[ОБЛАСТЬ ТЕХНИКИ]
[0001]
Настоящее изобретение относится к замещенным полициклическим производным пиридона, имеющим ингибирующую активность кэп-зависимой эндонуклеазы, их пролекарствам и фармацевтическим композициям, содержащим указанные соединения.
[УРОВЕНЬ ТЕХНИКИ]
[0002]
Грипп является острым респираторным инфекционным заболеванием, вызываемым при инфицировании вирусом гриппа. В Японии каждую зиму регистрируют миллионы пациентов с гриппоподобными заболеваниями, а грипп сопровождается высокой заболеваемостью и смертностью. Грипп является особенно опасным заболеванием в группах повышенного риска, таких как маленькие дети и пожилые люди, у пожилых людей высока вероятность возникновения осложнений при пневмонии, и во многих случаях у пожилых людей заболевание гриппом приводит к летальному исходу.
[0003]
Известны противогриппозные лекарственные средства, такие как симметрел (торговое название: амантадин) и флумадин (торговое название: римантадин), которые ингибируют процесс денуклеации вируса, и осельтамивир (торговое название: тамифлю) и занамивир (торговое название: реленза), которые являются ингибиторами нейраминидаз, подавляющими репликацию вируса и его высвобождение из клетки. Тем не менее, из-за опасений, связанных с возможностью появления резистентных штаммов и побочных эффектов, а также мировой эпидемии вируса гриппа нового типа с высокой патогенностью и смертностью, является желательным разработка противогриппозного лекарственного средства с новым механизмом действия.
[0004]
Поскольку кэп-зависимая эндонуклеаза, которая является ферментом, продуцируемым вирусом гриппа, необходима для пролиферации вируса и обладает вирусспецифической ферментативной активностью, которую не имеет хозяин, считается, что эндонуклеаза подходит в качестве мишени для противогриппозного лекарственного средства. Кэп-зависимая эндонуклеаза вируса гриппа имеет в качестве субстрата предшественник мРНК хозяина и проявляет активность эндонуклеазы с образованием фрагмента из 9-13 оснований, включая кэп-структуру (не включая количество оснований кэп-структуры). Указанный фрагмент действует, как праймер РНК-полимеразы вируса, и используется для синтеза мРНК, кодирующей вирусный белок. То есть считается, что вещество, которое ингибирует кэп-зависимую эндонуклеазу, ингибирует синтез вирусного белка путем ингибирования синтеза мРНК вируса и, как следствие, ингибирует пролиферацию вируса.
[0005]
В качестве соединений, которые ингибируют кэп-зависимую эндонуклеазу, описаны флутимид (патентный документ 1 и непатентные документы 1 и 2), 4-замещенная 2,4-диоксобутановая кислота (патентный документ 2 и непатентные документы 3 и 4), соединения, описанные в патентных документах 3-12, и т.п., но они еще не задействованы в клиническом применений в качестве противогриппозных лекарственных средств. В патентных документах 9 и 12 описаны соединения, имеющие структуру, сходную со структурой соединений согласно настоящему изобретению, но не описаны соединения, относящиеся к настоящему изобретению. Также в патентных документах 13-15 описаны соединения, имеющие структуру, сходную со структурой соединений согласно настоящему изобретению, в качестве соединений, обладающих ингибирующей активностью в отношении интегразы, тем не менее, в документах не описана кэп-зависимая эндонуклеаза. Кроме того, в патентных документах 16 и 17 описано изобретение, относящееся к соединениям, имеющим структуру, сходную со структурой соединений согласно настоящему изобретению, в качестве соединений, обладающих ингибирующей активностью кэп-зависимой эндонуклеазы, как описано в заявке, поданной заявителями, но не описаны соединения, относящиеся к настоящему изобретению.
[ДОКУМЕНТЫ СОГЛАСНО УРОВНЮ ТЕХНИКИ]
[ПАТЕНТНЫЕ ДОКУМЕНТЫ]
[0006]


[НЕПАТЕНТНЫЕ ДОКУМЕНТЫ]
[0007]
Непатентный документ V Tetrahedron Lett 1995, 36(12), 2005
Непатентный документ 2: Tetrahedron Lett 1995, 36(12), 2009
Непатентный документ 3: Antimicrobial Agents And Chemotherapy, декабрь 1994, стр. 2827-2837
Непатентный документ 4: Antimicrobial Agents And Chemotherapy, май 1996, стр. 1304-1307
[КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ]
[ЗАДАЧИ, РЕШАЕМЫЕ ИЗОБРЕТЕНИЕМ]
[0008]
Задачей настоящего изобретения является получение соединений, обладающих противовирусной активностью, в частности, ингибирующих развитие вируса гриппа. Другой задачей настоящего изобретения является получение пролекарства из соединений для применения для введения in vivo (например, перорального введения), эффективно абсорбирующегося в организме после введения и демонстрирующего высокий фармакологический эффект.
[СРЕДСТВА ДЛЯ РЕШЕНИЯ ЗАДАЧ]
[0009]
В настоящем изобретении предложены технические решения, представленные ниже.
(1) Соединение, представленное формулой (I):

или его фармацевтически приемлемая соль:
где
Р представляет собой водород или группу PR, образующую пролекарство:
А1 представляет собой CR1AR1B, S или О;
А2 представляет собой CR2AR2B, S или О;
А3 представляет собой CR3AR3B, S или О;
каждый из А4 независимо представляет собой CR4AR4B, S или О;
количество гетероатомов, среди атомов, образующих кольцо, которое состоит из А1, А2, А3, А4, атома азота, смежного с А1, и атома углерода, смежного с А4, составляет 1 или 2;
каждый из R1A и R1B независимо представляет собой водород, галоген-, алкил, галогеналкил, алкилокси или фенил;
каждый из R2A и R2B независимо представляет собой водород, галоген-, алкил, галогеналкил, алкилокси или фенил;
каждый из R3A и R3B независимо представляет собой водород, галоген-, алкил, галогеналкил, алкилокси или фенил;
каждый из R4A и R4B независимо представляет собой водород, галоген-, алкил, галогеналкил, алкилокси или фенил;
R3A и R3B совместно со смежным атомом углерода могут образовывать неароматический карбоцикл или неароматический гетероцикл;
X представляет собой CH2, S или О;
каждый из R1 независимо представляет собой галоген-, гидрокси, алкил, галогеналкил или алкилокси;
m представляет собой любое целое число от 0 до 2; и
n представляет собой любое целое число от 1 до 2;
при условии, что исключены следующие соединения:

где каждое из определений имеет такое же значение, как описано выше.
(2) Соединение по п. (1), где группа, представленная формулой:

где каждое из определений имеет такое же значение, как описано в п. (1), представляет собой группу, представленную формулой:

где каждый из R2, R3, R4 и R5 независимо представляет собой водород или фтор; количество атомов фтора среди R2, R3, R4 и R5 составляет 1 или 2, или его фармацевтически приемлемая соль.
(3) Соединение по п. (1), где группа, представленная формулой:

где каждое из определений имеет такое же значение, как описано в п. (1), представляет собой группу, представленную формулой:
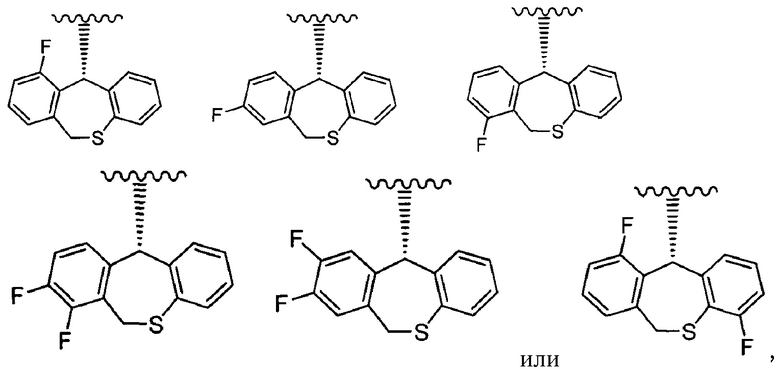
или его фармацевтически приемлемая соль.
(4) Соединение по любому из пп. (1)-(3), где группа, представленная формулой:

где каждое из определений имеет такое же значение, как описано в п. (1), представлена формулой:
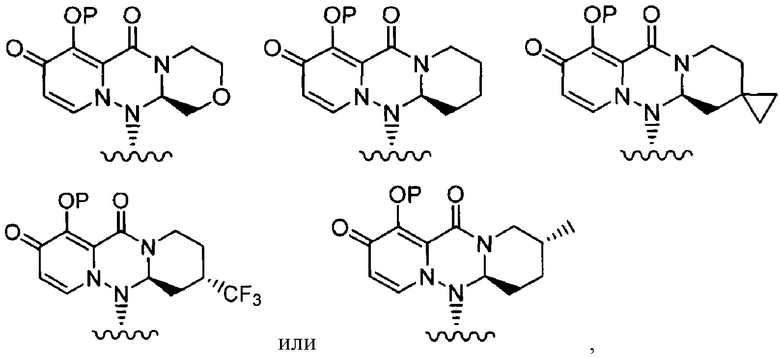
где каждое из определений имеет такое же значение, как описано в п. (1), или его фармацевтически приемлемая соль.
(5) Соединение по п. (1), представленное следующей формулой:
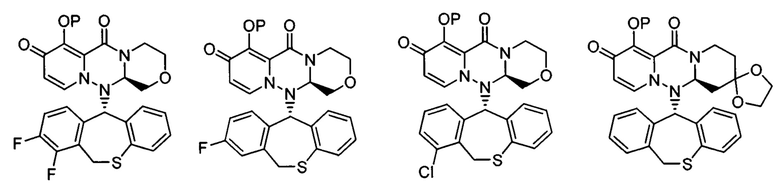




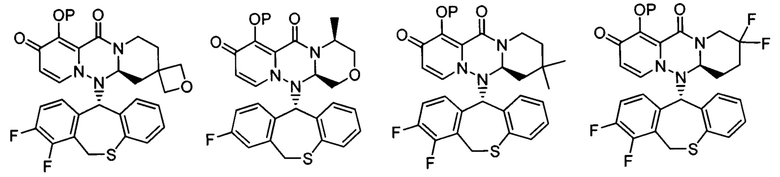



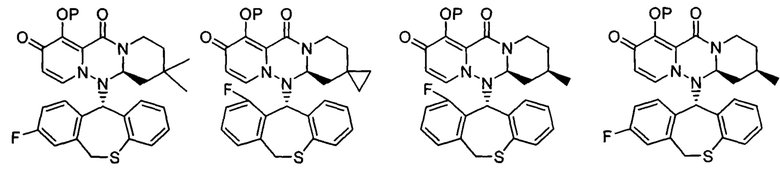

где каждое из определений имеет такое же значение, как описано в п. (1), или его фармацевтически приемлемая соль.
(6) Соединение по п. (1), представленное следующей формулой:
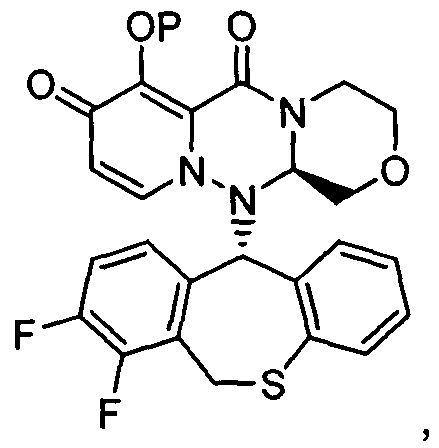
где каждое из определений имеет такое же значение, как описано в п. (1), или его фармацевтически приемлемая соль.
(7) Соединение по п. (1), представленное следующей формулой:
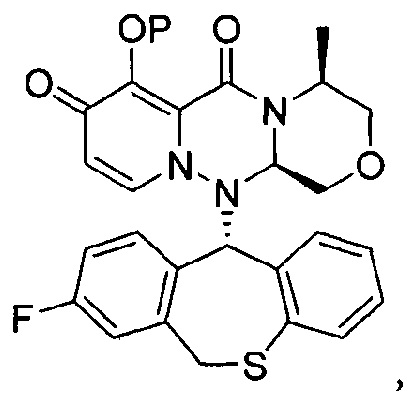
где каждое из определений имеет такое же значение, как описано в п. (1), или его фармацевтически приемлемая соль.
(8) Соединение по п. (1), представленное следующей формулой:

где каждое из определений имеет такое же значение, как описано в п. (1), или его фармацевтически приемлемая соль.
(9) Соединение по п. (1), представленное следующей формулой:

где каждое из определений имеет такое же значение, как описано в п. (1), или его фармацевтически приемлемая соль.
(10) Соединение по п. (1), представленное следующей формулой:

где каждое из определений имеет такое же значение, как описано в п. 1, или его фармацевтически приемлемая соль.
(11) Соединение, представленное следующей формулой:

где Р представляет собой водород или группу PR, образующую пролекарство, или его фармацевтически приемлемая соль.
(12) Соединение по любому из пп. (1)-(11) или его фармацевтически приемлемая соль,
где PR представляет собой группу, выбранную из следующих формул а)-ас):
a) -C(=O)-PR0,
b) -C(=O)-PR1,
c) -C(=O)-L-PR1,
d) -C(=O)-L-O-PR1,
e) -C(=O)-L-O-L-O-PR1,
f) -C(=O)-L-O-C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
j) -C(PR3)2-O-PR4,
k) -C(PR3)2-O-L-O-PR4,
l) -С(PR3)2-O-С(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
n) -C(PR3)2-O-C(=O)-N(-K)-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
p) -C(PR3)2-O-C(=O)-O-L-N(PR4)2,
q) -C(PR3)2-O-C(=O)-N(-K)-L-O-PR4,
r) -C(PR3)2-O-C(=O)-N(-K)-L-N(PR4)2,
s) -C(PR3)2-O-C(=O)-O-L-O-L-O-PR4,
t) -C(PR3)2-O-C(=O)-O-L-N(-K)-C(=O)-PR4,
u) -C(PR3)2-O-P(=O)(-PR5)2,
v) -C(PR3)2-PR6,
w) -C(=N+(PR7)2)(-N(PR7)2),
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
y) -C(PR3)2-N(-K)-C(=O)-O-PR2,
z) -P(=O)(-PR8)(-PR9),
aa) -S(=O)2-PR10,
ab) -PR11, и
ac) -C(PR3)2-C(PR3)2-O-PR2,
где L представляет собой линейный или разветвленный алкилен или линейный или разветвленный алкенилен;
K представляет собой водород или алкил, необязательно замещенный группой-заместителем А;
PR0 представляет собой алкил, необязательно замещенный группой-заместителем А, или алкенил, необязательно замещенный группой-заместителем А;
PR1 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, или алкилсульфанильную группу, необязательно замещенную группой-заместителем А;
PR2 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, гетероциклилалкил, необязательно замещенный группой-заместителем А, или триалкилсилил;
каждый из PR3 независимо представляет собой водород или алкил;
каждый из PR4 независимо представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, гетероциклилалкил, необязательно замещенный группой-заместителем А, или триалкилсилил;
каждый из PR5 независимо представляет собой гидрокси или OBn;
PR6 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
каждый из PR7 независимо представляет собой алкил, необязательно замещенный группой-заместителем А;
PR8 представляет собой алкокси, необязательно замещенный группой-заместителем А;
PR9 представляет собой алкокси, необязательно замещенный группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилокси, необязательно замещенный группой-заместителем А, гетероциклилокси, необязательно замещенный группой-заместителем А, карбоциклиламино, необязательно замещенный группой-заместителем А, или гетероциклиламино, необязательно замещенный группой-заместителем А;
PR8 и PR9 совместно со смежным атомом фосфора могут образовывать гетероцикл, необязательно замещенный группой-заместителем А;
PR10 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, или гетероциклилалкил, необязательно замещенный группой-заместителем А;
PR11 представляет собой алкил, необязательно замещенный группой-заместителем А, алкенил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
Группа-заместитель А представляет собой оксогруппу, алкил, гидроксиалкил, амино, алкиламино, карбоциклильную группу, гетероциклильную группу, карбоциклилалкил, алкилкарбонил, галоген-, гидрокси, карбокси, алкилкарбониламино, алкилкарбониламиноалкил, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкилоксикарбонилокси, алкиламинокарбонилокси, алкиламиноалкил, алкилокси, циано, нитро, азидо, алкилсульфонил, триалкилсилил или фосфо.
(13) Соединение по п. (12) или его фармацевтически приемлемая соль,
где PR представляет собой группу, выбранную из следующих формул:
a) -C(=O)-PR0,
b) -C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
v) -C(PR3)2-PR6,
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
y) -C(PR3)2-N(-K)-C(=O)-O-PR2, и
z) -P(=O)(-PR8)(-PR9),
где L представляет собой линейный или разветвленный алкилен;
K представляет собой водород или алкил, необязательно замещенный группой-заместителем А;
PR0 представляет собой алкил, необязательно замещенный группой-заместителем А;
PR1 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR2 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, или гетероциклилалкил, необязательно замещенный группой-заместителем А;
каждый из PR3 независимо представляет собой водород или алкил;
PR4 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR6 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR8 представляет собой алкокси, необязательно замещенный группой-заместителем А;
PR9 представляет собой алкокси, необязательно замещенный группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилокси, необязательно замещенный группой-заместителем А, гетероциклилокси, необязательно замещенный группой-заместителем А, карбоциклиламино, необязательно замещенный группой-заместителем А, или гетероциклиламино, необязательно замещенный группой-заместителем А; и
PR8 и PR9 совместно со смежным атомом фосфора могут образовывать гетероцикл, необязательно замещенный группой-заместителем А,
Группа-заместитель А представляет собой оксогруппу, алкил, алкиламино, карбоциклильную группу, гетероциклильную группу, алкилкарбонил, галоген-, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, циано, нитро, азидо, алкилсульфонил или триалкилсилил.
(14) Соединение, представленное следующей формулой:

или его фармацевтически приемлемая соль.
(15) Соединение, представленное следующей формулой:

или его фармацевтически приемлемая соль.
(16) Фармацевтическая композиция, содержащая соединение по любому из пп. (1)-(15) или его фармацевтически приемлемую соль.
(17) Фармацевтическая композиция по п. (16), которая проявляет противогриппозную активность.
(18) Фармацевтическая композиция по п. (16), которая проявляет ингибирующую активность кэп-зависимой эндонуклеазы.
(19) Способ лечения и/или предотвращения заболевания, вызванного вирусом, содержащим кэп-зависимую эндонуклеазу, включающий введение соединения по любому из пп. (1)-(15) или его фармацевтически приемлемой соли.
(20) Соединение по любому из пп. (1)-(15) или его фармацевтически приемлемая соль для лечения или предотвращения заболевания, вызванного вирусом, содержащим кэп-зависимую эндонуклеазу.
(21) Применение соединения по любому из пп. (1)-(15) или его фармацевтически приемлемой соли для производства терапевтического или профилактического средства для заболевания, вызванного вирусом, содержащим кэп-зависимую эндонуклеазу.
(22) Фармацевтическая композиция, содержащая соединение по любому из пп. (1)-(15) или его фармацевтически приемлемую соль, для перорального введения.
(23) Фармацевтическая композиция по п. (22), которая представляет собой таблетку, порошок, гранулу, капсулу, пилюлю, пленку, суспензию, эмульсию, эликсир, сироп, лимонад, спиртовой раствор, ароматическую воду, экстракт, отвар или настойку.
(24) Фармацевтическая композиция по п. (16), которая представляет собой таблетку, покрытую сахарной оболочкой, таблетку, покрытую пленочной оболочкой, таблетку, покрытую кишечнорастворимой оболочкой, таблетку с замедленным высвобождением, пастилку, подъязычную таблетку, защечную таблетку, жевательную таблетку, таблетку для рассасывания, сухой сироп, капсулу в мягкой оболочке, микрокапсулу или капсулу с замедленным высвобождением.
(25) Фармацевтическая композиция, содержащая соединение по любому из пп. (1)-(15) или его фармацевтически приемлемую соль, для парентерального введения.
(26) Фармацевтическая композиция по п. (25) для накожного применения, подкожного, внутривенного, внутриартериального, внутримышечного, внутрибрюшинного, чресслизистого, ингаляционного или трансназального введения, введения через глаза или внутреннее ухо или вагинального введения.
(27) Фармацевтическая композиция по п. (25) или п. (26), которая представляет собой раствор для инъекции, инфузионный раствор, глазные капли, капли для носа, ушные капли, аэрозоль, состав для ингаляции, лосьон, состав для пропитки, линимент, ополаскиватель для ротовой полости, клизму, мазь, пластырь, желе, крем, накладку, катаплазму, порошок для наружного применения или суппозиторий.
(28) Фармацевтическая композиция, содержащая соединение по любому из пп. (1)-(l5) или его фармацевтически приемлемую соль, для детей и пожилых пациентов.
(29) Фармацевтическая композиция, состоящая из комбинации соединения по любому из пп. (1)-(15) или его фармацевтически приемлемой соли и ингибитора нейраминидазы, ингибитора PHK-зависимой PHK-полимеразы, ингибитора белка М2, РВ2-специфического кэп-связывающего ингибитора, антитела к гемагглютинину или иммунологического агента.
(30) Фармацевтическая композиция, содержащая соединение по любому из пп. (1)-(15) или его фармацевтически приемлемую соль, для комбинированной терапии в комбинации с ингибитором нейраминидазы, ингибитором PHK-зависимой PHK-полимеразы, ингибитором белка М2, РВ2-специфическим кэп-связывающим ингибитором, антителом к гемагглютинину или иммунологическим агентом.
[0010]
В настоящем изобретении также предложен способ лечения или предотвращения инфекционного гриппозного заболевания с применением пролекарственного соединения и соединения, которое проявляет противогриппозную активность. В настоящем изобретении также предложено исходное соединение пролекарственного соединения. Исходное соединение является эффективным в качестве средства против гриппа или промежуточного соединения пролекарственного соединения.
[ТЕХНИЧЕСКИЙ РЕЗУЛЬТАТ ИЗОБРЕТЕНИЯ]
[0011]
Соединение согласно настоящему изобретению обладает ингибирующей активностью кэп-зависимой эндонуклеазы. Более предпочтительным соединением является пролекарство, и пролекарство превращается в исходное соединение, обладающее ингибирующей активностью кэп-зависимой эндонуклеазы, после введения in vivo и, таким образом, является эффективным в качестве терапевтического средства и/или профилактического средства для инфекционного гриппозного заболевания.
[КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ]
[0012]
[Фигура 1] На фигуре 1 представлен результат измерения концентрации соединения III-2 в плазме после перорального введения пролекарственного соединения II-6, исходным соединением которого является соединение III-2, крысам после приема пищи.
[Фигура 2] На фигуре 2 представлен результат измерения концентрации соединения II-6 в плазме после перорального введения пролекарственного соединения II-6, исходным соединением которого является соединение III-2, крысам после приема пищи.
[ЛУЧШИЙ ВАРИАНТ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ]
[0013]
Ниже объясняется значение каждого из терминов, применяемых в настоящем описании. Каждый из терминов используется в унифицированном смысле и используется в том же смысле при использовании отдельно или в комбинации с другим термином.
Термин "состоящий из" означает наличие только указанных компонентов.
Термин "содержащий" означает отсутствие ограничений указанными компонентами и не исключает наличие неописанных факторов.
[0014]
"Необязательно замещенный группой-заместителем А" означает, что фрагмент в произвольном положении может быть замещен одним, двумя или более одинаковыми или различными заместителями, выбранными из группы-заместителя А.
[0015]
"Пролекарство" в настоящем описании относится к соединению, представленному формулой (II) в следующей формуле реакцию
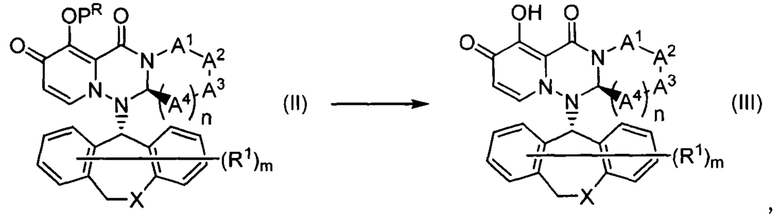
где каждый из символов является таким, как описано выше,
или его фармацевтически приемлемой соли, и обозначает соединение, демонстрирующее ингибирующую активность кэп-зависимой эндонуклеазы (CEN) и/или ингибирующее действие в отношении ЦПЭ при превращении в соединение, представленное формулой (III), в результате реакции разложения, вызванной ферментами, метаболизирующими лекарственное средство, гидролазами, кислотами желудочного сока, энтеробактериями и т.д., в физиологических условиях in vivo.
Пролекарство более предпочтительно обозначает соединение, для которого биодоступность и/или ППК (площадь под кривой зависимости концентрации в крови от времени) при введении in vivo улучшаются больше, чем для соединения, представленного формулой (III).
Следовательно, пролекарство эффективно абсорбируется в организме в желудке и/или кишечнике после введения in vivo (например, перорального введения), а затем превращается в соединение, представленное формулой (III). Таким образом, пролекарство предпочтительно проявляет более сильный эффект при лечении и/или предотвращении гриппа по сравнению с соединением, представленным формулой (III).
Один из вариантов реализации
"группы, представленной 
где каждое из определений имеет такое же значение, как описано в п. (1), представляет собой группу, представленную формулой:
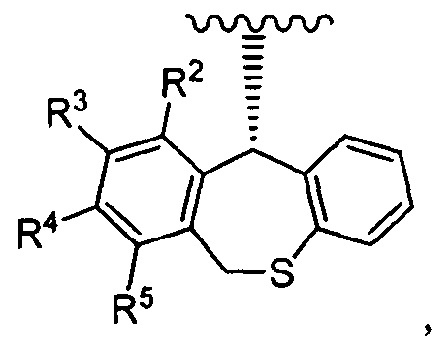
где каждый из R2, R3, R4 и R5 независимо представляет собой водород или фтор; количество атомов фтора среди R2, R3, R4 и R5 составляет 1 или 2.
Другой вариант реализации представляет собой группу, представленную формулой:

и группа, представленная формулой:
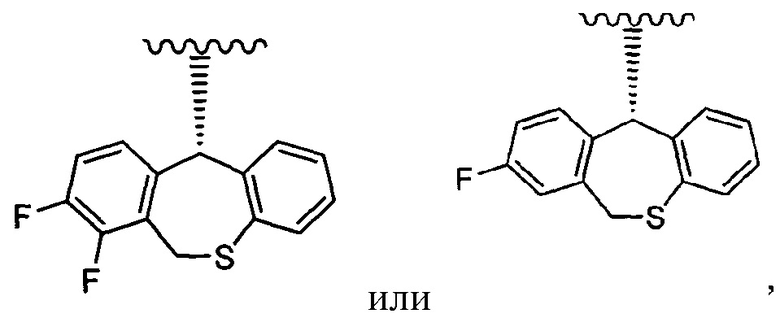
является предпочтительной, а группа, представленная формулой:
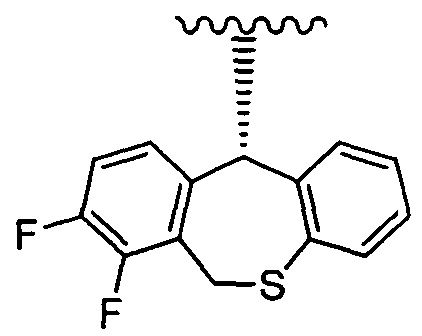
является особенно предпочтительной.
[0016]
В настоящем описании "группа PR, образующая пролекарство," относится к группе "PR" в формуле (II) в следующей формуле реакции:

[0017]
где каждый из символов является таким, как описано выше,
и группа -OPR превращается в группу -ОН в формуле (III) в результате реакции разложения, вызванной ферментами, метаболизирующими лекарственное средство, гидролазами, кислотами желудочного сока, энтеробактериями и т.д., в физиологических условиях in vivo.
"Группа PR, образующая пролекарство," более предпочтительно обозначает группу, которая улучшает биодоступность и/или ППК (площадь под кривой зависимости концентрации в крови от времени) соединения, представленного формулой (III), при введении в соединение, представленное формулой (III).
[0018]
Примеры группы PR, образующей пролекарство, включают группы, описанные в Prog. Med. 5: 2157-2161 (1985) и представленные в The British Library - "The world's Knowledge".
Группа "PR" в группе -OPR в формуле (I) или (II) может представлять собой группу, превращаемую в группу -ОН in vivo, и примеры предпочтительно включают группу, выбранную из следующих формул а)-ас).
a) -C(=O)-PR0,
b) -C(=O)-PR1,
c) -C(=O)-L-PR1,
d) -C(=O)-L-O-PR1,
e) -C(=O)-L-O-L-O-PR1,
f) -C(=O)-L-O-C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
j) -C(PR3)2-O-PR4,
k) -C(PR3)2-O-L-O-PR4,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
n) -C(PR3)2-O-C(=O)-N(-K)-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
p) -C(PR3)2-O-C(=O)-O-L-N(PR4)2,
q) -C(PR3)2-O-C(=O)-N(-K)-L-O-PR4,
r) -C(PR3)2-O-C(=O)-N(-K)-L-N(PR4)2,
s) -C(PR3)2-O-C(=O)-O-L-O-L-O-PR4,
t) -C(PR3)2-O-C(=O)-O-L-N(-K)-C(=O)-PR4,
u) -C(PR3)2-O-P(=O)(-PR5)2,
v) -C(PR3)2-PR6 (за исключением бензильной группы),
w) -C(=N+(PR7)2)(-N(PR7)2),
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
y) -C(PR3)2-N(-K)-C(=O)-O-PR2,
z) -P(=O)(-PR8)(-PR9),
aa) -S(=O)2-PR10,
ab) -PR11, и
ac)-C(PR3)2-C(PR3)2-O-PR2,
где L представляет собой линейный или разветвленный алкилен или линейный или разветвленный алкенилен;
K представляет собой водород или алкил, необязательно замещенный группой-заместителем А;
PR0 представляет собой алкил, необязательно замещенный группой-заместителем А, или алкенил, необязательно замещенный группой-заместителем А;
PR1 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, или алкилсульфанильную группу, необязательно замещенную группой-заместителем А;
PR2 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, гетероциклилалкил, необязательно замещенный группой-заместителем А, или триалкилсилил;
каждый из PR3 независимо представляет собой водород или алкил;
каждый из PR4 независимо представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, гетероциклилалкил, необязательно замещенный группой-заместителем А, или триалкилсилил;
каждый из PR5 независимо представляет собой гидрокси или OBn;
PR6 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
каждый из PR7 независимо представляет собой алкил, необязательно замещенный группой-заместителем А;
PR8 представляет собой алкокси, необязательно замещенный группой-заместителем А;
PR9 представляет собой алкокси, необязательно замещенный группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилокси, необязательно замещенный группой-заместителем А, гетероциклилокси, необязательно замещенный группой-заместителем А, карбоциклиламино, необязательно замещенный группой-заместителем А, или гетероциклиламино, необязательно замещенный группой-заместителем А;
PR8 и PR9 совместно со смежным атомом фосфора могут образовывать гетероцикл, необязательно замещенный группой-заместителем А;
PR10 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, или гетероциклилалкил, необязательно замещенный группой-заместителем А;
PR11 представляет собой алкил, необязательно замещенный группой-заместителем А, алкенил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А.
Группа-заместитель А представляет собой оксогруппу, алкил, гидроксиалкил, амино, алкиламино, карбоциклил, гетероциклил, карбоциклилалкил, алкилкарбонил, галоген-, гидрокси, карбокси, алкилкарбониламино, алкилкарбониламиноалкил, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкилоксикарбонилокси, алкиламинокарбонилокси, алкиламиноалкил, алкилокси, циано, нитро, азидо, алкилсульфонил, триалкилсилил или фосфо.
[0019]
Группа PR, образующая пролекарство, предпочтительно представляет собой группу, выбранную из следующих.
a) -C(=O)-PR0,
b) -C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
v) -C(PR3)2-PR6 (за исключением бензильной группы),
х) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
у) -C(PR3)2-N(-K)-C(=O)-O-PR2, и
z) -P(=O)(-PR8)(-PR9),
где L представляет собой линейный или разветвленный алкилен;
K представляет собой водород или алкил, необязательно замещенный группой-заместителем А;
PR0 представляет собой алкил, необязательно замещенный группой-заместителем А;
PR1 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
каждый из PR2 независимо представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, или гетероциклилалкил, необязательно замещенный группой-заместителем А;
каждый из PR3 независимо представляет собой водород или алкил;
каждый из PR4 независимо представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR6 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR8 представляет собой алкокси, необязательно замещенный группой-заместителем А;
PR9 представляет собой алкокси, необязательно замещенный группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилокси, необязательно замещенный группой-заместителем А, гетероциклилокси, необязательно замещенный группой-заместителем А, карбоциклиламино, необязательно замещенный группой-заместителем А, или гетероциклиламино, необязательно замещенный группой-заместителем А; и
PR8 и PR9 совместно со смежным атомом фосфора могут образовывать гетероцикл, необязательно замещенный группой-заместителем А.
Группа-заместитель А представляет собой оксогруппу, алкил, алкиламино, карбоциклил, гетероциклил, алкилкарбонил, галоген-, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил или триалкилсилил.
[0020]
"Превращенный в пролекарство" в настоящем описании означает, что, как показано в следующей формуле реакции:

[0021]
где каждый из символов является таким, как описано выше,
гидрокси в соединении формулы (III) или его фармацевтически приемлемой соли превращается в группу -OPR.
[0022]
"Исходное соединение" в настоящем описании обозначает соединение, которое является источником при получении "пролекарства", и/или соединение, которое высвобождается из "пролекарства" в результате реакции под воздействиемферментов, кислот желудочного сока и т.д., в физиологических условиях in vivo, и, в частности, обозначает соединение, представленное формулой (III), или его фармацевтически приемлемую соль или сольват.
[0023]
Термин "галоген-" включает атом фтора, атом хлора, атом брома и атом йода. Атом фтора и атом хлора являются особенно предпочтительными.
[0024]
Термин "алкил" включает C1-C15, предпочтительно C1-C10, более предпочтительно C1-C6 и более предпочтительно C1-С4 линейную или разветвленную углеводородную группу. Примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, н-гептил, изогептил, н-октил, изооктил, н-нонил, н-децил и т.п.
Предпочтительный вариант релизации "алкила" представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил или н-пентил. Более предпочтительный вариант релизации представляет собой метил, этил, н-пропил, изопропил или трет-бутил.
[0025]
Термин "алкенил" включает C2-С15, предпочтительно C2-C10, более предпочтительно C2-C6 и более предпочтительно C2-C4 линейную или разветвленную углеводородную группу, содержащую одну или более двойных связей в любом(ых) положении(ях). Примеры включают винил, аллил, пропенил, изопропенил, бутенил, изобутенил, пренил, бутадиенил, пентенил, изопентенил, пентадиенил, гексенил, изогексенил, гексадиенил, гептенил, октенил, ноненил, деценил, ундеценил, додеценил, тридеценил, тетрадеценил, пентадеценил и т.п.
Предпочтительный вариант релизации "алкенила" представляет собой винил, аллил, пропенил, изопропенил или бутенил.
[0026]
Термин "алкилен" включает C1-C15, предпочтительно C1-C10, более предпочтительно C1-C6 и более предпочтительно C1-С4 линейную или разветвленную двухвалентную углеводородную группу. Примеры включают метилен, этилен, триметилен, пропилен, тетраметилен, пентаметилен, гексаметилен и т.п.
[0027]
Термин "алкенилен" включает C2-C15, предпочтительно C2-C10, более предпочтительно C2-C6 и более предпочтительно C1-C4 линейную или разветвленную двухвалентную углеводородную группу, содержащую одну или более двойных связей в любом(ых) положении(ях). Примеры включают винилен, пренилен, бутенилен, пентенилен и т.п.
[0028]
Термин "гидроксиалкил" означает группу, в которой атом(ы) водорода, присоединенный(е) к атому(ам) углерода описанного выше "алкила", заменены на одну или более гидроксильные группы. Примеры включают гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 1-гидроксипропил, 2-гидроксипропил, 1,2-дигидроксиэтил и т.п.
Предпочтительный вариант релизации "гидроксиалкила" представляет собой гидроксиметил.
[0029]
Термин "алкилокси" означает группу, в которой описанный выше "алкил" связан с атомом кислорода. Примеры включают метилокси, этилокси, н-пропилокси, изопропилокси, н-бутилокси, трет-бутилокси, изобутилокси, втор-бутилокси, пентилокси, изопентилокси, гексилокси и т.п.
Предпочтительный вариант релизации "алкилокси" представляет собой метилокси, этилокси, н-пропилокси, изопропилокси или трет-бутилокси.
[0030]
Термин "галогеналкил" означает группу, в которой один или более "галогенов-", описанных выше, связаны с описанным выше «алкилом». Примеры включают монофторметил, монофторэтил, монофторпропил, 2,2,3,3,3-пентафторпропил, монохлорметил, трифторметил, трихлорметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил, 1,2-дибромэтил, 1,1,1-трифторпропан-2-ил и т.п.
Предпочтительный вариант релизации "галогеналкила" представляет собой трифторметил или трихлорметил.
[0031]
Термин "алкилкарбонил" означает группу, в которой описанный выше "алкил" связан с карбонильной группой. Примеры включают метилкарбонил, этилкарбонил, пропилкарбонил, изопропилкарбонил, трет-бутилкарбонил, изобутилкарбонил, втор-бутилкарбонил, пентилкарбонил, изопентилкарбонил, гексилкарбонил и т.п.
Предпочтительный вариант релизации "алкилкарбонила" представляет собой метилкарбонил, этилкарбонил или н-пропилкарбонил.
[0032]
Термин "алкиламино" означает группу, в которой один или два атома водорода, присоединенных к атому азота аминогруппы, заменены на описанный выше "алкил". Две алкильные группы могут являться одинаковыми или могут различаться. Примеры включают метиламино, этиламино, изопропиламино, диметиламино, диэтиламино, N,N-диизопропиламино, N-метил-N-этиламино, N-изопропил-N-этиламино и т.п.
Предпочтительный вариант релизации "алкиламино" представляет собой метиламино, этиламино, диметиламино или диэтиламино.
[0033]
Термин "алкиламиноалкил" означает группу, в которой описанный выше "алкиламино" связан с описанным выше «алкилом».
[0034]
Термин "алкиламинокарбонил" означает группу, в которой описанный выше "алкиламино" связан с карбонильной группой.
[0035]
Термин "алкиламинокарбонилокси" означает группу, в которой описанный выше "алкиламинокарбонил" связан с атомом кислорода.
[0036]
Термин "алкилкарбониламино" означает группу, в которой атом водорода, связанный с атомом азота аминогруппы, заменен на описанный выше "алкилкарбонил". Примеры включают метилкарбониламино, этилкарбониламино, пропилкарбониламино, изопропилкарбониламино, трет-бутилкарбониламино, изобутилкарбониламино, втор-бутилкарбониламино и т.п.
Предпочтительный вариант релизации "алкилкарбониламино" представляет собой метилкарбониламино или этилкарбониламино.
[0037]
Термин "алкилкарбонилокси" означает группу, в которой описанный выше "алкилкарбонил" связан с атомом кислорода. Примеры включают метилкарбонилокси, этилкарбонилокси, пропилкарбонилокси, изопропилкарбонилокси, трет-бутилкарбонилокси, изобутилкарбонилокси, втор-бутилкарбонилокси и т.п.
Предпочтительный вариант релизации "алкилкарбонилокси" представляет собой метилкарбонилокси или этилкарбонилокси.
[0038]
Термин "алкилкарбониламиноалкил" означает группу, в которой описанный выше "алкилкарбониламино" связан с описанным выше «алкилом».
[0039]
Термин "алкилоксикарбонил" означает группу, в которой описанный выше "алкилокси" связан с карбонильной группой. Примеры включают метилоксикарбонил, этилоксикарбонил, пропилоксикарбонил, изопропилоксикарбонил, трет-бутилоксикарбонил, изобутилоксикарбонил, втор-бутилоксикарбонил, пентилоксикарбонил, изобентилоксикарбонил, гексилоксикарбонил и т.п.
Предпочтительный вариант релизации "алкилоксикарбонила" представляет собой метилоксикарбонил, этилоксикарбонил или пропилоксикарбонил.
[0040]
Термин "алкилоксикарбонилалкил" означает группу, в которой описанный выше "алкилоксикарбонил" связан с описанным выше «алкилом».
[0041]
Термин "алкилоксикарбонилокси" означает группу, в которой описанный выше "алкилоксикарбонил" связан с атомом кислорода.
[0042]
Термин "алкилсульфанил" означает группу, в которой атом водорода, связанный с атомом серы сульфанильной группы, заменен на описанный выше "алкил". Примеры включают метилсульфанил, этилсульфанил, н-пропилсульфанил, изопропилсульфанил и т.п.
[0043]
Термин "алкилсульфонил" означает группу, в которой описанный выше "алкил" связан с сульфонильной группой. Примеры включают метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, трет-бутилсульфонил, изобутилсульфонил, втор-бутилсульфонил и т.п.
Предпочтительный вариант релизации "алкилсульфонила" представляет собой метилсульфонил или этилсульфонил.
[0044]
Термин "триалкилсилил" означает группу, в которой три описанных выше "алкила" связаны с атомом кремния. Три алкильные группы могут являться одинаковыми или могут различаться. Примеры включают триметилсилил, триэтилсилил, трет-бутилдиметилсилил и т.п.
[0045]
Термин "карбоциклильная группа" означает C3-С20, предпочтительно C3-C16, более предпочтительно С4-С12 циклическую углеводородную группу и включает ароматический карбоциклил и неароматический карбоциклил.
Термин "ароматический карбоциклил" означает циклическую ароматическую углеводородную группу, которая является моноциклической или полициклической, имеющей два или более колец. Примеры включают фенил, нафтил, антрил, фенантрил и т.п.
Предпочтительный вариант релизации "ароматического карбоциклила" представляет собой фенил, 1-нафтил или 2-нафтил. Другой вариант релизации "ароматического карбоциклила" представляет собой фенил,
Термин "неароматический карбоциклил" означает циклическую насыщенную углеводородную группу или циклическую ненасыщенную неароматическую углеводородную группу, которая является моноциклической или полициклической, имеющей два или более колец. Примеры "неароматического карбоциклила", который является полициклическим, имеющим два или более колец, включают конденсированную кольцевую группу, в которой неароматический карбоциклил, который является моноциклическим или полициклическим, имеющим два или более колец, конденсирован с кольцом описанного выше "ароматического карбоциклила".
Кроме того, примеры "неароматического карбоциклила" также включают группы, имеющие мостиковую связь, или группы, образующие спирокольцо, как показано ниже:

Неароматический карбоциклил, который является моноциклическим, предпочтительно представляет собой C3-C16, более предпочтительно C3-С12 и более предпочтительно C3-C8 карбоциклил. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклогексадиенил и т.п.
Примеры неароматического карбоциклила, который является полициклическим, имеющим два или более колец, включают инданил, инденил, аценафтил, тетрагидронафтил, флуоренил и т.п.
[0046]
Термин "карбоцикл" означает C3-С20, предпочтительно C3-C16, более предпочтительно C4-C12 циклический углеводород и включает ароматический карбоцикл и неароматический карбоцикл.
Термин "ароматический карбоцикл" означает циклический ароматический углеводород, который является моноциклическим или полициклическим, имеющим два или более колец. Примеры включают бензольное кольцо, нафталиновое кольцо, антраценовое кольцо, фенантреновое кольцо и т.п.
Предпочтительный вариант реализации "ароматического карбоцикла" представляет собой бензольное кольцо или нафталиновое кольцо. Другой вариант реализации "ароматического карбоцикла" представляет собой бензольное кольцо.
Термин "неароматический карбоцикл" означает насыщенный карбоцикл или ненасыщенный неароматический карбоцикл, который является моноциклическим или полициклическим, имеющим два или более колец. Примеры "неароматического карбоцикла", который является полициклическим, имеющим два или более колец, включают конденсированную кольцевую систему, в которой неароматический карбоцикл, который является моноциклическим или полициклическим, имеющим два или более колец, конденсирован с кольцом описанного выше "ароматического карбоцикла".
Кроме того, примеры "неароматического карбоцикла" также включают циклы, имеющие мостиковую связь, или циклы, образующие спирокольцо, как показано ниже:
Неароматический карбоцикл, который является моноциклическим, предпочтительно представляет собой C3-C16, более предпочтительно C3-C12 и более предпочтительно C3-C8 карбоцикл. Примеры включают циклопропан, циклобутан, циклопентан, циклогексан, циклогептан, циклооктан, циклононан, циклодекан, циклопропен, циклобутен, циклопентен, циклогексен, циклогептен, циклогександиен и т.п.
Примеры неароматического карбоцикла, который является полициклическим, содержащим два или более колец, включают индан, инден, аценафталин, тетрагидронафталин, флуорин и т.п.
[0047]
Термин "гетероциклильная группа" включает ароматический гетероциклил и неароматический гетероциклил, который содержит один или более гетероатомов, независимо выбранных из О, S и N.
Термин "ароматический гетероциклил" означает ароматический циклил, который является моноциклическим или полициклическим, имеющим два или более колец, содержащий один или более гетероатомов, независимо выбранных из О, S и N. Примеры "ароматического гетероциклила", который является полициклическим, имеющим два или более колец, включают конденсированную кольцевую группу, в которой ароматический гетероциклил, который является моноциклическим или полициклическим, имеющим два или более колец, конденсирован с кольцом описанного выше "ароматического карбоциклила".
Ароматический гетероциклил, который является моноциклическим, предпочтительно представляет собой 5-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают пирролил, имидазолил, пиразолил, пиридил, пиридазинил, пиримидинил, пиразинил, триазолил, триазинил, тетразолил, фурил, тиенил, изоксазолил, оксазолил, оксадиазолил, изотиазолил, тиазолил, тиадиазолил и т.п.
Примеры ароматического гетероциклила, который является бициклическим, включают индолил, изоиндолил, индазолил, индолизинил, хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, хиноксалинил, пуринил, птеридинил, бензимидазолил, бензизоксазолил, бензоксазолил, бензоксадиазолил, бензизотиазолил, бензотиазолил, бензотиадиазолил, бензофурил, изобензофурил, бензотиенил, бензотриазолил, имидазопиридил, триазолопиридил, имидазотиазолил, пиразинопиридазинил, оксазолопиридил, тиазолопиридил и т.п.
Примеры ароматического гетероциклила, который является полициклическим, имеющим три или более колец, включают карбазолил, акридинил, ксантенил, фенотиазинил, феноксатиинил, феноксазинил, дибензофурил и т.п.
Термин "неароматический гетероциклил" означает неароматический циклил, который является моноциклическим или полициклическим, имеющим два или более колец, содержащий один или более гетероатомов, независимо выбранных из О, S и N. Примеры "неароматического гетероциклила", который является полициклическим, имеющим два или более колец, включают конденсированную кольцевую группу, в которой неароматический гетероцикл, который является моноциклическим или полициклическим, имеющим два или более колец, конденсирован с кольцом описанного выше "ароматического карбоциклила", "неароматического карбоциклила" и/или "ароматического гетероциклила".
Кроме того, примеры "неароматического гетероциклила" также включают группы, имеющие мостиковую связь, или группы, образующие спирокольцо, как показано ниже:
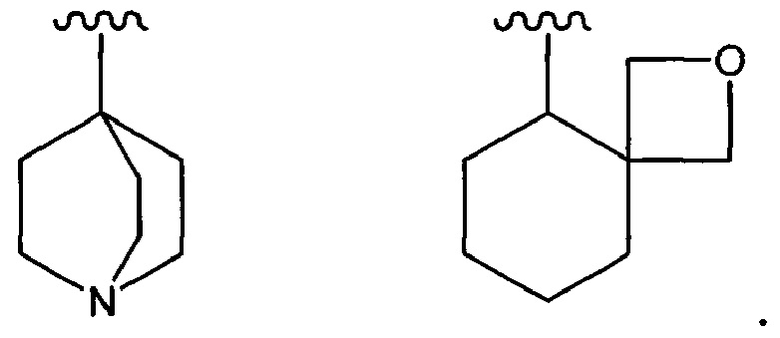
Неароматический гетероциклил, который является моноциклическим, предпочтительно представляет собой 3-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают диоксанил, тииранил, оксиранил, оксетанил, оксатиоланил, азетидинил, тианил, тиазолидинил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, пиразолинил, пиперидинил, пиперазинил, морфолинил, морфолино, тиоморфолинил, тиоморфолино, дигидропиридинил, тетрагидропиридинил, тетрагидрофурил, тетрагидропиранил, дигидротиазолинил, тетрагидротиазолинил, тетрагидроизотиазолинил, дигидрооксазинил, гексагидроазепинил, тетрагидродиазепинил, тетрагидропиридазинил, гексагидропиримидинил, диоксоланил, диоксазинил, азиридинил, диоксолинил, оксапанил, тиоланил, тиинил, тиазинил и т.п.
Примеры неароматического гетероциклила, который является полициклическим, имеющим два или более колец, включают индолинил, изоиндолинил, хроманил, изохроманил и т.п.
[0048]
Термин "гетероцикл" включает ароматический цикл и неароматический гетероцикл, содержащие один или более гетероатомов, независимо выбранных из О, S и N.
Термин "ароматический гетероцикл" означает ароматический цикл, который является моноциклическим или полициклическим, имеющим два или более колец, содержащий один или более гетероатомов, независимо выбранных из О, S и N. Примеры "ароматического гетероцикла", который является полициклическим, имеющим два или более колец, включают конденсированную кольцевую систему, в которой ароматический гетероцикл, который является моноциклическим или полициклическим, имеющим два или более колец, конденсирован с кольцом описанного выше "ароматического карбоцикла".
Ароматический гетероцикл, который является моноциклическим, предпочтительно представляет собой 5-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают пиррол, имидазол, пиразол, пиридин, пиридазин, пиримидин, пиразин, триазол, триазин, тетразол, фуран, тиофен, изоксазол, оксазол, оксадиазол, изотиазол, тиазол, тиадиазол и т.п.
Примеры ароматического гетероцикла, который является бициклическим, включают индолин, изоиндолин, индазорин, индолизин, хинолин, изохинолин, циннолин, фталазин, хиназолин, нафтиридин, хиноксалин, пурин, птеридин, бензимидазол, бензизоксазол, бензоксазол, бензоксадиазол, бензизотиазол, бензотиазол, бензотиадиазол, бензофуран, изобензофуран, бензотиофен, бензотриазол, имидазопиридин, триазолопиридин, имидазотиазол, пиразинопиридазин, оксазолопиридин, тиазолопиридин и т.п.
Примеры ароматического гетероцикла, который является полициклическим, имеющим три или более колец, включают карбазол, акридин, ксантен, фенотиазин, феноксатиин, феноксазин, дибензофуран и т.п.
Термин "неароматический гетероцикл" означает неароматический цикл, который является моноциклическим или полициклическим, имеющим два или более колец, содержащий один или более гетероатомов, независимо выбранных из О, S и N. Примеры "неароматического гетероцикла", который является полициклическим, имеющим два или более колец, включают конденсированную кольцевую систему, в которой неароматический гетероцикл, который является моноциклическим или полициклическим, имеющим два или более колец, конденсирован с кольцом описанного выше "ароматического карбоцикла", "неароматического карбоцикла" и/или "ароматического гетероцикла".
Кроме того, примеры "неароматического гетероцикла" также включают циклы, имеющие мостиковую связь, или циклы, образующие спирокольцо, как показано ниже:

Неароматический гетероцикл, который является моноциклическим, предпочтительно представляет собой 3-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают диоксан, тииран, оксиран, оксетан, оксатиолан, азетидин, тиан, тиазолидин, пирролидин, пирролин, имидазолидин, имидазолин, пиразолидин, пиразолин, пиперидин, пиперазин, морфолин, тиоморфолин, дигидропиридин, тетрагидропиридин, тетрагидрофуран, тетрагидропиран, дигидротиазолин, тетрагидротиазолин, тетрагидроизотиазолин, дигидрооксазин, гексагидроазепин, тетрагидродиазепин, тетрагидропиридазин, гексагидропиримидин, диоксолан, диоксазин, азиридин, диоксолин, оксепан, тиолан, тиазин и т.п.
Примеры неароматического гетероцикла, который является полициклическим, имеющим два или более колец, включают индолин, изоиндолин, хроман, изохроман и т.п.
[0049]
"Карбоцикл", как часть "карбоциклилалкила", "карбоциклилокси" или "карбоциклиламино", является таким же, как описанный выше "карбоцикл".
[0050]
"Гетероцикл", как часть "гетероциклилалкила", "гетероциклилокси" или "гетероциклиламино", является таким же, как описанный выше "гетероцикл".
[0051]
Настоящее изобретение отличается тем, что соединение, выделенное путем оптического разделения изомеров трициклических соединений, замещенных другой трициклической группой, улучшает инигибирующую активность кэп-зависимой эндонуклеазы.
[0052]
Настоящее изобретение также отличается тем, что настоящее соединение эффективно абсорбируется в организме после введения (например, перорального введения) и демонстрирует высокую эффективность благодаря введению группы PR, образующей пролекарство.
[0053]
Один или более атомов водорода, углерода и/или других атомов в соединениях согласно настоящему изобретению можно заменять на изотопы водорода, углерода и/или других атомов. Примеры изотопов включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, йода и хлора, такие как 2Н, 3H, 11C, 13С, 14C, 15N, 18O, 17O, 31P, 32P, 35S, 18F, 123I и 36Cl, соответственно. Соединения согласно настоящему изобретению включают соединения, содержащие указанные изотопы. Соединения, содержащие указанные выше изотопы, подходят для применения в качестве лекарственных средств и включают все радиоактивно меченые соединения из числа соединений согласно настоящему изобретению. "Способ введения радиоактивной метки" при получении "радиоактивно меченых соединений" охватывается настоящим изобретением, и "радиоактивно меченые соединения" подходят для применения для изучения фармакокинетики метаболизированных лекарственных средств, изучения связывания и/или в средствах для диагностики.
[0054]
Радиоактивно меченое соединение согласно настоящему изобретению можно получать с применением способов, хорошо известных в данной области. Например, меченое тритием соединение согласно настоящему изобретению можно получать путем введения трития в конкретное соединение согласно настоящему изобретению при помощи реакции каталитического галогенирования с применением трития. Указанный способ включает взаимодействие галогенированного подходящим образом соединения-предшественника соединения согласно настоящему изобретению с газообразным тритием в присутствии подходящего катализатора, такого как Pd/C, и в присутствии или при отсутствии основания. Другой подходящий способ получения меченого тритием соединения можно найти в "Isotopes in the Physical and Biomedical Sciences, Vol. 1, Labeled Compounds (Part A), Chapter 6 (1987)". 14C-меченое соединение можно получать с применением исходного вещества, содержащего 14C.
[0055]
Фармацевтически приемлемые соли соединений согласно настоящему изобретению включают, например, соли с щелочными металлами (например, литием, натрием, калием и т.п.), щелочноземельными металлами (например, кальцием, барием и т.п.), магнием, переходными металлами (например, цинком, железом и т.п.), аммонийные соли, соли с органическими основаниями (например, триметиламином, триэтиламином, дициклогексиламином, этаноламином, диэтаноламином, триэтаноламином, меглумином, этилендиамином, пиридином, пиколином, хинолином и т.п.) или аминокислотами, или соли с неорганическими кислотами (например, хлороводородной кислотой, серной кислотой, азотной кислотой, угольной кислотой, бромистоводородной кислотой, фосфорной кислотой, йодистоводородной кислотой и т.п.) или органическими кислотами (например, муравьиной кислотой, уксусной кислотой, пропионовой кислотой, трифторуксусной кислотой, лимонной кислотой, молочной кислотой, винной кислотой, щавелевой кислотой, малеиновой кислотой, фумаровой кислотой, миндальной кислотой, глутаровой кислотой, яблочной кислотой, бензойной кислотой, фталевой кислотой, аскорбиновой кислотой, бензолсульфокислотой, п-толуолсульфокислотой, метансульфокислотой, этансульфокислотой и т.п.). Главным образом включены соли с хлороводородной кислотой, серной кислотой, фосфорной кислотой, винной кислотой, метансульфокислотой и т.п. Указанные соли можно получать путем обычных способов.
[0056]
Соединения согласно настоящему изобретению и их фармацевтически приемлемые соли могут образовывать сольваты (например, гидраты и т.п.) и/или кристаллические полиморфы. Настоящее изобретение охватывает такие различные сольваты и кристаллические полиморфы. "Сольваты" могут представлять собой сольваты, в которых любое количество молекул растворителя (например, молекул воды и т.п.) координируются с соединениями согласно настоящему изобретению. Если соединения согласно настоящему изобретению или их фармацевтически приемлемые соли оставить на воздухе, соединения могут абсорбировать воду, что приведет к присоединению адсорбированной воды или образованию гидратов. Путем перекристаллизации соединений согласно настоящему изобретению или их фармацевтически приемлемых солей можно получать кристаллические полиморфы.
[0057]
Группа PR предпочтительно представляет собой группу, которая превращается в группу ОН под воздействием ферментов, метаболизирующих лекарственное средство, гидролаз, кислот желудочного сока и/или энтеробактерий после введения (например, перорального введения) in vivo.
[0058]
Примеры более предпочтительных вариантов реализации PR включают группы, выбранные из следующих формул а)-ас).
a) -C(=O)-PR0,
b) -C(=O)-PR1,
c) -C(=O)-L-PR1,
d) -C(=O)-L-O-PR1,
e) -C(=O)-L-O-L-O-PR1,
f) -C(=O)-L-O-C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
j) -C(PR3)2-OPR4,
k) -C(PR3)2-O-L-O-PR4,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
n) -C(PR3)2-O-C(=O)-N(-K)-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
p) -C(PR3)2-O-C(=O)-O-L-N(PR4)2,
q) -C(PR3)2-O-C(=O)-N(-K)-L-O-PR4,
r) -C(PR3)2-O-C(=O)-N(-K)-L-N(PR4)2,
s) -C(PR3)2-O-C(=O)-O-L-O-L-O-PR4,
t) -C(PR3)2-O-C(=O)-O-L-N(-K)-C(=O)-PR4,
u) -C(PR3)2-O-P(=O)(-PR5)2,
v) -C(PR3)2-PR6 (за исключением бензильной группы),
w) -C(=N+(PR7)2)(-N(PR7)2),
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
y) -C(PR3)2-N(-K)-C(=O)-O-PR2,
z) -P(=O)(-PR8)(-PR9),
aa) -S(=O)2-PR10,
ab) -PR11, и
ac) -C(PR3)2-C(PR3)2-O-PR2,
где L представляет собой линейный или разветвленный алкилен или линейный или разветвленный алкенилен;
K представляет собой водород или алкил, необязательно замещенный группой-заместителем А;
PR0 представляет собой алкил, необязательно замещенный группой-заместителем А, или алкенил, необязательно замещенный группой-заместителем А;
PR1 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, или алкилсульфанильную группу, необязательно замещенную группой-заместителем А;
PR2 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, гетероциклилалкил, необязательно замещенный группой-заместителем А, или триалкилсилил, необязательно замещенный группой-заместителем А;
каждый из PR3 независимо представляет собой водород или алкил;
каждый из PR4 независимо представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, гетероциклилалкил, необязательно замещенный группой-заместителем А, или триалкилсилил;
каждый из PR5 независимо представляет собой гидрокси или OBn;
PR6 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
каждый из PR7 независимо представляет собой алкил, необязательно замещенный группой-заместителем А;
PR8 представляет собой алкокси, необязательно замещенный группой-заместителем А;
PR9 представляет собой алкокси, необязательно замещенный группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилокси, необязательно замещенный группой-заместителем А, гетероциклилокси, необязательно замещенный группой-заместителем А, карбоциклиламино, необязательно замещенный группой-заместителем А, или гетероциклиламино, необязательно замещенный группой-заместителем А;
PR8 и PR9 совместно со смежным атомом фосфора могут образовывать гетероцикл, необязательно замещенный группой-заместителем А;
PR10 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, или гетероциклилалкил, необязательно замещенный группой-заместителем А;
PR11 представляет собой алкил, необязательно замещенный группой-заместителем А, алкенил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А.
Группа-заместитель А представляет собой оксогруппу, алкил, гидроксиалкил, амино, алкиламино, карбоциклил, гетероциклил, карбоциклилалкил, алкилкарбонил, галоген-, гидрокси, карбокси, алкилкарбониламино, алкилкарбониламиноалкил, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкилоксикарбонилокси, алкиламинокарбонилокси, алкиламиноалкил, алкилокси, циано, нитро, азидо, алкилсульфонил, триалкилсилил или фосфо.
[0059]
Примеры другого предпочтительного варианта реализации PR включают следующие группы.
a) -C(=O)-PR0,
b) -C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
v) -C(PR3)2-PR6 (за исключением бензильной группы),
х) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
у) -C(PR3)2-N(-K)-C(=O)-O-PR2, и
z) -P(=O)(-PR8)(-PR9),
где L представляет собой линейный или разветвленный алкилен;
K представляет собой водород или алкил, необязательно замещенный группой-заместителем А;
PR0 представляет собой алкил, необязательно замещенный группой-заместителем А;
PR1 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR2 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, гетероциклильную группу, необязательно замещенную группой-заместителем А, карбоциклилалкил, необязательно замещенный группой-заместителем А, или гетероциклилалкил, необязательно замещенный группой-заместителем А;
каждый из PR3 независимо представляет собой водород или алкил;
PR4 представляет собой алкил, необязательно замещенный группой-заместителем А, карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR6 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем А, или гетероциклильную группу, необязательно замещенную группой-заместителем А;
PR8 представляет собой алкокси, необязательно замещенный группой-заместителем А;
PR9 представляет собой алкокси, необязательно замещенный группой-заместителем А, алкиламино, необязательно замещенный группой-заместителем А, карбоциклилокси, необязательно замещенный группой-заместителем А, гетероциклилокси, необязательно замещенный группой-заместителем А, карбоциклиламино, необязательно замещенный группой-заместителем А, или гетероциклиламино, необязательно замещенный группой-заместителем А; и
PR8 и PR9 совместно со смежным атомом фосфора могут образовывать гетероцикл, необязательно замещенный группой-заместителем А.
Группа-заместитель А представляет собой оксогруппу, алкил, алкиламино, карбоциклил, гетероциклил, алкилкарбонил, галоген-, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил или триалкилсилил.
[0060]
Примеры другого варианта реализации предпочтительного заместителя PR включают следующие группы.

[0061]
(Способ получения соединения согласно настоящему изобретению)
Ниже приведен пример общего способа получения соединения согласно настоящему изобретению. Обработку с целью экстракции и очистки можно проводить при помощи обычного эксперимента из области органической химии.
Получение соединения согласно настоящему изобретению можно проводить при помощи процедур, известных в данной области.
[0062]
В качестве исходного вещества можно использовать коммерчески доступные соединения, соединения, описанные в настоящем описании, соединения, описанные в ссылках, приведенных в настоящем описании, и другие известные соединения.
Когда требуется получить соль соединения согласно настоящему изобретению, в случае, когда соединение согласно настоящему изобретению получают в форме соли, его можно очищать, как таковое, и, в случае, когда соединение согласно настоящему изобретению получают в свободной форме, соль можно получать путем обычного способа путем растворения или суспендирования соединения в подходящем органическом растворителе и добавления кислоты или основания.
Кроме того, в некоторых случаях соединение согласно настоящему изобретению и его фармацевтически приемлемая соль присутствуют в форме продуктов присоединения воды или других растворителей (гидрат или сольват), и указанные продукты присоединения включены в настоящее изобретение.
[0063]
В общем способе получения, а также в примерах сравнения, примерах и примерах получения промежуточных соединений значение каждой из аббревиатур является следующим.
Верх и низ "клиновидной связи" и "пунктирной клиновидной связи" указывают абсолютную конфигурацию.
[0064]
(Способ получения 1)

где Р1 представляет собой гидроксил-защитную группу; RP представляет собой ацеталь-защитную группу; L представляет собой уходящую группу; каждый из других символов является таким же, как описано выше.
Первая стадия
Соединение A3 можно получать путем добавления соединения А2 к соединению А1 в присутствии агента дегидратации-конденсации, такого как дициклогексилкарбодиимида, карбонилдиимидазола, дициклогексилкарбодиимидо-N-гидроксибензотриазола, хлорида 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния, 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфорной кислоты, WSO⋅HCl, HATU и т.п., в растворителе, таком как ДМФ, ТГФ, дихлорметан, ацетонитрил и т.п., или в смеси указанных растворителей, и проведения реакции при температуре от -20°C до 60°C, предпочтительно от -10°C до 40°C в течение времени от 0,1 часа до 24 часов, предпочтительно от 1 часа до 12 часов.
Альтернативно, соединение A3 можно получать путем добавления ацилирующего реагента, такого как дифенилхлорфосфат, тионилхлорид, оксалилхлорид и т.п., к соединению А1 в присутствии или при отсутствии основания, такого как пиридин, триэтиламин, диизопропилэтиламин, 1-метилимидазол и т.п., в присутствии растворителя, такого как ТГФ, диоксан, дихлорметан, ДМФ и т.п., с образованием, таким образом, хлорангидрида кислоты, добавления соединения А2, имеющего заместитель, соответствующий целевому соединению, и проведения реакции при температуре от -20°C до 60°C, предпочтительно от -10°C до 40°C в течение времени от 0,1 часа до 24 часов, предпочтительно от 0,5 часа до 12 часов.
Вторая стадия
Соединение А4 можно получать путем добавления карбоната калия, карбоната натрия и O-(2,4-динитрофенил)гидроксиламина к соединению A3 в присутствии растворителя, такого как ДМФ, ДМА, НМП, ТГФ и т.п., и проведения реакции при температуре от 10°C до 60°C, предпочтительно от 20°C до 40°C времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Третья стадия
Реакцию снятия ацеталь-защитной группы соединения А4 можно проводить путем общего способа, описанного в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons), и т.п. Затем получаемую альдегидную группу подвергают внутримолекулярной рекации с получением, таким образом, соединения А5.
Например, рацемат соединения А5 можно получать путем добавления уксусной кислоты и/или п-толуолсульфокислоты, метансульфокислоты и т.п. к соединению А4 в присутствии растворителя, такого как ДМФ, толуол, ТГФ и т.п., и проведения реакции при температуре от 10°C до 80°C, предпочтительно от 30°C до 60°C в течение времени от 0,5 часа до 12 часов, предпочтительно от 1 часа до 6 часов. Соединение А5 можно получать путем оптического разделения рацемата соединения А5 с применением СЖХ или ВЭЖХ (на хиральной колонке).
Четвертая стадия
Соединение А7 можно получать путем добавления соединения А6 и основания, такого как карбонат натрия, карбонат калия, карбонат цезия и т.п., к соединению А5 в присутствии растворителя, такого как ДМФ, ДМА, НМП, ТГФ и т.п., или смеси указанных растворителей и проведения реакции при температуре от 0°C до 60°C, предпочтительно от 10°C до 40°C в течение времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Альтернативно соединение А7 можно получать путем добавления соединения А6, Т3Р и метансульфокислоты или п-толуолсульфокислоты к соединению А5 в присутствии растворителя, такого как ДМФ, этилацетат, бутилацетат, 1,4-диоксан и т.п., или смеси указанных растворителей и проведения реакции при температуре от 40°C до 150°C, предпочтительно от 60°C до 120°C в течение времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Пятая стадия
Реакцию снятия гидроксил-защитной группы соединения А7 можно проводить путем общего способа, описанного в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons), и т.п.
Шестая стадия
Соединение (III) можно получать путем общего способа, включающего превращение гидроксильной группы соединения (II) в сложноэфирную группу или простую эфирную группу.
Например, можно применять способ, описанный в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons), Prog. Med. 5: 2157-2161 (1985), и представленный в Supplied by The British Library - "The world's Knowledge", и т.п.
(Способ получения 2)
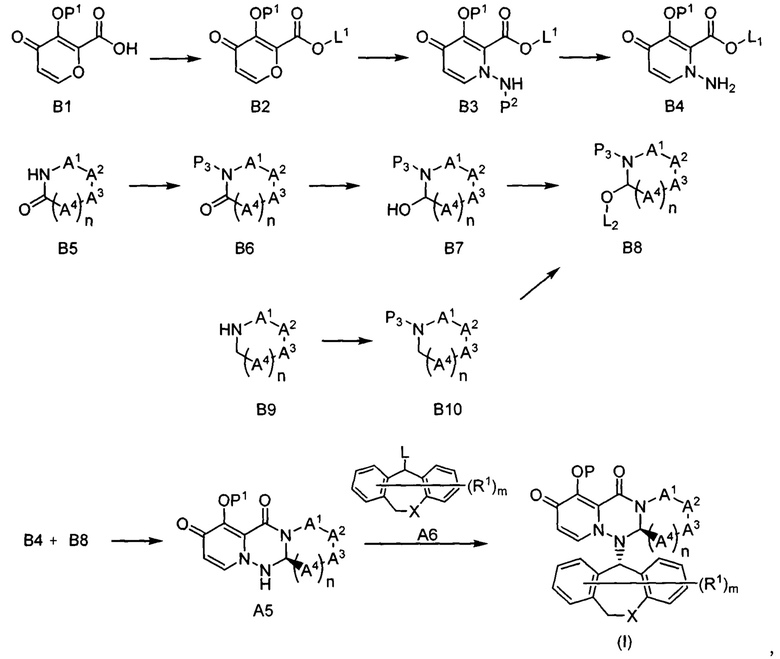
где Р2 представляет собой NH-защитную группу; L1 и L2 представляют собой уходящие группы; каждый из других символов является таким же, как описано выше.
Первая стадия
Соединение В2 можно получать путем добавления соединения А2 и галогенированного алкила, такого как метилиодид, к соединению В1 в присутствии основания, такого как диазабициклоундецен, в растворителе, таком как ДМФ, ТГФ, дихлорметан, ацетонитрил и т.п., или в смеси указанных растворителей и проведения реакции при температуре от -20°C до 60°C, предпочтительно от -10°C до 40°C в течение времени от 0,1 часа до 24 часов, предпочтительно от 1 часа до 24 часов.
Альтернативно соединение В2 можно получать путем добавления ацилирующего реагента, такого как дифенилхлорфосфат, тионилхлорид, оксалилхлорид и т.п., к соединению В1 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФ и т.п., или в смеси указанных растворителей, добавления спирта в присутствии основания, такого как пиридин, триэтиламин, диизопропилэтиламин, 1-метилимидазол и т.п., и проведения реакции при температуре от -20°C до 60°C, предпочтительно от -10°C до 40°C в течение времени от 0,1 часа до 24 часов, предпочтительно от 0,5 часа до 12 часов.
Вторая стадия
Соединение В3 можно получать путем добавления соли пиридиния и п-толуолсульфокислоты и гидразина, защищенного Boc-, и т.п. к соединению В2 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФ и т.п., или в смеси указанных растворителей и проведения реакции при температуре от 10°C до 150°C, предпочтительно от 40°C до 100°C в течение времени от 1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Третья стадия
Реакцию снятия амино-защитной группы соединения В3 можно проводить путем общего способа, описанного в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons), и т.п.
Четвертая стадия
Соединение В6 можно получать путем добавления основания, такого как н-бутиллитий и т.п., к соединению В5 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФ и т.п., или в смеси указанных растворителей с последующим добавлением алкилового эфира галогенмуравьиной кислоты и проведением реакции в течение времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Пятая стадия
Соединение В7 можно получать путем добавления восстановителя, такого как диизобутилалюминийгидрид лития и т.п., к соединению В6 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФ и т.п., или в смеси указанных растворителей и проведения реакции в течение времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Шестая стадия
Соединение В8 можно получать путем добавления п-толуолсульфокислоты или метансульфокислоты к соединению В7 в спирте и проведения реакции при температуре от 0°C до 100°C в течение времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Седьмая стадия
Соединение В10 можно получать путем добавления алкилового эфира галогенмуравьиной кислоты к соединению В9 в присутствии или при отсутствии основания, такого как пиридин, триэтиламин, диизопропилэтиламин, 1-метилимидазол и т.п., в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФ и т.п., или в смеси указанных растворителей и проведения реакции при температуре от -40°C до 40°C в течение времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Восьмая стадия
Соединение В8 можно получать путем погружения угольного электрода (анод) и платинового электрода (катод) в соединение В10 в растворителе, таком как спирт, в присутствии основания, такого как карбонат калия и перхлорат тетраэтиламмония, и промывания при постоянном токе 0,1~1,0 А при перемешивании в течение времени от 0,1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Девятая и десятая стадии
Соединение (I) можно получать из соединений В4 и В8 при помощи такого же способа, как способ, описанный в стадиях с третьей по шестую в способе получения 1.
[0065]
Соединение согласно настоящему изобретению проявляет ингибирующую активность кэп-зависимой эндонуклеазы и подходит для применения в качестве терапевтического средства и/или профилактического средства при гриппе.
[0066]
Соединение согласно настоящему изобретению не только проявляет ингибирующую активность кэп-зависимой эндонуклеазы, но и подходит для применения в качестве лекарственного средства и обладает любыми или всеми из следующих превосходных характеристик:
a) соединение является слабым ингибитором ферментов CYP (например, CYP1A2, CYP2C9, CYP2C19, CYP2D6, CYP3A4 и т.п.).
b) соединение демонстрирует хорошую фармакокинетику, такую как высокая биодоступность, умеренный клиренс и т.п.
c) соединение обладает высокой метаболической устойчивостью.
d) соединение соединение не проявляет необратимого ингибирующего дейстия в отношении ферментов CYP (например, CYP3A4) при условии измерения при концентрации, находящейся в пределах диапазона, описанного в настоящем изобретении.
e) соединение не проявляет мутагенности.
f) соединение связано с низким риском развития сердечно-сосудистых заболеваний.
g) соединение имеет высокую растворимость.
h) соединение не проявляет фототоксичности.
[0067]
С целью лечения упомянутых выше заболеваний у людей соединения согласно настоящему изобретению можно вводить перорально в виде порошка, гранулы, таблеток, капсул, пилюль, жидкости и т.п. или парентерально в виде инъекции, суппозиториев, лекарственного средства для чрескожного введения, лекарственной формы для ингаляции и т.п. При необходимости для получения фармацевтических препаратов эффективные дозы настоящих соединений можно смешивать со вспомогательными веществами, подходящими для лекарственной формы, такими как наполнители, связующие вещества, увлажнители, разрыхлители и смазывающие вещества. Для получения инъекции проводят стерилизацию с подходящим веществом-носителем.
Фармацевтическую композицию согласно настоящему изобретению можно вводить перорально или парентерально. Лекарственные формы, обычно применяемые для перорального введения, такие как таблетки, гранулы, порошки и капсулы, можно получать в соответствии с обычными способами. Для парентерального введения можно применять любую обычную подходящую лекарственную форму, такую как инъекция. Соединения согласно настоящему изобретению могут подходить для применения в качестве препаратов для перорального введения благодаря своей высокой способности к всасыванию в ротовой полости.
[0068]
При необходимости для получения фармацевтических композиций эффективные дозы соединений согласно настоящему изобретению можно смешивать с различными фармацевтическими вспомогательными веществами, подходящими для лекарственной формы, такими как наполнители, связующие вещества, разрыхлители и смазывающие вещества.
[0069]
Доза зависит от состояния заболевания, способа введения и возраста или веса пациента. Обычная доза для перорального введения для взрослых составляет от 0,1 до 100 мг/кг в день, предпочтительно от 1 до 20 мг/кг в день.
Дозу фармацевтической композиции согласно настоящему изобретению предпочтительно определяют, исходя из возраста и веса пациента, типа и тяжести заболевания, способа введения и т.п. Обычная доза для перорального введения для взрослых находится в диапазоне от 0,05 до 100 мг/кг в день, предпочтительно от 0,1 до 10 мг/кг в день. Доза для парентерального введения для взрослых значительно варьируется в зависимости от способа введения и обычно находится в диапазоне от 0,005 до 10 мг/кг в день, предпочтительно от 0,01 до 1 мг/кг в день. Дозу можно вводить один раз в день или можно разделить на несколько суточных доз.
[0070]
Соединение согласно настоящему изобретению можно применять в комбинации с другими лекарственными средствами и т.п. (далее в настоящей заявке именуемыми комбинированными лекарственными средствами) для повышения активности соединения, уменьшения дозы соединения и т.п. В случае лечения гриппа соединение также можно применять в сочетании с или в виде комбинированного состава с ингибитором нейраминидазы (например, осельтамивиром, занамивиром, перамивиром, Inabiru и т.п.); ингибитором PHK-зависимой PHK-полимеразы (например, фавипиравиром); ингибитором белка М2 (например, амантадином); РВ2-специфическим кэп-связывающим ингибитором (например, VX-787); антителом к гемагглютинину (например, МНАА4549А); иммунными агонистами (например, нитазоксанидом). В этом случае время введения соединения согласно настоящему изобретению и комбинированного лекарственного средства не ограничено. Их можно вводить пациенту, нуждающемуся в лечении, в одно время или в разное время. Кроме того, соединение согласно настоящему изобретению и комбинированное лекарственное средство можно вводить в виде двух или более составов, независимо содержащих каждый из активных ингредиентов, или в виде одного состава, содержащего все активные ингредиенты.
[0071]
Дозу комбинированных лекарственных средств можно выбирать в соответствии с клинической дозой. Отношения соединений согласно настоящему изобретению и совместно вводимых лекарственных средств в составе можно выбирать в соответствии с пациентом, нуждающимся в лечении, способом введения, заболеванием, подлежащим лечению, симптомами, комбинации лекарственных средств и т.п. Например, для введения пациенту 1 массовую долю соединения согласно настоящему изобретению можно применять в комбинации с 0,01-100 массовыми долями совместно вводимых лекарственных средств.
[0072]
Настоящее изобретение более подробно объяснено ниже посредством примеров, примеров сравнения, примеров получения промежуточных соединений, а также примеров испытаний согласно настоящему изобретению, но настоящее изобретение не ограничивается ими.
[0073]
ЯМР-анализ для каждого из примеров сравнения и примеров проводили при 300 МГц и измерения проводили с применением ДМСО⋅d6, CDCl3.
Термин "RT" представляет собой время удерживания при проведении ЖХ/МС: жидкостной хроматографии/масс-спектрометрии, и измерения проводили в следующих условиях.
(Условия измерения)
(1) Колонка: ACQUITY UPLC (зарегистрированная торговая марка) ВЕН С18 (1,7 мкм, i.d. 2,1×50 мм) (Waters)
Расход'- 0,8 мл/мин
Длина волны УФ-детектирования: 254 нм
Подвижная фаза: [А]: водный раствор, содержащий 0,1% муравьиной кислоты, [В]: раствор ацетонитрила, содержащий 0,1% муравьиной кислоты.
Градиент: линейный градиент от 5% до 100% растворителя [В] поддерживали в течение 3,5 минут и 100% растворителя [В] поддерживали в течение 0,5 минуты.
(2) Колонка: Shim-pack XR-ODS (2,2 мкм, i.d. 50×3,0 мм) (Shimadzu)
Расход: 1,6 мл/мин
Длина волны УФ-детектирования: 254 нм
Подвижная фаза: [А]: водный раствор, содержащий 0,1% муравьиной кислоты, [В]: - раствор ацетонитрила, содержащий 0,1% муравьиной кислоты.
Градиент: линейный градиент от 10% до 100% растворителя [В] поддерживали в течение 3 минут и 100% растворителя [В] поддерживали в течение 0,5 минуты.
[0074]
Пример сравнения 1

Первая стадия
К раствору соединения 1 (5,0 г, 49,5 ммоль) в ТГФ (100 мл) по каплям добавляли 1,62 моль/л раствор н-бутиллития в гексане (30,5 мл, 49,5 ммоль) при -78°С в атмосфере азота и смесь перемешивали при -78°С в течение 2 часов. По каплям добавляли раствор аллилхлорформиата (5,96 г, 49,5 ммоль) в ТГФ (20 мл) и смесь перемешивали при -78°С в течение 2 часов. Реакцию гасили насыщенным водным раствором хлорида аммония, смесь нагревали до комнатной температуры и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 2 (5,66 г, 62%).
1H-ЯМР (CDCl3) δ: 3,83 (t, J=8,0 Гц, 2Н), 3,92 (t, J=8,0 Гц, 2Н), 4,26 (s, 2H), 4,78 (d, J=8,0 Гц, 2Н), 5,30 (d, J=12,0 Гц, 1Н), 5,44 (d, J=16,0 Гц, 1Н), 5,93-6,03 (m, 1Н).
Вторая стадия
К раствору соединения 2 (6,6 г, 35,6 ммоль) в ТГФ (66 мл) по каплям добавляли 1,03 моль/л раствор DIBAL-H в гексане (45,0 мл, 46,3 ммоль) и смесь перемешивали при -78°С в течение 1 часа. Реакцию гасили ацетоном и к смеси добавляли водный раствор Сегнетовой соли. Смесь перемешивали и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 3 (6,21 г, 93%).
1H-ямр (CDCl3) δ: 3,44 (шир., 1Н), 3,50-3,64 (m, 2Н), 3,71 (шир., 1Н), 3,95 (d, J=8,0 Гц, 2Н), 4,64 (d, J=8,0 Гц, 2Н), 5,24 (d, J=12,0 Гц, 1Н), 5,40 (d, J=16,0 Гц, 1H), 5,47 (d, J=4 Гц, 1H), 5,87-6,00 (m, 1H).
Третья стадия
К раствору соединения 3 (6,2 г, 33,1 ммоль) в метаноле (65 мл) добавляли моногидрат п-толуолсульфокислоты (0,63 г, 3,31 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Реакцию гасили водным раствором гидрокарбоната натрия, смесь концентрировали и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 4 (5,77 г, 87%).
1H-ЯМР (CDCl3) δ: 3,34 (s, 3Н), 3,55 (шир., 2Н), 3,73-3,99 (m, 3Н), 4,64 (d, J=8,0 Гц, 2Н), 5,10-5,20 (m, 1H), 5,25 (d, J=8,0 Гц, 1H), 5,33 (d, J=16 Гц, 1H), 5,88-6,05 (m, 1H).
Четвертая стадия
К раствору соединения 5 (20,0 г, 81 ммоль) в ДМФ (100 мл) добавляли йодистый этил (22,8 г, 146 ммоль) и диазабициклоундецен (18,4 мл, 122 ммоль) и смесь перемешивали при комнатной температуре в течение ночи. Смесь погружали в 10% водный раствор хлорида аммония и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 6 (22,3 г, 100%).
1H-ЯМР (CDCl3) δ: 1,23 (t, J=8,0 Гц, 3Н), 4,28 (q, J=8,0 Гц, 2Н), 5,16 (s, 2Н), 6,57 (d, J=4,0 Гц, 1H), 7,28-7,48 (m, 5H), 8,21 (d, J=4,0 Гц, 1H).
Пятая стадия
К раствору соединения 6 (500 мг, 1,82 ммоль) в ДМА (5,0 мл) добавляли п-толуолсульфонат пиридиния (1,37 г, 5,47 ммоль) и Boc-гидразин (361 мг, 2,74 ммоль) и смесь перемешивали при 60°С в течение 14 часов. К смеси добавляли воду и смесь экстрагировали этилацетатом. Полученный органический слой промывали насыщенным водным раствором хлорида аммония и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью хлороформ-метанол) с получением соединения 7 (519 мг, 73%).
1H-ямр (CDCl3) δ: 1,24 (t, J=8,0 Гц, 3Н), 1,46 (s, 9H), 4,26 (q, J=8,0 Гц, 2Н), 5,28 (s, 2Н), 6,40 (d, J=8,0 Гц, 1H), 7,27-7,38 (m, 4Н), 7,40-7,45 (m, 2Н).
Шестая стадия
Соединение 7 (500 мг, 1,29 ммоль) растворяли в 4 моль/л растворе хлороводорода в этилацетате (5 мл) и смесь перемешивали при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении. К полученному остатку добавляли насыщенный водный раствор гидрокарбоната натрия и смесь экстрагировали дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 8 (369 мг, 99%).
1H-ямр (CDCl3) δ: 1,26 (t, J=8,0 Гц, 3Н), 4,31 (q, J=8,0 Гц, 2Н), 5,24 (s, 2Н), 6,47 (d, J=8,0, 1H), 7,28-7,44 (m, 5H), 7,64 (d, J=8,0, 1H).
Седьмая стадия
К раствору соединения 8 (365 мг, 1,27 ммоль) и соединения 4 (306 мг, 1,52 ммоль) в ацетонитриле (8 мл) по каплям добавляли хлорид олова (0,223 мл, 1,90 ммоль) при -25°С в атмосфере азота и смесь перемешивали при -25°С в течение 45 минут. Реакцию гасили насыщенным водным раствором гидрокарбоната натрия и к смеси добавляли дихлорметан. Смесь перемешивали при комнатной температуре и фильтровали через целит и фильтрат экстрагировали дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением неочищенного соединения 9. Полученное соединение 9 растворяли в ТГФ (8 мл), добавляли морфолин (1,10 мл, 12,7 ммоль) и тетракис(трифенилфосфин)палладий (146 мг, 0,127 ммоль) и смесь перемешивали при комнатной температуре в течение 2 часов. К смеси добавляли диэтиловый эфир (16 мл) и осажденное твердое вещество фильтровали и сушили с получением соединения 10 (418 мг, 100%).
1H-ЯМР (CDCl3) δ: 2,90-2,99 (m, 1Н), 3,13 (t, J=12,0 Гц, 1Н), 3,40-3,46 (m, 1Н), 4,00-4,08 (m, 1Н), 4,14 (d, J=12,0 Гц, 1Н), 5,07 (s, 2H), 6,22 (d, J=8,0 Гц, 1Н), 7,29-7,40 (m, 3Н), 7,56 (d, J=8,0 Гц, 2H), 7,71 (d, J=8,0 Гц, 1Н).
Восьмая стадия
К суспензии (R)-2-тетрагидропирослизевой кислоты (855 мг, 7,36 ммоль) и соединения 10 (2,00 г, 6,11 ммоль) в этилацетате (9 мл) добавляли пиридин (4,00 мл, 49,6 ммоль) и Т3Р (50% раствор в этилацетате, 11,0 мл, 18,5 ммоль) при комнатной температуре и смесь перемешивали в течение ночи. Осажденное твердое вещество отфильтровывали и промывали этилацетатом (4 мл) и этанолом (4 мл). Полученное твердое вещество суспендировали в этаноле (6 мл) и суспензию перемешивали при комнатной температуре в течение 6,5 часов. Суспензию фильтровали и полученное твердое вещество дважды промывали этанолом (2 мл) с получением соединения 11 (1,18 г, 45,4%).
1H-ЯМР (ДМСО) δ: 1,80-1,94 (m, 2H), 1,95-2,14 (m, 2H), 3,21-3,35-(m, 2H), 3,50-3,60 (m, 1Н), 3,70-3,82 (m, 3Н), 4,00-4,05 (m, 1Н), 4,32-4,38 (m, 1Н), 5,14 (dd, J=10,8 Гц, 21,6 Гц, 2H), 5,76-5,81 (m, 1Н), 6,29 (d; J=4,8 Гц, 1Н), 7,28-7,39 (m, 3Н), 7,48-7,54 (m, 2H), 7,64-7,75 (m, 1Н).
Девятая стадия
К суспензии соединения 11 (500 мг, 1,18 ммоль) в этаноле (3,5 мл) добавляли ДБУ (0,0035 мл, 0,023 ммоль) при комнатной температуре и смесь перемешивали в течение 30 минут. К полученной суспензии добавляли диизопропиловый эфир (6,5 мл) и смесь перемешивали при комнатной температуре в течение 30 минут. Осажденное твердое вещество отфильтровывали и дважды промывали этилацетатом (1,5 мл) с получением соединения i1 (346 мг, 89,9%).
1H-ЯМР (ДМСО) δ: 2,80-3,00 (m, 1Н), 3,10-3,18 (m, 1Н), 3,38-3,50 (m, 1Н), 3,98-4,08 (m, 2H), 4,10-4,20 (m, 1Н), 4,76-4,84 (m, 1Н), 5,04-5,14 (m, 2H), 6,22 (m, J=7,6 Гц, 1Н), 7,27-7,40 (m, 4H), 7,56-7,60 (m, 2H), 7,70 (d, J=7,6 Гц, 1Н).
[0075]
Пример сравнения 2
Первая стадия
К суспензии соединения 13 (8,0 г, 50,8 ммоль) в дихлорметане (120 мл) на водяной бане со льдом добавляли триэтиламин (17,6 мл, 127 ммоль) и по каплям добавляли аллилхлорформиат (6,44 мл, 60,9 ммоль) и смесь перемешивали при 0°С в течение 1 часа. К смеси добавляли воду и смесь экстрагировали дихлорметаном. Полученный органический слой промывали 5% водным раствором лимонной кислоты и насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 14 (10,1 г, 97%).
1H-ямр (CDCl3) δ: 1,96 (шир., 4Н), 3,62 (s, 4Н), 4,60 (s, 2H), 5,22 (d, J=12,0 Гц, 1Н), 5,30 (d, J=16,0 Гц, 1Н), 5,86-5,99 (m, 1Н).
Вторая стадия
В раствор соединения 14 (0,9 г, 4,39 ммоль), карбоната калия (60 мг, 0,44 ммоль) и перхлората тетраэтиламмония (50 мг, 0,22 ммоль) в метаноле (30 мл) погружали угольных электрод (анод) и платиновый электрод (катод) и смесь промывали при постоянном токе 0,1 А при перемешивании при комнатной температуре в течение 6 часов. К смеси добавляли этилацетат и воду и смесь экстрагировали этилацетатом. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 15 (992 мг, 96%).
1H-ЯМР (CDCl3) δ: 1,81-2,15 (m, 3Н), 2,39 (t, J=12,0 Гц, 1Н), 3,27 (s, 3H), 3,61 (s, 1Н), 4,11 (шир., 1Н), 4,61 (шир., 2H), 5,20-5,36 (m, 2H), 5,57 (шир., 1Н), 5,88-5,99 (m, 1Н).
Третья стадия
Соединение 16 получали при помощи такого же способа, как способ, описанный в седьмой и восьмой стадиях примера сравнения 1.
Четвертая стадия
Путем оптического разделения соединения 16 (870 мг, 2,41 ммоль) при помощи системы Waters SFC30 (Daicel CHIRALPAK IB, сжиженный углекислый газ-метанол) получали соединение i2 (270 мг, 31%).
Условия измерения
<Waters SFC30 System (SPRC4⋅5N406)>
Колонка: CHIRALPAK IB/SFC (5 мкм i.d. 250×4,6 мм) (DAICEL)
Расход: 8,0 мл/мин; Длина волны УФ-детектирования: 254 нм
Обратное давление: 100 бар
Подвижная фаза: [А]: сжиженный углекислый газ, [В]: метанол
Градиент: 5% растворителя [В] поддерживали в течение 1 минуты, линейный градиент от 5% до 40% растворителя [В] поддерживали в течение 6 минут, 40% растворителя [В] поддерживали в течение 2 минут и 5% растворителя [В] поддерживали в течение 1 минуты.
Время элюирования: 7,3 минут
Пример сравнения 3

Первая стадия
К раствору соединения 17 (4,00 г, 16,3 ммоль) в дихлорметане (40 мл) на ледяной бане добавляли оксалилдихлорид (1,56 мл, 17,9 ммоль) и ДМФ (0,013 мл, 0,162 ммоль) и смесь нагревали до комнатной температуры и перемешивали в течение 5 часов. Смесь концентрировали при пониженном давлении и полученный остаток растворяли в дихлорметане (40 мл), на ледяной бане добавляли 2,2,2-трифторэтанол (2,44 г, 24,4 ммоль), триэтиламин (4,50 мл, 32,5 ммоль) и 4-(диметиламино)пиридин (99,0 мг, 0,812 ммоль) и смесь нагревали до комнатной температуры и перемешивали в течение 1 часа. Смесь концентрировали при пониженном давлении, к полученному остатку добавляли 1 моль/л водный раствор хлороводородной кислоты и смесь экстрагировали этилацетатом. Полученный органический слой промывали 1 моль/л водным раствором хлороводородной кислоты и солевым раствором и сушили над безводным сульфатом магния с получением соединения 18 (5,33 г, 100%).
1H-ЯМР (CDCl3) δ: 4,64 (q, J=8,2 Гц, 2Н), 5,38 (s, 2H), 6,49 (d, J=5,6 Гц, 1Н), 7,30-7,38 (m, 3Н), 7,43-7,49 (m, 2H), 7,75 (d, J=5,6 Гц, 1Н).
Вторая и третья стадии
Соединение 20 получали при помощи такого же способа, как способ, описанный в пятой и шестой стадиях примера сравнения 1.
1H-ЯМР (CDCl3) δ: 4,55 (q, J=8,3 Гц, 2H), 5,18 (s, 2H), 5,29 (s, 2H), 6,37 (d, J=7,8 Гц, 1Н), 7,30-7,42 (m, 6H).
Четвертая и пятая стадии
Соединение 23 получали при помощи такого же способа, как способ, описанный в седьмой стадии примера сравнения 1.
ЖХ/МС (ИЭР): m/z=342,1 [М+Н]+, RT=1,00, 1,09 мин, способ (1)
Шестая стадия
К раствору соединения 23 (820 мг, 2,40 ммоль) в дихлорметане (16,5 мл) добавляли Boc2O (0,837 мл, 3,60 ммоль), триэтиламин (0,499 мл, 3,60 ммоль) и 4-(диметиламино)пиридин (44,0 мг, 0,360 ммоль) и смесь перемешивали при комнатной температуре в течение 3,5 часов. К смеси добавляли 1 моль/л водный раствор хлороводородной кислоты и смесь экстрагировали этилацетатом. Полученный органический слой промывали 1 моль/л водным раствором хлороводородной кислоты и солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью хлороформ-метанол) с получением соединения 24 (593 мг, 56%) и соединения i3 (170 мг, 16%).
Соединение 24: ЖХ/МС (ИЭР): m/z=441,9 [М+Н]+, RT=1,67 мин, способ (1)
Седьмая стадия
Соединение 24 (547 мг, 1,24 ммоль) растворяли в уксусной кислоте (5,5 мл) и смесь перемешивали при 80°С в течение 5 часов. Смесь концентрировали при пониженном давлении и полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью хлороформ-метанол) с получением соединения i3 (454 мг, 100%).
1H-ямр (CDCl3) δ: 1,46 (d, J=6,4 Гц, 3Н), 3,45 (dd, J=10,5, 10,5 Гц, 1Н), 3,55 (dd, J=11,7, 4,3 Гц, 1Н), 3,92 (dd, J=11,7, 3,6 Гц, 1Н), 3,95-4,01 (m, 2H), 4,76 (dq, J=13,9, 4,3 Гц, 1Н), 5,19 (d, J=10,2 Гц, 1Н), 5,22 (d, J=10,2 Гц, 1Н), 5,36 (d, J=12,9 Гц, 1Н), 6,28 (d, J=7,8 Гц, 1Н), 7,25 (d, J=7,8 Гц, 1Н), 7,28-7,36 (m, 3Н), 7,56-7,61 (m, 2H).
[0076]
Пример 1
Первая стадия
Соединение i1 (1100 г, 3360 ммоль) и 7,8-дифтор-6,11-дигидродибензотиепин-11-ол (977 г, 3697 ммоль) суспендировали в 50 мас. % растворе Т3Р в этилацетате (3208 г, 5041 ммоль) и этилацетате (1,1 л). К смеси добавляли метансульфокислоту (436 мл, 6721 ммоль) при комнатной температуре и смесь перемешивали при 70°С в течение 5,5 часов. К смеси на водяной бане со льдом добавляли воду и смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли ТГФ добавляли и смесь экстрагировали этилацетатом. Полученный органический слой промывали водой и 8% водным раствором гидрокарбоната натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток растворяли в ТГФ (5,5 л) и добавляли карбонат калия (790 г, 5713 ммоль). Смесь нагревали до 50°С, по каплям добавляли бензилбромид (240 мл, 2016 ммоль) и смесь перемешивали при 60°С в течение 8,5 часов. К смеси на водяной бане со льдом по каплям добавляли 2 моль/л водный раствор хлороводородной кислоты и смесь перемешивали при комнатной температуре в течение 10 минут и экстрагировали этилацетатом. Полученный органический слой промывали водой и 8% водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. Добавляли активированный уголь (Norit SX-2, 240 г), смесь фильтровали через целит и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат и гексан и осажденное твердое вещество отфильтровывали с получением соединения 25 (1019 г, 1776 ммоль, 53%).
1H-ямр (CDCl3) δ: 2,88 (1Н, t, J=11,2 Гц), 3,28-3,39 (2H, m), 3,72 (1Н, d, J=12,6 Гц), 3,86 (1Н, d, J=9,6 Гц), 4,03 (1Н, d, J=13,9 Гц), 4,45 (1Н, d, J=8,6 Гц), 4,67 (1Н, d, J=13,1 Гц), 5,19-5,26 (2H, m), 5,45 (1Н, d, J=10,9 Гц), 5,63 (1Н, d, J=10,9 Гц), 5,77 (1Н, d, J=7,6 Гц), 6,40 (1Н, d, J=7,8 Гц), 6,68 (1Н, t, J=6,9 Гц), 6,94-7,01 (2H, m), 7,03-7,12 (3Н, m), 7,29-7,38 (3Н, m), 7,61 (2H, d, J=7,1 Гц).
Вторая стадия
К раствору соединения 25 (1200 г, 2092 ммоль) в ДМА (3,6 л) добавляли хлорид лития (443 г, 10,5 моль) при комнатной температуре и смесь перемешивали при 80°С в течение 3 часов. К смеси на водяной бане со льдом добавляли ацетон (1,2 л), 0,5 моль/л водный раствор хлороводородной кислоты (6,0 л) и воду (2,4 л) и смесь перемешивали в течение 1 часа. Осажденное твердое вещество отфильтровывали. Полученное твердое вещество растворяли в хлороформе, добавляли изопропиловый эфир и осажденное твердое вещество отфильтровывали с получением соединения III-2 (950 г, 1965 ммоль, 94%).
1H-ямр (CDCl3) δ: 2,99 (1Н, dt, J=17,5, 6,8 Гц), 3,47 (1Н, td, J=11,9, 2,5 Гц), 3,60 (1Н, t, J=10,6 Гц), 3,81 (1Н, dd, J=11,9, 3,3 Гц), 3,96 (1Н, dd, J=11,0, 2,9 Гц), 4,07 (1Н, d, J=13,8 Гц), 4,58 (1Н, dd, J=10,0, 2,9 Гц), 4,67 (1Н, dd, J=13,5, 1,9 Гц), 5,26-5,30 (2H, m), 5,75 (1Н, d, J=7,8 Гц), 6,69 (1Н, d, J=7,7 Гц), 6,83-6,87 (1Н, m), 6,99-7,04 (2H, m), 7,07-7,15 (3Н, m).
Пример 2
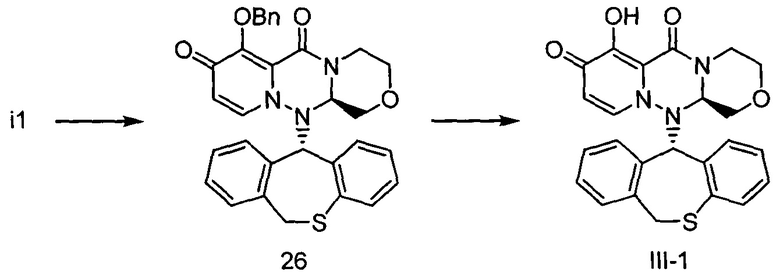
Первая стадия
Соединение i1 (400 мг, 1,22 ммоль) и 6,11-дигидродибензотиепин-11-ол (418 мг, 1,83 ммоль) растворяли в 50% растворе Т3Р в этилацетате (7,27 мл, 12,2 ммоль) и смесь перемешивали в герметичной колбе при 110°С в течение 1,5 часов. К смеси добавляли воду и смесь экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесями хлороформ-метанол и этилацетат-метанол) с получением соединения 26 (316 мг, 47%).
1H-ЯМР (CDCl3) δ: 2,86 (dd, J=11,4, 11,4 Гц, 1Н), 3,26-3,40 (m, 2Н), 3,55 (d, J=13,4 Гц, 1Н), 3,70 (d, J=10,4 Гц, 1Н), 3,86 (d, J=10,4 Гц, 1Н), 4,48 (d, J=9,5 Гц, 1Н), 4,66 (d, J=13,4 Гц, 1Н), 5,20 (s, 1Н), 5,43-5,50 (m, 2Н), 5,63 (d, J=10,9 Гц, 1Н), 5,79 (d, J=7,8 Гц, 1Н), 6,40 (d, J=7,7 Гц, 1Н), 6,62-6,69 (m, 1Н), 7,02-7,07 (m, 3Н), 7,18 (d, J=7,4 Гц, 1Н), 7,27-7,44 (m, 6H), 7,60-7,66 (m, 2Н).
Вторая стадия
Соединение III-1 получали при помощи такого же способа, как способ, описанный во второй стадии примера 1.
1H-ЯМР (CDCl3) δ: 2,98 (dd, J=13,0, 12,3 Гц, 1Н), 3,46 (dd, J=13,1, 10,0 Гц, 1Н), 3,55-3,63 (m, 2Н), 3,79 (d, J=11,4 Гц, 1Н), 3,96 (d, J=11,0 Гц, 1Н), 4,62-4,66 (m, 2Н), 5,26 (s, 1Н), 5,52 (d, J=13,4 Гц, 1Н), 5,75 (d, J=7,7 Гц, 1Н), 6,70 (d, J=7,7 Гц, 1Н), 6,79-6,85 (m, 1Н), 7,05-7,12 (m, 3Н), 7,23 (d, J=7,4 Гц, 1Н), 7,30 (t, J=7,3 Гц, 1Н), 7,36 (d, J=7,4 Гц, 1Н), 7,44 (t, J=7,4 Гц, 1Н).
Пример 3

Первая стадия
Соединение 27 (290 мг, 0,880 ммоль) и соединение i1 (240 мг, 0,733 ммоль) растворяли в 50% растворе Т3Р в этилацетате (2,4 мл) и смесь перемешивали в герметичной колбе при 100°С в течение 1,5 часов. К смеси добавляли воду и смесь экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью хлороформ-этилацетат-метанол) с получением соединения 28 (106 мг, 24%).
1H-ямр (CDCl3) δ: 2,37 (s, 3Н), 2,94-3,03 (m, 1Н), 3,15-3,23 (m, 1Н), 3,28 (t, J=10,4 Гц, 1Н), 3,58 (d, J=13,2 Гц, 1Н), 3,66 (dd, J=3,2 Гц, 11,6 Гц, 1Н), 3,84 (dd, J=2,8 Гц, 10,8 Гц, 1Н), 4,40-4,52 (m, 2Н), 5,49 (t, J=13,6 Гц, 2Н), 5,60 (d, J=10,4 Гц, 2Н), 5,78 (d, J=7,6 Гц, 1Н), 6,41 (d, J=7,2 Гц, 1Н), 6,66-6,71 (m, 1Н), 6,98-7,12 (m, 4H), 7,21 (d, J=7,6 Гц, 1Н), 7,30-7,42 (m, 4H), 7,56-7,61 (m, 2Н).
Вторая стадия
К раствору соединения 28 (100 мг, 0,168 ммоль) в метаноле (1 мл) добавляли 2 моль/л водный раствор гидроксида натрия (252 мкл, 0,504 ммоль) и смесь перемешивали при комнатной температуре в течение 1 часа. К смеси добавляли 2 моль/л водный раствор хлороводородной кислоты (0,3 мл) и смесь экстрагировали хлороформом. Полученный органический слой концентрировали при пониженном давлении. Полученный остаток растворяли в ДМА (1,0 мл), добавляли хлорид лития (35,6 мг, 0,839 ммоль) и смесь перемешивали при 100°С в течение 15 часов. Смесь очищали путем обращенно-фазовой колоночной хроматографии на силикагеле (смесью ацетонитрил-вода) с получением соединения III-24 (20 мг, 26%).
1H-ЯМР (CDCl3) δ: 3,09 (t, J=11,2 Гц, 1Н), 3,40-3,58 (m, 3H), 3,76 (d, J=10,8 Гц, 1Н), 3,91 (d, J=10,8 Гц, 1Н), 4,66 (d, J=13,2 Гц, 1Н), 4,73 (d, J=9,6 Гц, 1Н), 5,50 (d, J=13,6 Гц, 1Н), 5,79 (d, J=6,8 Гц, 1Н), 6,25 (s, 1Н), 6,61-6,70 (m, 2H), 6,79 (d, J=6,8 Гц, 1Н), 6,93-7,08 (m, 3H), 7,10-7,19 (m, 2H).
[0077]
Следующие примеры соединений получали из коммерчески доступных соединений или промежуточных соединений, описанных в примере сравнения, в соответствии с приведенными выше примерами.

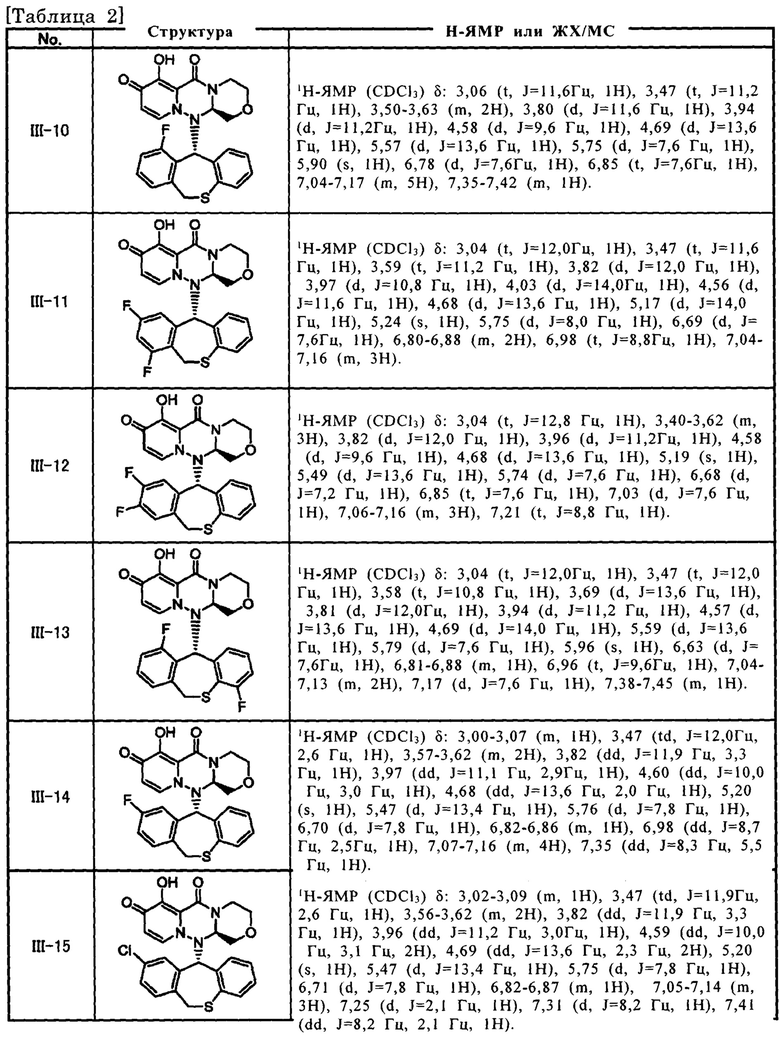

[0078]



[0079]
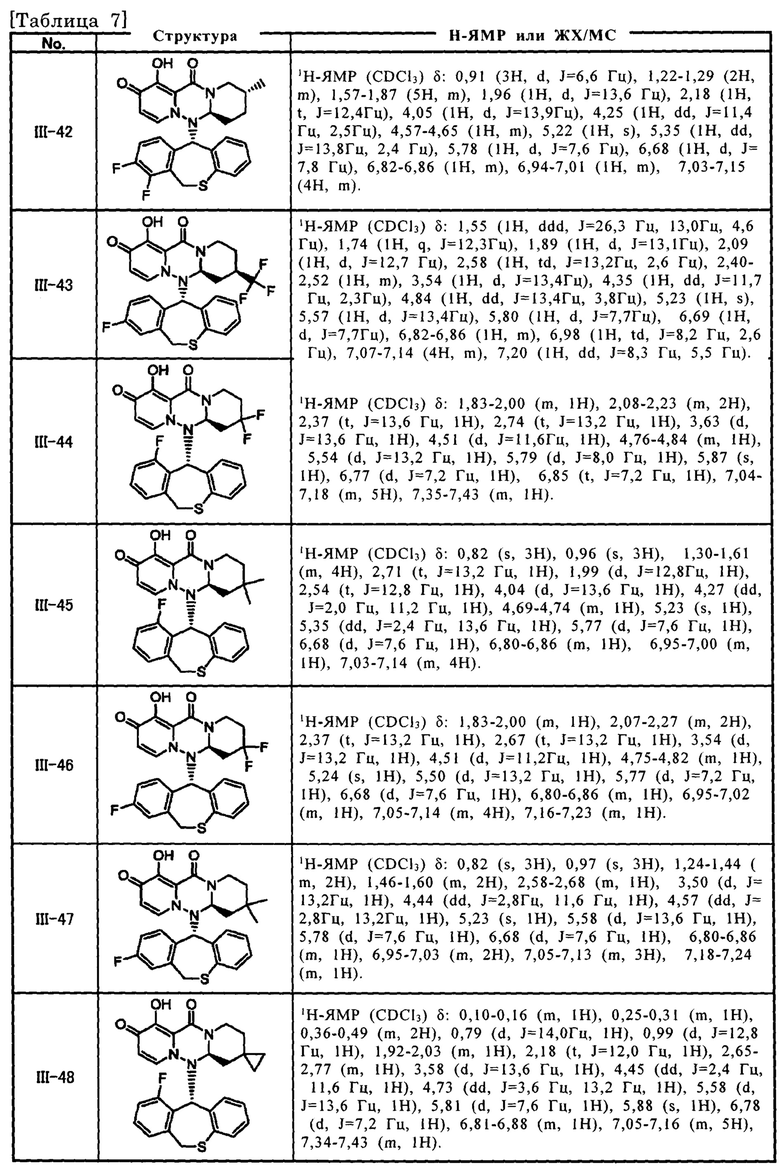


[0080]
Пример 4
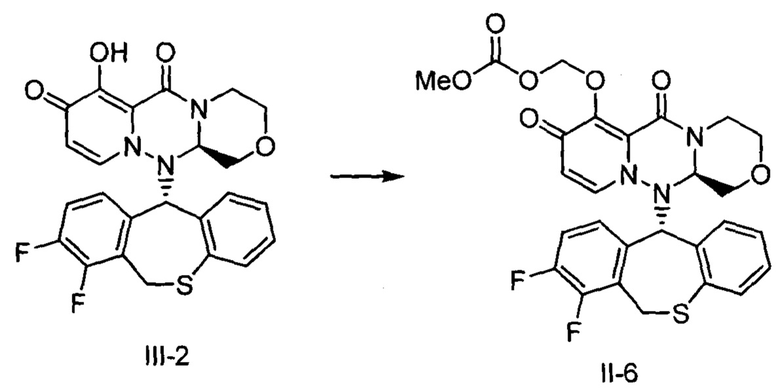
К суспензии соединения III-2 (1,00 г, 2,07 ммоль) в ДМА (5 мл) добавляли хлорметилметиловый эфир угольной кислоты (0,483 г, 3,10 ммоль), карбонат калия (0,572 г, 4,14 ммоль) и йодид калия (0,343 г, 2,07 ммоль) и смесь перемешивали при 50°С в течение 6 часов. К смеси добавляли ДМА (1 мл) и смесь перемешивали в течение 6 часов. Смесь охлаждали до комнатной температуры, добавляли ДМА (6 мл) и смесь перемешивали при 50°С в течение 5 минут. Смесь фильтровали. К полученному фильтрату добавляли 1 моль/л водный раствор хлороводородной кислоты (10 мл) и воду (4 мл) и смесь перемешивали в течение 1 часа. Осажденное твердое вещество отфильтровывали и сушили при пониженном давлении при 60°С в течение 3 часов с получением соединения II-6 (1,10 г, 1,93 ммоль, 93%).
1H-ЯМР (ДМСО-d6) δ: 2,91-2,98 (1H, m), 3,24-3,31 (1H, m), 3,44 (1H, t, J=10,4 Гц), 3,69 (1H, dd, J=11,5, 2,8 Гц), 3,73 (3Н, s), 4,00 (1H, dd, J=10,8, 2,9 Гц), 4,06 (1H, d, J=14,3 Гц), 4,40 (1H, d, J=11,8 Гц), 4,45 (1H, dd, J=9,9, 2,9 Гц), 5,42 (1H, dd, J=14,4, 1,8 Гц), 5,67 (1H, d, J=6,5 Гц), 5,72-5,75 (3Н, m), 6,83-6,87 (1H, m), 7,01 (1H, d, J=6,9 Гц), 7,09 (1H, dd, J=8,0, 1,1 Гц), 7,14-7,18 (1H, m), 7,23 (1H, d, J=7,8 Гц), 7,37-7,44 (2Н, m).
Пример 5

Первая стадия
К раствору хлорметилхлорформиата (300 мг, 2,33 ммоль) и соединения 30 (330 мг, 2,79 ммоль) в дихлорметане (6,0 мл) добавляли пиридин (207 мкл, 2,56 ммоль) при 0°С в атмосфере азота и смесь перемешивали при 0°С в течение 30 минут, нагревали до комнатной температуры и перемешивали в течение 1 часа. К смеси добавляли 2 моль/л водный раствор хлороводородной кислоты и смесь экстрагировали дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении с получением соединения 31 (440 мг, 90%).
1H-ЯМР (CDCl3) δ: 1,65 (s, 6Н), 3,77 (s, 3H), 5,71 (s, 2H).
Вторая стадия
Соединение III-2 (300 мг, 0,62 ммоль), карбонат калия (172 мг, 1,24 ммоль), йодид калия (103 мг, 0,62 ммоль) и соединение 31 (261 мг, 1,24 ммоль) растворяли в ДМА (3,0 мл) и смесь перемешивали при 80°С в течение 3 часов. К смеси добавляли 2 моль/л водный раствор хлороводородной кислоты и смесь экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью хлороформ-метанол) с получением соединения II-61 (350 мг, 86%).
1H-ЯМР (CDCl3) δ: 1,63 (s, 3H), 1,67 (s, 3H), 2,86-2,93 (m, 1H), 3,38-3,61 (m, 2H), 3,68-3,78 (m, 4H), 3,90-3,96 (m, 1H), 4,06 (d, J=14,0 Гц, 1H), 4,51 (dd, J=2,0 Гц, 9,6 Гц, 1H), 4,65 (d, J=12,4 Гц, 1H), 5,21 (d, J=14,4 Гц, 1H), 5,36 (s, 1H), 5,80-5,95 (m, 3H), 6,85-6,92 (m, 2H), 7,03-7,22 (m, 5H).
Пример 6

К раствору соединения III-2 (90 мг, 0,186 ммоль) в дихлорметане (2 мл) добавляли ангидрид уксусной кислоты (0,053 мл, 0,558 ммоль), триэтиламин (0,077 мл, 0,558 ммоль) и каталитическое количество ДМАП и смесь перемешивали при комнатной температуре в течение 2 часов. Смесь концентрировали при пониженном давлении и полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью хлороформ-метанол). К полученному раствору добавляли эфир и осажденное твердое вещество отфильтровывали с получением соединения II-4 (71 мг, 73%).
1H-ямр (CDCl3) δ: 2,46 (s, 3H), 2,88-2,99 (m, 1H), 3,35-3,50 (m, 1H), 3,60-3,65 (m, 1H), 3,75-3,83 (m, 1H), 3,90-4,00 (m, 1H), 4,05 (d, J=14,0 Гц, 1Н), 4,52-4,57 (m, 1H), 4,60-4,70 (m, 1H), 5,24-5,34 (m, 1H), 5,35 (s, 1H), 5,88 (d, J=7,6 Гц, 1Н), 6,85-6,82 (m, 1H), 6,90-7,05 (m, 2H), 7,06-7,20 (m, 4H)
ЖХ/МС (ИЭР): m/z=526,2 [M+H]+, RT=1,87 мин, способ (1)
[0081]
Пример 7

Первая стадия
К раствору трифосгена (300 мг, 2,54 ммоль) в дихлорметане (6,0 мл) добавляли пиридин (257 мкл, 3,17 ммоль) при 0°С в атмосфере азота и смесь перемешивали в течение 15 минут. К смеси добавляли раствор соединения 30 (377 мг, 1,27 ммоль) в дихлорметане (1,0 мл) и смесь перемешивали при 0°С в течение 15 минут, нагревали до комнатной температуры и перемешивали в течение 15 минут. Смесь концентрировали при пониженном давлении и добавляли этилацетат (4,0 мл), смесь фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения 32 (380 мг).
Вторая стадия
К раствору соединения III-2 (350 мг, 0,724 ммоль) в дихлорметане (3,5 мл) добавляли соединение 32 (196 мг, 1,09 ммоль) и триэтиламин (301 мкл, 2,17 ммоль) при 0°С и смесь перемешивали при 0°С в течение 30 минут. К смеси добавляли 2 моль/л водный раствор хлороводородной кислоты и смесь экстрагировали дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью хлороформ-метанол) с получением соединения II-65 (380 мг, 84%).
1H-ямр (CDCl3) δ: 1,73 (s, 3H), 1,77 (s, 3H), 2,90-2,99 (m, 1H), 3,37-3,43 (m, 1H), 3,57 (t, J=8,8 Гц, 1H), 3,76 (dd, J=2,8 Гц, 12,0 Гц, 1H), 3,81 (s, 3H), 3,94 (dd, J=2,8 Гц, 10,8 Гц, 1H), 4,05 (d, J=14,0 Гц, 1H), 4,55 (dd, J=2,8 Гц, 9,6 Гц, 1H), 4,65 (d, J=12,0 Гц, 1H), 5,28 (d, J=12,0 Гц, 1H), 5,34 (s, 1H), 5,89 (d, J=8,0 Гц, 1H), 6,86-6,95 (m, 2H), 7,03-7,15 (m, 5H).
Пример 8

К раствору соединения 33 (276 мг, 0,402 ммоль) в ТГФ (1 мл) на ледяной бане со льдом добавляли уксусную кислоту (121 мг, 2,01 ммоль) и 1 моль/л раствор ТБАФ в ТГФ (1,21 мл, 1,21 ммоль) и смесь перемешивали при комнатной температуре в течение 4 часов. Смесь концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью этилацетат-метанол) с получением соединения II-129 (179 мг, 78%).
ЖХ/МС (ИЭР): m/z=572,0 [М+Н]+, RT=1,74 мин, способ (2)
Пример 9

К раствору соединения III-2 (300 мг, 0,62 ммоль) в ДМФ (4 мл) добавляли карбонат калия (258 мг, 1,87 ммоль), 4-(хлорметил)фенилацетат (344 мг, 1,87 ммоль) и йодид натрия (139 мг, 1,87 ммоль) при комнатной температуре и смесь перемешивали при 65°С в течение 1 часа. К смеси добавляли воду и смесь экстрагировали этилацетатом. Полученный органический слой промывали водой, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью этилацетат-метанол) с получением соединения II-115 (120 мг, 31%).
ЖХ/МС (ИЭР): m/z=631,95 [М+Н]+, RT=2,07 мин, способ (2)
[0082]
Пример 10

К раствору соединения III-2 (150 мг, 0,31 ммоль) в дихлорметане (2 мл) добавляли 3 ммоль/г трифенилфосфин, нанесенный на полимер (310 мг, 0,93 ммоль), пиридин-4-илметанол (68 мг, 0,62 ммоль) и 40% раствор DEAD в толуоле (270 мг, 0,62 ммоль) при комнатной температуре и смесь перемешивали при комнатной температуре в течение 30 минут. Смесь очищали путем колоночной хроматографии на колонке для разделения аминокислот (смесью этилацетат-метанол) с получением соединения II-143 (63 мг, 35%).
ЖХ/МС (ИЭР): m/z=575,00 [М+Н]+, RT=1,43 мин, способ (2)
Пример 11

К раствору соединения III-2 (65 мг, 0,134 ммоль) в пиридине (0,8 мл) добавляли диметилкарбамоилхлорид (21,7 мг, 0,202 ммоль) и смесь перемешивали при 80°С в течение ночи. К смеси добавляли 1 моль/л водный раствор хлороводородной кислоты и смесь экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток затвердевали смесью этилацетат-гексан с получением соединения II-27 (65 мг, 87%).
1H-ЯМР (CDCl3) δ: 2,89 (t, J=11,2 Гц, 1Н), 2,99 (s, 1H), 3,01 (s, 3H), 3,18-3,26 (m, 4H), 3,45 (t, J=10,8 Гц, 1H), 3,59 (t, J=10,8 Гц, 1H), 3,70-3,80 (m, 1H), 3,90-3,98 (m, 1H), 4,03 (d, J=13,6 Гц, 1H), 4,50-4,70 (m, 2H), 5,21-5,35 (m, 2H), 5,82 (d, J=7,6 Гц, 1H), 6,91 (t, J=7,6 Гц, 1H), 7,00-7,20 (m, 6H).
Пример 12

К раствору этилфосфородихлоридата (135 мг, 0,829 ммоль) в дихлорметане (3 мл) добавляли гидрохлорид метилового эфира L-валина (139 мг, 0,829 ммоль), а затем по каплям добавляли раствор триэтиламина (168 мг, 1,66 ммоль) в дихлорметане (2 мл) при -78°С. Смесь перемешивали при комнатной температуре в течение 1 часа. Добавляли соединение III-2 (200 мг, 0,414 ммоль) и триэтиламин (126 мг, 1,25 ммоль) и смесь перемешивали при той же температуре в течение 6 часов. Смесь концентрировали и полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью этилацетат-метанол) с получением соединения II-55 (112 мг, 38%).
ЖХ/МС (ИЭР): m/z=705,05 [М+Н]+, RT=2,18 мин, способ (2)
[0083]
Пример 13

К раствору этилфосфородихлоридата (202 мг, 1,24 ммоль) в дихлорметане (3 мл) по каплям добавляли смесь триэтиламина (126 мг, 1,24 ммоль) и метилгликолят (112 мг, 1,24 ммоль) в дихлорметане (2 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. Добавляли соединение III-2 (200 мг, 0,414 ммоль) и триэтиламин (126 мг, 1,25 ммоль) и смесь перемешивали при той же температуре в течение 1 часа. Смесь концентрировали и полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью этилацетат-метанол) с получением соединения II-57 (143 мг, 52%).
ЖХ/МС (ИЭР): m/z=664,00 [М+Н]+, RT=1,93 мин, способ (2)
Пример 14

К раствору фосфорилхлорида (1,53 г, 10 ммоль) в дихлорметане (10 мл) по каплям добавляли смесь триэтиламина (2,12 г, 20,95 ммоль) и метилгликолят (1,89 мг, 21 ммоль) в дихлорметане (5 мл). Смесь перемешивали при комнатной температуре в течение 2 часов. К смеси (2 мл) добавляли соединение III-2 (200 мг, 0,414 ммоль) и триэтиламин (126 мг, 1,25 ммоль) и смесь перемешивали при той же температуре в течение 1 часа. Смесь концентрировали и полученный остаток очищали путем колоночной хроматографии на силикагеле (смесью этилацетат-метанол) с получением соединения II-58 (166 мг, 57%).
ЖХ/МС (ИЭР): m/z=707,90 [М+Н]+, RT=1,93 мин, способ (2)
[0084]
Следующие примеры соединений получали из коммерчески доступных соединений в соответствии с приведенными выше примерами.
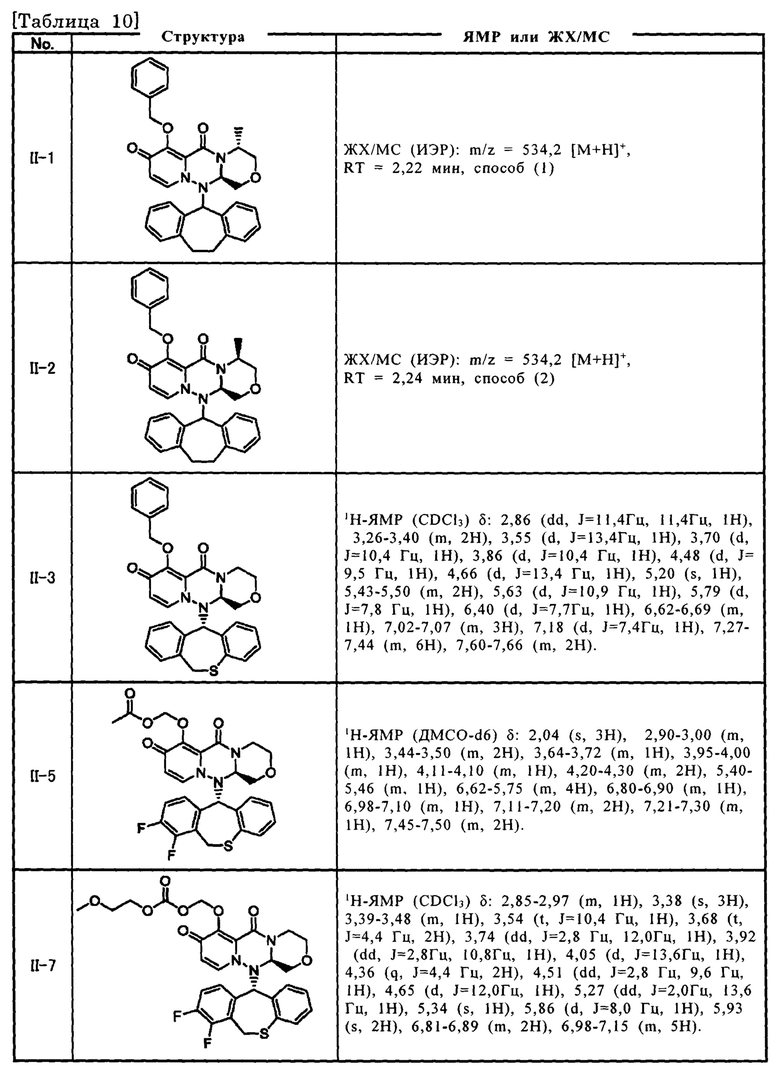


[0085]
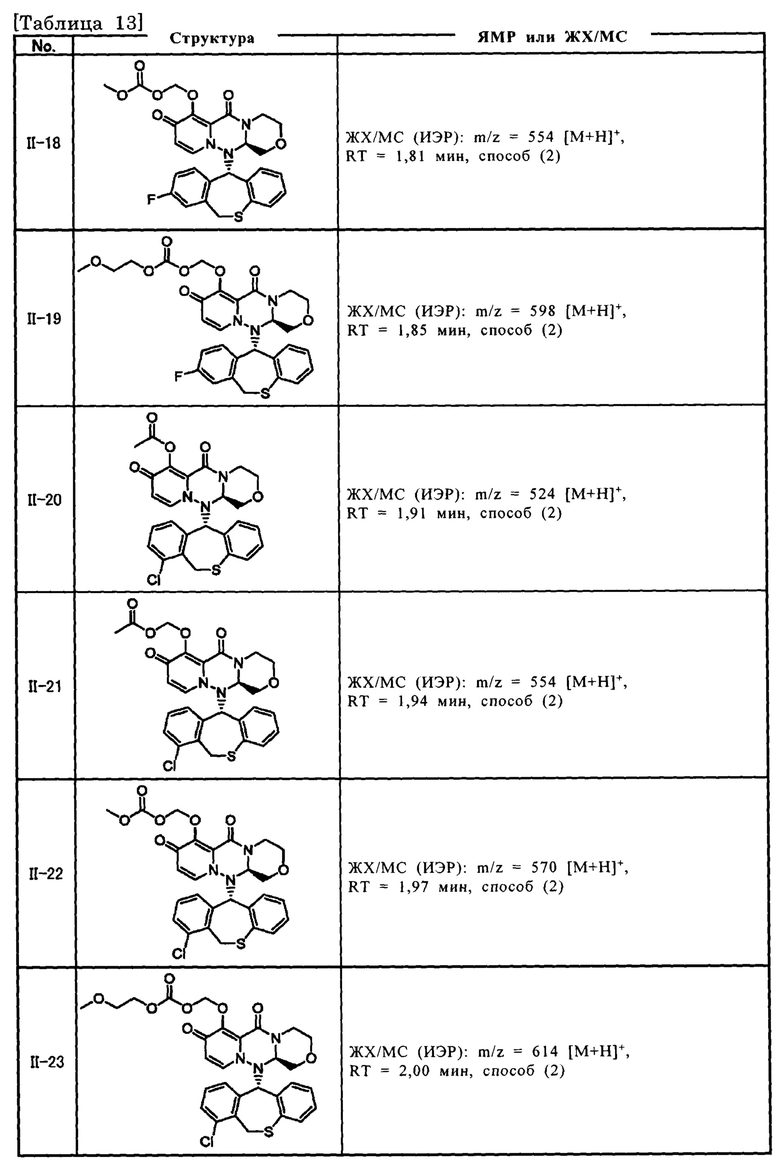


[0086]
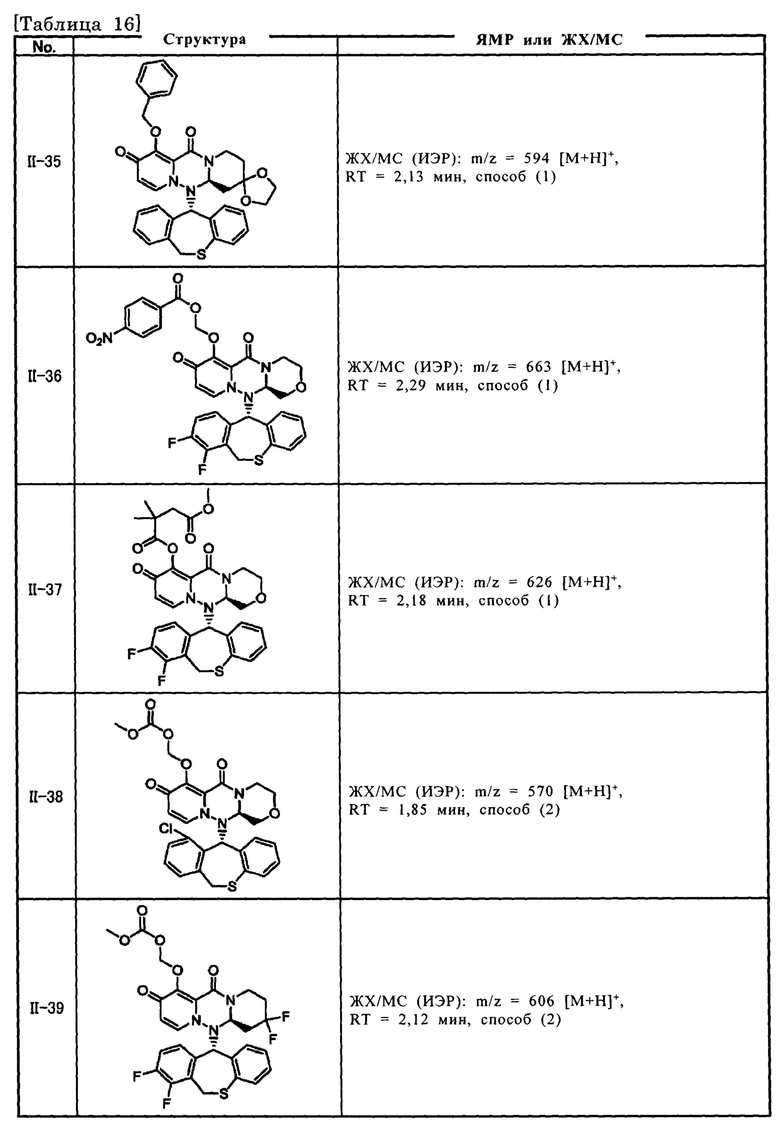


[0087]
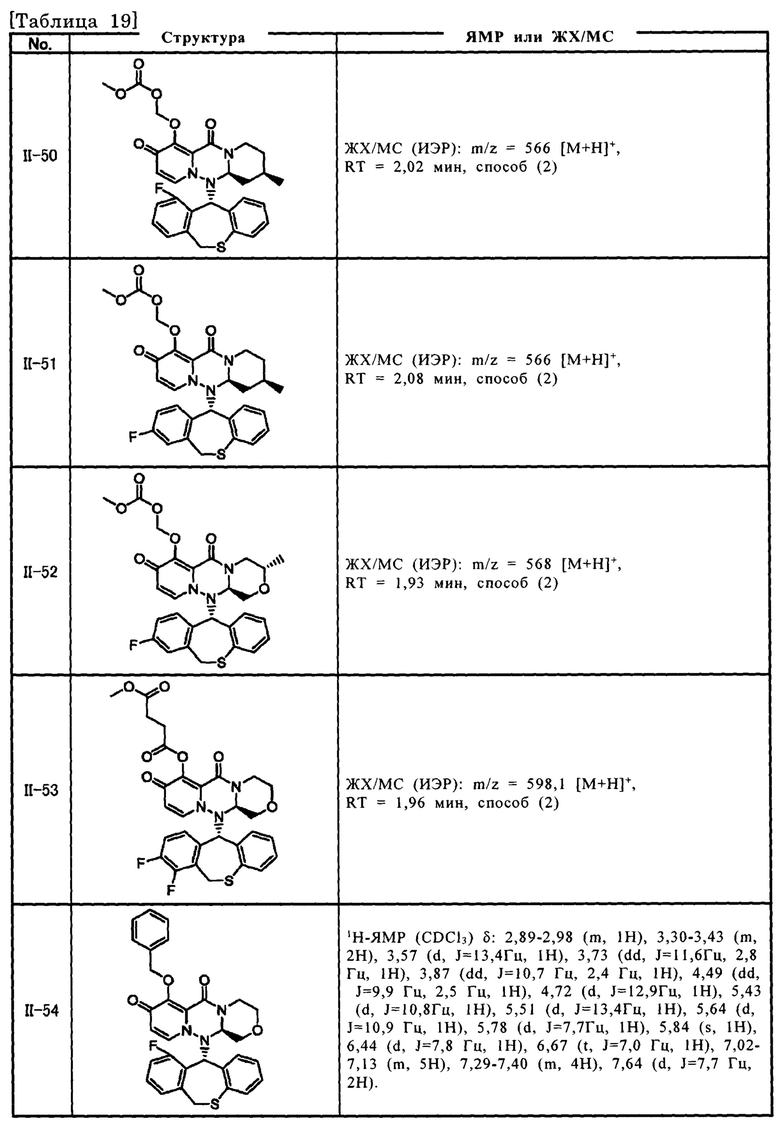

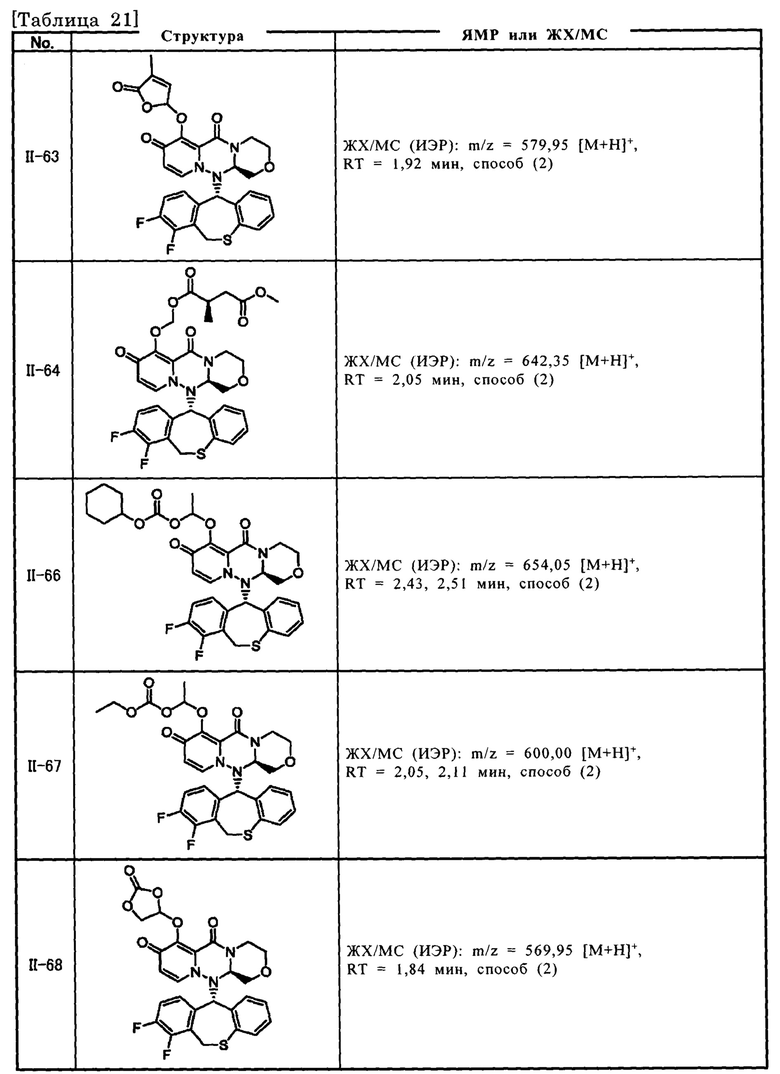
[0088]
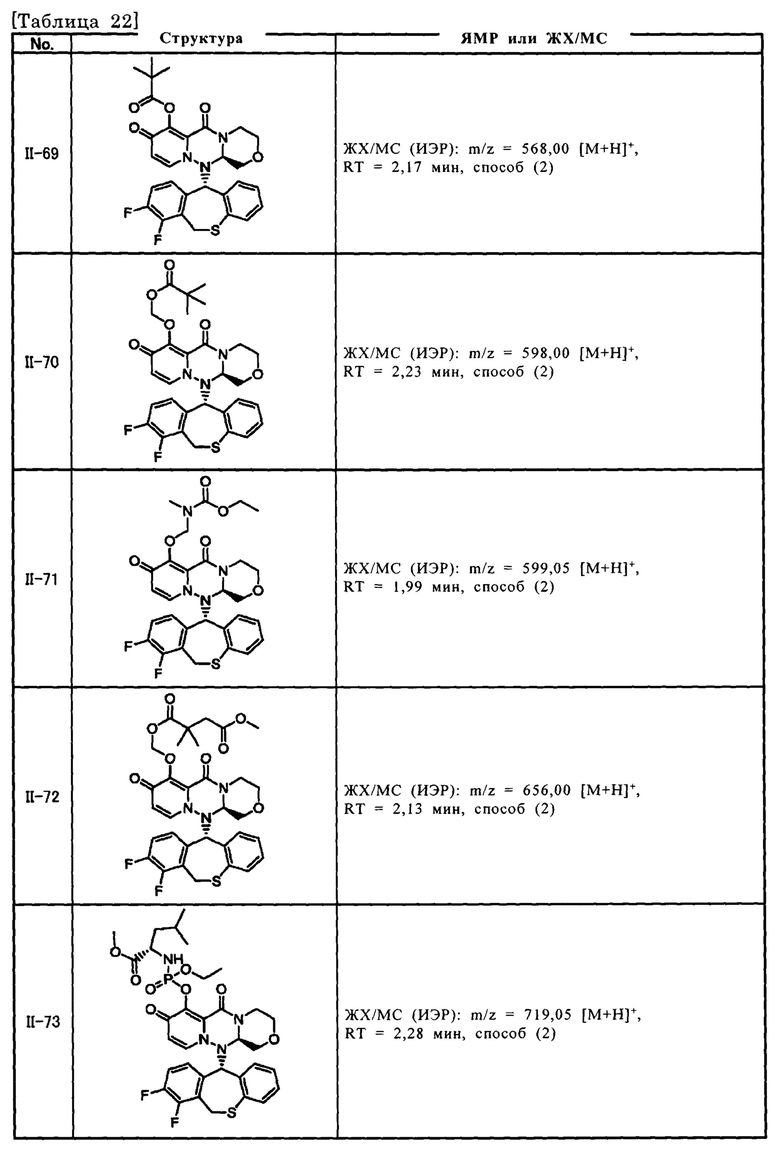


[0089]

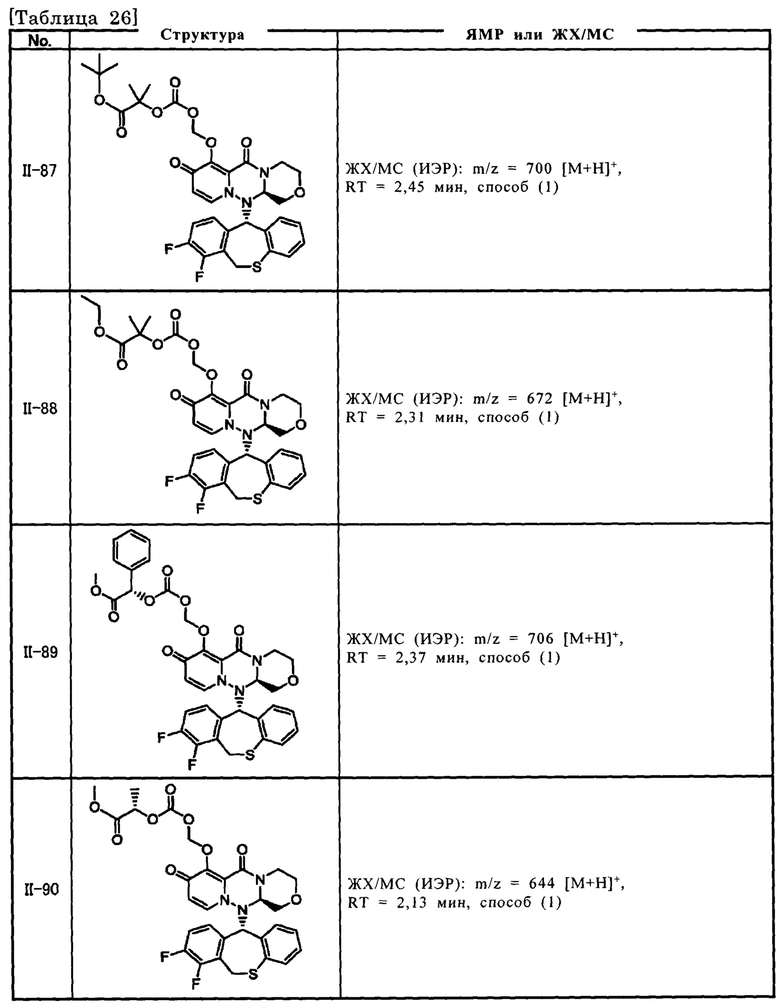
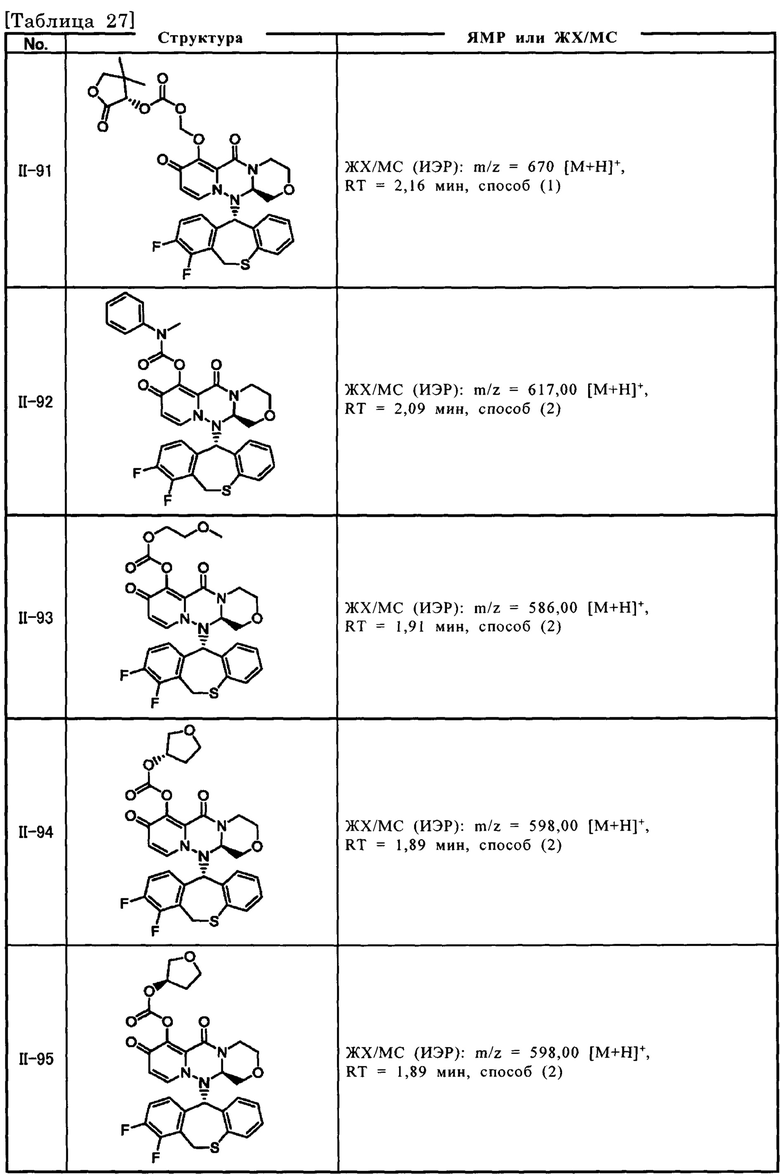
[0090]
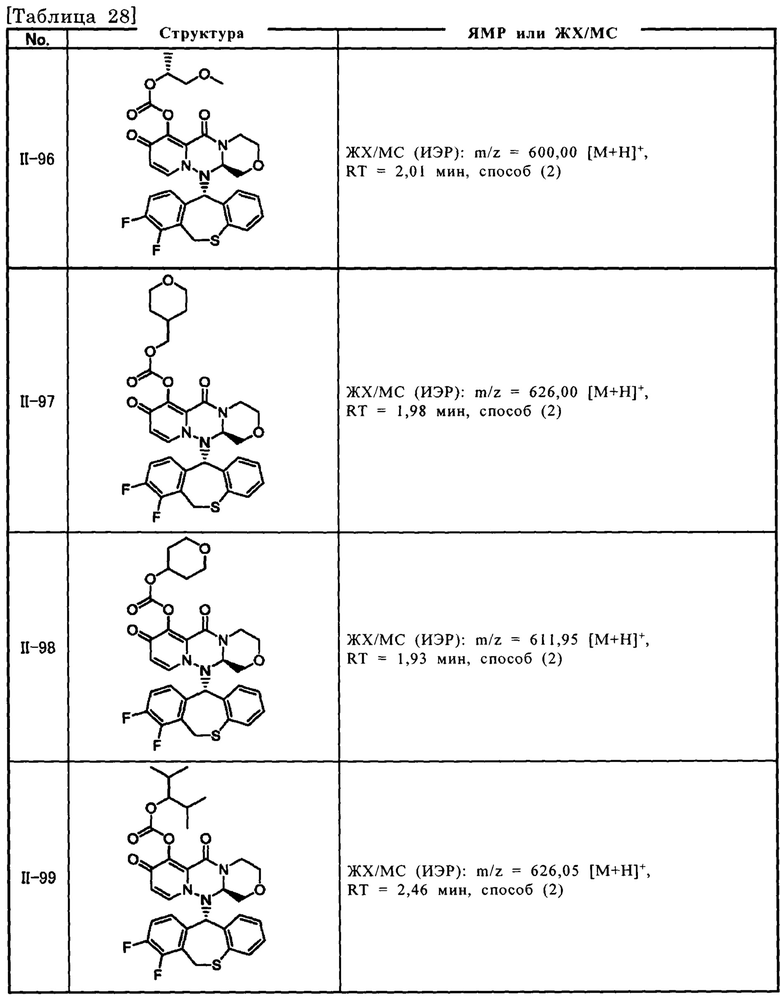


[0091]
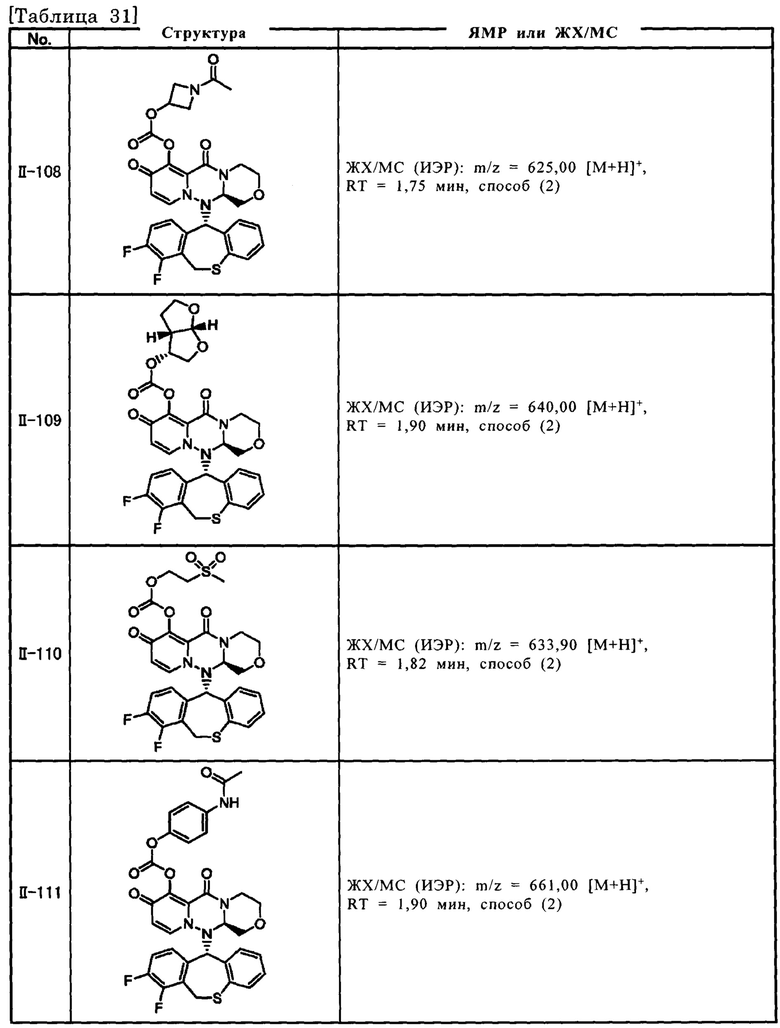
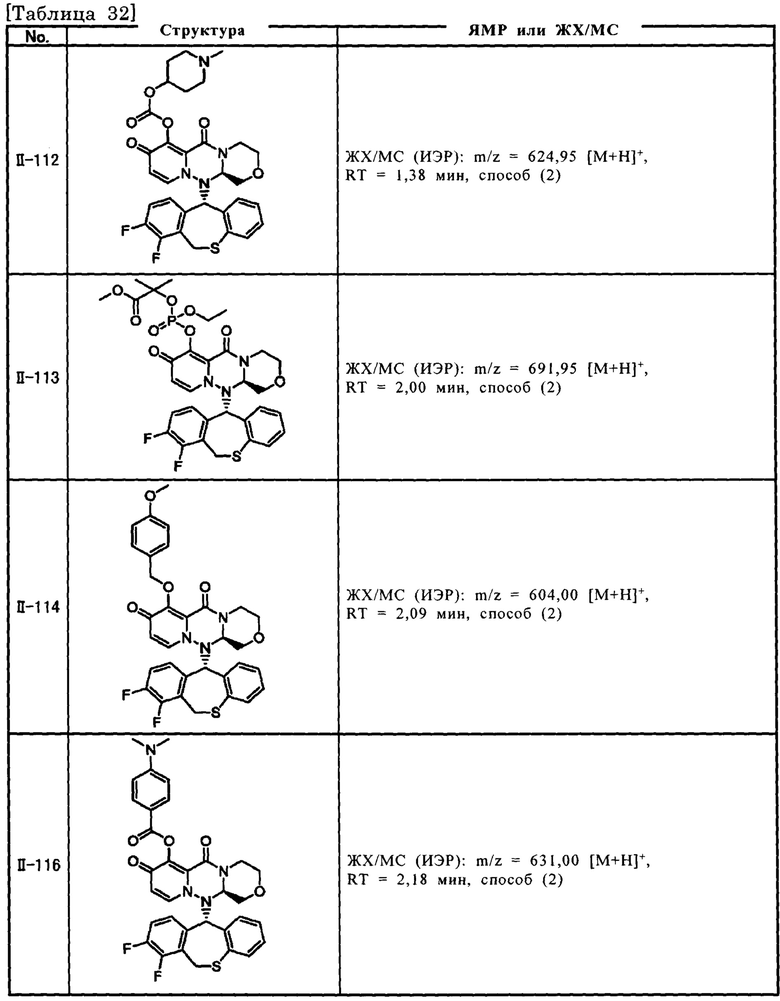


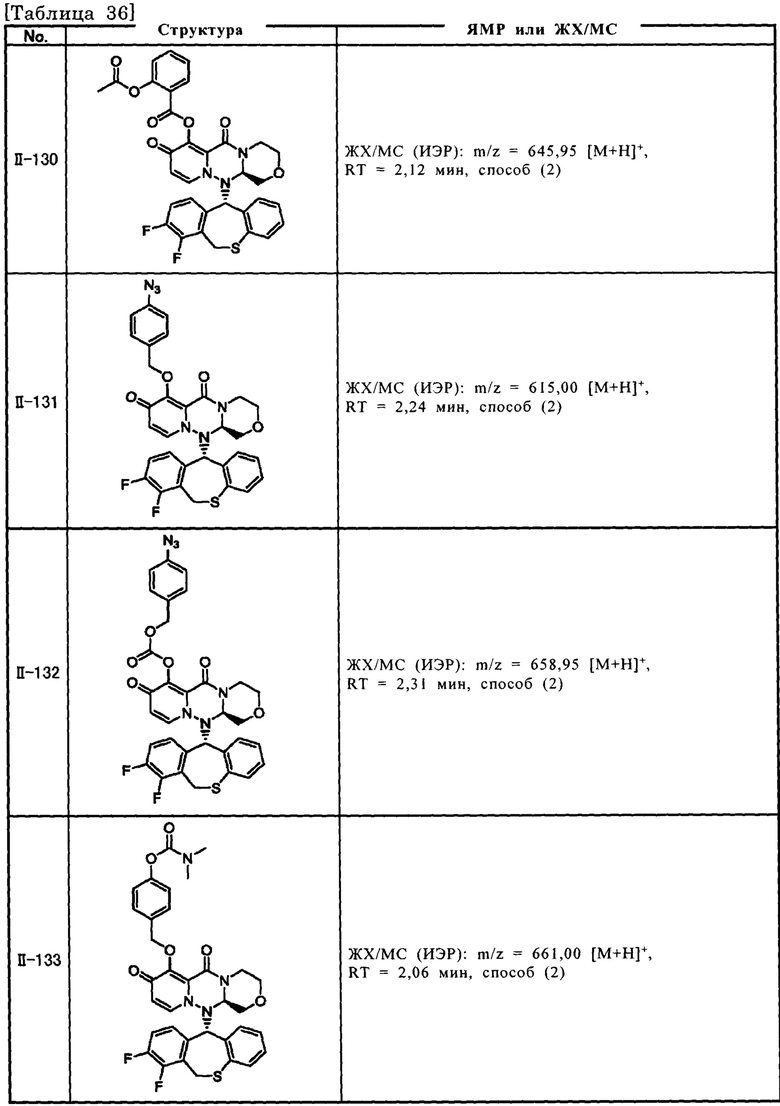
[0093]



[0094]
Соединения в соответствии с настоящим изобретением и/или соединения, которые являются исходными для соединений в соответствии с настоящим изобретением, подходят для борьбы с симптомами и/или заболеваниями, которые вызваны вирусом гриппа. Например, они подходят для лечения и/или предотвращения или ослабления симптомов, например, симптомов простуды, сопровождающих лихорадку, озноба, головной боли, мышечной боли, общего недомогания и т.п., симптомов воспаления дыхательных путей, таких как фарингалгия, выделения из носа, заложенность носа, кашель, мокрота и т.п., желудочно-кишечных симптомов, таких как боль в животе, тошнота, диарея и т.п., а также осложнений, сопровождающих вторичную инфекцию, таких как острая энцефалопатия и пневмония.
Поскольку соединения в соответствии с настоящим изобретением представляют собой пролекарства и, таким образом, обладают преимуществами, заключающимися в высокой всасываемости в ротовой полости, хорошей биодоступности, хорошем клиренсе и высокой способности прохождения через легкие, они могут являться превосходными лекарственными средствами.
Поскольку соединения, которые являются исходными для соединений в соответствии с настоящим изобретением, обладают такими свойствами, как высокая ингибирующая активность кэп-зависимой эндонуклеазы и высокая селективность к вирус-специфическому ферменту, они могут представлять собой лекарственные средства, характеризующиеся пониженными побочными эффектами.
Кроме того, поскольку соединения в соответствии с настоящим изобретением и/или соединения, которые являются исходными для соединений в соответствии с настоящим изобретением, также обладают преимуществами, заключающимися в высокой устойчивости к метаболизму, высокой растворимости, высокой всасываемости в ротовой полости, хорошей биодоступности, хорошем клиренсе, высокой способности прохождения через легкие, длительном времени полувыведения, высокой степени небелкового связывания, слабом ингибировании канала hERG, слабом ингибировании CYP, ингибирующем действии в отношении ЦПЭ (цитопатогенного эффекта) и/или отрицательном результате исследования на фототоксичность, тест Эймса и исследование на токсичность в отношении генов или токсичность, такую как поражение печени, демонстрируют отрицательные результаты. Таким образом, соединения в соответствии с настоящим изобретением могут являться превосходными лекарственными средствами.
[0095]
Соединения в соответствии с настоящим изобретением и/или соединения, которые являются исходными для соединений в соответствии с настоящим изобретением, можно вводить перорально или парентерально. В случае перорального введения настоящие соединения также можно применять в виде обычного препарата, например, в виде любой из лекарственных форм твердых препаратов, таких как таблетки, порошки, гранулы, капсулы и т.п.; растворов; маслянистых суспензий или жидких препаратов, таких как сиропы или эликсиры и т.п. В случае парентерального введения соединения в соответствии с настоящим изобретением можно применять в виде водных или маслянистых суспензий для инъекций или капель для носа. При их получении можно использовать обычные вспомогательные вещества, связующие вещества, смазывающие вещества, водные растворители, масляные растворители, эмульгаторы, суспендирующие агенты, консерванты, стабилизаторы и т.п. Фармацевтическую композицию согласно настоящему изобретению можно получать путем объединения (например, смешивания) терапевтически эффективного количества настоящего соединения с фармацевтически приемлемыми веществами-носителями или разбавителями.
Доза соединений в соответствии с настоящим изобретением может быть различной в зависимости от способа введения, возраста, веса и состояния пациента и типа заболевания, и в случае перорального введения взрослому обычно можно вводить от примерно 0,05 мг до 3000 мг, предпочтительно от примерно 0,1 мг до 1000 мг в день, при необходимости, в виде нескольких отдельных доз. Кроме того, в случае парентерального введения взрослому вводят от примерно 0,01 мг до 1000 мг, предпочтительно от примерно 0,05 мг до 500 мг в день.
[0096]
Пример исследования 1: Измерение ингибирующей активности кэп-зависимой эндонуклеазы (CEN)
1) Получение субстрата
Заказывали 30-мер РНК (5'-pp-[m2'-O]GAA UAU(-Cy3) GCA UCA CUA GUAAGC UUU GCU CUA-BHQ2-3' производства Japan Bio Services Co., LTD.), в котором G в концевом 5'-положении модифицирован дифосфатом, гидроксильная группа в 2'-положении модифицирована путем метоксилирования, шестой U от концевого 5'-положения помечен Cy3, и 3'-положение помечено BHQ2, и кэп-структуру добавляли при помощи системы ScriptCap производства EPICENTRE (продукт представлял собой m7G [5']-ррр-[5'] [m2'-O]GAA UAU(-Cy3) GCA UCA CUA GUA AGC UUU GCU CUA(-BHQ2)-3'). Его отделяли и очищали путем денатурирующего электрофореза в полиакриламидном геле и применяли в качестве субстрата.
2) Получение фермента
РНП получали из вирусной частицы при помощи стандартного способа (ссылочный документ: VIROLOGY(1976) 73, р327-338 OLGA М. ROCHOVANSKY). В частности, вирус A/WSN/33 (1×103 БОЕ/мл, 200 мкл) инокулировали в 10-дневном курином яйце с развивающимся эмбрионом. После инкубации при 37°С в течение 2 дней выделяли аллантоисную жидкость из куриного яйца. Вирусную частицу очищали путем ультрацентрифугирования с применением 20% раствора сахарозы, растворенной в TritonX-100 и лизолецитине, фракцию РНП (фракция, содержащая 50-70% глицерина) собирали путем ультрацентрифугирования с градиентом 30-70% плотности глицерина и применяли в качестве ферментного раствора (содержащего примерно 1 нМ комплекса РВ1-РВ2-РА).
3) Ферментная реакция
Ферментный реакционный раствор (2,5 мкл) (состав: 53 мМ Трис гидрохлорида (рН 7,8), 1 мМ MgCl2, 1,25 мМ дитиотреитола, 80 мМ NaCl, 12,5% глицерина, 0,15 мкл раствора фермента) вносили в 384-луночный планшет из полипропилена. Затем в планшет добавляли 0,5 мкл раствора исследуемого соединения, которое последовательно разбавляли диметилсульфоксидом (ДМСО). В качестве положительного контроля (PC) или отрицательного контроля (NC) в планшет добавляли 0,5 мкл ДМСО, соответственно. Содержимое каждого планшета интенсивно перемешивали. Затем для инициирования реакции добавляли 2 мкл раствора субстрата (1,4 нМ раствор субстратной РНК, 0,05% Tween20). После инкубации при комнатной температуре в течение 60 минут собирали 1 мкл реакционного раствора и добавляли к 10 мкл высокоочищенного деионизированного раствора формамида (содержащего стандарт GeneScan 120 Liz Size Standard в качестве маркера для калибровки по размерам производства Applied Biosystems (ABI)) для остановки реакции. В случае NC реакцию останавливали заранее путем добавления ЭДТК (4,5 мМ) перед инициированием реакции (все концентрации, описанные выше, являются
конечными концентрациями).
3) Измерение коэффициента ингибирования (значения IC50)
Раствор, в котором реакцию останавливали, грели при 85°С в течение 5 минут, быстро охлаждали во льду в течение 2 минут и анализировали при помощи генетического анализатора ABI PRIZM 3730. Пик кэп-зависимой эндонуклеазы количественно оценивали при помощи аналитического программного обеспечения ABI Genemapper, коэффициент ингибирования реакции с участием CEN (%) для исследуемого соединения получали путем принятия интенсивности флуоресценции для PC и NC, равной 0% ингибированию и 100% ингибированию, соответственно, значение IC50 получали при помощи программного обеспечения для аппроксимации кривых (XLfit2.0: модель 205 (производства IDBS) и т.п.). Значения IC50 для исследуемых веществ, являющихся исходными соединениями, представлены в таблице 39.
[0097]
Пример исследования 2: Исследование, подтверждающее ингибирующее действие на ЦПЭ
<Материал>
- 2% раствор FCS Е-МЕМ (полученный путем добавления канамицина и FCS к MEM (минимальной питательной среде) (Invitrogen))
- 0,5% раствор BSA Е-МЕМ (полученный путем добавления канамицина и BSA к MEM (минимальной питательной среде) (Invitrogen))
- HBSS (сбалансированный солевой раствор Хэнкса)
- Клетки MDBK
Клетки доводили до требуемого количества клеток (3×105/мл) 2% раствором FCS Е-МЕМ.
- Клетки MDCK
После двукратной промывки с применением HBSS клетки доводили до требуемого количества клеток (5×105/мл) 0,5% раствором BSA Е-МЕМ.
- Раствор трипсина
Трипсин из свиной поджелудочной железы (SIGMA) растворяли в ФБР(-) и фильтровали через 0,45 мкм фильтр.
- EnVision (PerkinElmer)
- Набор WST-8 (Kishida Chemical Co., Ltd.)
- 10% раствор ДСН
[0098]
<Последовательность операций>
- Разбавление и распределение исследуемого образца
В качестве питательной среды при использовании клеток MDBK применяли 2% раствор FCS Е-МЕМ и при использовании клеток MDCK применяли 0,5% раствор BSA Е-МЕМ. В дальнейшем в настоящем описании для разбавления вируса, клеток и исследуемого образца применяли ту же питательную среду.
Исследуемый образец заранее разбавляли питательной средой до требуемой концентрации, а затем проводили 2-5-кратное последовательное разбавление в 96-луночном планшете (50 мкл/лунка). Готовили два планшета, один для измерения активности против гриппа и один для измерения цитотоксичности. Каждый анализ проводили три раза для каждого лекарственного средства.
При использовании клеток MDCK к клеткам добавляли трипсин для получения конечной концентрации 3 мкг/мл только при измерении активности против гриппа.
- Разбавление и распределение вируса гриппа
Вирус гриппа заранее разбавляли питательной средой до требуемой концентрации и в количестве 50 мкл/лунка вносили в каждую лунку 96-луночного планшета, содержащего исследуемое вещество. Питательную среду в количестве 50 мкл/лунка вносили в каждую лунку планшета, содержащего исследуемое вещество, для измерения цитотоксичности.
- Разбавление и распределение клеток
Клетки, доведенные до требуемого количества клеток, в количестве 100 мкл/лунка вносили в каждую лунку 96-луночного планшета, содержащего исследуемый образец.
Полученный состав перемешивали при помощи мешалки для планшета и инкубировали в инкубаторе с атмосферой СO2 в течение 3 дней для измерения активности против гриппа и измерения цитотоксичности.
- Распределение WST-8
Клетки в 96-луночном планшете, который инкубировали в течение 3 дней, визуально исследовали под микроскопом и изучали внешний вид клеток и наличие или отсутствие кристаллов исследуемого соединения. Надосадочную жидкость удаляли, чтобы клетки не адсорбировались из планшета.
Набор WST-8 разбавляли в 10 раз питательной средой и в количестве 100 мкл вносили в каждую лунку планшета. После перемешивания при помощи мешалки для планшета клетки инкубировали в инкубаторе с атмосферой CO2 в течение времени от 1 до 3 часов.
После инкубации в каждую лунку планшета для измерения активности против гриппа вносили 10% раствор ДСН в количестве 10 мкл/лунка для инактивации вируса.
- Измерение коэффициента абсорбции
После перемешивания 96-луночного планшета коэффициент абсорбции измеряли на EnVision на двух длинах волн, 450 нм/620 нм.
[0099]
<Расчет значения каждого измеряемого параметра>
Значения рассчитывали с применением Microsoft Excel или программы, имеющей аналогичные расчетные и вычислительные функции, на основе следующих расчетных уравнений.
- Расчет эффективной ингибирующей концентрации для достижения гибели 50% клеток, инфицированных гриппом (ЕС50)
ЕС50=10Z
Z=(50% - высокий %)/(высокий % - низкий %)×{log(высокая конц.)-log(низкая конц.)}+log(высокая конц.)
[0100]
Результаты измерения для исследуемых веществ (соединения из примеров сравнения), являющихся исходными соединениями, в примере исследования 1 и примере исследования 2 приведены в таблице 39.
[0101]


Согласно приведенным выше результатам исходные соединения проявляют высокую ингибирующую активность кэп-зависимой эндонуклеазы (CEN) и/или сильное ингибирующее действие в отношении ЦПЭ и, таким образом, могут являться подходящими агентами для лечения и/или предотвращения симптомов и/или заболеваний, вызываемых при инфицировании вирусом гриппа.
[0102]
Ниже описаны примеры биологических исследований соединений согласно настоящему изобретению.
[0103]
Пример исследования 3: Исследование ингибирования CYP
С применением коммерчески доступных объединенных микросом печени человека и при использовании в качестве маркеров O-деэтилирования 7-этоксирезоруфина (CYP1A2), метилгидроксилирования толбутамида (CYP2C9), 4'-гидроксилирования мефенитоина (CYP2C19), O-деметилирования декстрометорфана (CYP2D6) и гидроксилирования терфенедина (CYP3A4), как типичных реакций метаболизма субстратов пяти основных форм фермента CYP у человека (CYP1A2, 2С9, 2С19, 2D6, 3А4), оценивали степень ингибирования каждого процесса образования метаболита соединением согласно настоящему изобретению.
[0104]
Использовали следующие условия реакции: субстрат, 0,5 мкмоль/л этоксирезоруфина (CYP1A2), 100 мкмоль/л толбутамида (CYP2C9), 50 мкмоль/л S-мефенитоинмефенитоина (CYP2C19), 5 мкмоль/л декстрометорфана (CYP2D6), 1 мкмоль/л терфенедина (CYP3A4); время реакции, 15 минут; температура реакции, 37°С; фермент, объединенные микросы печени человека 0,2 мг белка/мл; концентрация соединения согласно настоящему изобретению, 1, 5, 10, 20 мкмоль/л (четыре точки).
[0105]
Каждые пять видов субстратов, микросомы печени человека или соединение согласно настоящему изобретению в 50 ммоль/л буфере HEPES в качестве реакционного раствора в виде композиции, описанной выше, добавляли в 96-луночный планшет, в качестве кофактора добавляли НАДФН для инициирования реакций метаболизма, выступающих в качестве маркеров, и после инкубации при 37°С в течение 15 минут добавляли раствор метанол/ацетонитрил = 1/1 (об./об.) для прекращения реакции. После центрифугирования при 3000 об/мин в течение 15 минут резоруфин (метаболит CYP1A2) в надосадочной жидкости количественно оценивали при помощи флуоресцентного многофункционального счетчика, а гидроксид толтрибутамида (метаболит CYP2C9P), 4'-гидроксид мефенитоина (метаболит CYP2C19), декстрометорфан (метаболит CYP2D6) и терфенадиновый спирт (метаболит CYP3A4) количественно оценивали путем ЖХ/МС/МС.
[0106]
В качестве контроля (100%) принимали добавление в реакционную систему только ДМСО, который является растворителем, растворяющим соединение согласно настоящему изобретению, для каждой концентрации соединения согласно настоящему изобретению, добавленного в виде раствора, рассчитывали остаточную активность (%) и на основании обратного предположения при помощи логистической модели с применением концентрации и скорости ингибирования рассчитывали IC50.
(Результат)
Соединение III-2: пять видов >20 мкмоль/л
[0107]
Пример исследования 4: исследование биодоступности
Материалы и способы для проведения экспериментов по оценке всасывания в ротовой полости
(1) Экспериментальные животные: использовали мышей или крыс линии Спраг-Доули (SD).
(2) Условия разведения: мышам или крысам линии SD обеспечивали свободный доступ к твердому корму и стерилизованной водопроводной воде.
(3) Режим дозирования и принцип разделения на группы: пероральное введение и внутривенное введение проводили с применением заранее определенных дозировок. Разделение на группы осуществляли следующим образом. (Дозировку корректировали для каждого соединения)
Пероральное введение: от 1 до 30 мг/кг (n= от 2 до 3)
Внутривенное введение: от 0,5 до 10 мг/кг (n= от 2 до 3)
(4) Получение растворов для введения: пероральное введение проводили в виде раствора или суспензии. Внутривенное введение проводили после растворения.
(5) Способы введения: пероральное введение проводили исключительно в желудок при помощи зонда для перорального введения. Внутривенное введение проводили через хвостовую вену при помощи шприцев с иглой.
(6) Параметры оценки: кровь собирали последовательно и концентрацию соединения согласно настоящему изобретению в плазме измеряли путем ЖХ/МС/МС.
(7) Статистический анализ: для исследования изменений концентрации соединения согласно настоящему изобретению в плазме площадь под кривой зависимости концентрации в плазме от времени (ППК) рассчитывали при помощи программного обеспечения для применения нелинейного метода наименьших квадратов, WinNonlin (зарегистрированная торговая марка) и биодоступность (ВА) соединения согласно настоящему изобретению рассчитывали на основе ППК для группы с пероральным введением и группы с внутривенным введением.
(Результат)
Соединение II-6: 14,9%
Соединение III-2: 4,2%
Согласно приведенным выше результатам пролекарство обладает улучшенной биодоступностью по сравнению с исходным соединением.
Следовательно, соединение согласно настоящему изобретению обладает превосходной всасываемостью в ротовой полости и может являться подходящим агентом для лечения и/или предотвращения симптомов и/или заболеваний, вызываемых при инфицировании вирусом гриппа.
[0108]
Пример исследования 5: Исследование устойчивости к метаболизму
Соединение согласно настоящему изобретению подвергали воздействию в течение постоянного промежутка времени с применением коммерчески доступных объединенных микросом печени человека и уровень остаточного соединения вычисляли путем сравнения прореагировавшего образца и непрореагировавшего образца, таким образом, оценивая степень метаболизма в печени.
[0109]
Реакцию (окислительную реакцию) проводили при 37°С в течение 0 минут или 30 минут в присутствии 1 ммоль/л раствора НАДФН в 0,2 мл буфера (50 ммоль/л Трис-HCl рН 7,4, 150 ммоль/л хлорида калия, 10 ммоль/л хлорида магния), содержащего 0,5 мг белка/мл микросом печени человека. После проведения реакции 50 мкл реакционного раствора добавляли к 100 мкл раствора метанол/ацетонитрил = 1/1 (об./об.), перемешивали и центрифугировали при 3000 об/мин в течение 15 минут. Соединение согласно настоящему изобретению в надосадочной жидкости количественно оценивали путем ЖХ/МС/МС или твердофазной экстракции (ТФЭ)/МС и рассчитывали количество соединения согласно настоящему изобретению, оставшегося после проведения реакции, принимая количество соединения после проведения реакции в течение 0 минут за 100%. Реакцию гидролиза проводили при отсутствии НАДФН и реакцию глюконидирования проводили в присутствии 5 мМ раствора УДФ-глюкуроновой кислоты вместо НАДФН при помощи аналогичных операций.
(Результат) показан % ингибирования при 2 мкмоль/л исследуемого соединения.
Соединение III-2: 90,1%
[0110]
Пример исследования 6: флуоресцентное исследование ингибирования продуктами метаболизма CYP3A4
Флуоресцентное исследование ингибирования продуктами метаболизма CYP3A4 представляет собой исследование усиления ингибирования соединения согласно настоящему изобретению под воздействием CYP3A4 в результате метаболической реакции, и исследование проводили с применением фермента CYP3A4, экспрессированного в Escherichia coli, и с использованием реакции, в которой 7-бензилокситрифторметилкумарин (7-BFC) дебензилируется ферментом CYP3A4 с получением метаболита, 7-гидрокситрифторметилкумарина (HFC), испускающего флуоресцентное излучение, в качестве показателя.
[0111]
Использовали следующие условия реакции: субстрат, 5,6 мкмоль/л 7-BFC; время предварительной реакции, 0 или 30 минут; время реакции, 15 минут; температура реакции, 25°С (комнатная температура); содержание CYP3A4 (экспрессированного в Escherichia coli) во время предварительной реакции 62,5 пмоль/мл, во время реакции 6,25 пмоль/мл (при 10-кратном разбавлении); концентрация исследуемого лекарственного средства на основе соединения согласно настоящему изобретению, 0,625, 1,25, 2,5, 5, 10, 20 мкмоль/л (шесть точек).
[0112]
Фермент в буфере K-Pi (pH 7,4) и раствор соединения согласно настоящему изобретению в качестве предреакционного раствора добавляли в 96-луночный планшет с получением указанной выше предварительной реакционной композиции, часть состава переносили в другой 96-луночный планшет и разбавляли в 10 раз с применением субстрата и буфера K-Pi, добавляли НАДФН в качестве кофактора для инициирования реакции, выступающей в качестве показателя (без предварительной инкубации), и после протекания реакции в течение заранее установленного времени добавляли раствор ацетонитрил/0,5 моль/л Трис (трисгидроксиаминометан) = 4/1 (об./об.) для завершения реакции. Кроме того, к оставшемуся раствору для предварительной инкубации добавляли НАДФН для инициирования предварительной инкубации (с предварительной инкубацией) и после инкубации в течение заранее установленного времени часть раствора переносили в другой планшет и разбавляли в 10 раз с применением субстрата и буфера K-Pi для инициирования реакции, выступающей в качестве показателя. После прохождения реакции в течение заранее установленного времени добавляли раствор ацетонитрил / 0,5 моль/л Трис (трисгидроксиаминометан) = 4/1 (об./об.) для завершения реакции. Для планшета, в котором проводили каждую из реакций, выступающих в качестве показателя, при помощи флуоресцентного устройства для прочтения планшетов (возб. = 420 нм, исп. = 535 нм) измеряли значение флуоресценции для 7-HFC, который является метаболитом.
[0113]
В качестве контроля (100%) принимали добавление в реакционную систему только ДМСО, который является растворителем, растворяющим соединение согласно настоящему изобретению, для каждой концентрации соединения согласно настоящему изобретению, добавленного в виде раствора, рассчитывали остаточную активность (%) и на основании обратного предположения при помощи логистической модели с применением концентрации и скорости ингибирования рассчитывали IС50. При разнице между значениями IC50, составляющей 5 мкмоль/л или более, результат определяли, как (+), и при разнице, составляющей 3 мкмоль/л или менее, результат определяли, как (-).
(Результат)
Соединение III-2: (-)
[0114]
Пример исследования 7: Флуктуационный тест Эймса
Оценивали мутагенность соединений согласно настоящему изобретению.
20 мкл хранящейся в холоде брюшнотифозной палочки крыс (Salmonella typhimurium штамм ТА98, штамм ТА100) инокулировали в 10 мл жидкой питательной среды (2,5% питательный бульон Oxoid №2) и выращивали при 37°С в течение 10 часов, а затем интенсивно перемешивали. 9 мл бактериального раствора штамма ТА98 центрифугировали (2000 × g, 10 минут) для удаления питательного раствора. Бактерии суспендировали в 9 мл буфера Micro F (K2HPO4: 3,5 г/л, KH2PO4: 1 г/л, (NH4)2SO4: 1 г/л, дегидрат цитрата тринатрия: 0,25 г/л, MgSO4 ⋅ 7H2O: 0,1 г/л), суспензию добавляли к 110 мл среды для проведения воздействия (буфер Micro F, содержащий биотин: 8 мкг/мл, гистидин: 0,2 мкг/мл, глюкозу: 8 мг/мл). 3,16 мл бактериального раствора штамма ТА100 добавляли к 120 мл среды для проведения воздействия для получения исследуемого бактериального раствора. В каждом из случаев смешивали 12 мкл раствора соединения раствора согласно настоящему изобретению в ДМСО (2-3-кратное разведение в несколько этапов, начиная с максимальной дозы 50 мг/мл), где ДМСО выступает в качестве отрицательного контроля, и 50 мкг/мл раствора 4-нитрохинолин-1-оксида в ДМСО в случае штамма ТА98, 0,25 мкг/мл раствора 2-(2-фурил)-3-(5-нитро-2-фурил)акриламида в ДМСО в случае штамма ТА100 в условиях неметаболического активирования, 40 мкг/мл раствора 2-аминоантрацена в ДМСО в случае штамма ТА98, 20 мкг/мл раствора 2-аминоантрацена в ДМСО в случае штамма ТА100 в условиях метаболического активирования, выступающего в качестве положительного контроля, и 588 мкл исследуемого бактериального раствора (смешанного раствора 498 мкл исследуемого бактериального раствора и 90 мкл смеси S9 в условиях метаболического активирования) и выращивали при 37°С в течение 90 минут при интенсивном перемешивании. 460 мкл бактериального раствора, который подвергали воздействию соединения согласно настоящему изобретению, смешивали с 2300 мкл индикаторной среды (буфер Micro F, содержащий биотин: 8 мкг/мл, гистидин: 0,2 мкг/мл, глюкозу: 8 мг/мл, бромкрезолового пурпурного: 37,5 мкг/мл), вносили в 48 лунок планшета в количестве 50 мкл/лунка и выращивали стационарным способом при 37°С в течение 3 дней. Так как содержимое лунки, содержащей бактерии, которые обладают способностью к пролиферации в результате мутации гена фермента, синтезирующего аминокислоту (гистидин), меняет цвет с пурпурного на желтый из-за изменения рН, для каждой дозы подсчитывали количество лунок, содержащих бактерии, способные к пролиферации, которые окрасились в желтый цвет, из 48 лунок и результат оценивали по сравнению с группой отрицательного контроля. (-) означает, что мутагенность отрицательна, и (+) означает, что мутагенность положительна.
(Результат)
Соединение III-2: (-)
[0115]
Пример исследования 8: Исследование hERG
Для оценки риска удлинения интервала QT на электрокардиограмме при использовании соединения согласно настоящему изобретению изучали действие соединения согласно настоящему изобретению на K+ ток замедленного выпрямления (IKr), который играет важную роль в процессе реполяризации желудочков, с применением клеток НЕК293, экспрессирующих ген специфических калиевых каналов сердца человека (hERG).
После фиксации на клетке мембранного потенциала -80 мВ при помощи метода локальной фиксации потенциала на целой клетке с применением автоматизированной системы фиксации потенциала (PatchXpress 7000A, Axon Instruments Inc.) регистрировали IKr, индуцированный импульсной стимуляцией деполяризации при +40 мВ в течение 2 секунд и последующей импульсной стимуляцией реполяризации при -50 мВ в течение 2 секунд. После стабилизации генерируемого тока внеклеточный раствор (NaCl: 135 ммоль/л, KCl: 5,4 ммоль/л, NaH2PO4: 0,3 ммоль/л, CaCl2⋅2H2O: 1,8 ммоль/л, MgCl2⋅6H2O: 1 ммоль/л, глюкоза: 10 ммоль/л, HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфокислота): 10 ммоль/л, рН=7,4), в котором в целевой концентрации растворено соединение согласно настоящему изобретению, наносили на клетки при комнатной температуре в течение 10 минут. Используя зарегистрированный IKr, на основе текущего значения мембранного потенциала покоя измеряли абсолютное значение пикового следового тока при помощи программного обеспечения для анализа (DataXpress ver. 1, Molecular Devices Corporation). Кроме того, рассчитывали % ингибирования в соответствии с пиковым следовым током до нанесения соединения согласно настоящему изобретению и сравнивали с группой, в которой применяли только носитель (0,1% раствор диметилсульфоксида), для оценки влияния соединения согласно настоящему изобретению на IKr.
(Результат) показан % ингибирования при концентрации исследуемого соединения от 0,3 до 10 мкМ.
Соединение III-2: 7,9%
[0116]
Пример исследования 9: Исследование растворимости
Растворимость соединения согласно настоящему изобретению определяли при добавлении 1% раствора ДМСО. Готовили 10 ммоль/л раствор соединения в ДМСО и 2 мкл раствора соединения согласно настоящему изобретению добавляли к 198 мкл раствора JP-1 (к 2,0 г хлорида натрия и 7,0 мл хлороводородной кислоты добавляли воду до 1000 мл) и раствора JP-2 (1 объем воды добавляли к 1 объема раствора, который содержит 3,40 г дигидрофосфата калия и 3,55 г безводного моногидрофосфата натрия, до 1000 мл), соответственно. Смесь интенсивно перемешивали в течение 1 часа при комнатной температуре и фильтровали. Фильтрат разбавляли в 10 раз раствором метанол/вода = 1/1 (об./об.) и концентрацию соединения в фильтрате измеряли путем ЖХ/МС или ТФЭ/МС методом абсолютной калибровки.
(Результат)
Соединение III-2: 42,2 мкмоль/л
[0117]
Пример исследования 10: Исследование растворимости порошка
Подходящие количества соединения согласно настоящему изобретению помещали в ампулы и в каждую ампулу добавляли 200 мкл JP-1-ой жидкости (к 2,0 г хлорида натрия и 7,0 мл хлороводородной кислоты добавляли воду до 1000 мл), JP-2-ой жидкости (добавляли воду к 500 мл фосфатно-буферного раствора с рН 6,8) и смеси 20 ммоль/л раствора таурохолата натрия (ТСА) и JP-2-ой жидкости (к 1,08 г ТСА во JP-2-ой жидкости добавляли JP-2-ую жидкость до 100 мл). После полного растворения соединения добавляли подходящее количество соединения. После интенсивного перемешивания в течение 1 часа при 37°С смесь фильтровали и к 100 мкл каждого из фильтратов (двойное разбавление) добавляли 100 мкл метанола. При необходимости проводили дополнительное разбавление. После подтверждения наличия или отсутствия в ампулах пузырьков воздуха и осадка ампулы интенсивно перемешивали с герметичной пробкой. Концентрацию соединения определяли путем ВЭЖХ методом абсолютной калибровки.
(Результат)
Соединение III-2: раствор JP-1; 7,1 мкг/мл, раствор JP-2; 4,4 мкг/мл, 20 ммоль/л раствор TCA/JP-2; 16,1 мкг/мл
[0118]
Пример исследования 11: Тест Эймса
Тест Эймса проводили с использованием бактерий сальмонеллы (Salmonella typhimurium) ТА98, ТА100, ТА1535 и ТА1537 и Escherichia coli WP2uvrA в качестве исследуемых штаммов после метаболической активации в способе предварительной инкубации или без нее для проверки наличия или отсутствия мутагенности у соединений согласно настоящему изобретению.
(Результат)
Соединение III-2: (-)
[0119]
Пример исследования 12: исследование гемолиза на свету
Соединение согласно настоящему изобретению растворяли с получением целевых концентраций и смешивали с 2,5 об. %/об. % суспензией красных кровяных клеток, полученных из дефибринированной крови овцы, в микропланшете в концентрациях от 0,0008 до 0,1 мас. %/об. %. Смеси подвергали воздействую 10 Дж/см2 дозы УФ-облучения в диапазоне длин волн от 290 до 400 нм, УФА и УФВ с применением ультрафиолетовых флуоресцентных ламп, ламп GL20SE и FL20S-BLB производства Sankyo Denki Co., Ltd. и Panasonic Corporation, соответственно. После завершения облучения смеси центрифугировали и надосадочные жидкости смесей собирали и помещали в микропланшет. Фототоксичность оценивали путем измерения поглощения в надосадочных жидкостях при длинах волн 540 нм и 630 нм. Значения поглощения при длинах волн 540 нм и 630 нм использовали в качестве индикаторов повреждения биомембраны (% фотогемолиза) и гипероксидирования липидной мембраны (образование метгемоглобина), соответственно. Применяли следующие критерии фототоксичности. Соединение считали нефототоксичным (-), если наблюдали % фотогемолиза <10 и максимальное изменение поглощения при 630 нм (ΔOD)<0,05. Соединение считали нефототоксичным (+), если фотогемолиз составлял более 10%, и максимальное изменение поглощения при 630 нм (ΔOD) составляло более 0,05.
(Результат)
Соединение III-2: (-)
[0120]
На фигурах 1 и 2 показаны результаты измерения концентрации соединения III-2 и соединения II-6 в плазме после перорального введения пролекарства соединения II-6, исходное соединение которого представляет собой соединение III-2, крысам после приема пищи.
Кроме того, концентрация соединения II-6 во всех образцах плазмы находилась на уровне предела определения или ниже. Таким образом, было установлено, что пролекарство соединения II-6, исходное соединение которого представляет собой соединение III-2, незамедлительно превращается в соединение III-2 после введения in vivo (см. фигуру 2).
[0121]
На основе представленных выше результатов исследований было обнаружено, что соединение, превращенное в пролекарство, абсорбируется в организме после перорального введения и быстро превращается в исходное соединение в крови. Таким образом, соединение согласно настоящему изобретению может являться подходящим агентом для лечения и/или предотвращения симптомов и/или заболеваний, вызываемых при инфицировании вирусом гриппа.
[0122]
Пример исследования 13: Исследование внутривенного введения
Материалы и способ для проведения экспериментов по исследованию внутривенного введения
(1) Экспериментальные животные: использовали крыс линии SD.
(2) Условия разведения: крысам линии SD обеспечивали свободный доступ к зерну и стерилизованной водопроводной воде.
(3) Режим дозирования и принцип разделения на группы: внутривенное введение проводили с применением заранее определенных дозировок. Разделение на группы осуществляли следующим образом. (Дозировку корректировали для каждого соединения)
Внутривенное введение: 0,5-1 мг/кг (n=2-3)
(4) Получение раствора для введения: внутривенное введение проводили после растворения.
(5) Способ введения: внутривенное введение проводили через хвостовую вену при помощи шприцев с иглой.
(6) Параметры оценки: кровь собирали последовательно и концентрацию соединения согласно настоящему изобретению в плазме измеряли путем ЖХ/МС/МС.
(7) Статистический анализ: для исследования изменений концентрации соединения согласно настоящему изобретению в плазме рассчитывали общий клиренс (CLtot) и период полуэлиминации (t1/2, z) при помощи программного обеспечения для применения нелинейного метода наименьших квадратов, WinNonlin (R).
(Результат)
Соединение № III-2:
CLtot: 16,4 мл/мин/кг
t1/2, z: 3,4 часов
На основе приведенных выше результатов было установлено, что соединение III-2 представляет собой соединение, имеющее низкий общий клиренс и длительный период полувыведения.
Таким образом, соединение согласно настоящему изобретению обладает отличной устойчивостью и может являться подходящим агентом для лечения и/или предотвращения симптомов и/или заболеваний, вызываемых при инфицировании вирусом гриппа.
[0123]
Пример лекарственной формы
Следующие примеры лекарственных форм приведены только в качестве примеров и не ограничивают объем настоящего изобретения. Пример лекарственной формы 1: Таблетки
Смешивали соединения согласно настоящему изобретению, лактозу и стеарат кальция. Смесь измельчали, гранулировали и высушивали с получением гранул подходящего размера. Затем к гранулам добавляли стеарат кальция и смесь спрессовывали и формовали с получением таблеток.
[0124]
Пример лекарственной формы 2: Капсулы
Равномерно смешивали соединения согласно настоящему изобретению, лактозу и стеарат кальция с получением порошковых лекарственных средств в виде порошков или мелких гранул. Порошковые лекарственные средства вносили в капсульные емкости с получением капсул.
[0125]
Пример лекарственной формы 3: Гранулы
Равномерно смешивали соединения согласно настоящему изобретению, лактозу и стеарат кальция и смесь спрессовывали и формовали. Затем смесь измельчали, гранулировали и просеивали с получением гранул подходящего размера.
[0126]
Пример лекарственной формы 4: Таблетки, распадающиеся в ротовой полости
Соединения согласно настоящему изобретению и кристаллическую целлюлозу смешивали и гранулировали с получением таблеток, распадающихся в ротовой полости.
[0127]
Пример лекарственной формы 5: Сухие сиропы
Соединения согласно настоящему изобретению и лактозу смешивали, измельчали, гранулировали и просеивали с получением сухих сиропов из частиц подходящего размера.
[0128]
Пример лекарственной формы 6: Составы для инъекции
Смешивали соединения согласно настоящему изобретению и фосфатный буфер с получением состава для инъекции.
[0129]
Пример лекарственной формы 7: Составы для инфузии
Смешивали соединения согласно настоящему изобретению и фосфатный буфер с получением состава для инъекции.
[0130]
Пример лекарственной формы 8: Составы для ингаляции
Соединения согласно настоящему изобретению и лактозу смешивали и мелко измельчали с получением составов для ингаляции.
[0131]
Пример лекарственной формы 9: Мази
Смешивали соединения согласно настоящему изобретению и вазелин с получением мазей.
[0132]
Пример лекарственной формы 10: Накладки
Объединяли соединения согласно настоящему изобретению и подложку, такую как лейкопластырь и т.п., с получением накладок. [Промышленная применимость]
[0133]
Соединение согласно настоящему изобретению после абсорбции в организме проявляет ингибирующую активность кэп-зависимой эндонуклеазы (CEN). Соединение согласно настоящему изобретению может являться подходящим агентом для лечения и/или предотвращения симптомов и/или заболеваний, вызываемых при инфицировании вирусом гриппа.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ ДЛЯ ЛЕЧЕНИЯ ГРИППА, ХАРАКТЕРИЗУЮЩИЙСЯ ТЕМ, ЧТО В НЕМ ОБЪЕДИНЕНЫ ИНГИБИТОР КЭП-ЗАВИСИМОЙ ЭНДОНУКЛЕАЗЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО ПРОТИВ ГРИППА | 2016 |
|
RU2745071C2 |
| ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ, СОДЕРЖАЩИЕ ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПИРИДОНОВЫЕ ПРОИЗВОДНЫЕ И ИХ ПРОЛЕКАРСТВО | 2017 |
|
RU2727962C1 |
| СЕЛЕКТИВНЫЕ ИНГИБИТОРЫ ЭСТРОГЕНОВЫХ РЕЦЕПТОРОВ И ИХ ПРИМЕНЕНИЕ | 2017 |
|
RU2734501C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2571662C2 |
| ПРОТИВОВИРУСНЫЕ СОЕДИНЕНИЯ И ИХ ПРИМЕНЕНИЯ | 2010 |
|
RU2552533C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2808166C2 |
| НЕЙРОАКТИВНЫЕ СТЕРОИДЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2014 |
|
RU2684103C2 |
| ПРОИЗВОДНЫЕ БОРОНОВОЙ КИСЛОТЫ И ИХ ТЕРАПЕВТИЧЕСКИЕ ПРИМЕНЕНИЯ | 2017 |
|
RU2773346C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛА, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ ПРОТЕИНКИНАЗЫ | 2001 |
|
RU2340611C2 |
| ОКСИСТЕРОЛЫ И СПОСОБЫ ИХ ПРИМЕНЕНИЯ | 2016 |
|
RU2744267C2 |

Изобретение относится к соединению, представленному формулой (I), или его фармацевтически приемлемой соли. В формуле (I) P представляет собой водород или группу PR, выбранную из следующих формул: a) -C(=O)-PR0, b) -C(=O)-PR1, g) -C(=O)-O-PR2, h) -C(=O)-N(-K)(PR2), i) -C(=O)-O-L-O-PR2, l) -C(PR3)2-O-C(=O)-PR4, m) -C(PR3)2-O-C(=O)-O-PR4, v) -C(PR3)2-PR6, y) -C(PR3)2-N(-K)-C(=O)-O-PR2, z) -P(=O)(-PR8)(-PR9) и ab) -PR11, A1 представляет собой CR1AR1B; A2 представляет собой CR2AR2B; A3 представляет собой CR3AR3B, S или O; каждый из A4 независимо представляет собой CR4AR4B; количество гетероатомов среди атомов, образующих кольцо, которое состоит из A1, A2, A3, A4, атома азота, смежного с A1, и атома углерода, смежного с A4, составляет 1 или 2; группа, представленная формулой:  , представляет собой группу, представленную формулой:
, представляет собой группу, представленную формулой:  , значения остальных радикалов указаны в формуле изобретения. Изобретение также относится к индивидуальным соединениям. Технический результат: получены новые соединения формулы (I), ингибирующие активность кэп-зависимой эндонуклеазы и которые могут применяться при лечении вируса гриппа. 4 н. и 7 з.п. ф-лы, 2 ил., 40 табл., 39 пр.
, значения остальных радикалов указаны в формуле изобретения. Изобретение также относится к индивидуальным соединениям. Технический результат: получены новые соединения формулы (I), ингибирующие активность кэп-зависимой эндонуклеазы и которые могут применяться при лечении вируса гриппа. 4 н. и 7 з.п. ф-лы, 2 ил., 40 табл., 39 пр.

1. Соединение, представленное формулой (I):
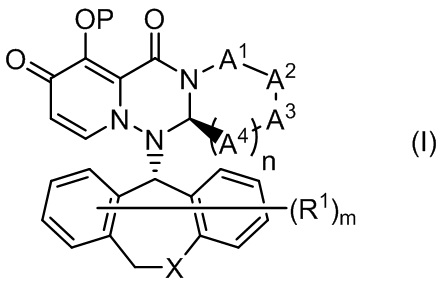 ,
,
или его фармацевтически приемлемая соль,
где
P представляет собой водород или группу PR, выбранную из следующих формул:
a) -C(=O)-PR0,
b) -C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
v) -C(PR3)2-PR6,
y) -C(PR3)2-N(-K)-C(=O)-O-PR2,
z) -P(=O)(-PR8)(-PR9) и
ab) -PR11, и
где L представляет собой линейный или разветвленный С1-6алкилен;
K представляет собой С1-6алкил;
PR0 представляет собой С1-6алкил, необязательно замещенный группой-заместителем A;
PR1 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем A, или гетероциклильную группу, необязательно замещенную группой-заместителем A;
PR2 представляет собой С1-6алкил, необязательно замещенный группой-заместителем A, карбоциклильную группу, необязательно замещенную группой-заместителем A, гетероциклильную группу, необязательно замещенную группой-заместителем A, или триС1-6алкилсилил;
каждый из PR3 независимо представляет собой водород или С1-6алкил;
каждый из PR4 независимо представляет собой С1-6алкил, необязательно замещенный группой-заместителем A, карбоциклильную группу, необязательно замещенную группой-заместителем A, или гетероциклильную группу, необязательно замещенную группой-заместителем A;
PR6 представляет собой карбоциклильную группу, необязательно замещенную группой-заместителем A, или гетероциклильную группу, необязательно замещенную группой-заместителем A;
PR8 представляет собой С1-6алкокси, необязательно замещенный группой-заместителем A;
PR9 представляет собой С1-6алкокси, необязательно замещенный группой-заместителем A, С1-6алкиламино, необязательно замещенный группой-заместителем A, гетероциклилокси, необязательно замещенный группой-заместителем A, или карбоциклиламино, необязательно замещенный группой-заместителем A;
PR11 представляет собой гетероциклильную группу, необязательно замещенную группой-заместителем A;
группа-заместитель A представляет собой оксогруппу, С1-6алкил, С1-6алкиламино, карбоциклильную группу, гетероциклильную группу, С1-6алкилкарбонил, гидрокси, С1-6алкилкарбониламино, С1-6алкилкарбонилокси, С1-6алкилоксикарбонил, С1-6алкилоксикарбонилС1-6алкил, С1-6алкиламинокарбонилокси, С1-6алкилокси, нитро, азидо или/и С1-6алкилсульфонил; и
где карбоциклильная группа означает карбоциклильную группу, имеющую от 3 до 6 атомов углерода, и
где гетероциклильная группа означает:
5- или 6-членную ароматическую гетероциклильную группу, содержащую в кольце 1-2 гетероатома, независимо выбранных из O, S и N; или
4-9-членную неароматическую гетероциклильную группу, содержащую в кольце 1-2 гетероатома, независимо выбранных из O, S и N; и
A1 представляет собой CR1AR1B;
A2 представляет собой CR2AR2B;
A3 представляет собой CR3AR3B, S или O;
каждый из A4 независимо представляет собой CR4AR4B;
количество гетероатомов среди атомов, образующих кольцо, которое состоит из A1, A2, A3, A4, атома азота, смежного с A1, и атома углерода, смежного с A4, составляет 1 или 2;
каждый из R1A и R1B независимо представляет собой водород или С1-6алкил;
каждый из R2A и R2B независимо представляет собой водород, галоген, С1-6алкил или галогенС1-6алкил;
каждый из R3A и R3B независимо представляет собой водород, галоген, С1-6алкил, галогенС1-6алкил или С1-6алкилокси;
каждый из R4A и R4B независимо представляет собой водород;
R3A и R3B совместно со смежным атомом углерода могут образовывать неароматический С3карбоцикл или 4-6-членный неароматический гетероцикл, содержащий 1-2 гетероатома, выбранных из О;
n представляет собой 1, и
где группа, представленная формулой:
 ,
,
представляет собой группу, представленную формулой:
 .
.
2. Соединение по п. 1, где группа, представленная формулой:
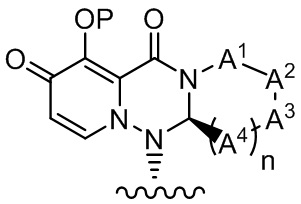 ,
,
представлена формулой:

 или
или  ,
,
где Р имеет такое же значение, как указано в п. 1, или его фармацевтически приемлемая соль.
3. Соединение по п. 1, представленное следующей формулой:

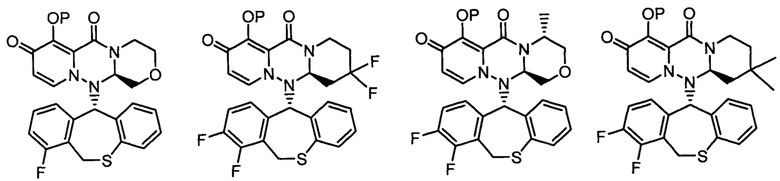
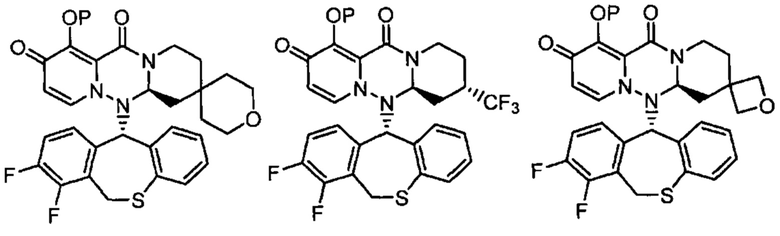



 или
или 
где P имеет такое же значение, как указано в п. 1, или его фармацевтически приемлемая соль.
4. Соединение по п. 1, представленное следующей формулой:
 или
или
где Р имеет такое же значение, как указано в п. 1, или его фармацевтически приемлемая соль.
5. Соединение по п. 1, представленное следующей формулой:
,
где Р имеет такое же значение, как указано в п. 1, или его фармацевтически приемлемая соль.
6. Соединение по п. 1, представленное следующей формулой:
,
где P имеет такое же значение, как указано в п. 1, или его фармацевтически приемлемая соль.
7. Соединение по п. 1, представленное следующей формулой:
 ,
,
где Р имеет такое же значение, как указано в п. 1, или его фармацевтически приемлемая соль.
8. Соединение по п. 1 или его фармацевтически приемлемая соль, где
PR представляет собой группу следующей формулы:
m) -C(PR3)2-O-C(=O)-O-PR4,
в которой
каждый PR3 независимо представляет собой водород или С1-6алкил; и
PR4 представляет собой С1-6алкил, необязательно замещенный группой-заместителем A, карбоциклил, необязательно замещенный группой-заместителем A, или гетероциклил, необязательно замещенный группой-заместителем A;
группа-заместитель A представляет собой оксогруппу, С1-6алкил, С1-6алкиламино, карбоциклильную группу, гетероциклильную группу, С1-6алкилкарбонил, гидрокси, С1-6алкилкарбониламино, С1-6алкилкарбонилокси, С1-6алкилоксикарбонил, С1-6алкилоксикарбонилС1-6алкил, С1-6алкиламинокарбонилокси, С1-6алкилокси, нитро, азидо или/и С1-6алкилсульфонил.
9. Соединение, представленное следующей формулой:
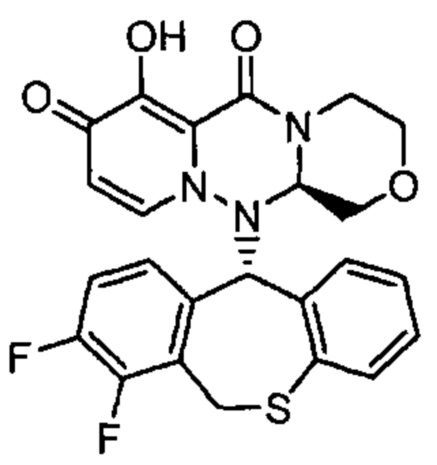 ,
,
или его фармацевтически приемлемая соль.
10. Соединение, представленное следующей формулой:
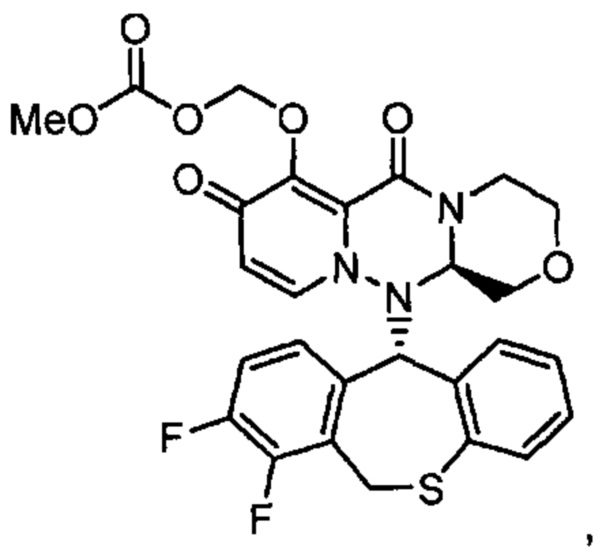 ,
,
или его фармацевтически приемлемая соль.
11. Соединение, представленное следующей формулой:
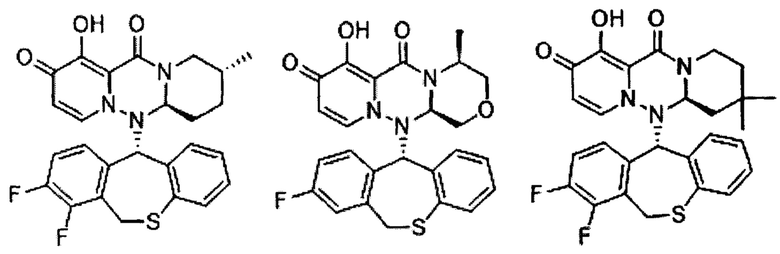 или ,
или ,
или его фармацевтически приемлемая соль.
| ДУШЕВОЕ УСТРОЙСТВО | 2013 |
|
RU2620436C2 |
| СПОСОБ РЕАЛИЗУЕМОГО В НЕПРЕРЫВНОМ РЕЖИМЕ ДЛИТЕЛЬНОГО ГЕТЕРОГЕННО КАТАЛИЗИРУЕМОГО ЧАСТИЧНОГО ДЕГИДРИРОВАНИЯ УГЛЕВОДОРОДОВ | 2007 |
|
RU2444400C2 |
| КОНДЕНСИРОВАННЫЕ ПРОИЗВОДНЫЕ ПИРИДАЗИНА И ЛЕКАРСТВЕННЫЕ ПРЕПАРАТЫ, СОДЕРЖАЩИЕ ДАННЫЕ СОЕДИНЕНИЯ В КАЧЕСТВЕ АКТИВНОГО ИНГРЕДИЕНТА | 2003 |
|
RU2292337C2 |
| WO 2006116764 A1, 02.11.2006 | |||
| WO 2014100323 A1, 26.06.2014 | |||
| ПРИМЕНЕНИЕ ПРОИЗВОДНОГО ГЛУТАРИМИДА ДЛЯ ТЕРАПИИ ЗАБОЛЕВАНИЙ, АССОЦИИРОВАННЫХ С АБЕРРАНТНОЙ АКТИВНОСТЬЮ ИНТЕРЛЕЙКИНА-6 | 2020 |
|
RU2774928C2 |

