Область техники, к которой относится изобретение
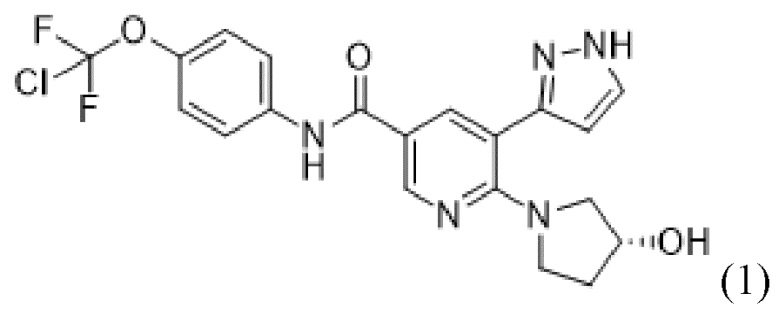
Настоящее изобретение относится к новому химическому способу синтеза соединения, представляющего собой N-[4-(хлордифторметокси)фенил]-6-[(3R)-3-гидроксипирролидин-1-ил]-5-(1H-пиразол-5-ил)пиридин-3-карбоксамид.
Предпосылки к созданию изобретения
Соединение, представляющее собой N-[4-(хлордифторметокси)фенил]-6-[(3R)-3-гидроксипирролидин-1-ил]-5-(1H-пиразол-5-ил)пиридин-3-карбоксамид, также называемое в данном документе соединением формулы (1),
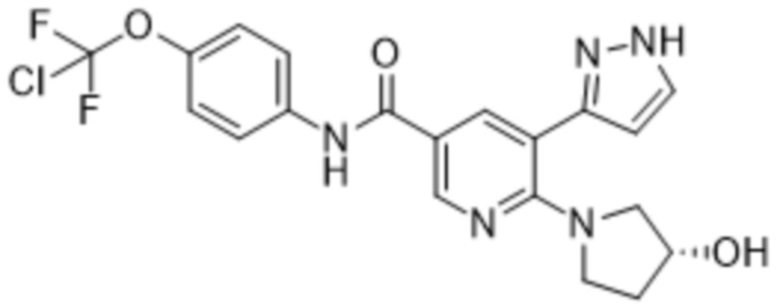 (1),
(1),
представляет собой ингибитор тирозинкиназы BCR-ABL (химерного белка кластерного региона точечного разрыва Абельсона). В WO 2013/171639 A1 представлены соединения формулы (1), пригодные для лечения заболеваний, которые отвечают на ингибирование ферментативной активности тирозинкиназы белка Абельсона (ABL1), связанного с геном Абельсона белка (ABL2) и связанных химерных белков, в частности, BCR-ABL1. Соединение формулы (1) также известно как (R)-N-(4-(хлордифторметокси)фенил)-6-(3-гидроксипирролидин-1-ил)-5-(1H-пиразол-5-ил)никотинамид, или асциминиб.
Соединение формулы (1), получение соединения формулы (1) и фармацевтических композиций соединения формулы (1) изначально описаны в WO 2013/171639 A1 в качестве примера 9.
Тем не менее, остается потребность в обеспечении улучшенных способов получения соединения формулы (1), которые являются более экономически эффективными, безопасными и более пригодными для полномасштабного промышленного изготовления.
Описание изобретения
Настоящее изобретение направлено на улучшенный синтез соединения формулы (1) и его очистку с применением менее вредных химических веществ и/или условий реакции, образованием меньшего количества отходов и обеспечением воспроизводимого способа, который проще осуществлять в промышленном масштабе. Настоящее изобретение также направлено на более эффективные средства получения соединения формулы (1) с более высоким выходом и с более высокой чистотой, которые образуют меньшее количество побочных продуктов, и для которых требуется более низкая загрузка катализатора по сравнению со способами, раскрытыми в предшествующем уровне техники.
В этом отношении настоящее изобретение представлено в следующих аспектах.
В соответствии с первым аспектом настоящего изобретения, предусмотрен способ получения соединения формулы (1),
(1),
или его соли, сольвата, стереоизомера, комплекса, сокристалла, сложного эфира или оксазолинового производного,
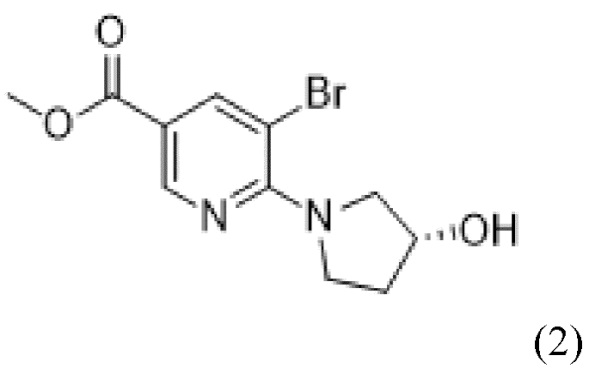
предусматривающий стадию осуществления реакции соединения формулы (2),
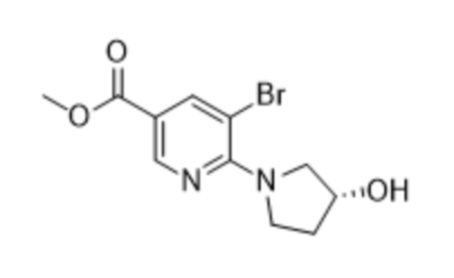 (2),
(2),
или его соли, сольвата, стереоизомера, комплекса, сокристалла, сложного эфира или оксазолинового производного;
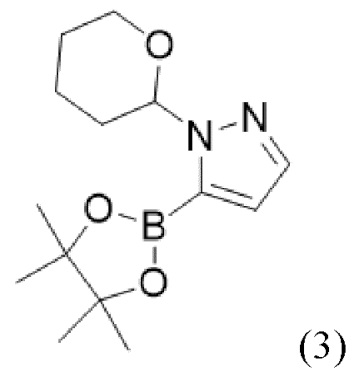
и соединения формулы (3),
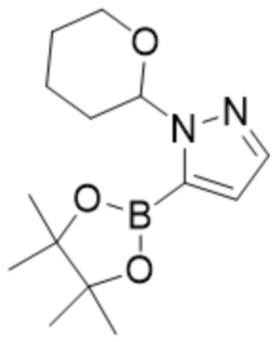 (3),
(3),
или пинаколового сложного эфира 1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-бороновой кислоты с солью щелочного металла или щелочной солью в присутствии катализатора на основе комплекса с металлом с получением соединения формулы (1) или его соли, сольвата, стереоизомера, комплекса, сокристалла, сложного эфира или оксазолинового производного, предпочтительно с получением соединения формулы (1) в его форме свободной карбоновой кислоты.
В данном способе, в соответствии с первым аспектом, катализатор на основе комплекса с металлом может представлять собой предшественник на основе металла и лиганд или предварительно образованный катализатор на основе комплекса с металлом, содержащий металл M и лиганд.
Металл M может быть выбран из Cu (меди) и Pd (палладия). Предпочтительно металл M представляет собой Pd.
Предшественник на основе металла может быть выбран из M(OAc)2, M2(dba)3, [M(C3H5)Cl]2 (димера хлорида аллил-металла), M(TFA)2, M(MeCN)2Cl2, MCl2, [(циннамил)MCl]2, [MCl]2 (хлорида металла) и M(acac)2. Предпочтительно предшественник на основе металла представляет собой [MCl]2 (хлорид металла).
Лиганд может быть выбран из Cy3P, (2-MeOPh)3P, P(tBu)2-n-PrSO2H, Q-phos (1,2,3,4,5-пентафенил-1′-(ди-трет-бутилфосфино)ферроцена), CataCXium ABn (ди-(1-адамантил)бензилфосфина), CataCXium A (ди-(1-адамантил)-н-бутилфосфина) и S-Phos (2-дициклогексилфосфино-2′,6′-диметоксибифенила). Предпочтительно лиганд представляет собой S-Phos.
Предварительно образованный катализатор на основе комплекса с металлом может состоять из металлов в качестве лигандов, указанных выше, или может быть выбран из [(o-tol)3P]2PdCl2, [t-Bu3PPdBr]2/Pd-113, (dtbpf)PdCl2/Pd-118, PEPPSI, PdCl2(PPh3)2, Pd(tBu2PhP)2, Pd(dppf)Cl2·CH2Cl2, [(t-Bu)3P]Pd(0), CataCXium C, Pd(tBu2PhP)2(Pd-122), Pd(dppf)Cl2·CH2Cl2 (Pd-106), (2-MeOPh)3P/ Pd2(dba)3 и PdCl2(Amphos)2/Pd-132. Предпочтительно предварительно образованный катализатор на основе комплекса с металлом выбран из (dtbpf)PdCl2 (Pd-118), Pd(tBu2PhP)2(Pd-122), Pd(dppf)Cl2·CH2Cl2 (Pd-106) и (2-MeOPh)3P/Pd2(dba)3.
Соль щелочного металла или щелочная соль может быть выбрана из Na2CO3, Cs2CO3, K3PO4, KF и K2CO3. Предпочтительно щелочная соль представляет собой K2CO3.
В WO 2013/171639 A1, пример 9, описан подобный способ на стадии 9.5, в соответствии с которым предварительно образованный катализатор на основе комплекса с металлом Pd(PPh3)2Cl2 и щелочную соль K3PO4 в толуоле применяют для получения промежуточного соединения, представляющего собой метил-6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотинат. Тем не менее, при увеличении масштаба до 3 кг, синтез обеспечивал в результате неполное преобразование исходных материалов, и для него требовалось дополнительное количество соединения формулы (3) и K3PO4 для завершения реакции, что делает его, таким образом, непригодным для крупномасштабного изготовления соединения формулы (1).
Предусмотренная в данном документе реакция обеспечивает высокую степень превращения [более 98% превращения соединения формулы (1)] и высокую чистоту [чистота более 96%, измеренная в качестве контроля в ходе процесса (IPC)]. Соединение формулы (1) получают с большим выходом (более 80%).
Преимущество данного способа по сравнению со способами из предшествующего уровня техники состоит в снижении количества образующихся побочных продуктов и загрузки катализатора, необходимого для более полного превращения исходных материалов. Следовательно, способ по настоящему изобретению является подходящим для увеличения масштаба для целей промышленного получения.
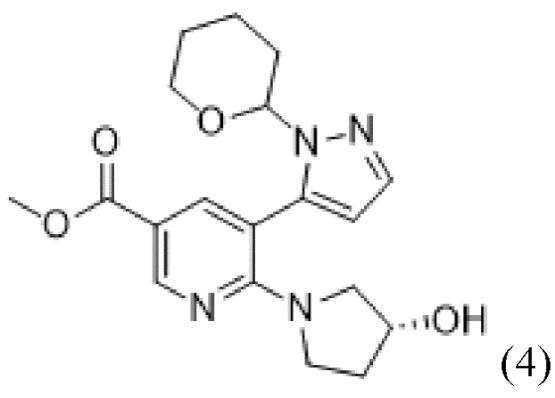
В соответствии со вторым аспектом настоящего изобретения, предусмотрен способ в соответствии с первым аспектом, дополнительно предусматривающий стадию осуществления реакции соединения формулы (4),
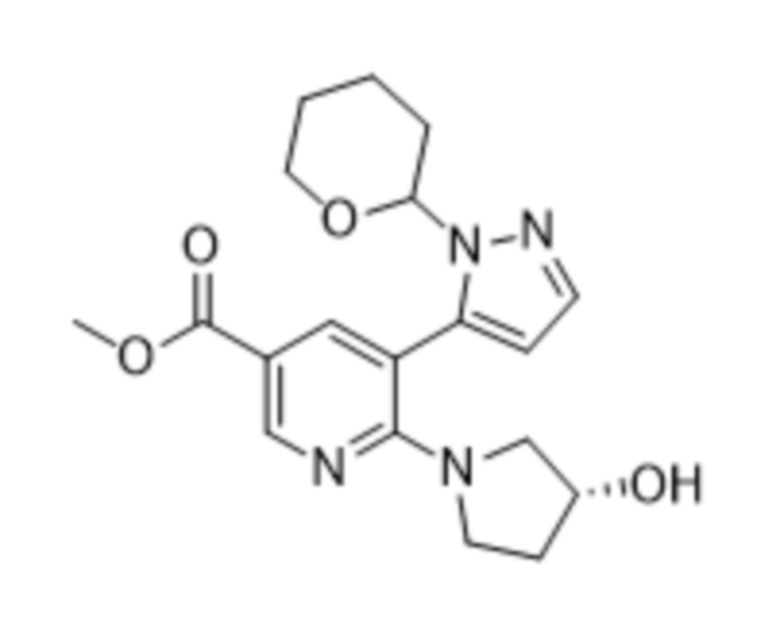 (4),
(4),
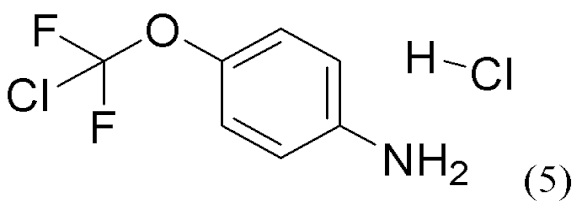
или его соли, сольвата, стереоизомера, комплекса, сокристалла, сложного эфира или оксазолинового производного, предпочтительно соединение формулы (4) находится в его форме свободного метилового сложного эфира; с соединением формулы (5),
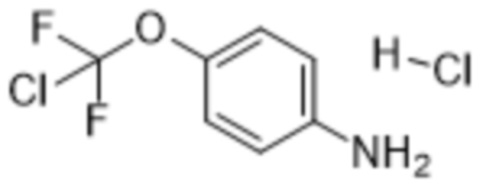 (5),
(5),
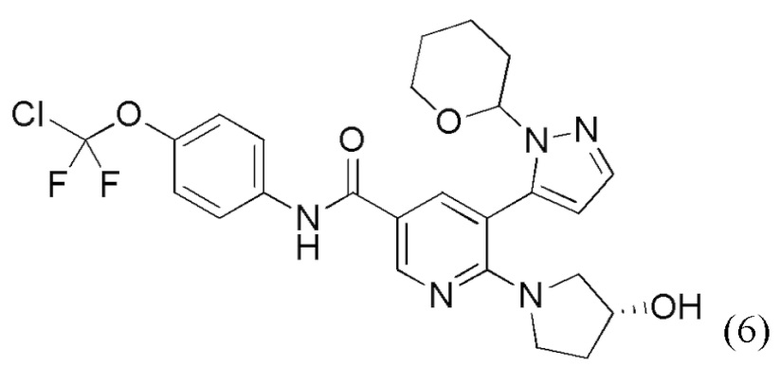
с получением соединения формулы (6),
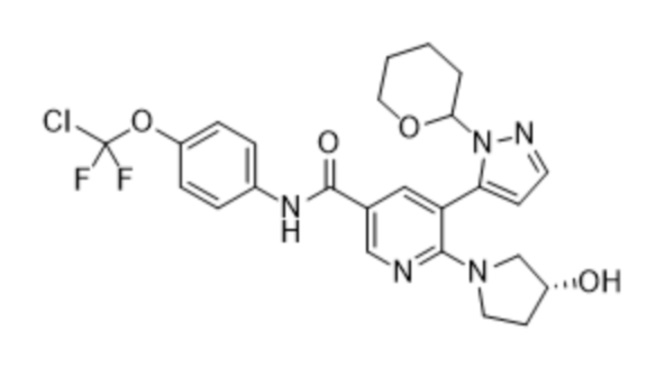 (6),
(6),
или его соли, сольвата, стереоизомера, комплекса, сокристалла, сложного эфира или оксазолинового производного, предпочтительно соединение формулы (6) находится в его форме свободной карбоновой кислоты.
В WO 2013/171639 A1, пример 9, описан подобный способ на альтернативной стадии 9.1, заключающийся в замещении соединения формулы (4) 6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотиновой кислотой в NMM и объединении HOBt·H2O и EDCI·HCl в THF. Тем не менее, было обнаружено, что 6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотиновую кислоту сложно кристаллизовать, экстрагировать и обеспечивать ее постоянный выход из реакции. Синтез также обеспечивал в результате трудноизвлекаемые примеси, которые трудно устранить, и которые отрицательно влияли на выход.
Преимущество способа, раскрытого в данном документе, по сравнению со способом из предшествующего уровня техники заключается в том, что он обеспечивает возможность избежать проблем, связанных с 6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотиновой кислотой, и обеспечивает постоянный выход соединения формулы (6). Следовательно, способ по настоящему изобретению является подходящим для увеличения масштаба для целей промышленного получения.
Примеры
Пример 1. Стадия D2+D3 -> D4
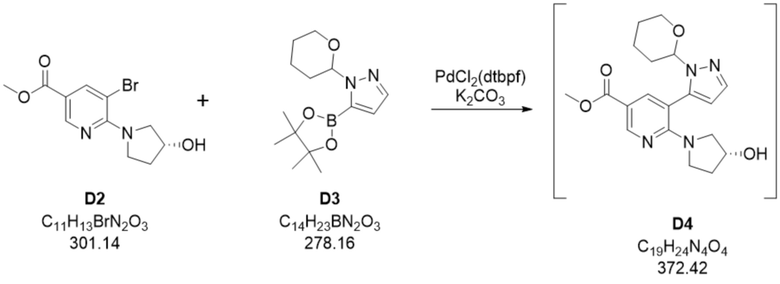
Основная процедура
В сухой сосуд добавляли смесь (R)-метил 5-бром-6-(3-гидроксипирролидин-1-ил)никотината (45,6 кг, D2), 1-1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-бороновой кислоты, сложного пинаколового эфира (50,5 кг, D3), и K2CO3 (41,8 кг) в толуоле (282 мл). Суспензию перемешивали и добавляли воду. Добавляли PdCl2(dtbpf) (500 г) и суспензию перемешивали при приблизительно 50°C до достижения полного превращения. После завершения реакции в реакционную смесь добавляли QuadraSil MP. Твердые остатки удаляли путем фильтрации через фильтр с активированным углем и остаток на фильтре промывали толуолом, питьевой водой и снова толуолом. Органическую и водную фазы разделяли и водный слой промывали толуолом. Объединенные органические слои промывали раствором хлорида натрия, высушивали с применением [Na2SO4] и выпаривали in situ с получением метил-6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотината (D4). По оценкам, выход D4 составлял приблизительно 54 кг D4/кг D2 (~ 95%).
Пример 2. Стадия D4+D5 -> D6
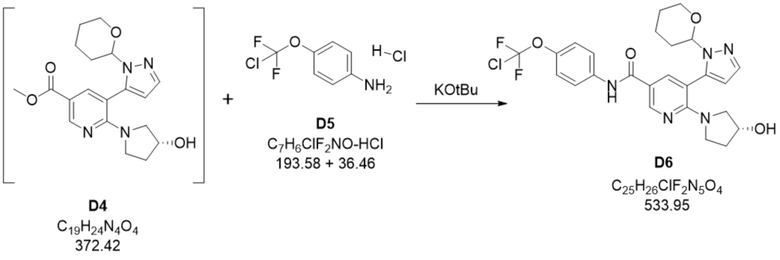
В пустой сосуд добавляли раствор гидроксида натрия (15,8 кг) и воду к смеси 31,6 кг 4-(хлордифторметокси)анилина-HCl (D5) в метилтетрагидрофуране (344 кг) и реакционную смесь перемешивали при примерно 25°С. Двухфазную смесь разделяли и органическую фазу дважды промывали водой. Органическую фазу концентрировали путем перегонки с последующим добавлением свежего метилтетрагидрофурана (2 × 148 кг) с получением концентрированного раствора 4-(хлордифторметокси)анилина в метилтетрагидрофуране.
В 10-20% раствор 660 кг метил-6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотината (D4) в толуоле добавляли метилтетрагидрофуран (239 кг) и раствор концентрировали путем перегонки. В данный концентрированный раствор добавляли 148 кг концентрированного раствора D5 в метилтетрагидрофуране. В полученную смесь дозировали 20% раствор трет-бутоксида калия (258 кг) в тетрагидрофуране (169 кг) при приблизительно 25°C. После завершения реакции добавляли водный раствор хлорида натрия (602 кг) и двухфазную смесь разделяли. Органическую фазу экстрагировали водным раствором хлорида натрия (602 кг). Органический слой фильтровали через фильтр с активированным углем. Растворитель заменяли путем перегонки с метилтетрагидрофурана на изопропанол. В данный раствор добавляли затравочные кристаллы N-(4-(хлордифторметокси)фенил)-6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотинамида (0,16 кг, D6) с последующим добавлением н-гептана (1274 кг). D6 собирали путем фильтрации, промывали смесью н-гептана и изопропанола и высушивали в вакууме. По оценкам, выход D6 составлял приблизительно 79-87% D6, в зависимости от количества загруженного D3.
Пример 3. Стадия D6 -> A1
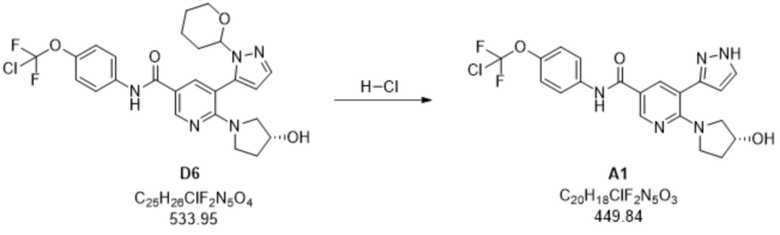
В суспензию N-(4-(хлордифторметокси)фенил)-6-((R)-3-гидроксипирролидин-1-ил)-5-(1-(тетрагидро-2H-пиран-2-ил)-1H-пиразол-5-ил)никотинамида (39,1 кг, D6) в метаноле (346 кг) добавляли 37% раствор HCl (9 кг) при 22°C (pH < 1). Затем прозрачный раствор перемешивали в течение 1 ч. при 22°C (IPC). Затем смесь гасили с помощью 30% NaOH (4 кг) (pH=10). В смесь добавляли воду и регулировали pH до 2,5-3,0 путем добавления 30% NaOH. Раствор фильтровали через фильтр с активированным углем и затем pH регулировали до 3,0-3,5 путем добавления еще 30% гидроксида натрия, после чего добавляли затравку N-(4-(хлордифторметокси)фенил)-6-(3-гидроксипирролидин-1-ил)-5-(1H-пиразол-5-ил)никотинамида (0,027 кг, A1). Окончательное регулирование pH до 7,5-9,0 осуществляли путем добавления прибл. 1% раствора гидроксида натрия, в результате чего продукт осаждался. Суспензию охлаждали до 10°C и перемешивали, после чего выделяли продукт A1 путем фильтрации, промывали смесью 4:1 вода/метанол и высушивали. По оценкам, выход A1 составлял приблизительно 76%.
Пример 4. A1 -> A1a
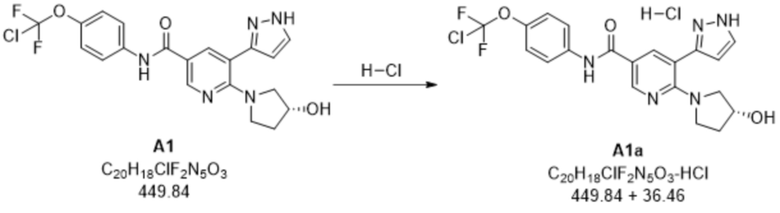
Нагревали смесь N-(4-(хлордифторметокси)фенил)-6-(3-гидроксипирролидин-1-ил)-5-(1H-пиразол-5-ил)никотинамида (29,2 кг, A1), метанола (190 кг) и 37% хлористоводородной кислоты (7 кг) до приблизительно 50°C и полученный раствор фильтровали. Первую порцию трет-бутилметилового эфира TBME (146 кг) и затравочные кристаллы гидрохлорида N-(4-(хлордифторметокси)фенил)-6-(3-гидроксипирролидин-1-ил)-5-(1H-пиразол-5-ил)никотинамида (0,26 кг, A1a) добавляли к фильтрату при приблизительно 50°C. Добавляли вторую порцию TBME (271 кг) и суспензию охлаждали до приблизительно 0°C и перемешивали с обеспечением завершения кристаллизации. Собирали A1a путем фильтрации, промывали смесью TBME (78 кг) и метанола (9 кг) и высушивали в вакууме. По оценкам, выход A1a составлял приблизительно 96%.
| название | год | авторы | номер документа |
|---|---|---|---|
| КРИСТАЛЛИЧЕСКИЕ ФОРМЫ N-[4-(ХЛОРДИФТОРМЕТОКСИ)ФЕНИЛ]-6-[(3R)-3-ГИДРОКСИПИРРОЛИДИН-1-ИЛ]-5-(1H-ПИРАЗОЛ-5-ИЛ)ПИРИДИН-3-КАРБОКСАМИДА | 2020 |
|
RU2836337C2 |
| СПОСОБ ПОЛУЧЕНИЯ АНТАГОНИСТОВ АНДРОГЕННОГО РЕЦЕПТОРА И ИХ ПРОМЕЖУТОЧНЫХ СОЕДИНЕНИЙ | 2016 |
|
RU2719590C2 |
| СПОСОБ ПОЛУЧЕНИЯ ИНГИБИТОРОВ 5-ЛИПОКСИГЕНАЗЫ, ИМЕЮЩИХ РАЗЛИЧАЮЩИЕСЯ ГЕТЕРОЦИКЛИЧЕСКИЕ СИСТЕМЫ | 2000 |
|
RU2177005C1 |
| СПОСОБ ПОЛУЧЕНИЯ 2-[(3R)-3-МЕТИЛМОРФОЛИН-4-ИЛ]-4-(1-МЕТИЛ-1H-ПИРАЗОЛ-5-ИЛ)-8-(1H-ПИРАЗОЛ-5-ИЛ)-1,7-НАФТИРИДИНА | 2019 |
|
RU2802512C2 |
| ПИРАЗОЛОХИНОЛИНОВОЕ ПРОИЗВОДНОЕ | 2012 |
|
RU2605096C2 |
| ПРОИЗВОДНЫЕ ПИРАЗОЛОХИНОЛИНОНА, ИХ ПОЛУЧЕНИЕ И ИХ ТЕРАПЕВТИЧЕСКОЕ ПРИМЕНЕНИЕ | 2012 |
|
RU2621037C2 |
| ПРОИЗВОДНЫЕ АЗАИНДАЗОЛА ИЛИ ДИАЗАИНДАЗОЛА В КАЧЕСТВЕ МЕДИКАМЕНТА | 2012 |
|
RU2600976C2 |
| СОЕДИНЕНИЕ, ОБЛАДАЮЩЕЕ АГОНИСТИЧЕСКОЙ АКТИВНОСТЬЮ В ОТНОШЕНИИ STING | 2019 |
|
RU2798265C2 |
| ИНГИБИТОРЫ СЕРИН/ТРЕОНИНОВЫХ КИНАЗ | 2013 |
|
RU2650501C2 |
| ГЕТЕРОЦИКЛИЧЕСКОЕ СОЕДИНЕНИЕ | 2018 |
|
RU2791533C2 |
Изобретение относится к способу получения соединения формулы (1) или его соли. Способ предусматривает стадию осуществления реакции соединения формулы (2) или его соли и соединения формулы (3) с солью щелочного металла или щелочной солью в присутствии катализатора на основе комплекса с металлом с получением соединения формулы (4) или его соли. Соль щелочного металла представляет собой K2CO3 и катализатором на основе комплекса с металлом является (dtbpf)PdCl2. Затем осуществляют стадию реакции соединения формулы (4) или его соли с соединением формулы (5) с получением соединения формулы (6) или его соли и стадию соединения формулы (6) с HCl с получением соединения формулы (1) или его соли. Предлагаемый способ позволяет получать соединение формулы (1) с более высоким выходом и более высокой чистотой с меньшим количеством побочных продуктов и при более низкой загрузке катализатора. Изобретение относится также к варианту указанного способа. 2 н.п. ф-лы, 4 пр.
1. Способ получения соединения формулы (1)
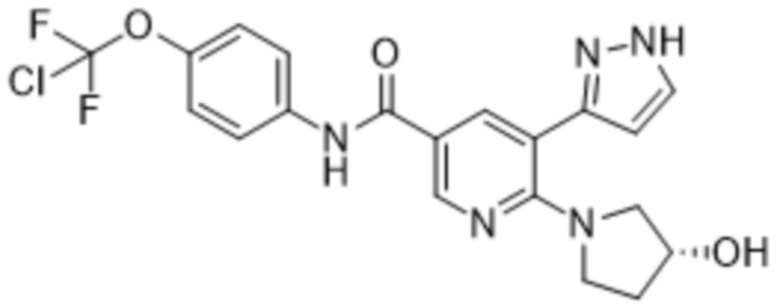 (1)
(1)
или его соли,
предусматривающий стадию осуществления реакции соединения формулы (2)
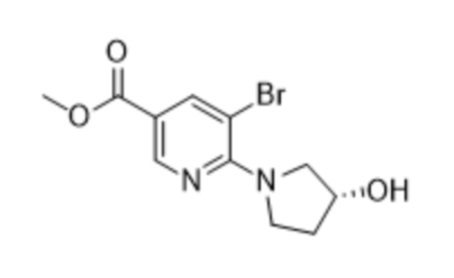 (2)
(2)
или его соли
и соединения формулы (3)
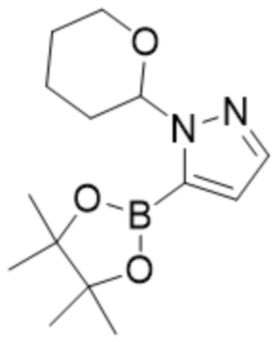 (3)
(3)
с солью щелочного металла или щелочной солью в присутствии катализатора на основе комплекса с металлом с получением соединения формулы (4) или его соли,
где соль щелочного металла представляет собой K2CO3 и катализатором на основе комплекса с металлом является (dtbpf)PdCl2;
стадию осуществления реакции соединения формулы (4)
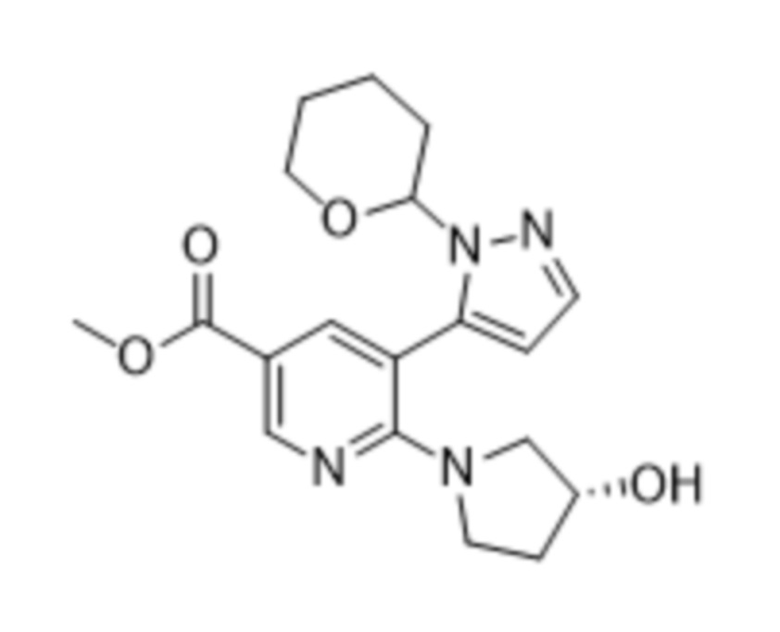 (4)
(4)
или его соли с соединением формулы (5)
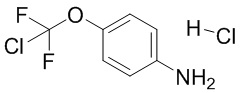 с получением соединения формулы (6)
с получением соединения формулы (6)
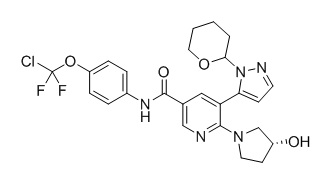 (6)
(6)
или его соли; и
стадию соединения формулы (6) с HCl с получением соединения формулы (1) или его соли.
2. Способ получения соединения формулы (1)
(1),
предусматривающий стадию осуществления реакции соединения формулы (2)
(2)
или его соли и соединения формулы (3)
(3)
с солью щелочного металла или щелочной солью в присутствии катализатора на основе комплекса с металлом с получением соединения формулы (4) или его соли,
где соль щелочного металла представляет собой K2CO3 и катализатором на основе комплекса с металлом является (dtbpf)PdCl2;
стадию осуществления реакции соединения формулы (4)
(4)
или его соли с соединением формулы (5)
с получением соединения формулы (6)
(6).
| Многоступенчатая активно-реактивная турбина | 1924 |
|
SU2013A1 |
| СОЕДИНЕНИЯ И КОМПОЗИЦИИ В КАЧЕСТВЕ ИНГИБИТОРОВ ITPKb | 2007 |
|
RU2425826C2 |
| ПРОИЗВОДНЫЕ АМИНОПИРАЗИНА И ЛЕКАРСТВЕННЫЕ СРЕДСТВА | 2010 |
|
RU2535217C2 |

