Область техники, к которой относится изобретение
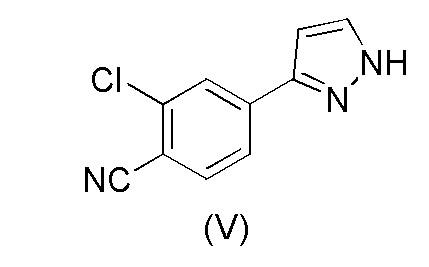
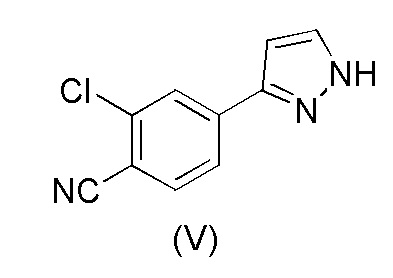
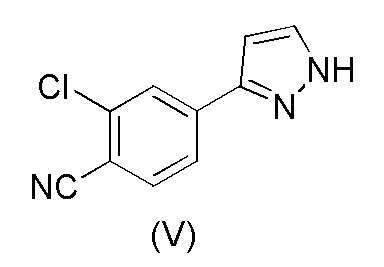
Настоящее изобретение относится к усовершенствованному способу получения карбоксамид-структурированных антагонистов андрогенного рецептора, таких как N-((S)-1-(3-(3-хлор-4-цианофенил)-1H-пиразол-1-ил)-пропан-2-ил)-5-(1-гидроксиэтил)-1H-пиразол-3-карбоксамид (1A), и их ключевых промежуточных соединений, таких как 2-хлор-4-(1H-пиразол-3-ил)бензонитрил (V).
Предпосылки создания изобретения
Соединение N-((S)-1-(3-(3-хлор-4-цианофенил)-1H-пиразол-1-ил)-пропан-2-ил)-5-(1-гидроксиэтил)-1H-пиразол-3-карбоксамид формулы (1A) и его производные раскрыты в WO 2011/051540, Соединение формулы (1A) и его производные являются сильными антагонистами андрогенного рецептора (АР), которые полезны в лечении рака, в частности, рака предстательной железы, и других заболеваний, при которых желателен АР-антагонизм.
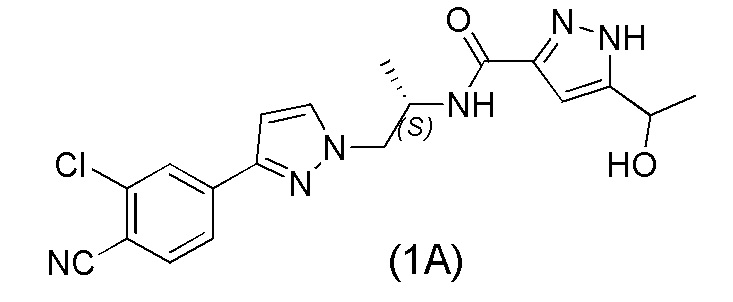
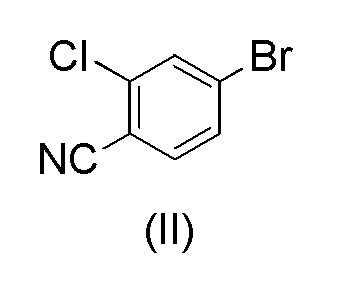
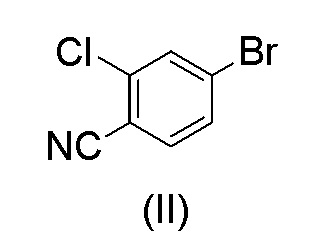
WO 2011/051540 раскрывает способ получения соединения формулы (1A) через 2-хлор-4-(1H-пиразол-3-ил)бензонитрильное промежуточное соединение формулы (V). Промежуточное соединение формулы (V) было получено, как показано на Схеме I:
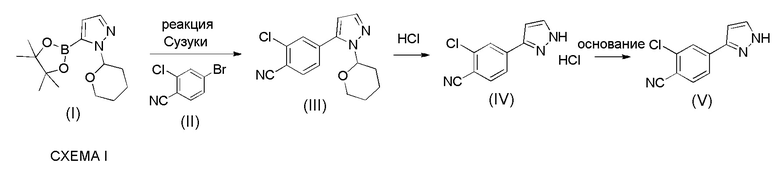
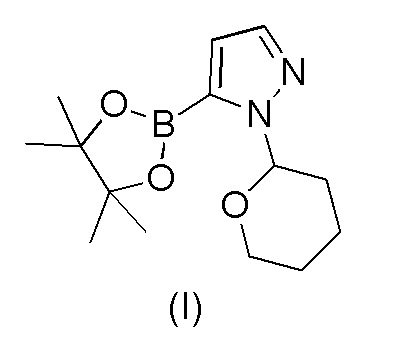
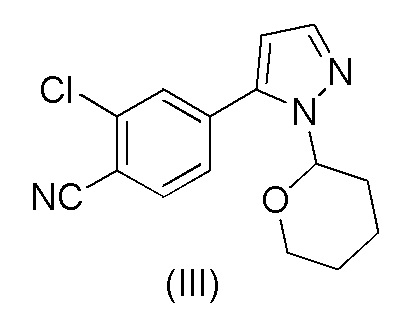
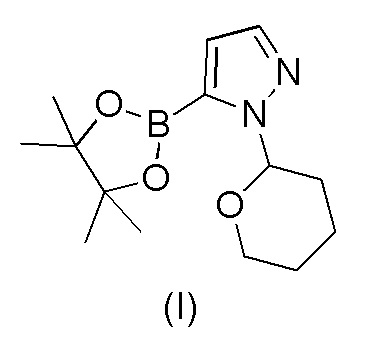
Этот способ включает взаимодействие пинаколового эфира 1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-бороновой кислоты (I) с 4-бром-2-хлорбензонитрилом (II) в реакции Сузуки с получением 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил) бензонитрила формулы (III). Реакцию Сузуки осуществляли в присутствии катализатора, такого как бис(трифенилфосфин)палладий(II) хлорид, и основания, такого как карбонат натрия, в растворителе THF-вода. После завершения реакции растворители отгоняли практически досуха и добавляли воду для осаждения соединения формулы (III). Выделенное соединение формулы (III) далее обрабатывали 10% раствором HCl в этаноле с получением соли 2-хлор-4-(1H-пиразол-3-ил)бензонитрил гидрохлорида формулы (IV), которую выделяли. В конце, получали 2-хлор-4-(1H-пиразол-3-ил)бензонитрил формулы (V) путем обработки соединения формулы (IV) гидроксидом натрия в растворителе вода-метанол.
Аналогичный способ получения соединения формулы (V) раскрыт в WO 2012/143599. Реакцию Сузуки осуществляли в растворителе THF-толуол-вода и также использовали катализатор фазового переноса (TBAB). Выделение соединения формулы (III) осуществляли добавлением воды и дистилляцией выделенной органической фазы практически досуха, с последующим добавлением этанола и фильтрованием кристаллического продукта. Выделенное соединение формулы (III) обрабатывали 10% раствором HCl в этаноле с получением соединения формулы (IV). Это соединение растворяли в метаноле для обработки активированным углем и целитом. Часть метанола отгоняли и добавляли воду и 50% раствор NaOH. После завершения реакции метанол отгоняли и добавляли воду для осаждения соединения формулы (V). Суммарный выход от всех трех стадий составил 84,5%.
Указанные выше способы имеют несколько недостатков. Количество дорогостоящего катализатора бис(трифенил фосфин)палладий(II) хлорида, необходимого для эффективного осуществления реакции Сузуки, очень высокое около 5 моль-%. Использование этанольного раствора HCl является нецелесообразным, и обработка для выделения продукта в этом способе является очень сложной из-за множества дистилляций досуха, поскольку такие процедуры трудно осуществлять в больших масштабах. Более того, несколько выделений делают процесс трудоемким и снижают выход.
Таким образом, существует потребность в более практичном и экономичном способе, который подходит для получения промежуточных соединений, таких как соединение формулы (V), в большом масштабе.
Сущность изобретения
Было обнаружено, что соединение формулы (V) может быть получено с использованием более практичного и экономичного способа и пригодного для использования в больших масштабах. В частности, можно значительно уменьшить количество дорогостоящего палладиевого катализатора и избежать трудоемких стадий дистилляции, а также использования этанольного раствора HCl. Кроме того, уменьшается количество стадий выделения, что даст более высокий выход. Также существенно снижены уровни палладиевых остатков, обнаруженных в конечном продукте.
Таким образом, настоящее изобретение обеспечивает способ получения 2-хлор-4-(1H-пиразол-3-ил)бензонитрила формулы (V),
включающий стадии
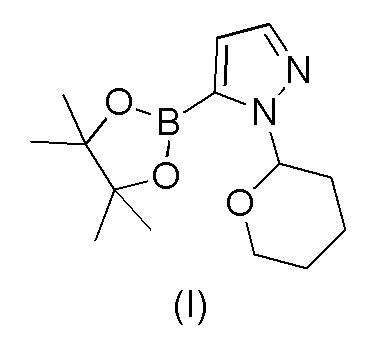
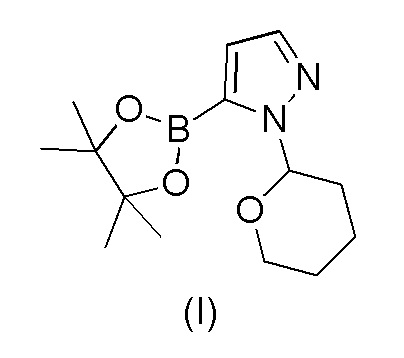
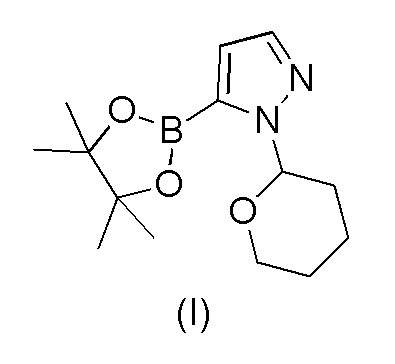
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
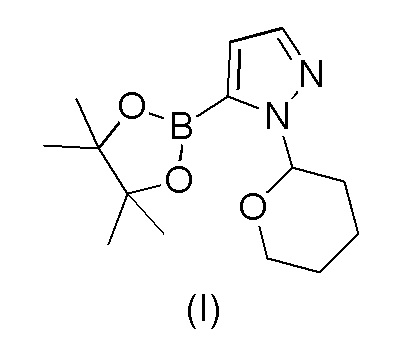
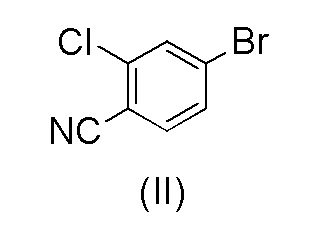
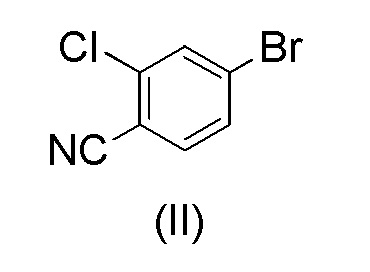
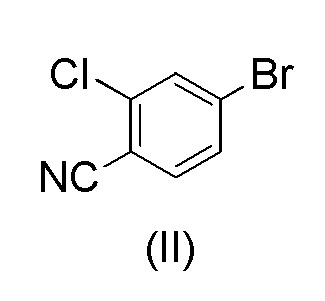
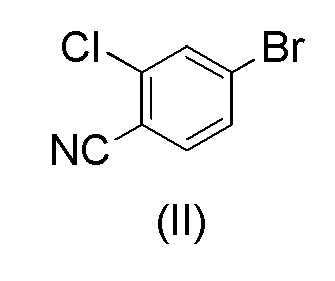
с 4-бром-2-хлорбензонитрилом формулы (II)
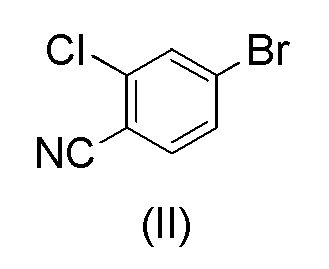
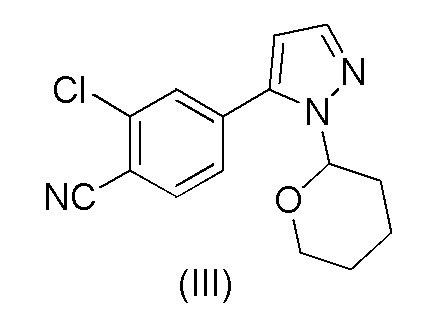
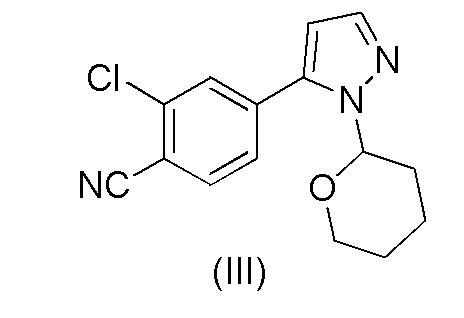
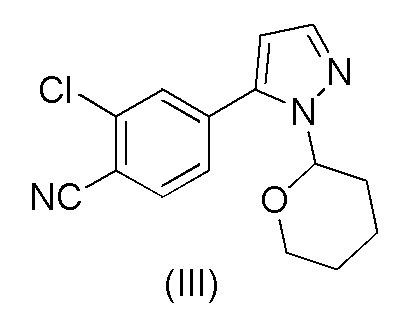
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода с образованием 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
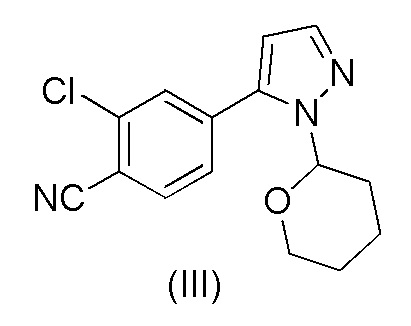
b) обработки соединения формулы (III) каталитическим количеством HCl в метанольном растворителе;
c) добавления основания для нейтрализации смеси; и
d) выделения соединения формулы (V).
В другом аспекте, настоящее изобретение обеспечивает способ получения 2-хлор-4-(1H-пиразол-3-ил)бензонитрила формулы (V)
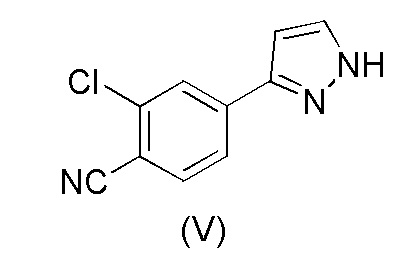 ,
,
включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
с 4-бром-2-хлорбензонитрилом формулы (II)
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода;
b) выделения ацетонитрильной фазы;
c) добавления воды к охлажденной ацетонитрильной фазе;
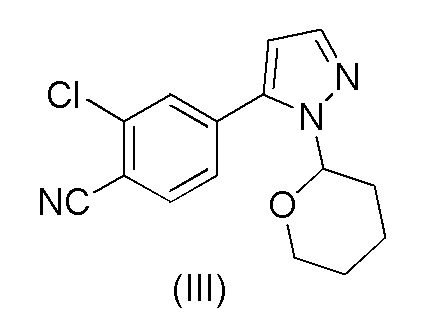
d) выделения осажденного 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
e) обработки соединения формулы (III) каталитическим количеством HCl в метанольном растворителе;
f) добавления основания для нейтрализации смеси;
g) добавления воды к смеси; и
h) выделения осажденного соединения формулы (V).
В еще одном аспекте, настоящее изобретение обеспечивает способ получения 2-хлор-4-(1H-пиразол-3-ил)бензонитрила формулы (V)
 ,
,
включающий стадии
a) обработки 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
каталитическим количеством HCl в метанольном растворителе;
b) добавления основания для нейтрализации смеси;
c) выделения осажденного соединения формулы (V).
В еще одном аспекте, настоящее изобретение обеспечивает способ получения 2-хлор-4-(1H-пиразол-3-ил)бензонитрила формулы (V)
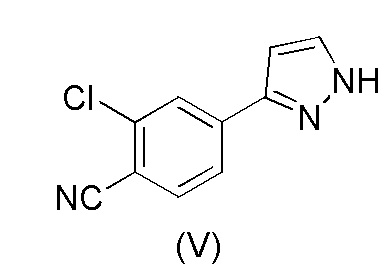 ,
,
включающий стадии
a) обработки 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
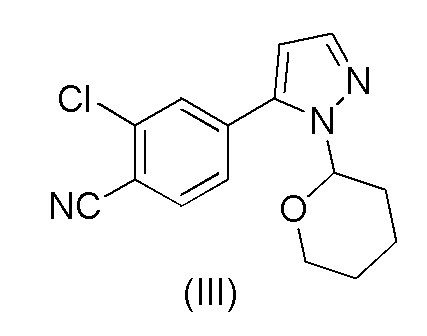
каталитическим количеством HCl в метанольном растворителе;
b) добавления основания для нейтрализации смеси;
c) добавления воды к смеси; и
d) выделения осажденного соединения формулы (V).
В еще одном аспекте, настоящее изобретение обеспечивает применение соединения формулы (V) в получении соединения формулы (1A), где соединение формулы (V) получают в соответствии с любым из способов, описанных выше.
В еще одном аспекте, настоящее изобретение обеспечивает способ получения 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
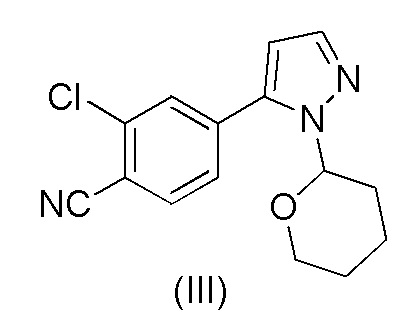 ,
,
включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
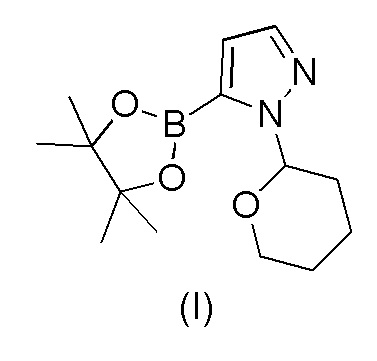
с 4-бром-2-хлорбензонитрилом формулы (II)
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода.
В еще одном аспекте, настоящее изобретение обеспечивает способ получения 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
 ,
,
включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
с 4-бром-2-хлорбензонитрилом формулы (II)
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода;
b)выделения ацетонитрильной фазы;
c) добавления воды к охлажденной ацетонитрильной фазе;
d) выделения осажденного соединения формулы (III).
В еще одном аспекте, настоящее изобретение обеспечивает применение соединения формулы (III) в получении соединения формулы (1A), где соединение формулы (III) получают в соответствии с любым из способов, описанных выше.
Подробное описание изобретения
Термин ʺмоль-% Pd(OAc)2ʺ, используемый в настоящей заявке, относится к указанному в процентах количеству катализатора Pd(OAc)2 (в молях), используемому на стадии реакции, по отношению к количеству исходного соединения (в молях). Например, если 0,005 моль Pd(OAc)2 используют на 1 моль бром-2-хлорбензонитрила на стадии реакция a), то моль-% Pd(OAc)2, используемого на стадии a), составляет (0,005/1) × 100 моль-%=0,5 моль-%.
Таутомерия: Поскольку атом водорода пиразольного кольца может существовать в таутомерном равновесии между 1- и 2-положением, специалисту в данной области будет понятно, что формулы и химические названия, раскрытые в настоящей заявке, включающие атом водорода в пиразольном кольце, включают таутомер соответствующего соединения. Например, химическое название, такое как ʺ2-хлор-4-(1H-пиразол-3-ил)бензонитрилʺ, и соответствующая формула (V) включает таутомер соединения, а именно ʺ2-хлор-4-(1H-пиразол-5-ил)бензонитрилʺ.
В соответствии с настоящим изобретением, 2-хлор-4-(1H-пиразол-3-ил)бензонитрил формулы (V)
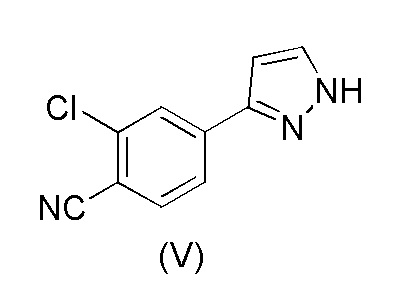
получают путем
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
с 4-бром-2-хлорбензонитрилом формулы (II)
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода с образованием 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
b) обработки соединения формулы (III) каталитическим количеством HCl в метанольном растворителе;
c) добавления основания для нейтрализации смеси; и
d) выделения соединения формулы (V).
В соответствии с настоящим изобретением, в частности, 2-хлор-4-(1H-пиразол-3-ил) бензонитрил формулы (V)
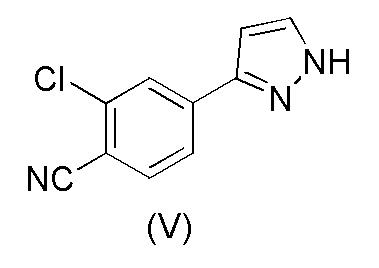
получают путем
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
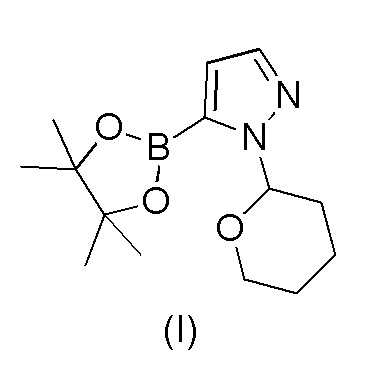
с 4-бром-2-хлорбензонитрилом формулы (II)
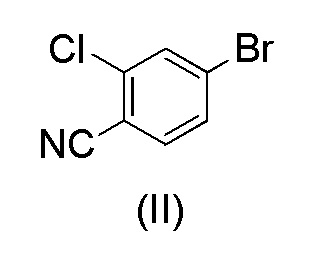
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода;
b) выделения ацетонитрильной фазы;
c) добавления воды к охлажденной ацетонитрильной фазе;
d) выделения осажденного 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
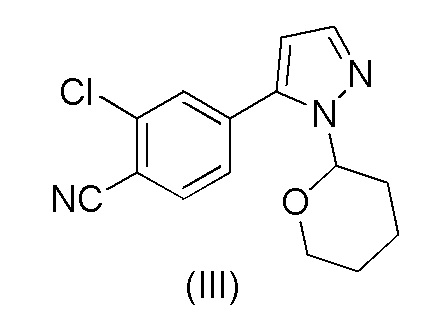
e) обработки соединения формулы (III) каталитическим количеством HCl в метанольном растворителе;
f) добавления основания для нейтрализации смеси;
g) добавления воды к смеси; и
h) выделения осажденного соединения формулы (V).
Было обнаружено, что при замене растворителя на стадии a) на смесь ацетонитрил-вода и катализатора на Pd(OAc)2 и трифенилфосфин количество дорогостоящего Pd катализатора может быть существенно уменьшено. В частности, количество Pd(OAc)2 на количество 4-бром-2-хлорбензонитрила формулы (II), которое необходимо для эффективного осуществления реакция Сузуки, составляет всего лишь от около 0,5 до около 2 моль-%, предпочтительно от около 0,6 до около 0,8 моль-%. Кроме того, после завершения реакции Сузуки растворитель ацетонитрил-вода образует две отдельные жидкие фазы, и выделение соединения формулы (III) из ацетонитрильной фазы происходит легко без необходимости каких-либо стадий дистилляции.
Соединения формулы (I) и (II) являются коммерчески доступными, или они могут быть получены в соответствии со способами, известными в данной области техники.
Для осуществления реакции Сузуки смесь ацетонитрила, воды, основания и 4-бром-2-хлорбензонитрила формулы (II) сначала можно кипятить с обратным холодильником в атмосфере азота в течение примерно 15-60 минут, например, около 30 минут. Реакцию предпочтительно осуществляют под потокои азота. Таким образом, воздух удаляют, например, путем кипячения с обратным холодильником, и заменяют азотом. В растворителе ацетонитрил-вода отношение ацетонитрила к воде обычно составляет от около 25:75 до около 75:25, предпочтительно от около 35:65 до около 65:35, более предпочтительно от около 40:60 до около 60:40, например, 50:50, по объему. Основание представляет собой подходящее неорганическое основание, предпочтительно карбонат калия.
Смесь затем соответствующим образом охлаждают до 60-70°C и добавляют Pd(OAc)2 и трифенилфосфин. Молярное отношение Pd(OAc)2 к трифенилфосфину для использования в этом споосбе составляет, соответственно, примерно 1:3. Количество Pd(OAc)2 в расчете на количества 4-бром-2-хлорбензонитрила формулы (II) обычно составляет от около 0,5 до около 2 моль-%, предпочтительно от около 0,6 до около 0,8 моль-%. Соединение формулы (I) можно растворить в ацетонитриле и медленно, например, в течение 0,5 часа добавлять к смеси. Реакционную смесь затем перемешивают при температуре от около 60 до около 75°C, предпочтительно при 70±3°C, в течение периода времени, достаточного для завершения реакции, обычно от около 1 до около 5 часов, например, в течение 2 часов. Образуются отдельные фазы воды и ацетонитрила, и водную фазу можно удалить из смеси соответствующим образом при температуре 65-70°C. На этой стадии к выделенной ацетонитрильной фазе можно добавить основание, такое как аммиачная вода (25%), для предотвращения возможного отсоединения тетрагидропиранильного кольца от соединения формулы (III). Затем можно осуществить осаждение соединения формулы (III) путем охлаждения смеси, например, до 20±5°C, и постепенного добавления воды к охлажденной смеси. Количество добавляемой воды составляет, соответственно, около 80-120%, например, около 100%, в расчете на объем ацетонитрильного растворителя. Смесь перемешивают при 20±5°C в течение некоторого периода времени для полного осаждения соединения формулы (III), например, в течение примерно 6-24 часов. Можно осуществить выделение осажденного продукта, например, путем фильтрования и промывку смесью ацетонитрил-вода и сушку, например, при пониженном давлении при около 50-60°C.
Кроме того, было обнаружено, что преобразование соединения формулы (III) в соединение формулы (V) можно осуществлять в одном реакторе без выделения соединения формулы (IV). Нет необходимости в стадиях дистилляции, и способ может быть осуществлен с использованием только каталитического количества 30% водного раствора HCl, что намного более практично, чем использование этанольного раствора HCl. Выделение соединения формулы (V) является простым, и способ в целом обеспечивает улучшенный выход.
Преобразование соединения формулы (III) в соединение формулы (V) можно осуществить путем смешивания соединения формулы (III), метанола и небольшого количества 30% раствора HCl (водный раствор) соответствующим образом при пониженной температуре, такой как 0-15°C, например 10±3°C. Количество HCl может составлять от около 0,05 до около 0,1, например, 0,08 мольных эквивалентов на один моль соединения формулы (III). Смесь перемешивают при вышеуказанной температуре в течение периода времени, необходимого для того, чтобы произошло отсоединение тетрагидропиранильного кольца, такого как 0,5-5 часов, например, в течение 2 часов. Затем к смеси добавляют основание, например, аммиачную воду (25%) при вышеуказанной температуре. После этого постепенно добавляют воду, например, при 10-20°C и смесь перемешивают, например, в течение 6-24 часов. Количество добавляемой воды подходяще составляет около 30-50%, например, 35-40% в расчете на объем метанольного растворителя. Осаждение соединения формулы (V) можно осуществить путем охлаждения смеси, например, до около 0-5°C и перемешивания при этой температуре в течение времени, достаточного для завершения осаждения, подходяще от около 1 до около 8 часов, например, от около 3 до около 5 часов. Осажденный продукт можно выделить, например, путем фильтрации с промывкой смесью 3:1 холодная вода:метанол и сушкой, например, при пониженном давлении при температуре 50-60°C.
Соединение формулы (1A) можно получить из соединения формулы (V), например, с использованием способов, описанных в WO 2011/051540 и WO 2012/143599. Например, в соответствии с одним вариантом осуществления, способ получения соединения формулы (1A) включает стадии
i) взаимодействия соединения формулы (V)
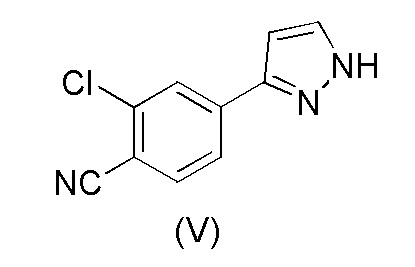
с соединением формулы (VI)
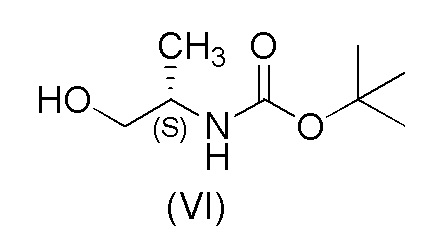
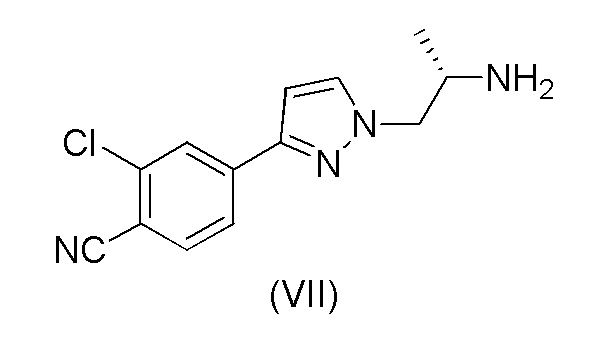
с получением соединения формулы (VII);
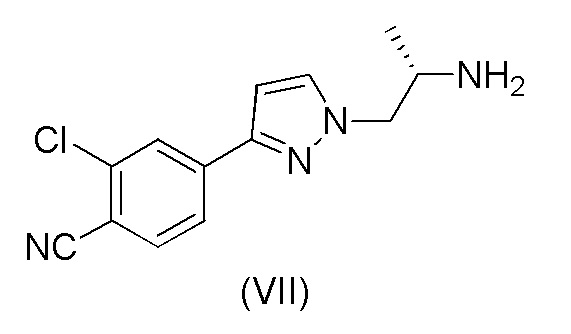
j) взаимодействия соединения формулы (VII) с соединением формулы (VIII)
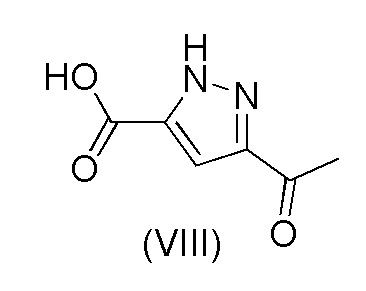
с получением соединения формулы (IX); и
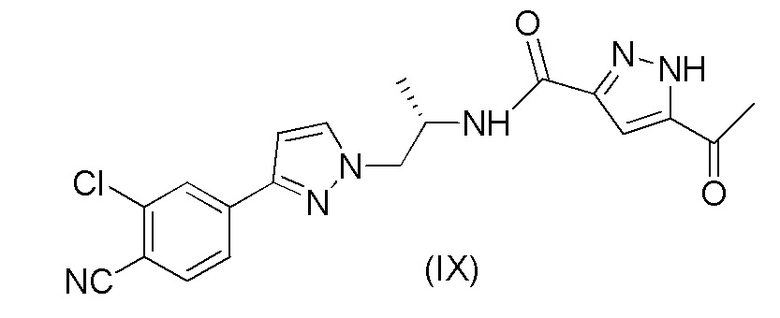
k) восстановления соединения формулы (IX) с получением соединения формулы (1A).
Реакцию на стадии i) можно осуществить, например, с использованием условий реакции Мицунобу, например, при комнатной температуре в присутствии трифенилфосфина и DIAD (диизопропил азодикарбоксилата) в подходящем растворителе, например, THF или EtOAc, с последующим снятием Boc защиты путем обработки при помощи HCl и в конце основанием, таким как NaOH.
Реакцию стадии j) можно осуществить при комнатной температуре в присутствии подходящей системы активирующего и связывающего вещества, такой как комбинация DIPEA (N,N-диизопропилэтиламин), EDCI (1-этил-3-(3-диметиламинопропил)карбодиимид) и безводного HOBt (1-гидрокси-бензотриазол) в подходящем растворителе, например, DCM. В качестве альтернативы, можно использовать HOBt, HBTU (O-(бензотриазол-1-ил)-N,N,N´,N´-тетраметилуроний гексафторфосфат). Альтернативно, комбинацию DIPEA и T3P (циклический ангидрид 1-пропанофосфоновой кислоты) можно использовать в качестве системы активирующего и связывающего вещества.
Реакцию стадии k) можно осуществить при комнатной температуре путем обработки соединения формулы (IX) восстановителем, например, борогидридом натрия в подходящем растворителе, например, этаноле, с последующей обработкой смеси водным раствором HCl.
Изобретение далее проиллюстрировано следующими неограничивающими примерами.
Пример 1. Получение 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
1-(Тетрагидро-2H-пиран-2-ил)-1H-пиразол (5 кг), THF (7,0 л) и толуол (28 л) смешивали при комнатной температуре (комн.темп.) в атмосфере азота. Смесь охлаждали до 0°C, по каплям добавляли n-BuLi (17,9 кг, 1,42 M в гексане) при 0-5°C в течение 2-3 часов и смесь перемешивали при 0-5°C в течение 1 часа. По каплям добавляли триизопропилборат (6,8 кг) при 0-5°C в течение 45 минут. Смесь доводили до комнатной температуры и перемешивали в течение 1-2 часов. В смесь при комнатной температуре порциями добавляли пинакол (3,88 кг) в течение 20-30 минут, с последующим перемешиванием в течение 45 минут. Смесь охлаждали до 0°C и по каплям добавляли уксусную кислоту (3,9 кг) в течение 30 минут при 0-5°C. Смесь доводили до комнатной температуры и поддерживали при этой температуре в течение 12-14 часов. Смесь затем охлаждали до 0°C и по каплям добавляли воду (20 л) при 0-5°C в течение 30 минут. Смесь доводили до комнатной температуры и перемешивали в течение 30 минут. Водный слой отделяли и экстрагировали толуолом (20 л). Объединенный органический слой промывали 10% раствором NaHCO3 (22 л), затем водой (20 л). Органический слой концентрировали при пониженном давлении при температуре ниже 60°C. Полученное неочищенное соединение затем подвергали перегонке с гептаном (7 л). К полученному остатку добавляли гептан (5 л) и смесь перемешивали при 0-5°C в течение 1-2 часов. Твердое вещество затем фильтровали, промывали холодным гептаном (5 л) и сушили при 25-30°C в течение 2-3 часов. Выход 6,2 кг (67.8%), ВЭЖХ чистота 99,8.
Пример 2. Получение 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила (III)
Загружали ацетонитрил (50 мл), воду (50 мл), карбонат калия * H2O (21,7 г, 2,07 экв.) и 4-бром-2-хлорбензонитрил (II) (14,0 г, 1,00 экв.). Смесь кипятили с обратным холодильником в атмосфере азота в течение примерно 0,5 часа. Смесь охлаждали до 60-70°C в защитной атмосфере азота. Под защитным слоем азота добавляли ацетат палладия(II) Pd(OAc)2 (0,10 г, 0,007 экв.) и трифенилфосфин (0,40 г, 0024 экв.). 1-(Тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (I) (21.0 г, 1.17 экв.) растворяли в ацетонитриле (30 мл). Воздух откачивали под вакуумом и заменяли азотом. Этот раствор добавляли к реакционной смеси в течение примерно 0,5 часа при 70±3°C. Реакционную смесь перемешивали в течение 2 часов при 70±3°C. Водную фазу отделяли и удаляли из реакционной смеси при 65-70°C. К реакционной смеси добавляли 2 мл аммиачной воды (25%) и смесь затем охлаждали до 20±5°C. Постепенно добавляли воду (80 мл) при 20±5°C. Смесь перемешивали в течение ночи при 20±5°C. Кристаллический продукт фильтровали и промывали дважды смесью ацетонитрил:вода 1:1 (20 мл). Продукт сушили при пониженном давлении при 50-60°C. Выход 17,18 г (92,3%). ВЭЖХ-чистота 99,8%.
Пример 3. Получение 2-хлор-4-(1H-пиразол-3-ил)бензонитрила (V)
Загружали 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрил (III) (10,0 г, 1,00 экв.) и метанол (40 мл). Добавляли 30% раствор HCl (0,3 мл, 0,08 экв.) при 10±3°C. Смесь перемешивали в течение 2 часов при 10±3°C. Добавляли аммиачную воду (25%) (3,0 мл, 1,1 экв.) при 10±5°C. Постепенно добавляли воду (15 мл) при 10-20°C. Смесь перемешивали в течение ночи при 20±5°C. Смесь затем охлаждали до 0-5°C и перемешивали в течение 4 часов при 0-5°C. Кристаллический продукт фильтровали и промывали смесью холодная вода:метанол 3:1 (30 мл) и сушили при 50-60°C. Выход 6,78 г (95,8%). ВЭЖХ-чистота 99,7%.
Пример 4. Получение (S)-4-(1-(2-аминопропил)-1H-пиразол-3-ил)-2-хлорбензонитрила (VII)
15 г (73,7 ммоль) 2-хлор-4-(1H-пиразол-3-ил)бензонитрила (V), 26,5 г (151 ммоль) (S)-трет-бутил-1-гидрокси пропан-2-илкарбамата (VI), трифенилфосфин (39,6 г, 151 ммоль) и 84 мл EtOAc помещали в реакционный сосуд в атмосфере азота. Смесь охлаждали до 10±5°C. Добавляли DIAD (29,7 мл, 151 ммоль) равномерно в течение 4 часов при перемешивании, поддерживая температуру при 10±10°C. Смесь нагревали до 20±5°C и перемешивали в течение ночи. Добавляли по каплям концентрированную HCl (31,1 мл, 295 ммоль) в течение 10-30 минут при перемешивании, поддерживая температуру при 30±5°C. Смесь перемешивали при 45±5°C до завершения реакции. Добавляли воду (82,5 мл) и температуру доводили до 35±5°C. Затем добавляли DCM (105 мл) и смесь энергично перемешивали в течение по меньшей мере 1 минуты и оставляли на 10 минут для разделения слоев. Органический слой выделяли и промывали 60 мл теплой воды. Водные фазы объединяли и промывали при помощи 75 мл DCM. После этого к водной фазе добавляли 75 мл DCM и 19,3 мл (125 ммоль) 25% раствора аммония (NH4OH). Уровень pH устанавливали выше 9 путем добавления 50% раствора NaOH и смесь перемешивали при 40±5°C до завершения реакции. Уровень pH устанавливали выше 9 путем добавления 50% раствора NaOH. Раствор фильтровали через целит при 35°C, слои разделяли и выделяли органическую фазу. DCM отгоняли при нормальном давлении до тех пор, пока не осталось 25 мл раствора. Добавляли 2-пропанол (4,65 мл) и температуру доводили до около 50°C. Затем добавляли 90 мл N-гептана в течение 1 часа. В раствор вносили затравку, когда было добавлено примерно 22 мл N-гептана. Смесь охлаждали до 0±5°C в течение 6 часов и затем перемешивали в течение ночи. Осажденный продукт выделяли путем фильтрации, промывали N-гептаном (30 мл) и сушили под вакуумом при 50°C. Выход 81,8%.
Пример 5. Получение 3-ацетил-1H-пиразол-5-карбоновой кислоты (VIII)
3-Ацетил-1H-пиразол-5-карбоксилат (5 г, 29,7 ммоль), воду (30 мл) и гидроксид натрия 48% (2,83 мл, 52,0 ммоль) осторожно добавляли в реакционный сосуд. Смесь нагревали до 60-65°C и перемешивали до завершения реакции. Смесь затем охлаждали до 50°C. 30% Раствор HCl (2,83 мл, 26,8 ммоль) добавляли при 50°C в течение 1 часа и в смесь вносили затравку по завершении добавления HCl. Смесь перемешивали в течение 2 часов. Затем добавляли 30% раствор HCl (2,451 мл, 23,19 ммоль) при 50°C в течение 3 часов с последующим перемешиванием при 50°C в течение 30 минут. Осажденный продукт выделяли путем фильтрации, промывали водой (5 мл) и затем метанолом (2,5 мл) и сушили под вакуумом при 60°C. Выход 92,8%.
Пример 6. Получение (S)-5-ацетил-N-(1-(3-(3-хлор-4-цианофенил)-1H-пиразол-1-ил)пропан-2-ил)-1H-пиразол-3-карбоксамида (IX)
6,80 г (44,1 ммоль) 3-ацетил-1H-пиразол-5-карбоновой кислоты (VIII), DCM (76 мл), 10,33 г (38,3 ммоль) (S)-4-(1-(2-аминопропил)-1H-пиразол-3-ил)-2-хлорбензонитрила (VII) и DIPEA (18,04 мл, 104 ммоль) помещали в реакционную колбу в атмосфере азота при около 20°C. Смесь охлаждали до 5°C. Затем добавляли в течение 2 часов 28,2 мл (49,9 ммоль) T3P (циклический ангидрид 1-пропанфосфоновой кислоты) в EtOAc (50%) при энергичном перемешивании при около 10°C. Смесь перемешивали при 10±3°C в течение ночи. Затем к смеси добавляли этанол (30 мл). Примерно 70 мл DCM затем отгоняли при около 60°C и в смесь вносили затравку при около 60°C, с последующим перемешиванием в течение 30 минут при этой температуре. Смесь воды (40 мл), 0,75 мл 30% раствора HCl воде и этанола (10 мл) затем добавляли в течение примерно 2 часов с последующим перемешиванием при 60±5°C в течение примерно 2 часов. Смесь охлаждали до 5-10°C в течение 4 часов с последующим перемешиванием при этой температуре в течение ночи. Осажденный продукт выделяли путем фильтрации, промывали при помощи 2 × 30 мл воды и 1 × 20 мл этанола и сушили под вакуумом при 60°C в течение ночи. Выход 87,5%.
Пример 7. Получение N-((S)-1-(3-(3-хлор-4-цианофенил)-1H-пиразол-1-ил)-пропан-2-ил)-5-(1-гидроксиэтил)-1H-пиразол-3-карбоксамида (IA)
100 мг (0,25 ммоль) (S)-5-ацетил-N-(1-(3-(3-хлор-4-цианофенил)-1H-пиразол-1-ил)пропан-2-ил)-1H-пиразол-3-карбоксамида (IX) и 5 мл EtOH помещали в реакционную колбу и медленно добавляли 19 мг (0,5 ммоль) боргидрида натрия в виде суспензии в EtOH. Реакционную смесь перемешивали в течение ночи до завершения реакции. Добавляли по каплям 0,5 мл воды и 1 мл 0,5 M раствора HCl. Раствор упаривали досуха и добавляли 20 мл DCM. Смесь промывали при помощи 10 мл 1 M раствора NaHCO3 и 10 мл воды с последующей сушкой над Na2SO4. После фильтрации и упаривания получали 76 мг продукта. Выход 76%.
| название | год | авторы | номер документа |
|---|---|---|---|
| ПРОИЗВОДНЫЕ ПИРАЗОЛОПИРИМИДИНА И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРОВ PDE10 | 2011 |
|
RU2543386C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ФЕНОКСИМЕТИЛЬНЫЕ ПРОИЗВОДНЫЕ | 2016 |
|
RU2746481C1 |
| ХИМИЧЕСКИЙ СПОСОБ | 2020 |
|
RU2820241C2 |
| ПРОИЗВОДНЫЕ АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА В КАЧЕСТВЕ МЕДИКАМЕНТА | 2012 |
|
RU2600976C2 |
| НОВЫЕ ПРОИЗВОДНЫЕ ТРИАЗОЛОПИРАЗИНА И ИХ ПРИМЕНЕНИЕ | 2013 |
|
RU2643361C2 |
| ПИРАЗОЛОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ | 2012 |
|
RU2605096C2 |
| СПОСОБ ПОЛУЧЕНИЯ КОНДЕНСИРОВАННОГО ТРИЦИКЛИЧЕСКОГО СОЕДИНЕНИЯ И СООТВЕТСТВУЮЩЕГО ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ | 2020 |
|
RU2785963C1 |
| ПРОИЗВОДНЫЕ N-[(1Н-ПИРАЗОЛ-1-ИЛ) АРИЛ]-1Н-ИНДОЛА ИЛИ 1Н-ИНДАЗОЛ-3-КАРБОКСАМИДА, ИХ ПОЛУЧЕНИЕ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ АНТАГОНИСТОВ P2Y12 | 2011 |
|
RU2572593C2 |
| ТРИЦИКЛИЧЕСКИЕ ТРИАЗОЛЬНЫЕ СОЕДИНЕНИЯ | 2014 |
|
RU2666728C2 |
Настоящее изобретение относится к усовершенствованному способу получения карбоксамид-структурированных антагонистов андрогенного рецептора (АР), таких как N-((S)-1-(3-(3-хлор-4-цианофенил)-1H-пиразол-1-ил)-пропан-2-ил)-5-(1-гидроксиэтил)-1H-пиразол-3-карбоксамид (1A), и их ключевых промежуточных соединений, таких как 2-хлор-4-(1H-пиразол-3-ил)бензонитрил (V). Антагонисты АР полезны при лечении рака, в частности рака предстательной железы, и других заболеваний, при которых желателен АР-антагонизм. 4 н. и 18 з.п. ф-лы, 7 пр.
1. Способ получения 2-хлор-4-(1H-пиразол-3-ил)бензонитрила формулы (V)
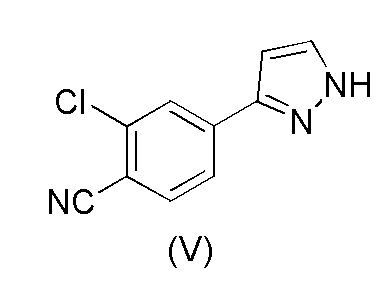 ,
,
включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
с 4-бром-2-хлорбензонитрилом формулы (II)
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода с образованием 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
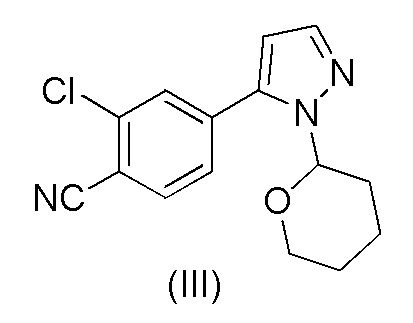
b) обработки соединения формулы (III) в метанольном растворителе каталитическим количеством HCl;
c) добавления основания для нейтрализации смеси; и
d) выделения соединения формулы (V).
2. Способ по п.1, включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
с 4-бром-2-хлорбензонитрилом формулы (II)
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода;
b) выделения ацетонитрильной фазы;
c) добавления воды к охлажденной ацетонитрильной фазе;
d) выделения осажденного 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
e) обработки соединения формулы (III) в метанольном растворителе каталитическим количеством HCl;
f) добавления основания для нейтрализации смеси;
g) добавления воды к смеси; и
h) выделения осажденного соединения формулы (V).
3. Способ по п.1 или 2, в котором количество Pd(OAc)2, используемое в ресчете на количество соединения формулы (II), на стадии a) составляет от около 0,5 до около 2, предпочтительно от около 0,6 до около 0,8 мол.%.
4. Способ по пп.1, 2 или 3, в котором молярное отношение Pd(OAc)2 к трифенилфосфину составляет 1:3.
5. Способ по любому из предшествующих пунктов, в котором основание представляет собой карбонат калия.
6. Способ по любому из предшествующих пунктов, в котором температура реакции на стадии a) составляет от около 60 до около 75°C, предпочтительно 70±3°C.
7. Способ по любому из предшествующих пунктов, в котором стадию a) осуществляют под потоком азота.
8. Способ по любому из пп.2-7, в котором основание добавляют к выделенной ацетонитрильной фазе перед стадией c).
9. Способ по п.8, в котором основание представляет собой аммиачную воду.
10. Способ по любому из пп.2-9, в котором температура смеси после стадии c) составляет 10-40°C, предпочтительно 20±5°C.
11. Способ по любому из предшествующих пунктов, в котором время реакции на стадии a) составляет 1-8 часов, предпочтительно 2-4 часа.
12. Способ по любому из пп.2-11, в котором количество HCl, используемое в расчете на количество соединения формулы (III), на стадии e) составляет от около 0,05 до около 0,2, предпочтительно от около 0,07 до около 0,10 молярных эквивалентов.
13. Способ по любому из пп.2-12, в котором температура реакции на стадии e) составляет 0-20°C, предпочтительно 10±5°C.
14. Способ по любому из пп.2-13, в котором время реакции на стадии e) составляет 1-8 часов, предпочтительно 2-4 часа.
15. Способ по любому из пп.2-14, в котором используемое основание на стадии f) представляет собой аммиачную воду.
16. Способ по любому из пп.2-15, в котором температура смеси после стадии g) составляет 10-20°C.
17. Способ по любому из пп.2-16, в котором выделение на стадии h) осуществляют при температуре 0-5°C.
18. Способ получения 2-хлор-4-(1H-пиразол-3-ил)бензонитрила формулы (V)
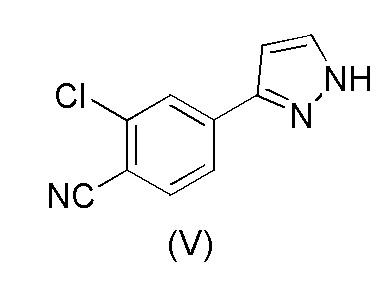 ,
,
включающий стадии
a) обработки 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
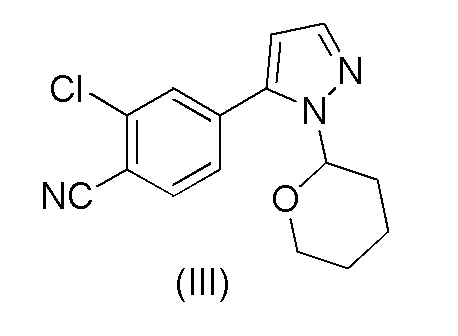
каталитическим количеством HCl в метанольном растворителе;
b) добавления основания для нейтрализации смеси;
c) выделения осажденного соединения формулы (V).
19. Способ по п.18, включающий стадии
a) обработки 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
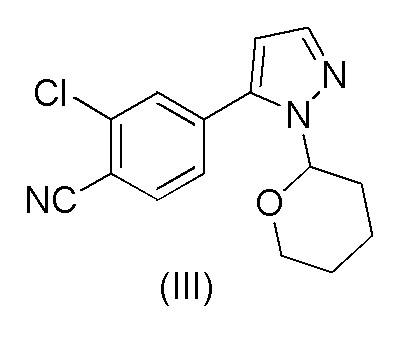
каталитическим количеством HCl в метанольном растворителе;
b) добавления основания для нейтрализации смеси;
c) добавления воды к смеси; и
d) выделения осажденного соединения формулы (V).
20. Способ получения 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
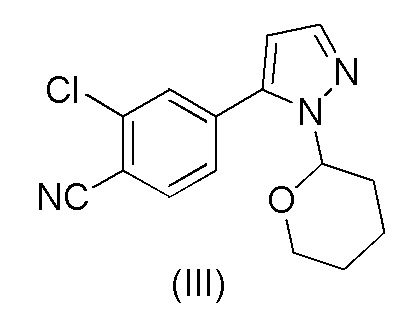 ,
,
включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
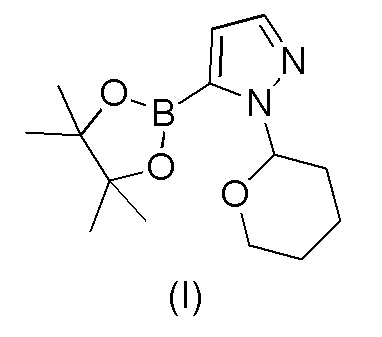
с 4-бром-2-хлорбензонитрилом формулы (II)
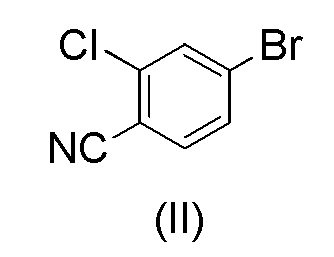
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода.
21. Способ по п.20, включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
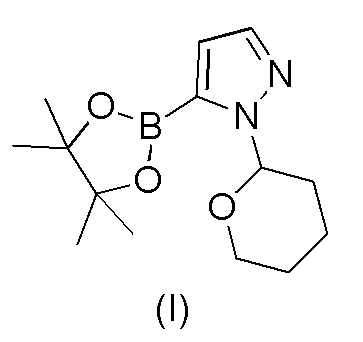
с 4-бром-2-хлорбензонитрилом формулы (II)
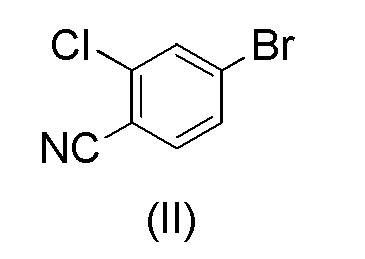
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода;
b) выделения ацетонитрильной фазы;
c) добавления воды к охлажденной ацетонитрильной фазе;
d) выделения осажденного соединения формулы (III).
22. Способ получения соединения формулы (1A)
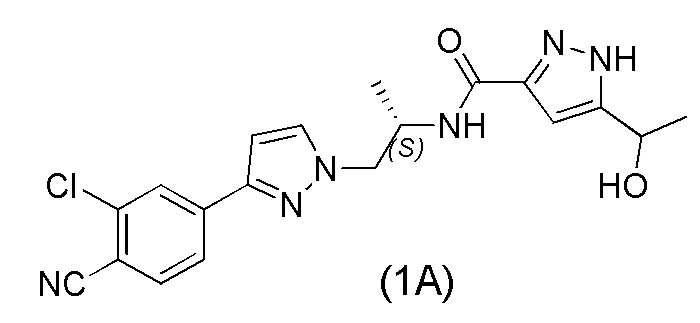 ,
,
включающий стадии
a) взаимодействия 1-(тетрагидро-2H-пиран-2-ил)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола формулы (I)
с 4-бром-2-хлорбензонитрилом формулы (II)
при повышенной температуре в присутствии Pd(OAc)2, трифенилфосфина и основания в растворителе ацетонитрил-вода с образованием 2-хлор-4-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)бензонитрила формулы (III)
b) обработки соединения формулы (III) в метанольном растворителе каталитическим количеством HCl;
c) добавления основания для нейтрализации смеси;
d) выделения соединения формулы (V),
i) взаимодействия соединения формулы (V)
с соединением формулы (VI)
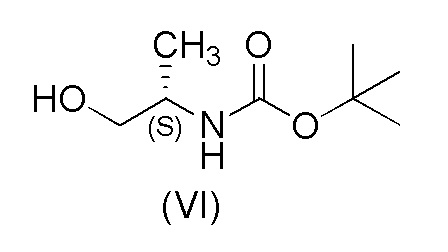
с получением соединения формулы (VII);
j) взаимодействия соединения формулы (VII) с соединением формулы (VIII)
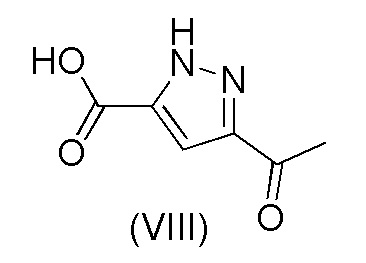
с получением соединения формулы (IX); и
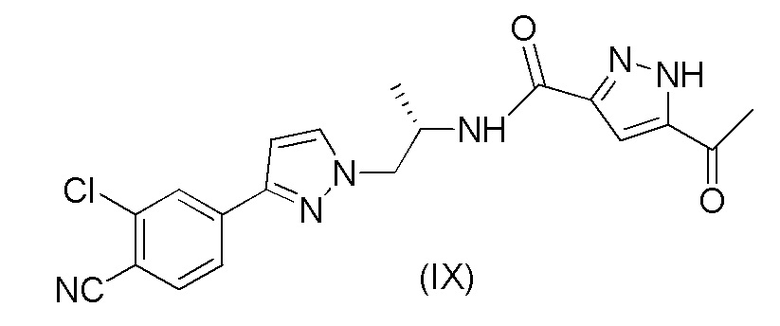
k) восстановления соединения формулы (IX) с получением соединения формулы (1A).
| ЕА 201270597 А1, 30.10.2012 | |||
| ЕА 201391560 А1, 28.02.2014. |

