 H), 7,02-7,00(d, 1 H), 6,89-6,83(br.s, 1 H), 6,66-6,64(t, J=6,00, 1 H), 3,05-3,02(s, 3 H)2,90-2,84(m, 1 H), 2,26(s, 3 H), 1,56-1,52(s, 3 H), 1,32- 1,29(m, 2 H), 0,90-0,79(m, 2 H). способ определения SFC: колонка Chiral: AS(250мм*30мм,5мкм); подвижная фаза :[0,1%NH3H2O EtOH];B%: 40%-40%, мин. Время удерживания соединения WX116 и WX117 составило 4,522 мин и 4,166 мин, соответственно, соотношение было 1:1.
[M+H]+
669,9
[M+H]+
[M+H]+
WX114
WX114:1H ЯМР (400 МГц, ДМСО-d6) δ 11,31(s, 1 H), 9,22(s, 1 H), 7,73(dd, J=10,4 Гц, J=2,0 Гц, 1 H), 7,53(d, J=8,8 Гц, 1 H), 7,43-7,38(m, 1 H), 7,36-7,30(m, 1 H), 7,12(d, J=7,6 Гц, 1 H), 6,89(t, J=8,4 Гц, 1 H), 3,08-3,04(m, 1 H), 3,02(s, 3 H), 2,24(s, 3 H), 2,07(s, 3 H), 1,45(s, 3 H), 1,27-1,15(m, 2 H), 1,01-0,85(m, 2 H). способ определения SFC:колонка: Chiralpak OJ-3 100 × 4,6 мм I.D., 3 мкм; подвижная фаза: A:CO2 B:метанол (0,05% DEA); градиент: от 5%B до 40%B при постоянной скорости за 4,5 минуты, 40%B в течение 2,5 минут, 5% B в течение 1 минуты; скорость потока: 2,8 мл/мин; температура колонки:40oC. Время удерживания соединения WX113 и WX114 составило 4,503 мин и 4,166 мин, соответственно, соотношение было 1:1.
[M+H]+
650,0
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+Na-HCl]+
[M+H]+
[M+H]+
[M+H-HCl]+
[M+H]+
WX095
1H ЯМР (400 МГц, CDCl3-d) δ 11,14(s, 1 H), 7,49 -7,42(m, 1 H), 7. 38-7,36(m, 1 H), 6,99-6,97(m, 1 H), 6,75-6,72(m, 1 H), 2,96-2,94(m, 1 H), 2,40(s, 3 H), 1,61(s, 3 H), 1,45(m, 9 H), 1,39-1,37(m, 2 H), 0,99(m, 2 H).
[M+H]+
677,9 [M+H]+
[M+Na]+
[M+Na]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[M+H]+
[579] Биологический эксперимент
[580] Метод биологического испытания 1: эксперимент MEK Lance Ultra
[581] Соединения серийно разводили в три раза до достижения десяти концентраций с двумя повторностями. Конечные исследуемые концентрации соединений составляли от 10 мкМ до 0,51 нМ. 0,07 нМ активированного MEK1 (Millipore # 14-429) и 2 нМ неактивированного ERK (Millipore # 14-515) смешивали с соединениями или ДМСО, смесь инкубировали при 23°C в течение 30 минут. Затем для инициирования реакции добавляли 50 нМ меченного ULight MBP (PerkinElmer # TRF0109-M) и 50 мкМ ATP (Invitrogen # PV3227) и смесь инкубировали при 23°C в течение 90 минут. После того как реакцию останавливали путем добавления ЭДТА в конечной концентрации 15 мМ, добавляли 2 нМ Eu-меченного антифосфорилированного антитела (PerkinElmer # TRF0201-M) и инкубировали в течение 1 часа. Данные сигнала флуоресценции (полоса возбуждения: 320 нм; полоса испускания: 665 нМ/615 нМ) определяли с помощью микропланшет-ридера Envision (PerkinElmer). Программное обеспечение XLfit5 использовали для анализа данных и составления диаграмм. Результаты эксперимента приведены в таблице 5.
[582] Метод биологического исследования 2: эксперимент по жизнеспособности клеток
[583] Клетки HT29 и A375 высевали в 96-луночный планшет для культивирования клеток с плотностью 40000 клеток/лунка и 20000 клеток/лунка, соответственно, и клетки культивировали в течение ночи. Соединения серийно разводили с градиентом 1:3, разведения добавляли в среду для культивирования клеток и инкубировали с клетками в инкубаторе при 37°С в течение 3 дней. 96-луночный планшет для культивирования клеток брали из инкубатора и уравновешивали при комнатной температуре в течение 30 минут, затем добавляли реагент Cell Titer-Glo (Promega Cat # G7573) в соотношении 1:2, и смесь тщательно перемешивали на шейкере в течение 2 минут, чтобы способствовать лизису клеток. Планшет для культивирования клеток инкубировали при комнатной температуре в течение 10 минут и затем считывали на микропланшет-ридере Envision (PerkinElmer). Программное обеспечение XLfit5 использовали для анализа данных и составления диаграмм. Результаты эксперимента приведены в таблице 5.
[584] Таблица 5: Результаты скрининг-теста in vitro для соединений согласно настоящему описанию
MEK
IC50 (нМ)
IC50 (нМ)
IC50 (нМ)
[585] Вывод: Данные биологических исследований демонстрируют, что все соединения настоящего описания обладают хорошей биологической активностью в отношении MEK и ингибирующей активностью в отношении роста опухолевых клеток.
[586] Метод биологического исследования 3: исследование фармакодинамики in vivo на модели опухоли путем подкожной ксенотрансплантации клеток НТ-29 у бестимусных мышей BALB/c
[587] Клетки HT-29 рака толстой кишки человека (ATCC-HTB-38) культивировали в монослое in vitro со средой McCoy 5a (Gibco, 1835937), дополненной 10% фетальной бычьей сывороткой, 100 ед/мл пенициллина и 100 мкг/мл стрептомицина, при 37°С в атмосфере 5% CO2. Клетки традиционным образом разделяли трипсином-ЭДТА и пересевали два раза в неделю. Когда насыщение клеток составляло 80-90%, клетки собирали, подсчитывали и инокулировали. 0,1 мл (5×106) клеток НТ-29 инокулировали подкожно в правую часть спины каждой бестимусной мыши. Когда средний объем опухоли достигал 100-180 мм3, животных группировали и начинали введение (QD, 14-21 день). Диаметр опухоли измеряли два раза в неделю с помощью штангенциркуля. Объем опухоли рассчитывали по формуле: V = 0.5a × b2, где a и b представляют длину и ширину, соответственно. Противоопухолевую активность соединений оценивали по TGI (%), который отражает степень ингибирования роста опухоли. Расчет TGI (%): TGI (%) = [(1 - (средний объем опухоли по окончании введения в группе лечения - средний объем опухоли в начале лечения в этой группе лечения))/(средний объем по окончании лечения в контрольной группе с растворителем - средний объем опухоли в начале лечения в контрольной группе с растворителем)] × 100%. Результаты эксперимента приведены в таблице 6..
[588] Таблица 6: Результаты исследования фармакодинамики:
0,35 мг/кг (12-21Д)
(5 дней введения и 2 дня перерыва)
[589] Вывод: Соединения согласно настоящему описанию обладают хорошей ингибирующей активностью в отношении роста опухоли.
[590] Метод биологического исследования 4. Фармакокинетические исследования соединений у мышей C57B /6.
[591] Экспериментальные материалы:
[592] Мыши C57BL/6 (самцы, 18-22 г, возраст 7-9 недель, Shanghai Lingchang)
[593] Процедура эксперимента:
[594] Фармакокинетические характеристики соединений у грызунов определяли после внутривенного и перорального введения соединений по стандартным протоколам. В эксперименте соединения-кандидаты были получены в виде прозрачных растворов и их внутривенно и перорально вводили крысам в однократной дозе. Растворитель для внутривенного и перорального введения представлял собой ДМСО, ПЭГ и воду в определенном соотношении или водные растворы Solutol, HPMC и SLS в определенном соотношении. Образцы цельной крови собирали в течение 24 часов, центрифугировали при 3000 об/мин в течение 15 минут и супернатанты выделяли для получения образцов плазмы. Раствор ацетонитрила, содержащий внутренний стандарт с объемом в 4 раза больше объема пробы плазмы, добавляли для осаждения белка. После центрифугирования супернатант собирали и добавляли равный объем воды. После центрифугирования супернатант собирали и вводили для количественного анализа концентрации лекарственного средства в крови методом ЖХ-МС/МС, и рассчитывали фармакокинетические параметры, например, пиковую концентрацию, время пика, клиренс, период полувыведения, площадь под кривой концентрация лекарственного средства-время, биодоступность и т. д.
[595] Экспериментальные результаты:
[596] Таблица 7: Результаты фармакокинетического исследования
выведения T1/2 (ч)
ППК (нМ.ч)
[597] Вывод. Соединения согласно настоящему описанию имеют хорошие фармакокинетические показатели у крыс.
| название | год | авторы | номер документа |
|---|---|---|---|
| Ингибитор CDK4/6 | 2017 |
|
RU2747311C2 |
| ПРОИЗВОДНОЕ С КОНДЕНСИРОВАННЫМ КОЛЬЦОМ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРА A | 2018 |
|
RU2748993C1 |
| ТИЕНОДИАЗЕПИНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРИМЕНЕНИЕ | 2018 |
|
RU2795005C2 |
| ЗАМЕЩЁННЫЕ ИНДОЛЬНЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ ПОНИЖАЮЩИХ РЕГУЛЯТОРОВ РЕЦЕПТОРОВ ЭСТРОГЕНА | 2017 |
|
RU2722441C2 |
| ТИОФЕНОВОЕ СОЕДИНЕНИЕ, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО ФАРМАЦЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2017 |
|
RU2709473C1 |
| АНАЛОГИ ЭХИНОКАНДИНА И СПОСОБ ИХ ПОЛУЧЕНИЯ | 2020 |
|
RU2832679C1 |
| АНАЛОГ БЕНЗОФУРАНА В КАЧЕСТВЕ ИНГИБИТОРА NS4B | 2015 |
|
RU2672257C2 |
| ПРОИЗВОДНЫЕ ПИРРОЛИДИНА В КАЧЕСТВЕ АГОНИСТОВ PPAR | 2017 |
|
RU2711991C1 |
| Тиазололактамные соединения в качестве ингибиторов ERK и их применение | 2020 |
|
RU2805569C1 |
| ТРИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ ЯНУС-КИНАЗЫ 1, ИХ КОМПОЗИЦИИ, СПОСОБЫ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ | 2019 |
|
RU2824349C2 |
Изобретение относится к соединению, представленному формулой (I), его фармацевтически приемлемой соли или его таутомеру. В формуле (I) n выбран из 0, 1 или 2; r выбран из 0, 1, 2 или 3; m выбран из 0 или 1; когда m представляет собой 0, тогда 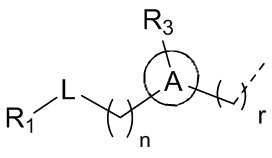 представляет собой H; кольцо A выбрано из фенила или 6-членного гетероарила; L выбран из одинарной связи, -S(=O)-, -S(=O)2-, -C(=O)-, -NH-, -NH-C(=O)-, -NH-C(=O)-O-, -NH-S(=O)2-, -NH-S(=O)-, -NH-C(=O)-NH- и
представляет собой H; кольцо A выбрано из фенила или 6-членного гетероарила; L выбран из одинарной связи, -S(=O)-, -S(=O)2-, -C(=O)-, -NH-, -NH-C(=O)-, -NH-C(=O)-O-, -NH-S(=O)2-, -NH-S(=O)-, -NH-C(=O)-NH- и  , где -NH-S(=O)2- необязательно замещен 1 R; R1 выбран из H, NH2, C1-6 алкила, 4-6-членного гетероциклоалкила, C3-6 циклоалкила и C1-3 гетероалкила, где каждый C1-6 алкил и C1-3 гетероалкил необязательно замещен 1, 2 R; R2 выбран из H, C1-6 алкила, C3-6 циклоалкила и 6-членного гетероциклоалкила, где C1-6 алкил необязательно замещен 1, 2 или 3 R; R3 выбран из H, F, Cl, Br, I, C1-3 алкила; R4 и R5 независимо выбраны из C1-6 алкила; R6 и R7 независимо выбраны из F, Cl, Br, I; R выбран из OH, NH2 и C1-3 гетероалкила, где NH2 необязательно замещен 1 R'; R' выбран из C1-3 алкила; каждый из «гетеро» в 6-членном гетероариле, 4-6-членном гетероциклоалкиле, C1-3 гетероалкиле независимо выбран из -NH-, N, -O-, -S(=O)2-, -S(=O)2-NH-, - C(=O)-NH-, -C(=O)-; в любом из вышеперечисленных случаев количество гетероатомов и гетероатомных групп независимо выбрано из 1, 2. Изобретение также относится к индивидуальным соединениям, к фамацевтической композиции и к применению соединения. Технический результат: получены новые соединения формулы (I), обладающие свойствами ингибитора MEK. 4 н. и 21 з.п. ф-лы, 7 табл., 83 пр.
, где -NH-S(=O)2- необязательно замещен 1 R; R1 выбран из H, NH2, C1-6 алкила, 4-6-членного гетероциклоалкила, C3-6 циклоалкила и C1-3 гетероалкила, где каждый C1-6 алкил и C1-3 гетероалкил необязательно замещен 1, 2 R; R2 выбран из H, C1-6 алкила, C3-6 циклоалкила и 6-членного гетероциклоалкила, где C1-6 алкил необязательно замещен 1, 2 или 3 R; R3 выбран из H, F, Cl, Br, I, C1-3 алкила; R4 и R5 независимо выбраны из C1-6 алкила; R6 и R7 независимо выбраны из F, Cl, Br, I; R выбран из OH, NH2 и C1-3 гетероалкила, где NH2 необязательно замещен 1 R'; R' выбран из C1-3 алкила; каждый из «гетеро» в 6-членном гетероариле, 4-6-членном гетероциклоалкиле, C1-3 гетероалкиле независимо выбран из -NH-, N, -O-, -S(=O)2-, -S(=O)2-NH-, - C(=O)-NH-, -C(=O)-; в любом из вышеперечисленных случаев количество гетероатомов и гетероатомных групп независимо выбрано из 1, 2. Изобретение также относится к индивидуальным соединениям, к фамацевтической композиции и к применению соединения. Технический результат: получены новые соединения формулы (I), обладающие свойствами ингибитора MEK. 4 н. и 21 з.п. ф-лы, 7 табл., 83 пр.
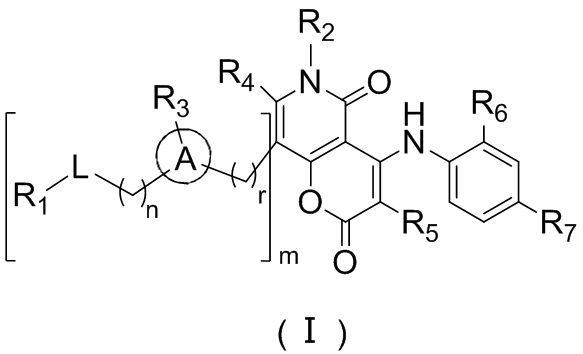
1. Соединение, представленное формулой (I), его фармацевтически приемлемая соль или его таутомер:
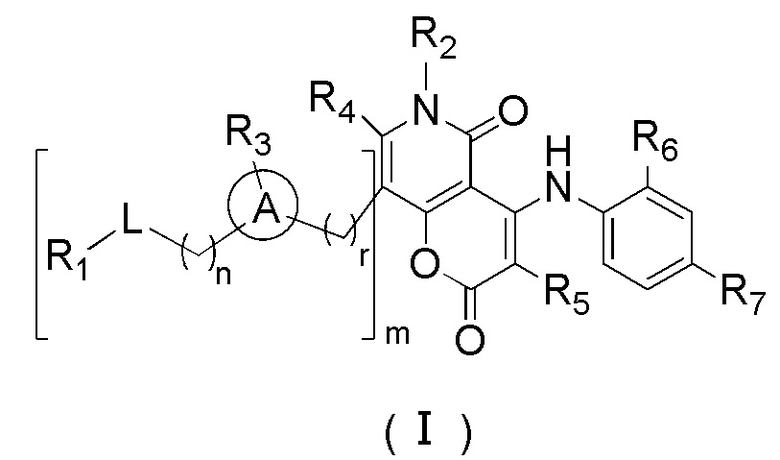
где
n выбран из 0, 1 или 2;
r выбран из 0, 1, 2 или 3;
m выбран из 0 или 1; когда m представляет собой 0, тогда 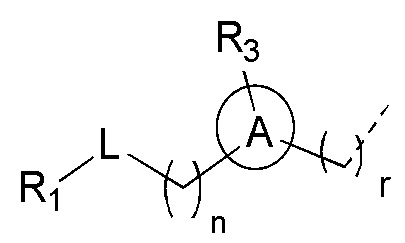 представляет собой H;
представляет собой H;
кольцо A выбрано из фенила или 6-членного гетероарила;
L выбран из одинарной связи, -S(=O)-, -S(=O)2-, -C(=O)-, -NH-, -NH-C(=O)-, -NH-C(=O)-O-, -NH-S(=O)2-, -NH-S(=O)-, -NH-C(=O)-NH- и  , где -NH-S(=O)2- необязательно замещен 1 R;
, где -NH-S(=O)2- необязательно замещен 1 R;
R1 выбран из H, NH2, C1-6 алкила, 4-6-членного гетероциклоалкила, C3-6 циклоалкила и C1-3 гетероалкила, где каждый C1-6 алкил и C1-3 гетероалкил необязательно замещен 1, 2 R;
R2 выбран из H, C1-6 алкила, C3-6 циклоалкила и 6-членного гетероциклоалкила, где C1-6 алкил необязательно замещен 1, 2 или 3 R;
R3 выбран из H, F, Cl, Br, I, C1-3 алкила;
R4 и R5 независимо выбраны из C1-6 алкила;
R6 и R7 независимо выбраны из F, Cl, Br, I;
R выбран из OH, NH2 и C1-3 гетероалкила, где NH2 необязательно замещен 1 R';
R' выбран из C1-3 алкила;
каждый из «гетеро» в 6-членном гетероариле, 4-6-членном гетероциклоалкиле, C1-3 гетероалкиле независимо выбран из -NH-, N, -O-, -S(=O)2-, -S(=O)2-NH-, - C(=O)-NH-, -C(=O)-;
в любом из вышеперечисленных случаев количество гетероатомов и гетероатомных групп независимо выбрано из 1, 2.
2. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 1, отличающееся тем, что R' выбран из CH3.
3. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 1, отличающееся тем, что R выбран из OH, NH2, C1-3 алкил-S(=O)2-NH-, C1-3 алкил-S(=O)2-, C1-3 алкил-C(=O)-NH- и C1-3 алкил-O-, где NH2 необязательно замещен 1 R'.
4. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 3, отличающееся тем, что R выбран из OH, NH2,  и
и 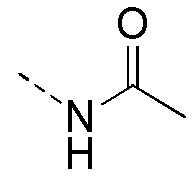 , где NH2 необязательно замещен 1 R'.
, где NH2 необязательно замещен 1 R'.
5. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 4, отличающееся тем, что R выбран из OH, NH2,  .
.
6. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, где кольцо A выбрано из фенила, пиридила или пиразинила.
7. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 6, отличающееся тем, что кольцо A выбрано из  , или
, или  .
.
8. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, где L выбран из одинарной связи, -NH-, 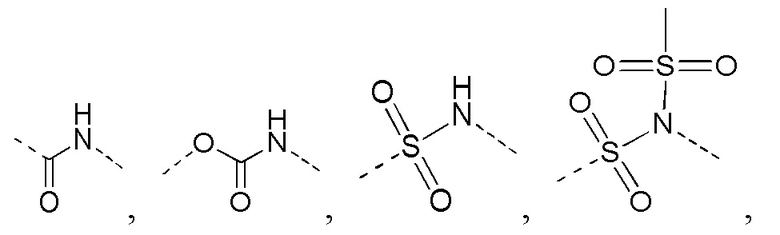
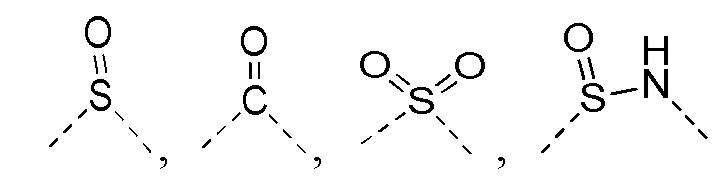 или
или  .
.
9. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что R1 выбран из H, NH2, метила, этила, изобутила, оксетанила, морфолинила, циклопропила и CH3-O-, где каждый метил, этил, изобутил и CH3-O- необязательно замещен 1, 2 R.
10. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что R1 выбран из H, NH2, Me, Et,  и
и  , где каждый Me, Et,
, где каждый Me, Et,  необязательно замещен 1, 2 R.
необязательно замещен 1, 2 R.
11. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что R1 выбран из H, NH2, CH3, Et, 
 или
или  .
.
12. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что R2 выбран из H, метила, этила, пропила, циклопропила и тетрагидропиранила, где каждый метил, этил, пропил необязательно замещен 1, 2 или 3 R.
13. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 12, отличающееся тем, что R2 выбран из H, CH3, 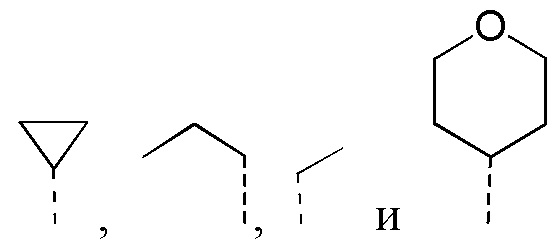 , где каждый CH3,
, где каждый CH3, 
 необязательно замещен 1, 2 или 3 R.
необязательно замещен 1, 2 или 3 R.
14. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 13, отличающееся тем, что R2 выбран из H, CH3, 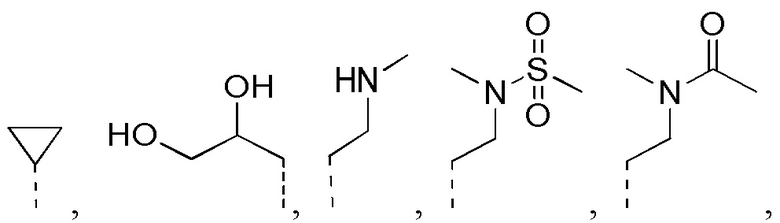
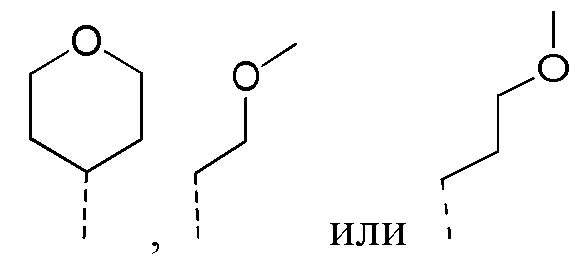 .
.
15. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что R3 выбран из H, F, Cl, Br, I, CH3.
16. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что R4 и R5 независимо выбраны из CH3, CH3CH2-.
17. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что структурное звено  выбрано из H, CH3, NH2,
выбрано из H, CH3, NH2, 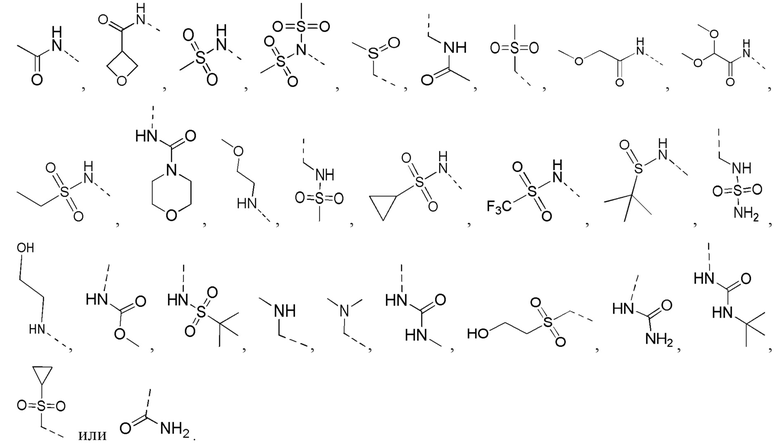
18. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, отличающееся тем, что структурное звено 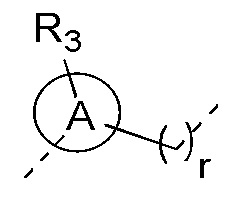 выбрано из
выбрано из  ,
,  или
или  .
.
19. Соединение, его фармацевтически приемлемая соль или его таутомер по любому из пп. 1-5, где соединение выбрано из
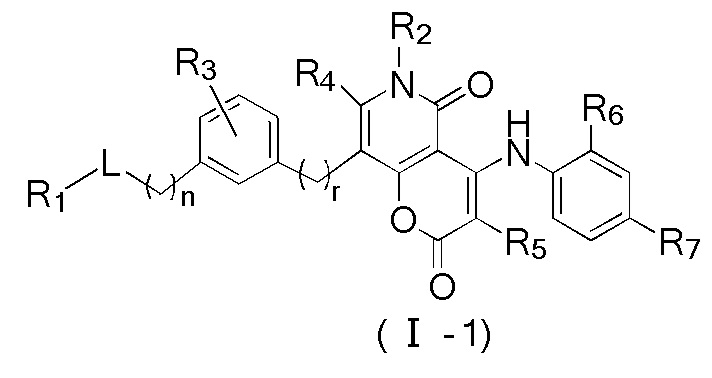
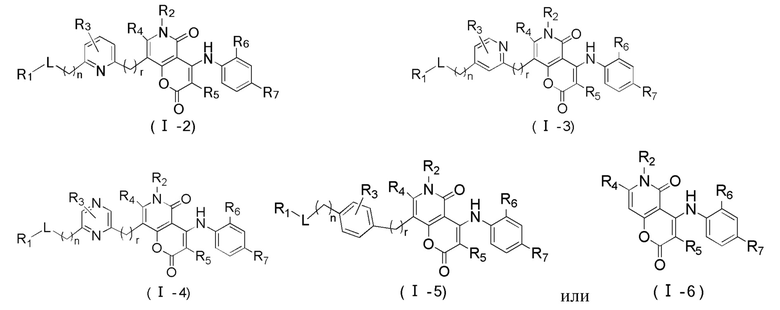
где R1, R2, R3, R4, R5, R6, R7, L, r и n такие, как определено в любом из пп. 1-5.
20. Соединение, его фармацевтически приемлемая соль или его таутомер, где соединение выбрано из
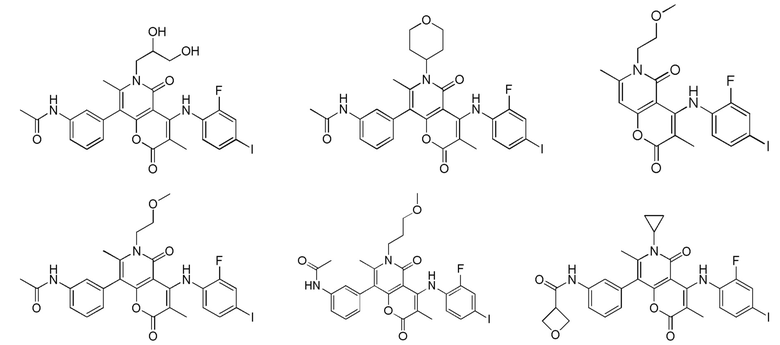
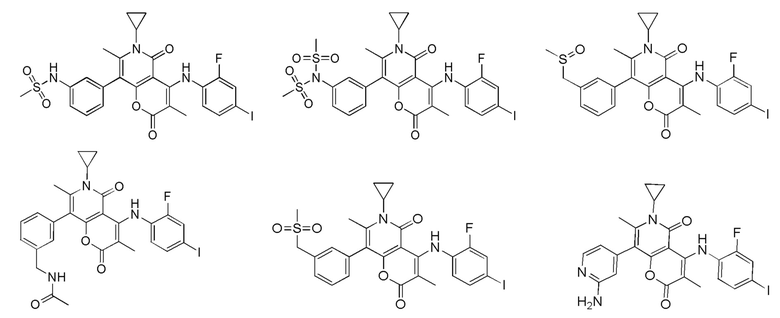
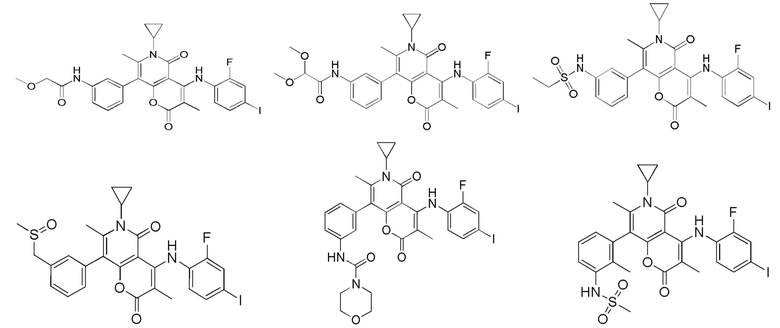
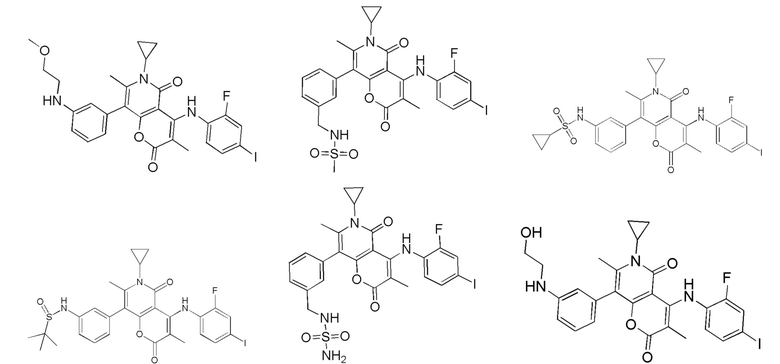
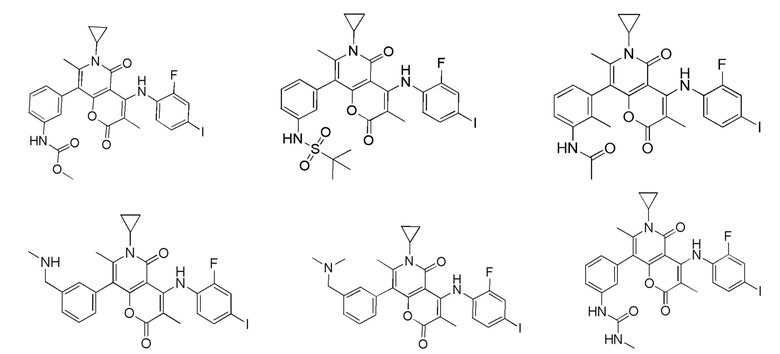
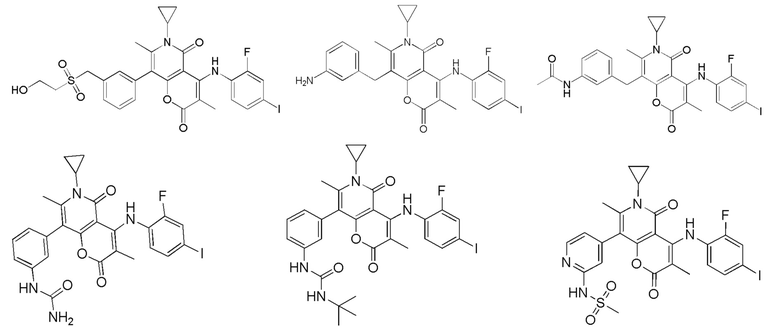
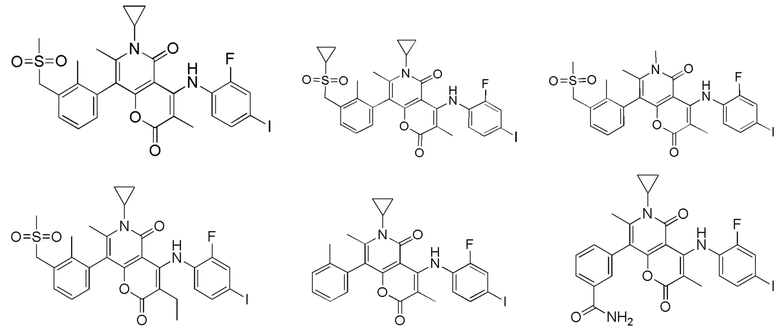
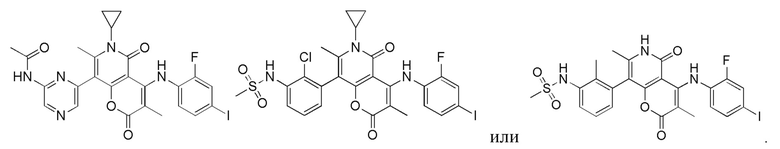
21. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 20, где соединение выбрано из
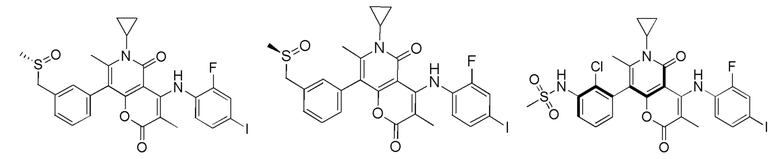
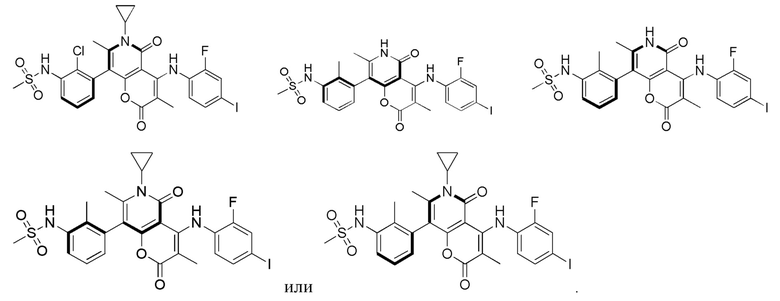
22. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 20, отличающееся тем, что соль выбрана из гидрохлорида или формиата.
23. Соединение, его фармацевтически приемлемая соль или его таутомер по п. 22, отличающееся тем, что гидрохлорид выбран из
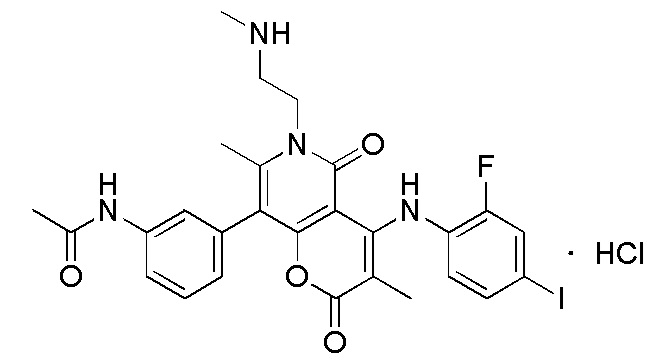 ,
, 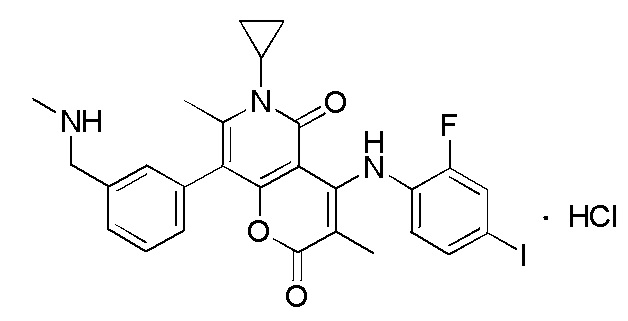 или
или 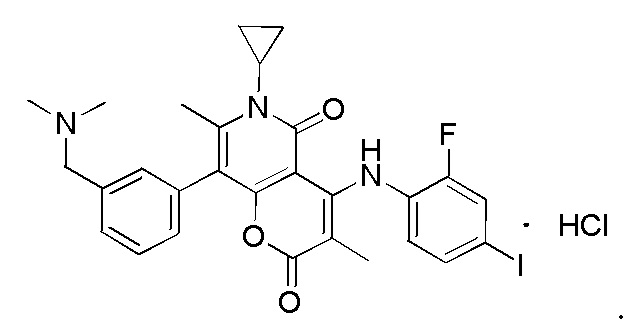
24. Фармацевтическая композиция, обладающая свойствами ингибитора MEK, содержащая терапевтически эффективное количество соединения или его фармацевтически приемлемой соли или таутомера по любому из пп. 1-23 в качестве активного ингредиента и фармацевтически приемлемый носитель.
25. Применение соединения или его фармацевтически приемлемой соли или таутомера по любому из пп. 1-23 для получения лекарственного средства для лечения заболевания, связанного с MEK.
| CN 105287509 A, 03.02.2016 | |||
| CN 106810558 A, 09.06.2017 | |||
| НОВОЕ ПРОИЗВОДНОЕ КУМАРИНА, ОБЛАДАЮЩЕЕ ПРОТИВООПУХОЛЕВОЙ АКТИВНОСТЬЮ | 2007 |
|
RU2428420C2 |
| Цоколь к электрической лампе накаливания | 1928 |
|
SU16312A1 |
| Станок для изготовления деревянных ниточных катушек из цилиндрических, снабженных осевым отверстием, заготовок | 1923 |
|
SU2008A1 |
| JP 2016155776 A, 01.09.2016. | |||

