Область техники
Настоящее изобретение относится к области аналитических исследований, и может быть использовано в медицинской диагностике и биохимических исследованиях для определения маркеров нейромедиаторного обмена в биологических жидкостях с высокими чувствительностью, селективностью и экспрессностью.
Уровень техники
Нейромедиаторный обмен осуществляется посредством метаболизма катехоламинов и составляет основу нервной медиации, как периферической, так и центральной нервной систем. Такие катехоламины (КА), такие как дофамин (ДА), адреналин (АД), норадреналин (НА) и их метаболиты (гомованилиновая и ванилилминдальная кислоты (ГВК и ВМК, соответственно)), норметанефрин (НМН), их предшественник L-ДОФА (3-(3,4-дигидроксифенил)-L-аланин), серотонин (5-гидрокситирамин, 5-ГТ) и его катаболит 5-гидроксииндол-3-уксусная кислота (5-ГИУК), являются биологически активными аминами, участвующими в регуляции нервной деятельности, сердечно-сосудистой системы, системы липопероксидации, в энергетическом обмене и сократительной способности миокарда, в микроциркуляции и снабжении тканей кислородом, в регуляции эмбриогенеза. В связи с тем, что количество катехоламинов и их метаболитов в норме и при патологии различно, их возможно использовать в качестве диагностических маркеров как при проведении фундаментальных исследований, так и в клинической практике. Уровень НА в плазме крови позволяет прогнозировать течение заболевания у пациентов с нарушениями в работе сердечно-сосудистой системы. Соотношение катехоламинов и метаболитов в моче используют для диагностики опухолевых нейроэндокринных (нейробластомы, карциномы, параганглиомы, феохромоцитомы, инсулиномы, соматостиномы, глюкогономы, гастринома и др.) и нейродегенеративных заболеваний (болезней Альцгеймера и Паркинсона). Кроме того, показано, что нарушения в биосинтезе и метаболизме катехоламинов могут возникать впоследствии действия ионизирующего излучения на организм в ходе лечения лучевой терапией.
Сложность определения катехоламинов в плазме крови и других биологических жидкостях на сегодняшний день обусловлена тем, что у здорового человека их концентрации очень низки (на уровне 1 нМ), а при различных патологических нарушениях их концентрация в биообъектах в ряде случаев снижается еще на порядок. При этом следует учитывать, что в крови они быстро окисляются моноаминоксидазами тромбоцитов, в связи с этим определение маркеров нейромедиаторного обмена в организме (особенно при развитии кризисных состояний) должно быть очень быстрым (в течение 15-30 мин). Современные инструментальные методы анализа, к сожалению, не решают эту проблему комплексно: высокая чувствительность и селективность метода высокоэффективной жидкостной хроматографии (ВЭЖХ) с масс-спектрометрическим детектированием не обеспечивает нужной экспрессности, а ферментативные биосенсоры и иммунохимические методы определения указанного класса аналитов не обеспечивают нужной чувствительности и воспроизводимости полученных результатов. В связи с этим, необходимо создание новых биораспознающих флуоресцентных индикаторных систем для разработки высокоэкспрессных, чувствительных и селективных методик для одновременного определения сразу нескольких катехоламинов и их метаболитов в большом количестве проб биологических жидкостей без предварительной пробоподготовки с целью диагностики различных заболеваний.
Из уровня техники известно решение, представленное в статье Н. Nohta, A. Mitsui, Y. Ohkura «High-performance liquid chromatographic determination of urinary catecholamines by direct pre-column fluorescence derivatization with 1,2-diphenylethylenediamine». J. Chromatogr. 1986. V. 380. P. 229-231, описывающее способ дериватизации KA и их метаболитов 0.1 М 1,2-дифенилэтилендиамином (ДЭД) в присутствии ферроцианида калия, катализирующего окисление растворенным кислородом воздуха молекул КА до КА-о-хинонов, которые в свою очередь взаимодействуют с дериватизирующим агентом (ДЭД) по реакции Михаэля. Конечным продуктом дериватизации КА с ДЭД являются 2-фенил(4,5-дигидропирроло)[2,3-f]бензоксазольные производные, обладающие более интенсивной флуоресценцией, чем К А и их метаболиты в видимой области спектра. Процесс проходит в смеси буферного раствора и метанола: 0.3 М CAPS-KOH (N-cyclohexyl-3-aminopropanesulfonic acid, N-циклогексил-3-аминопропансульфокислота) с рН 10: метанол (30:70 об. %). Окисление кислородом воздуха проходит в присутствии 20 мМ раствора ферроцианида K3[Fe(CN)6] в качестве катализатора, при температуре 50°С, в течение 20 мин.
Однако использование известного решения невозможно в биологических жидкостях из-за использования токсичного растворителя 70 об.% метанола, необходимости термостатирования реакционной смеси при повышенной температуре 50°С, длительности времени получения производных катехоламинов и их метаболитов. Кроме того, известный способ не позволяет селективно определять сразу несколько нейромедиаторов и не дает возможности их определения в большом количестве образцов.
Из уровня техники известно решение, представленное в автореферате на соискание степени кандидата химических наук по специальности «Аналитическая химия» 02.00.02. Москва. 2010. 22 с. К.В. Яблоцкий «Новые аспекты применения нативной и иммобилизованной пероксидазы хрена для определения ее ингибиторов и субстратов», описывающее способ дериватизации К А и их метаболитов 0.5 М бензиламином (БА) или дифенилэтилендиамином (ДЭД) в мицеллярной и водно-органической среде в присутствии пероксидазы из корней хрена (ПХ), катализирующей окисление пероксидом водорода молекул КА до КА-о-хинонов, которые в свою очередь взаимодействуют с дериватизирующим агентом по реакции Михаэля. Конечным продуктом дериватизации КА с БА или ДЭД являются 2-фенил(4,5-дигидропирроло)[2,3-f]бензоксазольные производные, обладающие в видимой области спектра более интенсивной флуоресценцией, чем сами КА и их метаболиты. Процесс проходит в 5 мМ растворе цетилтриметиламмоний бромида (ЦТМА) в 0.1 М CAPS-KOH с рН 11, в случае дериватизации БА или в 0.5 М буферном растворе глицин-KOH : диметилсульфоксид (ДМСО) (70:30 об. %) в случае дериватизации ДЭД, концентрация пероксида водорода - 1 мМ, в течение 30 мин (меньшее время дериватизации, 5 мин, удалось достигнуть только для НА), при температуре 37°С.
Однако и это решение имеет те же недостатки, что и приведенный выше аналог: невозможность использования в биологических объектах обусловливается наличием токсичного растворителя 30 об. % ДМСО, а также необходимостью термостатирования реакционной смеси при повышенной температуре 37°С и использования поверхностно-активных веществ (ПАВ). Известный способ также не позволяет селективно определять одновременно несколько катехоламинов и не дает возможности их определения в большом количестве образцов. Кроме того, ПХ используется в нативном, а не в иммобилизованном состоянии, что обуславливает сложность масштабирования методики и ее низкую воспроизводимость (93%).
Из уровня техники известно решение, представленное в патенте RU 2162228 «Способ одновременного определения свободных и конъюгированных форм катехоламинов, 5,4-диоксифенилаланина, 3,4-диоксифенилуксусной кислоты, 3,4-диоксифенилэтиленгликоля в плазме крови и моче», описывающее способ одновременного определения количества норадреналина, адреналина, дофамина, 3,4-диоксифенилаланина, 3,4-диоксифенилуксусной кислоты и 3,4-диоксифенилэтиленгликоля в плазме крови и моче. Процесс определения включает адсорбцию определяемых веществ из биологических жидкостей путем нанесения их на смолу Bio Rex 70, затем на активированную окись алюминия, элюирование 0.1 М HClO4 с последующим определением количества веществ по калибровочной кривой с помощью высокоэффективной жидкостной хроматографии (ВЭЖХ). Причем в качестве подвижной фазы при хроматографии используют 70 мМ раствор NaH2PO4, 10 мМ гептансульфоновой кислоты с pH 3 и общим временем хроматографирования - 10 мин.
Однако этот способ является трудоемким, требующим высококлассного оборудования, постоянного тока высокочистых растворителей и высококвалифицированного персонала из-за использования ВЭЖХ, необходимости длительной пробоподготовки биообъектов и компонентов подвижной фазы, применения стадий предконцентрирования, что увеличивает общее время анализа вплоть до 30-40 мин и возможность получения заниженных результатов из-за потерь анализируемых соединений. Также недостатком известного решения является использование сильных кислот - HClO4 и гептансульфоновой кислоты, что может вызвать деструкцию катехоламинов. Использование электрохимических детекторов с ВЭЖХ также неэффективно, так как потенциалы окисления катехоламинов очень близки, что снижает селективность известного способа определения катехоламинов и повышает вероятность получения ложных результатов.
Наиболее близким к заявляемому является решение, представленное в патенте RU 2546672 «Способ получения флуоресцирующих производных катехоламинов и их метаболитов методом дериватизации», раскрывающем способ получения интенсивно флуоресцирующих производных нейромедиаторов по реакции их ферментативной дериватизации. Наиболее интенсивный сигнал получают при проведении процесса в 0.1 М CAPS-КОН или глицин-КОН буферном растворе при концентрации ПХ: 0.01-1 мкМ; концентрации пероксида водорода: 100 мкМ, концентрации дериватизирующего агента (амина): 0.1-33 мМ; концентрации аналитов - катехоламинов и их метаболитов: 0.03-1 мкМ. В качестве дериватизирующего агента был использован бензиламин (БА) и дифенилэтилендиамин (ДЭД). Время реакции составляло 5 мин, при этом не использовалось длительное термостатирование реакционной смеси при повышенной температуре. Реакция проводилась в водной среде, в отсутствие токсичных растворителей (метанола, ДМСО).
Недостатком известного решения является невозможность его применения в анализе реальных биологических жидкостей (моче, плазме крови, церебральной жидкости и др.), особенно для диагностики заболеваний из-за отсутствия селективности описанного способа анализа. Продукты реакций дериватизации всех изучаемых аналитов имеют уширенные перекрывающиеся сигналы флуоресценции, что не позволяет ни качественно судить о присутствии, ни количественно определять содержание конкретных катехоламинов или их метаболитов, а, следовательно, ограничивает применимость ранее известного решения в одновременном определении нескольких маркеров нейромедиаторного обмена при их совместном присутствии в анализируемой пробе. Также в представленном решении невозможно экспрессно анализировать большое количество проб, поскольку для каждого единичного измерения необходимо проводить реакцию дериватизации, что вместе с измерением по времени занимает около 10 мин. Более того, для каждого анализа требуется значительный объем исследуемой пробы около 2-3 мл при том, что для достоверного количественного определения требуется три параллельных пробы, то есть нежелательно большие в случае анализа плазмы крови объемы 6-9 мл. Также необходим существенный расход фермента: 30-300 нмоль на единичный анализ. Кроме того, для постановки достоверного диагноза необходима информация о количественном содержании в анализируемом образце всей тройки катехоламинов: дофамина, норадреналина и адреналина, в многокомпонентной смеси (И.А. Веселова, Е.А. Сергеева, М.И. Македонская, О.Е. Еремина, С.Н. Калмыков, Т.Н. Шеховцова. «Методы определения маркеров нейромедиаторного обмена в целях клинической диагностики». Журн. аналит. химии. 2016. Т. 71. С. 1235-1249.), в то время как в известном решении описано определение только дофамина и адреналина в их индивидуальных растворах. Кроме того, в предложенном способе анализа фермент - ПХ - находится в растворе, т.е. в нативном, а не в иммобилизованном состоянии, что обусловливает относительные нестабильность и, следовательно, непродолжительные сроки хранения фермента (около 1 суток при 4°С), большой расход дорогостоящего фермента в пересчете на объем кюветы, а также низкую воспроизводимость методики (95%). Также описанную методику сложно масштабировать и затруднительно перенести на экспрессный анализ в большом количестве проб.
Раскрытие изобретения
Задачей настоящего изобретения является разработка способа одновременного определения нескольких маркеров нейромедиаторного обмена с низким пределом обнаружения, высокой селективностью, широким линейным диапазоном определяемых содержаний (концентраций) маркеров нейромедиаторного обмена, высокой воспроизводимостью и экспрессностью.
Заявляемое изобретение устраняет недостатки перечисленных аналогов.
Техническим результатом изобретения является возможность одновременного экспресс-определения сразу нескольких катехоламинов и их метаболитов: норадреналина, дофамина, адреналина, гомованилиновой и ванилилминдальной кислот, в их модельных растворах и пробах биологических жидкостей с достигаемыми воспроизводимостью результатов не менее 99% и временем анализа до 12.5 с на одну пробу.
Поставленная задача решается тест-системой для экспресс-определения низкомолекулярных маркеров нейромедиаторного обмена в образцах биологических жидкостей, представляющая собой планшет с нанесенной на поверхность лунок пленки хитозана с иммобилизованной пероксидазой растительного происхождения в количестве, достаточном для проведения реакции дериватизации при введении пробы биологической жидкости с дериватизирующим агентом в присутствии пероксида водорода, при этом пероксидазы содержится в количестве от 2 до 16 пмоль на 1 мм площади пленки, а хитозана от 15 до 60 мг на 1 мм площади пленки. Толщина пленки составляет от 0.5 до 1.5 мкм.
Предпочтительно в качестве пероксидазы растительного происхождения использовать пероксидазу из корней хрена, соевых бобов, арахиса, листьев табака, клубней батата и листьев масличных пальм, а хитозан с распределением молекулярных масс 50-220 кДа и степенью дезацетилирования не менее 75%.
Поставленная задача также решается способом получения заявляемой тест-системы, включающий приготовление водных растворов хитозана и пероксидазы растительного происхождения с последующим их нанесением на лунки планшета и высушиванием до образования пленки. Концентрация наносимого водного раствора хитозана составляет от 0.5 до 2 мас. %, а раствора пероксидазы от 15 до 20 мкМ. При этом для приготовления водного раствора хитозана используют водные растворы кислот с рН не менее 2 в количестве достаточном для растворения хитозана в воде, при этом предотвращающем реакцию сшивания хитозана.
Предпочтительно в качестве пероксидазы растительного происхождения использовать пероксидазу из корней хрена, соевых бобов, арахиса, листьев табака, клубней батата и листьев масличных пальм и хитозан с распределением молекулярных масс 50-220 кДа и степенью дезацетилирования не менее 75%.
Предпочтительно для растворения хитозана использовать водные растворы уксусной, соляной или угольной кислот.
Объемное соотношение растворов хитозана и пероксидазы при их нанесении на лунку составляет от 1:1 до 6:1.
Высушивание планшета с нанесенными растворами осуществляют при температуре до 23±3°С.
Способ экспресс-определения катехоламинов и их метаболитов в образцах биологических жидкостей, включающий нанесение пробы биологической жидкости на лунку заявляемой тест-системы, полученной заявляемым способом, с последующим внесением пероксида водорода и дериватизирующего агента в количестве обеспечивающем проведения реакции дериватизации с образованием флуоресцирующих производных; возбуждение флюоресценции в пробе с регистрацией полученных сигналов, и при выявлении максимума в характерном диапазоне для определяемого аналита делают вывод о его наличии в образце. При этом пробу биологической жидкости наносят в объеме от 10 до 30 мкл, и на 1 объемную часть пробы используют 3-5 об. частей дериватизирующего агента и 1-5 об. частей пероксида водорода. Для получения сигнала флуоресценции пробу облучают монохроматичным излучением с длиной волны в диапазоне 300-370 нм. Вывод о наличии в образце биологической жидкости норадреналина делают при выявлении максимума в диапазоне от 452 до 458 нм, дофамина - в диапазоне от 455 до 465 нм, адреналина - в диапазоне от 475 до 485 нм, ванилилминдальной кислоты - в диапазоне от 417 до 423 нм, гомованилиновой кислоты - в диапазоне от 422 до 428 нм, L-ДОФА - в диапазоне от 456 до 460 нм, норметанефрина - в диапазоне от 460 до 460 нм, серотонина - в диапазоне от 463 до 473 нм, 5-гидроксииндол-3-уксусной кислоты - в диапазоне от 470 до 480 нм.
Предпочтительно в качестве дериватизирующего агента использовать ароматические амины, а именно бензиламин или 1,2-дифенилэтилендиамин.
Для определения количественного содержания аналитов в образцах биологических жидкостей измеряют интенсивность полученного сигнала, вывод о количественном содержании делают по предварительно рассчитанным градуировочным зависимостям. Дополнительно проводят обработку полученных сигналов путем вычисления второй производной интенсивности флуоресцентных сигналов и при выявлении минимума в характерном диапазоне для определяемого аналита делают вывод о его наличии в образце: о наличии в образце биологической жидкости норадреналина делают при выявлении минимума в диапазоне от 452 до 458 нм, дофамина - в диапазоне от 455 до 465 нм, адреналина - в диапазоне от 475 до 485 нм, ванилилминдальной кислоты - в диапазоне от 417 до 423 нм, гомованилиновой кислоты - в диапазоне от 422 до 428 нм, L-ДОФА - в диапазоне от 456 до 460 нм, норметанефрина - в диапазоне от 460 до 460 нм, серотонина - в диапазоне от 463 до 473 нм, 5-гидроксииндол-3-уксусной кислоты - в диапазоне от 470 до 480 нм.
Краткое описание чертежей
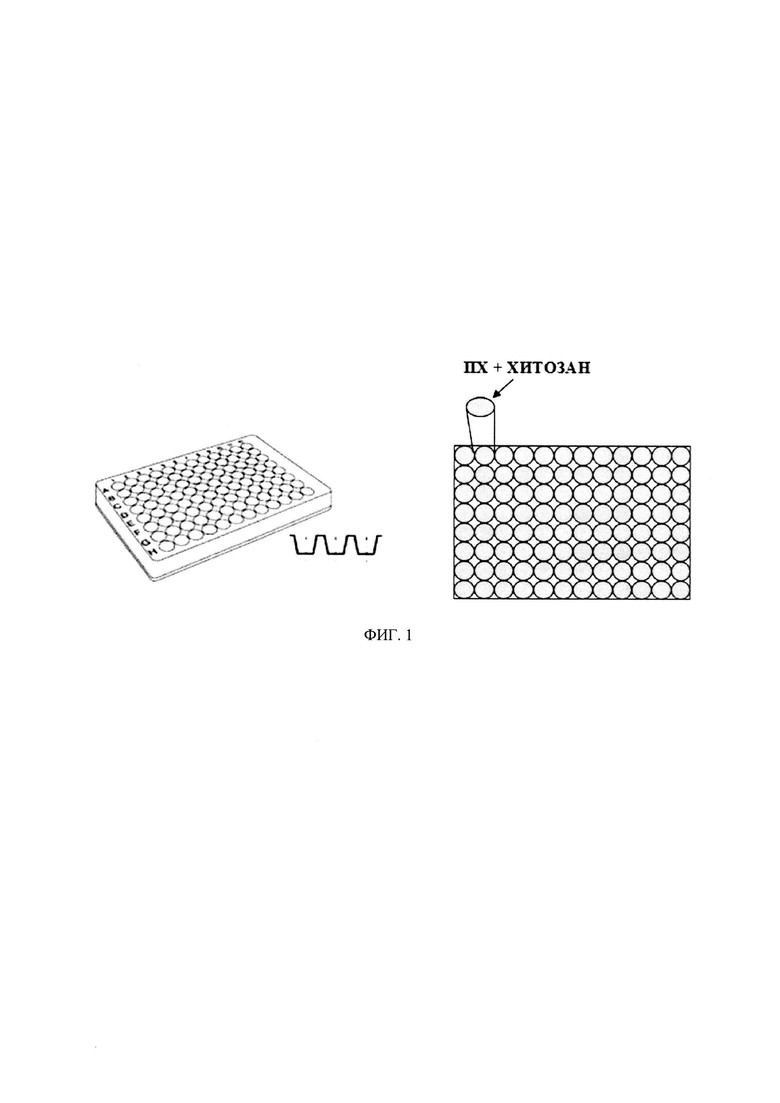
На фиг. 1 представлен микропланшетный оптический биосенсор, состоящий из 96-луночного полистирольного планшета в качестве подложки и оптически прозрачной пленки, состоящей из пероксидазы из корней хрена (ПХ) в слое хитозана, в качестве чувствительного слоя.
На фиг. 2 представлена схема формирования чувствительного слоя биосенсора. Позициями на фигурах обозначены: 1 - смесь для чувствительного слоя, состоящая из раствора ПХ и 1 мас. %-го раствора хитозана; 2 - 96-луночный полистирольный планшет в качестве подложки; 3 - формирование чувствительного слоя в каждой ячейке микропланшета; 4 - высушивание на воздухе; 5 - микропланшетный оптический биосенсор.
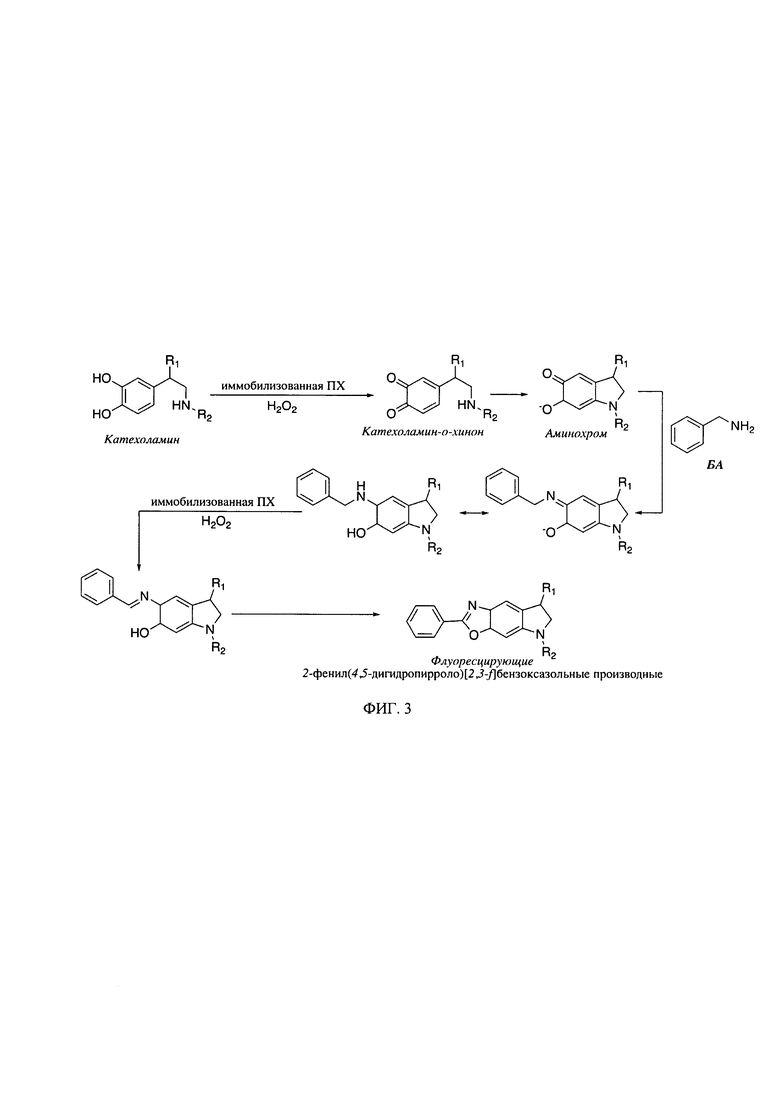
На фиг. 3 представлена схема образования флуоресцирующих производных КА (R1=Н и R2=Н для ДА, R1=ОН и R2=Н для НА, R1=ОН и R2=СН3 для АД) с использованием бензиламина (БА) в качестве дериватизирующего агента.
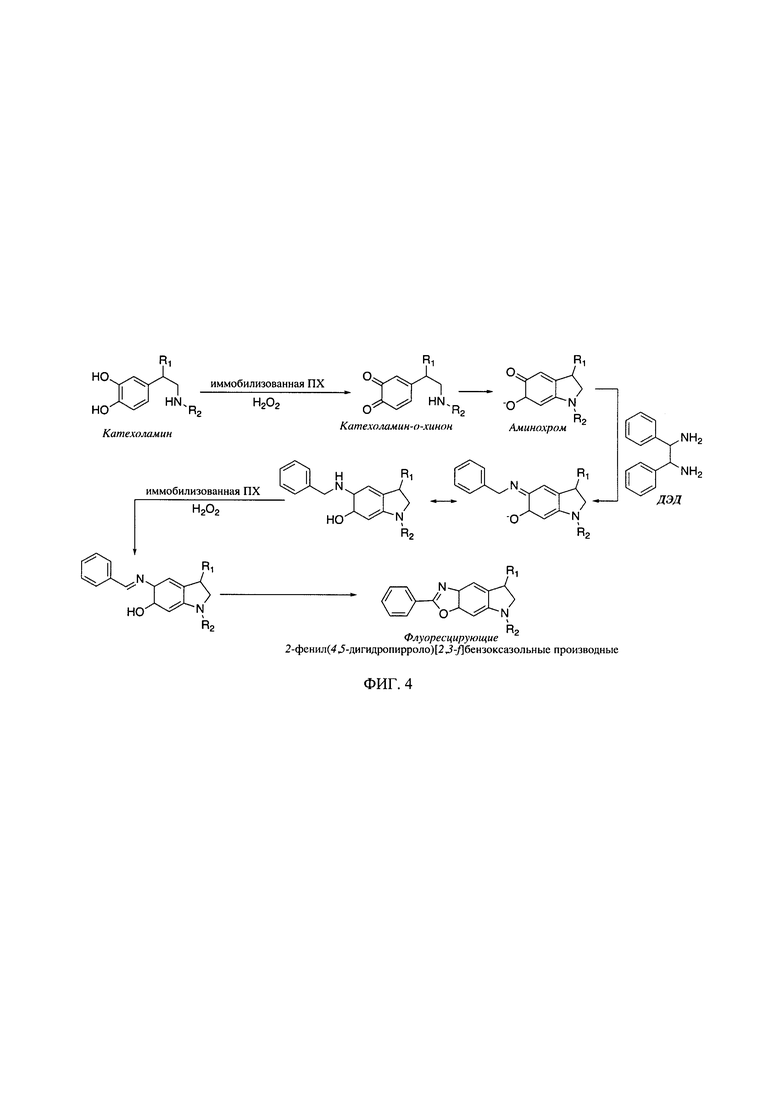
На фиг. 4 представлена схема образования флуоресцирующих производных КА (R1=Н и R2=Н для ДА, R1=ОН и R2=Н для НА, R1=ОН и R2=СН3 для АД) с использованием 7,2-дифенилэтилендиамина (ДЭД) в качестве дериватизирующего агента.
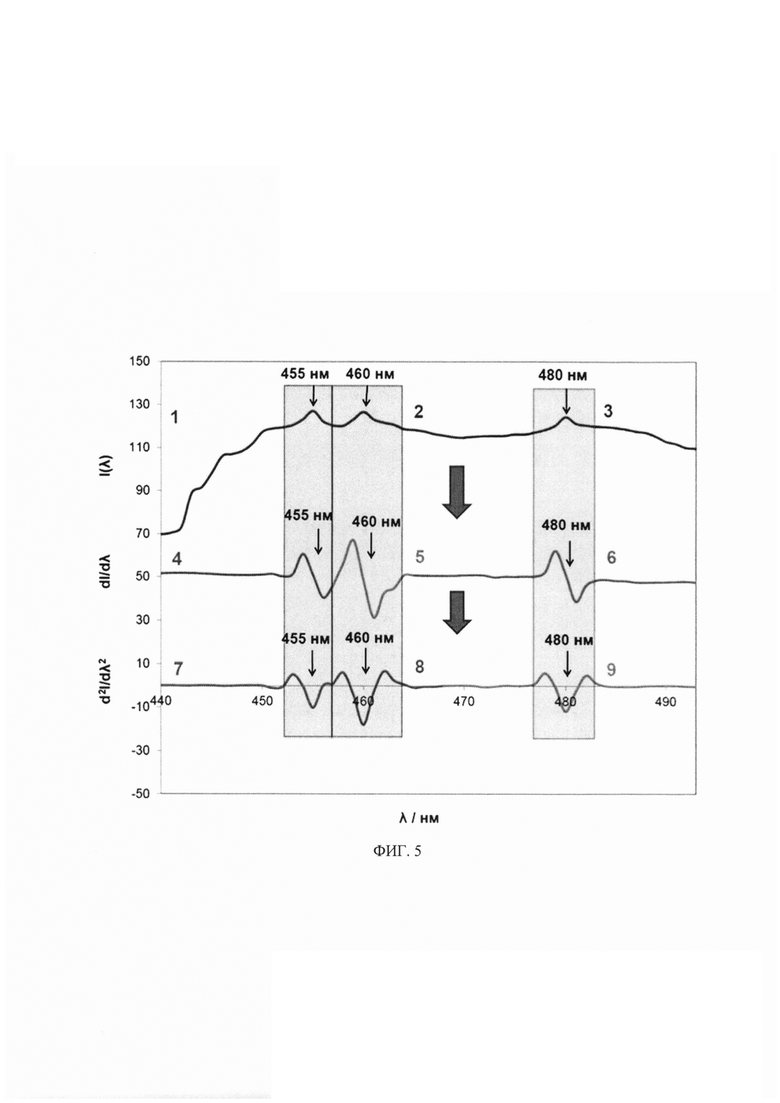
На фиг. 5 представлены: 1-3 спектры флуоресценции, производные спектры флуоресценции 4-6 первого и 7-9 второго порядка системы флуоресцирующих производных КА - НА / ДА / АД в соотношениях, аналогичных референсным концентрациям КА в моче здорового человека (0.1, 1.0 и 0.05 мкМ для НА, ДА, АД, соответственно). Позициями на фигурах обозначены: 1, 4, 7 - НА / ДА / АД с БА, λex=330 нм, λem=455 нм; 2, 5, 8 - НА / ДА / АД с ДЭД,=340 нм, λex=460 нм; 3, 6, 9 - НА / ДА / АД с БА, λex=356 нм, λem=480 нм.
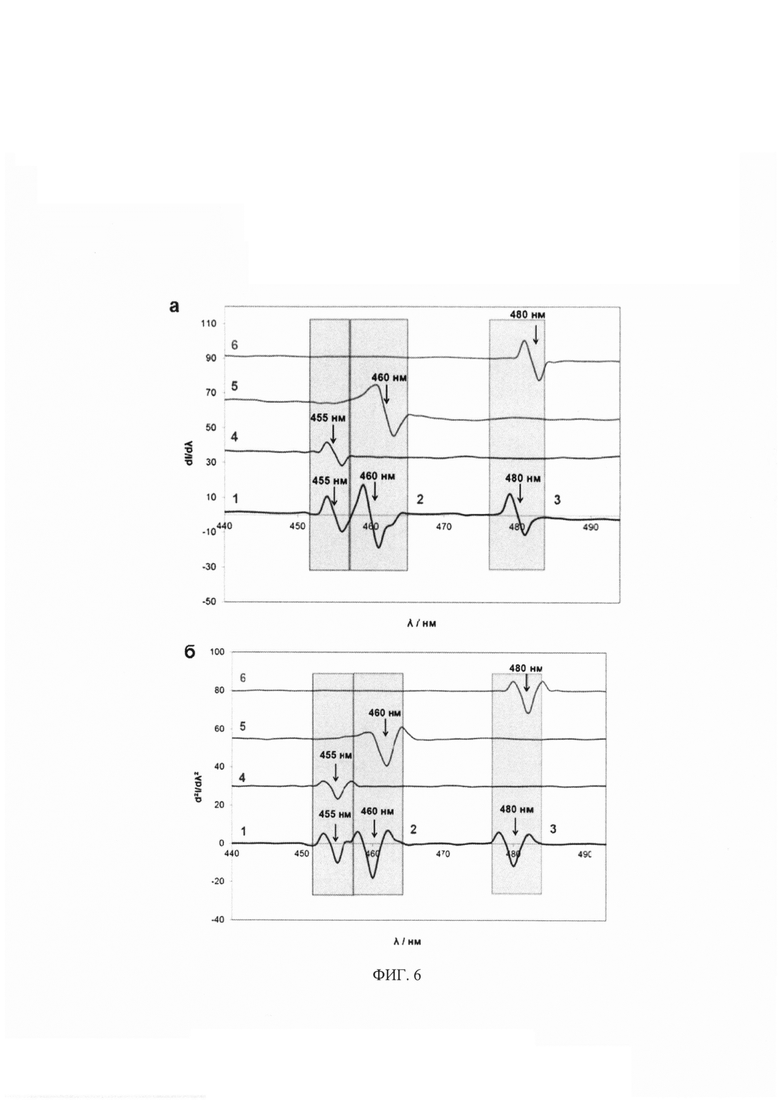
На фиг. 6 представлены: производные спектры флуоресценции первого (а) и второго (б) порядка системы флуоресцирующих производных КА - НА / ДА / АД в соотношениях, аналогичных референсным концентрациям КА в моче здорового человека (0.1, 1.0 и 0.05 мкМ для НА, ДА, АД, соответственно) и индивидуальных флуоресцирующих производных НА, ДА, АД. Позициями на фигурах обозначены: 1 - НА / ДА / АД с БА, λех=330 нм, λem=455 нм; 2 - НА / ДА / АД с ДЭД, λех=340 нм, λem=460 нм; 3 - НА / ДА / АД с БА, λех=356 нм, λem=480 нм; 4 - НА с БА, λех=330 нм, λem=455 нм; 5 - ДА с ДЭД, λех=340 нм, λem=460 нм; 6 - АД с БА, λех=356 нм, λem=480 нм.
Осуществление изобретения
Получение флуоресцирующих низкомолекулярных маркеров нейромедиаторного обмена - биогенных аминов методом дериватизации (фиг. 3, 4), включает окисление исходных соединений и их обработку образующими конденсированные структуры аминами, в присутствии биокатализатора - пероксидазы растительного происхождения, в частности пероксидазы из корней хрена (ПХ). Новизна и неочевидность при этом заключается в том, что окисление исходных соединений, катализируемое пероксидазой, проводят в присутствии малого количества фермента, например, на единичный анализ используется 75-500 пмоль ПХ. Использование микропланшетного оптического биосенсора на основе иммобилизованной в пленках хитозана ПХ (фиг. 2) позволяет увеличить воспроизводимость получаемых результатов до 99% и сроки хранения фермента до 6 месяцев при 4°С. При этом хитозан, природный полисахарид, является эффективной биосовместимой и нетоксичной матрицей и образует оптически прозрачные тонкие пленки. Биосенсор существенно упрощает процедуру массового экспрессного анализа - 96 проб за 20 мин, т.е. около 12.5 с на одну пробу. Иммобилизация фермента в пленках хитозана позволяет повысить удобство, экспрессность и воспроизводимость определения маркеров нейромедиаторного обмена в биологических жидкостях.
Необходимое количество хитозана (с различными по распределению молекулярными массами 50-220 кДа и степенью дезацетилирования не менее 75%) для получения 1 мас. %-го раствора растворяют в 0.1-1 об. %-й слабой кислоте или разбавленной сильной, преимущественно одноосновной, кислоте, например, уксусной кислоте, поскольку хитозан гидрофобен и водонерастворим, однако в присутствии небольших количеств даже слабых кислот происходит протонирование амино-групп хитозана, что делает его водорастворимым. Поэтому для растворения хитозана подойдет не только раствор уксусной, но и соляной, угольной и других кислот, рН которых не менее 2, т.е. в присутствии которых не происходит сшивки хитозана.
В ячейки планшета заливают полученные растворы хитозана и пероксидазы в объемном соотношении от 1:1 до 6:1 при нанесении на лунку. Так, например, в каждую ячейку 96-луночного полистирольного планшета (∅ 6.54 мм) заливают по 30 мкл 0.5-2 мас. %-го раствора хитозана и 5-30 мкл 15-20 мкМ раствора ПХ, при этом в качестве фермента, катализирующего процесс получения флуоресцирующих производных КА и их метаболитов, могут быть использованы пероксидазы (КФ 1.11.1.7) не только из корней хрена, но и из других источников растительного происхождения: соевых бобов, арахиса, листьев табака, клубней батата и листьев масличных пальм. Затем добавляли 0-25 мкл деионизированной воды таким образом, чтобы общий объем смеси составлял 60 мкл. При этом на один образец приходился малый расход фермента: 150-450 пмоль. Точная концентрация фермента устанавливалась фотометрически согласно инструкции производителя (для ПХ ε403=1.02⋅105 М-1⋅см-1 согласно данным производителя «Sigma-Aldrich»). Для образования тонких пленок полимера, толщиной 0.5-1.5 мкм, планшет с лунками высушивали не менее 12 ч на воздухе при комнатной температуре (фиг. 2). Планшеты с иммобилизованной ПХ хранили при температуре 4°С, при этом срок хранения фермента составлял до 6 месяцев.
В качестве планшета для изготовления тест-системы могут быть использованы любые цельные или сборные культуральные полимерные (например, полистирольные, полипропиленовые и др.) планшеты с любым количеством лунок, из которых наиболее применимыми являются 6, 12, 24, 48, 72, 96-луночные, подходящие для проведения флуоресцентного анализа, т.е. оптически непрозрачные: либо белые (абсолютно отражающие), либо черные (абсолютно поглощающие), при этом их химическая и термическая устойчивости не имеют значения, поскольку анализ проводится в водной среде при комнатной температуре.
Способ определения низкомолекулярных маркеров нейромедиаторного обмена (с молекулярной массой до 900 Да) с помощью оптического биосенсенсора, описанного в заявляемом изобретении, включает в себя последовательное введение компонентов реакции: на 1 объемную часть водного раствора нейромедиатора или биологической жидкости (мочи, крови) 10-15 частей глицин-КОН или CAPS-KOH буфера (0.1-0.5 М), 3-5 об. частей дериватизирующего агента (ДЭД или БА) и 1-5 об. частей пероксида водорода (фиг. 3, 4). После чего реакционную смесь тщательно перемешивали и измеряли интенсивность флуоресценции по истечении 5 мин.
Для осуществления высокоселективного одновременного определения сразу нескольких нейромедиаторов из одной пробы использована дополнительная математическая обработка интенсивности флуоресцентного сигнала путем взятия первой и второй производной по длине волны. Исходные зарегистрированные спектры флуоресценции (нулевого порядка) представляли собой зависимость интенсивности (I) испускания от длины волны (λ) (фиг. 5). Затем спектры нулевого порядка преобразовывались в соответствующие спектры производных первого и второго порядков. Спектр первой производной представлял собой график зависимости градиента кривой испускания (скорость изменения интенсивности с длиной волны, dI/dλ) от длины волны (фиг. 5). Спектр второй производной представлял собой график зависимости кривизны спектра испускания (d2I/dλ2) от длины волны. Первая (dI/dλ) и вторая производные (d2I/dλ2) при характерной длине волны для данного аналита линейно зависят от его концентрации. Производная спектрофлуориметрия может быть использована как для целей идентификации катехоламинов и их метаболитов, так и их количественного определения в многокомпонентных смесях, а также в тех случаях, когда имеется фоновая флуоресценция (например, для мочевины в образце мочи).
Направленный выбор дериватизирующего агента, длины волны возбуждения, использование микропланшетного биосенсора и производной спектрофлуориметрии позволяет разделять и определять сразу несколько КА и их метаболитов в смеси (фиг. 5, 6).
Наиболее интенсивный сигнал получают при проведении процесса в 0.1-0.5 М CAPS-КОН с рН 11 и глицин-КОН с рН 8 буферном растворе при концентрации ПХ: 0.1-10 мкМ; концентрации пероксида водорода: 0.01-1 мМ, концентрации дериватизирующего агента: 0.1-100 мМ; концентрации определяемых соединений -катехоламинов и их метаболитов: 0.005-5 мкМ.
Наибольшую интенсивность флуоресцентного сигнала получают при использовании в качестве дериватизирующих агентов бензиламина и дифенилэтилендиамина, а также при возбуждении флуоресценции в диапазоне λex=305-356 нм и регистрации аналитического сигнала в диапазоне λem=420-480 нм.
Наилучшую воспроизводимость (99%) и наиболее интенсивный флуоресцентный сигнал получают при иммобилизации ПХ в ячейках полистирольного планшета в 1 мас. %-м растворе хитозана и высушивании на воздухе в течение суток. Необходимое количество хитозана растворяют в 0.5 об.% уксусной кислоте. Полученный биосенсор сохраняет свою каталитическую активность в течение 6 месяцев при хранении в холодильнике при 4°С.
Предлагаемое изобретение также позволяет сократить время проведения единичного анализа с 10 мин до почти 12.5 с, что может быть использовано в массовых анализах биологических жидкостей в целях диагностики на молекулярном уровне нейродегенеративных заболеваний (деменций) и нейроэндокринных опухолей (карциномы, нейробластомы, параганглиомы, феохромоцитомы и др.) на ранних стадиях.
Качественное определение и вывод о присутствии того или иного нейромедиатора делались по наличию максимумов на спектрах флуоресценции и экстремумов на спектрах второй производной в диапазоне от 452 до 458 нм для норадреналина, в диапазоне от 475 до 485 нм для дофамина, в диапазоне от 455 до 465 нм для адреналина, в диапазоне от 417 до 423 нм для ванилилминдальной кислоты, в диапазоне от 422 до 428 нм для гомованилиновой кислоты. Количественное определение молекул нейромедиаторов в образце проводят с применением градуровочных зависимостей. Градуировочный график строится в координатах интенсивность флуоресцентного сигнала - концентрация нейромедиатора в максимуме характеристического пика. В целях установления неизвестного содержания катехоламинов и их метаболитов в растворе используют уравнения вида I=(а±Δа)×с+(b±Δb), либо dI/dλ=(а±Δа)×с+(b±Δb), либо d2I/dλ2=(а±Δа)×с+(b±Δb), в которые подставляется значение интенсивности флуоресцентного сигнала в максимуме характеристического пика (I, либо dI/dλ, либо d2I/dλ2), и вычисляется концентрация определяемого соединения (с). В настоящем изобретении приняты следующие обозначения и термины:
n - количество параллельных измерений,
Р - доверительная вероятность,
сн - концентрация аналита, минимальная из диапазона определяемых содержаний,
sr - относительное стандартное отклонение измерений,
r - коэффициент корреляции,
λem - длина волны испускания (эмиссии),
λех - длина волны возбуждения флуоресценции,
5-ГИУК - 5-гидроксииндол-3-уксусная кислота,
5-ГТ - 5-гидрокситриптамин (серотонин),
CAPS - N-циклогексил-3-аминопропансульфокислота (N-cyclohexyl-3-aminopro-panesulfonic acid),
L-ДОФА - 3-(3,4-дигидроксифенил)-L-аланин
АД - адреналин (эпинефрин, 4-[(1R)-1-гидрокси-2-(метиламино)этил]фенил-1,2-диол),
БА - бензиламин,
ВМК - ванилилминдальная кислота (2-гидрокси-2-(4-гидрокси-3-метоксифенил)уксусная кислота),
ВЭЖХ - высокоэффективная жидкостная хроматография,
ГВК - гомованилиновая кислота (4-гидрокси-3-метоксифенилуксусная кислота),
ДА - дофамин (допамин, 4-(2-аминоэтил)фенил-1,2-диол),
ДМСО - диметилсульфоксид,
ДЭД - 1,2-дифенилэтилендиамин,
ДОС - диапазон определяемых содержаний,
КА - катехоламин (катехоламины),
КФ - шифр классификации (кода) фермента, представляющий собой классификационный номер фермента по международной иерархической классификации,
НА - норадреналин (норэпинефрин, 4-[(1R)-2-амино-7-гидроксиэтил]фенил-1,2-диол),
НМН - норметанефрин (D,L-α-(аминометил)-4-гидрокси-3-метоксибензилметанол),
ПАВ - поверхностно-активное вещество,
ПО - предел обнаружения,
ПХ - пероксидаза из корней хрена,
ЦТМА - цетилтриметиламмоний бромид.
Химические символы / сокращения имеют свои обычные значения: °С (градус (градусы) Цельсия), нм (нанометр (нанометры)), мм (миллиметр (миллиметры)), мин (минута (минуты)), с (секунда (секунды)), мкл (микролитр (микролитры)), М (моль (моли) в литре), л (литр (литры)), мкл (микролитр (микролитры)), мг (миллиграмм (миллиграммы)), нмоль (наномоль (наномоли)), пмоль (пикомоль (пикомоли)), нМ (наномоль (наномоли) в литре), мкМ (микромоль (микромоли) в литре), мМ (миллимоль (миллимоли) в литре), % (об.%) (процент (проценты) по объему), мас. % (массовый процент (проценты)), В (вольт (вольты)).
Представленные ниже примеры конкретного осуществления изобретения приведены для предоставления специалистам в данной области техники полного описания проведения и применения анализа по изобретению, но не ограничивают предполагаемый авторами изобретения объем изобретения.
Все приведенные ниже реагенты и полистирольные планшеты являются коммерчески доступными. Все процедуры, если не оговорено особо, осуществляли при комнатной температуре или температуре окружающей среды, то есть в диапазоне от 18 до 25°С; в ходе всех экспериментов для приготовления водных растворов использовали деионизированную воду высокой чистоты, очищенную с использованием установки «Milli-Q», «Millipore»; спектры флуоресценции зарегистрированы на спектрофлуориметре «Cary Eclipse», «Agilent» с использованием приставки для микропланшетов, с мощностью лампы 800 В, с шириной входной и выходной щелей 5 или 10 нм; взвешивание препаратов проводили с точностью ± 0.02 мг на аналитических весах «Discovery», «OHAUS»; точные объемы жидкостей отбирали с использованием автоматических дозаторов «Eppendorf» с диапазонами объемов: 2-20, 10-100, 20-200, 100-1000 и 500-5000 мкл.
Пример 1. Микропланшетный оптический биосенсор.
Микропланшетный оптический биосенсор представляет собой 96-луночный полистирольный планшет с чувствительным слоем в виде тонких пленок ПХ в хитозане в каждой ячейке (∅ 6.54 мм) (фиг. 1). Необходимое количество хитозана растворяют в 0.5 об. %-й уксусной кислоте для получения 1 мас. %-го раствора. В ячейки планшета заливали по 30 мкл 1 мас. %-го раствора хитозана, 10 мкл 15 мкМ раствора ПХ и 20 мкл деионизированной воды. Для получения тонких пленок высушивали планшет в течение суток на воздухе при температуре 23±3°С (фиг. 2). Планшеты с иммобилизованной ПХ хранили при температуре 4°С.
Способ определения маркеров нейромедиаторного обмена с помощью оптического биосенсенсора, описанного в заявляемом изобретении, включает в себя последовательное введение компонентов реакции: буферного раствора, водного раствора нейромедиатора или биологической жидкости (мочи, крови), дериватизирующего агента (ДЭД или БА) и пероксида водорода (фиг. 3, 4); детектирование образующегося дериватизированного производного с помощью флуориметра с приставкой для планшетов, выбор длин волн возбуждения и испускания в соответствии с природой и структурой образующихся производных (λex=305-356 нм, λem=420-480 нм), с последующей математической обработкой флуоресцентных сигналов с использованием производной спектрофлуориметрии 1-го и 2-го порядка.
Для детектирования анализируемых веществ с концентрациями 0.005-5 мкМ используют жидкие пробы объемом 10-40 мкл путем их введения в реакционную систему в ячейку планшета с чувствительным слоем. Для обеспечения высокой скорости анализа при большом отношении сигнал / шум и отсутствии появления ложной спектральной информации мощность излучения составляла 800 В при ширине входной и выходной щелей 5 или 10 нм.
Пример 2. Определение норадреналина (НА) с концентрацией в интервале 0.025-0.5 мкМ по реакции его ферментативной дериватизации с ДЭД или БА.
Для определения НА был использован биосенсор из примера 1. В ячейку планшета с чувствительным слоем последовательно вводили 140 мкл 0.5 М глицин-КОН буферного раствора рН 8 (120 мкл 0.1 М CAPS-KOH буферного раствора, рН 11), 10 мкл 30 мкМ раствора НА, 30 мкл 20 мМ раствора ДЭД (40 мкл 0.2 М раствора БА) и 10 мкл 2.0 мМ раствора Н2О2. После чего реакционную смесь тщательно перемешивали и измеряли интенсивность флуоресценции по истечении 5 мин. Контрольный опыт проводили аналогично вышеописанной методике за исключением того, что вместо НА вводили воду. Для обеспечения высокой скорости анализа при большом отношении сигнал / шум и отсутствии появления ложной спектральной информации мощность излучения составляла 800 В при ширине входной и выходной щелей 5 или 10 нм. Возбуждение флуоресценции для регистрации сигналов в пробе с НА проводили при λem=330 нм. Делали вывод о количественном содержании НА по градуировочной зависимости, построенной для сигнала флуоресценции при λex=455 нм (табл. 1).
Градуировочный график строился в координатах интенсивность флуоресцентного сигнала - концентрация нейромедиатора в максимуме характеристического пика (фиг. 5). В диапазоне определяемых содержаний соблюдалась линейная зависимость между интенсивностью флуоресценции максимума пика на спектрах и концентрацией КА и их метаболитов. Прямолинейность графика сохраняется только в интервале диапазона определяемых содержаний, указанных в табл. 1. Нахождение значения концентраций нейромедиаторов в испытуемом растворе по градуировочным зависимостям ниже и выше диапазона определяемых содержаний возможно, но не рекомендуется из-за большой погрешности. В целях установления неизвестного содержания маркеров нейромедиаторного обмена в растворе используют уравнение вида I=(а±Δа)×с+(b±Δb), в которое подставляется значение интенсивности флуоресцентного сигнала в максимуме характеристического пика (I), и вычисляется концентрация определяемого соединения (с). Также для более чувствительного и селективного определения катехоламинов и их метаболитов градуировочный график строился в координатах вторая производная интенсивности флуоресцентного сигнала по длине волны в экстремуме характеристического пика - концентрация нейромедиаторного соединения (фиг. 6, б). В целях нахождения неизвестного содержания нейромедиатора в испытуемом растворе использовали уравнение вида d2I/dλ2=(а±Δа)×с+(b±Δb), в которое подставляли значение второй производной интенсивности флуоресцентного сигнала по длине волны (d2I/dλ2), и вычисляли концентрацию определяемого соединения (табл. 3).
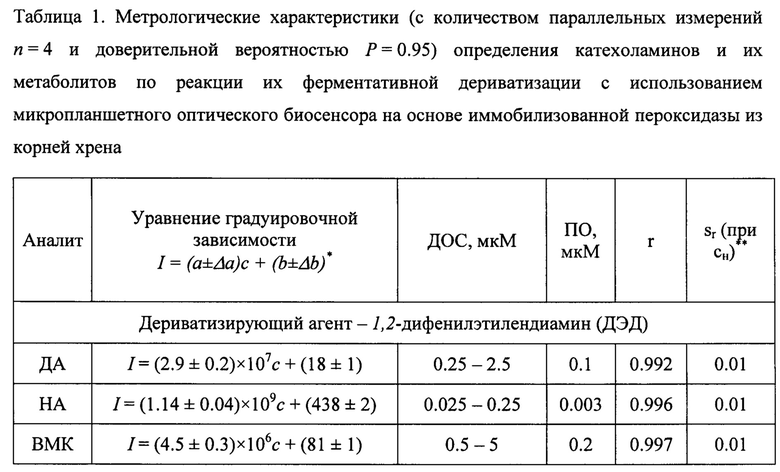
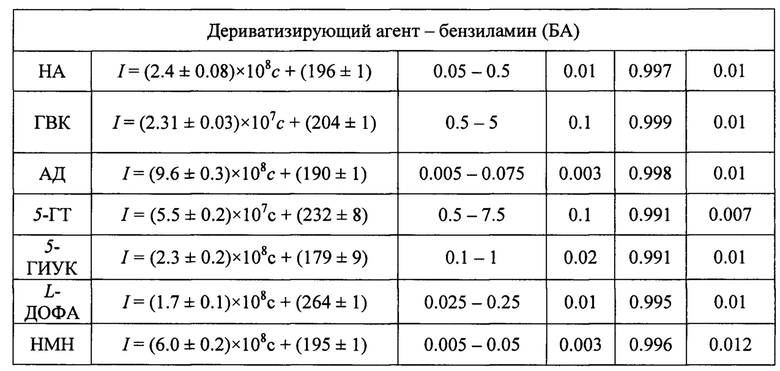
** - стандартное относительное отклонение для измерений при минимальной концентрации (с„) из диапазона определяемых содержаний
Пример 3. Определение дофамина (ДА) с концентрацией в интервале 0.25-2.5 мкМ по реакции его ферментативной дериватизации с ДЭД.
Анализ проводился аналогично примеру 2 с отличием в том, что в качестве дериватизирующего агента был использован ДЭД, а в качестве буфера - 0.5 М глицин-КОН рН 8.0. Возбуждение флуоресценции для регистрации сигналов в пробе с ДА проводили при λem=340 нм. Делали вывод о количественном содержании ДА по градуировочной зависимости, построенной для сигнала флуоресценции при λex=460 нм (табл. 1).
Пример 4. Определение ванилилминдальной кислоты (ВМК) с концентрацией в интервале 0.5-5 мкМ по реакции ее ферментативной дериватизации с ДЭД.
Анализ проводился аналогично примеру 2 с отличием в том, что в качестве дериватизирующего агента был использован ДЭД, а в качестве буфера - 0.5 М глицин-КОН рН 8.0. Возбуждение флуоресценции для регистрации сигналов в пробе с ВМК проводили при λem=305 нм. Делали вывод о количественном содержании ВМК по градуировочной зависимости, построенной для сигнала флуоресценции при λex=420 нм (табл. 1).
Пример 5. Определение гомованилиновой кислоты (ГВК) с концентрацией в интервале 0.5-5 мкМ по реакции ее ферментативной дериватизации с БА.
Анализ проводился аналогично примеру 2 с отличием в том, что в качестве дериватизирующего агента был использован БА, а в качестве буфера - 0.1 М CAPS-KOH рН 11.0. Возбуждение флуоресценции для регистрации сигналов в пробе с ГВК проводили при λem=315 нм. Делали вывод о количественном содержании ГВК по градуировочной зависимости, построенной для сигнала флуоресценции при λex=425 нм (табл. 1).
Пример 6. Определение адреналина (АД) с концентрацией в интервале 0.005-0.075 мкМ по реакции его ферментативной дериватизации с БА.
Анализ проводился аналогично примеру 2 с отличием в том, что в качестве дериватизирующего агента был использован БА, а в качестве буфера - 0.1 М CAPS-KOH рН 11. Возбуждение флуоресценции для регистрации сигналов в пробе с АД проводили при λem=356 нм. Делали вывод о количественном содержании ДА по градуировочной зависимости, построенной для сигнала флуоресценции при λex=480 нм (табл. 1).
Пример 7. Определение L-ДОФА с концентрацией в интервале 0.025 - 0.25 мкМ по реакции его ферментативной дериватизации с БА.
Анализ проводился аналогично примеру 6. Возбуждение флуоресценции для регистрации сигналов в пробе с L-ДОФА проводили при λem=335 нм. Делали вывод о количественном содержании L-ДОФА по градуировочной зависимости, построенной для сигнала флуоресценции при λех=458 нм (табл. 1).
Пример 8. Определение норметанефрина (НМН) с концентрацией в интервале 0.005-0.05 мкМ по реакции его ферментативной дериватизации с БА.
Анализ проводился аналогично примеру 6. Возбуждение флуоресценции для регистрации сигналов в пробе с НМН проводили при λem=340 нм. Делали вывод о количественном содержании НМН по градуировочной зависимости, построенной для сигнала флуоресценции при λех=463 нм (табл. 1).
Пример 9. Определение 5-гидроксииндол-3-уксусной кислоты (5-ГИУК) с концентрацией в интервале 0.1-1.0 мкМ по реакции ее ферментативной дериватизации с БА.
Анализ проводился аналогично примеру 6. Возбуждение флуоресценции для регистрации сигналов в пробе с 5-ГИУК проводили при λem=345 нм. Делали вывод о количественном содержании 5-ГИУК по градуировочной зависимости, построенной для сигнала флуоресценции при λех=475 нм (табл. 1).
Пример 10. Определение серотонина (5-гидрокситирамина, 5-ГТ) с концентрацией в интервале 0.5-7.5 мкМ по реакции его ферментативной дериватизации с БА.
Анализ проводился аналогично примеру 6. Возбуждение флуоресценции для регистрации сигналов в пробе с 5-ГТ проводили при λem=345 нм. Делали вывод о количественном содержании 5-ГТ по градуировочной зависимости, построенной для сигнала флуоресценции при λех=468 нм (табл. 1).
Пример 11. Определение ДА в моче здорового человека методом «введено-найдено» по реакции его ферментативной дериватизации с ДЭД.
Анализ проводился аналогично примеру 3 с отличием в том, что в качестве пробы вводили 10 мкл мочи (без предварительной пробоподготовки). О количественном содержании ДА судили по градуировочной зависимости (табл. 1). Полученные результаты согласовались с литературными данными о нормальных референсных содержаниях ДА в моче здорового человека (табл. 2) (Pussard Е., Neveux М., Guigueno N. Reference intervals for urinary catecholamines and metabolites from birth to adulthood. Clin. Biochem. 2009. V. 42. P. 536-539, Pagana K.D. Mosby's Manual of Diagnostic and Laboratory Tests. St. Louis: Mosby Inc., 1998.).

Пример 12. Определение НА, ДА, АД в смеси в тройной системе НА / ДА / АД по реакции их ферментативной дериватизации с ДЭД или БА.
Анализ проводился аналогично примеру 2 с отличием в том, что в качестве пробы вводили смесь, состоящую из НА, ДА и АД с концентрациями 0.1, 1.0 и 0.05 мкМ, соответственно.
На первом этапе регистрировали спектры флуоресценции индивидуальных производных катехоламинов и тройной системы с соотношениями компонентов, аналогичными для референсных содержаний катехоламинов в моче здорового человека (0.1, 1.0 и 0.05 мкМ для НА, ДА, АД, соответственно). На втором этапе, с использованием математической обработки данных, получали первые и вторые производные спектров флуоресценции системы катехоламинов и индивидуальных соединений (фиг. 5, 6). Математическая модель обработки спектров была аналогична модели производной спектрофотометрии. По спектрам первой и второй производной для разных концентраций индивидуальных соединений строили градуировочные зависимости и рассчитывали метрологические характеристики, чтобы сравнить их с метрологическими характеристиками для флуоресцентных спектров (табл. 3). Обнаружено, что ПО для предложенных методик, с использованием производных спектров, выше, но незначительно, однако дополнительно позволило использовать этот подход для обеспечения, главным образом, селективного, мультиплексного и, в то же время, высокочувствительного определения сразу нескольких катехоламинов в их сложной многокомпонентной смеси (фиг. 5, 6).
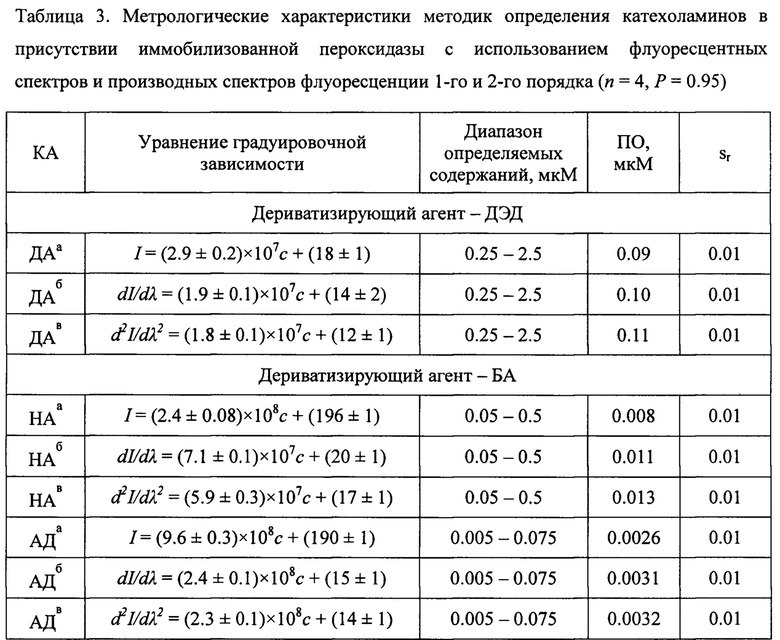
а - с использованием спектра флуоресценции флуоресцирующего производного КА;
б - с использованием производного спектра флуоресценции первого порядка флуоресцирующего производного КА;
в - с использованием производного спектра флуоресценции второго порядка флуоресцирующего производного КА.
Пример 13. Определение дофамина (ДА) с концентрацией в интервале 0.5-2.0 мкМ по реакции его ферментативной дериватизации с ДЭД с использованием пероксидазы из соевых бобов.
Анализ проводился аналогично примеру 3 с отличием в том, что в качестве пероксидазы растительного происхождения использовалась пероксидаза из соевых бобов. Возбуждение флуоресценции для регистрации сигналов в пробе с ДА проводили при λem=340 нм. Делали вывод о количественном содержании ДА по градуировочной зависимости, построенной для сигнала флуоресценции при λex=460 нм (табл. 4).

Как видно из приведенных примеров, предлагаемое изобретение решает задачу одновременного определения сразу нескольких маркеров нейромедиаторного обмена в большом количестве проб биологических жидкостей за счет получения флуоресцирующих производных катехоламинов и их метаболитов: норадреналина, дофамина, адреналина, гомованилиновой и ванилилминдальной кислот, а также других низкомолекулярных нейромедиаторов - биогенных аминов: L-ДОФА, норметанефрина, серотонина и 5-гидроксииндол-3-уксусной кислоты. Решена задача получения соответствующих более интенсивно флуоресцирующих производных за короткий промежуток времени (3-5 мин), что позволило проводить единичный анализ за промежуток времени до 12.5 с (96 проб за 20 мин) при одновременном уменьшении необходимого объема пробы биологической жидкости (например, плазмы крови) до 10-30 мкл за счет проведения анализа в лунке планшета. Кроме того, решена задача нестабильности раствора фермента, устранения мешающего влияния матрицы реальных образцов и возможности предконцентрирования целевых аналитов за счет использования тонкой пленки оптически прозрачного биосовместимого природного полимера - хитозана, в структуру которого иммобилизован фермент, катализирующий реакции окисления и дериватизации - пероксидаза. Обнаруженный эффект нивелирования матричного влияния на интенсивность, разрешение и стабильность регистрируемых сигналов благодаря оптически прозрачной пленке хитозана, способной крепиться в лунках планшета, иммобилизовать в своей структуре фермент, набухать при внесении водных растворов, осуществлять транспорт биогенных аминов - низкомолекулярных маркеров нейромедиаторного обмена, и предконцентрировать их, позволил осуществлять высокочувствительное и селективное определение катехоламинов и их метаболитов в биологических образцах малого объема. В совокупности перечисленные улучшения технологичности процесса за счет сокращения времени, отказа от нестабильного при хранении раствора фермента нагревания и термостатирования, использования токсичных или агрессивных органических растворителей и ПАВ, позволили повысить удобство и экспрессность единичного анализа (с 10 мин до 12.5 с) и воспроизводимость (с 93 до 99%) определения катехоламинов и их метаболитов в биообъектах, а применение производной спектрофлуориметрии позволяет повысить селективность, достоверность и правильность определения катехоламинов и их метаболитов вплоть до полного их разделения. Это позволяет использовать предлагаемое изобретение для определения молекул нейромедиаторов в биологических жидкостях с целью диагностики нейродегенеративных и нейроэнокринных заболеваний.
| название | год | авторы | номер документа |
|---|---|---|---|
| СПОСОБ ПОЛУЧЕНИЯ ФЛУОРЕСЦИРУЮЩИХ ПРОИЗВОДНЫХ КАТЕХОЛАМИНОВ И ИХ МЕТАБОЛИТОВ МЕТОДОМ ДЕРИВАТИЗАЦИИ | 2012 |
|
RU2546672C2 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КАТЕХОЛАМИНОВ И ИХ МЕТАБОЛИТОВ С ИСПОЛЬЗОВАНИЕМ ТВЕРДОФАЗНОГО ФЛУОРЕСЦЕНТНОГО БИОСЕНСОРА | 2013 |
|
RU2554499C2 |
| Способ подготовки проб мочи на принципах мицеллярной экстракции для определения содержания адреналина | 2022 |
|
RU2800474C1 |
| Способ определения производных катехоламинов в моче | 2018 |
|
RU2688184C1 |
| Способ определения ароматических микробных метаболитов в форме фенилкарбоновых кислот в сыворотке крови | 2017 |
|
RU2663571C1 |
| СПОСОБ ГЕНЕРАЦИИ ПЕРОКСИДА ВОДОРОДА НА ОСНОВЕ НАНО- И/ИЛИ МИКРОЧАСТИЦ ZnO2 ДЛЯ ПРИМЕНЕНИЯ В СПЕКТРОФОТОМЕТРИЧЕСКОМ И ЛЮМИНЕСЦЕНТНОМ АНАЛИЗЕ С УЧАСТИЕМ ПЕРОКСИДАЗЫ | 2022 |
|
RU2800949C1 |
| Способ количественного определения @ , @ -метил- @ -(3,4-диоксифенил)-аланина | 1984 |
|
SU1168831A1 |
| Способ определения массовых концентраций фенола и пирокатехина в крови методом высокоэффективной жидкостной хроматографии | 2022 |
|
RU2786509C1 |
| СПОСОБ ОПРЕДЕЛЕНИЯ КОНЦЕНТРАЦИИ ФЕНОЛА И ЕГО ФЛУОРЕСЦИРУЮЩИХ ПРОИЗВОДНЫХ В ВОДНЫХ СРЕДАХ | 1994 |
|
RU2091766C1 |
| Способ количественного определения салицилатов в плазме крови | 2016 |
|
RU2622996C1 |
Группа изобретений относится к биотехнологии и может быть использована в медицинской диагностике и биохимических исследованиях для количественного обнаружения катехоламинов и их метаболитов в биологических жидкостях. Предложены тест-система и способ для экспресс-определения низкомолекулярных маркеров нейромедиаторного обмена в образцах биологических жидкостей, а также способ получения тест-системы. Тест-система представляет собой планшет с нанесенной на поверхность лунок пленки хитозана с иммобилизованной пероксидазой растительного происхождения толщиной от 0,5 до 1,5 мкм. Пероксидаза содержится в количестве от 2 до 16 пмоль на 1 мм2 площади пленки, а хитозан - от 15 до 60 мг на 1 мм2 площади пленки. Хитозан имеет распределение молекулярных масс 50-220 кДа и степень дезацетилирования не менее 75%. Способ экспресс-определения катехоламинов и их метаболитов включает нанесение пробы биологической жидкости на лунку тест-системы с последующим внесением пероксида водорода и дериватизирующего агента с образованием флуоресцирующих производных, возбуждение флуоресценции в пробе с регистрацией полученных сигналов. Изобретения позволяют определять одновременно несколько маркеров нейромедиаторного обмена, выбранных из норадреналина, дофамина, адреналина и их метаболитов, выбранных из гомованилиновой и ванилилминдальной кислот, в большом количестве образцов биологических жидкостей с достигаемыми воспроизводимостью результатов не менее 99% и временем анализа до 12.5 с на одну пробу. 3 н. и 12 з.п. ф-лы, 6 ил., 4 табл., 13 пр.
1. Тест-система для экспресс-определения низкомолекулярных маркеров нейромедиаторного обмена в образцах биологических жидкостей, а именно катехоламинов и их метаболитов, представляющая собой планшет с нанесенной на поверхность лунок пленки хитозана с иммобилизованной пероксидазой растительного происхождения толщиной от 0,5 до 1,5 мкм, при этом пероксидаза содержится в количестве от 2 до 16 пмоль на 1 мм2 площади пленки, а хитозан - от 15 до 60 мг на 1 мм2 площади пленки, и хитозан имеет распределение молекулярных масс 50-220 кДа и степень дезацетилирования не менее 75%.
2 Тест-система по п. 1, характеризующаяся тем, что в качестве пероксидазы растительного происхождения используют пероксидазу из корней хрена, соевых бобов, арахиса, листьев табака, клубней батата и листьев масличных пальм.
3. Способ получения тест-системы по п. 1, включающий приготовление 0,5-2 мас. % водного растворов хитозана и 15-20 мкМ водного раствора пероксидазы растительного происхождения с последующим их нанесением на лунки планшета в объемном соотношении растворов хитозана и пероксидазы от 1:1 до 6:1 и высушиванием не менее 12 часов на воздухе при температуре до 23±3°С до образования пленки толщиной от 0,5 до 1,5 мкм и содержанием пероксидазы в количестве от 2 до 16 пмоль на 1 мм2 площади пленки и хитозана в количестве от 15 до 60 мг на 1 мм2 площади пленки, при этом используют хитозан с распределением молекулярных масс 50-220 кДа и степенью дезацетилирования не менее 75%.
4. Способ по п. 3, характеризующийся тем, что для приготовления водного раствора хитозана используют водные растворы кислот с рН не менее 2 в количестве, достаточном для растворения хитозана в воде, при этом предотвращающем реакцию сшивания хитозана.
5. Способ по п. 4, характеризующийся тем, что в качестве кислот используют уксусную, соляную, угольную.
6. Способ по п. 3, характеризующийся тем, что в качестве пероксидазы растительного происхождения используют пероксидазу из корней хрена, соевых бобов, арахиса, листьев табака, клубней батата и листьев масличных пальм.
7. Способ экспресс-определения катехоламинов и их метаболитов в образцах биологических жидкостей, включающий нанесение пробы биологической жидкости на лунку тест-системы по п. 1, полученной способом по п. 3, с последующим внесением пероксида водорода и дериватизирующего агента в количестве, обеспечивающем проведения реакции дериватизации с образованием флуоресцирующих производных, возбуждение флуоресценции в пробе с регистрацией полученных сигналов, и при выявлении максимума в характерном диапазоне для определяемого аналита делают вывод о его наличии в образце.
8. Способ по п. 7, характеризующийся тем, что вывод о наличии в образце биологической жидкости норадреналина делают при выявлении максимума в диапазоне от 452 до 458 нм, дофамина - в диапазоне от 455 до 465 нм, адреналина - в диапазоне от 475 до 485 нм, ванилилминдальной кислоты - в диапазоне от 417 до 423 нм, гомованилиновой кислоты - в диапазоне от 422 до 428 нм, L-ДОФА - в диапазоне от 456 до 460 нм, норметанефрина - в диапазоне от 460 до 463 нм, серотонина - в диапазоне от 463 до 473 нм, 5-гидроксииндол-3-уксусной кислоты - в диапазоне от 470 до 480 нм.
9. Способ по п. 7, характеризующийся тем, что в качестве дериватизирующего агента используют ароматические амины.
10. Способ по п. 9, характеризующийся тем, что в качестве ароматического амина используют бензил амин или 1,2-дифенилэтилендиамин.
11. Способ по п. 8, характеризующийся тем, что для определения количественного содержания аналитов в образцах биологических жидкостей измеряют интенсивность полученного сигнала, вывод о количественном содержании делают по предварительно рассчитанным градуировочным зависимостям.
12. Способ по п. 8, характеризующийся тем, что дополнительно проводят обработку полученных сигналов путем вычисления второй производной интенсивности флуоресцентных сигналов и при выявлении минимума в характерном диапазоне для определяемого аналита делают вывод о его наличии в образце: вывод о наличии в образце биологической жидкости норадреналина делают при выявлении минимума в диапазоне от 452 до 458 нм, дофамина - в диапазоне от 455 до 465 нм, адреналина - в диапазоне от 475 до 485 нм, ванилилминдальной кислоты - в диапазоне от 417 до 423 нм, гомованилиновой кислоты - в диапазоне от 422 до 428 нм, L-ДОФА - в диапазоне от 456 до 460 нм, норметанефрина - в диапазоне от 460 до 463 нм, серотонина - в диапазоне от 463 до 473 нм, 5-гидроксииндол-3-уксусной кислоты - в диапазоне от 470 до 480 нм.
13. Способ по п. 7, характеризующийся тем, что пробу биологической жидкости используют объемом 10-30 мкл.
14. Способ по п. 7, характеризующийся тем, что для получения сигнала флуоресценции пробу облучают монохроматичным излучением с длиной волны в диапазоне 300-370 нм.
15. Способ по п. 7, характеризующийся тем, что на 1 объемную часть пробы используют 3-5 об. частей дериватизирующего агента и 1-5 об. частей пероксида водорода.
| RODIONOV P | |||
| V | |||
| et al | |||
| "A solid-phase fluorescent biosensor for the determination of phenolic compounds and peroxides in samples with complex matrices" | |||
| Analytical and Bioanalytical Chemistry, 2013, 406(5), p.1531-1540 | |||
| Печь-кухня, могущая работать, как самостоятельно, так и в комбинации с разного рода нагревательными приборами | 1921 |
|
SU10A1 |
| VESELOVA I | |||
| A.et al | |||
| "Properties and analytical applications of the self-assembled complex { | |||

