Область применения
Настоящее изобретение распространяется на имидазохинолиновые соединения с сульфонамидным заместителем в положении 1 и на фармацевтические формулы, содержащие такие соединения. Еще один аспект настоящего изобретения связан с применением таких соединений в качестве иммуномодуляторов для индукции биосинтеза цитокинов у животных и для лечения болезней, в частности вирусных и онкологических.
Предпосылки изобретения
В первом достоверном сообщении о системе 1H-имидазо-[4,5-с]-хинолинового кольца, Backman et al., J. Org. Chem. 15, 1278-1284 (1950), описывается синтез 1-(6-метокси-8-хинолинил)-2-метил-1H-имидазо-[4,5-с]-хинолина с целью возможного применения антималярийного агента. Впоследствии сделаны многочисленные сообщения о синтезе различных замещенных 1H-имидазо-[4,5-с]-хинолинов. Например, в работе Jain et al., J. Med. Chem. 11, pp.87-92 (1968) сообщается о синтезе вещества 1-[2-(4-пиперидил)-этил]-1H-имидазо-[4,5-с]-хинолина в качестве потенциального противосудорожного и сердечно-сосудистого агента. Кроме того, Baranov et al., Chem. Abs. 85, 94362 (1976), сообщали о нескольких 2-оксоимидазо-[4,5-с]-хинолинах, и Berenyi et al., J. Heterocyclic Chem. 18, 1537-1540 (1981), сообщили о некоторых 2-оксоимидазо-[4,5-с]-хинолинах.
Позже было обнаружено, что некоторые 1H-имидазо-[4,5-с]-хинолин-4-амины и их 1- и 2-замещенные производные можно применять в качестве противовирусных, бронхорасширяющих и иммуномодулирующих средств. Они описываются, в частности, в патентах США № 4689338; 4698348; 4929624; 5037986; 5268376; 5346905 и 5389640, которые включены в настоящий документ по ссылке.
Продолжает вызывать интерес система имидазохинолинового кольца, как можно видеть, например, в документах WO 98/30562, ЕР 894797 и WO 00/09506. Так, в патенте ЕР 894797 описываются амидзамещенные имидазохинолиновые соединения, которые раскрываются как вещества, способные оказывать модулирующее действие на иммунный ответ, а в документе WO 00/09506 раскрываются имидазохинолиновые вещества, которые содержат сульфонамидную замещающую группу, в которой азот сульфонамида является частью насыщенного гетероциклического кольца. Несмотря на эти попытки, однако, сохраняется потребность в поиске новых соединений, способных модулировать иммунный ответ путем индукции биосинтеза цитокинов или через другие механизмы.
Краткое описание изобретения
Нами открыт новый класс соединений, которые способны индуцировать биосинтез цитокинов у животных. Таким образом, данное изобретение представляет соединения, соответствующие формуле I:
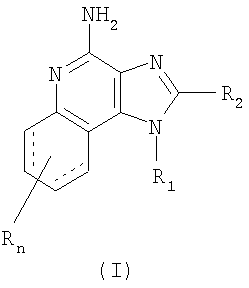
где R, R1 и R2 определяются, как описано ниже.
Соединения, соответствующие формуле I, могут действовать как модуляторы иммунного ответа благодаря их способности индуцировать биосинтез цитокинов и, с другой стороны, модулировать иммунный ответ при введении животным. Это делает такие соединения пригодными для лечения различных состояний, таких как вирусные болезни и опухоли, которые реагируют на подобные изменения иммунного ответа.
В одном из вариантов реализации данного изобретения рассматриваемые соединения выбираются из группы, состоящей из следующих веществ:
N-[4-(4-амино-2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид;
N-[4-(4-амино-2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид;
N-[4-(4-амино-2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид;
N-[4-(4-амино-2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид;
N-[4-(4-амино-2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид;
N-[4-{4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид;
N-[(4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид;
N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]метансульфонамид;
N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]бензолсульфонамид;
N-[4-(4-амино-2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид;
N-{8-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]октил}бензолсульфонамид;
N-{8-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]октил}метансульфонамид;
N-[8-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)октил]метансульфонамид;
N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]-5-(диметиламино)нафталин-1-сульфонамид;
N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]-4-метилбензолсульфонамид;
N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамид;
N-[8-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)октил]бензолсульфонамид;
N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}бензолсульфонамид;
N-[4-(4-амино-2-пентил-1H-имидазо[4,5-с]хинолин-1-ил)бутил]метансульфонамид;
N-[4-(4-амино-2-пентил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид;
N-[8-(4-амино-1H-имидазо-[4,5-с]-хинолин-1-ил)октил]метансульфонамид;
N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-пропил}-4-метилбензолсульфонамид;
N-[4-(4-амино-2-пентил-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид;
N-{3-[4-амино-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамид;
N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-2,2-диметилпропил}метансульфонамид;
N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-пропил}-5-(диметиламино)нафталин-1-сульфонамид;
N-[3-(4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]-метансульфонамид;
N-{3-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамид;
N-{3-[4-амино-2-(этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-c]-хинолин-1-ил]пропил}метансульфонамид;
N-{3-[4-амино-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил}метансульфонамид;
N-{4-[4-амино-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-1-ил]бутил}метансульфонамид;
N-[4-(4-амино-2-метил-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид-гидрохлорид;
N-[2-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)этил]-4-метилбензолсульфонамид;
N-[2-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)этил]метансульфонамид;
1-[4-(1,1-диоксидоизотиазолидин-2-ил)бутил]-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амин;
2-бутил-1-[4-(1,1-диоксидоизотиазолидин-2-ил)бутил]-1H-имидазо-[4,5-с]-хинолин-4-амин;
N-{2-[4-амино-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтил}-метансульфонамид;
N-[4-(4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]этансульфонамид;
1-(2-амино-2-метилпропил)-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-4-амин и
N-{4-[4-амино-2-(циклопропилметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]бутил}метансульфонамид;
или фармацевтически приемлемая соль этих веществ.
В особо предпочтительном варианте реализации данного изобретения таким веществом или солью является N-{2-[4-амино-2-(этоксиметил)-1H-имидазо[4,5-с]-хинолин-1-ил]-1,1-диметилэтил}метансульфонамид или фармацевтически приемлемая соль этого вещества. Помимо соответствующей формулы и низкой токсичности это соединение обладает неожиданно высокой активностью в отношении индукции интерлейкина IL-12, сопоставимой с интерферон-(α)-индуцирующей активностью.
Кроме того, в изобретении представлены фармацевтические композиции, содержащие терапевтически эффективное количество вещества или соли, соответствующих формуле I или вышеприведенным примерам, а также способы индукции биосинтеза цитокинов в организме животного, лечения вирусной инфекции и/или лечения онкологического заболевания в организме животного путем введения в организм животного эффективного количества вещества, соответствующего формуле I или вышеприведенным примерам, или его соли.
В дополнение к этому представлены способы синтеза соединений, соответствующих формуле I, и промежуточных веществ, используемых при синтезе этих соединений.
Подробное описание изобретения
Как упоминалось выше, в настоящем изобретении представлены соединения, соответствующие формуле I:
в которой R1 - -алкил-NR3-SO2-Х-R4, -алкенил-NR3-SO2-Х-R4 или -алкил-NR6-SO2-R7 ;
Х - ковалентная связь или -NR5-;
R4 - арил, гетероарил, гетероциклил, алкил или алкенил, каждый их которых может быть незамещенным или замещенным одним или несколькими заместителями, выбираемыми из группы, состоящей из следующих остатков:
- алкил;
- алкенил;
- арил;
- гетероарил;
- гетероциклил;
- замещенный арил;
- замещенный гетероарил;
- замещенный гетероциклил; -O-алкил;
-O-(алкил)0-1-арил;
-O-(алкил)0-1-замещенный арил;
-O-(алкил)0-1-гетероарил;
-O-(алкил)0-1-замещенный гетероарил; -O-(алкил)0-1-гетероциклил;
-O-(алкил)0-1-замещенный гетероциклил;
-СООН;
-СО-O-алкил;
-СО-алкил;
-S(O)0-2-алкил;
-S(O)0-2-(алкил)0-1-арил;
-S(O)0-2-(алкил)0-1-замещенныйарил;
-S(O)0-2-(алкил)0-1-гетероарил;
-S(O)0-2-(алкил)0-1-замещенный гетероарил;
-S(O)0-2-(алкил)0-1-гетероциклил;
-S(O)0-1-(алкил)0-1-замещенный гетероциклил;
-(алкил)0-1-NR3R3;
-(алкил)0-1-NR3-COO-алкил;
-(алкил)0-1-NR3-СО-алкил;
-(алкил)0-1-NR3-СО-арил;
-(алкил)0-1-NR3-CO-замещенный арил;
-(алкил)0-1-NR3-СО-гетероарил;
-(алкил)0-1-NR3-CO-замещенный гетероарил;
-N3;
- галоген;
- галоалкил;
- галоалкокси;
-СО-галоалкокси;
-NO2;
-CN;
-ОН;
-SH; и в случае алкила, алкенила, или гетероциклила - оксо;
R2 выбирается из группы, состоящей из следующих:
- водород;
- алкил;
- алкенил;
- арил;
- замещенный арил;
- гетероарил;
- замещенный гетероарил;
- алкил-O-алкил;
- алкил-O-алкенил и
- алкил или алкенил, замещенный одним или несколькими заместителями, выбираемыми из группы, включающей следующие остатки:
-ОН;
- галоген;
-N(R3)2;
-CO-N(R3)2;
-CO-C1-10алкил;
-CO-O-C1-10алкил;
-N3;
- арил;
- замещенный арил;
- гетероарил;
- замещенный гетероарил;
- гетероциклил;
- замещенный гетероциклил;
-СО-арил;
-CO-(замещенный арил);
-СО-гетероарил и
-CO-(замещенный гетероарил);
каждый R3 независимо выбирается из группы, включающей водород и C1-10 алкил;
R5 выбирается из группы, состоящей из водорода и C1-10алкила, или R4 и R5 могут соединяться с образованием 3-7-членного гетероциклического или замещенного гетероциклического кольца;
R6 выбирается из группы, состоящей из водорода и C1-10алкила;
R7 выбирается из группы, состоящей из водорода и C1-10алкила, где R6 и R7 соединяются с образованием 3-7-членного гетероциклического или замещенного гетероциклического кольца;
n - число от 0 до 4, и каждый представленный остаток R независимо выбирается из группы, включающей С1-10-алкил, C1-10-алкокси, галоген и трифторметил, или фармацевтически приемлемые соли этих веществ.
Синтез веществ
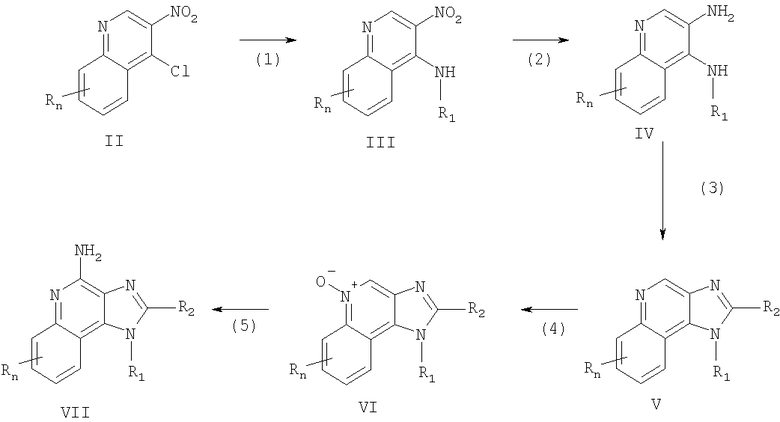
Имидазохинолины, представленные в настоящем изобретении, можно получить согласно Реакционной схеме I, где R, R1, R2 и n определяются, как описано выше.
На этапе (1) Реакционной схемы I 4-хлор-3-нитрохинолин, соответствующий формуле II вступает в реакцию с амином, соответствующим формуле R1NH2, где R1 определяется, как описано выше, с образованием 3-нитрохинолин-4-амина, соответствующего формуле III. Эту реакцию можно провести путем добавления амина к раствору вещества, соответствующего формуле II, в подходящем растворителе, таком как хлороформ или дихлорметан, и факультативно при нагревании. Многие хинолины, соответствующие формуле II, являются известными веществами (см., например, патент США 4689338 и ссылки, процитированные в настоящем документе).
На этапе (2) Реакционной схемы I 3-нитрохинолин-4-амин, соответствующий формуле III, восстанавливается с образованием хинолин-3,4-диамина, соответствующего формуле IV. Предпочтительно, чтобы восстановление проводилось с использованием стандартного гетерогенного катализатора гидрогенизации, такого как платина на углероде или палладий на углероде. Эту реакцию можно проводить стандартным способом на аппарате Парра в подходящем растворителе, например изопропиловом спирте или толуоле.
На этапе (3) Реакционной схемы I хинолин-3,4-диамин, соответствующий формуле IV, вступает в реакцию с карбоновой кислотой или ее эквивалентом с образованием 1H-имидазо-[4,5-с]-хинолина, соответствующего формуле V. В число веществ, эквивалентных карбоновым кислотам, входят галиды кислот, ортоэфиры и 1,1-диалкоксиалкил-алканоаты. Карбоновая кислота или ее эквивалент выбирается таким образом, чтобы в результате реакция получился требуемый R2 - заместитель в веществе, соответствующем формуле V. Например, триэтил-орто-формиат даст вещество, в котором R2 - это водород, а триэтил-орто-ацетат даст вещество, в котором R2 - это метильная группа. Реакцию можно проводить в отсутствие растворителя или в инертном растворителе, таком как толуол. Реакция проводится при нагревании, достаточном для того, чтобы удалить любой спирт или воду, которые являются побочными продуктами данной реакции.
На этапе (4) Реакционной схемы I 1H-имидазо-[4,5-с]-хинолин, соответствующий формуле V, окисляется с образованием 1H-имидазо-[4,5-с]-хинолин-5N-оксида, соответствующего формуле VI, с использованием стандартного окислителя, способного привести к образованию N-оксидов. Предпочтительными условиями для проведения реакции является реагирование раствора вещества, соответствующего формуле V, в хлороформе с 3-хлорпероксибензоевой кислотой при нормальных условиях.
На этапе (5) Реакционной схемы I 1H-имидазо-[4,5-с]-хинолин-5N-оксид, соответствующий формуле VI, аминируют с образованием 1H-имидазо-[4,5-с]-хинолин-4-амина, соответствующего формуле VII, который представляет собой подрод веществ, соответствующих формуле I. Этап (5) включает (i) проведение реакции вещества, соответствующего формуле VI, с ацилирующим агентом, а затем (ii) реакцию продукта с аминирующим агентом. Часть (i) этапа (5) включает реакцию N-оксида, соответствующего формуле VI, с ацилирующим агентом. К числу подходящих ацилирующих агентов относятся алкил- или арил-сульфонил-хлориды (например, бензолсульфонил-хлорид, метансульфонил-хлорид, п-толуолсульфонил-хлорид). Арилсульфонил-хлориды предпочтительны. пара-Толуолсульфонил-хлорид является наиболее предпочтительным. Часть (ii) этапа (5) включает реакцию продукта части (i) с избытком аминирующего агента. В число подходящих аминирующих агентов входят аммоний (например, в форме гидроксида аммония) и соли аммония (например, карбонат аммония, бикарбонат аммония, фосфат аммония). Предпочтительным является гидроксид аммония. Реакцию предпочтительно проводить путем растворения N-оксида, соответствующего формуле VI, в инертном растворителе, таком как дихлорметан, добавления к раствору аминирующего агента, а затем медленного добавления ацилирующего агента. Продукт или его фармацевтически приемлемую соль можно выделить с использованием стандартных способов.
В альтернативном варианте этап (5) можно провести путем (i) реакции N-оксида, соответствующего формуле VI, с изоцианатом, а затем (ii) гидролиза полученного продукта. Часть (i) включает реакцию N-оксида с изоцианатом, при которой группа изоцианата связывается с карбоксильной группой. В число предпочтительных изоцианатов входят трихлорацетил-изоцианат и ароил-изоцианаты, такие как бензоил-изоцианат. Реакцию изоцианата с N-оксидом проводят в практически безводных условиях путем добавления изоцианата к раствору N-оксида в инертном растворителе, таком как хлороформ или дихлорметан. Часть (ii) включает гидролиз продукта из части (i). Этот гидролиз можно провести стандартными способами, например, при нагревании в присутствии воды или низшего спирта, факультативно, в присутствии катализатора, такого как гидроксид щелочного металла или низший алкоксид.
Схема реакции I
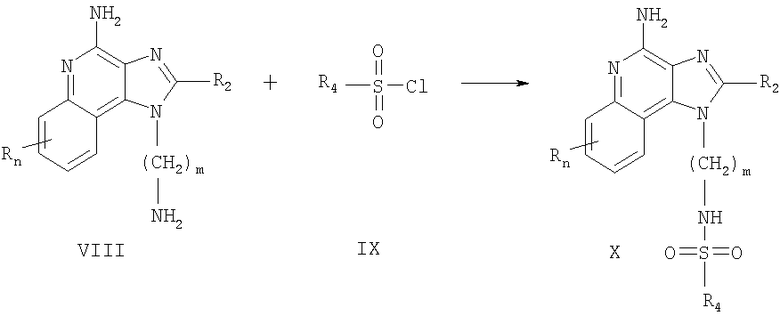
Вещества, которые представлены в настоящем изобретении и в которых заместитель R1 содержит сульфонамид, можно также получить согласно Схеме реакции II, в которой R, R1, R4 и n определяются, как описано выше, a m - это число от 1 до 20.
В Схеме реакции II аминоалкилзамещенный 1H-имидазо-[4,5-с]-хинолин-4-амин, соответствующий формуле VIII, вступает в реакцию с сульфонил-хлоридом, соответствующим формуле IX, с образованием соединения, соответствующего формуле X, которая описывает подрод соединений, соответствующих формуле I. Реакцию можно проводить при комнатной температуре в инертном растворителе, таком как дихлорметан, в присутствии основания, например, пиридина или N,N-диизопропилэтиламина. Многие 1H-имидазо-[4,5-с]-хинолин-4-амины, соответствующие формуле VIII, являются известными веществами, описанными, например в патенте США 6069149 (Namba); другие можно без особых сложностей получить с использованием известных способов синтеза. Многие сульфонил-хлориды, соответствующие формуле IX, являются коммерчески доступными; другие можно легко получить с использованием известных способов синтеза. Полученный продукт или его фармацевтически приемлемую соль можно выделить с использованием стандартных способов.
Схема реакции II
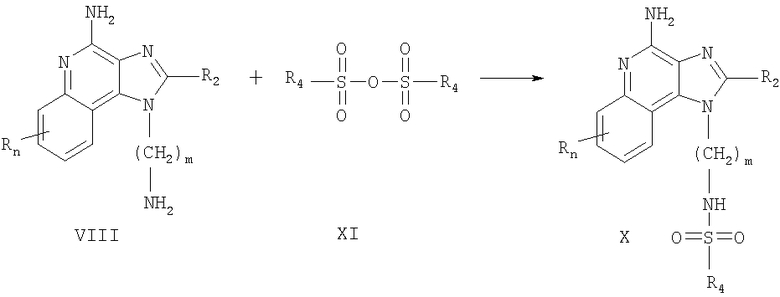
Соединения, которые представлены в настоящем изобретении и в которых заместитель R1 содержит сульфонамид, можно также получить согласно Схеме реакции III, в которой R, R2, R4 и n определяются, как описано выше, a m - это число от 1 до 20.
В Схеме реакции III аминоалкилзамещенный 1H-имидазо-[4,5-с]-хинолин-4-амин, соответствующий формуле VIII, вступает в реакцию с сульфоновым ангидридом, соответствующим формуле XI, с образованием соединения, соответствующего формуле X, которая описывает подрод соединений, соответствующих формуле I. Реакцию можно проводить при комнатной температуре в инертном растворителе, таком как дихлорметан, в присутствии основания, такого как пиридин или N,N-диизопропилэтиламин. В альтернативном варианте реакцию можно проводить при комнатной температуре в ацетонитриле. Многие сульфоновые ангидриды, соответствующие формуле XI, являются коммерчески доступными; другие можно легко получить с использованием известных способов синтеза. Продукт или его фармацевтически приемлемую соль можно выделить с использованием стандартных способов.
Схема реакции III
Третичные сульфонамиды, представленные в настоящем изобретении, можно получить согласно Реакционной схеме IV, в которой R, R2, R3, R4 и n определяются, как описано выше, a m - это число от 1 до 20.
В Схеме реакции IV 1H-имидазо-[4,5-с]-хинолинил-сульфонамид, соответствующий формуле X, вступает в реакцию с галидом, соответствующим формуле XII, с образованием соединения, соответствующего формуле XIII, которая описывает подрод соединений, соответствующих формуле I. Реакцию можно проводить при комнатной температуре путем добавления гидрида натрия к раствору вещества, соответствующего формуле X, в N,N-диметилформамиде с последующим добавлением галида. Многие галиды, соответствующие формуле XII, являются коммерчески доступными; другие можно легко получить с использованием известных способов синтеза. Полученный продукт или его фармацевтически приемлемую соль можно выделить с использованием стандартных способов.
Схема реакции IV
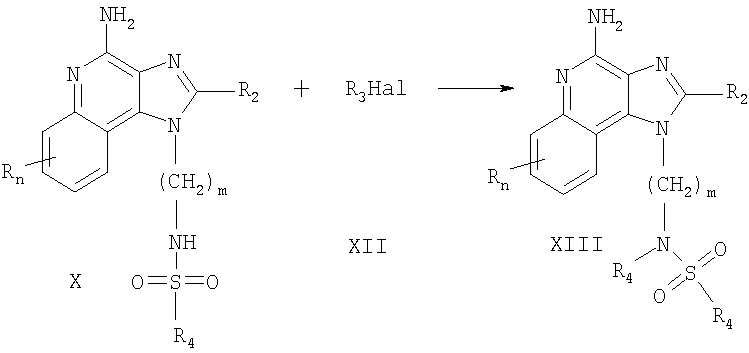
Соединения, которые представлены в настоящем изобретении и в которых R1 содержит сульфамидную группу, можно получить согласно Реакционной схеме V, в которой R, R2, R4, R5 и n определяются, как описано выше, a m - это число от 1 до 20.
На этапе (1) Схемы реакции V аминоалкилзамещенный 1Н-имидазо-[4,5-с]-хинолин-4-амин, соответствующий формуле VIII, вступает в реакцию с сульфурилхлоридом с образованием in situ сульфамоилхлорида, соответствующего формуле XIV. Реакцию можно проводить путем добавления раствора сульфурилхлорида в дихлорметане к раствору вещества, соответствующего формуле VIII, в дихлорметане в присутствии одного эквивалента 4-(диметиламино)пиридина. Эту реакцию предпочтительно проводить при низкой температуре (-78°С). Факультативно, после того как добавление завершено, реакционной смеси можно дать нагреться до комнатной температуры.
На этапе (2) Схемы реакции V амин, соответствующий формуле R5R4NH, вступает в реакцию с сульфамоилхлоридом, соответствующим формуле XIV, с образованием 1H-имидазо-[4,5-с]-хинолинилсульфамида, соответствующего формуле XV, которая описывает подрод соединений, соответствующих формуле I. Реакцию можно проводить путем добавления раствора, содержащего 2 эквивалента амина и 2 эквивалента триэтиламина в дихлорметане в реакционную смесь, полученную на этапе (1). Добавление предпочтительно проводить при низкой температуре (-78°С). После того как добавление завершено, реакционной смеси можно дать нагреться до комнатной температуры. Полученный продукт или его фармацевтически приемлемую соль можно выделить с использованием стандартных способов.
Схема реакции V
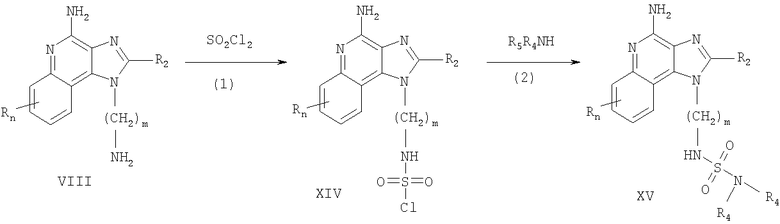
Тетрагидроимидазохинолины, представленные в настоящем изобретении, можно получить согласно Реакционной схеме VI, в которой R2, R3, R4, и R5 определяются, как описано выше, a m - это 1-20.
На этапе (1) Схемы реакции VI аминоалкилзамещенный 1Н-имидазо-[4,5-с]-хинолин-4-амин, соответствующий формуле XVI, восстанавливается с образованием аминоалкилзамещенного 6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амина, соответствующего формуле XVII. Предпочтительно, чтобы восстановление проводилось путем суспендирования или растворения вещества, соответствующего формуле XVI, в трифторуксусной кислоте, добавления каталитического количества оксида платины (IV) с последующей обработкой смеси водородом при повышенном давлении. Эту реакцию можно проводить стандартным способом на аппарате Парра. Полученный продукт или его соль можно выделить с использованием стандартных способов.
На этапе (2а) Схемы реакции VI аминоалкилзамещенный 6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амин, соответствующий формуле XVII, реагирует с образованием соединения, соответствующего формуле XVIII, которая описывает подрод соединений, соответствующих формуле I. Если R3 - это водород, то реакцию можно проводить в один этап в соответствии с способами, описанными в приведенных выше Схемах реакции II и III с использованием тетрагидроимидазохинолина, соответствующего формуле XVII, вместо имидазохинолина, соответствующего формуле VIII. Если R3 - заместитель, отличный от водорода, то реакцию можно проводить в два этапа, причем один этап проводится в соответствии с способами Реакционных схем II и III, а второй этап проводится в соответствии с способом из Реакции IV с использованием тетрагидроимидазохинолинового аналога имидазохинолина. Полученный продукт или его фармацевтически приемлемую соль можно выделить с использованием стандартных способов.
На этапе (2b) Схема реакции VI аминоалкилзамещенный 6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амин, соответствующий формуле XVII, реагирует с образованием соединения, соответствующего формуле XIX, которая описывает подрод соединений, соответствующих формуле I. Реакцию можно проводить в соответствии с способом, описанным в Реакционной схеме V, с использованием тетрагидроимидазохинолина, соответствующего формуле XVII, вместо имидазохинолина, соответствующего формуле VIII. Полученный продукт или его фармацевтически приемлемую соль можно выделить с использованием стандартных способов.
Схема реакции VI
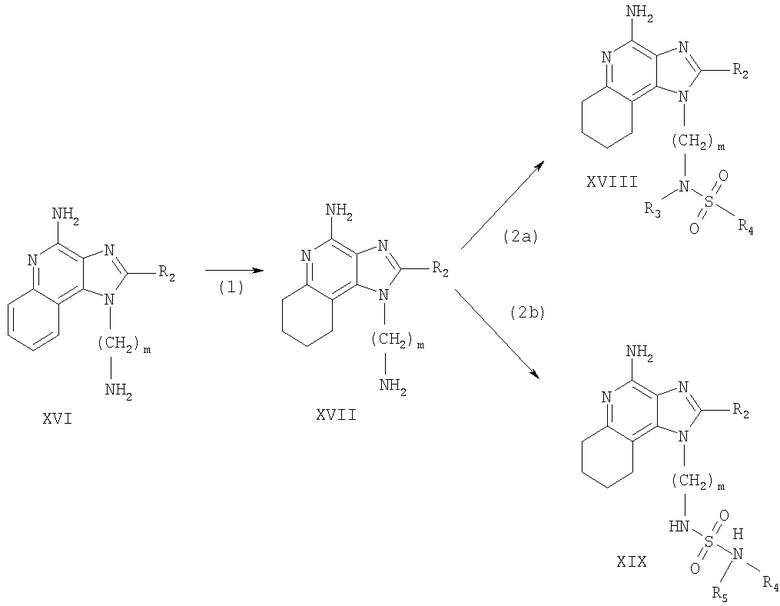
Тетрагидроимидазохинолины, представленные в настоящем изобретении, можно также получить согласно Реакционной схеме VII, в которой R, R2, R3, R4, R5 и n определяются, как описано выше, a m - это число от 1 до 20.
На этапе (1) Схемы реакции VII 6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолинил-трет-бутилкарбамат, соответствующий формуле XX, подвергается гидролизу с образованием аминоалкилзамещенного 6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амина, соответствующего формуле XXI. Реакцию можно проводить путем растворения вещества, соответствующего формуле XX, в смеси трифторуксусной кислоты и ацетонитрила и перемешивания при комнатной температуре. В альтернативном варианте вещество, соответствующее формуле XX, можно смешать с разбавленной соляной кислотой и нагреть на паровой бане. Тетрагидро-1H-имидазо-[4,5-с]-хинолинил-трет-бутилкарбаматы, соответствующие формуле XX, можно получить с использованием способа синтеза, раскрытого в Патенте США 5352784 (Nikolaides). Полученный продукт или его соль можно выделить с использованием стандартных способов.
Этапы (2а) и (2b) можно выполнять тем же способом, который указан в Реакционной схеме VI.
Схема реакции VII
Некоторые соединения, соответствующие формуле I, можно легко получить из других соединений, соответствующих формуле I. Например, вещества, в которых заместитель R4 содержит хлоралкильную группу, могут вступать в реакцию с амином с образованием R4-заместителя, замещенного вторичной или третичной аминогруппой; вещества, в которых заместитель R4 содержит нитрогруппу, могут быть восстановлены с образованием вещества, в котором заместитель R4 содержит первичный амин.
При использовании в рамках настоящего документа термины "алкил", "алкенил", "алкинил" и префикс "алк-" включают как прямоцепочечные, так и разветвленные группы, а также группы с циклической структурой, то есть циклоалкил и циклоалкенил. Если не указывается иное, эти группы содержат от 1 до 20 атомов углерода, причем алкенильные и алкинильные группы содержат от 2 до 20 атомов углерода. Предпочтительны группы длиной до 10 атомов углерода. Циклические группы могут быть моноциклическими или полициклическими и предпочтительно, чтобы количество атомов углерода в кольце составляло от 3 до 10. В качестве примеров циклических групп можно назвать циклопропил, циклопентил, циклогексил и адамантил.
Термин "галоалкил" включает группы, которые содержат в качестве заместителей один или несколько атомов галогенов, в том числе группы, в которых все доступные атомы водорода замещены атомами галогенов. Это справедливо также для групп, в названии которых содержится префикс "галоалк-". В качестве примеров галоалкильных групп можно назвать хлорметил, трифторметил и т.п.
Термин "арил" при использовании в рамках настоящего документа включает карбоциклические ароматические кольца или системы колец. В качестве примеров арильных групп можно назвать фенил, нафтил, бифенил, флуоренил и инденил. Термин "гетероарил" включает ароматические кольца или системы колец, которые содержат, по меньшей мере, один гетероатом (например, О, S, N) в составе кольца. Подходящими гетероарильными группами являются фурил, тиенил, пиридил, хинолинил, тетразолил, имидазо, пиразоло, тиазоло, оксазоло и так далее.
Термин "гетероциклил" включает неароматические кольца или системы колец, которые содержат, по меньшей мере, один кольцевой гетероатом (например, О, S, N). В качестве примеров гетероциклических групп можно назвать пирролидинил, тетрагидрофуранил, морфолинил, тиоморфолинил, пиперидинил, пиперазинил, тиазолидинил, имидазолидинил и так далее.
Если не указывается иное, термины "замещенный циклоалкил", "замещенный арил", "замещенный гетероарил" и "замещенный гетероциклил" указывают на то, что рассматриваемые кольца или системы колец являются дополнительно замещенными одним или несколькими заместителями, выбираемыми из группы, включающей алкил, алкокси, алкилтио, гидрокси, галоген, галоалкил, галоалкилкарбонил, галоалкокси (например, трифторметокси), нитро, алкилкарбонил, алкенилкарбонил, арилкарбонил, гетероарилкарбонил, арил, арилалкил, гетероарил, гетероарилалкил, гетероциклил, гетероциклоалкил, нитрил, алкоксикарбонил, алканоилокси, алканоилтио, а в случае циклоалкила и гетероциклила - оксо.
На структурных формулах веществ, представленных в настоящем изобретении, некоторые связи изображаются пунктирными линиями. Эти линии обозначают, что связи, изображаемые пунктирной линией, могут присутствовать или отсутствовать. В связи с этим, соединения, соответствующие формуле I, могут быть либо имидазохинолиновыми веществами или тетрагидроимидазохинолиновыми веществами.
Изобретение распространяется на все вещества, упоминаемые в настоящем документе, в любой из фармацевтически приемлемых форм, в том числе такие изомеры, как диастереомеры и энантиомеры, соли, сольваты, полиморфные модификации и так далее.
Фармацевтические композиции и биологическая активность
Фармацевтические композиции, представленные в настоящем изобретении, содержат терапевтически эффективное количество вещества, соответствующего формуле I, в комбинации с фармацевтически приемлемым носителем.
При использовании в рамках настоящего документа термин "терапевтически эффективное количество" означает такое количество вещества, которое достаточно для оказания терапевтического действия, такого как индукция цитокинов, противоопухолевое действие и/или противовирусное действие. Хотя точное количество активного вещества, используемого в фармацевтической композиции, представленного в настоящем изобретении, будет варьировать в зависимости от факторов, которые хорошо известны опытным специалистам, в том числе от физической и химической природы вещества, а также от природы носителя и назначенного режима введения, однако считается, что композиции, представленные в настоящем изобретении, содержат достаточно активного ингредиента для создания дозы от примерно 100 нг/кг до примерно 50 мг/кг, предпочтительно примерно от 10 мг/кг до примерно 5 мг вещества/кг веса субъекта. Можно использовать любые стандартные лекарственные формы, такие как таблетки, пилюли, формулы для парентерального введения, сиропы, кремы, мази, аэрозольные формулы, трансдермальные пластыри, трансмукозальные пластыри и другие.
В экспериментах, проведенных в соответствии с тестами, представленными ниже, показано, что вещества, представленные в настоящем изобретении, вызывают продукцию некоторых цитокинов. Эти результаты показывают, что эти вещества можно использовать в качестве модуляторов иммунного ответа, которые способны усиливать иммунный ответ самыми разными способами, что делает их пригодными для лечения ряда заболеваний.
В число цитокинов, синтез которых может быть индуцирован при введении веществ, представляемых в рамках настоящего изобретения, обычно включают интерферон-α (IFN-α) и фактор-α некроза опухолей (TNF-α), а также некоторые интерлейкины (IL). Цитокины, биосинтез которых фактор некроза опухолей-α веществами, представленными в настоящем изобретении, включают IFN-α, TNF-α, IL-1, 6, 10 и 12 и ряд других цитокинов. Среди многих других эффектов цитокины подавляют размножение вирусов и рост опухолевых клеток, что делает эти соединения полезными для лечения вирусных болезней и опухолей.
В дополнение к способности стимулировать продукцию цитокинов вещества, представленные в настоящем изобретении, оказывают влияние на другие аспекты врожденного иммунного ответа. Например, они могут стимулировать активность естественных клеток-киллеров - эффект, который может быть связан с индукцией цитокинов. Эти соединения могут также активировать макрофаги, что, в свою очередь, повышает секрецию оксида азота и продукцию дополнительных цитокинов. Кроме того, соединения могут вызывать пролиферацию и дифференциацию В-лимфоцитов.
Соединения, представленные в настоящем изобретении, оказывают также воздействие на приобретенный иммунный ответ. Например, хотя считается, что не существует прямого воздействия на Т-клетки или прямой индукции Т-клеточных цитокинов, однако при введении этих веществ продукция цитокина IFN-γ T-хелперами 1 типа (The1) индуцируется опосредованною, а продукция цитокинов IL-4, IL-5 и IL-13 Т-хелперами 2 типа (The2) подавляется. Такая активность означает, что данные соединения можно применять для лечения болезней, при которых требуется положительная регуляция Th1-ответа и/или отрицательная регуляция Th2-ответа. С точки зрения способности соединений, соответствующих формуле Ia, подавлять Th2-иммунный ответ, можно полагать, что эти соединения применимы при лечении аллергических болезней, например атопического дерматита, астмы, аллергии и аллергического ринита и системной красной волчанки; возможно их применение в качестве адъюванта к вакцинам для клеточного иммунитета и, по-видимому, в качестве средства для лечения рекуррентных грибковых заболеваний и хламидиозов.
Модулирующее действие на иммунный ответ, оказываемое этими веществами, делает их полезными для лечения широкого спектра заболеваний. Вследствие их способности индуцировать продукцию таких цитокинов, как IFN-α и/или TNF-α, соединения особенно полезными для лечения вирусных болезней и опухолей. Такая иммуномодулирующая активность позволяет предположить, что вещества, представленные в настоящем изобретении, можно применять для лечения болезней, включая, но не ограничиваясь, такие как вирусные болезни, в том числе генитальные бородавки; обычные бородавки; роговые бородавки; гепатит В; гепатит С; простой вирусный герпес типа I и типа II; контагиозный моллюск; ВИЧ; цитомегаловирус; вирус ветряной оспы; внутриэпителиальные опухоли, такие как внутриэпителиальная опухоль шейки матки; поражение вирусом папилломы человека (HPV) и ассоциированные опухоли; грибковые заболевания, например кандидоз, аспергиллез и криптококковый менингит; онкологические болезни, например, базально-клеточная карцинома, лейкоз ворсистых клеток, саркома Капоши, карцинома почечных клеток, плоскоклеточная карцинома, миелогенная лейкемия, множественная миелома, меланома, неходжкинская лимфома, кожная Т-клеточная лимфома и другие формы рака; паразитарные болезни, такие как пневмоцистоз, криптоспоридиоз, гистоплазмоз, токсоплазмоз, трипаносомная инфекция, лейшманиоз и бактериальные инфекции, например туберкулез и микобактериоз птиц. В число других заболеваний или состояний, которые можно лечить с использованием веществ, представленных в настоящем изобретении, входят экзема; эозинофилия; эссенциальная тромбоцитемия; проказа; множественный склероз; синдром Оммена; дискоидная волчанка; болезнь Боуэна; боуэноидный папулез, а также для улучшения состояния и стимуляции заживления ран, включая хронические раны.
В связи с этим данное изобретение представляет способ индукции биосинтеза цитокинов в организме животного, включающий введение эффективного количества вещества, соответствующего формуле I, в организм животного. Количество вещества, эффективное для индукции биосинтеза цитокинов, - это количество, достаточное для того, чтобы простимулировать один или несколько типов клеток, таких как моноциты, макрофаги, дендровидные клетки и В-клетки, продуцировать такое количество одного или нескольких цитокинов, таких, например, как IFN-α, TNF-α, IL-1, 6, 10 и 12, которое превышает исходный уровень для таких цитокинов. Точное количество будет меняться в зависимости от факторов, известных опытным специалистам, однако ожидаемая доза составляет примерно от 100 нг/кг до примерно 50 мг/кг, предпочтительно примерно от 10 мкг/кг до примерно 5 мг/кг. Кроме того, изобретение представляет способ лечения вирусных инфекций у животных и способ лечения онкологических болезней у животных, включающий введение эффективного количества вещества, соответствующего формуле I, в организм животного. Количество, эффективное для лечения или подавления вирусной инфекции, - это количество, которое вызывает снижение одного или нескольких проявлений вирусной инфекции, таких как вирусные поражения, вирусная нагрузка, скорость продукции вируса, и смертность по сравнению с контрольными животными, не получавшими этого вещества. Точное количество будет меняться в зависимости от факторов, известных опытным специалистам, однако ожидаемая доза составляет примерно от 100 нг/кг до примерно 50 мг/кг, предпочтительно от примерно 10 мкг/кг до примерно 5 мг/кг. Количество вещества, эффективное для лечения новообразований, - это количество, которое вызывает уменьшение размера опухоли или количества опухолевых очагов. И в этом случае точное количество будет меняться в зависимости от факторов, известных опытным специалистам, однако ожидаемая доза составляет примерно от 100 нг/кг до примерно 50 мг/кг, предпочтительно от примерно 10 мкг/кг до примерно 5 мг/кг.
Далее изобретение будет описано следующими примерами, которые представлены исключительно для иллюстрации и не подразумевают каких бы то ни было ограничений области применения изобретения.
Пример 1
N-[4-(4-Амино-2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид
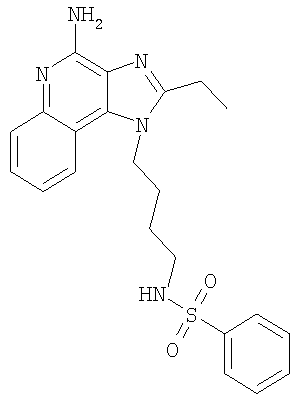
Триэтиламин (1,18 мл, 8,5 ммоль) добавляют к смеси 1-(4-аминобутил)-2-этил-1H-имидазо-[4,5-с]-хинолин-4-амина (2,00 г, 7,1 ммоль) и хлороформа (200 мл). Полученный раствор охлаждают на ацетон/ледяной бане в течение 10 минут. Бензолсульфонил-хлорид (0,90 мл, 8,5 ммоль) медленно добавляют в течение 5 минут. Через 45 минут добавляют 0,2 эквивалента триэтиламина. Через 6 часов реакционную смесь промывают соляным раствором (2×250 мл) и водой (1×100 мл), сушат над сульфатом магния, а затем концентрируют при пониженном давлении. Остаток перекристаллизовывают из N,N-диметилформамида. Перекристаллизованный материал и фильтрат перемешивают с метанолом. Оставшиеся твердые осадки отделяют способом фильтрации, объединяют, а затем сушат в сушильном аппарате Абдельхолдена в течение ночи с получением 0,80 г N-[4-(4-амино-2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида в виде белого твердого вещества, т.пл. 180,6-182,0°С. Анализ: Расчет для C22H25N5O2S·0,25H2O: %С, 61,73; %Н, 6,00; %N, 16,36.
Фактически: %С, 61,79; %Н, 6,04; %N, 16,43.
Пример 2
N-[4-(4-Амино-2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид
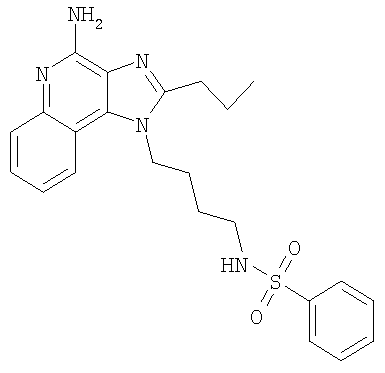
Часть А
трет-Бутил-4-(2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутилкарбамат (5,00 г, 13,1 ммоль) смешивали с соляной кислотой (50 мл 4,0 М в диоксане) и встряхивали в течение 1,5 часов. Реакционную смесь разбавляли дихлорметаном (~200 мл). Насыщенный раствор бикарбоната натрия добавляли до достижения рН 8. В водной фазе происходило образование осадка. Слои разделяли. Осадок в водном слое отделяли способом фильтрации, перемешивали с водой, а затем отделяли способом фильтрации с получением 3,6 г 4-(2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутан-1-амина.
Часть В
Материал из части А объединяли с хлороформом (600 мл) и подогревали до 40°С. Добавляли триэтиламин (3,48 мл, 25 ммоль) и получали раствор.
Добавляли бензолсульфонил-хлорид (1,60 мл, 12,5 ммоль). Эту реакционную смесь встряхивали при 40°С в течение ночи.
Реакционную смесь охлаждали до комнатной температуры, а затем концентрировали при пониженном давлении. Остаток размешивали в дихлорметане (~100 мл), промывали водой (3×125 мл), сушили над сульфатом магния, а затем концентрировали при пониженном давлении с получением 3,96 г N-[4-(2-пропил-1Н-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида в виде твердого кристаллического вещества желтого цвета, т.пл. 155,9-157,1°С.
Часть С
3-Хлорпероксибензоевую кислоту (896 мг 77% раствора) добавляли в течение 5 минут к раствору N-[4-(2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида (1,0 г, 2,4 ммоль) в хлороформе (100 мл). Через 2,5 часа добавляли дополнительно 0,1 эквивалента 3-хлорпероксибензоевой кислоты. Через 3 часа реакционную смесь выдерживали при низкой температуре в течение ночи. Реакционную смесь промывали насыщенным раствором бикарбоната натрия (3×150 мл), а затем концентрировали при пониженном давлении с получением 1,44 г сырого продукта. Этот материал перекристаллизовывали из метилацетата с получением 0,67 г 1-{4-[(фенилсульфонил)амино]бутил}-2-пропил-1Н-имидазо-[4,5-с]-хинолин-5N-оксида в виде твердого вещества коричневого цвета, т.пл. 203,8-205,2°С.
Часть D
Гидроксид аммония (3,5 мл 27% раствора) добавляли к смеси материала из части С и дихлорметана (15 мл). Через 10 минут медленно добавляли тозилхлорид (0,35 г) в течение 5 минут. Через 45 минут реакционную смесь оставляли при низкой температуре на выходные.
Добавляли дополнительно 35 мг тозилхлорида и реакционную смесь встряхивали в течение 1 часа. Отделяли органическую фазу, а затем промывали насыщенным раствором бикарбоната натрия (3×80 мл). В водной фазе происходило образование осадка. Этот материал выделяли способом фильтрации, а затем перекристаллизовывали из метилацетата. Полученное твердое вещество и фильтрат объединяли, растворяли в дихлорметане, содержащем небольшое количество метанола, а затем очищали способом колоночной хроматографии (силикагель, подвижная фаза - 10% метанол в дихлорметане). Полученный материал очищали способом колоночной хроматографии (силикагель, подвижная фаза - 0-7,5% метанола в дихлорметане). Этот материал перекристаллизовывали 3 раза из метилацетата с получением 42 мг N-[4-(4-амино-2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида в виде белого твердого вещества, т.пл. 158,8-160,8°С. Анализ: расчет для C23H27N5O2S-0,25 С3Н6О2: %С, 62,15; %Н, 6,22; %N, 15,59. Фактически: %С, 62,41; %Н, 5,91; %N, 15,41.
Пример 3
N-[4-(4-Амино-2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид
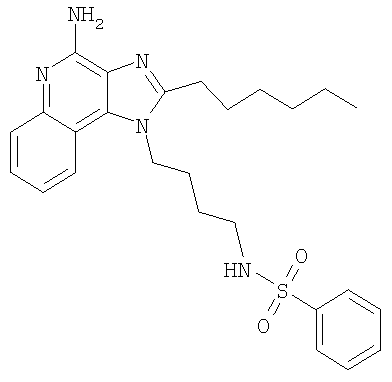
Часть А
С использованием общего способа, описанного в примере 2, часть А, трет-бутил-4-(2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутилкарбамат (33,85 г) подвергали гидролизу с получением 3,43 г 4-(2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)-бутан-1-амина в виде беловатого твердого вещества, т.пл. 172,2-174,2°С.
Часть В
С использованием общего способа, описанного в примере 2, часть В, за исключением того, что реакцию проводили при комнатной температуре, 4-(2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)-бутан-1-амин (1,20 г, 3,7 ммоль) реагировал с бензолсульфонил-хлоридом (429 мкл, 3,7 ммоль) с получением 0,75 г N-[4-(2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида в виде твердого вещества желтого цвета, т.пл. 137,0-138,1°С.
Часть С
С использованием общего способа, описанного в примере 2, часть С, N-[4-(2-гексил-1Н-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид (0,95 г, 2,0 ммоль) окисляли с получением 1,21 г сырого 1-{4-[(фенилсульфонил)амино]бутил}-2-гексил-1H-имидазо-[4,5-с]-хинолин-5N-оксида.
Часть D
С использованием общего способа, описанного в примере 2, часть D, материал из части С аминировали с получением 118 мг N-[4-(4-амино-2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида в виде беловатого кристаллического твердого вещества, т.пл. 84,8-85,4°С. Анализ: расчет для C26H33N5O2S·0,5H2O: %С, 63,91; %Н, 7,01; %N, 14,33. Фактически: %С, 63,63; %Н, 6,93; %N, 14,80.
Пример 4
N-[4-(4-Амино-2-пропил-1Н-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид
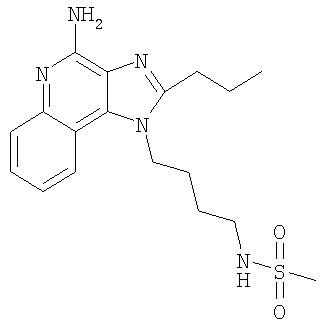
Часть А
С использованием общего способа, описанного в примере 2, часть В, за исключением того, что реакцию проводили при комнатной температуре, 4-(2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутан-1-амин (2,00 г, 7,1 ммоль) реагировал с метансульфонилхлоридом (1,65 мл, 21,3 ммоль) с получением 1,23 г N-[4-(2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде твердого вещества желтого цвета, т.пл. 133,2-134,6°С.
Часть В
С использованием общего способа, описанного в примере 2, часть С, N-[4-(2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид окисляли с получением 1,44 г сырого 1-{4-[(метилсульфонил)амино]бутил}-2-пропил-1H-имидазо-[4,5-с]-хинолин-5N-оксида в виде твердого вещества желтого цвета.
Часть С
С использованием общего способа, описанного в примере 2, часть D, материал из части В аминировали с получением 0,21 г N-[4-(4-амино-2-пропил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде беловатого кристаллического твердого вещества, т.пл. 186,5-187,9°С. Анализ: расчет для C18H25N5C2S·0,25Н2О: %С, 56,89; %Н, 6,76; %N, 18,43. Фактически: %С, 56,95; %Н, 6,89; %N, 18,13.
Пример 5
N-[4-(4-Амино-2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид

Часть А
С использованием общего способа, описанного в примере 2, часть А, трет-бутил-4-(2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутилкарбамат (20,69 г) подвергали гидролизу с получением 14,94 г 4-(2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутан-1-амина в виде беловатого твердого вещества, т.пл. 84,8-88,7°С.
Часть В
С использованием общего способа, описанного в примере 2, часть В, 4-(2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутан-1-амин (4,00 г, 14,9 ммоль) реагировал с метансульфонилхлоридом с получением 1,78 г N-[4-(2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде твердого вещества желтого цвета.
Часть С
С использованием общего способа, описанного в примере 2, часть С, материал из части В окисляли с получением ~ 2,00 г сырого 1-{4-[(метилсульфонил)амино]бутил}-2-этил-1H-имидазо-[4,5-с]-хинолин-5N-оксида.
Часть D
С использованием общего способа, описанного в примере 2, часть D, материал из части С обрабатывали с получением 0,42 г N-[4-(4-амино-2-этил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде белого твердого вещества, т.пл. 203,3-204,4°С. Анализ: расчет для C17H23N5O2S: %C, 56,49; %Н, 6,41; %N, 19,37. Фактически: %C, 56,21; %Н 6,36; %N, 19,09.
Пример 6
N-[4-(4-Амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид
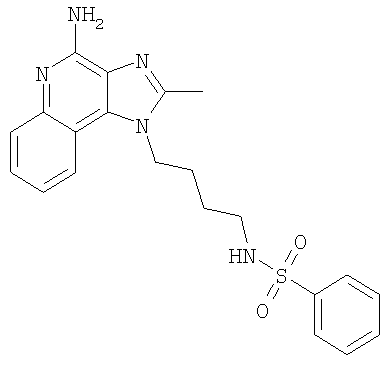
С использованием общего способа, описанного в примере 1, 1-(4-аминобутил)-2-метил-1H-имидазо-[4,5-с]-хинолин-4-амин (0,50 г, 1,9 ммоль) реагировал с бензолсульфонил-хлоридом (0,24 мл, 1,9 ммоль) с получением 0,38 г N-[4-(4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида в виде коричневых гранул, т.пл. 215,4-216,0°С. Анализ: расчет для C21H23N5O2S: %С, 61,59; %Н, 5,66; %N, 17,10. Фактически: %С, 61,24; %Н, 5,65; %N, 16,95.
Пример 7
N-[4-(4-Амино-2-метил-1Н-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид
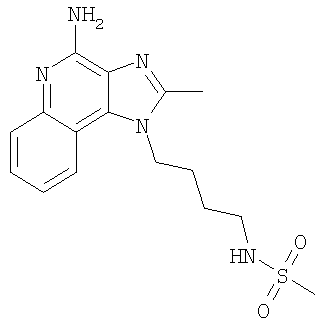
С использованием общего способа, описанного в примере 1,1-(4-аминобутил)-2-метил-1H-имидазо-[4,5-с]-хинолин-4-амин (1,00 г, 3,7 ммоль) реагировал с метансульфонилхлоридом (0,46 мл, 5,9 ммоль) с получением 0,16 г N-[4-(4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде беловатого твердого вещества, т.пл. 229,4-230,5°С. Анализ: расчет для C16H21N5O2S·0,25H2O: %C, 54,60; %Н, 6,16; %N, 19,90. Фактически: %C, 54,80; %Н, 6,24; %N, 19,58.
Пример 8
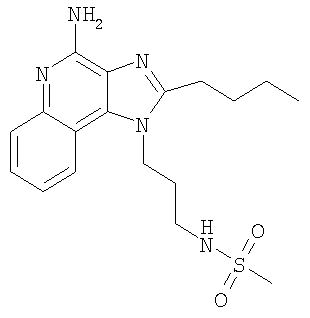
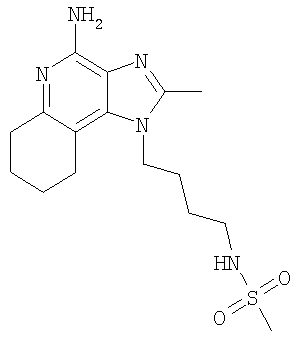
N-(4-Амино-2-бутил-1H-имидазо[4,5-с]-хинолин-1-ил)пропил]метансульфонамид
Часть А
трет-Бутил-3-(2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропилкарбамат (~80 г) растворяли в 1,4-диоксане (400 мл) при осторожном нагревании. Соляную кислоту (55 мл 4,0 М раствора в 1,4-диоксане) добавляли в виде одной порции и реакционную смесь нагревали до кипения.
Реакцию контролировали с помощью ВПЖХ. Добавляли дополнительную порцию кислоты (150-200 мл), и реакционную смесь кипятили с обратным холодильником до завершения реакции. Реакционную смесь охлаждали до комнатной температуры. Твердое вещество выделяли способом фильтрации с получением ~72 г 3-(2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропиламина гидрохлорида. Этот материал объединяли с материалом из предыдущего эксперимента, а затем растворяли в воде (400 мл).
Раствор нейтрализовали добавлением твердого карбоната калия. При рН 7 выпадал твердый осадок. Выпавший осадок отделяли способом фильтрации, а затем растворяли в воде (1500 мл). Уровень рН доводили до рН 10 добавлением твердого карбоната калия. Раствор экстрагировали хлороформом до тех пор, пока анализ способом ВПЖХ не показал, что в водном слое не осталось аминов. Органические слои объединяли, а затем концентрировали при пониженном давлении с получением 45 г 3-(2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропиламина.
Часть В
Триэтиламин (1,1 г, 10,6 ммоль) добавляли при перемешивании к раствору 3-(2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропиламина (2,00 г, 7,08 ммоль) в дихлорметане (~150 мл). Добавляли метансульфонил-хлорид (892 мг, 7,79 ммоль) и реакционную смесь встряхивали в атмосфере азота в течение ночи. Реакционную смесь промывали 1% водным раствором бикарбоната натрия (3×50 мл). Водные смывы экстрагировали дихлорметаном (2×20 мл). Органические фракции объединяли, сушили над сульфатом магния, а затем концентрировали при пониженном давлении с получением 1,89 г N-[3-(2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]метансульфонамида в виде светло-коричневого твердого вещества.
Часть С
С использованием общего способа, описанного в примере 2, часть С, материал из части В окисляли с получением 1,24 г N-[3-(2-бутил-5-оксидо-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]метансульфонамида.
Часть D
С использованием общего способа, описанного в примере 2, часть D, материал из части С обрабатывали с получением 690 мг N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]метансульфонамида в виде светло-бежевого твердого вещества, т.пл. 239,2-240,8°С. Анализ: расчет для
C18H25O2S: %C, 57,58; %Н, 6,71; %N, 18,65. Фактически: %C, 57,37; %Н, 6,78; %N, 18,42.
Пример 9
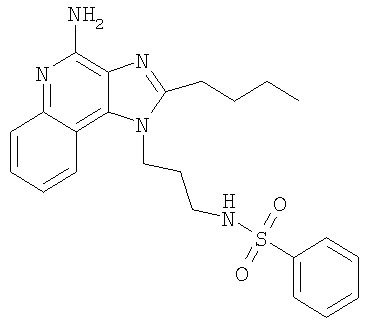
N-[3-(4-Амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]бензолсульфонамид
Часть А
С использованием общего способа, описанного в примере 8, часть В, 3-(2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропиламин (2,00 г, 7,08 ммоль) реагировал с бензолсульфонил-хлоридом (1,38 г, 7,79 ммоль) с получением 2,83 г N-[3-(2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]бензолсульфонамида в виде светло-красной пены.
Часть В
С использованием общего способа, описанного в примере 2, часть С, материал из части А окисляли с получением 3,28 г N-[3-(2-бутил-5-оксидо-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]бензолсульфонамида.
Часть С
С использованием общего способа, описанного в примере 2, часть D, материал из части В обрабатывали с получением 1,08 г N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]бензолсульфонамида в виде светло-бежевого твердого вещества, т.пл. 210,5-212,0°С. Анализ: расчет для
C23H27N5O2S: %С, 63,13; %Н, 6,22; %N, 16,01. Фактически: %С, 62,89; %Н, 6,16; %N, 15,74.
Пример 10
N-[4-(4-Амино-2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)-бутил]метансульфонамид
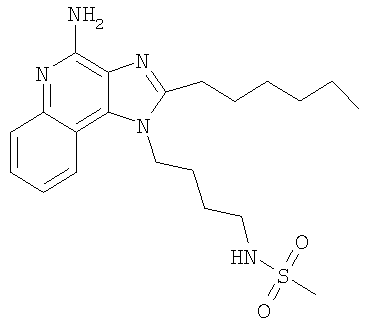
Часть А
С использованием общего способа, описанного в примере 1, 4-(2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутан-1-амин (1,00 г, 3,1 ммоль) реагировал с метансульфонил-хлоридом (0,48 мл, 6,2 ммоль) с получением 1,15 г N-[4-(2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде белого твердого вещества.
Часть В
С использованием общего способа, описанного в примере 2, часть С, N-[4-(2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид (1,47 г, 3,7 ммоль) окисляли с получением 3,78 г сырого 1-{4-[(метилсульфонил)амино]бутил}-2-гексил-1H-имидазо-[4,5-с]-хинолин-5N-оксида в виде желтого осадка.
Часть С
С использованием общего способа, описанного в примере 2, часть D, материал из части В обрабатывали с получением 0,28 г N-[4-(4-амино-2-гексил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде беловатого твердого вещества, т.пл. 170,2-171,1°С. Анализ: расчет для
C21H31N5O2S: %С, 60,40; %Н, 7,48; %N, 16,77. Фактически: %С, 59,97; %Н, 7,26; %N, 16,33.
Пример 11
N-{8-[4-Амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]октил}бензолсульфонамид
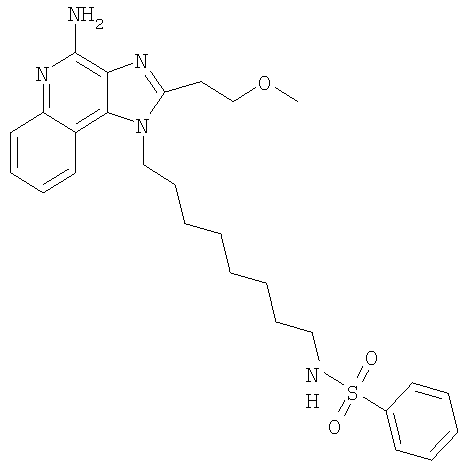
В атмосфере азота раствор 1-(8-аминооктил)-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амина (1,0 г, 2,7 ммоль) в дихлорметане (50 мл) охлаждали до 0°С. Добавляли триэтиламин (415 мкл, 2,98 ммоль), а затем бензолсульфонил-хлорид (345 мкл, 2,71 ммоль). Реакционной смеси давали медленно нагреться до комнатной температуры, а затем поддерживали ее в течение ночи. Реакционную смесь промывали водой, сушили над сульфатом магния, а затем концентрировали при пониженном давлении. Остаток очищали способом колоночной хроматографии (50 г силикагеля, подвижная фаза - 7,5% метанол в дихлорметане). Очищенный материал перекристаллизовывали из пропилацетата, растирали в порошок с гексанами, а затем сушили в вакуумном жаровом шкафу с получением 590 мг N-{8-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]октил}бензолсульфонамида в виде желтого порошка, т.пл. 146-149°С. Анализ: расчет для C27N35N5O3S: %С, 63,63; %Н, 6,92; %N, 13,74. Фактически: %С, 62,96; %Н, 7,03; %N, 13,09. По Карлу Фишеру содержание воды на уровне 0,16% или 0,045 моль.
1H ЯМР (300 МГц, ДМСО-d6) δ 8,01 (d, J=7,8 Гц, 1Н), 7,78 (m, 2H), 7,65-7,55 (m, 5H), 7,45 (m, 1H), 7,28 (m, 1H), 6,71 (s, 2H), 4,50 (m, 2H), 3,83 (m, 2H), 3,5 (broad s, 3H), 3,18 (m, 2H), 2,71 (m, 2H), 1,77 (m, 2H), 1,38-1,17 (m, 10H).
13C ЯМР (75 МГц, ДМСО-d6) δ 151,7, 151,3, 144,0, 141,0, 132,8, 132,6, 129,5, 127,0, 126,8, 125,9, 121,9, 120,4, 114,9, 70,5, 58,5, 45,3, 42,8, 30,0, 29,2, 28,8, 28,7, 27,5, 26,2, 26,1.
MC m/z 510 (M+H).
Пример 12
N-{8-[4-Амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]октил}метансульфонамид
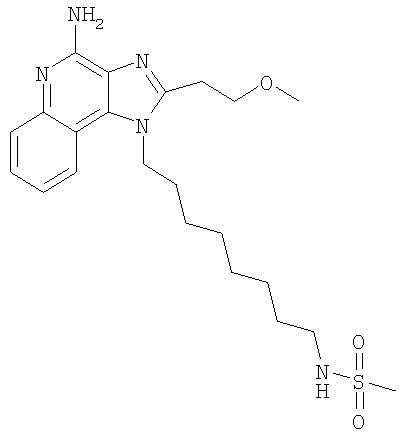
С использованием общего способа, описанного в примере 11, 1-(8-аминооктил)-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амин (800 мг, 2,17 ммоль) реагировал с метансульфонилхлоридом (172 мкл, 2,17 ммоль) с получением 720 мг N-{8-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]октил}метансульфонамида в виде желтого порошка, т.пл. 109-110°С. Анализ: расчет для C22H33N5O3S: %С, 59,04; %Н, 7,43; %N, 15,65. Фактически: %С, 58,78; %Н, 7,38; %N, 15,48.
1H ЯМР (300 МГц, ДМСО-d6) δ 8,01 (d, J-8,3 Гц, 1Н), 7,62 (d, J=8,3 Гц, 1H), 7,42 (m, 1H), 7,26 (m, 1H), 6,91 (m, 1H), 6,51 (s, 2H), 4,51 (t, J=7,3 Гц, 2Н), 3,83 (t, J=6,8 Гц, 2Н), 3,34 (s, 3H), 3,18 (t, J=6,8 Гц, 2Н), 2,89 (m, 2H), 2,86 (s, 3H), 1,80 (m, 2H), 1,27 (m, 10Н).
13С ЯМР (125 МГц, ДМСО-d6) δ 152,0, 151,0, 145,0, 132,6, 132,6, 126,7, 126,6, 121,56, 120,3, 115,1, 70,5, 58,5, 45,3, 42,8, 30,0, 29,7, 28,9, 28,8, 27,5, 26,4, 26,2. MC m/z 448 (M+1).
Пример 13
N-[8-(4-Амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)октил]метансульфонамид
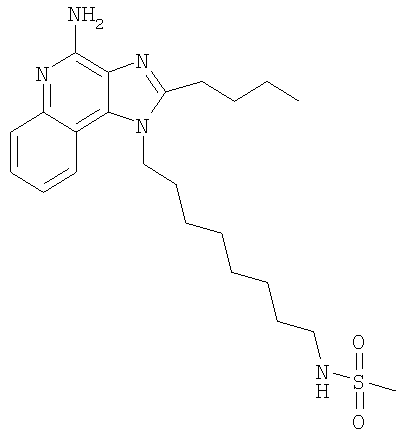
С использованием общего способа, описанного в примере 11, 1-(8-аминооктил)-2-бутил-1Н-имидазо-[4,5-с]-хинолин-4-амин (1,2 г, 3,26 ммоль) реагировал с метансульфонилхлоридом (260 мкл, 3,26 ммоль) с получением 0,70 г N-[8-(4-амино-2-бутил-1Н-имидазо-[4,5-с]-хинолин-1-ил)октил]метансульфонамида в виде желтовато-коричневого порошка, т.пл. 121-124°С. Анализ: расчет для C23H35N5O3S: %С, 61,99; %Н, 7,92; %N, 15,72. Фактически: %С, 62,01; %Н, 7,97; %N, 15,75.
1H ЯMP (300 МГц, ДМСО-d6) δ 8,01 (d, J=8,3 Гц, 1Н), 7,61 (dd, J=8,3, 1,0 Гц, 1H), 7,41 (dt, J=8,3 1,5 Гц, 1H), 7,25 (dt, J=8,3, 1,5 Гц, 1Н), 6,91 (t, J=4,9 Гц, 1H), 6,47 (s, 2H), 4,48 (t, J=7,3 Гц, 2Н), 2,90 (m, 4H), 2,86 (s, 3Н), 1,80 (m, 4H), 1,44 (m, 6H), 1,27 (m, 6H), 0,96 (t, J=7,3 Гц, 3Н).
13С ЯМР (500 МГц, ДМСО-d6) δ 153,3, 152,1, 145,1, 132,5, 126,8, 126,7, 126,6, 121,5, 120,2, 115,2, 45,1, 42,8, 39,6, 30,1, 30,0, 29,8, 28,9, 28,8, 26,5, 26,4, 26,2, 22,3, 14,1.
MC m/z 446 (M+1).
Пример 14
N-(4-Амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]-5-(диметиламино)нафталин-1-сульфонамид
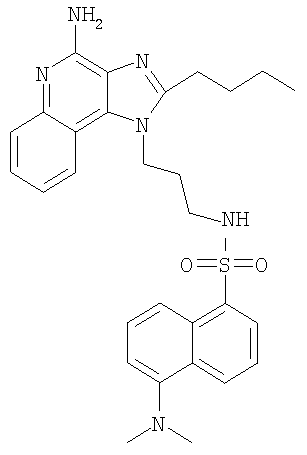
В атмосфере азота триэтиламин (765 мг, 7,56 ммоль) добавляли к раствору 1-(3-аминопропил)-2-бутил-1H-имидазо-[4,5-с]-хинолин-4-амина (1,5 г, 5,04 ммоль) в 1-метил-2-пирролидоне (75 мл). Добавляли раствор 5-диметиламино-1-нафталинсульфонил-хлорида (1,5 г, 5,55 ммоль) в 1-метил-2-пирролидоне. За ходом реакции следили по данным ВПЖХ, реакционную смесь смешивали с водой (500 мл) и доводили рН до 10 добавлением твердого карбоната калия. Полученный желтый осадок выделяли способом фильтрации, ополаскивали водой, а затем очищали способом колоночной хроматографии (силикагель, подвижная фаза - 1-5% метанол в хлороформе). Очищенный материал перекристаллизовывали из ацетонитрила с получением 1,76 г N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]-5-(диметиламино)нафталин-1-сульфонамида в виде твердого вещества, т.пл. 216,5-217,5°С. Анализ: расчет для C29H34N6O2S: %C, 65,64; %Н, 6,46; %N, 15,84. Фактически: %C, 65,52; %Н, 6,44; %N, 15,90.
Пример 15
N-[3-(4-Амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)-пропил]4-метилбензолсульфонамид
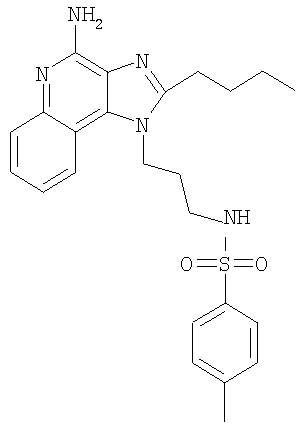
С использованием общего способа, описанного в примере 14, 1-(3-аминопропил)-2-бутил-1H-имидазо-[4,5-с]-хинолин-4-амин (1,5 г, 5,04 ммоль) реагировал с n-толуолсульфонил-хлоридом (1,08 г, 5,55 ммоль) с получением 1,57 г N-[3-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]-4-метилбензолсульфонамида в виде беловатого порошка, т.пл. 197,0-198,5°С. Анализ: расчет для C24H29N5O2S: %C, 63,83; %Н, 6,47; %N, 15,51. Фактически: %C, 63,68; %Н, 6,40; %N, 15,51.
Пример 16
N-{3-[4-Амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-3-ил]пропил}метансульфонамид
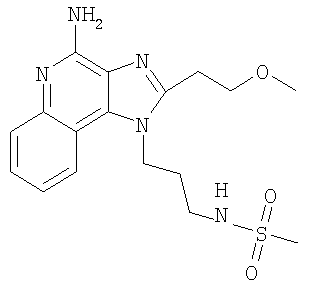
С использованием общего способа, описанного в примере 11,1-(3-аминопропил)-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амин (1,53 г, 5,11 ммоль) реагировал с метансульфонилхлоридом с получением 800 мг N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамида в виде светло-желтых игольчатых кристаллов, т.пл. 193-194°С. Анализ: расчет для C17H23N5O3S: %C, 54,09; %Н, 6,14; %N, 18,55. Фактически: %С, 54,09; %Н, 5,93; %N, 18,49.
Пример 17
N-[8-(4-Амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)октил]бензолсульфонамид
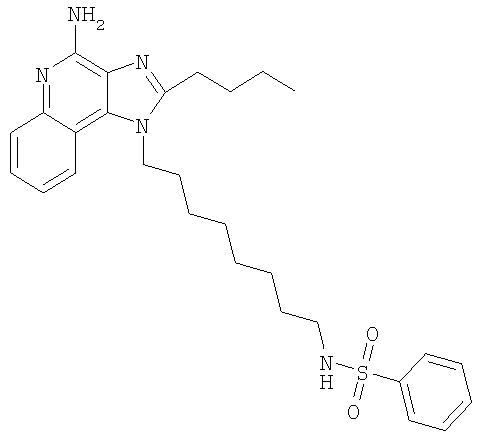
С использованием общего способа, описанного в примере 11, проводили реакцию 1-(8-аминооктил)-2-бутил-1H-имидазо-[4,5-с]-хинолин-4-амина (1,0 г, 2,72 ммоль) с бензолсульфонил-хлоридом (350 мкл, 2,72 ммоль) и получали 1,38 г N-[8-(4-амино-2-бутил-1H-имидазо-[4,5-c]-хинолин-1-ил)октил]бензолсульфонамида в виде беловатого порошка, т.пл. 143-144°С. Анализ: расчет для C28H37N5O2S: %C, 66,24; %Н, 7,35; %N, 13,79. Фактически: %C, 66,08; %Н, 7,25; %N, 13,72. Титрование по Карлу Фишеру показало содержание воды 0,23%.
1Н ЯMP (300 МГц, ДМСО-d6) δ 7,98 (d, J=7,8 Гц, 1H), 7,77 (m, 2H), 7,62-7,53 (m, 5H), 7,41 (m, 1H), 7,25 (m, 1H), 6,47 (s, 2H), 4,47 (m, 2H), 2,90 (m, 2H), 2,70 (q, J=6,3 Гц, 2H), 1,78 (m, 4H), 1,49-1,17 (m, 12H), 0,95 (t, J=7,3, 3H).
13C ЯМР (125 МГц, ДМСО-d6) δ 153,3, 152,0, 145,0, 141,0, 132,5, 129,5, 126,82, 126,76, 126,7, 126,6, 121,5, 120,3, 120,2, 115,1, 45,1, 42,8, 30,0, 29,2, 28,8, 28,7, 26,5, 26,2, 26,1, 22,3, 14,2, 14,1.
MC m/z 507 (M+1).
Пример 18
N-{3-[4-Амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил} бензолсульфонамид
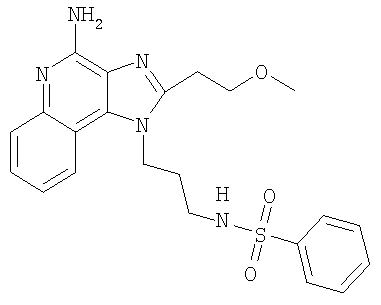
С использованием общего способа, описанного в примере 11, 1-(3-аминопропил)-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амин (1,53 г, 5,11 ммоль) реагировал с бензолсульфонил-хлоридом (993 мг, 5,62 ммоль) с получением 1,37 г N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}бензолсульфонамида в виде белого порошка, т.пл. 149-151°С. Анализ: расчет для C22H25N5O3S: %С, 60,12; %Н, 5,73; %N, 15,93. Фактически: %С, 60,40; %Н, 5,82; %N, 15,85.
Пример 19
N-[4-(4-Амино-2-пентил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид
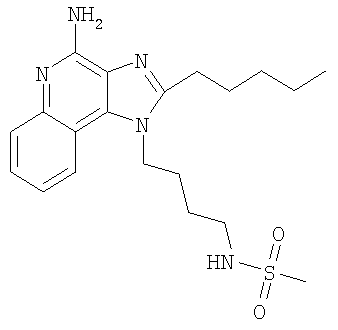
С использованием общего способа, описанного в примере 14, 1-(4-аминобутил)-2-пентил-1Н-имидазо-[4,5-с]-хинолин-4-амин (1,50 г, 4,6 ммоль) реагировал с метансульфонилхлоридом (0,57 мл, 7,4 ммоль) с получением 636 мг N-[4-(4-амино-2-пентил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде беловатого твердого вещества, т.пл. 136,8-138,1°С. Анализ: расчет для C20H29N5O2S: %C, 59,53; %Н, 7,24; %N, 17,35. Фактически: %C, 59,50; %Н, 7,31; %N, 16,80.
Пример 20
N-[4-(4-Амино-2-пентил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамид
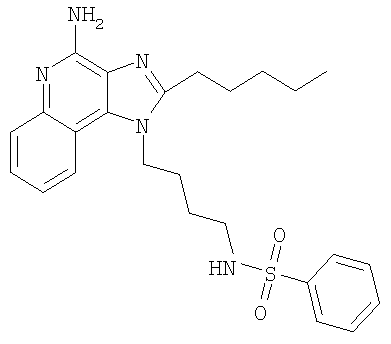
С использованием общего способа, описанного в примере 1, 1-(4-аминобутил)-2-пентил-1H-имидазо-[4,5-с]-хинолин-4-амин (1,00 г, 3,1 ммоль) реагировал с бензолсульфонил-хлоридом (0,51 мл, 4,0 ммоль) с получением 0,35 г N-[4-(4-амино-2-пентил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]бензолсульфонамида в виде желтого кристаллического твердого вещества. Анализ: расчет для C25H31N5O2S·0,5H2O: %С, 63,27; %Н, 6,80; %N, 14,76. Фактически: %С, 62,99; %Н, 6,61; %N, 14,42.
Пример 21
N-[8-(4-Амино-1H-имидазо-[4,5-с]-хинолин-1-ил)октил]метансульфонамид
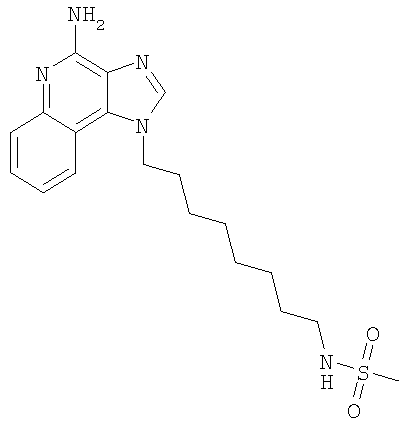
С использованием общего способа, описанного в примере 11, 1-(8-аминооктил)-1H-имидазо-[4,5-с]-хинолин-4-амин (3,85 ммоль) реагировал с метансульфонилхлоридом (310 мкл, 3,85 ммоль) с получением 0,43 г N-{8-[4-амино-1H-имидазо-[4,5-с]-хинолин-1-ил]октил}метансульфонамида в виде беловатого порошка, т.пл. 153-155°С. Анализ: расчет для C19H27N5O2S: %C, 58,59; %Н, 6,99; %N, 17,98; %S, 8,23. Фактически: %C, 58,40; %Н, 6,99; %N, 17,71; %S, 8,14.
1H ЯMP (300 МГц, ДМСО-d6) δ 8,20 (s, 1H), 8,03 (d, J=7,8 Гц, 1Н), 7,63 (d, J=8,3 Гц, 1H), 7,45 (m, 1H), 7,27 (m, 1H), 6,91 (m, 1H), 6,63 (d, 2H), 4,59 (m, 2H), 2,89 (m, 2H), 2,86 (s, 3H), 1,86 (m, 2H), 1,41-1,25 (m, 10H).
13С ЯМР (125 МГц, ДМСО-d6) δ 152,5, 145,2, 143,2, 132,0, 128,5, 127,1, 126,5, 121,6, 120,8, 115,2, 46,9, 42,8, 39,6, 30,0, 29,7, 28,81, 28,78, 26,4, 26,1.
MC m/z 390 (M+1).
Пример 22
N-{3-[4-Амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}-4-метилбензолсульфонамид

С использованием общего способа, описанного в примере 11, проводили реакцию между 1-(3-аминопропил)-2-(2-метоксиэтил)-1Н-имидазо-[4,5-с]-хинолин-4-амином (1,53 г, 5,11 ммоль) и n-толуолсульфонил-хлоридом (1,07 г, 5,62 ммоль) с получением 750 мг N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}-4-метилбензолсульфонамида в виде твердого вещества, т.пл. 189-191°С. Анализ: расчет для C23H27N5O3S·0,50H2O: %С, 59,72; %Н, 6,10; %N, 15,14. Фактически: %С, 59,73; %Н, 5,95; %N, 15,08.
Пример 23
N-[4-(4-Амино-2-пентил-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1 -ил)бутил]метансульфонамид
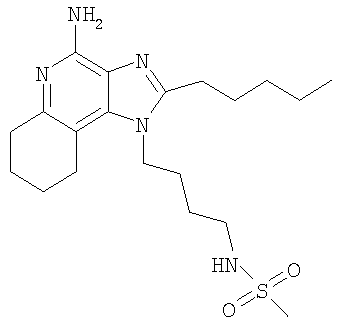
Раствор 1-(4-аминобутил)-2-пентил-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амина (1,50 г, 3,7 ммоль) в хлороформе (150 мл) охлаждали в ацетон/ледяной бане, затем медленно добавляли метансульфоновый ангидрид (0,79 г, 3,7 ммоль). Через 1,75 часа добавляли 0,018 г ангидрида. В 2,5 часа добавляли 0,079 г ангидрида. Через 3 часа реакционную смесь промывали 1% водным раствором карбоната натрия (3×150 мл). Органический слой сушили над сульфатом магния, а затем концентрировали при пониженном давлении с получением 2,2 г светло-желтого осадка. Остаток объединяли с 1% водным раствором карбоната натрия (200 мл) и доводили рН до 13 добавлением твердого карбоната натрия и 50% гидроксида натрия. Отделяли органическую фазу, промывали 1% водным раствором карбоната натрия (3×200 мл), сушили над сульфатом магния, а затем концентрировали при пониженном давлении с получением 2,18 г коричневого остатка. Этот материал смешивали с метилацетатом. Полученное твердое вещество выделяли с получением 1,25 г N-[4-(4-амино-2-пентил-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамида в виде белого твердого вещества, т.пл. 167,0-167,8°С. Анализ: расчет для C20H33N5O2S: %C, 58,94; %Н, 8,16; %N, 17,18. Фактически: %C, 58,79; %Н, 7,92; %N, 17,02.
Пример 24
N-{3-[4-Амино-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамид
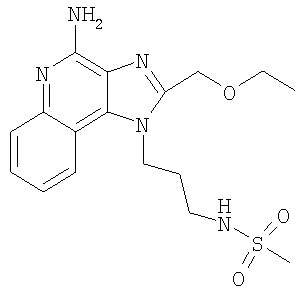
Смесь 1-(3-аминопропил)-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-4-амина (2,0 г, 6,7 ммоль), триэтиламина (1,5 мл, 15 ммоль) и ацетонитрила (75 мл) нагревали до получения раствора. Метансульфоновый ангидрид (1,28 г, 7,4 ммоль) добавляли в виде одной порции. Через 5 минут добавляли небольшое количество ангидрида. Реакционную смесь оставляли на встряхивателе на ночь. Реакционную смесь быстро разбавляли 1% водным раствором карбоната натрия. Водный слой экстрагировали хлороформом. Органические фракции сушили над сульфатом магния, фильтровали, а затем концентрировали при пониженном давлении. Остаток сушили в высоком вакууме в течение 3 часов и получали 2,73 г стекловидного твердого вещества. Этот материал перекристаллизовывали из метанола и получали 1,38 г N-{3-[4-амино-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамида, т.пл. 208,2-209,6°С.
Анализ: расчет для C17H23N5O3S: %C, 54,09; %Н, 6,14; %N, 18,55.
Фактически: %C, 53,97; %Н, 6,29; %N, 18,32.
Пример 25
N-(3-[4-Амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-2,2-диметилпропил}метансульфонамид

С использованием общего способа, описанного в примере 11, 1-(3-амино-2,2-диметилпропил)-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амин (0,22 г, 0,672 ммоль) реагировал с метансульфонилхлоридом (125 мкл) с получением 270 мг N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-2,2-диметилпропил}метансульфонамида в виде порошка кремового цвета, т.пл. 204,0-206,0°С. Анализ: расчет для C19H27N5O3S·0,50Н2О: %C, 55,05; %Н, 6,81; %N, 16,89; %S, 7,74. Фактически: %C, 55,10; %Н, 6,58; %N, 17,23; %S, 7,51. % Н2О: 2,17. Фактически: 2,28 (по Карлу Фишеру).
1H ЯMP (300 МГц, ДМСО-d6) δ 8,36 (d, J=8,3 Гц, 1Н), 7,59 (d, J=8,3 Гц, 1H), 7,38 (m, 2H), 7,20 (m, 1H), 6,49 (s, 2H), 4,81 (br s, 1H), 4,39 (br s, 1H), 3,82 (m, 2H), 3,27 (s, 3H), 3,19 (br s, 2H), 3,02 (d, J=6,8 Гц, 2Н), 2,94 (s, 3H), 0,82 (br s, 6H).
13С ЯМР (125 МГц, ДМСО-d6) δ 152,5, 152,0, 145,3, 133,9, 126,8, 126,7, 126,6, 121,5, 120,7, 115,8, 71,0, 58,5, 51,8, 51,5, 39,7, 39,0, 28,3, 24,4, 23,1. MC m/z 406 (М+Н).
Пример 26
N-{3-[4-Амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}-5-(диметиламино)нафталин-1-сульфонамид
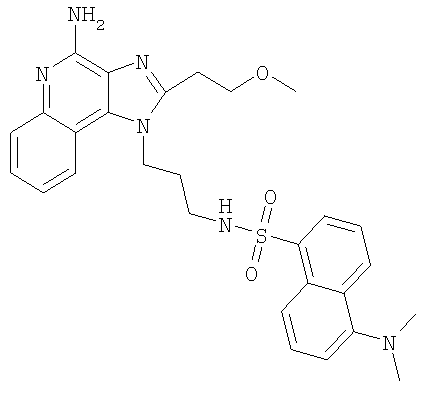
С использованием общего способа, описанного в примере 14, за исключением того, что в качестве растворителя использовали хлороформ, проводили реакцию между 1-(3-аминопропил)-2-(метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амином (1,53 г, 5,11 ммоль) и 5-диметиламино-1-нафталинсульфонил-хлоридом (5,87 ммоль) с получением 1,45 г N-{3-[4-амино-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}-5-(диметиламино)нафталин-1-сульфонамида в виде желтого твердого вещества, т.пл. 210-215°С. Анализ: расчет для C28H32N6O3S·1,50Н2О: %С, 60,09; %Н, 6,30; %N, 15,02. Фактически: %С, 59,89; %Н, 6,22; %N, 14,86.
Пример 27
N-[3-(4-Амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]метансульфонамид
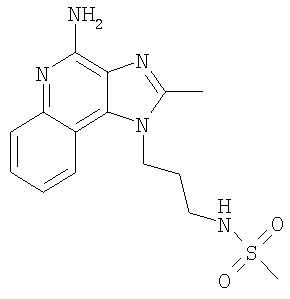
С использованием общего способа, описанного в примере 24, проводили реакцию между 1-(3-аминопропил)-2-метил-1Н-имидазо-[4,5-с]-хинолин-4-амином (2,0 г, 7,8 ммоль) и метансульфоновым ангидридом (1,49 г, 8,6 ммоль) с получением 1,2 г N-[3-(4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)пропил]метансульфонамида в виде твердого вещества, т.пл. 236,0-238,0°С. Анализ: расчет для C15H19N5O2S·0,25H2O: %С, 53,32; %Н, 5,82; %N, 20,72. Фактически: %С, 53,35; %Н, 5,72; %N, 20,57.
Пример 28
N-{3-[4-Амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамид
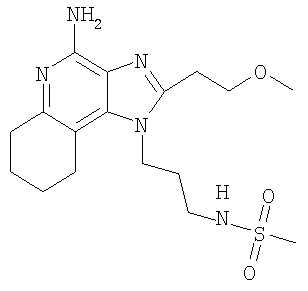
С использованием общего способа, описанного в примере 24, проводили реакцию между 1-(3-аминопропил)-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амином (2,0 г, 6,6 ммоль) и метансульфоновым ангидридом (1,26 г, 7,3 ммоль) с получением 630 мг N-{3-[4-амино-2-(2-метоксиэтил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамида в виде твердого вещества, т.пл. 150,0-152,0°С. Анализ: расчет для C17H27N5O3S: %С, 53,52; %Н, 7,13; %N, 18,36. Фактически: %С, 53,27; %Н, 7,12; %N, 18,37.
Пример 29
N-{3-[4-Амино-2-(этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамид
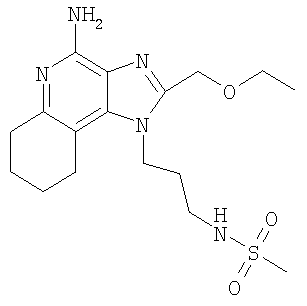
С использованием общего способа, описанного в примере 24, за исключением того, что хлороформ использовали вместо ацетонитрила, проводили реакцию между 1-(3-аминопропил)-2-(2-этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амином (2,6 г, 8,35 ммоль) и метансульфоновым ангидридом (3+ г) с получением 850 мг N-{3-[4-амино-2-(2-этоксиметил)-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамида в виде твердого вещества, т.пл. 212,0-214,0°С. Анализ: расчет для C17H27N5O3S: %С, 53,52; %Н, 7,13; %N, 18,36. Фактически: %С, 53,25; %Н, 7,16; %N, 18,09.
Пример 30
N-{3-[4-Амино-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил}метансульфонамид
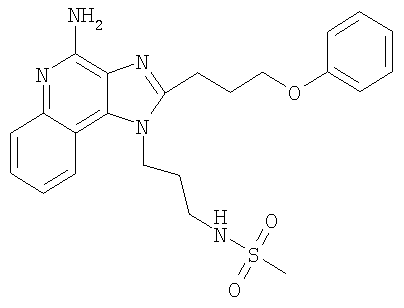
С использованием общего способа, описанного в примере 11, за исключением того, что хлороформ использовали вместо дихлорметана, проводили реакцию между 1-(3-аминопропил)-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-4-амином (2,00 г, 5,32 ммоль) и метансульфонилхлоридом (3+ г) с получением 1,38 г N-{3-[4-амино-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-1-ил]пропил)метансульфонамида в виде твердого вещества, т.пл. 176-178°С. Анализ: расчет для C23H27N5O3S: %С, 60,93; %Н, 6,00; %N, 15,44. Фактически: %С, 60,71; %Н, 5,98; %N, 15,45.
Пример 31
N-{4-[4-Амино-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-1-ил]бутил}метансульфонамид
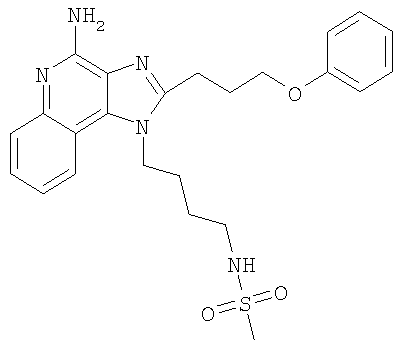
С использованием общего способа, описанного в примере 24, за исключением того, что вместо ацетонитрила использовали пиридин, проводили реакцию между 1-(3-аминобутил)-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-4-амином (2,00 г, 5,1 ммоль) и избытком метансульфонового ангидрида с получением 1,36 г N-{4-[4-амино-2-(3-феноксипропил)-1H-имидазо-[4,5-с]-хинолин-1-ил]бутил}метансульфонамида в виде твердого вещества, т.пл. 156,4-157,1°С. Анализ: расчет для C24H29N5O3S: %С, 60,48; %Н, 6,34; %N, 14,69. Фактически: %С, 60,75; %Н, 6,36; %N, 14,31.
Пример 32
N-[4-(4-Амино-2-метил-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид-гидрохлорид
С использованием общего способа, описанного в примере 23, проводили реакцию между 1-(4-аминобутил)-2-метил-6,7,8,9-тетрагидро-1H-имидазо-[4,5-с]-хинолин-4-амином (1,00 г, 3,7 ммоль) и метансульфоновым ангидридом (0,96 г, 5,5 ммоль) в присутствии триэтиламина (0,76 мл, 5,5 ммоль) с получением 0,55 г свободного основания требуемого продукта. Этот материал объединяли с метанолом (~20 мл), подогревали, давали остыть до комнатной температуры, а затем фильтровали, чтобы удалить небольшое количество нерастворимого материала. Объем фильтрата уменьшали до ~10 мл, а затем смешивали с 1 н. соляной кислотой (3 мл). Добавляли диэтиловый эфир (15 мл), а затем смесь концентрировали при пониженном давлении. Полученный остаток перемешивали с изопропиловым спиртом с получением белого твердого вещества, которое выделяли способом фильтрации, а затем сушили и получили 0,46 г N-[4-(4-амино-2-метил-6,7,8,9-тетрагидро-1Н-имидазо-[4,5-с]-хинолин-1-ил)бутил]метансульфонамид-гидрохлорида, т.пл. >250°С. Анализ: расчет для C16H25N5O2S·1,00 HCl·1,00 H2O: %С, 47,34; %Н, 6,95; %N, 17,25. Фактически: %С, 47,40; %Н, 6,49; %N, 17,22.
Пример 33
N-[2-(4-Амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)этил]-4-метилбензолсульфонамид
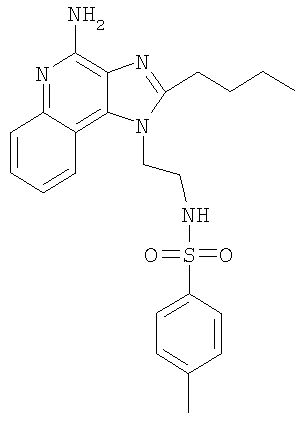
К охлажденному (0°С) раствору 1-(2-аминоэтил)-2-бутил-1H-имидазо-[4,5-с]-хинолин-4-амина (3,0 г, 10,6 ммоль) в 1-метил-2-пирролидоне (100 мл) добавляли триэтиламин (1,1 г, 15,9 ммоль). Раствор тозилхлорида (2,11 г, 11,1 ммоль) в 1-метил-2-пирролидоне (20 мл) добавляли медленно, по каплям. Реакционной смеси давали нагреться до комнатной температуры, и эту температуру поддерживали в течение ночи. Реакционную смесь вливали в воду (1500 мл) и доводили рН до 9. Белый осадок отделяли способом фильтрации, а затем перекристаллизовывали из ацетонитрила (60 мл) с получением 3,9 г N-[2-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)этил]-4-метилбензолсульфонамида, т.пл. 187,0-188,0°С. Анализ: расчет для C23H27N5O2S·0,3Н2О: %С, 62,29; %Н, 6,28; %N, 15,79. Фактически: %С, 62,52; %Н, 6,36; %N, 15,88.
Пример 34
N-[2-(4-Амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)этил]метансульфонамид
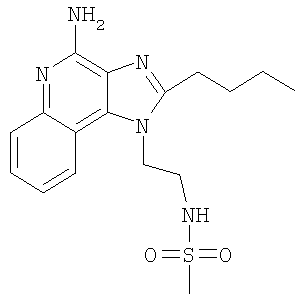
Метансульфонилхлорид (1,27 г, 11,1 ммоль) медленно добавляли к раствору 1-(2-аминоэтил)-2-бутил-1H-имидазо-[4,5-с]-хинолин-4-амина (3,0 г, 10,6 ммоль) в пиридине (60 мл). Реакционную смесь выдерживали при комнатной температуре в течение ночи, а затем концентрировали досуха. Остаток объединяли с теплым дихлорэтаном и водой, а затем фильтровали с образованием беловатого твердого вещества. Дихлорэтановый слой концентрировали с образованием беловатого твердого вещества. Оба твердых вещества объединяли, а затем перекристаллизовывали из N,N-диметилформамида с получением 1,1 г N-[2-(4-амино-2-бутил-1H-имидазо-[4,5-с]-хинолин-1-ил)этил]метансульфонамида в виде белого твердого вещества, т.пл. 210,0-211,0°С. Анализ: расчет для C17H23N5O2S: %C, 56,49; %Н, 6,41; %N, 19,37. Фактически: %C, 56,45; %Н, 6,49; %N, 19,50.
Пример 35
1-[4-(1,1-Диоксидоизотиазолидин-2-ил)бутил]-2-(2-метоксиэтил)-1Н-имидазо-[4,5-с]-хинолин-4-амин
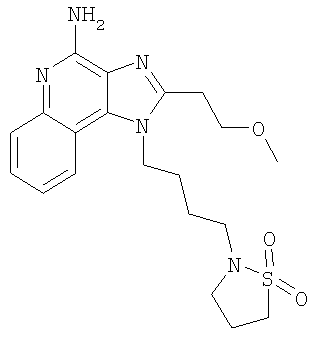
В атмосфере азота растворяли 1-(4-аминобутил)-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амин (500 мг, 1,6 ммоль) в дихлорметане (5 мл) и триэтиламине (0,33 мл, 2,4 ммоль). По каплям добавляли 3-хлорпропилсульфонил-хлорид (0,19 мл, 1,6 ммоль) и реакционную смесь встряхивали в течение 2 часов. Растворитель удаляли в вакууме. Остаток растворяли в N,N-диметилформамиде (5 мл) и добавляли 1,8-диазабицикло-[5,4,0]-ундец-7-ен (0,48 мл, 3,2 ммоль). Реакционную смесь встряхивали в течение 72 часов, а затем вливали в воду и экстрагировали дихлорметаном. Органический слой промывали водой, а затем соляным раствором; сушили (Na2SO4); декантировали и упаривали с получением сырого продукта в виде коричневого масла. Очистку проводили способом импульсной колоночной хроматографии (силикагель, градиент элюции метанол/дихлорметан от 100:0 до 94:6) с последующей перекристаллизацией из ацетонитрила с получением 289 мг 1-[4-(1,1-диоксидоизотиазолидин-2-ил)бутил]-2-(2-метоксиэтил)-1H-имидазо-[4,5-с]-хинолин-4-амина в виде желтого кристаллического твердого вещества, т.пл. 156,4-157,7°С.
1Н-ЯMP (500 МГц, ДМСО-d6) δ 8,04 (a, J=7,4 Гц, 1Н), 7,62 (dd, J=8,3,1,2 Гц, 1Н); 7,42 (ddd, J=8,2, 7,0, 1,2 Гц, 1Н), 7,26 (ddd, J=8,2, 7,0, 1,2 Гц, 1H), 6,48 (bs, 2H), 4,54 (t, J=7,6 Гц, 2Н), 3,84 (t, J=6,7 Гц, 2Н), 3,29 (s, 3H), 3,22-3,12 (m, 6H), 2,93 (t, J=6,6 Гц, 2H), 2,23-2,13 (m, 2H), 1,90-1,65 (m,4H).
13С-ЯМР (125 МГц, ДМСО-d6) δ 151,6, 150,6, 144,8, 132,2, 126,5, 126,3, 121,2, 120,0, 114,7, 70,2, 58,1, 46,5, 46,1, 44,5, 43,6, 27,1, 24,1, 18,3.
Анализ: расчет для C20H27N5O3S: %С, 57,53; %Н, 6,52; %N, 16,77; %S, 7,68. Фактически: %С, 57,52; %Н, 6,67; %N, 16,88; %S, 7,71.
Пример 36
2-Бутил-1-[4-(1,1-диоксидоизотиазолидин-2-ил)бутил]-1Н-имидазо-[4,5-с]-хинолин-4-амин

С использованием общего способа, описанного в примере 35, за исключением того, что вместо дихлорметана использовали 1-метил-2-пирролидон, проводили реакцию между 1-(4-аминобутил)-2-бутил-1H-имидазо-[4,5-с]-хинолин-4-амином (5,0 г, 16,0 ммоль) и 3-хлорпропансульфонилхлоридом (2,83 г, 16,0 ммоль) с получением 0,75 г 2-бутил-1-[4-(1,1-диоксидоизотиазолидин-2-ил)бутил]-1H-имидазо-[4,5-с]-хинолин-4-амина в виде белого твердого вещества, т.пл. 173,0-176,0°С.
1H-ЯMP (300 МГц, ДМСО-d6) δ 8,30 (d, J=8,1 Гц, 1Н), 7,62 (d, J=8,2 Гц, 1Н), 7,41 (t, J=7,6 Гц, 1Н), 7,26 (t, J=8,0 Гц, 1Н), 6,48 (bs, 2H), 4,51 (t, J=7,5 Гц, 2Н), 3,18-3,11 (m, 4H), 2,96-2,89 (m, 4H), 2,22-2,12 (m, 2H), 1,92-1,63 (m, 6Н), 1,45 (sextet, J=7,4 Гц, 2Н), 0,96 (t, J=7,3 Гц, 3Н).
13С-ЯМР (75 МГц, ДМСО-d6) δ 153,0, 151,7, 144,7, 132,2, 126,4, 126,2, 121,1, 120,0, 114,7, 46,5, 46,1, 44,3, 43,6, 29,7, 27,1, 26,1, 24,1, 22,0, 18,3, 13,8. MC (CI) m/e 416,2124 (416,2120 расчет для C21H30N5O2S, M+H). Анализ: расчет для C21H29N5O2S: %C, 60,70; %Н, 7,03; %N, 16,85; %S, 7,72. Фактически: %C, 60,67; %Н, 6,94; %N, 17,02; %S, 7,42.
Пример 37
N-{2-[4-Амино-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтил}метансульфонамид
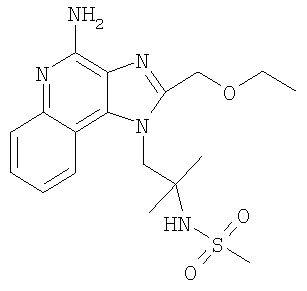
Часть А
Хорошо перемешанный раствор 4-хлор-3-нитрохинолина (17,3 г, 83,2 ммоль) в 200 мл безводного CH2Cl2, в атмосфере N2, обрабатывали триэтиламином (23,2 мл, 166,4 ммоль) и 1,2-диамино-2-метилпропаном (9,57 мл, 91,5 ммоль). После встряхивания в течение ночи реакционную смесь разбавляли, приливая 800 мл CHCl3, промывали водой (3×300 мл) и солевым раствором (300 мл). Органическую фракцию сушили над Na2SO4 и концентрировали с получением 2-метил-N1-(3-нитрохинолин-4-ил)пропан-1,2-диамина (21,0 г) в виде ярко-желтого твердого вещества.
Часть В
Раствор 2-метил-N1-(3-нитрохинолин-4-ил)пропан-1,2-диамин (2,60 г, 10,0 ммоль) в 50 мл THF, в атмосфере азота, охлаждали до 0°С и обрабатывали 10 мл 1 н. раствора NaOH. Затем к энергично перешиваемому раствору добавляли ди-трет-бутилдикарбонат (2,18 г, 10,0 ммоль).
После этого реакционную смесь оставляли нагреваться до комнатной температуры и встряхивали в течение ночи. Добавляли дополнительную порцию 400 мг ди-трет-бутилдикарбоната и встряхивание продолжали в течение 3 дней. Реакционную смесь после этого обрабатывали этилацетатом (200 мл) и промывали водой (2Х) и солевым раствором. Органическую фракцию сушили над Na2SO4 и концентрировали с получением желтого твердого вещества, которое обрабатывали 10% смесью этилацетат/гексаны. Твердое вещество выделяли способом фильтрации и сушили в вакууме в течение ночи с получением трет-бутил-1,1-диметил-2-[(3-нитрохинолин-4-ил)амино]этилкарбамата (2,80 г) в виде желтого порошка.
Часть С
Раствор трет-бутил-1,1-диметил-2-[(3-нитрохинолин-4-ил)амино]этилкарбамата (3,50 г, 9,72 ммоль) в 150 мл толуола обрабатывали 0,3 г 5% Pt на углероде и встряхивали в атмосфере Н2 (3 атм, 3 кг/см2) в течение 6 часов. Раствор затем фильтровали через слой целита и концентрировали с получением 3,04 г сырого трет-бутил-2-[(3-аминохинолин-4-ил]-1,1-диметилэтилкарбамата в виде светло-оранжевого пенистого вещества.
Часть D
Раствор трет-бутил-2-[(3-аминохинолин-4-ил]-1,1-диметилэтилкарбамата (3,04 г, 9,21 ммоль) в 50 мл CH2Cl2 охлаждали до 0°С и обрабатывали триэтиламином (1,41 мл, 10,13 ммоль) и этоксиацетилхлоридом (1,02 мл, 10,17 ммоль). Через 2 часа реакционную смесь концентрировали при пониженном давлении. Полученный сироп переносили в 100 мл этилового спирта и обрабатывали 4,5 мл триэтиламина. Раствор нагревали с обратным холодильником в течение ночи. Реакционную смесь концентрировали, переносили в 100 мл CH2Cl2 и промывали водой (2Х) и солевым раствором. Органическую фракцию сушили над Na2SO4 и концентрировали. Получившийся сироп очищали способом колоночной хроматографии (SiO2, 80% этилацетат/гексаны) с получением трет-бутил-2-[2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтилкарбамата (1,57 г) в виде пены персикового цвета.
Часть Е
Раствор трет-бутил-2-[2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтилкарбамата (1,57 г, 3,94 ммоль) в 30 мл CH2Cl2 обрабатывали 3-хлорпероксибензоевой кислотой (77%, 1,01 г, 4,57 ммоль). После встряхивания в течение 2 часов реакционную смесь обрабатывали 30 мл дополнительного CH2Cl2 и промывали 1% раствором Na2CO3 (2×30 мл), Н2О и солевым раствором. Органическую фракцию затем сушили над Na2SO4 и концентрировали с получением трет-бутил-2-[2-(2-(этоксиметил)-5-оксидо-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтилкарбамата (1,58 г) в виде светло-коричневой пены.
Часть F
Раствор трет-бутил-2-[2-(2-(этоксиметил)-5-оксидо-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтилкарбамата (1,57 г, 3,79 ммоль) в 20 мл 1,2-дихлорэтана нагревали до 70°С и обрабатывали 2 мл концентрированного раствора NH4OH. К энергично перемешиваемому раствору добавляли твердый n-толуолсульфонил-хлорид (795 мг, 4,17 ммоль). Затем реакционную смесь герметично закрывали в сосуде высокого давления и нагревание продолжали в течение 2 часов. Реакционную смесь после этого охлаждали и обрабатывали 50 мл CHCl3. Затем реакционную смесь промывали водой, 1% раствором Na2CO3 (3X) и солевым раствором. Органическую фракцию сушили над Na2SO4 и концентрировали с получением продукта в виде светло-коричневой маслянистой жидкости. Полученную жидкость очищали способом колоночной хроматографии (SiO2, 2-5% МеОН/CHCl3) с получением трет-бутил-2-[4-амино-2-(этоксиметил)-1Н-имилазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтилкарбамата (1,26 г) в виде светло-желтого пенистого вещества.
Часть G
трет-Бутил-2-[4-амино-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтилкарбамат (1,26 г, 3,05 ммоль) растворяли в 10 мл этилового спирта и обрабатывали 10 мл 2 М HCl в этиловом спирте. После нагревания с обратным холодильником в течение 2 часов реакционную смесь охлаждали и концентрировали при пониженном давлении. Полученное желтое твердое вещество растворяли в 50 мл
Н2О и экстрагировали CHCl3 (20 мл). Органический слой отбрасывали и водную фракцию подщелачивали (рН~12) добавлением концентрированного раствора NH4OH. Затем проводили экстракцию CHCl3 (4×20 мл) и объединенные органические фракции сушили с помощью Na2SO4 и концентрировали с получением 1-(2-амино-2-метилпропил)-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-4-амина (808 мг) в виде светло-коричневого порошка, т.пл. 161,0-162,0°С. МС m/z 314 (М+Н).
1Н ЯMP (300 МГц, d6-ДМСО) δ 8,30 (d, J=7,7 Гц, 1Н), 7,59 (dd, J=1,2, 8,3 Гц, 1Н), 7,40 (ddd, J=1,0, 7,2, 8,1 Гц, 1Н), 7,21 (ddd, J=1,2, 7,0, 8,2 Гц, 1H), 6,57 (s, 2H), 4,94 (br s, 2H), 4,61 (br s, 2H), 3,52 (q, J=7,0 Гц, 2Н), 1,61 (s, 2H), 1,31 (t, J=7,0 Гц, 3Н), 1,07 (s, 6H).
13С ЯМР (75 МГц, d6-ДМСО) δ 152,4, 151,1, 145,7, 134,3, 126,8, 126,7, 121,7, 120,8, 115,7, 65,6, 65,2, 55,8, 52,5, 29,2, 15,4.
Анализ: расчет для C17H23N5O: %C, 65,15; %Н, 7,40; %N, 22,35.
Фактически: %C, 65,04; %Н, 7,52; %N, 22,07.
Часть Н
1-(2-Амино-2-метилпропил)-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-4-амин (111 мг, 0,355 ммоль) растворяли в 5 мл безводного CH2Cl2 и охлаждали до 0°С в атмосфере азота. К перемешиваемому раствору добавляли Et3N (99 мкл, 0,71 ммоль) и метансульфонил-хлорид (28 мкл, 0,36 ммоль) и реакционной смеси давали нагреться до комнатной температуры в течение ночи. Реакцию после этого останавливали добавлением насыщенного раствора NaHCO3 (5 мл). Органический слой отделяли и промывали водой (2×5 мл) и солевым раствором, сушили над Na2SO4 и концентрировали при пониженном давлении с получением пенистого вещества бежево-коричневого цвета. После очистки способом колоночной хроматографии (SiO2, 2,5%-5% МеОН/CHCl3) получали N-{2-[4-амино-2-(этоксиметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтил}метансульфонамид (75 мг) в виде белого пенистого вещества,
т.пл. 105,0-110,0°С. МС m/z 392 (М+Н)+.
1Н ЯMP (300 МГц, CDCl3) δ 8,06 (dd, J=1,0, 8,3 Гц, 1Н), 7,79 (dd, J=1,1, 8,4 Гц, 1Н), 7,51 (ddd, J=1,3, 7,0, 8,4 Гц, 1Н), 7,31 (ddd, J=1,3, 7,0, 8,3 Гц, 1H), 5,90 (br s, 1H), 5,51 (br s, 2H), 4,96 (s, 2H), 4,92 (br s, 2H), 3,74 (q, J=7,0 Гц, 2H), 3,02 (s, 3Н), 1,55 (br s, 6H), 1,29 (t, J=7,0 Гц, 3Н).
13С ЯМР (75 МГц, CDCl3) δ 152,0, 150,8, 145,5, 135,2, 127,8, 127,6, 127,2, 122,2, 120,6, 116,0, 67,2, 65,4, 58,4, 55,8, 45,3, 26,6, 15,3.
Анализ: расчет для C18K25N5O3S·0,75H2O: С, 53,38; %Н, 6,60; %N, 17,29.
Фактически: %С, 53,49; %Н, 6,23; %N, 16,93.
Пример 38
N-[4-(4-Амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]этансульфонамид
С использованием общего способа, описанного в примере 1, проводили реакцию между 1-(4-аминобутил)-2-метил-1H-имидазо-[4,5-с]-хинолин-4-амином (1,00 г, 3,7 ммоль) и этансульфонил-хлоридом (2,11 мл, 22,3 ммоль) с получением 85 мг N-[4-(4-амино-2-метил-1H-имидазо-[4,5-с]-хинолин-1-ил)бутил]этансульфонамида в виде беловатого твердого вещества, т.пл. 210,7-211,6°С.
Пример 39
N-{4-[4-Амино-2-(циклопропилметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]бутил}метансульфонамид
С использованием общего способа, описанного в примере 38, часть В, за исключением того, что хлороформ использовали вместо дихлорметана, проводили реакцию между 1-(4-аминобутил)-2-(циклопропилметил)-1H-имидазо-[4,5-с]-хинолин-4-амином (1,00 г, 3,2 ммоль) и метансульфоновым ангидридом (1,29 г, 7,4 ммоль) с получением 0,42 г N-{4-[4-амино-2-(циклопропилметил)-1H-имидазо-[4,5-с]-хинолин-1-ил]бутил}метансульфонамида в виде твердого вещества коричневого цвета, т.пл. 199,7-200,7°С.
ИНДУКЦИЯ ЦИТОКИНОВ В КЛЕТКАХ ЧЕЛОВЕКА
Систему клеток крови человека in vitro использовали для оценки индукции цитокинов веществами, представленными в настоящем изобретении. Оценка этой активности основывается на измерении количества интерферона и фактора (α) некроза опухолей (IFN и TNF соответственно), секретируемого в культуральную среду, как описано в работе Testerman et. al. "Cytokine Induction by the Immunomodulators Imiquimod and S-27609", Journal of Leukocyte Biology, 58, 365-372 (сентябрь, 1995).
Подготовка клеток крови для культивирования
Цельную кровь собирали из вены у здоровых доноров в пробирки "vacutainer" с ЭДТА. Одноядерные клетки периферической крови (ОКПК) выделяли из цельной крови центрифугированием в градиенте плотности с использованием Histopaque®-1077 (Sigma Chemicals, г.Сент-Луис, штат Миссури, США). Выделенные ОКПК суспендировали при 3-4×106 клеток/мл в среде RPMI 1640, содержащей 10% фетальной сыворотки быка, 2 мМ L-глутамина и 1% раствор пенициллина/стрептомицина (RPMI complete). Суспензию ОКПК добавляли в стерильные плашки для культивирования с 48 ячейками с плоским дном (Costar, г.Кембридж, штат Мериленд, или Becton Dickinson Labware, г.Линкольн-Парк, штат Нью-Джерси, США), содержащий равный объем полной среды RPMI, содержащей испытуемое вещество.
Подготовка вещества
Соединения солюбилизируют в диметилсульфоксиде (ДМСО). Концентрация ДМСО не должна превышать конечной концентрации 1% для добавления в ячейки для культивирования.
Инкубация
Раствор испытуемого вещества добавляли при 60 мкм в первую ячейку, содержащую полную среду RPMI, и проводили последовательные (трехкратные или десятикратные) разведения. Затем в ячейки добавляли равные объемы суспензии ОКПК, создавая требуемый диапазон концентраций испытуемого вещества. Конечная концентрация суспензии ОКПК составляет 1,5-2×106 клеток/мл. Плашки накрывают стерильными пластиковыми крышками, осторожно смешивают, а затем инкубируют в течение 18 до 24 часов при 37°С в атмосфере 5% окиси углерода.
Разделение
После инкубации плашки центрифугируют в течение 5-10 минут при 1000 об/мин (~200 × g) при 4°С. Супернатант клеточной культуры собирают с помощью пипетки со стерильным полипропиленовым наконечником и переносят в стерильные полипропиленовые пробирки. Образцы хранят при температуре от -30 до -70°С до проведения анализа. Образцы анализируют на интерферон (α) и фактор-α некроза опухолей иммуноферментным способом ELISA.
Интерферон-α и фактор-α некроза опухолей - анализ способом ELISA
Концентрацию интерферона (а) определяют способом ELISA с использованием набора Human Multi-Species от компании PBL Biomedical Laboratories, г.Нью-Брунсуик, штат Нью-Джерси, США.
Концентрацию фактора-α некроза опухолей (TNF) определяют с использованием наборов для ELISA производства компаний Genzyme, г.Кембридж, Мериленд, США; R&D Systems, г.Миннеаполис, Миннесота, или Pharmingen, г.Сан-Диего, Калифорния, США.
В приведенной ниже таблице представлен список минимальных концентраций каждого вещества, которые, по результатам испытаний, способны индуцировать интерферон и минимальных концентраций каждого вещества, которые, по результатам испытаний, способны индуцировать фактор некроза опухолей. Значок ** указывает на то, что не отмечается индукции ни при какой из исследуемых концентраций (0,12, 0,37, 1,11, 3,33, 10 и 30 мкмоль). Значок *** указывает на то, что не обнаружено индукции ни при какой из исследуемых концентраций (0,0001, 0,001, 0,01, 0,1,1 и 10 мкмоль).
Настоящее изобретение описывается со ссылками на некоторые примеры и варианты реализации. Предшествующее подробное описание и примеры представлены лишь для прояснения понимания и не подразумевают никаких необязательных ограничений. Как очевидно опытным специалистам, много изменений может быть сделано в отношении описанных примеров без нарушения рамок и сути настоящего изобретения. Таким образом, рамки изобретения не следует ограничивать точными подробностями описываемых здесь композиций и структур, а скорее языком патентной формулы, представленной ниже.
| название | год | авторы | номер документа |
|---|---|---|---|
| ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2248975C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ И ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ НА ИХ ОСНОВЕ | 2002 |
|
RU2304143C2 |
| АМИДОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2000 |
|
RU2295343C2 |
| АРИЛЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЕ СОСТАВЫ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ВИРУСНОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ, СПОСОБЫ ЛЕЧЕНИЯ ОПУХОЛЕВОГО ЗАБОЛЕВАНИЯ НА ИХ ОСНОВЕ | 2001 |
|
RU2308456C2 |
| ГЕТЕРОАРИЛЬНЫЕ СОЕДИНЕНИЯ, СОДЕРЖАЩИЕ ИХ КОМПОЗИЦИИ И СПОСОБЫ ЛЕЧЕНИЯ С ПРИМЕНЕНИЕМ ЭТИХ СОЕДИНЕНИЙ | 2007 |
|
RU2478635C2 |
| ТИОЭФИРЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2001 |
|
RU2315049C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ | 2018 |
|
RU2768629C2 |
| КАРБАМИДЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНОВЫЕ ЭФИРЫ | 2001 |
|
RU2302418C2 |
| МОЧЕВИНОЗАМЕЩЕННЫЕ ИМИДАЗОХИНОЛИНЫ, ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ И СПОСОБ ИНДУКЦИИ БИОСИНТЕЗА ЦИТОКИНОВ НА ИХ ОСНОВЕ | 2000 |
|
RU2265020C2 |
| ЗАМЕЩЕННЫЕ ИМИДАЗОПИРИДИНИЛ-АМИНОПИРИДИНОВЫЕ СОЕДИНЕНИЯ, ПОЛЕЗНЫЕ ПРИ ЛЕЧЕНИИ РАКА | 2010 |
|
RU2619463C2 |
Изобретение относится к новому производному сульфонамидзамещенных имидазохинолинов, а именно к N-{2-[4-амино-2-(этоксиметил)-1Н-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтил}-метансульфонамиду и его фармацевтически приемлемым солям, а также к фармацевтической композиции на основе этого соединения.
Изобретение также относится к способу индукции биосинтеза цитокинов в организме животного и способам лечения вирусного и онкологического заболевания у животного с использованием указанного соединения и фармацевтической композиции на его основе. 5 н.п. ф-лы., 1 табл.
1. N-{2-[4-Амино-2-(этоксиметил)-1Н-имидазо-[4,5-с]-хинолин-1-ил]-1,1-диметилэтил}метансульфонамид или фармацевтически приемлемая соль этого вещества.
2. Фармацевтическая композиция, обладающая способностью индуцировать биосинтез цитокинов, содержащая терапевтически эффективное количество вещества или его соль по п.1 и фармацевтически приемлемый носитель.
3. Способ индукции биосинтеза цитокинов в организме животного, включающий введение эффективного количества вещества или его соли по п.1 в организм животного.
4. Способ лечения вирусного заболевания у животного, включающий введение эффективного количества вещества или его соли по п.1 в организм животного.
5. Способ лечения онкологического заболевания у животного, включающий введение эффективного количества вещества или его соли по п.1 в организм животного.
| US 6331559 А, 18.12.2001 | |||
| Механические сани | 1927 |
|
SU9506A1 |
| WO 9830562 A1, 16.07.1998 | |||
| ПРОИЗВОДНЫЕ ТЕТРАГИДРОИМИДАЗО [2,1-А]ИЗОХИНОЛИНА И ФАРМАЦЕВТИЧЕСКИЙ ПРЕПАРАТ НА ИХ ОСНОВЕ | 1998 |
|
RU2204560C2 |
| ПРОИЗВОДНОЕ 1,3-ДИГИДРО-2H-ИМИДАЗО/4,5-B/ХИНОЛИН-2-ОНА В КАЧЕСТВЕ ИНГИБИТОРА ФОСФОДИЭСТЕРАЗЫ, СПОСОБ ЕГО ПОЛУЧЕНИЯ, ПРОМЕЖУТОЧНЫЕ ДЛЯ НЕГО, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ И СПОСОБ ЕЕ ПОЛУЧЕНИЯ | 1992 |
|
RU2127273C1 |

