Область техники, к которой относится изобретение
Настоящее изобретение относится к фармацевтически активным пиразоло[1,5-a]пиразин-4-ил TYK2-лигандам и аналогам. Такие соединения полезны для ингибирования янус-киназ (JAK). Настоящее изобретение также относится к композициям, способам получения таких соединений, и к способам лечения и профилактики состояний, опосредованных JAK.
Предшествующий уровень техники
Протеинкиназы представляют собой семейства ферментов, катализирующих фосфорилирование специфических остатков в белках, в широком смысле классифицируемых на тирозин- и серин/треонин-киназы. Неприемлемая киназная активность, возникающая в результате мутации, сверхэкспресии или неприемлемой регуляции, дисрегуляции или дерегуляции, а также сверх- или недостаточной продукции факторов роста или цитокинов, вовлечена во многие заболевания, включая, но не ограничиваясь ими, рак, сердечно-сосудистые заболевания, аллергии, астму и другие респираторные заболевания, аутоиммунные заболевания, воспалительные заболевания, заболевания костей, метаболические расстройства, и неврологические и нейродегенеративные расстройства, такие как болезнь Альцгеймера. Неприемлемая киназная активность запускает ряд биологических клеточных реакций, связанных с ростом клеток, клеточной дифференцировкой, функцией клеток, выживаемостью, апоптозом и клеточной подвижностью, вовлеченных в вышеупомянутые и родственные заболевания.
Таким образом, протеинкиназы стали важным классом ферментов в качестве мишеней для терапевтического воздействия. В частности, JAK семейство клеточных белков тирозинкиназ (JAK1, JAK2, JAK3 и Tyk2) играет центральную роль в передаче сигналов цитокинов (Kisseleva et al., Gene, 2002, 285, 1; Yamaoka et al. Genome Biology 2004, 5, 253)). После связывания с их рецепторами цитокины активируют JAK, которые затем фосфорилируют цитокиновый рецептор, тем самым создавая стыковочные сайты для сигнальных молекул, в особенности, членов семейства переносчиков сигнала и активаторов транскрипции (STAT), которые в итоге приводят к экспрессии генов. Известно, что множество цитокинов активируют семейство JAK. Эти цитокины включают семейство интерферонов (IFN) (IFN-альфа, IFN-бета, IFN-омега, лимитин, IFN-гамма, IL-10, IL-19, IL-20, IL-22), семейство gp130 (IL-6, IL-11, OSM, LIF, CNTF, NNT-1/BSF-3, G-CSF, CT-1, лептин, IL-12, IL-23), семейство гамма C (IL-2, IL-7, TSLP, IL-9, IL-15, IL-21, IL-4, IL-13), семейство IL-3 (IL-3, IL-5, GM-CSF), одноцепочечное семейство (EPO, GH, PRL, TPO), рецепторные тирозинкиназы (EGF, PDGF, CSF-1, HGF) и рецепторы, сопряженные с G-белками (AT1).
Остается необходимость в новых соединениях, которые эффективно и избирательно ингибируют специфические JAK ферменты: в частности, TYK2. TYK2 является членом семейства JAK-киназ и имеет важное значение в передаче сигнала интерферонов типа I (включая IFNальфа, INFбета), IL-6, IL-10, IL-12 и IL-23 (Liang, Y. et al., Expert Opinion on Therapeutic Targets, 18, 5, 571-580 (2014)). Таким образом, TYK2 передает сигнал другим членам семейства JAK-киназ в следующих комбинациях: JAK1/JAK2, JAK1/JAK3, JAK1/TYK2, JAK2/TYK2 или JAK2/JAK2. Было показано, что TYK2 играет важную роль в дифференциации и функции множественных типов клеток, важных при воспалительном заболевании и аутоиммунном заболевании, включая естественные клетки-киллеры, В-клетки и типы Т-хелперов. Аберрантная экспрессия TYK2 связана с различными аутоиммунными или воспалительными состояниями. Модуляция иммунной активности посредством ингибирования TYK2 киназной активности может быть полезной при лечении различных иммунных нарушений (O'Shea JJ, Plenge R, Immunity, 36, 542-50 (2012); Murray, P.J., J. Immunol., 178, 2623-2629 (2007); Kisseleva, T., et al., Gene, 285, 1-24 (2002)), в то же время избегая JAK2-зависимой передачи сигнала эритропоэтина (ЕРО) и тромбопоэтина (TPO) (Neubauer H., et al., Cell, 93(3), 397-409 (1998); Parganas E., et al., Cell, 93(3), 385-95 (1998)).
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы I, имеющему структуру:

или его фармацевтически приемлемой соли, или фармацевтически приемлемому сольвату указанного соединения или фармацевтически приемлемой соли, где: A, A' и A'' представляют собой независимо O, C=O, C-R' или N-R'', где R' и R'' могут независимо быть H, амино, -NR7COR6, COR6, -CONR7R8, C1-C6 алкилом или гидрокси(C1-C6 алкил), и R'' может присутствовать или отсутствовать, и присутствует там, где позволяют правила валентности, и где не более чем один из A, A' и A'' представляет собой O или C=O; R0 и R независимо представляют собой H, Br, Cl, F или C1-C6 алкил; R1 представляет собой H, C1-C6 алкил или гидрокси(C1-C6 алкил); R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси, гидрокси(C1-C6 алкил), фенил(C1-C6 алкил), формила, гетероарила, гетероциклической группы, -COR6, -OCOR6, -COOR6, -NR7COR6, -CONR7R8 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил, -SO2NR7R8 и -SO2-R9, где R9 представляет собой C1-C6 алкил, C3-C8 циклоалкил, гетероарил или гетероциклическую группу; где каждый из указанных алкила, циклоалкила, гетероциклической группы или гетероарила может быть незамещенным или замещенным галогеном, циано, гидрокси или C1-C6 алкилом; X представляет собой C-R3 или N, где R3 может быть H или C1-C6 алкилом; R4 и R5 независимо представляют собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил); R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил, C1-C4 алкокси(C1-C6 алкил) или C3-C8 циклоалкил, указанный C1-C6 алкил необязательно замещен галогеном, CN или гидрокси; или, R7 и R8 вместе с атомом, связанным с ними, образуют 5- или 6-членное кольцо, указанное кольцо необязательно замещено галогеном, гидрокси, CN или C1-C6 алкилом; и n равен 0, 1, 2 или 3.
В других аспектах настоящее изобретение также предоставляет:
фармацевтические композиции, которые содержат фармацевтически приемлемый носитель и соединение формулы I;
способы лечения состояний или нарушений, включая воспаления, аутоиммунное заболевание, системную красную волчанку, волчаночный нефрит, дискоидную волчанку, кожную волчанку, волчанку центральной нервной системы, ревматоидный артрит, псориатический артрит, воспалительное заболевание кишечника, болезнь Крона, язвенный колит, астму, аллергическую астму, диабет типа I, полимиозит, дерматомиозит, интерферонопатию I типа, включая синдром Айкарди-Гутьереса и другие менделевские заболевания сверхэкспрессии интерферона I типа, рассеянный склероз, первично-прогрессирующий рассеянный склероз, рецидивирующе-ремиттирующий рассеянный склероз, первичный билиарный цирроз, известный также как первичный билиарный холангит, первичный склерозирующий холангит, аутоиммунный гепатит, неалкогольный стеатоз печени, неалкогольный стеатогепатит, псориаз, дерматомиозит, склеродермию, атопический дерматит, витилиго, круговую алопецию, спондилопатию, анкилозирующий спондилоартрит, болезнь Альцгеймера, нейровоспаление миозит, васкулит, пемфигус, болезнь Крона, волчанку, нефрит, псориаз, рассеянный склероз, большое депрессивное расстройство, аллергию, астму, болезнь Шегрена, синдром сухого глаза, отторжение трансплантата, рак, воспалительное заболевание кишечника, септический шок, кардиопульмональное нарушение, витилиго, алопецию, острое респираторное заболевание, анкилозирующий спондилоартрит, аутоиммунный гепатит, первичный склерозирующий холангит, первичный билиарный цирроз, болезнь Альцгеймера или кахексию, путем введения субъекту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы I или его фармацевтически приемлемой соли;
Способы лечения состояний или нарушений, включая атопический дерматит, экзему, псориаз, склеродермию, волчанку, прурит, усталость, другие зудящие состояния, аллергические реакции, включая аллергический дерматит у млекопитающих, аллергические заболевания лошадей, включая сверхчувствительность к укусам, летнюю экзему, сладкую чесотку у лошадей, запал, воспалительное заболевание дыхательных путей, рецидивирующую обструкцию дыхательных путей, гиперчувствительность дыхательных путей и хроническую обструктивную болезнь легких, путем введения млекопитающему, нуждающемуся в лечении, терапевтически эффективного количества соединения формулы I, или его фармацевтически приемлемой соли; и, способы получения соединений по настоящему изобретению.
Настоящее изобретение будет дополнительно понято из нижеследующего описания, приведенного только в качестве примера. Настоящее изобретение относится к классу производных пиразоло[1,5-a]пиразин-4-ила. В частности, настоящее изобретение относится к пиразоло[1,5-a]пиразин-4-иловым соединениям, полезным в качестве ингибиторов JAK и, в частности, TYK2. Хотя настоящее изобретение этим не ограничивается, понимание различных аспектов изобретения будет получена посредством следующего обсуждения и примеров.
Термин «алкил», отдельно или в комбинации, означает ациклическую насыщенную углеводородную группу формулы CnH2n+1, которая может быть линейной или разветвленной. Если не указано иное, алкильная группа содержит 1-6 атомов углерода. Содержание атомов углерода в алкиле и различных других углеводородсодержащих группах обозначено префиксом, обозначающим нижнее и верхнее число атомов углерода в группе, то есть префикс Ci-Cj указывает содержание атомов углерода целого числа "i" к содержанию атомов углерода целого числа "j", включительно. Так, например, C1-C6 алкил относится к алкилу с одним-шестью атомами углерода, включительно.
Термин «гидрокси», как используется в настоящем описании, означает группу ОН. Термин «гетероциклический» относится к насыщенному или частично насыщенному (т.е. неароматическому) гетероциклу, который содержит от трех до десяти кольцевых атомов, где один или несколько, предпочтительно один, два или три кольцевых атома, являются гетероатом(ами), выбранными из N, O и S, причем остаток является углеродом, и который может быть присоединен через кольцевой атом азота или кольцевой атом углерода. Аналогично, при замещении, заместитель может быть расположен на кольцевом атоме азота (если заместитель присоединен через атом углерода) или кольцевом атоме углерода (во всех случаях). Конкретные примеры включают оксиранил, азиридинил, оксетанил, азетидинил, тетрагидрофуранил, пирролидинил, тетрагидропиранил, пиперидинил, 1,4-диоксанил, морфолинил, пиперазинил, азепанил, оксепанил, оксазепанил и диазепинил.
Термин «арил» относится к ароматическому моноциклическому или бициклическому углеводороду, содержащему от шести до десяти кольцевых атомов углерода, которые могут быть присоединены через один из кольцевых атомов углерода. Аналогично, при замещении заместитель может быть расположен на кольцевом атоме углерода. Конкретные примеры включают, но не ограничиваются ими, фенил, толил, ксилил, триметилфенил и нафтил. Примеры арилзаместителей включают, но не ограничиваются ими, алкил, гидроксил, галоген, нитрил, алкокси, трифторметил, карбоксамидо, SO2Me, бензил и замещенный бензил.
Термин «гетероарил» относится к моновалентному ароматическому моноциклическому или бициклическому гетероциклу из пяти-десяти кольцевых атомов, где один или несколько, предпочтительно один, два или три кольцевых атома, являются гетероатом(ами), выбранными из N, О и S, остальные являются углеродом, и которые могут быть присоединены через кольцевой атом углерода или кольцевой атом азота с соответствующей валентностью. Аналогично, при замещении, заместитель может быть расположен на кольцевом атоме углерода или кольцевом атоме азота с соответствующей валентностью. Конкретные примеры включают, но не ограничиваются ими, тиенил, фуранил, пирролил, пиразолил, имидазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридил, пиридазинил, пиримидинил и пиразинил. Термин «циклоалкил» означает моноциклическую насыщенную углеводородную группу формулы CnH2n-1. Примеры включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклогексил и циклогептил. Если не указано иное, циклоалкильная группа содержит от 3 до 8 атомов углерода.
Термины «гало» и «галоген» относятся к фториду (F), хлориду (Cl), бромиду (Br) или иодиду (I).
Термин «млекопитающее» относится к человеку, сельскохозяйственным животным или животным-компаньонам.
Термин ʺживотное-компаньонʺ или ʺдомашние питомцыʺ относится к животным, которых содержат в качестве домашних любимцев или домашних животных. Примеры животных-компаньонов включают собак, кошек и грызунов, включая хомячков, морских свинок, песчанок и т.п., кроликов, хорьков и птиц.
Термин «сельскохозяйственные животные» относится к животным, которых разводят или выращивают в сельскохозяйственных условиях, для получения продуктов, таких как продукты питания или волокно, или для его труда. В некоторых вариантах сельскохозяйственные животные являются подходящими для потребления млекопитающими, например, человеком. Примерами сельскохозяйственных животных являются крупный рогатый скот, козы, лошади, свиньи, овцы, включая ягнят, и кролики, а также птицы, такие как куры, утки и индейки.
Термин «лечение» или «терапия» означает облегчение симптомов, связанных с заболеванием, нарушением или состоянием, или остановку дальнейшего прогрессирования или ухудшения этих симптомов. В зависимости от заболевания и состояния пациента термин «лечение», как используется в настоящем описании, может включать одно или несколько радикальных, паллиативных и профилактических лечений. Лечение может также включать введение фармацевтической композиции по настоящему изобретению в комбинации с другими видами терапии.
Термин «терапевтически эффективное» указывает на способность средства предотвращать или ослаблять тяжесть заболевания. Фраза «терапевтически эффективное» должно пониматься как эквивалентная фразе «эффективное для лечения, профилактики или ослабления», и обе предназначены для определения количества средства, которое будет достигать цели облегчения тяжести рака, сердечно-сосудистых заболеваний или боли и воспаления, и частоты случаев заболевания после лечения каждым средством отдельно.
«Фармацевтически приемлемый» означает подходящий для использования у млекопитающих, животных-компаньонов или сельскохозяйственных животных.
Если заместители описаны как «независимо выбранные» из группы, каждый заместитель выбирают независимо от другого. Поэтому каждый заместитель может быть идентичным или отличаться от другого заместителя(ей).
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым соединениям, которые являются модуляторами TYK2, полезными для лечения заболеваний и состояний, связанных с нарушением регуляции TYK2. Кроме того, настоящее изобретение относится к фармацевтическим композициям, содержащим такие модуляторы фермента JAK, а также к способам лечения и/или профилактики таких заболеваний и состояний. Соответственно, настоящее изобретение относится к соединению формулы I, представленному выше, имеющему структуру (I):
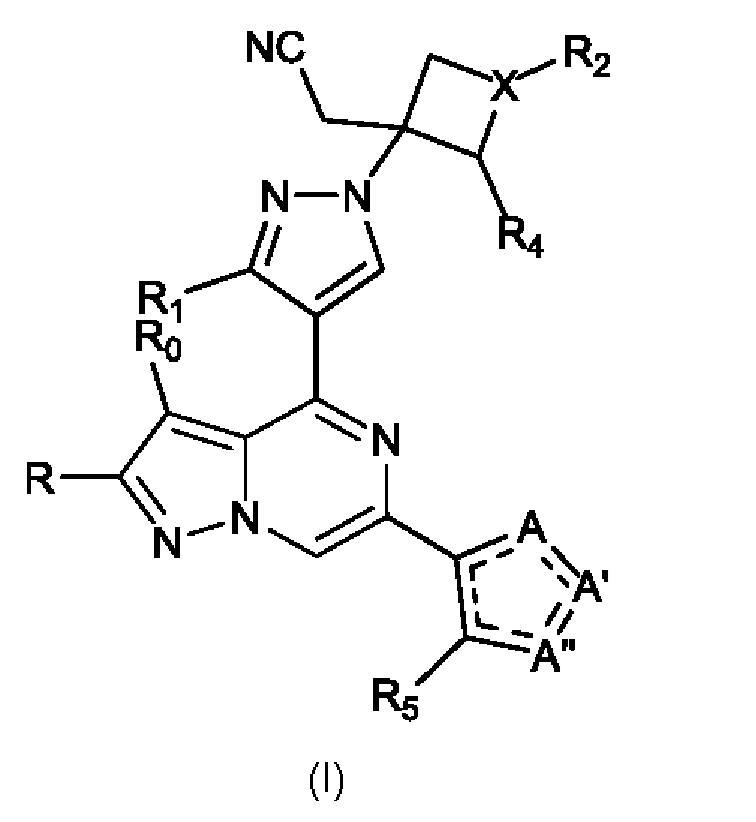
Изобретение также относится к соединению, имеющему структуру (Ia):
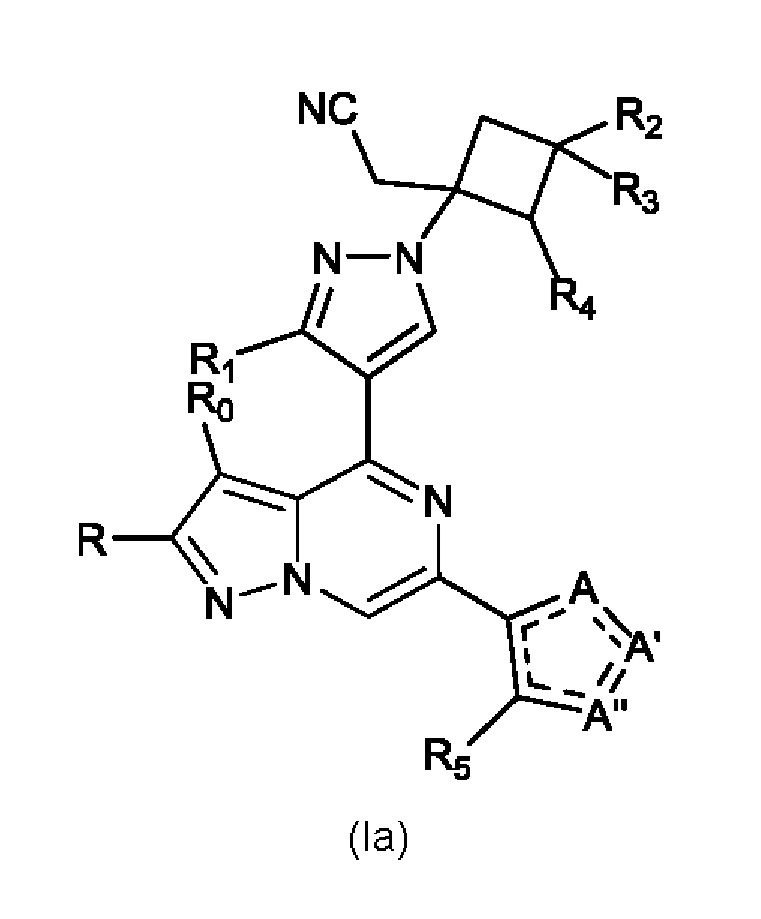
или его фармацевтически приемлемой соли, или фармацевтически приемлемому сольвату указанного соединения или фармацевтически приемлемой соли, где: A, A' и A'' представляют собой независимо O, C=O, C-R' или N-R'', где R' и R'' могут независимо быть H, амино, -NR7COR6, COR6, -CONR7R8, C1-C6 алкил- или гидрокси(C1-C6 алкил)-, и R'' может присутствовать или отсутствовать, и присутствует там, где позволяют правила валентности, и где не более чем один из A, A' и A'' представляет собой O или C=O; R0 и R независимо представляют собой H, Br, Cl, F или C1-C6 алкил; R1 представляет собой H, C1-C6 алкил или гидрокси(C1-C6 алкил)-; R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси-, гидрокси(C1-C6 алкил)-, фенил(C1-C6 алкил)-, формила, гетероарила, гетероциклической группы, -COR6, -OCOR6, -COOR6, -NR7COR6, -CONR7R8 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил, -SO2NR7R8 и -SO2-R9, где R9 представляет собой C1-C6 алкил, C3-C8 циклоалкил, гетероарил или гетероциклическую группу; где каждый из указанных алкила, циклоалкила, гетероциклической группы или гетероарила может быть незамещенным или замещенным галогеном, циано, гидрокси или C1-C6 алкилом; R3 может быть H или C1-C6 алкилом; R4 и R5 независимо представляют собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил); R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил, C1-C4 алкокси(C1-C6 алкил) или C3-C8 циклоалкил, указанный C1-C6 алкил необязательно замещен галогеном, CN или гидрокси; или, R7 и R8 вместе с атомом, связанным с ними, образуют 5- или 6-членное кольцо, указанное кольцо необязательно замещено галогеном, гидрокси, CN или C1-C6 алкилом; и n равен 0, 1, 2 или 3.
Кроме того, настоящее изобретение относится к соединению, имеющему структуру (Ib):
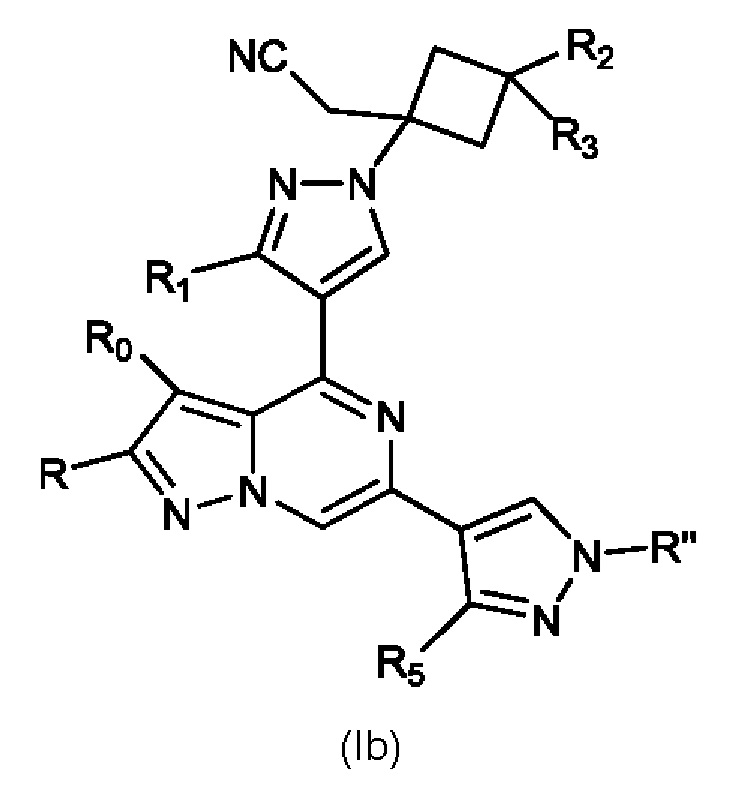
или его фармацевтически приемлемой соли, или фармацевтически приемлемому сольвату указанного соединения или фармацевтически приемлемой соли, где: R'' представляет собой H, -COR6, -CONR7R8, C1-C6 алкил- или гидрокси(C1-C6 алкил)-; R0 и R независимо представляют собой H, Br, Cl F или C1-C6 алкил; R1 представляет собой H, C1-C6 алкил или гидрокси(C1-C6 алкил); R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси, гидрокси(C1-C6 алкил), фенил(C1-C6 алкил), формила, гетероарила, гетероциклической группы, -COR6, -OCOR6, -COOR6, -NR7COR6, -CONR7R8 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил, -SO2NR7R8 и -SO2-R9, где R9 представляет собой C1-C6 алкил, C3-C8 циклоалкил, гетероарил или гетероциклическую группу; где каждый из указанных алкила, циклоалкила, гетероциклической группы или гетероарила может быть незамещенным или замещенным галогеном, циано, гидрокси или C1-C6 алкилом; R3 может быть H или C1-C6 алкилом; R5 представляет собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил); R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил, C1-C4 алкокси(C1-C6 алкил) или C3-C8 циклоалкил, указанный C1-C6 алкил необязательно замещен галогеном, CN или гидрокси; или, R7 и R8 вместе с атомом, связанным с ними, образуют 5- или 6-членное кольцо, указанное кольцо необязательно замещено галогеном, гидрокси, CN или C1-C6 алкилом; и n равен 0, 1, 2 или 3.
Изобретение также относится к соединению, имеющему структуру (Ic):
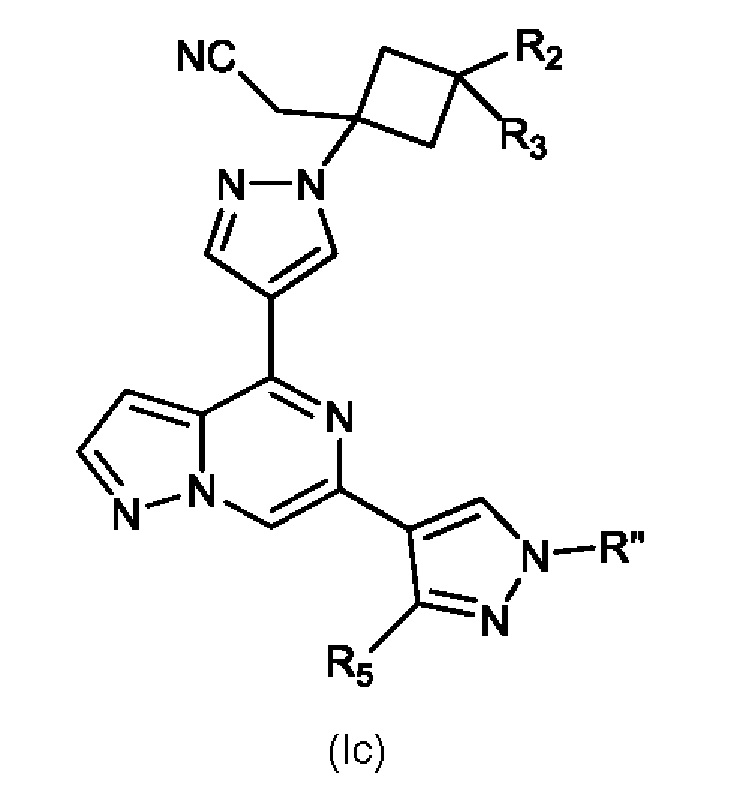
или его фармацевтически приемлемой соли, или фармацевтически приемлемому сольвату указанного соединения или фармацевтически приемлемой соли, где: R'' представляет собой H, -COR6, -CONR7R8, C1-C6 алкил или гидрокси(C1-C6 алкил); R2 выбран из группы, состоящей из H, C1-C6 алкил-, C1-C6 алкокси-, гидрокси(C1-C6 алкил)-, фенил(C1-C6 алкил)-, формила, гетероарила, гетероциклической группы, -COR6, -OCOR6, -COOR6, -NR7COR6, -CONR7R8 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил, -SO2NR7R8 и -SO2-R9, где R9 представляет собой C1-C6 алкил, C3-C8 циклоалкил, гетероарил или гетероциклическую группу; где каждый из указанных алкила, циклоалкила, гетероциклической группы или гетероарила может быть незамещенным или замещенным галогеном, циано, гидрокси или C1-C6 алкилом; R3 представляет собой H или C1-C6 алкил; R5 представляет собой H, амино, C1-C6 алкил- или гидрокси(C1-C6 алкил)-; R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил-, C1-C4 алкокси(C1-C6 алкил)- или C3-C8 циклоалкил, указанный C1-C6 алкил необязательно замещен галогеном, CN или гидрокси; или, R7 и R8 вместе с атомом, связанным с ними, образуют 5- или 6-членное кольцо, указанное кольцо необязательно замещено галогеном, гидрокси, CN или C1-C6 алкилом; и n равен 1, 2 или 3. В конкретном варианте осуществления настоящее изобретение относится к указанному соединению, где R'' представляет собой C1-C6 алкил и R5 представляет собой H.
Изобретение также относится к соединению, имеющему структуру (Id):
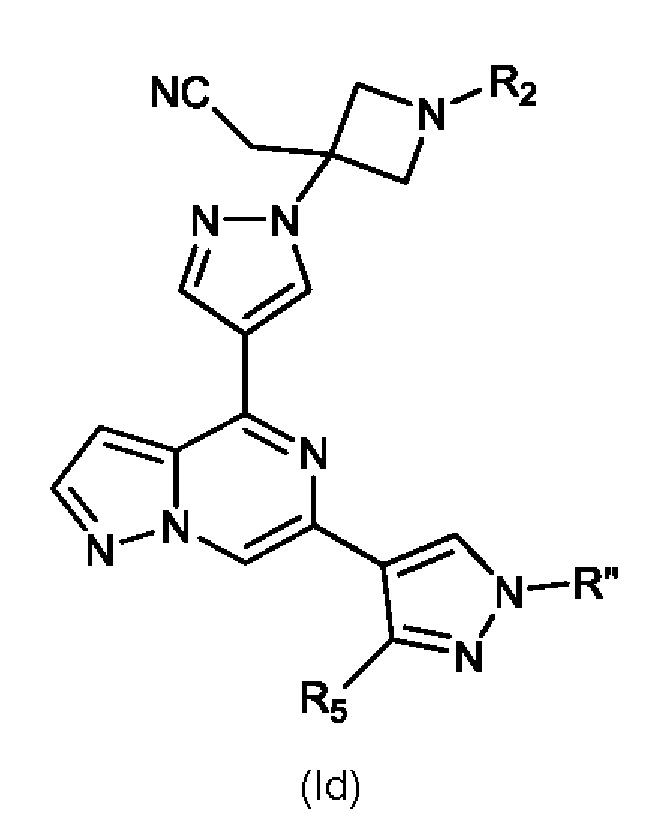
или его фармацевтически приемлемой соли, или фармацевтически приемлемому сольвату указанного соединения или фармацевтически приемлемой соли, где: R'' представляет собой H, -COR6, -CONR7R8, C1-C6 алкил или гидрокси(C1-C6 алкил); R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси, гидрокси(C1-C6 алкил), фенил(C1-C6 алкил), формила, гетероарила, гетероциклической группы, -COR6, -OCOR6, -COOR6, -NR7COR6, -CONR7R8 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил, -SO2NR7R8 и -SO2-R9, где R9 представляет собой C1-C6 алкил, C3-C8 циклоалкил, гетероарил или гетероциклическую группу; где каждый из указанных алкила, циклоалкила, гетероциклической группы или гетероарила может быть незамещенным или замещенным галогеном, циано, гидрокси или C1-C6 алкилом; R5 представляет собой H, амино, C1-C6 алкил- или гидрокси(C1-C6 алкил)-; R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил, C1-C4 алкокси(C1-C6 алкил) или C3-C8 циклоалкил, указанный C1-C6 алкил необязательно замещен галогеном, CN или гидрокси; или, R7 и R8 вместе с атомом, связанным с ними, образуют 5- или 6-членное кольцо, указанное кольцо необязательно замещено галогеном, гидрокси, CN или C1-C6 алкилом; и n равен 1, 2 или 3. В конкретном варианте осуществления настоящее изобретение относится к указанному соединению, где R'' представляет собой C1-C6 алкил и R5 представляет собой H.
Изобретение также относится к соединению, имеющему структуру (Ie):
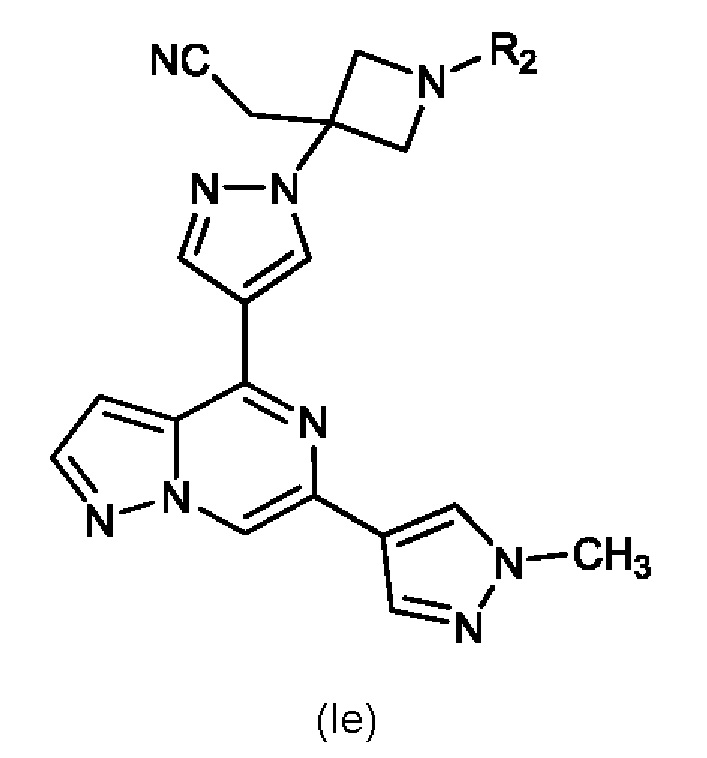
или его фармацевтически приемлемой соли, или фармацевтически приемлемому сольвату указанного соединения или фармацевтически приемлемой соли, где: R2 выбран из группы, состоящей из H, C1-C6 алкил-, C1-C6 алкокси-, гидрокси(C1-C6 алкил)-, фенил(C1-C6 алкил)-, формила, гетероарила, гетероциклической группы, -COR6, -OCOR6, -COOR6, -CONR7R8 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил, -SO2NR7R8 и -SO2-R', где R' представляет собой C1-C6 алкил, C3-C8 циклоалкил, гетероарил или гетероциклическую группу; где каждый из указанных алкила, циклоалкила, гетероциклической группы или гетероарила может быть незамещенным или замещенным галогеном, циано, гидрокси или C1-C6 алкилом; R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил-, C1-C4 алкокси(C1-C6 алкил)- или C3-C8 циклоалкил, указанный C1-C6 алкил необязательно замещен галогеном, CN или гидрокси; или, R7 и R8 вместе с атомом, связанным с ними, образуют 5- или 6-членное кольцо, указанное кольцо необязательно замещено галогеном, гидрокси, CN или C1-C6 алкилом; и n равен 1, 2 или 3. В конкретном варианте осуществления настоящее изобретение относится к указанному соединению, где R2 представляет собой -(CH2)n-W, где W представляет собой циано и n равен 1, 2 или 3.
Изобретение также относится к соединению, имеющему структуру (If):
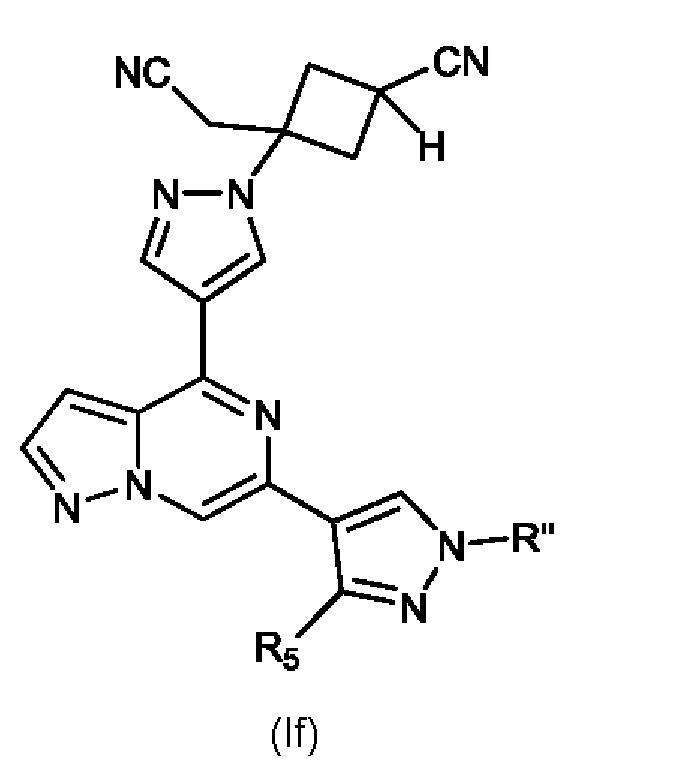
или его фармацевтически приемлемой соли, или фармацевтически приемлемому сольвату указанного соединения или фармацевтически приемлемой соли, где: R'' представляет собой H, -COR6, -CONR7R8, C1-C6 алкил- или гидрокси(C1-C6 алкил)-; R5 представляет собой H, амино, C1-C6 алкил- или гидрокси(C1-C6 алкил)-; R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил-, C1-C4 алкокси(C1-C6 алкил)- или C3-C8 циклоалкил, указанный C1-C6 алкил необязательно замещен галогеном, CN или гидрокси; или, R7 и R8 вместе с атомом, связанным с ними, образуют 5- или 6-членное кольцо, указанное кольцо необязательно замещено галогеном, гидрокси, CN или C1-C6 алкилом; и n равен 0, 1, 2 или 3. В конкретном варианте осуществления настоящее изобретение относится к указанному соединению, где R'' представляет собой C1-C6 алкил. В другом конкретном варианте осуществления изобретение относится к указанному соединению, где R'' представляет собой метил.
В некоторых предпочтительных вариантах осуществления изобретение относится к соединению, выбранному из группы, состоящей из:
(1r,3r)-3-(4-(6-(3-амино-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила;
2,2'-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1,3-диил)диацетонитрила;
2-((1s,3r)-1-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила;
5-(4-(1-((1s,3r)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксамида;
(1s,3s)-3-(цианометил)-3-(4-(6-(5-(гидроксиметил)изоксазол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
(1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
(1s,3s)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
(1r,3r)-3-(цианометил)-3-(4-(3-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
2-((1r,3s)-1-(4-(6-(3-амино-1H-пиразол-5-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила;
(1r,3r)-3-(цианометил)-3-(4-(6-(1-(гидроксиметил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила; и,
2-(1-этил-3-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила, или его фармацевтически приемлемой соли.
В некотором варианте осуществления изобретение относится к соединению, которое представляет собой (1r,3r)-3-(4-(6-(3-амино-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрил, или его фармацевтически приемлемой соли.
В некотором варианте осуществления изобретение относится к соединению, которое представляет собой 2,2'-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1,3-диил)диацетонитрил, или его фармацевтически приемлемой соли.
В некотором варианте осуществления изобретение относится к соединению, которое представляет собой 2-((1s,3r)-1-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил, или его фармацевтически приемлемой соли.
В некотором варианте осуществления изобретение относится к соединению, которое представляет собой 5-(4-(1-((1s,3r)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксамид, или его фармацевтически приемлемой соли.
В некотором варианте осуществления изобретение относится к соединению, которое представляет собой (1s,3s)-3-(цианометил)-3-(4-(6-(5-(гидроксиметил)изоксазол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил, или его фармацевтически приемлемой соли.
В некотором варианте осуществления изобретение относится к соединению, которое представляет собой (1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил, или его фармацевтически приемлемой соли.
В другом конкретном варианте осуществления изобретение относится к соединению, которое представляет собой (1s,3s)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил, или его фармацевтически приемлемой соли.
В еще одном варианте осуществления изобретение относится к соединению, которое представляет собой (1r,3r)-3-(цианометил)-3-(4-(3-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил, или его фармацевтически приемлемой соли.
В определенном другом варианте осуществления изобретение относится к соединению, которое представляет собой 2-((1r,3s)-1-(4-(6-(3-амино-1H-пиразол-5-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил, или его фармацевтически приемлемой соли.
В другом конкретном варианте осуществления изобретение относится к соединению, которое представляет собой 2-(1-этил-3-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил, или, его фармацевтически приемлемой соли.
Кроме того, настоящее изобретение относится к фармацевтической или ветеринарной композиции, содержащей соединение формулы I и Ia-f или его фармацевтически приемлемую соль, и фармацевтически приемлемый носитель.
Изобретение также относится к способу лечения заболевания или состояния, для которого показан ингибитор Tyk2, у субъекта, нуждающегося в таком лечении, включающему введение субъекту терапевтически эффективного количества соединения формулы I или Ia-f, или его фармацевтически приемлемой соли, или фармацевтически приемлемого сольвата указанного соединения или соли.
Изобретение также относится к способу лечения или профилактики нарушения или состояния, выбранного из аллергического ринита, заложенности носа, ринореи, круглогодичного ринита, воспаления носа, астмы всех типов, хронической обструктивной болезни легких, хронической или острой бронхоконстрикции, хронического бронхита, обструкции малых дыхательных путей, эмфиземы, хронической эозинофильной пневмонии, респираторного дистресс-синдрома у взрослых, обострения гиперреактивности дыхательных путей, связанное с другой лекарственной терапией, легочного сосудистого заболевания, легочной артериальной гипертензии, острого повреждения легких, бронхоэктаза, синусита, аллергического конъюнктивита, идиопатического легочного фиброза или атопического дерматита, включающему введение субъекту терапевтически эффективного количества соединения формулы I и Ia-f, или его фармацевтически приемлемой соли, или фармацевтически приемлемого сольвата указанного соединения или соли.
Изобретение также относится к способу лечения первичного билиарного цирроза, включающему введение субъекту терапевтически эффективного количества соединения формулы I или Ia-f, или его фармацевтически приемлемой соли, или фармацевтически приемлемого сольвата указанного соединения или соли.
Изобретение также относится к способу лечения заболевания или состояния, выбранного из воспаления, воспаления, аутоиммунного заболевания, системной красной волчанки, волчаночного нефрита, дискоидной волчанки, кожной волчанки, волчанки центральной нервной системы, ревматоидного артрита, псориатического артрита, воспалительного заболевания кишечника, болезни Крона, язвенного колита, астмы, аллергической астмы, диабета типа I, полимиозита, дерматомиозита, интерферонопатии I типа, включая синдром Айкарди-Гутьереса и другие менделевские заболевания сверхэкспрессии интерферона I типа, рассеянного склероза, первично-прогрессирующего рассеянного склероза, рецидивирующе-ремиттирующего рассеянного склероза, первичного билиарного цирроза, известного также как первичный билиарный холангит, первичного склерозирующего холангита, аутоиммунного гепатита, неалкогольного стеатоза печени, неалкогольного стеатогепатита, псориаза, дерматомиозита, склеродермии, атопического дерматита, витилиго, круговой алопеции, спондилопатии, анкилозирующего спондилоартрита, болезни Альцгеймера, нейровоспаления, включающему введение субъекту терапевтически эффективного количества соединения формулы I или Ia-f, или его фармацевтически приемлемой соли, или фармацевтически приемлемого сольвата указанного соединения или соли.
Изобретение также относится к способу лечения симптомов воспалительного или аутоиммунного заболевания, включая прурит и усталость.
В некоторых вариантах осуществления терапевтически эффективное количество, используемое в соответствии со способом, составляет от 0,01 мг/кг массы тела/день до 100 мг/кг массы тела/день. В некоторых других вариантах осуществления терапевтически эффективное количество, используемое в соответствии со способом составляет от 0,1 мг/кг массы тела/день до 10 мг/кг массы тела/день.
Соединения по изобретению, имеющие одну и ту же молекулярную формулу, но отличающиеся природой или последовательностью связывания их атомов или расположением их атомов в пространстве, называются «изомерами». Изомеры, которые отличаются расположением их атомов в пространстве, называются «стереоизомерами». Специалистам в данной области будет понятно, что соединение формулы I может существовать в виде цис- и транс-ахиральных диастереомеров.
В объем описанных соединений входят все изомеры (например, цис-, транс- или диастереомеры) соединений, описанных в настоящем документе отдельно, а также любые смеси. Все эти формы, включая энантиомеры, диастереомеры, цис, транс, син, анти, сольваты (включая гидраты), таутомеры и их смеси, включены в описанные соединения. Стереоизомерные смеси, например, смеси диастереомеров, могут быть разделены на их соответствующие изомеры известным способом с помощью подходящих методов разделения. Диастереомерные смеси, например, могут быть разделены на их индивидуальные диастереомеры посредством фракционной кристаллизации, хроматографии, распределения в растворителе и подобными методиками. Такое разделение может быть осуществлено на уровне одного из исходных соединений или в самом соединении формулы I. Энантиомеры могут быть разделены посредством образования диастереомерных солей, например, образованием соли с энантиомерно чистой хиральной кислотой, или посредством хроматографии, например, ВЭЖХ, с использованием хроматографических субстратов с хиральными лигандами. Настоящее изобретение включает все фармацевтически приемлемые изотопно-меченные соединения формулы I, в которых один или несколько атомов замещены атомами, имеющими такой же атомный номер, но атомную массу или массовое число, отличающиеся от атомной массы или массового числа, преобладающих в природе.
Примеры изотопов, подходящих для включения в соединения по изобретению, включают изотопы водорода, такие как 2H и 3H, углерода, такие как 11C, 13C и 14C, хлора, такие как 36Cl, фтора, такие как 18F, йода, такие как 123I и 125I, азота, такие как 13N и 15N, кислорода, такие как 15O, 17O и 18O, фосфора, такие как 32P, и и серы, такие как 35S.
Некоторые изотопно-меченные соединения формулы I, например, соединения, включающие радиоактивный изотоп, полезны в исследованиях распределения лекарственных средств и/или субстрата в тканях. Радиоактивные изотопы трития, т.е. 3H, и углерод-14, т.е. 14C, являются особенно полезными для этой цели с точки зрения легкости их включения и быстрого способа детекции.
Замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может обеспечить определенные терапевтические преимущества в результате большей метаболической стабильности, например, увеличение периода полураспада in vivo или требование пониженной дозы и, следовательно, может быть предпочтительным в некоторых случаях. Замещение позитрон-излучающими изотопами, такими 11C, 18F, 15O и 13N, может быть полезным при исследованиях методом позитронной эмиссионной томографии (PET) с целью изучения занятости рецепторов субстратом. Изотопно-меченые соединения формулы I обычно можно получить обычными способами, известными специалистам в данной области, или способами, аналогичными способам, описанным в сопутствующих примерах и получениях, с использованием подходящего изотопно-меченного реагента вместо применяемого ранее немеченого реагента.
В терапевтическом применении для лечения нарушений у млекопитающих соединение по настоящему изобретению или его фармацевтические композиции можно вводить перорально, парентерально, местно, ректально, трансмукозально или интестинально. Парентеральные введения включают непрямые инъекции, чтобы генерировать системный эффект, или прямые инъекции в пораженную область. Местные введения включают лечение кожи или органов, легко доступных при местном введении, например, глаза или уши. Это введение также включает трансдермальное введение, чтобы генерировать системный эффект. Ректальное введение включает форму суппозиториев. Предочтительными путями введения являются пероральный и парентеральный.
Фармацевтически приемлемые соли соединений формулы I и Ia-f включают его соли присоединения кислоты и основания. Подходящие соли присоединения кислоты получают из кислот, которые образуют нетоксичные соли. Примеры включают соли ацетат, адипат, аспартат, бензоат, безилат, бикарбонат/карбонат, бисульфат/сульфат, борат, камсилат, цитрат, цикламат, эдисилат, эзилат, формиат, фумарат, глюцептат, глюконат, глюкуронат, гексафторфосфат, гибензат, гидрохлорид/хлорид, гидробромид/бромид, гидройодид/йодид, изэтионат, лактат, малат, малеат, малонат, мезилат, метилсульфат, нафтилат, 2-напсилат, никотинат, нитрат, оротат, оксалат, пальмитат, памоат, фосфат/гидрофосфат/дигидрофосфат, пироглютамат, сахарат, стеарат, сукцинат, таннат, тартрат, тозилат, трифторацетат и ксинофоат.
Подходящие соли оснований получают из оснований, которые образуют нетоксичные соли. Примеры включают соли алюминия, аргинина, бензатина, кальция, холина, диэтиламина, диоламина, глицина, лизина, магния, меглумина, оламина, калия, натрия, трометамина и цинка.
Также могут быть получены гемисоли кислот и оснований, например, гемисульфатные и гемикальциевые соли. Обзор подходящих солей см. Handbook of Pharmaceutical Salts: Properties, Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002).
Фармацевтически приемлемые соли соединений формулы I и Ia-f могут быть получены, соответственно, одним или несколькими из трех способов:: (i) путем взаимодействия соединения формулы I и Ia-f с желаемой кислотой или основанием; (ii) путем удаления чувствительной к кислоте или основанию защитной группы с подходящего предшественника соединения формулы I и Ia-f или путем раскрытия кольца подходящего циклического предшественника, например, лактона или лактама, с использованием желаемой кислоты или основания; или (iii) путем превращения одной соли соединения формулы I и Ia-f в другую посредством взаимодействия с подходящей кислотой или основанием или с помощью подходящей ионообменной колонки. Все три взаимодействия, как правило, осуществляют в растворе. Полученную соль можно осадить и собрать фильтрованием или можно выделить путем выпаривания растворителя. Степень ионизации полученной соли может варьировать от полностью ионизированной до почти неионизированной.
Фармацевтические композиции по настоящему изобретению могут быть изготовлены способами, хорошо известными в данной области техники, например, посредством традиционного смешивания, растворения, гранулирования, дражирования, растирания в порошок, эмульгирования, инкапсулирования, включения, лиофилизации или распылительной сушки.
Фармацевтическая композиции для применения в соответствии с настоящим изобретением могут быть изготовлены общепринятыми методиками с использованием одного или более фармацевтически приемлемых носителей, включающих эксципиенты и вспомогательные вещества, которые облегчают переработку активного соединения в препараты, которые могут быть использованы фармацевтически. Подходящая лекарственная форма зависит от выбранного пути введения. Фармацевтически приемлемые эксципиенты и носители обычно известны специалистам в данной области и, таким образом, включены в настоящее изобретение. Такие эксципиенты и носители описаны, например, в Remington's Pharmaceutical Sciences, Mack Pub. Co., New Jersey (1991). Препараты по изобретению могут быть разработаны таким образом, чтобы иметь свойства быстрого действия, быстрого высобождения, длительного действия и непрерывного высвобождения. Таким образом, фармацевтические препараты также могут быть изготовлены для контролируемого высвобождения или для медленного высвобождения.
Фармацевтические композиции, подходящие для применения в настоящем изобретении, включают композиции, где активные ингредиенты содержатся в количестве, достаточном для достижения намеченной цели, т.е., контроле или лечении расстройств или заболеваний. Более конкретно, терапевтически эффективное количество означает количество соединения, эффективное для предупреждения, улучшения или ослабления симптомов/признаков заболевания или продления выживания субъекта, подвергаемого лечению.
Количество активного компонента, которое представляет собой соединение по настоящему изобретению, в фармацевтической композиции и ее стандартной лекарственной форме может широко варьироваться или регулироваться в зависимости от способа введения, эффективности конкретного соединения и желаемой концентрации. Определение терапевтически эффективного количества находится в компетенции специалистов в данной области техники. Как правило, количество активного компонента находится в диапазоне от 0,01% до 99% мас. композиции.
В общем, терапевтически эффективное количество дозировки активного компонента находится в диапазоне от примерно 0,01 до примерно 100 мг/кг массы тела/сутки, предпочтительно от примерно 0,1 до примерно 10 мг/кг массы тела/сутки, более предпочтительно от примерно 0,3 до 3 мг/кг массы тела/сутки, еще более предпочтительно от примерно 0,3 до 1,5 мг/кг массы тела/сутки. Следует понимать, что дозировки могут варьировать в зависимости от потребности каждого субъекта и тяжести нарушений или заболеваний, подлежащих лечению.
Требуемая доза может традиционно быть представлена в виде однократной дозы или в виде разделенных доз, вводимых через подходящие интервалы, например, в виде двух, трех, четырех или более субдоз в сутки. Субдоза сама по себе может быть дополнительно разделена, например, на несколько отдельных введений с произвольными интервалами, таких как несколько ингаляций из инсуффлятора, или путем внесения нескольких капель в глаз.
Также следует понимать, что начальная вводимая доза может быть увеличена больше верхнего уровня дозировки с целью быстрого достижения желаемой концентрации в плазме. С другой стороны, начальная доза может быть меньше оптимальной, и суточная доза может быть постепенно увеличена в течение курса лечения в зависимости от конкретной ситуации. При необходимости суточная доза также может быть разделена на несколько доз для введения, например, от двух до четырех раз в сутки.
Настоящее изобретение также относится к любому из применений, способов или композиций, как определено выше, где соединение формулы I или Ia-f или его фармацевтически приемлемая соль, или фармацевтически приемлемый сольват указанного соединения или соли, используют в комбинации с другим фармакологически активным соединением, в частности, с одним из функционально определенных классов или конкретных соединений, перечисленных ниже. Эти средства могут быть введены как часть одной или отдельных лекарственных форм, посредством одного и того же или разных путей введения и по одной или разным схемам введения согласно стандартной фармацевтической практике, известной специалисту в данной области.
Подходящие средства для использования в комбинированной терапии с соединением формулы I или Ia-f или его фармацевтически приемлемой солью, или фармацевтически приемлемым сольватом указанного соединения или солью, сульфасалазин, месалазин, преднизон, азатиоприн, инфликсимаб, адалимумаб, белимумаб, бецертолизумаб, натализумаб, ведолизумаб, гидрокортизон, будесонид, циклоспорин, такролимус, фексофенадин, 6-меркаптопурин, метотрексат, урсодеоксихолевая кислота, обетихолевая кислота, антигистамины, рифампин, преднизон, метотрексат, азатиоприн, циклофосфамид, гидроксихлорохин, мофетил, натрий-микофенолат, такролимус, лефлуномид, хлорохин и хинакрин, талидомид, ритуксан, НПВС, солумедрол, депомедрол и дексаметазон.
Другие подходящие средства для применения в комбинированной терапии с соединением формулы I или Ia-f или его фармацевтически приемлемой солью, или фармацевтически приемлемым сольватом указанного соединения или солью, включают: антагонисты белка, активирующего 5-липоксигеназу (FLAP); лейкотриеновые антагонисты (LTRA), включая антагонисты, например, LTB4, LTC4, LTD4, LTE4, CysLT1 или CysLT2, монтелукаст или зафирлукаст; антагонисты гистаминовых рецепторов, такие как, антагонист гистаминового рецептора типа 1 или антагонист гистаминового рецептора типа 2, например, лоратидин, фексофенадин, дезлоратидин, левоцетиризин, метаперилен или цетиризин; агонист α1-адренорецептора или агонист α1-адренорецептора, например фенилэфрин, метоксамин, оксиметазолин или метилнорефрин; антагонист мускаринового М3-рецептора, например, тиотропий или ипратропий; двойной антагонист мускаринового М3-рецептора/β2-агонист; ингибитор PDE, такой как ингибитор PDE3, ингибитор PDE4 или ингибитор PDE5, например, теофиллин, силденафил, варденафил, тадалафил, ибудиласт, циломиласт или рофлумиласт; кромогликат натрия или недокромил натрия; ингибитор циклооксигеназы (СОХ), такой как неселективный ингибитор (например, аспирин или ибупрофен) или селективный ингибитор (например, целекоксиб или валдекоксиб); глюкокортикостероид, например, флутиказон, мометазон, дексаметазон, преднизолон, будесонид, циклесонид или бекламетазон; противовоспалительное моноклональное антитело, например, инфликсимаб, адалимумаб, танезумаб, ранибизумаб, бевацизумаб или меполизумаб; β2-агонист, например, салметерол, альбутерол, сальбутамол, фенотерол или формотерол, особенно β2-агонист длительного действия; антагонист интегрина, например, натализумаб; ингибитор молекулы адгезии, такой как антагонист VLA-4; антагонист кининовых рецепторов B1 или B2; иммунодепрессивное средство, такой как ингибитор пути IgE (например, омализумаб) или циклоспорин; ингибитор матриксной металлопротеиназы (MMP), такой как ингибитор MMP-9 или MMP-12; антагонист рецептора тахикинина NK1, NK2 или NK3; ингибитор протеазы, такой как ингибитор эластазы, химазы или катепсина G; агонист рецептора аденозина A2a; агонист рецептора аденозина A2b; ингибитор урокиназы; агонист дофаминового рецептора (например, ропинирол), в частности, агонист дофаминового рецептора D2 (например, бромокриптин); модулятор пути NFκB, такой как ингибитор IKK; дополнительный модулятор сигнального пути цитокина, такой как ингибитор JAK-киназы, syk-киназы, p38-киназы, SPHK-1-киназы, Rho-киназы, EGF-R или MK-2; муколитическое, мукокинетическое или противокашлевое средство; антибиотик; противовирусное средство; вакцину; хемокин; блокатор эпителиального натриевого канала (ENaC) или ингибитор эпителиального натриевого канала (ENaC); агонист нуклеотидного рецептора, такой как агонист P2Y2; ингибитор тромбоксана; ниацин; ингибитор 5-липоксигеназы (5-LO), например, зилейтон; фактор адгезии, такой как VLAM, ICAM или ELAM; антагонист рецептора CRTH2 (DP2); антагонист простагландина D2 (DP1); ингибитор гематопоэтической простагландин-D2-синтазы (HPGDS); интерферон-β; растворимый человеческий TNF рецептор, например, этанерцепт; ингибитор HDAC; ингибитор фосфатидилинозитол-3-киназы гамма (PI3Kγ); ингибитор ингибиторы фосфатидилинозитол-3-киназы дельта (PI3Kδ); антагонист рецептора CXCR-1 или CXCR-2; ингибитор IRAK-4; и, ингибитор TLR-4 или TLR-9, включая фармацевтически приемлемые соли конкретно названных соединений и фармацевтически приемлемые сольваты указанных конкретно названных соединений и соли.
Соответственно, изобретение относится к способам лечения или профилактики заболевания, состояния или расстройства, связанного с JAK у субъекта, такого как человека или млекопитающее, не являющееся человеком, включающим введение эффективного количества одного или нескольких соединений, описанных в настоящем документе субъекту. Подходящие субъекты, которых можно лечить, включают домашних или диких животных, животных-компаньонов, таких как собаки, кошки, лошади и тому подобное; домашний скот, включая коров и других жвачных животных, свиней, домашних птиц, кроликов и тому подобное; приматов, например, обезьян, таких как макака-резус и яванские макаки (также известные как крабоеды или длиннохвостые), мартышки, игрунки, шимпанзе, макаки и им подобные; и грызунов, таких как крысы, мыши, песчанки, морские свинки и им подобные. В одном варианте осуществления соединение вводят в фармацевтически приемлемой форме, необязательно в фармацевтически приемлемом носителе.
Предполагают, что состояния, при которых избирательное нацеливание на JAK-путь или модуляция JAK-киназ, в частности, TYK2, является терапевтически полезным, включают, в частности, артрит, астму, аутоиммунные заболевания, злокачественные образования или опухоли, диабет, некоторые глазные заболевания, расстройства или состояния, воспаление, воспаления кишечника, аллергии или состояния, нейродегенеративные заболевания, псориаз, и отторжение трансплантата. Состояния, при которых полезно избирательное ингибирование TYK2, более подробно обсуждаются ниже.
Соответственно, соединение формулы I или Ia-f или его фармацевтически приемлемые соли и сольваты, и их фармацевтические композиции могут быть использованы для лечения множества состояний или заболеваний, таких как следующие:
Артрит, включая ревматоидный артрит, ювенильный артрит и псориатический артрит;
Аутоиммунные или воспалительные заболевания или расстройства, например, тиреоидит Хашимото, аутоиммунная гемолитическая анемия, аутоиммунный атрофический гастрит пернициозной анемии, аутоиммунный энцефаломиелит, аутоиммунный орхит, болезнь Гудпасчера, аутоиммунная тромбоцитопения, симпатическая офтальмия, тяжелая миастения, болезнь Грейвса, первичный билиарный цирроз, аутоиммунный гепатит, первичный склерозирующий холангит, хронический агрессивный гепатит, неалкогольный стеатоз печени, неалкогольный стеатогепатит язвенный колит и мембранная гломерулопатия, системная красная волчанка, ревматоидный артрит, псориатический артрит, синдром Шегрена, синдром Рейтера, полимиозит, дерматомиозит, интерферонопатии I типа, включая синдром Айкарди-Гутьереса и другие менделевские заболевания сверхэкспрессии интерферона I типа, системный склероз, узелковый полиартериит, рассеянный склероз, рецидивирующе-ремиттирующий рассеянный склероз, первично-прогрессирующий рассеянный склероз, вторично-прогрессирующий рассеянный склероз и буллезный пемфигоид, и другие аутоиммунные заболевания, которые могут быть на основе O-клеток (гуморальные) или Т-клеток, включая синдром Когана, анкилозирующий спондилоартрит, гранулематоз Вегенера, аутоиммунную алопецию, диабет I типа или юношеский диабет, или тиреоидит;
Рак или опухоли, включая рак пищевого/желудочно-кишечного тракта, рак толстой кишки, рак печени, рак кожи, включая мастоцитому и плоскоклеточную карциному, рак груди и молочной железы, рак яичников, рак предстательной железы, лимфому, лейкоз, включая острый миелоидный лейкоз и хронический миелоидный лейкоз, рак почки, рак легкого, рак мышцы, рак кости, рак мочевого пузыря, рак головного мозга, меланому, включая меланому ротовой полости и метастатическую меланому, саркому Капоши, миеломы, включая множественную миелому, миелопролиферативные заболевания, пролиферативную диабетическую ретинопатию или связанные с ангиогенезом заболевания, включая солидные опухоли;
Диабет, включая диабет типа I и осложнения при диабете;
Глазные заболевания, нарушения или состояния, включая аутоиммунные заболевания глаз, кератоконъюнктивит, весенний конъюнктивит, увеит, включая увеит, связанный с болезнью Бехчета, и вызванный линзами увеит, кератит, герпетический кератит, конический кератит, эпителиальную дистрофию роговицы, кератолейкому, пемфигоид конъюнктивы, разъедающую язву роговицы (язва Мурена), склерит, офтальмопатию Грейвса, синдром Фогта-Коянаги-Харада, сухой кератоконъюнктивит (синдром сухого глаза), фликтену, иридоциклит, саркоидоз, эндокринную офтальмопатию, симпатический офтальмит, аллергический конъюнктивит или окулярную неоваскуляризацию;
Кишечные воспаления, включая болезнь Крона, язвенный колит, воспалительное заболевание кишечника, целиакию, проктит, эозинофильный гастроэнтерит и мастоцитоз;
Нейродегенеративные заболевания, включая заболевание двигательных нейронов, болезнь Альцгеймера, болезнь Паркинсона, боковой амиотрофический склероз, болезнь Хантингтона, церебральную имшемию или нейродегенеративное заболевание, вызванное травматическим повреждением, инсульт, глутаматную нейротоксичность и гипоксию; ишемическое/реперфузионное повреждение при ударе, ишемию миокарда, почечную ишемию, сердечные приступы, сердечную гипертрофию, атеросклероз и артериосклероз, гипоксию органа или агрегацию тромбоцитов;
Кожные заболевания, состояния или расстройства, включая атопический дерматит, экзему, псориаз, склеродермию, прурит или другие зудящие состояния, витилиго, алопецию;
Аллергические реакции, включая аллергический дерматит у млекопитающих (включая аллергические заболевания лошадей, такие как сверхчувствительность к укусам), летнюю экзему, сладкую чесотку у лошадей, запал, воспалительное заболевание дыхательных путей, рецидивирующую обструкцию дыхательных путей, гиперчувствительность дыхательных путей или хроническую обструктивную болезнь легких;
Астма и другие обструктивные заболевания дыхательных путей, включая хроническую или запущенную астму, позднюю астму, бронхит, бронхиальную астму, аллергическую астму, наследственную астму, приобретенную астму или пылевую астму;
Отторжение трансплантата, включая отторжение трансплантата панкреатических островков, отторжение трансплантата костного мозга, реакция трансплантат против хозяина, отторжение трансплантата органа и клеток, такого как костный мозг, хрящ, роговица, сердце, межпозвоночный диск, островок, почка, конечность, печень, легкое, мышца, миобласт, нерв, поджелудочная железа, кожа, тонкая кишка или трахея, или ксенотрансплантация.
Химический синтез
Специалисту в данной области будет понятно, что экспериментальные условия, изложенные в приведенных ниже схемах, иллюстрируют подходящие условия для осуществления указанных преобразований и что может быть необходимо или желательно изменять определенные условия, используемые для получения соединений формулы (I). Кроме того, будет понятно, что может быть необходимо или желательно осуществить преобразования в другом порядке по сравнению с описанными в схемах или модифицировать одно или несколько преобразований для получения желаемого соединения по изобретению.
Все производные формулы (I) могут быть получены с помощью процедур, описанных в общих способах, представленных ниже, или путем их обычных модификаций. Настоящее изобретение также охватывает любой один или несколько из этих способов для получения производных формулы (I), в дополнение к любым новым промежуточным соединениям, используемым в них. Специалисту в данной области техники будет понятно, что следующие реакции можно нагревать термическим способом или с помощью микроволнового облучения.
Кроме того, будет понятно, что может быть необходимо или желательно осуществить преобразования в другом порядке по сравнению с описанными на схемах, или модифицировать одно или несколько преобразований для получения желаемого соединения по изобретению.
Приведенные ниже пути, в том числе упомянутые в примерах и получениях, иллюстрируют способы синтеза соединений формулы (I). Специалистам в данной области будет понятно, что соединения по изобретению и промежуточные соединения могут быть получены способами, отличными от тех, которые конкретно описаны в настоящем документе, например, путем адаптации способов, описанных в настоящем документе, например, способами, известными в данной области. Подходящими руководствами по синтезу, взаимопревращениям функциональных групп, использованию защитных групп и т.п., являются, например: ʺComprehensive Organic Transformationsʺ by RC Larock, VCH Publishers Inc. (1989); Advanced Organic Chemistryʺ by J. March, Wiley Interscience (1985); ʺDesigning Organic Synthesisʺ by S Warren, Wiley Interscience (1978); ʺOrganic Synthesis - The Disconnection Approachʺ by S Warren, Wiley Interscience (1982); ʺGuidebook to Organic Synthesisʺ by RK Mackie and DM Smith, Longman (1982); ʺProtective Groups in Organic Synthesisʺ by TW Greene and PGM Wuts, John Wiley and Sons, Inc. (1999); и ʺProtecting Groupsʺ by PJ, Kocienski, Georg Thieme Verlag (1994); и любые обновленные версии упомянутых стандартных работ.
Кроме того, специалистам в данной области техники понятно, что на любой стадии синтеза соединений по изобретению может оказаться необходимым или желательным защита одной или нескольких чувствительных групп с тем, чтобы предотвратить нежелательные побочные реакции. В частности, может быть необходимо или желательно защитить группы амино или карбоновой кислоты. Защитные группы, используемые при получении соединений по изобретению, могут быть использованы обычным способом. См., например, те, которые описаны в 'Greene's Protective Groups in Organic Synthesis' by Theodora W Greene and Peter G M Wuts, third edition, (John Wiley and Sons, 1999), в частности, главы 7 (ʺProtection for the Amino Groupʺ) и 5 (ʺProtection for the Carboxyl Groupʺ), которая включена в настоящее описание в качестве ссылки, где также описаны способы удаления таких групп.
В приведенных ниже общих способах синтеза, если не указано иное, заместители являются такими, как определено выше, со ссылкой на соединения формулы (I) выше.
Где даны соотношения растворителей, соотношения представлены по объему.
Соединения по изобретению могут быть получены любым способом, известным в данной области для получения соединений аналогичной структуры. В частности, соединения по изобретению могут быть получены с помощью процедур, описанных со ссылкой на приведенные ниже схемы, или с помощью конкретных способов, описанных в примерах, или с помощью аналогичных им процессов.
Специалисту в данной области будет понятно, что экспериментальные условия, изложенные в приведенных ниже схемах, иллюстрируют подходящие условия для осуществления указанных преобразований и что может быть необходимо или желательно изменять определенные условия, используемые для получения соединений формулы (I).
Квалифицированный специалист оценит, что экспериментальные условия, изложенные в нижеследующих схемах, иллюстрируют подходящие условия для осуществления показанных преобразований, и что может быть необходимо или желательно варьировать точные условия, используемые для получения соединений формулы (I).
Кроме того, специалистам в данной области техники понятно, что на любой стадии синтеза соединений по изобретению может оказаться необходимым или желательным защита одной или нескольких чувствительных групп с тем, чтобы предотвратить нежелательные побочные реакции. В частности, может быть необходимо или желательно защитить группы амино или карбоновой кислоты. Защитные группы, используемые при получении соединений по изобретению, могут быть использованы обычным способом. См., например, те, которые описаны в 'Greene's Protective Groups in Organic Synthesis' by Theodora W Greene and Peter G M Wuts, third edition, (John Wiley and Sons, 1999), в частности, главы 7 (ʺProtection for the Amino Groupʺ) и 5 (ʺProtection for the Carboxyl Groupʺ), которая включена в настоящее описание в качестве ссылки, где также описаны способы удаления таких групп.
Все производные формулы (I) могут быть получены с помощью процедур, описанных в общих способах, представленных ниже, или путем их обычных модификаций. Настоящее изобретение также охватывает любой один или несколько из этих способов для получения производных формулы (I), в дополнение к любым новым промежуточным соединениям, используемым в них. Специалисту в данной области техники будет понятно, что следующие реакции можно нагревать термическим способом или с помощью микроволнового облучения.
Кроме того, будет понятно, что может быть необходимо или желательно осуществить преобразования в другом порядке по сравнению с описанными в схемах или модифицировать одно или несколько преобразований для получения желаемого соединения по изобретению. Схемы являются репрезентативными для способов, полезных для синтеза соединений по настоящему изобретению. Они не должны каким-либо образом ограничивать объем изобретения.
Согласно первому способу соединения формулы I могут быть получены из соединений формул (A), (B), (C) и (D), как показано на схеме 1.
Схема 1
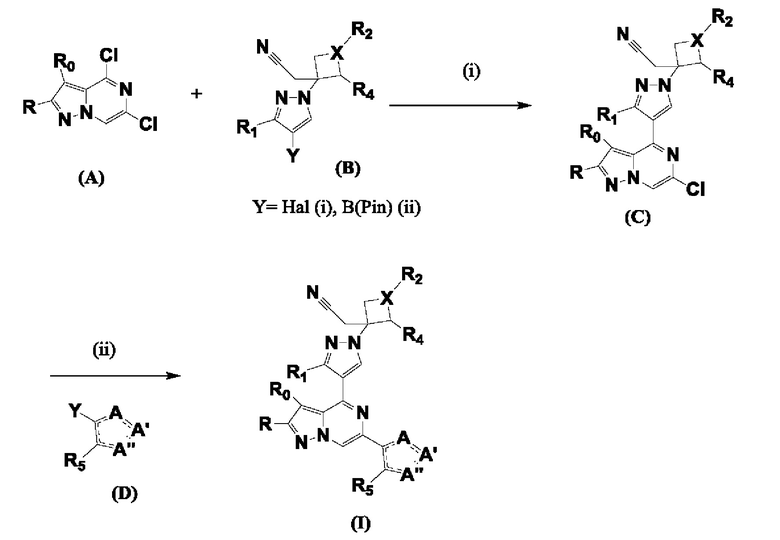
На схеме 1 соединение формулы Bi (где Y=Hal) превращают в соединение формулы Bii (Y=B(OR*)2) обработкой подходящим боронатом, таким как B2(Pin)2, в присутствии подходящего основания, такого как K2CO3, и подходящего катализатора, такого как Pd(dppf)Cl2, в подходящем растворителе, таком как диоксан. Специалист в данной области также знает, что альтернативные стратегии металлоорганического сочетания могут быть использованы с использованием альтернативных партнеров сочетания, металлов и комбинаций растворителей. Соединение формулы Bii получают и выделяют, как описано выше, или получают in situ без выделения в ходе стратегии последовательного перекрестного сочетания, которая хорошо понятна специалисту. Таким образом, соединение формулы Bii подвергают реакции кросс-сочетания с соединением формулы A в присутствии подходящего катализатора, такого как Pd(dppf)Cl2, с подходящим основанием, таким как K2CO3, в подходящем растворителе, таком как диоксан, при подходящей температуре от комнатной температуры до температуры кипения с обратным холодильником. Полученное соединение формулы C подвергают реакции кросс-сочетания с соединением формулы D, содержащим подходящую уходящую группу, такую как Bu3Sn или (Pin)2B, с подходящим металлическим катализатором, таким как Pd(PPh3)4, в подходящем растворителе, таком как MeCN при комнатной или повышенной температуре.
Согласно второму способу соединение формулы I также получают путем реакции металлоорганического перекрестного сочетания соединений формулы F с соединениями формулы B, схема 2.
Схема 2
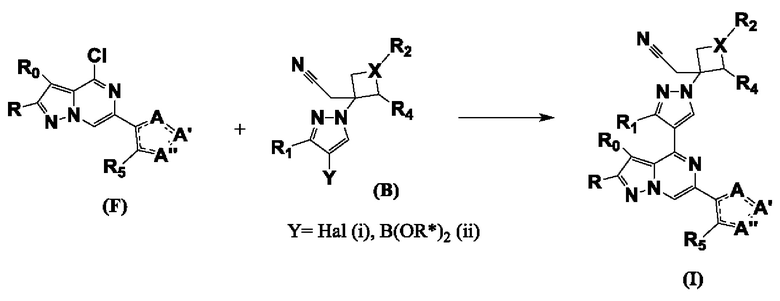
Соединение формулы Bi (где Y=Hal) превращают в соединение формулы Bii (Y=B(OR*)2) обработкой подходящим боронатом, таким как B(Pin)2, в присутствии подходящего основания, такого как K2CO3, и подходящего катализатора, такого как Pd(dppf)Cl2, в подходящем растворителе, таком как диоксан. Специалист в данной области также знает, что альтернативные стратегии металлоорганического сочетания могут быть использованы с использованием альтернативных партнеров сочетания, металлов и комбинаций растворителей. Соединение формулы Bii получают и выделяют, как описано выше, или получают in situ без выделения в ходе стратегии последовательного перекрестного сочетания, которая хорошо понятна специалисту. Таким образом, соединение формулы Bii подвергают реакции кросс-сочетания с соединением формулы F в присутствии подходящего катализатора, такого как Pd(dppf)Cl2, с подходящим основанием, таким как K2CO3, в подходящем растворителе, таком как диоксан, при подходящей температуре от комнатной температуры до температуры кипения с обратным холодильником.
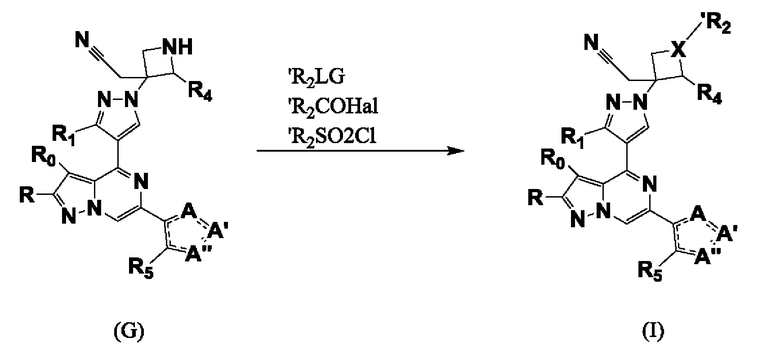
Согласно третьему способу соединение формулы I получают алкилированием, ацилированием, сульфонилированием и т.п. соединения формулы G, схема 3.
Схема 3
Алкилирование и ацилирование свободного NH
Согласно четвертому способу соединение формулы I получают реакцией присоединения Михаэля соединения формулы H к соединению формулы J в присутствии подходящего ненуклеофильного основания, такого как DBU, в подходящем растворителе, таком как MeCN при подходящей температуре, схема 4.
Схема 4
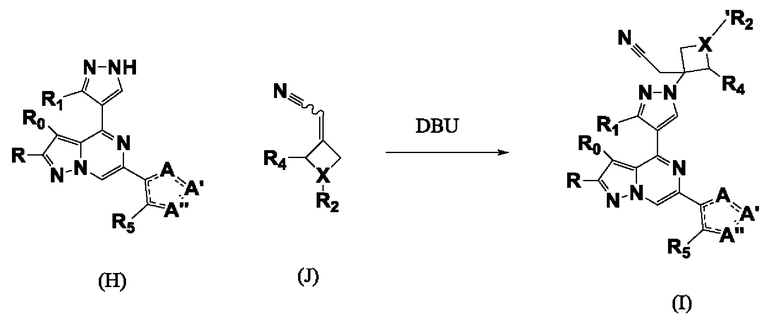
Схема 5 иллюстрирует способ получения соединений формулы F. Диэфир формулы Ei обрабатывают алкилирующим агентом формулы K и основанием, таким как K2CO3, в подходящем растворителе, таком как MeCN. Полученный диэфир формулы Eii затем циклизуют в присутствии NH4OAc в подходящем растворителе, таком как EtOH, при повышенной температуре, с получением бициклического гетероциклического соединения формулы Eiii. Соединение формулы Eiii гидролизуют до соединения формулы Eiv с подходящим основанием, таким как LiOH, в подходящем растворителе, таком как MeOH. Полученную карбоновую кислоту формулы Eiv термически декарбоксилируют в подходящем растворителе, таком как сульфолан при повышенной температуре, такой как 280°C. Полученное соединение формулы Ev хлорируют обработкой подходящим реагентом, таким как POCl3, в подходящем растворителе, таком как MeCN, при подходящей температуре, такой как температура кипения с обратным холодильником, для получения соединений формулы F.
Схема 5

Следующие схемы и письменные описания содержат общие сведения о получении соединений по изобретению. Соединения по изобретению могут быть получены любым способом, известным в данной области для получения соединений аналогичной структуры. В частности, соединения по изобретению могут быть получены с помощью процедур, описанных со ссылкой на приведенные ниже схемы, или с помощью конкретных способов, описанных в примерах, или с помощью аналогичных процессов с ними.
Специалисту в данной области будет понятно, что экспериментальные условия, изложенные в приведенных ниже схемах, иллюстрируют подходящие условия для осуществления указанных преобразований и что может быть необходимо или желательно изменять определенные условия, используемые для получения соединений формулы (I).
Кроме того, специалистам в данной области техники понятно, что на любой стадии синтеза соединений по изобретению может оказаться необходимым или желательным защита одной или нескольких чувствительных групп с тем, чтобы предотвратить нежелательные побочные реакции. В частности, может быть необходимо или желательно защитить группы амино или карбоновой кислоты. Защитные группы, используемые при получении соединений по изобретению, могут быть использованы обычным способом. См., например, те, которые описаны в Protective Groups in Organic Synthesis by Theodora W. Greene and Peter G. M. Wuts, 3rd edition, (John Wiley and Sons, 1999), в частности, главы 7 (ʺProtection for the Amino Groupʺ) и 5 (ʺProtection for the Carboxyl Groupʺ), которая включена в настоящее описание в качестве ссылки, где также описаны способы удаления таких групп.
Все производные формулы I и Ia-f могут быть получены с помощью процедур, описанных в общих способах, представленных ниже, или путем их обычных модификаций. Настоящее изобретение также охватывает любой один или несколько из этих способов для получения производных формулы (I), в дополнение к любым новым промежуточным соединениям, используемым в них. Специалисту в данной области техники будет понятно, что следующие реакции можно нагревать термическим способом или с помощью микроволнового облучения.
При осуществлении синтеза соединений по изобретению специалисту в данной области очевидна необходимость взятия образцов и анализа реакционных смесей до начала обработки с целью мониторинга развития реакций и заключение относительно того следует ли продолжить реакцию или образец готов для переработки с получением целевого продукта. Общие методы анализа реакционных смесей включают тонкослойную хроматографию (ТСХ), жидкостную хроматографию/масс-спектрометрию (LCMS) и ядерный магнитный резонанс (ЯМР).
Специалисту в данной области техники также очевидно, что соединения по изобретению могут быть получены в виде смесей диастереомеров или геометрических изомеров (например, цис и транс замещение по циклоалкановому кольцу). Эти изомеры могут быть разделены стандартными хроматографическими методиками, такими как хроматография на силикагеле с нормальной фазой, препаративная высокоэффективная жидкостная хроматография с обращенной фазой или сверхкритическая флюидная хроматография. Специалисту в данной области техники также очевидно, что некоторые соединения по изобретению являются хиральными и, следовательно, могут быть получены в виде рацемических или скалемических смесей энантиомеров. Несколько способов разделения энантиомеров доступны и хорошо известны специалистам в данной области техники. Предпочтительный способ для рутинного разделения энантиомеров представляет собой сверхкритическую флюидную хроматографию с использованием хиральной стационарной фазы.
Экспериментальный раздел
За исключением тех мест, где указано иное, реакции осуществляли в атмосфере азота. Хроматографию на силикагеле проводили с использованием силикагеля 250-400 меш, используя сжатый азот (примерно 10-15 фунт./кв.дюйм) для прогонки растворителя через колонку ("флэш-хроматография"). Где указано, растворы и реакционные смеси концентрировали с помощью роторного испарения под вакуумом.
Номенклатура в настоящем патенте написана так, как описано IUPAC (International Union of Pure and Applied Chemistry и с использованием ChemBioDraw Ultra 13,0, Perkin Elmer для получения названий.
Следующие неограничивающие получения и примеры иллюстрируют получение соединений и солей по настоящему изобретению. В примерах и получениях, которые приведены ниже, и в вышеупомянутых схемах, могут быть упомянуты следующие аббревиатуры, определения и аналитические процедуры. Другие аббревиатуры, общие в данной области, также могут быть использованы. Использовалась стандартная номенклатура IUPAC.
AcOH представляет собой уксусную кислоту;
водн. представляет собой водный;
Boc представляет собой трет-бутоксикарбонил;
шир. представляет собой широкий;
насыщенный солевой раствор представляет собой насыщенный раствор хлорида натрия в воде;
t-Bu представляет собой трет-бутил;
n-BuLi представляет собой н-бутиллитий;
°C представляет собой градусы Цельсия;
Cbz представляет собой карбобензилокси;
CDCl3 представляет собой дейтеро-хлороформ;
CDI представляет собой 1,1'-карбонилдиимидазол;
конц. представляет собой концентрированный (в отношении реагентов);
Cs2CO3 представляет собой карбонат цезия;
δ представляет собой химический сдвиг;
д представляет собой дублет;
DBU представляет собой 1,8-диазабицикло[5.4.0]ундец-7-ен;
DCM представляет собой дихлорметан;
DHP представляет собой 3,4-дигидро-2H-пиран;
DIPEA представляет собой N,N-диизопропилэтиламин;
DMAP представляет собой 4-диметиламинопиридин;
DMF представляет собой N,N-диметилформамид;
DMSO представляет собой диметилсульфоксид;
Et2O представляет собой диэтиловый эфир;
EtOAc представляет собой этилацетат;
EtOH представляет собой этанол;
(EtO)2P(O)CH2CN представляет собой диэтил(цианометил)фосфонат;
г представляет собой грамм;
GCMS представляет собой газовая хроматография масс-спектрометрия
HCl представляет собой хлористоводородную кислоту;
HCO2H представляет собой муравьиную кислоту;
ВЭЖХ представляет собой высокоэффективная жидкостная хроматография;
ч представляет собой часы;
H2SO4 представляет собой серную кислоту;
K2CO3 представляет собой карбонат калия;
KH2PO4 представляет собой калия дигидрофосфат
K2HPO4 представляет собой моногидрофосфат калия;
K3PO4 представляет собой фосфат калия (трехосновный);
KOAc представляет собой ацетат калия
л представляет собой литр;
LCMS представляет собой жидкостную хроматографию масс-спектрометрию;
LiBr представляет собой бромид лития;
LiOH представляет собой гидроксид лития;
м представляет собой мультиплет;
M представляет собой молярный;
MeCN представляет собой ацетонитрил;
MeOH представляет собой метанол;
мг представляет собой миллиграмм;
MgSO4 представляет собой магния сульфат;
MГц представляет собой мегагерц;
мин представляет собой минуты;
мл представляет собой миллилитр;
ммоль представляет собой миллимоль;
моль представляет собой моль;
MS m/z представляет собой масс-спектр ионный пик;
MTBE представляет собой метил трет-бутиловый эфир
NaBH(OAc)3 представляет собой триацетоксиборгидрид натрия;
Na2CO3 представляет собой карбонат натрия;
NaHCO3 представляет собой гидрокарбонат натрия;
NaH2PO4 представляет собой натрия дигидрофосфат;
Na2HPO4 представляет собой моногидрофосфат натрия;
NaI представляет собой йодид натрия;
NaIO4 представляет собой перйодат натрия;
NaOAc представляет собой ацетат натрия;
NaOCl представляет собой гипохлорит натрия;
NaOH представляет собой гидроксид натрия;
NH3 представляет собой аммиак;
NH4Cl представляет собой хлорид аммония;
NH4OH представляет собой гидроксид аммония;
NH4OAc представляет собой ацетат аммония;
ЯМР представляет собой ядерный магнитный резонанс;
OsO4 представляет собой осмий тетраоксид;
Pd/C представляет собой палладий на угле;
Pd(dppf)Cl2 представляет собой 1,1-бис(дифенилфосфино)ферроцен палладий(II)дихлорид (CAS: 72287-26-4);
Pd(dppf)Cl2.DCM представляет собой 1,1-бис(дифенилфосфино)ферроцен палладий(II)дихлорид; комплекс с дихлорметаном (CAS: 95464-05-4);
Pd(OAc)2 представляет собой ацетат палладия;
Pd(PPh3)4 представляет собой тетракис(трифенилфосфин)палладий;
PMB-Cl представляет собой (4-метокси)бензил хлорид;
POCl3 представляет собой оксихлорид фосфора(V);
ppm представляет собой частей на миллион;
фунт/кв. дюйм представляет собой фунтов на квадратный дюйм;
PTSA представляет собой пара-толуолсульфоновую кислоту
PyHBr3 представляет собой пиридин гидробромид пербромид
PyHCl представляет собой пиридин гидрохлорид
кв представляет собой квартет;
Rt представляет собой время удерживания;
Rh2(OAc)4 представляет собой родий(II) ацетат димер;
RuCl3 гидрат представляет собой рутений(III) хлорид гидрат;
с представляет собой синглет;
SOCl2 представляет собой тионилхлорид;
т представляет собой триплет;
TBAB представляет собой тетрабутиламмоний бромид
TEA представляет собой триэтиламин;
TFA представляет собой трифторуксусную кислоту;
THF представляет собой тетрагидрофуран;
TMSCl представляет собой хлортриметилсилан;
мкл представляет собой микролитр;
мкмоль представляет собой микромоль
XPhos Pd G2 представляет собой хлор(2-дициклогексилфосфино-2′,4′,6′-триизопропил-1,1′-бифенил)[2-(2′-амино-1,1′-бифенил)]палладий(II); CAS 1310584-14-5.
Спектры 1H ядерного магнитного резонанса (ЯМР) во всех случаях соответствовали предложенным структурам. Характеристические химические сдвиги (δ) приведены в частях на миллион в сторону слабого поля от тетраметилсилана с использованием обычных сокращений для обозначения основных пиков: например, с, синглет; д, дублет; т, триплет; кв, квартет; м, мультиплет; шир., широкий. Следующие аббревиатуры были использованы для обычных ЯМР-растворителей: CD3CN, дейтероацетонитрил; CDCl3, дейтерохлороформ; DMSO-d6, дейтеродиметилсульфоксид; и CD3OD, дейтерометанол. Там, где это необходимо, таутомеры могут регистрироваться в данных ЯМР; и некоторые способные к обмену протоны могут быть не видны.
Масс-спектры регистрировали с использованием ионизации электронным ударом (EI), электрораспылительной ионизации (ESI) или химической ионизации при атмосферном давлении (APCI). Наблюдаемые ионы описываются как MS m/z и могут быть положительными ионами соединения [M]+, соединения плюс протон [MH]+ или соединения плюс ион натрия [MNa]+. В некоторых случаях единственными наблюдаемыми ионами могут быть фрагменты-ионы, зарегистрированные как [MH-(фрагмент потерян)]+. В соответствующих случаях, зарегистрированные ионы определяют для изотопов хлора (35Cl и/или 37Cl), брома (79Br и/или 81Br) и олова (120Sn).
В тех случаях, когда для очистки соединений используют ТСХ, хроматографию или ВЭЖХ, специалист в данной области может выбрать любой подходящий растворитель или комбинацию растворителей для очистки желаемого соединения. Хроматографические разделения (за исключением ВЭЖХ) проводили с использованием силикагеля в качестве адсорбента, если не указано иное.
Все реакции проводили при непрерывном перемешивании в атмосфере азота или аргона, если не указано иное. В некоторых случаях реакционные смеси продували азотом или газообразным аргоном до начала реакции. В этих случаях газ азот или аргон барботировали через жидкую фазу смеси в течение приблизительного определенного времени. Используемыми растворителями были коммерческие безводные марки. Все исходные материалы были коммерчески доступными продуктами. В некоторых случаях идентификационный номер Chemical Abstracts Service (CAS) предоставляется для ясности. В некоторых случаях исходные материалы получали в соответствии с описанными в литературе процедурами, отмеченными звездочкой (*). Специалисту в данной области техники будет очевидно, что слово «концентрированный», как используется здесь, в целом относится к практике испарения растворителя при пониженном давлении, как правило, с использованием роторного испарителя.
Условия GCMS
Колонка: 12 м×0,2 мм, HP-1 Methyl Siloxane, 0,33 мкм слой, 1,0 мл/мин поток колонки.
Способы: 7,6 мин: Начальная температура термостата 105°C; 0,1 мин удерживание; 30°C/мин изменение температуры до 300°C конечная точка при 7,6 мин; или 7,6 мин: Начальная температура термостата 60°C; 0,1 мин удерживание; 40°C/мин изменение температуры до 320°C конечная точка при 7,6 мин; или 5,1 мин: Начальная температура термостата 40°C; 0,1 мин удерживание; 30°C/мин изменение температуры до 150°C конечная точка при 5,1 мин.
Параметры на входе GC: фронтальный вход, Сплит 30:1, He, давление 8 фунт/кв.дюйм, 250°C Инжектор, 33,9 мл/мин полный поток.
Настройка MSD: 230°C Исходная температура, 150°C температура Quad, 280°C температура Aux2
Объем пробы: 1,0 мкл
Системные компоненты: Agilent 5890 GC Oven с масс-селективным детектором Agilent 5973 Mass Selective Detector
Условия LCMS
Кислота: Waters Acquity HSS T3, 2,1 мм×50 мм, C18, 1,7 мкм; Температура колонки 60°C
Основание: Waters Acquity UPLC BEH, 2,1 мм×50 мм, C18, 1,8 мкм; Температура колонки 60°C
Подвижная фаза: A: 0,1% муравьиной кислоты в воде (об/об); Подвижная фаза B: 0,1% муравьиной кислоты в ацетонитриле (об/об).
Подвижная фаза A: 0,1% аммиака в воде (об/об); Подвижная фаза B: 0,1% аммиака в ацетонитриле (об/об)
Градиентные профили: 1,5 мин цикл: исходные условия: A-95%:B-5%; удерживание в начале от 0,0-0,1 мин; линейное изменение до A-5%:B-95% в течение 0,1-1,0 мин; удерживание A-5%:B-95% от 1,0-1,1 мин; вернуться к начальным условиям 1,1-1,5 мин
Способы очистки (СО)
Соединения примеров очищали в соответствии с одним из способов очистки (СО), упомянутых ниже, если не указано иначе:
Способ очистки A: Препаративная ВЭЖХ с использованием [Agella venusil ASB C18 150×21,2 мм×5 мкм, от 16% MeCN в воде (0,225% муравьиная кислота) до 36% MeCN в воде (0,225% муравьиная кислота)]
Способ очистки B: Препаративная ВЭЖХ с использованием [Phenomenex Gemini C18 250×21,2 мм×8 мкм или 150 мм×25 мм×5 мкм; от 16-55% MeCN в воде (0,1% аммиак) до 36-60% MeCN в воде (0,1% аммиак)]
Способ очистки C: [YMC -Actus Triart C18 150×30 мкм, от 24% MeCN в воде (0,1% аммиак) до 44% MeCN в воде (0,1% аммиак)]
Способ очистки D: Препаративная ВЭЖХ с использованием [Phenomenex Gemini C18 250×21,2 мм×8 мкм, от 25% MeCN в воде (аммиак pH=10) до 45% MeCN в воде (аммиак pH=10)], затем хиральная хроматография с использованием AS 250×25 мм В.Д. 20 мкM колонка, со сверхкритической CO2: EtOH или IPA (0,05% водный аммиак) 70:30 при 50-80 мл/мин
Способ очистки E: Препаративная ВЭЖХ с использованием [Phenomenex Gemini C18 250×21,2 мм×8 мкм, от 25% MeCN в воде (0,225% аммиак) до 45% MeCN в воде (0,225% аммиак), затем хиральная хроматография с использованием AD колонки 250 мм ×30 мм×20 мкм с подвижной фазой A: сверхкритическая CO2 и подвижной фазой B MeOH с 0,1% аммиак A:B 50:50 при 180 мл/мин
Способ очистки F: Колоночная хроматография на силикагеле, элюируя 100% DCM до 12% MeOH с 1% NH4OH.
Способ очистки G: Колоночная хроматография на силикагеле, элюируя 97:2:1 DCM:MeOH:NH3 с последующей препаративной ВЭЖХ.
Способ очистки H: Препаративная ВЭЖХ с использованием колонки: Waters XBridge C18 19 мм×100 мм, 5 мк; Подвижная фаза A: 0,03% гидроксид аммония в воде (об./об.); Подвижная фаза B: 0,03% гидроксид аммония в ацетонитриле (об./об.); от 5-20% B до 40-100% B при скорости потока 25 мл/мин.
Способ очистки I: Препаративная ВЭЖХ с использованием колонки: Waters Sunfire C18 19 мм×100 мм, 5 мк; Подвижная фаза A: 0,05% TFA в воде (об./об.); Подвижная фаза B: 0,05% TFA в ацетонитриле (об./об.); от 20% B до 40% B в 6,75 минут, затем до 100% B в 7 минут при скорости потока 30 мл/мин.
Удельное вращение
Удельные вращения основаны на уравнении [α]=(100⋅α)/(l⋅c) и представлены как единичные числа, где концентрация c представлена в г/100 мл, а длина пути l представлена в дециметрах. Единицы удельного вращения (град⋅мл)/(g⋅дм), являются неявными и не включаются в отчетную величину.
Получение 1
Этил 1-(цианометил)-1H-пиразол-3-карбоксилат
и
Этил 1-(цианометил)-1H-пиразол-5-карбоксилат
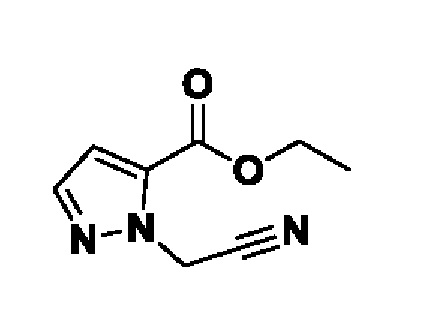
К суспензии Cs2CO3 (2100 г, 6,44 моль) в DMF (12 л) добавляли этил 1H-пиразол-3-карбоксилат (750 г, 5,36 моль), затем 2-хлорацетонитрил (450 г, 5,96 моль) и смесь перемешивали при примерно 25°C в течение примерно 16 часов. Реакционную смесь выливали в воду (12 л) и экстрагировали EtOAc (5×5 л). Объединенные экстракты EtOAc промывали насыщенным солевым раствором (2×5 л), сушили (Na2SO4) и концентрировали с получением остатка, который очищали хроматографией с получением этил 1-(цианометил)-1H-пиразол-3-карбоксилата (398 г, 39%) в виде желтого масла и этил 1-(цианометил)-1H-пиразол-5-карбоксилата (680 г). Этил 1-(цианометил)-1H-пиразол-5-карбоксилат растворяли в MTBE (15 л) и промывали насыщенным солевым раствором (3×5 л), сушили (Na2SO4) и концентрировали с получением соединения в виде желтого масла (489 г, 51%).
1H ЯМР (400 МГц, CDCl3) δ: 7,49 (с, 1 H), 6,82 (с, 1 H), 5,45 (с, 2 H), 4,29 (кв, 2 H), 1,29 (т, 3 H).
LCMS m/z=180,1 [MH]+
Получение 2
Этил 1-(2-амино-2-оксоэтил)-1H-пиразол-5-карбоксилат

Две идентичные реакции проводили параллельно.
К раствору этил 1-(цианометил)-1H-пиразол-5-карбоксилата (Получение 1, 235,5 г, 1,32 моль) в TFA (1,2 л) добавляли конц. H2SO4 (377 мл, 7,04 моль) при примерно 25°C. Реакционную смесь перемешивали при примерно 25°C в течение примерно 16 часов перед объединением с параллельной реакцией и концентрировали для удаления большей части TFA. Остаток выливали в ледяную воду (5 л) и экстрагировали EtOAc (5 л). Водную фазу дополнительно экстрагировали EtOAc (10×5 л) и объединенные экстракты EtOAc промывали насыщенным водн. NaHCO3 (2×10 л), сушили (Na2SO4) и концентрировали с получением указанного в заголовке соединения в виде желтого твердого вещества (477 г, 92%).
1H ЯМР (400 МГц, CDCl3) δ: 7,60 (с, 1 H), 6,93 (с, 1 H), 5,90 (шир.с, 1 H), 5,71 (шир.с, 1 H), 5,27 (с, 2 H), 4,35 (кв, 2 H), 1,37 (т, 3 H).
LCMS m/z=198,2 [MH]+
Получение 3
Пиразоло[1,5-а]пиразин-4,6(5H,7H)-дион
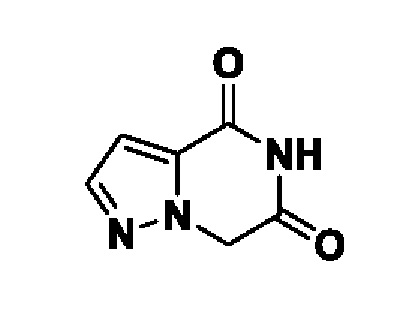
К раствору этил 1-(2-амино-2-оксоэтил)-1H-пиразол-5-карбоксилата (Получение 2, 466 г, 2,17 моль) в EtOH (56 л) добавляли NaOtBu (498 г, 5,20 моль) в THF (4 л) при примерно 25°C. Белая суспензия образовывалась во время добавления и затем смесь нагревали при примерно 70°C в течение примерно 16 часов. Реакционную смесь охлаждали до примерно 25°C и подкисляли до рН 6 с помощью 12 M водн. HCl (500 мл), что приводит к образованию белой суспензии. Смесь концентрировали с получением указанного в заголовке соединения (с примесями хлорида натрия) в виде желтого твердого вещества (783 г). Это использовали без дополнительной очистки.
1H ЯМР (400 МГц, DMSO-d6) δ: 11,82 (с, 1 H), 7,74 (с, 1 H), 6,97 (с, 1 H), 5,19 (с, 2 H).
LCMS m/z=152,1 [MH]+
Получение 4
4,6-Дихлорпиразоло[1,5-а]пиразин
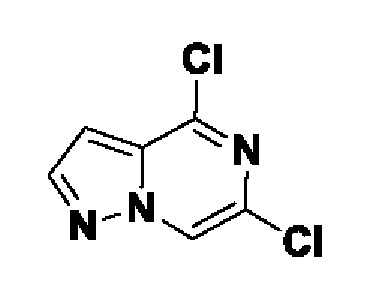
Три идентичные реакции проводили параллельно.
Пиразоло[1,5-а]пиразин-4,6(5H,7H)-дион (Получение 3, 278 г, 1,14 моль) добавляли к POCl3 (1,84 кг, 12 моль) при примерно 25°C, затем PyHCl (131 г, 1,14 моль). Реакционную смесь нагревали при примерно 120°C в течение примерно 16 часов. Реакционную смесь охлаждали примерно до 25°C и концентрировали для удаления большей части POCl3. Каждый остаток разбавляли EtOAc (2 л) и три экстракта EtOAc объединяли и выливали в 1 М вод. NaH2PO4 (7,5 л) при примерно 25°C и и фильтровали через слой целита. Фильтровальную лепешку промывали EtOAc (3×2 л) и все фильтраты объединяли и отделяли от водной фазы. Водную фазу экстрагировали MTBE (10 л). Объединенные экстракты EtOAc и MTBE промывали насыщенным солевым раствором (2×5 л), сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии и продукт растирали с петролейным эфиром (300 мл) и фильтровали. Фильтровальную лепешку сушили с получением указанного в заголовке соединения в виде белого твердого вещества (110 г, 22%).
1H ЯМР (400 МГц, CDCl3) δ: 8,42 (с, 1 H), 8,06 (с, 1 H), 6,93 (с, 1 H).
LCMS m/z=189,8 [MH]+ (37Cl изотоп)
Получение 5
1-(1-метил-1H-пиразол-4-ил)этан-1-он
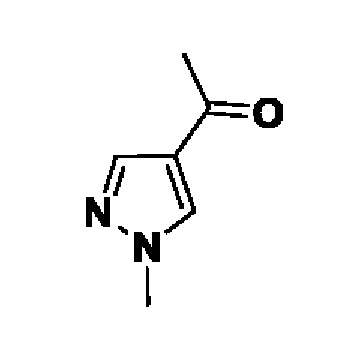
Две идентичные реакции проводили параллельно.
К смеси 1-метилпиразола (750 г, 9,16 моль) и уксусного ангидрида (1,7 кг, 16,67 моль) добавляли концентрированную H2SO4 (75 г, 0,75 моль) при примерно 20°C. Реакционную смесь нагревали при примерно 150°C в течение примерно 3 часов. После охлаждения две смеси объединяли, выливали в ледяную воду (15 л), доводили примерно до pH 10 20% водн. NaOH и экстрагировали DCM (4×10 л). Объединенные экстракты DCM сушили (Na2SO4) и концентрировали с получением указанного в заголовке соединения в виде коричневого масла (1240 г, 72%).
1H ЯМР (400 МГц, CDCl3) δ: 7,86 (с, 1 H), 7,84 (с, 1 H), 3,92 (с, 3 H), 2,40 (с, 3 H).
GCMS m/z=109,0 [M-CH3]+
Получение 6
2-бром-1-(1-метил-1H-пиразол-4-ил)этан-1-он
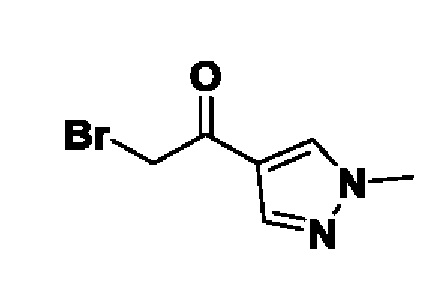
Две идентичные реакции проводили параллельно.
К раствору 1-(1-метил-1H-пиразол-4-ил)этан-1-она (Получение 5, 620 г, 5 моль) в DCM (12 л) и этаноле (3 л) добавляли PyHBr3 (1,6 кг, 5 моль) при примерно 15°C. Смесь перемешивали при примерно 15°C в течение примерно 18 часов. Две реакционные смеси объединяли, гасили водой (10 л), разделяли и водную фазу экстрагировали DCM (4×10 л). Объединенные экстракты DCM сушили (Na2SO4) и концентрировали для удаления примерно 69 л растворителя. Остаток разбавляли петролейным эфиром (5 л), перемешивали при примерно 15°C в течение примерно 30 мин и смесь фильтровали. Осадок сушили с получением указанного в заголовке соединения в виде желтого твердого вещества (1,73 кг, 85%).
1H ЯМР (400 МГц, CDCl3) δ: 7,97 (с, 1 H), 7,91 (с, 1 H), 4,17 (с, 2 H), 3,93 (с, 3 H).
LCMS m/z=203,1 [MH]+ (79Br изотоп)
Получение 7
диэтил 1-(2-(1-метил-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3,5-дикарбоксилат
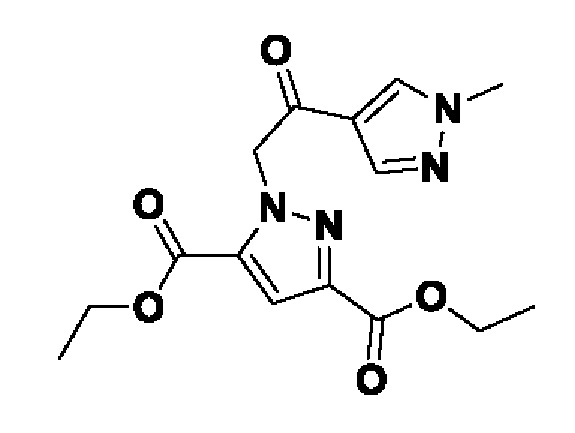
Две реакции проводили параллельно.
К смеси 2-бром-1-(1-метил-1H-пиразол-4-ил)этан-1-она (Получение 6; 500 г, 2,46 моль) и диэтил-1H-пиразол-3,5-дикарбоксилата (580 г, 2,73 моль) в DMF (8 л) добавляли Cs2CO3 (1050 г, 3,23 моль) при примерно 20°C. Через примерно 18 часов, две реакционные смеси объединяли, разбавляли водой (10 л) и экстрагировали DCM (3×10 л). Объединенные экстракты DCM сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (1,53 кг, 93%).
1H ЯМР (400 МГц, CDCl3) δ: 7,96 (с, 2 H), 7,46 (с, 1 H), 5,86 (с, 2 H), 4,45 (кв, 2 H), 4,32 (кв, 2 H), 3,99 (с, 3 H), 1,44 (т, 3 H), 1,36 (т, 3 H).
LCMS m/z=335,0 [MH]+
Получение 8
Этил 4-гидрокси-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоксилат

Три идентичные реакции проводили параллельно.
К раствору диэтил 1-(2-(1-метил-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3,5-дикарбоксилата (Получение 7; 510 г, 1,52 моль) в этаноле (6 л) добавляли NH4OAc (352 г, 4,57 моль) при примерно 20°C. Смесь нагревали в автоклаве при примерно 130°C в течение примерно 24 часов. Реакционную смесь охлаждали до примерно 50°C и объединяли и фильтровали. Осадок сушили с получением указанного в заголовке соединения (1090 г, 83%) в виде не совсем белого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ: 11,35 (шир.с, 1 H), 8,31 (с, 1 H), 8,20 (с, 1 H), 8,05 (с, 1 H), 7,38 (с, 1 H), 4,34 (кв, 2 H), 3,89 (с, 3 H), 1,33 (т, 3 H).
LCMS m/z=288,0 [MH]+
Получение 9
4-гидрокси-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоновая кислота
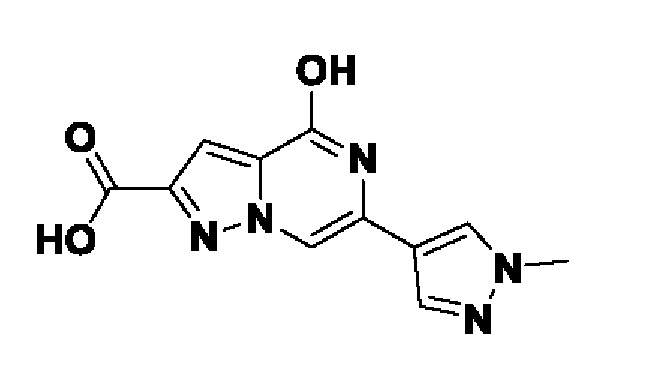
Две идентичные реакции проводили параллельно.
К суспензии этил 4-гидрокси-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоксилата (Получение 8, 545 г, 1,9 моль) в MeOH (10 л) добавляли 1 M водн. NaOH (5,75 л) при примерно 20°C. Через примерно 30 минут суспензия стала прозрачным раствором и перемешивание продолжали при примерно 20°C в течение примерно 18 часов. Реакционные смеси доводили примерно до рН 2 с помощью 12 M водн. HCl (650 мл), объединяли, и концентрировали для удаления большей части MeOH. Остаток фильтровали и осадок сушили с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (1040 г, 100%).
1H ЯМР (400 МГц, DMSO-d6) δ: 13,25 (шир.с, 1 H), 11,67 (с, 1 H), 8,34 (с, 1 H), 8,16 (с, 1 H), 8,06 (с, 1 H), 7,32 (с, 1 H), 3,88 (с, 3 H).
LCMS m/z=260,0 [MH]+
Получение 10
6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ол
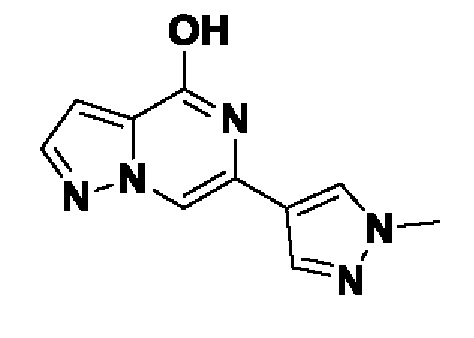
Пять идентичных реакций проводили параллельно.
4-Гидрокси-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоновую кислоту (Получение 9, 85 г, 0,328 моль) добавляли порциями к предварительно нагретому сульфолану (800 мл) при примерно 280°C. Пять реакционных смесей перемешивали при примерно 280°C в течение примерно 2 часов, охлаждали до примерно 25°C, и перемешивали в течение примерно 18 часов. Реакционные смеси объединяли и смесь очищали с помощью хроматографии, элюируя петролейным эфиром-EtOAc (10:1-0:1), затем DCM-MeOH (10:1) с получением указанного в заголовке соединения в виде желтого твердого вещества (490 г, 75%).
1H ЯМР (400 МГц, DMSO-d6) δ: 11,45 (с, 1 H), 8,28 (с, 1 H), 8,10 (с, 1 H), 8,04 (с, 1 H), 7,88 (с, 1 H), 6,99 (с, 1 H), 3,88 (с, 3 H).
LCMS m/z=216,0 [MH]+
Получение 11
4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин
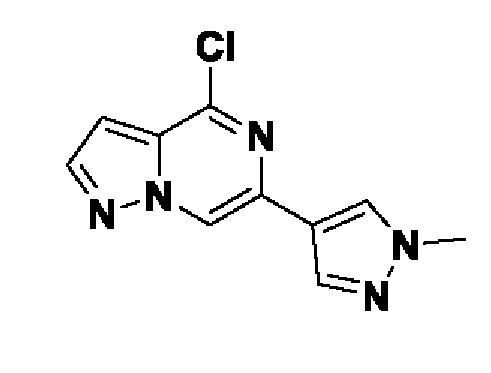
Две идентичные реакции проводили параллельно.
К суспензии 6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ола (Получение 10, 307 г, 1,43 моль) в MeCN (7,5 л) добавляли POCl3 (2006 г, 13 моль) при примерно 25°C. Смесь нагревали при примерно 85°C в течение примерно 48 часов. Реакционные смеси объединяли и фильтровали. Осадок промывали EtOAc и сушили в вакууме. Высушенный осадок очищали хроматографией с получением желтого твердого вещества, которое растворяли в DCM (15 л) и промывали 1 M водн. NaHCO3 (5 л). DCM концентрировали для удаления примерно 13 л растворителя и остаток разбавляли MTBE (2 л) и петролейным эфиром (2 л). Смесь фильтровали и осадок сушили с получением указанного в заголовке соединения в виде желтого твердого вещества (385 г, 58%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,22 (с, 1 H), 8,27 (с, 1 H), 8,21 (с, 1 H), 8,04 (с, 1 H), 7,02 (с, 1 H), 3,88 (с, 3 H).
LCMS m/z=233,8 [MH]+ (35Cl изотоп)
Получение 12
1-(4-Метоксибензил)-1H-пиразол-4-карбоновая кислота
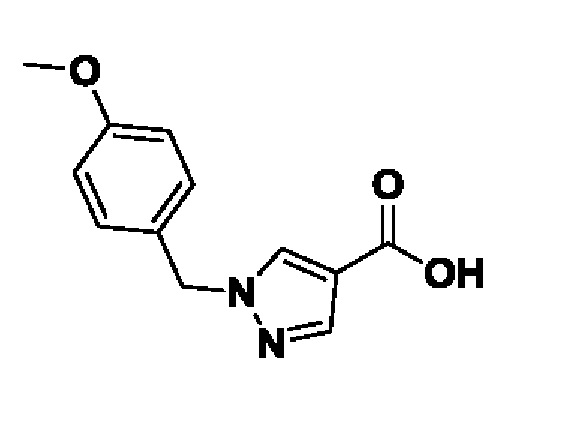
Часть 1: Три идентичные реакции проводили параллельно.
К перемешиваемому раствору этил 1H-пиразол-4-карбоксилата (16 г, 110 ммоль) в MeCN (160 мл) добавляли PMB-Cl (85,8 г, 548 ммоль) и K2CO3 (23,7 г, 171 ммоль) и смесь нагревали с обратным холодильником в течение примерно 18 часов. Три партии охлаждали, объединяли и фильтровали. Фильтрат концентрировали с получением этил 1-(4-метоксибензил)-1H-пиразол-4-карбоксилата в виде желтого масла, который использовали без дополнительной очистки
Часть 2: Три идентичные реакции проводили параллельно.
К перемешиваемому раствору неочищенного этил 1-(4-метоксибензил)-1H-пиразол-4-карбоксилата (часть 1, 50,0 г, 96 ммоль) в THF (150 мл) и MeOH (150 мл) добавляли раствор LiOH (10 г, 238 ммоль) в воде (75 мл). Смесь нагревали при примерно 60°C в течение примерно 18 часов. Три партии объединяли и выпаривали досуха. Остаток разбавляли водой (800 мл) и MeOH (150 мл) и промывали EtOAc (2×500 мл). Экстракты EtOAc отбрасывали и водный раствор подкисляли до значения рН приблизительно 2 с помощью 6 М водн. HCl и экстрагировали EtOAc (2×800 мл). Объединенные экстракты EtOAc промывали насыщенным солевым раствором (500 мл), сушили (Na2SO4) и концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества (33,0 г, 83% для двух стадий).
1H ЯМР (400 МГц, DMSO-d6) δ: 12,34 (шир.с, 1 H), 8,32 (с, 1 H), 7,79 (с, 1 H), 7,23 (м, 2 H), 6,89 (м, 2 H), 5,26 (с, 2 H), 3,72 (с, 3 H).
Получение 13
1-(4-Метоксибензил)-1H-пиразол-4-карбонил хлорид
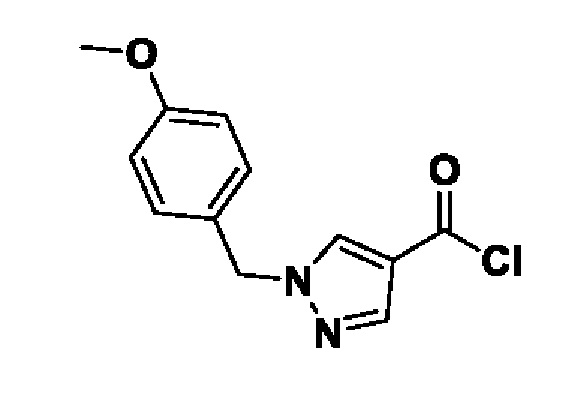
Раствор 1-(4-метоксибензил)-1H-пиразол-4-карбоновой кислоты (Получение 12, 25,0 г, 110 ммоль) в SOCl2 (40 мл) перемешивали при примерно 60°C в течение примерно 5 часов. Раствор концентрировали с получением указанного в заголовке соединения в виде коричневого масла (27,0 г, 100%), которое использовали без дополнительной очистки или характеризации.
Получение 14
N-метокси-1-(4-метоксибензил)-N-метил-1H-пиразол-4-карбоксамид
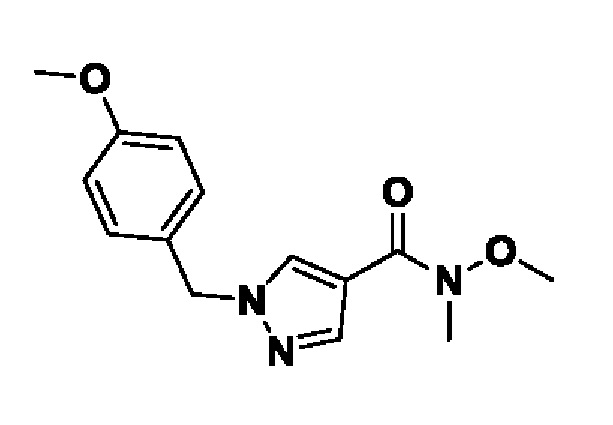
К раствору N,O-диметилгидроксиламин гидрохлорида (26,3 г, 269 ммоль) и TEA (131,0 г, 1,29 моль) в DCM (200 мл) медленно добавляли раствор 1-(4-метоксибензил)-1H-пиразол-4-карбонил хлорида (Получение 13, 27,0 г, 108 ммоль) в DCM (50 мл). После завершения добавления реакционную смесь перемешивали при примерно 20°C в течение примерно 5 часов. Смесь разбавляли DCM (150 мл) и водой (300 мл). Объединенные экстракты DCM промывали насыщенным солевым раствором, сушили (Na2SO4) и концентрировали с получением указанного в заголовке соединения (20,0 г, 67%) в виде коричневого масла, которое использовали без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ: 7,99 (с, 1 H), 7,90 (с, 1 H), 7,22 (д, 2 H), 6,89 (д, 2 H), 5,25 (с, 2 H), 3,81 (с, 3 H), 3,69 (с, 3 H), 3,31 (с, 3 H).
LCMS m/z=275,0 [MH]+
Получение 15
1-(1-(4-Метоксибензил)-1H-пиразол-4-ил)этан-1-он
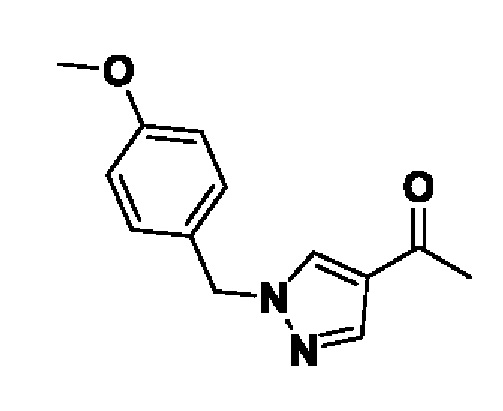
Две идентичные реакции проводили параллельно.
К раствору N-метокси-1-(4-метоксибензил)-N-метил-1H-пиразол-4-карбоксамида (Получение 14, 10,0 г, 36,3 ммоль) в THF (120 мл) добавляли по каплям 3 М метилмагнийбромид в эфире (24,2 мл) при примерно 0°C. Реакционную смесь нагревали до примерно 25°C и перемешивали в течение примерно 5 часов. Реакцию гасили добавлением насыщенного водн. NH4Cl (100 мл) и экстрагировали EtOAc (2×200 мл). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Концентрированные остатки обоих экспериментов объединяли и очищали хроматографией с получением указанного в заголовке соединения (10,0 г, 60%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ: 8,47 (с, 1 H), 7,91 (с, 1 H), 7,25 (м, 2 H), 6,90 (м, 2 H), 5,27 (с, 2 H), 3,72 (с, 3 H), 2,34 (с, 3 H).
LCMS m/z=231,7 [MH]+
Получение 16
2-бром-1-(1-(4-метоксибензил)-1H-пиразол-4-ил)этан-1-он
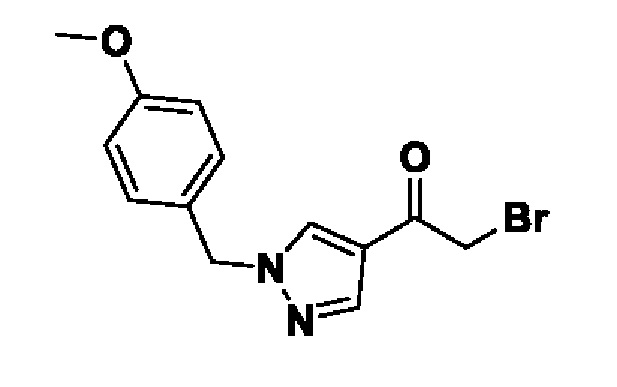
Две идентичные реакции проводили параллельно.
К раствору 1-(1-(4-метоксибензил)-1H-пиразол-4-ил)этан-1-она (Получение 15, 8,0 г, 34,7 ммоль) в DCM (96 мл) и EtOH (24 мл) добавляли PyHBr3 (13,3 г, 41,7 ммоль) при примерно 20°C. Реакционные смеси выдерживали при примерно 25°C в течение примерно 18 часов и гасили водой (100 мл) перед объединением и экстрагировали EtOAc (2×300 мл). Объединенные экстракты EtOAc промывали насыщенным солевым раствором, сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением желтого твердого вещества. Полученное вещество растирали в порошок с MTBE (100 мл) с получением указанного в заголовке соединения (15,0 г, 70%) в виде желтого твердого вещества. Дополнительный образец (5,0 г, 23%) не совсем чистого продукта получали в виде желтого твердого вещества путем концентрирования и растирания в порошок.
1H ЯМР (400 МГц, CDCl3) δ: 7,99 (с, 1 H), 7,89 (с, 1 H), 7,23 (м, 2 H), 6,92 (м, 2 H), 6,92 (м, 2 H), 5,25 (с, 2 H), 4,15 (с, 2 H), 3,81 (с, 3 H).
LCMS m/z=333,0 [MNa]+ (81Br изотоп)
Получение 17
Диметил 1-(2-(1-(4-метоксибензил)-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3,5-дикарбоксилат
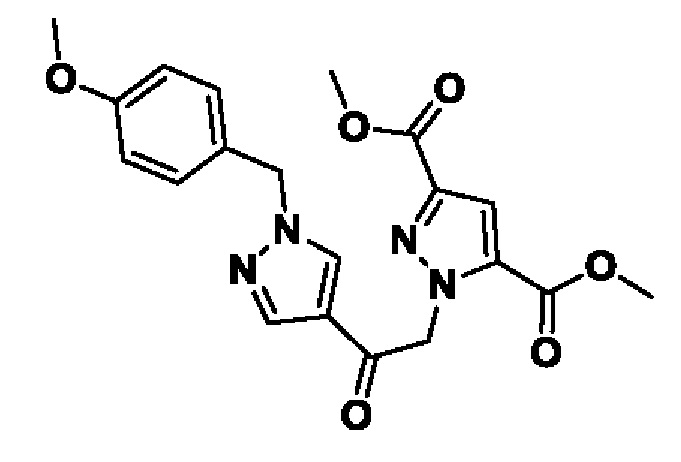
К смеси диметил 1H-пиразол-3,5-дикарбоксилата (1 г, 5 ммоль) и 2-бром-1-(1-(4-метоксибензил)-1H-пиразол-4-ил)этан-1-она (Получение 16, 2,18 г, 7,06 ммоль) в DMF (20 мл) добавляли Cs2CO3 (2,3 г, 7,06 ммоль) при примерно 20°C. Примерно через 2 дня, смесь концентрировали досуха. Остаток растворяли в DCM и промывали один раз насыщенным водн. NH4Cl. DCM концентрировали и остаток очищали с помощью хроматографии. Продукт перемешивали в EtOAc (20 мл) при примерно 20°C в течение примерно 18 часов. Полученное твердое вещество фильтровали и сушили с получением указанного в заголовке соединения (1,38 г, 60%).
1H ЯМР (400 МГц, CDCl3) δ: 7,96 (с, 1 H), 7,82 (с, 1 H), 7,42 (с, 1 H), 7,24 (д, 2 H), 6,93 (д, 2 H), 5,80 (с, 2 H), 5,27 (с, 2 H), 3,95 (с, 3 H), 3,84 (с, 3 H), 3,83 (с, 3 H).
LCMS m/z=413,1 [MH]+
Получение 18
Метил 4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоксилат

и
Этил 4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоксилат
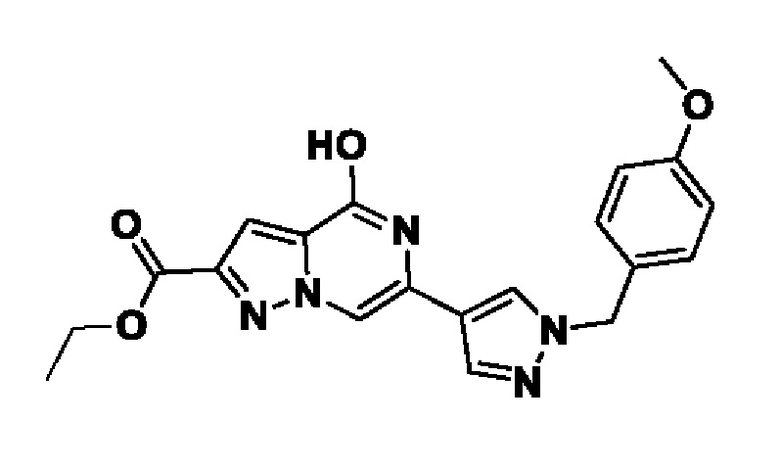
Шесть идентичных реакций проводили параллельно.
В каждый из шести флаконов добавляли диметил 1-(2-(1-(4-метоксибензил)-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3,5-дикарбоксилат (Получение 17, 300 мг, 0,73 ммоль), NH4OAc (336 мг, 4,37 ммоль) и EtOH (6 мл). Смеси нагревали при микроволновом облучении при примерно 150°C в течение примерно 2 часов, затем охлаждали до примерно 20°C, перемешивали в течение примерно 1 часа, и фильтровали. Объединенные твердые вещества сушили с получением смеси обоих указанных в заголовке соединений, которую использовали без дополнительной очистки на следующей стадии (1,61 г).
1H ЯМР (400 МГц, DMSO-d6) δ: 11,65 (шир.с, 1 H), 8,38 (с, 1 H), 8,17 (с, 1 H), 8,07 (с, 1 H), 7,36 (с, 1 H), 7,26 (д, 2 H), 6,93 (д, 2 H), 5,75 (с, 1 H), 5,28 (с, 2 H), 3,86 (с, 3 H), 3,74 (с, 3 H). Это метиловый эфир, который является основным компонентом.
LCMS m/z=380,1 [MH]+, 394,1 [MH]+
Получение 19
4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоновая кислота
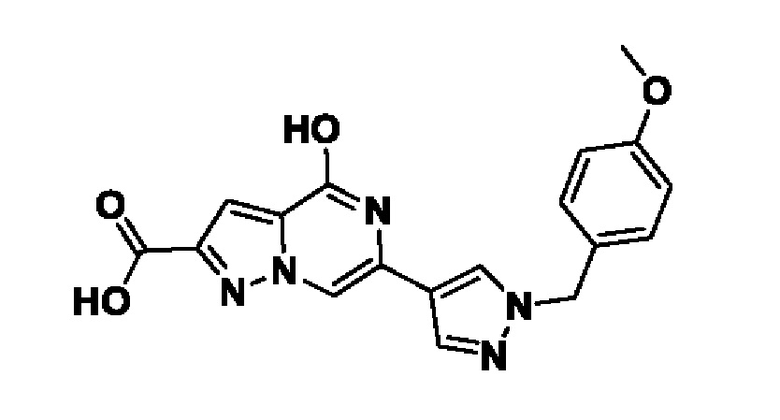
К раствору смеси метил 4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоксилата и этил 4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоксилата (Получение 18, 524 мг, примерно 1,38 ммоль) в MeOH (10 мл) добавляли 1 M водн. NaOH (4,83 мл). Смесь выдерживали при примерно 20°C в течение примерно 18 часов перед добавлением дополнительного количества 1 M водн. NaOH (1,38 мл). Смесь выдерживали при примерно 20°C в течение примерно 24 дополнительных часов. MeOH выпаривали и остаток разбавляли водой (2 мл) и перемешивали при примерно 40°C пока все твердые вещества не растворялись. Раствор подкисляли 12 M водн. HCl и перемешивали при примерно 0°C в течение примерно 10 мин. Образовавшийся осадок фильтровали и осадок промывали водой. Твердое вещество сушили в вакууме с получением указанного в заголовке соединения (487 мг, 96%).
1H ЯМР (400 МГц, DMSO-d6) δ: 11,65 (с, 1 H), 8,38 (с, 1 H), 8,18 (м, 1 H), 8,10 (с, 1 H), 7,25-7,32 (м, 4 H), 6,95 (м, 2 H), 5,28 (с, 2 H), 3,78 (с, 3 H).
LCMS m/z=366,0 [MH]+
Получение 20
6-(1-(4-Метоксибензил)-1H-пиразол-4-ил)пиразол[1,5-a]пиразол-4-ол
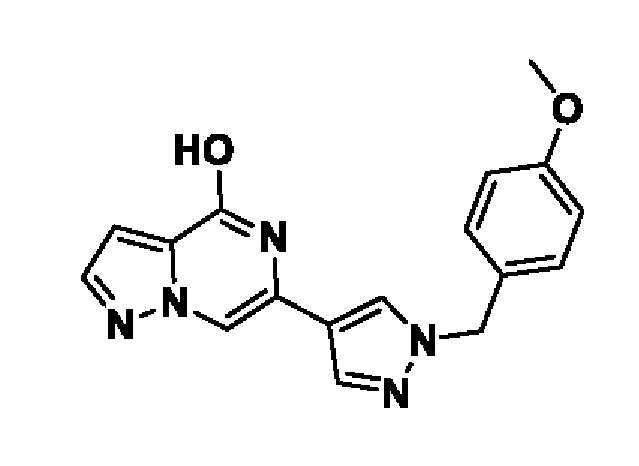
Три реакции проводили параллельно.
Образец 1:
4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразол[1,5-a]пиразин-2-карбоновую кислоту (Получение 19, 40 мг, 0,11 ммоль) нагревали при примерно 350°C в течение примерно 10 секунд до тех пор, пока не совсем белое твердое вещество не расплавится и не превратится в темно-коричневую жидкость.
Образец 2:
4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразол[1,5-a]пиразин-2-карбоновую кислоту (Получение 19, 120 мг, 0,33 ммоль) нагревали при примерно 350°C в течение примерно 15 секунд до тех пор, пока не совсем белое твердое вещество не расплавится и не превратится в темно-коричневую жидкость.
Образец 3:
4-гидрокси-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразол[1,5-a]пиразин-2-карбоновую кислоту (Получение 19, 310 мг, 0,85 ммоль) нагревали при примерно 350°C в течение примерно 15 секунд до тех пор, пока все не совсем белое твердое вещество не расплавится и не превратится в темно-коричневую жидкость.
Все три партии охлаждали, комбинировали и концентрировали дважды с толуолом с получением указанного в заголовке соединения, которое использовали без дополнительной очистки на следующей стадии.
1H ЯМР (400 МГц, DMSO-d6) δ: 11,43 (с, 1 H), 8,37 (с, 1 H), 8,11 (с, 1 H), 8,08 (с, 1 H), 7,88 (д, 1 H), 7,28 (д, 2 H), 6,99 (д, 1 H), 6,94 (д, 2 H), 5,28 (с, 2 H), 3,76 (с, 3 H).
LCMS m/z=322,1 [MH]+
Получение 21
4-хлор-6-(1-(4-метоксибензил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин
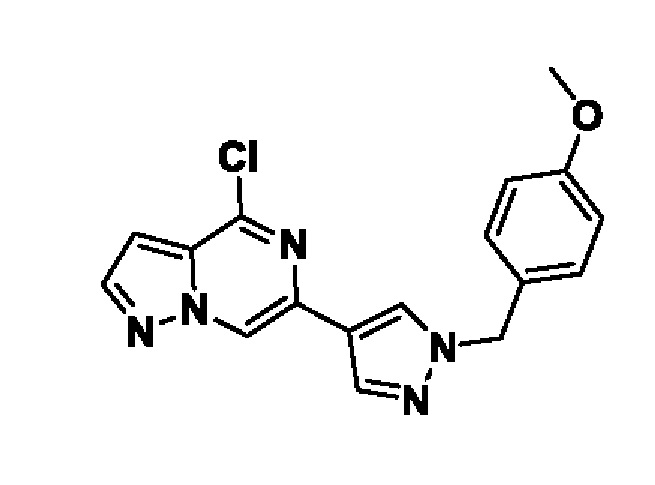
6-(1-(4-Метоксибензил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ол (Получение 20, 390 мг, 1,21 ммоль), PyHCl (143 мг, 1,21 ммоль) и POCl3 (10 мл) нагревали при примерно 120°C в течение примерно 18 часов. Смесь концентрировали и остаток обрабатывали водн. раствором NaH2PO4 для поддержания рН около 4. Полученный раствор перемешивали при примерно 20°C в течение примерно 10 мин и три раза экстрагировали DCM. Объединенные экстракты DCM сушили и концентрировали с получением указанного в заголовке соединения в виде коричневого твердого вещества (300 мг, 72%).
1H ЯМР (400 МГц, CDCl3) δ: 8,74 (с, 1 H), 8,50 (с, 1 H), 8,22 (м, 2 H), 7,45 (м, 2 H), 6,92-7,02 (м, 3 H), 3,88 (с, 2 H), 2,15 (с, 3 H).
LCMS m/z=340,0 [MH]+
Получение 22
трет-бутил 4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-карбоксилат
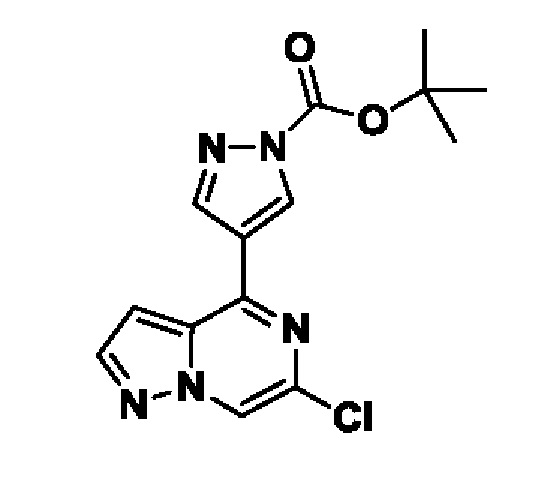
Раствор 4,6-дихлорпиразоло[1,5-а]пиразина (Получение 4, 700 мг, 3,72 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-карбоксилата (1100 мг, 3,72 ммоль), 2 M водн. K3PO4 (3 мл, 6 ммоль) в 1,4-диоксане (10,0 мл) продували аргоном в течение примерно 5 мин. К этому добавляли бис(три-трет-бутилфосфин)палладий(0) (96,1 мг, 0,19 ммоль) и реакцию выдерживали при примерно 20°C в течение примерно 18 часов. Растворитель концентрировали с получением янтарного остатка, который растворяли в DCM и очищали с помощью хроматографии с получением указанного в заголовке соединения (710 мг, 60%).
1H ЯМР (400 МГц, CDCl3) δ: 8,82 (м, 1 H), 8,44 (м, 3 H), 8,14 (с, 1 H), 1,60 (с, 9 H).
LCMS m/z=220,1 [MH-BOC]+
Получение 23
трет-бутил 4-(6-винилпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-карбоксилат
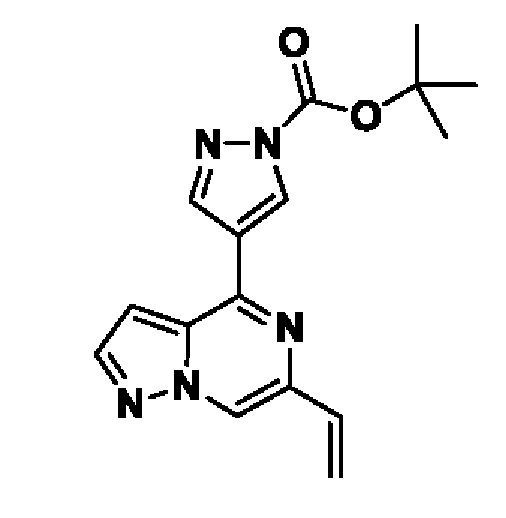
Раствор трет-бутил 4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-карбоксилата (Получение 22, 700 мг, 2,19 ммоль) и трибутил(винил)олова (694 мг, 2,19 ммоль) в 1,4-диоксане (30 мл) продували аргоном в течение примерно 5 мин, с последующим добавлением XPhos Pd G2 (344 мг, 0,44 ммоль). Смесь нагревали при примерно 55°C в течение примерно 18 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (449 мг, 66%).
1H ЯМР (400 МГц, CDCl3) δ: 8,79 (с, 1 H), 8,46 (с, 1 H), 8,26 (с, 1 H), 8,07 (д, 1 H), 6,95 (дд, 1 H), 6,77 (дд, 1 H), 6,43 (дд, 1 H), 5,52 (дд, 1 H), 1,74 (с, 9 H).
LCMS m/z=312,3 [MH]+
Получение 24
трет-бутил 4-(6-формилпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-карбоксилат
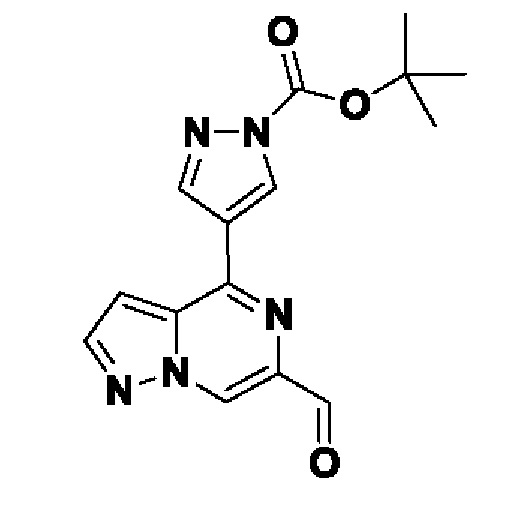
Раствор трет-бутил 4-(6-винилпиразоло[1,5-a]пиразин-4-ил)-1H-пиразол-1-карбоксилата (Получение 23; 446 мг, 1,43 ммоль) и 2,6-lutidine (767 мг, 7,16 ммоль) в 1,4-диоксане (9 мл) и воде (3 мл) охлаждали до примерно 0°C и добавляли NaIO4 (1530 мг, 7,16 ммоль) и 4% водн. раствор OsO4 (0,54 мл). Смеси давали нагреться до примерно 20°C в течение примерно 3 часов. Твердые вещества удаляли фильтрованием и промывали эфиром. Объединенный 1,4-диоксан и простой эфир концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (289 мг, 65%).
1H ЯМР (400 МГц, CDCl3) δ: 10,20 (с, 1 H), 9,01 (с, 1 H), 8,85 (с, 1 H), 8,50 (с, 1 H), 8,30 (д, 1 H), 7,08-7,12 (м, 1 H), 1,73 (с, 9 H).
LCMS m/z=314,2 [MH]+
Получение 25
трет-бутил €-4-(6-((гидроксиимино)метил)пиразин[1,5-a]пиразин-4-ил)-1H-пиразол-1-карбоксилат
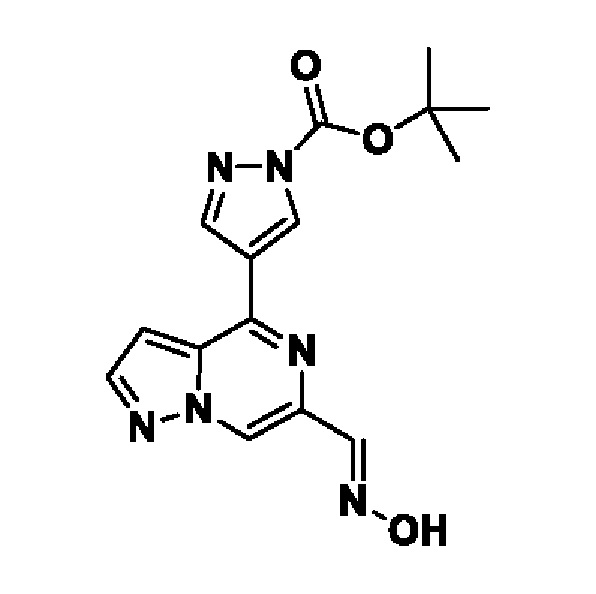
Гидроксиламин HCl (112 мг, 1,58 ммоль) добавляли к смеси трет-бутил 4-(6-формилпиразоло[1,5-a]пиразин-4-ил)-1H-пиразол-1-карбоксилата (Получение 24, 450 мг, 1,44 ммоль), и Na2CO3 (196 мг, 1,58 ммоль) в MeOH (20 мл). Смесь выдерживали при примерно 20°C в течение примерно 1,5 часа. Смесь концентрировали, добавляли воду (30 мл) и смесь перемешивали в течение примерно 5 мин прежде чем твердое вещество отфильтровали и высушили с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (325 мг, 69%).
1H ЯМР (400 МГц, CDCl3) δ: 10,46 (шир.с, 1 H), 9,53 (с, 1 H), 8,81 (с, 1 H), 8,46 (с, 1 H), 8,41 (с, 1 H), 8,19 (д, 1 H), 7,03 (д, 1 H), 1,73 (с, 9 H).
LCMS m/z=329,2 [MH]+
Получение 26
(3-(4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)изоксазол-5-ил)метанол
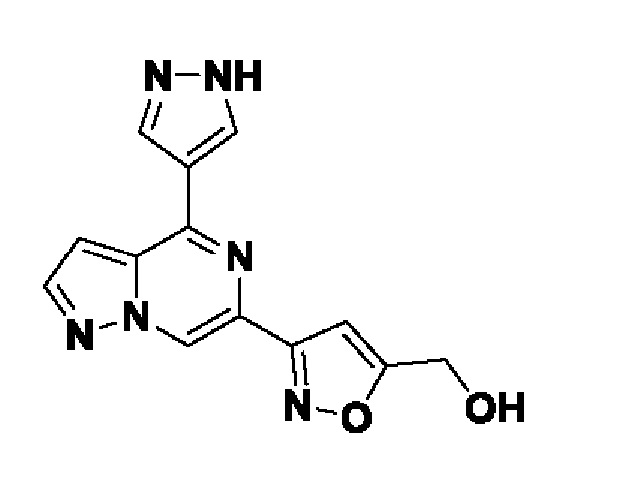
Раствор гипохлорита натрия (примерно 12%-15%, 0,19 мл, приблизительно 3,0 ммоль) добавляли по каплям к раствору трет-бутил (E)-4-(6-((гидроксиимино)метил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-карбоксилата (Получение 25, 200 мг, 0,61 ммоль) и пропаргилового спирта (171 мг, 3,05 ммоль) в DCM (5 мл) при примерно 0°C. Смеси давали нагреться до примерно 20°C в течение примерно 18 часов. Полученное твердое вещество фильтровали с получением указанного в заголовке соединения (115 мг, 67%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,14 (с, 1 H), 8,56 (с, 2 H), 8,31 (д, 1 H), 7,50 (д, 1 H), 7,09 (с, 1 H), 4,57-4,75 (м, 2 H).
LCMS m/z=283,1 [MH]+
Получение 27
3-(Цианометилен)циклобутан-1-карбонитрил
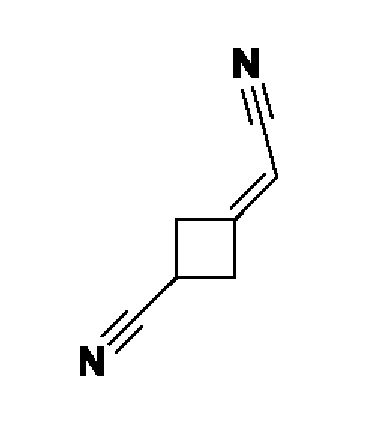
Раствор 3-оксоциклобутан-1-карбонитрила* (CAS: 20249-16-5, 14,5 г, 152 ммоль) в THF (250 мл) добавляли к смеси (EtO)2P(O)CH2CN (31,1 г, 175 ммоль), LiBr (19,9 г, 229 ммоль) и TEA (30,9 г, 305 ммоль) в THF (300 мл) при примерно 25°C. Примерно через 16 часов, смесь фильтровали и фильтрат концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде светло-желтого масла (16,01 г, 89%).
*См. Synthetic Communications 2005, 35, 657-662.
1H ЯМР (400 МГц, CD3CN) δ: 5,38 (с, 1 H), 3,30-3,43 (м, 2 H), 3,16-3,30 (м, 3 H).
LCMS m/z=119,1 [MH]+
Пример 1
(1s,3s)-3-(цианометил)-3-(4-(6-(5-(Гидроксиметил)изоксазол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
(цис-изомер)
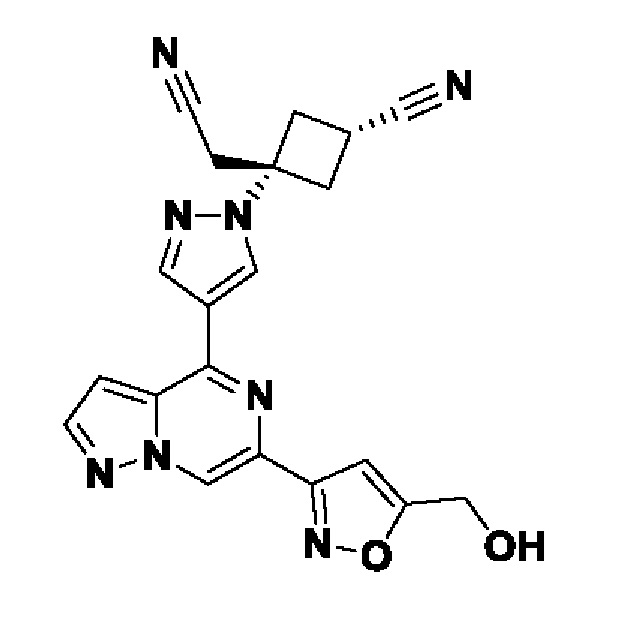
DBU (89,0 мг, 0,58 ммоль) добавляли к раствору (3-(4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)изоксазол-5-ил)метанола (Получение 26; 55,0 мг, 0,19 ммоль) и 3-(цианометилен)циклобутан-1-карбонитрила (Получение 27; 23,0 мг, 0,19 ммоль) в MeCN (4 мл). Реакционную смесь продували азотом и перемешивали при примерно 20°C в течение примерно 20 часов. Смесь разделяли между EtOAc (5 мл) и 1 M водн. NaH2PO4 (5 мл). EtOAc отделяли, сушили (Na2SO4), и концентрировали. Остаток очищали хроматографией с получением указанного в заголовке соединения (5 мг, 6%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,26 (м, 1 H), 8,92 (м, 1 H), 8,54 (м, 1 H), 8,40 (м, 1 H), 7,52 (м, 1 H), 7,14 (м, 1 H), 5,55 (шир.с, 1 H), 4,70 (с, 2 H), 3,60 (м, 3 H), 3,42-3,45 (м, 2 H), 2,78-2,83 (м, 2 H).
LCMS m/z=401,4 [MH]+
Получение 28
трет-бутил 3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)азетидин-1-карбоксилат

К раствору трет-бутил 3-(цианометилен)азетидин-1-карбоксилата (CAS 1153949-11-1, 7,00 г, 36,1 ммоль) в MeCN (100 мл) добавляли пинаколовый эфир 4-пиразолбороновой кислоты (7,71 г, 39,7 ммоль) и DBU (2,75 г, 18,0 ммоль) при примерно 25°C. Через примерно 18 часов, смесь концентрировали и остаток очищали колоночной хроматографией с получением указанного в заголовке соединения в виде белого твердого вещества (11 г, 78%).
1H ЯМР (400 МГц, CDCl3) δ: 7,92 (с, 1 H), 7,86 (с, 1 H), 4,40 (м, 2 H), 4,21 (м, 2 H), 3,52 (с, 2 H), 1,44 (с, 9 H), 1,32 (с, 12 H).
LC-MS m/z=333,0 [MH-C4H8]+
Получение 29
трет-бутил 3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)азетидин-1-карбоксилат
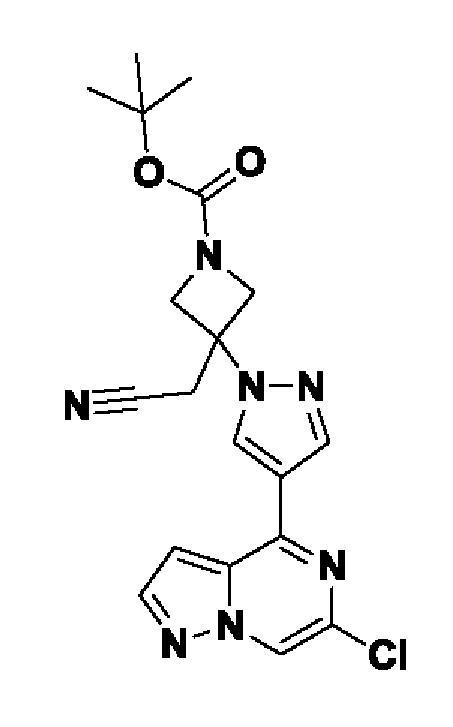
К раствору трет-бутил 3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)азетидин-1-карбоксилата (Получение 28, 362 мг, 0,93 ммоль) и 4,6-дихлорпиразоло[1,5-а]пиразина (Получение 4; 167 мг, 0,89 ммоль) в 1,4-диоксане (5 мл) добавляли 2 M водн. K3PO4 (1,40 мл) при примерно 25°C. Смесь продували аргоном в течение примерно 2 мин и добавляли бис(три-трет-бутилфосфин)палладий(0) (94,3 мг, 0,184 ммоль). Смесь перемешивали при примерно 20°C в течение примерно 2 часов. Смесь разбавляли DCM, разделяли, и водную фазу дважды экстрагировали DCM. Объединенные экстракты DCM сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде белого твердого вещества (295 мг, 75%).
1H ЯМР (400 МГц, CDCl3) δ: 8,42 (с, 1 H), 8,41 (с, 1 H), 8,31 (с, 1 H), 8,10 (д, 1 H), 7,02 (д, 1 H), 4,54 (д, 2 H), 4,31 (д, 3 H), 3,33 (с, 2 H), 1,49 (с, 9 H).
LCMS m/z=358,1 [MH-C4H8]+
Получение 30
2-(3-(4-(6-Хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-(циклопропилметил)азетидин-3-ил)ацетонитрил
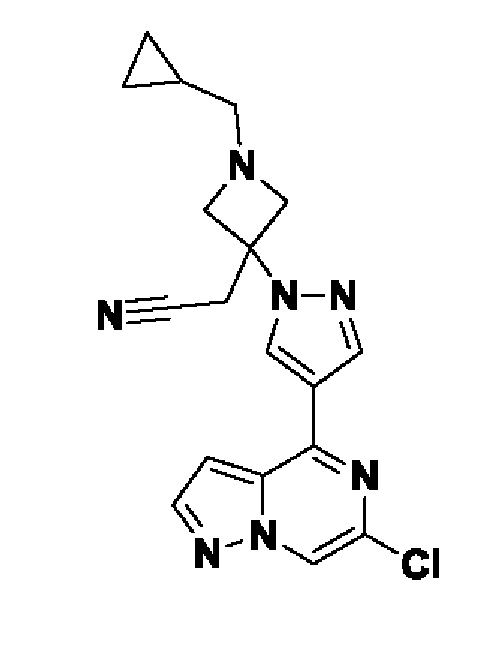
Часть 1
К раствору трет-бутил 3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)азетидин-1-карбоксилата (Получение 29, 0,56 г, 1,35 ммоль) в DCM (13,5 мл) добавляли TFA (7 мл) при примерно 25°C. Примерно через 4 часа, смесь концентрировали досуха с получением 2-(3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила в виде желтого твердого вещества (578 мг, примерно 100%), которое использовали без дополнительной очистки.
Часть 2
К раствору 2-(3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила (часть 1, 1,35 ммоль) и бромметилциклопропана (365 мг, 2,71 ммоль) в DMF (13,5 мл) добавляли TEA (548 мг, 5,41 ммоль) при примерно 25°C. Смесь нагревали при примерно 50°C в течение примерно 14 часов. Охлажденный раствор разбавляли водой (20 мл) и экстрагировали EtOAc (3×20 мл). Объединенные экстракты EtOAc промывали насыщенным солевым раствором (30 мл), сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (314 мг, 63%).
1H ЯМР (400 МГц, CDCl3) δ: 8,39 (с, 1 H), 8,37 (с, 1 H), 8,27 (с, 1 H), 8,08 (д, 1 H), 7,02 (д, 1 H), 3,78-3,84 (м, 2 H), 3,59 (д, 2 H), 3,42 (с, 2 H), 2,45 (д, 2 H), 0,77-0,87 (м, 1 H), 0,48-0,55 (м, 2 H), 0,14 (кв, 2 H).
LCMS m/z=367,9 [MH]+ (35Cl изотоп)
Получение 31
Этил 3-(трибутилстаннил)-1H-пиразол-5-карбоксилат
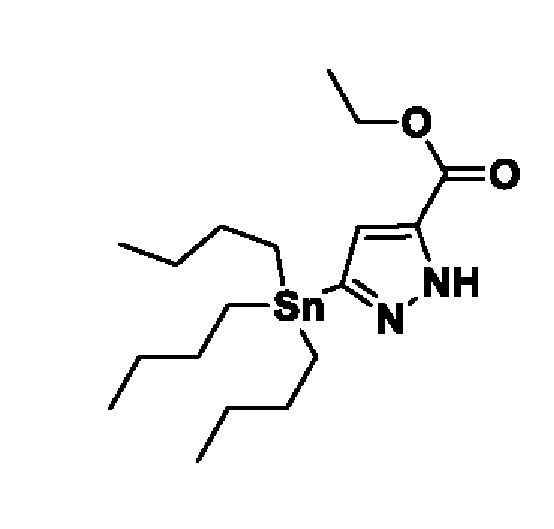
Этинилтрибутилстаннан (50 г, 158 ммоль) добавляли к раствору этилдиазоацетата (19,9 г, 175 ммоль) в толуоле (500 мл) при примерно 25°C. Раствор нагревали в течение примерно 16 часов при примерно 100°C, затем концентрировали с получением неочищенного продукта в виде желтого масла (110 г). Его объединяли с неочищенным продуктом из эквивалентной реакции, проводимой с этинилтрибутилстаннаном (22 г, 70 ммоль) и этилдиазоацетатом (8,76 г, 77 ммоль), и объединенные остатки очищали с помощью колоночной хроматографии на оксиде алюминия с получением указанного в заголовке соединения в виде желтого масла (42 г, 61%).
1H ЯМР (400 МГц, CDCl3) δ: 10,28 (шир.с, 1 H), 6,84-6,99 (м, 1 H), 4,41 (кв, 2 H), 1,47-1,67 (м, 6 H), 1,41 (т, 3 H), 1,28-1,39 (м, 6 H), 1,04-1,24 (м, 6 H), 0,90 (т, 9 H).
LCMS m/z 431,2 [MH]+ (120Sn изотоп).
Получение 32
(3-(Трибутилстаннил)-1H-пиразол-5-ил)метанол

Этил 3-(трибутилстаннил)-1H-пиразол-5-карбоксилат (Получение 31, 6000 мг, 13,98 ммоль) в THF (200 мл) добавляли к перемешиваемой суспензии литий алюминий гидрида (3108 мг, 83,9 ммоль) в THF (200 мл) при примерно -10°C. Примерно через 4 часа, смесь гасили декагидратом Na2SO4 при примерно -10°C пока не прекратится вскипание. Смесь фильтровали и фильтровальную лепешку промывали THF (500 мл) и DCM (5×500 мл). Комбинированные фильтраты концентрировали с получением указанного в заголовке соединения в виде белого твердого вещества (4460 мг, 82%).
1H ЯМР (400 МГц, CDCl3) δ: 10,35 (шир.с, 1 H), 6,28-6,43 (м, 1 H), 4,75 (с, 2 H), 1,49-1,60 (м, 6 H), 1,29-1,39 (м, 6 H), 1,08-1,15 (м, 6 H), 0,87-0,93 (м, 9 H).
LCMS m/z=388,9 [MH]+ (120Sn изотоп)
Пример 2
2-(1-(Циклопропилметил)-3-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил
К раствору 2-(3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-(циклопропилметил)азетидин-3-ил)ацетонитрила (Получение 30, 204 мг, 0,55 ммоль) и 3-(трибутилстаннил)-1H-пиразол-5-ил)метанола (Получение 32, 215 мг, 0,55 ммоль) добавляли XPhos Pd G2 (43,6 мг, 0,055 ммоль) в 1,4-диоксане (5,5 мл). Смесь нагревали при примерно 110°C в течение примерно 4 часов. Смесь выпаривали досуха и остаток объединяли с остатком из эквивалентной реакции с использованием 2-(3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-(циклопропилметил)азетидин-3-ил)ацетонитрила (Получение 30, 110 мг, 0,27 ммоль) и 3-(трибутилстаннил)-1H-пиразол-5-ил)метанола (Получение 32, 105 мг, 0,27 ммоль). Объединенные остатки очищали с помощью ВЭЖХ с получением указанного в заголовке соединения (112 мг, 31%).
1H ЯМР (400 МГц, CDCl3) δ: 8,66 (с, 1 H), 8,64-8,69 (м, 1 H), 8,38 (с, 1 H), 8,23 (с, 1 H), 8,06 (д, 1 H), 6,94 (д, 1 H), 6,67 (с, 1 H), 4,80 (с, 2 H), 3,80 (д, 2 H), 3,62 (д, 2 H), 3,41 (с, 2 H), 2,44 (д, 2 H), 0,76-0,86 (м, 1 H), 0,46-0,53 (м, 2 H), 0,10-0,16 (м, 2 H).
LCMS m/z 430,1 [MH]+
Получение 33
2-(3-(4-(6-Хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил
К раствору трет-бутил 3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)азетидин-1-карбоксилата (Получение 29, 485 мг, 1,17 ммоль) в DCM (20 мл) добавляли TFA (6 мл) при примерно 0°C. Смесь перемешивали при примерно 25°C в течение примерно 4 часов. Раствор концентрировали. Остаток доводили до рН около 9 с помощью конц. NH4OH (примерно 0,5 мл) и разделяли между водой (10 мл) и DCM (30 мл). Водный раствор экстрагировали DCM (3×30 мл). Объединенные экстракты DCM сушили (Na2SO4) и концентрировали с получением указанного в заголовке соединения в виде желтого твердого вещества (300 мг, 81%).
1H ЯМР (400 МГц, CD3OD) δ: 8,91 (с, 1 H), 8,72 (с, 1 H), 8,53 (с, 1 H), 8,20 (д, 1 H), 7,40 (д, 1 H), 4,90 (д, 2 H), 4,66 (д, 2 H), 3,72 (с, 2 H).
LCMS m/z 313,9 [MH]+ (35Cl изотоп)
Получение 34
2-(3-(4-(6-Хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-этилазетидин-3-ил)ацетонитрил
Ацетат натрия (314 мг, 3,82 ммоль) и ацетальдегид (842 мг, 19,1 ммоль) добавляли к раствору 2-(3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила (Получение 33, 120 мг, 0,38 ммоль) в MeOH (6 мл) и смесь перемешивали в течение примерно 4 часов. Затем добавляли NaBH(OAc)3 (243 мг, 1,15 ммоль) и смесь перемешивали в течение примерно 16 часов дольше при примерно 25°C. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (115 мг, 88%).
1H ЯМР (400 МГц, CDCl3) δ: 8,41 (с, 1 H), 8,39 (с, 1 H), 8,29 (с, 1 H), 8,10 (д, 1 H), 7,04 (д, 1 H), 3,79 (д, 2 H), 3,59 (д, 2 H), 3,43 (с, 2 H), 2,65 (кв, 2 H), 1,06 (т, 3 H).
LCMS m/z=342,1 [MH]+ (35Cl изотоп)
Пример 3
2-(1-Этил-3-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил
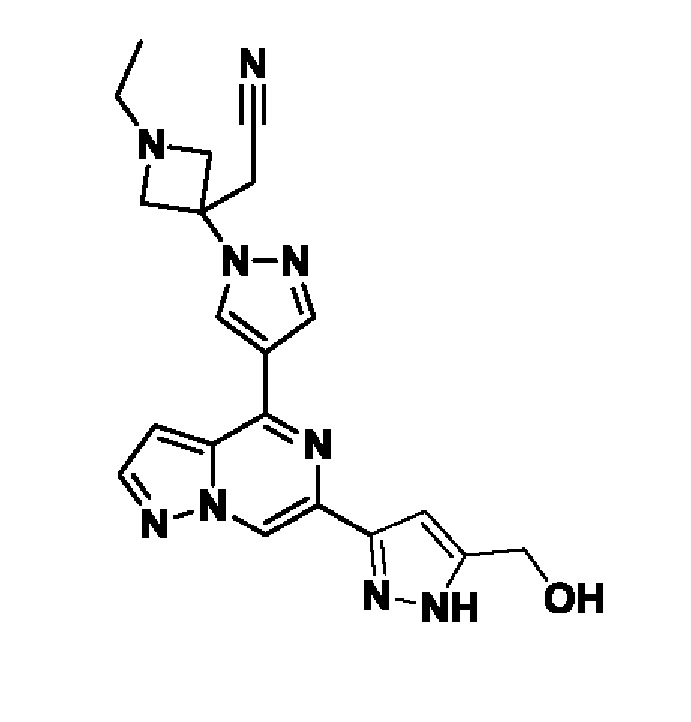
К раствору 2-(3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-этилазетидин-3-ил)ацетонитрила (Получение 34, 100 мг, 0,29 ммоль) в 1,4-диоксане (5 мл) добавляли 3-(трибутилстаннил)-1H-пиразол-5-ил)метанол (Получение 32, 136 мг, 0,35 ммоль) и XPhos Pd G2 (23,0 мг, 0,029 ммоль). Смесь нагревали при примерно 110°C в течение примерно 16 часов. Смесь концентрировали и остаток очищали с помощью хроматографии. Продукт далее очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде желтого твердого вещества (59 мг, 46%).
1H ЯМР (400 МГц, DMSO-d6) δ: 13,21 (с, 0,5 H), 12,93 (с, 0,5 H), 9,23 (с, 0,5 H), 9,07 (с, 1 H), 8,6-8,89 (м, 1 H), 8,69 (с, 0,5 H), 8,47 (с, 0,5 H), 8,26 (с, 1 H), 7,47-7,53 (м, 1 H), 6. - 9-6,94 (м, 1 H), 5,35 (с, 0,5 H), 5,35 (с, 0,5 H), 4,50-4,57 (м, 2 H), 3,68-3,71 (м, 2 H), 3,57-3,54 (м, 4 H), 3,17-3,16 (м, 0,5 H), 2,57-2,54 (м, 2 H), 0,96-0,93 (м, 3 H). Этот спектр согласуется с наличием различимых таутомеров.
LCMS m/z=404,3 [MH]+
Получение 35
2-Циклобутилиденацетонитрил
Смесь (EtO)2P(O)CH2CN (4,48 г, 25,2 ммоль), LiBr (1,96 г, 22,6 ммоль) и TEA (2,28 г, 22,6 ммоль) в сухом THF (40 мл) перемешивали при примерно 25°C в течение примерно 2 часов. К этому добавляли раствор циклобутанона (1,58 г, 22,6 ммоль) в THF (5 мл) при примерно 25°C. Примерно через 16 часов, смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (1,2 г, 57%).
1H ЯМР (400 МГц, CDCl3) δ: 5,11 (квин), 2,93-3,05 (м), 2,82-2,92 (м), 2,04-2,17 (м).
Получение 36
2-(1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил
К смеси 2-циклобутилиденацетонитрила (Получение 35, 200 мг, 2,15 ммоль) и пинаколового эфира 4-пиразолбороновой кислоты (458 мг, 2,36 ммоль) в MeCN (15 мл) добавляли DBU (981 мг, 6,44 ммоль). Реакционную смесь перемешивали при примерно 25°C в течение примерно 16 часов и затем нагревали до примерно 50°C в течение примерно 24 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (150 мг, 24%).
1H ЯМР (400 МГц, CDCl3) δ: м 7,89 (с, 1 H), 7,86 (с, 1 H), 3,09 (с, 2 H), 2,68-2,80 (м, 2 H), 2,45-2,55 (м, 2 H), 2,01-2,10 (м, 2 H), 1,33 (с, 12 H).
LCMS m/z=287,9 [MH]+
Пример 4
2-(1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил
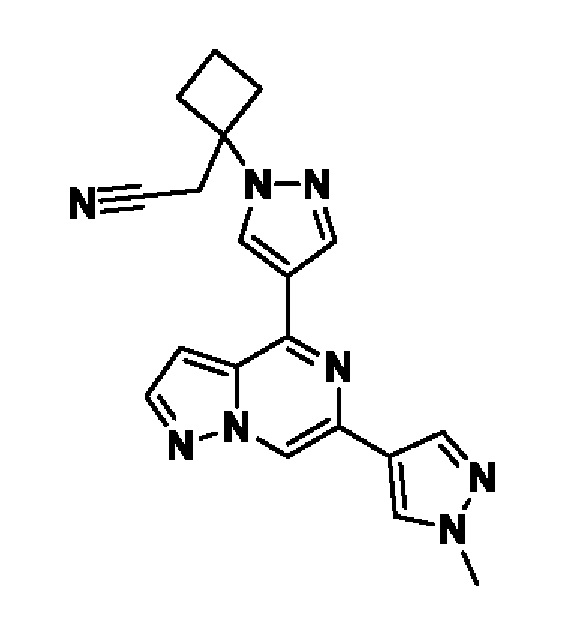
К смеси 2-(1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (Получение 36, 129 мг, 0,45 ммоль) и 4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 11, 100 мг, 0,43 ммоль) в 1,4-диоксане (4,3 мл) добавляли 2 M водн. K3PO4 (0,85 мл) и PdCl2(dppf) (15,7 мг, 0,021 ммоль). Смесь продували азотом в течение примерно 1 мин и перемешивали при примерно 80°C в течение примерно 16 часов. Реакционную смесь объединяли с эквивалентной реакцией, проведенной с использованием 2-(1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (Получение 36, 20 мг, 0,07 ммоль) и 4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 11, 19, мг, 0,083 ммоль), 2 M водн. K3PO4 (0,14 мл) и PdCl2(dppf) (2,5 мг, 0,0035 ммоль) в 1,4-диоксане (2 мл). Объединенные реакционные смеси концентрировали и остаток очищали с помощью хроматографии. Соединение дополнительно очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (22 мг, 12%).
1H ЯМР (400 МГц, CD3OD) δ: 8,68 (с, 1 H), 8,63 (с, 1 H), 8,39 (с, 1 H), 8,22 (с, 1 H), 8,07 (д, 1 H), 8,06 (с, 1 H), 7,21 (д, 1 H), 3,97 (с, 3 H), 3,94-3,99 (м, 1 H), 3,37 (с, 2 H), 3,35-3,39 (м, 1 H), 2,84-2,95 (м, 2 H), 2,54 (ддд, 2 H), 2,05-2,21 (м, 2 H).
LCMS m/z=358,9 [MH]+
Получение 37
2-((1r,3s)-1-(4-бром-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
(транс-изомер)
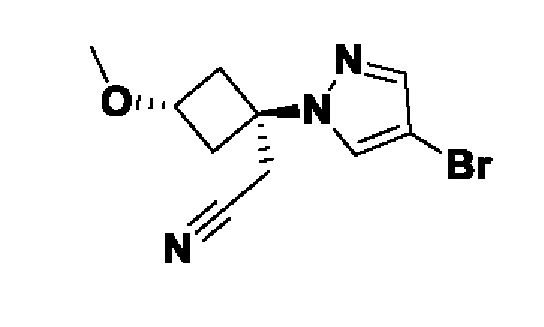
и
2-((1s,3r)-1-(4-бром-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
(цис-изомер).
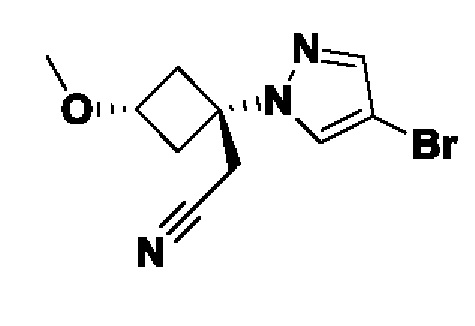
DBU (4,25 мл, 28,4 ммоль) добавляли к раствору 2-(3-метоксициклобутилиден)ацетонитрила (Получение 90, 3,50 г, 28,4 ммоль) и 4-бромпиразол (4,18 г, 28,4 ммоль) в MeCN (80 мл) при примерно 25°C. Через примерно 18 часов, смесь выливали в NaH2PO4 (17,04 г, 142 ммоль) в воде и фазы разделяли. Водный слой дважды экстрагировали EtOAc и объединенные экстракты EtOAc концентрировали. Избыток 4-бромпиразола удаляли с помощью хроматографии, элюируя DCM:THF (100:0-95:5). Вещество далее очищали с помощью хроматографии, элюируя эфир:гептан с получением 2-((1r,3s)-1-(4-бром-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила в виде белого твердого вещества (транс-изомер, 2,19 г, 28%)
1H ЯМР (400 МГц, CDCl3) δ: 7,62 (с, 1 H), 7,55 (с, 1 H), 3,99 (тт, 1 H), 3,30 (с, 3 H), 3,12 (с, 2 H), 2,96-3,04 (м, 2 H), 2,44-2,51 (м, 2 H).
LCMS m/z=270,0 [MH]+ (79Br изотоп)
и 2-((1s,3r)-1-(4-бром-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила в виде бесцветного масла (цис-изомер, 5,00 г, 65%).
1H ЯМР (400 МГц, CDCl3) δ: 7,60 (с, 1 H), 7,53 (с, 1 H), 4,00 (квин, 1 H), 3,29 (с, 3 H), 2,99 (с, 2 H), 2,85-2,96 (м, 2 H), 2,56-2,67 (м, 2 H).
LCMS m/z=270,0 [MH]+ (79Br изотоп)
Получение 38
2-((1r,3s)-3-метокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил.
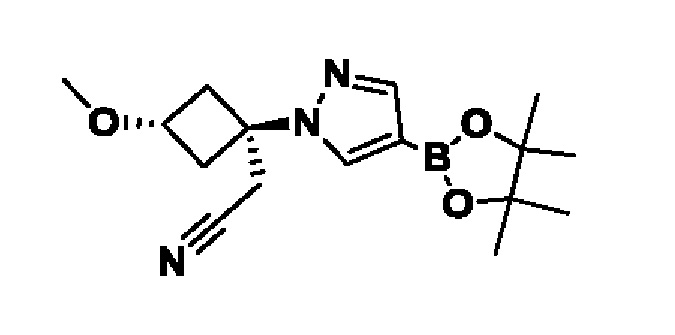
Смесь 2-((1r,3s)-1-(4-бром-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 37, транс-изомер, 3399 мг, 12,58 ммоль), бис(пинаколато)диборона (3510 мг, 13,8 ммоль) и ацетата калия (3700 мг, 37,7 ммоль) в 1,4-диоксане (33 мл) продували аргоном в течение примерно 5 мин, с последующим добавлением XPhos Pd G2 (1980 мг, 2,52 ммоль) при примерно 25°C. Смесь нагревали при примерно 65°C в течение примерно 4 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением твердого вещества. К этому твердому веществу добавляли EtOAc (10 мл) и гептан (40 мл) и смесь перемешивали при примерно 25°C в течение примерно 30 мин. Твердое вещество фильтровали и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (1,95 г, 49%).
1H ЯМР (500 МГц, CDCl3) δ: 7,91 (с, 1 H), 7,87 (с, 1 H), 3,98 (тт, 1 H), 3,28 (с, 3 H), 3,17 (с, 2 H), 2,98-3,07 (м, 2 H), 2,45-2,53 (м, 2 H), 1,31 (с, 12 H).
LCMS m/z=318,0 [MH]+
Получение 39
2-((1r,3s)-1-(4-(6-Хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
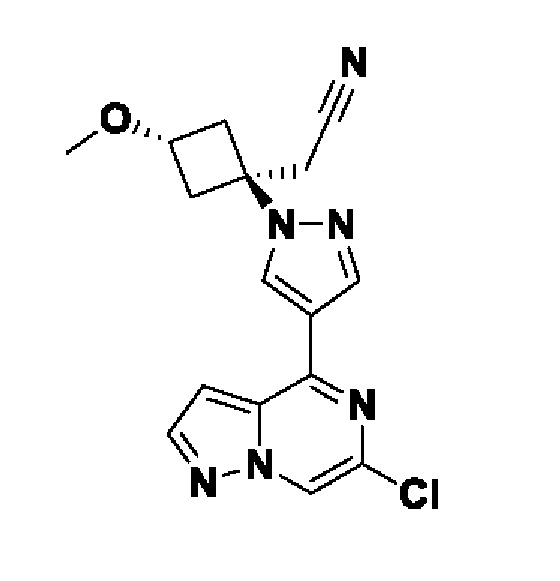
Раствор 2-((1r,3s)-3-метокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (Получение 38, 1950 мг, 6,15 ммоль), 4,6-дихлорпиразоло[1,5-а]пиразина (Получение 4, 1160 мг, 6,15 ммоль) и 2 M водн. K3PO4 (9,22 мл) в 1,4-диоксане (25 мл) продували аргоном в течение примерно 5 мин, с последующим добавлением бис(три-трет-бутилфосфин)палладия(0) (157 мг, 0,31 ммоль) при примерно 25°C. Примерно через 2 часа, смесь разбавляли EtOAc, фазы разделяли и водную фазу дважды экстрагировали DCM. Объединенные экстракты EtOAc и DCM сушили (Na2SO4) и концентрировали для получения твердого вещества, которое перекристаллизовывали из теплой (примерно 40°С) смеси DCM и гептана с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (1,12 г, 53%). Фильтрат концентрировали и очищали с помощью хроматографии с получением дополнительного указанного в заголовке соединения (1,01 г, 47%).
1H ЯМР (500 МГц, CDCl3) δ: m 8,39 (д, 1 H), 8,38 (с, 1 H), 8,28 (с, 1 H), 8,09 (д, 1 H), 7,03 (дд, 1 H), 4,04-4,10 (м, 1 H), 3,33 (с, 3 H), 3,25 (с, 2 H), 3,09-3,17 (м, 2 H), 2,53-2,61 (м, 2 H).
LCMS m/z=343,1 [MH]+ (35Cl изотоп)
Получение 40
3,5-дибром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол
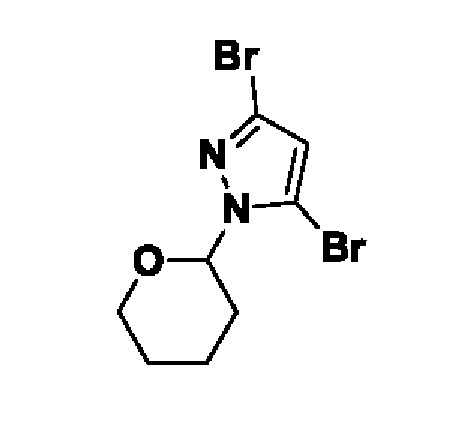
К раствору 3,5-дибромпиразола* (CAS: 67460-86-0, 18,0 г, 79,7 ммоль) и DHP (30 мл) добавляли CF3COOH (73 мг, 0,64 ммоль). Смесь нагревали при примерно 95°C в течение примерно 12 часов. Реакцию гасили NaOH (96 мг, 2,4 ммоль) и затем концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (11,5 г, 46%).
*См.: Justus Liebigs Annalen der Chemie 1959, 625, 55-65.
1H ЯМР (400 МГц, CDCl3) δ: 6,35 (с, 1 H), 5,42 (д, 1 H), 4,05 (м, 1 H), 3,65 (м, 1 H), 2,38-2,48 (м, 1 H), 2,11 (м, 1 H), 1,90 (м, 1 H), 1,62-1,77 (м, 3 H).
LCMS m/z=226,7 [MH-THP]+(79Br, 81Br изотоп)
Получение 41
3-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-карбоновая кислота
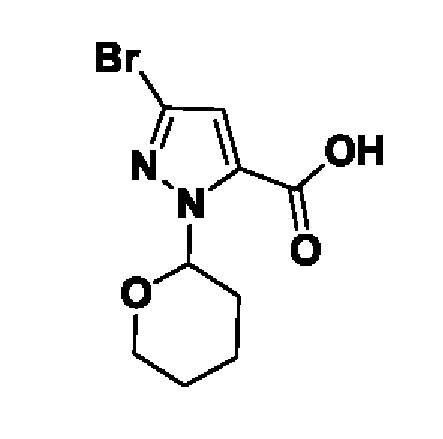
n-BuLi (2,5 M, 15,8 мл, 39,5 ммоль) добавляли по каплям к раствору 3,5-дибром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола (Получение 40, 9,4 г, 30,0 ммоль) в THF (87 мл) при примерно -78°C. Смесь выдерживали при примерно -78°C в течение примерно 2 часов. Раствор СО2 (полученный путем барботирования СО2 в безводный THF (100 мл) в течение 20 мин при примерно -70°С и перемешивания при этой температуре в течение примерно 1,5 часов) добавляли по каплям при поддержании внутренней температуры реакционной смеси ниже примерно -65°C. Смесь затем перемешивали при примерно -70°C в течение примерно 1 часа. Смесь доводили до значения рН примерно 4 с помощью 1 М водн. HCl при примерно 0°C и экстрагировали EtOAc (3×100 мл). Объединенные экстракты EtOAc промывали насыщенным солевым раствором (2×50 мл), сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (5,0 г, 60%).
1H ЯМР (400 МГц, DMSO-d6) δ: 6,99 (с, 1 H), 6,17 (дд, 1 H), 3,90 (д, 1 H), 3,49-3,66 (м, 2 H), 2,12-2,28 (м, 1 H), 1,91-2,04 (м, 1 H), 1,83-1,92 (м, 1 H), 1,58-1,70 (м, 1 H), 1,45-1,57 (м, 2 H).
Получение 42
трет-бутил (3-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)карбамат
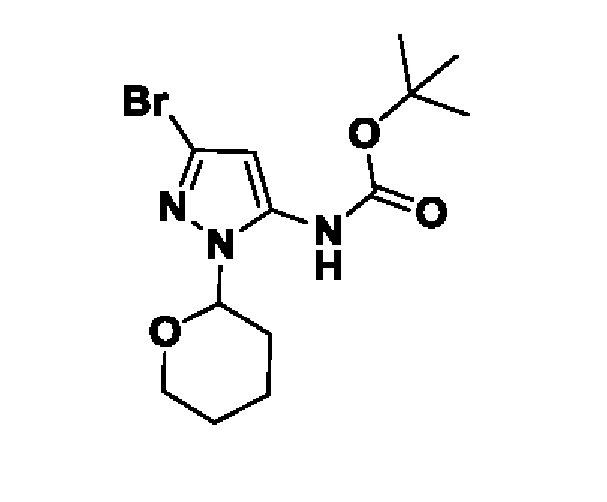
Дифенилфосфорил азид (10 г, 36,4 ммоль) добавляли к раствору 3-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-карбоновой кислоты (Получение 41, 5 г, 18,17 ммоль) и DIPEA (6,4 мл, 37,0 ммоль) в трет-бутаноле (60,6 мл). Смесь нагревали при примерно 45°C в течение примерно 30 мин, и затем нагревали с обратным холодильником в течение примерно 5 часов. Смесь разбавляли EtOAc (300 мл) и промывали насыщенным водн. NaHCO3 (3×100 мл), насыщенным солевым раствором (2×100 мл), сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде светло-желтого масла (3,36 г, 53%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,64 (шир.с, 1 H), 6,26 (с, 1 H), 5,44 (дд, 1 H), 3,84 (д, 1 H), 3,55-3,70 (м, 1 H), 2,08-2,20 (м, 1 H), 1,91-2,00 (м, 1 H), 1,75 (дд, 1 H), 1,54-1,64 (м, 1 H), 1,48-1,53 (м, 2 H), 1,46 (с, 9 H).
LCMS m/z=367,9 [MNa]+ (79Br изотоп)
Получение 43
3-бром-1-(тетрагидро-2H-пиран-2-ил)-5-(диBoc)-амино-1H-пиразол
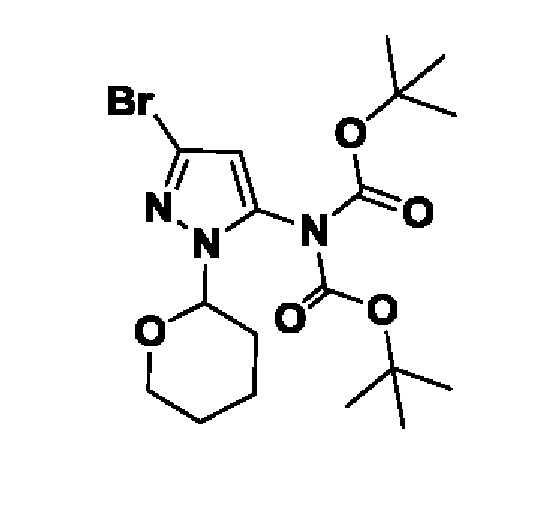
DMAP (27 мг, 0,22 ммоль) добавляли к раствору трет-бутил (3-бром-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)карбамата (Получение 42, 390 мг, 1,13 ммоль), ди-трет-бутил дикарбоната (492 мг, 2,25 ммоль) и TEA (0,47 мл, 3,38 ммоль) в DCM (4 мл) при примерно 20°C. Через примерно 18 часов, смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (450 мг, 89%)
1H ЯМР (400 МГц, CDCl3) δ: 6,42 (с, 1 H), 5,15 (м, 1 H), 4,02 (м, 1 H), 3,60 (м, 1 H), 2,40 (м, 1 H), 2,15 (м, 1 H), 1,88 (м, 1 H), 1,58-1,76 (м, 3 H), 1,48 (с, 18 H).
LCMS m/z=467,9 [MNa]+ (79Br изотоп)
Получение 44
2-((1r,3s)-1-(4-(6-(5-(диBoc)-амино-1-(тетрагидро-2H-пиран-2-ил)-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
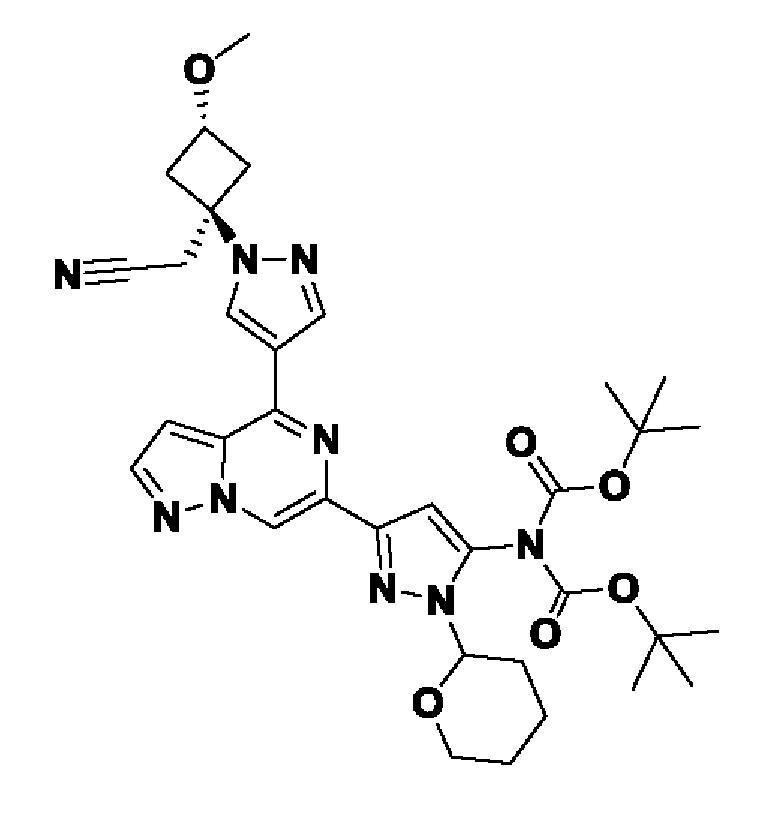
Смесь KOAc (110 мг, 1,06 ммоль), бис(пинаколато)диборона (164 мг, 0,64 ммоль), 3-бром-1-(тетрагидро-2H-пиран-2-ил)-5-(диBoc)-амино-1H-пиразола (Получение 43, 191 мг, 0,43 ммоль) и XPhos Pd G2 (55 мг, 0,07 ммоль) в 1,4-диоксане (3,5 мл) нагревали до примерно 65°C в течение примерно 3,5 часов. Смесь охлаждали до примерно 25°C и добавляли 2-((1r,3s)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метокси-циклобутил)ацетонитрил (Получение 39, 68 мг, 0,20 ммоль) и смесь продували азотом до добавления 2 M водн. K3PO4 (0,53 мл, 1,06 ммоль) и XPhos Pd G2 (55 мг, 0,07 ммоль). Смесь нагревали до примерно 80°C в течение примерно 3 часов. Реакцию гасили насыщенным солевым раствором и экстрагировали EtOAc. Экстракт EtOAc концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (91 мг, 32%).
1H ЯМР (400 МГц, CDCl3) δ: 9,08 (с, 1 H), 8,39 (с, 1 H), 8,33 (с, 1 H), 8,09 (д, 1 H), 6,97 (д, 1 H), 6,90 (с, 1 H), 5,26 (дд, 1 H), 4,02-4,11 (м, 2 H), 3,64 (т, 1 H), 3,34 (с, 3 H), 3,25 (с, 2 H), 3,12-3,20 (м, 2 H), 2,55-2,62 (м, 2 H), 2,17-2,25 (м, 1 H), 1,93 (дд, 1 H), 1,58-1,82 (м, 4 H), 1,45 (с, 18 H).
LCMS m/z=674,5 [MH]+
Получение 45
2-((1r,3s)-1-(4-(6-(5-амино-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
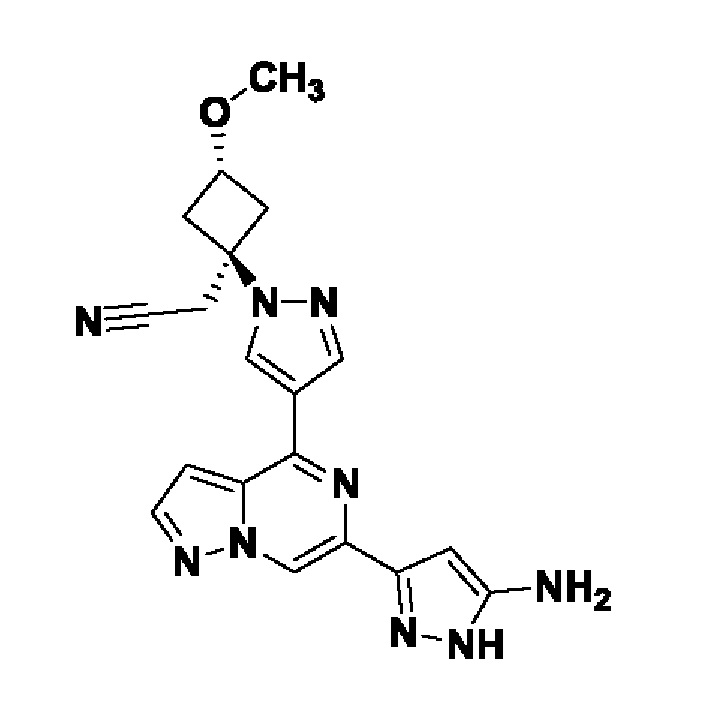
TFA (2 мл) добавляли к 2-((1r,3s)-1-(4-(6-(5-(диBoc)-амино-1-(тетрагидро-2H-пиран-2-ил)-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)-ацетонитрилу (Получение 44, 91 мг, 0,13 ммоль) в безводном DCM (1 мл) при примерно 20°C. Примерно через 1 час, смесь концентрировали. Добавляли DCM, затем насыщенный водн. NaHCO3 до тех пор, пока рН раствора не станет основным. Фазы разделяли и водную фазу дважды экстрагировали DCM. Объединенные экстракты DCM сушили (Na2SO4), концентрировали, и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (50 мг, 95%).
1H ЯМР (400 МГц, CDCl3) δ: 8,53 (с, 1 H), 8,40 (с, 1 H), 8,25 (с, 1 H), 8,06 (с, 1 H), 6,96 (шир.с, 1 H), 6,01 (шир.с, 1 H), 4,33 (шир.с, 1 H), 4,05 (м, 1 H), 3,31 (с, 3 H), 3,25 (с, 2 H), 3,11 (дд, 2 H), 2,55 (дд, 2 H).
LCMS m/z=390,3 [MH]+
Пример 5
N-(3-(4-(1-((1r,3s)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-5-ил)ацетамид
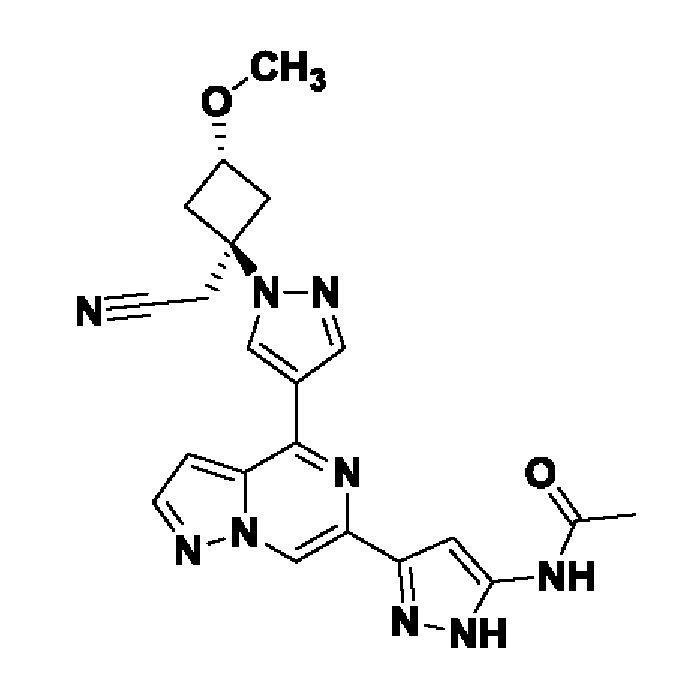
2-((1r,3s)-1-(4-(6-(5-амино-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метокси-циклобутил)ацетонитрил (Получение 45, 39 мг, 0,1 ммоль) помещали в реакционную пробирку, которую затем эвакуировали и заполняли три раза азотом. К этому добавляли безводный DCM (1 мл). Пробирку охлаждали до примерно 0°C перед до добавления N-метилморфолина (11 мг, 0,11 ммоль) и ацетилхлорида (8,6 мг, 0,11 ммоль). Смеси давали нагреться до примерно 25°C в течение примерно 18 часов. Смесь концентрировали и остаток растворяли в MeOH (2 мл). Добавляли K2CO3 (30 мг, 0,22 ммоль) при примерно 0°C. Примерно через 3 часа, смесь фильтровали через целит. Фильтрат концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (30 мг, 63%).
1H ЯМР (400 МГц, CD3OD) δ: 8,80 (с, 1 H), 8,75 (с, 1 H), 8,40 (с, 1 H), 8,09 (д, 1 H), 7,23 (д, 1 H), 6,88 (с, 1 H), 4,02-4,12 (м, 1 H), 3,81 (с, 3 H), 3,34 (с, 2 H), 3,15-3,24 (м, 2 H), 2,48-2,59 (м, 2 H), 2,21 (с, 3 H).
LCMS m/z=432,2 [MH]+
Получение 46
этил 5-(4-(1-((1r,3s)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксилат
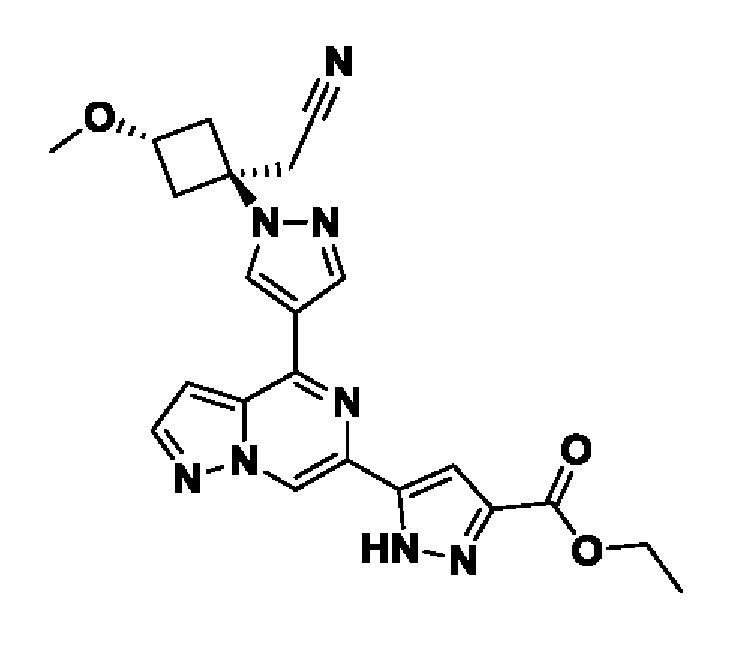
Раствор 2-((1r,3s)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 39, 200 мг, 0,58 ммоль) и этил 3-(трибутилстаннил)-1H-пиразол-5-карбоксилата (Получение 31, 300 мг, 0,70 ммоль) в 1,4-диоксане (5,8 мл) продували азотом и добавляли XPhos Pd G2 (45,9 мг, 0,058 ммоль) при примерно 25°C. Реакционную смесь нагревали при примерно 80°C в течение примерно 16 часов. Охлажденную смесь очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (220 мг, 84%).
1H ЯМР (400 МГц, DMSO-d6) δ: 14,09 (с, 1 H), 9,39 (с, 1 H), 9,10 (с, 1 H), 8,72 (с, 1 H), 8,30 (д, 1 H), 7,59 (д, 1 H), 7,53 (д, 1 H), 4,33 (кв, 2 H), 4,01 (дд, 1 H), 3,44 (с, 3 H), 3,22 (с, 2 H), 3,14-3,20 (м, 2 H), 2,43-2,48 (м, 2 H), 1,34 (т, 3 H).
LCMS m/z=447,2 [MH]+
Пример 6
5-(4-(1-((1r,3s)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-N-метил-1H-пиразол-3-карбоксамид

Раствор этил 5-(4-(1-((1r,3s)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксилата (Получение 46, 100 мг, 0,22 ммоль) в 30% MeNH2 в растворе EtOH (35 мл) герметизировали в пробирке для проведения реакций под микроволновым излучением при примерно 20°C. Примерно через 16 часов, смесь концентрировали и остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (62 мг, 58%).
1H ЯМР (400 МГц, CD3OD) δ: 9,00 (с, 1 H), 8,91 (с, 1 H), 8,57 (с, 1 H), 8,18 (с, 1 H), 7,37 (шир.с, 1 H), 7,26 (с, 1 H), 4,61 (с, 1 H), 4,04-4,15 (м, 1 H), 3,37 (с, 3 H), 3,33 (с, 2 H), 3,18-3,26 (м, 2 H), 2,95 (с, 3 H), 2,56 (дд, 2 H).
LCMS m/z=432,1 [M+H]+
Получение 47
трет-бутил 3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1-карбоксилат

В сосуд добавляли 4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин (Получение 11, 150 мг), трет-бутил 3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)азетидин-1-карбоксилат (Получение 28, 374 мг, 0,96 ммоль), 2 M водн. Na2CO3 (0,96 мл) и 1,4-диоксан (4 мл). Смесь продували аргоном в течение примерно 5 мин, с последующим добавлением PdCl2(dppf) (93,7 мг, 0,13 ммоль). Смесь нагревали при примерно 120°C в течение примерно 1 часа под микроволновым облучением. Смесь разбавляли EtOAc, промывали водой, сушили (Na2SO4), и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (233 мг, 79%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,03 (с, 1 H), 8,95 (с, 1 H), 8,53 (с, 1 H), 8,37 (с, 1 H), 8,20 (д, 1 H), 8,16 (с, 1 H), 7,46 (с, 1 H), 4,54 (д, 2 H), 4,25 (д, 3 H), 3,91 (с, 3 H), 3,67 (с, 2 H), 1,42 (с, 9 H).
LCMS m/z=460,2 [MH]+
Получение 48
2-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил

TFA (1 мл) добавляли к трет-бутил 3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1-карбоксилату (Получение 47, 233 мг, 0,51 ммоль) в DCM (5 мл) при примерно 25°C. Примерно через 90 мин, смесь концентрировали. Остаток концентрировали дважды с толуолом, затем сушили в вакууме. Остаток растворяли в MeOH и пропускали через слой карбонатной смолы на полимерной подложке. Элюированный раствор концентрировали с получением указанного в заголовке соединения (200 мг, примерно 100%), которое использовали без дополнительной очистки.
1H ЯМР (400 МГц, DMSO-d6) δ: 9,12 (с, 1 H), 9,06 (с, 1 H), 8,58 (с, 1 H), 8,39 (с, 1 H), 8,22 (д, 1 H), 8,17 (с, 1 H), 7,50 (д, 1 H), 4,68-4,77 (м, 2 H), 4,35-4,40 (м, 2 H), 3,93 (с, 2 H), 3,91 (с, 3 H).
LCMS m/z=360,5 [MH]+
Пример 7
2,2'-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1,3-диил)диацетонитрил
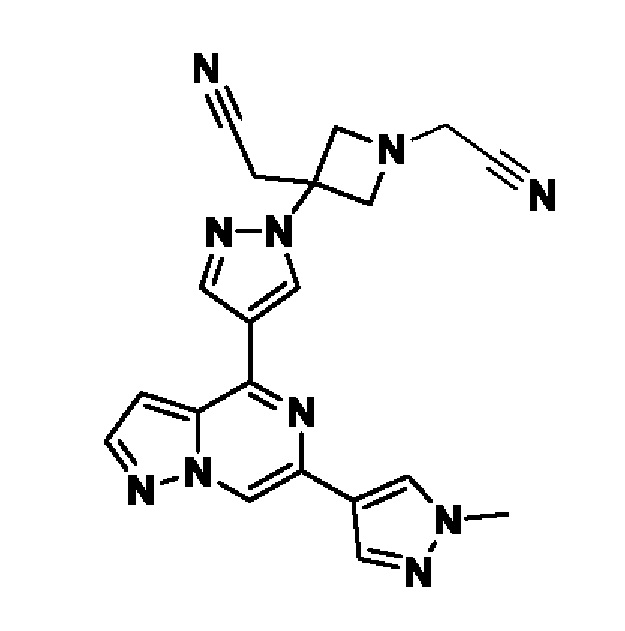
DIPEA (132 мкл, 0,76 ммоль) добавляли к раствору 2-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила (Получение 48, 91 мг, 0,25 ммоль) в DMF (2 мл). Добавляли бромацетонитрил (36 мг, 0,30 ммоль) при примерно 25°C. Примерно через 18 часов реакцию гасили конц. NH4OH и перемешивали при примерно 25°C в течение примерно 10 мин. Смесь концентрировали и остаток разбавляли DCM. DCM дважды промывали насыщенным водн. NH4Cl, затем дважды насыщенным водн. Na2CO3. DCM сушили (Na2SO4) и концентрировали. Остаток очищали хроматографией с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (68 мг, 68%).
1H ЯМР (400 МГц, CD3OD) δ: 8,77 (с, 1 H), 8,74 (с, 1 H), 8,45 (с, 1 H), 8,25 (с, 1 H), 8,08-8,10 (м, 2 H), 7,25 (д, 1 H), 4,01 (д, 2 H), 3,98 (с, 3 H), 3,86 (д, 2 H), 3,78 (с, 2 H), 3,54 (с, 2 H).
LCMS m/z=399,3 [MH]+
Пример 8
2-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-(метилсульфонил)азетидин-3-ил)ацетонитрил
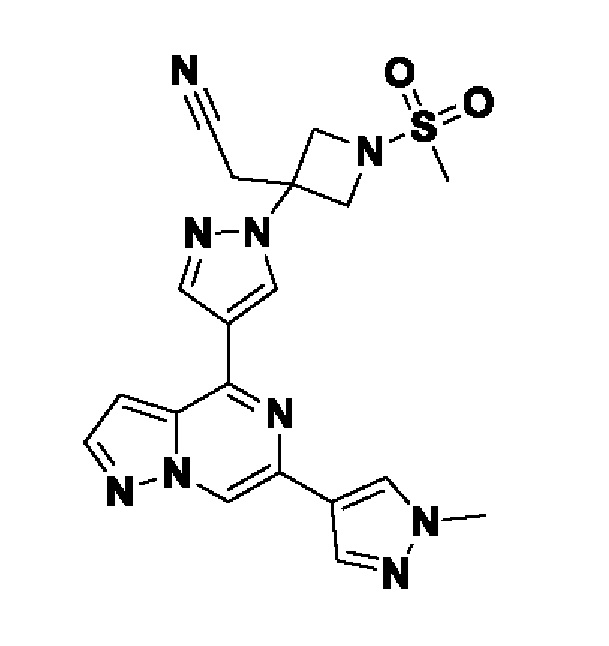
К раствору 2-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила (Получение 48, 120 мг, 0,27 ммоль) и TEA (331 мг, 3,27 ммоль) в DCM (20 мл) добавляли метансульфонил хлорид (313 мг, 2,73 ммоль) при примерно 0°C. Смесь затем перемешивали при примерно 10°C в течение примерно 1 часа до концентрирования. Остаток очищали с использованием ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (39 мг, 33%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,05 (с, 1 H), 8,99 (с, 1 H), 8,57 (с, 1 H), 8,37 (с, 1 H), 8,21 (д, 1 H), 8,17 (с, 1 H), 7,47 (д, 1 H), 4,65 (д, 2 H), 4,30 (д, 2 H), 3,91 (с, 3 H), 3,70 (с, 2 H), 3,14 (с, 3 H).
LCMS m/z=437,9 [MH]+
Пример 9
2-(1-(Циклопропилсульфонил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил
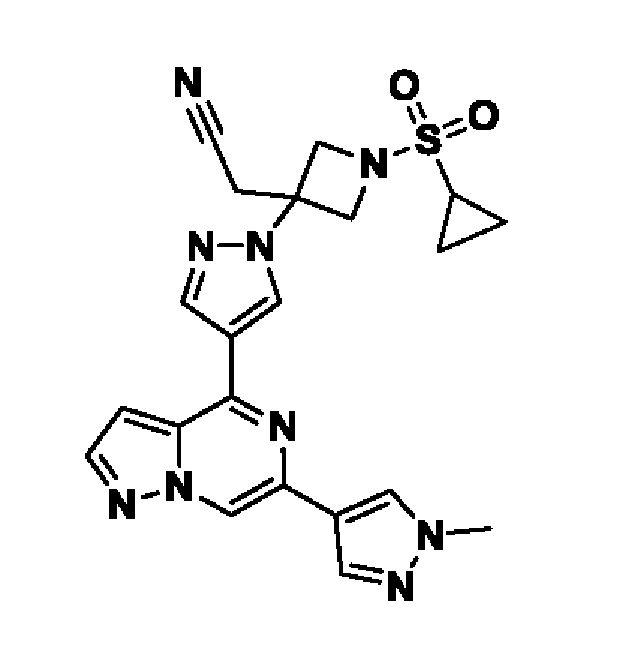
К смеси 2-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила (Получение 48, 100 мг, 0,25 ммоль) в DCM (10 мл) добавляли TEA (153 мг, 1,52 ммоль) и циклопропансульфонил хлорид (107 мг, 0,76 ммоль) при примерно 0°C. Смесь перемешивали при примерно 20°C в течение примерно 16 часов, затем концентрировали. Остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (60 мг, 46%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,05 (с, 1 H), 9,00 (с, 1 H), 8,57 (с, 1 H), 8,37 (с, 1 H), 8,21 (д, 1 H), 8,16 (с, 1 H), 7,46 (д, 1 H), 4,69 (д, 2 H), 4,32 (д, 2 H), 3,91 (с, 3 H), 3,70 (с, 2 H), 2,85 (м, 1 H), 0,96-1,09 (м, 4 H).
LCMS m/z=464,0 [MH]+
Пример 10
2-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-пропионилазетидин-3-ил)ацетонитрил
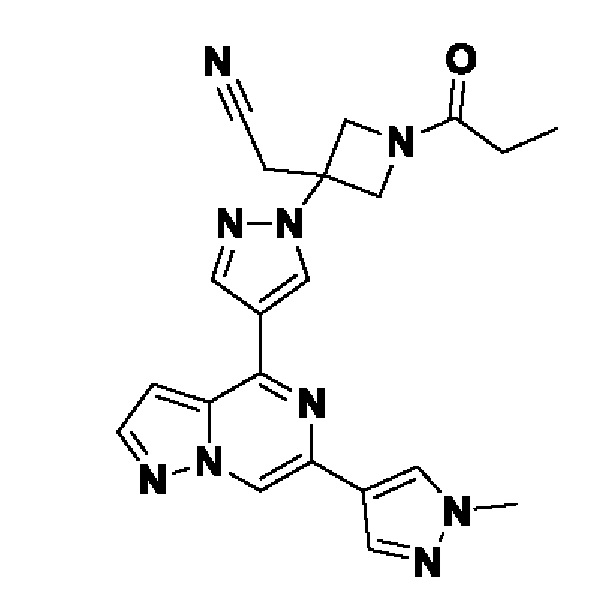
К раствору 2-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила (Получение 48, 90 мг, 0,23 ммоль) в DCM (5 мл) добавляли TEA (69 мг, 0,68 ммоль) и пропионовый ангидрид (59 мг, 0,45 ммоль). Реакционную смесь перемешивали при примерно 20°C в течение примерно 30 мин, затем концентрировали и остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (28 мг, 30%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,05 (с, 1 H), 8,97 (с, 1 H), 8,55 (с, 1 H), 8,37 (с, 1 H), 8,21 (д, 1 H), 8,16 (с, 1 H), 7,46 (д, 1 H), 4,80 (д, 1 H), 4,52 (дд, 2 H), 4,23 (д, 1 H), 3,91 (с, 3 H), 3,70 (с, 2 H), 2,16 (кв, 2 H), 0,99 (т, 3 H).
LCMS m/z 416,0 [MH]+
Получение 49
2-(3-(Бензилокси)циклобутилиден)ацетонитрил
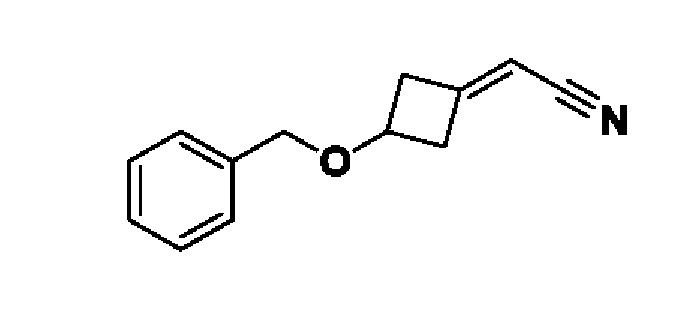
LiBr (0,270 г, 3,12 ммоль) помещали под вакуумом, затем заполняли азотом и добавляли THF (28 мл), затем (EtO)2POCH2CN (0,53 мл, 3,12 ммоль) и TEA (0,79 мл, 5,67 ммоль). Полученный раствор перемешивали при примерно 20°C в течение примерно 45 мин, затем добавляли раствор 3-(бензилокси)циклобутанона (500 мг, 2,84 ммоль) в сухом THF (3 мл). Примерно через 5 часов, смесь выливали в EtOAc (100 мл) и EtOAc три раза промывали насыщенным водн. NH4Cl (3×50 мл) и насыщенным солевым раствором (25 мл). Экстракт EtOAc сушили (MgSO4) и концентрировали. Остаток хроматографировали с получением указанного в заголовке соединения (480 мг, 85%).
1H ЯМР (400 МГц, CDCl3) δ: 7,29-7,43 (м, 5 H), 5,24 (квин, 1 H), 4,45-4,53 (м, 2 H), 4,19 (квин, 1 H), 3,18-3,29 (м, 1 H), 3,03-3,13 (м, 1 H), 2,86-3,00 (м, 2 H).
Получение 50
2-(3-Гидроксициклобутил)ацетонитрил
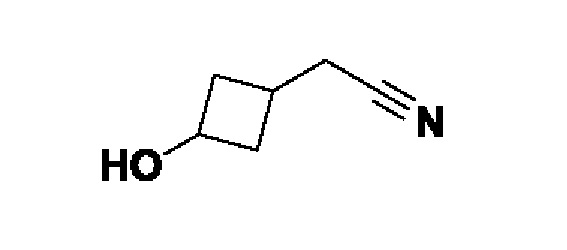
Гидроксид палладия на углероде (20% Pd, влажн, 430 мг) добавляли к раствору 2-(3-(бензилокси)циклобутилиден)ацетонитрила (Получение 49, 430 мг, 2,16 ммоль) в THF (6,5 мл). Смесь герметизировали под давлением водорода (100 фунт./кв. дюйм) в стальном реакторе и перемешивали при примерно 20°C в течение примерно 1 часа. Добавляли дополнительное количество гидроксида палладия на углероде (20% Pd, влажн, 430 мг) и смесь повторно герметизировали под давлением водорода (100 фунт./кв. дюйм) и перемешивали в течение примерно более 1,5 часов. Смесь разбавляли EtOAc, фильтровали через целит, и фильтрат концентрировали с получением указанного в заголовке соединения в виде бесцветного масла (240 мг, 100%) в виде смеси цис- и транс-изомеров.
изомер 1: 1H ЯМР (400 МГц, CDCl3) δ: 4,52 (квин, 1 H), 2,63-2,74 (м, 2 H), 2,19-2,28 (м, 4 H), 2,07-2,17 (м, 1 H), 1,83 (шир.с, 1 H).
изомер 2: 1H ЯМР (400 МГц, CDCl3) δ: 4,14-4,25 (м, 1 H), 2,55-2,64 (м, 2 H), 2,48 (дд, 4 H), 1,83 (шир.с, 1 H), 1,67-1,79 (м, 1 H).
Получение 51
2-(3-Оксоциклобутил)ацетонитрил
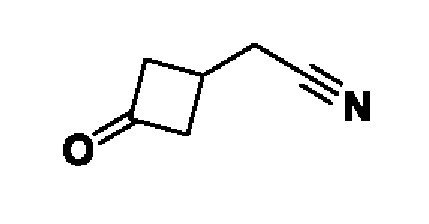
К раствору 2-(3-гидроксициклобутил)ацетонитрила (Получение 50, 50 мг, 0,45 ммоль) в сухом THF (1,8 мл) добавляли периодинан Десса-Мартина (CAS: 87413-09-0, 216 мг, 0,49 ммоль). Сосуд герметизировали в окружающей атмосфере и перемешивали при примерно 50°C в течение примерно 2 часов. Смесь разбавляли Et2O (10 мл), затем насыщенным водн. NaHCO3 (4 мл). Добавляли пентагидрат тиосульфата натрия (954 мг, 3,82 ммоль) и смесь энергично перемешивали в течение примерно 10 мин до полного растворения твердых веществ. Фазы разделяли и водную фазу экстрагировали еще раз Et2O. Объединенные экстракты Et2O сушили (MgSO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (27 мг, 55%).
1H ЯМР (400 МГц, CDCl3) δ: 3,23-3,37 (м, 2 H), 2,89-3,01 (м, 2 H), 2,75-2,89 (м, 1 H), 2,69 (д, 2 H).
13C ЯМР (101 МГц, CDCl3) δ: 203,6, 117,6, 52,3, 23,1, 20,6.
Получение 52
2-(3-(цианометил)циклобутилиден)ацетонитрил
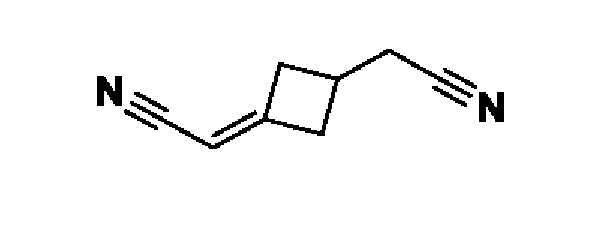
Указанное в заголовке соединение получали (75 мг, 89%) используя способ получения 35 с использованием 2-(3-оксоциклобутил)ацетонитрила (Получение 51, 70 мг, 0,64 ммоль), (EtO)2POCH2CN (213 мг, 1,15 ммоль), LiBr (100 мг, 1,15 ммоль) и TEA (0,27 мл, 1,92 ммоль). Указанное в заголовке соединение использовали непосредственно в следующей реакции (Пример 11 и 12).
1H ЯМР (400 МГц, CDCl3) δ: 5,25 (м, 1 H), 3,10-3,30 (м, 2 H), 2,80 (м, 3 H), 2,60 (м, 2 H).
GCMS m/z=132 [M]+
Получение 53
6-(1-метил-1H-пиразол-4-ил)-4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразин
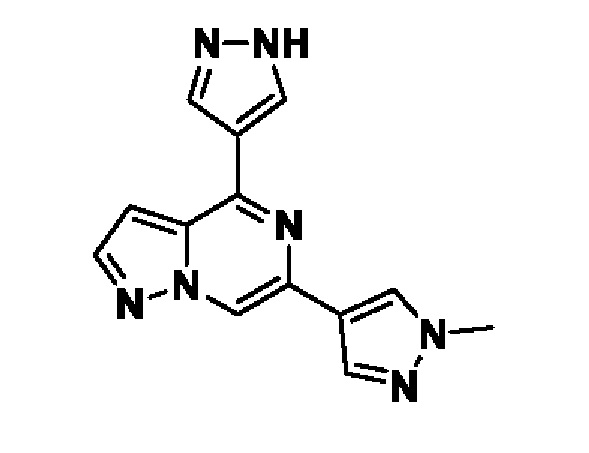
Pd(dppf)Cl2 (5,01 г, 6,85 ммоль) добавляли к смеси 4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 11, 8 г, 34 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-карбоксилата (12,1 г, 41,1 ммоль) и 2 M водн. K2CO3 (34,2 мл) в 1,4-диоксане (30 мл) и толуоле (30 мл) при примерно 10°C, в то время как поток азота барботировали через раствор. Смесь перемешивали при примерно 100°C в течение примерно 16 часов, затем выдерживали при примерно 10°C в течение примерно 48 часов. Смесь фильтровали и фильтрат концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде серого масла (6,3 г, 69%).
1H ЯМР (400 МГц, DMSO-d6) δ: 8,97 (с, 1 H), 8,72 (с, 1 H), 8,40 (с, 1 H), 8,35 (с, 1 H), 8,15 (д, 1 H), 8,12 (с, 1 H), 7,36 (д, 1 H), 3,91 (с, 3 H).
LCMS m/z=265,8 [MH]+
Пример 11
2,2'-((1s,3s)-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1,3-диил)диацетонитрил
(цис-изомер)
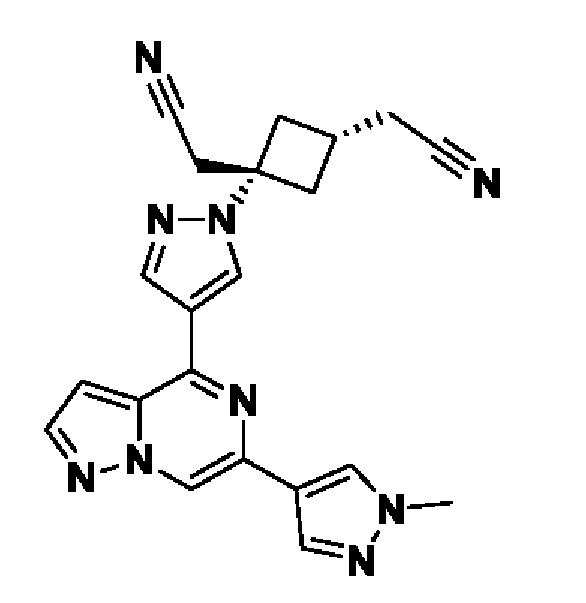
и
Пример 12
2,2'-((1r,3r)-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1,3-диил)диацетонитрил
(транс-изомер)
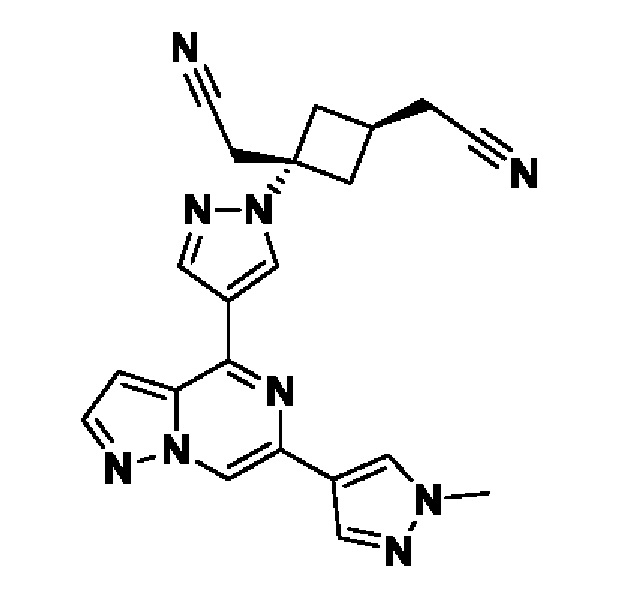
DBU (34 мг, 0,22 ммоль) добавляли к суспензии 2-(3-(цианометил)циклобутилиден)ацетонитрила (Получение 52, 32 мг, 0,24 ммоль) и 6-(1-метил-1H-пиразол-4-ил)-4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 53, 53 мг, 0,20 ммоль) в MeCN (4,8 мл). Смесь нагревали при примерно 50°C в течение примерно 16 часов, затем концентрировали. Остаток очищали с помощью хроматографии с получением смеси указанных в заголовке соединений 1:1 смеси цис/транс-изомеров (80 мг, 83%). Изомеры разделяли с помощью ВЭЖХ с получением 2,2'-((1s,3s)-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1,3-диил)диацетонитрила в виде твердого вещества (цис-изомер, 27 мг, 36%).
1H ЯМР (500 МГц, 9:1 CDCl3-CD3OD) δ: 8,45 (с, 1 H), 8,35 (с, 1 H), 8,25 (с, 1 H), 7,99 (д, 1 H), 7,95 (с, 1 H), 7,92 (с, 1 H), 6,93 (с, 1 H), 3,95 (с, 3 H), 3,72 (с, 2 H), 3,18 (с, 2 H), 2,81-2,91 (м, 2 H), 2,72-2,81 (м, 1 H), 2,72-2,81 (м, 1 H), 2,64-2,72 (м, 2 H), 2,61 (д, 2 H).
LCMS m/z=399,1 [MH] +
и 2,2'-((1r,3r)-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1,3-диил)диацетонитрила в виде твердого вещества (транс-изомер, 14 мг, 18%).
1H ЯМР (500 МГц, CDCl3) δ: 8,49 (с, 1 H), 8,46 (с, 1 H), 8,32 (с, 1 H), 8,05 (д, 1 H), 7,96 (м, 2 H), 6,95 (д, 1 H), 4,00 (с, 3 H), 3,75 (шир.с, 1 H), 3,15-3,25 (м, 2 H), 3,11 (с, 2 H), 2,88 (тт, 1 H), 2,64 (д, 2 H), 2,48-2,59 (м, 2 H).
LCMS m/z=399,1 [MH] +
Получение 54
2-(3-(Бензилокси)циклобутилиден)ацетонитрил
К раствору (EtO)2P(O)CH2CN (24,1 г, 136 ммоль) в сухом THF (500 мл) добавляли LiBr (11,8 г, 136 ммоль) и TEA (18,4 г, 182 ммоль) и полученную смесь перемешивали при примерно 20°C в течение примерно 1 часа. К этому добавляли 3-(бензилокси)циклобутан-1-он (16,00 г, 90,8 ммоль). Смесь перемешивали при примерно 20°C в течение примерно 20 часов. Смесь фильтровали и фильтрат концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бледно-желтой жидкости (18 г, 99%).
1H ЯМР (400 МГц, CDCl3) δ: 7,29-7,40 (м, 5 H), 5,24 (дт, 1 H), 4,45-4,53 (м, 2 H), 4,19 (квин, 1 H), 3,19-3,29 (м, 1 H), 3,03-3,13 (м, 1 H), 2,86-3,00 (м, 2 H).
LCMS m/z=200,1 [MH]+
Получение 55
2-(3-(Бензилокси)-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил
(смесь цис- и транс-изомеров)
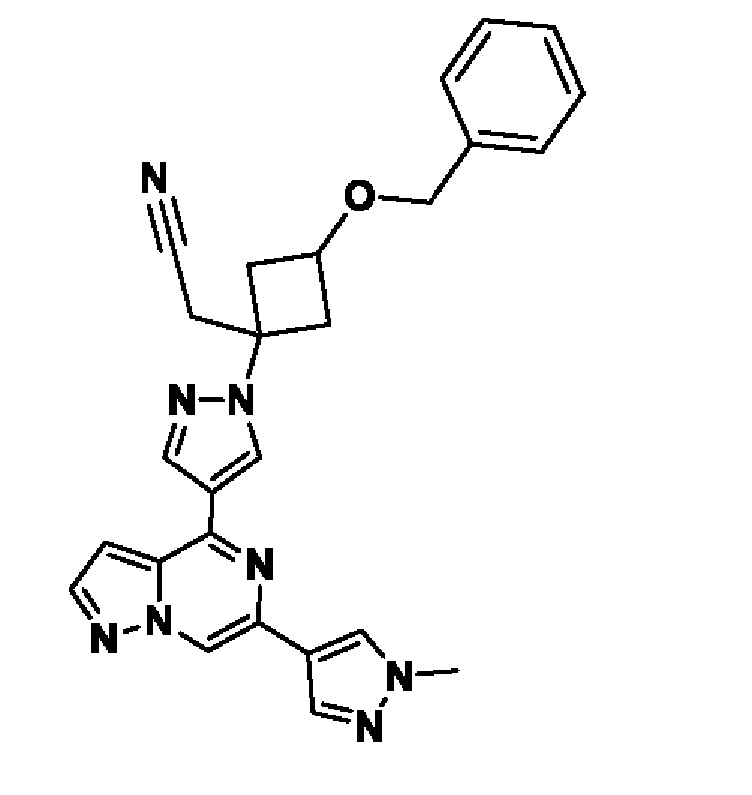
DBU (1,448 г, 9,4 ммоль) добавляли к раствору 6-(1-метил-1H-пиразол-4-ил)-4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 53, 313 мг, 1,18 ммоль) и 2-(3-(бензилокси)циклобутилиден)ацетонитрила (Получение 54, 470 мг, 2,36 ммоль) в MeCN (6,38 мл). Смесь перемешивали при примерно 20°C в течение примерно 18 часов, затем смесь выливали в водн. раствор NaH2PO4 для поддержания рН около 5. Смесь экстрагировали три раза DCM. Объединенные экстракты DCM концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (474 мг, 86%).
1H ЯМР (400 МГц, CD3OD) δ: 8,67-8,72 (м, 2 H), 8,63 (с, 1 H), 8,39 (с, 1 H), 8,22 (с, 1 H), 8,04-8,10 (м, 2 H), 7,25-7,42 (м, 5 H), 4,53 (с, 2 H), 4,24-4,34 (м, 1 H), 3,97 (с, 3 H), 3,29 (с, 2 H), 2,92-3,02 (м, 2 H), 2,77-2,88 (м, 2 H).
LCMS m/z=465,3 [MH]+
Пример 13
2-((1s,3r)-3-гидрокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил
(цис-изомер)
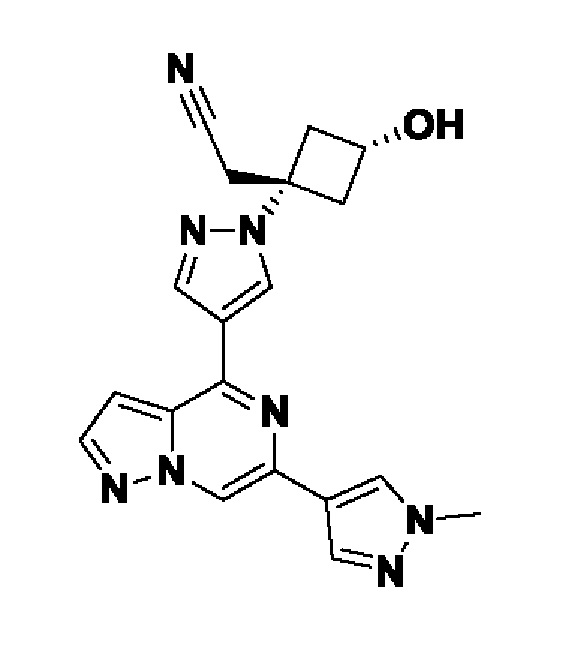
и
Пример 14 PF-06877900
2-((1r,3s)-3-гидрокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил
(транс-изомер)
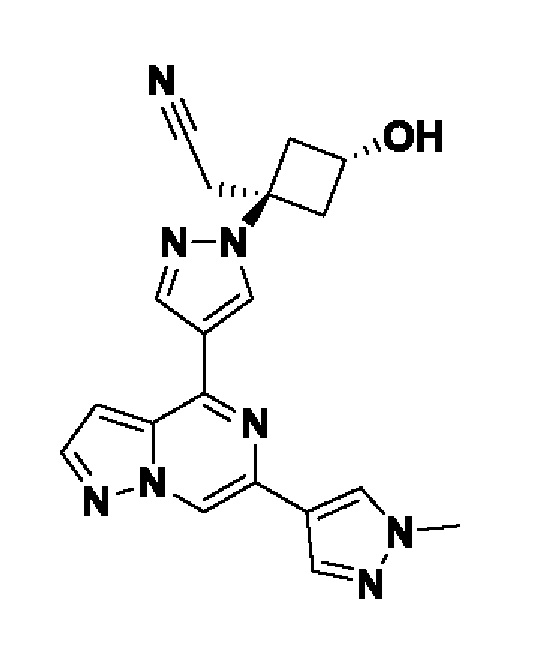
Иодид натрия сушили с помощью струйной воздушной сушилки при полном нагревании в течение 5 мин, затем охлаждли до примерно 25°С в атмосфере азота до использования. Высушенный NaI (1,21 г, 8,1 ммоль) добавляли при примерно 20°C к перемешиваемому раствору (2-(3-(бензилокси)-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (Получение 55, 377 мг, 0,81 ммоль) в MeCN (12 мл), затем TMSCl (1,05 мл, 8,1 ммоль). Смесь перемешивали при примерно 50°C в течение примерно 18 часов. Добавляли дополнительную порцию обоих NaI и TMSCl и смесь поддерживали при примерно 50°C в течение примерно 8 часов дополнительно. Охлажденную смесь выливали в ледяной насыщенный водн. NaHCO3, содержащий пентагидрат тиосульфата натрия (10,1 г, 40,6 ммоль) и три раза экстрагировали EtOAc. Объединенные экстракты EtOAc концентрировали с получением смеси двух указанных в заголовке соединений. Данную смесь очищали с помощью хроматографии с получением 2-((1s,3r)-3-гидрокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (транс-изомер, 80 мг, 26%)
1H ЯМР (400 МГц, CD3OD) δ: 8,72 (с, 1 H), 8,71 (с, 1 H), 8,42 (с, 1 H), 8,24 (с, 1 H), 8,05-8,10 (м, 2 H), 7,24 (д, 1 H), 4,34-4,44 (м, 1 H), 3,98 (с, 3 H), 3,35 (с, 2 H), 3,22-3,28 (м, 2 H), 2,49 (дд, 2 H).
LCMS m/z=375,4 [MH]+
и 2-((1r,3s)-3-гидрокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (цис-изомер, 180 мг, 59%).
1H ЯМР (400 МГц, CD3OD) δ: 8,67 (с, 1 H), 8,62 (с, 1 H), 8,38 (с, 1 H), 8,21 (с, 1 H), 8,07 (д, 1 H), 8,05 (с, 1 H), 8,04-8,06 (м, 1 H), 7,21 (д, 1 H), 4,41 (квин, 1 H), 3,97 (с, 3 H), 3,28 (с, 2 H), 2,95-3,04 (м, 2 H), 2,68-2,78 (м, 2 H).
LCMS m/z=375,4 [MH]+
Пример 15
2-((1r,3s)-3-метокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил
(транс-изомер)
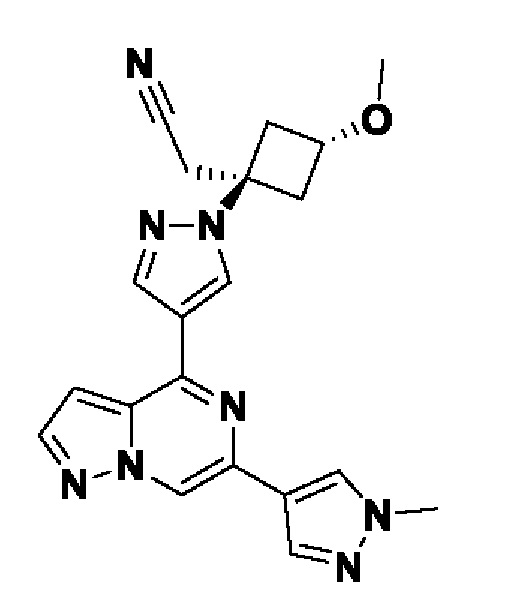
и
Пример 16
2-((1s,3r)-3-метокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил
(цис-изомер)
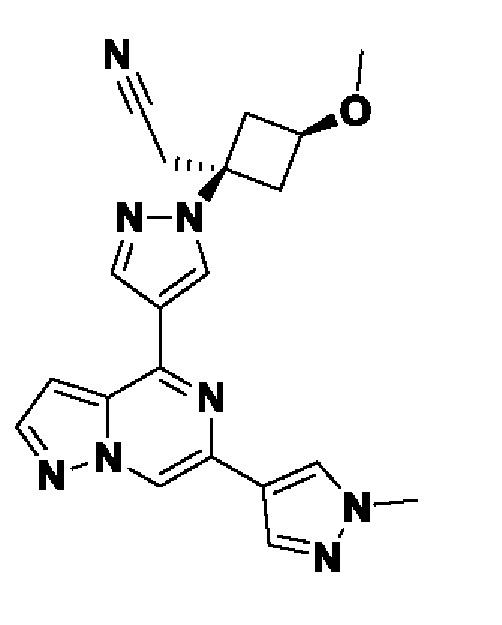
Раствор смеси 2-((1s,3r)-3-гидрокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила и 2-((1r,3s)-3-гидрокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (Примеры 13 и 14, смесь цис- и транс-изомеров, 48 мг, 0,13 ммоль) в THF (0,2 мл) последовательно обрабатывали тетрабутиламмоний бромидом (TBAB, 126 мг, 0,38 ммоль), 1 M водн. NaOH (1,02 мл) и диметилсульфатом (6 мкл). Реакционный сосуд запечатывали и энергично перемешивали при примерно 20°C в течение примерно 1 часа. Добавляли дополнительное количество диметилсульфата (14 мкл) и TBAB (10 мг) и реакционную смесь выдерживали при примерно 20°C в течение примерно 2 часов. Смесь экстрагировали три раза EtOAc и объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением смеси цис- и транс-изомеров. Дальнейшая очистка с помощью ВЭЖХ давала 2-((1r,3s)-3-метокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил в виде твердого вещества (транс-изомер, 10 мг, 20%)
1H ЯМР (400 МГц, CD3CN) δ: 8,61 (д, 1 H), 8,55 (с, 1 H), 8,38 (с, 1 H), 8,12 (с, 1 H), 8,05 (д, 1 H), 7,99 (с, 1 H), 7,13 (д, 1 H), 3,99-4,08 (м, 1 H), 3,93 (с, 3 H), 3,27 (с, 5 H), 3,13-3,21 (м, 2 H), 2,45-2,53 (м, 2 H).
LCMS m/z=389,1 [MH]+
и 2-((1s,3r)-3-метокси-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрил в виде твердого вещества (цис-изомер, 17 мг, 33%).
1H ЯМР (400 МГц, CD3CN) δ: 8,60 (д, 1 H), 8,49 (с, 1 H), 8,36 (с, 1 H), 8,12 (с, 1 H), 8,04 (д, 1 H), 7,98 (с, 1 H), 7,12 (дд, 1 H), 4,03 (квин, 1 H), 3,92 (с, 3 H), 3,26 (с, 3 H), 3,19 (с, 2 H), 2,89-2,97 (м, 2 H), 2,65-2,74 (м, 2 H).
LCMS m/z=389,1 [MH]+
Получение 56
Бензил (R)-2-метил-3-оксоазетидин-1-карбоксилат
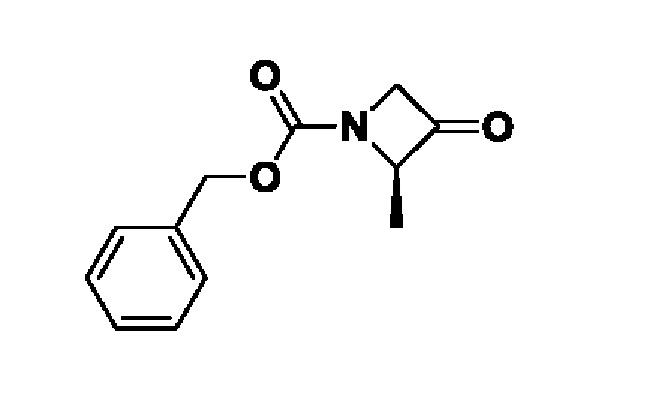
Часть 1
Раствор N,4-диметил-N-нитрозобензолсульфонамида (Diazald®, 21,25 г, 99,19 ммоль) в Et2O (150 мл) добавляли по каплям к раствору KOH (6 г, 106,9 ммоль) и 2-(2-этоксиэтокси)этанола (30 мл) и воды (10 мл) при примерно 70°C. Смесь нагревали при примерно 70°C и эфирный раствор диазометана собирали в виде желтой жидкости путем дистилляции (100 мл, по оценкам, содержит 66 ммоль диазометана) с использованием конденсатора сухой лед/ацетон и сразу же использовали в части 2.
Часть 2
Этилхлорформиат (2,430 г, 22,4 ммоль) добавляли по каплям к раствору Cbz-D-аланина (5,00 г, 22,4 ммоль) и TEA (2,27 мг, 22,4 ммоль) в THF (50 мл) при примерно -15°C. Реакционную смесь нагревали до примерно 0°C и раствор диазометана из части 1 (100 мл, примерно 66 ммоль) добавляли по каплям и перемешивали при примерно 20°C в течение примерно 16 часов. Реакционную смесь гасили водой (10 мл) и экстрагировали EtOAc (2×30 мл). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением диазокетонового промежуточного соединения (4,5 г, 81%) в виде белого твердого вещества. Вещество использовали в части 3.
Часть 3
К раствору диазокетонового промежуточного соединения части 2 (4,50 г, 18,2 ммоль) в DCM (450 мл) добавляли TEA (18 мг, 0,18 ммоль) и Rh2(OAc)4 (40 мг, 0,091 ммоль) при примерно 0°C. Смесь перемешивали в течение примерно 16 часов при примерно 25°C. Смесь гасили водой (50 мл) и фазу DCM отделяли и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде белого твердого вещества (1,20 г, 30%).
1H ЯМР (400 МГц, CDCl3) δ: 7,34-7,38 (м, 5 H), 5,18 (м, 2 H), 5,03 (м, 1 H), 4,65-4,81 (м, 2 H), 1,49 (д, 3 H).
GCMS m/z=219 [M]+
Получение 57
Бензил (R)-3-(цианометилен)-2-метилазетидин-1-карбоксилат
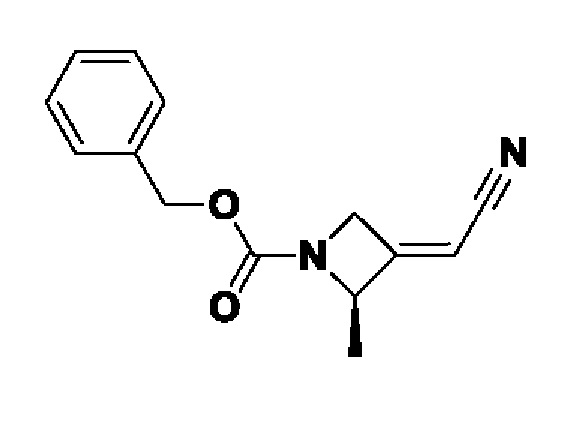
К смеси LiBr (166 мг, 1,92 ммоль) и TEA (277 мг, 2,74 ммоль) в THF (10 мл) добавляли (EtO)2P(O)CH2CN (339 мг, 1,92 ммоль) при примерно 25°C. Примерно через 2,5 часа, добавляли бензил (R)-2-метил-3-оксоазетидин-1-карбоксилат (Получение 56, 300 мг, 1,37 ммоль) в THF (2 мл) при примерно 25°C и смесь перемешивали в течение примерно 16 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде смеси олефиновых изомеров E/Z в виде бесцветного масла (290 мг, 87%).
1H ЯМР (400 МГц, CDCl3) δ: 7,33-7,40 (м, 5 H), 5,36 (м, 1 H), 5,10-5,17 (м, 2 H), 4,97 (м, 1 H), 4,63-4,77 (м, 2 H), 1,50-1,67 (м, 3 H).
Получение 58
Бензил (2R)-3-(цианометил)-2-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1-карбоксилат
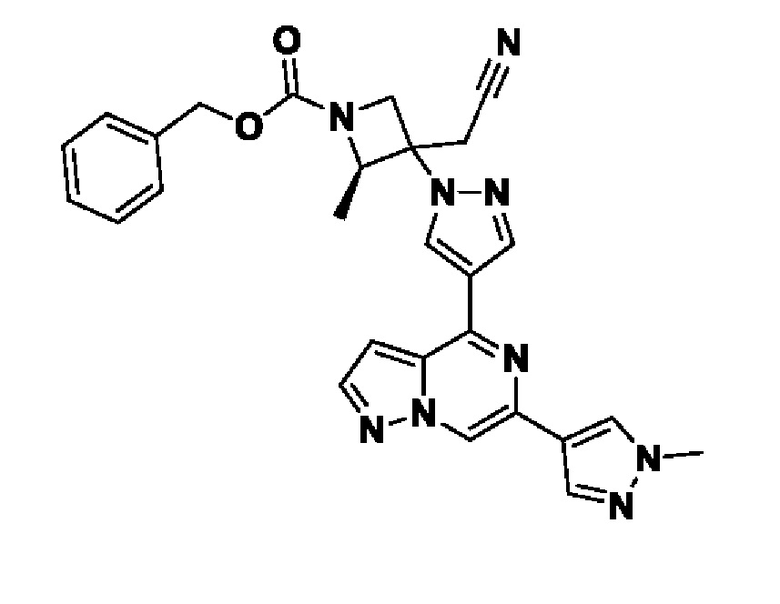
К раствору 6-(1-метил-1H-пиразол-4-ил)-4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 53, 268 мг, 1,01 ммоль) в MeCN (15 мл) добавляли бензил (R)-3-(цианометилен)-2-метилазетидин-1-карбоксилат (Получение 57, 294 мг, 1,21 ммоль) и DBU (77 мг, 0,51 ммоль) при примерно 15°C. Смесь перемешивали при примерно 25°C в течение приблизительно 7 часов, до разбавления EtOAc (50 мл) и промывали 1 M водн. лимонной кислотой (15 мл), затем насыщенным солевым раствором (15 мл). Экстракт EtOAc концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтой камеди (500 мг, 97%).
1H ЯМР (400 МГц, CDCl3) δ: 8,49 (с, 1 H), 8,33 (с, 1 H), 8,32 (с, 1 H), 8,05 (д, 1 H), 7,96 (с, 1 H), 7,95 (с, 1 H), 7,33-7,40 (м, 5 H), 6,93 (д, 2 H), 5,15 (с, 2 H), 4,87 (м, 1 H), 4,61 (д, 1 H), 4,26 (д, 1 H), 4,01 (с, 3 H), 3,27 (с, 2 H), 1,69 (д, 3 H).
LCMS m/z=530,1 [M+Na]+
Получение 59
2-((2R)-2-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил
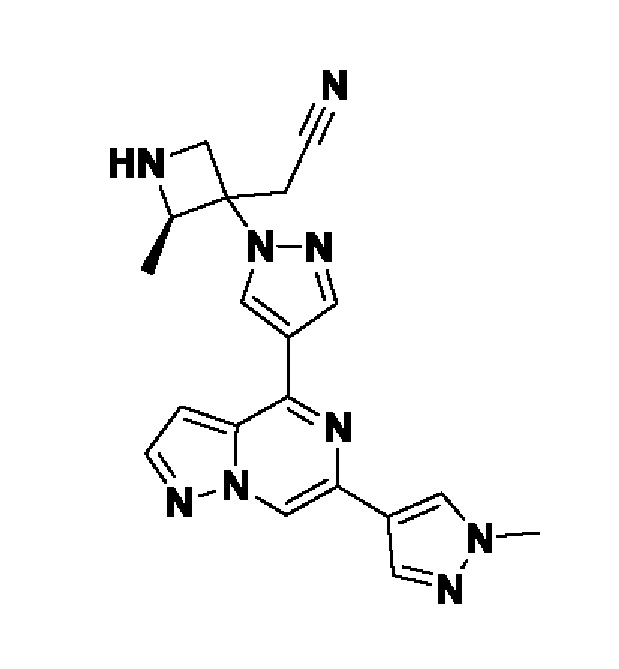
К смеси NaI (2,36 г, 15,8 ммоль) в MeCN (16 мл) добавляли TMSCl (2 мл, 15,8 ммоль) при примерно 0°C. Смесь перемешивали при примерно 15°C в течение примерно 4 часов. Раствор бензил (2R)-3-(цианометил)-2-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1-карбоксилата (Получение 58, 400 мг, 0,79 ммоль) в MeCN (4 мл) добавляли при примерно 0°C. Перемешивание продолжали при примерно 15°C в течение примерно 3 часов. Смесь охлаждали до примерно 10°C и гасили добавлением TEA (2 мл). Смесь концентрировали. Остаток растворяли в EtOAc (20 мл) и MeOH (2 мл) и присутствующие твердые вещества удаляли фильтрованием. Фильтрат концентрировали и остаток очищали с помощью ТСХ с получением указанного в заголовке соединения в виде желтого твердого вещества (200 мг, 68%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,06 (с, 1 H), 9,03 (с, 1 H), 8,59 (с, 1 H), 8,39 (с, 1 H), 8,22 (д, 1 H), 8,16 (с, 1 H), 7,47 (с, 1 H), 5,09 (м, 1 H), 4,74 (д, 1 H), 4,24 (д, 1 H), 3,91 (с, 3 H), 2,99 (с, 2 H), 1,65 (д, 3 H).
LCMS m/z=374,0 [MH]+
Пример 17
2-((2R,3S)-2-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-1-(2,2,2-трифторэтил)азетидин-3-ил)ацетонитрил
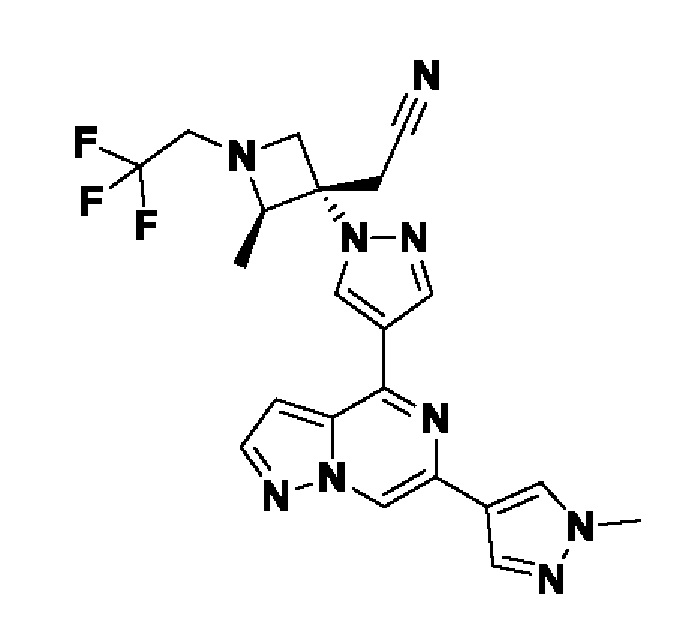
К раствору 2-((2R)-2-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила (Получение 59, 200 мг, 0,43 ммоль) в DMF (5 мл) добавляли 2,2,2-трифторэтил трифторметансульфонат (298 мг, 1,29 ммоль) и DIPEA (332 мг, 2,57 ммоль). Смесь перемешивали при примерно 10°C в течение примерно 36 часов, прежде чем разбавить EtOAc (30 мл) и промывали насыщенным солевым раствором (15 мл). Экстракт EtOAc концентрировали, остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде смеси диастереомеров (80 мг, 41%) в виде белого твердого вещества. Дальнейшая очистка ВЭЖХ дает указанное в заголовке соединение в виде белого твердого вещества (41,8 мг, 21%, 94,7% э.и.).
1H ЯМР (400 МГц, CD3CN) δ: 8,59 (с, 1 H), 8,45 (с, 1 H), 8,36 (с, 1 H), 8,10 (с, 1 H), 8,04 (д, 1 H), 7,98 (с, 1 H), 7,09-7,13 (м, 1 H), 3,92-3,96 (м, 1 H), 3,91 (с, 3 H), 3,84-3,89 (м, 1 H), 3,77-3,84 (м, 1 H), 3,38 (с, 2 H), 3,28-3,43 (м, 1 H), 3,15 (дкв, 1 H), 1,36 (д, 3 H).
LCMS m/z=456,2 [MH]+
Пример 18
2-((1r,3r)-1-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(1H-пиразол-5-ил)циклобутил)ацетонитрил
К раствору LiBr (70 мг, 0,81 ммоль), TEA (205 мкл, 1,47 ммоль) в THF (10 мл), добавляли (EtO)2P(O)CH2CN (143 мг, 0,81 ммоль). Смесь перемешивали при примерно 20°C в течение примерно 30 мин. Добавляли 3-(1H-пиразол-5-ил)циклобутан-1-он (Получение 99, 100 мг, 0,74 ммоль) и смесь выдерживали при примерно 20°C в течение примерно 18 часов. Около 30% реакционного раствора извлекали и добавляли 6-(1-метил-1H-пиразол-4-ил)-4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразин (Получение 53, 58 мг, 0,22 ммоль) и DBU (110 мкл, 0,73 ммоль) в MeCN (5 мл). Перемешивание при примерно 20°C поддерживали в течение примерно 20 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (6 мг, 6%).
1H ЯМР (500 МГц, CD3OD) δ: 8,85 (с, 1 H), 8,73 (с, 1 H), 8,46 (с, 1 H), 8,26 (с, 1 H), 8,10 (с, 2 H), 7,60 (шир.с, 1 H), 7,28 (д, 1 H), 6,32 (д, 1 H), 3,95-4,04 (м, 3 H), 3,69-3,78 (м, 1 H), 3,33-3,43 (м, 2 H), 2,77-2,89 (м, 2 H).
LCMS m/z=425,4 [MH]+
Пример 19
(1s,3s)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
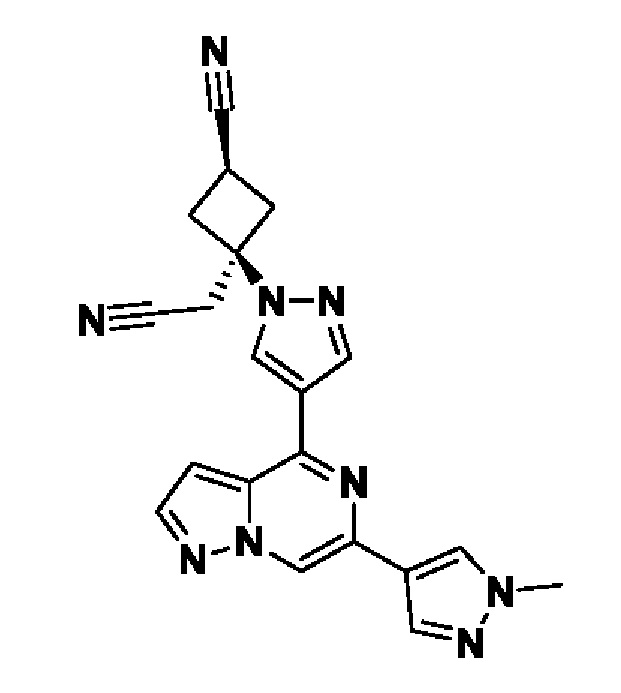
К раствору (1s,3s)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 91, 539 мг, 1,72 ммоль) и 4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 11, 350 мг, 1,5 ммоль) в 1,4-диоксане (7,5 мл) добавляли 2 M водн. K3PO4 (2,25 мл) при примерно 25°C. Смесь помещали под азот, затем добавляли XPhos Pd G2 (11,8 мг, 0,0150 ммоль). Смесь нагревали при примерно 40°C в течение примерно 6 ч, затем нагревали до примерно 80°C для растворения осажденного вещества. Водный слой удаляли, поддерживая температуру при примерно 80°C, затем к EtOH добавляли 1,4-диоксановую фазу (70 мл, заранее предварительно нагретую до примерно 50°C). Смесь перемешивали в течение примерно 10 мин при примерно 50°C перед тем как прекратить нагревание. Перемешивание при примерно 25°C продолжали в течение примерно 18 ч. Твердое вещество отфильтровали и промывали EtOH (2×25 мл) и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (483 мг, 84%).
Т.п. 217-220°C
1H ЯМР (400 МГц, DMSO-d6) δ: 9,02 (с, 1H), 8,87 (с, 1H), 8,51 (с, 1H), 8,37 (с, 1H), 8,20 (с, 1H), 8,16 (с, 1H), 7,45 (с, 1H), 3,92 (с, 3H), 3,65-3,59 (м, 1H), 3,57 (с, 2H), 3,26-3,16 (м, 2H), 2,88 (м, 2H).
13C ЯМР (101 МГц, DMSO-d6) δ: 145,21, 142,50, 140,09, 137,15, 133,80, 131,39, 129,54, 129,24, 121,94, 121,03, 120,33, 117,69, 114,55, 99,94, 59,62, 39,28, 36,90, 27,42, 14,64.
LCMS m/z=384,2 [MH]+
Пример 20
(1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
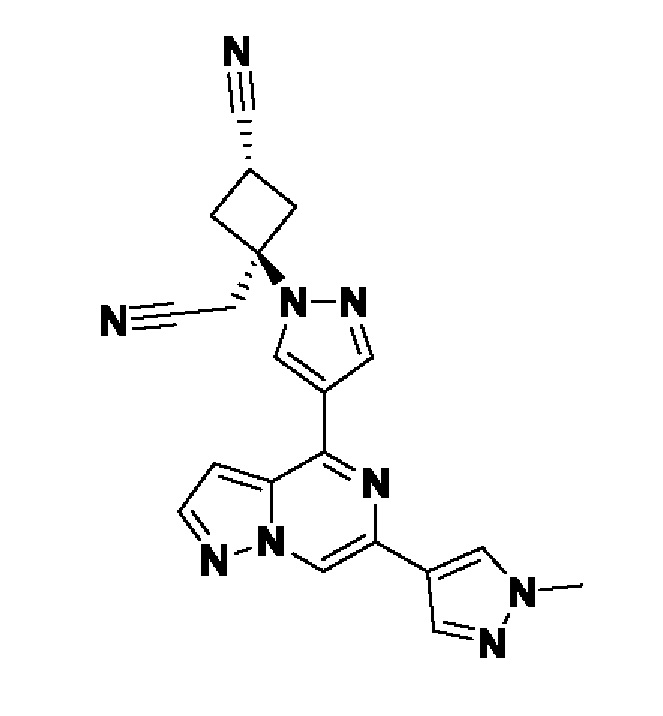
К раствору (1r,3r)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 91, 3,38 г, 10,8 ммоль) и 4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 11, 2,20 г, 9,4 ммоль) в 1,4-диоксане (47,1 мл) добавляли 2 M водн. K3PO4 (14,1 мл). Через смесь барботировали азот в течение примерно 5 мин при примерно 25°C, затем добавляли XPhos Pd G2 (37,0 мг, 0,047 ммоль). Смесь нагревали при примерно 40°C в течение примерно 18 ч, затем смесь нагревали до примерно 80°C для растворения осажденного вещества. Водный слой удаляли, поддерживая температуру при примерно 80°C, затем к EtOH добавляли 1,4-диоксановую фазу (471 мл, заранее предварительно нагретую до примерно 50°C). Смесь перемешивали в течение примерно 10 мин при примерно 50°C перед тем как прекратить нагревание. Перемешивание при примерно 25°C продолжали в течение примерно 6 ч. Твердое вещество фильтровали и промывали EtOH (2×25 мл), водой (2×50 мл) и EtOH (2×25 мл). Осадок сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (3,61 г, 74%).
Указанное в заголовке соединение (500 мг, 1,30 ммоль) нагревали в 1,4-диоксане (6,5 мл) при примерно 80°C пока все вещество не растворится. Добавляли 1,2-бис(дифенилфосфино)этан (7,8 мг, 0,019 ммоль) и нагревание при примерно 80°C продолжали в течение примерно 4 часов, затем к EtOH добавляли 1,4-диоксановую фазу (58,7 мл, заранее предварительно нагретую до примерно 50°C). Дополнительные 6,5 мл предварительно нагретого EtOH использовали для промывки реакционного сосуда. Смесь убирали с нагревания. Перемешивание при примерно 25°C продолжали в течение примерно 18 ч. Твердое вещество фильтровали и промывали EtOH (2×5 мл), водой (5 мл), затем EtOH (3×5 мл). Осадок сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (450 мг, 90%).
Температура плавления 213-215°C
1H ЯМР (400 МГц, DMSO-d6) δ: 9,02 (с, 1 H) 8,92 (с, 1 H) 8,52 (с, 1 H) 8,37 (с, 1 H) 8,19 (с, 1 H) 8,16 (с, 1 H) 7,45 (с, 1 H) 3,91 (с, 3 H) 3,55-3,65 (м, 1 H) 3,52 (с, 2 H) 3,22-3,38 (м, 2 H) 2,85-3,00 (м, 2 H).
13C ЯМР (126 МГц, DMSO-d6) δ: 145,17, 142,46, 140,20, 137,13, 133,78, 131,38, 129,82, 129,51, 122,28, 121,13, 120,33, 117,30, 114,54, 99,91, 61,51, 39,26, 36,26, 29,65, 16,05.
LCMS m/z=384,1 [MH]+.
Получение 60
(1r,3r)-3-(цианометил)-3-(3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
(транс-изомер)
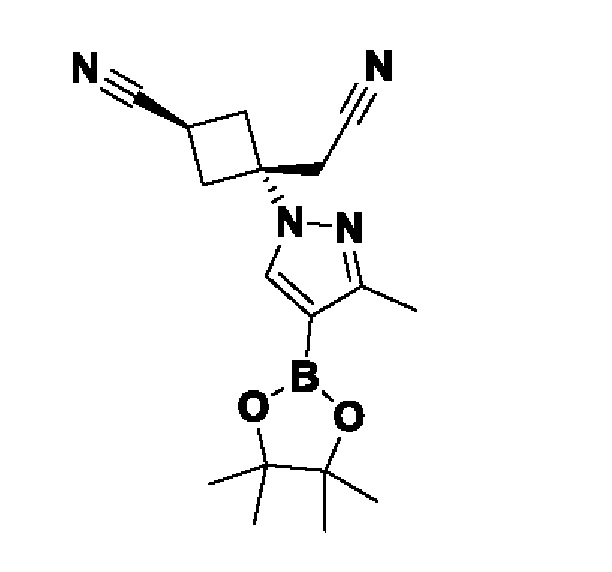
и
(1s,3s)-3-(цианометил)-3-(3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
(цис-изомер)
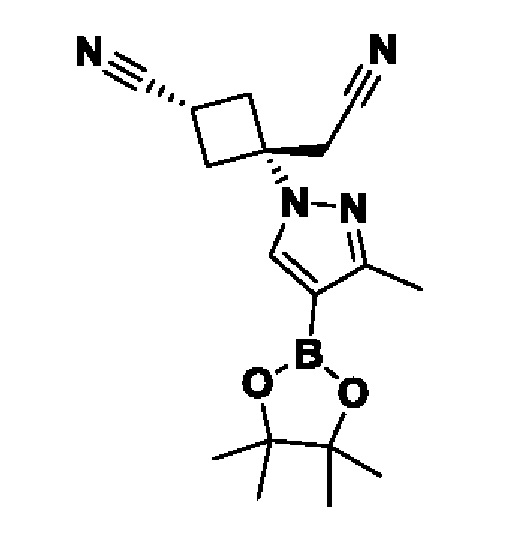
К раствору 3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (600 мг, 2,88 ммоль) и 3-(цианометилен)циклобутан-1-карбонитрила (Получение 27, 341 мг, 2,88 ммоль) в MeCN (28,8 мл) добавляли DBU (439 мг, 2,88 ммоль) при примерно 20°C. Через примерно 18 часов при примерно 20°C, смесь выливали в EtOAc и 10% водн. K2HPO4. EtOAc отделяли и водную фазу экстрагировали дважды EtOAc. Объединенные экстракты EtOAc промывали насыщенным солевым раствором, сушили (Na2SO4) и концентрировали. Остаток очищали с помощью колоночной хроматографии с получением (1r,3r)-3-(цианометил)-3-(3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (транс-изомер, 325 мг, 35%)
1H ЯМР (400 МГц, CDCl3) δ: 7,79 (с, 1 H), 3,17-3,28 (м, 3 H), 3,16 (с, 2 H), 2,81-2,89 (м, 2 H), 2,39 (с, 3 H), 1,32 (с, 12 H).
LCMS m/z=327,2 [MH]+
и (1s,3s)-3-(цианометил)-3-(3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (цис-изомер, 171 мг, 18%).
1H ЯМР (400 МГц, CDCl3) δ: 7,75 (с, 1 H), 3,18-3,28 (м, 1 H), 3,08-3,18 (м, 2 H), 3,05 (с, 2 H), 2,93-3,02 (м, 2 H), 2,39 (с, 3 H), 1,32 (с, 12 H).
LCMS m/z=327,2 [MH]+
Пример 21
(1r,3r)-3-(цианометил)-3-(3-метил-4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
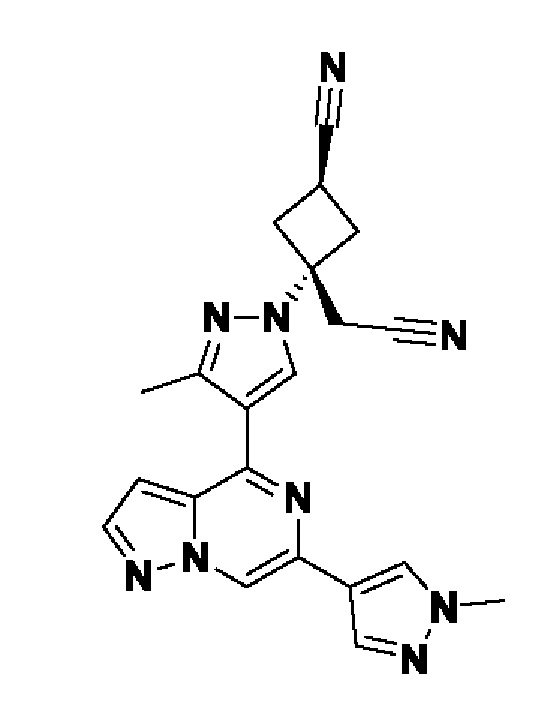
К раствору (1r,3r)-3-(цианометил)-3-(3-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 60, транс-изомер, 209 мг, 0,64 ммоль) в 1,4-диоксане (4,3 мл) добавляли 4-хлор-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин (Получение 11, 150 мг, 0,64 ммоль) и 2 M водн. K3PO4 (0,96 мл). Смесь продували азотом в течение примерно 5 мин, затем добавляли XPhos Pd G2 (101 мг, 0,13 ммоль). Смесь нагревали при примерно 40°C в течение примерно 2 часов, затем концентрировали. Остаток растворяли в EtOAc и EtOAc промывали водой. Водную фазу экстрагировали дважды EtOAc, затем один раз DCM. Объединенные экстракты EtOAc и DCM сушили (Na2SO4), концентрировали и остаток очищали с помощью хроматографии с получением твердого вещества, которое перекристаллизовывали из MeCN с получением указанного в заголовке соединения (120 мг, 47%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,00 (с, 1 H), 8,74 (с, 1 H), 8,26 (с, 1 H), 8,18 (с, 1 H), 8,08 (с, 1 H), 7,32 (с, 1 H), 3,91 (с, 3 H), 3,55 (квин, 1 H), 3,48 (с, 2 H), 3,22-3,30 (м, 2 H), 2,84-2,93 (м, 2 H), 2,63 (с, 3 H).
LCMS m/z=398,3 [MH]+
Получение 61
4-бром-1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразол
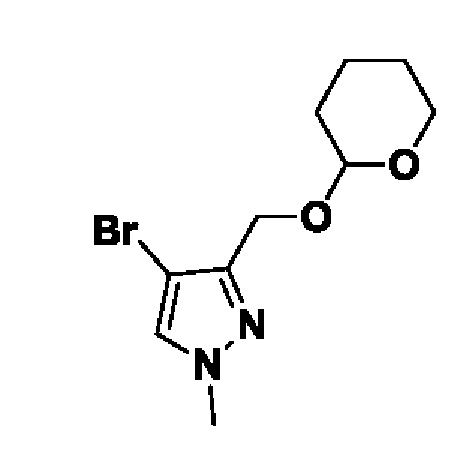
К раствору 4-бром-1-метил-1H-пиразол-3-метанола (720 мг, 3,77 ммоль) в THF (30 мл) добавляли DHP (951 мг, 11,3 ммоль) и PTSA (14 мг, 0,075 ммоль). Раствор перемешивали при примерно 50°C в течение примерно 16 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (1,0 г, 84%).
1H ЯМР (400 МГц, CDCl3) δ: 7,37 (с, 1 H), 4,81 (т, 1 H), 4,74 (д, 1 H), 4,46 (д, 1 H), 3,96-4,03 (м, 1 H), 3,88 (с, 3 H), 3,56-3,63 (м, 1 H), 1,80-1,93 (м, 1 H), 1,59-1,78 (м, 3 H), 1,48-1,57 (м, 2 H).
LCMS m/z=190,7 [MH-THP]+
Получение 62
(1s,3s)-3-(цианометил)-3-(4-(6-(1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
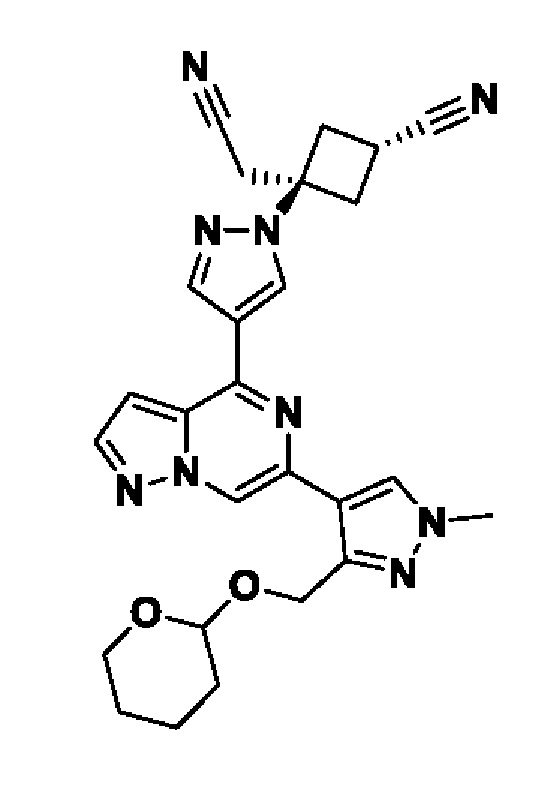
Часть 1
К раствору 4-бром-1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразола (Получение 61, 200 мг, 0,73 ммоль) в 1,4-диоксане (10 мл) добавляли KOAc (313 мг, 3,19 ммоль), и бис(пинаколато)диборон (405 мг, 1,59 ммоль). Смесь продували азотом в течение примерно 5 мин до добавления Pd(dppf)Cl2 (78 мг, 0,11 ммоль). Смесь нагревали при примерно 90°C в течение примерно 18 часов. Смесь концентрировали с получением неочищенного образца 1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола в виде черного масла (234 мг), который использовали в части 2 ниже без дополнительной очистки.
Часть 2
Смесь (1r,3r)-3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила (Получение 75, 85 мг, 0,25 ммоль), 1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (часть 1, 81 мг, 0,25 ммоль), и 2 M водн. K3PO4 (1,0 мл) в 1,4-диоксане (3,0 мл) продували аргоном в течение примерно 2 мин, после чего добавляли XPhos Pd G2 (39 мг, 0,050 ммоль). Реакционную смесь нагревали при примерно 45°C в течение примерно 45 мин. 1,4-Диоксановую фазу отделяли от водной фазы, которую дополнительно экстрагировали EtOAc (5 мл). Объединенные экстракты 1,4-диоксана и EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (50 мг, 40%).
1H ЯМР (400 МГц, CDCl3) δ: 8,92 (с, 1 H), 8,45 (с, 1 H), 8,33 (с, 1 H), 8,07-8,10 (м, 1 H), 8,00 (с, 1 H), 6,96 (д, 1 H), 5,02 (д, 1 H), 4,87-4,91 (м, 1 H), 4,70 (м, 1 H), 4,59 (д, 1 H), 4,51 (д, 1 H), 3,99 (с, 3 H), 3,51-3,62 (м, 2 H), 3,34-3,44 (м, 1 H), 3,29 (с, 2 H), 2,99 (м, 2 H), 1,80-1,96 (м, 3 H), 1,68-1,80 (м, 3 H).
LCMS m/z=498,3 [MH]+
Пример 22
(1r,3r)-3-(Цианометил)-3-(4-(6-(3-(гидроксиметил)-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
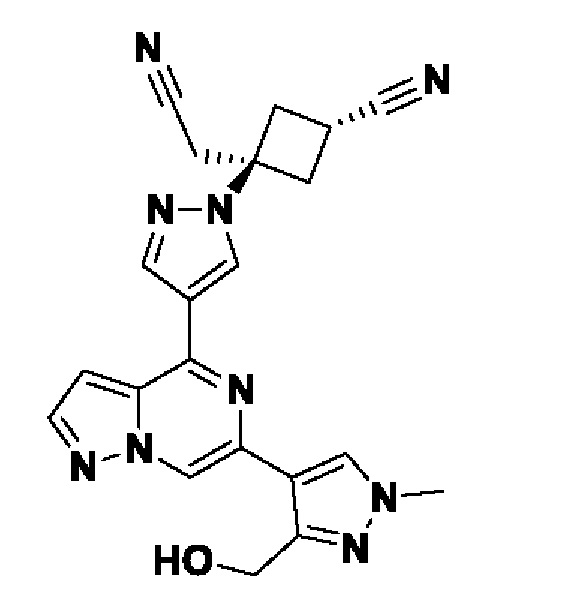
PTSA (10 мг, 0,052 ммоль) добавляли к раствору (1s,3s)-3-(цианометил)-3-(4-(6-(1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 62, 50 мг, 0,10 ммоль) в MeOH (10 мл). Реакционную смесь выдерживали при примерно 20°C в течение примерно 18 часов. Осажденное твердое вещество фильтровали с получением указанного в заголовке соединения (20 мг, 48%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,00 (с, 1 H), 8,91 (с, 1 H), 8,48 (с, 1 H), 8,35 (с, 1 H), 8,22 (д, 1 H), 7,47 (д, 1 H), 5,41 (шир.с, 1 H), 4,66 (с, 2 H), 3,88 (с, 3 H), 3,54-3,62 (м, 1 H), 3,52 (с, 2 H), 3,28-3,34 (м, 2 H), 2,90-2,98 (м, 2 H).
LCMS m/z=414,4 [MH]+
Получение 63
(1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-3-нитро-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
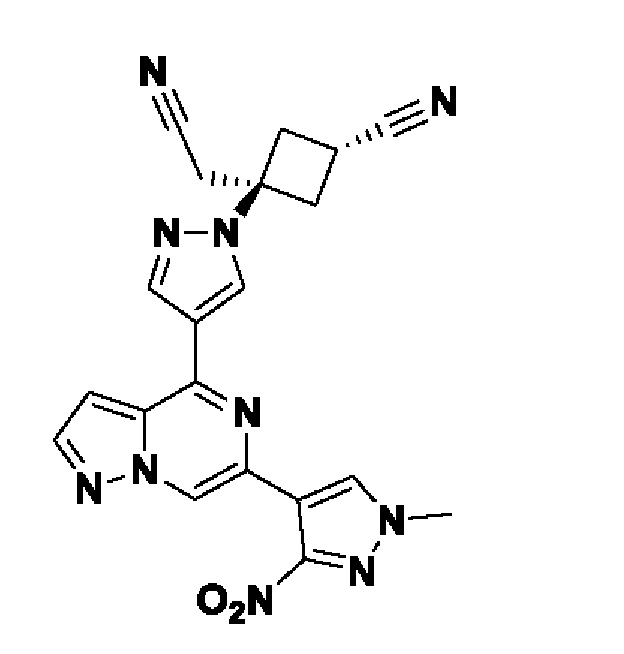
Часть 1
К раствору 4-бром-1-метил-3-нитро-1H-пиразола (200 мг, 0,97 ммоль) в 1,4-диоксане (10 мл) добавляли KOAc (285 мг, 2,90 ммоль), и бис(пинаколато)диборон (368 мг, 1,45 ммоль). Смесь продували азотом в течение примерно 5 мин, после чего добавляли Pd(dppf)Cl2 (70,8 мг, 0,097 ммоль). Смесь нагревали при примерно 90°C в течение примерно 18 ч. Смесь концентрировали с получением неочищенного 1-метил-3-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола в виде черного масла, которое использовали без дополнительной очистки в части 2.
Часть 2
Смесь (1r,3r)-3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила (Получение 75, 130 мг, 0,38 ммоль), 1-метил-3-нитро-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (часть 1, 97 мг, 0,38 ммоль), и 2 M водн. K3PO4 (2,0 мл) в 1,4-диоксане (6,0 мл) продували аргоном в течение примерно 2 мин, после чего добавляли XPhos Pd G2 (60,6 мг, 0,077 ммоль). Смесь нагревали при примерно 45°C в течение примерно 45 мин. 1,4-Диоксановую фазу отделяли от водной фазы, которую дополнительно экстрагировали EtOAc (10 мл). Объединенные экстракты 1,4-диоксана и EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (120 мг, 72%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,10 (с, 1 H), 8,90 (с, 1 H), 8,56 (с, 1 H), 8,43 (с, 1 H), 8,32 (д, 1 H), 7,56 (шир.с, 1 H), 4,05 (с, 3 H), 3,54-3,60 (м, 1 H), 3,51 (с, 2 H), 3,18-3,29 (м, 2 H), 2,88-2,98 (м, 2 H).
LCMS m/z=429,4 [MH]+
Пример 23
(1r,3r)-3-(4-(6-(3-амино-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрил
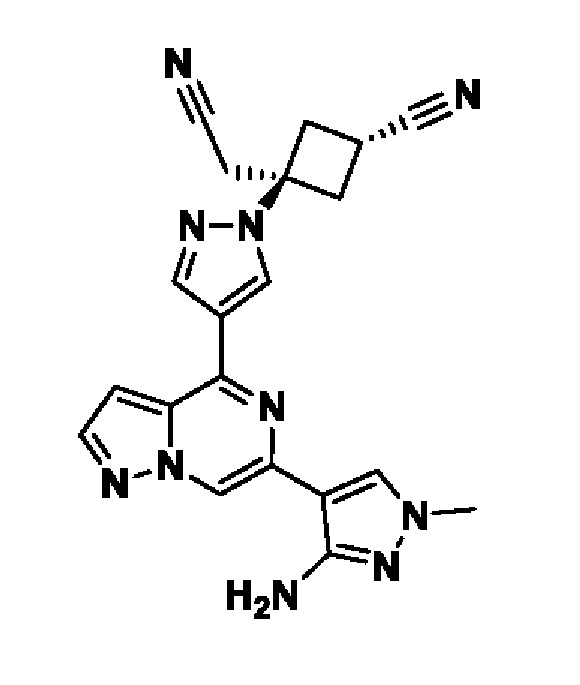
К раствору (1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-3-нитро-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 63, 120 мг, 0,28 ммоль) в EtOH (3 мл) и воде (0,5 мл) добавляли NH4Cl (90 мг, 1,68 ммоль) и железный порошок (50 мг, 0,90 ммоль). Реакционную смесь перемешивали при примерно 60°C в течение примерно 18 часов, после чего добавляли дополнительные порции порошка железа и NH4Cl. Смесь нагревали при примерно 100°C в течение примерно 3 часов. Смесь концентрировали и добавляли горячий EtOH (20 мл). Нерастворенные твердые вещества удаляли фильтрованием. Фильтрат концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде белого твердого вещества (10 мг, 9%).
1H ЯМР (400 МГц, CD3OD) δ: 8,74 (с, 1 H), 8,67 (с, 1 H), 8,40 (с, 1 H), 8,08 (д, 1 H), 7,94 (с, 1 H), 7,24 (д, 1 H), 3,77 (с, 3 H), 3,47-3,55 (м, 1 H), 3,43 (с, 2 H), 3,34-3,41 (м, 2 H), 2,98 (дд, 2 H).
LCMS m/z=399,4 [MH]+
Получение 64
3,5-дибром-1-метил-1H-пиразол
Раствор 3,5-дибромпиразола (6,5 г, 28,7 ммоль) в THF (30 мл) добавляли по каплям к охлаждаемой льдом суспензии NaH (2,88 г, 60% в минеральном масле, 71,9 ммоль) в THF (45 мл). Смесь перемешивали при примерно 0°C в течение примерно 1 часа. Добавляли иодметан (5,37 мл, 86,3 ммоль) и смесь перемешивали при примерно 0°C в течение примерно 3 часов, затем перемешивание продолжали при примерно 15°C в течение примерно 2 часов. Смесь выливали в насыщенный водн. NH4Cl (20 мл) и экстрагировали EtOAc (40 мл). Экстракт EtOAc промывали насыщенным солевым раствором (20 мл), сушили (Na2SO4), и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (5,5 г, 79%).
1H ЯМР (400 МГц, CDCl3) δ: 6,31 (с, 1 H), 3,87 (с, 3 H).
LCMS m/z=240,6 [MH]+ (79Br, 81Br изотоп)
Получение 65
3-бром-1-метил-1H-пиразол-5-карбоновая кислота
Раствор 3,5-дибром-1-метил-1H-пиразола (Получение 64, 3,0 г, 12,5 ммоль) в THF (30 мл) обрабатывали по каплям n-BuLi (2,5 M, 6,25 мл, 15,6 ммоль) при примерно -70°C. Примерно через 30 мин при этой температуре, раствор CO2 в THF (30 мл) добавляли по каплям, сохраняя при этом внутреннюю температуру ниже -65°C. Смесь перемешивали при этой температуре в течение примерно 1 часа. Затем реакционную смесь выливали в 1 M водн. HCl (50 мл) и смесь частично концентрировали для удаления большей части THF. Водный слой экстрагировали DCM (50 мл). Экстракт DCM сушили (Na2SO4) и концентрировали с получением указанного в заголовке соединения в виде желтого твердого вещества (2,1 г, 81%).
1HЯМР (400 МГц, DMSO-d6) δ: 13,70 (шир.с, 1H), 6,95 (с, 1H), 4,04 (с, 3H).
LCMS m/z=206,9 [MH]+ (81Br изотоп)
Получение 66
трет-бутил (3-бром-1-метил-1H-пиразол-5-ил)карбамат
Дифенилфосфорил азид (18,8 г, 68,5 ммоль) добавляли к раствору 5-бром-2-метил-2H-пиразол-3-карбоновой кислоты (Получение 65, 7,02 г, 34,2 ммоль) и DIPEA (11,9 мл, 68,5 ммоль) в трет-бутаноле (114 мл). Смесь перемешивали при примерно 45°C в течение примерно 30 мин, затем нагревали с обратным холодильником в течение примерно 5 часов. Охлажденную смесь разбавляли EtOAc (60 мл) и промывали насыщенным водн. NaHCO3 (2×30 мл) и насыщенным солевым раствором (20 мл). Экстракт EtOAc концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (4,80 г, 51%).
1H ЯМР (400 МГц, DMSO-d6) δ: 6,28 (шир.с, 1H), 6,18 (с, 1H), 3,73 (с, 3H), 1,50 (с, 9H).
LCMS m/z=221,7 [MH-C4H8]+ (81Br изотоп)
Получение 67
3-бром-1-метил-5-(диBoc)-амино-1H-пиразол

Ди-трет-бутил дикарбонат (1,58 г, 7,24 ммоль) добавляли к раствору трет-бутил (3-бром-1-метил-1H-пиразол-5-ил)карбамата (Получение 66, 2,0 г, 7,24 ммоль), TEA (4,04 мл, 29,0 ммоль), и DMAP (177 мг, 1,45 ммоль) в DCM (40 мл). Смесь перемешивали при примерно 20°C в течение примерно 18 часов. Добавляли воду (25 мл) и смесь экстрагировали DCM (2×30 мл). Объединенные экстракты DCM концентрировали и остаток очищали хроматографией с получением указанного в заголовке соединения (1,85 г, 67)%.
1H ЯМР (400 МГц, CDCl3) δ: 6,06-6,20 (м, 1 H), 3,64 (с, 3 H), 1,44 (с, 18 H).
LCMS m/z=378,2 [MH]+ (81Br изотоп)
Пример 24
2-((1r,3s)-1-(4-(6-(5-амино-1-метил-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
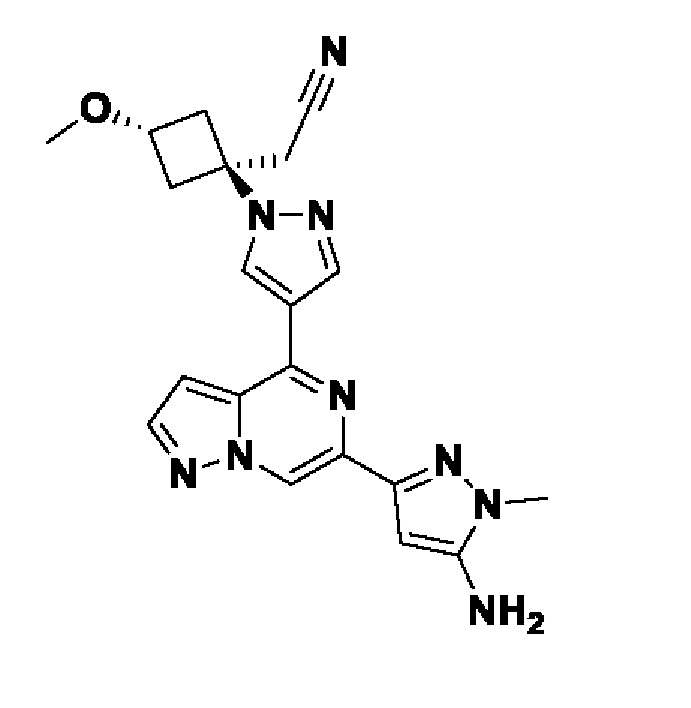
Часть 1
Смесь 3-бром-1-метил-5-(диBoc)-амино-1H-пиразола (Получение 67, 1200 мг, 3,19 ммоль), KOAc (988 мг, 9,57 ммоль) и бис(пинаколато)диборона (1210 мг, 4,78 ммоль) в 1,4-диоксане (15 мл) продували аргоном в течение примерно 5 минут до добавления XPhos Pd G2 (502 мг, 0,64 ммоль). Реакционную смесь нагревали при примерно 65°C в течение примерно 3,5 часов, затем добавляли 2-((1s,3r)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил (Получение 100, 1090 мг, 3,19 ммоль), 2 M водн. K3PO4 (4,78 мл) и XPhos Pd G2 (502 мг, 0,64 ммоль). Смесь снова продували аргоном, затем нагревали при примерно 80°C в течение примерно 1 часа. Добавляли EtOAc и фазы разделяли. Водную фазу экстрагировали дважды EtOAc и объединенные экстракты EtOAc концентрировали. Остаток очищали с помощью хроматографии с получением смеси моно- и ди-BOC промежуточных соединений (710 мг, 36%), которую использовали в части 2.
Часть 2
TFA (6 мл, 80 ммоль) добавляли к раствору моно- и ди-BOC промежуточных соединений части 1 (710 мг, 1,18 ммоль) в DCM (6 мл) при примерно 20°C. Примерно через 1 час, смесь концентрировали. Добавляли DCM, затем достаточное количество насыщенного водн. NaHCO3 для того, чтобы сделать рН раствора основным. Фазы разделяли и водную фазу экстрагировали дважды DCM. Объединенные экстракты DCM сушили (Na2SO4) и концентрировали. Эквивалентную реакцию с использованием соединения части 1 (220 мг, 0,36 ммоль) и TFA (2 мл, 30 ммоль) в DCM (2 мл) объединяли с веществом из вышеприведенной реакции и объединенные образцы очищали хроматографией с получением указанного в заголовке соединения в виде прозрачной камеди (470 мг, 99%).
1H ЯМР (400 МГц, DMSO-d6) δ: 8,81 (с, 1 H), 8,73 (с, 1 H), 8,40 (с, 1 H), 8,21 (д, 1 H), 7,44 (д, 1 H), 5,33 (шир.с, 2 H), 3,94-4,02 (м, 1 H), 3,62 (с, 3 H), 3,44 (с, 2 H), 3,21 (с, 3 H), 3,14-3,20 (м, 2 H), 2,39-2,45 (м, 2 H).
LCMS m/z=404,5 [MH]+
Получение 68
диэтил 4-бром-1H-пиразол-3,5-дикарбоксилат
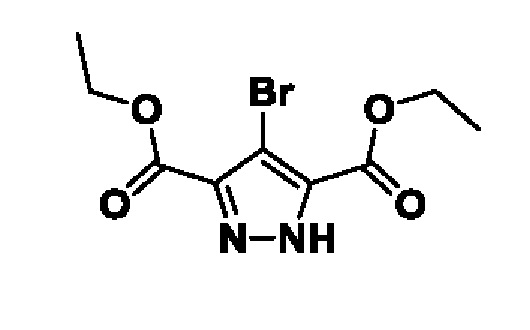
Смесь диэтилового эфира 1H-пиразол-3,5-дикарбоновой кислоты (4,0 г, 18,85 ммоль) и N-бромсукцинимида (4,03 г, 22,6 ммоль) в смеси конц. азотной кислоты и ледяной уксусной кислоты (12,0 мл, 5:95 об./об.) нагревали под микроволновым излучением при примерно 120°C в течение примерно 20 минут. В общей сложности 13,0 г (61,26 ммоль) исходного диэтилового эфира 1H-пиразол-3,5-дикарбоновой кислоты обрабатывали таким образом в параллельных партиях. Полученные коричневые неочищенные реакционные смеси объединяли, выливали в воду (260 мл), и обрабатывали достаточным количеством NaHCO3, чтобы сделать рН раствора основным. Смесь экстрагировали EtOAc (3×300 мл). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (17,0 г, 95%).
1H ЯМР (400 МГц, CDCl3) δ: 8,79 (шир.с, 1 H), 4,44 (кв, 4 H), 1,42 (т, 6 H).
LCMS m/z=290,7 [MH]+ (79Br изотоп)
Получение 69
Диэтил 4-бром-1-(2-(1-метил-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3,5-дикарбоксилат

Раствор диэтилового эфира 4-бром-1H-пиразол-3,5-дикарбоновой кислоты (Получение 68, 9,66 г, 33,18 ммоль) и 2-бром-1-(1-метил-1H-пиразол-4-ил)-этанона (Получение 6, 6,0 г, 29,55 ммоль) в MeCN (140 мл) обрабатывали K2CO3 (5,31 г, 38,40 ммоль) и реакционную смесь перемешивали при примерно 25°C в течение примерно 16 часов. Добавляли воду (100 мл) и смесь экстрагировали EtOAc (3×100 мл). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде белого твердого вещества (10,0 г, 82%).
1HЯМР (400 МГц, CDCl3) δ: 7,95 (с, 1 H), 7,92 (с, 1 H), 5,82 (с, 2 H), 4,43 (кв, 2 H), 4,36 (кв, 2 H), 3,96 (с, 3 H), 1,45 (т, 3 H), 1,35 (т, 3 H).
LCMS m/z=436,8 [MNa]+ (81Br изотоп)
Получение 70
Диэтил 4-метил-1-(2-(1-метил-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3,5-дикарбоксилат
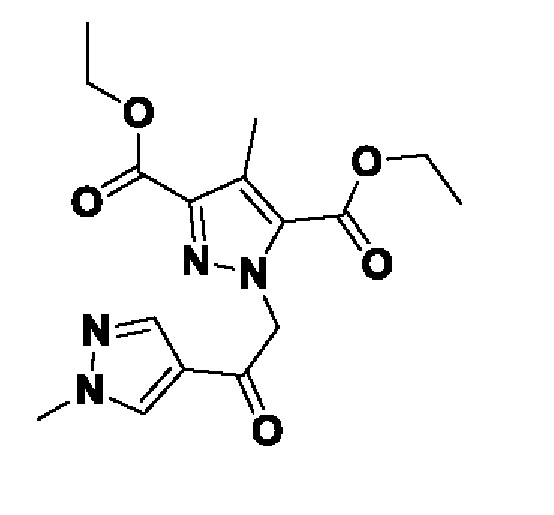
Смесь K2CO3 (11,0 г, 79,9 ммоль) и диэтилового эфира 4-бром-1H-[2-(1-метил-1H-пиразол-4-ил)-2-оксо-этил]-1H-пиразол-3,5-дикарбоновой кислоты (Получение 69, 11,0 г, 26,62 ммоль) в DMF (133,0 мл) продували азотом при примерно 25°C, после чего добавляли Pd(dppf)Cl2 (1950 мг, 2,66 ммоль) и триметилбороксин (10,0 г, 79,9 ммоль) и раствор нагревали при примерно 110°C в течение примерно 5 часов. Охлажденная реакционную смесь разбавляли EtOAc (100 мл) и полученную смесь промывали насыщенным солевым раствором (2×100 мл), сушили (Na2SO4), и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (5,45 г, 50%).
1H ЯМР (400 МГц, CDCl3) δ: 7,92 (с, 1 H), 7,90 (с, 1 H), 5,78 (с, 2 H), 4,41 (кв, 2 H), 4,28 (кв, 2 H), 3,95 (с, 3 H), 2,58 (с, 3 H), 1,39 (т, 3 H), 1,31 (т, 3 H).
LCMS m/z=348,9 [MH]+
Получение 71
Этил 4-гидрокси-3-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-2-карбоксилат
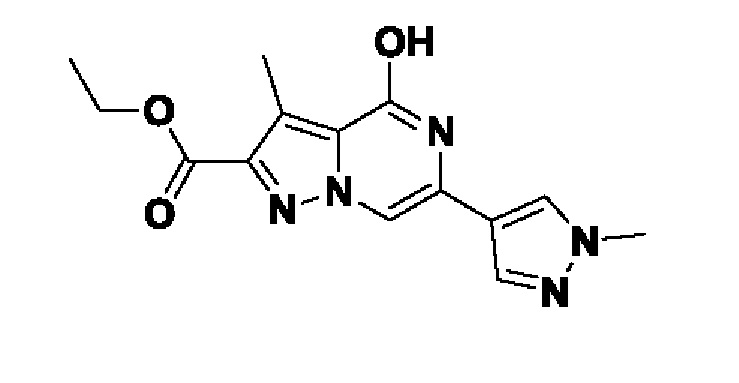
Диэтиловый эфир 4-метил-[2-(1-метил-1H-пиразол-4-ил)-2-оксо-этил]-1H-пиразол-3,5-дикарбоновой кислоты (Получение 70, 5,35 г, 15,36 ммоль) смешивали с сухим EtOH (15 мл) и концентрировали. Полученное твердое вещество растворяли в EtOH (40 мл) и добавляли NH4OAc (3,55 г, 46,1 ммоль). Реакционную смесь нагревали при примерно 130°C в автоклаве в течение примерно 8 часов. После охлаждения, смесь фильтровали и осадок сушили с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (3,40 г, 73%).
1H ЯМР (400 МГц, DMSO-d6) δ: 11,45 (шир.с, 1 H), 8,27 (с, 1 H), 8,08 (с, 1 H), 8,02 (с, 1 H), 4,31 (м, 2 H), 3,87 (с, 3 H), 2,61 (с, 3 H), 1,24-1,40 (м, 3 H).
LCMS m/z=301,8 [MH]+
Получение 72
4-гидрокси-3-метил-6-(1-метил-1H-пиразол-4-ил)-пиразоло[1,5-а]пиразин-2-карбоновая кислота
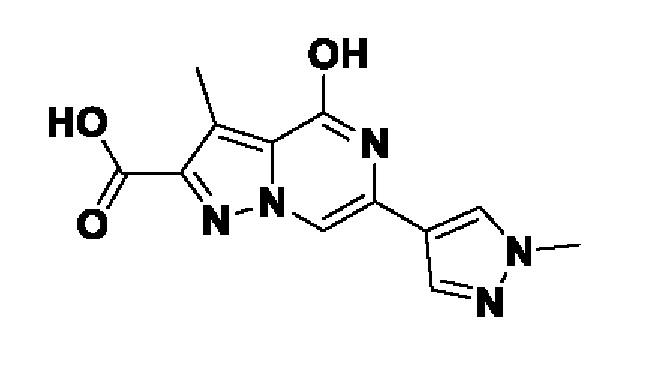
Моногидрат гидроксида лития (1,42 г, 33,9 ммоль) добавляли к суспензии этилового эфира 4-гидрокси-3-метил-6-(1-метил-1H-пиразол-4-ил)-пиразоло[1,5-а]пиразин-2-карбоновой кислоты (Получение 71, 3,40 г, 11,3 ммоль) в THF (50 мл), MeOH (50 мл) и воды (25 мл). Смесь перемешивали при примерно 60°C в течение примерно 4 часов. Охлажденную смесь концентрировали и добавляли воду (50 мл). Значение pH смеси доводили до примерно 2 путем добавления 12 M водн. HCl. Образовавшийся осадок фильтровали и промывали водой (50 мл), затем сушили в вакууме с получением указанного в заголовке соединения в виде твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ: 12,90-13,20 (шир.с, 1 H), 11,40 (с, 1 H), 8,30 (с, 1 H), 8,02 (м, 2 H), 3,90 (с, 3 H), 2,60 (с, 3 H).
LCMS m/z=301,8 [MH]+, 323,8 [MNa]+
Получение 73
3-метил-6-(1-метил-1H-пиразол-4-ил)-пиразоло[1,5-а]пиразин-4-ол
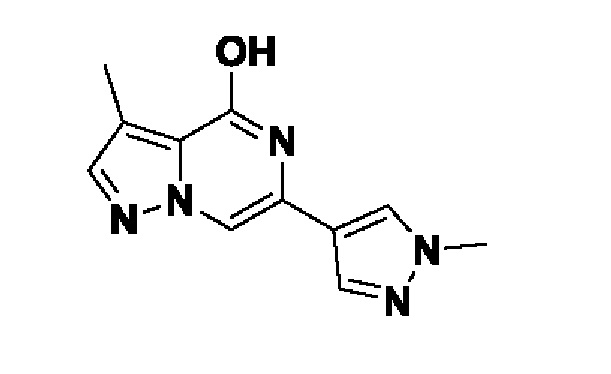
4-Гидрокси-3-метил-6-(1-метил-1H-пиразол-4-ил)-пиразоло[1,5-а]пиразин-2-карбоновую кислоту (Получение 72, 2,58 г, 9,44 ммоль) добавляли порциями к предварительно нагретому сульфолану (18,9 мл) при примерно 280°C. После завершения добавления, смесь перемешивали в течение дополнительного 1 часа при примерно 280°C. Охлажденную смесь очищали непосредственно при помощи хроматографии на силикагеле (элюирование петролейным эфиром:EtOAc (100:0-50:50), затем DCM:MeOH (91:9)) с получением указанного в заголовке соединения в виде желтого твердого вещества (1,50 г, 69%).
1HЯМР (400 МГц, DMSO-d6) δ: 11,20 (с, 1 H), 8,38 (с, 1 H), 8,02 (с, 1 H), 7,96 (с, 1 H), 7,70 (с, 1 H), 3,85 (с, 3 H), 2,40 (с, 3 H).
Получение 74
4-хлор-3-метил-6-(1-метил-1H-пиразол-4-ил)-пиразоло[1,5-а]пиразин

К суспензии 3-метил-6-(1-метил-1H-пиразол-4-ил)-пиразоло[1,5-а]пиразин-4-ола (Получение 73, 1,40 г, 6,11 ммоль) в MeCN (60,0 мл) добавляли POCl3 (4,68 г, 30,5 ммоль). Реакционную смесь нагревали при примерно 80°C в течение примерно 16 часов. После охлаждения смесь выливали в воду (200 мл) при примерно 25°C. Смесь доводили до значения рН примерно 9 с помощью добавления насыщенного водн. NaHCO3 (200 мл), затем экстрагировали EtOAc (5×100 мл). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии, затем дополнительно очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (163 мг, 11%).
1H ЯМР (400 МГц, CDCl3) δ: 8,37 (с, 1 H), 7,89 (с, 1 H), 7,85 (с, 1 H), 7,80 (с, 1 H), 3,96 (с, 3 H), 2,57 (с, 3 H).
LCMS m/z=247,7 [MH]+ (35Cl изотоп)
Пример 25
(1r,3r)-3-(Цианометил)-3-(4-(3-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
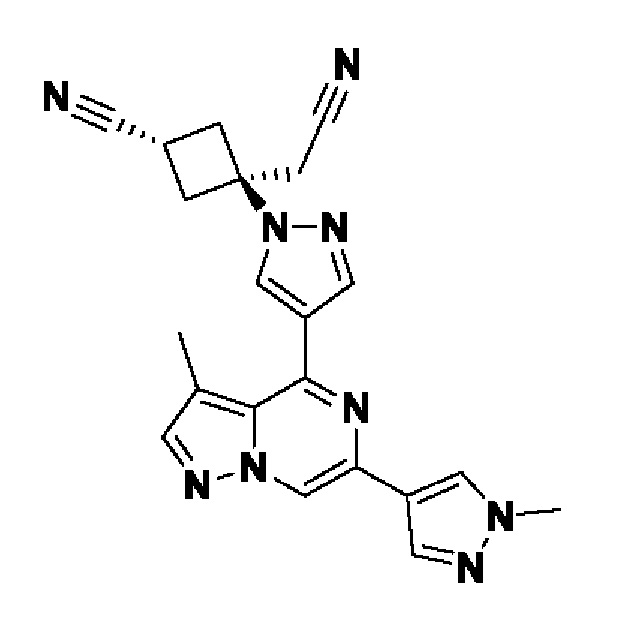
К раствору 4-хлор-3-метил-6-(1-метил-1H-пиразол-4-ил)-пиразоло[1,5-а]пиразина (Получение 74, 98 мг, 0,40 ммоль) и (1r,3r)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 91, 247 мг, 0,79 ммоль) в 1,4-диоксане (9 мл) добавляли 2 M водн. K3PO4 (0,59 мл) при примерно 25°C и смесь продували азотом в течение примерно 2 мин до добавления XPhos Pd G2 (62,3 мг, 0,079 ммоль). Смесь продували азотом в течение примерно 3 мин, затем нагревали при примерно 80°C в течение примерно 16 часов. Смесь фильтровали и концентрировали. Остаток очищали с помощью хроматографии, затем дополнительно очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (56 мг, 36%).
1H ЯМР (400 МГц, DMSO-d6): δ: 8,98 (с, 1 H), 8,94 (с, 1 H), 8,60 (с, 1 H), 8,27 (с, 1 H), 8,22 (с, 1 H), 8,10 (с, 1 H), 7,98 (с, 1 H), 3,90 (с, 3 H), 3,52-3,57 (м, 3 H), 3,19-3,26 (м, 2 H), 2,91-2,97 (м, 2 H), 3,06 (с, 3 H).
LCMS m/z=398,0 [MH]+
Получение 75
(1r,3r)-3-(4-(6-Хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрил
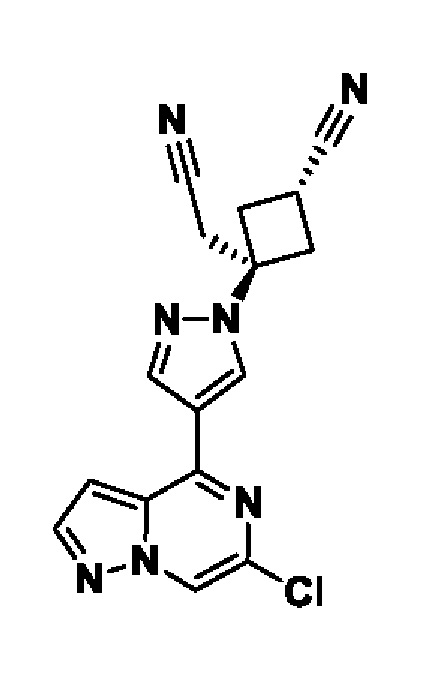
Смесь 4,6-дихлорпиразоло[1,5-а]пиразина (Получение 4, 350 мг, 1,86 ммоль), (1r,3r)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила, (Получение 91, транс-изомер, 581 мг, 1,86 ммоль) и 2 M водн. K3PO4 (2,79 мл) в 1,4-диоксане (10 мл) продували аргоном в течение примерно 5 мин, после чего добавляли бис(три-трет-бутилфосфин)палладий(0) (48,0 мг, 0,093 ммоль). Смесь выдерживали при примерно 25°C в течение примерно 2 часов, затем фильтровали. Осадок промывали Et2O и сушили. Фильтрат концентрировали, растирали в порошок с Et2O, фильтровали, промывали Et2O и сушили. Два осадка объединяли с получением указанного в заголовке соединения в виде белого твердого вещества (605 мг, 96%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,05 (д, 1 H), 8,93 (с, 1 H), 8,46 (с, 1 H), 8,32 (д, 1 H), 7,61 (д, 1 H), 3,55-3,62 (м, 1 H), 3,54 (с, 2 H), 3,24-3,32 (м, 2 H), 2,89-3,00 (м, 2 H).
LCMS m/z=338,2 [MH]+
Пример 26
(1r,3r)-3-(цианометил)-3-(4-(6-(5-метил-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
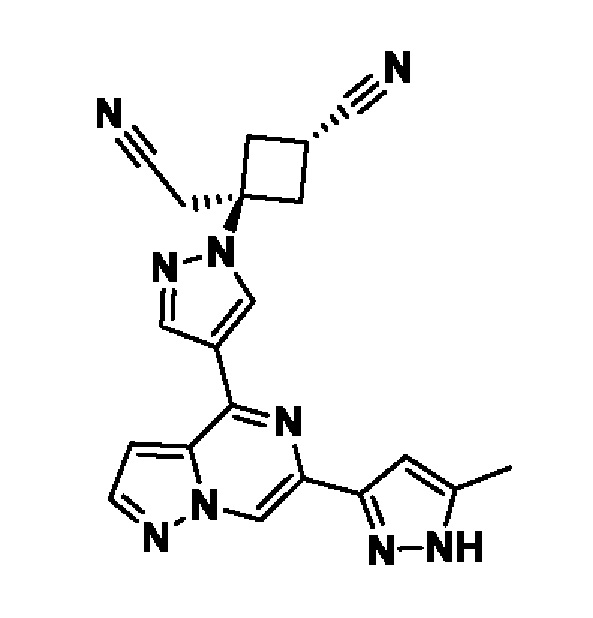
Часть 1
Смесь (1r,3r)-3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила (Получение 75, 85 мг, 0,25 ммоль), 3-метил-1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (Получение 93, 73 мг, 0,25 ммоль), и 2 M водн. K3PO4 (1,0 мл) в 1,4-диоксане (3,0 мл) продували аргоном в течение примерно 2 мин, после чего добавляли XPhos Pd G2 (39,6 мг, 0,050 ммоль). Смесь нагревали при примерно 45°C в течение примерно 45 мин перед охлаждением и концентрировали. Остаток очищали с помощью хроматографии с получением (1r,3r)-3-(цианометил)-3-(4-(6-(5-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (26 мг, 22%), который использовался непосредственно в части 2.
LCMS m/z=384,3 [MH-THP]+
Часть 2
TFA (1 мл) добавляли к раствору (1r,3r)-3-(цианометил)-3-(4-(6-(5-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (26 мг, 0,055 ммоль) в DCM (2 мл). Реакционную смесь нагревали до примерно 50°C в течение примерно 1 часа. Смесь концентрировали и остаток концентрировали дважды с толуолом (5 мл каждый раз). Остаток затем очищали хроматографией с получением твердого вещества, которое далее растирали в порошок с эфиром с получением указанного в заголовке соединения (8 мг, 38%).
1H ЯМР (400 МГц, CD3CN) δ: 8,87 (с, 1 H), 8,66 (с, 1 H), 8,48 (с, 1 H), 8,15 (д, 1 H), 7,23 (с, 1 H), 6,73 (с, 1 H), 3,46 (дд, 1 H), 3,36 (с, 2 H), 3,32-3,41 (м, 2 H), 2,98 (дд, 2 H), 2,37 (с, 3 H).
LCMS m/z=384,4 [MH]+
Пример 27
2-((1s,3r)-1-(4-(6-(5-(Гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
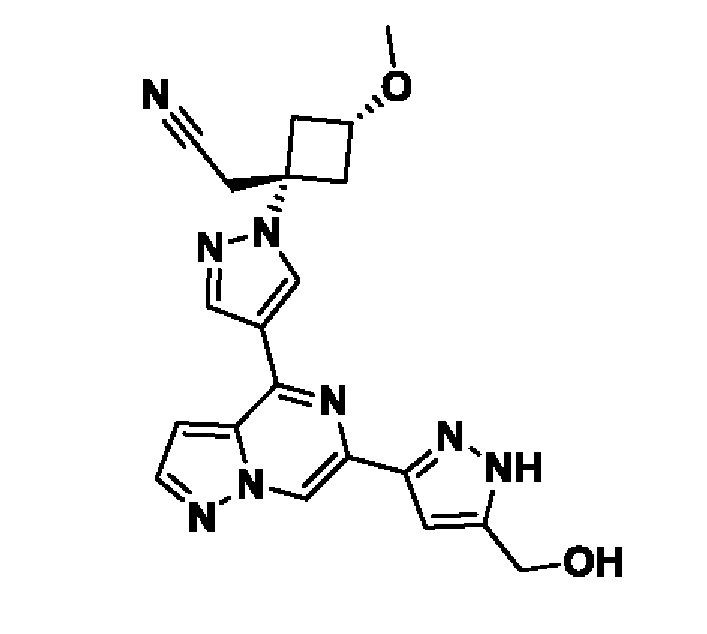
Раствор 2-((1s,3r)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 82, 100 мг, 0,29 ммоль) и (3-(трибутилстаннил)-1H-пиразол-5-ил)метанола (Получение 32, 113 мг, 0,29 ммоль) в 1,4-диоксане (2 мл) продували аргоном в течение 5 мин, с последующим добавлением XPhos Pd G2 (45,9 мг, 0,058 ммоль). Смесь нагревали при примерно 80°C в течение примерно 18 часов. Образовавшийся осадок фильтровали, промывали 1,4-диоксан и Et2O, и сушили с получением указанного в заголовке соединения в виде белого твердого вещества (62 мг, 53%).
1H ЯМР (400 МГц, CD3CN) δ: 8,86 (с, 1 H), 8,58 (с, 1 H), 8,44 (с, 1 H), 8,13 (д, 1 H), 7,21 (д, 1 H), 6,85 (с, 1 H), 4,65 (д, 2 H), 4,05 (квин, 1 H), 3,27 (с, 3 H), 3,21 (с, 2 H), 2,90-2,99 (м, 2 H), 2,67-2,76 (м, 2 H).
LCMS m/z=405,3 [MH]+
Пример 28
2-((1r,3s)-1-(4-(6-(5-(Гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
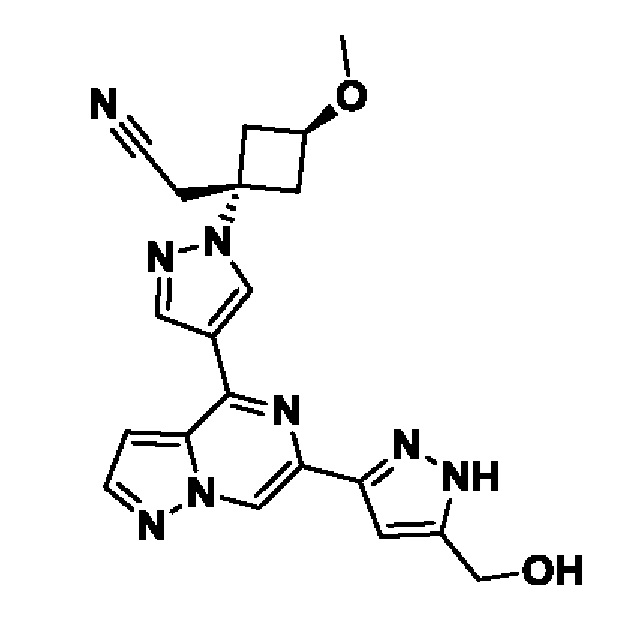
Раствор 2-((1r,3s)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 39; 1000 мг, 2,92 ммоль) и (3-(трибутилстаннил)-1H-пиразол-5-ил)метанола (Получение 32, 1130 мг, 2,92 ммоль) в 1,4-диоксане (2 мл) продували аргоном в течение 5 мин, с последующим добавлением XPhos Pd G2 (45,9 мг, 0,058 ммоль). Смесь нагревали при примерно 80°C в течение примерно 18 часов. Осадок фильтровали и затем очищали хроматографией с получением не совсем белого твердого вещества. Твердое вещество нагревали в достаточном количестве кипящего EtOH до полного растворения, и затем перемешивали в течение примерно 18 часов при примерно 20°C. Осадок фильтровали и сушили в вакууме с получением указанного в заголовке соединения в виде кристаллического твердого вещества (507 мг, 43%).
1H ЯМР (400 МГц, CD3CN) δ: 11,48 (шир. с.), 8,89 (с), 8,65 (с), 8,46 (с), 8,15 (д), 7,23 (с), 6,87 (с), 4,67 (д), 4,08 (квин), 3,31 (с), 3,30 (с), 3,15-3,25 (м), 2,53 (дд).
LCMS m/z=405,3 [MH]+
Получение 76
Этил 5-(трибутилстаннил)изоксазол-3-карбоксилат
TEA (0,69 мл, 4,95 ммоль) добавляли по каплям к охлажденному льдом раствору этил 2-хлор-2-(гидроксиимино)ацетата (500 мг, 3,30 ммоль) и этинилтрибутилстаннана (1040 мг, 3,30 ммоль) в Et2O (10 мл). Реакционную смесь нагревали до примерно 20°C и выдерживали в течение примерно 18 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (810 мг, 57%).
1H ЯМР (400 МГц, CDCl3) δ: 6,80 (с, 1 H), 6,77-6,84 (м, 1 H), 4,45 (кв, 2 H), 1,51-1,63 (м, 6 H), 1,43 (т, 3 H), 1,34 (дкв, 6 H), 1,15-1,26 (м, 6 H), 0,90 (т, 9 H).
LCMS m/z=454,2 [MNa]+ (120Sn изотоп)
Получение 77
Этил 5-(4-(1-((1r,3r)-3-циано-1-(цианометил)циклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)изоксазол-3-карбоксилат
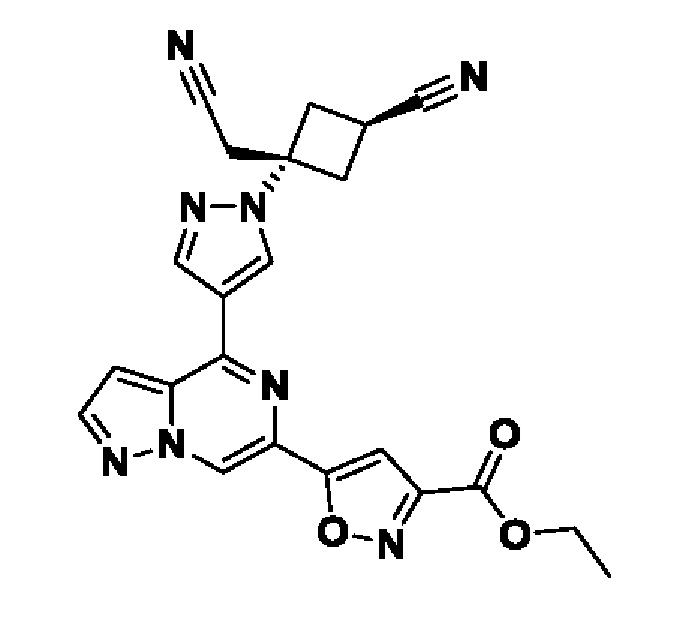
Раствор (1r,3r)-3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила (Получение 75, 110 мг, 0,33 ммоль) и этил 5-(трибутилстаннил)изоксазол-3-карбоксилата (Получение 76, 140 мг, 0,33 ммоль) в 1,4-диоксане (3 мл) продували аргоном в течение примерно 5 мин, с последующим добавлением XPhos Pd G2 (51,2 мг, 0,065 ммоль). Смесь нагревали при примерно 100°C в течение примерно 5 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (66 мг, 45%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,44 (с, 1 H), 9,02 (с, 1 H), 8,60 (с, 1 H), 8,42 (д, 1 H), 7,65 (д, 1 H), 7,62 (с, 1 H), 4,44 (кв, 2 H), 3,54-3,63 (м, 1 H), 3,53 (с, 2 H), 3,26-3,36 (м, 2 H), 2,90-2,98 (м, 2 H), 1,37 (т, 3 H).
LCMS m/z=443,3 [MH]+
Пример 29
5-(4-(1-((1r,3r)-3-Циано-1-(цианометил)циклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)изоксазол-3-карбоксамид
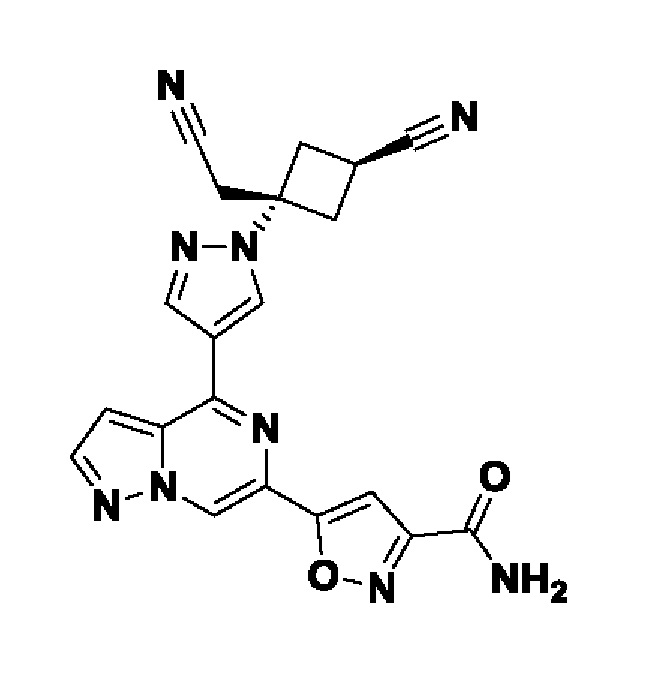
Раствор этил 5-(4-(1-((1r,3r)-3-циано-1-(цианометил)циклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)изоксазол-3-карбоксилата (Получение 77, 54 мг, 0,12 ммоль) в метаноле (3 мл) обрабатывали 7 M раствором газообразного аммиака в метаноле (2 мл, 14 ммоль). Реакционный сосуд герметично закрывали, и смесь нагревали при примерно 95°C в течение примерно 18 часов. После охлаждения осадок отфильтровывали, промывали EtOAc, затем Et2O, и сушили с получением указанного в заголовке соединения в виде белого твердого вещества (45 мг, 89%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,36 (с, 1 H), 9,01 (с, 1 H), 8,58 (с, 1 H), 8,41 (д, 1 H), 8,21 (шир.с, 1 H), 7,91 (шир.с, 1 H), 7,64 (д, 1 H), 7,50 (с, 1 H), 3,55-3,62 (м, 1 H), 3,53 (с, 2 H), 3,26-3,35 (м, 2 H), 2,90-2,98 (м, 2 H).
LCMS m/z=414,3 [MH]+
Получение 78
2-((1r,3s)-1-(4-(6-(5-(диBoc)-амино-1-(тетрагидро-2H-пиран-2-ил)-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
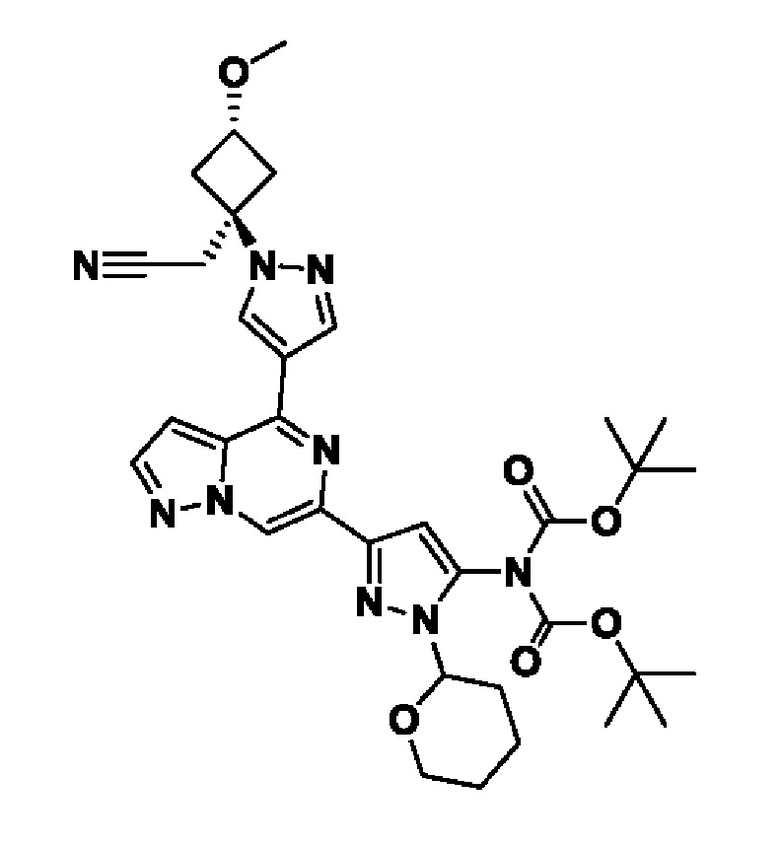
Смесь 3-бром-1-(тетрагидро-2H-пиран-2-ил)-5-(диBoc)-амино-1H-пиразола (Получение 43, 233 мг, 0,52 ммоль), KOAc (162 мг, 1,57 ммоль), и бис(пинаколато)диборона (199 мг, 0,78 ммоль) в 1,4-диоксане (4 мл) продували аргоном в течение примерно 5 мин, после чего добавляли XPhos Pd G2 (82,1 мг, 0,10 ммоль). Смесь нагревали при примерно 65°C в течение примерно 3,5 часов. После охлаждения примерно до 20°C, добавляли 2-((1r,3s)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3- метоксициклобутил)ацетонитрил (Получение 39, 125 мг, 0,36 ммоль), 2 M водн. K3PO4 (0,783 мл) и XPhos Pd G2 (82,1 мг, 0,10 ммоль). Смесь снова продували аргоном, затем нагревали при примерно 80°C в течение примерно 1 часа. После охлаждения, смесь разбавляли EtOAc и фазы разделяли. Водную фазу дважды экстрагировали EtOAc и объединенные экстракты EtOAc концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде прозрачного масла (220 мг, 62%).
1H ЯМР (400 МГц, CDCl3) δ: 9,08 (с, 1 H), 8,39 (с, 1 H), 8,33 (с, 1 H), 8,09 (д, 1 H), 6,97 (д, 1 H), 6,90 (с, 1 H), 5,26 (дд, 1 H), 4,03-4,11 (м, 2 H), 3,64 (т, 1 H), 3,34 (с, 3 H), 3,25 (с, 2 H), 3,11-3,19 (м, 2 H), 2,55-2,63 (м, 2 H), 2,21 (м, 1 H), 1,93 (дд, 1 H), 1,59-1,84 (м, 4 H), 1,45 (с, 18 H).
LCMS m/z=674,5 [MH]+
Пример 30
2-((1r,3s)-1-(4-(6-(3-Амино-1H-пиразол-5-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
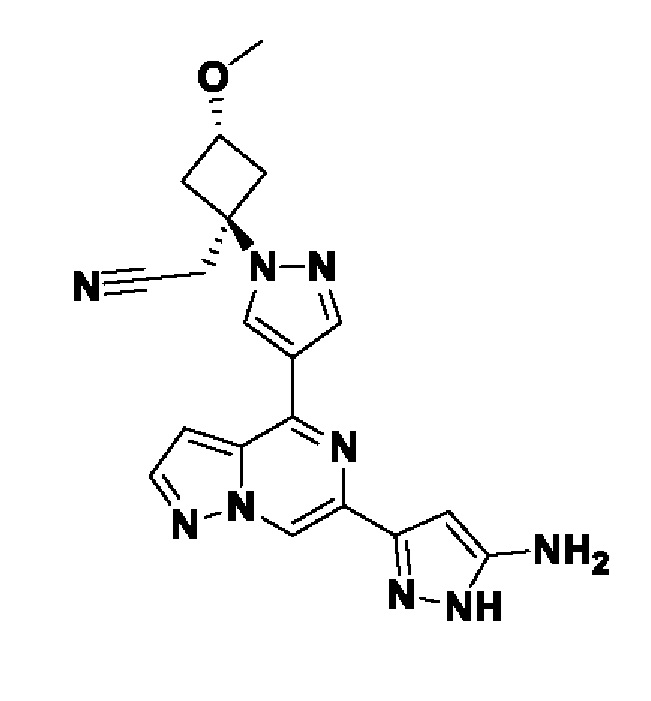
TFA (3 мл) добавляли к раствору 2-((1r,3s)-1-(4-(6-(5-(диBoc)-амино-1-(тетрагидро-2H-пиран-2-ил)-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 78, 220 мг, 0,33 ммоль) в DCM (2 мл) при примерно 20°C. Примерно через 1 час при этой температуре, смесь концентрировали и остаток растворяли в DCM и насыщенном водн. NaHCO3, обеспечивая, чтобы pH был основным. DCM отделяли и водную фазу дважды экстрагировали DCM. Объединенные экстракты DCM сушили (Na2SO4), концентрировали, и остаток очищали с помощью хроматографии, затем ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (17 мг, 13%).
1H ЯМР (400 МГц, CD3OD) δ: 8,89 (с, 1 H), 8,84 (с, 1 H), 8,54 (с, 1 H), 8,17-8,18 (м, 1 H), 7,34 (с, 1 H), 6,22 (с, 1 H), 4,09-4,12 (м, 1 H), 3,39 (с, 3 H), 3,22-3,27 (м, 2 H), 2,56-2,60 (м, 2 H).
LCMS m/z=390,3 [MH]+
Получение 79
(4-бром-1-метил-1H-пиразол-3-ил)метанол
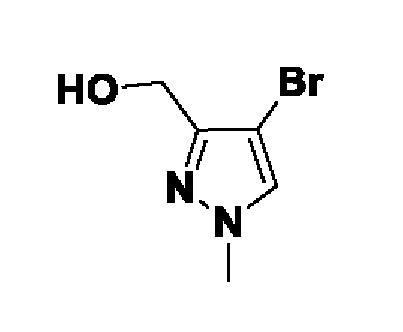
Боргидрид натрия (3,4 г, 90 ммоль) добавляли к раствору этил 4-бром-1-метил-1H-пиразол-3-карбоксилата (4,2 г, 18 ммоль) в безводном EtOH (150 мл) при примерно 5°C. Затем смесь нагревали при примерно 50°C в течение примерно 16 ч, после чего добавляли EtOAc (100 мл) и воду (100 мл). EtOAc отделяли, сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде белого твердого вещества (1,05 г, 31%).
1H ЯМР (400 МГц, DMSO-d6) δ: 7,84 (с, 1 H), 5,01 (тд, 1 H), 4,33 (д, 2 H), 3,77 (с, 3 H).
LCMS m/z=192,7 [MH]+ (81Br изотоп)
Получение 80
4-бром-1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразол
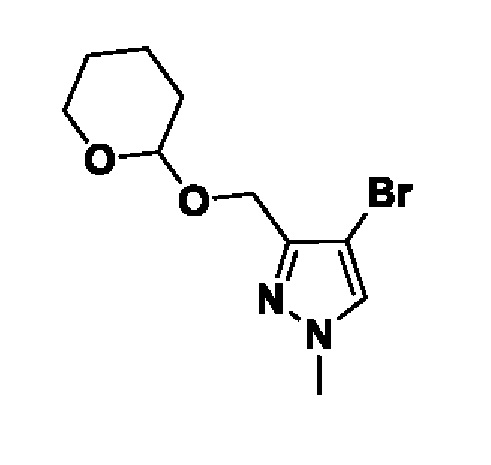
К раствору (4-бром-1-метил-1H-пиразол-3-ил)метанола (Получение 79, 1,00 мг, 5,23 ммоль) в THF (30 мл) добавляли DHP (1,32 г, 15,7 ммоль) и PTSA (19,9 мг, 0,10 ммоль). Раствор нагревали при примерно 50°C в течение примерно 16 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (1,02 г, 71%).
1H ЯМР (400 МГц, CDCl3) δ: 7,36 (с, 1 H), 4,80 (т, 1 H), 4,73 (д, 1 H), 4,45 (д, 1 H), 3,95-4,03 (м, 1 H), 3,87 (с, 3 H), 3,53-3,64 (м, 1 H), 1,78-1,93 (м, 1 H), 1,60-1,77 (м, 3 H), 1,45-1,60 (м, 2 H).
LCMS m/z=174,6 [MH-THP]+
Получение 81
1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол
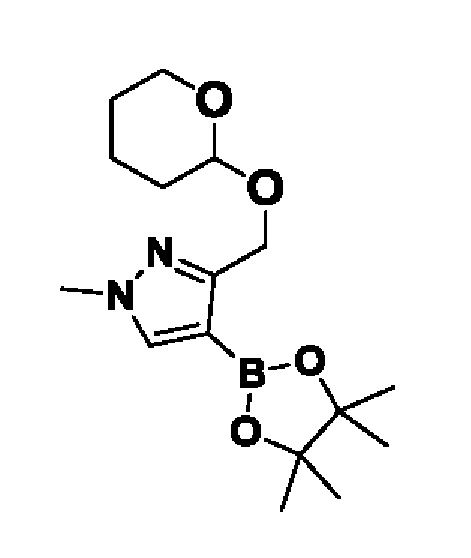
К раствору 4-бром-1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразола (Получение 80, 330 мг, 1,20 ммоль) в DMF (12 мл) добавляли бис(пинаколато)диборон (457 мг, 1,8 ммоль) и KOAc (353 мг, 3,6 ммоль). Смесь продували азотом в течение примерно 2 мин, после чего добавляли Pd(dppf)Cl2 (87,8 мг, 0,12 ммоль). Смесь нагревали при примерно 90°C в течение примерно 16 часов. Охлажденную смесь фильтровали через слой целита и фильтр промывали метанолом (15 мл). Фильтрат концентрировали с получением неочищенного указанного в заголовке соединения в виде темной камеди (1,17 г), которую использовали в следующем примере 31 без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ: 7,61 (с, 1 H), 4,75-4,77 (м, 1 H), 4,50-4,57 (м, 2 H), 4,03-4,10 (м, 1 H), 3,91-3,98 (м, 1 H), 3,89 (с, 3 H), 1,81-1,90 (м, 1 H), 1,61-1,76 (м, 3 H), 1,49-1,56 (м, 2 H), 1,30 (с, 12 H).
LCMS m/z=239,1 [MH-THP]+
Пример 31
2-((1r,3s)-1-(4-(6-(3-(Гидроксиметил)-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
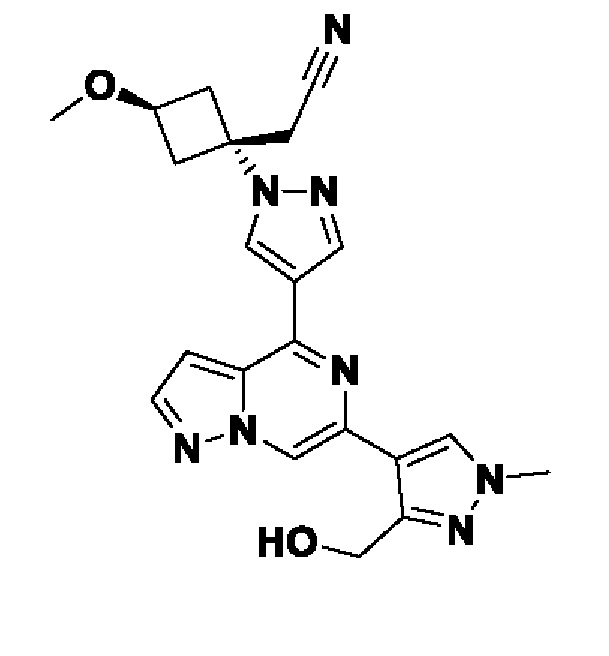
Часть 1
Смесь 2-((1r,3s)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метокси-циклобутил)ацетонитрила (Получение 39, 80 мг, 0,23 ммоль), 1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (Получение 81, 376 мг, 1,17 ммоль) и 2 M водн. K3PO4 (0,35 мл) в 1,4-диоксане (2,5 мл) обрабатывали XPhos Pd G2 (18,4 мг, 0,023 ммоль) и смесь продували азотом. Смесь нагревали при примерно 60°C в течение примерно 20 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтой камеди. Этот образец объединяли с продуктом из эквивалентной реакции, проведенной с использованием 2-((1r,3s)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 39, 120 мг, 0,35 ммоль), 1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (Получение 81, 564 мг, 1,75 ммоль), 2 M водн. K3PO4 (0,525 мл), и XPhos Pd G2 (13,8 мг, 0,023 ммоль) в 1,4-диоксане (3,5 мл) с получением 2-((1r,3s)-3-метокси-1-(4-(6-(1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила с общим выходом 150 мг (51%), который использовали в части 2 без дополнительной очистки.
Часть 2
TFA (1 мл) добавляли к охлаждаемому льдом раствору 2-((1r,3s)-3-метокси-1-(4-(6-(1-метил-3-(((тетрагидро-2H-пиран-2-ил)окси)метил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (часть 1, 150 мг, 0,30 ммоль) в DCM (3 мл). Смесь перемешивали при охлаждении на льду в течение примерно 2 часов, затем концентрировали. Остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде светло-желтого твердого вещества (53 мг, 38%).
1H ЯМР (400 МГц, CD3OD) δ: 8,87 (с, 1 H), 8,69 (с, 1 H), 8,40 (с, 1 H), 8,21 (с, 1 H), 8,13 (д, 1 H), 7,28 (д, 1 H), 4,83 (с, 2 H), 4,05-4,14 (м, 1 H), 3,96 (с, 3 H), 3,37 (с, 2 H), 3,34 (с, 3 H), 3,18-3,27 (м, 2 H), 2,53-2,62 (м, 2 H).
LCMS m/z=419,1 [MH]+
Получение 82
2-((1s,3r)-1-(4-(6-Хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
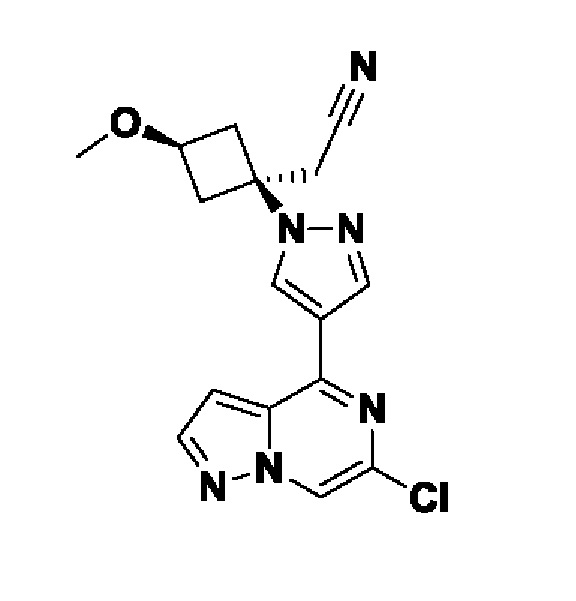
Часть 1
К раствору 2-((1s,3r)-1-(4-бром-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 37, цис-изомер, 400 мг, 1,48 ммоль) в толуоле (9 мл) добавляли бис(пинаколато)диборон (564 мг, 2,22 ммоль) и KOAc (436 мг, 4,44 ммоль). Смесь продували азотом в течение примерно 3 мин, после чего добавляли Pd(dppf)Cl2 (108 мг, 0,15 ммоль). Смесь снова продували азотом в течение примерно 3 мин, затем нагревали при примерно 110°C в течение примерно 16 часов. Охлажденную смесь концентрировали и остаток очищали с помощью хроматографии с получением неочищенного 2-((1s,3r)-3-метокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила в виде светло-желтого масла (500 мг), который использовали в части 2.
Часть 2
Раствор 2-((1s,3r)-3-метокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (часть 1, 470 мг, 1,48 ммоль) и 4,6-дихлорпиразоло[1,5-а]пиразина (Получение 4, 279 мг, 1,48 ммоль), и 2 M водн. K2CO3 (2,22 мл) в 1,4-диоксане (10 мл) продували азотом в течение примерно 2 мин, после чего добавляли Pd(dppf)Cl2 (108 мг, 0,15 ммоль). Смесь снова продували азотом в течение примерно 3 мин, затем нагревали при примерно 90°C в течение примерно 16 часов. Охлажденную смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (130 мг, 26%).
1H ЯМР (400 МГц, CDCl3) δ: 8,40 (с, 1 H), 8,35 (с, 1 H), 8,27 (с, 1 H), 8,08 (д, 1 H), 7,00 (с, 1 H), 4,05 (квин, 1 H), 3,30 (с, 3 H), 3,10 (с, 2 H), 3,00 (м, 2 H), 2,75 (м, 2 H).
LCMS m/z=343,2 [MH]+ (35Cl изотоп)
Получение 83
Этил 5-(4-(1-((1s,3r)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксилат
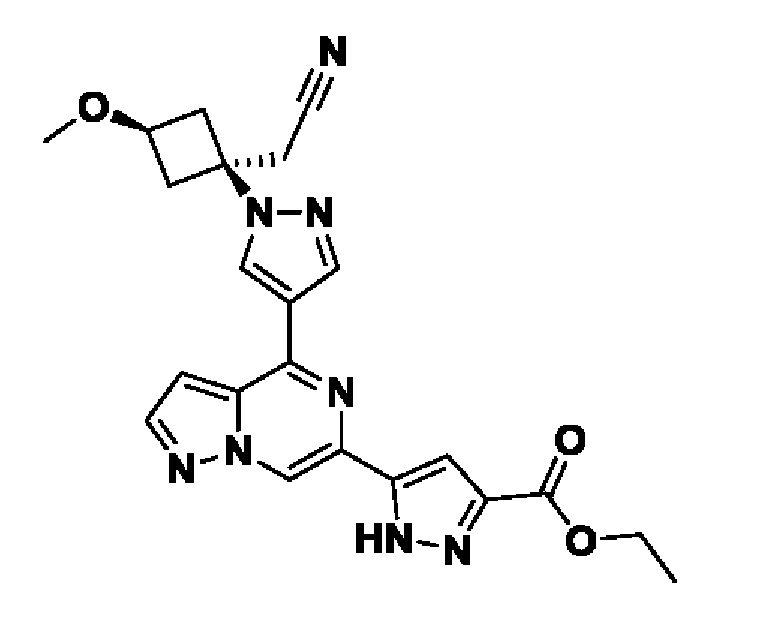
Раствор 2-((1s,3r)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 82, 120 мг, 0,35 ммоль) и этил 5-(трибутилстаннил)-1H-пиразол-3-карбоксилата (Получение 31, 150 мг, 0,35 ммоль) в 1,4-диоксане (5 мл) обрабатывали XPhos Pd G2 (13,8 мг, 0,017 ммоль) и смесь продували азотом в течение примерно 2 мин. Смесь нагревали при примерно 110°C в течение примерно 2 часов. Охлажденную смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (180 мг).
1H ЯМР (400 МГц, CDCl3) δ: 9,05 (шир.с, 1 H), 8,39 (с, 1 H), 8,32 (с, 1 H), 8,14 (с, 1 H), 7,65 (с, 1 H), 7,02 (с, 1 H), 6,88 (с, 1 H), 4,44-4,50 (м, 2 H), 4,08 (т, 1 H), 3,80 (с, 3 H), 3,13 (с, 2 H), 3,00-3,08 (м, 2 H), 2,75-2,83 (м, 2 H), 1,46 (т, 3 H).
LCMS m/z=447,1 [MH]+
Пример 32
5-(4-(1-((1s,3r)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксамид
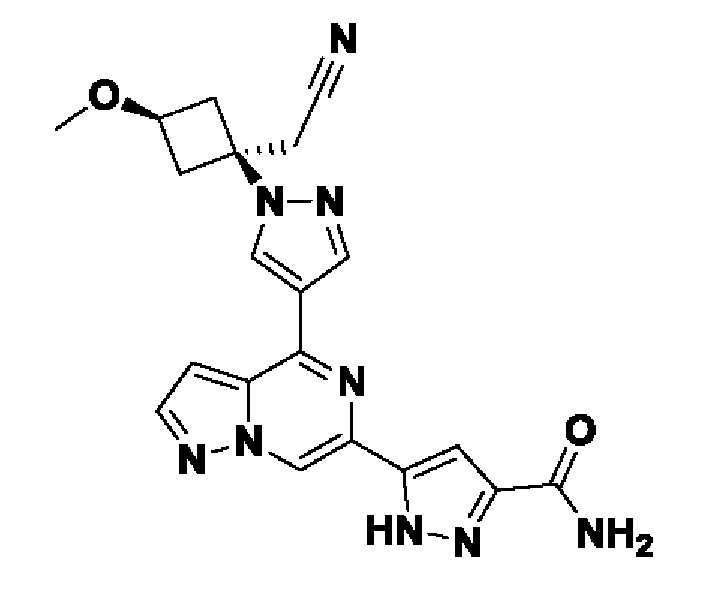
Раствор этил 5-(4-(1-((1s,3r)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксилата (Получение 83, 100 мг, 0,22 ммоль) в MeOH (2 мл) обрабатывали 4 M раствором газообразного аммиака в метаноле (1 мл). Реакционный сосуд герметично закрывали и смесь нагревали при примерно 60°C в течение примерно 16 часов. Смесь концентрировали и остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения (23 мг, 25%).
1H ЯМР (400 МГц, DMSO-d6) δ: 13,79 (шир.с, 1 H), 9,31 (шир.с, 1 H), 9,00 (шир.с, 1 H), 8,67 (шир.с, 1 H), 8,28 (д, 1 H), 7,56 (шир.с, 2 H), 7,39 (шир.с, 1 H), 7,25-7,35 (м, 1 H), 4,08 (квин, 1 H), 3,44 (с, 2 H), 3,21 (с, 3 H), 2,79-2,90 (м, 2 H), 2,63-2,73 (м, 3 H).
LCMS m/z=440,1 [MNa]+
Получение 84
2-(Тетрагидро-2H-пиран-2-ил)-2H-1,2,3-триазол
К смеси 1H-1,2,3-триазола (15 г, 220 ммоль) в DCM (724 мл) добавляли DHP (21,9 г, 261 ммоль) и PTSA (0,374 г, 2,17 ммоль). Смесь выдерживали при примерно 25°C в течение примерно 18 часов, после чего добавляли NaOH (96 мг, 2,39 ммоль). Смесь перемешивали при примерно 25°C в течение примерно 1 часа, затем фильтровали. Фильтрат концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (18 г, 54%).
1H ЯМР (400 МГц, CDCl3) δ: 7,68 (с, 2 H), 5,74 (дд, 1 H), 4,00-4,08 (м, 1 H), 3,69-3,81 (м, 1 H), 2,36-2,51 (м, 1 H), 2,02-2,20 (м, 2 H), 1,61-1,81 (м, 3 H).
Получение 85
2-(Тетрагидро-2H-пиран-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-1,2,3-триазол
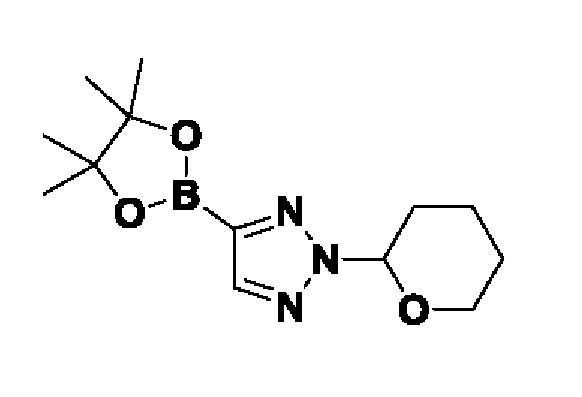
К раствору 4,4,5,5-тетраметил-1,3,2-диоксаборолана (24 г, 129 ммоль) в пентане (200 мл) добавляли 4,4'-ди-трет-бутил-2,2'-бипиридин (0,315 г, 1,18 ммоль) и (1,5-циклооктадиен)(метокси)иридий(I) димер (234 мг, 0,35 ммоль). Раствор быстро стал красного цвета, и наблюдалось выделение газа. Примерно через 15 мин, добавляли 2-(тетрагидро-2H-пиран-2-ил)-2H-1,2,3-триазол (Получение 84, 18 г, 117 ммоль). Смесь перемешивали при примерно 25°C в течение примерно 6 ч. Смесь концентрировали и остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного твердого вещества (26 г, 79%).
1H ЯМР (400 МГц, CDCl3) δ: 7,99 (с, 1 H), 5,81 (дд, 1 H), 4,07 (д, 1 H), 3,67-3,78 (м, 1 H), 2,41-2,55 (м, 1 H), 2,02-2,16 (м, 2 H), 1,60-1,80 (м, 3 H), 1,37 (с, 12 H).
LCMS m/z=113,9 [MH-C2H2N]+
Получение 86
(1r,3r)-3-(цианометил)-3-(4-(6-(2-(тетрагидро-2H-пиран-2-ил)-2H-1,2,3-триазол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
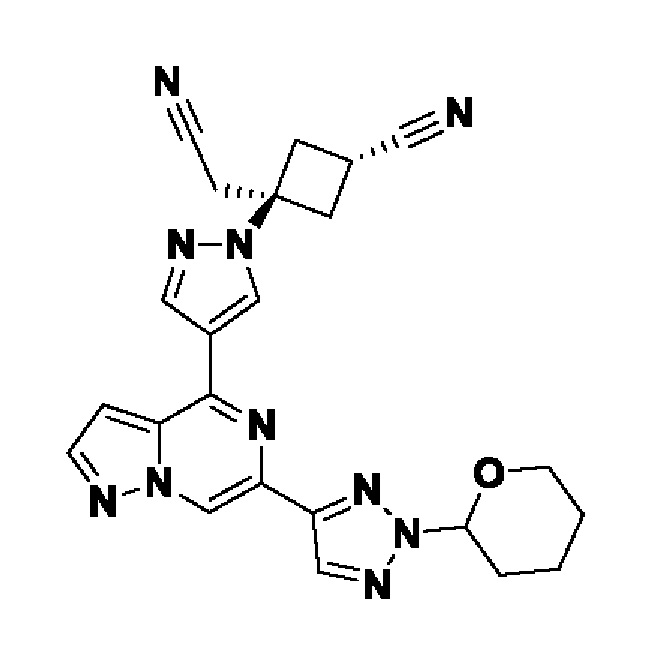
Смесь (1r,3r)-3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила (Получение 75, 300 мг, 0,89 ммоль), 2-(тетрагидро-2H-пиран-2-ил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2H-1,2,3-триазола (Получение 85, 248 мг, 0,89 ммоль), и 2 M водн. K3PO4 (1 мл) в 1,4-диоксане (4 мл) продували аргоном в течение примерно 5 мин, после чего добавляли XPhos Pd G2 (140 мг, 0,18 ммоль). Смесь нагревали при примерно 50°C в течение примерно 1 часа, затем охлаждали и разбавляли EtOAc. Фазы разделяли и водную фазу дважды экстрагировали DCM. Объединенные экстракты EtOAc и DCM сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде прозрачной камеди (384 мг, 95%).
1H ЯМР (400 МГц, CDCl3) δ: 9,02 (с, 1 H), 8,41 (с, 1 H), 8,35 (с, 1 H), 8,28 (с, 1 H), 8,13 (д, 1 H), 7,00 (д, 1 H), 5,81 (дд, 1 H), 4,06-4,12 (м, 1 H), 3,77-3,87 (м, 1 H), 3,35-3,47 (м, 3 H), 3,30 (с, 2 H), 2,95-3,04 (м, 2 H), 2,45-2,59 (м, 1 H), 2,12-2,24 (м, 2 H), 1,69-1,87 (м, 3 H).
LCMS m/z=455,3 [MH]+
Получение 87
(1r,3r)-3-(4-(6-(2H-1,2,3-триазол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрил
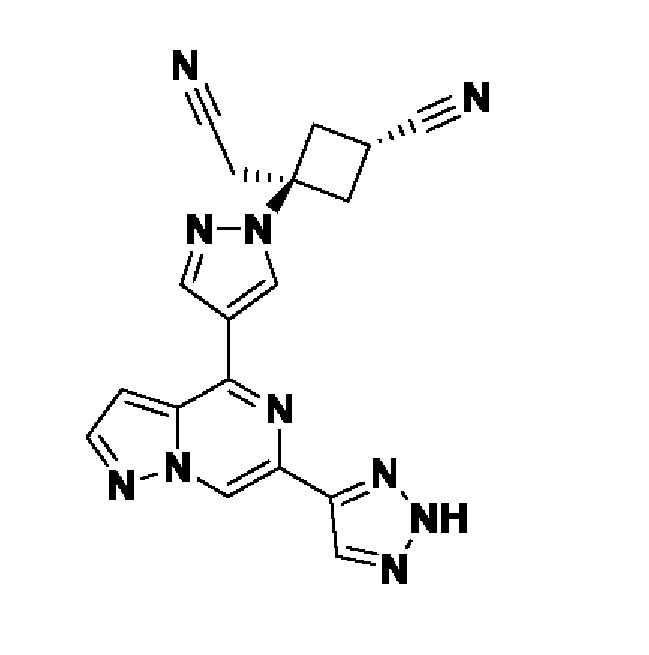
Суспензию (1r,3r)-3-(цианометил)-3-(4-(6-(2-(тетрагидро-2H-пиран-2-ил)-2H-1,2,3-триазол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 86, 384 мг, 0,84 ммоль) в MeOH (5 мл) обрабатывали PTSA (16,1 мг, 0,084 ммоль). Смесь нагревали при примерно 60°C в течение примерно 3,5 ч. Твердые вещества растворяли с образованием гомогенного раствора, после чего осаждалось белое твердое вещество. Нагревание продолжали в течение примерно 1 часа дополнительно. Смесь охлаждали до примерно 0°C в течение примерно 30 мин и фильтровали. Осадок промывали MeOH и сушили в вакууме с получением указанного в заголовке соединения в виде белого твердого вещества (204 мг, 65%).
1H ЯМР (400 МГц, DMSO-d6) δ: 9,05 (с, 1 H), 8,97 (с, 1 H), 8,72 (шир.с, 1 H), 8,57 (с, 1 H), 8,46 (с, 1 H), 8,30 (с, 1 H), 7,56 (с, 1 H), 3,55-3,62 (м, 1 H), 3,53 (с, 2 H), 3,22-3,30 (м, 2 H), 2,89-3,00 (м, 2 H).
LCMS m/z=371,3 [MH]+
Пример 33
(1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-1H-1,2,3-триазол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
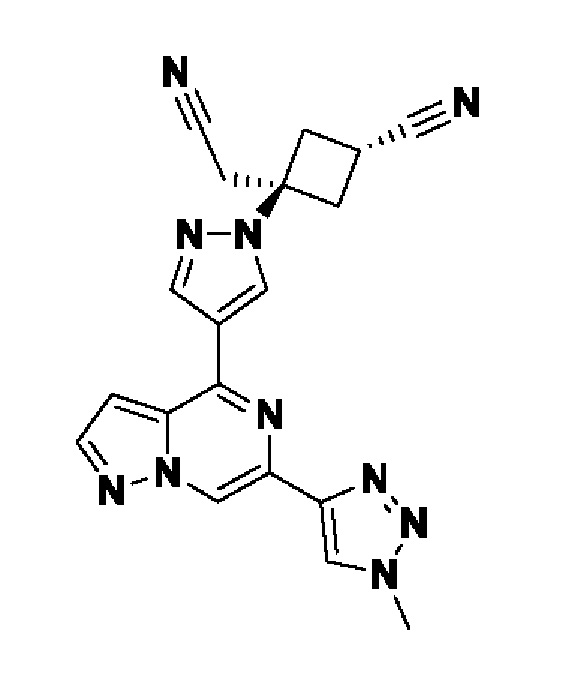
К смеси (1r,3r)-3-(4-(6-(2H-1,2,3-триазол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила (Получение 87, 100 мг, 0,27 ммоль) и K2CO3 (75 мг, 0,54 ммоль) в DMF (1 мл) добавляли иодметан (0,034 мл, 0,540 ммоль). Смесь перемешивали в течение примерно 2 часов при примерно 25°C. Смесь фильтровали и фильтровальную лепешку дважды промывали DCM. Фильтрат концентрировали и остаток очищали с помощью хроматографии, затем ВЭЖХ с получением указанного в заголовке соединения в виде белого твердого вещества (40 мг, 39%).
1H ЯМР (400 МГц, CDCl3) δ: 9,19 (с, 1 H), 8,51 (с, 1 H), 8,33 (с, 1 H), 8,30 (с, 1 H), 8,15 (д, 1 H), 7,03 (с, 1 H), 4,22 (с, 3 H), 3,35-3,46 (м, 3 H), 3,30 (с, 2 H), 2,99 (д, 2 H).
LCMS m/z=385,4 [MH]+
Пример 34
(1s,3s)-3-(цианометил)-1-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
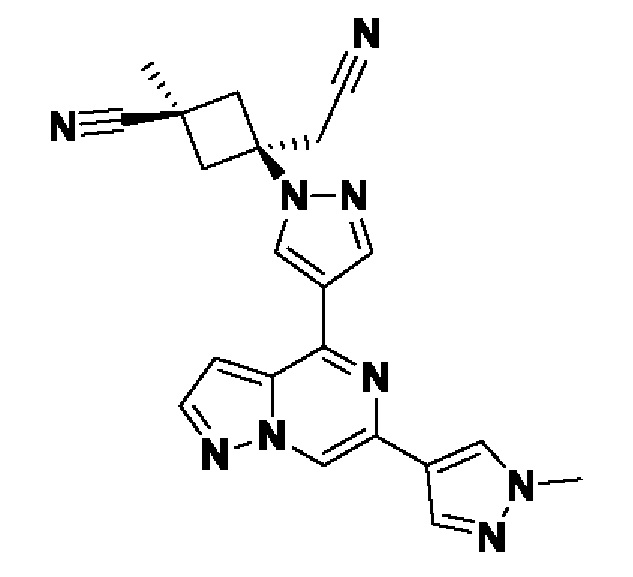
и
Пример 35
(1r,3r)-3-(цианометил)-1-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
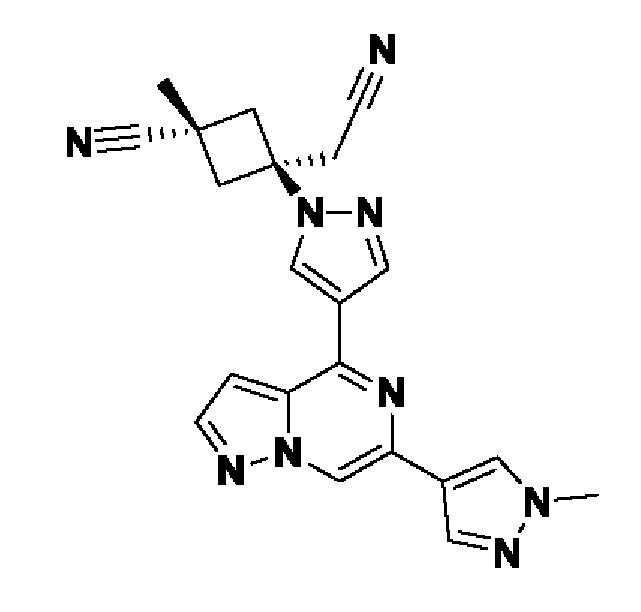
DBU (327 мг, 2,15 ммоль) добавляли к раствору 6-(1-метил-1H-пиразол-4-ил)-4-(1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 53, 190 мг, 0,72 ммоль) и 3-(цианометилен)-1-метилциклобутан-1-карбонитрила (Получение 89, 142 мг, 1,07 ммоль) в MeCN (15 мл) и смесь перемешивали при примерно 50°C в течение примерно 4 часов. Смесь концентрировали и остаток очищали с помощью хроматографии с получением остатка, который далее очищали с помощью TLC с получением смеси двух указанных в заголовке соединений (200 мг, 70%) в виде коричневого твердого вещества. Смесь изомеров разделяли с помощью ВЭЖХ с получением (1r,3r)-3-(цианометил)-1-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила в виде светло-розового твердого вещества (транс-изомер, 24 мг 8%)
1H ЯМР (400 МГц, CD3CN) δ: 8,36 (с, 1 H), 8,28 (с, 1 H), 8,26-8,31 (м, 1 H), 8,14 (с, 1 H), 7,86 (с, 1 H), 7,79 (д, 1 H), 7,73 (с, 1 H), 6,87 (д, 1 H), 3,66 (с, 3 H), 3,10 (с, 2 H), 2,80-2,92 (м, 4 H), 1,25 (с, 3 H).
LCMS m/z=398,0 [MH]+ и 420,0 [MNa]+
и (1s,3s)-3-(цианометил)-1-метил-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (цис-изомер, 24 мг, 8%).
1H ЯМР (400 МГц, CD3CN) δ: 8,68 (с, 1 H), 8,63 (с, 1 H), 8,44 (с, 1 H), 8,14 (с, 1 H), 8,05 (д, 1 H), 8,00 (с, 1 H), 7,15 (д, 1 H), 3,92 (с, 3 H), 3,47-3,55 (м, 2 H), 3,19 (с, 2 H), 2,74-2,82 (м, 2 H), 1,63 (с, 3 H).
LCMS m/z=398,0 [MH]+ и 419,9 [MNa]+
Получение 88
1-метил-3-оксоциклобутан-1-карбонитрил
Часть 1
К раствору диизопропиламина (1,30 г, 12,9 ммоль) в THF (30 мл) добавляли 2,5 M n-BuLi (5,15 мл) при примерно 0°C. Раствор перемешивали в течение примерно 30 мин при примерно 0°C перед охлаждением до примерно -78°C. Добавляли 3-метиленциклобутан-1-карбонитрил (1,00 г, 10,74 ммоль) и раствор перемешивали в течение примерно 1 часа при примерно -78°C. К раствору добавляли иодметан (1,98 г, 14,0 ммоль) при примерно -78°C, затем смеси давали нагреться до примерно 20°C и выдерживали при этой температуре в течение примерно 0,5 часов. Добавляли насыщенный водн. NH4Cl (30 мл) и смесь экстрагировали EtOAc (3×30 мл). Объединенные экстракты EtOAc концентрировали с получением 1-метил-3-метиленциклобутан-1-карбонитрила в виде бледно-желтого масла (1,1 г, 96%).
1H ЯМР (400 МГц, CDCl3) δ: 4,90-4,98 (м, 2 H), 3,23-3,35 (м, 2 H), 2,64-2,75 (м, 2 H), 1,55 (с, 3 H).
Часть 2
К смеси 1-метил-3-метиленциклобутан-1-карбонитрила (1,10 г, 10,3 ммоль) и RuCl3 гидрат (50,9 мг, 0,23 ммоль) в смеси DCM (20 мл), MeCN (20 мл) и воды (40 мл) добавляли NaIO4 (8,78 г, 41,1 ммоль) небольшими порциями при примерно 5°C. Смесь затем перемешивали при примерно 25°C в течение примерно 17 часов. Водную фазу отделяли и экстрагировали DCM (2×50 мл). Экстракты DCM объединяли с фазой DCM-MeCN и сушили (Na2SO4), затем фильтровали через приблизительно 10 г силикагеля. Силикагель промывали DCM (50 мл). Фильтрат концентрировали с получением указанного в заголовке соединения в виде коричневого масла (0,80 г, 71%).
1H ЯМР (400 МГц, CDCl3) δ: 3,71 (м, 2 H), 3,14 (м, 2 H), 1,71 (с, 3 H).
Получение 89
3-(Цианометилен)-1-метилциклобутан-1-карбонитрил
Смесь 1-метил-3-оксоциклобутан-1-карбонитрила (Получение 88, 0,80 г, 7,0 ммоль), (EtO)2P(O)CH2CN (1,43 г, 8,06 ммоль), LiBr (0,955 г, 11,0 ммоль) и TEA (1,48 г, 14,7 ммоль) в THF (20 мл) перемешивали при примерно 25°C в течение примерно 16 часов. Добавляли воду (30 мл) и смесь экстрагировали EtOAc (3×50 мл). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (0,65 г, 70%).
1H ЯМР (400 МГц, CDCl3) δ: 5,34 (м, 1 H), 3,47 (м, 2 H), 2,98 (м, 2 H), 1,60 (с, 3 H).
GCMS m/z=131 [M-H]+
Получение 90
2-(3-метоксициклобутилиден)ацетонитрил

Часть 1
Двенадцать идентичных реакций проводили параллельно следующим образом:
Для каждой реакции в 100 мл герметизированную пробирку загружали метоксиэтен (37 г, 637 ммоль), DIPEA (9,88 г, 76,4 ммоль) и ацетилхлорид (5 г, 60 ммоль) при примерно -30°C. Затем смесь нагревали при примерно 70°C в течение примерно 5 часов. Двенадцать реакций объединяли и промывали 1 M водн. HCl (2×100 мл), насыщенным водн. NaHCO3 (2×100 мл), сушили (Na2SO4) и концентрировали с получением неочищенного образца 3-метоксициклобутан-1-она в виде черного масла (27 г, 37%). Это вещество было примерно на 50% чистым, как оценивалось с помощью 1H ЯМР, и использовали без дополнительной очистки в части 2.
Часть 2
Две идентичные реакции проводили параллельно.
К смеси LiBr (13,4 г, 154 ммоль) и TEA (39 мл, 280 ммоль) в THF (200 мл) добавляли (EtO)2P(O)CH2CN (26 г, 147 ммоль) при примерно 0°C. Перемешивание продолжали при примерно 25°C в течение примерно 2 часов. К этой смеси добавляли раствор неочищенного 3-метоксициклобутан-1-она, полученного в части 1 (13 г, примерно 70 ммоль) в THF (40 мл) при примерно 0°C. Смесь затем перемешивали при примерно 25°C в течение примерно 16 часов. Две реакционные смеси объединяли и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде светло-желтого масла (9,2 г, 56%).
1H ЯМР (400 МГц, CDCl3) δ: 5,25 (м, 1 H), 4,05 (м, 1 H), 3,30 (с, 3 H), 3,25 (м,1 H), 3,10 (м, 1 H), 2,85 (м, 2 H).
LCMS m/z=124,08 [MH]+
Получение 91
(1r,3r)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
(транс-изомер)
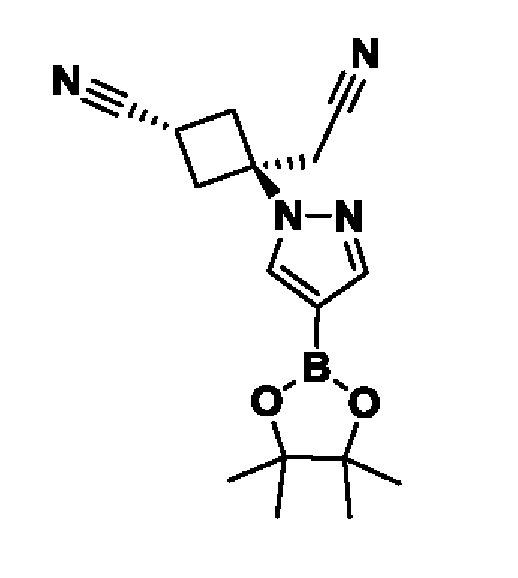
и
(1s,3s)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
(цис-изомер)
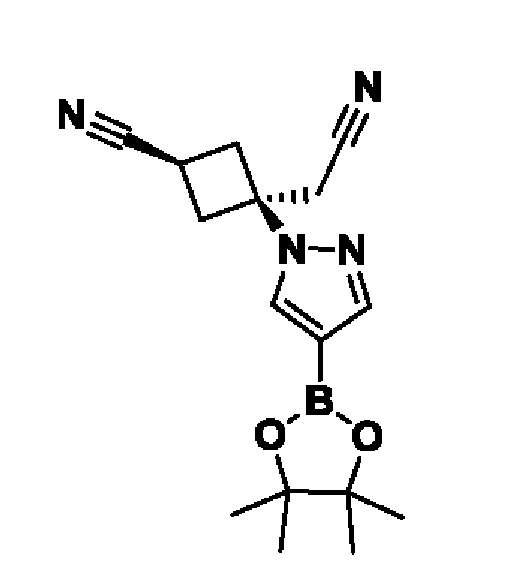
К раствору 4-(4,4,5,5,-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (1,3 кг, 6,67 моль) в MeCN (43 л) добавляли 3-цианометилен)циклобутан-1-карбонитрил (Получение 27, 953 г, 8 моль) и DBU (3,06 кг, 20,1 моль) при примерно 20°C. Перемешивание продолжали при примерно 20°C в течение примерно 16 часов. Смесь выливали в 1 M водн. KH2PO4 (10 л) и экстрагировали EtOAc (5×5 л). Объединенные экстракты EtOAc концентрировали и остаток очищали с помощью хроматографии с получением (1r,3r)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила в виде белого твердого вещества (транс-изомер, 610 г, 30%)
Температура плавления 137-140°C
1H ЯМР (400 МГц, CDCl3) δ: 7,90 (с, 1 H), 7,88 (с, 1 H), 3,21-3,28 (м, 3 H), 3,19 (с, 2 H), 2,86-2,94 (м, 2 H), 1,33 (с, 12 H).
13C ЯМР (101 МГц, CD3OD) δ: 147,54, 136,49, 122,49, 117,11, 84,96, 61,93, 37,52, 30,47, 25,28, 25,18, 17,21.
LCMS m/z=313,1 [MH]+
и (1s,3s)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила в виде не совсем белого твердого вещества (цис-изомер, 250 г, 12%).
Температура плавления 95-98°C
1H ЯМР (400 МГц, CDCl3) δ: 7,86 (с, 2 H), 3,24-3,31 (м, 1 H), 3,13-3,21 (м, 2 H), 3,07 (с, 2 H), 2,96-3,04 (м, 2 H), 1,34 (с, 12 H).
13C ЯМР (101 МГц, CD3OD) δ: 147,43, 135,94, 121,94, 117,37, 84,98, 75,96, 60,10, 37,95, 28,42, 25,29, 16,16.
LCMS m/z=313,1 [MH]+
Получение 92
5-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол
и
3-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол
Смесь 3-метилпиразола (1,00 г, 12,18 ммоль), DHP (1,54 г, 18,3 ммоль) и TFA (0,007 мл, 0,089 ммоль) нагревали при примерно 85°C в течение примерно 4 часов. Смесь охлаждали до примерно 20°C и добавляли NaH (60% в масле, 20 мг, 0,5 ммоль). Перемешивание при примерно 20°C продолжали в течение примерно 18 часов. Смесь концентрировали, остаток очищали с помощью хроматографии с получением 5-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола в виде масла (400 мг, 20%)
1H ЯМР (400 МГц, CDCl3) δ: 7,45 (д, 1 H), 6,05 (д, 1 H), 5,24-5,30 (м, 1 H), 4,00-4,08 (м, 1 H), 3,61-3,70 (м, 1 H), 2,41-2,55 (м, 1 H), 2,35 (с, 3 H), 2,08-2,18 (м, 1 H), 1,93-2,02 (м, 1 H), 1,65-1,77 (м, 2 H), 1,55-1,64 (м, 1 H).
GCMS m/z=166,1 [M]+
и 3-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола в виде масла (352 мг, 15%).
1H ЯМР (400 МГц, CDCl3) δ: 7,48 (д, 1 H), 6,08 (д, 1 H), 5,29 (дд, 1 H), 4,08 (дт, 1 H), 3,64-3,74 (м, 1 H), 2,30 (с, 3 H), 2,07-2,18 (м, 1 H), 1,98-2,07 (м, 2 H), 1,64-1,77 (м, 3 H).
GCMS m/z=166,1 [M]+
Получение 93
3-метил-1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол
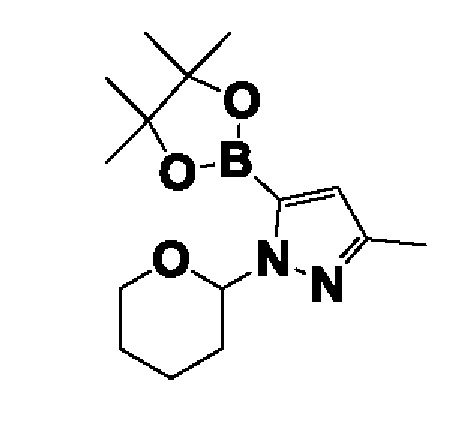
Раствор 2,5 M n-BuLi в гексане (0,44 мл, 1,10 ммоль) добавляли к раствору 3-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола (Получение 92, 200 мг, 1,10 ммоль) в THF (2 мл) при примерно -70°C. Смесь перемешивали при этой температуре в течение примерно 10 мин перед добавлением 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (214 мг, 1,15 ммоль). Смесь выдерживали при примерно -70°C в течение примерно дольше 1 часа, затем нагревали до примерно 20°C и концентрировали с получением указанного в заголовке соединения, смешанного с примерно 65% непрореагировавшего 3-метил-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразола. Эту смесь использовали без дополнительной очистки.
1H ЯМР (400 МГц, CDCl3) δ: 6,51 (с, 1 H), 5,76 (дд, 1 H), 4,01-4,12 (м, 1 H), 3,59-3,74 (м, 1 H), 2,37-2,51 (м, 2 H), 2,29 (с, 3 H), 1,97-2,18 (м, 1 H), 1,62-1,76 (м, 2 H), 1,47-1,62 (м, 1 H), 1,33 (с, 12 H).
GCMS m/z=292,2 [M]+
Получение 94
3-(Бензилокси)-N-метокси-N-метилциклобутанкарбоксамид
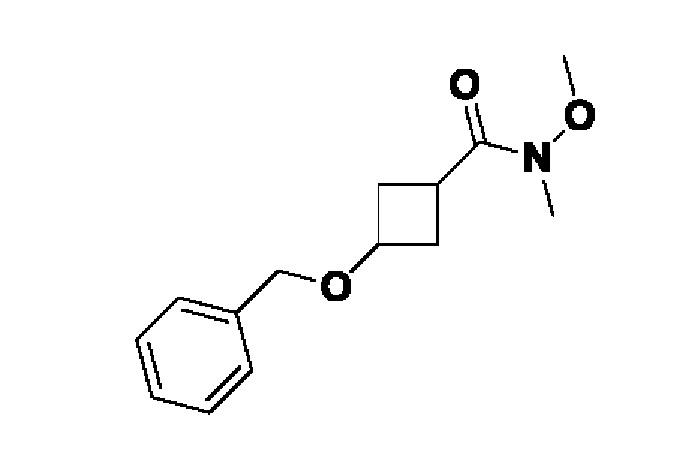
К раствору 3-(бензилокси)циклобутанкарбоновой кислоты (324 г, 1,57 моль) в DCM (1,5 л) добавляли CDI (280 г, 1,73 моль) по частям. Смесь нагревали с обратным холодильником в течение примерно 2 часов, после чего добавляли N,O-диметилгидроксиламин гидрохлорид (183,7 г, 1,88 моль) и TEA (261 мл, 1,88 моль). Смесь нагревали с обратным холодильником в течение примерно 3 часов далее, затем перемешивали при примерно 25°C в течение примерно 16 часов. DCM промывали насыщенным водн. K2СО3, сушили (K2СО3) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (298 г, 76%).
1H ЯМР (400 МГц, DMSO-d6) δ: 7,24-7,38 (м, 5 H), 4,37 (с, 2 H), 4,06-4,15 (м, 1 H), 3,61 (с, 3 H), 3,09 (с, 3 H), 2,96 (с, 1 H), 2,30-2,40 (м, 2 H), 2,09-2,22 (м, 1 H), 1,95-2,07 (м, 1 H).
Получение 95
1-[3-(Бензилокси)циклобутил]этанон
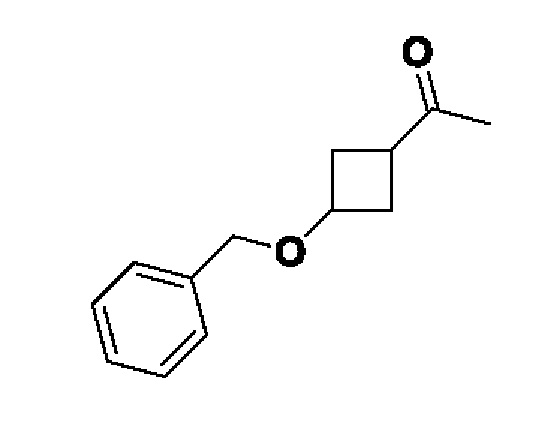
К раствору 3-(бензилокси)-N-метокси-N-метилциклобутанкарбоксамида (Получение 94, 298 г, 1,2 моль) в THF (1,5 л) добавляли по каплям раствор метилмагний бромида (1,3 моль) при примерно -20°C. После завершения добавления метилмагний бромида охлаждающую баню удаляли и смеси давали нагреться до примерно 25°C. Затем добавляли 0,5 M водн. HCl (2,5 л) и смесь экстрагировали Et2O (1 л+500 мл). Объединенные экстракты Et2O промывали водой, сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (194 г, 79%).
1H ЯМР (400 МГц, DMSO-d6) δ: 7,24-7,38 (м, 5 H), 4,35 (с, 2 H), 3,90-3,99 (м, 0,5 H), 3,16-3,24 (м, 0,5 H), 2,75-2,87 (м, 1 H), 2,31-2,40 (м, 2 H), 2,06-2,15 (м, 1 H), 2,04 (с, 1,5 H), 1,99 (с, 1,5 H), 1,87-1,97 (м, 1 H).
Получение 96
1-[3-(Бензилокси)циклобутил]-3-(диметиламино)проп-2-ен-1-он
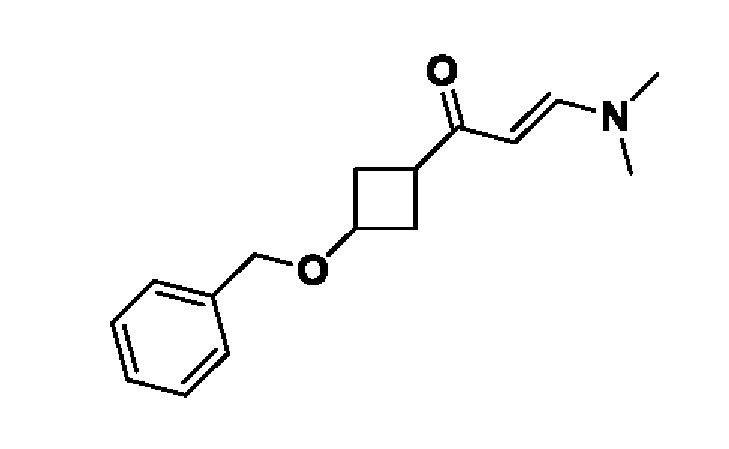
К раствору 1-[3-(бензилокси)циклобутил]этанона (Получение 95, 194 г, 0,95 моль) в DMF (500 мл) добавляли диметилформамид диметилацеталь (285 г, 2,4 моль). Смесь нагревали при примерно 110°С в течение примерно 12 часов. Смесь концентрировали с получением указанного в заголовке соединения (258 г), который использовали без очистки или дополнительной характеристики.
Получение 97
5-[3-(Бензилокси)циклобутил]-1H-пиразол
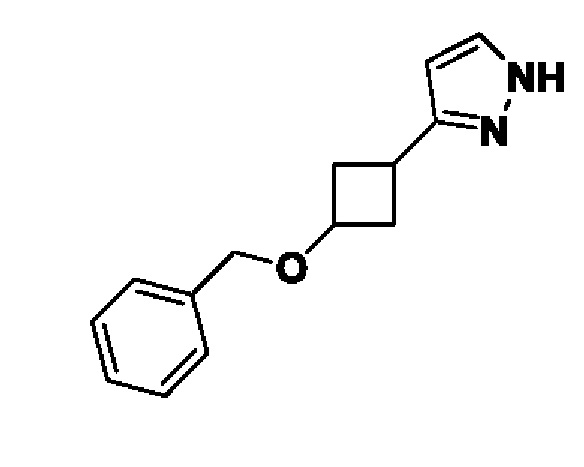
К раствору 1-[3-(бензилокси)циклобутил]-3-(диметиламино)проп-2-ен-1-она (Получение 96, 110 г, 0,42 моль) в MeOH (500 мл) добавляли гидразингидрат (30 г, 0,6 моль). Смесь нагревали с обратным холодильником в течение примерно 12 часов, затем концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (79 г, 82%), который использовали без очистки или дальнейшей характеристики.
Получение 98
3-(1H-пиразол-5-ил)циклобутанол
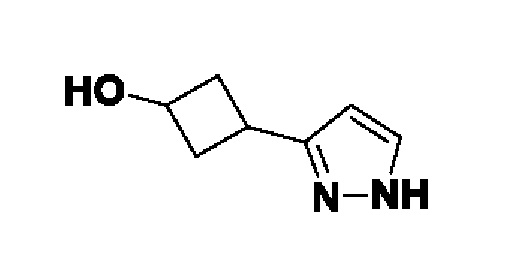
К раствору 5-[3-(бензилокси)циклобутил]-1H-пиразола (Получение 97, 79 г, 0,35 моль) в метаноле (800 мл) в 2 л сосуд для гидрирования добавляли палладий на угле (10% Pd, 16 г). Смесь герметизировали под давлением водорода (30 фунт/кв.дюйм) и смесь встряхивали при примерно 60°C в течение примерно 12 часов. Добавляли дополнительное количество палладия на угле (10% Pd, 16 г), и гидрирование продолжали в течение примерно 10 часов. Смесь фильтровали и фильтрат концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (46 г, 96%), которое использовали без очистки или дополнительной характеристики.
Получение 99
3-(1H-пиразол-5-ил)циклобутан-1-он
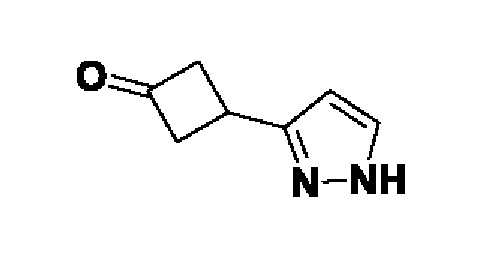
К раствору оксалилхлорида (7,8 мл, 0,09 моль) в DCM (50 мл) при примерно -78°C добавляли по каплям раствор DMSO (12,7 мл, 0,18 моль) в DCM (50 мл). Смесь перемешивали в течение примерно 30 мин, затем 3-(1H-пиразол-5-ил)циклобутанол (Получение 98, 13,7 г, 0,09 моль) добавляли по каплям при этой температуре. Полученную смесь выдерживали в течение примерно 30 мин, после чего добавляли по каплям TEA (25 мл, 0,18 моль). Охлаждающую баню удаляли и смеси давали нагреться до примерно 25°C и выдерживали при этой температуре в течение примерно 3 часов. Затем добавляли водный раствор K2CO3 (100 мл). DCM отделяли и водную фазу экстрагировали DCM. Объединенные экстракты DCM промывали водой, сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения (7,4 г, 55%) в виде белого твердого вещества.
1H ЯМР (400 МГц, DMSO-d6) δ: 12,56 (с, 1 H), 7,61 (с, 1 H), 6,21 (д, 1 H), 3,46-3,68 (м, 1 H), 3,37-3,46 (м, 2 H), 3,15-3,25 (м, 2 H).
LCMS m/z=137,1 [MH]+
Получение 100
2-((1r,3s)-1-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил
(транс-изомер)
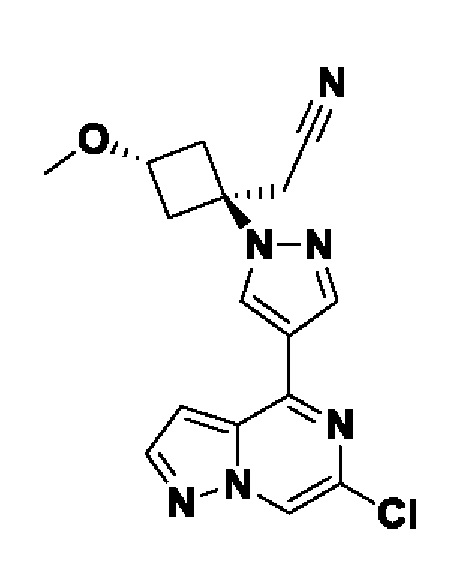
Часть 1
К раствору 2-((1r,3s)-1-(4-бром-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила (Получение 37, транс-изомер, 3399 мг, 12,58 ммоль) в 1,4-диоксане (33 мл) добавляли бис(пинаколато)диборон (3510 мг, 13,8 ммоль) и KOAc (3700 мг, 37,7 ммоль). Смесь продували аргоном в течение примерно 5 мин, после чего добавляли XPhos Pd G2 (1980 мг, 2,52 ммоль). Смесь нагревали при примерно 65°C в течение примерно 4 часов. Охлажденную смесь концентрировали и остаток очищали с помощью хроматографии. Продукт перемешивали с EtOAc (10 мл) при примерно 25°C, затем добавляли гептан (40 мл) и оставляли для кристаллизации в течение примерно 30 мин. Осадок фильтровали и сушили с получением 2-((1r,3s)-3-метокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила в виде белого твердого вещества (1950 мг, 49%).
1H ЯМР (400 МГц, CDCl3) δ: 7,91 (с, 1 H), 7,87 (с, 1 H), 3,98 (тт, 1 H), 3,29 (с, 3 H), 3,17 (с, 2 H), 2,97-3,07 (м, 2 H), 2,44-2,53 (м, 2 H), 1,33 (с, 12 H).
LCMS m/z=318,0 [MH]+
Часть 2
Раствор 2-((1r,3s)-3-метокси-1-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутил)ацетонитрила (часть 1, 1950 мг, 6,15 ммоль) и 4,6-дихлорпиразоло[1,5-а]пиразина (Получение 4, 1160 мг, 6,15 ммоль), и 2 M водн. K3PO4 (9,22 мл) в 1,4-диоксане (25 мл) продували аргоном в течение примерно 5 мин, после чего добавляли бис(три-трет-бутилфосфин)палладий(0) (157 мг, 0,31 ммоль). Смесь перемешивали при примерно 25°C в течение примерно 2 часов. Охлажденную смесь разбавляли EtOAc и фазы разделяли. Водную фазу дважды экстрагировали DCM. Объединенные экстракты EtOAc и DCM сушили (Na2SO4) и концентрировали. Остаток растворяли в DCM (20 мл) и нагревали при примерно 40°C до полного растворения, затем добавляли гептан (10 мл) и оставляли для кристаллизации в течение примерно 30 мин. Осадок фильтровали и сушили с получением указанного в заголовке соединения в виде не совсем белого твердого вещества (1120 мг, 53%). Фильтрат концентрировали и остаток очищали с помощью хроматографии с получением дополнительного указанного в заголовке соединения (640 мг, 30%).
1H ЯМР (500 МГц, CDCl3) δ: 8,39 (д, 1 H), 8,38 (с, 1 H), 8,28 (с, 1 H), 8,08 (д, 1 H), 7,03 (дд, 1 H), 4,03-4,10 (м, 1 H), 3,34 (с, 3 H), 3,25 (с, 2 H), 3,08-3,16 (м, 2 H), 2,53-2,60 (м, 2 H).
LCMS m/z=343,3 [MH]+ (35Cl изотоп)
Получение 101
Этил 5-метил-1-(2-(1-метил-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3-карбоксилат
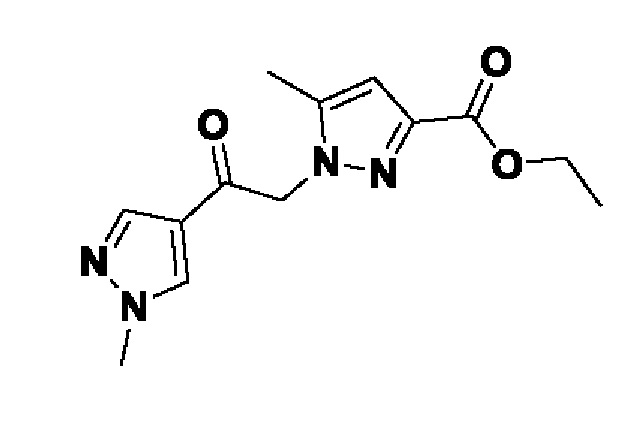
Этил 3-метил-1H-пиразол-5-карбоксилат (92 мг, 0,6 ммоль), 2-бром-1-(1-метил-1H-пиразол-4-ил)этан-1-он (Получение 6, 134 мг, 0,66 ммоль) и K2CO3 (104 мг, 0,75 ммоль). объединяли в MeCN (2 мл) и суспензию перемешивали при примерно 40°C в течение примерно 16 часов. Твердые вещества фильтровали и фильтрат концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого масла (125 мг, 5,5%).
1H ЯМР (400 МГц, CDCl3) δ: 7,86 (с, 2 H), 6,73 (с, 1 H), 5,65 (с, 2 H), 4,26 (кв, 2 H), 3,95 (с, 3 H), 2,32 (с, 3 H), 1,31 (т, 3 H).
LCMS m/z=277,1 [MH]+
Получение 102
2-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ол
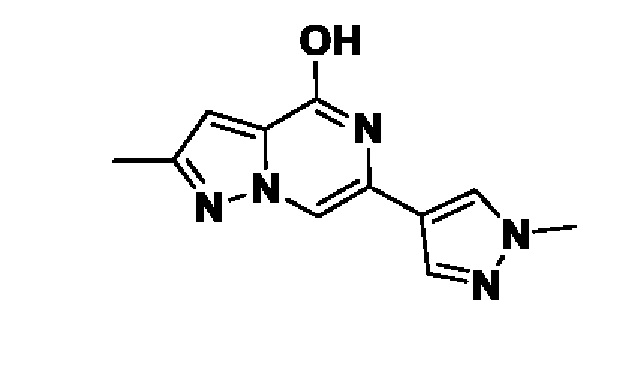
К раствору этил 5-метил-1-(2-(1-метил-1H-пиразол-4-ил)-2-оксоэтил)-1H-пиразол-3-карбоксилата (Получение 101, 125 мг, 0,45 ммоль) в EtOH (5 мл) добавляли NH4OAc (105 мг, 1,06 ммоль). Смесь нагревали при микроволновом облучении при примерно 105°C в течение примерно 4 часов. Смесь концентрировали и остаток растворяли в EtOH и концентрировали снова с получением указанного в заголовке соединения (110 мг, 85%).
1H ЯМР (400 МГц, DMSO-d6) δ: 8,25 (с, 1 H), 8,01 (с, 1 H), 7,97 (с, 1 H), 6,76 (с, 1 H), 3,87 (с, 3 H), 2,34 (с, 3 H).
LCMS m/z=230,0 [MH]+
Получение 103
4-хлор-2-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин
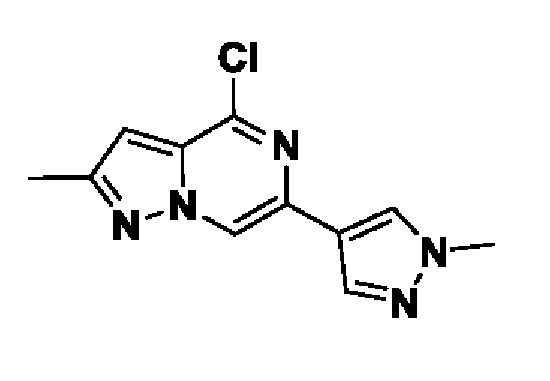
2-Метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ол (Получение 102) суспендировали в POCl3 и нагревали до примерно 120°C в течение примерно 6 часов. Раствор концентрировали, получая неочищенный образец указанного в заголовке соединения, который использовали на следующей стадии без дополнительной очистки.
LCMS m/z=248,0 [MH]+ (35Cl изотоп)
Пример 36
(1r,3r)-3-(цианометил)-3-(4-(2-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
(транс-изомер)
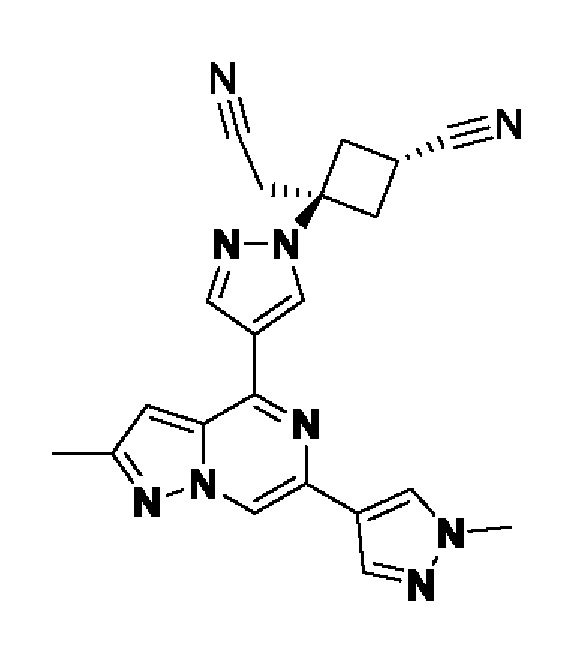
Смесь неочищенного 4-хлор-2-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразина (Получение 103, 100 мг, 0,40 ммоль), (1r,3r)-3-(цианометил)-3-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (Получение 91, 132 мг, 0,42 ммоль), Pd(dppf)Cl2 DCM (16,5 мг, 0,02 ммоль) и K2CO3 (167 мг, 1,21 ммоль) объединяли в смеси 1,4-диоксана (2,5 мл) и воды (0,5 мл). Смесь продували азотом в течение примерно 5 мин, затем нагревали при примерно 90°C в течение примерно 3 часов. Смесь концентрировали и остаток очищали с помощью хроматографии и ВЭЖХ с получением указанного в заголовке соединения (5,6 мг, 3% за две стадии).
1H ЯМР (400 МГц, CDCl3) δ: 8,37 (с, 1 H), 8,33 (с, 1 H), 8,30 (с, 1 H), 7,92 (с, 1 H), 7,90 (с, 1 H), 6,68 (с, 1 H), 3,99 (с, 3 H), 3,32-3,42 (м, 3 H), 3,27 (с, 2 H), 2,93-3,02 (м, 2 H), 2,56 (с, 3 H).
LCMS m/z=398,0 [MH]+
Получение 104
3-((4-Метоксибензил)окси)-1-метил-1H-пиразол
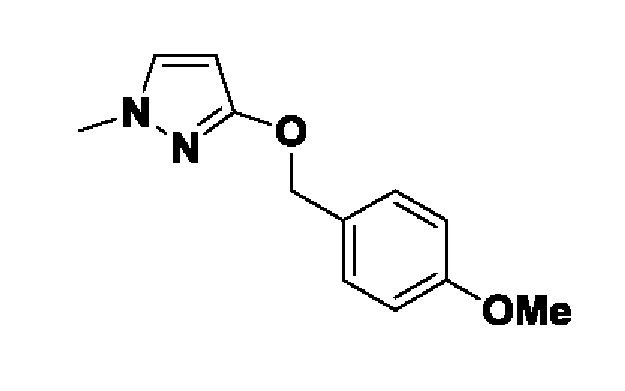
В 100 мл круглодонную колбу добавляли 1-метил-1H-пиразол-3-ол (1,40 г, 14,3 ммоль), DMF (30 мл), и K2CO3 (3,94 г, 28,5 ммоль). В заключение к смеси добавляли 4-метоксибензилхлорид (2,32 мл, 17,1 ммоль). Смесь нагревали при примерно 60°C в течение примерно 8 часов. Смесь затем разбавляли водой (80 мл) и экстрагировали EtOAc (60 мл×3). Объединенные экстракты EtOAc промывали насыщенным солевым раствором (60 мл×2), сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде бесцветного масла (2,6 г, 83%).
1H ЯМР (400 МГц, CDCl3) δ: 7,38 (д, 2 H), 7,13 (д, 1 H), 6,91 (д, 2 H), 5,64 (д, 1 H), 5,11 (с, 2 H), 3,82 (с, 3 H), 3,76 (с, 3 H).
LCMS m/z=218,9 [MH]+
Получение 105
4-Иод-3-((4-метоксибензил)окси)-1-метил-1H-пиразол

В 100 мл круглодонную колбу добавляли 3-((4-метоксибензил)окси)-1-метил-1H-пиразол (Получение 104, 1,0 г, 4,58 ммоль) и MeCN (20 мл), после чего к смеси добавляли церий аммония нитрат (1,51 г, 2,75 ммоль) и йод (698 мг, 2,75 ммоль). Коричневую смесь перемешивали при примерно 20°C в течение примерно 1 часа. Смесь гасили 5% водным бисульфитом натрия (50 мл) и экстрагировали EtOAc (40 мл×3). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде зеленого масла (800 мг, 51%).
1H ЯМР (400 МГц, CDCl3) δ: 7,40 (д, 2 H), 7,19 (с, 1 H), 6,91 (д, 2 H), 5,18 (с, 2 H), 3,82 (с, 3 H), 3,77 (с, 3 H).
Получение 106
3-((4-Метоксибензил)окси)-1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол
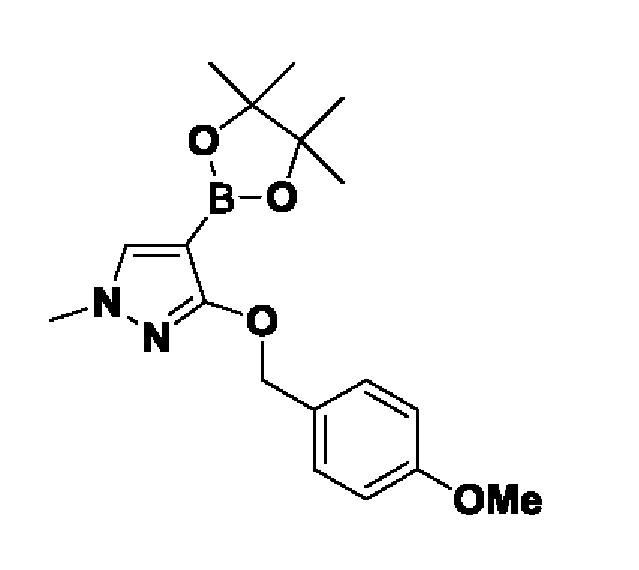
В 100 мл круглодонную колбу добавляли 4-иод-3-((4-метоксибензил)окси)-1-метил-1H-пиразол (Получение 105, 800 мг, 2,32 ммоль) и THF (16 мл). с последующим добавлением к смеси по каплям хлорида изопропилмагния (1,3 M в THF, 2,15 мл, 2,79 ммоль) при примерно -10°C. Смесь перемешивали при температуре между примерно -18°C и 10°C в течение примерно 45 мин. 2-Изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолан (649 мг, 3,49 ммоль) добавляли к смеси при примерно -10°C и смеси давали нагреться до примерно 15°C в течение примерно 1,5 часа. Дополнительное количество хлорида изопропилмагния (1,3 M в THF, 0,72 мл, 0,93 ммоль) и 2-изопропокси-4,4,5,5-тетраметил-1,3,2-диоксаборолана (216 мг, 1,16 ммоль) добавляли к смеси при примерно -15°C. Смеси давали нагреться до примерно 15°C в течение примерно 1 часа. Смесь разбавляли EtOAc (40 мл) и промывали насыщенным водным NH4Cl (30 мл), насыщенным солевым раствором (30 мл), сушили (Na2SO4), и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде белого твердого вещества (600 мг, 75%).
1H ЯМР (400 МГц, CDCl3) δ: 7,40-7,45 (м, 3 H), 6,88 (д, 2 H), 5,24 (с, 2 H), 3,81 (с, 3 H), 3,73 (с, 3 H), 1,31 (с, 12 H).
LCMS m/z=345,1 [MH]+
Получение 107
(1r,3r)-3-(цианометил)-3-(4-(6-(3-((4-метоксибензил)окси)-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил
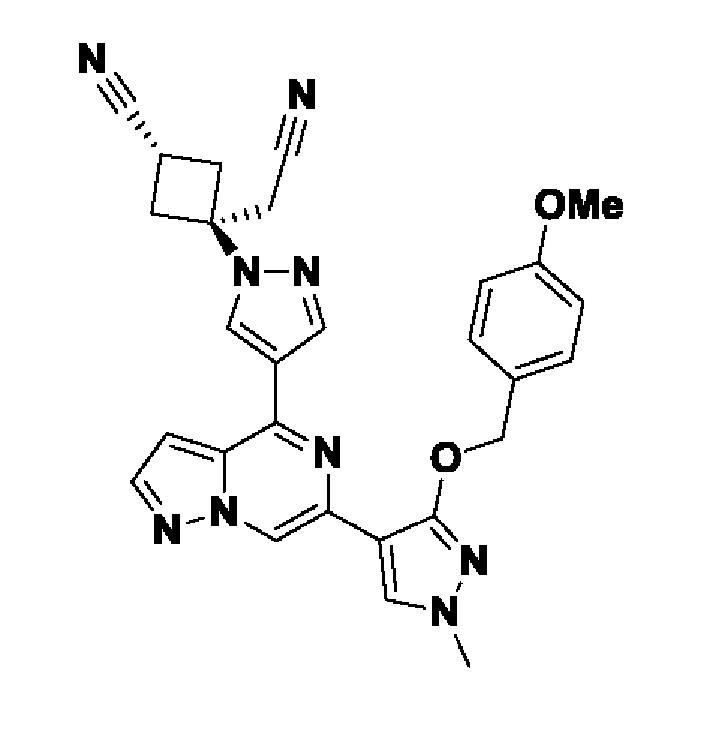
В 25 мл круглодонную колбу добавляли (1r,3r)-3-(4-(6-хлорпиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрил (Получение 75, 300 мг, 0,88 ммоль), 3-((4-метоксибензил)окси)-1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (Получение 106, 367 мг, 1,07 ммоль), диоксан (16 мл), XPhos Pd G2 (140 мг, 0,178 ммоль) и 2 M водн. K3PO4 (3,55 мл, 7,11 ммоль). Смесь помещали под азот, затем нагревали при примерно 60°C в течение примерно 4 часов. Смесь разбавляли водой (70 мл) и экстрагировали EtOAc (50 мл×3). Объединенные экстракты EtOAc сушили (Na2SO4) и концентрировали. Остаток очищали с помощью хроматографии с получением указанного в заголовке соединения в виде желтого твердого вещества (450 мг, 98%).
1H ЯМР (400 МГц, DMSO-d6) δ: 8,92 (с, 1 H), 8,57 (с, 1 H), 8,52 (с, 1 H), 8,31 (с, 1 H), 8,16 (д, 1 H), 7,50 (д, 2 H), 7,45 (с, 1 H), 7,00 (д, 2 H), 5,29 (с, 2 H), 3,95 (с, 3 H), 3,82 (с, 3 H), 3,55-3,59 (м, 1 H), 3,52 (с, 2 H), 3,25-3,32 (м, 2 H), 2,94 (дд, 2 H).
LCMS m/z=542,1 [MNa]+
Пример 37
(1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-3-оксо-2,3-дигидро-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил (транс-изомер)
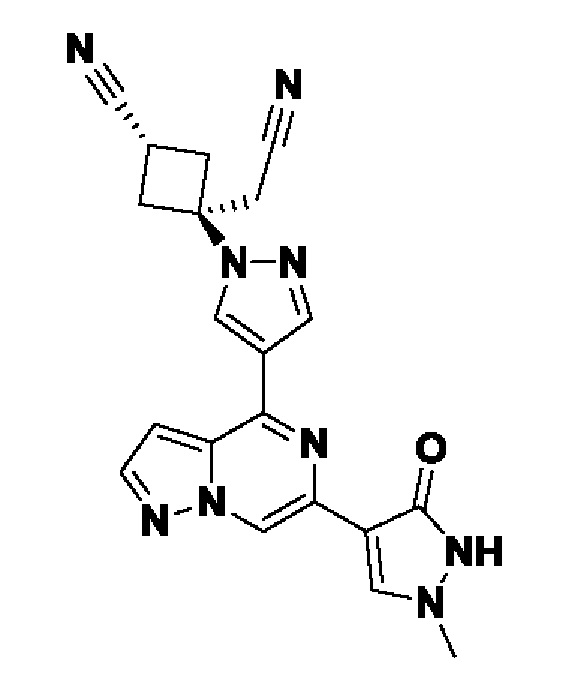
(1r,3r)-3-(цианометил)-3-(4-(6-(3-((4-метоксибензил)окси)-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил (Получение 107, 450 мг, 0,86 ммоль) и TFA (13 мл) перемешивали при примерно 10°C в течение примерно 4 часов. Смесь концентрировали и остаток разбавляли DCM (40 мл) и MeOH (40 мл) и нейтрализовали твердым NaHCO3. Смесь фильтровали. Фильтрат концентрировали и остаток очищали с помощью ВЭЖХ с получением указанного в заголовке соединения в виде желтого твердого вещества (54 мг, 16%).
1H ЯМР (400 МГц, DMSO-d6) δ: 10,76 (шир.с, 1 H), 8,92 (с, 1 H), 8,71 (с, 1 H), 8,51 (с, 1 H), 8,18 (д, 1 H), 8,16 (с, 1 H), 7,45 (д, 1 H), 3,73 (с, 3 H), 3,55-3,62 (м, 1 H), 3,53 (с, 2 H), 3,25-3,33 (м, 2 H), 2,90-2,99 (м, 2 H).
LCMS m/z=400,1 [MH]+
Биологическая оценка
Соединения по изобретению оценивали методами in vitro для определения их соответствующей способности ингибировать киназы JAK (TYK2, JAK1, JAK2, JAK3).
Формат анализа
Ингибирующую активность JAK человека определяли с использованием микрожидкостного анализа для мониторинга фосфорилирования синтетического пептида рекомбинантным человеческим киназным доменом каждого из четырех членов семейства JAK, JAK1, JAK2, JAK3 и TYK2. Реакционные смеси содержали 1 мкМ флуоресцентно меченого синтетического пептида, концентрация меньше кажущегося Km, и 1 мМ АТФ. Каждое условие анализа было оптимизировано для концентрации фермента и времени инкубации при комнатной температуре, чтобы получить конверсию 20%-30% фосфорилированного пептидного продукта. Реакции прекращали добавлением стоп-буфера, содержащего EDTA. Используя технологию смещения подвижности LabChip 3000 (Caliper Life Science), в каждом анализе реакции отбирали образец для определения уровня фосфорилирования. Эта технология, основанная на разделении, позволяет прямое обнаружение флюоресцентно меченных субстратов и продуктов. Разделения контролируют комбинацией вакуумного давления и напряженности электрического поля, оптимизированной для каждого пептидного субстрата.
Протокол анализа
Ферментативный анализ JAK Caliper при 1 мМ АТР
Соединения добавляли в 384-луночный планшет. Реакционные смеси содержали 10 мМ HEPES, рН 7,4, 10 мМ MgCl2, 0,01% BSA, 0,0005% Tween 20, 1 мM ATP и 1 мкМ пептидный субстрат. JAK1 и TYK2 анализы содержали 1 мкМ пептида IRStide (5FAM-KKSRGDYMTMQID), и JAK2 и JAK3 анализы содержали 1 мкМ пептида JAKtide (FITC-KGGEEEEYFELVKK). Анализы инициировали добавлением 20 нМ JAK1, 1 нМ JAK2, 1 нМ JAK или 1 нМ TYK2 фермента и инкубировали при комнатной температуре в течение трех часов для JAK1, 60 минут для JAK2, 75 минут для JAK3 или 135 минут для TYK2. Концентрации ферментов и время инкубирования оптимизировали для каждого нового препарата ферментов и слегка модифицировали в зависимости от времени для обеспечения 20-30% фосфорилирования. Анализы останавливали с помощью 15 мкл 180 мM HEPES, pH 7,4, 20 мM EDTA, и 0,2% покрывающего реагента 3. Аналитические планшеты помещали на прибор Caliper Life Science LC3000 и с каждой лунки отбирали образец с использованием подходящих условий разделения для измерения нефосфорилированного и фосфорилированного пептида.
Анализ данных
Данные были собраны с использованием программного обеспечения HTS Well Analyzer от Caliper Life Sciences. Выходные данные для анализа данных представляют собой процент преобразованного продукта, рассчитанный по высоте пика (уравнение 1).
Уравнение 1: %преобразованного продукта=100*((продукт)/(продукт+субстрат))
Процентный эффект при каждой концентрации соединений рассчитывали на основе положительной и отрицательной контрольной лунки, содержащейся в каждом аналитическом планшете (уравнение 2). Положительные контрольные лунки содержали насыщенную концентрацию контрольного соединения, которая вызывала уровень фосфорилирования, сравнимый с фоном (то есть полностью ингибировал JAK1, JAK2, JAK3 или TYK2). Отрицательные контрольные лунки содержали только DMSO (при той же концентрации, что и лунки, содержащие соединение), который использовался для определения исходной активности в анализе (то есть, неингибированный JAK1, JAK2, JAK3 или TYK2).
уравнение 2: %эффект=100*((лунка с образцом- отрицательный контроль)/(положительный контроль-отрицательный контроль))
Процентный эффект был нанесен на график в сравнении с концентрацией соединения. С использованием 4-параметрической логистической модели была построена неограниченная сигмоидная кривая и определена концентрация соединения, необходимая для 50% ингибирования (IC50) (уравнение 3).
уравнение 3:
y=((max - min)/(1+((x/IC50)^s)))+min
Где max представляет собой максимальную асимптоту (полное ингибирование), min представляет собой минимальную асимптоту (отсутствие ингибирования), s представляет собой коэффициент наклона. Значения IC50 представлены в нM для каждого соединения:
Таблица I. Данные JAK Caliper
IC50 (нM)
IC50 (нM)
IC50 (нM)
IC50 (нM)
Отобранные соединения оценивали на их способность ингибировать передачу сигналов IL-12 в анализе проточной цитометрии цельной крови человека. Сигналы IL-12 через TYK2 и JAK2.
Анализ человеческой цельной крови (HWB) IL-12 индуцированного фосфорилирования STAT4
Тестируемые образцы готовили в виде 30 мМ исходных растворов в DMSO. Делали 11-точечный 2,5 ряд разведений в DMSO с верхней концентрацией 10 мМ. Дальнейшее разведение производили путем добавления 4 мкл вышеописанных растворов тестируемых образцов в 96 мкл PBS с верхней концентрацией 400 мкМ. Человеческую цельную кровь собирали у здоровых доноров через прокол вены в пробирки для сбора образцов Vacutainer, содержащие гепарин натрия (Catalog No. 366480; Becton Dickinson, Franklin Lakes, NJ). Кровь нагревали до 37°С перед использованием. Человеческую цельную кровь разделяли на аликвоты (90 мл/лунку) в 96-луночных планшетах с глубокими лунками с V-образным дном и обрабатывали соединениями в 11 различных концентрациях (0,2% DMSO окончательный) при 37°С в течение 60 минут. За этим следовала нагррузка IL-12 (5 мл/лунка; конечный, 5 нг/мл) в течение 15 минут. Образцы обрабатывали теплым 1X буфером Lyse/Fix (700 мл/лунку) для прекращения активации и дополнительно инкубировали при 37°C в течение 20 минут для лизиса эритроцитов. Планшеты центрифугировали при 300×g в течение 5 минут, супернатант отсасывали и клетки промывали 800 мл на лунку окрашивающим буфером (PBS, содержащий 0,5% фетальной бычьей сыворотки и 0,01% азида натрия). Промытый клеточный осадок ресуспендировали с 350 мл/лунку предварительно охлажденного 90% метанола и инкубировали при 4°С в течение 30 минут. Планшеты центрифугировали при 300×g в течение 5 минут, супернатант, содержащий 90% метанол удаляли, и клетки промывали 800 мл/лунку окрашивающим буфером. Клеточные осадки ресуспендировали в окрашивающем буфере, содержащем анти-pSTAT4-AlexaFluor647 (разведение 1-150, 150 мл/лунку) и инкубировали при комнатной температуре в темноте в течение ночи.
Образцы переносили в 96-луночные планшеты с U-образным дном, и проточный цитометрический анализ проводили на FACSCalibur или LSRFortessa, оборудованных HTS для автоматической подачи проб из планшетов (BD Biosciences). Популяцию лимфоцитов гейтировали для анализа гистограммы pSTAT4. Фоновую флуоресценцию определяли с использованием нестимулированных клеток, и ʺворотаʺ были установлены у основания пика для включения ~0,5% популяции клеток, дающих сигнал выше порогового значения. Статистический анализ гистограммы осуществляли с использованием программного обеспечения CellQuestÔ Pro версия 5.2.1 (BD Biosciences) или FACSDiva версия 6.2 (BD Biosciences). Относительную единицу флуоресценции (RFU), которая измеряет уровень фосфо-STAT4, рассчитывали путем умножения процента положительной популяции и ее средней флуоресценции. Данные из 11 концентраций соединения (в одну повторность для каждой концентрации) нормализовали как процент контроля на основе формулы:
% контроля =100×(A-B)/(C-B)
где A представляет собой RFU из лунок, содержащих соединение и IL-12, B представляет собой RFU из лунок без IL-12 и соединения (минимальная флуоресценция) и C представляет собой RFU из лунок, содержащих только IL-12 (максимальная флуоресценция). Кривые ингибирования и значения IC50 определяли с использованием программного обеспечения Prism версия 5 (GraphPad, La Jolla, CA).
Таблица II. Данные человеческой цельной крови IL-12.
| название | год | авторы | номер документа |
|---|---|---|---|
| БИЦИКЛИЧЕСКИЕ ГЕТЕРОЦИКЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ МЕК | 2009 |
|
RU2509078C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИДИНА В КАЧЕСТВЕ ИНГИБИТОРОВ КИНАЗЫ 1β-ДРЕНЕРГИЧЕСКОГО РЕЦЕПТОРА | 2006 |
|
RU2415855C2 |
| БОРСОДЕРЖАЩИЕ ИНГИБИТОРЫ PDE4 | 2019 |
|
RU2793936C2 |
| АНИЛИДЫ АМИНОКИСЛОТ В КАЧЕСТВЕ НИЗКОМОЛЕКУЛЯРНЫХ МОДУЛЯТОРОВ IL-17 | 2019 |
|
RU2815505C2 |
| ГЕТЕРОАРОМАТИЧЕСКИЕ МОДУЛЯТОРЫ РЕТИНОЛ-СВЯЗАННОГО ОРФАННОГО РЕЦЕПТОРА ГАММА | 2017 |
|
RU2771280C2 |
| ПРОИЗВОДНЫЕ АМИНОХИНАЗОЛИНА В КАЧЕСТВЕ P2X3 ИНГИБИТОРОВ | 2020 |
|
RU2821714C2 |
| ЗАМЕЩЕННЫЕ ГЕТЕРОЦИКЛИЧЕСКИЕ СУЛЬФОНАМИДНЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ В КАЧЕСТВЕ МОДУЛЯТОРОВ TRPA 1 | 2014 |
|
RU2675792C2 |
| ПРОИЗВОДНЫЕ ПИРИДОПИРИМИДИНОВ В КАЧЕСТВЕ ИНГИБИТОРОВ P2X | 2020 |
|
RU2833037C2 |
| НЕСТЕРОИДНЫЕ МОДУЛЯТОРЫ ГЛЮКОКОРТИОДИДНЫХ РЕЦЕПТОРОВ ДЛЯ МЕСТНОЙ ДОСТАВКИ ЛЕКАРСТВ | 2016 |
|
RU2731618C2 |
| ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ, ИСПОЛЬЗУЕМЫЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDK1 | 2010 |
|
RU2615130C2 |
Изобретение относится к соединению, имеющему структуру (I), или его фармацевтически приемлемой соли, где A, A' и A'' представляют собой независимо O, C=O, C-R' или N-R'', где R' и R'' могут независимо быть H, амино, -NR7COR6, -CONR7R8, C1-C6 алкилом или гидрокси(C1-C6 алкил)-, и R'' может присутствовать или отсутствовать и присутствует там, где позволяют правила валентности, и где не более чем один из A, A' и A'' представляет собой O; R0 и R независимо представляют собой H или C1-C6 алкил; R1 представляет собой H; R2 выбран из группы, состоящей из H, C1-C6 алкила-, C1-C6 алкокси-, фенил(C1-C6 алкила)-, пиразола, -COR6 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил и -SO2-R9, где R9 представляет собой C1-C6 алкил; где каждый из указанных алкила и циклоалкила может быть незамещенным или замещенным галогеном; X представляет собой C-R3 или N, где R3 может быть H или C1-C6 алкилом; R4 и R5 независимо представляют собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил)-; R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил или C3-C8 циклоалкил и n равен 0, 1, 2 или 3. Производные пиразоло[1,5-a]пиразин-4-ила обладают ингибирующим действием в отношении янус-киназ (JAK). 23 з.п. ф-лы, 2 табл., 37 пр.
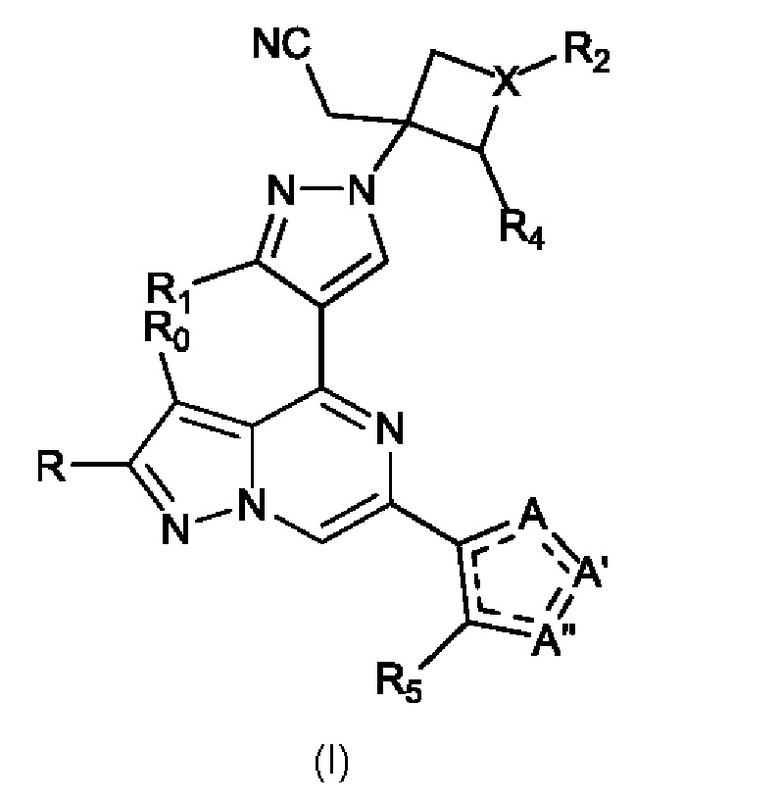
1. Соединение, имеющее структуру (I):
или его фармацевтически приемлемая соль, где:
A, A' и A'' представляют собой независимо O, C=O, C-R' или N-R'', где R' и R'' могут независимо быть H, амино, -NR7COR6, -CONR7R8, C1-C6 алкилом или гидрокси(C1-C6 алкил)-, и R'' может присутствовать или отсутствовать и присутствует там, где позволяют правила валентности, и где не более чем один из A, A' и A'' представляет собой O;
R0 и R независимо представляют собой H или C1-C6 алкил;
R1 представляет собой H;
R2 выбран из группы, состоящей из H, C1-C6 алкила-, C1-C6 алкокси-, фенил(C1-C6 алкила)-, пиразола, -COR6 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил и -SO2-R9, где R9 представляет собой C1-C6 алкил; где каждый из указанных алкила и циклоалкила может быть незамещенным или замещенным галогеном;
X представляет собой C-R3 или N, где R3 может быть H или C1-C6 алкилом;
R4 и R5 независимо представляют собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил)-;
R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил или C3-C8 циклоалкил;
и n равен 0, 1, 2 или 3.
2. Соединение по п.1, имеющее структуру (Ia):
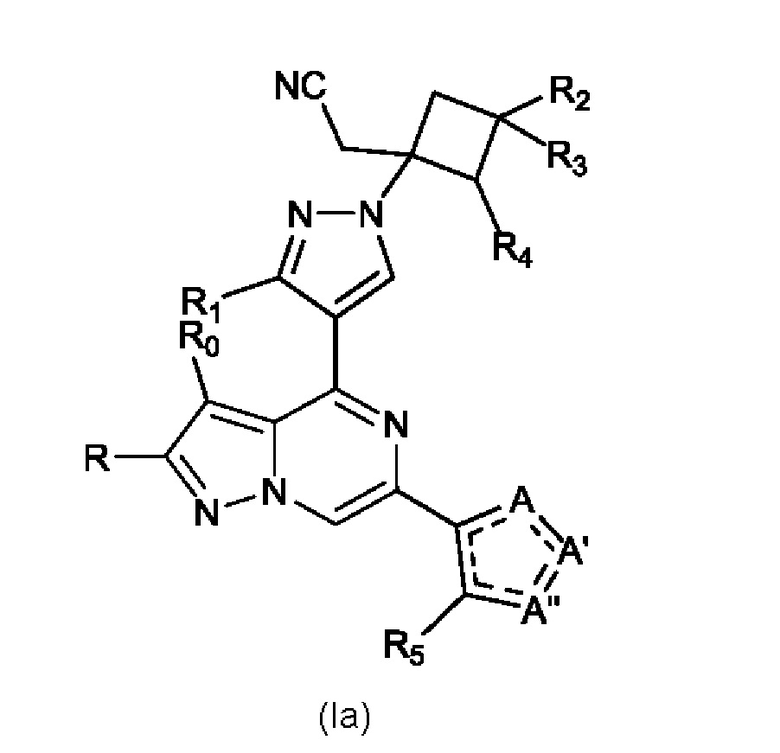
или его фармацевтически приемлемая соль, где:
A, A' и A'' представляют собой независимо O, C=O, C-R' или N-R'', где R' и R'' могут независимо быть H, амино, -NR7COR6, -CONR7R8, C1-C6 алкилом или гидрокси(C1-C6 алкил), и R'' может присутствовать или отсутствовать и присутствует там, где позволяют правила валентности, и где не более чем один из A, A' и A'' представляет собой O;
R0 и R независимо представляют собой H или C1-C6 алкил;
R1 представляет собой H;
R2 выбран из группы, состоящей из H, C1-C6 алкила-, C1-C6 алкокси-, фенил(C1-C6 алкила)-, пиразола, -COR6 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил и -SO2-R9, где R9 представляет собой C1-C6 алкил; где каждый из указанных алкила и циклоалкила может быть незамещенным или замещенным галогеном;
R3 может быть H или C1-C6 алкилом;
R4 и R5 независимо представляют собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил)-;
R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил или C3-C8 циклоалкил;
и n равен 0, 1, 2 или 3.
3. Соединение по п.1, имеющее структуру (Ib):
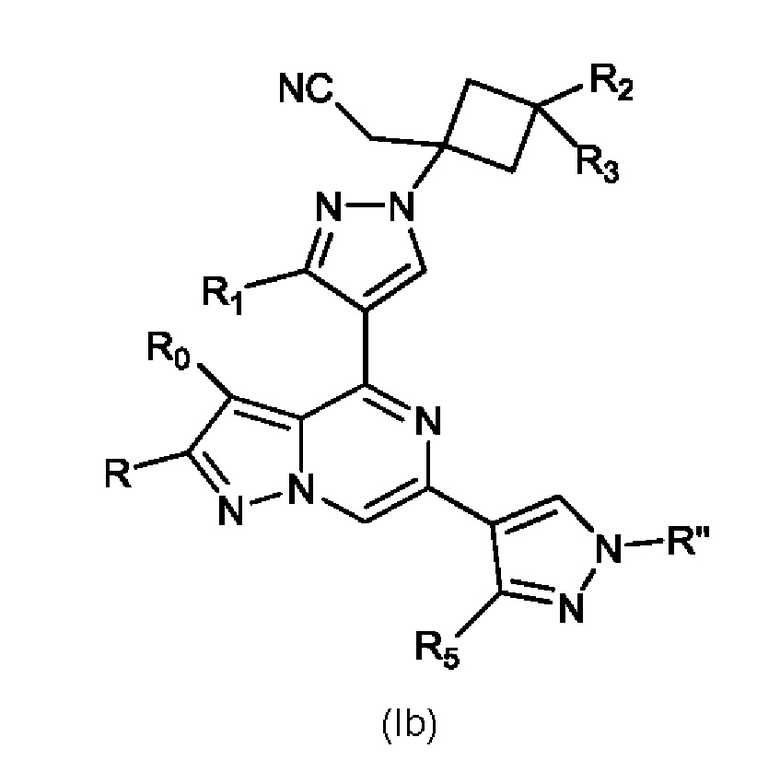
или его фармацевтически приемлемая соль, где:
R'' представляет собой H, -CONR7R8, C1-C6 алкил или гидрокси(C1-C6 алкил)-;
R0 и R независимо представляют собой H или C1-C6 алкил;
R1 представляет собой H;
R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси, фенил(C1-C6 алкила), пиразола, -COR6 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил и -SO2-R9, где R9 представляет собой C1-C6 алкил; где каждый из указанных алкила и циклоалкила может быть незамещенным или замещенным галогеном;
R3 может быть H или C1-C6 алкилом;
R5 независимо представляет собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил);
R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил или C3-C8 циклоалкил;
и n равен 0, 1, 2 или 3.
4. Соединение по п.3, где Rʺ представляет собой C1-C6 алкил и R5 представляет собой H.
5. Соединение по п.1, имеющее структуру (Ic):
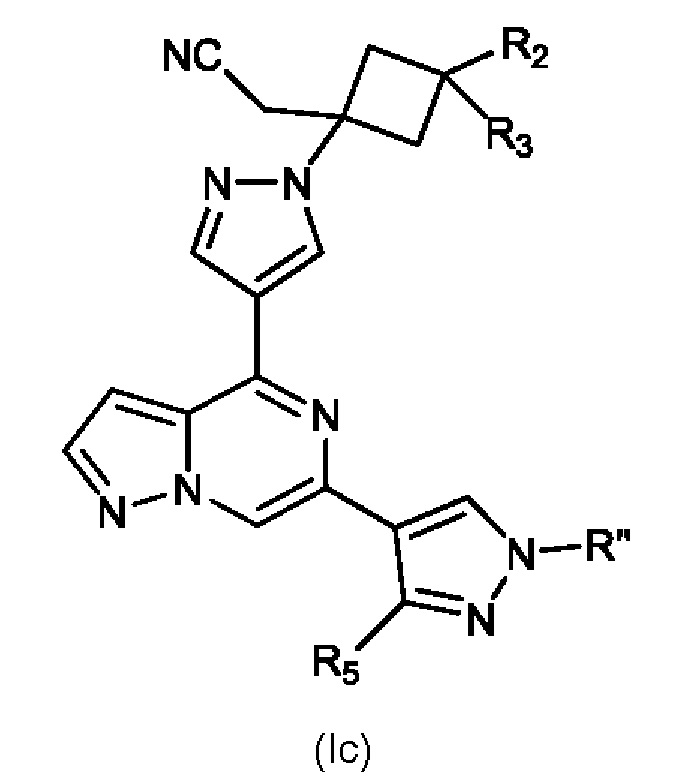
или его фармацевтически приемлемая соль, где:
R'' представляет собой H, -CONR7R8, C1-C6 алкил- или гидрокси(C1-C6 алкил)-;
R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси-, фенил(C1-C6 алкила-), пиразола, -COR6 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил и -SO2-R9, где R9 представляет собой C1-C6 алкил; где каждый из указанных алкила и циклоалкила может быть незамещенным или замещенным галогеном;
R3 представляет собой H или C1-C6 алкил;
R5 представляет собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил)-;
R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил или C3-C8 циклоалкил;
и n равен 0, 1, 2 или 3.
6. Соединение по п.5, где R'' представляет собой C1-C6 алкил и R5 представляет собой H.
7. Соединение по п.1, имеющее структуру (Id):
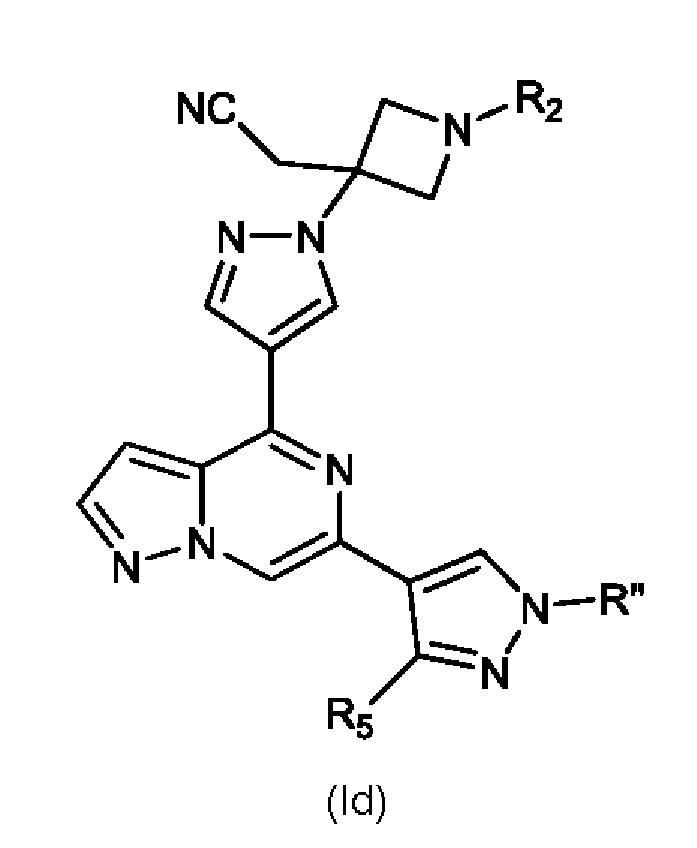
или его фармацевтически приемлемая соль, где:
R'' представляет собой H, -CONR7R8, C1-C6 алкил или гидрокси(C1-C6 алкил)-;
R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси-, фенил(C1-C6 алкила)-, пиразола, -COR6 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил- и -SO2-R9, где R9 представляет собой C1-C6 алкил; где каждый из указанных алкила и циклоалкила может быть незамещенным или замещенным галогеном;
R5 представляет собой H, амино, C1-C6 алкил- или гидрокси(C1-C6 алкил)-;
R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил или C3-C8 циклоалкил;
и n равен 0, 1, 2 или 3.
8. Соединение по п.7, где R'' представляет собой C1-C6 алкил и R5 представляет собой H.
9. Соединение по п.1, имеющее структуру (Ie):
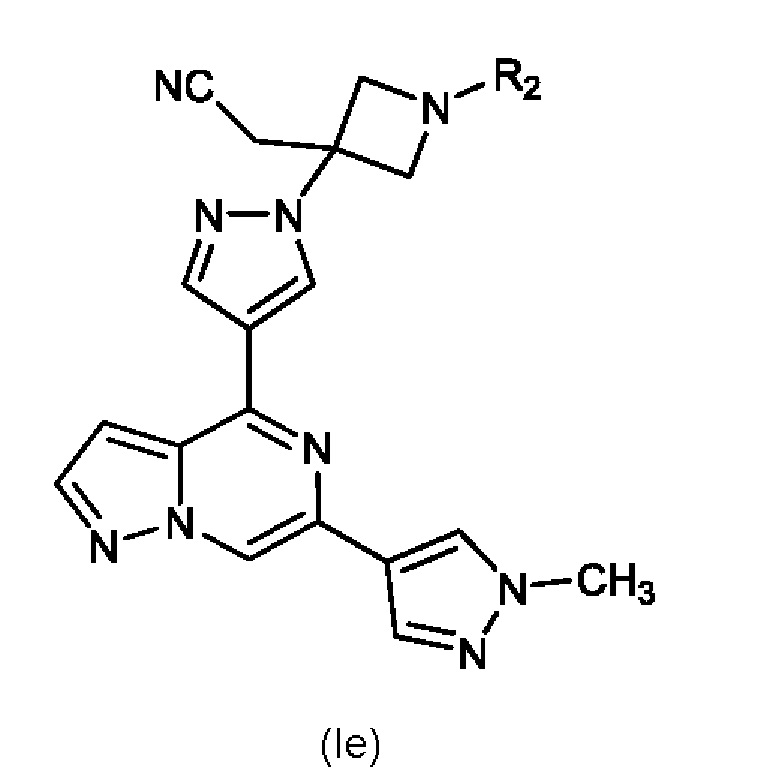
или его фармацевтически приемлемая соль, где:
R2 выбран из группы, состоящей из H, C1-C6 алкила, C1-C6 алкокси-, фенил(C1-C6 алкила)-, пиразола, -COR6 и -(CH2)n-W, где W представляет собой циано, гидрокси, C3-C8 циклоалкил и -SO2-R9, где R9 представляет собой C1-C6 алкил; где каждый из указанных алкила и циклоалкила может быть незамещенным или замещенным галогеном;
R6, R7 и R8, каждый независимо, представляют собой H, C1-C6 алкил или C3-C8 циклоалкил;
и n равен 0, 1, 2 или 3.
10. Соединение по п.9, где R2 представляет собой -(CH2)n-W, где W представляет собой циано и n равен 1, 2 или 3.
11. Соединение по п.1, имеющее структуру (If):
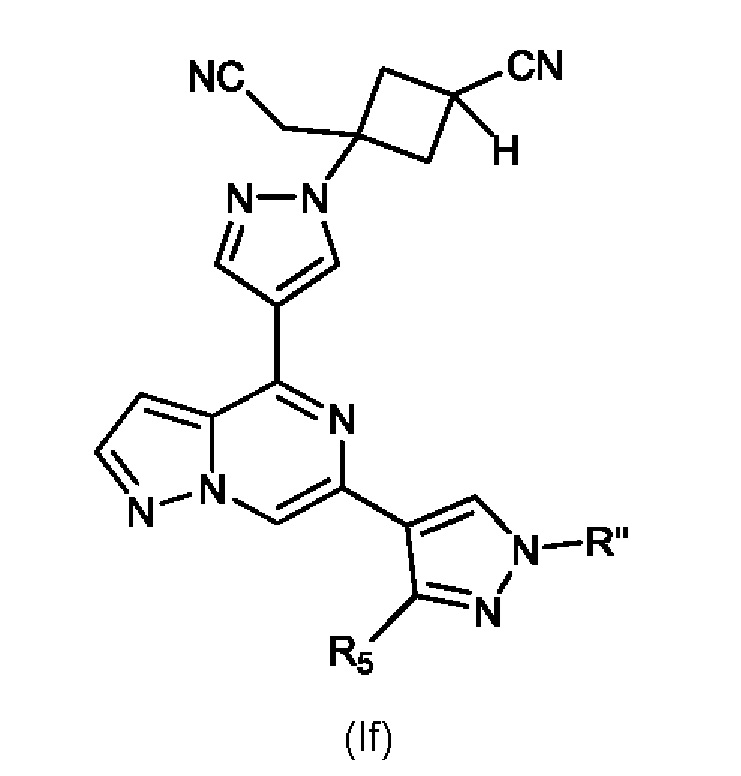
или его фармацевтически приемлемая соль, где:
R'' представляет собой H, C1-C6 алкил или гидрокси(C1-C6 алкил)-; и
R5 представляет собой H, амино, C1-C6 алкил или гидрокси(C1-C6 алкил)-.
12. Соединение по п.11, где R'' представляет собой C1-C6 алкил и R5 представляет собой H.
13. Соединение по п.1, выбранное из группы, состоящей из:
(1r,3r)-3-(4-(6-(3-амино-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрила;
2,2'-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1,3-диил)диацетонитрила;
2-((1s,3r)-1-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила;
5-(4-(1-((1s,3r)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксамида;
(1s,3s)-3-(цианометил)-3-(4-(6-(5-(гидроксиметил)изоксазол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
(1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
(1s,3s)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
(1r,3r)-3-(цианометил)-3-(4-(3-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила;
2-((1r,3s)-1-(4-(6-(3-амино-1H-пиразол-5-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрила;
2-(1-этил-3-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрила;
(1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-3-оксо-2,3-дигидро-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрила (транс-изомер);
или его фармацевтически приемлемая соль.
14. Соединение по п.1, где соединение представляет собой (1r,3r)-3-(4-(6-(3-амино-1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-(цианометил)циклобутан-1-карбонитрил или его фармацевтически приемлемую соль.
15. Соединение по п.1, где соединение представляет собой 2,2'-(3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-1,3-диил)диацетонитрил или его фармацевтически приемлемую соль.
16. Соединение по п.1, где соединение представляет собой 2-((1s,3r)-1-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил или его фармацевтически приемлемую соль.
17. Соединение по п.1, где соединение представляет собой 5-(4-(1-((1s,3r)-1-(цианометил)-3-метоксициклобутил)-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-6-ил)-1H-пиразол-3-карбоксамид или его фармацевтически приемлемую соль.
18. Соединение по п.1, где соединение представляет собой (1s,3s)-3-(цианометил)-3-(4-(6-(5-(гидроксиметил)изоксазол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил или его фармацевтически приемлемую соль.
19. Соединение по п.1, где соединение представляет собой (1r,3r)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил или его фармацевтически приемлемую соль.
20. Соединение по п.1, где соединение представляет собой (1s,3s)-3-(цианометил)-3-(4-(6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил или его фармацевтически приемлемую соль.
21. Соединение по п.1, где соединение представляет собой (1r,3r)-3-(цианометил)-3-(4-(3-метил-6-(1-метил-1H-пиразол-4-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)циклобутан-1-карбонитрил или его фармацевтически приемлемую соль.
22. Соединение по п.1, где соединение представляет собой 2-((1r,3s)-1-(4-(6-(3-амино-1H-пиразол-5-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)-3-метоксициклобутил)ацетонитрил или его фармацевтически приемлемую соль.
23. Соединение по п.1, где соединение представляет собой 2-(1-этил-3-(4-(6-(5-(гидроксиметил)-1H-пиразол-3-ил)пиразоло[1,5-а]пиразин-4-ил)-1H-пиразол-1-ил)азетидин-3-ил)ацетонитрил или его фармацевтически приемлемую соль.
| Способ приготовления лака | 1924 |
|
SU2011A1 |
| RU 2014118954 А, 20.11.2015 | |||
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| MENET CHRISTEL J | |||
| et al.: "Advance in the discovery of selective JAK inhibitors", PROGRESS IN MEDICINAL CHEMISTRY, 2013, vol.52, p.153-223. | |||

