Настоящее изобретение относится к гетерофазному сополимеру пропилена с низким модулем упругости при растяжении, к способу получения такого гетерофазный сополимера пропилена и изделиям, полученным из них.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ
В области упаковки и пленок усиливается тенденция использования пластиковых контейнеров. Очень часто в этих областях используют полиолефиновые материалы.
Одним из основных свойств, требуемых в области упаковок и пленок, являются хорошие механические свойства, такие как мягкость и гибкость, необходимые для применений в области упаковок.
В частности, гетерофазный сополимер пропилена, который обычно имеет модуль упругости при растяжении в пределах около 1000 МПа или более, может быть адаптирован для каучукоподобных материалов с очень низкими модулями растяжения около 20 МПа, которые подходят для тонкостенной упаковки, предназначенной для софт тач (приятный на ощупь) продуктов, такие как герметично закупориваемые упаковки, гибкие тубы и аналогичное им.
В настоящее время основным источником коммерчески доступных продуктов на основе мягкого полипропилена является способ Catalloy™-process от Lyondell-Basell, в котором гетерофазный сополимер пропилена получают в присутствии специфического катализатора Циглера-Натта в последовательности из вплоть до четырех последовательных газофазных реакторов для максимального производства каучука. Эти каучуки коммерчески доступны от Lyondell-Basell под торговой маркой «Adflex». Однако из-за ограничений процесса полимеризации свойства этих каучуков в определенной степени ограничены, по существу в отношении их твердости и трудной достижимости более высоких скоростей течения расплава.
Следовательно, в данной области техники продолжает существовать потребность в гетерофазных пропиленовых полимерных каучуках с высокой гибкостью (то есть низким модулем упругости при растяжении), которые также показывают низкую твердость (например, твердость по Шору D), которые могут быть получены в широких пределах MFR2.
Неожиданно было обнаружено, что применение катализаторов с единым центром полимеризации на металле в многостадийных процессах полимеризации гетерофазных полимеров пропилена позволяет получить таковые с низким модулем упругости при растяжении, низкой твердостью по Шору D с широкими пределами MFR2.
КРАТКОЕ ОПИСАНИЕ
Настоящее изобретение относится к композиции гетерофазного полипропилена, содержащей каучуковое полимерное основание, которое содержит матричную фазу и эластомерную фазу, диспергированную в ней, где матричную фазу и эластомерную фазу полимеризуют в присутствии катализатора с единым центром полимеризации на металле; и
где каучуковое полимерное основание содержит:
(A) от 20 до 55 мас.% кристаллической фракции (CF), как измерено при использовании Crystex QC в трихлорбензоле, который представляет фракцию гомополимера пропилена или фракцию сополимера из пропиленовых мономерных единиц и сомономерных единиц из этилена или альфа-олефинов с от 4 до 12 атомов углерода с содержанием сомономерных единиц вплоть до 6,0 мас.%; и
(B) от 45 до 80 мас.% растворимой фракции (SF), как измерено при использовании Crystex QC в трихлорбензоле, которая представляет сополимер из пропиленовых мономерных единиц и сомономерных единиц из этилена или альфа-олефинов с 4-12 атомами углерода, с содержанием сомономерных единиц от 17 до 55 мас.% и характеристической вязкостью iV от 1,2 до 7,0 дл/г,
где композиция гетерофазного полипропилена имеет модуль упругости при растяжении не более чем 700 МПа.
В другом аспекте настоящее изобретение относится к способу получения композиции гетерофазного полипропилена, как указано выше или ниже, включающему стадии:
a) полимеризации пропилена и необязательно сомономерных единиц, выбранных из этилена или альфа-олефинов с от 4 до 12 атомов углерода в первом реакторе полимеризации в присутствии катализатора с единым центром полимеризации на металле с получением первой полимеризационной смеси, содержащей первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле;
b) перемещения первой полимеризационной смеси во второй реактор полимеризации;
c) полимеризации пропилена и сомономерных единиц, выбранных из этилена или альфа-олефинов с от 4 до 12 атомов углерода, в указанный второй реактор полимеризации в присутствии указанного катализатора с единым центром полимеризации на металле с получением второй полимеризационной смеси, содержащей второй сополимер пропилена, указанный первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле, где массовое соотношение указанного первого гомо- или сополимера пропилена к указанному второму сополимеру пропилена составляет в пределах от 20:80 до 55:45;
d) удаления указанной второй полимеризационной смеси из указанного второго реактора полимеризации ; и
e) компаундирования указанной второй полимеризационной смеси необязательно с добавлением добавок с получением композиции гетерофазного полипропилена.
Дополнительно, настоящее изобретение относится к изделию, содержащему композицию гетерофазного полипропилена, как указано выше или ниже.
Дополнительно, настоящее изобретение относится к применению композиции гетерофазного полипропилена, как указано выше или ниже, для получения изделия.
ОПРЕДЕЛЕНИЯ
Гетерофазный полипропилен представляет сополимер на основе пропилена с кристаллической матричной фазой, которая может представлять гомополимер пропилена или неупорядоченный сополимер пропилена с по меньшей мере одним сомономером альфа-олефина, и эластомерную фазу, диспергированную в ней. Эластомерная фаза может представлять сополимер пропилена с высоким содержанием сомономера, который распределен неравномерно в полимерной цепи, и распределен в богатой сомономерами блочной структуре и богатой пропиленом блочной структуре.
Гетерофазный полипропилен, как правило, отличается от однофазного сополимера пропилена, тем что демонстрирует две различные температуры стеклования Tg, которые относятся к матричной фазе и эластомерной фазе.
Гомополимер пропилена представляет полимер, который по существу состоит из пропиленовых мономерных единиц. В виду примесей, по существу, при время коммерческих процессах полимеризации гомополимер пропилена может содержать вплоть до 0,1 мол.% сомономерных единиц, предпочтительно вплоть до 0,05 мол.% сомономерных единиц и наиболее предпочтительно вплоть до 0,01 мол.% сомономерных единиц.
Неупорядоченный сополимер пропилена представляет сополимер из пропиленовых мономерных единиц и сомономерных единиц, предпочтительно выбранный из этилена и C4-C12 альфа-олефинов, в которых сомономерные единицы распределены случайным образом в полимерной цепи. Неупорядоченный сополимер пропилена может содержать сомономерные единицы из одного или более сомономера, отличающегося числом атомов углерода.
Каучуковое полимерное основание понимается, как совокупность всех полимерных компонентов, присутствующих в композиции гетерофазного полипропилена, как указано выше или ниже. Каучуковое полимерное основание может состоять только из матричной фазы и эластомерной фазы. Однако каучуковое полимерное основание может содержать дополнительные полимерные компоненты, иные, чем матричная фаза и эластомерная фаза. Каучуковое полимерное основание предпочтительно представляет каучуковое полипропиленовое основание, которое подразумевает, что большая часть массы каучукового полимерного основания состоит из гомо- или сополимеров пропилена.
Если ясно не указано иное, то последующие количества приведены в % по массе (мас.%).
ФИГУРЫ
Фиг. 1a - демонстрирует принципиальную схему устройства CRYSTEX QC instrument.
Фиг. 1b - демонстрирует диаграмму элюирования приведенного в качестве примера образца сополимера этилена-пропилена и полученных растворимых и кристаллических фракций в TREF колонке (колонка заполнена инертным материалом, например, стеклянными шариками) (смотрите, Del Hierro, P.; Ortin, A.; Monrabal, B.; 'Soluble Fraction Analysis in polypropylene, The Column Advanstar Publications, February 2014, pp. 18-23).
Фиг. 2 - демонстрирует график модуля твердости по Шору D примеров IE1-4, CE5-6 и RE7-19.
ПОДРОБНОЕ ОПИСАНИЕ
Композиция гетерофазного полипропилена
Композиция гетерофазного полипропилена по настоящему изобретению содержит каучуковое полимерное основание, которое содержит матричную фазу и эластомерную фазу, диспергированную в ней.
Помимо каучукового полимерного основания композиция гетерофазного полипропилена может содержать одну или более добавку в количестве от 0,0 вплоть до 5,0 мас.% гетерофазного полипропилена от общей массы композиции. Одну или более добавку, выбранную из группы, состоящей из агентов, снижающих трение, агентов против слипания, УФ стабилизаторов, поглотителей кислот, антиоксидантов, альфа и/или бета-нуклеирующих агентов, антистатических агентов, пигментов, аналогичного им. Такие добавки хорошо известны специалисту в области техники, к которой относится настоящее изобретение.
Как правило, эти добавки добавляют в количествах от 100 до 2,000 частей на миллион для каждого компонента.
Одна или более добавка может быть добавлена в каучуковое полимерное основание на стадии смешивания после полимеризации матричной фазы и эластомерной фазы.
Следовательно, одна или более добавка может быть добавлена в каучуковое полимерное основание в форме мастербатчей, в которых одна или более добавка смешана с полимерным носителем в концентрированных формах. Любой необязательный полимерный носитель рассчитывают по количеству добавок от общей массы всей композиции гетерофазного полипропилена.
Композиция гетерофазного полипропилена имеет модуль упругости при растяжении не более чем 700 МПа, предпочтительно не более чем 600 МПа, более предпочтительно не более чем 500 МПа, еще более предпочтительно не более чем 450 Мпа и наиболее предпочтительно не более чем 400 МПа.
Нижний предел модуля упругости при растяжении композиции гетерофазного полипропилена, как правило, не ограничивается и может составлять такую малую величину, как 1 МПа, предпочтительно такую малую, как 5 МПа, более предпочтительно такую малую, как 10 МПа.
Гетерофазный полипропилен по настоящему изобретению не только демонстрирует высокую гибкость, также предпочтительно высокую мягкость.
Следовательно, композиция гетерофазного полипропилена предпочтительно имеет твердость по Шору D не более чем 50, более предпочтительно не более чем 45 и наиболее предпочтительно не более чем 40. Нижний предел твердости по Шору D композиции гетерофазного полипропилена, как правило, не ограничивается и может составлять такую малую, как 10, предпочтительно такую малую, как 12 и наиболее предпочтительно такую малую, как 15.
Дополнительно композиция гетерофазного полипропилена предпочтительно имеет твердость по Шору A не более чем 100. Нижний предел твердости по Шору A композиции гетерофазного полипропилена, как правило, не ограничивается и может составлять такую малую, как 50, предпочтительно такую малую, как 60 и наиболее предпочтительно такую малую, как 65.
Было обнаружено, что чем ниже модуль упругости при растяжении композиции гетерофазного полипропилена, тем также ниже твердость по Шору D и A.
Следовательно, для композиции гетерофазного полипропилена с модулем упругости при растяжении вплоть до 100 МПа твердость по Шору D предпочтительно представляет менее чем 30, более предпочтительно не более чем 28. Нижний предел может составлять такую малую, как 10.
Твердость по Шору A композиции гетерофазного полипропилена с модулем упругости при растяжении вплоть до 100 МПа предпочтительно представляет менее чем 95, более предпочтительно не более чем 92. Нижний предел может составлять такую малую, как 50.
Для композиции гетерофазного полипропилена с модулем упругости при растяжении от 100 МПа до 200 МПа твердость по Шору D предпочтительно составляет менее чем 40, более предпочтительно не более чем 35. Нижний предел может составлять такую малую, как 15.
Твердость по Шору A композиции гетерофазного полипропилена с модулем упругости при растяжении от 100 МПа до 200 МПа предпочтительно составляет менее чем 100, более предпочтительно не более чем 95. Нижний предел может составлять такую малую, как 60.
Для композиции гетерофазного полипропилена с модулем упругости при растяжении от 200 МПа до 400 МПа твердость по Шору D предпочтительно составляет менее чем 45, более предпочтительно менее чем 40. Нижний предел может составлять такую малую, как 20.
Твердость по Шору A композиции гетерофазного полипропилена с модулем упругости при растяжении от 200 МПа до 400 МПа предпочтительно составляет менее чем 100. Нижний предел может составлять такую малую, как 75.
Скорость течения расплава MFR2 (230°C, 2,16 кг) композиции гетерофазного полипропилена по существу не ограничена. Следовательно, скорость течения расплава MFR2 (230°C, 2,16 кг) композиции гетерофазного полипропилена предпочтительно представляет в пределах от 0,05 до 100 г/10 минут, более предпочтительно в пределах от 0,10 до 70 г/10 минут, такую как в пределах от 0,15 до 50 г/10 минут.
Композиция гетерофазного полипропилена предпочтительно имеет общее содержание сомономерных единиц, как определено при использовании 13C ЯМР спектроскопии от 6,0 до 35,0 мас.%, более предпочтительно от 7,5 до 30,0 мас.% и наиболее предпочтительно от 10,0 до 27,0 мас.% гетерофазного полипропилена от общей массы композиции.
Предпочтительно композиция гетерофазного полипропилена имеет общую характеристическую вязкость iV, как определено в декалине при 135°C, от 1,2 до 7,0 дл/г, более предпочтительно от 1,5 до 5,5 дл/г и наиболее предпочтительно от 1,7 до 5,0 дл/г.
Дополнительно, композиция гетерофазного полипропилена имеет динамический модуль упругости G', как определено при использовании динамического механического анализа (DMA) при 23°C, от 20 МПа до 300 МПа, более предпочтительно от 30 МПа до 250 МПа и наиболее предпочтительно от 35 МПа до 200 МПа.
Композиция гетерофазного полипропилена предпочтительно имеет температуру плавления, как определено при использовании дифференциальной сканирующей калориметрии (DSC), от 135°C до 165°C, более предпочтительно от 140°C до 160°C и наиболее предпочтительно от 145°C до 158°C.
Дополнительно, композиция гетерофазного полипропилена имеет по меньшей мере две температуры стеклования Tg1 и Tg2, как определено при использовании динамического механического анализа (DMA).
Tg1 относится к эластомерной фазе и предпочтительно представляет в пределах от -75°C до -20°C, более предпочтительно в пределах от -70°C до -25°C и наиболее предпочтительно в пределах от -65°C до -30°C.
Tg2 относится к матричной фазе и предпочтительно представляет в пределах от -15°C до 15°C, более предпочтительно в пределах от -10°C до 10°C и наиболее предпочтительно в пределах от -5°C до 5°C.
Каучуковое полимерное основание:
Каучуковое полимерное основание содержит матричную фазу и эластомерную фазу, диспергированную в ней. Матричная фаза и эластомерная фаза обе полимеризуют в присутствии катализатора с единым центром полимеризации на металле.
Каучуковое полимерное основание может содержать дополнительные полимерные компоненты, иные чем матричная фаза и эластомерная фаза. Эти дополнительные полимерные компоненты могут быть введены в каучуковое полимерное основание, либо in-situ смешиванием, то есть, во время полимеризации дополнительные полимерные компоненты на одной или более стадии полимеризации многостадийного процесса полимеризации полимеризуют в присутствии друг друга, либо на стадии пост-полимеризационного смешивания, такого как сухое смешивание или смешивание в расплаве.
Однако, в предпочтительном варианте осуществления настоящего изобретения каучуковое полимерное основание состоит из матричной фазы и эластомерной фазы.
Сомономерные единицы каучукового полимерного основания предпочтительно выбирают из этилена и альфа-олефинов с от 4 до 12 атомов углерода. По существу, предпочтительными являются сомономерные единицы, выбранные из этилена, 1-бутена, 1-гексена и 1-октена. Наиболее предпочтительным является этилен.
Сомономерные единицы каучукового полимерного основания могут быть выбраны из одних или более, чем одних сомономерных единиц, выбранных из этилена и альфа-олефинов с от 4 до 12 атомов углерода.
Следовательно, эластомерная фаза может содержать те же самые сомономерные единицы, что и матричная фаза, или может содержать сомономерные единицы, отличные от таковых матричной фазы.
Предпочтительно сомономерные единицы каучукового основания выбирают из одного вида сомономерных единиц. Как следствие, сомономерные единицы матричной фазы и эластомерной фазы представляют аналогичные.
В предпочтительном варианте осуществления настоящего изобретения матричная фаза и эластомерная фаза содержат только пропиленовые мономерные единицы и этиленовые сомономерные единицы.
В композиции гетерофазного полипропилена матричная фаза и эластомерная фаза, как правило, не могут быть полностью отделены друг от друга. В области техники, к которой относится настоящее изобретение, известно несколько способов, чтобы охарактеризовать матричную фазу и эластомерную фазу композиции гетерофазного полипропилена. Одним из способов является экстракция фракции, которая состоит из большей части эластомерной фазы с ксилолом, таким образом, отделяя фракцию, растворимую в холодном ксилоле (XCS), от фракции, нерастворимой в холодном ксилоле (XCI).
XCS фракция содержит большую часть эластомерной фазы и только меньшую часть матричной фазы, при этом XCI фракция содержит большую часть матричная фаза и только меньшую часть эластомерной фазы. Экстракция ксилолом по существу подходит для композиции гетерофазного полипропилена с высококриталлической матричной фазой, такой как матричная фаза гомополимера пропилена или матричная фаза неупорядоченного сополимера пропилена с низким содержанием сомономера не более чем около 3 мас.%. Для композиции гетерофазного полипропилена с матричной фазой неупорядоченного сополимера пропилена с содержанием сомономера не более чем около 3 мас.% содержание матричной фазы в XCS фракции настолько высокое (около 5 мас.% или выше), что XCS фракция не может быть надлежащим образом использована для характеристики эластомерной фазы композиции гетерофазного полипропилена, в виду большого количества матричной фазы в XCS фракции.
Другим способом является отделение кристаллической фракции и растворимой фракции при использовании метода CRYSTEX QC с использованием трихлорбензола (TCB) в качестве растворителя. Этот метод описан ниже в части методов измерения. В этом способе кристаллическую фракцию (CF) и растворимую фракцию (SF) отделяют друг от друга. Кристаллическая фракция (CF) содержит большую часть матричной фазы и только меньшую часть эластомерной фазы, а растворимая фракция (SF) содержит большую часть эластомерной фазы и только меньшую часть матричной фазы. Этот способ больше подходит для характеристики кристаллической фазы и эластомерной фазы композиции гетерофазного полипропилена с матричной фазой неупорядоченного сополимера пропилена с содержанием сомономера не более чем около 3 мас.%, поскольку при использовании метода CRYSTEX QC отделяется только небольшое количество матричной фазы в растворимую фракцию (SF).
В виду различий в методах отделения экстракцией ксилолом и методе CRYSTEX, свойства фракций XCS/XCI с одной стороны и кристаллической/растворимой фракций (CF/SF) с другой стороны не совсем одинаковы, это означает, что количества матричной фазы и эластомерной фазы могут отличаться наряду со свойствами.
Каучуковое полимерное основание предпочтительно имеет содержание XCS фракции от 40 мас.% до 75 мас.%, более предпочтительно от 45 мас.% до 70 мас.% и наиболее предпочтительно от 47 мас.% до 67 мас.% от общей массы каучукового полимерного основания.
Предпочтительно XCS фракция имеет содержание сомономера от 10 мас.% до 45 мас.%, более предпочтительно от 15 мас.% до 40 мас.% и наиболее предпочтительно от 17 мас.% до 35 мас.% мономерных единиц в XCS фазе от общей массы.
Следовательно, оставшееся количество мономерных единиц XCS фракции, составляющее вплоть до 100 мас.%, представляет пропиленовые мономерные единицы.
Сомономерные единиц предпочтительно выбирают из одних или более сомономерных единиц, выбранных из этилена и альфа-олефинов с от 4 до 12 атомов углерода, более предпочтительно сомономерные единицы выбирают из этилена, 1-бутена, 1-гексена и 1-октена.
Предпочтительно XCS фаза состоит только из одного вида сомономерных единиц, как указано выше.
В одном по существу предпочтительном варианте осуществления настоящего изобретения сомономерные единицы такой XCS фракции представляют этиленовые сомономерные единицы.
Дополнительно, XCS фракция предпочтительно имеет характеристическую вязкость iV от 1,0 дл/г до 7,0 дл/г, более предпочтительно от 1,3 дл/г до 5,0 дл/г и наиболее предпочтительно от 1,5 дл/г до 4,0 дл/г.
XCI фракция предпочтительно присутствует в каучуковом основании в количестве от 25 мас.% до 60 мас.%, более предпочтительно от 30 мас.% до 55 мас.% и наиболее предпочтительно от 33 мас.% до 53 мас.% от общей массы каучукового полимерного основания.
Предпочтительно XCI фракция имеет содержание сомономера от 0,1 мас.% до 15 мас.%, более предпочтительно от 1,5 мас.% до 12 мас.% и наиболее предпочтительно от 1,7 мас.% до 10 мас.% от общей массы мономерных единиц во XCI фракции.
Следовательно, оставшееся количество мономерных единиц XCI фракции, составляющее вплоть до 100 мас.%, представляет пропиленовых мономерные единицы.
Сомономерные единицы XCI фракции предпочтительно выбирают из одних или более сомономерных единиц, выбранных из этилена и альфа-олефинов с от 4 до 12 атомов углерода, более предпочтительно сомономерные единицы выбирают из этилена, 1-бутена, 1-гексена и 1-октена.
Предпочтительно XCI фракция состоит только из одного вида сомономерных единиц, как указано выше.
В одном по существу предпочтительном варианте осуществления настоящего изобретения сомономерные единицы такой XCI фракции представляют этиленовые сомономерные единицы.
CRYSTEX QC измерение кристаллической фракции:
CRYSTEX QC измерение кристаллической фракции (CF), предпочтительно присутствующей в каучуковом полимерном основании в количестве от 20 мас.% до менее чем 55 мас.%, более предпочтительно от 25 мас.% до 53 мас.%, наиболее предпочтительно от 30 мас.% до 52 мас.% от общей массы каучукового полимерного основания.
Предпочтительно кристаллическая фракция (CF) имеет содержание сомономера вплоть до 6,0 мас.%, такое как от 1,1 мас.% до 6,0 мас.%, более предпочтительно от 1,5 мас.% до 5,0 мас.% и наиболее предпочтительно от 1,7 мас.% до 4,0 мас.% от общей массы мономерных единиц в кристаллической фракции (CF).
Следовательно, оставшееся количество мономерных единиц кристаллической фракции (CF), составляющее вплоть до 100 мас.%, представляет пропиленовые мономерные единицы.
Сомономерные единицы кристаллической фракции (CF) предпочтительно выбирают из одних или более сомономерных единиц, выбранных из этилена и альфа-олефинов с от 4 до 12 атомов углерода, более предпочтительно сомономерные единицы выбирают из этилена, 1-бутена, 1-гексена и 1-октена.
Предпочтительно кристаллическая фракция (CF) состоит только из одного вида сомономерных единиц, как указано выше.
В одном по существу предпочтительном варианте осуществления настоящего изобретения сомономерные единицы такой кристаллической фракции (CF) представляют этиленовые сомономерные единицы.
Дополнительно, кристаллическая фракция (CF) предпочтительно имеет характеристическую вязкость iV от 1,0 дл/г до 7,0 дл/г, более предпочтительно от 1,3 дл/г до 5,0 дл/г и наиболее предпочтительно от 1,4 дл/г до 4,0 дл/г, такую как от 1,5 дл/г до 3,5 дл/г.
CRYSTEX QC измерение растворимой фракции (SF):
CRYSTEX QC измерение растворимой фракции (SF), предпочтительно присутствующей в каучуковом полимерном основании в количестве от более чем 45 мас.% до 80 мас.%, более предпочтительно от 47 мас.% до 75 мас.%, наиболее предпочтительно от 48 мас.% до 70 мас.% от общей массы каучукового полимерного основания.
Растворимая фракция (SF) имеет содержание сомономера от 17 мас.% до 55 мас.%, более предпочтительно от 19 мас.% до 52 мас.% и наиболее предпочтительно от 20 мас.% до 50 мас.% от общей массы мономерных единиц в растворимой фракции (SF).
Следовательно, оставшееся количество мономерных единиц растворимой фракции (SF) кристаллической фракции, составляющее вплоть до 100 мас.%, представляет пропиленовые мономерные единицы.
Сомономерные единицы растворимой фракции (SF) предпочтительно выбирают из одних или более сомономерных единиц, выбранных из этилена и альфа-олефинов с от 4 до 12 атомов углерода, более предпочтительно сомономерные единицы выбирают из этилена, 1-бутен, 1-гексен и 1-октен.
Предпочтительно растворимая фракция (SF) состоит только из одного вида сомономерных единиц, как указано выше.
В одном по существу предпочтительном варианте осуществления настоящего изобретения сомономерные единицы растворимой фракции (SF) представляют этиленовые сомономерные единицы.
Дополнительно растворимая фракция (SF) имеет характеристическую вязкость iV от 1,2 дл/г до 7,0 дл/г, предпочтительно от 1,5 дл/г до 6,0 дл/г и наиболее предпочтительно от 1,7 дл/г до 5,0 дл/г, такую как от 1,8 дл/г до 4,5 дл/г.
Предпочтительно соотношение характеристической вязкости растворимой фракции (SF) к характеристической вязкости кристаллической фракции (CF), iVSF / iVCF, составляет от 1,6 до 1,8, более предпочтительно от 1,7 до 1,6 и наиболее предпочтительно от 1,8 до 1,5.
Предпочтительно соотношение характеристической вязкости растворимой фракции (SF) к характеристической вязкости композиции гетерофазного полипропилена, iVSF / iVHECO, составляет от 1,6 до 1,7, более предпочтительно от 1,7 до 1,4 и наиболее предпочтительно от 1,8 до 1,3.
Способ
В другом аспекте настоящее изобретение также относится к способу получения композиция гетерофазного полипропилена, как указано выше или ниже, включающему стадии:
a) полимеризации пропилена и необязательно сомономерных единиц, выбранных из этилена или альфа-олефинов с от 4 до 12 атомов углерода, в первом реакторе полимеризации в присутствии катализатора с единым центром полимеризации на металле с получением первой полимеризационной смеси, содержащей первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле;
b) перемещения первой полимеризационной смеси во второй реактор полимеризации;
c) полимеризации пропилена и сомономерных единиц, выбранных из этилена или альфа-олефинов с от 4 до 12 атомов углерода в указанном втором реакторе полимеризации в присутствии указанного катализатора с единым центром полимеризации на металле с получением второй полимеризационной смеси, содержащей второй сополимер пропилена, указанный первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле, где массовое соотношение указанного первого гомо- или сополимера пропилена к указанному второму сополимеру пропилена составляет в пределах от 20:80 до 55:45;
d) удаления указанной второй полимеризационной смеси из указанного второго реактора полимеризации ; и
e) компаундирования указанной второй полимеризационной смеси необязательно с добавлением добавок с получением композиции гетерофазного полипропилена.
Полимеризация:
Предпочтительно в способе по настоящему изобретению матричную фазу каучукового полимерного основания полимеризуют перед эластомерной фазой каучукового полимерного основания.
Следовательно, предпочтительно матричная фаза образует первый гомо- или сополимер пропилена, полимеризованный на стадии a) способа, а эластомерная фаза образует второй сополимер пропилена, полимеризованный на стадии c) способа.
Специалисту в области техники, к которой относится настоящее изобретение, понятно, что первый гомо- или сополимер пропилена предпочтительно отражает матричную фазу, как правило, не идентичную кристаллической фракции (CF) в CRYSTEX QC измерении, и что второй сополимер пропилена предпочтительно отражает эластомерную фазу, как правило, не идентичную растворимой фракции (SF) в CRYSTEX QC измерении.
Стадия a) способа может быть проведена в единственном реакторе полимеризации. В указанном варианте осуществления настоящего изобретения матричная фаза представляет унимодальный гомо- или сополимер пропилена.
Стадия a) способа также может быть проведена в двух или более реакторах полимеризации, таких как 2, 3 или 4 реакторах полимеризации, наиболее предпочтительно 2 реакторах полимеризации, соединенных в серию.
Это означает, что в первом реакторе полимеризации стадии a) способа полимеризуют первую часть первого гомо- или сополимера пропилена в присутствии катализатора с единым центром полимеризации на металле с получением первой части первой полимеризационной смеси, содержащей первую часть первого гомо- или сополимера пропилена и катализатор с единым центром полимеризации на металле, перемещение первой части первой полимеризационной смеси во второй реактор полимеризации стадии a) способа и полимеризацию второй части первого гомо- или сополимера пропилена в присутствии катализатора с единым центром полимеризации на металле в присутствии указанной первой части первого гомо- или сополимера пропилена с получением второй части первой полимеризационной смеси, содержащей первую и вторую часть первого гомо- или сополимера пропилена и катализатор с единым центром полимеризации на металле.
Эти стадии способа могут быть повторены дополнительно в одном или более дополнительном последующем реакторе(ах) полимеризации стадии a) способа.
В другом варианте осуществления настоящего изобретения вторая часть первой полимеризационной смеси отражает первую полимеризационную смесь стадии a) способа, которую затем перемещают во второй реактор полимеризации стадии b)способа.
Условия полимеризации в первом, втором и необязательно последующих реакторах(ы) полимеризации стадии a) способа могут быть сопоставимы. В указанном варианте осуществления настоящего изобретения матричная фаза представляет унимодальный гомо- или сополимер пропилена.
В качестве альтернативы, условия полимеризации в первом, втором и необязательно последующем реакторе(ах) полимеризации стадии a) способа могут отличаться друг от друга, по существу по одной или более температуре полимеризации, давлению полимеризация, подаче сомономера или подаче агента передачи цепи. В указанном варианте осуществления настоящего изобретения матричная фаза представляет мультимодальный гомо- или сополимер пропилена. В случае, когда используют два реактора полимеризации в серии указанного варианта осуществления настоящего изобретения, матричная фаза представляет бимодальный гомо- или сополимер пропилена.
В указанном варианте осуществления настоящего изобретения возможно проводить полимеризацию гомополимера пропилена в одном или более реакторе полимеризации и неупорядоченного сополимера пропилена в одном или более реакторе полимеризации. В указанном варианте осуществления настоящего изобретения матричная фаза представляет мультимодальный сополимер пропилена, содержащий фракцию гомополимера пропилена и фракцию неупорядоченного сополимера пропилена. По существу предпочтительно гомополимер пропилена полимеризуют в одном реакторе полимеризации, и неупорядоченный сополимер пропилена полимеризуют в другом реакторе полимеризации из последовательности из двух реакторов для полимеризации матричной фазы с одной фракцией гомополимера пропилена и одной фракцией неупорядоченного сополимера пропилена.
Нет особого предпочтения в последовательности полимеризации фракций матричной фазы.
Предпочтительно в способе по настоящему изобретению эластомерную фазу каучукового полимерного основания полимеризуют после и в присутствии матричной фазы каучукового полимерного основания.
Следовательно, предпочтительно эластомерная фаза образует второй сополимер пропилена, полимеризованный на стадии способа c), а матричная фаза образует первый гомо- или сополимер пропилена, полимеризованный на стадии способа a).
Стадия c) способа может быть проведена в единственном реакторе полимеризации. В указанном варианте осуществления настоящего изобретения эластомерная фаза представляет унимодальный сополимер пропилена.
Стадия c) способа также может быть проведена в двух или более реакторах полимеризации, таких как 2, 3 или 4 реакторах полимеризации, наиболее предпочтительно 2 реакторах полимеризации, соединенных в серию.
Это означает, что в первом реакторе полимеризации стадии c) способа первую часть второго сополимера пропилена полимеризуют в присутствии катализатора с единым центром полимеризации на металле с получением первой части второй полимеризационной смеси, содержащей первая часть второго сополимера пропилена, первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле, перемещение первой части второй полимеризационной смеси во второй реактор полимеризации стадии c) способа и полимеризации второй части второго сополимера пропилена в присутствии катализатора с единым центром полимеризации на металле в присутствии указанного первой части второго сополимера пропилена с получением второй части второй полимеризационной смеси, содержащей первую и вторую часть второго сополимера пропилена, первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле.
Эти стадии способа могут быть повторены дополнительно в одном или более дополнительном последующем реакторе(ах) полимеризации стадии c) способа.
В другом варианте осуществления настоящего изобретения вторая часть второй полимеризационной смеси отражает вторую полимеризационную смесь стадии c) способа, которую затем удаляют из второго реактора полимеризации стадии c) способа на стадию d) способа.
Условия полимеризации в первом, втором и необязательно последующем реакторе(ах) полимеризации стадии c) способа могут быть сопоставимы. В указанном варианте осуществления настоящего изобретения эластомерная фаза представляет унимодальный сополимер пропилена.
В качестве альтернативы, условия полимеризации в первом, втором и необязательно последующем реакторе(ах) полимеризации стадии c) способа могут отличаться друг от друга, по существу по одной или более температуре полимеризации, давлению полимеризация, подаче сомономера или подаче агента передачи цепи. В указанном варианте осуществления настоящего изобретения эластомерная фаза представляет мультимодальный сополимер пропилена. В случае, когда используют два реактора полимеризации в серии указанного варианта осуществления настоящего изобретения, эластомерная фаза представляет бимодальный сополимер пропилена.
В указанном варианте осуществления настоящего изобретения возможно проводить полимеризацию сополимера пропиленаs с различными сомономерами в двух или более реакторах полимеризации. В указанном варианте осуществления настоящего изобретения эластомерная фаза представляет мультимодальный сополимер пропилена, содержащий фракцию сополимера пропилена с одним сомономером и фракцией сополимера пропилена с другим сосмономером.
Нет особого предпочтения в последовательности полимеризации фракций эластомерной фазы.
Предпочтительно первый реактор полимеризации, такой как циркуляционный реактор, работает в массе, и все последующие реакторы полимеризации, предпочтительно включая необязательный второй и последующий реактор(ы) полимеризации стадии a) способа, работают в газовой фазе.
Предпочтительно стадии полимеризации способа по настоящему изобретению проводят в реакторе полимеризации в массе, предпочтительно циркуляционном реакторе, последующем одном или более, таком как 1, 2, 3 или 4, предпочтительно 1 или 2 газофазных реакторах, соединенных в серию.
Стадии a) способа также может предшествовать стадия предварительной полимеризации. В указанном варианте осуществления настоящего изобретения предпочтительно стадии полимеризации способа по настоящему изобретению проводят в реакторе предварительной полимеризации с последующим реактором полимеризации в массе, предпочтительно циркуляционном реакторе, с последующим одним или более, таким как 1, 2, 3 или 4, предпочтительно 1 или 2 газофазных реакторах, соединенных в серию.
Условия полимеризации, такие как температура полимеризации, давление полимеризации, подача сомономера, подача агента передачи цепи или время пребывания на разных стадиях полимеризации, по существу не ограничены. Специалисту в области техники, к которой относится настоящее изобретение, известно, как регулировать эти условия полимеризации, чтобы регулировать свойства первого гомо- или сополимера пропилена и второго сополимера пропилена.
Время пребывания на стадиях a) и c) способа предпочтительно выбирают так, чтобы массовое соотношение первого гомо- или сополимера пропилена ко второму сополимеру пропилена составляло в пределах от 20:80 до 55:45.
Предпочтительно массовое соотношение указанного первого гомо- или сополимера пропилена к указанному второму сополимеру пропилена составляет в пределах от 25:75 до 53:47, более предпочтительно в пределах от 30:70 до 50:50, наиболее предпочтительно в пределах от 35:65 до 47:53.
Предпочтительно стадии полимеризации способа по настоящему изобретению проводят при использовании «циркуляционно-газофазного» способа, такого, как разработанный Borealis, известный как BORSTARTM technology. Примеры такого способа описаны в EP 0 887 379, WO 92/12182, WO 2004/000899, WO 2004/111095, WO 99/24478, WO 99/24479 и WO 00/68315. В этих патентных заявках также описаны подходящие условия полимеризации. Другим подходящим способом является суспензионно-газофазный, называемый Spheripol™ process.
Катализатор с единым центром полимеризации на металле:
Композицию гетерофазного полипропилена по настоящему изобретению полимеризуют в присутствии катализатора с единым центром полимеризации на металле.
Катализатор, используемый в настоящем изобретении, может быть использован в бесподложечной форме (форме без внешнего носителя) или в твердой форме. Однако катализатор может быть использован в виде гетерогенного (твердого) катализатора.
Как правило, количество используемого катализатора зависит от природы катализатора, типа реактора, условий и заданных свойств композиции полипропилена.
Катализатор по настоящему изобретению в твердой форме, предпочтительно в форме твердых частиц, может быть, как нанесен на подложку в виде внешнего материала-носителя, такого как оксид кремния или оксид алюминия, или по существу в предпочтительном варианте осуществления настоящего изобретения - свободен от внешнего носителя, однако все еще находится в твердой форме. Например, твердый катализатор может быть получен при использовании способа, в котором:
(a) получают эмульсионную систему жидкость/жидкость, указанная эмульсионная система жидкость/жидкость содержит раствор компонентов катализатора (i) и (ii), диспергированных в растворителе, с получением, таким образом, диспергированных капель; и
(b) твердые частицы получают отверждением указанных диспергированных капель.
Идеальным получением катализатора является получение (i) комплекса, например, формулы (I), и (ii) сокатализатора; формированием эмульсионной системы жидкость/жидкость, которая содержит раствор каталитических компонентов (i), и диспергированием в растворителе (ii), и отверждением указанных диспергированных капель с получением твердых частиц.
Свободный от внешнего носителя означает, что катализатор не содержит внешнюю подложку, такую как неорганическая подложка, например, оксид кремния или оксид алюминия, или органический полимерный материал-носитель.
В приведенных ниже определениях используемый в описании настоящей патентной заявки термин группа C1-20 нециклического углеводородного остатка/гидрокарбильная группа (hydrocarbyl group) включает C1-20 алкил, C2-20 алкенил, C2-20 алкинил, C3-20 циклоалкил, C3-20 циклоалкенил, C6-20 арильные группы, C7-20 алкиларильные группы или C7-20 арилалкильные группы или, конечно же, смеси из этих групп, таких как циклоалкил, замещенный алкилом. Линейные и разветвленные группы нециклического углеводородного остатка / гидрокарбильные группы не могут содержать циклические единицы. Алифатические группы нециклического углеводородного остатка / гидрокарбильные группы не могут содержать арильные группы.
Если ясно не указано иное, предпочтительные группы C1-20 нециклического углеводородного остатка / гидрокарбильные группы представляют C1-20 алкил, C4-20 циклоалкил, C5-20 циклоалкил-алкильные группы, C7-20 алкиларильные группы, C7-20 арилалкильные группы или C6-20 арильные группы, по существу C1-10 алкильные группы, C6-10 арильные группы, или C7-12 арилалкильные группы, например, C1-8 алкильные группы. Наиболее предпочтительными группами нециклического углеводородного остатка/ гидрокарбильными группами являются метильная, этильная, пропильная, изопропильная, третбутильная, изобутильная, C5-6-циклоалкильная, циклогексилметильная, фенильная или бензильная.
Используемый в описании настоящей патентной заявки термин галогено- включает фторо-, хлоро-, бромо- и йодогруппы, в частности хлорогруппы, когда он относится к определению комплекса.
Степень окисления иона металла главным образом определяется природой указанного иона металла и стабильностью индивидуальных степеней окисления каждого иона металла.
Следует принимать во внимание, что в комплексах ион металла M соединен координационно лигандами X, удовлетворяя, таким образом, валентности иона металла и заполняя его доступные координационные узлы. Природа этих σ-лигандов может сильно варьировать.
Активность катализатора в настоящей патентной заявке определяют, как количество полученного полимера/г к катализатору/ч. Используемый в описании настоящей патентной заявки термин продуктивность также иногда используют для указания активности катализатора, хотя в описании настоящей патентной заявки он обозначает количество полимера, полученного на единицу массы катализатора.
Катализатор с единым центром полимеризации на металле предпочтительно представляет металлоценовый. Получение металлоценового катализатора может быть проведено согласно или аналогично методам, известным из литературы, и известным специалисту в области техники, к которой относится настоящее изобретение. Указанные металлоцены, как правило, несут по меньшей мере один органический лиганд, как правило 1, 2 или 3, например, 1 или 2, который представляет η- связанный с металлом, например, η2-6-лиганд, такой как η5-лиганд. Предпочтительно металлоцен представляет переходный металл Группы 4-6, соответственно, титаноцен, цирконоцен или гафноцен, который содержит по меньшей мере один η5-лиганд, который является необязательно замещенным циклопентадиенилом, необязательно замещенным инденилом, необязательно замещенным тетрагидроинденилом или необязательно замещенным флуоренилом.
Металлоценовое соединение может иметь формулу I
(Cp)mTnMAq (I)
где
каждый Cp независимо представляет ненасыщенный или насыщенный и/или слитый гомо- или гетероциклопентадиенил лиганд, например, замещенный или незамещенный циклопентадиенильный, замещенный или незамещенный инденильный или замещенный или незамещенный флуорениловый лиганд; необязательно один или более заместитель(и), предпочтительно выбираемый из галогена, нециклического углеводородного остатка/гидрокарбила (например, C1-C20 алкила, C2-C20 алкенила, C1-C20 алкинила, C3-C12 циклоалкила, C6-C20 арила или C7-C20 арилалкила), C3-C12 циклоалкила, который содержит 1, 2, 3 или 4 гетероатома в кольцевой функциональной группе, C6-C20 -гетероарила, C1-C20-галоалкила, -SiR"3 , -OSiR"3, -SR", -PR"2 или -NR"2, где каждый R" независимо представляет водород или нециклический углеводородный остаток / гидрокарбил, например, C1-C20 алкил, C2-C20 алкенил, C2-C20 алкинил, C3-C12 циклоалкил или C6-C20 арил; или, например, в случае -NR"2, два заместителя R" могут образовывать кольцо, например, пяти или шестичленное кольцо вместе с атомом азота, с которым они соединены.
T представляет мостик из 1-3 атомов, например, мостик из 1-2 C-атомов или 1-2 гетероатомов, где гетероатом(ы) может представлять, например, Si, Ge и/или O атом(ы), при этом каждый из мостиковых атомов независимо может нести заместители, такие как C1-C20-алкил, три(C1-C20-алкил)силил, три(C1-C20-алкил)силокси или C6-C20-арил); или мостик из 1-3, например, одного или двух гетероатомов, таких как атом(ы) кремния, германия и/или кислорода, например, -SiR1 2-, где каждый R1 независимо представляет C1-C20-алкил, C6-C20-арил или три(C1-C20-алкил)силильный остаток, такой как триметилсилильный остаток.
M - переходный металл группы 4-6, такой как группа 4, например Ti, Zr или Hf.
Каждый A независимо представляет сигма-лиганд, такой как H, галоген, C1-C20 алкил, C1-C20 алкокси, C2-C20 алкенил, C1-C20 алкинил, C3-C12 циклоалкил, C6-C20 арил, C6-C20 арилокси, C7-C20 арилалкил, C7-C20 арилалкенил, -CH2-Y, где Y представляет C6-20-арил, C6-20-гетероарил, C1-20-алкокси, C6-20-арилокси, -NR"2, - SiR"3 или-OSiR"3, -SR", -PR"3, -SiR"3, -OSiR"3 или -NR"2; где каждый R" независимо представляет водород или нециклический углеводородный остаток / гидрокарбил, например, C1-C20 алкил, C2-C20 алкенил, C2-C20 алкинил, C3-C12 циклоалкил или C6-C20 арил; или, например, в случае -NR"2, два заместителя R" могут образовывать кольцо, например, пяти или шестичленное кольцо вместе с атомом азота, с которым они соединены.
Каждая из указанных выше кольцевых функциональных групп, как таковая или как часть кольцевой функциональной группы в качестве заместителя для Cp, A, R" или R<1>, может быть дополнительно замещена, например, C1-C20-алкилом, который может содержать атомы Si и/или O;
n представляет 1 или 2, например 1,
m представляет 1, 2 или 3, например 1 или 2,
q представляет 1, 2 или 3, например 2 или 3, где m+q равно валентности M.
Следовательно, настоящее изобретение, как правило, применимо к стереоспецифическому катализатору с единым центром полимеризации на металле, композиции гетерофазного полипропилена, предпочтительно полученному в присутствии металлоцена с формулой (I).
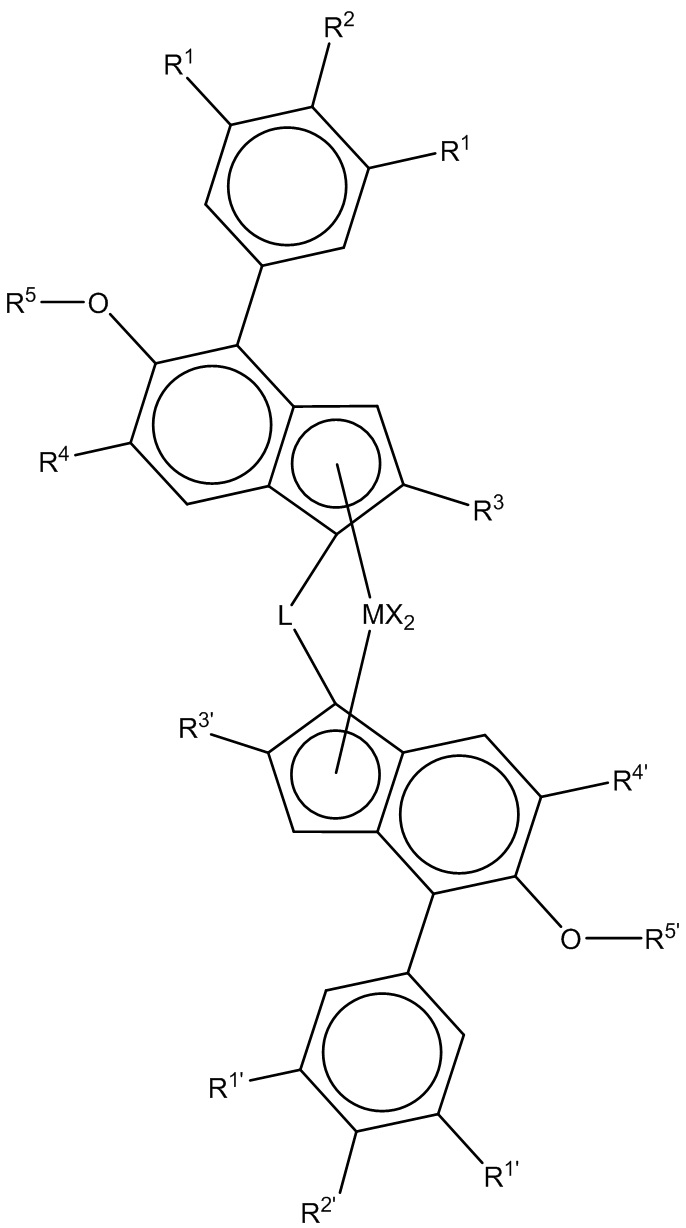
(Формула (I)),
где
M представляет цирконий или гафний;
каждый X независимо представляет лиганд сигма-донора;
L представляет двухвалентный мостик, выбранный из -R'2C-, -R'2C-CR'2-, -R'2Si-, -R'2Si-SiR'2-, -R'2Ge-, где каждый R' независимо представляет атом водорода или группу C1-C20- нециклического углеводородного остатка/гидрокарбильную группу, необязательно содержащую один или более гетероатом из групп 14-16 Периодической таблицы, или атомы фтора, или необязательно две R' группы вместе взятые образуют кольцо;
R1 и R1´ каждый независимо представляет водород, C5-C10-арил или группу -CH(Rx)2 где каждый Rx независимо представляет H или группу C1-10 нециклического углеводородного остатка/гидрокарбильную группу, и необязательно два Rx вместе взятые образуют кольцо,
R2 и R2´ каждый независимо представляет водород, C5-C10-арил или группу -C(Ry)3,
где каждый Ry независимо представляет H или группу C1-10 нециклического углеводородного остатка / гидрокарбильную группу, или необязательно две или три Ry группы вместе взятые образуют кольцо,
при этом по меньшей мере одно из R1 или R2 и одно из R1´ или R2´ отличается от водорода и
при этом R2 вместе с одним из R1, наряду с R2´ вместе с одним из R1´ могут являться частью дополнительного моно- или мультициклического кольца, конденсированного с фенильным кольцом;
R3 и R3´ каждый независимо представляет линейную группу C1-C6 нециклического углеводородного остатка/гидрокарбильную группу или разветвленную или циклическую группу C4 - C10 нециклического углеводородного остатка/гидрокарбильную группу (cyclic C4 to C10 hydrocarbyl group), при этом группы представляют неразветвленные в α-позиции;
R4 и R4´ каждый независимо представляет третичную группу C4-C10 нециклического углеводородного остатка/гидрокарбильную группу;
R5 и R5´ каждый независимо представляет линейную или разветвленную C1-C10 алкильную группу или C5-C10-арильную группу, и
(ii) сокатализатор, содержащий соединение металла группы 13.
Комплексы могут представлять асимметричные или симметричные. Асимметричные просто означает, что два инденильных лиганда, образующие металлоцен, различны, то есть, каждый инденильный лиганд несет ряд заместителей, которые либо химически отличаются, либо находятся в разных положениях относительно другого инденильного лиганда. Симметричные комплексы основаны на двух одинаковых инденильных лигандах.
Предпочтительно комплексы, используемые в настоящем изобретении, представляют симметричные.
Комплексы представляют хиральные, рцемические металлоцены, соединенные бисинденильными мостиками. Металлоценовые комплексы представляют, как C2-симметричные или C1-симметричные. В случае, когда они представляют C1-симметричные, они могут сохранять псевдо-C2-симметрию, поскольку они сохраняют C2-симметрию в непосредственной близости от металлического центра, хотя не на периферии лиганда. За счет их химической природы обе, и мезо форма, и рацемическая энантиомерная пара (в случае C2-симметричных комплексов) или анти, и син энантиомерные пары (в случае C1-симметричных комплексов) образуются во время синтеза из комплексов. В этом контексте рацемический или рацемический анти- означает, что два инденильных лиганда ориентированы в противоположных направлениях относительно плоскости циклопентадиенил-металл-циклопентадиенила, тогда как мезо или рацемический син означает, что два инденильных лиганда ориентированы в одном направлении относительно плоскости циклопентадиенил-металл-циклопентадиенила, как показано на Фигуре ниже.
Формула (I) включает в объем все эти конфигурации.
Предпочтительно металлоцены используют в качестве рацемических или рацемических-анти изомеров. Следовательно, в идеале по меньшей мере 95,0 мол.%, такое как по меньшей мере 98,0 мол.%, по существу по меньшей мере 99,0 мол.% металлоценов находится в форме рацемических или рацемических-анти изомеров.
В катализаторах применяют следующие предпочтения:
M.
M представляет цирконий или гафний, предпочтительно цирконий.
В приведенных ниже определениях термин группа нециклического углеводородного остатка/гидрокарбильная группа (hydrocarbyl group) включает алкильные группы, алкенильные группы, алкинильные группы, циклоалкильные группы, циклоалкенильные группы, арильные группы, алкиларильные группы или арилалкильные группы или, конечно, смеси из этих групп, такие как циклоалкил, замещенный алкилом.
Каждый X независимо представляет лиганд сигма-донора.
Следовательно, каждый X может представлять идентичный или отличающийся, и предпочтительно представляет атом водорода, атом галогена, линейную или разветвленную, циклическую или ациклическую C1-20-алкильную или -алкокси группу, C6-20-арильную группу, C7-20-алкиларильную группу или C7-20-арилалкильную группу; необязательно содержащий один или более гетероатом групп 14-16 Периодической таблицы.
Используемый в описании настоящей патентной заявки термин галогено- включает фторо-, хлоро-, бромо- и йодогруппы, в частности хлорогруппы.
Используемый в описании настоящей патентной заявки термин гетероатомы, принадлежащие к группам 14-16 Периодической таблицы, включают, например, Si, N, O или S.
Более предпочтительно каждый X независимо представляет атом водорода, атом галогена, линейную или разветвленную C1-6-алкильную или C1-6-алкокси группу, фенильную или бензильную группу.
Еще более предпочтительно каждый X независимо представляет атом галогена, линейную или разветвленную C1-4-алкильную или C1-4-алкокси группу, фенильную или бензильную группу.
Наиболее предпочтительно каждый X независимо представляет хлорогруппу, бензильную или метильную группу.
Предпочтительно обе X группы представляют идентичные.
Наиболее предпочтительными вариантами для обеих X групп являются две хлорогруппы, две метильные или две бензильные группы.
L представляет двухвалентный мостик, выбранный из -R'2C-, -R'2C-CR'2-, -R'2Si-, -R'2Si-SiR'2-, -R'2Ge-, где каждый R' независимо представляет атом водорода или группу C1-C20- нециклического углеводородного остатка/ гидрокарбильную группу, необязательно содержащую один или более гетероатом из групп 14-16 Периодической таблицы или атомы фтора, или необязательно две R' группы вместе взятые образуют кольцо.
Используемый в описании настоящей патентной заявки термин гетероатомы, принадлежащие к группам 14-16 Периодической таблицы, включают, например, Si, N, O или S.
Предпочтительно L представляет диметилсилил, метилциклогексилсилил (то есть Me-Si-циклогексил), этилен или метилен.
R1 и R1´ каждый независимо представляет водород, C5-C10-арил или группу -CH(Rx)2, где каждый Rx независимо представляет H или группу C1-10 нециклического углеводородного остатка/гидрокарбильную группу, и необязательно два Rx вместе взятые образуют кольцо,
R2 и R2´ каждый независимо представляет водород, C5-C10-арил или группу -C(Ry)3, где каждый Ry независимо представляет H или группу C1-10 нециклического углеводородного остатка/ гидрокарбильную группу, или необязательно две или три Ry группы вместе взятые образуют кольцо.
По меньшей мере одно из R1 или R2 и одно из R1´ или R2´ отличается от водорода. Это означает, что фенильные группы в позиции 4 обоих инденильных лигандов замещены по меньшей мере одним заместителем, отличающимся от водорода.
Следовательно, фенильные группы в позиции 4 обоих инденильных лигандов могут быть замещены одним, двумя или тремя заместителями от водорода.
В другом варианте осуществления настоящего изобретенияs R2 вместе с одним из R1, наряду с R2´ вместе с одним из R1´ могут являться частью дополнительного моно- или мультициклического кольца, конденсированного с фенильным кольцом. Новое кольцо предпочтительно представляет 5 или 6 членное, или группы предпочтительно образуют два новых кольца, такие как одно дополнительное пяти членное или шести членное кольцо.
Новое кольцо или кольца могут представлять алифатические или ароматические.
Таким образом, могут быть образованы группы, такие как 2-нафтил, 5- или 6-(инданил), 5- или 6-(1,1-диалкил-1H-инденил), 6-(1,2,3,4-тетрагидронафтил), 6-(1,1,4,4-tetraметил-1,2,3,4-тетрагидронафтил), 5- или 6-(N-алкил-индолил), 5- или 6-(N-алкилиндоленил) , 2- или 3-(N-алкилкарбазолил), 5- или 6-бензотиофенил.
Предпочтительно R1 и R1´ представляют идентичные и представляют, либо водород, либо группу -CH(Rx)2 где каждый Rx независимо представляет, либо H, либо группу C1-3 нециклического углеводородного остатка/гидрокарбильную группу.
Более предпочтительно R1 и R1´ представляют, либо водород, либо группу -CH(Rx)2, где каждый Rx представляет H, то есть, группа представляет метил.
Предпочтительно R2 и R2´ также представляют идентичные и представляют, либо водород, либо группу -C(Ry)3, где каждый Ry представляет, либо H, либо группу C1-3 нециклического углеводородного остатка/гидрокарбильную группу.
Более предпочтительно R2 и R2´ оба представляют, либо водород, либо группу-C(Ry)3, где каждый Ry представляет C1-алкильную группу, то есть, группу, представляющую трет-бутильную группу.
По существу предпочтительно в комплексе формулы (I), либо R1 и R1´ или R2 и R2´ представляют водород.
В таком случае фенильные группы в позиции 4 обоих инденильных лигандов обе замещены, либо в позиции 4´ фенильной группы или в позиции 3´и 5´фенильной группы.
В объем притязаний настоящей патентной заявки входят две отличающиеся 4-фенильные группы (например, 3,5-диметилфенил на одном индене и 3,5-ди-этилфенил на другом) или идентичные. В качестве альтернативы, два 3,5-заместителя в каждой 4-фенильной группе могут быть отличающимися (например, 3-метил-5-пропил) или идентичными.
Предпочтительно если два 3,5-заместителя в каждой фенильной группе представляют идентичные. Предпочтительно если две 4-позиции фенильных групп представляют идентичные. Более предпочтительно 4-фенильные группы представляют идентичные у обоих лигандов, и оба 3,5-заместителя представляют идентичные.
Еще более предпочтительно фенильные группы в позиции 4 инденильных лигандов, либо оба представляют 3,5-диметил-фенил (3,5-Me2Ph) группу, либо оба представляют 4-трет-бутил-фенильную (4-tBu-Ph) группу.
R3 и R3´ каждый независимо представляет линейную группу C1 - C6 нециклического углеводородного остатка/гидрокарбильную группу (hydrocarbyl group) или разветвленную или циклическую группу C4 - C10 нециклического углеводородного остатка/ гидрокарбильную группу / (cyclic C4 to C10 hydrocarbyl group), при этом группы представляют неразветвленные в α-позиции.
Подходящими примерами линейного C1-C6 нециклического углеводородного остатка/гидрокарбила являются алкильные группы, такие как метил, этил, n-пропил, n-бутил, n-пропил и n-гексил.
Подходящими примерами разветвленных или циклических групп C4-C10 нециклического углеводородного остатка / гидрокарбильных групп / (cyclic C4 to C10 hydrocarbyl group), которые не разветвлены в α-позиции, представляют бензил, изо-бутил, изопентил, изогексил, 2-(циклогексилметил), аналогичное им.
Предпочтительно R3 и R3´ представляют C1-C4 алкильную группу, более предпочтительно C1 - C2 алкильную группу и еще более предпочтительно метильную группу.
R3 и R3´ может представлять идентичный или отличающийся, предпочтительно они идентичны.
R4 и R4´ каждый независимо представляет третичную группу C4-C10 нециклического углеводородного остатка / гидрокарбильную группу.
Подходящие примеры третичных групп C4-C10 нециклического углеводородного остатка/гидрокарбильных групп представляют трет-бутил, 1-адамантил, 1,1-диметилбензил и аналогичное им.
Предпочтительно R4 и R4´ представляют третичную C4-C6 алкильную группу, более предпочтительно трет-бутил.
R4 и R4´ может представлять идентичный или отличающийся, предпочтительно они идентичны.
R5 и R5´ каждый независимо представляет линейную или разветвленную C1-C10 алкильную группу или C5-C10-арильную группу.
Предпочтительно R5 и R5´ каждый независимо представляет линейную или разветвленную C1 - C6 алкильную группу или фенильную группу и более предпочтительно линейную C1 - C4 алкильную группу.
Еще более предпочтительно R5 и R5´ представляют идентичные и наиболее предпочтительно R5 и R5´ оба представляют метил.
Конкретные соединения включают:
рац-Me2Si(2-Me-4-(3,5-Me2Ph)-5-OMe-6-tBu-Ind)2ZrCl2 и
рац-Me2Si(2-Me-4-(4-tBu-Ph)-5-OMe-6-tBu-Ind)2ZrCl2
Другой подходящий катализатор с единым центром полимеризации на металле для полимеризации композиции гетерофазного полипропилена по настоящему изобретению содержит (i) металлоцен с формулой (II)
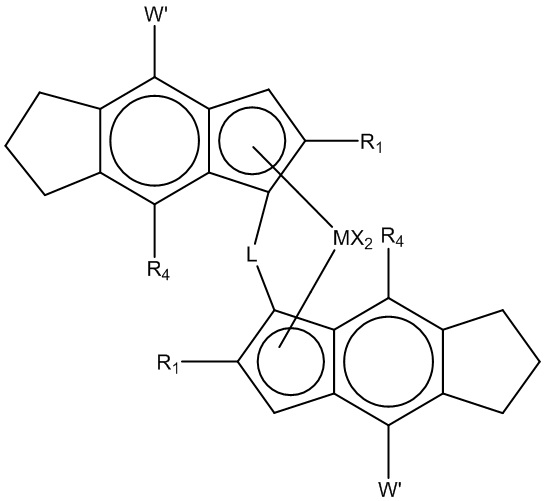
(Формула (II))
где
M представляет Zr или Hf;
каждый R1 представляет линейный или разветвленный C1-10 алкил;
L представляет этилен или Si(R6)2;
R6 представляет C1-10 алкил, C6-10-арил, C7-12-алкиларил, или C7-12-арилалкил;
каждый X представляет атом водорода, C1-6-алкокси, атом галогена или R группу;
R представляет C1-10 алкил,
каждый R4 представляет H или C1-3-алкил;
W' представляет фенил, пиридил, тиоофенил или фурил, необязательно замещенный вплоть до 2-мя группами R5;
каждый R5 представляет C1-10-алкильную, или две соседние R5 группы вместе взятые образуют фенильное кольцо, слитое с W', и
(ii) по меньшей мере, алюмоксан или соединение, способное образовывать алкилметаллоценовый катион.
Такие структуры описаны, например, в WO 2006/097497 или WO 2011/076780.
Пост-полимеризационная обработка
После удаления второй полимеризационной смеси из второго реактора полимеризации вторую полимеризационную смесь компаундируют с получением композиции гетерофазного полипропилена.
Следовательно, вторая полимеризационная смесь может быть смешана с добавками, как указано выше, и необязательно с другими полимерными компонентами.
Перед компаундированием вторая полимеризационная смесь может быть обработана при использовании традиционных пост-полимеризационных обработок, известных из области техники, к которой относится настоящее изобретение, таких как деактивация катализатора, отделение реагентов, пост-полимеризационная реакция полимера, таких как висбрекинг или нуклеирование (зародышеобразование).
Эти пост-полимеризационные обработки хорошо известны специалисту в области техники, к которой относится настоящее изобретение.
Компаундирование может быть проведено в миксерах или экструдере при использовании подходящих условий компаундирования.
Предпочтительно композицию гетерофазного полипропилена получают компаундированием и гранулированием.
Изделие
Дополнительно настоящее изобретение относится к изделию, содержащему композицию гетерофазного полипропилена, как приведено в описании настоящей патентной заявки, или применению композиции гетерофазного полипропилена, как приведено в описании настоящей патентной заявки, для получения изделия.
Согласно одному варианту осуществления настоящего изобретения, указанную композицию гетерофазного полипропилена используют для получения пленок, экструдированных изделий, изделий, полученных литьем в форме с раздувом, или изделий, полученных литьем под давлением, таких как пакетики или мешки, гибкие трубы или гибкие тубы, складные/разборные транспортировочные контейнеры, наряду с компонентами или изделиями для внешней или внутренней отделки автомобилей и изделия, используемые с внешней стороны или внутри автомобиля, такие как элементы приборных досок, облицовка дверей, консоли и изделия для облицовки, обшивки, обивки отделки салона автомобиля.
Изделие предпочтительно получают при использовании любого известного традиционного способа получения, подходящего для термопластичных полимеров, такого как литье под давлением, литье экструзионно-раздувным формованием, литье под давлением с раздувом и ориентированием или экструзия пленок с поливом.
ПРИМЕРЫ
1. Методы измерения
a) Скорость течения расплава (MFR2)
Скорость течения расплава представляет количество полимера в граммах, которое тестирующее устройство, стандартизованное согласно ISO 1133, экструдирует в течение 10 минут при определенной температуре и определенной нагрузке.
Скорость течения расплава MFR2 пропиленового полимера измеряют при температуре 230°C с нагрузкой 2,16 кг (MFR230/2,16), согласно ISO 1133.
Скорость течения расплава MFR2 пластомера на основе этилена измеряют при температуре 190°C с нагрузкой 2,16 кг (MFR190/2,16), согласно ISO 1133.
Скорость течения расплава MFR2 композиции полипропилена измеряют при температуре 230°C с нагрузкой 2,16 кг (MFR230/2,16), согласно ISO 1133.
b) Плотность
Плотность измеряют согласно ISO 1183D. Образцы получают при использовании прессования в формах согласно ISO 1872-2:2007.
c) Содержание сомономера
Количественную спектроскопию ядерно-магнитного резонанса (ЯМР) используют для количественной оценки содержания сомономера в полимерах.
Количественный анализ содержания сомономера в сополимерах поли(пропилена с этиленом)
Количественный анализ 13C{1H}ЯМР спектра записывают в состоянии раствора при использовании ЯМР спектрометра Bruker Advance III 400, работающего на частотах в пределах от 400,15 до 100,62 МГц для 1H и 13C, соответственно. Весь спектр записывают при использовании 13C оптимизированного 10 мм датчика измерения линейных величин при расширенном диапазоне температур при 125°C при использовании во всей пневматике газообразного азота. Около 200 мг материала растворяют в 3 мл 1,2-тетрахлорэтана-d2 (TCE-d2) с хром-(III)-ацетилацетонатом (Cr(acac)3) с получением в результате 65 мМ раствора релаксационного агента в растворителе {8}. Для обеспечения однородности раствора после получения начального образца в термоблоке ампулу для ЯМР спектроскопии дополнительно нагревают в печи с круглым вращающимся подом в течение по меньшей мере 1 часа. При установке в магнит ампулу подвергают воздействию 10 Гц. Такая схема была выбрана в первую очередь в виду необходимости высокого разрешения для определения регулярности молекулярной структуры. Создали стандартное одноимпульсное возбуждение без использования NOE, с оптимизированным углом наклона, с 1 секундной задержкой повтора и двух уровневой схемой развязки WALTZ16 {3, 4}. Всего для спектра потребовалось 6144 (6k) импульсов.
Провели количественный анализ на основе 13C{1H} ЯМР спектра с определенным средним значением и определили соответствующие количественные значения при использовании интеграла с использованием специальных собственных компьютерных программ. Для сополимеров этилен-пропилена все химические сдвиги косвенно указывают на центральную метиленовую группу этиленового блока (EEE) при 30,00 частей на миллион при использовании химического сдвига в растворителе. Этот подход позволяет провести сравнение с эталоном даже при отсутствии структурной единицы. Наблюдались характерные сигналы, соответствующие встраиванию этилена {7}.
Провели количественную оценку фракции сомономера при использовании способа Wang et. al. {6} путем интеграции множества сигналов всей спектральной области 13C{1H} спектра. Этот способ был выбран за его точность, надежность и возможность при необходимости объяснить присутствие региодефектов. Интегральные области незначительно регулируют для повышения применяемости к широким пределам содержания сомономеров.
Для систем, где наблюдается только соединенный в блоки этилен в PPEPP последовательностях, использовали способ Wang et. al., модифицированный для снижения влияния областей ненулевых интегралов, которые, как известно, отсутствуют. Такой подход снижает переоценку содержания этилена для такой системы и позволяет снизить число областей, используемых для определения абсолютного содержания этилена:
E = 0,5(Sββ + Sβγ + Sβγ + 0,5(Sαβ + Sαγ))
При использовании этого ряда областей соответствующее интегральное уравнение становится:
E = 0,5(IH +IG + 0,5(IC + ID))
Используются те же обозначения, что и в статье Wang et. al. {6}. Уравнения, использованные для определения абсолютного содержания пропилена, не модифицировали.
Молярный процент сомономера, введенного в полимер, рассчитывают по молярной фракции согласно:
E [мол. %] = 100 * fE
Массовый процент сомономера, введенного в полимер, рассчитывают по молярной фракции согласно:
E [мас.%] = 100 * (fE * 28,06) / ((fE * 28,06) + ((1-fE) * 42,08))
Библиографические ссылки:
1) Busico, V., Cipullo, R., Prog. Polym. Sci. 26 (2001) 443.
2) Busico, V., Cipullo, R., Monaco, G., Vacatello, M., Segre, A.L., Macroмольecules 30 (1997) 6251.
3) Zhou, Z., Kuemmerle, R., Qiu, X., Redwine, D., Cong, R., Taha, A., Baugh, D. Winniford, B., J. Mag. Reson. 187 (2007) 225.
4) Busico, V., Carbonniere, P., Cipullo, R., Pellecchia, R., Severn, J., Talarico, G., Macroмоль. Rapid Commun. 2007, 28, 1128.
5) Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253.
6) Wang, W-J., Zhu, S., Macroмольecules 33 (2000), 1157.
7) Cheng, H. N., Macroмольecules 17 (1984), 1950,
8) Singh, G., Kothari, A., Gupta, V., Polymer Testing 28 5 (2009), 475.
9) Kakugo, M., Naito, Y., Mizunuma, K., Miyatake, T. Macroмольecules 15 (1982) 1150,
10) Randall, J. Macroмоль. Sci., Rev. Macroмоль. Chem. Phys. 1989, C29, 201.
11) Resconi, L., Cavallo, L., Fait, A., Piemontesi, F., Chem. Rev. 2000, 100, 1253.
d) Дифференциальная сканирующая калориметрия (DSC анализ), температура плавления (Tm) и температура кристаллизации (Tc):
Измерения провели при использовании дифференциальной сканирующей калориметрии (DSC) TA Instrument Q2000 при использовании образцов от 5 до 7 мг. DSC проводят согласно ISO ISO 11357/ part 3 /method C2 в цикле нагревание /охлаждение/ нагревание при показателе сканирования 10°C/минуту при температуре в пределах от -30 до +225°C.
Температуру кристаллизации и теплоту кристаллизации (Hc) определили по стадии охлаждения, при этом температуру плавления и теплоту плавления (Hf) определяют на второй стадии нагревания.
e) Анализ Crystex QC
Метод кристаллической и растворимой фракций
Анализируют кристаллическую (CF) и растворимую фракцию (SF) полипропилена (PP), а также содержание сомономера и характеристическую вязкость соответствующих фракций при использовании CRYSTEX QC, Polymer Char (Valencia, Spain).
на Фигуре 1а приведено схематическое представление прибора CRYSTEX QC. Кристаллическая и аморфная фракции разделяются посредством температурных циклов растворения при 160°C, кристаллизации при 40°C и повторного растворения в 1,2,4-трихлорбензоле (1,2,4-TCB) при 160°C, как показано на Фигуре 1b. Количественная оценка SF и CF и определение содержания этилена (C2) проводится при использовании инфракрасного детектора (IR4) и онлайн-2-капиллярного вискозиметра, который используется для определения очевидной вязкости (IV).
IR4 детектор представляет собой детектор с несколькими длинами волн, детектирующий ИК-поглощение в двух разных диапазонах (CH3 и CH2) для определения концентрации и содержания этилена в сополимерах этилена-полипропилена. Детектор IR4 калибруют при использовании серии из 8 сополимеров EP с известным содержанием этилена в пределах от 2 до 69 мас.% (Определено 13C-ЯМР) и различной концентрацией от 2 до 13 мг / мл для каждого использованного сополимера EP, используемого для калибровки.
Количество растворимой фракции (SF) и кристаллической фракции (CF) коррелирует посредством калибровки XS с количеством «Фракции, растворимой в холодном ксилоле» (XCS), и, соответственно, фракции ксилола, нерастворимой в холодной воде (XCI), определенной согласно стандартному гравиметрическому методу, согласно ISO16152, калибровка XS проводится тестированием различных сополимеров EP с содержанием XS в диапазоне 2-31 мас.%.
Характеристическую вязкость (IV) исходного сополимера EP и его растворимую и кристаллическую фракции определяют при использовании онлайнового 2-капиллярного вискозиметра и соотносят с соответствующими показателями IV, определенными стандартным методом в декалине согласно ISO 1628. Калибровка достигается различными EP PP сополимерами с IV = 2-4 дл/г.
Образец композиции полипропилена для анализа взвешивают в концентрациях от 10 мг/мл до 20 мг/мл. После автоматического заполнения флакона 1,2,4-ТХБ, содержащего 250 мг/л 2,6-трет-бутил-4-метилфенола (ВНТ) в качестве антиоксиданта, образец растворяют при 160°С до полного растворения, обычно в течение 60 мин при постоянном перемешивании 800 об / мин.
Как показано на Фигурах 1a и b, определенный объем раствора образца впрыскивается в колонку, заполненную инертным носителем, где происходит кристаллизация образца и отделение растворимой фракции от кристаллической части. Этот процесс повторяется два раза. Во время первого впрыска весь образец измеряют при высокой температуре, определяя IV [дл/г] и C2 [мас.%] композиции PP. Во время второго впрыска измеряют растворимую фракцию (при низкой температуре) и кристаллическую фракцию (при высокой температуре) с циклом кристаллизации (мас.% SF, мас.% C2, IV).
EP означает сополимер этилена и пропилена.
PP означает полипропилен.
На Фигуре 1a приведена принципиальная схема прибора CRYSTEX QC.
На Фигуре 1b приведена диаграмма элюирования приведенного в качестве примера образца этилен-пропиленового сополимера и полученных растворимых и кристаллических фракций в колонке TREF (колонка, заполненная инертным материалом, например стеклянными шариками) (смотрите, Del Hierro, P.; Ortin, A.; Monrabal, B.; 'Soluble Fрацtion Analysis in polypropylene, The Column Advanstar Publications, February 2014. Pages 18-23).
f) Содержание фракции, растворимой в холодном ксилоле (XCS)
измеряют при 25°C согласно ISO 16152, first edition; 2005-07-01.
g) Характеристическую вязкость (iV)
измеряют согласно DIN ISO 1628/1, October 1999 в декалине при 135°C.
h) Температура стеклования (Tg), Динамический модуль упругости (G')
Температуру стеклования Tg и динамический модуль упругости G' определяют при использовании динамического механического анализа (DMA) согласно ISO 6721-7. Измерения проводят в режиме крутильных колебаний при использовании образцов, полученных литьем с прессованием (40×10×1 мм) при температуре от -100°C до +150°C со скоростью нагревания 2°C/минуту и частотой 1 Гц.
i) Молекулярные массы (Mw, Mn), распределение молекулярной массы (MWD)
Средние молекулярные массы (Mw, Mn), распределение молекулярной массы (MWD), их широту описывают при использовании индекса полидисперности PDI= Mw/Mn (где Mn представляет среднечисловую молекулярную массу, а Mw представляет среднемассовую молекулярную массу) определяют при использовании гельпроникающей хроматографии (GPC) согласно ISO 16014-4 2003 и ASTM D 6474-99.
Используют устройство Waters Alliance GPCV 2000 с рефрактометрическим детектором и он-лайн вискозиметром при использовании колонок 2 x GMHXL-HT и 1x G7000HXL-HT TSK-gel от Tosoh Bioscience и 1,2,4-трихлорбензола (TCB, стабилизированный 200 мг/л 2,6-ди третбутил-4-метил-фенолом) в качестве растворителя при температуре 145 °C и постоянной скорости потока 1 мл/минуту. Для анализа инжектируют 209,5 μл образца раствора. Колонку калибруют (согласно ISO 16014-2:2003) при использовании универсальной калибровки по узким 15 MWD стандартам полистирола (PS) в пределах от 1 кг/моль до 12 000 кг/моль. Для PS, PE и PP используют константы Марка-Хаувинка согласно ASTM D 6474-99. Все образцы получают, растворяя в пределах от 0,5 - 4,0 мг полимера в 4 мл (при 140°C) стабилизированного TCB (такой же, как мобильная фаза) и выдерживают максимально в течение 3 часов при максимально 160°C с непрерывным мягким перемешиванием перед забором образцов в GPC устройство.
k) Модуль упругости при изгибе
Модуль упругости при изгибе определяют согласно ISO 178 при тестовой скорости 2 мм/минуту и усилии 100 N, при этом длина между опорами составляет 64 мм, тестовые образцы имеют размер 80x10x4 мм³ (длина x ширина x толщина), полученных литьем под давлением согласно EN ISO 1873-2.
l) Тест на предел прочности при растяжении:
Тест на предел прочности при растяжении (модуль упругости при растяжении, предел прочности при растяжении до разрыва и удлинение при растяжении до разрыва) проводят при 23 °C согласно ISO 527-1 (скорость ползуна 1 мм/минуту) при использовании образцов типа 5A согласно ISO 527-2(1B), вырезанных из пластин толщиной 2мм, полученных литьем с прессованием.
m) твердость по Шору
Твердость по Шору A и твердость по Шору D определяют согласно ISO 868 при использовании пластин 2 мм толщиной, полученных литьем с прессованием, при этом постоянную нагрузку выдерживают в течение 15 секунд.
2. Катализаторы
MAO приобретен от Chemtura и используют, как 30 мас.% раствора в толуоле.
В качестве сурфактантов использовали 1H,1H-Перфтор(2-метил-3-оксагексан-1-ол) (CAS 26537-88-2), приобретенный у/от Unimatec, которые высушивают над активированными молекулярными ситами (2 раза) и дегазируют барботированием аргоном перед использованием (S2).
Гексадекафтор-1,3-диметилциклогексан (PFC) (CAS number 335-27-3) получают из коммерческих источников и высушивают над активированными молекулярными ситами (2 раза) и дегазируют барботированием аргоном перед использованием. Пропилен предоставлен от Borealis и надлежащим образом очищен перед использованием. Триэтилалюминия приобретен от Crompton и используют в чистой форме. Водород предоставлен от AGA и очищен перед использованием.
Все манипуляции с химическими реагентами и химические реакции проводились в атмосфере инертного газа при использовании технологии Schlenk и технологии перчаточного бокса, высушенной в печи стеклянной посудой, шприцами, иглами или канюлями.
a) MC-1
Синтез металлоцена IC1
2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-индан-1-он
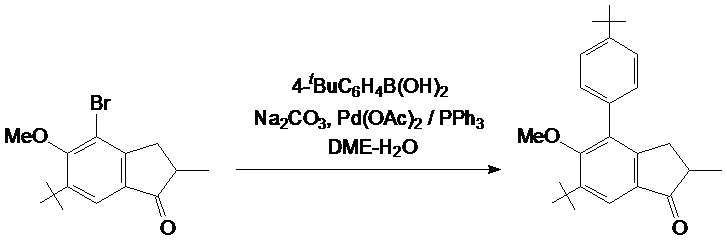
Смесь из 31,1 г (100 ммоль) 2-метил-4-бром-5-метокси-6-трет-бутил-индан-1-она, 25,0 г (140 ммоль) 4-трет-бутилфенил борной кислоты, 29,4 г (280 ммоль) Na2CO3, 1,35 г (6,00 ммоль, 6 мол. %) Pd(OAc)2, и 3,15 г (12,0 ммоль, 12 мол. %) PPh3 в 130 мл воды и 380 мл DME дефлегмируют в течение 6 часов в атмосфере аргона. Полученную смесь упаривают досуха. К остатку добавляли 500 мл дихлорметана и 500 мл воды. Органический слой отделяют, водный слой дополнительно экстрагируют 100 мл дихлорметана. Комбинированный органический экстракт сушат над Na2SO4, выпаривают досуха, и технический продукт выделяли при использовании флэш-хроматографии на силикагеле 60 (40-63 μм; элюент: гексаны-дихлорметан = 2:1, объем.). Технический продукт рекристаллизуют из n-гексана с получением 29,1 г (81%) белого твердого вещества.
Аналитический расчет для C25H32O2: C, 82,37; H, 8,85. Обнаружено: C, 82,26; H, 8,81.
1H ЯМР (CDCl3): δ 7,74 (s, 1H, 7-H в индениле), 7,48 (d, J = 8,0 Гц, 2H, 2,6-H в C6H4tBu), 7,33 (d, J = 8,0 Гц, 2H, 3,5-H в C6H4tBu), 3,27 (s, 3H, OMe), 3.15 (dd, J = 17,3 Гц, J = 7,7 Гц, 1H, 3-H в индан-1-оне), 2,67-2,59 (m, 1H, 2-H в индан-1-оне), 2,48 (dd, J = 17,3 Гц, J = 3,7 Гц, 3'-H в индан-1-оне), 1,42 (s, 9H, tBu в C6H4tBu), 1,38 (s, 9H, 6-tBu в индан-1-оне), 1,25 (d, J = 7,3 Гц, 3H, 2-Me в индан-1-оне).
2-метил-5-трет-бутил-6-метокси-7-(4-трет-бутилфенил)-1H-инден
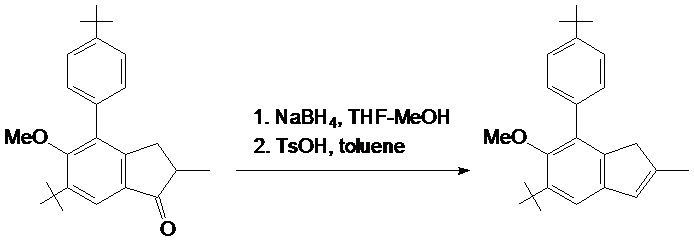
Раствор 28,9 г (79,2 ммоль) 2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-индан-1-она в 400 мл THF охладили до 5°C и добавили 5,00 г (132 ммоль) NaBH4. Далее к этой смеси по каплям добавили 100 мл метанола при интенсивном перемешивании в течение около 7 часов при 5°C. Полученную в результате смесь выпаривают досуха и остаток разделяют между 500 мл дихлорметана и 1000 мл 0,5 M HCl. Органический слой отделяют, водный слой дополнительно экстрагируют 100 мл дихлорметана. Комбинированный органический экстракт выпаривают досуха с получением бесцветного масла. В раствор из этого масла в 500 мл толуола добавили 1,0 г TsOH. Полученную смесь дефлегмируют при использовании головки Дина-Старка в течение 15 минут и затем охлаждают до комнатной температуры при использовании водяной бани. Полученный в результате красноватый раствор промывают 10% водным Na2CO3, органический слой отделяют, водный сой экстрагируют 2x100 мл дихлорметана. Комбинированный органический экстракт сушат над K2CO3 и затем пропускают через подложку с силикагелем 60 (40-63 μм). Силикагелевую подложку дополнительно промывают 50 мл дихлорметана. Комбинированный органический элюат выпаривают досуха с получением желтоватой кристаллической массы. Продукт выделяют рекристаллизацией этой массы из 150 мл горячего n-гексана. Кристаллы осаждают при 5°C и собирают высушенными в вакууме. Эта процедура привела к получению 23,8 г белого крупнокристаллического 2-метил-5-трет-бутил-6-метокси-7-(4-трет-бутилфенил)-1H-индена. Маточный раствор выпаривают досуха и остаток рекристаллизуют из 20 мл горячего n-гексана аналогичным образом. Эта процедура привела к получению дополнительных 2,28 г продукта. Следовательно, общий выход титульного продукта (title product) составил 26,1 г (95%).
Аналитический расчет для C25H32O: C, 86,15; H, 9,25. Обнаружено: C, 86,24; H, 9,40,
1H ЯМР (CDCl3): δ 7,44 (d, J = 8,5 Гц, 2H, 2,6-H в C6H4tBu), 7,40 (d, J = 8,5 Гц, 2H, 3,5-H в C6H4tBu), 7,21 (s, 1H, 4-H в индениле), 6,43 (m, 1H, 3-H в индениле), 3,20 (s, 3H, OMe), 3.15 (s, 2H, 1-H в индениле), 2,05 (s, 3H, 2-Me в индениле), 1,43 (s, 9H, 5-tBu в индениле), 1,37 (s, 9H, tBu в C6H4tBu).
Бис[2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил]диметилсилан
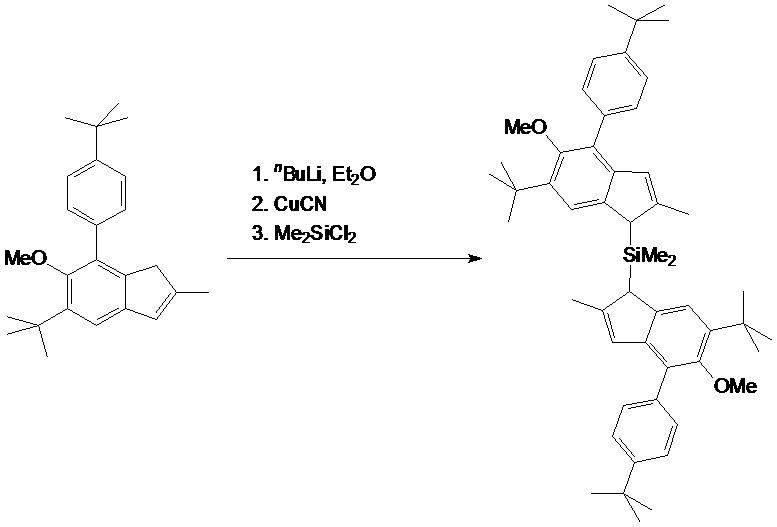
20,6 мл (50,06 ммоль) 2,43 M nBuLi в гексане добавили одной порцией в раствор 17,43 г (50,01 ммоль) 2-метил-5-трет-бутил-7-(4-трет-бутилфенил)-6-метокси-1H-индена в 300 мл простого эфира при -50°C. Эту смесь перемешивали в течение ночи при комнатной температуре, затем полученный в результате желтый раствор с большим количеством желтого преципитата охладили до -60°C, и добавили 225 мг CuCN. Полученную смесь перемешивали в течение 30 минут при -25°C, и затем добавили одной порцией 3,23 г (25,03 ммоль) дихлордиметилсилана. Далее эту смесь перемешивали в течение ночи при комнатной температуре. Раствор профильтровали через подложку из силикагеля (40-63 μм), которую дополнительно промыли 2×50 мл дихлорметана. Комбинированный фильтрат выпарили при пониженном давлении, и остаток высушили в вакууме при повышенной температуре. Эта процедура привела к получению 18,76 г (около 100%, чистота около 85%) бис[2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил]диметилсилана (Около 7:3 смесь диастероизомеров) в виде белого порошка.
1H ЯМР (CDCl3): δ 7,50-7,39 (m, 4H), 7,32 and 7,25 (2s, sum (суммарный) 1H), 6,48 и 6,46 (2s, sum 1H), 3,61 и 3,58(2s, sum 1H), 3,21 (s, 3H), 2,12 и 2,06 (2s, sum 3H), 1,43, 1,42, 1,39 и 1,38 (4s, sum 18H), -0,18 and -0,19 (2s, sum 3H).
13C{1H} ЯМР (CDCl3): δ 155,50, 149,45, 147,55, 147,20, 143,70, 139,37, 137,09, 135,22, 135,19, 129,74, 127,26, 126,01, 125,94, 125,04, 120,58, 120,36, 60,48, 47,42, 47,16, 35,15, 34,56, 31,47, 31,27, 31,20, 17,75, -4,92, -5,22, -5,32.
Рац-диметилсиландиил-бис[2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-инден-1-ил]цирконий дихлорид
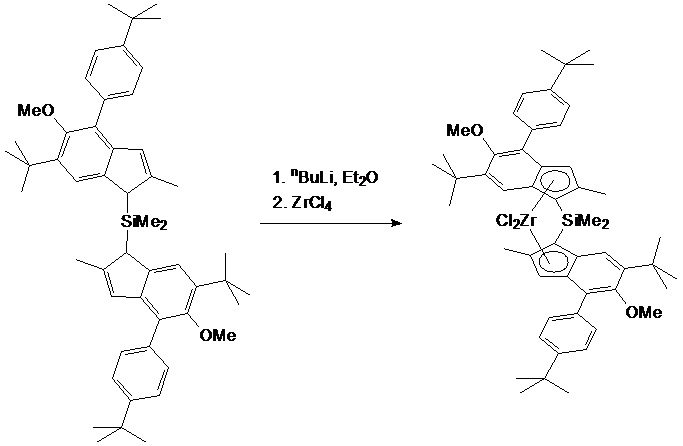
19,0 мл (46,17 ммоль) 2,43 M nBuLi в гексане добавили одной порцией в раствор 17,3 г (22,97 ммоль) бис[2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-1H-инден-1-ил]диметилсилана в 320 мл простого эфира, охладили до -60°C. Эту смесь перемешивали в течение ночи при комнатной температуре, затем полученный в результате желтый раствор с большим количеством желтого преципитата охладили до -60°C, и добавили 5,36 г (23,0 ммоль) ZrCl4. Реакционную смесь перемешивали в течение 24 часов при комнатной температуре с получением оранжевого раствора с большим количеством оранжевого преципитата. Этот преципитата профильтровали (G4), нагрели с 300 мл метилциклогексана, и полученную суспензию профильтровали, пока она была горячей от LiCl через стеклянную фритту (G4). Желтый порошок осаждали в течение ночи при комнатной температуре из профильтрованного (G3) фильтрата, и затем высушили в вакууме. Эта процедура привела к получению 3,98 г рац-комплекса, загрязненного около 3% мезо-формы. Эту смесь растворили в 40 мл горячего толуола, полученный раствор выпарили в вакууме - Около 10 мл. Желтый порошок, осажденный при комнатной температуре, профильтровали (G3) и затем высушили в вакууме с получением 3,41 г (16%) чистого рац-диметилсиландиил-бис[2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-инден-1-ил]циркония дихлорида (содержание мезо-формы <1%). Эфирный маточный раствор выпарили досуха, и остаток растворили в 100 мл теплого толуола. Это раствор профильтровали через стеклянную фритту (G4), и полученный фильтрат выпарили - Около 40 мл. Желтый порошок, осажденный из этого раствора при комнатной температуре сразу же профильтровали и высушили в вакууме с получением 2,6 г. Около 5 к 1 смеси рац- и мезо-цирконоценов (в пользу рац-). Все маточные растворы скомбинировали, выпарили до объема около 20 мл, и остаток измельчили в порошок с 100 мл n-гексана. Полученный оранжевый порошок собрали и высушили в вакууме. Эта процедура привела к получению 5,8 г смеси из рац- и мезо-цирконоценов. Следовательно, общий выход рац- и мезо-цирконоценов, выделенных при этом синтезе составил 11,81 г (56%).
Рац-диметилсиландиил-бис[2-метил-4-(4-трет-бутилфенил)-5-метокси-6-трет-бутил-инден-1-ил]цирконий дихлорид.
Аналитический расчет для C52H66Cl2O2SiZr: C, 68,39; H, 7,28. Обнаружено: C, 68,70; H, 7,43.
1H ЯМР (CDCl3): δ 7,63-7,52 (m, 2H), 7,50 (s, 1H), 7,44 (d, J = 8,1 Гц, 2H), 6,63 (s, 1H), 3,39 (s, 3H), 2,16 (s, 3H), 1,38 (s, 9H), 1,33 (s, 9H), 1,29 (s, 3H).
13C{1H} ЯМР (CDCl3): δ 160,00, 150,16, 144,25, 135,07, 133,79, 133,70, 129,25, 127,08, 125,39, 123,09, 121,32, 120,81, 81,57, 62,61, 35,78, 34,61, 31,39, 30,33, 18,37, 2,41.
Получение катализатора MC-1
Внутри перчаточного бокса 86,4 мг сухого и дегазированного сурфактанта S2 смешали с 2 мл 30 мас.% Chemtura MAO в колбе с септой и оставили реагировать в течение ночи. На следующий день 69,3 мг металлоцена IC1 (0,076 ммоль, 1 эквивалент) растворили в 4 мл 30 мас.% Chemtura MAO раствора в другой колбе с септой и оставили при перемешивании внутри печаточного бокса. Через 60 минут 1 мл MAO/раствор сурфактанта и 4 мл Раствора MAO-металлоцена последовательно добавили в 50 мл стеклянный реактор для эмульгирования, содержащий 40 мл PFC при -10 °C, снабженный верхней мешалкой (скорость перемешивания 600 оборотов в минуту). Красная эмульсия образуется сразу же и ее перемешивают в течение 15 минут при -10°C / 600 оборотов в минуту. Затем эмульсию перемещали через 2/4 тефлоновую трубку в 100 мл горячего PFC при 90°C, и перемешивают при 600 оборотов в минуту до момента завершения перемещения, и затем скорость снижают до 300 оборотов в минуту. После 15 минут перемешивания масляную баню удаляют и выключают мешалку. Катализатор оставляют отстаиваться для осаждения поверх PFC и через 35 минут растворитель отводят. Оставшийся катализатор высушивают в течение 2 часов при 50 °C в потоке аргона. В результате получили 0,75 г красного свободносыпучего порошка was.
b) MC-2
Синтез металлоцена IC2
(3,5-Диметилфенил)борная кислота
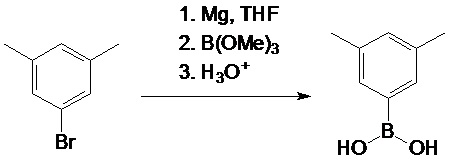
Получили раствор 3,5-диметилфенилмагний бромида из раствора 190,3 г (1,03 моль) 1-бром-3,5-диметилбензола в 1000 мл THF и 32 г (1,32 моль, 28% избыток) стружек магния, который охладили до -78°C, и добавили одной порцией 104 г (1,0 моль) триметилбората. Полученную в результате гетерогенную смесь перемешивали в течение ночи при комнатной температуре. Сложный борный эфир гидролизовали постепенным аккуратным добавлением 1200 мл 2 M HCl. Добавили 500 мл простого диэтилового эфира, органический слой отделяют, а водный слой дополнительно экстрагируют 2 x 500 мл простого диэтилового эфира. Комбинированный органический экстракт сушат над Na2SO4 и затем выпаривают досуха с получением белой массы. Последнюю измельчили в порошок с 200 мл n-гексана, профильтровали через стеклянную фритту (G3), и высушили преципитат в вакууме. Эта процедура привела к получению 114,6 г (74%) (3,5-диметилфенил)борной кислоты.
Аналитический расчет для C8H11BO2: C, 64,06; H, 7,39. Обнаружено: C, 64,38; H, 7,72.
1H ЯМР (DMSO-d6): δ 7,38 (s, 2H), 7,00 (s, 1H), 3,44(very br.s, 2H), 2,24 (s, 6H).
6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метилиндан-1-он
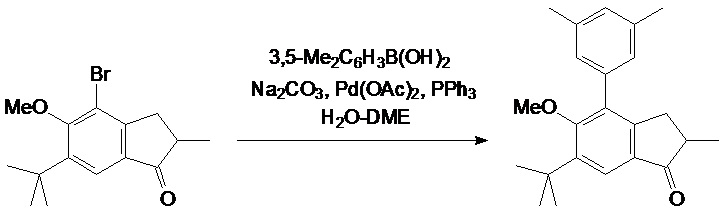
Смесь из 49,14 г (157,9 ммоль) 4-бром-6-трет-бутил-5-метокси-2-метилиндан-1-она, 29,6 г (197,4 ммоль, 1,25 экв.) (3,5-диметилфенил)борной кислоты, 45,2 г (427 ммоль) Na2CO3, 1,87 г (8,3 ммоль, 5 мол.%) Pd(OAc)2, 4,36 г (16,6 ммоль, 10 мол.%) PPh3, 200 мл воды, и 500 мл 1,2-диметоксиэтана дефлегмируют в течение 6,5 часов. DME выпарили при использовании ротационного испарителя, в остаток добавили 600 мл воды и 700 мл дихлорметана. Органический слой отделяют, а водный слой дополнительно экстрагируют 200 мл дихлорметана. Комбинированный экстракт сушат над K2CO3 и затем выпаривают досуха с получением черного масла. Технический продукт очистили при использовании флэш-хроматографии на силикагеле 60 (40-63 μм, гексан-дихлорметан = 1:1, объем., в таком случае, 1:3, объем.) с получением 48,43 г (91%) 6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метилиндан-1-она в виде коричневатого масла.
Аналитический расчет для C23H28O2: C, 82,10; H, 8,39. Обнаружено: C, 82,39; H, 8,52.
1H ЯМР (CDCl3): δ 7,73 (s, 1H), 7,02 (s, 1H), 7,01 (s, 2H), 3,32 (s, 3H), 3,13 (dd, J = 17,5 Гц, J = 7,8 Гц, 1H), 2,68-2,57 (m, 1H), 2,44 (dd, J = 17,5 Гц, J = 3.9 Гц), 2,36 (s, 6H), 1,42 (s, 9H), 1,25 (d, J = 7,5 Гц, 3H).
13C{1H} ЯМР (CDCl3): δ 208,90, 163,50, 152,90, 143,32, 138,08, 136,26, 132,68, 130,84, 129,08, 127,18, 121,30, 60,52, 42,17, 35,37, 34,34, 30,52, 21,38, 16,40.
5-трет-бутил-7-(3,5-диметилфенил)-6-метокси-2-метил-1H-инден
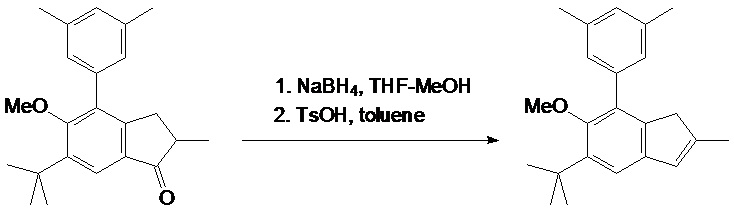
Добавили 8,2 г (217 ммоль) NaBH4 в раствор 48,43 г (143,9 ммоль) 6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метилиндан-1-она в 300 мл THF, охлажденного до 5°C. Затем в эту смесь покапельно добавили 150 мл метанола при интенсивном перемешивании в течение около 7 часов при 5°C. Полученную в результате смесь выпаривают досуха, и остаток разделяют между 500 мл дихлорметана и 500 мл 2 M HCl. Органический слой отделяют, водный слой дополнительно экстрагируют 100 мл дихлорметана. Комбинированный органический экстракт выпаривают досуха с получением немного желтоватого масла. В раствор из этого масла в 600 мл толуола добавили 400 мг TsOH, эту смесь дефлегмируют при использовании головки Дина-Старка в течение 10 минут и затем охлаждают до комнатной температуры при использовании водяной бани. Полученный раствор промыли 10% Na2CO3, органический слой отделяют, водный сой экстрагируют 150 мл дихлорметана. Комбинированный органический экстракт сушат над K2CO3 и затем пропускают через слой силикагеля 60 (40-63 μм). Слой силикагеля дополнительно промывают 100 мл дихлорметана. Комбинированный органический элюат выпарили досуха, и полученное в результате масло высушили в вакууме при повышенной температуре. Эта процедура привела к получению 45,34 г (98%) 5-трет-бутил-7-(3,5-диметилфенил)-6-метокси-2-метил-1H-индена, который далее используют без дополнительной очистки.
Аналитический расчет для C23H28O: C, 86,20; H, 8,81. Обнаружено: C, 86,29; H, 9,07.
1H ЯМР (CDCl3): δ 7,20 (s, 1H), 7,08 (br.s, 2H), 6,98 (br.s, 1H), 6,42 (m, 1H), 3,25 (s, 3H), 3,11 (s, 2H), 2,36 (s, 6H), 2,06 (s, 3H), 1,43 (s, 9H).
13C{1H} ЯМР (CDCl3): δ 154,20, 145,22, 141,78, 140,82, 140,64, 138,30, 137,64, 131,80, 128,44, 127,18, 126,85, 116,98, 60,65, 42,80, 35,12, 31,01, 21,41, 16,65.
Бис[6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метил-1H-инден-1-ил]диметилсилан
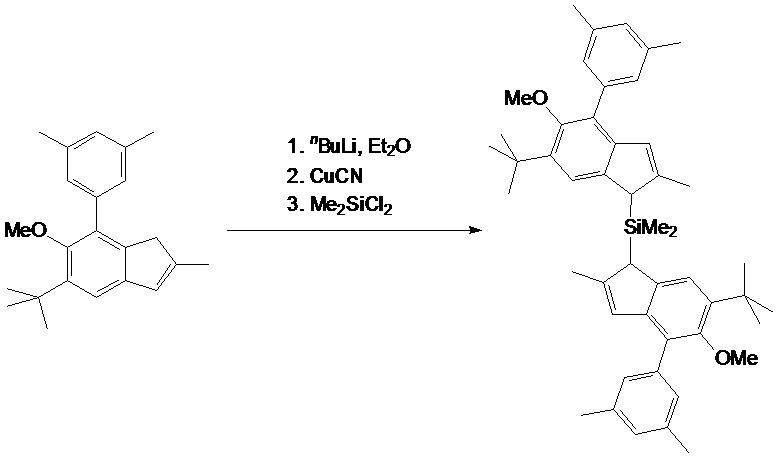
28,0 мл (70 ммоль) 2,5 M nBuLi в гексане добавили одной порцией в раствор 22,36 г (69,77 ммоль) 5-трет-бутил-7-(3,5-диметилфенил)-6-метокси-2-метил-1H-индена в 350 мл простого эфира при -50°C. Эту смесь перемешивали в течение ночи при комнатной температуре, затем полученный в результате оранжевый раствор с большим количеством желтого преципитата охладили до -60°C (при этой температуре преципитат почти полностью исчезает), и добавили 400 мг CuCN. Полученную в результате смесь перемешивают в течение 30 минут при -25°C, и затем добавили одной порцией 4,51 г (34,95 ммоль) дихлордиметилсилана. Эту смесь перемешивали в течение ночи при комнатной температуре, затем профильтровали через подложку из силикагеля 60 (40-63 μм), которую дополнительно промыли 2 x 50 мл дихлорметана. Комбинированный фильтрат выпарили при пониженном давлении, и остаток высушили в вакууме при повышенной температуре. Эта процедура привела к получению 24,1 г (99%) бис[6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метил-1H-инден-1-ил]диметилсилана (>90% чистота при проведении ЯМР, около 3:1 смесь стереоизомеров) в виде желтоватого стекла, которое далее используют без дополнительной очистки.
1H ЯМР (CDCl3): δ 7,49, 7,32, 7,23, 7,11, 6,99 (5s, sum 8H), 6,44 and 6,43 (2s, sum 2H), 3,67, 3,55 (2s, sum 2H), 3,27, 3,26 (2s, sum 6H), 2,38 (s, 12H), 2,13 (s, 6H), 1,43 (s, 18H), -0,13, -0,18, -0,24 (3s, sum 6H).
13C{1H} ЯМР (CDCl3): δ 155,29, 147,57, 147,23, 143,63, 139,37, 139,26, 138,19, 137,51, 137,03, 128,24, 127,90, 127,47, 126,01, 125,89, 120,53, 120,34, 60,51, 47,35, 47,16, 35,14, 31,28, 31,20, 21,44, 17,94, 17,79, -4,84, -4,89, -5,84.
Рац-диметилсиландиил-бис[2-метил-4-(3,5-диметилфенил)-5-метокси-6-трет-бутил-инден-1-ил]цирконий дихлорид (IE2)
27,7 мл (69,3 ммоль) 2,5 M nBuLi в гексане добавили одной порцией в раствор 24,1 г (34,53 ммоль) бис[6-трет-бутил-4-(3,5-диметилфенил)-5-метокси-2-метил-1H-инден-1-ил]диметилсилана (полученный выше) в 350 мл простого диэтилового эфира, охладили до -50°C. Эту смесь перемешивали в течение ночи при комнатной температуре, затем полученный в результате желтый раствор с большим количеством желтого преципитата охладили до -50°C, и добавили 8,05 г (34,54 ммоль) ZrCl4. Реакционную смесь перемешивали в течение 24 часов при комнатной температуре с получением красновато-оранжевого раствора, содержащего некоторое количество преципитата. Эту смесь выпарили досуха. Остаток нагрели с 200 мл толуола, и полученную суспензию профильтровали, пока она была горячей, через стеклянную фритту (G4). Фильтрат выпарили до 70 мл, и затем добавили 50 мл гексана. Кристаллы, осажденные из этого раствора в течение ночи при комнатной температуре, собрали, промыли 25 мл гексана, и высушили в вакууме. Эта процедура привела к получению 4,01 г чистого рац-цирконоцена. Маточный раствор выпарили - Около 50 мл, и добавили 50 мл гексана. Оранжевые кристаллы, осажденные из этого раствора в течение ночи при комнатной температуре, собрали и затем высушили в вакууме. Эта процедура привела к получению 2,98 г рац-цирконоцена. Снова маточный раствор выпарили почти досуха и добавили 50 мл гексана. Оранжевые кристаллы, осажденные из этого раствора в течение ночи при -30°C, собрали и высушили в вакууме. Эта процедура привела к получению 3,14 г рац-цирконоцена. Следовательно, общий выход рац-цирконоцена, выделенного при этом синтезе составил 10,13 г (34%).
Рац-IC2.
Аналитический расчет для C48H58Cl2O2SiZr: C, 67,26; H, 6,82. Обнаружено: C, 67,42; H, 6,99.
1H ЯМР (CDCl3): δ 7,49 (s, 1H), 7,23 (very br.s (очень уширенный), 2H), 6,96 (s, 1H), 6,57 (s, 1H), 3,44 (s,3H), 2,35 (s, 6H), 2,15 (s, 3H), 1,38 (s, 9H), 1,27 (s, 3H).
13C{1H} ЯМР (CDCl3): δ 159,78, 144,04, 137,87, 136,85, 134,89, 133,86, 128,85, 127,39, 127,05, 122,91, 121,18, 120,80, 81,85, 62,66, 35,76, 30,38, 21,48, 18,35, 2,41.
Получение катализатора MC-2
Внутри перчаточного бокса 86,2 мг сухого и дегазированного сурфактанта (S2) смешали с 2 мл 30 мас.% Chemtura MAO в колбе с септой и оставили реагировать в течение ночи. На следующий день 65,1 мг металлоцена IC2 (0,076 ммоль, 1 эквивалент) растворили в 4 мл 30 мас.% Chemtura MAO раствора в другой колбе с септой и оставили при перемешивании внутри печаточного бокса. Через 60 минут 1 мл MAO/раствор сурфактанта и 4 мл раствора MAO-металлоцена последовательно добавили в 50 мл стеклянный реактор для эмульгирования, содержащий 40 мл PFC при -10°C, снабженный верхней мешалкой (скорость перемешивания 600 оборотов в минуту). Красная эмульсия образуется сразу же, и ее перемешивают в течение 15 минут при -10°C / 600 оборотов в минуту. Затем эмульсию перемещали через 2/4 тефлоновую трубку в 100 мл горячего PFC при 90°C, и перемешивают при 600 оборотов в минуту до момента завершения перемещения, и затем и затем скорость снижают до 300 оборотов в минуту. После 15 минут перемешивания масляную баню удаляют и выключают мешалку. Катализатор оставляют отстаиваться для осаждения поверх PFC и через 35 минут растворитель отводят. Оставшийся катализатор высушивают в течение 2 часов при 50°C в потоке аргона. В результате получили 0,79 г красного свободносыпучего порошка.
c) MC-3
Синтез металлоцена IC3
(рац-μ-{бис-[η5-2-метил-4-(4-трет-бутилфенил)-1,5,6,7-тетрагидро-s-индацен-1-ил]диметилсиландиил}дихлорцирконий), полученный, как описано в WO2006/097497A1.
Его 1H ЯМР спектр соответствует представленному в указанной патентной заявке.
Получение катализатора MC-3
Внутри перчаточного бокса 85,7 мг сухого и дегазированного сурфактанта S2 смешали с 2 мл 30 мас.% Chemtura MAO в колбе с септой и оставили реагировать в течение ночи. На следующий день 38,0 мг металлоцена IC-3 (0,051 ммоль, 1 эквивалент) растворили в 4 мл 30 мас.% Chemtura MAO раствора в другой колбе с септой и оставили для перемешивания в перчаточном боксе.
Через 60 минут 1 мл раствора сурфактанта и 4 мл раствора MAO-металлоцен последовательно добавили в 50 мл стеклянный реактор для эмульгирования, содержащий 40 мл PFC при -10°C, снабженный верхней мешалкой (скорость перемешивания 600 оборотов в минуту). Общее количество MAO составляет 5 мл (300 эквивалентов). Красная эмульсия образуется сразу же и ее перемешивают в течение 15 минут при -10°C / 600 оборотов в минуту. Затем эмульсию перемещали через 2/4 тефлоновую трубку в 100 мл горячего PFC при 90°C, и перемешивают при 600 оборотов в минуту до момента завершения перемещения, и затем скорость снижают до 300 оборотов в минуту. После 15 минут перемешивания, масляную баню удаляют и выключают мешалку. Катализатор оставляют отстаиваться осаждения поверх PFC и через 35 минут растворитель отводят. Оставшийся катализатор высушивают в течение 2 часов при 50°C в потоке аргона. В результате получили 0,66 г красного свободносыпучего порошка.
d) MC-4
Синтез металлоцена IC-4
Получили аналогично металлоцену IC-3.
Получение катализатора MC-4
Внутри перчаточного бокса 85,7 мг сухого и дегазированного сурфактанта S2 смешали с 2 мл 30 мас.% Chemtura MAO в колбе с септой и оставили реагировать в течение ночи. На следующий день 58,1 мг металлоцена IC-4 (0,051 ммоль, 1 эквивалент) растворили 4 мл 30 мас.% Chemtura MAO раствора в другой колбе с септой и оставили для перемешивания в перчаточном боксе.
Через 60 минут 1 мл раствора сурфактанта и 4 мл раствора MAO-металлоцен последовательно добавили в в 50 мл стеклянном реакторе для эмульгирования, содержащим 40 мл при -10°C, снабженном верхней мешалкой (скорость перемешивания 600 оборотов в минуту). Общее количество MAO составляет 5 мл (300 эквивалентов). Красная эмульсия образуется сразу же и ее перемешивают в течение 15 минут при -10°C / 600 оборотов в минуту. Затем эмульсию перемещали через 2/4 тефлоновую трубку в 100 мл горячего PFC при 90°C, и перемешивают при 600 оборотов в минуту до момента завершения перемещения, и затем скорость снижают до 300 оборотов в минуту. После 15 минут перемешивания масляную баню удаляют и выключают мешалку. Катализатор оставляют отстаиваться для осаждения поверх PFC и через 35 минут растворитель отводят. Оставшийся катализатор высушивают в течение 2 часов при 50°C в потоке аргона. В результате получили 0,60 г красного свободносыпучего порошка.
3. Процедура офлайн предварительной полимеризации
Офлайн предварительно полимеризованные катализаторы подвергли офлайн предварительной полимеризации согласно следующей процедуре:
Катализаторы подвергли предварительной полимеризации согласно следующей процедуре: экспериментальную офлайн предварительную полимеризацию провели в 125 мл реакторе высокого давления, снабженном линиями подачи газа и верхней мешалкой. Сухой и дегазированный перфтор-1,3-диметилциклогексан (15 см3) и заданное количество прошедшего предварительную полимеризацию катализатора загрузили в реактор внутри перчаточного бокса и закупорили реактор. Затем реактор вынули из перчаточного бокса и поместили в охлаждающую водяную баню с поддержанием температуры 25°С. Верхняя мешалка и питающие линии были соединены и установлена скорость перемешивания 450 оборотов в минуту. Суммарное давление в реакторе повысили до около 5 бар избыточного давления и поддержали постоянным подачей пропилена через регулятор массового расхода до достижения целевой степени полимеризации. Реакцию останавливали мгновенным испарением летучих компонентов. Реактор открыли внутри перчаточного бокса, и содержание перелили в стеклянную емкость. Перфтор-1,3-диметилциклогексан выпаривали до постоянной массы для получения в результате прошедшего предварительную полимеризацию катализатора.
Таблица 1: Офлайн предварительная полимеризация
4. Получение примеров
a) Примеры по настоящему изобретению IE1-4
Гетерофазные сополимеры пропилена по Примерам по настоящему изобретению IE1-IE4 полимеризовали в 20 л реакторе, следуя следующим процедурам:
Стадия 1: предварительная полимеризация и гомополимеризация в массе
20 л реактор из нержавеющей стали, содержащий пропилен при 0,4 бар избыточного давления, заполнили 4430 г пропилена. В реактор ввели триэтилалюминий (1,6 мл 0,6 моль/л раствора в гептане) с добавлением 240 г пропилена. Раствор перемешали при 20°C и 250 оборотов в минуту в течение по меньшей мере 20 минут. Катализатор вводили, как описано далее. Заданное количество твердого прошедшего предварительную полимеризацию катализатора загрузили в 5 мл колбу из нержавеющей стали внутри перчаточного бокса и поверх первой колбы установили вторую 5 мл колбу, содержащую 4 мл n-гептана под давлением 10 бар избыточного давления азота. Эту систему подачи катализатора установили на порт поверх автоклава. Клапан между двумя колбами открыли и твердый катализатор проконтактировал с гептаном под давлением азота в течение 2 с, далее провели мгновенное испарение в реакторе 240 г пропилена. Предварительную полимеризацию провели в течение 10 минут. В конце стадии предварительной полимеризации температуру повысили до 80°C. Когда температура внутри реактора достигла 71°C, добавили 1,5 NL H2 через регулятор массового расхода в течение одной минуты. В течение полимеризации температуру в реакторе поддерживали постоянной при 80°C. Время полимеризации 37 минут измеряли, начиная с того момента, когда внутренняя температура реактора снизилась на 2°C ниже заданной температуры полимеризации.
Стадия 2: газофазная сополимеризация этилена и пропилена
По окончании стадии полимеризации в массе скорость мешалки снизили до 50 оборотов в минуту и снизили давление до 0,3 бар избыточного давления удалением мономеров. Затем в реактор ввели триэтилалюминий (0,80 мл 0,62 моль/л раствора в гептане) добавлением 240 г пропилена. Далее снова снижают давление до 0,3 бар избыточного давления удалением мономеров. Скорость мешалки установили на 180 оборотов в минуту и температуру реактора установили на 70 °C. Затем давление в реакторе повысили до 20 бар избыточного давления подачей газовой смеси C3/C2 с подачей C2/C3 0,25 масса/масса. Температуру поддерживали постоянной при использовании термостата, и давление поддерживали постоянным путем подачи через регулятор массового расхода газовой смеси C2/C3 композиции, соответствующей целевой полимерной композиции, до момента окончания установленного времени для этой стадии 67 минут.
Затем реактор охладили до около 30°C и провели мгновенное испарение летучих компонентов. После мгновенного испарения реактор 3 раза продували N2 и однократно проводили цикл вакуум/N2, продукт удалили и сушили в течение ночи в вытяжном шкафу. В 100 г полимера добавили Irganox B225 (раствор в ацетоне, поставщик: BASF SE), стеарат кальция (Cas No 1592-23-0, поставщик Faci) и Irgastab FS 042 (поставщик. BASF SE) в количествах, приведенных в Таблице 2 ниже и сушили в течение ночи в вытяжном шкафу с последующей 2 часовой сушкой в вакуумном сушильном шкафу при 60°C.
Таблица 2: Композиции по Примерам по настоящему изобретению IE1-4
b) Сравнительные примеры CE5-6
Гомополимер пропилена Сравнительных примеров CE5 и CE6 полимеризовали в 20 л реакторе, следуя следующим процедурам:
Стадия 1: Предварительная полимеризация и гомополимеризация в массе
20 л реактор из нержавеющей стали, содержащий пропилен при 0,4 бар избыточного давления, заполнили 4455 г пропилена. В реактор ввели триэтилалюминий (0,8 мл 0,62 моль/л раствора в гептане) добавлением 250 г пропилена. Раствор перемешали при 20°C и 250 оборотов в минуту в течение по меньшей мере 20 минут. Катализатор вводили, как описано далее. Заданное количество твердого прошедшего предварительную полимеризацию катализатора загрузили в 5 мл колбу из нержавеющей стали внутри перчаточного бокса и поверх первой колбы установили вторую 5 мл колбу, содержащую 4 мл n-гептана под давлением 10 бар избыточного давления азота. Эту систему подачи катализатора установили на порт поверх автоклава. Клапан между двумя колбами открыли и твердый катализатор проконтактировал с гептаном под давлением азота в течение 2 с, далее провели мгновенное испарение в реакторе 250 г пропилена. Предварительную полимеризацию провели в течение 10 минут. В конце стадии предварительной полимеризации температуру повысили до 80°C. Когда температура внутри реактора достигла 71°C, добавили 2,0 NL H2 через регулятор массового расхода в течение одной минуты. В течение полимериазции температуру в реакторе поддерживали постоянной 80°C. Время полимеризации 37 минут измеряли, начиная с того момента, когда внутренняя температура реактора снизилась на 2°C ниже заданной температуры полимеризации.
Стадия 2: газофазная гомополимеризация
По окончании стадии полимеризации в массе, скорость мешалки снизили до 50 оборотов в минуту, и давление снизили до 23 бар избыточного давления удалением мономера. После чего скорость мешалки установили на 180 оборотов в минуту, а температуру установили на 80°C и давление установили 35,7 бар избыточного давления. Через регулятор массового расхода подавали 1,5 NL водорода за одну минуту. Во время газофазной гомополимеризации оба показателя, и давление, и температура поддердивались постоянными при использовании регулятора массового расхода (подача в г пропилена) и термостата в течение 40 минут.
Затем реактор охладили до около 30°C и провели мгновенное испарение летучих компонентов. После мгновенного испарения реактор 3 раза продували N2 и однократно проводили цикл вакуум/N2, продукт удалили и сушили в течение ночи в вытяжном шкафу. 45 г гомополимера пропилена компаундировали с 55 г Queo 8201 или Vistamaxx 6102, как приведено в Таблица 2 ниже, с получением композиции гетерофазного полипропилена и сушили в течение ночи в вытяжном шкафу с последующей 2 часовой сушкой в вакуумном сушильном шкафу при 60°C.
Queo™ 8201 представляет пластомер 1-октена на основе этилена, полученный в процессе полимеризации в растворе при использовании металлоценнового катализатора с MFR2 (2,16 кг, 190°C) 1,1 г/10 минут и плотностью 883 кг/м³, коммерчески доступен от Borealis AG, Austria.
Vistamaxx™ 6102 представляет пластомер этилена на основе пропилена, полученный в процессе полимеризации в растворе при использовании металлоценнового катализатора с MFR2 (2,16 кг, 230°C) 1,4 г/10 минут и плотностью 862 кг/м³, коммерчески доступен от Exxon Mobil, USA.
Компаундирование Сравнительных примеров провели в двух шнековом экструдере PRISM TSE16 при 200-220°C с последующим отверждением расплавленной стренги в водяной бане и гранулированием стренги.
Таблица 3: Композиции Сравнительных примеров CE5-6
5. Свойства композици по Примерам
Примеры IE1-4 и CE5-6 имеют следующие свойства, приведенные в Таблице 4.
Таблица 4: Свойства примеров IE1-4 и CE5-6
н.о. не определяли
Следующие каучуки использовали в качестве Контрольных примеров RE7-RE11, они представляют современные мягкие термопластичные полиолефины на основе пропилена (TPO), полученные в присутствии катализатора Циглера-Наттапри при использовании собственного процесса LyondellBasell's Catalloy, и все они коммерчески доступны от LyondellBasell, Italy, под торговой маркой Adflex.
Дополнительно, в качестве Контрольного примера RE12 используют каучук, представляющий гетерофазный сополимер пропилена с матричной фазой из неупорядоченного сополимера этилена и пропилена, и эластомерной фазой из этилена и пропилена, который получили по примеру E1 WO 2014/075973 A1 в присутствии катализатора Циглера-Натта. Для RE12 дополнительно к модулю упругости при изгибе определяли модуль упругости при растяжении (TM) 484 МПа. Свойства этих каучуков приведены ниже в Таблице 5
Таблица 5: Свойства Контрольных примеров RE7-11
дл/г
н.о. не определяли
Как видно из приведенных выше в Таблицах 4 и 5 свойств, композиция гетерофазного полипропилена по настоящему изобретению (IE1-4) демонстрирует улучшенную мягкость по сравнению с композицией гетерофазного полипропилена Сравнительных примеров CE5-6, Контрольных примеров RE7-11 и Контрольного примера RE12. Это видно из того, что композиции Примеров по настоящему изобретению демонстрируют более низкую твердость по Шору D при таком же модуле упругости при растяжении (модуль упругости при изгибе для RE7-11). По сравнению с Контрольным примером RE12 композиции Примеров по настоящему изобретению демонстрируют более низкую твердость по Шору D вместе с более низким модулем упругости при растяжении.
Фиг. 2 иллюстрирует указанную улучшенную мягкость в графике модулей Твердости по Шору D. Следовательно, для примеров IE1-4 и CE5-6 приведен модуль упругости при растяжении, для примеров RE7-11 приведен модуль упругости при изгибе.
Следовательно, ромбовидные точки показывают отношение модуль упругости при растяжении - Твердость по Шору D примеров по настоящему изобретению IE1-4. Точки квадратной формы показывают отношение модуль упругости при растяжении - Твердость по Шору D Сравнительных примеров CE5-6. Точки треугольной формы показывают отношение модуль упругости при изгибе - Твердость по Шору D Контрольных примеров RE7-11.
| название | год | авторы | номер документа |
|---|---|---|---|
| МНОГОСЛОЙНАЯ ПОЛИМЕРНАЯ ПЛЕНКА | 2014 |
|
RU2635599C2 |
| Полупроводящая полипропиленовая композиция | 2021 |
|
RU2832967C1 |
| КОМПОЗИЦИЯ ПРОПИЛЕНА С ПОВЫШЕННОЙ УДАРНОЙ ПРОЧНОСТЬЮ ПРИ НИЗКОЙ ТЕМПЕРАТУРЕ | 2014 |
|
RU2648673C2 |
| АРМИРОВАННАЯ ВОЛОКНАМИ КОМПОЗИЦИЯ ПОЛИПРОПИЛЕНА С БОЛЬШИМ УДЛИНЕНИЕМ ПРИ РАЗРЫВЕ | 2016 |
|
RU2684109C1 |
| КОМПОЗИЦИЯ ГЕТЕРОФАЗНОГО ПОЛИПРОПИЛЕНА С УЛУЧШЕННЫМ БАЛАНСОМ СВОЙСТВ | 2019 |
|
RU2754224C1 |
| ГЕТЕРОФАЗНЫЙ СОПОЛИМЕР ПРОПИЛЕНА С ВЫСОКОЙ ТЕМПЕРАТУРОЙ ПЛАВЛЕНИЯ | 2015 |
|
RU2654696C2 |
| КОМПОЗИЦИЯ ПОЛИПРОПИЛЕНА, АРМИРОВАННОГО ВОЛОКНАМИ С ВЫСОКОЙ ТЕКУЧЕСТЬЮ | 2013 |
|
RU2588568C2 |
| КОМПОЗИЦИЯ ПОЛИПРОПИЛЕНА | 2018 |
|
RU2745620C1 |
| Гетерофазный сополимер | 2014 |
|
RU2668075C2 |
| ОГНЕЗАЩИТНАЯ ПОЛИПРОПИЛЕНОВАЯ КОМПОЗИЦИЯ | 2016 |
|
RU2667288C1 |
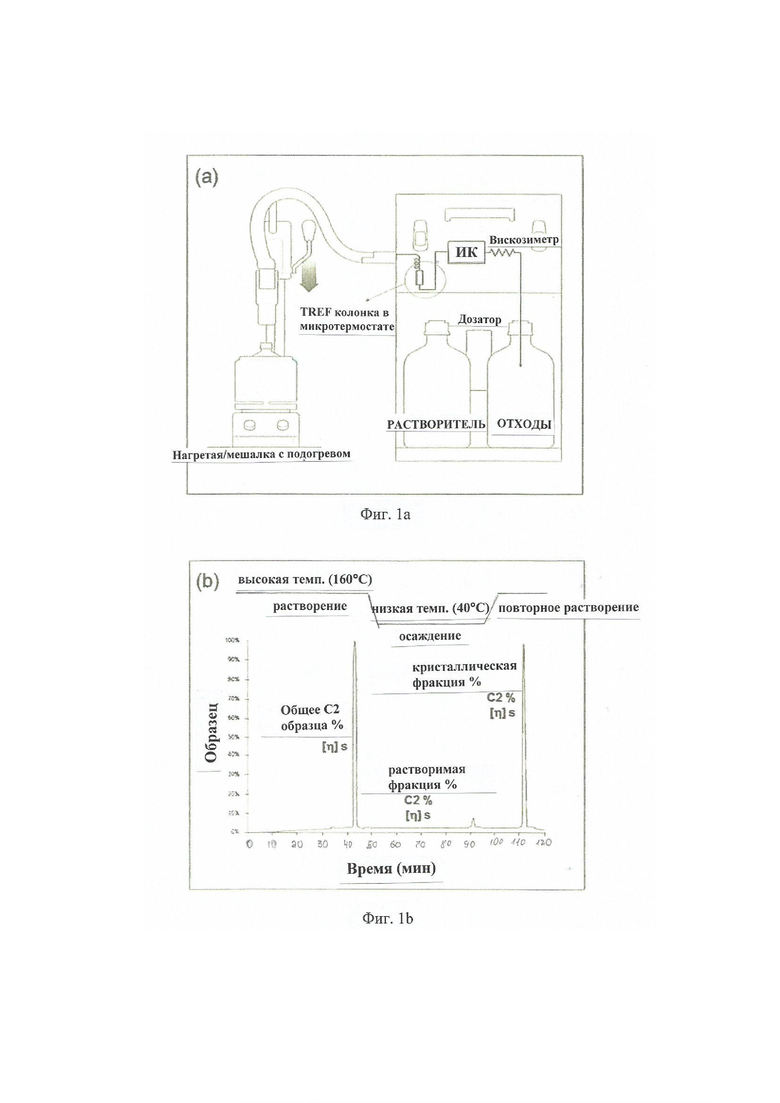
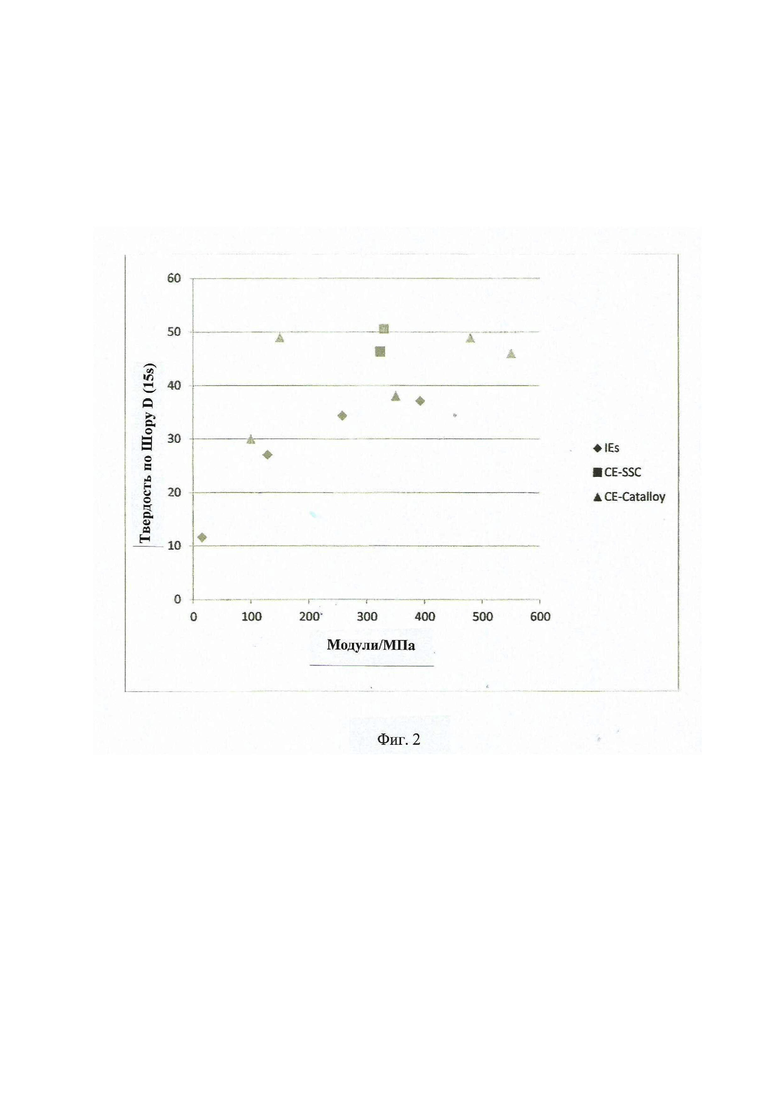
Настоящее изобретение относится к композиции гетерофазного полипропилена для получения изделий, содержащей каучуковое полимерное основание, которое содержит матричную фазу и эластомерную фазу, диспергированную в ней, где матричную фазу и эластомерную фазу полимеризуют в присутствии катализатора с единым центром полимеризации на металле; и где каучуковое полимерное основание содержит: (A) от 20 до 55 мас.% кристаллической фракции (CF), как измерено при использовании Crystex QC в трихлорбензоле, которая представляет фракцию гомополимера пропилена или фракцию сополимера из пропиленовых мономерных единиц и сомономерных единиц из этилена или альфа-олефинов с от 4 до 12 атомов углерода с содержанием сомономерных единиц вплоть до 6,0 мас.%; и (B) от 45 до 80 мас.% растворимой фракции (SF), как измерено при использовании Crystex QC в трихлорбензоле, которая представляет сополимер из пропиленовых мономерных единиц и сомономерных единиц из этилена или альфа-олефинов с 4-12 атомами углерода, с содержанием сомономерных единиц от 17 до 55 мас.% и характеристической вязкостью iV от 1,2 до 7,0 дл/г, где композиция гетерофазного полипропилена имеет модуль упругости при растяжении не более чем 700 МПа. 4 н. и 11 з.п. ф-лы, 2 ил., 5 табл.
1. Композиция гетерофазного полипропилена для получения изделий, содержащая каучуковое полимерное основание, которое содержит матричную фазу и эластомерную фазу, диспергированную в ней, где матричную фазу и эластомерную фазу полимеризуют в присутствии катализатора с единым центром полимеризации на металле; и
где каучуковое полимерное основание содержит:
(A) от 20 до 55 мас.% кристаллической фракции (CF), как измерено при использовании Crystex QC в трихлорбензоле, которая представляет фракцию гомополимера пропилена или фракцию сополимера из пропиленовых мономерных единиц и сомономерных единиц из этилена или альфа-олефинов с от 4 до 12 атомов углерода с содержанием сомономерных единиц вплоть до 6,0 мас.%; и
(B) от 45 до 80 мас.% растворимой фракции (SF), как измерено при использовании Crystex QC в трихлорбензоле, которая представляет сополимер из пропиленовых мономерных единиц и сомономерных единиц из этилена или альфа-олефинов с 4-12 атомами углерода, с содержанием сомономерных единиц от 17 до 55 мас.% и характеристической вязкостью iV от 1,2 до 7,0 дл/г,
где композиция гетерофазного полипропилена имеет модуль упругости при растяжении не более чем 700 МПа.
2. Композиция гетерофазного полипропилена по п. 1 с твердостью по Шору D не более чем 50.
3. Композиция гетерофазного полипропилена по п. 1 или 2 с твердостью по Шору A не более чем 100.
4. Композиция гетерофазного полипропилена по любому из предшествующих пунктов, с общим содержанием сомономерных единиц от 6,0 до 35,0 мас.% от общей массы композиции гетерофазного полипропилена.
5. Композиция гетерофазного полипропилена по любому из предшествующих пунктов с общей характеристической вязкостью iV от 1,2 до 7,0 дл/г.
6. Композиция гетерофазного полипропилена по любому из предшествующих пунктов с температурой плавления от 135°C до 165°C.
7. Композиция гетерофазного полипропилена по любому из предшествующих пунктов со скоростью течения расплава MFR2 (230°C, 2,16 кг) от 0,05 до 100 г/10 минут.
8. Композиция гетерофазного полипропилена по любому из предшествующих пунктов, где кристаллическая фракция (CF) имеет характеристическую вязкость iV от 1,0 до 7,0 дл/г.
9. Композиция гетерофазного полипропилена по любому из предшествующих пунктов, где кристаллическая фракция (CF) представляет фракцию сополимера из пропиленовых мономерных единиц и этиленовых сомономерных единиц.
10. Композиция гетерофазного полипропилена по любому из предшествующих пунктов, где растворимая фракция (SF) представляет фракцию сополимера из пропиленовых мономерных единиц и этиленовых сомономерных единиц.
11. Способ получения композиции гетерофазного полипропилена по любому из предшествующих пунктов, включающий стадии:
a) полимеризации пропилена и необязательно сомономерных единиц, выбранных из этилена или альфа-олефинов с от 4 до 12 атомов углерода, в первом реакторе полимеризации в присутствии катализатора с единым центром полимеризации на металле с получением первой полимеризационной смеси, содержащей первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле;
b) перемещения первой полимеризационной смеси во второй реактор полимеризации;
c) полимеризации пропилена и сомономерных единиц, выбранных из этилена или альфа-олефинов с от 4 до 12 атомов углерода, в указанном втором реакторе полимеризации в присутствии указанного катализатора с единым центром полимеризации на металле с получением второй полимеризационной смеси, содержащей второй сополимер пропилена, указанный первый гомо- или сополимер пропилена и катализатор с единым центром полимеризации на металле, где массовое соотношение указанного первого гомо- или сополимера пропилена к указанному второму сополимеру пропилена составляет в пределах от 20:80 до 55:45;
d) удаления указанной второй полимеризационной смеси из указанного второго реактора полимеризации; и
e) компаундирования указанной второй полимеризационной смеси необязательно с добавлением добавок с получением композиции гетерофазного полипропилена.
12. Способ по п. 11, где катализатор с единым центром полимеризации на металле содержит (i) Комплекс с формулой (I)
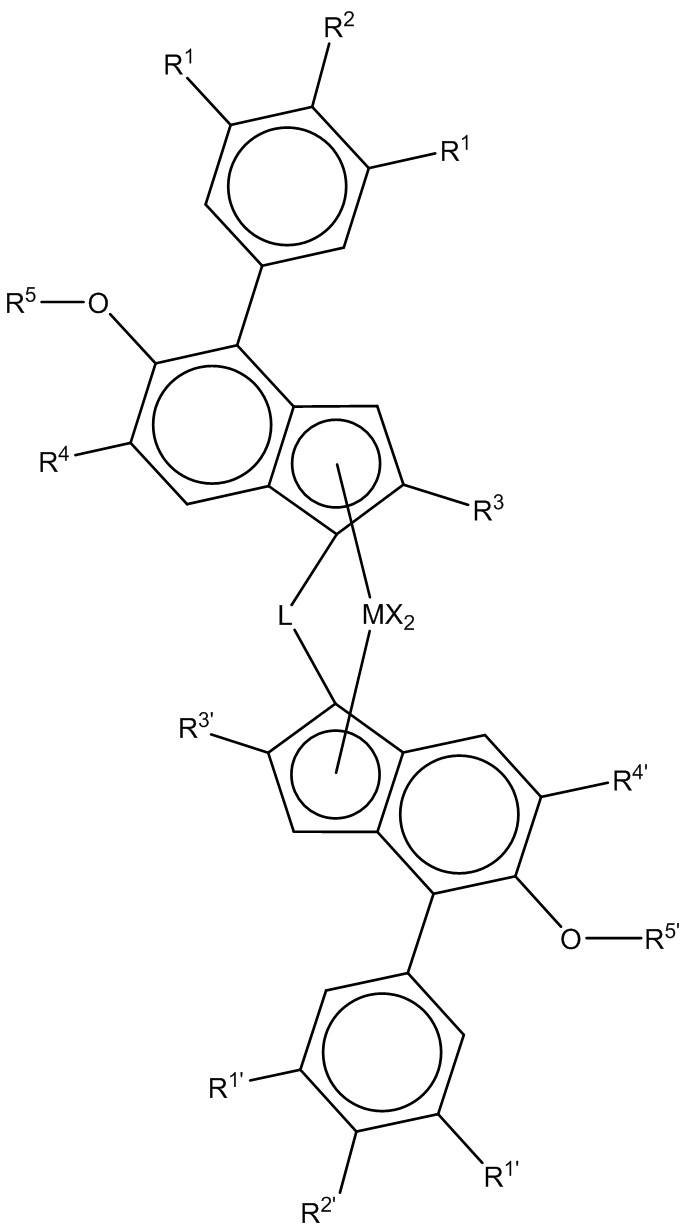 (Формула (I)),
(Формула (I)),
где
M представляет цирконий или гафний;
каждый X независимо представляет лиганд сигма-донора;
L представляет двухвалентный мостик, выбранный из -R'2C-, -R'2C-CR'2-, -R'2Si-, -R'2Si-SiR'2-, -R'2Ge-, где каждый R' независимо представляет атом водорода или C1-C20 гидрокарбильную группу, необязательно содержащую один или более гетероатом из групп 14-16 Периодической таблицы или атомы фтора, или необязательно две R' группы вместе взятые образуют кольцо;
R1 и R1´ каждый независимо представляет водород, C5-C10-арил или группу -CH(Rx)2, где каждый Rx независимо представляет H или C1-10 гидрокарбильную группу, и необязательно два Rx вместе взятые образуют кольцо,
R2 и R2´ каждый независимо представляет водород, C5-C10-арил или группу –C(Ry)3,
где каждый Ry независимо представляет H или C1-10 гидрокарбильную группу, или необязательно две или три Ry группы вместе взятые образуют кольцо,
при этом по меньшей мере одно из R1 или R2 и одно из R1´ или R2´ отличается от водорода и
при этом R2 вместе с одним из R1, наряду с R2´ вместе с одним из R1´ могут являться частью дополнительного моно- или мультициклического кольца, конденсированного с фенильным кольцом;
R3 и R3´ каждый независимо представляет линейную C1-C6 гидрокарбильную группу или разветвленную или циклическую C4-C10 гидрокарбильную группу, при этом группы представляют неразветвленные в α-позиции;
R4 и R4´ каждый независимо представляет третичную C4-C10 гидрокарбильную группу;
R5 и R5´ каждый независимо представляет линейную или разветвленную C1-C10 алкильную группу или C5-C10-арильную группу и
(ii) сокатализатор, содержащий соединение металла группы 13.
13. Способ по п. 11, где катализатор с единым центром полимеризации на металле содержит (i) Комплекс с формулой (II)
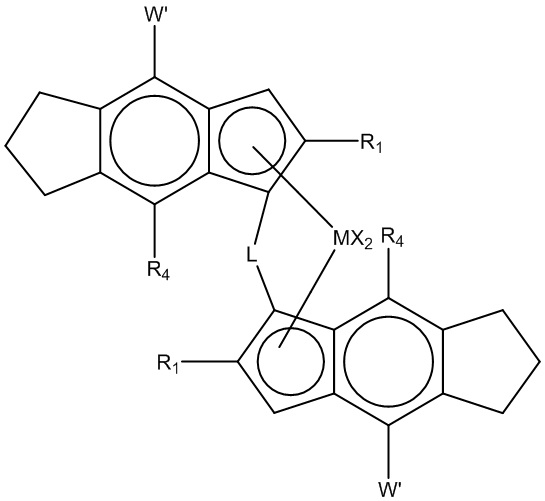 (Формула (II))
(Формула (II))
где
M представляет Zr или Hf;
каждый R1 представляет линейный или разветвленный C1-10 алкил;
L представляет этилен или Si(R6)2;
R6 представляет C1-10 алкил, C6-10-арил, C7-12-алкиларил или C7-12-арилалкил;
каждый X представляет атом водорода, C1-6-алкокси, атом галогена или R группу;
R представляет C1-10 алкил;
каждый R4 представляет H или C1-3-алкил;
W' представляет фенил, пиридил, тиоoфенил или фурил, необязательно замещенный вплоть до 2-мя группами R5;
каждый R5 представляет C1-10-алкил, или две соседние R5 группы вместе взятые образуют фенильное кольцо, слитое с W', и
(ii) по меньшей мере, алюмоксан или соединение, способное образовывать алкилметаллоценовый катион.
14. Изделие, содержащее композицию гетерофазного полипропилена по любому из пп. 1-10.
15. Применение композиции гетерофазного полипропилена по любому из пп. 1-10 для получения изделия.
| Устройство для закрепления лыж на раме мотоциклов и велосипедов взамен переднего колеса | 1924 |
|
SU2015A1 |
| ИНТЕГРИРОВАННОЕ УСТРОЙСТВО ОПОЗНАВАНИЯ ВОЗДУШНЫХ ЦЕЛЕЙ | 2010 |
|
RU2452975C1 |
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| СМЕСИТЕЛЬ | 1991 |
|
RU2048185C1 |
| ГЕТЕРОФАЗНЫЙ ПОЛИПРОПИЛЕН С ХАРАКТЕРИСТИКАМИ ВЫСОКОЙ УДАРНОЙ ВЯЗКОСТИ | 2008 |
|
RU2446181C1 |

