ОБЛАСТЬ ТЕХНИКИ
[0001]
Настоящее изобретение относится к замещенным полициклическим производным пиридона, обладающим ингибирующей активностью в отношении кэп-зависимой эндонуклеазы, их пролекарствам и содержащим их фармацевтическим композициям.
УРОВЕНЬ ТЕХНИКИ
[0002]
Грипп представляет собой острое респираторное инфекционное заболевание, вызванное инфекцией вируса гриппа. В Японии каждую зиму сообщается о миллионах больных гриппом людей, при этом грипп сопровождается высокой заболеваемостью и смертностью. Грипп является особенно опасным для групп высокого риска, таких как младенцы и пожилые люди, у пожилых людей высока частота осложнений с пневмонией, и в данной группе часто встречаются смертельные случаи.
[0003]
В качестве препаратов против гриппа известны симметрел (торговое название Амантадин) и флумадин (торговое название Римантадин), которые ингибируют процесс денуклеации вируса, осельтамивир (торговое название: Тамифлю) и занамивир (торговое название: Реленза), которые являются ингибиторами нейраминидазы, подавляющими размножение и высвобождение вируса из клетки. Однако ввиду опасности появления устойчивых штаммов и возникновения побочных эффектов, а также всемирной эпидемии вируса гриппа нового типа, имеющего высокую патогенность и вызывающего высокую смертность, необходимо разработать препарат против гриппа, обладающий новым механизмом действия.
[0004]
Кэп-зависимая эндонуклеаза представляет собой фермент, полученный из вируса гриппа; она имеет важное значение для пролиферации вируса и обладает вирусоспецифической ферментативной активностью, которой не обладает хозяин, поэтому полагают, что указанная эндонуклеаза является подходящей в качестве мишени для лекарственного средства против гриппа. Кэп-зависимая эндонуклеаза вируса гриппа использует в качестве субстрата предшественник мРНК хозяина, и ее эндонуклеазная активность позволяет получать фрагмент из 9-13 оснований, включая кэп-структуру (не включая ряд оснований кэп-структуры). Этот фрагмент функционирует в качестве праймера вирусной РНК-полимеразы и используется для синтеза мРНК, кодирующей вирусный белок. Таким образом, считается, что вещество, которое ингибирует кэп-зависимую эндонуклеазу, ингибирует синтез вирусного белка путем ингибирования синтеза вирусной мРНК и в результате ингибирует пролиферацию вируса.
[0005]
В качестве веществ, которые ингибируют кэп-зависимую эндонуклеазу, описаны флутиимид (патентный документ 1 и непатентные документы 1 и 2), 4-замещенная 2,4-диоксобутановая кислота (патентный документ 2 и непатентные документы 3 и 4), соединения, описанные в патентных документах 3-12, и тому подобные, но указанные вещества еще не доведены до клинического применения в качестве лекарственных средств против гриппа. В патентных документах 9 и 12 описаны соединения, имеющие сходную с применяемыми в настоящем изобретении соединениями структуру, однако соединения, применяемые в настоящем изобретении, в указанных документах не описаны. Кроме того, в патентных документах 13-15 описаны соединения, имеющие структуру, аналогичную той, которая применяется в настоящем изобретении, в качестве соединений, обладающих ингибирующей активностью в отношении интегразы, однако в указанных документах кэп-зависимая эндонуклеаза не описана. Помимо этого, в патентных документах 16 и 17 описано изобретение, относящееся к соединениям, имеющим структуру, аналогичную той, которая применяется в настоящем изобретении, в качестве соединений, обладающих ингибирующей активностью в отношении кэп-зависимой эндонуклеазы, которое подано заявителями, но в них не описаны соединения, применяемые в настоящем изобретении.
ДОКУМЕНТЫ, ХАРАКТЕРИЗУЮЩИЕ ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
ПАТЕНТНЫЕ ДОКУМЕНТЫ
[0006]
Патентный документ 1: GB 2280435
Патентный документ 2: US 5475109
Патентный документ 3: US 20130090300
Патентный документ 4: WO 2013/057251
Патентный документ 5: WO 2013/174930
Патентный документ 6: WO 2014/023691
Патентный документ 7: WO 2014/043252
Патентный документ 8: WO 2014/074926
Патентный документ 9: WO 2014/108406
Патентный документ 10: WO 2014/108407
Патентный документ 11: WO 2014/108408
Патентный документ 12: WO 2015/038655
Патентный документ 13: WO 2005/016927
Патентный документ 14: WO 2006/066414
Патентный документ 15: WO 2007/049675
Патентный документ 16: WO 2010/147068
Патентный документ 17: WO 2012/039414
НЕПАТЕНТНЫЕ ДОКУМЕНТЫ
[0007]
Непатентный документ 1: Tetrahedron Lett 1995, 36(12), 2005
Непатентный документ 2: Tetrahedron Lett 1995, 36(12), 2009
Непатентный документ 3: Antimicrobial Agents And Chemotherapy, Dec. 1994, p. 2827-2837
Непатентный документ 4: Antimicrobial Agents And Chemotherapy, May 1996, p. 1304-1307
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
ЗАДАЧИ, РЕШАЕМЫЕ В НАСТОЯЩЕМ ИЗОБРЕТЕНИИ
[0008]
Задача настоящего изобретения состоит в том, чтобы обеспечить фармацевтическую композицию, содержащую соединения, обладающие противовирусной активностью, в частности ингибирующие рост активности вируса гриппа. Другая задача настоящего изобретения состоит в обеспечении фармацевтической композиции, содержащая пролекарство, полученное из соединений, применяемой для введения in vivo (например, для перорального введения), которая эффективно всасывается в организм после введения и демонстрирует высокий фармакологический эффект. Еще одна задача состоит в том, чтобы обеспечить фармацевтическую композицию, сокращающую время облегчения симптомов гриппа.
СРЕДСТВА ДЛЯ РЕШЕНИЯ УКАЗАННЫХ ЗАДАЧ
[0009]
Согласно настоящему изобретению предложены изобретения, представленные ниже.
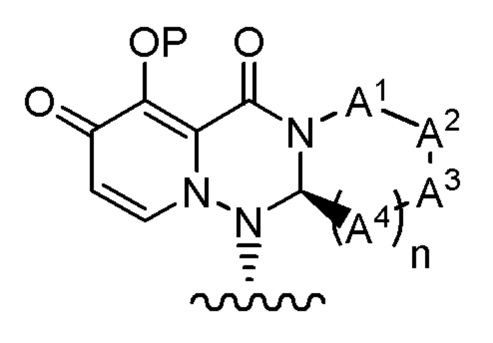
(1) Фармацевтическая композиция, содержащая соединение, представленное формулой (I), или его фармацевтически приемлемую соль:

где
Р представляет собой водород или группу для образования пролекарства;
А1 представляет собой CR1AR1B, S или О;
А2 представляет собой CR2AR2B, S или О;
А3 представляет собой CR3AR3B, S или О;
каждый А4 независимо представляет собой CR4AR4B, S или О;
число гетероатомов среди атомов, составляющих кольцо, которое состоит из А1, А2, А3, А4, атома азота, соседнего с А1, и атома углерода, соседнего с А4, равно 1 или 2;
каждый R1A и R1B независимо представляет собой водород, галоген, алкил, галогеналкил, алкилокси или фенил;
каждый R2A и R2B независимо представляет собой водород, галоген, алкил, галогеналкил, алкилокси или фенил;
каждый R3A и R3B независимо представляет собой водород, галоген, алкил, галогеналкил, алкилокси или фенил;
каждый R4A и R4B независимо представляет собой водород, галоген, алкил, галогеналкил, алкилокси или фенил;
R3A и R3B могут быть объединены с соседним атомом углерода с образованием неароматического карбоцикла или неароматического гетероцикла;
R1 представляет собой фтор;
m представляет собой целое число от 1 до 2; и
n представляет собой целое число от 1 до 2.
(2) Фармацевтическая композиция в соответствии с (1), содержащая соединение или его фармацевтически приемлемую соль, где указанная группа, представленная формулой:
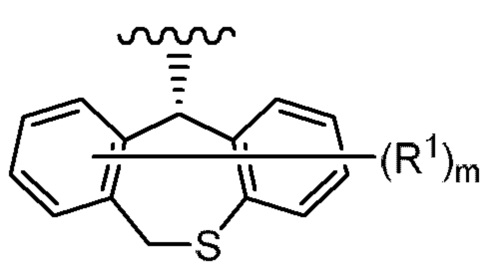
где каждая переменная имеет значения, описанные в (1),
представляет собой группу, представленную формулой:

где каждый R2, R3, R4 и R5 независимо представляет собой водород или фтор; число атомов фтора из числа R2, R3, R4 и R5 равно 1 или 2.
(3) Фармацевтическая композиция в соответствии с (1), содержащая соединение или его фармацевтически приемлемую соль, причем указанная группа, представленная формулой:

где каждая переменная имеет значения, описанные в (1),
представляет собой группу, представленную формулой:


(4) Фармацевтическая композиция в соответствии с любым из (1)-(3), содержащая соединение или его фармацевтически приемлемую соль, причем указанная группа представленная формулой:
где каждая переменная имеет значения, описанные в (1),
представляет собой группу, представленную формулой:

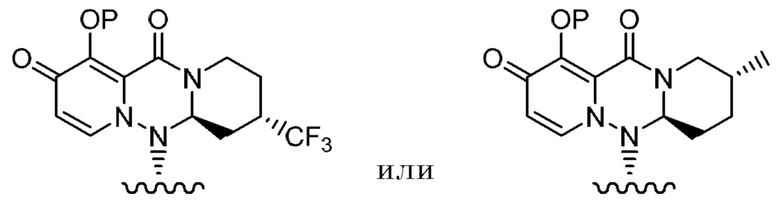
где каждая переменная имеет значения, описанные в (1).
(5) Фармацевтическая композиция в соответствии с (1), содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

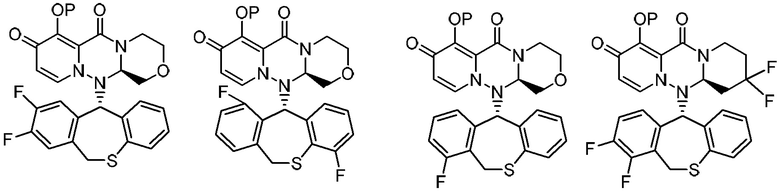
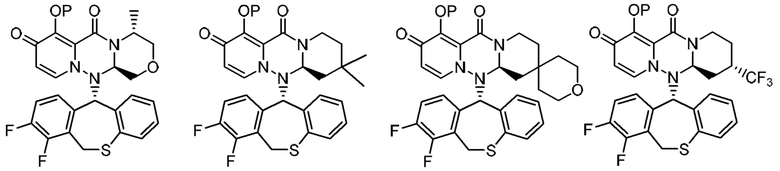



где каждая переменная имеет значения, описанные в (1).
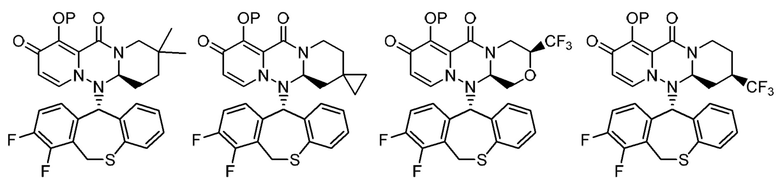
(6) Фармацевтическая композиция в соответствии с (1), содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

где каждая переменная имеет значения, описанные в (1).
(7) Фармацевтическая композиция в соответствии с (1), содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

где каждая переменная имеет значения, описанные в (1).
(8) Фармацевтическая композиция в соответствии с (1), содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

где каждая переменная имеет значения, описанные в (1).
(9) Фармацевтическая композиция в соответствии с (1), содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:
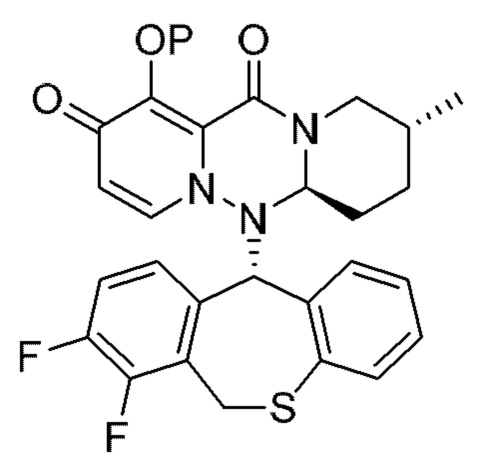
где каждая переменная имеет значения, описанные в (1).
(10) Фармацевтическая композиция в соответствии с (1), содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:
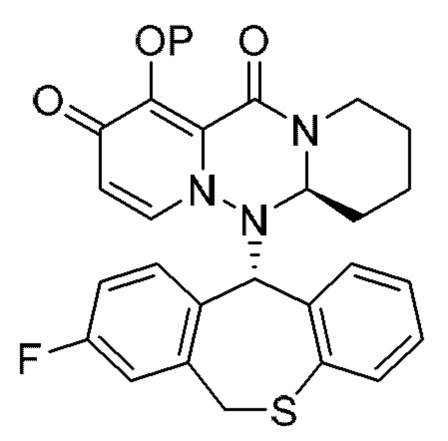
где каждая переменная имеет значение, описанное в (1).
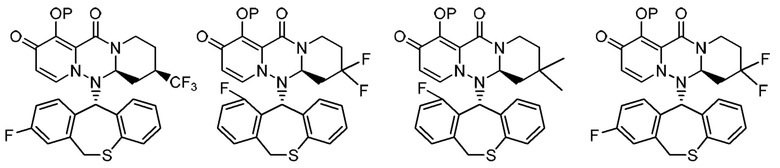
(11) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

где каждая переменная имеет значение, описанное в (1).
(12) Фармацевтическая композиция в соответствии с любым из (1)-(11), содержащая соединение или его фармацевтически приемлемую соль,
причем группа для образования пролекарства представляет собой группу, выбранную из следующей формулы а)-ас):
a) -C(=O)-PR0,
b) -C(=O)-PR1,
c) -C(=O)-L-PR1,
d) -C(=O)-L-O-PR1,
e) -C(=O)-L-O-L-O-PR1,
f) -C(=O)-L-O-C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
j) -C(PR3)2-O-PR4,
k) -C(PR3)2-O-L-O-PR4,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
n) -C(PR3)2-O-C(=O)-N(-K)-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
p) -C(PR3)2-O-C(=O)-O-L-N(PR4)2,
q) -C(PR3)2-O-C(=O)-N(-K)-L-O-PR4,
r) -C(PR3)2-O-C(=O)-N(-K)-L-N(PR4)2,
s) -C(PR3)2-O-C(=O)-O-L-O-L-O-PR4,
t) -C(PR3)2-O-C(=O)-O-L-N(-K)-C(=O)-PR4,
u) -C(PR3)2-O-P(=O)(-PR5)2,
v) -C(PR3)2-PR6,
w) -C(=N+(PR7)2)(-N(PR7)2),
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
у) -C(PR3)2-N(-K)-C(=O)-O-PR2,
z) -P(=O)(-PR8)(-PR9),
aa) -S(=O)2-PR10,
ab) -PR11 и
ac) -C(PR3)2-C(PR3)2-O-PR2,
где L представляет собой неразветвленный или разветвленный алкилен, или неразветвленный или разветвленный алкенилен;
K представляет собой водород или алкил, необязательно замещенный замещающей группой А;
PR0 представляет собой алкил, необязательно замещенный замещающей группой А, или алкенил, необязательно замещенный замещающей группой А;
PR1 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, или алкилсульфанил, необязательно замещенный замещающей группой А;
PR2 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, гетероциклилалкил, необязательно замещенный замещающей группой А, или триалкилсилил;
каждый PR3 независимо представляет собой водород или алкил;
каждый PR4 независимо представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, гетероциклилалкил, необязательно замещенный замещающей группой А, или триалкилсилил;
каждый PR5 независимо представляет собой гидрокси или OBn;
PR6 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
каждый PR7 независимо представляет собой алкил, необязательно замещенный замещающей группой А;
PR8 представляет собой алкилокси, необязательно замещенный замещающей группой А;
PR9 представляет собой алкилокси, необязательно замещенный замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилокси, необязательно замещенный замещающей группой А, гетероциклилокси, необязательно замещенный замещающей группой А, карбоциклиламино, необязательно замещенный замещающей группой А, или гетероциклиламино, необязательно замещенный замещающей группой А;
PR8 и PR9 могут быть объединены с соседним атомом фосфора с образованием гетероцикла, необязательно замещенного замещающей группой А;
PR10 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А или гетероциклилалкил, необязательно замещенный замещающей группой А; и
PR11 представляет собой алкил, необязательно замещенный замещающей группой А, алкенил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
Замещающая группа А; оксо, алкил, гидроксиалкил, амино, алкиламино, карбоциклическая группа, гетероциклическая группа, карбоциклилалкил, алкилкарбонил, галоген, гидрокси, карбокси, алкилкарбониламино, алкилкарбониламиноалкил, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкилоксикарбонилокси, алкиламинокарбонилокси, алкиламиноалкил, алкилокси, циано, нитро, азидо, алкилсульфонил, триалкилсилил и фосфо.
(13) Фармацевтическая композиция в соответствии с (12), содержащая соединение или его фармацевтически приемлемую соль,
причем указанная группа для образования пролекарства представляет собой группу, выбранную из следующей формулы:
a) -C(=O)-PR0,
b) -C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
v) -C(PR3)2-PR6,
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
y) -C(PR3)2-N(-K)-C(=O)-O-PR2 и
z) -P(=O)(-PR8)(-PR9),
где L представляет собой неразветвленный или разветвленный алкилен;
K представляет собой водород или алкил, необязательно замещенный замещающей группой А;
PR0 представляет собой алкил, необязательно замещенный замещающей группой А;
PR1 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR2 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, или гетероциклилалкил, необязательно замещенный замещающей группой А;
каждый PR3 независимо представляет собой водород или алкил;
PR4 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR6 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR8 представляет собой алкилокси, необязательно замещенный замещающей группой А;
PR9 представляет собой алкилокси, необязательно замещенный замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилокси, необязательно замещенный замещающей группой А, гетероциклилокси, необязательно замещенный замещающей группой А, карбоциклиламино, необязательно замещенный замещающей группой А, или гетероциклиламино, необязательно замещенный замещающей группой А; и
PR8 и PR9 могут быть объединены с соседним атомом фосфора с образованием гетероцикла, необязательно замещенного замещающей группой А;
Замещающая группа А; оксо, алкил, алкиламино, карбоциклическая группа, гетероциклическая группа, алкилкарбонил, галоген, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил и триалкилсилил. (13-1) Фармацевтическая композиция в соответствии с (12), содержащая соединение или его фармацевтически приемлемую соль,
причем указанная группа для образования пролекарства представляет собой группу, выбранную из следующей формулы:
a) -C(=O)-PR0,
b) -C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
y) -C(PR3)2-N(-K)-C(=O)-O-PR2 и
z) -P(=O)(-PR8)(-PR9),
где L представляет собой неразветвленный или разветвленный алкилен;
K представляет собой водород или алкил, необязательно замещенный замещающей группой А;
PR0 представляет собой алкил, необязательно замещенный замещающей группой А;
PR1 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR2 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, или гетероциклилалкил, необязательно замещенный замещающей группой А;
каждый PR3 независимо представляет собой водород или алкил;
PR4 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR6 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR8 представляет собой алкилокси, необязательно замещенный замещающей группой А;
PR9 представляет собой алкилокси, необязательно замещенный замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилокси, необязательно замещенный замещающей группой А, гетероциклилокси, необязательно замещенный замещающей группой А, карбоциклиламино, необязательно замещенный замещающей группой А или гетероциклиламино, необязательно замещенный замещающей группой А; и
PR8 и PR9 могут быть объединены с соседним атомом фосфора с образованием гетероцикла, необязательно замещенного замещающей группой А;
Замещающая группа А; оксо, алкил, алкиламино, карбоциклил, гетероциклил, алкилкарбонил, галоген, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил и триалкилсилил.
(14) Фармацевтическая композиция в соответствии с (12), содержащая соединение или его фармацевтически приемлемую соль,
причем указанная группа для образования пролекарства имеет следующую формулу:
m) -C(PR3)2-O-C(=O)-O-PR4
где каждый PR3 независимо представляет собой водород или алкил; и
PR4 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
Замещающая группа А; оксо, алкил, алкиламино, карбоциклическая группа, гетероциклическая группа, алкилкарбонил, галоген, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил и триалкилсилил.
(15) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:
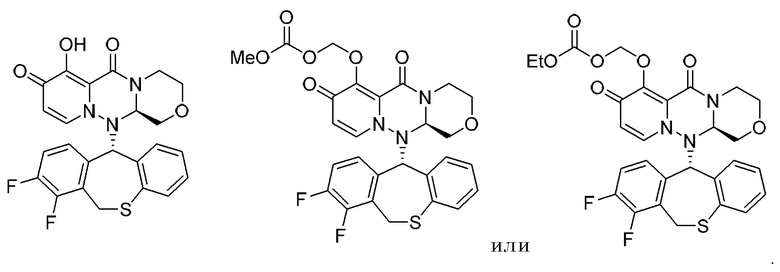
(16) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

(17) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

(18) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:
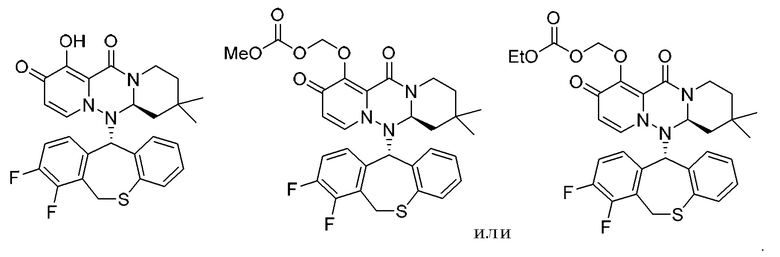
(19) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

(20) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:
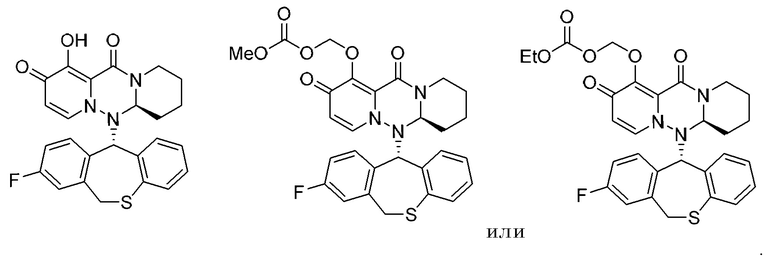
(21) Фармацевтическая композиция, содержащая соединение, представленное следующей формулой, или его фармацевтически приемлемую соль:

(22) Фармацевтическая композиция в соответствии с любым из (1)-(21), которая представляет собой противовирусный агент.
(23) Фармацевтическая композиция в соответствии с любым из (1)-(21), которая представляет собой ингибитор кэп-зависимой эндонуклеазы.
(24) Фармацевтическая композиция в соответствии с любым из (1)-(21), которая применяется для сокращения времени до облегчения симптомов гриппа.
(24-1) Способ сокращения времени до облегчения симптомов гриппа, характеризующийся введением соединения, любого из (1)-(21), или его фармацевтически приемлемой соли.
(24-2) Способ сокращения времени до облегчения симптомов гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа, характеризующийся введением соединения, любого из (1)-(21), или его фармацевтически приемлемой соли.
(24-3) Соединение, любое из (1)-(21), или его фармацевтически приемлемая соль для сокращения времени до облегчения симптомов гриппа.
(24-4) Соединение, любое из (1)-(21), или его фармацевтически приемлемая соль для сокращения времени до облегчения симптомов гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа.
(25) Фармацевтическая композиция в соответствии с любым из (1)-(21), которая применяется для ослабления воздействия вируса гриппа.
(25-1) Способ ослабления воздействия вируса гриппа, характеризующийся введением соединения, любого из (1)-(21), или его фармацевтически приемлемой соли.
(25-2) Способ ослабления воздействия вируса гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа, характеризующийся введением соединения, любого из (1)-(21), или его фармацевтически приемлемой соли.
(25-3) Соединение, любое из (1)-(21), или его фармацевтически приемлемая соль, для ослабления воздействия вируса гриппа.
(25-4) Соединение, любое из (1)-(21), или его фармацевтически приемлемая соль, для ослабления воздействия вируса гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа.
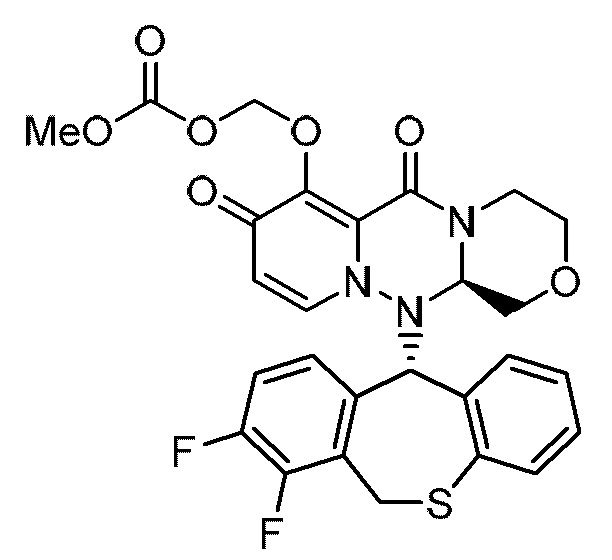
(26) Кристалл соединения следующей формулы:
(27) Кристалл в соответствии с (26), имеющий два или более пиков в дифракционных углах (2θ), выбранных из 8.6±0.2°, 14.1±0.2°, 17.4±0.2°, 20.0±0.2°, 24.0±0.2°, 26.3±0.2°, 29.6±0.2°, и 35.4±0.2° на рентгеновской порошковой дифрактограмме.
(28) Кристалл в соответствии с (26), имеющий пики в дифракционных углах (2θ): 8.6±0.2°, 14.1±0.2°, 17.4±0.2°, 20.0±0.2°, 24.0±0.2°, 26.3±0.2°, 29.6±0.2° и 35.4±0.2° на рентгеновской порошковой дифрактограмме.
(29) Кристалл в соответствии с (26), при этом рентгеновская порошковая дифрактограмма указанного кристалла по существу идентична такой, как показано на фигуре 3.
(30) Фармацевтическая композиция, содержащая кристалл, любой из (26)-(29).
(31) Фармацевтическая композиция в соответствии с (30), которая представляет собой противовирусный агент.
(32) Фармацевтическая композиция в соответствии с (30), которая представляет собой ингибитор кэп-зависимой эндонуклеазы.
(33) Фармацевтическая композиция, содержащая кристалл, любой из (26)-(29), которая используется для сокращения времени до облегчения симптомов гриппа.
(33-1) Способ сокращения времени до облегчения симптомов гриппа, характеризующийся введением кристалла, любого из (26)-(29).
(33-2) Способ сокращения времени до облегчения симптомов гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа, характеризующийся введением кристалла, любого из (26)-(29).
(33-3) Кристалл в соответствии с любым из (26)-(29) для сокращения времени до облегчения симптомов гриппа.
(33-4) Кристалл в соответствии с любым из (26)-(29) для сокращения времени до облегчения симптомов гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа.
(34) Фармацевтическая композиция, содержащая кристалл, любой из (26)-(29), который используется для ослабления воздействия вируса гриппа.
(34-1) Способ ослабления воздействия вируса гриппа, характеризующийся введением кристалла, любого из (26)-(29).
(34-2) Способ ослабления воздействия вируса гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа, характеризующийся введением кристалла, любого из (26)-(29).
(34-3) Кристалл в соответствии с любым из (26)-(29) для ослабления воздействия вируса гриппа.
(35) Фармацевтическая композиция в соответствии с любым из (1)-(21) и (30)-(34) для перорального введения.
(36) Фармацевтическая композиция в соответствии с (35), которая представляет собой таблетку, порошок, гранулу, капсулу, пилюлю, пленку, суспензию, эмульсию, эликсир, сироп, лимонад, эссенцию, ароматическую воду, экстракт, отвар или настойку.
(37) Фармацевтическая композиция в соответствии с (35), которая представляет собой таблетку с сахарным покрытием, таблетку с пленочным покрытием, таблетку, покрытую кишечнорастворимой оболочкой, таблетку с замедленным высвобождением, таблетку-пастилку, подъязычную таблетку, защечную таблетку, жевательную таблетку, таблетку для рассасывания, сухой сироп, мягкую капсулу, микрокапсулу или капсулу с замедленным высвобождением.
(38) Фармацевтическая композиция в соответствии с любым из (1)-(21) и (30)-(34) для парентерального введения.
(39) Фармацевтическая композиция в соответствии с (38) для накожного, подкожного, внутривенного, внутриартериального, внутримышечного, внутрибрюшинного, чресслизистого, ингаляционного, трансназального, офтальмологического, внутриушного или вагинального введения.
(40) Фармацевтическая композиция в соответствии с (38) или (39), которая представляет собой инъекцию, инфузию, глазные капли, капли в нос, ушные капли, аэрозоль, ингаляцию, лосьон, пропитку, линимент, жидкость для полоскания рта, клизму, мазь, пластырь, желе, крем, пластырь, припарку, порошок для наружного применения или суппозиторий.
(41) Фармацевтическая композиция в соответствии с любым из (1)-(21) и (30)-(34) для педиатрического или гериатрического пациента.
(42) Фармацевтическая композиция, состоящая из комбинации фармацевтической композиции в соответствии с любым из (1)-(21) и (30)-(34) и ингибитора нейраминидазы, ингибитора РНК-зависимой РНК-полимеразы, ингибитора белка М2, РВ2 кэп-связанного ингибитора, анти-НА-антитела или иммунологического агента.
(43) Фармацевтическая композиция, содержащая фармацевтическую композицию в соответствии с любым из (1)-(21) и (30)-(34) для комбинированной терапии с ингибитором нейраминидазы, ингибитором РНК-зависимой РНК-полимеразы, ингибитором белка М2, РВ2 кэп-связанным ингибитором, анти-НА-антителом или иммунологическим агентом.
Следует отметить, что пункт (13) и выше охватывает пункт (13-1), представленный выше.
[0010]
В настоящем изобретении предложен способ лечения или предупреждении инфекционного заболевания вируса гриппа с применением соединений (исходных соединений и/или пролекарственных соединений), применяемых в настоящем изобретении, и фармацевтических композиций, применяемых в настоящем изобретении. Исходные соединения эффективны в качестве противогриппозных агентов или промежуточных соединений для пролекарственных соединений.
РЕЗУЛЬТАТ ИЗОБРЕТЕНИЯ
[0011]
Соединения (исходные соединения и/или пролекарства), применяемые в настоящем изобретении, обладают ингибирующей активностью в отношении кэп-зависимой эндонуклеазы. Более предпочтительным соединением является пролекарство, преобразующееся в исходное соединение, обладающее ингибирующей активностью в отношении кэп-зависимой эндонуклеазы in vivo после введения, таким образом, указанное пролекарство является эффективным в качестве терапевтического агента и/или предотвращающего агента против инфекционного заболевания вируса гриппа.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
[0012]
[Фигура 1] На фигуре 1 представлен результат измерения концентрации в плазме соединения III-2 после перорального введения крысе не натощак пролекарственного соединения II-6, исходное соединение которого представляет
собой соединение III-2.
[Фигура 2] На фигуре 2 представлен результат измерения концентрации в плазме соединения II-6 после перорального введения крысе не натощак пролекарственного соединения II-6, исходное соединение которого представляет собой соединение III-2.
[Фигура 3] На фигуре 3 представлены рентгеновские порошковые дифрактограммы кристаллов формы I соединения II-6. По горизонтальной оси отложены углы в градусах 2θ, а по вертикальной оси - интенсивность.
[Фигура 4] На фигуре 4 представлены рентгеновские порошковые дифрактограммы кристаллов формы II соединения II-6.
[Фигура 5] На фигуре 5 представлены рентгеновские порошковые дифрактограммы кристаллов формы III соединения II-6.
НАИЛУЧШИЙ ВАРИАНТ ОСУЩЕСТВЛЕНИЯ НАСТОЯЩЕГО ИЗОБРЕТЕНИЯ
[0013]
Значение каждого термина, применяемого в настоящем описании, объясняется ниже. Каждый термин применяется в унифицированном смысле и применяется в одном и том же смысле при применении отдельно или при применении в сочетании с другим термином.
Термин «состоящий из» означает включающий только компоненты.
Термин «содержащий» означает, что нет ограничения в отношении компонентов и нет исключения неописанных факторов.
[0014]
«Необязательно замещенный замещающей группой А» означает, что произвольное положение может быть замещено одним, двумя или более одинаковыми или различными заместителями, выбранными из замещающей группы А.
[0015]
«Пролекарство» в настоящем описании относится к соединению, представленному формулой (II), в следующей реакционной формуле:

где каждый символ имеет значение, указанное выше,
или его фармацевтически приемлемой соли, и означает соединение, демонстрирующее ингибирующую активность в отношении кэп-зависимой эндонуклеазы (CEN) и/или эффект по подавлению СРЕ (цитопатического эффекта) путем превращения в соединение, представленное формулой (III), реакцией разложения, обусловленной ферментами, метаболизирующими лекарственное средство, гидролазами, желудочными кислотами, энтеробактериями и т.д. в физиологических условиях in vivo.
Пролекарство более предпочтительно означает соединение, у которого биодоступность и/или AUC (площадь под кривой концентрации в крови) при введении in vivo значительно больше, чем у соединения, представленного формулой (III).
Следовательно, пролекарство эффективно всасывается в организм в желудке и/или кишечнике после введения in vivo (например, пероральное введение), затем превращается в соединение, представленное формулой (III). Таким образом, пролекарство предпочтительно проявляет эффект лечения и/или предотвращения гриппа выше, чем соединение, представленное формулой (III).
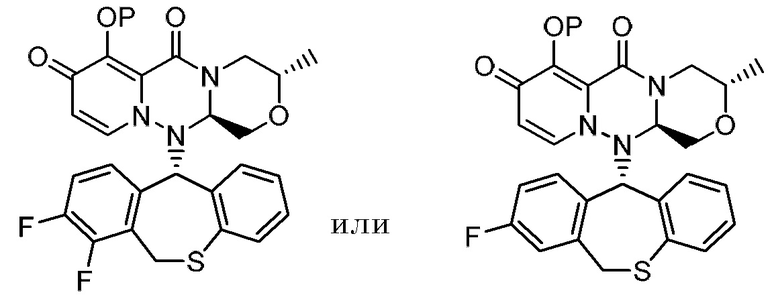
В одном из вариантов реализации «группа, представленная
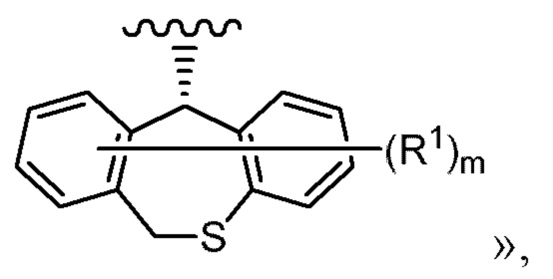
где каждая переменная имеет значение, описанное в (1), представляет собой группу, представленную формулой:

где каждый R2, R3, R4 и R5 независимо представляет собой водород или фтор; число атомов фтора из числа R2, R3, R4 и R5 равно 1 или 2.
Другой вариант реализации представляет собой группу, представленную формулой:
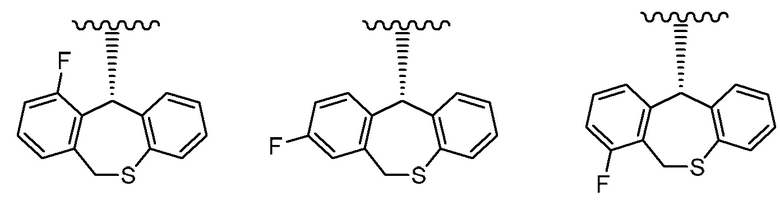
, и предпочтительно группу, представленную формулой:

, и особенно предпочтительно группу, представленную формулой:

[0016]
«Группа для образования пролекарства» в настоящем описании относится к «PR» группе в формуле (II) в следующей реакционной формуле:

[0017]
где каждый символ имеет значение, указанное выше,
и -OPR группа преобразуется в -ОН группу в формуле (III) реакцией разложения, вызванной ферментами, метаболизирующими лекарственное средство, гидролазами, желудочными кислотами, энтеробактериями и т.д. в физиологических условиях in vivo.
Термин «группа для образования пролекарства» более предпочтительно означает группу, которая улучшает биодоступность и/или AUC (площадь под кривой
концентрации в крови) соединения, представленного формулой (III), и присоединена
к соединению, представленному формулой (III).
[0018]
Примеры группы для образования пролекарства включают группы, описанные в Prog. Med. 5: 2157-2161 (1985) и в предоставленном Британской библиотекой - «The world's Knowledge".
«Группа для образования пролекарства» в группе -OPR в формуле (I) или (II) может представлять собой группу, преобразующуюся в -ОН группу in vivo, и примеры предпочтительно включают в себя группу, выбранную из следующих формул а)-ас).
a) -C(=O)-PR0,
b) -C(=O)-PR1,
c) -C(=O)-L-PR1,
d) -C(=O)-L-O-PR1,
e) -C(=O)-L-O-L-O-PR1,
f) -C(=O)-L-O-C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
j) -C(PR3)2-O-PR4,
k) -C(PR3)2-O-L-O-PR4,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
n) -C(PR3)2-O-C(=O)-N(-K)-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
p) -C(PR3)2-O-C(=O)-O-L-N(PR4)2,
q) -C(PR3)2-O-C(=O)-N(-K)-L-O-PR4,
r) -C(PR3)2-O-C(=O)-N(-K)-L-N(PR4)2,
s) -C(PR3)2-O-C(=O)-O-L-O-L-O-PR4,
t) -C(PR3)2-O-C(=O)-O-L-N(-K)-C(=O)-PR4,
u) -C(PR3)2-O-P(=O)(-PR5)2,
v) -C(PR3)2-PR6,
w) -C(=N+(PR7)2)(-N(PR7)2),
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
у) -C(PR3)2-N(-K)-C(=O)-O-PR2,
z) -P(=O)(-PR8)(-PR9),
aa) -S(=O)2-PR10,
ab) -PR11 и
ac) -C(PR3)2-C(PR3)2-O-PR2,
где L представляет собой неразветвленный или разветвленный алкилен, или неразветвленный или разветвленный алкенилен;
K представляет собой водород или алкил, необязательно замещенный замещающей группой А;
PR0 представляет собой алкил, необязательно замещенный замещающей группой А, или алкенил, необязательно замещенный замещающей группой А;
PR1 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, или алкилсульфанил, необязательно замещенный замещающей группой А;
PR2 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, гетероциклилалкил, необязательно замещенный замещающей группой А или триалкилсилил;
каждый PR3 независимо представляет собой водород или алкил;
каждый PR4 независимо представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, гетероциклилалкил, необязательно замещенный замещающей группой А, или триалкилсилил;
каждый PR5 независимо представляет собой гидрокси или OBn;
PR6 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
каждый PR7 независимо представляет собой алкил, необязательно замещенный замещающей группой А;
PR8 представляет собой алкилокси, необязательно замещенный замещающей группой А;
PR9 представляет собой алкилокси, необязательно замещенный замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилокси, необязательно замещенный замещающей группой А, гетероциклилокси, необязательно замещенный замещающей группой А, карбоциклиламино, необязательно замещенный замещающей группой А или гетероциклиламино, необязательно замещенный замещающей группой А;
PR8 и PR9 могут быть объединены с соседним атомом фосфора с образованием гетероцикла, необязательно замещенного замещающей группой А;
PR10 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А или гетероциклилалкил, необязательно замещенный замещающей группой А; и
PR11 представляет собой алкил, необязательно замещенный замещающей группой А, алкенил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
Замещающая группа А; оксо, алкил, гидроксиалкил, амино, алкиламино, карбоциклил, гетероциклил, карбоциклилалкил, алкилкарбонил, галоген, гидрокси, карбокси, алкилкарбониламино, алкилкарбониламиноалкил, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкилоксикарбонилокси, алкиламинокарбонилокси, алкиламиноалкил, алкилокси, циано, нитро, азидо, алкилсульфонил, триалкилсилил и фосфо.
[0019]
Термин «преобразующуюся в пролекарство» в настоящем описании означает, что гидроксигруппа в формуле (III) или ее фармацевтически приемлемая соль преобразуется в -OPR группу, как представлено на следующей схеме реакции:

[0020]
где каждый символ имеет значение, указанное выше.
[0021]
Термин «исходное соединение» в настоящем описании означает соединение, которое представляет собой источник до синтеза «пролекарства», и/или соединение, высвобождаемое из «пролекарства» путем реакции с ферментами, желудочной кислотой и т.п. в физиологических условиях in vivo, и в частности означает соединение, представленное формулой (III), или его фармацевтически приемлемую соль, или его сольват.
[0022]
Термин «галоген» включает атом фтора, атом хлора, атом брома и атом иода. Атом фтора и атом хлора особенно предпочтительны.
[0023]
Термин «алкил» включает линейную или разветвленную углеводородную группу С1-С15, предпочтительно С1-С10, более предпочтительно С1-С6 и еще более предпочтительно С1-С4. Примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил, изопентил, неопентил, н-гексил, изогексил, н-гептил, изогептил, н-октил, изооктил, н-нонил, н-децил и тому подобное.
В предпочтительном варианте реализации «алкил» представляет собой метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил или н-пентил. В более предпочтительном варианте реализации - представляет собой метил, этил, н-пропил, изопропил или трет-бутил.
[0024]
Термин «алкенил» включает линейную или разветвленную углеводородную группу С2-С15, предпочтительно С2-С10, более предпочтительно С2-С6 и еще более предпочтительно С2-С4, имеющую одну или более двойную связь(и) при любом положении(ях). Примеры включают винил, аллил, пропенил, изопропенил, бутенил, изобутенил, пренил, бутадиенил, пентенил, изопентенил, пентадиенил, гексенил, изогексенил, гексадиенил, гептенил, октенил, ноненил, деценил, ундеценил, додеценил, тридеценил, тетрадеценил, пентадеценил и тому подобное.
В предпочтительном варианте реализации «алкенил» представляет собой винил, аллил, пропенил, изопропенил или бутенил.
[0025]
Термин «алкилен» включает линейную или разветвленную бивалентную углеводородную группу С1-С15, предпочтительно С1-С10, более предпочтительно С1-С6 и еще более предпочтительно С1-С4. Примеры включают метилен, этилен, триметилен, пропилен, тетраметилен, пентаметилен, гексаметилен и тому подобное.
[0026]
Термин «алкенилен» включает линейную или разветвленную бивалентную углеводородную группу С2-С15, предпочтительно С2-С10, более предпочтительно С2-С6 и еще более предпочтительно С2-С4, имеющую одну или более двойную связь(и) при любом положении(ях). Примеры включают винилен, пренилен, бутенилен, пентенилен и тому подобное.
[0027]
Термин «гидроксиалкил» означает группу, где одна или более гидроксильная группа(ы) заменяет атом(ы) водорода, присоединенный(ые) к атому(ам) углерода указанного выше «алкила». Примеры включают гидроксиметил, 1-гидроксиэтил, 2-гидроксиэтил, 1-гидроксипропил, 2-гидроксипропил, 1,2-гидроксиэтил и тому подобное.
В предпочтительном варианте реализации «гидроксиалкил» представляет собой гидроксиметил.
[0028]
Термин «алкилокси» означает группу, где указанный выше «алкил» связан с атомом кислорода. Примеры включают метилокси, этилокси, n-пропилокси, изопропилокси, н-бутилокси, трет-бутилокси, изобутилокси, втор-бутилокси, пентилокси, изопентилокси, гексилокси и тому подобное.
В предпочтительном варианте реализации «алкилокси» представляет собой метилокси, этилокси, н-пропилокси, изопропилокси или трет-бутилокси.
[0029]
Термин «галогеналкил» означает группу, где один или более «галоген», описанный выше, связан с указанным выше «алкилом». Примеры включают монофторметил, монофторэтил, монофторпропил, 2,2,3,3,3-пентафторпропил, монохлорметил, трифторметил, трихлорметил, 2,2,2-трифторэтил, 2,2,2-трихлорэтил, 1,2-дибромэтил, 1,1,1-трифторпропан-2-ил и тому подобное.
В предпочтительном варианте реализации «галогеналкил» представляет собой трифторметил или трихлорметил.
[0030]
Термин «алкилкарбонил» означает группу, где указанный выше «алкил» связан с карбонильной группой. Примеры включают метилкарбонил, этилкарбонил, пропилкарбонил, изопропилкарбонил, трет-бутилкарбонил, изобутилкарбонил, втор-бутилкарбонил, пентилкарбонил, изопентилкарбонил, гексилкарбонил и тому подобное.
В предпочтительном варианте реализации «алкилкарбонил» представляет собой метилкарбонил, этилкарбонил или н-пропилкарбонил.
[0031]
Термин «алкиламино» означает группу, где один или два атома водорода, присоединенные к атому азота в аминогруппе, заменены посредством указанного выше «алкила». Две алкильные группы могут быть одинаковые или разные. Примеры включают метиламино, этиламино, изопропиламино, диметиламино, диэтиламино, N,N-диизопропиламино, N-метил-N-этиламино, N-изопропил-N-этиламино и тому подобное.
В предпочтительном варианте реализации «алкиламино» представляет собой метиламино, этиламино, диметиламино или диэтиламино.
[0032]
Термин «алкиламиноалкил» означает группу, где указанный выше «алкиламино» связан с указанным выше «алкилом».
[0033]
Термин «алкиламинокарбонил» означает группу, где указанный выше «алкиламино» связан с карбонильной группой.
[0034]
Термин «алкиламинокарбонилокси» означает группу, где указанный выше «алкиламинокарбонил» связан с атомом кислорода.
[0035]
Термин «алкилкарбониламино» означает группу, где указанный выше «алкилкарбонил» заменяет атом водорода, связанный с атомом азота аминогруппы. Примеры включают метилкарбониламино, этилкарбониламино, пропилкарбониламино, изопропилкарбониламино, трет-бутилкарбониламино, изобутилкарбониламино, втор-бутилкарбониламино и тому подобное.
В предпочтительном варианте реализации «алкилкарбониламино» представляет собой метилкарбониламино или этилкарбониламино.
[0036]
Термин «алкилкарбонилокси» означает группу, где указанный выше «алкилкарбонил» связан с атомом кислорода. Примеры включают метилкарбонилокси, этилкарбонилокси, пропилкарбонилокси, изопропилкарбонилокси, трет-бутилкарбонилокси, изобутилкарбонилокси, втор-бутилкарбонилокси и тому подобное.
В предпочтительном варианте реализации «алкилкарбонилокси» представляет собой метилкарбонилокси или этилкарбонилокси.
[0037]
Термин «алкилкарбониламиноалкил» означает группу, где указанный выше «алкилкарбониламино» связан с указанным выше «алкилом».
[0038]
Термин «алкилоксикарбонил» означает группу, где указанный выше «алкилокси» связан с карбонильной группой. Примеры включают метилоксикарбонил, этилоксикарбонил, пропилоксикарбонил, изопропилоксикарбонил, трет-бутилоксикарбонил, изобутилоксикарбонил, втор-бутилоксикарбонил, пентилоксикарбонил, изопентилоксикарбонил, гексилоксикарбонил и тому подобное.
В предпочтительном варианте реализации «алкилоксикарбонил» представляет собой метилоксикарбонил, этилоксикарбонил или пропилоксикарбонил.
[0039]
Термин «алкилоксикарбонилалкил» означает группу, где указанный выше «алкилоксикарбонил» связан с указанным выше «алкилом».
[0040]
Термин «алкилоксикарбонилокси» означает группу, где указанный выше «алкилоксикарбонил» связан с атомом кислорода.
[0041]
Термин «алкилсульфанил» означает группу, где указанный выше «алкил» заменяет атом водорода, связанный с атомом серы сульфанильной группы. Примеры включают метилсульфанил, этилсульфанил, н-пропилсульфанил, изопропилсульфанил и тому подобное.
[0042]
Термин «алкилсульфонил» означает группу, где указанный выше «алкил» связан с сульфонильной группой. Примеры включают метилсульфонил, этилсульфонил, пропилсульфонил, изопропилсульфонил, трет-бутилсульфонил, изобутилсульфонил, втор-бутилсульфонил и тому подобное.
В предпочтительном варианте реализации «алкилсульфонил» представляет собой метилсульфонил или этилсульфонил.
[0043]
Термин «триалкилсилил» означает группу, где три указанных выше «алкила» связаны с атомом кремния. Три алкильные группы могут быть одинаковые или разные. Примеры включают триметилсилил, триэтилсилил, трет-бутилдиметилсилил и тому подобное.
[0044]
Термин «карбоциклическая группа» означает циклическую углеводородную группу С3-С20, предпочтительно С3-С16, более предпочтительно С4-С12, и включает ароматический карбоциклил и неароматический карбоциклил.
Термин «ароматический карбоциклил» означает циклическую ароматическую углеводородную группу, которая представляет собой моноцикл или полицикл, имеющий два или более колец. Примеры включают фенил, нафтил, антрил, фенантрил и тому подобное.
В предпочтительном варианте реализации «ароматический карбоциклил» представляет собой фенил, 1-нафтил или 2-нафтил. В другом варианте реализации «ароматический карбоциклил» представляет собой фенил,
Термин «неароматический карбоциклил» означает циклическую насыщенную углеводородную группу или циклическую ненасыщенную неароматическую углеводородную группу, которая представляет собой моноцикл или полицикл, имеющий два или более колец. Примеры «неароматического карбоциклила», который представляет собой полицикл, имеющий два или более колец, включают конденсированную циклическую группу, при этом неароматический карбоциклил, который представляет собой моноцикл или полицикл, имеющий два или более колец, конденсирован с кольцом указанного выше «ароматического карбоциклила».
Помимо этого, примеры «неароматического карбоциклила» также включают группу, имеющую мостик, или группу с образованием спирокольца следующим образом:
Неароматический карбоциклил, который представляет собой моноцикл, предпочтительно представляет собой карбоциклил С3-С16, более предпочтительно С3-С12 и еще более предпочтительно С3-С8. Примеры включают циклопропил, циклобутил, циклопентил, циклогексил, циклогептил, циклооктил, циклононил, циклодецил, циклопропенил, циклобутенил, циклопентенил, циклогексенил, циклогептенил, циклогексадиенил и тому подобное.
Примеры неароматического карбоциклила, который представляет собой полицикл, имеющий два или более колец, включают инданил, инденил, аценафтил, тетрагидронафтил, флуоренил и тому подобное.
[0045]
Термин «карбоцикл» означает циклический углеводород С3-С20, предпочтительно С3-С16, более предпочтительно С4-С12 и включает ароматический карбоцикл и неароматический карбоцикл.
Термин «ароматический карбоцикл» означает циклический ароматический углеводород, который представляет собой моноцикл или полицикл, имеющий два или более колец. Примеры включают бензольное кольцо, нафталиновое кольцо, антраценовое кольцо, фенантреновое кольцо и тому подобное.
Примерами предпочтительных вариантов реализации «ароматического карбоцикла» являются бензольное кольцо и нафталиновое кольцо. В другом варианте реализации «ароматический карбоцикл» представляет собой бензольное кольцо.
Термин «неароматический карбоцикл» означает насыщенный карбоцикл или ненасыщенный неароматический карбоцикл, который представляет собой моноцикл или полицикл, имеющий два или более колец. Примеры «неароматического карбоцикла», который представляет собой полицикл, имеющий два или более колец, включают конденсированное кольцо, при этом неароматический карбоцикл, который представляет собой моноцикл или полицикл, имеющий два или более колец, конденсирован с кольцом указанного выше «ароматического карбоцикла».
Помимо этого, примеры «неароматического карбоцикла» также включают цикл, имеющий мостик, или цикл с образованием спирокольца следующим образом:

Неароматический карбоцикл, который представляет собой моноцикл, предпочтительно представляет собой карбоцикл С3-С16, более предпочтительно С3-С12 и еще более предпочтительно С3-С8. Примеры включают циклопропан, циклобутан, циклопентан, циклогексан, циклогептан, циклооктан, циклононан, циклодекан, циклопропен, циклобутен, циклопентен, циклогексен, циклогептен, циклогексадиен и тому подобное.
Примеры неароматического карбоцикла, который представляет собой полицикл, имеющий два или более кольца, включают индан, инден, аценафтален, тетрагидронафтален, флуорин и тому подобное.
[0046]
Термин «гетероциклическая группа» включает ароматический циклил и неароматический гетероциклил, который содержит один или более гетероатом(ов), независимо выбранный(х) из О, S и N.
Термин «ароматический гетероциклил» означает ароматический циклил, который представляет собой моноцикл или полицикл, имеющий два или более колец, содержащий один или более гетероатом(ов), независимо выбранный(х) из О, S и N.
Примеры «ароматического гетероциклила», который представляет собой полицикл, имеющий два или более кольца, включают конденсированную кольцевую группу, при этом ароматический гетероциклил, который представляет собой моноцикл или полицикл, имеющий два или более кольца, конденсирован с кольцом указанного выше «ароматического карбоциклила».
Ароматический гетероциклил, который представляет собой моноцикл, предпочтительно представляет собой 5-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают пирролил, имидазолил, пиразолил, пиридил, пиридазинил, пиримидинил, пиразинил, триазолил, триазинил, тетразолил, фурил, тиенил, изоксазолил, оксазолил, оксадиазолил, изотиазолил, тиазолил, тиадиазолил и тому подобное.
Примеры ароматического гетероциклила, который представляет собой бицикл, включают индолил, изоиндолил, индазолил, индолизинил, хинолинил, изохинолинил, циннолинил, фталазинил, хиназолинил, нафтиридинил, хиноксалинил, пуринил, птеридинил, бензимидазолил, бензизоксазолил, бензоксазолил, бензоксадиазолил, бензизотиазолил, бензотиазолил, бензотиадиазолил, бензофурил, изобензофурил, бензотиенил, бензотриазолил, имидазопиридил, триазолопиридил, имидазотиазолил, пиразинопиридазинил, оксазолопиридил, тиазолопиридил и тому подобное.
Примеры ароматического гетероциклила, который представляет собой полицикл, имеющий три или более колец, включают карбазолил, акридинил, ксантенил, фенотиазинил, феноксатиинил, феноксазинил, дибензофурил и тому подобное.
Термин «неароматический гетероциклил» означает неароматический циклил, который представляет собой моноцикл или полицикл, имеющий два или более кольца, содержащий один или более гетероатом(ов), независимо выбранный(х) из О, S и N.
Примеры «неароматического гетероциклила», который представляет собой полицикл, имеющий два или более кольца, включают конденсированную кольцевую группу, при этом неароматический гетероцикл, который представляет собой моноцикл или полицикл, имеющий два или более колец, конденсирован с кольцом указанного выше «ароматического карбоциклила», «неароматического карбоциклила» и/или «ароматического гетероциклила».
Помимо этого, примеры «неароматического гетероциклила» также включают группу, имеющую мостик, или группа с образованием спирокольца следующим образом:

Неароматический гетероциклил, который представляет собой моноцикл, предпочтительно представляет собой 3-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают диоксанил, трииранил, оксиранил, оксетанил, оксатиоланил, азетидинил, тианил, тиазолидинил, пирролидинил, пирролинил, имидазолидинил, имидазолинил, пиразолидинил, пиразолинил, пиперидинил, пиперазинил, морфолинил, морфолино, тиоморфолинил, тиоморфолино, дигидропиридинил, тетрагидропиридинил, тетрагидрофурил, тетрагидропиранил, дигидротиазолинил, тетрагидротиазолинил, тетрагидроизотиазолинил, дигидрооксазинил, гексагидроазепинил, тетрагидродиазепинил, тетрагидропиридазинил, гексагидропиримидинил, диоксоланил, диоксазинил, азиридинил, диоксолинил, оксепанил, тиоланил, тиинил, тиазинил и тому подобное.
Примеры неароматического гетероциклила, который представляет собой полицикл, имеющий два или более кольца, включают индолинил, изоиндолинил, хроманил, изохроманил и тому подобное.
[0047]
Термин «гетероцикл» включает ароматический цикл и неароматический гетероцикл, который содержит один или более гетероатом(ов), независимо выбранный(х) из О, S и N.
Термин «ароматический гетероцикл» означает ароматический цикл, который представляет собой моноцикл или полицикл, имеющий два или более кольца, содержащий один или более гетероатом(ов), независимо выбранный(х) из О, S и N.
Примеры «ароматического гетероцикла», который представляет собой полицикл, имеющий два или более кольца, включают конденсированное кольцо, при этом ароматический гетероцикл, который представляет собой моноцикл или полицикл, имеющий два или более кольца, конденсирован с кольцом указанного выше «ароматического карбоцикла».
Ароматический гетероцикл, который представляет собой моноцикл, предпочтительно представляет собой 5-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают пиррол, имидазол, пиразол, пиридин, пиридазин, пиримидин, пиразин, триазол, триазин, тетразол, фуран, тиофен, изоксазол, оксазол, оксадиазол, изотиазол, тиазол, тиадиазол и тому подобное.
Примеры ароматического гетероцикла, который представляет собой бицикл, включают индолин, изоиндолин, индазорин, индолизин, хинолин, изохинолин, циннолин, фталазин, хиназолин, нафтиридин, хиноксалин, пурин, птеридин, бензимидазол, бензизоксазол, бензоксазол, бензоксадиазол, бензизотиазол, бензотиазол, бензотиадиазол, бензофуран, изобензофуран, бензотиофен, бензотриазол, имидазопиридин, триазолопиридин, имидазотиазол, пиразинопиридазин, оксазолопиридин, тиазолопиридин и тому подобное.
Примеры ароматического гетероцикла, который представляет собой полицикл, имеющий три и более кольца, включают карбазол, акридин, ксантен, фенотиазин, феноксатиин, феноксазин, дибензофуран и тому подобное.
Термин «неароматический гетероцикл» означает неароматический цикл, который представляет собой моноцикл или полицикл, имеющий два или более кольца, содержащий один или более гетероатом(ов), независимо выбранный(х) из О, S и N.
Примеры «неароматического гетероцикла», который представляет собой полицикл, имеющий два или более кольца, включают конденсированную связь, при этом неароматический гетероцикл, который представляет собой моноцикл или полицикл, имеющий два или более кольца(ец), конденсирован с кольцом указанного выше «ароматического карбоцикла», «неароматического карбоцикла» и/или «ароматического гетероцикла».
Помимо этого, примеры «неароматического гетероцикла» также включают цикл, имеющий мостик, или цикл с образованием спирокольца следующим образом:
Неароматический гетероцикл, который представляет собой моноцикл, предпочтительно представляет собой 3-8-членное и более предпочтительно 5-6-членное кольцо. Примеры включают диоксан, трииран, оксиран, оксетан, оксатиолан, азетидин, тиан, тиазолидин, пирролидин, пирролин, имидазолидин, имидазолин, пиразолидин, пиразолин, пиперидин, пиперазин, морфолин, тиоморфолин, дигидропиридин, тетрагидропиридин, тетрагидрофуран, тетрагидропиран, дигидротиазолин, тетрагидротиазолин, тетрагидроизотиазолин, дигидрооксазин, гексагидроазепин, тетрагидродиазепин, тетрагидропиридазин, гексагидропиримидин, диоксолан, диоксазин, азиридин, диоксолин, оксепан, тиолан, тиазин и тому подобное.
Примеры неароматического гетероцикла, который представляет собой полицикл, имеющий два или более кольца, включают индолин, изоиндолин, хроман, изохроман и тому подобное.
[0048]
«Карбоциклическая» часть «карбоциклилалкила», «карбоциклилокси» или «карбоциклиламино» представляет собой то же самое, что указанный выше «карбоцикл».
[0049]
«Гетероциклическая» часть «гетероциклилалкила», «гетероциклилокси» или «гетероциклиламино» представляет собой то же самое, что указанный выше «гетероцикл».
[0050]
Соединения, применяемые в настоящем изобретении, характеризуются тем, что соединения, выделенные с помощью оптического разделения трициклических соединений, замещенных другой трициклической группой, улучшают ингибирующую активность в отношении кэп-зависимой эндонуклеазы.
[0051]
Соединения, применяемые в настоящем изобретении, также характеризуются тем, что соединения эффективно абсорбируются в организме после введения (например, перорального введения) и демонстрируют высокую эффективность при введении группы с образованием пролекарства.
[0052]
Один или более атомов водорода, углерода и/или других атомов в соединениях, применяемых в настоящем изобретении, могут быть заменены изотопами водорода, углерода и/или других атомов, соответственно. Примеры изотопов включают водород, углерод, азот, кислород, фосфор, серу, фтор, иод и хлор, такие как 2Н, 3Н, 11С, 13С, 14С, 15N, 18O, 17O, 31Р, 32Р, 35S, 18F, 123I и 36Cl, соответственно. Соединения, применяемые в настоящем изобретении, включают соединения, замещенные этими изотопами. Соединения, замещенные указанными выше изотопами, пригодны для применения в качестве лекарственных средств и включают все радиоактивно меченные соединения, применяемые в настоящем изобретении. «Способ метки радиоактивным изотопом» при получении «радиоактивно меченных соединений» охватывается настоящим изобретением, и «радиоактивно меченные соединения» пригодны для применения при исследованиях фармакокинетики метаболизируемых лекарственных средств, исследованиях связывающей способности и/или в диагностических целях.
[0053]
Радиоактивно меченное соединение, применяемое в настоящем изобретении, может быть получено с применением хорошо известных способов в этой области техники. Например, меченное тритием соединение, применяемое в настоящем изобретении, может быть получено введением трития в определенное соединение, применяемое в настоящем изобретении, вследствие каталитической реакции дегалогенирования с применением трития. Этот способ включает взаимодействие соответствующим образом галогенированного предшественника соединения, применяемого в настоящем изобретении, с тритиевым газом в присутствии подходящего катализатора, такого как Pd/C, и в присутствии или в отсутствие основания. Другой подходящий способ получения меченного тритием соединения см. «Isotopes in the Physical and Biomedical Sciences, Vol. 1, Labeled Compounds (Part A), Chapter 6 (1987)». 14С-меченое соединение может быть получено путем применения исходного материала, содержащего 14С.
[0054]
Фармацевтически приемлемые соли соединений, применяемых в настоящем изобретении, включают, например, соли со щелочным металлом (например, литием, натрием, калием или тому подобное), щелочноземельным металлом (например, кальцием, барием или тому подобное), магнием, переходными металлами (например, цинком, железом или тому подобное), аммонием, органическими основаниями (например, триметиламином, триэтиламином, дициклогексиламином, этаноламином, диэтаноламином, триэтаноламином, меглумином, этилендиамином, пиридином, пиколином, хинолином или тому подобное) или аминокислотами, или соли с неорганическими кислотами (например, соляной кислотой, серной кислотой, азотной кислотой, углекислотой, бромоводородной кислотой, фосфорной кислотой, иодоводородной кислотой или тому подобное) или органическими кислотами (например, муравьиной кислотой, уксусной кислотой, пропионовой кислотой, трифторуксусной кислотой, лимонной кислотой, молочной кислотой, винной кислотой, щавелевой кислотой, малеиновой кислотой, фумаровой кислотой, миндальной кислотой, глутаровой кислотой, яблочной кислотой, бензойной кислотой, фталевой кислотой, аскорбиновой кислотой, бензолсульфоновой кислотой, п-толуолсульфоновой кислотой, метансульфоновой кислотой, этансульфоновой кислотой или тому подобное). Главным образом включены соли с соляной кислотой, серной кислотой, фосфорной кислотой, винной кислотой, метансульфоновой кислотой и тому подобное. Эти соли могут быть получены обычными способами.
[0055]
Соединения, применяемые в настоящем изобретении, или их фармацевтически приемлемые соли могут образовывать сольваты (например, гидраты или тому подобное) и/или кристаллические полиморфы. Настоящее изобретение охватывает указанные различные сольваты и кристаллические полиморфы. «Сольваты» могут быть такими, в которых любое число молекул растворителя (например, молекул воды или тому подобное) координируется с соединениями, применяемыми в настоящем изобретении. Когда соединения, применяемые в настоящем изобретении, или их фармацевтически приемлемые соли оставляют на открытом воздухе, соединения могут поглощать воду, что приводит к присоединению адсорбированной воды или образованию гидратов. Перекристаллизация соединений, применяемых в настоящем изобретении, или их фармацевтически приемлемых солей может привести к получению кристаллических полиморф.
[0056]
Группа для образования пролекарства предпочтительно представляет собой группу, преобразующуюся в ОН-группу под действием ферментов, метаболизирующих лекарственное средство, гидролаз, желудочных кислот и/или энтеробактерий, после введения in vivo (например, перорального введения).
[0057]
Примеры наиболее предпочтительного варианта реализации группы для образования пролекарства включают группу, выбранную из следующей формулы а)-ас).
a) -C(=O)-PR0,
b) -C(=O)-PR1,
c) -C(=O)-L-PR1,
d) -C(=O)-L-O-PR1,
e) -C(=O)-L-O-L-O-PR1,
f) -C(=O)-L-O-C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
j) -C(PR3)2-O-PR4,
k) -C(PR3)2-O-L-O-PR4,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
n) -C(PR3)2-O-C(=O)-N(-K)-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
p) -C(PR3)2-O-C(=O)-O-L-N(PR4)2,
q) -C(PR3)2-O-C(=O)-N(-K)-L-O-PR4,
r) -C(PR3)2-O-C(=O)-N(-K)-L-N(PR4)2,
s) -C(PR3)2-O-C(=O)-O-L-O-L-O-PR4,
t) -C(PR3)2-O-C(=O)-O-L-N(-K)-C(=O)-PR4,
u) -C(PR3)2-O-P(=O)(-PR5)2,
v) -C(PR3)2-PR6,
w) -C(=N+(PR7)2)(-N(PR7)2),
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
у) -C(PR3)2-N(-K)-C(=O)-O-PR2,
z) -P(=O)(-PR8)(-PR9),
aa) -S(=O)2-PR10,
ab) -PR11 и
ac) -C(PR3)2-C(PR3)2-O-PR2,
где L представляет собой неразветвленный или разветвленный алкилен, или неразветвленный или разветвленный алкенилен;
K представляет собой водород или алкил, необязательно замещенный замещающей группой А;
PR0 представляет собой алкил, необязательно замещенный замещающей группой А, или алкенил, необязательно замещенный замещающей группой А;
PR1 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, или алкилсульфанил, необязательно замещенный замещающей группой А;
PR2 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, гетероциклилалкил, необязательно замещенный замещающей группой А, или триалкилсилил, необязательно замещенный замещающей группой А;
каждый PR3 независимо представляет собой водород или алкил;
каждый PR4 независимо представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, гетероциклилалкил, необязательно замещенный замещающей группой А, или триалкилсилил;
каждый PR5 независимо представляет собой гидрокси или OBn;
PR6 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
каждый PR7 независимо представляет собой алкил, необязательно замещенный замещающей группой А;
PR8 представляет собой алкилокси, необязательно замещенный замещающей группой А;
PR9 представляет собой алкилокси, необязательно замещенный замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилокси, необязательно замещенный замещающей группой А, гетероциклилокси, необязательно замещенный замещающей группой А, карбоциклиламино, необязательно замещенный замещающей группой А, или гетероциклиламино, необязательно замещенный замещающей группой А;
PR8 и PR9 могут быть объединены с соседним атомом фосфора с образованием гетероцикла, необязательно замещенного замещающей группой А;
PR10 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, или гетероциклилалкил, необязательно замещенный замещающей группой А; и
PR11 представляет собой алкил, необязательно замещенный замещающей группой А, алкенил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
Замещающая группа А; оксо, алкил, гидроксиалкил, амино, алкиламино, карбоциклил, гетероциклил, карбоциклилалкил, алкилкарбонил, галоген, гидрокси, карбокси, алкилкарбониламино, алкилкарбониламиноалкил, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкилоксикарбонилокси, алкиламинокарбонилокси, алкиламиноалкил, алкилокси, циано, нитро, азидо, алкилсульфонил, триалкилсилил и фосфо. [0058]
Примеры более предпочтительного варианта реализации группы для
образования пролекарства включают следующие группы.
a) -C(=O)-PR0,
b) -C(=O)-PR1,
g) -C(=O)-O-PR2,
h) -C(=O)-N(-K)(PR2),
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4,
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
v) -C(PR3)2-PR6,
x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2,
у) -C(PR3)2-N(-K)-C(=O)-O-PR2 и
z) -P(=O)(-PR8)(-PR9),
где L представляет собой неразветвленный или разветвленный алкилен;
K представляет собой водород или алкил, необязательно замещенный замещающей группой А;
PR0 представляет собой алкил, необязательно замещенный замещающей группой А;
PR1 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR2 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, гетероциклическую группу, необязательно замещенную замещающей группой А, карбоциклилалкил, необязательно замещенный замещающей группой А, или гетероциклилалкил, необязательно замещенный замещающей группой А;
каждый PR3 независимо представляет собой водород или алкил;
PR4 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR6 представляет собой карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
PR8 представляет собой алкилокси, необязательно замещенный замещающей группой А;
PR9 представляет собой алкилокси, необязательно замещенный замещающей группой А, алкиламино, необязательно замещенный замещающей группой А, карбоциклилокси, необязательно замещенный замещающей группой А, гетероциклилокси, необязательно замещенный замещающей группой А, карбоциклиламино, необязательно замещенный замещающей группой А, или гетероциклиламино, необязательно замещенный замещающей группой А; и
PR8 и PR9 могут быть объединены с соседним атомом фосфора с образованием гетероцикла, необязательно замещенного замещающей группой А;
Замещающая группа А; оксо, алкил, алкиламино, карбоциклил, гетероциклил, алкилкарбонил, галоген, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил и триалкилсилил.
Среди указанных выше групп для образования пролекарства, a) -C(=O)-PR0, b) -C(=O)-PR1, g) -C(=O)-O-PR2, h) -C(=O)-N(-K)(PR2), i) -C(=O)-O-L-O-PR2, l) -C(PR3)2-O-C(=O)-PR4, m) -C(PR3)2-O-C(=O)-O-PR4, o) -C(PR3)2-O-C(=O)-O-L-O-PR4, x) -C(PR3)2-C(PR3)2-C(=O)-O-PR2, у) -C(PR3)2-N(-K)-C(=O)-O-PR2 являются предпочтительными.
В частности, в наиболее предпочтительном варианте реализации группа для образования пролекарства имеет следующую группу.
m) -C(PR3)2-O-C(=O)-O-PR4
где каждый PR3 независимо представляет собой водород или алкил; и PR4 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А;
Замещающая группа А; оксо, алкил, алкиламино, карбоциклил, гетероциклил, алкилкарбонил, галоген, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил и триалкилсилил.
Вариант реализации PR3 каждый независимо представляет собой водород или алкил, и предпочтительно водород.
Вариант реализации PR4 представляет собой алкил, необязательно замещенный замещающей группой А, карбоциклическую группу, необязательно замещенную замещающей группой А, или гетероциклическую группу, необязательно замещенную замещающей группой А, и предпочтительно метил, этил или тому подобное. Вариант реализации замещающей группы А включает оксо, алкил, алкиламино, карбоциклил, гетероциклил, алкилкарбонил, галоген, гидрокси, алкилкарбониламино, алкилкарбонилокси, алкилоксикарбонил, алкилоксикарбонилалкил, алкиламинокарбонилокси, алкилокси, нитро, азидо, алкилсульфонил и триалкилсилил, и предпочтительно фтор, хлор, гидроксил, метил и этил.
[0059]
Примеры другого варианта реализации предпочтительного заместителя группы для образования пролекарства включают следующие группы.

Примеры другого варианта реализации предпочтительного заместителя группы для образования пролекарства включают следующие группы.
[0060]
Примеры варианта реализации особенно предпочтительного заместителя группы для образования пролекарства включают следующие группы.

[0061]
(Способ получения соединения согласно настоящему изобретению)
Общий способ получения соединения, применяемого в настоящем изобретении, приведен ниже в качестве примера. Что касается экстракции и очистки, то может быть проведена обработка, которая выполняется в обычном эксперименте органической химии.
Синтез соединения, применяемого в настоящем изобретении, может быть осуществлен в соответствии с методиками, известными в данной области техники.
[0062]
В качестве исходного материала соединения могут быть использованы коммерчески доступные соединения, соединения, описанные в настоящем описании, соединения, описанные в ссылках, цитируемых в настоящем описании, и другие известные соединения.
Если согласно настоящему изобретению необходимо получить соль соединения, то в случае, когда соединение, применяемое в настоящем изобретении, получают в форме соли, оно может быть очищено, как оно есть, и, в случае, когда соединение, используемое в настоящем изобретении, получают в свободной форме, соль может быть получена обычным способом растворением или суспендированием соединения в подходящем органическом растворителе и добавлением кислоты или основания.
Помимо этого, соединение, применяемое в настоящем изобретении, и его фармацевтически приемлемая соль в некоторых случаях присутствуют в форме аддуктов с водой или различными растворителями (гидрат или сольват), и эти аддукты применяются в настоящем изобретении.
[0063]
В общем способе синтеза, а также в примерах и в примерах синтеза промежуточных соединений, значение каждого сокращения представляет собой следующее.
Boc: трет-бутоксикарбонил
DBU: диазабициклоундецен
ДМА: N,N-диметилацетамид
ДМФА: N,N-диметилформамид
HATU: O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилуроний гексафторфосфат
NMP: N-метилпирролидон
OBn: бензилокси
ТГФ: тетрагидрофуран
Т3Р: пропилфосфоновый ангидрид
WSC⋅HCl: N-этил-N'-(3-диметиламинопропил)карбодиимид гидрохлорид
Верх и низ «клина» и «ломаного линейного клина» означают абсолютную конфигурацию.
[0064]
(Способ получения 1)
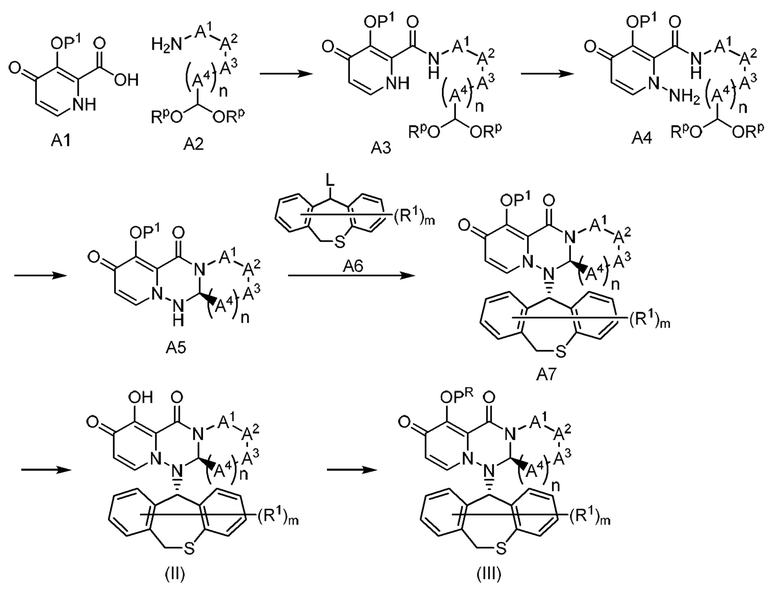
где P1 представляет собой гидроксильную защитную группу; RP представляет собой ацетальную защитную группу; L представляет собой уходящую группу; каждый другой символ имеет то же значение, что и выше.
Первая стадия
Соединение A3 может быть получено добавлением соединения А2 к соединению А1 в присутствии дегидратирующе-конденсирующего агента, такого как дициклогексилкарбодиимид, карбонилдиимидазол, дициклогексилкарбодиимидо-N-гидроксибензотриазол, 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолина хлорид, 2-(7-аза-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфорной кислоты, WSC⋅HCl, HATU и т.д., в растворителе, таком как ДМФА, ТГФ, дихлорметан, ацетонитрил и т.д. или в их смешанном растворителе, и проведением реакции при температуре от -20°С до 60°С, предпочтительно от -10°С до 40°С, в течение от 0.1 часа до 24 часов, предпочтительно от 1 часа до 12 часов.
В качестве альтернативы, соединение A3 может быть получено добавлением ацилирующего реагента, такого как дифенилхлорфосфат, тионилхлорид, оксалилхлорид и т.д., к соединению А1 в присутствии или в отсутствие основания, такого как пиридин, триэтиламин, диизопропилэтиламин, 1-метилимидазол и т.д., в присутствии растворителя, такого как ТГФ, диоксан, дихлорметан, ДМФА и т.д., тем самым образуя хлорангидрид, и добавлением соединения А2, имеющего заместитель, соответствующий целевому соединению, и проведением реакции при температуре от -20°С до 60°С, предпочтительно от -10°С до 40°С, в течение от 0.1 часа до 24 часов, предпочтительно от 0.5 часа до 12 часов.
Вторая стадия
Соединение А4 может быть получено добавлением карбоната калия, карбоната натрия и O-(2,4-динитрофенил)гидроксиламина к соединению A3 в присутствии растворителя, такого как ДМФА, ДМА, NMP, ТГФ и т.д., и проведением реакции при температуре от 10°С до 60°С, предпочтительно от 20°С до 40°С, в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Третья стадия
Реакцию снятия защиты ацетальной защитной группы соединения А4 можно проводить общим способом, описанным в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons) и т.п. После указанной реакции генерируемая альдегидная группа подвергается внутримолекулярной реакции, таким образом, может быть получено соединение А5.
Например, рацемат соединения А5 может быть получен добавлением уксусной кислоты и/или пара-толуолсульфоновой кислоты, метансульфоновой кислоты и т.д., к соединению А4 в присутствии растворителя, такого как ДМФА, толуол, ТГФ и т.д., и проведением реакции при температуре от 10°С до 80°С, предпочтительно от 30°С до 60°С, в течение от 0.5 часа до 12 часов, предпочтительно от 1 часа до 6 часов. Соединение А5 может быть получено оптическим разделением рацемата соединения А5 с помощью СФХ (сверхкритическая флюидная хроматография) или ВЭЖХ (на хиральной колонке).
Четвертая стадия
Соединение А7 может быть получено добавлением соединения А6 и основания, такого как карбонат натрия, карбонат калия, карбонат цезия и т.д., к соединению А5 в присутствии растворителя, такого как ДМФА, ДМА, NMP, ТГФ и т.д., или их смешанного растворителя, и проведением реакции при температуре от 0°С до 60°С, предпочтительно от 10°С до 40°С, в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
В качестве альтернативы, соединение А7 может быть получено добавлением соединения А6 и Т3Р, метансульфоновой кислоты или пара-толуолсульфоновой кислоты к соединению А5 в присутствии растворителя, такого как ДМФА, этилацетат, бутилацетат, 1,4-диоксан и т.д., или их смешанного растворителя, и проведением реакции при температуре от 40°С до 150°С, предпочтительно от 60°С до 120°С, в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Пятая стадия
Реакцию снятия защиты гидроксильной защитной группы соединения А7 можно проводить общим способом, описанным в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons) и т.д.
Шестая стадия
Соединение (III) может быть получено общим способом, включающим преобразование гидроксильной группы соединения (II) в группу сложного эфира или группу простого эфира.
Например, может быть использован способ, описанный в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons), Prog. Med. 5: 2157-2161 (1985) и предоставленный Британской библиотекой - «The world's Knowledge» и т.д.
(Способ получения 2)
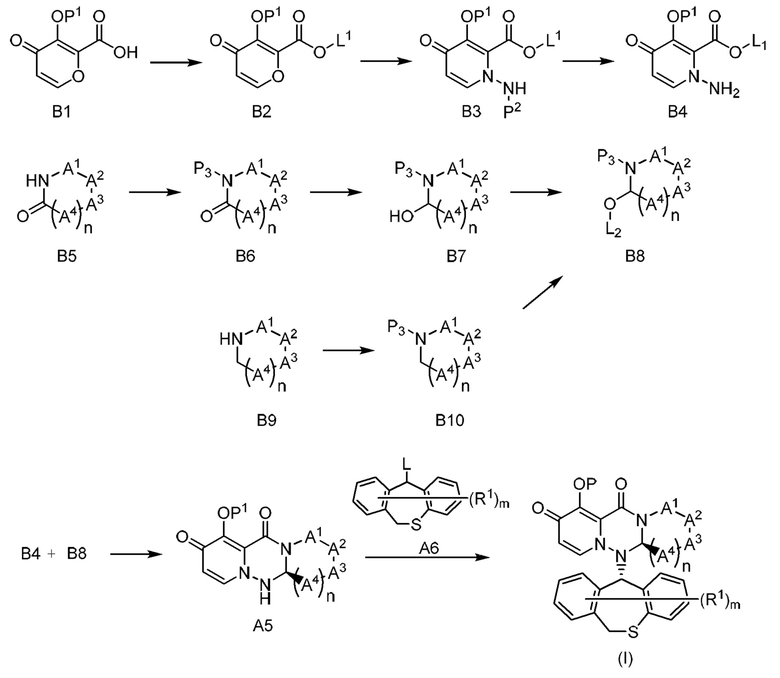
где Р2 представляет собой NH-защитную группу; L1 и L2 представляют собой уходящую группу; каждый другой символ имеет то же значение, что и выше.
Первая стадия
Соединение В2 может быть получено добавлением соединения А2 и галогенированного алкила, такого как метилиодид, к соединению В1 в присутствии основания, такого как диазабициклоундецен, в растворителе, таком как ДМФА, ТГФ, дихлорметан, ацетонитрил и т.д., или в их смешанном растворителе, и проведением реакции при температуре от -20°С до 60°С, предпочтительно от -10°С до 40°С, в течение от 0.1 часа до 24 часов, предпочтительно от 1 часа до 24 часов.
В качестве альтернативы, соединение В2 может быть получено добавлением ацилирующего реагента, такого как дифенилхлорфосфат, тионилхлорид, оксалилхлорид и т.д., к соединению В1 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФА и т.д., или в их смешанном растворителе, и добавлением спирта в присутствии основания, такого как пиридин, триэтиламин, диизопропилэтиламин, 1-метилимидазол и т.д., и проведением реакции при температуре от -20°С до 60°С, предпочтительно от -10°С до 40°С, в течение от 0.1 часа до 24 часов, предпочтительно от 0.5 часа до 12 часов.
Вторая стадия
Соединение В3 может быть получено добавлением пара-толуолсульфоновой кислоты пиридиния и гидразина, защищенного BOc и т.д., к соединению В2 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФА и т.д., или в их смешанном растворителе, и проведением реакции при температуре от 10°С до 150°С, предпочтительно от 40°С до 100°С, в течение от 1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Третья стадия
Реакцию снятия защиты защитной аминогруппы соединения В3 можно проводить общим способом, описанным в Protective Groups in Organic Synthesis, Theodora W Green (John Wiley & Sons) и т.д.
Четвертая стадия
Соединение В6 может быть получено добавлением основания, такого как н-бутиллитий и т.д., к соединению В5 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФА и т.д., или в их смешанном растворителе, и затем добавлением алкила галогенмуравьиной кислоты и проведением реакции в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Пятая стадия
Соединение В7 может быть получено добавлением восстановителя, такого как лития диизобутилалюминийгидрид и т.д., к соединению В6 в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФА и т.д., или в их смешанном растворителе, и проведением реакции в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Шестая стадия
Соединение В8 может быть получено добавлением пара-толуолсульфоновой кислоты или метансульфоновой кислоты к соединению В7 в спирте и проведением реакции при температуре от 0°С до 100°С в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Седьмая стадия
Соединение В10 может быть получено добавлением алкила галогенмуравьиной кислоты к соединению В9 в присутствии или в отсутствие основания, такого как пиридин, триэтиламин, диизопропилэтиламин, 1-метилимидазол и т.д., в растворителе, таком как ТГФ, диоксан, дихлорметан, ДМФА и т.д., или в их смешанном растворителе, и проведением реакции при температуре от -40°С до 40°С в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Восьмая стадия
Соединение В8 может быть получено погружением углеродного электрода (анода) и платинового электрода (катода) в соединение В10 в растворителе, таком как спирт, в присутствии основания, такого как карбонат калия и тетраэтиламмония перхлорат, и пропусканием постоянного тока силой 0.1~1.0 А при перемешивании в течение от 0.1 часа до 48 часов, предпочтительно от 1 часа до 24 часов.
Девятая-десятая стадии
Соединение (I) может быть получено из соединений В4 и В8 таким же образом, как и на стадиях с третьей по шестую при получении 1.
[0065]
Соединения, применяемые в настоящем изобретении, обладают ингибирующей активностью в отношении кэп-зависимой эндонуклеазы и являются подходящими для применения в качестве терапевтических или профилактических агентов против гриппа.
[0066]
Соединения (исходные соединения и/или пролекарства), применяемые в настоящем изобретении, пригодны для применения при симптомах и/или заболеваниях, которые вызывает вирус гриппа. Например, они пригодны для применения для лечения, предотвращения и/или улучшения состояния при симптомах: простудных симптомах, сопровождающих лихорадку, ознобе, головной боли, мышечной или суставной боли, усталости и т.д., симптомах воспаления дыхательных путей, таких как боль в горле, выделения из носа, заложенность носа, кашель, мокрота и т.д., симптомах со стороны желудочно-кишечного тракта, таких как боль в животе, рвота, диарея и т.д., и, кроме того, осложнениях, сопровождающих вторичную инфекцию, таких как острая энцефалопатия и пневмония. То есть соединения, применяемые в настоящем изобретении, пригодны для лечения и/или предотвращения инфекционных заболеваний, вызванных вирусом гриппа.
Соединения, применяемые в настоящем изобретении, эффективны для сокращения времени до облегчения симптомов гриппа. Например, они могут сократить время до облегчения симптомов гриппа примерно от 20 до 40 часов, предпочтительно примерно от 25 до 30 часов. В частности, они могут сократить время, пока не уменьшатся «кашель», «боль в горле», «головная боль», «заложенность носа», «лихорадка или озноб», «боль в мышцах или суставах» и «усталость». В частности, они подходят для сокращения времени до тех пор, пока не уменьшатся «заложенность носа», «боль в мышцах или суставах», «усталость», «лихорадка или озноб» и «головная боль». Кроме того, они пригодны для сокращения времени, пока не уменьшатся «заложенность носа» и «боль в мышцах или суставах».
Кроме того, поскольку соединения (исходные соединения и/или пролекарства), применяемые в настоящем изобретении, уменьшают количество вируса гриппа в организме в течение короткого периода времени, они могут найти отличное фармацевтическое применение для лечения и/или предотвращения инфекционных заболеваний вируса гриппа. После введения соединений, применяемых в настоящем изобретении, эффект уменьшения количества вируса гриппа в организме наблюдается в течение 72 часов, предпочтительно в течение 48 часов, более предпочтительно в течение 24 часов, и ожидается более ранний терапевтический эффект по сравнению с другими лекарственными средствами.
[0067]
Кроме того, соединения, применяемые в настоящем изобретении, имеют применение в качестве лекарственных средств.
Например, поскольку соединения (пролекарства), используемые в настоящем изобретении, имеют преимущества, заключающиеся в высокой пероральной абсорбируемости, хорошей биодоступности, хорошем клиренсе и высокой легочной транзитивности, они могут быть отличными лекарственными средствами.
Поскольку соединения (исходные соединения), применяемые в настоящем изобретении, обладают такими эффектами, как высокая ингибирующая активность в отношении кэп-зависимой эндонуклеазы и высокая селективность, обусловленная специфическим для вируса ферментом, они могут представлять собой лекарственные средства с уменьшенными побочными эффектами.
Кроме того, соединения (исходные соединения и/или пролекарства), применяемые в настоящем изобретении, так же имеют преимущества, такие как высокая стабильность метаболизма, высокая растворимость, высокая всасываемость при пероральном приеме, хорошая биодоступность, хороший клиренс, высокая легочная транзитивность, длинный период полураспада, высокая скорость связывания небелков, низкое ингибирование hERG-каналов, низкое ингибирование CYP, определяется эффект ингибирования СРЕ (цитопатический эффект), и/или проявляются отрицательные эффекты в тесте на фототоксичность, в тесте Эймса и тесте на генную токсичность, или не вызывается токсичность, такая как повреждение печени. Таким образом, фармацевтическая композиция по настоящему изобретению может быть отличным лекарственным средством.
[0068]
Соединения (исходные соединения и/или пролекарства), применяемые в настоящем изобретении, могут быть введены перорально или парентерально. В случае перорального введения соединения, применяемые в настоящем изобретении, также могут быть применены в качестве обычного препарата, например, в качестве любой лекарственной формы твердых препаратов, таких как таблетки, порошки, гранулы, капсулы и т.д.; растворы; маслянистые суспензии; или жидкие препараты, такие как сиропы или эликсиры и т.д. В случае парентерального введения соединения, применяемые в настоящем изобретении, могут быть применены в виде водной или масляной инъекционной суспензии или капель в нос. При их получении могут произвольно применяться обычные вспомогательные вещества, связующие вещества, смазывающие вещества, водные растворители, маслянистые растворители, эмульгаторы, суспендирующие агенты, консерванты, стабилизаторы и т.д. Фармацевтическая композиция согласно настоящему изобретению может быть получена объединением (например, смешиванием) терапевтически и/или профилактически эффективного количества соединения, применяемого в настоящем изобретении, с фармацевтически приемлемыми носителями или разбавителями. Соединения, применяемые в настоящем изобретении, могут быть подходящим образом применимы в качестве пероральных препаратов из-за их высокой пероральной абсорбируемости.
Доза соединений, применяемых в настоящем изобретении, различается в зависимости от способа введения, возраста, массы и состояния пациента, а также от типа заболевания и, как правило, в случае перорального введения составляет для взрослого примерно от 0.05 мг до 3000 мг, предпочтительно примерно от 0.1 мг до 1000 мг, более предпочтительно примерно от 10 мг до 80 мг, еще более предпочтительно примерно от 10 до 40 мг в день, если необходимо, в виде разделенных доз. В другом варианте реализации в случае взрослых могут быть введены примерно 40 мг или 80 мг в одной дозе. В случае детей могут быть введены примерно от 5 до 40 мг в одной дозе в зависимости от массы тела. Помимо этого, в случае парентерального введения для взрослого вводят примерно от 0.01 мг до 1000 мг, предпочтительно примерно от 0.05 мг до 500 мг, или примерно от 1 мг до 80 мг в день. Доза может быть введена один раз в день или может быть разделена на несколько доз в день.
[0069]
Соединения, применяемые в настоящем изобретении, могут быть применены в комбинации с другими лекарственными средствами или тому подобным (в дальнейшем именуемые комбинированными лекарственными средствами) для увеличения активности соединений, уменьшения дозы соединений или тому подобного. В случае лечения гриппа соединения могут быть применены в сочетании с или в сдвоенном составе с ингибитором нейраминидазы (например, осельтамивиром, занамивиром, перамивиром, инабиру и тому подобном); также возможны варианты с ингибитором РНК-зависимой РНК-полимеразы (например, фавипиравир); ингибитором белка М2 (например, амантадин); РВ2 кэп-зависимым ингибитором (например, VX-787); анти-НА антителом (например, МНАА4549А); иммунными агонистами (например, нитазоксанидом). В этом случае время введения соединения, применяемого в настоящем изобретении, и комбинированного лекарственного средства не ограничены. Их можно вводить субъектам, подлежащим лечению, одновременно или в разное время. Кроме того, комбинированное лекарственное средство с соединением, применяемым в настоящем изобретении, может быть введено в виде двух или более составов, независимо включающих каждый активный ингредиент, или в виде одного состава, включающего каждый активный ингредиент.
[0070]
Доза для комбинированных лекарственных средств может быть соответствующим образом выбрана с учетом клинической дозы. Соотношение компонентов соединений, применяемых в настоящем изобретении, и лекарственных средств, вводимых совместно, может быть соответствующим образом выбрано в зависимости от подлежащего лечению субъекта, пути введения, подлежащего лечению заболевания, симптомов, комбинации лекарственных средств и тому подобного. Для введения человеку, например, 1 массовая часть соединений, применяемых в настоящем изобретении, может применяться в комбинации с 0.01-100 массовых частей совместно вводимых лекарственных средств.
[0071]
Настоящее изобретение будет объяснено более подробно ниже посредством примеров, примеров промежуточного синтеза, а также тестовых примеров настоящего изобретения, но настоящее изобретение ими не ограничивается.
[0072]
ЯМР-анализ, полученный в каждом примере, проводили при 300 МГц, измерения проводили с использованием ДМСО-d6, CDCl3.
Термин RT представляет собой время удерживания при ЖХ/МС: жидкостная хроматография/масс-спектрометрия, и измерения проводили в следующих условиях.
(Условия измерения)
(1) Колонка: ACQUITY UPLC (зарегистрированная торговая марка) ВЕН С18 (1.7 мкм внутр.д. 2.1x50 мм) (Waters)
Скорость потока: 0.8 мл/мин
Длина волны УФ-детектирования: 254 нм
Подвижная фаза: [А]: 0.1% раствор муравьиной кислоты в воде, [В]: 0.1% раствор муравьиной кислоты в ацетонитриле
Градиент: линейный градиент от 5% до 100% растворителя [В] выполняли за 3.5 минуты, и 100% растворителя [В] выдерживали в течение 0.5 минуты.
(2) Колонка: Shim-pack XR-ODS (2.2 мкм, внутр.д.50x3.0 мм) (Shimadzu)
Скорость потока: 1.6 мл/мин
Длина волны УФ-детектирования: 254 нм
Подвижная фаза: [А]: 0.1% раствор муравьиной кислоты в воде, [В]: 0.1% раствор муравьиной кислоты в ацетонитриле
Градиент: линейный градиент от 10% до 100% растворителя [В] выполняли за 3 минуты, и 100% растворителя [В] выдерживали в течение 0.5 минуты.
[0073]
Измерение рентгеновской порошковой дифрактограммы
Ренгеновская порошковая дифракция кристаллов, полученных в каждом примере, была измерена в соответствии с методом рентгеновской порошковой дифракции, описанным в общих тестах, процессах и аппаратуре Японской фармакопеи. Условия измерения приведены ниже.
[0074]
(Прибор)
MinFlex 600 RINT-TTR III, производства Rigaku Corporation
(Способ работы)
Детектор: Высокоскоростной 1-мерный детектор (D/Tec Ultra 2) и регулируемая режущая кромка
Метод измерения: метод отражения
Тип источника света: Cu трубка
Применяемая длина волны: излучение CuKα
Ток трубки: 10 мА или 15 мА
Напряжение трубки: 30 кВ или 40 кВ
Кювета для образца: алюминий или стекло
Угол падения рентгеновского излучения (θ): 3-4°, ширина выборки: 0.01° или
угол падения рентгеновского излучения (θ): 4-40°, ширина выборки: 0.02°
Как правило, угол дифракции (2θ) в рентгеновской порошковой дифракции может иметь погрешность в пределах диапазона ±0.2°, и, таким образом, значение угла дифракции также охватывает диапазон примерно ±0.2° от числового значения. Соответственно, настоящее изобретение охватывает не только кристалл, пиковые углы дифракции которого в рентгеновской порошковой дифракции полностью совпадают, но также и кристалл, пиковые углы дифракции которого соответствуют с погрешностью примерно ±0.2°.
[0075]
Пример 1-1: Способ получения соединения i1

Первая стадия
К раствору соединения 1 (5.0 г, 49.5 ммоль) в ТГФ (100 мл) по каплям добавляли раствор 1.62 моль/л н-бутиллития в гексане (30.5 мл, 49.5 ммоль) при -78°С в атмосфере азота и перемешивали смесь при -78°С в течение 2 часов. К нему по каплям добавляли раствор аллилхлорформиата (5.96 г, 49.5 ммоль) в ТГФ (20 мл) и перемешивали смесь при -78°С в течение 2 часов. Смесь гасили насыщенным водным раствором хлорида аммония, нагревали до комнатной температуры и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 2 (5.66 г, 62%).
1Н-ЯМР(CDCl3)δ:3.83 (t, J=8.0 Гц, 2Н), 3.92 (t, J=8.0 Гц, 2Н), 4.26 (s, 2Н), 4.78 (d, J=8.0 Гц, 2Н), 5.30 (d, J=12.0 Гц, 1H), 5.44 (d, J=16.0 Гц, 1H), 5.93-6.03 (m, 1H),
Вторая стадия
К раствору соединения 2 (6.6 г, 35.6 ммоль) в ТГФ (66 мл) по каплям добавляли раствор 1.03 моль/л DIBAL-H (диизобутилалюминий-гидрид) в гексане (45.0 мл, 46.3 ммоль) и перемешивали смесь при -78°С в течение 1 часа. Смесь гасили ацетоном, к ней добавляли водный раствор соли Рошелля. Смесь перемешивали и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 3 (6.21 г, 93%).
1Н-ЯМР(CDCl3)δ:3.44 (br, 1H), 3.50-3.64 (m, 2Н), 3.71 (br, 1H), 3.95 (d, J=8.0 Гц, 2Н), 4.64 (d, J=8.0 Гц, 2Н), 5.24 (d, J=12.0 Гц, 1H), 5.40 (d, J=16.0 Гц, 1H), 5.47 (d, J=4 Гц, 1Н), 5.87-6.00 (m, 1H)
Третья стадия
К раствору соединения 3 (6.2 г, 33.1 ммоль) в метаноле (65 мл) добавляли моногидрат п-толуолсульфоновой кислоты (0.63 г, 3.31 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Смесь гасили водным раствором гидрокарбоната натрия, концентрировали и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 4 (5.77 г, 87%).
1Н-ЯМР(CDCl3)δ:3.34 (s, 3Н), 3.55 (br, 2Н), 3.73-3.99 (m, 3Н), 4.64 (d, J=8.0 Гц, 2Н), 5.10-5.20 (m, 1Н), 5.25 (d, J=8.0 Гц, 1H), 5.33 (d, J=16 Гц, 1H), 5.88-6.05 (m, 1H)
Четвертая стадия
К раствору соединения 5 (20.0 г, 81 ммоль) в ДМФА (100 мл) добавляли этилиодид (22.8 г, 146 ммоль) и диазабициклоундецен (18.4 мл, 122 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Смесь вливали в 10% водный раствор хлорида аммония и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 6 (22.3 г, 100%).
1Н-ЯМР(CDCl3)δ:1.23 (t, J=8.0 Гц, 3Н), 4.28 (q, J=8.0 Гц, 2Н), 5.16 (s, 2Н), 6.57 (d, J=4.0 Гц, 1H), 7.28-7.48 (m, 5Н), 8.21 (d, J=4.0 Гц, 1H).
Пятая стадия
К раствору соединения 6 (500 мг, 1.82 ммоль) в ДМА (5.0 мл) добавляли п-толуолсульфонат пиридиния (1.37 г, 5.47 ммоль) и Вос-гидразин (361 мг, 2.74 ммоль) и перемешивали смесь при 60°С в течение 14 часов. К смеси добавляли воду и экстрагировали смесь этилацетатом. Полученный органический слой промывали насыщенным водным раствором хлорида аммония и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 7 (519 мг, 73%).
1Н-ЯМР(CDCl3)δ:1.24 (t, J=8.0 Гц, 3Н), 1.46 (s, 9Н), 4.26 (q, J=8.0 Гц, 2Н), 5.28 (s, 2Н), 6.40 (d, J=8.0 Гц, 1H), 7.27-7.38 (m, 4Н), 7.40-7.45 (m, 2Н).
Шестая стадия
Соединение 7 (500 мг, 1.29 ммоль) растворяли в растворе 4 моль/л хлористого водорода в этилацетате (5 мл) и перемешивали смесь при комнатной температуре в течение 1 часа. Смесь концентрировали при пониженном давлении. К полученному остатку добавляли насыщенный водный раствор гидрокарбоната натрия и экстрагировали смесь дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 8 (369 мг, 99%).
1Н-ЯМР(CDCl3)δ:1.26 (t, J=8.0 Гц, 3Н), 4.31 (q, J=8.0 Гц, 2Н), 5.24 (s, 2Н), 6.47 (d, J=8.0, 1H), 7.28-7.44 (m, 5H), 7.64 (d, J=8.0, 1H).
Седьмая стадия
К раствору соединения 8 (365 мг, 1.27 ммоль) и соединения 4 (306 мг, 1.52 ммоль) в ацетонитриле (8 мл) по каплям добавляли хлорид олова (0.223 мл, 1.90 ммоль) при -25°С в атмосфере азота и перемешивали смесь при -25°С в течение 45 минут. Смесь гасили насыщенным водным раствором гидрокарбоната натрия и добавляли к ней дихлорметан. Перемешивали смесь при комнатной температуре и фильтровали через Celite и экстрагировали фильтрат дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая неочищенное соединение 9. Полученное соединение 9 растворяли в ТГФ (8 мл), добавляли к нему морфолин (1.10 мл, 12.7 ммоль) и тетракис(трифенилфосфин)палладий (146 мг, 0.127 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. К смеси добавляли диэтиловый эфир (16 мл) и выпавшее в осадок твердое вещество фильтровали и сушили, получая соединение 10 (418 мг, 100%).
1Н-ЯМР(CDCl3)δ:2.90-2.99 (m, 1Н), 3.13 (t, J=12.0 Гц, 1Н), 3.40-3.46 (m, 1Н), 4.00-4.08 (m, 1H), 4.14 (d, J=12.0 Гц, 1H), 5.07 (s, 2Н), 6.22 (d, J=8.0 Гц, 1H), 7.29-7.40 (m, 3Н), 7.56 (d, J=8.0 Гц, 2Н), 7.71 (d, J=8.0 Гц, 1H)
Восьмая стадия
К суспензии (R)-2-тетрагидрофуровой кислоты (855 мг, 7.36 ммоль) и соединения 10 (2.00 г, 6.11 ммоль) в этилацетате (9 мл) добавляли пиридин (4.00 мл, 49.6 ммоль) и Т3Р (50% в этилацетате, 11.0 мл, 18.5 ммоль) при комнатной температуре и перемешивали смесь в течение ночи. Выпавшее в осадок твердое вещество фильтровали и промывали этилацетатом (4 мл) и этанолом (4 мл). Полученное твердое вещество суспендировали в этаноле (6 мл) и перемешивали суспензию при комнатной температуре в течение 6.5 часов. Суспензию фильтровали, и полученное твердое вещество промывали этанолом (2 мл) дважды, получая соединение 11 (1.18 г, 45.4%).
1Н-ЯМР (ДМСО)δ: 1.80-1.94(m, 2Н), 1.95-2.14(m, 2Н), 3.21-3.35-(m, 2Н), 3.50-3.60(m, 1H), 3.70-3.82(m, 3Н), 4.00-4.05(m, 1H), 4.32-4.38(m, 1H), 5.14(dd, J=10.8 Гц, 21.6 Гц, 2Н), 5.76-5.81(m, 1H), 6.29(d; J=4.8 Гц, 1H), 7.28-7.39(m, 3Н), 7.48-7.54(m, 2Н), 7.64-7.75(m, 1H)
Девятая стадия
К суспензии соединения 11 (500 мг, 1.18 ммоль) в этаноле (3.5 мл) добавляли DBU (0.0035 мл, 0.023 ммоль) при комнатной температуре и перемешивали смесь в течение 30 минут.К полученной суспензии добавляли диизопропиловый эфир (6.5 мл) и перемешивали смесь при комнатной температуре в течение 30 минут. Выпавшее в осадок твердое вещество фильтровали и промывали этилацетатом (1.5 мл) дважды, получая соединение i1 (346 мг, 89.9%).
1Н-ЯМР (ДМСО)δ: 2.80-3.00(m, 1H), 3.10-3.18(m, 1Н), 3.38-3.50(m, 1Н), 3.98-4.08(m, 2Н), 4.10-4.20(m, 1Н), 4.76-4.84(m, 1Н), 5.04-5.14(m, 2Н), 6.22(m, J=7.6 Гц, 1H), 7.27-7.40(m, 4Н), 7.56-7.60(m, 2Н), 7.70(d, J=7.6 Гц, 1H)
[0076]
Пример 1-2: Способ получения соединения i2

Первая стадия
К суспензии соединения 13 (8.0 г, 50.8 ммоль) в дихлорметане (120 мл) добавляли триэтиламин (17.6 мл, 127 ммоль) в бане с ледяной водой, к смеси по каплям добавляли аллилхлорформиат (6.44 мл, 60.9 ммоль) и перемешивали смесь при 0°С в течение 1 часа. К смеси добавляли воду и экстрагировали смесь дихлорметаном. Полученный органический слой промывали 5% водным раствором лимонной кислоты и насыщенным водным раствором гидрокарбоната натрия, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 14 (10.1 г, 97%).
1Н-ЯМР (CDCl3)δ:1.96 (br, 4Н), 3.62 (s, 4Н), 4.60 (s, 2Н), 5.22 (d, J=12.0 Гц, 1H), 5.30 (d, J=16.0 Гц, 1H), 5.86-5.99 (m, 1H)
Вторая стадия
В раствор соединения 14 (0.9 г, 4.39 ммоль), карбоната калия (60 мг, 0.44 ммоль) и перхлората тетраэтиламмония (50 мг, 0.22 ммоль) в метаноле (30 мл) погружали углеродный электрод (анод) и платиновый электрод (катод) и через смесь пропускали постоянный ток 0.1 А при перемешивании при комнатной температуре в течение 6 часов. К смеси добавляли этилацетат и воду и экстрагировали смесь этилацетатом. Полученный органический слой сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 15 (992 мг, 96%).
1Н-ЯМР (CDCl3)δ:1.81-2.15 (m, 3Н), 2.39 (t, J=12.0 Гц, 1H), 3.27 (s, 3Н), 3.61 (s, 1H), 4.11 (br, 1H), 4.61 (br, 2H), 5.20-5.36 (m, 2H), 5.57 (br, 1H), 5.88-5.99 (m, 1H)
Third стадия
Соединение 16 получали таким же способом, как на седьмой и восьмой стадиях в примере 1-1.
Четвертая стадия
Оптическое разделение соединения 16 (870 мг, 2.41 ммоль) с помощью Waters SFC30 System (Daicel CHIRALPAK IB, сжиженный диоксид углерода-метанол) дает соединение i2 (270 мг, 31%).
Условия анализа
<Waters SFC30 System>
Колонка: CHIRALPAK IB/SFC (5 мкм, внутр.д.250x4.6 мм) (DAICEL)
Скорость потока: 8.0 мл/мин; Длина волны УФ-детектирования: 254 нм
Обратное давление: 100 бар
Подвижная фаза: [А]: сжиженный диоксид углерода, [В]: метанол
Градиент: 5% растворителя [В] выдерживали в течение 1 минуты, линейный градиент от 5% до 40% растворителя [В] проводили в течение 6 минут, 40% растворителя [В] выдерживали в течение 2 минут, и 5% растворителя [В] выдерживали в течение 1 минуты.
Время элюирования: 7.3 минуты
[0077]
Пример 1-3: Способ получения соединения i3

Первая стадия
К раствору соединения 17 (4.00 г, 16.3 ммоль) в дихлорметане (40 мл) добавляли оксалилдихлорид (1.56 мл, 17.9 ммоль) и ДМФА (0.013 мл, 0.162 ммоль) в бане со льдом, нагревали смесь до комнатной температуры и перемешивали в течение 5 часов. Смесь концентрировали при пониженном давлении, и полученный остаток растворяли в дихлорметане (40 мл), к нему добавляли 2,2,2-трифторэтанол (2.44 г, 24.4 ммоль), триэтиламин (4.50 мл, 32.5 ммоль) и 4-(диметиламино)пиридин (99.0 мг, 0.812 ммоль) в бане со льдом, нагревали смесь до комнатной температуры и перемешивали в течение 1 часа. Смесь концентрировали при пониженном давлении, к полученному остатку добавляли 1 моль/л водный раствор соляной кислоты и экстрагировали смесь этилацетатом. Полученный органический слой промывали 1 моль/л водным раствором соляной кислоты и солевым раствором, сушили над безводным сульфатом магния, получая соединение 18 (5.33 г, 100%).
1Н-ЯМР (CDCl3)δ: 4.64 (q, J=8.2 Гц, 2Н), 5.38 (s, 2Н), 6.49 (d, J=5.6 Гц, 1H), 7.30-7.38 (m, 3Н), 7.43-7.49 (m, 2H), 7.75 (d, J=5.6 Гц, 1H).
Вторая и третья стадии
Соединение 20 получали таким же способом, как на пятой и шестой стадиях в примере 1-1.
1Н-ЯМР (CDCl3)δ: 4.55 (q, J=8.3 Гц, 2Н), 5.18 (s, 2Н), 5.29 (s, 2Н), 6.37 (d, J=7.8 Гц, 1Н), 7.30-7.42 (m, 6H).
Четвертая и пятая стадии
Соединение 23 получали таким же способом, как и на седьмой стадии в примере 1-1.
ЖХ/МС (ИЭР):m/z=342.1 [М+Н]+, RT=1.00,1.09 мин, способ (1)
Шестая стадия
К раствору соединения 23 (820 мг, 2.40 ммоль) в дихлорметане (16.5 мл) добавляли Вос2О (0.837 мл, 3.60 ммоль), триэтиламин (0.499 мл, 3.60 ммоль) и 4-(диметиламино)пиридин (44.0 мг, 0.360 ммоль) и перемешивали смесь при комнатной температуре в течение 3.5 часов. К смеси добавляли 1 моль/л водный раствор соляной кислоты и экстрагировали смесь этилацетатом. Полученный органический слой промывали 1 моль/л водным раствором соляной кислоты и солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 24 (593 мг, 56%) и соединение i3 (170 мг, 16%).
Соединение 24: ЖХ/МС (ИЭР):m/z=441.9 [М+Н]+, RT=1.67 мин, способ (1)
Седьмая стадия
Соединение 24 (547 мг, 1.24 ммоль) растворяли в уксусной кислоте (5.5 мл) и перемешивали смесь при 80°С в течение 5 часов. Смесь концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение i3 (454 мг, 100%).
1Н-ЯМР (CDCl3)δ: 1.46 (d, J=6.4 Гц, 3Н), 3.45 (dd, J=10.5, 10.5 Гц, 1H), 3.55 (dd, J=11.7, 4.3 Гц, 1H), 3.92 (dd, J=11.7, 3.6 Гц, 1H), 3.95-4.01 (m, 2H), 4.76 (dq, J=13.9, 4.3 Гц, 1H), 5.19 (d, J=10.2 Гц, 1H), 5.22 (d, J=10.2 Гц, 1H), 5.36 (d, J=12.9 Гц, 1H), 6.28 (d, J=7.8 Гц, 1H), 7.25 (d, J=7.8 Гц, 1H), 7.28-7.36 (m, 3H), 7.56-7.61 (m, 2H).
[0078]
Пример 1-4: Способ получения соединения III-2

Первая стадия
Соединение i1 (1100 г, 3360 ммоль) и 7,8-дифтор-6,11-дигидродибензотиепин-11-ол (977 г, 3697 ммоль) суспендировали в растворе 50 мас. % Т3Р в этилацетате (3208 г, 5041 ммоль) и этилацетате (1.1 л). К смеси добавляли метансульфоновую кислоту (436 мл, 6721 ммоль) при комнатной температуре и перемешивали смесь при 70°С в течение 5.5 часов. К смеси добавляли воду в бане с ледяной водой и перемешивали смесь при комнатной температуре в течение 1 часа. Добавляли к ней ТГФ и экстрагировали смесь этилацетатом. Полученный органический слой промывали водой и 8% водным раствором гидрокарбоната натрия, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток растворяли в ТГФ (5.5 л) и добавляли к нему карбонат калия (790 г, 5713 ммоль). Смесь нагревали до 50°С, по каплям добавляли к ней бензилбромид (240 мл, 2016 ммоль) и перемешивали смесь при 60°С в течение 8.5 часов. К смеси по каплям добавляли 2 моль/л водный раствор соляной кислоты в бане с ледяной водой, перемешивали смесь при комнатной температуре в течение 10 минут и экстрагировали этилацетатом. Полученный органический слой промывали водой и 8% водным раствором гидрокарбоната натрия и сушили над безводным сульфатом магния. К нему добавляли активированный уголь (Norit SX-2, 240 г), фильтровали смесь через Celite и фильтрат концентрировали при пониженном давлении. К полученному остатку добавляли этилацетат и гексан, и осажденное твердое вещество отфильтровывали, получая соединение 25 (1019 г, 1776 ммоль, 53%).
1Н-ЯМР (CDCl)δ: 2.88 (1H, t, J=11.2 Гц), 3.28-3.39 (2Н, 1H), 3.72 (1H, d, J=12.6 Гц), 3.86 (1Н, d, J=9.6 Гц), 4.03 (1H, d, J=13.9 Гц), 4.45 (1H, d, J=8.6 Гц), 4.67 (1H, d, J=13.1 Гц), 5.19-5.26 (2H, m), 5.45 (1H, d, J=10.9 Гц), 5.63 (1H, d, J=10.9 Гц), 5.77 (1H, d, J=7.6 Гц), 6.40 (1H, d, J=7.8 Гц), 6.68 (1H, t, J=6.9 Гц), 6.94-7.01 (2H, m), 7.03-7.12 (3H, m), 7.29-7.38 (3H, m), 7.61 (2H, d, J=7.1 Гц).
Вторая стадия
К раствору соединения 25 (1200 г, 2092 ммоль) в ДМА (3.6 л) добавляли хлорид лития (443 г, 10.5 моль) при комнатной температуре и перемешивали смесь при 80°С в течение 3 часов. К смеси добавляли ацетон (1.2 л), 0.5 моль/л водный раствор соляной кислоты (6.0 л) и воду (2.4 л) в бане с ледяной водой и перемешивали смесь в течение 1 часа. Выпавшее в осадок твердое вещество фильтровали. Полученное твердое вещество растворяли в хлороформе, к нему добавляли изопропиловый эфир и выпавшее в осадок твердое вещество фильтровали, получая соединение III-2 (950 г, 1965 ммоль, 94%).
1Н-ЯМР (CDCl3)δ: 2.99 (1H, dt, J=17.5, 6.8 Гц), 3.47 (1H, td, J=11.9, 2.5 Гц), 3.60 (1H, t, J=10.6 Гц), 3.81 (1H, dd, J=11.9, 3.3 Гц), 3.96 (1H, dd, J=11.0, 2.9 Гц), 4.07 (1H, d, J=13.8 Гц), 4.58 (1H, dd, J=10.0, 2.9 Гц), 4.67 (1H, dd, J=13.5, 1.9 Гц), 5.26-5.30 (2Н, m), 5.75 (1H, d, J=7.8 Гц), 6.69 (1H, d, J=7.7 Гц), 6.83-6.87 (1H, m), 6.99-7.04 (2Н, m), 7.07-7.15 (3Н, m).
[0079]
Пример 2: Способ получения соединения III-42

Первая стадия
Соединение 34 (947 мг, 5.56 ммоль) растворяли в толуоле (8 мл), добавляли к нему триэтиламин (0.848 мл, 6.12 ммоль) и перемешивали смесь при комнатной температуре в течение 30 минут. Добавляли к ней дифенилфосфоноазид (1.32 мл, 6.12 ммоль), перемешивали смесь при 80°С в течение 1 часа, добавляли к ней (9Н-флуорен-9-ил)метанол (5.46 г, 27.8 ммоль) и нагревали смесь с обратным холодильником при 120°С в течение 1 часа. Смесь охлаждали до комнатной температуры и добавляли к ней насыщенный водный раствор гидрокарбоната натрия, чтобы остановить реакцию. Реакционный раствор экстрагировали этилацетатом, и полученный органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-гексан), получая соединение 35 (1.5 г, 74%).
1Н-ЯМР (CDCl3) δ:7.77 (2Н, d, J=7.3 Гц), 7.60 (2Н, d, J=7.3 Гц), 7.41-7.39 (2Н, m), 7.32-7.30 (2Н, m), 5.10-5.07 (1H, m), 4.75 (1H, brs), 4.41 (2Н, d, J=6.6 Гц), 4.22 (1H, t, J=6.6 Гц), 3.15-3.12 (1H, m), 3.04-3.01 (1H, m), 2.04-1.96 (2Н, m), 1.68 (3Н, s), 1.62 (3Н, s), 1.39-1.37 (1Н, m), 1.17-1.15 (1H, m), 0.90 (3Н, d, J=6.6 Гц), 0.87-0.83 (1H, 1H)
Вторая стадия
Соединение 35 (204 мг, 0.561 ммоль) растворяли в смешанном растворе диоксана (3 мл) и воды (1.5 мл), затем добавляли к нему дигидрат осмата(VI) калия (10.3 мг, 0.028 ммоль) и периодат натрия (360 мг, 1.68 ммоль) и перемешивали смесь при комнатной температуре в течение ночи. Реакцию останавливали 10% водным раствором тиосульфата натрия, реакционный раствор экстрагировали этилацетатом и полученный органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении, получая неочищенный продукт. Неочищенный продукт растворяли в метаноле (4 мл), добавляли к нему моногидрат тосиновой кислоты (13.8 мг, 0.072 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. Реакцию останавливали добавлением насыщенного водного раствора гидрокарбоната натрия, реакционный раствор экстрагировали этилацетатом и полученный органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), получая диастереомерную смесь соединения 36 (115 мг, 58%).
1Н-ЯМР (CDCl3) δ:7.80-7.73 (2Н, m), 7.60-7.54 (2Н, m), 7.45-7.28 (4Н, m), 5.36 (0.5Н, s), 4.95 (0.5 Н, s), 4.62-4.55 (1H, m), 4.48 (1H, d, J=6.6 Гц), 4.30-4.20 (1H, m), 3.90-3.82 (0.5Н, m), 3.74-3.65 (m, 0.5Н), 3.20 (1.5Н, s), 2.90 (1.5Н, s), 2.57 (0.5Н, t, J=11.4 Гц), 2.47 (0.5H, t, J=11.4 Гц), 1.90-1.70 (1H, m), 1.60-1.30 (2.5H, m), 0.93-0.80 (3.5H, m)
Третья стадия
Соединение 36 (115 мг, 0.33 ммоль) и метил 1-амино-3-(бензилокси)-4-оксо-1,4-дигидропиридин-2-карбоксилата (50 мг, 0.182 ммоль) растворяли в ацетонитриле (3 мл) и охлаждали смесь до -30°С. Добавляли к ней тетрахлорид олова (0.032 мл, 0.27 ммоль) и перемешивали смесь в течение 4 часов. К реакционному раствору добавляли насыщенный раствор гидрокарбоната натрия и экстрагировали реакционный раствор метиленхлоридом. Полученный органический слой промывали насыщенным водным раствором хлорида натрия, сушили над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая диастереомерную смесь соединения 37 (115 мг, 59%).
ЖХ/МС (ИЭР):m/z=595 [М+Н]+, RT=2.49, 2.63 мин, способ (1)
Четвертая стадия
Соединение 37 (20.5 г, 34.5 ммоль) растворяли в ТГФ (400 мл), добавляли к нему пиперидин (68.4 мл, 691 ммоль) и перемешивали смесь при комнатной температуре в течение 2 часов. Реакционный раствор разбавляли диэтиловым эфиром (500 мл) и полученные осадки фильтровали с получением неочищенного продукта.
Неочищенный продукт растворяли в этаноле (200 мл), добавляли к нему DBU (5.06 мл, 33.6 ммоль) и перемешивали смесь при 80°С в течение 2 часов. Реакционный раствор концентрировали и полученные твердые вещества перекристаллизовывали из ТГФ, получая соединение 38 (6.5 г, 57%).
ЖХ/МС (H3P):m/z=340 [М+Н]+, RT=1.29 мин, способ (1)
Пятая стадия
Соединение 38 (2.0 г, 5.89 ммоль) и 7,8-дифтор-6,11-дигидродибензо[b,е]тиепин-11-ол (2.34 г, 8.84 ммоль) растворяли в растворе Т3Р в этилацетате (18 мл) и перемешивали смесь в закрытой пробирке при 110°С в течение 1,5 часов. Реакцию останавливали добавлением воды, реакционный раствор экстрагировали этилацетатом и полученный органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из смеси хлороформ-гексан, получая соединение 39 (1.1 г, 32%).
ЖХ/МС (ИЭР):m/z=586 [М+Н]+, RT=2.46 мин, способ (1)
Шестая стадия
Соединение 39 (1.1 г, 1.89 ммоль) растворяли в диметилацетамиде (10 мл), добавляли к нему хлорид лития (398 мг, 9.39 ммоль) и перемешивали смесь при 120°С. Реакционный раствор разбавляли этилацетатом, и полученный органический слой промывали 2 моль/л соляной кислотой, сушили безводным сульфатом натрия и концентрировали при пониженном давлении, получая соединение III-42 (555 мг, 59.6%).
1Н-ЯМР (CDCl3) δ: 7.15-7.03 (4Н, m), 7.01-6.94 (1Н, m), 6.86-6.82 (1H, m), 6.68 (1Н, d, J=7.8 Гц), 5.78 (1H, d, J=7.6 Гц), 5.35 (1H, dd, J=13.8, 2.4 Гц), 5.22 (1H, s), 4.65-4.57 (1H, m), 4.25 (1H, dd, J=11.4, 2.5 Гц), 4.05 (1H, d, J=13.9 Гц), 2.18 (1H, t, J=12.4 Гц), 1.96 (1Н, d, J=13.6 Гц), 1.87-1.57 (5Н, m), 1.29-1.22 (2Н, m), 0.91 (3Н, d, J=6.6 Гц).
ЖХ/МС (ИЭР):m/z=497 [М+Н]+, RT=2.16 мин, способ (1)
[0080]
Пример 3: Способ получения соединения 42

Первая стадия
Соединение 40 (1.00 г, 13.3 ммоль) растворяли в ТГФ (100 мл), добавляли к нему 60% гидрид натрия (0.59 г, 14.7 ммоль) при комнатной температуре и перемешивали смесь в токе азота при комнатной температуре в течение 30 минут. Добавляли к ней этилхлорацетат (1.4 мл, 13.3 ммоль) и перемешивали смесь в токе азота при комнатной температуре в течение 30 минут и при 90°С в течение 3 часов. После концентрирования при пониженном давлении к остатку добавляли ТГФ (40 мл), затем к нему добавляли 60% гидрид натрия (0.59 г, 14.7 ммоль) и перемешивали смесь в токе азота при комнатной температуре в течение 30 минут. К смеси по каплям добавляли аллилхлорформиат и перемешивали смесь при комнатной температуре в течение 3 часов. Добавляли к ней насыщенный водный раствор хлорида аммония (30 мл) и экстрагировали реакционный раствор этилацетатом (150 мл). Полученный органический слой промывали водой (50 мл) и солевым раствором (100 мл) и сушили над безводным сульфатом магния, полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), получая соединение 41 (0.96 г, 36%).
1Н-ЯМР (CDCl3) δ: 1.42 (d, J=6.5 Гц, 3Н), 3.77-3.89 (m, 2Н), 4.19 (d, J=17.4 Гц, 1H), 4.27-4.36 (m, 2H), 4.74-4.83 (m, 2H), 5.31 (dd, J=10.4, 1.4 Гц, 1H), 5.46 (dd, J=17.2, 1.4 Гц, 1H), 5.98 (dddd, J=17.2, 10.4, 5.6, 5.6 Гц, 1H).
ЖХ/МС (ИЭР):m/z=199.8 [M+H]+, способ (1)
Вторая стадия
Соединение 41 (2.69 г, 13.5 ммоль) растворяли в ТГФ (30 мл) в атмосфере азота и охлаждали до -78°С сухим льдом-ацетоном. К нему по каплям добавляли раствор 1.02 моль/л DIBAL-H в гексане (17.2 мл, 17.6 ммоль) и перемешивали смесь при -78°С в течение 1 часа. К ней добавляли водный раствор соли Рошелля, перемешивали смесь и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток растворяли в метаноле (30 мл), добавляли к нему моногидрат п-толуолсульфоновой кислоты (0.244 г, 1.28 ммоль) и перемешивали смесь при комнатной температуре в течение 7 часов. Смесь гасили водным раствором гидрокарбоната натрия и экстрагировали этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-гексан), получая соединение 42 (2.43 г, 88%).
1Н-ЯМР (CDCl3) δ: 1.42 (d, J=7.0 Гц, 3Н), 3.34 (s, 3Н), 3.53 (dd, J=12.0, 2.3 Гц, 1H), 3.64 (dd, J=11.5, 3.8 Гц, 1H), 3.76 (d, J=11.4 Гц, 1H), 4.01 (d, J=12.0 Гц, 1H), 4.06-4.12 (m, 1H), 4.65 (d, J=5.4 Гц, 2H), 5.14 (br s, 1H), 5.24 (dd, J=10.4, 1.3 Гц, 2H), 5.33 (dd, J=17.3, 1.4 Гц, 2H), 5.95 (ddd, J=22.6, 10.7, 5.5 Гц, 1H).
[0081]
Пример 4: Способ получения соединения 50

Первая стадия
Соединение 43 (4.00 г, 16.3 ммоль) растворяли в дихлорметане (40 мл), затем к нему по каплям добавляли оксалилдихлорид (1.56 мл, 17.9 ммоль) и ДМФА (0.013 мл, 0.162 ммоль) в бане со льдом, нагревали смесь до комнатной температуры и перемешивали в течение 5 часов. Смесь концентрировали при пониженном давлении, и полученный остаток растворяли в дихлорметане (40 мл), затем к нему по каплям добавляли 2,2,2-трифторэтанол (2.44 г, 24.4 ммоль), триэтиламин (4.50 мл, 32.5 ммоль) и 4-(диметиламино)пиридин (99.0 мг, 0.812 ммоль) в бане со льдом, нагревали смесь до комнатной температуры и перемешивали в течение 1 часа. Смесь концентрировали при пониженном давлении, к полученному остатку добавляли 1 моль/л водный раствор соляной кислоты (100 мл) и экстрагировали смесь этилацетатом (200 мл). Полученный органический слой промывали 1 моль/л водным раствором соляной кислоты (100 мл) и солевым раствором (100 мл), сушили над безводным сульфатом магния, получая соединение 44 (5.33 г, 100%).
1Н-ЯМР (CDCl3) δ: 4.64 (q, J=8.2 Гц, 2Н), 5.38 (s, 2Н), 6.49 (d, J=5.6 Гц, 1H), 7.30-7.38 (m, 3Н), 7.43-7.49 (m, 2Н), 7.75 (d, J=5.6 Гц, 1H).
Вторая стадия
Соединение 44 (5.33 г, 16.2 ммоль) растворяли в ДМФА (55 мл), затем добавляли к нему Вос-гидразин (1.93 г, 14.6 ммоль) и PPTS (12.2 г, 0.162 ммоль) и перемешивали смесь при 60°С в течение 16 часов. К реакционному раствору добавляли воду (100 мл) и экстрагировали реакционный раствор этилацетатом (300 мл). Полученный органический слой промывали водой (100 мл) и солевым раствором (100 мл) и сушили над безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), получая соединение 45 (1.59 г, 22%).
1Н-ЯМР (CDCl3) δ: 1.45 (s, 9Н), 4.51 (q, J=8.2 Гц, 2Н), 5.29 (s, 2Н), 6.42 (d, J=7.9 Гц, 1H), 7.28-7.37 (m, 4Н), 7.39-7.43 (m, 2Н), 7.68 (brs, 1H).
Третья стадия
Соединение 45 (1.59 г, 3.59 ммоль) растворяли в 4 моль/л хлористого водорода в этилацетате (16 мл) и перемешивали смесь при комнатной температуре в течение 1,5 часов. К реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия (100 мл), экстрагировали реакционный раствор дихлорметаном (200 мл) и сушили над безводным сульфатом магния, получая соединение 46 (1.18 г, 96%).
1Н-ЯМР (CDCl3) δ: 4.55 (q, J=8.3 Гц, 2Н), 5.18 (s, 2Н), 5.29 (s, 2Н), 6.37 (d, J=7.8 Гц, 1H), 7.30-7.42 (m, 6Н).
Четвертая стадия
Соединение 46 (1.18 г, 3.45 ммоль) и соединение 42 (890 мг, 4.14 ммоль) растворяли в ацетонитриле (24 мл), к раствору по каплям добавляли тетрахлорид олова (0.607 мл, 5.17 ммоль) при -30°С и перемешивали смесь при -30°С в течение 1 часа. К реакционному раствору добавляли насыщенный водный раствор гидрокарбоната натрия (200 мл) и дихлорметан (200 мл), и нежелательные вещества отфильтровывали. Полученный органический слой промывали солевым раствором (100 мл) и сушили над безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 47 (1.22 г, 67%).
ЖХ/МС (ИЭР):m/z=525.9 [М+Н]+, RT=2.02 мин, способ (1)
Пятая стадия
Соединение 47 (1.15 г, 2.19 ммоль) растворяли в ТГФ (23 мл), затем добавляли к нему в атмосфере азота морфолин (0.953 мл, 10.9 ммоль) и тетракис(трифенилфосфин)палладий (126 мг, 0.109 ммоль) и перемешивали смесь при комнатной температуре в течение 7.5 часов. Смесь концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 48 (890 мг, колич.).
ЖХ/МС (ИЭР):m/z=342.1 [М+Н]+, RT=1.00, 1.09 мин, способ (1)
Шестая стадия
Соединение 48 (820 мг, 2.40 ммоль) растворяли в дихлорметане (16.5 мл), затем добавляли к нему Вос2О (0.837 мл, 3.60 ммоль), триэтиламин (0.499 мл, 3.60 ммоль) и 4-(диметиламино)пиридин (44.0 мг, 0.360 ммоль) и перемешивали смесь при комнатной температуре в течение 3.5 часов. К реакционному раствору добавляли 1 моль/л водный раствор соляной кислоты (50 мл) и экстрагировали смесь этилацетатом (125 мл). Полученный органический слой промывали 1 моль/л водным раствором соляной кислоты (50 мл) и солевым раствором (50 мл) и сушили над безводным сульфатом натрия. Полученный органический слой концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 49 (593 мг, 56%) и соединение 50 (170 мг, 16%).
ЖХ/МС (ИЭР):m/z=441.9 [М+Н]+, RT=1.67 мин, способ (1)
Седьмая стадия
Соединение 49 (547 мг, 1.24 ммоль) растворяли в уксусной кислоте (5.5 мл) и перемешивали смесь при 80°С в течение 5 часов. Смесь концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 50 (454 мг, колич.).
1Н-ЯМР (CDCl3) δ: 1.46 (d, J=6.4 Гц, 3Н), 3.45 (dd, J=10.5, 10.5 Гц, 1H), 3.55 (dd, J=11.7, 4.3 Гц, 1H), 3.92 (dd, J=11.7, 3.6 Гц, 1H), 3.95-4.01 (m, 2Н), 4.76 (dq, J=13.9, 4.3 Гц, 1H), 5.19 (d, J=10.2 Гц, 1H), 5.22 (d, J=10.2 Гц, 1H), 5.36 (d, J=12.9 Гц, 1H), 6.28 (d, J=7.8 Гц, 1H), 7.25 (d, J=7.8 Гц, 1H), 7.28-7.36 (1H, 3Н), 7.56-7.61 (m, 2Н).
[0082]
Пример 5: Способ получения соединения 55
Первая стадия
К суспензии соединения 51 (19.2 г, 77.8 ммоль) и карбоната калия (16.13 г, 117 ммоль) в ацетоне (190 мл) добавляли тиофенол (8.01 мл, 78 ммоль) и перемешивали смесь при 40°С в течение 1 часа. Реакционный раствор охлаждали до 25°С и добавляли к нему этилацетат и воду. Смесь экстрагировали этилацетатом, полученный органический слой дважды промывали водой и концентрировали при пониженном давлении, получая соединение 52.
1Н-ЯМР (CDCl3) δ: 3.93 (3Н, s), 4.93 (2Н, s), 7.03-7.07 (1Н, m), 7.18-7.35 (6Н, m), 7.93-8.06 (1Н, m).
Вторая стадия
К раствору соединения 52 (21.5 г, 77.8 ммоль) в метаноле (60 мл) и ТГФ (40 мл) по каплям добавляли раствор 2 моль/л гидроксида натрия (97.0 мл, 195 ммоль) на ледяной бане и реакционный раствор оставляли стоять в течение всей ночи. Реакционный раствор концентрировали при пониженном давлении и добавляли к нему воду. Полученный водный слой дважды промывали гексаном. Водный слой подкисляли 6 моль/л соляной кислотой и дважды экстрагировали этилацетатом. Полученный органический слой сушили сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток кристаллизовали из смеси этилацетат/гексан, получая соединение 53 в виде кристаллов (7.7 г).
1Н-ЯМР (CDCl3) δ: 4.40 (2Н, s), 6.81-6.84 (1Н, m), 7.07-7.32 (6Н, m), 7.86-7.90 (1Н, m).
Третья стадия
К полифосфорной кислоте (200 г, 29.4 ммоль) добавляли соединение 53 (7.70 г, 29.4 ммоль) при 60°С, нагревали реакционную смесь до 140°С и перемешивали в течение 1 часа. Реакционный раствор охлаждали до 40°С и добавляли к нему воду в бане со льдом. Суспензию фильтровали, и фильтрат промывали водой. Добавляли к фильтрату этилацетат и полученный органический слой промывали водой и солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток кристаллизовали из смеси этилацетат/гексан, получая соединение 54 в виде кристаллов (3.6 г).
1Н-ЯМР (CDCl3) δ: 4.03 (2Н, s), 6.92-7.06 (2Н, m), 7.26-7.40 (4Н, m), 7.67 (1H, dd, J=5.5 Гц, J=8.0 Гц), 8.25 (1H, d, J=8.0 Гц).
Четвертая стадия
К раствору соединения 54 (3.60 г, 14.7 ммоль) в метаноле (14 мл) и ТГФ (28 мл) добавляли гидроборат натрия (613 мг, 16.2 ммоль) в ледяной бане. Реакционный раствор перемешивали при комнатной температуре в течение 30 минут. К нему добавляли воду и оставляли реакционный раствор стоять в течение всей ночи. Концентрировали реакционный раствор при пониженном давлении и добавляли к концентрату этилацетат и воду. Смесь экстрагировали. Полученный органический слой промывали солевым раствором, сушили над сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (метиленхлорид), получая соединение 55 (1.7 г) в виде кристаллов.
1Н-ЯМР (CDCl3) δ: 4.15 (1H, d, J=14.0 Гц), 4.57 (1H, d, J=14.0 Гц), 6.09 (1H, s), 6.91-6.93 (2Н, m), 7.10-7.17 (3Н, m), 7.39 (1H, dd, J=5.5 Гц, J=8.0 Гц), 7.48 (1H, d, J=8.0 Гц).
[0083]
Пример 6: Способ получения соединения Ш-30
Первая стадия
Соединение 50 (154.0 мг, 0.451 ммоль) и соединение 55 (117 мг, 0.474 ммоль) растворяли в растворе Т3Р в этилацетате (1.5 мл) и перемешивали смесь в герметичной пробирке при 100°С в течение 4 часов. Реакцию останавливали добавлением воды, реакционный раствор экстрагировали этилацетатом и полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из смеси хлороформ-гексан, получая соединение 56 (70.2 мг, 27%).
ЖХ/МС (ИЭР):m/z=570 [М+Н]+, RT=2.18 мин, способ (1)
Вторая стадия
Соединение 56 (70.2 мг, 0.123 ммоль) растворяли в ДМА (1 мл), добавляли к нему хлорид лития (52.2 г, 1.23 ммоль) и перемешивали смесь при 100°С в течение 4 часов. Смесь охлаждали до комнатной температуры и добавляли к ней 1 моль/л водный раствор соляной кислоты, чтобы остановить реакцию. Реакционный раствор экстрагировали этилацетатом, полученный органический слой промывали 1 моль/л соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученные твердые вещества промывали этилацетатом, получая соединение III-30 (28.6 мг, 48%).
1Н-ЯМР (CDCl3) δ: 1.78 (d, J=7.2 Гц, 3Н), 3.26-3.32 (m, 1Н), 3.44-3.60 (1H, 3Н), 3.72 (dd, J=11.7, 2.6 Гц, 1H), 3.94 (dd, J=11.2, 2.9 Гц, 1H), 4.42 (dd, J=9.9, 2.8 Гц, 1H), 5.29 (s, 1H), 5.54 (d, J=13.6 Гц, 1H), 5.76 (d, J=7.8 Гц, 1H), 6.71 (d, J=7.7 Гц, 1H), 6.81-6.86 (m, 1H), 6.96-7.04 (m, 2H), 7.07-7.11 (m, 3H), 7.23-7.25 (m, 1H).
ЖХ/МС (ИЭР):m/z=480 [M+H]+, RT=1.87 мин, способ (1)
[0084]
Пример 7: Способ получения соединения III-54
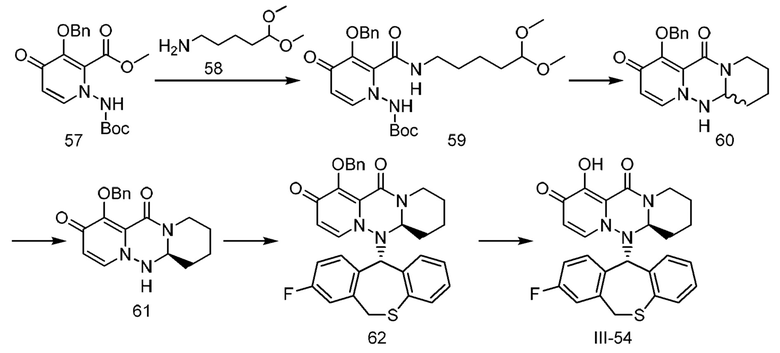
Первая стадия
Соединение 57 (60.6 г, 0.162 моль) растворяли в ТГФ (122 мл), затем добавляли к нему соединение 58 (Journal of Organic Chemistry, 80(20), 9868-9880; 2015) (50 г, 0.34 моль) и DBU (2.6 г, 17 ммоль) и перемешивали смесь при 60°С в течение 48 часов. Реакционный раствор концентрировали и очищали с помощью колоночной хроматографии на силикагеле (дихлорметан-метанол), получая соединение 59 (73.2 г, 93.3%).
Вторая стадия
Соединение 59 (73.2 г, 0.15 моль) растворяли в смешанном растворителе ацетонитрила (580 мл) и воды (145 мл), добавляли к нему метансульфоновую кислоту (43.1 г, 0.45 моль) и перемешивали смесь при 60°С в течение 20 часов. Смесь концентрировали при пониженном давлении до тех пор, пока остаток не составил примерно 300 мл. Добавляли к нему этилацетат (250 мл) и водный раствор карбоната натрия (500 мл), рН смеси доводили до 8 и перемешивали смесь в течение 30 минут. Осадки отфильтровывали и промывали смешанным растворителем этилацетата-гексана и воды и сушили, получая соединение 60 (33 г, 68%).
1H ЯМР (400 МГц, d-ДМСО) δ:7.70 (d, J=7.6 Гц, 1H), 7.55 (d, J=7.1 Гц, 2Н), 7.40-7.22 (m, 4Н), 6.21 (d, J=7.6 Гц, 1H), 5.05 (s, 2Н), 4.59 (d, J=3.6 Гц, 1H), 4.39-4.27 (m, 1H), 2.80-2.65 (m, 1H), 1.99-1.87 (m, 1H), 1.78 (s, 2Н), 1.51-1.15 (m, 3Н).
Третья стадия
Оптическое разделение соединения 60 (4.0 г, 12.3 ммоль) с помощью Waters SFC30 System (Daicel CHIRALPAK IB, сжиженный диоксид углерода-метанол) дает соединение 61 (1.79 г, 45%).
Условия анализа
<Waters SFC30 System>
Колонка: CHIRALPAK IB/SFC (5 мкм, внутр.д. 250×4.6 мм) (DAICEL)
Скорость потока: 8.0 мл/мин; Длина волны УФ-детектирования: 254 нм
Обратное давление: 100 бар
Подвижная фаза: [А]: сжиженный диоксид углерода, [В]: метанол
Градиент: 5% растворителя [В] выдерживали в течение 1 минуты, линейный градиент от 5% до 40% растворителя [В] проводили в течение 6 минут, 40% растворителя [В] выдерживали в течение 2 минут, и 5% растворителя [В] выдерживали в течение 1 минуты.
Время элюирования: 7.9 минут
Четвертая стадия
Соединение 61 (1.76 г, 5.41 ммоль) и 8-фтор-6,11-дигидродибензо[b,е]тиепин-11-ол (1.99 г, 8.11 ммоль) растворяли в Т3Р (50%, раствор этилацетата, 16 мл) и перемешивали смесь в герметичной пробирке при 100°С в течение 3 часов. Добавляли к ней этилацетат и воду, смесь нейтрализовали насыщенным водным раствором гидрокарбоната натрия и экстрагировали водный слой этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток растворяли в ТГФ (15 мл), затем добавляли к нему карбонат калия (1.49 г, 10.8 ммоль) и бензилбромид (0.321 мл) и нагревали смесь с обратным холодильником в течение 6 часов. Далее добавляли к ней 1-метилпиперазин (0.30 мл) и нагревали смесь с обратным холодильником в течение 1 часа. Реакционный раствор экстрагировали этилацетатом. Полученный органический слой промывали 2 моль/л соляной кислотой и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-ацетон), получая соединение 62 (1.15 г, 38.4%).
ЖХ/МС (ИЭР):m/z=554 [М+Н]+, RT=2.24 мин, способ (1)
Пятая стадия
Соединение 62 (1.0 г, 1.81 моль) и хлорид лития (383 мг, 9.03 ммоль) растворяли в ДМА и перемешивали смесь при 100°С в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры, добавляли к нему ацетон (5 мл), по каплям добавляли к нему 1 моль/л водный раствор соляной кислоты (40 мл) и перемешивали смесь при комнатной температуре в течение 15 минут. Полученные белые твердые вещества фильтровали, промывали 50% водным раствором ацетона и сушили, получая соединение III-54 (760 мг, 91%).
1Н-ЯМР(СDСl3)δ:1.47-2.05(m, 6Н), 2.50-2.58(m, 1Н), 3.51(d, J=12.0 Гц, 1Н), 4.26-4.31(m, 1Н), 4.68-4.74(m, 1H), 5.22(s, 1H), 5.62(d, J=13.6 Гц, 1H), 5.77(d, J=7.6 Гц, 1H), 6.68(d, J=7.6 Гц, 1Н), 6.80-6.82(m, 1H), 6.88-7.02(m, 1Н), 7.03-7.15(m, 5Н)
ЖХ/МС (ИЭР):m/z=464.2 [М+Н]+, RT=1.92 мин, способ (1)
[0085]
Пример 8: Способ получения соединения III-20
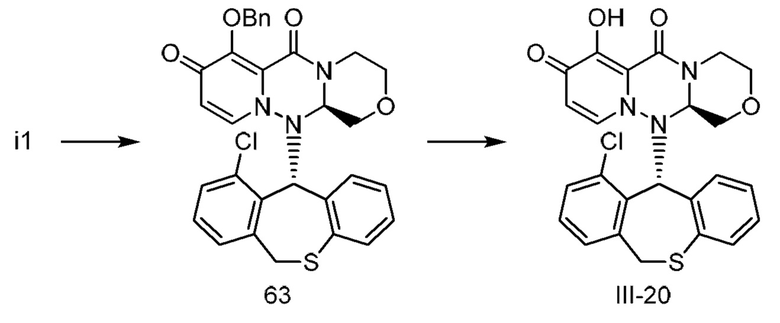
Первая стадия
Соединение i1 (500 мг, 1.53 ммоль) и 10-хлор-6,11-дигидродибензо[b,е]тиепин-11-ол (602 мг, 2.29 ммоль) растворяли в Т3Р (50%, раствор этилацетата, 5 мл) и перемешивали смесь в герметичной пробирке при 105°С в течение 1,5 часов. Реакционный раствор экстрагировали этилацетатом, полученный органический слой промывали водой и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-этилацетат-метанол), получая соединение 63 (210 мг, 24%).
1Н-ЯМР(СDСl3)δ: 2.96 (t, J=12.0 Гц, 1H), 3.30-3.43 (m, 2Н), 3.53 (d, J=13.2 Гц, 1H), 3.73 (d, J=11.6 Гц, 1H), 3.86 (d, J=10.8 Гц, 1H), 4.42 (d, J=7.2 Гц, 1H), 4.71 (d, J=13.6 Гц, 1H), 5.42 (d, J=10.8 Гц, 1H), 5.57-5.65 (m, 2Н), 5.79 (d, J=7.6 Гц, 1H), 6.18 (s, 1H), 6.47 (d, J=7.2 Гц, 1H), 6.67 (t, J=6.8 Гц, 1H), 7.00-7.14 (1H, 3Н), 7.22-7.40 (m, 6Н), 7.64 (d, J=7.2 Гц, 2Н).
Вторая стадия
Соединение 63 (210 мг, 0.368 ммоль) растворяли в ДМА (3 мл), добавляли к нему хлорид лития (78 мг, 1.84 ммоль) и перемешивали смесь при 100°С в течение 4 часов. Реакционный раствор экстрагировали этилацетатом, полученный органический слой промывали 1 моль/л соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток перекристаллизовывали из смеси дихлорметан-диэтиловый эфир, получая соединение III-20 (124 мг, 70%).
1Н-ЯМР(СDСl3)δ: 3.09 (t, J=12.8 Гц, 1H), 3.48 (t, J=11.6 Гц, 1H), 3.55-3.62 (m, 2Н), 3.81 (d, J=11.6 Гц, 1H), 3.93 (d, J=10.8 Гц, 1H), 4.53 (d, J=9.6 Гц, 1H), 4.69 (d, J=13.2 Гц, 1H), 5.68 (d, J=12.8 Гц, 1H), 5.76 (d, J=6.8 Гц, 1H), 6.26 (s, 1H), 6.80-6.88 (m, 2Н), 7.05-7.15 (m, 3Н), 7.24-7.28 (m, 1H), 7.34 (t, J=7.6 Гц, 1H), 7.39 (d, J=8.0 Гц, 1H).
[0086]
Пример 9: Способ получения соединения III-33

Первая стадия
Соединение 64 (3.54 г, 14.9 ммоль) растворяли в дихлорметане (70 мл) и по каплям добавляли к нему DIBAL-H (1 моль/л, 16.4 мл, 16.4 ммоль) при -78°С. Перемешивали смесь при -78°С в течение 1 часа, реакцию гасили метанолом и температуру смеси повышали до комнатной температуры. Реакционную смесь экстрагировали дихлорметаном, полученный органический слой промывали 2 моль/л соляной кислотой и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток растворяли в метаноле (35 мл), добавляли к нему хлорид аммония (80 мг, 1.49 ммоль) и нагревали смесь с обратным холодильником в течение 1 часа. Реакционную смесь экстрагировали диэтиловым эфиром, и полученный органический слой промывали насыщенным водным раствором гидрокарбоната натрия и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 65 (2.73 г, 76%).
1Н-ЯМР(СDСl3)δ: 0.96(s, 6Н), 1.53(d, J=5.2 Гц, 2Н), 1.85-1.90(m, 2Н), 3.31(s, 6Н), 3.38-3.43(m, 2Н), 4.44(t, J=5.2 Гц, 1H)
Вторая стадия
Соединение 65 (2.7 г, 11.3 ммоль) растворяли в ацетоне (50 мл), затем добавляли к нему карбонат калия (5.46 г), изоиндолин-1,3-дион (2.49 г, 16.9 ммоль) и бромид тетрабутиламмония (1.09 г, 3.39 ммоль) и нагревали смесь с обратным холодильником в течение 6 часов. Реакционный раствор фильтровали и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (гексан-этилацетат), получая соединение 66 (1.1 г, 32%).
1Н-ЯМР(СDСl3)δ: 1.04(s, 6Н), 1.58-1.65(m, 4Н), 3.32(s, 6Н), 3.65-3.75(m, 2Н), 4.52(t, J=5.2 Гц, 1Н), 7.65-7.75 (m, 2Н), 7.80-7.90(m, 2Н)
Третья стадия
Соединение 66 (1.14 г, 3.73 ммоль) растворяли в смеси этанол-вода (1:1, 4 мл), добавляли к раствору моногидрат гидразина (374 мг, 7.47 ммоль) и перемешивали смесь при 60°С в течение 5 часов. Реакционный раствор экстрагировали дихлорметаном, и полученный органический слой промывали 1 моль/л водным раствором гидроксида натрия и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 67 (750 мг, 100%).
1Н-ЯМР(СDСl3)δ: 0.93(s, 6Н), 1.38-1.45(m, 2Н), 1.52(d, J=5.2 Гц, 2Н), 2.65-2.75(m, 2Н), 3.30(s, 6Н), 4.46 (t, J=5.2 Гц, 1H), 5.30(s, 2Н)
Четвертая стадия
Соединение 57 (300 мг, 0.80 ммоль) растворяли в ТГФ (0.6 мл), затем добавляли к нему соединение 67 (351 мг, 2.00 ммоль) и DBU (12 мкл, 0.08 ммоль) и перемешивали смесь при 60°С в течение 18 часов. Реакционный раствор концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 68 (440 мг, 80%).
1Н-ЯМР(СDСl3)δ: 0.90(s, 6Н), 1.20-1.32(m, 2Н), 1.44(s, 9Н), 1.47(d, J=5.2 Гц, 2Н), 3.11-3.25(m, 2Н), 3.26(s, 6Н), 4.41 (t, J=5.2 Гц, 1H), 5.28(s, 2Н), 6.38 (d, J=8.0, 1H), 6.87(br, 1H), 7.29-7.40(m, 6H), 8.49(br, 1H)
Пятая стадия
Соединение 68 (210 мг, 0.41 ммоль) растворяли в смешанном растворителе ацетонитрила (1.8 мл) и воды (310 мкл), добавляли к раствору метансульфоновую кислоту (79 мкл, 1.22 ммоль) и перемешивали смесь при 60°С в течение 6 часов. Смесь охлаждали до комнатной температуры, рН смеси доводили до 7 2 моль/л водным раствором гидроксида натрия и экстрагировали реакционный раствор дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение 69 (55 мг, 49%).
1Н-ЯМР(СDСl3)δ: 0.95(s, 3Н), 0.98(s, 3Н), 1.40-1.45(m, 2Н), 1.53-1.65(m, 2Н), 2.80-2.89(m, 1H), 4.05-4.15(m, 1H), 4.40-4.48(m, 1H), 5.14-5.31 (m, 2Н), 5.48(d, J=12.4 Гц, 1H), 6.32 (d, J=7.6 Гц, 1H), 7.25-7.40(m, 4Н), 7.59(d, J=6.8 Гц, 2Н)
Шестая стадия
Оптическое разделение соединения 69 (6.69 г, 18.9 ммоль) с помощью Waters SFC30 System (Daicel CHIRALPAK IB, сжиженный диоксид углерода-метанол) дает соединение 70 (3.40 г, 50%). Условия анализа<Waters SFC30 System>
Колонка: CHIRALPAK IB/SFC (5 мкм, внутр.д. 250×4.6 мм) (DAICEL) Скорость потока: 8.0 мл/мин; Длина волны УФ-детектирования: 254 нм Обратное давление: 100 бар
Подвижная фаза: [А]: сжиженный диоксид углерода, [В]: метанол
Градиент: 5% растворителя [В] выдерживали в течение 1 минуты, линейный градиент от 5% до 40% растворителя [В] проводили в течение 6 минут, 40% растворителя [В] выдерживали в течение 2 минут, и 5% растворителя [В] выдерживали в течение 1 минуты.
Время элюирования: 7.67 минут Седьмая стадия
Соединение 70 (2.0 г, 5.66 ммоль) и 7,8-дифтор-6,11-дигидродибензо[Ь,е]тиепин-11-ол (1.94 г, 7.36 ммоль) растворяли в ТЗР (50% этилацетат, 20 мл) и перемешивали смесь в герметичной пробирке при 105°С в течение 2.5 часов. Реакционный раствор экстрагировали этилацетатом, и полученный органический слой промывали водой и солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-ацетон), получая диастереомерную смесь (2.84 г).
Перекристаллизовывали ее из этилацетата, получая соединение 71 (1.26 г, 37%).
1Н-ЯМР(CDCl3)δ: 0.80(s, 3Н), 0.91(s, 3Н), 1.20-1.45(m, 4Н), 2.49-2.58(m, 1H),v4.02(d, J=13.6 Гц, 1H), 4.25-4.33(m, 1H), 4.55-4.65(m, 1Н), 5.19(s, 1H), 5.24-5.30(m, 1Н), 5.47-5.59(m, 2Н), 5.80(d, J=7.6 Гц, 1H), 6.42(d, J=8.0 Гц, 1H), 6.65-6.71(m, 1H), 6.95-7.12(m, 5Н), 7.27-7.39(m, 3Н), 7.61(d, J=6.8 Гц, 2Н)
Восьмая стадия
Соединение 71 (980 мг, 1.63 ммоль) растворяли в ДМА (8 мл), добавляли к нему хлорид лития (346 мг, 8.17 ммоль) и перемешивали смесь при 100°С в течение 3 часов. Реакционный раствор экстрагировали этилацетатом, полученный органический слой промывали 2 моль/л соляной кислотой и солевым раствором, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток кристаллизовали из этилацетата, получая соединение III-33 (780 мг, 94%).
1Н-ЯМР(CDCl3)δ: 0.85(s, 3Н), 0.97(s, 3Н), 1.34-2.00(m, 4Н), 2.62-2.66(m, 1H), 4.05(d, J=13.6 Гц, 1H), 4.40-4.48(m, 1H), 4.56-4.63(m, 1H), 5.24(s, 1H), 5.30-5.35(m, 1Н), 5.80(d, J=7.6 Гц, 1H), 6.68(d, J=7.6 Гц, 1H), 6.78-6.90(m, 1H), 6.95-7.15(m, 4Н), 7.16-7.22(m, 1H)
ЖХ/МС (ИЭР):m/z=510.2 [М+Н]+, RT=2.25 мин, способ (1)
[0087]
Следующие примеры соединений были синтезированы из коммерчески доступных соединений или промежуточных соединений, подходящим образом синтезированных из коммерчески доступных соединений в соответствии с приведенными выше примерами.





[0088]

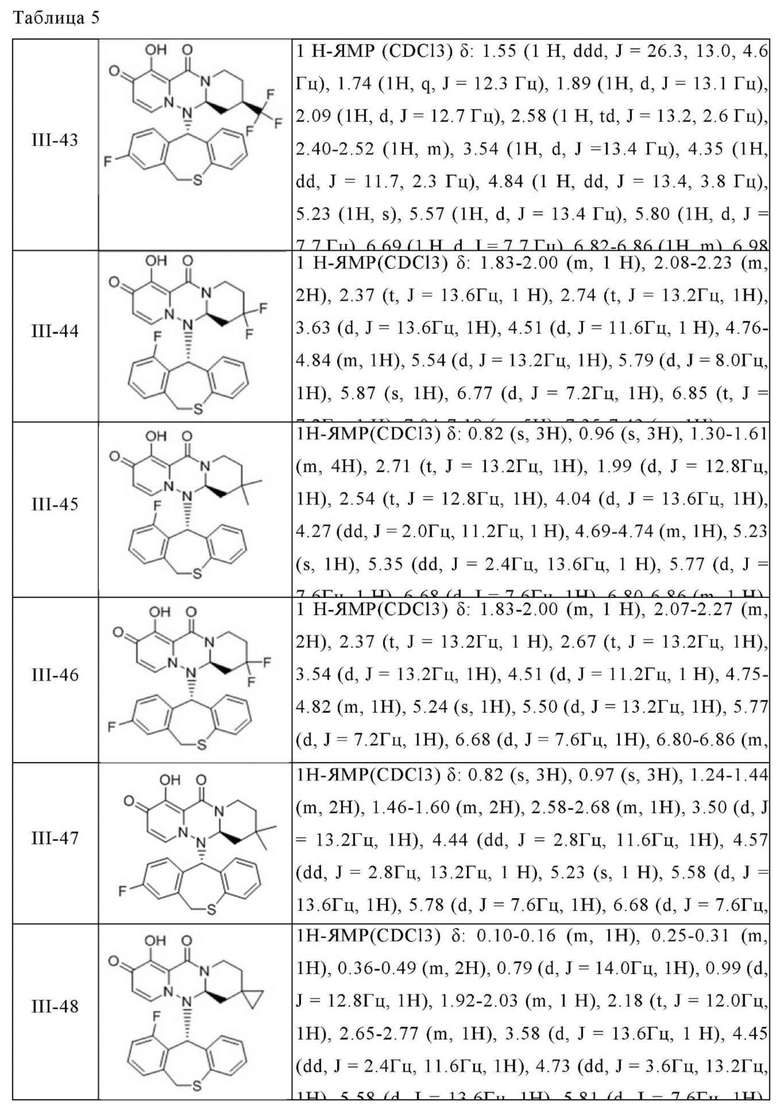


[0089]
Пример 10: Способ получения соединения II-6

К соединению III-2 (4.0 г, 8.3 ммоль) добавляли карбонат калия (1483.4 мг, 10.7 ммоль), иодид калия (549.5 мг, 3.3 ммоль), тетрагидрофуран (33.1 г), N,N-диметилацетамид (3.8 г) и воду (80.3 мг) и полученную смесь перемешивали. Повышали температуру до 60°С и добавляли хлорметилметилкарбонат (1758.9 мг, 14.2 ммоль). Смесь перемешивали при 60°С в течение 9 часов и охлаждали до 20°С. Добавляли уксусную кислоту (822.0 мг), 2-пропанол (3.1 г) и воду (20.0 г) и дважды проводили экстракцию смеси тетрагидрофураном (1.8 г, 8.9 г). Полученный органический слой концентрировали при пониженном давлении отгонкой растворителя до массы жидкости примерно 32 г. Температуру повышали до 45°С, добавляли 2-пропанол (1.6 г) и охлаждали смесь до 20°С. Добавляли водный раствор ацетата натрия, полученный из ацетата натрия (339.0 мг) и воды (46.0 г), и охлаждали смесь до 5°С. Перемешивали смесь при 5°С в течение 3 часов и отфильтровывали полученные белые с бледно-желтоватым оттенком осадки. Промывали полученные твердые вещества смешанным раствором 2-пропанола (4.7 г) и воды (6.0 г) и снова промывали твердые вещества 2-пропанолом (6.3 г). Добавляли диметилсульфоксид (30.9 г) к полученным белым с бледно-желтоватым оттенком твердым веществам и перемешивали смесь. Температуру повышали до 60°С и добавляли смешанный раствор диметилсульфоксида (2.2 г) и воды (4.8 г). Далее добавляли смешанный раствор диметилсульфоксида (19.9 г) и воды (28.4 г) и охлаждали смесь до 20°С. Перемешивали смесь при 20°С в течение 3 часов и полученные белые осадки фильтровали. Промывали полученные твердые вещества смешанным раствором диметилсульфоксида (8.0 г) и воды (4.8 г) и снова промывали твердые вещества водой (12.0 г). Полученные твердые вещества высушивали с получением белых кристаллов (форма I) соединения II-6 (4.21 г).
1H-ЯМР (ДМСО-D6) δ: 2.91-2.98 (1H, m), 3.24-3.31 (1Н, m), 3.44 (1Н, t, J=10.4 Гц), 3.69 (1Н, dd, J=11.5, 2.8 Гц), 3.73 (3Н, s), 4.00 (1H, dd, J=10.8, 2.9 Гц), 4.06 (1H, d, J=14.3 Гц), 4.40 (1H, d, J=11.8 Гц), 4.45 (1H, dd, J=9.9, 2.9 Гц), 5.42 (1H, dd, J=14.4, 1.8 Гц), 5.67 (1H, d, J=6.5 Гц), 5.72-5.75 (3Н, m), 6.83-6.87 (1H, m), 7.01 (1H, d, J=6.9 Гц), 7.09 (1H, dd, J=8.0, 1.1 Гц), 7.14-7.18 (1H, m), 7.23 (1H, d, J=7.8 Гц), 7.37-7.44 (2H, m).
Рентгеновская порошковая дифракция 2θ(°): 8.6±0.2°, 14.1±0.2°, 17.4±0.2°, 20.0±0.2°, 24.0±0.2°, 26.3±0.2°, 29.6±0.2°, и 35.4±0.2°.
На фигуре 3 показаны результаты рентгеновской порошковой дифракции кристаллов формы I соединения II-6.
[0090]
Пример 11: Способ получения соединения II-61

Первая стадия
К раствору хлорметилхлорформиата (300 мг, 2.33 ммоль) и соединения 73 (330 мг, 2.79 ммоль) в дихлорметане (6.0 мл) добавляли пиридин (207 мкл, 2.56 ммоль) при 0°С в атмосфере азота и перемешивали смесь при 0°С в течение 30 минут, нагревали до комнатной температуры и перемешивали в течение 1 часа. К смеси добавляли 2 моль/л водный раствор соляной кислоты и экстрагировали смесь дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении, получая соединение 74 (440 мг, 90%).
1Н-ЯМР(CDCl3)δ:1.65 (s, 6Н), 3.77 (s, 3Н), 5.71 (s, 2Н).
Вторая стадия
Соединение III-2 (300 мг, 0.62 ммоль), карбонат калия (172 мг, 1.24 ммоль), иодид калия (103 мг, 0.62 ммоль) и соединение 74 (261 мг, 1.24 ммоль) растворяли в ДМА (3.0 мл) и перемешивали смесь при 80°С в течение 3 часов. К смеси добавляли 2 моль/л водный раствор соляной кислоты и экстрагировали смесь этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение II-61 (350 мг, 86%).
1Н-ЯМР(CDCl3)δ:1.63 (s, 3Н), 1.67 (s, 3Н), 2.86-2.93 (m, 1Н), 3.38-3.61 (m, 2Н), 3.68-3.78 (m, 4Н), 3.90-3.96 (m, 1H), 4.06 (d, J=14.0 Гц, 1H), 4.51 (dd, J=2.0 Гц, 9.6 Гц, 1H), 4.65 (d, J=12.4 Гц, 1H), 5.21 (d, J=14.4 Гц, 1H), 5.36 (s, 1H), 5.80-5.95 (m, 3Н), 6.85-6.92 (m, 2Н), 7.03-7.22 (m, 5Н).
[0091]
Пример 12: Способ получения соединения II-4

К раствору соединения III-2 (90 мг, 0.186 ммоль) в дихлорметане (2 мл) добавляли уксусный ангидрид (0.053 мл, 0.558 ммоль), триэтиламин (0.077 мл, 0.558 ммоль) и каталитическое количество DMAP и перемешивали смесь при комнатной температуре в течение 2 часов. Смесь концентрировали при пониженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол). К полученному раствору добавляли эфир и выпавшее в осадок твердое вещество фильтровали, получая соединение II-4 (71 мг, 73%).
1H-ЯМР(CDCl3)δ:2.46(s, 3Н), 2.88-2.99(m, 1Н), 3.35-3.50(m 1Н), 3.60-3.65(m, 1Н), 3.75-3.83(m, 1Н), 3.90-4.00(m, 1Н), 4.05(d, J=14.0 Гц, 1H), 4.52-4.57(m, 1H), 4.60-4.70(m, 1H), 5.24-5.34(m, 1H), 5.35(s, 1H), 5.88(d, J=7.6 Гц, 1H), 6.85-6.82(m, 1H), 6.90-7.05(m, 2Н), 7.06-7.20(m, 4Н)
ЖХ/МС (ИЭР):m/z=526.2 [М+Н]+, RT=1.87 мин, способ (1)
[0092]
Пример 13: Способ получения соединения II-65
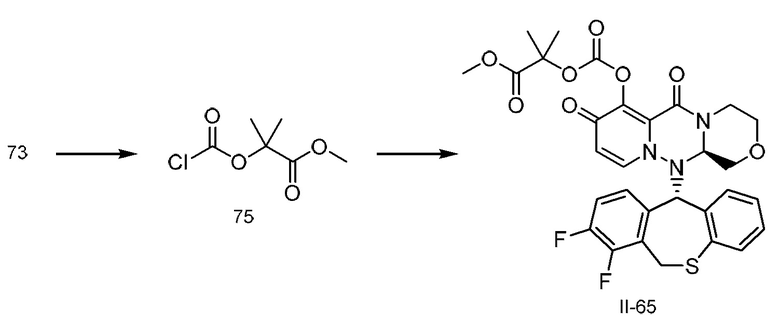
Первая стадия
К раствору трифосгена (300 мг, 2.54 ммоль) в дихлорметане (6.0 мл) добавляли пиридин (257 мкл, 3.17 ммоль) при 0°С в атмосфере азота и перемешивали смесь в течение 15 минут. К смеси добавляли раствор соединения 73 (377 мг, 1.27 ммоль) в дихлорметане (1.0 мл) и перемешивали смесь при 0°С в течение 15 минут, нагревали до комнатной температуры и перемешивали в течение 15 минут. Смесь концентрировали при пониженном давлении, добавляли к ней этилацетат (4.0 мл) и фильтровали смесь. Фильтрат концентрировали при пониженном давлении, получая соединение 75 (380 мг).
Вторая стадия
К раствору соединения III-2 (350 мг, 0.724 ммоль) в дихлорметане (3.5 мл) добавляли соединение 75 (196 мг, 1.09 ммоль) и триэтиламин (301 мкл, 2.17 ммоль) при 0°С и перемешивали смесь при 0°С в течение 30 минут. К смеси добавляли 2 моль/л водный раствор соляной кислоты и экстрагировали смесь дихлорметаном. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (хлороформ-метанол), получая соединение II-65 (380 мг, 84%).
1Н-ЯМР(CDCl3)δ:1.73 (s, 3Н), 1.77 (s, 3Н), 2.90-2.99 (m, 1H), 3.37-3.43 (m, 1H), 3.57 (t, J=8.8 Гц, 1H), 3.76 (dd, J=2.8 Гц, 12.0 Гц, 1H), 3.81 (s, 3Н), 3.94 (dd, J=2.8 Гц, 10.8 Гц, 1Н), 4.05 (d, J=14.0 Гц, 1H), 4.55 (dd, J=2.8 Гц, 9.6 Гц, 1H), 4.65 (d, J=12.0 Гц, 1H), 5.28 (d, J=12.0 Гц, 1H), 5.34 (s, 1H), 5.89 (d, J=8.0 Гц, 1H), 6.86-6.95 (m, 2Н), 7.03-7.15 (m, 5Н).
[0093]
Пример 14: Способ получения соединения II-129
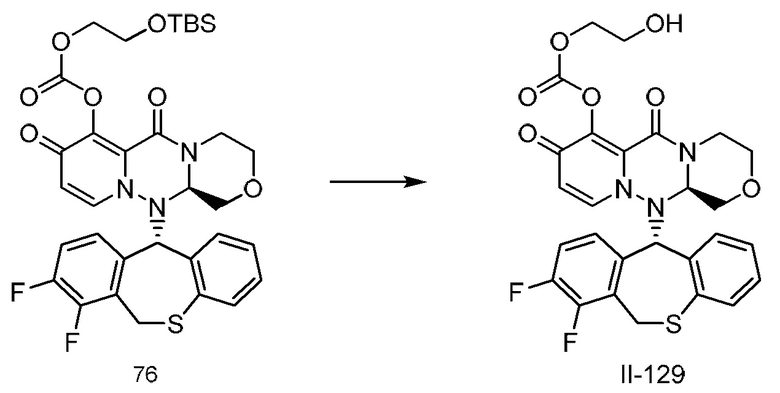
К раствору соединения 76 (276 мг, 0.402 ммоль) в ТГФ (1 мл) добавляли уксусную кислоту (121 мг, 2.01 ммоль) и раствор 1 моль/л TBAF в ТГФ (1.21 мл, 1.21 ммоль) в бане с ледяной водой и перемешивали смесь при комнатной температуре в течение 4 часов. Смесь концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), получая соединение II-129 (179 мг, 78%).
ЖХ/МС (ИЭР):m/z=572.0 [М+Н]+, RT=1.74 мин, способ (2)
[0094]
Пример 15: Способ получения соединения II-115

К раствору соединения III-2 (300 мг, 0.62 ммоль) в ДМФА (4 мл) добавляли карбонат калия (258 мг, 1.87 ммоль), 4-(хлорметил)фенилацетат (344 мг, 1.87 ммоль) и иодид натрия (139 мг, 1.87 ммоль) при комнатной температуре и перемешивали смесь при 65°С в течение 1 часа. К смеси добавляли воду и экстрагировали смесь этилацетатом. Полученный органический слой промывали водой, сушили над безводным сульфатом натрия и концентрировали при пониженном давлении. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), получая соединение II-115 (120 мг, 31%).
ЖХ/МС (ИЭР):m/z=631.95 [М+Н]+, RT=2.07 мин, способ (2)
[0095]
Пример 16: Способ получения соединения II-143
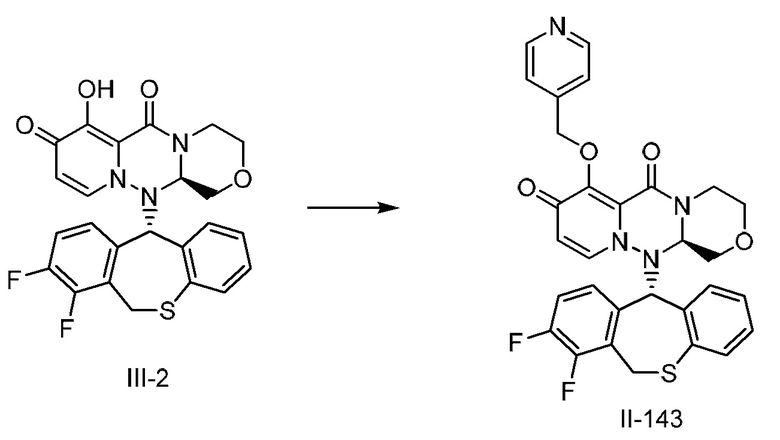
К раствору соединения III-2 (150 мг, 0.31 ммоль) в дихлорметане (2 мл) добавляли 3 ммоль/г трифенилфосфин, нанесенный на полимер (310 мг, 0.93 ммоль), пиридин-4-илметанол (68 мг, 0.62 ммоль) и 40% DEAD в толуоле (270 мг, 0.62 ммоль) при комнатной температуре и перемешивали смесь при комнатной температуре в течение 30 минут. Смесь очищали с помощью колоночной хроматографии на аминогруппе (этилацетат-метанол), получая соединение II-143 (63 мг, 35%).
ЖХ/МС (ИЭР):m/z=575.00 [М+Н]+, RT=1.43 мин, способ (2)
[0096]
Пример 17: Способ получения соединения II-27

К раствору соединения III-2 (65 мг, 0.134 ммоль) в пиридине (0.8 мл) добавляли диметилкарбамоилхлорид (21.7 мг, 0.202 ммоль) и перемешивали смесь при 80°С в течение ночи. К смеси добавляли 1 моль/л водный раствор соляной кислоты и экстрагировали смесь этилацетатом. Полученный органический слой промывали солевым раствором, сушили над безводным сульфатом магния и концентрировали при пониженном давлении. Полученный остаток затвердевал в смеси этилацетат-гексан с получением соединения II-27 (65 мг, 87%).
1Н-ЯМР(CDCl3)δ:2.89 (t, J=11.2 Гц, 1H), 2.99 (s, 1H), 3.01 (s, 3Н), 3.18-3.26 (m, 4Н), 3.45 (t, J=10.8 Гц, 1H), 3.59 (t, J=10.8 Гц, 1H), 3.70-3.80 (m, 1H), 3.90-3.98 (m, 1H), 4.03 (d, J=13.6 Гц, 1H), 4.50-4.70 (m, 2Н), 5.21-5.35 (m, 2Н), 5.82 (d, J=7.6 Гц, 1H), 6.91 (t, J=7.6 Гц, 1H), 7.00-7.20 (m, 6Н).
[0097]
Пример 18: Способ получения соединения II-55

К раствору этилфосфордихлоридата (135 мг, 0.829 ммоль) в дихлорметане (3 мл) добавляли гидрохлорид метилового эфира L-валина (139 мг, 0.829 ммоль) и затем по каплям добавляли раствор триэтиламина (168 мг, 1.66 ммоль) в дихлорметане (2 мл) при -78°С. Перемешивали смесь при комнатной температуре в течение 1 часа. Добавляли к ней соединение III-2 (200 мг, 0.414 ммоль) и триэтиламин (126 мг, 1.25 ммоль) и перемешивали смесь при той же температуре в течение 6 часов. Смесь концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), получая соединение II-55 (112 мг, 38%).
ЖХ/МС (ИЭР):m/z=705.05 [М+Н]+, RT=2.18 мин, способ (2)
[0098]
Пример 19: Способ получения соединения II-57

К раствору этилфосфордихлоридата (202 мг, 1.24 ммоль) в дихлорметане (3 мл) по каплям добавляли смесь триэтиламина (126 мг, 1.24 ммоль) и метилгликолята (112 мг, 1.24 ммоль) в дихлорметане (2 мл). Перемешивали смесь при комнатной температуре в течение 2 часов. Добавляли к ней соединение III-2 (200 мг, 0.414 ммоль) и триэтиламин (126 мг, 1.25 ммоль) и перемешивали смесь при той же температуре в течение 1 часа. Смесь концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), получая соединение II-57 (143 мг, 52%).
ЖХ/МС (ИЭР):m/z=664.00 [М+Н]+, RT=1.93 мин, способ (2)
[0099]
Пример 20: Способ получения соединения II-58
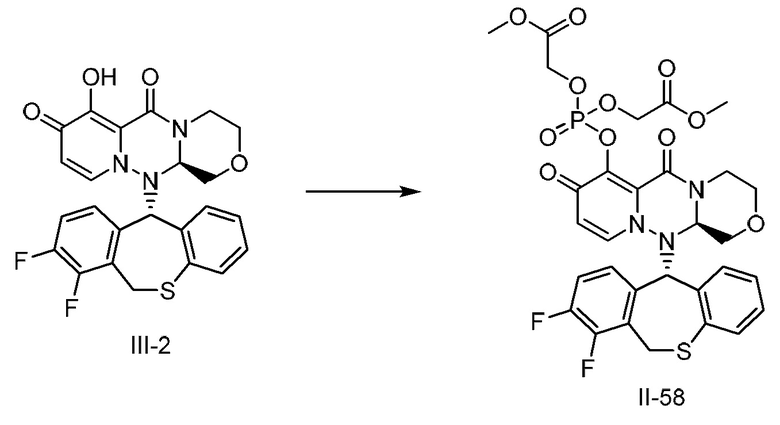
К раствору фосфорилхлорида (1.53 г, 10 ммоль) в дихлорметане (10 мл) по каплям добавляли смесь триэтиламина (2.12 г, 20.95 ммоль) и метилгликолята (1.89 мг, 21 ммоль) в дихлорметане (5 мл). Перемешивали смесь при комнатной температуре в течение 2 часов. К смеси (2 мл) добавляли соединение III-2 (200 мг, 0.414 ммоль) и триэтиламин (126 мг, 1.25 ммоль) и перемешивали смесь при той же температуре в течение 1 часа. Смесь концентрировали, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), получая соединение II-58 (166 мг, 57%).
ЖХ/МС (ИЭР):m/z=707.90 [М+Н]+, RT=1.93 мин, способ (2)
[0100]
Следующие иллюстративные соединения были синтезированы из коммерчески доступных соединений или промежуточных соединений, подходящим образом синтезированных из коммерчески доступных соединений в соответствии с приведенными выше примерами.
[0101]


[0102]

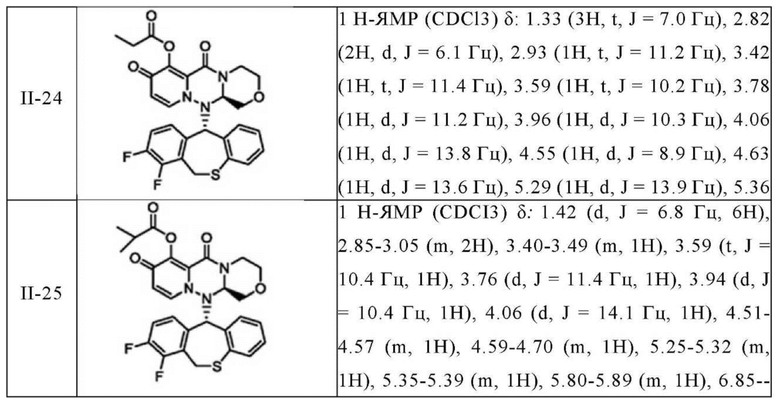
[0103]
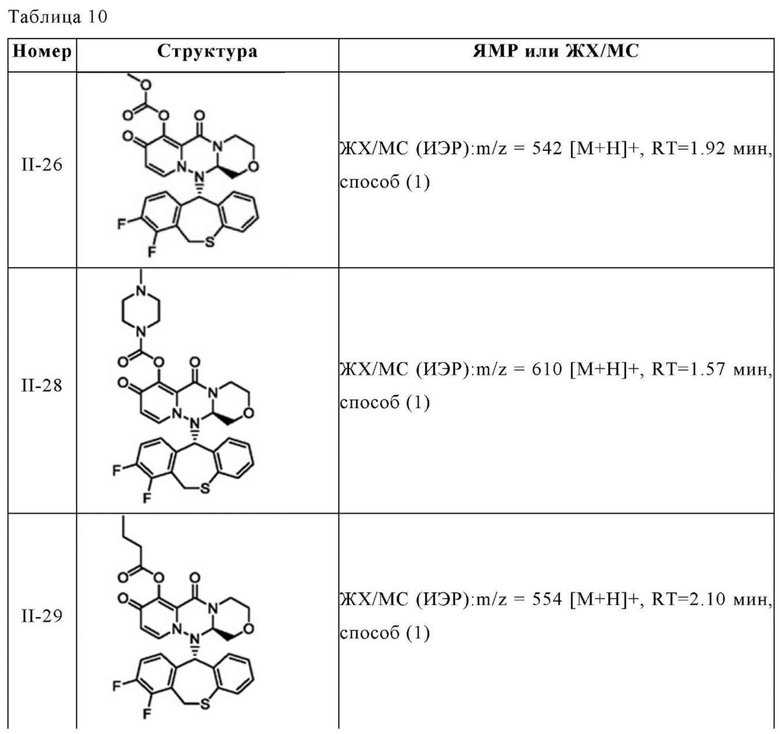

[0104]


[0105]


[0106]
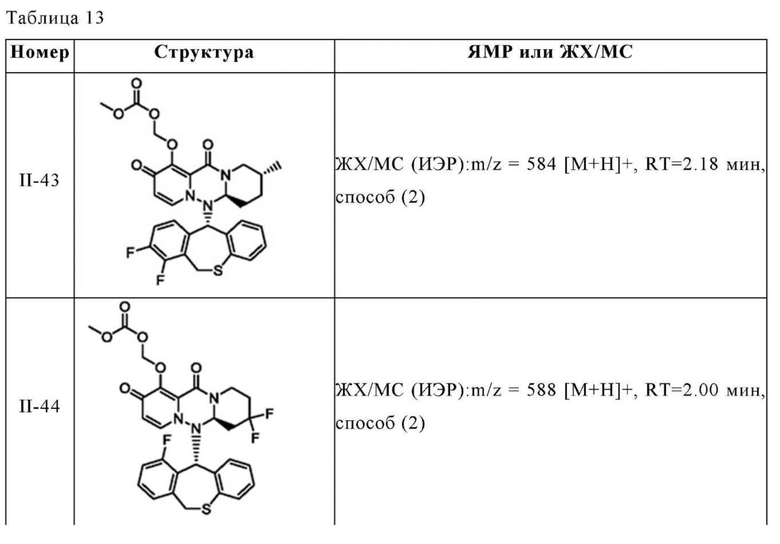
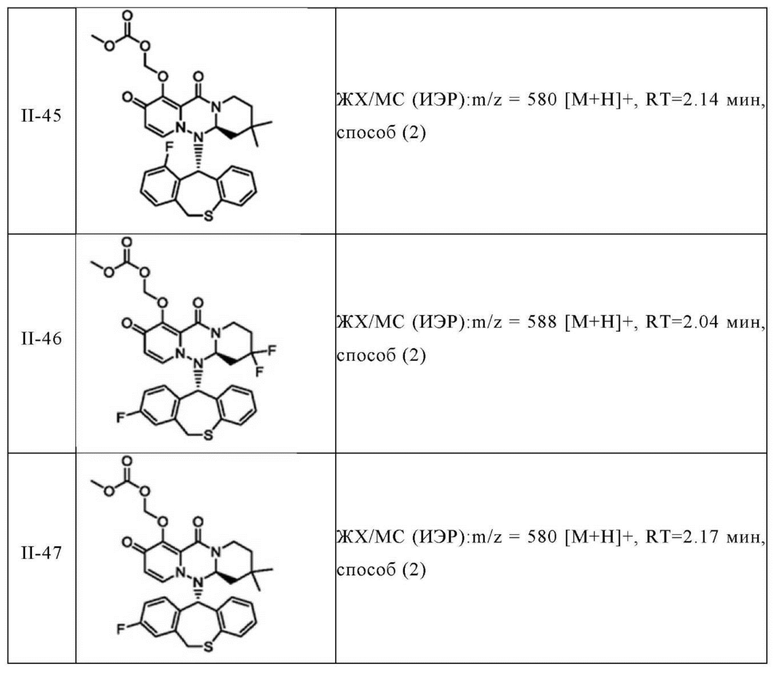
[0107]
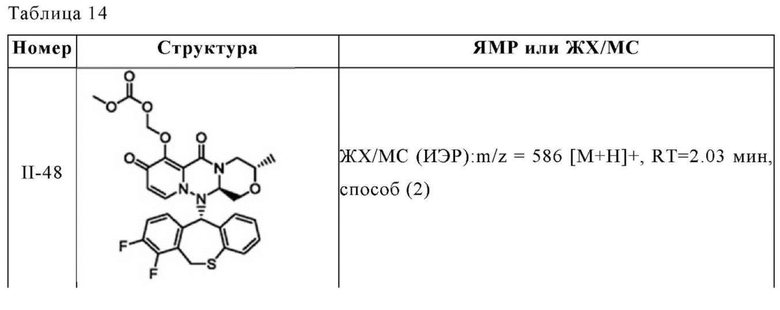

[0108]

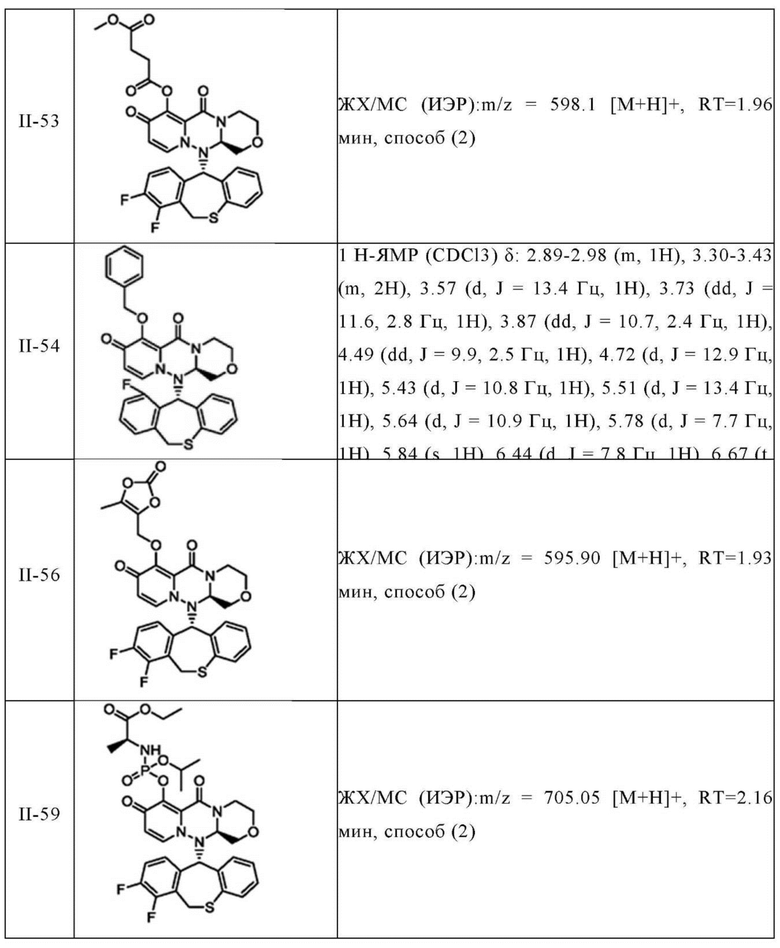
[0109]


[0110]
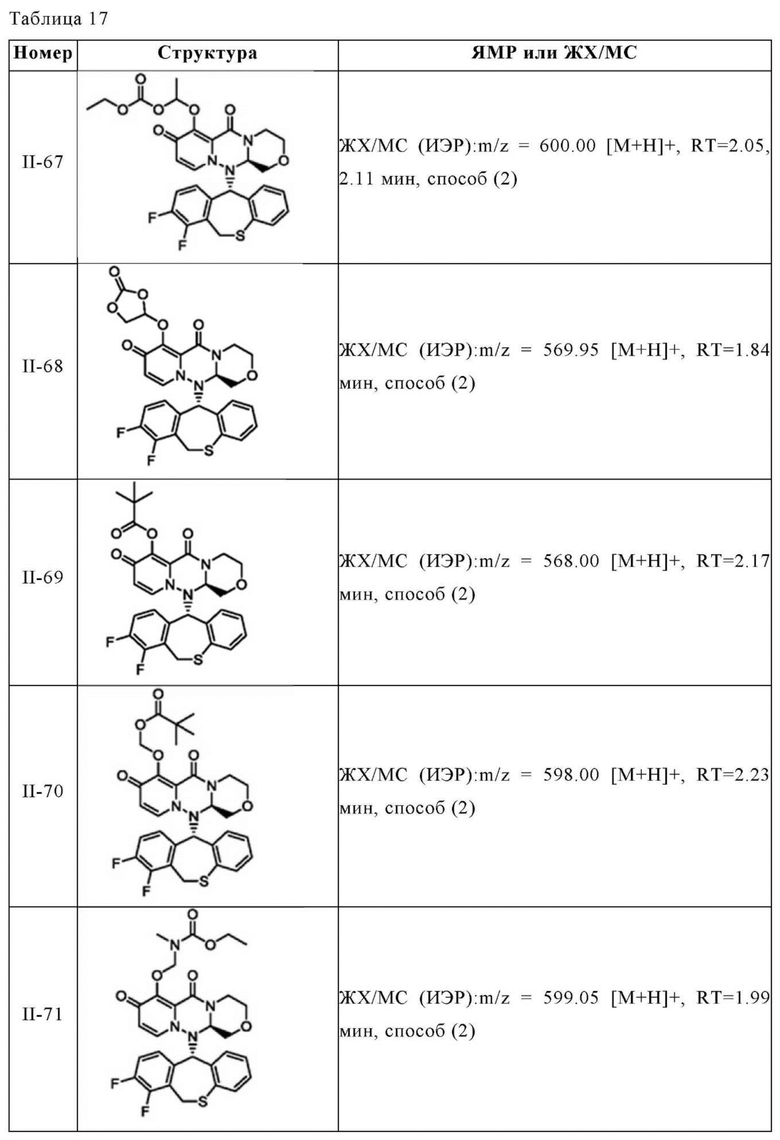
[0111]

[0112]

[0113]

[0114]

[0115]

[0116]
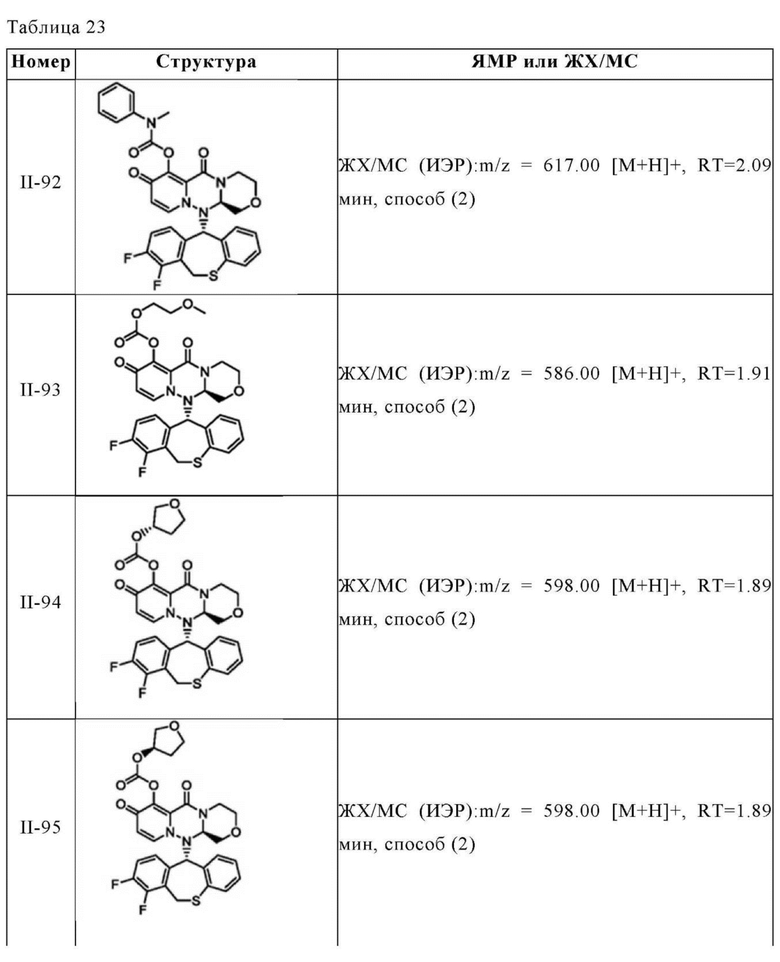

[0117]


[0118]
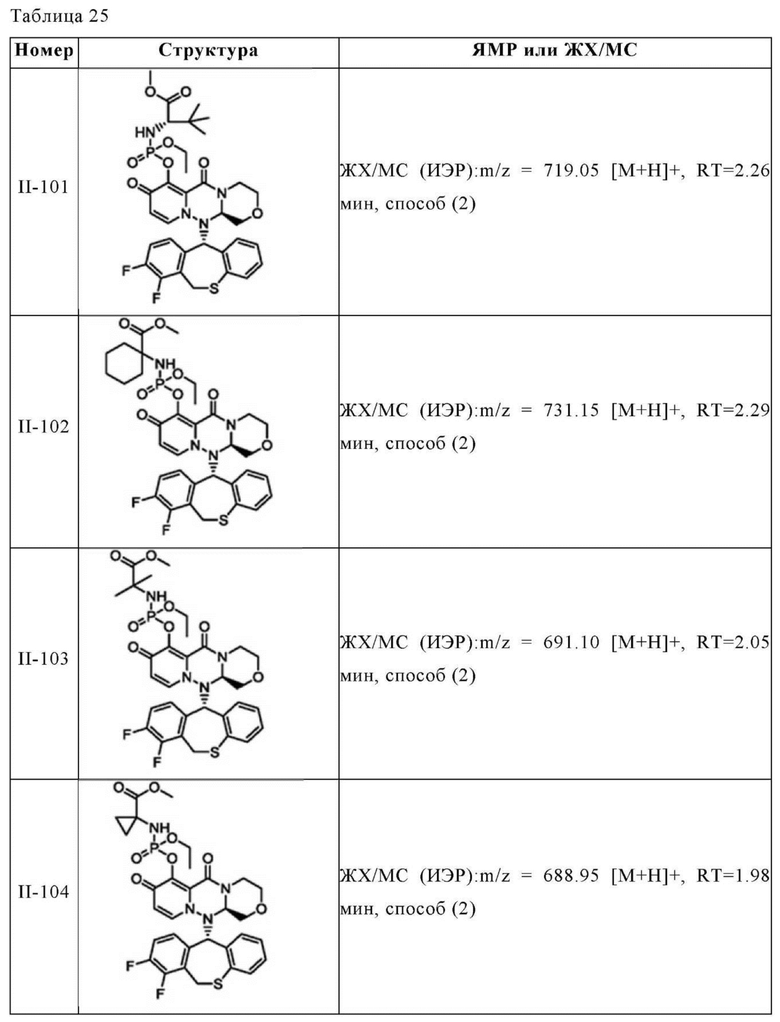
[0119]
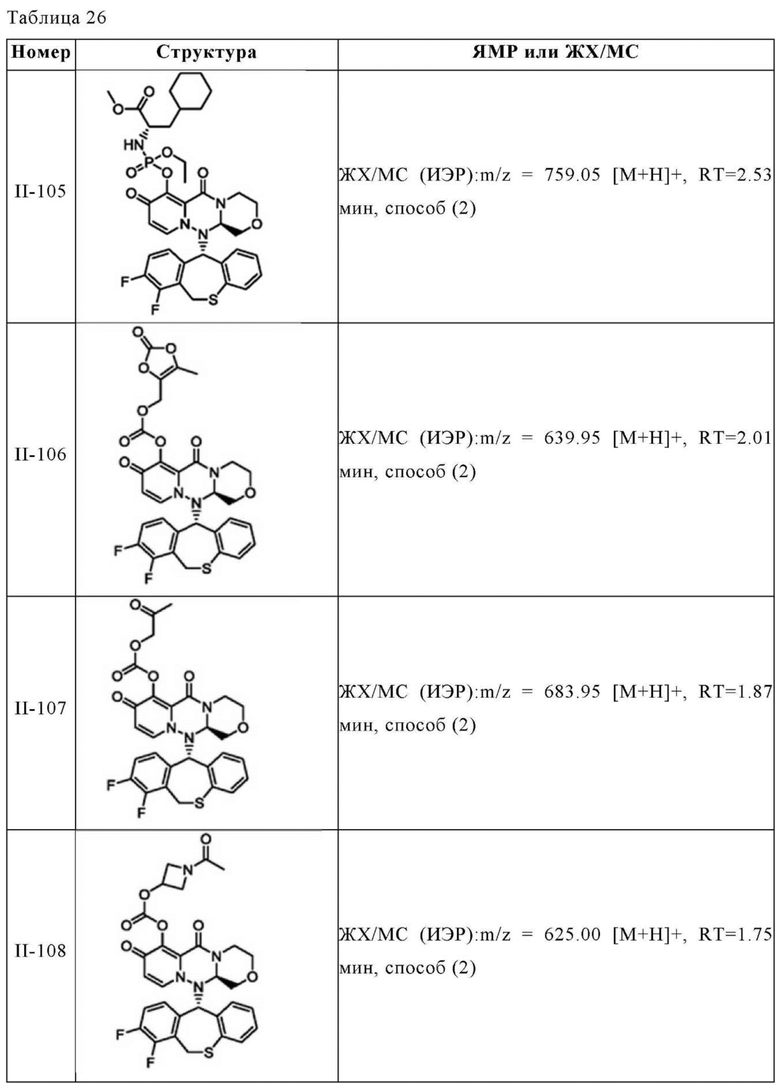
[0120]

[0121]

[0122]

[0123]

[0124]
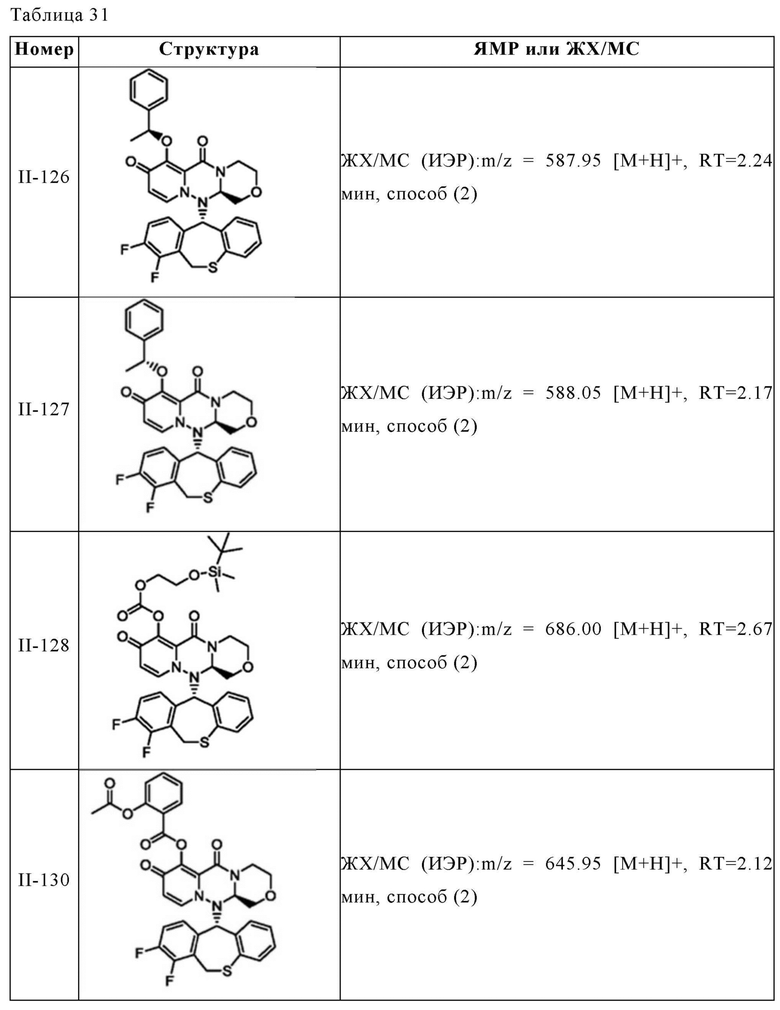
[0125]
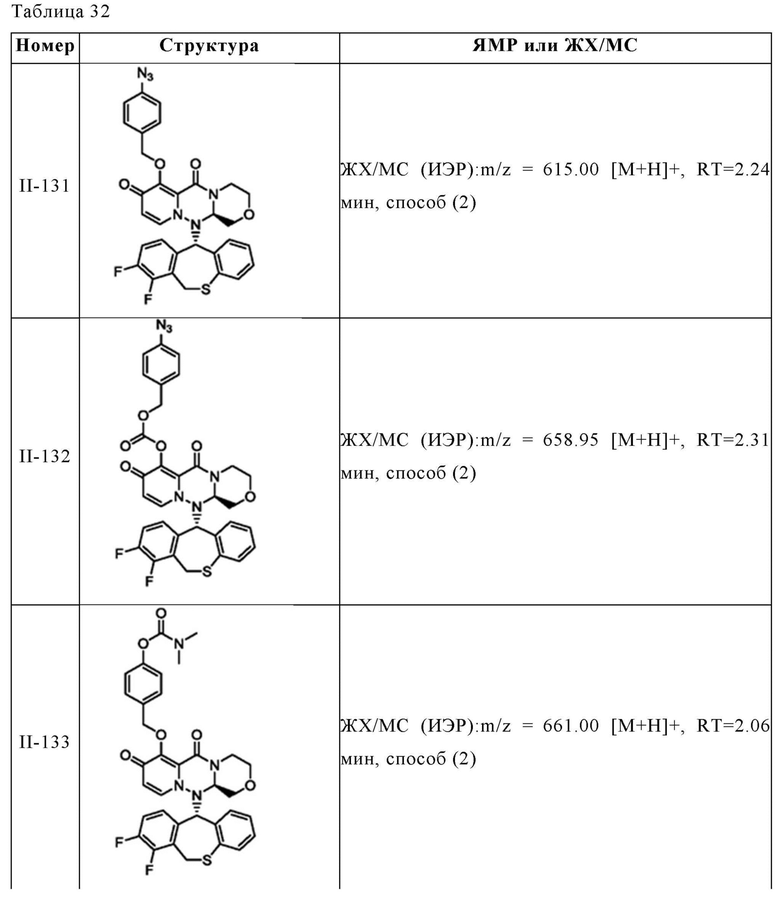

[0126]


[0127]

[0128]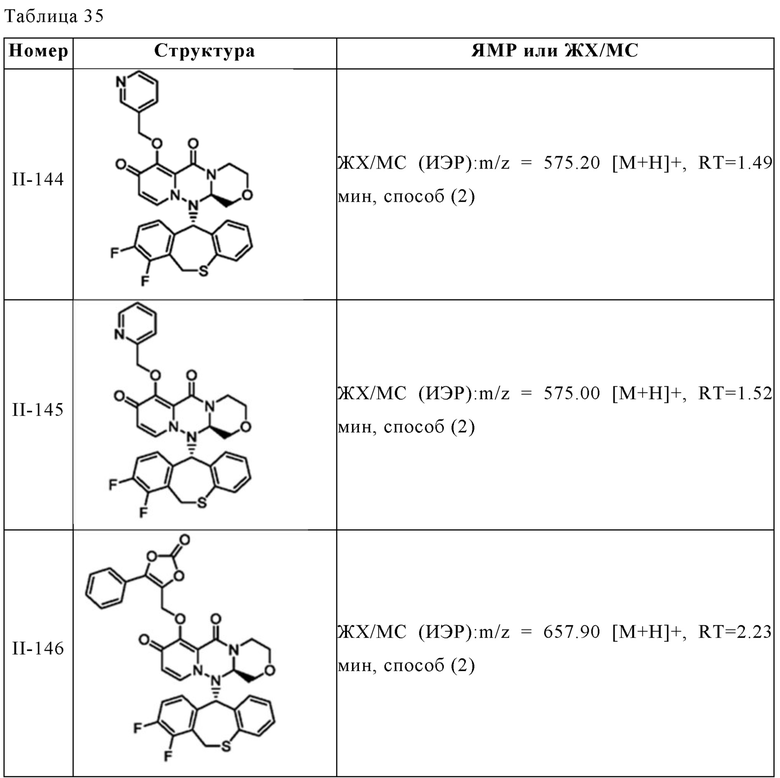
[0129]
Следующие соединения также могут быть синтезированы вышеуказанными способами.

[0130]
Пример 21: Способ получения кристаллов формы II соединения II-6
К соединению II-6 (10.00 г) добавляли ацетонитрил (50 мл) и воду (5 мл), нагревали соединение до растворения и добавляли к нему воду (95 мл). Раствор перемешивали в течение 10 минут при комнатной температуре и выпавшие кристаллы фильтровали. Полученные кристаллы подвергали проточной сушке, получая кристаллы формы II (9.04 г) соединения II-6.
На фигуре 4 показаны результаты рентгеновской порошковой дифракции кристаллов формы II соединения II-6.
[0131]
Пример 22: Способ получения кристаллов формы III соединения II-6
К соединению II-6 (10.00 г) добавляли мэтилацетат (400 мл) и нагревали соединение до растворения. Метилацетат (примерно 230 мл) из раствора концентрировали при пониженном давлении, перемешивали раствор в течение 70 минут при комнатной температуре и фильтровали выпавшие кристаллы. Полученные кристаллы подвергали проточной сушке, получая кристаллы формы III (7.87 г) соединения II-6.
На фигуре 5 показаны результаты рентгеновской порошковой дифракции кристаллов формы III соединения II-6.
[0132]
Примеры биологических испытаний для соединений, применяемых в настоящем изобретении, описаны ниже.
[0133]
Тестовый пример 1: Измерение ингибирующей активности в отношении кэп-зависимой эндонуклеазы (CEN)
1) Получение субстрата
Приобретали 30-мерную РНК (5'-pp-[m2'-O]GAA UAU(-Cy3) GCA UCA CUA GUA AGC UUU GCU CUA-BHQ2-3' производства компании Japan Bio Services Co., LTD.), в которой G на 5'-конце дифосфатмодифицирован, гидроксигруппа в положении 2' метоксилмодифицирована, U-шестая с 5'-конца помечена Су3, 3'-конец помечен BHQ2, и добавляли кэп-структуру с использованием системы ScriptCap, изготовленной EPICENTRE (продукт m7G [5']-ррр-[5'] [m2'-O]GAA UAU(-Cy3) GCA UCA CUA GUA AGC UUU GCU CUA(-BHQ2)-3'). Указанную РНК выделяли и очищали с помощью электрофореза в денатурированном полиакриламидном геле и использовали в качестве субстрата.
2) Получение фермента
РНП получали из вирусной частицы с использованием стандартного метода (Справочный документ: VIROLOGY(1976) 73, р327-338 OLGA М. ROCHOVANSKY). В частности, вирус A/WSN/33 (1 х 103 БОЕ/мл, 200 мкл) инокулировали в 10-дневное куриное яйцо с эмбрионом. После инкубации при 37°С в течение 2 дней извлекали аллантоисную жидкость куриного яйца. Вирусную частицу очищали ультрацентрифугированием с использованием 20% сахарозы, солюбилизировали с использованием TritonX-100 и лизолецитина, а фракцию РНП (50-70% глицериновая фракция) собирали ультрацентрифугированием с использованием градиента плотности глицерина 30-70% и использовали в качестве раствора фермента (содержащего примерно 1 нмоль РВ1-РВ2-РА комплекса).
3) Ферментативная реакция
Ферментативный реакционный раствор (2.5 мкл) (состав: 53 ммоль трис-гидрохлорида (рН 7.8), 1 ммоль MgCl2, 1.25 ммоль дитиотреитола, 80 ммоль NaCl, 12.5% глицерин, раствор фермента 0.15 мкл) помещали в 384-луночный планшет из полипропилена. Затем в планшет добавляли 0.5 мкл раствора тестируемого соединения, который последовательно разбавляли диметилсульфоксидом (ДМСО). В качестве положительного контроля (ПК) или отрицательного контроля (ОК) в планшет добавляли 0.5 мкл ДМСО, соответственно. Каждый планшет хорошо перемешивали. Затем для инициирования реакции добавляли 2 мкл раствора субстрата (1.4 нмоль субстрата РНК, 0.05% Tween20). После инкубации при комнатной температуре в течение 60 минут собирали 1 мкл реакционного раствора и для того, чтобы остановить реакцию, добавляли его к 10 мкл раствора высокоочищенного деионизированного формамида (содержащего стандарт размера GeneScan 120 Liz Size Standard в качестве калибровочного маркера: производства Applied Biosystems (ABI)). Для ОК реакцию останавливали заранее, добавляя ЭДТА (4.5 ммоль) до начала реакции (все концентрации, описанные выше, представляют собой конечные концентрации).
4) Измерение коэффициента ингибирования (значение IC50)
Раствор, в котором реакция была остановлена, нагревали при 85°С в течение 5 минут, быстро охлаждали на льду в течение 2 минут и анализировали с помощью генетического анализатора ABI PRIZM 3730. Пик продукта кэп-зависимой эндонуклеазы количественно определяли с помощью программного обеспечения для анализа ABI Genemapper, получали коэффициент ингибирования реакции CEN (%) тестируемого соединения, устанавливая интенсивности флуоресценции ПК и НК равными 0% ингибирования и 100% ингибирования, соответственно, получали значение IC50 с использованием программного обеспечения для подгонки кривой (XLfit2.0: Model 205 (производства IDBS) и т.д.). Значения IC50 тестируемых веществ, являющихся исходным соединением, приведены в таблице 39.
[0134]
Тестовый пример 2: подтверждение действия по подавлению СРЕ
<Материал>
• 2% FCS Е-МЕМ (приготовленный добавлением канамицина и FCS к MEM (минимальная необходимая среда) (Invitrogen))
• 0.5% BSA Е-МЕМ (приготовленный добавлением канамицина и BSA к MEM (минимальная необходимая среда) (Invitrogen))
• HBSS (сбалансированный солевой раствор Хэнкса)
• Клетки MDBK
Клетки доводили до подходящего количества клеток (3×105/мл) с помощью 2% FCS Е-МЕМ.
• Клетки MDCK
После двукратной промывки клетки HBSS доводили до соответствующего количества клеток (5×105/мл) с помощью 0.5% BSA Е-МЕМ.
• Раствор трипсина
Трипсин из поджелудочной железы свиньи (SIGMA) растворяли в PBS(-) и фильтровали через фильтр 0.45 мкм.
• En Vision (PerkinElmer)
• Набор WST-8 (Kishida Chemical Co., Ltd.)
• Раствор 10% SDS
[0135]
<Порядок работы>
• Разведение и распределение тестируемого образца
В качестве культуральной среды применяли 2% FCS Е-МЕМ при применении клеток MDBK и применяли 0.5% BSA Е-МЕМ при применении клеток MDCK. Далее применялась одна и та же культуральная среда для разведения вируса, клеток и тестируемого образца.
Тестовый образец заранее разбавляли культуральной средой до соответствующей концентрации и затем производили серийное разведение в 2-5 раз на 96-луночном планшете (50 мкл/лунку). Были подготовлены два планшета, один для измерения активности против гриппа и другой для измерения цитотоксичности. Каждый анализ был выполнен трижды для каждого лекарственного средства.
При применении клеток MDCK к клеткам добавляли трипсин до конечной концентрации 3 мкг/мл только для измерения активности против гриппа.
• Разбавление и распределение вируса гриппа
Вирус гриппа заранее разбавляли культуральной средой до соответствующей концентрации и помещали каждые 50 мкл/лунку в 96-луночный планшет, содержащий тестируемое вещество. Для измерения цитотоксичности каждые 50 мкл/лунку культуральной среды помещали в лунку, содержащую тестируемое вещество.
• Разбавление и распределение клеток
В лунки 96-луночного планшета, содержащего тестируемый образец, вносили по 100 мкл/лунку клеток, количество которых в указанном объеме было доведено до соответствующего значения.
Содержимое планшета перемешивали с помощью планшетного миксера и инкубировали в инкубаторе с CO2 в течение 3 дней для измерения активности против гриппа и измерения цитотоксичности.
• Распределение WST-8
Клетки в 96-луночном планшете, которые инкубировали в течение 3 дней, оценивали визуально под микроскопом и проверяли внешний вид клеток, наличие или отсутствие кристаллов исследуемого вещества. Супернатант удаляли таким образом, чтобы клетки не абсорбировались из лунки.
Набор WST-8 разводили в 10 раз культуральной средой и помещали по 100 мкл в каждую лунку. После смешивания с помощью планшетного миксера клетки инкубировали в инкубаторе с CO2 в течение 1-3 часов.
После инкубации вносили по 10 мкл/лунку 10% раствора SDS для инактивации вируса для последующего измерения активности против гриппа.
• Измерение поглощения
Содержимое 96-луночного планшета перемешивали и измеряли поглощение с помощью En Vision при двух длинах волн 450 нм/620 нм.
[0136]
<Вычисление каждого значения элемента измерения>
Значение было рассчитано с использованием Microsoft Excel или программы, имеющей эквивалентные возможности вычисления и обработки, на основе следующего уравнения расчета.
• Расчет эффективной ингибирующей концентрации для достижения гибели 50% инфицированных клеток, зараженных гриппом (ЕС50)
ЕС50=10Z
Z = (50% - высокий %) / (высокий % - низкий %) × {log(высокая конц.) - log§(низкая конц.)} + log(высокая конц.)
[0137]
Для исходных соединений, применяемых в настоящем изобретении, результаты измерений согласно тестовому примеру 1 и тестовому примеру 2 показаны ниже.
[0138]


Приведенные выше результаты позволяют сделать вывод о том, что исходные соединения, применяемые в настоящем изобретении, проявляют высокую ингибирующую активность в отношении кэп-зависимой эндонуклеазы (CEN) и/или высокий ингибирующий эффект в отношении СРЕ и, таким образом, могут быть подходящим средством для лечения и/или предотвращения симптомов и/или заболевания, вызванного заражением вирусом гриппа.
[0139]
Тестовый пример 3: тест CYP-ингибирования
Оценивали степень ингибирования каждого количества вырабатываемого метаболита соединением, применяемым в настоящем изобретении, используя коммерчески доступные объединенные микросомы печени человека и используя в качестве маркеров О-деэтилирование 7-этоксирезоруфина (CYP1A2), метилгидроксилирование толбутамида (CYP2C9), 4'-гидроксилирование мефенитоина (CYP2C19), О-деметилирование декстрометорфана (CYP2D6) и гидроксилирование терфенидина (CYP3A4) в качестве типичных реакций метаболизма субстрата основных пяти форм фермента CYP человека (CYP1A2, 2С9, 2С19, 2D6, 3А4).
[0140]
Условия реакции были следующими: субстрат, 0.5 мкмоль/л этоксирезоруфина (CYP1A2), 100 мкмоль/л толбутамида (CYP2C9), 50 мкмоль/л S-мефенитоина (CYP2C19), 5 мкмоль/л декстрометорфана (CYP2D6), 1 мкмоль/л терфенидина (CYP3A4); время реакции, 15 минут; температура реакции, 37°С; фермент, объединенная человеческая печеночная микросома 0.2 мг белка/мл; концентрация соединения, применяемого в настоящем изобретении, 1, 5, 10, 20 мкмоль/л (четыре точки).
[0141]
В 96-луночный планшет с композицией, описанной выше, вносили каждый из пяти видов субстратов, микросомы печени человека или соединение, применяемое в настоящем изобретении, в буфере Hepes с концентрацией 50 ммоль/л в качестве реакционного раствора, добавляли NADPH в качестве кофактора для инициирования метаболических реакций, являющихся маркерами, и после инкубации при 37°С в течение 15 минут добавляли раствор метанол/ацетонитрил = 1/1 (об./об.) для остановки реакции. После центрифугирования при 3000 об/мин в течение 15 минут количественно определяли резоруфин (метаболит CYP1A2) в супернатанте с помощью флуоресцентного счетчика с несколькими метками и определяли количественно гидроксид толтрибутимида (метаболит CYP2C9P), 4' гидроксид мефенитоина (метаболит CYP2C19), декстрометорфан (метаболит CYP2D6) и терфенадиновый спирт (метаболит CYP3A4) с помощью ЖХ/МС/МС.
[0142]
В качестве контроля (100%) было принято добавление в реакционную систему только ДМСО, являющегося растворителем, растворяющим соединение, применяемое в настоящем изобретении, рассчитывали остаточную активность (%) для каждой концентрации соединения, применяемого в настоящем изобретении, добавленного в виде раствора, и рассчитывали IC50 путем обратного предположения методом логистической регрессии с использованием концентрации и степени ингибирования.
(Результат)
Соединение III-2: пять видов >20 мкмоль/л
[0143]
Тестовый пример 4: тест ВА (на биодоступность)
Материалы и способы для экспериментов по оценке абсорбции при пероральном введении
(1) Экспериментальные животные: использовали мышей или крыс SD.
(2) Условия выращивания: мыши или крысы SD имели свободный доступ к твердой пище и стерилизованной водопроводной воде.
(3) Установка дозировки и группирование: выполняли пероральное и внутривенное введение с заранее определенной дозировкой. Распределение по группам выполняли, как показано ниже. (Дозировку изменяли для каждого соединения)
Пероральное введение 1-30 мг/кг (n=2-3)
Внутривенное введение 0.5-10 мг/кг (n=2-3)
(4) Приготовление растворов для введения: Пероральное введение осуществляли в виде раствора или суспензии. Внутривенное введение проводили после солюбилизации.
(5) Способы введения: Пероральное введение было выполнено в обязательном порядке в желудок. Внутривенное введение проводилось в хвостовую вену шприцами с иглой.
(6) Элементы оценки: Собирали последовательно кровь и измеряли концентрацию в плазме соединения, применяемого в настоящем изобретении, с помощью ЖХ/МС/МС.
(7) Статистический анализ: Для изменения концентрации в плазме соединения, применяемого в настоящем изобретении, рассчитывали площадь под кривой зависимости концентрации в плазме от времени (AUC) с помощью программы нелинейного метода наименьших квадратов, WinNonlin (зарегистрированный товарный знак), и рассчитывали биодоступность (ВА) соединения, применяемого в настоящем изобретении, по AUC группы перорального введения и группы внутривенного введения.
(Результат)
Соединение II-6: 14.9%
Соединение III-2: 4.2%
На основании вышеуказанных результатов можно сделать вывод о том, что пролекарство улучшило биодоступность в отличие от исходного соединения.
Таким образом, соединение, применяемое в настоящем изобретении, обладает превосходной пероральной всасываемостью и может быть полезным средством для лечения и/или предотвращения симптомов и/или заболеваний, вызванных инфекцией вируса гриппа.
[0144]
Тестовый пример 5: Тест на стабильность метаболизма
Используя коммерчески доступные объединенные микросомы печени человека, соединение, применяемое в настоящем изобретении, подвергали реакции в течение постоянного времени и рассчитывали долю остатка путем сопоставления прореагировавшего образца и непрореагировавшего образца, таким образом, оценивали степень метаболизма в печени.
[0145]
Реакцию проводили (окислительная реакция) при 37°С в течение 0 минут или 30 минут в присутствии 1 ммоль/л NADPH в 0.2 мл буфера (50 ммоль/л трис-HCl рН 7.4, 150 ммоль/л хлорида калия, 10 ммоль/л хлорида магния), содержащего 0.5 мг белка/мл микросом печени человека. После реакции добавляли 50 мкл реакционного раствора к 100 мкл смеси метанол/ацетонитрил = 1/1 (об./об.), перемешивали и центрифугировали при 3000 об/мин в течение 15 минут. Соединение, применяемое в настоящем изобретении, в супернатанте количественно определяли с помощью ЖХ/МС/МС или твердофазной экстракции (ТФЭ)/МС и рассчитывали оставшееся после реакции количество соединения, применяемого в настоящем изобретении, принимая количество соединения при времени реакции 0 минут за 100%. Реакцию гидролиза проводили в отсутствие NADPH, а реакцию глюкуронидирования проводили в присутствии 5 ммоль UDP- глюкуроновой кислоты вместо NADPH с последующими аналогичными операциями.
(Результат) % ингибирования показан при 2 мкмоль/л тестового соединения.
Соединение III-2: 90.1%
[0146]
Тестовый пример 6: флуоресцентный MBI тест CYP3A4
Флуоресцентный MBI тест CYP3A4 представляет собой тест для изучения усиления ингибирования CYP3A4 соединения, применяемого в настоящем изобретении, реакцией метаболизма, и указанный тест проводили с использованием в качестве фермента CYP3A4, экспрессируемого в Escherichia coli, и с использованием в качестве индекса реакции, в которой 7-бензилокситрифторметилкумарин (7-BFC) дебензилируется ферментом CYP3A4 с образованием метаболита, 7-гидрокситрифторметилкумарина (ГФУ), излучающего флуоресцентный свет.
[0147]
Условия реакции были следующими: субстрат, 5.6 мкмоль/л 7-BFC; время до реакции, 0 или 30 минут; время реакции, 15 минут; температура реакции, 25°С (комнатная температура); содержание CYP3A4 (выраженное в Escherichia coli) до реакции 62.5 пмоль/мл, при реакции 6.25 пмоль/мл (при 10-кратном разбавлении); концентрация тестируемого лекарственного средства, применяемого в настоящем изобретении, 0.625, 1.25, 2.5, 5, 10, 20 мкмоль/л (шесть точек).
[0148]
Фермент в буфере K-Pi (рН 7.4) и раствор соединения, применяемого в настоящем изобретении, в качестве дореакционного раствора добавляли в 96-луночный планшет с вышеуказанной дореакционной композицией, часть его переносили в другой 96-луночный планшет с тем, чтобы разбавить его 1/10 субстратом и буфером K-Pi, добавляли NADPH в качестве кофактора для инициирования реакции в качестве показателя (без предварительной инкубации) и добавляли после заданного времени реакции ацетонитрил/0.5 моль/л трис(тригидроксиаминометан) = 4/1 (об./об.), чтобы остановить реакцию. Помимо этого, добавляли NADPH к оставшемуся преинкубационному раствору, чтобы инициировать преинкубацию (с преинкубацией), и после предварительно определенного времени преинкубации часть переносили в другую лунку с тем, чтобы разбавить ее 1/10 субстратом и буфером K-Pi для инициирования реакции в качестве индекса. После заданного времени реакции добавляли ацетонитрил/0.5 моль/л трис(тригидроксиаминометан) = 4/1 (об./об.), чтобы остановить реакцию. Для планшета, на котором была проведена каждая индексная реакция, измеряли флуоресцентное значение 7-ГФУ, которое является метаболитом, с помощью флуоресцентного планшет-ридера (возбуждение = 420 нм, эмиссия = 535 нм).
[0149]
Для контроля (100%) в реакционную систему вносили только ДМСО, который представляет собой растворитель, растворяющий соединение, применяемое в настоящем изобретении, рассчитывали оставшуюся активность (%) при каждой концентрации соединения, применяемого в настоящем изобретении, добавленного в виде раствора, и рассчитывали IC50 путем обратного предположения с помощью логистической регрессии с использованием концентрации и степени ингибирования. Если различие между значениями IC50 составляет 5 мкмоль/л или более, ставили (+), а если разница составляет 3 мкмоль/л или менее, ставили (-).
(Результат)
Соединение III-2: (-)
[0150]
Тестовый пример 7: Флуктуационный тест Эймса
Оценивали мутагенность соединений, применяемых в настоящем изобретении.
Инокулировали 20 мкл хранящегося в замороженном состоянии крысиного брюшного тифа (штамм Salmonella typhimurium ТА98, штамм ТА100) в 10 мл жидкой питательной среды (2.5% питательный бульон Oxoid №2) и культивировали перед встряхиванием при 37°С в течение 10 часов. Центрифугировали 9 мл бактериального раствора штамма ТА98 (2000 × г, 10 минут) для удаления раствора для культивирования. Бактерии суспендировали в 9 мл буфера Micro F (K2HPO4: 3.5 г/л, KH2PO4: 1 г/л, (NH4)2SO4: 1 г/л, дегидрат тринатрийцитрата: 0.25 г/л, MgSO4 ⋅ 7Н2О: 0.1 г/л), добавляли суспензию к 110 мл среды экспонирования (буфер Micro F, содержащий биотин: 8 мкг/мл, гистидин: 0.2 мкг/мл, глюкозу: 8 мг/мл). Добавляли штамм ТА100 к 120 мл среды экспонирования относительно 3.16 мл бактериального раствора для приготовления тестируемого бактериального раствора. Смешивали по 12 мкл ДМСО раствора соединения, применяемого в настоящем изобретении, (разведение в несколько стадий от максимальной дозы 50 мг/мл при соотношении 2 к 3), ДМСО в качестве отрицательного контроля и 50 мкг/мл ДМСО раствора 4-нитрохинолин-1-оксида для штамма ТА98, 0.25 мкг/мл ДМСО раствора 2-(2-фурил)-3-(5-нитро-2-фурил)акриламида для штамма ТА100 в условиях активации метаболизма, 40 мкг/мл ДМСО раствора 2-аминоантрацена для штамма ТА98, 20 мкг/мл ДМСО раствора 2-аминоантрацена для штамма ТА100 в условиях активации метаболизма в качестве положительного контроля и 588 мкл тестового бактериального раствора (смешанный раствор 498 мкл тестового бактериального раствора и 90 мкл смеси S9 в условиях, стимулирующих метаболизм) и культивировали при встряхивании при 37°С в течение 90 минут. Смешивали 460 мкл бактериального раствора, подвергнутого воздействию соединения, применяемого в настоящем изобретении, с 2300 мкл индикаторной среды (Micro F-буфер, содержащий биотин: 8 мкг/мл, гистидин: 0.2 мкг/мл, глюкозу: 8 мг/мл, бромкрезол пурпурный: 37.5 мкг/мл), распределяли каждые 50 мкл в микропланшет 48 лунок/дозу и подвергали стационарному культивированию при 37°С в течение 3 дней. Поскольку лунка, содержащая бактерию, которая приобрела способность к пролиферации благодаря мутации гена, кодирующего фермент, синтезирующий аминокислоту (гистидин), изменяет цвет с пурпурного на желтый из-за изменения рН, подсчитывали число лунок с пролиферацией бактерий, содержимое которых стала желтым, из 48 лунок на дозу, и проводили оценку путем сравнения с контрольной группой. (-) означает, что мутагенность отрицательная, а (+) - положительная.
(Результат)
Соединение III-2: (-)
[0151]
Тестовый пример 8: Тест с hERG
В целях оценки риска продления интервала QT электрокардиограммы из-за соединения, применяемого в настоящем изобретении, было изучено влияние соединения, применяемого в настоящем изобретении, на ток K + задержанного выпрямления (IKr), который играет важную роль в процессе реполяризации желудочков, с использованием клеток HEK293, экспрессирующих канал, связанный с геном человека human ether-a-go-go related gene (hERG).
После того как клетка удерживалась при мембранном потенциале -80 мВ методом цельноклеточного зажима с использованием автоматической системы зажима (PatchXpress 7000А, Axon Instruments Inc.), регистрировали 1 кг, индуцированный путем стимуляции импульсом деполяризации при +40 мВ в течение 2 секунд и далее регистрировали импульсную стимуляцию реполяризации при -50 мВ в течение 2 секунд. После стабилизации генерируемого тока наносили на клетку внеклеточный раствор (NaCl: 135 ммоль/л, KСl: 5.4 ммоль/л, NaH2PO4: 0.3 ммоль/л, СаСl2 ⋅ 2Н2О: 1.8 ммоль/л, MgCl2 ⋅ 6Н2О: 1 ммоль/л, глюкоза: 10 ммоль/л, HEPES (4-(2-гидроксиэтил)-1-пиперазинэтансульфоновая кислота): 10 ммоль/л, рН=7.4), в котором было растворено соединение, применяемое в настоящем изобретении, в целевой концентрации, при комнатной температуре на 10 минут. На основе записи 1 кг измеряли абсолютное значение тока концевого пика, принимая во внимание значения тока при потенциале мембраны в покое, с использованием программного обеспечения для анализа (DataXpress ver. 1, Molecular Devices Corporation). Кроме того, рассчитывали % ингибирования относительно тока концевого пика перед применением соединения, применяемого в настоящем изобретении, и сравнивали с группой, применяющей носитель (0,1% раствор диметилсульфоксида), для оценки влияния соединения, используемого в настоящем изобретении на 1 кг.
(Результат) % ингибирования приведен при 0.3-10 мкмоль тестового соединения.
Соединение III-2: 7.9%
[0152]
Тестовый пример 9: Тест на растворимость
Растворимость соединения, применяемого в настоящем изобретении, определяли в условиях добавления 1% ДМСО. Готовили 10 ммоль/л раствор соединения с ДМСО и добавляли 2 мкл раствора соединения, применяемого в настоящем изобретении, соответственно, к 198 мкл раствора JP-1 (к 2.0 г хлорида натрия и 7.0 мл соляной кислоты добавляли воду до достижения 1000 мл) и раствора JP-2 (к 1 объему раствора, который содержал 3,40 г дигидрофосфата калия и 3,55 г безводного динатрийгидрофосфата добавляли 1 объем воды до достижения 1000 мл). Смесь встряхивали в течение 1 часа при комнатной температуре и фильтровали. Фильтрат разбавляли в десять раз смесью метанол/вода = 1/1(об./об.) и измеряли концентрацию соединения в фильтрате с помощью ЖХ/МС или ТФЭ/МС методом абсолютной калибровки.
(Результат)
Соединение III-2: 42.2 мкмоль/л
[0153]
Тестовый пример 10: Тест на растворимость в порошкообразном состоянии
Соответствующие количества соединения, применяемого в настоящем изобретении, помещали во флаконы и в каждый флакон вносили 200 мкл жидкости JP-1 (к 2.0 г хлорида натрия в 7.0 мл соляной кислоты добавляли воду до достижения 1000 мл), жидкости JP-2 (к 500 мл фосфатного буферного раствора с рН 6.8 добавляли воду) и 20 ммоль/л таурохолата натрия (ТСА)/JР-2-й жидкости (к 1.08 г ТСА в жидкости JP-2 добавляли жидкость JP-2 до достижения 100 мл). Когда соединение полностью растворялось, добавляли соответствующее количество соединения. После встряхивания в течение 1 часа при 37°С смесь фильтровали и к 100 мкл каждого фильтрата добавляли 100 мкл метанола (двойное разбавление). Увеличение разведения менялось при необходимости. Удостоверившись в отсутствии пузырьков воздуха и осадков во флаконах, встряхивали флаконы с плотной пробкой. Концентрацию соединения определяли с помощью ВЭЖХ методом абсолютной калибровки.
(Результат)
Соединение III-2: раствор JP-1; 7.1 мкг/мл, раствор JP-2; 4.4 мкг/мл, 20 ммоль/л TCA/JP-2 раствор; 16.1 мкг/мл
[0154]
Тестовый пример 11: Тест Эймса
Тест Эймса проводили с применением сальмонелл (Salmonella typhimurium) ТА 98, ТА100, ТА1535 и ТА1537 и Escherichia coli WP2uvrA в качестве тестовых штаммов с метаболической активацией или без нее в методе преинкубации для проверки наличия или отсутствия генной мутагенности соединений, применяемых в настоящем изобретении.
(Результат)
Соединение III-2: (-)
[0155]
Тестовый пример 12: Тест на легкий гемолиз
Соединение, применяемое в настоящем изобретении, растворяли в целевых концентрациях и смешивали с 2.5 об./об.% суспензией эритроцитов, приготовленной из дефибринированной крови овец, на микропланшете в концентрациях от 0.0008 до 0.1 масс./об.%. Смеси подвергали воздействию 10° Дж/см2 УФ-излучения в диапазоне длин волн от 290 до 400 нм, УФА и УФБ с использованием ультрафиолетовых люминесцентных ламп, ламп GL20SE и FL20S-BLB производства Sankyo Denki Co., Ltd. и Panasonic Corporation, соответственно. После завершения облучения смеси центрифугировали, и супернатант смеси собирали и помещали на микропланшет. Фототоксичность оценивали путем измерения поглощения при длине волны 540 нм и 630 нм в супернатанте. Данные по оптической плотности при длине волны 540 нм и 630 нм были использованы в качестве индикаторов биомембранного повреждения (% фотогемолиза) и гиперокисления липидной мембраны (образование метгемоглобина), соответственно. Критерии фототоксичности были следующими; Было принято, что фототоксичности нет (-), если наблюдается фотогемолиз <10% и максимальное изменение поглощения при 630 нм (ΔOD) <0.05. Было принято, что фототоксичность есть (+), если фотогемолиз составляет более 10% и максимальное изменение поглощения при 630 нм (ΔOD) составляет более 0.05.
(Результат)
Соединение III-2: (-)
[0156]
Тестовый пример 13: Переход концентрации в плазме
Была измерена концентрация в плазме соединения III-2 и соединения II-6 после перорального введения крысам не натощак пролекарства соединения II-6, исходным соединением которого было соединение III-2. Результат показан на фигурах 1 и 2.
Концентрация соединения II-6 во всех образцах плазмы была на уровне предела определения или менее. Таким образом, было обнаружено, что пролекарственное соединение II-6, исходное соединение которого представляло собой соединение III-2, быстро превращалось в соединение III-2 in vivo после введения (см. фигуру 2).
[0157]
На основании указанных выше результатов тестов было выявлено, что соединение, превращенное в пролекарство, всасывалось в организм после перорального введения и быстро превращалось в исходное соединение в крови. Таким образом, соединения (исходные соединения и/или пролекарства), применяемые в настоящем изобретении, могут быть пригодны для применения для лечения и/или предотвращения симптомов и/или заболеваний, вызванных инфекцией вируса гриппа.
[0158]
Тестовый пример 14: Тест на внутривенное введение
Исследуемые экспериментальные материалы и тестирование способа внутривенного введения
(1) Подопытные животные: использовались крысы SD.
(2) Условия содержания: сухой корм и стерилизованную водопроводную воду скармливали крысам SD без ограничений.
(3) Дозировка и группирование: заданную дозировку вводили внутривенно. Группы были установлены, как описано далее. (Дозировку варьировали для каждого соединения)
Внутривенное введение 0.5-1 мг/кг (n=2-3)
(4) Приготовление раствора для введения: внутривенное введение проводили после солюбилизации.
(5) Способ введения: внутривенное введение проводили с помощью шприца с иглой в хвостовую вену.
(6) Критерий оценки: кровь собирали с течением времени, и измеряли концентрацию в плазме соединения, применяемого в настоящем изобретении, с использованием ЖХ/МС/МС.
(7) Статистический анализ: в отношении изменения концентрации в плазме соединения, применяемого в настоящем изобретении, рассчитывали общий клиренс тела (CLtot) и период полувыведения (t1/2, z) с использованием нелинейной программы наименьших квадратов WinNonlin (зарегистрированная торговая марка).
(Результаты)
Соединение № III-2:
CLtot: 16.4 мл/мин/кг
t1/2, z: 3.4 часа
Из указанных выше результатов было обнаружено, что соединение III-2 представляет собой соединение, имеющее низкий общий клиренс тела и длительный период полураспада.
Таким образом, соединение, применяемое в настоящем изобретении, имеет превосходную стойкость и может быть пригодно для применения для лечения и/или предотвращения симптомов и/или заболеваний, вызванных инфекцией вируса гриппа.
[0159]
Тестовый пример 15: Клинический тест
Эффективность и безопасность однократного перорального введения исследуемого лекарственного средства (активный ингредиент (соединение II-6): 10 мг, 20 мг, 40 мг) пациентам, инфицированным вирусом гриппа, оценивали с помощью рандомизированного плацебо-контролируемого двойного слепого сравнительного исследования. Что касается первичного критерия оценки, то субъекты сами оценивали эффективность исследуемого лекарственного средства по сравнению с плацебо по 4-балльной шкале [0: нет, 1: легкая, 2: средняя, 3: сильная] в отношении времени ослабления симптомов гриппа (время от начала введения исследуемого препарата до облегчения семи симптомов гриппа («кашель», «боль в горле», «головная боль», «заложенность носа», «лихорадка или озноб», «боль в мышцах или суставах» и «усталость»)).
В качестве субъектов были выбраны пациенты, которые удовлетворяли всем следующим критериям.
(a) Пациенты мужского или женского пола в возрасте 20 лет или старше и моложе 65 лет
(b) Пациенты, удовлетворяющие всем следующим критериям, у которых диагностировано заболевание инфекцией вируса гриппа
- Положительный результат при быстрой диагностике гриппа [экспресс-тест на антиген (RAT)] на основе мазка из носа или горла
- Температура тела (подмышечная температура) 38.0°С или выше
- Наличие одного или более симптомов, умеренных или сильных, среди следующих системных симптомов и респираторных симптомов, обусловленных инфекционным заболеванием вирусом гриппа
- Системные симптомы (головная боль, лихорадка или озноб, мышечная или суставная боль, усталость)
- Респираторные симптомы (кашель, боль в горле, заложенность носа)
(С) Пациенты в течение 48 часов после начала заболевания (при регистрации)
Определение начала заболевания представляет собой любое наблюдение из следующих.
- Когда температура тела увеличивалась в первый раз (повышение по меньшей мере на 1°С от нормальной температуры)
- Когда развивались один или более системных симптомов и респираторных симптомов.
[0160]
Способ введения исследуемого лекарственного средства
(i) Тестируемое лекарственное средство
Таблетка с 10 мг соединения II-6: таблетка от белого до бледно-желтоватого цвета, круглая, покрытая пленочной оболочкой, содержащая 10 мг соединения II-6
Таблетка с 20 мг соединения II-6: таблетка от белого до бледно-желтоватого цвета, эллиптическая, покрытая пленочной оболочкой, содержащая 20 мг соединения II-6
(ii) Плацебо или контрольное лекарственное средство
Плацебо для таблетки с 10 мг соединения II-6: таблетка, не отличимая от таблетки с 10 мг соединения II-6
Плацебо для таблетки с 20 мг соединения II-6: таблетка, не отличимая от таблетки с 20 мг соединения II-6
[0161]
Дозировка и способ введения
Приемлемые субъекты были случайным образом распределены в соотношении 1:1:1:1 по группам: группа соединения II-6 10 мг, группа соединения II-6 20 мг, группа соединения II-6 40 мг и группа плацебо. В первый день субъекты получали однократное пероральное введение всего 3 таблеток: таблеток соединения II-6 и/или таблеток плацебо, в комбинации, указанной в следующей таблице.
Исследуемое лекарственное средство для каждой группы введения

[0162]
Основной критерий оценки эффективности
Основным критерием оценки эффективности является время ослабления симптомов гриппа (время ослабления симптомов).
Это время от начала введения до ослабления симптомов гриппа. Облегчение симптомов гриппа относится к тем случаям, когда все семь симптомов гриппа (кашель, боль в горле, головная боль, заложенность носа, лихорадка или озноб, мышечная или суставная боль, усталость) становятся «0: нет» или «1: легкими» в дневнике пациента, который ведет субъект, и это состояние сохраняется, по меньшей мере, в течение 21.5 часа (24 часа - 10%).
[0163]
Вторичный критерий оценки эффективности
Вторичным критерием оценки эффективности является следующее. (1) Время облегчения каждого симптома гриппа
Это время от начала введения до ослабления каждого симптома гриппа. Под облегчением симптома понимается, когда целевой оценкой становится «0: нет» или «1: легкий», и это состояние сохраняется по меньшей мере в течение 21,5 часа (24 часа - 10%).
[0164]
Анализ первичного критерия оценки
Что касается времени облегчения симптомов гриппа, которое является первичным критерием оценки, описаны первичный анализ и вторичный анализ. В дополнение к группе ITTI (intent-to-treat infected - совокупность всех пациентов, первоначально отобранных для исследования) первичный анализ был также выполнен в группе PPS (Per-Protocol Set - совокупность пациентов, выполнивших требования протокола) для анализа чувствительности. Другие анализы были выполнены только в группе ITTI.
(1) Первичный анализ
С помощью модели пропорционального риска Кокса были рассчитаны коэффициент риска, 95% доверительный интервал и значение Р для каждой группы введения относительно группы плацебо, при расчетах использовали: в качестве отклика - время ослабления симптомов гриппа, в качестве фиксированных эффектов - группы введения и в качестве ковариат - текущую привычку к курению и общий балл семи симптомов гриппа, которые являются факторами распределения, на исходном уровне до введения. Чтобы предотвратить увеличение вероятности ошибки типа I из-за многократного выполнения теста, значение Р корректировали методом Хоммеля.
(2) Вторичный анализ
Группу плацебо и каждую исследуемую группу, которой вводили исследуемое лекарственное средство, сравнивали с помощью стратифицированного обобщенного критерия Вилкоксона, используя в качестве ответа - время ослабления симптомов гриппа, в качестве независимых переменных - группы введения и в качестве факторов стратификации - категорию общего балла (11 баллов или менее, 12 баллов или более) семи симптомов гриппа до введения и привычку к курению, которые являются факторами распределения.
Кроме того, для каждой группы была построена кривая выживаемости Каплана-Мейера для расчета среднего времени ослабления симптомов гриппа и 95%-ного доверительного интервала. Для расчета доверительного интервала был использован метод Гринвуда.
[0165]
Анализ вторичного критерия оценки
(1) Время до облегчения каждого симптома гриппа
Был выполнен тот же анализ, что и для первичного критерия оценки, причем в качестве отклика оценивалось время до облегчения каждого симптома гриппа. При этом случаи, когда симптом до введения был «0: нет» или «1: слабый», были исключены из цели анализа.
[0166]
(1) Результаты первичного критерия оценки (время облегчения симптомов гриппа)
Из 400 случайно отобранных пациентов тест завершили 389 пациентов (98 пациентов (98%) в группе, получавшей 10 мг, 95 пациентов (95%) в группе, получавшей 20 мг, 99 пациентов (99%) в группе, получавшей 40 мг, и 97 пациентов (97%) в группе плацебо). Что касается первичного критерия оценки, популяция ITTI (случаи, когда вводили исследуемое лекарственное средство, и инфекция вируса гриппа была подтверждена) состояла из 400 пациентов.
Случаи, соответствующие установленному протоколу, включали 368 пациентов (89 пациентов (89%) в группе, получавшей 10 мг, 92 пациента (92%) в группе, получавшей 20 мг, 96 пациентов (96%) в группе, получавшей 40 мг, и 91 пациент (91%) в группе плацебо). Что касается популяции ITTI в каждой группе, то по результатам теста быстрого обнаружения антигена было установлено, что от 75% до 79% пациентов были заражены вирусом гриппа А, и от 21% до 25% пациентов были заражены вирусом гриппа В.
Результаты анализа приведены в следующих таблицах.
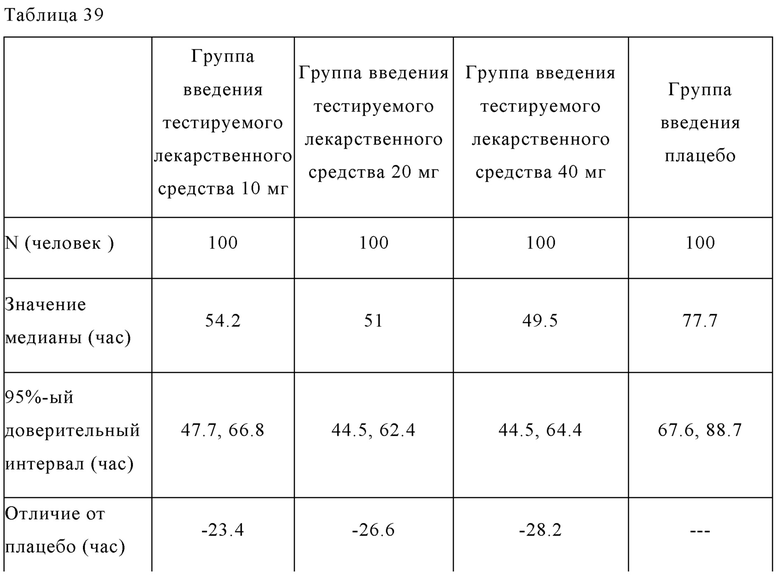
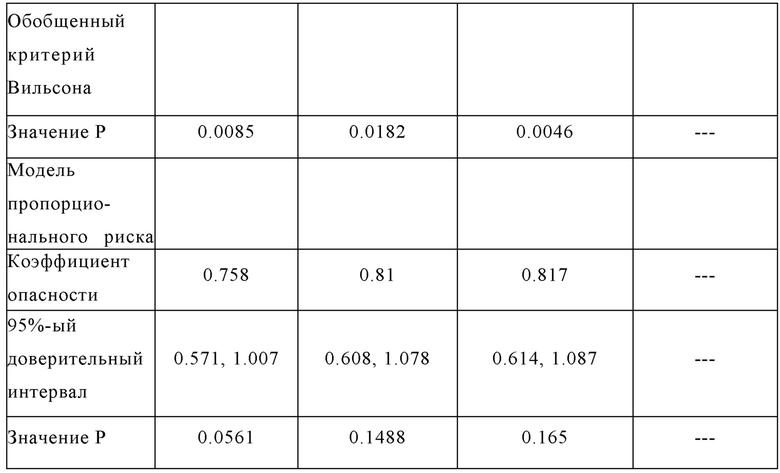
Первичный критерий оценки этого теста, то есть среднее время до ослабления симптомов, составил 54.2 часа в группе, получавшей 10 мг (95% ДИ (доверительный интервал): 47.7, 66.8), 51.0 часа в группе, получавшей 20 мг (95% ДИ: 47.7, 66.8), 49.5 часа в группе, получавшей 40 мг (95% ДИ: 44.5, 64.4), и 77.7 часа в группе плацебо (95% ДИ: 67.6, 88.7).
[0167]
(2) Время до облегчения каждого из семи симптомов
В следующих таблицах показаны результаты анализа времени до облегчения каждого из семи симптомов гриппа («кашель», «боль в горле», «головная боль», «заложенность носа», «лихорадка или озноб», «мышечная или суставная боль» «усталость»).
(i) Время до облегчения симптома «заложенность носа»
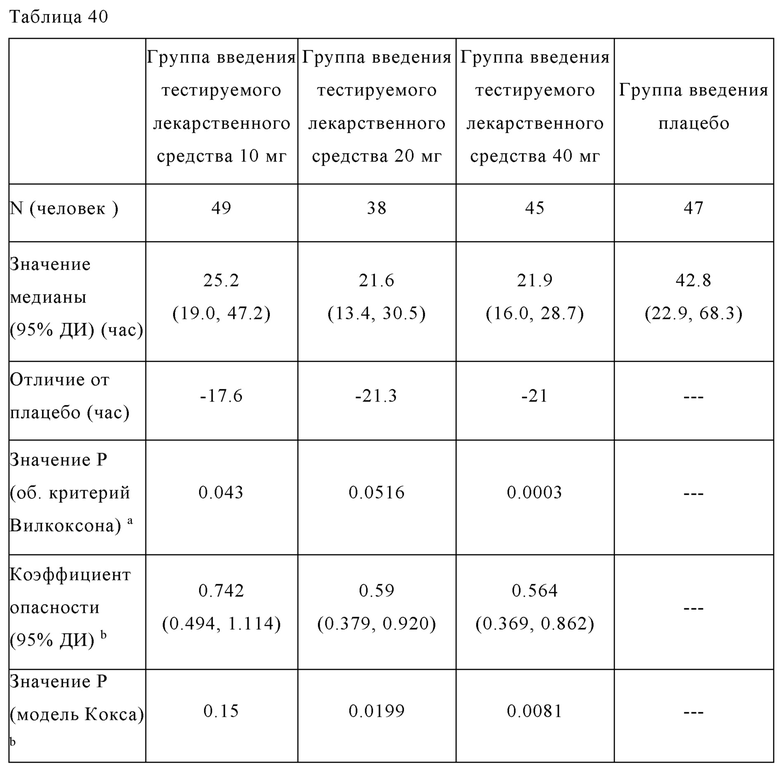
(ii) Время до облегчения симптома «мышечная или суставная боль»

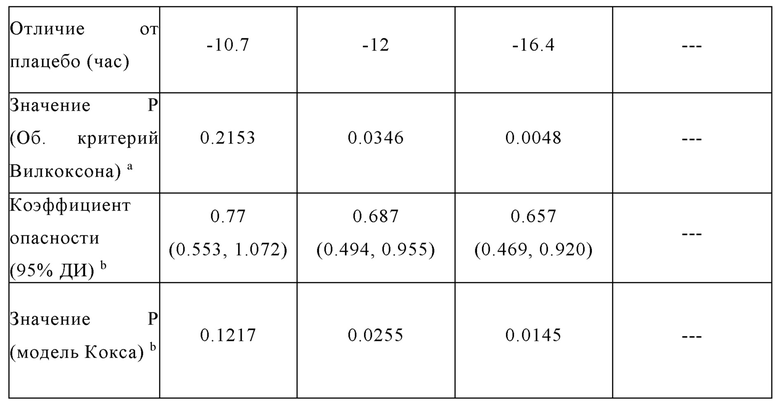
(iii) Время до облегчения симптома «усталость»

(iv) Время до облегчения симптома «лихорадка или озноб»
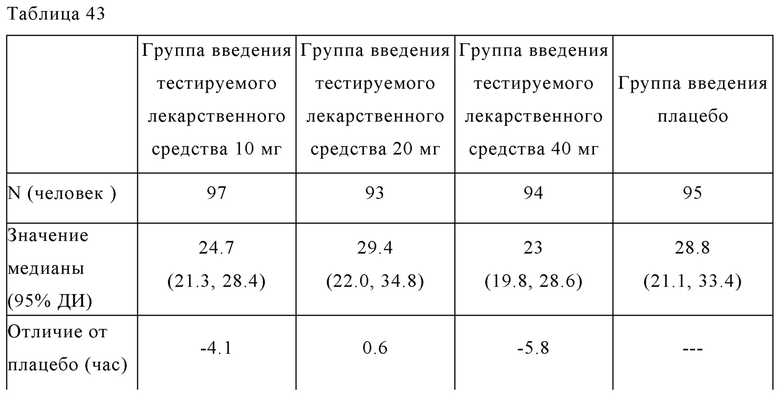

(v) Время до облегчения симптома «головная боль»
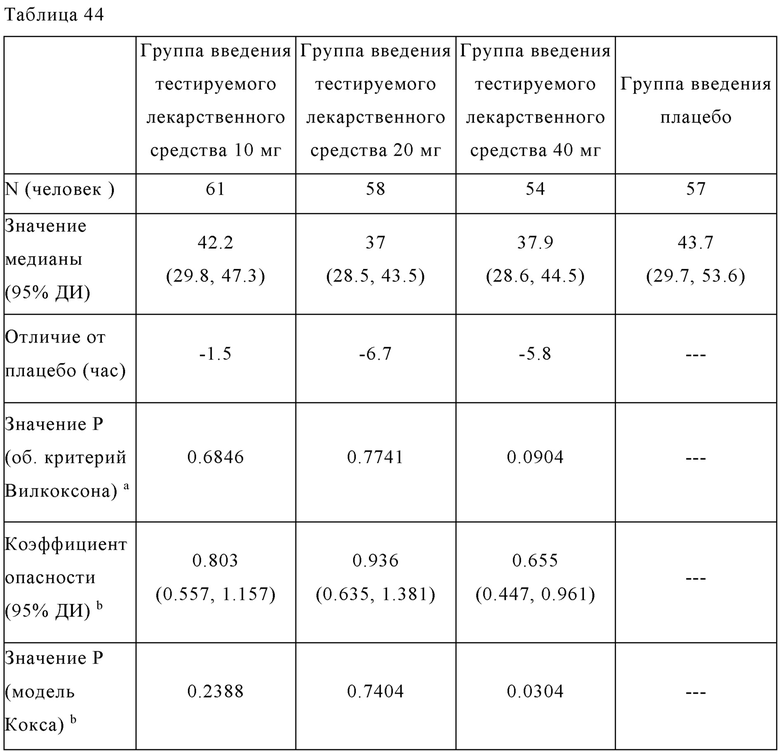
(vi) Время до облегчения симптома «кашель»

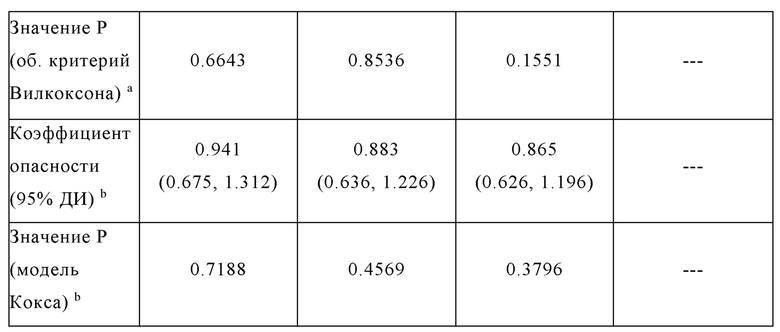
(vii) Время до облегчения симптома «боль в горле»

Анализ с использованием модели пропорционального риска Кокса показал, что в группе, получавшей 40 мг, по сравнению с группой, принимавшей плацебо, наблюдалось значительное уменьшение времени до облегчения следующих 5 симптомов: «заложенность носа», «мышечная или суставная боль», «усталость», «лихорадка или озноб» и «головная боль». Например, что касается 2 симптомов, таких как «заложенность носа» и «мышечная или суставная боль», среднее время до облегчения этих симптомов составило 21.0 часа и 16.4 часа, соответственно, и оно было короче в группе, получавшей 40 мг, по сравнению с группой плацебо.
Статистически значимые различия наблюдались также в группе, получавшей 10 мг, и в группе, получавшей 20 мг, в отношении следующих симптомов: «мышечная или суставная боль», «заложенность носа» и «лихорадка или озноб».
[0168]
Тестовый пример 16: Клинический тест (Ph3: взрослые и подростки)
Эффективность и безопасность однократного перорального введения исследуемого лекарственного средства (активный ингредиент (соединение II-6): 40 мг, 80 мг) пациентам, инфицированным вирусом гриппа, оценивались в рандомизированном двойном слепом сравнительном исследовании по сравнению с введением пациентам 75 мг осельтамивира, назначенного два раза в день в течение 5 дней, или плацебо. Что касается первичного критерия оценки, субъекты сами оценивали эффективность исследуемого лекарственного средства по сравнению с плацебо по 4-балльной шкале [0: нет, 1: легкая, 2: средняя, 3: сильная] в отношении времени ослабления симптомов гриппа (время от начала введения исследуемого лекарственного средства до облегчения семи симптомов гриппа («кашель», «боль в горле», «головная боль», «заложенность носа», «лихорадка или озноб», «боль в мышцах или суставах» и «усталость»)).
Кроме того, что касается вторичного критерия оценки эффективности, эффективность и побочные эффекты исследуемого лекарственного средства оценивали в соответствии с титром вируса гриппа с применением мазка из носа или горла.
В качестве субъектов были выбраны пациенты, которые удовлетворяли всем следующим критериям.
(a) Пациенты мужского или женского пола в возрасте 12 лет или старше и моложе 65 лет
(b) Пациенты, удовлетворяющие всем следующим критериям, у которых было диагностировано инфекционное заболевание вирусом гриппа
- Температура тела (подмышечная температура) 38.0°С или выше
- Наличие одного или нескольких умеренных или более тяжелых симптомов среди следующих системных симптомов и респираторных симптомов вследствие инфекционного заболевания вирусом гриппа
- Системные симптомы (головная боль, лихорадка или озноб, мышечная или суставная боль, усталость)
- Респираторные симптомы (кашель, боль в горле, заложенность носа)
(с) Пациенты в течение 48 часов после начала заболевания (при регистрации)
Определение начала заболевания представляет собой любое наблюдение из следующих.
- Когда температура тела увеличилась в первый раз (увеличение, по крайней мере, на 1°С от нормальной температуры)
- Когда развился какой-либо один или более системных симптомов и респираторных симптомов
[0169]
Способ введения исследуемого лекарственного средства
(i) Тестируемое лекарственное средство
Таблетка 20 мг соединения II-6
(ii) Плацебо или контрольное лекарственное средство
Плацебо для таблетки 20 мг соединения II-6
Капсула 75 мг осельтамивира
Плацебо для таблетки 75 мг осельтамивира: капсула, неотличимая от капсулы 75 мг осельтамивира
[0170]
Дозировка и способ введения
Подходящие пациенты в возрасте от 20 до 64 лет были случайным образом распределены по следующим группам: группа, получавшая однократное введение соединения II-6 (40 или 80 мг в зависимости от массы тела), группа, получавшая 75 мг осельтамивира два раза в день в течение 5 дней, и группа плацебо в соотношении 2:2:1.
Подходящие пациенты в возрасте от 12 до 19 лет были случайным образом распределены в по следующим группам: группа, получавшая однократное введение соединения II-6 (40 или 80 мг в зависимости от массы тела), и группу, получавшую плацебо в соотношении 2:1.
Дозировка соединения II-6 составляла 40 мг для субъектов массой менее 80 кг и 80 мг для субъектов массой 80 кг или более.
Исследуемое лекарственное средство для каждой группы введения
[Группа соединения II-6]
День 1:
Таблетки 20 мг соединения II-6 вводили перорально (2 таблетки или 4 таблетки в зависимости от массы тела). Капсулы плацебо для осельтамивира вводили перорально два раза в день (утром, вечером), по одной капсуле на введение.
День 2 - день 5:
Капсулы плацебо для осельтамивира вводили перорально два раза в день (утром, вечером), по одной капсуле на введение.
[Группа осельтамивира]
День 1:
Таблетки плацебо для соединения II-6 вводили перорально (2 таблетки или 4 таблетки в зависимости от массы тела). Капсулы по 75 мг осельтамивира вводили перорально два раза в день (утром, вечером), по одной капсуле на введение.
День 2 - день 5:
Капсулы по 75 мг осельтамивира вводили перорально два раза в день (утром, вечером), по одной капсуле на введение.
[Группа плацебо]
День 1:
Таблетки плацебо для соединения II-6 вводили перорально (2 таблетки или 4 таблетки в зависимости от массы тела). Капсулы плацебо для осельтамивира вводили перорально два раза в день (утром, вечером), по одной капсуле на введение.
День 2 - день 5:
Капсулы плацебо для осельтамивира вводили перорально два раза в день (утром, вечером), по одной капсуле на введение.
«День 1» означает первый день введения, а «день 2 - день 5» означает дни от второго до пятого, считая с первого дня введения.
[0171]
Основной критерий оценки эффективности
Основным критерием оценки эффективности является время облегчения симптомов гриппа (время облегчения симптомов гриппа).
Это время от начала введения до ослабления симптомов гриппа. Облегчение симптомов гриппа относится к тем случаям, когда все семь симптомов гриппа (кашель, боль в горле, головная боль, заложенность носа, лихорадка или озноб, мышечная или суставная боль, усталость) становятся «0: нет» или «1: легкими» в дневнике пациента, который ведет субъект, и это состояние сохраняется, по меньшей мере, в течение 21,5 часа (24 часа - 10%).
[0172]
Вторичный критерий оценки эффективности
Вторичный критерий оценки эффективности представляет собой следующее.
(1) Доля пациентов с положительным титром вируса гриппа в каждой точке
(2) Количество изменений в титре вируса от базовой линии в каждой точке
(3) Время до прекращения выделения вируса на основе титра вируса
(4) Частота побочных эффектов
[0173]
Титр вируса измеряли следующим образом.
(1) Клетки MDCK-SIAT1, высеянные в 96-луночный микропланшет с плоским дном, культивировали в инкубаторе с 5% СО2 при 37±1°С в течение 1 дня.
(2) Стандартный штамм (вирус гриппа AH3N2, A/Victoria/361/2011, условия хранения: -80°С, происхождение: Национальный институт инфекционных заболеваний), образец (собранный у пациентов в ходе фазы III клинических испытаний соединения II-6 и хранившийся в ультранизкотемпературной морозильной камере) и среду для контроля клеток разбавляли в 101 - 107 раз методом серийного 10-кратного разведения.
(3) После того, как расположение клеток в форме листа было подтверждено под инвертированным микроскопом, среду удаляли и добавляли новую среду по 100 мкл/лунку.
(4) Среду удаляли.
(5) Каждый из образцов (от 100 до 107), приготовленных по п. (2) выше, инокулировали по 100 мкл/лунку, используя 4 лунки на образец.
(6) Проводили центробежную адсорбцию при комнатной температуре при 1000 об/мин в течение 30 минут.
(7) После центрифугирования удаляли среду и промывали один раз клетки новой средой.
(8) Добавляли новую среду по 100 мкл/лунку.
(9) Инкубацию проводили в инкубаторе с 5% СО2 при 33±1°С в течение 3 дней.
(10) После инкубации оценивали цитопатический эффект (СРЕ) под инвертированным микроскопом.
[0174]
Способ определения наличия положительного титра вируса
Предел обнаружения определяли как положительный, если при измерении указанным выше методом измерения титра вируса наблюдалось его превышение.
[0175]
Анализ по первичному критерию оценки
Относительно времени облегчения симптомов гриппа, которое является первичным критерием оценки, описываются первичный анализ и вторичный анализ. Первичный анализ был выполнен на группе ITTI.
(1) Первичный анализ
Для пациентов в возрасте от 12 до 64 лет сравнивали группу плацебо и исследуемую группу, которой вводили исследуемое лекарственное средство, с помощью стратифицированного обобщенного теста Вилкоксона с использованием общего балла семи симптомов гриппа до введения (11 баллов или менее, 12 баллов или более) и регионов (Япония/Азия, другие регионы) в качестве факторов стратификации.
Кроме того, для каждой группы была построена кривая выживаемости Каплана-Мейера для расчета среднего времени ослабления симптомов гриппа и его 95%-ного доверительного интервала, а также разницы во времени смягчения симптомов гриппа между группами и его 95%-ный доверительный интервал.
(2) Вторичный анализ
Для пациентов в возрасте от 20 до 64 лет сравнивали время ослабления симптомов гриппа между группой соединения II-6 и группой осельтамивира тем же методом, что и первичный анализ.
[0176]
Анализ по вторичному критерию оценки
Проводили сравнение группы соединения II-6 с группой плацебо, а также группы соединения II-6 с группой осельтамивира (возрастная группа от 20 до 64 лет) по следующим вторичным критериям оценки эффективности.
(1) Доля пациентов с положительным титром вируса гриппа в различные моменты времени
В анализ были включены только пациенты, имеющие титр вируса, равный или превышающий предел определения до начала введения при посещении 1. При каждом посещении применяли тест Мантеля-Хензеля с использованием в качестве факторов стратификации общего балла семи симптомов гриппа до введения и регионов, и сравнивали долю пациентов с положительным титром вируса между двумя группами.
(2) Количество изменений в титре вируса по сравнению с исходным уровнем в различные моменты времени
В анализ были включены только пациенты, имеющие титр вируса до начала введения при посещении 1. При каждом посещении применялся тест Ван Элтерена с использованием в качестве факторов стратификации общего балла семи симптомов гриппа до введения и регионов, и сравнивали величину изменения титра вируса гриппа по сравнению с исходным уровнем между двумя группами.
(3) Время до прекращения выделения вируса на основе титра вируса
В анализ были включены только пациенты, имеющие титр вируса, равный или превышающий предел определения до начала введения при посещении 1. Применяли стратифицированный обобщенный тест Вилкоксона с использованием в качестве факторов стратификации общего балла семи симптомов гриппа до введения и регионов.
(4) Частота побочных эффектов
Подсчитывали количество эпизодов побочных эффектов и количество пациентов с побочным эффектом для каждой группы введения.
[0177]
(1) Результаты первичного критерия оценки (время облегчения симптомов гриппа)
Из 1436 случайно выбранных пациентов тестирование завершили 1366 пациентов (578 пациентов в группе, которой вводили соединение II-6 по 40 мг или 80 мг, 498 пациентов в группе, которой вводили осельтамивир, и 290 пациентов в группе плацебо). Что касается первичного критерия оценки, случаи ITTI составляли 1064 пациентов (случаи, когда были соблюдены правила GCP (надлежащая клиническая практика), вводился исследуемый препарат, и инфекция вируса гриппа была подтверждена).
Случаи, соответствующие установленному протоколу, составили 990 пациентов (427 пациентов в группе, которой вводили соединение II-6 по 40 или 80 мг, 351 пациент в группе, которой вводили осельтамивир, и 212 пациентов в группе, получавшей плацебо).
Результаты анализа приведены в следующей таблице.

В группе ITTI: в группе соединения II-6 время ослабления симптомов гриппа (медиана) (95% ДИ) составило 53.7 часа (95% ДИ: 49.5, 58.5), тогда как в группе плацебо - 80.2 часа (95% ДИ: 72.6, 87.1), разница между группой соединения II-6 и группой плацебо составила -26.5 часа. Время ослабления симптомов гриппа в группе соединения II-6 было значительно короче, чем в группе плацебо в первичном анализе с использованием стратифицированного обобщенного критерия Вилкоксона (р<.0001).
В подгруппе пациентов в возрасте 20 лет или старше и моложе 65 лет время ослабления симптомов гриппа составило 53.5 часа (95% ДИ: 48.0, 58.5), тогда как в группе осельтамивира - 53.8 часа (95% CI: 50.2, 56.4), разница между группой соединения II-6 и группой осельтамивира составила -0.3 часы. В стратифицированном генерализованном тесте Вилкоксона не наблюдалось значительного различия между временами ослабления симптомов гриппа в группе соединения II-6 и группе осельтамивира.
[0178]
Анализ вторичного критерия оценки
(1) Доля пациентов с положительным титром вируса гриппа в различных точках Результаты анализа приведены в следующей таблице.


Доля пациентов, имеющих положительный титр вируса, была значительно ниже в группе соединения II-6, чем в группе плацебо, в день 2 (тест Мантеля-Хензеля: р<.0001), и более того, значительно ниже в группе соединения II-6, чем в группе плацебо, в день 3 (р<.0001). В подгруппе пациентов в возрасте 20 лет или старше и моложе 65 лет доля пациентов, имеющих положительный титр вируса, была значительно ниже в группе соединения II-6, чем в группе осельтамивира, на день 2 и день 3 (р<.0001).
[0179]
(2) Количество изменений в титре вируса по сравнению с исходным уровнем в различных точках
Результаты анализа приведены в следующей таблице.

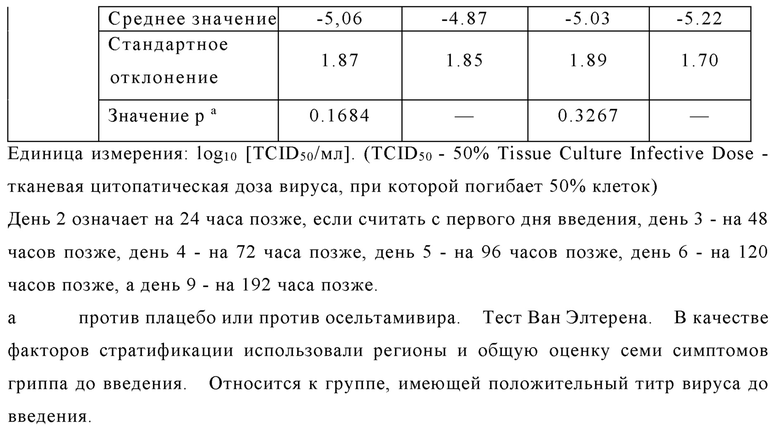
Титр вируса значительно снизился в группе соединения II-6 по сравнению с группой плацебо на 2-й день, и более того, значительно снизился по сравнению с группой плацебо на 3-й день (тест Ван Элтерена: р<.0001). В подгруппе пациентов в возрасте 20 лет или старше и моложе 65 лет титр вируса значительно снизился в группе соединения II-6 по сравнению с группой осельтамивира на день 2 и день 3 (р<.0001).
[0180]
(3) Время до прекращения выделения вируса на основе титра вируса
Результаты анализа приведены в следующей таблице.


Время (медиана) до прекращения выделения вируса на основе титра вируса в группе соединения II-6 составило 24.0 часа, тогда как в группе плацебо - 96.0 часов, и время до прекращения выделения вируса было значительно короче в группе соединения II-6, чем в группе плацебо (стратифицированной обобщенный критерий Вилкоксона: р<.0001). Время до прекращения выделения вируса в подгруппе пациентов в возрасте 20 лет или старше и моложе 65 лет в группе соединения II-6 составило 24.0 часа и в группе осельтамивира 72.0 часа, и время до прекращения выделения вируса было значительно короче в группе соединения II-6, чем в группе осельтамивира (р<.0001).
[0181]
(4) Частота нежелательных явлений
Не сообщается о серьезных нежелательных явлениях, причинно-следственные связи которых нельзя отрицать. Нежелательные явления, причинно-следственную связь которых нельзя отрицать, в группе соединения II-6 произошли у 27 пациентов из 610 пациентов (4.4%, 37 эпизодов), в группе плацебо - у 12 пациентов из 309 пациентов (3.9%, 19 эпизодов) и в группе осельтамивира - у 43 пациентов из 513 пациентов (8.4%, 53 эпизода). Не наблюдалось статистически значимой разницы между случаями в группе соединения II-6 и в группе плацебо (точный критерий Фишера, двустороннее значение Р: 0.8627). Однако частота встречаемости в группе соединения II-6 была значительно ниже, чем в группе осельтамивира (точный критерий Фишера, двустороннее значение Р: 0.0088).
[0182]
Тестовый пример 17: Клинический тест (Ph3: дети)
Была проведена оценка эффективности и безопасности однократного перорального введения исследуемого лекарственного средства (активный ингредиент (соединение II-6): 5 мг, 10 мг, 20 мг, 40 мг) пациентам, инфицированным вирусом гриппа. Что касается первичного критерия оценки, опекуны или субъекты сами проводили оценки и измерения, касающиеся времени облегчения симптомов гриппа (время от начала введения исследуемого лекарственного средства до облегчения симптомов гриппа («кашель», «насморк/заложенность носа» и «лихорадка»)), для оценки эффективности исследуемого лекарственного средства.
«Кашель» и «насморк/заложенность носа» оценивались по 4-балльной шкале [0: нет, 1: слабый, 2: умеренный, 3: сильный].
В качестве субъектов были выбраны пациенты, которые удовлетворяли всем следующим критериям.
(a) Пациенты мужского или женского пола в возрасте 6 месяцев или старше и младше 12 лет
(b) Пациенты, удовлетворяющие всем следующим критериям, у которых диагностировано наличие вируса гриппа
- Положительный результат при быстрой диагностике гриппа [экспресс-тест на антиген (RAT)] на основе мазка из носа или горла
- Температура тела (подмышечная температура) 38.0°С или выше
- Наличие одного или нескольких умеренных или более тяжелых симптомов среди респираторных симптомов вследствие инфекционного заболевания вирусом гриппа у пациентов в возрасте 7 лет и старше
(c) Пациенты в течение 48 часов после начала заболевания (при регистрации). Начало заболевания определяется первым подтверждением температуры тела, превышающей 37.5°С.
(d) Пациенты, имеющие массу тела 5 кг или более.
[0183]
Способ введения исследуемого лекарственного средства (i)
Тестируемое лекарственное средство
Таблетка 5 мг соединения II-6: половина таблетки 10 мг соединения II-6
Таблетка 10 мг соединения II-6
Таблетка 20 мг соединения II-6
[0184]
Дозировка и способ введения
Пациенты получали однократное пероральное введение в день 1 в дозе, рассчитанной на основе массы тела (см. таблицу ниже).

[0185]
Основной критерий оценки эффективности
Основной критерий оценки эффективности представляет собой время облегчения симптомов гриппа (время облегчения симптомов гриппа).
Это время от начала применения до ослабления симптомов гриппа. Облегчение симптома гриппа относится ко времени, когда а и b, значения которых описаны ниже, удовлетворяются с начала введения, и это клиническое состояние сохраняется по меньшей мере в течение 21,5 часа (24 часа - 10%).
a. «Кашель» и «насморк/заложенность носа» представляют собой оба «0: нет» или «1: легкий» в дневнике пациента
b. Температура тела (подмышечная температура) ниже, чем 37.5°С
[0186]
Анализ первичного критерия оценки
Что касается времени облегчения симптомов гриппа, который является первичным критерием оценки, описан первичный анализ. Первичный анализ был выполнен на группе ITTI.
(1) Первичный анализ
Для расчета среднего времени полного облегчения симптомов гриппа и его 95%-ного доверительного интервала была построена кривая Каплана-Мейера времени облегчения симптомов гриппа («кашель», «насморк/заложенность носа» и «лихорадка») (время облегчения симптомов гриппа). Пациенты, у которых симптомы гриппа не были полностью облегчены в течение периода наблюдения, рассматривались отдельно (censored).
[0187]
(1) Результаты первичного критерия оценки (время облегчения симптомов гриппа)
Что касается первичного критерия оценки, в него было вовлечено 103 пациента. Время (медиана) ослабления симптомов гриппа в группе ITTI составило 44.6 часов (95% ДИ: 38.9, 62.5).
[0188]
Пример состава
Следующие примеры составов приведены только в качестве примера и не предназначены для ограничения объема настоящего изобретения.
Пример состава 1: Таблетки
Смешивают соединения, применяемые в настоящем изобретении, лактозу и стеарат кальция. Смесь измельчают, гранулируют и сушат, получая гранулы подходящего размера. Затем к гранулам добавляют стеарат кальция, смесь прессуют и формуют, получая таблетки.
[0189]
Пример состава 2: Капсулы
Равномерно смешивают соединения, применяемые в настоящем изобретении, лактозу и стеарат кальция, получая порошковые лекарственные средства в форме порошков или мелких гранул. Порошковые лекарства помещаются в капсульные контейнеры, получая капсулы.
[0190]
Пример состава 3: Гранулы
Равномерно смешивают соединения, применяемые в настоящем изобретении, лактозу и стеарат кальция, смесь прессуют и формуют. Затем смесь измельчают, гранулируют и просеивают, получая гранулы подходящего размера.
[0191]
Пример состава 4: Перорально распадающиеся таблетки
Смешивают соединения, применяемые в настоящем изобретении, и кристаллическую целлюлозу, гранулируют и изготавливают таблетки, получая перорально распадающиеся таблетки.
[0192]
Пример состава 5: Сухие сиропы
Смешивают соединения, применяемые в настоящем изобретении, и лактозу, измельчают, гранулируют и просеивают, получая подходящих размеров сухие сиропы.
[0193]
Пример состава 6: Инъекции
Смешивают соединения, применяемые в настоящем изобретении, и фосфатный буфер, получая инъекцию.
[0194]
Пример состава 7: Инфузии
Смешивают соединения, применяемые в настоящем изобретении, и фосфатный буфер, получая раствор для инфузии.
[0195]
Пример состава 8: Ингаляции
Смешивают соединение, применяемое в настоящем изобретении, и лактозу, тонко измельчают, получая ингаляции.
[0196]
Пример состава 9: Мази
Смешивают соединения, применяемые в настоящем изобретении, и вазелин, получая мази.
[0197]
Пример состава 10: Пластыри
Смешивают соединения, применяемые в настоящем изобретении, и основу, такую как лейкопластырь или тому подобное, получая пластыри.
[Промышленная применимость]
[0198]
Исходные соединения, применяемые в настоящем изобретении, обладают ингибирующей активностью в отношении кэп-зависимой эндонуклеазы (CEN) после всасывания в организме. Соединения (исходные соединения и/или пролекарства), применяемые в настоящем изобретении, могут быть подходящими агентами для применения для лечения и/или предотвращения симптомов и/или заболевания, вызванного инфекцией вируса гриппа. Фармацевтическая композиция эффективна для сокращения времени до облегчения симптомов гриппа, а также для применения в целях лечения и/или предотвращения инфекционного заболевания, вызванного вирусом гриппа.
| название | год | авторы | номер документа |
|---|---|---|---|
| ЛЕКАРСТВЕННЫЙ ПРЕПАРАТ ДЛЯ ЛЕЧЕНИЯ ГРИППА, ХАРАКТЕРИЗУЮЩИЙСЯ ТЕМ, ЧТО В НЕМ ОБЪЕДИНЕНЫ ИНГИБИТОР КЭП-ЗАВИСИМОЙ ЭНДОНУКЛЕАЗЫ И ЛЕКАРСТВЕННОЕ СРЕДСТВО ПРОТИВ ГРИППА | 2016 |
|
RU2745071C2 |
| ЗАМЕЩЕННЫЕ ПОЛИЦИКЛИЧЕСКИЕ ПРОИЗВОДНЫЕ ПИРИДОНА И ИХ ПРОЛЕКАРСТВА | 2016 |
|
RU2712275C2 |
| 1,4,5-ТРИЗАМЕЩЕННЫЕ ПРОИЗВОДНЫЕ ИМИДАЗОЛА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 1996 |
|
RU2196139C2 |
| ПРОТИВОВИРУСНЫЕ КОМПОЗИЦИИ ШИРОКОГО СПЕКТРА ДЕЙСТВИЯ И СПОСОБЫ | 2018 |
|
RU2813125C2 |
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2815493C2 |
| АРИЛ-N-АРИЛЬНЫЕ ПРОИЗВОДНЫЕ ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2020 |
|
RU2821414C2 |
| Противовирусные агенты и их применение | 2015 |
|
RU2730453C2 |
| ПРОИЗВОДНЫЕ ФЕНИЛ-N-ХИНОЛИНА ДЛЯ ЛЕЧЕНИЯ РНК-ВИРУСНОЙ ИНФЕКЦИИ | 2019 |
|
RU2805064C2 |
| ИМИДАЗОХИНОЛИНОВЫЕ СОЕДИНЕНИЯ | 2005 |
|
RU2415857C2 |
| ПРОЛЕКАРСТВЕННАЯ ФОРМА ЗАМЕЩЕННОГО ПОЛИЦИКЛИЧЕСКОГО ПРОИЗВОДНОГО КАРБАМОИЛПИРИДОНА | 2011 |
|
RU2608519C2 |
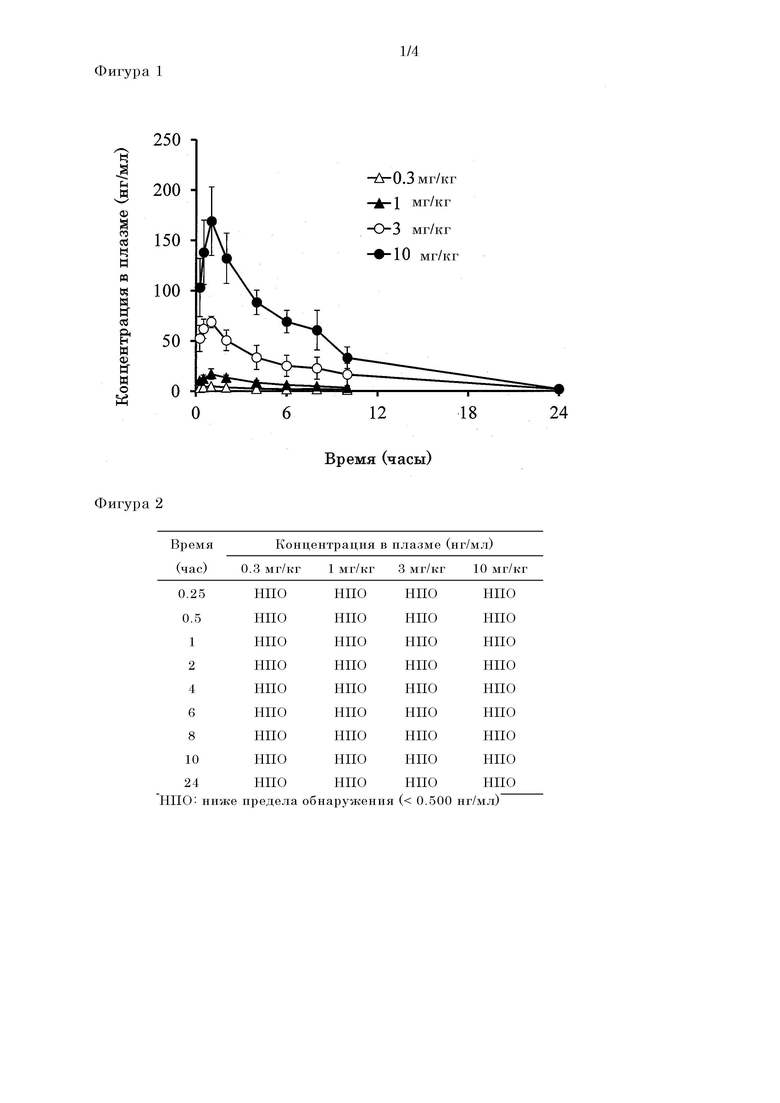

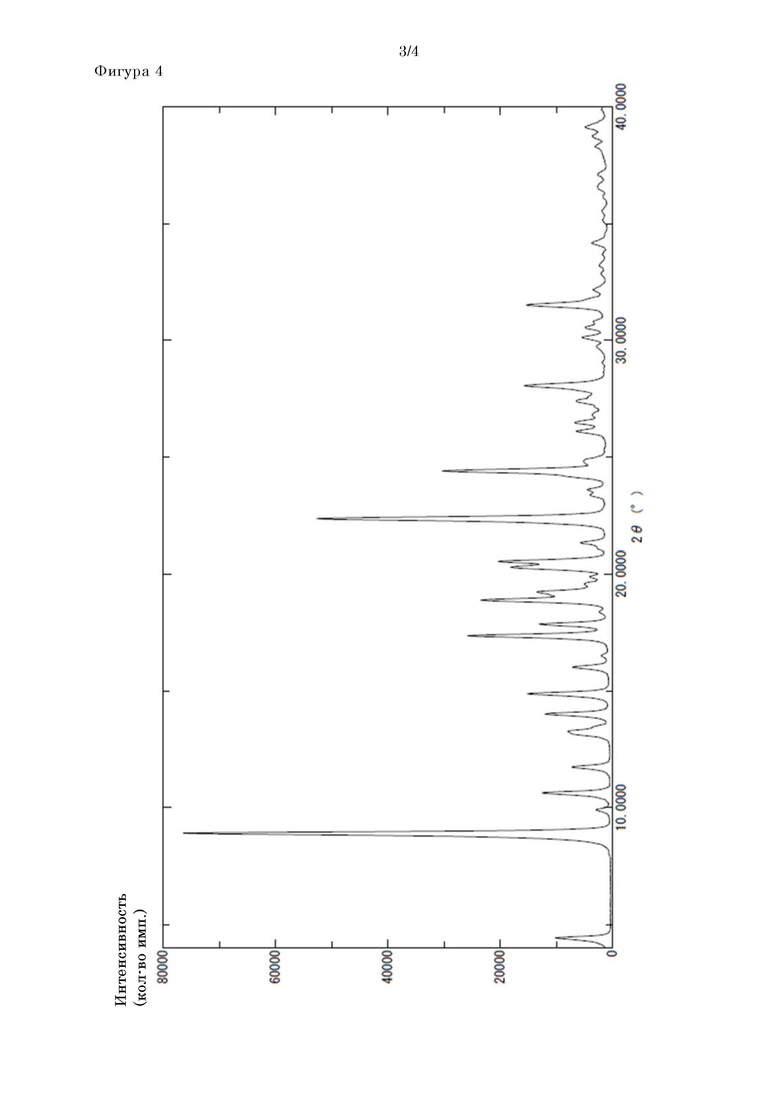
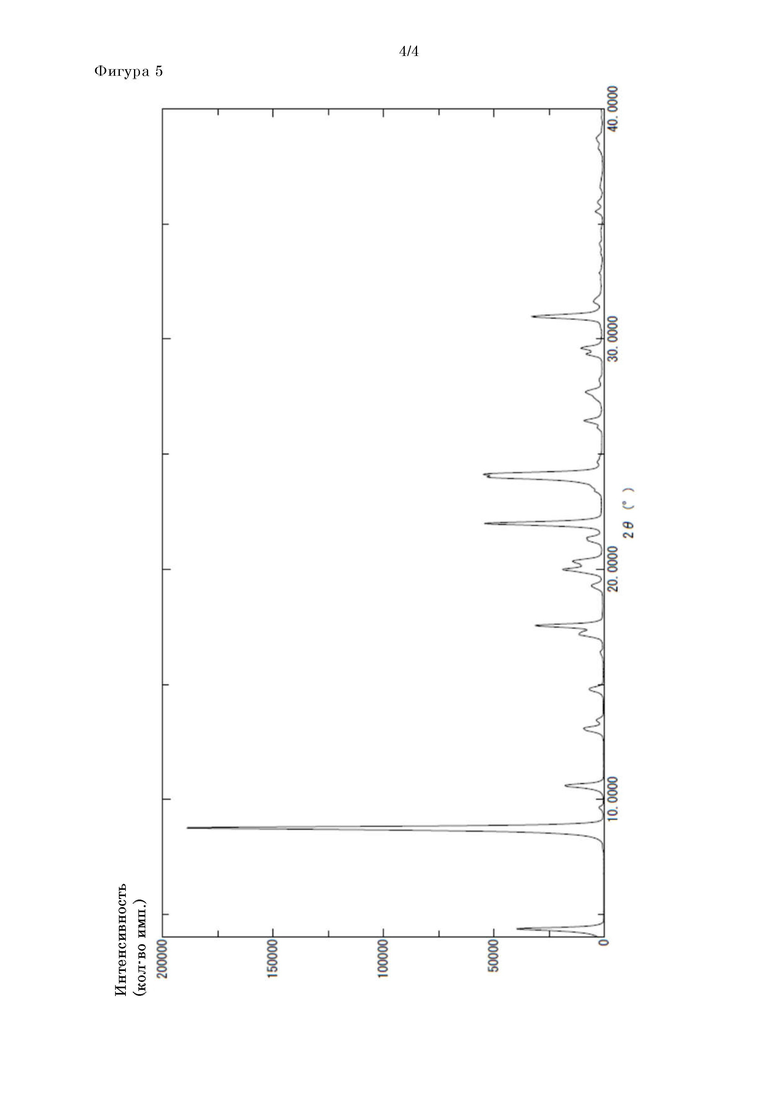
Изобретение относится к применению соединения для лечения и/или предупреждения инфекционного заболевания вируса гриппа, где указанное соединение имеет следующую формулу:
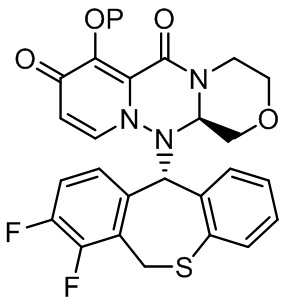
где P представляет собой водород или группу, выбранную из следующих формул:
a) -C(=O)-PR0, g) -C(=O)-O-PR2, i) -C(=O)-O-L-O-PR2, l) -C(PR3)2-O-C(=O)-PR4, m) -C(PR3)2-O-C(=O)-O-PR4 и o) -C(PR3)2-O-C(=O)-O-L-O-PR4, где L представляет собой неразветвленный или разветвленный алкилен; PR0 представляет собой алкил; PR2 представляет собой алкил; PR3 представляет собой водород; и PR4 представляет собой алкил. 11 н. и 29 з.п. ф-лы., 5 ил., 51 табл., 22 пр.
1. Применение соединения для сокращения времени до облегчения симптомов гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа, где указанное соединение имеет следующую формулу:

или его фармацевтически приемлемую соль,
где P представляет собой водород или группу, выбранную из следующих формул:
a) -C(=O)-PR0,
g) -C(=O)-O-PR2,
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4 и
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
где L представляет собой неразветвленный или разветвленный алкилен;
PR0 представляет собой алкил;
PR2 представляет собой алкил;
PR3 представляет собой водород; и
PR4 представляет собой алкил.
2. Применение соединения для ослабления воздействия вируса гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа,
где указанное соединение имеет следующую формулу:
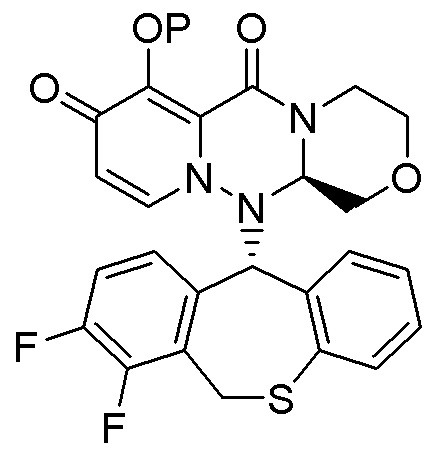
или его фармацевтически приемлемую соль,
где P представляет собой водород или группу, выбранную из следующих формул:
a) -C(=O)-PR0,
g) -C(=O)-O-PR2,
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4 и
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
где L представляет собой неразветвленный или разветвленный алкилен;
PR0 представляет собой алкил;
PR2 представляет собой алкил;
PR3 представляет собой водород; и
PR4 представляет собой алкил.
3. Применение по п. 1 или 2, отличающееся тем, что указанное соединение представляет собой следующее:
 ,
, 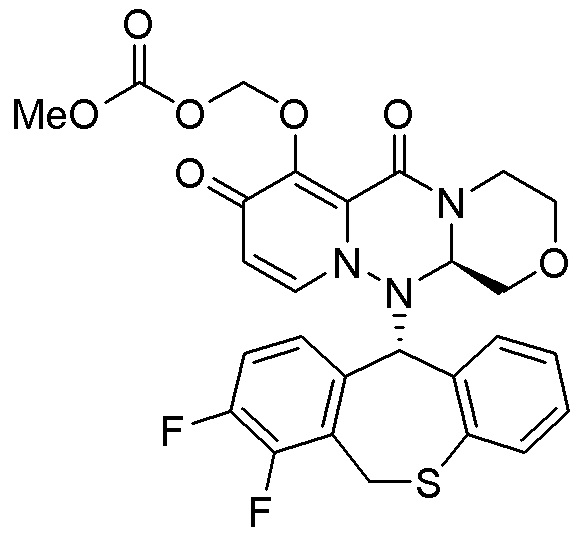 или
или 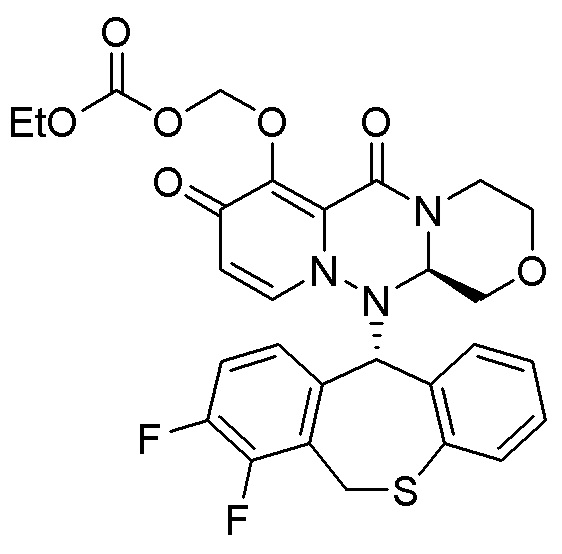
или его фармацевтически приемлемая соль.
4. Применение по п. 3, отличающееся тем, что указанное соединение представляет собой следующее:
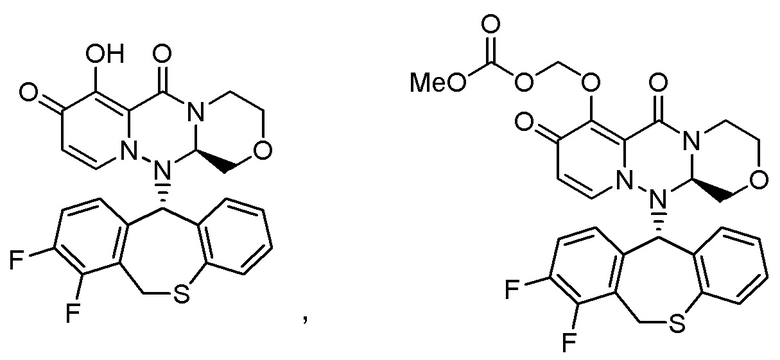
или его фармацевтически приемлемую соль.
5. Применение по п. 3, отличающееся тем, что указанное соединение представляет собой следующее:

или его фармацевтически приемлемую соль.
6. Применение по любому из пп. 1-5, отличающееся тем, что указанное соединение вводят перорально в одной дозе.
7. Применение по п. 6, отличающееся тем, что в случае взрослых вводят 40 мг или 80 мг указанного соединения в одной дозе.
8. Применение по п. 6, отличающееся тем, что в случае детей вводят от 5 до 40 мг указанного соединения в одной дозе в зависимости от массы тела.
9. Применение соединения для получения лекарственного средства для сокращения времени до облегчения симптомов гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа, где указанное соединение имеет следующую формулу:
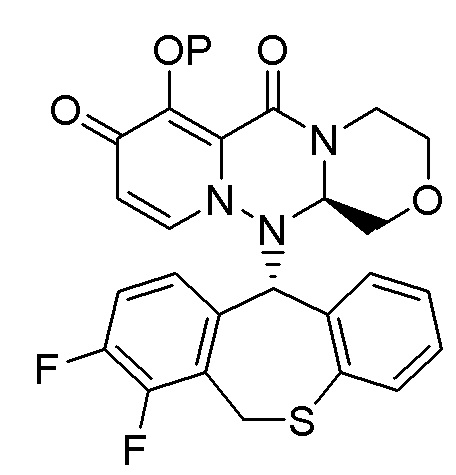
или его фармацевтически приемлемую соль,
где P представляет собой водород или группу, выбранную из следующих формул:
a) -C(=O)-PR0,
g) -C(=O)-O-PR2,
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4 и
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
где L представляет собой неразветвленный или разветвленный алкилен;
PR0 представляет собой алкил;
PR2 представляет собой алкил;
PR3 представляет собой водород; и
PR4 представляет собой алкил.
10. Применение соединения для получения лекарственного средства для ослабления воздействия вируса гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа,
где указанное соединение имеет следующую формулу:

или его фармацевтически приемлемую соль,
где P представляет собой водород или группу, выбранную из следующих формул:
a) -C(=O)-PR0,
g) -C(=O)-O-PR2,
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4 и
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
где L представляет собой неразветвленный или разветвленный алкилен;
PR0 представляет собой алкил;
PR2 представляет собой алкил;
PR3 представляет собой водород; и
PR4 представляет собой алкил.
11. Применение по п. 9 или 10, отличающееся тем, что указанное соединение представляет собой следующее:
 ,
, 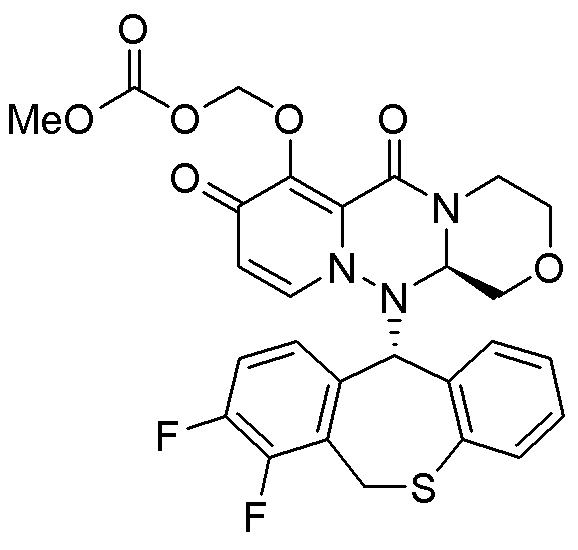 или
или 
или его фармацевтически приемлемая соль.
12. Применение по п. 11, отличающееся тем, что указанное соединение представляет собой следующее:
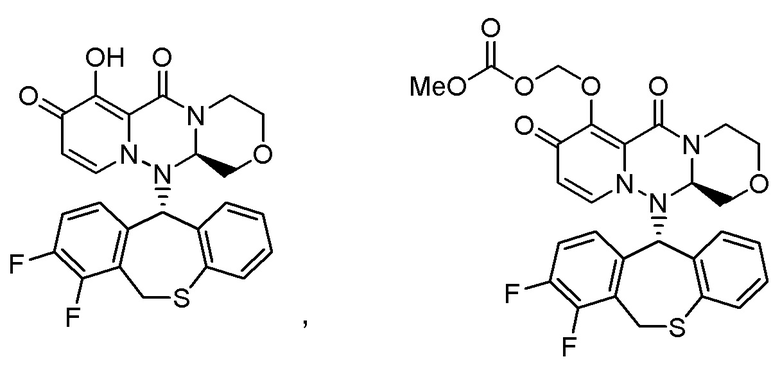
или его фармацевтически приемлемую соль.
13. Применение по п. 12, отличающееся тем, что указанное соединение представляет собой следующее:

или его фармацевтически приемлемую соль.
14. Применение по любому из пп. 9-13, отличающееся тем, что указанное соединение вводят перорально в одной дозе.
15. Применение по п. 14, отличающееся тем, что в случае взрослых вводят 40 мг или 80 мг указанного соединения в одной дозе.
16. Применение по п. 14, отличающееся тем, что в случае детей вводят от 5 до 40 мг указанного соединения в одной дозе в зависимости от массы тела.
17. Способ ослабления воздействия вируса гриппа, включающий введение человеку эффективного количества соединения следующей формулы:
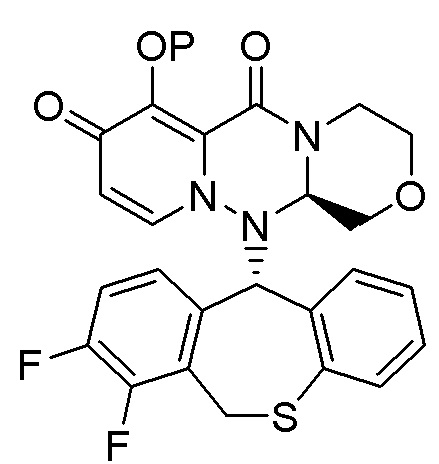
или его фармацевтической приемлемой соли,
где P представляет собой водород или группу, выбранную из следующих формул:
a) -C(=O)-PR0,
g) -C(=O)-O-PR2,
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4 и
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
где L представляет собой неразветвленный или разветвленный алкилен;
PR0 представляет собой алкил;
PR2 представляет собой алкил;
PR3 представляет собой водород; и
PR4 представляет собой алкил.
18. Способ по п. 17, отличающийся тем, что указанное соединение представляет собой следующее:
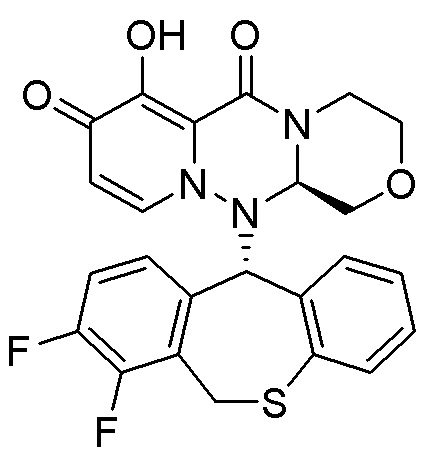 ,
, 
или его фармацевтически приемлемая соль.
19. Способ по п. 18, отличающийся тем, что указанное соединение представляет собой следующее:

или его фармацевтически приемлемую соль.
20. Способ по п. 19, отличающийся тем, что указанное соединение представляет собой следующее:

или его фармацевтически приемлемую соль.
21. Способ по любому из пп. 17-20, отличающийся тем, что указанное соединение вводят перорально в одной дозе.
22. Способ по п. 21, отличающийся тем, что в случае взрослых вводят 40 мг или 80 мг указанного соединения в одной дозе.
23. Способ по п. 21, отличающийся тем, что в случае детей вводят от 5 до 40 мг указанного соединения в одной дозе в зависимости от массы тела.
24. Противогриппозный агент, содержащий соединение следующей формулы:

или его фармацевтически приемлемую соль,
где P представляет собой водород или группу, выбранную из следующих формул:
a) -C(=O)-PR0,
g) -C(=O)-O-PR2,
i) -C(=O)-O-L-O-PR2,
l) -C(PR3)2-O-C(=O)-PR4,
m) -C(PR3)2-O-C(=O)-O-PR4 и
o) -C(PR3)2-O-C(=O)-O-L-O-PR4,
где L представляет собой неразветвленный или разветвленный алкилен;
PR0 представляет собой алкил;
PR2 представляет собой алкил;
PR3 представляет собой водород; и
PR4 представляет собой алкил.
25. Противогриппозный агент по п. 24, отличающийся тем, что указанное соединение представляет собой следующее:

или его фармацевтически приемлемая соль.
26. Противогриппозный агент по п. 25, отличающийся тем, что указанное соединение представляет собой следующее:
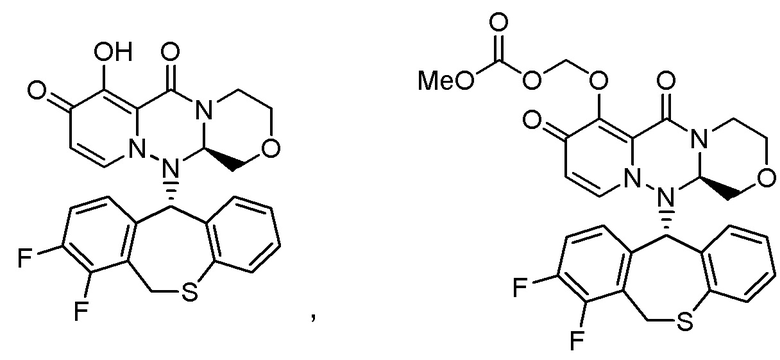
или его фармацевтически приемлемую соль.
27. Противогриппозный агент по п. 26, отличающийся тем, что указанное соединение представляет собой следующее:
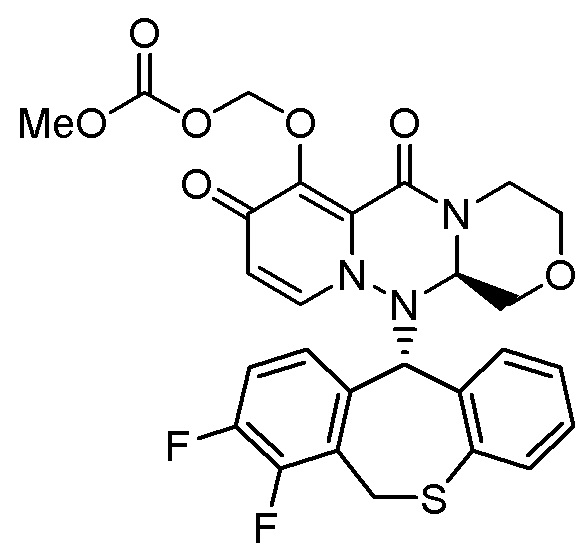
или его фармацевтически приемлемую соль.
28. Кристалл соединения следующей формулы:
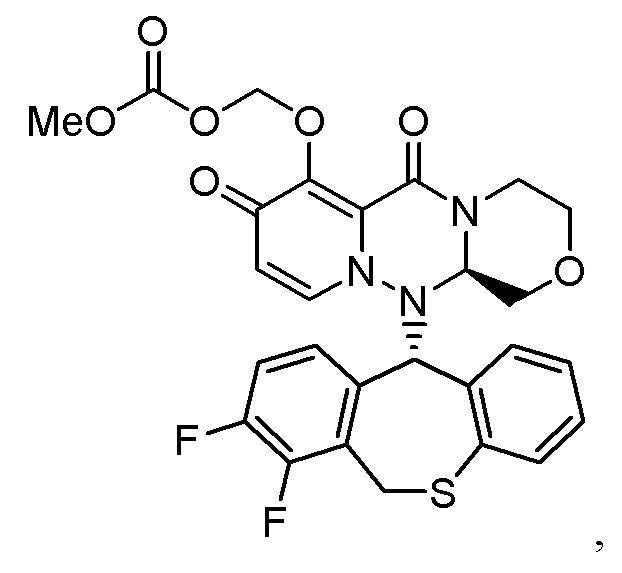
где указанный кристалл имеет два или более пиков на рентгеновской порошковой дифрактограмме, при углах дифракции (2θ), выбранных из пиков 8,6±0,2°, 14,1±0,2°, 17.4±0,2°, 20,0±0,2°, 24,0±0,2°, 26,3±0,2°, 29,6±0,2°и 35,4±0,2° .
29. Кристалл по п. 28, где указанный кристалл имеет следующие пики на рентгеновской порошковой дифрактограмме при углах дифракции (2θ): 8,6±0,2°, 14,1±0,2°, 17,4±0,2°, 20,0±0,2°, 24,0±0,2°, 26,3±0,2°, 29,6±0,2° и 35,4±0,2°.
30. Кристалл по п. 28, где указанный кристалл имеет рентгеновскую порошковую дифрактограмму, по существу идентичную представленной на фигуре 3.
31. Фармацевтическая композиция, обладающая ингибирующей активностью в отношении кэп-зависимой эндонуклеазы, содержащая эффективное количество кристалла по любому из пп. 28-30 и фармацевтически приемлемые носители или разбавители.
32. Применение кристалла по любому из пп. 28-30 для сокращения времени до облегчения симптомов гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа.
33. Применение кристалла по любому из пп. 28-30 для ослабления воздействия вируса гриппа при лечении и/или предупреждении инфекционного заболевания вируса гриппа.
34. Применение по п. 32 или 33, отличающееся тем, что указанный кристалл вводят перорально в одной дозе.
35. Применение по п. 34, отличающееся тем, что в случае взрослых вводят 40 мг или 80 мг указанного кристалла в одной дозе.
36. Применение по п. 35, отличающееся тем, что в случае детей вводят от 5 до 40 мг указанного кристалла в одной дозе в зависимости от массы тела.
37. Способ ослабления воздействия вируса гриппа, включающий введение человеку эффективного количества кристалла по любому из пп. 28-30.
38. Способ по п. 37, отличающийся тем, что указанный кристалл вводят перорально в одной дозе.
39. Способ по п. 38, отличающийся тем, что в случае взрослых вводят примерно 40 мг или 80 мг кристалла в одной дозе.
40. Способ по п. 39, отличающийся тем, что в случае детей вводят от 5 до 40 мг кристалла в одной дозе в зависимости от массы тела.
| WO 2012039414 A1, 29.03.2012 | |||
| WO 2010147068 A1, 23.12.2010 | |||
| RU 2181357 C2, 20.04.2002. |

